User login
Americans support the right to affordable health care
Despite the rhetorical winds blowing out of Washington, 92% of Americans believe that they have the right to affordable health care, according to a recent survey by the Commonwealth Fund.
Political affiliation, it turns out, does not appear to determine support for such a right. Democrats aged 19-64 years voiced their support to the tune of 99% in favor of a right to affordable care, compared with 82% of Republicans and 92% of independents, the Commonwealth Fund said in a survey brief released March 1.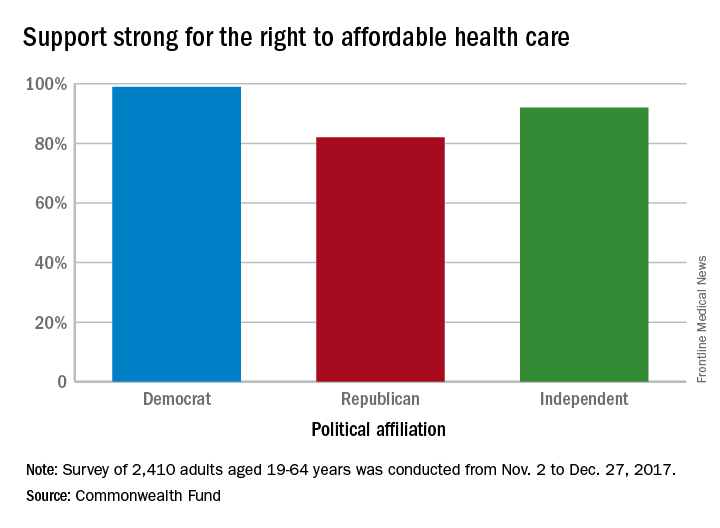
“This survey’s finding that strong majorities of U.S. adults, regardless of party affiliation, believe that all Americans should have a right to affordable health care suggests there may be popular support for a discussion over our preferred path,” the report’s authors wrote.
The survey showed that 36% of those who have health care insurance through the Affordable Care Act marketplaces are pessimistic about their chances of keeping that coverage, compared with 27% of those with Medicaid and 9% of adults with employer-sponsored health benefits.
Among those who lacked confidence about maintaining their coverage, the largest proportion (32%) of respondents believe that they will lose it because the “Trump administration will not carry out the law” and 19% think that they won’t be able to afford it in the future, they said.
“ Such a shift also would provide a more stable regulatory environment for insurers participating in both the marketplaces and Medicaid,” according to the report.
The Commonwealth Fund’s sixth Affordable Care Act Tracking Survey was conducted by the research firm SSRS between Nov. 2 and Dec. 27, 2017, with responses from 2,410 adults aged 19-64 years. The overall margin of error was ±2.7 percentage points at the 95% confidence level.
Despite the rhetorical winds blowing out of Washington, 92% of Americans believe that they have the right to affordable health care, according to a recent survey by the Commonwealth Fund.
Political affiliation, it turns out, does not appear to determine support for such a right. Democrats aged 19-64 years voiced their support to the tune of 99% in favor of a right to affordable care, compared with 82% of Republicans and 92% of independents, the Commonwealth Fund said in a survey brief released March 1.
“This survey’s finding that strong majorities of U.S. adults, regardless of party affiliation, believe that all Americans should have a right to affordable health care suggests there may be popular support for a discussion over our preferred path,” the report’s authors wrote.
The survey showed that 36% of those who have health care insurance through the Affordable Care Act marketplaces are pessimistic about their chances of keeping that coverage, compared with 27% of those with Medicaid and 9% of adults with employer-sponsored health benefits.
Among those who lacked confidence about maintaining their coverage, the largest proportion (32%) of respondents believe that they will lose it because the “Trump administration will not carry out the law” and 19% think that they won’t be able to afford it in the future, they said.
“ Such a shift also would provide a more stable regulatory environment for insurers participating in both the marketplaces and Medicaid,” according to the report.
The Commonwealth Fund’s sixth Affordable Care Act Tracking Survey was conducted by the research firm SSRS between Nov. 2 and Dec. 27, 2017, with responses from 2,410 adults aged 19-64 years. The overall margin of error was ±2.7 percentage points at the 95% confidence level.
Despite the rhetorical winds blowing out of Washington, 92% of Americans believe that they have the right to affordable health care, according to a recent survey by the Commonwealth Fund.
Political affiliation, it turns out, does not appear to determine support for such a right. Democrats aged 19-64 years voiced their support to the tune of 99% in favor of a right to affordable care, compared with 82% of Republicans and 92% of independents, the Commonwealth Fund said in a survey brief released March 1.
“This survey’s finding that strong majorities of U.S. adults, regardless of party affiliation, believe that all Americans should have a right to affordable health care suggests there may be popular support for a discussion over our preferred path,” the report’s authors wrote.
The survey showed that 36% of those who have health care insurance through the Affordable Care Act marketplaces are pessimistic about their chances of keeping that coverage, compared with 27% of those with Medicaid and 9% of adults with employer-sponsored health benefits.
Among those who lacked confidence about maintaining their coverage, the largest proportion (32%) of respondents believe that they will lose it because the “Trump administration will not carry out the law” and 19% think that they won’t be able to afford it in the future, they said.
“ Such a shift also would provide a more stable regulatory environment for insurers participating in both the marketplaces and Medicaid,” according to the report.
The Commonwealth Fund’s sixth Affordable Care Act Tracking Survey was conducted by the research firm SSRS between Nov. 2 and Dec. 27, 2017, with responses from 2,410 adults aged 19-64 years. The overall margin of error was ±2.7 percentage points at the 95% confidence level.
Aesthetic procedures becoming more popular in skin of color patients
in a presentation at the Caribbean Dermatology Symposium.
In 2015, ethnic minority patients accounted for 25% of aesthetic procedures in the United States, up from 20% in 2010, according to data from the American Society for Aesthetic Plastic Surgery, said Dr. Alexis, chair of dermatology and director of the Skin of Color Center at Mount Sinai St. Luke’s and Mount Sinai West hospitals in New York.
Chemical peels can be used successfully to treat a range of conditions in skin of color patients, including postinflammatory hyperpigmentation, acne, melasma, textural irregularities, and pseudofolliculitis barbae. They also can be used for skin brightening, said Dr. Alexis, who recommended a chemical peel protocol of salicylic acid, glycolic acid, or Jessner’s every 2-4 weeks. “Consider hydroquinone 4% concurrently to enhance efficacy for treating hyperpigmentation and to prevent postinflammatory hyperpigmentation,” he said. Patients on retinoids should discontinue them for 1 week prior to a chemical peel, he added.
Dr. Alexis shared several treatment pearls to promote successful peels in skin of color patients:
- Salicylic acid: Resist the urge to overapply and “titrate according to patient tolerability.” The endpoint of a salicylic acid peel is white precipitate, not frost; cool compresses can be used for patient comfort and for later removal of the white precipitate.
- Glycolic acid: Stick to a contact time of 2-4 minutes to avoid epidermolysis. “Completely neutralize all areas of application to avoid overpeeling.”
- Trichloroacetic acid (TCA): TCA carries a greater risk of dyspigmentation, and should be reserved for patients who have not been successfully treated with salicylic or glycolic acid; a 10%-15% concentration of TCA, applied conservatively, is recommended.
Regardless of the type of chemical, potential pitfalls of peels in patients of color include using too much product, allowing too long of an application time, and applying the chemical to an inflamed or excoriated area, Dr. Alexis said. Patients who don’t discontinue retinoids before a peel are at increased risk of developing erosions or crusting, he added.
Dr. Alexis disclosed relationships with Allergan, BioPharmX, Dermira, Galderma, Novan, Novartis, RXi, Unilever, and Valeant.
Global Academy and this news organization are owned by the same parent company.
in a presentation at the Caribbean Dermatology Symposium.
In 2015, ethnic minority patients accounted for 25% of aesthetic procedures in the United States, up from 20% in 2010, according to data from the American Society for Aesthetic Plastic Surgery, said Dr. Alexis, chair of dermatology and director of the Skin of Color Center at Mount Sinai St. Luke’s and Mount Sinai West hospitals in New York.
Chemical peels can be used successfully to treat a range of conditions in skin of color patients, including postinflammatory hyperpigmentation, acne, melasma, textural irregularities, and pseudofolliculitis barbae. They also can be used for skin brightening, said Dr. Alexis, who recommended a chemical peel protocol of salicylic acid, glycolic acid, or Jessner’s every 2-4 weeks. “Consider hydroquinone 4% concurrently to enhance efficacy for treating hyperpigmentation and to prevent postinflammatory hyperpigmentation,” he said. Patients on retinoids should discontinue them for 1 week prior to a chemical peel, he added.
Dr. Alexis shared several treatment pearls to promote successful peels in skin of color patients:
- Salicylic acid: Resist the urge to overapply and “titrate according to patient tolerability.” The endpoint of a salicylic acid peel is white precipitate, not frost; cool compresses can be used for patient comfort and for later removal of the white precipitate.
- Glycolic acid: Stick to a contact time of 2-4 minutes to avoid epidermolysis. “Completely neutralize all areas of application to avoid overpeeling.”
- Trichloroacetic acid (TCA): TCA carries a greater risk of dyspigmentation, and should be reserved for patients who have not been successfully treated with salicylic or glycolic acid; a 10%-15% concentration of TCA, applied conservatively, is recommended.
Regardless of the type of chemical, potential pitfalls of peels in patients of color include using too much product, allowing too long of an application time, and applying the chemical to an inflamed or excoriated area, Dr. Alexis said. Patients who don’t discontinue retinoids before a peel are at increased risk of developing erosions or crusting, he added.
Dr. Alexis disclosed relationships with Allergan, BioPharmX, Dermira, Galderma, Novan, Novartis, RXi, Unilever, and Valeant.
Global Academy and this news organization are owned by the same parent company.
in a presentation at the Caribbean Dermatology Symposium.
In 2015, ethnic minority patients accounted for 25% of aesthetic procedures in the United States, up from 20% in 2010, according to data from the American Society for Aesthetic Plastic Surgery, said Dr. Alexis, chair of dermatology and director of the Skin of Color Center at Mount Sinai St. Luke’s and Mount Sinai West hospitals in New York.
Chemical peels can be used successfully to treat a range of conditions in skin of color patients, including postinflammatory hyperpigmentation, acne, melasma, textural irregularities, and pseudofolliculitis barbae. They also can be used for skin brightening, said Dr. Alexis, who recommended a chemical peel protocol of salicylic acid, glycolic acid, or Jessner’s every 2-4 weeks. “Consider hydroquinone 4% concurrently to enhance efficacy for treating hyperpigmentation and to prevent postinflammatory hyperpigmentation,” he said. Patients on retinoids should discontinue them for 1 week prior to a chemical peel, he added.
Dr. Alexis shared several treatment pearls to promote successful peels in skin of color patients:
- Salicylic acid: Resist the urge to overapply and “titrate according to patient tolerability.” The endpoint of a salicylic acid peel is white precipitate, not frost; cool compresses can be used for patient comfort and for later removal of the white precipitate.
- Glycolic acid: Stick to a contact time of 2-4 minutes to avoid epidermolysis. “Completely neutralize all areas of application to avoid overpeeling.”
- Trichloroacetic acid (TCA): TCA carries a greater risk of dyspigmentation, and should be reserved for patients who have not been successfully treated with salicylic or glycolic acid; a 10%-15% concentration of TCA, applied conservatively, is recommended.
Regardless of the type of chemical, potential pitfalls of peels in patients of color include using too much product, allowing too long of an application time, and applying the chemical to an inflamed or excoriated area, Dr. Alexis said. Patients who don’t discontinue retinoids before a peel are at increased risk of developing erosions or crusting, he added.
Dr. Alexis disclosed relationships with Allergan, BioPharmX, Dermira, Galderma, Novan, Novartis, RXi, Unilever, and Valeant.
Global Academy and this news organization are owned by the same parent company.
EXPERT ANALYSIS FROM THE CARIBBEAN DERMATOLOGY SYMPOSIUM
Demand, not need, may drive further expansion of telepsychiatry
TAMPA – The growth of telepsychiatry has been driven largely by needs of access, particularly in rural areas without specialists. But telemedicine is convenient, and those growing up with computers, smartphones, and other technology are going to demand this type of access to their clinicians, according to a leader of a course on telepsychiatry at the annual meeting of the American College of Psychiatrists.
“Digital natives – the consumers – are going to drive the use of technology more and more. They are used to videoconferencing. They want to see their doctors over video. They want to communicate via text and email. They want that convenience, and they are much more comfortable with it,” said James (Jay) H. Shore, MD, director of telemedicine at the Johnson Depression Center at the University of Colorado Denver.
Meanwhile, telepsychiatry is evolving, allowing for more sophisticated approaches and expanded applications.
“When we started doing video conferencing technologies, we basically were taking what we do in person and just doing that over video,” Dr. Shore said. “Where we are now, .”
A prolific author on the topic of telepsychiatry and long involved in this practice, Dr. Shore has said that the widespread introduction of fiber optic networks and other technological advances over the last 15 years has advanced all forms of digital technology. These are enabling and will likely accelerate synergies possible with integration of different platforms, such as electronic health records, patient portals, videoconferencing, and various methods of communication.
In his own experience, which includes providing remote services from his office in Denver to native populations in Alaska, he has discovered some unexpected advantages to telepsychiatry. For example, some victims recounting histories of domestic abuse feel more secure during videoconferencing than during a face-to-face interview, facilitating capture of a complete history. In general, he now prefers telepsychiatry in those situations.
As telepsychiatry advances, it will be increasingly integrated into hybrid models of care that involve communicating with both the patient and other clinicians over multiple platforms (for example, in-person, video, patient portals). This is not just relevant to patients in a geographically distant facility. With greater acceptance and integration, videoconferencing will be part of this mix of communication tools that might also include in-person consultations. The goal will be to use the most convenient communication strategies to coordinate the diagnosis, a treatment plan, and follow-up.
“The neat thing about telepsychiatry is really the virtual teaming models that we can create,” Dr. Shore said. However, he acknowledged that this type of team participation requires an adjustment in reimbursement models for psychiatrists that traditionally have centered on psychopharmacology. The problem with the models limited to prescription writing is that they “do not tap into the psychiatrist’s leadership of the mental health team, knowledge of human behavior, and they are not, at least for me, as personally rewarding.”
He believes that the growing array of technologies contained in telepsychiatry will increase opportunities for psychiatrists in a host of such settings such as crisis management in emergency care settings or coordination of psychiatric care in residential treatment settings.
The expansion of telemedicine already is reflected in the growing number of companies marketing services directly to consumers. Dr. Shore listed several offering virtual health care that may contribute to both acceptance and demand for medical care delivered digitally. Although telepsychiatry already is associated with many effective applications, Dr. Shore reiterated that consumer demand will be a driver for further expansion of telemedicine in general.
He also emphasized that change involving digital advances in psychiatry is inevitable. According to Dr. Shore, artificial intelligence, virtual reality treatments, and social networking are among potential tools for altering care. Inside and outside of medicine, the pace of change driven by advances in digital exchange of information has been and is expected to continue to be brisk.
“Then there is the technology that is going to disrupt us all that we can’t see coming,” Dr. Shore said. “It is being invented right now in somebody’s garage in Palo Alto.”
Dr. Shore reported that he is chief medical officer of AccessCare Services, which provides telehealth services and technologies.
TAMPA – The growth of telepsychiatry has been driven largely by needs of access, particularly in rural areas without specialists. But telemedicine is convenient, and those growing up with computers, smartphones, and other technology are going to demand this type of access to their clinicians, according to a leader of a course on telepsychiatry at the annual meeting of the American College of Psychiatrists.
“Digital natives – the consumers – are going to drive the use of technology more and more. They are used to videoconferencing. They want to see their doctors over video. They want to communicate via text and email. They want that convenience, and they are much more comfortable with it,” said James (Jay) H. Shore, MD, director of telemedicine at the Johnson Depression Center at the University of Colorado Denver.
Meanwhile, telepsychiatry is evolving, allowing for more sophisticated approaches and expanded applications.
“When we started doing video conferencing technologies, we basically were taking what we do in person and just doing that over video,” Dr. Shore said. “Where we are now, .”
A prolific author on the topic of telepsychiatry and long involved in this practice, Dr. Shore has said that the widespread introduction of fiber optic networks and other technological advances over the last 15 years has advanced all forms of digital technology. These are enabling and will likely accelerate synergies possible with integration of different platforms, such as electronic health records, patient portals, videoconferencing, and various methods of communication.
In his own experience, which includes providing remote services from his office in Denver to native populations in Alaska, he has discovered some unexpected advantages to telepsychiatry. For example, some victims recounting histories of domestic abuse feel more secure during videoconferencing than during a face-to-face interview, facilitating capture of a complete history. In general, he now prefers telepsychiatry in those situations.
As telepsychiatry advances, it will be increasingly integrated into hybrid models of care that involve communicating with both the patient and other clinicians over multiple platforms (for example, in-person, video, patient portals). This is not just relevant to patients in a geographically distant facility. With greater acceptance and integration, videoconferencing will be part of this mix of communication tools that might also include in-person consultations. The goal will be to use the most convenient communication strategies to coordinate the diagnosis, a treatment plan, and follow-up.
“The neat thing about telepsychiatry is really the virtual teaming models that we can create,” Dr. Shore said. However, he acknowledged that this type of team participation requires an adjustment in reimbursement models for psychiatrists that traditionally have centered on psychopharmacology. The problem with the models limited to prescription writing is that they “do not tap into the psychiatrist’s leadership of the mental health team, knowledge of human behavior, and they are not, at least for me, as personally rewarding.”
He believes that the growing array of technologies contained in telepsychiatry will increase opportunities for psychiatrists in a host of such settings such as crisis management in emergency care settings or coordination of psychiatric care in residential treatment settings.
The expansion of telemedicine already is reflected in the growing number of companies marketing services directly to consumers. Dr. Shore listed several offering virtual health care that may contribute to both acceptance and demand for medical care delivered digitally. Although telepsychiatry already is associated with many effective applications, Dr. Shore reiterated that consumer demand will be a driver for further expansion of telemedicine in general.
He also emphasized that change involving digital advances in psychiatry is inevitable. According to Dr. Shore, artificial intelligence, virtual reality treatments, and social networking are among potential tools for altering care. Inside and outside of medicine, the pace of change driven by advances in digital exchange of information has been and is expected to continue to be brisk.
“Then there is the technology that is going to disrupt us all that we can’t see coming,” Dr. Shore said. “It is being invented right now in somebody’s garage in Palo Alto.”
Dr. Shore reported that he is chief medical officer of AccessCare Services, which provides telehealth services and technologies.
TAMPA – The growth of telepsychiatry has been driven largely by needs of access, particularly in rural areas without specialists. But telemedicine is convenient, and those growing up with computers, smartphones, and other technology are going to demand this type of access to their clinicians, according to a leader of a course on telepsychiatry at the annual meeting of the American College of Psychiatrists.
“Digital natives – the consumers – are going to drive the use of technology more and more. They are used to videoconferencing. They want to see their doctors over video. They want to communicate via text and email. They want that convenience, and they are much more comfortable with it,” said James (Jay) H. Shore, MD, director of telemedicine at the Johnson Depression Center at the University of Colorado Denver.
Meanwhile, telepsychiatry is evolving, allowing for more sophisticated approaches and expanded applications.
“When we started doing video conferencing technologies, we basically were taking what we do in person and just doing that over video,” Dr. Shore said. “Where we are now, .”
A prolific author on the topic of telepsychiatry and long involved in this practice, Dr. Shore has said that the widespread introduction of fiber optic networks and other technological advances over the last 15 years has advanced all forms of digital technology. These are enabling and will likely accelerate synergies possible with integration of different platforms, such as electronic health records, patient portals, videoconferencing, and various methods of communication.
In his own experience, which includes providing remote services from his office in Denver to native populations in Alaska, he has discovered some unexpected advantages to telepsychiatry. For example, some victims recounting histories of domestic abuse feel more secure during videoconferencing than during a face-to-face interview, facilitating capture of a complete history. In general, he now prefers telepsychiatry in those situations.
As telepsychiatry advances, it will be increasingly integrated into hybrid models of care that involve communicating with both the patient and other clinicians over multiple platforms (for example, in-person, video, patient portals). This is not just relevant to patients in a geographically distant facility. With greater acceptance and integration, videoconferencing will be part of this mix of communication tools that might also include in-person consultations. The goal will be to use the most convenient communication strategies to coordinate the diagnosis, a treatment plan, and follow-up.
“The neat thing about telepsychiatry is really the virtual teaming models that we can create,” Dr. Shore said. However, he acknowledged that this type of team participation requires an adjustment in reimbursement models for psychiatrists that traditionally have centered on psychopharmacology. The problem with the models limited to prescription writing is that they “do not tap into the psychiatrist’s leadership of the mental health team, knowledge of human behavior, and they are not, at least for me, as personally rewarding.”
He believes that the growing array of technologies contained in telepsychiatry will increase opportunities for psychiatrists in a host of such settings such as crisis management in emergency care settings or coordination of psychiatric care in residential treatment settings.
The expansion of telemedicine already is reflected in the growing number of companies marketing services directly to consumers. Dr. Shore listed several offering virtual health care that may contribute to both acceptance and demand for medical care delivered digitally. Although telepsychiatry already is associated with many effective applications, Dr. Shore reiterated that consumer demand will be a driver for further expansion of telemedicine in general.
He also emphasized that change involving digital advances in psychiatry is inevitable. According to Dr. Shore, artificial intelligence, virtual reality treatments, and social networking are among potential tools for altering care. Inside and outside of medicine, the pace of change driven by advances in digital exchange of information has been and is expected to continue to be brisk.
“Then there is the technology that is going to disrupt us all that we can’t see coming,” Dr. Shore said. “It is being invented right now in somebody’s garage in Palo Alto.”
Dr. Shore reported that he is chief medical officer of AccessCare Services, which provides telehealth services and technologies.
REPORTING FROM THE COLLEGE 2018
Acute cardiorenal syndrome: Mechanisms and clinical implications
As the heart goes, so go the kidneys—and vice versa. Cardiac and renal function are intricately interdependent, and failure of either organ causes injury to the other in a vicious circle of worsening function.1
Here, we discuss acute cardiorenal syndrome, ie, acute exacerbation of heart failure leading to acute kidney injury, a common cause of hospitalization and admission to the intensive care unit. We examine its clinical definition, pathophysiology, hemodynamic derangements, clues that help in diagnosing it, and its treatment.
A GROUP OF LINKED DISORDERS

Two types of acute cardiac dysfunction
Although these definitions offer a good general description, further clarification of the nature of organ dysfunction is needed. Acute renal dysfunction can be unambiguously defined using the AKIN (Acute Kidney Injury Network) and RIFLE (risk, injury, failure, loss of kidney function, and end-stage kidney disease) classifications.3 Acute cardiac dysfunction, on the other hand, is an ambiguous term that encompasses 2 clinically and pathophysiologically distinct conditions: cardiogenic shock and acute heart failure.
Cardiogenic shock is characterized by a catastrophic compromise of cardiac pump function leading to global hypoperfusion severe enough to cause systemic organ damage.4 The cardiac index at which organs start to fail varies in different cases, but a value of less than 1.8 L/min/m2 is typically used to define cardiogenic shock.4
Acute heart failure, on the other hand, is defined as gradually or rapidly worsening signs and symptoms of congestive heart failure due to worsening pulmonary or systemic congestion.5 Hypervolemia is the hallmark of acute heart failure, whereas patients with cardiogenic shock may be hypervolemic, normovolemic, or hypovolemic. Although cardiac output may be mildly reduced in some cases of acute heart failure, systemic perfusion is enough to maintain organ function.
These two conditions cause renal injury by distinct mechanisms and have entirely different therapeutic implications. As we discuss later, reduced renal perfusion due to renal venous congestion is now believed to be the major hemodynamic mechanism of renal injury in acute heart failure. On the other hand, in cardiogenic shock, renal perfusion is reduced due to a critical decline of cardiac pump function.
The ideal definition of acute cardiorenal syndrome should describe a distinct pathophysiology of the syndrome and offer distinct therapeutic options that counteract it. Hence, we propose that renal injury from cardiogenic shock should not be included in its definition, an approach that has been adopted in some of the recent reviews as well.6 Our discussion of acute cardiorenal syndrome is restricted to renal injury caused by acute heart failure.
PATHOPHYSIOLOGY OF ACUTE CARDIORENAL SYNDROME
Multiple mechanisms have been implicated in the pathophysiology of cardiorenal syndrome.7,8
Sympathetic hyperactivity is a compensatory mechanism in heart failure and may be aggravated if cardiac output is further reduced. Its effects include constriction of afferent and efferent arterioles, causing reduced renal perfusion and increased tubular sodium and water reabsorption.7
The renin-angiotensin-aldosterone system is activated in patients with stable congestive heart failure and may be further stimulated in a state of reduced renal perfusion, which is a hallmark of acute cardiorenal syndrome. Its activation can cause further salt and water retention.
However, direct hemodynamic mechanisms likely play the most important role and have obvious diagnostic and therapeutic implications.
Elevated venous pressure, not reduced cardiac output, drives kidney injury
The classic view was that renal dysfunction in acute heart failure is caused by reduced renal blood flow due to failing cardiac pump function. Cardiac output may be reduced in acute heart failure for various reasons, such as atrial fibrillation, myocardial infarction, or other processes, but reduced cardiac output has a minimal role, if any, in the pathogenesis of renal injury in acute heart failure.
As evidence of this, acute heart failure is not always associated with reduced cardiac output.5 Even if the cardiac index (cardiac output divided by body surface area) is mildly reduced, renal blood flow is largely unaffected, thanks to effective renal autoregulatory mechanisms. Not until the mean arterial pressure falls below 70 mm Hg do these mechanisms fail and renal blood flow starts to drop.9 Hence, unless cardiac performance is compromised enough to cause cardiogenic shock, renal blood flow usually does not change significantly with mild reduction in cardiac output.
Hanberg et al10 performed a post hoc analysis of the Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheter Effectiveness (ESCAPE) trial, in which 525 patients with advanced heart failure underwent pulmonary artery catheterization to measure their cardiac index. The authors found no association between the cardiac index and renal function in these patients.
How venous congestion impairs the kidney
In view of the current clinical evidence, the focus has shifted to renal venous congestion. According to Poiseuille’s law, blood flow through the kidneys depends on the pressure gradient—high pressure on the arterial side, low pressure on the venous side.8 Increased renal venous pressure causes reduced renal perfusion pressure, thereby affecting renal perfusion. This is now recognized as an important hemodynamic mechanism of acute cardiorenal syndrome.
Renal congestion can also affect renal function through indirect mechanisms. For example, it can cause renal interstitial edema that may then increase the intratubular pressure, thereby reducing the transglomerular pressure gradient.11

Firth et al,14 in experiments in animals, found that increasing the renal venous pressure above 18.75 mm Hg significantly reduced the glomerular filtration rate, which completely resolved when renal venous pressure was restored to basal levels.
Mullens et al,15 in a study of 145 patients admitted with acute heart failure, reported that 58 (40%) developed acute kidney injury. Pulmonary artery catheterization revealed that elevated central venous pressure, rather than reduced cardiac index, was the primary hemodynamic factor driving renal dysfunction.
DIAGNOSIS AND CLINICAL ASSESSMENT
Patients with acute cardiorenal syndrome present with clinical features of pulmonary or systemic congestion (or both) and acute kidney injury.
Elevated left-sided pressures are usually but not always associated with elevated right-sided pressures. In a study of 1,000 patients with advanced heart failure, a pulmonary capillary wedge pressure of 22 mm Hg or higher had a positive predictive value of 88% for a right atrial pressure of 10 mm Hg or higher.16 Hence, the clinical presentation may vary depending on the location (pulmonary, systemic, or both) and degree of congestion.
Symptoms of pulmonary congestion include worsening exertional dyspnea and orthopnea; bilateral crackles may be heard on physical examination if pulmonary edema is present.
Systemic congestion can cause significant peripheral edema and weight gain. Jugular venous distention may be noted. Oliguria may be present due to renal dysfunction; patients on maintenance diuretic therapy often note its lack of efficacy.
Signs of acute heart failure
Wang et al,17 in a meta-analysis of 22 studies, concluded that the features that most strongly suggested acute heart failure were:
- History of paroxysmal nocturnal dyspnea
- A third heart sound
- Evidence of pulmonary venous congestion on chest radiography.
Features that most strongly suggested the patient did not have acute heart failure were:
- Absence of exertional dyspnea
- Absence of rales
- Absence of radiographic evidence of cardiomegaly.
Patients may present without some of these classic clinical features, and the diagnosis of acute heart failure may be challenging. For example, even if left-sided pressures are very high, pulmonary edema may be absent because of pulmonary vascular remodeling in chronic heart failure.18 Pulmonary artery catheterization reveals elevated cardiac filling pressures and can be used to guide therapy, but clinical evidence argues against its routine use.19
Urine electrolytes (fractional excretion of sodium < 1% and fractional excretion of urea < 35%) often suggest a prerenal form of acute kidney injury, since the hemodynamic derangements in acute cardiorenal syndrome reduce renal perfusion.
Biomarkers of cell-cycle arrest such as urine insulinlike growth factor-binding protein 7 and tissue inhibitor of metalloproteinase 2 have recently been shown to identify patients with acute heart failure at risk of developing acute cardiorenal syndrome.20
Acute cardiorenal syndrome vs renal injury due to hypovolemia
The major alternative in the differential diagnosis of acute cardiorenal syndrome is renal injury due to hypovolemia. Patients with stable heart failure usually have mild hypervolemia at baseline, but they can become hypovolemic due to overaggressive diuretic therapy, severe diarrhea, or other causes.
Although the fluid status of patients in these 2 conditions is opposite, they can be difficult to distinguish. In both conditions, urine electrolytes suggest a prerenal acute kidney injury. A history of recent fluid losses or diuretic overuse may help identify hypovolemia. If available, analysis of the recent trend in weight can be vital in making the right diagnosis.
Misdiagnosis of acute cardiorenal syndrome as hypovolemia-induced acute kidney injury can be catastrophic. If volume depletion is erroneously judged to be the cause of acute kidney injury, fluid administration can further worsen both cardiac and renal function. This can perpetuate the vicious circle that is already in play. Lack of renal recovery may invite further fluid administration.
TREATMENT
Fluid removal with diuresis or ultrafiltration is the cornerstone of treatment. Other treatments such as inotropes are reserved for patients with resistant disease.
Diuretics
The goal of therapy in acute cardiorenal syndrome is to achieve aggressive diuresis, typically using intravenous diuretics. Loop diuretics are the most potent class of diuretics and are the first-line drugs for this purpose. Other classes of diuretics can be used in conjunction with loop diuretics; however, using them by themselves is neither effective nor recommended.
Resistance to diuretics at usual doses is common in patients with acute cardiorenal syndrome. Several mechanisms contribute to diuretic resistance in these patients.21
Oral bioavailability of diuretics may be reduced due to intestinal edema.
Diuretic pharmacokinetics are significantly deranged in cardiorenal syndrome. All diuretics except mineralocorticoid antagonists (ie, spironolactone and eplerenone) act on targets on the luminal side of renal tubules, but are highly protein-bound and are hence not filtered at the glomerulus. Loop diuretics, thiazides, and carbonic anhydrase inhibitors are secreted in the proximal convoluted tubule via the organic anion transporter,22 whereas epithelial sodium channel inhibitors (amiloride and triamterene) are secreted via the organic cation transporter 2.23 In renal dysfunction, various uremic toxins accumulate in the body and compete with diuretics for secretion into the proximal convoluted tubule via these transporters.24
Finally, activation of the sympathetic nervous system and renin-angiotensin-aldosterone system leads to increased tubular sodium and water retention, thereby also blunting the diuretic response.
Diuretic dosage. In patients whose creatinine clearance is less than 15 mL/min, only 10% to 20% as much loop diuretic is secreted into the renal tubule as in normal individuals.25 This effect warrants dose adjustment of diuretics during uremia.

Continuous infusion or bolus? Continuous infusion of loop diuretics is another strategy to optimize drug delivery. Compared with bolus therapy, continuous infusion provides more sustained and uniform drug delivery and prevents postdiuretic sodium retention.
The Diuretic Optimization Strategies Evaluation (DOSE) trial compared the efficacy and safety of continuous vs bolus furosemide therapy in 308 patients admitted with acute decompensated heart failure.26 There was no difference in symptom control or net fluid loss at 72 hours in either group. Other studies have shown more diuresis with continuous infusion than with a similarly dosed bolus regimen.27 However, definitive clinical evidence is lacking at this point to support routine use of continuous loop diuretic therapy.
Combination diuretic therapy. Sequential nephron blockade with combination diuretic therapy is an important therapeutic strategy against diuretic resistance. Notably, urine output-guided diuretic therapy has been shown to be superior to standard diuretic therapy.28 Such therapeutic protocols may employ combination diuretic therapy as a next step when the desired diuretic response is not obtained with high doses of loop diuretic monotherapy.
The desired diuretic response depends on the clinical situation. For example, in patients with severe congestion, we would like the net fluid output to be at least 2 to 3 L more than the fluid intake after the first 24 hours. Sometimes, patients in the intensive care unit are on several essential drug infusions, so that their net intake amounts to 1 to 2 L. In these patients, the desired urine output would be even more than in patients not on these drug infusions.
Loop diuretics block sodium reabsorption at the thick ascending loop of Henle. This disrupts the countercurrent exchange mechanism and reduces renal medullary interstitial osmolarity; these effects prevent water reabsorption. However, the unresorbed sodium can be taken up by the sodium-chloride cotransporter and the epithelial sodium channel in the distal nephron, thereby blunting the diuretic effect. This is the rationale for combining loop diuretics with thiazides or potassium-sparing diuretics.
Similarly, carbonic anhydrase inhibitors (eg, acetazolamide) reduce sodium reabsorption from the proximal convoluted tubule, but most of this sodium is then reabsorbed distally. Hence, the combination of a loop diuretic and acetazolamide can also have a synergistic diuretic effect.
The most popular combination is a loop diuretic plus a thiazide, although no large-scale placebo-controlled trials have been performed.29 Metolazone (a thiazidelike diuretic) is typically used due to its low cost and availability.30 Metolazone has also been shown to block sodium reabsorption at the proximal tubule, which may contribute to its synergistic effect. Chlorothiazide is available in an intravenous formulation and has a faster onset of action than metolazone. However, studies have failed to detect any benefit of one over the other.31
The potential benefit of combining a loop diuretic with acetazolamide is a lower tendency to develop metabolic alkalosis, a potential side effect of loop diuretics and thiazides. Although data are limited, a recent study showed that adding acetazolamide to bumetanide led to significantly increased natriuresis.32
In the Aldosterone Targeted Neurohormonal Combined With Natriuresis Therapy in Heart Failure (ATHENA-HF) trial, adding spironolactone in high doses to usual therapy was not found to cause any significant change in N-terminal pro-B-type natriuretic peptide level or net urine output.33
Ultrafiltration
Venovenous ultrafiltration (or aquapheresis) employs an extracorporeal circuit, similar to the one used in hemodialysis, which removes iso-osmolar fluid at a fixed rate.34 Newer ultrafiltration systems are more portable, can be used with peripheral venous access, and require minimal nursing supervision.35
Although ultrafiltration seems an attractive alternative to diuresis in acute heart failure, studies have been inconclusive. The Cardiorenal Rescue Study in Acute Decompensated Heart Failure (CARRESS-HF) trial compared ultrafiltration and diuresis in 188 patients with acute heart failure and acute cardiorenal syndrome.36 Diuresis, performed according to an algorithm, was found to be superior to ultrafiltration in terms of a bivariate end point of change in weight and change in serum creatinine level at 96 hours. However, the level of cystatin C is thought to be a more accurate indicator of renal function, and the change in cystatin C level from baseline did not differ between the two treatment groups. Also, the ultrafiltration rate was 200 mL per hour, which, some argue, may have been excessive and may have caused intravascular depletion.
Although the ideal rate of fluid removal is unknown, it should be individualized and adjusted based on the patient’s renal function, volume status, and hemodynamic status. The initial rate should be based on the degree of fluid overload and the anticipated plasma refill rate from the interstitial fluid.37 For example, a malnourished patient may have low serum oncotic pressure and hence have low plasma refill upon ultrafiltration. Disturbance of this delicate balance between the rates of ultrafiltration and plasma refill may lead to intravascular volume contraction.
In summary, although ultrafiltration is a valuable alternative to diuretics in resistant cases, its use as a primary decongestive therapy cannot be endorsed in view of the current data.
Inotropes
Inotropes such as dobutamine and milrinone are typically used in cases of cardiogenic shock to maintain organ perfusion. There is a physiologic rationale to using inotropes in acute cardiorenal syndrome as well, especially when the aforementioned strategies fail to overcome diuretic resistance.7
Inotropes increase cardiac output, improve renal blood flow, improve right ventricular output, and thereby relieve systemic congestion. These hemodynamic effects may improve renal perfusion and response to diuretics. However, clinical evidence to support this is lacking.
The Renal Optimization Strategies Evaluation (ROSE) trial enrolled 360 patients with acute heart failure and renal dysfunction. Adding dopamine in a low dose (2 μg/kg/min) to diuretic therapy had no significant effect on 72-hour cumulative urine output or renal function as measured by cystatin C levels.38 However, acute kidney injury was not identified in this trial, and the renal function of many of these patients may have been at its baseline when they were admitted. In other words, this trial did not necessarily include patients with acute kidney injury along with acute heart failure. Hence, it did not necessarily include patients with acute cardiorenal syndrome.

Vasodilators
Vasodilators such as nitroglycerin, sodium nitroprusside, and hydralazine are commonly used in patients with acute heart failure, although the clinical evidence supporting their use is weak.
Physiologically, arterial dilation reduces afterload and can help relieve pulmonary congestion, and venodilation increases capacitance and reduces preload. In theory, venodilators such as nitroglycerin can relieve renal venous congestion in patients with acute cardiorenal syndrome, thereby improving renal perfusion.
However, the use of vasodilators is often limited by their adverse effects, the most important being hypotension. This is especially relevant in light of recent data identifying reduction in blood pressure during treatment of acute heart failure as an independent risk factor for worsening renal function.39,40 It is important to note that in these studies, changes in cardiac index did not affect the propensity for developing worsening renal function. The precise mechanism of this finding is unclear but it is plausible that systemic vasodilation redistributes the cardiac output to nonrenal tissues, thereby overriding the renal autoregulatory mechanisms that are normally employed in low output states.
Preventive strategies
Various strategies can be used to prevent acute cardiorenal syndrome. An optimal outpatient diuretic regimen to avoid hypervolemia is essential. Patients with advanced congestive heart failure should be followed up closely in dedicated heart failure clinics until their diuretic regimen is optimized. Patients should be advised to check their weight on a regular basis and seek medical advice if they notice an increase in their weight or a reduction in their urine output.
TAKE-HOME POINTS
- A robust clinical definition of cardiorenal syndrome is lacking. Hence, recognition of this condition can be challenging.
- Volume overload is central to its pathogenesis, and accurate assessment of volume status is critical.
- Renal venous congestion is the major mechanism of type 1 cardiorenal syndrome.
- Misdiagnosis can have devastating consequences, as it may lead to an opposite therapeutic approach.
- Fluid removal by various strategies is the mainstay of treatment.
- Temporary inotropic support should be saved for the last resort.
- Geisberg C, Butler J. Addressing the challenges of cardiorenal syndrome. Cleve Clin J Med 2006; 73:485–491.
- House AA, Anand I, Bellomo R, et al. Definition and classification of cardio-renal syndromes: workgroup statements from the 7th ADQI Consensus Conference. Nephrol Dial Transplant 2010; 25:1416–1420.
- Chang CH, Lin CY, Tian YC, et al. Acute kidney injury classification: comparison of AKIN and RIFLE criteria. Shock 2010; 33:247-252.
- Reynolds HR, Hochman JS. Cardiogenic shock: current concepts and improving outcomes. Circulation 2008; 117:686–697.
- Gheorghiade M, Pang PS. Acute heart failure syndromes. J Am Coll Cardiol 2009; 53:557–573.
- ter Maaten JM, Valente MA, Damman K, et al. Diuretic response in acute heart failure—pathophysiology, evaluation, and therapy. Nat Rev Cardiol 2015; 12:184–192.
- Hatamizadeh P, Fonarow GC, Budoff MJ, Darabian S, Kovesdy CP, Kalantar-Zadeh K. Cardiorenal syndrome: pathophysiology and potential targets for clinical management. Nat Rev Nephrol 2013; 9:99–111.
- Bock JS, Gottlieb SS. Cardiorenal syndrome: new perspectives. Circulation 2010; 121:2592–2600.
- Burke M, Pabbidi MR, Farley J, et al. Molecular mechanisms of renal blood flow autoregulation. Curr Vasc Pharmacol 2014; 12:845–858.
- Hanberg JS, Sury K, Wilson FP, et al. Reduced cardiac index is not the dominant driver of renal dysfunction in heart failure. J Am Coll Cardiol 2016; 67:2199–2208.
- Afsar B, Ortiz A, Covic A, et al. Focus on renal congestion in heart failure. Clin Kidney J 2016; 9:39–47.
- Verbrugge FH, Dupont M, Steels P, et al. Abdominal contributions to cardiorenal dysfunction in congestive heart failure. J Am Coll Cardiol 2013; 62:485–495.
- Mullens W, Abrahams Z, Skouri HN, et al. Elevated intra-abdominal pressure in acute decompensated heart failure: a potential contributor to worsening renal function? J Am Coll Cardiol 2008; 51:300–306.
- Firth JD, Raine AE, Ledingham JG. Raised venous pressure: a direct cause of renal sodium retention in oedema? Lancet 1988; 1:1033–1035.
- Mullens W, Abrahams Z, Francis GS, et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol 2009; 53:589–596.
- Drazner MH, Hamilton MA, Fonarow G, et al. Relationship between right and left-sided filling pressures in 1000 patients with advanced heart failure. J Heart Lung Transplant 1999; 18:1126–1132.
- Wang CS, FitzGerald JM, Schulzer M, et al. Does this dyspneic patient in the emergency department have congestive heart failure? JAMA 2005; 294:1944–1956.
- Gehlbach BK, Geppert E. The pulmonary manifestations of left heart failure. Chest 2004; 125:669–682.
- Binanay C, Califf RM, Hasselblad V, et al. Evaluation study of congestive heart failure and pulmonary artery catheterization effectiveness: the ESCAPE trial. JAMA 2005; 294:1625–1633.
- Schanz M, Shi J , Wasser C , Alscher MD, Kimmel M. Urinary [TIMP-2] × [IGFBP7] for risk prediction of acute kidney injury in decompensated heart failure. Clin Cardiol 2017; doi.org/10.1002/clc.22683.
- Bowman BN, Nawarskas JJ, Anderson JR. Treating diuretic resistance: an overview. Cardiol Rev 2016; 24:256–260.
- Uwai Y, Saito H, Hashimoto Y, Inui KI. Interaction and transport of thiazide diuretics, loop diuretics, and acetazolamide via rat renal organic anion transporter rOAT1. J Pharmacol Exp Ther 2000; 295:261–265.
- Hacker K, Maas R, Kornhuber J, et al. Substrate-dependent inhibition of the human organic cation transporter OCT2: a comparison of metformin with experimental substrates. PLoS One 2015; 10:e0136451.
- Schophuizen CM, Wilmer MJ, Jansen J, et al. Cationic uremic toxins affect human renal proximal tubule cell functioning through interaction with the organic cation transporter. Pflugers Arch 2013; 465:1701–1714.
- Brater DC. Diuretic therapy. N Engl J Med 1998; 339:387–395.
- Felker GM, Lee KL, Bull DA, et al. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med 2011; 364:797–805.
- Thomson MR, Nappi JM, Dunn SP, Hollis IB, Rodgers JE, Van Bakel AB. Continuous versus intermittent infusion of furosemide in acute decompensated heart failure. J Card Fail 2010; 16:188–193.
- Grodin JL, Stevens SR, de Las Fuentes L, et al. Intensification of medication therapy for cardiorenal syndrome in acute decompensated heart failure. J Card Fail 2016; 22:26–32.
- Ng TM, Konopka E, Hyderi AF, et al. Comparison of bumetanide- and metolazone-based diuretic regimens to furosemide in acute heart failure. J Cardiovasc Pharmacol Ther 2013; 18:345–353.
- Sica DA. Metolazone and its role in edema management. Congest Heart Fail 2003; 9:100–105.
- Moranville MP, Choi S, Hogg J, Anderson AS, Rich JD. Comparison of metolazone versus chlorothiazide in acute decompensated heart failure with diuretic resistance. Cardiovasc Ther 2015; 33:42–49.
- Verbrugge FH, Dupont M, Bertrand PB, et al. Determinants and impact of the natriuretic response to diuretic therapy in heart failure with reduced ejection fraction and volume overload. Acta Cardiol 2015; 70:265–373.
- Butler J, Anstrom KJ, Felker GM, et al. Efficacy and safety of spironolactone in acute heart failure: the ATHENA-HF randomized clinical trial. JAMA Cardiol 2017 Jul 12. doi: 10.1001/jamacardio.2017.2198. [Epub ahead of print]
- Pourafshar N, Karimi A, Kazory A. Extracorporeal ultrafiltration therapy for acute decompensated heart failure. Expert Rev Cardiovasc Ther 2016; 14:5–13.
- Jaski BE, Ha J, Denys BG, et al. Peripherally inserted veno-venous ultrafiltration for rapid treatment of volume overloaded patients. J Card Fail 2003; 9:227–231.
- Jaski BE, Ha J, Denys BG, Lamba S, Trupp RJ, Abraham WT. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med 2012; 367:2296–2304.
- Kazory A. Cardiorenal syndrome: ultrafiltration therapy for heart failure—trials and tribulations. Clin J Am Soc Nephrol 2013; 8:1816–1828.
- Chen HH, Anstrom KJ, Givertz MM, et al. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA 2013; 310:2533–2543.
- Testani JM, Coca SG, McCauley BD, et al. Impact of changes in blood pressure during the treatment of acute decompensated heart failure on renal and clinical outcomes. Eur J Heart Fail 2011; 13:877–884.
- Dupont M, Mullens W, Finucan M, et al. Determinants of dynamic changes in serum creatinine in acute decompensated heart failure: the importance of blood pressure reduction during treatment. Eur J Heart Fail 2013; 15:433–440.
As the heart goes, so go the kidneys—and vice versa. Cardiac and renal function are intricately interdependent, and failure of either organ causes injury to the other in a vicious circle of worsening function.1
Here, we discuss acute cardiorenal syndrome, ie, acute exacerbation of heart failure leading to acute kidney injury, a common cause of hospitalization and admission to the intensive care unit. We examine its clinical definition, pathophysiology, hemodynamic derangements, clues that help in diagnosing it, and its treatment.
A GROUP OF LINKED DISORDERS
Two types of acute cardiac dysfunction
Although these definitions offer a good general description, further clarification of the nature of organ dysfunction is needed. Acute renal dysfunction can be unambiguously defined using the AKIN (Acute Kidney Injury Network) and RIFLE (risk, injury, failure, loss of kidney function, and end-stage kidney disease) classifications.3 Acute cardiac dysfunction, on the other hand, is an ambiguous term that encompasses 2 clinically and pathophysiologically distinct conditions: cardiogenic shock and acute heart failure.
Cardiogenic shock is characterized by a catastrophic compromise of cardiac pump function leading to global hypoperfusion severe enough to cause systemic organ damage.4 The cardiac index at which organs start to fail varies in different cases, but a value of less than 1.8 L/min/m2 is typically used to define cardiogenic shock.4
Acute heart failure, on the other hand, is defined as gradually or rapidly worsening signs and symptoms of congestive heart failure due to worsening pulmonary or systemic congestion.5 Hypervolemia is the hallmark of acute heart failure, whereas patients with cardiogenic shock may be hypervolemic, normovolemic, or hypovolemic. Although cardiac output may be mildly reduced in some cases of acute heart failure, systemic perfusion is enough to maintain organ function.
These two conditions cause renal injury by distinct mechanisms and have entirely different therapeutic implications. As we discuss later, reduced renal perfusion due to renal venous congestion is now believed to be the major hemodynamic mechanism of renal injury in acute heart failure. On the other hand, in cardiogenic shock, renal perfusion is reduced due to a critical decline of cardiac pump function.
The ideal definition of acute cardiorenal syndrome should describe a distinct pathophysiology of the syndrome and offer distinct therapeutic options that counteract it. Hence, we propose that renal injury from cardiogenic shock should not be included in its definition, an approach that has been adopted in some of the recent reviews as well.6 Our discussion of acute cardiorenal syndrome is restricted to renal injury caused by acute heart failure.
PATHOPHYSIOLOGY OF ACUTE CARDIORENAL SYNDROME
Multiple mechanisms have been implicated in the pathophysiology of cardiorenal syndrome.7,8
Sympathetic hyperactivity is a compensatory mechanism in heart failure and may be aggravated if cardiac output is further reduced. Its effects include constriction of afferent and efferent arterioles, causing reduced renal perfusion and increased tubular sodium and water reabsorption.7
The renin-angiotensin-aldosterone system is activated in patients with stable congestive heart failure and may be further stimulated in a state of reduced renal perfusion, which is a hallmark of acute cardiorenal syndrome. Its activation can cause further salt and water retention.
However, direct hemodynamic mechanisms likely play the most important role and have obvious diagnostic and therapeutic implications.
Elevated venous pressure, not reduced cardiac output, drives kidney injury
The classic view was that renal dysfunction in acute heart failure is caused by reduced renal blood flow due to failing cardiac pump function. Cardiac output may be reduced in acute heart failure for various reasons, such as atrial fibrillation, myocardial infarction, or other processes, but reduced cardiac output has a minimal role, if any, in the pathogenesis of renal injury in acute heart failure.
As evidence of this, acute heart failure is not always associated with reduced cardiac output.5 Even if the cardiac index (cardiac output divided by body surface area) is mildly reduced, renal blood flow is largely unaffected, thanks to effective renal autoregulatory mechanisms. Not until the mean arterial pressure falls below 70 mm Hg do these mechanisms fail and renal blood flow starts to drop.9 Hence, unless cardiac performance is compromised enough to cause cardiogenic shock, renal blood flow usually does not change significantly with mild reduction in cardiac output.
Hanberg et al10 performed a post hoc analysis of the Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheter Effectiveness (ESCAPE) trial, in which 525 patients with advanced heart failure underwent pulmonary artery catheterization to measure their cardiac index. The authors found no association between the cardiac index and renal function in these patients.
How venous congestion impairs the kidney
In view of the current clinical evidence, the focus has shifted to renal venous congestion. According to Poiseuille’s law, blood flow through the kidneys depends on the pressure gradient—high pressure on the arterial side, low pressure on the venous side.8 Increased renal venous pressure causes reduced renal perfusion pressure, thereby affecting renal perfusion. This is now recognized as an important hemodynamic mechanism of acute cardiorenal syndrome.
Renal congestion can also affect renal function through indirect mechanisms. For example, it can cause renal interstitial edema that may then increase the intratubular pressure, thereby reducing the transglomerular pressure gradient.11
Firth et al,14 in experiments in animals, found that increasing the renal venous pressure above 18.75 mm Hg significantly reduced the glomerular filtration rate, which completely resolved when renal venous pressure was restored to basal levels.
Mullens et al,15 in a study of 145 patients admitted with acute heart failure, reported that 58 (40%) developed acute kidney injury. Pulmonary artery catheterization revealed that elevated central venous pressure, rather than reduced cardiac index, was the primary hemodynamic factor driving renal dysfunction.
DIAGNOSIS AND CLINICAL ASSESSMENT
Patients with acute cardiorenal syndrome present with clinical features of pulmonary or systemic congestion (or both) and acute kidney injury.
Elevated left-sided pressures are usually but not always associated with elevated right-sided pressures. In a study of 1,000 patients with advanced heart failure, a pulmonary capillary wedge pressure of 22 mm Hg or higher had a positive predictive value of 88% for a right atrial pressure of 10 mm Hg or higher.16 Hence, the clinical presentation may vary depending on the location (pulmonary, systemic, or both) and degree of congestion.
Symptoms of pulmonary congestion include worsening exertional dyspnea and orthopnea; bilateral crackles may be heard on physical examination if pulmonary edema is present.
Systemic congestion can cause significant peripheral edema and weight gain. Jugular venous distention may be noted. Oliguria may be present due to renal dysfunction; patients on maintenance diuretic therapy often note its lack of efficacy.
Signs of acute heart failure
Wang et al,17 in a meta-analysis of 22 studies, concluded that the features that most strongly suggested acute heart failure were:
- History of paroxysmal nocturnal dyspnea
- A third heart sound
- Evidence of pulmonary venous congestion on chest radiography.
Features that most strongly suggested the patient did not have acute heart failure were:
- Absence of exertional dyspnea
- Absence of rales
- Absence of radiographic evidence of cardiomegaly.
Patients may present without some of these classic clinical features, and the diagnosis of acute heart failure may be challenging. For example, even if left-sided pressures are very high, pulmonary edema may be absent because of pulmonary vascular remodeling in chronic heart failure.18 Pulmonary artery catheterization reveals elevated cardiac filling pressures and can be used to guide therapy, but clinical evidence argues against its routine use.19
Urine electrolytes (fractional excretion of sodium < 1% and fractional excretion of urea < 35%) often suggest a prerenal form of acute kidney injury, since the hemodynamic derangements in acute cardiorenal syndrome reduce renal perfusion.
Biomarkers of cell-cycle arrest such as urine insulinlike growth factor-binding protein 7 and tissue inhibitor of metalloproteinase 2 have recently been shown to identify patients with acute heart failure at risk of developing acute cardiorenal syndrome.20
Acute cardiorenal syndrome vs renal injury due to hypovolemia
The major alternative in the differential diagnosis of acute cardiorenal syndrome is renal injury due to hypovolemia. Patients with stable heart failure usually have mild hypervolemia at baseline, but they can become hypovolemic due to overaggressive diuretic therapy, severe diarrhea, or other causes.
Although the fluid status of patients in these 2 conditions is opposite, they can be difficult to distinguish. In both conditions, urine electrolytes suggest a prerenal acute kidney injury. A history of recent fluid losses or diuretic overuse may help identify hypovolemia. If available, analysis of the recent trend in weight can be vital in making the right diagnosis.
Misdiagnosis of acute cardiorenal syndrome as hypovolemia-induced acute kidney injury can be catastrophic. If volume depletion is erroneously judged to be the cause of acute kidney injury, fluid administration can further worsen both cardiac and renal function. This can perpetuate the vicious circle that is already in play. Lack of renal recovery may invite further fluid administration.
TREATMENT
Fluid removal with diuresis or ultrafiltration is the cornerstone of treatment. Other treatments such as inotropes are reserved for patients with resistant disease.
Diuretics
The goal of therapy in acute cardiorenal syndrome is to achieve aggressive diuresis, typically using intravenous diuretics. Loop diuretics are the most potent class of diuretics and are the first-line drugs for this purpose. Other classes of diuretics can be used in conjunction with loop diuretics; however, using them by themselves is neither effective nor recommended.
Resistance to diuretics at usual doses is common in patients with acute cardiorenal syndrome. Several mechanisms contribute to diuretic resistance in these patients.21
Oral bioavailability of diuretics may be reduced due to intestinal edema.
Diuretic pharmacokinetics are significantly deranged in cardiorenal syndrome. All diuretics except mineralocorticoid antagonists (ie, spironolactone and eplerenone) act on targets on the luminal side of renal tubules, but are highly protein-bound and are hence not filtered at the glomerulus. Loop diuretics, thiazides, and carbonic anhydrase inhibitors are secreted in the proximal convoluted tubule via the organic anion transporter,22 whereas epithelial sodium channel inhibitors (amiloride and triamterene) are secreted via the organic cation transporter 2.23 In renal dysfunction, various uremic toxins accumulate in the body and compete with diuretics for secretion into the proximal convoluted tubule via these transporters.24
Finally, activation of the sympathetic nervous system and renin-angiotensin-aldosterone system leads to increased tubular sodium and water retention, thereby also blunting the diuretic response.
Diuretic dosage. In patients whose creatinine clearance is less than 15 mL/min, only 10% to 20% as much loop diuretic is secreted into the renal tubule as in normal individuals.25 This effect warrants dose adjustment of diuretics during uremia.
Continuous infusion or bolus? Continuous infusion of loop diuretics is another strategy to optimize drug delivery. Compared with bolus therapy, continuous infusion provides more sustained and uniform drug delivery and prevents postdiuretic sodium retention.
The Diuretic Optimization Strategies Evaluation (DOSE) trial compared the efficacy and safety of continuous vs bolus furosemide therapy in 308 patients admitted with acute decompensated heart failure.26 There was no difference in symptom control or net fluid loss at 72 hours in either group. Other studies have shown more diuresis with continuous infusion than with a similarly dosed bolus regimen.27 However, definitive clinical evidence is lacking at this point to support routine use of continuous loop diuretic therapy.
Combination diuretic therapy. Sequential nephron blockade with combination diuretic therapy is an important therapeutic strategy against diuretic resistance. Notably, urine output-guided diuretic therapy has been shown to be superior to standard diuretic therapy.28 Such therapeutic protocols may employ combination diuretic therapy as a next step when the desired diuretic response is not obtained with high doses of loop diuretic monotherapy.
The desired diuretic response depends on the clinical situation. For example, in patients with severe congestion, we would like the net fluid output to be at least 2 to 3 L more than the fluid intake after the first 24 hours. Sometimes, patients in the intensive care unit are on several essential drug infusions, so that their net intake amounts to 1 to 2 L. In these patients, the desired urine output would be even more than in patients not on these drug infusions.
Loop diuretics block sodium reabsorption at the thick ascending loop of Henle. This disrupts the countercurrent exchange mechanism and reduces renal medullary interstitial osmolarity; these effects prevent water reabsorption. However, the unresorbed sodium can be taken up by the sodium-chloride cotransporter and the epithelial sodium channel in the distal nephron, thereby blunting the diuretic effect. This is the rationale for combining loop diuretics with thiazides or potassium-sparing diuretics.
Similarly, carbonic anhydrase inhibitors (eg, acetazolamide) reduce sodium reabsorption from the proximal convoluted tubule, but most of this sodium is then reabsorbed distally. Hence, the combination of a loop diuretic and acetazolamide can also have a synergistic diuretic effect.
The most popular combination is a loop diuretic plus a thiazide, although no large-scale placebo-controlled trials have been performed.29 Metolazone (a thiazidelike diuretic) is typically used due to its low cost and availability.30 Metolazone has also been shown to block sodium reabsorption at the proximal tubule, which may contribute to its synergistic effect. Chlorothiazide is available in an intravenous formulation and has a faster onset of action than metolazone. However, studies have failed to detect any benefit of one over the other.31
The potential benefit of combining a loop diuretic with acetazolamide is a lower tendency to develop metabolic alkalosis, a potential side effect of loop diuretics and thiazides. Although data are limited, a recent study showed that adding acetazolamide to bumetanide led to significantly increased natriuresis.32
In the Aldosterone Targeted Neurohormonal Combined With Natriuresis Therapy in Heart Failure (ATHENA-HF) trial, adding spironolactone in high doses to usual therapy was not found to cause any significant change in N-terminal pro-B-type natriuretic peptide level or net urine output.33
Ultrafiltration
Venovenous ultrafiltration (or aquapheresis) employs an extracorporeal circuit, similar to the one used in hemodialysis, which removes iso-osmolar fluid at a fixed rate.34 Newer ultrafiltration systems are more portable, can be used with peripheral venous access, and require minimal nursing supervision.35
Although ultrafiltration seems an attractive alternative to diuresis in acute heart failure, studies have been inconclusive. The Cardiorenal Rescue Study in Acute Decompensated Heart Failure (CARRESS-HF) trial compared ultrafiltration and diuresis in 188 patients with acute heart failure and acute cardiorenal syndrome.36 Diuresis, performed according to an algorithm, was found to be superior to ultrafiltration in terms of a bivariate end point of change in weight and change in serum creatinine level at 96 hours. However, the level of cystatin C is thought to be a more accurate indicator of renal function, and the change in cystatin C level from baseline did not differ between the two treatment groups. Also, the ultrafiltration rate was 200 mL per hour, which, some argue, may have been excessive and may have caused intravascular depletion.
Although the ideal rate of fluid removal is unknown, it should be individualized and adjusted based on the patient’s renal function, volume status, and hemodynamic status. The initial rate should be based on the degree of fluid overload and the anticipated plasma refill rate from the interstitial fluid.37 For example, a malnourished patient may have low serum oncotic pressure and hence have low plasma refill upon ultrafiltration. Disturbance of this delicate balance between the rates of ultrafiltration and plasma refill may lead to intravascular volume contraction.
In summary, although ultrafiltration is a valuable alternative to diuretics in resistant cases, its use as a primary decongestive therapy cannot be endorsed in view of the current data.
Inotropes
Inotropes such as dobutamine and milrinone are typically used in cases of cardiogenic shock to maintain organ perfusion. There is a physiologic rationale to using inotropes in acute cardiorenal syndrome as well, especially when the aforementioned strategies fail to overcome diuretic resistance.7
Inotropes increase cardiac output, improve renal blood flow, improve right ventricular output, and thereby relieve systemic congestion. These hemodynamic effects may improve renal perfusion and response to diuretics. However, clinical evidence to support this is lacking.
The Renal Optimization Strategies Evaluation (ROSE) trial enrolled 360 patients with acute heart failure and renal dysfunction. Adding dopamine in a low dose (2 μg/kg/min) to diuretic therapy had no significant effect on 72-hour cumulative urine output or renal function as measured by cystatin C levels.38 However, acute kidney injury was not identified in this trial, and the renal function of many of these patients may have been at its baseline when they were admitted. In other words, this trial did not necessarily include patients with acute kidney injury along with acute heart failure. Hence, it did not necessarily include patients with acute cardiorenal syndrome.
Vasodilators
Vasodilators such as nitroglycerin, sodium nitroprusside, and hydralazine are commonly used in patients with acute heart failure, although the clinical evidence supporting their use is weak.
Physiologically, arterial dilation reduces afterload and can help relieve pulmonary congestion, and venodilation increases capacitance and reduces preload. In theory, venodilators such as nitroglycerin can relieve renal venous congestion in patients with acute cardiorenal syndrome, thereby improving renal perfusion.
However, the use of vasodilators is often limited by their adverse effects, the most important being hypotension. This is especially relevant in light of recent data identifying reduction in blood pressure during treatment of acute heart failure as an independent risk factor for worsening renal function.39,40 It is important to note that in these studies, changes in cardiac index did not affect the propensity for developing worsening renal function. The precise mechanism of this finding is unclear but it is plausible that systemic vasodilation redistributes the cardiac output to nonrenal tissues, thereby overriding the renal autoregulatory mechanisms that are normally employed in low output states.
Preventive strategies
Various strategies can be used to prevent acute cardiorenal syndrome. An optimal outpatient diuretic regimen to avoid hypervolemia is essential. Patients with advanced congestive heart failure should be followed up closely in dedicated heart failure clinics until their diuretic regimen is optimized. Patients should be advised to check their weight on a regular basis and seek medical advice if they notice an increase in their weight or a reduction in their urine output.
TAKE-HOME POINTS
- A robust clinical definition of cardiorenal syndrome is lacking. Hence, recognition of this condition can be challenging.
- Volume overload is central to its pathogenesis, and accurate assessment of volume status is critical.
- Renal venous congestion is the major mechanism of type 1 cardiorenal syndrome.
- Misdiagnosis can have devastating consequences, as it may lead to an opposite therapeutic approach.
- Fluid removal by various strategies is the mainstay of treatment.
- Temporary inotropic support should be saved for the last resort.
As the heart goes, so go the kidneys—and vice versa. Cardiac and renal function are intricately interdependent, and failure of either organ causes injury to the other in a vicious circle of worsening function.1
Here, we discuss acute cardiorenal syndrome, ie, acute exacerbation of heart failure leading to acute kidney injury, a common cause of hospitalization and admission to the intensive care unit. We examine its clinical definition, pathophysiology, hemodynamic derangements, clues that help in diagnosing it, and its treatment.
A GROUP OF LINKED DISORDERS
Two types of acute cardiac dysfunction
Although these definitions offer a good general description, further clarification of the nature of organ dysfunction is needed. Acute renal dysfunction can be unambiguously defined using the AKIN (Acute Kidney Injury Network) and RIFLE (risk, injury, failure, loss of kidney function, and end-stage kidney disease) classifications.3 Acute cardiac dysfunction, on the other hand, is an ambiguous term that encompasses 2 clinically and pathophysiologically distinct conditions: cardiogenic shock and acute heart failure.
Cardiogenic shock is characterized by a catastrophic compromise of cardiac pump function leading to global hypoperfusion severe enough to cause systemic organ damage.4 The cardiac index at which organs start to fail varies in different cases, but a value of less than 1.8 L/min/m2 is typically used to define cardiogenic shock.4
Acute heart failure, on the other hand, is defined as gradually or rapidly worsening signs and symptoms of congestive heart failure due to worsening pulmonary or systemic congestion.5 Hypervolemia is the hallmark of acute heart failure, whereas patients with cardiogenic shock may be hypervolemic, normovolemic, or hypovolemic. Although cardiac output may be mildly reduced in some cases of acute heart failure, systemic perfusion is enough to maintain organ function.
These two conditions cause renal injury by distinct mechanisms and have entirely different therapeutic implications. As we discuss later, reduced renal perfusion due to renal venous congestion is now believed to be the major hemodynamic mechanism of renal injury in acute heart failure. On the other hand, in cardiogenic shock, renal perfusion is reduced due to a critical decline of cardiac pump function.
The ideal definition of acute cardiorenal syndrome should describe a distinct pathophysiology of the syndrome and offer distinct therapeutic options that counteract it. Hence, we propose that renal injury from cardiogenic shock should not be included in its definition, an approach that has been adopted in some of the recent reviews as well.6 Our discussion of acute cardiorenal syndrome is restricted to renal injury caused by acute heart failure.
PATHOPHYSIOLOGY OF ACUTE CARDIORENAL SYNDROME
Multiple mechanisms have been implicated in the pathophysiology of cardiorenal syndrome.7,8
Sympathetic hyperactivity is a compensatory mechanism in heart failure and may be aggravated if cardiac output is further reduced. Its effects include constriction of afferent and efferent arterioles, causing reduced renal perfusion and increased tubular sodium and water reabsorption.7
The renin-angiotensin-aldosterone system is activated in patients with stable congestive heart failure and may be further stimulated in a state of reduced renal perfusion, which is a hallmark of acute cardiorenal syndrome. Its activation can cause further salt and water retention.
However, direct hemodynamic mechanisms likely play the most important role and have obvious diagnostic and therapeutic implications.
Elevated venous pressure, not reduced cardiac output, drives kidney injury
The classic view was that renal dysfunction in acute heart failure is caused by reduced renal blood flow due to failing cardiac pump function. Cardiac output may be reduced in acute heart failure for various reasons, such as atrial fibrillation, myocardial infarction, or other processes, but reduced cardiac output has a minimal role, if any, in the pathogenesis of renal injury in acute heart failure.
As evidence of this, acute heart failure is not always associated with reduced cardiac output.5 Even if the cardiac index (cardiac output divided by body surface area) is mildly reduced, renal blood flow is largely unaffected, thanks to effective renal autoregulatory mechanisms. Not until the mean arterial pressure falls below 70 mm Hg do these mechanisms fail and renal blood flow starts to drop.9 Hence, unless cardiac performance is compromised enough to cause cardiogenic shock, renal blood flow usually does not change significantly with mild reduction in cardiac output.
Hanberg et al10 performed a post hoc analysis of the Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheter Effectiveness (ESCAPE) trial, in which 525 patients with advanced heart failure underwent pulmonary artery catheterization to measure their cardiac index. The authors found no association between the cardiac index and renal function in these patients.
How venous congestion impairs the kidney
In view of the current clinical evidence, the focus has shifted to renal venous congestion. According to Poiseuille’s law, blood flow through the kidneys depends on the pressure gradient—high pressure on the arterial side, low pressure on the venous side.8 Increased renal venous pressure causes reduced renal perfusion pressure, thereby affecting renal perfusion. This is now recognized as an important hemodynamic mechanism of acute cardiorenal syndrome.
Renal congestion can also affect renal function through indirect mechanisms. For example, it can cause renal interstitial edema that may then increase the intratubular pressure, thereby reducing the transglomerular pressure gradient.11
Firth et al,14 in experiments in animals, found that increasing the renal venous pressure above 18.75 mm Hg significantly reduced the glomerular filtration rate, which completely resolved when renal venous pressure was restored to basal levels.
Mullens et al,15 in a study of 145 patients admitted with acute heart failure, reported that 58 (40%) developed acute kidney injury. Pulmonary artery catheterization revealed that elevated central venous pressure, rather than reduced cardiac index, was the primary hemodynamic factor driving renal dysfunction.
DIAGNOSIS AND CLINICAL ASSESSMENT
Patients with acute cardiorenal syndrome present with clinical features of pulmonary or systemic congestion (or both) and acute kidney injury.
Elevated left-sided pressures are usually but not always associated with elevated right-sided pressures. In a study of 1,000 patients with advanced heart failure, a pulmonary capillary wedge pressure of 22 mm Hg or higher had a positive predictive value of 88% for a right atrial pressure of 10 mm Hg or higher.16 Hence, the clinical presentation may vary depending on the location (pulmonary, systemic, or both) and degree of congestion.
Symptoms of pulmonary congestion include worsening exertional dyspnea and orthopnea; bilateral crackles may be heard on physical examination if pulmonary edema is present.
Systemic congestion can cause significant peripheral edema and weight gain. Jugular venous distention may be noted. Oliguria may be present due to renal dysfunction; patients on maintenance diuretic therapy often note its lack of efficacy.
Signs of acute heart failure
Wang et al,17 in a meta-analysis of 22 studies, concluded that the features that most strongly suggested acute heart failure were:
- History of paroxysmal nocturnal dyspnea
- A third heart sound
- Evidence of pulmonary venous congestion on chest radiography.
Features that most strongly suggested the patient did not have acute heart failure were:
- Absence of exertional dyspnea
- Absence of rales
- Absence of radiographic evidence of cardiomegaly.
Patients may present without some of these classic clinical features, and the diagnosis of acute heart failure may be challenging. For example, even if left-sided pressures are very high, pulmonary edema may be absent because of pulmonary vascular remodeling in chronic heart failure.18 Pulmonary artery catheterization reveals elevated cardiac filling pressures and can be used to guide therapy, but clinical evidence argues against its routine use.19
Urine electrolytes (fractional excretion of sodium < 1% and fractional excretion of urea < 35%) often suggest a prerenal form of acute kidney injury, since the hemodynamic derangements in acute cardiorenal syndrome reduce renal perfusion.
Biomarkers of cell-cycle arrest such as urine insulinlike growth factor-binding protein 7 and tissue inhibitor of metalloproteinase 2 have recently been shown to identify patients with acute heart failure at risk of developing acute cardiorenal syndrome.20
Acute cardiorenal syndrome vs renal injury due to hypovolemia
The major alternative in the differential diagnosis of acute cardiorenal syndrome is renal injury due to hypovolemia. Patients with stable heart failure usually have mild hypervolemia at baseline, but they can become hypovolemic due to overaggressive diuretic therapy, severe diarrhea, or other causes.
Although the fluid status of patients in these 2 conditions is opposite, they can be difficult to distinguish. In both conditions, urine electrolytes suggest a prerenal acute kidney injury. A history of recent fluid losses or diuretic overuse may help identify hypovolemia. If available, analysis of the recent trend in weight can be vital in making the right diagnosis.
Misdiagnosis of acute cardiorenal syndrome as hypovolemia-induced acute kidney injury can be catastrophic. If volume depletion is erroneously judged to be the cause of acute kidney injury, fluid administration can further worsen both cardiac and renal function. This can perpetuate the vicious circle that is already in play. Lack of renal recovery may invite further fluid administration.
TREATMENT
Fluid removal with diuresis or ultrafiltration is the cornerstone of treatment. Other treatments such as inotropes are reserved for patients with resistant disease.
Diuretics
The goal of therapy in acute cardiorenal syndrome is to achieve aggressive diuresis, typically using intravenous diuretics. Loop diuretics are the most potent class of diuretics and are the first-line drugs for this purpose. Other classes of diuretics can be used in conjunction with loop diuretics; however, using them by themselves is neither effective nor recommended.
Resistance to diuretics at usual doses is common in patients with acute cardiorenal syndrome. Several mechanisms contribute to diuretic resistance in these patients.21
Oral bioavailability of diuretics may be reduced due to intestinal edema.
Diuretic pharmacokinetics are significantly deranged in cardiorenal syndrome. All diuretics except mineralocorticoid antagonists (ie, spironolactone and eplerenone) act on targets on the luminal side of renal tubules, but are highly protein-bound and are hence not filtered at the glomerulus. Loop diuretics, thiazides, and carbonic anhydrase inhibitors are secreted in the proximal convoluted tubule via the organic anion transporter,22 whereas epithelial sodium channel inhibitors (amiloride and triamterene) are secreted via the organic cation transporter 2.23 In renal dysfunction, various uremic toxins accumulate in the body and compete with diuretics for secretion into the proximal convoluted tubule via these transporters.24
Finally, activation of the sympathetic nervous system and renin-angiotensin-aldosterone system leads to increased tubular sodium and water retention, thereby also blunting the diuretic response.
Diuretic dosage. In patients whose creatinine clearance is less than 15 mL/min, only 10% to 20% as much loop diuretic is secreted into the renal tubule as in normal individuals.25 This effect warrants dose adjustment of diuretics during uremia.
Continuous infusion or bolus? Continuous infusion of loop diuretics is another strategy to optimize drug delivery. Compared with bolus therapy, continuous infusion provides more sustained and uniform drug delivery and prevents postdiuretic sodium retention.
The Diuretic Optimization Strategies Evaluation (DOSE) trial compared the efficacy and safety of continuous vs bolus furosemide therapy in 308 patients admitted with acute decompensated heart failure.26 There was no difference in symptom control or net fluid loss at 72 hours in either group. Other studies have shown more diuresis with continuous infusion than with a similarly dosed bolus regimen.27 However, definitive clinical evidence is lacking at this point to support routine use of continuous loop diuretic therapy.
Combination diuretic therapy. Sequential nephron blockade with combination diuretic therapy is an important therapeutic strategy against diuretic resistance. Notably, urine output-guided diuretic therapy has been shown to be superior to standard diuretic therapy.28 Such therapeutic protocols may employ combination diuretic therapy as a next step when the desired diuretic response is not obtained with high doses of loop diuretic monotherapy.
The desired diuretic response depends on the clinical situation. For example, in patients with severe congestion, we would like the net fluid output to be at least 2 to 3 L more than the fluid intake after the first 24 hours. Sometimes, patients in the intensive care unit are on several essential drug infusions, so that their net intake amounts to 1 to 2 L. In these patients, the desired urine output would be even more than in patients not on these drug infusions.
Loop diuretics block sodium reabsorption at the thick ascending loop of Henle. This disrupts the countercurrent exchange mechanism and reduces renal medullary interstitial osmolarity; these effects prevent water reabsorption. However, the unresorbed sodium can be taken up by the sodium-chloride cotransporter and the epithelial sodium channel in the distal nephron, thereby blunting the diuretic effect. This is the rationale for combining loop diuretics with thiazides or potassium-sparing diuretics.
Similarly, carbonic anhydrase inhibitors (eg, acetazolamide) reduce sodium reabsorption from the proximal convoluted tubule, but most of this sodium is then reabsorbed distally. Hence, the combination of a loop diuretic and acetazolamide can also have a synergistic diuretic effect.
The most popular combination is a loop diuretic plus a thiazide, although no large-scale placebo-controlled trials have been performed.29 Metolazone (a thiazidelike diuretic) is typically used due to its low cost and availability.30 Metolazone has also been shown to block sodium reabsorption at the proximal tubule, which may contribute to its synergistic effect. Chlorothiazide is available in an intravenous formulation and has a faster onset of action than metolazone. However, studies have failed to detect any benefit of one over the other.31
The potential benefit of combining a loop diuretic with acetazolamide is a lower tendency to develop metabolic alkalosis, a potential side effect of loop diuretics and thiazides. Although data are limited, a recent study showed that adding acetazolamide to bumetanide led to significantly increased natriuresis.32
In the Aldosterone Targeted Neurohormonal Combined With Natriuresis Therapy in Heart Failure (ATHENA-HF) trial, adding spironolactone in high doses to usual therapy was not found to cause any significant change in N-terminal pro-B-type natriuretic peptide level or net urine output.33
Ultrafiltration
Venovenous ultrafiltration (or aquapheresis) employs an extracorporeal circuit, similar to the one used in hemodialysis, which removes iso-osmolar fluid at a fixed rate.34 Newer ultrafiltration systems are more portable, can be used with peripheral venous access, and require minimal nursing supervision.35
Although ultrafiltration seems an attractive alternative to diuresis in acute heart failure, studies have been inconclusive. The Cardiorenal Rescue Study in Acute Decompensated Heart Failure (CARRESS-HF) trial compared ultrafiltration and diuresis in 188 patients with acute heart failure and acute cardiorenal syndrome.36 Diuresis, performed according to an algorithm, was found to be superior to ultrafiltration in terms of a bivariate end point of change in weight and change in serum creatinine level at 96 hours. However, the level of cystatin C is thought to be a more accurate indicator of renal function, and the change in cystatin C level from baseline did not differ between the two treatment groups. Also, the ultrafiltration rate was 200 mL per hour, which, some argue, may have been excessive and may have caused intravascular depletion.
Although the ideal rate of fluid removal is unknown, it should be individualized and adjusted based on the patient’s renal function, volume status, and hemodynamic status. The initial rate should be based on the degree of fluid overload and the anticipated plasma refill rate from the interstitial fluid.37 For example, a malnourished patient may have low serum oncotic pressure and hence have low plasma refill upon ultrafiltration. Disturbance of this delicate balance between the rates of ultrafiltration and plasma refill may lead to intravascular volume contraction.
In summary, although ultrafiltration is a valuable alternative to diuretics in resistant cases, its use as a primary decongestive therapy cannot be endorsed in view of the current data.
Inotropes
Inotropes such as dobutamine and milrinone are typically used in cases of cardiogenic shock to maintain organ perfusion. There is a physiologic rationale to using inotropes in acute cardiorenal syndrome as well, especially when the aforementioned strategies fail to overcome diuretic resistance.7
Inotropes increase cardiac output, improve renal blood flow, improve right ventricular output, and thereby relieve systemic congestion. These hemodynamic effects may improve renal perfusion and response to diuretics. However, clinical evidence to support this is lacking.
The Renal Optimization Strategies Evaluation (ROSE) trial enrolled 360 patients with acute heart failure and renal dysfunction. Adding dopamine in a low dose (2 μg/kg/min) to diuretic therapy had no significant effect on 72-hour cumulative urine output or renal function as measured by cystatin C levels.38 However, acute kidney injury was not identified in this trial, and the renal function of many of these patients may have been at its baseline when they were admitted. In other words, this trial did not necessarily include patients with acute kidney injury along with acute heart failure. Hence, it did not necessarily include patients with acute cardiorenal syndrome.
Vasodilators
Vasodilators such as nitroglycerin, sodium nitroprusside, and hydralazine are commonly used in patients with acute heart failure, although the clinical evidence supporting their use is weak.
Physiologically, arterial dilation reduces afterload and can help relieve pulmonary congestion, and venodilation increases capacitance and reduces preload. In theory, venodilators such as nitroglycerin can relieve renal venous congestion in patients with acute cardiorenal syndrome, thereby improving renal perfusion.
However, the use of vasodilators is often limited by their adverse effects, the most important being hypotension. This is especially relevant in light of recent data identifying reduction in blood pressure during treatment of acute heart failure as an independent risk factor for worsening renal function.39,40 It is important to note that in these studies, changes in cardiac index did not affect the propensity for developing worsening renal function. The precise mechanism of this finding is unclear but it is plausible that systemic vasodilation redistributes the cardiac output to nonrenal tissues, thereby overriding the renal autoregulatory mechanisms that are normally employed in low output states.
Preventive strategies
Various strategies can be used to prevent acute cardiorenal syndrome. An optimal outpatient diuretic regimen to avoid hypervolemia is essential. Patients with advanced congestive heart failure should be followed up closely in dedicated heart failure clinics until their diuretic regimen is optimized. Patients should be advised to check their weight on a regular basis and seek medical advice if they notice an increase in their weight or a reduction in their urine output.
TAKE-HOME POINTS
- A robust clinical definition of cardiorenal syndrome is lacking. Hence, recognition of this condition can be challenging.
- Volume overload is central to its pathogenesis, and accurate assessment of volume status is critical.
- Renal venous congestion is the major mechanism of type 1 cardiorenal syndrome.
- Misdiagnosis can have devastating consequences, as it may lead to an opposite therapeutic approach.
- Fluid removal by various strategies is the mainstay of treatment.
- Temporary inotropic support should be saved for the last resort.
- Geisberg C, Butler J. Addressing the challenges of cardiorenal syndrome. Cleve Clin J Med 2006; 73:485–491.
- House AA, Anand I, Bellomo R, et al. Definition and classification of cardio-renal syndromes: workgroup statements from the 7th ADQI Consensus Conference. Nephrol Dial Transplant 2010; 25:1416–1420.
- Chang CH, Lin CY, Tian YC, et al. Acute kidney injury classification: comparison of AKIN and RIFLE criteria. Shock 2010; 33:247-252.
- Reynolds HR, Hochman JS. Cardiogenic shock: current concepts and improving outcomes. Circulation 2008; 117:686–697.
- Gheorghiade M, Pang PS. Acute heart failure syndromes. J Am Coll Cardiol 2009; 53:557–573.
- ter Maaten JM, Valente MA, Damman K, et al. Diuretic response in acute heart failure—pathophysiology, evaluation, and therapy. Nat Rev Cardiol 2015; 12:184–192.
- Hatamizadeh P, Fonarow GC, Budoff MJ, Darabian S, Kovesdy CP, Kalantar-Zadeh K. Cardiorenal syndrome: pathophysiology and potential targets for clinical management. Nat Rev Nephrol 2013; 9:99–111.
- Bock JS, Gottlieb SS. Cardiorenal syndrome: new perspectives. Circulation 2010; 121:2592–2600.
- Burke M, Pabbidi MR, Farley J, et al. Molecular mechanisms of renal blood flow autoregulation. Curr Vasc Pharmacol 2014; 12:845–858.
- Hanberg JS, Sury K, Wilson FP, et al. Reduced cardiac index is not the dominant driver of renal dysfunction in heart failure. J Am Coll Cardiol 2016; 67:2199–2208.
- Afsar B, Ortiz A, Covic A, et al. Focus on renal congestion in heart failure. Clin Kidney J 2016; 9:39–47.
- Verbrugge FH, Dupont M, Steels P, et al. Abdominal contributions to cardiorenal dysfunction in congestive heart failure. J Am Coll Cardiol 2013; 62:485–495.
- Mullens W, Abrahams Z, Skouri HN, et al. Elevated intra-abdominal pressure in acute decompensated heart failure: a potential contributor to worsening renal function? J Am Coll Cardiol 2008; 51:300–306.
- Firth JD, Raine AE, Ledingham JG. Raised venous pressure: a direct cause of renal sodium retention in oedema? Lancet 1988; 1:1033–1035.
- Mullens W, Abrahams Z, Francis GS, et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol 2009; 53:589–596.
- Drazner MH, Hamilton MA, Fonarow G, et al. Relationship between right and left-sided filling pressures in 1000 patients with advanced heart failure. J Heart Lung Transplant 1999; 18:1126–1132.
- Wang CS, FitzGerald JM, Schulzer M, et al. Does this dyspneic patient in the emergency department have congestive heart failure? JAMA 2005; 294:1944–1956.
- Gehlbach BK, Geppert E. The pulmonary manifestations of left heart failure. Chest 2004; 125:669–682.
- Binanay C, Califf RM, Hasselblad V, et al. Evaluation study of congestive heart failure and pulmonary artery catheterization effectiveness: the ESCAPE trial. JAMA 2005; 294:1625–1633.
- Schanz M, Shi J , Wasser C , Alscher MD, Kimmel M. Urinary [TIMP-2] × [IGFBP7] for risk prediction of acute kidney injury in decompensated heart failure. Clin Cardiol 2017; doi.org/10.1002/clc.22683.
- Bowman BN, Nawarskas JJ, Anderson JR. Treating diuretic resistance: an overview. Cardiol Rev 2016; 24:256–260.
- Uwai Y, Saito H, Hashimoto Y, Inui KI. Interaction and transport of thiazide diuretics, loop diuretics, and acetazolamide via rat renal organic anion transporter rOAT1. J Pharmacol Exp Ther 2000; 295:261–265.
- Hacker K, Maas R, Kornhuber J, et al. Substrate-dependent inhibition of the human organic cation transporter OCT2: a comparison of metformin with experimental substrates. PLoS One 2015; 10:e0136451.
- Schophuizen CM, Wilmer MJ, Jansen J, et al. Cationic uremic toxins affect human renal proximal tubule cell functioning through interaction with the organic cation transporter. Pflugers Arch 2013; 465:1701–1714.
- Brater DC. Diuretic therapy. N Engl J Med 1998; 339:387–395.
- Felker GM, Lee KL, Bull DA, et al. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med 2011; 364:797–805.
- Thomson MR, Nappi JM, Dunn SP, Hollis IB, Rodgers JE, Van Bakel AB. Continuous versus intermittent infusion of furosemide in acute decompensated heart failure. J Card Fail 2010; 16:188–193.
- Grodin JL, Stevens SR, de Las Fuentes L, et al. Intensification of medication therapy for cardiorenal syndrome in acute decompensated heart failure. J Card Fail 2016; 22:26–32.
- Ng TM, Konopka E, Hyderi AF, et al. Comparison of bumetanide- and metolazone-based diuretic regimens to furosemide in acute heart failure. J Cardiovasc Pharmacol Ther 2013; 18:345–353.
- Sica DA. Metolazone and its role in edema management. Congest Heart Fail 2003; 9:100–105.
- Moranville MP, Choi S, Hogg J, Anderson AS, Rich JD. Comparison of metolazone versus chlorothiazide in acute decompensated heart failure with diuretic resistance. Cardiovasc Ther 2015; 33:42–49.
- Verbrugge FH, Dupont M, Bertrand PB, et al. Determinants and impact of the natriuretic response to diuretic therapy in heart failure with reduced ejection fraction and volume overload. Acta Cardiol 2015; 70:265–373.
- Butler J, Anstrom KJ, Felker GM, et al. Efficacy and safety of spironolactone in acute heart failure: the ATHENA-HF randomized clinical trial. JAMA Cardiol 2017 Jul 12. doi: 10.1001/jamacardio.2017.2198. [Epub ahead of print]
- Pourafshar N, Karimi A, Kazory A. Extracorporeal ultrafiltration therapy for acute decompensated heart failure. Expert Rev Cardiovasc Ther 2016; 14:5–13.
- Jaski BE, Ha J, Denys BG, et al. Peripherally inserted veno-venous ultrafiltration for rapid treatment of volume overloaded patients. J Card Fail 2003; 9:227–231.
- Jaski BE, Ha J, Denys BG, Lamba S, Trupp RJ, Abraham WT. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med 2012; 367:2296–2304.
- Kazory A. Cardiorenal syndrome: ultrafiltration therapy for heart failure—trials and tribulations. Clin J Am Soc Nephrol 2013; 8:1816–1828.
- Chen HH, Anstrom KJ, Givertz MM, et al. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA 2013; 310:2533–2543.
- Testani JM, Coca SG, McCauley BD, et al. Impact of changes in blood pressure during the treatment of acute decompensated heart failure on renal and clinical outcomes. Eur J Heart Fail 2011; 13:877–884.
- Dupont M, Mullens W, Finucan M, et al. Determinants of dynamic changes in serum creatinine in acute decompensated heart failure: the importance of blood pressure reduction during treatment. Eur J Heart Fail 2013; 15:433–440.
- Geisberg C, Butler J. Addressing the challenges of cardiorenal syndrome. Cleve Clin J Med 2006; 73:485–491.
- House AA, Anand I, Bellomo R, et al. Definition and classification of cardio-renal syndromes: workgroup statements from the 7th ADQI Consensus Conference. Nephrol Dial Transplant 2010; 25:1416–1420.
- Chang CH, Lin CY, Tian YC, et al. Acute kidney injury classification: comparison of AKIN and RIFLE criteria. Shock 2010; 33:247-252.
- Reynolds HR, Hochman JS. Cardiogenic shock: current concepts and improving outcomes. Circulation 2008; 117:686–697.
- Gheorghiade M, Pang PS. Acute heart failure syndromes. J Am Coll Cardiol 2009; 53:557–573.
- ter Maaten JM, Valente MA, Damman K, et al. Diuretic response in acute heart failure—pathophysiology, evaluation, and therapy. Nat Rev Cardiol 2015; 12:184–192.
- Hatamizadeh P, Fonarow GC, Budoff MJ, Darabian S, Kovesdy CP, Kalantar-Zadeh K. Cardiorenal syndrome: pathophysiology and potential targets for clinical management. Nat Rev Nephrol 2013; 9:99–111.
- Bock JS, Gottlieb SS. Cardiorenal syndrome: new perspectives. Circulation 2010; 121:2592–2600.
- Burke M, Pabbidi MR, Farley J, et al. Molecular mechanisms of renal blood flow autoregulation. Curr Vasc Pharmacol 2014; 12:845–858.
- Hanberg JS, Sury K, Wilson FP, et al. Reduced cardiac index is not the dominant driver of renal dysfunction in heart failure. J Am Coll Cardiol 2016; 67:2199–2208.
- Afsar B, Ortiz A, Covic A, et al. Focus on renal congestion in heart failure. Clin Kidney J 2016; 9:39–47.
- Verbrugge FH, Dupont M, Steels P, et al. Abdominal contributions to cardiorenal dysfunction in congestive heart failure. J Am Coll Cardiol 2013; 62:485–495.
- Mullens W, Abrahams Z, Skouri HN, et al. Elevated intra-abdominal pressure in acute decompensated heart failure: a potential contributor to worsening renal function? J Am Coll Cardiol 2008; 51:300–306.
- Firth JD, Raine AE, Ledingham JG. Raised venous pressure: a direct cause of renal sodium retention in oedema? Lancet 1988; 1:1033–1035.
- Mullens W, Abrahams Z, Francis GS, et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol 2009; 53:589–596.
- Drazner MH, Hamilton MA, Fonarow G, et al. Relationship between right and left-sided filling pressures in 1000 patients with advanced heart failure. J Heart Lung Transplant 1999; 18:1126–1132.
- Wang CS, FitzGerald JM, Schulzer M, et al. Does this dyspneic patient in the emergency department have congestive heart failure? JAMA 2005; 294:1944–1956.
- Gehlbach BK, Geppert E. The pulmonary manifestations of left heart failure. Chest 2004; 125:669–682.
- Binanay C, Califf RM, Hasselblad V, et al. Evaluation study of congestive heart failure and pulmonary artery catheterization effectiveness: the ESCAPE trial. JAMA 2005; 294:1625–1633.
- Schanz M, Shi J , Wasser C , Alscher MD, Kimmel M. Urinary [TIMP-2] × [IGFBP7] for risk prediction of acute kidney injury in decompensated heart failure. Clin Cardiol 2017; doi.org/10.1002/clc.22683.
- Bowman BN, Nawarskas JJ, Anderson JR. Treating diuretic resistance: an overview. Cardiol Rev 2016; 24:256–260.
- Uwai Y, Saito H, Hashimoto Y, Inui KI. Interaction and transport of thiazide diuretics, loop diuretics, and acetazolamide via rat renal organic anion transporter rOAT1. J Pharmacol Exp Ther 2000; 295:261–265.
- Hacker K, Maas R, Kornhuber J, et al. Substrate-dependent inhibition of the human organic cation transporter OCT2: a comparison of metformin with experimental substrates. PLoS One 2015; 10:e0136451.
- Schophuizen CM, Wilmer MJ, Jansen J, et al. Cationic uremic toxins affect human renal proximal tubule cell functioning through interaction with the organic cation transporter. Pflugers Arch 2013; 465:1701–1714.
- Brater DC. Diuretic therapy. N Engl J Med 1998; 339:387–395.
- Felker GM, Lee KL, Bull DA, et al. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med 2011; 364:797–805.
- Thomson MR, Nappi JM, Dunn SP, Hollis IB, Rodgers JE, Van Bakel AB. Continuous versus intermittent infusion of furosemide in acute decompensated heart failure. J Card Fail 2010; 16:188–193.
- Grodin JL, Stevens SR, de Las Fuentes L, et al. Intensification of medication therapy for cardiorenal syndrome in acute decompensated heart failure. J Card Fail 2016; 22:26–32.
- Ng TM, Konopka E, Hyderi AF, et al. Comparison of bumetanide- and metolazone-based diuretic regimens to furosemide in acute heart failure. J Cardiovasc Pharmacol Ther 2013; 18:345–353.
- Sica DA. Metolazone and its role in edema management. Congest Heart Fail 2003; 9:100–105.
- Moranville MP, Choi S, Hogg J, Anderson AS, Rich JD. Comparison of metolazone versus chlorothiazide in acute decompensated heart failure with diuretic resistance. Cardiovasc Ther 2015; 33:42–49.
- Verbrugge FH, Dupont M, Bertrand PB, et al. Determinants and impact of the natriuretic response to diuretic therapy in heart failure with reduced ejection fraction and volume overload. Acta Cardiol 2015; 70:265–373.
- Butler J, Anstrom KJ, Felker GM, et al. Efficacy and safety of spironolactone in acute heart failure: the ATHENA-HF randomized clinical trial. JAMA Cardiol 2017 Jul 12. doi: 10.1001/jamacardio.2017.2198. [Epub ahead of print]
- Pourafshar N, Karimi A, Kazory A. Extracorporeal ultrafiltration therapy for acute decompensated heart failure. Expert Rev Cardiovasc Ther 2016; 14:5–13.
- Jaski BE, Ha J, Denys BG, et al. Peripherally inserted veno-venous ultrafiltration for rapid treatment of volume overloaded patients. J Card Fail 2003; 9:227–231.
- Jaski BE, Ha J, Denys BG, Lamba S, Trupp RJ, Abraham WT. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med 2012; 367:2296–2304.
- Kazory A. Cardiorenal syndrome: ultrafiltration therapy for heart failure—trials and tribulations. Clin J Am Soc Nephrol 2013; 8:1816–1828.
- Chen HH, Anstrom KJ, Givertz MM, et al. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA 2013; 310:2533–2543.
- Testani JM, Coca SG, McCauley BD, et al. Impact of changes in blood pressure during the treatment of acute decompensated heart failure on renal and clinical outcomes. Eur J Heart Fail 2011; 13:877–884.
- Dupont M, Mullens W, Finucan M, et al. Determinants of dynamic changes in serum creatinine in acute decompensated heart failure: the importance of blood pressure reduction during treatment. Eur J Heart Fail 2013; 15:433–440.
KEY POINTS
- Acute cardiorenal syndrome is the acute worsening of renal function due to acute decompensated heart failure.
- The most important mechanism of acute cardiorenal syndrome is now believed to be systemic congestion leading to increased renal venous pressure, which in turn reduces renal perfusion.
- The major alternative in the differential diagnosis of acute cardiorenal syndrome is renal injury due to hypovolemia. Differentiating the 2 may be challenging if signs of systemic and pulmonary congestion are not obvious.
- Diuretic resistance is common in acute cardiorenal syndrome but may be overcome by using higher doses of diuretics and combinations of diuretics that block reabsorption at different segments of the renal tubules.
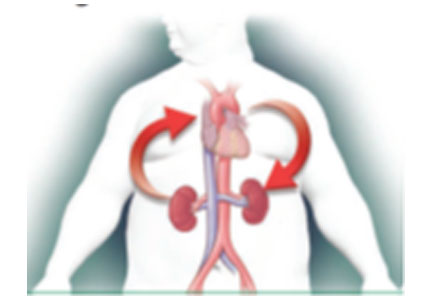
Hemodynamically, the kidney is at the heart of cardiorenal syndrome
In heart failure, the heart and the kidneys share a rocky relationship. Cardiac dysfunction can heighten renal dysfunction and vice versa—appropriately dubbed “cardiorenal syndrome.”
Although classically defined by a reduction in the glomerular filtration rate (GFR),1 cardiorenal syndrome also encompasses complex neurohormonal, pharmacologic, and metabolic interactions affecting or affected by both glomerular and tubular function. Unfortunately, all of these maladaptive processes occur in heart failure and perpetuate a vicious circle of continued dual-organ dysfunction.
The central insult here is hemodynamic disarray from acute or chronic cardiac dysfunction, which can directly influence glomerular function. However, to understand the hemodynamic ramifications for glomerular function, we focus on the determinants of glomerular filtration.
DETERMINANTS OF GFR
The GFR is the rate of fluid flow between the glomerular capillaries and the Bowman capsule and is classically represented by the following equations2:
GFR = Kf × (PG – PB – πG + πB)
Kf = N × Lp × S
Kf is the filtration constant, N the number of functional nephrons, Lp the hydraulic conductivity of the glomerular capillary, S the filtration area, PG the hydrostatic pressure in the glomerular capillaries, PB the hydrostatic pressure in the Bowman capsule, and πG and πB the colloid osmotic pressures within the glomerular capillaries and Bowman space, respectively.
Based on this relationship, the GFR is reduced when PG is reduced in the setting of hypovolemia, hypotension, or renin-angiotensin system antagonist use or when PB is increased in the setting of elevated central venous pressure or elevated abdominal pressure—all common in heart failure. With this understanding, one would assume that strategies to increase PG (improve perfusion) and reduce PB (reduce congestion) might ameliorate ongoing renal dysfunction and improve the GFR in heart failure.
In this issue, Thind et al3 highlight the impact of hemodynamic derangements in heart failure with acute cardiorenal syndrome and provide an overview of its treatment. They review the complex relationship between progressive cardiac failure translating into accelerated neurohormonal responses (increases in sympathetic nervous system and renin-angiotensin-aldosterone system activation) and the impact of increased central venous pressure on progressive renal dysfunction. They also provide an overview of efforts to mitigate cardiorenal syndrome, after careful appraisal of volume status, through diuretic-mediated decongestion with aggressive use of loop diuretics (either in isolation or in the form of sequential nephron blockade with a thiazide or acetazolamide), and they highlight the lingering uncertainty regarding inotrope use.
VENOUS CONGESTION VS DECREASED CARDIAC OUTPUT
Returning to the GFR equation, it is clear that an imbalance in PG and PB can worsen glomerular function. Because cardiac dysfunction can lead to both venous congestion and decreased cardiac output, this leads to the question, “Of these, which is the more important driver of this imbalance and its effects on renal function?”
A compelling argument can be made for each side. On one hand, experiments over a half-century old in human models of venous congestion highlighted the profound impact of elevated venous pressure, which decreases electrolyte excretion (sodium included) and diminishes urine flow.4,5 This has been replicated in more-contemporary decompensated heart failure cohorts in which worsening renal function was more closely associated with elevated central venous pressure rather than cardiac output.6,7 On the other hand, early landmark experiments and more recent cohorts with heart failure have also shown that reductions in effective arterial blood volume, renal blood flow, and cardiac output are also associated with reductions in GFR.5,8,9
How then shall we reconcile whether cardiorenal syndrome is a “backward failure” (from central venous pressure) or a “forward failure” (from decreased perfusion) phenomenon?
The answer is complicated and is likely “both,” with the major component being increased central venous pressure. To understand this construct, we must first exclude frank cardiogenic shock—when the hydraulic function of the heart fails to provide enough flow, leading to a catastrophic drop in mean arterial pressure that supersedes the kidney’s ability to autoregulate renal blood flow.10,11
In patients with chronic heart failure and congestion who are not in shock, historical observations suggest that both intra-abdominal pressure (which increases renal venous pressure) and central venous pressure lead to reduced renal blood flow and increased renal vasomotor resistance (increase in afferent, intrarenal, and efferent vascular tone).12–14 More recent observations from epidemiologic studies have largely replicated these findings. Central venous pressure remains essential to impacting renal function in heart failure,6,15 and the impact of cardiac output on renal function remains uncertain.16
The relationship of intracardiac hemodynamics may also play a role in modifying renal function. Several reports recently described the relationship between both right- and left-sided filling pressures as being associated with worse renal function in heart failure.17–19 Patients with a disproportionately higher right atrial pressure to pulmonary capillary wedge pressure have higher serum creatinine during and after decongestive therapies. Therefore, the concept of “right-sided heart failure” expands beyond the simple representation of “backward congestion” at the level of venous return. In fact, a higher ratio of right atrial pressure to pulmonary capillary wedge pressure may point to an inability of the venous and pulmonary circulations to provide adequate left ventricular preload. Therefore, a relatively underfilled left ventricle in the face of biventricular dysfunction may result in worsening renal function.
TREATMENT IS CHALLENGING
The treatment of cardiorenal syndrome is challenging. It is often accompanied by heightened azotemia, diuretic resistance, electrolyte abnormalities, and a spectrum of hemodynamic disarray. As Thind et al point out, there is, unfortunately, no firmly established treatment. While “sequential nephron blockade” (pharmacologically blocking multiple sites on the nephron simultaneously) is theoretically promising, there are no rigorously studied therapeutic strategies with proven efficacy.
On the other hand, mechanical removal of isotonic fluid with ultrafiltration showed early promise in decompensated heart failure, but enthusiasm diminished with results from the Cardiorenal Rescue Study in Acute Decompensated Heart Failure (CARRESS-HF) trial.20 Ultrafiltration was roughly equivalent to aggressive pharmacologic therapy for fluid loss, was associated with higher serum creatinine levels, and was more challenging to administer.
Equally uncertain is the benefit of inotropic or vasoactive therapy, which directly alters cardiac hemodynamics. Low-dose dopamine or low-dose nesiritide is of no benefit toward enhancement of decongestion or renal protection when added to standard diuretic therapy.21 Furthermore, routine use of inotropes is fraught with more arrhythmias and hypotension and is associated with dismal long-term outcomes.22,23
Alternative therapies that act directly on renal physiology—eg, rolofylline, a selective adenosine A1 receptor antagonist that may enhance renal blood flow, augment natriuresis, and break diuretic resistance—have been similarly disappointing.24
With so much uncertainty, more investigation into novel treatments for cardiorenal syndrome is clearly warranted.
However, because venous congestion is the hemodynamic hallmark of acute cardiorenal syndrome (increasing PB), reducing central venous pressure remains the cornerstone treatment for cardiorenal syndrome. Additionally, efforts to preserve renal perfusion and avoid hypotension are prudent to maintain glomerular capillary hydrostatic pressure (PG).
In light of these considerations, there is no “one size fits all” for the treatment of cardiorenal syndrome. Treatment should be based on thoughtful individualized strategies tailored to the underlying cardiorenal pathophysiology, and with the understanding that the kidney is at the heart of the matter.
- House AA, Anand I, Bellomo R, et al; Acute Dialysis Quality Initiative Consensus Group. Definition and classification of cardio-renal syndromes: workgroup statements from the 7th ADQI Consensus Conference. Nephrol Dial Transplant 2010; 25:1416–1420.
- Tucker BJ, Blantz RC. An analysis of the determinants of nephron filtration rate. Am J Physiol 1977; 232:F477–F483.
- Thind GS, Loehrke M, Wilt JL. Acute cardiorenal syndrome: mechanisms and clinical implications. Cleve Clin J Med 2018; 85:231–239.
- Wilkins RW, Tinsley CM, Culbertson JW, et al. The effects of venous congestion of the limbs upon renal clearances and the excretion of water and salt. I. Studies in normal subjects and in hypertensive patients before and after splanchnicectomy. J Clin Invest 1953; 32:1101–1116.
- Judson WE, Hatcher JD, Hollander W, Halperin MH, Wilkins RW. The effects of venous congestion of the limbs and phlebotomy upon renal clearances and the excretion of water and salt. II. Studies in patients with congestive failure. J Clin Invest 1955; 34:1591–1599.
- Mullens W, Abrahams Z, Francis GS, et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol 2009; 53:589–596.
- Damman K, van Deursen VM, Navis G, Voors AA, van Veldhuisen DJ, Hillege HL. Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol 2009; 53:582–588.
- Ljungman S, Laragh JH, Cody RJ. Role of the kidney in congestive heart failure. Relationship of cardiac index to kidney function. Drugs 1990; 39(suppl 4):10–21; discussion 22–24.
- Damman K, Navis G, Smilde TD, et al. Decreased cardiac output, venous congestion and the association with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail 2007; 9:872–878.
- Fincke R, Hochman JS, Lowe AM, et al. Cardiac power is the strongest hemodynamic correlate of mortality in cardiogenic shock: a report from the SHOCK trial registry. J Am Coll Cardiol 2004; 44:340–348.
- Adams PL, Adams FF, Bell PD, Navar LG. Impaired renal blood flow autoregulation in ischemic acute renal failure. Kidney Int 1980; 18:68–76.
- Maxwell MH, Breed ES, Schwartz IL. Renal venous pressure in chronic congestive heart failure. J Clin Invest 1950; 29:342–348.
- Blake WD, Wégria R, Keating RP, Ward HP. Effect of increased renal venous pressure on renal function. Am J Physiol 1949; 157:1–13.
- Bradley SE, Bradley GP. The effect of increased intra-abdominal pressure on renal function in man. J Clin Invest 1947; 26:1010–1022.
- Mullens W, Abrahams Z, Skouri HN, et al. Elevated intra-abdominal pressure in acute decompensated heart failure: a potential contributor to worsening renal function? J Am Coll Cardiol 2008; 51:300–306.
- Hanberg JS, Sury K, Wilson FP, et al. Reduced cardiac index is not the dominant driver of renal dysfunction in heart failure. J Am Coll Cardiol 2016; 67:2199–2208.
- Drazner MH, Brown RN, Kaiser PA, et al. Relationship of right- and left-sided filling pressures in patients with advanced heart failure: a 14-year multi-institutional analysis. J Heart Lung Transplant 2012; 31:67–72.
- Drazner MH, Velez-Martinez M, Ayers CR, et al. Relationship of right- to left-sided ventricular filling pressures in advanced heart failure: insights from the ESCAPE trial. Circ Heart Fail 2013; 6:264–270.
- Grodin JL, Drazner MH, Dupont M, et al. A disproportionate elevation in right ventricular filling pressure, in relation to left ventricular filling pressure, is associated with renal impairment and increased mortality in advanced decompensated heart failure. Am Heart J 2015; 169:806–812.
- Bart BA, Goldsmith SR, Lee KL, et al, for the Heart Failure Clinical Research Network. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med 2012; 367:2296–2304.
- Chen HH, Anstrom KJ, Givertz MM, et al; NHLBI Heart Failure Clinical Research Network. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA 2013; 310:2533–2543.
- Gorodeski EZ, Chu EC, Reese JR, Shishehbor MH, Hsich E, Starling RC. Prognosis on chronic dobutamine or milrinone infusions for stage D heart failure. Circ Heart Fail 2009; 2:320–324.
- Cuffe MS, Califf RM, Adams KF Jr, et al, for the Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME-CHF) Investigators. Short-term intravenous milrinone for acute exacerbation of chronic heart failure: a randomized controlled trial. JAMA 2002; 287:1541–1547.
- Massie BM, O’Connor CM, Metra M, et al, for the PROTECT Investigators and Committees. Rolofylline, an adenosine A1-receptor antagonist, in acute heart failure. N Engl J Med 2010; 363:1419–1428.
In heart failure, the heart and the kidneys share a rocky relationship. Cardiac dysfunction can heighten renal dysfunction and vice versa—appropriately dubbed “cardiorenal syndrome.”
Although classically defined by a reduction in the glomerular filtration rate (GFR),1 cardiorenal syndrome also encompasses complex neurohormonal, pharmacologic, and metabolic interactions affecting or affected by both glomerular and tubular function. Unfortunately, all of these maladaptive processes occur in heart failure and perpetuate a vicious circle of continued dual-organ dysfunction.
The central insult here is hemodynamic disarray from acute or chronic cardiac dysfunction, which can directly influence glomerular function. However, to understand the hemodynamic ramifications for glomerular function, we focus on the determinants of glomerular filtration.
DETERMINANTS OF GFR
The GFR is the rate of fluid flow between the glomerular capillaries and the Bowman capsule and is classically represented by the following equations2:
GFR = Kf × (PG – PB – πG + πB)
Kf = N × Lp × S
Kf is the filtration constant, N the number of functional nephrons, Lp the hydraulic conductivity of the glomerular capillary, S the filtration area, PG the hydrostatic pressure in the glomerular capillaries, PB the hydrostatic pressure in the Bowman capsule, and πG and πB the colloid osmotic pressures within the glomerular capillaries and Bowman space, respectively.
Based on this relationship, the GFR is reduced when PG is reduced in the setting of hypovolemia, hypotension, or renin-angiotensin system antagonist use or when PB is increased in the setting of elevated central venous pressure or elevated abdominal pressure—all common in heart failure. With this understanding, one would assume that strategies to increase PG (improve perfusion) and reduce PB (reduce congestion) might ameliorate ongoing renal dysfunction and improve the GFR in heart failure.
In this issue, Thind et al3 highlight the impact of hemodynamic derangements in heart failure with acute cardiorenal syndrome and provide an overview of its treatment. They review the complex relationship between progressive cardiac failure translating into accelerated neurohormonal responses (increases in sympathetic nervous system and renin-angiotensin-aldosterone system activation) and the impact of increased central venous pressure on progressive renal dysfunction. They also provide an overview of efforts to mitigate cardiorenal syndrome, after careful appraisal of volume status, through diuretic-mediated decongestion with aggressive use of loop diuretics (either in isolation or in the form of sequential nephron blockade with a thiazide or acetazolamide), and they highlight the lingering uncertainty regarding inotrope use.
VENOUS CONGESTION VS DECREASED CARDIAC OUTPUT
Returning to the GFR equation, it is clear that an imbalance in PG and PB can worsen glomerular function. Because cardiac dysfunction can lead to both venous congestion and decreased cardiac output, this leads to the question, “Of these, which is the more important driver of this imbalance and its effects on renal function?”
A compelling argument can be made for each side. On one hand, experiments over a half-century old in human models of venous congestion highlighted the profound impact of elevated venous pressure, which decreases electrolyte excretion (sodium included) and diminishes urine flow.4,5 This has been replicated in more-contemporary decompensated heart failure cohorts in which worsening renal function was more closely associated with elevated central venous pressure rather than cardiac output.6,7 On the other hand, early landmark experiments and more recent cohorts with heart failure have also shown that reductions in effective arterial blood volume, renal blood flow, and cardiac output are also associated with reductions in GFR.5,8,9
How then shall we reconcile whether cardiorenal syndrome is a “backward failure” (from central venous pressure) or a “forward failure” (from decreased perfusion) phenomenon?
The answer is complicated and is likely “both,” with the major component being increased central venous pressure. To understand this construct, we must first exclude frank cardiogenic shock—when the hydraulic function of the heart fails to provide enough flow, leading to a catastrophic drop in mean arterial pressure that supersedes the kidney’s ability to autoregulate renal blood flow.10,11
In patients with chronic heart failure and congestion who are not in shock, historical observations suggest that both intra-abdominal pressure (which increases renal venous pressure) and central venous pressure lead to reduced renal blood flow and increased renal vasomotor resistance (increase in afferent, intrarenal, and efferent vascular tone).12–14 More recent observations from epidemiologic studies have largely replicated these findings. Central venous pressure remains essential to impacting renal function in heart failure,6,15 and the impact of cardiac output on renal function remains uncertain.16
The relationship of intracardiac hemodynamics may also play a role in modifying renal function. Several reports recently described the relationship between both right- and left-sided filling pressures as being associated with worse renal function in heart failure.17–19 Patients with a disproportionately higher right atrial pressure to pulmonary capillary wedge pressure have higher serum creatinine during and after decongestive therapies. Therefore, the concept of “right-sided heart failure” expands beyond the simple representation of “backward congestion” at the level of venous return. In fact, a higher ratio of right atrial pressure to pulmonary capillary wedge pressure may point to an inability of the venous and pulmonary circulations to provide adequate left ventricular preload. Therefore, a relatively underfilled left ventricle in the face of biventricular dysfunction may result in worsening renal function.
TREATMENT IS CHALLENGING
The treatment of cardiorenal syndrome is challenging. It is often accompanied by heightened azotemia, diuretic resistance, electrolyte abnormalities, and a spectrum of hemodynamic disarray. As Thind et al point out, there is, unfortunately, no firmly established treatment. While “sequential nephron blockade” (pharmacologically blocking multiple sites on the nephron simultaneously) is theoretically promising, there are no rigorously studied therapeutic strategies with proven efficacy.
On the other hand, mechanical removal of isotonic fluid with ultrafiltration showed early promise in decompensated heart failure, but enthusiasm diminished with results from the Cardiorenal Rescue Study in Acute Decompensated Heart Failure (CARRESS-HF) trial.20 Ultrafiltration was roughly equivalent to aggressive pharmacologic therapy for fluid loss, was associated with higher serum creatinine levels, and was more challenging to administer.
Equally uncertain is the benefit of inotropic or vasoactive therapy, which directly alters cardiac hemodynamics. Low-dose dopamine or low-dose nesiritide is of no benefit toward enhancement of decongestion or renal protection when added to standard diuretic therapy.21 Furthermore, routine use of inotropes is fraught with more arrhythmias and hypotension and is associated with dismal long-term outcomes.22,23
Alternative therapies that act directly on renal physiology—eg, rolofylline, a selective adenosine A1 receptor antagonist that may enhance renal blood flow, augment natriuresis, and break diuretic resistance—have been similarly disappointing.24
With so much uncertainty, more investigation into novel treatments for cardiorenal syndrome is clearly warranted.
However, because venous congestion is the hemodynamic hallmark of acute cardiorenal syndrome (increasing PB), reducing central venous pressure remains the cornerstone treatment for cardiorenal syndrome. Additionally, efforts to preserve renal perfusion and avoid hypotension are prudent to maintain glomerular capillary hydrostatic pressure (PG).
In light of these considerations, there is no “one size fits all” for the treatment of cardiorenal syndrome. Treatment should be based on thoughtful individualized strategies tailored to the underlying cardiorenal pathophysiology, and with the understanding that the kidney is at the heart of the matter.
In heart failure, the heart and the kidneys share a rocky relationship. Cardiac dysfunction can heighten renal dysfunction and vice versa—appropriately dubbed “cardiorenal syndrome.”
Although classically defined by a reduction in the glomerular filtration rate (GFR),1 cardiorenal syndrome also encompasses complex neurohormonal, pharmacologic, and metabolic interactions affecting or affected by both glomerular and tubular function. Unfortunately, all of these maladaptive processes occur in heart failure and perpetuate a vicious circle of continued dual-organ dysfunction.
The central insult here is hemodynamic disarray from acute or chronic cardiac dysfunction, which can directly influence glomerular function. However, to understand the hemodynamic ramifications for glomerular function, we focus on the determinants of glomerular filtration.
DETERMINANTS OF GFR
The GFR is the rate of fluid flow between the glomerular capillaries and the Bowman capsule and is classically represented by the following equations2:
GFR = Kf × (PG – PB – πG + πB)
Kf = N × Lp × S
Kf is the filtration constant, N the number of functional nephrons, Lp the hydraulic conductivity of the glomerular capillary, S the filtration area, PG the hydrostatic pressure in the glomerular capillaries, PB the hydrostatic pressure in the Bowman capsule, and πG and πB the colloid osmotic pressures within the glomerular capillaries and Bowman space, respectively.
Based on this relationship, the GFR is reduced when PG is reduced in the setting of hypovolemia, hypotension, or renin-angiotensin system antagonist use or when PB is increased in the setting of elevated central venous pressure or elevated abdominal pressure—all common in heart failure. With this understanding, one would assume that strategies to increase PG (improve perfusion) and reduce PB (reduce congestion) might ameliorate ongoing renal dysfunction and improve the GFR in heart failure.
In this issue, Thind et al3 highlight the impact of hemodynamic derangements in heart failure with acute cardiorenal syndrome and provide an overview of its treatment. They review the complex relationship between progressive cardiac failure translating into accelerated neurohormonal responses (increases in sympathetic nervous system and renin-angiotensin-aldosterone system activation) and the impact of increased central venous pressure on progressive renal dysfunction. They also provide an overview of efforts to mitigate cardiorenal syndrome, after careful appraisal of volume status, through diuretic-mediated decongestion with aggressive use of loop diuretics (either in isolation or in the form of sequential nephron blockade with a thiazide or acetazolamide), and they highlight the lingering uncertainty regarding inotrope use.
VENOUS CONGESTION VS DECREASED CARDIAC OUTPUT
Returning to the GFR equation, it is clear that an imbalance in PG and PB can worsen glomerular function. Because cardiac dysfunction can lead to both venous congestion and decreased cardiac output, this leads to the question, “Of these, which is the more important driver of this imbalance and its effects on renal function?”
A compelling argument can be made for each side. On one hand, experiments over a half-century old in human models of venous congestion highlighted the profound impact of elevated venous pressure, which decreases electrolyte excretion (sodium included) and diminishes urine flow.4,5 This has been replicated in more-contemporary decompensated heart failure cohorts in which worsening renal function was more closely associated with elevated central venous pressure rather than cardiac output.6,7 On the other hand, early landmark experiments and more recent cohorts with heart failure have also shown that reductions in effective arterial blood volume, renal blood flow, and cardiac output are also associated with reductions in GFR.5,8,9
How then shall we reconcile whether cardiorenal syndrome is a “backward failure” (from central venous pressure) or a “forward failure” (from decreased perfusion) phenomenon?
The answer is complicated and is likely “both,” with the major component being increased central venous pressure. To understand this construct, we must first exclude frank cardiogenic shock—when the hydraulic function of the heart fails to provide enough flow, leading to a catastrophic drop in mean arterial pressure that supersedes the kidney’s ability to autoregulate renal blood flow.10,11
In patients with chronic heart failure and congestion who are not in shock, historical observations suggest that both intra-abdominal pressure (which increases renal venous pressure) and central venous pressure lead to reduced renal blood flow and increased renal vasomotor resistance (increase in afferent, intrarenal, and efferent vascular tone).12–14 More recent observations from epidemiologic studies have largely replicated these findings. Central venous pressure remains essential to impacting renal function in heart failure,6,15 and the impact of cardiac output on renal function remains uncertain.16
The relationship of intracardiac hemodynamics may also play a role in modifying renal function. Several reports recently described the relationship between both right- and left-sided filling pressures as being associated with worse renal function in heart failure.17–19 Patients with a disproportionately higher right atrial pressure to pulmonary capillary wedge pressure have higher serum creatinine during and after decongestive therapies. Therefore, the concept of “right-sided heart failure” expands beyond the simple representation of “backward congestion” at the level of venous return. In fact, a higher ratio of right atrial pressure to pulmonary capillary wedge pressure may point to an inability of the venous and pulmonary circulations to provide adequate left ventricular preload. Therefore, a relatively underfilled left ventricle in the face of biventricular dysfunction may result in worsening renal function.
TREATMENT IS CHALLENGING
The treatment of cardiorenal syndrome is challenging. It is often accompanied by heightened azotemia, diuretic resistance, electrolyte abnormalities, and a spectrum of hemodynamic disarray. As Thind et al point out, there is, unfortunately, no firmly established treatment. While “sequential nephron blockade” (pharmacologically blocking multiple sites on the nephron simultaneously) is theoretically promising, there are no rigorously studied therapeutic strategies with proven efficacy.
On the other hand, mechanical removal of isotonic fluid with ultrafiltration showed early promise in decompensated heart failure, but enthusiasm diminished with results from the Cardiorenal Rescue Study in Acute Decompensated Heart Failure (CARRESS-HF) trial.20 Ultrafiltration was roughly equivalent to aggressive pharmacologic therapy for fluid loss, was associated with higher serum creatinine levels, and was more challenging to administer.
Equally uncertain is the benefit of inotropic or vasoactive therapy, which directly alters cardiac hemodynamics. Low-dose dopamine or low-dose nesiritide is of no benefit toward enhancement of decongestion or renal protection when added to standard diuretic therapy.21 Furthermore, routine use of inotropes is fraught with more arrhythmias and hypotension and is associated with dismal long-term outcomes.22,23
Alternative therapies that act directly on renal physiology—eg, rolofylline, a selective adenosine A1 receptor antagonist that may enhance renal blood flow, augment natriuresis, and break diuretic resistance—have been similarly disappointing.24
With so much uncertainty, more investigation into novel treatments for cardiorenal syndrome is clearly warranted.
However, because venous congestion is the hemodynamic hallmark of acute cardiorenal syndrome (increasing PB), reducing central venous pressure remains the cornerstone treatment for cardiorenal syndrome. Additionally, efforts to preserve renal perfusion and avoid hypotension are prudent to maintain glomerular capillary hydrostatic pressure (PG).
In light of these considerations, there is no “one size fits all” for the treatment of cardiorenal syndrome. Treatment should be based on thoughtful individualized strategies tailored to the underlying cardiorenal pathophysiology, and with the understanding that the kidney is at the heart of the matter.
- House AA, Anand I, Bellomo R, et al; Acute Dialysis Quality Initiative Consensus Group. Definition and classification of cardio-renal syndromes: workgroup statements from the 7th ADQI Consensus Conference. Nephrol Dial Transplant 2010; 25:1416–1420.
- Tucker BJ, Blantz RC. An analysis of the determinants of nephron filtration rate. Am J Physiol 1977; 232:F477–F483.
- Thind GS, Loehrke M, Wilt JL. Acute cardiorenal syndrome: mechanisms and clinical implications. Cleve Clin J Med 2018; 85:231–239.
- Wilkins RW, Tinsley CM, Culbertson JW, et al. The effects of venous congestion of the limbs upon renal clearances and the excretion of water and salt. I. Studies in normal subjects and in hypertensive patients before and after splanchnicectomy. J Clin Invest 1953; 32:1101–1116.
- Judson WE, Hatcher JD, Hollander W, Halperin MH, Wilkins RW. The effects of venous congestion of the limbs and phlebotomy upon renal clearances and the excretion of water and salt. II. Studies in patients with congestive failure. J Clin Invest 1955; 34:1591–1599.
- Mullens W, Abrahams Z, Francis GS, et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol 2009; 53:589–596.
- Damman K, van Deursen VM, Navis G, Voors AA, van Veldhuisen DJ, Hillege HL. Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol 2009; 53:582–588.
- Ljungman S, Laragh JH, Cody RJ. Role of the kidney in congestive heart failure. Relationship of cardiac index to kidney function. Drugs 1990; 39(suppl 4):10–21; discussion 22–24.
- Damman K, Navis G, Smilde TD, et al. Decreased cardiac output, venous congestion and the association with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail 2007; 9:872–878.
- Fincke R, Hochman JS, Lowe AM, et al. Cardiac power is the strongest hemodynamic correlate of mortality in cardiogenic shock: a report from the SHOCK trial registry. J Am Coll Cardiol 2004; 44:340–348.
- Adams PL, Adams FF, Bell PD, Navar LG. Impaired renal blood flow autoregulation in ischemic acute renal failure. Kidney Int 1980; 18:68–76.
- Maxwell MH, Breed ES, Schwartz IL. Renal venous pressure in chronic congestive heart failure. J Clin Invest 1950; 29:342–348.
- Blake WD, Wégria R, Keating RP, Ward HP. Effect of increased renal venous pressure on renal function. Am J Physiol 1949; 157:1–13.
- Bradley SE, Bradley GP. The effect of increased intra-abdominal pressure on renal function in man. J Clin Invest 1947; 26:1010–1022.
- Mullens W, Abrahams Z, Skouri HN, et al. Elevated intra-abdominal pressure in acute decompensated heart failure: a potential contributor to worsening renal function? J Am Coll Cardiol 2008; 51:300–306.
- Hanberg JS, Sury K, Wilson FP, et al. Reduced cardiac index is not the dominant driver of renal dysfunction in heart failure. J Am Coll Cardiol 2016; 67:2199–2208.
- Drazner MH, Brown RN, Kaiser PA, et al. Relationship of right- and left-sided filling pressures in patients with advanced heart failure: a 14-year multi-institutional analysis. J Heart Lung Transplant 2012; 31:67–72.
- Drazner MH, Velez-Martinez M, Ayers CR, et al. Relationship of right- to left-sided ventricular filling pressures in advanced heart failure: insights from the ESCAPE trial. Circ Heart Fail 2013; 6:264–270.
- Grodin JL, Drazner MH, Dupont M, et al. A disproportionate elevation in right ventricular filling pressure, in relation to left ventricular filling pressure, is associated with renal impairment and increased mortality in advanced decompensated heart failure. Am Heart J 2015; 169:806–812.
- Bart BA, Goldsmith SR, Lee KL, et al, for the Heart Failure Clinical Research Network. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med 2012; 367:2296–2304.
- Chen HH, Anstrom KJ, Givertz MM, et al; NHLBI Heart Failure Clinical Research Network. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA 2013; 310:2533–2543.
- Gorodeski EZ, Chu EC, Reese JR, Shishehbor MH, Hsich E, Starling RC. Prognosis on chronic dobutamine or milrinone infusions for stage D heart failure. Circ Heart Fail 2009; 2:320–324.
- Cuffe MS, Califf RM, Adams KF Jr, et al, for the Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME-CHF) Investigators. Short-term intravenous milrinone for acute exacerbation of chronic heart failure: a randomized controlled trial. JAMA 2002; 287:1541–1547.
- Massie BM, O’Connor CM, Metra M, et al, for the PROTECT Investigators and Committees. Rolofylline, an adenosine A1-receptor antagonist, in acute heart failure. N Engl J Med 2010; 363:1419–1428.
- House AA, Anand I, Bellomo R, et al; Acute Dialysis Quality Initiative Consensus Group. Definition and classification of cardio-renal syndromes: workgroup statements from the 7th ADQI Consensus Conference. Nephrol Dial Transplant 2010; 25:1416–1420.
- Tucker BJ, Blantz RC. An analysis of the determinants of nephron filtration rate. Am J Physiol 1977; 232:F477–F483.
- Thind GS, Loehrke M, Wilt JL. Acute cardiorenal syndrome: mechanisms and clinical implications. Cleve Clin J Med 2018; 85:231–239.
- Wilkins RW, Tinsley CM, Culbertson JW, et al. The effects of venous congestion of the limbs upon renal clearances and the excretion of water and salt. I. Studies in normal subjects and in hypertensive patients before and after splanchnicectomy. J Clin Invest 1953; 32:1101–1116.
- Judson WE, Hatcher JD, Hollander W, Halperin MH, Wilkins RW. The effects of venous congestion of the limbs and phlebotomy upon renal clearances and the excretion of water and salt. II. Studies in patients with congestive failure. J Clin Invest 1955; 34:1591–1599.
- Mullens W, Abrahams Z, Francis GS, et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol 2009; 53:589–596.
- Damman K, van Deursen VM, Navis G, Voors AA, van Veldhuisen DJ, Hillege HL. Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol 2009; 53:582–588.
- Ljungman S, Laragh JH, Cody RJ. Role of the kidney in congestive heart failure. Relationship of cardiac index to kidney function. Drugs 1990; 39(suppl 4):10–21; discussion 22–24.
- Damman K, Navis G, Smilde TD, et al. Decreased cardiac output, venous congestion and the association with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail 2007; 9:872–878.
- Fincke R, Hochman JS, Lowe AM, et al. Cardiac power is the strongest hemodynamic correlate of mortality in cardiogenic shock: a report from the SHOCK trial registry. J Am Coll Cardiol 2004; 44:340–348.
- Adams PL, Adams FF, Bell PD, Navar LG. Impaired renal blood flow autoregulation in ischemic acute renal failure. Kidney Int 1980; 18:68–76.
- Maxwell MH, Breed ES, Schwartz IL. Renal venous pressure in chronic congestive heart failure. J Clin Invest 1950; 29:342–348.
- Blake WD, Wégria R, Keating RP, Ward HP. Effect of increased renal venous pressure on renal function. Am J Physiol 1949; 157:1–13.
- Bradley SE, Bradley GP. The effect of increased intra-abdominal pressure on renal function in man. J Clin Invest 1947; 26:1010–1022.
- Mullens W, Abrahams Z, Skouri HN, et al. Elevated intra-abdominal pressure in acute decompensated heart failure: a potential contributor to worsening renal function? J Am Coll Cardiol 2008; 51:300–306.
- Hanberg JS, Sury K, Wilson FP, et al. Reduced cardiac index is not the dominant driver of renal dysfunction in heart failure. J Am Coll Cardiol 2016; 67:2199–2208.
- Drazner MH, Brown RN, Kaiser PA, et al. Relationship of right- and left-sided filling pressures in patients with advanced heart failure: a 14-year multi-institutional analysis. J Heart Lung Transplant 2012; 31:67–72.
- Drazner MH, Velez-Martinez M, Ayers CR, et al. Relationship of right- to left-sided ventricular filling pressures in advanced heart failure: insights from the ESCAPE trial. Circ Heart Fail 2013; 6:264–270.
- Grodin JL, Drazner MH, Dupont M, et al. A disproportionate elevation in right ventricular filling pressure, in relation to left ventricular filling pressure, is associated with renal impairment and increased mortality in advanced decompensated heart failure. Am Heart J 2015; 169:806–812.
- Bart BA, Goldsmith SR, Lee KL, et al, for the Heart Failure Clinical Research Network. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med 2012; 367:2296–2304.
- Chen HH, Anstrom KJ, Givertz MM, et al; NHLBI Heart Failure Clinical Research Network. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA 2013; 310:2533–2543.
- Gorodeski EZ, Chu EC, Reese JR, Shishehbor MH, Hsich E, Starling RC. Prognosis on chronic dobutamine or milrinone infusions for stage D heart failure. Circ Heart Fail 2009; 2:320–324.
- Cuffe MS, Califf RM, Adams KF Jr, et al, for the Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME-CHF) Investigators. Short-term intravenous milrinone for acute exacerbation of chronic heart failure: a randomized controlled trial. JAMA 2002; 287:1541–1547.
- Massie BM, O’Connor CM, Metra M, et al, for the PROTECT Investigators and Committees. Rolofylline, an adenosine A1-receptor antagonist, in acute heart failure. N Engl J Med 2010; 363:1419–1428.

Which test for CAD should be used in patients with left bundle branch block?
A 62-year-old woman with hypertension and type 2 diabetes mellitus has been experiencing shortness of breath on exertion and chest discomfort for 2 months. Her hypertension has been suboptimally controlled, and her most recent hemoglobin A1c measurement was 7.0%. She has never smoked and has no family history of premature coronary artery disease (CAD). She is otherwise well and walks for 30 minutes 3 times per week. A 12-lead electrocardiogram demonstrated normal sinus rhythm with left bundle branch block. Her physician suspects she has CAD. What testing does this patient need?
LIMITED DATA, GUIDELINES
For clinicians investigating suspected obstructive CAD in patients with left bundle branch block on resting electrocardiography, the data and guidelines are limited regarding the optimal noninvasive tests and how to interpret them.
Here, we present a practical review of the diagnostic utility of exercise stress electrocardiography, exercise stress echocardiography, dobutamine stress echocardiography, nuclear myocardial perfusion imaging, and computed tomographic (CT) angiography for assessing suspected obstructive CAD in patients with resting left bundle branch block.
WHAT IS LEFT BUNDLE BRANCH BLOCK?
In left bundle branch block, as the name implies, electrical conduction along the left bundle branch is blocked or delayed. Ventricular activation therefore begins in the right ventricle and the right side of the interventricular septum.1 Transseptal activation from the right ventricle to the left ventricle is slow, because it is transmyocardial.1 Left ventricular basal and posterolateral wall segments become activated last.1 Due to delay in the onset of left ventricular contraction, ventricular contraction is dyssynchronous. Classically, interventricular septal motion during systole has been described as paradoxical, with anterior septal motion.2–4

On electrocardiography, the QRS duration is widened (≥ 120 ms), with a distinctive morphology as shown in Figure 1. Left bundle branch block makes it difficult to accurately assess for dynamic ST-segment changes with exercise, rendering exercise stress electrocardiography a suboptimal test for obstructive CAD if left bundle branch block is present.
LEFT BUNDLE BRANCH BLOCK AND RISK OF DEATH
Although left bundle branch block can be an isolated finding, it can also be associated with underlying obstructive CAD5 or cardiomyopathy.6 When it occurs at rest, the risk of death from a cardiovascular event is 3 to 4 times higher.7 However, the exact incidence of significant obstructive CAD in asymptomatic patients with incidentally detected left bundle branch block is unknown.
Acute left bundle branch block accompanying acute myocardial infarction is associated with a high risk of death. Hindman et al,8 in a 1978 multicenter study, described 432 patients with acute myocardial infarction and left or right bundle branch block. In the 163 patients who had left bundle branch block, the in-hospital mortality rate was 24% and the 1-year mortality rate was 32%.
Freedman et al9 in 1987 reviewed 15,609 patients with chronic CAD who underwent coronary angiography, of whom 522 had left or right bundle branch block. During a follow-up of nearly 5 years, 2,386 patients died. The actuarial probability of death at 2 years in patients with left bundle branch block was more than 5 times that of patients without it (P < .0001).
During 18 years of observation in the Framingham study,10 55 participants developed left bundle branch block, at a mean age at onset of 62. Twenty-six (48%) of these participants developed clinically significant CAD or heart failure coincident with or subsequent to the onset of left bundle branch block. Fifty percent of the participants who developed left bundle branch block died of cardiovascular disease within 10 years of its onset.
EXERCISE STRESS ELECTROCARDIOGRAPHY
Exercise stress electrocardiography, although valuable for assessing functional capacity, cannot be used to diagnose obstructive CAD in patients with left bundle branch block.11
EXERCISE STRESS ECHOCARDIOGRAPHY
Exercise stress echocardiography is proven and widely used for assessing myocardial ischemia in patients with suspected obstructive CAD. But the data are limited on its diagnostic utility in patients with left bundle branch block. Until recently, recommendations for its use in this situation were based on only 1 small study.12
Peteiro et al12 in 2000 described 35 patients who underwent exercise stress echocardiography and coronary angiography. Detection of wall-motion abnormalities had high sensitivity (76%), specificity (83%), and diagnostic accuracy (80%).
Of note, 8 (23%) of the patients could not achieve at least 85% of the maximum predicted heart rate, and for them, the study was not diagnostic for ischemia. (Technically, the study is said to be nondiagnostic when the patient fails to achieve the target heart rate of at least 85% of the maximum predicted heart rate.)
Additionally, 18 of the 35 patients—over half—had a decrease in left ventricular ejection fraction in response to exercise. These 18 patients included 12 of the 17 patients with obstructive CAD and 6 of the 18 patients without obstructive CAD.12 It is unclear whether a significant proportion of these 18 patients would have been otherwise categorized as having a globally abnormal left ventricular contractile response to exercise according to contemporary (2007) reporting standards.13
Xu et al14,15 in 2016 examined the diagnostic utility of exercise stress echocardiography in assessing suspected obstructive CAD in 191 patients with resting left bundle branch block; 17 patients who failed to achieve a heart rate of at least 85% of the age-predicted maximum heart rate were excluded. Of the remaining 174 patients, 82 demonstrated a normal left ventricular contractile response to exercise and 92 had an abnormal response. In the abnormal group, 70 patients had a globally abnormal response, and 22 patients had a regional ischemic response. Of those who had a globally abnormal left ventricular contractile response who subsequently underwent angiography, only 30% were found to have obstructive CAD.
Although the sensitivity of exercise stress echocardiography was high (94%), its specificity and diagnostic accuracy were poor (specificity 21%, diagnostic accuracy 52%).14,15 These results suggest that for patients with resting left bundle branch block undergoing exercise stress echocardiography, obstructive CAD cannot be reliably diagnosed in those who develop a globally abnormal left ventricular contractile response. Therefore, an alternative imaging strategy should be considered.
DOBUTAMINE STRESS ECHOCARDIOGRAPHY
The evidence base for dobutamine stress echocardiography in patients with left bundle branch block is more robust than that for exercise stress echocardiography.
Geleijnse et al1 studied 64 patients with left bundle branch block undergoing dobutamine stress echocardiography who also underwent coronary angiography. Dobutamine stress echocardiography was moderately sensitive for detecting anterior and posterior myocardial wall ischemia (60% and 67%, respectively). Its specificity and diagnostic accuracy were high, at 94% and 98%, respectively.
Yanik et al16 studied 30 patients with left bundle branch block undergoing both dobutamine stress echocardiography and coronary angiography. The sensitivity of dobutamine stress echocardiography for identifying ischemia in the left anterior descending territory was 82%, the specificity was 95%, and the diagnostic accuracy was 90%. For identifying ischemia in the circumflex and right coronary artery territories, the sensitivity was 88%, specificity 96%, and accuracy 93%.
Mairesse et al17 studied 24 patients with left bundle branch block undergoing dobutamine stress echocardiography, myocardial perfusion tomography, and coronary angiography. Dobutamine stress echocardiography performed well in detecting ischemia in the left anterior descending territory, with a sensitivity of 83%, specificity 92%, and diagnostic accuracy 87%.
Of note, the available data come from very small studies published more than 15 years ago, and pharmacologic stress testing cannot provide the very important prognostic information derived from treadmill testing.
NUCLEAR MYOCARDIAL PERFUSION IMAGING

Exercise nuclear single-photon emission computed tomography (SPECT) myocardial perfusion imaging in patients with left bundle branch block is challenging, due to the development of septal perfusion defects at rest and during exercise in the absence of obstructive disease in the left anterior descending artery (Figure 2).18,19 Asynchronous contraction of the septum, with resulting compression of the septal arteries, decreased flow demands to the septal region, and attenuation artifacts are possible explanations for this phenomenon.20
Pharmacologic stress has been reported to improve the diagnostic accuracy of SPECT myocardial perfusion imaging.21
Biagini et al,21 in a meta-analysis of noninvasive techniques for diagnosing CAD in patients with left bundle branch block, found 1,785 patients from 39 studies who underwent nuclear myocardial perfusion imaging (48.8% with exercise, 41.9% with pharmacologic stress). Overall, sensitivity was high for both exercise and pharmacologic stress (92.9% and 88.5%). However, the reported specificity with exercise stress was significantly lower than with pharmacologic stress (23.3% vs 74.2%, P < .01).
Nuclear positron-emission tomography (PET) may further improve the diagnostic utility of nuclear myocardial perfusion imaging in patients with left bundle branch block. In a study of 440 patients with left bundle branch block undergoing myocardial perfusion imaging, 67 underwent PET and 373 underwent SPECT.22 Possible septal perfusion artifacts were significantly less common with PET than with SPECT (1.5% vs 19.3%, P < .001).
CT ANGIOGRAPHY
CT angiography has a high sensitivity and specificity for detecting significant obstructive CAD.23,24 Machines with 320 detector rows have been reported to have a sensitivity of 94% and specificity of 87% for detecting significant CAD and are not affected by resting left bundle branch block.25
Of note, coronary artery calcification increases in older patients, especially those age 65 and older,26 and this confers a higher likelihood of “bystander” CAD. Significant coronary artery calcification limits the diagnostic accuracy of multidetector cardiac CT. Additionally, the detection of bystander CAD leads to positive findings of uncertain clinical significance.
CURRENT GUIDELINES
Exercise stress echocardiography
American College of Cardiology Foundation/American Heart Association guidelines for diagnosis and management of patients with stable ischemic heart disease recommend exercise stress echocardiography for patients with an intermediate to high pretest probability of ischemic heart disease who have an uninterpretable electrocardiogram and at least moderate physical functioning or no disabling comorbidity (class 1 indication, level of evidence B).11
Current American Society of Echocardiography guidelines also support exercise stress echocardiography as an appropriate test for suspected obstructive CAD in patients with resting left bundle branch block.27 However, this recommendation is based on limited data.
Pharmacologic stress nuclear myocardial perfusion imaging
American Society of Nuclear Cardiology guidelines endorse pharmacologic stress nuclear myocardial perfusion imaging using coronary vasodilators for evaluating suspected obstructive CAD in patients with resting left bundle branch block.28,29
THE POSSIBLE HARMS OF TESTING
Although current guidelines recommend it, recent data show that exercise stress echocardiography has poor specificity and diagnostic accuracy for significant obstructive CAD in patients with resting left bundle branch block. And performing this test in patients with left bundle branch block may result in further downstream investigations.
Based on limited data from a small number of studies published more than 15 years ago, dobutamine stress echocardiography has moderate sensitivity and specificity for significant CAD in patients with resting left bundle branch block. However, this test does not provide functional information about the patient’s exercise performance.
Pharmacologic stress nuclear myocardial perfusion imaging using coronary vasodilators is an appropriate investigation strategy. However, radiation exposure is a limitation.30
CT angiography can assess for significant obstructive CAD in patients with resting left bundle branch block. However, its diagnostic accuracy can be affected by coronary calcification in older patients. Additionally, each scan is associated with a small amount of radiation exposure,31 and a small number of patients will have a true contrast allergy.32
CLINICAL BOTTOM LINE

For patients with typical ischemic symptoms and new left bundle branch block on electrocardiography, specialist cardiology consultation should be sought, with consideration given to proceeding directly to coronary angiography. For stable outpatients, we propose the following diagnostic approach (Figure 3).
Exercise stress echocardiography is recommended by current guidelines, but it cannot reliably detect significant obstructive CAD in patients with resting left bundle branch block—its specificity and diagnostic accuracy are poor.14,15 Alternative imaging strategies include CT angiography, pharmacologic nuclear myocardial perfusion imaging using coronary vasodilators, and dobutamine stress echocardiography.
For investigating suspected obstructive CAD in patients with resting left bundle branch block, we propose CT angiography as the first-line imaging test for patients under age 65 and pharmacologic stress nuclear myocardial perfusion imaging using coronary vasodilators or dobutamine stress echocardiography for those age 65 and older. For patients who cannot tolerate contrast due to renal impairment or who have a true contrast allergy, pharmacologic nuclear myocardial perfusion imaging using coronary vasodilators and dobutamine stress echocardiography may be used as alternatives.
- Geleijnse ML, Vigna C, Kasprzak JD, et al. Usefulness and limitations of dobutamine-atropine stress echocardiography for the diagnosis of coronary artery disease in patients with left bundle branch block. A multicentre study. Eur Heart J 2000; 21:1666–1673.
- Dillon JC, Chang S, Feigenbaum H. Echocardiographic manifestations of left bundle branch block. Circulation 1974; 49:876–880.
- Abbasi AS, Eber LM, Macalpin RN, Kattus AA. Paradoxical motion of interventricular septum in left bundle branch block. Circulation 1974; 49:423–427.
- McDonald IG. Echocardiographic demonstration of abnormal motion of the interventricular septum in left bundle branch block. Circulation 1973; 48:272–280.
- Bouzas-Mosquera A, Peteiro J, Alvarez-García N, et al. Prognostic value of exercise echocardiography in patients with left bundle branch block. JACC Cardiovasc Imaging 2009; 2:251–259.
- Vaillant C, Martins RP, Donal E, et al. Resolution of left bundle branch block-induced cardiomyopathy by cardiac resynchronization therapy. J Am Coll Cardiol 2013; 61:1089–1095.
- Schneider JF, Thomas HE Jr, Sorlie P, Kreger BE, McNamara PM, Kannel WB. Comparative features of newly acquired left and right bundle branch block in the general population: the Framingham study. Am J Cardiol 1981; 47:931–940.
- Hindman MC, Wagner GS, JaRo M, et al. The clinical significance of bundle branch block complicating acute myocardial infarction. Circulation 1978; 58:689–699.
- Freedman RA, Alderman EL, Sheffield LT, Saporito M, Fisher LD. Bundle branch block in patients with chronic coronary artery disease: angiographic correlates and prognostic significance. J Am Coll Cardiol 1987; 10:73–80.
- Schneider JF, Thomas HE Jr, Kreger BE, McNamara PM, Kannel WB. Newly acquired left bundle-branch block: the Framingham study. Ann Intern Med 1979; 90:303–310.
- Fihn SD, Gardin JM, Abrams J, et al. 2012 ACCF/AHA/ACP/AATS/PCNA/SCAI/STS guideline for the diagnosis and management of patients with stable ischemic heart disease: executive summary. J Am Coll Cardiol 2012; 60:2564–2603.
- Peteiro J, Monserrat L, Martinez D, Castro-Beiras A. Accuracy of exercise echocardiography to detect coronary artery disease in left bundle branch block unassociated with either acute or healed myocardial infarction. Am J Cardiol 2000; 85:890–893, A9.
- Pellikka PA, Nagueh SF, Elhendy AA, Kuehl CA, Sawada SG; American Society of Echocardiography. American Society of Echocardiography recommendations for performance, interpretation, and application of stress echocardiography. J Am Soc Echocardiogr 2007; 20:1021–1041.
- Xu B, Dobson L, Mottram P, Moir S. Is exercise stress echocardiography useful in patients with suspected obstructive coronary artery disease who have resting left bundle branch block? J Am Coll Cardiol 2016; 67:1570.
- Xu B, Dobson L, Mottram P, Nasis A, Cameron J, Moir S. Is exercise stress echocardiography useful in patients with suspected obstructive coronary artery disease who have resting left bundle branch block? Clin Cardiol 2018; in press.
- Yanik A, Yetkin E, Senen K, et al. Value of dobutamine stress echocardiography for diagnosis of coronary artery disease in patients with left bundle branch. Coron Artery Dis 2000; 11:545–548.
- Mairesse GH, Marwick TH, Arnese M, et al. Improved identification of coronary artery disease in patients with left bundle branch block by use of dobutamine stress echocardiography and comparison with myocardial perfusion tomography. Am J Cardiol 1995; 76:321–325.
- Vaduganathan P, He ZX, Raghavan C, Mahmarian JJ, Verani MS. Detection of left anterior descending coronary artery stenosis in patients with left bundle branch block: exercise, adenosine or dobutamine imaging? J Am Coll Cardiol 1996; 28:543–550.
- Jazmati B, Sadaniantz A, Emaus SP, Heller GV. Exercise thallium-201 imaging in complete left bundle branch block and the prevalence of septal perfusion defects. Am J Cardiol 1991; 67:46–49.
- Hasegawa S, Sakata Y, Ishikura F, et al. Mechanism for abnormal thallium-201 myocardial scintigraphy in patients with left bundle branch block in the absence of angiographic coronary artery disease. Ann Nucl Med 1999; 13:253–259.
- Biagini E, Shaw LJ, Poldermans D, et al. Accuracy of non-invasive techniques for diagnosis of coronary artery disease and prediction of cardiac events in patients with left bundle branch block: a meta-analysis. Eur J Nucl Med Mol Imaging 2006; 33:1442–1451.
- Cremer P, Brunken R, Menon V, Cerqueira M, Jaber W. Septal perfusion abnormalities are common in regadenoson SPECT myocardial perfusion imaging (MPI) but not PET MPI in patients with left bundle branch block (LBBB). J Am Coll Cardiol 2015; 65:A1148.
- Arbab-Zadeh A, Miller JM, Rochitte CE, et al. Diagnostic accuracy of computed tomography coronary angiography according to pre-test probability of coronary artery disease and severity of coronary arterial calcification. The CORE-64 (Coronary Artery Evaluation Using 64-Row Multidetector Computed Tomography Angiography) International Multicenter Study. J Am Coll Cardiol 2012; 59:379–387.
- Chow BJ, Abraham A, Wells GA, et al. Diagnostic accuracy and impact of computed tomographic coronary angiography on utilization of invasive coronary angiography. Circ Cardiovasc Imaging 2009; 2:16–23.
- Nasis A, Leung MC, Antonis PR, et al. Diagnostic accuracy of noninvasive coronary angiography with 320-detector row computed tomography. Am J Cardiol 2010; 106:1429–1435.
- Whelton SP, Silverman MG, McEvoy JW, et al. Predictors of long-term healthy arterial aging: coronary artery calcium nondevelopment in the MESA study. JACC Cardiovasc Imaging 2015; 8:1393–1400.
- Douglas PS, Garcia MJ, Haines DE, et al. ACCF/ASE/AHA/ASNC/HFSA/HRS/SCAI/SCCM/SCCT/SCMR 2011 appropriate use criteria for echocardiography. J Am Soc Echocardiogr 2011; 24:229–267.
- Henzlova MJ, Duvall WL, Einstein AJ, Travin MI, Verberne HJ. ASNC imaging guidelines for SPECT nuclear cardiology procedures: Stress, protocols, and tracers. J Nucl Cardiol 2016; 23:606–639.
- Wolk MJ, Bailey SR, Doherty JU, et al. ACCF/AHA/ASE/ASNC/HFSA/HRS/SCAI/SCCT/SCMR/STS 2013 multimodality appropriate use criteria for the detection and risk assessment of stable ischemic heart disease. J Am Coll Cardiol 2014; 63:380–406.
- Cerqueira MD, Allman KC, Ficaro EP, et al. Recommendations for reducing radiation exposure in myocardial perfusion imaging. J Nucl Cardiol 2010; 17:709–718.
- Halliburton SS, Abbara S, Chen MY, et al; Society of Cardiovascular Computed Tomography. SCCT guidelines on radiation dose and dose-optimization strategies in cardiovascular CT. J Cardiovasc Comput Tomogr 2011; 5:198–224.
- Wang CL, Cohan RH, Ellis JH, Caoili EM, Wang G, Francis IR. Frequency, outcome, and appropriateness of treatment of nonionic iodinated contrast media reactions. AJR Am J Roentgenol 2008; 191:409–415.
A 62-year-old woman with hypertension and type 2 diabetes mellitus has been experiencing shortness of breath on exertion and chest discomfort for 2 months. Her hypertension has been suboptimally controlled, and her most recent hemoglobin A1c measurement was 7.0%. She has never smoked and has no family history of premature coronary artery disease (CAD). She is otherwise well and walks for 30 minutes 3 times per week. A 12-lead electrocardiogram demonstrated normal sinus rhythm with left bundle branch block. Her physician suspects she has CAD. What testing does this patient need?
LIMITED DATA, GUIDELINES
For clinicians investigating suspected obstructive CAD in patients with left bundle branch block on resting electrocardiography, the data and guidelines are limited regarding the optimal noninvasive tests and how to interpret them.
Here, we present a practical review of the diagnostic utility of exercise stress electrocardiography, exercise stress echocardiography, dobutamine stress echocardiography, nuclear myocardial perfusion imaging, and computed tomographic (CT) angiography for assessing suspected obstructive CAD in patients with resting left bundle branch block.
WHAT IS LEFT BUNDLE BRANCH BLOCK?
In left bundle branch block, as the name implies, electrical conduction along the left bundle branch is blocked or delayed. Ventricular activation therefore begins in the right ventricle and the right side of the interventricular septum.1 Transseptal activation from the right ventricle to the left ventricle is slow, because it is transmyocardial.1 Left ventricular basal and posterolateral wall segments become activated last.1 Due to delay in the onset of left ventricular contraction, ventricular contraction is dyssynchronous. Classically, interventricular septal motion during systole has been described as paradoxical, with anterior septal motion.2–4
On electrocardiography, the QRS duration is widened (≥ 120 ms), with a distinctive morphology as shown in Figure 1. Left bundle branch block makes it difficult to accurately assess for dynamic ST-segment changes with exercise, rendering exercise stress electrocardiography a suboptimal test for obstructive CAD if left bundle branch block is present.
LEFT BUNDLE BRANCH BLOCK AND RISK OF DEATH
Although left bundle branch block can be an isolated finding, it can also be associated with underlying obstructive CAD5 or cardiomyopathy.6 When it occurs at rest, the risk of death from a cardiovascular event is 3 to 4 times higher.7 However, the exact incidence of significant obstructive CAD in asymptomatic patients with incidentally detected left bundle branch block is unknown.
Acute left bundle branch block accompanying acute myocardial infarction is associated with a high risk of death. Hindman et al,8 in a 1978 multicenter study, described 432 patients with acute myocardial infarction and left or right bundle branch block. In the 163 patients who had left bundle branch block, the in-hospital mortality rate was 24% and the 1-year mortality rate was 32%.
Freedman et al9 in 1987 reviewed 15,609 patients with chronic CAD who underwent coronary angiography, of whom 522 had left or right bundle branch block. During a follow-up of nearly 5 years, 2,386 patients died. The actuarial probability of death at 2 years in patients with left bundle branch block was more than 5 times that of patients without it (P < .0001).
During 18 years of observation in the Framingham study,10 55 participants developed left bundle branch block, at a mean age at onset of 62. Twenty-six (48%) of these participants developed clinically significant CAD or heart failure coincident with or subsequent to the onset of left bundle branch block. Fifty percent of the participants who developed left bundle branch block died of cardiovascular disease within 10 years of its onset.
EXERCISE STRESS ELECTROCARDIOGRAPHY
Exercise stress electrocardiography, although valuable for assessing functional capacity, cannot be used to diagnose obstructive CAD in patients with left bundle branch block.11
EXERCISE STRESS ECHOCARDIOGRAPHY
Exercise stress echocardiography is proven and widely used for assessing myocardial ischemia in patients with suspected obstructive CAD. But the data are limited on its diagnostic utility in patients with left bundle branch block. Until recently, recommendations for its use in this situation were based on only 1 small study.12
Peteiro et al12 in 2000 described 35 patients who underwent exercise stress echocardiography and coronary angiography. Detection of wall-motion abnormalities had high sensitivity (76%), specificity (83%), and diagnostic accuracy (80%).
Of note, 8 (23%) of the patients could not achieve at least 85% of the maximum predicted heart rate, and for them, the study was not diagnostic for ischemia. (Technically, the study is said to be nondiagnostic when the patient fails to achieve the target heart rate of at least 85% of the maximum predicted heart rate.)
Additionally, 18 of the 35 patients—over half—had a decrease in left ventricular ejection fraction in response to exercise. These 18 patients included 12 of the 17 patients with obstructive CAD and 6 of the 18 patients without obstructive CAD.12 It is unclear whether a significant proportion of these 18 patients would have been otherwise categorized as having a globally abnormal left ventricular contractile response to exercise according to contemporary (2007) reporting standards.13
Xu et al14,15 in 2016 examined the diagnostic utility of exercise stress echocardiography in assessing suspected obstructive CAD in 191 patients with resting left bundle branch block; 17 patients who failed to achieve a heart rate of at least 85% of the age-predicted maximum heart rate were excluded. Of the remaining 174 patients, 82 demonstrated a normal left ventricular contractile response to exercise and 92 had an abnormal response. In the abnormal group, 70 patients had a globally abnormal response, and 22 patients had a regional ischemic response. Of those who had a globally abnormal left ventricular contractile response who subsequently underwent angiography, only 30% were found to have obstructive CAD.
Although the sensitivity of exercise stress echocardiography was high (94%), its specificity and diagnostic accuracy were poor (specificity 21%, diagnostic accuracy 52%).14,15 These results suggest that for patients with resting left bundle branch block undergoing exercise stress echocardiography, obstructive CAD cannot be reliably diagnosed in those who develop a globally abnormal left ventricular contractile response. Therefore, an alternative imaging strategy should be considered.
DOBUTAMINE STRESS ECHOCARDIOGRAPHY
The evidence base for dobutamine stress echocardiography in patients with left bundle branch block is more robust than that for exercise stress echocardiography.
Geleijnse et al1 studied 64 patients with left bundle branch block undergoing dobutamine stress echocardiography who also underwent coronary angiography. Dobutamine stress echocardiography was moderately sensitive for detecting anterior and posterior myocardial wall ischemia (60% and 67%, respectively). Its specificity and diagnostic accuracy were high, at 94% and 98%, respectively.
Yanik et al16 studied 30 patients with left bundle branch block undergoing both dobutamine stress echocardiography and coronary angiography. The sensitivity of dobutamine stress echocardiography for identifying ischemia in the left anterior descending territory was 82%, the specificity was 95%, and the diagnostic accuracy was 90%. For identifying ischemia in the circumflex and right coronary artery territories, the sensitivity was 88%, specificity 96%, and accuracy 93%.
Mairesse et al17 studied 24 patients with left bundle branch block undergoing dobutamine stress echocardiography, myocardial perfusion tomography, and coronary angiography. Dobutamine stress echocardiography performed well in detecting ischemia in the left anterior descending territory, with a sensitivity of 83%, specificity 92%, and diagnostic accuracy 87%.
Of note, the available data come from very small studies published more than 15 years ago, and pharmacologic stress testing cannot provide the very important prognostic information derived from treadmill testing.
NUCLEAR MYOCARDIAL PERFUSION IMAGING
Exercise nuclear single-photon emission computed tomography (SPECT) myocardial perfusion imaging in patients with left bundle branch block is challenging, due to the development of septal perfusion defects at rest and during exercise in the absence of obstructive disease in the left anterior descending artery (Figure 2).18,19 Asynchronous contraction of the septum, with resulting compression of the septal arteries, decreased flow demands to the septal region, and attenuation artifacts are possible explanations for this phenomenon.20
Pharmacologic stress has been reported to improve the diagnostic accuracy of SPECT myocardial perfusion imaging.21
Biagini et al,21 in a meta-analysis of noninvasive techniques for diagnosing CAD in patients with left bundle branch block, found 1,785 patients from 39 studies who underwent nuclear myocardial perfusion imaging (48.8% with exercise, 41.9% with pharmacologic stress). Overall, sensitivity was high for both exercise and pharmacologic stress (92.9% and 88.5%). However, the reported specificity with exercise stress was significantly lower than with pharmacologic stress (23.3% vs 74.2%, P < .01).
Nuclear positron-emission tomography (PET) may further improve the diagnostic utility of nuclear myocardial perfusion imaging in patients with left bundle branch block. In a study of 440 patients with left bundle branch block undergoing myocardial perfusion imaging, 67 underwent PET and 373 underwent SPECT.22 Possible septal perfusion artifacts were significantly less common with PET than with SPECT (1.5% vs 19.3%, P < .001).
CT ANGIOGRAPHY
CT angiography has a high sensitivity and specificity for detecting significant obstructive CAD.23,24 Machines with 320 detector rows have been reported to have a sensitivity of 94% and specificity of 87% for detecting significant CAD and are not affected by resting left bundle branch block.25
Of note, coronary artery calcification increases in older patients, especially those age 65 and older,26 and this confers a higher likelihood of “bystander” CAD. Significant coronary artery calcification limits the diagnostic accuracy of multidetector cardiac CT. Additionally, the detection of bystander CAD leads to positive findings of uncertain clinical significance.
CURRENT GUIDELINES
Exercise stress echocardiography
American College of Cardiology Foundation/American Heart Association guidelines for diagnosis and management of patients with stable ischemic heart disease recommend exercise stress echocardiography for patients with an intermediate to high pretest probability of ischemic heart disease who have an uninterpretable electrocardiogram and at least moderate physical functioning or no disabling comorbidity (class 1 indication, level of evidence B).11
Current American Society of Echocardiography guidelines also support exercise stress echocardiography as an appropriate test for suspected obstructive CAD in patients with resting left bundle branch block.27 However, this recommendation is based on limited data.
Pharmacologic stress nuclear myocardial perfusion imaging
American Society of Nuclear Cardiology guidelines endorse pharmacologic stress nuclear myocardial perfusion imaging using coronary vasodilators for evaluating suspected obstructive CAD in patients with resting left bundle branch block.28,29
THE POSSIBLE HARMS OF TESTING
Although current guidelines recommend it, recent data show that exercise stress echocardiography has poor specificity and diagnostic accuracy for significant obstructive CAD in patients with resting left bundle branch block. And performing this test in patients with left bundle branch block may result in further downstream investigations.
Based on limited data from a small number of studies published more than 15 years ago, dobutamine stress echocardiography has moderate sensitivity and specificity for significant CAD in patients with resting left bundle branch block. However, this test does not provide functional information about the patient’s exercise performance.
Pharmacologic stress nuclear myocardial perfusion imaging using coronary vasodilators is an appropriate investigation strategy. However, radiation exposure is a limitation.30
CT angiography can assess for significant obstructive CAD in patients with resting left bundle branch block. However, its diagnostic accuracy can be affected by coronary calcification in older patients. Additionally, each scan is associated with a small amount of radiation exposure,31 and a small number of patients will have a true contrast allergy.32
CLINICAL BOTTOM LINE
For patients with typical ischemic symptoms and new left bundle branch block on electrocardiography, specialist cardiology consultation should be sought, with consideration given to proceeding directly to coronary angiography. For stable outpatients, we propose the following diagnostic approach (Figure 3).
Exercise stress echocardiography is recommended by current guidelines, but it cannot reliably detect significant obstructive CAD in patients with resting left bundle branch block—its specificity and diagnostic accuracy are poor.14,15 Alternative imaging strategies include CT angiography, pharmacologic nuclear myocardial perfusion imaging using coronary vasodilators, and dobutamine stress echocardiography.
For investigating suspected obstructive CAD in patients with resting left bundle branch block, we propose CT angiography as the first-line imaging test for patients under age 65 and pharmacologic stress nuclear myocardial perfusion imaging using coronary vasodilators or dobutamine stress echocardiography for those age 65 and older. For patients who cannot tolerate contrast due to renal impairment or who have a true contrast allergy, pharmacologic nuclear myocardial perfusion imaging using coronary vasodilators and dobutamine stress echocardiography may be used as alternatives.
A 62-year-old woman with hypertension and type 2 diabetes mellitus has been experiencing shortness of breath on exertion and chest discomfort for 2 months. Her hypertension has been suboptimally controlled, and her most recent hemoglobin A1c measurement was 7.0%. She has never smoked and has no family history of premature coronary artery disease (CAD). She is otherwise well and walks for 30 minutes 3 times per week. A 12-lead electrocardiogram demonstrated normal sinus rhythm with left bundle branch block. Her physician suspects she has CAD. What testing does this patient need?
LIMITED DATA, GUIDELINES
For clinicians investigating suspected obstructive CAD in patients with left bundle branch block on resting electrocardiography, the data and guidelines are limited regarding the optimal noninvasive tests and how to interpret them.
Here, we present a practical review of the diagnostic utility of exercise stress electrocardiography, exercise stress echocardiography, dobutamine stress echocardiography, nuclear myocardial perfusion imaging, and computed tomographic (CT) angiography for assessing suspected obstructive CAD in patients with resting left bundle branch block.
WHAT IS LEFT BUNDLE BRANCH BLOCK?
In left bundle branch block, as the name implies, electrical conduction along the left bundle branch is blocked or delayed. Ventricular activation therefore begins in the right ventricle and the right side of the interventricular septum.1 Transseptal activation from the right ventricle to the left ventricle is slow, because it is transmyocardial.1 Left ventricular basal and posterolateral wall segments become activated last.1 Due to delay in the onset of left ventricular contraction, ventricular contraction is dyssynchronous. Classically, interventricular septal motion during systole has been described as paradoxical, with anterior septal motion.2–4
On electrocardiography, the QRS duration is widened (≥ 120 ms), with a distinctive morphology as shown in Figure 1. Left bundle branch block makes it difficult to accurately assess for dynamic ST-segment changes with exercise, rendering exercise stress electrocardiography a suboptimal test for obstructive CAD if left bundle branch block is present.
LEFT BUNDLE BRANCH BLOCK AND RISK OF DEATH
Although left bundle branch block can be an isolated finding, it can also be associated with underlying obstructive CAD5 or cardiomyopathy.6 When it occurs at rest, the risk of death from a cardiovascular event is 3 to 4 times higher.7 However, the exact incidence of significant obstructive CAD in asymptomatic patients with incidentally detected left bundle branch block is unknown.
Acute left bundle branch block accompanying acute myocardial infarction is associated with a high risk of death. Hindman et al,8 in a 1978 multicenter study, described 432 patients with acute myocardial infarction and left or right bundle branch block. In the 163 patients who had left bundle branch block, the in-hospital mortality rate was 24% and the 1-year mortality rate was 32%.
Freedman et al9 in 1987 reviewed 15,609 patients with chronic CAD who underwent coronary angiography, of whom 522 had left or right bundle branch block. During a follow-up of nearly 5 years, 2,386 patients died. The actuarial probability of death at 2 years in patients with left bundle branch block was more than 5 times that of patients without it (P < .0001).
During 18 years of observation in the Framingham study,10 55 participants developed left bundle branch block, at a mean age at onset of 62. Twenty-six (48%) of these participants developed clinically significant CAD or heart failure coincident with or subsequent to the onset of left bundle branch block. Fifty percent of the participants who developed left bundle branch block died of cardiovascular disease within 10 years of its onset.
EXERCISE STRESS ELECTROCARDIOGRAPHY
Exercise stress electrocardiography, although valuable for assessing functional capacity, cannot be used to diagnose obstructive CAD in patients with left bundle branch block.11
EXERCISE STRESS ECHOCARDIOGRAPHY
Exercise stress echocardiography is proven and widely used for assessing myocardial ischemia in patients with suspected obstructive CAD. But the data are limited on its diagnostic utility in patients with left bundle branch block. Until recently, recommendations for its use in this situation were based on only 1 small study.12
Peteiro et al12 in 2000 described 35 patients who underwent exercise stress echocardiography and coronary angiography. Detection of wall-motion abnormalities had high sensitivity (76%), specificity (83%), and diagnostic accuracy (80%).
Of note, 8 (23%) of the patients could not achieve at least 85% of the maximum predicted heart rate, and for them, the study was not diagnostic for ischemia. (Technically, the study is said to be nondiagnostic when the patient fails to achieve the target heart rate of at least 85% of the maximum predicted heart rate.)
Additionally, 18 of the 35 patients—over half—had a decrease in left ventricular ejection fraction in response to exercise. These 18 patients included 12 of the 17 patients with obstructive CAD and 6 of the 18 patients without obstructive CAD.12 It is unclear whether a significant proportion of these 18 patients would have been otherwise categorized as having a globally abnormal left ventricular contractile response to exercise according to contemporary (2007) reporting standards.13
Xu et al14,15 in 2016 examined the diagnostic utility of exercise stress echocardiography in assessing suspected obstructive CAD in 191 patients with resting left bundle branch block; 17 patients who failed to achieve a heart rate of at least 85% of the age-predicted maximum heart rate were excluded. Of the remaining 174 patients, 82 demonstrated a normal left ventricular contractile response to exercise and 92 had an abnormal response. In the abnormal group, 70 patients had a globally abnormal response, and 22 patients had a regional ischemic response. Of those who had a globally abnormal left ventricular contractile response who subsequently underwent angiography, only 30% were found to have obstructive CAD.
Although the sensitivity of exercise stress echocardiography was high (94%), its specificity and diagnostic accuracy were poor (specificity 21%, diagnostic accuracy 52%).14,15 These results suggest that for patients with resting left bundle branch block undergoing exercise stress echocardiography, obstructive CAD cannot be reliably diagnosed in those who develop a globally abnormal left ventricular contractile response. Therefore, an alternative imaging strategy should be considered.
DOBUTAMINE STRESS ECHOCARDIOGRAPHY
The evidence base for dobutamine stress echocardiography in patients with left bundle branch block is more robust than that for exercise stress echocardiography.
Geleijnse et al1 studied 64 patients with left bundle branch block undergoing dobutamine stress echocardiography who also underwent coronary angiography. Dobutamine stress echocardiography was moderately sensitive for detecting anterior and posterior myocardial wall ischemia (60% and 67%, respectively). Its specificity and diagnostic accuracy were high, at 94% and 98%, respectively.
Yanik et al16 studied 30 patients with left bundle branch block undergoing both dobutamine stress echocardiography and coronary angiography. The sensitivity of dobutamine stress echocardiography for identifying ischemia in the left anterior descending territory was 82%, the specificity was 95%, and the diagnostic accuracy was 90%. For identifying ischemia in the circumflex and right coronary artery territories, the sensitivity was 88%, specificity 96%, and accuracy 93%.
Mairesse et al17 studied 24 patients with left bundle branch block undergoing dobutamine stress echocardiography, myocardial perfusion tomography, and coronary angiography. Dobutamine stress echocardiography performed well in detecting ischemia in the left anterior descending territory, with a sensitivity of 83%, specificity 92%, and diagnostic accuracy 87%.
Of note, the available data come from very small studies published more than 15 years ago, and pharmacologic stress testing cannot provide the very important prognostic information derived from treadmill testing.
NUCLEAR MYOCARDIAL PERFUSION IMAGING
Exercise nuclear single-photon emission computed tomography (SPECT) myocardial perfusion imaging in patients with left bundle branch block is challenging, due to the development of septal perfusion defects at rest and during exercise in the absence of obstructive disease in the left anterior descending artery (Figure 2).18,19 Asynchronous contraction of the septum, with resulting compression of the septal arteries, decreased flow demands to the septal region, and attenuation artifacts are possible explanations for this phenomenon.20
Pharmacologic stress has been reported to improve the diagnostic accuracy of SPECT myocardial perfusion imaging.21
Biagini et al,21 in a meta-analysis of noninvasive techniques for diagnosing CAD in patients with left bundle branch block, found 1,785 patients from 39 studies who underwent nuclear myocardial perfusion imaging (48.8% with exercise, 41.9% with pharmacologic stress). Overall, sensitivity was high for both exercise and pharmacologic stress (92.9% and 88.5%). However, the reported specificity with exercise stress was significantly lower than with pharmacologic stress (23.3% vs 74.2%, P < .01).
Nuclear positron-emission tomography (PET) may further improve the diagnostic utility of nuclear myocardial perfusion imaging in patients with left bundle branch block. In a study of 440 patients with left bundle branch block undergoing myocardial perfusion imaging, 67 underwent PET and 373 underwent SPECT.22 Possible septal perfusion artifacts were significantly less common with PET than with SPECT (1.5% vs 19.3%, P < .001).
CT ANGIOGRAPHY
CT angiography has a high sensitivity and specificity for detecting significant obstructive CAD.23,24 Machines with 320 detector rows have been reported to have a sensitivity of 94% and specificity of 87% for detecting significant CAD and are not affected by resting left bundle branch block.25
Of note, coronary artery calcification increases in older patients, especially those age 65 and older,26 and this confers a higher likelihood of “bystander” CAD. Significant coronary artery calcification limits the diagnostic accuracy of multidetector cardiac CT. Additionally, the detection of bystander CAD leads to positive findings of uncertain clinical significance.
CURRENT GUIDELINES
Exercise stress echocardiography
American College of Cardiology Foundation/American Heart Association guidelines for diagnosis and management of patients with stable ischemic heart disease recommend exercise stress echocardiography for patients with an intermediate to high pretest probability of ischemic heart disease who have an uninterpretable electrocardiogram and at least moderate physical functioning or no disabling comorbidity (class 1 indication, level of evidence B).11
Current American Society of Echocardiography guidelines also support exercise stress echocardiography as an appropriate test for suspected obstructive CAD in patients with resting left bundle branch block.27 However, this recommendation is based on limited data.
Pharmacologic stress nuclear myocardial perfusion imaging
American Society of Nuclear Cardiology guidelines endorse pharmacologic stress nuclear myocardial perfusion imaging using coronary vasodilators for evaluating suspected obstructive CAD in patients with resting left bundle branch block.28,29
THE POSSIBLE HARMS OF TESTING
Although current guidelines recommend it, recent data show that exercise stress echocardiography has poor specificity and diagnostic accuracy for significant obstructive CAD in patients with resting left bundle branch block. And performing this test in patients with left bundle branch block may result in further downstream investigations.
Based on limited data from a small number of studies published more than 15 years ago, dobutamine stress echocardiography has moderate sensitivity and specificity for significant CAD in patients with resting left bundle branch block. However, this test does not provide functional information about the patient’s exercise performance.
Pharmacologic stress nuclear myocardial perfusion imaging using coronary vasodilators is an appropriate investigation strategy. However, radiation exposure is a limitation.30
CT angiography can assess for significant obstructive CAD in patients with resting left bundle branch block. However, its diagnostic accuracy can be affected by coronary calcification in older patients. Additionally, each scan is associated with a small amount of radiation exposure,31 and a small number of patients will have a true contrast allergy.32
CLINICAL BOTTOM LINE
For patients with typical ischemic symptoms and new left bundle branch block on electrocardiography, specialist cardiology consultation should be sought, with consideration given to proceeding directly to coronary angiography. For stable outpatients, we propose the following diagnostic approach (Figure 3).
Exercise stress echocardiography is recommended by current guidelines, but it cannot reliably detect significant obstructive CAD in patients with resting left bundle branch block—its specificity and diagnostic accuracy are poor.14,15 Alternative imaging strategies include CT angiography, pharmacologic nuclear myocardial perfusion imaging using coronary vasodilators, and dobutamine stress echocardiography.
For investigating suspected obstructive CAD in patients with resting left bundle branch block, we propose CT angiography as the first-line imaging test for patients under age 65 and pharmacologic stress nuclear myocardial perfusion imaging using coronary vasodilators or dobutamine stress echocardiography for those age 65 and older. For patients who cannot tolerate contrast due to renal impairment or who have a true contrast allergy, pharmacologic nuclear myocardial perfusion imaging using coronary vasodilators and dobutamine stress echocardiography may be used as alternatives.
- Geleijnse ML, Vigna C, Kasprzak JD, et al. Usefulness and limitations of dobutamine-atropine stress echocardiography for the diagnosis of coronary artery disease in patients with left bundle branch block. A multicentre study. Eur Heart J 2000; 21:1666–1673.
- Dillon JC, Chang S, Feigenbaum H. Echocardiographic manifestations of left bundle branch block. Circulation 1974; 49:876–880.
- Abbasi AS, Eber LM, Macalpin RN, Kattus AA. Paradoxical motion of interventricular septum in left bundle branch block. Circulation 1974; 49:423–427.
- McDonald IG. Echocardiographic demonstration of abnormal motion of the interventricular septum in left bundle branch block. Circulation 1973; 48:272–280.
- Bouzas-Mosquera A, Peteiro J, Alvarez-García N, et al. Prognostic value of exercise echocardiography in patients with left bundle branch block. JACC Cardiovasc Imaging 2009; 2:251–259.
- Vaillant C, Martins RP, Donal E, et al. Resolution of left bundle branch block-induced cardiomyopathy by cardiac resynchronization therapy. J Am Coll Cardiol 2013; 61:1089–1095.
- Schneider JF, Thomas HE Jr, Sorlie P, Kreger BE, McNamara PM, Kannel WB. Comparative features of newly acquired left and right bundle branch block in the general population: the Framingham study. Am J Cardiol 1981; 47:931–940.
- Hindman MC, Wagner GS, JaRo M, et al. The clinical significance of bundle branch block complicating acute myocardial infarction. Circulation 1978; 58:689–699.
- Freedman RA, Alderman EL, Sheffield LT, Saporito M, Fisher LD. Bundle branch block in patients with chronic coronary artery disease: angiographic correlates and prognostic significance. J Am Coll Cardiol 1987; 10:73–80.
- Schneider JF, Thomas HE Jr, Kreger BE, McNamara PM, Kannel WB. Newly acquired left bundle-branch block: the Framingham study. Ann Intern Med 1979; 90:303–310.
- Fihn SD, Gardin JM, Abrams J, et al. 2012 ACCF/AHA/ACP/AATS/PCNA/SCAI/STS guideline for the diagnosis and management of patients with stable ischemic heart disease: executive summary. J Am Coll Cardiol 2012; 60:2564–2603.
- Peteiro J, Monserrat L, Martinez D, Castro-Beiras A. Accuracy of exercise echocardiography to detect coronary artery disease in left bundle branch block unassociated with either acute or healed myocardial infarction. Am J Cardiol 2000; 85:890–893, A9.
- Pellikka PA, Nagueh SF, Elhendy AA, Kuehl CA, Sawada SG; American Society of Echocardiography. American Society of Echocardiography recommendations for performance, interpretation, and application of stress echocardiography. J Am Soc Echocardiogr 2007; 20:1021–1041.
- Xu B, Dobson L, Mottram P, Moir S. Is exercise stress echocardiography useful in patients with suspected obstructive coronary artery disease who have resting left bundle branch block? J Am Coll Cardiol 2016; 67:1570.
- Xu B, Dobson L, Mottram P, Nasis A, Cameron J, Moir S. Is exercise stress echocardiography useful in patients with suspected obstructive coronary artery disease who have resting left bundle branch block? Clin Cardiol 2018; in press.
- Yanik A, Yetkin E, Senen K, et al. Value of dobutamine stress echocardiography for diagnosis of coronary artery disease in patients with left bundle branch. Coron Artery Dis 2000; 11:545–548.
- Mairesse GH, Marwick TH, Arnese M, et al. Improved identification of coronary artery disease in patients with left bundle branch block by use of dobutamine stress echocardiography and comparison with myocardial perfusion tomography. Am J Cardiol 1995; 76:321–325.
- Vaduganathan P, He ZX, Raghavan C, Mahmarian JJ, Verani MS. Detection of left anterior descending coronary artery stenosis in patients with left bundle branch block: exercise, adenosine or dobutamine imaging? J Am Coll Cardiol 1996; 28:543–550.
- Jazmati B, Sadaniantz A, Emaus SP, Heller GV. Exercise thallium-201 imaging in complete left bundle branch block and the prevalence of septal perfusion defects. Am J Cardiol 1991; 67:46–49.
- Hasegawa S, Sakata Y, Ishikura F, et al. Mechanism for abnormal thallium-201 myocardial scintigraphy in patients with left bundle branch block in the absence of angiographic coronary artery disease. Ann Nucl Med 1999; 13:253–259.
- Biagini E, Shaw LJ, Poldermans D, et al. Accuracy of non-invasive techniques for diagnosis of coronary artery disease and prediction of cardiac events in patients with left bundle branch block: a meta-analysis. Eur J Nucl Med Mol Imaging 2006; 33:1442–1451.
- Cremer P, Brunken R, Menon V, Cerqueira M, Jaber W. Septal perfusion abnormalities are common in regadenoson SPECT myocardial perfusion imaging (MPI) but not PET MPI in patients with left bundle branch block (LBBB). J Am Coll Cardiol 2015; 65:A1148.
- Arbab-Zadeh A, Miller JM, Rochitte CE, et al. Diagnostic accuracy of computed tomography coronary angiography according to pre-test probability of coronary artery disease and severity of coronary arterial calcification. The CORE-64 (Coronary Artery Evaluation Using 64-Row Multidetector Computed Tomography Angiography) International Multicenter Study. J Am Coll Cardiol 2012; 59:379–387.
- Chow BJ, Abraham A, Wells GA, et al. Diagnostic accuracy and impact of computed tomographic coronary angiography on utilization of invasive coronary angiography. Circ Cardiovasc Imaging 2009; 2:16–23.
- Nasis A, Leung MC, Antonis PR, et al. Diagnostic accuracy of noninvasive coronary angiography with 320-detector row computed tomography. Am J Cardiol 2010; 106:1429–1435.
- Whelton SP, Silverman MG, McEvoy JW, et al. Predictors of long-term healthy arterial aging: coronary artery calcium nondevelopment in the MESA study. JACC Cardiovasc Imaging 2015; 8:1393–1400.
- Douglas PS, Garcia MJ, Haines DE, et al. ACCF/ASE/AHA/ASNC/HFSA/HRS/SCAI/SCCM/SCCT/SCMR 2011 appropriate use criteria for echocardiography. J Am Soc Echocardiogr 2011; 24:229–267.
- Henzlova MJ, Duvall WL, Einstein AJ, Travin MI, Verberne HJ. ASNC imaging guidelines for SPECT nuclear cardiology procedures: Stress, protocols, and tracers. J Nucl Cardiol 2016; 23:606–639.
- Wolk MJ, Bailey SR, Doherty JU, et al. ACCF/AHA/ASE/ASNC/HFSA/HRS/SCAI/SCCT/SCMR/STS 2013 multimodality appropriate use criteria for the detection and risk assessment of stable ischemic heart disease. J Am Coll Cardiol 2014; 63:380–406.
- Cerqueira MD, Allman KC, Ficaro EP, et al. Recommendations for reducing radiation exposure in myocardial perfusion imaging. J Nucl Cardiol 2010; 17:709–718.
- Halliburton SS, Abbara S, Chen MY, et al; Society of Cardiovascular Computed Tomography. SCCT guidelines on radiation dose and dose-optimization strategies in cardiovascular CT. J Cardiovasc Comput Tomogr 2011; 5:198–224.
- Wang CL, Cohan RH, Ellis JH, Caoili EM, Wang G, Francis IR. Frequency, outcome, and appropriateness of treatment of nonionic iodinated contrast media reactions. AJR Am J Roentgenol 2008; 191:409–415.
- Geleijnse ML, Vigna C, Kasprzak JD, et al. Usefulness and limitations of dobutamine-atropine stress echocardiography for the diagnosis of coronary artery disease in patients with left bundle branch block. A multicentre study. Eur Heart J 2000; 21:1666–1673.
- Dillon JC, Chang S, Feigenbaum H. Echocardiographic manifestations of left bundle branch block. Circulation 1974; 49:876–880.
- Abbasi AS, Eber LM, Macalpin RN, Kattus AA. Paradoxical motion of interventricular septum in left bundle branch block. Circulation 1974; 49:423–427.
- McDonald IG. Echocardiographic demonstration of abnormal motion of the interventricular septum in left bundle branch block. Circulation 1973; 48:272–280.
- Bouzas-Mosquera A, Peteiro J, Alvarez-García N, et al. Prognostic value of exercise echocardiography in patients with left bundle branch block. JACC Cardiovasc Imaging 2009; 2:251–259.
- Vaillant C, Martins RP, Donal E, et al. Resolution of left bundle branch block-induced cardiomyopathy by cardiac resynchronization therapy. J Am Coll Cardiol 2013; 61:1089–1095.
- Schneider JF, Thomas HE Jr, Sorlie P, Kreger BE, McNamara PM, Kannel WB. Comparative features of newly acquired left and right bundle branch block in the general population: the Framingham study. Am J Cardiol 1981; 47:931–940.
- Hindman MC, Wagner GS, JaRo M, et al. The clinical significance of bundle branch block complicating acute myocardial infarction. Circulation 1978; 58:689–699.
- Freedman RA, Alderman EL, Sheffield LT, Saporito M, Fisher LD. Bundle branch block in patients with chronic coronary artery disease: angiographic correlates and prognostic significance. J Am Coll Cardiol 1987; 10:73–80.
- Schneider JF, Thomas HE Jr, Kreger BE, McNamara PM, Kannel WB. Newly acquired left bundle-branch block: the Framingham study. Ann Intern Med 1979; 90:303–310.
- Fihn SD, Gardin JM, Abrams J, et al. 2012 ACCF/AHA/ACP/AATS/PCNA/SCAI/STS guideline for the diagnosis and management of patients with stable ischemic heart disease: executive summary. J Am Coll Cardiol 2012; 60:2564–2603.
- Peteiro J, Monserrat L, Martinez D, Castro-Beiras A. Accuracy of exercise echocardiography to detect coronary artery disease in left bundle branch block unassociated with either acute or healed myocardial infarction. Am J Cardiol 2000; 85:890–893, A9.
- Pellikka PA, Nagueh SF, Elhendy AA, Kuehl CA, Sawada SG; American Society of Echocardiography. American Society of Echocardiography recommendations for performance, interpretation, and application of stress echocardiography. J Am Soc Echocardiogr 2007; 20:1021–1041.
- Xu B, Dobson L, Mottram P, Moir S. Is exercise stress echocardiography useful in patients with suspected obstructive coronary artery disease who have resting left bundle branch block? J Am Coll Cardiol 2016; 67:1570.
- Xu B, Dobson L, Mottram P, Nasis A, Cameron J, Moir S. Is exercise stress echocardiography useful in patients with suspected obstructive coronary artery disease who have resting left bundle branch block? Clin Cardiol 2018; in press.
- Yanik A, Yetkin E, Senen K, et al. Value of dobutamine stress echocardiography for diagnosis of coronary artery disease in patients with left bundle branch. Coron Artery Dis 2000; 11:545–548.
- Mairesse GH, Marwick TH, Arnese M, et al. Improved identification of coronary artery disease in patients with left bundle branch block by use of dobutamine stress echocardiography and comparison with myocardial perfusion tomography. Am J Cardiol 1995; 76:321–325.
- Vaduganathan P, He ZX, Raghavan C, Mahmarian JJ, Verani MS. Detection of left anterior descending coronary artery stenosis in patients with left bundle branch block: exercise, adenosine or dobutamine imaging? J Am Coll Cardiol 1996; 28:543–550.
- Jazmati B, Sadaniantz A, Emaus SP, Heller GV. Exercise thallium-201 imaging in complete left bundle branch block and the prevalence of septal perfusion defects. Am J Cardiol 1991; 67:46–49.
- Hasegawa S, Sakata Y, Ishikura F, et al. Mechanism for abnormal thallium-201 myocardial scintigraphy in patients with left bundle branch block in the absence of angiographic coronary artery disease. Ann Nucl Med 1999; 13:253–259.
- Biagini E, Shaw LJ, Poldermans D, et al. Accuracy of non-invasive techniques for diagnosis of coronary artery disease and prediction of cardiac events in patients with left bundle branch block: a meta-analysis. Eur J Nucl Med Mol Imaging 2006; 33:1442–1451.
- Cremer P, Brunken R, Menon V, Cerqueira M, Jaber W. Septal perfusion abnormalities are common in regadenoson SPECT myocardial perfusion imaging (MPI) but not PET MPI in patients with left bundle branch block (LBBB). J Am Coll Cardiol 2015; 65:A1148.
- Arbab-Zadeh A, Miller JM, Rochitte CE, et al. Diagnostic accuracy of computed tomography coronary angiography according to pre-test probability of coronary artery disease and severity of coronary arterial calcification. The CORE-64 (Coronary Artery Evaluation Using 64-Row Multidetector Computed Tomography Angiography) International Multicenter Study. J Am Coll Cardiol 2012; 59:379–387.
- Chow BJ, Abraham A, Wells GA, et al. Diagnostic accuracy and impact of computed tomographic coronary angiography on utilization of invasive coronary angiography. Circ Cardiovasc Imaging 2009; 2:16–23.
- Nasis A, Leung MC, Antonis PR, et al. Diagnostic accuracy of noninvasive coronary angiography with 320-detector row computed tomography. Am J Cardiol 2010; 106:1429–1435.
- Whelton SP, Silverman MG, McEvoy JW, et al. Predictors of long-term healthy arterial aging: coronary artery calcium nondevelopment in the MESA study. JACC Cardiovasc Imaging 2015; 8:1393–1400.
- Douglas PS, Garcia MJ, Haines DE, et al. ACCF/ASE/AHA/ASNC/HFSA/HRS/SCAI/SCCM/SCCT/SCMR 2011 appropriate use criteria for echocardiography. J Am Soc Echocardiogr 2011; 24:229–267.
- Henzlova MJ, Duvall WL, Einstein AJ, Travin MI, Verberne HJ. ASNC imaging guidelines for SPECT nuclear cardiology procedures: Stress, protocols, and tracers. J Nucl Cardiol 2016; 23:606–639.
- Wolk MJ, Bailey SR, Doherty JU, et al. ACCF/AHA/ASE/ASNC/HFSA/HRS/SCAI/SCCT/SCMR/STS 2013 multimodality appropriate use criteria for the detection and risk assessment of stable ischemic heart disease. J Am Coll Cardiol 2014; 63:380–406.
- Cerqueira MD, Allman KC, Ficaro EP, et al. Recommendations for reducing radiation exposure in myocardial perfusion imaging. J Nucl Cardiol 2010; 17:709–718.
- Halliburton SS, Abbara S, Chen MY, et al; Society of Cardiovascular Computed Tomography. SCCT guidelines on radiation dose and dose-optimization strategies in cardiovascular CT. J Cardiovasc Comput Tomogr 2011; 5:198–224.
- Wang CL, Cohan RH, Ellis JH, Caoili EM, Wang G, Francis IR. Frequency, outcome, and appropriateness of treatment of nonionic iodinated contrast media reactions. AJR Am J Roentgenol 2008; 191:409–415.
KEY POINTS
- Although current guidelines recommend exercise stress echocardiography, it cannot reliably detect significant obstructive CAD in patients who have left bundle branch block at rest.
- CT angiography is the first-line imaging test for these patients if they are under age 65. For those 65 and older, the first-line test is either pharmacologic stress nuclear myocardial perfusion imaging with coronary vasodilators or dobutamine stress echocardiography.
- For patients who cannot tolerate CT contrast due to renal impairment or who have a true contrast allergy, pharmacologic nuclear myocardial perfusion imaging using coronary vasodilators and dobutamine stress echocardiography can be alternatives.
Alzheimer dementia: Starting, stopping drug therapy
Alzheimer disease is the most common form of dementia. In 2016, an estimated 5.2 million Americans age 65 and older had Alzheimer disease. The prevalence is projected to increase to 13.8 million by 2050, including 7 million people age 85 and older.1
Although no cure for dementia exists, several cognition-enhancing drugs have been approved by the US Food and Drug Administration (FDA) to treat the symptoms of Alzheimer dementia. The purpose of these drugs is to stabilize cognitive and functional status, with a secondary benefit of potentially reducing behavioral problems associated with dementia.
CURRENTLY APPROVED DRUGS

Two classes of drugs are approved to treat Alzheimer disease: cholinesterase inhibitors and an N-methyl-d-aspartate (NMDA) receptor antagonist (Table 1).
Cholinesterase inhibitors
The cholinesterase inhibitors act by reversibly binding and inactivating acetylcholinesterase, consequently increasing the time the neurotransmitter acetylcholine remains in the synaptic cleft. The 3 FDA-approved cholinesterase inhibitors are donepezil, galantamine, and rivastigmine. Tacrine, the first approved cholinesterase inhibitor, was removed from the US market after reports of severe hepatic toxicity.2
The clinical efficacy of cholinesterase inhibitors in improving cognitive function has been shown in several randomized controlled trials.3–10 However, benefits were generally modest, and some trials used questionable methodology, leading experts to challenge the overall efficacy of these agents.

All 3 drugs are approved for mild to moderate Alzheimer disease (stages 4–6 on the Global Deterioration Scale; Table 2)11,12; only donepezil is approved for severe Alzheimer disease. Rivastigmine has an added indication for treating mild to moderate dementia associated with Parkinson disease. Cholinesterase inhibitors are often used off-label to treat other forms of dementia such as vascular dementia, mixed dementia, and dementia with Lewy bodies.13
NMDA receptor antagonist
Memantine, currently the only FDA-approved NMDA receptor antagonist, acts by reducing neuronal calcium ion influx and its associated excitation and toxicity. Memantine is approved for moderate to severe Alzheimer disease.
Combination therapy
Often, these 2 classes of medications are prescribed in combination. In a randomized controlled trial that added memantine to stable doses of donepezil, patients had significantly better clinical response on combination therapy than on cholinesterase inhibitor monotherapy.14
In December 2014, the FDA approved a capsule formulation combining donepezil and memantine to treat symptoms of Alzheimer dementia. However, no novel pharmacologic treatment for Alzheimer disease has been approved since 2003. Furthermore, recently Pfizer announced a plan to eliminate 300 research positions aimed at finding new drugs to treat Alzheimer disease and Parkinson disease.15
CONSIDERATIONS WHEN STARTING COGNITIVE ENHANCERS
Cholinesterase inhibitors

Adverse effects of cholinesterase inhibitors are generally mild and well tolerated and subside within 1 to 2 weeks. Gastrointestinal effects are common, primarily diarrhea, nausea, and vomiting. They are transient but can occur in about 20% of patients (Table 3).
Other potential adverse effects include bradycardia, syncope, rhabdomyolysis, neuroleptic malignant syndrome, and esophageal rupture. Often, the side-effect profile helps determine which patients are appropriate candidates for these medications.
As expected, higher doses of donepezil (23 mg vs 5–10 mg) are associated with higher rates of nausea, diarrhea, and vomiting.
Dosing. The cholinesterase inhibitors should be slowly titrated to minimize side effects. Starting at the lowest dose and maintaining it for 4 weeks allows sufficient time for transient side effects to abate. Some patients may require a longer titration period.
As the dose is escalated, the probability of side effects may increase. If they do not subside, dose reduction with maintenance at the next lower dose is appropriate.
Gastrointestinal effects. Given the adverse gastrointestinal effects associated with this class of medications, patients experiencing significant anorexia and weight loss should generally avoid cholinesterase inhibitors. However, the rivastigmine patch, a transdermal formulation, is an alternative for patients who experience gastrointestinal side effects.
Bradycardia risk. Patients with significant bradycardia or who are taking medications that lower the heart rate may experience a worsening of their bradycardia or associated symptoms if they take a cholinesterase inhibitor. Syncope from bradycardia is a significant concern, especially in patients already at risk of falls or fracture due to osteoporosis.
NMDA receptor antagonist
The side-effect profile of memantine is generally more favorable than that of cholinesterase inhibitors. In clinical trials, it has been better tolerated with fewer adverse effects than placebo, with the exception of an increased incidence of dizziness, confusion, and delusions.16,17
Caution is required when treating patients with renal impairment. In patients with a creatinine clearance of 5 to 29 mL/min, the recommended maximum total daily dose is 10 mg (twice-daily formulation) or 14 mg (once-daily formulation).
Off-label use to treat behavioral problems
These medications have been used off-label to treat behavioral problems associated with dementia. A systematic review and meta-analysis showed cholinesterase inhibitor therapy had a statistically significant effect in reducing the severity of behavioral problems.18 Unfortunately, the number of dropouts increased in the active-treatment groups.
Patients with behavioral problems associated with dementia with Lewy bodies may experience a greater response to cholinesterase inhibitors than those with Alzheimer disease.19 Published post hoc analyses suggest that patients with moderate to severe Alzheimer disease receiving memantine therapy have less severe agitation, aggression, irritability, and other behavioral disturbances compared with those on placebo.20,21 However, systematic reviews have not found that memantine has a clinically significant effect on neuropsychiatric symptoms of dementia.18,22,23
Combination therapy
In early randomized controlled trials, adding memantine to a cholinesterase inhibitor provided additional cognitive benefit in patients with Alzheimer disease.15,24 However, a more recent randomized controlled trial did not show significant benefits for combined memantine and donepezil vs donepezil alone in moderate to severe dementia.25
In patients who had mild to moderate Alzheimer disease at 14 Veterans Affairs medical centers who were already on cholinesterase inhibitor treatment, adding memantine did not show benefit. However, the group receiving alpha-tocopherol (vitamin E) showed slower functional decline than those on placebo.26 Cognition and function are not expected to improve with memantine.
CONSIDERATIONS WHEN STOPPING COGNITIVE ENHANCERS
The cholinesterase inhibitors are usually prescribed early in the course of dementia, and some patients take these drugs for years, although no studies have investigated benefit or risk beyond 1 year. It is generally recommended that cholinesterase inhibitor therapy be assessed periodically, eg, every 3 to 6 months, for perceived cognitive benefits and adverse gastrointestinal effects.
These medications should be stopped if the desired effects—stabilizing cognitive and functional status—are not perceived within a reasonable time, such as 12 weeks. In some cases, stopping cholinesterase inhibitor therapy may cause negative effects on cognition and neuropsychiatric symptoms.27
Deciding whether benefit has occurred during a trial of cholinesterase inhibitors often requires input and observations from the family and caregivers. Soliciting this information is key for practitioners to determine the correct treatment approach for each patient.
Although some patients with moderately severe disease experience clinical benefits from cholinesterase inhibitor therapy, it is reasonable to consider discontinuing therapy when a patient has progressed to advanced dementia with loss of functional independence, thus making the use of the therapy—ie, to preserve functional status—less relevant. Results from a randomized discontinuation trial of cholinesterase inhibitors in institutionalized patients with moderate to severe dementia suggest that discontinuation is safe and well tolerated in most of these patients.28
Abruptly stopping high-dose cholinesterase inhibitors is not recommended. Most clinical trials tapered these medications over 2 to 4 weeks. Patients taking the maximum dose of a cholinesterase inhibitor should have the dose reduced to the next lowest dose for 2 weeks before the dose is reduced further or stopped completely.
CONSIDERATIONS FOR OTHER DEMENTIA THERAPY
Behavioral and psychiatric problems often accompany dementia; however, no drugs are approved to treat these symptoms in patients with Alzheimer disease. Nonpharmacologic interventions are recommended as the initial treatment.29 Some practitioners prescribe psychotropic drugs off-label for Alzheimer disease, but most clinical trials have not found these therapies to be very effective for psychiatric symptoms associated with Alzheimer disease.30,31
Recently, a randomized controlled trial of dextromethorphan-quinidine showed mild reduction in agitation in patients with Alzheimer disease, but there were significant increases in falls, dizziness, and diarrhea.32
Patients prescribed medications for behavioral and psychological symptoms of dementia should be assessed every 3 to 6 months to determine if the medications have been effective in reducing the symptoms they were meant to reduce. If there has been no clear reduction in the target behaviors, a trial off the drug should be initiated, with careful monitoring to see if the target behavior changes. Dementia-related behaviors may worsen off the medication, but a lower dose may be found to be as effective as a higher dose. As dementia advances, behaviors initially encountered during one stage may diminish or abate.
In a long-term care setting, a gradual dose-reduction trial of psychotropic medications should be conducted every year to determine if the medications are still necessary.33 This should be considered during routine management and follow-up of patients with dementia-associated behavioral problems.
REASONABLE TO TRY
Cognitive enhancers have been around for more than 10 years and are reasonable to try in patients with Alzheimer disease. All the available drugs are FDA-approved for reducing dementia symptoms associated with mild to moderate Alzheimer disease; donepezil and memantine are also approved for severe Alzheimer disease, either in combination or as monotherapy.
When selecting a cognitive enhancer, practitioners need to consider the potential for adverse effects. And if a cholinesterase inhibitor is prescribed, it is important to periodically assess for perceived cognitive benefits and adverse gastrointestinal effects. The NMDA receptor antagonist has a more favorable side effect profile. Combining the drugs is also an option.
Similarly, patients prescribed psychotropic medications for behavioral problems related to dementia should be reassessed to determine if the dose could be reduced or eliminated, particularly if targeted behaviors have not responded to the treatment or the dementia has advanced.
For patients on cognitive enhancers, discontinuation should be considered when the dementia advances to the point where the patient is totally dependent for all basic activities of daily living, and the initial intended purpose of these medications—preservation of cognitive and functional status—is no longer achievable.
- Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013; 80:1778–1783.
- Watkins PB, Zimmerman HJ, Knapp MJ, et al. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA 1994; 271:992–998.
- Courtney C, Farrell D, Gray R, et al. Long-term donepezil treatment in 565 patients with Alzheimer’s disease (AD2000): randomised double-blind trial. Lancet 2004; 363:2105–2115.
- Wang J, Yu JT, Wang HF, et al. Pharmacological treatment of neuropsychiatric symptoms in Alzheimer’s disease: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2015; 86:101–109.
- Raina P, Santaguida P, Ismaila A, et al. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Ann Intern Med 2008; 148:379–397.
- Lanctot KL, Hermann N, Yau KK, et al. Efficacy and safety of cholinesterase inhibitors in Alzheimer’s disease: a meta-analysis. CMAJ 2003; 169:557–564.
- Qaseem A, Snow V, Cross JT Jr, et al. Current pharmacologic treatment of dementia: a clinical practice guideline from the American College of Physicians and the American Academy of Family Physicians. Ann Intern Med 2008; 148:370–378.
- Trinh NH, Hoblyn J, Mohanty S, Yaffe K. Efficacy of cholinesterase inhibitors in the treatment of neuropsychiatric symptoms and functional impairment in Alzheimer disease: a meta-analysis. JAMA 2003; 289:210–216.
- Kaduszkiewicz H, Zimmermann T, Beck-Bornholdt HP, van den Bussche H. Cholinesterase inhibitors for patients with Alzheimer’s disease: systematic review of randomised clinical trials. BMJ 2005; 331:321–327.
- Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev 2006; 1:CD005593.
- Reisberg B, Ferris SH, de Leon MJ, Crook T. The Global Deterioration Scale for assessment of primary degenerative dementia. Am J Psychiatry 1982; 139:1136–1139.
- Mitchell SL. Advanced dementia. N Engl J Med 2015; 372:2533–2540.
- Rolinski M, Fox C, Maidment I, McShane R. Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson’s disease dementia and cognitive impairment in Parkinson’s disease. Cochrane Database Syst Rev 2012; 3:CD006504.
- Tariot PN, Farlow MR, Grossberg GT, et al. Memantine treatment in patients with moderate to severe Alzheimer’s disease already receiving donepezil: a randomized controlled trial. JAMA 2004; 291:317–324.
- Reuters Staff. Pfizer ends research for new Alzheimer’s, Parkinson’s drugs. January 7, 2018. https://www.reuters.com/article/us-pfizer-alzheimers/pfizer-ends-research-for-new-alzheimers-parkinsons-drugs-idUSKBN1EW0TN. Accessed February 2, 2018.
- Aerosa SA, Sherriff F, McShane R. Memantine for dementia. Cochrane Database Syst Rev 2005 Jul 20;(3):CD003154.
- Rossom R, Adityanjee, Dysken M. Efficacy and tolerability of memantine in the treatment of dementia. Am J Geriatr Pharmacother 2004; 2:303–312.
- Wang J, Yu JT, Wang HF, et al. Pharmacological treatment of neuropsychiatric symptoms in Alzheimer’s disease: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2015; 86:101–109.
- McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomized, double-blind, placebo-controlled international study. Lancet 2000; 356:2031–2036.
- Cummings JL, Schneider E, Tariot PN, Graham SM, Memantine MEM-MD-02 Study Group. Behavioral effects of memantine in Alzheimer disease patients receiving donepezil treatment. Neurology 2006; 67:57–63.
- Wilcock GK, Ballard CG, Cooper JA, Loft H. Memantine for agitation/aggression and psychosis in moderately severe to severe Alzheimer’s disease: a pooled analysis of 3 studies. J Clin Psychiatry 2008; 69:341–348.
- Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA 2005; 293:596–608.
- McShane R, Areosa Sastre A, Minakaran N. Memantine for dementia. Cochrane Database Syst Rev 2006; 2:CD003154.
- Reisberg B, Doody R, Stoffler A, et al. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med 2003; 348:1333–1341.
- Howard R, McShane R, Lindesay J, et al. Donepezil and memantine for moderate-to-severe Alzheimer’s disease. N Engl J Med 2012; 366:893–903.
- Dysken MW, Sano M, Asthana S, et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA 2014; 311:33–44.
- O’Regan J, Lanctot KL, Mazereeuw G, Herrmann N. Cholinesterase inhibitor discontinuation in patients with Alzheimer’s disease: a meta-analysis of randomized controlled trials. J Clin Psychiatry 2015; 76:e1424–e1431.
- Herrmann N, O’Reagan J, Ruthirahukhan M, et al. A randomized placebo-controlled discontinuation study of cholinesterase inhibitors in institutionalized patients with moderate to severe Alzheimer disease. J Am Med Dir Assoc 2016; 17:142–174.
- Gitlin LN, Kales HC, Lyketsos CG. Nonpharmacological management of behavioral symptoms in dementia. JAMA 2012; 308:2020–2029.
- Schwab W, Messinger-Rapport B, Franco K. Psychiatric symptoms of dementia: treatable, but no silver bullet. Cleve Clin J Med 2009; 76:167–174.
- Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA 2005; 293:596–608.
- Cummings JL, Lyketsos CG, Peskind ER, et al. Effects of dextromethorphan-quinidine on agitation in patients with Alzheimer disease dementia: a randomized clinical trial. JAMA 2015; 314:1242–1254.
- Centers for Medicare and Medicaid Services. Dementia care in nursing homes: clarification to Appendix P State Operations Manual (SOM) and Appendix PP in the SOM for F309—quality of care and F329—unnecessary drugs. https://www.cms.gov/Medicare/Provider-Enrollment-and-Certification/SurveyCertificationGenInfo/Downloads/Survey-and-Cert-Letter-13-35.pdf. Accessed February 1, 2018.
Alzheimer disease is the most common form of dementia. In 2016, an estimated 5.2 million Americans age 65 and older had Alzheimer disease. The prevalence is projected to increase to 13.8 million by 2050, including 7 million people age 85 and older.1
Although no cure for dementia exists, several cognition-enhancing drugs have been approved by the US Food and Drug Administration (FDA) to treat the symptoms of Alzheimer dementia. The purpose of these drugs is to stabilize cognitive and functional status, with a secondary benefit of potentially reducing behavioral problems associated with dementia.
CURRENTLY APPROVED DRUGS
Two classes of drugs are approved to treat Alzheimer disease: cholinesterase inhibitors and an N-methyl-d-aspartate (NMDA) receptor antagonist (Table 1).
Cholinesterase inhibitors
The cholinesterase inhibitors act by reversibly binding and inactivating acetylcholinesterase, consequently increasing the time the neurotransmitter acetylcholine remains in the synaptic cleft. The 3 FDA-approved cholinesterase inhibitors are donepezil, galantamine, and rivastigmine. Tacrine, the first approved cholinesterase inhibitor, was removed from the US market after reports of severe hepatic toxicity.2
The clinical efficacy of cholinesterase inhibitors in improving cognitive function has been shown in several randomized controlled trials.3–10 However, benefits were generally modest, and some trials used questionable methodology, leading experts to challenge the overall efficacy of these agents.
All 3 drugs are approved for mild to moderate Alzheimer disease (stages 4–6 on the Global Deterioration Scale; Table 2)11,12; only donepezil is approved for severe Alzheimer disease. Rivastigmine has an added indication for treating mild to moderate dementia associated with Parkinson disease. Cholinesterase inhibitors are often used off-label to treat other forms of dementia such as vascular dementia, mixed dementia, and dementia with Lewy bodies.13
NMDA receptor antagonist
Memantine, currently the only FDA-approved NMDA receptor antagonist, acts by reducing neuronal calcium ion influx and its associated excitation and toxicity. Memantine is approved for moderate to severe Alzheimer disease.
Combination therapy
Often, these 2 classes of medications are prescribed in combination. In a randomized controlled trial that added memantine to stable doses of donepezil, patients had significantly better clinical response on combination therapy than on cholinesterase inhibitor monotherapy.14
In December 2014, the FDA approved a capsule formulation combining donepezil and memantine to treat symptoms of Alzheimer dementia. However, no novel pharmacologic treatment for Alzheimer disease has been approved since 2003. Furthermore, recently Pfizer announced a plan to eliminate 300 research positions aimed at finding new drugs to treat Alzheimer disease and Parkinson disease.15
CONSIDERATIONS WHEN STARTING COGNITIVE ENHANCERS
Cholinesterase inhibitors
Adverse effects of cholinesterase inhibitors are generally mild and well tolerated and subside within 1 to 2 weeks. Gastrointestinal effects are common, primarily diarrhea, nausea, and vomiting. They are transient but can occur in about 20% of patients (Table 3).
Other potential adverse effects include bradycardia, syncope, rhabdomyolysis, neuroleptic malignant syndrome, and esophageal rupture. Often, the side-effect profile helps determine which patients are appropriate candidates for these medications.
As expected, higher doses of donepezil (23 mg vs 5–10 mg) are associated with higher rates of nausea, diarrhea, and vomiting.
Dosing. The cholinesterase inhibitors should be slowly titrated to minimize side effects. Starting at the lowest dose and maintaining it for 4 weeks allows sufficient time for transient side effects to abate. Some patients may require a longer titration period.
As the dose is escalated, the probability of side effects may increase. If they do not subside, dose reduction with maintenance at the next lower dose is appropriate.
Gastrointestinal effects. Given the adverse gastrointestinal effects associated with this class of medications, patients experiencing significant anorexia and weight loss should generally avoid cholinesterase inhibitors. However, the rivastigmine patch, a transdermal formulation, is an alternative for patients who experience gastrointestinal side effects.
Bradycardia risk. Patients with significant bradycardia or who are taking medications that lower the heart rate may experience a worsening of their bradycardia or associated symptoms if they take a cholinesterase inhibitor. Syncope from bradycardia is a significant concern, especially in patients already at risk of falls or fracture due to osteoporosis.
NMDA receptor antagonist
The side-effect profile of memantine is generally more favorable than that of cholinesterase inhibitors. In clinical trials, it has been better tolerated with fewer adverse effects than placebo, with the exception of an increased incidence of dizziness, confusion, and delusions.16,17
Caution is required when treating patients with renal impairment. In patients with a creatinine clearance of 5 to 29 mL/min, the recommended maximum total daily dose is 10 mg (twice-daily formulation) or 14 mg (once-daily formulation).
Off-label use to treat behavioral problems
These medications have been used off-label to treat behavioral problems associated with dementia. A systematic review and meta-analysis showed cholinesterase inhibitor therapy had a statistically significant effect in reducing the severity of behavioral problems.18 Unfortunately, the number of dropouts increased in the active-treatment groups.
Patients with behavioral problems associated with dementia with Lewy bodies may experience a greater response to cholinesterase inhibitors than those with Alzheimer disease.19 Published post hoc analyses suggest that patients with moderate to severe Alzheimer disease receiving memantine therapy have less severe agitation, aggression, irritability, and other behavioral disturbances compared with those on placebo.20,21 However, systematic reviews have not found that memantine has a clinically significant effect on neuropsychiatric symptoms of dementia.18,22,23
Combination therapy
In early randomized controlled trials, adding memantine to a cholinesterase inhibitor provided additional cognitive benefit in patients with Alzheimer disease.15,24 However, a more recent randomized controlled trial did not show significant benefits for combined memantine and donepezil vs donepezil alone in moderate to severe dementia.25
In patients who had mild to moderate Alzheimer disease at 14 Veterans Affairs medical centers who were already on cholinesterase inhibitor treatment, adding memantine did not show benefit. However, the group receiving alpha-tocopherol (vitamin E) showed slower functional decline than those on placebo.26 Cognition and function are not expected to improve with memantine.
CONSIDERATIONS WHEN STOPPING COGNITIVE ENHANCERS
The cholinesterase inhibitors are usually prescribed early in the course of dementia, and some patients take these drugs for years, although no studies have investigated benefit or risk beyond 1 year. It is generally recommended that cholinesterase inhibitor therapy be assessed periodically, eg, every 3 to 6 months, for perceived cognitive benefits and adverse gastrointestinal effects.
These medications should be stopped if the desired effects—stabilizing cognitive and functional status—are not perceived within a reasonable time, such as 12 weeks. In some cases, stopping cholinesterase inhibitor therapy may cause negative effects on cognition and neuropsychiatric symptoms.27
Deciding whether benefit has occurred during a trial of cholinesterase inhibitors often requires input and observations from the family and caregivers. Soliciting this information is key for practitioners to determine the correct treatment approach for each patient.
Although some patients with moderately severe disease experience clinical benefits from cholinesterase inhibitor therapy, it is reasonable to consider discontinuing therapy when a patient has progressed to advanced dementia with loss of functional independence, thus making the use of the therapy—ie, to preserve functional status—less relevant. Results from a randomized discontinuation trial of cholinesterase inhibitors in institutionalized patients with moderate to severe dementia suggest that discontinuation is safe and well tolerated in most of these patients.28
Abruptly stopping high-dose cholinesterase inhibitors is not recommended. Most clinical trials tapered these medications over 2 to 4 weeks. Patients taking the maximum dose of a cholinesterase inhibitor should have the dose reduced to the next lowest dose for 2 weeks before the dose is reduced further or stopped completely.
CONSIDERATIONS FOR OTHER DEMENTIA THERAPY
Behavioral and psychiatric problems often accompany dementia; however, no drugs are approved to treat these symptoms in patients with Alzheimer disease. Nonpharmacologic interventions are recommended as the initial treatment.29 Some practitioners prescribe psychotropic drugs off-label for Alzheimer disease, but most clinical trials have not found these therapies to be very effective for psychiatric symptoms associated with Alzheimer disease.30,31
Recently, a randomized controlled trial of dextromethorphan-quinidine showed mild reduction in agitation in patients with Alzheimer disease, but there were significant increases in falls, dizziness, and diarrhea.32
Patients prescribed medications for behavioral and psychological symptoms of dementia should be assessed every 3 to 6 months to determine if the medications have been effective in reducing the symptoms they were meant to reduce. If there has been no clear reduction in the target behaviors, a trial off the drug should be initiated, with careful monitoring to see if the target behavior changes. Dementia-related behaviors may worsen off the medication, but a lower dose may be found to be as effective as a higher dose. As dementia advances, behaviors initially encountered during one stage may diminish or abate.
In a long-term care setting, a gradual dose-reduction trial of psychotropic medications should be conducted every year to determine if the medications are still necessary.33 This should be considered during routine management and follow-up of patients with dementia-associated behavioral problems.
REASONABLE TO TRY
Cognitive enhancers have been around for more than 10 years and are reasonable to try in patients with Alzheimer disease. All the available drugs are FDA-approved for reducing dementia symptoms associated with mild to moderate Alzheimer disease; donepezil and memantine are also approved for severe Alzheimer disease, either in combination or as monotherapy.
When selecting a cognitive enhancer, practitioners need to consider the potential for adverse effects. And if a cholinesterase inhibitor is prescribed, it is important to periodically assess for perceived cognitive benefits and adverse gastrointestinal effects. The NMDA receptor antagonist has a more favorable side effect profile. Combining the drugs is also an option.
Similarly, patients prescribed psychotropic medications for behavioral problems related to dementia should be reassessed to determine if the dose could be reduced or eliminated, particularly if targeted behaviors have not responded to the treatment or the dementia has advanced.
For patients on cognitive enhancers, discontinuation should be considered when the dementia advances to the point where the patient is totally dependent for all basic activities of daily living, and the initial intended purpose of these medications—preservation of cognitive and functional status—is no longer achievable.
Alzheimer disease is the most common form of dementia. In 2016, an estimated 5.2 million Americans age 65 and older had Alzheimer disease. The prevalence is projected to increase to 13.8 million by 2050, including 7 million people age 85 and older.1
Although no cure for dementia exists, several cognition-enhancing drugs have been approved by the US Food and Drug Administration (FDA) to treat the symptoms of Alzheimer dementia. The purpose of these drugs is to stabilize cognitive and functional status, with a secondary benefit of potentially reducing behavioral problems associated with dementia.
CURRENTLY APPROVED DRUGS
Two classes of drugs are approved to treat Alzheimer disease: cholinesterase inhibitors and an N-methyl-d-aspartate (NMDA) receptor antagonist (Table 1).
Cholinesterase inhibitors
The cholinesterase inhibitors act by reversibly binding and inactivating acetylcholinesterase, consequently increasing the time the neurotransmitter acetylcholine remains in the synaptic cleft. The 3 FDA-approved cholinesterase inhibitors are donepezil, galantamine, and rivastigmine. Tacrine, the first approved cholinesterase inhibitor, was removed from the US market after reports of severe hepatic toxicity.2
The clinical efficacy of cholinesterase inhibitors in improving cognitive function has been shown in several randomized controlled trials.3–10 However, benefits were generally modest, and some trials used questionable methodology, leading experts to challenge the overall efficacy of these agents.
All 3 drugs are approved for mild to moderate Alzheimer disease (stages 4–6 on the Global Deterioration Scale; Table 2)11,12; only donepezil is approved for severe Alzheimer disease. Rivastigmine has an added indication for treating mild to moderate dementia associated with Parkinson disease. Cholinesterase inhibitors are often used off-label to treat other forms of dementia such as vascular dementia, mixed dementia, and dementia with Lewy bodies.13
NMDA receptor antagonist
Memantine, currently the only FDA-approved NMDA receptor antagonist, acts by reducing neuronal calcium ion influx and its associated excitation and toxicity. Memantine is approved for moderate to severe Alzheimer disease.
Combination therapy
Often, these 2 classes of medications are prescribed in combination. In a randomized controlled trial that added memantine to stable doses of donepezil, patients had significantly better clinical response on combination therapy than on cholinesterase inhibitor monotherapy.14
In December 2014, the FDA approved a capsule formulation combining donepezil and memantine to treat symptoms of Alzheimer dementia. However, no novel pharmacologic treatment for Alzheimer disease has been approved since 2003. Furthermore, recently Pfizer announced a plan to eliminate 300 research positions aimed at finding new drugs to treat Alzheimer disease and Parkinson disease.15
CONSIDERATIONS WHEN STARTING COGNITIVE ENHANCERS
Cholinesterase inhibitors
Adverse effects of cholinesterase inhibitors are generally mild and well tolerated and subside within 1 to 2 weeks. Gastrointestinal effects are common, primarily diarrhea, nausea, and vomiting. They are transient but can occur in about 20% of patients (Table 3).
Other potential adverse effects include bradycardia, syncope, rhabdomyolysis, neuroleptic malignant syndrome, and esophageal rupture. Often, the side-effect profile helps determine which patients are appropriate candidates for these medications.
As expected, higher doses of donepezil (23 mg vs 5–10 mg) are associated with higher rates of nausea, diarrhea, and vomiting.
Dosing. The cholinesterase inhibitors should be slowly titrated to minimize side effects. Starting at the lowest dose and maintaining it for 4 weeks allows sufficient time for transient side effects to abate. Some patients may require a longer titration period.
As the dose is escalated, the probability of side effects may increase. If they do not subside, dose reduction with maintenance at the next lower dose is appropriate.
Gastrointestinal effects. Given the adverse gastrointestinal effects associated with this class of medications, patients experiencing significant anorexia and weight loss should generally avoid cholinesterase inhibitors. However, the rivastigmine patch, a transdermal formulation, is an alternative for patients who experience gastrointestinal side effects.
Bradycardia risk. Patients with significant bradycardia or who are taking medications that lower the heart rate may experience a worsening of their bradycardia or associated symptoms if they take a cholinesterase inhibitor. Syncope from bradycardia is a significant concern, especially in patients already at risk of falls or fracture due to osteoporosis.
NMDA receptor antagonist
The side-effect profile of memantine is generally more favorable than that of cholinesterase inhibitors. In clinical trials, it has been better tolerated with fewer adverse effects than placebo, with the exception of an increased incidence of dizziness, confusion, and delusions.16,17
Caution is required when treating patients with renal impairment. In patients with a creatinine clearance of 5 to 29 mL/min, the recommended maximum total daily dose is 10 mg (twice-daily formulation) or 14 mg (once-daily formulation).
Off-label use to treat behavioral problems
These medications have been used off-label to treat behavioral problems associated with dementia. A systematic review and meta-analysis showed cholinesterase inhibitor therapy had a statistically significant effect in reducing the severity of behavioral problems.18 Unfortunately, the number of dropouts increased in the active-treatment groups.
Patients with behavioral problems associated with dementia with Lewy bodies may experience a greater response to cholinesterase inhibitors than those with Alzheimer disease.19 Published post hoc analyses suggest that patients with moderate to severe Alzheimer disease receiving memantine therapy have less severe agitation, aggression, irritability, and other behavioral disturbances compared with those on placebo.20,21 However, systematic reviews have not found that memantine has a clinically significant effect on neuropsychiatric symptoms of dementia.18,22,23
Combination therapy
In early randomized controlled trials, adding memantine to a cholinesterase inhibitor provided additional cognitive benefit in patients with Alzheimer disease.15,24 However, a more recent randomized controlled trial did not show significant benefits for combined memantine and donepezil vs donepezil alone in moderate to severe dementia.25
In patients who had mild to moderate Alzheimer disease at 14 Veterans Affairs medical centers who were already on cholinesterase inhibitor treatment, adding memantine did not show benefit. However, the group receiving alpha-tocopherol (vitamin E) showed slower functional decline than those on placebo.26 Cognition and function are not expected to improve with memantine.
CONSIDERATIONS WHEN STOPPING COGNITIVE ENHANCERS
The cholinesterase inhibitors are usually prescribed early in the course of dementia, and some patients take these drugs for years, although no studies have investigated benefit or risk beyond 1 year. It is generally recommended that cholinesterase inhibitor therapy be assessed periodically, eg, every 3 to 6 months, for perceived cognitive benefits and adverse gastrointestinal effects.
These medications should be stopped if the desired effects—stabilizing cognitive and functional status—are not perceived within a reasonable time, such as 12 weeks. In some cases, stopping cholinesterase inhibitor therapy may cause negative effects on cognition and neuropsychiatric symptoms.27
Deciding whether benefit has occurred during a trial of cholinesterase inhibitors often requires input and observations from the family and caregivers. Soliciting this information is key for practitioners to determine the correct treatment approach for each patient.
Although some patients with moderately severe disease experience clinical benefits from cholinesterase inhibitor therapy, it is reasonable to consider discontinuing therapy when a patient has progressed to advanced dementia with loss of functional independence, thus making the use of the therapy—ie, to preserve functional status—less relevant. Results from a randomized discontinuation trial of cholinesterase inhibitors in institutionalized patients with moderate to severe dementia suggest that discontinuation is safe and well tolerated in most of these patients.28
Abruptly stopping high-dose cholinesterase inhibitors is not recommended. Most clinical trials tapered these medications over 2 to 4 weeks. Patients taking the maximum dose of a cholinesterase inhibitor should have the dose reduced to the next lowest dose for 2 weeks before the dose is reduced further or stopped completely.
CONSIDERATIONS FOR OTHER DEMENTIA THERAPY
Behavioral and psychiatric problems often accompany dementia; however, no drugs are approved to treat these symptoms in patients with Alzheimer disease. Nonpharmacologic interventions are recommended as the initial treatment.29 Some practitioners prescribe psychotropic drugs off-label for Alzheimer disease, but most clinical trials have not found these therapies to be very effective for psychiatric symptoms associated with Alzheimer disease.30,31
Recently, a randomized controlled trial of dextromethorphan-quinidine showed mild reduction in agitation in patients with Alzheimer disease, but there were significant increases in falls, dizziness, and diarrhea.32
Patients prescribed medications for behavioral and psychological symptoms of dementia should be assessed every 3 to 6 months to determine if the medications have been effective in reducing the symptoms they were meant to reduce. If there has been no clear reduction in the target behaviors, a trial off the drug should be initiated, with careful monitoring to see if the target behavior changes. Dementia-related behaviors may worsen off the medication, but a lower dose may be found to be as effective as a higher dose. As dementia advances, behaviors initially encountered during one stage may diminish or abate.
In a long-term care setting, a gradual dose-reduction trial of psychotropic medications should be conducted every year to determine if the medications are still necessary.33 This should be considered during routine management and follow-up of patients with dementia-associated behavioral problems.
REASONABLE TO TRY
Cognitive enhancers have been around for more than 10 years and are reasonable to try in patients with Alzheimer disease. All the available drugs are FDA-approved for reducing dementia symptoms associated with mild to moderate Alzheimer disease; donepezil and memantine are also approved for severe Alzheimer disease, either in combination or as monotherapy.
When selecting a cognitive enhancer, practitioners need to consider the potential for adverse effects. And if a cholinesterase inhibitor is prescribed, it is important to periodically assess for perceived cognitive benefits and adverse gastrointestinal effects. The NMDA receptor antagonist has a more favorable side effect profile. Combining the drugs is also an option.
Similarly, patients prescribed psychotropic medications for behavioral problems related to dementia should be reassessed to determine if the dose could be reduced or eliminated, particularly if targeted behaviors have not responded to the treatment or the dementia has advanced.
For patients on cognitive enhancers, discontinuation should be considered when the dementia advances to the point where the patient is totally dependent for all basic activities of daily living, and the initial intended purpose of these medications—preservation of cognitive and functional status—is no longer achievable.
- Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013; 80:1778–1783.
- Watkins PB, Zimmerman HJ, Knapp MJ, et al. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA 1994; 271:992–998.
- Courtney C, Farrell D, Gray R, et al. Long-term donepezil treatment in 565 patients with Alzheimer’s disease (AD2000): randomised double-blind trial. Lancet 2004; 363:2105–2115.
- Wang J, Yu JT, Wang HF, et al. Pharmacological treatment of neuropsychiatric symptoms in Alzheimer’s disease: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2015; 86:101–109.
- Raina P, Santaguida P, Ismaila A, et al. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Ann Intern Med 2008; 148:379–397.
- Lanctot KL, Hermann N, Yau KK, et al. Efficacy and safety of cholinesterase inhibitors in Alzheimer’s disease: a meta-analysis. CMAJ 2003; 169:557–564.
- Qaseem A, Snow V, Cross JT Jr, et al. Current pharmacologic treatment of dementia: a clinical practice guideline from the American College of Physicians and the American Academy of Family Physicians. Ann Intern Med 2008; 148:370–378.
- Trinh NH, Hoblyn J, Mohanty S, Yaffe K. Efficacy of cholinesterase inhibitors in the treatment of neuropsychiatric symptoms and functional impairment in Alzheimer disease: a meta-analysis. JAMA 2003; 289:210–216.
- Kaduszkiewicz H, Zimmermann T, Beck-Bornholdt HP, van den Bussche H. Cholinesterase inhibitors for patients with Alzheimer’s disease: systematic review of randomised clinical trials. BMJ 2005; 331:321–327.
- Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev 2006; 1:CD005593.
- Reisberg B, Ferris SH, de Leon MJ, Crook T. The Global Deterioration Scale for assessment of primary degenerative dementia. Am J Psychiatry 1982; 139:1136–1139.
- Mitchell SL. Advanced dementia. N Engl J Med 2015; 372:2533–2540.
- Rolinski M, Fox C, Maidment I, McShane R. Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson’s disease dementia and cognitive impairment in Parkinson’s disease. Cochrane Database Syst Rev 2012; 3:CD006504.
- Tariot PN, Farlow MR, Grossberg GT, et al. Memantine treatment in patients with moderate to severe Alzheimer’s disease already receiving donepezil: a randomized controlled trial. JAMA 2004; 291:317–324.
- Reuters Staff. Pfizer ends research for new Alzheimer’s, Parkinson’s drugs. January 7, 2018. https://www.reuters.com/article/us-pfizer-alzheimers/pfizer-ends-research-for-new-alzheimers-parkinsons-drugs-idUSKBN1EW0TN. Accessed February 2, 2018.
- Aerosa SA, Sherriff F, McShane R. Memantine for dementia. Cochrane Database Syst Rev 2005 Jul 20;(3):CD003154.
- Rossom R, Adityanjee, Dysken M. Efficacy and tolerability of memantine in the treatment of dementia. Am J Geriatr Pharmacother 2004; 2:303–312.
- Wang J, Yu JT, Wang HF, et al. Pharmacological treatment of neuropsychiatric symptoms in Alzheimer’s disease: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2015; 86:101–109.
- McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomized, double-blind, placebo-controlled international study. Lancet 2000; 356:2031–2036.
- Cummings JL, Schneider E, Tariot PN, Graham SM, Memantine MEM-MD-02 Study Group. Behavioral effects of memantine in Alzheimer disease patients receiving donepezil treatment. Neurology 2006; 67:57–63.
- Wilcock GK, Ballard CG, Cooper JA, Loft H. Memantine for agitation/aggression and psychosis in moderately severe to severe Alzheimer’s disease: a pooled analysis of 3 studies. J Clin Psychiatry 2008; 69:341–348.
- Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA 2005; 293:596–608.
- McShane R, Areosa Sastre A, Minakaran N. Memantine for dementia. Cochrane Database Syst Rev 2006; 2:CD003154.
- Reisberg B, Doody R, Stoffler A, et al. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med 2003; 348:1333–1341.
- Howard R, McShane R, Lindesay J, et al. Donepezil and memantine for moderate-to-severe Alzheimer’s disease. N Engl J Med 2012; 366:893–903.
- Dysken MW, Sano M, Asthana S, et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA 2014; 311:33–44.
- O’Regan J, Lanctot KL, Mazereeuw G, Herrmann N. Cholinesterase inhibitor discontinuation in patients with Alzheimer’s disease: a meta-analysis of randomized controlled trials. J Clin Psychiatry 2015; 76:e1424–e1431.
- Herrmann N, O’Reagan J, Ruthirahukhan M, et al. A randomized placebo-controlled discontinuation study of cholinesterase inhibitors in institutionalized patients with moderate to severe Alzheimer disease. J Am Med Dir Assoc 2016; 17:142–174.
- Gitlin LN, Kales HC, Lyketsos CG. Nonpharmacological management of behavioral symptoms in dementia. JAMA 2012; 308:2020–2029.
- Schwab W, Messinger-Rapport B, Franco K. Psychiatric symptoms of dementia: treatable, but no silver bullet. Cleve Clin J Med 2009; 76:167–174.
- Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA 2005; 293:596–608.
- Cummings JL, Lyketsos CG, Peskind ER, et al. Effects of dextromethorphan-quinidine on agitation in patients with Alzheimer disease dementia: a randomized clinical trial. JAMA 2015; 314:1242–1254.
- Centers for Medicare and Medicaid Services. Dementia care in nursing homes: clarification to Appendix P State Operations Manual (SOM) and Appendix PP in the SOM for F309—quality of care and F329—unnecessary drugs. https://www.cms.gov/Medicare/Provider-Enrollment-and-Certification/SurveyCertificationGenInfo/Downloads/Survey-and-Cert-Letter-13-35.pdf. Accessed February 1, 2018.
- Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013; 80:1778–1783.
- Watkins PB, Zimmerman HJ, Knapp MJ, et al. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA 1994; 271:992–998.
- Courtney C, Farrell D, Gray R, et al. Long-term donepezil treatment in 565 patients with Alzheimer’s disease (AD2000): randomised double-blind trial. Lancet 2004; 363:2105–2115.
- Wang J, Yu JT, Wang HF, et al. Pharmacological treatment of neuropsychiatric symptoms in Alzheimer’s disease: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2015; 86:101–109.
- Raina P, Santaguida P, Ismaila A, et al. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Ann Intern Med 2008; 148:379–397.
- Lanctot KL, Hermann N, Yau KK, et al. Efficacy and safety of cholinesterase inhibitors in Alzheimer’s disease: a meta-analysis. CMAJ 2003; 169:557–564.
- Qaseem A, Snow V, Cross JT Jr, et al. Current pharmacologic treatment of dementia: a clinical practice guideline from the American College of Physicians and the American Academy of Family Physicians. Ann Intern Med 2008; 148:370–378.
- Trinh NH, Hoblyn J, Mohanty S, Yaffe K. Efficacy of cholinesterase inhibitors in the treatment of neuropsychiatric symptoms and functional impairment in Alzheimer disease: a meta-analysis. JAMA 2003; 289:210–216.
- Kaduszkiewicz H, Zimmermann T, Beck-Bornholdt HP, van den Bussche H. Cholinesterase inhibitors for patients with Alzheimer’s disease: systematic review of randomised clinical trials. BMJ 2005; 331:321–327.
- Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev 2006; 1:CD005593.
- Reisberg B, Ferris SH, de Leon MJ, Crook T. The Global Deterioration Scale for assessment of primary degenerative dementia. Am J Psychiatry 1982; 139:1136–1139.
- Mitchell SL. Advanced dementia. N Engl J Med 2015; 372:2533–2540.
- Rolinski M, Fox C, Maidment I, McShane R. Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson’s disease dementia and cognitive impairment in Parkinson’s disease. Cochrane Database Syst Rev 2012; 3:CD006504.
- Tariot PN, Farlow MR, Grossberg GT, et al. Memantine treatment in patients with moderate to severe Alzheimer’s disease already receiving donepezil: a randomized controlled trial. JAMA 2004; 291:317–324.
- Reuters Staff. Pfizer ends research for new Alzheimer’s, Parkinson’s drugs. January 7, 2018. https://www.reuters.com/article/us-pfizer-alzheimers/pfizer-ends-research-for-new-alzheimers-parkinsons-drugs-idUSKBN1EW0TN. Accessed February 2, 2018.
- Aerosa SA, Sherriff F, McShane R. Memantine for dementia. Cochrane Database Syst Rev 2005 Jul 20;(3):CD003154.
- Rossom R, Adityanjee, Dysken M. Efficacy and tolerability of memantine in the treatment of dementia. Am J Geriatr Pharmacother 2004; 2:303–312.
- Wang J, Yu JT, Wang HF, et al. Pharmacological treatment of neuropsychiatric symptoms in Alzheimer’s disease: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2015; 86:101–109.
- McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomized, double-blind, placebo-controlled international study. Lancet 2000; 356:2031–2036.
- Cummings JL, Schneider E, Tariot PN, Graham SM, Memantine MEM-MD-02 Study Group. Behavioral effects of memantine in Alzheimer disease patients receiving donepezil treatment. Neurology 2006; 67:57–63.
- Wilcock GK, Ballard CG, Cooper JA, Loft H. Memantine for agitation/aggression and psychosis in moderately severe to severe Alzheimer’s disease: a pooled analysis of 3 studies. J Clin Psychiatry 2008; 69:341–348.
- Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA 2005; 293:596–608.
- McShane R, Areosa Sastre A, Minakaran N. Memantine for dementia. Cochrane Database Syst Rev 2006; 2:CD003154.
- Reisberg B, Doody R, Stoffler A, et al. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med 2003; 348:1333–1341.
- Howard R, McShane R, Lindesay J, et al. Donepezil and memantine for moderate-to-severe Alzheimer’s disease. N Engl J Med 2012; 366:893–903.
- Dysken MW, Sano M, Asthana S, et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA 2014; 311:33–44.
- O’Regan J, Lanctot KL, Mazereeuw G, Herrmann N. Cholinesterase inhibitor discontinuation in patients with Alzheimer’s disease: a meta-analysis of randomized controlled trials. J Clin Psychiatry 2015; 76:e1424–e1431.
- Herrmann N, O’Reagan J, Ruthirahukhan M, et al. A randomized placebo-controlled discontinuation study of cholinesterase inhibitors in institutionalized patients with moderate to severe Alzheimer disease. J Am Med Dir Assoc 2016; 17:142–174.
- Gitlin LN, Kales HC, Lyketsos CG. Nonpharmacological management of behavioral symptoms in dementia. JAMA 2012; 308:2020–2029.
- Schwab W, Messinger-Rapport B, Franco K. Psychiatric symptoms of dementia: treatable, but no silver bullet. Cleve Clin J Med 2009; 76:167–174.
- Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA 2005; 293:596–608.
- Cummings JL, Lyketsos CG, Peskind ER, et al. Effects of dextromethorphan-quinidine on agitation in patients with Alzheimer disease dementia: a randomized clinical trial. JAMA 2015; 314:1242–1254.
- Centers for Medicare and Medicaid Services. Dementia care in nursing homes: clarification to Appendix P State Operations Manual (SOM) and Appendix PP in the SOM for F309—quality of care and F329—unnecessary drugs. https://www.cms.gov/Medicare/Provider-Enrollment-and-Certification/SurveyCertificationGenInfo/Downloads/Survey-and-Cert-Letter-13-35.pdf. Accessed February 1, 2018.
KEY POINTS
- In 2016, an estimated 5.2 million Americans age 65 and older had Alzheimer disease; by 2050, the prevalence is expected to be 13.8 million.
- Cognitive enhancers (cholinesterase inhibitors and an N-methyl-d-aspartate receptor antagonist) have shown modest efficacy in preserving cognitive function.
- When evaluating therapy with a cognitive enhancer, practitioners need to consider the potential adverse effects, especially gastrointestinal effects with cholinesterase inhibitors.
- Discontinuation should be considered when the dementia reaches the advanced stage and the initial intended purpose of these drugs is no longer achievable.

Primary livedo reticularis of the abdomen

Livedo reticularis can be the manifestation of a wide range of conditions: hematologic and hypercoagulable states, embolic events, connective tissue disease, infection, vasculitis, malignancy, neurologic and endocrine conditions, and medication effects.1 Our patient had no recent history of vascular procedures or peripheral eosinophilia to suggest cholesterol embolization, and he had not recently started taking any new medications. His current medications included aspirin 81 mg, atorvastatin 40 mg, amlodipine 10 mg, and insulin glargine 20 units. Tests for cryoglobulin and antiphospholipid antibodies were negative. There was no evidence of malignancy, and evaluations for infectious and autoimmune diseases were negative.
Biopsy study of a skin lesion showed features consistent with livedo reticularis, with no evidence of vasculitis. The lesions resolved without definitive therapy by hospital day 3. This, in addition to other features of the lesions (eg, uniformity, unbroken reticular segments) and the extensive negative workup for systemic disease, suggested primary livedo reticularis.
CAUSES, TYPES, SUBTYPES
Livedo reticularis results from changes in the cutaneous microvasculature, composed of central arterioles that drain into an interconnecting, netlike venous plexus.1,2 Conditions such as arteriolar deoxygenation and venous plexus venodilation that result in a prominent venous plexus can give rise to clinical livedo reticularis.3

Primary livedo reticularis is thought to occur from spontaneous arteriolar vasospasm. It is a diagnosis of exclusion. An evaluation for underlying disease is important, as livedo reticularis can be associated with the range of conditions listed above.
In our patient, methamphetamine was considered a possible cause, but the findings of livedo reticularis were delayed and persisted longer than expected if they were drug-related.
Livedo racemosa
Distinguishing livedo reticularis from livedo racemosa is important. Livedo racemosa is always secondary and is often associated with antiphospholipid syndrome. It is present in 25% of cases of primary antiphospholipid syndrome and in up to 70% of cases of antiphospholipid syndrome associated with systemic lupus erythematosus.5 The reticular pattern of livedo racemosa is permanent and often has irregular and incomplete segments of reticular lattice, with a distribution that is more generalized, involving the trunk or buttocks (or both) in addition to the extremities.3,4 Consequently, a thorough history and physical examination are needed to guide additional workup.
- Gibbs MB, English JC 3rd, Zirwas MJ. Livedo reticularis: an update. J Am Acad Dermatol 2005; 52:1009–1019.
- Kraemer M, Linden D, Berlit P. The spectrum of differential diagnosis in neurological patients with livedo reticularis and livedo racemosa. A literature review. J Neurol 2005; 252:1155–1166.
- Uthman IW, Khamashta MA. Livedo racemosa: a striking dermatological sign for the antiphospholipid syndrome. J Rheumatol 2006; 33:2379–2382.
- Dean SM. Livedo reticularis and related disorders. Curr Treat Options Cardiovasc Med 2011; 13:179–191.
- Chadachan V, Dean SM, Eberhardt RT. Cutaneous changes in peripheral arterial vascular disease. In: Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Education; 2012.
Livedo reticularis can be the manifestation of a wide range of conditions: hematologic and hypercoagulable states, embolic events, connective tissue disease, infection, vasculitis, malignancy, neurologic and endocrine conditions, and medication effects.1 Our patient had no recent history of vascular procedures or peripheral eosinophilia to suggest cholesterol embolization, and he had not recently started taking any new medications. His current medications included aspirin 81 mg, atorvastatin 40 mg, amlodipine 10 mg, and insulin glargine 20 units. Tests for cryoglobulin and antiphospholipid antibodies were negative. There was no evidence of malignancy, and evaluations for infectious and autoimmune diseases were negative.
Biopsy study of a skin lesion showed features consistent with livedo reticularis, with no evidence of vasculitis. The lesions resolved without definitive therapy by hospital day 3. This, in addition to other features of the lesions (eg, uniformity, unbroken reticular segments) and the extensive negative workup for systemic disease, suggested primary livedo reticularis.
CAUSES, TYPES, SUBTYPES
Livedo reticularis results from changes in the cutaneous microvasculature, composed of central arterioles that drain into an interconnecting, netlike venous plexus.1,2 Conditions such as arteriolar deoxygenation and venous plexus venodilation that result in a prominent venous plexus can give rise to clinical livedo reticularis.3
Primary livedo reticularis is thought to occur from spontaneous arteriolar vasospasm. It is a diagnosis of exclusion. An evaluation for underlying disease is important, as livedo reticularis can be associated with the range of conditions listed above.
In our patient, methamphetamine was considered a possible cause, but the findings of livedo reticularis were delayed and persisted longer than expected if they were drug-related.
Livedo racemosa
Distinguishing livedo reticularis from livedo racemosa is important. Livedo racemosa is always secondary and is often associated with antiphospholipid syndrome. It is present in 25% of cases of primary antiphospholipid syndrome and in up to 70% of cases of antiphospholipid syndrome associated with systemic lupus erythematosus.5 The reticular pattern of livedo racemosa is permanent and often has irregular and incomplete segments of reticular lattice, with a distribution that is more generalized, involving the trunk or buttocks (or both) in addition to the extremities.3,4 Consequently, a thorough history and physical examination are needed to guide additional workup.
Livedo reticularis can be the manifestation of a wide range of conditions: hematologic and hypercoagulable states, embolic events, connective tissue disease, infection, vasculitis, malignancy, neurologic and endocrine conditions, and medication effects.1 Our patient had no recent history of vascular procedures or peripheral eosinophilia to suggest cholesterol embolization, and he had not recently started taking any new medications. His current medications included aspirin 81 mg, atorvastatin 40 mg, amlodipine 10 mg, and insulin glargine 20 units. Tests for cryoglobulin and antiphospholipid antibodies were negative. There was no evidence of malignancy, and evaluations for infectious and autoimmune diseases were negative.
Biopsy study of a skin lesion showed features consistent with livedo reticularis, with no evidence of vasculitis. The lesions resolved without definitive therapy by hospital day 3. This, in addition to other features of the lesions (eg, uniformity, unbroken reticular segments) and the extensive negative workup for systemic disease, suggested primary livedo reticularis.
CAUSES, TYPES, SUBTYPES
Livedo reticularis results from changes in the cutaneous microvasculature, composed of central arterioles that drain into an interconnecting, netlike venous plexus.1,2 Conditions such as arteriolar deoxygenation and venous plexus venodilation that result in a prominent venous plexus can give rise to clinical livedo reticularis.3
Primary livedo reticularis is thought to occur from spontaneous arteriolar vasospasm. It is a diagnosis of exclusion. An evaluation for underlying disease is important, as livedo reticularis can be associated with the range of conditions listed above.
In our patient, methamphetamine was considered a possible cause, but the findings of livedo reticularis were delayed and persisted longer than expected if they were drug-related.
Livedo racemosa
Distinguishing livedo reticularis from livedo racemosa is important. Livedo racemosa is always secondary and is often associated with antiphospholipid syndrome. It is present in 25% of cases of primary antiphospholipid syndrome and in up to 70% of cases of antiphospholipid syndrome associated with systemic lupus erythematosus.5 The reticular pattern of livedo racemosa is permanent and often has irregular and incomplete segments of reticular lattice, with a distribution that is more generalized, involving the trunk or buttocks (or both) in addition to the extremities.3,4 Consequently, a thorough history and physical examination are needed to guide additional workup.
- Gibbs MB, English JC 3rd, Zirwas MJ. Livedo reticularis: an update. J Am Acad Dermatol 2005; 52:1009–1019.
- Kraemer M, Linden D, Berlit P. The spectrum of differential diagnosis in neurological patients with livedo reticularis and livedo racemosa. A literature review. J Neurol 2005; 252:1155–1166.
- Uthman IW, Khamashta MA. Livedo racemosa: a striking dermatological sign for the antiphospholipid syndrome. J Rheumatol 2006; 33:2379–2382.
- Dean SM. Livedo reticularis and related disorders. Curr Treat Options Cardiovasc Med 2011; 13:179–191.
- Chadachan V, Dean SM, Eberhardt RT. Cutaneous changes in peripheral arterial vascular disease. In: Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Education; 2012.
- Gibbs MB, English JC 3rd, Zirwas MJ. Livedo reticularis: an update. J Am Acad Dermatol 2005; 52:1009–1019.
- Kraemer M, Linden D, Berlit P. The spectrum of differential diagnosis in neurological patients with livedo reticularis and livedo racemosa. A literature review. J Neurol 2005; 252:1155–1166.
- Uthman IW, Khamashta MA. Livedo racemosa: a striking dermatological sign for the antiphospholipid syndrome. J Rheumatol 2006; 33:2379–2382.
- Dean SM. Livedo reticularis and related disorders. Curr Treat Options Cardiovasc Med 2011; 13:179–191.
- Chadachan V, Dean SM, Eberhardt RT. Cutaneous changes in peripheral arterial vascular disease. In: Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Education; 2012.
Hypertension in older adults: What is the target blood pressure?
We should aim for a standard office systolic pressure lower than 130 mm Hg in most adults age 65 and older if the patient can take multiple antihypertensive medications and be followed closely for adverse effects.
This recommendation is part of the 2017 hypertension guideline from the American College of Cardiology and American Heart Association.1 This new guideline advocates drug treatment of hypertension to a target less than 130/80 mm Hg for patients of all ages for secondary prevention of cardiovascular disease, and for primary prevention in those at high risk (ie, an estimated 10-year risk of atherosclerotic cardiovascular disease of 10% or higher). The target blood pressure for those at lower risk is less than 140/90 mm Hg.
There are multiple tools to estimate the 10-year risk. All tools incorporate major predictors such as age, blood pressure, cholesterol profile, and other markers, depending on the tool. Although risk increases with age, the tools are inaccurate once the patient is approximately 80 years of age.
The recommendation for older adults omits a target diastolic pressure, since treating elevated systolic pressure has more data supporting it than treating elevated diastolic blood pressure in older people. These recommendations apply only to older adults who can walk and are living in the community, not in an institution, and includes the subset of older adults who have mild cognitive impairment and frailty. The goals of treatment should be patient-centered.
DATA BEHIND THE GUIDELINE: THE SPRINT TRIAL
The Systolic Blood Pressure Intervention Trial (SPRINT)2 enrolled 9,361 patients who, to enter, had to be at least 50 years old (the mean age was 67.9), have a systolic blood pressure of 130 to 180 mm Hg (the mean was 139.7 mm Hg), and be at risk of cardiovascular disease due to chronic kidney disease, clinical or subclinical cardiovascular disease, a 10-year Framingham risk score of at least 15%, or age 75 or older. They had few comorbidities, and patients with diabetes mellitus or prior stroke were excluded. The objective was to see if intensive blood pressure treatment reduced the incidence of adverse cardiovascular outcomes compared with standard control.
The participants were randomized to either an intensive treatment goal of systolic pressure less than 120 mm Hg or a standard treatment goal of less than 140 mm Hg. Investigators chose drugs and doses according to their clinical judgment. The study protocol called for blood pressure measurement using an untended automated cuff, which probably resulted in systolic pressure readings 5 to 10 mm Hg lower than with typical methods used in the office.3
The intensive treatment group achieved a mean systolic pressure of 121.5 mm Hg, which required an average of 3 drugs. In contrast, the standard treatment group achieved a systolic pressure of 136.2 mm Hg, which required an average of 1.9 drugs.
Due to an absolute risk reduction in cardiovascular events and mortality, SPRINT was discontinued early after a median follow-up of 3.3 years. In the entire cohort, 61 patients needed to be treated intensively to prevent 1 cardiovascular event, and 90 needed to be treated intensively to prevent 1 death.2
Favorable outcomes in the oldest subgroup
The oldest patients in the SPRINT trial tolerated the intensive treatment as well as the youngest.2,4
Exploratory analysis of the subgroup of patients age 75 and older, who constituted 28% of the patients in the trial, demonstrated significant benefit from intensive treatment. In this subgroup, 27 patients needed to be treated aggressively (compared with standard treatment) to prevent 1 cardiovascular event, and 41 needed to be treated intensively to prevent 1 death.4 The lower numbers needing to be treated in the older subgroup than in the overall trial reflect the higher absolute risk in this older population.
Serious adverse events were more common with intensive treatment than with standard treatment in the subgroup of older patients who were frail.4 Emergency department visits or serious adverse events were more likely when gait speed (a measure of frailty) was missing from the medical record in the intensive treatment group compared with the standard treatment group. Hyponatremia (serum sodium level < 130 mmol/L) was more likely in the intensively treated group than in the standard treatment group. Although the rate of falls was higher in the oldest subgroup than in the overall SPRINT population, within this subgroup the rate of injurious falls resulting in an emergency department visit was lower with intensive treatment than with standard treatment (11.6% vs 14.1%, P = .04).4
Most of the oldest patients scored below the nominal cutoff for normal (26 points)5 on the 30-point Montreal Cognitive Assessment, and about one-quarter scored below 19, which may be consistent with a major neurocognitive disorder.6
The SPRINT investigators validated a frailty scale in the study patients and found that the most frail benefited from intensive blood pressure control, as did the slowest walkers.
SPRINT results do not apply to very frail, sick patients
For older patients with hypertension, a high burden of comorbidity, and a limited life expectancy, the 2017 guidelines defer treatment decisions to clinical judgment and patient preference.
There have been no randomized trials of blood pressure management for older adults with substantial comorbidities or dementia. The “frail” older adults in the SPRINT trial were still living in the community, without dementia. The intensively treated frail older adults had more serious adverse events than with standard treatment. Those who were documented as being unable to walk at the time of enrollment also had more serious adverse events. Institutionalized older adults and nonambulatory adults in the community would likely have even higher rates of serious adverse events with intensive treatment than the SPRINT patients, and there is concern for excessive adverse effects from intensive blood pressure control in more debilitated older patients.
DOES TREATING HIGH BLOOD PRESSURE PREVENT FRAILTY OR DEMENTIA?
Aging without frailty is an important goal of geriatric care and is likely related to cardiovascular health.7 An older adult who becomes slower physically or mentally, with diminished strength and energy, is less likely to be able to live independently.
Would treating systolic blood pressure to a target of 120 to 130 mm Hg reduce the risk of prefrailty or frailty? Unfortunately, the 3-year SPRINT follow-up of the adults age 75 and older did not show any effect of intensive treatment on gait speed or mobility limitation.8 It is possible that the early termination of the study limited outcomes.
Regarding cognition, the new guidelines say that lowering blood pressure in adults with hypertension to prevent cognitive decline and dementia is reasonable, giving it a class IIa (moderate) recommendation, but they do not offer a particular blood pressure target.
Two systematic reviews of randomized placebo-controlled trials9,10 suggested that pharmacologic treatment of hypertension reduces the progression of cognitive impairment. The trials did not use an intensive treatment goal.
The impact of intensive treatment of hypertension (to a target of 120–130 mm Hg) on the development or progression of cognitive impairment is not known at this time. The SPRINT Memory and Cognition in Decreased Hypertension analysis may shed light on the effect of intensive treatment of blood pressure on the incidence of dementia, although the early termination of SPRINT may limit its conclusions as well.
GOALS SHOULD BE PATIENT-CENTERED
The new hypertension guideline gives clinicians 2 things to think about when treating hypertensive, ambulatory, noninstitutionalized, nondemented older adults, including those age 75 and older:
- Older adults tolerate intensive blood pressure treatment as well as standard treatment. In particular, the fall rate is not increased and may even be less with intensive treatment.
- Older adults have better cardiovascular outcomes with blood pressure less than 130 mm Hg than with higher levels.
Adherence to the new guidelines would require many older adults without significant multimorbidity to take 3 drugs and undergo more frequent monitoring. This burden may align with the goals of care for many older adults. However, data do not exist to prove a benefit from intensive blood pressure control in debilitated elderly patients, and there may be harm. Lowering the medication burden may be a more important goal than lowering the pressure for this population. Blood pressure targets and hypertension management should reflect patient-centered goals of care.
- Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2017. Epub ahead of print.
- SPRINT Research Group; Wright JT Jr, Williamson JD, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373:2103–2116.
- Bakris GL. The implications of blood pressure measurement methods on treatment targets for blood pressure. Circulation 2016; 134:904–905.
- Williamson JD, Supiano MA, Applegate WB, et al; SPRINT Research Group. Intensive vs standard blood pressure control and cardiovascular disease outcomes in adults aged ≥ 75 years: a randomized clinical trial. JAMA 2016; 315:2673–2682.
- Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005; 53:695–699.
- Borland E, Nagga K, Nilsson PM, Minthon L, Nilsson ED, Palmqvist S. The Montreal Cognitive Assessment: normative data from a large Swedish population-based cohort. J Alzheimers Dis 2017; 59:893–901.
- Graciani A, Garcia-Esquinas E, Lopez-Garcia E, Banegas JR, Rodriguez-Artalejo F. Ideal cardiovascular health and risk of frailty in older adults. Circ Cardiovasc Qual Outcomes 2016; 9:239–245.
- Odden MC, Peralta CA, Berlowitz DR, et al; Systolic Blood Pressure Intervention Trial (SPRINT) Research Group. Effect of intensive blood pressure control on gait speed and mobility limitation in adults 75 years or older: a randomized clinical trial. JAMA Intern Med 2017; 177:500–507.
- Tully PJ, Hanon O, Cosh S, Tzourio C. Diuretic antihypertensive drugs and incident dementia risk: a systematic review, meta-analysis and meta-regression of prospective studies. J Hypertens 2016; 34:1027–1035.
- Rouch L, Cestac P, Hanon O, et al. Antihypertensive drugs, prevention of cognitive decline and dementia: a systematic review of observational studies, randomized controlled trials and meta-analyses, with discussion of potential mechanisms. CNS Drugs 2015; 29:113–130.
We should aim for a standard office systolic pressure lower than 130 mm Hg in most adults age 65 and older if the patient can take multiple antihypertensive medications and be followed closely for adverse effects.
This recommendation is part of the 2017 hypertension guideline from the American College of Cardiology and American Heart Association.1 This new guideline advocates drug treatment of hypertension to a target less than 130/80 mm Hg for patients of all ages for secondary prevention of cardiovascular disease, and for primary prevention in those at high risk (ie, an estimated 10-year risk of atherosclerotic cardiovascular disease of 10% or higher). The target blood pressure for those at lower risk is less than 140/90 mm Hg.
There are multiple tools to estimate the 10-year risk. All tools incorporate major predictors such as age, blood pressure, cholesterol profile, and other markers, depending on the tool. Although risk increases with age, the tools are inaccurate once the patient is approximately 80 years of age.
The recommendation for older adults omits a target diastolic pressure, since treating elevated systolic pressure has more data supporting it than treating elevated diastolic blood pressure in older people. These recommendations apply only to older adults who can walk and are living in the community, not in an institution, and includes the subset of older adults who have mild cognitive impairment and frailty. The goals of treatment should be patient-centered.
DATA BEHIND THE GUIDELINE: THE SPRINT TRIAL
The Systolic Blood Pressure Intervention Trial (SPRINT)2 enrolled 9,361 patients who, to enter, had to be at least 50 years old (the mean age was 67.9), have a systolic blood pressure of 130 to 180 mm Hg (the mean was 139.7 mm Hg), and be at risk of cardiovascular disease due to chronic kidney disease, clinical or subclinical cardiovascular disease, a 10-year Framingham risk score of at least 15%, or age 75 or older. They had few comorbidities, and patients with diabetes mellitus or prior stroke were excluded. The objective was to see if intensive blood pressure treatment reduced the incidence of adverse cardiovascular outcomes compared with standard control.
The participants were randomized to either an intensive treatment goal of systolic pressure less than 120 mm Hg or a standard treatment goal of less than 140 mm Hg. Investigators chose drugs and doses according to their clinical judgment. The study protocol called for blood pressure measurement using an untended automated cuff, which probably resulted in systolic pressure readings 5 to 10 mm Hg lower than with typical methods used in the office.3
The intensive treatment group achieved a mean systolic pressure of 121.5 mm Hg, which required an average of 3 drugs. In contrast, the standard treatment group achieved a systolic pressure of 136.2 mm Hg, which required an average of 1.9 drugs.
Due to an absolute risk reduction in cardiovascular events and mortality, SPRINT was discontinued early after a median follow-up of 3.3 years. In the entire cohort, 61 patients needed to be treated intensively to prevent 1 cardiovascular event, and 90 needed to be treated intensively to prevent 1 death.2
Favorable outcomes in the oldest subgroup
The oldest patients in the SPRINT trial tolerated the intensive treatment as well as the youngest.2,4
Exploratory analysis of the subgroup of patients age 75 and older, who constituted 28% of the patients in the trial, demonstrated significant benefit from intensive treatment. In this subgroup, 27 patients needed to be treated aggressively (compared with standard treatment) to prevent 1 cardiovascular event, and 41 needed to be treated intensively to prevent 1 death.4 The lower numbers needing to be treated in the older subgroup than in the overall trial reflect the higher absolute risk in this older population.
Serious adverse events were more common with intensive treatment than with standard treatment in the subgroup of older patients who were frail.4 Emergency department visits or serious adverse events were more likely when gait speed (a measure of frailty) was missing from the medical record in the intensive treatment group compared with the standard treatment group. Hyponatremia (serum sodium level < 130 mmol/L) was more likely in the intensively treated group than in the standard treatment group. Although the rate of falls was higher in the oldest subgroup than in the overall SPRINT population, within this subgroup the rate of injurious falls resulting in an emergency department visit was lower with intensive treatment than with standard treatment (11.6% vs 14.1%, P = .04).4
Most of the oldest patients scored below the nominal cutoff for normal (26 points)5 on the 30-point Montreal Cognitive Assessment, and about one-quarter scored below 19, which may be consistent with a major neurocognitive disorder.6
The SPRINT investigators validated a frailty scale in the study patients and found that the most frail benefited from intensive blood pressure control, as did the slowest walkers.
SPRINT results do not apply to very frail, sick patients
For older patients with hypertension, a high burden of comorbidity, and a limited life expectancy, the 2017 guidelines defer treatment decisions to clinical judgment and patient preference.
There have been no randomized trials of blood pressure management for older adults with substantial comorbidities or dementia. The “frail” older adults in the SPRINT trial were still living in the community, without dementia. The intensively treated frail older adults had more serious adverse events than with standard treatment. Those who were documented as being unable to walk at the time of enrollment also had more serious adverse events. Institutionalized older adults and nonambulatory adults in the community would likely have even higher rates of serious adverse events with intensive treatment than the SPRINT patients, and there is concern for excessive adverse effects from intensive blood pressure control in more debilitated older patients.
DOES TREATING HIGH BLOOD PRESSURE PREVENT FRAILTY OR DEMENTIA?
Aging without frailty is an important goal of geriatric care and is likely related to cardiovascular health.7 An older adult who becomes slower physically or mentally, with diminished strength and energy, is less likely to be able to live independently.
Would treating systolic blood pressure to a target of 120 to 130 mm Hg reduce the risk of prefrailty or frailty? Unfortunately, the 3-year SPRINT follow-up of the adults age 75 and older did not show any effect of intensive treatment on gait speed or mobility limitation.8 It is possible that the early termination of the study limited outcomes.
Regarding cognition, the new guidelines say that lowering blood pressure in adults with hypertension to prevent cognitive decline and dementia is reasonable, giving it a class IIa (moderate) recommendation, but they do not offer a particular blood pressure target.
Two systematic reviews of randomized placebo-controlled trials9,10 suggested that pharmacologic treatment of hypertension reduces the progression of cognitive impairment. The trials did not use an intensive treatment goal.
The impact of intensive treatment of hypertension (to a target of 120–130 mm Hg) on the development or progression of cognitive impairment is not known at this time. The SPRINT Memory and Cognition in Decreased Hypertension analysis may shed light on the effect of intensive treatment of blood pressure on the incidence of dementia, although the early termination of SPRINT may limit its conclusions as well.
GOALS SHOULD BE PATIENT-CENTERED
The new hypertension guideline gives clinicians 2 things to think about when treating hypertensive, ambulatory, noninstitutionalized, nondemented older adults, including those age 75 and older:
- Older adults tolerate intensive blood pressure treatment as well as standard treatment. In particular, the fall rate is not increased and may even be less with intensive treatment.
- Older adults have better cardiovascular outcomes with blood pressure less than 130 mm Hg than with higher levels.
Adherence to the new guidelines would require many older adults without significant multimorbidity to take 3 drugs and undergo more frequent monitoring. This burden may align with the goals of care for many older adults. However, data do not exist to prove a benefit from intensive blood pressure control in debilitated elderly patients, and there may be harm. Lowering the medication burden may be a more important goal than lowering the pressure for this population. Blood pressure targets and hypertension management should reflect patient-centered goals of care.
We should aim for a standard office systolic pressure lower than 130 mm Hg in most adults age 65 and older if the patient can take multiple antihypertensive medications and be followed closely for adverse effects.
This recommendation is part of the 2017 hypertension guideline from the American College of Cardiology and American Heart Association.1 This new guideline advocates drug treatment of hypertension to a target less than 130/80 mm Hg for patients of all ages for secondary prevention of cardiovascular disease, and for primary prevention in those at high risk (ie, an estimated 10-year risk of atherosclerotic cardiovascular disease of 10% or higher). The target blood pressure for those at lower risk is less than 140/90 mm Hg.
There are multiple tools to estimate the 10-year risk. All tools incorporate major predictors such as age, blood pressure, cholesterol profile, and other markers, depending on the tool. Although risk increases with age, the tools are inaccurate once the patient is approximately 80 years of age.
The recommendation for older adults omits a target diastolic pressure, since treating elevated systolic pressure has more data supporting it than treating elevated diastolic blood pressure in older people. These recommendations apply only to older adults who can walk and are living in the community, not in an institution, and includes the subset of older adults who have mild cognitive impairment and frailty. The goals of treatment should be patient-centered.
DATA BEHIND THE GUIDELINE: THE SPRINT TRIAL
The Systolic Blood Pressure Intervention Trial (SPRINT)2 enrolled 9,361 patients who, to enter, had to be at least 50 years old (the mean age was 67.9), have a systolic blood pressure of 130 to 180 mm Hg (the mean was 139.7 mm Hg), and be at risk of cardiovascular disease due to chronic kidney disease, clinical or subclinical cardiovascular disease, a 10-year Framingham risk score of at least 15%, or age 75 or older. They had few comorbidities, and patients with diabetes mellitus or prior stroke were excluded. The objective was to see if intensive blood pressure treatment reduced the incidence of adverse cardiovascular outcomes compared with standard control.
The participants were randomized to either an intensive treatment goal of systolic pressure less than 120 mm Hg or a standard treatment goal of less than 140 mm Hg. Investigators chose drugs and doses according to their clinical judgment. The study protocol called for blood pressure measurement using an untended automated cuff, which probably resulted in systolic pressure readings 5 to 10 mm Hg lower than with typical methods used in the office.3
The intensive treatment group achieved a mean systolic pressure of 121.5 mm Hg, which required an average of 3 drugs. In contrast, the standard treatment group achieved a systolic pressure of 136.2 mm Hg, which required an average of 1.9 drugs.
Due to an absolute risk reduction in cardiovascular events and mortality, SPRINT was discontinued early after a median follow-up of 3.3 years. In the entire cohort, 61 patients needed to be treated intensively to prevent 1 cardiovascular event, and 90 needed to be treated intensively to prevent 1 death.2
Favorable outcomes in the oldest subgroup
The oldest patients in the SPRINT trial tolerated the intensive treatment as well as the youngest.2,4
Exploratory analysis of the subgroup of patients age 75 and older, who constituted 28% of the patients in the trial, demonstrated significant benefit from intensive treatment. In this subgroup, 27 patients needed to be treated aggressively (compared with standard treatment) to prevent 1 cardiovascular event, and 41 needed to be treated intensively to prevent 1 death.4 The lower numbers needing to be treated in the older subgroup than in the overall trial reflect the higher absolute risk in this older population.
Serious adverse events were more common with intensive treatment than with standard treatment in the subgroup of older patients who were frail.4 Emergency department visits or serious adverse events were more likely when gait speed (a measure of frailty) was missing from the medical record in the intensive treatment group compared with the standard treatment group. Hyponatremia (serum sodium level < 130 mmol/L) was more likely in the intensively treated group than in the standard treatment group. Although the rate of falls was higher in the oldest subgroup than in the overall SPRINT population, within this subgroup the rate of injurious falls resulting in an emergency department visit was lower with intensive treatment than with standard treatment (11.6% vs 14.1%, P = .04).4
Most of the oldest patients scored below the nominal cutoff for normal (26 points)5 on the 30-point Montreal Cognitive Assessment, and about one-quarter scored below 19, which may be consistent with a major neurocognitive disorder.6
The SPRINT investigators validated a frailty scale in the study patients and found that the most frail benefited from intensive blood pressure control, as did the slowest walkers.
SPRINT results do not apply to very frail, sick patients
For older patients with hypertension, a high burden of comorbidity, and a limited life expectancy, the 2017 guidelines defer treatment decisions to clinical judgment and patient preference.
There have been no randomized trials of blood pressure management for older adults with substantial comorbidities or dementia. The “frail” older adults in the SPRINT trial were still living in the community, without dementia. The intensively treated frail older adults had more serious adverse events than with standard treatment. Those who were documented as being unable to walk at the time of enrollment also had more serious adverse events. Institutionalized older adults and nonambulatory adults in the community would likely have even higher rates of serious adverse events with intensive treatment than the SPRINT patients, and there is concern for excessive adverse effects from intensive blood pressure control in more debilitated older patients.
DOES TREATING HIGH BLOOD PRESSURE PREVENT FRAILTY OR DEMENTIA?
Aging without frailty is an important goal of geriatric care and is likely related to cardiovascular health.7 An older adult who becomes slower physically or mentally, with diminished strength and energy, is less likely to be able to live independently.
Would treating systolic blood pressure to a target of 120 to 130 mm Hg reduce the risk of prefrailty or frailty? Unfortunately, the 3-year SPRINT follow-up of the adults age 75 and older did not show any effect of intensive treatment on gait speed or mobility limitation.8 It is possible that the early termination of the study limited outcomes.
Regarding cognition, the new guidelines say that lowering blood pressure in adults with hypertension to prevent cognitive decline and dementia is reasonable, giving it a class IIa (moderate) recommendation, but they do not offer a particular blood pressure target.
Two systematic reviews of randomized placebo-controlled trials9,10 suggested that pharmacologic treatment of hypertension reduces the progression of cognitive impairment. The trials did not use an intensive treatment goal.
The impact of intensive treatment of hypertension (to a target of 120–130 mm Hg) on the development or progression of cognitive impairment is not known at this time. The SPRINT Memory and Cognition in Decreased Hypertension analysis may shed light on the effect of intensive treatment of blood pressure on the incidence of dementia, although the early termination of SPRINT may limit its conclusions as well.
GOALS SHOULD BE PATIENT-CENTERED
The new hypertension guideline gives clinicians 2 things to think about when treating hypertensive, ambulatory, noninstitutionalized, nondemented older adults, including those age 75 and older:
- Older adults tolerate intensive blood pressure treatment as well as standard treatment. In particular, the fall rate is not increased and may even be less with intensive treatment.
- Older adults have better cardiovascular outcomes with blood pressure less than 130 mm Hg than with higher levels.
Adherence to the new guidelines would require many older adults without significant multimorbidity to take 3 drugs and undergo more frequent monitoring. This burden may align with the goals of care for many older adults. However, data do not exist to prove a benefit from intensive blood pressure control in debilitated elderly patients, and there may be harm. Lowering the medication burden may be a more important goal than lowering the pressure for this population. Blood pressure targets and hypertension management should reflect patient-centered goals of care.
- Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2017. Epub ahead of print.
- SPRINT Research Group; Wright JT Jr, Williamson JD, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373:2103–2116.
- Bakris GL. The implications of blood pressure measurement methods on treatment targets for blood pressure. Circulation 2016; 134:904–905.
- Williamson JD, Supiano MA, Applegate WB, et al; SPRINT Research Group. Intensive vs standard blood pressure control and cardiovascular disease outcomes in adults aged ≥ 75 years: a randomized clinical trial. JAMA 2016; 315:2673–2682.
- Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005; 53:695–699.
- Borland E, Nagga K, Nilsson PM, Minthon L, Nilsson ED, Palmqvist S. The Montreal Cognitive Assessment: normative data from a large Swedish population-based cohort. J Alzheimers Dis 2017; 59:893–901.
- Graciani A, Garcia-Esquinas E, Lopez-Garcia E, Banegas JR, Rodriguez-Artalejo F. Ideal cardiovascular health and risk of frailty in older adults. Circ Cardiovasc Qual Outcomes 2016; 9:239–245.
- Odden MC, Peralta CA, Berlowitz DR, et al; Systolic Blood Pressure Intervention Trial (SPRINT) Research Group. Effect of intensive blood pressure control on gait speed and mobility limitation in adults 75 years or older: a randomized clinical trial. JAMA Intern Med 2017; 177:500–507.
- Tully PJ, Hanon O, Cosh S, Tzourio C. Diuretic antihypertensive drugs and incident dementia risk: a systematic review, meta-analysis and meta-regression of prospective studies. J Hypertens 2016; 34:1027–1035.
- Rouch L, Cestac P, Hanon O, et al. Antihypertensive drugs, prevention of cognitive decline and dementia: a systematic review of observational studies, randomized controlled trials and meta-analyses, with discussion of potential mechanisms. CNS Drugs 2015; 29:113–130.
- Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2017. Epub ahead of print.
- SPRINT Research Group; Wright JT Jr, Williamson JD, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373:2103–2116.
- Bakris GL. The implications of blood pressure measurement methods on treatment targets for blood pressure. Circulation 2016; 134:904–905.
- Williamson JD, Supiano MA, Applegate WB, et al; SPRINT Research Group. Intensive vs standard blood pressure control and cardiovascular disease outcomes in adults aged ≥ 75 years: a randomized clinical trial. JAMA 2016; 315:2673–2682.
- Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005; 53:695–699.
- Borland E, Nagga K, Nilsson PM, Minthon L, Nilsson ED, Palmqvist S. The Montreal Cognitive Assessment: normative data from a large Swedish population-based cohort. J Alzheimers Dis 2017; 59:893–901.
- Graciani A, Garcia-Esquinas E, Lopez-Garcia E, Banegas JR, Rodriguez-Artalejo F. Ideal cardiovascular health and risk of frailty in older adults. Circ Cardiovasc Qual Outcomes 2016; 9:239–245.
- Odden MC, Peralta CA, Berlowitz DR, et al; Systolic Blood Pressure Intervention Trial (SPRINT) Research Group. Effect of intensive blood pressure control on gait speed and mobility limitation in adults 75 years or older: a randomized clinical trial. JAMA Intern Med 2017; 177:500–507.
- Tully PJ, Hanon O, Cosh S, Tzourio C. Diuretic antihypertensive drugs and incident dementia risk: a systematic review, meta-analysis and meta-regression of prospective studies. J Hypertens 2016; 34:1027–1035.
- Rouch L, Cestac P, Hanon O, et al. Antihypertensive drugs, prevention of cognitive decline and dementia: a systematic review of observational studies, randomized controlled trials and meta-analyses, with discussion of potential mechanisms. CNS Drugs 2015; 29:113–130.

A 67-year-old woman with bilateral hand numbness
A 67-year-old woman presents to the emergency department after 8 weeks of progressive numbness and tingling in both hands, involving all fingers. The numbness has increased in severity in the last 3 days. She also has occasional numbness around her mouth. She reports no numbness in her feet.
She says she underwent thyroid surgery twice for thyroid cancer 10 years ago. Her medical history also includes type 2 diabetes mellitus (diagnosed 1 year ago), hypertension, dyslipidemia, and diastolic heart failure (diagnosed 5 years ago).
Her current medications are:
- Metformin 1 g twice a day
- Candesartan 16 mg once a day
- Atorvastatin 20 mg once a day
- Furosemide 40 mg twice a day
- Levothyroxine 100 μg per day
- Calcium carbonate 1,500 mg twice a day
- A vitamin D tablet twice a day, which she has not taken for the last 2 months.
She admits she has not been taking her medications regularly because she has been feeling depressed.
On physical examination, she is alert and oriented but appears anxious. She is not in respiratory distress. Her blood pressure is 150/90 mm Hg and her pulse is 92 beats per minute and regular. There is a thyroidectomy scar on the anterior neck. Her jugular venous pressure is not elevated. Her heart sounds are normal without extra sounds. She has no pulmonary rales and no lower-extremity edema.
The Phalen test and Tinel test for carpal tunnel syndrome are negative in both hands. Using a Katz hand diagram, the patient reports tingling and numbness in all fingers, both palms, and the dorsum of both hands. Tapping the area over the facial nerve does not elicit twitching of the facial muscles (ie, no Chvostek sign), but compression of the upper arm elicits carpal spasm (ie, positive Trousseau sign). There is no evidence of motor weakness in her hands. The rest of the physical examination is unremarkable.
POSSIBLE CAUSES OF NUMBNESS
1. Based on the initial evaluation, which of the following is the most likely cause of our patient’s bilateral hand numbness?
- Hypocalcemia due to primary hypoparathyroidism
- Carpal tunnel syndrome due to primary hypothyroidism
- Diabetic peripheral neuropathy
- Vitamin B12 deficiency due to metformin
- Hypocalcemia due to low serum calcitonin
All the conditions above except low serum calcitonin can cause bilateral hand paresthesia. Our patient most likely has hypocalcemia due to primary hypoparathyroidism.
Hypocalcemia
In our patient, bilateral hand numbness and perioral numbness after stopping vitamin D and a positive Trousseau sign strongly suggest hypocalcemia. The classic physical findings in patients with hypocalcemia are the Trousseau sign and the Chvostek sign. The Trousseau sign is elicited by inflating a blood pressure cuff above the systolic blood pressure for 3 minutes and observing for ischemia-induced carpopedal spasm, wrist and metacarpophalangeal joint flexion, thumb adduction, and interphalangeal joint extension. The Chvostek sign is elicited by tapping over the area of the facial nerve below the zygoma in front of the tragus, resulting in ipsilateral twitching of facial muscles.
Although the Trousseau sign is more sensitive and specific than the Chvostek sign, neither is pathognomonic for hypocalcemia.1 The Chvostek sign has been reported to be negative in 30% of patients with hypocalcemia and positive in 10% of normocalcemic individuals.1 The Trousseau sign, however, is present in 94% of hypocalcemic patients vs 1% of normocalcemic individuals.2
Primary hypoparathyroidism secondary to thyroidectomy. Postsurgical hypoparathyroidism is the most common cause of primary hypoparathyroidism. It results from ischemic injury or accidental removal of the parathyroid glands during anterior neck surgery.3,4 The consequent hypocalcemia can be transient, intermittent, or permanent. Permanent postsurgical hypoparathyroidism is defined as persistent hypocalcemia with insufficient parathyroid hormone (PTH) for more than 12 months after neck surgery; however, some consider 6 months to be enough to define the condition.5–7
The incidence of postsurgical hypoparathyroidism varies considerably with the extent of thyroid surgery and the experience of the surgeon.6,8 In the hands of experienced surgeons, permanent hypoparathyroidism occurs in fewer than 1% of patients after total thyroidectomy, whereas the rate may be higher than 6% with less-experienced surgeons.5,9 Other risk factors for postsurgical hypoparathyroidism include female sex, autoimmune thyroid disease, pregnancy, and lactation.5
Pseudohypoparathyroidism is a group of disorders characterized by renal resistance to PTH, leading to hypocalcemia, hyperphosphatemia, and elevated serum PTH. It is also associated with phenotypic features such as short stature and short fourth metacarpal bones.
Calcitonin deficiency. Calcitonin is a polypeptide hormone secreted from the parafollicular (C) cells of the thyroid gland. After total thyroidectomy, calcitonin levels are expected to be reduced. However, the role of calcitonin in humans is unclear. One study has shown that calcitonin is possibly a vestigial hormone, given that no calcitonin-related disorders (excess or deficiency) have been reported in humans.10
Carpal tunnel syndrome due to hypothyroidism
Our patient also could have primary hypothyroidism as a result of thyroidectomy. Hypothyroidism can cause bilateral hand numbness due to carpal tunnel syndrome, which is mediated by mucopolysaccharide deposition and synovial membrane swelling.11 One study reported that 29% of patients with hypothyroidism had carpal tunnel syndrome.12 Symptoms of carpal tunnel syndrome in hypothyroid patients may occur despite thyroid replacement therapy.13

Carpal tunnel syndrome is a clinical diagnosis. Patients usually experience hand paresthesia in the distribution of the median nerve. Provocative physical tests for carpal tunnel syndrome include the Tinel test, the Phalen test, and the Katz hand diagram, which is considered the best of the 3 tests.14,15 Based on how the patient marks the location and type of symptoms on the diagram, carpal tunnel syndrome is rated as classic, probable, possible, or unlikely (Table 1).14,16,17 The sensitivity of a classic or probable diagram ranges from 64% to 80%, while the specificity ranges from 73% to 90%.14,15
Carpal tunnel syndrome is less likely to be the cause of our patient’s symptoms, as her Katz hand diagram indicates only “possible” carpal tunnel syndrome. Her perioral numbness and positive Trousseau sign make hypocalcemia a more likely cause.
Diabetic peripheral neuropathy
Sensory peripheral neuropathy is a recognized complication of diabetes mellitus. However, neuropathy in diabetic patients most commonly manifests initially as distal symmetrical ascending neuropathy starting in the lower extremities.18 Therefore, diabetic peripheral neuropathy is less likely in this patient since her symptoms are limited to her hands.
Vitamin B12 deficiency
Metformin-induced vitamin B12 deficiency is another possible cause of peripheral neuropathy. It might be secondary to metformin-induced changes in intrinsic factor levels and small-intestine motility with resultant bacterial overgrowth, as well as inhibition of vitamin B12 absorption in the terminal ileum.19
However, metformin-induced vitamin B12 deficiency is not the most likely cause of our patient’s neuropathy, since she has been taking this drug for only 1 year. Vitamin B12 deficiency with consequent peripheral neuropathy is more likely in patients taking metformin in high doses for 10 or more years.20
Laboratory results and electrocardiography

Table 2 shows the patient’s initial laboratory results. Of note, her serum calcium level is 5.7 mg/dL (reference range 8.9–10.1). Electrocardiography in the emergency department shows:
- Prolonged PR interval (23 msec)
- Wide QRS complexes (13 msec)
- Flat T waves
- Prolonged corrected QT interval (475 msec)
- Occasional premature ventricular complexes.
CLINICAL MANIFESTATIONS OF HYPOCALCEMIA
2. Which of the following is not a manifestation of hypocalcemia?
- Tonic-clonic seizures
- Cyanosis
- Cardiac ventricular arrhythmias
- Acute pancreatitis
- Depression

Hypocalcemia can cause a wide range of clinical manifestations (Table 3), the extent and severity of which depend on the severity of hypocalcemia and how quickly it develops. The more acute the hypocalcemia, the more severe the manifestations.21
Tetany can cause seizures
Hypocalcemia is characterized by neuromuscular hyperexcitability, manifested clinically by tetany.22 Manifestations of tetany are numerous and include acral paresthesia, perioral numbness, muscle cramps, carpopedal spasm, and seizures. Tetany is the hallmark of hypocalcemia regardless of etiology. However, certain causes are associated with peculiar clinical manifestations. For example, chronic primary hypoparathyroidism may be associated with basal ganglia calcifications that can result in parkinsonism, other extrapyramidal disorders, and dementia (Table 4).6

Airway spasm can be fatal
A serious manifestation of acute severe hypocalcemia is spasm of the glottis muscles, which may cause cyanosis and, if untreated, death.21
Ventricular arrhythmias
Another potential fatal complication of acute severe hypocalcemia is polymorphic ventricular tachycardia due to prolongation of the QT interval, which is readily identified with electrocardiography.23
Hypocalcemia does not cause pancreatitis
Hypercalcemia, rather than hypocalcemia, may cause acute pancreatitis.24 Conversely, acute pancreatitis may cause hypocalcemia due to precipitation of calcium in the abdominal cavity.25
Psychiatric manifestations
In addition to depression, hypocalcemia is associated with psychiatric manifestations including anxiety, confusion, and emotional instability.
STEPS TO DIAGNOSIS OF HYPOCALCEMIA
First step: Confirm true hypocalcemia
Calcium circulates in the blood in 3 forms: bound to albumin (40% to 45%), bound to anions (10% to 15%), and free (ionized) (45%). Although ionized calcium is the active form, most laboratories report total serum calcium.
Since changes in serum albumin concentration affect the total serum calcium level, it is imperative to correct the measured serum calcium to the serum albumin concentration. Each 1-g/dL decrease in serum albumin lowers the total serum calcium by 0.8 mg/dL. Thus:
Corrected serum calcium (mg/dL) =
measured total serum calcium (mg/dL) +
0.8 (4 − serum albumin [g/dL]).
If the patient’s serum calcium level remains low when corrected for serum albumin, he or she has true hypocalcemia, which implies a low ionized serum calcium. Conversely, pseudohypocalcemia means that the measured calcium level is low but the corrected serum calcium is normal.
Using this formula, our patient’s corrected calcium level is calculated as 5.7 + 0.8 (4 – 3.2) = 6.3 mg/dL, indicating true hypocalcemia.
PHOSPHATE IS OFTEN HIGH WHEN CALCIUM IS LOW
In addition to hypocalcemia, our patient has an elevated phosphate level (Table 2).
3. Which of the following hypocalcemic disorders is not associated with hyperphosphatemia?
- End-stage renal disease
- Primary hypoparathyroidism
- Pseudohypoparathyroidism
- Vitamin D3 deficiency
- Rhabdomyolysis
Vitamin D deficiency is not associated with hyperphosphatemia.
Second step in evaluating hypocalcemia: Check phosphate, magnesium, creatinine

The major causes of hypocalcemia can be categorized according to the serum phosphate level: high vs normal or low (Table 5).
High-phosphate, low-calcium states. In the absence of concurrent end-stage renal disease and an excessive phosphate load, primary hypoparathyroidism is the most likely cause of hypocalcemia associated with hyperphosphatemia.
PTH increases serum ionized calcium by26,27:
- Increasing bone resorption
- Increasing reabsorption of calcium from the distal renal tubules
- Increasing the activity of 1-alpha-hydroxylase, responsible for conversion of 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3 (the most biologically active vitamin D metabolite); 1,25-dihydroxyvitamin D increases the absorption of calcium and phosphate from the intestine.
Conversely, PTH decreases reabsorption of phosphate from proximal renal tubules, resulting in hypophosphatemia. Therefore, low serum PTH (primary hypoparathyroidism) or a PTH-resistant state (pseudohypoparathyroidism) results in hypocalcemia and hyperphosphatemia.26,27
Both end-stage renal disease and rhabdomyolysis are associated with high serum phosphate levels. The kidney normally excretes excess dietary phosphate to maintain phosphate homeostasis; however, this is impaired in end-stage renal disease, leading to hyperphosphatemia. In rhabdomyolysis, it is mainly the transcellular shift of phosphate into the extracellular space from myocyte injury that raises phosphate levels.
Normal- or low-phosphate, low calcium states. Hypocalcemia can also result from vitamin D deficiency, but this cause is associated with a low or normal serum phosphate level. In such cases, hypocalcemia causes secondary hyperparathyroidism with consequent renal phosphate loss and, thus, hypophosphatemia.27
Third step: Check serum intact PTH and 25-hydroxyvitamin D levels
Hypocalcemia stimulates secretion of PTH. Therefore, hypocalcemia with elevated serum PTH is caused by disorders that do not impair PTH secretion, including chronic renal failure and vitamin D deficiency (Table 5). Conversely, hypocalcemia with low or normal serum PTH levels suggests primary hypoparathyroidism.
Our patient’s serum PTH level is 20 ng/mL, which is within the reference range. This does not discount the diagnosis of primary hypoparathyroidism. Although most patients with primary hypoparathyroidism have low or undetectable serum PTH levels, some have normal PTH levels if some degree of PTH production is preserved.5,7,28–30 In these patients, the remaining functioning parathyroid tissue is not enough to maintain a normal serum calcium level, resulting in hypocalcemia. As a result, hypocalcemia stimulates the remaining parathyroid tissue to its maximum output, producing PTH levels usually within the lower or middle-normal range.30 In such patients, the terms parathyroid insufficiency and relative primary hypoparathyroidism are more precise than primary hypoparathyroidism.
Postsurgical hypoparathyroidism with an inappropriately normal PTH level is usually seen in patients with disorders that impair intestinal calcium absorption or bone resorption.31 In our patient’s case, the “normal” serum PTH level is likely due to maximal stimulation of remaining functioning parathyroid tissue by severe hypocalcemia, which is a result of her discontinuation of calcium and calcitriol therapy and her vitamin D deficiency.
CASE RESUMED: NO RESPONSE TO INTRAVENOUS CALCIUM GLUCONATE
The patient is given 2 10-mL ampules of 10% calcium gluconate diluted in 100 mL of 5% dextrose in water over 20 minutes intravenously. Electrocardiographic monitoring is continued. Two hours later, her measured serum calcium is only 5.8 mg/dL, with no improvement in her symptoms.
A continuous infusion of calcium gluconate is started: 12 ampules of calcium gluconate are added to 380 mL of 5% dextrose in water and infused at 40 mL/hour (infused rate of elemental calcium = 1.3 mg/kg/hour); 3 hours later, her measured serum calcium level is still only 5.8 mg//dL; at 6 hours it is 5.9 mg/dL, and her symptoms have not improved.
4. Which of the following is the most appropriate next step?
- Change the calcium gluconate to calcium chloride
- Increase the infusion rate to 1.5 mg of elemental calcium/kg/hour
- Give a bolus of 2 10-mL ampules of 10% calcium gluconate intravenously over 1 minute
- Give additional oral calcium tablets
- Check the serum magnesium level
Treatment of hypocalcemia can involve intravenous or oral calcium therapy.
Intravenous calcium is indicated for patients with any of the following6,32:
- Moderate to severe neuromuscular irritability (eg, acral paresthesia, carpopedal spasm, prolonged QT interval, seizures, laryngospasm, bronchospasm)
- Acute hypocalcemia with corrected serum calcium level less than 7.6 mg/dL, even if the patient is asymptomatic
- Cardiac failure.
One 10-mL ampule of 10% calcium gluconate contains 93 mg of elemental calcium; 1 or 2 ampules are typically diluted in 50 to 100 mL of 5% dextrose in water and infused slowly over 15 to 20 minutes. Rapid administration of intravenous calcium is contraindicated, as it may produce cardiac arrhythmias and possibly cardiac arrest. Therefore, intravenous calcium should be given slowly while continuing electrocardiographic monitoring.33
Since the effect of 1 ampule of calcium gluconate lasts only 2 to 3 hours, most patients with symptomatic hypocalcemia require continuous intravenous calcium infusion. The recommended dose of infused elemental calcium is 0.5 to 1.5 mg/kg/hour.34 Several ampules are added to 500 to 1,000 mL of 5% dextrose in water or 0.9% normal saline and infused at a rate appropriate for the patient’s corrected calcium and symptoms.
Oral calcium and vitamin D supplements can be given initially to patients with a corrected serum calcium level of 7.6 mg/dL or greater, with or without mild symptoms, if they can tolerate oral intake. However, this is not the treatment of choice for resistant acute hypocalcemia, as in this case.
Calcium chloride has no advantages over calcium gluconate. Further, it can be associated with local irritation and may result in tissue necrosis if extravasation occurs.35
Increasing the infusion rate of calcium gluconate to the maximum recommended dose may improve the patient’s ionized calcium level and symptoms somewhat. However, it is not the best option for this patient, given that she did not respond to 2 ampules of calcium gluconate followed by continuous infusion of 1.3 mg/kg/hour for 6 hours.
Calcium gluconate bolus. Similarly, giving the patient an additional 2 ampules of calcium gluconate over 1 minute would not be recommended, as rapid administration of intravenous calcium gluconate (eg, over 1 minute) is contraindicated.
Check magnesium
If hypocalcemia persists despite intravenous calcium therapy, as in our patient, further investigation or action is required. An important cause of persistent hypocalcemia is severe hypomagnesemia. Severe hypomagnesemia (serum magnesium < 0.8 mg/dL) causes resistant hypocalcemia by several mechanisms:
- Inducing PTH resistance32,36,37
- Decreasing PTH secretion32,36
- Decreasing calcitriol production.
The decrease in calcitriol production is a direct effect of hypomagnesemia, but it is also an indirect effect of low PTH secretion, which inhibits the enzyme 1-alpha-hydroxylase. Thus, conversion of 25-hydroxyvitamin D3 to calcitriol is impaired, leading to low calcitriol production.
Our patient could have hypomagnesemia due to furosemide use and uncontrolled diabetes mellitus. Hypocalcemia resistant to calcium therapy may occasionally respond to magnesium therapy even if the serum magnesium level is normal. This may be due to depleted intracellular magnesium salt levels.6,38 Rarely, severe hypermagnesemia can also be associated with hypocalcemia due to inhibition of PTH secretion.37,39
CASE RESUMED
Our patient’s serum magnesium level is 0.6 mg/dL (reference range 1.7–2.4 mg/dL). She is given 2 g of magnesium sulfate in 60 mL of 0.9% normal saline infused over 1 hour, followed by a continuous infusion of magnesium sulfate (12 g diluted in 250 mL of 0.9% normal saline, infused over 24 hours). On repeat testing 4 hours later, her serum magnesium level is 0.7 mg/dL, and at 8 hours later it is 0.9 mg/dL. She is subsequently started on oral magnesium oxide 600 mg per day. The magnesium sulfate infusion is continued for another 24 hours.
PREVENTING HYPERCALCIURIA
Patients with low PTH (primary hypoparathyroidism) may have hypercalciuria due to decreased renal tubular calcium reabsorption. Two important measures can minimize hypercalciuria in such patients:
- Keeping the serum calcium level in the low-normal range4,5,40
- Giving a thiazide diuretic (eg, hydrochlorothiazide 12.5–50 mg daily) with a low-salt diet.41,42
A thiazide diuretic is usually started once the 24-hour urine calcium reaches 250 mg.6 Thiazides are thought to enhance both proximal and distal renal tubular calcium reabsorption.43,44
PRIMARY HYPOPARATHYROIDISM: LONG-TERM MANAGEMENT
Long-term management of primary hypoparathyroidism requires calcium and vitamin D supplementation.
Calcium supplements. The most commonly prescribed calcium preparations are calcium carbonate and calcium citrate (containing 40% and 20% elemental calcium, respectively). Calcium carbonate, which is less expensive than calcium citrate, binds with phosphate intake and requires an acidic environment for absorption, and so it is better absorbed when taken with meals. Because calcium citrate does not require an acidic environment for absorption, it is the calcium preparation of choice for patients on proton pump inhibitors, or patients with achlorhydria or constipation.45 Calcium doses vary widely, with most hypoparathyroid patients requiring 1 to 2 g of elemental calcium daily.6
Vitamin D supplements. To promote intestinal absorption, calcium is combined with vitamin D in a fixed-dose preparation given in divided doses.46 Calcitriol (1,25-dihydroxyvitamin D3) is the most active metabolite of vitamin D, with rapid onset and offset of action, and it is the preferred form of vitamin D therapy for patients with hypoparathyroidism. If calcitriol is not available or is not affordable, alphacalcidol (1-alpha-hydroxyvitamin D3) is another option. This is a synthetic analogue of vitamin D that is already hyroxylated at the C1 position. After oral intake, it is hydroxylated in the liver to form calcitriol.
Since renal production of calcitriol is PTH-dependent, in hypoparathyroidism the conversion of 25-hydroxyvitamin D3 to calcitriol is limited. Therefore, vitamin D3 (cholecalciferol) and vitamin D2 (ergocalciferol) are not the preferred forms of vitamin D for such patients. However, either can be added to calcitriol, as they may have extraskeletal benefits.7
CASE CONCLUDED
Our patient presented with primary parathyroid insufficiency associated with vitamin D deficiency. Therefore, in addition to calcitriol and calcium combined with vitamin D in a fixed-dose preparation, her management included vitamin D3 for her vitamin D deficiency.
She was discharged on these medications. At a follow-up visit 3 weeks later, her measured serum calcium level was 8.6 mg/dL. She reported gradual resolution of her symptoms. She was also referred to a psychiatrist for her depression.
TAKE-HOME POINTS
- Hypocalcemia causes neuromuscular excitability, manifested clinically by tetany.
- Common causes of hypocalcemia include vitamin D deficiency, hypomagnesemia, renal failure, and primary hypoparathyroidism.
- The first step in evaluating hypocalcemia is to correct the measured serum calcium to the serum albumin concentration.
- Laboratory testing for hypocalcemia should include serum phosphorus, magnesium, creatinine, PTH, and 25-hydroxyvitamin D3.
- Primary hypoparathyroidism is characterized by hypocalcemia, hyperphosphatemia, and low serum PTH.
- Moderate to severe manifestations of hypo-
calcemia and acute hypocalcemia (< 7.6 mg/dL), even if asymptomatic, warrant intravenous calcium therapy. - Correction of hypomagnesemia is essential to treat hypocalcemia, especially if resistant to intravenous calcium therapy.
- The goal of chronic management of primary hypoparathyroidism includes correcting the serum calcium level to a low-normal range, the serum phosphorus level to an upper-normal range, and prevention of hypercalciuria.
Acknowledgments: The authors wish to thank Mr. Michael Edward Tierney of the School of Medicine, University of Sydney, Australia, for his linguistic editing of the manuscript.
- Jesus JE, Landry A. Images in clinical medicine. Chvostek’s and Trousseau’s signs. N Engl J Med 2012; 367:e15.
- Urbano FL. Signs of hypocalcemia: Chvostek’s and Trousseau’s. Hosp Physician 2000; 36:43–45.
- Chisthi MM, Nair RS, Kuttanchettiyar KG, Yadev I. Mechanisms behind post-thyroidectomy hypocalcemia: interplay of calcitonin, parathormone, and albumin—a prospective study. J Invest Surg 2017; 30:217–225.
- Shoback DM, Bilezikian JP, Costa AG, et al. Presentation of hypoparathyroidism: etiologies and clinical features. J Clin Endocrinol Metab 2016; 101:2300–2312.
- Stack BC Jr, Bimston DN, Bodenner DL, et al. American Association of Clinical Endocrinologists and American College of Endocrinology disease state clinical review: postoperative hypoparathyroidism—definitions and management. Endocr Pract 2015; 21:674–685.
- Shoback D. Clinical practice. Hypoparathyroidism. N Engl J Med 2008; 359:391–403.
- Abate EG, Clarke BL. Review of hypoparathyroidism. Front Endocrinol (Lausanne) 2017; 7:172.
- Coimbra C, Monteiro F, Oliveira P, Ribeiro L, de Almeida MG, Condé A. Hypoparathyroidism following thyroidectomy: predictive factors. Acta Otorrinolaringol Esp 2017; 68:106–111.
- Thomusch O, Machens A, Sekulla C, Ukkat J, Brauckhoff M, Dralle H. The impact of surgical technique on postoperative hypoparathyroidism in bilateral thyroid surgery: a multivariate analysis of 5846 consecutive patients. Surgery 2003; 133:180–185.
- Hirsch PF, Lester GE, Talmage RV. Calcitonin, an enigmatic hormone: does it have a function? J Musculoskelet Neuronal Interact 2001; 1:299–305.
- Karne SS, Bhalerao NS. Carpal tunnel syndrome in hypothyroidism. J Clin Diagn Res 2016; 10:OC36–OC38.
- Duyff RF, Van den Bosch J, Laman DM, van Loon BJ, Linssen WH. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68:750–755.
- Palumbo CF, Szabo RM, Olmsted SL. The effects of hypothyroidism and thyroid replacement on the development of carpal tunnel syndrome. J Hand Surg Am 2000; 25:734–739.
- Katz JN, Stirrat CR, Larson MG, Fossel AH, Eaton HM, Liang MH. A self-administered hand symptom diagram for the diagnosis and epidemiologic study of carpal tunnel syndrome. J Rheumatol 1990; 17:1495–1498.
- Katz JN, Stirrat CR. A self-administered hand diagram for the diagnosis of carpal tunnel syndrome. J Hand Surg Am 1990; 15:360–363.
- Calfee RP, Dale AM, Ryan D, Descatha A, Franzblau A, Evanoff B. Performance of simplified scoring systems for hand diagrams in carpal tunnel syndrome screening. J Hand Surg Am 2012; 37:10–17.
- D’Arcy CA, McGee S. The rational clinical examination. Does this patient have carpal tunnel syndrome? JAMA 2000; 283:3110–3117.
- Marchettini P, Lacerenza M, Mauri E, Marangoni C. Painful peripheral neuropathies. Curr Neuropharmacol 2006; 4:175–181.
- Kibirige D, Mwebaze R. Vitamin B12 deficiency among patients with diabetes mellitus: is routine screening and supplementation justified? J Diabetes Metab Disord 2013;12:17.
- Akinlade KS, Agbebaku SO, Rahamon SK, Balogun WO. Vitamin B12 levels in patients with type 2 diabetes mellitus on metformin. Ann Ib Postgrad Med 2015; 13:79–83.
- Tohme JF, Bilezikian JP. Hypocalcemic emergencies. Endocrinol Metab Clin North Am 1993; 22:363–375.
- Macefield G, Burke D. Paraesthesiae and tetany induced by voluntary hyperventilation. Increased excitability of human cutaneous and motor axons. Brain 1991; 114:527–540.
- Benoit SR, Mendelsohn AB, Nourjah P, Staffa JA, Graham DJ. Risk factors for prolonged QTc among US adults: Third National Health and Nutrition Examination Survey. Eur J Cardiovasc Prev Rehabil 2005; 12:363–368.
- Khoo TK, Vege SS, Abu-Lebdeh HS, Ryu E, Nadeem S, Wermers RA. Acute pancreatitis in primary hyperparathyroidism: a population-based study. J Clin Endocrinol Metab 2009; 94:2115–2118.
- McKay C, Beastall GH, Imrie CW, Baxter JN. Circulating intact parathyroid hormone levels in acute pancreatitis. Br J Surg 1994; 81:357–360.
- Talmage RV, Mobley HT. Calcium homeostasis: reassessment of the actions of parathyroid hormone. Gen Comp Endocrinol 2008; 156:1–8.
- Friedman PA, Gesek FA. Calcium transport in renal epithelial cells. Am J Physiol 1993; 264:F181–F198.
- Jensen PV, Jelstrup SM, Homøe P. Long-term outcomes after total thyroidectomy. Dan Med J 2015; 62:A5156.
- Ritter K, Elfenbein D, Schneider DF, Chen H, Sippel RS. Hypoparathyroidism after total thyroidectomy: incidence and resolution. J Surg Res 2015; 197:348–353.
- Promberger R, Ott J, Kober F, Karik M, Freissmuth M, Hermann M. Normal parathyroid hormone levels do not exclude permanent hypoparathyroidism after thyroidectomy. Thyroid 2011; 21:145–150.
- Lorente-Poch L, Sancho JJ, Muñoz-Nova JL, Sánchez-Velázquez P, Sitges-Serra A. Defining the syndromes of parathyroid failure after total thyroidectomy. Gland Surgery 2015; 4:82–90.
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ 2008; 336:1298–1302.
- Tohme JF, Bilezikian JP. Diagnosis and treatment of hypocalcemic emergencies. Endocrinologist 1996; 6:10–18.
- Carroll R, Matfin G. Endocrine and metabolic emergencies: hypocalcaemia. Ther Adv Endocrinol Metab 2010; 1:29–33.
- Kim MP, Raho VJ, Mak J, Kaynar AM. Skin and soft tissue necrosis from calcium chloride in a deicer. J Emerg Med 2007; 32:41–44.
- Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med 2005; 20:3–17.
- Cholst IN, Steinberg SF, Tropper PJ, Fox HE, Segre GV, Bilezikian JP. The influence of hypermagnesemia on serum calcium and parathyroid hormone levels in human subjects. N Engl J Med 1984; 310:1221–1225.
- Ryzen E, Nelson TA, Rude RK. Low blood mononuclear cell magnesium content and hypocalcemia in normomagnesemic patients. West J Med 1987; 147:549–553.
- Koontz SL, Friedman SA, Schwartz ML. Symptomatic hypocalcemia after tocolytic therapy with magnesium sulfate and nifedipine. Am J Obstet Gynecol 2004; 190:1773–1776.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Porter RH, Cox BG, Heaney D, Hostetter TH, Stinebaugh BJ, Suki WN. Treatment of hypoparathyroid patients with chlorthalidone. N Engl J Med 1978; 298:577–581.
- Clarke BL, Brown EM, Collins MT, et al. Epidemiology and diagnosis of hypoparathyroidism. J Clin Endocrinol Metab 2016; 101:2284–2299.
- Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 2005; 115:1651–1658.
- Costanzo LS. Localization of diuretic action in microperfused rat distal tubules: Ca and Na transport. Am J Physiol 1985; 248:F527–F535.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Scotti A, Bianchini C, Abbiati G, Marzo A. Absorption of calcium administered alone or in fixed combination with vitamin D to healthy volunteers. Arzneimittelforschung 2001; 51:493–500.
A 67-year-old woman presents to the emergency department after 8 weeks of progressive numbness and tingling in both hands, involving all fingers. The numbness has increased in severity in the last 3 days. She also has occasional numbness around her mouth. She reports no numbness in her feet.
She says she underwent thyroid surgery twice for thyroid cancer 10 years ago. Her medical history also includes type 2 diabetes mellitus (diagnosed 1 year ago), hypertension, dyslipidemia, and diastolic heart failure (diagnosed 5 years ago).
Her current medications are:
- Metformin 1 g twice a day
- Candesartan 16 mg once a day
- Atorvastatin 20 mg once a day
- Furosemide 40 mg twice a day
- Levothyroxine 100 μg per day
- Calcium carbonate 1,500 mg twice a day
- A vitamin D tablet twice a day, which she has not taken for the last 2 months.
She admits she has not been taking her medications regularly because she has been feeling depressed.
On physical examination, she is alert and oriented but appears anxious. She is not in respiratory distress. Her blood pressure is 150/90 mm Hg and her pulse is 92 beats per minute and regular. There is a thyroidectomy scar on the anterior neck. Her jugular venous pressure is not elevated. Her heart sounds are normal without extra sounds. She has no pulmonary rales and no lower-extremity edema.
The Phalen test and Tinel test for carpal tunnel syndrome are negative in both hands. Using a Katz hand diagram, the patient reports tingling and numbness in all fingers, both palms, and the dorsum of both hands. Tapping the area over the facial nerve does not elicit twitching of the facial muscles (ie, no Chvostek sign), but compression of the upper arm elicits carpal spasm (ie, positive Trousseau sign). There is no evidence of motor weakness in her hands. The rest of the physical examination is unremarkable.
POSSIBLE CAUSES OF NUMBNESS
1. Based on the initial evaluation, which of the following is the most likely cause of our patient’s bilateral hand numbness?
- Hypocalcemia due to primary hypoparathyroidism
- Carpal tunnel syndrome due to primary hypothyroidism
- Diabetic peripheral neuropathy
- Vitamin B12 deficiency due to metformin
- Hypocalcemia due to low serum calcitonin
All the conditions above except low serum calcitonin can cause bilateral hand paresthesia. Our patient most likely has hypocalcemia due to primary hypoparathyroidism.
Hypocalcemia
In our patient, bilateral hand numbness and perioral numbness after stopping vitamin D and a positive Trousseau sign strongly suggest hypocalcemia. The classic physical findings in patients with hypocalcemia are the Trousseau sign and the Chvostek sign. The Trousseau sign is elicited by inflating a blood pressure cuff above the systolic blood pressure for 3 minutes and observing for ischemia-induced carpopedal spasm, wrist and metacarpophalangeal joint flexion, thumb adduction, and interphalangeal joint extension. The Chvostek sign is elicited by tapping over the area of the facial nerve below the zygoma in front of the tragus, resulting in ipsilateral twitching of facial muscles.
Although the Trousseau sign is more sensitive and specific than the Chvostek sign, neither is pathognomonic for hypocalcemia.1 The Chvostek sign has been reported to be negative in 30% of patients with hypocalcemia and positive in 10% of normocalcemic individuals.1 The Trousseau sign, however, is present in 94% of hypocalcemic patients vs 1% of normocalcemic individuals.2
Primary hypoparathyroidism secondary to thyroidectomy. Postsurgical hypoparathyroidism is the most common cause of primary hypoparathyroidism. It results from ischemic injury or accidental removal of the parathyroid glands during anterior neck surgery.3,4 The consequent hypocalcemia can be transient, intermittent, or permanent. Permanent postsurgical hypoparathyroidism is defined as persistent hypocalcemia with insufficient parathyroid hormone (PTH) for more than 12 months after neck surgery; however, some consider 6 months to be enough to define the condition.5–7
The incidence of postsurgical hypoparathyroidism varies considerably with the extent of thyroid surgery and the experience of the surgeon.6,8 In the hands of experienced surgeons, permanent hypoparathyroidism occurs in fewer than 1% of patients after total thyroidectomy, whereas the rate may be higher than 6% with less-experienced surgeons.5,9 Other risk factors for postsurgical hypoparathyroidism include female sex, autoimmune thyroid disease, pregnancy, and lactation.5
Pseudohypoparathyroidism is a group of disorders characterized by renal resistance to PTH, leading to hypocalcemia, hyperphosphatemia, and elevated serum PTH. It is also associated with phenotypic features such as short stature and short fourth metacarpal bones.
Calcitonin deficiency. Calcitonin is a polypeptide hormone secreted from the parafollicular (C) cells of the thyroid gland. After total thyroidectomy, calcitonin levels are expected to be reduced. However, the role of calcitonin in humans is unclear. One study has shown that calcitonin is possibly a vestigial hormone, given that no calcitonin-related disorders (excess or deficiency) have been reported in humans.10
Carpal tunnel syndrome due to hypothyroidism
Our patient also could have primary hypothyroidism as a result of thyroidectomy. Hypothyroidism can cause bilateral hand numbness due to carpal tunnel syndrome, which is mediated by mucopolysaccharide deposition and synovial membrane swelling.11 One study reported that 29% of patients with hypothyroidism had carpal tunnel syndrome.12 Symptoms of carpal tunnel syndrome in hypothyroid patients may occur despite thyroid replacement therapy.13
Carpal tunnel syndrome is a clinical diagnosis. Patients usually experience hand paresthesia in the distribution of the median nerve. Provocative physical tests for carpal tunnel syndrome include the Tinel test, the Phalen test, and the Katz hand diagram, which is considered the best of the 3 tests.14,15 Based on how the patient marks the location and type of symptoms on the diagram, carpal tunnel syndrome is rated as classic, probable, possible, or unlikely (Table 1).14,16,17 The sensitivity of a classic or probable diagram ranges from 64% to 80%, while the specificity ranges from 73% to 90%.14,15
Carpal tunnel syndrome is less likely to be the cause of our patient’s symptoms, as her Katz hand diagram indicates only “possible” carpal tunnel syndrome. Her perioral numbness and positive Trousseau sign make hypocalcemia a more likely cause.
Diabetic peripheral neuropathy
Sensory peripheral neuropathy is a recognized complication of diabetes mellitus. However, neuropathy in diabetic patients most commonly manifests initially as distal symmetrical ascending neuropathy starting in the lower extremities.18 Therefore, diabetic peripheral neuropathy is less likely in this patient since her symptoms are limited to her hands.
Vitamin B12 deficiency
Metformin-induced vitamin B12 deficiency is another possible cause of peripheral neuropathy. It might be secondary to metformin-induced changes in intrinsic factor levels and small-intestine motility with resultant bacterial overgrowth, as well as inhibition of vitamin B12 absorption in the terminal ileum.19
However, metformin-induced vitamin B12 deficiency is not the most likely cause of our patient’s neuropathy, since she has been taking this drug for only 1 year. Vitamin B12 deficiency with consequent peripheral neuropathy is more likely in patients taking metformin in high doses for 10 or more years.20
Laboratory results and electrocardiography
Table 2 shows the patient’s initial laboratory results. Of note, her serum calcium level is 5.7 mg/dL (reference range 8.9–10.1). Electrocardiography in the emergency department shows:
- Prolonged PR interval (23 msec)
- Wide QRS complexes (13 msec)
- Flat T waves
- Prolonged corrected QT interval (475 msec)
- Occasional premature ventricular complexes.
CLINICAL MANIFESTATIONS OF HYPOCALCEMIA
2. Which of the following is not a manifestation of hypocalcemia?
- Tonic-clonic seizures
- Cyanosis
- Cardiac ventricular arrhythmias
- Acute pancreatitis
- Depression
Hypocalcemia can cause a wide range of clinical manifestations (Table 3), the extent and severity of which depend on the severity of hypocalcemia and how quickly it develops. The more acute the hypocalcemia, the more severe the manifestations.21
Tetany can cause seizures
Hypocalcemia is characterized by neuromuscular hyperexcitability, manifested clinically by tetany.22 Manifestations of tetany are numerous and include acral paresthesia, perioral numbness, muscle cramps, carpopedal spasm, and seizures. Tetany is the hallmark of hypocalcemia regardless of etiology. However, certain causes are associated with peculiar clinical manifestations. For example, chronic primary hypoparathyroidism may be associated with basal ganglia calcifications that can result in parkinsonism, other extrapyramidal disorders, and dementia (Table 4).6
Airway spasm can be fatal
A serious manifestation of acute severe hypocalcemia is spasm of the glottis muscles, which may cause cyanosis and, if untreated, death.21
Ventricular arrhythmias
Another potential fatal complication of acute severe hypocalcemia is polymorphic ventricular tachycardia due to prolongation of the QT interval, which is readily identified with electrocardiography.23
Hypocalcemia does not cause pancreatitis
Hypercalcemia, rather than hypocalcemia, may cause acute pancreatitis.24 Conversely, acute pancreatitis may cause hypocalcemia due to precipitation of calcium in the abdominal cavity.25
Psychiatric manifestations
In addition to depression, hypocalcemia is associated with psychiatric manifestations including anxiety, confusion, and emotional instability.
STEPS TO DIAGNOSIS OF HYPOCALCEMIA
First step: Confirm true hypocalcemia
Calcium circulates in the blood in 3 forms: bound to albumin (40% to 45%), bound to anions (10% to 15%), and free (ionized) (45%). Although ionized calcium is the active form, most laboratories report total serum calcium.
Since changes in serum albumin concentration affect the total serum calcium level, it is imperative to correct the measured serum calcium to the serum albumin concentration. Each 1-g/dL decrease in serum albumin lowers the total serum calcium by 0.8 mg/dL. Thus:
Corrected serum calcium (mg/dL) =
measured total serum calcium (mg/dL) +
0.8 (4 − serum albumin [g/dL]).
If the patient’s serum calcium level remains low when corrected for serum albumin, he or she has true hypocalcemia, which implies a low ionized serum calcium. Conversely, pseudohypocalcemia means that the measured calcium level is low but the corrected serum calcium is normal.
Using this formula, our patient’s corrected calcium level is calculated as 5.7 + 0.8 (4 – 3.2) = 6.3 mg/dL, indicating true hypocalcemia.
PHOSPHATE IS OFTEN HIGH WHEN CALCIUM IS LOW
In addition to hypocalcemia, our patient has an elevated phosphate level (Table 2).
3. Which of the following hypocalcemic disorders is not associated with hyperphosphatemia?
- End-stage renal disease
- Primary hypoparathyroidism
- Pseudohypoparathyroidism
- Vitamin D3 deficiency
- Rhabdomyolysis
Vitamin D deficiency is not associated with hyperphosphatemia.
Second step in evaluating hypocalcemia: Check phosphate, magnesium, creatinine
The major causes of hypocalcemia can be categorized according to the serum phosphate level: high vs normal or low (Table 5).
High-phosphate, low-calcium states. In the absence of concurrent end-stage renal disease and an excessive phosphate load, primary hypoparathyroidism is the most likely cause of hypocalcemia associated with hyperphosphatemia.
PTH increases serum ionized calcium by26,27:
- Increasing bone resorption
- Increasing reabsorption of calcium from the distal renal tubules
- Increasing the activity of 1-alpha-hydroxylase, responsible for conversion of 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3 (the most biologically active vitamin D metabolite); 1,25-dihydroxyvitamin D increases the absorption of calcium and phosphate from the intestine.
Conversely, PTH decreases reabsorption of phosphate from proximal renal tubules, resulting in hypophosphatemia. Therefore, low serum PTH (primary hypoparathyroidism) or a PTH-resistant state (pseudohypoparathyroidism) results in hypocalcemia and hyperphosphatemia.26,27
Both end-stage renal disease and rhabdomyolysis are associated with high serum phosphate levels. The kidney normally excretes excess dietary phosphate to maintain phosphate homeostasis; however, this is impaired in end-stage renal disease, leading to hyperphosphatemia. In rhabdomyolysis, it is mainly the transcellular shift of phosphate into the extracellular space from myocyte injury that raises phosphate levels.
Normal- or low-phosphate, low calcium states. Hypocalcemia can also result from vitamin D deficiency, but this cause is associated with a low or normal serum phosphate level. In such cases, hypocalcemia causes secondary hyperparathyroidism with consequent renal phosphate loss and, thus, hypophosphatemia.27
Third step: Check serum intact PTH and 25-hydroxyvitamin D levels
Hypocalcemia stimulates secretion of PTH. Therefore, hypocalcemia with elevated serum PTH is caused by disorders that do not impair PTH secretion, including chronic renal failure and vitamin D deficiency (Table 5). Conversely, hypocalcemia with low or normal serum PTH levels suggests primary hypoparathyroidism.
Our patient’s serum PTH level is 20 ng/mL, which is within the reference range. This does not discount the diagnosis of primary hypoparathyroidism. Although most patients with primary hypoparathyroidism have low or undetectable serum PTH levels, some have normal PTH levels if some degree of PTH production is preserved.5,7,28–30 In these patients, the remaining functioning parathyroid tissue is not enough to maintain a normal serum calcium level, resulting in hypocalcemia. As a result, hypocalcemia stimulates the remaining parathyroid tissue to its maximum output, producing PTH levels usually within the lower or middle-normal range.30 In such patients, the terms parathyroid insufficiency and relative primary hypoparathyroidism are more precise than primary hypoparathyroidism.
Postsurgical hypoparathyroidism with an inappropriately normal PTH level is usually seen in patients with disorders that impair intestinal calcium absorption or bone resorption.31 In our patient’s case, the “normal” serum PTH level is likely due to maximal stimulation of remaining functioning parathyroid tissue by severe hypocalcemia, which is a result of her discontinuation of calcium and calcitriol therapy and her vitamin D deficiency.
CASE RESUMED: NO RESPONSE TO INTRAVENOUS CALCIUM GLUCONATE
The patient is given 2 10-mL ampules of 10% calcium gluconate diluted in 100 mL of 5% dextrose in water over 20 minutes intravenously. Electrocardiographic monitoring is continued. Two hours later, her measured serum calcium is only 5.8 mg/dL, with no improvement in her symptoms.
A continuous infusion of calcium gluconate is started: 12 ampules of calcium gluconate are added to 380 mL of 5% dextrose in water and infused at 40 mL/hour (infused rate of elemental calcium = 1.3 mg/kg/hour); 3 hours later, her measured serum calcium level is still only 5.8 mg//dL; at 6 hours it is 5.9 mg/dL, and her symptoms have not improved.
4. Which of the following is the most appropriate next step?
- Change the calcium gluconate to calcium chloride
- Increase the infusion rate to 1.5 mg of elemental calcium/kg/hour
- Give a bolus of 2 10-mL ampules of 10% calcium gluconate intravenously over 1 minute
- Give additional oral calcium tablets
- Check the serum magnesium level
Treatment of hypocalcemia can involve intravenous or oral calcium therapy.
Intravenous calcium is indicated for patients with any of the following6,32:
- Moderate to severe neuromuscular irritability (eg, acral paresthesia, carpopedal spasm, prolonged QT interval, seizures, laryngospasm, bronchospasm)
- Acute hypocalcemia with corrected serum calcium level less than 7.6 mg/dL, even if the patient is asymptomatic
- Cardiac failure.
One 10-mL ampule of 10% calcium gluconate contains 93 mg of elemental calcium; 1 or 2 ampules are typically diluted in 50 to 100 mL of 5% dextrose in water and infused slowly over 15 to 20 minutes. Rapid administration of intravenous calcium is contraindicated, as it may produce cardiac arrhythmias and possibly cardiac arrest. Therefore, intravenous calcium should be given slowly while continuing electrocardiographic monitoring.33
Since the effect of 1 ampule of calcium gluconate lasts only 2 to 3 hours, most patients with symptomatic hypocalcemia require continuous intravenous calcium infusion. The recommended dose of infused elemental calcium is 0.5 to 1.5 mg/kg/hour.34 Several ampules are added to 500 to 1,000 mL of 5% dextrose in water or 0.9% normal saline and infused at a rate appropriate for the patient’s corrected calcium and symptoms.
Oral calcium and vitamin D supplements can be given initially to patients with a corrected serum calcium level of 7.6 mg/dL or greater, with or without mild symptoms, if they can tolerate oral intake. However, this is not the treatment of choice for resistant acute hypocalcemia, as in this case.
Calcium chloride has no advantages over calcium gluconate. Further, it can be associated with local irritation and may result in tissue necrosis if extravasation occurs.35
Increasing the infusion rate of calcium gluconate to the maximum recommended dose may improve the patient’s ionized calcium level and symptoms somewhat. However, it is not the best option for this patient, given that she did not respond to 2 ampules of calcium gluconate followed by continuous infusion of 1.3 mg/kg/hour for 6 hours.
Calcium gluconate bolus. Similarly, giving the patient an additional 2 ampules of calcium gluconate over 1 minute would not be recommended, as rapid administration of intravenous calcium gluconate (eg, over 1 minute) is contraindicated.
Check magnesium
If hypocalcemia persists despite intravenous calcium therapy, as in our patient, further investigation or action is required. An important cause of persistent hypocalcemia is severe hypomagnesemia. Severe hypomagnesemia (serum magnesium < 0.8 mg/dL) causes resistant hypocalcemia by several mechanisms:
- Inducing PTH resistance32,36,37
- Decreasing PTH secretion32,36
- Decreasing calcitriol production.
The decrease in calcitriol production is a direct effect of hypomagnesemia, but it is also an indirect effect of low PTH secretion, which inhibits the enzyme 1-alpha-hydroxylase. Thus, conversion of 25-hydroxyvitamin D3 to calcitriol is impaired, leading to low calcitriol production.
Our patient could have hypomagnesemia due to furosemide use and uncontrolled diabetes mellitus. Hypocalcemia resistant to calcium therapy may occasionally respond to magnesium therapy even if the serum magnesium level is normal. This may be due to depleted intracellular magnesium salt levels.6,38 Rarely, severe hypermagnesemia can also be associated with hypocalcemia due to inhibition of PTH secretion.37,39
CASE RESUMED
Our patient’s serum magnesium level is 0.6 mg/dL (reference range 1.7–2.4 mg/dL). She is given 2 g of magnesium sulfate in 60 mL of 0.9% normal saline infused over 1 hour, followed by a continuous infusion of magnesium sulfate (12 g diluted in 250 mL of 0.9% normal saline, infused over 24 hours). On repeat testing 4 hours later, her serum magnesium level is 0.7 mg/dL, and at 8 hours later it is 0.9 mg/dL. She is subsequently started on oral magnesium oxide 600 mg per day. The magnesium sulfate infusion is continued for another 24 hours.
PREVENTING HYPERCALCIURIA
Patients with low PTH (primary hypoparathyroidism) may have hypercalciuria due to decreased renal tubular calcium reabsorption. Two important measures can minimize hypercalciuria in such patients:
- Keeping the serum calcium level in the low-normal range4,5,40
- Giving a thiazide diuretic (eg, hydrochlorothiazide 12.5–50 mg daily) with a low-salt diet.41,42
A thiazide diuretic is usually started once the 24-hour urine calcium reaches 250 mg.6 Thiazides are thought to enhance both proximal and distal renal tubular calcium reabsorption.43,44
PRIMARY HYPOPARATHYROIDISM: LONG-TERM MANAGEMENT
Long-term management of primary hypoparathyroidism requires calcium and vitamin D supplementation.
Calcium supplements. The most commonly prescribed calcium preparations are calcium carbonate and calcium citrate (containing 40% and 20% elemental calcium, respectively). Calcium carbonate, which is less expensive than calcium citrate, binds with phosphate intake and requires an acidic environment for absorption, and so it is better absorbed when taken with meals. Because calcium citrate does not require an acidic environment for absorption, it is the calcium preparation of choice for patients on proton pump inhibitors, or patients with achlorhydria or constipation.45 Calcium doses vary widely, with most hypoparathyroid patients requiring 1 to 2 g of elemental calcium daily.6
Vitamin D supplements. To promote intestinal absorption, calcium is combined with vitamin D in a fixed-dose preparation given in divided doses.46 Calcitriol (1,25-dihydroxyvitamin D3) is the most active metabolite of vitamin D, with rapid onset and offset of action, and it is the preferred form of vitamin D therapy for patients with hypoparathyroidism. If calcitriol is not available or is not affordable, alphacalcidol (1-alpha-hydroxyvitamin D3) is another option. This is a synthetic analogue of vitamin D that is already hyroxylated at the C1 position. After oral intake, it is hydroxylated in the liver to form calcitriol.
Since renal production of calcitriol is PTH-dependent, in hypoparathyroidism the conversion of 25-hydroxyvitamin D3 to calcitriol is limited. Therefore, vitamin D3 (cholecalciferol) and vitamin D2 (ergocalciferol) are not the preferred forms of vitamin D for such patients. However, either can be added to calcitriol, as they may have extraskeletal benefits.7
CASE CONCLUDED
Our patient presented with primary parathyroid insufficiency associated with vitamin D deficiency. Therefore, in addition to calcitriol and calcium combined with vitamin D in a fixed-dose preparation, her management included vitamin D3 for her vitamin D deficiency.
She was discharged on these medications. At a follow-up visit 3 weeks later, her measured serum calcium level was 8.6 mg/dL. She reported gradual resolution of her symptoms. She was also referred to a psychiatrist for her depression.
TAKE-HOME POINTS
- Hypocalcemia causes neuromuscular excitability, manifested clinically by tetany.
- Common causes of hypocalcemia include vitamin D deficiency, hypomagnesemia, renal failure, and primary hypoparathyroidism.
- The first step in evaluating hypocalcemia is to correct the measured serum calcium to the serum albumin concentration.
- Laboratory testing for hypocalcemia should include serum phosphorus, magnesium, creatinine, PTH, and 25-hydroxyvitamin D3.
- Primary hypoparathyroidism is characterized by hypocalcemia, hyperphosphatemia, and low serum PTH.
- Moderate to severe manifestations of hypo-
calcemia and acute hypocalcemia (< 7.6 mg/dL), even if asymptomatic, warrant intravenous calcium therapy. - Correction of hypomagnesemia is essential to treat hypocalcemia, especially if resistant to intravenous calcium therapy.
- The goal of chronic management of primary hypoparathyroidism includes correcting the serum calcium level to a low-normal range, the serum phosphorus level to an upper-normal range, and prevention of hypercalciuria.
Acknowledgments: The authors wish to thank Mr. Michael Edward Tierney of the School of Medicine, University of Sydney, Australia, for his linguistic editing of the manuscript.
A 67-year-old woman presents to the emergency department after 8 weeks of progressive numbness and tingling in both hands, involving all fingers. The numbness has increased in severity in the last 3 days. She also has occasional numbness around her mouth. She reports no numbness in her feet.
She says she underwent thyroid surgery twice for thyroid cancer 10 years ago. Her medical history also includes type 2 diabetes mellitus (diagnosed 1 year ago), hypertension, dyslipidemia, and diastolic heart failure (diagnosed 5 years ago).
Her current medications are:
- Metformin 1 g twice a day
- Candesartan 16 mg once a day
- Atorvastatin 20 mg once a day
- Furosemide 40 mg twice a day
- Levothyroxine 100 μg per day
- Calcium carbonate 1,500 mg twice a day
- A vitamin D tablet twice a day, which she has not taken for the last 2 months.
She admits she has not been taking her medications regularly because she has been feeling depressed.
On physical examination, she is alert and oriented but appears anxious. She is not in respiratory distress. Her blood pressure is 150/90 mm Hg and her pulse is 92 beats per minute and regular. There is a thyroidectomy scar on the anterior neck. Her jugular venous pressure is not elevated. Her heart sounds are normal without extra sounds. She has no pulmonary rales and no lower-extremity edema.
The Phalen test and Tinel test for carpal tunnel syndrome are negative in both hands. Using a Katz hand diagram, the patient reports tingling and numbness in all fingers, both palms, and the dorsum of both hands. Tapping the area over the facial nerve does not elicit twitching of the facial muscles (ie, no Chvostek sign), but compression of the upper arm elicits carpal spasm (ie, positive Trousseau sign). There is no evidence of motor weakness in her hands. The rest of the physical examination is unremarkable.
POSSIBLE CAUSES OF NUMBNESS
1. Based on the initial evaluation, which of the following is the most likely cause of our patient’s bilateral hand numbness?
- Hypocalcemia due to primary hypoparathyroidism
- Carpal tunnel syndrome due to primary hypothyroidism
- Diabetic peripheral neuropathy
- Vitamin B12 deficiency due to metformin
- Hypocalcemia due to low serum calcitonin
All the conditions above except low serum calcitonin can cause bilateral hand paresthesia. Our patient most likely has hypocalcemia due to primary hypoparathyroidism.
Hypocalcemia
In our patient, bilateral hand numbness and perioral numbness after stopping vitamin D and a positive Trousseau sign strongly suggest hypocalcemia. The classic physical findings in patients with hypocalcemia are the Trousseau sign and the Chvostek sign. The Trousseau sign is elicited by inflating a blood pressure cuff above the systolic blood pressure for 3 minutes and observing for ischemia-induced carpopedal spasm, wrist and metacarpophalangeal joint flexion, thumb adduction, and interphalangeal joint extension. The Chvostek sign is elicited by tapping over the area of the facial nerve below the zygoma in front of the tragus, resulting in ipsilateral twitching of facial muscles.
Although the Trousseau sign is more sensitive and specific than the Chvostek sign, neither is pathognomonic for hypocalcemia.1 The Chvostek sign has been reported to be negative in 30% of patients with hypocalcemia and positive in 10% of normocalcemic individuals.1 The Trousseau sign, however, is present in 94% of hypocalcemic patients vs 1% of normocalcemic individuals.2
Primary hypoparathyroidism secondary to thyroidectomy. Postsurgical hypoparathyroidism is the most common cause of primary hypoparathyroidism. It results from ischemic injury or accidental removal of the parathyroid glands during anterior neck surgery.3,4 The consequent hypocalcemia can be transient, intermittent, or permanent. Permanent postsurgical hypoparathyroidism is defined as persistent hypocalcemia with insufficient parathyroid hormone (PTH) for more than 12 months after neck surgery; however, some consider 6 months to be enough to define the condition.5–7
The incidence of postsurgical hypoparathyroidism varies considerably with the extent of thyroid surgery and the experience of the surgeon.6,8 In the hands of experienced surgeons, permanent hypoparathyroidism occurs in fewer than 1% of patients after total thyroidectomy, whereas the rate may be higher than 6% with less-experienced surgeons.5,9 Other risk factors for postsurgical hypoparathyroidism include female sex, autoimmune thyroid disease, pregnancy, and lactation.5
Pseudohypoparathyroidism is a group of disorders characterized by renal resistance to PTH, leading to hypocalcemia, hyperphosphatemia, and elevated serum PTH. It is also associated with phenotypic features such as short stature and short fourth metacarpal bones.
Calcitonin deficiency. Calcitonin is a polypeptide hormone secreted from the parafollicular (C) cells of the thyroid gland. After total thyroidectomy, calcitonin levels are expected to be reduced. However, the role of calcitonin in humans is unclear. One study has shown that calcitonin is possibly a vestigial hormone, given that no calcitonin-related disorders (excess or deficiency) have been reported in humans.10
Carpal tunnel syndrome due to hypothyroidism
Our patient also could have primary hypothyroidism as a result of thyroidectomy. Hypothyroidism can cause bilateral hand numbness due to carpal tunnel syndrome, which is mediated by mucopolysaccharide deposition and synovial membrane swelling.11 One study reported that 29% of patients with hypothyroidism had carpal tunnel syndrome.12 Symptoms of carpal tunnel syndrome in hypothyroid patients may occur despite thyroid replacement therapy.13
Carpal tunnel syndrome is a clinical diagnosis. Patients usually experience hand paresthesia in the distribution of the median nerve. Provocative physical tests for carpal tunnel syndrome include the Tinel test, the Phalen test, and the Katz hand diagram, which is considered the best of the 3 tests.14,15 Based on how the patient marks the location and type of symptoms on the diagram, carpal tunnel syndrome is rated as classic, probable, possible, or unlikely (Table 1).14,16,17 The sensitivity of a classic or probable diagram ranges from 64% to 80%, while the specificity ranges from 73% to 90%.14,15
Carpal tunnel syndrome is less likely to be the cause of our patient’s symptoms, as her Katz hand diagram indicates only “possible” carpal tunnel syndrome. Her perioral numbness and positive Trousseau sign make hypocalcemia a more likely cause.
Diabetic peripheral neuropathy
Sensory peripheral neuropathy is a recognized complication of diabetes mellitus. However, neuropathy in diabetic patients most commonly manifests initially as distal symmetrical ascending neuropathy starting in the lower extremities.18 Therefore, diabetic peripheral neuropathy is less likely in this patient since her symptoms are limited to her hands.
Vitamin B12 deficiency
Metformin-induced vitamin B12 deficiency is another possible cause of peripheral neuropathy. It might be secondary to metformin-induced changes in intrinsic factor levels and small-intestine motility with resultant bacterial overgrowth, as well as inhibition of vitamin B12 absorption in the terminal ileum.19
However, metformin-induced vitamin B12 deficiency is not the most likely cause of our patient’s neuropathy, since she has been taking this drug for only 1 year. Vitamin B12 deficiency with consequent peripheral neuropathy is more likely in patients taking metformin in high doses for 10 or more years.20
Laboratory results and electrocardiography
Table 2 shows the patient’s initial laboratory results. Of note, her serum calcium level is 5.7 mg/dL (reference range 8.9–10.1). Electrocardiography in the emergency department shows:
- Prolonged PR interval (23 msec)
- Wide QRS complexes (13 msec)
- Flat T waves
- Prolonged corrected QT interval (475 msec)
- Occasional premature ventricular complexes.
CLINICAL MANIFESTATIONS OF HYPOCALCEMIA
2. Which of the following is not a manifestation of hypocalcemia?
- Tonic-clonic seizures
- Cyanosis
- Cardiac ventricular arrhythmias
- Acute pancreatitis
- Depression
Hypocalcemia can cause a wide range of clinical manifestations (Table 3), the extent and severity of which depend on the severity of hypocalcemia and how quickly it develops. The more acute the hypocalcemia, the more severe the manifestations.21
Tetany can cause seizures
Hypocalcemia is characterized by neuromuscular hyperexcitability, manifested clinically by tetany.22 Manifestations of tetany are numerous and include acral paresthesia, perioral numbness, muscle cramps, carpopedal spasm, and seizures. Tetany is the hallmark of hypocalcemia regardless of etiology. However, certain causes are associated with peculiar clinical manifestations. For example, chronic primary hypoparathyroidism may be associated with basal ganglia calcifications that can result in parkinsonism, other extrapyramidal disorders, and dementia (Table 4).6
Airway spasm can be fatal
A serious manifestation of acute severe hypocalcemia is spasm of the glottis muscles, which may cause cyanosis and, if untreated, death.21
Ventricular arrhythmias
Another potential fatal complication of acute severe hypocalcemia is polymorphic ventricular tachycardia due to prolongation of the QT interval, which is readily identified with electrocardiography.23
Hypocalcemia does not cause pancreatitis
Hypercalcemia, rather than hypocalcemia, may cause acute pancreatitis.24 Conversely, acute pancreatitis may cause hypocalcemia due to precipitation of calcium in the abdominal cavity.25
Psychiatric manifestations
In addition to depression, hypocalcemia is associated with psychiatric manifestations including anxiety, confusion, and emotional instability.
STEPS TO DIAGNOSIS OF HYPOCALCEMIA
First step: Confirm true hypocalcemia
Calcium circulates in the blood in 3 forms: bound to albumin (40% to 45%), bound to anions (10% to 15%), and free (ionized) (45%). Although ionized calcium is the active form, most laboratories report total serum calcium.
Since changes in serum albumin concentration affect the total serum calcium level, it is imperative to correct the measured serum calcium to the serum albumin concentration. Each 1-g/dL decrease in serum albumin lowers the total serum calcium by 0.8 mg/dL. Thus:
Corrected serum calcium (mg/dL) =
measured total serum calcium (mg/dL) +
0.8 (4 − serum albumin [g/dL]).
If the patient’s serum calcium level remains low when corrected for serum albumin, he or she has true hypocalcemia, which implies a low ionized serum calcium. Conversely, pseudohypocalcemia means that the measured calcium level is low but the corrected serum calcium is normal.
Using this formula, our patient’s corrected calcium level is calculated as 5.7 + 0.8 (4 – 3.2) = 6.3 mg/dL, indicating true hypocalcemia.
PHOSPHATE IS OFTEN HIGH WHEN CALCIUM IS LOW
In addition to hypocalcemia, our patient has an elevated phosphate level (Table 2).
3. Which of the following hypocalcemic disorders is not associated with hyperphosphatemia?
- End-stage renal disease
- Primary hypoparathyroidism
- Pseudohypoparathyroidism
- Vitamin D3 deficiency
- Rhabdomyolysis
Vitamin D deficiency is not associated with hyperphosphatemia.
Second step in evaluating hypocalcemia: Check phosphate, magnesium, creatinine
The major causes of hypocalcemia can be categorized according to the serum phosphate level: high vs normal or low (Table 5).
High-phosphate, low-calcium states. In the absence of concurrent end-stage renal disease and an excessive phosphate load, primary hypoparathyroidism is the most likely cause of hypocalcemia associated with hyperphosphatemia.
PTH increases serum ionized calcium by26,27:
- Increasing bone resorption
- Increasing reabsorption of calcium from the distal renal tubules
- Increasing the activity of 1-alpha-hydroxylase, responsible for conversion of 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3 (the most biologically active vitamin D metabolite); 1,25-dihydroxyvitamin D increases the absorption of calcium and phosphate from the intestine.
Conversely, PTH decreases reabsorption of phosphate from proximal renal tubules, resulting in hypophosphatemia. Therefore, low serum PTH (primary hypoparathyroidism) or a PTH-resistant state (pseudohypoparathyroidism) results in hypocalcemia and hyperphosphatemia.26,27
Both end-stage renal disease and rhabdomyolysis are associated with high serum phosphate levels. The kidney normally excretes excess dietary phosphate to maintain phosphate homeostasis; however, this is impaired in end-stage renal disease, leading to hyperphosphatemia. In rhabdomyolysis, it is mainly the transcellular shift of phosphate into the extracellular space from myocyte injury that raises phosphate levels.
Normal- or low-phosphate, low calcium states. Hypocalcemia can also result from vitamin D deficiency, but this cause is associated with a low or normal serum phosphate level. In such cases, hypocalcemia causes secondary hyperparathyroidism with consequent renal phosphate loss and, thus, hypophosphatemia.27
Third step: Check serum intact PTH and 25-hydroxyvitamin D levels
Hypocalcemia stimulates secretion of PTH. Therefore, hypocalcemia with elevated serum PTH is caused by disorders that do not impair PTH secretion, including chronic renal failure and vitamin D deficiency (Table 5). Conversely, hypocalcemia with low or normal serum PTH levels suggests primary hypoparathyroidism.
Our patient’s serum PTH level is 20 ng/mL, which is within the reference range. This does not discount the diagnosis of primary hypoparathyroidism. Although most patients with primary hypoparathyroidism have low or undetectable serum PTH levels, some have normal PTH levels if some degree of PTH production is preserved.5,7,28–30 In these patients, the remaining functioning parathyroid tissue is not enough to maintain a normal serum calcium level, resulting in hypocalcemia. As a result, hypocalcemia stimulates the remaining parathyroid tissue to its maximum output, producing PTH levels usually within the lower or middle-normal range.30 In such patients, the terms parathyroid insufficiency and relative primary hypoparathyroidism are more precise than primary hypoparathyroidism.
Postsurgical hypoparathyroidism with an inappropriately normal PTH level is usually seen in patients with disorders that impair intestinal calcium absorption or bone resorption.31 In our patient’s case, the “normal” serum PTH level is likely due to maximal stimulation of remaining functioning parathyroid tissue by severe hypocalcemia, which is a result of her discontinuation of calcium and calcitriol therapy and her vitamin D deficiency.
CASE RESUMED: NO RESPONSE TO INTRAVENOUS CALCIUM GLUCONATE
The patient is given 2 10-mL ampules of 10% calcium gluconate diluted in 100 mL of 5% dextrose in water over 20 minutes intravenously. Electrocardiographic monitoring is continued. Two hours later, her measured serum calcium is only 5.8 mg/dL, with no improvement in her symptoms.
A continuous infusion of calcium gluconate is started: 12 ampules of calcium gluconate are added to 380 mL of 5% dextrose in water and infused at 40 mL/hour (infused rate of elemental calcium = 1.3 mg/kg/hour); 3 hours later, her measured serum calcium level is still only 5.8 mg//dL; at 6 hours it is 5.9 mg/dL, and her symptoms have not improved.
4. Which of the following is the most appropriate next step?
- Change the calcium gluconate to calcium chloride
- Increase the infusion rate to 1.5 mg of elemental calcium/kg/hour
- Give a bolus of 2 10-mL ampules of 10% calcium gluconate intravenously over 1 minute
- Give additional oral calcium tablets
- Check the serum magnesium level
Treatment of hypocalcemia can involve intravenous or oral calcium therapy.
Intravenous calcium is indicated for patients with any of the following6,32:
- Moderate to severe neuromuscular irritability (eg, acral paresthesia, carpopedal spasm, prolonged QT interval, seizures, laryngospasm, bronchospasm)
- Acute hypocalcemia with corrected serum calcium level less than 7.6 mg/dL, even if the patient is asymptomatic
- Cardiac failure.
One 10-mL ampule of 10% calcium gluconate contains 93 mg of elemental calcium; 1 or 2 ampules are typically diluted in 50 to 100 mL of 5% dextrose in water and infused slowly over 15 to 20 minutes. Rapid administration of intravenous calcium is contraindicated, as it may produce cardiac arrhythmias and possibly cardiac arrest. Therefore, intravenous calcium should be given slowly while continuing electrocardiographic monitoring.33
Since the effect of 1 ampule of calcium gluconate lasts only 2 to 3 hours, most patients with symptomatic hypocalcemia require continuous intravenous calcium infusion. The recommended dose of infused elemental calcium is 0.5 to 1.5 mg/kg/hour.34 Several ampules are added to 500 to 1,000 mL of 5% dextrose in water or 0.9% normal saline and infused at a rate appropriate for the patient’s corrected calcium and symptoms.
Oral calcium and vitamin D supplements can be given initially to patients with a corrected serum calcium level of 7.6 mg/dL or greater, with or without mild symptoms, if they can tolerate oral intake. However, this is not the treatment of choice for resistant acute hypocalcemia, as in this case.
Calcium chloride has no advantages over calcium gluconate. Further, it can be associated with local irritation and may result in tissue necrosis if extravasation occurs.35
Increasing the infusion rate of calcium gluconate to the maximum recommended dose may improve the patient’s ionized calcium level and symptoms somewhat. However, it is not the best option for this patient, given that she did not respond to 2 ampules of calcium gluconate followed by continuous infusion of 1.3 mg/kg/hour for 6 hours.
Calcium gluconate bolus. Similarly, giving the patient an additional 2 ampules of calcium gluconate over 1 minute would not be recommended, as rapid administration of intravenous calcium gluconate (eg, over 1 minute) is contraindicated.
Check magnesium
If hypocalcemia persists despite intravenous calcium therapy, as in our patient, further investigation or action is required. An important cause of persistent hypocalcemia is severe hypomagnesemia. Severe hypomagnesemia (serum magnesium < 0.8 mg/dL) causes resistant hypocalcemia by several mechanisms:
- Inducing PTH resistance32,36,37
- Decreasing PTH secretion32,36
- Decreasing calcitriol production.
The decrease in calcitriol production is a direct effect of hypomagnesemia, but it is also an indirect effect of low PTH secretion, which inhibits the enzyme 1-alpha-hydroxylase. Thus, conversion of 25-hydroxyvitamin D3 to calcitriol is impaired, leading to low calcitriol production.
Our patient could have hypomagnesemia due to furosemide use and uncontrolled diabetes mellitus. Hypocalcemia resistant to calcium therapy may occasionally respond to magnesium therapy even if the serum magnesium level is normal. This may be due to depleted intracellular magnesium salt levels.6,38 Rarely, severe hypermagnesemia can also be associated with hypocalcemia due to inhibition of PTH secretion.37,39
CASE RESUMED
Our patient’s serum magnesium level is 0.6 mg/dL (reference range 1.7–2.4 mg/dL). She is given 2 g of magnesium sulfate in 60 mL of 0.9% normal saline infused over 1 hour, followed by a continuous infusion of magnesium sulfate (12 g diluted in 250 mL of 0.9% normal saline, infused over 24 hours). On repeat testing 4 hours later, her serum magnesium level is 0.7 mg/dL, and at 8 hours later it is 0.9 mg/dL. She is subsequently started on oral magnesium oxide 600 mg per day. The magnesium sulfate infusion is continued for another 24 hours.
PREVENTING HYPERCALCIURIA
Patients with low PTH (primary hypoparathyroidism) may have hypercalciuria due to decreased renal tubular calcium reabsorption. Two important measures can minimize hypercalciuria in such patients:
- Keeping the serum calcium level in the low-normal range4,5,40
- Giving a thiazide diuretic (eg, hydrochlorothiazide 12.5–50 mg daily) with a low-salt diet.41,42
A thiazide diuretic is usually started once the 24-hour urine calcium reaches 250 mg.6 Thiazides are thought to enhance both proximal and distal renal tubular calcium reabsorption.43,44
PRIMARY HYPOPARATHYROIDISM: LONG-TERM MANAGEMENT
Long-term management of primary hypoparathyroidism requires calcium and vitamin D supplementation.
Calcium supplements. The most commonly prescribed calcium preparations are calcium carbonate and calcium citrate (containing 40% and 20% elemental calcium, respectively). Calcium carbonate, which is less expensive than calcium citrate, binds with phosphate intake and requires an acidic environment for absorption, and so it is better absorbed when taken with meals. Because calcium citrate does not require an acidic environment for absorption, it is the calcium preparation of choice for patients on proton pump inhibitors, or patients with achlorhydria or constipation.45 Calcium doses vary widely, with most hypoparathyroid patients requiring 1 to 2 g of elemental calcium daily.6
Vitamin D supplements. To promote intestinal absorption, calcium is combined with vitamin D in a fixed-dose preparation given in divided doses.46 Calcitriol (1,25-dihydroxyvitamin D3) is the most active metabolite of vitamin D, with rapid onset and offset of action, and it is the preferred form of vitamin D therapy for patients with hypoparathyroidism. If calcitriol is not available or is not affordable, alphacalcidol (1-alpha-hydroxyvitamin D3) is another option. This is a synthetic analogue of vitamin D that is already hyroxylated at the C1 position. After oral intake, it is hydroxylated in the liver to form calcitriol.
Since renal production of calcitriol is PTH-dependent, in hypoparathyroidism the conversion of 25-hydroxyvitamin D3 to calcitriol is limited. Therefore, vitamin D3 (cholecalciferol) and vitamin D2 (ergocalciferol) are not the preferred forms of vitamin D for such patients. However, either can be added to calcitriol, as they may have extraskeletal benefits.7
CASE CONCLUDED
Our patient presented with primary parathyroid insufficiency associated with vitamin D deficiency. Therefore, in addition to calcitriol and calcium combined with vitamin D in a fixed-dose preparation, her management included vitamin D3 for her vitamin D deficiency.
She was discharged on these medications. At a follow-up visit 3 weeks later, her measured serum calcium level was 8.6 mg/dL. She reported gradual resolution of her symptoms. She was also referred to a psychiatrist for her depression.
TAKE-HOME POINTS
- Hypocalcemia causes neuromuscular excitability, manifested clinically by tetany.
- Common causes of hypocalcemia include vitamin D deficiency, hypomagnesemia, renal failure, and primary hypoparathyroidism.
- The first step in evaluating hypocalcemia is to correct the measured serum calcium to the serum albumin concentration.
- Laboratory testing for hypocalcemia should include serum phosphorus, magnesium, creatinine, PTH, and 25-hydroxyvitamin D3.
- Primary hypoparathyroidism is characterized by hypocalcemia, hyperphosphatemia, and low serum PTH.
- Moderate to severe manifestations of hypo-
calcemia and acute hypocalcemia (< 7.6 mg/dL), even if asymptomatic, warrant intravenous calcium therapy. - Correction of hypomagnesemia is essential to treat hypocalcemia, especially if resistant to intravenous calcium therapy.
- The goal of chronic management of primary hypoparathyroidism includes correcting the serum calcium level to a low-normal range, the serum phosphorus level to an upper-normal range, and prevention of hypercalciuria.
Acknowledgments: The authors wish to thank Mr. Michael Edward Tierney of the School of Medicine, University of Sydney, Australia, for his linguistic editing of the manuscript.
- Jesus JE, Landry A. Images in clinical medicine. Chvostek’s and Trousseau’s signs. N Engl J Med 2012; 367:e15.
- Urbano FL. Signs of hypocalcemia: Chvostek’s and Trousseau’s. Hosp Physician 2000; 36:43–45.
- Chisthi MM, Nair RS, Kuttanchettiyar KG, Yadev I. Mechanisms behind post-thyroidectomy hypocalcemia: interplay of calcitonin, parathormone, and albumin—a prospective study. J Invest Surg 2017; 30:217–225.
- Shoback DM, Bilezikian JP, Costa AG, et al. Presentation of hypoparathyroidism: etiologies and clinical features. J Clin Endocrinol Metab 2016; 101:2300–2312.
- Stack BC Jr, Bimston DN, Bodenner DL, et al. American Association of Clinical Endocrinologists and American College of Endocrinology disease state clinical review: postoperative hypoparathyroidism—definitions and management. Endocr Pract 2015; 21:674–685.
- Shoback D. Clinical practice. Hypoparathyroidism. N Engl J Med 2008; 359:391–403.
- Abate EG, Clarke BL. Review of hypoparathyroidism. Front Endocrinol (Lausanne) 2017; 7:172.
- Coimbra C, Monteiro F, Oliveira P, Ribeiro L, de Almeida MG, Condé A. Hypoparathyroidism following thyroidectomy: predictive factors. Acta Otorrinolaringol Esp 2017; 68:106–111.
- Thomusch O, Machens A, Sekulla C, Ukkat J, Brauckhoff M, Dralle H. The impact of surgical technique on postoperative hypoparathyroidism in bilateral thyroid surgery: a multivariate analysis of 5846 consecutive patients. Surgery 2003; 133:180–185.
- Hirsch PF, Lester GE, Talmage RV. Calcitonin, an enigmatic hormone: does it have a function? J Musculoskelet Neuronal Interact 2001; 1:299–305.
- Karne SS, Bhalerao NS. Carpal tunnel syndrome in hypothyroidism. J Clin Diagn Res 2016; 10:OC36–OC38.
- Duyff RF, Van den Bosch J, Laman DM, van Loon BJ, Linssen WH. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68:750–755.
- Palumbo CF, Szabo RM, Olmsted SL. The effects of hypothyroidism and thyroid replacement on the development of carpal tunnel syndrome. J Hand Surg Am 2000; 25:734–739.
- Katz JN, Stirrat CR, Larson MG, Fossel AH, Eaton HM, Liang MH. A self-administered hand symptom diagram for the diagnosis and epidemiologic study of carpal tunnel syndrome. J Rheumatol 1990; 17:1495–1498.
- Katz JN, Stirrat CR. A self-administered hand diagram for the diagnosis of carpal tunnel syndrome. J Hand Surg Am 1990; 15:360–363.
- Calfee RP, Dale AM, Ryan D, Descatha A, Franzblau A, Evanoff B. Performance of simplified scoring systems for hand diagrams in carpal tunnel syndrome screening. J Hand Surg Am 2012; 37:10–17.
- D’Arcy CA, McGee S. The rational clinical examination. Does this patient have carpal tunnel syndrome? JAMA 2000; 283:3110–3117.
- Marchettini P, Lacerenza M, Mauri E, Marangoni C. Painful peripheral neuropathies. Curr Neuropharmacol 2006; 4:175–181.
- Kibirige D, Mwebaze R. Vitamin B12 deficiency among patients with diabetes mellitus: is routine screening and supplementation justified? J Diabetes Metab Disord 2013;12:17.
- Akinlade KS, Agbebaku SO, Rahamon SK, Balogun WO. Vitamin B12 levels in patients with type 2 diabetes mellitus on metformin. Ann Ib Postgrad Med 2015; 13:79–83.
- Tohme JF, Bilezikian JP. Hypocalcemic emergencies. Endocrinol Metab Clin North Am 1993; 22:363–375.
- Macefield G, Burke D. Paraesthesiae and tetany induced by voluntary hyperventilation. Increased excitability of human cutaneous and motor axons. Brain 1991; 114:527–540.
- Benoit SR, Mendelsohn AB, Nourjah P, Staffa JA, Graham DJ. Risk factors for prolonged QTc among US adults: Third National Health and Nutrition Examination Survey. Eur J Cardiovasc Prev Rehabil 2005; 12:363–368.
- Khoo TK, Vege SS, Abu-Lebdeh HS, Ryu E, Nadeem S, Wermers RA. Acute pancreatitis in primary hyperparathyroidism: a population-based study. J Clin Endocrinol Metab 2009; 94:2115–2118.
- McKay C, Beastall GH, Imrie CW, Baxter JN. Circulating intact parathyroid hormone levels in acute pancreatitis. Br J Surg 1994; 81:357–360.
- Talmage RV, Mobley HT. Calcium homeostasis: reassessment of the actions of parathyroid hormone. Gen Comp Endocrinol 2008; 156:1–8.
- Friedman PA, Gesek FA. Calcium transport in renal epithelial cells. Am J Physiol 1993; 264:F181–F198.
- Jensen PV, Jelstrup SM, Homøe P. Long-term outcomes after total thyroidectomy. Dan Med J 2015; 62:A5156.
- Ritter K, Elfenbein D, Schneider DF, Chen H, Sippel RS. Hypoparathyroidism after total thyroidectomy: incidence and resolution. J Surg Res 2015; 197:348–353.
- Promberger R, Ott J, Kober F, Karik M, Freissmuth M, Hermann M. Normal parathyroid hormone levels do not exclude permanent hypoparathyroidism after thyroidectomy. Thyroid 2011; 21:145–150.
- Lorente-Poch L, Sancho JJ, Muñoz-Nova JL, Sánchez-Velázquez P, Sitges-Serra A. Defining the syndromes of parathyroid failure after total thyroidectomy. Gland Surgery 2015; 4:82–90.
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ 2008; 336:1298–1302.
- Tohme JF, Bilezikian JP. Diagnosis and treatment of hypocalcemic emergencies. Endocrinologist 1996; 6:10–18.
- Carroll R, Matfin G. Endocrine and metabolic emergencies: hypocalcaemia. Ther Adv Endocrinol Metab 2010; 1:29–33.
- Kim MP, Raho VJ, Mak J, Kaynar AM. Skin and soft tissue necrosis from calcium chloride in a deicer. J Emerg Med 2007; 32:41–44.
- Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med 2005; 20:3–17.
- Cholst IN, Steinberg SF, Tropper PJ, Fox HE, Segre GV, Bilezikian JP. The influence of hypermagnesemia on serum calcium and parathyroid hormone levels in human subjects. N Engl J Med 1984; 310:1221–1225.
- Ryzen E, Nelson TA, Rude RK. Low blood mononuclear cell magnesium content and hypocalcemia in normomagnesemic patients. West J Med 1987; 147:549–553.
- Koontz SL, Friedman SA, Schwartz ML. Symptomatic hypocalcemia after tocolytic therapy with magnesium sulfate and nifedipine. Am J Obstet Gynecol 2004; 190:1773–1776.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Porter RH, Cox BG, Heaney D, Hostetter TH, Stinebaugh BJ, Suki WN. Treatment of hypoparathyroid patients with chlorthalidone. N Engl J Med 1978; 298:577–581.
- Clarke BL, Brown EM, Collins MT, et al. Epidemiology and diagnosis of hypoparathyroidism. J Clin Endocrinol Metab 2016; 101:2284–2299.
- Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 2005; 115:1651–1658.
- Costanzo LS. Localization of diuretic action in microperfused rat distal tubules: Ca and Na transport. Am J Physiol 1985; 248:F527–F535.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Scotti A, Bianchini C, Abbiati G, Marzo A. Absorption of calcium administered alone or in fixed combination with vitamin D to healthy volunteers. Arzneimittelforschung 2001; 51:493–500.
- Jesus JE, Landry A. Images in clinical medicine. Chvostek’s and Trousseau’s signs. N Engl J Med 2012; 367:e15.
- Urbano FL. Signs of hypocalcemia: Chvostek’s and Trousseau’s. Hosp Physician 2000; 36:43–45.
- Chisthi MM, Nair RS, Kuttanchettiyar KG, Yadev I. Mechanisms behind post-thyroidectomy hypocalcemia: interplay of calcitonin, parathormone, and albumin—a prospective study. J Invest Surg 2017; 30:217–225.
- Shoback DM, Bilezikian JP, Costa AG, et al. Presentation of hypoparathyroidism: etiologies and clinical features. J Clin Endocrinol Metab 2016; 101:2300–2312.
- Stack BC Jr, Bimston DN, Bodenner DL, et al. American Association of Clinical Endocrinologists and American College of Endocrinology disease state clinical review: postoperative hypoparathyroidism—definitions and management. Endocr Pract 2015; 21:674–685.
- Shoback D. Clinical practice. Hypoparathyroidism. N Engl J Med 2008; 359:391–403.
- Abate EG, Clarke BL. Review of hypoparathyroidism. Front Endocrinol (Lausanne) 2017; 7:172.
- Coimbra C, Monteiro F, Oliveira P, Ribeiro L, de Almeida MG, Condé A. Hypoparathyroidism following thyroidectomy: predictive factors. Acta Otorrinolaringol Esp 2017; 68:106–111.
- Thomusch O, Machens A, Sekulla C, Ukkat J, Brauckhoff M, Dralle H. The impact of surgical technique on postoperative hypoparathyroidism in bilateral thyroid surgery: a multivariate analysis of 5846 consecutive patients. Surgery 2003; 133:180–185.
- Hirsch PF, Lester GE, Talmage RV. Calcitonin, an enigmatic hormone: does it have a function? J Musculoskelet Neuronal Interact 2001; 1:299–305.
- Karne SS, Bhalerao NS. Carpal tunnel syndrome in hypothyroidism. J Clin Diagn Res 2016; 10:OC36–OC38.
- Duyff RF, Van den Bosch J, Laman DM, van Loon BJ, Linssen WH. Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J Neurol Neurosurg Psychiatry 2000; 68:750–755.
- Palumbo CF, Szabo RM, Olmsted SL. The effects of hypothyroidism and thyroid replacement on the development of carpal tunnel syndrome. J Hand Surg Am 2000; 25:734–739.
- Katz JN, Stirrat CR, Larson MG, Fossel AH, Eaton HM, Liang MH. A self-administered hand symptom diagram for the diagnosis and epidemiologic study of carpal tunnel syndrome. J Rheumatol 1990; 17:1495–1498.
- Katz JN, Stirrat CR. A self-administered hand diagram for the diagnosis of carpal tunnel syndrome. J Hand Surg Am 1990; 15:360–363.
- Calfee RP, Dale AM, Ryan D, Descatha A, Franzblau A, Evanoff B. Performance of simplified scoring systems for hand diagrams in carpal tunnel syndrome screening. J Hand Surg Am 2012; 37:10–17.
- D’Arcy CA, McGee S. The rational clinical examination. Does this patient have carpal tunnel syndrome? JAMA 2000; 283:3110–3117.
- Marchettini P, Lacerenza M, Mauri E, Marangoni C. Painful peripheral neuropathies. Curr Neuropharmacol 2006; 4:175–181.
- Kibirige D, Mwebaze R. Vitamin B12 deficiency among patients with diabetes mellitus: is routine screening and supplementation justified? J Diabetes Metab Disord 2013;12:17.
- Akinlade KS, Agbebaku SO, Rahamon SK, Balogun WO. Vitamin B12 levels in patients with type 2 diabetes mellitus on metformin. Ann Ib Postgrad Med 2015; 13:79–83.
- Tohme JF, Bilezikian JP. Hypocalcemic emergencies. Endocrinol Metab Clin North Am 1993; 22:363–375.
- Macefield G, Burke D. Paraesthesiae and tetany induced by voluntary hyperventilation. Increased excitability of human cutaneous and motor axons. Brain 1991; 114:527–540.
- Benoit SR, Mendelsohn AB, Nourjah P, Staffa JA, Graham DJ. Risk factors for prolonged QTc among US adults: Third National Health and Nutrition Examination Survey. Eur J Cardiovasc Prev Rehabil 2005; 12:363–368.
- Khoo TK, Vege SS, Abu-Lebdeh HS, Ryu E, Nadeem S, Wermers RA. Acute pancreatitis in primary hyperparathyroidism: a population-based study. J Clin Endocrinol Metab 2009; 94:2115–2118.
- McKay C, Beastall GH, Imrie CW, Baxter JN. Circulating intact parathyroid hormone levels in acute pancreatitis. Br J Surg 1994; 81:357–360.
- Talmage RV, Mobley HT. Calcium homeostasis: reassessment of the actions of parathyroid hormone. Gen Comp Endocrinol 2008; 156:1–8.
- Friedman PA, Gesek FA. Calcium transport in renal epithelial cells. Am J Physiol 1993; 264:F181–F198.
- Jensen PV, Jelstrup SM, Homøe P. Long-term outcomes after total thyroidectomy. Dan Med J 2015; 62:A5156.
- Ritter K, Elfenbein D, Schneider DF, Chen H, Sippel RS. Hypoparathyroidism after total thyroidectomy: incidence and resolution. J Surg Res 2015; 197:348–353.
- Promberger R, Ott J, Kober F, Karik M, Freissmuth M, Hermann M. Normal parathyroid hormone levels do not exclude permanent hypoparathyroidism after thyroidectomy. Thyroid 2011; 21:145–150.
- Lorente-Poch L, Sancho JJ, Muñoz-Nova JL, Sánchez-Velázquez P, Sitges-Serra A. Defining the syndromes of parathyroid failure after total thyroidectomy. Gland Surgery 2015; 4:82–90.
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ 2008; 336:1298–1302.
- Tohme JF, Bilezikian JP. Diagnosis and treatment of hypocalcemic emergencies. Endocrinologist 1996; 6:10–18.
- Carroll R, Matfin G. Endocrine and metabolic emergencies: hypocalcaemia. Ther Adv Endocrinol Metab 2010; 1:29–33.
- Kim MP, Raho VJ, Mak J, Kaynar AM. Skin and soft tissue necrosis from calcium chloride in a deicer. J Emerg Med 2007; 32:41–44.
- Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med 2005; 20:3–17.
- Cholst IN, Steinberg SF, Tropper PJ, Fox HE, Segre GV, Bilezikian JP. The influence of hypermagnesemia on serum calcium and parathyroid hormone levels in human subjects. N Engl J Med 1984; 310:1221–1225.
- Ryzen E, Nelson TA, Rude RK. Low blood mononuclear cell magnesium content and hypocalcemia in normomagnesemic patients. West J Med 1987; 147:549–553.
- Koontz SL, Friedman SA, Schwartz ML. Symptomatic hypocalcemia after tocolytic therapy with magnesium sulfate and nifedipine. Am J Obstet Gynecol 2004; 190:1773–1776.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Porter RH, Cox BG, Heaney D, Hostetter TH, Stinebaugh BJ, Suki WN. Treatment of hypoparathyroid patients with chlorthalidone. N Engl J Med 1978; 298:577–581.
- Clarke BL, Brown EM, Collins MT, et al. Epidemiology and diagnosis of hypoparathyroidism. J Clin Endocrinol Metab 2016; 101:2284–2299.
- Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 2005; 115:1651–1658.
- Costanzo LS. Localization of diuretic action in microperfused rat distal tubules: Ca and Na transport. Am J Physiol 1985; 248:F527–F535.
- Brandi ML, Bilezikian JP, Shoback D, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016; 101:2273–2283.
- Scotti A, Bianchini C, Abbiati G, Marzo A. Absorption of calcium administered alone or in fixed combination with vitamin D to healthy volunteers. Arzneimittelforschung 2001; 51:493–500.