User login
Front-line BTK inhibitor+anti-CD20 antibody tops chemoimmunotherapy in CLL, reveals meta-analysis
Key clinical point: Compared with chemoimmunotherapy (CIT), Bruton tyrosine kinase inhibitor (BTKi) therapy combined with anti-CD20 antibody therapy improves clinical outcomes in patients with treatment-naive chronic lymphocytic leukemia (CLL) without causing increased toxicity.
Major finding: Patients receiving BTKi+anti-CD20 antibody vs CIT had significantly prolonged progression-free survival (hazard ratio 0.25; 95% CI 0.15-0.42) and higher objective response rates (risk ratio [RR] 1.16; 95% CI 1.13-1.20) and a comparable risk for grade ≥3 adverse events (RR 1.04; 95% CI 0.92-1.17).
Study details: The data come from a meta-analysis of four randomized controlled trials involving 1479 patients with treatment-naive CLL who had been randomized to receive CIT or BTKi+anti-CD20 antibody therapy.
Disclosures: This study was partly supported by the Ministry of Science and Technology, Taiwan, and Taipei Medical University, Taiwan. The authors declared no conflicts of interest.
Source: Nguyen TT et al. Efficacy and safety of Bruton tyrosine kinase inhibitor plus anti-CD20 antibody therapy compared with chemoimmunotherapy as front-line treatment for chronic lymphocytic leukemia: A systematic review and meta-analysis of randomized controlled trials. J Immunother. 2023 (May 23). doi: 10.1097/CJI.0000000000000471
Key clinical point: Compared with chemoimmunotherapy (CIT), Bruton tyrosine kinase inhibitor (BTKi) therapy combined with anti-CD20 antibody therapy improves clinical outcomes in patients with treatment-naive chronic lymphocytic leukemia (CLL) without causing increased toxicity.
Major finding: Patients receiving BTKi+anti-CD20 antibody vs CIT had significantly prolonged progression-free survival (hazard ratio 0.25; 95% CI 0.15-0.42) and higher objective response rates (risk ratio [RR] 1.16; 95% CI 1.13-1.20) and a comparable risk for grade ≥3 adverse events (RR 1.04; 95% CI 0.92-1.17).
Study details: The data come from a meta-analysis of four randomized controlled trials involving 1479 patients with treatment-naive CLL who had been randomized to receive CIT or BTKi+anti-CD20 antibody therapy.
Disclosures: This study was partly supported by the Ministry of Science and Technology, Taiwan, and Taipei Medical University, Taiwan. The authors declared no conflicts of interest.
Source: Nguyen TT et al. Efficacy and safety of Bruton tyrosine kinase inhibitor plus anti-CD20 antibody therapy compared with chemoimmunotherapy as front-line treatment for chronic lymphocytic leukemia: A systematic review and meta-analysis of randomized controlled trials. J Immunother. 2023 (May 23). doi: 10.1097/CJI.0000000000000471
Key clinical point: Compared with chemoimmunotherapy (CIT), Bruton tyrosine kinase inhibitor (BTKi) therapy combined with anti-CD20 antibody therapy improves clinical outcomes in patients with treatment-naive chronic lymphocytic leukemia (CLL) without causing increased toxicity.
Major finding: Patients receiving BTKi+anti-CD20 antibody vs CIT had significantly prolonged progression-free survival (hazard ratio 0.25; 95% CI 0.15-0.42) and higher objective response rates (risk ratio [RR] 1.16; 95% CI 1.13-1.20) and a comparable risk for grade ≥3 adverse events (RR 1.04; 95% CI 0.92-1.17).
Study details: The data come from a meta-analysis of four randomized controlled trials involving 1479 patients with treatment-naive CLL who had been randomized to receive CIT or BTKi+anti-CD20 antibody therapy.
Disclosures: This study was partly supported by the Ministry of Science and Technology, Taiwan, and Taipei Medical University, Taiwan. The authors declared no conflicts of interest.
Source: Nguyen TT et al. Efficacy and safety of Bruton tyrosine kinase inhibitor plus anti-CD20 antibody therapy compared with chemoimmunotherapy as front-line treatment for chronic lymphocytic leukemia: A systematic review and meta-analysis of randomized controlled trials. J Immunother. 2023 (May 23). doi: 10.1097/CJI.0000000000000471
R-High-CHOP/CHASER/LEED with auto-PBSCT provides favorable long-term survival outcomes in untreated MCL
Key clinical point: High-dose rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-High-CHOP)/cyclophosphamide, high-dose cytarabine, dexamethasone, etoposide, and rituximab (CHASER) followed by high-dose chemotherapy (HDC) with melphalan, cyclophosphamide, etoposide, and dexamethasone (LEED), and autologous peripheral blood stem cell transplantation (auto-PBSCT) provided favorable long-term survival outcomes in younger patients with untreated advanced mantle cell lymphoma (MCL).
Major finding: At 6-year median follow-up, the 5- and 8-year progression-free survival rates were 52.3% (95% CI 36.7%-65.7%) and 17.1% (95% CI 5.2%-34.7%), respectively, and the overall survival rates were 75.0% (95% CI 59.4%-85.3%) and 69.0% (95% CI 52.3%-80.9%), respectively. The incidence of secondary malignancies (11.1%) and continuous relapses was high.
Study details: This final analysis of the JCOG0406 study included 45 patients age ≤ 65 years with untreated advanced MCL who received R-High-CHOP/CHASER followed by HDC with LEED and auto-PBSCT.
Disclosures: This study was supported by Health and Labour Sciences Research Grants (Japan) and others. S Nakamura declared being an Editorial Board Member of Cancer Science. Some authors reported ties with various organizations.
Source: Ogura M et al. Long-term follow-up after R-High CHOP/CHASER/LEED with Auto-PBSCT in untreated mantle cell lymphoma-Final analysis of JCOG0406. Cancer Sci. 2023 (May 26). doi: 10.1111/cas.15849
Key clinical point: High-dose rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-High-CHOP)/cyclophosphamide, high-dose cytarabine, dexamethasone, etoposide, and rituximab (CHASER) followed by high-dose chemotherapy (HDC) with melphalan, cyclophosphamide, etoposide, and dexamethasone (LEED), and autologous peripheral blood stem cell transplantation (auto-PBSCT) provided favorable long-term survival outcomes in younger patients with untreated advanced mantle cell lymphoma (MCL).
Major finding: At 6-year median follow-up, the 5- and 8-year progression-free survival rates were 52.3% (95% CI 36.7%-65.7%) and 17.1% (95% CI 5.2%-34.7%), respectively, and the overall survival rates were 75.0% (95% CI 59.4%-85.3%) and 69.0% (95% CI 52.3%-80.9%), respectively. The incidence of secondary malignancies (11.1%) and continuous relapses was high.
Study details: This final analysis of the JCOG0406 study included 45 patients age ≤ 65 years with untreated advanced MCL who received R-High-CHOP/CHASER followed by HDC with LEED and auto-PBSCT.
Disclosures: This study was supported by Health and Labour Sciences Research Grants (Japan) and others. S Nakamura declared being an Editorial Board Member of Cancer Science. Some authors reported ties with various organizations.
Source: Ogura M et al. Long-term follow-up after R-High CHOP/CHASER/LEED with Auto-PBSCT in untreated mantle cell lymphoma-Final analysis of JCOG0406. Cancer Sci. 2023 (May 26). doi: 10.1111/cas.15849
Key clinical point: High-dose rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-High-CHOP)/cyclophosphamide, high-dose cytarabine, dexamethasone, etoposide, and rituximab (CHASER) followed by high-dose chemotherapy (HDC) with melphalan, cyclophosphamide, etoposide, and dexamethasone (LEED), and autologous peripheral blood stem cell transplantation (auto-PBSCT) provided favorable long-term survival outcomes in younger patients with untreated advanced mantle cell lymphoma (MCL).
Major finding: At 6-year median follow-up, the 5- and 8-year progression-free survival rates were 52.3% (95% CI 36.7%-65.7%) and 17.1% (95% CI 5.2%-34.7%), respectively, and the overall survival rates were 75.0% (95% CI 59.4%-85.3%) and 69.0% (95% CI 52.3%-80.9%), respectively. The incidence of secondary malignancies (11.1%) and continuous relapses was high.
Study details: This final analysis of the JCOG0406 study included 45 patients age ≤ 65 years with untreated advanced MCL who received R-High-CHOP/CHASER followed by HDC with LEED and auto-PBSCT.
Disclosures: This study was supported by Health and Labour Sciences Research Grants (Japan) and others. S Nakamura declared being an Editorial Board Member of Cancer Science. Some authors reported ties with various organizations.
Source: Ogura M et al. Long-term follow-up after R-High CHOP/CHASER/LEED with Auto-PBSCT in untreated mantle cell lymphoma-Final analysis of JCOG0406. Cancer Sci. 2023 (May 26). doi: 10.1111/cas.15849
Tafasitamab+lenalidomide provides survival benefit over pola-BR and R2 in relapsed or refractory DLBCL
Key clinical point: Compared with polatuzumab vedotin+bendamustine+rituximab (pola-BR) and rituximab+lenalidomide (R2), tafasitamab+lenalidomide provided a significant survival benefit in patients with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL).
Major finding: A significant overall survival (OS) benefit was observed with tafasitamab+lenalidomide vs pola-BR (hazard ratio [HR] 0.441; P = .034) and R2 (HR 0.435; P = .012). However, the OS was similar between the tafasitamab+lenalidomide and CD19-chimeric antigen receptor T-cell groups (CAR-T; HR 0.953; P = .892).
Study details: This expanded analysis of the RE-MIND2 study of propensity score-matched transplant-ineligible patients with R/R DLBCL treated with ≥2 systemic therapies who received tafasitamab+lenalidomide in the L-MIND trial and those who received pola-BR (24 pairs), R2 (33 pairs), or CAR-T (37 pairs) from an observational cohort.
Disclosures: This study was sponsored by MorphoSys AG. Some authors declared serving as consultants, advisory board members, or speakers and receiving research funding, honoraria, or travel support from MorphoSys and others. Four authors declared being employees of or holding equities in MorphoSys.
Source: Nowakowski GS et al. RE-MIND2: Comparative effectiveness of tafasitamab plus lenalidomide versus polatuzumab vedotin/bendamustine/rituximab (pola‑BR), CAR‑T therapies, and lenalidomide/rituximab (R2) based on real‑world data in patients with relapsed/refractory diffuse large B‑cell lymphoma. Ann Hematol. 2023;102:1773-1787 (May 12). doi: 10.1007/s00277-023-05196-4
Key clinical point: Compared with polatuzumab vedotin+bendamustine+rituximab (pola-BR) and rituximab+lenalidomide (R2), tafasitamab+lenalidomide provided a significant survival benefit in patients with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL).
Major finding: A significant overall survival (OS) benefit was observed with tafasitamab+lenalidomide vs pola-BR (hazard ratio [HR] 0.441; P = .034) and R2 (HR 0.435; P = .012). However, the OS was similar between the tafasitamab+lenalidomide and CD19-chimeric antigen receptor T-cell groups (CAR-T; HR 0.953; P = .892).
Study details: This expanded analysis of the RE-MIND2 study of propensity score-matched transplant-ineligible patients with R/R DLBCL treated with ≥2 systemic therapies who received tafasitamab+lenalidomide in the L-MIND trial and those who received pola-BR (24 pairs), R2 (33 pairs), or CAR-T (37 pairs) from an observational cohort.
Disclosures: This study was sponsored by MorphoSys AG. Some authors declared serving as consultants, advisory board members, or speakers and receiving research funding, honoraria, or travel support from MorphoSys and others. Four authors declared being employees of or holding equities in MorphoSys.
Source: Nowakowski GS et al. RE-MIND2: Comparative effectiveness of tafasitamab plus lenalidomide versus polatuzumab vedotin/bendamustine/rituximab (pola‑BR), CAR‑T therapies, and lenalidomide/rituximab (R2) based on real‑world data in patients with relapsed/refractory diffuse large B‑cell lymphoma. Ann Hematol. 2023;102:1773-1787 (May 12). doi: 10.1007/s00277-023-05196-4
Key clinical point: Compared with polatuzumab vedotin+bendamustine+rituximab (pola-BR) and rituximab+lenalidomide (R2), tafasitamab+lenalidomide provided a significant survival benefit in patients with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL).
Major finding: A significant overall survival (OS) benefit was observed with tafasitamab+lenalidomide vs pola-BR (hazard ratio [HR] 0.441; P = .034) and R2 (HR 0.435; P = .012). However, the OS was similar between the tafasitamab+lenalidomide and CD19-chimeric antigen receptor T-cell groups (CAR-T; HR 0.953; P = .892).
Study details: This expanded analysis of the RE-MIND2 study of propensity score-matched transplant-ineligible patients with R/R DLBCL treated with ≥2 systemic therapies who received tafasitamab+lenalidomide in the L-MIND trial and those who received pola-BR (24 pairs), R2 (33 pairs), or CAR-T (37 pairs) from an observational cohort.
Disclosures: This study was sponsored by MorphoSys AG. Some authors declared serving as consultants, advisory board members, or speakers and receiving research funding, honoraria, or travel support from MorphoSys and others. Four authors declared being employees of or holding equities in MorphoSys.
Source: Nowakowski GS et al. RE-MIND2: Comparative effectiveness of tafasitamab plus lenalidomide versus polatuzumab vedotin/bendamustine/rituximab (pola‑BR), CAR‑T therapies, and lenalidomide/rituximab (R2) based on real‑world data in patients with relapsed/refractory diffuse large B‑cell lymphoma. Ann Hematol. 2023;102:1773-1787 (May 12). doi: 10.1007/s00277-023-05196-4
First-line rituximab+bendamustine+cytarabine combination shows long-term efficacy in the elderly with MCL
Key clinical point: The rituximab, bendamustine, and low-dose cytarabine (R-BAC) regimen, not succeeded by maintenance therapy, induces long-term responses in previously untreated elderly patients with mantle cell lymphoma (MCL).
Major finding: After a median follow-up of 86 months, the median overall survival (OS) and progression-free survival (PFS) were not reached; the 7-year PFS and OS rates were 55% (95% CI 41%-67%) and 63% (95% CI 49%-74%), respectively. The 7-year duration of response rate among the 52 responding patients was 59% (95% CI 44%-71%). No signal of late toxicity was reported.
Study details: Findings are from a long-term analysis of the FIL-RBAC500 trial that included 57 previously untreated elderly patients with MCL who received the R-BAC regimen.
Disclosures: This study was supported by Progetto di Ricerca Sanitaria Finalizzata (Italy) grants and other sources. Some authors declared serving as advisory board members or consultants and receiving research funding, speaker honoraria, or travel expenses and accommodations from various sources.
Source: Tisi MC et al. Long term follow-up of rituximab plus bendamustine and cytarabine (R-BAC) in elderly patients with newly diagnosed MCL. Blood Adv. 2023 (May 12). doi: 10.1182/bloodadvances.2023009744
Key clinical point: The rituximab, bendamustine, and low-dose cytarabine (R-BAC) regimen, not succeeded by maintenance therapy, induces long-term responses in previously untreated elderly patients with mantle cell lymphoma (MCL).
Major finding: After a median follow-up of 86 months, the median overall survival (OS) and progression-free survival (PFS) were not reached; the 7-year PFS and OS rates were 55% (95% CI 41%-67%) and 63% (95% CI 49%-74%), respectively. The 7-year duration of response rate among the 52 responding patients was 59% (95% CI 44%-71%). No signal of late toxicity was reported.
Study details: Findings are from a long-term analysis of the FIL-RBAC500 trial that included 57 previously untreated elderly patients with MCL who received the R-BAC regimen.
Disclosures: This study was supported by Progetto di Ricerca Sanitaria Finalizzata (Italy) grants and other sources. Some authors declared serving as advisory board members or consultants and receiving research funding, speaker honoraria, or travel expenses and accommodations from various sources.
Source: Tisi MC et al. Long term follow-up of rituximab plus bendamustine and cytarabine (R-BAC) in elderly patients with newly diagnosed MCL. Blood Adv. 2023 (May 12). doi: 10.1182/bloodadvances.2023009744
Key clinical point: The rituximab, bendamustine, and low-dose cytarabine (R-BAC) regimen, not succeeded by maintenance therapy, induces long-term responses in previously untreated elderly patients with mantle cell lymphoma (MCL).
Major finding: After a median follow-up of 86 months, the median overall survival (OS) and progression-free survival (PFS) were not reached; the 7-year PFS and OS rates were 55% (95% CI 41%-67%) and 63% (95% CI 49%-74%), respectively. The 7-year duration of response rate among the 52 responding patients was 59% (95% CI 44%-71%). No signal of late toxicity was reported.
Study details: Findings are from a long-term analysis of the FIL-RBAC500 trial that included 57 previously untreated elderly patients with MCL who received the R-BAC regimen.
Disclosures: This study was supported by Progetto di Ricerca Sanitaria Finalizzata (Italy) grants and other sources. Some authors declared serving as advisory board members or consultants and receiving research funding, speaker honoraria, or travel expenses and accommodations from various sources.
Source: Tisi MC et al. Long term follow-up of rituximab plus bendamustine and cytarabine (R-BAC) in elderly patients with newly diagnosed MCL. Blood Adv. 2023 (May 12). doi: 10.1182/bloodadvances.2023009744
First-line fixed-duration ibrutinib+venetoclax shows promise against high-risk CLL
Key clinical point: First-line ibrutinib+venetoclax led to high response and survival rates in patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) regardless of high-risk genomic features generally associated with poor outcomes.
Major finding: Overall response and 36-month overall survival rates were >95% regardless of high-risk features. Patients with and without high-risk features had similar complete response (61% [95% CI 53%-70%] and 53% [95% CI 41%-65%], respectively) and 36-month progression-free survival (88% [95% CI 81%-93%] and 92% [95% CI 82%-97%], respectively) rates.
Study details: This post hoc analysis of the CAPTIVATE trial analyzed the pooled data of 195 patients with previously untreated CLL/SLL and known status of high-risk features [del(17p), TP53 mutation, and unmutated immunoglobulin heavy chain] treated with fixed-duration ibrutinib+venetoclax, of which 129 had ≥1 high-risk feature.
Disclosures: This study was funded by Pharmacyclics LLC, an AbbVie Company. Some authors declared receiving grants, personal fees, or other support from Pharmacyclics, AbbVie, or others. Two authors declared being employees of Pharmacyclics or AbbVie or having stock or other ownership interests in AbbVie.
Source: Allan JN et al. Outcomes in patients with high-risk features after fixed-duration ibrutinib plus venetoclax: Phase II CAPTIVATE study in first-line chronic lymphocytic leukemia. Clin Cancer Res. 2023 (Jun 7). doi: 10.1158/1078-0432.CCR-22-2779
Key clinical point: First-line ibrutinib+venetoclax led to high response and survival rates in patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) regardless of high-risk genomic features generally associated with poor outcomes.
Major finding: Overall response and 36-month overall survival rates were >95% regardless of high-risk features. Patients with and without high-risk features had similar complete response (61% [95% CI 53%-70%] and 53% [95% CI 41%-65%], respectively) and 36-month progression-free survival (88% [95% CI 81%-93%] and 92% [95% CI 82%-97%], respectively) rates.
Study details: This post hoc analysis of the CAPTIVATE trial analyzed the pooled data of 195 patients with previously untreated CLL/SLL and known status of high-risk features [del(17p), TP53 mutation, and unmutated immunoglobulin heavy chain] treated with fixed-duration ibrutinib+venetoclax, of which 129 had ≥1 high-risk feature.
Disclosures: This study was funded by Pharmacyclics LLC, an AbbVie Company. Some authors declared receiving grants, personal fees, or other support from Pharmacyclics, AbbVie, or others. Two authors declared being employees of Pharmacyclics or AbbVie or having stock or other ownership interests in AbbVie.
Source: Allan JN et al. Outcomes in patients with high-risk features after fixed-duration ibrutinib plus venetoclax: Phase II CAPTIVATE study in first-line chronic lymphocytic leukemia. Clin Cancer Res. 2023 (Jun 7). doi: 10.1158/1078-0432.CCR-22-2779
Key clinical point: First-line ibrutinib+venetoclax led to high response and survival rates in patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) regardless of high-risk genomic features generally associated with poor outcomes.
Major finding: Overall response and 36-month overall survival rates were >95% regardless of high-risk features. Patients with and without high-risk features had similar complete response (61% [95% CI 53%-70%] and 53% [95% CI 41%-65%], respectively) and 36-month progression-free survival (88% [95% CI 81%-93%] and 92% [95% CI 82%-97%], respectively) rates.
Study details: This post hoc analysis of the CAPTIVATE trial analyzed the pooled data of 195 patients with previously untreated CLL/SLL and known status of high-risk features [del(17p), TP53 mutation, and unmutated immunoglobulin heavy chain] treated with fixed-duration ibrutinib+venetoclax, of which 129 had ≥1 high-risk feature.
Disclosures: This study was funded by Pharmacyclics LLC, an AbbVie Company. Some authors declared receiving grants, personal fees, or other support from Pharmacyclics, AbbVie, or others. Two authors declared being employees of Pharmacyclics or AbbVie or having stock or other ownership interests in AbbVie.
Source: Allan JN et al. Outcomes in patients with high-risk features after fixed-duration ibrutinib plus venetoclax: Phase II CAPTIVATE study in first-line chronic lymphocytic leukemia. Clin Cancer Res. 2023 (Jun 7). doi: 10.1158/1078-0432.CCR-22-2779
Depression or anxiety shortens the survival of DLBCL patients
Key clinical point: The preexistence of depression, anxiety, or both is associated with shorter survival in older patients with diffuse large B-cell lymphoma (DLBCL).
Major finding: At a median follow-up of 2.0 years, patients with depression, anxiety, or both vs without any mental disorder had significantly lower 5-year overall survival rates (27.0% vs 37.4%; hazard ratio [HR] 1.37; 95% CI 1.29-1.44), with those with preexisting depression vs without any mental disorder having the worst rate (23.4% vs 38.0%; HR 1.37; P < .0001).
Study details: This retrospective cohort study analyzed the data of 13,244 patients age ≥ 67 years with DLBCL from the Surveillance, Epidemiology, and End Results-Medicare (SEER) registry, of which 2094 had depression, anxiety, or both at the time of their DLBCL diagnosis.
Disclosures: This study was funded by the American Society of Hematology and others. Some authors declared participating on Data Safety Monitoring or Advisory Boards or receiving financial support, royalties or licenses, or payments or honoraria for lectures, presentations, speakers’ bureaus, manuscript writing, or educational events from various organizations.
Source: Kuczmarski TM et al. Mental health disorders and survival among older patients with diffuse large B-cell lymphoma in the USA: A population-based study. Lancet Haematol. 2023 (Jun 1). doi: 10.1016/S2352-3026(23)00094-7
Key clinical point: The preexistence of depression, anxiety, or both is associated with shorter survival in older patients with diffuse large B-cell lymphoma (DLBCL).
Major finding: At a median follow-up of 2.0 years, patients with depression, anxiety, or both vs without any mental disorder had significantly lower 5-year overall survival rates (27.0% vs 37.4%; hazard ratio [HR] 1.37; 95% CI 1.29-1.44), with those with preexisting depression vs without any mental disorder having the worst rate (23.4% vs 38.0%; HR 1.37; P < .0001).
Study details: This retrospective cohort study analyzed the data of 13,244 patients age ≥ 67 years with DLBCL from the Surveillance, Epidemiology, and End Results-Medicare (SEER) registry, of which 2094 had depression, anxiety, or both at the time of their DLBCL diagnosis.
Disclosures: This study was funded by the American Society of Hematology and others. Some authors declared participating on Data Safety Monitoring or Advisory Boards or receiving financial support, royalties or licenses, or payments or honoraria for lectures, presentations, speakers’ bureaus, manuscript writing, or educational events from various organizations.
Source: Kuczmarski TM et al. Mental health disorders and survival among older patients with diffuse large B-cell lymphoma in the USA: A population-based study. Lancet Haematol. 2023 (Jun 1). doi: 10.1016/S2352-3026(23)00094-7
Key clinical point: The preexistence of depression, anxiety, or both is associated with shorter survival in older patients with diffuse large B-cell lymphoma (DLBCL).
Major finding: At a median follow-up of 2.0 years, patients with depression, anxiety, or both vs without any mental disorder had significantly lower 5-year overall survival rates (27.0% vs 37.4%; hazard ratio [HR] 1.37; 95% CI 1.29-1.44), with those with preexisting depression vs without any mental disorder having the worst rate (23.4% vs 38.0%; HR 1.37; P < .0001).
Study details: This retrospective cohort study analyzed the data of 13,244 patients age ≥ 67 years with DLBCL from the Surveillance, Epidemiology, and End Results-Medicare (SEER) registry, of which 2094 had depression, anxiety, or both at the time of their DLBCL diagnosis.
Disclosures: This study was funded by the American Society of Hematology and others. Some authors declared participating on Data Safety Monitoring or Advisory Boards or receiving financial support, royalties or licenses, or payments or honoraria for lectures, presentations, speakers’ bureaus, manuscript writing, or educational events from various organizations.
Source: Kuczmarski TM et al. Mental health disorders and survival among older patients with diffuse large B-cell lymphoma in the USA: A population-based study. Lancet Haematol. 2023 (Jun 1). doi: 10.1016/S2352-3026(23)00094-7
Phase 3 data favor axi-cel over standard care in high-risk large B-cell lymphoma
Key clinical point: Compared with the current standard-of-care chemoimmunotherapy, second-line axicabtagene ciloleucel (axi-cel) significantly prolongs the overall survival of patients with early relapsed or refractory large B-cell lymphoma (LBCL).
Major finding: At a median follow-up of 47.2 months, patients receiving axi-cel vs standard care had a significantly longer median overall survival (not reached vs 31.1 months; hazard ratio 0.73; P = .03) and an absolute improvement in overall survival (8.6 percentage points at 4 years). No new treatment-related deaths were reported since the primary event-free survival analysis.
Study details: This primary overall survival analysis of the phase 3 ZUMA-7 trial included 359 adults with LBCL (refractory to or relapsed after first-line treatment) who were randomly assigned to receive axi-cel (n = 180) or standard care (n = 179).
Disclosures: This study was funded by Kite Pharma. Some authors, including the lead author, declared serving as advisory board members, consultants, or speakers for; receiving research support, speaker fees, travel expenses, or honoraria from; or owning stock or stock options in various sources, including Kite.
Source: Westin JR et al. Survival with axicabtagene ciloleucel in large B-cell lymphoma. N Engl J Med. 2023 (Jun 5). doi: 10.1056/NEJMoa2301665
Key clinical point: Compared with the current standard-of-care chemoimmunotherapy, second-line axicabtagene ciloleucel (axi-cel) significantly prolongs the overall survival of patients with early relapsed or refractory large B-cell lymphoma (LBCL).
Major finding: At a median follow-up of 47.2 months, patients receiving axi-cel vs standard care had a significantly longer median overall survival (not reached vs 31.1 months; hazard ratio 0.73; P = .03) and an absolute improvement in overall survival (8.6 percentage points at 4 years). No new treatment-related deaths were reported since the primary event-free survival analysis.
Study details: This primary overall survival analysis of the phase 3 ZUMA-7 trial included 359 adults with LBCL (refractory to or relapsed after first-line treatment) who were randomly assigned to receive axi-cel (n = 180) or standard care (n = 179).
Disclosures: This study was funded by Kite Pharma. Some authors, including the lead author, declared serving as advisory board members, consultants, or speakers for; receiving research support, speaker fees, travel expenses, or honoraria from; or owning stock or stock options in various sources, including Kite.
Source: Westin JR et al. Survival with axicabtagene ciloleucel in large B-cell lymphoma. N Engl J Med. 2023 (Jun 5). doi: 10.1056/NEJMoa2301665
Key clinical point: Compared with the current standard-of-care chemoimmunotherapy, second-line axicabtagene ciloleucel (axi-cel) significantly prolongs the overall survival of patients with early relapsed or refractory large B-cell lymphoma (LBCL).
Major finding: At a median follow-up of 47.2 months, patients receiving axi-cel vs standard care had a significantly longer median overall survival (not reached vs 31.1 months; hazard ratio 0.73; P = .03) and an absolute improvement in overall survival (8.6 percentage points at 4 years). No new treatment-related deaths were reported since the primary event-free survival analysis.
Study details: This primary overall survival analysis of the phase 3 ZUMA-7 trial included 359 adults with LBCL (refractory to or relapsed after first-line treatment) who were randomly assigned to receive axi-cel (n = 180) or standard care (n = 179).
Disclosures: This study was funded by Kite Pharma. Some authors, including the lead author, declared serving as advisory board members, consultants, or speakers for; receiving research support, speaker fees, travel expenses, or honoraria from; or owning stock or stock options in various sources, including Kite.
Source: Westin JR et al. Survival with axicabtagene ciloleucel in large B-cell lymphoma. N Engl J Med. 2023 (Jun 5). doi: 10.1056/NEJMoa2301665
Pirtobrutinib offers a promising treatment option for covalent BTK-inhibitor pretreated MCL
Key clinical point: Pirtobrutinib demonstrated durable efficacy and a favorable safety profile in patients with covalent Bruton tyrosine kinase inhibitor (cBTKi) pretreated relapsed or refractory mantle cell lymphoma (MCL).
Major finding: The overall response rate was 57.8% (95% CI 46.9%-68.1%), with the complete response rate being 20.0%. At a median follow-up of 12 months, the median duration of response was 21.6 (95% CI 7.5-not reached) months. Grade ≥3 treatment-related adverse events were not frequent, with neutropenia (8.5%) being the most common.
Study details: This multicenter phase 1/2 BRUIN trial included 90 cBTKi pretreated patients with relapsed or refractory MCL in the primary efficacy cohort who received 25-300 mg and 200 mg pirtobrutinib once daily orally in the phases 1 and 2 of the trial, respectively.
Disclosures: This study was sponsored by Loxo Oncology Inc., a wholly owned subsidiary of Eli Lilly and Company. Some authors reported ties with Eli Lilly and others. Seven authors declared being employees of or stockholders in Eli Lilly.
Source: Wang ML et al. Pirtobrutinib in covalent BTK-inhibitor pre-treated mantle cell lymphoma. J Clin Oncol. 2023 (May 16). doi: 10.1200/JCO.23.00562
Key clinical point: Pirtobrutinib demonstrated durable efficacy and a favorable safety profile in patients with covalent Bruton tyrosine kinase inhibitor (cBTKi) pretreated relapsed or refractory mantle cell lymphoma (MCL).
Major finding: The overall response rate was 57.8% (95% CI 46.9%-68.1%), with the complete response rate being 20.0%. At a median follow-up of 12 months, the median duration of response was 21.6 (95% CI 7.5-not reached) months. Grade ≥3 treatment-related adverse events were not frequent, with neutropenia (8.5%) being the most common.
Study details: This multicenter phase 1/2 BRUIN trial included 90 cBTKi pretreated patients with relapsed or refractory MCL in the primary efficacy cohort who received 25-300 mg and 200 mg pirtobrutinib once daily orally in the phases 1 and 2 of the trial, respectively.
Disclosures: This study was sponsored by Loxo Oncology Inc., a wholly owned subsidiary of Eli Lilly and Company. Some authors reported ties with Eli Lilly and others. Seven authors declared being employees of or stockholders in Eli Lilly.
Source: Wang ML et al. Pirtobrutinib in covalent BTK-inhibitor pre-treated mantle cell lymphoma. J Clin Oncol. 2023 (May 16). doi: 10.1200/JCO.23.00562
Key clinical point: Pirtobrutinib demonstrated durable efficacy and a favorable safety profile in patients with covalent Bruton tyrosine kinase inhibitor (cBTKi) pretreated relapsed or refractory mantle cell lymphoma (MCL).
Major finding: The overall response rate was 57.8% (95% CI 46.9%-68.1%), with the complete response rate being 20.0%. At a median follow-up of 12 months, the median duration of response was 21.6 (95% CI 7.5-not reached) months. Grade ≥3 treatment-related adverse events were not frequent, with neutropenia (8.5%) being the most common.
Study details: This multicenter phase 1/2 BRUIN trial included 90 cBTKi pretreated patients with relapsed or refractory MCL in the primary efficacy cohort who received 25-300 mg and 200 mg pirtobrutinib once daily orally in the phases 1 and 2 of the trial, respectively.
Disclosures: This study was sponsored by Loxo Oncology Inc., a wholly owned subsidiary of Eli Lilly and Company. Some authors reported ties with Eli Lilly and others. Seven authors declared being employees of or stockholders in Eli Lilly.
Source: Wang ML et al. Pirtobrutinib in covalent BTK-inhibitor pre-treated mantle cell lymphoma. J Clin Oncol. 2023 (May 16). doi: 10.1200/JCO.23.00562
Erythematous Dermal Facial Plaques in a Neutropenic Patient
THE DIAGNOSIS: Neutrophilic Eccrine Hidradenitis
A biopsy from the left preauricular cheek demonstrated dermal neutrophilic inflammation around eccrine coils with focal necrosis (Figure). No notable diffuse dermal neutrophilic infiltrate was present, ruling out Sweet syndrome, and no notable interstitial neutrophilic infiltrate was present, making cellulitis and erysipelas less likely; panculture of tissue also was negative.1,2 Atypical cells in the deep dermis were positive for CD163 and negative for CD117, CD34, CD123, and myeloperoxidase, consistent with a diagnosis of neutrophilic eccrine hidradenitis (NEH) and reactive histiocytes.3 Treatment with oral prednisone resulted in rapid improvement of symptoms.
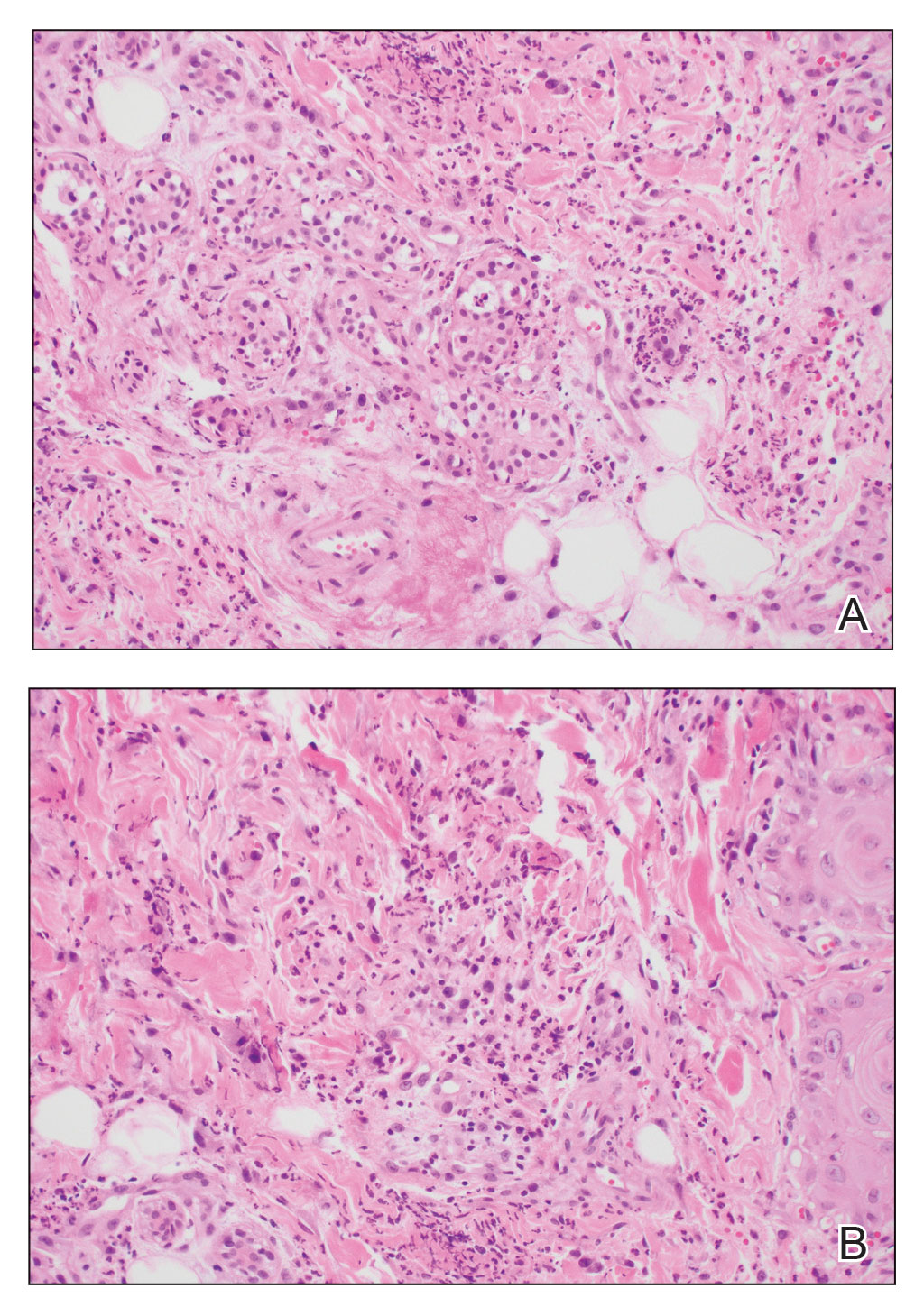
Neutrophilic eccrine hidradenitis is a rare reactive neutrophilic dermatosis characterized by eccrine gland involvement. This benign and self-limited condition presents as asymmetric erythematous papules and plaques.2 Among 8 granulocytopenic patients with neutrophilic dermatoses, 5 were diagnosed with NEH.4 Although first identified in 1982, NEH remains poorly understood.2 Initial theories suggested that NEH developed due to cytotoxic substances secreted in sweat glands causing necrosis and neutrophil chemotaxis; however, chemotherapy exposure cannot be linked to every case of NEH. Neutrophilic eccrine hidradenitis can be extremely difficult to differentiate clinically from conditions such as cellulitis and Sweet syndrome.
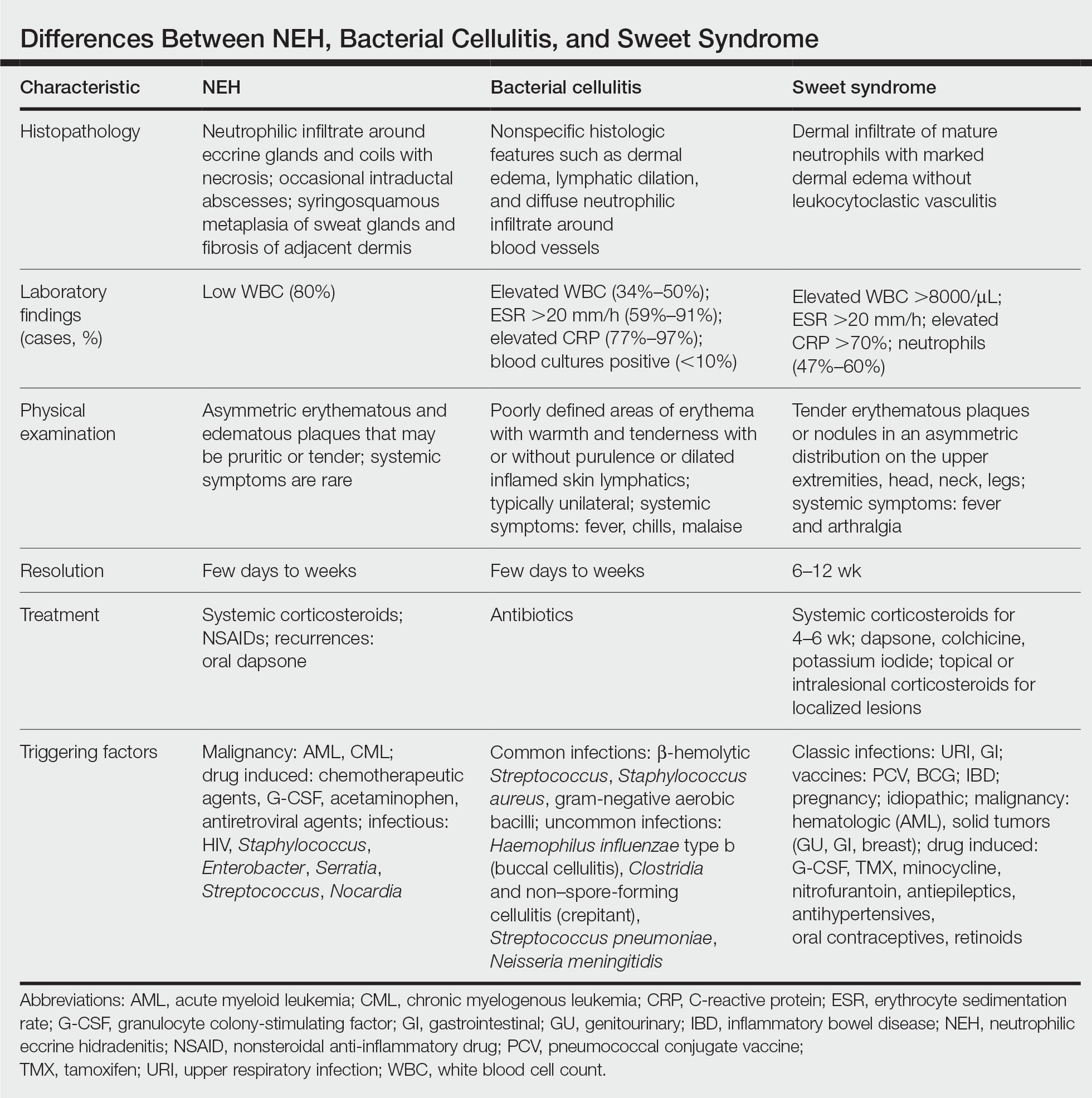
A patient history can be helpful in identifying triggering factors. Neutrophilic eccrine hidradenitis most commonly is associated with malignant, drug-induced, or infectious triggers, while Sweet syndrome has a broad range of associations including infections, vaccines, inflammatory bowel disease, pregnancy, malignancy, and drug-induced etiologies (Table).1 On average, NEH presents 10 days after chemotherapy induction, with 70% of cases presenting after the first chemotherapy cycle.5 Bacterial cellulitis or erysipelas have an infectious etiology, and patients may report symptoms such as fever, chills, or malaise. Immunosuppressed patients are at greater risk for infection; therefore, clinical signs of infection in a granulocytopenic patient should be addressed urgently.
Physical examination may have limited value in differentiating between these diagnoses, as neutrophilic dermatoses notoriously mimic infection. Cutaneous lesions can appear as pruritic or tender erythematous plaques, papules, or nodules in these conditions, though cellulitis and erysipelas tend to be unilateral and may have associated purulence or inflamed skin lymphatics. Given the potential for misdiagnosis, approaching patients with a broad differential can be helpful. In our patient, the differential diagnosis included Sweet syndrome, NEH, bacterial cellulitis, erysipelas, leukemia cutis, sarcoid, and eosinophilic cellulitis.
Leukemia cutis refers to the infiltration of neoplastic leukocytes in the skin and often occurs in patients with peripheral leukemia, most often acute myeloid leukemia or chronic lymphocytic leukemia. Patients with leukemia cutis often have a worse prognosis, as this finding signifies extramedullary spread of disease.6 Clinically, lesions can appear similar to those seen in our patient, though they typically are not symptomatic, can be nodular, tend to exhibit a violaceous hue, and occasionally may be hemorrhagic. Wells syndrome (also known as eosinophilic cellulitis) is an inflammatory dermatosis that presents as painful or pruritic, edematous and erythematous plaques.7,8 A green hue on resolution is present in some cases and may help clinicians differentiate this disease from mimickers.7 Often, eosinophilic cellulitis is misdiagnosed as bacterial cellulitis and treated with antibiotics. The presence of systemic symptoms such as fever or arthralgia is more typical of bacterial cellulitis, erysipelas, eosinophilic cellulitis, or Sweet syndrome than of NEH.1 Additionally, inflammatory markers (ie, C-reactive protein) and white blood cell counts tend to be elevated in bacterial cellulitis and Sweet syndrome, while leukopenia often is seen in NEH.
Histopathology is crucial in distinguishing these disease etiologies. Neutrophilic eccrine hidradenitis is diagnosed by the characteristic neutrophilic infiltrate and necrosis surrounding eccrine glands and coils. There also may be occasional intraductal abscesses and syringosquamous metaplasia of the sweat glands along with fibrosis of the adjacent dermis. In contrast, histologic sections of Sweet syndrome show numerous mature neutrophils infiltrating the dermis with marked papillary dermal edema. The histopathology of bacterial cellulitis and erysipelas is less specific, but common features include dermal edema, lymphatic dilation, and a diffuse neutrophilic infiltrate surrounding blood vessels. Pathogenic organisms may be seen on histopathology but are not required for the diagnosis of bacterial cellulitis or erysipelas.2 Additionally, blood and tissue culture can assist in identification of both the source of infection and the causative organism, but cultures may not always be positive.
Comparatively, the histopathologic features of eosinophilic cellulitis include dermal edema, eosinophilic infiltration, and flame figures that form when eosinophils degranulate and coat collagen fibers with major basic protein. Flame figures are characteristic but not pathognomonic for eosinophilic cellulitis.7 The histopathology of leukemia cutis varies based on the leukemia classification; generally, in acute myeloid leukemia the infiltrate is composed of neoplastic cells in the early stages of development that are positive for myeloid markers such as myeloperoxidase. Atypical and immature granulocytes within the leukocytic infiltrate differentiate this condition from the other diagnoses. Treatment may entail chemotherapy or radiotherapy, and this diagnosis generally carries the worst prognosis of all the conditions in the differential.6
Differentiating between these conditions is important in guiding treatment, especially in patients with febrile neutropenia. Unnecessary steroids in immunosuppressed patients can be dangerous, especially if the patient has an infection such as bacterial cellulitis. Furthermore, unwarranted antibiotic use for noninfectious conditions may expose patients to substantial side effects and not improve the condition. Neutrophilic eccrine hidradenitis typically is self-limited and treated symptomatically with systemic corticosteroids and nonsteroidal anti-inflammatory drugs.3 Sweet syndrome often requires a longer course of oral steroids. Leukemia cutis worsens as the leukemia advances, and treatment of the underlying malignancy is the most effective treatment.9
Early and accurate recognition of the diagnosis can prevent harmful diagnostic delay, unnecessary antibiotic use, or extended steroid taper in neutropenic patients. Appreciating the differences between these diagnoses can assist clinicians in investigating and tailoring a broad differential to specific patient presentations, which is especially critical when considering common mimickers for life-threatening conditions.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses. J Am Acad Dermatol. 2018;79:987-1006. doi:10.1016/j.jaad.2017.11.0642
- Srivastava M, Scharf S, Meehan SA, et al. Neutrophilic eccrine hidradenitis masquerading as facial cellulitis. J Am Acad Dermatol. 2007;56:693-696. doi:10.1016/j.jaad.2006.07.032
- Copaescu AM, Castilloux JF, Chababi-Atallah M, et al. A classic clinical case: neutrophilic eccrine hidradenitis. Case Rep Dermatol. 2013; 5:340-346. doi:10.1159/000356229
- Aractingi S, Mallet V, Pinquier L, et al. Neutrophilic dermatoses during granulocytopenia. Arch Dermatol. 1995;131:1141-1145.
- Cohen PR. Neutrophilic dermatoses occurring in oncology patients. Int J Dermatol. 2007;46:106-111. doi:10.1111/j.1365-4632.2006.02605.x
- Wang CX, Pusic I, Anadkat MJ. Association of leukemia cutis with survival in acute myeloid leukemia. JAMA Dermatol. 2019;155:826. doi:10.1001/jamadermatol.2019.0052
- Chung CL, Cusack CA. Wells syndrome: an enigmatic and therapeutically challenging disease. J Drugs Dermatol. 2006;5:908-911.
- Räßler F, Lukács J, Elsner P. Treatment of eosinophilic cellulitis (Wells syndrome): a systematic review. J Eur Acad Dermatol Venereol. 2016;30:1465-1479. doi:10.1111/jdv.13706
- Hobbs LK, Carr PC, Gru AA, et al. Case and review: cutaneous involvement by chronic neutrophilic leukemia vs Sweet syndrome: a diagnostic dilemma. J Cutan Pathol. 2021;48:644-649. doi:10.1111 /cup.13925
THE DIAGNOSIS: Neutrophilic Eccrine Hidradenitis
A biopsy from the left preauricular cheek demonstrated dermal neutrophilic inflammation around eccrine coils with focal necrosis (Figure). No notable diffuse dermal neutrophilic infiltrate was present, ruling out Sweet syndrome, and no notable interstitial neutrophilic infiltrate was present, making cellulitis and erysipelas less likely; panculture of tissue also was negative.1,2 Atypical cells in the deep dermis were positive for CD163 and negative for CD117, CD34, CD123, and myeloperoxidase, consistent with a diagnosis of neutrophilic eccrine hidradenitis (NEH) and reactive histiocytes.3 Treatment with oral prednisone resulted in rapid improvement of symptoms.
Neutrophilic eccrine hidradenitis is a rare reactive neutrophilic dermatosis characterized by eccrine gland involvement. This benign and self-limited condition presents as asymmetric erythematous papules and plaques.2 Among 8 granulocytopenic patients with neutrophilic dermatoses, 5 were diagnosed with NEH.4 Although first identified in 1982, NEH remains poorly understood.2 Initial theories suggested that NEH developed due to cytotoxic substances secreted in sweat glands causing necrosis and neutrophil chemotaxis; however, chemotherapy exposure cannot be linked to every case of NEH. Neutrophilic eccrine hidradenitis can be extremely difficult to differentiate clinically from conditions such as cellulitis and Sweet syndrome.
A patient history can be helpful in identifying triggering factors. Neutrophilic eccrine hidradenitis most commonly is associated with malignant, drug-induced, or infectious triggers, while Sweet syndrome has a broad range of associations including infections, vaccines, inflammatory bowel disease, pregnancy, malignancy, and drug-induced etiologies (Table).1 On average, NEH presents 10 days after chemotherapy induction, with 70% of cases presenting after the first chemotherapy cycle.5 Bacterial cellulitis or erysipelas have an infectious etiology, and patients may report symptoms such as fever, chills, or malaise. Immunosuppressed patients are at greater risk for infection; therefore, clinical signs of infection in a granulocytopenic patient should be addressed urgently.
Physical examination may have limited value in differentiating between these diagnoses, as neutrophilic dermatoses notoriously mimic infection. Cutaneous lesions can appear as pruritic or tender erythematous plaques, papules, or nodules in these conditions, though cellulitis and erysipelas tend to be unilateral and may have associated purulence or inflamed skin lymphatics. Given the potential for misdiagnosis, approaching patients with a broad differential can be helpful. In our patient, the differential diagnosis included Sweet syndrome, NEH, bacterial cellulitis, erysipelas, leukemia cutis, sarcoid, and eosinophilic cellulitis.
Leukemia cutis refers to the infiltration of neoplastic leukocytes in the skin and often occurs in patients with peripheral leukemia, most often acute myeloid leukemia or chronic lymphocytic leukemia. Patients with leukemia cutis often have a worse prognosis, as this finding signifies extramedullary spread of disease.6 Clinically, lesions can appear similar to those seen in our patient, though they typically are not symptomatic, can be nodular, tend to exhibit a violaceous hue, and occasionally may be hemorrhagic. Wells syndrome (also known as eosinophilic cellulitis) is an inflammatory dermatosis that presents as painful or pruritic, edematous and erythematous plaques.7,8 A green hue on resolution is present in some cases and may help clinicians differentiate this disease from mimickers.7 Often, eosinophilic cellulitis is misdiagnosed as bacterial cellulitis and treated with antibiotics. The presence of systemic symptoms such as fever or arthralgia is more typical of bacterial cellulitis, erysipelas, eosinophilic cellulitis, or Sweet syndrome than of NEH.1 Additionally, inflammatory markers (ie, C-reactive protein) and white blood cell counts tend to be elevated in bacterial cellulitis and Sweet syndrome, while leukopenia often is seen in NEH.
Histopathology is crucial in distinguishing these disease etiologies. Neutrophilic eccrine hidradenitis is diagnosed by the characteristic neutrophilic infiltrate and necrosis surrounding eccrine glands and coils. There also may be occasional intraductal abscesses and syringosquamous metaplasia of the sweat glands along with fibrosis of the adjacent dermis. In contrast, histologic sections of Sweet syndrome show numerous mature neutrophils infiltrating the dermis with marked papillary dermal edema. The histopathology of bacterial cellulitis and erysipelas is less specific, but common features include dermal edema, lymphatic dilation, and a diffuse neutrophilic infiltrate surrounding blood vessels. Pathogenic organisms may be seen on histopathology but are not required for the diagnosis of bacterial cellulitis or erysipelas.2 Additionally, blood and tissue culture can assist in identification of both the source of infection and the causative organism, but cultures may not always be positive.
Comparatively, the histopathologic features of eosinophilic cellulitis include dermal edema, eosinophilic infiltration, and flame figures that form when eosinophils degranulate and coat collagen fibers with major basic protein. Flame figures are characteristic but not pathognomonic for eosinophilic cellulitis.7 The histopathology of leukemia cutis varies based on the leukemia classification; generally, in acute myeloid leukemia the infiltrate is composed of neoplastic cells in the early stages of development that are positive for myeloid markers such as myeloperoxidase. Atypical and immature granulocytes within the leukocytic infiltrate differentiate this condition from the other diagnoses. Treatment may entail chemotherapy or radiotherapy, and this diagnosis generally carries the worst prognosis of all the conditions in the differential.6
Differentiating between these conditions is important in guiding treatment, especially in patients with febrile neutropenia. Unnecessary steroids in immunosuppressed patients can be dangerous, especially if the patient has an infection such as bacterial cellulitis. Furthermore, unwarranted antibiotic use for noninfectious conditions may expose patients to substantial side effects and not improve the condition. Neutrophilic eccrine hidradenitis typically is self-limited and treated symptomatically with systemic corticosteroids and nonsteroidal anti-inflammatory drugs.3 Sweet syndrome often requires a longer course of oral steroids. Leukemia cutis worsens as the leukemia advances, and treatment of the underlying malignancy is the most effective treatment.9
Early and accurate recognition of the diagnosis can prevent harmful diagnostic delay, unnecessary antibiotic use, or extended steroid taper in neutropenic patients. Appreciating the differences between these diagnoses can assist clinicians in investigating and tailoring a broad differential to specific patient presentations, which is especially critical when considering common mimickers for life-threatening conditions.
THE DIAGNOSIS: Neutrophilic Eccrine Hidradenitis
A biopsy from the left preauricular cheek demonstrated dermal neutrophilic inflammation around eccrine coils with focal necrosis (Figure). No notable diffuse dermal neutrophilic infiltrate was present, ruling out Sweet syndrome, and no notable interstitial neutrophilic infiltrate was present, making cellulitis and erysipelas less likely; panculture of tissue also was negative.1,2 Atypical cells in the deep dermis were positive for CD163 and negative for CD117, CD34, CD123, and myeloperoxidase, consistent with a diagnosis of neutrophilic eccrine hidradenitis (NEH) and reactive histiocytes.3 Treatment with oral prednisone resulted in rapid improvement of symptoms.
Neutrophilic eccrine hidradenitis is a rare reactive neutrophilic dermatosis characterized by eccrine gland involvement. This benign and self-limited condition presents as asymmetric erythematous papules and plaques.2 Among 8 granulocytopenic patients with neutrophilic dermatoses, 5 were diagnosed with NEH.4 Although first identified in 1982, NEH remains poorly understood.2 Initial theories suggested that NEH developed due to cytotoxic substances secreted in sweat glands causing necrosis and neutrophil chemotaxis; however, chemotherapy exposure cannot be linked to every case of NEH. Neutrophilic eccrine hidradenitis can be extremely difficult to differentiate clinically from conditions such as cellulitis and Sweet syndrome.
A patient history can be helpful in identifying triggering factors. Neutrophilic eccrine hidradenitis most commonly is associated with malignant, drug-induced, or infectious triggers, while Sweet syndrome has a broad range of associations including infections, vaccines, inflammatory bowel disease, pregnancy, malignancy, and drug-induced etiologies (Table).1 On average, NEH presents 10 days after chemotherapy induction, with 70% of cases presenting after the first chemotherapy cycle.5 Bacterial cellulitis or erysipelas have an infectious etiology, and patients may report symptoms such as fever, chills, or malaise. Immunosuppressed patients are at greater risk for infection; therefore, clinical signs of infection in a granulocytopenic patient should be addressed urgently.
Physical examination may have limited value in differentiating between these diagnoses, as neutrophilic dermatoses notoriously mimic infection. Cutaneous lesions can appear as pruritic or tender erythematous plaques, papules, or nodules in these conditions, though cellulitis and erysipelas tend to be unilateral and may have associated purulence or inflamed skin lymphatics. Given the potential for misdiagnosis, approaching patients with a broad differential can be helpful. In our patient, the differential diagnosis included Sweet syndrome, NEH, bacterial cellulitis, erysipelas, leukemia cutis, sarcoid, and eosinophilic cellulitis.
Leukemia cutis refers to the infiltration of neoplastic leukocytes in the skin and often occurs in patients with peripheral leukemia, most often acute myeloid leukemia or chronic lymphocytic leukemia. Patients with leukemia cutis often have a worse prognosis, as this finding signifies extramedullary spread of disease.6 Clinically, lesions can appear similar to those seen in our patient, though they typically are not symptomatic, can be nodular, tend to exhibit a violaceous hue, and occasionally may be hemorrhagic. Wells syndrome (also known as eosinophilic cellulitis) is an inflammatory dermatosis that presents as painful or pruritic, edematous and erythematous plaques.7,8 A green hue on resolution is present in some cases and may help clinicians differentiate this disease from mimickers.7 Often, eosinophilic cellulitis is misdiagnosed as bacterial cellulitis and treated with antibiotics. The presence of systemic symptoms such as fever or arthralgia is more typical of bacterial cellulitis, erysipelas, eosinophilic cellulitis, or Sweet syndrome than of NEH.1 Additionally, inflammatory markers (ie, C-reactive protein) and white blood cell counts tend to be elevated in bacterial cellulitis and Sweet syndrome, while leukopenia often is seen in NEH.
Histopathology is crucial in distinguishing these disease etiologies. Neutrophilic eccrine hidradenitis is diagnosed by the characteristic neutrophilic infiltrate and necrosis surrounding eccrine glands and coils. There also may be occasional intraductal abscesses and syringosquamous metaplasia of the sweat glands along with fibrosis of the adjacent dermis. In contrast, histologic sections of Sweet syndrome show numerous mature neutrophils infiltrating the dermis with marked papillary dermal edema. The histopathology of bacterial cellulitis and erysipelas is less specific, but common features include dermal edema, lymphatic dilation, and a diffuse neutrophilic infiltrate surrounding blood vessels. Pathogenic organisms may be seen on histopathology but are not required for the diagnosis of bacterial cellulitis or erysipelas.2 Additionally, blood and tissue culture can assist in identification of both the source of infection and the causative organism, but cultures may not always be positive.
Comparatively, the histopathologic features of eosinophilic cellulitis include dermal edema, eosinophilic infiltration, and flame figures that form when eosinophils degranulate and coat collagen fibers with major basic protein. Flame figures are characteristic but not pathognomonic for eosinophilic cellulitis.7 The histopathology of leukemia cutis varies based on the leukemia classification; generally, in acute myeloid leukemia the infiltrate is composed of neoplastic cells in the early stages of development that are positive for myeloid markers such as myeloperoxidase. Atypical and immature granulocytes within the leukocytic infiltrate differentiate this condition from the other diagnoses. Treatment may entail chemotherapy or radiotherapy, and this diagnosis generally carries the worst prognosis of all the conditions in the differential.6
Differentiating between these conditions is important in guiding treatment, especially in patients with febrile neutropenia. Unnecessary steroids in immunosuppressed patients can be dangerous, especially if the patient has an infection such as bacterial cellulitis. Furthermore, unwarranted antibiotic use for noninfectious conditions may expose patients to substantial side effects and not improve the condition. Neutrophilic eccrine hidradenitis typically is self-limited and treated symptomatically with systemic corticosteroids and nonsteroidal anti-inflammatory drugs.3 Sweet syndrome often requires a longer course of oral steroids. Leukemia cutis worsens as the leukemia advances, and treatment of the underlying malignancy is the most effective treatment.9
Early and accurate recognition of the diagnosis can prevent harmful diagnostic delay, unnecessary antibiotic use, or extended steroid taper in neutropenic patients. Appreciating the differences between these diagnoses can assist clinicians in investigating and tailoring a broad differential to specific patient presentations, which is especially critical when considering common mimickers for life-threatening conditions.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses. J Am Acad Dermatol. 2018;79:987-1006. doi:10.1016/j.jaad.2017.11.0642
- Srivastava M, Scharf S, Meehan SA, et al. Neutrophilic eccrine hidradenitis masquerading as facial cellulitis. J Am Acad Dermatol. 2007;56:693-696. doi:10.1016/j.jaad.2006.07.032
- Copaescu AM, Castilloux JF, Chababi-Atallah M, et al. A classic clinical case: neutrophilic eccrine hidradenitis. Case Rep Dermatol. 2013; 5:340-346. doi:10.1159/000356229
- Aractingi S, Mallet V, Pinquier L, et al. Neutrophilic dermatoses during granulocytopenia. Arch Dermatol. 1995;131:1141-1145.
- Cohen PR. Neutrophilic dermatoses occurring in oncology patients. Int J Dermatol. 2007;46:106-111. doi:10.1111/j.1365-4632.2006.02605.x
- Wang CX, Pusic I, Anadkat MJ. Association of leukemia cutis with survival in acute myeloid leukemia. JAMA Dermatol. 2019;155:826. doi:10.1001/jamadermatol.2019.0052
- Chung CL, Cusack CA. Wells syndrome: an enigmatic and therapeutically challenging disease. J Drugs Dermatol. 2006;5:908-911.
- Räßler F, Lukács J, Elsner P. Treatment of eosinophilic cellulitis (Wells syndrome): a systematic review. J Eur Acad Dermatol Venereol. 2016;30:1465-1479. doi:10.1111/jdv.13706
- Hobbs LK, Carr PC, Gru AA, et al. Case and review: cutaneous involvement by chronic neutrophilic leukemia vs Sweet syndrome: a diagnostic dilemma. J Cutan Pathol. 2021;48:644-649. doi:10.1111 /cup.13925
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses. J Am Acad Dermatol. 2018;79:987-1006. doi:10.1016/j.jaad.2017.11.0642
- Srivastava M, Scharf S, Meehan SA, et al. Neutrophilic eccrine hidradenitis masquerading as facial cellulitis. J Am Acad Dermatol. 2007;56:693-696. doi:10.1016/j.jaad.2006.07.032
- Copaescu AM, Castilloux JF, Chababi-Atallah M, et al. A classic clinical case: neutrophilic eccrine hidradenitis. Case Rep Dermatol. 2013; 5:340-346. doi:10.1159/000356229
- Aractingi S, Mallet V, Pinquier L, et al. Neutrophilic dermatoses during granulocytopenia. Arch Dermatol. 1995;131:1141-1145.
- Cohen PR. Neutrophilic dermatoses occurring in oncology patients. Int J Dermatol. 2007;46:106-111. doi:10.1111/j.1365-4632.2006.02605.x
- Wang CX, Pusic I, Anadkat MJ. Association of leukemia cutis with survival in acute myeloid leukemia. JAMA Dermatol. 2019;155:826. doi:10.1001/jamadermatol.2019.0052
- Chung CL, Cusack CA. Wells syndrome: an enigmatic and therapeutically challenging disease. J Drugs Dermatol. 2006;5:908-911.
- Räßler F, Lukács J, Elsner P. Treatment of eosinophilic cellulitis (Wells syndrome): a systematic review. J Eur Acad Dermatol Venereol. 2016;30:1465-1479. doi:10.1111/jdv.13706
- Hobbs LK, Carr PC, Gru AA, et al. Case and review: cutaneous involvement by chronic neutrophilic leukemia vs Sweet syndrome: a diagnostic dilemma. J Cutan Pathol. 2021;48:644-649. doi:10.1111 /cup.13925
A 50-year-old woman undergoing cytarabine induction therapy for acute myeloid leukemia developed tender, erythematous, dermal plaques on the nasal dorsum, left medial eyebrow, left preauricular cheek, and right cheek. The rash erupted 7 days after receiving the cytarabine induction regimen. She had a fever (temperature, 39.9 °C [103.8 °F]) and also was neutropenic.
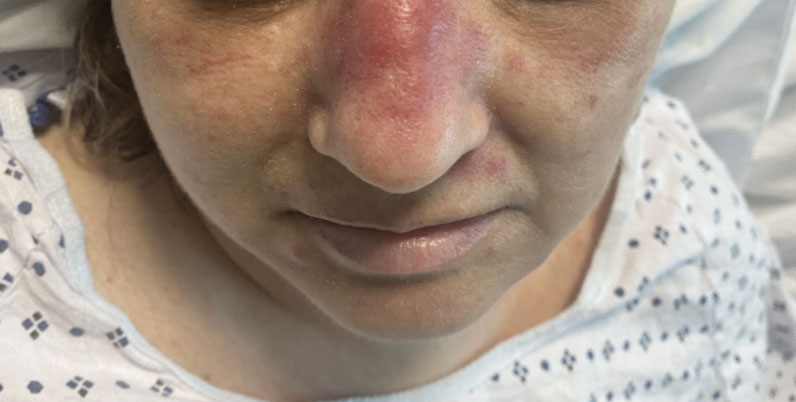
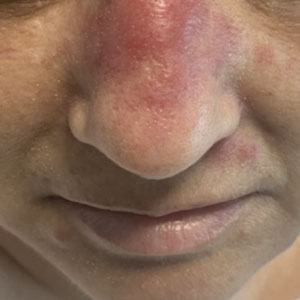
CMML: GM-CSF inhibitor lenzilumab shows early promise
There is currently no international standard of care for patients with CMML, but given its overlap with other myelodysplastic and myeloproliferative syndromes, CMML is usually treated with the hypomethylating agent azacitidine (Vidaza, Onureg), which is associated with objective response rates of 40%-50% and a complete response rate of less than 20%. Alternatively, some patients are treated with the antimetabolite hydroxurea in the palliative setting.
CMML is “insidious, it’s rare, but we think the incidence is increasing because more patients are now getting sequencing done by their doctors, and therapy [related] cases, patients that have survived chemo in the last 10 years, can also develop this disease,” said Daniel Thomas, MD, PhD, from the South Australian Health and Medical Research Institute, Adelaide, in an interview.
Dr. Thomas is a co-investigator of the ongoing phase 2/3 PREACH-M trial, which is testing a novel strategy of treating CMML with mutations in the RAS pathway with a combination of azacitidine and the investigational antibody lenzilumab, which is a targeted inhibitor of granulocyte-macrophage colony-stimulating factor (GM-CSF).
Preliminary results from the trial, reported at the European Hematology Association (EHA) annual meeting, showed that among 10 patients with CMML bearing mutations in the RAS pathway, the combination was associated with durable decreases in monocyte counts, increases in platelet counts and hemoglobin levels, and reductions in both spleen size and C-reactive protein level.
Targeting GM-CSF
More than 90% of cases of CMML carry somatic mutations that are thought to be leukemogenic, with an estimated 46%-60% of cases having mutations in TET2, a tumor suppressor, and an estimated 40% having mutations in KRAS, NRAS, or CBL, all of which are involved in cellular proliferation, and which, research suggests, are sensitive to GM-CSF inhibition.
“I was very surprised that the RAS-mutant arm – so, patients that have KRAS, NRAS, or CBL mutations – are just responding beautifully to [lenzilumab], ” Dr. Thomas said.
“It’s [in the] early days, but if what we’re seeing is durable across the next 10 patients, then I think we’re looking at a game changer,” he added.
Cameron Durrant, MD, DRCOG, MRCGP, chairman and CEO of lenzilumab’s maker Humanigen, said in an interview that the development of the antibody for CMML was spurred in part by research from investigators at the Mayo Clinic, showing that patients with mutations that increased sensitivity to GM-CSF seemed to have better clinical outcomes when the growth factor was blocked.
In addition, Dr. Durrant said, preclinical research from investigators at the Moffitt Cancer Center, Tampa, found that myeloid and monocytic progenitors “fed” on GM-CSF and were sensitive to GM-CSF signal inhibition.
“The biological idea that’s being explored here in the clinic in this study is that by blocking, or starving, if you will, those cells of that food, then you can prevent this overgrowth of certain blood cells that lead to chronic myelomonocytic leukemia,” he said.
PREACH-M details
Lenzilumab is an engineered human immunoglobulin G1-kappa monoclonal antibody with high affinity for human GM-CSF.
In the open label, nonrandomized PREACH-M trial, 72 patients with CMML were enrolled and were assigned to receive 24 monthly cycles of therapy depending on mutational status.
Patients with RAS pathway mutations were assigned to receive azacitidine delivered subcutaneously 75 mg/m2 for 7 days, plus intravenous lenzilumab 552 mg on days 1 and 15 of cycle 1 and on day 1 only of all subsequent cycles.
Patients with TET2 mutations only were assigned to receive azacitidine on the same schedule, plus IV sodium ascorbate 30 g for 7 days, with the first dose 15 g, and subsequent doses 30 g if there is no evidence of tumor lysis syndrome. Following IV administration, patients continue on oral sodium ascorbate 1.1 g on all other days.
The primary endpoint of complete and partial responses any time during the first 12 cycles is planned for reporting at the annual meeting of the American Society of Hematology in December, Dr. Thomas said.
At EHA 2023, the investigators reported available data on 10 patients enrolled in the lenzilumab arm and one enrolled in the azacitidine-sodium ascorbate arm.
Among patients in the lenzilumab arm there was a 5.1-fold decrease in monocyte counts (P = .03) and 2.4-fold decrease in blast counts (P = .04) at 12 months of follow-up.
In addition there was a trend toward increased platelet counts over baseline at 12 months, a significant increase in blood hemoglobin concentration (P = .024), a significant reduction in spleen size (P = .03) and a trend toward lower levels of the inflammatory marker C-reactive protein.
There were 21 grade 3 or 4 adverse events reported, of which 5 were deemed to be possibly related to lenzilumab.
Dr. Thomas told this news organization that the investigators have been “pleasantly surprised” at how well patients tolerated the monoclonal antibody.
“We haven’t had any infusion reactions, we haven’t had any pulmonary alveolar proteinosis, [and] we haven’t had any fevers from the infusion, from the antibody,” he said.
There were some instances of neutropenia and thrombocytopenia that the investigators think may have been related to azacitidine, he noted.
The study is sponsored by the National Health and Medical Research Council of Australia. Dr. Thomas reported no relevant financial relationships. Dr. Durrant is an employee and director of Humanigen.
A version of this article first appeared on Medscape.com.
There is currently no international standard of care for patients with CMML, but given its overlap with other myelodysplastic and myeloproliferative syndromes, CMML is usually treated with the hypomethylating agent azacitidine (Vidaza, Onureg), which is associated with objective response rates of 40%-50% and a complete response rate of less than 20%. Alternatively, some patients are treated with the antimetabolite hydroxurea in the palliative setting.
CMML is “insidious, it’s rare, but we think the incidence is increasing because more patients are now getting sequencing done by their doctors, and therapy [related] cases, patients that have survived chemo in the last 10 years, can also develop this disease,” said Daniel Thomas, MD, PhD, from the South Australian Health and Medical Research Institute, Adelaide, in an interview.
Dr. Thomas is a co-investigator of the ongoing phase 2/3 PREACH-M trial, which is testing a novel strategy of treating CMML with mutations in the RAS pathway with a combination of azacitidine and the investigational antibody lenzilumab, which is a targeted inhibitor of granulocyte-macrophage colony-stimulating factor (GM-CSF).
Preliminary results from the trial, reported at the European Hematology Association (EHA) annual meeting, showed that among 10 patients with CMML bearing mutations in the RAS pathway, the combination was associated with durable decreases in monocyte counts, increases in platelet counts and hemoglobin levels, and reductions in both spleen size and C-reactive protein level.
Targeting GM-CSF
More than 90% of cases of CMML carry somatic mutations that are thought to be leukemogenic, with an estimated 46%-60% of cases having mutations in TET2, a tumor suppressor, and an estimated 40% having mutations in KRAS, NRAS, or CBL, all of which are involved in cellular proliferation, and which, research suggests, are sensitive to GM-CSF inhibition.
“I was very surprised that the RAS-mutant arm – so, patients that have KRAS, NRAS, or CBL mutations – are just responding beautifully to [lenzilumab], ” Dr. Thomas said.
“It’s [in the] early days, but if what we’re seeing is durable across the next 10 patients, then I think we’re looking at a game changer,” he added.
Cameron Durrant, MD, DRCOG, MRCGP, chairman and CEO of lenzilumab’s maker Humanigen, said in an interview that the development of the antibody for CMML was spurred in part by research from investigators at the Mayo Clinic, showing that patients with mutations that increased sensitivity to GM-CSF seemed to have better clinical outcomes when the growth factor was blocked.
In addition, Dr. Durrant said, preclinical research from investigators at the Moffitt Cancer Center, Tampa, found that myeloid and monocytic progenitors “fed” on GM-CSF and were sensitive to GM-CSF signal inhibition.
“The biological idea that’s being explored here in the clinic in this study is that by blocking, or starving, if you will, those cells of that food, then you can prevent this overgrowth of certain blood cells that lead to chronic myelomonocytic leukemia,” he said.
PREACH-M details
Lenzilumab is an engineered human immunoglobulin G1-kappa monoclonal antibody with high affinity for human GM-CSF.
In the open label, nonrandomized PREACH-M trial, 72 patients with CMML were enrolled and were assigned to receive 24 monthly cycles of therapy depending on mutational status.
Patients with RAS pathway mutations were assigned to receive azacitidine delivered subcutaneously 75 mg/m2 for 7 days, plus intravenous lenzilumab 552 mg on days 1 and 15 of cycle 1 and on day 1 only of all subsequent cycles.
Patients with TET2 mutations only were assigned to receive azacitidine on the same schedule, plus IV sodium ascorbate 30 g for 7 days, with the first dose 15 g, and subsequent doses 30 g if there is no evidence of tumor lysis syndrome. Following IV administration, patients continue on oral sodium ascorbate 1.1 g on all other days.
The primary endpoint of complete and partial responses any time during the first 12 cycles is planned for reporting at the annual meeting of the American Society of Hematology in December, Dr. Thomas said.
At EHA 2023, the investigators reported available data on 10 patients enrolled in the lenzilumab arm and one enrolled in the azacitidine-sodium ascorbate arm.
Among patients in the lenzilumab arm there was a 5.1-fold decrease in monocyte counts (P = .03) and 2.4-fold decrease in blast counts (P = .04) at 12 months of follow-up.
In addition there was a trend toward increased platelet counts over baseline at 12 months, a significant increase in blood hemoglobin concentration (P = .024), a significant reduction in spleen size (P = .03) and a trend toward lower levels of the inflammatory marker C-reactive protein.
There were 21 grade 3 or 4 adverse events reported, of which 5 were deemed to be possibly related to lenzilumab.
Dr. Thomas told this news organization that the investigators have been “pleasantly surprised” at how well patients tolerated the monoclonal antibody.
“We haven’t had any infusion reactions, we haven’t had any pulmonary alveolar proteinosis, [and] we haven’t had any fevers from the infusion, from the antibody,” he said.
There were some instances of neutropenia and thrombocytopenia that the investigators think may have been related to azacitidine, he noted.
The study is sponsored by the National Health and Medical Research Council of Australia. Dr. Thomas reported no relevant financial relationships. Dr. Durrant is an employee and director of Humanigen.
A version of this article first appeared on Medscape.com.
There is currently no international standard of care for patients with CMML, but given its overlap with other myelodysplastic and myeloproliferative syndromes, CMML is usually treated with the hypomethylating agent azacitidine (Vidaza, Onureg), which is associated with objective response rates of 40%-50% and a complete response rate of less than 20%. Alternatively, some patients are treated with the antimetabolite hydroxurea in the palliative setting.
CMML is “insidious, it’s rare, but we think the incidence is increasing because more patients are now getting sequencing done by their doctors, and therapy [related] cases, patients that have survived chemo in the last 10 years, can also develop this disease,” said Daniel Thomas, MD, PhD, from the South Australian Health and Medical Research Institute, Adelaide, in an interview.
Dr. Thomas is a co-investigator of the ongoing phase 2/3 PREACH-M trial, which is testing a novel strategy of treating CMML with mutations in the RAS pathway with a combination of azacitidine and the investigational antibody lenzilumab, which is a targeted inhibitor of granulocyte-macrophage colony-stimulating factor (GM-CSF).
Preliminary results from the trial, reported at the European Hematology Association (EHA) annual meeting, showed that among 10 patients with CMML bearing mutations in the RAS pathway, the combination was associated with durable decreases in monocyte counts, increases in platelet counts and hemoglobin levels, and reductions in both spleen size and C-reactive protein level.
Targeting GM-CSF
More than 90% of cases of CMML carry somatic mutations that are thought to be leukemogenic, with an estimated 46%-60% of cases having mutations in TET2, a tumor suppressor, and an estimated 40% having mutations in KRAS, NRAS, or CBL, all of which are involved in cellular proliferation, and which, research suggests, are sensitive to GM-CSF inhibition.
“I was very surprised that the RAS-mutant arm – so, patients that have KRAS, NRAS, or CBL mutations – are just responding beautifully to [lenzilumab], ” Dr. Thomas said.
“It’s [in the] early days, but if what we’re seeing is durable across the next 10 patients, then I think we’re looking at a game changer,” he added.
Cameron Durrant, MD, DRCOG, MRCGP, chairman and CEO of lenzilumab’s maker Humanigen, said in an interview that the development of the antibody for CMML was spurred in part by research from investigators at the Mayo Clinic, showing that patients with mutations that increased sensitivity to GM-CSF seemed to have better clinical outcomes when the growth factor was blocked.
In addition, Dr. Durrant said, preclinical research from investigators at the Moffitt Cancer Center, Tampa, found that myeloid and monocytic progenitors “fed” on GM-CSF and were sensitive to GM-CSF signal inhibition.
“The biological idea that’s being explored here in the clinic in this study is that by blocking, or starving, if you will, those cells of that food, then you can prevent this overgrowth of certain blood cells that lead to chronic myelomonocytic leukemia,” he said.
PREACH-M details
Lenzilumab is an engineered human immunoglobulin G1-kappa monoclonal antibody with high affinity for human GM-CSF.
In the open label, nonrandomized PREACH-M trial, 72 patients with CMML were enrolled and were assigned to receive 24 monthly cycles of therapy depending on mutational status.
Patients with RAS pathway mutations were assigned to receive azacitidine delivered subcutaneously 75 mg/m2 for 7 days, plus intravenous lenzilumab 552 mg on days 1 and 15 of cycle 1 and on day 1 only of all subsequent cycles.
Patients with TET2 mutations only were assigned to receive azacitidine on the same schedule, plus IV sodium ascorbate 30 g for 7 days, with the first dose 15 g, and subsequent doses 30 g if there is no evidence of tumor lysis syndrome. Following IV administration, patients continue on oral sodium ascorbate 1.1 g on all other days.
The primary endpoint of complete and partial responses any time during the first 12 cycles is planned for reporting at the annual meeting of the American Society of Hematology in December, Dr. Thomas said.
At EHA 2023, the investigators reported available data on 10 patients enrolled in the lenzilumab arm and one enrolled in the azacitidine-sodium ascorbate arm.
Among patients in the lenzilumab arm there was a 5.1-fold decrease in monocyte counts (P = .03) and 2.4-fold decrease in blast counts (P = .04) at 12 months of follow-up.
In addition there was a trend toward increased platelet counts over baseline at 12 months, a significant increase in blood hemoglobin concentration (P = .024), a significant reduction in spleen size (P = .03) and a trend toward lower levels of the inflammatory marker C-reactive protein.
There were 21 grade 3 or 4 adverse events reported, of which 5 were deemed to be possibly related to lenzilumab.
Dr. Thomas told this news organization that the investigators have been “pleasantly surprised” at how well patients tolerated the monoclonal antibody.
“We haven’t had any infusion reactions, we haven’t had any pulmonary alveolar proteinosis, [and] we haven’t had any fevers from the infusion, from the antibody,” he said.
There were some instances of neutropenia and thrombocytopenia that the investigators think may have been related to azacitidine, he noted.
The study is sponsored by the National Health and Medical Research Council of Australia. Dr. Thomas reported no relevant financial relationships. Dr. Durrant is an employee and director of Humanigen.
A version of this article first appeared on Medscape.com.
FROM EHA 2023