User login
WCD: Dapsone gel effective for acne in women of color
VANCOUVER – Dapsone gel 5% proved effective and well tolerated for facial acne in women with skin of color in a multicenter pilot study.
The study was conducted because even though dapsone gel 5% (Aczone) is approved for the treatment of acne on the strength of two pivotal randomized, double-blind clinical trials totaling more than 3,000 patients, scant data exist on the topical agent’s performance in women with skin of color, Dr. Andrew F. Alexis explained at the World Congress of Dermatology.

He presented an open-label, seven-center, 12-week, single-arm study involving 68 women of color – three-quarters of whom were black – who treated their facial acne with dapsone gel 5% twice daily as monotherapy.
Participants averaged a mean baseline score of 2.6 on the 0-4 Global Acne Assessment Score (GAAS), with a mean total of 50 inflammatory and noninflammatory acne lesions on the face.
The primary endpoint was change in GAAS at 12 weeks, although patients also were formally assessed at 2 and 6 weeks. The average reduction in GAAS was 8.8% at 2 weeks, 20% at 6 weeks, and 39% at 12 weeks. At week 12, 43% of the women were categorized as responders, meaning they had a GAAS of 0 (meaning no acne lesions) or 1 (indicating mild disease), reported Dr. Alexis of Mt. Sinai Hospital in New York.
Total lesion counts dropped steadily throughout the 12-week trial: by 16% from baseline to week 2, 30% at week 6, and 52% at week 12. Inflammatory lesions responded best, with a 65% reduction in number at week 12.
Patient-reported outcomes on the validated, 17-item Acne Symptom and Impact Scale were favorable: Reductions of roughly 50% were documented over 12 weeks on the scale’s two domains, acne signs and quality of life impact.
No clinically meaningful treatment-related adverse events were reported in the study, although a handful of women reported trace levels of redness, burning, dryness, and/or oiliness.
Acne is more common among African American than white women. In a large epidemiologic study of adolescent and adult women, the prevalence of acne vulgaris was 37% in African Americans, compared with 24% in whites (J. Eur. Acad. Dermatol. Venereol. 2011;25:1054-60).
Dr. Alexis’ study was sponsored by Allergan. He reported serving as a consultant to and receiving research grants from the company.
VANCOUVER – Dapsone gel 5% proved effective and well tolerated for facial acne in women with skin of color in a multicenter pilot study.
The study was conducted because even though dapsone gel 5% (Aczone) is approved for the treatment of acne on the strength of two pivotal randomized, double-blind clinical trials totaling more than 3,000 patients, scant data exist on the topical agent’s performance in women with skin of color, Dr. Andrew F. Alexis explained at the World Congress of Dermatology.
He presented an open-label, seven-center, 12-week, single-arm study involving 68 women of color – three-quarters of whom were black – who treated their facial acne with dapsone gel 5% twice daily as monotherapy.
Participants averaged a mean baseline score of 2.6 on the 0-4 Global Acne Assessment Score (GAAS), with a mean total of 50 inflammatory and noninflammatory acne lesions on the face.
The primary endpoint was change in GAAS at 12 weeks, although patients also were formally assessed at 2 and 6 weeks. The average reduction in GAAS was 8.8% at 2 weeks, 20% at 6 weeks, and 39% at 12 weeks. At week 12, 43% of the women were categorized as responders, meaning they had a GAAS of 0 (meaning no acne lesions) or 1 (indicating mild disease), reported Dr. Alexis of Mt. Sinai Hospital in New York.
Total lesion counts dropped steadily throughout the 12-week trial: by 16% from baseline to week 2, 30% at week 6, and 52% at week 12. Inflammatory lesions responded best, with a 65% reduction in number at week 12.
Patient-reported outcomes on the validated, 17-item Acne Symptom and Impact Scale were favorable: Reductions of roughly 50% were documented over 12 weeks on the scale’s two domains, acne signs and quality of life impact.
No clinically meaningful treatment-related adverse events were reported in the study, although a handful of women reported trace levels of redness, burning, dryness, and/or oiliness.
Acne is more common among African American than white women. In a large epidemiologic study of adolescent and adult women, the prevalence of acne vulgaris was 37% in African Americans, compared with 24% in whites (J. Eur. Acad. Dermatol. Venereol. 2011;25:1054-60).
Dr. Alexis’ study was sponsored by Allergan. He reported serving as a consultant to and receiving research grants from the company.
VANCOUVER – Dapsone gel 5% proved effective and well tolerated for facial acne in women with skin of color in a multicenter pilot study.
The study was conducted because even though dapsone gel 5% (Aczone) is approved for the treatment of acne on the strength of two pivotal randomized, double-blind clinical trials totaling more than 3,000 patients, scant data exist on the topical agent’s performance in women with skin of color, Dr. Andrew F. Alexis explained at the World Congress of Dermatology.
He presented an open-label, seven-center, 12-week, single-arm study involving 68 women of color – three-quarters of whom were black – who treated their facial acne with dapsone gel 5% twice daily as monotherapy.
Participants averaged a mean baseline score of 2.6 on the 0-4 Global Acne Assessment Score (GAAS), with a mean total of 50 inflammatory and noninflammatory acne lesions on the face.
The primary endpoint was change in GAAS at 12 weeks, although patients also were formally assessed at 2 and 6 weeks. The average reduction in GAAS was 8.8% at 2 weeks, 20% at 6 weeks, and 39% at 12 weeks. At week 12, 43% of the women were categorized as responders, meaning they had a GAAS of 0 (meaning no acne lesions) or 1 (indicating mild disease), reported Dr. Alexis of Mt. Sinai Hospital in New York.
Total lesion counts dropped steadily throughout the 12-week trial: by 16% from baseline to week 2, 30% at week 6, and 52% at week 12. Inflammatory lesions responded best, with a 65% reduction in number at week 12.
Patient-reported outcomes on the validated, 17-item Acne Symptom and Impact Scale were favorable: Reductions of roughly 50% were documented over 12 weeks on the scale’s two domains, acne signs and quality of life impact.
No clinically meaningful treatment-related adverse events were reported in the study, although a handful of women reported trace levels of redness, burning, dryness, and/or oiliness.
Acne is more common among African American than white women. In a large epidemiologic study of adolescent and adult women, the prevalence of acne vulgaris was 37% in African Americans, compared with 24% in whites (J. Eur. Acad. Dermatol. Venereol. 2011;25:1054-60).
Dr. Alexis’ study was sponsored by Allergan. He reported serving as a consultant to and receiving research grants from the company.
AT WCD 2015
Key clinical point: Dapsone gel 5% is effective and well tolerated for treatment of facial acne in women with skin of color.
Major finding: Women of color experienced a mean 39% reduction in Global Acne Assessment Scores after 12 weeks of self-treatment of facial acne using dapsone gel 5% twice daily as monotherapy.
Data source: This was a 68-patient, open-label, seven-site, single-arm, 12-week study.
Disclosures: The study was sponsored by Allergan. Dr. Andrew F. Alexis reported serving as a consultant to and receiving research grants from the company.

Gout increases risk of vascular disease and events
My late father, H. H. Samson, an anesthesiologist, was convinced that hyperuricemia and over consumption of sugar were instigating factors for vascular disease and published on the subject (S. Afr. Med. J. 1978 Oct 7;54:590-1.) It has only taken some 30-odd years to prove him right! - Dr. Russell Samson, medical editor, Vascular Specialist.
Gout’s association with a host of vascular events was confirmed in a new study that explored the links between the inflammatory condition and coronary artery disease, peripheral vascular disease, and cerebrovascular events.
Though both men and women with gout were at increased risk for vascular events overall, the association appeared strongest for women. Dr. Lorna Clarson of Keele (England) University and her associates drew these conclusions from a retrospective cohort study of men and women with an incident diagnosis of gout (Ann. Rheum. Dis. 2015;74:642-7).
Gout, caused by the deposition of uric acid crystals in joints, is characterized by acute flares of intensely painful and inflamed joints. However, the state of hyperuricemia that predisposes patients to acute attacks of gout may precede the first attack by years, and may persist between flares. The proinflammatory course of the natural history of gout has increasingly been recognized as a potential contributor to vascular disease.
The precise mechanism by which gout may increase vascular risk has not been identified. Dr. Clarson and associates noted that in addition to the acute and chronic inflammation associated with gout and hyperuricemia, serum uric acid may have a more direct effect on vascular health, as urate crystal deposition on vessel walls may promote vascular damage.
To clarify gout’s impact on vascular risk, Dr. Clarson and her associates used the Clinical Practice Datalink, a large United Kingdom health database, to compare 8,366 patients with gout to 39,766 age- and sex-matched controls. None of those studied had a baseline history of vascular disease, and all were aged 50 or older.
Careful accounting for covariates was accomplished by multivariate analysis that took into account sex, age, body mass index, tobacco and alcohol consumption, statin or aspirin use, and any history of hypertension, dyslipidemia, or chronic kidney disease. In addition, the study employed the composite Charlson Comorbidity Index, which weights 19 comorbid conditions – including diabetes – to arrive at a single score that captures many risk factors. Patients in the cohort were tracked until their first vascular event, or until death or loss to follow-up.
To assess the incidence of vascular events, the study noted the first recording in the medical record of any events signaling vascular disease. These included angina or myocardial infarction, transient ischemic attack and stroke, and a range of diagnoses associated with peripheral vascular disease.
Final analysis after accounting for the many covariates tracked in the study showed increased risk for vascular events for those with gout, with a definite difference between the sexes. For men, gout predicted an increased risk of any vascular event (hazard ratio, 1.06; 95% confidence interval, 1.01–1.12) and of coronary heart disease and peripheral vascular disease.
For women, gout predicted an increased risk of all vascular events (HR, 1.25; 95% CI, 1.15-1.35) except myocardial infarction and cerebrovascular disease overall. Further, the degree of increased risk of vascular events was greater for women than for men with gout (P < .001 for intersex difference).
Noting that “clinical management of gout in primary care is suboptimal,” Dr. Clarson and her colleagues urged greater attention to screening for vascular risk in those diagnosed with gout; these individuals comprise a significant population of over 8 million people in the United States. Regarding the sex differences unearthed in their study,
Dr. Clarson and her associates observed that “both gout and vascular disease have historically been considered diseases of men ... [M]ore attention should be paid to prompt and reliable diagnosis of gout, followed by optimal management in female patients, including serious consideration of vascular risk reduction.”
In an editorial accompanying Dr. Clarson’s report (Ann. Rheum. Dis. 2015;74:631-4),Dr. Jasvinder Singh, commented that Dr. Clarson and colleagues’ study is limited by its lack of validation of gout and cardiac diagnoses and the self-selection bias inherent in using primary care registries, rather than a true population-based cohort.
Patients older than 35 or 40 with gout should be screened and followed with lipid profiles, hemoglobin A1c levels, blood pressure levels, and smoking status, and should undergo an assessment of other lifestyle factors that may impact cardiovascular risk, added Dr Singh, who a professor of medicine in the division of clinical immunology and rheumatology at the University of Alabama, Birmingham.
Recognizing gout’s contribution to cardiac risk, and managing both the disease and the associated risk factors, will be a key task for primary care doctors, rheumatologists, and cardiologists going forward, Dr. Singh concluded.
The United Kingdom’s National School for Primary Research funded the study. The authors reported no relevant disclosures. Dr. Singh reported receiving research and travel grants from Takeda and Savient, and consultant fees from Takeda, Savient, Regeneron, and Allergan.
My late father, H. H. Samson, an anesthesiologist, was convinced that hyperuricemia and over consumption of sugar were instigating factors for vascular disease and published on the subject (S. Afr. Med. J. 1978 Oct 7;54:590-1.) It has only taken some 30-odd years to prove him right! - Dr. Russell Samson, medical editor, Vascular Specialist.
Gout’s association with a host of vascular events was confirmed in a new study that explored the links between the inflammatory condition and coronary artery disease, peripheral vascular disease, and cerebrovascular events.
Though both men and women with gout were at increased risk for vascular events overall, the association appeared strongest for women. Dr. Lorna Clarson of Keele (England) University and her associates drew these conclusions from a retrospective cohort study of men and women with an incident diagnosis of gout (Ann. Rheum. Dis. 2015;74:642-7).
Gout, caused by the deposition of uric acid crystals in joints, is characterized by acute flares of intensely painful and inflamed joints. However, the state of hyperuricemia that predisposes patients to acute attacks of gout may precede the first attack by years, and may persist between flares. The proinflammatory course of the natural history of gout has increasingly been recognized as a potential contributor to vascular disease.
The precise mechanism by which gout may increase vascular risk has not been identified. Dr. Clarson and associates noted that in addition to the acute and chronic inflammation associated with gout and hyperuricemia, serum uric acid may have a more direct effect on vascular health, as urate crystal deposition on vessel walls may promote vascular damage.
To clarify gout’s impact on vascular risk, Dr. Clarson and her associates used the Clinical Practice Datalink, a large United Kingdom health database, to compare 8,366 patients with gout to 39,766 age- and sex-matched controls. None of those studied had a baseline history of vascular disease, and all were aged 50 or older.
Careful accounting for covariates was accomplished by multivariate analysis that took into account sex, age, body mass index, tobacco and alcohol consumption, statin or aspirin use, and any history of hypertension, dyslipidemia, or chronic kidney disease. In addition, the study employed the composite Charlson Comorbidity Index, which weights 19 comorbid conditions – including diabetes – to arrive at a single score that captures many risk factors. Patients in the cohort were tracked until their first vascular event, or until death or loss to follow-up.
To assess the incidence of vascular events, the study noted the first recording in the medical record of any events signaling vascular disease. These included angina or myocardial infarction, transient ischemic attack and stroke, and a range of diagnoses associated with peripheral vascular disease.
Final analysis after accounting for the many covariates tracked in the study showed increased risk for vascular events for those with gout, with a definite difference between the sexes. For men, gout predicted an increased risk of any vascular event (hazard ratio, 1.06; 95% confidence interval, 1.01–1.12) and of coronary heart disease and peripheral vascular disease.
For women, gout predicted an increased risk of all vascular events (HR, 1.25; 95% CI, 1.15-1.35) except myocardial infarction and cerebrovascular disease overall. Further, the degree of increased risk of vascular events was greater for women than for men with gout (P < .001 for intersex difference).
Noting that “clinical management of gout in primary care is suboptimal,” Dr. Clarson and her colleagues urged greater attention to screening for vascular risk in those diagnosed with gout; these individuals comprise a significant population of over 8 million people in the United States. Regarding the sex differences unearthed in their study,
Dr. Clarson and her associates observed that “both gout and vascular disease have historically been considered diseases of men ... [M]ore attention should be paid to prompt and reliable diagnosis of gout, followed by optimal management in female patients, including serious consideration of vascular risk reduction.”
In an editorial accompanying Dr. Clarson’s report (Ann. Rheum. Dis. 2015;74:631-4),Dr. Jasvinder Singh, commented that Dr. Clarson and colleagues’ study is limited by its lack of validation of gout and cardiac diagnoses and the self-selection bias inherent in using primary care registries, rather than a true population-based cohort.
Patients older than 35 or 40 with gout should be screened and followed with lipid profiles, hemoglobin A1c levels, blood pressure levels, and smoking status, and should undergo an assessment of other lifestyle factors that may impact cardiovascular risk, added Dr Singh, who a professor of medicine in the division of clinical immunology and rheumatology at the University of Alabama, Birmingham.
Recognizing gout’s contribution to cardiac risk, and managing both the disease and the associated risk factors, will be a key task for primary care doctors, rheumatologists, and cardiologists going forward, Dr. Singh concluded.
The United Kingdom’s National School for Primary Research funded the study. The authors reported no relevant disclosures. Dr. Singh reported receiving research and travel grants from Takeda and Savient, and consultant fees from Takeda, Savient, Regeneron, and Allergan.
My late father, H. H. Samson, an anesthesiologist, was convinced that hyperuricemia and over consumption of sugar were instigating factors for vascular disease and published on the subject (S. Afr. Med. J. 1978 Oct 7;54:590-1.) It has only taken some 30-odd years to prove him right! - Dr. Russell Samson, medical editor, Vascular Specialist.
Gout’s association with a host of vascular events was confirmed in a new study that explored the links between the inflammatory condition and coronary artery disease, peripheral vascular disease, and cerebrovascular events.
Though both men and women with gout were at increased risk for vascular events overall, the association appeared strongest for women. Dr. Lorna Clarson of Keele (England) University and her associates drew these conclusions from a retrospective cohort study of men and women with an incident diagnosis of gout (Ann. Rheum. Dis. 2015;74:642-7).
Gout, caused by the deposition of uric acid crystals in joints, is characterized by acute flares of intensely painful and inflamed joints. However, the state of hyperuricemia that predisposes patients to acute attacks of gout may precede the first attack by years, and may persist between flares. The proinflammatory course of the natural history of gout has increasingly been recognized as a potential contributor to vascular disease.
The precise mechanism by which gout may increase vascular risk has not been identified. Dr. Clarson and associates noted that in addition to the acute and chronic inflammation associated with gout and hyperuricemia, serum uric acid may have a more direct effect on vascular health, as urate crystal deposition on vessel walls may promote vascular damage.
To clarify gout’s impact on vascular risk, Dr. Clarson and her associates used the Clinical Practice Datalink, a large United Kingdom health database, to compare 8,366 patients with gout to 39,766 age- and sex-matched controls. None of those studied had a baseline history of vascular disease, and all were aged 50 or older.
Careful accounting for covariates was accomplished by multivariate analysis that took into account sex, age, body mass index, tobacco and alcohol consumption, statin or aspirin use, and any history of hypertension, dyslipidemia, or chronic kidney disease. In addition, the study employed the composite Charlson Comorbidity Index, which weights 19 comorbid conditions – including diabetes – to arrive at a single score that captures many risk factors. Patients in the cohort were tracked until their first vascular event, or until death or loss to follow-up.
To assess the incidence of vascular events, the study noted the first recording in the medical record of any events signaling vascular disease. These included angina or myocardial infarction, transient ischemic attack and stroke, and a range of diagnoses associated with peripheral vascular disease.
Final analysis after accounting for the many covariates tracked in the study showed increased risk for vascular events for those with gout, with a definite difference between the sexes. For men, gout predicted an increased risk of any vascular event (hazard ratio, 1.06; 95% confidence interval, 1.01–1.12) and of coronary heart disease and peripheral vascular disease.
For women, gout predicted an increased risk of all vascular events (HR, 1.25; 95% CI, 1.15-1.35) except myocardial infarction and cerebrovascular disease overall. Further, the degree of increased risk of vascular events was greater for women than for men with gout (P < .001 for intersex difference).
Noting that “clinical management of gout in primary care is suboptimal,” Dr. Clarson and her colleagues urged greater attention to screening for vascular risk in those diagnosed with gout; these individuals comprise a significant population of over 8 million people in the United States. Regarding the sex differences unearthed in their study,
Dr. Clarson and her associates observed that “both gout and vascular disease have historically been considered diseases of men ... [M]ore attention should be paid to prompt and reliable diagnosis of gout, followed by optimal management in female patients, including serious consideration of vascular risk reduction.”
In an editorial accompanying Dr. Clarson’s report (Ann. Rheum. Dis. 2015;74:631-4),Dr. Jasvinder Singh, commented that Dr. Clarson and colleagues’ study is limited by its lack of validation of gout and cardiac diagnoses and the self-selection bias inherent in using primary care registries, rather than a true population-based cohort.
Patients older than 35 or 40 with gout should be screened and followed with lipid profiles, hemoglobin A1c levels, blood pressure levels, and smoking status, and should undergo an assessment of other lifestyle factors that may impact cardiovascular risk, added Dr Singh, who a professor of medicine in the division of clinical immunology and rheumatology at the University of Alabama, Birmingham.
Recognizing gout’s contribution to cardiac risk, and managing both the disease and the associated risk factors, will be a key task for primary care doctors, rheumatologists, and cardiologists going forward, Dr. Singh concluded.
The United Kingdom’s National School for Primary Research funded the study. The authors reported no relevant disclosures. Dr. Singh reported receiving research and travel grants from Takeda and Savient, and consultant fees from Takeda, Savient, Regeneron, and Allergan.

Transcription factor promotes MM progression

rim of a bone spicule (pink)
New research indicates that multiple myeloma (MM) cells can “disguise” themselves as bone cells to elude the immune system, a trick that enables MM progression.
Investigators found evidence suggesting that MM cells mimic bone-marrow-resident cells by expressing bone-related genes, and this process is driven by overexpression of Runx2, a transcription factor that regulates bone formation.
“[R]unx2 overexpression can give multiple myeloma cells a bone-cell-like phenotype,” said Yang Yang, MD, PhD, of the University of Alabama at Birmingham.
“When the multiple myeloma cells come to the new bone sites, the bone immune cells think, ‘This is one of our neighbor cells,’ and therefore do not eliminate them. The bone immune cells do not recognize these cells as strangers.”
Dr Yang and her colleagues explained this phenomenon in Blood.
The investigators first conducted in vitro experiments and found that Runx2 expression in MM cells does not affect proliferation, but it does increase the cells’ invasiveness.
The team then used molecular genetic techniques to increase or decrease the expression of Runx2 in MM cells in vivo. They found that Runx2 overexpression promoted tumor growth and progression in mice. And mice with decreased Runx2 expression had less tumor growth and disease spread than control mice.
Further investigation revealed that Runx2 overexpression activates the Akt/β-catenin/survivin signaling pathway in MM cells. This is a different pathway than the one activated by Runx2 in solid tumors.
Downstream of the signaling pathway, Runx2 overexpression led to overexpression of bone-related genes, including genes expressed by osteoblasts, osteoclasts, and osteocytes.
Overexpression of Runx2 also enhanced secretion of soluble factors—including cytokines and growth factors—that aid tumor progression and metastasis.
In their final experiments, the investigators looked at Runx2 expression in human samples.
The team compared samples from 14 healthy bone marrow donors, 35 MM patients, and 11 patients with monoclonal gammopathy of undetermined significance (MGUS). Runx2 levels were significantly higher in MM cells than in plasma cells from normal and MGUS samples.
The investigators also assessed Runx2 expression in a larger group of 351 newly diagnosed MM patients. Runx2 levels were significantly higher in patients who had a high risk of early disease-related death. The risk of death was determined by an existing gene expression profile test.
“This suggests that Runx2 levels in myeloma cells may be a gene predictor of a patient’s prognosis, good or bad,” Dr Yang said.
She and her colleagues also believe that targeting Runx2 expression could be a feasible strategy for treating aggressive MM. ![]()
rim of a bone spicule (pink)
New research indicates that multiple myeloma (MM) cells can “disguise” themselves as bone cells to elude the immune system, a trick that enables MM progression.
Investigators found evidence suggesting that MM cells mimic bone-marrow-resident cells by expressing bone-related genes, and this process is driven by overexpression of Runx2, a transcription factor that regulates bone formation.
“[R]unx2 overexpression can give multiple myeloma cells a bone-cell-like phenotype,” said Yang Yang, MD, PhD, of the University of Alabama at Birmingham.
“When the multiple myeloma cells come to the new bone sites, the bone immune cells think, ‘This is one of our neighbor cells,’ and therefore do not eliminate them. The bone immune cells do not recognize these cells as strangers.”
Dr Yang and her colleagues explained this phenomenon in Blood.
The investigators first conducted in vitro experiments and found that Runx2 expression in MM cells does not affect proliferation, but it does increase the cells’ invasiveness.
The team then used molecular genetic techniques to increase or decrease the expression of Runx2 in MM cells in vivo. They found that Runx2 overexpression promoted tumor growth and progression in mice. And mice with decreased Runx2 expression had less tumor growth and disease spread than control mice.
Further investigation revealed that Runx2 overexpression activates the Akt/β-catenin/survivin signaling pathway in MM cells. This is a different pathway than the one activated by Runx2 in solid tumors.
Downstream of the signaling pathway, Runx2 overexpression led to overexpression of bone-related genes, including genes expressed by osteoblasts, osteoclasts, and osteocytes.
Overexpression of Runx2 also enhanced secretion of soluble factors—including cytokines and growth factors—that aid tumor progression and metastasis.
In their final experiments, the investigators looked at Runx2 expression in human samples.
The team compared samples from 14 healthy bone marrow donors, 35 MM patients, and 11 patients with monoclonal gammopathy of undetermined significance (MGUS). Runx2 levels were significantly higher in MM cells than in plasma cells from normal and MGUS samples.
The investigators also assessed Runx2 expression in a larger group of 351 newly diagnosed MM patients. Runx2 levels were significantly higher in patients who had a high risk of early disease-related death. The risk of death was determined by an existing gene expression profile test.
“This suggests that Runx2 levels in myeloma cells may be a gene predictor of a patient’s prognosis, good or bad,” Dr Yang said.
She and her colleagues also believe that targeting Runx2 expression could be a feasible strategy for treating aggressive MM. ![]()
rim of a bone spicule (pink)
New research indicates that multiple myeloma (MM) cells can “disguise” themselves as bone cells to elude the immune system, a trick that enables MM progression.
Investigators found evidence suggesting that MM cells mimic bone-marrow-resident cells by expressing bone-related genes, and this process is driven by overexpression of Runx2, a transcription factor that regulates bone formation.
“[R]unx2 overexpression can give multiple myeloma cells a bone-cell-like phenotype,” said Yang Yang, MD, PhD, of the University of Alabama at Birmingham.
“When the multiple myeloma cells come to the new bone sites, the bone immune cells think, ‘This is one of our neighbor cells,’ and therefore do not eliminate them. The bone immune cells do not recognize these cells as strangers.”
Dr Yang and her colleagues explained this phenomenon in Blood.
The investigators first conducted in vitro experiments and found that Runx2 expression in MM cells does not affect proliferation, but it does increase the cells’ invasiveness.
The team then used molecular genetic techniques to increase or decrease the expression of Runx2 in MM cells in vivo. They found that Runx2 overexpression promoted tumor growth and progression in mice. And mice with decreased Runx2 expression had less tumor growth and disease spread than control mice.
Further investigation revealed that Runx2 overexpression activates the Akt/β-catenin/survivin signaling pathway in MM cells. This is a different pathway than the one activated by Runx2 in solid tumors.
Downstream of the signaling pathway, Runx2 overexpression led to overexpression of bone-related genes, including genes expressed by osteoblasts, osteoclasts, and osteocytes.
Overexpression of Runx2 also enhanced secretion of soluble factors—including cytokines and growth factors—that aid tumor progression and metastasis.
In their final experiments, the investigators looked at Runx2 expression in human samples.
The team compared samples from 14 healthy bone marrow donors, 35 MM patients, and 11 patients with monoclonal gammopathy of undetermined significance (MGUS). Runx2 levels were significantly higher in MM cells than in plasma cells from normal and MGUS samples.
The investigators also assessed Runx2 expression in a larger group of 351 newly diagnosed MM patients. Runx2 levels were significantly higher in patients who had a high risk of early disease-related death. The risk of death was determined by an existing gene expression profile test.
“This suggests that Runx2 levels in myeloma cells may be a gene predictor of a patient’s prognosis, good or bad,” Dr Yang said.
She and her colleagues also believe that targeting Runx2 expression could be a feasible strategy for treating aggressive MM. ![]()
Older cancer patients under-utilize advanced care planning

Photo courtesy of NCI
and Mathews Media Group
Survey results suggest the use of aggressive treatment at the end of life is on the rise among older cancer patients, and these patients often fail to employ advanced care planning measures.
Researchers reviewed nearly 2000 surveys of people whose loved ones died of cancer and found that, from 2000 to 2012, there was a 51% increase in reports that patients received “all care possible” at the end of life.
There was a 22% increase in power-of-attorney assignment over the same period, but the use of living wills and discussions about end-of-life preferences decreased slightly.
“Although more cancer patients are assigning power-of-attorney privileges to someone they know and trust to make their medical decisions when they can’t, this practice may be the least helpful among advanced care planning tactics because it may be least associated with treatment intensity at the end of life,” said Amol Narang, MD, of The Johns Hopkins Hospital in Baltimore, Maryland.
Dr Narang and his colleagues described this research in JAMA Oncology.
The team analyzed survey data from 1985 next-of-kin surrogates of cancer patients. The patients were older than 50 years of age, had taken part in the Health and Retirement Study, and died between 2000 and 2012.
The data included in-depth “exit” interviews conducted with the surrogates after a patient died. Seventy-nine percent of exit survey respondents said they were the primary decision-maker in the patient’s medical care.
The data showed a significant increase in power-of-attorney assignments, from 52% in 2000 to 74% in 2012 (P=0.03). But the use of living wills decreased slightly, from 49% to 40% (P=0.63), as did discussions about end-of-life preferences, which fell from 68% to 60% (P=0.62).
“We found that many cancer patients still do not communicate their preferences for end-of-life care, despite the potential benefits to patients’ quality of life and caregiver bereavement,” Dr Narang said.
Survey results also suggested a significant increase in the percentage of patients who received “all care possible” at the end of life, from 7% in 2000 to 58% in 2012 (P=0.004). But there was no significant change in the rates of terminal hospitalizations, which fell from 29% to 27% (P=0.70).
The researchers found that granting power-of-attorney privileges significantly decreased the odds that patients would die in the hospital as opposed to hospice or their homes (adjusted odds ratio [AOR]=0.70, P<0.05). However, granting power of attorney was not associated with a significant change in limiting or withholding treatment at the end of life (AOR=1.52).
On the other hand, patients who created living wills or had end-of-life discussions were significantly more likely than their peers to limit or withhold certain treatments. The AOR was 2.51 for living wills (P<0.001) and 1.93 for end-of-life discussions (P=0.002).
The researchers said they observed the same trends regardless of who completed the exit survey.
Dr Narang noted that this study had its limitations. The survey questions were subjective, answers could have been hampered by a respondent’s lapse in memory, and answers could be biased by a respondent’s desire to meet social norms.
“But we were looking at trends over time,” he said, “so respondents’ bias would not likely change over time.” ![]()
Photo courtesy of NCI
and Mathews Media Group
Survey results suggest the use of aggressive treatment at the end of life is on the rise among older cancer patients, and these patients often fail to employ advanced care planning measures.
Researchers reviewed nearly 2000 surveys of people whose loved ones died of cancer and found that, from 2000 to 2012, there was a 51% increase in reports that patients received “all care possible” at the end of life.
There was a 22% increase in power-of-attorney assignment over the same period, but the use of living wills and discussions about end-of-life preferences decreased slightly.
“Although more cancer patients are assigning power-of-attorney privileges to someone they know and trust to make their medical decisions when they can’t, this practice may be the least helpful among advanced care planning tactics because it may be least associated with treatment intensity at the end of life,” said Amol Narang, MD, of The Johns Hopkins Hospital in Baltimore, Maryland.
Dr Narang and his colleagues described this research in JAMA Oncology.
The team analyzed survey data from 1985 next-of-kin surrogates of cancer patients. The patients were older than 50 years of age, had taken part in the Health and Retirement Study, and died between 2000 and 2012.
The data included in-depth “exit” interviews conducted with the surrogates after a patient died. Seventy-nine percent of exit survey respondents said they were the primary decision-maker in the patient’s medical care.
The data showed a significant increase in power-of-attorney assignments, from 52% in 2000 to 74% in 2012 (P=0.03). But the use of living wills decreased slightly, from 49% to 40% (P=0.63), as did discussions about end-of-life preferences, which fell from 68% to 60% (P=0.62).
“We found that many cancer patients still do not communicate their preferences for end-of-life care, despite the potential benefits to patients’ quality of life and caregiver bereavement,” Dr Narang said.
Survey results also suggested a significant increase in the percentage of patients who received “all care possible” at the end of life, from 7% in 2000 to 58% in 2012 (P=0.004). But there was no significant change in the rates of terminal hospitalizations, which fell from 29% to 27% (P=0.70).
The researchers found that granting power-of-attorney privileges significantly decreased the odds that patients would die in the hospital as opposed to hospice or their homes (adjusted odds ratio [AOR]=0.70, P<0.05). However, granting power of attorney was not associated with a significant change in limiting or withholding treatment at the end of life (AOR=1.52).
On the other hand, patients who created living wills or had end-of-life discussions were significantly more likely than their peers to limit or withhold certain treatments. The AOR was 2.51 for living wills (P<0.001) and 1.93 for end-of-life discussions (P=0.002).
The researchers said they observed the same trends regardless of who completed the exit survey.
Dr Narang noted that this study had its limitations. The survey questions were subjective, answers could have been hampered by a respondent’s lapse in memory, and answers could be biased by a respondent’s desire to meet social norms.
“But we were looking at trends over time,” he said, “so respondents’ bias would not likely change over time.” ![]()
Photo courtesy of NCI
and Mathews Media Group
Survey results suggest the use of aggressive treatment at the end of life is on the rise among older cancer patients, and these patients often fail to employ advanced care planning measures.
Researchers reviewed nearly 2000 surveys of people whose loved ones died of cancer and found that, from 2000 to 2012, there was a 51% increase in reports that patients received “all care possible” at the end of life.
There was a 22% increase in power-of-attorney assignment over the same period, but the use of living wills and discussions about end-of-life preferences decreased slightly.
“Although more cancer patients are assigning power-of-attorney privileges to someone they know and trust to make their medical decisions when they can’t, this practice may be the least helpful among advanced care planning tactics because it may be least associated with treatment intensity at the end of life,” said Amol Narang, MD, of The Johns Hopkins Hospital in Baltimore, Maryland.
Dr Narang and his colleagues described this research in JAMA Oncology.
The team analyzed survey data from 1985 next-of-kin surrogates of cancer patients. The patients were older than 50 years of age, had taken part in the Health and Retirement Study, and died between 2000 and 2012.
The data included in-depth “exit” interviews conducted with the surrogates after a patient died. Seventy-nine percent of exit survey respondents said they were the primary decision-maker in the patient’s medical care.
The data showed a significant increase in power-of-attorney assignments, from 52% in 2000 to 74% in 2012 (P=0.03). But the use of living wills decreased slightly, from 49% to 40% (P=0.63), as did discussions about end-of-life preferences, which fell from 68% to 60% (P=0.62).
“We found that many cancer patients still do not communicate their preferences for end-of-life care, despite the potential benefits to patients’ quality of life and caregiver bereavement,” Dr Narang said.
Survey results also suggested a significant increase in the percentage of patients who received “all care possible” at the end of life, from 7% in 2000 to 58% in 2012 (P=0.004). But there was no significant change in the rates of terminal hospitalizations, which fell from 29% to 27% (P=0.70).
The researchers found that granting power-of-attorney privileges significantly decreased the odds that patients would die in the hospital as opposed to hospice or their homes (adjusted odds ratio [AOR]=0.70, P<0.05). However, granting power of attorney was not associated with a significant change in limiting or withholding treatment at the end of life (AOR=1.52).
On the other hand, patients who created living wills or had end-of-life discussions were significantly more likely than their peers to limit or withhold certain treatments. The AOR was 2.51 for living wills (P<0.001) and 1.93 for end-of-life discussions (P=0.002).
The researchers said they observed the same trends regardless of who completed the exit survey.
Dr Narang noted that this study had its limitations. The survey questions were subjective, answers could have been hampered by a respondent’s lapse in memory, and answers could be biased by a respondent’s desire to meet social norms.
“But we were looking at trends over time,” he said, “so respondents’ bias would not likely change over time.” ![]()
How B-cell lymphoma evades NK cells

Image by Joshua Strokes
Researchers say they have determined how lymphoma cells evade natural killer (NK) cells, and this discovery has revealed potential solutions to the problem.
The team found that NK-cell activation and a second, “triggering” event are both necessary for NK cells to exhibit cytotoxicity in the presence of B-cell lymphoma.
Previous research demonstrated this 2-step process in vitro. Now, researchers have shown that it occurs in vivo.
Dr Ralph Mocikat, of Helmholtz Zentrum München in Munich, Germany, and his colleagues described this research in the European Journal of Immunology.
The team conducted experiments using transplantable tumors, a λ-myc-transgenic model of endogenously arising lymphoma that mimics human Burkitt lymphoma, and mice deficient in the NK group 2 D (NKG2D) receptor.
The experiments showed that NK cells could eliminate lymphoma cells after receiving 2 signals. The first was NK-cell activation, which gave rise to IFN-γ expression.
The researchers found that NK cells could be activated in the presence of MHC class Ilow tumor cells or by injecting bone marrow-derived dendritic cells. Previous research had shown that interleukin 2 (IL-2) and IL-15 could activate NK cells.
The second step involved the NKG2D receptor and its ligands. NKG2D ligands are located on the surface of tumor cells and bind to NK cells. The researchers found that, if these ligands are down-regulated, the NK cells cannot carry out cytotoxic activity.
However, the team found they could increase NKG2D ligand levels. They introduced bortezomib to the tumor cell line 291 and saw a roughly 4-fold increase in NKG2D ligand levels.
“Our results show that the transfer of NK cells is a possible strategic option to treat B-cell lymphoma,” Dr Mocikat said. “According to our findings, this therapeutic approach can be optimized when transferred NK cells are already activated in vitro prior to their injection, thus bypassing the missing activation potential in the tumor microenvironment. An additional injection of IFN-γ or of antibodies against IL-10 could further support the immune activity.” ![]()
Image by Joshua Strokes
Researchers say they have determined how lymphoma cells evade natural killer (NK) cells, and this discovery has revealed potential solutions to the problem.
The team found that NK-cell activation and a second, “triggering” event are both necessary for NK cells to exhibit cytotoxicity in the presence of B-cell lymphoma.
Previous research demonstrated this 2-step process in vitro. Now, researchers have shown that it occurs in vivo.
Dr Ralph Mocikat, of Helmholtz Zentrum München in Munich, Germany, and his colleagues described this research in the European Journal of Immunology.
The team conducted experiments using transplantable tumors, a λ-myc-transgenic model of endogenously arising lymphoma that mimics human Burkitt lymphoma, and mice deficient in the NK group 2 D (NKG2D) receptor.
The experiments showed that NK cells could eliminate lymphoma cells after receiving 2 signals. The first was NK-cell activation, which gave rise to IFN-γ expression.
The researchers found that NK cells could be activated in the presence of MHC class Ilow tumor cells or by injecting bone marrow-derived dendritic cells. Previous research had shown that interleukin 2 (IL-2) and IL-15 could activate NK cells.
The second step involved the NKG2D receptor and its ligands. NKG2D ligands are located on the surface of tumor cells and bind to NK cells. The researchers found that, if these ligands are down-regulated, the NK cells cannot carry out cytotoxic activity.
However, the team found they could increase NKG2D ligand levels. They introduced bortezomib to the tumor cell line 291 and saw a roughly 4-fold increase in NKG2D ligand levels.
“Our results show that the transfer of NK cells is a possible strategic option to treat B-cell lymphoma,” Dr Mocikat said. “According to our findings, this therapeutic approach can be optimized when transferred NK cells are already activated in vitro prior to their injection, thus bypassing the missing activation potential in the tumor microenvironment. An additional injection of IFN-γ or of antibodies against IL-10 could further support the immune activity.” ![]()
Image by Joshua Strokes
Researchers say they have determined how lymphoma cells evade natural killer (NK) cells, and this discovery has revealed potential solutions to the problem.
The team found that NK-cell activation and a second, “triggering” event are both necessary for NK cells to exhibit cytotoxicity in the presence of B-cell lymphoma.
Previous research demonstrated this 2-step process in vitro. Now, researchers have shown that it occurs in vivo.
Dr Ralph Mocikat, of Helmholtz Zentrum München in Munich, Germany, and his colleagues described this research in the European Journal of Immunology.
The team conducted experiments using transplantable tumors, a λ-myc-transgenic model of endogenously arising lymphoma that mimics human Burkitt lymphoma, and mice deficient in the NK group 2 D (NKG2D) receptor.
The experiments showed that NK cells could eliminate lymphoma cells after receiving 2 signals. The first was NK-cell activation, which gave rise to IFN-γ expression.
The researchers found that NK cells could be activated in the presence of MHC class Ilow tumor cells or by injecting bone marrow-derived dendritic cells. Previous research had shown that interleukin 2 (IL-2) and IL-15 could activate NK cells.
The second step involved the NKG2D receptor and its ligands. NKG2D ligands are located on the surface of tumor cells and bind to NK cells. The researchers found that, if these ligands are down-regulated, the NK cells cannot carry out cytotoxic activity.
However, the team found they could increase NKG2D ligand levels. They introduced bortezomib to the tumor cell line 291 and saw a roughly 4-fold increase in NKG2D ligand levels.
“Our results show that the transfer of NK cells is a possible strategic option to treat B-cell lymphoma,” Dr Mocikat said. “According to our findings, this therapeutic approach can be optimized when transferred NK cells are already activated in vitro prior to their injection, thus bypassing the missing activation potential in the tumor microenvironment. An additional injection of IFN-γ or of antibodies against IL-10 could further support the immune activity.” ![]()
AYAs with cancer receive aggressive EOL care

patient and her father
Photo by Rhoda Baer
In a retrospective study, a majority of adolescents and young adults (AYAs) with terminal cancer received aggressive end-of-life (EOL) care.
Investigators looked at the use of intensive care, emergency room visits, chemotherapy use, and hospitalization among more than 600 AYAs with cancer who were treated at Kaiser Permanente in California.
Nearly 70% of patients made use of at least one of these measures in the last month of their life.
The investigators noted that their findings, which were published in JAMA Oncology, may not reflect care for the wider US population. But the study does suggest a need for more research into whether this
pattern reflects AYA cancer patients’ preferences for EOL care.
“A young person facing the end of life is a particularly difficult issue,” said study author Jennifer Mack, MD, MPH, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
“While use of aggressive measures might be an informed decision by young people who would do anything they could to live longer, some interventions come with a cost, which is a poorer quality of life. This study raises questions about what kind of care they’re getting and how we can get them to the best quality of life at the end of their lives.”
The study included 633 patients, ages 15 to 39, who died of cancer between 2001 and 2010. The patients, who received care at Kaiser Permanente Southern California, had either been diagnosed with stage IV cancer or had a recurrence of stage I-III cancer. An initial review of a subset of 111 patients showed that death had been anticipated in 98% of cases.
The most common cancer diagnosis was gastrointestinal cancer (17%), while other common diagnoses were breast cancer (15%), genitourinary cancers (11%), leukemia (14%), and lymphoma (10%).
The investigators measured the use of 4 aggressive treatment measures—intensive care, emergency room visits, chemotherapy, and hospitalization—in patients’ last month of life.
Overall, 68% of patients (449/663) received at least one of these medically intensive EOL care measures. Eleven percent of patients (72/663) received chemotherapy, 22% (144/663) were admitted to the intensive care unit, 22% (147/663) had more than one emergency department visit, and 62% (413/663) were hospitalized.
Rates of hospitalization were higher among patients diagnosed with stage IV disease (66%) than among patients with stage I to III disease—66% and 58%, respectively (P=0.04).
The percentage of patients who received at least one medically intensive EOL care measure was higher in the stage IV cohort as well—71% and 63%, respectively (P=0.03). But there were no significant differences between the cohorts with regard to the other measures.
The investigators said these findings suggest the need to better understand EOL care preferences and decision-making in AYAs with cancer.
“We should think about talking with younger patients earlier about their prognoses, identifying their preferences, and working with them to deliver care that reflects those preferences,” Dr Mack said. “It may be that aggressive care is what they want, but they may end up on this pathway without thoughtful conversation and may be without recognition that they are dying.” ![]()
patient and her father
Photo by Rhoda Baer
In a retrospective study, a majority of adolescents and young adults (AYAs) with terminal cancer received aggressive end-of-life (EOL) care.
Investigators looked at the use of intensive care, emergency room visits, chemotherapy use, and hospitalization among more than 600 AYAs with cancer who were treated at Kaiser Permanente in California.
Nearly 70% of patients made use of at least one of these measures in the last month of their life.
The investigators noted that their findings, which were published in JAMA Oncology, may not reflect care for the wider US population. But the study does suggest a need for more research into whether this
pattern reflects AYA cancer patients’ preferences for EOL care.
“A young person facing the end of life is a particularly difficult issue,” said study author Jennifer Mack, MD, MPH, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
“While use of aggressive measures might be an informed decision by young people who would do anything they could to live longer, some interventions come with a cost, which is a poorer quality of life. This study raises questions about what kind of care they’re getting and how we can get them to the best quality of life at the end of their lives.”
The study included 633 patients, ages 15 to 39, who died of cancer between 2001 and 2010. The patients, who received care at Kaiser Permanente Southern California, had either been diagnosed with stage IV cancer or had a recurrence of stage I-III cancer. An initial review of a subset of 111 patients showed that death had been anticipated in 98% of cases.
The most common cancer diagnosis was gastrointestinal cancer (17%), while other common diagnoses were breast cancer (15%), genitourinary cancers (11%), leukemia (14%), and lymphoma (10%).
The investigators measured the use of 4 aggressive treatment measures—intensive care, emergency room visits, chemotherapy, and hospitalization—in patients’ last month of life.
Overall, 68% of patients (449/663) received at least one of these medically intensive EOL care measures. Eleven percent of patients (72/663) received chemotherapy, 22% (144/663) were admitted to the intensive care unit, 22% (147/663) had more than one emergency department visit, and 62% (413/663) were hospitalized.
Rates of hospitalization were higher among patients diagnosed with stage IV disease (66%) than among patients with stage I to III disease—66% and 58%, respectively (P=0.04).
The percentage of patients who received at least one medically intensive EOL care measure was higher in the stage IV cohort as well—71% and 63%, respectively (P=0.03). But there were no significant differences between the cohorts with regard to the other measures.
The investigators said these findings suggest the need to better understand EOL care preferences and decision-making in AYAs with cancer.
“We should think about talking with younger patients earlier about their prognoses, identifying their preferences, and working with them to deliver care that reflects those preferences,” Dr Mack said. “It may be that aggressive care is what they want, but they may end up on this pathway without thoughtful conversation and may be without recognition that they are dying.” ![]()
patient and her father
Photo by Rhoda Baer
In a retrospective study, a majority of adolescents and young adults (AYAs) with terminal cancer received aggressive end-of-life (EOL) care.
Investigators looked at the use of intensive care, emergency room visits, chemotherapy use, and hospitalization among more than 600 AYAs with cancer who were treated at Kaiser Permanente in California.
Nearly 70% of patients made use of at least one of these measures in the last month of their life.
The investigators noted that their findings, which were published in JAMA Oncology, may not reflect care for the wider US population. But the study does suggest a need for more research into whether this
pattern reflects AYA cancer patients’ preferences for EOL care.
“A young person facing the end of life is a particularly difficult issue,” said study author Jennifer Mack, MD, MPH, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
“While use of aggressive measures might be an informed decision by young people who would do anything they could to live longer, some interventions come with a cost, which is a poorer quality of life. This study raises questions about what kind of care they’re getting and how we can get them to the best quality of life at the end of their lives.”
The study included 633 patients, ages 15 to 39, who died of cancer between 2001 and 2010. The patients, who received care at Kaiser Permanente Southern California, had either been diagnosed with stage IV cancer or had a recurrence of stage I-III cancer. An initial review of a subset of 111 patients showed that death had been anticipated in 98% of cases.
The most common cancer diagnosis was gastrointestinal cancer (17%), while other common diagnoses were breast cancer (15%), genitourinary cancers (11%), leukemia (14%), and lymphoma (10%).
The investigators measured the use of 4 aggressive treatment measures—intensive care, emergency room visits, chemotherapy, and hospitalization—in patients’ last month of life.
Overall, 68% of patients (449/663) received at least one of these medically intensive EOL care measures. Eleven percent of patients (72/663) received chemotherapy, 22% (144/663) were admitted to the intensive care unit, 22% (147/663) had more than one emergency department visit, and 62% (413/663) were hospitalized.
Rates of hospitalization were higher among patients diagnosed with stage IV disease (66%) than among patients with stage I to III disease—66% and 58%, respectively (P=0.04).
The percentage of patients who received at least one medically intensive EOL care measure was higher in the stage IV cohort as well—71% and 63%, respectively (P=0.03). But there were no significant differences between the cohorts with regard to the other measures.
The investigators said these findings suggest the need to better understand EOL care preferences and decision-making in AYAs with cancer.
“We should think about talking with younger patients earlier about their prognoses, identifying their preferences, and working with them to deliver care that reflects those preferences,” Dr Mack said. “It may be that aggressive care is what they want, but they may end up on this pathway without thoughtful conversation and may be without recognition that they are dying.” ![]()
FDA will strengthen heart attack, stroke risk warnings for all NSAIDs
The Food and Drug Administration has taken new action to strengthen existing warning labels about the increased risk of heart attack or stroke with the use of prescription and over-the-counter nonaspirin nonsteroidal anti-inflammatory drugs.
In a July 9 drug safety communication, the agency did not provide the exact language that will be used on NSAID labels, but said that they “will be revised to reflect” information describing that:
• The risk of heart attack or stroke can occur as early as the first weeks of using an NSAID.
• The risk may increase with longer use and at higher doses of the NSAID.

• The drugs can increase the risk of heart attack or stroke even in patients without heart disease or risk factors for heart disease, but patients with heart disease or risk factors for it have a greater likelihood of heart attack or stroke following NSAID use.
• Treatment with NSAIDs following a first heart attack increases the risk of death in the first year after the heart attack, compared with patients who were not treated with NSAIDs after their first heart attack.
• NSAID use increases the risk of heart failure.
The new wording will also note that although newer information may suggest that the risk for heart attack or stroke is not the same for all NSAIDs, it “is not sufficient for us to determine that the risk of any particular NSAID is definitely higher or lower than that of any other particular NSAID.”
*The update to NSAID labels follows the recommendations given by panel members from a joint meeting of the FDA’s Arthritis Advisory Committee and the Drug Safety and Risk Management Advisory Committee in February 2014 in which there was a split vote (14-11) that was slightly in favor of rewording the warning labeling for NSAIDs in regard to the drug class’s current labeling, which implies that the cardiovascular thrombotic risk is not substantial with short treatment courses. At that meeting, the panelists also voted 16-9 that there were not enough data to suggest that naproxen presented a substantially lower risk of CV events than did either ibuprofen or selective NSAIDs, such as cyclooxygenase-2 inhibitors.
The FDA made its decision based on a comprehensive review of the data presented during that meeting.
*Correction, 7/16/2015: An earlier version of this story misstated the FDA panels’ recommendation for labeling changes.
The Food and Drug Administration has taken new action to strengthen existing warning labels about the increased risk of heart attack or stroke with the use of prescription and over-the-counter nonaspirin nonsteroidal anti-inflammatory drugs.
In a July 9 drug safety communication, the agency did not provide the exact language that will be used on NSAID labels, but said that they “will be revised to reflect” information describing that:
• The risk of heart attack or stroke can occur as early as the first weeks of using an NSAID.
• The risk may increase with longer use and at higher doses of the NSAID.
• The drugs can increase the risk of heart attack or stroke even in patients without heart disease or risk factors for heart disease, but patients with heart disease or risk factors for it have a greater likelihood of heart attack or stroke following NSAID use.
• Treatment with NSAIDs following a first heart attack increases the risk of death in the first year after the heart attack, compared with patients who were not treated with NSAIDs after their first heart attack.
• NSAID use increases the risk of heart failure.
The new wording will also note that although newer information may suggest that the risk for heart attack or stroke is not the same for all NSAIDs, it “is not sufficient for us to determine that the risk of any particular NSAID is definitely higher or lower than that of any other particular NSAID.”
*The update to NSAID labels follows the recommendations given by panel members from a joint meeting of the FDA’s Arthritis Advisory Committee and the Drug Safety and Risk Management Advisory Committee in February 2014 in which there was a split vote (14-11) that was slightly in favor of rewording the warning labeling for NSAIDs in regard to the drug class’s current labeling, which implies that the cardiovascular thrombotic risk is not substantial with short treatment courses. At that meeting, the panelists also voted 16-9 that there were not enough data to suggest that naproxen presented a substantially lower risk of CV events than did either ibuprofen or selective NSAIDs, such as cyclooxygenase-2 inhibitors.
The FDA made its decision based on a comprehensive review of the data presented during that meeting.
*Correction, 7/16/2015: An earlier version of this story misstated the FDA panels’ recommendation for labeling changes.
The Food and Drug Administration has taken new action to strengthen existing warning labels about the increased risk of heart attack or stroke with the use of prescription and over-the-counter nonaspirin nonsteroidal anti-inflammatory drugs.
In a July 9 drug safety communication, the agency did not provide the exact language that will be used on NSAID labels, but said that they “will be revised to reflect” information describing that:
• The risk of heart attack or stroke can occur as early as the first weeks of using an NSAID.
• The risk may increase with longer use and at higher doses of the NSAID.
• The drugs can increase the risk of heart attack or stroke even in patients without heart disease or risk factors for heart disease, but patients with heart disease or risk factors for it have a greater likelihood of heart attack or stroke following NSAID use.
• Treatment with NSAIDs following a first heart attack increases the risk of death in the first year after the heart attack, compared with patients who were not treated with NSAIDs after their first heart attack.
• NSAID use increases the risk of heart failure.
The new wording will also note that although newer information may suggest that the risk for heart attack or stroke is not the same for all NSAIDs, it “is not sufficient for us to determine that the risk of any particular NSAID is definitely higher or lower than that of any other particular NSAID.”
*The update to NSAID labels follows the recommendations given by panel members from a joint meeting of the FDA’s Arthritis Advisory Committee and the Drug Safety and Risk Management Advisory Committee in February 2014 in which there was a split vote (14-11) that was slightly in favor of rewording the warning labeling for NSAIDs in regard to the drug class’s current labeling, which implies that the cardiovascular thrombotic risk is not substantial with short treatment courses. At that meeting, the panelists also voted 16-9 that there were not enough data to suggest that naproxen presented a substantially lower risk of CV events than did either ibuprofen or selective NSAIDs, such as cyclooxygenase-2 inhibitors.
The FDA made its decision based on a comprehensive review of the data presented during that meeting.
*Correction, 7/16/2015: An earlier version of this story misstated the FDA panels’ recommendation for labeling changes.

Rosai-Dorfman Disease
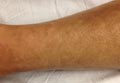
Rosai-Dorfman disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy, is a rare benign histioproliferative disorder of unknown etiology.1 Clinically, it is most frequently characterized by massive painless cervical lymphadenopathy with other systemic manifestations, including fever, night sweats, and weight loss. Accompanying laboratory findings include leukocytosis with neutrophilia, elevated erythrocyte sedimentation rate, and polyclonal hypergammaglobulinemia. Extranodal involvement has been noted in more than 40% of cases, and cutaneous lesions represent the most common form of extranodal disease.2 Cutaneous RDD is a distinct and rare entity limited to the skin without lymphadenopathy or other extracutaneous involvement.3 Patients with cutaneous RDD typically present with papules and plaques that can grow to form nodules with satellite lesions that resolve into fibrotic plaques before spontaneous regression.4
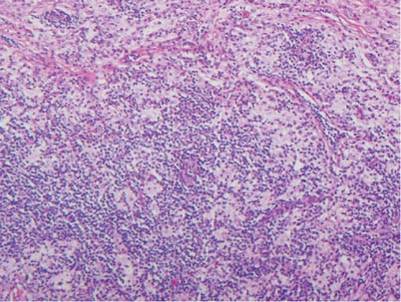
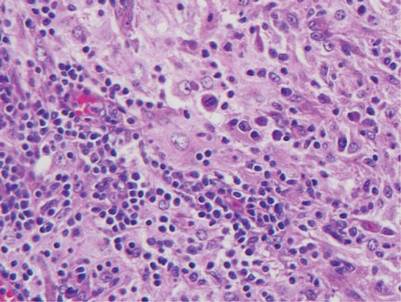
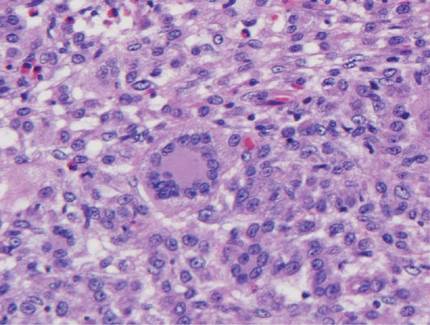
Histologic examination of cutaneous lesions of RDD reveals a dense nodular dermal and often subcutaneous infiltrate of characteristic large polygonal histiocytes termed Rosai-Dorfman cells, which feature abundant pale to eosinophilic cytoplasm, indistinct borders, and large vesicular nuclei with prominent nucleoli (Figure 1).4,5 Some multinucleate forms may be seen. These Rosai-Dorfman cells display positive staining for CD68 and S-100, and negative staining for CD1a on immunohistochemistry. Lymphocytes and plasma cells often are admixed with the Rosai-Dorfman cells, and neutrophils and eosinophils also may be present in the infiltrate.4 The histologic hallmark of RDD is emperipolesis, a phenomenon whereby inflammatory cells such as lymphocytes and plasma cells reside intact within the cytoplasm of histiocytes (Figure 2).5
 |  |
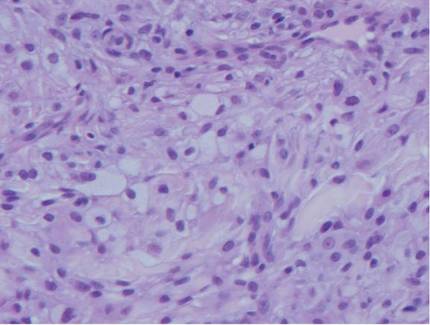
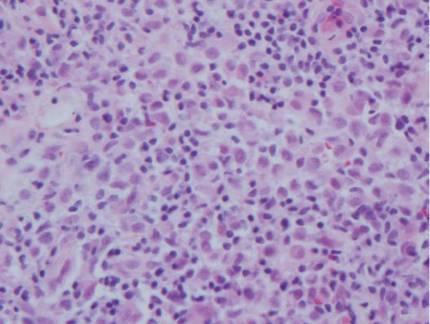
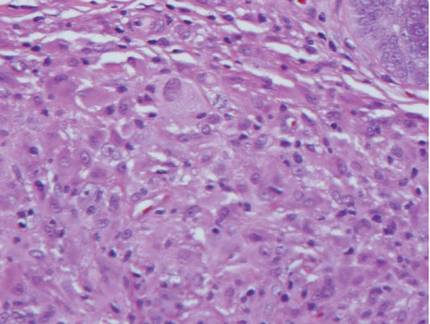
The histologic differential diagnosis of cutaneous lesions of RDD includes other histiocytic and xanthomatous diseases, including eruptive xanthoma, juvenile xanthogranuloma, Langerhans cell histiocytosis, and solitary reticulohistiocytoma, which should not display emperipolesis. Eruptive xanthomas display collections of foamy histiocytes in the dermis and typically contain extracellular lipid. They may contain infiltrates of lymphocytes (Figure 3). Juvenile xanthogranuloma also features a dense infiltrate of histiocytes in the papillary and reticular dermis but distinctly shows Touton giant cells and lipidization of histiocytes (Figure 4). Both eruptive xanthomas and juvenile xanthogranulomas typically stain negatively for S-100. Langerhans cell histiocytosis is histologically characterized by a dermal infiltrate of Langerhans cells that have their own distinctive morphologic features. They are uniformly ovoid with abundant eosinophilic cytoplasm. Their nuclei are smaller than those of Rosai-Dorfman cells and have a kidney bean shape with inconspicuous nucleoli (Figure 5). Epidermotropism of these cells can be observed. Immunohistochemically, Langerhans cell histiocytosis typically is S-100 positive, CD1a positive, and langerin positive. Reticulohistiocytoma features histiocytes that have a characteristic dusty rose or ground glass cytoplasm with two-toned darker and lighter areas (Figure 6). Reticulohistiocytoma cells stain positively for CD68 but typically stain negatively for both CD1a and S-100.
 |  | ||
 |  |
1. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
2. Foucar E, Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): a review of the entity. Semin Diagn Pathol. 1990;7:19-73.
3. Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002;24:385-391.
4. Wang KH, Chen WY, Lie HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
5. Chu P, LeBoit PE. Histologic features of cutaneous sinus histiocytosis (Rosai-Dorfman disease): study of cases both with and without systemic involvement. J Cutan Pathol. 1992;19:201-206.
Rosai-Dorfman disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy, is a rare benign histioproliferative disorder of unknown etiology.1 Clinically, it is most frequently characterized by massive painless cervical lymphadenopathy with other systemic manifestations, including fever, night sweats, and weight loss. Accompanying laboratory findings include leukocytosis with neutrophilia, elevated erythrocyte sedimentation rate, and polyclonal hypergammaglobulinemia. Extranodal involvement has been noted in more than 40% of cases, and cutaneous lesions represent the most common form of extranodal disease.2 Cutaneous RDD is a distinct and rare entity limited to the skin without lymphadenopathy or other extracutaneous involvement.3 Patients with cutaneous RDD typically present with papules and plaques that can grow to form nodules with satellite lesions that resolve into fibrotic plaques before spontaneous regression.4
Histologic examination of cutaneous lesions of RDD reveals a dense nodular dermal and often subcutaneous infiltrate of characteristic large polygonal histiocytes termed Rosai-Dorfman cells, which feature abundant pale to eosinophilic cytoplasm, indistinct borders, and large vesicular nuclei with prominent nucleoli (Figure 1).4,5 Some multinucleate forms may be seen. These Rosai-Dorfman cells display positive staining for CD68 and S-100, and negative staining for CD1a on immunohistochemistry. Lymphocytes and plasma cells often are admixed with the Rosai-Dorfman cells, and neutrophils and eosinophils also may be present in the infiltrate.4 The histologic hallmark of RDD is emperipolesis, a phenomenon whereby inflammatory cells such as lymphocytes and plasma cells reside intact within the cytoplasm of histiocytes (Figure 2).5
| |
The histologic differential diagnosis of cutaneous lesions of RDD includes other histiocytic and xanthomatous diseases, including eruptive xanthoma, juvenile xanthogranuloma, Langerhans cell histiocytosis, and solitary reticulohistiocytoma, which should not display emperipolesis. Eruptive xanthomas display collections of foamy histiocytes in the dermis and typically contain extracellular lipid. They may contain infiltrates of lymphocytes (Figure 3). Juvenile xanthogranuloma also features a dense infiltrate of histiocytes in the papillary and reticular dermis but distinctly shows Touton giant cells and lipidization of histiocytes (Figure 4). Both eruptive xanthomas and juvenile xanthogranulomas typically stain negatively for S-100. Langerhans cell histiocytosis is histologically characterized by a dermal infiltrate of Langerhans cells that have their own distinctive morphologic features. They are uniformly ovoid with abundant eosinophilic cytoplasm. Their nuclei are smaller than those of Rosai-Dorfman cells and have a kidney bean shape with inconspicuous nucleoli (Figure 5). Epidermotropism of these cells can be observed. Immunohistochemically, Langerhans cell histiocytosis typically is S-100 positive, CD1a positive, and langerin positive. Reticulohistiocytoma features histiocytes that have a characteristic dusty rose or ground glass cytoplasm with two-toned darker and lighter areas (Figure 6). Reticulohistiocytoma cells stain positively for CD68 but typically stain negatively for both CD1a and S-100.
| | ||
| |
Rosai-Dorfman disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy, is a rare benign histioproliferative disorder of unknown etiology.1 Clinically, it is most frequently characterized by massive painless cervical lymphadenopathy with other systemic manifestations, including fever, night sweats, and weight loss. Accompanying laboratory findings include leukocytosis with neutrophilia, elevated erythrocyte sedimentation rate, and polyclonal hypergammaglobulinemia. Extranodal involvement has been noted in more than 40% of cases, and cutaneous lesions represent the most common form of extranodal disease.2 Cutaneous RDD is a distinct and rare entity limited to the skin without lymphadenopathy or other extracutaneous involvement.3 Patients with cutaneous RDD typically present with papules and plaques that can grow to form nodules with satellite lesions that resolve into fibrotic plaques before spontaneous regression.4
Histologic examination of cutaneous lesions of RDD reveals a dense nodular dermal and often subcutaneous infiltrate of characteristic large polygonal histiocytes termed Rosai-Dorfman cells, which feature abundant pale to eosinophilic cytoplasm, indistinct borders, and large vesicular nuclei with prominent nucleoli (Figure 1).4,5 Some multinucleate forms may be seen. These Rosai-Dorfman cells display positive staining for CD68 and S-100, and negative staining for CD1a on immunohistochemistry. Lymphocytes and plasma cells often are admixed with the Rosai-Dorfman cells, and neutrophils and eosinophils also may be present in the infiltrate.4 The histologic hallmark of RDD is emperipolesis, a phenomenon whereby inflammatory cells such as lymphocytes and plasma cells reside intact within the cytoplasm of histiocytes (Figure 2).5
| |
The histologic differential diagnosis of cutaneous lesions of RDD includes other histiocytic and xanthomatous diseases, including eruptive xanthoma, juvenile xanthogranuloma, Langerhans cell histiocytosis, and solitary reticulohistiocytoma, which should not display emperipolesis. Eruptive xanthomas display collections of foamy histiocytes in the dermis and typically contain extracellular lipid. They may contain infiltrates of lymphocytes (Figure 3). Juvenile xanthogranuloma also features a dense infiltrate of histiocytes in the papillary and reticular dermis but distinctly shows Touton giant cells and lipidization of histiocytes (Figure 4). Both eruptive xanthomas and juvenile xanthogranulomas typically stain negatively for S-100. Langerhans cell histiocytosis is histologically characterized by a dermal infiltrate of Langerhans cells that have their own distinctive morphologic features. They are uniformly ovoid with abundant eosinophilic cytoplasm. Their nuclei are smaller than those of Rosai-Dorfman cells and have a kidney bean shape with inconspicuous nucleoli (Figure 5). Epidermotropism of these cells can be observed. Immunohistochemically, Langerhans cell histiocytosis typically is S-100 positive, CD1a positive, and langerin positive. Reticulohistiocytoma features histiocytes that have a characteristic dusty rose or ground glass cytoplasm with two-toned darker and lighter areas (Figure 6). Reticulohistiocytoma cells stain positively for CD68 but typically stain negatively for both CD1a and S-100.
| | ||
| |
1. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
2. Foucar E, Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): a review of the entity. Semin Diagn Pathol. 1990;7:19-73.
3. Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002;24:385-391.
4. Wang KH, Chen WY, Lie HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
5. Chu P, LeBoit PE. Histologic features of cutaneous sinus histiocytosis (Rosai-Dorfman disease): study of cases both with and without systemic involvement. J Cutan Pathol. 1992;19:201-206.
1. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
2. Foucar E, Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): a review of the entity. Semin Diagn Pathol. 1990;7:19-73.
3. Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002;24:385-391.
4. Wang KH, Chen WY, Lie HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
5. Chu P, LeBoit PE. Histologic features of cutaneous sinus histiocytosis (Rosai-Dorfman disease): study of cases both with and without systemic involvement. J Cutan Pathol. 1992;19:201-206.
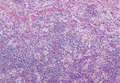
Sticks and stones: Navigating patient satisfaction scores
There is an old saying, “Sticks and stones may break my bones, but words will never hurt me.” When it comes to our patients’ impressions of us, nothing could be farther from the truth. As a matter of fact, their words, expressed in post-discharge patient satisfaction surveys, can play a tremendous role in the financial stability of our hospitals.
Throughout the years, hospitals have employed a wide variety of methods to evaluate their patients’ experiences, if only to improve their service and strengthen their brand. The Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) initiative has taken patient satisfaction to a new level by creating the first national, standardized survey tool to measure patients’ perspectives of the care they received while hospitalized. In addition, HCAHPS was designed to provide objective, meaningful comparisons of hospitals, and these comparisons are publicly reported, which increases transparency of the quality of care provided to hospitalized patients.
The survey is administered between 48 hours and 6 weeks post discharge to a random sample of adult patients by mail, telephone, mail and telephone, or Interactive Voice Response (IVR). Discharged patients are asked 27 questions about their recent hospitalization including communication with doctors and nurses, pain management, discharge information, communication about medication, overall hospital rating, and whether they would recommend the hospital to others.
Four times per year, the Centers for Medicaid & Medicare Services publishes HCAHPS scores of participating hospitals on the Hospital Compare website (www.hospitalcompare.hhs.gov), though the survey is not restricted to Medicare patients. Patients can pick and choose which hospitals they like, and which ones they would avoid like the plague.
Of course, it’s not realistic to think that we are going to please all of our patients all of the time, but this initiative does have the potential to create a new sense of accountability, as well as competitiveness for hospital systems and providers alike. No one wants to be at the bottom of the pack.
So, how do we increase our scores? Many models and companies claim to help improve patient satisfaction. Just do an Internet search. Keep in mind, what works well for one group may be ineffective for another.
For instance, 5-minute per patient multidisciplinary bedside rounding – including the provider, nurse, pharmacist, and case manager – may be easy to implement and skyrocket patient satisfaction in some institutions. In others, getting appropriate staffing may be prohibitive. Regardless of the approach that may be right for your group, it is important to keep in mind that the tide of health care is ever changing. Patients are demanding, and receiving, a bigger role in their health care. We all want to be in the forefront, not at the tail end, of that tide.
Dr. Hester is a hospitalist at Baltimore-Washington Medical Center in Glen Burnie, Md. She is the creator of the Patient Whiz, a patient-engagement app for iOS. Reach her at healthsavvy@aol.com.
There is an old saying, “Sticks and stones may break my bones, but words will never hurt me.” When it comes to our patients’ impressions of us, nothing could be farther from the truth. As a matter of fact, their words, expressed in post-discharge patient satisfaction surveys, can play a tremendous role in the financial stability of our hospitals.
Throughout the years, hospitals have employed a wide variety of methods to evaluate their patients’ experiences, if only to improve their service and strengthen their brand. The Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) initiative has taken patient satisfaction to a new level by creating the first national, standardized survey tool to measure patients’ perspectives of the care they received while hospitalized. In addition, HCAHPS was designed to provide objective, meaningful comparisons of hospitals, and these comparisons are publicly reported, which increases transparency of the quality of care provided to hospitalized patients.
The survey is administered between 48 hours and 6 weeks post discharge to a random sample of adult patients by mail, telephone, mail and telephone, or Interactive Voice Response (IVR). Discharged patients are asked 27 questions about their recent hospitalization including communication with doctors and nurses, pain management, discharge information, communication about medication, overall hospital rating, and whether they would recommend the hospital to others.
Four times per year, the Centers for Medicaid & Medicare Services publishes HCAHPS scores of participating hospitals on the Hospital Compare website (www.hospitalcompare.hhs.gov), though the survey is not restricted to Medicare patients. Patients can pick and choose which hospitals they like, and which ones they would avoid like the plague.
Of course, it’s not realistic to think that we are going to please all of our patients all of the time, but this initiative does have the potential to create a new sense of accountability, as well as competitiveness for hospital systems and providers alike. No one wants to be at the bottom of the pack.
So, how do we increase our scores? Many models and companies claim to help improve patient satisfaction. Just do an Internet search. Keep in mind, what works well for one group may be ineffective for another.
For instance, 5-minute per patient multidisciplinary bedside rounding – including the provider, nurse, pharmacist, and case manager – may be easy to implement and skyrocket patient satisfaction in some institutions. In others, getting appropriate staffing may be prohibitive. Regardless of the approach that may be right for your group, it is important to keep in mind that the tide of health care is ever changing. Patients are demanding, and receiving, a bigger role in their health care. We all want to be in the forefront, not at the tail end, of that tide.
Dr. Hester is a hospitalist at Baltimore-Washington Medical Center in Glen Burnie, Md. She is the creator of the Patient Whiz, a patient-engagement app for iOS. Reach her at healthsavvy@aol.com.
There is an old saying, “Sticks and stones may break my bones, but words will never hurt me.” When it comes to our patients’ impressions of us, nothing could be farther from the truth. As a matter of fact, their words, expressed in post-discharge patient satisfaction surveys, can play a tremendous role in the financial stability of our hospitals.
Throughout the years, hospitals have employed a wide variety of methods to evaluate their patients’ experiences, if only to improve their service and strengthen their brand. The Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) initiative has taken patient satisfaction to a new level by creating the first national, standardized survey tool to measure patients’ perspectives of the care they received while hospitalized. In addition, HCAHPS was designed to provide objective, meaningful comparisons of hospitals, and these comparisons are publicly reported, which increases transparency of the quality of care provided to hospitalized patients.
The survey is administered between 48 hours and 6 weeks post discharge to a random sample of adult patients by mail, telephone, mail and telephone, or Interactive Voice Response (IVR). Discharged patients are asked 27 questions about their recent hospitalization including communication with doctors and nurses, pain management, discharge information, communication about medication, overall hospital rating, and whether they would recommend the hospital to others.
Four times per year, the Centers for Medicaid & Medicare Services publishes HCAHPS scores of participating hospitals on the Hospital Compare website (www.hospitalcompare.hhs.gov), though the survey is not restricted to Medicare patients. Patients can pick and choose which hospitals they like, and which ones they would avoid like the plague.
Of course, it’s not realistic to think that we are going to please all of our patients all of the time, but this initiative does have the potential to create a new sense of accountability, as well as competitiveness for hospital systems and providers alike. No one wants to be at the bottom of the pack.
So, how do we increase our scores? Many models and companies claim to help improve patient satisfaction. Just do an Internet search. Keep in mind, what works well for one group may be ineffective for another.
For instance, 5-minute per patient multidisciplinary bedside rounding – including the provider, nurse, pharmacist, and case manager – may be easy to implement and skyrocket patient satisfaction in some institutions. In others, getting appropriate staffing may be prohibitive. Regardless of the approach that may be right for your group, it is important to keep in mind that the tide of health care is ever changing. Patients are demanding, and receiving, a bigger role in their health care. We all want to be in the forefront, not at the tail end, of that tide.
Dr. Hester is a hospitalist at Baltimore-Washington Medical Center in Glen Burnie, Md. She is the creator of the Patient Whiz, a patient-engagement app for iOS. Reach her at healthsavvy@aol.com.

What Is Your Diagnosis? Eosinophilic Fasciitis
|
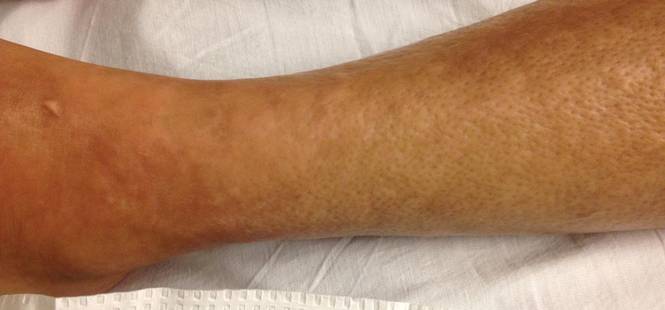
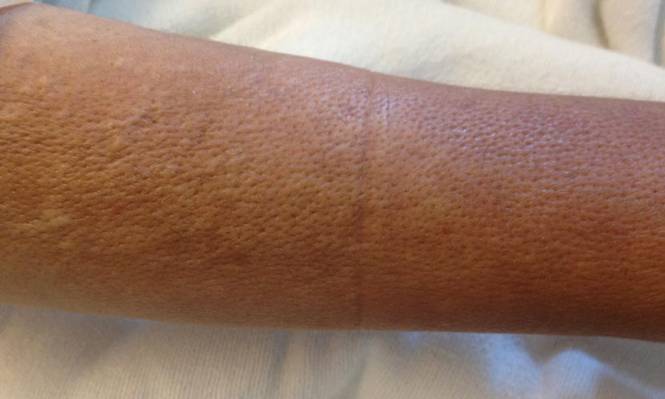
A 43-year-old woman presented with pain and paresthesia of the bilateral legs of 3 months’ duration with skin tightness and discoloration, which she attributed to a car accident that occurred 7 months prior. She also reported abdominal pain, shortness of breath, fever, double vision, dysphagia, voice changes, temperature sensitivity, and hair loss. The patient underwent outpatient steroid injections with limited symptomatic relief. She denied any antecedent exposure to vinyl chloride, rapeseed oil, or L-tryptophan. Physical examination revealed thickened skin on the bilateral legs (top), reddish discoloration of the feet, decreased sensation to light touch, and edema of the ankles and wrists, as well as a peau d’orange appearance of the skin on the arms (bottom), legs, and abdomen.
The Diagnosis: Eosinophilic Fasciitis
Eosinophilic fasciitis is a rare autoimmune disease of uncertain etiology first described by Shulman1 in 1974. It is similar in presentation and is perhaps related to scleroderma. Classic differentiating features include a peculiar peau d’orange appearance, peripheral eosinophilia, and lack of Raynaud phenomenon, thus it is regarded as a unique disease.2 Despite the name of the disease, eosinophilia has been known to be absent in later stages of eosinophilic fasciitis.1

On physical examination, “prayer and groove signs” can sometimes be evident.3 Although it was not initially observed in our case, a groove sign was noted on the left forearm on a second inspection (Figure 1). In contrast with systemic sclerosis, visceral involvement rarely is seen with eosinophilic fasciitis. There are, however, 3 major exceptions to this rule. First, there can be widespread nerve deficits, esophageal dysmotility, and nonspecific electromyography findings (ie, denervation, reinnervation, fasciculations).4 There also can be a concomitant hematologic disorder or Hashimoto thyroiditis.5 Because eosinophilic fasciitis has been associated with monoclonal gammopathy, which our patient also demonstrated, it is important to conduct a workup with serum or urine protein electrophoresis. If the test is negative, it should be followed up with an immunofixation assay or serum light chain assays.
Some proposed risk factors for eosinophilic fasciitis include trauma, extensive exercise, and Borrelia burgdorferi infection, but many cases have none of these associations.8 Although not firmly proven in the literature, there have been reports of eosinophilic fasciitis after isolated trauma.5 A causal link could not be established between our patient’s car accident and eosinophilic fasciitis, but the coincidence was notable.
The treatment of eosinophilic fasciitis is similar to scleroderma. Corticosteroids are effective in most cases and recovery often occurs with monotherapy.5 Case series have demonstrated efficacy in adding methotrexate, azathioprine, colchicine, and hydroxychloroquine in refractory patients.2,9 Our patient demonstrated a good response with a combination of prednisone and methotrexate. Relapses have been known to occur.2
A punch biopsy obtained from the right arm showed thickened acellular collagen bundles throughout the dermis and extending into the underlying subcutis. There also was obliteration of adnexal structures, loss of perieccrine fat, and sparse perivascular and interstitial lymphoplasmacytic infiltrate (Figure 2), consistent with a sclerosing disorder such as scleroderma or eosinophilic fasciitis.

A complete blood cell count revealed eosinophil levels of 12.5% (reference range, 2.7%). Rheumatologic workup was negative for antinuclear antibody, double-stranded DNA, thyroid-stimulating hormone, anticentromere antibodies, and Scl-70 autoantibodies. Computed tomography of the chest and pelvis revealed a thickened patulous esophagus. Endoscopy showed dysmotility of the lower esophagus. At this point the differential diagnosis included scleredema versus eosinophilic fasciitis, and the patient was started on oral prednisone 60 mg daily. She showed rapid improvement in sclerosis, joint mobility, and ability to swallow. Magnetic resonance imaging was then performed and revealed thickening and contrast enhancement of the forearm fascia, particularly along the distal aspect, confirming a diagnosis of eosinophilic fasciitis. Further workup including immunofixation assay and serum light chain assays were performed, revealing IgG λ hypergammaglobulinemia. She was then additionally treated with oral methotrexate 15 mg weekly. Due to the rapid improvement of symptoms on oral prednisone over 2 weeks, the peripheral eosinophilia, the magnetic resonance imaging findings, and the results of skin biopsy, a diagnosis of eosinophilic fasciitis was heavily favored over scleroderma and scleredema.
1. Shulman L. Diffuse fasciitis with hypergammaglobulinemia and eosinophilia in a new syndrome. J. Rheumatol. 1974;1(suppl):46.
2. Lakhanpal S, Ginsburg WW, Michet CJ, et al. Eosinophilic fasciitis: clinical spectrum and therapeutic response in 52 cases. Semin Arthritis Rheum. 1988;17:221-231.
3. Servy A, Clerici T, Malines C, et al. Eosinophilic fasciitis: a rare skin sclerosis. Pathol Res Int. 2010;2011:716935.
4. Satsangi J, Donaghy M. Multifocal peripheral neuropathy in eosinophilic fasciitis. J Neurol. 1992;239:91-92.
5. Antic M, Lautenschlager S, Itin PH. Eosinophilic fasciitis 30 years after—what do we really know? Dermatology. 2006;213:93-101.
6. Doyle JA, Ginsburg WW. Eosinophilic fasciitis. Med Clin North Am. 1989;73:1157-1166.
7. Naschitz JE, Yeshurun D, Miselevich I, et al. Colitis and pericarditis in a patient with eosinophilic fasciitis—a contribution to the multisystem nature of eosinophilic fasciitis. J Rheumatol. 1989;16:688-692.
8. Haustein UF. Scleroderma and pseudoscleroderma: uncommon presentations. Clin Dermatol. 2005;23:480-490.
9. Lebeaux D, Francès C, Barete S, et al. Eosinophilic fasciitis (Shulman disease): new insights into the therapeutic management from a series of 34 patients. Rheumatology. 2012;51:557-561.
|
A 43-year-old woman presented with pain and paresthesia of the bilateral legs of 3 months’ duration with skin tightness and discoloration, which she attributed to a car accident that occurred 7 months prior. She also reported abdominal pain, shortness of breath, fever, double vision, dysphagia, voice changes, temperature sensitivity, and hair loss. The patient underwent outpatient steroid injections with limited symptomatic relief. She denied any antecedent exposure to vinyl chloride, rapeseed oil, or L-tryptophan. Physical examination revealed thickened skin on the bilateral legs (top), reddish discoloration of the feet, decreased sensation to light touch, and edema of the ankles and wrists, as well as a peau d’orange appearance of the skin on the arms (bottom), legs, and abdomen.
The Diagnosis: Eosinophilic Fasciitis
Eosinophilic fasciitis is a rare autoimmune disease of uncertain etiology first described by Shulman1 in 1974. It is similar in presentation and is perhaps related to scleroderma. Classic differentiating features include a peculiar peau d’orange appearance, peripheral eosinophilia, and lack of Raynaud phenomenon, thus it is regarded as a unique disease.2 Despite the name of the disease, eosinophilia has been known to be absent in later stages of eosinophilic fasciitis.1
On physical examination, “prayer and groove signs” can sometimes be evident.3 Although it was not initially observed in our case, a groove sign was noted on the left forearm on a second inspection (Figure 1). In contrast with systemic sclerosis, visceral involvement rarely is seen with eosinophilic fasciitis. There are, however, 3 major exceptions to this rule. First, there can be widespread nerve deficits, esophageal dysmotility, and nonspecific electromyography findings (ie, denervation, reinnervation, fasciculations).4 There also can be a concomitant hematologic disorder or Hashimoto thyroiditis.5 Because eosinophilic fasciitis has been associated with monoclonal gammopathy, which our patient also demonstrated, it is important to conduct a workup with serum or urine protein electrophoresis. If the test is negative, it should be followed up with an immunofixation assay or serum light chain assays.
Some proposed risk factors for eosinophilic fasciitis include trauma, extensive exercise, and Borrelia burgdorferi infection, but many cases have none of these associations.8 Although not firmly proven in the literature, there have been reports of eosinophilic fasciitis after isolated trauma.5 A causal link could not be established between our patient’s car accident and eosinophilic fasciitis, but the coincidence was notable.
The treatment of eosinophilic fasciitis is similar to scleroderma. Corticosteroids are effective in most cases and recovery often occurs with monotherapy.5 Case series have demonstrated efficacy in adding methotrexate, azathioprine, colchicine, and hydroxychloroquine in refractory patients.2,9 Our patient demonstrated a good response with a combination of prednisone and methotrexate. Relapses have been known to occur.2
A punch biopsy obtained from the right arm showed thickened acellular collagen bundles throughout the dermis and extending into the underlying subcutis. There also was obliteration of adnexal structures, loss of perieccrine fat, and sparse perivascular and interstitial lymphoplasmacytic infiltrate (Figure 2), consistent with a sclerosing disorder such as scleroderma or eosinophilic fasciitis.
A complete blood cell count revealed eosinophil levels of 12.5% (reference range, 2.7%). Rheumatologic workup was negative for antinuclear antibody, double-stranded DNA, thyroid-stimulating hormone, anticentromere antibodies, and Scl-70 autoantibodies. Computed tomography of the chest and pelvis revealed a thickened patulous esophagus. Endoscopy showed dysmotility of the lower esophagus. At this point the differential diagnosis included scleredema versus eosinophilic fasciitis, and the patient was started on oral prednisone 60 mg daily. She showed rapid improvement in sclerosis, joint mobility, and ability to swallow. Magnetic resonance imaging was then performed and revealed thickening and contrast enhancement of the forearm fascia, particularly along the distal aspect, confirming a diagnosis of eosinophilic fasciitis. Further workup including immunofixation assay and serum light chain assays were performed, revealing IgG λ hypergammaglobulinemia. She was then additionally treated with oral methotrexate 15 mg weekly. Due to the rapid improvement of symptoms on oral prednisone over 2 weeks, the peripheral eosinophilia, the magnetic resonance imaging findings, and the results of skin biopsy, a diagnosis of eosinophilic fasciitis was heavily favored over scleroderma and scleredema.
|
A 43-year-old woman presented with pain and paresthesia of the bilateral legs of 3 months’ duration with skin tightness and discoloration, which she attributed to a car accident that occurred 7 months prior. She also reported abdominal pain, shortness of breath, fever, double vision, dysphagia, voice changes, temperature sensitivity, and hair loss. The patient underwent outpatient steroid injections with limited symptomatic relief. She denied any antecedent exposure to vinyl chloride, rapeseed oil, or L-tryptophan. Physical examination revealed thickened skin on the bilateral legs (top), reddish discoloration of the feet, decreased sensation to light touch, and edema of the ankles and wrists, as well as a peau d’orange appearance of the skin on the arms (bottom), legs, and abdomen.
The Diagnosis: Eosinophilic Fasciitis
Eosinophilic fasciitis is a rare autoimmune disease of uncertain etiology first described by Shulman1 in 1974. It is similar in presentation and is perhaps related to scleroderma. Classic differentiating features include a peculiar peau d’orange appearance, peripheral eosinophilia, and lack of Raynaud phenomenon, thus it is regarded as a unique disease.2 Despite the name of the disease, eosinophilia has been known to be absent in later stages of eosinophilic fasciitis.1
On physical examination, “prayer and groove signs” can sometimes be evident.3 Although it was not initially observed in our case, a groove sign was noted on the left forearm on a second inspection (Figure 1). In contrast with systemic sclerosis, visceral involvement rarely is seen with eosinophilic fasciitis. There are, however, 3 major exceptions to this rule. First, there can be widespread nerve deficits, esophageal dysmotility, and nonspecific electromyography findings (ie, denervation, reinnervation, fasciculations).4 There also can be a concomitant hematologic disorder or Hashimoto thyroiditis.5 Because eosinophilic fasciitis has been associated with monoclonal gammopathy, which our patient also demonstrated, it is important to conduct a workup with serum or urine protein electrophoresis. If the test is negative, it should be followed up with an immunofixation assay or serum light chain assays.
Some proposed risk factors for eosinophilic fasciitis include trauma, extensive exercise, and Borrelia burgdorferi infection, but many cases have none of these associations.8 Although not firmly proven in the literature, there have been reports of eosinophilic fasciitis after isolated trauma.5 A causal link could not be established between our patient’s car accident and eosinophilic fasciitis, but the coincidence was notable.
The treatment of eosinophilic fasciitis is similar to scleroderma. Corticosteroids are effective in most cases and recovery often occurs with monotherapy.5 Case series have demonstrated efficacy in adding methotrexate, azathioprine, colchicine, and hydroxychloroquine in refractory patients.2,9 Our patient demonstrated a good response with a combination of prednisone and methotrexate. Relapses have been known to occur.2
A punch biopsy obtained from the right arm showed thickened acellular collagen bundles throughout the dermis and extending into the underlying subcutis. There also was obliteration of adnexal structures, loss of perieccrine fat, and sparse perivascular and interstitial lymphoplasmacytic infiltrate (Figure 2), consistent with a sclerosing disorder such as scleroderma or eosinophilic fasciitis.
A complete blood cell count revealed eosinophil levels of 12.5% (reference range, 2.7%). Rheumatologic workup was negative for antinuclear antibody, double-stranded DNA, thyroid-stimulating hormone, anticentromere antibodies, and Scl-70 autoantibodies. Computed tomography of the chest and pelvis revealed a thickened patulous esophagus. Endoscopy showed dysmotility of the lower esophagus. At this point the differential diagnosis included scleredema versus eosinophilic fasciitis, and the patient was started on oral prednisone 60 mg daily. She showed rapid improvement in sclerosis, joint mobility, and ability to swallow. Magnetic resonance imaging was then performed and revealed thickening and contrast enhancement of the forearm fascia, particularly along the distal aspect, confirming a diagnosis of eosinophilic fasciitis. Further workup including immunofixation assay and serum light chain assays were performed, revealing IgG λ hypergammaglobulinemia. She was then additionally treated with oral methotrexate 15 mg weekly. Due to the rapid improvement of symptoms on oral prednisone over 2 weeks, the peripheral eosinophilia, the magnetic resonance imaging findings, and the results of skin biopsy, a diagnosis of eosinophilic fasciitis was heavily favored over scleroderma and scleredema.
1. Shulman L. Diffuse fasciitis with hypergammaglobulinemia and eosinophilia in a new syndrome. J. Rheumatol. 1974;1(suppl):46.
2. Lakhanpal S, Ginsburg WW, Michet CJ, et al. Eosinophilic fasciitis: clinical spectrum and therapeutic response in 52 cases. Semin Arthritis Rheum. 1988;17:221-231.
3. Servy A, Clerici T, Malines C, et al. Eosinophilic fasciitis: a rare skin sclerosis. Pathol Res Int. 2010;2011:716935.
4. Satsangi J, Donaghy M. Multifocal peripheral neuropathy in eosinophilic fasciitis. J Neurol. 1992;239:91-92.
5. Antic M, Lautenschlager S, Itin PH. Eosinophilic fasciitis 30 years after—what do we really know? Dermatology. 2006;213:93-101.
6. Doyle JA, Ginsburg WW. Eosinophilic fasciitis. Med Clin North Am. 1989;73:1157-1166.
7. Naschitz JE, Yeshurun D, Miselevich I, et al. Colitis and pericarditis in a patient with eosinophilic fasciitis—a contribution to the multisystem nature of eosinophilic fasciitis. J Rheumatol. 1989;16:688-692.
8. Haustein UF. Scleroderma and pseudoscleroderma: uncommon presentations. Clin Dermatol. 2005;23:480-490.
9. Lebeaux D, Francès C, Barete S, et al. Eosinophilic fasciitis (Shulman disease): new insights into the therapeutic management from a series of 34 patients. Rheumatology. 2012;51:557-561.
1. Shulman L. Diffuse fasciitis with hypergammaglobulinemia and eosinophilia in a new syndrome. J. Rheumatol. 1974;1(suppl):46.
2. Lakhanpal S, Ginsburg WW, Michet CJ, et al. Eosinophilic fasciitis: clinical spectrum and therapeutic response in 52 cases. Semin Arthritis Rheum. 1988;17:221-231.
3. Servy A, Clerici T, Malines C, et al. Eosinophilic fasciitis: a rare skin sclerosis. Pathol Res Int. 2010;2011:716935.
4. Satsangi J, Donaghy M. Multifocal peripheral neuropathy in eosinophilic fasciitis. J Neurol. 1992;239:91-92.
5. Antic M, Lautenschlager S, Itin PH. Eosinophilic fasciitis 30 years after—what do we really know? Dermatology. 2006;213:93-101.
6. Doyle JA, Ginsburg WW. Eosinophilic fasciitis. Med Clin North Am. 1989;73:1157-1166.
7. Naschitz JE, Yeshurun D, Miselevich I, et al. Colitis and pericarditis in a patient with eosinophilic fasciitis—a contribution to the multisystem nature of eosinophilic fasciitis. J Rheumatol. 1989;16:688-692.
8. Haustein UF. Scleroderma and pseudoscleroderma: uncommon presentations. Clin Dermatol. 2005;23:480-490.
9. Lebeaux D, Francès C, Barete S, et al. Eosinophilic fasciitis (Shulman disease): new insights into the therapeutic management from a series of 34 patients. Rheumatology. 2012;51:557-561.