User login
AAP: MenB vaccines are safe for healthy adolescents, young adults
The serotype B meningococcal vaccines MenB-FHbp and MenB-4C are safe and can be administered to healthy people aged 10-25 years, according to a policy statement from the American Academy of Pediatrics Committee on Infectious Diseases.
The AAP recommends that people older than 10 years at increased risk for serogroup B meningococcal disease (category A) should receive MenB vaccines regularly. Category A includes people with persistent complement component deficiencies, people with anatomic or functional asplenia, and healthy people at increased risk because of a disease outbreak.

Young adults aged 16-23 years old may receive a vaccination, but it is not routinely recommended (category B), with a preferred vaccination age between 16 and 18 years.
Annual incidence of serogroup B meningococcal disease in people aged 11-24 years in the United States is about 50-60 cases per year, and a routine vaccination program would prevent 15-29 cases and 2-5 deaths per year, the researchers noted. The cost of routine vaccination in the general population would range from $3.7 million per quality-adjusted life year (QALY) to $9.4 million per QALY.
Both MenB-FHbp and MenB-4C have been safely administered in clinical trials, with no deaths related to either vaccine. Data on duration of immunogenicity and proportion of MenB strains covered by vaccines in different geographic regions remain incomplete, and both vaccine manufacturers must complete postmarketing studies to determine overall vaccine effectiveness.
“Pediatricians are encouraged to discuss the availability of the MenB vaccines with families. Discussion should include the low incidence of MenB disease and the unknown efficacy of the vaccines... The treating clinician should discuss the benefits, risks, and costs with patients and their families and then work with them to determine what is in their best interest,” the AAP committee noted.
Find the full study in Pediatrics (doi: 10.1542/peds.2016-1890).
The serotype B meningococcal vaccines MenB-FHbp and MenB-4C are safe and can be administered to healthy people aged 10-25 years, according to a policy statement from the American Academy of Pediatrics Committee on Infectious Diseases.
The AAP recommends that people older than 10 years at increased risk for serogroup B meningococcal disease (category A) should receive MenB vaccines regularly. Category A includes people with persistent complement component deficiencies, people with anatomic or functional asplenia, and healthy people at increased risk because of a disease outbreak.
Young adults aged 16-23 years old may receive a vaccination, but it is not routinely recommended (category B), with a preferred vaccination age between 16 and 18 years.
Annual incidence of serogroup B meningococcal disease in people aged 11-24 years in the United States is about 50-60 cases per year, and a routine vaccination program would prevent 15-29 cases and 2-5 deaths per year, the researchers noted. The cost of routine vaccination in the general population would range from $3.7 million per quality-adjusted life year (QALY) to $9.4 million per QALY.
Both MenB-FHbp and MenB-4C have been safely administered in clinical trials, with no deaths related to either vaccine. Data on duration of immunogenicity and proportion of MenB strains covered by vaccines in different geographic regions remain incomplete, and both vaccine manufacturers must complete postmarketing studies to determine overall vaccine effectiveness.
“Pediatricians are encouraged to discuss the availability of the MenB vaccines with families. Discussion should include the low incidence of MenB disease and the unknown efficacy of the vaccines... The treating clinician should discuss the benefits, risks, and costs with patients and their families and then work with them to determine what is in their best interest,” the AAP committee noted.
Find the full study in Pediatrics (doi: 10.1542/peds.2016-1890).
The serotype B meningococcal vaccines MenB-FHbp and MenB-4C are safe and can be administered to healthy people aged 10-25 years, according to a policy statement from the American Academy of Pediatrics Committee on Infectious Diseases.
The AAP recommends that people older than 10 years at increased risk for serogroup B meningococcal disease (category A) should receive MenB vaccines regularly. Category A includes people with persistent complement component deficiencies, people with anatomic or functional asplenia, and healthy people at increased risk because of a disease outbreak.
Young adults aged 16-23 years old may receive a vaccination, but it is not routinely recommended (category B), with a preferred vaccination age between 16 and 18 years.
Annual incidence of serogroup B meningococcal disease in people aged 11-24 years in the United States is about 50-60 cases per year, and a routine vaccination program would prevent 15-29 cases and 2-5 deaths per year, the researchers noted. The cost of routine vaccination in the general population would range from $3.7 million per quality-adjusted life year (QALY) to $9.4 million per QALY.
Both MenB-FHbp and MenB-4C have been safely administered in clinical trials, with no deaths related to either vaccine. Data on duration of immunogenicity and proportion of MenB strains covered by vaccines in different geographic regions remain incomplete, and both vaccine manufacturers must complete postmarketing studies to determine overall vaccine effectiveness.
“Pediatricians are encouraged to discuss the availability of the MenB vaccines with families. Discussion should include the low incidence of MenB disease and the unknown efficacy of the vaccines... The treating clinician should discuss the benefits, risks, and costs with patients and their families and then work with them to determine what is in their best interest,” the AAP committee noted.
Find the full study in Pediatrics (doi: 10.1542/peds.2016-1890).
FROM PEDIATRICS

Investigator-Reported Efficacy of Azelaic Acid Foam 15% in Patients With Papulopustular Rosacea: Secondary Efficacy Outcomes From a Randomized, Controlled, Double-blind, Phase 3 Trial
Papulopustular rosacea (PPR) is characterized by centrofacial papules, pustules, erythema, and occasionally telangiectasia.1,2 A myriad of factors, including genetic predisposition3 and environmental triggers,4 have been associated with dysregulated inflammatory responses,5 contributing to the disease pathogenesis and symptoms. Inflammation associated with PPR may decrease skin barrier function, increase transepidermal water loss, and reduce stratum corneum hydration,6,7 resulting in heightened skin sensitivity, pain, burning, and/or stinging.5,8
Azelaic acid (AzA), which historically has only been available in gel or cream formulations, is well established for the treatment of rosacea9; however, these formulations have been associated with application-site adverse events (AEs)(eg, burning, erythema, irritation), limited cosmetic acceptability, and reduced compliance or efficacy.10
For select skin conditions, active agents delivered in foam vehicles may offer superior tolerability with improved outcomes.11 An AzA foam 15% formulation was approved for the treatment of mild to moderate PPR. Primary outcomes from a phase 3 trial demonstrated the efficacy and safety of AzA foam in improving inflammatory lesion counts (ILCs) and disease severity in participants with PPR. The trial also evaluated additional secondary end points, including the effect of AzA foam on erythema, inflammatory lesions, treatment response, and other manifestations of PPR.12 The current study evaluated investigator-reported efficacy outcomes for these secondary end points for AzA foam 15% versus vehicle foam.
Methods
Study Design
This phase 3 multicenter, randomized, double-blind, vehicle-controlled, parallel-group clinical trial was conducted from September 2012 to January 2014 at 48 US study centers comparing the efficacy of AzA foam versus vehicle foam in patients with PPR. Eligible participants were 18 years and older with PPR rated as moderate or severe according to investigator global assessment (IGA), plus 12 to 50 inflammatory lesions and persistent erythema with or without telangiectasia. Exclusion criteria included known nonresponse to AzA, current or prior use (within 6 weeks of randomization) of noninvestigational products to treat rosacea, and presence of other dermatoses that could interfere with rosacea evaluation.
Participants were randomized into the AzA foam or vehicle group (1:1 ratio). The study medication was applied in 0.5-g doses twice daily until the end of treatment (EoT) at 12 weeks. Efficacy and safety parameters were evaluated at baseline and at 4, 8, and 12 weeks of treatment, and at a follow-up visit 4 weeks after EoT (week 16).
Results for the coprimary efficacy end points—therapeutic success rate according to IGA and nominal change in ILC—were previously reported.12
Investigator-Reported Secondary Efficacy Outcomes
The secondary efficacy end points were grouped change in erythema rating, grouped change in telangiectasia rating, grouped change in IGA score, therapeutic response rate according to IGA, percentage change in ILC from baseline, and facial skin color rating at EoT.
Grouped change for all secondary end points was measured as improved, no change, or worsened relative to baseline. For grouped change in erythema and telangiectasia ratings, a participant was considered improved if the rating at the postbaseline visit was lower than the baseline rating, no change if the postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline. For grouped change in IGA score, a participant was considered improved if a responder showed at least a 1-step improvement postbaseline compared to baseline, no change if postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline.
For the therapeutic response rate, a participant was considered a treatment responder if the IGA score improved from baseline and resulted in clear, minimal, or mild disease severity at EoT.
Safety
Adverse events also were assessed.
Statistical Analyses
Secondary efficacy and safety end points were assessed for all randomized participants who were dispensed the study medication. Missing data were imputed using last observation carried forward.
For the percentage change in ILC from baseline, therapeutic response rate, and grouped change in erythema rating, confirmatory analyses were conducted in a hierarchical manner (in the order listed), with testing stopped as soon as a null hypothesis of superior treatment effect could not be rejected. Analyses without significance level were exploratory. The Cochran-Mantel-Haenszel van Elteren test stratified by study center was used for grouped change in erythema rating (1-tailed, 2.5%) and IGA score (2-tailed, 5%); Wilcoxon rank sum tests also were performed. Percentage change in ILC from baseline was evaluated using the Student t test and F test of analysis of covariance (1-tailed, 2.5%). Therapeutic response rate was evaluated using the Cochran-Mantel-Haenszel van Elteren test stratified by study center and the Pearson χ2 test. Facial skin color and grouped change in telangiectasia rating were evaluated using the Wilcoxon rank sum test.
Adverse events beginning or worsening after the first dose of the study drug were considered treatment emergent and were coded using the Medical Dictionary for Regulatory Activities (MedDRA) Version 16.1. Statistical analyses were performed using SAS software version 9.2.
Results
Study Participants
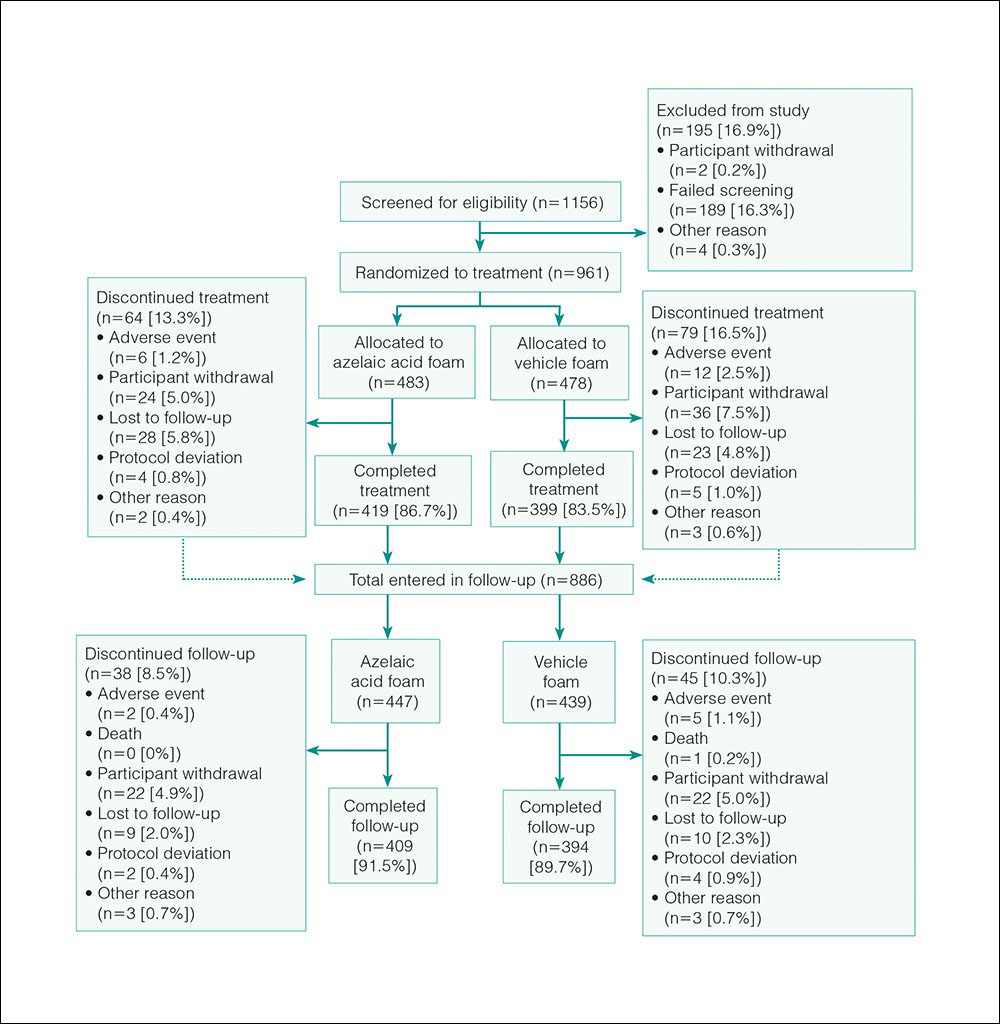
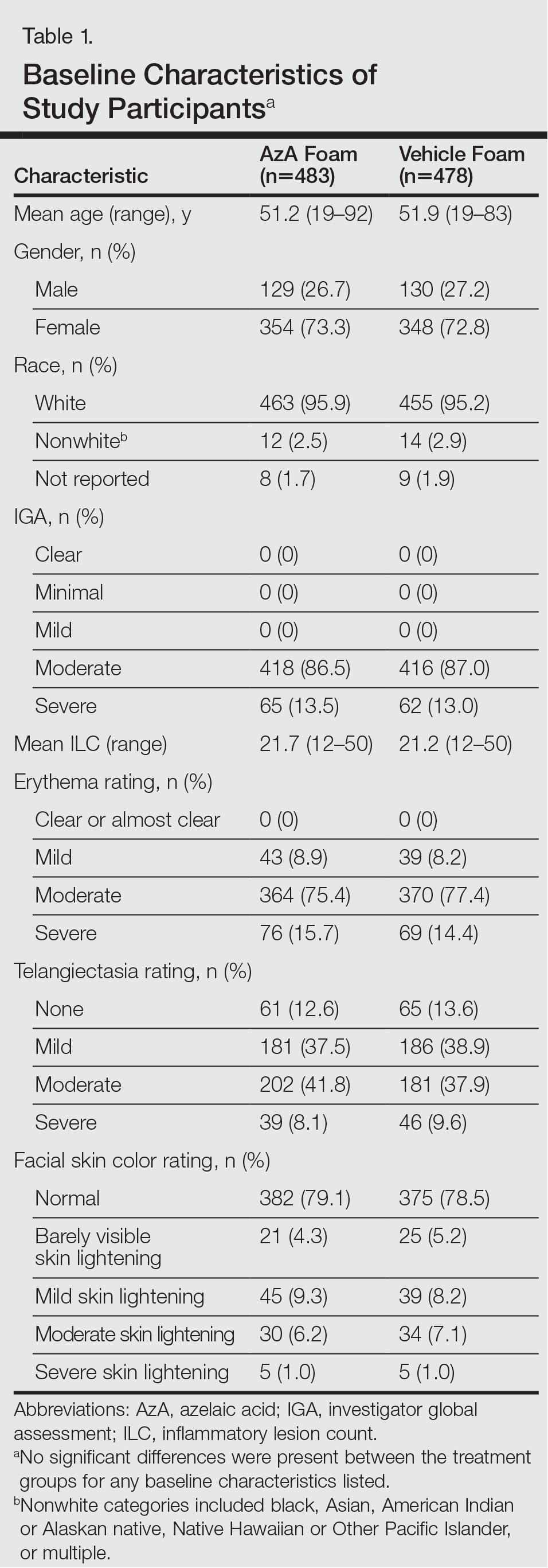
The study included 961 total participants; 483 were randomized to the AzA foam group and 478 to the vehicle group (Figure 1). Overall, 803 participants completed follow-up; however, week 16 results for the efficacy outcomes include data for 4 additional patients (2 per study arm) who did not formally meet all requirements for follow-up completion. The mean age was 51.5 years, and the majority of the participants were white and female (Table 1). Most participants (86.8%) had moderate PPR at baseline, with the remaining rated as having severe disease (13.2%). The majority (76.4%) had more than 14 inflammatory lesions with moderate (76.4%) or severe (15.1%) erythema at baseline.
Efficacy
Significantly more participants in the AzA group than in the vehicle group showed an improved erythema rating at EoT (61.5% vs 51.3%; P<.001)(Figure 2), with more participants in the AzA group showing improvement at weeks 4 (P=.022) and 8 (P=.002).
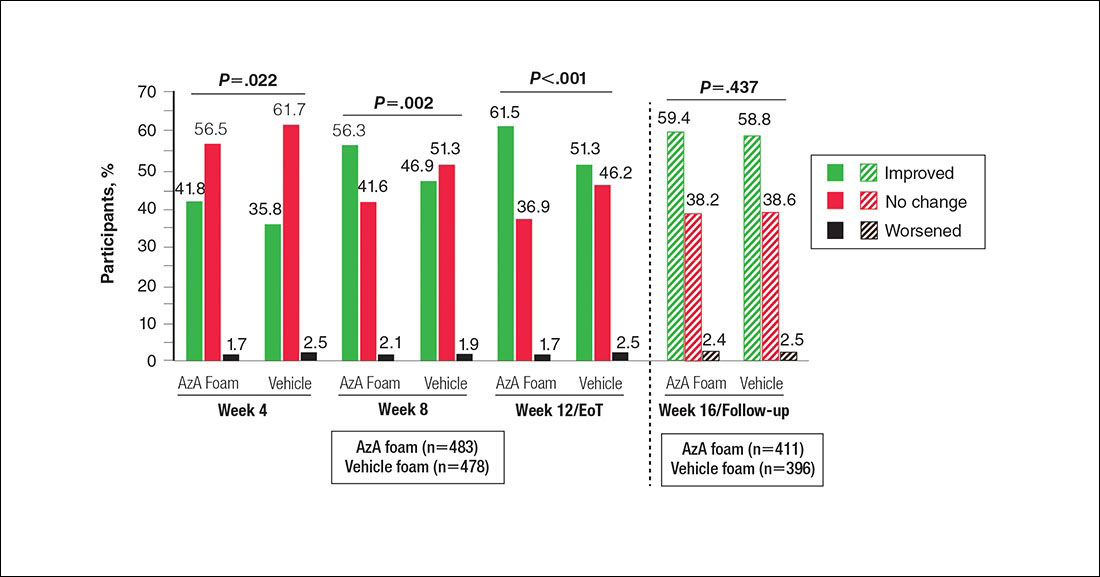
A significantly greater mean percentage reduction in ILC from baseline to EoT was observed in the AzA group versus the vehicle group (61.6% vs 50.8%; P<.001)(Figure 3), and between-group differences were observed at week 4 (P<.001), week 8 (P=.003), and week 16 (end of study/follow-up)(P=.002).
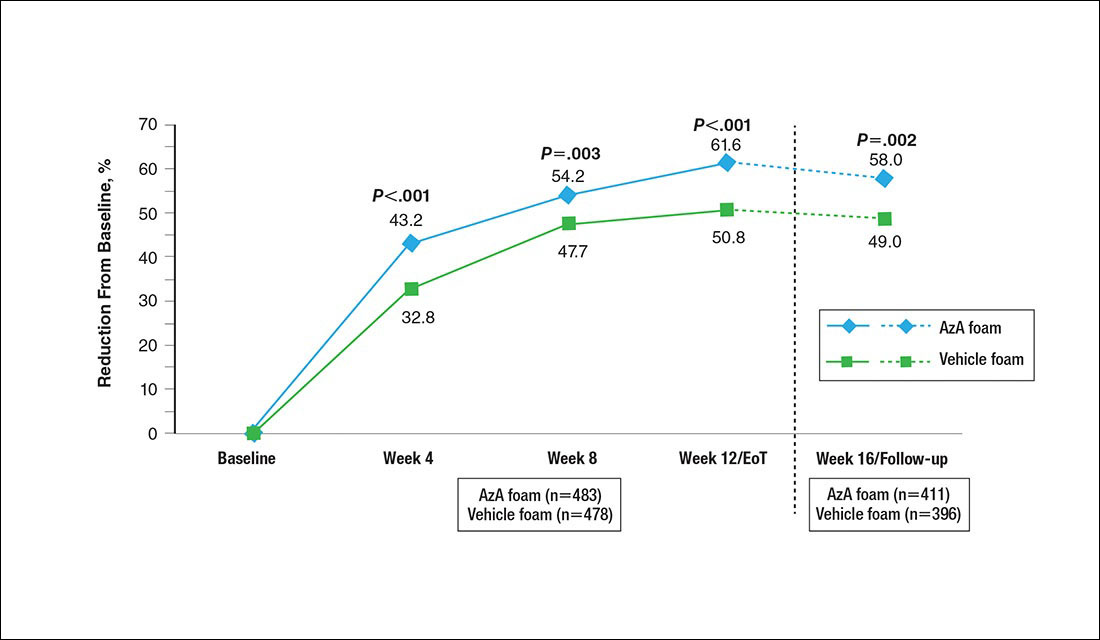
A significantly higher proportion of participants treated with AzA foam versus vehicle were considered responders at week 12/EoT (66.3% vs 54.4%; P<.001)(Figure 4). Differences in responder rate also were observed at week 4 (P=.026) and week 8 (P=.026).
No study drug was administered between week 12/EoT and week 16/follow-up; last observation carried forward was not applied to week 16/follow-up analysis. AzA indicates azelaic acid; IGA, investigator global assessment.
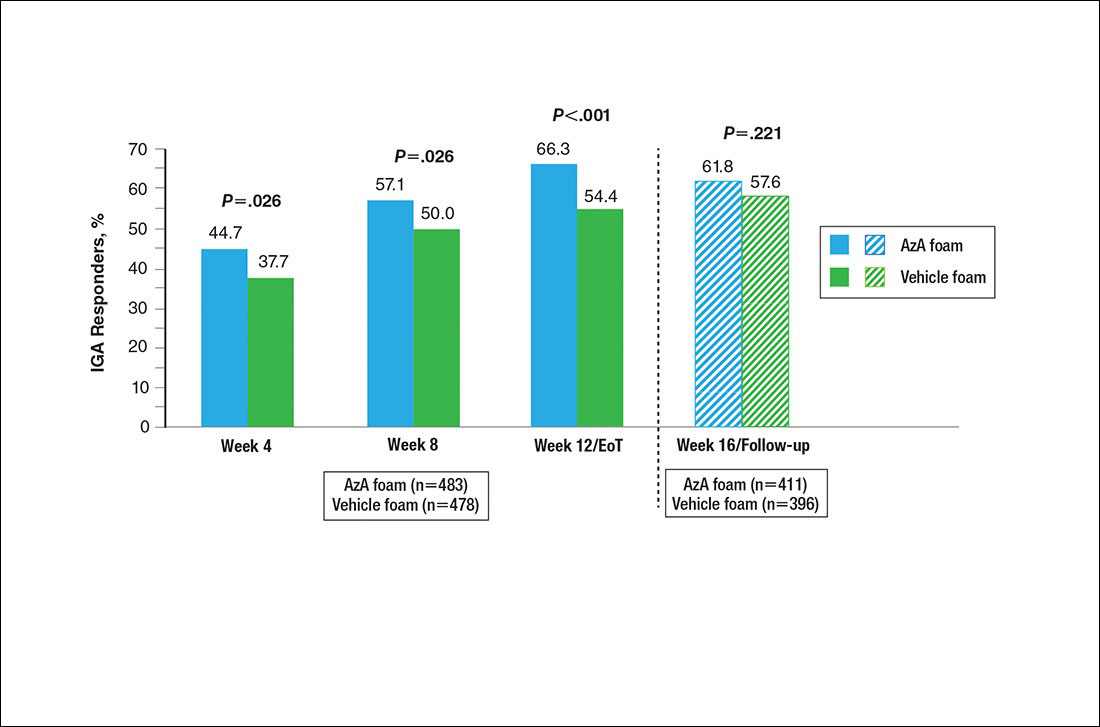
Differences in grouped change in IGA score were observed between groups at every evaluation during the treatment phase (Figure 5). Specifically, IGA score was improved at week 12/EoT relative to baseline in 71.2% of participants in the AzA group versus 58.8% in the vehicle group (P<.001).
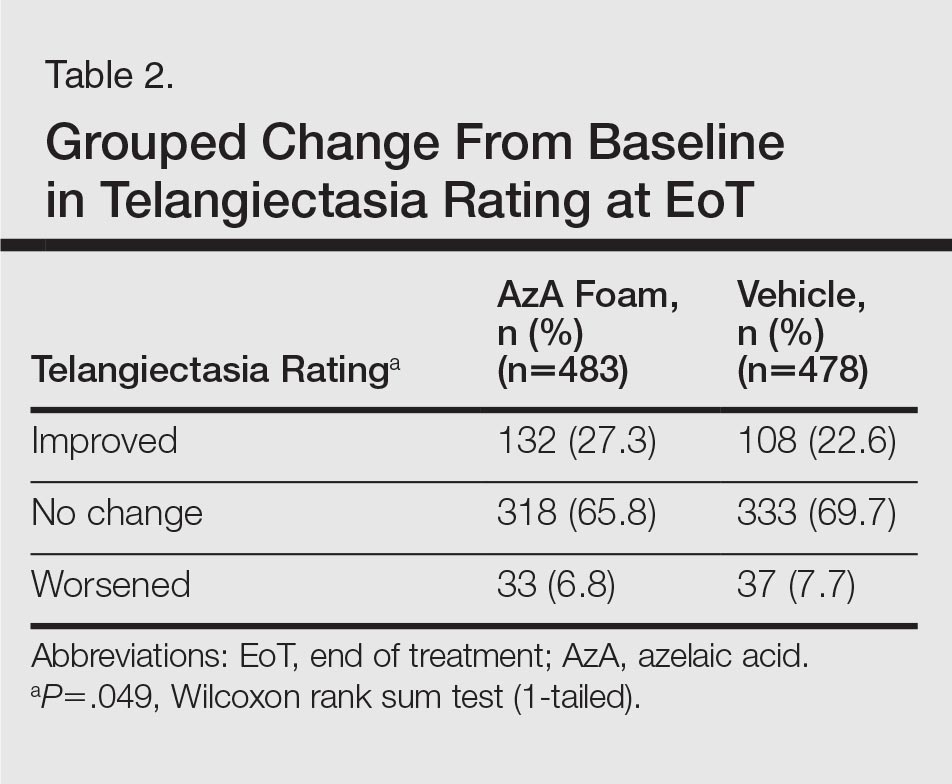
For grouped change in telangiectasia rating at EoT, the majority of participants in both treatment groups showed no change (Table 2). Regarding facial skin color, the majority of participants in both the AzA and vehicle treatment groups (80.1% and 78.7%, respectively) showed normal skin color compared to nontreated skin EoT; no between-group differences were detected for facial skin color rating (P=.315, Wilcoxon rank sum test).
Safety
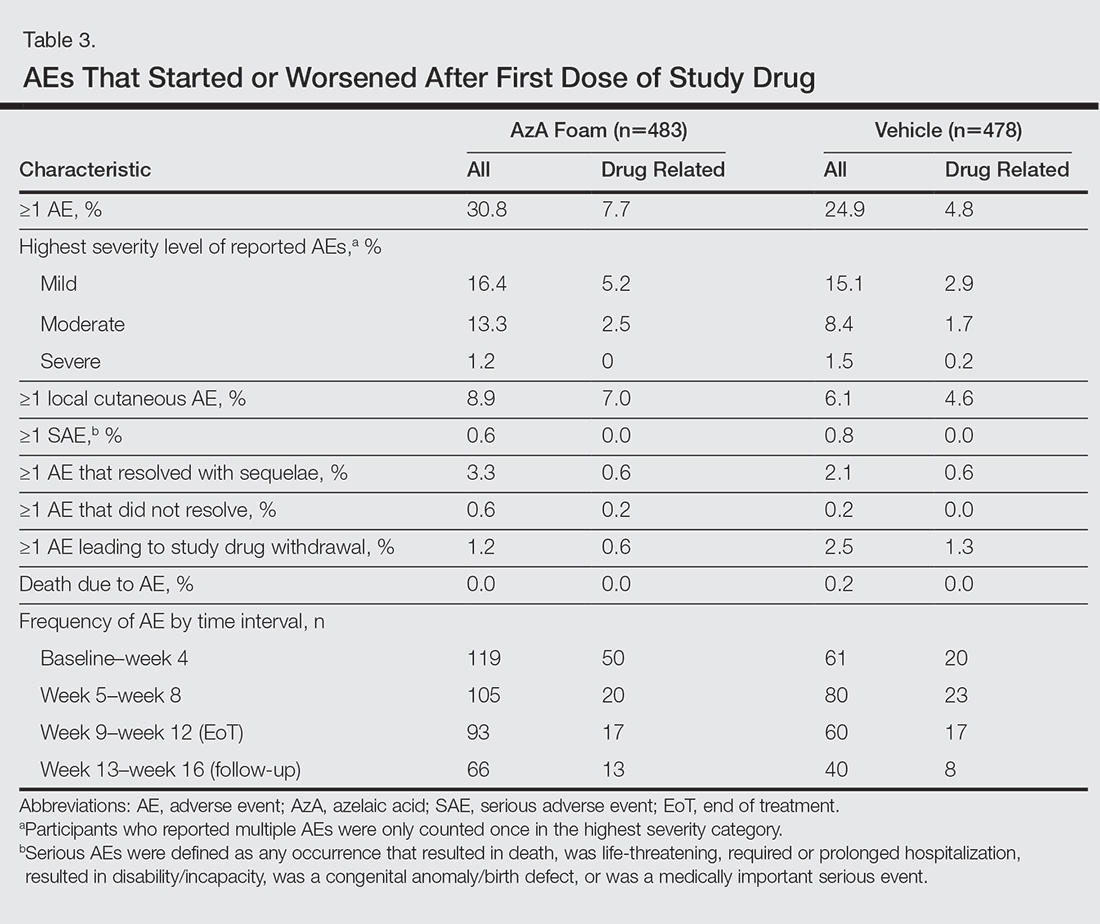
The incidence of drug-related AEs was greater in the AzA group than the vehicle group (7.7% vs 4.8%)(Table 3). Drug-related AEs occurring in at least 1% of the AzA group were pain at application site (eg, tenderness, stinging, burning)(AzA group, 3.5%; vehicle group, 1.3%), application-site pruritus (1.4% vs 0.4%), and application-site dryness (1.0% vs 0.6%). A single drug-related AE of severe intensity (ie, application-site dermatitis) was observed in the vehicle group; all other drug-related AEs were mild or moderate. The incidence of withdrawals due to AEs was lower in the AzA group than the vehicle group (1.2% vs 2.5%). This AE profile correlated with a treatment compliance (the percentage of expected doses that were actually administered) of 97.0% in the AzA group and 95.9% in the vehicle group. One participant in the vehicle group died due to head trauma unrelated to administration of the study drug.
Comment
The results of this study further support the efficacy of AzA foam for the treatment of PPR. The percentage reduction in ILC was consistent with nominal decreases in ILC, a coprimary efficacy end point of this study.12 Almost two-thirds of participants treated with AzA foam achieved a therapeutic response, indicating that many participants who did not strictly achieve the primary outcome of therapeutic success nevertheless attained notable reductions in disease severity. The number of participants who showed any improvement on the IGA scale increased throughout the course of treatment (63.8% AzA foam vs 55.0% vehicle at week 8) up to EoT (71.2% vs 58.8%)(Figure 5). In addition, the number of participants showing any improvement at week 8 (63.8% AzA foam vs 55.0% vehicle)(Figure 5) was comparable to the number of participants achieving therapeutic response at week 12/EoT (66.3% vs 54.4%)(Figure 4). These data suggest that increasing time of treatment increases the likelihood of achieving better results.
Erythema also appeared to respond to AzA foam, with 10.2% more participants in the AzA group demonstrating improvement at week 12/EoT compared to vehicle. The difference in grouped change in erythema rating also was statistically significant and favored AzA foam, sustained up to 4 weeks after EoT.
The outcomes for percentage change in ILC, therapeutic response rate, and grouped change in erythema rating consequently led to the rejection of all 3 null hypotheses in hierarchical confirmatory analyses, underscoring the benefits of AzA foam treatment.
The therapeutic effects of AzA foam were apparent at the first postbaseline evaluation and persisted throughout treatment. Differences favoring AzA foam were observed at every on-treatment evaluation for grouped change in erythema rating, percentage change in ILC, therapeutic response rate, and grouped change in IGA score. Symptoms showed minimal resurgence after treatment cessation, and there were no signs of disease flare-up within the 4 weeks of observational follow-up. In addition, the percentage reduction in ILC remained higher in the AzA foam group during follow-up.
These results also show that AzA foam was well tolerated with a low incidence of discontinuation because of drug-related AEs. No serious drug-related AEs were reported for this study or in the preceding phase 2 trial.12,13 Although not directly evaluated, the low incidence of cutaneous AEs suggests that AzA foam may be better tolerated than prior formulations of AzA14,15 and correlates with high compliance observed during the study.12 Azelaic acid foam appeared to have minimal to no effect on skin color, with more than 88% of participants reporting barely visible or no skin lightening.
Interestingly, the vehicle foam showed appreciable efficacy independent of AzA. Improvements in erythema were recorded in approximately half of the vehicle group at week 12/EoT. A similar proportion attained a therapeutic response, and ILC was reduced by 50.8% at week 12/EoT. Comparable results also were evident in the vehicle group for the primary end points of this study.12 Vehicles in dermatologic trials frequently exert effects on diseased skin16,17 via a skin care regimen effect (eg, moisturization and other vehicle-related effects that may improve skin barrier integrity and function) and thus should not be regarded as placebo controls. The mechanism underlying this efficacy may be due to the impact of vehicle composition on skin barrier integrity and transepidermal water loss.18 The hydrophilic emulsion or other constituents of AzA foam (eg, fatty alcohols) may play a role.
A notable strength of our study is detailed clinical characterization using carefully chosen parameters and preplanned analyses that complement the primary end points. As the latter are often driven by regulatory requirements, opportunities to characterize other outcomes of interest to clinicians may be missed. The additional analyses reported here hopefully will aid dermatologists in both assessing the role of AzA foam in the treatment armamentarium for PPR and counseling patients.
Because participants with lighter skin pigmentation dominated our study population, the impact of AzA foam among patients with darker skin complexions is unknown. Although AzA is unlikely to cause hypopigmentation in normal undiseased skin, patients should be monitored for early signs of hypopigmentation.19,20 Our data also do not allow assessment of the differential effect, if any, of AzA foam on erythema of different etiologies in PPR, as corresponding information was not collected in the trial.
Conclusion
Azelaic acid foam 15% combines a well-established treatment of PPR with new vehicle technology to deliver effective therapy across multiple disease dimensions. In addition, the vehicle foam appears to demonstrate inherent therapeutic properties independent of AzA. The availability of this novel, efficacious, and well-tolerated option for PPR has the potential to improve patient care, reduce disease burden, and minimize unnecessary costs through increased tolerability and compliance.21
Acknowledgment
Editorial support through inVentiv Medical Communications (New York, New York) was provided by Bayer Pharmaceuticals.
- Tan J, Berg M. Rosacea: current state of epidemiology. J Am Acad Dermatol. 2013;69(6, suppl 1):S27-S35.
- Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the classification and staging of rosacea. J Am Acad Dermatol. 2002;46:584-587.
- Chang AL, Raber I, Xu J, et al. Assessment of the genetic basis of rosacea by genome-wide association study. J Invest Dermatol. 2015;135:1548-1555.
- Abram K, Silm H, Maaroos HI, et al. Risk factors associated with rosacea. J Eur Acad Dermatol Venereol. 2010;24:565-571.
- Yamasaki K, Di Nardo A, Bardan A, et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med. 2007;13:975-980.
- Yamasaki K, Kanada K, Macleod DT, et al. TLR2 expression is increased in rosacea and stimulates enhanced serine protease production by keratinocytes. J Invest Dermatol. 2011;131:688-697.
- Darlenski R, Kazandjieva J, Tsankov N, et al. Acute irritant threshold correlates with barrier function, skin hydration and contact hypersensitivity in atopic dermatitis and rosacea. Exp Dermatol. 2013;22:752-753.
- Del Rosso JQ, Levin J. The clinical relevance of maintaining the functional integrity of the stratum corneum in both healthy and disease-affected skin. J Clin Aesthet Dermatol. 2011;4:22-42.
- van Zuuren EJ, Kramer SF, Carter BR, et al. Effective and evidence-based management strategies for rosacea: summary of a Cochrane systematic review. Br J Dermatol. 2011;165:760-781.
- Tan X, Feldman SR, Chang J, et al. Topical drug delivery systems in dermatology: a review of patient adherence issues. Expert Opin Drug Deliv. 2012;9:1263-1271.
- Stein L. Clinical studies of a new vehicle formulation for topical corticosteroids in the treatment of psoriasis. J Am Acad Dermatol. 2005;53(1, suppl 1):S39-S49.
- Draelos ZD, Elewski BE, Harper JC, et al. A phase 3 randomized, double-blind, vehicle-controlled trial of azelaic acid foam 15% in the treatment of papulopustular rosacea. Cutis. 2015;96:54-61.
- Draelos ZD, Elewski B, Staedtler G, et al. Azelaic acid foam 15% in the treatment of papulopustular rosacea: a randomized, double-blind, vehicle-controlled study. Cutis. 2013;92:306-317.
- Finacea gel [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2016.
- Elewski BE, Fleischer AB Jr, Pariser DM. A comparison of 15% azelaic acid gel and 0.75% metronidazole gel in the topical treatment of papulopustular rosacea: results of a randomized trial. Arch Dermatol. 2003;139:1444-1450.
- Daniels R, Knie U. Galenics of dermal products—vehicles, properties and drug release. J Dtsch Dermatol Ges. 2007;5:367-383.
- Shamsudin N, Fleischer AB Jr. Vehicle or placebo? Investigators use incorrect terminology in randomized controlled trials half of the time: a systematic review of randomized controlled trials published in three major dermatology journals. J Drugs Dermatol. 2010;9:1221-1226.
- Del Rosso JQ, Thiboutot D, Gallo R, et al. Consensus recommendations from the American Acne & Rosacea Society on the management of rosacea, part 2: a status report on topical agents. Cutis. 2013;92:277-284.
- Finacea foam [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2015.
- Solano F, Briganti S, Picardo M, et al. Hypopigmenting agents: an updated review on biological, chemical and clinical aspects. Pigment Cell Res. 2006;19:550-571.
- Hammarstrom B, Wessling A, Nilsson JL. Pharmaceutical care for patients with skin diseases: a campaign year at Swedish pharmacies. J Clin Pharm Ther. 1995;20:327-334.
Papulopustular rosacea (PPR) is characterized by centrofacial papules, pustules, erythema, and occasionally telangiectasia.1,2 A myriad of factors, including genetic predisposition3 and environmental triggers,4 have been associated with dysregulated inflammatory responses,5 contributing to the disease pathogenesis and symptoms. Inflammation associated with PPR may decrease skin barrier function, increase transepidermal water loss, and reduce stratum corneum hydration,6,7 resulting in heightened skin sensitivity, pain, burning, and/or stinging.5,8
Azelaic acid (AzA), which historically has only been available in gel or cream formulations, is well established for the treatment of rosacea9; however, these formulations have been associated with application-site adverse events (AEs)(eg, burning, erythema, irritation), limited cosmetic acceptability, and reduced compliance or efficacy.10
For select skin conditions, active agents delivered in foam vehicles may offer superior tolerability with improved outcomes.11 An AzA foam 15% formulation was approved for the treatment of mild to moderate PPR. Primary outcomes from a phase 3 trial demonstrated the efficacy and safety of AzA foam in improving inflammatory lesion counts (ILCs) and disease severity in participants with PPR. The trial also evaluated additional secondary end points, including the effect of AzA foam on erythema, inflammatory lesions, treatment response, and other manifestations of PPR.12 The current study evaluated investigator-reported efficacy outcomes for these secondary end points for AzA foam 15% versus vehicle foam.
Methods
Study Design
This phase 3 multicenter, randomized, double-blind, vehicle-controlled, parallel-group clinical trial was conducted from September 2012 to January 2014 at 48 US study centers comparing the efficacy of AzA foam versus vehicle foam in patients with PPR. Eligible participants were 18 years and older with PPR rated as moderate or severe according to investigator global assessment (IGA), plus 12 to 50 inflammatory lesions and persistent erythema with or without telangiectasia. Exclusion criteria included known nonresponse to AzA, current or prior use (within 6 weeks of randomization) of noninvestigational products to treat rosacea, and presence of other dermatoses that could interfere with rosacea evaluation.
Participants were randomized into the AzA foam or vehicle group (1:1 ratio). The study medication was applied in 0.5-g doses twice daily until the end of treatment (EoT) at 12 weeks. Efficacy and safety parameters were evaluated at baseline and at 4, 8, and 12 weeks of treatment, and at a follow-up visit 4 weeks after EoT (week 16).
Results for the coprimary efficacy end points—therapeutic success rate according to IGA and nominal change in ILC—were previously reported.12
Investigator-Reported Secondary Efficacy Outcomes
The secondary efficacy end points were grouped change in erythema rating, grouped change in telangiectasia rating, grouped change in IGA score, therapeutic response rate according to IGA, percentage change in ILC from baseline, and facial skin color rating at EoT.
Grouped change for all secondary end points was measured as improved, no change, or worsened relative to baseline. For grouped change in erythema and telangiectasia ratings, a participant was considered improved if the rating at the postbaseline visit was lower than the baseline rating, no change if the postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline. For grouped change in IGA score, a participant was considered improved if a responder showed at least a 1-step improvement postbaseline compared to baseline, no change if postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline.
For the therapeutic response rate, a participant was considered a treatment responder if the IGA score improved from baseline and resulted in clear, minimal, or mild disease severity at EoT.
Safety
Adverse events also were assessed.
Statistical Analyses
Secondary efficacy and safety end points were assessed for all randomized participants who were dispensed the study medication. Missing data were imputed using last observation carried forward.
For the percentage change in ILC from baseline, therapeutic response rate, and grouped change in erythema rating, confirmatory analyses were conducted in a hierarchical manner (in the order listed), with testing stopped as soon as a null hypothesis of superior treatment effect could not be rejected. Analyses without significance level were exploratory. The Cochran-Mantel-Haenszel van Elteren test stratified by study center was used for grouped change in erythema rating (1-tailed, 2.5%) and IGA score (2-tailed, 5%); Wilcoxon rank sum tests also were performed. Percentage change in ILC from baseline was evaluated using the Student t test and F test of analysis of covariance (1-tailed, 2.5%). Therapeutic response rate was evaluated using the Cochran-Mantel-Haenszel van Elteren test stratified by study center and the Pearson χ2 test. Facial skin color and grouped change in telangiectasia rating were evaluated using the Wilcoxon rank sum test.
Adverse events beginning or worsening after the first dose of the study drug were considered treatment emergent and were coded using the Medical Dictionary for Regulatory Activities (MedDRA) Version 16.1. Statistical analyses were performed using SAS software version 9.2.
Results
Study Participants
The study included 961 total participants; 483 were randomized to the AzA foam group and 478 to the vehicle group (Figure 1). Overall, 803 participants completed follow-up; however, week 16 results for the efficacy outcomes include data for 4 additional patients (2 per study arm) who did not formally meet all requirements for follow-up completion. The mean age was 51.5 years, and the majority of the participants were white and female (Table 1). Most participants (86.8%) had moderate PPR at baseline, with the remaining rated as having severe disease (13.2%). The majority (76.4%) had more than 14 inflammatory lesions with moderate (76.4%) or severe (15.1%) erythema at baseline.
Efficacy
Significantly more participants in the AzA group than in the vehicle group showed an improved erythema rating at EoT (61.5% vs 51.3%; P<.001)(Figure 2), with more participants in the AzA group showing improvement at weeks 4 (P=.022) and 8 (P=.002).
A significantly greater mean percentage reduction in ILC from baseline to EoT was observed in the AzA group versus the vehicle group (61.6% vs 50.8%; P<.001)(Figure 3), and between-group differences were observed at week 4 (P<.001), week 8 (P=.003), and week 16 (end of study/follow-up)(P=.002).
A significantly higher proportion of participants treated with AzA foam versus vehicle were considered responders at week 12/EoT (66.3% vs 54.4%; P<.001)(Figure 4). Differences in responder rate also were observed at week 4 (P=.026) and week 8 (P=.026).
No study drug was administered between week 12/EoT and week 16/follow-up; last observation carried forward was not applied to week 16/follow-up analysis. AzA indicates azelaic acid; IGA, investigator global assessment.
Differences in grouped change in IGA score were observed between groups at every evaluation during the treatment phase (Figure 5). Specifically, IGA score was improved at week 12/EoT relative to baseline in 71.2% of participants in the AzA group versus 58.8% in the vehicle group (P<.001).
For grouped change in telangiectasia rating at EoT, the majority of participants in both treatment groups showed no change (Table 2). Regarding facial skin color, the majority of participants in both the AzA and vehicle treatment groups (80.1% and 78.7%, respectively) showed normal skin color compared to nontreated skin EoT; no between-group differences were detected for facial skin color rating (P=.315, Wilcoxon rank sum test).
Safety
The incidence of drug-related AEs was greater in the AzA group than the vehicle group (7.7% vs 4.8%)(Table 3). Drug-related AEs occurring in at least 1% of the AzA group were pain at application site (eg, tenderness, stinging, burning)(AzA group, 3.5%; vehicle group, 1.3%), application-site pruritus (1.4% vs 0.4%), and application-site dryness (1.0% vs 0.6%). A single drug-related AE of severe intensity (ie, application-site dermatitis) was observed in the vehicle group; all other drug-related AEs were mild or moderate. The incidence of withdrawals due to AEs was lower in the AzA group than the vehicle group (1.2% vs 2.5%). This AE profile correlated with a treatment compliance (the percentage of expected doses that were actually administered) of 97.0% in the AzA group and 95.9% in the vehicle group. One participant in the vehicle group died due to head trauma unrelated to administration of the study drug.
Comment
The results of this study further support the efficacy of AzA foam for the treatment of PPR. The percentage reduction in ILC was consistent with nominal decreases in ILC, a coprimary efficacy end point of this study.12 Almost two-thirds of participants treated with AzA foam achieved a therapeutic response, indicating that many participants who did not strictly achieve the primary outcome of therapeutic success nevertheless attained notable reductions in disease severity. The number of participants who showed any improvement on the IGA scale increased throughout the course of treatment (63.8% AzA foam vs 55.0% vehicle at week 8) up to EoT (71.2% vs 58.8%)(Figure 5). In addition, the number of participants showing any improvement at week 8 (63.8% AzA foam vs 55.0% vehicle)(Figure 5) was comparable to the number of participants achieving therapeutic response at week 12/EoT (66.3% vs 54.4%)(Figure 4). These data suggest that increasing time of treatment increases the likelihood of achieving better results.
Erythema also appeared to respond to AzA foam, with 10.2% more participants in the AzA group demonstrating improvement at week 12/EoT compared to vehicle. The difference in grouped change in erythema rating also was statistically significant and favored AzA foam, sustained up to 4 weeks after EoT.
The outcomes for percentage change in ILC, therapeutic response rate, and grouped change in erythema rating consequently led to the rejection of all 3 null hypotheses in hierarchical confirmatory analyses, underscoring the benefits of AzA foam treatment.
The therapeutic effects of AzA foam were apparent at the first postbaseline evaluation and persisted throughout treatment. Differences favoring AzA foam were observed at every on-treatment evaluation for grouped change in erythema rating, percentage change in ILC, therapeutic response rate, and grouped change in IGA score. Symptoms showed minimal resurgence after treatment cessation, and there were no signs of disease flare-up within the 4 weeks of observational follow-up. In addition, the percentage reduction in ILC remained higher in the AzA foam group during follow-up.
These results also show that AzA foam was well tolerated with a low incidence of discontinuation because of drug-related AEs. No serious drug-related AEs were reported for this study or in the preceding phase 2 trial.12,13 Although not directly evaluated, the low incidence of cutaneous AEs suggests that AzA foam may be better tolerated than prior formulations of AzA14,15 and correlates with high compliance observed during the study.12 Azelaic acid foam appeared to have minimal to no effect on skin color, with more than 88% of participants reporting barely visible or no skin lightening.
Interestingly, the vehicle foam showed appreciable efficacy independent of AzA. Improvements in erythema were recorded in approximately half of the vehicle group at week 12/EoT. A similar proportion attained a therapeutic response, and ILC was reduced by 50.8% at week 12/EoT. Comparable results also were evident in the vehicle group for the primary end points of this study.12 Vehicles in dermatologic trials frequently exert effects on diseased skin16,17 via a skin care regimen effect (eg, moisturization and other vehicle-related effects that may improve skin barrier integrity and function) and thus should not be regarded as placebo controls. The mechanism underlying this efficacy may be due to the impact of vehicle composition on skin barrier integrity and transepidermal water loss.18 The hydrophilic emulsion or other constituents of AzA foam (eg, fatty alcohols) may play a role.
A notable strength of our study is detailed clinical characterization using carefully chosen parameters and preplanned analyses that complement the primary end points. As the latter are often driven by regulatory requirements, opportunities to characterize other outcomes of interest to clinicians may be missed. The additional analyses reported here hopefully will aid dermatologists in both assessing the role of AzA foam in the treatment armamentarium for PPR and counseling patients.
Because participants with lighter skin pigmentation dominated our study population, the impact of AzA foam among patients with darker skin complexions is unknown. Although AzA is unlikely to cause hypopigmentation in normal undiseased skin, patients should be monitored for early signs of hypopigmentation.19,20 Our data also do not allow assessment of the differential effect, if any, of AzA foam on erythema of different etiologies in PPR, as corresponding information was not collected in the trial.
Conclusion
Azelaic acid foam 15% combines a well-established treatment of PPR with new vehicle technology to deliver effective therapy across multiple disease dimensions. In addition, the vehicle foam appears to demonstrate inherent therapeutic properties independent of AzA. The availability of this novel, efficacious, and well-tolerated option for PPR has the potential to improve patient care, reduce disease burden, and minimize unnecessary costs through increased tolerability and compliance.21
Acknowledgment
Editorial support through inVentiv Medical Communications (New York, New York) was provided by Bayer Pharmaceuticals.
Papulopustular rosacea (PPR) is characterized by centrofacial papules, pustules, erythema, and occasionally telangiectasia.1,2 A myriad of factors, including genetic predisposition3 and environmental triggers,4 have been associated with dysregulated inflammatory responses,5 contributing to the disease pathogenesis and symptoms. Inflammation associated with PPR may decrease skin barrier function, increase transepidermal water loss, and reduce stratum corneum hydration,6,7 resulting in heightened skin sensitivity, pain, burning, and/or stinging.5,8
Azelaic acid (AzA), which historically has only been available in gel or cream formulations, is well established for the treatment of rosacea9; however, these formulations have been associated with application-site adverse events (AEs)(eg, burning, erythema, irritation), limited cosmetic acceptability, and reduced compliance or efficacy.10
For select skin conditions, active agents delivered in foam vehicles may offer superior tolerability with improved outcomes.11 An AzA foam 15% formulation was approved for the treatment of mild to moderate PPR. Primary outcomes from a phase 3 trial demonstrated the efficacy and safety of AzA foam in improving inflammatory lesion counts (ILCs) and disease severity in participants with PPR. The trial also evaluated additional secondary end points, including the effect of AzA foam on erythema, inflammatory lesions, treatment response, and other manifestations of PPR.12 The current study evaluated investigator-reported efficacy outcomes for these secondary end points for AzA foam 15% versus vehicle foam.
Methods
Study Design
This phase 3 multicenter, randomized, double-blind, vehicle-controlled, parallel-group clinical trial was conducted from September 2012 to January 2014 at 48 US study centers comparing the efficacy of AzA foam versus vehicle foam in patients with PPR. Eligible participants were 18 years and older with PPR rated as moderate or severe according to investigator global assessment (IGA), plus 12 to 50 inflammatory lesions and persistent erythema with or without telangiectasia. Exclusion criteria included known nonresponse to AzA, current or prior use (within 6 weeks of randomization) of noninvestigational products to treat rosacea, and presence of other dermatoses that could interfere with rosacea evaluation.
Participants were randomized into the AzA foam or vehicle group (1:1 ratio). The study medication was applied in 0.5-g doses twice daily until the end of treatment (EoT) at 12 weeks. Efficacy and safety parameters were evaluated at baseline and at 4, 8, and 12 weeks of treatment, and at a follow-up visit 4 weeks after EoT (week 16).
Results for the coprimary efficacy end points—therapeutic success rate according to IGA and nominal change in ILC—were previously reported.12
Investigator-Reported Secondary Efficacy Outcomes
The secondary efficacy end points were grouped change in erythema rating, grouped change in telangiectasia rating, grouped change in IGA score, therapeutic response rate according to IGA, percentage change in ILC from baseline, and facial skin color rating at EoT.
Grouped change for all secondary end points was measured as improved, no change, or worsened relative to baseline. For grouped change in erythema and telangiectasia ratings, a participant was considered improved if the rating at the postbaseline visit was lower than the baseline rating, no change if the postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline. For grouped change in IGA score, a participant was considered improved if a responder showed at least a 1-step improvement postbaseline compared to baseline, no change if postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline.
For the therapeutic response rate, a participant was considered a treatment responder if the IGA score improved from baseline and resulted in clear, minimal, or mild disease severity at EoT.
Safety
Adverse events also were assessed.
Statistical Analyses
Secondary efficacy and safety end points were assessed for all randomized participants who were dispensed the study medication. Missing data were imputed using last observation carried forward.
For the percentage change in ILC from baseline, therapeutic response rate, and grouped change in erythema rating, confirmatory analyses were conducted in a hierarchical manner (in the order listed), with testing stopped as soon as a null hypothesis of superior treatment effect could not be rejected. Analyses without significance level were exploratory. The Cochran-Mantel-Haenszel van Elteren test stratified by study center was used for grouped change in erythema rating (1-tailed, 2.5%) and IGA score (2-tailed, 5%); Wilcoxon rank sum tests also were performed. Percentage change in ILC from baseline was evaluated using the Student t test and F test of analysis of covariance (1-tailed, 2.5%). Therapeutic response rate was evaluated using the Cochran-Mantel-Haenszel van Elteren test stratified by study center and the Pearson χ2 test. Facial skin color and grouped change in telangiectasia rating were evaluated using the Wilcoxon rank sum test.
Adverse events beginning or worsening after the first dose of the study drug were considered treatment emergent and were coded using the Medical Dictionary for Regulatory Activities (MedDRA) Version 16.1. Statistical analyses were performed using SAS software version 9.2.
Results
Study Participants
The study included 961 total participants; 483 were randomized to the AzA foam group and 478 to the vehicle group (Figure 1). Overall, 803 participants completed follow-up; however, week 16 results for the efficacy outcomes include data for 4 additional patients (2 per study arm) who did not formally meet all requirements for follow-up completion. The mean age was 51.5 years, and the majority of the participants were white and female (Table 1). Most participants (86.8%) had moderate PPR at baseline, with the remaining rated as having severe disease (13.2%). The majority (76.4%) had more than 14 inflammatory lesions with moderate (76.4%) or severe (15.1%) erythema at baseline.
Efficacy
Significantly more participants in the AzA group than in the vehicle group showed an improved erythema rating at EoT (61.5% vs 51.3%; P<.001)(Figure 2), with more participants in the AzA group showing improvement at weeks 4 (P=.022) and 8 (P=.002).
A significantly greater mean percentage reduction in ILC from baseline to EoT was observed in the AzA group versus the vehicle group (61.6% vs 50.8%; P<.001)(Figure 3), and between-group differences were observed at week 4 (P<.001), week 8 (P=.003), and week 16 (end of study/follow-up)(P=.002).
A significantly higher proportion of participants treated with AzA foam versus vehicle were considered responders at week 12/EoT (66.3% vs 54.4%; P<.001)(Figure 4). Differences in responder rate also were observed at week 4 (P=.026) and week 8 (P=.026).
No study drug was administered between week 12/EoT and week 16/follow-up; last observation carried forward was not applied to week 16/follow-up analysis. AzA indicates azelaic acid; IGA, investigator global assessment.
Differences in grouped change in IGA score were observed between groups at every evaluation during the treatment phase (Figure 5). Specifically, IGA score was improved at week 12/EoT relative to baseline in 71.2% of participants in the AzA group versus 58.8% in the vehicle group (P<.001).
For grouped change in telangiectasia rating at EoT, the majority of participants in both treatment groups showed no change (Table 2). Regarding facial skin color, the majority of participants in both the AzA and vehicle treatment groups (80.1% and 78.7%, respectively) showed normal skin color compared to nontreated skin EoT; no between-group differences were detected for facial skin color rating (P=.315, Wilcoxon rank sum test).
Safety
The incidence of drug-related AEs was greater in the AzA group than the vehicle group (7.7% vs 4.8%)(Table 3). Drug-related AEs occurring in at least 1% of the AzA group were pain at application site (eg, tenderness, stinging, burning)(AzA group, 3.5%; vehicle group, 1.3%), application-site pruritus (1.4% vs 0.4%), and application-site dryness (1.0% vs 0.6%). A single drug-related AE of severe intensity (ie, application-site dermatitis) was observed in the vehicle group; all other drug-related AEs were mild or moderate. The incidence of withdrawals due to AEs was lower in the AzA group than the vehicle group (1.2% vs 2.5%). This AE profile correlated with a treatment compliance (the percentage of expected doses that were actually administered) of 97.0% in the AzA group and 95.9% in the vehicle group. One participant in the vehicle group died due to head trauma unrelated to administration of the study drug.
Comment
The results of this study further support the efficacy of AzA foam for the treatment of PPR. The percentage reduction in ILC was consistent with nominal decreases in ILC, a coprimary efficacy end point of this study.12 Almost two-thirds of participants treated with AzA foam achieved a therapeutic response, indicating that many participants who did not strictly achieve the primary outcome of therapeutic success nevertheless attained notable reductions in disease severity. The number of participants who showed any improvement on the IGA scale increased throughout the course of treatment (63.8% AzA foam vs 55.0% vehicle at week 8) up to EoT (71.2% vs 58.8%)(Figure 5). In addition, the number of participants showing any improvement at week 8 (63.8% AzA foam vs 55.0% vehicle)(Figure 5) was comparable to the number of participants achieving therapeutic response at week 12/EoT (66.3% vs 54.4%)(Figure 4). These data suggest that increasing time of treatment increases the likelihood of achieving better results.
Erythema also appeared to respond to AzA foam, with 10.2% more participants in the AzA group demonstrating improvement at week 12/EoT compared to vehicle. The difference in grouped change in erythema rating also was statistically significant and favored AzA foam, sustained up to 4 weeks after EoT.
The outcomes for percentage change in ILC, therapeutic response rate, and grouped change in erythema rating consequently led to the rejection of all 3 null hypotheses in hierarchical confirmatory analyses, underscoring the benefits of AzA foam treatment.
The therapeutic effects of AzA foam were apparent at the first postbaseline evaluation and persisted throughout treatment. Differences favoring AzA foam were observed at every on-treatment evaluation for grouped change in erythema rating, percentage change in ILC, therapeutic response rate, and grouped change in IGA score. Symptoms showed minimal resurgence after treatment cessation, and there were no signs of disease flare-up within the 4 weeks of observational follow-up. In addition, the percentage reduction in ILC remained higher in the AzA foam group during follow-up.
These results also show that AzA foam was well tolerated with a low incidence of discontinuation because of drug-related AEs. No serious drug-related AEs were reported for this study or in the preceding phase 2 trial.12,13 Although not directly evaluated, the low incidence of cutaneous AEs suggests that AzA foam may be better tolerated than prior formulations of AzA14,15 and correlates with high compliance observed during the study.12 Azelaic acid foam appeared to have minimal to no effect on skin color, with more than 88% of participants reporting barely visible or no skin lightening.
Interestingly, the vehicle foam showed appreciable efficacy independent of AzA. Improvements in erythema were recorded in approximately half of the vehicle group at week 12/EoT. A similar proportion attained a therapeutic response, and ILC was reduced by 50.8% at week 12/EoT. Comparable results also were evident in the vehicle group for the primary end points of this study.12 Vehicles in dermatologic trials frequently exert effects on diseased skin16,17 via a skin care regimen effect (eg, moisturization and other vehicle-related effects that may improve skin barrier integrity and function) and thus should not be regarded as placebo controls. The mechanism underlying this efficacy may be due to the impact of vehicle composition on skin barrier integrity and transepidermal water loss.18 The hydrophilic emulsion or other constituents of AzA foam (eg, fatty alcohols) may play a role.
A notable strength of our study is detailed clinical characterization using carefully chosen parameters and preplanned analyses that complement the primary end points. As the latter are often driven by regulatory requirements, opportunities to characterize other outcomes of interest to clinicians may be missed. The additional analyses reported here hopefully will aid dermatologists in both assessing the role of AzA foam in the treatment armamentarium for PPR and counseling patients.
Because participants with lighter skin pigmentation dominated our study population, the impact of AzA foam among patients with darker skin complexions is unknown. Although AzA is unlikely to cause hypopigmentation in normal undiseased skin, patients should be monitored for early signs of hypopigmentation.19,20 Our data also do not allow assessment of the differential effect, if any, of AzA foam on erythema of different etiologies in PPR, as corresponding information was not collected in the trial.
Conclusion
Azelaic acid foam 15% combines a well-established treatment of PPR with new vehicle technology to deliver effective therapy across multiple disease dimensions. In addition, the vehicle foam appears to demonstrate inherent therapeutic properties independent of AzA. The availability of this novel, efficacious, and well-tolerated option for PPR has the potential to improve patient care, reduce disease burden, and minimize unnecessary costs through increased tolerability and compliance.21
Acknowledgment
Editorial support through inVentiv Medical Communications (New York, New York) was provided by Bayer Pharmaceuticals.
- Tan J, Berg M. Rosacea: current state of epidemiology. J Am Acad Dermatol. 2013;69(6, suppl 1):S27-S35.
- Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the classification and staging of rosacea. J Am Acad Dermatol. 2002;46:584-587.
- Chang AL, Raber I, Xu J, et al. Assessment of the genetic basis of rosacea by genome-wide association study. J Invest Dermatol. 2015;135:1548-1555.
- Abram K, Silm H, Maaroos HI, et al. Risk factors associated with rosacea. J Eur Acad Dermatol Venereol. 2010;24:565-571.
- Yamasaki K, Di Nardo A, Bardan A, et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med. 2007;13:975-980.
- Yamasaki K, Kanada K, Macleod DT, et al. TLR2 expression is increased in rosacea and stimulates enhanced serine protease production by keratinocytes. J Invest Dermatol. 2011;131:688-697.
- Darlenski R, Kazandjieva J, Tsankov N, et al. Acute irritant threshold correlates with barrier function, skin hydration and contact hypersensitivity in atopic dermatitis and rosacea. Exp Dermatol. 2013;22:752-753.
- Del Rosso JQ, Levin J. The clinical relevance of maintaining the functional integrity of the stratum corneum in both healthy and disease-affected skin. J Clin Aesthet Dermatol. 2011;4:22-42.
- van Zuuren EJ, Kramer SF, Carter BR, et al. Effective and evidence-based management strategies for rosacea: summary of a Cochrane systematic review. Br J Dermatol. 2011;165:760-781.
- Tan X, Feldman SR, Chang J, et al. Topical drug delivery systems in dermatology: a review of patient adherence issues. Expert Opin Drug Deliv. 2012;9:1263-1271.
- Stein L. Clinical studies of a new vehicle formulation for topical corticosteroids in the treatment of psoriasis. J Am Acad Dermatol. 2005;53(1, suppl 1):S39-S49.
- Draelos ZD, Elewski BE, Harper JC, et al. A phase 3 randomized, double-blind, vehicle-controlled trial of azelaic acid foam 15% in the treatment of papulopustular rosacea. Cutis. 2015;96:54-61.
- Draelos ZD, Elewski B, Staedtler G, et al. Azelaic acid foam 15% in the treatment of papulopustular rosacea: a randomized, double-blind, vehicle-controlled study. Cutis. 2013;92:306-317.
- Finacea gel [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2016.
- Elewski BE, Fleischer AB Jr, Pariser DM. A comparison of 15% azelaic acid gel and 0.75% metronidazole gel in the topical treatment of papulopustular rosacea: results of a randomized trial. Arch Dermatol. 2003;139:1444-1450.
- Daniels R, Knie U. Galenics of dermal products—vehicles, properties and drug release. J Dtsch Dermatol Ges. 2007;5:367-383.
- Shamsudin N, Fleischer AB Jr. Vehicle or placebo? Investigators use incorrect terminology in randomized controlled trials half of the time: a systematic review of randomized controlled trials published in three major dermatology journals. J Drugs Dermatol. 2010;9:1221-1226.
- Del Rosso JQ, Thiboutot D, Gallo R, et al. Consensus recommendations from the American Acne & Rosacea Society on the management of rosacea, part 2: a status report on topical agents. Cutis. 2013;92:277-284.
- Finacea foam [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2015.
- Solano F, Briganti S, Picardo M, et al. Hypopigmenting agents: an updated review on biological, chemical and clinical aspects. Pigment Cell Res. 2006;19:550-571.
- Hammarstrom B, Wessling A, Nilsson JL. Pharmaceutical care for patients with skin diseases: a campaign year at Swedish pharmacies. J Clin Pharm Ther. 1995;20:327-334.
- Tan J, Berg M. Rosacea: current state of epidemiology. J Am Acad Dermatol. 2013;69(6, suppl 1):S27-S35.
- Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the classification and staging of rosacea. J Am Acad Dermatol. 2002;46:584-587.
- Chang AL, Raber I, Xu J, et al. Assessment of the genetic basis of rosacea by genome-wide association study. J Invest Dermatol. 2015;135:1548-1555.
- Abram K, Silm H, Maaroos HI, et al. Risk factors associated with rosacea. J Eur Acad Dermatol Venereol. 2010;24:565-571.
- Yamasaki K, Di Nardo A, Bardan A, et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med. 2007;13:975-980.
- Yamasaki K, Kanada K, Macleod DT, et al. TLR2 expression is increased in rosacea and stimulates enhanced serine protease production by keratinocytes. J Invest Dermatol. 2011;131:688-697.
- Darlenski R, Kazandjieva J, Tsankov N, et al. Acute irritant threshold correlates with barrier function, skin hydration and contact hypersensitivity in atopic dermatitis and rosacea. Exp Dermatol. 2013;22:752-753.
- Del Rosso JQ, Levin J. The clinical relevance of maintaining the functional integrity of the stratum corneum in both healthy and disease-affected skin. J Clin Aesthet Dermatol. 2011;4:22-42.
- van Zuuren EJ, Kramer SF, Carter BR, et al. Effective and evidence-based management strategies for rosacea: summary of a Cochrane systematic review. Br J Dermatol. 2011;165:760-781.
- Tan X, Feldman SR, Chang J, et al. Topical drug delivery systems in dermatology: a review of patient adherence issues. Expert Opin Drug Deliv. 2012;9:1263-1271.
- Stein L. Clinical studies of a new vehicle formulation for topical corticosteroids in the treatment of psoriasis. J Am Acad Dermatol. 2005;53(1, suppl 1):S39-S49.
- Draelos ZD, Elewski BE, Harper JC, et al. A phase 3 randomized, double-blind, vehicle-controlled trial of azelaic acid foam 15% in the treatment of papulopustular rosacea. Cutis. 2015;96:54-61.
- Draelos ZD, Elewski B, Staedtler G, et al. Azelaic acid foam 15% in the treatment of papulopustular rosacea: a randomized, double-blind, vehicle-controlled study. Cutis. 2013;92:306-317.
- Finacea gel [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2016.
- Elewski BE, Fleischer AB Jr, Pariser DM. A comparison of 15% azelaic acid gel and 0.75% metronidazole gel in the topical treatment of papulopustular rosacea: results of a randomized trial. Arch Dermatol. 2003;139:1444-1450.
- Daniels R, Knie U. Galenics of dermal products—vehicles, properties and drug release. J Dtsch Dermatol Ges. 2007;5:367-383.
- Shamsudin N, Fleischer AB Jr. Vehicle or placebo? Investigators use incorrect terminology in randomized controlled trials half of the time: a systematic review of randomized controlled trials published in three major dermatology journals. J Drugs Dermatol. 2010;9:1221-1226.
- Del Rosso JQ, Thiboutot D, Gallo R, et al. Consensus recommendations from the American Acne & Rosacea Society on the management of rosacea, part 2: a status report on topical agents. Cutis. 2013;92:277-284.
- Finacea foam [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2015.
- Solano F, Briganti S, Picardo M, et al. Hypopigmenting agents: an updated review on biological, chemical and clinical aspects. Pigment Cell Res. 2006;19:550-571.
- Hammarstrom B, Wessling A, Nilsson JL. Pharmaceutical care for patients with skin diseases: a campaign year at Swedish pharmacies. J Clin Pharm Ther. 1995;20:327-334.
Practice Points
- Papulopustular rosacea (PPR) is a common chronic inflammatory dermatosis.
- A novel hydrophilic foam formulation of azelaic acid (AzA) was approved for the treatment of PPR.
- In addition to effectively treating papules and pustules, AzA foam also may reduce rosacea-associated erythema.
- The unique AzA foam vehicle may improve epidermal barrier integrity and function, thereby offering patients a distinct topical approach to rosacea management.
Pediatric questionnaire sorts out psychosocial effects of skin conditions
MINNEAPOLIS – A new screening tool may help dermatologists address the psychosocial issues relating to appearance and body image in children and adolescents.
The Pediatric Dermatology Psychosocial Screen (PDPS), is being developed as a standardized tool to evaluate psychosocial stress related to birthmarks, skin diseases, and conditions affecting pigmentation or hair growth. Elizabeth Tocci, MD, and her colleagues, who have been involved with the development of the PDPS, envision it as a useful tool to provide support for pediatric dermatology patients and to help dermatologists decide when mental health consults are warranted in their pediatric patients.
Dr. Tocci, a resident in dermatology at Roger Williams Medical Center, Providence, R.I., and her colleagues described the tool and initial testing results in a poster session at the annual meeting of the Society for Pediatric Dermatology.
The PDPS is a refinement of a pilot survey, created by the coauthors in consultation with experts in neurodermatitis and body dysmorphic disorder (BDD). Following preliminary validity analysis of the pilot questionnaire, a revised PDPS was administered to 105 children, aged 8-19 years, who were patients at a pediatric dermatology clinic. In addition to completing the PDPS, they also filled out psychological questionnaires that assessed for depression, self-esteem, and social problems.
The PDPS asks general questions about the skin diagnosis and any treatments the patient may have used, such as over-the-counter products, prescription medications, and procedures, as well as the use of makeup. In addition, the PDPS asks what social and psychological supports or online resources the patient might have tried, including support groups and appointments with school counselors or mental health providers.
Psychosocial aspects of the skin condition are explored by asking how upset patients are about social sequelae of having a visible skin condition, and whether they are asked about the condition and whether they are made fun of, stared at, or avoided because of the condition. Other questions pertain to whether they notice others’ skin, are hyperobservant of their own skin condition, or feel their popularity and their willingness to date are affected by their skin condition.
Respondent resiliency as it relates to the skin condition is explored by asking whether the respondent found it difficult to move on after a negative social interaction related to the skin condition, and how long the feeling of upset persisted after a negative incident.
Of the 105 surveys, 87 were complete enough to allow analysis. The analysis showed that higher self-reported resiliency was associated with higher positive scores on other psychosocial factors, such as self-esteem, body image, fewer negative and more positive social supports, less self-consciousness, less negative affect, and less BDD. “Self-reported resilience was a significant predictor and determinant of all the psychosocial factors measured,” Dr. Tocci and her associates wrote.
Results indicate that the PDPS is useful to evaluate children and teens in a busy clinic setting, and is “an excellent self-reporting tool for measuring resilience versus psychosocial distress,” they added.
The test is not yet available; the next steps include refining the length and wording of the PDPS, with further validation and testing.
Dr. Tocci and her collaborators reported no conflicts of interest.
On Twitter @karioakes
MINNEAPOLIS – A new screening tool may help dermatologists address the psychosocial issues relating to appearance and body image in children and adolescents.
The Pediatric Dermatology Psychosocial Screen (PDPS), is being developed as a standardized tool to evaluate psychosocial stress related to birthmarks, skin diseases, and conditions affecting pigmentation or hair growth. Elizabeth Tocci, MD, and her colleagues, who have been involved with the development of the PDPS, envision it as a useful tool to provide support for pediatric dermatology patients and to help dermatologists decide when mental health consults are warranted in their pediatric patients.
Dr. Tocci, a resident in dermatology at Roger Williams Medical Center, Providence, R.I., and her colleagues described the tool and initial testing results in a poster session at the annual meeting of the Society for Pediatric Dermatology.
The PDPS is a refinement of a pilot survey, created by the coauthors in consultation with experts in neurodermatitis and body dysmorphic disorder (BDD). Following preliminary validity analysis of the pilot questionnaire, a revised PDPS was administered to 105 children, aged 8-19 years, who were patients at a pediatric dermatology clinic. In addition to completing the PDPS, they also filled out psychological questionnaires that assessed for depression, self-esteem, and social problems.
The PDPS asks general questions about the skin diagnosis and any treatments the patient may have used, such as over-the-counter products, prescription medications, and procedures, as well as the use of makeup. In addition, the PDPS asks what social and psychological supports or online resources the patient might have tried, including support groups and appointments with school counselors or mental health providers.
Psychosocial aspects of the skin condition are explored by asking how upset patients are about social sequelae of having a visible skin condition, and whether they are asked about the condition and whether they are made fun of, stared at, or avoided because of the condition. Other questions pertain to whether they notice others’ skin, are hyperobservant of their own skin condition, or feel their popularity and their willingness to date are affected by their skin condition.
Respondent resiliency as it relates to the skin condition is explored by asking whether the respondent found it difficult to move on after a negative social interaction related to the skin condition, and how long the feeling of upset persisted after a negative incident.
Of the 105 surveys, 87 were complete enough to allow analysis. The analysis showed that higher self-reported resiliency was associated with higher positive scores on other psychosocial factors, such as self-esteem, body image, fewer negative and more positive social supports, less self-consciousness, less negative affect, and less BDD. “Self-reported resilience was a significant predictor and determinant of all the psychosocial factors measured,” Dr. Tocci and her associates wrote.
Results indicate that the PDPS is useful to evaluate children and teens in a busy clinic setting, and is “an excellent self-reporting tool for measuring resilience versus psychosocial distress,” they added.
The test is not yet available; the next steps include refining the length and wording of the PDPS, with further validation and testing.
Dr. Tocci and her collaborators reported no conflicts of interest.
On Twitter @karioakes
MINNEAPOLIS – A new screening tool may help dermatologists address the psychosocial issues relating to appearance and body image in children and adolescents.
The Pediatric Dermatology Psychosocial Screen (PDPS), is being developed as a standardized tool to evaluate psychosocial stress related to birthmarks, skin diseases, and conditions affecting pigmentation or hair growth. Elizabeth Tocci, MD, and her colleagues, who have been involved with the development of the PDPS, envision it as a useful tool to provide support for pediatric dermatology patients and to help dermatologists decide when mental health consults are warranted in their pediatric patients.
Dr. Tocci, a resident in dermatology at Roger Williams Medical Center, Providence, R.I., and her colleagues described the tool and initial testing results in a poster session at the annual meeting of the Society for Pediatric Dermatology.
The PDPS is a refinement of a pilot survey, created by the coauthors in consultation with experts in neurodermatitis and body dysmorphic disorder (BDD). Following preliminary validity analysis of the pilot questionnaire, a revised PDPS was administered to 105 children, aged 8-19 years, who were patients at a pediatric dermatology clinic. In addition to completing the PDPS, they also filled out psychological questionnaires that assessed for depression, self-esteem, and social problems.
The PDPS asks general questions about the skin diagnosis and any treatments the patient may have used, such as over-the-counter products, prescription medications, and procedures, as well as the use of makeup. In addition, the PDPS asks what social and psychological supports or online resources the patient might have tried, including support groups and appointments with school counselors or mental health providers.
Psychosocial aspects of the skin condition are explored by asking how upset patients are about social sequelae of having a visible skin condition, and whether they are asked about the condition and whether they are made fun of, stared at, or avoided because of the condition. Other questions pertain to whether they notice others’ skin, are hyperobservant of their own skin condition, or feel their popularity and their willingness to date are affected by their skin condition.
Respondent resiliency as it relates to the skin condition is explored by asking whether the respondent found it difficult to move on after a negative social interaction related to the skin condition, and how long the feeling of upset persisted after a negative incident.
Of the 105 surveys, 87 were complete enough to allow analysis. The analysis showed that higher self-reported resiliency was associated with higher positive scores on other psychosocial factors, such as self-esteem, body image, fewer negative and more positive social supports, less self-consciousness, less negative affect, and less BDD. “Self-reported resilience was a significant predictor and determinant of all the psychosocial factors measured,” Dr. Tocci and her associates wrote.
Results indicate that the PDPS is useful to evaluate children and teens in a busy clinic setting, and is “an excellent self-reporting tool for measuring resilience versus psychosocial distress,” they added.
The test is not yet available; the next steps include refining the length and wording of the PDPS, with further validation and testing.
Dr. Tocci and her collaborators reported no conflicts of interest.
On Twitter @karioakes
EXPERT ANALYSIS FROM THE SPD ANNUAL MEETING
Some psoriasis patients benefit from switching anti-TNF agents
Psoriasis patients may have more success with a second tumor necrosis factor (TNF) antagonist after failure with a first, report Paul S. Yamauchi, MD, PhD, and coauthors.
Investigators analyzed 15 studies evaluating the efficacy of switching TNF antagonists after primary or secondary failure. Response rates at 24 weeks for a second antagonist were 30%-74% for a 75% improvement in Psoriasis Area and Severity Index score, and 20%-70% for achieving a Physician Global Assessment score of 0/1. Mean improvements in Dermatology Life Quality Index ranged from –3.5 to –13, Dr. Yamauchi and colleagues reported.

Patients who experienced secondary failure with initial treatment generally achieved better responses than those with primary failure, the authors said.
Though response rates to a second anti-TNF agent were lower than for a first, “a substantial proportion of patients in every study achieved treatment success,” they added.
Read the full article in the Journal of the American Academy of Dermatology.
Psoriasis patients may have more success with a second tumor necrosis factor (TNF) antagonist after failure with a first, report Paul S. Yamauchi, MD, PhD, and coauthors.
Investigators analyzed 15 studies evaluating the efficacy of switching TNF antagonists after primary or secondary failure. Response rates at 24 weeks for a second antagonist were 30%-74% for a 75% improvement in Psoriasis Area and Severity Index score, and 20%-70% for achieving a Physician Global Assessment score of 0/1. Mean improvements in Dermatology Life Quality Index ranged from –3.5 to –13, Dr. Yamauchi and colleagues reported.
Patients who experienced secondary failure with initial treatment generally achieved better responses than those with primary failure, the authors said.
Though response rates to a second anti-TNF agent were lower than for a first, “a substantial proportion of patients in every study achieved treatment success,” they added.
Read the full article in the Journal of the American Academy of Dermatology.
Psoriasis patients may have more success with a second tumor necrosis factor (TNF) antagonist after failure with a first, report Paul S. Yamauchi, MD, PhD, and coauthors.
Investigators analyzed 15 studies evaluating the efficacy of switching TNF antagonists after primary or secondary failure. Response rates at 24 weeks for a second antagonist were 30%-74% for a 75% improvement in Psoriasis Area and Severity Index score, and 20%-70% for achieving a Physician Global Assessment score of 0/1. Mean improvements in Dermatology Life Quality Index ranged from –3.5 to –13, Dr. Yamauchi and colleagues reported.
Patients who experienced secondary failure with initial treatment generally achieved better responses than those with primary failure, the authors said.
Though response rates to a second anti-TNF agent were lower than for a first, “a substantial proportion of patients in every study achieved treatment success,” they added.
Read the full article in the Journal of the American Academy of Dermatology.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY

An Update on Neurotoxin Products and Administration Methods
The first botulinum neurotoxin (BoNT) approved by the US Food and Drug Administration (FDA) was onabotulinumtoxinA in 1989 for the treatment of strabismus and blepharospasm. It was not until 1992, however, that the aesthetic benefits of BoNT were first reported in the medical literature by Carruthers and Carruthers,1 and a cosmetic indication was not approved by the FDA until 2002. Since that time, the popularity of BoNT products has grown rapidly with a nearly 6500% increase in popularity from 1997 to 2015 in addition to the introduction of a variety of new BoNT formulations to the market.2 It is estimated by the American Society for Aesthetic Plastic Surgery that there were at least 4,000,000 BoNT injections performed in 2015 alone, making it the most popular nonsurgical aesthetic procedure available.2 As the demand for minimally invasive cosmetic procedures continues to increase, we will continue to see the introduction of additional formulations of BoNT products as well as novel administration techniques and delivery devices. In this article, we provide an update on current and upcoming BoNT products and also review the literature on novel administration methods based on studies published from January 1, 2014, to December 31, 2015.
Current Products
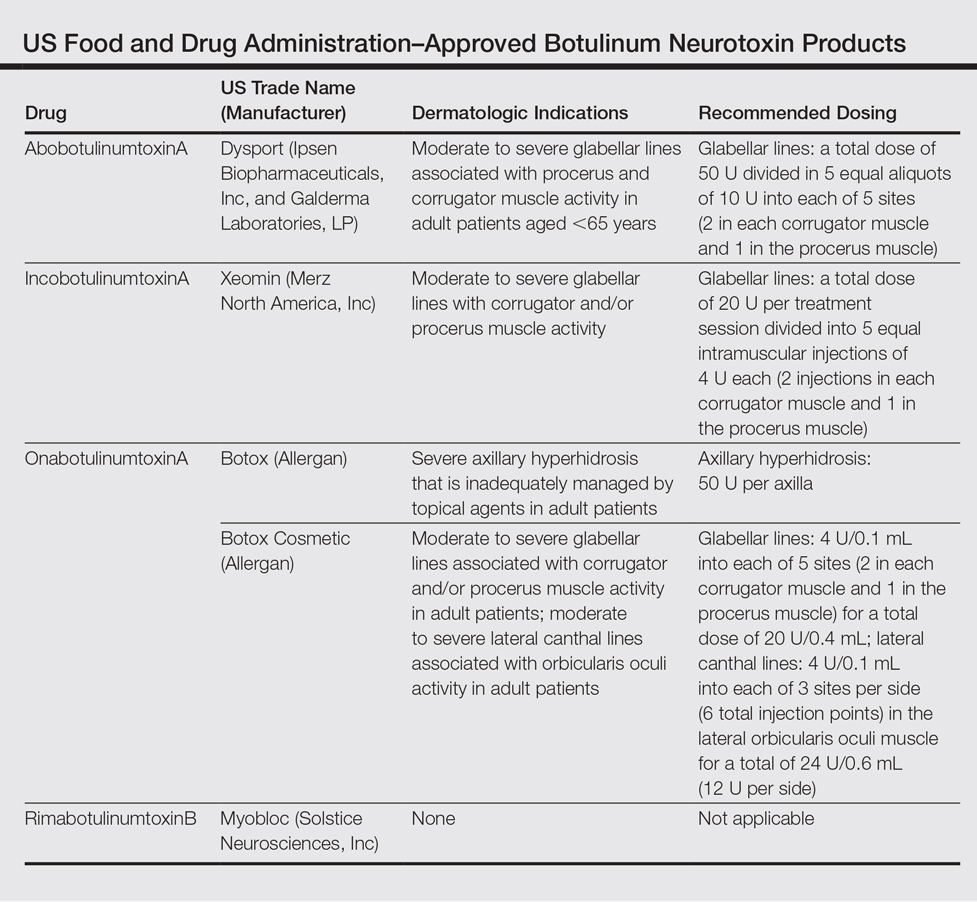
To date, there are only 4 FDA-approved formulations of BoNT available for clinical use (eg, cervical dystonia, strabismus, blepharospasm, headache, urinary incontinence) in the United States: abobotulinumtoxinA, incobotulinumtoxinA, onabotulinumtoxinA, and rimabotulinumtoxinB.The FDA-approved dermatologic indications (eg, moderate to severe glabellar or canthal lines, severe axillary hyperhidrosis) for these products are provided in the Table. On a global scale, there are several other commonly utilized formulations of BoNT, including a Korean serotype resembling onabotulinumtoxinA and a Chinese botulinum toxin type A.3 Although there is some evidence to demonstrate comparable efficacy and safety of these latter products, the literature is relatively lacking in comparison to the FDA-approved products.4,5
Upcoming Products
Currently, there are several new BoNT formulations being studied for clinical use. RT 002 (Revance Therapeutics, Inc) is a novel injectable formulation of onabotulinumtoxinA that consists of the purified neurotoxin in combination with patented TransMTS peptides that have been shown to provide high-binding avidity for the neurotoxin, and thus the product is designed to reduce diffusion to adjacent muscles and diminish unwanted effects. With a reduced level of neurotoxin dissemination, it is theorized that a higher administration of targeted doses can be injected, which may lead to a longer duration of desired effects.6 A clinical pilot study done to establish the safety and efficacy of RT 002 for treatment of moderate to severe glabellar lines evaluated 4 equally sized cohorts of 12 participants, each receiving single-dose administration of RT 002 ranging in potency equivalent to 25 U, 50 U, 75 U, and 100 U of abobotulinumtoxinA as determined by the gelatin phosphate method.6 It was concluded that RT 002 is both safe and efficacious with an extended duration of action, with a median duration of effect of 7 months observed in the highest dose group (dose equivalent to 100 U of abobotulinumtoxinA). Notably, 80% of all 48 participants maintained a minimum 1-point improvement in investigator-determined glabellar line severity scores at the 6-month time point and 60% achieved wrinkle scores of none or mild at 6 months posttreatment.6
DWP 450 (Daewoong Pharmaceutical Co, Ltd) is derived from the wild-type Clostridium botulinum and is reported to be of higher purity than standard onabotulinumtoxinA. An initial 16-week pilot study demonstrated that 20 U of DWP 450 is noninferior and of comparable efficacy and safety to 20 U of onabotulinumtoxinA in the treatment of glabellar lines.7
NTC (Botulax [Hugel, Inc]) is the name of the toxin derived from the C botulinum strain CBFC26, which has already been approved in many Asian, European, and Latin American countries for the treatment of blepharospasm. This formulation has demonstrated noninferiority to onabotulinumtoxinA at equivalent 20-U doses for the treatment of moderate to severe glabellar lines in a double-blind, randomized, multicenter, phase 3 trial of 272 participants with a 16-week follow-up.8
MT 10109L (Medytox Inc) is a unique product in that it is distributed as a liquid type A botulinum toxin rather than the standard freeze-dried formulation; thus, a major advantage of this product is its convenience, as it does not need reconstitution or dilution prior to administration. In a double-blind, randomized, active drug–controlled, phase 3 study of 168 participants, it was determined that MT 10109L (20 U) is comparable in efficacy to onabotulinumtoxinA (20 U) for the treatment of moderate to severe glabellar lines. No significant difference was seen between the 2 treatment groups when glabellar lines were assessed at rest at 4 and 16 weeks after treatment, but a significantly greater improvement in glabellar lines was seen at maximum frown in the MT 10109L group at the 16-week follow-up (P=.0064).9
Administration Techniques
With regard to safe and effective BoNT product administration techniques, a variety of studies have provided insight into optimal practice methods. A 2015 expert consensus statement formed by an American Society for Dermatologic Surgery task force reviewed data from 42 papers and unanimously determined that for all current type A BoNT products available in the United States, a vial of BoNT reconstituted appropriately for the purpose of facial injections can be reconstituted at least 4 weeks prior to administration without contamination risk or decrease in efficacy and that multiple patients can be treated with the same vial.Although the statement was not explicit on whether or not preserved or unpreserved saline is to be used, it is considered routine practice to use preservative-containing saline to reconstitute BoNT, as it has been shown to reduce patient discomfort and is not associated with adverse effects.10
Pain Minimization
With respect to minimizing the pain associated with BoNT injections, several studies have assessed administration techniques to minimize patient discomfort. A split-face, double-blind study of 20 participants demonstrated that the use of a 32-gauge needle has a significantly greater chance of reducing clinically significant levels of pain as compared to a 30-gauge needle when performing facial injections (P=.04). Overall, however, injections of the face and arms were on average only nominally and not significantly more painful with 30-gauge needles compared to 32-gauge needles.11
Another technique that has been found to reduce patient discomfort is the application of cold packs prior to injection. A study of patients with chronic facial palsy observed a significant reduction in pain with the administration of a cold (3°C–5°C) gel pack for 1 minute compared to a room temperature (20°C) gel pack prior to the administration of onabotulinumtoxinA into the platysma (P<.001).12 In the matter of injection with rimabotulinumtoxinB, which has been shown to be considerably more painful to receive than its more popularly administered counterpart onabotulinumtoxinA, a split-face pilot study examined the effect of increasing the pH of rimabotulinumtoxinB to 7.5 with sodium bicarbonate from the usual pH of 5.6.13,14 Pain was reported to be considerably less in the higher pH group and no reduction of efficacy was seen over the 10-week follow-up period.14
Delivery Methods
Several preliminary studies also have examined novel delivery techniques to identify minimally painful yet effective methods for administering BoNT. It has been reported that standard BoNT formulations are not effective as topical agents in a comparison study in which onabotulinumtoxinA injection was compared to topically applied onabotulinumtoxinA.15 However, a follow-up prospective study by the same authors has demonstrated efficacy of topical onabotulinumtoxinA following pretreatment with a fractional ablative CO2 laser for treatment of crow’s-feet. In this randomized, split-face, controlled trial (N=10), participants were first pretreated with topical lidocaine 30% before receiving a single pass of fractional ablative CO2 laser with no overlap and a pulse energy of 100 mJ. Within 60 seconds of laser treatment, participants then received either 100 U of abobotulinumtoxinA diluted in 0.1 mL of saline or simple normal saline applied topically. A clinically significant improvement in periorbital wrinkles was seen both at 1-week and 1-month posttreatment in the laser and onabotulinumtoxinA–treated group compared to the laser and topical saline–treated group (P<.02).15
Another unique administration method studied, albeit with less successful results, involves the use of iontophoresis to deliver BoNT painlessly in a transdermal fashion with the assistance of an electrical current.16 This prospective, randomized, assessor-blinded, split-axilla, controlled trial of 11 participants compared the effectiveness of administering onabotulinumtoxinA via iontophoresis to traditional injection with onabotulinumtoxinA (250 U). Iontophoresis was accomplished with a single electrode pad soaked with 250 U of onabotulinumtoxinA applied directly to the axilla and a second electrode pad soaked in 0.9% saline applied to the hand to complete the circuit. An alternating electrical current was slowly increased for 30 minutes to a maximum current of 15 mA with a voltage of 12 V. Among the 11 participants recruited, the side treated with traditional injection showed a significantly greater percentage reduction in baseline sweating at the 1-week, 1-month, and 6-month posttreatment evaluations compared to iontophoresis (84%, 76%, and 50%, respectively vs 73%, 22%, and 32%, respectively)(P<.05). Despite being less efficacious than standard injection therapy, participants reported that iontophoresis delivery was significantly less painful (P<.05).16
A high-pressure oxygen delivery device, which utilizes a powerful jet of microdroplets containing water, the drug, air, and oxygen to deliver medication onto the skin surface, also has been studied as a means of delivery of BoNT in a minimally painful manner. In this study, the device was used to assess the efficacy of transdermal delivery of BoNT via jet nebulization in the treatment of primary palmar, plantar, and axillary hyperhidrosis.17 The 20 participants included in the study were randomized to receive either a combination of lidocaine and onabotulinumtoxinA (50 U) administered through the device or lidocaine delivered through the device followed by multiple transcutaneous injections of onabotulinumtoxinA (100 U). Both treatments significantly reduced sweating compared to baseline as measured by a visual analogue scale at 3-month follow-up (P<.001), but the combination delivery of lidocaine and onabotulinumtoxinA via the device resulted in significantly less procedure-related pain and sweating (P<.001) as well as significantly greater patient satisfaction (P<.001).17
Optimizing Aesthetic Outcomes
A frequent concern of patients receiving BoNT for cosmetic purposes is a desire to avoid a “frozen” or expressionless look. As such, many clinicians have attempted a variety of techniques to achieve more natural aesthetic results. One such method is known as the multipoint and multilevel injection technique, which consists of utilizing multiple injection sites at varying depths (intramuscular, subcutaneous, or intradermal) and doses (2–6 U) depending on the degree of contractility of the targeted muscle. In a preliminary study of 223 participants using this technique with a total dose of 125 U of abobotulinumtoxinA, good and natural results were reported with perseveration of facial emotion in all participants in addition to a mean overall satisfaction rate of 6.4 of 7 on the Facial Line Treatment Satisfaction Questionnaire with the maximum satisfaction rating possible reported in 66% of cases.18 It also has been postulated that injection depth of BoNT can affect brow elevation whereupon deeper injection depths can result in inactivation of the brow depressors and allow for increased elevation of the eyebrows. This technique has been applied in attempts to correct brow height asymmetry. However, a prospective, split-face study of 23 women suggested that this method is not effective.19 Participants received 64 U of onabotulinumtoxinA via 16 injection sites in the glabella, forehead, and lateral canthal area with either all deep or all shallow injections depending on the side treated and whether brow-lift was desired. Results at 4 weeks posttreatment showed no significant difference in brow height, and it was concluded that eyebrow depressor muscles cannot be accurately targeted with deep injection into the muscle belly for correction of eyebrow height discrepancies.19 Conversely, a 5-year retrospective, nonrandomized study of 227 patients with 563 treatments utilizing a “microdroplet” technique reported success at selectively targeting the eyebrow depressors while leaving the brow elevators unaffected.20 Here, a total dose of 33 U of onabotulinumtoxinA was administered via microdroplets of 10 to 20 μL, each with more than 60 to 100 injections into the brow, glabella, and crow’s-feet area. This method of injection resulted in a statistically significant improvement of forehead lines and brow ptosis and furrowing at follow-up between 10 and 45 days after treatment (P<.0001). Additionally, average brow height was significantly increased from 24.6 mm to 25 mm after treatment (P=.02).20
Conclusion
The use of BoNT products for both on- and off-label cosmetic and medical indications has rapidly grown over the past 2 decades. As demonstrated in this review, a variety of promising new products and delivery techniques are being developed. Given the rise in popularity of BoNT products among both physicians and consumers, clinicians should be aware of the current data and ongoing research.
- Carruthers JD, Carruthers JA. Treatment of glabellar frown lines with C. botulinum-A exotoxin. J Dermatol Surg Oncol. 1992;18:17-21.
- American Society for Aesthetic Plastic Surgery. Cosmetic Surgery National Data Bank statistics. American Society for Aesthetic Plastic Surgery website. http://www.surgery.org/sites/default/files/ASAPS-Stats2015.pdf. Accessed June 12, 2016.
- Walker TJ, Dayan SH. Comparison and overview of currently available neurotoxins. J Clin Aesthet Dermatol. 2014;7:31-39.
- Feng Z, Sun Q, He L, et al. Optimal dosage of botulinum toxin type A for treatment of glabellar frown lines: efficacy and safety in a clinical trial. Dermatol Surg. 2015;41(suppl 1):S56-S63.
- Jiang HY, Chen S, Zhou J, et al. Diffusion of two botulinum toxins type A on the forehead: double-blinded, randomized, controlled study. Dermatol Surg. 2014;40:184-192.
- Garcia-Murray E, Velasco Villasenor ML, Acevedo B, et al. Safety and efficacy of RT002, an injectable botulinum toxin type A, for treating glabellar lines: results of a phase 1/2, open-label, sequential dose-escalation study. Dermatol Surg. 2015;41(suppl 1):S47-S55.
- Won CH, Kim HK, Kim BJ, et al. Comparative trial of a novel botulinum neurotoxin type A versus onabotulinumtoxinA in the treatment of glabellar lines: a multicenter, randomized, double-blind, active-controlled study. Int J Dermatol. 2015;54:227-234.
- Kim BJ, Kwon HH, Park SY, et al. Double-blind, randomized non-inferiority trial of a novel botulinum toxin A processed from the strain CBFC26, compared with onabotulinumtoxin A in the treatment of glabellar lines. J Eur Acad Dermatol Venereol. 2014;28:1761-1767.
- Kim JE, Song EJ, Choi GS, et al. The efficacy and safety of liquid-type botulinum toxin type A for the management of moderate to severe glabellar frown lines. Plast Reconstr Surg. 2015;135:732-741.
- Alam M, Bolotin D, Carruthers J, et al. Consensus statement regarding storage and reuse of previously reconstituted neuromodulators. Dermatol Surg. 2015;41:321-326.
- Alam M, Geisler A, Sadhwani D, et al. Effect of needle size on pain perception in patients treated with botulinum toxin type A injections: a randomized clinical trial. JAMA Dermatol. 2015;151:1194-1199.
- Pucks N, Thomas A, Hallam MJ, et al. Cutaneous cooling to manage botulinum toxin injection-associated pain in patients with facial palsy: a randomised controlled trial. J Plast Reconstr Aesthet Surg. 2015;68:1701-1705.
- Kranz G, Sycha T, Voller B, et al. Pain sensation during intradermal injections of three different botulinum toxin preparations in different doses and dilutions. Dermatol Surg. 2006;32:886-890.
- Lowe PL, Lowe NJ. Botulinum toxin type B: pH change reduces injection pain, retains efficacy. Dermatol Surg. 2014;40:1328-1333.
- Mahmoud BH, Burnett C, Ozog D. Prospective randomized controlled study to determine the effect of topical application of botulinum toxin A for crow’s feet after treatment with ablative fractional CO2 laser. Dermatol Surg. 2015;41(suppl 1):S75-S81.
- Montaser-Kouhsari L, Zartab H, Fanian F, et al. Comparison of intradermal injection with iontophoresis of abo-botulinum toxin A for the treatment of primary axillary hyperhidrosis: a randomized, controlled trial. J Dermatolog Treat. 2014;25:337-341.
- Iannitti T, Palmieri B, Aspiro A, et al. A preliminary study of painless and effective transdermal botulinum toxin A delivery by jet nebulization for treatment of primary hyperhidrosis. Drug Des Devel Ther. 2014;8:931-935.
- Iozzo I, Tengattini V, Antonucci VA. Multipoint and multilevel injection technique of botulinum toxin A in facial aesthetics. J Cosmet Dermatol. 2014;13:135-142.
- Sneath J, Humphrey S, Carruthers A, et al. Injecting botulinum toxin at different depths is not effective for the correction of eyebrow asymmetry. Dermatol Surg. 2015;41(suppl 1):S82-S87.
- Steinsapir KD, Rootman D, Wulc A, et al. Cosmetic microdroplet botulinum toxin A forehead lift: a new treatment paradigm. Ophthal Plast Reconstr Surg. 2015;31:263-268.
The first botulinum neurotoxin (BoNT) approved by the US Food and Drug Administration (FDA) was onabotulinumtoxinA in 1989 for the treatment of strabismus and blepharospasm. It was not until 1992, however, that the aesthetic benefits of BoNT were first reported in the medical literature by Carruthers and Carruthers,1 and a cosmetic indication was not approved by the FDA until 2002. Since that time, the popularity of BoNT products has grown rapidly with a nearly 6500% increase in popularity from 1997 to 2015 in addition to the introduction of a variety of new BoNT formulations to the market.2 It is estimated by the American Society for Aesthetic Plastic Surgery that there were at least 4,000,000 BoNT injections performed in 2015 alone, making it the most popular nonsurgical aesthetic procedure available.2 As the demand for minimally invasive cosmetic procedures continues to increase, we will continue to see the introduction of additional formulations of BoNT products as well as novel administration techniques and delivery devices. In this article, we provide an update on current and upcoming BoNT products and also review the literature on novel administration methods based on studies published from January 1, 2014, to December 31, 2015.
Current Products
To date, there are only 4 FDA-approved formulations of BoNT available for clinical use (eg, cervical dystonia, strabismus, blepharospasm, headache, urinary incontinence) in the United States: abobotulinumtoxinA, incobotulinumtoxinA, onabotulinumtoxinA, and rimabotulinumtoxinB.The FDA-approved dermatologic indications (eg, moderate to severe glabellar or canthal lines, severe axillary hyperhidrosis) for these products are provided in the Table. On a global scale, there are several other commonly utilized formulations of BoNT, including a Korean serotype resembling onabotulinumtoxinA and a Chinese botulinum toxin type A.3 Although there is some evidence to demonstrate comparable efficacy and safety of these latter products, the literature is relatively lacking in comparison to the FDA-approved products.4,5
Upcoming Products
Currently, there are several new BoNT formulations being studied for clinical use. RT 002 (Revance Therapeutics, Inc) is a novel injectable formulation of onabotulinumtoxinA that consists of the purified neurotoxin in combination with patented TransMTS peptides that have been shown to provide high-binding avidity for the neurotoxin, and thus the product is designed to reduce diffusion to adjacent muscles and diminish unwanted effects. With a reduced level of neurotoxin dissemination, it is theorized that a higher administration of targeted doses can be injected, which may lead to a longer duration of desired effects.6 A clinical pilot study done to establish the safety and efficacy of RT 002 for treatment of moderate to severe glabellar lines evaluated 4 equally sized cohorts of 12 participants, each receiving single-dose administration of RT 002 ranging in potency equivalent to 25 U, 50 U, 75 U, and 100 U of abobotulinumtoxinA as determined by the gelatin phosphate method.6 It was concluded that RT 002 is both safe and efficacious with an extended duration of action, with a median duration of effect of 7 months observed in the highest dose group (dose equivalent to 100 U of abobotulinumtoxinA). Notably, 80% of all 48 participants maintained a minimum 1-point improvement in investigator-determined glabellar line severity scores at the 6-month time point and 60% achieved wrinkle scores of none or mild at 6 months posttreatment.6
DWP 450 (Daewoong Pharmaceutical Co, Ltd) is derived from the wild-type Clostridium botulinum and is reported to be of higher purity than standard onabotulinumtoxinA. An initial 16-week pilot study demonstrated that 20 U of DWP 450 is noninferior and of comparable efficacy and safety to 20 U of onabotulinumtoxinA in the treatment of glabellar lines.7
NTC (Botulax [Hugel, Inc]) is the name of the toxin derived from the C botulinum strain CBFC26, which has already been approved in many Asian, European, and Latin American countries for the treatment of blepharospasm. This formulation has demonstrated noninferiority to onabotulinumtoxinA at equivalent 20-U doses for the treatment of moderate to severe glabellar lines in a double-blind, randomized, multicenter, phase 3 trial of 272 participants with a 16-week follow-up.8
MT 10109L (Medytox Inc) is a unique product in that it is distributed as a liquid type A botulinum toxin rather than the standard freeze-dried formulation; thus, a major advantage of this product is its convenience, as it does not need reconstitution or dilution prior to administration. In a double-blind, randomized, active drug–controlled, phase 3 study of 168 participants, it was determined that MT 10109L (20 U) is comparable in efficacy to onabotulinumtoxinA (20 U) for the treatment of moderate to severe glabellar lines. No significant difference was seen between the 2 treatment groups when glabellar lines were assessed at rest at 4 and 16 weeks after treatment, but a significantly greater improvement in glabellar lines was seen at maximum frown in the MT 10109L group at the 16-week follow-up (P=.0064).9
Administration Techniques
With regard to safe and effective BoNT product administration techniques, a variety of studies have provided insight into optimal practice methods. A 2015 expert consensus statement formed by an American Society for Dermatologic Surgery task force reviewed data from 42 papers and unanimously determined that for all current type A BoNT products available in the United States, a vial of BoNT reconstituted appropriately for the purpose of facial injections can be reconstituted at least 4 weeks prior to administration without contamination risk or decrease in efficacy and that multiple patients can be treated with the same vial.Although the statement was not explicit on whether or not preserved or unpreserved saline is to be used, it is considered routine practice to use preservative-containing saline to reconstitute BoNT, as it has been shown to reduce patient discomfort and is not associated with adverse effects.10
Pain Minimization
With respect to minimizing the pain associated with BoNT injections, several studies have assessed administration techniques to minimize patient discomfort. A split-face, double-blind study of 20 participants demonstrated that the use of a 32-gauge needle has a significantly greater chance of reducing clinically significant levels of pain as compared to a 30-gauge needle when performing facial injections (P=.04). Overall, however, injections of the face and arms were on average only nominally and not significantly more painful with 30-gauge needles compared to 32-gauge needles.11
Another technique that has been found to reduce patient discomfort is the application of cold packs prior to injection. A study of patients with chronic facial palsy observed a significant reduction in pain with the administration of a cold (3°C–5°C) gel pack for 1 minute compared to a room temperature (20°C) gel pack prior to the administration of onabotulinumtoxinA into the platysma (P<.001).12 In the matter of injection with rimabotulinumtoxinB, which has been shown to be considerably more painful to receive than its more popularly administered counterpart onabotulinumtoxinA, a split-face pilot study examined the effect of increasing the pH of rimabotulinumtoxinB to 7.5 with sodium bicarbonate from the usual pH of 5.6.13,14 Pain was reported to be considerably less in the higher pH group and no reduction of efficacy was seen over the 10-week follow-up period.14
Delivery Methods
Several preliminary studies also have examined novel delivery techniques to identify minimally painful yet effective methods for administering BoNT. It has been reported that standard BoNT formulations are not effective as topical agents in a comparison study in which onabotulinumtoxinA injection was compared to topically applied onabotulinumtoxinA.15 However, a follow-up prospective study by the same authors has demonstrated efficacy of topical onabotulinumtoxinA following pretreatment with a fractional ablative CO2 laser for treatment of crow’s-feet. In this randomized, split-face, controlled trial (N=10), participants were first pretreated with topical lidocaine 30% before receiving a single pass of fractional ablative CO2 laser with no overlap and a pulse energy of 100 mJ. Within 60 seconds of laser treatment, participants then received either 100 U of abobotulinumtoxinA diluted in 0.1 mL of saline or simple normal saline applied topically. A clinically significant improvement in periorbital wrinkles was seen both at 1-week and 1-month posttreatment in the laser and onabotulinumtoxinA–treated group compared to the laser and topical saline–treated group (P<.02).15
Another unique administration method studied, albeit with less successful results, involves the use of iontophoresis to deliver BoNT painlessly in a transdermal fashion with the assistance of an electrical current.16 This prospective, randomized, assessor-blinded, split-axilla, controlled trial of 11 participants compared the effectiveness of administering onabotulinumtoxinA via iontophoresis to traditional injection with onabotulinumtoxinA (250 U). Iontophoresis was accomplished with a single electrode pad soaked with 250 U of onabotulinumtoxinA applied directly to the axilla and a second electrode pad soaked in 0.9% saline applied to the hand to complete the circuit. An alternating electrical current was slowly increased for 30 minutes to a maximum current of 15 mA with a voltage of 12 V. Among the 11 participants recruited, the side treated with traditional injection showed a significantly greater percentage reduction in baseline sweating at the 1-week, 1-month, and 6-month posttreatment evaluations compared to iontophoresis (84%, 76%, and 50%, respectively vs 73%, 22%, and 32%, respectively)(P<.05). Despite being less efficacious than standard injection therapy, participants reported that iontophoresis delivery was significantly less painful (P<.05).16
A high-pressure oxygen delivery device, which utilizes a powerful jet of microdroplets containing water, the drug, air, and oxygen to deliver medication onto the skin surface, also has been studied as a means of delivery of BoNT in a minimally painful manner. In this study, the device was used to assess the efficacy of transdermal delivery of BoNT via jet nebulization in the treatment of primary palmar, plantar, and axillary hyperhidrosis.17 The 20 participants included in the study were randomized to receive either a combination of lidocaine and onabotulinumtoxinA (50 U) administered through the device or lidocaine delivered through the device followed by multiple transcutaneous injections of onabotulinumtoxinA (100 U). Both treatments significantly reduced sweating compared to baseline as measured by a visual analogue scale at 3-month follow-up (P<.001), but the combination delivery of lidocaine and onabotulinumtoxinA via the device resulted in significantly less procedure-related pain and sweating (P<.001) as well as significantly greater patient satisfaction (P<.001).17
Optimizing Aesthetic Outcomes
A frequent concern of patients receiving BoNT for cosmetic purposes is a desire to avoid a “frozen” or expressionless look. As such, many clinicians have attempted a variety of techniques to achieve more natural aesthetic results. One such method is known as the multipoint and multilevel injection technique, which consists of utilizing multiple injection sites at varying depths (intramuscular, subcutaneous, or intradermal) and doses (2–6 U) depending on the degree of contractility of the targeted muscle. In a preliminary study of 223 participants using this technique with a total dose of 125 U of abobotulinumtoxinA, good and natural results were reported with perseveration of facial emotion in all participants in addition to a mean overall satisfaction rate of 6.4 of 7 on the Facial Line Treatment Satisfaction Questionnaire with the maximum satisfaction rating possible reported in 66% of cases.18 It also has been postulated that injection depth of BoNT can affect brow elevation whereupon deeper injection depths can result in inactivation of the brow depressors and allow for increased elevation of the eyebrows. This technique has been applied in attempts to correct brow height asymmetry. However, a prospective, split-face study of 23 women suggested that this method is not effective.19 Participants received 64 U of onabotulinumtoxinA via 16 injection sites in the glabella, forehead, and lateral canthal area with either all deep or all shallow injections depending on the side treated and whether brow-lift was desired. Results at 4 weeks posttreatment showed no significant difference in brow height, and it was concluded that eyebrow depressor muscles cannot be accurately targeted with deep injection into the muscle belly for correction of eyebrow height discrepancies.19 Conversely, a 5-year retrospective, nonrandomized study of 227 patients with 563 treatments utilizing a “microdroplet” technique reported success at selectively targeting the eyebrow depressors while leaving the brow elevators unaffected.20 Here, a total dose of 33 U of onabotulinumtoxinA was administered via microdroplets of 10 to 20 μL, each with more than 60 to 100 injections into the brow, glabella, and crow’s-feet area. This method of injection resulted in a statistically significant improvement of forehead lines and brow ptosis and furrowing at follow-up between 10 and 45 days after treatment (P<.0001). Additionally, average brow height was significantly increased from 24.6 mm to 25 mm after treatment (P=.02).20
Conclusion
The use of BoNT products for both on- and off-label cosmetic and medical indications has rapidly grown over the past 2 decades. As demonstrated in this review, a variety of promising new products and delivery techniques are being developed. Given the rise in popularity of BoNT products among both physicians and consumers, clinicians should be aware of the current data and ongoing research.
The first botulinum neurotoxin (BoNT) approved by the US Food and Drug Administration (FDA) was onabotulinumtoxinA in 1989 for the treatment of strabismus and blepharospasm. It was not until 1992, however, that the aesthetic benefits of BoNT were first reported in the medical literature by Carruthers and Carruthers,1 and a cosmetic indication was not approved by the FDA until 2002. Since that time, the popularity of BoNT products has grown rapidly with a nearly 6500% increase in popularity from 1997 to 2015 in addition to the introduction of a variety of new BoNT formulations to the market.2 It is estimated by the American Society for Aesthetic Plastic Surgery that there were at least 4,000,000 BoNT injections performed in 2015 alone, making it the most popular nonsurgical aesthetic procedure available.2 As the demand for minimally invasive cosmetic procedures continues to increase, we will continue to see the introduction of additional formulations of BoNT products as well as novel administration techniques and delivery devices. In this article, we provide an update on current and upcoming BoNT products and also review the literature on novel administration methods based on studies published from January 1, 2014, to December 31, 2015.
Current Products
To date, there are only 4 FDA-approved formulations of BoNT available for clinical use (eg, cervical dystonia, strabismus, blepharospasm, headache, urinary incontinence) in the United States: abobotulinumtoxinA, incobotulinumtoxinA, onabotulinumtoxinA, and rimabotulinumtoxinB.The FDA-approved dermatologic indications (eg, moderate to severe glabellar or canthal lines, severe axillary hyperhidrosis) for these products are provided in the Table. On a global scale, there are several other commonly utilized formulations of BoNT, including a Korean serotype resembling onabotulinumtoxinA and a Chinese botulinum toxin type A.3 Although there is some evidence to demonstrate comparable efficacy and safety of these latter products, the literature is relatively lacking in comparison to the FDA-approved products.4,5
Upcoming Products
Currently, there are several new BoNT formulations being studied for clinical use. RT 002 (Revance Therapeutics, Inc) is a novel injectable formulation of onabotulinumtoxinA that consists of the purified neurotoxin in combination with patented TransMTS peptides that have been shown to provide high-binding avidity for the neurotoxin, and thus the product is designed to reduce diffusion to adjacent muscles and diminish unwanted effects. With a reduced level of neurotoxin dissemination, it is theorized that a higher administration of targeted doses can be injected, which may lead to a longer duration of desired effects.6 A clinical pilot study done to establish the safety and efficacy of RT 002 for treatment of moderate to severe glabellar lines evaluated 4 equally sized cohorts of 12 participants, each receiving single-dose administration of RT 002 ranging in potency equivalent to 25 U, 50 U, 75 U, and 100 U of abobotulinumtoxinA as determined by the gelatin phosphate method.6 It was concluded that RT 002 is both safe and efficacious with an extended duration of action, with a median duration of effect of 7 months observed in the highest dose group (dose equivalent to 100 U of abobotulinumtoxinA). Notably, 80% of all 48 participants maintained a minimum 1-point improvement in investigator-determined glabellar line severity scores at the 6-month time point and 60% achieved wrinkle scores of none or mild at 6 months posttreatment.6
DWP 450 (Daewoong Pharmaceutical Co, Ltd) is derived from the wild-type Clostridium botulinum and is reported to be of higher purity than standard onabotulinumtoxinA. An initial 16-week pilot study demonstrated that 20 U of DWP 450 is noninferior and of comparable efficacy and safety to 20 U of onabotulinumtoxinA in the treatment of glabellar lines.7
NTC (Botulax [Hugel, Inc]) is the name of the toxin derived from the C botulinum strain CBFC26, which has already been approved in many Asian, European, and Latin American countries for the treatment of blepharospasm. This formulation has demonstrated noninferiority to onabotulinumtoxinA at equivalent 20-U doses for the treatment of moderate to severe glabellar lines in a double-blind, randomized, multicenter, phase 3 trial of 272 participants with a 16-week follow-up.8
MT 10109L (Medytox Inc) is a unique product in that it is distributed as a liquid type A botulinum toxin rather than the standard freeze-dried formulation; thus, a major advantage of this product is its convenience, as it does not need reconstitution or dilution prior to administration. In a double-blind, randomized, active drug–controlled, phase 3 study of 168 participants, it was determined that MT 10109L (20 U) is comparable in efficacy to onabotulinumtoxinA (20 U) for the treatment of moderate to severe glabellar lines. No significant difference was seen between the 2 treatment groups when glabellar lines were assessed at rest at 4 and 16 weeks after treatment, but a significantly greater improvement in glabellar lines was seen at maximum frown in the MT 10109L group at the 16-week follow-up (P=.0064).9
Administration Techniques
With regard to safe and effective BoNT product administration techniques, a variety of studies have provided insight into optimal practice methods. A 2015 expert consensus statement formed by an American Society for Dermatologic Surgery task force reviewed data from 42 papers and unanimously determined that for all current type A BoNT products available in the United States, a vial of BoNT reconstituted appropriately for the purpose of facial injections can be reconstituted at least 4 weeks prior to administration without contamination risk or decrease in efficacy and that multiple patients can be treated with the same vial.Although the statement was not explicit on whether or not preserved or unpreserved saline is to be used, it is considered routine practice to use preservative-containing saline to reconstitute BoNT, as it has been shown to reduce patient discomfort and is not associated with adverse effects.10
Pain Minimization
With respect to minimizing the pain associated with BoNT injections, several studies have assessed administration techniques to minimize patient discomfort. A split-face, double-blind study of 20 participants demonstrated that the use of a 32-gauge needle has a significantly greater chance of reducing clinically significant levels of pain as compared to a 30-gauge needle when performing facial injections (P=.04). Overall, however, injections of the face and arms were on average only nominally and not significantly more painful with 30-gauge needles compared to 32-gauge needles.11
Another technique that has been found to reduce patient discomfort is the application of cold packs prior to injection. A study of patients with chronic facial palsy observed a significant reduction in pain with the administration of a cold (3°C–5°C) gel pack for 1 minute compared to a room temperature (20°C) gel pack prior to the administration of onabotulinumtoxinA into the platysma (P<.001).12 In the matter of injection with rimabotulinumtoxinB, which has been shown to be considerably more painful to receive than its more popularly administered counterpart onabotulinumtoxinA, a split-face pilot study examined the effect of increasing the pH of rimabotulinumtoxinB to 7.5 with sodium bicarbonate from the usual pH of 5.6.13,14 Pain was reported to be considerably less in the higher pH group and no reduction of efficacy was seen over the 10-week follow-up period.14
Delivery Methods
Several preliminary studies also have examined novel delivery techniques to identify minimally painful yet effective methods for administering BoNT. It has been reported that standard BoNT formulations are not effective as topical agents in a comparison study in which onabotulinumtoxinA injection was compared to topically applied onabotulinumtoxinA.15 However, a follow-up prospective study by the same authors has demonstrated efficacy of topical onabotulinumtoxinA following pretreatment with a fractional ablative CO2 laser for treatment of crow’s-feet. In this randomized, split-face, controlled trial (N=10), participants were first pretreated with topical lidocaine 30% before receiving a single pass of fractional ablative CO2 laser with no overlap and a pulse energy of 100 mJ. Within 60 seconds of laser treatment, participants then received either 100 U of abobotulinumtoxinA diluted in 0.1 mL of saline or simple normal saline applied topically. A clinically significant improvement in periorbital wrinkles was seen both at 1-week and 1-month posttreatment in the laser and onabotulinumtoxinA–treated group compared to the laser and topical saline–treated group (P<.02).15
Another unique administration method studied, albeit with less successful results, involves the use of iontophoresis to deliver BoNT painlessly in a transdermal fashion with the assistance of an electrical current.16 This prospective, randomized, assessor-blinded, split-axilla, controlled trial of 11 participants compared the effectiveness of administering onabotulinumtoxinA via iontophoresis to traditional injection with onabotulinumtoxinA (250 U). Iontophoresis was accomplished with a single electrode pad soaked with 250 U of onabotulinumtoxinA applied directly to the axilla and a second electrode pad soaked in 0.9% saline applied to the hand to complete the circuit. An alternating electrical current was slowly increased for 30 minutes to a maximum current of 15 mA with a voltage of 12 V. Among the 11 participants recruited, the side treated with traditional injection showed a significantly greater percentage reduction in baseline sweating at the 1-week, 1-month, and 6-month posttreatment evaluations compared to iontophoresis (84%, 76%, and 50%, respectively vs 73%, 22%, and 32%, respectively)(P<.05). Despite being less efficacious than standard injection therapy, participants reported that iontophoresis delivery was significantly less painful (P<.05).16
A high-pressure oxygen delivery device, which utilizes a powerful jet of microdroplets containing water, the drug, air, and oxygen to deliver medication onto the skin surface, also has been studied as a means of delivery of BoNT in a minimally painful manner. In this study, the device was used to assess the efficacy of transdermal delivery of BoNT via jet nebulization in the treatment of primary palmar, plantar, and axillary hyperhidrosis.17 The 20 participants included in the study were randomized to receive either a combination of lidocaine and onabotulinumtoxinA (50 U) administered through the device or lidocaine delivered through the device followed by multiple transcutaneous injections of onabotulinumtoxinA (100 U). Both treatments significantly reduced sweating compared to baseline as measured by a visual analogue scale at 3-month follow-up (P<.001), but the combination delivery of lidocaine and onabotulinumtoxinA via the device resulted in significantly less procedure-related pain and sweating (P<.001) as well as significantly greater patient satisfaction (P<.001).17
Optimizing Aesthetic Outcomes
A frequent concern of patients receiving BoNT for cosmetic purposes is a desire to avoid a “frozen” or expressionless look. As such, many clinicians have attempted a variety of techniques to achieve more natural aesthetic results. One such method is known as the multipoint and multilevel injection technique, which consists of utilizing multiple injection sites at varying depths (intramuscular, subcutaneous, or intradermal) and doses (2–6 U) depending on the degree of contractility of the targeted muscle. In a preliminary study of 223 participants using this technique with a total dose of 125 U of abobotulinumtoxinA, good and natural results were reported with perseveration of facial emotion in all participants in addition to a mean overall satisfaction rate of 6.4 of 7 on the Facial Line Treatment Satisfaction Questionnaire with the maximum satisfaction rating possible reported in 66% of cases.18 It also has been postulated that injection depth of BoNT can affect brow elevation whereupon deeper injection depths can result in inactivation of the brow depressors and allow for increased elevation of the eyebrows. This technique has been applied in attempts to correct brow height asymmetry. However, a prospective, split-face study of 23 women suggested that this method is not effective.19 Participants received 64 U of onabotulinumtoxinA via 16 injection sites in the glabella, forehead, and lateral canthal area with either all deep or all shallow injections depending on the side treated and whether brow-lift was desired. Results at 4 weeks posttreatment showed no significant difference in brow height, and it was concluded that eyebrow depressor muscles cannot be accurately targeted with deep injection into the muscle belly for correction of eyebrow height discrepancies.19 Conversely, a 5-year retrospective, nonrandomized study of 227 patients with 563 treatments utilizing a “microdroplet” technique reported success at selectively targeting the eyebrow depressors while leaving the brow elevators unaffected.20 Here, a total dose of 33 U of onabotulinumtoxinA was administered via microdroplets of 10 to 20 μL, each with more than 60 to 100 injections into the brow, glabella, and crow’s-feet area. This method of injection resulted in a statistically significant improvement of forehead lines and brow ptosis and furrowing at follow-up between 10 and 45 days after treatment (P<.0001). Additionally, average brow height was significantly increased from 24.6 mm to 25 mm after treatment (P=.02).20
Conclusion
The use of BoNT products for both on- and off-label cosmetic and medical indications has rapidly grown over the past 2 decades. As demonstrated in this review, a variety of promising new products and delivery techniques are being developed. Given the rise in popularity of BoNT products among both physicians and consumers, clinicians should be aware of the current data and ongoing research.
- Carruthers JD, Carruthers JA. Treatment of glabellar frown lines with C. botulinum-A exotoxin. J Dermatol Surg Oncol. 1992;18:17-21.
- American Society for Aesthetic Plastic Surgery. Cosmetic Surgery National Data Bank statistics. American Society for Aesthetic Plastic Surgery website. http://www.surgery.org/sites/default/files/ASAPS-Stats2015.pdf. Accessed June 12, 2016.
- Walker TJ, Dayan SH. Comparison and overview of currently available neurotoxins. J Clin Aesthet Dermatol. 2014;7:31-39.
- Feng Z, Sun Q, He L, et al. Optimal dosage of botulinum toxin type A for treatment of glabellar frown lines: efficacy and safety in a clinical trial. Dermatol Surg. 2015;41(suppl 1):S56-S63.
- Jiang HY, Chen S, Zhou J, et al. Diffusion of two botulinum toxins type A on the forehead: double-blinded, randomized, controlled study. Dermatol Surg. 2014;40:184-192.
- Garcia-Murray E, Velasco Villasenor ML, Acevedo B, et al. Safety and efficacy of RT002, an injectable botulinum toxin type A, for treating glabellar lines: results of a phase 1/2, open-label, sequential dose-escalation study. Dermatol Surg. 2015;41(suppl 1):S47-S55.
- Won CH, Kim HK, Kim BJ, et al. Comparative trial of a novel botulinum neurotoxin type A versus onabotulinumtoxinA in the treatment of glabellar lines: a multicenter, randomized, double-blind, active-controlled study. Int J Dermatol. 2015;54:227-234.
- Kim BJ, Kwon HH, Park SY, et al. Double-blind, randomized non-inferiority trial of a novel botulinum toxin A processed from the strain CBFC26, compared with onabotulinumtoxin A in the treatment of glabellar lines. J Eur Acad Dermatol Venereol. 2014;28:1761-1767.
- Kim JE, Song EJ, Choi GS, et al. The efficacy and safety of liquid-type botulinum toxin type A for the management of moderate to severe glabellar frown lines. Plast Reconstr Surg. 2015;135:732-741.
- Alam M, Bolotin D, Carruthers J, et al. Consensus statement regarding storage and reuse of previously reconstituted neuromodulators. Dermatol Surg. 2015;41:321-326.
- Alam M, Geisler A, Sadhwani D, et al. Effect of needle size on pain perception in patients treated with botulinum toxin type A injections: a randomized clinical trial. JAMA Dermatol. 2015;151:1194-1199.
- Pucks N, Thomas A, Hallam MJ, et al. Cutaneous cooling to manage botulinum toxin injection-associated pain in patients with facial palsy: a randomised controlled trial. J Plast Reconstr Aesthet Surg. 2015;68:1701-1705.
- Kranz G, Sycha T, Voller B, et al. Pain sensation during intradermal injections of three different botulinum toxin preparations in different doses and dilutions. Dermatol Surg. 2006;32:886-890.
- Lowe PL, Lowe NJ. Botulinum toxin type B: pH change reduces injection pain, retains efficacy. Dermatol Surg. 2014;40:1328-1333.
- Mahmoud BH, Burnett C, Ozog D. Prospective randomized controlled study to determine the effect of topical application of botulinum toxin A for crow’s feet after treatment with ablative fractional CO2 laser. Dermatol Surg. 2015;41(suppl 1):S75-S81.
- Montaser-Kouhsari L, Zartab H, Fanian F, et al. Comparison of intradermal injection with iontophoresis of abo-botulinum toxin A for the treatment of primary axillary hyperhidrosis: a randomized, controlled trial. J Dermatolog Treat. 2014;25:337-341.
- Iannitti T, Palmieri B, Aspiro A, et al. A preliminary study of painless and effective transdermal botulinum toxin A delivery by jet nebulization for treatment of primary hyperhidrosis. Drug Des Devel Ther. 2014;8:931-935.
- Iozzo I, Tengattini V, Antonucci VA. Multipoint and multilevel injection technique of botulinum toxin A in facial aesthetics. J Cosmet Dermatol. 2014;13:135-142.
- Sneath J, Humphrey S, Carruthers A, et al. Injecting botulinum toxin at different depths is not effective for the correction of eyebrow asymmetry. Dermatol Surg. 2015;41(suppl 1):S82-S87.
- Steinsapir KD, Rootman D, Wulc A, et al. Cosmetic microdroplet botulinum toxin A forehead lift: a new treatment paradigm. Ophthal Plast Reconstr Surg. 2015;31:263-268.
- Carruthers JD, Carruthers JA. Treatment of glabellar frown lines with C. botulinum-A exotoxin. J Dermatol Surg Oncol. 1992;18:17-21.
- American Society for Aesthetic Plastic Surgery. Cosmetic Surgery National Data Bank statistics. American Society for Aesthetic Plastic Surgery website. http://www.surgery.org/sites/default/files/ASAPS-Stats2015.pdf. Accessed June 12, 2016.
- Walker TJ, Dayan SH. Comparison and overview of currently available neurotoxins. J Clin Aesthet Dermatol. 2014;7:31-39.
- Feng Z, Sun Q, He L, et al. Optimal dosage of botulinum toxin type A for treatment of glabellar frown lines: efficacy and safety in a clinical trial. Dermatol Surg. 2015;41(suppl 1):S56-S63.
- Jiang HY, Chen S, Zhou J, et al. Diffusion of two botulinum toxins type A on the forehead: double-blinded, randomized, controlled study. Dermatol Surg. 2014;40:184-192.
- Garcia-Murray E, Velasco Villasenor ML, Acevedo B, et al. Safety and efficacy of RT002, an injectable botulinum toxin type A, for treating glabellar lines: results of a phase 1/2, open-label, sequential dose-escalation study. Dermatol Surg. 2015;41(suppl 1):S47-S55.
- Won CH, Kim HK, Kim BJ, et al. Comparative trial of a novel botulinum neurotoxin type A versus onabotulinumtoxinA in the treatment of glabellar lines: a multicenter, randomized, double-blind, active-controlled study. Int J Dermatol. 2015;54:227-234.
- Kim BJ, Kwon HH, Park SY, et al. Double-blind, randomized non-inferiority trial of a novel botulinum toxin A processed from the strain CBFC26, compared with onabotulinumtoxin A in the treatment of glabellar lines. J Eur Acad Dermatol Venereol. 2014;28:1761-1767.
- Kim JE, Song EJ, Choi GS, et al. The efficacy and safety of liquid-type botulinum toxin type A for the management of moderate to severe glabellar frown lines. Plast Reconstr Surg. 2015;135:732-741.
- Alam M, Bolotin D, Carruthers J, et al. Consensus statement regarding storage and reuse of previously reconstituted neuromodulators. Dermatol Surg. 2015;41:321-326.
- Alam M, Geisler A, Sadhwani D, et al. Effect of needle size on pain perception in patients treated with botulinum toxin type A injections: a randomized clinical trial. JAMA Dermatol. 2015;151:1194-1199.
- Pucks N, Thomas A, Hallam MJ, et al. Cutaneous cooling to manage botulinum toxin injection-associated pain in patients with facial palsy: a randomised controlled trial. J Plast Reconstr Aesthet Surg. 2015;68:1701-1705.
- Kranz G, Sycha T, Voller B, et al. Pain sensation during intradermal injections of three different botulinum toxin preparations in different doses and dilutions. Dermatol Surg. 2006;32:886-890.
- Lowe PL, Lowe NJ. Botulinum toxin type B: pH change reduces injection pain, retains efficacy. Dermatol Surg. 2014;40:1328-1333.
- Mahmoud BH, Burnett C, Ozog D. Prospective randomized controlled study to determine the effect of topical application of botulinum toxin A for crow’s feet after treatment with ablative fractional CO2 laser. Dermatol Surg. 2015;41(suppl 1):S75-S81.
- Montaser-Kouhsari L, Zartab H, Fanian F, et al. Comparison of intradermal injection with iontophoresis of abo-botulinum toxin A for the treatment of primary axillary hyperhidrosis: a randomized, controlled trial. J Dermatolog Treat. 2014;25:337-341.
- Iannitti T, Palmieri B, Aspiro A, et al. A preliminary study of painless and effective transdermal botulinum toxin A delivery by jet nebulization for treatment of primary hyperhidrosis. Drug Des Devel Ther. 2014;8:931-935.
- Iozzo I, Tengattini V, Antonucci VA. Multipoint and multilevel injection technique of botulinum toxin A in facial aesthetics. J Cosmet Dermatol. 2014;13:135-142.
- Sneath J, Humphrey S, Carruthers A, et al. Injecting botulinum toxin at different depths is not effective for the correction of eyebrow asymmetry. Dermatol Surg. 2015;41(suppl 1):S82-S87.
- Steinsapir KD, Rootman D, Wulc A, et al. Cosmetic microdroplet botulinum toxin A forehead lift: a new treatment paradigm. Ophthal Plast Reconstr Surg. 2015;31:263-268.
Practice Points
- Botulinum neurotoxin (BoNT) injection is the most popular nonsurgical aesthetic procedure available of which there are currently 4 products approved by the US Food and Drug Administration.
- A variety of new BoNT products with unique properties and formulations are currently being studied, some of which are already available for clinical use in foreign markets.
- Administration technique and novel product delivery methods also can be utilized to minimize pain and maximize aesthetic outcomes.
Hospitals increase CRE risk when they share patients
The more hospitals share patients, the more likely they are to have a problem with carbapenem-resistant Enterobacteriaceae (CRE), especially if long-term acute care hospitals (LTACHs) are in the mix, according to a state-wide investigation from Illinois.
Greater hospital centrality was independently associated with higher rates overall, and sharing four or more patients with a long-term acute care hospital (LTACH) in the 3-month study window doubled the rate of CRE cases.
Although it’s possible that was because of chance (P = 0.11), the link between LTACHs and CRE “is consistent with prior analyses that have shown the central role LTACHs have in” spreading the organism, said the researchers, led by Michael Ray of the Illinois Department of Public Health (Clin Infect Dis. 2016 Aug 2. pii: ciw461).
Patients often spend weeks in LTACH facilities for ongoing, serious health problems. The severity of illness, long stay, and sometimes chronic antibiotic use increase the risk of CRE exposure, and the team found that many LTACH patients are colonized.
“These findings have immediate public health implications. … Early interventions should be focused on the most connected facilities, as well as those with strong connections to LTACHs.” When one hospital has an outbreak, facilities that share its patients need to swing into action screening new admissions and taking other steps to prevent regional spread, the team said.
Meanwhile, “state-wide patient-sharing data, which are now increasingly available through sources like the Healthcare Cost and Utilization Project, provide an important way to assess hospital risk of CRE exposure based on its position in regional patient-sharing networks,” they noted. “Public health can play a critical role in identifying tightly connected hospitals and educating personnel at such facilities about their risk and need for enhanced infection control interventions.”
The team came to their conclusions after linking Illinois’ drug-resistant organisms registry with admissions data for 185 hospitals. About half reported at least one CRE case over 3 months, with a mean of 3.5 cases per hospital.
There was an average of 64 patient-sharing connections per facility, with a minimum of one connection and a maximum of 145 connections. Each additional patient two hospitals shared corresponded to a 3% increase in the CRE rate in urban facilities and a 6% increase in rural ones. The investigators didn’t explain the discrepancy, except to note that rural areas don’t have LTACHs.
Almost two-thirds of hospitals reporting CRE were in Chicago-area counties; almost half had shared at least one patient with an LTACH, and 21% had shared four or more.
CRE cases were an average of 64 years old, and equally distributed between men and women and black and white patients.
The Centers for Disease Control and Prevention funded the work. The authors had no disclosures.
The more hospitals share patients, the more likely they are to have a problem with carbapenem-resistant Enterobacteriaceae (CRE), especially if long-term acute care hospitals (LTACHs) are in the mix, according to a state-wide investigation from Illinois.
Greater hospital centrality was independently associated with higher rates overall, and sharing four or more patients with a long-term acute care hospital (LTACH) in the 3-month study window doubled the rate of CRE cases.
Although it’s possible that was because of chance (P = 0.11), the link between LTACHs and CRE “is consistent with prior analyses that have shown the central role LTACHs have in” spreading the organism, said the researchers, led by Michael Ray of the Illinois Department of Public Health (Clin Infect Dis. 2016 Aug 2. pii: ciw461).
Patients often spend weeks in LTACH facilities for ongoing, serious health problems. The severity of illness, long stay, and sometimes chronic antibiotic use increase the risk of CRE exposure, and the team found that many LTACH patients are colonized.
“These findings have immediate public health implications. … Early interventions should be focused on the most connected facilities, as well as those with strong connections to LTACHs.” When one hospital has an outbreak, facilities that share its patients need to swing into action screening new admissions and taking other steps to prevent regional spread, the team said.
Meanwhile, “state-wide patient-sharing data, which are now increasingly available through sources like the Healthcare Cost and Utilization Project, provide an important way to assess hospital risk of CRE exposure based on its position in regional patient-sharing networks,” they noted. “Public health can play a critical role in identifying tightly connected hospitals and educating personnel at such facilities about their risk and need for enhanced infection control interventions.”
The team came to their conclusions after linking Illinois’ drug-resistant organisms registry with admissions data for 185 hospitals. About half reported at least one CRE case over 3 months, with a mean of 3.5 cases per hospital.
There was an average of 64 patient-sharing connections per facility, with a minimum of one connection and a maximum of 145 connections. Each additional patient two hospitals shared corresponded to a 3% increase in the CRE rate in urban facilities and a 6% increase in rural ones. The investigators didn’t explain the discrepancy, except to note that rural areas don’t have LTACHs.
Almost two-thirds of hospitals reporting CRE were in Chicago-area counties; almost half had shared at least one patient with an LTACH, and 21% had shared four or more.
CRE cases were an average of 64 years old, and equally distributed between men and women and black and white patients.
The Centers for Disease Control and Prevention funded the work. The authors had no disclosures.
The more hospitals share patients, the more likely they are to have a problem with carbapenem-resistant Enterobacteriaceae (CRE), especially if long-term acute care hospitals (LTACHs) are in the mix, according to a state-wide investigation from Illinois.
Greater hospital centrality was independently associated with higher rates overall, and sharing four or more patients with a long-term acute care hospital (LTACH) in the 3-month study window doubled the rate of CRE cases.
Although it’s possible that was because of chance (P = 0.11), the link between LTACHs and CRE “is consistent with prior analyses that have shown the central role LTACHs have in” spreading the organism, said the researchers, led by Michael Ray of the Illinois Department of Public Health (Clin Infect Dis. 2016 Aug 2. pii: ciw461).
Patients often spend weeks in LTACH facilities for ongoing, serious health problems. The severity of illness, long stay, and sometimes chronic antibiotic use increase the risk of CRE exposure, and the team found that many LTACH patients are colonized.
“These findings have immediate public health implications. … Early interventions should be focused on the most connected facilities, as well as those with strong connections to LTACHs.” When one hospital has an outbreak, facilities that share its patients need to swing into action screening new admissions and taking other steps to prevent regional spread, the team said.
Meanwhile, “state-wide patient-sharing data, which are now increasingly available through sources like the Healthcare Cost and Utilization Project, provide an important way to assess hospital risk of CRE exposure based on its position in regional patient-sharing networks,” they noted. “Public health can play a critical role in identifying tightly connected hospitals and educating personnel at such facilities about their risk and need for enhanced infection control interventions.”
The team came to their conclusions after linking Illinois’ drug-resistant organisms registry with admissions data for 185 hospitals. About half reported at least one CRE case over 3 months, with a mean of 3.5 cases per hospital.
There was an average of 64 patient-sharing connections per facility, with a minimum of one connection and a maximum of 145 connections. Each additional patient two hospitals shared corresponded to a 3% increase in the CRE rate in urban facilities and a 6% increase in rural ones. The investigators didn’t explain the discrepancy, except to note that rural areas don’t have LTACHs.
Almost two-thirds of hospitals reporting CRE were in Chicago-area counties; almost half had shared at least one patient with an LTACH, and 21% had shared four or more.
CRE cases were an average of 64 years old, and equally distributed between men and women and black and white patients.
The Centers for Disease Control and Prevention funded the work. The authors had no disclosures.
FROM CLINICAL INFECTIOUS DISEASES
Key clinical point: The more hospitals share patients, the more likely they are to have a problem with CRE, especially if long-term acute care hospitals are in the mix.
Major finding: Sharing four or more patients with a long-term acute care hospital in the 3-month study window doubled the rate of CRE cases (P = 0.11).
Data source: 185 Illinois hospitals.
Disclosures: The Centers for Disease Control and Prevention funded the work. The authors had no disclosures.
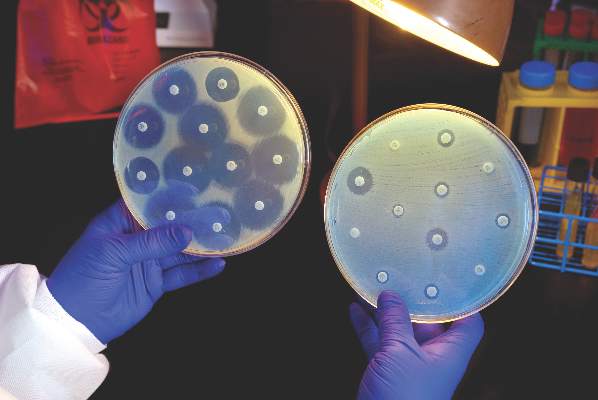
Proper Wound Management: How to Work With Patients
What does your patient need to know at the first visit?
A thorough patient history is imperative for proper diagnosis of wounds, thus detailed information on the onset, duration, temporality, modifying factors, symptoms, and attempted treatments should be provided. Associated comorbidities that may influence wound healing, such as diabetes mellitus or connective tissue diseases, must be considered when formulating a treatment regimen. Patients should disclose current medications, as certain medications (eg, vascular endothelial growth factor inhibitors) may decrease vascularization or soft tissue matrix regeneration, further complicating the wound healing process. All patients should have a basic understanding of the cause of their wound to have realistic expectations of the prognosis.
What are your go-to treatments?
Treatment ultimately depends on the cause of the wound. In general, proper healing requires a wound bed that is well vascularized and moistened without devitalized tissue or bacterial colonization. Wound dressings should be utilized to reduce dead space, control exudate, prevent bacterial overgrowth, and ensure proper fluid balance. Maintaining good overall health promotes proper healing. Thus, any relevant underlying medical conditions should be properly managed (eg, glycemic control for diabetic patients, management of fluid overload in patients with congestive heart failure).
When treating wounds, it is important to consider several factors. Although all wounds are colonized with microbes, not all wounds are infected. Thus, antibiotic therapy is not necessary for all wounds and should only be used to treat wounds that are clinically infected. Rule out pyoderma gangrenosum prior to wound debridement, as the associated pathergic response will notably worsen the ulcer. Wound dressings have an impact on the speed of wound healing, strength of repaired skin, and cosmetic appearance. Because no single dressing is perfect for all wounds, physicians should use their discretion when determining the type of wound dressing necessary.
Certain wounds require specific treatments. Off-loading and compression dressings/garments are the main components involved in the treatment of pressure ulcers. Protective wound care in conjunction with glycemic control is imperative for diabetic ulcers. Often, the causes of wounds are multifactorial and may complicate treatment. For instance, it is important to confirm that there is no associated arterial insufficiency before treating venous insufficiency with compression. Furthermore, patients with diabetic ulcers in association with venous insufficiency often have minimal response to hyperbaric oxygen treatment.
Several agents have been implicated to improve wound healing. Timolol, a topically applied beta-blocker, may promote keratinocyte migration and epithelialization of chronic refractory wounds. Recombinant human growth factors, most notably becaplermin (a platelet-derived growth factor), have been developed to promote cellular proliferation and angiogenesis, thereby improving healing of chronic wounds. Wounds that have devitalized tissue or contamination require debridement prior to further management.
How do you keep patients compliant with treatment?
Because recurrence is a common complication of chronic wounds, it is imperative that patients understand the importance of preventive care and follow-up appointments. Additionally, an open patient-physician dialogue may help address potential lifestyle limitations that may complicate wound care treatment. For instance, home care arrangement may be necessary to assist certain patient populations with wound care management.
What do you do if they refuse treatment?
Ultimately, it is hard to enforce treatment if the patient refuses. However, in my experience practicing dermatology, I have found it to be uncommon for patients to refuse treatment without a particular reason. If a patient refuses treatment, try to understand why and then try to alleviate any concerns by clarifying misconceptions and/or recommending alternative therapies.
What resources do you recommend to patients for more information?
Consult the American Academy of Dermatology website (https://www.aad.org/File%20Library/Unassigned/Wound-Dressings_Online-BF-DIR-Summer-2016--FINAL.pdf) for more information.
Additional resources include:
- Diabetic Wound Care (Source: American Podiatric Medical Association)(http://www.apma.org/Learn/FootHealth.cfm?ItemNumber=981)
- Pyoderma Gangrenosum (Source: Wound Care Centers)(http://www.woundcarecenters.org/article/wound-types/pyoderma-gangrenosum)
- Take the Pressure Off: A Patient Guide for Preventing and Treating Pressure Ulcers (Source: Association for the Advancement of Wound Care)(http://aawconline.org/wp-content/uploads/2012/04/Take-the-Pressure-Off.pdf)
- Wound Healing Society (http://woundheal.org/home.aspx)
What does your patient need to know at the first visit?
A thorough patient history is imperative for proper diagnosis of wounds, thus detailed information on the onset, duration, temporality, modifying factors, symptoms, and attempted treatments should be provided. Associated comorbidities that may influence wound healing, such as diabetes mellitus or connective tissue diseases, must be considered when formulating a treatment regimen. Patients should disclose current medications, as certain medications (eg, vascular endothelial growth factor inhibitors) may decrease vascularization or soft tissue matrix regeneration, further complicating the wound healing process. All patients should have a basic understanding of the cause of their wound to have realistic expectations of the prognosis.
What are your go-to treatments?
Treatment ultimately depends on the cause of the wound. In general, proper healing requires a wound bed that is well vascularized and moistened without devitalized tissue or bacterial colonization. Wound dressings should be utilized to reduce dead space, control exudate, prevent bacterial overgrowth, and ensure proper fluid balance. Maintaining good overall health promotes proper healing. Thus, any relevant underlying medical conditions should be properly managed (eg, glycemic control for diabetic patients, management of fluid overload in patients with congestive heart failure).
When treating wounds, it is important to consider several factors. Although all wounds are colonized with microbes, not all wounds are infected. Thus, antibiotic therapy is not necessary for all wounds and should only be used to treat wounds that are clinically infected. Rule out pyoderma gangrenosum prior to wound debridement, as the associated pathergic response will notably worsen the ulcer. Wound dressings have an impact on the speed of wound healing, strength of repaired skin, and cosmetic appearance. Because no single dressing is perfect for all wounds, physicians should use their discretion when determining the type of wound dressing necessary.
Certain wounds require specific treatments. Off-loading and compression dressings/garments are the main components involved in the treatment of pressure ulcers. Protective wound care in conjunction with glycemic control is imperative for diabetic ulcers. Often, the causes of wounds are multifactorial and may complicate treatment. For instance, it is important to confirm that there is no associated arterial insufficiency before treating venous insufficiency with compression. Furthermore, patients with diabetic ulcers in association with venous insufficiency often have minimal response to hyperbaric oxygen treatment.
Several agents have been implicated to improve wound healing. Timolol, a topically applied beta-blocker, may promote keratinocyte migration and epithelialization of chronic refractory wounds. Recombinant human growth factors, most notably becaplermin (a platelet-derived growth factor), have been developed to promote cellular proliferation and angiogenesis, thereby improving healing of chronic wounds. Wounds that have devitalized tissue or contamination require debridement prior to further management.
How do you keep patients compliant with treatment?
Because recurrence is a common complication of chronic wounds, it is imperative that patients understand the importance of preventive care and follow-up appointments. Additionally, an open patient-physician dialogue may help address potential lifestyle limitations that may complicate wound care treatment. For instance, home care arrangement may be necessary to assist certain patient populations with wound care management.
What do you do if they refuse treatment?
Ultimately, it is hard to enforce treatment if the patient refuses. However, in my experience practicing dermatology, I have found it to be uncommon for patients to refuse treatment without a particular reason. If a patient refuses treatment, try to understand why and then try to alleviate any concerns by clarifying misconceptions and/or recommending alternative therapies.
What resources do you recommend to patients for more information?
Consult the American Academy of Dermatology website (https://www.aad.org/File%20Library/Unassigned/Wound-Dressings_Online-BF-DIR-Summer-2016--FINAL.pdf) for more information.
Additional resources include:
- Diabetic Wound Care (Source: American Podiatric Medical Association)(http://www.apma.org/Learn/FootHealth.cfm?ItemNumber=981)
- Pyoderma Gangrenosum (Source: Wound Care Centers)(http://www.woundcarecenters.org/article/wound-types/pyoderma-gangrenosum)
- Take the Pressure Off: A Patient Guide for Preventing and Treating Pressure Ulcers (Source: Association for the Advancement of Wound Care)(http://aawconline.org/wp-content/uploads/2012/04/Take-the-Pressure-Off.pdf)
- Wound Healing Society (http://woundheal.org/home.aspx)
What does your patient need to know at the first visit?
A thorough patient history is imperative for proper diagnosis of wounds, thus detailed information on the onset, duration, temporality, modifying factors, symptoms, and attempted treatments should be provided. Associated comorbidities that may influence wound healing, such as diabetes mellitus or connective tissue diseases, must be considered when formulating a treatment regimen. Patients should disclose current medications, as certain medications (eg, vascular endothelial growth factor inhibitors) may decrease vascularization or soft tissue matrix regeneration, further complicating the wound healing process. All patients should have a basic understanding of the cause of their wound to have realistic expectations of the prognosis.
What are your go-to treatments?
Treatment ultimately depends on the cause of the wound. In general, proper healing requires a wound bed that is well vascularized and moistened without devitalized tissue or bacterial colonization. Wound dressings should be utilized to reduce dead space, control exudate, prevent bacterial overgrowth, and ensure proper fluid balance. Maintaining good overall health promotes proper healing. Thus, any relevant underlying medical conditions should be properly managed (eg, glycemic control for diabetic patients, management of fluid overload in patients with congestive heart failure).
When treating wounds, it is important to consider several factors. Although all wounds are colonized with microbes, not all wounds are infected. Thus, antibiotic therapy is not necessary for all wounds and should only be used to treat wounds that are clinically infected. Rule out pyoderma gangrenosum prior to wound debridement, as the associated pathergic response will notably worsen the ulcer. Wound dressings have an impact on the speed of wound healing, strength of repaired skin, and cosmetic appearance. Because no single dressing is perfect for all wounds, physicians should use their discretion when determining the type of wound dressing necessary.
Certain wounds require specific treatments. Off-loading and compression dressings/garments are the main components involved in the treatment of pressure ulcers. Protective wound care in conjunction with glycemic control is imperative for diabetic ulcers. Often, the causes of wounds are multifactorial and may complicate treatment. For instance, it is important to confirm that there is no associated arterial insufficiency before treating venous insufficiency with compression. Furthermore, patients with diabetic ulcers in association with venous insufficiency often have minimal response to hyperbaric oxygen treatment.
Several agents have been implicated to improve wound healing. Timolol, a topically applied beta-blocker, may promote keratinocyte migration and epithelialization of chronic refractory wounds. Recombinant human growth factors, most notably becaplermin (a platelet-derived growth factor), have been developed to promote cellular proliferation and angiogenesis, thereby improving healing of chronic wounds. Wounds that have devitalized tissue or contamination require debridement prior to further management.
How do you keep patients compliant with treatment?
Because recurrence is a common complication of chronic wounds, it is imperative that patients understand the importance of preventive care and follow-up appointments. Additionally, an open patient-physician dialogue may help address potential lifestyle limitations that may complicate wound care treatment. For instance, home care arrangement may be necessary to assist certain patient populations with wound care management.
What do you do if they refuse treatment?
Ultimately, it is hard to enforce treatment if the patient refuses. However, in my experience practicing dermatology, I have found it to be uncommon for patients to refuse treatment without a particular reason. If a patient refuses treatment, try to understand why and then try to alleviate any concerns by clarifying misconceptions and/or recommending alternative therapies.
What resources do you recommend to patients for more information?
Consult the American Academy of Dermatology website (https://www.aad.org/File%20Library/Unassigned/Wound-Dressings_Online-BF-DIR-Summer-2016--FINAL.pdf) for more information.
Additional resources include:
- Diabetic Wound Care (Source: American Podiatric Medical Association)(http://www.apma.org/Learn/FootHealth.cfm?ItemNumber=981)
- Pyoderma Gangrenosum (Source: Wound Care Centers)(http://www.woundcarecenters.org/article/wound-types/pyoderma-gangrenosum)
- Take the Pressure Off: A Patient Guide for Preventing and Treating Pressure Ulcers (Source: Association for the Advancement of Wound Care)(http://aawconline.org/wp-content/uploads/2012/04/Take-the-Pressure-Off.pdf)
- Wound Healing Society (http://woundheal.org/home.aspx)

Combined OCs remain a good choice for teen acne
MINNEAPOLIS – Whether a young female patient has a refractory flare of inflammatory acne, or has a condition that can predispose to androgen excess, using a hormonal approach can be an effective management tool for controlling adolescent acne.
During a presentation at the annual meeting of the Society for Pediatric Dermatology, Dr. Diane Thiboutot outlined tips and tricks for optimizing hormonal therapy for acne in teens, and referred to the new acne treatment guidelines from the American Academy of Dermatology, which clarify when to treat with hormones, which to choose, and when further testing might be indicated.

The full range of hormonal therapy options for acne can include oral contraceptives, which block ovarian hormone production; antiandrogens such as spironolactone, and the less commonly used flutamide, which blocks the effects of androgen on the skin; and glucocorticoids, which block adrenal production.
The 2016 guidelines recommend oral contraceptives as an effective treatment for inflammatory acne in females (J Am Acad Dermatol. 2016 May;74[5]; 945-973.e33). Combined oral contraceptives (COCs) reduce serum androgens, and reduce free testosterone by increasing sex hormone binding globulin production, thus reducing sebum production. “The only things that really decrease sebum are oral contraceptives in women, and isotretinoin,” said Dr. Thiboutot, professor of dermatology at Penn State University, Hershey.
For most female adolescents with acne, hormonal testing is not indicated. The AAD guidelines recommend laboratory evaluation for younger patients with acne who have clinical signs of androgen excess, such as early onset body odor and axillary and/or pubic hair, accelerated growth, advanced bone age, or early genital maturation. Just obtaining a hand film for bone age and mapping growth against a growth chart can be a good initial screening tool when considering whether to perform hormonal testing, she noted.
For postpubertal females in whom polycystic ovary syndrome (PCOS) or other hyperandrogenic states are suspected, hormonal testing is indicated in the presence of the clinical signs of infrequent menses and infertility, hirsutism, truncal obesity, androgenetic alopecia, polycystic ovaries, or clitoromegaly.
In searching for an endocrine disorder, Dr. Thiboutot recommends checking total and free testosterone, luteinizing hormone/follicle stimulating hormone ratio, 17-hydroxyprogesterone levels, and dehydroepiandrosterone (DHEA-S) levels. These tests should be performed at least 6 weeks after the patient has been off hormonal contraception, and should be done during the menstrual period, or during the week prior to menses, in order to avoid ovulation-related hormonal changes.
Lab findings consistent with congenital adrenal hyperplasia include elevated serum DHEA-S, together with elevated 17-hydroxyprogesterone or testosterone. A PCOS diagnosis can be made in adolescent females if there is clinical or laboratory evidence of hyperandrogenism with concomitant persistent oligomenorrhea.
Acne related to hyperandrogenism may respond well to oral contraceptives, but COCs can also be an effective alternative to repeated courses of isotretinoin and antibiotics, as well as an effective adjunct to topical therapy, Dr. Thiboutot said.
When beginning a patient on oral contraceptives, it’s not necessary to perform a pelvic exam or obtain a Pap smear before initiating the COC, but it is important to obtain a thorough medical history and an accurate blood pressure measurement at the outset, she noted. The World Health Organization (WHO) has established recommendations outlining contraindications to COC use, also identifying populations in whom COCs should be used with caution, and who should be monitored.
Headaches are a condition frequently seen among healthy teens and young women, and one for which the WHO advises caution. There are concerns that women with migraines may be at increased risk of stroke if they take COCs, but the overall risk is low, and the American College of Obstetricians and Gynecologists (ACOG) advises that COCs can be considered for women younger than 35 with migraines if they have no focal neurologic signs, are nonsmokers, and are otherwise healthy, Dr. Thiboutot added.
A large Food and Drug Administration–sponsored retrospective cohort study examined the risk of venous thromboembolism in contraceptive users. In April 2012, the FDA concluded that though the risk of blood clots may be higher for those on hormonal contraception methods than for those who are not using them, the risk of blood clots during pregnancy and the postpartum period is higher than the thromboembolism risk for contraceptive users.
Regarding the potential for antibiotics to reduce contraceptive efficacy, Dr. Thiboutot said,“it’s okay to use oral contraceptives with antibiotics. There’s a lot of misunderstanding about antibiotics and combined oral contraceptives.” She cited an ACOG practice bulletin that reported that only rifampin has been shown to reduce serum steroid levels when taken with oral contraceptives (Obstet Gynecol. 2006 Jun;107[6]:1453-72).
According to the 2016 AAD guidelines, the use of oral glucocorticoids may be appropriate over the short term when initiating therapy for severe inflammatory acne. “Pharmacokinetic studies have not demonstrated decreased oral contraceptive levels with common antibiotics,” Dr. Thiboutot said.
Spironolactone, according to the new guidelines, is useful for acne in select females. Spironolactone is an androgen receptor and 5a-reductase blocker, and its antiandrogen effects can improve acne. Many patients do well with 25-50 mg twice daily, though breast tenderness and menstrual irregularities are commonly seen side effects, she noted. If a woman taking spironolactone becomes pregnant, there’s a risk of hypospadias for a male fetus.
Though spironolactone carries a boxed warning because of tumorigenicity observed in animal studies, Dr. Thiboutot said that a large Danish study searched for any association between breast, uterine, or ovarian cancers and spironolactone use. Among the 2.3 million women studied, no increased association was seen (Cancer Epidemiol. 2013 Dec;37:870-5).
She also noted that there’s “low usefulness in monitoring potassium levels in young healthy women on spironolactone.” She cited a study that compared 974 healthy young women taking spironolactone with 1,165 women who were not on spironolactone, which found that the hyperkalemia rate of 0.72% among those on spironolactone was equivalent to the 0.76% baseline rate of hyperkalemia in the young, healthy female population (JAMA Dermatol. 2015;151[9];941-944).
Oral corticosteroids for acne, Dr. Thiboutot said, should be reserved to quiet a severe bout of inflammatory acne while standard therapies are being initiated.
She reported being an investigator or a consultant for a number of pharmaceutical companies.
On Twitter @karioakes
MINNEAPOLIS – Whether a young female patient has a refractory flare of inflammatory acne, or has a condition that can predispose to androgen excess, using a hormonal approach can be an effective management tool for controlling adolescent acne.
During a presentation at the annual meeting of the Society for Pediatric Dermatology, Dr. Diane Thiboutot outlined tips and tricks for optimizing hormonal therapy for acne in teens, and referred to the new acne treatment guidelines from the American Academy of Dermatology, which clarify when to treat with hormones, which to choose, and when further testing might be indicated.
The full range of hormonal therapy options for acne can include oral contraceptives, which block ovarian hormone production; antiandrogens such as spironolactone, and the less commonly used flutamide, which blocks the effects of androgen on the skin; and glucocorticoids, which block adrenal production.
The 2016 guidelines recommend oral contraceptives as an effective treatment for inflammatory acne in females (J Am Acad Dermatol. 2016 May;74[5]; 945-973.e33). Combined oral contraceptives (COCs) reduce serum androgens, and reduce free testosterone by increasing sex hormone binding globulin production, thus reducing sebum production. “The only things that really decrease sebum are oral contraceptives in women, and isotretinoin,” said Dr. Thiboutot, professor of dermatology at Penn State University, Hershey.
For most female adolescents with acne, hormonal testing is not indicated. The AAD guidelines recommend laboratory evaluation for younger patients with acne who have clinical signs of androgen excess, such as early onset body odor and axillary and/or pubic hair, accelerated growth, advanced bone age, or early genital maturation. Just obtaining a hand film for bone age and mapping growth against a growth chart can be a good initial screening tool when considering whether to perform hormonal testing, she noted.
For postpubertal females in whom polycystic ovary syndrome (PCOS) or other hyperandrogenic states are suspected, hormonal testing is indicated in the presence of the clinical signs of infrequent menses and infertility, hirsutism, truncal obesity, androgenetic alopecia, polycystic ovaries, or clitoromegaly.
In searching for an endocrine disorder, Dr. Thiboutot recommends checking total and free testosterone, luteinizing hormone/follicle stimulating hormone ratio, 17-hydroxyprogesterone levels, and dehydroepiandrosterone (DHEA-S) levels. These tests should be performed at least 6 weeks after the patient has been off hormonal contraception, and should be done during the menstrual period, or during the week prior to menses, in order to avoid ovulation-related hormonal changes.
Lab findings consistent with congenital adrenal hyperplasia include elevated serum DHEA-S, together with elevated 17-hydroxyprogesterone or testosterone. A PCOS diagnosis can be made in adolescent females if there is clinical or laboratory evidence of hyperandrogenism with concomitant persistent oligomenorrhea.
Acne related to hyperandrogenism may respond well to oral contraceptives, but COCs can also be an effective alternative to repeated courses of isotretinoin and antibiotics, as well as an effective adjunct to topical therapy, Dr. Thiboutot said.
When beginning a patient on oral contraceptives, it’s not necessary to perform a pelvic exam or obtain a Pap smear before initiating the COC, but it is important to obtain a thorough medical history and an accurate blood pressure measurement at the outset, she noted. The World Health Organization (WHO) has established recommendations outlining contraindications to COC use, also identifying populations in whom COCs should be used with caution, and who should be monitored.
Headaches are a condition frequently seen among healthy teens and young women, and one for which the WHO advises caution. There are concerns that women with migraines may be at increased risk of stroke if they take COCs, but the overall risk is low, and the American College of Obstetricians and Gynecologists (ACOG) advises that COCs can be considered for women younger than 35 with migraines if they have no focal neurologic signs, are nonsmokers, and are otherwise healthy, Dr. Thiboutot added.
A large Food and Drug Administration–sponsored retrospective cohort study examined the risk of venous thromboembolism in contraceptive users. In April 2012, the FDA concluded that though the risk of blood clots may be higher for those on hormonal contraception methods than for those who are not using them, the risk of blood clots during pregnancy and the postpartum period is higher than the thromboembolism risk for contraceptive users.
Regarding the potential for antibiotics to reduce contraceptive efficacy, Dr. Thiboutot said,“it’s okay to use oral contraceptives with antibiotics. There’s a lot of misunderstanding about antibiotics and combined oral contraceptives.” She cited an ACOG practice bulletin that reported that only rifampin has been shown to reduce serum steroid levels when taken with oral contraceptives (Obstet Gynecol. 2006 Jun;107[6]:1453-72).
According to the 2016 AAD guidelines, the use of oral glucocorticoids may be appropriate over the short term when initiating therapy for severe inflammatory acne. “Pharmacokinetic studies have not demonstrated decreased oral contraceptive levels with common antibiotics,” Dr. Thiboutot said.
Spironolactone, according to the new guidelines, is useful for acne in select females. Spironolactone is an androgen receptor and 5a-reductase blocker, and its antiandrogen effects can improve acne. Many patients do well with 25-50 mg twice daily, though breast tenderness and menstrual irregularities are commonly seen side effects, she noted. If a woman taking spironolactone becomes pregnant, there’s a risk of hypospadias for a male fetus.
Though spironolactone carries a boxed warning because of tumorigenicity observed in animal studies, Dr. Thiboutot said that a large Danish study searched for any association between breast, uterine, or ovarian cancers and spironolactone use. Among the 2.3 million women studied, no increased association was seen (Cancer Epidemiol. 2013 Dec;37:870-5).
She also noted that there’s “low usefulness in monitoring potassium levels in young healthy women on spironolactone.” She cited a study that compared 974 healthy young women taking spironolactone with 1,165 women who were not on spironolactone, which found that the hyperkalemia rate of 0.72% among those on spironolactone was equivalent to the 0.76% baseline rate of hyperkalemia in the young, healthy female population (JAMA Dermatol. 2015;151[9];941-944).
Oral corticosteroids for acne, Dr. Thiboutot said, should be reserved to quiet a severe bout of inflammatory acne while standard therapies are being initiated.
She reported being an investigator or a consultant for a number of pharmaceutical companies.
On Twitter @karioakes
MINNEAPOLIS – Whether a young female patient has a refractory flare of inflammatory acne, or has a condition that can predispose to androgen excess, using a hormonal approach can be an effective management tool for controlling adolescent acne.
During a presentation at the annual meeting of the Society for Pediatric Dermatology, Dr. Diane Thiboutot outlined tips and tricks for optimizing hormonal therapy for acne in teens, and referred to the new acne treatment guidelines from the American Academy of Dermatology, which clarify when to treat with hormones, which to choose, and when further testing might be indicated.
The full range of hormonal therapy options for acne can include oral contraceptives, which block ovarian hormone production; antiandrogens such as spironolactone, and the less commonly used flutamide, which blocks the effects of androgen on the skin; and glucocorticoids, which block adrenal production.
The 2016 guidelines recommend oral contraceptives as an effective treatment for inflammatory acne in females (J Am Acad Dermatol. 2016 May;74[5]; 945-973.e33). Combined oral contraceptives (COCs) reduce serum androgens, and reduce free testosterone by increasing sex hormone binding globulin production, thus reducing sebum production. “The only things that really decrease sebum are oral contraceptives in women, and isotretinoin,” said Dr. Thiboutot, professor of dermatology at Penn State University, Hershey.
For most female adolescents with acne, hormonal testing is not indicated. The AAD guidelines recommend laboratory evaluation for younger patients with acne who have clinical signs of androgen excess, such as early onset body odor and axillary and/or pubic hair, accelerated growth, advanced bone age, or early genital maturation. Just obtaining a hand film for bone age and mapping growth against a growth chart can be a good initial screening tool when considering whether to perform hormonal testing, she noted.
For postpubertal females in whom polycystic ovary syndrome (PCOS) or other hyperandrogenic states are suspected, hormonal testing is indicated in the presence of the clinical signs of infrequent menses and infertility, hirsutism, truncal obesity, androgenetic alopecia, polycystic ovaries, or clitoromegaly.
In searching for an endocrine disorder, Dr. Thiboutot recommends checking total and free testosterone, luteinizing hormone/follicle stimulating hormone ratio, 17-hydroxyprogesterone levels, and dehydroepiandrosterone (DHEA-S) levels. These tests should be performed at least 6 weeks after the patient has been off hormonal contraception, and should be done during the menstrual period, or during the week prior to menses, in order to avoid ovulation-related hormonal changes.
Lab findings consistent with congenital adrenal hyperplasia include elevated serum DHEA-S, together with elevated 17-hydroxyprogesterone or testosterone. A PCOS diagnosis can be made in adolescent females if there is clinical or laboratory evidence of hyperandrogenism with concomitant persistent oligomenorrhea.
Acne related to hyperandrogenism may respond well to oral contraceptives, but COCs can also be an effective alternative to repeated courses of isotretinoin and antibiotics, as well as an effective adjunct to topical therapy, Dr. Thiboutot said.
When beginning a patient on oral contraceptives, it’s not necessary to perform a pelvic exam or obtain a Pap smear before initiating the COC, but it is important to obtain a thorough medical history and an accurate blood pressure measurement at the outset, she noted. The World Health Organization (WHO) has established recommendations outlining contraindications to COC use, also identifying populations in whom COCs should be used with caution, and who should be monitored.
Headaches are a condition frequently seen among healthy teens and young women, and one for which the WHO advises caution. There are concerns that women with migraines may be at increased risk of stroke if they take COCs, but the overall risk is low, and the American College of Obstetricians and Gynecologists (ACOG) advises that COCs can be considered for women younger than 35 with migraines if they have no focal neurologic signs, are nonsmokers, and are otherwise healthy, Dr. Thiboutot added.
A large Food and Drug Administration–sponsored retrospective cohort study examined the risk of venous thromboembolism in contraceptive users. In April 2012, the FDA concluded that though the risk of blood clots may be higher for those on hormonal contraception methods than for those who are not using them, the risk of blood clots during pregnancy and the postpartum period is higher than the thromboembolism risk for contraceptive users.
Regarding the potential for antibiotics to reduce contraceptive efficacy, Dr. Thiboutot said,“it’s okay to use oral contraceptives with antibiotics. There’s a lot of misunderstanding about antibiotics and combined oral contraceptives.” She cited an ACOG practice bulletin that reported that only rifampin has been shown to reduce serum steroid levels when taken with oral contraceptives (Obstet Gynecol. 2006 Jun;107[6]:1453-72).
According to the 2016 AAD guidelines, the use of oral glucocorticoids may be appropriate over the short term when initiating therapy for severe inflammatory acne. “Pharmacokinetic studies have not demonstrated decreased oral contraceptive levels with common antibiotics,” Dr. Thiboutot said.
Spironolactone, according to the new guidelines, is useful for acne in select females. Spironolactone is an androgen receptor and 5a-reductase blocker, and its antiandrogen effects can improve acne. Many patients do well with 25-50 mg twice daily, though breast tenderness and menstrual irregularities are commonly seen side effects, she noted. If a woman taking spironolactone becomes pregnant, there’s a risk of hypospadias for a male fetus.
Though spironolactone carries a boxed warning because of tumorigenicity observed in animal studies, Dr. Thiboutot said that a large Danish study searched for any association between breast, uterine, or ovarian cancers and spironolactone use. Among the 2.3 million women studied, no increased association was seen (Cancer Epidemiol. 2013 Dec;37:870-5).
She also noted that there’s “low usefulness in monitoring potassium levels in young healthy women on spironolactone.” She cited a study that compared 974 healthy young women taking spironolactone with 1,165 women who were not on spironolactone, which found that the hyperkalemia rate of 0.72% among those on spironolactone was equivalent to the 0.76% baseline rate of hyperkalemia in the young, healthy female population (JAMA Dermatol. 2015;151[9];941-944).
Oral corticosteroids for acne, Dr. Thiboutot said, should be reserved to quiet a severe bout of inflammatory acne while standard therapies are being initiated.
She reported being an investigator or a consultant for a number of pharmaceutical companies.
On Twitter @karioakes
EXPERT ANALYSIS FROM THE SPD ANNUAL MEETING

Hat-Wearing Patterns in Spectators Attending Baseball Games: A 10-Year Retrospective Comparison
Spectators at baseball games may be exposed to excess solar UV radiation (UVR), which has been linked to the development of both melanoma and nonmelanoma skin cancers.1,2 Although baseball hats traditionally are worn to demonstrate team support, they also may provide some sun protection for the head and face where skin cancers are commonly found.
The importance of protecting the skin from solar UVR has led to sun-protection programs and community education as well as efforts to evaluate the impact of these programs. Major League Baseball (MLB) has partnered with the American Academy of Dermatology since 1999 to promote the importance of sun protection and raise skin cancer awareness through its Play Sun Smart program.3 A study conducted 10 years ago (N=2030) evaluated hat use in spectators at MLB games and noted that less than half of all spectators in seating sections exposed to direct sunlight wore hats.4 The purpose of the current study was to evaluate how public education about sun protection has impacted the use of hats by spectators at MLB games in 2015 compared to the prior study in 2006.
Methods
Data were collected during a 3-game series (2 day games, 1 night game) in August 2015 in New York, New York. During one of the day games, 18,000 fans received a free wide-brimmed hat. High-resolution digital photographs of seating sections were obtained using a camera with a 300-mm lens. Using the same methodology as the prior study,4 sunny and shaded seating sections were photographed during all 3 games (Figure). Photographs of each section were analyzed by an independent reviewer using a high-resolution computer screen. Spectators wearing head coverings—baseball hats, visors, or hats with circumferential brims—were defined as using hats. The number of spectators wearing hats versus not wearing hats was recorded for all identical sections of interest. Bleacher seating was analyzed separately, as spectators presumably knew in advance of the continuous direct sun exposure during day games, and a subset of young children in the bleachers (<10 years of age) also was assessed. A continuously sunny section also was evaluated at the second and sixth innings to see if hats were presumably purchased during exposure. Statistical significance was determined using χ2 tests with P<.05 indicating statistical significance.

Results
This analysis consisted of 3539 spectators. In both the sunny and shaded sections of a day game, there were more spectators wearing hats (49% and 37%, respectively)(P<.001) than in the same sections at night games (35% and 29%, respectively)(Table 1). During the day game, more spectators wore hats in the sunny section than in the adjacent shaded section (49% vs 37%; P<.001). Analysis of the same 2 sections during the night game revealed no significant differences.
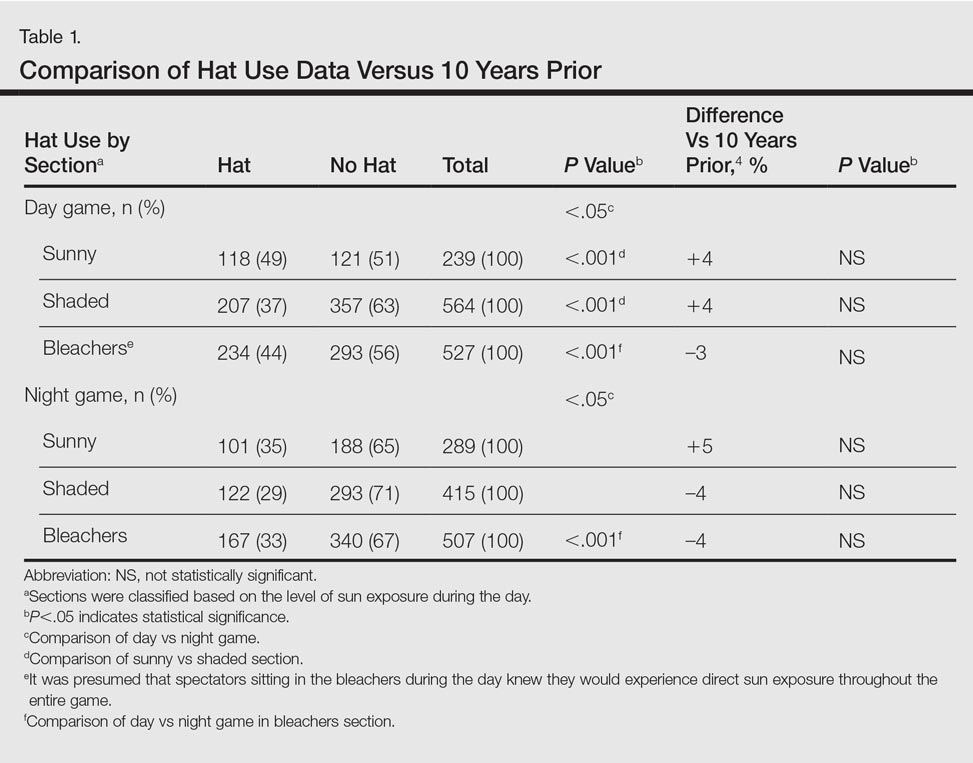
Spectators sitting in the bleachers during a day game who presumably knew to anticipate direct sun exposure showed no significant differences in hat-wearing patterns versus the sunny section (44% vs 49%) but were more likely to wear hats compared to those sitting in the bleachers at the night game (44% vs 33%)(P<.001)(Table 1). There was no significant difference in the number of hats worn by spectators in the sunny section in the second inning (43%) versus the same section after continuous sun exposure at the sixth inning (44%)(Table 2). Significantly more children seated in the bleachers during the day game wore hats compared to adults in the same section (64% vs 42%; P<.001)(Table 3). During the hat giveaway day, significantly more spectators wore hats (the majority of which were the free giveaway hats) across all sections studied (P<.001)(Table 4).
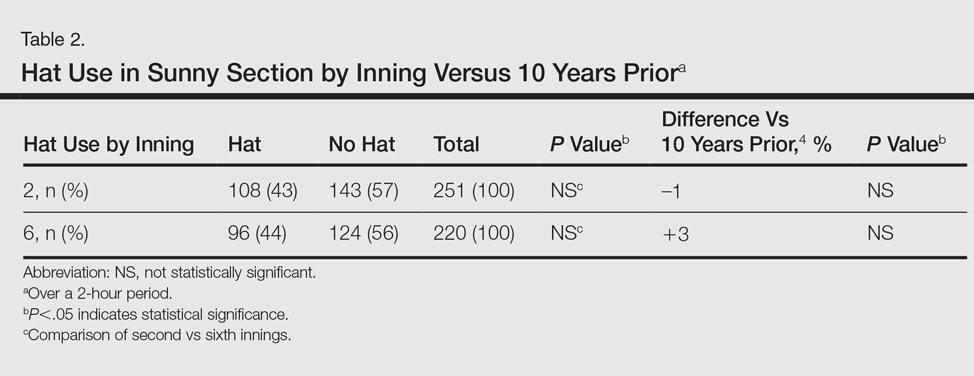


Comment
More than 23 million spectators attended daytime MLB games in 2015, with millions more attending minor league and amateur events.5Although sun-protection messages tend to be well understood and received by society, many choose to ignore them.6
In partnership with the American Academy of Dermatology, the MLB’s Play Sun Smart program has promoted UVR risk awareness at sporting events since 1999.3 Those affiliated with MLB teams also receive annual skin cancer screenings in conjunction with a public education effort in May of each season. However, despite the years of sun-protection education, our study found that less than half of attendees wore hats for UVR protection. In fact, there were no significant differences noted across all of the hat-wearing parameters studied (day vs night game, sunny vs shaded section, sunny section over course of game) between the current study compared to the results from 10 years prior4 (Tables 1 and 2). For spectators in the bleacher section, even presumably knowing in advance that seating would be in the sun did not significantly increase hat-wearing behavior. Although skin cancer rates continue to rise, hat-wearing trends remain stable, revealing a concerning trend.
Increased availability of sunscreen has led to improved sun-protective behaviors in many populations.7 In our study, the free hat giveaway had the greatest impact on hat wearing, which suggests that improved availability and access to hats can lead to an important opportunity for sun-protection programs to partner with hat manufacturers to augment their use and protective impact.
Sun avoidance during childhood and adolescence has been shown to decrease the risk for melanoma.1 Young children had the highest rate of hat usage in the current study, possibly due to parental example or dictates. Research has shown the importance of role models in promoting sun safety to young children,8,9 so perhaps use of hats by parents or MLB players contributed to the hat-wearing behavior observed in this subpopulation.
Given the limited change observed in hat-wearing behaviors over the last decade, a knowledge and behavioral gap appears to exist that may be able to be exploited to enhance future sun protection. Also, based on our findings, the MLB and other sun-protection education campaigns may wish to augment their UVR protective messages by offering hat giveaways, which appear to have a notable impact.
Acknowledgment
The authors thank Jessie Skapik, BS (New York, New York), for her independent review of the spectator photographs.
References
1. Rigel DS. Cutaneous ultraviolet exposure and its relationship to the development of skin cancer. J Am Acad Dermatol. 2008;58(5, suppl 2):S129-S132.
2. Lim HW, James WD, Rigel DS, et al. Adverse effects of ultraviolet radiation from the use of indoor tanning equipment: time to ban the tan. J Am Acad Dermatol. 2011;64:893-902.
3. Play Sun Smart. American Academy of Dermatology website. https://www.aad.org/public/spot-skin-cancer/programs/play-sun-smart. Accessed August 25, 2016.
4. Rigel AS, Lebwohl MG. Hat-wearing patterns in persons attending baseball games. J Am Acad Dermatol. 2006;54:918-919.
5. MLB attendance report - 2016. ESPN website. www.espn.go.com/mlb/attendance. Accessed May 20, 2016.
6. Turner D, Harrison SL, Buettner P, et al. Does being a “SunSmart School” influence hat-wearing compliance? an ecological study of hat-wearing rates at Australian primary schools in a region of high sun exposure [published online December 29, 2013]. Prev Med. 2014;60:107-114.
7. Dubas LE, Adams BB. Sunscreen use and availability among female collegiate athletes [published online February 3, 2012]. J Am Acad Dermatol. 2012;67:876.e1-876.e6.
8. O’Riodran DL, Geller AC, Brooks DR, et al. Sunburn reduction through parental role modeling and sunscreen vigilance. J Pediatr. 2003;142:67-72.
9. Turrisi R, Hillhouse J, Heavin S, et al. Examination of the short-term efficacy of a parent-based intervention to prevent skin cancer. J Behav Med. 2004;27:393-412.
Spectators at baseball games may be exposed to excess solar UV radiation (UVR), which has been linked to the development of both melanoma and nonmelanoma skin cancers.1,2 Although baseball hats traditionally are worn to demonstrate team support, they also may provide some sun protection for the head and face where skin cancers are commonly found.
The importance of protecting the skin from solar UVR has led to sun-protection programs and community education as well as efforts to evaluate the impact of these programs. Major League Baseball (MLB) has partnered with the American Academy of Dermatology since 1999 to promote the importance of sun protection and raise skin cancer awareness through its Play Sun Smart program.3 A study conducted 10 years ago (N=2030) evaluated hat use in spectators at MLB games and noted that less than half of all spectators in seating sections exposed to direct sunlight wore hats.4 The purpose of the current study was to evaluate how public education about sun protection has impacted the use of hats by spectators at MLB games in 2015 compared to the prior study in 2006.
Methods
Data were collected during a 3-game series (2 day games, 1 night game) in August 2015 in New York, New York. During one of the day games, 18,000 fans received a free wide-brimmed hat. High-resolution digital photographs of seating sections were obtained using a camera with a 300-mm lens. Using the same methodology as the prior study,4 sunny and shaded seating sections were photographed during all 3 games (Figure). Photographs of each section were analyzed by an independent reviewer using a high-resolution computer screen. Spectators wearing head coverings—baseball hats, visors, or hats with circumferential brims—were defined as using hats. The number of spectators wearing hats versus not wearing hats was recorded for all identical sections of interest. Bleacher seating was analyzed separately, as spectators presumably knew in advance of the continuous direct sun exposure during day games, and a subset of young children in the bleachers (<10 years of age) also was assessed. A continuously sunny section also was evaluated at the second and sixth innings to see if hats were presumably purchased during exposure. Statistical significance was determined using χ2 tests with P<.05 indicating statistical significance.
Results
This analysis consisted of 3539 spectators. In both the sunny and shaded sections of a day game, there were more spectators wearing hats (49% and 37%, respectively)(P<.001) than in the same sections at night games (35% and 29%, respectively)(Table 1). During the day game, more spectators wore hats in the sunny section than in the adjacent shaded section (49% vs 37%; P<.001). Analysis of the same 2 sections during the night game revealed no significant differences.
Spectators sitting in the bleachers during a day game who presumably knew to anticipate direct sun exposure showed no significant differences in hat-wearing patterns versus the sunny section (44% vs 49%) but were more likely to wear hats compared to those sitting in the bleachers at the night game (44% vs 33%)(P<.001)(Table 1). There was no significant difference in the number of hats worn by spectators in the sunny section in the second inning (43%) versus the same section after continuous sun exposure at the sixth inning (44%)(Table 2). Significantly more children seated in the bleachers during the day game wore hats compared to adults in the same section (64% vs 42%; P<.001)(Table 3). During the hat giveaway day, significantly more spectators wore hats (the majority of which were the free giveaway hats) across all sections studied (P<.001)(Table 4).
Comment
More than 23 million spectators attended daytime MLB games in 2015, with millions more attending minor league and amateur events.5Although sun-protection messages tend to be well understood and received by society, many choose to ignore them.6
In partnership with the American Academy of Dermatology, the MLB’s Play Sun Smart program has promoted UVR risk awareness at sporting events since 1999.3 Those affiliated with MLB teams also receive annual skin cancer screenings in conjunction with a public education effort in May of each season. However, despite the years of sun-protection education, our study found that less than half of attendees wore hats for UVR protection. In fact, there were no significant differences noted across all of the hat-wearing parameters studied (day vs night game, sunny vs shaded section, sunny section over course of game) between the current study compared to the results from 10 years prior4 (Tables 1 and 2). For spectators in the bleacher section, even presumably knowing in advance that seating would be in the sun did not significantly increase hat-wearing behavior. Although skin cancer rates continue to rise, hat-wearing trends remain stable, revealing a concerning trend.
Increased availability of sunscreen has led to improved sun-protective behaviors in many populations.7 In our study, the free hat giveaway had the greatest impact on hat wearing, which suggests that improved availability and access to hats can lead to an important opportunity for sun-protection programs to partner with hat manufacturers to augment their use and protective impact.
Sun avoidance during childhood and adolescence has been shown to decrease the risk for melanoma.1 Young children had the highest rate of hat usage in the current study, possibly due to parental example or dictates. Research has shown the importance of role models in promoting sun safety to young children,8,9 so perhaps use of hats by parents or MLB players contributed to the hat-wearing behavior observed in this subpopulation.
Given the limited change observed in hat-wearing behaviors over the last decade, a knowledge and behavioral gap appears to exist that may be able to be exploited to enhance future sun protection. Also, based on our findings, the MLB and other sun-protection education campaigns may wish to augment their UVR protective messages by offering hat giveaways, which appear to have a notable impact.
Acknowledgment
The authors thank Jessie Skapik, BS (New York, New York), for her independent review of the spectator photographs.
Spectators at baseball games may be exposed to excess solar UV radiation (UVR), which has been linked to the development of both melanoma and nonmelanoma skin cancers.1,2 Although baseball hats traditionally are worn to demonstrate team support, they also may provide some sun protection for the head and face where skin cancers are commonly found.
The importance of protecting the skin from solar UVR has led to sun-protection programs and community education as well as efforts to evaluate the impact of these programs. Major League Baseball (MLB) has partnered with the American Academy of Dermatology since 1999 to promote the importance of sun protection and raise skin cancer awareness through its Play Sun Smart program.3 A study conducted 10 years ago (N=2030) evaluated hat use in spectators at MLB games and noted that less than half of all spectators in seating sections exposed to direct sunlight wore hats.4 The purpose of the current study was to evaluate how public education about sun protection has impacted the use of hats by spectators at MLB games in 2015 compared to the prior study in 2006.
Methods
Data were collected during a 3-game series (2 day games, 1 night game) in August 2015 in New York, New York. During one of the day games, 18,000 fans received a free wide-brimmed hat. High-resolution digital photographs of seating sections were obtained using a camera with a 300-mm lens. Using the same methodology as the prior study,4 sunny and shaded seating sections were photographed during all 3 games (Figure). Photographs of each section were analyzed by an independent reviewer using a high-resolution computer screen. Spectators wearing head coverings—baseball hats, visors, or hats with circumferential brims—were defined as using hats. The number of spectators wearing hats versus not wearing hats was recorded for all identical sections of interest. Bleacher seating was analyzed separately, as spectators presumably knew in advance of the continuous direct sun exposure during day games, and a subset of young children in the bleachers (<10 years of age) also was assessed. A continuously sunny section also was evaluated at the second and sixth innings to see if hats were presumably purchased during exposure. Statistical significance was determined using χ2 tests with P<.05 indicating statistical significance.
Results
This analysis consisted of 3539 spectators. In both the sunny and shaded sections of a day game, there were more spectators wearing hats (49% and 37%, respectively)(P<.001) than in the same sections at night games (35% and 29%, respectively)(Table 1). During the day game, more spectators wore hats in the sunny section than in the adjacent shaded section (49% vs 37%; P<.001). Analysis of the same 2 sections during the night game revealed no significant differences.
Spectators sitting in the bleachers during a day game who presumably knew to anticipate direct sun exposure showed no significant differences in hat-wearing patterns versus the sunny section (44% vs 49%) but were more likely to wear hats compared to those sitting in the bleachers at the night game (44% vs 33%)(P<.001)(Table 1). There was no significant difference in the number of hats worn by spectators in the sunny section in the second inning (43%) versus the same section after continuous sun exposure at the sixth inning (44%)(Table 2). Significantly more children seated in the bleachers during the day game wore hats compared to adults in the same section (64% vs 42%; P<.001)(Table 3). During the hat giveaway day, significantly more spectators wore hats (the majority of which were the free giveaway hats) across all sections studied (P<.001)(Table 4).
Comment
More than 23 million spectators attended daytime MLB games in 2015, with millions more attending minor league and amateur events.5Although sun-protection messages tend to be well understood and received by society, many choose to ignore them.6
In partnership with the American Academy of Dermatology, the MLB’s Play Sun Smart program has promoted UVR risk awareness at sporting events since 1999.3 Those affiliated with MLB teams also receive annual skin cancer screenings in conjunction with a public education effort in May of each season. However, despite the years of sun-protection education, our study found that less than half of attendees wore hats for UVR protection. In fact, there were no significant differences noted across all of the hat-wearing parameters studied (day vs night game, sunny vs shaded section, sunny section over course of game) between the current study compared to the results from 10 years prior4 (Tables 1 and 2). For spectators in the bleacher section, even presumably knowing in advance that seating would be in the sun did not significantly increase hat-wearing behavior. Although skin cancer rates continue to rise, hat-wearing trends remain stable, revealing a concerning trend.
Increased availability of sunscreen has led to improved sun-protective behaviors in many populations.7 In our study, the free hat giveaway had the greatest impact on hat wearing, which suggests that improved availability and access to hats can lead to an important opportunity for sun-protection programs to partner with hat manufacturers to augment their use and protective impact.
Sun avoidance during childhood and adolescence has been shown to decrease the risk for melanoma.1 Young children had the highest rate of hat usage in the current study, possibly due to parental example or dictates. Research has shown the importance of role models in promoting sun safety to young children,8,9 so perhaps use of hats by parents or MLB players contributed to the hat-wearing behavior observed in this subpopulation.
Given the limited change observed in hat-wearing behaviors over the last decade, a knowledge and behavioral gap appears to exist that may be able to be exploited to enhance future sun protection. Also, based on our findings, the MLB and other sun-protection education campaigns may wish to augment their UVR protective messages by offering hat giveaways, which appear to have a notable impact.
Acknowledgment
The authors thank Jessie Skapik, BS (New York, New York), for her independent review of the spectator photographs.
References
1. Rigel DS. Cutaneous ultraviolet exposure and its relationship to the development of skin cancer. J Am Acad Dermatol. 2008;58(5, suppl 2):S129-S132.
2. Lim HW, James WD, Rigel DS, et al. Adverse effects of ultraviolet radiation from the use of indoor tanning equipment: time to ban the tan. J Am Acad Dermatol. 2011;64:893-902.
3. Play Sun Smart. American Academy of Dermatology website. https://www.aad.org/public/spot-skin-cancer/programs/play-sun-smart. Accessed August 25, 2016.
4. Rigel AS, Lebwohl MG. Hat-wearing patterns in persons attending baseball games. J Am Acad Dermatol. 2006;54:918-919.
5. MLB attendance report - 2016. ESPN website. www.espn.go.com/mlb/attendance. Accessed May 20, 2016.
6. Turner D, Harrison SL, Buettner P, et al. Does being a “SunSmart School” influence hat-wearing compliance? an ecological study of hat-wearing rates at Australian primary schools in a region of high sun exposure [published online December 29, 2013]. Prev Med. 2014;60:107-114.
7. Dubas LE, Adams BB. Sunscreen use and availability among female collegiate athletes [published online February 3, 2012]. J Am Acad Dermatol. 2012;67:876.e1-876.e6.
8. O’Riodran DL, Geller AC, Brooks DR, et al. Sunburn reduction through parental role modeling and sunscreen vigilance. J Pediatr. 2003;142:67-72.
9. Turrisi R, Hillhouse J, Heavin S, et al. Examination of the short-term efficacy of a parent-based intervention to prevent skin cancer. J Behav Med. 2004;27:393-412.
References
1. Rigel DS. Cutaneous ultraviolet exposure and its relationship to the development of skin cancer. J Am Acad Dermatol. 2008;58(5, suppl 2):S129-S132.
2. Lim HW, James WD, Rigel DS, et al. Adverse effects of ultraviolet radiation from the use of indoor tanning equipment: time to ban the tan. J Am Acad Dermatol. 2011;64:893-902.
3. Play Sun Smart. American Academy of Dermatology website. https://www.aad.org/public/spot-skin-cancer/programs/play-sun-smart. Accessed August 25, 2016.
4. Rigel AS, Lebwohl MG. Hat-wearing patterns in persons attending baseball games. J Am Acad Dermatol. 2006;54:918-919.
5. MLB attendance report - 2016. ESPN website. www.espn.go.com/mlb/attendance. Accessed May 20, 2016.
6. Turner D, Harrison SL, Buettner P, et al. Does being a “SunSmart School” influence hat-wearing compliance? an ecological study of hat-wearing rates at Australian primary schools in a region of high sun exposure [published online December 29, 2013]. Prev Med. 2014;60:107-114.
7. Dubas LE, Adams BB. Sunscreen use and availability among female collegiate athletes [published online February 3, 2012]. J Am Acad Dermatol. 2012;67:876.e1-876.e6.
8. O’Riodran DL, Geller AC, Brooks DR, et al. Sunburn reduction through parental role modeling and sunscreen vigilance. J Pediatr. 2003;142:67-72.
9. Turrisi R, Hillhouse J, Heavin S, et al. Examination of the short-term efficacy of a parent-based intervention to prevent skin cancer. J Behav Med. 2004;27:393-412.
Practice Points
- With less than half of attendees wearing hats to Major League Baseball games, there has been limited change in hat-wearing behavior over the last decade, possibly due to a knowledge or behavioral gap.
- Improved availability and access to hats can lead to improved sun-protective behaviors.

Mass administration of malaria drugs may cut morbidity during Ebola outbreaks
Mass administration of malaria chemoprevention during Ebola virus disease outbreaks may reduce cases of fever, according to a study published in PLOS ONE.
During the October-December 2014 Ebola virus disease (EVD) outbreak in Liberia, health care services were limited, negatively impacting malaria treatment. Hoping to reduce malaria-associated morbidity, investigators targeted four neighborhoods in Monrovia, Liberia – with a total population of 551,971 – for a mass drug administration (MDA) of malaria chemoprevention. MDA participants were divided into two treatment rounds, with 102,372 households verified as receiving treatment with the drug combination artesunate/amodiaquine by community leaders and a malaria committee in round 1, and 103,497 households verified in round 2.
Incidences of self-reported fever episodes declined significantly after round 1 (1.5%), compared with the month prior to round 1 (4.2%) (P < .0001). Self-reported fever incidences in children younger than 5 years of age (6.9%) and in older household members (3.8%) both decreased, to 1.1% and 1.6%, respectively, after round 1 of the MDA.
The researchers also found that self-reported fever was 4.9% lower after round 1 in household members who took a full course of artesunate/amodiaquine malaria chemoprevention (ASAQ-CP) but only 0.6% lower among household members who did not start or not complete a full course of ASAQ-CP. Still, reported incidence of fever declined in both groups, although the risk difference (RD) was significantly larger among the group that took part in the ASAQ-CP course (P < .001).
“Despite high acceptance and coverage of the MDA and the small impact of side effects, initiation of malaria chemoprevention was low, possibly due to health messaging and behavior in the pre-Ebola outbreak period and the ongoing lack of health care services,” researchers concluded. “Combining MDAs during Ebola outbreaks with longer-term interventions to prevent malaria and to improve access to health care might reduce the proportion of respondents saving their treatment for future malaria episodes.”
Read the full study in PLOS ONE (doi: 10.1371/journal.pone.0161311).
Mass administration of malaria chemoprevention during Ebola virus disease outbreaks may reduce cases of fever, according to a study published in PLOS ONE.
During the October-December 2014 Ebola virus disease (EVD) outbreak in Liberia, health care services were limited, negatively impacting malaria treatment. Hoping to reduce malaria-associated morbidity, investigators targeted four neighborhoods in Monrovia, Liberia – with a total population of 551,971 – for a mass drug administration (MDA) of malaria chemoprevention. MDA participants were divided into two treatment rounds, with 102,372 households verified as receiving treatment with the drug combination artesunate/amodiaquine by community leaders and a malaria committee in round 1, and 103,497 households verified in round 2.
Incidences of self-reported fever episodes declined significantly after round 1 (1.5%), compared with the month prior to round 1 (4.2%) (P < .0001). Self-reported fever incidences in children younger than 5 years of age (6.9%) and in older household members (3.8%) both decreased, to 1.1% and 1.6%, respectively, after round 1 of the MDA.
The researchers also found that self-reported fever was 4.9% lower after round 1 in household members who took a full course of artesunate/amodiaquine malaria chemoprevention (ASAQ-CP) but only 0.6% lower among household members who did not start or not complete a full course of ASAQ-CP. Still, reported incidence of fever declined in both groups, although the risk difference (RD) was significantly larger among the group that took part in the ASAQ-CP course (P < .001).
“Despite high acceptance and coverage of the MDA and the small impact of side effects, initiation of malaria chemoprevention was low, possibly due to health messaging and behavior in the pre-Ebola outbreak period and the ongoing lack of health care services,” researchers concluded. “Combining MDAs during Ebola outbreaks with longer-term interventions to prevent malaria and to improve access to health care might reduce the proportion of respondents saving their treatment for future malaria episodes.”
Read the full study in PLOS ONE (doi: 10.1371/journal.pone.0161311).
Mass administration of malaria chemoprevention during Ebola virus disease outbreaks may reduce cases of fever, according to a study published in PLOS ONE.
During the October-December 2014 Ebola virus disease (EVD) outbreak in Liberia, health care services were limited, negatively impacting malaria treatment. Hoping to reduce malaria-associated morbidity, investigators targeted four neighborhoods in Monrovia, Liberia – with a total population of 551,971 – for a mass drug administration (MDA) of malaria chemoprevention. MDA participants were divided into two treatment rounds, with 102,372 households verified as receiving treatment with the drug combination artesunate/amodiaquine by community leaders and a malaria committee in round 1, and 103,497 households verified in round 2.
Incidences of self-reported fever episodes declined significantly after round 1 (1.5%), compared with the month prior to round 1 (4.2%) (P < .0001). Self-reported fever incidences in children younger than 5 years of age (6.9%) and in older household members (3.8%) both decreased, to 1.1% and 1.6%, respectively, after round 1 of the MDA.
The researchers also found that self-reported fever was 4.9% lower after round 1 in household members who took a full course of artesunate/amodiaquine malaria chemoprevention (ASAQ-CP) but only 0.6% lower among household members who did not start or not complete a full course of ASAQ-CP. Still, reported incidence of fever declined in both groups, although the risk difference (RD) was significantly larger among the group that took part in the ASAQ-CP course (P < .001).
“Despite high acceptance and coverage of the MDA and the small impact of side effects, initiation of malaria chemoprevention was low, possibly due to health messaging and behavior in the pre-Ebola outbreak period and the ongoing lack of health care services,” researchers concluded. “Combining MDAs during Ebola outbreaks with longer-term interventions to prevent malaria and to improve access to health care might reduce the proportion of respondents saving their treatment for future malaria episodes.”
Read the full study in PLOS ONE (doi: 10.1371/journal.pone.0161311).
FROM PLOS ONE