User login
Soft Tissue Sarcoma: Diagnosis and Treatment
Introduction
Soft tissue sarcomas (STSs) are rare adult tumors, with 3.4 new cases per 100,000 persons or 12,310 expected new cases in 2016.1 Sarcomas are a heterogeneous collection of tumors that affect fat, muscle, nerve, nerve sheath, vascular, and connective tissues. There are more than 50 histological subtypes that comprise this diverse category of tumors. Treatment varies by stage, with limb-sparing surgery representing the mainstay of curative-intent treatment. Radiation and chemotherapy may also be considered depending on the size, grade, and location of the tumor. Survival rates have been stagnant until recently, with a disease-specific survival hovering around 65%.1 Given the complexity of these cases, all patients ideally should be evaluated and treated by a multidisciplinary team at an institution with extensive experience treating STS.2
Epidemiology and Classification
The most common STS subtypes are gastrointestinal stromal tumor (GIST), undifferentiate pleomorphic sarcoma (previously referred to as malignant fibrous histiocytoma), liposarcoma, leiomyosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, and unclassified sarcoma.3 Liposarcoma is one of the most common subtypes, comprising 20% of all STSs; it is subdivided into well-differentiated/dedifferentiated liposarcomas, myxoid/round cell liposarcomas, and pleomorphic liposarcomas. Well-differentiated liposarcomas tend to occur in the retroperitoneum and limbs, while both myxoid and round cell as well as pleomorphic liposarcomas more commonly originate on the limbs. Histology varies based on subtype and ranges from mature-appearing adipocytes and fibroblasts to undifferentiated cells with minimal lipogenic differentiation.4
Leiomyosarcomas are smooth muscle tumors and are usually located in the retroperitoneum, but have also been associated with peripheral soft tissue and vasculature. Typical histology ranges from well-defined areas of spindle-shaped cells to poorly differentiated anaplastic spindle cells.5,6 Synovial sarcomas are a distinct type of STS that can show epithelial differentiation and account for 5% of adult STSs. The extremities are the most common presenting location (90%).7
Rhabdomyosarcomas are skeletal muscle tumors and are further subdivided into embryonal, alveolar, and pleomorphic subtypes. Embryonal histology ranges from primitive mesenchymal-appearing cells to highly differentiated muscle cells. Alveolar rhabdomyosarcoma has the worst prognosis of the subtypes and consists of round cells with high nuclear-to-chromatin ratios that form “glandular-like” or “alveolar” spaces.8 Pleomorphic rhabdomyosarcomas are composed of rhabdomyoblasts that can affect many different locations, but most commonly present on the lower extremities.9
Malignant peripheral nerve sheath tumor (MPNST) comprises 5% to 10% of all STSs. These tumors are associated with neurofibromatosis type 1 (NF-1), with 25% to 50% of tumors occurring in NF-1 patients. Additionally, most patients have a truncating lesion in the NF1 gene on chromosome 17.10 Anghileri et al in their single institution analysis of 205 patients with MPNSTs found the 2 most common presenting sites were the trunk and extremities. Histologically, these tumors have dense fascicles of spindle cells.10
GISTs are the most common STS of the gastrointestinal (GI) tract. Previously, GISTs were classified as smooth muscle tumors and were not accounted for in the literature as a separate entity distinct from leiomyomas, leiomyoblastomas, and leiomyosarcomas.11 GISTs are found throughout the GI tract: the most common sites are the stomach (60%) and small intestine (30%). Less common sites include duodenum (4%–5%), esophagus (1%), rectum (1%–2%), and appendix (< 0.2%).12 GISTs can be spindle cell, epithelioid, or mesenchymal tumors. Immunohistochemically, GISTs are KIT (CD117) positive. Other cell markers that are also commonly positive include CD34 (60%–70%) and smooth muscle actin (SMA) (25%).11 The majority of GISTs (80%) have an activating c-KIT gene mutation. The most common mutation site is exon 11, with less common c-KIT gene mutations also occurring at exon 9 or 13. Not all GISTs have KIT mutations. The second most common mutation is the PDGFRA mutation (5%–10% of GISTs).2 A minority of GISTs are negative for both KIT and PDGFRA mutations. These tumors were previously called wild-type, but as the majority have either a succinate dehydrogenase (SDH) loss of function or loss of SDHB protein expression, they are now referred to as SDH-deficient GISTs.2 GISTs vary in aggressiveness from incidental to aggressive. Typically, small intestine and rectal GISTs are more aggressive than gastric GISTs. Both size and mitotic rate help to predict the metastatic potential of the tumor. Tumors less than 2 cm in size and having a mitotic rate of less than 5 per 50 high-power fields (hpf) have the lowest risk of metastases, while tumors greater than 5 cm and with more than 5 mitoses per 50 hpf have the highest rates of metastases.12
Angiosarcomas are rare tumors comprising 4% of all STSs. Although they can occur in any site, the majority are cutaneous and occur most frequently in the head and neck regions. These tumors are either of vascular or lymphatic origin and are comprised of abnormal, pleomorphic, malignant endothelial cells. The most useful immunohistochemical markers include von Willebrand factor, CD31, and Ulex europaeus agglutinin 1. The majority of these tumors occur sporadically; however, radiation exposure, chronic lymphedema, and certain toxins including vinyl chloride and thorium dioxide are known risk factors.13
Undifferentiated sarcomas have no specific features and typically consist of primitive mesenchymal cells.
Clinical Evaluation
› Case Presentation
Initial Presentation and History
A 55-year-old man presents to his primary care physician with a painless mass in his anterior thigh. The mass has been present for the past 3 months and he believes that it is enlarging. The patient has a history of well-controlled hypertension and hyperlipidemia. His medications include atorvastatin and hydrochlorothiazide. He has no known drug allergies. Family history is notable for diabetes and hypertension. He drinks 4 to 5 alcoholic drinks a week and he is a former smoker. He quit smoking in his 30s and only smoked intermittently prior to quitting. He denies any illicit drug use. He works as a high school principal. Currently, he feels well. His review of systems is otherwise noncontributory.
Physical Examination
On physical exam, he is afebrile with a blood pressure of 132/75 mm Hg, respiratory rate of 10 breaths/min, and oxygen saturation of 99% on room air. He is a well appearing, overweight male. His head and neck exam is unremarkable. Lung exam reveals clear breath sounds, and cardiac exam reveals a regular rate and rhythm. His abdomen is obese, soft, and without hepatosplenomegaly. There is a large, fixed mass on the anterior lateral aspect of his right thigh. He has no appreciable lymphadenopathy. His neurological exam is unremarkable.
• What are risk factors for sarcoma?
There are few known risk factors for sarcoma. Established risks factors include prior radiation therapy, chronic lymphedema, viruses, and genetic cancer syndromes including Li-Fraumeni syndrome, hereditary retinoblastoma, and NF-1. Other environmental exposures include phenoxyacetic acids and chlorophenols.14 The majority of cases are sporadic, with only a minority of patients having one of these known risk factors.15 Up to one third of sarcomas have a specific translocation and are driven by fusion oncogenes (
• What is the typical presentation for sarcomas?
A painless mass is the most typical presenting symptom. Size at presentation varies based on location, with extremity and head and neck locations typically presenting at smaller sizes than retroperitoneal tumors.14 Patients may experience pain and numbness as the mass enlarges and impinges on surrounding structures including nerves and vasculature. The vast majority of patients are without systemic symptoms.
• How is sarcoma staged?
The American Joint Committee on Cancer (AJCC) staging system is the most widely used staging system in the United States. The latest AJCC manual was updated in 2010 to include a 3-tiered grading system where the tumor is classified according to tumor size, lymph node involvement, metastases, and grade at time of diagnosis (Table 2 and Table 3). Additionally, tumor depth in relation to deep fascia is also taken into account, with superficial tumors being assigned a designation of “a” and deep tumors a designation of “b.”
Previously, 2 of the most widely used grading systems were the National Cancer Institute (NCI) and French Federation of Cancer Centers Sarcoma Group (FNCLCC) systems, both 3-tier grading systems. The main components that determine the NCI grade are the tumor’s histologic type and location and the amount of tumor necrosis. The FNCLCC system evaluation focuses on tumor differentiation, mitotic rate, and amount of tumor necrosis. A study that compared the NCI and FNCLCC grading systems found that FNCLCC was a better predictor of mortality and distant metastasis.16 Previously, the AJCC was a 4-tier grading system, but the 2010 version was updated to the 3-tier FNCLCC grading system. Additionally, the AJCC system has reclassified single lymph node disease as stage III as it confers better survival than metastatic disease.17 It is important that pathology be evaluated by a sarcoma specialist as disagreements with regard to histologic subtype and grade are common.18,19
• What are the most important prognostic factors?
Prognostic factors include grade, size, and presence of metastases at presentation. Best survival is associated with low-grade, small tumors with no metastases at time of diagnosis.14
• What imaging should be considered?
Imaging should be undertaken to help differentiate between benign and malignant lesions. Ideally, it should be undertaken before a biopsy is planned as the imaging can be used to plan biopsy as well as provide invaluable prognostic information. There are several imaging modalities that should be considered during the preliminary work-up and staging of STSs. Conventional imaging includes magnetic resonance imaging (MRI) of the original tumor site; computed tomography (CT) to evaluate for pulmonary metastases and, depending on location, liver metastases; and in the case of small, low-grade tumors, chest radiography. MRI is considered the test of choice for soft tissue masses and can help delineate benign masses such as hematomas, lipomas, and hemangiomas from sarcomas.20 It is difficult to compare the accuracy of positron emission tomography (PET)/CT to CT and MRI because most studies have evaluated PET/CT in parallel with CT and MRI.21 Tateishi et al compared the accuracy of conventional imaging, PET/CT, and PET/CT combined with conventional imaging at determining the TNM staging for 117 patients. They found that conventional imaging correctly classified 77% of patients, PET alone correctly classified 70%, PET/CT correctly classified 83%, and PET/CT combined with conventional imaging correctly staged 87%.22
• Which subtypes are most likely to metastasize?
Although the vast majority of sarcomas spread hematogenously, 3 have a propensity to spread lymphogenously: epithelioid sarcoma, rhabdomyosarcoma, and clear-cell sarcoma. Additionally, certain subtypes are more likely to metastasize: leiomyosarcomas, synovial sarcomas, neurogenic sarcomas, rhabdomyosarcomas, and epithelioid sarcomas.23 Sarcomas metastasize to the lungs more frequently than to the liver. The metastatic pattern is defined primarily by sarcoma subtype and site of primary tumor. Sarcomas rarely metastasize to the brain (~1%).
Management
› Case Continued
The patient undergoes an ultrasound to better visualize the mass. Given the heterogeneous character of the mass, he is referred for an MRI to evaluate the mass and a CT scan of the chest, abdomen, and pelvis to evaluate for distant metastases. MRI reveals a 5.1 cm × 4.6 cm heterogeneous mass invading the superficial fascia of the rectus femoris muscle. No suspicious lymph nodes or other masses are identified on imaging. The patient next undergoes an image-guided core needle biopsy. Pathology from that procedure is consistent with a stage III, T2bNxMx, grade 3, dedifferentiated liposarcoma.
• What is the best management approach for this patient?
Surgery
Surgery is the mainstay of treatment for STS. Patients with the best prognosis are those who undergo complete resection with negative surgical margins.24,25 Goal tumor-free margin is 1 to 3 cm.26 Complete resection confers the best long-term survival. Both local and metastatic recurrence is higher in patients with incomplete resection and positive margins.24,25 In a study that analyzed 2084 localized primary STSs, patients with negative margins had a local recurrence rate of 15% versus a rate of 28% in patients with positive margins. This translated into higher 5-year local recurrence-free survival for patients with negative surgical margins (82%) compared to patients with positive margins (65%).27 Another study similarly found that patients with negative margins at referral to their institution who underwent postoperative radiation had high local control rates of 93% (95% confidence interval [CI] 87% to 97%) at 5, 10, and 15 years.26 Although radiation improves local control, neither preoperative or postoperative radiation has been shown to improve progression-free or overall survival.28 Other factors that are associated with risk of recurrence are tumor location, history of previous recurrence, age of patient, histopathology, tumor grade, and tumor size. Approximately 40% to 50% of patients with high-grade tumors (defined as size > 5 cm, deep location, and high grade) will develop distant metastases.29
Zagars et al found that positive or uncertain resection margin had a relative risk of local recurrence of 2.0 (95% CI 1.3 to 3.1; P = 0.002), and presentation with locally recurrent disease (vs new tumor) had a relative risk of local recurrence of 2.0 (95% CI 1.2 to 3.4; P = 0.013).26 Patients with STS of head and neck and deep trunk have higher recurrence rates than those with superficial trunk and extremity STS. A single-institution retrospective review demonstrated that patients with completely resectable retroperitoneal sarcomas have longer median survival (103 months) compared to patients with incompletely resected abdominal sarcomas (18 months).25Rosenberg and colleagues compared amputation to limb-sparing surgery and radiation.24 Their prospective analysis of 65 patients found no difference in disease-free and overall survival between the 2 treatment groups.The limb-sparing treatment group had higher rates of local recurrence, which was highly correlated with positive surgical margins on pathology.24 Evidence from this and similar studies has resulted in radical amputations being replaced by conservative limb-sparing procedures and radiation therapy. In those found to have positive margins, re-resection is an option for some. Patients who undergo re-resection have higher local control rates than patients with positive margins who do not undergo re-resection. The 5-year control rate for patients who undergo re-resection is 85% (95% CI 80% to 89%) compared to 78% (95% CI 71% to 83%) for those who do not undergo re-resection. Similarly, patients who undergo re-resection have lower rates of metastases at 5, 10, and 15 years as well as higher 5-, 10-, and 15-year disease-free survival rates.26
› Case Continued
The patient is referred for limb-sparing surgery after presentation at a multidisciplinary tumor board. Prior to undergoing resection of the tumor, he is also referred to radiation-oncology to discuss the risks and benefits of combination radiotherapy and surgery as opposed to surgical resection alone.
• What is the evidence for radiation therapy?
Radiation THERAPY
Radiation therapy is used in the preoperative, intraoperative, and postoperative settings to reduce the risk of local recurrence. There are several options for radiation, including external beam radiation therapy (EBRT), intraoperative radiation, and brachytherapy. A newer strategy, intensity-modulated radiation therapy (IMRT), utilizes 3-dimensional modeling to reduce radiation dosages. Overall there are no differences in overall survival or local recurrence rates between preoperative and postoperative radiation in STS.28
The rationale behind preoperative radiation is that it reduces seeding of tumor cells, especially at the time of surgery.30 Additionally, for EBRT, preoperative radiation has smaller field sizes and lower radiation doses. It can also help to reduce the size of the tumor prior to resection. Intraoperative radiation is often paired with preoperative radiation as a boost dose given only to the area of residual tumor.
Suit et al reviewed patients treated at a single institution with limb-sparing surgery and different radiation strategies. Local control rates between preoperative and postoperative radiation groups were not statistically significant. Local recurrence was linked to grade and size of the tumor in both groups. The authors did note, however, that the preoperative radiation group tended to have larger tumor sizes at baseline compared to the patients who received postoperative radiation.30 A study that compared 190 patients who received preoperative and postoperative EBRT or brachytherapy (primary end point was wound complications, and local control was a secondary end point) showed a trend towards greater local control with preoperative radiation; however, the preoperative radiation group had significantly more wound complications compared to the postoperative radiation group.31
Yang et al found that postoperative EBRT decreases rates of local recurrence compared to surgery alone in high-grade extremity sarcomas.32 However, there were no differences in rates of distant metastases and overall survival between the 2 treatment groups. Similarly, in patients with low-grade sarcoma, there were fewer local recurrences in those who received EBRT and surgery as compared to surgery alone.32 Another study that evaluated 164 patients who received either adjuvant brachytherapy or no further therapy after complete resection found that brachytherapy reduced local recurrence in high-grade sarcomas. No difference in local recurrence rates was found in patients with low-grade sarcomas, nor was a significant difference found in the rates of distant metastases and overall survival between the 2 treatment groups.33 With regards to IMRT, a single institution cohort experience with 41 patients who received IMRT following limb-sparing surgery had similar local control rates when compared to historical controls.34
› Case Continued
After discussion of the risks and benefits of radiation therapy, the patient opts for preoperative radiation prior to resection of his liposarcoma. He receives 50 Gy of EBRT prior to undergoing resection. Resection results in R1 margin consistent with microscopic disease. He receives 16 Gy of EBRT as a boost after recovery from his resection.2
• What is the evidence for neoadjuvant and adjuvant chemotherapy for stage I tumors?
Chemotherapy
Localized Sarcoma
For localized sarcoma, limb-sparing resection with or without radiation forms the backbone of treatment. Studies have evaluated chemotherapy in both the neoadjuvant and adjuvant settings, with the vast majority of studies evaluating doxorubicin-based chemotherapy regimens in the adjuvant settings. Due to the rare nature of sarcomas, most studies are not sufficiently powered to detect significant benefit from chemotherapy. Several trials evaluating chemotherapy regimens in the neoadjuvant and adjuvant settings needed to be terminated prematurely due to inadequate enrollment into the study.35,36
For stage IA (T1a-Tb, N0, M0, low grade) tumors, no additional therapy is recommended after limb-sparing surgery with appropriate surgical margins. For stage IB (T2a-2b, N0, M0, low grade) tumors with insufficient margins, re-resection and radiation therapy should be considered, while for stage IIA (T1a-1b, N0, M0, G2-3) tumors preoperative or postoperative radiation therapy is recommended.2 Studies have not found benefit of adjuvant chemotherapy in these low-grade, stage I tumors in terms of progression-free survival and overall survival.37
• At what stage should chemotherapy be considered?
For stage IIb and stage III tumors, surgery and radiation therapy again form the backbone of therapy; however, neoadjuvant and adjuvant chemotherapy are also recommended as considerations. Anthracycline-based chemotherapy with either single-agent doxorubicin or doxorubicin and ifosfamide in combination are considered first-line chemotherapy agents in locally advanced STS.2,29,37
Evidence regarding the efficacy of both neoadjuvant and adjuvant chemotherapy regimens in the setting of locally advanced high-grade STS has been mixed. The Sarcoma Meta-analysis Collaboration evaluated 14 trials of doxorubicin-based adjuvant chemotherapy and found a trend towards overall survival in the treatment groups that received chemotherapy.37 All trials included in the meta-analysis compared patients with localized resectable soft-tissue sarcomas who were randomized to either adjuvant chemotherapy or no adjuvant chemotherapy after limb-sparing surgery with or without radiation therapy. None of the individual trials showed a significant benefit, and all trials had large confidence intervals; however, the meta-analysis showed significant benefit in the chemotherapy treatment groups with regard to local recurrence, distant recurrence, and progression-free survival. No significant difference in overall survival was found.37 Pervais et al updated the Sarcoma Meta-analysis Collaboration’s 1997 meta-analysis with the inclusion of 4 new trials that evaluated doxorubicin combined with ifosfamide and found that both patients who received doxorubicin-based regimens or doxorubicin with ifosfamide had significant decreases in distant and overall recurrences. Only the trials that utilized doxorubicin and ifosfamide had an improved overall survival that was statistically significant (hazard ratio 0.56 [95% CI 0.36 to 0.85]; P = 0.01).29 Although no significant heterogeneity was found among the trials included in either meta-analysis, a variety of sarcomas were included in each clinical trial evaluated. Given the extremely small number of each sarcoma subtype present in each trial, subgroup analysis is difficult and prone to inaccuracies. As a result, it is not known if certain histological subtypes are more or less responsive to chemotherapy.37–39
One randomized controlled trial evaluated neoadjuvant chemotherapy in high-risk sarcomas defined as tumors greater than 8 cm or grade II/III tumors. This study evaluated doxorubicin and ifosfamide and found no significant difference in disease-free and overall survival in the neoadjuvant therapy group compared to the control group.35 There remains controversy in the literature with regards to adjuvant chemotherapy. Many oncologists offer adjuvant chemotherapy to patients with certain stage III subtypes. Examples of subtypes that may be offered adjuvant therapy include myxoid liposarcomas, synovial sarcomas, and leiomyosarcomas.2 With regards to how many cycles of chemotherapy should be considered, a noninferiority study compared 3 cycles of epirubicin and ifosfamide to 5 cycles of epirubicin and ifosfamide in patients with high-risk locally advanced adult STSs. Three cycles of preoperative epirubicin and ifosfamide was found to be noninferior to 5 cycles with regards to overall survival.38
• What is this patient’s risk for recurrence?
The patient is at intermediate risk for recurrence. Numerous studies have demonstrated that tumor size, grade, and location are the most important factors to determine risk of recurrence, with larger size, higher grades, and deeper locations being associated with higher risk of recurrence. In an analysis of 1041 patients with STS of the extremities, high grade was the most important risk factor for distant metastases.39 The highest risk of recurrence is within the first 2 years. Given that the patient’s initial tumor was located in the extremity, he is more likely to have a distant metastasis as his site of recurrence; individuals with retroperitoneal tumors and visceral tumors are more likely to recur locally.40 For STSs of the extremity, distant metastases determine overall survival, whereas patients with retroperitoneal sarcomas can die from complications of local metastases.41 Once a patient develops distant metastases, the most important prognostic factor is the size of the tumor, with tumors larger than 10 cm having a relative risk of 1.5 (95% CI 1.0 to 2.0).39
• What are the recommendations for surveillance?
Surveillance recommendations are based on the stage of the sarcoma. Stage I tumors are the least likely to recur either locally or distally. As a result, it is recommended that stage I tumors be followed with history and physical exam every 3 to 6 months for the first 2 to 3 years, and then annually after the first 2 to 3 years. Chest x-rays should be considered every 6 to 12 months.2 For stage II–IV tumors, history and physical exam is recommended every 3 to 6 months for the first 2 to 3 years. Chest and distant metastases imaging should also be performed every 3 to 6 months during this time frame. For the next 2 years, history and physical exam and imaging are recommended every 6 months. After the first 4 to 5 years, annual follow-up is recommended.2
A study that followed 141 patients with primary extremity STSs for a median interval of 49 months found that high-grade tumors were most likely to recur during the first 2 years, with 20% of their patients recurring locally and 40% recurring distally. Chest x-rays performed during surveillance follow-up found distant lung metastases in 36 asymptomatic patients and had a positive predictive value of 92%, a negative predictive value of 97%, and a quality-adjusted life-year of $30,000.40,41 No laboratory testing was found to aid in detection of recurrence.
› Case Continued
The patient does well for 1 year. With physical therapy, he regains most of the strength and coordination of the lower extremity. He is followed every 3 months with chest x-rays and a MRI of the thigh for the first year. On his fourth follow-up clinic visit, he describes increased dysp-nea on exertion over the previous few weeks and is found to have multiple lung metastases in both lungs on chest x-ray. He undergoes further evaluation for metastases and is not found to have any other metastatic lesions. Bronchoscopy and biopsy of 1 of the lung nodules confirms recurrent dedifferentiated liposarcoma.
• Should this patient undergo metastectomy?
An analysis of 3149 patients with STS treated at Memorial Sloan-Kettering who developed lung metastases found that patients with pulmonary metastases have survival rates of 25%. The most important prognostic factor for survival was complete resection of all metastases.42 For stage IV disease, surgery is used only in certain instances. In instances where tumor is more localized or limited, removal of metastases or metastectomy can play a role in management.2
› Case Continued
Because the patient’s metastases are limited to the lungs, he is referred for metastectomy. He undergoes wedge resection for definitive diagnosis but it is not possible to completely resect all of the metastases. He is thus referred to a medical oncologist to discuss his treatment options.
• What are treatment options for unresectable or metastatic disease?
Metastatic Disease
Unlike local and locally advanced disease, chemotherapy forms the backbone of treatment in stage IV disease. Doxorubicin and olaratumab or doxorubicin and ifosfamide in combination are considered first line in metastatic disease. Response rates for single-agent doxorubicin range from 16% to 27%, while phase 2 and phase 3 studies of doxorubicin and ifosfamide have found response rates ranging from 18% to 36%.43 In addition, the effectiveness of doxorubicin and ifosfamide phase 2 and 3 trials varied. Edmonson et al found a tumor regression rate of 34% for doxorubicin and ifosfamide as compared to 20% for doxorubicin alone.44 In comparison, Santoro et al found a response rate of 21.3% for doxorubicin alone and 25.2% for doxorubicin and ifosfamide.45 Neither study found increased survival benefit for doxorubicin and ifosfamide when compared to doxorubicin alone. In a Cochrane review evaluating randomized trials that compared doxorubicin and combination chemotherapy regimens, response rates varied from 14% for doxorubicin in combination with streptomycin to 34% for doxorubicin and ifosfamide. Most trials did not show a significant benefit for combination therapies when compared to doxorubicin alone.43 Mean survival with doxorubicin or doxorubicin and ifosfamide is 12 months. High rates of recurrence highlight the need for additional chemotherapy regimens.
The newest approved agent is olaratumab, a monoclonal antibody that binds platelet-derived growth factor receptor alpha and prevents receptor activation. A phase 1-b and phase 2 trial evaluated patients with locally advanced and metastatic STS and randomly assigned them to either olaratumab and doxorubicin or doxorubicin alone.46 Progression-free survival for olaratumab/doxorubicin was 6.6 months (95% CI 4.1 to 8.3) compared to 4.1 months (95% CI 2.8 to 5.4) for doxorubicin alone. The objective response rate was 18.2% (95% CI 9.8 to 29.6) for olaratumab/doxorubicin compared to 7.5% (95% CI 2.5 to 6.6) for doxorubicin alone. Furthermore, the median overall survival for olaratumab plus doxorubicin was 26.5 months (95% CI 20.9 to 31.7) compared to 14.7 months for doxorubicin alone (95% CI 5.5 to 26.0). Impressively, this improved response was notable across histological types. Furthermore, patients who had previously been treated with more than 1 regimen and those who were treatment naïve had similar response rates.46
• What are second-line treatment options?
Doxorubicin has been used in combination with several other agents including dacarbazine (DTIC) as well as DTIC and ifosfamide (MAID). Borden et al evaluated patients with metastatic STS and randomly assigned the patients to either doxorubicin or doxorubicin and DTIC. Combination therapy demonstrated better tumor response than doxorubicin alone: 30% complete or partial response for combination therapy and 18% for doxorubicin alone.47 However, Omura et al found similar rates of efficacy between doxorubicin and combination doxorubicin and DTIC in women with recurrent or nonresectable uterine sarcomas.48 MAID has never been directly compared in a randomized trial to doxorubicin alone. In a study that compared MAID to doxorubicin and DTIC (AD) in patients with unresectable or metastatic sarcomas, MAID had superior response rates (32% versus 17%), but there was no difference with regards to overall survival (mean survival of 12.5 months).49
Several additional regimens have undergone evaluation in metastatic and recurrent STSs. Gemcitabine has been used both as a single agent and as part of combination therapy in many studies. Studies with gemcitabine in combination with either docetaxel or DTIC have been the most efficacious. In a phase 2 trial, patients with metastatic STS were randomly assigned to either gemcitabine alone or gemcitabine and docetaxel. Combination therapy had a higher response rate (16% versus 8%) and longer overall survival (17.9 months versus 11.5 months) than gemcitabine alone.50 Furthermore, a phase 2 trial of gemcitabine and docetaxel in patients with unresectable leiomyosarcoma showed an overall response rate of 56%, with 3 complete and 15 partial responses among the 34 patients enrolled in the study.51 A phase 2 trial randomly assigned patients with unresectable or metastatic STS to either DTIC or combination gemcitabine and DTIC.52 Gemcitabine-DTIC had a superior progression-free survival at 3 months (56% [95% CI 43% to 69%]) as compared to DTIC alone (37% [95% CI 23.5% to 50%]). Furthermore, mean progression-free survival and overall survival were improved in the gemcitabine-DTIC group (4.2 months and 16.8 months) as compared to the DTIC group (2.0 months and 8.2 months).52 DTIC has a single-agent response rate of 16%, but has been shown to be particularly effective in the setting of leiomyosarcomas.49
• Does response to treatment regimens differ by histologic subtype?
The majority of STS trials include many different histologic subtypes. Given the rarity of sarcomas as a whole, many trials have had difficulty recruiting adequate numbers of patients to have sufficient power to definitely determine if the treatment under investigation has clinical benefit. Furthermore, the patients recruited have been heterogeneous with regard to subtype. Many older studies hypothesized that the efficacy of chemotherapeutic agents vary based on histologic subtype; however, for most subtypes the number of individuals included in those trials was too low to evaluate efficacy based on subtype.
Some exceptions exist, however. For example, both gemcitabine-DTIC and gemcitabine-docetaxel have been found to be particularly effective in the treatment of leiomyosarcomas.50,52 Additionally, a retrospective study found a 51% overall response rate for patients with myxoid liposarcomas treated with trabectedin.53 Studies of patients with angiosarcoma treated with paclitaxel have demonstrated response rates of 43% and 53%.54,55
• What are the newest approved and investigational agents?
A recently approved agent is trabectedin, a tris tetrahydroisoquinoline alkaloid isolated from ascidians that binds to the minor groove of DNA and causes disruptions in the cell cycle. Samuels et al reported data from a single-arm, open-label expanded access trial that evaluated patients with advanced metastatic sarcomas.56 In this study, patients with liposarcomas and leiomyosarcomas had an objective response rate of 6.9% (95% CI 4.8 to 9.6) as compared to a rate of 5.9% (95% CI 4.4 to 7.8) for all assessable patients. Median survival was 11.9 months for all patients, with improved median survivals for liposarcoma and leiomyosarcomas of 16.2 months (95% CI 14.1 to 19.5) compared to 8.4 months (95% CI 7.1 to 10.7 months) for other subtypes.56
Schöffski et al evaluated eribulin, a chemotherapeutic agent that affects microtubule dynamics, in a phase 2 trial of patients with progressive or high-grade STS with progression on previous chemotherapy. They found a median progression-free survival of 2.6 months (95% CI 1.7 to 6.2) for adipocytic sarcoma, 2.9 months (95% CI 2.4 to 4.6) for leiomyosarcoma, 2.6 months (95% CI 2.3 to 4.3) for synovial sarcoma, and 2.1 months (95% CI 1.4 to 2.9) for other sarcomas.57
Van der Graaf and colleagues randomly assigned patients with metastatic nonadipocytic STS to pazopanib or placebo in a phase 3 trial. Pazopanib is a small-molecule endothelial growth factor inhibitor with activity against vascular endothelial growth factors 1, 2, and 3 as well as platelet-derived growth factors. Median progression-free survival was 4.6 months (95% CI 3.7 to 4.8) with pazopanib compared to 1.6 months (95% CI 0.9 to 1.8) with placebo.58 Adipocytic sarcomas (liposarcomas) were excluded from the trial because phase 2 trials had found a lower rate of progression-free survival (26%) for them compared to other subtypes.
• What are the most common toxicities associated with the approved and investigational chemotherapeutic agents?
Toxicities were seen with each of the regimens studied and were common in the randomized trials, with higher rates of toxicities in the combination chemotherapy regimens. The most common toxicities are myelosuppression, nausea, and vomiting. In the doxorubicin trials, the most common toxicities were myelosuppression, nausea, and vomiting.44
Ifosfamide both as an individual agent and in combination with doxorubicin has higher rates and higher grades of toxicity than doxorubicin alone. Myelosuppression is the most common toxicity associated with ifosfamide, and the most commonly affected cell line is leukocytes.44 Combination doxorubicin and ifosfamide also had high rates of nausea and vomiting (95%) and alopecia (100%).35Neutropenia is the most common toxicity associated with gemcitabine and dacarbazine, while their most common nonhematologic toxicities are fatigue and nausea.52,59 Trabectedin’s most common toxicities are nausea (29%), neutropenia (24%), and fatigue (23%). It has also been shown to cause increased alkaline phosphatase (20%) and alanine aminotransferase (19%) levels.56 In a phase 2 study of eribulin, 50% of patients had neutropenia, and other toxicities included fatigue, alopecia, nausea, sensory neuropathy, and thrombocytopenia.57 Pazopanib is generally well tolerated; the most common toxicities are fatigue (65%), diarrhea (58%), nausea (54%), and hypertension (41%).58 Higher rates of neutropenia, mucositis, nausea, vomiting, diarrhea, and transfusion reactions were seen with olaratumab and doxorubicin compared to doxorubicin alone in phase 1b and 2 studies.46
› Case Continued
Given his poor prognosis with unresectable metastatic undifferentiated liposarcoma, the patient considers a clinical trial prior to undergoing combined therapy with doxorubicin and ifosfamide. He tolerates therapy well with stable disease at 6 months.
Conclusion
STSs are a heterogeneous collection of rare tumors. Low-grade, localized tumors have the best prognosis, and patients who undergo complete resection have the best long-term survival. Due to the rarity of STSs, trials often have limited enrollment, and little progress has been made with regards to treatment and survival rates for metastatic and unresectable disease. All patients should be evaluated and treated at specialized sarcoma centers. This case highlights the need for continued research and clinical trials to improve overall survival of patients with sarcoma. TSJ
CORRESPONDENCE
Ashley Pariser, MD, Resident, Department of Medicine, Northwestern University Feinberg School of Medicine Chicago, IL. Accepted for publication Jan/Feb 2017; Hosp Phys; Vol. 12, Part1
References
1. American Cancer Society. Cancer facts and figures 2016. American Cancer Society Web site. www.cancer.org/acs/groups/content/@research/documents/document/acspc-047079.pdf. Accessed December 20, 2016.
2. National Comprehensive Cancer Network. NCCN clinical guidelines in oncology: soft tissue sarcoma. 2016
3. Coindre J, Terrier P, Guillou L, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001;91:1914–26.
4. Dei Tos A. Liposarcoma: new entities and evolving concepts. Ann Diagn Pathol 2000;4: 252–66.
5. Wile AG, Evans HL, Romsdahl MM. Leiomyosarcoma of soft tissue: a clinicopathologic study. Cancer 1981;48:1022–32.
6. Hashimoto H, Daimaru Y, Tsuneyoshi M, Enjoji M. Leiomyosarcoma of the external soft tissues. A clinicopathologic, immunohistochemical, and electron microscopic study. Cancer 1986;57:2077–88
7. Fisher C. Synovial sarcoma. Ann Diagn Pathol 1998;2:401–21.
8. Newton WA Jr, Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer 1995;76:1073–85.
9. Furlong MA. Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol. 2001;14:595–603.

10. Anghileri M, Miceli R, Fiore M. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer 2006;107:1065–74.
11. Miettinen M, Lasota J. Gastrointestinal stromal tumors–definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Archive 2001;438:1–12.
12. Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006;23:70–83.
13. Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol 2010;11:983–91.
14. Cormier JN, Pollock RE. Soft tissue sarcomas. CA Cancer J Clin 2004;54:94–109.
15. Penel N, Grosjean J, Robin YM, et al. Frequency of certain established risk factors in soft tissue sarcomas in adults: a prospective descriptive study of 658 cases. Sarcoma 2008;2008:459386.
16. Guillou L, Coindre JM, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 1997;15:350–62.
17. Maki RG, Moraco N, Antonescu CR, et al. Toward better soft tissue sarcoma staging: building on American joint committee on cancer staging systems versions 6 and 7. Ann Surg Oncol 2013;20:3377–83.
18. Shiraki M, Enterline HT, Brooks JJ, et al. Pathologic analysis of advanced adult soft tissue sarcomas, bone sarcomas, and mesotheliomas. The Eastern Cooperative Oncology Group (ECOG) experience. Cancer 1989;64:484–90.
19. Presant CA, Russell WO, Alexander RW, Fu YS. Soft-tissue and bone sarcoma histopathology peer review: The frequency of disagreement in diagnosis and the need for second pathology opinions. The Southeastern Cancer Study Group experience. J Clin Oncol 1986; 4:1658–61.
20. Sundaram M, McLeod RA. MR imaging of tumor and tumorlike lesions of bone and soft tissue. AJR Am J Roentgenol 1990;155:817–24.
21. Ioannidis JP, Lau J. 18F-FDG PET for the diagnosis and grading of soft-tissue sarcoma: a meta-analysis. J Nucl Med 2003;44:717–24.
22. Tateishi U, Yamaguchi U, Seki K, et al. Bone and soft-tissue sarcoma: preoperative staging with fluorine 18 fluorodeoxyglucose PET/CT and conventional imaging. Radiology 2007;245:839–47.
23. Zagars GK, Ballo MT, Pisters PW, et al. Prognostic factors for patients with localized soft-tissue sarcoma treated with conservation surgery and radiation therapy: an analysis of 1225 patients. Cancer 2003;97:2530–43
24. Rosenberg S, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 1982;196:305–14.
25. Lewis J, Leung D, Woodruff J, et al. Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg 1998;288:355–65.
26. Zagars GK, Ballo MT, Pisters PW, et al. Surgical margins and reresection in the management of patients with soft tissue sarcoma using conservative surgery and radiation therapy. Cancer 2003;97:2544–53.
27. Stojadinovic A, Leung DH, Hoos A. Analysis of the prognostic significance of microscopic margins in 2,084 localized primary adult soft tisusse sarcomas. Ann Surg 2002;235:424–34.
28. O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
29. Pervaiz N, Colterjohn N, Farrokhyar F, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008;113:573–81.
30. Suit HD, Mankin HJ, Wood WC, Proppe KH. Preoperative, intraoperative, and postoperative radiation in the treatment of primary soft tissue sarcoma. Cancer 1985;55:2659–67
31. O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
32. Yang J, Chang A, Baker A, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 1998;16:197–203.
33. Pisters PW, Harrison LB, Leung DH, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 1996;14:859–68.
34. Alektiar KM, Brennan MF, Healey JH, Singer S. Impact of intensity-modulated radiation therapy on local control in primary soft-tissue sarcoma of the extremity. J Clin Oncol 2008;26:3440–5.
35. Gortzak E, Azzarelli A, Buesa J, et al. A randomized phase II study on neo-adjuvant chemotherapy for ‘high-risk’ adult soft-tissue sarcoma. Eur J Cancer 2001;37:1096–1103.
36. Fakhari N, Ebm C, Kostler WJ, et al. Intensified adjuvant IFADIC chemotherapy in combination with radiotherapy versus radiotherapy alone for soft tissue sarcoma: long-term follow-up of a prospective randomized feasibility trial. Wein Klin Wochenschr 2010;122:614–9.
37. Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Lancet 1997;350:1647–54.
38. Gronchi A, Frustaci S, Mercuri M, et al. Short, full-dose adjuvant chemotherapy in high-risk adult soft tissue sarcomas: a randomized clinical trial from the Italian Sarcoma Group and the Spanish Sarcoma Group. J Clin Oncol 2012;30:850–56.
39. Pisters PW, Leung DH, Woodruff J. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996;14:1679–89.
40. Whooley B, Gibbs J, Mooney M. Primary Extremity Sarcoma: What is the Appropriate Follow-up? Annals of Surg Oncol 2000; 7: 9-14.
41. Whooley BP, Mooney MN, Gibbs JF, Graybill WG. Effective follow-up strategies in soft tissue sarcoma. Sem Surg Oncol 1999;17:83–87.
42. Billingsley KG, Burt ME, Jara E, et al. Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival. Ann Surg 1999;229:602–10.
43. Bramwell VH, Anderson D, Charette ML; Sarcoma Disease Site Group. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev 2003;(3):CD003293.
44. Edmonson J, Ryan L, Blum R. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 1993;11:1269–75.
45. Santoro A, Tursz T, Mouridsen H. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 1995;13:1537–45.
46. Tap WD, Jones RL, Van Tine B, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet 2016;388:488–97.
47. Borden EC, Amato DA, Rosenbaum C, et al. Randomized comparison of three adriamycin regimens for metastatic soft tissue sarcomas. J Clin Oncol 1987;5:840–50.
48. Omura GA, Major FJ, Blessing JA, et al. A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 1983;52:626–32.
49. Antman K, Crowley J, Balcerzak SP, et al. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol 1993;11:1276–85.
50. Maki R, Wathen K, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 2007; 25: 2755–63.
51. Hensley ML, Maki R, Venkatraman E, et al. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol 2002;12:2824–31.
52. Garcia-del-Muro X, Lopez-Pousa A, Maurel J, et al. Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: a Spanish Group for Research on Sarcomas study. J Clin Oncol 2011;29:2528–33.
53. Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol 2007;7:595–602.
54. Italiano A, Cioffi A, Penel N, et al. Comparison of doxorubicin and weekly paclitaxel efficacy in metastatic angiosarcomas. Cancer 2012;118:3330–6.
55. Penel N, Italiano A, Ray-Coquard I, et al. Metastatic angiosarcomas: doxorubicin-based regimens, weekly paclitaxel and metastasectomy significantly improve outcome. Ann Oncol 2012;23:517–23.
56. Samuels BL, Chawla S, Patel S, et al. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol 2013;24:1703–9.
57. Schöffski P, Ray-Coquard IL, Cioffi A, et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histolical subtypes. Lancet 2011;11:1045–52.
58. Van der Graaf W, Blay JY, Chawla S, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomized, double-blind, placebo-controlled phase 3 trial. Lancet 2012;379:1879–86.
59. Dileo P, Morgan JA, Zahrieh D, et al. Gemcitabine and vinorelbine combination chemotherapy for patients with advanced soft tissue sarcomas: results of a phase II trial. Cancer 2007;109:1863–9.
Introduction
Soft tissue sarcomas (STSs) are rare adult tumors, with 3.4 new cases per 100,000 persons or 12,310 expected new cases in 2016.1 Sarcomas are a heterogeneous collection of tumors that affect fat, muscle, nerve, nerve sheath, vascular, and connective tissues. There are more than 50 histological subtypes that comprise this diverse category of tumors. Treatment varies by stage, with limb-sparing surgery representing the mainstay of curative-intent treatment. Radiation and chemotherapy may also be considered depending on the size, grade, and location of the tumor. Survival rates have been stagnant until recently, with a disease-specific survival hovering around 65%.1 Given the complexity of these cases, all patients ideally should be evaluated and treated by a multidisciplinary team at an institution with extensive experience treating STS.2
Epidemiology and Classification
The most common STS subtypes are gastrointestinal stromal tumor (GIST), undifferentiate pleomorphic sarcoma (previously referred to as malignant fibrous histiocytoma), liposarcoma, leiomyosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, and unclassified sarcoma.3 Liposarcoma is one of the most common subtypes, comprising 20% of all STSs; it is subdivided into well-differentiated/dedifferentiated liposarcomas, myxoid/round cell liposarcomas, and pleomorphic liposarcomas. Well-differentiated liposarcomas tend to occur in the retroperitoneum and limbs, while both myxoid and round cell as well as pleomorphic liposarcomas more commonly originate on the limbs. Histology varies based on subtype and ranges from mature-appearing adipocytes and fibroblasts to undifferentiated cells with minimal lipogenic differentiation.4
Leiomyosarcomas are smooth muscle tumors and are usually located in the retroperitoneum, but have also been associated with peripheral soft tissue and vasculature. Typical histology ranges from well-defined areas of spindle-shaped cells to poorly differentiated anaplastic spindle cells.5,6 Synovial sarcomas are a distinct type of STS that can show epithelial differentiation and account for 5% of adult STSs. The extremities are the most common presenting location (90%).7
Rhabdomyosarcomas are skeletal muscle tumors and are further subdivided into embryonal, alveolar, and pleomorphic subtypes. Embryonal histology ranges from primitive mesenchymal-appearing cells to highly differentiated muscle cells. Alveolar rhabdomyosarcoma has the worst prognosis of the subtypes and consists of round cells with high nuclear-to-chromatin ratios that form “glandular-like” or “alveolar” spaces.8 Pleomorphic rhabdomyosarcomas are composed of rhabdomyoblasts that can affect many different locations, but most commonly present on the lower extremities.9
Malignant peripheral nerve sheath tumor (MPNST) comprises 5% to 10% of all STSs. These tumors are associated with neurofibromatosis type 1 (NF-1), with 25% to 50% of tumors occurring in NF-1 patients. Additionally, most patients have a truncating lesion in the NF1 gene on chromosome 17.10 Anghileri et al in their single institution analysis of 205 patients with MPNSTs found the 2 most common presenting sites were the trunk and extremities. Histologically, these tumors have dense fascicles of spindle cells.10
GISTs are the most common STS of the gastrointestinal (GI) tract. Previously, GISTs were classified as smooth muscle tumors and were not accounted for in the literature as a separate entity distinct from leiomyomas, leiomyoblastomas, and leiomyosarcomas.11 GISTs are found throughout the GI tract: the most common sites are the stomach (60%) and small intestine (30%). Less common sites include duodenum (4%–5%), esophagus (1%), rectum (1%–2%), and appendix (< 0.2%).12 GISTs can be spindle cell, epithelioid, or mesenchymal tumors. Immunohistochemically, GISTs are KIT (CD117) positive. Other cell markers that are also commonly positive include CD34 (60%–70%) and smooth muscle actin (SMA) (25%).11 The majority of GISTs (80%) have an activating c-KIT gene mutation. The most common mutation site is exon 11, with less common c-KIT gene mutations also occurring at exon 9 or 13. Not all GISTs have KIT mutations. The second most common mutation is the PDGFRA mutation (5%–10% of GISTs).2 A minority of GISTs are negative for both KIT and PDGFRA mutations. These tumors were previously called wild-type, but as the majority have either a succinate dehydrogenase (SDH) loss of function or loss of SDHB protein expression, they are now referred to as SDH-deficient GISTs.2 GISTs vary in aggressiveness from incidental to aggressive. Typically, small intestine and rectal GISTs are more aggressive than gastric GISTs. Both size and mitotic rate help to predict the metastatic potential of the tumor. Tumors less than 2 cm in size and having a mitotic rate of less than 5 per 50 high-power fields (hpf) have the lowest risk of metastases, while tumors greater than 5 cm and with more than 5 mitoses per 50 hpf have the highest rates of metastases.12
Angiosarcomas are rare tumors comprising 4% of all STSs. Although they can occur in any site, the majority are cutaneous and occur most frequently in the head and neck regions. These tumors are either of vascular or lymphatic origin and are comprised of abnormal, pleomorphic, malignant endothelial cells. The most useful immunohistochemical markers include von Willebrand factor, CD31, and Ulex europaeus agglutinin 1. The majority of these tumors occur sporadically; however, radiation exposure, chronic lymphedema, and certain toxins including vinyl chloride and thorium dioxide are known risk factors.13
Undifferentiated sarcomas have no specific features and typically consist of primitive mesenchymal cells.
Clinical Evaluation
› Case Presentation
Initial Presentation and History
A 55-year-old man presents to his primary care physician with a painless mass in his anterior thigh. The mass has been present for the past 3 months and he believes that it is enlarging. The patient has a history of well-controlled hypertension and hyperlipidemia. His medications include atorvastatin and hydrochlorothiazide. He has no known drug allergies. Family history is notable for diabetes and hypertension. He drinks 4 to 5 alcoholic drinks a week and he is a former smoker. He quit smoking in his 30s and only smoked intermittently prior to quitting. He denies any illicit drug use. He works as a high school principal. Currently, he feels well. His review of systems is otherwise noncontributory.
Physical Examination
On physical exam, he is afebrile with a blood pressure of 132/75 mm Hg, respiratory rate of 10 breaths/min, and oxygen saturation of 99% on room air. He is a well appearing, overweight male. His head and neck exam is unremarkable. Lung exam reveals clear breath sounds, and cardiac exam reveals a regular rate and rhythm. His abdomen is obese, soft, and without hepatosplenomegaly. There is a large, fixed mass on the anterior lateral aspect of his right thigh. He has no appreciable lymphadenopathy. His neurological exam is unremarkable.
• What are risk factors for sarcoma?
There are few known risk factors for sarcoma. Established risks factors include prior radiation therapy, chronic lymphedema, viruses, and genetic cancer syndromes including Li-Fraumeni syndrome, hereditary retinoblastoma, and NF-1. Other environmental exposures include phenoxyacetic acids and chlorophenols.14 The majority of cases are sporadic, with only a minority of patients having one of these known risk factors.15 Up to one third of sarcomas have a specific translocation and are driven by fusion oncogenes (
• What is the typical presentation for sarcomas?
A painless mass is the most typical presenting symptom. Size at presentation varies based on location, with extremity and head and neck locations typically presenting at smaller sizes than retroperitoneal tumors.14 Patients may experience pain and numbness as the mass enlarges and impinges on surrounding structures including nerves and vasculature. The vast majority of patients are without systemic symptoms.
• How is sarcoma staged?
The American Joint Committee on Cancer (AJCC) staging system is the most widely used staging system in the United States. The latest AJCC manual was updated in 2010 to include a 3-tiered grading system where the tumor is classified according to tumor size, lymph node involvement, metastases, and grade at time of diagnosis (Table 2 and Table 3). Additionally, tumor depth in relation to deep fascia is also taken into account, with superficial tumors being assigned a designation of “a” and deep tumors a designation of “b.”
Previously, 2 of the most widely used grading systems were the National Cancer Institute (NCI) and French Federation of Cancer Centers Sarcoma Group (FNCLCC) systems, both 3-tier grading systems. The main components that determine the NCI grade are the tumor’s histologic type and location and the amount of tumor necrosis. The FNCLCC system evaluation focuses on tumor differentiation, mitotic rate, and amount of tumor necrosis. A study that compared the NCI and FNCLCC grading systems found that FNCLCC was a better predictor of mortality and distant metastasis.16 Previously, the AJCC was a 4-tier grading system, but the 2010 version was updated to the 3-tier FNCLCC grading system. Additionally, the AJCC system has reclassified single lymph node disease as stage III as it confers better survival than metastatic disease.17 It is important that pathology be evaluated by a sarcoma specialist as disagreements with regard to histologic subtype and grade are common.18,19
• What are the most important prognostic factors?
Prognostic factors include grade, size, and presence of metastases at presentation. Best survival is associated with low-grade, small tumors with no metastases at time of diagnosis.14
• What imaging should be considered?
Imaging should be undertaken to help differentiate between benign and malignant lesions. Ideally, it should be undertaken before a biopsy is planned as the imaging can be used to plan biopsy as well as provide invaluable prognostic information. There are several imaging modalities that should be considered during the preliminary work-up and staging of STSs. Conventional imaging includes magnetic resonance imaging (MRI) of the original tumor site; computed tomography (CT) to evaluate for pulmonary metastases and, depending on location, liver metastases; and in the case of small, low-grade tumors, chest radiography. MRI is considered the test of choice for soft tissue masses and can help delineate benign masses such as hematomas, lipomas, and hemangiomas from sarcomas.20 It is difficult to compare the accuracy of positron emission tomography (PET)/CT to CT and MRI because most studies have evaluated PET/CT in parallel with CT and MRI.21 Tateishi et al compared the accuracy of conventional imaging, PET/CT, and PET/CT combined with conventional imaging at determining the TNM staging for 117 patients. They found that conventional imaging correctly classified 77% of patients, PET alone correctly classified 70%, PET/CT correctly classified 83%, and PET/CT combined with conventional imaging correctly staged 87%.22
• Which subtypes are most likely to metastasize?
Although the vast majority of sarcomas spread hematogenously, 3 have a propensity to spread lymphogenously: epithelioid sarcoma, rhabdomyosarcoma, and clear-cell sarcoma. Additionally, certain subtypes are more likely to metastasize: leiomyosarcomas, synovial sarcomas, neurogenic sarcomas, rhabdomyosarcomas, and epithelioid sarcomas.23 Sarcomas metastasize to the lungs more frequently than to the liver. The metastatic pattern is defined primarily by sarcoma subtype and site of primary tumor. Sarcomas rarely metastasize to the brain (~1%).
Management
› Case Continued
The patient undergoes an ultrasound to better visualize the mass. Given the heterogeneous character of the mass, he is referred for an MRI to evaluate the mass and a CT scan of the chest, abdomen, and pelvis to evaluate for distant metastases. MRI reveals a 5.1 cm × 4.6 cm heterogeneous mass invading the superficial fascia of the rectus femoris muscle. No suspicious lymph nodes or other masses are identified on imaging. The patient next undergoes an image-guided core needle biopsy. Pathology from that procedure is consistent with a stage III, T2bNxMx, grade 3, dedifferentiated liposarcoma.
• What is the best management approach for this patient?
Surgery
Surgery is the mainstay of treatment for STS. Patients with the best prognosis are those who undergo complete resection with negative surgical margins.24,25 Goal tumor-free margin is 1 to 3 cm.26 Complete resection confers the best long-term survival. Both local and metastatic recurrence is higher in patients with incomplete resection and positive margins.24,25 In a study that analyzed 2084 localized primary STSs, patients with negative margins had a local recurrence rate of 15% versus a rate of 28% in patients with positive margins. This translated into higher 5-year local recurrence-free survival for patients with negative surgical margins (82%) compared to patients with positive margins (65%).27 Another study similarly found that patients with negative margins at referral to their institution who underwent postoperative radiation had high local control rates of 93% (95% confidence interval [CI] 87% to 97%) at 5, 10, and 15 years.26 Although radiation improves local control, neither preoperative or postoperative radiation has been shown to improve progression-free or overall survival.28 Other factors that are associated with risk of recurrence are tumor location, history of previous recurrence, age of patient, histopathology, tumor grade, and tumor size. Approximately 40% to 50% of patients with high-grade tumors (defined as size > 5 cm, deep location, and high grade) will develop distant metastases.29
Zagars et al found that positive or uncertain resection margin had a relative risk of local recurrence of 2.0 (95% CI 1.3 to 3.1; P = 0.002), and presentation with locally recurrent disease (vs new tumor) had a relative risk of local recurrence of 2.0 (95% CI 1.2 to 3.4; P = 0.013).26 Patients with STS of head and neck and deep trunk have higher recurrence rates than those with superficial trunk and extremity STS. A single-institution retrospective review demonstrated that patients with completely resectable retroperitoneal sarcomas have longer median survival (103 months) compared to patients with incompletely resected abdominal sarcomas (18 months).25Rosenberg and colleagues compared amputation to limb-sparing surgery and radiation.24 Their prospective analysis of 65 patients found no difference in disease-free and overall survival between the 2 treatment groups.The limb-sparing treatment group had higher rates of local recurrence, which was highly correlated with positive surgical margins on pathology.24 Evidence from this and similar studies has resulted in radical amputations being replaced by conservative limb-sparing procedures and radiation therapy. In those found to have positive margins, re-resection is an option for some. Patients who undergo re-resection have higher local control rates than patients with positive margins who do not undergo re-resection. The 5-year control rate for patients who undergo re-resection is 85% (95% CI 80% to 89%) compared to 78% (95% CI 71% to 83%) for those who do not undergo re-resection. Similarly, patients who undergo re-resection have lower rates of metastases at 5, 10, and 15 years as well as higher 5-, 10-, and 15-year disease-free survival rates.26
› Case Continued
The patient is referred for limb-sparing surgery after presentation at a multidisciplinary tumor board. Prior to undergoing resection of the tumor, he is also referred to radiation-oncology to discuss the risks and benefits of combination radiotherapy and surgery as opposed to surgical resection alone.
• What is the evidence for radiation therapy?
Radiation THERAPY
Radiation therapy is used in the preoperative, intraoperative, and postoperative settings to reduce the risk of local recurrence. There are several options for radiation, including external beam radiation therapy (EBRT), intraoperative radiation, and brachytherapy. A newer strategy, intensity-modulated radiation therapy (IMRT), utilizes 3-dimensional modeling to reduce radiation dosages. Overall there are no differences in overall survival or local recurrence rates between preoperative and postoperative radiation in STS.28
The rationale behind preoperative radiation is that it reduces seeding of tumor cells, especially at the time of surgery.30 Additionally, for EBRT, preoperative radiation has smaller field sizes and lower radiation doses. It can also help to reduce the size of the tumor prior to resection. Intraoperative radiation is often paired with preoperative radiation as a boost dose given only to the area of residual tumor.
Suit et al reviewed patients treated at a single institution with limb-sparing surgery and different radiation strategies. Local control rates between preoperative and postoperative radiation groups were not statistically significant. Local recurrence was linked to grade and size of the tumor in both groups. The authors did note, however, that the preoperative radiation group tended to have larger tumor sizes at baseline compared to the patients who received postoperative radiation.30 A study that compared 190 patients who received preoperative and postoperative EBRT or brachytherapy (primary end point was wound complications, and local control was a secondary end point) showed a trend towards greater local control with preoperative radiation; however, the preoperative radiation group had significantly more wound complications compared to the postoperative radiation group.31
Yang et al found that postoperative EBRT decreases rates of local recurrence compared to surgery alone in high-grade extremity sarcomas.32 However, there were no differences in rates of distant metastases and overall survival between the 2 treatment groups. Similarly, in patients with low-grade sarcoma, there were fewer local recurrences in those who received EBRT and surgery as compared to surgery alone.32 Another study that evaluated 164 patients who received either adjuvant brachytherapy or no further therapy after complete resection found that brachytherapy reduced local recurrence in high-grade sarcomas. No difference in local recurrence rates was found in patients with low-grade sarcomas, nor was a significant difference found in the rates of distant metastases and overall survival between the 2 treatment groups.33 With regards to IMRT, a single institution cohort experience with 41 patients who received IMRT following limb-sparing surgery had similar local control rates when compared to historical controls.34
› Case Continued
After discussion of the risks and benefits of radiation therapy, the patient opts for preoperative radiation prior to resection of his liposarcoma. He receives 50 Gy of EBRT prior to undergoing resection. Resection results in R1 margin consistent with microscopic disease. He receives 16 Gy of EBRT as a boost after recovery from his resection.2
• What is the evidence for neoadjuvant and adjuvant chemotherapy for stage I tumors?
Chemotherapy
Localized Sarcoma
For localized sarcoma, limb-sparing resection with or without radiation forms the backbone of treatment. Studies have evaluated chemotherapy in both the neoadjuvant and adjuvant settings, with the vast majority of studies evaluating doxorubicin-based chemotherapy regimens in the adjuvant settings. Due to the rare nature of sarcomas, most studies are not sufficiently powered to detect significant benefit from chemotherapy. Several trials evaluating chemotherapy regimens in the neoadjuvant and adjuvant settings needed to be terminated prematurely due to inadequate enrollment into the study.35,36
For stage IA (T1a-Tb, N0, M0, low grade) tumors, no additional therapy is recommended after limb-sparing surgery with appropriate surgical margins. For stage IB (T2a-2b, N0, M0, low grade) tumors with insufficient margins, re-resection and radiation therapy should be considered, while for stage IIA (T1a-1b, N0, M0, G2-3) tumors preoperative or postoperative radiation therapy is recommended.2 Studies have not found benefit of adjuvant chemotherapy in these low-grade, stage I tumors in terms of progression-free survival and overall survival.37
• At what stage should chemotherapy be considered?
For stage IIb and stage III tumors, surgery and radiation therapy again form the backbone of therapy; however, neoadjuvant and adjuvant chemotherapy are also recommended as considerations. Anthracycline-based chemotherapy with either single-agent doxorubicin or doxorubicin and ifosfamide in combination are considered first-line chemotherapy agents in locally advanced STS.2,29,37
Evidence regarding the efficacy of both neoadjuvant and adjuvant chemotherapy regimens in the setting of locally advanced high-grade STS has been mixed. The Sarcoma Meta-analysis Collaboration evaluated 14 trials of doxorubicin-based adjuvant chemotherapy and found a trend towards overall survival in the treatment groups that received chemotherapy.37 All trials included in the meta-analysis compared patients with localized resectable soft-tissue sarcomas who were randomized to either adjuvant chemotherapy or no adjuvant chemotherapy after limb-sparing surgery with or without radiation therapy. None of the individual trials showed a significant benefit, and all trials had large confidence intervals; however, the meta-analysis showed significant benefit in the chemotherapy treatment groups with regard to local recurrence, distant recurrence, and progression-free survival. No significant difference in overall survival was found.37 Pervais et al updated the Sarcoma Meta-analysis Collaboration’s 1997 meta-analysis with the inclusion of 4 new trials that evaluated doxorubicin combined with ifosfamide and found that both patients who received doxorubicin-based regimens or doxorubicin with ifosfamide had significant decreases in distant and overall recurrences. Only the trials that utilized doxorubicin and ifosfamide had an improved overall survival that was statistically significant (hazard ratio 0.56 [95% CI 0.36 to 0.85]; P = 0.01).29 Although no significant heterogeneity was found among the trials included in either meta-analysis, a variety of sarcomas were included in each clinical trial evaluated. Given the extremely small number of each sarcoma subtype present in each trial, subgroup analysis is difficult and prone to inaccuracies. As a result, it is not known if certain histological subtypes are more or less responsive to chemotherapy.37–39
One randomized controlled trial evaluated neoadjuvant chemotherapy in high-risk sarcomas defined as tumors greater than 8 cm or grade II/III tumors. This study evaluated doxorubicin and ifosfamide and found no significant difference in disease-free and overall survival in the neoadjuvant therapy group compared to the control group.35 There remains controversy in the literature with regards to adjuvant chemotherapy. Many oncologists offer adjuvant chemotherapy to patients with certain stage III subtypes. Examples of subtypes that may be offered adjuvant therapy include myxoid liposarcomas, synovial sarcomas, and leiomyosarcomas.2 With regards to how many cycles of chemotherapy should be considered, a noninferiority study compared 3 cycles of epirubicin and ifosfamide to 5 cycles of epirubicin and ifosfamide in patients with high-risk locally advanced adult STSs. Three cycles of preoperative epirubicin and ifosfamide was found to be noninferior to 5 cycles with regards to overall survival.38
• What is this patient’s risk for recurrence?
The patient is at intermediate risk for recurrence. Numerous studies have demonstrated that tumor size, grade, and location are the most important factors to determine risk of recurrence, with larger size, higher grades, and deeper locations being associated with higher risk of recurrence. In an analysis of 1041 patients with STS of the extremities, high grade was the most important risk factor for distant metastases.39 The highest risk of recurrence is within the first 2 years. Given that the patient’s initial tumor was located in the extremity, he is more likely to have a distant metastasis as his site of recurrence; individuals with retroperitoneal tumors and visceral tumors are more likely to recur locally.40 For STSs of the extremity, distant metastases determine overall survival, whereas patients with retroperitoneal sarcomas can die from complications of local metastases.41 Once a patient develops distant metastases, the most important prognostic factor is the size of the tumor, with tumors larger than 10 cm having a relative risk of 1.5 (95% CI 1.0 to 2.0).39
• What are the recommendations for surveillance?
Surveillance recommendations are based on the stage of the sarcoma. Stage I tumors are the least likely to recur either locally or distally. As a result, it is recommended that stage I tumors be followed with history and physical exam every 3 to 6 months for the first 2 to 3 years, and then annually after the first 2 to 3 years. Chest x-rays should be considered every 6 to 12 months.2 For stage II–IV tumors, history and physical exam is recommended every 3 to 6 months for the first 2 to 3 years. Chest and distant metastases imaging should also be performed every 3 to 6 months during this time frame. For the next 2 years, history and physical exam and imaging are recommended every 6 months. After the first 4 to 5 years, annual follow-up is recommended.2
A study that followed 141 patients with primary extremity STSs for a median interval of 49 months found that high-grade tumors were most likely to recur during the first 2 years, with 20% of their patients recurring locally and 40% recurring distally. Chest x-rays performed during surveillance follow-up found distant lung metastases in 36 asymptomatic patients and had a positive predictive value of 92%, a negative predictive value of 97%, and a quality-adjusted life-year of $30,000.40,41 No laboratory testing was found to aid in detection of recurrence.
› Case Continued
The patient does well for 1 year. With physical therapy, he regains most of the strength and coordination of the lower extremity. He is followed every 3 months with chest x-rays and a MRI of the thigh for the first year. On his fourth follow-up clinic visit, he describes increased dysp-nea on exertion over the previous few weeks and is found to have multiple lung metastases in both lungs on chest x-ray. He undergoes further evaluation for metastases and is not found to have any other metastatic lesions. Bronchoscopy and biopsy of 1 of the lung nodules confirms recurrent dedifferentiated liposarcoma.
• Should this patient undergo metastectomy?
An analysis of 3149 patients with STS treated at Memorial Sloan-Kettering who developed lung metastases found that patients with pulmonary metastases have survival rates of 25%. The most important prognostic factor for survival was complete resection of all metastases.42 For stage IV disease, surgery is used only in certain instances. In instances where tumor is more localized or limited, removal of metastases or metastectomy can play a role in management.2
› Case Continued
Because the patient’s metastases are limited to the lungs, he is referred for metastectomy. He undergoes wedge resection for definitive diagnosis but it is not possible to completely resect all of the metastases. He is thus referred to a medical oncologist to discuss his treatment options.
• What are treatment options for unresectable or metastatic disease?
Metastatic Disease
Unlike local and locally advanced disease, chemotherapy forms the backbone of treatment in stage IV disease. Doxorubicin and olaratumab or doxorubicin and ifosfamide in combination are considered first line in metastatic disease. Response rates for single-agent doxorubicin range from 16% to 27%, while phase 2 and phase 3 studies of doxorubicin and ifosfamide have found response rates ranging from 18% to 36%.43 In addition, the effectiveness of doxorubicin and ifosfamide phase 2 and 3 trials varied. Edmonson et al found a tumor regression rate of 34% for doxorubicin and ifosfamide as compared to 20% for doxorubicin alone.44 In comparison, Santoro et al found a response rate of 21.3% for doxorubicin alone and 25.2% for doxorubicin and ifosfamide.45 Neither study found increased survival benefit for doxorubicin and ifosfamide when compared to doxorubicin alone. In a Cochrane review evaluating randomized trials that compared doxorubicin and combination chemotherapy regimens, response rates varied from 14% for doxorubicin in combination with streptomycin to 34% for doxorubicin and ifosfamide. Most trials did not show a significant benefit for combination therapies when compared to doxorubicin alone.43 Mean survival with doxorubicin or doxorubicin and ifosfamide is 12 months. High rates of recurrence highlight the need for additional chemotherapy regimens.
The newest approved agent is olaratumab, a monoclonal antibody that binds platelet-derived growth factor receptor alpha and prevents receptor activation. A phase 1-b and phase 2 trial evaluated patients with locally advanced and metastatic STS and randomly assigned them to either olaratumab and doxorubicin or doxorubicin alone.46 Progression-free survival for olaratumab/doxorubicin was 6.6 months (95% CI 4.1 to 8.3) compared to 4.1 months (95% CI 2.8 to 5.4) for doxorubicin alone. The objective response rate was 18.2% (95% CI 9.8 to 29.6) for olaratumab/doxorubicin compared to 7.5% (95% CI 2.5 to 6.6) for doxorubicin alone. Furthermore, the median overall survival for olaratumab plus doxorubicin was 26.5 months (95% CI 20.9 to 31.7) compared to 14.7 months for doxorubicin alone (95% CI 5.5 to 26.0). Impressively, this improved response was notable across histological types. Furthermore, patients who had previously been treated with more than 1 regimen and those who were treatment naïve had similar response rates.46
• What are second-line treatment options?
Doxorubicin has been used in combination with several other agents including dacarbazine (DTIC) as well as DTIC and ifosfamide (MAID). Borden et al evaluated patients with metastatic STS and randomly assigned the patients to either doxorubicin or doxorubicin and DTIC. Combination therapy demonstrated better tumor response than doxorubicin alone: 30% complete or partial response for combination therapy and 18% for doxorubicin alone.47 However, Omura et al found similar rates of efficacy between doxorubicin and combination doxorubicin and DTIC in women with recurrent or nonresectable uterine sarcomas.48 MAID has never been directly compared in a randomized trial to doxorubicin alone. In a study that compared MAID to doxorubicin and DTIC (AD) in patients with unresectable or metastatic sarcomas, MAID had superior response rates (32% versus 17%), but there was no difference with regards to overall survival (mean survival of 12.5 months).49
Several additional regimens have undergone evaluation in metastatic and recurrent STSs. Gemcitabine has been used both as a single agent and as part of combination therapy in many studies. Studies with gemcitabine in combination with either docetaxel or DTIC have been the most efficacious. In a phase 2 trial, patients with metastatic STS were randomly assigned to either gemcitabine alone or gemcitabine and docetaxel. Combination therapy had a higher response rate (16% versus 8%) and longer overall survival (17.9 months versus 11.5 months) than gemcitabine alone.50 Furthermore, a phase 2 trial of gemcitabine and docetaxel in patients with unresectable leiomyosarcoma showed an overall response rate of 56%, with 3 complete and 15 partial responses among the 34 patients enrolled in the study.51 A phase 2 trial randomly assigned patients with unresectable or metastatic STS to either DTIC or combination gemcitabine and DTIC.52 Gemcitabine-DTIC had a superior progression-free survival at 3 months (56% [95% CI 43% to 69%]) as compared to DTIC alone (37% [95% CI 23.5% to 50%]). Furthermore, mean progression-free survival and overall survival were improved in the gemcitabine-DTIC group (4.2 months and 16.8 months) as compared to the DTIC group (2.0 months and 8.2 months).52 DTIC has a single-agent response rate of 16%, but has been shown to be particularly effective in the setting of leiomyosarcomas.49
• Does response to treatment regimens differ by histologic subtype?
The majority of STS trials include many different histologic subtypes. Given the rarity of sarcomas as a whole, many trials have had difficulty recruiting adequate numbers of patients to have sufficient power to definitely determine if the treatment under investigation has clinical benefit. Furthermore, the patients recruited have been heterogeneous with regard to subtype. Many older studies hypothesized that the efficacy of chemotherapeutic agents vary based on histologic subtype; however, for most subtypes the number of individuals included in those trials was too low to evaluate efficacy based on subtype.
Some exceptions exist, however. For example, both gemcitabine-DTIC and gemcitabine-docetaxel have been found to be particularly effective in the treatment of leiomyosarcomas.50,52 Additionally, a retrospective study found a 51% overall response rate for patients with myxoid liposarcomas treated with trabectedin.53 Studies of patients with angiosarcoma treated with paclitaxel have demonstrated response rates of 43% and 53%.54,55
• What are the newest approved and investigational agents?
A recently approved agent is trabectedin, a tris tetrahydroisoquinoline alkaloid isolated from ascidians that binds to the minor groove of DNA and causes disruptions in the cell cycle. Samuels et al reported data from a single-arm, open-label expanded access trial that evaluated patients with advanced metastatic sarcomas.56 In this study, patients with liposarcomas and leiomyosarcomas had an objective response rate of 6.9% (95% CI 4.8 to 9.6) as compared to a rate of 5.9% (95% CI 4.4 to 7.8) for all assessable patients. Median survival was 11.9 months for all patients, with improved median survivals for liposarcoma and leiomyosarcomas of 16.2 months (95% CI 14.1 to 19.5) compared to 8.4 months (95% CI 7.1 to 10.7 months) for other subtypes.56
Schöffski et al evaluated eribulin, a chemotherapeutic agent that affects microtubule dynamics, in a phase 2 trial of patients with progressive or high-grade STS with progression on previous chemotherapy. They found a median progression-free survival of 2.6 months (95% CI 1.7 to 6.2) for adipocytic sarcoma, 2.9 months (95% CI 2.4 to 4.6) for leiomyosarcoma, 2.6 months (95% CI 2.3 to 4.3) for synovial sarcoma, and 2.1 months (95% CI 1.4 to 2.9) for other sarcomas.57
Van der Graaf and colleagues randomly assigned patients with metastatic nonadipocytic STS to pazopanib or placebo in a phase 3 trial. Pazopanib is a small-molecule endothelial growth factor inhibitor with activity against vascular endothelial growth factors 1, 2, and 3 as well as platelet-derived growth factors. Median progression-free survival was 4.6 months (95% CI 3.7 to 4.8) with pazopanib compared to 1.6 months (95% CI 0.9 to 1.8) with placebo.58 Adipocytic sarcomas (liposarcomas) were excluded from the trial because phase 2 trials had found a lower rate of progression-free survival (26%) for them compared to other subtypes.
• What are the most common toxicities associated with the approved and investigational chemotherapeutic agents?
Toxicities were seen with each of the regimens studied and were common in the randomized trials, with higher rates of toxicities in the combination chemotherapy regimens. The most common toxicities are myelosuppression, nausea, and vomiting. In the doxorubicin trials, the most common toxicities were myelosuppression, nausea, and vomiting.44
Ifosfamide both as an individual agent and in combination with doxorubicin has higher rates and higher grades of toxicity than doxorubicin alone. Myelosuppression is the most common toxicity associated with ifosfamide, and the most commonly affected cell line is leukocytes.44 Combination doxorubicin and ifosfamide also had high rates of nausea and vomiting (95%) and alopecia (100%).35Neutropenia is the most common toxicity associated with gemcitabine and dacarbazine, while their most common nonhematologic toxicities are fatigue and nausea.52,59 Trabectedin’s most common toxicities are nausea (29%), neutropenia (24%), and fatigue (23%). It has also been shown to cause increased alkaline phosphatase (20%) and alanine aminotransferase (19%) levels.56 In a phase 2 study of eribulin, 50% of patients had neutropenia, and other toxicities included fatigue, alopecia, nausea, sensory neuropathy, and thrombocytopenia.57 Pazopanib is generally well tolerated; the most common toxicities are fatigue (65%), diarrhea (58%), nausea (54%), and hypertension (41%).58 Higher rates of neutropenia, mucositis, nausea, vomiting, diarrhea, and transfusion reactions were seen with olaratumab and doxorubicin compared to doxorubicin alone in phase 1b and 2 studies.46
› Case Continued
Given his poor prognosis with unresectable metastatic undifferentiated liposarcoma, the patient considers a clinical trial prior to undergoing combined therapy with doxorubicin and ifosfamide. He tolerates therapy well with stable disease at 6 months.
Conclusion
STSs are a heterogeneous collection of rare tumors. Low-grade, localized tumors have the best prognosis, and patients who undergo complete resection have the best long-term survival. Due to the rarity of STSs, trials often have limited enrollment, and little progress has been made with regards to treatment and survival rates for metastatic and unresectable disease. All patients should be evaluated and treated at specialized sarcoma centers. This case highlights the need for continued research and clinical trials to improve overall survival of patients with sarcoma. TSJ
CORRESPONDENCE
Ashley Pariser, MD, Resident, Department of Medicine, Northwestern University Feinberg School of Medicine Chicago, IL. Accepted for publication Jan/Feb 2017; Hosp Phys; Vol. 12, Part1
Introduction
Soft tissue sarcomas (STSs) are rare adult tumors, with 3.4 new cases per 100,000 persons or 12,310 expected new cases in 2016.1 Sarcomas are a heterogeneous collection of tumors that affect fat, muscle, nerve, nerve sheath, vascular, and connective tissues. There are more than 50 histological subtypes that comprise this diverse category of tumors. Treatment varies by stage, with limb-sparing surgery representing the mainstay of curative-intent treatment. Radiation and chemotherapy may also be considered depending on the size, grade, and location of the tumor. Survival rates have been stagnant until recently, with a disease-specific survival hovering around 65%.1 Given the complexity of these cases, all patients ideally should be evaluated and treated by a multidisciplinary team at an institution with extensive experience treating STS.2
Epidemiology and Classification
The most common STS subtypes are gastrointestinal stromal tumor (GIST), undifferentiate pleomorphic sarcoma (previously referred to as malignant fibrous histiocytoma), liposarcoma, leiomyosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, and unclassified sarcoma.3 Liposarcoma is one of the most common subtypes, comprising 20% of all STSs; it is subdivided into well-differentiated/dedifferentiated liposarcomas, myxoid/round cell liposarcomas, and pleomorphic liposarcomas. Well-differentiated liposarcomas tend to occur in the retroperitoneum and limbs, while both myxoid and round cell as well as pleomorphic liposarcomas more commonly originate on the limbs. Histology varies based on subtype and ranges from mature-appearing adipocytes and fibroblasts to undifferentiated cells with minimal lipogenic differentiation.4
Leiomyosarcomas are smooth muscle tumors and are usually located in the retroperitoneum, but have also been associated with peripheral soft tissue and vasculature. Typical histology ranges from well-defined areas of spindle-shaped cells to poorly differentiated anaplastic spindle cells.5,6 Synovial sarcomas are a distinct type of STS that can show epithelial differentiation and account for 5% of adult STSs. The extremities are the most common presenting location (90%).7
Rhabdomyosarcomas are skeletal muscle tumors and are further subdivided into embryonal, alveolar, and pleomorphic subtypes. Embryonal histology ranges from primitive mesenchymal-appearing cells to highly differentiated muscle cells. Alveolar rhabdomyosarcoma has the worst prognosis of the subtypes and consists of round cells with high nuclear-to-chromatin ratios that form “glandular-like” or “alveolar” spaces.8 Pleomorphic rhabdomyosarcomas are composed of rhabdomyoblasts that can affect many different locations, but most commonly present on the lower extremities.9
Malignant peripheral nerve sheath tumor (MPNST) comprises 5% to 10% of all STSs. These tumors are associated with neurofibromatosis type 1 (NF-1), with 25% to 50% of tumors occurring in NF-1 patients. Additionally, most patients have a truncating lesion in the NF1 gene on chromosome 17.10 Anghileri et al in their single institution analysis of 205 patients with MPNSTs found the 2 most common presenting sites were the trunk and extremities. Histologically, these tumors have dense fascicles of spindle cells.10
GISTs are the most common STS of the gastrointestinal (GI) tract. Previously, GISTs were classified as smooth muscle tumors and were not accounted for in the literature as a separate entity distinct from leiomyomas, leiomyoblastomas, and leiomyosarcomas.11 GISTs are found throughout the GI tract: the most common sites are the stomach (60%) and small intestine (30%). Less common sites include duodenum (4%–5%), esophagus (1%), rectum (1%–2%), and appendix (< 0.2%).12 GISTs can be spindle cell, epithelioid, or mesenchymal tumors. Immunohistochemically, GISTs are KIT (CD117) positive. Other cell markers that are also commonly positive include CD34 (60%–70%) and smooth muscle actin (SMA) (25%).11 The majority of GISTs (80%) have an activating c-KIT gene mutation. The most common mutation site is exon 11, with less common c-KIT gene mutations also occurring at exon 9 or 13. Not all GISTs have KIT mutations. The second most common mutation is the PDGFRA mutation (5%–10% of GISTs).2 A minority of GISTs are negative for both KIT and PDGFRA mutations. These tumors were previously called wild-type, but as the majority have either a succinate dehydrogenase (SDH) loss of function or loss of SDHB protein expression, they are now referred to as SDH-deficient GISTs.2 GISTs vary in aggressiveness from incidental to aggressive. Typically, small intestine and rectal GISTs are more aggressive than gastric GISTs. Both size and mitotic rate help to predict the metastatic potential of the tumor. Tumors less than 2 cm in size and having a mitotic rate of less than 5 per 50 high-power fields (hpf) have the lowest risk of metastases, while tumors greater than 5 cm and with more than 5 mitoses per 50 hpf have the highest rates of metastases.12
Angiosarcomas are rare tumors comprising 4% of all STSs. Although they can occur in any site, the majority are cutaneous and occur most frequently in the head and neck regions. These tumors are either of vascular or lymphatic origin and are comprised of abnormal, pleomorphic, malignant endothelial cells. The most useful immunohistochemical markers include von Willebrand factor, CD31, and Ulex europaeus agglutinin 1. The majority of these tumors occur sporadically; however, radiation exposure, chronic lymphedema, and certain toxins including vinyl chloride and thorium dioxide are known risk factors.13
Undifferentiated sarcomas have no specific features and typically consist of primitive mesenchymal cells.
Clinical Evaluation
› Case Presentation
Initial Presentation and History
A 55-year-old man presents to his primary care physician with a painless mass in his anterior thigh. The mass has been present for the past 3 months and he believes that it is enlarging. The patient has a history of well-controlled hypertension and hyperlipidemia. His medications include atorvastatin and hydrochlorothiazide. He has no known drug allergies. Family history is notable for diabetes and hypertension. He drinks 4 to 5 alcoholic drinks a week and he is a former smoker. He quit smoking in his 30s and only smoked intermittently prior to quitting. He denies any illicit drug use. He works as a high school principal. Currently, he feels well. His review of systems is otherwise noncontributory.
Physical Examination
On physical exam, he is afebrile with a blood pressure of 132/75 mm Hg, respiratory rate of 10 breaths/min, and oxygen saturation of 99% on room air. He is a well appearing, overweight male. His head and neck exam is unremarkable. Lung exam reveals clear breath sounds, and cardiac exam reveals a regular rate and rhythm. His abdomen is obese, soft, and without hepatosplenomegaly. There is a large, fixed mass on the anterior lateral aspect of his right thigh. He has no appreciable lymphadenopathy. His neurological exam is unremarkable.
• What are risk factors for sarcoma?
There are few known risk factors for sarcoma. Established risks factors include prior radiation therapy, chronic lymphedema, viruses, and genetic cancer syndromes including Li-Fraumeni syndrome, hereditary retinoblastoma, and NF-1. Other environmental exposures include phenoxyacetic acids and chlorophenols.14 The majority of cases are sporadic, with only a minority of patients having one of these known risk factors.15 Up to one third of sarcomas have a specific translocation and are driven by fusion oncogenes (
• What is the typical presentation for sarcomas?
A painless mass is the most typical presenting symptom. Size at presentation varies based on location, with extremity and head and neck locations typically presenting at smaller sizes than retroperitoneal tumors.14 Patients may experience pain and numbness as the mass enlarges and impinges on surrounding structures including nerves and vasculature. The vast majority of patients are without systemic symptoms.
• How is sarcoma staged?
The American Joint Committee on Cancer (AJCC) staging system is the most widely used staging system in the United States. The latest AJCC manual was updated in 2010 to include a 3-tiered grading system where the tumor is classified according to tumor size, lymph node involvement, metastases, and grade at time of diagnosis (Table 2 and Table 3). Additionally, tumor depth in relation to deep fascia is also taken into account, with superficial tumors being assigned a designation of “a” and deep tumors a designation of “b.”
Previously, 2 of the most widely used grading systems were the National Cancer Institute (NCI) and French Federation of Cancer Centers Sarcoma Group (FNCLCC) systems, both 3-tier grading systems. The main components that determine the NCI grade are the tumor’s histologic type and location and the amount of tumor necrosis. The FNCLCC system evaluation focuses on tumor differentiation, mitotic rate, and amount of tumor necrosis. A study that compared the NCI and FNCLCC grading systems found that FNCLCC was a better predictor of mortality and distant metastasis.16 Previously, the AJCC was a 4-tier grading system, but the 2010 version was updated to the 3-tier FNCLCC grading system. Additionally, the AJCC system has reclassified single lymph node disease as stage III as it confers better survival than metastatic disease.17 It is important that pathology be evaluated by a sarcoma specialist as disagreements with regard to histologic subtype and grade are common.18,19
• What are the most important prognostic factors?
Prognostic factors include grade, size, and presence of metastases at presentation. Best survival is associated with low-grade, small tumors with no metastases at time of diagnosis.14
• What imaging should be considered?
Imaging should be undertaken to help differentiate between benign and malignant lesions. Ideally, it should be undertaken before a biopsy is planned as the imaging can be used to plan biopsy as well as provide invaluable prognostic information. There are several imaging modalities that should be considered during the preliminary work-up and staging of STSs. Conventional imaging includes magnetic resonance imaging (MRI) of the original tumor site; computed tomography (CT) to evaluate for pulmonary metastases and, depending on location, liver metastases; and in the case of small, low-grade tumors, chest radiography. MRI is considered the test of choice for soft tissue masses and can help delineate benign masses such as hematomas, lipomas, and hemangiomas from sarcomas.20 It is difficult to compare the accuracy of positron emission tomography (PET)/CT to CT and MRI because most studies have evaluated PET/CT in parallel with CT and MRI.21 Tateishi et al compared the accuracy of conventional imaging, PET/CT, and PET/CT combined with conventional imaging at determining the TNM staging for 117 patients. They found that conventional imaging correctly classified 77% of patients, PET alone correctly classified 70%, PET/CT correctly classified 83%, and PET/CT combined with conventional imaging correctly staged 87%.22
• Which subtypes are most likely to metastasize?
Although the vast majority of sarcomas spread hematogenously, 3 have a propensity to spread lymphogenously: epithelioid sarcoma, rhabdomyosarcoma, and clear-cell sarcoma. Additionally, certain subtypes are more likely to metastasize: leiomyosarcomas, synovial sarcomas, neurogenic sarcomas, rhabdomyosarcomas, and epithelioid sarcomas.23 Sarcomas metastasize to the lungs more frequently than to the liver. The metastatic pattern is defined primarily by sarcoma subtype and site of primary tumor. Sarcomas rarely metastasize to the brain (~1%).
Management
› Case Continued
The patient undergoes an ultrasound to better visualize the mass. Given the heterogeneous character of the mass, he is referred for an MRI to evaluate the mass and a CT scan of the chest, abdomen, and pelvis to evaluate for distant metastases. MRI reveals a 5.1 cm × 4.6 cm heterogeneous mass invading the superficial fascia of the rectus femoris muscle. No suspicious lymph nodes or other masses are identified on imaging. The patient next undergoes an image-guided core needle biopsy. Pathology from that procedure is consistent with a stage III, T2bNxMx, grade 3, dedifferentiated liposarcoma.
• What is the best management approach for this patient?
Surgery
Surgery is the mainstay of treatment for STS. Patients with the best prognosis are those who undergo complete resection with negative surgical margins.24,25 Goal tumor-free margin is 1 to 3 cm.26 Complete resection confers the best long-term survival. Both local and metastatic recurrence is higher in patients with incomplete resection and positive margins.24,25 In a study that analyzed 2084 localized primary STSs, patients with negative margins had a local recurrence rate of 15% versus a rate of 28% in patients with positive margins. This translated into higher 5-year local recurrence-free survival for patients with negative surgical margins (82%) compared to patients with positive margins (65%).27 Another study similarly found that patients with negative margins at referral to their institution who underwent postoperative radiation had high local control rates of 93% (95% confidence interval [CI] 87% to 97%) at 5, 10, and 15 years.26 Although radiation improves local control, neither preoperative or postoperative radiation has been shown to improve progression-free or overall survival.28 Other factors that are associated with risk of recurrence are tumor location, history of previous recurrence, age of patient, histopathology, tumor grade, and tumor size. Approximately 40% to 50% of patients with high-grade tumors (defined as size > 5 cm, deep location, and high grade) will develop distant metastases.29
Zagars et al found that positive or uncertain resection margin had a relative risk of local recurrence of 2.0 (95% CI 1.3 to 3.1; P = 0.002), and presentation with locally recurrent disease (vs new tumor) had a relative risk of local recurrence of 2.0 (95% CI 1.2 to 3.4; P = 0.013).26 Patients with STS of head and neck and deep trunk have higher recurrence rates than those with superficial trunk and extremity STS. A single-institution retrospective review demonstrated that patients with completely resectable retroperitoneal sarcomas have longer median survival (103 months) compared to patients with incompletely resected abdominal sarcomas (18 months).25Rosenberg and colleagues compared amputation to limb-sparing surgery and radiation.24 Their prospective analysis of 65 patients found no difference in disease-free and overall survival between the 2 treatment groups.The limb-sparing treatment group had higher rates of local recurrence, which was highly correlated with positive surgical margins on pathology.24 Evidence from this and similar studies has resulted in radical amputations being replaced by conservative limb-sparing procedures and radiation therapy. In those found to have positive margins, re-resection is an option for some. Patients who undergo re-resection have higher local control rates than patients with positive margins who do not undergo re-resection. The 5-year control rate for patients who undergo re-resection is 85% (95% CI 80% to 89%) compared to 78% (95% CI 71% to 83%) for those who do not undergo re-resection. Similarly, patients who undergo re-resection have lower rates of metastases at 5, 10, and 15 years as well as higher 5-, 10-, and 15-year disease-free survival rates.26
› Case Continued
The patient is referred for limb-sparing surgery after presentation at a multidisciplinary tumor board. Prior to undergoing resection of the tumor, he is also referred to radiation-oncology to discuss the risks and benefits of combination radiotherapy and surgery as opposed to surgical resection alone.
• What is the evidence for radiation therapy?
Radiation THERAPY
Radiation therapy is used in the preoperative, intraoperative, and postoperative settings to reduce the risk of local recurrence. There are several options for radiation, including external beam radiation therapy (EBRT), intraoperative radiation, and brachytherapy. A newer strategy, intensity-modulated radiation therapy (IMRT), utilizes 3-dimensional modeling to reduce radiation dosages. Overall there are no differences in overall survival or local recurrence rates between preoperative and postoperative radiation in STS.28
The rationale behind preoperative radiation is that it reduces seeding of tumor cells, especially at the time of surgery.30 Additionally, for EBRT, preoperative radiation has smaller field sizes and lower radiation doses. It can also help to reduce the size of the tumor prior to resection. Intraoperative radiation is often paired with preoperative radiation as a boost dose given only to the area of residual tumor.
Suit et al reviewed patients treated at a single institution with limb-sparing surgery and different radiation strategies. Local control rates between preoperative and postoperative radiation groups were not statistically significant. Local recurrence was linked to grade and size of the tumor in both groups. The authors did note, however, that the preoperative radiation group tended to have larger tumor sizes at baseline compared to the patients who received postoperative radiation.30 A study that compared 190 patients who received preoperative and postoperative EBRT or brachytherapy (primary end point was wound complications, and local control was a secondary end point) showed a trend towards greater local control with preoperative radiation; however, the preoperative radiation group had significantly more wound complications compared to the postoperative radiation group.31
Yang et al found that postoperative EBRT decreases rates of local recurrence compared to surgery alone in high-grade extremity sarcomas.32 However, there were no differences in rates of distant metastases and overall survival between the 2 treatment groups. Similarly, in patients with low-grade sarcoma, there were fewer local recurrences in those who received EBRT and surgery as compared to surgery alone.32 Another study that evaluated 164 patients who received either adjuvant brachytherapy or no further therapy after complete resection found that brachytherapy reduced local recurrence in high-grade sarcomas. No difference in local recurrence rates was found in patients with low-grade sarcomas, nor was a significant difference found in the rates of distant metastases and overall survival between the 2 treatment groups.33 With regards to IMRT, a single institution cohort experience with 41 patients who received IMRT following limb-sparing surgery had similar local control rates when compared to historical controls.34
› Case Continued
After discussion of the risks and benefits of radiation therapy, the patient opts for preoperative radiation prior to resection of his liposarcoma. He receives 50 Gy of EBRT prior to undergoing resection. Resection results in R1 margin consistent with microscopic disease. He receives 16 Gy of EBRT as a boost after recovery from his resection.2
• What is the evidence for neoadjuvant and adjuvant chemotherapy for stage I tumors?
Chemotherapy
Localized Sarcoma
For localized sarcoma, limb-sparing resection with or without radiation forms the backbone of treatment. Studies have evaluated chemotherapy in both the neoadjuvant and adjuvant settings, with the vast majority of studies evaluating doxorubicin-based chemotherapy regimens in the adjuvant settings. Due to the rare nature of sarcomas, most studies are not sufficiently powered to detect significant benefit from chemotherapy. Several trials evaluating chemotherapy regimens in the neoadjuvant and adjuvant settings needed to be terminated prematurely due to inadequate enrollment into the study.35,36
For stage IA (T1a-Tb, N0, M0, low grade) tumors, no additional therapy is recommended after limb-sparing surgery with appropriate surgical margins. For stage IB (T2a-2b, N0, M0, low grade) tumors with insufficient margins, re-resection and radiation therapy should be considered, while for stage IIA (T1a-1b, N0, M0, G2-3) tumors preoperative or postoperative radiation therapy is recommended.2 Studies have not found benefit of adjuvant chemotherapy in these low-grade, stage I tumors in terms of progression-free survival and overall survival.37
• At what stage should chemotherapy be considered?
For stage IIb and stage III tumors, surgery and radiation therapy again form the backbone of therapy; however, neoadjuvant and adjuvant chemotherapy are also recommended as considerations. Anthracycline-based chemotherapy with either single-agent doxorubicin or doxorubicin and ifosfamide in combination are considered first-line chemotherapy agents in locally advanced STS.2,29,37
Evidence regarding the efficacy of both neoadjuvant and adjuvant chemotherapy regimens in the setting of locally advanced high-grade STS has been mixed. The Sarcoma Meta-analysis Collaboration evaluated 14 trials of doxorubicin-based adjuvant chemotherapy and found a trend towards overall survival in the treatment groups that received chemotherapy.37 All trials included in the meta-analysis compared patients with localized resectable soft-tissue sarcomas who were randomized to either adjuvant chemotherapy or no adjuvant chemotherapy after limb-sparing surgery with or without radiation therapy. None of the individual trials showed a significant benefit, and all trials had large confidence intervals; however, the meta-analysis showed significant benefit in the chemotherapy treatment groups with regard to local recurrence, distant recurrence, and progression-free survival. No significant difference in overall survival was found.37 Pervais et al updated the Sarcoma Meta-analysis Collaboration’s 1997 meta-analysis with the inclusion of 4 new trials that evaluated doxorubicin combined with ifosfamide and found that both patients who received doxorubicin-based regimens or doxorubicin with ifosfamide had significant decreases in distant and overall recurrences. Only the trials that utilized doxorubicin and ifosfamide had an improved overall survival that was statistically significant (hazard ratio 0.56 [95% CI 0.36 to 0.85]; P = 0.01).29 Although no significant heterogeneity was found among the trials included in either meta-analysis, a variety of sarcomas were included in each clinical trial evaluated. Given the extremely small number of each sarcoma subtype present in each trial, subgroup analysis is difficult and prone to inaccuracies. As a result, it is not known if certain histological subtypes are more or less responsive to chemotherapy.37–39
One randomized controlled trial evaluated neoadjuvant chemotherapy in high-risk sarcomas defined as tumors greater than 8 cm or grade II/III tumors. This study evaluated doxorubicin and ifosfamide and found no significant difference in disease-free and overall survival in the neoadjuvant therapy group compared to the control group.35 There remains controversy in the literature with regards to adjuvant chemotherapy. Many oncologists offer adjuvant chemotherapy to patients with certain stage III subtypes. Examples of subtypes that may be offered adjuvant therapy include myxoid liposarcomas, synovial sarcomas, and leiomyosarcomas.2 With regards to how many cycles of chemotherapy should be considered, a noninferiority study compared 3 cycles of epirubicin and ifosfamide to 5 cycles of epirubicin and ifosfamide in patients with high-risk locally advanced adult STSs. Three cycles of preoperative epirubicin and ifosfamide was found to be noninferior to 5 cycles with regards to overall survival.38
• What is this patient’s risk for recurrence?
The patient is at intermediate risk for recurrence. Numerous studies have demonstrated that tumor size, grade, and location are the most important factors to determine risk of recurrence, with larger size, higher grades, and deeper locations being associated with higher risk of recurrence. In an analysis of 1041 patients with STS of the extremities, high grade was the most important risk factor for distant metastases.39 The highest risk of recurrence is within the first 2 years. Given that the patient’s initial tumor was located in the extremity, he is more likely to have a distant metastasis as his site of recurrence; individuals with retroperitoneal tumors and visceral tumors are more likely to recur locally.40 For STSs of the extremity, distant metastases determine overall survival, whereas patients with retroperitoneal sarcomas can die from complications of local metastases.41 Once a patient develops distant metastases, the most important prognostic factor is the size of the tumor, with tumors larger than 10 cm having a relative risk of 1.5 (95% CI 1.0 to 2.0).39
• What are the recommendations for surveillance?
Surveillance recommendations are based on the stage of the sarcoma. Stage I tumors are the least likely to recur either locally or distally. As a result, it is recommended that stage I tumors be followed with history and physical exam every 3 to 6 months for the first 2 to 3 years, and then annually after the first 2 to 3 years. Chest x-rays should be considered every 6 to 12 months.2 For stage II–IV tumors, history and physical exam is recommended every 3 to 6 months for the first 2 to 3 years. Chest and distant metastases imaging should also be performed every 3 to 6 months during this time frame. For the next 2 years, history and physical exam and imaging are recommended every 6 months. After the first 4 to 5 years, annual follow-up is recommended.2
A study that followed 141 patients with primary extremity STSs for a median interval of 49 months found that high-grade tumors were most likely to recur during the first 2 years, with 20% of their patients recurring locally and 40% recurring distally. Chest x-rays performed during surveillance follow-up found distant lung metastases in 36 asymptomatic patients and had a positive predictive value of 92%, a negative predictive value of 97%, and a quality-adjusted life-year of $30,000.40,41 No laboratory testing was found to aid in detection of recurrence.
› Case Continued
The patient does well for 1 year. With physical therapy, he regains most of the strength and coordination of the lower extremity. He is followed every 3 months with chest x-rays and a MRI of the thigh for the first year. On his fourth follow-up clinic visit, he describes increased dysp-nea on exertion over the previous few weeks and is found to have multiple lung metastases in both lungs on chest x-ray. He undergoes further evaluation for metastases and is not found to have any other metastatic lesions. Bronchoscopy and biopsy of 1 of the lung nodules confirms recurrent dedifferentiated liposarcoma.
• Should this patient undergo metastectomy?
An analysis of 3149 patients with STS treated at Memorial Sloan-Kettering who developed lung metastases found that patients with pulmonary metastases have survival rates of 25%. The most important prognostic factor for survival was complete resection of all metastases.42 For stage IV disease, surgery is used only in certain instances. In instances where tumor is more localized or limited, removal of metastases or metastectomy can play a role in management.2
› Case Continued
Because the patient’s metastases are limited to the lungs, he is referred for metastectomy. He undergoes wedge resection for definitive diagnosis but it is not possible to completely resect all of the metastases. He is thus referred to a medical oncologist to discuss his treatment options.
• What are treatment options for unresectable or metastatic disease?
Metastatic Disease
Unlike local and locally advanced disease, chemotherapy forms the backbone of treatment in stage IV disease. Doxorubicin and olaratumab or doxorubicin and ifosfamide in combination are considered first line in metastatic disease. Response rates for single-agent doxorubicin range from 16% to 27%, while phase 2 and phase 3 studies of doxorubicin and ifosfamide have found response rates ranging from 18% to 36%.43 In addition, the effectiveness of doxorubicin and ifosfamide phase 2 and 3 trials varied. Edmonson et al found a tumor regression rate of 34% for doxorubicin and ifosfamide as compared to 20% for doxorubicin alone.44 In comparison, Santoro et al found a response rate of 21.3% for doxorubicin alone and 25.2% for doxorubicin and ifosfamide.45 Neither study found increased survival benefit for doxorubicin and ifosfamide when compared to doxorubicin alone. In a Cochrane review evaluating randomized trials that compared doxorubicin and combination chemotherapy regimens, response rates varied from 14% for doxorubicin in combination with streptomycin to 34% for doxorubicin and ifosfamide. Most trials did not show a significant benefit for combination therapies when compared to doxorubicin alone.43 Mean survival with doxorubicin or doxorubicin and ifosfamide is 12 months. High rates of recurrence highlight the need for additional chemotherapy regimens.
The newest approved agent is olaratumab, a monoclonal antibody that binds platelet-derived growth factor receptor alpha and prevents receptor activation. A phase 1-b and phase 2 trial evaluated patients with locally advanced and metastatic STS and randomly assigned them to either olaratumab and doxorubicin or doxorubicin alone.46 Progression-free survival for olaratumab/doxorubicin was 6.6 months (95% CI 4.1 to 8.3) compared to 4.1 months (95% CI 2.8 to 5.4) for doxorubicin alone. The objective response rate was 18.2% (95% CI 9.8 to 29.6) for olaratumab/doxorubicin compared to 7.5% (95% CI 2.5 to 6.6) for doxorubicin alone. Furthermore, the median overall survival for olaratumab plus doxorubicin was 26.5 months (95% CI 20.9 to 31.7) compared to 14.7 months for doxorubicin alone (95% CI 5.5 to 26.0). Impressively, this improved response was notable across histological types. Furthermore, patients who had previously been treated with more than 1 regimen and those who were treatment naïve had similar response rates.46
• What are second-line treatment options?
Doxorubicin has been used in combination with several other agents including dacarbazine (DTIC) as well as DTIC and ifosfamide (MAID). Borden et al evaluated patients with metastatic STS and randomly assigned the patients to either doxorubicin or doxorubicin and DTIC. Combination therapy demonstrated better tumor response than doxorubicin alone: 30% complete or partial response for combination therapy and 18% for doxorubicin alone.47 However, Omura et al found similar rates of efficacy between doxorubicin and combination doxorubicin and DTIC in women with recurrent or nonresectable uterine sarcomas.48 MAID has never been directly compared in a randomized trial to doxorubicin alone. In a study that compared MAID to doxorubicin and DTIC (AD) in patients with unresectable or metastatic sarcomas, MAID had superior response rates (32% versus 17%), but there was no difference with regards to overall survival (mean survival of 12.5 months).49
Several additional regimens have undergone evaluation in metastatic and recurrent STSs. Gemcitabine has been used both as a single agent and as part of combination therapy in many studies. Studies with gemcitabine in combination with either docetaxel or DTIC have been the most efficacious. In a phase 2 trial, patients with metastatic STS were randomly assigned to either gemcitabine alone or gemcitabine and docetaxel. Combination therapy had a higher response rate (16% versus 8%) and longer overall survival (17.9 months versus 11.5 months) than gemcitabine alone.50 Furthermore, a phase 2 trial of gemcitabine and docetaxel in patients with unresectable leiomyosarcoma showed an overall response rate of 56%, with 3 complete and 15 partial responses among the 34 patients enrolled in the study.51 A phase 2 trial randomly assigned patients with unresectable or metastatic STS to either DTIC or combination gemcitabine and DTIC.52 Gemcitabine-DTIC had a superior progression-free survival at 3 months (56% [95% CI 43% to 69%]) as compared to DTIC alone (37% [95% CI 23.5% to 50%]). Furthermore, mean progression-free survival and overall survival were improved in the gemcitabine-DTIC group (4.2 months and 16.8 months) as compared to the DTIC group (2.0 months and 8.2 months).52 DTIC has a single-agent response rate of 16%, but has been shown to be particularly effective in the setting of leiomyosarcomas.49
• Does response to treatment regimens differ by histologic subtype?
The majority of STS trials include many different histologic subtypes. Given the rarity of sarcomas as a whole, many trials have had difficulty recruiting adequate numbers of patients to have sufficient power to definitely determine if the treatment under investigation has clinical benefit. Furthermore, the patients recruited have been heterogeneous with regard to subtype. Many older studies hypothesized that the efficacy of chemotherapeutic agents vary based on histologic subtype; however, for most subtypes the number of individuals included in those trials was too low to evaluate efficacy based on subtype.
Some exceptions exist, however. For example, both gemcitabine-DTIC and gemcitabine-docetaxel have been found to be particularly effective in the treatment of leiomyosarcomas.50,52 Additionally, a retrospective study found a 51% overall response rate for patients with myxoid liposarcomas treated with trabectedin.53 Studies of patients with angiosarcoma treated with paclitaxel have demonstrated response rates of 43% and 53%.54,55
• What are the newest approved and investigational agents?
A recently approved agent is trabectedin, a tris tetrahydroisoquinoline alkaloid isolated from ascidians that binds to the minor groove of DNA and causes disruptions in the cell cycle. Samuels et al reported data from a single-arm, open-label expanded access trial that evaluated patients with advanced metastatic sarcomas.56 In this study, patients with liposarcomas and leiomyosarcomas had an objective response rate of 6.9% (95% CI 4.8 to 9.6) as compared to a rate of 5.9% (95% CI 4.4 to 7.8) for all assessable patients. Median survival was 11.9 months for all patients, with improved median survivals for liposarcoma and leiomyosarcomas of 16.2 months (95% CI 14.1 to 19.5) compared to 8.4 months (95% CI 7.1 to 10.7 months) for other subtypes.56
Schöffski et al evaluated eribulin, a chemotherapeutic agent that affects microtubule dynamics, in a phase 2 trial of patients with progressive or high-grade STS with progression on previous chemotherapy. They found a median progression-free survival of 2.6 months (95% CI 1.7 to 6.2) for adipocytic sarcoma, 2.9 months (95% CI 2.4 to 4.6) for leiomyosarcoma, 2.6 months (95% CI 2.3 to 4.3) for synovial sarcoma, and 2.1 months (95% CI 1.4 to 2.9) for other sarcomas.57
Van der Graaf and colleagues randomly assigned patients with metastatic nonadipocytic STS to pazopanib or placebo in a phase 3 trial. Pazopanib is a small-molecule endothelial growth factor inhibitor with activity against vascular endothelial growth factors 1, 2, and 3 as well as platelet-derived growth factors. Median progression-free survival was 4.6 months (95% CI 3.7 to 4.8) with pazopanib compared to 1.6 months (95% CI 0.9 to 1.8) with placebo.58 Adipocytic sarcomas (liposarcomas) were excluded from the trial because phase 2 trials had found a lower rate of progression-free survival (26%) for them compared to other subtypes.
• What are the most common toxicities associated with the approved and investigational chemotherapeutic agents?
Toxicities were seen with each of the regimens studied and were common in the randomized trials, with higher rates of toxicities in the combination chemotherapy regimens. The most common toxicities are myelosuppression, nausea, and vomiting. In the doxorubicin trials, the most common toxicities were myelosuppression, nausea, and vomiting.44
Ifosfamide both as an individual agent and in combination with doxorubicin has higher rates and higher grades of toxicity than doxorubicin alone. Myelosuppression is the most common toxicity associated with ifosfamide, and the most commonly affected cell line is leukocytes.44 Combination doxorubicin and ifosfamide also had high rates of nausea and vomiting (95%) and alopecia (100%).35Neutropenia is the most common toxicity associated with gemcitabine and dacarbazine, while their most common nonhematologic toxicities are fatigue and nausea.52,59 Trabectedin’s most common toxicities are nausea (29%), neutropenia (24%), and fatigue (23%). It has also been shown to cause increased alkaline phosphatase (20%) and alanine aminotransferase (19%) levels.56 In a phase 2 study of eribulin, 50% of patients had neutropenia, and other toxicities included fatigue, alopecia, nausea, sensory neuropathy, and thrombocytopenia.57 Pazopanib is generally well tolerated; the most common toxicities are fatigue (65%), diarrhea (58%), nausea (54%), and hypertension (41%).58 Higher rates of neutropenia, mucositis, nausea, vomiting, diarrhea, and transfusion reactions were seen with olaratumab and doxorubicin compared to doxorubicin alone in phase 1b and 2 studies.46
› Case Continued
Given his poor prognosis with unresectable metastatic undifferentiated liposarcoma, the patient considers a clinical trial prior to undergoing combined therapy with doxorubicin and ifosfamide. He tolerates therapy well with stable disease at 6 months.
Conclusion
STSs are a heterogeneous collection of rare tumors. Low-grade, localized tumors have the best prognosis, and patients who undergo complete resection have the best long-term survival. Due to the rarity of STSs, trials often have limited enrollment, and little progress has been made with regards to treatment and survival rates for metastatic and unresectable disease. All patients should be evaluated and treated at specialized sarcoma centers. This case highlights the need for continued research and clinical trials to improve overall survival of patients with sarcoma. TSJ
CORRESPONDENCE
Ashley Pariser, MD, Resident, Department of Medicine, Northwestern University Feinberg School of Medicine Chicago, IL. Accepted for publication Jan/Feb 2017; Hosp Phys; Vol. 12, Part1
References
1. American Cancer Society. Cancer facts and figures 2016. American Cancer Society Web site. www.cancer.org/acs/groups/content/@research/documents/document/acspc-047079.pdf. Accessed December 20, 2016.
2. National Comprehensive Cancer Network. NCCN clinical guidelines in oncology: soft tissue sarcoma. 2016
3. Coindre J, Terrier P, Guillou L, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001;91:1914–26.
4. Dei Tos A. Liposarcoma: new entities and evolving concepts. Ann Diagn Pathol 2000;4: 252–66.
5. Wile AG, Evans HL, Romsdahl MM. Leiomyosarcoma of soft tissue: a clinicopathologic study. Cancer 1981;48:1022–32.
6. Hashimoto H, Daimaru Y, Tsuneyoshi M, Enjoji M. Leiomyosarcoma of the external soft tissues. A clinicopathologic, immunohistochemical, and electron microscopic study. Cancer 1986;57:2077–88
7. Fisher C. Synovial sarcoma. Ann Diagn Pathol 1998;2:401–21.
8. Newton WA Jr, Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer 1995;76:1073–85.
9. Furlong MA. Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol. 2001;14:595–603.
10. Anghileri M, Miceli R, Fiore M. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer 2006;107:1065–74.
11. Miettinen M, Lasota J. Gastrointestinal stromal tumors–definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Archive 2001;438:1–12.
12. Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006;23:70–83.
13. Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol 2010;11:983–91.
14. Cormier JN, Pollock RE. Soft tissue sarcomas. CA Cancer J Clin 2004;54:94–109.
15. Penel N, Grosjean J, Robin YM, et al. Frequency of certain established risk factors in soft tissue sarcomas in adults: a prospective descriptive study of 658 cases. Sarcoma 2008;2008:459386.
16. Guillou L, Coindre JM, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 1997;15:350–62.
17. Maki RG, Moraco N, Antonescu CR, et al. Toward better soft tissue sarcoma staging: building on American joint committee on cancer staging systems versions 6 and 7. Ann Surg Oncol 2013;20:3377–83.
18. Shiraki M, Enterline HT, Brooks JJ, et al. Pathologic analysis of advanced adult soft tissue sarcomas, bone sarcomas, and mesotheliomas. The Eastern Cooperative Oncology Group (ECOG) experience. Cancer 1989;64:484–90.
19. Presant CA, Russell WO, Alexander RW, Fu YS. Soft-tissue and bone sarcoma histopathology peer review: The frequency of disagreement in diagnosis and the need for second pathology opinions. The Southeastern Cancer Study Group experience. J Clin Oncol 1986; 4:1658–61.
20. Sundaram M, McLeod RA. MR imaging of tumor and tumorlike lesions of bone and soft tissue. AJR Am J Roentgenol 1990;155:817–24.
21. Ioannidis JP, Lau J. 18F-FDG PET for the diagnosis and grading of soft-tissue sarcoma: a meta-analysis. J Nucl Med 2003;44:717–24.
22. Tateishi U, Yamaguchi U, Seki K, et al. Bone and soft-tissue sarcoma: preoperative staging with fluorine 18 fluorodeoxyglucose PET/CT and conventional imaging. Radiology 2007;245:839–47.
23. Zagars GK, Ballo MT, Pisters PW, et al. Prognostic factors for patients with localized soft-tissue sarcoma treated with conservation surgery and radiation therapy: an analysis of 1225 patients. Cancer 2003;97:2530–43
24. Rosenberg S, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 1982;196:305–14.
25. Lewis J, Leung D, Woodruff J, et al. Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg 1998;288:355–65.
26. Zagars GK, Ballo MT, Pisters PW, et al. Surgical margins and reresection in the management of patients with soft tissue sarcoma using conservative surgery and radiation therapy. Cancer 2003;97:2544–53.
27. Stojadinovic A, Leung DH, Hoos A. Analysis of the prognostic significance of microscopic margins in 2,084 localized primary adult soft tisusse sarcomas. Ann Surg 2002;235:424–34.
28. O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
29. Pervaiz N, Colterjohn N, Farrokhyar F, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008;113:573–81.
30. Suit HD, Mankin HJ, Wood WC, Proppe KH. Preoperative, intraoperative, and postoperative radiation in the treatment of primary soft tissue sarcoma. Cancer 1985;55:2659–67
31. O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
32. Yang J, Chang A, Baker A, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 1998;16:197–203.
33. Pisters PW, Harrison LB, Leung DH, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 1996;14:859–68.
34. Alektiar KM, Brennan MF, Healey JH, Singer S. Impact of intensity-modulated radiation therapy on local control in primary soft-tissue sarcoma of the extremity. J Clin Oncol 2008;26:3440–5.
35. Gortzak E, Azzarelli A, Buesa J, et al. A randomized phase II study on neo-adjuvant chemotherapy for ‘high-risk’ adult soft-tissue sarcoma. Eur J Cancer 2001;37:1096–1103.
36. Fakhari N, Ebm C, Kostler WJ, et al. Intensified adjuvant IFADIC chemotherapy in combination with radiotherapy versus radiotherapy alone for soft tissue sarcoma: long-term follow-up of a prospective randomized feasibility trial. Wein Klin Wochenschr 2010;122:614–9.
37. Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Lancet 1997;350:1647–54.
38. Gronchi A, Frustaci S, Mercuri M, et al. Short, full-dose adjuvant chemotherapy in high-risk adult soft tissue sarcomas: a randomized clinical trial from the Italian Sarcoma Group and the Spanish Sarcoma Group. J Clin Oncol 2012;30:850–56.
39. Pisters PW, Leung DH, Woodruff J. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996;14:1679–89.
40. Whooley B, Gibbs J, Mooney M. Primary Extremity Sarcoma: What is the Appropriate Follow-up? Annals of Surg Oncol 2000; 7: 9-14.
41. Whooley BP, Mooney MN, Gibbs JF, Graybill WG. Effective follow-up strategies in soft tissue sarcoma. Sem Surg Oncol 1999;17:83–87.
42. Billingsley KG, Burt ME, Jara E, et al. Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival. Ann Surg 1999;229:602–10.
43. Bramwell VH, Anderson D, Charette ML; Sarcoma Disease Site Group. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev 2003;(3):CD003293.
44. Edmonson J, Ryan L, Blum R. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 1993;11:1269–75.
45. Santoro A, Tursz T, Mouridsen H. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 1995;13:1537–45.
46. Tap WD, Jones RL, Van Tine B, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet 2016;388:488–97.
47. Borden EC, Amato DA, Rosenbaum C, et al. Randomized comparison of three adriamycin regimens for metastatic soft tissue sarcomas. J Clin Oncol 1987;5:840–50.
48. Omura GA, Major FJ, Blessing JA, et al. A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 1983;52:626–32.
49. Antman K, Crowley J, Balcerzak SP, et al. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol 1993;11:1276–85.
50. Maki R, Wathen K, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 2007; 25: 2755–63.
51. Hensley ML, Maki R, Venkatraman E, et al. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol 2002;12:2824–31.
52. Garcia-del-Muro X, Lopez-Pousa A, Maurel J, et al. Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: a Spanish Group for Research on Sarcomas study. J Clin Oncol 2011;29:2528–33.
53. Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol 2007;7:595–602.
54. Italiano A, Cioffi A, Penel N, et al. Comparison of doxorubicin and weekly paclitaxel efficacy in metastatic angiosarcomas. Cancer 2012;118:3330–6.
55. Penel N, Italiano A, Ray-Coquard I, et al. Metastatic angiosarcomas: doxorubicin-based regimens, weekly paclitaxel and metastasectomy significantly improve outcome. Ann Oncol 2012;23:517–23.
56. Samuels BL, Chawla S, Patel S, et al. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol 2013;24:1703–9.
57. Schöffski P, Ray-Coquard IL, Cioffi A, et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histolical subtypes. Lancet 2011;11:1045–52.
58. Van der Graaf W, Blay JY, Chawla S, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomized, double-blind, placebo-controlled phase 3 trial. Lancet 2012;379:1879–86.
59. Dileo P, Morgan JA, Zahrieh D, et al. Gemcitabine and vinorelbine combination chemotherapy for patients with advanced soft tissue sarcomas: results of a phase II trial. Cancer 2007;109:1863–9.
References
1. American Cancer Society. Cancer facts and figures 2016. American Cancer Society Web site. www.cancer.org/acs/groups/content/@research/documents/document/acspc-047079.pdf. Accessed December 20, 2016.
2. National Comprehensive Cancer Network. NCCN clinical guidelines in oncology: soft tissue sarcoma. 2016
3. Coindre J, Terrier P, Guillou L, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001;91:1914–26.
4. Dei Tos A. Liposarcoma: new entities and evolving concepts. Ann Diagn Pathol 2000;4: 252–66.
5. Wile AG, Evans HL, Romsdahl MM. Leiomyosarcoma of soft tissue: a clinicopathologic study. Cancer 1981;48:1022–32.
6. Hashimoto H, Daimaru Y, Tsuneyoshi M, Enjoji M. Leiomyosarcoma of the external soft tissues. A clinicopathologic, immunohistochemical, and electron microscopic study. Cancer 1986;57:2077–88
7. Fisher C. Synovial sarcoma. Ann Diagn Pathol 1998;2:401–21.
8. Newton WA Jr, Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer 1995;76:1073–85.
9. Furlong MA. Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol. 2001;14:595–603.
10. Anghileri M, Miceli R, Fiore M. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer 2006;107:1065–74.
11. Miettinen M, Lasota J. Gastrointestinal stromal tumors–definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Archive 2001;438:1–12.
12. Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006;23:70–83.
13. Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol 2010;11:983–91.
14. Cormier JN, Pollock RE. Soft tissue sarcomas. CA Cancer J Clin 2004;54:94–109.
15. Penel N, Grosjean J, Robin YM, et al. Frequency of certain established risk factors in soft tissue sarcomas in adults: a prospective descriptive study of 658 cases. Sarcoma 2008;2008:459386.
16. Guillou L, Coindre JM, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 1997;15:350–62.
17. Maki RG, Moraco N, Antonescu CR, et al. Toward better soft tissue sarcoma staging: building on American joint committee on cancer staging systems versions 6 and 7. Ann Surg Oncol 2013;20:3377–83.
18. Shiraki M, Enterline HT, Brooks JJ, et al. Pathologic analysis of advanced adult soft tissue sarcomas, bone sarcomas, and mesotheliomas. The Eastern Cooperative Oncology Group (ECOG) experience. Cancer 1989;64:484–90.
19. Presant CA, Russell WO, Alexander RW, Fu YS. Soft-tissue and bone sarcoma histopathology peer review: The frequency of disagreement in diagnosis and the need for second pathology opinions. The Southeastern Cancer Study Group experience. J Clin Oncol 1986; 4:1658–61.
20. Sundaram M, McLeod RA. MR imaging of tumor and tumorlike lesions of bone and soft tissue. AJR Am J Roentgenol 1990;155:817–24.
21. Ioannidis JP, Lau J. 18F-FDG PET for the diagnosis and grading of soft-tissue sarcoma: a meta-analysis. J Nucl Med 2003;44:717–24.
22. Tateishi U, Yamaguchi U, Seki K, et al. Bone and soft-tissue sarcoma: preoperative staging with fluorine 18 fluorodeoxyglucose PET/CT and conventional imaging. Radiology 2007;245:839–47.
23. Zagars GK, Ballo MT, Pisters PW, et al. Prognostic factors for patients with localized soft-tissue sarcoma treated with conservation surgery and radiation therapy: an analysis of 1225 patients. Cancer 2003;97:2530–43
24. Rosenberg S, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 1982;196:305–14.
25. Lewis J, Leung D, Woodruff J, et al. Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg 1998;288:355–65.
26. Zagars GK, Ballo MT, Pisters PW, et al. Surgical margins and reresection in the management of patients with soft tissue sarcoma using conservative surgery and radiation therapy. Cancer 2003;97:2544–53.
27. Stojadinovic A, Leung DH, Hoos A. Analysis of the prognostic significance of microscopic margins in 2,084 localized primary adult soft tisusse sarcomas. Ann Surg 2002;235:424–34.
28. O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
29. Pervaiz N, Colterjohn N, Farrokhyar F, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008;113:573–81.
30. Suit HD, Mankin HJ, Wood WC, Proppe KH. Preoperative, intraoperative, and postoperative radiation in the treatment of primary soft tissue sarcoma. Cancer 1985;55:2659–67
31. O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
32. Yang J, Chang A, Baker A, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 1998;16:197–203.
33. Pisters PW, Harrison LB, Leung DH, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 1996;14:859–68.
34. Alektiar KM, Brennan MF, Healey JH, Singer S. Impact of intensity-modulated radiation therapy on local control in primary soft-tissue sarcoma of the extremity. J Clin Oncol 2008;26:3440–5.
35. Gortzak E, Azzarelli A, Buesa J, et al. A randomized phase II study on neo-adjuvant chemotherapy for ‘high-risk’ adult soft-tissue sarcoma. Eur J Cancer 2001;37:1096–1103.
36. Fakhari N, Ebm C, Kostler WJ, et al. Intensified adjuvant IFADIC chemotherapy in combination with radiotherapy versus radiotherapy alone for soft tissue sarcoma: long-term follow-up of a prospective randomized feasibility trial. Wein Klin Wochenschr 2010;122:614–9.
37. Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Lancet 1997;350:1647–54.
38. Gronchi A, Frustaci S, Mercuri M, et al. Short, full-dose adjuvant chemotherapy in high-risk adult soft tissue sarcomas: a randomized clinical trial from the Italian Sarcoma Group and the Spanish Sarcoma Group. J Clin Oncol 2012;30:850–56.
39. Pisters PW, Leung DH, Woodruff J. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996;14:1679–89.
40. Whooley B, Gibbs J, Mooney M. Primary Extremity Sarcoma: What is the Appropriate Follow-up? Annals of Surg Oncol 2000; 7: 9-14.
41. Whooley BP, Mooney MN, Gibbs JF, Graybill WG. Effective follow-up strategies in soft tissue sarcoma. Sem Surg Oncol 1999;17:83–87.
42. Billingsley KG, Burt ME, Jara E, et al. Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival. Ann Surg 1999;229:602–10.
43. Bramwell VH, Anderson D, Charette ML; Sarcoma Disease Site Group. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev 2003;(3):CD003293.
44. Edmonson J, Ryan L, Blum R. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 1993;11:1269–75.
45. Santoro A, Tursz T, Mouridsen H. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 1995;13:1537–45.
46. Tap WD, Jones RL, Van Tine B, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet 2016;388:488–97.
47. Borden EC, Amato DA, Rosenbaum C, et al. Randomized comparison of three adriamycin regimens for metastatic soft tissue sarcomas. J Clin Oncol 1987;5:840–50.
48. Omura GA, Major FJ, Blessing JA, et al. A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 1983;52:626–32.
49. Antman K, Crowley J, Balcerzak SP, et al. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol 1993;11:1276–85.
50. Maki R, Wathen K, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 2007; 25: 2755–63.
51. Hensley ML, Maki R, Venkatraman E, et al. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol 2002;12:2824–31.
52. Garcia-del-Muro X, Lopez-Pousa A, Maurel J, et al. Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: a Spanish Group for Research on Sarcomas study. J Clin Oncol 2011;29:2528–33.
53. Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol 2007;7:595–602.
54. Italiano A, Cioffi A, Penel N, et al. Comparison of doxorubicin and weekly paclitaxel efficacy in metastatic angiosarcomas. Cancer 2012;118:3330–6.
55. Penel N, Italiano A, Ray-Coquard I, et al. Metastatic angiosarcomas: doxorubicin-based regimens, weekly paclitaxel and metastasectomy significantly improve outcome. Ann Oncol 2012;23:517–23.
56. Samuels BL, Chawla S, Patel S, et al. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol 2013;24:1703–9.
57. Schöffski P, Ray-Coquard IL, Cioffi A, et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histolical subtypes. Lancet 2011;11:1045–52.
58. Van der Graaf W, Blay JY, Chawla S, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomized, double-blind, placebo-controlled phase 3 trial. Lancet 2012;379:1879–86.
59. Dileo P, Morgan JA, Zahrieh D, et al. Gemcitabine and vinorelbine combination chemotherapy for patients with advanced soft tissue sarcomas: results of a phase II trial. Cancer 2007;109:1863–9.
Breakthrough cancer gene assay approved, CMS proposes coverage
The Food and Drug Administration approved a new genetic sequencing test that detects mutations across 324 genes in tumor biopsy specimens with an accuracy of 94.6%.
The FoundationOne CDx (F1CDx) test from Foundation Medicine “can identify which patients with any of five tumor types” – non–small-cell lung cancer, melanoma, breast cancer, colorectal cancer, or ovarian cancer – “may benefit from 15 different FDA-approved targeted treatment options,” as well as clinical trial eligibility, “with one test report, avoiding duplicative biopsies,” the agency said in a statement.
On the same day as the approval, the Centers for Medicare & Medicaid Services proposed nationwide coverage for Medicare beneficiaries with recurrent or metastatic disease. CMS is accepting public comments on the proposal for 30 days. The cost of the test is $5,800.
F1CDx went through the FDA and CMS Parallel Review Program, in which the agencies review medical devices concurrently to help reduce the time between approval and Medicare coverage.
F1CDx reads the order of nucleotides on DNA isolated from biopsy specimens to detect a range of genetic anomalies, including base substitutions, insertion and deletion alterations, copy number alterations, and select gene rearrangements, as well as genomic signatures including microsatellite instability and tumor mutational burden. Clinical performance was established by comparing the F1CDx to previously approved tests.
The Food and Drug Administration approved a new genetic sequencing test that detects mutations across 324 genes in tumor biopsy specimens with an accuracy of 94.6%.
The FoundationOne CDx (F1CDx) test from Foundation Medicine “can identify which patients with any of five tumor types” – non–small-cell lung cancer, melanoma, breast cancer, colorectal cancer, or ovarian cancer – “may benefit from 15 different FDA-approved targeted treatment options,” as well as clinical trial eligibility, “with one test report, avoiding duplicative biopsies,” the agency said in a statement.
On the same day as the approval, the Centers for Medicare & Medicaid Services proposed nationwide coverage for Medicare beneficiaries with recurrent or metastatic disease. CMS is accepting public comments on the proposal for 30 days. The cost of the test is $5,800.
F1CDx went through the FDA and CMS Parallel Review Program, in which the agencies review medical devices concurrently to help reduce the time between approval and Medicare coverage.
F1CDx reads the order of nucleotides on DNA isolated from biopsy specimens to detect a range of genetic anomalies, including base substitutions, insertion and deletion alterations, copy number alterations, and select gene rearrangements, as well as genomic signatures including microsatellite instability and tumor mutational burden. Clinical performance was established by comparing the F1CDx to previously approved tests.
The Food and Drug Administration approved a new genetic sequencing test that detects mutations across 324 genes in tumor biopsy specimens with an accuracy of 94.6%.
The FoundationOne CDx (F1CDx) test from Foundation Medicine “can identify which patients with any of five tumor types” – non–small-cell lung cancer, melanoma, breast cancer, colorectal cancer, or ovarian cancer – “may benefit from 15 different FDA-approved targeted treatment options,” as well as clinical trial eligibility, “with one test report, avoiding duplicative biopsies,” the agency said in a statement.
On the same day as the approval, the Centers for Medicare & Medicaid Services proposed nationwide coverage for Medicare beneficiaries with recurrent or metastatic disease. CMS is accepting public comments on the proposal for 30 days. The cost of the test is $5,800.
F1CDx went through the FDA and CMS Parallel Review Program, in which the agencies review medical devices concurrently to help reduce the time between approval and Medicare coverage.
F1CDx reads the order of nucleotides on DNA isolated from biopsy specimens to detect a range of genetic anomalies, including base substitutions, insertion and deletion alterations, copy number alterations, and select gene rearrangements, as well as genomic signatures including microsatellite instability and tumor mutational burden. Clinical performance was established by comparing the F1CDx to previously approved tests.
Indurated Plaque on the Eyebrow
The Diagnosis: Microcystic Adnexal Carcinoma
Microcystic adnexal carcinoma (MAC) is a rare, low-grade adnexal carcinoma consisting of both ductal and pilar differentiation.1 It typically presents in young to middle-aged adults as a flesh-colored or yellow indurated plaque on the upper lip, medial cheek, or chin. Histologically, MACs exhibit a biphasic pattern consisting of epithelial islands of cords and lumina creating tadpolelike ducts intermixed with basaloid nests (quiz image). Keratin horn cysts are common superficially. A dense red sclerotic stroma is seen interspersed between the ducts and epithelial islands creating a "paisley tie" appearance. The lesion displays an infiltrative pattern and can be deeply invasive, extending down to the fat and muscle (quiz image, inset). Perineural invasion is common. Atypia, when present, is minimal or mild and mitoses are rare. Although this tumor's histologic pattern appears aggressive in nature, it lacks immunohistochemical staining such as p53, Ki-67, bcl-2, and c-erbB-2 that correlate with malignant behavior.2 A common diagnostic pitfall is examination of a superficial biopsy in which an MAC may be mistakenly identified as another entity.
Syringomas are benign adnexal neoplasms with ductal differentiation.3 They are more common in women, especially those of Asian descent, and in patients with Down syndrome. They typically present as multiple small, firm, flesh-colored papules in the periorbital area or upper trunk. Histologically, syringomas also display comma-shaped tubules and ducts with a tadpolelike appearance and a dense red stroma creating a paisley tie-like pattern. Ductal cells have an abundant pink cytoplasm. Syringomas are well-circumscribed and more superficial than MACs without an infiltrative pattern. They lack mitotic activity or perineural invasion (Figure 1).

Desmoplastic trichoepithelioma (DTE) is a benign follicular neoplasm.4 It presents in adulthood with a female predominance. Clinically, it appears as a solitary flesh-colored to yellow annular plaque with raised borders and a depressed central area, often on the medial cheek. Histologically, DTEs are well-circumscribed with narrow branching cords lined with polygonal cells. A dense red stroma in combination with the epithelioid aggregates also creates the paisley tie-like pattern in this lesion. Retraction between collagen bundles within the stroma can be seen, helping distinguish this lesion from a morpheaform basal cell carcinoma (BCC), which has retraction between the epithelium and stroma. Immunohistochemistry also can be a useful tool to help differentiate DTEs from morpheaform BCCs in that sparse cytokeratin 20-positive Merkel cells can be seen within the basaloid islands of DTE but not BCC.5 Also seen with DTEs are numerous keratin horn cysts that commonly are filled with dystrophic calcifications. Cellular atypia and mitoses are not seen (Figure 2). Compared to MACs, DTEs lack abundant ductal structures and also contain papillary mesenchymal bodies and a more fibroblast-rich stroma.
Morpheaform BCC is an aggressive subtype of BCC. It presents as a scarlike plaque that gradually expands. Thin infiltrating strands of basaloid cells are seen haphazardly throughout a pink sclerotic stroma. Tadpolelike basaloid islands and rarely horn cysts can be seen scattered superficially, creating the paisley tie-like pattern. This lesion is more infiltrating than a syringoma or a DTE, and perineural invasion is common. Retraction is uncommon, but when present, it is seen between the epithelial cords and adjacent stroma (Figure 3).
Trichoadenoma is another benign neoplasm of follicular differentiation.6 It typically presents as a dome-shaped papule or plaque on the head or neck. Histologically it displays numerous dilated cystic spaces that reflect its origin from isthmic and infundibular differentiation. There is no attachment to the overlying epidermis. It can be distinguished from MAC, DTE, and syringoma due to a lack of basaloid aggregates and only a small number of non-cyst-forming epithelial cells (Figure 4).
- Nickoloff BJ, Fleischmann HE, Carmel J. Microcystic adnexal carcinoma: immunohistologic observations suggesting dual (pilar and eccrine) differentiation. Arch Dermatol. 1986;122:290-294.
- Smith KJ, Williams J, Corbett D, et al. Microcystic adnexal carcinoma: an immunohistochemical study including markers of proliferation and apoptosis. Am J Surg Pathol. 2001;25:464-471.
- Hashimoto K, Lever WF. Histogenesis of skin appendage tumors. Arch Dermatol. 1969;100:356-369.
- Brownstein MH, Shapiro L. Desmoplastic trichoepithelioma. Cancer. 1977;40:2979-2986.
- Hartschuh W, Schulz T. Merkel cells are integral constituents of desmoplastic trichoepithelioma: an immunohistochemical and electron microscopy study. J Cutan Pathol. 1995;22:413-421.
- Rahbari H, Mehregan A, Pinkus A. Trichoadenoma of Nikolowski. J Cutan Pathol. 1977;4:90-98.
The Diagnosis: Microcystic Adnexal Carcinoma
Microcystic adnexal carcinoma (MAC) is a rare, low-grade adnexal carcinoma consisting of both ductal and pilar differentiation.1 It typically presents in young to middle-aged adults as a flesh-colored or yellow indurated plaque on the upper lip, medial cheek, or chin. Histologically, MACs exhibit a biphasic pattern consisting of epithelial islands of cords and lumina creating tadpolelike ducts intermixed with basaloid nests (quiz image). Keratin horn cysts are common superficially. A dense red sclerotic stroma is seen interspersed between the ducts and epithelial islands creating a "paisley tie" appearance. The lesion displays an infiltrative pattern and can be deeply invasive, extending down to the fat and muscle (quiz image, inset). Perineural invasion is common. Atypia, when present, is minimal or mild and mitoses are rare. Although this tumor's histologic pattern appears aggressive in nature, it lacks immunohistochemical staining such as p53, Ki-67, bcl-2, and c-erbB-2 that correlate with malignant behavior.2 A common diagnostic pitfall is examination of a superficial biopsy in which an MAC may be mistakenly identified as another entity.
Syringomas are benign adnexal neoplasms with ductal differentiation.3 They are more common in women, especially those of Asian descent, and in patients with Down syndrome. They typically present as multiple small, firm, flesh-colored papules in the periorbital area or upper trunk. Histologically, syringomas also display comma-shaped tubules and ducts with a tadpolelike appearance and a dense red stroma creating a paisley tie-like pattern. Ductal cells have an abundant pink cytoplasm. Syringomas are well-circumscribed and more superficial than MACs without an infiltrative pattern. They lack mitotic activity or perineural invasion (Figure 1).
Desmoplastic trichoepithelioma (DTE) is a benign follicular neoplasm.4 It presents in adulthood with a female predominance. Clinically, it appears as a solitary flesh-colored to yellow annular plaque with raised borders and a depressed central area, often on the medial cheek. Histologically, DTEs are well-circumscribed with narrow branching cords lined with polygonal cells. A dense red stroma in combination with the epithelioid aggregates also creates the paisley tie-like pattern in this lesion. Retraction between collagen bundles within the stroma can be seen, helping distinguish this lesion from a morpheaform basal cell carcinoma (BCC), which has retraction between the epithelium and stroma. Immunohistochemistry also can be a useful tool to help differentiate DTEs from morpheaform BCCs in that sparse cytokeratin 20-positive Merkel cells can be seen within the basaloid islands of DTE but not BCC.5 Also seen with DTEs are numerous keratin horn cysts that commonly are filled with dystrophic calcifications. Cellular atypia and mitoses are not seen (Figure 2). Compared to MACs, DTEs lack abundant ductal structures and also contain papillary mesenchymal bodies and a more fibroblast-rich stroma.
Morpheaform BCC is an aggressive subtype of BCC. It presents as a scarlike plaque that gradually expands. Thin infiltrating strands of basaloid cells are seen haphazardly throughout a pink sclerotic stroma. Tadpolelike basaloid islands and rarely horn cysts can be seen scattered superficially, creating the paisley tie-like pattern. This lesion is more infiltrating than a syringoma or a DTE, and perineural invasion is common. Retraction is uncommon, but when present, it is seen between the epithelial cords and adjacent stroma (Figure 3).
Trichoadenoma is another benign neoplasm of follicular differentiation.6 It typically presents as a dome-shaped papule or plaque on the head or neck. Histologically it displays numerous dilated cystic spaces that reflect its origin from isthmic and infundibular differentiation. There is no attachment to the overlying epidermis. It can be distinguished from MAC, DTE, and syringoma due to a lack of basaloid aggregates and only a small number of non-cyst-forming epithelial cells (Figure 4).
The Diagnosis: Microcystic Adnexal Carcinoma
Microcystic adnexal carcinoma (MAC) is a rare, low-grade adnexal carcinoma consisting of both ductal and pilar differentiation.1 It typically presents in young to middle-aged adults as a flesh-colored or yellow indurated plaque on the upper lip, medial cheek, or chin. Histologically, MACs exhibit a biphasic pattern consisting of epithelial islands of cords and lumina creating tadpolelike ducts intermixed with basaloid nests (quiz image). Keratin horn cysts are common superficially. A dense red sclerotic stroma is seen interspersed between the ducts and epithelial islands creating a "paisley tie" appearance. The lesion displays an infiltrative pattern and can be deeply invasive, extending down to the fat and muscle (quiz image, inset). Perineural invasion is common. Atypia, when present, is minimal or mild and mitoses are rare. Although this tumor's histologic pattern appears aggressive in nature, it lacks immunohistochemical staining such as p53, Ki-67, bcl-2, and c-erbB-2 that correlate with malignant behavior.2 A common diagnostic pitfall is examination of a superficial biopsy in which an MAC may be mistakenly identified as another entity.
Syringomas are benign adnexal neoplasms with ductal differentiation.3 They are more common in women, especially those of Asian descent, and in patients with Down syndrome. They typically present as multiple small, firm, flesh-colored papules in the periorbital area or upper trunk. Histologically, syringomas also display comma-shaped tubules and ducts with a tadpolelike appearance and a dense red stroma creating a paisley tie-like pattern. Ductal cells have an abundant pink cytoplasm. Syringomas are well-circumscribed and more superficial than MACs without an infiltrative pattern. They lack mitotic activity or perineural invasion (Figure 1).
Desmoplastic trichoepithelioma (DTE) is a benign follicular neoplasm.4 It presents in adulthood with a female predominance. Clinically, it appears as a solitary flesh-colored to yellow annular plaque with raised borders and a depressed central area, often on the medial cheek. Histologically, DTEs are well-circumscribed with narrow branching cords lined with polygonal cells. A dense red stroma in combination with the epithelioid aggregates also creates the paisley tie-like pattern in this lesion. Retraction between collagen bundles within the stroma can be seen, helping distinguish this lesion from a morpheaform basal cell carcinoma (BCC), which has retraction between the epithelium and stroma. Immunohistochemistry also can be a useful tool to help differentiate DTEs from morpheaform BCCs in that sparse cytokeratin 20-positive Merkel cells can be seen within the basaloid islands of DTE but not BCC.5 Also seen with DTEs are numerous keratin horn cysts that commonly are filled with dystrophic calcifications. Cellular atypia and mitoses are not seen (Figure 2). Compared to MACs, DTEs lack abundant ductal structures and also contain papillary mesenchymal bodies and a more fibroblast-rich stroma.
Morpheaform BCC is an aggressive subtype of BCC. It presents as a scarlike plaque that gradually expands. Thin infiltrating strands of basaloid cells are seen haphazardly throughout a pink sclerotic stroma. Tadpolelike basaloid islands and rarely horn cysts can be seen scattered superficially, creating the paisley tie-like pattern. This lesion is more infiltrating than a syringoma or a DTE, and perineural invasion is common. Retraction is uncommon, but when present, it is seen between the epithelial cords and adjacent stroma (Figure 3).
Trichoadenoma is another benign neoplasm of follicular differentiation.6 It typically presents as a dome-shaped papule or plaque on the head or neck. Histologically it displays numerous dilated cystic spaces that reflect its origin from isthmic and infundibular differentiation. There is no attachment to the overlying epidermis. It can be distinguished from MAC, DTE, and syringoma due to a lack of basaloid aggregates and only a small number of non-cyst-forming epithelial cells (Figure 4).
- Nickoloff BJ, Fleischmann HE, Carmel J. Microcystic adnexal carcinoma: immunohistologic observations suggesting dual (pilar and eccrine) differentiation. Arch Dermatol. 1986;122:290-294.
- Smith KJ, Williams J, Corbett D, et al. Microcystic adnexal carcinoma: an immunohistochemical study including markers of proliferation and apoptosis. Am J Surg Pathol. 2001;25:464-471.
- Hashimoto K, Lever WF. Histogenesis of skin appendage tumors. Arch Dermatol. 1969;100:356-369.
- Brownstein MH, Shapiro L. Desmoplastic trichoepithelioma. Cancer. 1977;40:2979-2986.
- Hartschuh W, Schulz T. Merkel cells are integral constituents of desmoplastic trichoepithelioma: an immunohistochemical and electron microscopy study. J Cutan Pathol. 1995;22:413-421.
- Rahbari H, Mehregan A, Pinkus A. Trichoadenoma of Nikolowski. J Cutan Pathol. 1977;4:90-98.
- Nickoloff BJ, Fleischmann HE, Carmel J. Microcystic adnexal carcinoma: immunohistologic observations suggesting dual (pilar and eccrine) differentiation. Arch Dermatol. 1986;122:290-294.
- Smith KJ, Williams J, Corbett D, et al. Microcystic adnexal carcinoma: an immunohistochemical study including markers of proliferation and apoptosis. Am J Surg Pathol. 2001;25:464-471.
- Hashimoto K, Lever WF. Histogenesis of skin appendage tumors. Arch Dermatol. 1969;100:356-369.
- Brownstein MH, Shapiro L. Desmoplastic trichoepithelioma. Cancer. 1977;40:2979-2986.
- Hartschuh W, Schulz T. Merkel cells are integral constituents of desmoplastic trichoepithelioma: an immunohistochemical and electron microscopy study. J Cutan Pathol. 1995;22:413-421.
- Rahbari H, Mehregan A, Pinkus A. Trichoadenoma of Nikolowski. J Cutan Pathol. 1977;4:90-98.
A 52-year-old woman presented with an indurated plaque on the right lateral eyebrow that had been slowly enlarging over the last 4 months.
DDSEP® 8 Quick Quiz - December 2017 Question 2
Correct Answer: E
Rationale
On serial imaging, two worrisome features have developed in the pancreas cyst, i.e., an enhancing mural nodule and dilation of the main pancreatic duct. These features are high-risk stigmata, and therefore surgical resection is recommended. EUS FNA can be considered but is unlikely to change management if cytology is negative. Radiologic surveillance is not appropriate unless the patient refuses surgery.
Reference
1. Tanaka M., Fernández-del Castillo C., Adsay V., et al. International consensus guidelines 2012 for the management of IPMN and MCN of the pancreas. Pancreatology. 2012;12(3):183-97.
Correct Answer: E
Rationale
On serial imaging, two worrisome features have developed in the pancreas cyst, i.e., an enhancing mural nodule and dilation of the main pancreatic duct. These features are high-risk stigmata, and therefore surgical resection is recommended. EUS FNA can be considered but is unlikely to change management if cytology is negative. Radiologic surveillance is not appropriate unless the patient refuses surgery.
Reference
1. Tanaka M., Fernández-del Castillo C., Adsay V., et al. International consensus guidelines 2012 for the management of IPMN and MCN of the pancreas. Pancreatology. 2012;12(3):183-97.
Correct Answer: E
Rationale
On serial imaging, two worrisome features have developed in the pancreas cyst, i.e., an enhancing mural nodule and dilation of the main pancreatic duct. These features are high-risk stigmata, and therefore surgical resection is recommended. EUS FNA can be considered but is unlikely to change management if cytology is negative. Radiologic surveillance is not appropriate unless the patient refuses surgery.
Reference
1. Tanaka M., Fernández-del Castillo C., Adsay V., et al. International consensus guidelines 2012 for the management of IPMN and MCN of the pancreas. Pancreatology. 2012;12(3):183-97.
A 55-year-old man was diagnosed with a 3.1-cm cyst in the tail of the pancreas 2 years ago. He had an endoscopic ultrasound–guided fine-needle aspiration at that time and approximately 2 cc of mucinous fluid were aspirated; cyst fluid CEA (carcinoembryonic antigen) was 790 ng/mL and cytology showed a paucicellular specimen with abundant extracellular mucin. The patient was asymptomatic and opted for radiologic surveillance with MRI. On his most recent MRI, the cyst size is currently 3.4 cm. In addition, the MRI notes the presence of an enhancing nodule in the wall of the cyst measuring 5 mm and the pancreatic duct in the tail is mildly dilated to 5 mm. He continues to be asymptomatic and in good health.
Early inguinal hernia linked to schizophrenia
PARIS – One of the most unexpected and intriguing new developments in the field of schizophrenia has to be the discovery that the risk of the disease is significantly increased in men who were diagnosed with inguinal hernia before they were 13 years old.
“I think this is interesting because inguinal hernia in boys has to do with fibroblasts producing abnormal collagen structure,” according to Kristina Melkersson, MD, PhD, who presented her study findings at the annual congress of the European College of Neuropsychopharmacology.
She first detected a signal for a potential relationship in an earlier, small interview study in which she noticed that men with schizophrenia were more likely to have a history of inguinal hernia surgery than did men in the general population. This prompted her to try to confirm this preliminary observation in a large Swedish registry-based cohort study.
Among the nearly 1.3 million Swedes born during 1987-1999, there were 20,705 who were diagnosed with inguinal hernia before age 13 years. During a median 9.9 years of follow-up starting at age 13 years, 1,294 of these individuals were diagnosed with schizophrenia or schizoaffective disorder at a mean age of 21.4 years.
Among men, a history of inguinal hernia diagnosed before age 13 years was associated with a 56% increase in subsequent risk of schizophrenia or schizoaffective disorder, compared with men without such a history.
Women with a history of having inguinal hernia before age 13 years were at 16% increased risk; however, this modest increase in risk was not statistically significant, possibly because of small numbers. Inguinal hernia is 25 times more common in men than women.
Dr. Melkersson reported having no financial conflicts of interest regarding her study, which was supported by a grant from the Swedish Society of Medicine.
PARIS – One of the most unexpected and intriguing new developments in the field of schizophrenia has to be the discovery that the risk of the disease is significantly increased in men who were diagnosed with inguinal hernia before they were 13 years old.
“I think this is interesting because inguinal hernia in boys has to do with fibroblasts producing abnormal collagen structure,” according to Kristina Melkersson, MD, PhD, who presented her study findings at the annual congress of the European College of Neuropsychopharmacology.
She first detected a signal for a potential relationship in an earlier, small interview study in which she noticed that men with schizophrenia were more likely to have a history of inguinal hernia surgery than did men in the general population. This prompted her to try to confirm this preliminary observation in a large Swedish registry-based cohort study.
Among the nearly 1.3 million Swedes born during 1987-1999, there were 20,705 who were diagnosed with inguinal hernia before age 13 years. During a median 9.9 years of follow-up starting at age 13 years, 1,294 of these individuals were diagnosed with schizophrenia or schizoaffective disorder at a mean age of 21.4 years.
Among men, a history of inguinal hernia diagnosed before age 13 years was associated with a 56% increase in subsequent risk of schizophrenia or schizoaffective disorder, compared with men without such a history.
Women with a history of having inguinal hernia before age 13 years were at 16% increased risk; however, this modest increase in risk was not statistically significant, possibly because of small numbers. Inguinal hernia is 25 times more common in men than women.
Dr. Melkersson reported having no financial conflicts of interest regarding her study, which was supported by a grant from the Swedish Society of Medicine.
PARIS – One of the most unexpected and intriguing new developments in the field of schizophrenia has to be the discovery that the risk of the disease is significantly increased in men who were diagnosed with inguinal hernia before they were 13 years old.
“I think this is interesting because inguinal hernia in boys has to do with fibroblasts producing abnormal collagen structure,” according to Kristina Melkersson, MD, PhD, who presented her study findings at the annual congress of the European College of Neuropsychopharmacology.
She first detected a signal for a potential relationship in an earlier, small interview study in which she noticed that men with schizophrenia were more likely to have a history of inguinal hernia surgery than did men in the general population. This prompted her to try to confirm this preliminary observation in a large Swedish registry-based cohort study.
Among the nearly 1.3 million Swedes born during 1987-1999, there were 20,705 who were diagnosed with inguinal hernia before age 13 years. During a median 9.9 years of follow-up starting at age 13 years, 1,294 of these individuals were diagnosed with schizophrenia or schizoaffective disorder at a mean age of 21.4 years.
Among men, a history of inguinal hernia diagnosed before age 13 years was associated with a 56% increase in subsequent risk of schizophrenia or schizoaffective disorder, compared with men without such a history.
Women with a history of having inguinal hernia before age 13 years were at 16% increased risk; however, this modest increase in risk was not statistically significant, possibly because of small numbers. Inguinal hernia is 25 times more common in men than women.
Dr. Melkersson reported having no financial conflicts of interest regarding her study, which was supported by a grant from the Swedish Society of Medicine.
AT THE ECNP CONGRESS
Key clinical point:
Major finding: Swedish boys diagnosed with inguinal hernia before age 13 years were 56% more likely to be diagnosed with schizophrenia or schizoaffective disorder later in life.
Data source: This retrospective cohort study included nearly 1.3 million Swedes, 20,705 of whom were diagnosed with an inguinal hernia before age 13 years.
Disclosures: The study was supported by a grant from the Swedish Society of Medicine. The presenter reported having no financial conflicts.
DDSEP® 8 Quick quiz - December 2017 Question 1
Correct Answer: C
Rationale
Mixed connective tissue disease can be associated with atrophy of the smooth muscle of the gut, like scleroderma. In the esophagus, this can manifest as a hypotensive lower esophageal sphincter and impaired esophageal smooth muscle peristalsis; in extreme cases, there is absent contractility in the esophagus. This contributes to impaired esophageal clearance of refluxed material, leading to prolonged acid residence times in the esophagus and severe reflux esophagitis. Many patients with mixed connective tissue disease have overlap Sjogren’s syndrome, reducing salivary neutralization of esophageal mucosal acidification and further contributing to esophagitis. While esophageal body motor function can be suboptimal in diabetes mellitus and Barrett’s esophagus, the mechanism of hypomotility is not smooth muscle atrophy and fibrosis. Polymyositis can affect skeletal muscle of the proximal esophagus, but not the smooth muscle. Lichen planus affects mucosa but not muscle.
Reference
1. Savarino E., Mei F., Parodi A., et al. Gastrointestinal motility disorder assessment in systemic sclerosis. Rheumatology (Oxford). 2013 Jun;52(6):1095-100.
2. Langdon P.C., Mulcahy K., Shepherd K.L., et al. Pharyngeal dysphagia in inflammatory muscle diseases resulting from impaired suprahyoid musculature. Dysphagia. 2012 Sep;27(3):408-17.
Correct Answer: C
Rationale
Mixed connective tissue disease can be associated with atrophy of the smooth muscle of the gut, like scleroderma. In the esophagus, this can manifest as a hypotensive lower esophageal sphincter and impaired esophageal smooth muscle peristalsis; in extreme cases, there is absent contractility in the esophagus. This contributes to impaired esophageal clearance of refluxed material, leading to prolonged acid residence times in the esophagus and severe reflux esophagitis. Many patients with mixed connective tissue disease have overlap Sjogren’s syndrome, reducing salivary neutralization of esophageal mucosal acidification and further contributing to esophagitis. While esophageal body motor function can be suboptimal in diabetes mellitus and Barrett’s esophagus, the mechanism of hypomotility is not smooth muscle atrophy and fibrosis. Polymyositis can affect skeletal muscle of the proximal esophagus, but not the smooth muscle. Lichen planus affects mucosa but not muscle.
Reference
1. Savarino E., Mei F., Parodi A., et al. Gastrointestinal motility disorder assessment in systemic sclerosis. Rheumatology (Oxford). 2013 Jun;52(6):1095-100.
2. Langdon P.C., Mulcahy K., Shepherd K.L., et al. Pharyngeal dysphagia in inflammatory muscle diseases resulting from impaired suprahyoid musculature. Dysphagia. 2012 Sep;27(3):408-17.
Correct Answer: C
Rationale
Mixed connective tissue disease can be associated with atrophy of the smooth muscle of the gut, like scleroderma. In the esophagus, this can manifest as a hypotensive lower esophageal sphincter and impaired esophageal smooth muscle peristalsis; in extreme cases, there is absent contractility in the esophagus. This contributes to impaired esophageal clearance of refluxed material, leading to prolonged acid residence times in the esophagus and severe reflux esophagitis. Many patients with mixed connective tissue disease have overlap Sjogren’s syndrome, reducing salivary neutralization of esophageal mucosal acidification and further contributing to esophagitis. While esophageal body motor function can be suboptimal in diabetes mellitus and Barrett’s esophagus, the mechanism of hypomotility is not smooth muscle atrophy and fibrosis. Polymyositis can affect skeletal muscle of the proximal esophagus, but not the smooth muscle. Lichen planus affects mucosa but not muscle.
Reference
1. Savarino E., Mei F., Parodi A., et al. Gastrointestinal motility disorder assessment in systemic sclerosis. Rheumatology (Oxford). 2013 Jun;52(6):1095-100.
2. Langdon P.C., Mulcahy K., Shepherd K.L., et al. Pharyngeal dysphagia in inflammatory muscle diseases resulting from impaired suprahyoid musculature. Dysphagia. 2012 Sep;27(3):408-17.
Which of the following conditions is associated with smooth muscle atrophy impairing esophageal clearance, contributing to prolonged esophageal acid contact and reflux esophagitis?
Self-harm on rise in U.S. among girls aged 10-14
The rate of self-inflicted injuries has increased significantly among young girls since 2009, according to a study of emergency department visits for self-inflicted injuries from 2001 to 2015.
In a research letter, Melissa C. Mercado, PhD, and her associates reached that conclusion based on data from 43,138 emergency department visits for self-inflicted injury among young people aged 10-24 years, which were captured by the National Electronic Injury Surveillance System–All Injury Program (JAMA. 2017;318[19]:1931-3. doi: 10.1001/jama.2017.13317).
From 2001 to 2008, the overall weighted, age-adjusted rate of self-inflicted injury showed no statistically significant trend upward or downward, reported Dr. Mercado of the National Center for Injury Prevention and Control, Atlanta, and her associates. From 2009 to 2015, however, the rate increased by a significant 5.7% per year, reaching 303.7 per 100,000 population in 2015, compared with 201.6 in 2001.
This increase was even more pronounced among girls, rising by 8.4% per year from 2008 to 2015 in all females but by 18.8% per year in those aged 10-14 years. In adolescent females aged 15-19, the rate of self-inflicted injury rose 7.2% per year from 2008 to 2015. In young women aged 20-24 years, the rate rose 2% per year from 2001 to 2015.
Meanwhile, the rates of self-inflicted injury for males were stable across all time periods and age groups.
“Self-inflicted injury is one of the strongest risk factors for suicide – the second-leading cause of death among those aged 10 to 24 years during 2015,” Dr. Mercado and her coauthors wrote.
The most common method of self-inflicted injury for females was poisoning. As with the overall rates of injury in females, the rates of this method of harm were stable until 2007, then increased by 5.3% until 2015. Self-inflicted injuries among females using a sharp object increased by 7.1% each year from 2001 to 2015, but the rates of blunt-object injuries were stable from 2006 to 2015.
The authors wrote that the finding of an increase in self-harm among females was consistent with youth suicide data, which also show an increase after 2006, particularly among girls and female adolescents aged 10-14 years.
Dr. Mercado and her associates called for the implementation of evidence-based, comprehensive suicide and self-harm prevention strategies. “These strategies include strengthening access to and delivery of care for suicidal youth within health systems and creating protective environments, promoting youth connectedness, teaching coping and problem-solving skills, and identifying and supporting at-risk youth within communities.”
The study was conducted under the auspices of the National Center for Injury Prevention and Control, which is part of the Centers for Disease Control and Prevention. The findings, however, do not necessarily represent the views of the CDC. No conflicts of interest were declared.
The rate of self-inflicted injuries has increased significantly among young girls since 2009, according to a study of emergency department visits for self-inflicted injuries from 2001 to 2015.
In a research letter, Melissa C. Mercado, PhD, and her associates reached that conclusion based on data from 43,138 emergency department visits for self-inflicted injury among young people aged 10-24 years, which were captured by the National Electronic Injury Surveillance System–All Injury Program (JAMA. 2017;318[19]:1931-3. doi: 10.1001/jama.2017.13317).
From 2001 to 2008, the overall weighted, age-adjusted rate of self-inflicted injury showed no statistically significant trend upward or downward, reported Dr. Mercado of the National Center for Injury Prevention and Control, Atlanta, and her associates. From 2009 to 2015, however, the rate increased by a significant 5.7% per year, reaching 303.7 per 100,000 population in 2015, compared with 201.6 in 2001.
This increase was even more pronounced among girls, rising by 8.4% per year from 2008 to 2015 in all females but by 18.8% per year in those aged 10-14 years. In adolescent females aged 15-19, the rate of self-inflicted injury rose 7.2% per year from 2008 to 2015. In young women aged 20-24 years, the rate rose 2% per year from 2001 to 2015.
Meanwhile, the rates of self-inflicted injury for males were stable across all time periods and age groups.
“Self-inflicted injury is one of the strongest risk factors for suicide – the second-leading cause of death among those aged 10 to 24 years during 2015,” Dr. Mercado and her coauthors wrote.
The most common method of self-inflicted injury for females was poisoning. As with the overall rates of injury in females, the rates of this method of harm were stable until 2007, then increased by 5.3% until 2015. Self-inflicted injuries among females using a sharp object increased by 7.1% each year from 2001 to 2015, but the rates of blunt-object injuries were stable from 2006 to 2015.
The authors wrote that the finding of an increase in self-harm among females was consistent with youth suicide data, which also show an increase after 2006, particularly among girls and female adolescents aged 10-14 years.
Dr. Mercado and her associates called for the implementation of evidence-based, comprehensive suicide and self-harm prevention strategies. “These strategies include strengthening access to and delivery of care for suicidal youth within health systems and creating protective environments, promoting youth connectedness, teaching coping and problem-solving skills, and identifying and supporting at-risk youth within communities.”
The study was conducted under the auspices of the National Center for Injury Prevention and Control, which is part of the Centers for Disease Control and Prevention. The findings, however, do not necessarily represent the views of the CDC. No conflicts of interest were declared.
The rate of self-inflicted injuries has increased significantly among young girls since 2009, according to a study of emergency department visits for self-inflicted injuries from 2001 to 2015.
In a research letter, Melissa C. Mercado, PhD, and her associates reached that conclusion based on data from 43,138 emergency department visits for self-inflicted injury among young people aged 10-24 years, which were captured by the National Electronic Injury Surveillance System–All Injury Program (JAMA. 2017;318[19]:1931-3. doi: 10.1001/jama.2017.13317).
From 2001 to 2008, the overall weighted, age-adjusted rate of self-inflicted injury showed no statistically significant trend upward or downward, reported Dr. Mercado of the National Center for Injury Prevention and Control, Atlanta, and her associates. From 2009 to 2015, however, the rate increased by a significant 5.7% per year, reaching 303.7 per 100,000 population in 2015, compared with 201.6 in 2001.
This increase was even more pronounced among girls, rising by 8.4% per year from 2008 to 2015 in all females but by 18.8% per year in those aged 10-14 years. In adolescent females aged 15-19, the rate of self-inflicted injury rose 7.2% per year from 2008 to 2015. In young women aged 20-24 years, the rate rose 2% per year from 2001 to 2015.
Meanwhile, the rates of self-inflicted injury for males were stable across all time periods and age groups.
“Self-inflicted injury is one of the strongest risk factors for suicide – the second-leading cause of death among those aged 10 to 24 years during 2015,” Dr. Mercado and her coauthors wrote.
The most common method of self-inflicted injury for females was poisoning. As with the overall rates of injury in females, the rates of this method of harm were stable until 2007, then increased by 5.3% until 2015. Self-inflicted injuries among females using a sharp object increased by 7.1% each year from 2001 to 2015, but the rates of blunt-object injuries were stable from 2006 to 2015.
The authors wrote that the finding of an increase in self-harm among females was consistent with youth suicide data, which also show an increase after 2006, particularly among girls and female adolescents aged 10-14 years.
Dr. Mercado and her associates called for the implementation of evidence-based, comprehensive suicide and self-harm prevention strategies. “These strategies include strengthening access to and delivery of care for suicidal youth within health systems and creating protective environments, promoting youth connectedness, teaching coping and problem-solving skills, and identifying and supporting at-risk youth within communities.”
The study was conducted under the auspices of the National Center for Injury Prevention and Control, which is part of the Centers for Disease Control and Prevention. The findings, however, do not necessarily represent the views of the CDC. No conflicts of interest were declared.
FROM JAMA
Key clinical point: Rates of self-inflicted injury rose significantly in young women between 2009 and 2015, particularly in those aged 10-14 years.
Major finding: The rate of emergency department visits for self-inflicted injury rose 18.8% per year from 2009 to 2015 in females aged 10-14 years.
Data source: Analysis of data from 43,138 emergency department visits of young people aged 10-24 years for self-inflicted injury.
Disclosures: The study was conducted under the auspices of the Centers for Disease Control and Prevention, but the findings do not necessarily represent the views of the CDC. No conflicts of interest were declared.
Delving into the details
Editor’s note: The Society of Hospital Medicine’s (SHM’s) Physician in Training Committee launched a scholarship program in 2015 for medical students to help transform health care and revolutionize patient care. The program has been expanded for the 2017-2018 year, offering two options for students to receive funding and engage in scholarly work during their first, second, and third years of medical school. As a part of the longitudinal (18-month) program, recipients are required to write about their experience on a monthly basis.
For my research project, we are looking to develop a tool that would use data from within 24 hours of a patient’s admission to the hospital to predict whether they will require post-acute care placement after discharge. While I have often been summarizing my project with this broad one-liner, in the last two weeks I have been delving more into the details of what exactly we mean by “data from within 24 hours of a patient’s admission.”
We are going through each of the variables systematically to take into account prior literature on how they were treated in other studies, as well as the practical limitations imposed by the data-gathering within our own system to choose how these values will be selected for each admission. My mentor Dr. Eduard Vasilevskis is helping me with making these decisions, based on the prototype model that was the inspiration for this project. Once we have identified all of the details of each variable we want to track, Dr. Jesse Ehrenfeld will be facilitating our use of the database.
Certainly this project has helped illuminate not only research-specific hurdles, but also underscores the fundamental difficulty of clinical decision-making in the first 24 hours of a patient’s admission. With data changing rapidly and sometimes incomplete data, clinicians need to quickly make care decisions that can impact a lot more than the patient’s post-discharge destination.
We anticipate that once we’ve made these choices, there will be further choices to make about how to treat these variables in the analysis. We hope to have the assistance of an experienced statistician to help guide us in making those decisions.
Monisha Bhatia, a native of Nashville, Tenn., is a fourth-year medical student at Vanderbilt University in Nashville. She is hoping to pursue either a residency in internal medicine or a combined internal medicine/emergency medicine program. Prior to medical school, she completed a JD/MPH program at Boston University, and she hopes to use her legal training in working with regulatory authorities to improve access to health care for all Americans.
Editor’s note: The Society of Hospital Medicine’s (SHM’s) Physician in Training Committee launched a scholarship program in 2015 for medical students to help transform health care and revolutionize patient care. The program has been expanded for the 2017-2018 year, offering two options for students to receive funding and engage in scholarly work during their first, second, and third years of medical school. As a part of the longitudinal (18-month) program, recipients are required to write about their experience on a monthly basis.
For my research project, we are looking to develop a tool that would use data from within 24 hours of a patient’s admission to the hospital to predict whether they will require post-acute care placement after discharge. While I have often been summarizing my project with this broad one-liner, in the last two weeks I have been delving more into the details of what exactly we mean by “data from within 24 hours of a patient’s admission.”
We are going through each of the variables systematically to take into account prior literature on how they were treated in other studies, as well as the practical limitations imposed by the data-gathering within our own system to choose how these values will be selected for each admission. My mentor Dr. Eduard Vasilevskis is helping me with making these decisions, based on the prototype model that was the inspiration for this project. Once we have identified all of the details of each variable we want to track, Dr. Jesse Ehrenfeld will be facilitating our use of the database.
Certainly this project has helped illuminate not only research-specific hurdles, but also underscores the fundamental difficulty of clinical decision-making in the first 24 hours of a patient’s admission. With data changing rapidly and sometimes incomplete data, clinicians need to quickly make care decisions that can impact a lot more than the patient’s post-discharge destination.
We anticipate that once we’ve made these choices, there will be further choices to make about how to treat these variables in the analysis. We hope to have the assistance of an experienced statistician to help guide us in making those decisions.
Monisha Bhatia, a native of Nashville, Tenn., is a fourth-year medical student at Vanderbilt University in Nashville. She is hoping to pursue either a residency in internal medicine or a combined internal medicine/emergency medicine program. Prior to medical school, she completed a JD/MPH program at Boston University, and she hopes to use her legal training in working with regulatory authorities to improve access to health care for all Americans.
Editor’s note: The Society of Hospital Medicine’s (SHM’s) Physician in Training Committee launched a scholarship program in 2015 for medical students to help transform health care and revolutionize patient care. The program has been expanded for the 2017-2018 year, offering two options for students to receive funding and engage in scholarly work during their first, second, and third years of medical school. As a part of the longitudinal (18-month) program, recipients are required to write about their experience on a monthly basis.
For my research project, we are looking to develop a tool that would use data from within 24 hours of a patient’s admission to the hospital to predict whether they will require post-acute care placement after discharge. While I have often been summarizing my project with this broad one-liner, in the last two weeks I have been delving more into the details of what exactly we mean by “data from within 24 hours of a patient’s admission.”
We are going through each of the variables systematically to take into account prior literature on how they were treated in other studies, as well as the practical limitations imposed by the data-gathering within our own system to choose how these values will be selected for each admission. My mentor Dr. Eduard Vasilevskis is helping me with making these decisions, based on the prototype model that was the inspiration for this project. Once we have identified all of the details of each variable we want to track, Dr. Jesse Ehrenfeld will be facilitating our use of the database.
Certainly this project has helped illuminate not only research-specific hurdles, but also underscores the fundamental difficulty of clinical decision-making in the first 24 hours of a patient’s admission. With data changing rapidly and sometimes incomplete data, clinicians need to quickly make care decisions that can impact a lot more than the patient’s post-discharge destination.
We anticipate that once we’ve made these choices, there will be further choices to make about how to treat these variables in the analysis. We hope to have the assistance of an experienced statistician to help guide us in making those decisions.
Monisha Bhatia, a native of Nashville, Tenn., is a fourth-year medical student at Vanderbilt University in Nashville. She is hoping to pursue either a residency in internal medicine or a combined internal medicine/emergency medicine program. Prior to medical school, she completed a JD/MPH program at Boston University, and she hopes to use her legal training in working with regulatory authorities to improve access to health care for all Americans.
Targeted therapies forge ahead in multiple breast cancer subtypes
As our understanding of the biology of breast cancer has improved, treatment has become increasingly personalized. Targeted therapies continue to significantly improve patient outcomes in multiple subtypes, with several recent drug approvals. Here, we discuss some of these latest developments.
A disease of many faces
Clinically speaking, breast cancers can be divided into at least 5 subtypes on the basis of the genes they express (Figure 1). The luminal subtypes make up the largest proportion and are characterized by the expression of hormone receptor (HR) genes. Luminal A tumors are negative for human epidermal growth factor receptor 2 (HER2; HER2-negative), whereas luminal B tumors often co-express the HER2 genes.1
The remainder of HER2-positive patients fall into the HER2-enriched category, in which HER2 expression is the defining characteristic. Basal-like tumors, meanwhile, represent the most heterogeneous subtype, overlapping to a large extent with tumors dubbed “triple-negative” because of their lack of either HER2 or ESR1 and PGR gene expression. The fifth subtype is known as normal breast-like and remains poorly characterized.
In recent years, there have been significant advancements in the genomic characterization of breast cancer that have begun to provide a more comprehensive understanding of the driver molecular mechanisms, which has helped to explain some of the limitations of current targeted approaches and to reveal new possible treatments, with a shift toward increasingly personalized strategies.2
HER2: what’s neu?
An estimated 18%-20% of breast tumors are HER2 positive, displaying amplification of the HER2/neu gene or overexpression of its protein product.3 Historically, HER2 positivity correlated with a highly aggressive and metastatic form of disease, conferring poor prognosis.4,5 The HER2-targeted monoclonal antibody (mAb), trastuzumab serves as a prime example of the power of personalized medicine. Evidence suggests that trastuzumab has altered the natural history of HER2-positive breast cancer, such that trastuzumab-treated patients with HER2-positive breast cancer now have a better prognosis than do patients with HER2-negative disease.6,7
Several additional HER2-targeted drugs have joined trastuzumab on the market, including other mAbs, small molecule tyrosine kinase inhibitors (TKIs), and an antibody–drug conjugate that combines the specificity of a mAb with the anti-tumor potency of a cytotoxic drug. These drugs have further improved patient outcomes in both early and advanced disease settings (Table 1).
The most recent regulatory approval was for neratinib, a potent TKI inhibiting all members of the HER protein family. On the basis of the phase 3 ExteNET study, neratinib was granted approval by the US Food and Drug Administration (FDA) for extended adjuvant treatment of patients with HER2-positive, early-stage breast cancer previously treated with trastuzumab. In a 5-year analysis of the study, invasive disease-free survival (DFS) was 90.4% with neratinib, compared with 87.9% with placebo (hazard ratio [HR], 0.74; P = .017).8,9
The tide of advancements in HER2-targeted therapy looks set to continue in the coming years as potentially practice-changing data emerges from ongoing clinical trials and, as the patent on trastuzumab has expired, a number of biosimilars, such as MYL-1401O have the potential to help patients who may not have access to trastuzumab.10
One of the biggest remaining challenges is identifying drugs that can effectively treat patients with brain metastases because the blood–brain barrier presents an impediment to the delivery of effective concentrations of anticancer drugs. Initially, it was hoped that the small molecule inhibitors lapatinib and neratinib could cross the blood–brain barrier and may be more effective in patients with brain metastases, but that hypothesis has not borne out in randomized clinical trials.11
Tucatinib (ONT-380) has shown significant promise in this respect. In a phase 1 trial, ONT-380 had significant efficacy in patients with and without central nervous system metastases; the overall response rate (ORR) in the CNS was 36%. ONT-380 is also notable for its specificity for HER2, without significant inhibition of HER1 and EGFR, which could translate into a better toxicity profile.12
Doubling down on resistant tumors
Since the success of HER2-targeted therapy is limited by the development of resistance, there has been significant interest in assessing the potential of dual HER2 blockade, exploiting the unique mechanisms of action of different drugs in combination therapy, and ensuring more complete inhibition of the HER2 pathway. Although numerous different combinations have been tested, a double antibody combination has proved most effective.
In fact, dual HER2 targeting with trastuzumab and pertuzumab in combination with chemotherapy has replaced a trastuzumab-chemotherapy regimen as the new standard of care in the metastatic setting. A 6-month improvement in progression-free survival (PFS) sealed FDA approval for the combination and in a recently published final analysis of the trial overall survival (OS) was also improved to a level unprecedented in the first-line setting.13,14The double antibody combination has also been successful in the neoadjuvant setting. Approval followed the results of the phase 2 NeoSphere trial, in which the combination was associated with a significant improvement in pathologic complete response (pCR) rate, a measure that acts as a surrogate for improved survival in the neoadjuvant setting. In a 5-year analysis of the NeoSphere trial, improved pCR did indeed translate into improved PFS and DFS.15,16
The results of the phase 3 APHINITY trial evaluating this combination in the adjuvant setting have been hotly anticipated. In a presentation at the 2017 American Society of Clinical Oncology (ASCO) meeting in June, the study authors reported that in 4,085 patients with operable HER2-positive disease, it significantly reduced the risk of disease recurrence or death compared with trastuzumab and chemotherapy alone.17
There is an ongoing effort to determine if it is possible to de-escalate treatment by removing the chemotherapy component. At least in the neoadjuvant setting, pCR rates in the chemotherapy-free arms of several studies suggest that a proportion of patients might benefit from this strategy15,18,19 and the challenge now is to identify them. To that end, the phase 2 PAMELA trial identified the HER2-enriched subtype as a strong predictor of response to neoadjuvant dual blockade (lapatinib and trastuzumab) without chemotherapy. The pCR rate was 40.6% for the combination in patients with the HER2-enriched subtype of breast cancer and only 10% in patients with non–HER2-enriched tumors.20
Targeting resistance to endocrine therapy
Another coup for personalized medicine in breast cancer is the treatment of hormone receptor–positive cases with endocrine therapy, which has become the cornerstone of treatment in the metastatic and adjuvant settings. Those drugs are designed to block the growth-stimulating effects of the estrogen and progesterone hormones on tumor cells. They include the selective estrogen receptor (ER) modulator tamoxifen, aromatase inhibitors (AIs) such as letrozole, anastrozole, and exemestane, which work by blocking the activity of the aromatase enzyme that converts androgens into estrogens, and the selective estrogen-receptor down-regulator fulvestrant.
As with HER2-targeted therapy, patients treated with endocrine therapy often develop resistance. Activation of alternate signaling cascades, such as the P13K–Akt–mTOR (phosphatidylinositol-3-kinase–Akt–mammalian target of rapamycin) pathway, or downstream targets of ER signaling, including the cyclin-dependent kinases, CDK4 and CDK6, have emerged as important mechanisms of resistance.21,22
Drugs directed against these secondary targets, aimed to enhance the efficacy of endocrine therapies, have shown significant promise (Table 2). The mTOR inhibitor everolimus received FDA approval in 2012 in combination with exemestane for the treatment of advanced HR-positive, HER2-negative breast cancer.23 More recently, everolimus has also proven effective in combination with either fulvestrant or letrozole, according to the phase 2 PrECOG 0102 and BOLERO-4 studies, both doubling PFS compared with endocrine therapy alone.24,25
Buparlisib is an oral reversible pan-PI3K inhibitor, and the results of the first phase 3 trial of this drug in metastatic breast cancer (MBC) were recently reported. Among 1,147 postmenopausal women with HR-positive, HER2-negative MBC that progressed on or after AI therapy, the combination of buparlisib and fulvestrant prolonged PFS compared with fulvestrant alone (median PFS, 6.9 vs 5 months; HR,0.78; P < .001). However, Novartis, which was developing buparlisib, reported that the combination will not be pursued further due to increased toxicity.26
Two other PI3K inhibitors are currently in phase 3 clinical trials; taselisib and alpelisib, both selective PI3K-alpha inhibitors. The results of a phase 1 dose-escalation study of taselisib were recently published and the ORR among patients with PIK3CA-mutant solid tumors was 36%, including responses in 4 patients with breast cancer.27 Meanwhile, alpelisib has also demonstrated early promise in combination with both letrozole and fulvestrant in patients with ER-positive MBC refractory to endocrine therapy. In combination with letrozole, the clinical benefit rate was 35% overall (44% in patients with PIK3CA mutations, compared with 20% in patients with wild-type PIK3CA status). The combination of alpesilib and fulvestrant produced an ORR of 27%, and both combinations were well tolerated.28,29
Another exciting therapeutic avenue is CDK4 and CDK6 inhibitors. These proteins are critical regulators of cell cycle progression, ensuring transition from G1 to S phase occurs at the appropriate time. The CDK pathway is also a downstream target of ER activation and, unsurprisingly, aberrant expression of the proteins involved in this pathway is commonly observed in breast tumors.
Palbociclib became the first FDA-approved member of this drug class, receiving accelerated approval in patients with HR-positive, HER2-negative metastatic breast cancer, in combination with letrozole in 2015. This became full regulatory approval in combination with any AI earlier this year, following the phase 3 PALOMA-3 study, in which the combination of palbociclib and fulvestrant (accelerated approval was based upon a trial testing palbociclib and letrozole) improved PFS by 5 months (HR, 0.46; P < .0001).30
In addition, a second CDK4/6 inhibitor hit the market this year. Ribociclib demonstrated a significant PFS benefit in combination with letrozole; median PFS was 25.3 months, compared with 16 months for letrozole alone, translating to a 44% reduction in the risk of disease progression or death.31
Abemaciclib, which has greater selectivity for CDK4 than its predecessors, also appears to be heading towards approval. It was granted priority review by the FDA based on data from the MONARCH-2 trial, showing a significant improvement in PFS for the combination of abemaciclib and fulvestrant (median PFS, 16.4 vs 9.3 months for fulvestrant alone; HR, 0.553; P < .001).32
Teasing out ‘HER2-positive’ subtypes
Until recently, “HER2-positive” and “HR-positive” tumors have been treated as separate subtypes, despite the fact that about half of HER2-positive tumors fall into the luminal A subtype and are also HR-positive. Patients were typically treated with HER2-targeted therapy regardless of their endocrine status because of the aggressive nature of HER2-positive disease.
Increasingly, researchers are reconsidering this view, especially as several studies have shown differential response rates to HER2-targeted therapy in HR-positive compared with HR-negative patients and accumulating evidence suggests that there is significant crosstalk between the HER2 and HR pathways, which may be responsible for the development of resistance with both treatment paradigms.
Findings from several studies have shown a benefit to combining HER2-targeted and hormonal therapies in patients with luminal (HR-positive), HER2-positive disease. In the metastatic setting, the results of the phase 2 PERTAIN study, presented at the 2017 ASCO annual meeting suggest that dual HER2 blockade could prove even more effective. The addition of pertuzumab to a combination of trastuzumab and an AI improved PFS by more than 3 months (median PFS, 19.89 vs 15.8 months; HR, 0.65; P = .007).33
The clinical application of these combinations may be limited by the additional cost – several studies have suggested that they are not cost effective – and toxicity, but have served to drive the development of new clinical trial designs as the importance of considering luminal and nonluminal HER2-positive tumors has become increasingly apparent.
PARP inhibitors make a dent in BRCA1/2-mutated cancers
The most renowned breast cancer genes, BRCA1 and BRCA2 are present in about 5%-10% of all breast cancers. They play a central role in the homologous recombination pathway that fixes double-strand breaks in the DNA. Genome sequencing studies have revealed that the presence of the BRCA1/2 genes and other DNA repair defects is highest among patients with the basal-like subtype of breast cancer, in particular those who have triple-negative disease.34,35
This type of breast cancer has proved stubbornly resistant to efforts to improve patient outcomes with targeted therapies. BRCA1/2 mutations and other DNA repair defects that confer a so-called BRCAness phenotype, render tumor cells dependent on other pathways for DNA repair and there has been considerable interest in therapeutically exploiting this through the development of inhibitors of the poly(ADP-ribose) polymerase (PARP) enzyme, which is involved in the repair of single-strand breaks in the DNA. The double damage to DNA repair mechanisms through PARP inhibition in patients with BRCA1/2-mutant tumors proves overwhelming to cancerous cells.
Despite more than a decade of investigation in breast cancer, PARP inhibitors have yet to yield any FDA-approved treatment options. That may be set to change imminently, following the success of olaparib (Table 3). In the first randomized phase 3 trial of a PARP inhibitor in breast cancer (OlympiAD), olaparib was compared with standard chemotherapy in patients with BRCA1/2-mutated MBC who had received up to 2 previous lines of chemotherapy. Olaparib reduced the risk of disease progression by 42% compared with standard chemotherapy and was well tolerated.36
The novel PARP inhibitor talazoparib, which is the most potent to date, is also demonstrating significant efficacy in clinical trials. The results of the phase 2 ABRAZO trial were presented at the ASCO annual meeting. Two cohorts were treated; the first included 49 patients who had responded to their last platinum-containing regimen for metastatic disease and progressed more than 8 weeks after last platinum dose and the other included 35 patients previously treated with 3 or more nonplatinum regimens for metastatic disease. ORR was 28% across the 2 cohorts; 23% and 33% in BRCA1- and BRCA2-mutant carriers, respectively; and 26% in patients with triple-negative breast cancer.37 PARP inhibition is not faring so well in early-stage triple-negative disease; a phase 3 trial of veliparib in combination with chemotherapy did not meet its primary endpoint.38
As our understanding of the biology of breast cancer has improved, treatment has become increasingly personalized. Targeted therapies continue to significantly improve patient outcomes in multiple subtypes, with several recent drug approvals. Here, we discuss some of these latest developments.
A disease of many faces
Clinically speaking, breast cancers can be divided into at least 5 subtypes on the basis of the genes they express (Figure 1). The luminal subtypes make up the largest proportion and are characterized by the expression of hormone receptor (HR) genes. Luminal A tumors are negative for human epidermal growth factor receptor 2 (HER2; HER2-negative), whereas luminal B tumors often co-express the HER2 genes.1
The remainder of HER2-positive patients fall into the HER2-enriched category, in which HER2 expression is the defining characteristic. Basal-like tumors, meanwhile, represent the most heterogeneous subtype, overlapping to a large extent with tumors dubbed “triple-negative” because of their lack of either HER2 or ESR1 and PGR gene expression. The fifth subtype is known as normal breast-like and remains poorly characterized.
In recent years, there have been significant advancements in the genomic characterization of breast cancer that have begun to provide a more comprehensive understanding of the driver molecular mechanisms, which has helped to explain some of the limitations of current targeted approaches and to reveal new possible treatments, with a shift toward increasingly personalized strategies.2
HER2: what’s neu?
An estimated 18%-20% of breast tumors are HER2 positive, displaying amplification of the HER2/neu gene or overexpression of its protein product.3 Historically, HER2 positivity correlated with a highly aggressive and metastatic form of disease, conferring poor prognosis.4,5 The HER2-targeted monoclonal antibody (mAb), trastuzumab serves as a prime example of the power of personalized medicine. Evidence suggests that trastuzumab has altered the natural history of HER2-positive breast cancer, such that trastuzumab-treated patients with HER2-positive breast cancer now have a better prognosis than do patients with HER2-negative disease.6,7
Several additional HER2-targeted drugs have joined trastuzumab on the market, including other mAbs, small molecule tyrosine kinase inhibitors (TKIs), and an antibody–drug conjugate that combines the specificity of a mAb with the anti-tumor potency of a cytotoxic drug. These drugs have further improved patient outcomes in both early and advanced disease settings (Table 1).
The most recent regulatory approval was for neratinib, a potent TKI inhibiting all members of the HER protein family. On the basis of the phase 3 ExteNET study, neratinib was granted approval by the US Food and Drug Administration (FDA) for extended adjuvant treatment of patients with HER2-positive, early-stage breast cancer previously treated with trastuzumab. In a 5-year analysis of the study, invasive disease-free survival (DFS) was 90.4% with neratinib, compared with 87.9% with placebo (hazard ratio [HR], 0.74; P = .017).8,9
The tide of advancements in HER2-targeted therapy looks set to continue in the coming years as potentially practice-changing data emerges from ongoing clinical trials and, as the patent on trastuzumab has expired, a number of biosimilars, such as MYL-1401O have the potential to help patients who may not have access to trastuzumab.10
One of the biggest remaining challenges is identifying drugs that can effectively treat patients with brain metastases because the blood–brain barrier presents an impediment to the delivery of effective concentrations of anticancer drugs. Initially, it was hoped that the small molecule inhibitors lapatinib and neratinib could cross the blood–brain barrier and may be more effective in patients with brain metastases, but that hypothesis has not borne out in randomized clinical trials.11
Tucatinib (ONT-380) has shown significant promise in this respect. In a phase 1 trial, ONT-380 had significant efficacy in patients with and without central nervous system metastases; the overall response rate (ORR) in the CNS was 36%. ONT-380 is also notable for its specificity for HER2, without significant inhibition of HER1 and EGFR, which could translate into a better toxicity profile.12
Doubling down on resistant tumors
Since the success of HER2-targeted therapy is limited by the development of resistance, there has been significant interest in assessing the potential of dual HER2 blockade, exploiting the unique mechanisms of action of different drugs in combination therapy, and ensuring more complete inhibition of the HER2 pathway. Although numerous different combinations have been tested, a double antibody combination has proved most effective.
In fact, dual HER2 targeting with trastuzumab and pertuzumab in combination with chemotherapy has replaced a trastuzumab-chemotherapy regimen as the new standard of care in the metastatic setting. A 6-month improvement in progression-free survival (PFS) sealed FDA approval for the combination and in a recently published final analysis of the trial overall survival (OS) was also improved to a level unprecedented in the first-line setting.13,14The double antibody combination has also been successful in the neoadjuvant setting. Approval followed the results of the phase 2 NeoSphere trial, in which the combination was associated with a significant improvement in pathologic complete response (pCR) rate, a measure that acts as a surrogate for improved survival in the neoadjuvant setting. In a 5-year analysis of the NeoSphere trial, improved pCR did indeed translate into improved PFS and DFS.15,16
The results of the phase 3 APHINITY trial evaluating this combination in the adjuvant setting have been hotly anticipated. In a presentation at the 2017 American Society of Clinical Oncology (ASCO) meeting in June, the study authors reported that in 4,085 patients with operable HER2-positive disease, it significantly reduced the risk of disease recurrence or death compared with trastuzumab and chemotherapy alone.17
There is an ongoing effort to determine if it is possible to de-escalate treatment by removing the chemotherapy component. At least in the neoadjuvant setting, pCR rates in the chemotherapy-free arms of several studies suggest that a proportion of patients might benefit from this strategy15,18,19 and the challenge now is to identify them. To that end, the phase 2 PAMELA trial identified the HER2-enriched subtype as a strong predictor of response to neoadjuvant dual blockade (lapatinib and trastuzumab) without chemotherapy. The pCR rate was 40.6% for the combination in patients with the HER2-enriched subtype of breast cancer and only 10% in patients with non–HER2-enriched tumors.20
Targeting resistance to endocrine therapy
Another coup for personalized medicine in breast cancer is the treatment of hormone receptor–positive cases with endocrine therapy, which has become the cornerstone of treatment in the metastatic and adjuvant settings. Those drugs are designed to block the growth-stimulating effects of the estrogen and progesterone hormones on tumor cells. They include the selective estrogen receptor (ER) modulator tamoxifen, aromatase inhibitors (AIs) such as letrozole, anastrozole, and exemestane, which work by blocking the activity of the aromatase enzyme that converts androgens into estrogens, and the selective estrogen-receptor down-regulator fulvestrant.
As with HER2-targeted therapy, patients treated with endocrine therapy often develop resistance. Activation of alternate signaling cascades, such as the P13K–Akt–mTOR (phosphatidylinositol-3-kinase–Akt–mammalian target of rapamycin) pathway, or downstream targets of ER signaling, including the cyclin-dependent kinases, CDK4 and CDK6, have emerged as important mechanisms of resistance.21,22
Drugs directed against these secondary targets, aimed to enhance the efficacy of endocrine therapies, have shown significant promise (Table 2). The mTOR inhibitor everolimus received FDA approval in 2012 in combination with exemestane for the treatment of advanced HR-positive, HER2-negative breast cancer.23 More recently, everolimus has also proven effective in combination with either fulvestrant or letrozole, according to the phase 2 PrECOG 0102 and BOLERO-4 studies, both doubling PFS compared with endocrine therapy alone.24,25
Buparlisib is an oral reversible pan-PI3K inhibitor, and the results of the first phase 3 trial of this drug in metastatic breast cancer (MBC) were recently reported. Among 1,147 postmenopausal women with HR-positive, HER2-negative MBC that progressed on or after AI therapy, the combination of buparlisib and fulvestrant prolonged PFS compared with fulvestrant alone (median PFS, 6.9 vs 5 months; HR,0.78; P < .001). However, Novartis, which was developing buparlisib, reported that the combination will not be pursued further due to increased toxicity.26
Two other PI3K inhibitors are currently in phase 3 clinical trials; taselisib and alpelisib, both selective PI3K-alpha inhibitors. The results of a phase 1 dose-escalation study of taselisib were recently published and the ORR among patients with PIK3CA-mutant solid tumors was 36%, including responses in 4 patients with breast cancer.27 Meanwhile, alpelisib has also demonstrated early promise in combination with both letrozole and fulvestrant in patients with ER-positive MBC refractory to endocrine therapy. In combination with letrozole, the clinical benefit rate was 35% overall (44% in patients with PIK3CA mutations, compared with 20% in patients with wild-type PIK3CA status). The combination of alpesilib and fulvestrant produced an ORR of 27%, and both combinations were well tolerated.28,29
Another exciting therapeutic avenue is CDK4 and CDK6 inhibitors. These proteins are critical regulators of cell cycle progression, ensuring transition from G1 to S phase occurs at the appropriate time. The CDK pathway is also a downstream target of ER activation and, unsurprisingly, aberrant expression of the proteins involved in this pathway is commonly observed in breast tumors.
Palbociclib became the first FDA-approved member of this drug class, receiving accelerated approval in patients with HR-positive, HER2-negative metastatic breast cancer, in combination with letrozole in 2015. This became full regulatory approval in combination with any AI earlier this year, following the phase 3 PALOMA-3 study, in which the combination of palbociclib and fulvestrant (accelerated approval was based upon a trial testing palbociclib and letrozole) improved PFS by 5 months (HR, 0.46; P < .0001).30
In addition, a second CDK4/6 inhibitor hit the market this year. Ribociclib demonstrated a significant PFS benefit in combination with letrozole; median PFS was 25.3 months, compared with 16 months for letrozole alone, translating to a 44% reduction in the risk of disease progression or death.31
Abemaciclib, which has greater selectivity for CDK4 than its predecessors, also appears to be heading towards approval. It was granted priority review by the FDA based on data from the MONARCH-2 trial, showing a significant improvement in PFS for the combination of abemaciclib and fulvestrant (median PFS, 16.4 vs 9.3 months for fulvestrant alone; HR, 0.553; P < .001).32
Teasing out ‘HER2-positive’ subtypes
Until recently, “HER2-positive” and “HR-positive” tumors have been treated as separate subtypes, despite the fact that about half of HER2-positive tumors fall into the luminal A subtype and are also HR-positive. Patients were typically treated with HER2-targeted therapy regardless of their endocrine status because of the aggressive nature of HER2-positive disease.
Increasingly, researchers are reconsidering this view, especially as several studies have shown differential response rates to HER2-targeted therapy in HR-positive compared with HR-negative patients and accumulating evidence suggests that there is significant crosstalk between the HER2 and HR pathways, which may be responsible for the development of resistance with both treatment paradigms.
Findings from several studies have shown a benefit to combining HER2-targeted and hormonal therapies in patients with luminal (HR-positive), HER2-positive disease. In the metastatic setting, the results of the phase 2 PERTAIN study, presented at the 2017 ASCO annual meeting suggest that dual HER2 blockade could prove even more effective. The addition of pertuzumab to a combination of trastuzumab and an AI improved PFS by more than 3 months (median PFS, 19.89 vs 15.8 months; HR, 0.65; P = .007).33
The clinical application of these combinations may be limited by the additional cost – several studies have suggested that they are not cost effective – and toxicity, but have served to drive the development of new clinical trial designs as the importance of considering luminal and nonluminal HER2-positive tumors has become increasingly apparent.
PARP inhibitors make a dent in BRCA1/2-mutated cancers
The most renowned breast cancer genes, BRCA1 and BRCA2 are present in about 5%-10% of all breast cancers. They play a central role in the homologous recombination pathway that fixes double-strand breaks in the DNA. Genome sequencing studies have revealed that the presence of the BRCA1/2 genes and other DNA repair defects is highest among patients with the basal-like subtype of breast cancer, in particular those who have triple-negative disease.34,35
This type of breast cancer has proved stubbornly resistant to efforts to improve patient outcomes with targeted therapies. BRCA1/2 mutations and other DNA repair defects that confer a so-called BRCAness phenotype, render tumor cells dependent on other pathways for DNA repair and there has been considerable interest in therapeutically exploiting this through the development of inhibitors of the poly(ADP-ribose) polymerase (PARP) enzyme, which is involved in the repair of single-strand breaks in the DNA. The double damage to DNA repair mechanisms through PARP inhibition in patients with BRCA1/2-mutant tumors proves overwhelming to cancerous cells.
Despite more than a decade of investigation in breast cancer, PARP inhibitors have yet to yield any FDA-approved treatment options. That may be set to change imminently, following the success of olaparib (Table 3). In the first randomized phase 3 trial of a PARP inhibitor in breast cancer (OlympiAD), olaparib was compared with standard chemotherapy in patients with BRCA1/2-mutated MBC who had received up to 2 previous lines of chemotherapy. Olaparib reduced the risk of disease progression by 42% compared with standard chemotherapy and was well tolerated.36
The novel PARP inhibitor talazoparib, which is the most potent to date, is also demonstrating significant efficacy in clinical trials. The results of the phase 2 ABRAZO trial were presented at the ASCO annual meeting. Two cohorts were treated; the first included 49 patients who had responded to their last platinum-containing regimen for metastatic disease and progressed more than 8 weeks after last platinum dose and the other included 35 patients previously treated with 3 or more nonplatinum regimens for metastatic disease. ORR was 28% across the 2 cohorts; 23% and 33% in BRCA1- and BRCA2-mutant carriers, respectively; and 26% in patients with triple-negative breast cancer.37 PARP inhibition is not faring so well in early-stage triple-negative disease; a phase 3 trial of veliparib in combination with chemotherapy did not meet its primary endpoint.38
As our understanding of the biology of breast cancer has improved, treatment has become increasingly personalized. Targeted therapies continue to significantly improve patient outcomes in multiple subtypes, with several recent drug approvals. Here, we discuss some of these latest developments.
A disease of many faces
Clinically speaking, breast cancers can be divided into at least 5 subtypes on the basis of the genes they express (Figure 1). The luminal subtypes make up the largest proportion and are characterized by the expression of hormone receptor (HR) genes. Luminal A tumors are negative for human epidermal growth factor receptor 2 (HER2; HER2-negative), whereas luminal B tumors often co-express the HER2 genes.1
The remainder of HER2-positive patients fall into the HER2-enriched category, in which HER2 expression is the defining characteristic. Basal-like tumors, meanwhile, represent the most heterogeneous subtype, overlapping to a large extent with tumors dubbed “triple-negative” because of their lack of either HER2 or ESR1 and PGR gene expression. The fifth subtype is known as normal breast-like and remains poorly characterized.
In recent years, there have been significant advancements in the genomic characterization of breast cancer that have begun to provide a more comprehensive understanding of the driver molecular mechanisms, which has helped to explain some of the limitations of current targeted approaches and to reveal new possible treatments, with a shift toward increasingly personalized strategies.2
HER2: what’s neu?
An estimated 18%-20% of breast tumors are HER2 positive, displaying amplification of the HER2/neu gene or overexpression of its protein product.3 Historically, HER2 positivity correlated with a highly aggressive and metastatic form of disease, conferring poor prognosis.4,5 The HER2-targeted monoclonal antibody (mAb), trastuzumab serves as a prime example of the power of personalized medicine. Evidence suggests that trastuzumab has altered the natural history of HER2-positive breast cancer, such that trastuzumab-treated patients with HER2-positive breast cancer now have a better prognosis than do patients with HER2-negative disease.6,7
Several additional HER2-targeted drugs have joined trastuzumab on the market, including other mAbs, small molecule tyrosine kinase inhibitors (TKIs), and an antibody–drug conjugate that combines the specificity of a mAb with the anti-tumor potency of a cytotoxic drug. These drugs have further improved patient outcomes in both early and advanced disease settings (Table 1).
The most recent regulatory approval was for neratinib, a potent TKI inhibiting all members of the HER protein family. On the basis of the phase 3 ExteNET study, neratinib was granted approval by the US Food and Drug Administration (FDA) for extended adjuvant treatment of patients with HER2-positive, early-stage breast cancer previously treated with trastuzumab. In a 5-year analysis of the study, invasive disease-free survival (DFS) was 90.4% with neratinib, compared with 87.9% with placebo (hazard ratio [HR], 0.74; P = .017).8,9
The tide of advancements in HER2-targeted therapy looks set to continue in the coming years as potentially practice-changing data emerges from ongoing clinical trials and, as the patent on trastuzumab has expired, a number of biosimilars, such as MYL-1401O have the potential to help patients who may not have access to trastuzumab.10
One of the biggest remaining challenges is identifying drugs that can effectively treat patients with brain metastases because the blood–brain barrier presents an impediment to the delivery of effective concentrations of anticancer drugs. Initially, it was hoped that the small molecule inhibitors lapatinib and neratinib could cross the blood–brain barrier and may be more effective in patients with brain metastases, but that hypothesis has not borne out in randomized clinical trials.11
Tucatinib (ONT-380) has shown significant promise in this respect. In a phase 1 trial, ONT-380 had significant efficacy in patients with and without central nervous system metastases; the overall response rate (ORR) in the CNS was 36%. ONT-380 is also notable for its specificity for HER2, without significant inhibition of HER1 and EGFR, which could translate into a better toxicity profile.12
Doubling down on resistant tumors
Since the success of HER2-targeted therapy is limited by the development of resistance, there has been significant interest in assessing the potential of dual HER2 blockade, exploiting the unique mechanisms of action of different drugs in combination therapy, and ensuring more complete inhibition of the HER2 pathway. Although numerous different combinations have been tested, a double antibody combination has proved most effective.
In fact, dual HER2 targeting with trastuzumab and pertuzumab in combination with chemotherapy has replaced a trastuzumab-chemotherapy regimen as the new standard of care in the metastatic setting. A 6-month improvement in progression-free survival (PFS) sealed FDA approval for the combination and in a recently published final analysis of the trial overall survival (OS) was also improved to a level unprecedented in the first-line setting.13,14The double antibody combination has also been successful in the neoadjuvant setting. Approval followed the results of the phase 2 NeoSphere trial, in which the combination was associated with a significant improvement in pathologic complete response (pCR) rate, a measure that acts as a surrogate for improved survival in the neoadjuvant setting. In a 5-year analysis of the NeoSphere trial, improved pCR did indeed translate into improved PFS and DFS.15,16
The results of the phase 3 APHINITY trial evaluating this combination in the adjuvant setting have been hotly anticipated. In a presentation at the 2017 American Society of Clinical Oncology (ASCO) meeting in June, the study authors reported that in 4,085 patients with operable HER2-positive disease, it significantly reduced the risk of disease recurrence or death compared with trastuzumab and chemotherapy alone.17
There is an ongoing effort to determine if it is possible to de-escalate treatment by removing the chemotherapy component. At least in the neoadjuvant setting, pCR rates in the chemotherapy-free arms of several studies suggest that a proportion of patients might benefit from this strategy15,18,19 and the challenge now is to identify them. To that end, the phase 2 PAMELA trial identified the HER2-enriched subtype as a strong predictor of response to neoadjuvant dual blockade (lapatinib and trastuzumab) without chemotherapy. The pCR rate was 40.6% for the combination in patients with the HER2-enriched subtype of breast cancer and only 10% in patients with non–HER2-enriched tumors.20
Targeting resistance to endocrine therapy
Another coup for personalized medicine in breast cancer is the treatment of hormone receptor–positive cases with endocrine therapy, which has become the cornerstone of treatment in the metastatic and adjuvant settings. Those drugs are designed to block the growth-stimulating effects of the estrogen and progesterone hormones on tumor cells. They include the selective estrogen receptor (ER) modulator tamoxifen, aromatase inhibitors (AIs) such as letrozole, anastrozole, and exemestane, which work by blocking the activity of the aromatase enzyme that converts androgens into estrogens, and the selective estrogen-receptor down-regulator fulvestrant.
As with HER2-targeted therapy, patients treated with endocrine therapy often develop resistance. Activation of alternate signaling cascades, such as the P13K–Akt–mTOR (phosphatidylinositol-3-kinase–Akt–mammalian target of rapamycin) pathway, or downstream targets of ER signaling, including the cyclin-dependent kinases, CDK4 and CDK6, have emerged as important mechanisms of resistance.21,22
Drugs directed against these secondary targets, aimed to enhance the efficacy of endocrine therapies, have shown significant promise (Table 2). The mTOR inhibitor everolimus received FDA approval in 2012 in combination with exemestane for the treatment of advanced HR-positive, HER2-negative breast cancer.23 More recently, everolimus has also proven effective in combination with either fulvestrant or letrozole, according to the phase 2 PrECOG 0102 and BOLERO-4 studies, both doubling PFS compared with endocrine therapy alone.24,25
Buparlisib is an oral reversible pan-PI3K inhibitor, and the results of the first phase 3 trial of this drug in metastatic breast cancer (MBC) were recently reported. Among 1,147 postmenopausal women with HR-positive, HER2-negative MBC that progressed on or after AI therapy, the combination of buparlisib and fulvestrant prolonged PFS compared with fulvestrant alone (median PFS, 6.9 vs 5 months; HR,0.78; P < .001). However, Novartis, which was developing buparlisib, reported that the combination will not be pursued further due to increased toxicity.26
Two other PI3K inhibitors are currently in phase 3 clinical trials; taselisib and alpelisib, both selective PI3K-alpha inhibitors. The results of a phase 1 dose-escalation study of taselisib were recently published and the ORR among patients with PIK3CA-mutant solid tumors was 36%, including responses in 4 patients with breast cancer.27 Meanwhile, alpelisib has also demonstrated early promise in combination with both letrozole and fulvestrant in patients with ER-positive MBC refractory to endocrine therapy. In combination with letrozole, the clinical benefit rate was 35% overall (44% in patients with PIK3CA mutations, compared with 20% in patients with wild-type PIK3CA status). The combination of alpesilib and fulvestrant produced an ORR of 27%, and both combinations were well tolerated.28,29
Another exciting therapeutic avenue is CDK4 and CDK6 inhibitors. These proteins are critical regulators of cell cycle progression, ensuring transition from G1 to S phase occurs at the appropriate time. The CDK pathway is also a downstream target of ER activation and, unsurprisingly, aberrant expression of the proteins involved in this pathway is commonly observed in breast tumors.
Palbociclib became the first FDA-approved member of this drug class, receiving accelerated approval in patients with HR-positive, HER2-negative metastatic breast cancer, in combination with letrozole in 2015. This became full regulatory approval in combination with any AI earlier this year, following the phase 3 PALOMA-3 study, in which the combination of palbociclib and fulvestrant (accelerated approval was based upon a trial testing palbociclib and letrozole) improved PFS by 5 months (HR, 0.46; P < .0001).30
In addition, a second CDK4/6 inhibitor hit the market this year. Ribociclib demonstrated a significant PFS benefit in combination with letrozole; median PFS was 25.3 months, compared with 16 months for letrozole alone, translating to a 44% reduction in the risk of disease progression or death.31
Abemaciclib, which has greater selectivity for CDK4 than its predecessors, also appears to be heading towards approval. It was granted priority review by the FDA based on data from the MONARCH-2 trial, showing a significant improvement in PFS for the combination of abemaciclib and fulvestrant (median PFS, 16.4 vs 9.3 months for fulvestrant alone; HR, 0.553; P < .001).32
Teasing out ‘HER2-positive’ subtypes
Until recently, “HER2-positive” and “HR-positive” tumors have been treated as separate subtypes, despite the fact that about half of HER2-positive tumors fall into the luminal A subtype and are also HR-positive. Patients were typically treated with HER2-targeted therapy regardless of their endocrine status because of the aggressive nature of HER2-positive disease.
Increasingly, researchers are reconsidering this view, especially as several studies have shown differential response rates to HER2-targeted therapy in HR-positive compared with HR-negative patients and accumulating evidence suggests that there is significant crosstalk between the HER2 and HR pathways, which may be responsible for the development of resistance with both treatment paradigms.
Findings from several studies have shown a benefit to combining HER2-targeted and hormonal therapies in patients with luminal (HR-positive), HER2-positive disease. In the metastatic setting, the results of the phase 2 PERTAIN study, presented at the 2017 ASCO annual meeting suggest that dual HER2 blockade could prove even more effective. The addition of pertuzumab to a combination of trastuzumab and an AI improved PFS by more than 3 months (median PFS, 19.89 vs 15.8 months; HR, 0.65; P = .007).33
The clinical application of these combinations may be limited by the additional cost – several studies have suggested that they are not cost effective – and toxicity, but have served to drive the development of new clinical trial designs as the importance of considering luminal and nonluminal HER2-positive tumors has become increasingly apparent.
PARP inhibitors make a dent in BRCA1/2-mutated cancers
The most renowned breast cancer genes, BRCA1 and BRCA2 are present in about 5%-10% of all breast cancers. They play a central role in the homologous recombination pathway that fixes double-strand breaks in the DNA. Genome sequencing studies have revealed that the presence of the BRCA1/2 genes and other DNA repair defects is highest among patients with the basal-like subtype of breast cancer, in particular those who have triple-negative disease.34,35
This type of breast cancer has proved stubbornly resistant to efforts to improve patient outcomes with targeted therapies. BRCA1/2 mutations and other DNA repair defects that confer a so-called BRCAness phenotype, render tumor cells dependent on other pathways for DNA repair and there has been considerable interest in therapeutically exploiting this through the development of inhibitors of the poly(ADP-ribose) polymerase (PARP) enzyme, which is involved in the repair of single-strand breaks in the DNA. The double damage to DNA repair mechanisms through PARP inhibition in patients with BRCA1/2-mutant tumors proves overwhelming to cancerous cells.
Despite more than a decade of investigation in breast cancer, PARP inhibitors have yet to yield any FDA-approved treatment options. That may be set to change imminently, following the success of olaparib (Table 3). In the first randomized phase 3 trial of a PARP inhibitor in breast cancer (OlympiAD), olaparib was compared with standard chemotherapy in patients with BRCA1/2-mutated MBC who had received up to 2 previous lines of chemotherapy. Olaparib reduced the risk of disease progression by 42% compared with standard chemotherapy and was well tolerated.36
The novel PARP inhibitor talazoparib, which is the most potent to date, is also demonstrating significant efficacy in clinical trials. The results of the phase 2 ABRAZO trial were presented at the ASCO annual meeting. Two cohorts were treated; the first included 49 patients who had responded to their last platinum-containing regimen for metastatic disease and progressed more than 8 weeks after last platinum dose and the other included 35 patients previously treated with 3 or more nonplatinum regimens for metastatic disease. ORR was 28% across the 2 cohorts; 23% and 33% in BRCA1- and BRCA2-mutant carriers, respectively; and 26% in patients with triple-negative breast cancer.37 PARP inhibition is not faring so well in early-stage triple-negative disease; a phase 3 trial of veliparib in combination with chemotherapy did not meet its primary endpoint.38
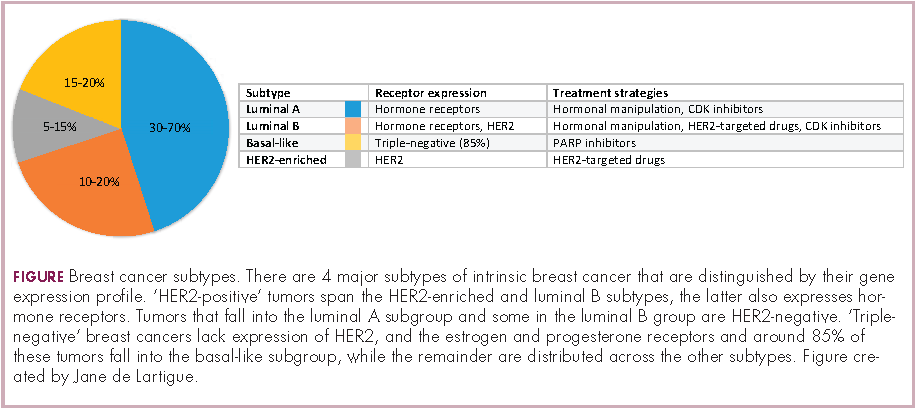
The effect of centralizing breast cancer care in an urban public hospital
When cancer care is centralized in a comprehensive fashion, the quality of care and the outcomes improve.1,2 Unfortunately, because of the medical insurance structure in New York City, most patients of lower socioeconomic status do not receive their cancer care in such dedicated cancer centers. In New York City, the majority of the underserved vulnerable populations – that is, those without health insurance – receive their care from the public hospital system known as NYC Health and Hospitals. Cancer care in this system is not centralized and may result in fragmented implementation of various modalities of treatment. In addition, because there is no centralized care, needs such as early screening and prevention programs are often not addressed. This problem was evident in Queens in 2000 and before when many patients with late-stage cancers were presenting for cancer care. Queens, which is one of the 5 boroughs of New York City, has more than 2.3 million residents. It has 2 public hospitals, Elmhurst Hospital Center and Queens Hospital Center (QHC). In 2001, the plan was devised for the establishment of a cancer center at QHC, mainly because of the high rate of late-stage cancers that were being seen at presentation and recognition of the need for more comprehensive care. In 2002, the Queens Cancer Center (QCC) began to see patients. QCC is a single facility that provides medical, surgical, radiation, gynecologic, and urologic oncology all in one area of the QHC.
This study is an investigation of the possible impact on care for breast cancer patients of low socioeconomic status who were treated at a comprehensive cancer center, with specific consideration of the change or improvement in treatment modalities and outcomes. Data on treatment modalities and outcomes of cancer patients who were treated at the QHC during 2000, before the QCC was set up, were compared with data of patients treated during 2008 (2008 was selected because we have 5-year survival data for those patients). The public hospital system treats all patients regardless of their ability to pay, so the majority of patients in the system are of lower socioeconomic status. In addition, 92% of the patients seen QHC are from a minority population. These are the populations that tend to have a worse prognosis and often are not given optimal treatment.3 The payer mix of patients in the public hospital system is different than that of private hospitals. Most of the patients present at the hospital with no insurance and if they are diagnosed with cancer they may be converted to emergency Medicaid. About 10% of patients will not be converted because of their document status.
Patients and methods
We used the Queens Hospital Tumor Registry to identify the patients who had been diagnosed with and treated for breast cancer in 2000 and 2008. The electronic medical records were reviewed, and in the case of the 2000-year patients, the written charts were also reviewed. The study was approved by the Mount Sinai institutional review board. It was not necessary to obtain patient consent because it was a retrospective study.
Only patients diagnosed with stage 0, I, II, or III breast cancer who received their treatment at QHC were included in the study. Patients who were seen in consultation at QHC but not treated there were excluded. Statistics were done using the 2x2 chi-squared SPSS analysis; a P value of .05 was considered significant. The survival data was analyzed using SAS.
Results
There were 24 evaluable patients in 2000 and 78 evaluable patients in 2008 who had stage 0, I, II, or III primary breast cancer and were treated at QHC. The average age of the patients in 2000 was 53.5 years and 54.7 years in 2008. The mean age for both groups was 55 years. The patients were ethnically diverse in both groups with 46% black, 17% Hispanic, 25% ethnic Asian Indian, and 6% white (Figure 1).
The payer mix in 2000 was 9 patients (37.5%) self-pay, 7 (29%) Medicaid, and 8 (33%) Medicare. In 2008, 11 patients (14%) were self-pay, 46 (59%) Medicaid, 11 (14%) Medicare, and 10 (13%) were private insurance. In 2000, there were 3 (12%) patients with stage 0 disease, 5 (21%) with stage I; 9 (37.5%) with stage II, and 7 (29%) with stage III. In 2008 there were 28 (36%) patients with stage 0 disease, 15 (19%) with stage I, 17 (22%) with stage II, and 18 (23%) with stage III (Figure 2).
None of those values are statistically different. In 2000, 2 of the 24 patients had lumpectomies (partial mastectomy) and the rest had mastectomies. In 2008, 39 (50%) patients had mastectomy and 39 (50%) had lumpectomies (Figure 3). This was a statistically significant difference.
Radiation was given to both patients with lumpectomy in the 2000 group. In the 2008 group, all patients with lumpectomies were evaluated for radiation, and 6 of them did not receive radiation for the following reasons: 3 had very small foci of ductal carcinoma in situ (DCIS) and were treated with hormone therapy and no radiation; 1 patient had a lumpectomy for stage 1 cancer and also did not get radiation therapy because of a low oncotype and very small lesion; 2 patients were older than 70 years and had DCIS and were treated with tamoxifen alone as per NCCN Guidelines for women in that age group. The rest of the patients with lumpectomies received postoperative radiation.
Hormone and HER2 (human epidermal growth factor receptor 2) status was obtained on all patients. For the 2000 patients, 71% had 1 hormone receptor–positive (estrogen receptor [ER] or progesterone receptor [PR]), 21% were triple negative (ER-PR and HER2-neu), and 42% had HER2-neu–positive tumors. For the 2008, patients 65% were positive for 1 hormone receptor (ER or PR), 28% were triple negative (ER-PR and HER2-neu), and 7% had HER2-neu-positive tumors.
All patients were offered chemotherapy and hormone therapy if appropriate, as per NCCN guidelines. If a patient’s tumor was found to be HER2-positive, then the chemotherapy regimen would include the use of trastuzumab in both groups.
The 5-year survival for the 2008 stage III patients was 73.7%, compared with 14.2% for the 2000 stage III patients. The only deaths in the 2008 group were in patients with stage III disease. In the 2000 group, 4 of the 5 patients with stage III cancer died, and 33% of patients with stage I or II either died or were lost to follow-up before 5 years. This survival difference is significant by a chi-square and Wilcoxon analysis, with a P value of .01.
In 2000, 86% of patients with cancer were termed self-pay, that is, they had no insurance and they were not converted to emergency Medicaid. In 2008, 16% of patients were self-pay, and the rest were converted to Medicaid. In 2000, fewer than 2% of patients had commercial insurance, compared with 9% in 2008.
Discussion
There have been numerous studies reporting on disparities in the treatment of patients with breast cancer based on race or socioeconomic status.4-18 Many studies have shown inferior survival for black women with breast cancer, but it is not entirely clear whether these differences are the result of the quality of medical care received or biologic differences.14,19 A moderately large study from a metropolitan medical center in Detroit showed no difference in survival in their patients based on race when all of the patients received equal treatments.15 A meta-analysis of survival in black and white breast cancer patients showed that the black women had significantly poorer outcomes.19
Findings from a recent study showed that patients of lower socioeconomic status are more likely to undergo mastectomy than breast conserving therapy.20 The study, which identified 727,927 patients with early-stage breast cancer during 1998-2011, found that the rate of breast conservation increased from 54% to 59% during that time period and that there were significant barriers to women receiving breast-conserving therapy based on their type of insurance and having a lower socioeconomic status.20
The treatment of breast cancer is best delivered in a multimodality setting, but many inner-city public hospitals do not have such a facility for their patients. QHC is the only public hospital in New York City that has established a comprehensive cancer center. The patient population of QHC is overwhelmingly of minority origin (only 5% of patients are white). In addition, it is a safety net hospital, so no patient is turned away because they cannot pay, and most patients are of lower socioeconomic status and do not have insurance. The purpose of the cancer center was to provide a single site at which our patients could receive all their treatment. It was to ensure that our patients had easy access to care and treatment during all phases of their disease trajectory and did not “fall through the cracks” of the system. Those goals were addressed by having all of the center’s physicians in one place. Physicians involved in care included medical, surgical, and radiation oncologists, a gynecologic oncologist, a genitourinary oncologist, and a thoracic surgery oncologist. The support groups organized for the cancer patients included 3 oncology social workers, an oncology navigator, a nutritionist, a pastoral care supporter, and an oncology psychologist, all located in the same area. All of the clerical and financial aspects of care were also placed within the center. This made the experience as seamless as possible for both the patients and the treating physicians. A “survivors clinic” was established so the cancer patients could be seen by integrated primary care providers to address all noncancer-related health issues such as hypertension, diabetes, or heart disease. Finally, a robust clinical oncology research team was established in the same location. The research included several protocols for new drug treatments for breast cancer from pharmaceutical companies as well as the multi-institutional oncology groups.
Part of the mission of the cancer center was to reach out into the community of Queens to provide education about early detection, cancer prevention, and other public health issues such as tobacco cessation. We established a close working relationship with the Queens Public Library System to connect with their users and dispense information about cancer care and early detection. The Queens Library system is the largest in the United States, and everyone who lives in Queens has easy access to one of its 63 branch libraries. We arranged several lectures about breast cancer awareness in some of the branch libraries. We also procured a mobile mammogram unit for free screening events at the lectures, especially in neighborhoods with a large number residents who were of lower socioeconomic status.
To study the possible effect of these changes on our patients with breast cancer, we compared 2 groups of patients. One group was from the year 2000, a year before the cancer center was opened. The other was from the year 2008, the last year we could get real 5-year survival statistics. We explored how establishing the cancer center might have changed the patients’ stage at diagnosis, care, treatment modalities such as type of surgery, and outcomes. It is difficult to compare these 2 groups because of differences in the patients’ cancers, such as their receptor status, as well as differences in treatment options between the two time periods. However, we had no other way to compare the data to see if there were any trends.
There was a migration to earlier-stage cancer at diagnosis during the 6-year period after the cancer center was opened. It is likely that the educational sessions that were done in the community contributed to this migration. We also saw an increase in the number of mammograms done, from 6,300 in 2000 to 8,800 in 2008. This increase in screening also could account for more patients being identified with earlier-stage disease and might be attributable to the community education through the outreach programs.
As a quality control method, the cancer center has been evaluated by the Commission on Cancer every 3 years. At the 2013 evaluation, we received the Gold Commendation – the highest possible recognition for having 8 out of 8 commendations – and a 3-year accreditation.
There was a notable increase in the use of lumpectomy over mastectomy after the establishment of the cancer center, possibly due to the addition of 2 surgical oncologists to the cancer center’s care team. The integration of multimodiality care for each patient may also have increased the use of breast-conserving surgery.
There was a significant increase from 2000 to 2008 in the survival of patients treated for stage III breast cancer. New drugs and new patterns of adjuvant care might have been partly responsible for that change. The establishment of the comprehensive cancer center with access to new protocols ensured that patients received state-of-the-art cancer treatment. Moreover, the facility addressed all aspects of patient care throughout the disease trajectory by including designated social workers, psychologists, a nutritionist, pastoral care, and patient and survivor support groups to ensure that patients would keep coming to the center for their therapy, with no delays and very little loss to follow-up.
Most patients without insurance were able to acquire emergency Medicaid through the cancer center. This was done by having 2 financial counselors who met with every patient and who could facilitate access to Medicaid as needed. As a result of that, the percentage of patients with no coverage went from 86% in 2000 to 16% in 2008. Before this system was set up, patients who were designated self-pay would pay a fee as low as $15 for each visit and received thousands of dollars’ worth of care. Thus, by forming a cancer center and facilitating patient access to Medicaid, we were able to save money for this public institution because of the gain in revenue from Medicaid.
Our findings suggest that the development of comprehensive cancer centers within inner-city health systems can ensure better treatment for patients of lower socioeconomic status. We present evidence that this may result in increased survival, more sophisticated surgical options, and better patient quality of life. Moreover, this can be achieved while effectively increasing revenue for the public hospitals. Correcting the inequality of access to care and better therapeutic options by setting up comprehensive cancer centers could contribute to improved parity of outcomes for underserved populations.
The author acknowledges the statistical help of Brian Altonen, MPH.
1. Kesson EM, Allardice GM, George WD, Morrison DS. Effects of multidisciplinary team working on breast cancer survival: retrospective, comparative, interventional cohort study of 13,722 women. BMJ. 2012;344:e2718.
2. Vrijens F, Stordeur S, Beirens K, Devriese S, Van Eycken E, Vlayen J. Effect of hospital volume on processes of care and 5-year survival after breast cancer: a population-based study on 25000 women. Breast. 2012;21(3):261-266.
3. Bradley CJ, Given CW, Roberts C. Race, socioeconomic status and breast cancer treatment and survival. J Natl Cancer Inst. 2002;94(7):490-496.
4. Wheeler SB, Hayes-Reeder KE, Carey LA. Disparities in breast cancer treatment and outcomes: biological, social, and health system determinants and opportunities for research. Oncologist. 2013;18:986-993.
5. Ward E, Jemal A, Cokkinides V, et al. Cancer disparities by race/ethnicity and socioeconomic status. CA Cancer J Clin. 2004;54:78-93.
6. Chen F, Puig M, Yermilov I, et al. Using breast cancer quality indicators in a vulnerable population. Cancer. 2011;117:3311-3321.
7. Banerjee M, George J, Yee C, Hryniuk W, Schwartz K. Disentangling the effects of race on breast cancer treatment. Cancer. 2007;110:2169-2177.
8. Freedman RA, He Y, Winer EP, Keating NL. Trends in racial and age disparities in definitive local therapy of early-stage breast cancer. J Clin Oncol. 2009;27:713-719.
9. Bickell NA, Shastri K, Fei K, et al. A tracking and feedback registry to reduce racial disparities in breast cancer care. J Natl Cancer Inst. 2008;100:1717-1723.
19. Bickell NA, Wang JJ, Oluwole S, et al. Missed opportunities: Racial disparities in adjuvant breast cancer treatment. J Clin Oncol. 2006;24:1357-1362.
11. Harper S, Lynch J, Meersman SC, Breen N, Davis WW, Reichman MC. Trends in area-socioeconomic and race-ethnic disparities in breast cancer incidence, stage at diagnosis, screening, mortality, and survival among women ages 50 years and over (1987-2005). Cancer Epidemiol Biomarkers Prev. 2009;18:121-131.
12. Ward E, Halpern M, Schrag N, et al. Association of insurance with cancer care utilization and outcomes. CA Cancer J Clin. 2008;58:9-31.
13. Naik AM, Joseph K, Harris M, Davis C, Shapiro R, Hiotis KL. Indigent breast cancer patients among all racial and ethnic groups present with more advanced disease compared with nationally reported date. Am J Surg. 2003;186:400-403.
14. Hersman DL, Unger JM, Barlow WE, et al. Treatment quality and outcomes of African American versus white breast cancer patients: retrospective analysis of southwest oncology studies S8814/S8897. J Clin Oncol. 2009;27: 2157-2162.
15
16. Brawley OW. Disaggregating the effects of race and poverty on breast cancer outcomes. J Natl Cancer Inst. 2002;94:471-473.
17. Baquet CR, Commiskey P. Socioeconomic factors and breast carcinoma in multicultural women. Cancer. 2000;88:1256-1264.
18. Cross C, Harris J, Recht A. Race, socioeconomic status, and breast carcinoma in the US. Cancer. 2002;95:1988-1999.
19. Newman LA, Griffith KA, Jatoi I, Simon MS, Crowe JP, Colditz GA. Meta-analysis of survival in African American and white American patients with breast cancer: Ethnicity compared with socioeconomic status. J Clin Oncol. 2006;24:1342-1349.
20. Lautner M, Lin H, Shen Y, et al. Disparities in the use of breast-conserving therapy among patients with early-stage breast cancer. JAMA. 2015;150:778-786.
When cancer care is centralized in a comprehensive fashion, the quality of care and the outcomes improve.1,2 Unfortunately, because of the medical insurance structure in New York City, most patients of lower socioeconomic status do not receive their cancer care in such dedicated cancer centers. In New York City, the majority of the underserved vulnerable populations – that is, those without health insurance – receive their care from the public hospital system known as NYC Health and Hospitals. Cancer care in this system is not centralized and may result in fragmented implementation of various modalities of treatment. In addition, because there is no centralized care, needs such as early screening and prevention programs are often not addressed. This problem was evident in Queens in 2000 and before when many patients with late-stage cancers were presenting for cancer care. Queens, which is one of the 5 boroughs of New York City, has more than 2.3 million residents. It has 2 public hospitals, Elmhurst Hospital Center and Queens Hospital Center (QHC). In 2001, the plan was devised for the establishment of a cancer center at QHC, mainly because of the high rate of late-stage cancers that were being seen at presentation and recognition of the need for more comprehensive care. In 2002, the Queens Cancer Center (QCC) began to see patients. QCC is a single facility that provides medical, surgical, radiation, gynecologic, and urologic oncology all in one area of the QHC.
This study is an investigation of the possible impact on care for breast cancer patients of low socioeconomic status who were treated at a comprehensive cancer center, with specific consideration of the change or improvement in treatment modalities and outcomes. Data on treatment modalities and outcomes of cancer patients who were treated at the QHC during 2000, before the QCC was set up, were compared with data of patients treated during 2008 (2008 was selected because we have 5-year survival data for those patients). The public hospital system treats all patients regardless of their ability to pay, so the majority of patients in the system are of lower socioeconomic status. In addition, 92% of the patients seen QHC are from a minority population. These are the populations that tend to have a worse prognosis and often are not given optimal treatment.3 The payer mix of patients in the public hospital system is different than that of private hospitals. Most of the patients present at the hospital with no insurance and if they are diagnosed with cancer they may be converted to emergency Medicaid. About 10% of patients will not be converted because of their document status.
Patients and methods
We used the Queens Hospital Tumor Registry to identify the patients who had been diagnosed with and treated for breast cancer in 2000 and 2008. The electronic medical records were reviewed, and in the case of the 2000-year patients, the written charts were also reviewed. The study was approved by the Mount Sinai institutional review board. It was not necessary to obtain patient consent because it was a retrospective study.
Only patients diagnosed with stage 0, I, II, or III breast cancer who received their treatment at QHC were included in the study. Patients who were seen in consultation at QHC but not treated there were excluded. Statistics were done using the 2x2 chi-squared SPSS analysis; a P value of .05 was considered significant. The survival data was analyzed using SAS.
Results
There were 24 evaluable patients in 2000 and 78 evaluable patients in 2008 who had stage 0, I, II, or III primary breast cancer and were treated at QHC. The average age of the patients in 2000 was 53.5 years and 54.7 years in 2008. The mean age for both groups was 55 years. The patients were ethnically diverse in both groups with 46% black, 17% Hispanic, 25% ethnic Asian Indian, and 6% white (Figure 1).
The payer mix in 2000 was 9 patients (37.5%) self-pay, 7 (29%) Medicaid, and 8 (33%) Medicare. In 2008, 11 patients (14%) were self-pay, 46 (59%) Medicaid, 11 (14%) Medicare, and 10 (13%) were private insurance. In 2000, there were 3 (12%) patients with stage 0 disease, 5 (21%) with stage I; 9 (37.5%) with stage II, and 7 (29%) with stage III. In 2008 there were 28 (36%) patients with stage 0 disease, 15 (19%) with stage I, 17 (22%) with stage II, and 18 (23%) with stage III (Figure 2).
None of those values are statistically different. In 2000, 2 of the 24 patients had lumpectomies (partial mastectomy) and the rest had mastectomies. In 2008, 39 (50%) patients had mastectomy and 39 (50%) had lumpectomies (Figure 3). This was a statistically significant difference.
Radiation was given to both patients with lumpectomy in the 2000 group. In the 2008 group, all patients with lumpectomies were evaluated for radiation, and 6 of them did not receive radiation for the following reasons: 3 had very small foci of ductal carcinoma in situ (DCIS) and were treated with hormone therapy and no radiation; 1 patient had a lumpectomy for stage 1 cancer and also did not get radiation therapy because of a low oncotype and very small lesion; 2 patients were older than 70 years and had DCIS and were treated with tamoxifen alone as per NCCN Guidelines for women in that age group. The rest of the patients with lumpectomies received postoperative radiation.
Hormone and HER2 (human epidermal growth factor receptor 2) status was obtained on all patients. For the 2000 patients, 71% had 1 hormone receptor–positive (estrogen receptor [ER] or progesterone receptor [PR]), 21% were triple negative (ER-PR and HER2-neu), and 42% had HER2-neu–positive tumors. For the 2008, patients 65% were positive for 1 hormone receptor (ER or PR), 28% were triple negative (ER-PR and HER2-neu), and 7% had HER2-neu-positive tumors.
All patients were offered chemotherapy and hormone therapy if appropriate, as per NCCN guidelines. If a patient’s tumor was found to be HER2-positive, then the chemotherapy regimen would include the use of trastuzumab in both groups.
The 5-year survival for the 2008 stage III patients was 73.7%, compared with 14.2% for the 2000 stage III patients. The only deaths in the 2008 group were in patients with stage III disease. In the 2000 group, 4 of the 5 patients with stage III cancer died, and 33% of patients with stage I or II either died or were lost to follow-up before 5 years. This survival difference is significant by a chi-square and Wilcoxon analysis, with a P value of .01.
In 2000, 86% of patients with cancer were termed self-pay, that is, they had no insurance and they were not converted to emergency Medicaid. In 2008, 16% of patients were self-pay, and the rest were converted to Medicaid. In 2000, fewer than 2% of patients had commercial insurance, compared with 9% in 2008.
Discussion
There have been numerous studies reporting on disparities in the treatment of patients with breast cancer based on race or socioeconomic status.4-18 Many studies have shown inferior survival for black women with breast cancer, but it is not entirely clear whether these differences are the result of the quality of medical care received or biologic differences.14,19 A moderately large study from a metropolitan medical center in Detroit showed no difference in survival in their patients based on race when all of the patients received equal treatments.15 A meta-analysis of survival in black and white breast cancer patients showed that the black women had significantly poorer outcomes.19
Findings from a recent study showed that patients of lower socioeconomic status are more likely to undergo mastectomy than breast conserving therapy.20 The study, which identified 727,927 patients with early-stage breast cancer during 1998-2011, found that the rate of breast conservation increased from 54% to 59% during that time period and that there were significant barriers to women receiving breast-conserving therapy based on their type of insurance and having a lower socioeconomic status.20
The treatment of breast cancer is best delivered in a multimodality setting, but many inner-city public hospitals do not have such a facility for their patients. QHC is the only public hospital in New York City that has established a comprehensive cancer center. The patient population of QHC is overwhelmingly of minority origin (only 5% of patients are white). In addition, it is a safety net hospital, so no patient is turned away because they cannot pay, and most patients are of lower socioeconomic status and do not have insurance. The purpose of the cancer center was to provide a single site at which our patients could receive all their treatment. It was to ensure that our patients had easy access to care and treatment during all phases of their disease trajectory and did not “fall through the cracks” of the system. Those goals were addressed by having all of the center’s physicians in one place. Physicians involved in care included medical, surgical, and radiation oncologists, a gynecologic oncologist, a genitourinary oncologist, and a thoracic surgery oncologist. The support groups organized for the cancer patients included 3 oncology social workers, an oncology navigator, a nutritionist, a pastoral care supporter, and an oncology psychologist, all located in the same area. All of the clerical and financial aspects of care were also placed within the center. This made the experience as seamless as possible for both the patients and the treating physicians. A “survivors clinic” was established so the cancer patients could be seen by integrated primary care providers to address all noncancer-related health issues such as hypertension, diabetes, or heart disease. Finally, a robust clinical oncology research team was established in the same location. The research included several protocols for new drug treatments for breast cancer from pharmaceutical companies as well as the multi-institutional oncology groups.
Part of the mission of the cancer center was to reach out into the community of Queens to provide education about early detection, cancer prevention, and other public health issues such as tobacco cessation. We established a close working relationship with the Queens Public Library System to connect with their users and dispense information about cancer care and early detection. The Queens Library system is the largest in the United States, and everyone who lives in Queens has easy access to one of its 63 branch libraries. We arranged several lectures about breast cancer awareness in some of the branch libraries. We also procured a mobile mammogram unit for free screening events at the lectures, especially in neighborhoods with a large number residents who were of lower socioeconomic status.
To study the possible effect of these changes on our patients with breast cancer, we compared 2 groups of patients. One group was from the year 2000, a year before the cancer center was opened. The other was from the year 2008, the last year we could get real 5-year survival statistics. We explored how establishing the cancer center might have changed the patients’ stage at diagnosis, care, treatment modalities such as type of surgery, and outcomes. It is difficult to compare these 2 groups because of differences in the patients’ cancers, such as their receptor status, as well as differences in treatment options between the two time periods. However, we had no other way to compare the data to see if there were any trends.
There was a migration to earlier-stage cancer at diagnosis during the 6-year period after the cancer center was opened. It is likely that the educational sessions that were done in the community contributed to this migration. We also saw an increase in the number of mammograms done, from 6,300 in 2000 to 8,800 in 2008. This increase in screening also could account for more patients being identified with earlier-stage disease and might be attributable to the community education through the outreach programs.
As a quality control method, the cancer center has been evaluated by the Commission on Cancer every 3 years. At the 2013 evaluation, we received the Gold Commendation – the highest possible recognition for having 8 out of 8 commendations – and a 3-year accreditation.
There was a notable increase in the use of lumpectomy over mastectomy after the establishment of the cancer center, possibly due to the addition of 2 surgical oncologists to the cancer center’s care team. The integration of multimodiality care for each patient may also have increased the use of breast-conserving surgery.
There was a significant increase from 2000 to 2008 in the survival of patients treated for stage III breast cancer. New drugs and new patterns of adjuvant care might have been partly responsible for that change. The establishment of the comprehensive cancer center with access to new protocols ensured that patients received state-of-the-art cancer treatment. Moreover, the facility addressed all aspects of patient care throughout the disease trajectory by including designated social workers, psychologists, a nutritionist, pastoral care, and patient and survivor support groups to ensure that patients would keep coming to the center for their therapy, with no delays and very little loss to follow-up.
Most patients without insurance were able to acquire emergency Medicaid through the cancer center. This was done by having 2 financial counselors who met with every patient and who could facilitate access to Medicaid as needed. As a result of that, the percentage of patients with no coverage went from 86% in 2000 to 16% in 2008. Before this system was set up, patients who were designated self-pay would pay a fee as low as $15 for each visit and received thousands of dollars’ worth of care. Thus, by forming a cancer center and facilitating patient access to Medicaid, we were able to save money for this public institution because of the gain in revenue from Medicaid.
Our findings suggest that the development of comprehensive cancer centers within inner-city health systems can ensure better treatment for patients of lower socioeconomic status. We present evidence that this may result in increased survival, more sophisticated surgical options, and better patient quality of life. Moreover, this can be achieved while effectively increasing revenue for the public hospitals. Correcting the inequality of access to care and better therapeutic options by setting up comprehensive cancer centers could contribute to improved parity of outcomes for underserved populations.
The author acknowledges the statistical help of Brian Altonen, MPH.
When cancer care is centralized in a comprehensive fashion, the quality of care and the outcomes improve.1,2 Unfortunately, because of the medical insurance structure in New York City, most patients of lower socioeconomic status do not receive their cancer care in such dedicated cancer centers. In New York City, the majority of the underserved vulnerable populations – that is, those without health insurance – receive their care from the public hospital system known as NYC Health and Hospitals. Cancer care in this system is not centralized and may result in fragmented implementation of various modalities of treatment. In addition, because there is no centralized care, needs such as early screening and prevention programs are often not addressed. This problem was evident in Queens in 2000 and before when many patients with late-stage cancers were presenting for cancer care. Queens, which is one of the 5 boroughs of New York City, has more than 2.3 million residents. It has 2 public hospitals, Elmhurst Hospital Center and Queens Hospital Center (QHC). In 2001, the plan was devised for the establishment of a cancer center at QHC, mainly because of the high rate of late-stage cancers that were being seen at presentation and recognition of the need for more comprehensive care. In 2002, the Queens Cancer Center (QCC) began to see patients. QCC is a single facility that provides medical, surgical, radiation, gynecologic, and urologic oncology all in one area of the QHC.
This study is an investigation of the possible impact on care for breast cancer patients of low socioeconomic status who were treated at a comprehensive cancer center, with specific consideration of the change or improvement in treatment modalities and outcomes. Data on treatment modalities and outcomes of cancer patients who were treated at the QHC during 2000, before the QCC was set up, were compared with data of patients treated during 2008 (2008 was selected because we have 5-year survival data for those patients). The public hospital system treats all patients regardless of their ability to pay, so the majority of patients in the system are of lower socioeconomic status. In addition, 92% of the patients seen QHC are from a minority population. These are the populations that tend to have a worse prognosis and often are not given optimal treatment.3 The payer mix of patients in the public hospital system is different than that of private hospitals. Most of the patients present at the hospital with no insurance and if they are diagnosed with cancer they may be converted to emergency Medicaid. About 10% of patients will not be converted because of their document status.
Patients and methods
We used the Queens Hospital Tumor Registry to identify the patients who had been diagnosed with and treated for breast cancer in 2000 and 2008. The electronic medical records were reviewed, and in the case of the 2000-year patients, the written charts were also reviewed. The study was approved by the Mount Sinai institutional review board. It was not necessary to obtain patient consent because it was a retrospective study.
Only patients diagnosed with stage 0, I, II, or III breast cancer who received their treatment at QHC were included in the study. Patients who were seen in consultation at QHC but not treated there were excluded. Statistics were done using the 2x2 chi-squared SPSS analysis; a P value of .05 was considered significant. The survival data was analyzed using SAS.
Results
There were 24 evaluable patients in 2000 and 78 evaluable patients in 2008 who had stage 0, I, II, or III primary breast cancer and were treated at QHC. The average age of the patients in 2000 was 53.5 years and 54.7 years in 2008. The mean age for both groups was 55 years. The patients were ethnically diverse in both groups with 46% black, 17% Hispanic, 25% ethnic Asian Indian, and 6% white (Figure 1).
The payer mix in 2000 was 9 patients (37.5%) self-pay, 7 (29%) Medicaid, and 8 (33%) Medicare. In 2008, 11 patients (14%) were self-pay, 46 (59%) Medicaid, 11 (14%) Medicare, and 10 (13%) were private insurance. In 2000, there were 3 (12%) patients with stage 0 disease, 5 (21%) with stage I; 9 (37.5%) with stage II, and 7 (29%) with stage III. In 2008 there were 28 (36%) patients with stage 0 disease, 15 (19%) with stage I, 17 (22%) with stage II, and 18 (23%) with stage III (Figure 2).
None of those values are statistically different. In 2000, 2 of the 24 patients had lumpectomies (partial mastectomy) and the rest had mastectomies. In 2008, 39 (50%) patients had mastectomy and 39 (50%) had lumpectomies (Figure 3). This was a statistically significant difference.
Radiation was given to both patients with lumpectomy in the 2000 group. In the 2008 group, all patients with lumpectomies were evaluated for radiation, and 6 of them did not receive radiation for the following reasons: 3 had very small foci of ductal carcinoma in situ (DCIS) and were treated with hormone therapy and no radiation; 1 patient had a lumpectomy for stage 1 cancer and also did not get radiation therapy because of a low oncotype and very small lesion; 2 patients were older than 70 years and had DCIS and were treated with tamoxifen alone as per NCCN Guidelines for women in that age group. The rest of the patients with lumpectomies received postoperative radiation.
Hormone and HER2 (human epidermal growth factor receptor 2) status was obtained on all patients. For the 2000 patients, 71% had 1 hormone receptor–positive (estrogen receptor [ER] or progesterone receptor [PR]), 21% were triple negative (ER-PR and HER2-neu), and 42% had HER2-neu–positive tumors. For the 2008, patients 65% were positive for 1 hormone receptor (ER or PR), 28% were triple negative (ER-PR and HER2-neu), and 7% had HER2-neu-positive tumors.
All patients were offered chemotherapy and hormone therapy if appropriate, as per NCCN guidelines. If a patient’s tumor was found to be HER2-positive, then the chemotherapy regimen would include the use of trastuzumab in both groups.
The 5-year survival for the 2008 stage III patients was 73.7%, compared with 14.2% for the 2000 stage III patients. The only deaths in the 2008 group were in patients with stage III disease. In the 2000 group, 4 of the 5 patients with stage III cancer died, and 33% of patients with stage I or II either died or were lost to follow-up before 5 years. This survival difference is significant by a chi-square and Wilcoxon analysis, with a P value of .01.
In 2000, 86% of patients with cancer were termed self-pay, that is, they had no insurance and they were not converted to emergency Medicaid. In 2008, 16% of patients were self-pay, and the rest were converted to Medicaid. In 2000, fewer than 2% of patients had commercial insurance, compared with 9% in 2008.
Discussion
There have been numerous studies reporting on disparities in the treatment of patients with breast cancer based on race or socioeconomic status.4-18 Many studies have shown inferior survival for black women with breast cancer, but it is not entirely clear whether these differences are the result of the quality of medical care received or biologic differences.14,19 A moderately large study from a metropolitan medical center in Detroit showed no difference in survival in their patients based on race when all of the patients received equal treatments.15 A meta-analysis of survival in black and white breast cancer patients showed that the black women had significantly poorer outcomes.19
Findings from a recent study showed that patients of lower socioeconomic status are more likely to undergo mastectomy than breast conserving therapy.20 The study, which identified 727,927 patients with early-stage breast cancer during 1998-2011, found that the rate of breast conservation increased from 54% to 59% during that time period and that there were significant barriers to women receiving breast-conserving therapy based on their type of insurance and having a lower socioeconomic status.20
The treatment of breast cancer is best delivered in a multimodality setting, but many inner-city public hospitals do not have such a facility for their patients. QHC is the only public hospital in New York City that has established a comprehensive cancer center. The patient population of QHC is overwhelmingly of minority origin (only 5% of patients are white). In addition, it is a safety net hospital, so no patient is turned away because they cannot pay, and most patients are of lower socioeconomic status and do not have insurance. The purpose of the cancer center was to provide a single site at which our patients could receive all their treatment. It was to ensure that our patients had easy access to care and treatment during all phases of their disease trajectory and did not “fall through the cracks” of the system. Those goals were addressed by having all of the center’s physicians in one place. Physicians involved in care included medical, surgical, and radiation oncologists, a gynecologic oncologist, a genitourinary oncologist, and a thoracic surgery oncologist. The support groups organized for the cancer patients included 3 oncology social workers, an oncology navigator, a nutritionist, a pastoral care supporter, and an oncology psychologist, all located in the same area. All of the clerical and financial aspects of care were also placed within the center. This made the experience as seamless as possible for both the patients and the treating physicians. A “survivors clinic” was established so the cancer patients could be seen by integrated primary care providers to address all noncancer-related health issues such as hypertension, diabetes, or heart disease. Finally, a robust clinical oncology research team was established in the same location. The research included several protocols for new drug treatments for breast cancer from pharmaceutical companies as well as the multi-institutional oncology groups.
Part of the mission of the cancer center was to reach out into the community of Queens to provide education about early detection, cancer prevention, and other public health issues such as tobacco cessation. We established a close working relationship with the Queens Public Library System to connect with their users and dispense information about cancer care and early detection. The Queens Library system is the largest in the United States, and everyone who lives in Queens has easy access to one of its 63 branch libraries. We arranged several lectures about breast cancer awareness in some of the branch libraries. We also procured a mobile mammogram unit for free screening events at the lectures, especially in neighborhoods with a large number residents who were of lower socioeconomic status.
To study the possible effect of these changes on our patients with breast cancer, we compared 2 groups of patients. One group was from the year 2000, a year before the cancer center was opened. The other was from the year 2008, the last year we could get real 5-year survival statistics. We explored how establishing the cancer center might have changed the patients’ stage at diagnosis, care, treatment modalities such as type of surgery, and outcomes. It is difficult to compare these 2 groups because of differences in the patients’ cancers, such as their receptor status, as well as differences in treatment options between the two time periods. However, we had no other way to compare the data to see if there were any trends.
There was a migration to earlier-stage cancer at diagnosis during the 6-year period after the cancer center was opened. It is likely that the educational sessions that were done in the community contributed to this migration. We also saw an increase in the number of mammograms done, from 6,300 in 2000 to 8,800 in 2008. This increase in screening also could account for more patients being identified with earlier-stage disease and might be attributable to the community education through the outreach programs.
As a quality control method, the cancer center has been evaluated by the Commission on Cancer every 3 years. At the 2013 evaluation, we received the Gold Commendation – the highest possible recognition for having 8 out of 8 commendations – and a 3-year accreditation.
There was a notable increase in the use of lumpectomy over mastectomy after the establishment of the cancer center, possibly due to the addition of 2 surgical oncologists to the cancer center’s care team. The integration of multimodiality care for each patient may also have increased the use of breast-conserving surgery.
There was a significant increase from 2000 to 2008 in the survival of patients treated for stage III breast cancer. New drugs and new patterns of adjuvant care might have been partly responsible for that change. The establishment of the comprehensive cancer center with access to new protocols ensured that patients received state-of-the-art cancer treatment. Moreover, the facility addressed all aspects of patient care throughout the disease trajectory by including designated social workers, psychologists, a nutritionist, pastoral care, and patient and survivor support groups to ensure that patients would keep coming to the center for their therapy, with no delays and very little loss to follow-up.
Most patients without insurance were able to acquire emergency Medicaid through the cancer center. This was done by having 2 financial counselors who met with every patient and who could facilitate access to Medicaid as needed. As a result of that, the percentage of patients with no coverage went from 86% in 2000 to 16% in 2008. Before this system was set up, patients who were designated self-pay would pay a fee as low as $15 for each visit and received thousands of dollars’ worth of care. Thus, by forming a cancer center and facilitating patient access to Medicaid, we were able to save money for this public institution because of the gain in revenue from Medicaid.
Our findings suggest that the development of comprehensive cancer centers within inner-city health systems can ensure better treatment for patients of lower socioeconomic status. We present evidence that this may result in increased survival, more sophisticated surgical options, and better patient quality of life. Moreover, this can be achieved while effectively increasing revenue for the public hospitals. Correcting the inequality of access to care and better therapeutic options by setting up comprehensive cancer centers could contribute to improved parity of outcomes for underserved populations.
The author acknowledges the statistical help of Brian Altonen, MPH.
1. Kesson EM, Allardice GM, George WD, Morrison DS. Effects of multidisciplinary team working on breast cancer survival: retrospective, comparative, interventional cohort study of 13,722 women. BMJ. 2012;344:e2718.
2. Vrijens F, Stordeur S, Beirens K, Devriese S, Van Eycken E, Vlayen J. Effect of hospital volume on processes of care and 5-year survival after breast cancer: a population-based study on 25000 women. Breast. 2012;21(3):261-266.
3. Bradley CJ, Given CW, Roberts C. Race, socioeconomic status and breast cancer treatment and survival. J Natl Cancer Inst. 2002;94(7):490-496.
4. Wheeler SB, Hayes-Reeder KE, Carey LA. Disparities in breast cancer treatment and outcomes: biological, social, and health system determinants and opportunities for research. Oncologist. 2013;18:986-993.
5. Ward E, Jemal A, Cokkinides V, et al. Cancer disparities by race/ethnicity and socioeconomic status. CA Cancer J Clin. 2004;54:78-93.
6. Chen F, Puig M, Yermilov I, et al. Using breast cancer quality indicators in a vulnerable population. Cancer. 2011;117:3311-3321.
7. Banerjee M, George J, Yee C, Hryniuk W, Schwartz K. Disentangling the effects of race on breast cancer treatment. Cancer. 2007;110:2169-2177.
8. Freedman RA, He Y, Winer EP, Keating NL. Trends in racial and age disparities in definitive local therapy of early-stage breast cancer. J Clin Oncol. 2009;27:713-719.
9. Bickell NA, Shastri K, Fei K, et al. A tracking and feedback registry to reduce racial disparities in breast cancer care. J Natl Cancer Inst. 2008;100:1717-1723.
19. Bickell NA, Wang JJ, Oluwole S, et al. Missed opportunities: Racial disparities in adjuvant breast cancer treatment. J Clin Oncol. 2006;24:1357-1362.
11. Harper S, Lynch J, Meersman SC, Breen N, Davis WW, Reichman MC. Trends in area-socioeconomic and race-ethnic disparities in breast cancer incidence, stage at diagnosis, screening, mortality, and survival among women ages 50 years and over (1987-2005). Cancer Epidemiol Biomarkers Prev. 2009;18:121-131.
12. Ward E, Halpern M, Schrag N, et al. Association of insurance with cancer care utilization and outcomes. CA Cancer J Clin. 2008;58:9-31.
13. Naik AM, Joseph K, Harris M, Davis C, Shapiro R, Hiotis KL. Indigent breast cancer patients among all racial and ethnic groups present with more advanced disease compared with nationally reported date. Am J Surg. 2003;186:400-403.
14. Hersman DL, Unger JM, Barlow WE, et al. Treatment quality and outcomes of African American versus white breast cancer patients: retrospective analysis of southwest oncology studies S8814/S8897. J Clin Oncol. 2009;27: 2157-2162.
15
16. Brawley OW. Disaggregating the effects of race and poverty on breast cancer outcomes. J Natl Cancer Inst. 2002;94:471-473.
17. Baquet CR, Commiskey P. Socioeconomic factors and breast carcinoma in multicultural women. Cancer. 2000;88:1256-1264.
18. Cross C, Harris J, Recht A. Race, socioeconomic status, and breast carcinoma in the US. Cancer. 2002;95:1988-1999.
19. Newman LA, Griffith KA, Jatoi I, Simon MS, Crowe JP, Colditz GA. Meta-analysis of survival in African American and white American patients with breast cancer: Ethnicity compared with socioeconomic status. J Clin Oncol. 2006;24:1342-1349.
20. Lautner M, Lin H, Shen Y, et al. Disparities in the use of breast-conserving therapy among patients with early-stage breast cancer. JAMA. 2015;150:778-786.
1. Kesson EM, Allardice GM, George WD, Morrison DS. Effects of multidisciplinary team working on breast cancer survival: retrospective, comparative, interventional cohort study of 13,722 women. BMJ. 2012;344:e2718.
2. Vrijens F, Stordeur S, Beirens K, Devriese S, Van Eycken E, Vlayen J. Effect of hospital volume on processes of care and 5-year survival after breast cancer: a population-based study on 25000 women. Breast. 2012;21(3):261-266.
3. Bradley CJ, Given CW, Roberts C. Race, socioeconomic status and breast cancer treatment and survival. J Natl Cancer Inst. 2002;94(7):490-496.
4. Wheeler SB, Hayes-Reeder KE, Carey LA. Disparities in breast cancer treatment and outcomes: biological, social, and health system determinants and opportunities for research. Oncologist. 2013;18:986-993.
5. Ward E, Jemal A, Cokkinides V, et al. Cancer disparities by race/ethnicity and socioeconomic status. CA Cancer J Clin. 2004;54:78-93.
6. Chen F, Puig M, Yermilov I, et al. Using breast cancer quality indicators in a vulnerable population. Cancer. 2011;117:3311-3321.
7. Banerjee M, George J, Yee C, Hryniuk W, Schwartz K. Disentangling the effects of race on breast cancer treatment. Cancer. 2007;110:2169-2177.
8. Freedman RA, He Y, Winer EP, Keating NL. Trends in racial and age disparities in definitive local therapy of early-stage breast cancer. J Clin Oncol. 2009;27:713-719.
9. Bickell NA, Shastri K, Fei K, et al. A tracking and feedback registry to reduce racial disparities in breast cancer care. J Natl Cancer Inst. 2008;100:1717-1723.
19. Bickell NA, Wang JJ, Oluwole S, et al. Missed opportunities: Racial disparities in adjuvant breast cancer treatment. J Clin Oncol. 2006;24:1357-1362.
11. Harper S, Lynch J, Meersman SC, Breen N, Davis WW, Reichman MC. Trends in area-socioeconomic and race-ethnic disparities in breast cancer incidence, stage at diagnosis, screening, mortality, and survival among women ages 50 years and over (1987-2005). Cancer Epidemiol Biomarkers Prev. 2009;18:121-131.
12. Ward E, Halpern M, Schrag N, et al. Association of insurance with cancer care utilization and outcomes. CA Cancer J Clin. 2008;58:9-31.
13. Naik AM, Joseph K, Harris M, Davis C, Shapiro R, Hiotis KL. Indigent breast cancer patients among all racial and ethnic groups present with more advanced disease compared with nationally reported date. Am J Surg. 2003;186:400-403.
14. Hersman DL, Unger JM, Barlow WE, et al. Treatment quality and outcomes of African American versus white breast cancer patients: retrospective analysis of southwest oncology studies S8814/S8897. J Clin Oncol. 2009;27: 2157-2162.
15
16. Brawley OW. Disaggregating the effects of race and poverty on breast cancer outcomes. J Natl Cancer Inst. 2002;94:471-473.
17. Baquet CR, Commiskey P. Socioeconomic factors and breast carcinoma in multicultural women. Cancer. 2000;88:1256-1264.
18. Cross C, Harris J, Recht A. Race, socioeconomic status, and breast carcinoma in the US. Cancer. 2002;95:1988-1999.
19. Newman LA, Griffith KA, Jatoi I, Simon MS, Crowe JP, Colditz GA. Meta-analysis of survival in African American and white American patients with breast cancer: Ethnicity compared with socioeconomic status. J Clin Oncol. 2006;24:1342-1349.
20. Lautner M, Lin H, Shen Y, et al. Disparities in the use of breast-conserving therapy among patients with early-stage breast cancer. JAMA. 2015;150:778-786.