User login
Diagnosis and treatment of hyperkalemia
Hyperkalemia is common in patients with cardiovascular disease. Its consequences can be severe and life-threatening, and its management and prevention require a multidisciplinary approach that entails reducing intake of high-potassium foods, adjusting medications that cause hyperkalemia, and adding medications that reduce the plasma potassium concentration. With this approach, patients at high risk can receive the cardiorenal benefits of drugs that block the renin-angiotensin-aldosterone system without developing hyperkalemia.
98% OF POTASSIUM IS INSIDE CELLS
The body of a typical 70-kg man contains about 3,500 mmol of potassium, 98% of which is in the intracellular space; the remaining 2% is in the extracellular space. This large intracellular-to-extracellular gradient determines the cell voltage and explains why disorders in plasma potassium give rise to manifestations in excitable tissues such as the heart and nervous system.
The most important determinants of potassium distribution between the intracellular and extracellular space are insulin and beta-adrenergic receptor stimulation.
Maintenance of total-body potassium content is primarily the job of the kidneys, with a small contribution by the gastrointestinal tract.1,2 Hyperkalemia is most commonly encountered in patients with decreased kidney function.
The normal kidney can secrete a large amount of potassium, making hyperkalemia uncommon in the absence of kidney disease. This large capacity may have evolved to handle the diet of Paleolithic humans, which contained 4 times as much potassium as contemporary diets.3,4 With the onset of agriculture, dietary intake of potassium has progressively declined while sodium intake has risen. A popular theory suggests this mismatch between the modern diet and the nutritional requirements encoded in the human genome during evolution may contribute to chronic diseases such as hypertension, stroke, kidney stones, and bone disease.5
MANY POTENTIAL CAUSES OF HYPERKALEMIA

Causes of hyperkalemia are outlined in Table 1. Shifting of potassium from the cells to the extracellular space is a cause of transient hyperkalemia, while chronic hyperkalemia indicates an impairment in renal potassium secretion. The following discussion is a guide to the approach to the hyperkalemic patient.
Is the patient’s hyperkalemia really pseudohyperkalemia?
Pseudohyperkalemia, an artifact of measurement, occurs due to mechanical release of potassium from cells during phlebotomy or specimen processing.6 This diagnosis is made when the serum potassium concentration exceeds the plasma potassium concentration by more than 0.5 mmol/L, and should be considered when hyperkalemia occurs in the absence of a clinical risk factor. Fist-clenching, application of a tight-fitting tourniquet, or use of small-bore needles during phlebotomy can all cause pseudohyperkalemia.
Mechanism of pseudohyperkalemia. Since serum is the liquid part of blood remaining after coagulation, release of potassium from cells injured during the process of coagulation raises the potassium level in the serum. Plasma is the cell-free part of blood that has been treated with anticoagulants; it has no cells that can be injured and release potassium. Thus, the serum potassium level will be higher than that in the plasma.
Reverse pseudohyperkalemia, in contrast, occurs when the plasma potassium level is falsely elevated but the serum value is normal. This situation has been described in hematologic disorders characterized by pronounced leukocytosis in which malignant cells are prone to lysis with minimal mechanical stress due to increased fragility or altered sodium-potassium ATPase pump activity.7 This phenomenon is unusual but occurs because the cells are so fragile.
A spurious increase in plasma potassium concentration along with a low plasma calcium concentration raises the possibility of calcium chelation and release of potassium in a sample tube contaminated with the anticoagulant ethylenediaminetetraacetic acid.
Is there increased potassium intake?
Increased potassium intake is a potential cause of hyperkalemia in patients with decreased kidney function or adrenal disease.
Foods naturally rich in potassium include bananas (a medium-sized banana contains 451 mg or 12 mmol of potassium) and potatoes (844 mg or 22 mmol in a large baked potato with skin). Other potassium-rich foods are melons, citrus juice, and avocados. Less-obvious food sources include raw coconut juice (potassium concentration 44.3 mmol/L) and noni juice (56 mmol/L).
Salt substitutes, recommended to hypertensive patients with chronic kidney disease, can be a hidden source of dietary potassium.
Clay ingestion is a potential cause of dyskalemia. White clay consumption causes hypokalemia due to potassium binding in the gastrointestinal tract. Red clay or river bed clay, on the other hand, is enriched in potassium (100 mmol of potassium in 100 g of clay) and can cause life-threatening hyperkalemia in patients with chronic kidney disease.8
Eating burnt match heads. Some individuals chew and ingest burnt match heads, a condition called cautopyreiophagia. In one reported case,9 this activity contributed an additional 80 mmol of daily potassium intake in a dialysis patient, resulting in a plasma potassium concentration of 8 mmol/L.
Is the hyperkalemia the result of a cellular shift?
Acute hyperkalemia can be the result of redistribution of cellular potassium. Shifting of as little as 2% of the body’s potassium from the intracellular to the extracellular space can double the plasma potassium concentration.
Tissue injury. Hyperkalemia frequently occurs in diseases that cause tissue injury such as rhabdomyolysis, trauma, massive hemolysis, and tumor lysis.
Insulin deficiency. Insulin and catecholamines are major regulators of potassium distribution within the body. After a meal, release of insulin not only regulates the plasma glucose concentration, it also causes potassium to move into cells until the kidneys have had sufficient time to excrete the dietary potassium load and reestablish total-body potassium content.
Exercise, beta-blockers. During exercise, potassium is released from skeletal muscle cells and accumulates in the interstitial compartment, where it exerts a vasodilatory effect. The simultaneous increase in circulating catecholamines regulates this release by promoting cell potassium uptake through beta-adrenergic receptor stimulation.
Metabolic acidosis can facilitate exit (ie, shift) of potassium from cells, but this effect depends on the type of acidosis. Hyperchloremic normal anion gap acidosis (mineral acidosis) most commonly causes this effect due to the relative impermeability of the cell membrane to the chloride anion. As hydrogen ions move into the cell due to accumulation of ammonium chloride or hydrogen chloride, electrical neutrality is maintained by potassium exit.
In contrast, organic acidosis (due to lactic, beta-hydroxybutyric, or methylmalonic acid) tends not to cause a potassium shift, since most organic anions readily cross the cell membrane along with hydrogen. Lactic acidosis is often associated with potassium shift, but this effect is due to loss of cell integrity as a result of cell ischemia. The hyperkalemia typically present on admission in patients with diabetic ketoacidosis is the result of insulin deficiency and hypertonicity and not the underlying organic acidosis.10
Hypertonic states can cause hyperkalemia due to cell shift. For example, hyperglycemia, as in diabetic ketoacidosis, pulls water from the intracellular into the extracellular compartment, thereby concentrating intracellular potassium and creating a more favorable gradient for potassium efflux through membrane channels. This same effect can occur in neurosurgical patients given large amounts of hypertonic mannitol. Repetitive doses of immunoglobulin can lead to extracellular accumulation of sorbitol, maltose, or sucrose, since these sugars are added to the preparations to prevent immunoglobulin aggregation.11
Is a disturbance in renal potassium excretion present?
Sustained hyperkalemia is more commonly associated with decreases in renal potassium excretion than with a cellular shift. In most instances the clinician can distinguish between cell shift and impaired renal excretion based on the available clinical data.
The transtubular potassium gradient has been used to determine whether there is a disturbance in renal potassium excretion and to assess renal potassium handling.12

This calculation is based on the assumption that only water is reabsorbed past the cortical collecting duct, and not solutes. It has fallen out of favor since we have found this assumption to be incorrect; a large amount of urea is reabsorbed daily in the downstream medullary collecting duct as a result of intrarenal recycling of urea.
The one situation in which the transtubular potassium gradient may be of use is determining whether hyperkalemia is a result of low aldosterone levels as opposed to aldosterone resistance. One can compare the transtubular potassium gradient before and after a physiologic dose (0.05 mg) of 9-alpha fludrocortisone. An increase of more than 6 over a 4-hour period favors aldosterone deficiency, whereas smaller changes would indicate aldosterone resistance.
24-hour potassium excretion, spot urine potassium-creatinine ratio. A better way to assess renal potassium handling is to measure the amount of potassium in a 24-hour urine collection or determine a spot urine potassium-creatinine ratio. A 24-hour urinary potassium excretion of less than 15 mmol or a potassium-creatinine ratio less than 1 suggests an extrarenal cause of hypokalemia. A ratio greater than 20 would be an appropriate renal response to hyperkalemia.
One or more of 3 abnormalities should be considered in the hyperkalemic patient with impaired renal excretion of potassium:
- Decreased distal delivery of sodium
- Mineralocorticoid deficiency
- Abnormal cortical collecting tubule function.13
Decreased distal delivery of sodium
Under normal circumstances, potassium is freely filtered across the glomerulus and then mostly reabsorbed in the proximal tubule and thick ascending limb. Potassium secretion begins in the distal convoluted tubule and increases in magnitude into the collecting duct. Tubular secretion is the component of potassium handling that varies and is regulated according to physiologic needs.
In acute kidney injury, the rapid decline in glomerular filtration rate and reduction in functioning nephron mass lead to decreased distal potassium secretion.
Hyperkalemia is a frequent problem when oliguria is present, since the reduction in distal delivery of sodium and water further impairs potassium secretion. Patients with oliguric acute kidney injury are more likely to have a more severe underlying disease state, and therefore tissue breakdown and catabolism further increase the risk of hyperkalemia.
In contrast, in nonoliguric patients, the renal injury tends to be less severe, and enough sodium and water are usually delivered distally to prevent hyperkalemia.
In chronic kidney disease, nephron dropout and reduction in collecting tubule mass also lead to a global decline in distal potassium secretion. However, this is countered by an increased capacity of the remaining individual nephrons for potassium secretion. High flow, increased distal sodium delivery, and increased activity and number of sodium-potassium ATPase pumps in the remaining nephrons account for this increased secretory capacity.14 As renal function declines over time, colonic potassium secretion progressively increases.15
These adaptive changes help to keep the plasma potassium concentration within the normal range until the glomerular filtration rate falls to less than 10 or 15 mL/min. Development of hyperkalemia with more modest reductions in the glomerular filtration rate suggest decreased mineralocorticoid activity or a specific lesion of the tubule.
Mineralocorticoid deficiency

Aldosterone deficiency can occur alone or in combination with decreased cortisol levels. Destruction of the adrenal glands is suggested when both hormones are reduced. Enzyme defects in cortisol metabolism can result in either isolated deficiency of aldosterone or adrenogenital syndromes associated with decreased mineralocorticoid activity.
Heparin administration leads to a reversible defect in adrenal synthesis of aldosterone. Drugs that block the stimulatory effect of angiotensin II on the zona glomerulosa cells of the adrenal gland will lower aldosterone.
Renin-angiotensin-aldosterone system blockers. Angiotensin-converting enzyme inhibitors block the formation of angiotensin II, whereas angiotensin II receptor blockers prevent angiotensin II from binding to its adrenal receptor. The direct renin inhibitor aliskiren lowers angiotensin II levels by blocking the enzymatic activity of renin and lowers the circulating levels of both angiotensin I and II.16
The syndrome of hyporeninemic hypoaldosteronism is a common cause of hyperkalemia in patients who have a glomerular filtration rate between 40 and 60 mL/min. Diabetic nephropathy and interstitial renal disease are the most common clinical entities associated with this syndrome.10 Other causes include analgesic nephropathy, urinary tract obstruction, sickle cell disease, systemic lupus erythematosus, and amyloidosis.
Nonsteroidal anti-inflammatory drugs can cause hyperkalemia by suppressing renin release and reducing delivery of sodium to the distal nephron.18
Calcineurin inhibitors impair potassium secretion by suppressing renin release and by direct tubular effects.19
Beta-blockers. Beta-1 and to a lesser extent beta-2 receptor blockade can also result in a hyporeninemic state.
Distal tubular defect
Hyperkalemia can result from interstitial renal diseases that specifically affect the distal nephron. In this setting, the glomerular filtration rate is only mildly reduced, and circulating aldosterone levels are normal.
Renal transplant, lupus erythematosus, amyloidosis, urinary obstruction, and sickle cell disease are conditions in which an impairment in renin release may coexist with a defect in tubular secretion.
Potassium-sparing diuretics impair the ability of the cortical collecting tubule to secrete potassium. Specifically, amiloride and triamterene inhibit sodium reabsorption mediated by the epithelial sodium channel located on the apical membrane of the principal cell. This effect abolishes the lumen’s negative potential and thereby removes a driving force for potassium secretion.
Trimethoprim and pentamidine cause similar effects.
Spironolactone and eplerenone compete with aldosterone at the level of the mineralocorticoid receptor and can result in hyperkalemia.
Drospirenone, a non-testosterone-derived progestin contained in certain oral contraceptives, possesses mineralocorticoid-blocking effects similar to those of spironolactone.
The plasma potassium level should be monitored when these drugs are prescribed in patients receiving potassium supplements, renin-angiotensin-aldosterone system blockers, or nonsteroidal anti-inflammatory drugs.20
CLINICAL FEATURES OF HYPERKALEMIA
Neuromuscular manifestations of hyperkalemia include paresthesias and fasciculations in the arms and legs. Severe elevation in potassium can give rise to an ascending paralysis with eventual flaccid quadriplegia. Typically, the trunk, head, and respiratory muscles are spared, and respiratory failure is rare.
Cardiac signs

Hyperkalemia has depolarizing effects on the heart that are manifested by changes in the electrocardiogram (Figure 2). The progressive changes of hyperkalemia are classically listed as:
- Peaked T waves that are tall, narrow, and symmetrical and can occasionally be confused with the hyperacute T-wave change associated with an ST-segment elevation myocardial infarction.21 However, in the latter condition, the T waves tend to be more broad-based and asymmetric in shape.
- ST-segment depression
- Widening of the PR interval
- Widening of the QRS interval
- Loss of the P wave
- A sine-wave pattern—an ominous development and a harbinger of impending ventricular fibrillation and asystole.
The plasma potassium concentration often correlates poorly with cardiac manifestations. In a retrospective review, only 16 of 90 cases met strict criteria for electrocardiographic changes reflective of hyperkalemia (defined as new peaked and symmetric T waves that resolved on follow-up).22 In 13 of these cases, the electrocardiogram was interpreted as showing no T-wave changes even when read by a cardiologist. In addition, electrocardiographic criteria for hyperkalemia were noted in only 1 of 14 patients who manifested arrhythmias or cardiac arrest attributed to increased plasma potassium concentration.
TREATMENT OF ACUTE HYPERKALEMIA
The treatment of hyperkalemia depends on the magnitude of increase in the plasma potassium concentration and the presence or absence of electrocardiographic changes or neuromuscular symptoms.23 Acute treatment is indicated for marked electrocardiographic changes and severe muscle weakness.
Intravenous calcium rapidly normalizes membrane excitability by antagonizing the potassium-induced decrease in membrane excitability but does not alter the plasma potassium concentration.
Insulin lowers the plasma potassium concentration by promoting its entry into cells. To avoid hypoglycemia, 10 units of short-acting insulin should be accompanied by a 50-g infusion of glucose, increased to 60 g if 20 units of insulin are given.24
Beta-2 receptor agonists produce a similar effect. The shift of potassium into cells with insulin and beta-2-adrenergic receptor stimulation is brought about by increases in sodium-potassium ATPase pump activity, primarily in skeletal muscle cells.
Sodium bicarbonate, in the absence of acidosis, lowers the plasma potassium concentration only slightly. It should be reserved for hyperkalemic patients who have coexisting metabolic acidosis after the patient has received insulin and glucose, an adrenergic agent, and calcium.
These acute treatments need to be followed by therapies designed to lower the total body potassium content such as diuretics, potassium-binding drugs, and dialysis.
TREATMENT OF CHRONIC HYPERKALEMIA
Review medications. Once the diagnosis of hyperkalemia has been made, the initial approach should be to review the patient’s medications and make every effort to discontinue drugs that can impair renal potassium excretion.16 Patients should be asked about their use of over-the-counter nonsteroidal anti-inflammatory drugs and herbal remedies, since herbs may be a hidden source of dietary potassium.
Dietary counseling. Patients should be instructed to reduce their dietary intake of potassium and to avoid salt substitutes that contain potassium.

Diuretic therapy is beneficial in minimizing hyperkalemia in patients with chronic kidney disease. Thiazide and loop diuretics enhance renal potassium excretion by increasing flow and delivery of sodium to the collecting duct. Thiazide diuretics are effective when the estimated glomerular filtration rate is greater than 30 mL/min, while loop diuretics should be used in patients with more severe renal insufficiency (Table 2).
Sodium bicarbonate is an effective agent to minimize increases in the plasma potassium concentration in patients with chronic kidney disease and metabolic acidosis. This drug increases renal potassium excretion by increasing distal sodium delivery and shifts potassium into cells as the acidosis is corrected. The likelihood of developing volume overload as a complication of sodium bicarbonate administration can be minimized with effective diuretic therapy.
Avoiding hyperkalemia if renin-angiotensin-aldosterone system blockers are needed
Renin-angiotensin-aldosterone system blockers can be problematic, as these drugs cause hyperkalemia, often in the very patients who derive the greatest cardiovascular benefit from them.16 A number of steps can reduce the risk of hyperkalemia and allow these drugs to be used.
The initial dose should be low and the plasma potassium should be measured within 1 to 2 weeks after drug initiation. If the potassium level is normal, the dose can be titrated upwards with remeasurement of the plasma potassium after each dose titration. If the plasma potassium concentration rises to 5.5 mmol/L, in some cases lowering the dose will reduce the potassium concentration and allow the patient to remain on the drug.
In patients at risk of hyperkalemia, angiotensin II receptor blockers and direct renin inhibitors should be used with the same caution as angiotensin-converting enzyme inhibitors.
If the plasma potassium concentration exceeds 5.5 mmol/L despite the above precautions, one can consider using a potassium-binding drug (see below) before deciding to avoid renin-angiotensin-aldosterone system blockers.
Sodium polystyrene sulfonate binds potassium in the gastrointestinal tract in exchange for sodium and has been used to manage hyperkalemia. This drug is most commonly given along with sorbitol as a therapy for acute hyperkalemia. Although the drug is widely used, most of the potassium-lowering effect is due to an increase in stool volume caused by sorbitol.25,26 In addition, long-term use is poorly tolerated, and the drug has been linked to gastrointestinal toxicity in rare cases.
Patiromer and sodium zirconium cyclosilicate are two new potassium-binding drugs that have been shown to be effective in reducing plasma potassium concentration in the setting of ongoing use of renin-angiotensin-aldosterone system blockers.
Patiromer is a nonabsorbed polymer approved for clinical use to treat hyperkalemia. The drug binds potassium in exchange for calcium in the gastrointestinal tract, predominantly in the colon, and lowers the plasma potassium concentration in a dose-dependent manner, with the greatest reduction in those with higher starting values.27,28
Patiromer effectively controlled plasma potassium concentrations in a 1-year randomized trial in high-risk patients on renin-angiotensin-aldosterone system blockers.29 The main adverse events in clinical trials have been constipation and hypomagnesemia, which required magnesium replacement in a small number of patients, but overall, the drug is well tolerated.
Sodium zirconium cyclosilicate is a nonabsorbed microporous compound that binds potassium in exchange for sodium throughout the gastrointestinal tract. It has been found effective in lowering plasma potassium concentration in a dose-dependent fashion in high-risk patients, most of whom were receiving renin-angiotensin-aldosterone system blockers.30–32 Adverse events were generally comparable to those with placebo in clinical trials; however, edema occurred more frequently when higher doses were used. This drug is not yet approved for clinical use.
- Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ 2016; 40:480–490.
- Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 2015; 10:1050–1060.
- Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. N Engl J Med 1985; 312:283–289.
- Sebastian A, Frassetto LA, Sellmeyer DE, Morris RC Jr. The evolution-informed optimal dietary potassium intake of human beings greatly exceeds current and recommended intakes. Semin Nephrol 2006; 26:447–453.
- Palmer BF, Clegg DJ. Achieving the benefits of a high potassium, Paleolithic diet, without the toxicity. Mayo Clin Proc 2016; 91:496–508.
- Liamis G, Liberopoulos E, Barkas F, Elisaf M. Spurious electrolyte disorders: a diagnostic challenge for clinicians. Am J Nephrol 2013; 38:50–57.
- Mansoor S, Holtzman N, Emadi A. Reverse pseudohyperkalemia: an important clinical entity in chronic lymphocytic leukemia. Case Rep Hematol 2015; 2015:930379.
- Gelfand M, Zarate A, Knepshield J. Geophagia. A cause of life-threatening hyperkalemia in patients with chronic renal failure. JAMA 1975; 234:738–740.
- Abu-Hamdan D, Sondheimer J, Mahajan S. Cautopyreiophagia. Cause of life-threatening hyperkalemia in a patient undergoing hemodialysis. Am J Med 1985; 79:517–519.
- Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med 2015; 373:548–559.
- Daphnis E, Stylianou K, Alexandrakis M, et al. Acute renal failure, translocational hyponatremia and hyperkalemia following intravenous immunoglobulin therapy. Nephron Clin Pract 2007; 106:c143–c148.
- Choi M, Ziyadeh F. The utility of the transtubular potassium gradient in the evaluation of hyperkalemia. J Am Soc Nephrol 2008; 19:424–426.
- Palmer BF. A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis 2010; 56:387–393.
- Stanton BA. Renal potassium transport: morphological and functional adaptations. Am J Physiol 1989; 257:R989–R997.
- Hayes CP Jr, McLeod ME, Robinson RR. An extravenal mechanism for the maintenance of potassium balance in severe chronic renal failure. Trans Assoc Am Physicians 1967; 80:207–216.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Palmer BF. Renal dysfunction complicating treatment of hypertension. N Engl J Med 2002; 347:1256–1261.
- Palmer BF. Renal complications associated with use of nonsteroidal anti-inflammatory agents. J Investig Med 1995; 43:516–533.
- Hoorn E, Walsh S, McCormick J, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 2011; 17:1304–1309.
- Bird ST, Pepe SR, Etminan M, Liu X, Brophy JM, Delaney JA. The association between drospirenone and hyperkalemia: a comparative-safety study. BMC Clin Pharmacol 2011; 11:23.
- Wang K. Images in clinical medicine. “Pseudoinfarction” pattern due to hyperkalemia. N Engl J Med 2004; 351:593.
- Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol 2008; 3:324–330.
- Weisberg LS. Management of severe hyperkalemia. Crit Care Med 2008; 36:3246–3251.
- Harel Z, Kamel KS. Optimal dose and method of administration of intravenous insulin in the management of emergency hyperkalemia: a systematic review. PLoS One 2016; 11:e0154963.
- Sterns RH, Rojas M, Bernstein P, Chennupati S. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol 2010; 21:733–735.
- Emmett M, Hootkins RE, Fine KD, Santa Ana CA, Porter JL, Fordtran JS. Effect of three laxatives and a cation exchange resin on fecal sodium and potassium excretion. Gastroenterology 1995; 108:752–760.
- Bushinsky DA, Spiegel DM, Gross C, et al. Effect of patiromer on urinary ion excretion in healthy adults. Clin J Am Soc Nephrol 2016; 11:1769–1776.
- Weir MR, Bakris GL, Bushinsky DA, et al; OPAL-HK Investigators. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med 2015; 372:211–221.
- Bakris GL, Pitt B, Weir MR, et al; AMETHYST-DN Investigators. Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: the AMETHYST-DN randomized clinical trial. JAMA 2015; 314:151–161.
- Kosiborod M, Rasmussen HS, Lavin P, et al. Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia. The HARMONIZE randomized clinical trial. JAMA 2014; 312:2223–2233.
- Packham DK, Rasmussen HS, Lavin PT, et al. Sodium zirconium cyclosilicate in hyperkalemia. N Engl J Med 2015; 372:222–231.
- Anker SD, Kosiborod M, Zannad F, et al. Maintenance of serum potassium with sodium zirconium cyclosilicate (ZS-9) in heart failure patients: results from a phase 3 randomized, double-blind, placebo-controlled trial. Eur J Heart Fail 2015; 17:1050–1056.
Hyperkalemia is common in patients with cardiovascular disease. Its consequences can be severe and life-threatening, and its management and prevention require a multidisciplinary approach that entails reducing intake of high-potassium foods, adjusting medications that cause hyperkalemia, and adding medications that reduce the plasma potassium concentration. With this approach, patients at high risk can receive the cardiorenal benefits of drugs that block the renin-angiotensin-aldosterone system without developing hyperkalemia.
98% OF POTASSIUM IS INSIDE CELLS
The body of a typical 70-kg man contains about 3,500 mmol of potassium, 98% of which is in the intracellular space; the remaining 2% is in the extracellular space. This large intracellular-to-extracellular gradient determines the cell voltage and explains why disorders in plasma potassium give rise to manifestations in excitable tissues such as the heart and nervous system.
The most important determinants of potassium distribution between the intracellular and extracellular space are insulin and beta-adrenergic receptor stimulation.
Maintenance of total-body potassium content is primarily the job of the kidneys, with a small contribution by the gastrointestinal tract.1,2 Hyperkalemia is most commonly encountered in patients with decreased kidney function.
The normal kidney can secrete a large amount of potassium, making hyperkalemia uncommon in the absence of kidney disease. This large capacity may have evolved to handle the diet of Paleolithic humans, which contained 4 times as much potassium as contemporary diets.3,4 With the onset of agriculture, dietary intake of potassium has progressively declined while sodium intake has risen. A popular theory suggests this mismatch between the modern diet and the nutritional requirements encoded in the human genome during evolution may contribute to chronic diseases such as hypertension, stroke, kidney stones, and bone disease.5
MANY POTENTIAL CAUSES OF HYPERKALEMIA
Causes of hyperkalemia are outlined in Table 1. Shifting of potassium from the cells to the extracellular space is a cause of transient hyperkalemia, while chronic hyperkalemia indicates an impairment in renal potassium secretion. The following discussion is a guide to the approach to the hyperkalemic patient.
Is the patient’s hyperkalemia really pseudohyperkalemia?
Pseudohyperkalemia, an artifact of measurement, occurs due to mechanical release of potassium from cells during phlebotomy or specimen processing.6 This diagnosis is made when the serum potassium concentration exceeds the plasma potassium concentration by more than 0.5 mmol/L, and should be considered when hyperkalemia occurs in the absence of a clinical risk factor. Fist-clenching, application of a tight-fitting tourniquet, or use of small-bore needles during phlebotomy can all cause pseudohyperkalemia.
Mechanism of pseudohyperkalemia. Since serum is the liquid part of blood remaining after coagulation, release of potassium from cells injured during the process of coagulation raises the potassium level in the serum. Plasma is the cell-free part of blood that has been treated with anticoagulants; it has no cells that can be injured and release potassium. Thus, the serum potassium level will be higher than that in the plasma.
Reverse pseudohyperkalemia, in contrast, occurs when the plasma potassium level is falsely elevated but the serum value is normal. This situation has been described in hematologic disorders characterized by pronounced leukocytosis in which malignant cells are prone to lysis with minimal mechanical stress due to increased fragility or altered sodium-potassium ATPase pump activity.7 This phenomenon is unusual but occurs because the cells are so fragile.
A spurious increase in plasma potassium concentration along with a low plasma calcium concentration raises the possibility of calcium chelation and release of potassium in a sample tube contaminated with the anticoagulant ethylenediaminetetraacetic acid.
Is there increased potassium intake?
Increased potassium intake is a potential cause of hyperkalemia in patients with decreased kidney function or adrenal disease.
Foods naturally rich in potassium include bananas (a medium-sized banana contains 451 mg or 12 mmol of potassium) and potatoes (844 mg or 22 mmol in a large baked potato with skin). Other potassium-rich foods are melons, citrus juice, and avocados. Less-obvious food sources include raw coconut juice (potassium concentration 44.3 mmol/L) and noni juice (56 mmol/L).
Salt substitutes, recommended to hypertensive patients with chronic kidney disease, can be a hidden source of dietary potassium.
Clay ingestion is a potential cause of dyskalemia. White clay consumption causes hypokalemia due to potassium binding in the gastrointestinal tract. Red clay or river bed clay, on the other hand, is enriched in potassium (100 mmol of potassium in 100 g of clay) and can cause life-threatening hyperkalemia in patients with chronic kidney disease.8
Eating burnt match heads. Some individuals chew and ingest burnt match heads, a condition called cautopyreiophagia. In one reported case,9 this activity contributed an additional 80 mmol of daily potassium intake in a dialysis patient, resulting in a plasma potassium concentration of 8 mmol/L.
Is the hyperkalemia the result of a cellular shift?
Acute hyperkalemia can be the result of redistribution of cellular potassium. Shifting of as little as 2% of the body’s potassium from the intracellular to the extracellular space can double the plasma potassium concentration.
Tissue injury. Hyperkalemia frequently occurs in diseases that cause tissue injury such as rhabdomyolysis, trauma, massive hemolysis, and tumor lysis.
Insulin deficiency. Insulin and catecholamines are major regulators of potassium distribution within the body. After a meal, release of insulin not only regulates the plasma glucose concentration, it also causes potassium to move into cells until the kidneys have had sufficient time to excrete the dietary potassium load and reestablish total-body potassium content.
Exercise, beta-blockers. During exercise, potassium is released from skeletal muscle cells and accumulates in the interstitial compartment, where it exerts a vasodilatory effect. The simultaneous increase in circulating catecholamines regulates this release by promoting cell potassium uptake through beta-adrenergic receptor stimulation.
Metabolic acidosis can facilitate exit (ie, shift) of potassium from cells, but this effect depends on the type of acidosis. Hyperchloremic normal anion gap acidosis (mineral acidosis) most commonly causes this effect due to the relative impermeability of the cell membrane to the chloride anion. As hydrogen ions move into the cell due to accumulation of ammonium chloride or hydrogen chloride, electrical neutrality is maintained by potassium exit.
In contrast, organic acidosis (due to lactic, beta-hydroxybutyric, or methylmalonic acid) tends not to cause a potassium shift, since most organic anions readily cross the cell membrane along with hydrogen. Lactic acidosis is often associated with potassium shift, but this effect is due to loss of cell integrity as a result of cell ischemia. The hyperkalemia typically present on admission in patients with diabetic ketoacidosis is the result of insulin deficiency and hypertonicity and not the underlying organic acidosis.10
Hypertonic states can cause hyperkalemia due to cell shift. For example, hyperglycemia, as in diabetic ketoacidosis, pulls water from the intracellular into the extracellular compartment, thereby concentrating intracellular potassium and creating a more favorable gradient for potassium efflux through membrane channels. This same effect can occur in neurosurgical patients given large amounts of hypertonic mannitol. Repetitive doses of immunoglobulin can lead to extracellular accumulation of sorbitol, maltose, or sucrose, since these sugars are added to the preparations to prevent immunoglobulin aggregation.11
Is a disturbance in renal potassium excretion present?
Sustained hyperkalemia is more commonly associated with decreases in renal potassium excretion than with a cellular shift. In most instances the clinician can distinguish between cell shift and impaired renal excretion based on the available clinical data.
The transtubular potassium gradient has been used to determine whether there is a disturbance in renal potassium excretion and to assess renal potassium handling.12
This calculation is based on the assumption that only water is reabsorbed past the cortical collecting duct, and not solutes. It has fallen out of favor since we have found this assumption to be incorrect; a large amount of urea is reabsorbed daily in the downstream medullary collecting duct as a result of intrarenal recycling of urea.
The one situation in which the transtubular potassium gradient may be of use is determining whether hyperkalemia is a result of low aldosterone levels as opposed to aldosterone resistance. One can compare the transtubular potassium gradient before and after a physiologic dose (0.05 mg) of 9-alpha fludrocortisone. An increase of more than 6 over a 4-hour period favors aldosterone deficiency, whereas smaller changes would indicate aldosterone resistance.
24-hour potassium excretion, spot urine potassium-creatinine ratio. A better way to assess renal potassium handling is to measure the amount of potassium in a 24-hour urine collection or determine a spot urine potassium-creatinine ratio. A 24-hour urinary potassium excretion of less than 15 mmol or a potassium-creatinine ratio less than 1 suggests an extrarenal cause of hypokalemia. A ratio greater than 20 would be an appropriate renal response to hyperkalemia.
One or more of 3 abnormalities should be considered in the hyperkalemic patient with impaired renal excretion of potassium:
- Decreased distal delivery of sodium
- Mineralocorticoid deficiency
- Abnormal cortical collecting tubule function.13
Decreased distal delivery of sodium
Under normal circumstances, potassium is freely filtered across the glomerulus and then mostly reabsorbed in the proximal tubule and thick ascending limb. Potassium secretion begins in the distal convoluted tubule and increases in magnitude into the collecting duct. Tubular secretion is the component of potassium handling that varies and is regulated according to physiologic needs.
In acute kidney injury, the rapid decline in glomerular filtration rate and reduction in functioning nephron mass lead to decreased distal potassium secretion.
Hyperkalemia is a frequent problem when oliguria is present, since the reduction in distal delivery of sodium and water further impairs potassium secretion. Patients with oliguric acute kidney injury are more likely to have a more severe underlying disease state, and therefore tissue breakdown and catabolism further increase the risk of hyperkalemia.
In contrast, in nonoliguric patients, the renal injury tends to be less severe, and enough sodium and water are usually delivered distally to prevent hyperkalemia.
In chronic kidney disease, nephron dropout and reduction in collecting tubule mass also lead to a global decline in distal potassium secretion. However, this is countered by an increased capacity of the remaining individual nephrons for potassium secretion. High flow, increased distal sodium delivery, and increased activity and number of sodium-potassium ATPase pumps in the remaining nephrons account for this increased secretory capacity.14 As renal function declines over time, colonic potassium secretion progressively increases.15
These adaptive changes help to keep the plasma potassium concentration within the normal range until the glomerular filtration rate falls to less than 10 or 15 mL/min. Development of hyperkalemia with more modest reductions in the glomerular filtration rate suggest decreased mineralocorticoid activity or a specific lesion of the tubule.
Mineralocorticoid deficiency
Aldosterone deficiency can occur alone or in combination with decreased cortisol levels. Destruction of the adrenal glands is suggested when both hormones are reduced. Enzyme defects in cortisol metabolism can result in either isolated deficiency of aldosterone or adrenogenital syndromes associated with decreased mineralocorticoid activity.
Heparin administration leads to a reversible defect in adrenal synthesis of aldosterone. Drugs that block the stimulatory effect of angiotensin II on the zona glomerulosa cells of the adrenal gland will lower aldosterone.
Renin-angiotensin-aldosterone system blockers. Angiotensin-converting enzyme inhibitors block the formation of angiotensin II, whereas angiotensin II receptor blockers prevent angiotensin II from binding to its adrenal receptor. The direct renin inhibitor aliskiren lowers angiotensin II levels by blocking the enzymatic activity of renin and lowers the circulating levels of both angiotensin I and II.16
The syndrome of hyporeninemic hypoaldosteronism is a common cause of hyperkalemia in patients who have a glomerular filtration rate between 40 and 60 mL/min. Diabetic nephropathy and interstitial renal disease are the most common clinical entities associated with this syndrome.10 Other causes include analgesic nephropathy, urinary tract obstruction, sickle cell disease, systemic lupus erythematosus, and amyloidosis.
Nonsteroidal anti-inflammatory drugs can cause hyperkalemia by suppressing renin release and reducing delivery of sodium to the distal nephron.18
Calcineurin inhibitors impair potassium secretion by suppressing renin release and by direct tubular effects.19
Beta-blockers. Beta-1 and to a lesser extent beta-2 receptor blockade can also result in a hyporeninemic state.
Distal tubular defect
Hyperkalemia can result from interstitial renal diseases that specifically affect the distal nephron. In this setting, the glomerular filtration rate is only mildly reduced, and circulating aldosterone levels are normal.
Renal transplant, lupus erythematosus, amyloidosis, urinary obstruction, and sickle cell disease are conditions in which an impairment in renin release may coexist with a defect in tubular secretion.
Potassium-sparing diuretics impair the ability of the cortical collecting tubule to secrete potassium. Specifically, amiloride and triamterene inhibit sodium reabsorption mediated by the epithelial sodium channel located on the apical membrane of the principal cell. This effect abolishes the lumen’s negative potential and thereby removes a driving force for potassium secretion.
Trimethoprim and pentamidine cause similar effects.
Spironolactone and eplerenone compete with aldosterone at the level of the mineralocorticoid receptor and can result in hyperkalemia.
Drospirenone, a non-testosterone-derived progestin contained in certain oral contraceptives, possesses mineralocorticoid-blocking effects similar to those of spironolactone.
The plasma potassium level should be monitored when these drugs are prescribed in patients receiving potassium supplements, renin-angiotensin-aldosterone system blockers, or nonsteroidal anti-inflammatory drugs.20
CLINICAL FEATURES OF HYPERKALEMIA
Neuromuscular manifestations of hyperkalemia include paresthesias and fasciculations in the arms and legs. Severe elevation in potassium can give rise to an ascending paralysis with eventual flaccid quadriplegia. Typically, the trunk, head, and respiratory muscles are spared, and respiratory failure is rare.
Cardiac signs
Hyperkalemia has depolarizing effects on the heart that are manifested by changes in the electrocardiogram (Figure 2). The progressive changes of hyperkalemia are classically listed as:
- Peaked T waves that are tall, narrow, and symmetrical and can occasionally be confused with the hyperacute T-wave change associated with an ST-segment elevation myocardial infarction.21 However, in the latter condition, the T waves tend to be more broad-based and asymmetric in shape.
- ST-segment depression
- Widening of the PR interval
- Widening of the QRS interval
- Loss of the P wave
- A sine-wave pattern—an ominous development and a harbinger of impending ventricular fibrillation and asystole.
The plasma potassium concentration often correlates poorly with cardiac manifestations. In a retrospective review, only 16 of 90 cases met strict criteria for electrocardiographic changes reflective of hyperkalemia (defined as new peaked and symmetric T waves that resolved on follow-up).22 In 13 of these cases, the electrocardiogram was interpreted as showing no T-wave changes even when read by a cardiologist. In addition, electrocardiographic criteria for hyperkalemia were noted in only 1 of 14 patients who manifested arrhythmias or cardiac arrest attributed to increased plasma potassium concentration.
TREATMENT OF ACUTE HYPERKALEMIA
The treatment of hyperkalemia depends on the magnitude of increase in the plasma potassium concentration and the presence or absence of electrocardiographic changes or neuromuscular symptoms.23 Acute treatment is indicated for marked electrocardiographic changes and severe muscle weakness.
Intravenous calcium rapidly normalizes membrane excitability by antagonizing the potassium-induced decrease in membrane excitability but does not alter the plasma potassium concentration.
Insulin lowers the plasma potassium concentration by promoting its entry into cells. To avoid hypoglycemia, 10 units of short-acting insulin should be accompanied by a 50-g infusion of glucose, increased to 60 g if 20 units of insulin are given.24
Beta-2 receptor agonists produce a similar effect. The shift of potassium into cells with insulin and beta-2-adrenergic receptor stimulation is brought about by increases in sodium-potassium ATPase pump activity, primarily in skeletal muscle cells.
Sodium bicarbonate, in the absence of acidosis, lowers the plasma potassium concentration only slightly. It should be reserved for hyperkalemic patients who have coexisting metabolic acidosis after the patient has received insulin and glucose, an adrenergic agent, and calcium.
These acute treatments need to be followed by therapies designed to lower the total body potassium content such as diuretics, potassium-binding drugs, and dialysis.
TREATMENT OF CHRONIC HYPERKALEMIA
Review medications. Once the diagnosis of hyperkalemia has been made, the initial approach should be to review the patient’s medications and make every effort to discontinue drugs that can impair renal potassium excretion.16 Patients should be asked about their use of over-the-counter nonsteroidal anti-inflammatory drugs and herbal remedies, since herbs may be a hidden source of dietary potassium.
Dietary counseling. Patients should be instructed to reduce their dietary intake of potassium and to avoid salt substitutes that contain potassium.
Diuretic therapy is beneficial in minimizing hyperkalemia in patients with chronic kidney disease. Thiazide and loop diuretics enhance renal potassium excretion by increasing flow and delivery of sodium to the collecting duct. Thiazide diuretics are effective when the estimated glomerular filtration rate is greater than 30 mL/min, while loop diuretics should be used in patients with more severe renal insufficiency (Table 2).
Sodium bicarbonate is an effective agent to minimize increases in the plasma potassium concentration in patients with chronic kidney disease and metabolic acidosis. This drug increases renal potassium excretion by increasing distal sodium delivery and shifts potassium into cells as the acidosis is corrected. The likelihood of developing volume overload as a complication of sodium bicarbonate administration can be minimized with effective diuretic therapy.
Avoiding hyperkalemia if renin-angiotensin-aldosterone system blockers are needed
Renin-angiotensin-aldosterone system blockers can be problematic, as these drugs cause hyperkalemia, often in the very patients who derive the greatest cardiovascular benefit from them.16 A number of steps can reduce the risk of hyperkalemia and allow these drugs to be used.
The initial dose should be low and the plasma potassium should be measured within 1 to 2 weeks after drug initiation. If the potassium level is normal, the dose can be titrated upwards with remeasurement of the plasma potassium after each dose titration. If the plasma potassium concentration rises to 5.5 mmol/L, in some cases lowering the dose will reduce the potassium concentration and allow the patient to remain on the drug.
In patients at risk of hyperkalemia, angiotensin II receptor blockers and direct renin inhibitors should be used with the same caution as angiotensin-converting enzyme inhibitors.
If the plasma potassium concentration exceeds 5.5 mmol/L despite the above precautions, one can consider using a potassium-binding drug (see below) before deciding to avoid renin-angiotensin-aldosterone system blockers.
Sodium polystyrene sulfonate binds potassium in the gastrointestinal tract in exchange for sodium and has been used to manage hyperkalemia. This drug is most commonly given along with sorbitol as a therapy for acute hyperkalemia. Although the drug is widely used, most of the potassium-lowering effect is due to an increase in stool volume caused by sorbitol.25,26 In addition, long-term use is poorly tolerated, and the drug has been linked to gastrointestinal toxicity in rare cases.
Patiromer and sodium zirconium cyclosilicate are two new potassium-binding drugs that have been shown to be effective in reducing plasma potassium concentration in the setting of ongoing use of renin-angiotensin-aldosterone system blockers.
Patiromer is a nonabsorbed polymer approved for clinical use to treat hyperkalemia. The drug binds potassium in exchange for calcium in the gastrointestinal tract, predominantly in the colon, and lowers the plasma potassium concentration in a dose-dependent manner, with the greatest reduction in those with higher starting values.27,28
Patiromer effectively controlled plasma potassium concentrations in a 1-year randomized trial in high-risk patients on renin-angiotensin-aldosterone system blockers.29 The main adverse events in clinical trials have been constipation and hypomagnesemia, which required magnesium replacement in a small number of patients, but overall, the drug is well tolerated.
Sodium zirconium cyclosilicate is a nonabsorbed microporous compound that binds potassium in exchange for sodium throughout the gastrointestinal tract. It has been found effective in lowering plasma potassium concentration in a dose-dependent fashion in high-risk patients, most of whom were receiving renin-angiotensin-aldosterone system blockers.30–32 Adverse events were generally comparable to those with placebo in clinical trials; however, edema occurred more frequently when higher doses were used. This drug is not yet approved for clinical use.
Hyperkalemia is common in patients with cardiovascular disease. Its consequences can be severe and life-threatening, and its management and prevention require a multidisciplinary approach that entails reducing intake of high-potassium foods, adjusting medications that cause hyperkalemia, and adding medications that reduce the plasma potassium concentration. With this approach, patients at high risk can receive the cardiorenal benefits of drugs that block the renin-angiotensin-aldosterone system without developing hyperkalemia.
98% OF POTASSIUM IS INSIDE CELLS
The body of a typical 70-kg man contains about 3,500 mmol of potassium, 98% of which is in the intracellular space; the remaining 2% is in the extracellular space. This large intracellular-to-extracellular gradient determines the cell voltage and explains why disorders in plasma potassium give rise to manifestations in excitable tissues such as the heart and nervous system.
The most important determinants of potassium distribution between the intracellular and extracellular space are insulin and beta-adrenergic receptor stimulation.
Maintenance of total-body potassium content is primarily the job of the kidneys, with a small contribution by the gastrointestinal tract.1,2 Hyperkalemia is most commonly encountered in patients with decreased kidney function.
The normal kidney can secrete a large amount of potassium, making hyperkalemia uncommon in the absence of kidney disease. This large capacity may have evolved to handle the diet of Paleolithic humans, which contained 4 times as much potassium as contemporary diets.3,4 With the onset of agriculture, dietary intake of potassium has progressively declined while sodium intake has risen. A popular theory suggests this mismatch between the modern diet and the nutritional requirements encoded in the human genome during evolution may contribute to chronic diseases such as hypertension, stroke, kidney stones, and bone disease.5
MANY POTENTIAL CAUSES OF HYPERKALEMIA
Causes of hyperkalemia are outlined in Table 1. Shifting of potassium from the cells to the extracellular space is a cause of transient hyperkalemia, while chronic hyperkalemia indicates an impairment in renal potassium secretion. The following discussion is a guide to the approach to the hyperkalemic patient.
Is the patient’s hyperkalemia really pseudohyperkalemia?
Pseudohyperkalemia, an artifact of measurement, occurs due to mechanical release of potassium from cells during phlebotomy or specimen processing.6 This diagnosis is made when the serum potassium concentration exceeds the plasma potassium concentration by more than 0.5 mmol/L, and should be considered when hyperkalemia occurs in the absence of a clinical risk factor. Fist-clenching, application of a tight-fitting tourniquet, or use of small-bore needles during phlebotomy can all cause pseudohyperkalemia.
Mechanism of pseudohyperkalemia. Since serum is the liquid part of blood remaining after coagulation, release of potassium from cells injured during the process of coagulation raises the potassium level in the serum. Plasma is the cell-free part of blood that has been treated with anticoagulants; it has no cells that can be injured and release potassium. Thus, the serum potassium level will be higher than that in the plasma.
Reverse pseudohyperkalemia, in contrast, occurs when the plasma potassium level is falsely elevated but the serum value is normal. This situation has been described in hematologic disorders characterized by pronounced leukocytosis in which malignant cells are prone to lysis with minimal mechanical stress due to increased fragility or altered sodium-potassium ATPase pump activity.7 This phenomenon is unusual but occurs because the cells are so fragile.
A spurious increase in plasma potassium concentration along with a low plasma calcium concentration raises the possibility of calcium chelation and release of potassium in a sample tube contaminated with the anticoagulant ethylenediaminetetraacetic acid.
Is there increased potassium intake?
Increased potassium intake is a potential cause of hyperkalemia in patients with decreased kidney function or adrenal disease.
Foods naturally rich in potassium include bananas (a medium-sized banana contains 451 mg or 12 mmol of potassium) and potatoes (844 mg or 22 mmol in a large baked potato with skin). Other potassium-rich foods are melons, citrus juice, and avocados. Less-obvious food sources include raw coconut juice (potassium concentration 44.3 mmol/L) and noni juice (56 mmol/L).
Salt substitutes, recommended to hypertensive patients with chronic kidney disease, can be a hidden source of dietary potassium.
Clay ingestion is a potential cause of dyskalemia. White clay consumption causes hypokalemia due to potassium binding in the gastrointestinal tract. Red clay or river bed clay, on the other hand, is enriched in potassium (100 mmol of potassium in 100 g of clay) and can cause life-threatening hyperkalemia in patients with chronic kidney disease.8
Eating burnt match heads. Some individuals chew and ingest burnt match heads, a condition called cautopyreiophagia. In one reported case,9 this activity contributed an additional 80 mmol of daily potassium intake in a dialysis patient, resulting in a plasma potassium concentration of 8 mmol/L.
Is the hyperkalemia the result of a cellular shift?
Acute hyperkalemia can be the result of redistribution of cellular potassium. Shifting of as little as 2% of the body’s potassium from the intracellular to the extracellular space can double the plasma potassium concentration.
Tissue injury. Hyperkalemia frequently occurs in diseases that cause tissue injury such as rhabdomyolysis, trauma, massive hemolysis, and tumor lysis.
Insulin deficiency. Insulin and catecholamines are major regulators of potassium distribution within the body. After a meal, release of insulin not only regulates the plasma glucose concentration, it also causes potassium to move into cells until the kidneys have had sufficient time to excrete the dietary potassium load and reestablish total-body potassium content.
Exercise, beta-blockers. During exercise, potassium is released from skeletal muscle cells and accumulates in the interstitial compartment, where it exerts a vasodilatory effect. The simultaneous increase in circulating catecholamines regulates this release by promoting cell potassium uptake through beta-adrenergic receptor stimulation.
Metabolic acidosis can facilitate exit (ie, shift) of potassium from cells, but this effect depends on the type of acidosis. Hyperchloremic normal anion gap acidosis (mineral acidosis) most commonly causes this effect due to the relative impermeability of the cell membrane to the chloride anion. As hydrogen ions move into the cell due to accumulation of ammonium chloride or hydrogen chloride, electrical neutrality is maintained by potassium exit.
In contrast, organic acidosis (due to lactic, beta-hydroxybutyric, or methylmalonic acid) tends not to cause a potassium shift, since most organic anions readily cross the cell membrane along with hydrogen. Lactic acidosis is often associated with potassium shift, but this effect is due to loss of cell integrity as a result of cell ischemia. The hyperkalemia typically present on admission in patients with diabetic ketoacidosis is the result of insulin deficiency and hypertonicity and not the underlying organic acidosis.10
Hypertonic states can cause hyperkalemia due to cell shift. For example, hyperglycemia, as in diabetic ketoacidosis, pulls water from the intracellular into the extracellular compartment, thereby concentrating intracellular potassium and creating a more favorable gradient for potassium efflux through membrane channels. This same effect can occur in neurosurgical patients given large amounts of hypertonic mannitol. Repetitive doses of immunoglobulin can lead to extracellular accumulation of sorbitol, maltose, or sucrose, since these sugars are added to the preparations to prevent immunoglobulin aggregation.11
Is a disturbance in renal potassium excretion present?
Sustained hyperkalemia is more commonly associated with decreases in renal potassium excretion than with a cellular shift. In most instances the clinician can distinguish between cell shift and impaired renal excretion based on the available clinical data.
The transtubular potassium gradient has been used to determine whether there is a disturbance in renal potassium excretion and to assess renal potassium handling.12
This calculation is based on the assumption that only water is reabsorbed past the cortical collecting duct, and not solutes. It has fallen out of favor since we have found this assumption to be incorrect; a large amount of urea is reabsorbed daily in the downstream medullary collecting duct as a result of intrarenal recycling of urea.
The one situation in which the transtubular potassium gradient may be of use is determining whether hyperkalemia is a result of low aldosterone levels as opposed to aldosterone resistance. One can compare the transtubular potassium gradient before and after a physiologic dose (0.05 mg) of 9-alpha fludrocortisone. An increase of more than 6 over a 4-hour period favors aldosterone deficiency, whereas smaller changes would indicate aldosterone resistance.
24-hour potassium excretion, spot urine potassium-creatinine ratio. A better way to assess renal potassium handling is to measure the amount of potassium in a 24-hour urine collection or determine a spot urine potassium-creatinine ratio. A 24-hour urinary potassium excretion of less than 15 mmol or a potassium-creatinine ratio less than 1 suggests an extrarenal cause of hypokalemia. A ratio greater than 20 would be an appropriate renal response to hyperkalemia.
One or more of 3 abnormalities should be considered in the hyperkalemic patient with impaired renal excretion of potassium:
- Decreased distal delivery of sodium
- Mineralocorticoid deficiency
- Abnormal cortical collecting tubule function.13
Decreased distal delivery of sodium
Under normal circumstances, potassium is freely filtered across the glomerulus and then mostly reabsorbed in the proximal tubule and thick ascending limb. Potassium secretion begins in the distal convoluted tubule and increases in magnitude into the collecting duct. Tubular secretion is the component of potassium handling that varies and is regulated according to physiologic needs.
In acute kidney injury, the rapid decline in glomerular filtration rate and reduction in functioning nephron mass lead to decreased distal potassium secretion.
Hyperkalemia is a frequent problem when oliguria is present, since the reduction in distal delivery of sodium and water further impairs potassium secretion. Patients with oliguric acute kidney injury are more likely to have a more severe underlying disease state, and therefore tissue breakdown and catabolism further increase the risk of hyperkalemia.
In contrast, in nonoliguric patients, the renal injury tends to be less severe, and enough sodium and water are usually delivered distally to prevent hyperkalemia.
In chronic kidney disease, nephron dropout and reduction in collecting tubule mass also lead to a global decline in distal potassium secretion. However, this is countered by an increased capacity of the remaining individual nephrons for potassium secretion. High flow, increased distal sodium delivery, and increased activity and number of sodium-potassium ATPase pumps in the remaining nephrons account for this increased secretory capacity.14 As renal function declines over time, colonic potassium secretion progressively increases.15
These adaptive changes help to keep the plasma potassium concentration within the normal range until the glomerular filtration rate falls to less than 10 or 15 mL/min. Development of hyperkalemia with more modest reductions in the glomerular filtration rate suggest decreased mineralocorticoid activity or a specific lesion of the tubule.
Mineralocorticoid deficiency
Aldosterone deficiency can occur alone or in combination with decreased cortisol levels. Destruction of the adrenal glands is suggested when both hormones are reduced. Enzyme defects in cortisol metabolism can result in either isolated deficiency of aldosterone or adrenogenital syndromes associated with decreased mineralocorticoid activity.
Heparin administration leads to a reversible defect in adrenal synthesis of aldosterone. Drugs that block the stimulatory effect of angiotensin II on the zona glomerulosa cells of the adrenal gland will lower aldosterone.
Renin-angiotensin-aldosterone system blockers. Angiotensin-converting enzyme inhibitors block the formation of angiotensin II, whereas angiotensin II receptor blockers prevent angiotensin II from binding to its adrenal receptor. The direct renin inhibitor aliskiren lowers angiotensin II levels by blocking the enzymatic activity of renin and lowers the circulating levels of both angiotensin I and II.16
The syndrome of hyporeninemic hypoaldosteronism is a common cause of hyperkalemia in patients who have a glomerular filtration rate between 40 and 60 mL/min. Diabetic nephropathy and interstitial renal disease are the most common clinical entities associated with this syndrome.10 Other causes include analgesic nephropathy, urinary tract obstruction, sickle cell disease, systemic lupus erythematosus, and amyloidosis.
Nonsteroidal anti-inflammatory drugs can cause hyperkalemia by suppressing renin release and reducing delivery of sodium to the distal nephron.18
Calcineurin inhibitors impair potassium secretion by suppressing renin release and by direct tubular effects.19
Beta-blockers. Beta-1 and to a lesser extent beta-2 receptor blockade can also result in a hyporeninemic state.
Distal tubular defect
Hyperkalemia can result from interstitial renal diseases that specifically affect the distal nephron. In this setting, the glomerular filtration rate is only mildly reduced, and circulating aldosterone levels are normal.
Renal transplant, lupus erythematosus, amyloidosis, urinary obstruction, and sickle cell disease are conditions in which an impairment in renin release may coexist with a defect in tubular secretion.
Potassium-sparing diuretics impair the ability of the cortical collecting tubule to secrete potassium. Specifically, amiloride and triamterene inhibit sodium reabsorption mediated by the epithelial sodium channel located on the apical membrane of the principal cell. This effect abolishes the lumen’s negative potential and thereby removes a driving force for potassium secretion.
Trimethoprim and pentamidine cause similar effects.
Spironolactone and eplerenone compete with aldosterone at the level of the mineralocorticoid receptor and can result in hyperkalemia.
Drospirenone, a non-testosterone-derived progestin contained in certain oral contraceptives, possesses mineralocorticoid-blocking effects similar to those of spironolactone.
The plasma potassium level should be monitored when these drugs are prescribed in patients receiving potassium supplements, renin-angiotensin-aldosterone system blockers, or nonsteroidal anti-inflammatory drugs.20
CLINICAL FEATURES OF HYPERKALEMIA
Neuromuscular manifestations of hyperkalemia include paresthesias and fasciculations in the arms and legs. Severe elevation in potassium can give rise to an ascending paralysis with eventual flaccid quadriplegia. Typically, the trunk, head, and respiratory muscles are spared, and respiratory failure is rare.
Cardiac signs
Hyperkalemia has depolarizing effects on the heart that are manifested by changes in the electrocardiogram (Figure 2). The progressive changes of hyperkalemia are classically listed as:
- Peaked T waves that are tall, narrow, and symmetrical and can occasionally be confused with the hyperacute T-wave change associated with an ST-segment elevation myocardial infarction.21 However, in the latter condition, the T waves tend to be more broad-based and asymmetric in shape.
- ST-segment depression
- Widening of the PR interval
- Widening of the QRS interval
- Loss of the P wave
- A sine-wave pattern—an ominous development and a harbinger of impending ventricular fibrillation and asystole.
The plasma potassium concentration often correlates poorly with cardiac manifestations. In a retrospective review, only 16 of 90 cases met strict criteria for electrocardiographic changes reflective of hyperkalemia (defined as new peaked and symmetric T waves that resolved on follow-up).22 In 13 of these cases, the electrocardiogram was interpreted as showing no T-wave changes even when read by a cardiologist. In addition, electrocardiographic criteria for hyperkalemia were noted in only 1 of 14 patients who manifested arrhythmias or cardiac arrest attributed to increased plasma potassium concentration.
TREATMENT OF ACUTE HYPERKALEMIA
The treatment of hyperkalemia depends on the magnitude of increase in the plasma potassium concentration and the presence or absence of electrocardiographic changes or neuromuscular symptoms.23 Acute treatment is indicated for marked electrocardiographic changes and severe muscle weakness.
Intravenous calcium rapidly normalizes membrane excitability by antagonizing the potassium-induced decrease in membrane excitability but does not alter the plasma potassium concentration.
Insulin lowers the plasma potassium concentration by promoting its entry into cells. To avoid hypoglycemia, 10 units of short-acting insulin should be accompanied by a 50-g infusion of glucose, increased to 60 g if 20 units of insulin are given.24
Beta-2 receptor agonists produce a similar effect. The shift of potassium into cells with insulin and beta-2-adrenergic receptor stimulation is brought about by increases in sodium-potassium ATPase pump activity, primarily in skeletal muscle cells.
Sodium bicarbonate, in the absence of acidosis, lowers the plasma potassium concentration only slightly. It should be reserved for hyperkalemic patients who have coexisting metabolic acidosis after the patient has received insulin and glucose, an adrenergic agent, and calcium.
These acute treatments need to be followed by therapies designed to lower the total body potassium content such as diuretics, potassium-binding drugs, and dialysis.
TREATMENT OF CHRONIC HYPERKALEMIA
Review medications. Once the diagnosis of hyperkalemia has been made, the initial approach should be to review the patient’s medications and make every effort to discontinue drugs that can impair renal potassium excretion.16 Patients should be asked about their use of over-the-counter nonsteroidal anti-inflammatory drugs and herbal remedies, since herbs may be a hidden source of dietary potassium.
Dietary counseling. Patients should be instructed to reduce their dietary intake of potassium and to avoid salt substitutes that contain potassium.
Diuretic therapy is beneficial in minimizing hyperkalemia in patients with chronic kidney disease. Thiazide and loop diuretics enhance renal potassium excretion by increasing flow and delivery of sodium to the collecting duct. Thiazide diuretics are effective when the estimated glomerular filtration rate is greater than 30 mL/min, while loop diuretics should be used in patients with more severe renal insufficiency (Table 2).
Sodium bicarbonate is an effective agent to minimize increases in the plasma potassium concentration in patients with chronic kidney disease and metabolic acidosis. This drug increases renal potassium excretion by increasing distal sodium delivery and shifts potassium into cells as the acidosis is corrected. The likelihood of developing volume overload as a complication of sodium bicarbonate administration can be minimized with effective diuretic therapy.
Avoiding hyperkalemia if renin-angiotensin-aldosterone system blockers are needed
Renin-angiotensin-aldosterone system blockers can be problematic, as these drugs cause hyperkalemia, often in the very patients who derive the greatest cardiovascular benefit from them.16 A number of steps can reduce the risk of hyperkalemia and allow these drugs to be used.
The initial dose should be low and the plasma potassium should be measured within 1 to 2 weeks after drug initiation. If the potassium level is normal, the dose can be titrated upwards with remeasurement of the plasma potassium after each dose titration. If the plasma potassium concentration rises to 5.5 mmol/L, in some cases lowering the dose will reduce the potassium concentration and allow the patient to remain on the drug.
In patients at risk of hyperkalemia, angiotensin II receptor blockers and direct renin inhibitors should be used with the same caution as angiotensin-converting enzyme inhibitors.
If the plasma potassium concentration exceeds 5.5 mmol/L despite the above precautions, one can consider using a potassium-binding drug (see below) before deciding to avoid renin-angiotensin-aldosterone system blockers.
Sodium polystyrene sulfonate binds potassium in the gastrointestinal tract in exchange for sodium and has been used to manage hyperkalemia. This drug is most commonly given along with sorbitol as a therapy for acute hyperkalemia. Although the drug is widely used, most of the potassium-lowering effect is due to an increase in stool volume caused by sorbitol.25,26 In addition, long-term use is poorly tolerated, and the drug has been linked to gastrointestinal toxicity in rare cases.
Patiromer and sodium zirconium cyclosilicate are two new potassium-binding drugs that have been shown to be effective in reducing plasma potassium concentration in the setting of ongoing use of renin-angiotensin-aldosterone system blockers.
Patiromer is a nonabsorbed polymer approved for clinical use to treat hyperkalemia. The drug binds potassium in exchange for calcium in the gastrointestinal tract, predominantly in the colon, and lowers the plasma potassium concentration in a dose-dependent manner, with the greatest reduction in those with higher starting values.27,28
Patiromer effectively controlled plasma potassium concentrations in a 1-year randomized trial in high-risk patients on renin-angiotensin-aldosterone system blockers.29 The main adverse events in clinical trials have been constipation and hypomagnesemia, which required magnesium replacement in a small number of patients, but overall, the drug is well tolerated.
Sodium zirconium cyclosilicate is a nonabsorbed microporous compound that binds potassium in exchange for sodium throughout the gastrointestinal tract. It has been found effective in lowering plasma potassium concentration in a dose-dependent fashion in high-risk patients, most of whom were receiving renin-angiotensin-aldosterone system blockers.30–32 Adverse events were generally comparable to those with placebo in clinical trials; however, edema occurred more frequently when higher doses were used. This drug is not yet approved for clinical use.
- Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ 2016; 40:480–490.
- Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 2015; 10:1050–1060.
- Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. N Engl J Med 1985; 312:283–289.
- Sebastian A, Frassetto LA, Sellmeyer DE, Morris RC Jr. The evolution-informed optimal dietary potassium intake of human beings greatly exceeds current and recommended intakes. Semin Nephrol 2006; 26:447–453.
- Palmer BF, Clegg DJ. Achieving the benefits of a high potassium, Paleolithic diet, without the toxicity. Mayo Clin Proc 2016; 91:496–508.
- Liamis G, Liberopoulos E, Barkas F, Elisaf M. Spurious electrolyte disorders: a diagnostic challenge for clinicians. Am J Nephrol 2013; 38:50–57.
- Mansoor S, Holtzman N, Emadi A. Reverse pseudohyperkalemia: an important clinical entity in chronic lymphocytic leukemia. Case Rep Hematol 2015; 2015:930379.
- Gelfand M, Zarate A, Knepshield J. Geophagia. A cause of life-threatening hyperkalemia in patients with chronic renal failure. JAMA 1975; 234:738–740.
- Abu-Hamdan D, Sondheimer J, Mahajan S. Cautopyreiophagia. Cause of life-threatening hyperkalemia in a patient undergoing hemodialysis. Am J Med 1985; 79:517–519.
- Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med 2015; 373:548–559.
- Daphnis E, Stylianou K, Alexandrakis M, et al. Acute renal failure, translocational hyponatremia and hyperkalemia following intravenous immunoglobulin therapy. Nephron Clin Pract 2007; 106:c143–c148.
- Choi M, Ziyadeh F. The utility of the transtubular potassium gradient in the evaluation of hyperkalemia. J Am Soc Nephrol 2008; 19:424–426.
- Palmer BF. A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis 2010; 56:387–393.
- Stanton BA. Renal potassium transport: morphological and functional adaptations. Am J Physiol 1989; 257:R989–R997.
- Hayes CP Jr, McLeod ME, Robinson RR. An extravenal mechanism for the maintenance of potassium balance in severe chronic renal failure. Trans Assoc Am Physicians 1967; 80:207–216.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Palmer BF. Renal dysfunction complicating treatment of hypertension. N Engl J Med 2002; 347:1256–1261.
- Palmer BF. Renal complications associated with use of nonsteroidal anti-inflammatory agents. J Investig Med 1995; 43:516–533.
- Hoorn E, Walsh S, McCormick J, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 2011; 17:1304–1309.
- Bird ST, Pepe SR, Etminan M, Liu X, Brophy JM, Delaney JA. The association between drospirenone and hyperkalemia: a comparative-safety study. BMC Clin Pharmacol 2011; 11:23.
- Wang K. Images in clinical medicine. “Pseudoinfarction” pattern due to hyperkalemia. N Engl J Med 2004; 351:593.
- Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol 2008; 3:324–330.
- Weisberg LS. Management of severe hyperkalemia. Crit Care Med 2008; 36:3246–3251.
- Harel Z, Kamel KS. Optimal dose and method of administration of intravenous insulin in the management of emergency hyperkalemia: a systematic review. PLoS One 2016; 11:e0154963.
- Sterns RH, Rojas M, Bernstein P, Chennupati S. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol 2010; 21:733–735.
- Emmett M, Hootkins RE, Fine KD, Santa Ana CA, Porter JL, Fordtran JS. Effect of three laxatives and a cation exchange resin on fecal sodium and potassium excretion. Gastroenterology 1995; 108:752–760.
- Bushinsky DA, Spiegel DM, Gross C, et al. Effect of patiromer on urinary ion excretion in healthy adults. Clin J Am Soc Nephrol 2016; 11:1769–1776.
- Weir MR, Bakris GL, Bushinsky DA, et al; OPAL-HK Investigators. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med 2015; 372:211–221.
- Bakris GL, Pitt B, Weir MR, et al; AMETHYST-DN Investigators. Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: the AMETHYST-DN randomized clinical trial. JAMA 2015; 314:151–161.
- Kosiborod M, Rasmussen HS, Lavin P, et al. Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia. The HARMONIZE randomized clinical trial. JAMA 2014; 312:2223–2233.
- Packham DK, Rasmussen HS, Lavin PT, et al. Sodium zirconium cyclosilicate in hyperkalemia. N Engl J Med 2015; 372:222–231.
- Anker SD, Kosiborod M, Zannad F, et al. Maintenance of serum potassium with sodium zirconium cyclosilicate (ZS-9) in heart failure patients: results from a phase 3 randomized, double-blind, placebo-controlled trial. Eur J Heart Fail 2015; 17:1050–1056.
- Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ 2016; 40:480–490.
- Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 2015; 10:1050–1060.
- Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. N Engl J Med 1985; 312:283–289.
- Sebastian A, Frassetto LA, Sellmeyer DE, Morris RC Jr. The evolution-informed optimal dietary potassium intake of human beings greatly exceeds current and recommended intakes. Semin Nephrol 2006; 26:447–453.
- Palmer BF, Clegg DJ. Achieving the benefits of a high potassium, Paleolithic diet, without the toxicity. Mayo Clin Proc 2016; 91:496–508.
- Liamis G, Liberopoulos E, Barkas F, Elisaf M. Spurious electrolyte disorders: a diagnostic challenge for clinicians. Am J Nephrol 2013; 38:50–57.
- Mansoor S, Holtzman N, Emadi A. Reverse pseudohyperkalemia: an important clinical entity in chronic lymphocytic leukemia. Case Rep Hematol 2015; 2015:930379.
- Gelfand M, Zarate A, Knepshield J. Geophagia. A cause of life-threatening hyperkalemia in patients with chronic renal failure. JAMA 1975; 234:738–740.
- Abu-Hamdan D, Sondheimer J, Mahajan S. Cautopyreiophagia. Cause of life-threatening hyperkalemia in a patient undergoing hemodialysis. Am J Med 1985; 79:517–519.
- Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med 2015; 373:548–559.
- Daphnis E, Stylianou K, Alexandrakis M, et al. Acute renal failure, translocational hyponatremia and hyperkalemia following intravenous immunoglobulin therapy. Nephron Clin Pract 2007; 106:c143–c148.
- Choi M, Ziyadeh F. The utility of the transtubular potassium gradient in the evaluation of hyperkalemia. J Am Soc Nephrol 2008; 19:424–426.
- Palmer BF. A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis 2010; 56:387–393.
- Stanton BA. Renal potassium transport: morphological and functional adaptations. Am J Physiol 1989; 257:R989–R997.
- Hayes CP Jr, McLeod ME, Robinson RR. An extravenal mechanism for the maintenance of potassium balance in severe chronic renal failure. Trans Assoc Am Physicians 1967; 80:207–216.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Palmer BF. Renal dysfunction complicating treatment of hypertension. N Engl J Med 2002; 347:1256–1261.
- Palmer BF. Renal complications associated with use of nonsteroidal anti-inflammatory agents. J Investig Med 1995; 43:516–533.
- Hoorn E, Walsh S, McCormick J, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 2011; 17:1304–1309.
- Bird ST, Pepe SR, Etminan M, Liu X, Brophy JM, Delaney JA. The association between drospirenone and hyperkalemia: a comparative-safety study. BMC Clin Pharmacol 2011; 11:23.
- Wang K. Images in clinical medicine. “Pseudoinfarction” pattern due to hyperkalemia. N Engl J Med 2004; 351:593.
- Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol 2008; 3:324–330.
- Weisberg LS. Management of severe hyperkalemia. Crit Care Med 2008; 36:3246–3251.
- Harel Z, Kamel KS. Optimal dose and method of administration of intravenous insulin in the management of emergency hyperkalemia: a systematic review. PLoS One 2016; 11:e0154963.
- Sterns RH, Rojas M, Bernstein P, Chennupati S. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol 2010; 21:733–735.
- Emmett M, Hootkins RE, Fine KD, Santa Ana CA, Porter JL, Fordtran JS. Effect of three laxatives and a cation exchange resin on fecal sodium and potassium excretion. Gastroenterology 1995; 108:752–760.
- Bushinsky DA, Spiegel DM, Gross C, et al. Effect of patiromer on urinary ion excretion in healthy adults. Clin J Am Soc Nephrol 2016; 11:1769–1776.
- Weir MR, Bakris GL, Bushinsky DA, et al; OPAL-HK Investigators. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med 2015; 372:211–221.
- Bakris GL, Pitt B, Weir MR, et al; AMETHYST-DN Investigators. Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: the AMETHYST-DN randomized clinical trial. JAMA 2015; 314:151–161.
- Kosiborod M, Rasmussen HS, Lavin P, et al. Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia. The HARMONIZE randomized clinical trial. JAMA 2014; 312:2223–2233.
- Packham DK, Rasmussen HS, Lavin PT, et al. Sodium zirconium cyclosilicate in hyperkalemia. N Engl J Med 2015; 372:222–231.
- Anker SD, Kosiborod M, Zannad F, et al. Maintenance of serum potassium with sodium zirconium cyclosilicate (ZS-9) in heart failure patients: results from a phase 3 randomized, double-blind, placebo-controlled trial. Eur J Heart Fail 2015; 17:1050–1056.
KEY POINTS
- Exclude pseudohyperkalemia in patients who have a normal electrocardiogram and no risk factors for the development of hyperkalemia.
- Decreased distal delivery of sodium, reduced mineralocorticoid levels or activity, and a distal tubular defect are causes of impaired renal potassium secretion.
- Medical conditions and medications that alter the renin-angiotensin-aldosterone system can give rise to hyperkalemia.
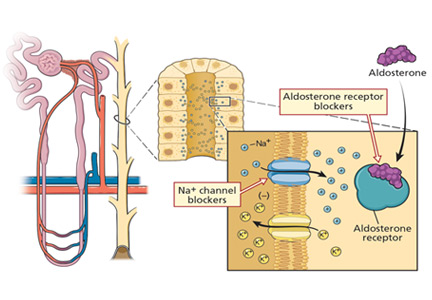
Diagnostic value of the physical examination in patients with dyspnea
Laennec’s stethoscope has survived more than 200 years, much longer than some of his contemporaries predicted. But will it survive the challenge of bedside ultrasonography and other technologic advances?
The physical examination, with its roots extending at least as far back as Hippocrates, may be at a crossroads as the mainstay of diagnosis. Physical signs can be subjective and lack sensitivity and specificity. Modern imaging and laboratory studies may already be more trusted.
If the physical examination is to survive, it must be accurate, reproducible, and efficient. Needed is a simple, evidence-based approach to the physical examination that enhances its diagnostic accuracy while maintaining bedside efficiency.
Here, we analyze the accuracy of the physical signs that are most effective in the clinical diagnosis of 4 common cardiopulmonary conditions that often present with dyspnea: pneumonia, pleural effusion, chronic obstructive pulmonary disease (COPD), and congestive heart failure.
LIKELIHOOD RATIOS
To grasp the significance of physical findings, it is necessary to understand the concept of likelihood ratios, which are widely accepted measures of the accuracy of a test or clinical finding.1,2 The positive likelihood ratio is the probability of a disease being present when the test is positive or the clinical finding is present, while the negative likelihood ratio is the probability that the disease is present when the test is negative or the clinical finding is absent. They are calculated as follows1:
Positive likelihood ratio = sensitivity / (1 – specificity)
Negative likelihood ratio = (1 – sensitivity) / specificity

Table 1 shows how the likelihood ratio of a test changes the posttest probability that a condition is present or absent, according to an analysis by McGee.2
STANDARDIZED TERMINOLOGY

PNEUMONIA
Pneumonia is a common disease, with more than 2 million cases annually in the United States. It is most often diagnosed by standard chest radiography, although computed tomography can identify it earlier and with higher sensitivity and specificity.5 The amount of published data on physical examination findings in pneumonia is surprisingly small.
Asymmetry in chest expansion: Specific, reproducible, but not sensitive
The physical finding with the highest positive likelihood ratio for diagnosing pneumonia is asymmetry in chest expansion.6,7

In a 1984 study of 1,819 patients presenting to an emergency department with acute cough, Diehr et al6 evaluated several physical signs of pneumonia. Asymmetric chest expansion had a specificity and positive predictive value of 100%, but its sensitivity was only 4.3%. Thus, it is not a good screening test, but it is a good diagnostic or confirmatory test. From these numbers, Metlay et al8 calculated that the positive likelihood ratio was infinity and the negative likelihood ratio was 0.96.
McGee,7 on the other hand, calculated the positive likelihood ratio of asymmetric chest expansion at 44.1. McGee also found chest expansion to be a highly reproducible finding, with an interobserver agreement kappa score of 0.85.7 (A kappa score of 1.0 would indicate perfect interobserver agreement.) Interestingly, chest radiographs interpreted for pulmonary infiltrates have an interobserver kappa score of only 0.38.7 Further studies of this physical sign could shed more light upon this area of uncertainty.
Other signs of pneumonia
None of the other physical signs studied for the diagnosis of pneumonia has as high a positive likelihood ratio as asymmetric chest expansion.6–12
Egophony is a high-pitched or nasal quality of the patient’s voice heard on auscultation over lung tissue that is consolidated or fibrosed, due to enhanced transmission of high-frequency sound across fluid. It is often described as the “E-to-A change.” Although listening for egophony is widely done and easy to do, we calculate that this sign has a positive likelihood ratio of only 6.8 based on pooled data from 3 trials with a total of 3,245 patients.6,10,11
Faring less favorably, in descending order of diagnostic accuracy, are:
Percussion dullness (positive likelihood ratio 5.7 based on 4 studies with 3,653 patients)6,10–12
Bronchophony or bronchial breath sounds (positive likelihood ratio 3.3 based on 1,118 patients)10
Crackles have long been taught as a common physical finding in pneumonia. Bohadana et al pointed out that “crackle” can be defined acoustically but does not suggest any means or site of generation.4 Pooled data from 4 studies in 3,647 patients6,10–12 result in a positive likelihood ratio for crackles in the diagnosis of pneumonia of only 3.2.
Diminished breath sounds (positive likelihood ratio 2.5 based on 3 studies with 1,828 patients).10–12
Consider pneumonia signs in combination
These physical examination maneuvers are time-honored and part of the rite of training for medical students and residents. As we have shown, they are not extremely helpful as individual tests in diagnosing pneumonia; however, they may be useful when used in combination as a clinical prediction rule or diagnostic algorithm. These rules often have higher diagnostic accuracy but drawbacks of taking more time and not being easily reproducible.

PLEURAL EFFUSION
Pleural effusion commonly occurs in patients with congestive heart failure, pneumonia, and malignancies. The following are signs of effusion.
Dullness to percussion had a positive likelihood ratio of 5.7 from pooled data from 3 studies analyzed by Wong et al.13
Asymmetric chest expansion, in a study by Kalantri et al,14 had a positive likelihood ratio of 8.1 and a negative likelihood ratio of 0.29, the latter making it a reasonably good test to help rule out a pleural effusion.
Negative signs. Since a pleural effusion is an abnormal fluid collection in the pleural space and not the lung parenchyma, one would not expect it to cause loud breath sounds, adventitious sounds, or vocal resonance. Since these 3 findings emanate from the lung, their absence would be expected to support the presence of a pleural effusion.
Tactile fremitus, also known as vocal fremitus, is the vibration felt on the chest wall while the patient is speaking. Traditionally, the patient says “ninety-nine” as the examiner feels for asymmetry in vibration. A consolidation such as pneumonia increases the vibration, while fluid in a pleural effusion diminishes it.

DIAGNOSTIC ALGORITHM FOR PNEUMONIA OR PLEURAL EFFUSION
Patients presenting with cough or dyspnea will most likely be evaluated for pneumonia and pleural effusion, among other diagnoses. We propose the following physical examination strategy in this setting.
First, evaluate the patient for asymmetric chest expansion. The positive likelihood ratio for this sign is excellent for pneumonia (44.1) and moderate for pleural effusion (8.1); therefore, both conditions are possible with a positive test.
Second, percuss the chest. Dullness to percussion has a low positive likelihood ratio for pneumonia but a moderate one for pleural effusion.13 The absence of this sign is only modest in excluding a pleural effusion (negative likelihood ratio 0.31 in pooled data analyzed by Wong et al).13
Third, auscultate the chest to elicit normal, diminished, or adventitious breath sounds. Diminished breath sounds may be noted in both conditions, but vocal resonance (egophony or bronchophony) and tactile fremitus should not be present directly over a pleural effusion. Either vocal resonance or tactile fremitus in a patient with asymmetric chest expansion would strongly support the diagnosis of pneumonia.

Figure 2 summarizes our proposed diagnostic algorithm for pneumonia and pleural effusion.
CHRONIC OBSTRUCTIVE PULMONARY DISEASE
COPD imposes a heavy burden on public health worldwide in terms of cost and mortality. It is the third leading cause of death in the United States, after heart disease and cancer.15
Spirometry remains the gold standard for diagnosis. The Global Initiative for Chronic Obstructive Lung Disease standard for diagnosing COPD was the better of 2 spirometry test results, showing a forced expiratory volume in 1 second (FEV1) and FEV1/forced vital capacity ratio less than 70%.16
Unfortunately, there is little evidence that physical signs aid in the early diagnosis of COPD, as physical signs of airflow limitation may not manifest until lung function is substantially impaired.17,18
Early inspiratory crackles had a positive likelihood ratio of 14.6 based on 2 small studies.19,20
Percussion dullness over the left sternal border in the fifth intercostal space should be present in the normal situation and is known as cardiac dullness. Absent cardiac dullness had a positive likelihood ratio of 16 and a negative likelihood ratio of 0.8 for diagnosing COPD in a study in 92 patients with a history of smoking or self-reported COPD.21 The kappa score was 0.49, signifying moderate interobserver agreement.
A combined strategy using the history and physical examination may have the highest diagnostic accuracy. Many of these combinations are too cumbersome for practical clinical use. However, 1 of them is based on only 3 questions21:
- Has the patient smoked for more than 70-pack years?
- Has the patient been previously diagnosed with chronic bronchitis or emphysema?
- Are breath sounds diminished in intensity?
Answering yes to 2 of these questions gives a positive likelihood ratio of a diagnosis of COPD of 33.5.
Early detection of COPD may improve outcomes and lower healthcare costs and thus would be clinically useful. Unfortunately, a diagnostic approach using the history and physical in the early diagnosis of COPD remains uncertain at this time.
CONGESTIVE HEART FAILURE
The clinical presentation of acute congestive heart failure has much in common with pneumonia, pleural effusion, and COPD.
Echocardiography, the gold standard for diagnosis, is costly and may not be immediately available for most patients evaluated for cardiorespiratory complaints. The American College of Cardiology reports the cost of standard echocardiography to be between $1,000 and $2,000.22 A physical examination approach in the assessment of dyspnea can be very useful.
Height of jugular venous distention approximates central venous pressure
Assessing the central venous pressure by estimating the vertical height of distention of the right internal or external jugular vein is validated and easily reproducible.23,24 The use of the external jugular vein is supported by correlation with catheter-measured central venous pressure in critically ill patients.25,26 The central venous pressure reflects the right atrial pressure, and in the absence of tricuspid stenosis, the right ventricular end-diastolic pressure. An elevation in central venous pressure can be seen in patients with congestive heart failure, pulmonary hypertension, and pulmonary valve stenosis.
The right side is preferred due to its anatomically direct route to the heart. In contrast, the left internal jugular vein crosses the mediastinum and can be compressed by the aorta, causing a false elevation.

In summary, an elevated jugular venous pressure on examination is a good test to rule in an elevated central venous pressure, and its absence is a good sign in ruling out an elevated central venous pressure. When using jugular venous pressure specifically for the diagnosis of congestive heart failure with reduced ejection fraction (ie, ejection fraction < 50%), the positive likelihood ratio is 6.3 based on 3 studies.25–27
Heart failure with preserved ejection fraction has not been well studied for physical examination. The Irbesartan in Heart Failure with Preserved Ejection Fraction Trial (I-Preserve)28 looked only at the sensitivity of elevated jugular venous pressure in 4,128 patients, which was 8%. Specificity was not reported.
The abdominojugular reflux
Another way to gauge the jugular venous pressure is to examine the neck veins while firmly pressing on the mid-abdomen for 10 to 15 seconds to look for the abdominojugular reflux, also known as the hepatojugular reflux. An increase in the jugular venous pressure of 3 cm from baseline constitutes a positive abdominojugular reflux. It has a positive likelihood ratio of 8.0 and a negative likelihood ratio of 0.3 for the diagnosis of congestive heart failure by the assessment of end-diastolic pressure of the left ventricle (Table 5).29–31
The abdominojugular reflux is a much more reliable test than examination of neck veins for jugular venous pressure. The interobserver agreement for examining neck veins has a wide range of kappa scores (0.08–0.81), whereas the abdominojugular reflux has a very high kappa score of 0.92.7 Interestingly, chest radiography showing interstitial edema has a kappa of 0.83.7
Displaced apical impulse
An evaluation of the apical impulse of the heart is also a very good and quick test in the examination of patients suspected of having congestive heart failure. An abnormal finding is defined by an apical impulse displaced laterally (to the left of the midclavicular line).
Using data from several studies,32–35 a displaced apical impulse has a positive likelihood ratio of 10.3. The absence of this finding, however, is not very good for ruling out congestive heart failure, with a negative likelihood ratio of 0.7. Interobserver agreement is moderate to excellent (kappa score 0.43–0.86).7
A third heart sound
Auscultation to assess the third heart sound is much more difficult. A systematic review found that likelihood ratios vary widely and confidence intervals are wide.36 Interobserver agreement also varies widely (kappa scores –0.17 to 0.84).7 In a primary care study,37 a third heart sound had a very low sensitivity (4.3%) but a specificity of 99.8%.
Therefore, we are uncertain about a conclusion for this physical finding based on the concern for wide ranges in likelihood ratio and poor interobserver reliability.
PHYSICAL EXAMINATION STILL HAS A FUTURE
The physical examination has a long and distinguished place in the history of medicine. Technologic advances have changed the manner in which clinicians practice the art of healing. Modern technology in US healthcare has become a double-edged sword, with many benefits as well as detriments.3 Reproducibility and accuracy are paramount for the physical examination to remain a core component of medical diagnosis. Advances in the diagnostic accuracy of laboratory and imaging studies challenge the importance of the physical examination. However, we firmly believe that the traditional techniques have stood the test of time and have a future in the clinical practice of medicine.
Acknowledgments: The authors thank Ruby Marr, MD, Mohammed Nabhan, MD, Rajiv Doddamani, MD, and Sohaib Galani, MD, for their important contributions to this article, which included research assistance and editorial advice.
- Lang TA, Secic M. Chapter 10. Determining the presence or absence of disease. Reporting the characteristics of diagnostic tests. In: Lang TC, Secic M. How to Report Statistics in Medicine. Annotated Guidelines for Authors, Editors, and Reviewers. Philadelphia, PA, American College of Physicians, 1997:147–169.
- McGee S. Simplifying likelihood ratios. J Gen Intern Med 2002; 17:647–650.
- Mikami R, Murao M, Cugell DW, et al. International symposium on lung sounds. Synopsis of proceedings. Chest 1987; 92:342–345.
- Bohadana A, Izbicki G, Kraman SS. Fundamentals of lung auscultation. N Engl J Med 2014; 370:744–751.
- Heussel CP, Kauczor HU, Ullmann AJ. Pneumonia in neutropenic patients. Eur Radiol 2004; 14:256–271.
- Diehr P, Wood RW, Bushyhead J, Krueger L, Wolcott B, Tompkins RK. Prediction of pneumonia in outpatients with acute cough—a statistical approach. J Chronic Dis 1984; 37:215–225.
- McGee S. Evidence-Based Physical Diagnosis. 4th ed. Philadelphia, PA: Elsevier; 2017.
- Metlay JP, Kapoor WN, Fine MJ. Does this patient have community-acquired pneumonia? Diagnosing pneumonia by history and physical examination. JAMA 1997; 278:1440–1445.
- Melbye H, Straume B, Aasebo U, Brox J. The diagnosis of adult pneumonia in general practice. The diagnostic value of history, physical examination and some blood tests. Scand J Prim Health Care 1988; 6:111–117.
- Heckerling PS, Tape TG, Wigton RS, et al. Clinical prediction rule for pulmonary infiltrates. Ann Intern Med 1990; 113:664–670.
- Gennis P, Gallagher J, Falvo C, Baker S, Than W. Clinical criteria for the detection of pneumonia in adults: guidelines for ordering chest roentgenograms in the emergency department. J Emerg Med 1989; 7:263–268.
- Melbye H, Straume B, Aasebo U, Dale K. Diagnosis of pneumonia in adults in general practice. Relative importance of typical symptoms and abnormal chest signs evaluated against a radiographic reference standard. Scand J Prim Health Care 1992; 10:226–233.
- Wong CL, Holroyd-Leduc J, Straus SE. Does this patient have a pleural effusion? JAMA 2009; 301:309–317.
- Kalantri S, Joshi R, Lokhande T, et al. Accuracy and reliability of physical signs in the diagnosis of pleural effusion. Respir Med 2007; 101:431–438.
- Heron M. Deaths: leading causes for 2014. National Vital Statistics Reports 2016; 65(5) June 30, 2016. www.cdc.gov/nchs/data/nvsr/nvsr65/nvsr65_05.pdf. Accessed October 20, 2017.
- Global Initiative for Chronic Obstructive Lung Disease. Pocket guide to COPD diagnosis, management, and prevention. http://goldcopd.org/wp-content/uploads/2016/12/wms-GOLD-2017-Pocket-Guide.pdf. Accessed November 13, 2017.
- Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet 2004; 364:613–620.
- Oshaug K, Halvorsen PA, Melbye H. Should chest examination be reinstated in the early diagnosis of chronic obstructive pulmonary disease? Int J Chron Obstruct Pulmon Dis 2013; 8:369–377.
- Bettencourt PE, Del Bono EA, Spiegelman D, Hertzmark E, Murphy RL Jr. Clinical utility of chest auscultation in common pulmonary diseases. Am J Respir Crit Care Med 1994; 150:1291–1297.
- Nath AR, Capel LH. Inspiratory crackles and mechanical events of breathing. Thorax 1974; 29:695–698.
- Badgett RG, Tanaka DJ, Hunt DK, et al. Can moderate chronic obstructive pulmonary disease be diagnosed by historical and physical findings alone? Am J Med 1993; 94:188–196.
- ABIM Foundation. Choosing wisely. Echocardiograms for heart valve disease. www.choosingwisely.org. Accessed November 13, 2017.
- Davison R, Cannon R. Estimation of central venous pressure by examination of jugular veins. Am Heart J 1974; 87:279–282.
- Ducas J, Magder S, McGregor M. Validity of the hepatojugular reflux as a clinical test for congestive heart failure. Am J Cardiol 1983; 52:1299–1303.
- Vinayak AG, Levitt J, Gehlbach B, Pohlman AS, Hall JB, Kress JP. Usefulness of the external jugular vein examination in detecting abnormal central venous pressure in critically ill patients. Arch Intern Med 2006; 166:2132–2137.
- Sankoff J, Zidulka A. Non-invasive method for the rapid assessment of central venous pressure: description and validation by a single examiner. West J Emerg Med 2008; 9:201–205.
- Davie AP, Francis CM, Caruana L, Sutherland GR, McMurray JJ. Assessing diagnosis in heart failure: which features are any use? QJM 1997; 90:335–339.
- Kristensen SL, Mogensen UM, Jhund PS, et al. Clinical and echocardiographic characeristics and cardiovascular outcomes according to diabetes status in patients with heart failure and preserved ejection fraction. A report from the Irbesartan in Heart Failure with Preserved Ejection Fraction Trial (I-Preserve). Circulation 2017; https://doi.org/10.1161/CIRCULATIONAHA.116.024593. Accessed November 1, 2017.
- Butman SM, Ewy GA, Standen JR, Kern KB, Hahn E. Bedside cardiovascular examination in patients with severe chronic heart failure: importance of rest or inducible jugular venous distension. J Am Coll Cardiol 1993; 22:968–974.
- Sochowski RA, Dubbin JD, Naqvi SZ. Clinical and hemodynamic assessment of the hepatojugular reflux. Am J Cardiol 1990; 66:1002–1006.
- Ewy GA. The abdominojugular test: technique and hemodynamic correlates. Ann Intern Med 1988; 109:456–460.
- Gadsboll N, Hoilund-Carlsen PF, Nielsen GG, et al. Symptoms and signs of heart failure in patients with myocardial infarction: reproducibility and relationship to chest X-ray, radionuclide ventriculography and right heart catheterization. Eur Heart J 1989; 10:1017–1028.
- Fahey T, Jeyaseelan S, McCowan C, et al. Diagnosis of left ventricular systolic dysfunction (LVSD): development and validation of a clinical prediction rule in primary care. Fam Pract 2007; 24:628–635.
- Gadsboll N, Hoilund-Carlsen PF, Nielsen GG, et al. Interobserver agreement and accuracy of bedside estimation of right and left ventricular ejection fraction in acute myocardial infarction. Am J Cardiol 1989; 63:1301–1307.
- Mattleman SJ, Hakki AH, Iskandrian AS, Segal BL, Kane SA. Reliability of bedside evaluation in determining left ventricular function: correlation with left ventricular ejection fraction determined by radionuclide ventriculography. J Am Coll Cardiol 1983; 1:417–420.
- Madhok V, Falk G, Rogers A, Struthers AD, Sullivan FM, Fahey T. The accuracy of symptoms, signs and diagnostic tests in the diagnosis of left ventricular dysfunction in primary care: a diagnostic accuracy systematic review. BMC Fam Pract 2008; 9:56.
- Kelder JC, Cramer MJ, van Wijngaarden J, et al. The diagnostic value of physical examination and additional testing in primary care patients with suspected heart failure. Circulation 2011; 124:2865–2873.
Laennec’s stethoscope has survived more than 200 years, much longer than some of his contemporaries predicted. But will it survive the challenge of bedside ultrasonography and other technologic advances?
The physical examination, with its roots extending at least as far back as Hippocrates, may be at a crossroads as the mainstay of diagnosis. Physical signs can be subjective and lack sensitivity and specificity. Modern imaging and laboratory studies may already be more trusted.
If the physical examination is to survive, it must be accurate, reproducible, and efficient. Needed is a simple, evidence-based approach to the physical examination that enhances its diagnostic accuracy while maintaining bedside efficiency.
Here, we analyze the accuracy of the physical signs that are most effective in the clinical diagnosis of 4 common cardiopulmonary conditions that often present with dyspnea: pneumonia, pleural effusion, chronic obstructive pulmonary disease (COPD), and congestive heart failure.
LIKELIHOOD RATIOS
To grasp the significance of physical findings, it is necessary to understand the concept of likelihood ratios, which are widely accepted measures of the accuracy of a test or clinical finding.1,2 The positive likelihood ratio is the probability of a disease being present when the test is positive or the clinical finding is present, while the negative likelihood ratio is the probability that the disease is present when the test is negative or the clinical finding is absent. They are calculated as follows1:
Positive likelihood ratio = sensitivity / (1 – specificity)
Negative likelihood ratio = (1 – sensitivity) / specificity
Table 1 shows how the likelihood ratio of a test changes the posttest probability that a condition is present or absent, according to an analysis by McGee.2
STANDARDIZED TERMINOLOGY
PNEUMONIA
Pneumonia is a common disease, with more than 2 million cases annually in the United States. It is most often diagnosed by standard chest radiography, although computed tomography can identify it earlier and with higher sensitivity and specificity.5 The amount of published data on physical examination findings in pneumonia is surprisingly small.
Asymmetry in chest expansion: Specific, reproducible, but not sensitive
The physical finding with the highest positive likelihood ratio for diagnosing pneumonia is asymmetry in chest expansion.6,7
In a 1984 study of 1,819 patients presenting to an emergency department with acute cough, Diehr et al6 evaluated several physical signs of pneumonia. Asymmetric chest expansion had a specificity and positive predictive value of 100%, but its sensitivity was only 4.3%. Thus, it is not a good screening test, but it is a good diagnostic or confirmatory test. From these numbers, Metlay et al8 calculated that the positive likelihood ratio was infinity and the negative likelihood ratio was 0.96.
McGee,7 on the other hand, calculated the positive likelihood ratio of asymmetric chest expansion at 44.1. McGee also found chest expansion to be a highly reproducible finding, with an interobserver agreement kappa score of 0.85.7 (A kappa score of 1.0 would indicate perfect interobserver agreement.) Interestingly, chest radiographs interpreted for pulmonary infiltrates have an interobserver kappa score of only 0.38.7 Further studies of this physical sign could shed more light upon this area of uncertainty.
Other signs of pneumonia
None of the other physical signs studied for the diagnosis of pneumonia has as high a positive likelihood ratio as asymmetric chest expansion.6–12
Egophony is a high-pitched or nasal quality of the patient’s voice heard on auscultation over lung tissue that is consolidated or fibrosed, due to enhanced transmission of high-frequency sound across fluid. It is often described as the “E-to-A change.” Although listening for egophony is widely done and easy to do, we calculate that this sign has a positive likelihood ratio of only 6.8 based on pooled data from 3 trials with a total of 3,245 patients.6,10,11
Faring less favorably, in descending order of diagnostic accuracy, are:
Percussion dullness (positive likelihood ratio 5.7 based on 4 studies with 3,653 patients)6,10–12
Bronchophony or bronchial breath sounds (positive likelihood ratio 3.3 based on 1,118 patients)10
Crackles have long been taught as a common physical finding in pneumonia. Bohadana et al pointed out that “crackle” can be defined acoustically but does not suggest any means or site of generation.4 Pooled data from 4 studies in 3,647 patients6,10–12 result in a positive likelihood ratio for crackles in the diagnosis of pneumonia of only 3.2.
Diminished breath sounds (positive likelihood ratio 2.5 based on 3 studies with 1,828 patients).10–12
Consider pneumonia signs in combination
These physical examination maneuvers are time-honored and part of the rite of training for medical students and residents. As we have shown, they are not extremely helpful as individual tests in diagnosing pneumonia; however, they may be useful when used in combination as a clinical prediction rule or diagnostic algorithm. These rules often have higher diagnostic accuracy but drawbacks of taking more time and not being easily reproducible.
PLEURAL EFFUSION
Pleural effusion commonly occurs in patients with congestive heart failure, pneumonia, and malignancies. The following are signs of effusion.
Dullness to percussion had a positive likelihood ratio of 5.7 from pooled data from 3 studies analyzed by Wong et al.13
Asymmetric chest expansion, in a study by Kalantri et al,14 had a positive likelihood ratio of 8.1 and a negative likelihood ratio of 0.29, the latter making it a reasonably good test to help rule out a pleural effusion.
Negative signs. Since a pleural effusion is an abnormal fluid collection in the pleural space and not the lung parenchyma, one would not expect it to cause loud breath sounds, adventitious sounds, or vocal resonance. Since these 3 findings emanate from the lung, their absence would be expected to support the presence of a pleural effusion.
Tactile fremitus, also known as vocal fremitus, is the vibration felt on the chest wall while the patient is speaking. Traditionally, the patient says “ninety-nine” as the examiner feels for asymmetry in vibration. A consolidation such as pneumonia increases the vibration, while fluid in a pleural effusion diminishes it.
DIAGNOSTIC ALGORITHM FOR PNEUMONIA OR PLEURAL EFFUSION
Patients presenting with cough or dyspnea will most likely be evaluated for pneumonia and pleural effusion, among other diagnoses. We propose the following physical examination strategy in this setting.
First, evaluate the patient for asymmetric chest expansion. The positive likelihood ratio for this sign is excellent for pneumonia (44.1) and moderate for pleural effusion (8.1); therefore, both conditions are possible with a positive test.
Second, percuss the chest. Dullness to percussion has a low positive likelihood ratio for pneumonia but a moderate one for pleural effusion.13 The absence of this sign is only modest in excluding a pleural effusion (negative likelihood ratio 0.31 in pooled data analyzed by Wong et al).13
Third, auscultate the chest to elicit normal, diminished, or adventitious breath sounds. Diminished breath sounds may be noted in both conditions, but vocal resonance (egophony or bronchophony) and tactile fremitus should not be present directly over a pleural effusion. Either vocal resonance or tactile fremitus in a patient with asymmetric chest expansion would strongly support the diagnosis of pneumonia.
Figure 2 summarizes our proposed diagnostic algorithm for pneumonia and pleural effusion.
CHRONIC OBSTRUCTIVE PULMONARY DISEASE
COPD imposes a heavy burden on public health worldwide in terms of cost and mortality. It is the third leading cause of death in the United States, after heart disease and cancer.15
Spirometry remains the gold standard for diagnosis. The Global Initiative for Chronic Obstructive Lung Disease standard for diagnosing COPD was the better of 2 spirometry test results, showing a forced expiratory volume in 1 second (FEV1) and FEV1/forced vital capacity ratio less than 70%.16
Unfortunately, there is little evidence that physical signs aid in the early diagnosis of COPD, as physical signs of airflow limitation may not manifest until lung function is substantially impaired.17,18
Early inspiratory crackles had a positive likelihood ratio of 14.6 based on 2 small studies.19,20
Percussion dullness over the left sternal border in the fifth intercostal space should be present in the normal situation and is known as cardiac dullness. Absent cardiac dullness had a positive likelihood ratio of 16 and a negative likelihood ratio of 0.8 for diagnosing COPD in a study in 92 patients with a history of smoking or self-reported COPD.21 The kappa score was 0.49, signifying moderate interobserver agreement.
A combined strategy using the history and physical examination may have the highest diagnostic accuracy. Many of these combinations are too cumbersome for practical clinical use. However, 1 of them is based on only 3 questions21:
- Has the patient smoked for more than 70-pack years?
- Has the patient been previously diagnosed with chronic bronchitis or emphysema?
- Are breath sounds diminished in intensity?
Answering yes to 2 of these questions gives a positive likelihood ratio of a diagnosis of COPD of 33.5.
Early detection of COPD may improve outcomes and lower healthcare costs and thus would be clinically useful. Unfortunately, a diagnostic approach using the history and physical in the early diagnosis of COPD remains uncertain at this time.
CONGESTIVE HEART FAILURE
The clinical presentation of acute congestive heart failure has much in common with pneumonia, pleural effusion, and COPD.
Echocardiography, the gold standard for diagnosis, is costly and may not be immediately available for most patients evaluated for cardiorespiratory complaints. The American College of Cardiology reports the cost of standard echocardiography to be between $1,000 and $2,000.22 A physical examination approach in the assessment of dyspnea can be very useful.
Height of jugular venous distention approximates central venous pressure
Assessing the central venous pressure by estimating the vertical height of distention of the right internal or external jugular vein is validated and easily reproducible.23,24 The use of the external jugular vein is supported by correlation with catheter-measured central venous pressure in critically ill patients.25,26 The central venous pressure reflects the right atrial pressure, and in the absence of tricuspid stenosis, the right ventricular end-diastolic pressure. An elevation in central venous pressure can be seen in patients with congestive heart failure, pulmonary hypertension, and pulmonary valve stenosis.
The right side is preferred due to its anatomically direct route to the heart. In contrast, the left internal jugular vein crosses the mediastinum and can be compressed by the aorta, causing a false elevation.
In summary, an elevated jugular venous pressure on examination is a good test to rule in an elevated central venous pressure, and its absence is a good sign in ruling out an elevated central venous pressure. When using jugular venous pressure specifically for the diagnosis of congestive heart failure with reduced ejection fraction (ie, ejection fraction < 50%), the positive likelihood ratio is 6.3 based on 3 studies.25–27
Heart failure with preserved ejection fraction has not been well studied for physical examination. The Irbesartan in Heart Failure with Preserved Ejection Fraction Trial (I-Preserve)28 looked only at the sensitivity of elevated jugular venous pressure in 4,128 patients, which was 8%. Specificity was not reported.
The abdominojugular reflux
Another way to gauge the jugular venous pressure is to examine the neck veins while firmly pressing on the mid-abdomen for 10 to 15 seconds to look for the abdominojugular reflux, also known as the hepatojugular reflux. An increase in the jugular venous pressure of 3 cm from baseline constitutes a positive abdominojugular reflux. It has a positive likelihood ratio of 8.0 and a negative likelihood ratio of 0.3 for the diagnosis of congestive heart failure by the assessment of end-diastolic pressure of the left ventricle (Table 5).29–31
The abdominojugular reflux is a much more reliable test than examination of neck veins for jugular venous pressure. The interobserver agreement for examining neck veins has a wide range of kappa scores (0.08–0.81), whereas the abdominojugular reflux has a very high kappa score of 0.92.7 Interestingly, chest radiography showing interstitial edema has a kappa of 0.83.7
Displaced apical impulse
An evaluation of the apical impulse of the heart is also a very good and quick test in the examination of patients suspected of having congestive heart failure. An abnormal finding is defined by an apical impulse displaced laterally (to the left of the midclavicular line).
Using data from several studies,32–35 a displaced apical impulse has a positive likelihood ratio of 10.3. The absence of this finding, however, is not very good for ruling out congestive heart failure, with a negative likelihood ratio of 0.7. Interobserver agreement is moderate to excellent (kappa score 0.43–0.86).7
A third heart sound
Auscultation to assess the third heart sound is much more difficult. A systematic review found that likelihood ratios vary widely and confidence intervals are wide.36 Interobserver agreement also varies widely (kappa scores –0.17 to 0.84).7 In a primary care study,37 a third heart sound had a very low sensitivity (4.3%) but a specificity of 99.8%.
Therefore, we are uncertain about a conclusion for this physical finding based on the concern for wide ranges in likelihood ratio and poor interobserver reliability.
PHYSICAL EXAMINATION STILL HAS A FUTURE
The physical examination has a long and distinguished place in the history of medicine. Technologic advances have changed the manner in which clinicians practice the art of healing. Modern technology in US healthcare has become a double-edged sword, with many benefits as well as detriments.3 Reproducibility and accuracy are paramount for the physical examination to remain a core component of medical diagnosis. Advances in the diagnostic accuracy of laboratory and imaging studies challenge the importance of the physical examination. However, we firmly believe that the traditional techniques have stood the test of time and have a future in the clinical practice of medicine.
Acknowledgments: The authors thank Ruby Marr, MD, Mohammed Nabhan, MD, Rajiv Doddamani, MD, and Sohaib Galani, MD, for their important contributions to this article, which included research assistance and editorial advice.
Laennec’s stethoscope has survived more than 200 years, much longer than some of his contemporaries predicted. But will it survive the challenge of bedside ultrasonography and other technologic advances?
The physical examination, with its roots extending at least as far back as Hippocrates, may be at a crossroads as the mainstay of diagnosis. Physical signs can be subjective and lack sensitivity and specificity. Modern imaging and laboratory studies may already be more trusted.
If the physical examination is to survive, it must be accurate, reproducible, and efficient. Needed is a simple, evidence-based approach to the physical examination that enhances its diagnostic accuracy while maintaining bedside efficiency.
Here, we analyze the accuracy of the physical signs that are most effective in the clinical diagnosis of 4 common cardiopulmonary conditions that often present with dyspnea: pneumonia, pleural effusion, chronic obstructive pulmonary disease (COPD), and congestive heart failure.
LIKELIHOOD RATIOS
To grasp the significance of physical findings, it is necessary to understand the concept of likelihood ratios, which are widely accepted measures of the accuracy of a test or clinical finding.1,2 The positive likelihood ratio is the probability of a disease being present when the test is positive or the clinical finding is present, while the negative likelihood ratio is the probability that the disease is present when the test is negative or the clinical finding is absent. They are calculated as follows1:
Positive likelihood ratio = sensitivity / (1 – specificity)
Negative likelihood ratio = (1 – sensitivity) / specificity
Table 1 shows how the likelihood ratio of a test changes the posttest probability that a condition is present or absent, according to an analysis by McGee.2
STANDARDIZED TERMINOLOGY
PNEUMONIA
Pneumonia is a common disease, with more than 2 million cases annually in the United States. It is most often diagnosed by standard chest radiography, although computed tomography can identify it earlier and with higher sensitivity and specificity.5 The amount of published data on physical examination findings in pneumonia is surprisingly small.
Asymmetry in chest expansion: Specific, reproducible, but not sensitive
The physical finding with the highest positive likelihood ratio for diagnosing pneumonia is asymmetry in chest expansion.6,7
In a 1984 study of 1,819 patients presenting to an emergency department with acute cough, Diehr et al6 evaluated several physical signs of pneumonia. Asymmetric chest expansion had a specificity and positive predictive value of 100%, but its sensitivity was only 4.3%. Thus, it is not a good screening test, but it is a good diagnostic or confirmatory test. From these numbers, Metlay et al8 calculated that the positive likelihood ratio was infinity and the negative likelihood ratio was 0.96.
McGee,7 on the other hand, calculated the positive likelihood ratio of asymmetric chest expansion at 44.1. McGee also found chest expansion to be a highly reproducible finding, with an interobserver agreement kappa score of 0.85.7 (A kappa score of 1.0 would indicate perfect interobserver agreement.) Interestingly, chest radiographs interpreted for pulmonary infiltrates have an interobserver kappa score of only 0.38.7 Further studies of this physical sign could shed more light upon this area of uncertainty.
Other signs of pneumonia
None of the other physical signs studied for the diagnosis of pneumonia has as high a positive likelihood ratio as asymmetric chest expansion.6–12
Egophony is a high-pitched or nasal quality of the patient’s voice heard on auscultation over lung tissue that is consolidated or fibrosed, due to enhanced transmission of high-frequency sound across fluid. It is often described as the “E-to-A change.” Although listening for egophony is widely done and easy to do, we calculate that this sign has a positive likelihood ratio of only 6.8 based on pooled data from 3 trials with a total of 3,245 patients.6,10,11
Faring less favorably, in descending order of diagnostic accuracy, are:
Percussion dullness (positive likelihood ratio 5.7 based on 4 studies with 3,653 patients)6,10–12
Bronchophony or bronchial breath sounds (positive likelihood ratio 3.3 based on 1,118 patients)10
Crackles have long been taught as a common physical finding in pneumonia. Bohadana et al pointed out that “crackle” can be defined acoustically but does not suggest any means or site of generation.4 Pooled data from 4 studies in 3,647 patients6,10–12 result in a positive likelihood ratio for crackles in the diagnosis of pneumonia of only 3.2.
Diminished breath sounds (positive likelihood ratio 2.5 based on 3 studies with 1,828 patients).10–12
Consider pneumonia signs in combination
These physical examination maneuvers are time-honored and part of the rite of training for medical students and residents. As we have shown, they are not extremely helpful as individual tests in diagnosing pneumonia; however, they may be useful when used in combination as a clinical prediction rule or diagnostic algorithm. These rules often have higher diagnostic accuracy but drawbacks of taking more time and not being easily reproducible.
PLEURAL EFFUSION
Pleural effusion commonly occurs in patients with congestive heart failure, pneumonia, and malignancies. The following are signs of effusion.
Dullness to percussion had a positive likelihood ratio of 5.7 from pooled data from 3 studies analyzed by Wong et al.13
Asymmetric chest expansion, in a study by Kalantri et al,14 had a positive likelihood ratio of 8.1 and a negative likelihood ratio of 0.29, the latter making it a reasonably good test to help rule out a pleural effusion.
Negative signs. Since a pleural effusion is an abnormal fluid collection in the pleural space and not the lung parenchyma, one would not expect it to cause loud breath sounds, adventitious sounds, or vocal resonance. Since these 3 findings emanate from the lung, their absence would be expected to support the presence of a pleural effusion.
Tactile fremitus, also known as vocal fremitus, is the vibration felt on the chest wall while the patient is speaking. Traditionally, the patient says “ninety-nine” as the examiner feels for asymmetry in vibration. A consolidation such as pneumonia increases the vibration, while fluid in a pleural effusion diminishes it.
DIAGNOSTIC ALGORITHM FOR PNEUMONIA OR PLEURAL EFFUSION
Patients presenting with cough or dyspnea will most likely be evaluated for pneumonia and pleural effusion, among other diagnoses. We propose the following physical examination strategy in this setting.
First, evaluate the patient for asymmetric chest expansion. The positive likelihood ratio for this sign is excellent for pneumonia (44.1) and moderate for pleural effusion (8.1); therefore, both conditions are possible with a positive test.
Second, percuss the chest. Dullness to percussion has a low positive likelihood ratio for pneumonia but a moderate one for pleural effusion.13 The absence of this sign is only modest in excluding a pleural effusion (negative likelihood ratio 0.31 in pooled data analyzed by Wong et al).13
Third, auscultate the chest to elicit normal, diminished, or adventitious breath sounds. Diminished breath sounds may be noted in both conditions, but vocal resonance (egophony or bronchophony) and tactile fremitus should not be present directly over a pleural effusion. Either vocal resonance or tactile fremitus in a patient with asymmetric chest expansion would strongly support the diagnosis of pneumonia.
Figure 2 summarizes our proposed diagnostic algorithm for pneumonia and pleural effusion.
CHRONIC OBSTRUCTIVE PULMONARY DISEASE
COPD imposes a heavy burden on public health worldwide in terms of cost and mortality. It is the third leading cause of death in the United States, after heart disease and cancer.15
Spirometry remains the gold standard for diagnosis. The Global Initiative for Chronic Obstructive Lung Disease standard for diagnosing COPD was the better of 2 spirometry test results, showing a forced expiratory volume in 1 second (FEV1) and FEV1/forced vital capacity ratio less than 70%.16
Unfortunately, there is little evidence that physical signs aid in the early diagnosis of COPD, as physical signs of airflow limitation may not manifest until lung function is substantially impaired.17,18
Early inspiratory crackles had a positive likelihood ratio of 14.6 based on 2 small studies.19,20
Percussion dullness over the left sternal border in the fifth intercostal space should be present in the normal situation and is known as cardiac dullness. Absent cardiac dullness had a positive likelihood ratio of 16 and a negative likelihood ratio of 0.8 for diagnosing COPD in a study in 92 patients with a history of smoking or self-reported COPD.21 The kappa score was 0.49, signifying moderate interobserver agreement.
A combined strategy using the history and physical examination may have the highest diagnostic accuracy. Many of these combinations are too cumbersome for practical clinical use. However, 1 of them is based on only 3 questions21:
- Has the patient smoked for more than 70-pack years?
- Has the patient been previously diagnosed with chronic bronchitis or emphysema?
- Are breath sounds diminished in intensity?
Answering yes to 2 of these questions gives a positive likelihood ratio of a diagnosis of COPD of 33.5.
Early detection of COPD may improve outcomes and lower healthcare costs and thus would be clinically useful. Unfortunately, a diagnostic approach using the history and physical in the early diagnosis of COPD remains uncertain at this time.
CONGESTIVE HEART FAILURE
The clinical presentation of acute congestive heart failure has much in common with pneumonia, pleural effusion, and COPD.
Echocardiography, the gold standard for diagnosis, is costly and may not be immediately available for most patients evaluated for cardiorespiratory complaints. The American College of Cardiology reports the cost of standard echocardiography to be between $1,000 and $2,000.22 A physical examination approach in the assessment of dyspnea can be very useful.
Height of jugular venous distention approximates central venous pressure
Assessing the central venous pressure by estimating the vertical height of distention of the right internal or external jugular vein is validated and easily reproducible.23,24 The use of the external jugular vein is supported by correlation with catheter-measured central venous pressure in critically ill patients.25,26 The central venous pressure reflects the right atrial pressure, and in the absence of tricuspid stenosis, the right ventricular end-diastolic pressure. An elevation in central venous pressure can be seen in patients with congestive heart failure, pulmonary hypertension, and pulmonary valve stenosis.
The right side is preferred due to its anatomically direct route to the heart. In contrast, the left internal jugular vein crosses the mediastinum and can be compressed by the aorta, causing a false elevation.
In summary, an elevated jugular venous pressure on examination is a good test to rule in an elevated central venous pressure, and its absence is a good sign in ruling out an elevated central venous pressure. When using jugular venous pressure specifically for the diagnosis of congestive heart failure with reduced ejection fraction (ie, ejection fraction < 50%), the positive likelihood ratio is 6.3 based on 3 studies.25–27
Heart failure with preserved ejection fraction has not been well studied for physical examination. The Irbesartan in Heart Failure with Preserved Ejection Fraction Trial (I-Preserve)28 looked only at the sensitivity of elevated jugular venous pressure in 4,128 patients, which was 8%. Specificity was not reported.
The abdominojugular reflux
Another way to gauge the jugular venous pressure is to examine the neck veins while firmly pressing on the mid-abdomen for 10 to 15 seconds to look for the abdominojugular reflux, also known as the hepatojugular reflux. An increase in the jugular venous pressure of 3 cm from baseline constitutes a positive abdominojugular reflux. It has a positive likelihood ratio of 8.0 and a negative likelihood ratio of 0.3 for the diagnosis of congestive heart failure by the assessment of end-diastolic pressure of the left ventricle (Table 5).29–31
The abdominojugular reflux is a much more reliable test than examination of neck veins for jugular venous pressure. The interobserver agreement for examining neck veins has a wide range of kappa scores (0.08–0.81), whereas the abdominojugular reflux has a very high kappa score of 0.92.7 Interestingly, chest radiography showing interstitial edema has a kappa of 0.83.7
Displaced apical impulse
An evaluation of the apical impulse of the heart is also a very good and quick test in the examination of patients suspected of having congestive heart failure. An abnormal finding is defined by an apical impulse displaced laterally (to the left of the midclavicular line).
Using data from several studies,32–35 a displaced apical impulse has a positive likelihood ratio of 10.3. The absence of this finding, however, is not very good for ruling out congestive heart failure, with a negative likelihood ratio of 0.7. Interobserver agreement is moderate to excellent (kappa score 0.43–0.86).7
A third heart sound
Auscultation to assess the third heart sound is much more difficult. A systematic review found that likelihood ratios vary widely and confidence intervals are wide.36 Interobserver agreement also varies widely (kappa scores –0.17 to 0.84).7 In a primary care study,37 a third heart sound had a very low sensitivity (4.3%) but a specificity of 99.8%.
Therefore, we are uncertain about a conclusion for this physical finding based on the concern for wide ranges in likelihood ratio and poor interobserver reliability.
PHYSICAL EXAMINATION STILL HAS A FUTURE
The physical examination has a long and distinguished place in the history of medicine. Technologic advances have changed the manner in which clinicians practice the art of healing. Modern technology in US healthcare has become a double-edged sword, with many benefits as well as detriments.3 Reproducibility and accuracy are paramount for the physical examination to remain a core component of medical diagnosis. Advances in the diagnostic accuracy of laboratory and imaging studies challenge the importance of the physical examination. However, we firmly believe that the traditional techniques have stood the test of time and have a future in the clinical practice of medicine.
Acknowledgments: The authors thank Ruby Marr, MD, Mohammed Nabhan, MD, Rajiv Doddamani, MD, and Sohaib Galani, MD, for their important contributions to this article, which included research assistance and editorial advice.
- Lang TA, Secic M. Chapter 10. Determining the presence or absence of disease. Reporting the characteristics of diagnostic tests. In: Lang TC, Secic M. How to Report Statistics in Medicine. Annotated Guidelines for Authors, Editors, and Reviewers. Philadelphia, PA, American College of Physicians, 1997:147–169.
- McGee S. Simplifying likelihood ratios. J Gen Intern Med 2002; 17:647–650.
- Mikami R, Murao M, Cugell DW, et al. International symposium on lung sounds. Synopsis of proceedings. Chest 1987; 92:342–345.
- Bohadana A, Izbicki G, Kraman SS. Fundamentals of lung auscultation. N Engl J Med 2014; 370:744–751.
- Heussel CP, Kauczor HU, Ullmann AJ. Pneumonia in neutropenic patients. Eur Radiol 2004; 14:256–271.
- Diehr P, Wood RW, Bushyhead J, Krueger L, Wolcott B, Tompkins RK. Prediction of pneumonia in outpatients with acute cough—a statistical approach. J Chronic Dis 1984; 37:215–225.
- McGee S. Evidence-Based Physical Diagnosis. 4th ed. Philadelphia, PA: Elsevier; 2017.
- Metlay JP, Kapoor WN, Fine MJ. Does this patient have community-acquired pneumonia? Diagnosing pneumonia by history and physical examination. JAMA 1997; 278:1440–1445.
- Melbye H, Straume B, Aasebo U, Brox J. The diagnosis of adult pneumonia in general practice. The diagnostic value of history, physical examination and some blood tests. Scand J Prim Health Care 1988; 6:111–117.
- Heckerling PS, Tape TG, Wigton RS, et al. Clinical prediction rule for pulmonary infiltrates. Ann Intern Med 1990; 113:664–670.
- Gennis P, Gallagher J, Falvo C, Baker S, Than W. Clinical criteria for the detection of pneumonia in adults: guidelines for ordering chest roentgenograms in the emergency department. J Emerg Med 1989; 7:263–268.
- Melbye H, Straume B, Aasebo U, Dale K. Diagnosis of pneumonia in adults in general practice. Relative importance of typical symptoms and abnormal chest signs evaluated against a radiographic reference standard. Scand J Prim Health Care 1992; 10:226–233.
- Wong CL, Holroyd-Leduc J, Straus SE. Does this patient have a pleural effusion? JAMA 2009; 301:309–317.
- Kalantri S, Joshi R, Lokhande T, et al. Accuracy and reliability of physical signs in the diagnosis of pleural effusion. Respir Med 2007; 101:431–438.
- Heron M. Deaths: leading causes for 2014. National Vital Statistics Reports 2016; 65(5) June 30, 2016. www.cdc.gov/nchs/data/nvsr/nvsr65/nvsr65_05.pdf. Accessed October 20, 2017.
- Global Initiative for Chronic Obstructive Lung Disease. Pocket guide to COPD diagnosis, management, and prevention. http://goldcopd.org/wp-content/uploads/2016/12/wms-GOLD-2017-Pocket-Guide.pdf. Accessed November 13, 2017.
- Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet 2004; 364:613–620.
- Oshaug K, Halvorsen PA, Melbye H. Should chest examination be reinstated in the early diagnosis of chronic obstructive pulmonary disease? Int J Chron Obstruct Pulmon Dis 2013; 8:369–377.
- Bettencourt PE, Del Bono EA, Spiegelman D, Hertzmark E, Murphy RL Jr. Clinical utility of chest auscultation in common pulmonary diseases. Am J Respir Crit Care Med 1994; 150:1291–1297.
- Nath AR, Capel LH. Inspiratory crackles and mechanical events of breathing. Thorax 1974; 29:695–698.
- Badgett RG, Tanaka DJ, Hunt DK, et al. Can moderate chronic obstructive pulmonary disease be diagnosed by historical and physical findings alone? Am J Med 1993; 94:188–196.
- ABIM Foundation. Choosing wisely. Echocardiograms for heart valve disease. www.choosingwisely.org. Accessed November 13, 2017.
- Davison R, Cannon R. Estimation of central venous pressure by examination of jugular veins. Am Heart J 1974; 87:279–282.
- Ducas J, Magder S, McGregor M. Validity of the hepatojugular reflux as a clinical test for congestive heart failure. Am J Cardiol 1983; 52:1299–1303.
- Vinayak AG, Levitt J, Gehlbach B, Pohlman AS, Hall JB, Kress JP. Usefulness of the external jugular vein examination in detecting abnormal central venous pressure in critically ill patients. Arch Intern Med 2006; 166:2132–2137.
- Sankoff J, Zidulka A. Non-invasive method for the rapid assessment of central venous pressure: description and validation by a single examiner. West J Emerg Med 2008; 9:201–205.
- Davie AP, Francis CM, Caruana L, Sutherland GR, McMurray JJ. Assessing diagnosis in heart failure: which features are any use? QJM 1997; 90:335–339.
- Kristensen SL, Mogensen UM, Jhund PS, et al. Clinical and echocardiographic characeristics and cardiovascular outcomes according to diabetes status in patients with heart failure and preserved ejection fraction. A report from the Irbesartan in Heart Failure with Preserved Ejection Fraction Trial (I-Preserve). Circulation 2017; https://doi.org/10.1161/CIRCULATIONAHA.116.024593. Accessed November 1, 2017.
- Butman SM, Ewy GA, Standen JR, Kern KB, Hahn E. Bedside cardiovascular examination in patients with severe chronic heart failure: importance of rest or inducible jugular venous distension. J Am Coll Cardiol 1993; 22:968–974.
- Sochowski RA, Dubbin JD, Naqvi SZ. Clinical and hemodynamic assessment of the hepatojugular reflux. Am J Cardiol 1990; 66:1002–1006.
- Ewy GA. The abdominojugular test: technique and hemodynamic correlates. Ann Intern Med 1988; 109:456–460.
- Gadsboll N, Hoilund-Carlsen PF, Nielsen GG, et al. Symptoms and signs of heart failure in patients with myocardial infarction: reproducibility and relationship to chest X-ray, radionuclide ventriculography and right heart catheterization. Eur Heart J 1989; 10:1017–1028.
- Fahey T, Jeyaseelan S, McCowan C, et al. Diagnosis of left ventricular systolic dysfunction (LVSD): development and validation of a clinical prediction rule in primary care. Fam Pract 2007; 24:628–635.
- Gadsboll N, Hoilund-Carlsen PF, Nielsen GG, et al. Interobserver agreement and accuracy of bedside estimation of right and left ventricular ejection fraction in acute myocardial infarction. Am J Cardiol 1989; 63:1301–1307.
- Mattleman SJ, Hakki AH, Iskandrian AS, Segal BL, Kane SA. Reliability of bedside evaluation in determining left ventricular function: correlation with left ventricular ejection fraction determined by radionuclide ventriculography. J Am Coll Cardiol 1983; 1:417–420.
- Madhok V, Falk G, Rogers A, Struthers AD, Sullivan FM, Fahey T. The accuracy of symptoms, signs and diagnostic tests in the diagnosis of left ventricular dysfunction in primary care: a diagnostic accuracy systematic review. BMC Fam Pract 2008; 9:56.
- Kelder JC, Cramer MJ, van Wijngaarden J, et al. The diagnostic value of physical examination and additional testing in primary care patients with suspected heart failure. Circulation 2011; 124:2865–2873.
- Lang TA, Secic M. Chapter 10. Determining the presence or absence of disease. Reporting the characteristics of diagnostic tests. In: Lang TC, Secic M. How to Report Statistics in Medicine. Annotated Guidelines for Authors, Editors, and Reviewers. Philadelphia, PA, American College of Physicians, 1997:147–169.
- McGee S. Simplifying likelihood ratios. J Gen Intern Med 2002; 17:647–650.
- Mikami R, Murao M, Cugell DW, et al. International symposium on lung sounds. Synopsis of proceedings. Chest 1987; 92:342–345.
- Bohadana A, Izbicki G, Kraman SS. Fundamentals of lung auscultation. N Engl J Med 2014; 370:744–751.
- Heussel CP, Kauczor HU, Ullmann AJ. Pneumonia in neutropenic patients. Eur Radiol 2004; 14:256–271.
- Diehr P, Wood RW, Bushyhead J, Krueger L, Wolcott B, Tompkins RK. Prediction of pneumonia in outpatients with acute cough—a statistical approach. J Chronic Dis 1984; 37:215–225.
- McGee S. Evidence-Based Physical Diagnosis. 4th ed. Philadelphia, PA: Elsevier; 2017.
- Metlay JP, Kapoor WN, Fine MJ. Does this patient have community-acquired pneumonia? Diagnosing pneumonia by history and physical examination. JAMA 1997; 278:1440–1445.
- Melbye H, Straume B, Aasebo U, Brox J. The diagnosis of adult pneumonia in general practice. The diagnostic value of history, physical examination and some blood tests. Scand J Prim Health Care 1988; 6:111–117.
- Heckerling PS, Tape TG, Wigton RS, et al. Clinical prediction rule for pulmonary infiltrates. Ann Intern Med 1990; 113:664–670.
- Gennis P, Gallagher J, Falvo C, Baker S, Than W. Clinical criteria for the detection of pneumonia in adults: guidelines for ordering chest roentgenograms in the emergency department. J Emerg Med 1989; 7:263–268.
- Melbye H, Straume B, Aasebo U, Dale K. Diagnosis of pneumonia in adults in general practice. Relative importance of typical symptoms and abnormal chest signs evaluated against a radiographic reference standard. Scand J Prim Health Care 1992; 10:226–233.
- Wong CL, Holroyd-Leduc J, Straus SE. Does this patient have a pleural effusion? JAMA 2009; 301:309–317.
- Kalantri S, Joshi R, Lokhande T, et al. Accuracy and reliability of physical signs in the diagnosis of pleural effusion. Respir Med 2007; 101:431–438.
- Heron M. Deaths: leading causes for 2014. National Vital Statistics Reports 2016; 65(5) June 30, 2016. www.cdc.gov/nchs/data/nvsr/nvsr65/nvsr65_05.pdf. Accessed October 20, 2017.
- Global Initiative for Chronic Obstructive Lung Disease. Pocket guide to COPD diagnosis, management, and prevention. http://goldcopd.org/wp-content/uploads/2016/12/wms-GOLD-2017-Pocket-Guide.pdf. Accessed November 13, 2017.
- Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet 2004; 364:613–620.
- Oshaug K, Halvorsen PA, Melbye H. Should chest examination be reinstated in the early diagnosis of chronic obstructive pulmonary disease? Int J Chron Obstruct Pulmon Dis 2013; 8:369–377.
- Bettencourt PE, Del Bono EA, Spiegelman D, Hertzmark E, Murphy RL Jr. Clinical utility of chest auscultation in common pulmonary diseases. Am J Respir Crit Care Med 1994; 150:1291–1297.
- Nath AR, Capel LH. Inspiratory crackles and mechanical events of breathing. Thorax 1974; 29:695–698.
- Badgett RG, Tanaka DJ, Hunt DK, et al. Can moderate chronic obstructive pulmonary disease be diagnosed by historical and physical findings alone? Am J Med 1993; 94:188–196.
- ABIM Foundation. Choosing wisely. Echocardiograms for heart valve disease. www.choosingwisely.org. Accessed November 13, 2017.
- Davison R, Cannon R. Estimation of central venous pressure by examination of jugular veins. Am Heart J 1974; 87:279–282.
- Ducas J, Magder S, McGregor M. Validity of the hepatojugular reflux as a clinical test for congestive heart failure. Am J Cardiol 1983; 52:1299–1303.
- Vinayak AG, Levitt J, Gehlbach B, Pohlman AS, Hall JB, Kress JP. Usefulness of the external jugular vein examination in detecting abnormal central venous pressure in critically ill patients. Arch Intern Med 2006; 166:2132–2137.
- Sankoff J, Zidulka A. Non-invasive method for the rapid assessment of central venous pressure: description and validation by a single examiner. West J Emerg Med 2008; 9:201–205.
- Davie AP, Francis CM, Caruana L, Sutherland GR, McMurray JJ. Assessing diagnosis in heart failure: which features are any use? QJM 1997; 90:335–339.
- Kristensen SL, Mogensen UM, Jhund PS, et al. Clinical and echocardiographic characeristics and cardiovascular outcomes according to diabetes status in patients with heart failure and preserved ejection fraction. A report from the Irbesartan in Heart Failure with Preserved Ejection Fraction Trial (I-Preserve). Circulation 2017; https://doi.org/10.1161/CIRCULATIONAHA.116.024593. Accessed November 1, 2017.
- Butman SM, Ewy GA, Standen JR, Kern KB, Hahn E. Bedside cardiovascular examination in patients with severe chronic heart failure: importance of rest or inducible jugular venous distension. J Am Coll Cardiol 1993; 22:968–974.
- Sochowski RA, Dubbin JD, Naqvi SZ. Clinical and hemodynamic assessment of the hepatojugular reflux. Am J Cardiol 1990; 66:1002–1006.
- Ewy GA. The abdominojugular test: technique and hemodynamic correlates. Ann Intern Med 1988; 109:456–460.
- Gadsboll N, Hoilund-Carlsen PF, Nielsen GG, et al. Symptoms and signs of heart failure in patients with myocardial infarction: reproducibility and relationship to chest X-ray, radionuclide ventriculography and right heart catheterization. Eur Heart J 1989; 10:1017–1028.
- Fahey T, Jeyaseelan S, McCowan C, et al. Diagnosis of left ventricular systolic dysfunction (LVSD): development and validation of a clinical prediction rule in primary care. Fam Pract 2007; 24:628–635.
- Gadsboll N, Hoilund-Carlsen PF, Nielsen GG, et al. Interobserver agreement and accuracy of bedside estimation of right and left ventricular ejection fraction in acute myocardial infarction. Am J Cardiol 1989; 63:1301–1307.
- Mattleman SJ, Hakki AH, Iskandrian AS, Segal BL, Kane SA. Reliability of bedside evaluation in determining left ventricular function: correlation with left ventricular ejection fraction determined by radionuclide ventriculography. J Am Coll Cardiol 1983; 1:417–420.
- Madhok V, Falk G, Rogers A, Struthers AD, Sullivan FM, Fahey T. The accuracy of symptoms, signs and diagnostic tests in the diagnosis of left ventricular dysfunction in primary care: a diagnostic accuracy systematic review. BMC Fam Pract 2008; 9:56.
- Kelder JC, Cramer MJ, van Wijngaarden J, et al. The diagnostic value of physical examination and additional testing in primary care patients with suspected heart failure. Circulation 2011; 124:2865–2873.
KEY POINTS
- Asymmetrical chest expansion, diminished breath sounds, egophony, bronchophony, and tactile fremitus can be used in combination to accurately diagnose pneumonia and pleural effusion.
- No physical sign performs with a high degree of accuracy for diagnosing early-stage chronic obstructive pulmonary disease.
- Inspiratory crackles, diminished breath sounds, and cardiac dullness have high diagnostic value for advanced obstructive airway disease.
- Congestive heart failure can be diagnosed at the bedside by examining the jugular veins and palpating the point of maximal intensity.
Is it time to abandon fasting for routine lipid testing?
Yes. The time has come to change the way we think about fasting before routine lipid testing. We now have robust evidence supporting the routine use of nonfasting lipid testing. Fasting lipid testing is rarely needed, but may be considered for patients with very high triglycerides or before starting treatment in patients with genetic lipid disorders. For most patients, nonfasting lipid testing is appropriate: it is evidence-based, safe, valid, and convenient. More widespread adoption of this strategy by US healthcare providers would improve quality of care and patient and clinician satisfaction.
GUIDELINES HAVE CHANGED
In 2014, the US Department of Veterans Affairs practice guidelines recommended nonfasting lipid testing for cardiovascular risk assessment.1 Other recent clinical guidelines and expert consensus statements from Europe and Canada now also recommend nonfasting lipid testing for most routine clinical evaluations.
Physiologically, we spend most of our lives in the nonfasting state, yet fasting lipid testing was standard practice advocated by earlier clinical guidelines. The rationale for fasting before measuring lipids was to reduce variability and to allow for a more accurate derivation of the low-density lipoprotein cholesterol (LDL-C) concentration using the Friedewald formula. There was also concern that an increase in triglyceride concentrations after consuming a fatty meal would reduce the validity of the results. However, numerous studies have found that the increase in plasma triglycerides after normal food intake is much less than that during a fat-tolerance test, making this less of a concern for most patients.2,3
In addition, recent studies suggest that postprandial effects do not diminish and may even strengthen the risk associations of lipids with cardiovascular disease, in particular for triglycerides.4 Moreover, in certain patients, such as those with metabolic syndrome, diabetes mellitus, or certain genetic abnormalities, fasting can mask abnormalities in triglyceride-rich lipid metabolism, which may only be detected when triglycerides are measured in a nonfasting state. Nonfasting measurements may identify patients with elevated residual risk despite optimal guideline-based treatment.

Recently published recommendations for nonfasting lipid testing for routine assessments are summarized in Table 1.1,5–11
EFFECTS OF THE POSTPRANDIAL STATE ON LIPID LEVELS AND RISK ASSESSMENT
A common concern for clinicians who do not routinely order nonfasting lipid testing is the potential variability in lipid levels and interpretation of these values for treatment decisions. But in most circumstances the differences between fasting and nonfasting measurements are small and are not clinically relevant. Differences in high-density lipoprotein cholesterol (HDL-C) are negligible; slightly lower levels are seen (up to −8 mg/dL) for nonfasting total cholesterol, LDL-C, and non-HDL-C compared with fasting levels; and differences are modest (up to 25 mg/dL higher) for triglycerides.5 These data should reassure clinicians who rely on lipid levels to guide management decisions.9
Cardiovascular risk assessment
Current algorithms for assessing risk of cardiovascular disease use total cholesterol and HDL-C, not triglycerides or LDL-C. Hence, eating has no effect on the risk estimates.
For clinicians who prefer an absolute lipid target for managing risk in patients on lipid-modifying therapy, a nonfasting LDL-C or non-HDL-C (or apolipoprotein B) may be used. The non-HDL-C level is a better risk marker than LDL-C, particularly in patients with low LDL-C or with triglyceride levels of 200 mg/dL or higher.12 Treatment goals for non-HDL-C are 30 mg/dL higher than for LDL-C (fasting or nonfasting). In addition, for these patients with low LDL-C or high triglycerides, a new LDL-C calculation method has more consistent results for fasting and nonfasting values than the commonly used Friedewald calculation.12
EVIDENCE SUPPORTING NONFASTING LIPID TESTING
The adequacy of nonfasting lipid testing for general screening for cardiovascular disease has been verified in large prospective studies over the past several decades.2,13,14 These studies evaluated cardiovascular event and mortality rates and found consistent associations of nonfasting lipid levels with cardiovascular disease. Studies that included both fasting and nonfasting patient populations found similar or occasionally even greater cardiovascular risk associations for nonfasting lipid measurements (including for LDL-C and triglycerides) compared with fasting lipid measurements.
The Emerging Risk Factors Collaboration14 reviewed the data from 68 studies in more than 300,000 people and found that the relationship between lipid levels and incident cardiovascular events was just as strong when nonfasting lipid values were used. In fact, at least 3 large statin trials reviewed (a total of 43,000 people) used nonfasting lipids.14
Genetic studies using mendelian randomization have also linked nonfasting triglyceride levels (and remnant cholesterol) to an increased risk of cardiovascular events and of death from any cause.15,16
Therefore, the evidence overall suggests that nonfasting lipid measurements are acceptable with respect to risk assessment, and indeed may be preferred in most instances, especially in patients with an atherogenic metabolic milieu that may otherwise be masked by the fasting state.
OTHER BENEFITS OF NONFASTING LIPID TESTING
Nonfasting lipid panels are more economical and safer for certain groups, such as elderly or diabetic patients. A pilot study17 found that up to 27.1% of patients with diabetes reported experiencing a fasting-evoked hypoglycemic event en route to testing because of fasting for blood work. These events are vastly underreported and add to patient morbidity that can easily be avoided by adopting nonfasting lipid testing.
No study has assessed the cost-effectiveness of fasting vs nonfasting lipid testing. It is common for patients to present for their office appointment without having obtained a fasting lipid panel simply because they forgot to fast and were turned away by the laboratory. Thus, management decisions during the visit are often deferred, and patients must return to the laboratory and reschedule follow-up visits. This is inefficient, increases outpatient waiting times, and also potentially deprives others of access to needed care. Laboratory workflow can also suffer from an influx of early morning visits for fasting tests, decreasing system efficiency. Decreased efficiency in multiple levels of the healthcare system leads to increased costs, burden on healthcare providers, and decreased patient and physician satisfaction.
GETTING WITH THE GUIDELINES
The 2002 National Cholesterol Education Program expert panel report18 and the 2013 joint cholesterol guidelines of the American College of Cardiology and the American Heart Association9 both recommended that initial screening should involve fasting lipid testing, but they also allowed measuring nonfasting total cholesterol, HDL-C, and non-HDL-C.18 And internationally, there has been a shift in practice recommendations toward nonfasting lipids over the past 10 years (Table 1).
In 2014, the US Department of Veterans Affairs, the UK National Clinical Guideline Centre, and the Joint British Societies said that fasting is no longer needed for routine testing.10 In 2016, the European Atherosclerosis Society and the European Federation of Clinical Chemistry and Laboratory Medicine recommended nonfasting lipid testing as the standard of care and provided clinically useful cut points for both fasting and nonfasting lipid measurements.5
In most guidelines, the threshold for elevated nonfasting triglycerides was defined as 175 mg/dL (≥ 2 mmol/L) or greater, and this level has been validated prospectively in a large study of US women.5,19 Repeat measurement of fasting triglycerides may be considered when nonfasting levels are greater than 400 mg/dL,5 although there is no consensus in the guidelines regarding when or if fasting triglycerides should be remeasured. (In the Danish experience,5 only 10% of patients have required repeat fasting values). In addition, the 2016 Canadian Hypertension Education Program guidelines6 removed fasting as a requirement. The 2016 Canadian Cardiovascular Society dyslipidemia guidelines7 reported that nonfasting lipid testing is a suitable alternative to fasting. Furthermore, the most recent revision of the European Society of Cardiology dyslipidemia guidelines8 acknowledged that nonfasting lipid panels are acceptable for screening and management of patients without severe hypertriglyceridemia or those with extremely low LDL-C levels.
LIMITATIONS OF THE EVIDENCE
To date, no study has assessed the predictive value of fasting vs nonfasting lipid measurements in the same individuals, and there have been no randomized outcomes trials or cost-effectiveness analyses. Ethnic variations in lipoproteins and nonfasting status also need to be investigated as nonfasting lipid testing becomes more universally accepted.
TAKE-HOME POINTS
- Robust evidence supports the routine use of nonfasting lipid testing, with fasting panels reserved potentially for patients with very high triglycerides and before starting treatment in those with genetic lipid disorders.
- For most patients, nonfasting tests are evidence-based, safe, valid, and convenient.
- More widespread adoption of this strategy by US healthcare providers would improve both quality of care and patient-clinician satisfaction.
- US Department of Veterans Affairs. VA/DoD Clinical Practice Guidelines: the management of dyslipidemia for cardiovascular risk reduction (lipids). 2014. www.healthquality.va.gov/guidelines/CD/lipids. Accessed October 18, 2017.
- Langsted A, Freiberg JJ, Nordestgaard BG. Fasting and nonfasting lipid levels: influence of normal food intake on lipids, lipoproteins, apolipoproteins, and cardiovascular risk prediction. Circulation 2008; 118:2047–2056.
- Langsted A, Nordestgaard BG. Nonfasting lipids, lipoproteins, and apolipoproteins in individuals with and without diabetes: 58 434 individuals from the Copenhagen General Population Study. Clin Chem 2011; 57:482–489.
- Rifai N, Young IS, Nordestgaard BG, et al. Nonfasting sample for the determination of routine lipid profile: is it an idea whose time has come? Clin Chem 2016; 62:428–435.
- Nordestgaard BG, Langsted A, Mora S, et al; European Atherosclerosis Society (EAS) and the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) joint consensus initiative. Fasting is not routinely required for determination of a lipid profile: clinical and laboratory implications including flagging at desirable concentration cut-points-a joint consensus statement from the European Atherosclerosis Society and European Federation of Clinical Chemistry and Laboratory Medicine. Eur Heart J 2016; 37:1944–1958.
- Leung AA, Nerenberg K, Daskalopoulou SS, et al; CHEP Guidelines Task Force. Hypertension Canada’s 2016 Canadian Hypertension Education Program guidelines for blood pressure measurement, diagnosis, assessment of risk, prevention, and treatment of hypertension. Can J Cardiol 2016; 32:569–588.
- Anderson TJ, Gregoire J, Pearson GJ, et al. 2016 Canadian Cardiovascular Society guidelines for the management of dyslipidemia for the prevention of cardiovascular disease in the adult. Can J Cardiol 2016; 32:1263–1282.
- Catapano AL, Graham I, De Backer G, et al; Authors/Task Force Members; Additional Contributor. 2016 ESC/EAS guidelines for the management of dyslipidaemias. Eur Heart J 2016; 37:2999–3058.
- Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129(suppl):S1–S45.
- National Institute for Health and Care Excellence (NICE). Cardiovascular disease: risk assessment and reduction, including lipid modification. Clinical guideline CG181. Published July 2014. Updated September 2016. www.nice.org.uk/guidance/cg181. Accessed October 18, 2017.
- Jellinger PS, Handelsman Y, Rosenblit PD, et al. American Association of Clinical Endocrinologists and American College of Endocrinology guidelines for management of dyslipidemia and prevention of cardiovascular disease. Endocr Pract 2017; 23(suppl 2):1–87.
- Martin SS, Blaha MJ, Elshazly MB, et al. Friedewald-estimated versus directly measured low-density lipoprotein cholesterol and treatment implications. J Am Coll Cardiol 2013; 62:732–739.
- Mora S, Rifai N, Buring JE, Ridker PM. Fasting compared with nonfasting lipids and apolipoproteins for predicting incident cardiovascular events. Circulation 2008; 118:993–1001.
- Emerging Risk Factors Collaboration; Di Angelantonio E, Sarwar N, Perry P, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009; 302:1993–2000.
- Varbo A, Benn M, Tybjaerg-Hansen A, Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol 2013; 61:427–436.
- Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjærg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med 2014; 371:32–41.
- Aldasouqi S, Corser W, Abela G, et al. Fasting for lipid profiles poses a high risk of hypoglycemia in patients with diabetes: a pilot prevalence study in clinical practice. Int J Clin Med 2016; 7:1653–1667.
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- White KT, Moorthy MV, Akinkuolie AO, et al. Identifying an optimal cutpoint for the diagnosis of hypertriglyceridemia in the nonfasting state. Clin Chem 2015; 61:1156–1163.
Yes. The time has come to change the way we think about fasting before routine lipid testing. We now have robust evidence supporting the routine use of nonfasting lipid testing. Fasting lipid testing is rarely needed, but may be considered for patients with very high triglycerides or before starting treatment in patients with genetic lipid disorders. For most patients, nonfasting lipid testing is appropriate: it is evidence-based, safe, valid, and convenient. More widespread adoption of this strategy by US healthcare providers would improve quality of care and patient and clinician satisfaction.
GUIDELINES HAVE CHANGED
In 2014, the US Department of Veterans Affairs practice guidelines recommended nonfasting lipid testing for cardiovascular risk assessment.1 Other recent clinical guidelines and expert consensus statements from Europe and Canada now also recommend nonfasting lipid testing for most routine clinical evaluations.
Physiologically, we spend most of our lives in the nonfasting state, yet fasting lipid testing was standard practice advocated by earlier clinical guidelines. The rationale for fasting before measuring lipids was to reduce variability and to allow for a more accurate derivation of the low-density lipoprotein cholesterol (LDL-C) concentration using the Friedewald formula. There was also concern that an increase in triglyceride concentrations after consuming a fatty meal would reduce the validity of the results. However, numerous studies have found that the increase in plasma triglycerides after normal food intake is much less than that during a fat-tolerance test, making this less of a concern for most patients.2,3
In addition, recent studies suggest that postprandial effects do not diminish and may even strengthen the risk associations of lipids with cardiovascular disease, in particular for triglycerides.4 Moreover, in certain patients, such as those with metabolic syndrome, diabetes mellitus, or certain genetic abnormalities, fasting can mask abnormalities in triglyceride-rich lipid metabolism, which may only be detected when triglycerides are measured in a nonfasting state. Nonfasting measurements may identify patients with elevated residual risk despite optimal guideline-based treatment.
Recently published recommendations for nonfasting lipid testing for routine assessments are summarized in Table 1.1,5–11
EFFECTS OF THE POSTPRANDIAL STATE ON LIPID LEVELS AND RISK ASSESSMENT
A common concern for clinicians who do not routinely order nonfasting lipid testing is the potential variability in lipid levels and interpretation of these values for treatment decisions. But in most circumstances the differences between fasting and nonfasting measurements are small and are not clinically relevant. Differences in high-density lipoprotein cholesterol (HDL-C) are negligible; slightly lower levels are seen (up to −8 mg/dL) for nonfasting total cholesterol, LDL-C, and non-HDL-C compared with fasting levels; and differences are modest (up to 25 mg/dL higher) for triglycerides.5 These data should reassure clinicians who rely on lipid levels to guide management decisions.9
Cardiovascular risk assessment
Current algorithms for assessing risk of cardiovascular disease use total cholesterol and HDL-C, not triglycerides or LDL-C. Hence, eating has no effect on the risk estimates.
For clinicians who prefer an absolute lipid target for managing risk in patients on lipid-modifying therapy, a nonfasting LDL-C or non-HDL-C (or apolipoprotein B) may be used. The non-HDL-C level is a better risk marker than LDL-C, particularly in patients with low LDL-C or with triglyceride levels of 200 mg/dL or higher.12 Treatment goals for non-HDL-C are 30 mg/dL higher than for LDL-C (fasting or nonfasting). In addition, for these patients with low LDL-C or high triglycerides, a new LDL-C calculation method has more consistent results for fasting and nonfasting values than the commonly used Friedewald calculation.12
EVIDENCE SUPPORTING NONFASTING LIPID TESTING
The adequacy of nonfasting lipid testing for general screening for cardiovascular disease has been verified in large prospective studies over the past several decades.2,13,14 These studies evaluated cardiovascular event and mortality rates and found consistent associations of nonfasting lipid levels with cardiovascular disease. Studies that included both fasting and nonfasting patient populations found similar or occasionally even greater cardiovascular risk associations for nonfasting lipid measurements (including for LDL-C and triglycerides) compared with fasting lipid measurements.
The Emerging Risk Factors Collaboration14 reviewed the data from 68 studies in more than 300,000 people and found that the relationship between lipid levels and incident cardiovascular events was just as strong when nonfasting lipid values were used. In fact, at least 3 large statin trials reviewed (a total of 43,000 people) used nonfasting lipids.14
Genetic studies using mendelian randomization have also linked nonfasting triglyceride levels (and remnant cholesterol) to an increased risk of cardiovascular events and of death from any cause.15,16
Therefore, the evidence overall suggests that nonfasting lipid measurements are acceptable with respect to risk assessment, and indeed may be preferred in most instances, especially in patients with an atherogenic metabolic milieu that may otherwise be masked by the fasting state.
OTHER BENEFITS OF NONFASTING LIPID TESTING
Nonfasting lipid panels are more economical and safer for certain groups, such as elderly or diabetic patients. A pilot study17 found that up to 27.1% of patients with diabetes reported experiencing a fasting-evoked hypoglycemic event en route to testing because of fasting for blood work. These events are vastly underreported and add to patient morbidity that can easily be avoided by adopting nonfasting lipid testing.
No study has assessed the cost-effectiveness of fasting vs nonfasting lipid testing. It is common for patients to present for their office appointment without having obtained a fasting lipid panel simply because they forgot to fast and were turned away by the laboratory. Thus, management decisions during the visit are often deferred, and patients must return to the laboratory and reschedule follow-up visits. This is inefficient, increases outpatient waiting times, and also potentially deprives others of access to needed care. Laboratory workflow can also suffer from an influx of early morning visits for fasting tests, decreasing system efficiency. Decreased efficiency in multiple levels of the healthcare system leads to increased costs, burden on healthcare providers, and decreased patient and physician satisfaction.
GETTING WITH THE GUIDELINES
The 2002 National Cholesterol Education Program expert panel report18 and the 2013 joint cholesterol guidelines of the American College of Cardiology and the American Heart Association9 both recommended that initial screening should involve fasting lipid testing, but they also allowed measuring nonfasting total cholesterol, HDL-C, and non-HDL-C.18 And internationally, there has been a shift in practice recommendations toward nonfasting lipids over the past 10 years (Table 1).
In 2014, the US Department of Veterans Affairs, the UK National Clinical Guideline Centre, and the Joint British Societies said that fasting is no longer needed for routine testing.10 In 2016, the European Atherosclerosis Society and the European Federation of Clinical Chemistry and Laboratory Medicine recommended nonfasting lipid testing as the standard of care and provided clinically useful cut points for both fasting and nonfasting lipid measurements.5
In most guidelines, the threshold for elevated nonfasting triglycerides was defined as 175 mg/dL (≥ 2 mmol/L) or greater, and this level has been validated prospectively in a large study of US women.5,19 Repeat measurement of fasting triglycerides may be considered when nonfasting levels are greater than 400 mg/dL,5 although there is no consensus in the guidelines regarding when or if fasting triglycerides should be remeasured. (In the Danish experience,5 only 10% of patients have required repeat fasting values). In addition, the 2016 Canadian Hypertension Education Program guidelines6 removed fasting as a requirement. The 2016 Canadian Cardiovascular Society dyslipidemia guidelines7 reported that nonfasting lipid testing is a suitable alternative to fasting. Furthermore, the most recent revision of the European Society of Cardiology dyslipidemia guidelines8 acknowledged that nonfasting lipid panels are acceptable for screening and management of patients without severe hypertriglyceridemia or those with extremely low LDL-C levels.
LIMITATIONS OF THE EVIDENCE
To date, no study has assessed the predictive value of fasting vs nonfasting lipid measurements in the same individuals, and there have been no randomized outcomes trials or cost-effectiveness analyses. Ethnic variations in lipoproteins and nonfasting status also need to be investigated as nonfasting lipid testing becomes more universally accepted.
TAKE-HOME POINTS
- Robust evidence supports the routine use of nonfasting lipid testing, with fasting panels reserved potentially for patients with very high triglycerides and before starting treatment in those with genetic lipid disorders.
- For most patients, nonfasting tests are evidence-based, safe, valid, and convenient.
- More widespread adoption of this strategy by US healthcare providers would improve both quality of care and patient-clinician satisfaction.
Yes. The time has come to change the way we think about fasting before routine lipid testing. We now have robust evidence supporting the routine use of nonfasting lipid testing. Fasting lipid testing is rarely needed, but may be considered for patients with very high triglycerides or before starting treatment in patients with genetic lipid disorders. For most patients, nonfasting lipid testing is appropriate: it is evidence-based, safe, valid, and convenient. More widespread adoption of this strategy by US healthcare providers would improve quality of care and patient and clinician satisfaction.
GUIDELINES HAVE CHANGED
In 2014, the US Department of Veterans Affairs practice guidelines recommended nonfasting lipid testing for cardiovascular risk assessment.1 Other recent clinical guidelines and expert consensus statements from Europe and Canada now also recommend nonfasting lipid testing for most routine clinical evaluations.
Physiologically, we spend most of our lives in the nonfasting state, yet fasting lipid testing was standard practice advocated by earlier clinical guidelines. The rationale for fasting before measuring lipids was to reduce variability and to allow for a more accurate derivation of the low-density lipoprotein cholesterol (LDL-C) concentration using the Friedewald formula. There was also concern that an increase in triglyceride concentrations after consuming a fatty meal would reduce the validity of the results. However, numerous studies have found that the increase in plasma triglycerides after normal food intake is much less than that during a fat-tolerance test, making this less of a concern for most patients.2,3
In addition, recent studies suggest that postprandial effects do not diminish and may even strengthen the risk associations of lipids with cardiovascular disease, in particular for triglycerides.4 Moreover, in certain patients, such as those with metabolic syndrome, diabetes mellitus, or certain genetic abnormalities, fasting can mask abnormalities in triglyceride-rich lipid metabolism, which may only be detected when triglycerides are measured in a nonfasting state. Nonfasting measurements may identify patients with elevated residual risk despite optimal guideline-based treatment.
Recently published recommendations for nonfasting lipid testing for routine assessments are summarized in Table 1.1,5–11
EFFECTS OF THE POSTPRANDIAL STATE ON LIPID LEVELS AND RISK ASSESSMENT
A common concern for clinicians who do not routinely order nonfasting lipid testing is the potential variability in lipid levels and interpretation of these values for treatment decisions. But in most circumstances the differences between fasting and nonfasting measurements are small and are not clinically relevant. Differences in high-density lipoprotein cholesterol (HDL-C) are negligible; slightly lower levels are seen (up to −8 mg/dL) for nonfasting total cholesterol, LDL-C, and non-HDL-C compared with fasting levels; and differences are modest (up to 25 mg/dL higher) for triglycerides.5 These data should reassure clinicians who rely on lipid levels to guide management decisions.9
Cardiovascular risk assessment
Current algorithms for assessing risk of cardiovascular disease use total cholesterol and HDL-C, not triglycerides or LDL-C. Hence, eating has no effect on the risk estimates.
For clinicians who prefer an absolute lipid target for managing risk in patients on lipid-modifying therapy, a nonfasting LDL-C or non-HDL-C (or apolipoprotein B) may be used. The non-HDL-C level is a better risk marker than LDL-C, particularly in patients with low LDL-C or with triglyceride levels of 200 mg/dL or higher.12 Treatment goals for non-HDL-C are 30 mg/dL higher than for LDL-C (fasting or nonfasting). In addition, for these patients with low LDL-C or high triglycerides, a new LDL-C calculation method has more consistent results for fasting and nonfasting values than the commonly used Friedewald calculation.12
EVIDENCE SUPPORTING NONFASTING LIPID TESTING
The adequacy of nonfasting lipid testing for general screening for cardiovascular disease has been verified in large prospective studies over the past several decades.2,13,14 These studies evaluated cardiovascular event and mortality rates and found consistent associations of nonfasting lipid levels with cardiovascular disease. Studies that included both fasting and nonfasting patient populations found similar or occasionally even greater cardiovascular risk associations for nonfasting lipid measurements (including for LDL-C and triglycerides) compared with fasting lipid measurements.
The Emerging Risk Factors Collaboration14 reviewed the data from 68 studies in more than 300,000 people and found that the relationship between lipid levels and incident cardiovascular events was just as strong when nonfasting lipid values were used. In fact, at least 3 large statin trials reviewed (a total of 43,000 people) used nonfasting lipids.14
Genetic studies using mendelian randomization have also linked nonfasting triglyceride levels (and remnant cholesterol) to an increased risk of cardiovascular events and of death from any cause.15,16
Therefore, the evidence overall suggests that nonfasting lipid measurements are acceptable with respect to risk assessment, and indeed may be preferred in most instances, especially in patients with an atherogenic metabolic milieu that may otherwise be masked by the fasting state.
OTHER BENEFITS OF NONFASTING LIPID TESTING
Nonfasting lipid panels are more economical and safer for certain groups, such as elderly or diabetic patients. A pilot study17 found that up to 27.1% of patients with diabetes reported experiencing a fasting-evoked hypoglycemic event en route to testing because of fasting for blood work. These events are vastly underreported and add to patient morbidity that can easily be avoided by adopting nonfasting lipid testing.
No study has assessed the cost-effectiveness of fasting vs nonfasting lipid testing. It is common for patients to present for their office appointment without having obtained a fasting lipid panel simply because they forgot to fast and were turned away by the laboratory. Thus, management decisions during the visit are often deferred, and patients must return to the laboratory and reschedule follow-up visits. This is inefficient, increases outpatient waiting times, and also potentially deprives others of access to needed care. Laboratory workflow can also suffer from an influx of early morning visits for fasting tests, decreasing system efficiency. Decreased efficiency in multiple levels of the healthcare system leads to increased costs, burden on healthcare providers, and decreased patient and physician satisfaction.
GETTING WITH THE GUIDELINES
The 2002 National Cholesterol Education Program expert panel report18 and the 2013 joint cholesterol guidelines of the American College of Cardiology and the American Heart Association9 both recommended that initial screening should involve fasting lipid testing, but they also allowed measuring nonfasting total cholesterol, HDL-C, and non-HDL-C.18 And internationally, there has been a shift in practice recommendations toward nonfasting lipids over the past 10 years (Table 1).
In 2014, the US Department of Veterans Affairs, the UK National Clinical Guideline Centre, and the Joint British Societies said that fasting is no longer needed for routine testing.10 In 2016, the European Atherosclerosis Society and the European Federation of Clinical Chemistry and Laboratory Medicine recommended nonfasting lipid testing as the standard of care and provided clinically useful cut points for both fasting and nonfasting lipid measurements.5
In most guidelines, the threshold for elevated nonfasting triglycerides was defined as 175 mg/dL (≥ 2 mmol/L) or greater, and this level has been validated prospectively in a large study of US women.5,19 Repeat measurement of fasting triglycerides may be considered when nonfasting levels are greater than 400 mg/dL,5 although there is no consensus in the guidelines regarding when or if fasting triglycerides should be remeasured. (In the Danish experience,5 only 10% of patients have required repeat fasting values). In addition, the 2016 Canadian Hypertension Education Program guidelines6 removed fasting as a requirement. The 2016 Canadian Cardiovascular Society dyslipidemia guidelines7 reported that nonfasting lipid testing is a suitable alternative to fasting. Furthermore, the most recent revision of the European Society of Cardiology dyslipidemia guidelines8 acknowledged that nonfasting lipid panels are acceptable for screening and management of patients without severe hypertriglyceridemia or those with extremely low LDL-C levels.
LIMITATIONS OF THE EVIDENCE
To date, no study has assessed the predictive value of fasting vs nonfasting lipid measurements in the same individuals, and there have been no randomized outcomes trials or cost-effectiveness analyses. Ethnic variations in lipoproteins and nonfasting status also need to be investigated as nonfasting lipid testing becomes more universally accepted.
TAKE-HOME POINTS
- Robust evidence supports the routine use of nonfasting lipid testing, with fasting panels reserved potentially for patients with very high triglycerides and before starting treatment in those with genetic lipid disorders.
- For most patients, nonfasting tests are evidence-based, safe, valid, and convenient.
- More widespread adoption of this strategy by US healthcare providers would improve both quality of care and patient-clinician satisfaction.
- US Department of Veterans Affairs. VA/DoD Clinical Practice Guidelines: the management of dyslipidemia for cardiovascular risk reduction (lipids). 2014. www.healthquality.va.gov/guidelines/CD/lipids. Accessed October 18, 2017.
- Langsted A, Freiberg JJ, Nordestgaard BG. Fasting and nonfasting lipid levels: influence of normal food intake on lipids, lipoproteins, apolipoproteins, and cardiovascular risk prediction. Circulation 2008; 118:2047–2056.
- Langsted A, Nordestgaard BG. Nonfasting lipids, lipoproteins, and apolipoproteins in individuals with and without diabetes: 58 434 individuals from the Copenhagen General Population Study. Clin Chem 2011; 57:482–489.
- Rifai N, Young IS, Nordestgaard BG, et al. Nonfasting sample for the determination of routine lipid profile: is it an idea whose time has come? Clin Chem 2016; 62:428–435.
- Nordestgaard BG, Langsted A, Mora S, et al; European Atherosclerosis Society (EAS) and the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) joint consensus initiative. Fasting is not routinely required for determination of a lipid profile: clinical and laboratory implications including flagging at desirable concentration cut-points-a joint consensus statement from the European Atherosclerosis Society and European Federation of Clinical Chemistry and Laboratory Medicine. Eur Heart J 2016; 37:1944–1958.
- Leung AA, Nerenberg K, Daskalopoulou SS, et al; CHEP Guidelines Task Force. Hypertension Canada’s 2016 Canadian Hypertension Education Program guidelines for blood pressure measurement, diagnosis, assessment of risk, prevention, and treatment of hypertension. Can J Cardiol 2016; 32:569–588.
- Anderson TJ, Gregoire J, Pearson GJ, et al. 2016 Canadian Cardiovascular Society guidelines for the management of dyslipidemia for the prevention of cardiovascular disease in the adult. Can J Cardiol 2016; 32:1263–1282.
- Catapano AL, Graham I, De Backer G, et al; Authors/Task Force Members; Additional Contributor. 2016 ESC/EAS guidelines for the management of dyslipidaemias. Eur Heart J 2016; 37:2999–3058.
- Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129(suppl):S1–S45.
- National Institute for Health and Care Excellence (NICE). Cardiovascular disease: risk assessment and reduction, including lipid modification. Clinical guideline CG181. Published July 2014. Updated September 2016. www.nice.org.uk/guidance/cg181. Accessed October 18, 2017.
- Jellinger PS, Handelsman Y, Rosenblit PD, et al. American Association of Clinical Endocrinologists and American College of Endocrinology guidelines for management of dyslipidemia and prevention of cardiovascular disease. Endocr Pract 2017; 23(suppl 2):1–87.
- Martin SS, Blaha MJ, Elshazly MB, et al. Friedewald-estimated versus directly measured low-density lipoprotein cholesterol and treatment implications. J Am Coll Cardiol 2013; 62:732–739.
- Mora S, Rifai N, Buring JE, Ridker PM. Fasting compared with nonfasting lipids and apolipoproteins for predicting incident cardiovascular events. Circulation 2008; 118:993–1001.
- Emerging Risk Factors Collaboration; Di Angelantonio E, Sarwar N, Perry P, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009; 302:1993–2000.
- Varbo A, Benn M, Tybjaerg-Hansen A, Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol 2013; 61:427–436.
- Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjærg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med 2014; 371:32–41.
- Aldasouqi S, Corser W, Abela G, et al. Fasting for lipid profiles poses a high risk of hypoglycemia in patients with diabetes: a pilot prevalence study in clinical practice. Int J Clin Med 2016; 7:1653–1667.
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- White KT, Moorthy MV, Akinkuolie AO, et al. Identifying an optimal cutpoint for the diagnosis of hypertriglyceridemia in the nonfasting state. Clin Chem 2015; 61:1156–1163.
- US Department of Veterans Affairs. VA/DoD Clinical Practice Guidelines: the management of dyslipidemia for cardiovascular risk reduction (lipids). 2014. www.healthquality.va.gov/guidelines/CD/lipids. Accessed October 18, 2017.
- Langsted A, Freiberg JJ, Nordestgaard BG. Fasting and nonfasting lipid levels: influence of normal food intake on lipids, lipoproteins, apolipoproteins, and cardiovascular risk prediction. Circulation 2008; 118:2047–2056.
- Langsted A, Nordestgaard BG. Nonfasting lipids, lipoproteins, and apolipoproteins in individuals with and without diabetes: 58 434 individuals from the Copenhagen General Population Study. Clin Chem 2011; 57:482–489.
- Rifai N, Young IS, Nordestgaard BG, et al. Nonfasting sample for the determination of routine lipid profile: is it an idea whose time has come? Clin Chem 2016; 62:428–435.
- Nordestgaard BG, Langsted A, Mora S, et al; European Atherosclerosis Society (EAS) and the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) joint consensus initiative. Fasting is not routinely required for determination of a lipid profile: clinical and laboratory implications including flagging at desirable concentration cut-points-a joint consensus statement from the European Atherosclerosis Society and European Federation of Clinical Chemistry and Laboratory Medicine. Eur Heart J 2016; 37:1944–1958.
- Leung AA, Nerenberg K, Daskalopoulou SS, et al; CHEP Guidelines Task Force. Hypertension Canada’s 2016 Canadian Hypertension Education Program guidelines for blood pressure measurement, diagnosis, assessment of risk, prevention, and treatment of hypertension. Can J Cardiol 2016; 32:569–588.
- Anderson TJ, Gregoire J, Pearson GJ, et al. 2016 Canadian Cardiovascular Society guidelines for the management of dyslipidemia for the prevention of cardiovascular disease in the adult. Can J Cardiol 2016; 32:1263–1282.
- Catapano AL, Graham I, De Backer G, et al; Authors/Task Force Members; Additional Contributor. 2016 ESC/EAS guidelines for the management of dyslipidaemias. Eur Heart J 2016; 37:2999–3058.
- Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129(suppl):S1–S45.
- National Institute for Health and Care Excellence (NICE). Cardiovascular disease: risk assessment and reduction, including lipid modification. Clinical guideline CG181. Published July 2014. Updated September 2016. www.nice.org.uk/guidance/cg181. Accessed October 18, 2017.
- Jellinger PS, Handelsman Y, Rosenblit PD, et al. American Association of Clinical Endocrinologists and American College of Endocrinology guidelines for management of dyslipidemia and prevention of cardiovascular disease. Endocr Pract 2017; 23(suppl 2):1–87.
- Martin SS, Blaha MJ, Elshazly MB, et al. Friedewald-estimated versus directly measured low-density lipoprotein cholesterol and treatment implications. J Am Coll Cardiol 2013; 62:732–739.
- Mora S, Rifai N, Buring JE, Ridker PM. Fasting compared with nonfasting lipids and apolipoproteins for predicting incident cardiovascular events. Circulation 2008; 118:993–1001.
- Emerging Risk Factors Collaboration; Di Angelantonio E, Sarwar N, Perry P, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009; 302:1993–2000.
- Varbo A, Benn M, Tybjaerg-Hansen A, Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol 2013; 61:427–436.
- Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjærg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med 2014; 371:32–41.
- Aldasouqi S, Corser W, Abela G, et al. Fasting for lipid profiles poses a high risk of hypoglycemia in patients with diabetes: a pilot prevalence study in clinical practice. Int J Clin Med 2016; 7:1653–1667.
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- White KT, Moorthy MV, Akinkuolie AO, et al. Identifying an optimal cutpoint for the diagnosis of hypertriglyceridemia in the nonfasting state. Clin Chem 2015; 61:1156–1163.
It’s time to consider pharmacotherapy for obesity
The article in this issue by Bersoux et al on pharmacotherapy to manage obesity1 is apropos in light of a recent study2 showing that patients are filling 15 times more prescriptions for antidiabetic medications (excluding insulin) than for antiobesity drugs. What makes this finding significant is that nearly 3 times more adults meet the criteria for use of antiobesity drugs than for antidiabetic drugs—116 million vs 30 million, respectively.
This underuse of antiobesity medications has been noted in other studies. In 1 study,3 only about 2% of adults eligible for weight-loss drug therapy received a prescription. Conversely, about 86% of adults diagnosed with diabetes received antidiabetic medications.3
WEIGHT LOSS: IT'S IMPORTANT
This underuse of weight-loss drugs occurs despite our understanding that obesity is a risk factor for developing diabetes and that weight loss in obese patients reduces the risk.
The landmark Diabetes Prevention Program study found that even modest weight loss of 7% reduced the risk of developing diabetes by 58% in overweight and prediabetic individuals.4 Additionally, a 5% to 10% weight loss can lead to significant improvements in many comorbidities, including diabetes, hyperlipidemia, hypertension, sleep apnea, and fatty liver disease.
Antiobesity medications can help patients achieve weight-loss goals, especially if lifestyle and behavioral modifications alone have been unsuccessful. Data show that these drugs result in an average weight loss of 5% to 15% when added to diet and exercise.
BARRIERS TO PRESCRIBING WEIGHT-LOSS DRUGS
Why are practitioners reluctant to prescribe these drugs despite the worsening obesity epidemic and despite knowing that obesity is a risk factor for diabetes? Many of us who practice obesity medicine believe there are several reasons.
One barrier is the misconception that obesity does not warrant treatment with weight-loss medications, even though most practitioners will readily admit that patients cannot achieve effective, durable, and meaningful weight loss with behavioral changes and lifestyle modifications alone.
Other barriers stem from issues such as time constraints in the office, lack of training to treat this condition, and not enough data on the newer chronic weight-loss medications. And there are stringent requirements for patient follow-up once a medication has been initiated. Finally, it’s often difficult to obtain insurance coverage.
Addressing the barriers
Of these, I believe the biggest barrier for busy practitioners is finding the time and effort they need to devote to prescribing weight-loss medications. There are ways to address these issues.
Regarding time constraints, practitioners can discuss weight loss at follow-up visits and refer patients to obesity specialists. Regarding gaps in training and knowledge of obesity management, there are consensus guidelines for the identification, evaluation, and treatment of the overweight or obese individual.5–7 Guidelines provide extensive information on the pharmacologic treatment of obesity. These resources provide valuable evidence-based recommendations on how to manage this chronic disease.
ARMED WITH INFORMATION, PHARMACOLOGIC OPTIONS
Bersoux et al provide another valuable resource for clinical use of weight-loss drugs.1 They accurately review the available medications, their mechanisms of action, dosing, efficacy, side effect profiles, and clinical indications. Their review is comprehensive in every aspect of this drug class.
This is important information for practitioners to have when considering prescribing antiobesity medications. It is especially important for primary care practitioners because of the large number of obese or overweight patients they treat.
Drug options have expanded
We did not always have this many drugs to choose from. As Bersoux et al note, practitioners had limited options for weight-loss medications during the 1990s and early 2000s, and several of those had to be taken off the market because of serious side effects. Then between 2012 and 2014, the US Food and Drug Administration approved 4 new medications, giving us a total of 6 weight-loss drugs. Those approvals greatly increased the available drug treatments, giving us much-needed options beyond lifestyle and behavioral modifications.
Although it is widely accepted that antiobesity drugs are underused, the study by Thomas et al was the first to quantify the extent of underuse, especially for the newer chronic weight-loss drugs.2 Their data show that only about 19% of antiobesity prescriptions were for the newer drugs while 74% were for the older but short-term medication phentermine.
Bersoux et al seem to encourage primary care physicians, or anyone caring for overweight or obese patients, to consider prescribing these treatments if nonpharmacologic options are unsuccessful. I agree with this concept because there are not enough specialists to care for the more than 116 million individuals who are potential candidates for antiobesity medications.
THE TIME HAS COME
This new class of medications has been strongly endorsed by the most prestigious organizations and societies involved in developing treatment guidelines for the overweight or obese patient. It is time for everyone who sees overweight or obese patients in daily practice to consider adopting chronic weight-loss medications as adjunctive therapy if lifestyle and behavioral strategies are ineffective.
- Bersoux S, Byun TH, Chaliki SS, Poole KJ Jr. Pharmacotherapy for obesity: what you need to know. Cleve Clin J Med 2017; 84:951–958.
- Thomas CE, Mauer EA, Shukla AP, Rathi S, Aronne LJ. Low adoption of weight loss medications: a comparison of prescribing patterns of antiobesity pharmacotherapies and SGLT2s. Obesity 2016; 24:1955–1961.
- Samaranayake NR, Ong KL, Leung RY, Cheung BM. Management of obesity in the National Health and Nutrition Examination Survey (NHANES), 2007–2008. Ann Epidemiol 2012; 22:349–353.
- Knowler WC, Fowler SE, Hamman RF, et al; for the Diabetes Prevention Program Research Group. 10-year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet 2009; 374:1677–1686.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. J Am Coll Cardiol 2014; 63:2985–3023.
- American Association of Clinical Endocrinologists. AACE/ACE algorithm for the medical care of patients with obesity.
https://www.aace.com/files/final-appendix.pdf. Accessed September 20, 2017.
- Obesity Medicine Association. Obesity algorithm: 2016-2017.
https://obesitymedicine.org/obesity-algorithm/. Accessed October 3, 2017.
The article in this issue by Bersoux et al on pharmacotherapy to manage obesity1 is apropos in light of a recent study2 showing that patients are filling 15 times more prescriptions for antidiabetic medications (excluding insulin) than for antiobesity drugs. What makes this finding significant is that nearly 3 times more adults meet the criteria for use of antiobesity drugs than for antidiabetic drugs—116 million vs 30 million, respectively.
This underuse of antiobesity medications has been noted in other studies. In 1 study,3 only about 2% of adults eligible for weight-loss drug therapy received a prescription. Conversely, about 86% of adults diagnosed with diabetes received antidiabetic medications.3
WEIGHT LOSS: IT'S IMPORTANT
This underuse of weight-loss drugs occurs despite our understanding that obesity is a risk factor for developing diabetes and that weight loss in obese patients reduces the risk.
The landmark Diabetes Prevention Program study found that even modest weight loss of 7% reduced the risk of developing diabetes by 58% in overweight and prediabetic individuals.4 Additionally, a 5% to 10% weight loss can lead to significant improvements in many comorbidities, including diabetes, hyperlipidemia, hypertension, sleep apnea, and fatty liver disease.
Antiobesity medications can help patients achieve weight-loss goals, especially if lifestyle and behavioral modifications alone have been unsuccessful. Data show that these drugs result in an average weight loss of 5% to 15% when added to diet and exercise.
BARRIERS TO PRESCRIBING WEIGHT-LOSS DRUGS
Why are practitioners reluctant to prescribe these drugs despite the worsening obesity epidemic and despite knowing that obesity is a risk factor for diabetes? Many of us who practice obesity medicine believe there are several reasons.
One barrier is the misconception that obesity does not warrant treatment with weight-loss medications, even though most practitioners will readily admit that patients cannot achieve effective, durable, and meaningful weight loss with behavioral changes and lifestyle modifications alone.
Other barriers stem from issues such as time constraints in the office, lack of training to treat this condition, and not enough data on the newer chronic weight-loss medications. And there are stringent requirements for patient follow-up once a medication has been initiated. Finally, it’s often difficult to obtain insurance coverage.
Addressing the barriers
Of these, I believe the biggest barrier for busy practitioners is finding the time and effort they need to devote to prescribing weight-loss medications. There are ways to address these issues.
Regarding time constraints, practitioners can discuss weight loss at follow-up visits and refer patients to obesity specialists. Regarding gaps in training and knowledge of obesity management, there are consensus guidelines for the identification, evaluation, and treatment of the overweight or obese individual.5–7 Guidelines provide extensive information on the pharmacologic treatment of obesity. These resources provide valuable evidence-based recommendations on how to manage this chronic disease.
ARMED WITH INFORMATION, PHARMACOLOGIC OPTIONS
Bersoux et al provide another valuable resource for clinical use of weight-loss drugs.1 They accurately review the available medications, their mechanisms of action, dosing, efficacy, side effect profiles, and clinical indications. Their review is comprehensive in every aspect of this drug class.
This is important information for practitioners to have when considering prescribing antiobesity medications. It is especially important for primary care practitioners because of the large number of obese or overweight patients they treat.
Drug options have expanded
We did not always have this many drugs to choose from. As Bersoux et al note, practitioners had limited options for weight-loss medications during the 1990s and early 2000s, and several of those had to be taken off the market because of serious side effects. Then between 2012 and 2014, the US Food and Drug Administration approved 4 new medications, giving us a total of 6 weight-loss drugs. Those approvals greatly increased the available drug treatments, giving us much-needed options beyond lifestyle and behavioral modifications.
Although it is widely accepted that antiobesity drugs are underused, the study by Thomas et al was the first to quantify the extent of underuse, especially for the newer chronic weight-loss drugs.2 Their data show that only about 19% of antiobesity prescriptions were for the newer drugs while 74% were for the older but short-term medication phentermine.
Bersoux et al seem to encourage primary care physicians, or anyone caring for overweight or obese patients, to consider prescribing these treatments if nonpharmacologic options are unsuccessful. I agree with this concept because there are not enough specialists to care for the more than 116 million individuals who are potential candidates for antiobesity medications.
THE TIME HAS COME
This new class of medications has been strongly endorsed by the most prestigious organizations and societies involved in developing treatment guidelines for the overweight or obese patient. It is time for everyone who sees overweight or obese patients in daily practice to consider adopting chronic weight-loss medications as adjunctive therapy if lifestyle and behavioral strategies are ineffective.
The article in this issue by Bersoux et al on pharmacotherapy to manage obesity1 is apropos in light of a recent study2 showing that patients are filling 15 times more prescriptions for antidiabetic medications (excluding insulin) than for antiobesity drugs. What makes this finding significant is that nearly 3 times more adults meet the criteria for use of antiobesity drugs than for antidiabetic drugs—116 million vs 30 million, respectively.
This underuse of antiobesity medications has been noted in other studies. In 1 study,3 only about 2% of adults eligible for weight-loss drug therapy received a prescription. Conversely, about 86% of adults diagnosed with diabetes received antidiabetic medications.3
WEIGHT LOSS: IT'S IMPORTANT
This underuse of weight-loss drugs occurs despite our understanding that obesity is a risk factor for developing diabetes and that weight loss in obese patients reduces the risk.
The landmark Diabetes Prevention Program study found that even modest weight loss of 7% reduced the risk of developing diabetes by 58% in overweight and prediabetic individuals.4 Additionally, a 5% to 10% weight loss can lead to significant improvements in many comorbidities, including diabetes, hyperlipidemia, hypertension, sleep apnea, and fatty liver disease.
Antiobesity medications can help patients achieve weight-loss goals, especially if lifestyle and behavioral modifications alone have been unsuccessful. Data show that these drugs result in an average weight loss of 5% to 15% when added to diet and exercise.
BARRIERS TO PRESCRIBING WEIGHT-LOSS DRUGS
Why are practitioners reluctant to prescribe these drugs despite the worsening obesity epidemic and despite knowing that obesity is a risk factor for diabetes? Many of us who practice obesity medicine believe there are several reasons.
One barrier is the misconception that obesity does not warrant treatment with weight-loss medications, even though most practitioners will readily admit that patients cannot achieve effective, durable, and meaningful weight loss with behavioral changes and lifestyle modifications alone.
Other barriers stem from issues such as time constraints in the office, lack of training to treat this condition, and not enough data on the newer chronic weight-loss medications. And there are stringent requirements for patient follow-up once a medication has been initiated. Finally, it’s often difficult to obtain insurance coverage.
Addressing the barriers
Of these, I believe the biggest barrier for busy practitioners is finding the time and effort they need to devote to prescribing weight-loss medications. There are ways to address these issues.
Regarding time constraints, practitioners can discuss weight loss at follow-up visits and refer patients to obesity specialists. Regarding gaps in training and knowledge of obesity management, there are consensus guidelines for the identification, evaluation, and treatment of the overweight or obese individual.5–7 Guidelines provide extensive information on the pharmacologic treatment of obesity. These resources provide valuable evidence-based recommendations on how to manage this chronic disease.
ARMED WITH INFORMATION, PHARMACOLOGIC OPTIONS
Bersoux et al provide another valuable resource for clinical use of weight-loss drugs.1 They accurately review the available medications, their mechanisms of action, dosing, efficacy, side effect profiles, and clinical indications. Their review is comprehensive in every aspect of this drug class.
This is important information for practitioners to have when considering prescribing antiobesity medications. It is especially important for primary care practitioners because of the large number of obese or overweight patients they treat.
Drug options have expanded
We did not always have this many drugs to choose from. As Bersoux et al note, practitioners had limited options for weight-loss medications during the 1990s and early 2000s, and several of those had to be taken off the market because of serious side effects. Then between 2012 and 2014, the US Food and Drug Administration approved 4 new medications, giving us a total of 6 weight-loss drugs. Those approvals greatly increased the available drug treatments, giving us much-needed options beyond lifestyle and behavioral modifications.
Although it is widely accepted that antiobesity drugs are underused, the study by Thomas et al was the first to quantify the extent of underuse, especially for the newer chronic weight-loss drugs.2 Their data show that only about 19% of antiobesity prescriptions were for the newer drugs while 74% were for the older but short-term medication phentermine.
Bersoux et al seem to encourage primary care physicians, or anyone caring for overweight or obese patients, to consider prescribing these treatments if nonpharmacologic options are unsuccessful. I agree with this concept because there are not enough specialists to care for the more than 116 million individuals who are potential candidates for antiobesity medications.
THE TIME HAS COME
This new class of medications has been strongly endorsed by the most prestigious organizations and societies involved in developing treatment guidelines for the overweight or obese patient. It is time for everyone who sees overweight or obese patients in daily practice to consider adopting chronic weight-loss medications as adjunctive therapy if lifestyle and behavioral strategies are ineffective.
- Bersoux S, Byun TH, Chaliki SS, Poole KJ Jr. Pharmacotherapy for obesity: what you need to know. Cleve Clin J Med 2017; 84:951–958.
- Thomas CE, Mauer EA, Shukla AP, Rathi S, Aronne LJ. Low adoption of weight loss medications: a comparison of prescribing patterns of antiobesity pharmacotherapies and SGLT2s. Obesity 2016; 24:1955–1961.
- Samaranayake NR, Ong KL, Leung RY, Cheung BM. Management of obesity in the National Health and Nutrition Examination Survey (NHANES), 2007–2008. Ann Epidemiol 2012; 22:349–353.
- Knowler WC, Fowler SE, Hamman RF, et al; for the Diabetes Prevention Program Research Group. 10-year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet 2009; 374:1677–1686.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. J Am Coll Cardiol 2014; 63:2985–3023.
- American Association of Clinical Endocrinologists. AACE/ACE algorithm for the medical care of patients with obesity.
https://www.aace.com/files/final-appendix.pdf. Accessed September 20, 2017.
- Obesity Medicine Association. Obesity algorithm: 2016-2017.
https://obesitymedicine.org/obesity-algorithm/. Accessed October 3, 2017.
- Bersoux S, Byun TH, Chaliki SS, Poole KJ Jr. Pharmacotherapy for obesity: what you need to know. Cleve Clin J Med 2017; 84:951–958.
- Thomas CE, Mauer EA, Shukla AP, Rathi S, Aronne LJ. Low adoption of weight loss medications: a comparison of prescribing patterns of antiobesity pharmacotherapies and SGLT2s. Obesity 2016; 24:1955–1961.
- Samaranayake NR, Ong KL, Leung RY, Cheung BM. Management of obesity in the National Health and Nutrition Examination Survey (NHANES), 2007–2008. Ann Epidemiol 2012; 22:349–353.
- Knowler WC, Fowler SE, Hamman RF, et al; for the Diabetes Prevention Program Research Group. 10-year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet 2009; 374:1677–1686.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. J Am Coll Cardiol 2014; 63:2985–3023.
- American Association of Clinical Endocrinologists. AACE/ACE algorithm for the medical care of patients with obesity.
https://www.aace.com/files/final-appendix.pdf. Accessed September 20, 2017.
- Obesity Medicine Association. Obesity algorithm: 2016-2017.
https://obesitymedicine.org/obesity-algorithm/. Accessed October 3, 2017.

To have not and then to have: A challenging immune paradox

The successful interplay between the host defense system and infectious invaders depends on controlling the tissue damage that ensues from both the infection and the resultant inflammatory response. Even though an underactive immune system predisposes to unusual and potentially severe infections, an overly vigorous host response to infection can be as destructive as the infection itself. We can improve the outcome of some infections by introducing potent anti-inflammatory and immunosuppressive therapy concurrent with appropriate anti-infective therapy. What initially seemed counterintuitive has become the standard of care in the treatment of bacterial and mycobacterial meningitis and severe Pneumocystis and bacterial pneumonias, and favorable data are accruing in other infections such as bacterial arthritis.
A twist on the above scenario can occur when an immunosuppressed patient with a partially controlled indolent infection has his or her immune system suddenly normalized due to successful treatment of the underlying cause of their immunodeficiency. This treatment may be the introduction of successful antiretroviral therapy against human immunodeficiency virus (HIV), effective therapy of an immunosuppressing infection like tuberculosis, or withdrawal of an immunosuppressive anti-tumor necrosis factor (anti-TNF) drug. In this scenario, where the immune system is rapidly reconstituted and concurrently activated by the presence of persistent antigenic challenge or immunostimulatory molecules, a vigorous and clinically counterproductive inflammatory response may ensue, causing “collateral damage” to normal tissue. This immune reactivation syndrome may include fever, sweats, adenitis, and local tissue destruction at the site of infectious agents and associated phlogistic breakdown products. The result of this robust, tissue-injurious inflammatory response can be particularly devastating if it occurs in the brain or the retina, and may cause diagnostic confusion.
The trigger for this regional and systemic inflammatory response is multifactorial. It includes the newly recovered responsiveness to high levels of circulating cytokines, reaction to immune-stimulating fatty acids and other molecules released from dying mycobacteria (perhaps akin to the Jarisch-Herxheimer reaction to rapidly dying spirochetes), and possibly an over-vigorous “rebooting” immune system if an appropriate regulatory cell network is yet to be reconstituted.
In this issue of the Journal, Hara et al provide images from a patient appropriately treated for tuberculosis who experienced continued systemic symptoms of infection with the appearance of new pulmonary lesions. The trigger was the withdrawal of the infliximab (anti-TNF) therapy he was taking for ulcerative colitis, which at face value might be expected to facilitate the successful treatment of his tuberculosis. This seemingly paradoxical reaction has been well described with the successful treatment of HIV-infected patients coinfected with mycobacteria (tuberculous or nontuberculous), cytomegalovirus, and herpes-associated Kaposi sarcoma and zoster. But as in this instructive description of a patient with an immune reactivation syndrome, it also occurs in the setting of non-HIV reversibly immunosuppressed patients.1,2 The syndrome is often recognized 1 to 2 months after immune reconstitution and the initiation of anti-infective therapy.
The treatment of this paradoxical reaction is (not so paradoxically) the administration of corticosteroids or other immunosuppressive drugs. The efficacy of corticosteroids has been demonstrated in a small placebo-controlled trial3 as well as in clinical practice. The mechanism driving this reaction may not be the same for all infections, and thus steroids may not be ideal treatment for all patients. There are reports of using infliximab to temper the immune reactivation syndrome in some patients who did not respond to corticosteroids.
There is no definitive confirmatory test for immune reactivation syndrome. And certainly in the case of known mycobacterial infection, we must ensure the absence of drug resistance and that the appropriate antibiotics are being used, and that no additional infection is present and untreated by the antimycobacterial therapy. While lymphocytosis and an overly robust tuberculin skin test response have been described in patients with tuberculosis experiencing an immune reactivation syndrome, this “paradoxical reaction” remains a clinical diagnosis, worth considering in the appropriate setting.
- Carvalho AC, De Iaco G, Saleri N, et al. Paradoxical reaction during tuberculosis treatment in HIV-seronegative patients. Clin Infect Dis 2006; 42:893–895.
- Garcia Vidal C, Rodríguez Fernández S, Martínez Lacasa J, et al. Paradoxical response to antituberculous therapy in infliximab-treated patients with disseminated tuberculosis. Clin Infect Dis 2005; 40:756–759.
- Meintjes G, Wilkinson RJ, Morroni C, et al. Randomized placebo-controlled trial of prednisone for paradoxical TB-associated immune reconstitution inflammatory syndrome. AIDS (London, England) 2010; 24:2381–2390.
The successful interplay between the host defense system and infectious invaders depends on controlling the tissue damage that ensues from both the infection and the resultant inflammatory response. Even though an underactive immune system predisposes to unusual and potentially severe infections, an overly vigorous host response to infection can be as destructive as the infection itself. We can improve the outcome of some infections by introducing potent anti-inflammatory and immunosuppressive therapy concurrent with appropriate anti-infective therapy. What initially seemed counterintuitive has become the standard of care in the treatment of bacterial and mycobacterial meningitis and severe Pneumocystis and bacterial pneumonias, and favorable data are accruing in other infections such as bacterial arthritis.
A twist on the above scenario can occur when an immunosuppressed patient with a partially controlled indolent infection has his or her immune system suddenly normalized due to successful treatment of the underlying cause of their immunodeficiency. This treatment may be the introduction of successful antiretroviral therapy against human immunodeficiency virus (HIV), effective therapy of an immunosuppressing infection like tuberculosis, or withdrawal of an immunosuppressive anti-tumor necrosis factor (anti-TNF) drug. In this scenario, where the immune system is rapidly reconstituted and concurrently activated by the presence of persistent antigenic challenge or immunostimulatory molecules, a vigorous and clinically counterproductive inflammatory response may ensue, causing “collateral damage” to normal tissue. This immune reactivation syndrome may include fever, sweats, adenitis, and local tissue destruction at the site of infectious agents and associated phlogistic breakdown products. The result of this robust, tissue-injurious inflammatory response can be particularly devastating if it occurs in the brain or the retina, and may cause diagnostic confusion.
The trigger for this regional and systemic inflammatory response is multifactorial. It includes the newly recovered responsiveness to high levels of circulating cytokines, reaction to immune-stimulating fatty acids and other molecules released from dying mycobacteria (perhaps akin to the Jarisch-Herxheimer reaction to rapidly dying spirochetes), and possibly an over-vigorous “rebooting” immune system if an appropriate regulatory cell network is yet to be reconstituted.
In this issue of the Journal, Hara et al provide images from a patient appropriately treated for tuberculosis who experienced continued systemic symptoms of infection with the appearance of new pulmonary lesions. The trigger was the withdrawal of the infliximab (anti-TNF) therapy he was taking for ulcerative colitis, which at face value might be expected to facilitate the successful treatment of his tuberculosis. This seemingly paradoxical reaction has been well described with the successful treatment of HIV-infected patients coinfected with mycobacteria (tuberculous or nontuberculous), cytomegalovirus, and herpes-associated Kaposi sarcoma and zoster. But as in this instructive description of a patient with an immune reactivation syndrome, it also occurs in the setting of non-HIV reversibly immunosuppressed patients.1,2 The syndrome is often recognized 1 to 2 months after immune reconstitution and the initiation of anti-infective therapy.
The treatment of this paradoxical reaction is (not so paradoxically) the administration of corticosteroids or other immunosuppressive drugs. The efficacy of corticosteroids has been demonstrated in a small placebo-controlled trial3 as well as in clinical practice. The mechanism driving this reaction may not be the same for all infections, and thus steroids may not be ideal treatment for all patients. There are reports of using infliximab to temper the immune reactivation syndrome in some patients who did not respond to corticosteroids.
There is no definitive confirmatory test for immune reactivation syndrome. And certainly in the case of known mycobacterial infection, we must ensure the absence of drug resistance and that the appropriate antibiotics are being used, and that no additional infection is present and untreated by the antimycobacterial therapy. While lymphocytosis and an overly robust tuberculin skin test response have been described in patients with tuberculosis experiencing an immune reactivation syndrome, this “paradoxical reaction” remains a clinical diagnosis, worth considering in the appropriate setting.
The successful interplay between the host defense system and infectious invaders depends on controlling the tissue damage that ensues from both the infection and the resultant inflammatory response. Even though an underactive immune system predisposes to unusual and potentially severe infections, an overly vigorous host response to infection can be as destructive as the infection itself. We can improve the outcome of some infections by introducing potent anti-inflammatory and immunosuppressive therapy concurrent with appropriate anti-infective therapy. What initially seemed counterintuitive has become the standard of care in the treatment of bacterial and mycobacterial meningitis and severe Pneumocystis and bacterial pneumonias, and favorable data are accruing in other infections such as bacterial arthritis.
A twist on the above scenario can occur when an immunosuppressed patient with a partially controlled indolent infection has his or her immune system suddenly normalized due to successful treatment of the underlying cause of their immunodeficiency. This treatment may be the introduction of successful antiretroviral therapy against human immunodeficiency virus (HIV), effective therapy of an immunosuppressing infection like tuberculosis, or withdrawal of an immunosuppressive anti-tumor necrosis factor (anti-TNF) drug. In this scenario, where the immune system is rapidly reconstituted and concurrently activated by the presence of persistent antigenic challenge or immunostimulatory molecules, a vigorous and clinically counterproductive inflammatory response may ensue, causing “collateral damage” to normal tissue. This immune reactivation syndrome may include fever, sweats, adenitis, and local tissue destruction at the site of infectious agents and associated phlogistic breakdown products. The result of this robust, tissue-injurious inflammatory response can be particularly devastating if it occurs in the brain or the retina, and may cause diagnostic confusion.
The trigger for this regional and systemic inflammatory response is multifactorial. It includes the newly recovered responsiveness to high levels of circulating cytokines, reaction to immune-stimulating fatty acids and other molecules released from dying mycobacteria (perhaps akin to the Jarisch-Herxheimer reaction to rapidly dying spirochetes), and possibly an over-vigorous “rebooting” immune system if an appropriate regulatory cell network is yet to be reconstituted.
In this issue of the Journal, Hara et al provide images from a patient appropriately treated for tuberculosis who experienced continued systemic symptoms of infection with the appearance of new pulmonary lesions. The trigger was the withdrawal of the infliximab (anti-TNF) therapy he was taking for ulcerative colitis, which at face value might be expected to facilitate the successful treatment of his tuberculosis. This seemingly paradoxical reaction has been well described with the successful treatment of HIV-infected patients coinfected with mycobacteria (tuberculous or nontuberculous), cytomegalovirus, and herpes-associated Kaposi sarcoma and zoster. But as in this instructive description of a patient with an immune reactivation syndrome, it also occurs in the setting of non-HIV reversibly immunosuppressed patients.1,2 The syndrome is often recognized 1 to 2 months after immune reconstitution and the initiation of anti-infective therapy.
The treatment of this paradoxical reaction is (not so paradoxically) the administration of corticosteroids or other immunosuppressive drugs. The efficacy of corticosteroids has been demonstrated in a small placebo-controlled trial3 as well as in clinical practice. The mechanism driving this reaction may not be the same for all infections, and thus steroids may not be ideal treatment for all patients. There are reports of using infliximab to temper the immune reactivation syndrome in some patients who did not respond to corticosteroids.
There is no definitive confirmatory test for immune reactivation syndrome. And certainly in the case of known mycobacterial infection, we must ensure the absence of drug resistance and that the appropriate antibiotics are being used, and that no additional infection is present and untreated by the antimycobacterial therapy. While lymphocytosis and an overly robust tuberculin skin test response have been described in patients with tuberculosis experiencing an immune reactivation syndrome, this “paradoxical reaction” remains a clinical diagnosis, worth considering in the appropriate setting.
- Carvalho AC, De Iaco G, Saleri N, et al. Paradoxical reaction during tuberculosis treatment in HIV-seronegative patients. Clin Infect Dis 2006; 42:893–895.
- Garcia Vidal C, Rodríguez Fernández S, Martínez Lacasa J, et al. Paradoxical response to antituberculous therapy in infliximab-treated patients with disseminated tuberculosis. Clin Infect Dis 2005; 40:756–759.
- Meintjes G, Wilkinson RJ, Morroni C, et al. Randomized placebo-controlled trial of prednisone for paradoxical TB-associated immune reconstitution inflammatory syndrome. AIDS (London, England) 2010; 24:2381–2390.
- Carvalho AC, De Iaco G, Saleri N, et al. Paradoxical reaction during tuberculosis treatment in HIV-seronegative patients. Clin Infect Dis 2006; 42:893–895.
- Garcia Vidal C, Rodríguez Fernández S, Martínez Lacasa J, et al. Paradoxical response to antituberculous therapy in infliximab-treated patients with disseminated tuberculosis. Clin Infect Dis 2005; 40:756–759.
- Meintjes G, Wilkinson RJ, Morroni C, et al. Randomized placebo-controlled trial of prednisone for paradoxical TB-associated immune reconstitution inflammatory syndrome. AIDS (London, England) 2010; 24:2381–2390.

Pharmacotherapy for obesity: What you need to know
Weight-loss drugs are not magic pills, but they can help patients lose about 10 to 25 more pounds than they otherwise could, when used in a program that includes diet, exercise, and other lifestyle changes.

HALF OF ADULTS MAY BE OBESE BY 2030
Obesity is a major public health challenge in the United States, with nearly 37% of adults classified as obese.1 The prevalence has increased more than 75% since 1980,2 and it is estimated that 51% of US adults will be obese by 2030.3 Obesity is the second-leading cause of preventable deaths, after smoking.4
Obesity increases the risk of many chronic medical conditions, including type 2 diabetes mellitus, heart disease, hypertension, stroke, nonalcoholic fatty liver disease, osteoarthritis, and cancers of the breast, colon, endometrium, and kidney.5
WHEN IS DRUG THERAPY INDICATED?
Guidelines from the major obesity societies recommend that all weight-loss programs have a lifestyle component that includes a low-calorie diet, increased physical activity, and behavioral therapy, to which pharmacotherapy may be added as an adjunct.6–8
Weight-loss medications are indicated for patients who have a body mass index (BMI) of at least 30 kg/m2 or who have obesity-associated comorbidities and a BMI of at least 27 kg/m2. However, the best results are achieved when pharmacotherapy is combined with lifestyle modification.9

HISTORY OF WEIGHT-LOSS DRUGS: NOT A PRETTY PICTURE
The earliest drugs to induce weight loss, which worked mainly by increasing metabolism, included thyroid hormone, amphetamines (which also suppress appetite), and dinitrophenol (a pesticide). Adverse reactions limited their usefulness: cardiovascular effects with thyroid hormones, abuse potential with amphetamines, and neuropathy and cataracts with dinitrophenol.
Researchers then looked to drugs that could suppress appetite like amphetamines do, but without the potential for abuse. Medications that increased levels of norepinephrine and serotonin, both by increasing release and decreasing reuptake of these neuromodulators, had some success. But again, serious adverse effects occurred, and several drugs had to be withdrawn from the market.
The most publicized of these withdrawals was for the combination fenfluramine and phentermine (“fen-phen”) and its cousin dexfenfluramine (Redux). Up to 30% of patients taking fenfluramine-phentermine developed echocardiographic evidence of valvular heart disease.11 Fenfluramine also increased the risk of pulmonary hypertension. These findings led to the 1997 withdrawal of these drugs from the US market.
Sibutramine (Meridia), a norepinephrine and serotonin reuptake inhibitor, was approved for weight loss in 1997. Increases in blood pressure and heart rate were noted in the initial trial,12 and then a postmarketing study found increased rates of nonfatal myocardial infarction and stroke in patients with preexisting cardiovascular disease or diabetes mellitus.13 Based on these results, sibutramine was withdrawn from both US and European markets.
Rimonabant (Acomplia, Zimulti), a cannabinoid-receptor inhibitor, was approved in Europe in 2006, but its approval was withdrawn just 2 years later because of increased suicidality in a postmarketing study.14 It was never approved for use in the United States.
NORADRENERGIC SYMPATHOMIMETICS: FOR SHORT-TERM USE
Several noradrenergic sympathomimetic drugs are FDA-approved for short-term weight loss, but phentermine is by far the most commonly prescribed drug in this class. In fact, it is the most commonly prescribed drug for obesity in the United States.15
Phentermine
Phentermine is an atypical amphetamine analogue that suppresses appetite by norepinephrine agonism in the central nervous system. The FDA approved it for short-term weight management in 1959, and its use became widespread in the 1960s, followed by decades of popularity.
Dosage. Phentermine is prescribed at an oral dose of 15, 30, or 37.5 mg daily, either before breakfast or 1 to 2 hours after. It is a schedule IV controlled substance, based on its similarity to amphetamine. (The 5 US controlled substance schedules range from schedule I, which includes heroin, amphetamine, and cannabis, to schedule V, which includes cough syrups containing no more than 200 mg of codeine per 100 mL.) However, concerns about addiction and dependence with phentermine are largely unfounded, and abrupt cessation of the drug has not been shown to cause amphetamine-like withdrawal.16
Adverse effects. Common adverse reactions include nervousness, insomnia, and dry mouth, but these effects tend to wane with continued use.
Contraindications. Cardiovascular disease is a contraindication to phentermine because of concerns about increased blood pressure and pulse rate, although these concerns seem to be more theoretic than observed.16 Other contraindications include hyperthyroidism, glaucoma, agitation, a history of drug abuse, pregnancy, breastfeeding, and current or recent use of a monoamine oxidase inhibitor. No serious adverse events have been reported in trials of phentermine.
Efficacy. In a pooled analysis of 6 trials lasting 2 to 24 weeks completed between 1975 and 1999, phentermine-treated patients lost an average of 3.6 kg more weight than placebo recipients.17 More than 80% of study participants were women.
In a 36-week study in 108 women,18 participants lost a mean of 12.2 kg with continuous phentermine use, 13.0 kg with intermittent use (4 weeks on, 4 weeks off; the difference was not significant), and 4.8 kg with placebo.
Minimal data exist on long-term efficacy of phentermine monotherapy.
DRUGS FOR LONG-TERM THERAPY
Orlistat
Orlistat was approved as a prescription drug (Xenical, 120 mg) in 1999 and as an over-the-counter medication (Alli, 60 mg) in 2007.
Orlistat works by inhibiting pancreatic and gastric lipase, causing incomplete hydrolysis of ingested fat, thereby increasing fecal fat excretion in a dose-dependent manner. It is a good choice for weight-loss drug therapy because of its safe cardiovascular risk profile and beneficial effects on lipid levels. However, its long-term effect on weight is only modest.19,20
Dosage. The dosage for prescription orlistat is 120 mg 3 times per day, in addition to a low-fat diet (< 30% of daily calories from fat). To prevent potential deficiencies of fat-soluble vitamins, a daily multivitamin supplement is recommended, but it should not be taken with meals.
Efficacy. In a 2014 systematic review, 35% to 73% of patients treated with orlistat 120 mg had lost at least 5% of their body weight at 1 year, and 14% to 41% had lost at least 10%.21 At the end of the second year, orlistat-treated patients had lost about 3.3 kg more than placebo recipients.
In a randomized trial,22 4 years of treatment with orlistat vs placebo led to a significant (37.3%) risk reduction in the incidence of type 2 diabetes mellitus in obese participants, as well as significant improvements in cardiovascular risk factors. Mean weight loss at 1 year was significantly greater with orlistat than with placebo (10.6 vs 6.2 kg), and it remained greater at 4 years (5.8 vs 3.0 kg; P < .001).
Adverse effects. Long-term orlistat use is hampered by adverse reactions. A population-based, retrospective cohort analysis showed that fewer than 10% of patients were still using it at 1 year, and only 2% were using it at 2 years, although reasons for discontinuation were not reported.23
Adverse reactions are predominantly gastrointestinal, attributed to the high content of undigested fat in stools. Patients who do not limit their dietary fat intake are affected the most. Other reported adverse reactions include hepatotoxicity and oxalate-induced nephropathy.
Orlistat has been reported to interfere with some drugs, particularly those that are lipophilic. Drugs that should be closely monitored with orlistat are warfarin, amiodarone, cyclosporine, certain antiepileptic drugs, and levothyroxine.
Phentermine-topiramate
The combination of phentermine and topiramate was approved by the FDA in 2012 and is available under the brand name Qsymia.
Topiramate had been approved for treating seizure disorder in 1996 and as migraine prophylaxis in 2004. It is not approved as monotherapy for obesity; however, patients taking it for seizures or for psychiatric disorders (eg, binge eating, borderline personality disorder) have reported weight loss during treatment.
How topiramate promotes weight loss is not known. Proposed mechanisms include taste inhibition by carbonic anhydrase, influences on gamma-aminobutyric acid transmission causing appetite suppression, sensitization of insulin activity, and adiponectin secretion in the peripheral tissues.24,25
Phentermine-topiramate therapy has an advantage over monotherapy because lower doses of each medication can be used to achieve the same benefit, thus avoiding dose-related adverse reactions.
Dosage. Phentermine-topiramate is available in capsules containing 3.75/23, 7.5/46, 11.25/69, and 15/92 mg. The recommended starting dosage is 3.75/23 mg/day for 14 days, increasing to 7.5/46 mg/day. If patients do not lose at least 3% of their body weight after 12 weeks, the dose can be increased to 11.25/69 mg daily for 14 days, followed by 15/92 mg daily.26 Phentermine-topiramate is a schedule IV controlled substance with a low potential for abuse and dependence.
Efficacy. Approval of phentermine-topiramate for treating obesity was primarily based on 3 clinical trials.27–29 In 1 of these trials,28 at 1 year, patients had lost 9.9 kg with the medium dose and 12.9 kg with the high dose.
Adverse effects. Phentermine-topiramate was well tolerated in the trials. The most commonly reported adverse reactions were dry mouth, dizziness, constipation, insomnia, dysgeusia, paresthesia, and increased resting heart rate.28,29 Acute myopia and angle-closure glaucoma also have been reported with topiramate.30 Topiramate monotherapy has been associated with dose-dependent neuropsychiatric adverse effects, including memory symptoms and depression. However, across all 3 trials of phentermine-topiramate therapy, symptoms of depression improved over time, and no significant increase in suicide risk was identified.27–29
Recommended monitoring for patients on phentermine-topiramate includes a blood chemistry panel, resting heart rate, blood pressure, and depression screening.
Because topiramate has teratogenic potential (craniofacial abnormalities), it is labeled as pregnancy category X (contraindicated). A negative pregnancy test is needed before women of childbearing age take the drug and monthly thereafter. Women should be counseled to use effective birth control. A home pregnancy test is an alternative to laboratory testing, but this option should be left to the prescribing clinician’s judgment and be based on reliability of the test and patient compliance.
Lorcaserin
Lorcaserin (Belviq) was approved by the FDA in 2012 for chronic weight management. It suppresses appetite by activating the serotonin 2C receptor in the brain. Because it is selective for the 2C receptor, it does not appear to have the same detrimental effects on heart valves as occurred with less-selective serotonergic agents such as fenfluramine and dexfenfluramine.31
Dosage. The recommended dosage for lorcaserin is 10 mg twice daily. Lorcaserin is a schedule IV controlled substance because of studies that showed increases in positive subjective measures such as euphoria in patients taking the drug. The incidence of euphoria was similar to that seen with zolpidem.32
Efficacy. Lorcaserin was approved on the basis of 2 trials in nondiabetic obese and overweight adults who did not have diabetes but who had a weight-related condition,33,34 and in a third trial in obese and overweight adults with type 2 diabetes mellitus who were taking oral hypoglycemic agents.35 In these trials, lorcaserin use resulted in a modest 4.7- to 5.8-kg weight loss compared with 1.6 to 2.2 kg in the placebo group.33–35 There was a high dropout rate in all 3 of these studies (33% to 45% of participants).
A pilot study that added phentermine to lorcaserin yielded double the weight loss from lorcaserin alone.36 This drug combination warrants further investigation.
Contraindications. Lorcaserin should not be given to patients who have severe renal insufficiency (creatinine clearance < 30 mL/min) or severe hepatic impairment, or who are pregnant.
Adverse effects. Common adverse reactions include dry mouth, dizziness, somnolence, headache, and gastrointestinal disturbances (nausea, constipation, or diarrhea).37
Patients with type 2 diabetes mellitus should be monitored for hypoglycemia.
Lorcaserin should be used with extreme caution in patients taking other serotonergic agents because of the risk of the serotonin syndrome.
A theoretic potential for increased risk of breast cancer also exists with lorcaserin. When rats were given supraphysiologic doses of lorcaserin (more than 50 times higher than recommended in humans), fibroadenomas and adenocarcinomas occurred at higher rates.38 Breast cancer data were not reported in the 3 randomized trials discussed above.33–35
Naltrexone-bupropion
The combination of naltrexone and bupropion was approved by the FDA in 2014 under the brand name Contrave. Both drugs are approved for monotherapy in conditions other than obesity.
Naltrexone is a mu opioid receptor antagonist approved to treat alcohol and opioid dependency. Bupropion is a dopamine-norepinephrine reuptake inhibitor approved to treat depression and to help with smoking cessation. Combining the drugs produces weight loss and metabolic benefits through effects on 2 areas of the brain that regulate food intake: the hypothalamus (appetite) and the mesolimbic dopamine circuit (reward system).
Dosage. Naltrexone-bupropion comes as an extended-release tablet of 8/90 mg. The maintenance dose of 2 tablets twice daily is reached at week 4 through a specific dose-titration regimen (Table 1). The dose should be adjusted if patients have renal or hepatic impairment or if they are also taking a CYP2B6 inhibitor.
Efficacy. FDA approval was based on the results of 4 clinical trials.39–42 Using a modified intention-to-treat analysis, Yanovski and Yanovski43 calculated that at 1 year, placebo-subtracted mean weight loss was 4.6% (4.9 kg), and mean total weight loss was 6.8% (7.3 kg) across the studies. Attrition rates, however, were high, ranging from 42% to 50%.
Cardiometabolic effects in 2 of the trials40,41 included decreased waist circumference, triglyceride levels, and C-reactive protein levels, and increased high-density lipoprotein levels at the initial dose. At the maintenance dose, additional lowering of fasting plasma insulin and glucose levels occurred along with lower levels of the homeostatic model assessment of insulin resistance. In the COR-Diabetes Study Group trial, patients with type 2 diabetes mellitus had decreased hemoglobin A1c levels without an increase in hypoglycemia and an increased likelihood of reaching the target hemoglobin A1c level below 7%.39
Contraindications. Naltrexone-bupropion is contraindicated for patients who have uncontrolled hypertension, seizure disorder, eating disorder, or end-stage renal failure; who are pregnant; or who have been treated with a monoamine oxidase inhibitor within 14 days. It should not be used with other bupropion-containing products or in patients who have taken opioids chronically or have acute opiate withdrawal.
Because of its bupropion component, this product carries an FDA black-box warning about possible suicidal thoughts and behaviors and neuropsychiatric reactions.
Adverse effects. The adverse reactions most commonly associated with naltrexone-bupropion were nausea (32.5%), constipation (19.2%), headache (17.6%), vomiting (10.7%), dizziness (9.9%), insomnia (9.2%), dry mouth (8.1%), and diarrhea (7.1%).44
Liraglutide
Liraglutide, previously FDA-approved to treat type 2 diabetes mellitus under the brand name Victoza, received approval in 2014 in a higher-dose formulation (Saxenda) to treat obesity.
Liraglutide is a glucagon-like peptide-1 receptor agonist that stimulates glucose-dependent insulin release from the pancreatic islet cells, slows gastric emptying, regulates postprandial glucagon, and reduces food intake.
Dosage. Liraglutide is given as a once-daily injection in the abdomen, thigh, or arm. The initial dosage is 0.6 mg daily for the first week and can be titrated up by 0.6 mg weekly to a target dose of 3 mg daily. If a patient does not lose 4% of baseline body weight after 16 weeks on the target dose, the drug should be discontinued because it is unlikely to lead to clinically significant weight loss.
Efficacy. Liraglutide for weight management (3 mg once daily) was evaluated in a large (N = 3,731), randomized, double-blind, placebo-controlled international trial.45 Participants did not have diabetes mellitus, but 60% had prediabetes. Liraglutide or placebo was given for 56 weeks, along with lifestyle counseling. At the end of the study, the liraglutide group had lost a mean of 8.4 kg vs 2.8 kg in the placebo group. Additionally, 63% of the liraglutide group lost at least 5% of body weight vs 27% in the placebo group, and 33% lost at least 10% of body weight vs 10% in the placebo group.
A 2-year extension found systolic blood pressure decreased with no change in pulse, and the prevalence of prediabetes and metabolic syndrome decreased by 52% and 59%, respectively.46 At 2 years, mean scores for physical function, self-esteem, and work had improved more in the liraglutide group than the placebo group.47
Adverse effects. The most common adverse reactions with liraglutide were nausea, vomiting, diarrhea, constipation, hypoglycemia, and loss of appetite. In most cases, nausea and vomiting were tolerable, transient, and associated with greater weight loss but not with decreased quality-of-life scores. Serious adverse reactions included pancreatitis, gallbladder disease, renal impairment, and suicidal thoughts.
CHOOSING A DRUG
For obese patients, when lifestyle modifications do not result in the desired weight loss, pharmacotherapy is an option. Practitioners have several FDA-approved options for weight management. Because of evidence that these drugs can postpone the onset of other complications and improve metabolic and cardiovascular parameters, they should be considered.
In phase 3 trials, these drugs caused modest weight loss of 5% to 10% of body weight. More weight was lost with the combination of phentermine-topiramate than with the other drugs.
In a 2016 meta-analysis, these drugs were associated with at least 5% weight reduction compared with placebo.48 Phentermine-topiramate and liraglutide were most likely to produce at least a 5% weight loss, while liraglutide and naltrexone-bupropion were most likely to be discontinued because of adverse events. Combination drugs may have the advantages of synergistic effects on weight loss and fewer adverse reactions because lower doses of the individual drug components are used.
Response to therapy with most of these drugs should be evaluated at 12 weeks on the maintenance dose. If less than 5% weight loss has been achieved, the medication should be discontinued.
Adverse-effect profiles, drug interactions, abuse, misuse, and overdose potential should be considered when prescribing these drugs. Weight-loss drugs are contraindicated in pregnancy because they offer no potential benefit to a pregnant woman and may harm the fetus.
The development of new drugs and better drug combinations is expected to provide more effective therapeutic strategies, which are essential for combating the obesity epidemic.
- Ogden CL, Carroll MD, Fryar CD, Flegal KM. Prevalence of obesity among adults and youth: United States, 2011-2014. NCHS Data Brief 2015; 219:1–8.
- Yanovski SZ, Yanovski JA. Obesity. N Engl J Med 2002; 346:591–602.
- Finkelstein EA, Khavjou OA, Thompson H, et al. Obesity and severe obesity forecasts through 2030. Am J Prev Med 2012; 42:563–570.
- Hill JO, Wyatt H. Outpatient management of obesity: a primary care perspective. Obes Res 2002; 10(suppl 2):124S–130S.
- US Department of Health and Human Services. National Institute of Diabetes and Digestive and Kidney Diseases. Overweight and obesity statistics. www.niddk.nih.gov/health-information/health-statistics/Pages/overweight-obesity-statistics.aspx#overweight. Accessed October 10, 2017.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Obesity Society. J Am Coll Cardiol 2014; 63:2985–3023.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Obesity Society. Circulation 2014; 129(suppl 2):S102–S138.
- American Association of Clinical Endocrinologists. AACE/ACE algorithm for the medical care of patients with obesity. www.aace.com/files/guidelines/ObesityAlgorithm.pdf. Accessed July 25, 2017.
- Wadden TA, Berkowitz RI, Womble LG, et al. Randomized trial of lifestyle modification and pharmacotherapy for obesity. N Engl J Med 2005; 353:2111–2120.
- Mechanick JI, Youdim A, Jones DB, et al. Clinical practice guidelines for the perioperative nutritional, metabolic, and nonsurgical support of the bariatric surgery patient, 2013 update: cosponsored by American Association of Clinical Endocrinologists, the Obesity Society, and American Society for Metabolic and Bariatric Surgery. Surg Obes Relat Dis 2013; 9:159–191.
- Connolly HM, Crary JL, McGoon MD, et al. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med 1997; 337:581–588.
- Kim SH, Lee YM, Jee SH, et al. Effect of sibutramine on weight loss and blood pressure: a meta-analysis of controlled trials. Obes Res 2003; 11:1116–1123.
- James WP, Caterson ID, Coutinho W, et al; SCOUT Investigators. Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. N Engl J Med 2010; 363:905–917.
- Nissen SE, Nicholls SJ, Wolski K, et al; STRADIVARIUS Investigators. Effect of rimonabant on progression of atherosclerosis in patients with abdominal obesity and coronary artery disease: the STRADIVARIUS randomized controlled trial. JAMA 2008; 299:1547–1560.
- Ryan DH, Bray GA. Pharmacologic treatment options for obesity: what is old is new again. Curr Hypertens Rep 2013; 15:182–189.
- Hendricks EJ, Greenway FL, Westman EC, Gupta AK. Blood pressure and heart rate effects, weight loss and maintenance during long-term phentermine pharmacotherapy for obesity. Obesity (Silver Spring) 2011; 19:2351–2360.
- Li Z, Maglione M, Tu W, et al. Meta-analysis: pharmacologic treatment of obesity. Ann Intern Med 2005; 142:532–546.
- Munro JF, MacCuish AC, Wilson EM, Duncan LJ. Comparison of continuous and intermittent anorectic therapy in obesity. Br Med J 1968; 1:352–354.
- Hauptman J, Lucas C, Boldrin MN, Collins H, Segal KR. Orlistat in the long-term treatment of obesity in primary care settings. Arch Fam Med 2000; 9:160–167.
- Rossner S, Sjostrom L, Noack R, Meinders AE, Noseda G. Weight loss, weight maintenance, and improved cardiovascular risk factors after 2 years treatment with orlistat for obesity. European Orlistat Obesity Study Group. Obes Res 2000; 8:49–61.
- Yanovski SZ, Yanovski JA. Long-term drug treatment for obesity: a systematic and clinical review. JAMA 2014; 311:74–86.
- Torgerson JS, Hauptman J, Boldrin MN, Sjostrom L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 2004; 27:155–161.
- Padwal R, Kezouh A, Levine M, Etminan M. Long-term persistence with orlistat and sibutramine in a population-based cohort. Int J Obes (Lond) 2007; 31:1567–1570.
- Xiong GL, Gadde KM. Combination phentermine-topiramate for obesity treatment in primary care: a review. Postgrad Med 2014; 126:110–116.
- Pucci A, Finer N. New medications for treatment of obesity: metabolic and cardiovascular effects. Can J Cardiol 2015; 31:142–152.
- Smith SM, Meyer M, Trinkley KE. Phentermine-topiramate for the treatment of obesity. Ann Pharmacother 2013; 47:340–349.
- Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine-topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring) 2012; 20:330–342.
- Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet 2011; 377:1341–1352.
- Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine-topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr 2012; 95:297–308.
- Richa S, Yazbek JC. Ocular adverse effects of common psychotropic agents: a review. CNS Drugs 2010; 24:501–526.
- Weissman NJ, Sanchez M, Koch GG, Smith SR, Shanahan WR, Anderson CM. Echocardiographic assessment of cardiac valvular regurgitation with lorcaserin from analysis of 3 phase 3 clinical trials. Circ Cardiovasc Imaging 2013; 6:560–567.
- US Department of Justice Drug Enforcement Administration. Schedules of controlled substances: placement of lorcaserin into Schedule IV. Federal Register 2013; 78:26701–26705.
- Smith SR, Weissman NJ, Anderson CM, et al; Behavioral Modification and Lorcaserin for Overweight and Obesity Management (BLOOM) Study Group. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med 2010; 363:245–256.
- Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab 2011; 96:3067–3077.
- O’Neil PM, Smith SR, Weissman NJ, et al. Randomized placebo-controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM study. Obesity (Silver Spring) 2012; 20:1426–1436.
- Kumar RB, Aronne LJ. Efficacy comparison of medications approved for chronic weight management. Obesity (Silver Spring) 2015; 23(suppl 1):S4–S7.
- Chan EW, He Y, Chui CS, Wong AY, Lau WC, Wong IC. Efficacy and safety of lorcaserin in obese adults: a meta-analysis of 1-year randomized controlled trials (RCTs) and narrative review on short-term RCTs. Obes Rev 2013; 14:383–392.
- Miller LE. Lorcaserin for weight loss: insights into US Food and Drug Administration approval. J Acad Nutr Diet 2013; 113:25–30.
- Hollander P, Gupta AK, Plodkowski R, et al; COR-Diabetes Study Group. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care 2013; 36:4022–4029.
- Apovian CM, Aronne L, Rubino D, et al; COR-II Study Group. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Spring) 2013; 21:935–943.
- Greenway FL, Fujioka K, Plodkowski RA, et al; COR-I Study Group. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2010; 376:595–605.
- Wadden TA, Foreyt JP, Foster GD, et al. Weight loss with naltrexone SR/bupropion SR combination therapy as an adjunct to behavior modification: the COR-BMOD trial. Obesity (Silver Spring) 2011; 19:110–120.
- Yanovski SZ, Yanovski JA. Naltrexone extended-release plus bupropion extended-release for treatment of obesity. JAMA 2015; 313:1213–1214.
- Contrave (naltrexone HC1 and bupropion HC1) extended release tablets [package insert]. Orexigen Therapeutics, 2017. https://contrave.com/wp-content/uploads/2017/05/Contrave_PI.pdf. Accessed November 7, 2017.
- Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med 2015; 373:11–22.
- Astrup A, Carraro R, Finer N, et al; NN8022-1807 Investigators. Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int J Obes (Lond) 2012; 36:843–854.
- Lean ME, Carraro R, Finer N, et al; NN8022-1807 Investigators. Tolerability of nausea and vomiting and associations with weight loss in a randomized trial of liraglutide in obese, non-diabetic adults. Int J Obes (Lond) 2014; 38:689–697.
- Khera R, Murad MH, Chandar AK, et al. Association of pharmacological treatments for obesity with weight loss and adverse events: a systematic review and meta-analysis. JAMA 2016; 315:2424–2434.
Weight-loss drugs are not magic pills, but they can help patients lose about 10 to 25 more pounds than they otherwise could, when used in a program that includes diet, exercise, and other lifestyle changes.
HALF OF ADULTS MAY BE OBESE BY 2030
Obesity is a major public health challenge in the United States, with nearly 37% of adults classified as obese.1 The prevalence has increased more than 75% since 1980,2 and it is estimated that 51% of US adults will be obese by 2030.3 Obesity is the second-leading cause of preventable deaths, after smoking.4
Obesity increases the risk of many chronic medical conditions, including type 2 diabetes mellitus, heart disease, hypertension, stroke, nonalcoholic fatty liver disease, osteoarthritis, and cancers of the breast, colon, endometrium, and kidney.5
WHEN IS DRUG THERAPY INDICATED?
Guidelines from the major obesity societies recommend that all weight-loss programs have a lifestyle component that includes a low-calorie diet, increased physical activity, and behavioral therapy, to which pharmacotherapy may be added as an adjunct.6–8
Weight-loss medications are indicated for patients who have a body mass index (BMI) of at least 30 kg/m2 or who have obesity-associated comorbidities and a BMI of at least 27 kg/m2. However, the best results are achieved when pharmacotherapy is combined with lifestyle modification.9
HISTORY OF WEIGHT-LOSS DRUGS: NOT A PRETTY PICTURE
The earliest drugs to induce weight loss, which worked mainly by increasing metabolism, included thyroid hormone, amphetamines (which also suppress appetite), and dinitrophenol (a pesticide). Adverse reactions limited their usefulness: cardiovascular effects with thyroid hormones, abuse potential with amphetamines, and neuropathy and cataracts with dinitrophenol.
Researchers then looked to drugs that could suppress appetite like amphetamines do, but without the potential for abuse. Medications that increased levels of norepinephrine and serotonin, both by increasing release and decreasing reuptake of these neuromodulators, had some success. But again, serious adverse effects occurred, and several drugs had to be withdrawn from the market.
The most publicized of these withdrawals was for the combination fenfluramine and phentermine (“fen-phen”) and its cousin dexfenfluramine (Redux). Up to 30% of patients taking fenfluramine-phentermine developed echocardiographic evidence of valvular heart disease.11 Fenfluramine also increased the risk of pulmonary hypertension. These findings led to the 1997 withdrawal of these drugs from the US market.
Sibutramine (Meridia), a norepinephrine and serotonin reuptake inhibitor, was approved for weight loss in 1997. Increases in blood pressure and heart rate were noted in the initial trial,12 and then a postmarketing study found increased rates of nonfatal myocardial infarction and stroke in patients with preexisting cardiovascular disease or diabetes mellitus.13 Based on these results, sibutramine was withdrawn from both US and European markets.
Rimonabant (Acomplia, Zimulti), a cannabinoid-receptor inhibitor, was approved in Europe in 2006, but its approval was withdrawn just 2 years later because of increased suicidality in a postmarketing study.14 It was never approved for use in the United States.
NORADRENERGIC SYMPATHOMIMETICS: FOR SHORT-TERM USE
Several noradrenergic sympathomimetic drugs are FDA-approved for short-term weight loss, but phentermine is by far the most commonly prescribed drug in this class. In fact, it is the most commonly prescribed drug for obesity in the United States.15
Phentermine
Phentermine is an atypical amphetamine analogue that suppresses appetite by norepinephrine agonism in the central nervous system. The FDA approved it for short-term weight management in 1959, and its use became widespread in the 1960s, followed by decades of popularity.
Dosage. Phentermine is prescribed at an oral dose of 15, 30, or 37.5 mg daily, either before breakfast or 1 to 2 hours after. It is a schedule IV controlled substance, based on its similarity to amphetamine. (The 5 US controlled substance schedules range from schedule I, which includes heroin, amphetamine, and cannabis, to schedule V, which includes cough syrups containing no more than 200 mg of codeine per 100 mL.) However, concerns about addiction and dependence with phentermine are largely unfounded, and abrupt cessation of the drug has not been shown to cause amphetamine-like withdrawal.16
Adverse effects. Common adverse reactions include nervousness, insomnia, and dry mouth, but these effects tend to wane with continued use.
Contraindications. Cardiovascular disease is a contraindication to phentermine because of concerns about increased blood pressure and pulse rate, although these concerns seem to be more theoretic than observed.16 Other contraindications include hyperthyroidism, glaucoma, agitation, a history of drug abuse, pregnancy, breastfeeding, and current or recent use of a monoamine oxidase inhibitor. No serious adverse events have been reported in trials of phentermine.
Efficacy. In a pooled analysis of 6 trials lasting 2 to 24 weeks completed between 1975 and 1999, phentermine-treated patients lost an average of 3.6 kg more weight than placebo recipients.17 More than 80% of study participants were women.
In a 36-week study in 108 women,18 participants lost a mean of 12.2 kg with continuous phentermine use, 13.0 kg with intermittent use (4 weeks on, 4 weeks off; the difference was not significant), and 4.8 kg with placebo.
Minimal data exist on long-term efficacy of phentermine monotherapy.
DRUGS FOR LONG-TERM THERAPY
Orlistat
Orlistat was approved as a prescription drug (Xenical, 120 mg) in 1999 and as an over-the-counter medication (Alli, 60 mg) in 2007.
Orlistat works by inhibiting pancreatic and gastric lipase, causing incomplete hydrolysis of ingested fat, thereby increasing fecal fat excretion in a dose-dependent manner. It is a good choice for weight-loss drug therapy because of its safe cardiovascular risk profile and beneficial effects on lipid levels. However, its long-term effect on weight is only modest.19,20
Dosage. The dosage for prescription orlistat is 120 mg 3 times per day, in addition to a low-fat diet (< 30% of daily calories from fat). To prevent potential deficiencies of fat-soluble vitamins, a daily multivitamin supplement is recommended, but it should not be taken with meals.
Efficacy. In a 2014 systematic review, 35% to 73% of patients treated with orlistat 120 mg had lost at least 5% of their body weight at 1 year, and 14% to 41% had lost at least 10%.21 At the end of the second year, orlistat-treated patients had lost about 3.3 kg more than placebo recipients.
In a randomized trial,22 4 years of treatment with orlistat vs placebo led to a significant (37.3%) risk reduction in the incidence of type 2 diabetes mellitus in obese participants, as well as significant improvements in cardiovascular risk factors. Mean weight loss at 1 year was significantly greater with orlistat than with placebo (10.6 vs 6.2 kg), and it remained greater at 4 years (5.8 vs 3.0 kg; P < .001).
Adverse effects. Long-term orlistat use is hampered by adverse reactions. A population-based, retrospective cohort analysis showed that fewer than 10% of patients were still using it at 1 year, and only 2% were using it at 2 years, although reasons for discontinuation were not reported.23
Adverse reactions are predominantly gastrointestinal, attributed to the high content of undigested fat in stools. Patients who do not limit their dietary fat intake are affected the most. Other reported adverse reactions include hepatotoxicity and oxalate-induced nephropathy.
Orlistat has been reported to interfere with some drugs, particularly those that are lipophilic. Drugs that should be closely monitored with orlistat are warfarin, amiodarone, cyclosporine, certain antiepileptic drugs, and levothyroxine.
Phentermine-topiramate
The combination of phentermine and topiramate was approved by the FDA in 2012 and is available under the brand name Qsymia.
Topiramate had been approved for treating seizure disorder in 1996 and as migraine prophylaxis in 2004. It is not approved as monotherapy for obesity; however, patients taking it for seizures or for psychiatric disorders (eg, binge eating, borderline personality disorder) have reported weight loss during treatment.
How topiramate promotes weight loss is not known. Proposed mechanisms include taste inhibition by carbonic anhydrase, influences on gamma-aminobutyric acid transmission causing appetite suppression, sensitization of insulin activity, and adiponectin secretion in the peripheral tissues.24,25
Phentermine-topiramate therapy has an advantage over monotherapy because lower doses of each medication can be used to achieve the same benefit, thus avoiding dose-related adverse reactions.
Dosage. Phentermine-topiramate is available in capsules containing 3.75/23, 7.5/46, 11.25/69, and 15/92 mg. The recommended starting dosage is 3.75/23 mg/day for 14 days, increasing to 7.5/46 mg/day. If patients do not lose at least 3% of their body weight after 12 weeks, the dose can be increased to 11.25/69 mg daily for 14 days, followed by 15/92 mg daily.26 Phentermine-topiramate is a schedule IV controlled substance with a low potential for abuse and dependence.
Efficacy. Approval of phentermine-topiramate for treating obesity was primarily based on 3 clinical trials.27–29 In 1 of these trials,28 at 1 year, patients had lost 9.9 kg with the medium dose and 12.9 kg with the high dose.
Adverse effects. Phentermine-topiramate was well tolerated in the trials. The most commonly reported adverse reactions were dry mouth, dizziness, constipation, insomnia, dysgeusia, paresthesia, and increased resting heart rate.28,29 Acute myopia and angle-closure glaucoma also have been reported with topiramate.30 Topiramate monotherapy has been associated with dose-dependent neuropsychiatric adverse effects, including memory symptoms and depression. However, across all 3 trials of phentermine-topiramate therapy, symptoms of depression improved over time, and no significant increase in suicide risk was identified.27–29
Recommended monitoring for patients on phentermine-topiramate includes a blood chemistry panel, resting heart rate, blood pressure, and depression screening.
Because topiramate has teratogenic potential (craniofacial abnormalities), it is labeled as pregnancy category X (contraindicated). A negative pregnancy test is needed before women of childbearing age take the drug and monthly thereafter. Women should be counseled to use effective birth control. A home pregnancy test is an alternative to laboratory testing, but this option should be left to the prescribing clinician’s judgment and be based on reliability of the test and patient compliance.
Lorcaserin
Lorcaserin (Belviq) was approved by the FDA in 2012 for chronic weight management. It suppresses appetite by activating the serotonin 2C receptor in the brain. Because it is selective for the 2C receptor, it does not appear to have the same detrimental effects on heart valves as occurred with less-selective serotonergic agents such as fenfluramine and dexfenfluramine.31
Dosage. The recommended dosage for lorcaserin is 10 mg twice daily. Lorcaserin is a schedule IV controlled substance because of studies that showed increases in positive subjective measures such as euphoria in patients taking the drug. The incidence of euphoria was similar to that seen with zolpidem.32
Efficacy. Lorcaserin was approved on the basis of 2 trials in nondiabetic obese and overweight adults who did not have diabetes but who had a weight-related condition,33,34 and in a third trial in obese and overweight adults with type 2 diabetes mellitus who were taking oral hypoglycemic agents.35 In these trials, lorcaserin use resulted in a modest 4.7- to 5.8-kg weight loss compared with 1.6 to 2.2 kg in the placebo group.33–35 There was a high dropout rate in all 3 of these studies (33% to 45% of participants).
A pilot study that added phentermine to lorcaserin yielded double the weight loss from lorcaserin alone.36 This drug combination warrants further investigation.
Contraindications. Lorcaserin should not be given to patients who have severe renal insufficiency (creatinine clearance < 30 mL/min) or severe hepatic impairment, or who are pregnant.
Adverse effects. Common adverse reactions include dry mouth, dizziness, somnolence, headache, and gastrointestinal disturbances (nausea, constipation, or diarrhea).37
Patients with type 2 diabetes mellitus should be monitored for hypoglycemia.
Lorcaserin should be used with extreme caution in patients taking other serotonergic agents because of the risk of the serotonin syndrome.
A theoretic potential for increased risk of breast cancer also exists with lorcaserin. When rats were given supraphysiologic doses of lorcaserin (more than 50 times higher than recommended in humans), fibroadenomas and adenocarcinomas occurred at higher rates.38 Breast cancer data were not reported in the 3 randomized trials discussed above.33–35
Naltrexone-bupropion
The combination of naltrexone and bupropion was approved by the FDA in 2014 under the brand name Contrave. Both drugs are approved for monotherapy in conditions other than obesity.
Naltrexone is a mu opioid receptor antagonist approved to treat alcohol and opioid dependency. Bupropion is a dopamine-norepinephrine reuptake inhibitor approved to treat depression and to help with smoking cessation. Combining the drugs produces weight loss and metabolic benefits through effects on 2 areas of the brain that regulate food intake: the hypothalamus (appetite) and the mesolimbic dopamine circuit (reward system).
Dosage. Naltrexone-bupropion comes as an extended-release tablet of 8/90 mg. The maintenance dose of 2 tablets twice daily is reached at week 4 through a specific dose-titration regimen (Table 1). The dose should be adjusted if patients have renal or hepatic impairment or if they are also taking a CYP2B6 inhibitor.
Efficacy. FDA approval was based on the results of 4 clinical trials.39–42 Using a modified intention-to-treat analysis, Yanovski and Yanovski43 calculated that at 1 year, placebo-subtracted mean weight loss was 4.6% (4.9 kg), and mean total weight loss was 6.8% (7.3 kg) across the studies. Attrition rates, however, were high, ranging from 42% to 50%.
Cardiometabolic effects in 2 of the trials40,41 included decreased waist circumference, triglyceride levels, and C-reactive protein levels, and increased high-density lipoprotein levels at the initial dose. At the maintenance dose, additional lowering of fasting plasma insulin and glucose levels occurred along with lower levels of the homeostatic model assessment of insulin resistance. In the COR-Diabetes Study Group trial, patients with type 2 diabetes mellitus had decreased hemoglobin A1c levels without an increase in hypoglycemia and an increased likelihood of reaching the target hemoglobin A1c level below 7%.39
Contraindications. Naltrexone-bupropion is contraindicated for patients who have uncontrolled hypertension, seizure disorder, eating disorder, or end-stage renal failure; who are pregnant; or who have been treated with a monoamine oxidase inhibitor within 14 days. It should not be used with other bupropion-containing products or in patients who have taken opioids chronically or have acute opiate withdrawal.
Because of its bupropion component, this product carries an FDA black-box warning about possible suicidal thoughts and behaviors and neuropsychiatric reactions.
Adverse effects. The adverse reactions most commonly associated with naltrexone-bupropion were nausea (32.5%), constipation (19.2%), headache (17.6%), vomiting (10.7%), dizziness (9.9%), insomnia (9.2%), dry mouth (8.1%), and diarrhea (7.1%).44
Liraglutide
Liraglutide, previously FDA-approved to treat type 2 diabetes mellitus under the brand name Victoza, received approval in 2014 in a higher-dose formulation (Saxenda) to treat obesity.
Liraglutide is a glucagon-like peptide-1 receptor agonist that stimulates glucose-dependent insulin release from the pancreatic islet cells, slows gastric emptying, regulates postprandial glucagon, and reduces food intake.
Dosage. Liraglutide is given as a once-daily injection in the abdomen, thigh, or arm. The initial dosage is 0.6 mg daily for the first week and can be titrated up by 0.6 mg weekly to a target dose of 3 mg daily. If a patient does not lose 4% of baseline body weight after 16 weeks on the target dose, the drug should be discontinued because it is unlikely to lead to clinically significant weight loss.
Efficacy. Liraglutide for weight management (3 mg once daily) was evaluated in a large (N = 3,731), randomized, double-blind, placebo-controlled international trial.45 Participants did not have diabetes mellitus, but 60% had prediabetes. Liraglutide or placebo was given for 56 weeks, along with lifestyle counseling. At the end of the study, the liraglutide group had lost a mean of 8.4 kg vs 2.8 kg in the placebo group. Additionally, 63% of the liraglutide group lost at least 5% of body weight vs 27% in the placebo group, and 33% lost at least 10% of body weight vs 10% in the placebo group.
A 2-year extension found systolic blood pressure decreased with no change in pulse, and the prevalence of prediabetes and metabolic syndrome decreased by 52% and 59%, respectively.46 At 2 years, mean scores for physical function, self-esteem, and work had improved more in the liraglutide group than the placebo group.47
Adverse effects. The most common adverse reactions with liraglutide were nausea, vomiting, diarrhea, constipation, hypoglycemia, and loss of appetite. In most cases, nausea and vomiting were tolerable, transient, and associated with greater weight loss but not with decreased quality-of-life scores. Serious adverse reactions included pancreatitis, gallbladder disease, renal impairment, and suicidal thoughts.
CHOOSING A DRUG
For obese patients, when lifestyle modifications do not result in the desired weight loss, pharmacotherapy is an option. Practitioners have several FDA-approved options for weight management. Because of evidence that these drugs can postpone the onset of other complications and improve metabolic and cardiovascular parameters, they should be considered.
In phase 3 trials, these drugs caused modest weight loss of 5% to 10% of body weight. More weight was lost with the combination of phentermine-topiramate than with the other drugs.
In a 2016 meta-analysis, these drugs were associated with at least 5% weight reduction compared with placebo.48 Phentermine-topiramate and liraglutide were most likely to produce at least a 5% weight loss, while liraglutide and naltrexone-bupropion were most likely to be discontinued because of adverse events. Combination drugs may have the advantages of synergistic effects on weight loss and fewer adverse reactions because lower doses of the individual drug components are used.
Response to therapy with most of these drugs should be evaluated at 12 weeks on the maintenance dose. If less than 5% weight loss has been achieved, the medication should be discontinued.
Adverse-effect profiles, drug interactions, abuse, misuse, and overdose potential should be considered when prescribing these drugs. Weight-loss drugs are contraindicated in pregnancy because they offer no potential benefit to a pregnant woman and may harm the fetus.
The development of new drugs and better drug combinations is expected to provide more effective therapeutic strategies, which are essential for combating the obesity epidemic.
Weight-loss drugs are not magic pills, but they can help patients lose about 10 to 25 more pounds than they otherwise could, when used in a program that includes diet, exercise, and other lifestyle changes.
HALF OF ADULTS MAY BE OBESE BY 2030
Obesity is a major public health challenge in the United States, with nearly 37% of adults classified as obese.1 The prevalence has increased more than 75% since 1980,2 and it is estimated that 51% of US adults will be obese by 2030.3 Obesity is the second-leading cause of preventable deaths, after smoking.4
Obesity increases the risk of many chronic medical conditions, including type 2 diabetes mellitus, heart disease, hypertension, stroke, nonalcoholic fatty liver disease, osteoarthritis, and cancers of the breast, colon, endometrium, and kidney.5
WHEN IS DRUG THERAPY INDICATED?
Guidelines from the major obesity societies recommend that all weight-loss programs have a lifestyle component that includes a low-calorie diet, increased physical activity, and behavioral therapy, to which pharmacotherapy may be added as an adjunct.6–8
Weight-loss medications are indicated for patients who have a body mass index (BMI) of at least 30 kg/m2 or who have obesity-associated comorbidities and a BMI of at least 27 kg/m2. However, the best results are achieved when pharmacotherapy is combined with lifestyle modification.9
HISTORY OF WEIGHT-LOSS DRUGS: NOT A PRETTY PICTURE
The earliest drugs to induce weight loss, which worked mainly by increasing metabolism, included thyroid hormone, amphetamines (which also suppress appetite), and dinitrophenol (a pesticide). Adverse reactions limited their usefulness: cardiovascular effects with thyroid hormones, abuse potential with amphetamines, and neuropathy and cataracts with dinitrophenol.
Researchers then looked to drugs that could suppress appetite like amphetamines do, but without the potential for abuse. Medications that increased levels of norepinephrine and serotonin, both by increasing release and decreasing reuptake of these neuromodulators, had some success. But again, serious adverse effects occurred, and several drugs had to be withdrawn from the market.
The most publicized of these withdrawals was for the combination fenfluramine and phentermine (“fen-phen”) and its cousin dexfenfluramine (Redux). Up to 30% of patients taking fenfluramine-phentermine developed echocardiographic evidence of valvular heart disease.11 Fenfluramine also increased the risk of pulmonary hypertension. These findings led to the 1997 withdrawal of these drugs from the US market.
Sibutramine (Meridia), a norepinephrine and serotonin reuptake inhibitor, was approved for weight loss in 1997. Increases in blood pressure and heart rate were noted in the initial trial,12 and then a postmarketing study found increased rates of nonfatal myocardial infarction and stroke in patients with preexisting cardiovascular disease or diabetes mellitus.13 Based on these results, sibutramine was withdrawn from both US and European markets.
Rimonabant (Acomplia, Zimulti), a cannabinoid-receptor inhibitor, was approved in Europe in 2006, but its approval was withdrawn just 2 years later because of increased suicidality in a postmarketing study.14 It was never approved for use in the United States.
NORADRENERGIC SYMPATHOMIMETICS: FOR SHORT-TERM USE
Several noradrenergic sympathomimetic drugs are FDA-approved for short-term weight loss, but phentermine is by far the most commonly prescribed drug in this class. In fact, it is the most commonly prescribed drug for obesity in the United States.15
Phentermine
Phentermine is an atypical amphetamine analogue that suppresses appetite by norepinephrine agonism in the central nervous system. The FDA approved it for short-term weight management in 1959, and its use became widespread in the 1960s, followed by decades of popularity.
Dosage. Phentermine is prescribed at an oral dose of 15, 30, or 37.5 mg daily, either before breakfast or 1 to 2 hours after. It is a schedule IV controlled substance, based on its similarity to amphetamine. (The 5 US controlled substance schedules range from schedule I, which includes heroin, amphetamine, and cannabis, to schedule V, which includes cough syrups containing no more than 200 mg of codeine per 100 mL.) However, concerns about addiction and dependence with phentermine are largely unfounded, and abrupt cessation of the drug has not been shown to cause amphetamine-like withdrawal.16
Adverse effects. Common adverse reactions include nervousness, insomnia, and dry mouth, but these effects tend to wane with continued use.
Contraindications. Cardiovascular disease is a contraindication to phentermine because of concerns about increased blood pressure and pulse rate, although these concerns seem to be more theoretic than observed.16 Other contraindications include hyperthyroidism, glaucoma, agitation, a history of drug abuse, pregnancy, breastfeeding, and current or recent use of a monoamine oxidase inhibitor. No serious adverse events have been reported in trials of phentermine.
Efficacy. In a pooled analysis of 6 trials lasting 2 to 24 weeks completed between 1975 and 1999, phentermine-treated patients lost an average of 3.6 kg more weight than placebo recipients.17 More than 80% of study participants were women.
In a 36-week study in 108 women,18 participants lost a mean of 12.2 kg with continuous phentermine use, 13.0 kg with intermittent use (4 weeks on, 4 weeks off; the difference was not significant), and 4.8 kg with placebo.
Minimal data exist on long-term efficacy of phentermine monotherapy.
DRUGS FOR LONG-TERM THERAPY
Orlistat
Orlistat was approved as a prescription drug (Xenical, 120 mg) in 1999 and as an over-the-counter medication (Alli, 60 mg) in 2007.
Orlistat works by inhibiting pancreatic and gastric lipase, causing incomplete hydrolysis of ingested fat, thereby increasing fecal fat excretion in a dose-dependent manner. It is a good choice for weight-loss drug therapy because of its safe cardiovascular risk profile and beneficial effects on lipid levels. However, its long-term effect on weight is only modest.19,20
Dosage. The dosage for prescription orlistat is 120 mg 3 times per day, in addition to a low-fat diet (< 30% of daily calories from fat). To prevent potential deficiencies of fat-soluble vitamins, a daily multivitamin supplement is recommended, but it should not be taken with meals.
Efficacy. In a 2014 systematic review, 35% to 73% of patients treated with orlistat 120 mg had lost at least 5% of their body weight at 1 year, and 14% to 41% had lost at least 10%.21 At the end of the second year, orlistat-treated patients had lost about 3.3 kg more than placebo recipients.
In a randomized trial,22 4 years of treatment with orlistat vs placebo led to a significant (37.3%) risk reduction in the incidence of type 2 diabetes mellitus in obese participants, as well as significant improvements in cardiovascular risk factors. Mean weight loss at 1 year was significantly greater with orlistat than with placebo (10.6 vs 6.2 kg), and it remained greater at 4 years (5.8 vs 3.0 kg; P < .001).
Adverse effects. Long-term orlistat use is hampered by adverse reactions. A population-based, retrospective cohort analysis showed that fewer than 10% of patients were still using it at 1 year, and only 2% were using it at 2 years, although reasons for discontinuation were not reported.23
Adverse reactions are predominantly gastrointestinal, attributed to the high content of undigested fat in stools. Patients who do not limit their dietary fat intake are affected the most. Other reported adverse reactions include hepatotoxicity and oxalate-induced nephropathy.
Orlistat has been reported to interfere with some drugs, particularly those that are lipophilic. Drugs that should be closely monitored with orlistat are warfarin, amiodarone, cyclosporine, certain antiepileptic drugs, and levothyroxine.
Phentermine-topiramate
The combination of phentermine and topiramate was approved by the FDA in 2012 and is available under the brand name Qsymia.
Topiramate had been approved for treating seizure disorder in 1996 and as migraine prophylaxis in 2004. It is not approved as monotherapy for obesity; however, patients taking it for seizures or for psychiatric disorders (eg, binge eating, borderline personality disorder) have reported weight loss during treatment.
How topiramate promotes weight loss is not known. Proposed mechanisms include taste inhibition by carbonic anhydrase, influences on gamma-aminobutyric acid transmission causing appetite suppression, sensitization of insulin activity, and adiponectin secretion in the peripheral tissues.24,25
Phentermine-topiramate therapy has an advantage over monotherapy because lower doses of each medication can be used to achieve the same benefit, thus avoiding dose-related adverse reactions.
Dosage. Phentermine-topiramate is available in capsules containing 3.75/23, 7.5/46, 11.25/69, and 15/92 mg. The recommended starting dosage is 3.75/23 mg/day for 14 days, increasing to 7.5/46 mg/day. If patients do not lose at least 3% of their body weight after 12 weeks, the dose can be increased to 11.25/69 mg daily for 14 days, followed by 15/92 mg daily.26 Phentermine-topiramate is a schedule IV controlled substance with a low potential for abuse and dependence.
Efficacy. Approval of phentermine-topiramate for treating obesity was primarily based on 3 clinical trials.27–29 In 1 of these trials,28 at 1 year, patients had lost 9.9 kg with the medium dose and 12.9 kg with the high dose.
Adverse effects. Phentermine-topiramate was well tolerated in the trials. The most commonly reported adverse reactions were dry mouth, dizziness, constipation, insomnia, dysgeusia, paresthesia, and increased resting heart rate.28,29 Acute myopia and angle-closure glaucoma also have been reported with topiramate.30 Topiramate monotherapy has been associated with dose-dependent neuropsychiatric adverse effects, including memory symptoms and depression. However, across all 3 trials of phentermine-topiramate therapy, symptoms of depression improved over time, and no significant increase in suicide risk was identified.27–29
Recommended monitoring for patients on phentermine-topiramate includes a blood chemistry panel, resting heart rate, blood pressure, and depression screening.
Because topiramate has teratogenic potential (craniofacial abnormalities), it is labeled as pregnancy category X (contraindicated). A negative pregnancy test is needed before women of childbearing age take the drug and monthly thereafter. Women should be counseled to use effective birth control. A home pregnancy test is an alternative to laboratory testing, but this option should be left to the prescribing clinician’s judgment and be based on reliability of the test and patient compliance.
Lorcaserin
Lorcaserin (Belviq) was approved by the FDA in 2012 for chronic weight management. It suppresses appetite by activating the serotonin 2C receptor in the brain. Because it is selective for the 2C receptor, it does not appear to have the same detrimental effects on heart valves as occurred with less-selective serotonergic agents such as fenfluramine and dexfenfluramine.31
Dosage. The recommended dosage for lorcaserin is 10 mg twice daily. Lorcaserin is a schedule IV controlled substance because of studies that showed increases in positive subjective measures such as euphoria in patients taking the drug. The incidence of euphoria was similar to that seen with zolpidem.32
Efficacy. Lorcaserin was approved on the basis of 2 trials in nondiabetic obese and overweight adults who did not have diabetes but who had a weight-related condition,33,34 and in a third trial in obese and overweight adults with type 2 diabetes mellitus who were taking oral hypoglycemic agents.35 In these trials, lorcaserin use resulted in a modest 4.7- to 5.8-kg weight loss compared with 1.6 to 2.2 kg in the placebo group.33–35 There was a high dropout rate in all 3 of these studies (33% to 45% of participants).
A pilot study that added phentermine to lorcaserin yielded double the weight loss from lorcaserin alone.36 This drug combination warrants further investigation.
Contraindications. Lorcaserin should not be given to patients who have severe renal insufficiency (creatinine clearance < 30 mL/min) or severe hepatic impairment, or who are pregnant.
Adverse effects. Common adverse reactions include dry mouth, dizziness, somnolence, headache, and gastrointestinal disturbances (nausea, constipation, or diarrhea).37
Patients with type 2 diabetes mellitus should be monitored for hypoglycemia.
Lorcaserin should be used with extreme caution in patients taking other serotonergic agents because of the risk of the serotonin syndrome.
A theoretic potential for increased risk of breast cancer also exists with lorcaserin. When rats were given supraphysiologic doses of lorcaserin (more than 50 times higher than recommended in humans), fibroadenomas and adenocarcinomas occurred at higher rates.38 Breast cancer data were not reported in the 3 randomized trials discussed above.33–35
Naltrexone-bupropion
The combination of naltrexone and bupropion was approved by the FDA in 2014 under the brand name Contrave. Both drugs are approved for monotherapy in conditions other than obesity.
Naltrexone is a mu opioid receptor antagonist approved to treat alcohol and opioid dependency. Bupropion is a dopamine-norepinephrine reuptake inhibitor approved to treat depression and to help with smoking cessation. Combining the drugs produces weight loss and metabolic benefits through effects on 2 areas of the brain that regulate food intake: the hypothalamus (appetite) and the mesolimbic dopamine circuit (reward system).
Dosage. Naltrexone-bupropion comes as an extended-release tablet of 8/90 mg. The maintenance dose of 2 tablets twice daily is reached at week 4 through a specific dose-titration regimen (Table 1). The dose should be adjusted if patients have renal or hepatic impairment or if they are also taking a CYP2B6 inhibitor.
Efficacy. FDA approval was based on the results of 4 clinical trials.39–42 Using a modified intention-to-treat analysis, Yanovski and Yanovski43 calculated that at 1 year, placebo-subtracted mean weight loss was 4.6% (4.9 kg), and mean total weight loss was 6.8% (7.3 kg) across the studies. Attrition rates, however, were high, ranging from 42% to 50%.
Cardiometabolic effects in 2 of the trials40,41 included decreased waist circumference, triglyceride levels, and C-reactive protein levels, and increased high-density lipoprotein levels at the initial dose. At the maintenance dose, additional lowering of fasting plasma insulin and glucose levels occurred along with lower levels of the homeostatic model assessment of insulin resistance. In the COR-Diabetes Study Group trial, patients with type 2 diabetes mellitus had decreased hemoglobin A1c levels without an increase in hypoglycemia and an increased likelihood of reaching the target hemoglobin A1c level below 7%.39
Contraindications. Naltrexone-bupropion is contraindicated for patients who have uncontrolled hypertension, seizure disorder, eating disorder, or end-stage renal failure; who are pregnant; or who have been treated with a monoamine oxidase inhibitor within 14 days. It should not be used with other bupropion-containing products or in patients who have taken opioids chronically or have acute opiate withdrawal.
Because of its bupropion component, this product carries an FDA black-box warning about possible suicidal thoughts and behaviors and neuropsychiatric reactions.
Adverse effects. The adverse reactions most commonly associated with naltrexone-bupropion were nausea (32.5%), constipation (19.2%), headache (17.6%), vomiting (10.7%), dizziness (9.9%), insomnia (9.2%), dry mouth (8.1%), and diarrhea (7.1%).44
Liraglutide
Liraglutide, previously FDA-approved to treat type 2 diabetes mellitus under the brand name Victoza, received approval in 2014 in a higher-dose formulation (Saxenda) to treat obesity.
Liraglutide is a glucagon-like peptide-1 receptor agonist that stimulates glucose-dependent insulin release from the pancreatic islet cells, slows gastric emptying, regulates postprandial glucagon, and reduces food intake.
Dosage. Liraglutide is given as a once-daily injection in the abdomen, thigh, or arm. The initial dosage is 0.6 mg daily for the first week and can be titrated up by 0.6 mg weekly to a target dose of 3 mg daily. If a patient does not lose 4% of baseline body weight after 16 weeks on the target dose, the drug should be discontinued because it is unlikely to lead to clinically significant weight loss.
Efficacy. Liraglutide for weight management (3 mg once daily) was evaluated in a large (N = 3,731), randomized, double-blind, placebo-controlled international trial.45 Participants did not have diabetes mellitus, but 60% had prediabetes. Liraglutide or placebo was given for 56 weeks, along with lifestyle counseling. At the end of the study, the liraglutide group had lost a mean of 8.4 kg vs 2.8 kg in the placebo group. Additionally, 63% of the liraglutide group lost at least 5% of body weight vs 27% in the placebo group, and 33% lost at least 10% of body weight vs 10% in the placebo group.
A 2-year extension found systolic blood pressure decreased with no change in pulse, and the prevalence of prediabetes and metabolic syndrome decreased by 52% and 59%, respectively.46 At 2 years, mean scores for physical function, self-esteem, and work had improved more in the liraglutide group than the placebo group.47
Adverse effects. The most common adverse reactions with liraglutide were nausea, vomiting, diarrhea, constipation, hypoglycemia, and loss of appetite. In most cases, nausea and vomiting were tolerable, transient, and associated with greater weight loss but not with decreased quality-of-life scores. Serious adverse reactions included pancreatitis, gallbladder disease, renal impairment, and suicidal thoughts.
CHOOSING A DRUG
For obese patients, when lifestyle modifications do not result in the desired weight loss, pharmacotherapy is an option. Practitioners have several FDA-approved options for weight management. Because of evidence that these drugs can postpone the onset of other complications and improve metabolic and cardiovascular parameters, they should be considered.
In phase 3 trials, these drugs caused modest weight loss of 5% to 10% of body weight. More weight was lost with the combination of phentermine-topiramate than with the other drugs.
In a 2016 meta-analysis, these drugs were associated with at least 5% weight reduction compared with placebo.48 Phentermine-topiramate and liraglutide were most likely to produce at least a 5% weight loss, while liraglutide and naltrexone-bupropion were most likely to be discontinued because of adverse events. Combination drugs may have the advantages of synergistic effects on weight loss and fewer adverse reactions because lower doses of the individual drug components are used.
Response to therapy with most of these drugs should be evaluated at 12 weeks on the maintenance dose. If less than 5% weight loss has been achieved, the medication should be discontinued.
Adverse-effect profiles, drug interactions, abuse, misuse, and overdose potential should be considered when prescribing these drugs. Weight-loss drugs are contraindicated in pregnancy because they offer no potential benefit to a pregnant woman and may harm the fetus.
The development of new drugs and better drug combinations is expected to provide more effective therapeutic strategies, which are essential for combating the obesity epidemic.
- Ogden CL, Carroll MD, Fryar CD, Flegal KM. Prevalence of obesity among adults and youth: United States, 2011-2014. NCHS Data Brief 2015; 219:1–8.
- Yanovski SZ, Yanovski JA. Obesity. N Engl J Med 2002; 346:591–602.
- Finkelstein EA, Khavjou OA, Thompson H, et al. Obesity and severe obesity forecasts through 2030. Am J Prev Med 2012; 42:563–570.
- Hill JO, Wyatt H. Outpatient management of obesity: a primary care perspective. Obes Res 2002; 10(suppl 2):124S–130S.
- US Department of Health and Human Services. National Institute of Diabetes and Digestive and Kidney Diseases. Overweight and obesity statistics. www.niddk.nih.gov/health-information/health-statistics/Pages/overweight-obesity-statistics.aspx#overweight. Accessed October 10, 2017.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Obesity Society. J Am Coll Cardiol 2014; 63:2985–3023.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Obesity Society. Circulation 2014; 129(suppl 2):S102–S138.
- American Association of Clinical Endocrinologists. AACE/ACE algorithm for the medical care of patients with obesity. www.aace.com/files/guidelines/ObesityAlgorithm.pdf. Accessed July 25, 2017.
- Wadden TA, Berkowitz RI, Womble LG, et al. Randomized trial of lifestyle modification and pharmacotherapy for obesity. N Engl J Med 2005; 353:2111–2120.
- Mechanick JI, Youdim A, Jones DB, et al. Clinical practice guidelines for the perioperative nutritional, metabolic, and nonsurgical support of the bariatric surgery patient, 2013 update: cosponsored by American Association of Clinical Endocrinologists, the Obesity Society, and American Society for Metabolic and Bariatric Surgery. Surg Obes Relat Dis 2013; 9:159–191.
- Connolly HM, Crary JL, McGoon MD, et al. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med 1997; 337:581–588.
- Kim SH, Lee YM, Jee SH, et al. Effect of sibutramine on weight loss and blood pressure: a meta-analysis of controlled trials. Obes Res 2003; 11:1116–1123.
- James WP, Caterson ID, Coutinho W, et al; SCOUT Investigators. Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. N Engl J Med 2010; 363:905–917.
- Nissen SE, Nicholls SJ, Wolski K, et al; STRADIVARIUS Investigators. Effect of rimonabant on progression of atherosclerosis in patients with abdominal obesity and coronary artery disease: the STRADIVARIUS randomized controlled trial. JAMA 2008; 299:1547–1560.
- Ryan DH, Bray GA. Pharmacologic treatment options for obesity: what is old is new again. Curr Hypertens Rep 2013; 15:182–189.
- Hendricks EJ, Greenway FL, Westman EC, Gupta AK. Blood pressure and heart rate effects, weight loss and maintenance during long-term phentermine pharmacotherapy for obesity. Obesity (Silver Spring) 2011; 19:2351–2360.
- Li Z, Maglione M, Tu W, et al. Meta-analysis: pharmacologic treatment of obesity. Ann Intern Med 2005; 142:532–546.
- Munro JF, MacCuish AC, Wilson EM, Duncan LJ. Comparison of continuous and intermittent anorectic therapy in obesity. Br Med J 1968; 1:352–354.
- Hauptman J, Lucas C, Boldrin MN, Collins H, Segal KR. Orlistat in the long-term treatment of obesity in primary care settings. Arch Fam Med 2000; 9:160–167.
- Rossner S, Sjostrom L, Noack R, Meinders AE, Noseda G. Weight loss, weight maintenance, and improved cardiovascular risk factors after 2 years treatment with orlistat for obesity. European Orlistat Obesity Study Group. Obes Res 2000; 8:49–61.
- Yanovski SZ, Yanovski JA. Long-term drug treatment for obesity: a systematic and clinical review. JAMA 2014; 311:74–86.
- Torgerson JS, Hauptman J, Boldrin MN, Sjostrom L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 2004; 27:155–161.
- Padwal R, Kezouh A, Levine M, Etminan M. Long-term persistence with orlistat and sibutramine in a population-based cohort. Int J Obes (Lond) 2007; 31:1567–1570.
- Xiong GL, Gadde KM. Combination phentermine-topiramate for obesity treatment in primary care: a review. Postgrad Med 2014; 126:110–116.
- Pucci A, Finer N. New medications for treatment of obesity: metabolic and cardiovascular effects. Can J Cardiol 2015; 31:142–152.
- Smith SM, Meyer M, Trinkley KE. Phentermine-topiramate for the treatment of obesity. Ann Pharmacother 2013; 47:340–349.
- Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine-topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring) 2012; 20:330–342.
- Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet 2011; 377:1341–1352.
- Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine-topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr 2012; 95:297–308.
- Richa S, Yazbek JC. Ocular adverse effects of common psychotropic agents: a review. CNS Drugs 2010; 24:501–526.
- Weissman NJ, Sanchez M, Koch GG, Smith SR, Shanahan WR, Anderson CM. Echocardiographic assessment of cardiac valvular regurgitation with lorcaserin from analysis of 3 phase 3 clinical trials. Circ Cardiovasc Imaging 2013; 6:560–567.
- US Department of Justice Drug Enforcement Administration. Schedules of controlled substances: placement of lorcaserin into Schedule IV. Federal Register 2013; 78:26701–26705.
- Smith SR, Weissman NJ, Anderson CM, et al; Behavioral Modification and Lorcaserin for Overweight and Obesity Management (BLOOM) Study Group. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med 2010; 363:245–256.
- Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab 2011; 96:3067–3077.
- O’Neil PM, Smith SR, Weissman NJ, et al. Randomized placebo-controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM study. Obesity (Silver Spring) 2012; 20:1426–1436.
- Kumar RB, Aronne LJ. Efficacy comparison of medications approved for chronic weight management. Obesity (Silver Spring) 2015; 23(suppl 1):S4–S7.
- Chan EW, He Y, Chui CS, Wong AY, Lau WC, Wong IC. Efficacy and safety of lorcaserin in obese adults: a meta-analysis of 1-year randomized controlled trials (RCTs) and narrative review on short-term RCTs. Obes Rev 2013; 14:383–392.
- Miller LE. Lorcaserin for weight loss: insights into US Food and Drug Administration approval. J Acad Nutr Diet 2013; 113:25–30.
- Hollander P, Gupta AK, Plodkowski R, et al; COR-Diabetes Study Group. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care 2013; 36:4022–4029.
- Apovian CM, Aronne L, Rubino D, et al; COR-II Study Group. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Spring) 2013; 21:935–943.
- Greenway FL, Fujioka K, Plodkowski RA, et al; COR-I Study Group. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2010; 376:595–605.
- Wadden TA, Foreyt JP, Foster GD, et al. Weight loss with naltrexone SR/bupropion SR combination therapy as an adjunct to behavior modification: the COR-BMOD trial. Obesity (Silver Spring) 2011; 19:110–120.
- Yanovski SZ, Yanovski JA. Naltrexone extended-release plus bupropion extended-release for treatment of obesity. JAMA 2015; 313:1213–1214.
- Contrave (naltrexone HC1 and bupropion HC1) extended release tablets [package insert]. Orexigen Therapeutics, 2017. https://contrave.com/wp-content/uploads/2017/05/Contrave_PI.pdf. Accessed November 7, 2017.
- Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med 2015; 373:11–22.
- Astrup A, Carraro R, Finer N, et al; NN8022-1807 Investigators. Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int J Obes (Lond) 2012; 36:843–854.
- Lean ME, Carraro R, Finer N, et al; NN8022-1807 Investigators. Tolerability of nausea and vomiting and associations with weight loss in a randomized trial of liraglutide in obese, non-diabetic adults. Int J Obes (Lond) 2014; 38:689–697.
- Khera R, Murad MH, Chandar AK, et al. Association of pharmacological treatments for obesity with weight loss and adverse events: a systematic review and meta-analysis. JAMA 2016; 315:2424–2434.
- Ogden CL, Carroll MD, Fryar CD, Flegal KM. Prevalence of obesity among adults and youth: United States, 2011-2014. NCHS Data Brief 2015; 219:1–8.
- Yanovski SZ, Yanovski JA. Obesity. N Engl J Med 2002; 346:591–602.
- Finkelstein EA, Khavjou OA, Thompson H, et al. Obesity and severe obesity forecasts through 2030. Am J Prev Med 2012; 42:563–570.
- Hill JO, Wyatt H. Outpatient management of obesity: a primary care perspective. Obes Res 2002; 10(suppl 2):124S–130S.
- US Department of Health and Human Services. National Institute of Diabetes and Digestive and Kidney Diseases. Overweight and obesity statistics. www.niddk.nih.gov/health-information/health-statistics/Pages/overweight-obesity-statistics.aspx#overweight. Accessed October 10, 2017.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Obesity Society. J Am Coll Cardiol 2014; 63:2985–3023.
- Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Obesity Society. Circulation 2014; 129(suppl 2):S102–S138.
- American Association of Clinical Endocrinologists. AACE/ACE algorithm for the medical care of patients with obesity. www.aace.com/files/guidelines/ObesityAlgorithm.pdf. Accessed July 25, 2017.
- Wadden TA, Berkowitz RI, Womble LG, et al. Randomized trial of lifestyle modification and pharmacotherapy for obesity. N Engl J Med 2005; 353:2111–2120.
- Mechanick JI, Youdim A, Jones DB, et al. Clinical practice guidelines for the perioperative nutritional, metabolic, and nonsurgical support of the bariatric surgery patient, 2013 update: cosponsored by American Association of Clinical Endocrinologists, the Obesity Society, and American Society for Metabolic and Bariatric Surgery. Surg Obes Relat Dis 2013; 9:159–191.
- Connolly HM, Crary JL, McGoon MD, et al. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med 1997; 337:581–588.
- Kim SH, Lee YM, Jee SH, et al. Effect of sibutramine on weight loss and blood pressure: a meta-analysis of controlled trials. Obes Res 2003; 11:1116–1123.
- James WP, Caterson ID, Coutinho W, et al; SCOUT Investigators. Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. N Engl J Med 2010; 363:905–917.
- Nissen SE, Nicholls SJ, Wolski K, et al; STRADIVARIUS Investigators. Effect of rimonabant on progression of atherosclerosis in patients with abdominal obesity and coronary artery disease: the STRADIVARIUS randomized controlled trial. JAMA 2008; 299:1547–1560.
- Ryan DH, Bray GA. Pharmacologic treatment options for obesity: what is old is new again. Curr Hypertens Rep 2013; 15:182–189.
- Hendricks EJ, Greenway FL, Westman EC, Gupta AK. Blood pressure and heart rate effects, weight loss and maintenance during long-term phentermine pharmacotherapy for obesity. Obesity (Silver Spring) 2011; 19:2351–2360.
- Li Z, Maglione M, Tu W, et al. Meta-analysis: pharmacologic treatment of obesity. Ann Intern Med 2005; 142:532–546.
- Munro JF, MacCuish AC, Wilson EM, Duncan LJ. Comparison of continuous and intermittent anorectic therapy in obesity. Br Med J 1968; 1:352–354.
- Hauptman J, Lucas C, Boldrin MN, Collins H, Segal KR. Orlistat in the long-term treatment of obesity in primary care settings. Arch Fam Med 2000; 9:160–167.
- Rossner S, Sjostrom L, Noack R, Meinders AE, Noseda G. Weight loss, weight maintenance, and improved cardiovascular risk factors after 2 years treatment with orlistat for obesity. European Orlistat Obesity Study Group. Obes Res 2000; 8:49–61.
- Yanovski SZ, Yanovski JA. Long-term drug treatment for obesity: a systematic and clinical review. JAMA 2014; 311:74–86.
- Torgerson JS, Hauptman J, Boldrin MN, Sjostrom L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 2004; 27:155–161.
- Padwal R, Kezouh A, Levine M, Etminan M. Long-term persistence with orlistat and sibutramine in a population-based cohort. Int J Obes (Lond) 2007; 31:1567–1570.
- Xiong GL, Gadde KM. Combination phentermine-topiramate for obesity treatment in primary care: a review. Postgrad Med 2014; 126:110–116.
- Pucci A, Finer N. New medications for treatment of obesity: metabolic and cardiovascular effects. Can J Cardiol 2015; 31:142–152.
- Smith SM, Meyer M, Trinkley KE. Phentermine-topiramate for the treatment of obesity. Ann Pharmacother 2013; 47:340–349.
- Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine-topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring) 2012; 20:330–342.
- Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet 2011; 377:1341–1352.
- Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine-topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr 2012; 95:297–308.
- Richa S, Yazbek JC. Ocular adverse effects of common psychotropic agents: a review. CNS Drugs 2010; 24:501–526.
- Weissman NJ, Sanchez M, Koch GG, Smith SR, Shanahan WR, Anderson CM. Echocardiographic assessment of cardiac valvular regurgitation with lorcaserin from analysis of 3 phase 3 clinical trials. Circ Cardiovasc Imaging 2013; 6:560–567.
- US Department of Justice Drug Enforcement Administration. Schedules of controlled substances: placement of lorcaserin into Schedule IV. Federal Register 2013; 78:26701–26705.
- Smith SR, Weissman NJ, Anderson CM, et al; Behavioral Modification and Lorcaserin for Overweight and Obesity Management (BLOOM) Study Group. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med 2010; 363:245–256.
- Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab 2011; 96:3067–3077.
- O’Neil PM, Smith SR, Weissman NJ, et al. Randomized placebo-controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM study. Obesity (Silver Spring) 2012; 20:1426–1436.
- Kumar RB, Aronne LJ. Efficacy comparison of medications approved for chronic weight management. Obesity (Silver Spring) 2015; 23(suppl 1):S4–S7.
- Chan EW, He Y, Chui CS, Wong AY, Lau WC, Wong IC. Efficacy and safety of lorcaserin in obese adults: a meta-analysis of 1-year randomized controlled trials (RCTs) and narrative review on short-term RCTs. Obes Rev 2013; 14:383–392.
- Miller LE. Lorcaserin for weight loss: insights into US Food and Drug Administration approval. J Acad Nutr Diet 2013; 113:25–30.
- Hollander P, Gupta AK, Plodkowski R, et al; COR-Diabetes Study Group. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care 2013; 36:4022–4029.
- Apovian CM, Aronne L, Rubino D, et al; COR-II Study Group. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Spring) 2013; 21:935–943.
- Greenway FL, Fujioka K, Plodkowski RA, et al; COR-I Study Group. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2010; 376:595–605.
- Wadden TA, Foreyt JP, Foster GD, et al. Weight loss with naltrexone SR/bupropion SR combination therapy as an adjunct to behavior modification: the COR-BMOD trial. Obesity (Silver Spring) 2011; 19:110–120.
- Yanovski SZ, Yanovski JA. Naltrexone extended-release plus bupropion extended-release for treatment of obesity. JAMA 2015; 313:1213–1214.
- Contrave (naltrexone HC1 and bupropion HC1) extended release tablets [package insert]. Orexigen Therapeutics, 2017. https://contrave.com/wp-content/uploads/2017/05/Contrave_PI.pdf. Accessed November 7, 2017.
- Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med 2015; 373:11–22.
- Astrup A, Carraro R, Finer N, et al; NN8022-1807 Investigators. Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int J Obes (Lond) 2012; 36:843–854.
- Lean ME, Carraro R, Finer N, et al; NN8022-1807 Investigators. Tolerability of nausea and vomiting and associations with weight loss in a randomized trial of liraglutide in obese, non-diabetic adults. Int J Obes (Lond) 2014; 38:689–697.
- Khera R, Murad MH, Chandar AK, et al. Association of pharmacological treatments for obesity with weight loss and adverse events: a systematic review and meta-analysis. JAMA 2016; 315:2424–2434.
KEY POINTS
- Weight-loss drugs should only be used in combination with lifestyle modification.
- Preparations that combine 2 drugs have greater weight-loss benefits and better side-effect profiles.
- Weight-loss drugs should be discontinued if substantial (5%) weight loss has not occurred by 12 weeks.
- All weight-loss drugs are contraindicated in pregnancy.

Drug reaction or metastatic lung cancer?
A 76-year-old man with ulcerative colitis presented with a 1-week history of low-grade fever and progressive dyspnea. He was taking infliximab for the ulcerative colitis. He was known to be negative for human immunodeficiency virus.
. The left lung appeared normal (B).")

Since the M tuberculosis cultured from his lung proved to be sensitive to the antituberculosis drugs, we suspected that the nodules were a paradoxical reaction to the drug therapy, and thus we continued the treatment because of the continued low-grade fever. After 9 months of therapy, the fever had resolved and the nodules had disappeared, confirming our suspicion of a paradoxical reaction. The number of lymphocytes gradually increased during drug therapy.
Paradoxical reaction during tuberculosis treatment is defined as a worsening of pre-existing lesions or as the emergence of new lesions during appropriate therapy.1,2 The diagnosis is sometimes difficult, since new lesions can resemble other lung diseases. However, a paradoxical reaction involving randomly distributed nodules is rare and radiographically resembles metastatic lung cancer. Clinicians should be aware of this type of reaction in patients on tuberculosis therapy.
- Cheng SL, Wang HC, Yang PC. Paradoxical response during anti-tuberculosis treatment in HIV-negative patients with pulmonary tuberculosis. Int J Tuberc Lung Dis 2007; 11:1290–1295.
- Narita M, Ashkin D, Hollender ES, Pitchenik AE. Paradoxical worsening of tuberculosis following antiretroviral therapy in patients with AIDS. Am J Respir Crit Care Med 1998; 158:157–161.
A 76-year-old man with ulcerative colitis presented with a 1-week history of low-grade fever and progressive dyspnea. He was taking infliximab for the ulcerative colitis. He was known to be negative for human immunodeficiency virus.
Since the M tuberculosis cultured from his lung proved to be sensitive to the antituberculosis drugs, we suspected that the nodules were a paradoxical reaction to the drug therapy, and thus we continued the treatment because of the continued low-grade fever. After 9 months of therapy, the fever had resolved and the nodules had disappeared, confirming our suspicion of a paradoxical reaction. The number of lymphocytes gradually increased during drug therapy.
Paradoxical reaction during tuberculosis treatment is defined as a worsening of pre-existing lesions or as the emergence of new lesions during appropriate therapy.1,2 The diagnosis is sometimes difficult, since new lesions can resemble other lung diseases. However, a paradoxical reaction involving randomly distributed nodules is rare and radiographically resembles metastatic lung cancer. Clinicians should be aware of this type of reaction in patients on tuberculosis therapy.
A 76-year-old man with ulcerative colitis presented with a 1-week history of low-grade fever and progressive dyspnea. He was taking infliximab for the ulcerative colitis. He was known to be negative for human immunodeficiency virus.
Since the M tuberculosis cultured from his lung proved to be sensitive to the antituberculosis drugs, we suspected that the nodules were a paradoxical reaction to the drug therapy, and thus we continued the treatment because of the continued low-grade fever. After 9 months of therapy, the fever had resolved and the nodules had disappeared, confirming our suspicion of a paradoxical reaction. The number of lymphocytes gradually increased during drug therapy.
Paradoxical reaction during tuberculosis treatment is defined as a worsening of pre-existing lesions or as the emergence of new lesions during appropriate therapy.1,2 The diagnosis is sometimes difficult, since new lesions can resemble other lung diseases. However, a paradoxical reaction involving randomly distributed nodules is rare and radiographically resembles metastatic lung cancer. Clinicians should be aware of this type of reaction in patients on tuberculosis therapy.
- Cheng SL, Wang HC, Yang PC. Paradoxical response during anti-tuberculosis treatment in HIV-negative patients with pulmonary tuberculosis. Int J Tuberc Lung Dis 2007; 11:1290–1295.
- Narita M, Ashkin D, Hollender ES, Pitchenik AE. Paradoxical worsening of tuberculosis following antiretroviral therapy in patients with AIDS. Am J Respir Crit Care Med 1998; 158:157–161.
- Cheng SL, Wang HC, Yang PC. Paradoxical response during anti-tuberculosis treatment in HIV-negative patients with pulmonary tuberculosis. Int J Tuberc Lung Dis 2007; 11:1290–1295.
- Narita M, Ashkin D, Hollender ES, Pitchenik AE. Paradoxical worsening of tuberculosis following antiretroviral therapy in patients with AIDS. Am J Respir Crit Care Med 1998; 158:157–161.
The Leser-Trélat sign
An 85-year-old woman presented with night sweats, dry cough, and an unintended 30-pound weight loss over the preceding 6 months. She also reported the sudden onset of “itchy moles” on her back.


KERATOSES AND MALIGNANCY
The Leser-Trélat sign is the sudden development of multiple pruritic seborrheic keratoses, often associated with malignancy.1–4 Roughly half of these associated malignancies are adenocarcinomas, most commonly of the stomach, breast, colon, or rectum. However, it can be seen in other malignancies, including lymphoma, leukemia, and squamous cell carcinoma, as in this case.
Eruption of seborrheic keratoses has also been observed with benign neoplasms, pregnancy, human immunodeficiency virus infections, and the use of adalimumab, which indicates that the Leser-Trélat sign is not very specific. Despite these concerns, the eruption of multiple seborrheic keratoses should continue to trigger the thought of an internal malignancy in the differential diagnosis.
- Ehst BD, Minzer-Conzetti K, Swerdlin A, Devere TS. Cutaneous manifestations of internal malignancy. Curr Probl Surg 2010; 47:384–445.
- Schwartz RA. Sign of Leser-Trélat. J Am Acad Dermatol 1996; 35:88–95.
- Ellis DL, Yates RA. Sign of Leser-Trélat. Clin Dermatol 1993; 11:141–148.
- Thiers BH, Sahn RE, Callen JP. Cutaneous manifestations of internal malignancy. CA Cancer J Clin 2009; 59:73–98.
An 85-year-old woman presented with night sweats, dry cough, and an unintended 30-pound weight loss over the preceding 6 months. She also reported the sudden onset of “itchy moles” on her back.
KERATOSES AND MALIGNANCY
The Leser-Trélat sign is the sudden development of multiple pruritic seborrheic keratoses, often associated with malignancy.1–4 Roughly half of these associated malignancies are adenocarcinomas, most commonly of the stomach, breast, colon, or rectum. However, it can be seen in other malignancies, including lymphoma, leukemia, and squamous cell carcinoma, as in this case.
Eruption of seborrheic keratoses has also been observed with benign neoplasms, pregnancy, human immunodeficiency virus infections, and the use of adalimumab, which indicates that the Leser-Trélat sign is not very specific. Despite these concerns, the eruption of multiple seborrheic keratoses should continue to trigger the thought of an internal malignancy in the differential diagnosis.
An 85-year-old woman presented with night sweats, dry cough, and an unintended 30-pound weight loss over the preceding 6 months. She also reported the sudden onset of “itchy moles” on her back.
KERATOSES AND MALIGNANCY
The Leser-Trélat sign is the sudden development of multiple pruritic seborrheic keratoses, often associated with malignancy.1–4 Roughly half of these associated malignancies are adenocarcinomas, most commonly of the stomach, breast, colon, or rectum. However, it can be seen in other malignancies, including lymphoma, leukemia, and squamous cell carcinoma, as in this case.
Eruption of seborrheic keratoses has also been observed with benign neoplasms, pregnancy, human immunodeficiency virus infections, and the use of adalimumab, which indicates that the Leser-Trélat sign is not very specific. Despite these concerns, the eruption of multiple seborrheic keratoses should continue to trigger the thought of an internal malignancy in the differential diagnosis.
- Ehst BD, Minzer-Conzetti K, Swerdlin A, Devere TS. Cutaneous manifestations of internal malignancy. Curr Probl Surg 2010; 47:384–445.
- Schwartz RA. Sign of Leser-Trélat. J Am Acad Dermatol 1996; 35:88–95.
- Ellis DL, Yates RA. Sign of Leser-Trélat. Clin Dermatol 1993; 11:141–148.
- Thiers BH, Sahn RE, Callen JP. Cutaneous manifestations of internal malignancy. CA Cancer J Clin 2009; 59:73–98.
- Ehst BD, Minzer-Conzetti K, Swerdlin A, Devere TS. Cutaneous manifestations of internal malignancy. Curr Probl Surg 2010; 47:384–445.
- Schwartz RA. Sign of Leser-Trélat. J Am Acad Dermatol 1996; 35:88–95.
- Ellis DL, Yates RA. Sign of Leser-Trélat. Clin Dermatol 1993; 11:141–148.
- Thiers BH, Sahn RE, Callen JP. Cutaneous manifestations of internal malignancy. CA Cancer J Clin 2009; 59:73–98.
Big heart, small ring
A 58-year-old man presents with a 1-year history of chronic daytime fatigue, low libido, and difficulty achieving erections. He is upset: his wife suspects him of having an extramarital affair because, in addition to problems with his sexual performance, he has not been wearing his wedding ring. The patient explains that the ring has become too small for his finger and that he has never cheated on his wife. His wife has also been complaining that he snores loudly at night.
The patient works as an accountant. He has no known allergies to medications and takes no medications or supplements. He has no surgical history. He has never smoked tobacco or abused illicit drugs. He drinks a glass of wine once a week.
His father died at age 78 of a myocardial infarction; his 86-year-old mother has hypertension. He has no siblings. His 28-year-old biological son is healthy.
Physical examination
His temperature is 97.9°F (36.6°C), blood pressure 150/90 mm Hg, heart rate 80 per minute, respiratory rate 12 per minute, and oxygen saturation 98% on room air. His height is 5 feet 11 inches (180 cm), weight 250 lb (113 kg), and body mass index 35 kg/m2.
His forehead is wide with deep creases, his jaw, nose, and lower lip are prominent, and his tongue, hands, and feet are large. He has mild thyromegaly with no palpable nodules.
On cardiac examination, his point of maximal impulse is 3 cm lateral to the left midclavicular line in the fifth intercostal space; he has normal S1 and S2 with no murmurs, rubs, or gallops. The lungs are clear on auscultation. His abdomen is soft, nontender, and nondistended; the liver is palpated 2 cm below the costal margin. His extremities are not edematous.
LABORATORY TESTING
1. In addition to a complete blood cell count and comprehensive metabolic panel, which is the most appropriate test to order?
- Growth hormone (GH) level
- Insulin-like growth factor 1 (IGF-1) level
- GH and IGF-1 levels
- IGF-2 level
Acromegaly, an overview
The patient’s history of snoring and daytime fatigue suggests obstructive sleep apnea, which together with his enlarging ring finger size, wide forehead with deep creases, prominent jaw, nose, and lower lip, and enlarged thyroid, heart, and liver suggests acromegaly.

In most cases, acromegaly is caused by a GH-secreting pituitary adenoma. Rare causes include hypothalamic tumors that secrete GH-releasing hormone (GHRH) and ectopic secretion of GHRH or GH.1 Pseudoacromegaly, a mimic, is characterized by acromegalic features without hypersecretion of GH and with normal IGF-1 levels.4
The prevalence of acromegaly is 36 to 60 cases per million, and its annual incidence is 3 to 4 per million.5
With this patient’s presentation, the most appropriate next step is to order an IGF-1 level to screen for acromegaly.
GH secretion is pulsatile, IGF-1 secretion is not
GH is synthesized and stored in somatotroph cells, which account for more than 50% of pituitary hormone-secreting cells.6 Three hormones regulate synthesis and secretion of GH: GHRH, ghrelin, and somatostatin.7 GH secretion is pulsatile, with minimal basal secretion dependent on sex, age, neurotransmitters, exercise, and stress.7 It exerts its physiologic effects through an interaction with the GH receptor, a single-chain transmembrane glycoprotein.8,9
A GH-secreting adenoma develops when pituitary somatotroph cells undergo a monoclonal expansion. Mutations of various genes such as GNAS, PRKAR1A, and AIP are suspected of triggering such expansion. Disruption of the MENIN gene leads to multiple endocrine neoplasia syndrome 1, a combination of pituitary adenoma, pancreatic tumor, and primary hyperparathyroidism.9 The pattern of cytoplasmic keratin in somatotroph cells defines 2 histologic subtypes: densely granulated and sparsely granulated. The latter subtype is associated with more-invasive lesions that are seen more often in younger patients and are less responsive to somatostatin ligand therapy.10
GH induces transcription of IGF-1, mostly in the liver. In contrast to GH, IGF-1 secretion is not pulsatile, and therefore IGF-1 can be measured more reliably in serum, and the results can be interpreted according to age- and sex-adjusted reference ranges.
The IGF-1 level is a very sensitive test, but it is not very specific. It can be falsely elevated in pregnancy, in patients on estrogen replacement therapy, and in late adolescence.11 In addition, it may be difficult to interpret the IGF-1 level in the setting of malnutrition, severe hyperglycemia, renal or hepatic failure, and hypothyroidism.11,12
Nonpulsatile secretion and high sensitivity make the IGF-1 level the screening test of choice for acromegaly.9,12 In contrast, because of the pulsatile nature of GH synthesis, one cannot rely on a random GH level alone to detect the hormone’s hypersecretion.
IGF-2 has no role in acromegaly
IGF-2, produced mainly by the liver, plays an important role in promoting fetal growth. IGF-2 may induce hypoglycemia when secreted by some mesenchymal tumors.13 This hormone has no role in the pathogenesis of acromegaly and should not be measured in this patient.
CASE CONTINUED: FURTHER TESTING
The patient’s IGF-1 level is 590 ng/mL; the reference range for his age and sex is 68 to 245 ng/mL.
A sleep study confirms obstructive sleep apnea, and the patient is started on continuous positive airway pressure at night, with some reduction of his fatigue.
2. What is the most appropriate next step?
- Order magnetic resonance imaging (MRI) of the pituitary with gadolinium contrast
- Perform a GH suppression test with a 75-g oral glucose load
- Perform a GH stimulation test
- Refer the patient to a neurosurgeon for a consultation
The most appropriate next step is a GH suppression test, performed by measuring the plasma GH level 2 hours after giving 75 g of glucose by mouth. This confirmatory test is necessary because the IGF-1 level can be falsely elevated. The normal response to an oral glucose challenge is suppression of the GH level to below 1 μg/L. Failure to suppress GH confirms the diagnosis of acromegaly.14
A GH stimulation test with insulin-induced hypoglycemia or with GHRH-arginine would be appropriate if GH deficiency were suspected rather than hypersecretion.
Imaging of the pituitary with MRI before obtaining biochemical confirmation of the diagnosis of acromegaly may mislead the physician because MRI does not determine the functional status of a pituitary tumor. Correct treatment of a pituitary tumor depends on whether the tumor causes hypersecretion or deficiency of any pituitary hormones.
Referral to a neurosurgeon for a consultation is premature until a biochemical diagnosis of acromegaly is made and a pituitary adenoma is subsequently demonstrated by imaging.
3. The patient’s GH level is 10 μg/L 2 hours after oral administration of 75 g of glucose. What is the most appropriate next step?
- Radiography of the skull to image the pituitary at a low cost
- MRI of the pituitary with contrast after making sure the patient’s renal function is normal
- MRI of the pituitary without contrast
- Computed tomography of the head
The next step is MRI of the pituitary with contrast (gadolinium) after obtaining blood urea nitrogen and creatinine measurements to make sure the patient’s renal function is normal.14
Gadolinium contrast is contraindicated in patients with severely reduced renal function (glomerular filtration rate < 30 mL/min/1.73 m2) because of the risk of nephrogenic systemic fibrosis. In such a case, MRI without contrast would be appropriate.
MRI is the most sensitive imaging test for detecting a pituitary adenoma, as it can detect tumors as small as 2 mm. A pituitary macroadenoma (> 10 mm in diameter) is detected in more than 75% of patients with acromegaly at diagnosis. The tumor often invades one or both cavernous sinuses or extends to the suprasellar region, possibly impinging on the optic chiasm.15
If MRI is contraindicated, computed tomography of the head should be performed.
CASE CONTINUED: IMAGING
The patient’s comprehensive metabolic panel is normal, but his fasting plasma glucose is 135 mg/dL (reference range 74–99). Pituitary MRI with contrast shows a 3-cm pituitary adenoma with suprasellar extension, impinging on the optic chiasm and invading the right cavernous sinus.
4. In addition to repeating the fasting plasma glucose and measuring hemoglobin A1c, what is the most appropriate next step in managing this patient?
- Measure the prolactin, morning serum cortisol, total testosterone, follicle-stimulating hormone (FSH), luteinizing hormone (LH), thyroid-stimulating hormone (TSH), and free thyroxine (T4) levels; refer the patient to an ophthalmologist for a formal evaluation of visual fields
- Measure these hormone levels; perform a gross evaluation of the visual fields and refer the patient to an ophthalmologist only if visual field deficits are found on the gross examination
- Measure these hormone levels; refer the patient to an ophthalmologist only if he complains of vision changes
- Do not order any additional tests; instruct the patient to call the office if he develops any vision changes
This patient should have all of these hormones measured. In addition, given that his macroadenoma is impinging on the optic chiasm, he should be referred to an ophthalmologist for a formal evaluation of visual fields even if the latter are intact on gross examination and even if the patient does not complain of any visual changes.
Abnormalities of hormones other than GH and IGF-1 in acromegaly
Secretion of pituitary hormones other than GH and IGF-1 must be assessed.
Prolactin. GH-secreting tumors also secrete prolactin in up to one-third of patients, with the resulting hyperprolactinemia contributing to hypogonadism.11 Prolactin hypersecretion should be distinguished from hyperprolactinemia caused by pituitary stalk compression, which may be evident on MRI.
Measuring the serum prolactin level with 1:100 dilution to counteract the “hook effect” may unmask severe hyperprolactinemia due to a large macroprolactinoma. (The hook effect occurs when the prolactin level is so high that there is not enough antibody in the assay to bind both ends of all the prolactin molecules present, causing the reading to be falsely low.).
Cortisol, T4, testosterone. Patients with acromegaly may develop central adrenal insufficiency, central hypothyroidism, and central hypogonadism; these hormonal deficits may occur in isolation or in combination.
Also, patients should be assessed for comorbidities such as colon cancer (all patients with acromegaly require a colonoscopy, as acromegaly raises the risk of colon cancer), diabetes mellitus, hypertension, cardiomyopathy, and sleep apnea.16
Visual field loss may be insidious

The diagnostic and treatment algorithm for acromegaly is summarized in Figure 1.
CASE CONTINUED: LABORATORY VALUES, TREATMENT OPTIONS
Our patient’s repeat fasting plasma glucose is 137 mg/dL; his hemoglobin A1c is 7.3%, consistent with diabetes mellitus secondary to acromegaly. Other laboratory values:
- Morning cortisol level 15 μg/dL (reference range 5.3–22.5),
- Prolactin 23 ng/mL, confirmed with 1:100 dilution (4.0–15.2)
- Total testosterone 59 ng/dL (193–824)
- LH 2.1 mIU/mL (1.8–10.8)
- FSH 3.0 mIU/mL (1.5–12.4)
- TSH 2.5 mIU/L (0.5–4.5)
- Free T4 1.3 ng/dL (0.9–1.7).
The patient is started on metformin 500 mg by mouth twice a day, counseled on a healthy diet, and informed that his diabetes may be a complication of his acromegaly. He is anxious to learn how his acromegaly can be treated.
5. What treatment would you recommend for the patient’s acromegaly?
- Medical treatment first, then transsphenoidal resection of the pituitary macroadenoma if medical treatment fails
- Medical treatment first, radiotherapy if medical treatment fails, and transsphenoidal resection of the pituitary macroadenoma as a last resort
- Transsphenoidal resection of the pituitary macroadenoma first, medical treatment if surgery fails, and radiotherapy if both surgery and medical treatment fail
- Taking a safe, conservative approach, monitoring IGF-1 levels frequently; starting medical treatment if acromegaly does not go into remission in 1 year
The initial treatment of choice for most patients with acromegaly is resection of the pituitary tumor.
A transsphenoidal approach is used for most patients; only rarely is craniotomy necessary. Endoscopic and microsurgical techniques reduce postoperative morbidity.17 Postoperative complications include symptoms related to the transsphenoidal approach (nasal congestion, sinusitis, epistaxis), cerebrospinal fluid leak, hemorrhage, meningitis, stroke, visual impairment, vascular damage, transient or permanent diabetes insipidus, and hypopituitarism. The surgical mortality rate is less than 0.5%.18,19
Successful resection of a pituitary tumor would lead to normalization of the IGF-1 level, a drop of the GH level to below 1 μg/L, and relief of the effect of the tumor pressing against other structures. An IGF-1 level and a random GH level should be obtained 12 weeks after the surgery.14 If the GH level is higher than 1 μg/L, a GH suppression test with a 75-g oral glucose load should be performed.14 MRI of the sella turcica should be done 12 weeks after surgery to visualize residual tumor and adjacent structures.14
A large tumor size, suprasellar extension, and high preoperative levels of IGF-1 and GH are associated with a lack of surgical success; however, surgical debulking should still be considered in patients with a low chance for surgical cure to improve the probability of achieving biochemical remission with postoperative medical and radiologic therapy.20
Medical therapy can be the initial treatment if the patient refuses surgery or if surgery is contraindicated because of severe comorbidities or because structural features of the tumor confer a high surgical risk (eg, if the adenoma encases the cavernous portion of a carotid artery).13 Medical therapy may shrink the tumor in some patients and may thereby make surgical resection easier and more likely to be successful.
Radiotherapy is usually reserved for patients whose tumors recur or persist postoperatively and who are resistant to or intolerant of medical therapy.14 The soft tissue changes caused by acromegaly may regress with treatment to some degree, but they are not likely to resolve completely; the bone changes do not regress.
CASE CONTINUED: MEDICAL TREATMENT
Three months after transsphenoidal resection of his pituitary macroadenoma, our patient’s laboratory values are as follows:
- IGF-1 400 ng/mL
- Morning cortisol 20 μg/dL
- Testosterone 95 ng/dL
- LH 2.1 mU/mL
- FSH 3.7 mU/mL
- Prolactin 12 ng/mL
- TSH 2.3 mIU/L
- Free T4 1.2 ng/dL
- Basic metabolic panel normal.
The patient denies frequent urination or increased thirst. Repeat MRI of the pituitary with contrast shows a residual 1.3-cm adenoma with no suprasellar extension.
6. What is the best next treatment choice for the patient?
- A GH receptor antagonist (pegvisomant)
- A somatostatin receptor ligand (SRL) such as octreotide
- Cabergoline (a dopamine agonist)
- A combination of an SRL and pegvisomant
An SRL such as octreotide would be the best choice for this patient.
The medical options for acromegaly are SRLs, pegvisomant, and cabergoline.21–23 The Endocrine Society guidelines recommend either an SRL or pegvisomant as the initial adjuvant medical therapy in patients with persistent disease after surgery.14 However, pegvisomant is much more expensive than any SRL, so an SRL would be a better choice in this patient. Also, pegvisomant does not suppress tumor growth, in contrast to SRLs, so SRLs are preferred in patients with large tumors abutting the optic chiasm.14
SRLs are used as primary therapy in patients who cannot be cured by surgery, have extensive cavernous sinus invasion, have no chiasmal compression, or are poor surgical candidates.


The medical treatment of acromegaly is summarized in Table 2.14,15 Side effects of the medications used to treat acromegaly are summarized in Table 3.14
CASE CONTINUED: RADIOTHERAPY
The patient is treated with octreotide, and the dose is subsequently titrated upward. His central hypogonadism is treated with testosterone gel. After 3 months, his IGF-1 level decreases to 190 ng/mL, the total testosterone increases to 450 ng/dL, and the hemoglobin A1c decreases to 5.9%.
The patient asks if stereotactic radiotherapy, which he read about on the Internet, can cure his acromegaly so that he can avoid the monthly octreotide injections.
7. Which statement best describes radiotherapy’s therapeutic effect in acromegaly?
- Stereotactic radiotherapy is more effective than medical therapy and should be used as a second-line treatment after surgery
- Stereotactic radiotherapy is less effective than conventional radiotherapy
- Stereotactic radiotherapy leads to stability or a decrease in the size of the GH-secreting tumor in 93% to 100% of patients in 5 to 10 years and to biochemical remission in 40% to 60% of patients at 5 years
- Stereotactic radiotherapy causes hypopituitarism in no more than 1% of patients
Stereotactic radiotherapy leads to stability or a decrease in the size of the GH-secreting tumor in 93% to 100% of patients in 5 to 10 years and biochemical remission in 40% to 60% of patients at 5 years.24,25
Hypopituitarism develops in up to 50% of patients at 5 years, and its incidence increases with the duration of follow-up.24 The risk of other complications is low (0% to 5% for new visual deficits, cranial nerve damage, or brain radionecrosis, and 0% to 1% for secondary brain tumors).24
Conventional radiotherapy has fallen out of favor because it is associated with an increased risk of death (mainly from stroke) independent of IGF-1 and GH levels, and a higher rate of complications than stereotactic radiotherapy.14,16 Radiotherapy is reserved for postsurgical treatment of patients with recurrent or persistent tumors who are resistant to or cannot tolerate medical therapy; it is the third-line treatment.24
Given that our patient responded to the medical therapy and tolerated it well and given the high risk of hypopituitarism associated with stereotactic radiotherapy, the latter would not be appropriate for the patient.
His fatigue has diminished further and his sexual performance has improved. He is still married and his wife no longer suspects him of infidelity.
KEY POINTS
- IGF-1 is the screening test of choice in a patient with signs and symptoms of acromegaly.
- A growth hormone suppression test with a 75-g oral glucose load is the gold standard test for confirmation of the diagnosis of acromegaly in patients with an elevated IGF-1 level.
- Transsphenoidal resection of the growth hormone-secreting pituitary macroadenoma is the initial treatment of choice for acromegaly.
- Patients with residual or recurrent growth hormone-secreting pituitary macroadenoma can be treated with somatostatin receptor ligands, a growth hormone receptor antagonist (pegvisomant), and a dopamine agonist cabergoline.
- Radiotherapy is reserved for postsurgical treatment of patients with recurrent or persistent tumors who are resistant to or intolerant of medical therapy. Stereotactic radiotherapy has largely replaced conventional radiotherapy.
- Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest 2009; 119:3189–3202.
- Molitch ME. Clinical manifestations of acromegaly. Endocrinol Metab Clin North Am 1992; 21:597–614.
- Dineen R, Stewart PM, Sherlock M. Acromegaly. QJM 2017; 110:411–420.
- Yacub A, Yaqub N. Insulin-mediated pseudoacromegaly: a case report and review of the literature. W V Med J 2008; 104:12–15.
- Mestron A, Webb SM, Astorga R, et al. Epidemiology, clinical characteristics, outcome, morbidity and mortality in acromegaly based on the Spanish Acromegaly Registry (Registro Espanol de Acromegalia, REA). Eur J Endocrinol 2004; 151:439–446.
- Zhu X, Lin CR, Prefontaine CG, Tollkuhn J, Rosenfeld MG. Genetic control of pituitary development and hypopituitarism. Curr Opin Genet Dev 2005; 15:332–340.
- Tannenbaum GS, Epelbaum J, Bowers CY. Interrelationship between the novel peptide ghrelin and somatostatin/growth hormone-releasing hormone in regulation of pulsatile growth hormone secretion. Endocrinology 2003; 144:967–974.
- Lanning NJ, Carter-Su C. Recent advances in growth hormone signaling. Rev Endocr Metab Disord 2006; 7:225–235.
- Colao A, Ferone D, Marzullo P, Lombardi G. Systemic complications of acromegaly: epidemiology, pathogenesis, and management. Endocr Rev 2004; 25:102–152.
- Larkin S, Reddy R, Karavitaki N, Cudlip S, Wass J, Ansorge O. Granulation pattern, but not GSP or GHR mutation, is associated with clinical characteristics in somatostatin-naive patients with somatotroph adenomas. Eur J Endocrinol 2013; 168:491–499.
- Dineen R, Stewart PM, Sherlock M. Acromegaly. QJM 2017; 110:411–420.
- Peacey SR, Toogood AA, Veldhuis JD, Thorner MO, Shalet SM. The relationship between 24-hour growth hormone secretion and insulin-like growth factor I in patients with successfully treated acromegaly: impact of surgery or radiotherapy. J Clin Endocrinol Metab 2001; 86:259–266.
- Livingstone C. IGF2 and cancer. Endocr Relat Cancer 2013; 20:R321–R339.
- Katznelson L, Laws ER Jr, Melmed S, et al. Acromegaly: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2014; 99:3933–3951.
- Melmed S. Acromegaly. N Engl J Med 2006; 355:2558–2573.
- Melmed S, Casanueva FF, Klibanski A, et al. A consensus on the diagnosis and treatment of acromegaly complications. Pituitary 2013; 16:294–302.
- Marquez Y, Tuchman A, Zada G. Surgery and radiosurgery for acromegaly: a review of indications, operative techniques, outcomes, and complications. Int J Endocrinol 2012; 2012: 386401.
- Jane JA Jr, Starke RM, Elzoghby MA, et al. Endoscopic transsphenoidal surgery for acromegaly: remission using modern criteria, complications, and predictors of outcome. J Clin Endocrinol Metab 2011; 96:2732–2740.
- Cappabianca P, Cavallo LM, Colao A, de Divitiis E. Surgical complications associated with the endoscopic endonasal transsphenoidal approach for pituitary adenomas. J Neurosurg 2002; 97:293–298.
- Nomikos P, Buchfelder M, Fahlbusch R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical “cure.” Eur J Endocrinol 2005; 152:379–387.
- Howlett TA, Willis D, Walker G, Wass JA, Trainer PJ; UK Acromegaly Register Study Group (UKAR-3). Control of growth hormone and IGF1 in patients with acromegaly in the UK: responses to medical treatment with somatostatin analogues and dopamine agonists. Clin Endocrinol (Oxf) 2013; 79:689–699.
- Katznelson L. Pegvisomant for the treatment of acromegaly-translation of clinical trials into clinical practice. Nat Clin Pract Endocrinol Metab 2007; 3:514–515.
- Freda PU, Reyes CM, Nuruzzaman AT, Sundeen RE, Khandji AG, Post KD. Cabergoline therapy of growth hormone & growth hormone/prolactin secreting pituitary tumors. Pituitary 2004; 7:21–30.
- Castinetti F, Morange I, Dufour H, Regis J, Brue T. Radiotherapy and radiosurgery in acromegaly. Pituitary 2009; 12:3–10.
- Gheorghiu ML. Updates in outcomes of stereotactic radiation therapy in acromegaly. Pituitary 2017; 20:154–168.
A 58-year-old man presents with a 1-year history of chronic daytime fatigue, low libido, and difficulty achieving erections. He is upset: his wife suspects him of having an extramarital affair because, in addition to problems with his sexual performance, he has not been wearing his wedding ring. The patient explains that the ring has become too small for his finger and that he has never cheated on his wife. His wife has also been complaining that he snores loudly at night.
The patient works as an accountant. He has no known allergies to medications and takes no medications or supplements. He has no surgical history. He has never smoked tobacco or abused illicit drugs. He drinks a glass of wine once a week.
His father died at age 78 of a myocardial infarction; his 86-year-old mother has hypertension. He has no siblings. His 28-year-old biological son is healthy.
Physical examination
His temperature is 97.9°F (36.6°C), blood pressure 150/90 mm Hg, heart rate 80 per minute, respiratory rate 12 per minute, and oxygen saturation 98% on room air. His height is 5 feet 11 inches (180 cm), weight 250 lb (113 kg), and body mass index 35 kg/m2.
His forehead is wide with deep creases, his jaw, nose, and lower lip are prominent, and his tongue, hands, and feet are large. He has mild thyromegaly with no palpable nodules.
On cardiac examination, his point of maximal impulse is 3 cm lateral to the left midclavicular line in the fifth intercostal space; he has normal S1 and S2 with no murmurs, rubs, or gallops. The lungs are clear on auscultation. His abdomen is soft, nontender, and nondistended; the liver is palpated 2 cm below the costal margin. His extremities are not edematous.
LABORATORY TESTING
1. In addition to a complete blood cell count and comprehensive metabolic panel, which is the most appropriate test to order?
- Growth hormone (GH) level
- Insulin-like growth factor 1 (IGF-1) level
- GH and IGF-1 levels
- IGF-2 level
Acromegaly, an overview
The patient’s history of snoring and daytime fatigue suggests obstructive sleep apnea, which together with his enlarging ring finger size, wide forehead with deep creases, prominent jaw, nose, and lower lip, and enlarged thyroid, heart, and liver suggests acromegaly.
In most cases, acromegaly is caused by a GH-secreting pituitary adenoma. Rare causes include hypothalamic tumors that secrete GH-releasing hormone (GHRH) and ectopic secretion of GHRH or GH.1 Pseudoacromegaly, a mimic, is characterized by acromegalic features without hypersecretion of GH and with normal IGF-1 levels.4
The prevalence of acromegaly is 36 to 60 cases per million, and its annual incidence is 3 to 4 per million.5
With this patient’s presentation, the most appropriate next step is to order an IGF-1 level to screen for acromegaly.
GH secretion is pulsatile, IGF-1 secretion is not
GH is synthesized and stored in somatotroph cells, which account for more than 50% of pituitary hormone-secreting cells.6 Three hormones regulate synthesis and secretion of GH: GHRH, ghrelin, and somatostatin.7 GH secretion is pulsatile, with minimal basal secretion dependent on sex, age, neurotransmitters, exercise, and stress.7 It exerts its physiologic effects through an interaction with the GH receptor, a single-chain transmembrane glycoprotein.8,9
A GH-secreting adenoma develops when pituitary somatotroph cells undergo a monoclonal expansion. Mutations of various genes such as GNAS, PRKAR1A, and AIP are suspected of triggering such expansion. Disruption of the MENIN gene leads to multiple endocrine neoplasia syndrome 1, a combination of pituitary adenoma, pancreatic tumor, and primary hyperparathyroidism.9 The pattern of cytoplasmic keratin in somatotroph cells defines 2 histologic subtypes: densely granulated and sparsely granulated. The latter subtype is associated with more-invasive lesions that are seen more often in younger patients and are less responsive to somatostatin ligand therapy.10
GH induces transcription of IGF-1, mostly in the liver. In contrast to GH, IGF-1 secretion is not pulsatile, and therefore IGF-1 can be measured more reliably in serum, and the results can be interpreted according to age- and sex-adjusted reference ranges.
The IGF-1 level is a very sensitive test, but it is not very specific. It can be falsely elevated in pregnancy, in patients on estrogen replacement therapy, and in late adolescence.11 In addition, it may be difficult to interpret the IGF-1 level in the setting of malnutrition, severe hyperglycemia, renal or hepatic failure, and hypothyroidism.11,12
Nonpulsatile secretion and high sensitivity make the IGF-1 level the screening test of choice for acromegaly.9,12 In contrast, because of the pulsatile nature of GH synthesis, one cannot rely on a random GH level alone to detect the hormone’s hypersecretion.
IGF-2 has no role in acromegaly
IGF-2, produced mainly by the liver, plays an important role in promoting fetal growth. IGF-2 may induce hypoglycemia when secreted by some mesenchymal tumors.13 This hormone has no role in the pathogenesis of acromegaly and should not be measured in this patient.
CASE CONTINUED: FURTHER TESTING
The patient’s IGF-1 level is 590 ng/mL; the reference range for his age and sex is 68 to 245 ng/mL.
A sleep study confirms obstructive sleep apnea, and the patient is started on continuous positive airway pressure at night, with some reduction of his fatigue.
2. What is the most appropriate next step?
- Order magnetic resonance imaging (MRI) of the pituitary with gadolinium contrast
- Perform a GH suppression test with a 75-g oral glucose load
- Perform a GH stimulation test
- Refer the patient to a neurosurgeon for a consultation
The most appropriate next step is a GH suppression test, performed by measuring the plasma GH level 2 hours after giving 75 g of glucose by mouth. This confirmatory test is necessary because the IGF-1 level can be falsely elevated. The normal response to an oral glucose challenge is suppression of the GH level to below 1 μg/L. Failure to suppress GH confirms the diagnosis of acromegaly.14
A GH stimulation test with insulin-induced hypoglycemia or with GHRH-arginine would be appropriate if GH deficiency were suspected rather than hypersecretion.
Imaging of the pituitary with MRI before obtaining biochemical confirmation of the diagnosis of acromegaly may mislead the physician because MRI does not determine the functional status of a pituitary tumor. Correct treatment of a pituitary tumor depends on whether the tumor causes hypersecretion or deficiency of any pituitary hormones.
Referral to a neurosurgeon for a consultation is premature until a biochemical diagnosis of acromegaly is made and a pituitary adenoma is subsequently demonstrated by imaging.
3. The patient’s GH level is 10 μg/L 2 hours after oral administration of 75 g of glucose. What is the most appropriate next step?
- Radiography of the skull to image the pituitary at a low cost
- MRI of the pituitary with contrast after making sure the patient’s renal function is normal
- MRI of the pituitary without contrast
- Computed tomography of the head
The next step is MRI of the pituitary with contrast (gadolinium) after obtaining blood urea nitrogen and creatinine measurements to make sure the patient’s renal function is normal.14
Gadolinium contrast is contraindicated in patients with severely reduced renal function (glomerular filtration rate < 30 mL/min/1.73 m2) because of the risk of nephrogenic systemic fibrosis. In such a case, MRI without contrast would be appropriate.
MRI is the most sensitive imaging test for detecting a pituitary adenoma, as it can detect tumors as small as 2 mm. A pituitary macroadenoma (> 10 mm in diameter) is detected in more than 75% of patients with acromegaly at diagnosis. The tumor often invades one or both cavernous sinuses or extends to the suprasellar region, possibly impinging on the optic chiasm.15
If MRI is contraindicated, computed tomography of the head should be performed.
CASE CONTINUED: IMAGING
The patient’s comprehensive metabolic panel is normal, but his fasting plasma glucose is 135 mg/dL (reference range 74–99). Pituitary MRI with contrast shows a 3-cm pituitary adenoma with suprasellar extension, impinging on the optic chiasm and invading the right cavernous sinus.
4. In addition to repeating the fasting plasma glucose and measuring hemoglobin A1c, what is the most appropriate next step in managing this patient?
- Measure the prolactin, morning serum cortisol, total testosterone, follicle-stimulating hormone (FSH), luteinizing hormone (LH), thyroid-stimulating hormone (TSH), and free thyroxine (T4) levels; refer the patient to an ophthalmologist for a formal evaluation of visual fields
- Measure these hormone levels; perform a gross evaluation of the visual fields and refer the patient to an ophthalmologist only if visual field deficits are found on the gross examination
- Measure these hormone levels; refer the patient to an ophthalmologist only if he complains of vision changes
- Do not order any additional tests; instruct the patient to call the office if he develops any vision changes
This patient should have all of these hormones measured. In addition, given that his macroadenoma is impinging on the optic chiasm, he should be referred to an ophthalmologist for a formal evaluation of visual fields even if the latter are intact on gross examination and even if the patient does not complain of any visual changes.
Abnormalities of hormones other than GH and IGF-1 in acromegaly
Secretion of pituitary hormones other than GH and IGF-1 must be assessed.
Prolactin. GH-secreting tumors also secrete prolactin in up to one-third of patients, with the resulting hyperprolactinemia contributing to hypogonadism.11 Prolactin hypersecretion should be distinguished from hyperprolactinemia caused by pituitary stalk compression, which may be evident on MRI.
Measuring the serum prolactin level with 1:100 dilution to counteract the “hook effect” may unmask severe hyperprolactinemia due to a large macroprolactinoma. (The hook effect occurs when the prolactin level is so high that there is not enough antibody in the assay to bind both ends of all the prolactin molecules present, causing the reading to be falsely low.).
Cortisol, T4, testosterone. Patients with acromegaly may develop central adrenal insufficiency, central hypothyroidism, and central hypogonadism; these hormonal deficits may occur in isolation or in combination.
Also, patients should be assessed for comorbidities such as colon cancer (all patients with acromegaly require a colonoscopy, as acromegaly raises the risk of colon cancer), diabetes mellitus, hypertension, cardiomyopathy, and sleep apnea.16
Visual field loss may be insidious
The diagnostic and treatment algorithm for acromegaly is summarized in Figure 1.
CASE CONTINUED: LABORATORY VALUES, TREATMENT OPTIONS
Our patient’s repeat fasting plasma glucose is 137 mg/dL; his hemoglobin A1c is 7.3%, consistent with diabetes mellitus secondary to acromegaly. Other laboratory values:
- Morning cortisol level 15 μg/dL (reference range 5.3–22.5),
- Prolactin 23 ng/mL, confirmed with 1:100 dilution (4.0–15.2)
- Total testosterone 59 ng/dL (193–824)
- LH 2.1 mIU/mL (1.8–10.8)
- FSH 3.0 mIU/mL (1.5–12.4)
- TSH 2.5 mIU/L (0.5–4.5)
- Free T4 1.3 ng/dL (0.9–1.7).
The patient is started on metformin 500 mg by mouth twice a day, counseled on a healthy diet, and informed that his diabetes may be a complication of his acromegaly. He is anxious to learn how his acromegaly can be treated.
5. What treatment would you recommend for the patient’s acromegaly?
- Medical treatment first, then transsphenoidal resection of the pituitary macroadenoma if medical treatment fails
- Medical treatment first, radiotherapy if medical treatment fails, and transsphenoidal resection of the pituitary macroadenoma as a last resort
- Transsphenoidal resection of the pituitary macroadenoma first, medical treatment if surgery fails, and radiotherapy if both surgery and medical treatment fail
- Taking a safe, conservative approach, monitoring IGF-1 levels frequently; starting medical treatment if acromegaly does not go into remission in 1 year
The initial treatment of choice for most patients with acromegaly is resection of the pituitary tumor.
A transsphenoidal approach is used for most patients; only rarely is craniotomy necessary. Endoscopic and microsurgical techniques reduce postoperative morbidity.17 Postoperative complications include symptoms related to the transsphenoidal approach (nasal congestion, sinusitis, epistaxis), cerebrospinal fluid leak, hemorrhage, meningitis, stroke, visual impairment, vascular damage, transient or permanent diabetes insipidus, and hypopituitarism. The surgical mortality rate is less than 0.5%.18,19
Successful resection of a pituitary tumor would lead to normalization of the IGF-1 level, a drop of the GH level to below 1 μg/L, and relief of the effect of the tumor pressing against other structures. An IGF-1 level and a random GH level should be obtained 12 weeks after the surgery.14 If the GH level is higher than 1 μg/L, a GH suppression test with a 75-g oral glucose load should be performed.14 MRI of the sella turcica should be done 12 weeks after surgery to visualize residual tumor and adjacent structures.14
A large tumor size, suprasellar extension, and high preoperative levels of IGF-1 and GH are associated with a lack of surgical success; however, surgical debulking should still be considered in patients with a low chance for surgical cure to improve the probability of achieving biochemical remission with postoperative medical and radiologic therapy.20
Medical therapy can be the initial treatment if the patient refuses surgery or if surgery is contraindicated because of severe comorbidities or because structural features of the tumor confer a high surgical risk (eg, if the adenoma encases the cavernous portion of a carotid artery).13 Medical therapy may shrink the tumor in some patients and may thereby make surgical resection easier and more likely to be successful.
Radiotherapy is usually reserved for patients whose tumors recur or persist postoperatively and who are resistant to or intolerant of medical therapy.14 The soft tissue changes caused by acromegaly may regress with treatment to some degree, but they are not likely to resolve completely; the bone changes do not regress.
CASE CONTINUED: MEDICAL TREATMENT
Three months after transsphenoidal resection of his pituitary macroadenoma, our patient’s laboratory values are as follows:
- IGF-1 400 ng/mL
- Morning cortisol 20 μg/dL
- Testosterone 95 ng/dL
- LH 2.1 mU/mL
- FSH 3.7 mU/mL
- Prolactin 12 ng/mL
- TSH 2.3 mIU/L
- Free T4 1.2 ng/dL
- Basic metabolic panel normal.
The patient denies frequent urination or increased thirst. Repeat MRI of the pituitary with contrast shows a residual 1.3-cm adenoma with no suprasellar extension.
6. What is the best next treatment choice for the patient?
- A GH receptor antagonist (pegvisomant)
- A somatostatin receptor ligand (SRL) such as octreotide
- Cabergoline (a dopamine agonist)
- A combination of an SRL and pegvisomant
An SRL such as octreotide would be the best choice for this patient.
The medical options for acromegaly are SRLs, pegvisomant, and cabergoline.21–23 The Endocrine Society guidelines recommend either an SRL or pegvisomant as the initial adjuvant medical therapy in patients with persistent disease after surgery.14 However, pegvisomant is much more expensive than any SRL, so an SRL would be a better choice in this patient. Also, pegvisomant does not suppress tumor growth, in contrast to SRLs, so SRLs are preferred in patients with large tumors abutting the optic chiasm.14
SRLs are used as primary therapy in patients who cannot be cured by surgery, have extensive cavernous sinus invasion, have no chiasmal compression, or are poor surgical candidates.
The medical treatment of acromegaly is summarized in Table 2.14,15 Side effects of the medications used to treat acromegaly are summarized in Table 3.14
CASE CONTINUED: RADIOTHERAPY
The patient is treated with octreotide, and the dose is subsequently titrated upward. His central hypogonadism is treated with testosterone gel. After 3 months, his IGF-1 level decreases to 190 ng/mL, the total testosterone increases to 450 ng/dL, and the hemoglobin A1c decreases to 5.9%.
The patient asks if stereotactic radiotherapy, which he read about on the Internet, can cure his acromegaly so that he can avoid the monthly octreotide injections.
7. Which statement best describes radiotherapy’s therapeutic effect in acromegaly?
- Stereotactic radiotherapy is more effective than medical therapy and should be used as a second-line treatment after surgery
- Stereotactic radiotherapy is less effective than conventional radiotherapy
- Stereotactic radiotherapy leads to stability or a decrease in the size of the GH-secreting tumor in 93% to 100% of patients in 5 to 10 years and to biochemical remission in 40% to 60% of patients at 5 years
- Stereotactic radiotherapy causes hypopituitarism in no more than 1% of patients
Stereotactic radiotherapy leads to stability or a decrease in the size of the GH-secreting tumor in 93% to 100% of patients in 5 to 10 years and biochemical remission in 40% to 60% of patients at 5 years.24,25
Hypopituitarism develops in up to 50% of patients at 5 years, and its incidence increases with the duration of follow-up.24 The risk of other complications is low (0% to 5% for new visual deficits, cranial nerve damage, or brain radionecrosis, and 0% to 1% for secondary brain tumors).24
Conventional radiotherapy has fallen out of favor because it is associated with an increased risk of death (mainly from stroke) independent of IGF-1 and GH levels, and a higher rate of complications than stereotactic radiotherapy.14,16 Radiotherapy is reserved for postsurgical treatment of patients with recurrent or persistent tumors who are resistant to or cannot tolerate medical therapy; it is the third-line treatment.24
Given that our patient responded to the medical therapy and tolerated it well and given the high risk of hypopituitarism associated with stereotactic radiotherapy, the latter would not be appropriate for the patient.
His fatigue has diminished further and his sexual performance has improved. He is still married and his wife no longer suspects him of infidelity.
KEY POINTS
- IGF-1 is the screening test of choice in a patient with signs and symptoms of acromegaly.
- A growth hormone suppression test with a 75-g oral glucose load is the gold standard test for confirmation of the diagnosis of acromegaly in patients with an elevated IGF-1 level.
- Transsphenoidal resection of the growth hormone-secreting pituitary macroadenoma is the initial treatment of choice for acromegaly.
- Patients with residual or recurrent growth hormone-secreting pituitary macroadenoma can be treated with somatostatin receptor ligands, a growth hormone receptor antagonist (pegvisomant), and a dopamine agonist cabergoline.
- Radiotherapy is reserved for postsurgical treatment of patients with recurrent or persistent tumors who are resistant to or intolerant of medical therapy. Stereotactic radiotherapy has largely replaced conventional radiotherapy.
A 58-year-old man presents with a 1-year history of chronic daytime fatigue, low libido, and difficulty achieving erections. He is upset: his wife suspects him of having an extramarital affair because, in addition to problems with his sexual performance, he has not been wearing his wedding ring. The patient explains that the ring has become too small for his finger and that he has never cheated on his wife. His wife has also been complaining that he snores loudly at night.
The patient works as an accountant. He has no known allergies to medications and takes no medications or supplements. He has no surgical history. He has never smoked tobacco or abused illicit drugs. He drinks a glass of wine once a week.
His father died at age 78 of a myocardial infarction; his 86-year-old mother has hypertension. He has no siblings. His 28-year-old biological son is healthy.
Physical examination
His temperature is 97.9°F (36.6°C), blood pressure 150/90 mm Hg, heart rate 80 per minute, respiratory rate 12 per minute, and oxygen saturation 98% on room air. His height is 5 feet 11 inches (180 cm), weight 250 lb (113 kg), and body mass index 35 kg/m2.
His forehead is wide with deep creases, his jaw, nose, and lower lip are prominent, and his tongue, hands, and feet are large. He has mild thyromegaly with no palpable nodules.
On cardiac examination, his point of maximal impulse is 3 cm lateral to the left midclavicular line in the fifth intercostal space; he has normal S1 and S2 with no murmurs, rubs, or gallops. The lungs are clear on auscultation. His abdomen is soft, nontender, and nondistended; the liver is palpated 2 cm below the costal margin. His extremities are not edematous.
LABORATORY TESTING
1. In addition to a complete blood cell count and comprehensive metabolic panel, which is the most appropriate test to order?
- Growth hormone (GH) level
- Insulin-like growth factor 1 (IGF-1) level
- GH and IGF-1 levels
- IGF-2 level
Acromegaly, an overview
The patient’s history of snoring and daytime fatigue suggests obstructive sleep apnea, which together with his enlarging ring finger size, wide forehead with deep creases, prominent jaw, nose, and lower lip, and enlarged thyroid, heart, and liver suggests acromegaly.
In most cases, acromegaly is caused by a GH-secreting pituitary adenoma. Rare causes include hypothalamic tumors that secrete GH-releasing hormone (GHRH) and ectopic secretion of GHRH or GH.1 Pseudoacromegaly, a mimic, is characterized by acromegalic features without hypersecretion of GH and with normal IGF-1 levels.4
The prevalence of acromegaly is 36 to 60 cases per million, and its annual incidence is 3 to 4 per million.5
With this patient’s presentation, the most appropriate next step is to order an IGF-1 level to screen for acromegaly.
GH secretion is pulsatile, IGF-1 secretion is not
GH is synthesized and stored in somatotroph cells, which account for more than 50% of pituitary hormone-secreting cells.6 Three hormones regulate synthesis and secretion of GH: GHRH, ghrelin, and somatostatin.7 GH secretion is pulsatile, with minimal basal secretion dependent on sex, age, neurotransmitters, exercise, and stress.7 It exerts its physiologic effects through an interaction with the GH receptor, a single-chain transmembrane glycoprotein.8,9
A GH-secreting adenoma develops when pituitary somatotroph cells undergo a monoclonal expansion. Mutations of various genes such as GNAS, PRKAR1A, and AIP are suspected of triggering such expansion. Disruption of the MENIN gene leads to multiple endocrine neoplasia syndrome 1, a combination of pituitary adenoma, pancreatic tumor, and primary hyperparathyroidism.9 The pattern of cytoplasmic keratin in somatotroph cells defines 2 histologic subtypes: densely granulated and sparsely granulated. The latter subtype is associated with more-invasive lesions that are seen more often in younger patients and are less responsive to somatostatin ligand therapy.10
GH induces transcription of IGF-1, mostly in the liver. In contrast to GH, IGF-1 secretion is not pulsatile, and therefore IGF-1 can be measured more reliably in serum, and the results can be interpreted according to age- and sex-adjusted reference ranges.
The IGF-1 level is a very sensitive test, but it is not very specific. It can be falsely elevated in pregnancy, in patients on estrogen replacement therapy, and in late adolescence.11 In addition, it may be difficult to interpret the IGF-1 level in the setting of malnutrition, severe hyperglycemia, renal or hepatic failure, and hypothyroidism.11,12
Nonpulsatile secretion and high sensitivity make the IGF-1 level the screening test of choice for acromegaly.9,12 In contrast, because of the pulsatile nature of GH synthesis, one cannot rely on a random GH level alone to detect the hormone’s hypersecretion.
IGF-2 has no role in acromegaly
IGF-2, produced mainly by the liver, plays an important role in promoting fetal growth. IGF-2 may induce hypoglycemia when secreted by some mesenchymal tumors.13 This hormone has no role in the pathogenesis of acromegaly and should not be measured in this patient.
CASE CONTINUED: FURTHER TESTING
The patient’s IGF-1 level is 590 ng/mL; the reference range for his age and sex is 68 to 245 ng/mL.
A sleep study confirms obstructive sleep apnea, and the patient is started on continuous positive airway pressure at night, with some reduction of his fatigue.
2. What is the most appropriate next step?
- Order magnetic resonance imaging (MRI) of the pituitary with gadolinium contrast
- Perform a GH suppression test with a 75-g oral glucose load
- Perform a GH stimulation test
- Refer the patient to a neurosurgeon for a consultation
The most appropriate next step is a GH suppression test, performed by measuring the plasma GH level 2 hours after giving 75 g of glucose by mouth. This confirmatory test is necessary because the IGF-1 level can be falsely elevated. The normal response to an oral glucose challenge is suppression of the GH level to below 1 μg/L. Failure to suppress GH confirms the diagnosis of acromegaly.14
A GH stimulation test with insulin-induced hypoglycemia or with GHRH-arginine would be appropriate if GH deficiency were suspected rather than hypersecretion.
Imaging of the pituitary with MRI before obtaining biochemical confirmation of the diagnosis of acromegaly may mislead the physician because MRI does not determine the functional status of a pituitary tumor. Correct treatment of a pituitary tumor depends on whether the tumor causes hypersecretion or deficiency of any pituitary hormones.
Referral to a neurosurgeon for a consultation is premature until a biochemical diagnosis of acromegaly is made and a pituitary adenoma is subsequently demonstrated by imaging.
3. The patient’s GH level is 10 μg/L 2 hours after oral administration of 75 g of glucose. What is the most appropriate next step?
- Radiography of the skull to image the pituitary at a low cost
- MRI of the pituitary with contrast after making sure the patient’s renal function is normal
- MRI of the pituitary without contrast
- Computed tomography of the head
The next step is MRI of the pituitary with contrast (gadolinium) after obtaining blood urea nitrogen and creatinine measurements to make sure the patient’s renal function is normal.14
Gadolinium contrast is contraindicated in patients with severely reduced renal function (glomerular filtration rate < 30 mL/min/1.73 m2) because of the risk of nephrogenic systemic fibrosis. In such a case, MRI without contrast would be appropriate.
MRI is the most sensitive imaging test for detecting a pituitary adenoma, as it can detect tumors as small as 2 mm. A pituitary macroadenoma (> 10 mm in diameter) is detected in more than 75% of patients with acromegaly at diagnosis. The tumor often invades one or both cavernous sinuses or extends to the suprasellar region, possibly impinging on the optic chiasm.15
If MRI is contraindicated, computed tomography of the head should be performed.
CASE CONTINUED: IMAGING
The patient’s comprehensive metabolic panel is normal, but his fasting plasma glucose is 135 mg/dL (reference range 74–99). Pituitary MRI with contrast shows a 3-cm pituitary adenoma with suprasellar extension, impinging on the optic chiasm and invading the right cavernous sinus.
4. In addition to repeating the fasting plasma glucose and measuring hemoglobin A1c, what is the most appropriate next step in managing this patient?
- Measure the prolactin, morning serum cortisol, total testosterone, follicle-stimulating hormone (FSH), luteinizing hormone (LH), thyroid-stimulating hormone (TSH), and free thyroxine (T4) levels; refer the patient to an ophthalmologist for a formal evaluation of visual fields
- Measure these hormone levels; perform a gross evaluation of the visual fields and refer the patient to an ophthalmologist only if visual field deficits are found on the gross examination
- Measure these hormone levels; refer the patient to an ophthalmologist only if he complains of vision changes
- Do not order any additional tests; instruct the patient to call the office if he develops any vision changes
This patient should have all of these hormones measured. In addition, given that his macroadenoma is impinging on the optic chiasm, he should be referred to an ophthalmologist for a formal evaluation of visual fields even if the latter are intact on gross examination and even if the patient does not complain of any visual changes.
Abnormalities of hormones other than GH and IGF-1 in acromegaly
Secretion of pituitary hormones other than GH and IGF-1 must be assessed.
Prolactin. GH-secreting tumors also secrete prolactin in up to one-third of patients, with the resulting hyperprolactinemia contributing to hypogonadism.11 Prolactin hypersecretion should be distinguished from hyperprolactinemia caused by pituitary stalk compression, which may be evident on MRI.
Measuring the serum prolactin level with 1:100 dilution to counteract the “hook effect” may unmask severe hyperprolactinemia due to a large macroprolactinoma. (The hook effect occurs when the prolactin level is so high that there is not enough antibody in the assay to bind both ends of all the prolactin molecules present, causing the reading to be falsely low.).
Cortisol, T4, testosterone. Patients with acromegaly may develop central adrenal insufficiency, central hypothyroidism, and central hypogonadism; these hormonal deficits may occur in isolation or in combination.
Also, patients should be assessed for comorbidities such as colon cancer (all patients with acromegaly require a colonoscopy, as acromegaly raises the risk of colon cancer), diabetes mellitus, hypertension, cardiomyopathy, and sleep apnea.16
Visual field loss may be insidious
The diagnostic and treatment algorithm for acromegaly is summarized in Figure 1.
CASE CONTINUED: LABORATORY VALUES, TREATMENT OPTIONS
Our patient’s repeat fasting plasma glucose is 137 mg/dL; his hemoglobin A1c is 7.3%, consistent with diabetes mellitus secondary to acromegaly. Other laboratory values:
- Morning cortisol level 15 μg/dL (reference range 5.3–22.5),
- Prolactin 23 ng/mL, confirmed with 1:100 dilution (4.0–15.2)
- Total testosterone 59 ng/dL (193–824)
- LH 2.1 mIU/mL (1.8–10.8)
- FSH 3.0 mIU/mL (1.5–12.4)
- TSH 2.5 mIU/L (0.5–4.5)
- Free T4 1.3 ng/dL (0.9–1.7).
The patient is started on metformin 500 mg by mouth twice a day, counseled on a healthy diet, and informed that his diabetes may be a complication of his acromegaly. He is anxious to learn how his acromegaly can be treated.
5. What treatment would you recommend for the patient’s acromegaly?
- Medical treatment first, then transsphenoidal resection of the pituitary macroadenoma if medical treatment fails
- Medical treatment first, radiotherapy if medical treatment fails, and transsphenoidal resection of the pituitary macroadenoma as a last resort
- Transsphenoidal resection of the pituitary macroadenoma first, medical treatment if surgery fails, and radiotherapy if both surgery and medical treatment fail
- Taking a safe, conservative approach, monitoring IGF-1 levels frequently; starting medical treatment if acromegaly does not go into remission in 1 year
The initial treatment of choice for most patients with acromegaly is resection of the pituitary tumor.
A transsphenoidal approach is used for most patients; only rarely is craniotomy necessary. Endoscopic and microsurgical techniques reduce postoperative morbidity.17 Postoperative complications include symptoms related to the transsphenoidal approach (nasal congestion, sinusitis, epistaxis), cerebrospinal fluid leak, hemorrhage, meningitis, stroke, visual impairment, vascular damage, transient or permanent diabetes insipidus, and hypopituitarism. The surgical mortality rate is less than 0.5%.18,19
Successful resection of a pituitary tumor would lead to normalization of the IGF-1 level, a drop of the GH level to below 1 μg/L, and relief of the effect of the tumor pressing against other structures. An IGF-1 level and a random GH level should be obtained 12 weeks after the surgery.14 If the GH level is higher than 1 μg/L, a GH suppression test with a 75-g oral glucose load should be performed.14 MRI of the sella turcica should be done 12 weeks after surgery to visualize residual tumor and adjacent structures.14
A large tumor size, suprasellar extension, and high preoperative levels of IGF-1 and GH are associated with a lack of surgical success; however, surgical debulking should still be considered in patients with a low chance for surgical cure to improve the probability of achieving biochemical remission with postoperative medical and radiologic therapy.20
Medical therapy can be the initial treatment if the patient refuses surgery or if surgery is contraindicated because of severe comorbidities or because structural features of the tumor confer a high surgical risk (eg, if the adenoma encases the cavernous portion of a carotid artery).13 Medical therapy may shrink the tumor in some patients and may thereby make surgical resection easier and more likely to be successful.
Radiotherapy is usually reserved for patients whose tumors recur or persist postoperatively and who are resistant to or intolerant of medical therapy.14 The soft tissue changes caused by acromegaly may regress with treatment to some degree, but they are not likely to resolve completely; the bone changes do not regress.
CASE CONTINUED: MEDICAL TREATMENT
Three months after transsphenoidal resection of his pituitary macroadenoma, our patient’s laboratory values are as follows:
- IGF-1 400 ng/mL
- Morning cortisol 20 μg/dL
- Testosterone 95 ng/dL
- LH 2.1 mU/mL
- FSH 3.7 mU/mL
- Prolactin 12 ng/mL
- TSH 2.3 mIU/L
- Free T4 1.2 ng/dL
- Basic metabolic panel normal.
The patient denies frequent urination or increased thirst. Repeat MRI of the pituitary with contrast shows a residual 1.3-cm adenoma with no suprasellar extension.
6. What is the best next treatment choice for the patient?
- A GH receptor antagonist (pegvisomant)
- A somatostatin receptor ligand (SRL) such as octreotide
- Cabergoline (a dopamine agonist)
- A combination of an SRL and pegvisomant
An SRL such as octreotide would be the best choice for this patient.
The medical options for acromegaly are SRLs, pegvisomant, and cabergoline.21–23 The Endocrine Society guidelines recommend either an SRL or pegvisomant as the initial adjuvant medical therapy in patients with persistent disease after surgery.14 However, pegvisomant is much more expensive than any SRL, so an SRL would be a better choice in this patient. Also, pegvisomant does not suppress tumor growth, in contrast to SRLs, so SRLs are preferred in patients with large tumors abutting the optic chiasm.14
SRLs are used as primary therapy in patients who cannot be cured by surgery, have extensive cavernous sinus invasion, have no chiasmal compression, or are poor surgical candidates.
The medical treatment of acromegaly is summarized in Table 2.14,15 Side effects of the medications used to treat acromegaly are summarized in Table 3.14
CASE CONTINUED: RADIOTHERAPY
The patient is treated with octreotide, and the dose is subsequently titrated upward. His central hypogonadism is treated with testosterone gel. After 3 months, his IGF-1 level decreases to 190 ng/mL, the total testosterone increases to 450 ng/dL, and the hemoglobin A1c decreases to 5.9%.
The patient asks if stereotactic radiotherapy, which he read about on the Internet, can cure his acromegaly so that he can avoid the monthly octreotide injections.
7. Which statement best describes radiotherapy’s therapeutic effect in acromegaly?
- Stereotactic radiotherapy is more effective than medical therapy and should be used as a second-line treatment after surgery
- Stereotactic radiotherapy is less effective than conventional radiotherapy
- Stereotactic radiotherapy leads to stability or a decrease in the size of the GH-secreting tumor in 93% to 100% of patients in 5 to 10 years and to biochemical remission in 40% to 60% of patients at 5 years
- Stereotactic radiotherapy causes hypopituitarism in no more than 1% of patients
Stereotactic radiotherapy leads to stability or a decrease in the size of the GH-secreting tumor in 93% to 100% of patients in 5 to 10 years and biochemical remission in 40% to 60% of patients at 5 years.24,25
Hypopituitarism develops in up to 50% of patients at 5 years, and its incidence increases with the duration of follow-up.24 The risk of other complications is low (0% to 5% for new visual deficits, cranial nerve damage, or brain radionecrosis, and 0% to 1% for secondary brain tumors).24
Conventional radiotherapy has fallen out of favor because it is associated with an increased risk of death (mainly from stroke) independent of IGF-1 and GH levels, and a higher rate of complications than stereotactic radiotherapy.14,16 Radiotherapy is reserved for postsurgical treatment of patients with recurrent or persistent tumors who are resistant to or cannot tolerate medical therapy; it is the third-line treatment.24
Given that our patient responded to the medical therapy and tolerated it well and given the high risk of hypopituitarism associated with stereotactic radiotherapy, the latter would not be appropriate for the patient.
His fatigue has diminished further and his sexual performance has improved. He is still married and his wife no longer suspects him of infidelity.
KEY POINTS
- IGF-1 is the screening test of choice in a patient with signs and symptoms of acromegaly.
- A growth hormone suppression test with a 75-g oral glucose load is the gold standard test for confirmation of the diagnosis of acromegaly in patients with an elevated IGF-1 level.
- Transsphenoidal resection of the growth hormone-secreting pituitary macroadenoma is the initial treatment of choice for acromegaly.
- Patients with residual or recurrent growth hormone-secreting pituitary macroadenoma can be treated with somatostatin receptor ligands, a growth hormone receptor antagonist (pegvisomant), and a dopamine agonist cabergoline.
- Radiotherapy is reserved for postsurgical treatment of patients with recurrent or persistent tumors who are resistant to or intolerant of medical therapy. Stereotactic radiotherapy has largely replaced conventional radiotherapy.
- Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest 2009; 119:3189–3202.
- Molitch ME. Clinical manifestations of acromegaly. Endocrinol Metab Clin North Am 1992; 21:597–614.
- Dineen R, Stewart PM, Sherlock M. Acromegaly. QJM 2017; 110:411–420.
- Yacub A, Yaqub N. Insulin-mediated pseudoacromegaly: a case report and review of the literature. W V Med J 2008; 104:12–15.
- Mestron A, Webb SM, Astorga R, et al. Epidemiology, clinical characteristics, outcome, morbidity and mortality in acromegaly based on the Spanish Acromegaly Registry (Registro Espanol de Acromegalia, REA). Eur J Endocrinol 2004; 151:439–446.
- Zhu X, Lin CR, Prefontaine CG, Tollkuhn J, Rosenfeld MG. Genetic control of pituitary development and hypopituitarism. Curr Opin Genet Dev 2005; 15:332–340.
- Tannenbaum GS, Epelbaum J, Bowers CY. Interrelationship between the novel peptide ghrelin and somatostatin/growth hormone-releasing hormone in regulation of pulsatile growth hormone secretion. Endocrinology 2003; 144:967–974.
- Lanning NJ, Carter-Su C. Recent advances in growth hormone signaling. Rev Endocr Metab Disord 2006; 7:225–235.
- Colao A, Ferone D, Marzullo P, Lombardi G. Systemic complications of acromegaly: epidemiology, pathogenesis, and management. Endocr Rev 2004; 25:102–152.
- Larkin S, Reddy R, Karavitaki N, Cudlip S, Wass J, Ansorge O. Granulation pattern, but not GSP or GHR mutation, is associated with clinical characteristics in somatostatin-naive patients with somatotroph adenomas. Eur J Endocrinol 2013; 168:491–499.
- Dineen R, Stewart PM, Sherlock M. Acromegaly. QJM 2017; 110:411–420.
- Peacey SR, Toogood AA, Veldhuis JD, Thorner MO, Shalet SM. The relationship between 24-hour growth hormone secretion and insulin-like growth factor I in patients with successfully treated acromegaly: impact of surgery or radiotherapy. J Clin Endocrinol Metab 2001; 86:259–266.
- Livingstone C. IGF2 and cancer. Endocr Relat Cancer 2013; 20:R321–R339.
- Katznelson L, Laws ER Jr, Melmed S, et al. Acromegaly: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2014; 99:3933–3951.
- Melmed S. Acromegaly. N Engl J Med 2006; 355:2558–2573.
- Melmed S, Casanueva FF, Klibanski A, et al. A consensus on the diagnosis and treatment of acromegaly complications. Pituitary 2013; 16:294–302.
- Marquez Y, Tuchman A, Zada G. Surgery and radiosurgery for acromegaly: a review of indications, operative techniques, outcomes, and complications. Int J Endocrinol 2012; 2012: 386401.
- Jane JA Jr, Starke RM, Elzoghby MA, et al. Endoscopic transsphenoidal surgery for acromegaly: remission using modern criteria, complications, and predictors of outcome. J Clin Endocrinol Metab 2011; 96:2732–2740.
- Cappabianca P, Cavallo LM, Colao A, de Divitiis E. Surgical complications associated with the endoscopic endonasal transsphenoidal approach for pituitary adenomas. J Neurosurg 2002; 97:293–298.
- Nomikos P, Buchfelder M, Fahlbusch R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical “cure.” Eur J Endocrinol 2005; 152:379–387.
- Howlett TA, Willis D, Walker G, Wass JA, Trainer PJ; UK Acromegaly Register Study Group (UKAR-3). Control of growth hormone and IGF1 in patients with acromegaly in the UK: responses to medical treatment with somatostatin analogues and dopamine agonists. Clin Endocrinol (Oxf) 2013; 79:689–699.
- Katznelson L. Pegvisomant for the treatment of acromegaly-translation of clinical trials into clinical practice. Nat Clin Pract Endocrinol Metab 2007; 3:514–515.
- Freda PU, Reyes CM, Nuruzzaman AT, Sundeen RE, Khandji AG, Post KD. Cabergoline therapy of growth hormone & growth hormone/prolactin secreting pituitary tumors. Pituitary 2004; 7:21–30.
- Castinetti F, Morange I, Dufour H, Regis J, Brue T. Radiotherapy and radiosurgery in acromegaly. Pituitary 2009; 12:3–10.
- Gheorghiu ML. Updates in outcomes of stereotactic radiation therapy in acromegaly. Pituitary 2017; 20:154–168.
- Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest 2009; 119:3189–3202.
- Molitch ME. Clinical manifestations of acromegaly. Endocrinol Metab Clin North Am 1992; 21:597–614.
- Dineen R, Stewart PM, Sherlock M. Acromegaly. QJM 2017; 110:411–420.
- Yacub A, Yaqub N. Insulin-mediated pseudoacromegaly: a case report and review of the literature. W V Med J 2008; 104:12–15.
- Mestron A, Webb SM, Astorga R, et al. Epidemiology, clinical characteristics, outcome, morbidity and mortality in acromegaly based on the Spanish Acromegaly Registry (Registro Espanol de Acromegalia, REA). Eur J Endocrinol 2004; 151:439–446.
- Zhu X, Lin CR, Prefontaine CG, Tollkuhn J, Rosenfeld MG. Genetic control of pituitary development and hypopituitarism. Curr Opin Genet Dev 2005; 15:332–340.
- Tannenbaum GS, Epelbaum J, Bowers CY. Interrelationship between the novel peptide ghrelin and somatostatin/growth hormone-releasing hormone in regulation of pulsatile growth hormone secretion. Endocrinology 2003; 144:967–974.
- Lanning NJ, Carter-Su C. Recent advances in growth hormone signaling. Rev Endocr Metab Disord 2006; 7:225–235.
- Colao A, Ferone D, Marzullo P, Lombardi G. Systemic complications of acromegaly: epidemiology, pathogenesis, and management. Endocr Rev 2004; 25:102–152.
- Larkin S, Reddy R, Karavitaki N, Cudlip S, Wass J, Ansorge O. Granulation pattern, but not GSP or GHR mutation, is associated with clinical characteristics in somatostatin-naive patients with somatotroph adenomas. Eur J Endocrinol 2013; 168:491–499.
- Dineen R, Stewart PM, Sherlock M. Acromegaly. QJM 2017; 110:411–420.
- Peacey SR, Toogood AA, Veldhuis JD, Thorner MO, Shalet SM. The relationship between 24-hour growth hormone secretion and insulin-like growth factor I in patients with successfully treated acromegaly: impact of surgery or radiotherapy. J Clin Endocrinol Metab 2001; 86:259–266.
- Livingstone C. IGF2 and cancer. Endocr Relat Cancer 2013; 20:R321–R339.
- Katznelson L, Laws ER Jr, Melmed S, et al. Acromegaly: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2014; 99:3933–3951.
- Melmed S. Acromegaly. N Engl J Med 2006; 355:2558–2573.
- Melmed S, Casanueva FF, Klibanski A, et al. A consensus on the diagnosis and treatment of acromegaly complications. Pituitary 2013; 16:294–302.
- Marquez Y, Tuchman A, Zada G. Surgery and radiosurgery for acromegaly: a review of indications, operative techniques, outcomes, and complications. Int J Endocrinol 2012; 2012: 386401.
- Jane JA Jr, Starke RM, Elzoghby MA, et al. Endoscopic transsphenoidal surgery for acromegaly: remission using modern criteria, complications, and predictors of outcome. J Clin Endocrinol Metab 2011; 96:2732–2740.
- Cappabianca P, Cavallo LM, Colao A, de Divitiis E. Surgical complications associated with the endoscopic endonasal transsphenoidal approach for pituitary adenomas. J Neurosurg 2002; 97:293–298.
- Nomikos P, Buchfelder M, Fahlbusch R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical “cure.” Eur J Endocrinol 2005; 152:379–387.
- Howlett TA, Willis D, Walker G, Wass JA, Trainer PJ; UK Acromegaly Register Study Group (UKAR-3). Control of growth hormone and IGF1 in patients with acromegaly in the UK: responses to medical treatment with somatostatin analogues and dopamine agonists. Clin Endocrinol (Oxf) 2013; 79:689–699.
- Katznelson L. Pegvisomant for the treatment of acromegaly-translation of clinical trials into clinical practice. Nat Clin Pract Endocrinol Metab 2007; 3:514–515.
- Freda PU, Reyes CM, Nuruzzaman AT, Sundeen RE, Khandji AG, Post KD. Cabergoline therapy of growth hormone & growth hormone/prolactin secreting pituitary tumors. Pituitary 2004; 7:21–30.
- Castinetti F, Morange I, Dufour H, Regis J, Brue T. Radiotherapy and radiosurgery in acromegaly. Pituitary 2009; 12:3–10.
- Gheorghiu ML. Updates in outcomes of stereotactic radiation therapy in acromegaly. Pituitary 2017; 20:154–168.
Transforming the delivery of cardiovascular care: Research and innovation in the Heart & Vascular Institute

Supplement Editor:
Umesh Khot, MD
Contents
Results of the GLAGOV trial
Steven E. Nissen and Stephen J. Nicholls
Trends in cardiovascular risk profiles
Samir Kapadia
Expanding indications for TAVR: The preferred procedure in intermediate-risk patients?
David L. Brown
CABG: A continuing evolution
Faisal Bakaeen
Improving the safety and efficacy of robotically assisted mitral valve surgery
Stephanie Mick
Aortic replacement in cardiac surgery
Eric E. Roselli
Supplement Editor:
Umesh Khot, MD
Contents
Results of the GLAGOV trial
Steven E. Nissen and Stephen J. Nicholls
Trends in cardiovascular risk profiles
Samir Kapadia
Expanding indications for TAVR: The preferred procedure in intermediate-risk patients?
David L. Brown
CABG: A continuing evolution
Faisal Bakaeen
Improving the safety and efficacy of robotically assisted mitral valve surgery
Stephanie Mick
Aortic replacement in cardiac surgery
Eric E. Roselli
Supplement Editor:
Umesh Khot, MD
Contents
Results of the GLAGOV trial
Steven E. Nissen and Stephen J. Nicholls
Trends in cardiovascular risk profiles
Samir Kapadia
Expanding indications for TAVR: The preferred procedure in intermediate-risk patients?
David L. Brown
CABG: A continuing evolution
Faisal Bakaeen
Improving the safety and efficacy of robotically assisted mitral valve surgery
Stephanie Mick
Aortic replacement in cardiac surgery
Eric E. Roselli
