User login
Atypical Intrathoracic Manifestations of Metastatic Prostate Cancer: A Case Series
Atypical Intrathoracic Manifestations of Metastatic Prostate Cancer: A Case Series
Prostate cancer is the most common noncutaneous cancer in men, accounting for 29% of all incident cancer cases.1 Typically, prostate cancer metastasizes to bone and regional lymph nodes.2 However, intrathoracic manifestation may occur. This report presents 3 cases of rare intrathoracic manifestations of metastatic prostate cancer with a review of the current literature.
CASE PRESENTATIONS
Case 1
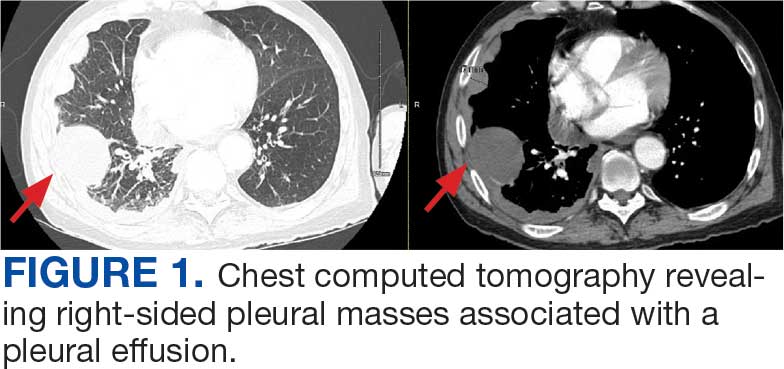
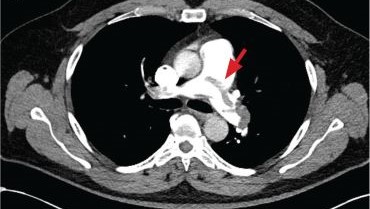
A 71-year-old male who was an active smoker and a long-standing employment as a plumber was diagnosed with rectal cancer in 2022. He completed neoadjuvant capecitabine and radiation therapy followed by a rectosigmoidectomy. Several weeks after surgery, the patient presented to the emergency department (ED) with a dry cough and worsening shortness of breath. Point-of-care ultrasound of the lungs revealed a moderate right pleural effusion with several nodular pleural masses. A chest computed tomography (CT) confirmed these findings (Figure 1). A CT of the abdomen and pelvis revealed prostatomegaly with the medial lobe of the prostate protruding into the bladder; however, no enlarged retroperitoneal, mesenteric or pelvic lymph nodes were noted. The patient underwent a right pleural fluid drainage and pleural mass biopsy. Pleural mass histomorphology as well as immunohistochemical (IHC) stains were consistent with metastatic prostate adenocarcinoma. The pleural fluid cytology also was consistent with metastatic prostate adenocarcinoma.
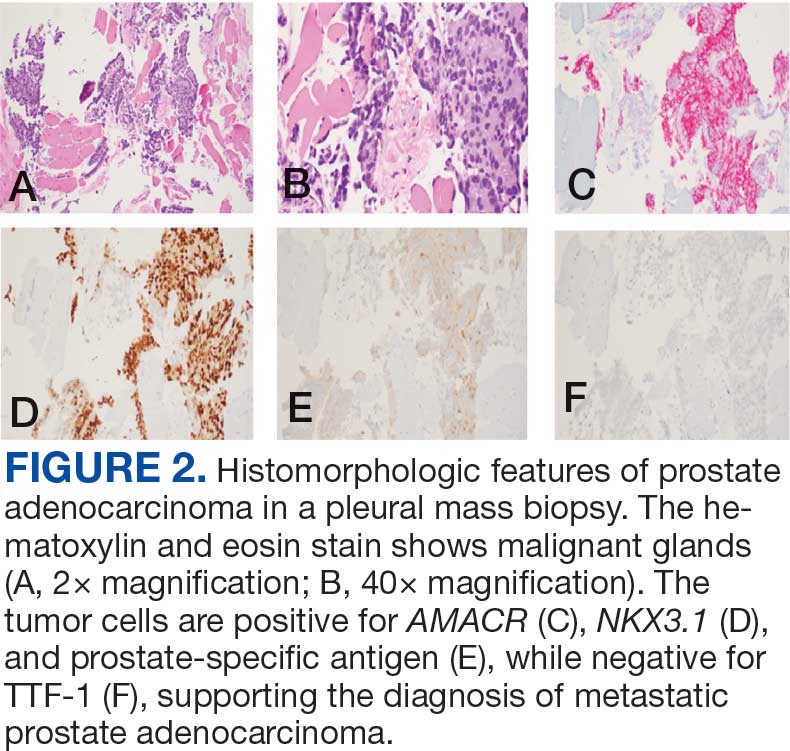
Immunohistochemistry showed weak positive staining for prostate-specific NK3 homeobox 1 gene (NKX3.1), alpha-methylacyl-CoA racemase gene (AMACR), and prosaposin, and negative transcription termination factor (TTF-1), keratin-7 (CK7), and prosaposin, and negative transcription termination factor (TTF-1), keratin-7 (CK7), keratin-20, and caudal type homeobox 2 gene (CDX2) (Figure 2) 2). The patient's prostate-specific antigen (PSA) was found to be elevated at 33.9 ng/mL (reference range, < 4 ng/mL).
Case 2
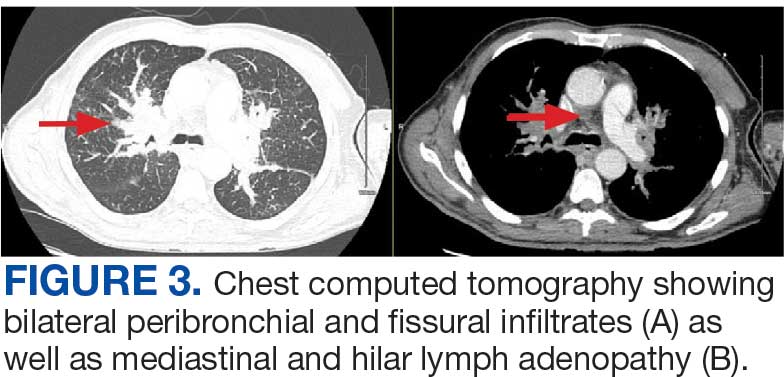
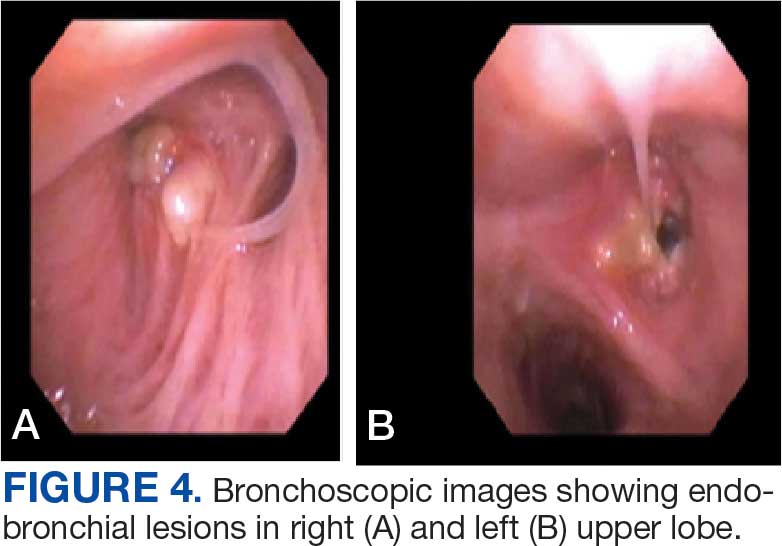
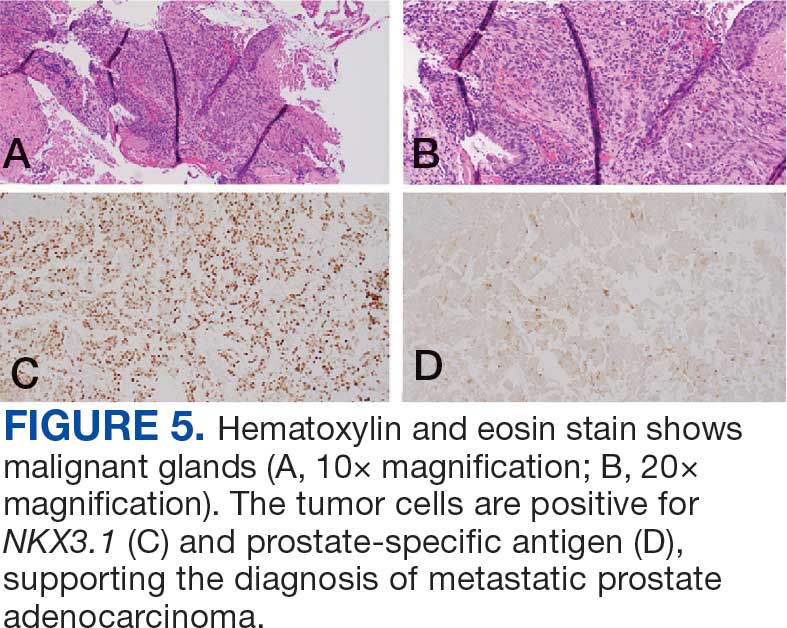
A 71-year-old male with a history of alcohol use disorder and a 30-year smoking history presented to the ED with worsening dyspnea on exertion. The patient’s baseline exercise tolerance decreased to walking for only 1 block. He reported unintentional weight loss of about 30 pounds over the prior year, no recent respiratory infections, no prior breathing problems, and no personal or family history of cancer. Chest CT revealed findings of bilateral peribronchial opacities as well as mediastinal and hilar lymphadenopathy (Figure 3). The patient developed hypoxic respiratory failure necessitating intubation, mechanical ventilation, and management in the medical intensive care unit, where he was treated for postobstructive pneumonia. Fiberoptic bronchoscopy revealed endobronchial lesions in the right and left upper lobe that were partially obstructing the airway (Figure 4).
The endobronchial masses were debulked using forceps, and samples were sent for surgical pathology evaluation. Staging was completed using linear endobronchial ultrasound, which revealed an enlarged subcarinal lymph node (S7). The surgical pathology of the endobronchial mass and the subcarinal lymph node cytology were consistent with metastatic adenocarcinoma of the prostate. The tumor cells were positive for AE1/AE3, PSA, and NKX3.1, but were negative for CK7 and TTF-1 (Figure 5). Further imaging revealed an enlarged heterogeneous prostate gland, prominent pelvic nodes, and left retroperitoneal lymphadenopathy, as well as sclerotic foci within the T10 vertebral body and right inferior pubic ramus. PSA was also found to be significantly elevated at 700 ng/mL.
Case 3
An 80-year-old male veteran with a history of prostate cancer and recently diagnosed T2N1M0 head and neck squamous cell carcinoma was referred to the Pulmonary service for evaluation of a pulmonary nodule. His medical history was notable for prostate cancer diagnosed 12 years earlier, with an unknown Gleason score. Initial treatment included prostatectomy followed by whole pelvic radiation therapy a year after, due to elevated PSA in surveillance monitoring. This treatment led to remission. After establishing remission for > 10 years, the patient was started on low-dose testosterone replacement therapy to address complications of radiation therapy, namely hypogonadism.
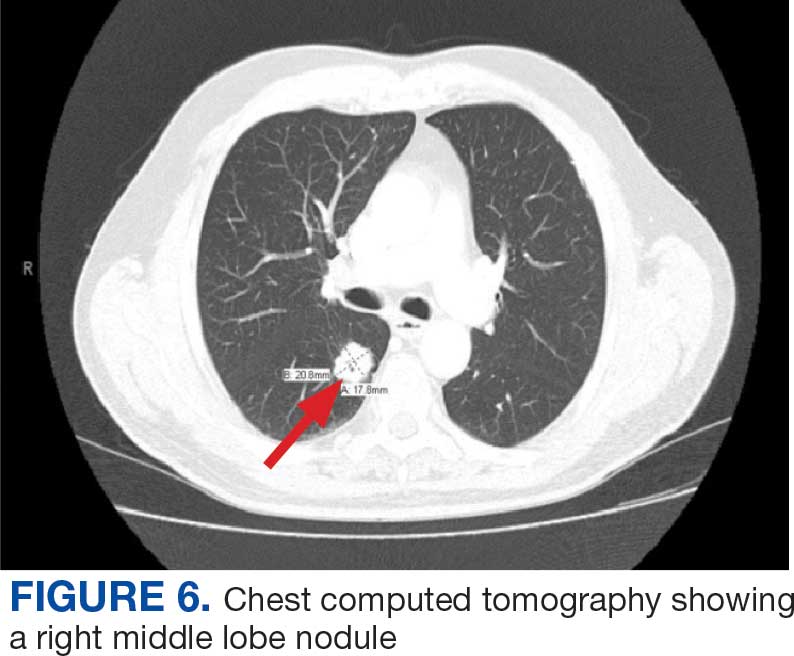
On evaluation, a chest CT was significant for a large 2-cm right middle lobe nodule (Figure 6). At that time, PSA was noted to be borderline elevated at 4.2 ng/mL, and whole-body imaging did not reveal any lesions elsewhere, specifically no bone metastasis. Biopsies of the right middle lobe lung nodule revealed adenocarcinoma consistent with metastatic prostate cancer. Testosterone therapy was promptly discontinued.
The patient initially refused androgen deprivation therapy owing to the antiandrogenic adverse effects. However, subsequent chest CTs revealed growing lung nodules, which convinced him to proceed with androgen deprivation therapy followed by palliative radiation, and chemotherapy and management of malignant pleural effusion with indwelling small bore pleural catheter for about 10 years. He died from COVID-19 during the pandemic.
DISCUSSION
These cases highlight the importance of including prostate cancer in the differential diagnoses of male patients with intrathoracic abnormalities, even in the absence of metastasis to the more common sites. In a large cohort study of 74,826 patients with metastatic prostate cancer, Gandaglia et al found that the most frequent sites of metastasis were bone (84.0%) and distant lymph nodes (10.6%).2 However, thoracic involvement was observed in 9.1% of cases, with isolated thoracic metastasis being rare. The cases described in this report exemplify exceptionally uncommon occurrences within that 9.1%.
Pleural metastases, as observed in Case 1, are a particularly rare manifestation. In a 10-year retrospective assessment, Vinjamoori et al discovered pleural nodules or masses in only 6 of 82 patients (7.3%) with atypical metastases.3 Adrenal and liver metastases accounted for 15% and 37% of cases with atypical distribution. As such, isolated pleural disease is rare even in atypical presentations.3
As seen in Case 2, endobronchial metastases producing airway obstruction are also rare, with the most common primary cancers associated with endobronchial metastasis being breast, colon, and renal cancer.4 The available literature on this presentation is confined to case reports. Hameed et al reported a case of synchronous biopsy-proven endobronchial metastasis from prostate cancer.5 These cases highlight the importance of maintaining a high level of clinical awareness when encountering endobronchial lesions in patients with prostate cancer.
Case 3 presents a unique situation of lung metastases without any involvement of the bones. It is well known—and was confirmed by Heidenreich et al—that lung metastases in prostate adenocarcinoma usually coincide with extensive osseous disease.6 This instance highlights the importance of watchful monitoring for unusual patterns of cancer recurrence.
Immunohistochemistry stains that are specific to prostate cancer include antibodies against PSA. Prostate-specific membrane antigen is another marker that is far more present in malignant than in benign prostate tissue.
The NKX3.1 gene encodes a homeobox protein, which is a transcription factor and tumor suppressor. In prostate cancer, there is loss of heterozygosity of the gene and stains for the IHC antibody to NKX3.1.7
On the other hand, lung cells stain positive for TTF-1, which is produced by surfactant-producing type 2 pneumocytes and club cells in the lung. Antibodies to TTF-1, a common IHC stain, are used to identify adenocarcinoma of lung origin and may carry a prognostic value.7
The immunohistochemistry profiles, specifically the presence of prostate-specific markers such as PSA and NKX3.1, played a vital role in making the diagnosis.
In Case 1, weak TTF-1 positivity was noted, an unusual finding in metastatic prostate adenocarcinoma. Marak et al documented a rare case of TTF-1–positive metastatic prostate cancer, illustrating the potential for diagnostic confusion with primary lung malignancies.8
The 3 cases described in this report demonstrate the importance of clinical consideration, serial follow-up of PSA levels, using more prostate-specific positron emission tomography tracers (eg, Pylarify) alongside traditional imaging, and tissue biopsy to detect unusual metastases.
CONCLUSIONS
Although thoracic metastases from prostate cancer are rare, these presentations highlight the importance of clinical awareness regarding atypical cases. Pleural disease, endobronchial lesions, and isolated pulmonary nodules might be the first clinical manifestation of metastatic prostate cancer. A high index of suspicion, appropriate imaging, and judicious use of immunohistochemistry are important to ensure accurate diagnosis and optimal patient management.
- Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74(1):12-49. doi:10.3322/caac.21820
- Gandaglia G, Abdollah F, Schiffmann J, et al. Distribution of metastatic sites in patients with prostate cancer: a population-based analysis. Prostate. 2014;74(2):210-216. doi:10.1002/pros.22742
- Vinjamoori AH, Jagannathan JP, Shinagare AB, et al. Atypical metastases from prostate cancer: 10-year experience at a single institution. AJR Am J Roentgenol. 2012;199(2):367-372. doi:10.2214/AJR.11.7533
- Salud A, Porcel JM, Rovirosa A, Bellmunt J. Endobronchial metastatic disease: analysis of 32 cases. J Surg Oncol. 1996;62(4):249-252. doi:10.1002/(SICI)1096- 9098(199608)62:4<249::AID-JSO4>3.0.CO;2-6
- Hameed M, Haq IU, Yousaf M, Hussein M, Rashid U, Al-Bozom I. Endobronchial metastases secondary to prostate cancer: a case report and literature review. Respir Med Case Rep. 2020;32:101326. doi:10.1016/j.rmcr.2020.101326
- Heidenreich A, Bastian PJ, Bellmunt J, et al; for the European Association of Urology. EAU guidelines on prostate cancer. Part II: treatment of advanced, relapsing, and castration- resistant prostate cancer. Eur Urol. 2014;65(2):467- 479. doi:10.1016/j.eururo.2013.11.002
- Schallenberg S, Dernbach G, Dragomir MP, et al. TTF-1 status in early-stage lung adenocarcinoma is an independent predictor of relapse and survival superior to tumor grading. Eur J Cancer. 2024;197:113474. doi:10.1016/j.ejca.2023.113474
- Marak C, Guddati AK, Ashraf A, Smith J, Kaushik P. Prostate adenocarcinoma with atypical immunohistochemistry presenting with a Cheerio sign. AIM Clinical Cases. 2023;1:e220508. doi:10.7326/aimcc.2022.0508
Prostate cancer is the most common noncutaneous cancer in men, accounting for 29% of all incident cancer cases.1 Typically, prostate cancer metastasizes to bone and regional lymph nodes.2 However, intrathoracic manifestation may occur. This report presents 3 cases of rare intrathoracic manifestations of metastatic prostate cancer with a review of the current literature.
CASE PRESENTATIONS
Case 1
A 71-year-old male who was an active smoker and a long-standing employment as a plumber was diagnosed with rectal cancer in 2022. He completed neoadjuvant capecitabine and radiation therapy followed by a rectosigmoidectomy. Several weeks after surgery, the patient presented to the emergency department (ED) with a dry cough and worsening shortness of breath. Point-of-care ultrasound of the lungs revealed a moderate right pleural effusion with several nodular pleural masses. A chest computed tomography (CT) confirmed these findings (Figure 1). A CT of the abdomen and pelvis revealed prostatomegaly with the medial lobe of the prostate protruding into the bladder; however, no enlarged retroperitoneal, mesenteric or pelvic lymph nodes were noted. The patient underwent a right pleural fluid drainage and pleural mass biopsy. Pleural mass histomorphology as well as immunohistochemical (IHC) stains were consistent with metastatic prostate adenocarcinoma. The pleural fluid cytology also was consistent with metastatic prostate adenocarcinoma.
Immunohistochemistry showed weak positive staining for prostate-specific NK3 homeobox 1 gene (NKX3.1), alpha-methylacyl-CoA racemase gene (AMACR), and prosaposin, and negative transcription termination factor (TTF-1), keratin-7 (CK7), and prosaposin, and negative transcription termination factor (TTF-1), keratin-7 (CK7), keratin-20, and caudal type homeobox 2 gene (CDX2) (Figure 2) 2). The patient's prostate-specific antigen (PSA) was found to be elevated at 33.9 ng/mL (reference range, < 4 ng/mL).
Case 2
A 71-year-old male with a history of alcohol use disorder and a 30-year smoking history presented to the ED with worsening dyspnea on exertion. The patient’s baseline exercise tolerance decreased to walking for only 1 block. He reported unintentional weight loss of about 30 pounds over the prior year, no recent respiratory infections, no prior breathing problems, and no personal or family history of cancer. Chest CT revealed findings of bilateral peribronchial opacities as well as mediastinal and hilar lymphadenopathy (Figure 3). The patient developed hypoxic respiratory failure necessitating intubation, mechanical ventilation, and management in the medical intensive care unit, where he was treated for postobstructive pneumonia. Fiberoptic bronchoscopy revealed endobronchial lesions in the right and left upper lobe that were partially obstructing the airway (Figure 4).
The endobronchial masses were debulked using forceps, and samples were sent for surgical pathology evaluation. Staging was completed using linear endobronchial ultrasound, which revealed an enlarged subcarinal lymph node (S7). The surgical pathology of the endobronchial mass and the subcarinal lymph node cytology were consistent with metastatic adenocarcinoma of the prostate. The tumor cells were positive for AE1/AE3, PSA, and NKX3.1, but were negative for CK7 and TTF-1 (Figure 5). Further imaging revealed an enlarged heterogeneous prostate gland, prominent pelvic nodes, and left retroperitoneal lymphadenopathy, as well as sclerotic foci within the T10 vertebral body and right inferior pubic ramus. PSA was also found to be significantly elevated at 700 ng/mL.
Case 3
An 80-year-old male veteran with a history of prostate cancer and recently diagnosed T2N1M0 head and neck squamous cell carcinoma was referred to the Pulmonary service for evaluation of a pulmonary nodule. His medical history was notable for prostate cancer diagnosed 12 years earlier, with an unknown Gleason score. Initial treatment included prostatectomy followed by whole pelvic radiation therapy a year after, due to elevated PSA in surveillance monitoring. This treatment led to remission. After establishing remission for > 10 years, the patient was started on low-dose testosterone replacement therapy to address complications of radiation therapy, namely hypogonadism.
On evaluation, a chest CT was significant for a large 2-cm right middle lobe nodule (Figure 6). At that time, PSA was noted to be borderline elevated at 4.2 ng/mL, and whole-body imaging did not reveal any lesions elsewhere, specifically no bone metastasis. Biopsies of the right middle lobe lung nodule revealed adenocarcinoma consistent with metastatic prostate cancer. Testosterone therapy was promptly discontinued.
The patient initially refused androgen deprivation therapy owing to the antiandrogenic adverse effects. However, subsequent chest CTs revealed growing lung nodules, which convinced him to proceed with androgen deprivation therapy followed by palliative radiation, and chemotherapy and management of malignant pleural effusion with indwelling small bore pleural catheter for about 10 years. He died from COVID-19 during the pandemic.
DISCUSSION
These cases highlight the importance of including prostate cancer in the differential diagnoses of male patients with intrathoracic abnormalities, even in the absence of metastasis to the more common sites. In a large cohort study of 74,826 patients with metastatic prostate cancer, Gandaglia et al found that the most frequent sites of metastasis were bone (84.0%) and distant lymph nodes (10.6%).2 However, thoracic involvement was observed in 9.1% of cases, with isolated thoracic metastasis being rare. The cases described in this report exemplify exceptionally uncommon occurrences within that 9.1%.
Pleural metastases, as observed in Case 1, are a particularly rare manifestation. In a 10-year retrospective assessment, Vinjamoori et al discovered pleural nodules or masses in only 6 of 82 patients (7.3%) with atypical metastases.3 Adrenal and liver metastases accounted for 15% and 37% of cases with atypical distribution. As such, isolated pleural disease is rare even in atypical presentations.3
As seen in Case 2, endobronchial metastases producing airway obstruction are also rare, with the most common primary cancers associated with endobronchial metastasis being breast, colon, and renal cancer.4 The available literature on this presentation is confined to case reports. Hameed et al reported a case of synchronous biopsy-proven endobronchial metastasis from prostate cancer.5 These cases highlight the importance of maintaining a high level of clinical awareness when encountering endobronchial lesions in patients with prostate cancer.
Case 3 presents a unique situation of lung metastases without any involvement of the bones. It is well known—and was confirmed by Heidenreich et al—that lung metastases in prostate adenocarcinoma usually coincide with extensive osseous disease.6 This instance highlights the importance of watchful monitoring for unusual patterns of cancer recurrence.
Immunohistochemistry stains that are specific to prostate cancer include antibodies against PSA. Prostate-specific membrane antigen is another marker that is far more present in malignant than in benign prostate tissue.
The NKX3.1 gene encodes a homeobox protein, which is a transcription factor and tumor suppressor. In prostate cancer, there is loss of heterozygosity of the gene and stains for the IHC antibody to NKX3.1.7
On the other hand, lung cells stain positive for TTF-1, which is produced by surfactant-producing type 2 pneumocytes and club cells in the lung. Antibodies to TTF-1, a common IHC stain, are used to identify adenocarcinoma of lung origin and may carry a prognostic value.7
The immunohistochemistry profiles, specifically the presence of prostate-specific markers such as PSA and NKX3.1, played a vital role in making the diagnosis.
In Case 1, weak TTF-1 positivity was noted, an unusual finding in metastatic prostate adenocarcinoma. Marak et al documented a rare case of TTF-1–positive metastatic prostate cancer, illustrating the potential for diagnostic confusion with primary lung malignancies.8
The 3 cases described in this report demonstrate the importance of clinical consideration, serial follow-up of PSA levels, using more prostate-specific positron emission tomography tracers (eg, Pylarify) alongside traditional imaging, and tissue biopsy to detect unusual metastases.
CONCLUSIONS
Although thoracic metastases from prostate cancer are rare, these presentations highlight the importance of clinical awareness regarding atypical cases. Pleural disease, endobronchial lesions, and isolated pulmonary nodules might be the first clinical manifestation of metastatic prostate cancer. A high index of suspicion, appropriate imaging, and judicious use of immunohistochemistry are important to ensure accurate diagnosis and optimal patient management.
Prostate cancer is the most common noncutaneous cancer in men, accounting for 29% of all incident cancer cases.1 Typically, prostate cancer metastasizes to bone and regional lymph nodes.2 However, intrathoracic manifestation may occur. This report presents 3 cases of rare intrathoracic manifestations of metastatic prostate cancer with a review of the current literature.
CASE PRESENTATIONS
Case 1
A 71-year-old male who was an active smoker and a long-standing employment as a plumber was diagnosed with rectal cancer in 2022. He completed neoadjuvant capecitabine and radiation therapy followed by a rectosigmoidectomy. Several weeks after surgery, the patient presented to the emergency department (ED) with a dry cough and worsening shortness of breath. Point-of-care ultrasound of the lungs revealed a moderate right pleural effusion with several nodular pleural masses. A chest computed tomography (CT) confirmed these findings (Figure 1). A CT of the abdomen and pelvis revealed prostatomegaly with the medial lobe of the prostate protruding into the bladder; however, no enlarged retroperitoneal, mesenteric or pelvic lymph nodes were noted. The patient underwent a right pleural fluid drainage and pleural mass biopsy. Pleural mass histomorphology as well as immunohistochemical (IHC) stains were consistent with metastatic prostate adenocarcinoma. The pleural fluid cytology also was consistent with metastatic prostate adenocarcinoma.
Immunohistochemistry showed weak positive staining for prostate-specific NK3 homeobox 1 gene (NKX3.1), alpha-methylacyl-CoA racemase gene (AMACR), and prosaposin, and negative transcription termination factor (TTF-1), keratin-7 (CK7), and prosaposin, and negative transcription termination factor (TTF-1), keratin-7 (CK7), keratin-20, and caudal type homeobox 2 gene (CDX2) (Figure 2) 2). The patient's prostate-specific antigen (PSA) was found to be elevated at 33.9 ng/mL (reference range, < 4 ng/mL).
Case 2
A 71-year-old male with a history of alcohol use disorder and a 30-year smoking history presented to the ED with worsening dyspnea on exertion. The patient’s baseline exercise tolerance decreased to walking for only 1 block. He reported unintentional weight loss of about 30 pounds over the prior year, no recent respiratory infections, no prior breathing problems, and no personal or family history of cancer. Chest CT revealed findings of bilateral peribronchial opacities as well as mediastinal and hilar lymphadenopathy (Figure 3). The patient developed hypoxic respiratory failure necessitating intubation, mechanical ventilation, and management in the medical intensive care unit, where he was treated for postobstructive pneumonia. Fiberoptic bronchoscopy revealed endobronchial lesions in the right and left upper lobe that were partially obstructing the airway (Figure 4).
The endobronchial masses were debulked using forceps, and samples were sent for surgical pathology evaluation. Staging was completed using linear endobronchial ultrasound, which revealed an enlarged subcarinal lymph node (S7). The surgical pathology of the endobronchial mass and the subcarinal lymph node cytology were consistent with metastatic adenocarcinoma of the prostate. The tumor cells were positive for AE1/AE3, PSA, and NKX3.1, but were negative for CK7 and TTF-1 (Figure 5). Further imaging revealed an enlarged heterogeneous prostate gland, prominent pelvic nodes, and left retroperitoneal lymphadenopathy, as well as sclerotic foci within the T10 vertebral body and right inferior pubic ramus. PSA was also found to be significantly elevated at 700 ng/mL.
Case 3
An 80-year-old male veteran with a history of prostate cancer and recently diagnosed T2N1M0 head and neck squamous cell carcinoma was referred to the Pulmonary service for evaluation of a pulmonary nodule. His medical history was notable for prostate cancer diagnosed 12 years earlier, with an unknown Gleason score. Initial treatment included prostatectomy followed by whole pelvic radiation therapy a year after, due to elevated PSA in surveillance monitoring. This treatment led to remission. After establishing remission for > 10 years, the patient was started on low-dose testosterone replacement therapy to address complications of radiation therapy, namely hypogonadism.
On evaluation, a chest CT was significant for a large 2-cm right middle lobe nodule (Figure 6). At that time, PSA was noted to be borderline elevated at 4.2 ng/mL, and whole-body imaging did not reveal any lesions elsewhere, specifically no bone metastasis. Biopsies of the right middle lobe lung nodule revealed adenocarcinoma consistent with metastatic prostate cancer. Testosterone therapy was promptly discontinued.
The patient initially refused androgen deprivation therapy owing to the antiandrogenic adverse effects. However, subsequent chest CTs revealed growing lung nodules, which convinced him to proceed with androgen deprivation therapy followed by palliative radiation, and chemotherapy and management of malignant pleural effusion with indwelling small bore pleural catheter for about 10 years. He died from COVID-19 during the pandemic.
DISCUSSION
These cases highlight the importance of including prostate cancer in the differential diagnoses of male patients with intrathoracic abnormalities, even in the absence of metastasis to the more common sites. In a large cohort study of 74,826 patients with metastatic prostate cancer, Gandaglia et al found that the most frequent sites of metastasis were bone (84.0%) and distant lymph nodes (10.6%).2 However, thoracic involvement was observed in 9.1% of cases, with isolated thoracic metastasis being rare. The cases described in this report exemplify exceptionally uncommon occurrences within that 9.1%.
Pleural metastases, as observed in Case 1, are a particularly rare manifestation. In a 10-year retrospective assessment, Vinjamoori et al discovered pleural nodules or masses in only 6 of 82 patients (7.3%) with atypical metastases.3 Adrenal and liver metastases accounted for 15% and 37% of cases with atypical distribution. As such, isolated pleural disease is rare even in atypical presentations.3
As seen in Case 2, endobronchial metastases producing airway obstruction are also rare, with the most common primary cancers associated with endobronchial metastasis being breast, colon, and renal cancer.4 The available literature on this presentation is confined to case reports. Hameed et al reported a case of synchronous biopsy-proven endobronchial metastasis from prostate cancer.5 These cases highlight the importance of maintaining a high level of clinical awareness when encountering endobronchial lesions in patients with prostate cancer.
Case 3 presents a unique situation of lung metastases without any involvement of the bones. It is well known—and was confirmed by Heidenreich et al—that lung metastases in prostate adenocarcinoma usually coincide with extensive osseous disease.6 This instance highlights the importance of watchful monitoring for unusual patterns of cancer recurrence.
Immunohistochemistry stains that are specific to prostate cancer include antibodies against PSA. Prostate-specific membrane antigen is another marker that is far more present in malignant than in benign prostate tissue.
The NKX3.1 gene encodes a homeobox protein, which is a transcription factor and tumor suppressor. In prostate cancer, there is loss of heterozygosity of the gene and stains for the IHC antibody to NKX3.1.7
On the other hand, lung cells stain positive for TTF-1, which is produced by surfactant-producing type 2 pneumocytes and club cells in the lung. Antibodies to TTF-1, a common IHC stain, are used to identify adenocarcinoma of lung origin and may carry a prognostic value.7
The immunohistochemistry profiles, specifically the presence of prostate-specific markers such as PSA and NKX3.1, played a vital role in making the diagnosis.
In Case 1, weak TTF-1 positivity was noted, an unusual finding in metastatic prostate adenocarcinoma. Marak et al documented a rare case of TTF-1–positive metastatic prostate cancer, illustrating the potential for diagnostic confusion with primary lung malignancies.8
The 3 cases described in this report demonstrate the importance of clinical consideration, serial follow-up of PSA levels, using more prostate-specific positron emission tomography tracers (eg, Pylarify) alongside traditional imaging, and tissue biopsy to detect unusual metastases.
CONCLUSIONS
Although thoracic metastases from prostate cancer are rare, these presentations highlight the importance of clinical awareness regarding atypical cases. Pleural disease, endobronchial lesions, and isolated pulmonary nodules might be the first clinical manifestation of metastatic prostate cancer. A high index of suspicion, appropriate imaging, and judicious use of immunohistochemistry are important to ensure accurate diagnosis and optimal patient management.
- Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74(1):12-49. doi:10.3322/caac.21820
- Gandaglia G, Abdollah F, Schiffmann J, et al. Distribution of metastatic sites in patients with prostate cancer: a population-based analysis. Prostate. 2014;74(2):210-216. doi:10.1002/pros.22742
- Vinjamoori AH, Jagannathan JP, Shinagare AB, et al. Atypical metastases from prostate cancer: 10-year experience at a single institution. AJR Am J Roentgenol. 2012;199(2):367-372. doi:10.2214/AJR.11.7533
- Salud A, Porcel JM, Rovirosa A, Bellmunt J. Endobronchial metastatic disease: analysis of 32 cases. J Surg Oncol. 1996;62(4):249-252. doi:10.1002/(SICI)1096- 9098(199608)62:4<249::AID-JSO4>3.0.CO;2-6
- Hameed M, Haq IU, Yousaf M, Hussein M, Rashid U, Al-Bozom I. Endobronchial metastases secondary to prostate cancer: a case report and literature review. Respir Med Case Rep. 2020;32:101326. doi:10.1016/j.rmcr.2020.101326
- Heidenreich A, Bastian PJ, Bellmunt J, et al; for the European Association of Urology. EAU guidelines on prostate cancer. Part II: treatment of advanced, relapsing, and castration- resistant prostate cancer. Eur Urol. 2014;65(2):467- 479. doi:10.1016/j.eururo.2013.11.002
- Schallenberg S, Dernbach G, Dragomir MP, et al. TTF-1 status in early-stage lung adenocarcinoma is an independent predictor of relapse and survival superior to tumor grading. Eur J Cancer. 2024;197:113474. doi:10.1016/j.ejca.2023.113474
- Marak C, Guddati AK, Ashraf A, Smith J, Kaushik P. Prostate adenocarcinoma with atypical immunohistochemistry presenting with a Cheerio sign. AIM Clinical Cases. 2023;1:e220508. doi:10.7326/aimcc.2022.0508
- Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74(1):12-49. doi:10.3322/caac.21820
- Gandaglia G, Abdollah F, Schiffmann J, et al. Distribution of metastatic sites in patients with prostate cancer: a population-based analysis. Prostate. 2014;74(2):210-216. doi:10.1002/pros.22742
- Vinjamoori AH, Jagannathan JP, Shinagare AB, et al. Atypical metastases from prostate cancer: 10-year experience at a single institution. AJR Am J Roentgenol. 2012;199(2):367-372. doi:10.2214/AJR.11.7533
- Salud A, Porcel JM, Rovirosa A, Bellmunt J. Endobronchial metastatic disease: analysis of 32 cases. J Surg Oncol. 1996;62(4):249-252. doi:10.1002/(SICI)1096- 9098(199608)62:4<249::AID-JSO4>3.0.CO;2-6
- Hameed M, Haq IU, Yousaf M, Hussein M, Rashid U, Al-Bozom I. Endobronchial metastases secondary to prostate cancer: a case report and literature review. Respir Med Case Rep. 2020;32:101326. doi:10.1016/j.rmcr.2020.101326
- Heidenreich A, Bastian PJ, Bellmunt J, et al; for the European Association of Urology. EAU guidelines on prostate cancer. Part II: treatment of advanced, relapsing, and castration- resistant prostate cancer. Eur Urol. 2014;65(2):467- 479. doi:10.1016/j.eururo.2013.11.002
- Schallenberg S, Dernbach G, Dragomir MP, et al. TTF-1 status in early-stage lung adenocarcinoma is an independent predictor of relapse and survival superior to tumor grading. Eur J Cancer. 2024;197:113474. doi:10.1016/j.ejca.2023.113474
- Marak C, Guddati AK, Ashraf A, Smith J, Kaushik P. Prostate adenocarcinoma with atypical immunohistochemistry presenting with a Cheerio sign. AIM Clinical Cases. 2023;1:e220508. doi:10.7326/aimcc.2022.0508
Atypical Intrathoracic Manifestations of Metastatic Prostate Cancer: A Case Series
Atypical Intrathoracic Manifestations of Metastatic Prostate Cancer: A Case Series

The Need for a Multidisciplinary Approach for Successful High-Risk Pulmonary Embolism Treatment
The Need for a Multidisciplinary Approach for Successful High-Risk Pulmonary Embolism Treatment
Pulmonary embolism (PE) is a common cause of morbidity and mortality in the general population.1 The incidence of PE has been reported to range from 39 to 115 per 100,000 persons per year and has remained stable.2 Although mortality rates have declined, they remain high.3 The clinical presentation is nonspecific, making diagnosis and management challenging. A crucial and difficult aspect in the management of patients with PE is weighing the risks vs benefits of treatment, including thrombolytic therapy and other invasive procedures, which carry inherent risks. These factors have led to the development of PE response teams (PERTs) in some hospitals to implement effective multidisciplinary protocols that facilitate prompt diagnosis, management, and follow-up.4
CASE PRESENTATIONS
Case 1
New onset seizures and cardiac arrest in the treatment of saddle PE. A 54-year-old male who worked as a draftsman and truck driver with a history of hypertension and nephrolithiasis presented to the emergency department (ED) with progressive shortness of breath for 2 weeks. On the morning of ED presentation the patient experienced an episode of severe shortness of breath, lightheadedness, and chest pressure. He reported no other symptoms such as palpitations, nausea, vomiting, abdominal discomfort, or extremity pain or swelling. He reported no recent travel, immunization, falls, or surgery. Upon evaluation, the patient was found to be in no acute distress, with stable vital signs and laboratory results except for 2 elevated results: > 20 μg/mL D-dimer (reference range, < 0.5 μg/mL) and N-terminal prohormone brain natriuretic peptide (proBNP) level, 3455 pg/mL (reference range, < 125 pg/mL for patients aged < 75 years). Electrocardiogram showed T-wave inversions in leads V2 to V4. Imaging revealed a saddle PE and left popliteal deep venous thrombosis (Figure 1). The patient received an anticoagulation loading dose and was started on heparin drip upon admission to the medical intensive care unit (MICU) for further management and monitoring. The Interventional Radiology Service recommended full anticoagulation with consideration of reperfusion therapies if deterioration developed.
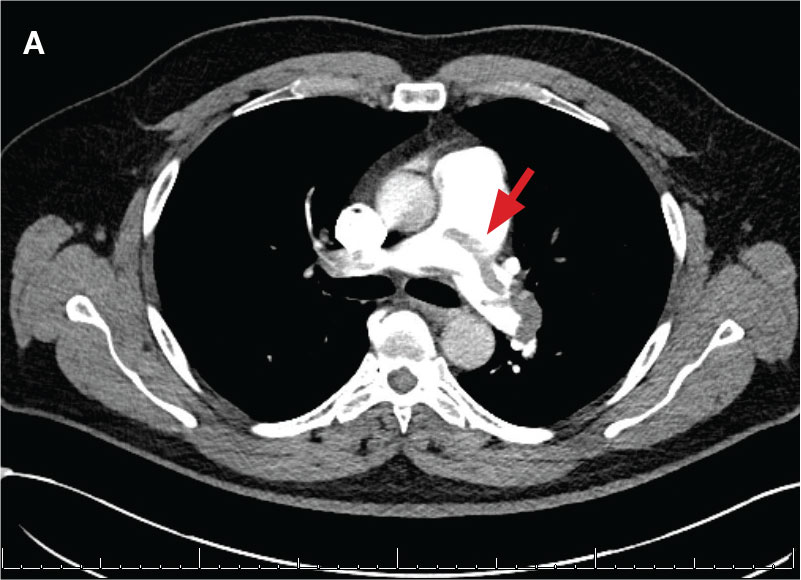
indicated by arrows in the pulmonary trunk extending to the left pulmonary artery (A),
and obliterating right pulmonary artery and branches of left pulmonary artery (B).
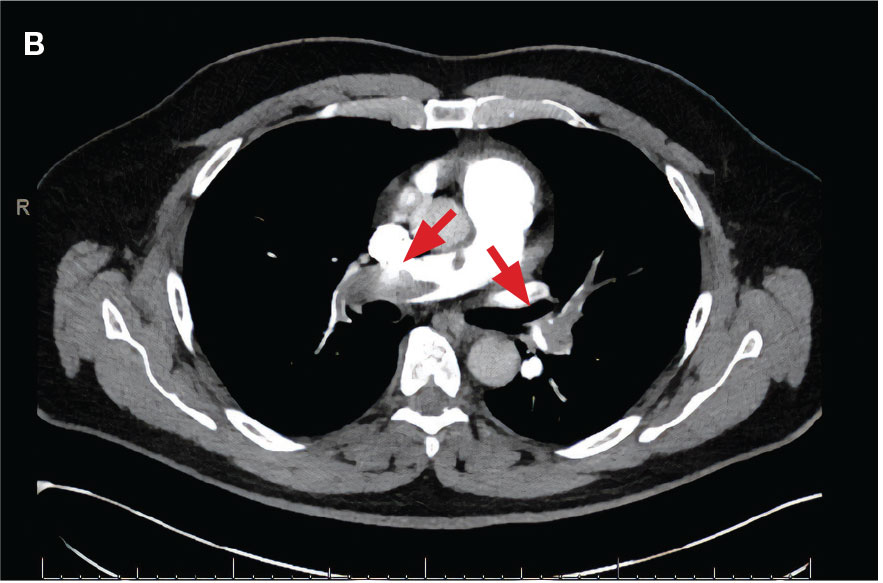
indicated by arrows in the pulmonary trunk extending to the left pulmonary artery (A),
and obliterating right pulmonary artery and branches of left pulmonary artery (B).
While in the MICU, point-of-care ultrasound findings were confirmed with official echocardiogram by the cardiology service, which demonstrated a preserved ejection fraction of 60% to 65%, a D-shaped left ventricle with septal wall hypokinesis secondary to right heart strain (Figure 2), a markedly elevated right ventricular systolic pressure (RVSP) of 73 mm Hg, and a mean pulmonary artery pressure (mPAP) of 38 mm Hg. The patient’s blood pressure progressively decreased, heart rate increased, and he required increased oxygen supplementation. The case was discussed with the Pharmacy Service, and since the patient had no contraindications to thrombolytic therapy, the appropriate dosage was calculated and 100 mg intravenous (IV) tissue plasminogen activator (tPA) was administered over 2 hours.
flattening and deviation to left in direction (A) and septal deviation to left with
formation of D-sign (B).
flattening and deviation to left in direction (A) and septal deviation to left with
formation of D-sign (B).
About 40 minutes into tPA infusion, the patient suddenly experienced marked shortness of breath, diaphoresis, and anxiety with seizure-like involuntary movements; as a result, the infusion was stopped. He also had episodes of posturing, mental status decline, and briefly going in and out of consciousness, which lasted about 3 minutes before he lost consciousness and pulse. High-quality advanced cardiac life support was initiated, followed by endotracheal intubation. Despite a secured airway and return of spontaneous circulation, the patient remained hypotensive and continued to have seizure-like activity.
The patient was administered a total of 8 mg of lorazepam, sedated with propofol, initiated at 5 μg/kg/min, titrated to stop seizure activity at 15μg/kg/min, and later maintained at 10 μg/kg/min, for a RASS of -1, and started on norepinephrine 0.1 μg/kg/min for acute stabilization. Head computed tomography without contrast showed no acute intracranial pathology as etiology of seizures. Seizure etiology differential at this time was broad; however, hypoxemia due to PE and medication adverse effects were strongly suspected.
The patient’s condition improved, and vasopressor therapy was tapered off the next day. Four days later, the patient was weaned from mechanical ventilation and transferred to the step-down unit. Echocardiogram obtained 48 hours after tPA infusion showed essentially normal left ventricular function (60%-65%), a RVSP of 17 mm Hg and mPAP of 13 mm Hg. The patient’s ProBNP levels markedly decreased to 137 pg/mL. Postextubation, the neurologic examination was at baseline. The Neurology Service recommended temporary treatment with levetiracetam, 1000 mg every 12 hours, and the Hematology Service recommended transitioning to direct oral anticoagulation with follow-up. The patient presented significant clinical and respiratory improvement and was referred for home-based physical rehabilitation as recommended by the physical medicine and rehabilitation service before being discharged.
Case 2
Localized tPA infusion for bilateral PEs via infusion catheters. A 91-year-old male with no history of smoking and a medical history of hypertension, diabetes mellitus, prostate cancer (> 20 years postradiotherapy) and severe osteoarthritis was receiving treatment in the medical ward for medication-induced liver injury secondary to an antibiotic for a urinary tract infection. During the night the patient developed hypotension (86/46 mm Hg), shortness of breath, tachypnea, desaturation, nonradiating retrosternal chest pain, and tachycardia. The hypotension resolved after a 500-mL 0.9 normal saline bolus, and hypoxemia improved with supplemental oxygen via Venturi mask. Chest computed tomography angiography was performed immediately and revealed extensive bilateral acute PE, located most proximally in the right main pulmonary artery (PA) and on the left in the proximal lobar branches, with associated right heart strain. The patient was started on IV heparin with a bolus of 5000 units (80 u/kg) followed by a drip with a partial thromboplastin time goal of 62-103 seconds and transferred to MICU.
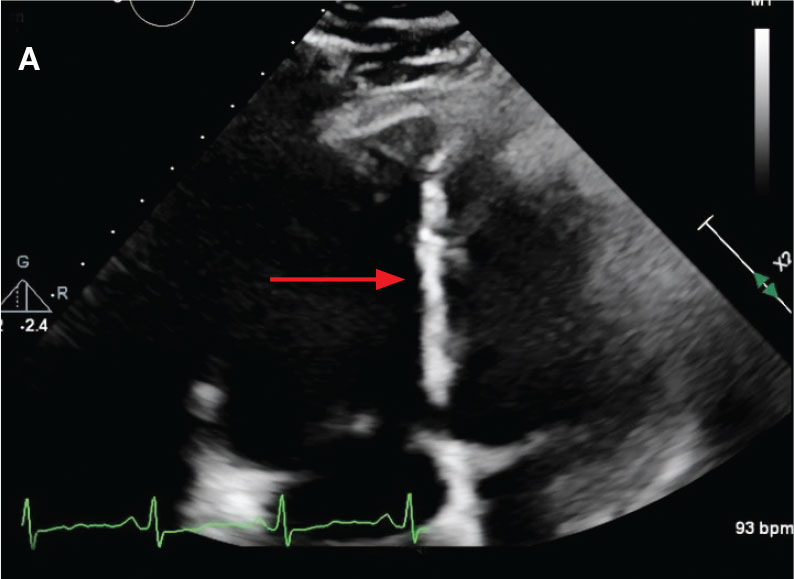
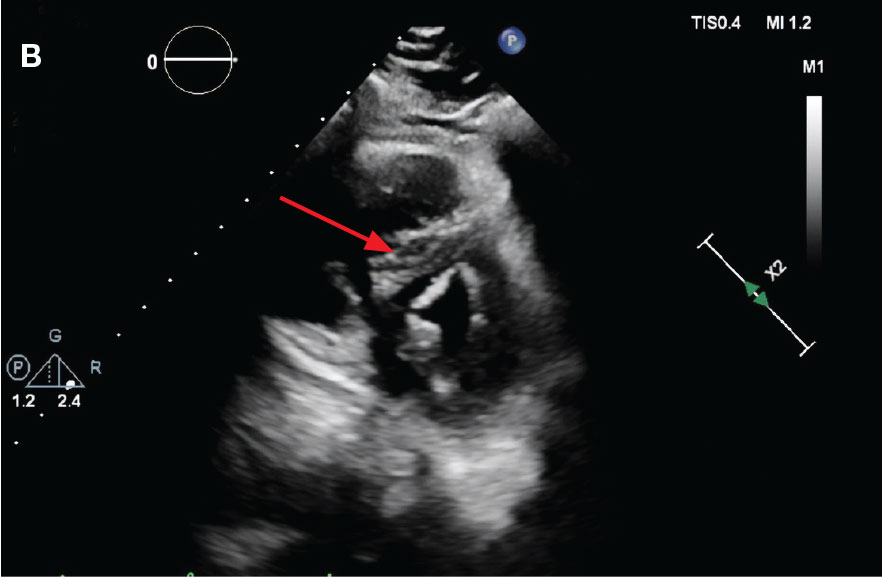
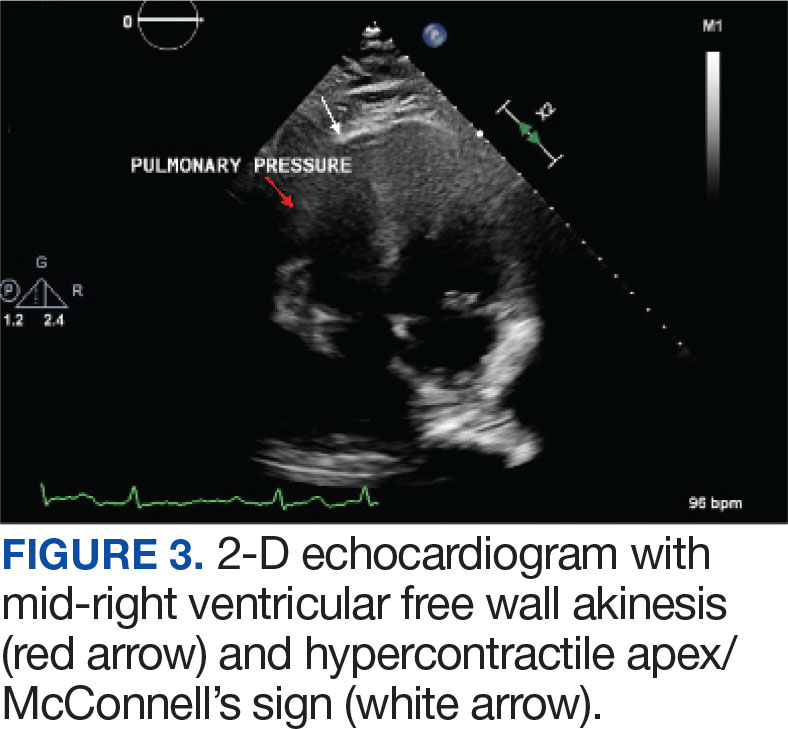
Laboratory findings were notable for proBNP that increased from 115 pg/mL to 4470 pg/mL (reference range, < 450 pg/mL for patients aged 75 years) and elevated troponin levels at 218 ng/L to 295 ng/L (reference range, < 22 ng/L), exhibiting chemical evidence of right heart strain. Initial echocardiogram showed mid-right ventricular free wall akinesis with a hypercontractile apex, suggestive of PE (McConnell’s sign) (Figure 3). Interventional Radiology Service was consulted and recommended tPA infusion given that the patient had bilateral PEs and stable blood pressure.
Pulmonary angiogram showed elevated pressures in the right PA of 64/21 mm Hg and the left PA pressures of 63/20 mm Hg. Mechanical disruption of the larger right lower PA thrombus was achieved via a pigtail catheter followed by bilateral catheter bolus infusions of 2 mg tPA (alteplase) and a continuous tPA infusion 0.5 mg/h for 24 hours, in conjunction with a heparin infusion.
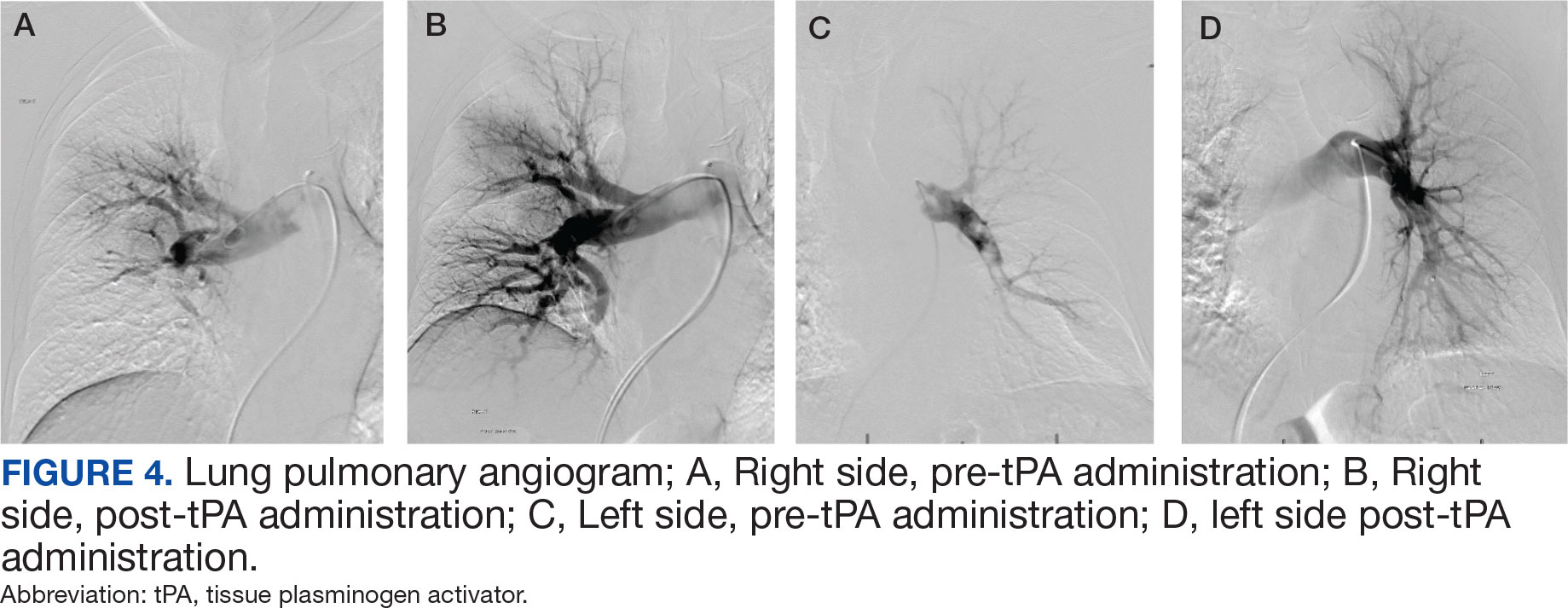
After 24 hours of tPA infusion, the catheters were removed, with posttreatment pulmonary angiography demonstrating right and left PA pressures of 42/15 mm Hg and 40/16 mm Hg, respectively. Pre- and postlocalized tPA infusion treatment images are provided for visual comparison (Figure 4). An echocardiogram performed after tPA infusion showed no signs of pulmonary hypertension. The Hematology Service provided recommendations regarding anticoagulation, and after completion of tPA infusion, the patient was transitioned to an unfractioned heparin infusion and subsequently to direct oral anticoagulation prior to transfer back to the medical ward, hemodynamically stable and asymptomatic.
DISCUSSION
PE management can be a straightforward decision when the patient meets criteria for hemodynamic instability, or with small PE burden. In contrast, management can be more challenging in intermediate-risk (submassive) PE when patients remain hemodynamically stable but show signs of cardiopulmonary stress, such as right heart strain, elevated troponins, or increased proBNP levels.2 In these situations, case-by- case evaluation is warranted. A PERT can assess the most beneficial treatment approach by considering factors such as right ventricular dysfunction, hemodynamic status, clot burden, and clinical deterioration despite appropriate anticoagulation. The evidence supporting the benefits these organized teams can provide is growing. These case reports emphasize the need for a multidisciplinary and systematic approach in these complex cases, especially in the management of intermediate-risk PE patients.
Currently, the Veterans Affairs Caribbean Healthcare System does not have an organized PERT, although a multidisciplinary approach was applied in the management of these patients. A systematic, structured team could have decreased time to interventions and alleviated the burden of physician decision-making. Having such a team would streamline the diagnostic pathway for patients presenting from a ward or emergency department with suspected PE.
We present 2 cases of patients found to have a high clot burden from PEs. The patients were initially hemodynamically stable (intermediate-risk PE), but later required systemic or localized thrombolysis due to hemodynamic deterioration despite adequate anticoagulation. Despite similar diagnoses and etiologies, these patients were successfully managed using different approaches, yielding positive outcomes. This reflects the complexity and variability in diagnosing and managing intermediate-risk PE in patients with different comorbidities and clot burden effects. In Case 1, our multidisciplinary approach was obtained via consults to selected services such as interventional radiology, cardiology, and direct involvement of pharmacy. An organized PERT conceivably would have allowed quicker discussions among these services, including hematology, to provide recommendations and collaborative support upon the patient’s arrival to the ED. Additionally, with a PERT team, a systematic approach to these patients could have allowed for an earlier official echocardiogram report for evaluation of right heart strain and develop an adequate therapeutic plan in a timely manner.
In Case 2, consultation with the Interventional Radiology Service yielded a better therapeutic plan, utilizing localized tPA infusion for this older adult patient with increased risk of bleeding with systemic tPA infusion. Having a PERT presents an opportunity to optimize PE management through early recognition, diagnosis, and treatment by institutional consensus from an interdisciplinary team.5,6 These response teams may improve outcomes and prognosis for patients with PE, especially where diagnosis and management is not clear.
The definite etiology of seizure activity in the first case pre- and postcardiac arrest, in the context of no acute intracranial process, remains unknown. Reports have emerged about postreperfusion seizures in acute ischemic stroke, as well as cases of seizures masquerading as PE as the primary presentation. 7,8 However, there were no reports of patients developing seizures post tPA infusion for the treatment of PE. This report may shed light into possible complications secondary to tPA infusion, raising awareness among physicians and encouraging further investigation into its possible etiologies.
CONCLUSIONS
Management of PE can be challenging in patients that meet criteria for intermediate risk. PERTs are a tool that allow for a multidisciplinary, standardized and systematic approach with a diagnostic and treatment algorithm that conceivably would result in a better consensus and therapeutic approach.
- Thompson BT, Kabrhel C. Epidemiology and pathogenesis of acute pulmonary embolism in adults. UpToDate. Wolters Kluwer. Updated December 4, 2023. Accessed February 26, 2025. https://www.uptodate.cn/contents/epidemiology-and-pathogenesis-of-acute-pulmonary-embolism-in-adults
- Kulka HC, Zeller A, Fornaro J, Wuillemin WA, Konstantinides S, Christ M. Acute pulmonary embolism– its diagnosis and treatment from a multidisciplinary viewpoint. Dtsch Arztebl Int. 2021;118(37):618-628. doi:10.3238/arztebl.m2021.0226
- Zghouzi M, Mwansa H, Shore S, et al. Sex, racial, and geographic disparities in pulmonary embolism-related mortality nationwide. Ann Am Thorac Soc. 2023;20(11):1571-1577. doi:10.1513/AnnalsATS.202302-091OC
- Channick RN. The pulmonary embolism response team: why and how? Semin Respir Crit Care Med. 2021;42(2):212-217. doi:10.1055/s-0041-1722963
- Rosovsky R, Zhao K, Sista A, Rivera-Lebron B, Kabrhel C. Pulmonary embolism response teams: purpose, evidence for efficacy, and future research directions. Res Pract Thromb Haemost. 2019;3(3):315-330. doi:10.1002/rth2.12216
- Glazier JJ, Patiño-Velasquez S, Oviedo C. The pulmonary embolism response team: rationale, operation, and outcomes. Int J Angiol. 2022;31(3):198-202. doi:10.1055/s-0042-1750328
- Lekoubou A, Fox J, Ssentongo P. Incidence and association of reperfusion therapies with poststroke seizures: a systematic review and meta-analysis. Stroke. 2020;51(9):2715-2723.doi:10.1161/STROKEAHA.119. 028899
- Alemany M, Nuñez A, Falip M, et al. Acute symptomatic seizures and epilepsy after mechanical thrombectomy. A prospective long-term follow-up study. Seizure. 2021;89:5-9. doi:10.1016/j.seizure.2021.04.011
Pulmonary embolism (PE) is a common cause of morbidity and mortality in the general population.1 The incidence of PE has been reported to range from 39 to 115 per 100,000 persons per year and has remained stable.2 Although mortality rates have declined, they remain high.3 The clinical presentation is nonspecific, making diagnosis and management challenging. A crucial and difficult aspect in the management of patients with PE is weighing the risks vs benefits of treatment, including thrombolytic therapy and other invasive procedures, which carry inherent risks. These factors have led to the development of PE response teams (PERTs) in some hospitals to implement effective multidisciplinary protocols that facilitate prompt diagnosis, management, and follow-up.4
CASE PRESENTATIONS
Case 1
New onset seizures and cardiac arrest in the treatment of saddle PE. A 54-year-old male who worked as a draftsman and truck driver with a history of hypertension and nephrolithiasis presented to the emergency department (ED) with progressive shortness of breath for 2 weeks. On the morning of ED presentation the patient experienced an episode of severe shortness of breath, lightheadedness, and chest pressure. He reported no other symptoms such as palpitations, nausea, vomiting, abdominal discomfort, or extremity pain or swelling. He reported no recent travel, immunization, falls, or surgery. Upon evaluation, the patient was found to be in no acute distress, with stable vital signs and laboratory results except for 2 elevated results: > 20 μg/mL D-dimer (reference range, < 0.5 μg/mL) and N-terminal prohormone brain natriuretic peptide (proBNP) level, 3455 pg/mL (reference range, < 125 pg/mL for patients aged < 75 years). Electrocardiogram showed T-wave inversions in leads V2 to V4. Imaging revealed a saddle PE and left popliteal deep venous thrombosis (Figure 1). The patient received an anticoagulation loading dose and was started on heparin drip upon admission to the medical intensive care unit (MICU) for further management and monitoring. The Interventional Radiology Service recommended full anticoagulation with consideration of reperfusion therapies if deterioration developed.
indicated by arrows in the pulmonary trunk extending to the left pulmonary artery (A),
and obliterating right pulmonary artery and branches of left pulmonary artery (B).
indicated by arrows in the pulmonary trunk extending to the left pulmonary artery (A),
and obliterating right pulmonary artery and branches of left pulmonary artery (B).
While in the MICU, point-of-care ultrasound findings were confirmed with official echocardiogram by the cardiology service, which demonstrated a preserved ejection fraction of 60% to 65%, a D-shaped left ventricle with septal wall hypokinesis secondary to right heart strain (Figure 2), a markedly elevated right ventricular systolic pressure (RVSP) of 73 mm Hg, and a mean pulmonary artery pressure (mPAP) of 38 mm Hg. The patient’s blood pressure progressively decreased, heart rate increased, and he required increased oxygen supplementation. The case was discussed with the Pharmacy Service, and since the patient had no contraindications to thrombolytic therapy, the appropriate dosage was calculated and 100 mg intravenous (IV) tissue plasminogen activator (tPA) was administered over 2 hours.
flattening and deviation to left in direction (A) and septal deviation to left with
formation of D-sign (B).
flattening and deviation to left in direction (A) and septal deviation to left with
formation of D-sign (B).
About 40 minutes into tPA infusion, the patient suddenly experienced marked shortness of breath, diaphoresis, and anxiety with seizure-like involuntary movements; as a result, the infusion was stopped. He also had episodes of posturing, mental status decline, and briefly going in and out of consciousness, which lasted about 3 minutes before he lost consciousness and pulse. High-quality advanced cardiac life support was initiated, followed by endotracheal intubation. Despite a secured airway and return of spontaneous circulation, the patient remained hypotensive and continued to have seizure-like activity.
The patient was administered a total of 8 mg of lorazepam, sedated with propofol, initiated at 5 μg/kg/min, titrated to stop seizure activity at 15μg/kg/min, and later maintained at 10 μg/kg/min, for a RASS of -1, and started on norepinephrine 0.1 μg/kg/min for acute stabilization. Head computed tomography without contrast showed no acute intracranial pathology as etiology of seizures. Seizure etiology differential at this time was broad; however, hypoxemia due to PE and medication adverse effects were strongly suspected.
The patient’s condition improved, and vasopressor therapy was tapered off the next day. Four days later, the patient was weaned from mechanical ventilation and transferred to the step-down unit. Echocardiogram obtained 48 hours after tPA infusion showed essentially normal left ventricular function (60%-65%), a RVSP of 17 mm Hg and mPAP of 13 mm Hg. The patient’s ProBNP levels markedly decreased to 137 pg/mL. Postextubation, the neurologic examination was at baseline. The Neurology Service recommended temporary treatment with levetiracetam, 1000 mg every 12 hours, and the Hematology Service recommended transitioning to direct oral anticoagulation with follow-up. The patient presented significant clinical and respiratory improvement and was referred for home-based physical rehabilitation as recommended by the physical medicine and rehabilitation service before being discharged.
Case 2
Localized tPA infusion for bilateral PEs via infusion catheters. A 91-year-old male with no history of smoking and a medical history of hypertension, diabetes mellitus, prostate cancer (> 20 years postradiotherapy) and severe osteoarthritis was receiving treatment in the medical ward for medication-induced liver injury secondary to an antibiotic for a urinary tract infection. During the night the patient developed hypotension (86/46 mm Hg), shortness of breath, tachypnea, desaturation, nonradiating retrosternal chest pain, and tachycardia. The hypotension resolved after a 500-mL 0.9 normal saline bolus, and hypoxemia improved with supplemental oxygen via Venturi mask. Chest computed tomography angiography was performed immediately and revealed extensive bilateral acute PE, located most proximally in the right main pulmonary artery (PA) and on the left in the proximal lobar branches, with associated right heart strain. The patient was started on IV heparin with a bolus of 5000 units (80 u/kg) followed by a drip with a partial thromboplastin time goal of 62-103 seconds and transferred to MICU.
Laboratory findings were notable for proBNP that increased from 115 pg/mL to 4470 pg/mL (reference range, < 450 pg/mL for patients aged 75 years) and elevated troponin levels at 218 ng/L to 295 ng/L (reference range, < 22 ng/L), exhibiting chemical evidence of right heart strain. Initial echocardiogram showed mid-right ventricular free wall akinesis with a hypercontractile apex, suggestive of PE (McConnell’s sign) (Figure 3). Interventional Radiology Service was consulted and recommended tPA infusion given that the patient had bilateral PEs and stable blood pressure.
Pulmonary angiogram showed elevated pressures in the right PA of 64/21 mm Hg and the left PA pressures of 63/20 mm Hg. Mechanical disruption of the larger right lower PA thrombus was achieved via a pigtail catheter followed by bilateral catheter bolus infusions of 2 mg tPA (alteplase) and a continuous tPA infusion 0.5 mg/h for 24 hours, in conjunction with a heparin infusion.
After 24 hours of tPA infusion, the catheters were removed, with posttreatment pulmonary angiography demonstrating right and left PA pressures of 42/15 mm Hg and 40/16 mm Hg, respectively. Pre- and postlocalized tPA infusion treatment images are provided for visual comparison (Figure 4). An echocardiogram performed after tPA infusion showed no signs of pulmonary hypertension. The Hematology Service provided recommendations regarding anticoagulation, and after completion of tPA infusion, the patient was transitioned to an unfractioned heparin infusion and subsequently to direct oral anticoagulation prior to transfer back to the medical ward, hemodynamically stable and asymptomatic.
DISCUSSION
PE management can be a straightforward decision when the patient meets criteria for hemodynamic instability, or with small PE burden. In contrast, management can be more challenging in intermediate-risk (submassive) PE when patients remain hemodynamically stable but show signs of cardiopulmonary stress, such as right heart strain, elevated troponins, or increased proBNP levels.2 In these situations, case-by- case evaluation is warranted. A PERT can assess the most beneficial treatment approach by considering factors such as right ventricular dysfunction, hemodynamic status, clot burden, and clinical deterioration despite appropriate anticoagulation. The evidence supporting the benefits these organized teams can provide is growing. These case reports emphasize the need for a multidisciplinary and systematic approach in these complex cases, especially in the management of intermediate-risk PE patients.
Currently, the Veterans Affairs Caribbean Healthcare System does not have an organized PERT, although a multidisciplinary approach was applied in the management of these patients. A systematic, structured team could have decreased time to interventions and alleviated the burden of physician decision-making. Having such a team would streamline the diagnostic pathway for patients presenting from a ward or emergency department with suspected PE.
We present 2 cases of patients found to have a high clot burden from PEs. The patients were initially hemodynamically stable (intermediate-risk PE), but later required systemic or localized thrombolysis due to hemodynamic deterioration despite adequate anticoagulation. Despite similar diagnoses and etiologies, these patients were successfully managed using different approaches, yielding positive outcomes. This reflects the complexity and variability in diagnosing and managing intermediate-risk PE in patients with different comorbidities and clot burden effects. In Case 1, our multidisciplinary approach was obtained via consults to selected services such as interventional radiology, cardiology, and direct involvement of pharmacy. An organized PERT conceivably would have allowed quicker discussions among these services, including hematology, to provide recommendations and collaborative support upon the patient’s arrival to the ED. Additionally, with a PERT team, a systematic approach to these patients could have allowed for an earlier official echocardiogram report for evaluation of right heart strain and develop an adequate therapeutic plan in a timely manner.
In Case 2, consultation with the Interventional Radiology Service yielded a better therapeutic plan, utilizing localized tPA infusion for this older adult patient with increased risk of bleeding with systemic tPA infusion. Having a PERT presents an opportunity to optimize PE management through early recognition, diagnosis, and treatment by institutional consensus from an interdisciplinary team.5,6 These response teams may improve outcomes and prognosis for patients with PE, especially where diagnosis and management is not clear.
The definite etiology of seizure activity in the first case pre- and postcardiac arrest, in the context of no acute intracranial process, remains unknown. Reports have emerged about postreperfusion seizures in acute ischemic stroke, as well as cases of seizures masquerading as PE as the primary presentation. 7,8 However, there were no reports of patients developing seizures post tPA infusion for the treatment of PE. This report may shed light into possible complications secondary to tPA infusion, raising awareness among physicians and encouraging further investigation into its possible etiologies.
CONCLUSIONS
Management of PE can be challenging in patients that meet criteria for intermediate risk. PERTs are a tool that allow for a multidisciplinary, standardized and systematic approach with a diagnostic and treatment algorithm that conceivably would result in a better consensus and therapeutic approach.
Pulmonary embolism (PE) is a common cause of morbidity and mortality in the general population.1 The incidence of PE has been reported to range from 39 to 115 per 100,000 persons per year and has remained stable.2 Although mortality rates have declined, they remain high.3 The clinical presentation is nonspecific, making diagnosis and management challenging. A crucial and difficult aspect in the management of patients with PE is weighing the risks vs benefits of treatment, including thrombolytic therapy and other invasive procedures, which carry inherent risks. These factors have led to the development of PE response teams (PERTs) in some hospitals to implement effective multidisciplinary protocols that facilitate prompt diagnosis, management, and follow-up.4
CASE PRESENTATIONS
Case 1
New onset seizures and cardiac arrest in the treatment of saddle PE. A 54-year-old male who worked as a draftsman and truck driver with a history of hypertension and nephrolithiasis presented to the emergency department (ED) with progressive shortness of breath for 2 weeks. On the morning of ED presentation the patient experienced an episode of severe shortness of breath, lightheadedness, and chest pressure. He reported no other symptoms such as palpitations, nausea, vomiting, abdominal discomfort, or extremity pain or swelling. He reported no recent travel, immunization, falls, or surgery. Upon evaluation, the patient was found to be in no acute distress, with stable vital signs and laboratory results except for 2 elevated results: > 20 μg/mL D-dimer (reference range, < 0.5 μg/mL) and N-terminal prohormone brain natriuretic peptide (proBNP) level, 3455 pg/mL (reference range, < 125 pg/mL for patients aged < 75 years). Electrocardiogram showed T-wave inversions in leads V2 to V4. Imaging revealed a saddle PE and left popliteal deep venous thrombosis (Figure 1). The patient received an anticoagulation loading dose and was started on heparin drip upon admission to the medical intensive care unit (MICU) for further management and monitoring. The Interventional Radiology Service recommended full anticoagulation with consideration of reperfusion therapies if deterioration developed.
indicated by arrows in the pulmonary trunk extending to the left pulmonary artery (A),
and obliterating right pulmonary artery and branches of left pulmonary artery (B).
indicated by arrows in the pulmonary trunk extending to the left pulmonary artery (A),
and obliterating right pulmonary artery and branches of left pulmonary artery (B).
While in the MICU, point-of-care ultrasound findings were confirmed with official echocardiogram by the cardiology service, which demonstrated a preserved ejection fraction of 60% to 65%, a D-shaped left ventricle with septal wall hypokinesis secondary to right heart strain (Figure 2), a markedly elevated right ventricular systolic pressure (RVSP) of 73 mm Hg, and a mean pulmonary artery pressure (mPAP) of 38 mm Hg. The patient’s blood pressure progressively decreased, heart rate increased, and he required increased oxygen supplementation. The case was discussed with the Pharmacy Service, and since the patient had no contraindications to thrombolytic therapy, the appropriate dosage was calculated and 100 mg intravenous (IV) tissue plasminogen activator (tPA) was administered over 2 hours.
flattening and deviation to left in direction (A) and septal deviation to left with
formation of D-sign (B).
flattening and deviation to left in direction (A) and septal deviation to left with
formation of D-sign (B).
About 40 minutes into tPA infusion, the patient suddenly experienced marked shortness of breath, diaphoresis, and anxiety with seizure-like involuntary movements; as a result, the infusion was stopped. He also had episodes of posturing, mental status decline, and briefly going in and out of consciousness, which lasted about 3 minutes before he lost consciousness and pulse. High-quality advanced cardiac life support was initiated, followed by endotracheal intubation. Despite a secured airway and return of spontaneous circulation, the patient remained hypotensive and continued to have seizure-like activity.
The patient was administered a total of 8 mg of lorazepam, sedated with propofol, initiated at 5 μg/kg/min, titrated to stop seizure activity at 15μg/kg/min, and later maintained at 10 μg/kg/min, for a RASS of -1, and started on norepinephrine 0.1 μg/kg/min for acute stabilization. Head computed tomography without contrast showed no acute intracranial pathology as etiology of seizures. Seizure etiology differential at this time was broad; however, hypoxemia due to PE and medication adverse effects were strongly suspected.
The patient’s condition improved, and vasopressor therapy was tapered off the next day. Four days later, the patient was weaned from mechanical ventilation and transferred to the step-down unit. Echocardiogram obtained 48 hours after tPA infusion showed essentially normal left ventricular function (60%-65%), a RVSP of 17 mm Hg and mPAP of 13 mm Hg. The patient’s ProBNP levels markedly decreased to 137 pg/mL. Postextubation, the neurologic examination was at baseline. The Neurology Service recommended temporary treatment with levetiracetam, 1000 mg every 12 hours, and the Hematology Service recommended transitioning to direct oral anticoagulation with follow-up. The patient presented significant clinical and respiratory improvement and was referred for home-based physical rehabilitation as recommended by the physical medicine and rehabilitation service before being discharged.
Case 2
Localized tPA infusion for bilateral PEs via infusion catheters. A 91-year-old male with no history of smoking and a medical history of hypertension, diabetes mellitus, prostate cancer (> 20 years postradiotherapy) and severe osteoarthritis was receiving treatment in the medical ward for medication-induced liver injury secondary to an antibiotic for a urinary tract infection. During the night the patient developed hypotension (86/46 mm Hg), shortness of breath, tachypnea, desaturation, nonradiating retrosternal chest pain, and tachycardia. The hypotension resolved after a 500-mL 0.9 normal saline bolus, and hypoxemia improved with supplemental oxygen via Venturi mask. Chest computed tomography angiography was performed immediately and revealed extensive bilateral acute PE, located most proximally in the right main pulmonary artery (PA) and on the left in the proximal lobar branches, with associated right heart strain. The patient was started on IV heparin with a bolus of 5000 units (80 u/kg) followed by a drip with a partial thromboplastin time goal of 62-103 seconds and transferred to MICU.
Laboratory findings were notable for proBNP that increased from 115 pg/mL to 4470 pg/mL (reference range, < 450 pg/mL for patients aged 75 years) and elevated troponin levels at 218 ng/L to 295 ng/L (reference range, < 22 ng/L), exhibiting chemical evidence of right heart strain. Initial echocardiogram showed mid-right ventricular free wall akinesis with a hypercontractile apex, suggestive of PE (McConnell’s sign) (Figure 3). Interventional Radiology Service was consulted and recommended tPA infusion given that the patient had bilateral PEs and stable blood pressure.
Pulmonary angiogram showed elevated pressures in the right PA of 64/21 mm Hg and the left PA pressures of 63/20 mm Hg. Mechanical disruption of the larger right lower PA thrombus was achieved via a pigtail catheter followed by bilateral catheter bolus infusions of 2 mg tPA (alteplase) and a continuous tPA infusion 0.5 mg/h for 24 hours, in conjunction with a heparin infusion.
After 24 hours of tPA infusion, the catheters were removed, with posttreatment pulmonary angiography demonstrating right and left PA pressures of 42/15 mm Hg and 40/16 mm Hg, respectively. Pre- and postlocalized tPA infusion treatment images are provided for visual comparison (Figure 4). An echocardiogram performed after tPA infusion showed no signs of pulmonary hypertension. The Hematology Service provided recommendations regarding anticoagulation, and after completion of tPA infusion, the patient was transitioned to an unfractioned heparin infusion and subsequently to direct oral anticoagulation prior to transfer back to the medical ward, hemodynamically stable and asymptomatic.
DISCUSSION
PE management can be a straightforward decision when the patient meets criteria for hemodynamic instability, or with small PE burden. In contrast, management can be more challenging in intermediate-risk (submassive) PE when patients remain hemodynamically stable but show signs of cardiopulmonary stress, such as right heart strain, elevated troponins, or increased proBNP levels.2 In these situations, case-by- case evaluation is warranted. A PERT can assess the most beneficial treatment approach by considering factors such as right ventricular dysfunction, hemodynamic status, clot burden, and clinical deterioration despite appropriate anticoagulation. The evidence supporting the benefits these organized teams can provide is growing. These case reports emphasize the need for a multidisciplinary and systematic approach in these complex cases, especially in the management of intermediate-risk PE patients.
Currently, the Veterans Affairs Caribbean Healthcare System does not have an organized PERT, although a multidisciplinary approach was applied in the management of these patients. A systematic, structured team could have decreased time to interventions and alleviated the burden of physician decision-making. Having such a team would streamline the diagnostic pathway for patients presenting from a ward or emergency department with suspected PE.
We present 2 cases of patients found to have a high clot burden from PEs. The patients were initially hemodynamically stable (intermediate-risk PE), but later required systemic or localized thrombolysis due to hemodynamic deterioration despite adequate anticoagulation. Despite similar diagnoses and etiologies, these patients were successfully managed using different approaches, yielding positive outcomes. This reflects the complexity and variability in diagnosing and managing intermediate-risk PE in patients with different comorbidities and clot burden effects. In Case 1, our multidisciplinary approach was obtained via consults to selected services such as interventional radiology, cardiology, and direct involvement of pharmacy. An organized PERT conceivably would have allowed quicker discussions among these services, including hematology, to provide recommendations and collaborative support upon the patient’s arrival to the ED. Additionally, with a PERT team, a systematic approach to these patients could have allowed for an earlier official echocardiogram report for evaluation of right heart strain and develop an adequate therapeutic plan in a timely manner.
In Case 2, consultation with the Interventional Radiology Service yielded a better therapeutic plan, utilizing localized tPA infusion for this older adult patient with increased risk of bleeding with systemic tPA infusion. Having a PERT presents an opportunity to optimize PE management through early recognition, diagnosis, and treatment by institutional consensus from an interdisciplinary team.5,6 These response teams may improve outcomes and prognosis for patients with PE, especially where diagnosis and management is not clear.
The definite etiology of seizure activity in the first case pre- and postcardiac arrest, in the context of no acute intracranial process, remains unknown. Reports have emerged about postreperfusion seizures in acute ischemic stroke, as well as cases of seizures masquerading as PE as the primary presentation. 7,8 However, there were no reports of patients developing seizures post tPA infusion for the treatment of PE. This report may shed light into possible complications secondary to tPA infusion, raising awareness among physicians and encouraging further investigation into its possible etiologies.
CONCLUSIONS
Management of PE can be challenging in patients that meet criteria for intermediate risk. PERTs are a tool that allow for a multidisciplinary, standardized and systematic approach with a diagnostic and treatment algorithm that conceivably would result in a better consensus and therapeutic approach.
- Thompson BT, Kabrhel C. Epidemiology and pathogenesis of acute pulmonary embolism in adults. UpToDate. Wolters Kluwer. Updated December 4, 2023. Accessed February 26, 2025. https://www.uptodate.cn/contents/epidemiology-and-pathogenesis-of-acute-pulmonary-embolism-in-adults
- Kulka HC, Zeller A, Fornaro J, Wuillemin WA, Konstantinides S, Christ M. Acute pulmonary embolism– its diagnosis and treatment from a multidisciplinary viewpoint. Dtsch Arztebl Int. 2021;118(37):618-628. doi:10.3238/arztebl.m2021.0226
- Zghouzi M, Mwansa H, Shore S, et al. Sex, racial, and geographic disparities in pulmonary embolism-related mortality nationwide. Ann Am Thorac Soc. 2023;20(11):1571-1577. doi:10.1513/AnnalsATS.202302-091OC
- Channick RN. The pulmonary embolism response team: why and how? Semin Respir Crit Care Med. 2021;42(2):212-217. doi:10.1055/s-0041-1722963
- Rosovsky R, Zhao K, Sista A, Rivera-Lebron B, Kabrhel C. Pulmonary embolism response teams: purpose, evidence for efficacy, and future research directions. Res Pract Thromb Haemost. 2019;3(3):315-330. doi:10.1002/rth2.12216
- Glazier JJ, Patiño-Velasquez S, Oviedo C. The pulmonary embolism response team: rationale, operation, and outcomes. Int J Angiol. 2022;31(3):198-202. doi:10.1055/s-0042-1750328
- Lekoubou A, Fox J, Ssentongo P. Incidence and association of reperfusion therapies with poststroke seizures: a systematic review and meta-analysis. Stroke. 2020;51(9):2715-2723.doi:10.1161/STROKEAHA.119. 028899
- Alemany M, Nuñez A, Falip M, et al. Acute symptomatic seizures and epilepsy after mechanical thrombectomy. A prospective long-term follow-up study. Seizure. 2021;89:5-9. doi:10.1016/j.seizure.2021.04.011
- Thompson BT, Kabrhel C. Epidemiology and pathogenesis of acute pulmonary embolism in adults. UpToDate. Wolters Kluwer. Updated December 4, 2023. Accessed February 26, 2025. https://www.uptodate.cn/contents/epidemiology-and-pathogenesis-of-acute-pulmonary-embolism-in-adults
- Kulka HC, Zeller A, Fornaro J, Wuillemin WA, Konstantinides S, Christ M. Acute pulmonary embolism– its diagnosis and treatment from a multidisciplinary viewpoint. Dtsch Arztebl Int. 2021;118(37):618-628. doi:10.3238/arztebl.m2021.0226
- Zghouzi M, Mwansa H, Shore S, et al. Sex, racial, and geographic disparities in pulmonary embolism-related mortality nationwide. Ann Am Thorac Soc. 2023;20(11):1571-1577. doi:10.1513/AnnalsATS.202302-091OC
- Channick RN. The pulmonary embolism response team: why and how? Semin Respir Crit Care Med. 2021;42(2):212-217. doi:10.1055/s-0041-1722963
- Rosovsky R, Zhao K, Sista A, Rivera-Lebron B, Kabrhel C. Pulmonary embolism response teams: purpose, evidence for efficacy, and future research directions. Res Pract Thromb Haemost. 2019;3(3):315-330. doi:10.1002/rth2.12216
- Glazier JJ, Patiño-Velasquez S, Oviedo C. The pulmonary embolism response team: rationale, operation, and outcomes. Int J Angiol. 2022;31(3):198-202. doi:10.1055/s-0042-1750328
- Lekoubou A, Fox J, Ssentongo P. Incidence and association of reperfusion therapies with poststroke seizures: a systematic review and meta-analysis. Stroke. 2020;51(9):2715-2723.doi:10.1161/STROKEAHA.119. 028899
- Alemany M, Nuñez A, Falip M, et al. Acute symptomatic seizures and epilepsy after mechanical thrombectomy. A prospective long-term follow-up study. Seizure. 2021;89:5-9. doi:10.1016/j.seizure.2021.04.011
The Need for a Multidisciplinary Approach for Successful High-Risk Pulmonary Embolism Treatment
The Need for a Multidisciplinary Approach for Successful High-Risk Pulmonary Embolism Treatment
Endoscopic Sleeve Gastroplasty is an Effective Treatment for Obesity in a Veteran With Metabolic and Psychiatric Comorbidities
Endoscopic Sleeve Gastroplasty is an Effective Treatment for Obesity in a Veteran With Metabolic and Psychiatric Comorbidities
Obesity is a growing worldwide epidemic with significant implications for individual health and public health care costs. It is also associated with several medical conditions, including diabetes, cardiovascular disease, cancer, and mental health disorders.1 Comprehensive lifestyle intervention is a first-line therapy for obesity consisting of dietary and exercise interventions. Despite initial success, long-term results and durability of weight loss with lifestyle modifications are limited. 2 Bariatric surgery, including sleeve gastrectomy and gastric bypass surgery, is a more invasive approach that is highly effective in weight loss. However, these operations are not reversible, and patients may not be eligible for or may not desire surgery. Overall, bariatric surgery is widely underutilized, with < 1% of eligible patients ultimately undergoing surgery.3,4
Endoscopic bariatric therapies are increasingly popular procedures that address the need for additional treatments for obesity among individuals who have not had success with lifestyle changes and are not surgical candidates. The most common procedure is the endoscopic sleeve gastroplasty (ESG), which applies full-thickness sutures in the stomach to reduce gastric volume, delay gastric emptying, and limit food intake while keeping the fundus intact compared with sleeve gastrectomy. This procedure is typically considered in patients with body mass index (BMI) ≥ 30, who do not qualify for or do not want traditional bariatric surgery. The literature supports robust outcomes after ESG, with studies demonstrating significant and sustained total body weight loss of up to 14% to 16% at 5 years and significant improvement in ≥ 1 metabolic comorbidities in 80% of patients.5,6 ESG adverse events (AEs) include abdominal pain, nausea, and vomiting that are typically self-limited to 1 week. Rarer but more serious AEs include bleeding, perforation, or infection, and occur in 2% of cases based on large trial data.5,7
Although the weight loss benefits of ESG are well established, to date, there are limited data on the effects of endoscopic bariatric therapies like ESG on mental health conditions. Here, we describe a case of a veteran with a history of mental health disorders that prevented him from completing bariatric surgery. The patient underwent ESG and had a successful clinical course.
CASE PRESENTATION
A 59-year-old male veteran with a medical history of class III obesity (42.4 BMI), obstructive sleep apnea, hypothyroidism, hypertension, type 2 diabetes mellitus, and a large ventral hernia was referred to the MOVE! (Management of Overweight/ Obese Veterans Everywhere!) multidisciplinary high-intensity weight loss program at the US Department of Veterans Affairs (VA) West Los Angeles VA Medical Center (WLAVAMC). His psychiatric history included generalized anxiety disorder, posttraumatic stress disorder (PTSD), and panic disorder, managed by the Psychiatry Service and treated with sertraline 25 mg daily, lorazepam 0.5 mg twice daily, and hydroxyzine 20 mg nightly. He had previously implemented lifestyle changes and attended MOVE! classes and nutrition coaching for 1 year but was unsuccessful in losing weight. He had also tried liraglutide 3 mg daily for weight loss but was unable to tolerate it and reported worsening medication-related anxiety.
The patient declined further weight loss pharmacotherapy and was referred to bariatric surgery. He was scheduled for a surgical sleeve gastrectomy. However, on the day he arrived at the hospital for surgery, he developed severe anxiety and had a panic attack, and it was canceled. Due to his mental health issues, he was no longer comfortable proceeding with surgery and was left without other options for obesity treatment. The veteran was extremely disappointed because the ventral hernia caused significant quality of life impairment, limited his ability to exercise, and caused him embarrassment in public settings. The hernia could not be surgically repaired until there was significant weight loss.
A bariatric endoscopy program within the Division of Gastroenterology was developed and implemented at the WLAVAMC in February 2023 in conjunction with MOVE! The patient was referred for consideration of an endoscopic weight loss procedure. He was determined to be a suitable candidate for ESG based on his BMI being > 40 and personal preference not to proceed with surgery to lose enough weight to qualify for hernia repair. The veteran underwent an endoscopy, which showed normal anatomy and gastric mucosa. ESG was performed in standard fashion (Figure).8 Three vertical lines were made using argon plasma coagulation from the incisura to 2 cm below the gastroesophageal junction along the anterior, posterior, and greater curvature of the stomach to mark the area for endoscopic suture placement. Starting at the incisura, 7 full-thickness sutures were placed to create a volume reduction plication, with preservation of the fundus. The patient did well postprocedure with no immediate or delayed AEs and was discharged home the same day.
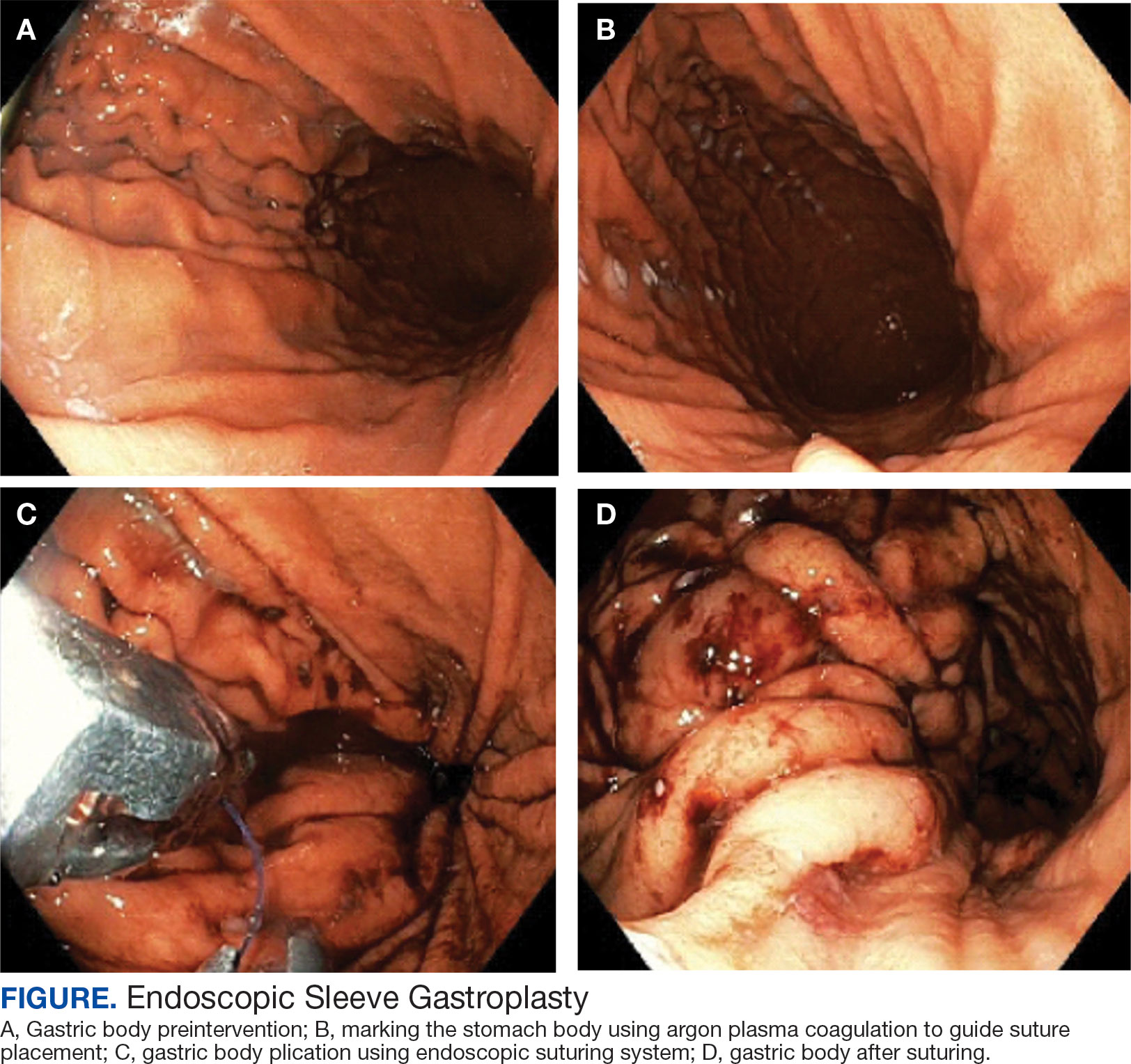
Follow-up
The veteran followed a gradual dietary advancement from a clear liquid diet to pureed and soft texture food. The patient’s weight dropped from 359 lbs preprocedure to 304 lbs 6 months postprocedure, a total body weight loss (TWBL) of 15.3%. At 12 months the veteran weighed 299 lbs (16.7% TBWL). He also had notable improvements in metabolic parameters. His systolic blood pressure decreased from ≥ 140 mm Hg to 120 to 130 mm Hg and hemoglobin A1c dropped from 7.0% to 6.3%. Remarkably, his psychiatrist noted significant improvement in his overall mental health. The veteran reported complete cessation of panic attacks since the ESG, improvements in PTSD and anxiety, and was able to discontinue lorazepam and decrease his dose of sertraline to 12.5 mg daily. He reported feeling more energetic and goal-oriented with increased clarity of thought. Perhaps the most significant outcome was that after the 55-lb weight loss at 6 months, the patient was eligible to undergo ventral hernia surgical repair, which had previously contributed to shame and social isolation. This, in turn, improved his quality of life, allowed him to start walking again, up to 8 miles daily, and to feel comfortable again going out in public settings.
DISCUSSION
Bariatric surgeries are an effective method of achieving weight loss and improving obesity-related comorbidities. However, only a small percentage of individuals with obesity are candidates for bariatric surgery. Given the dramatic increase in the prevalence of obesity, other options are needed. Specifically, within the VA, an estimated 80% of veterans are overweight or obese, but only about 500 bariatric surgeries are performed annually.9 With the need for additional weight loss therapies, VA programs are starting to offer endoscopic bariatric procedures as an alternative option. This may be a desirable choice for patients with obesity (BMI > 30), with or without associated metabolic comorbidities, who need more aggressive intervention beyond dietary and lifestyle changes and are either not interested in or not eligible for bariatric surgery or weight loss medications.
Although there is evidence that metabolic comorbidities are associated with obesity, there has been less research on obesity and mental health comorbidities such as depression and anxiety. These psychiatric conditions may even be more common among patients seeking weight loss procedures and more prominent in certain groups such as veterans, which may ultimately exclude these patients from bariatric surgery.10 Prior studies suggest that bariatric surgery can reduce the severity of depression and, to a lesser extent, anxiety symptoms at 2 years following the initial surgery; however, there is limited literature describing the impact of weight loss procedure on panic disorders.11-14 We suspect that a weight loss procedure such as ESG may have indirectly improved the veteran’s mood disorder due to the weight loss it induced, increasing the ability to exercise, quality of sleep, and participation in public settings.
This case highlights a veteran who did not tolerate weight loss medication and had severe anxiety and PTSD that prevented him from going through with bariatric surgery. He then underwent an endoscopic weight loss procedure. The ESG helped him successfully achieve significant weight loss, increase his physical activity, reduce his anxiety and panic disorder, and overall, significantly improve his quality of life. More than 1 year after the procedure, the patient has sustained improvements in his psychiatric and emotional health along with durable weight loss, maintaining > 15% of his total weight lost. Additional studies are needed to further understand the prevalence and long-term outcomes of mental health comorbidities, as well as weight loss outcomes in this group of patients who undergo endoscopic bariatric procedures.
CONCLUSIONS
We describe a case of a veteran with severe obesity and significant psychiatric comorbidities that prevented him from undergoing bariatric surgery, who underwent an ESG. This procedure led to significant weight loss, improvement of metabolic parameters, reduction in anxiety and PTSD, and enhancement of his quality of life. This case emphasizes the unique advantages of ESG and supports the expansion of endoscopic bariatric programs in the VA.
- Ritchie SA, Connell JM. The link between abdominal obesity, metabolic syndrome and cardiovascular disease. Nutr Metab Cardiovasc Dis. 2007;17(4):319-326. doi:10.1016/j.numecd.2006.07.005
- Bray GA, Kim KK, Wilding JPH; World Obesity Federation. Obesity: a chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes Rev. 2017;18(7):715-723. doi:10.1111/obr.12551
- Imbus JR, Voils CI, Funk LM. Bariatric surgery barriers: a review using andersen’s model of health services use. Surg Obes Relat Dis. 2018;14(3):404-412. doi:10.1016/j.soard.2017.11.012
- Dawes AJ, Maggard-Gibbons M, Maher AR, et al. Mental health conditions among patients seeking and undergoing bariatric surgery: a meta-analysis. JAMA. 2016;315(2):150- 163. doi:10.1001/jama.2015.18118
- Abu Dayyeh BK, Bazerbachi F, Vargas EJ, et al.. Endoscopic sleeve gastroplasty for treatment of class 1 and 2 obesity (MERIT): a prospective, multicentre, randomised trial. Lancet. 2022;400(10350):441-451. doi:10.1016/S0140-6736(22)01280-6
- Matteo MV, Bove V, Ciasca G, et al. Success predictors of endoscopic sleeve gastroplasty. Obes Surg. 2024;34(5):1496-1504. doi:10.1007/s11695-024-07109-4
- Maselli DB, Hoff AC, Kucera A, et al. Endoscopic sleeve gastroplasty in class III obesity: efficacy, safety, and durability outcomes in 404 consecutive patients. World J Gastrointest Endosc. 2023;15(6):469-479. doi:10.4253/wjge.v15.i6.469
- Kumar N, Abu Dayyeh BK, Lopez-Nava Breviere G, et al. Endoscopic sutured gastroplasty: procedure evolution from first-in-man cases through current technique. Surg Endosc. 2018;32(4):2159-2164. doi:10.1007/s00464-017-5869-2
- Maggard-Gibbons M, Shekelle PG, Girgis MD, et al. Endoscopic Bariatric Interventions versus lifestyle interventions or surgery for weight loss in patients with obesity: a systematic review and meta-analysis. Department of Veterans Affairs (US); 2022. https://www.ncbi.nlm.nih.gov/books/NBK587943/
- Maggard Gibbons MA, Maher AM, Dawes AJ, et al. Psychological clearance for bariatric surgery: a systematic review. VA-ESP project #05-2262014.
- van Hout GC, Verschure SK, van Heck GL. Psychosocial predictors of success following bariatric surgery. Obes Surg. 2005;15(4):552-560. doi:10.1381/0960892053723484
- Hudson JI, Hiripi E, Pope HG Jr, Kessler RC. The prevalence and correlates of eating disorders in the national comorbidity survey replication. Biol Psychiatry. 2007;61(3):348-358. doi:10.1016/j.biopsych.2006.03.040
- Aylward L, Lilly C, Konsor M, et al. How soon do depression and anxiety symptoms improve after bariatric surgery?. Healthcare (Basel). 2023;11(6):862. doi:10.3390/healthcare11060862
- Law S, Dong S, Zhou F, Zheng D, Wang C, Dong Z. Bariatric surgery and mental health outcomes: an umbrella review. Front Endocrinol (Lausanne). 2023;14:1283621. doi:10.3389/fendo.2023.1283621
Obesity is a growing worldwide epidemic with significant implications for individual health and public health care costs. It is also associated with several medical conditions, including diabetes, cardiovascular disease, cancer, and mental health disorders.1 Comprehensive lifestyle intervention is a first-line therapy for obesity consisting of dietary and exercise interventions. Despite initial success, long-term results and durability of weight loss with lifestyle modifications are limited. 2 Bariatric surgery, including sleeve gastrectomy and gastric bypass surgery, is a more invasive approach that is highly effective in weight loss. However, these operations are not reversible, and patients may not be eligible for or may not desire surgery. Overall, bariatric surgery is widely underutilized, with < 1% of eligible patients ultimately undergoing surgery.3,4
Endoscopic bariatric therapies are increasingly popular procedures that address the need for additional treatments for obesity among individuals who have not had success with lifestyle changes and are not surgical candidates. The most common procedure is the endoscopic sleeve gastroplasty (ESG), which applies full-thickness sutures in the stomach to reduce gastric volume, delay gastric emptying, and limit food intake while keeping the fundus intact compared with sleeve gastrectomy. This procedure is typically considered in patients with body mass index (BMI) ≥ 30, who do not qualify for or do not want traditional bariatric surgery. The literature supports robust outcomes after ESG, with studies demonstrating significant and sustained total body weight loss of up to 14% to 16% at 5 years and significant improvement in ≥ 1 metabolic comorbidities in 80% of patients.5,6 ESG adverse events (AEs) include abdominal pain, nausea, and vomiting that are typically self-limited to 1 week. Rarer but more serious AEs include bleeding, perforation, or infection, and occur in 2% of cases based on large trial data.5,7
Although the weight loss benefits of ESG are well established, to date, there are limited data on the effects of endoscopic bariatric therapies like ESG on mental health conditions. Here, we describe a case of a veteran with a history of mental health disorders that prevented him from completing bariatric surgery. The patient underwent ESG and had a successful clinical course.
CASE PRESENTATION
A 59-year-old male veteran with a medical history of class III obesity (42.4 BMI), obstructive sleep apnea, hypothyroidism, hypertension, type 2 diabetes mellitus, and a large ventral hernia was referred to the MOVE! (Management of Overweight/ Obese Veterans Everywhere!) multidisciplinary high-intensity weight loss program at the US Department of Veterans Affairs (VA) West Los Angeles VA Medical Center (WLAVAMC). His psychiatric history included generalized anxiety disorder, posttraumatic stress disorder (PTSD), and panic disorder, managed by the Psychiatry Service and treated with sertraline 25 mg daily, lorazepam 0.5 mg twice daily, and hydroxyzine 20 mg nightly. He had previously implemented lifestyle changes and attended MOVE! classes and nutrition coaching for 1 year but was unsuccessful in losing weight. He had also tried liraglutide 3 mg daily for weight loss but was unable to tolerate it and reported worsening medication-related anxiety.
The patient declined further weight loss pharmacotherapy and was referred to bariatric surgery. He was scheduled for a surgical sleeve gastrectomy. However, on the day he arrived at the hospital for surgery, he developed severe anxiety and had a panic attack, and it was canceled. Due to his mental health issues, he was no longer comfortable proceeding with surgery and was left without other options for obesity treatment. The veteran was extremely disappointed because the ventral hernia caused significant quality of life impairment, limited his ability to exercise, and caused him embarrassment in public settings. The hernia could not be surgically repaired until there was significant weight loss.
A bariatric endoscopy program within the Division of Gastroenterology was developed and implemented at the WLAVAMC in February 2023 in conjunction with MOVE! The patient was referred for consideration of an endoscopic weight loss procedure. He was determined to be a suitable candidate for ESG based on his BMI being > 40 and personal preference not to proceed with surgery to lose enough weight to qualify for hernia repair. The veteran underwent an endoscopy, which showed normal anatomy and gastric mucosa. ESG was performed in standard fashion (Figure).8 Three vertical lines were made using argon plasma coagulation from the incisura to 2 cm below the gastroesophageal junction along the anterior, posterior, and greater curvature of the stomach to mark the area for endoscopic suture placement. Starting at the incisura, 7 full-thickness sutures were placed to create a volume reduction plication, with preservation of the fundus. The patient did well postprocedure with no immediate or delayed AEs and was discharged home the same day.
Follow-up
The veteran followed a gradual dietary advancement from a clear liquid diet to pureed and soft texture food. The patient’s weight dropped from 359 lbs preprocedure to 304 lbs 6 months postprocedure, a total body weight loss (TWBL) of 15.3%. At 12 months the veteran weighed 299 lbs (16.7% TBWL). He also had notable improvements in metabolic parameters. His systolic blood pressure decreased from ≥ 140 mm Hg to 120 to 130 mm Hg and hemoglobin A1c dropped from 7.0% to 6.3%. Remarkably, his psychiatrist noted significant improvement in his overall mental health. The veteran reported complete cessation of panic attacks since the ESG, improvements in PTSD and anxiety, and was able to discontinue lorazepam and decrease his dose of sertraline to 12.5 mg daily. He reported feeling more energetic and goal-oriented with increased clarity of thought. Perhaps the most significant outcome was that after the 55-lb weight loss at 6 months, the patient was eligible to undergo ventral hernia surgical repair, which had previously contributed to shame and social isolation. This, in turn, improved his quality of life, allowed him to start walking again, up to 8 miles daily, and to feel comfortable again going out in public settings.
DISCUSSION
Bariatric surgeries are an effective method of achieving weight loss and improving obesity-related comorbidities. However, only a small percentage of individuals with obesity are candidates for bariatric surgery. Given the dramatic increase in the prevalence of obesity, other options are needed. Specifically, within the VA, an estimated 80% of veterans are overweight or obese, but only about 500 bariatric surgeries are performed annually.9 With the need for additional weight loss therapies, VA programs are starting to offer endoscopic bariatric procedures as an alternative option. This may be a desirable choice for patients with obesity (BMI > 30), with or without associated metabolic comorbidities, who need more aggressive intervention beyond dietary and lifestyle changes and are either not interested in or not eligible for bariatric surgery or weight loss medications.
Although there is evidence that metabolic comorbidities are associated with obesity, there has been less research on obesity and mental health comorbidities such as depression and anxiety. These psychiatric conditions may even be more common among patients seeking weight loss procedures and more prominent in certain groups such as veterans, which may ultimately exclude these patients from bariatric surgery.10 Prior studies suggest that bariatric surgery can reduce the severity of depression and, to a lesser extent, anxiety symptoms at 2 years following the initial surgery; however, there is limited literature describing the impact of weight loss procedure on panic disorders.11-14 We suspect that a weight loss procedure such as ESG may have indirectly improved the veteran’s mood disorder due to the weight loss it induced, increasing the ability to exercise, quality of sleep, and participation in public settings.
This case highlights a veteran who did not tolerate weight loss medication and had severe anxiety and PTSD that prevented him from going through with bariatric surgery. He then underwent an endoscopic weight loss procedure. The ESG helped him successfully achieve significant weight loss, increase his physical activity, reduce his anxiety and panic disorder, and overall, significantly improve his quality of life. More than 1 year after the procedure, the patient has sustained improvements in his psychiatric and emotional health along with durable weight loss, maintaining > 15% of his total weight lost. Additional studies are needed to further understand the prevalence and long-term outcomes of mental health comorbidities, as well as weight loss outcomes in this group of patients who undergo endoscopic bariatric procedures.
CONCLUSIONS
We describe a case of a veteran with severe obesity and significant psychiatric comorbidities that prevented him from undergoing bariatric surgery, who underwent an ESG. This procedure led to significant weight loss, improvement of metabolic parameters, reduction in anxiety and PTSD, and enhancement of his quality of life. This case emphasizes the unique advantages of ESG and supports the expansion of endoscopic bariatric programs in the VA.
Obesity is a growing worldwide epidemic with significant implications for individual health and public health care costs. It is also associated with several medical conditions, including diabetes, cardiovascular disease, cancer, and mental health disorders.1 Comprehensive lifestyle intervention is a first-line therapy for obesity consisting of dietary and exercise interventions. Despite initial success, long-term results and durability of weight loss with lifestyle modifications are limited. 2 Bariatric surgery, including sleeve gastrectomy and gastric bypass surgery, is a more invasive approach that is highly effective in weight loss. However, these operations are not reversible, and patients may not be eligible for or may not desire surgery. Overall, bariatric surgery is widely underutilized, with < 1% of eligible patients ultimately undergoing surgery.3,4
Endoscopic bariatric therapies are increasingly popular procedures that address the need for additional treatments for obesity among individuals who have not had success with lifestyle changes and are not surgical candidates. The most common procedure is the endoscopic sleeve gastroplasty (ESG), which applies full-thickness sutures in the stomach to reduce gastric volume, delay gastric emptying, and limit food intake while keeping the fundus intact compared with sleeve gastrectomy. This procedure is typically considered in patients with body mass index (BMI) ≥ 30, who do not qualify for or do not want traditional bariatric surgery. The literature supports robust outcomes after ESG, with studies demonstrating significant and sustained total body weight loss of up to 14% to 16% at 5 years and significant improvement in ≥ 1 metabolic comorbidities in 80% of patients.5,6 ESG adverse events (AEs) include abdominal pain, nausea, and vomiting that are typically self-limited to 1 week. Rarer but more serious AEs include bleeding, perforation, or infection, and occur in 2% of cases based on large trial data.5,7
Although the weight loss benefits of ESG are well established, to date, there are limited data on the effects of endoscopic bariatric therapies like ESG on mental health conditions. Here, we describe a case of a veteran with a history of mental health disorders that prevented him from completing bariatric surgery. The patient underwent ESG and had a successful clinical course.
CASE PRESENTATION
A 59-year-old male veteran with a medical history of class III obesity (42.4 BMI), obstructive sleep apnea, hypothyroidism, hypertension, type 2 diabetes mellitus, and a large ventral hernia was referred to the MOVE! (Management of Overweight/ Obese Veterans Everywhere!) multidisciplinary high-intensity weight loss program at the US Department of Veterans Affairs (VA) West Los Angeles VA Medical Center (WLAVAMC). His psychiatric history included generalized anxiety disorder, posttraumatic stress disorder (PTSD), and panic disorder, managed by the Psychiatry Service and treated with sertraline 25 mg daily, lorazepam 0.5 mg twice daily, and hydroxyzine 20 mg nightly. He had previously implemented lifestyle changes and attended MOVE! classes and nutrition coaching for 1 year but was unsuccessful in losing weight. He had also tried liraglutide 3 mg daily for weight loss but was unable to tolerate it and reported worsening medication-related anxiety.
The patient declined further weight loss pharmacotherapy and was referred to bariatric surgery. He was scheduled for a surgical sleeve gastrectomy. However, on the day he arrived at the hospital for surgery, he developed severe anxiety and had a panic attack, and it was canceled. Due to his mental health issues, he was no longer comfortable proceeding with surgery and was left without other options for obesity treatment. The veteran was extremely disappointed because the ventral hernia caused significant quality of life impairment, limited his ability to exercise, and caused him embarrassment in public settings. The hernia could not be surgically repaired until there was significant weight loss.
A bariatric endoscopy program within the Division of Gastroenterology was developed and implemented at the WLAVAMC in February 2023 in conjunction with MOVE! The patient was referred for consideration of an endoscopic weight loss procedure. He was determined to be a suitable candidate for ESG based on his BMI being > 40 and personal preference not to proceed with surgery to lose enough weight to qualify for hernia repair. The veteran underwent an endoscopy, which showed normal anatomy and gastric mucosa. ESG was performed in standard fashion (Figure).8 Three vertical lines were made using argon plasma coagulation from the incisura to 2 cm below the gastroesophageal junction along the anterior, posterior, and greater curvature of the stomach to mark the area for endoscopic suture placement. Starting at the incisura, 7 full-thickness sutures were placed to create a volume reduction plication, with preservation of the fundus. The patient did well postprocedure with no immediate or delayed AEs and was discharged home the same day.
Follow-up
The veteran followed a gradual dietary advancement from a clear liquid diet to pureed and soft texture food. The patient’s weight dropped from 359 lbs preprocedure to 304 lbs 6 months postprocedure, a total body weight loss (TWBL) of 15.3%. At 12 months the veteran weighed 299 lbs (16.7% TBWL). He also had notable improvements in metabolic parameters. His systolic blood pressure decreased from ≥ 140 mm Hg to 120 to 130 mm Hg and hemoglobin A1c dropped from 7.0% to 6.3%. Remarkably, his psychiatrist noted significant improvement in his overall mental health. The veteran reported complete cessation of panic attacks since the ESG, improvements in PTSD and anxiety, and was able to discontinue lorazepam and decrease his dose of sertraline to 12.5 mg daily. He reported feeling more energetic and goal-oriented with increased clarity of thought. Perhaps the most significant outcome was that after the 55-lb weight loss at 6 months, the patient was eligible to undergo ventral hernia surgical repair, which had previously contributed to shame and social isolation. This, in turn, improved his quality of life, allowed him to start walking again, up to 8 miles daily, and to feel comfortable again going out in public settings.
DISCUSSION
Bariatric surgeries are an effective method of achieving weight loss and improving obesity-related comorbidities. However, only a small percentage of individuals with obesity are candidates for bariatric surgery. Given the dramatic increase in the prevalence of obesity, other options are needed. Specifically, within the VA, an estimated 80% of veterans are overweight or obese, but only about 500 bariatric surgeries are performed annually.9 With the need for additional weight loss therapies, VA programs are starting to offer endoscopic bariatric procedures as an alternative option. This may be a desirable choice for patients with obesity (BMI > 30), with or without associated metabolic comorbidities, who need more aggressive intervention beyond dietary and lifestyle changes and are either not interested in or not eligible for bariatric surgery or weight loss medications.
Although there is evidence that metabolic comorbidities are associated with obesity, there has been less research on obesity and mental health comorbidities such as depression and anxiety. These psychiatric conditions may even be more common among patients seeking weight loss procedures and more prominent in certain groups such as veterans, which may ultimately exclude these patients from bariatric surgery.10 Prior studies suggest that bariatric surgery can reduce the severity of depression and, to a lesser extent, anxiety symptoms at 2 years following the initial surgery; however, there is limited literature describing the impact of weight loss procedure on panic disorders.11-14 We suspect that a weight loss procedure such as ESG may have indirectly improved the veteran’s mood disorder due to the weight loss it induced, increasing the ability to exercise, quality of sleep, and participation in public settings.
This case highlights a veteran who did not tolerate weight loss medication and had severe anxiety and PTSD that prevented him from going through with bariatric surgery. He then underwent an endoscopic weight loss procedure. The ESG helped him successfully achieve significant weight loss, increase his physical activity, reduce his anxiety and panic disorder, and overall, significantly improve his quality of life. More than 1 year after the procedure, the patient has sustained improvements in his psychiatric and emotional health along with durable weight loss, maintaining > 15% of his total weight lost. Additional studies are needed to further understand the prevalence and long-term outcomes of mental health comorbidities, as well as weight loss outcomes in this group of patients who undergo endoscopic bariatric procedures.
CONCLUSIONS
We describe a case of a veteran with severe obesity and significant psychiatric comorbidities that prevented him from undergoing bariatric surgery, who underwent an ESG. This procedure led to significant weight loss, improvement of metabolic parameters, reduction in anxiety and PTSD, and enhancement of his quality of life. This case emphasizes the unique advantages of ESG and supports the expansion of endoscopic bariatric programs in the VA.
- Ritchie SA, Connell JM. The link between abdominal obesity, metabolic syndrome and cardiovascular disease. Nutr Metab Cardiovasc Dis. 2007;17(4):319-326. doi:10.1016/j.numecd.2006.07.005
- Bray GA, Kim KK, Wilding JPH; World Obesity Federation. Obesity: a chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes Rev. 2017;18(7):715-723. doi:10.1111/obr.12551
- Imbus JR, Voils CI, Funk LM. Bariatric surgery barriers: a review using andersen’s model of health services use. Surg Obes Relat Dis. 2018;14(3):404-412. doi:10.1016/j.soard.2017.11.012
- Dawes AJ, Maggard-Gibbons M, Maher AR, et al. Mental health conditions among patients seeking and undergoing bariatric surgery: a meta-analysis. JAMA. 2016;315(2):150- 163. doi:10.1001/jama.2015.18118
- Abu Dayyeh BK, Bazerbachi F, Vargas EJ, et al.. Endoscopic sleeve gastroplasty for treatment of class 1 and 2 obesity (MERIT): a prospective, multicentre, randomised trial. Lancet. 2022;400(10350):441-451. doi:10.1016/S0140-6736(22)01280-6
- Matteo MV, Bove V, Ciasca G, et al. Success predictors of endoscopic sleeve gastroplasty. Obes Surg. 2024;34(5):1496-1504. doi:10.1007/s11695-024-07109-4
- Maselli DB, Hoff AC, Kucera A, et al. Endoscopic sleeve gastroplasty in class III obesity: efficacy, safety, and durability outcomes in 404 consecutive patients. World J Gastrointest Endosc. 2023;15(6):469-479. doi:10.4253/wjge.v15.i6.469
- Kumar N, Abu Dayyeh BK, Lopez-Nava Breviere G, et al. Endoscopic sutured gastroplasty: procedure evolution from first-in-man cases through current technique. Surg Endosc. 2018;32(4):2159-2164. doi:10.1007/s00464-017-5869-2
- Maggard-Gibbons M, Shekelle PG, Girgis MD, et al. Endoscopic Bariatric Interventions versus lifestyle interventions or surgery for weight loss in patients with obesity: a systematic review and meta-analysis. Department of Veterans Affairs (US); 2022. https://www.ncbi.nlm.nih.gov/books/NBK587943/
- Maggard Gibbons MA, Maher AM, Dawes AJ, et al. Psychological clearance for bariatric surgery: a systematic review. VA-ESP project #05-2262014.
- van Hout GC, Verschure SK, van Heck GL. Psychosocial predictors of success following bariatric surgery. Obes Surg. 2005;15(4):552-560. doi:10.1381/0960892053723484
- Hudson JI, Hiripi E, Pope HG Jr, Kessler RC. The prevalence and correlates of eating disorders in the national comorbidity survey replication. Biol Psychiatry. 2007;61(3):348-358. doi:10.1016/j.biopsych.2006.03.040
- Aylward L, Lilly C, Konsor M, et al. How soon do depression and anxiety symptoms improve after bariatric surgery?. Healthcare (Basel). 2023;11(6):862. doi:10.3390/healthcare11060862
- Law S, Dong S, Zhou F, Zheng D, Wang C, Dong Z. Bariatric surgery and mental health outcomes: an umbrella review. Front Endocrinol (Lausanne). 2023;14:1283621. doi:10.3389/fendo.2023.1283621
- Ritchie SA, Connell JM. The link between abdominal obesity, metabolic syndrome and cardiovascular disease. Nutr Metab Cardiovasc Dis. 2007;17(4):319-326. doi:10.1016/j.numecd.2006.07.005
- Bray GA, Kim KK, Wilding JPH; World Obesity Federation. Obesity: a chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes Rev. 2017;18(7):715-723. doi:10.1111/obr.12551
- Imbus JR, Voils CI, Funk LM. Bariatric surgery barriers: a review using andersen’s model of health services use. Surg Obes Relat Dis. 2018;14(3):404-412. doi:10.1016/j.soard.2017.11.012
- Dawes AJ, Maggard-Gibbons M, Maher AR, et al. Mental health conditions among patients seeking and undergoing bariatric surgery: a meta-analysis. JAMA. 2016;315(2):150- 163. doi:10.1001/jama.2015.18118
- Abu Dayyeh BK, Bazerbachi F, Vargas EJ, et al.. Endoscopic sleeve gastroplasty for treatment of class 1 and 2 obesity (MERIT): a prospective, multicentre, randomised trial. Lancet. 2022;400(10350):441-451. doi:10.1016/S0140-6736(22)01280-6
- Matteo MV, Bove V, Ciasca G, et al. Success predictors of endoscopic sleeve gastroplasty. Obes Surg. 2024;34(5):1496-1504. doi:10.1007/s11695-024-07109-4
- Maselli DB, Hoff AC, Kucera A, et al. Endoscopic sleeve gastroplasty in class III obesity: efficacy, safety, and durability outcomes in 404 consecutive patients. World J Gastrointest Endosc. 2023;15(6):469-479. doi:10.4253/wjge.v15.i6.469
- Kumar N, Abu Dayyeh BK, Lopez-Nava Breviere G, et al. Endoscopic sutured gastroplasty: procedure evolution from first-in-man cases through current technique. Surg Endosc. 2018;32(4):2159-2164. doi:10.1007/s00464-017-5869-2
- Maggard-Gibbons M, Shekelle PG, Girgis MD, et al. Endoscopic Bariatric Interventions versus lifestyle interventions or surgery for weight loss in patients with obesity: a systematic review and meta-analysis. Department of Veterans Affairs (US); 2022. https://www.ncbi.nlm.nih.gov/books/NBK587943/
- Maggard Gibbons MA, Maher AM, Dawes AJ, et al. Psychological clearance for bariatric surgery: a systematic review. VA-ESP project #05-2262014.
- van Hout GC, Verschure SK, van Heck GL. Psychosocial predictors of success following bariatric surgery. Obes Surg. 2005;15(4):552-560. doi:10.1381/0960892053723484
- Hudson JI, Hiripi E, Pope HG Jr, Kessler RC. The prevalence and correlates of eating disorders in the national comorbidity survey replication. Biol Psychiatry. 2007;61(3):348-358. doi:10.1016/j.biopsych.2006.03.040
- Aylward L, Lilly C, Konsor M, et al. How soon do depression and anxiety symptoms improve after bariatric surgery?. Healthcare (Basel). 2023;11(6):862. doi:10.3390/healthcare11060862
- Law S, Dong S, Zhou F, Zheng D, Wang C, Dong Z. Bariatric surgery and mental health outcomes: an umbrella review. Front Endocrinol (Lausanne). 2023;14:1283621. doi:10.3389/fendo.2023.1283621
Endoscopic Sleeve Gastroplasty is an Effective Treatment for Obesity in a Veteran With Metabolic and Psychiatric Comorbidities
Endoscopic Sleeve Gastroplasty is an Effective Treatment for Obesity in a Veteran With Metabolic and Psychiatric Comorbidities
Anti-Tumor Necrosis Factor Treatment for Glomerulopathy: Case Report and Review of Literature
Podocytes are terminally differentiated, highly specialized cells located in juxtaposition to the basement membrane over the abluminal surfaces of endothelial cells within the glomerular tuft. This triad structure is the site of the filtration barrier, which forms highly delicate and tightly regulated architecture to carry out the ultrafiltration function of the kidney.1 The filtration barrier is characterized by foot processes that are connected by specialized junctions called slit diaphragms.
Insults to components of the filtration barrier can initiate cascading events and perpetuate structural alterations that may eventually result in sclerotic changes.2 Common causes among children include minimal change disease (MCD) with the collapse of foot processes resulting in proteinuria, Alport syndrome due to mutation of collagen fibers within the basement membrane leading to hematuria and proteinuria, immune complex mediated nephropathy following common infections or autoimmune diseases, and focal segmental glomerulosclerosis (FSGS) that can show variable histopathology toward eventual glomerular scarring.3,4 These children often clinically have minimal, if any, signs of systemic inflammation.3-5 This has been a limiting factor for the commitment to immunomodulatory treatment, except for steroids for the treatment of MCD.6 Although prolonged steroid treatment may be efficacious, adverse effects are significant in a growing child. Alternative treatments, such as tacrolimus and rituximab have been suggested as second-line steroid-sparing agents.7,8 Not uncommonly, however, these cases are managed by supportive measures only during the progression of the natural course of the disease, which may eventually lead to renal failure, requiring transplant for survival.8,9
This case report highlights a child with a variant of uncertain significance (VUS) in genes involved in Alport syndrome and FSGS who developed an abrupt onset of proteinuria and hematuria after a respiratory illness. To our knowledge, he represents the youngest case demonstrating the benefit of targeted treatment against tumor necrosis factor-α (TNF-α) for glomerulopathy using biologic response modifiers.
Case Description
This is currently a 7-year-old male patient who was born at 39 weeks gestation to gravida 3 para 3 following induced labor due to elevated maternal blood pressure. During the first 2 years of life, his growth and development were normal and his immunizations were up to date. The patient's medical history included upper respiratory tract infections (URIs), respiratory syncytial virus, as well as 3 bouts of pneumonia and multiple otitis media that resulted in 18 rounds of antibiotics. The child was also allergic to nuts and milk protein. The patient’s parents are of Northern European and Native American descent. There is no known family history of eye, ear, or kidney diseases.
Renal concerns were first noted at the age of 2 years and 6 months when he presented to an emergency department in Fall 2019 (week 0) for several weeks of intermittent dark-colored urine. His mother reported that the discoloration recently progressed in intensity to cola-colored, along with the onset of persistent vomiting without any fever or diarrhea. On physical examination, the patient had normal vitals: weight 14.8 kg (68th percentile), height 91 cm (24th percentile), and body surface area 0.6 m2. There was no edema, rash, or lymphadenopathy, but he appeared pale.
The patient’s initial laboratory results included: complete blood count with white blood cells (WBC) 10 x 103/L (reference range, 4.5-13.5 x 103/L); differential lymphocytes 69%; neutrophils 21%; hemoglobin 10 g/dL (reference range, 12-16 g/dL); hematocrit, 30%; (reference range, 37%-45%); platelets 437 103/L (reference range, 150-450 x 103/L); serum creatinine 0.46 mg/dL (reference range, 0.5-0.9 mg/dL); and albumin 3.1 g/dL (reference range, 3.5-5.2 g/dL). Serum electrolyte levels and liver enzymes were normal. A urine analysis revealed 3+ protein and 3+ blood with dysmorphic red blood cells (RBC) and RBC casts without WBC. The patient's spot urine protein-to-creatinine ratio was 4.3 and his renal ultrasound was normal. The patient was referred to Nephrology.
During the next 2 weeks, his protein-to-creatinine ratio progressed to 5.9 and serum albumin fell to 2.7 g/dL. His urine remained red colored, and a microscopic examination with RBC > 500 and WBC up to 10 on a high powered field. His workup was negative for antinuclear antibodies, antineutrophil cytoplasmic antibody, antistreptolysin-O (ASO) and anti-DNase B. Serum C3 was low at 81 mg/dL (reference range, 90-180 mg/dL), C4 was 13.3 mg/dL (reference range, 10-40 mg/dL), and immunoglobulin G was low at 452 mg/dL (reference range 719-1475 mg/dL). A baseline audiology test revealed normal hearing.
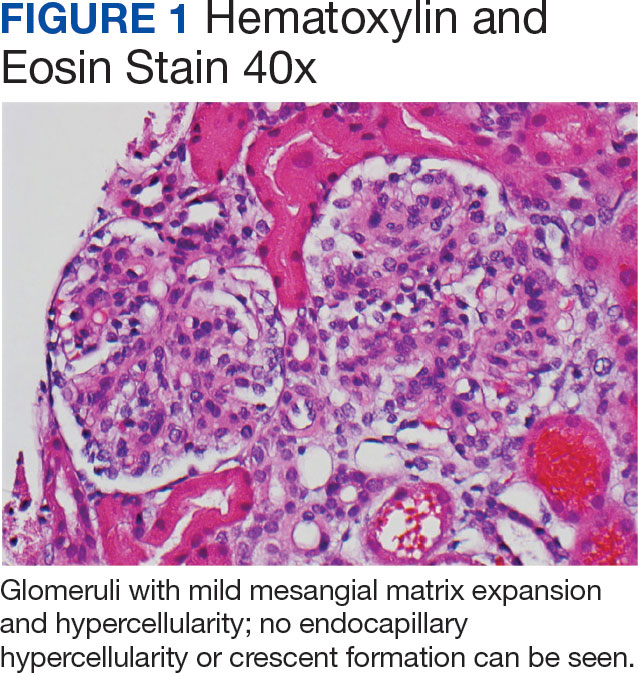
Percutaneous renal biopsy yielded about 12 glomeruli, all exhibiting mild mesangial matrix expansion and hypercellularity (Figure 1). One glomerulus had prominent parietal epithelial cells without endocapillary hypercellularity or crescent formation. There was no interstitial fibrosis or tubular atrophy. Immunofluorescence studies showed no evidence of immune complex deposition with negative staining for immunoglobulin heavy and light chains, C3 and C1q. Staining for α 2 and α 5 units of collagen was normal. Electron microscopy showed patchy areas of severe basement membrane thinning with frequent foci of mild to moderate lamina densa splitting and associated visceral epithelial cell foot process effacement (Figure 2).
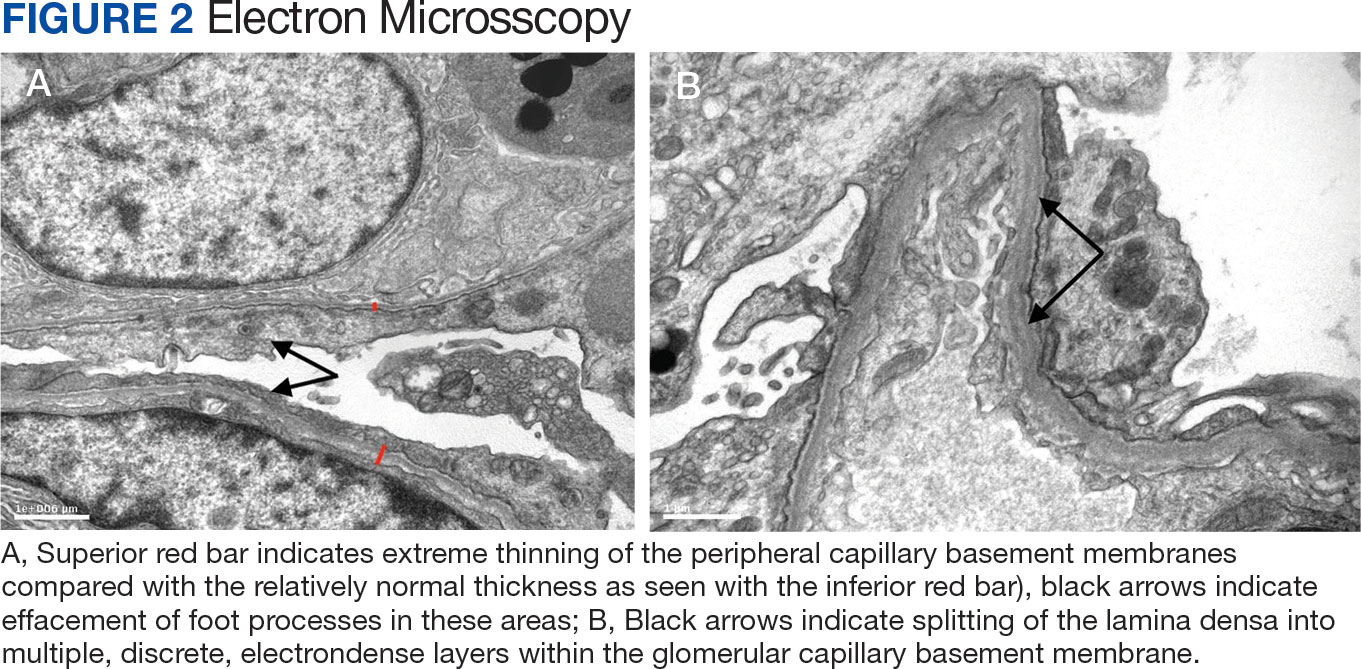
These were reported as concerning findings for possible Alport syndrome by 3 independent pathology teams. The genetic testing was submitted at a commercial laboratory to screen 17 mutations, including COL4A3, COL4A4, and COL4A5. Results showed the presence of a heterozygous VUS in the COL4A4 gene (c.1055C > T; p.Pro352Leu; dbSNP ID: rs371717486; PolyPhen-2: Probably Damaging; SIFT: Deleterious) as well as the presence of a heterozygous VUS in TRPC6 gene (c2463A>T; p.Lys821Asn; dbSNP ID: rs199948731; PolyPhen-2: Benign; SIFT: Tolerated). Further genetic investigation by whole exome sequencing on approximately 20,000 genes through MNG Laboratories showed a new heterozygous VUS in the OSGEP gene [c.328T>C; p.Cys110Arg]. Additional studies ruled out mitochondrial disease, CoQ10 deficiency, and metabolic disorders upon normal findings for mitochondrial DNA, urine amino acids, plasma acylcarnitine profile, orotic acid, ammonia, and homocysteine levels.
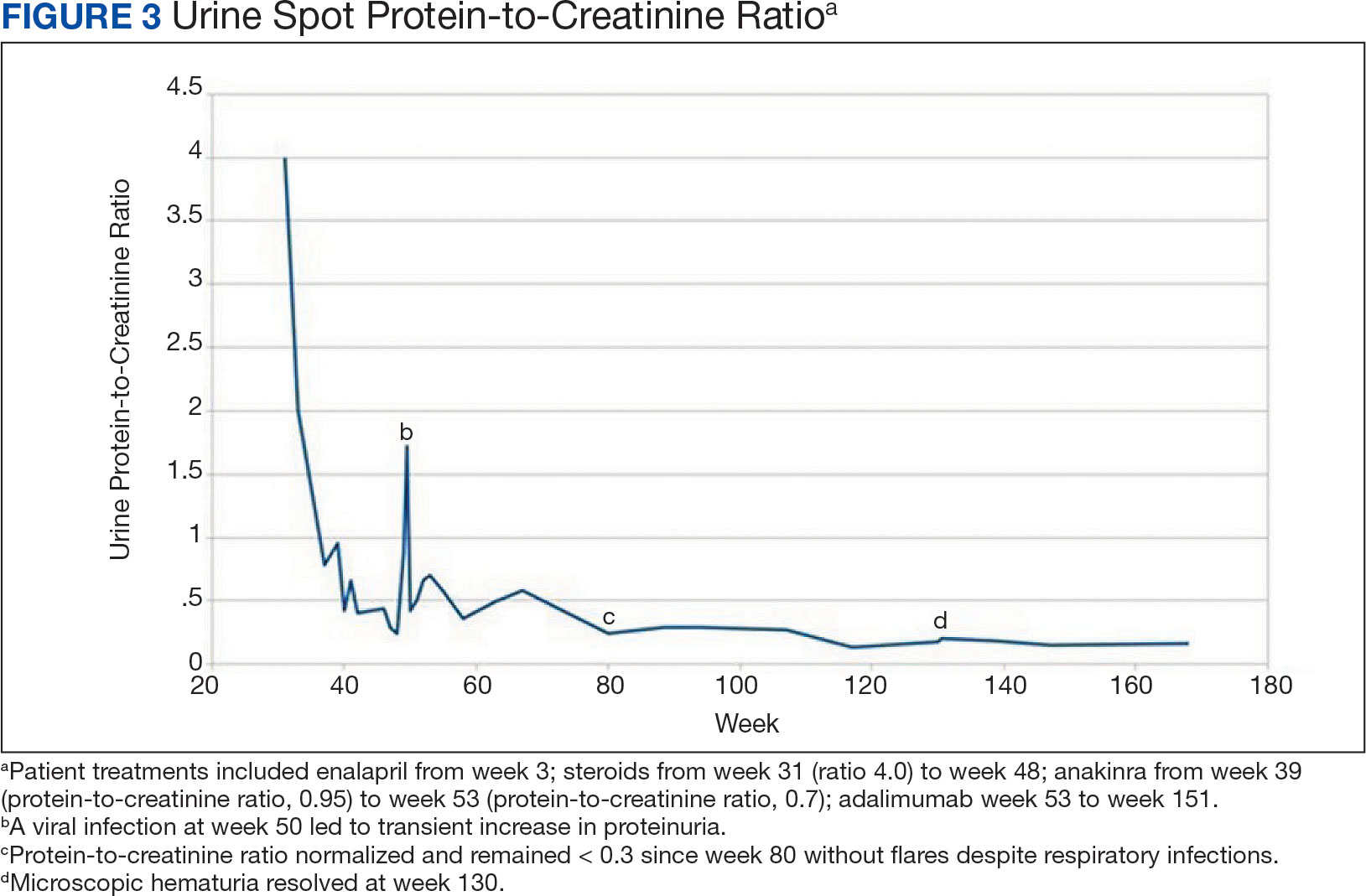
Figure 3 summarizes the patient’s treatment response during 170 weeks of follow-up (Fall 2019 to Summer 2023). The patient was started on enalapril 0.6 mg/kg daily at week 3, which continued throughout treatment. Following a rheumatology consult at week 30, the patient was started on prednisolone 3 mg/mL to assess the role of inflammation through the treatment response. An initial dose of 2 mg/kg daily (9 mL) for 1 month was followed by every other day treatment that was tapered off by week 48. To control mild but noticeably increasing proteinuria in the interim, subcutaneous anakinra 50 mg (3 mg/kg daily) was added as a steroid
DISCUSSION
This case describes a child with rapidly progressive proteinuria and hematuria following a URI who was found to have VUS mutations in 3 different genes associated with chronic kidney disease. Serology tests on the patient were negative for streptococcal antibodies and antinuclear antibodies, ruling out poststreptococcal glomerulonephritis, or systemic lupus erythematosus. His renal biopsy findings were concerning for altered podocytes, mesangial cells, and basement membrane without inflammatory infiltrate, immune complex, complements, immunoglobulin A, or vasculopathy. His blood inflammatory markers, erythrocyte sedimentation rate, C-reactive protein, and ferritin were normal when his care team initiated daily steroids.
Overall, the patient’s clinical presentation and histopathology findings were suggestive of Alport syndrome or thin basement membrane nephropathy with a high potential to progress into FSGS.10-12 Alport syndrome affects 1 in 5000 to 10,000 children annually due to S-linked inheritance of COL4A5, or autosomal recessive inheritance of COL4A3 or COL4A4 genes. It presents with hematuria and hearing loss.10 Our patient had a single copy COL4A4 gene mutation that was classified as VUS. He also had 2 additional VUS affecting the TRPC6 and OSGEP genes. TRPC6 gene mutation can be associated with FSGS through autosomal dominant inheritance. Both COL4A4 and TRPC6 gene mutations were paternally inherited. Although the patient’s father not having renal disease argues against the clinical significance of these findings, there is literature on the potential role of heterozygous COL4A4 variant mimicking thin basement membrane nephropathy that can lead to renal impairment upon copresence of superimposed conditions.13 The patient’s rapidly progressing hematuria and changes in the basement membrane were worrisome for emerging FSGS. Furthermore, VUS of TRPC6 has been reported in late onset autosomal dominant FSGS and can be associated with early onset steroid-resistant nephrotic syndrome (NS) in children.14 This concern was voiced by 3 nephrology consultants during the initial evaluation, leading to the consensus that steroid treatment for podocytopathy would not alter the patient’s long-term outcomes (ie, progression to FSGS).
Immunomodulation
Our rationale for immunomodulatory treatment was based on the abrupt onset of renal concerns following a URI, suggesting the importance of an inflammatory trigger causing altered homeostasis in a genetically susceptible host. Preclinical models show that microbial products such as lipopolysaccharides can lead to podocytopathy by several mechanisms through activation of toll-like receptor signaling. It can directly cause apoptosis by downregulation of the intracellular Akt survival pathway.15 Lipopolysaccharide can also activate the NF-αB pathway and upregulate the production of interleukin-1 (IL-1) and TNF-α in mesangial cells.16,17
Both cytokines can promote mesangial cell proliferation.18 Through autocrine and paracrine mechanisms, proinflammatory cytokines can further perpetuate somatic tissue changes and contribute to the development of podocytopathy. For instance, TNF-α can promote podocyte injury and proteinuria by downregulation of the slit diaphragm protein expression (ie, nephrin, ezrin, or podocin), and disruption of podocyte cytoskeleton.19,20 TNF-α promotes the influx and activation of macrophages and inflammatory cells. It is actively involved in chronic alterations within the glomeruli by the upregulation of matrix metalloproteases by integrins, as well as activation of myofibroblast progenitors and extracellular matrix deposition in crosstalk with transforming growth factor and other key mediators.17,21,22
For the patient described in this case report, initial improvement on steroids encouraged the pursuit of additional treatment to downregulate inflammatory pathways within the glomerular milieu. However, within the COVID-19 environment, escalating the patient’s treatment using traditional immunomodulators (ie, calcineurin inhibitors or mycophenolate mofetil) was not favored due to the risk of infection. Initially, anakinra, a recombinant IL-1 receptor antagonist, was preferred as a steroid-sparing agent for its short life and safety profile during the pandemic. At first, the patient responded well to anakinra and was allowed a steroid wean when the dose was titrated up to 6 mg/kg daily. However, anakinra did not prevent the escalation of proteinuria following a URI. After the treatment was changed to adalimumab, a fully humanized monoclonal antibody to TNF-α, the patient continued to improve and reach full remission despite experiencing a cold and the flu in the following months.
Literature Review
There is a paucity of literature on applications of biological response modifiers for idiopathic NS and FSGS.23,24 Angeletti and colleagues reported that 3 patients with severe long-standing FSGS benefited from anakinra 4 mg/kg daily to reduce proteinuria and improve kidney function. All the patients had positive C3 staining in renal biopsy and treatment response, which supported the role of C3a in inducing podocyte injury through upregulated expression of IL-1 and IL-1R.23 Trachtman and colleagues reported on the phase II FONT trial that included 14 of 21 patients aged < 18 years with advanced FSGS who were treated with adalimumab 24 mg/m2, or ≤ 40 mg every other week.24 Although, during a 6-month period, none of the 7 patients met the endpoint of reduced proteinuria by ≥ 50%, and the authors suggested that careful patient selection may improve the treatment response in future trials.24
A recent study involving transcriptomics on renal tissue samples combined with available pathology (fibrosis), urinary markers, and clinical characteristics on 285 patients with MCD or FSGS from 3 different continents identified 3 distinct clusters. Patients with evidence of activated kidney TNF pathway (n = 72, aged > 18 years) were found to have poor clinical outcomes.25 The study identified 2 urine markers associated with the TNF pathway (ie, tissue inhibitor of metalloproteinases-1 and monocyte chemoattractant protein-1), which aligns with the preclinical findings previously mentioned.25
Conclusions
The patient’s condition in this case illustrates the complex nature of biologically predetermined cascading events in the emergence of glomerular disease upon environmental triggers under the influence of genetic factors.
Chronic kidney disease affects 7.7% of veterans annually, illustrating the need for new therapeutics.26 Based on our experience and literature review, upregulation of TNF-α is a root cause of glomerulopathy; further studies are warranted to evaluate the efficacy of anti-TNF biologic response modifiers for the treatment of these patients. Long-term postmarketing safety profile and steroid-sparing properties of adalimumab should allow inclusion of pediatric cases in future trials. Results may also contribute to identifying new predictive biomarkers related to the basement membrane when combined with precision nephrology to further advance patient selection and targeted treatment.25,27
Acknowledgments
The authors thank the patient’s mother for providing consent to allow publication of this case report.
1. Arif E, Nihalani D. Glomerular filtration barrier assembly: an insight. Postdoc J. 2013;1(4):33-45.
2. Garg PA. Review of podocyte biology. Am J Nephrol. 2018;47(suppl 1):3-13. doi:10.1159/000481633SUPPL
3. Warady BA, Agarwal R, Bangalore S, et al. Alport syndrome classification and management. Kidney Med. 2020;2(5):639-649. doi:10.1016/j.xkme.2020.05.014
4. Angioi A, Pani A. FSGS: from pathogenesis to the histological lesion. J Nephrol. 2016;29(4):517-523. doi:10.1007/s40620-016-0333-2
5. Roca N, Martinez C, Jatem E, Madrid A, Lopez M, Segarra A. Activation of the acute inflammatory phase response in idiopathic nephrotic syndrome: association with clinicopathological phenotypes and with response to corticosteroids. Clin Kidney J. 2021;14(4):1207-1215. doi:10.1093/ckj/sfaa247
6. Vivarelli M, Massella L, Ruggiero B, Emma F. Minimal change disease. Clin J Am Soc Nephrol. 2017;12(2):332-345.
7. Medjeral-Thomas NR, Lawrence C, Condon M, et al. Randomized, controlled trial of tacrolimus and prednisolone monotherapy for adults with De Novo minimal change disease: a multicenter, randomized, controlled trial. Clin J Am Soc Nephrol. 2020;15(2):209-218. doi:10.2215/CJN.06290420
8. Ye Q, Lan B, Liu H, Persson PB, Lai EY, Mao J. A critical role of the podocyte cytoskeleton in the pathogenesis of glomerular proteinuria and autoimmune podocytopathies. Acta Physiol (Oxf). 2022;235(4):e13850. doi:10.1111/apha.13850
9. Trautmann A, Schnaidt S, Lipska-Ziμtkiewicz BS, et al. Long-term outcome of steroid-resistant nephrotic syndrome in children. J Am Soc Nephrol. 2017;28:3055-3065. doi:10.1681/ASN.2016101121
10. Kashtan CE, Gross O. Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults-an update for 2020. Pediatr Nephrol. 2021;36(3):711-719. doi:10.1007/s00467-020-04819-6
11. Savige J, Rana K, Tonna S, Buzza M, Dagher H, Wang YY. Thin basement membrane nephropathy. Kidney Int. 2003;64(4):1169-78. doi:10.1046/j.1523-1755.2003.00234.x
12. Rosenberg AZ, Kopp JB. Focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2017; 12(3):502-517. doi:10.2215/CJN.05960616
13. Savige J. Should we diagnose autosomal dominant Alport syndrome when there is a pathogenic heterozygous COL4A3 or COL4A4 variant? Kidney Int Rep. 2018;3(6):1239-1241. doi:10.1016/j.ekir.2018.08.002
14. Gigante M, Caridi G, Montemurno E, et al. TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin J Am Soc Nephrol. 2011;6(7):1626-1634. doi:10.2215/CJN.07830910
15. Saurus P, Kuusela S, Lehtonen E, et al. Podocyte apoptosis is prevented by blocking the toll-like receptor pathway. Cell Death Dis. 2015;6(5):e1752. doi:10.1038/cddis.2015.125
16. Baud L, Oudinet JP, Bens M, et al. Production of tumor necrosis factor by rat mesangial cells in response to bacterial lipopolysaccharide. Kidney Int. 1989;35(5):1111-1118. doi:10.1038/ki.1989.98
17. White S, Lin L, Hu K. NF-κB and tPA signaling in kidney and other diseases. Cells. 2020;9(6):1348. doi:10.3390/cells9061348
18. Tesch GH, Lan HY, Atkins RC, Nikolic-Paterson DJ. Role of interleukin-1 in mesangial cell proliferation and matrix deposition in experimental mesangioproliferative nephritis. Am J Pathol. 1997;151(1):141-150.
19. Lai KN, Leung JCK, Chan LYY, et al. Podocyte injury induced by mesangial-derived cytokines in IgA Nephropathy. Nephrol Dial Transplant. 2009;24(1):62-72. doi:10.1093/ndt/gfn441
20. Saleem MA, Kobayashi Y. Cell biology and genetics of minimal change disease. F1000 Res. 2016;5: F1000 Faculty Rev-412. doi:10.12688/f1000research.7300.1
21. Kim KP, Williams CE, Lemmon CA. Cell-matrix interactions in renal fibrosis. Kidney Dial. 2022;2(4):607-624. doi:10.3390/kidneydial2040055
22. Zvaifler NJ. Relevance of the stroma and epithelial-mesenchymal transition (EMT) for the rheumatic diseases. Arthritis Res Ther. 2006;8(3):210. doi:10.1186/ar1963
23. Angeletti A, Magnasco A, Trivelli A, et al. Refractory minimal change disease and focal segmental glomerular sclerosis treated with Anakinra. Kidney Int Rep. 2021;7(1):121-124. doi:10.1016/j.ekir.2021.10.018
24. Trachtman H, Vento S, Herreshoff E, et al. Efficacy of galactose and adalimumab in patients with resistant focal segmental glomerulosclerosis: report of the font clinical trial group. BMC Nephrol. 2015;16:111. doi:10.1186/s12882-015-0094-5
25. Mariani LH, Eddy S, AlAkwaa FM, et al. Precision nephrology identified tumor necrosis factor activation variability in minimal change disease and focal segmental glomerulosclerosis. Kidney Int. 2023;103(3):565-579. doi:10.1016/j.kint.2022.10.023
26. Korshak L, Washington DL, Powell J, Nylen E, Kokkinos P. Kidney Disease in Veterans. US Dept of Veterans Affairs, Office of Health Equity. Updated May 13, 2020. Accessed June 28, 2024. https://www.va.gov/HEALTHEQUITY/Kidney_Disease_In_Veterans.asp
27. Malone AF, Phelan PJ, Hall G, et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014;86(6):1253-1259. doi:10.1038/ki.2014.305
Podocytes are terminally differentiated, highly specialized cells located in juxtaposition to the basement membrane over the abluminal surfaces of endothelial cells within the glomerular tuft. This triad structure is the site of the filtration barrier, which forms highly delicate and tightly regulated architecture to carry out the ultrafiltration function of the kidney.1 The filtration barrier is characterized by foot processes that are connected by specialized junctions called slit diaphragms.
Insults to components of the filtration barrier can initiate cascading events and perpetuate structural alterations that may eventually result in sclerotic changes.2 Common causes among children include minimal change disease (MCD) with the collapse of foot processes resulting in proteinuria, Alport syndrome due to mutation of collagen fibers within the basement membrane leading to hematuria and proteinuria, immune complex mediated nephropathy following common infections or autoimmune diseases, and focal segmental glomerulosclerosis (FSGS) that can show variable histopathology toward eventual glomerular scarring.3,4 These children often clinically have minimal, if any, signs of systemic inflammation.3-5 This has been a limiting factor for the commitment to immunomodulatory treatment, except for steroids for the treatment of MCD.6 Although prolonged steroid treatment may be efficacious, adverse effects are significant in a growing child. Alternative treatments, such as tacrolimus and rituximab have been suggested as second-line steroid-sparing agents.7,8 Not uncommonly, however, these cases are managed by supportive measures only during the progression of the natural course of the disease, which may eventually lead to renal failure, requiring transplant for survival.8,9
This case report highlights a child with a variant of uncertain significance (VUS) in genes involved in Alport syndrome and FSGS who developed an abrupt onset of proteinuria and hematuria after a respiratory illness. To our knowledge, he represents the youngest case demonstrating the benefit of targeted treatment against tumor necrosis factor-α (TNF-α) for glomerulopathy using biologic response modifiers.
Case Description
This is currently a 7-year-old male patient who was born at 39 weeks gestation to gravida 3 para 3 following induced labor due to elevated maternal blood pressure. During the first 2 years of life, his growth and development were normal and his immunizations were up to date. The patient's medical history included upper respiratory tract infections (URIs), respiratory syncytial virus, as well as 3 bouts of pneumonia and multiple otitis media that resulted in 18 rounds of antibiotics. The child was also allergic to nuts and milk protein. The patient’s parents are of Northern European and Native American descent. There is no known family history of eye, ear, or kidney diseases.
Renal concerns were first noted at the age of 2 years and 6 months when he presented to an emergency department in Fall 2019 (week 0) for several weeks of intermittent dark-colored urine. His mother reported that the discoloration recently progressed in intensity to cola-colored, along with the onset of persistent vomiting without any fever or diarrhea. On physical examination, the patient had normal vitals: weight 14.8 kg (68th percentile), height 91 cm (24th percentile), and body surface area 0.6 m2. There was no edema, rash, or lymphadenopathy, but he appeared pale.
The patient’s initial laboratory results included: complete blood count with white blood cells (WBC) 10 x 103/L (reference range, 4.5-13.5 x 103/L); differential lymphocytes 69%; neutrophils 21%; hemoglobin 10 g/dL (reference range, 12-16 g/dL); hematocrit, 30%; (reference range, 37%-45%); platelets 437 103/L (reference range, 150-450 x 103/L); serum creatinine 0.46 mg/dL (reference range, 0.5-0.9 mg/dL); and albumin 3.1 g/dL (reference range, 3.5-5.2 g/dL). Serum electrolyte levels and liver enzymes were normal. A urine analysis revealed 3+ protein and 3+ blood with dysmorphic red blood cells (RBC) and RBC casts without WBC. The patient's spot urine protein-to-creatinine ratio was 4.3 and his renal ultrasound was normal. The patient was referred to Nephrology.
During the next 2 weeks, his protein-to-creatinine ratio progressed to 5.9 and serum albumin fell to 2.7 g/dL. His urine remained red colored, and a microscopic examination with RBC > 500 and WBC up to 10 on a high powered field. His workup was negative for antinuclear antibodies, antineutrophil cytoplasmic antibody, antistreptolysin-O (ASO) and anti-DNase B. Serum C3 was low at 81 mg/dL (reference range, 90-180 mg/dL), C4 was 13.3 mg/dL (reference range, 10-40 mg/dL), and immunoglobulin G was low at 452 mg/dL (reference range 719-1475 mg/dL). A baseline audiology test revealed normal hearing.
Percutaneous renal biopsy yielded about 12 glomeruli, all exhibiting mild mesangial matrix expansion and hypercellularity (Figure 1). One glomerulus had prominent parietal epithelial cells without endocapillary hypercellularity or crescent formation. There was no interstitial fibrosis or tubular atrophy. Immunofluorescence studies showed no evidence of immune complex deposition with negative staining for immunoglobulin heavy and light chains, C3 and C1q. Staining for α 2 and α 5 units of collagen was normal. Electron microscopy showed patchy areas of severe basement membrane thinning with frequent foci of mild to moderate lamina densa splitting and associated visceral epithelial cell foot process effacement (Figure 2).
These were reported as concerning findings for possible Alport syndrome by 3 independent pathology teams. The genetic testing was submitted at a commercial laboratory to screen 17 mutations, including COL4A3, COL4A4, and COL4A5. Results showed the presence of a heterozygous VUS in the COL4A4 gene (c.1055C > T; p.Pro352Leu; dbSNP ID: rs371717486; PolyPhen-2: Probably Damaging; SIFT: Deleterious) as well as the presence of a heterozygous VUS in TRPC6 gene (c2463A>T; p.Lys821Asn; dbSNP ID: rs199948731; PolyPhen-2: Benign; SIFT: Tolerated). Further genetic investigation by whole exome sequencing on approximately 20,000 genes through MNG Laboratories showed a new heterozygous VUS in the OSGEP gene [c.328T>C; p.Cys110Arg]. Additional studies ruled out mitochondrial disease, CoQ10 deficiency, and metabolic disorders upon normal findings for mitochondrial DNA, urine amino acids, plasma acylcarnitine profile, orotic acid, ammonia, and homocysteine levels.
Figure 3 summarizes the patient’s treatment response during 170 weeks of follow-up (Fall 2019 to Summer 2023). The patient was started on enalapril 0.6 mg/kg daily at week 3, which continued throughout treatment. Following a rheumatology consult at week 30, the patient was started on prednisolone 3 mg/mL to assess the role of inflammation through the treatment response. An initial dose of 2 mg/kg daily (9 mL) for 1 month was followed by every other day treatment that was tapered off by week 48. To control mild but noticeably increasing proteinuria in the interim, subcutaneous anakinra 50 mg (3 mg/kg daily) was added as a steroid
DISCUSSION
This case describes a child with rapidly progressive proteinuria and hematuria following a URI who was found to have VUS mutations in 3 different genes associated with chronic kidney disease. Serology tests on the patient were negative for streptococcal antibodies and antinuclear antibodies, ruling out poststreptococcal glomerulonephritis, or systemic lupus erythematosus. His renal biopsy findings were concerning for altered podocytes, mesangial cells, and basement membrane without inflammatory infiltrate, immune complex, complements, immunoglobulin A, or vasculopathy. His blood inflammatory markers, erythrocyte sedimentation rate, C-reactive protein, and ferritin were normal when his care team initiated daily steroids.
Overall, the patient’s clinical presentation and histopathology findings were suggestive of Alport syndrome or thin basement membrane nephropathy with a high potential to progress into FSGS.10-12 Alport syndrome affects 1 in 5000 to 10,000 children annually due to S-linked inheritance of COL4A5, or autosomal recessive inheritance of COL4A3 or COL4A4 genes. It presents with hematuria and hearing loss.10 Our patient had a single copy COL4A4 gene mutation that was classified as VUS. He also had 2 additional VUS affecting the TRPC6 and OSGEP genes. TRPC6 gene mutation can be associated with FSGS through autosomal dominant inheritance. Both COL4A4 and TRPC6 gene mutations were paternally inherited. Although the patient’s father not having renal disease argues against the clinical significance of these findings, there is literature on the potential role of heterozygous COL4A4 variant mimicking thin basement membrane nephropathy that can lead to renal impairment upon copresence of superimposed conditions.13 The patient’s rapidly progressing hematuria and changes in the basement membrane were worrisome for emerging FSGS. Furthermore, VUS of TRPC6 has been reported in late onset autosomal dominant FSGS and can be associated with early onset steroid-resistant nephrotic syndrome (NS) in children.14 This concern was voiced by 3 nephrology consultants during the initial evaluation, leading to the consensus that steroid treatment for podocytopathy would not alter the patient’s long-term outcomes (ie, progression to FSGS).
Immunomodulation
Our rationale for immunomodulatory treatment was based on the abrupt onset of renal concerns following a URI, suggesting the importance of an inflammatory trigger causing altered homeostasis in a genetically susceptible host. Preclinical models show that microbial products such as lipopolysaccharides can lead to podocytopathy by several mechanisms through activation of toll-like receptor signaling. It can directly cause apoptosis by downregulation of the intracellular Akt survival pathway.15 Lipopolysaccharide can also activate the NF-αB pathway and upregulate the production of interleukin-1 (IL-1) and TNF-α in mesangial cells.16,17
Both cytokines can promote mesangial cell proliferation.18 Through autocrine and paracrine mechanisms, proinflammatory cytokines can further perpetuate somatic tissue changes and contribute to the development of podocytopathy. For instance, TNF-α can promote podocyte injury and proteinuria by downregulation of the slit diaphragm protein expression (ie, nephrin, ezrin, or podocin), and disruption of podocyte cytoskeleton.19,20 TNF-α promotes the influx and activation of macrophages and inflammatory cells. It is actively involved in chronic alterations within the glomeruli by the upregulation of matrix metalloproteases by integrins, as well as activation of myofibroblast progenitors and extracellular matrix deposition in crosstalk with transforming growth factor and other key mediators.17,21,22
For the patient described in this case report, initial improvement on steroids encouraged the pursuit of additional treatment to downregulate inflammatory pathways within the glomerular milieu. However, within the COVID-19 environment, escalating the patient’s treatment using traditional immunomodulators (ie, calcineurin inhibitors or mycophenolate mofetil) was not favored due to the risk of infection. Initially, anakinra, a recombinant IL-1 receptor antagonist, was preferred as a steroid-sparing agent for its short life and safety profile during the pandemic. At first, the patient responded well to anakinra and was allowed a steroid wean when the dose was titrated up to 6 mg/kg daily. However, anakinra did not prevent the escalation of proteinuria following a URI. After the treatment was changed to adalimumab, a fully humanized monoclonal antibody to TNF-α, the patient continued to improve and reach full remission despite experiencing a cold and the flu in the following months.
Literature Review
There is a paucity of literature on applications of biological response modifiers for idiopathic NS and FSGS.23,24 Angeletti and colleagues reported that 3 patients with severe long-standing FSGS benefited from anakinra 4 mg/kg daily to reduce proteinuria and improve kidney function. All the patients had positive C3 staining in renal biopsy and treatment response, which supported the role of C3a in inducing podocyte injury through upregulated expression of IL-1 and IL-1R.23 Trachtman and colleagues reported on the phase II FONT trial that included 14 of 21 patients aged < 18 years with advanced FSGS who were treated with adalimumab 24 mg/m2, or ≤ 40 mg every other week.24 Although, during a 6-month period, none of the 7 patients met the endpoint of reduced proteinuria by ≥ 50%, and the authors suggested that careful patient selection may improve the treatment response in future trials.24
A recent study involving transcriptomics on renal tissue samples combined with available pathology (fibrosis), urinary markers, and clinical characteristics on 285 patients with MCD or FSGS from 3 different continents identified 3 distinct clusters. Patients with evidence of activated kidney TNF pathway (n = 72, aged > 18 years) were found to have poor clinical outcomes.25 The study identified 2 urine markers associated with the TNF pathway (ie, tissue inhibitor of metalloproteinases-1 and monocyte chemoattractant protein-1), which aligns with the preclinical findings previously mentioned.25
Conclusions
The patient’s condition in this case illustrates the complex nature of biologically predetermined cascading events in the emergence of glomerular disease upon environmental triggers under the influence of genetic factors.
Chronic kidney disease affects 7.7% of veterans annually, illustrating the need for new therapeutics.26 Based on our experience and literature review, upregulation of TNF-α is a root cause of glomerulopathy; further studies are warranted to evaluate the efficacy of anti-TNF biologic response modifiers for the treatment of these patients. Long-term postmarketing safety profile and steroid-sparing properties of adalimumab should allow inclusion of pediatric cases in future trials. Results may also contribute to identifying new predictive biomarkers related to the basement membrane when combined with precision nephrology to further advance patient selection and targeted treatment.25,27
Acknowledgments
The authors thank the patient’s mother for providing consent to allow publication of this case report.
Podocytes are terminally differentiated, highly specialized cells located in juxtaposition to the basement membrane over the abluminal surfaces of endothelial cells within the glomerular tuft. This triad structure is the site of the filtration barrier, which forms highly delicate and tightly regulated architecture to carry out the ultrafiltration function of the kidney.1 The filtration barrier is characterized by foot processes that are connected by specialized junctions called slit diaphragms.
Insults to components of the filtration barrier can initiate cascading events and perpetuate structural alterations that may eventually result in sclerotic changes.2 Common causes among children include minimal change disease (MCD) with the collapse of foot processes resulting in proteinuria, Alport syndrome due to mutation of collagen fibers within the basement membrane leading to hematuria and proteinuria, immune complex mediated nephropathy following common infections or autoimmune diseases, and focal segmental glomerulosclerosis (FSGS) that can show variable histopathology toward eventual glomerular scarring.3,4 These children often clinically have minimal, if any, signs of systemic inflammation.3-5 This has been a limiting factor for the commitment to immunomodulatory treatment, except for steroids for the treatment of MCD.6 Although prolonged steroid treatment may be efficacious, adverse effects are significant in a growing child. Alternative treatments, such as tacrolimus and rituximab have been suggested as second-line steroid-sparing agents.7,8 Not uncommonly, however, these cases are managed by supportive measures only during the progression of the natural course of the disease, which may eventually lead to renal failure, requiring transplant for survival.8,9
This case report highlights a child with a variant of uncertain significance (VUS) in genes involved in Alport syndrome and FSGS who developed an abrupt onset of proteinuria and hematuria after a respiratory illness. To our knowledge, he represents the youngest case demonstrating the benefit of targeted treatment against tumor necrosis factor-α (TNF-α) for glomerulopathy using biologic response modifiers.
Case Description
This is currently a 7-year-old male patient who was born at 39 weeks gestation to gravida 3 para 3 following induced labor due to elevated maternal blood pressure. During the first 2 years of life, his growth and development were normal and his immunizations were up to date. The patient's medical history included upper respiratory tract infections (URIs), respiratory syncytial virus, as well as 3 bouts of pneumonia and multiple otitis media that resulted in 18 rounds of antibiotics. The child was also allergic to nuts and milk protein. The patient’s parents are of Northern European and Native American descent. There is no known family history of eye, ear, or kidney diseases.
Renal concerns were first noted at the age of 2 years and 6 months when he presented to an emergency department in Fall 2019 (week 0) for several weeks of intermittent dark-colored urine. His mother reported that the discoloration recently progressed in intensity to cola-colored, along with the onset of persistent vomiting without any fever or diarrhea. On physical examination, the patient had normal vitals: weight 14.8 kg (68th percentile), height 91 cm (24th percentile), and body surface area 0.6 m2. There was no edema, rash, or lymphadenopathy, but he appeared pale.
The patient’s initial laboratory results included: complete blood count with white blood cells (WBC) 10 x 103/L (reference range, 4.5-13.5 x 103/L); differential lymphocytes 69%; neutrophils 21%; hemoglobin 10 g/dL (reference range, 12-16 g/dL); hematocrit, 30%; (reference range, 37%-45%); platelets 437 103/L (reference range, 150-450 x 103/L); serum creatinine 0.46 mg/dL (reference range, 0.5-0.9 mg/dL); and albumin 3.1 g/dL (reference range, 3.5-5.2 g/dL). Serum electrolyte levels and liver enzymes were normal. A urine analysis revealed 3+ protein and 3+ blood with dysmorphic red blood cells (RBC) and RBC casts without WBC. The patient's spot urine protein-to-creatinine ratio was 4.3 and his renal ultrasound was normal. The patient was referred to Nephrology.
During the next 2 weeks, his protein-to-creatinine ratio progressed to 5.9 and serum albumin fell to 2.7 g/dL. His urine remained red colored, and a microscopic examination with RBC > 500 and WBC up to 10 on a high powered field. His workup was negative for antinuclear antibodies, antineutrophil cytoplasmic antibody, antistreptolysin-O (ASO) and anti-DNase B. Serum C3 was low at 81 mg/dL (reference range, 90-180 mg/dL), C4 was 13.3 mg/dL (reference range, 10-40 mg/dL), and immunoglobulin G was low at 452 mg/dL (reference range 719-1475 mg/dL). A baseline audiology test revealed normal hearing.
Percutaneous renal biopsy yielded about 12 glomeruli, all exhibiting mild mesangial matrix expansion and hypercellularity (Figure 1). One glomerulus had prominent parietal epithelial cells without endocapillary hypercellularity or crescent formation. There was no interstitial fibrosis or tubular atrophy. Immunofluorescence studies showed no evidence of immune complex deposition with negative staining for immunoglobulin heavy and light chains, C3 and C1q. Staining for α 2 and α 5 units of collagen was normal. Electron microscopy showed patchy areas of severe basement membrane thinning with frequent foci of mild to moderate lamina densa splitting and associated visceral epithelial cell foot process effacement (Figure 2).
These were reported as concerning findings for possible Alport syndrome by 3 independent pathology teams. The genetic testing was submitted at a commercial laboratory to screen 17 mutations, including COL4A3, COL4A4, and COL4A5. Results showed the presence of a heterozygous VUS in the COL4A4 gene (c.1055C > T; p.Pro352Leu; dbSNP ID: rs371717486; PolyPhen-2: Probably Damaging; SIFT: Deleterious) as well as the presence of a heterozygous VUS in TRPC6 gene (c2463A>T; p.Lys821Asn; dbSNP ID: rs199948731; PolyPhen-2: Benign; SIFT: Tolerated). Further genetic investigation by whole exome sequencing on approximately 20,000 genes through MNG Laboratories showed a new heterozygous VUS in the OSGEP gene [c.328T>C; p.Cys110Arg]. Additional studies ruled out mitochondrial disease, CoQ10 deficiency, and metabolic disorders upon normal findings for mitochondrial DNA, urine amino acids, plasma acylcarnitine profile, orotic acid, ammonia, and homocysteine levels.
Figure 3 summarizes the patient’s treatment response during 170 weeks of follow-up (Fall 2019 to Summer 2023). The patient was started on enalapril 0.6 mg/kg daily at week 3, which continued throughout treatment. Following a rheumatology consult at week 30, the patient was started on prednisolone 3 mg/mL to assess the role of inflammation through the treatment response. An initial dose of 2 mg/kg daily (9 mL) for 1 month was followed by every other day treatment that was tapered off by week 48. To control mild but noticeably increasing proteinuria in the interim, subcutaneous anakinra 50 mg (3 mg/kg daily) was added as a steroid
DISCUSSION
This case describes a child with rapidly progressive proteinuria and hematuria following a URI who was found to have VUS mutations in 3 different genes associated with chronic kidney disease. Serology tests on the patient were negative for streptococcal antibodies and antinuclear antibodies, ruling out poststreptococcal glomerulonephritis, or systemic lupus erythematosus. His renal biopsy findings were concerning for altered podocytes, mesangial cells, and basement membrane without inflammatory infiltrate, immune complex, complements, immunoglobulin A, or vasculopathy. His blood inflammatory markers, erythrocyte sedimentation rate, C-reactive protein, and ferritin were normal when his care team initiated daily steroids.
Overall, the patient’s clinical presentation and histopathology findings were suggestive of Alport syndrome or thin basement membrane nephropathy with a high potential to progress into FSGS.10-12 Alport syndrome affects 1 in 5000 to 10,000 children annually due to S-linked inheritance of COL4A5, or autosomal recessive inheritance of COL4A3 or COL4A4 genes. It presents with hematuria and hearing loss.10 Our patient had a single copy COL4A4 gene mutation that was classified as VUS. He also had 2 additional VUS affecting the TRPC6 and OSGEP genes. TRPC6 gene mutation can be associated with FSGS through autosomal dominant inheritance. Both COL4A4 and TRPC6 gene mutations were paternally inherited. Although the patient’s father not having renal disease argues against the clinical significance of these findings, there is literature on the potential role of heterozygous COL4A4 variant mimicking thin basement membrane nephropathy that can lead to renal impairment upon copresence of superimposed conditions.13 The patient’s rapidly progressing hematuria and changes in the basement membrane were worrisome for emerging FSGS. Furthermore, VUS of TRPC6 has been reported in late onset autosomal dominant FSGS and can be associated with early onset steroid-resistant nephrotic syndrome (NS) in children.14 This concern was voiced by 3 nephrology consultants during the initial evaluation, leading to the consensus that steroid treatment for podocytopathy would not alter the patient’s long-term outcomes (ie, progression to FSGS).
Immunomodulation
Our rationale for immunomodulatory treatment was based on the abrupt onset of renal concerns following a URI, suggesting the importance of an inflammatory trigger causing altered homeostasis in a genetically susceptible host. Preclinical models show that microbial products such as lipopolysaccharides can lead to podocytopathy by several mechanisms through activation of toll-like receptor signaling. It can directly cause apoptosis by downregulation of the intracellular Akt survival pathway.15 Lipopolysaccharide can also activate the NF-αB pathway and upregulate the production of interleukin-1 (IL-1) and TNF-α in mesangial cells.16,17
Both cytokines can promote mesangial cell proliferation.18 Through autocrine and paracrine mechanisms, proinflammatory cytokines can further perpetuate somatic tissue changes and contribute to the development of podocytopathy. For instance, TNF-α can promote podocyte injury and proteinuria by downregulation of the slit diaphragm protein expression (ie, nephrin, ezrin, or podocin), and disruption of podocyte cytoskeleton.19,20 TNF-α promotes the influx and activation of macrophages and inflammatory cells. It is actively involved in chronic alterations within the glomeruli by the upregulation of matrix metalloproteases by integrins, as well as activation of myofibroblast progenitors and extracellular matrix deposition in crosstalk with transforming growth factor and other key mediators.17,21,22
For the patient described in this case report, initial improvement on steroids encouraged the pursuit of additional treatment to downregulate inflammatory pathways within the glomerular milieu. However, within the COVID-19 environment, escalating the patient’s treatment using traditional immunomodulators (ie, calcineurin inhibitors or mycophenolate mofetil) was not favored due to the risk of infection. Initially, anakinra, a recombinant IL-1 receptor antagonist, was preferred as a steroid-sparing agent for its short life and safety profile during the pandemic. At first, the patient responded well to anakinra and was allowed a steroid wean when the dose was titrated up to 6 mg/kg daily. However, anakinra did not prevent the escalation of proteinuria following a URI. After the treatment was changed to adalimumab, a fully humanized monoclonal antibody to TNF-α, the patient continued to improve and reach full remission despite experiencing a cold and the flu in the following months.
Literature Review
There is a paucity of literature on applications of biological response modifiers for idiopathic NS and FSGS.23,24 Angeletti and colleagues reported that 3 patients with severe long-standing FSGS benefited from anakinra 4 mg/kg daily to reduce proteinuria and improve kidney function. All the patients had positive C3 staining in renal biopsy and treatment response, which supported the role of C3a in inducing podocyte injury through upregulated expression of IL-1 and IL-1R.23 Trachtman and colleagues reported on the phase II FONT trial that included 14 of 21 patients aged < 18 years with advanced FSGS who were treated with adalimumab 24 mg/m2, or ≤ 40 mg every other week.24 Although, during a 6-month period, none of the 7 patients met the endpoint of reduced proteinuria by ≥ 50%, and the authors suggested that careful patient selection may improve the treatment response in future trials.24
A recent study involving transcriptomics on renal tissue samples combined with available pathology (fibrosis), urinary markers, and clinical characteristics on 285 patients with MCD or FSGS from 3 different continents identified 3 distinct clusters. Patients with evidence of activated kidney TNF pathway (n = 72, aged > 18 years) were found to have poor clinical outcomes.25 The study identified 2 urine markers associated with the TNF pathway (ie, tissue inhibitor of metalloproteinases-1 and monocyte chemoattractant protein-1), which aligns with the preclinical findings previously mentioned.25
Conclusions
The patient’s condition in this case illustrates the complex nature of biologically predetermined cascading events in the emergence of glomerular disease upon environmental triggers under the influence of genetic factors.
Chronic kidney disease affects 7.7% of veterans annually, illustrating the need for new therapeutics.26 Based on our experience and literature review, upregulation of TNF-α is a root cause of glomerulopathy; further studies are warranted to evaluate the efficacy of anti-TNF biologic response modifiers for the treatment of these patients. Long-term postmarketing safety profile and steroid-sparing properties of adalimumab should allow inclusion of pediatric cases in future trials. Results may also contribute to identifying new predictive biomarkers related to the basement membrane when combined with precision nephrology to further advance patient selection and targeted treatment.25,27
Acknowledgments
The authors thank the patient’s mother for providing consent to allow publication of this case report.
1. Arif E, Nihalani D. Glomerular filtration barrier assembly: an insight. Postdoc J. 2013;1(4):33-45.
2. Garg PA. Review of podocyte biology. Am J Nephrol. 2018;47(suppl 1):3-13. doi:10.1159/000481633SUPPL
3. Warady BA, Agarwal R, Bangalore S, et al. Alport syndrome classification and management. Kidney Med. 2020;2(5):639-649. doi:10.1016/j.xkme.2020.05.014
4. Angioi A, Pani A. FSGS: from pathogenesis to the histological lesion. J Nephrol. 2016;29(4):517-523. doi:10.1007/s40620-016-0333-2
5. Roca N, Martinez C, Jatem E, Madrid A, Lopez M, Segarra A. Activation of the acute inflammatory phase response in idiopathic nephrotic syndrome: association with clinicopathological phenotypes and with response to corticosteroids. Clin Kidney J. 2021;14(4):1207-1215. doi:10.1093/ckj/sfaa247
6. Vivarelli M, Massella L, Ruggiero B, Emma F. Minimal change disease. Clin J Am Soc Nephrol. 2017;12(2):332-345.
7. Medjeral-Thomas NR, Lawrence C, Condon M, et al. Randomized, controlled trial of tacrolimus and prednisolone monotherapy for adults with De Novo minimal change disease: a multicenter, randomized, controlled trial. Clin J Am Soc Nephrol. 2020;15(2):209-218. doi:10.2215/CJN.06290420
8. Ye Q, Lan B, Liu H, Persson PB, Lai EY, Mao J. A critical role of the podocyte cytoskeleton in the pathogenesis of glomerular proteinuria and autoimmune podocytopathies. Acta Physiol (Oxf). 2022;235(4):e13850. doi:10.1111/apha.13850
9. Trautmann A, Schnaidt S, Lipska-Ziμtkiewicz BS, et al. Long-term outcome of steroid-resistant nephrotic syndrome in children. J Am Soc Nephrol. 2017;28:3055-3065. doi:10.1681/ASN.2016101121
10. Kashtan CE, Gross O. Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults-an update for 2020. Pediatr Nephrol. 2021;36(3):711-719. doi:10.1007/s00467-020-04819-6
11. Savige J, Rana K, Tonna S, Buzza M, Dagher H, Wang YY. Thin basement membrane nephropathy. Kidney Int. 2003;64(4):1169-78. doi:10.1046/j.1523-1755.2003.00234.x
12. Rosenberg AZ, Kopp JB. Focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2017; 12(3):502-517. doi:10.2215/CJN.05960616
13. Savige J. Should we diagnose autosomal dominant Alport syndrome when there is a pathogenic heterozygous COL4A3 or COL4A4 variant? Kidney Int Rep. 2018;3(6):1239-1241. doi:10.1016/j.ekir.2018.08.002
14. Gigante M, Caridi G, Montemurno E, et al. TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin J Am Soc Nephrol. 2011;6(7):1626-1634. doi:10.2215/CJN.07830910
15. Saurus P, Kuusela S, Lehtonen E, et al. Podocyte apoptosis is prevented by blocking the toll-like receptor pathway. Cell Death Dis. 2015;6(5):e1752. doi:10.1038/cddis.2015.125
16. Baud L, Oudinet JP, Bens M, et al. Production of tumor necrosis factor by rat mesangial cells in response to bacterial lipopolysaccharide. Kidney Int. 1989;35(5):1111-1118. doi:10.1038/ki.1989.98
17. White S, Lin L, Hu K. NF-κB and tPA signaling in kidney and other diseases. Cells. 2020;9(6):1348. doi:10.3390/cells9061348
18. Tesch GH, Lan HY, Atkins RC, Nikolic-Paterson DJ. Role of interleukin-1 in mesangial cell proliferation and matrix deposition in experimental mesangioproliferative nephritis. Am J Pathol. 1997;151(1):141-150.
19. Lai KN, Leung JCK, Chan LYY, et al. Podocyte injury induced by mesangial-derived cytokines in IgA Nephropathy. Nephrol Dial Transplant. 2009;24(1):62-72. doi:10.1093/ndt/gfn441
20. Saleem MA, Kobayashi Y. Cell biology and genetics of minimal change disease. F1000 Res. 2016;5: F1000 Faculty Rev-412. doi:10.12688/f1000research.7300.1
21. Kim KP, Williams CE, Lemmon CA. Cell-matrix interactions in renal fibrosis. Kidney Dial. 2022;2(4):607-624. doi:10.3390/kidneydial2040055
22. Zvaifler NJ. Relevance of the stroma and epithelial-mesenchymal transition (EMT) for the rheumatic diseases. Arthritis Res Ther. 2006;8(3):210. doi:10.1186/ar1963
23. Angeletti A, Magnasco A, Trivelli A, et al. Refractory minimal change disease and focal segmental glomerular sclerosis treated with Anakinra. Kidney Int Rep. 2021;7(1):121-124. doi:10.1016/j.ekir.2021.10.018
24. Trachtman H, Vento S, Herreshoff E, et al. Efficacy of galactose and adalimumab in patients with resistant focal segmental glomerulosclerosis: report of the font clinical trial group. BMC Nephrol. 2015;16:111. doi:10.1186/s12882-015-0094-5
25. Mariani LH, Eddy S, AlAkwaa FM, et al. Precision nephrology identified tumor necrosis factor activation variability in minimal change disease and focal segmental glomerulosclerosis. Kidney Int. 2023;103(3):565-579. doi:10.1016/j.kint.2022.10.023
26. Korshak L, Washington DL, Powell J, Nylen E, Kokkinos P. Kidney Disease in Veterans. US Dept of Veterans Affairs, Office of Health Equity. Updated May 13, 2020. Accessed June 28, 2024. https://www.va.gov/HEALTHEQUITY/Kidney_Disease_In_Veterans.asp
27. Malone AF, Phelan PJ, Hall G, et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014;86(6):1253-1259. doi:10.1038/ki.2014.305
1. Arif E, Nihalani D. Glomerular filtration barrier assembly: an insight. Postdoc J. 2013;1(4):33-45.
2. Garg PA. Review of podocyte biology. Am J Nephrol. 2018;47(suppl 1):3-13. doi:10.1159/000481633SUPPL
3. Warady BA, Agarwal R, Bangalore S, et al. Alport syndrome classification and management. Kidney Med. 2020;2(5):639-649. doi:10.1016/j.xkme.2020.05.014
4. Angioi A, Pani A. FSGS: from pathogenesis to the histological lesion. J Nephrol. 2016;29(4):517-523. doi:10.1007/s40620-016-0333-2
5. Roca N, Martinez C, Jatem E, Madrid A, Lopez M, Segarra A. Activation of the acute inflammatory phase response in idiopathic nephrotic syndrome: association with clinicopathological phenotypes and with response to corticosteroids. Clin Kidney J. 2021;14(4):1207-1215. doi:10.1093/ckj/sfaa247
6. Vivarelli M, Massella L, Ruggiero B, Emma F. Minimal change disease. Clin J Am Soc Nephrol. 2017;12(2):332-345.
7. Medjeral-Thomas NR, Lawrence C, Condon M, et al. Randomized, controlled trial of tacrolimus and prednisolone monotherapy for adults with De Novo minimal change disease: a multicenter, randomized, controlled trial. Clin J Am Soc Nephrol. 2020;15(2):209-218. doi:10.2215/CJN.06290420
8. Ye Q, Lan B, Liu H, Persson PB, Lai EY, Mao J. A critical role of the podocyte cytoskeleton in the pathogenesis of glomerular proteinuria and autoimmune podocytopathies. Acta Physiol (Oxf). 2022;235(4):e13850. doi:10.1111/apha.13850
9. Trautmann A, Schnaidt S, Lipska-Ziμtkiewicz BS, et al. Long-term outcome of steroid-resistant nephrotic syndrome in children. J Am Soc Nephrol. 2017;28:3055-3065. doi:10.1681/ASN.2016101121
10. Kashtan CE, Gross O. Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults-an update for 2020. Pediatr Nephrol. 2021;36(3):711-719. doi:10.1007/s00467-020-04819-6
11. Savige J, Rana K, Tonna S, Buzza M, Dagher H, Wang YY. Thin basement membrane nephropathy. Kidney Int. 2003;64(4):1169-78. doi:10.1046/j.1523-1755.2003.00234.x
12. Rosenberg AZ, Kopp JB. Focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2017; 12(3):502-517. doi:10.2215/CJN.05960616
13. Savige J. Should we diagnose autosomal dominant Alport syndrome when there is a pathogenic heterozygous COL4A3 or COL4A4 variant? Kidney Int Rep. 2018;3(6):1239-1241. doi:10.1016/j.ekir.2018.08.002
14. Gigante M, Caridi G, Montemurno E, et al. TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin J Am Soc Nephrol. 2011;6(7):1626-1634. doi:10.2215/CJN.07830910
15. Saurus P, Kuusela S, Lehtonen E, et al. Podocyte apoptosis is prevented by blocking the toll-like receptor pathway. Cell Death Dis. 2015;6(5):e1752. doi:10.1038/cddis.2015.125
16. Baud L, Oudinet JP, Bens M, et al. Production of tumor necrosis factor by rat mesangial cells in response to bacterial lipopolysaccharide. Kidney Int. 1989;35(5):1111-1118. doi:10.1038/ki.1989.98
17. White S, Lin L, Hu K. NF-κB and tPA signaling in kidney and other diseases. Cells. 2020;9(6):1348. doi:10.3390/cells9061348
18. Tesch GH, Lan HY, Atkins RC, Nikolic-Paterson DJ. Role of interleukin-1 in mesangial cell proliferation and matrix deposition in experimental mesangioproliferative nephritis. Am J Pathol. 1997;151(1):141-150.
19. Lai KN, Leung JCK, Chan LYY, et al. Podocyte injury induced by mesangial-derived cytokines in IgA Nephropathy. Nephrol Dial Transplant. 2009;24(1):62-72. doi:10.1093/ndt/gfn441
20. Saleem MA, Kobayashi Y. Cell biology and genetics of minimal change disease. F1000 Res. 2016;5: F1000 Faculty Rev-412. doi:10.12688/f1000research.7300.1
21. Kim KP, Williams CE, Lemmon CA. Cell-matrix interactions in renal fibrosis. Kidney Dial. 2022;2(4):607-624. doi:10.3390/kidneydial2040055
22. Zvaifler NJ. Relevance of the stroma and epithelial-mesenchymal transition (EMT) for the rheumatic diseases. Arthritis Res Ther. 2006;8(3):210. doi:10.1186/ar1963
23. Angeletti A, Magnasco A, Trivelli A, et al. Refractory minimal change disease and focal segmental glomerular sclerosis treated with Anakinra. Kidney Int Rep. 2021;7(1):121-124. doi:10.1016/j.ekir.2021.10.018
24. Trachtman H, Vento S, Herreshoff E, et al. Efficacy of galactose and adalimumab in patients with resistant focal segmental glomerulosclerosis: report of the font clinical trial group. BMC Nephrol. 2015;16:111. doi:10.1186/s12882-015-0094-5
25. Mariani LH, Eddy S, AlAkwaa FM, et al. Precision nephrology identified tumor necrosis factor activation variability in minimal change disease and focal segmental glomerulosclerosis. Kidney Int. 2023;103(3):565-579. doi:10.1016/j.kint.2022.10.023
26. Korshak L, Washington DL, Powell J, Nylen E, Kokkinos P. Kidney Disease in Veterans. US Dept of Veterans Affairs, Office of Health Equity. Updated May 13, 2020. Accessed June 28, 2024. https://www.va.gov/HEALTHEQUITY/Kidney_Disease_In_Veterans.asp
27. Malone AF, Phelan PJ, Hall G, et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014;86(6):1253-1259. doi:10.1038/ki.2014.305
Three Anomalies and a Complication: Ruptured Noncoronary Sinus of Valsalva Aneurysm, Atrial Septal Aneurysm, and Patent Foramen Ovale
A 53 year-old white male with a past medical history of hypertension, hyperlipidemia, and former tobacco use was referred to the Dayton VAMC in Ohio for symptoms that included shortness of breath and a recent abnormal stress test. The patient reported no history of known coronary artery disease (CAD), congestive heart failure, or other cardiovascular diseases. The patient also reported no recent fever, bacterial blood infection, syphilis infection, recreational drug use, or chest trauma.
A physical examination was remarkable for grade 3/6 continuous murmur at the 5th interspace to the left of the sternum and a loud “pistol shot” sound heard over the femoral artery. The patient had jugular venous distension and 2+ leg edema bilaterally. His vital signs were normal, and laboratory blood tests showed normal hemoglobin level and kidney function.
An electrocardiogram showed nonspecific ST segment changes and a transthoracic echocardiogram (TTE) revealed a high-velocity jet in the right atrium (RA) above the tricuspid valve concerning for sinus of Valsalva aneurysm (SVA). 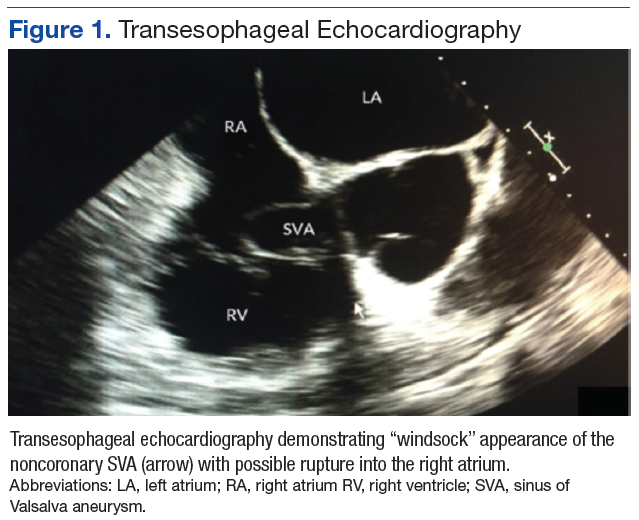
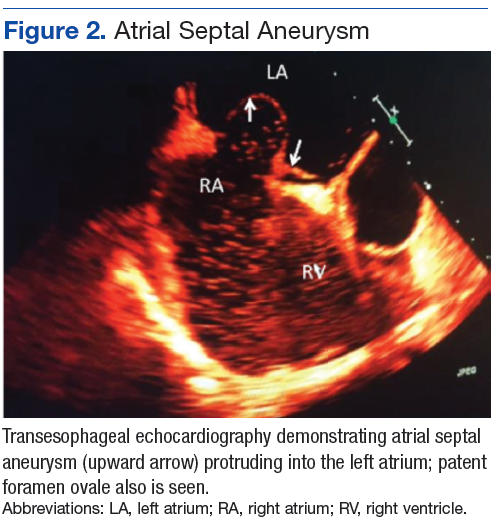
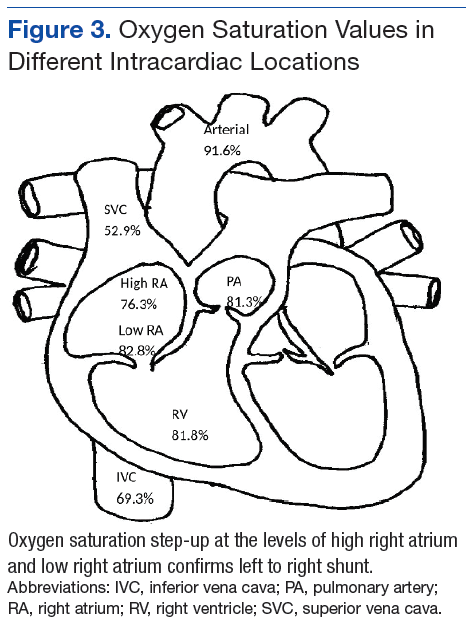
Right heart catheterization revealed elevated RA pressures with positive shunt study showing oxygen saturation step-up in the RA (Figure 3). Left heart hemodynamic measurement from an aortic approach to the distal part of the noncoronary cusp SVA revealed an RA pressure-tracing pattern consistent with rupture of the noncoronary SVA into the RA (Figure 4). 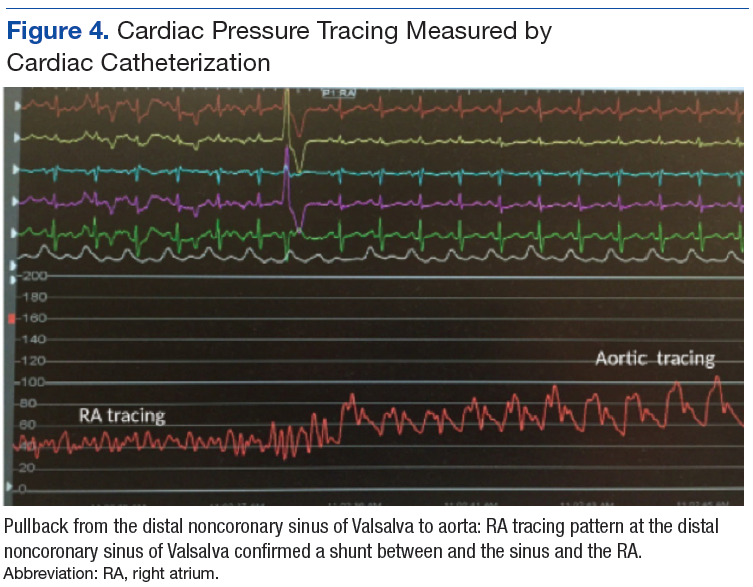
The primary diagnosis was of acute heart failure secondary to ruptured aneurysm of the noncoronary SVA into RA. The patient also received a secondary diagnosis of atrial septal aneurysm and PFO.
Treatment & Outcome
The patient was treated with aggressive diuresis and responded well to therapy. Considering the high mortality rate associated with a ruptured SVA, the patient was referred to a tertiary care center for surgical evaluation. He underwent repair of aorto-right atrial communication with a Cormatrix patch (Roswell, GA) from the aortic side and with primary closure from the right atrial side with resection of the windsock tract; coronary artery bypass graft x1 with right internal mammary artery to the right coronary artery; closure of the PFO with the Cormatrix patch.
The postoperative TEE confirmed preserved LV and RV function, no shunts, no aortic or tricuspid insufficiency. Biopsy of the tissue resected showed intimal fibroplasia. A TTE completed 1 year after surgery showed normal valvular function and without any structural abnormalities. The patient had improvement in symptoms and an uneventful year after surgical intervention followed by 24 session of cardiac rehabilitation.
Discussion
Sinus of Valsalva aneurysm is a dilation of the aortic wall between the aortic valve and the sinotubular junction that is caused by the lack of continuity between the middle layer of the aortic wall and the aortic valve.1 Cases of SVA are rare cardiac anomalies with prevalence of 1% in patients undergoing open-heart surgery.2 Between 65% and 85% of SVA cases originate from the right coronary sinus, 10% to 20% from the noncoronary sinus, and < 5% from the left coronary sinus.3
Sinus of Valsalva aneurysm is usually congenital, although cases associated with syphilis, bacterial endocarditis, trauma, Behçet disease, and aortic dissection have been reported. Structural defects associated with congenital SVAs include ventricular septal defect, bicuspid aortic valve, and aortic regurgitation. It is less commonly associated with pulmonary stenosis, coarctation of the aorta, patent ductus arteriosus, tricuspid regurgitation, and atrial septal defects.
The most common complication of the SVA is rupture into another cardiac chamber, frequently the right ventricle (60%) or RA (29%) and less frequently into left atrium (6%), left ventricle (4%), or pericardium (1%).1 Patients with ruptured SVA mainly develop dyspnea and chest pain, but cough, fatigue, peripheral edema, and continuous murmur have been reported.1
Atrial septal aneurysm is an uncommon finding in adults, with an incidence of 2.2 % in the general population, and it is often associated with atrial septal defect and PFO.1,4 Although ASA formation can be secondary to interatrial differences in pressures, it can be a primary malformation involving the region of the fossa ovalis or the entire atrial septum.4 Atrial septal aneurysm may be an isolated anomaly, but often is found in association with other structural cardiac anomalies, including SVA and PFO.4,5
Conclusion
Although coexistence of SVA and ASA has been reported previously, the case reported here, a ruptured noncoronary SVA that was associated with a large ASA and a PFO, has not been previously documented in the English literature. This patient’s anomalies are most likely congenital in origin. Progressive dyspnea and chest pain in the presence of a continuous loud murmur should raise the suspicion of ruptured sinus of Valsalva. Although no significant aortic regurgitation was noted on echocardiography, the pistol shot sound heard over the femoral artery was believed to be due to the rapid diastolic runoff into the RA through the ruptured SVA.
The significant increase in the RA pressure made the ASA and PFO more prominent. A TEE, left and right heart catheterizations with shunt study are vital for the diagnosis of SVA. If left untreated, SVA has an ominous prognosis. Surgical repair of ruptured SVA has an accepted risk and good prognosis with 10-year survival rate of 90%, whereas the mean survival of untreated ruptured SVA is about 4 years.6,7 Hence, the patient in this study was referred to a tertiary care center for surgical intervention.
1. Galicia-Tornell MM, Marín-Solís B, Mercado-Astorga O, Espinoza-Anguiano S, Martínez-Martínez M, Villalpando-Mendoza E. Sinus of Valsalva aneurysm with rupture. Case report and literature review. Cir Cir. 2009;77(6):441-445.
2. Takach TJ, Reul GJ, Duncan JM, et al. Sinus of Valsalva aneurysm or fistula: management and outcome. Ann Thorac Surg. 1999;68(5):1573-1577.
3. Meier JH, Seward JB, Miller FA Jr, Oh JK, Enriquez-Sarano M. Aneurysms in the left ventricular outflow tract: clinical presentation, causes, and echocardiographic features. J Am Soc Echocardiogr. 1998;11(7):729-745.
4. Mügge A, Daniel WG, Angermann C et al. Atrial septal aneurysm in adult patients: a multicenter study using transthoracic and transesophageal echocardiography. Circulation. 1995;91(11):2785-2792.
5. Silver MD, Dorsey JS. Aneurysms of the septum primum in adults. Arch Pathol Lab Med. 1978;102(2):62-65.
6. Wang ZJ, Zou CW, Li DC, et al. Surgical repair of sinus of Valsalva aneurysm in Asian patients. Ann Thorac Surg. 2007;84(1):156-160.
7. Yan F, Huo Q, Qiao J, Murat V, Ma SF. Surgery for sinus of valsalva aneurysm: 27-year experience with 100 patients. Asian Cardiovasc Thorac Ann. 2008;16(5):361-365.
A 53 year-old white male with a past medical history of hypertension, hyperlipidemia, and former tobacco use was referred to the Dayton VAMC in Ohio for symptoms that included shortness of breath and a recent abnormal stress test. The patient reported no history of known coronary artery disease (CAD), congestive heart failure, or other cardiovascular diseases. The patient also reported no recent fever, bacterial blood infection, syphilis infection, recreational drug use, or chest trauma.
A physical examination was remarkable for grade 3/6 continuous murmur at the 5th interspace to the left of the sternum and a loud “pistol shot” sound heard over the femoral artery. The patient had jugular venous distension and 2+ leg edema bilaterally. His vital signs were normal, and laboratory blood tests showed normal hemoglobin level and kidney function.
An electrocardiogram showed nonspecific ST segment changes and a transthoracic echocardiogram (TTE) revealed a high-velocity jet in the right atrium (RA) above the tricuspid valve concerning for sinus of Valsalva aneurysm (SVA).
Right heart catheterization revealed elevated RA pressures with positive shunt study showing oxygen saturation step-up in the RA (Figure 3). Left heart hemodynamic measurement from an aortic approach to the distal part of the noncoronary cusp SVA revealed an RA pressure-tracing pattern consistent with rupture of the noncoronary SVA into the RA (Figure 4).
The primary diagnosis was of acute heart failure secondary to ruptured aneurysm of the noncoronary SVA into RA. The patient also received a secondary diagnosis of atrial septal aneurysm and PFO.
Treatment & Outcome
The patient was treated with aggressive diuresis and responded well to therapy. Considering the high mortality rate associated with a ruptured SVA, the patient was referred to a tertiary care center for surgical evaluation. He underwent repair of aorto-right atrial communication with a Cormatrix patch (Roswell, GA) from the aortic side and with primary closure from the right atrial side with resection of the windsock tract; coronary artery bypass graft x1 with right internal mammary artery to the right coronary artery; closure of the PFO with the Cormatrix patch.
The postoperative TEE confirmed preserved LV and RV function, no shunts, no aortic or tricuspid insufficiency. Biopsy of the tissue resected showed intimal fibroplasia. A TTE completed 1 year after surgery showed normal valvular function and without any structural abnormalities. The patient had improvement in symptoms and an uneventful year after surgical intervention followed by 24 session of cardiac rehabilitation.
Discussion
Sinus of Valsalva aneurysm is a dilation of the aortic wall between the aortic valve and the sinotubular junction that is caused by the lack of continuity between the middle layer of the aortic wall and the aortic valve.1 Cases of SVA are rare cardiac anomalies with prevalence of 1% in patients undergoing open-heart surgery.2 Between 65% and 85% of SVA cases originate from the right coronary sinus, 10% to 20% from the noncoronary sinus, and < 5% from the left coronary sinus.3
Sinus of Valsalva aneurysm is usually congenital, although cases associated with syphilis, bacterial endocarditis, trauma, Behçet disease, and aortic dissection have been reported. Structural defects associated with congenital SVAs include ventricular septal defect, bicuspid aortic valve, and aortic regurgitation. It is less commonly associated with pulmonary stenosis, coarctation of the aorta, patent ductus arteriosus, tricuspid regurgitation, and atrial septal defects.
The most common complication of the SVA is rupture into another cardiac chamber, frequently the right ventricle (60%) or RA (29%) and less frequently into left atrium (6%), left ventricle (4%), or pericardium (1%).1 Patients with ruptured SVA mainly develop dyspnea and chest pain, but cough, fatigue, peripheral edema, and continuous murmur have been reported.1
Atrial septal aneurysm is an uncommon finding in adults, with an incidence of 2.2 % in the general population, and it is often associated with atrial septal defect and PFO.1,4 Although ASA formation can be secondary to interatrial differences in pressures, it can be a primary malformation involving the region of the fossa ovalis or the entire atrial septum.4 Atrial septal aneurysm may be an isolated anomaly, but often is found in association with other structural cardiac anomalies, including SVA and PFO.4,5
Conclusion
Although coexistence of SVA and ASA has been reported previously, the case reported here, a ruptured noncoronary SVA that was associated with a large ASA and a PFO, has not been previously documented in the English literature. This patient’s anomalies are most likely congenital in origin. Progressive dyspnea and chest pain in the presence of a continuous loud murmur should raise the suspicion of ruptured sinus of Valsalva. Although no significant aortic regurgitation was noted on echocardiography, the pistol shot sound heard over the femoral artery was believed to be due to the rapid diastolic runoff into the RA through the ruptured SVA.
The significant increase in the RA pressure made the ASA and PFO more prominent. A TEE, left and right heart catheterizations with shunt study are vital for the diagnosis of SVA. If left untreated, SVA has an ominous prognosis. Surgical repair of ruptured SVA has an accepted risk and good prognosis with 10-year survival rate of 90%, whereas the mean survival of untreated ruptured SVA is about 4 years.6,7 Hence, the patient in this study was referred to a tertiary care center for surgical intervention.
A 53 year-old white male with a past medical history of hypertension, hyperlipidemia, and former tobacco use was referred to the Dayton VAMC in Ohio for symptoms that included shortness of breath and a recent abnormal stress test. The patient reported no history of known coronary artery disease (CAD), congestive heart failure, or other cardiovascular diseases. The patient also reported no recent fever, bacterial blood infection, syphilis infection, recreational drug use, or chest trauma.
A physical examination was remarkable for grade 3/6 continuous murmur at the 5th interspace to the left of the sternum and a loud “pistol shot” sound heard over the femoral artery. The patient had jugular venous distension and 2+ leg edema bilaterally. His vital signs were normal, and laboratory blood tests showed normal hemoglobin level and kidney function.
An electrocardiogram showed nonspecific ST segment changes and a transthoracic echocardiogram (TTE) revealed a high-velocity jet in the right atrium (RA) above the tricuspid valve concerning for sinus of Valsalva aneurysm (SVA).
Right heart catheterization revealed elevated RA pressures with positive shunt study showing oxygen saturation step-up in the RA (Figure 3). Left heart hemodynamic measurement from an aortic approach to the distal part of the noncoronary cusp SVA revealed an RA pressure-tracing pattern consistent with rupture of the noncoronary SVA into the RA (Figure 4).
The primary diagnosis was of acute heart failure secondary to ruptured aneurysm of the noncoronary SVA into RA. The patient also received a secondary diagnosis of atrial septal aneurysm and PFO.
Treatment & Outcome
The patient was treated with aggressive diuresis and responded well to therapy. Considering the high mortality rate associated with a ruptured SVA, the patient was referred to a tertiary care center for surgical evaluation. He underwent repair of aorto-right atrial communication with a Cormatrix patch (Roswell, GA) from the aortic side and with primary closure from the right atrial side with resection of the windsock tract; coronary artery bypass graft x1 with right internal mammary artery to the right coronary artery; closure of the PFO with the Cormatrix patch.
The postoperative TEE confirmed preserved LV and RV function, no shunts, no aortic or tricuspid insufficiency. Biopsy of the tissue resected showed intimal fibroplasia. A TTE completed 1 year after surgery showed normal valvular function and without any structural abnormalities. The patient had improvement in symptoms and an uneventful year after surgical intervention followed by 24 session of cardiac rehabilitation.
Discussion
Sinus of Valsalva aneurysm is a dilation of the aortic wall between the aortic valve and the sinotubular junction that is caused by the lack of continuity between the middle layer of the aortic wall and the aortic valve.1 Cases of SVA are rare cardiac anomalies with prevalence of 1% in patients undergoing open-heart surgery.2 Between 65% and 85% of SVA cases originate from the right coronary sinus, 10% to 20% from the noncoronary sinus, and < 5% from the left coronary sinus.3
Sinus of Valsalva aneurysm is usually congenital, although cases associated with syphilis, bacterial endocarditis, trauma, Behçet disease, and aortic dissection have been reported. Structural defects associated with congenital SVAs include ventricular septal defect, bicuspid aortic valve, and aortic regurgitation. It is less commonly associated with pulmonary stenosis, coarctation of the aorta, patent ductus arteriosus, tricuspid regurgitation, and atrial septal defects.
The most common complication of the SVA is rupture into another cardiac chamber, frequently the right ventricle (60%) or RA (29%) and less frequently into left atrium (6%), left ventricle (4%), or pericardium (1%).1 Patients with ruptured SVA mainly develop dyspnea and chest pain, but cough, fatigue, peripheral edema, and continuous murmur have been reported.1
Atrial septal aneurysm is an uncommon finding in adults, with an incidence of 2.2 % in the general population, and it is often associated with atrial septal defect and PFO.1,4 Although ASA formation can be secondary to interatrial differences in pressures, it can be a primary malformation involving the region of the fossa ovalis or the entire atrial septum.4 Atrial septal aneurysm may be an isolated anomaly, but often is found in association with other structural cardiac anomalies, including SVA and PFO.4,5
Conclusion
Although coexistence of SVA and ASA has been reported previously, the case reported here, a ruptured noncoronary SVA that was associated with a large ASA and a PFO, has not been previously documented in the English literature. This patient’s anomalies are most likely congenital in origin. Progressive dyspnea and chest pain in the presence of a continuous loud murmur should raise the suspicion of ruptured sinus of Valsalva. Although no significant aortic regurgitation was noted on echocardiography, the pistol shot sound heard over the femoral artery was believed to be due to the rapid diastolic runoff into the RA through the ruptured SVA.
The significant increase in the RA pressure made the ASA and PFO more prominent. A TEE, left and right heart catheterizations with shunt study are vital for the diagnosis of SVA. If left untreated, SVA has an ominous prognosis. Surgical repair of ruptured SVA has an accepted risk and good prognosis with 10-year survival rate of 90%, whereas the mean survival of untreated ruptured SVA is about 4 years.6,7 Hence, the patient in this study was referred to a tertiary care center for surgical intervention.
1. Galicia-Tornell MM, Marín-Solís B, Mercado-Astorga O, Espinoza-Anguiano S, Martínez-Martínez M, Villalpando-Mendoza E. Sinus of Valsalva aneurysm with rupture. Case report and literature review. Cir Cir. 2009;77(6):441-445.
2. Takach TJ, Reul GJ, Duncan JM, et al. Sinus of Valsalva aneurysm or fistula: management and outcome. Ann Thorac Surg. 1999;68(5):1573-1577.
3. Meier JH, Seward JB, Miller FA Jr, Oh JK, Enriquez-Sarano M. Aneurysms in the left ventricular outflow tract: clinical presentation, causes, and echocardiographic features. J Am Soc Echocardiogr. 1998;11(7):729-745.
4. Mügge A, Daniel WG, Angermann C et al. Atrial septal aneurysm in adult patients: a multicenter study using transthoracic and transesophageal echocardiography. Circulation. 1995;91(11):2785-2792.
5. Silver MD, Dorsey JS. Aneurysms of the septum primum in adults. Arch Pathol Lab Med. 1978;102(2):62-65.
6. Wang ZJ, Zou CW, Li DC, et al. Surgical repair of sinus of Valsalva aneurysm in Asian patients. Ann Thorac Surg. 2007;84(1):156-160.
7. Yan F, Huo Q, Qiao J, Murat V, Ma SF. Surgery for sinus of valsalva aneurysm: 27-year experience with 100 patients. Asian Cardiovasc Thorac Ann. 2008;16(5):361-365.
1. Galicia-Tornell MM, Marín-Solís B, Mercado-Astorga O, Espinoza-Anguiano S, Martínez-Martínez M, Villalpando-Mendoza E. Sinus of Valsalva aneurysm with rupture. Case report and literature review. Cir Cir. 2009;77(6):441-445.
2. Takach TJ, Reul GJ, Duncan JM, et al. Sinus of Valsalva aneurysm or fistula: management and outcome. Ann Thorac Surg. 1999;68(5):1573-1577.
3. Meier JH, Seward JB, Miller FA Jr, Oh JK, Enriquez-Sarano M. Aneurysms in the left ventricular outflow tract: clinical presentation, causes, and echocardiographic features. J Am Soc Echocardiogr. 1998;11(7):729-745.
4. Mügge A, Daniel WG, Angermann C et al. Atrial septal aneurysm in adult patients: a multicenter study using transthoracic and transesophageal echocardiography. Circulation. 1995;91(11):2785-2792.
5. Silver MD, Dorsey JS. Aneurysms of the septum primum in adults. Arch Pathol Lab Med. 1978;102(2):62-65.
6. Wang ZJ, Zou CW, Li DC, et al. Surgical repair of sinus of Valsalva aneurysm in Asian patients. Ann Thorac Surg. 2007;84(1):156-160.
7. Yan F, Huo Q, Qiao J, Murat V, Ma SF. Surgery for sinus of valsalva aneurysm: 27-year experience with 100 patients. Asian Cardiovasc Thorac Ann. 2008;16(5):361-365.
Following the Hyperkalemia Trail: A Case Report of ECG Changes and Treatment Responses
Following the Hyperkalemia Trail: A Case Report of ECG Changes and Treatment Responses
Hyperkalemia involves elevated serum potassium levels (> 5.0 mEq/L) and represents an important electrolyte disturbance due to its potentially severe consequences, including cardiac effects that can lead to dysrhythmia and even asystole and death.1,2 In a US Medicare population, the prevalence of hyperkalemia has been estimated at 2.7% and is associated with substantial health care costs.3 The prevalence is even more marked in patients with preexisting conditions such as chronic kidney disease (CKD) and heart failure.4,5
Hyperkalemia can result from multiple factors, including impaired renal function, adrenal disease, adverse drug reactions of angiotensin-converting enzyme inhibitors (ACEIs) and other medications, and heritable mutations.6 Hyperkalemia poses a considerable clinical risk, associated with adverse outcomes such as myocardial infarction and increased mortality in patients with CKD.5,7,8 Electrocardiographic (ECG) changes associated with hyperkalemia play a vital role in guiding clinical decisions and treatment strategies.9 Understanding the pathophysiology, risk factors, and consequences of hyperkalemia, as well as the significance of ECG changes in its management, is essential for health care practitioners.
Case Presentation
An 81-year-old Hispanic man with a history of hypertension, hypothyroidism, gout, and CKD stage 3B presented to the emergency department with progressive weakness resulting in falls and culminating in an inability to ambulate independently. Additional symptoms included nausea, diarrhea, and myalgia. His vital signs were notable for a pulse of 41 beats/min. The physical examination was remarkable for significant weakness of the bilateral upper extremities, inability to bear his own weight, and bilateral lower extremity edema. His initial ECG upon arrival showed bradycardia with wide QRS, absent P waves, and peaked T waves (Figure 1a). These findings differed from his baseline ECG taken 1 year earlier, which showed sinus rhythm with premature atrial complexes and an old right bundle branch block (Figure 1b).
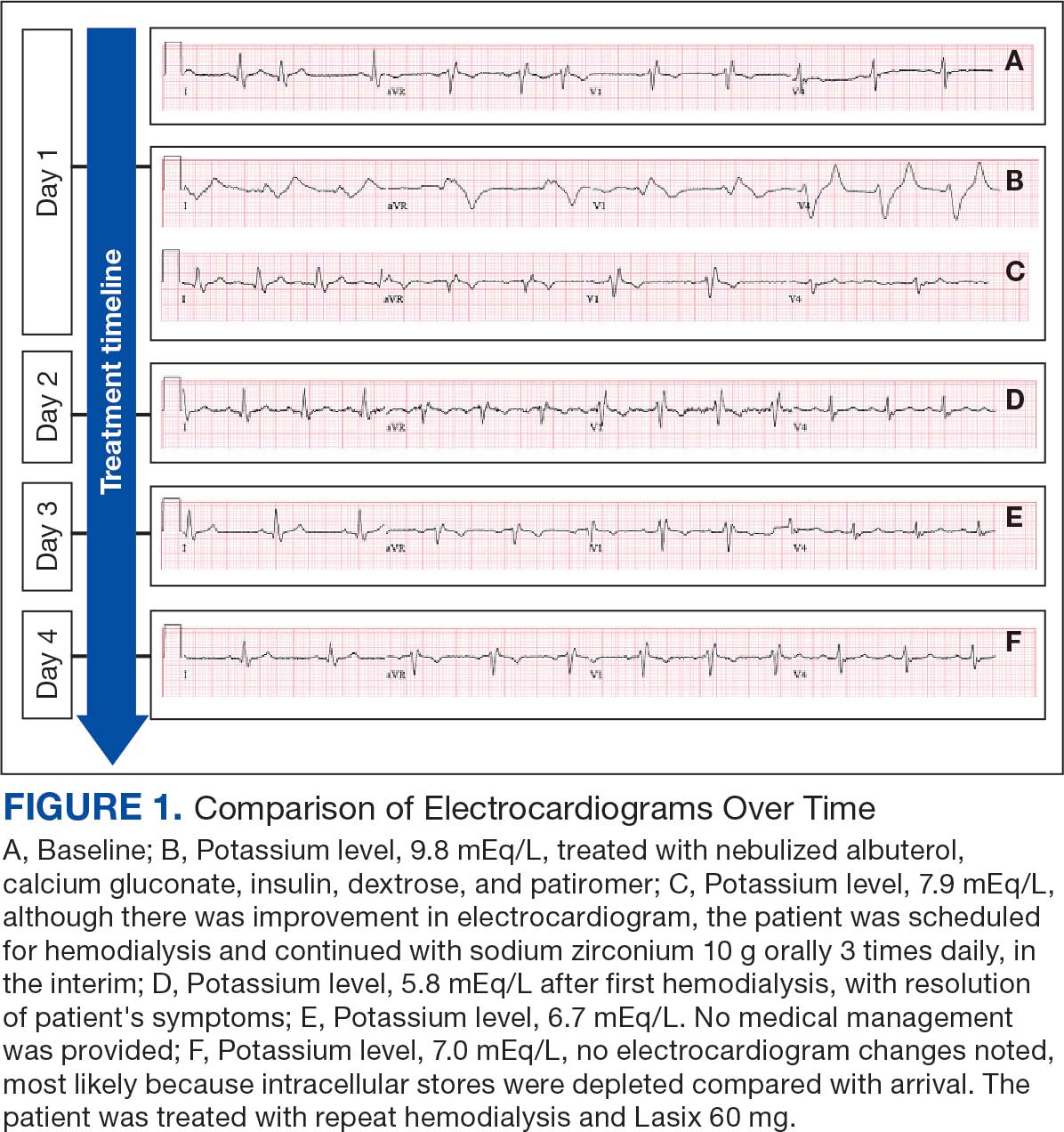
Medication review revealed that the patient was currently prescribed 100 mg allopurinol daily, 2.5 mg amlodipine daily, 10 mg atorvastatin at bedtime, 4 mg doxazosin daily, 112 mcg levothyroxine daily, 100 mg losartan daily, 25 mg metoprolol daily, and 0.4 mg tamsulosin daily. The patient had also been taking over-the-counter indomethacin for knee pain.
Based on the ECG results, he was treated with 0.083%/6 mL nebulized albuterol, 4.65 Mq/250 mL saline solution intravenous (IV) calcium gluconate, 10 units IV insulin with concomitant 50%/25 mL IV dextrose and 8.4 g of oral patiromer suspension. IV furosemide was held due to concern for renal function. The decision to proceed with hemodialysis was made. Repeat laboratory tests were performed, and an ECG obtained after treatment initiation but prior to hemodialysis demonstrated improvement of rate and T wave shortening (Figure 1c). The serum potassium level dropped from 9.8 mEq/L to 7.9 mEq/L (reference range, 3.5-5.0 mEq/L) (Table 1).
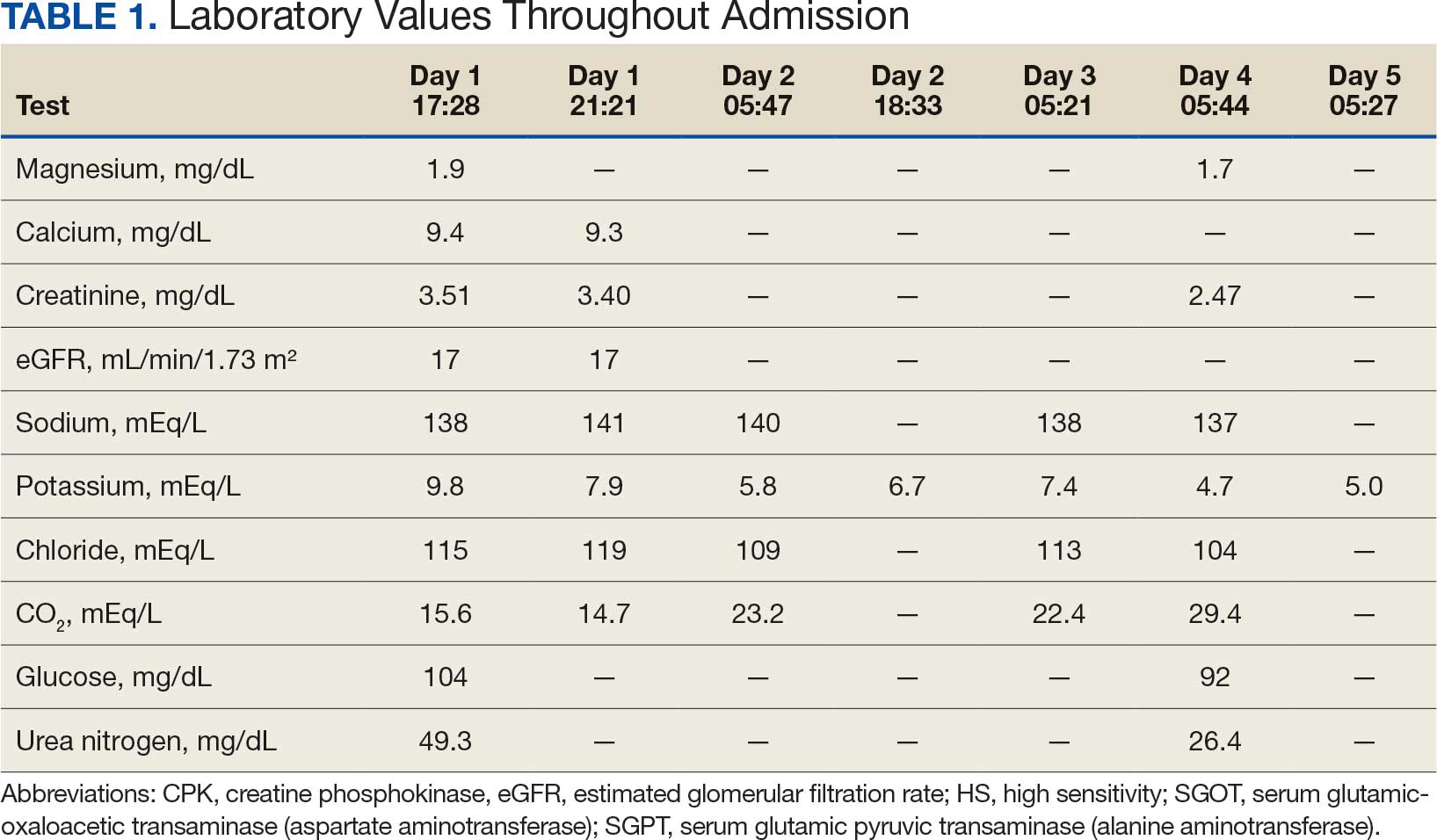
In addition to hemodialysis, sodium zirconium 10 g orally 3 times daily was added. Laboratory test results and an ECG was performed after dialysis continued to demonstrate improvement (Figure 1d). The patient’s potassium level decreased to 5.8 mEq/L, with the ECG demonstrating stability of heart rate and further improvement of the PR interval, QRS complex, and T waves.
Despite the established treatment regimen, potassium levels again rose to 6.7 mEq/L, but there were no significant changes in the ECG, and thus no medication changes were made (Figure 1e). Subsequent monitoring demonstrated a further increase in potassium to 7.4 mEq/L, with an ECG demonstrating a return to the baseline of 1 year prior. The patient underwent hemodialysis again and was given oral furosemide 60 mg every 12 hours. The potassium concentration after dialysis decreased to 4.7 mEq/L and remained stable, not going above 5.0 mEq/L on subsequent monitoring. The patient had resolution of all symptoms and was discharged.
Discussion
We have described in detail the presentation of each pathology and mechanisms of each treatment, starting with the patient’s initial condition that brought him to the emergency room—muscle weakness. Skeletal muscle weakness is a common manifestation of hyperkalemia, occurring in 20% to 40% of cases, and is more prevalent in severe elevations of potassium. Rarely, the weakness can progress to flaccid paralysis of the patient’s extremities and, in extreme cases, the diaphragm.
Muscle weakness progression occurs in a manner that resembles Guillain-Barré syndrome, starting in the lower extremities and ascending toward the upper extremities.10 This is known as secondary hyperkalemic periodic paralysis. Hyperkalemia lowers the transmembrane gradient in neurons, leading to neuronal depolarization independent of the degree of hyperkalemia. If the degree of hyperkalemia is large enough, this depolarization inactivates voltage-gated sodium channels, making neurons refractory to excitation. Electromyographical studies have shown reduction in the compounded muscle action potential.11 The transient nature of this paralysis is reflected by rapid correction of weakness and paralysis when the electrolyte disorder is corrected.
The patient in this case also presented with bradycardia. The ECG manifestations of hyperkalemia can include atrial asystole, intraventricular conduction disturbances, peaked T waves, and widened QRS complexes. However, some patients with renal insufficiency may not exhibit ECG changes despite significantly elevated serum potassium levels.12
The severity of hyperkalemia is crucial in determining the associated ECG changes, with levels > 6.0 mEq/L presenting with abnormalities.13 ECG findings alone may not always accurately reflect the severity of hyperkalemia, as up to 60% of patients with potassium levels > 6.0 mEq/L may not show ECG changes.14 Additionally, extreme hyperkalemia can lead to inconsistent ECG findings, making it challenging to rely solely on ECG for diagnosis and monitoring.8 The level of potassium that causes these effects varies widely through patient populations.
The main mechanism by which hyperkalemia affects the heart’s conduction system is through voltage differences across the conduction fibers and eventual steady-state inactivation of sodium channels. This combination of mechanisms shortens the action potential duration, allowing more cardiomyocytes to undergo synchronized depolarization. This amalgamation of cardiomyocytes repolarizing can be reflected on ECGs as peaked T waves. As the action potential decreases, there is a period during which cardiomyocytes are prone to tachyarrhythmias and ventricular fibrillation.
A reduced action potential may lead to increased rates of depolarization and thus conduction, which in some scenarios may increase heart rate. As the levels of potassium rise, intracellular accumulation impedes the entry of sodium by decreasing the cation gradient across the cell membrane. This effectively slows the sinus nodes and prolongs the QRS by slowing the overall propagation of action potentials. By this mechanism, conduction delays, blocks, or asystole are manifested. The patient in this case showed conduction delays, peaked T waves, and disappearance of P waves when he first arrived.
Hyperkalemia Treatment
Hyperkalemia develops most commonly due to acute or chronic kidney diseases, as was the case with this patient. The patient’s hyperkalemia was also augmented by the use of nonsteroidal anti-inflammatory drugs (NSAIDs), which can directly affect renal function. A properly functioning kidney is responsible for excretion of up to 90% of ingested potassium, while the remainder is excreted through the gastrointestinal (GI) tract. Definitive treatment of hyperkalemia is mitigated primarily through these 2 organ systems. The treatment also includes transitory mechanisms of potassium reduction. The goal of each method is to preserve the action potential of cardiomyocytes and myocytes. This patient presented with acute symptomatic hyperkalemia and received various medications to acutely, transitorily, and definitively treat it.
Initial therapy included calcium gluconate, which functions to stabilize the myocardial cell membrane. Hyperkalemia decreases the resting membrane action potential of excitable cells and predisposes them to early depolarization and thus dysrhythmias. Calcium decreases the threshold potential across cells and offsets the overall gradient back to near normal levels.15 Calcium can be delivered through calcium gluconate or calcium chloride. Calcium chloride is not preferred because extravasation can cause pain, blistering and tissue ischemia. Central venous access is required, potentially delaying prompt treatment. Calcium acts rapidly after administration—within 1 to 3 minutes—but only lasts 30 to 60 minutes.16 Administration of calcium gluconate can be repeated as often as necessary, but patients must be monitored for adverse effects of calcium such as nausea, abdominal pain, polydipsia, polyuria, muscle weakness, and paresthesia. Care must be taken when patients are taking digoxin, because calcium may potentiate toxicity.17 Although calcium provides immediate benefits it does little to correct the underlying cause; other medications are required to remove potassium from the body.
Two medication classes have been proven to shift potassium intracellularly. The first are β-2 agonists, such as albuterol/levalbuterol, and the second is insulin. Both work through sodium-potassium-ATPase in a direct manner. β-2 agonists stimulate sodium-potassium-ATPase to move more potassium intracellularly, but these effects have been seen only with high doses of albuterol, typically 4× the standard dose of 0.5 mg in nebulized solutions to achieve decreases in potassium of 0.3 to 0.6 mEq/L, although some trials have reported decreases of 0.62 to 0.98 mEq/L.15,18 These potassium-lowering effects of β-2 agonist are modest, but can be seen 20 to 30 minutes after administration and persist up to 1 to 2 hours. β-2 agonists are also readily affected by β blockers, which may reduce or negate the desired effect in hyperkalemia. For these reasons, a β-2 agonist should not be given as monotherapy and should be provided as an adjuvant to more independent therapies such as insulin. Insulin binds to receptors on muscle cells and increases the quantity of sodium-potassium-ATPase and glucose transporters. With this increase in influx pumps, surrounding tissues with higher resting membrane potentials can absorb the potassium load, thereby protecting cardiomyocytes.
Potassium Removal
Three methods are currently available to remove potassium from the body: GI excretion, renal excretion, and direct removal from the bloodstream. Under normal physiologic conditions, the kidneys account for about 90% of the body’s ability to remove potassium. Loop diuretics facilitate the removal of potassium by increasing urine production and have an additional potassium-wasting effect. Although the onset of action of loop diuretics is typically 30 to 60 minutes after oral administration, their effect can last for several hours. In this patient, furosemide was introduced later in the treatment plan to manage recurring hyperkalemia by enhancing renal potassium excretion.
Potassium binders such as patiromer act in the GI tract, effectively reducing serum potassium levels although with a slower onset of action than furosemide, generally taking hours to days to exert its effect. Both medications illustrate a tailored approach to managing potassium levels, adapted to the evolving needs and renal function of the patient. The last method is using hemodialysis—by far the most rapid method to remove potassium, but also the most invasive. The different methods of treating hyperkalemia are summarized in Table 2. This patient required multiple days of hemodialysis to completely correct the electrolyte disorder. Upon discharge, the patient continued oral furosemide 40 mg daily and eventually discontinued hemodialysis due to stable renal function.
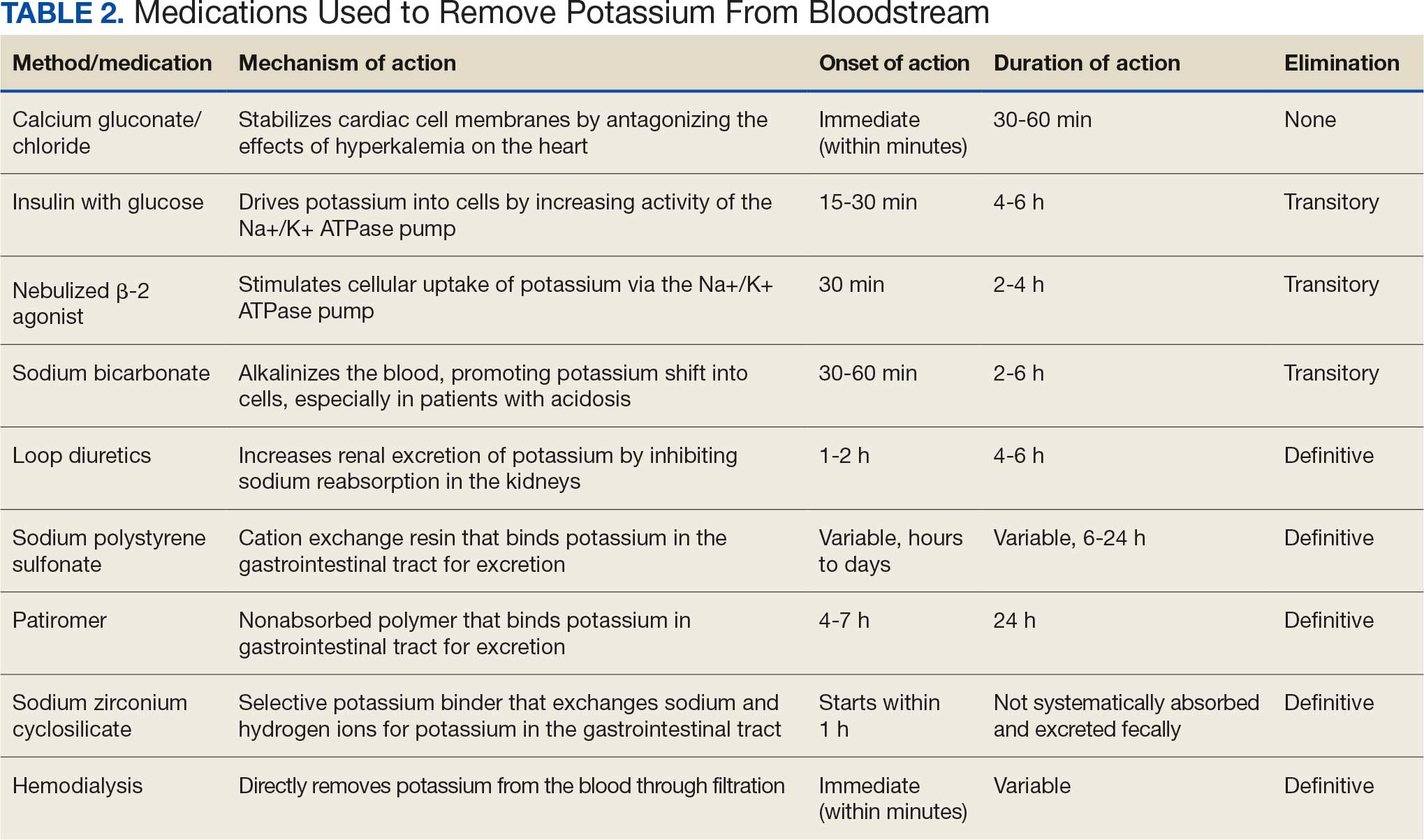
Often, after correcting an inciting event, potassium stores in the body eventually stabilize and do not require additional follow-up. Patients prone to hyperkalemia should be thoroughly educated on medications to avoid (NSAIDs, ACEIs/ARBs, trimethoprim), an adequate low potassium diet, and symptoms that may warrant medical attention.19
Conclusions
This case illustrates the importance of recognizing the spectrum of manifestations of hyperkalemia, which ranged from muscle weakness to cardiac dysrhythmias. Management strategies for the patient included stabilization of cardiac membranes, potassium shifting, and potassium removal, each tailored to the patient’s individual clinical findings.
The case further illustrates the critical role of continuous monitoring and dynamic adjustment of therapeutic strategies in response to evolving clinical and laboratory findings. The initial and subsequent ECGs, alongside laboratory tests, were instrumental in guiding the adjustments needed in the treatment regimen, ensuring both the efficacy and safety of the interventions. This proactive approach can mitigate the risk of recurrent hyperkalemia and its complications.
- Youn JH, McDonough AA. Recent advances in understanding integrative control of potassium homeostasis. Annu Rev Physiol. 2009;71:381-401. doi:10.1146/annurev.physiol.010908.163241 2.
- Simon LV, Hashmi MF, Farrell MW. Hyperkalemia. In: StatPearls. StatPearls Publishing; September 4, 2023. Accessed October 22, 2025.
- Mu F, Betts KA, Woolley JM, et al. Prevalence and economic burden of hyperkalemia in the United States Medicare population. Curr Med Res Opin. 2020;36:1333-1341. doi:10.1080/03007995.2020.1775072
- Loutradis C, Tolika P, Skodra A, et al. Prevalence of hyperkalemia in diabetic and non-diabetic patients with chronic kidney disease: a nested case-control study. Am J Nephrol. 2015;42:351-360. doi:10.1159/000442393
- Grodzinsky A, Goyal A, Gosch K, et al. Prevalence and prognosis of hyperkalemia in patients with acute myocardial infarction. Am J Med. 2016;129:858-865. doi:10.1016/j.amjmed.2016.03.008
- Hunter RW, Bailey MA. Hyperkalemia: pathophysiology, risk factors and consequences. Nephrol Dial Transplant. 2019;34(suppl 3):iii2-iii11. doi:10.1093/ndt/gfz206
- Luo J, Brunelli SM, Jensen DE, Yang A. Association between serum potassium and outcomes in patients with reduced kidney function. Clin J Am Soc Nephrol. 2016;11:90-100. doi:10.2215/CJN.01730215
- Montford JR, Linas S. How dangerous is hyperkalemia? J Am Soc Nephrol. 2017;28:3155-3165. doi:10.1681/ASN.2016121344
- Mattu A, Brady WJ, Robinson DA. Electrocardiographic manifestations of hyperkalemia. Am J Emerg Med. 2000;18:721-729. doi:10.1053/ajem.2000.7344
- Kimmons LA, Usery JB. Acute ascending muscle weakness secondary to medication-induced hyperkalemia. Case Rep Med. 2014;2014:789529. doi:10.1155/2014/789529
- Naik KR, Saroja AO, Khanpet MS. Reversible electrophysiological abnormalities in acute secondary hyperkalemic paralysis. Ann Indian Acad Neurol. 2012;15:339-343. doi:10.4103/0972-2327.104354
- Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol. 2008;3:324-330. doi:10.2215/CJN.04611007
- Larivée NL, Michaud JB, More KM, Wilson JA, Tennankore KK. Hyperkalemia: prevalence, predictors and emerging treatments. Cardiol Ther. 2023;12:35-63. doi:10.1007/s40119-022-00289-z
- Shingarev R, Allon M. A physiologic-based approach to the treatment of acute hyperkalemia. Am J Kidney Dis. 2010;56:578-584. doi:10.1053/j.ajkd.2010.03.014
- Parham WA, Mehdirad AA, Biermann KM, Fredman CS. Hyperkalemia revisited. Tex Heart Inst J. 2006;33:40-47.
- Ng KE, Lee CS. Updated treatment options in the management of hyperkalemia. U.S. Pharmacist. February 16, 2017. Accessed October 1, 2025. www.uspharmacist.com/article/updated-treatment-options-in-the-management-of-hyperkalemia
- Quick G, Bastani B. Prolonged asystolic hyperkalemic cardiac arrest with no neurologic sequelae. Ann Emerg Med. 1994;24:305-311. doi:10.1016/s0196-0644(94)70144-x 18.
- Allon M, Dunlay R, Copkney C. Nebulized albuterol for acute hyperkalemia in patients on hemodialysis. Ann Intern Med. 1989;110:426-429. doi:10.7326/0003-4819-110-6-42619.
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024;105(4 suppl):S117-S314. doi:10.1016/j.kint.2023.10.018
Hyperkalemia involves elevated serum potassium levels (> 5.0 mEq/L) and represents an important electrolyte disturbance due to its potentially severe consequences, including cardiac effects that can lead to dysrhythmia and even asystole and death.1,2 In a US Medicare population, the prevalence of hyperkalemia has been estimated at 2.7% and is associated with substantial health care costs.3 The prevalence is even more marked in patients with preexisting conditions such as chronic kidney disease (CKD) and heart failure.4,5
Hyperkalemia can result from multiple factors, including impaired renal function, adrenal disease, adverse drug reactions of angiotensin-converting enzyme inhibitors (ACEIs) and other medications, and heritable mutations.6 Hyperkalemia poses a considerable clinical risk, associated with adverse outcomes such as myocardial infarction and increased mortality in patients with CKD.5,7,8 Electrocardiographic (ECG) changes associated with hyperkalemia play a vital role in guiding clinical decisions and treatment strategies.9 Understanding the pathophysiology, risk factors, and consequences of hyperkalemia, as well as the significance of ECG changes in its management, is essential for health care practitioners.
Case Presentation
An 81-year-old Hispanic man with a history of hypertension, hypothyroidism, gout, and CKD stage 3B presented to the emergency department with progressive weakness resulting in falls and culminating in an inability to ambulate independently. Additional symptoms included nausea, diarrhea, and myalgia. His vital signs were notable for a pulse of 41 beats/min. The physical examination was remarkable for significant weakness of the bilateral upper extremities, inability to bear his own weight, and bilateral lower extremity edema. His initial ECG upon arrival showed bradycardia with wide QRS, absent P waves, and peaked T waves (Figure 1a). These findings differed from his baseline ECG taken 1 year earlier, which showed sinus rhythm with premature atrial complexes and an old right bundle branch block (Figure 1b).
Medication review revealed that the patient was currently prescribed 100 mg allopurinol daily, 2.5 mg amlodipine daily, 10 mg atorvastatin at bedtime, 4 mg doxazosin daily, 112 mcg levothyroxine daily, 100 mg losartan daily, 25 mg metoprolol daily, and 0.4 mg tamsulosin daily. The patient had also been taking over-the-counter indomethacin for knee pain.
Based on the ECG results, he was treated with 0.083%/6 mL nebulized albuterol, 4.65 Mq/250 mL saline solution intravenous (IV) calcium gluconate, 10 units IV insulin with concomitant 50%/25 mL IV dextrose and 8.4 g of oral patiromer suspension. IV furosemide was held due to concern for renal function. The decision to proceed with hemodialysis was made. Repeat laboratory tests were performed, and an ECG obtained after treatment initiation but prior to hemodialysis demonstrated improvement of rate and T wave shortening (Figure 1c). The serum potassium level dropped from 9.8 mEq/L to 7.9 mEq/L (reference range, 3.5-5.0 mEq/L) (Table 1).
In addition to hemodialysis, sodium zirconium 10 g orally 3 times daily was added. Laboratory test results and an ECG was performed after dialysis continued to demonstrate improvement (Figure 1d). The patient’s potassium level decreased to 5.8 mEq/L, with the ECG demonstrating stability of heart rate and further improvement of the PR interval, QRS complex, and T waves.
Despite the established treatment regimen, potassium levels again rose to 6.7 mEq/L, but there were no significant changes in the ECG, and thus no medication changes were made (Figure 1e). Subsequent monitoring demonstrated a further increase in potassium to 7.4 mEq/L, with an ECG demonstrating a return to the baseline of 1 year prior. The patient underwent hemodialysis again and was given oral furosemide 60 mg every 12 hours. The potassium concentration after dialysis decreased to 4.7 mEq/L and remained stable, not going above 5.0 mEq/L on subsequent monitoring. The patient had resolution of all symptoms and was discharged.
Discussion
We have described in detail the presentation of each pathology and mechanisms of each treatment, starting with the patient’s initial condition that brought him to the emergency room—muscle weakness. Skeletal muscle weakness is a common manifestation of hyperkalemia, occurring in 20% to 40% of cases, and is more prevalent in severe elevations of potassium. Rarely, the weakness can progress to flaccid paralysis of the patient’s extremities and, in extreme cases, the diaphragm.
Muscle weakness progression occurs in a manner that resembles Guillain-Barré syndrome, starting in the lower extremities and ascending toward the upper extremities.10 This is known as secondary hyperkalemic periodic paralysis. Hyperkalemia lowers the transmembrane gradient in neurons, leading to neuronal depolarization independent of the degree of hyperkalemia. If the degree of hyperkalemia is large enough, this depolarization inactivates voltage-gated sodium channels, making neurons refractory to excitation. Electromyographical studies have shown reduction in the compounded muscle action potential.11 The transient nature of this paralysis is reflected by rapid correction of weakness and paralysis when the electrolyte disorder is corrected.
The patient in this case also presented with bradycardia. The ECG manifestations of hyperkalemia can include atrial asystole, intraventricular conduction disturbances, peaked T waves, and widened QRS complexes. However, some patients with renal insufficiency may not exhibit ECG changes despite significantly elevated serum potassium levels.12
The severity of hyperkalemia is crucial in determining the associated ECG changes, with levels > 6.0 mEq/L presenting with abnormalities.13 ECG findings alone may not always accurately reflect the severity of hyperkalemia, as up to 60% of patients with potassium levels > 6.0 mEq/L may not show ECG changes.14 Additionally, extreme hyperkalemia can lead to inconsistent ECG findings, making it challenging to rely solely on ECG for diagnosis and monitoring.8 The level of potassium that causes these effects varies widely through patient populations.
The main mechanism by which hyperkalemia affects the heart’s conduction system is through voltage differences across the conduction fibers and eventual steady-state inactivation of sodium channels. This combination of mechanisms shortens the action potential duration, allowing more cardiomyocytes to undergo synchronized depolarization. This amalgamation of cardiomyocytes repolarizing can be reflected on ECGs as peaked T waves. As the action potential decreases, there is a period during which cardiomyocytes are prone to tachyarrhythmias and ventricular fibrillation.
A reduced action potential may lead to increased rates of depolarization and thus conduction, which in some scenarios may increase heart rate. As the levels of potassium rise, intracellular accumulation impedes the entry of sodium by decreasing the cation gradient across the cell membrane. This effectively slows the sinus nodes and prolongs the QRS by slowing the overall propagation of action potentials. By this mechanism, conduction delays, blocks, or asystole are manifested. The patient in this case showed conduction delays, peaked T waves, and disappearance of P waves when he first arrived.
Hyperkalemia Treatment
Hyperkalemia develops most commonly due to acute or chronic kidney diseases, as was the case with this patient. The patient’s hyperkalemia was also augmented by the use of nonsteroidal anti-inflammatory drugs (NSAIDs), which can directly affect renal function. A properly functioning kidney is responsible for excretion of up to 90% of ingested potassium, while the remainder is excreted through the gastrointestinal (GI) tract. Definitive treatment of hyperkalemia is mitigated primarily through these 2 organ systems. The treatment also includes transitory mechanisms of potassium reduction. The goal of each method is to preserve the action potential of cardiomyocytes and myocytes. This patient presented with acute symptomatic hyperkalemia and received various medications to acutely, transitorily, and definitively treat it.
Initial therapy included calcium gluconate, which functions to stabilize the myocardial cell membrane. Hyperkalemia decreases the resting membrane action potential of excitable cells and predisposes them to early depolarization and thus dysrhythmias. Calcium decreases the threshold potential across cells and offsets the overall gradient back to near normal levels.15 Calcium can be delivered through calcium gluconate or calcium chloride. Calcium chloride is not preferred because extravasation can cause pain, blistering and tissue ischemia. Central venous access is required, potentially delaying prompt treatment. Calcium acts rapidly after administration—within 1 to 3 minutes—but only lasts 30 to 60 minutes.16 Administration of calcium gluconate can be repeated as often as necessary, but patients must be monitored for adverse effects of calcium such as nausea, abdominal pain, polydipsia, polyuria, muscle weakness, and paresthesia. Care must be taken when patients are taking digoxin, because calcium may potentiate toxicity.17 Although calcium provides immediate benefits it does little to correct the underlying cause; other medications are required to remove potassium from the body.
Two medication classes have been proven to shift potassium intracellularly. The first are β-2 agonists, such as albuterol/levalbuterol, and the second is insulin. Both work through sodium-potassium-ATPase in a direct manner. β-2 agonists stimulate sodium-potassium-ATPase to move more potassium intracellularly, but these effects have been seen only with high doses of albuterol, typically 4× the standard dose of 0.5 mg in nebulized solutions to achieve decreases in potassium of 0.3 to 0.6 mEq/L, although some trials have reported decreases of 0.62 to 0.98 mEq/L.15,18 These potassium-lowering effects of β-2 agonist are modest, but can be seen 20 to 30 minutes after administration and persist up to 1 to 2 hours. β-2 agonists are also readily affected by β blockers, which may reduce or negate the desired effect in hyperkalemia. For these reasons, a β-2 agonist should not be given as monotherapy and should be provided as an adjuvant to more independent therapies such as insulin. Insulin binds to receptors on muscle cells and increases the quantity of sodium-potassium-ATPase and glucose transporters. With this increase in influx pumps, surrounding tissues with higher resting membrane potentials can absorb the potassium load, thereby protecting cardiomyocytes.
Potassium Removal
Three methods are currently available to remove potassium from the body: GI excretion, renal excretion, and direct removal from the bloodstream. Under normal physiologic conditions, the kidneys account for about 90% of the body’s ability to remove potassium. Loop diuretics facilitate the removal of potassium by increasing urine production and have an additional potassium-wasting effect. Although the onset of action of loop diuretics is typically 30 to 60 minutes after oral administration, their effect can last for several hours. In this patient, furosemide was introduced later in the treatment plan to manage recurring hyperkalemia by enhancing renal potassium excretion.
Potassium binders such as patiromer act in the GI tract, effectively reducing serum potassium levels although with a slower onset of action than furosemide, generally taking hours to days to exert its effect. Both medications illustrate a tailored approach to managing potassium levels, adapted to the evolving needs and renal function of the patient. The last method is using hemodialysis—by far the most rapid method to remove potassium, but also the most invasive. The different methods of treating hyperkalemia are summarized in Table 2. This patient required multiple days of hemodialysis to completely correct the electrolyte disorder. Upon discharge, the patient continued oral furosemide 40 mg daily and eventually discontinued hemodialysis due to stable renal function.
Often, after correcting an inciting event, potassium stores in the body eventually stabilize and do not require additional follow-up. Patients prone to hyperkalemia should be thoroughly educated on medications to avoid (NSAIDs, ACEIs/ARBs, trimethoprim), an adequate low potassium diet, and symptoms that may warrant medical attention.19
Conclusions
This case illustrates the importance of recognizing the spectrum of manifestations of hyperkalemia, which ranged from muscle weakness to cardiac dysrhythmias. Management strategies for the patient included stabilization of cardiac membranes, potassium shifting, and potassium removal, each tailored to the patient’s individual clinical findings.
The case further illustrates the critical role of continuous monitoring and dynamic adjustment of therapeutic strategies in response to evolving clinical and laboratory findings. The initial and subsequent ECGs, alongside laboratory tests, were instrumental in guiding the adjustments needed in the treatment regimen, ensuring both the efficacy and safety of the interventions. This proactive approach can mitigate the risk of recurrent hyperkalemia and its complications.
Hyperkalemia involves elevated serum potassium levels (> 5.0 mEq/L) and represents an important electrolyte disturbance due to its potentially severe consequences, including cardiac effects that can lead to dysrhythmia and even asystole and death.1,2 In a US Medicare population, the prevalence of hyperkalemia has been estimated at 2.7% and is associated with substantial health care costs.3 The prevalence is even more marked in patients with preexisting conditions such as chronic kidney disease (CKD) and heart failure.4,5
Hyperkalemia can result from multiple factors, including impaired renal function, adrenal disease, adverse drug reactions of angiotensin-converting enzyme inhibitors (ACEIs) and other medications, and heritable mutations.6 Hyperkalemia poses a considerable clinical risk, associated with adverse outcomes such as myocardial infarction and increased mortality in patients with CKD.5,7,8 Electrocardiographic (ECG) changes associated with hyperkalemia play a vital role in guiding clinical decisions and treatment strategies.9 Understanding the pathophysiology, risk factors, and consequences of hyperkalemia, as well as the significance of ECG changes in its management, is essential for health care practitioners.
Case Presentation
An 81-year-old Hispanic man with a history of hypertension, hypothyroidism, gout, and CKD stage 3B presented to the emergency department with progressive weakness resulting in falls and culminating in an inability to ambulate independently. Additional symptoms included nausea, diarrhea, and myalgia. His vital signs were notable for a pulse of 41 beats/min. The physical examination was remarkable for significant weakness of the bilateral upper extremities, inability to bear his own weight, and bilateral lower extremity edema. His initial ECG upon arrival showed bradycardia with wide QRS, absent P waves, and peaked T waves (Figure 1a). These findings differed from his baseline ECG taken 1 year earlier, which showed sinus rhythm with premature atrial complexes and an old right bundle branch block (Figure 1b).
Medication review revealed that the patient was currently prescribed 100 mg allopurinol daily, 2.5 mg amlodipine daily, 10 mg atorvastatin at bedtime, 4 mg doxazosin daily, 112 mcg levothyroxine daily, 100 mg losartan daily, 25 mg metoprolol daily, and 0.4 mg tamsulosin daily. The patient had also been taking over-the-counter indomethacin for knee pain.
Based on the ECG results, he was treated with 0.083%/6 mL nebulized albuterol, 4.65 Mq/250 mL saline solution intravenous (IV) calcium gluconate, 10 units IV insulin with concomitant 50%/25 mL IV dextrose and 8.4 g of oral patiromer suspension. IV furosemide was held due to concern for renal function. The decision to proceed with hemodialysis was made. Repeat laboratory tests were performed, and an ECG obtained after treatment initiation but prior to hemodialysis demonstrated improvement of rate and T wave shortening (Figure 1c). The serum potassium level dropped from 9.8 mEq/L to 7.9 mEq/L (reference range, 3.5-5.0 mEq/L) (Table 1).
In addition to hemodialysis, sodium zirconium 10 g orally 3 times daily was added. Laboratory test results and an ECG was performed after dialysis continued to demonstrate improvement (Figure 1d). The patient’s potassium level decreased to 5.8 mEq/L, with the ECG demonstrating stability of heart rate and further improvement of the PR interval, QRS complex, and T waves.
Despite the established treatment regimen, potassium levels again rose to 6.7 mEq/L, but there were no significant changes in the ECG, and thus no medication changes were made (Figure 1e). Subsequent monitoring demonstrated a further increase in potassium to 7.4 mEq/L, with an ECG demonstrating a return to the baseline of 1 year prior. The patient underwent hemodialysis again and was given oral furosemide 60 mg every 12 hours. The potassium concentration after dialysis decreased to 4.7 mEq/L and remained stable, not going above 5.0 mEq/L on subsequent monitoring. The patient had resolution of all symptoms and was discharged.
Discussion
We have described in detail the presentation of each pathology and mechanisms of each treatment, starting with the patient’s initial condition that brought him to the emergency room—muscle weakness. Skeletal muscle weakness is a common manifestation of hyperkalemia, occurring in 20% to 40% of cases, and is more prevalent in severe elevations of potassium. Rarely, the weakness can progress to flaccid paralysis of the patient’s extremities and, in extreme cases, the diaphragm.
Muscle weakness progression occurs in a manner that resembles Guillain-Barré syndrome, starting in the lower extremities and ascending toward the upper extremities.10 This is known as secondary hyperkalemic periodic paralysis. Hyperkalemia lowers the transmembrane gradient in neurons, leading to neuronal depolarization independent of the degree of hyperkalemia. If the degree of hyperkalemia is large enough, this depolarization inactivates voltage-gated sodium channels, making neurons refractory to excitation. Electromyographical studies have shown reduction in the compounded muscle action potential.11 The transient nature of this paralysis is reflected by rapid correction of weakness and paralysis when the electrolyte disorder is corrected.
The patient in this case also presented with bradycardia. The ECG manifestations of hyperkalemia can include atrial asystole, intraventricular conduction disturbances, peaked T waves, and widened QRS complexes. However, some patients with renal insufficiency may not exhibit ECG changes despite significantly elevated serum potassium levels.12
The severity of hyperkalemia is crucial in determining the associated ECG changes, with levels > 6.0 mEq/L presenting with abnormalities.13 ECG findings alone may not always accurately reflect the severity of hyperkalemia, as up to 60% of patients with potassium levels > 6.0 mEq/L may not show ECG changes.14 Additionally, extreme hyperkalemia can lead to inconsistent ECG findings, making it challenging to rely solely on ECG for diagnosis and monitoring.8 The level of potassium that causes these effects varies widely through patient populations.
The main mechanism by which hyperkalemia affects the heart’s conduction system is through voltage differences across the conduction fibers and eventual steady-state inactivation of sodium channels. This combination of mechanisms shortens the action potential duration, allowing more cardiomyocytes to undergo synchronized depolarization. This amalgamation of cardiomyocytes repolarizing can be reflected on ECGs as peaked T waves. As the action potential decreases, there is a period during which cardiomyocytes are prone to tachyarrhythmias and ventricular fibrillation.
A reduced action potential may lead to increased rates of depolarization and thus conduction, which in some scenarios may increase heart rate. As the levels of potassium rise, intracellular accumulation impedes the entry of sodium by decreasing the cation gradient across the cell membrane. This effectively slows the sinus nodes and prolongs the QRS by slowing the overall propagation of action potentials. By this mechanism, conduction delays, blocks, or asystole are manifested. The patient in this case showed conduction delays, peaked T waves, and disappearance of P waves when he first arrived.
Hyperkalemia Treatment
Hyperkalemia develops most commonly due to acute or chronic kidney diseases, as was the case with this patient. The patient’s hyperkalemia was also augmented by the use of nonsteroidal anti-inflammatory drugs (NSAIDs), which can directly affect renal function. A properly functioning kidney is responsible for excretion of up to 90% of ingested potassium, while the remainder is excreted through the gastrointestinal (GI) tract. Definitive treatment of hyperkalemia is mitigated primarily through these 2 organ systems. The treatment also includes transitory mechanisms of potassium reduction. The goal of each method is to preserve the action potential of cardiomyocytes and myocytes. This patient presented with acute symptomatic hyperkalemia and received various medications to acutely, transitorily, and definitively treat it.
Initial therapy included calcium gluconate, which functions to stabilize the myocardial cell membrane. Hyperkalemia decreases the resting membrane action potential of excitable cells and predisposes them to early depolarization and thus dysrhythmias. Calcium decreases the threshold potential across cells and offsets the overall gradient back to near normal levels.15 Calcium can be delivered through calcium gluconate or calcium chloride. Calcium chloride is not preferred because extravasation can cause pain, blistering and tissue ischemia. Central venous access is required, potentially delaying prompt treatment. Calcium acts rapidly after administration—within 1 to 3 minutes—but only lasts 30 to 60 minutes.16 Administration of calcium gluconate can be repeated as often as necessary, but patients must be monitored for adverse effects of calcium such as nausea, abdominal pain, polydipsia, polyuria, muscle weakness, and paresthesia. Care must be taken when patients are taking digoxin, because calcium may potentiate toxicity.17 Although calcium provides immediate benefits it does little to correct the underlying cause; other medications are required to remove potassium from the body.
Two medication classes have been proven to shift potassium intracellularly. The first are β-2 agonists, such as albuterol/levalbuterol, and the second is insulin. Both work through sodium-potassium-ATPase in a direct manner. β-2 agonists stimulate sodium-potassium-ATPase to move more potassium intracellularly, but these effects have been seen only with high doses of albuterol, typically 4× the standard dose of 0.5 mg in nebulized solutions to achieve decreases in potassium of 0.3 to 0.6 mEq/L, although some trials have reported decreases of 0.62 to 0.98 mEq/L.15,18 These potassium-lowering effects of β-2 agonist are modest, but can be seen 20 to 30 minutes after administration and persist up to 1 to 2 hours. β-2 agonists are also readily affected by β blockers, which may reduce or negate the desired effect in hyperkalemia. For these reasons, a β-2 agonist should not be given as monotherapy and should be provided as an adjuvant to more independent therapies such as insulin. Insulin binds to receptors on muscle cells and increases the quantity of sodium-potassium-ATPase and glucose transporters. With this increase in influx pumps, surrounding tissues with higher resting membrane potentials can absorb the potassium load, thereby protecting cardiomyocytes.
Potassium Removal
Three methods are currently available to remove potassium from the body: GI excretion, renal excretion, and direct removal from the bloodstream. Under normal physiologic conditions, the kidneys account for about 90% of the body’s ability to remove potassium. Loop diuretics facilitate the removal of potassium by increasing urine production and have an additional potassium-wasting effect. Although the onset of action of loop diuretics is typically 30 to 60 minutes after oral administration, their effect can last for several hours. In this patient, furosemide was introduced later in the treatment plan to manage recurring hyperkalemia by enhancing renal potassium excretion.
Potassium binders such as patiromer act in the GI tract, effectively reducing serum potassium levels although with a slower onset of action than furosemide, generally taking hours to days to exert its effect. Both medications illustrate a tailored approach to managing potassium levels, adapted to the evolving needs and renal function of the patient. The last method is using hemodialysis—by far the most rapid method to remove potassium, but also the most invasive. The different methods of treating hyperkalemia are summarized in Table 2. This patient required multiple days of hemodialysis to completely correct the electrolyte disorder. Upon discharge, the patient continued oral furosemide 40 mg daily and eventually discontinued hemodialysis due to stable renal function.
Often, after correcting an inciting event, potassium stores in the body eventually stabilize and do not require additional follow-up. Patients prone to hyperkalemia should be thoroughly educated on medications to avoid (NSAIDs, ACEIs/ARBs, trimethoprim), an adequate low potassium diet, and symptoms that may warrant medical attention.19
Conclusions
This case illustrates the importance of recognizing the spectrum of manifestations of hyperkalemia, which ranged from muscle weakness to cardiac dysrhythmias. Management strategies for the patient included stabilization of cardiac membranes, potassium shifting, and potassium removal, each tailored to the patient’s individual clinical findings.
The case further illustrates the critical role of continuous monitoring and dynamic adjustment of therapeutic strategies in response to evolving clinical and laboratory findings. The initial and subsequent ECGs, alongside laboratory tests, were instrumental in guiding the adjustments needed in the treatment regimen, ensuring both the efficacy and safety of the interventions. This proactive approach can mitigate the risk of recurrent hyperkalemia and its complications.
- Youn JH, McDonough AA. Recent advances in understanding integrative control of potassium homeostasis. Annu Rev Physiol. 2009;71:381-401. doi:10.1146/annurev.physiol.010908.163241 2.
- Simon LV, Hashmi MF, Farrell MW. Hyperkalemia. In: StatPearls. StatPearls Publishing; September 4, 2023. Accessed October 22, 2025.
- Mu F, Betts KA, Woolley JM, et al. Prevalence and economic burden of hyperkalemia in the United States Medicare population. Curr Med Res Opin. 2020;36:1333-1341. doi:10.1080/03007995.2020.1775072
- Loutradis C, Tolika P, Skodra A, et al. Prevalence of hyperkalemia in diabetic and non-diabetic patients with chronic kidney disease: a nested case-control study. Am J Nephrol. 2015;42:351-360. doi:10.1159/000442393
- Grodzinsky A, Goyal A, Gosch K, et al. Prevalence and prognosis of hyperkalemia in patients with acute myocardial infarction. Am J Med. 2016;129:858-865. doi:10.1016/j.amjmed.2016.03.008
- Hunter RW, Bailey MA. Hyperkalemia: pathophysiology, risk factors and consequences. Nephrol Dial Transplant. 2019;34(suppl 3):iii2-iii11. doi:10.1093/ndt/gfz206
- Luo J, Brunelli SM, Jensen DE, Yang A. Association between serum potassium and outcomes in patients with reduced kidney function. Clin J Am Soc Nephrol. 2016;11:90-100. doi:10.2215/CJN.01730215
- Montford JR, Linas S. How dangerous is hyperkalemia? J Am Soc Nephrol. 2017;28:3155-3165. doi:10.1681/ASN.2016121344
- Mattu A, Brady WJ, Robinson DA. Electrocardiographic manifestations of hyperkalemia. Am J Emerg Med. 2000;18:721-729. doi:10.1053/ajem.2000.7344
- Kimmons LA, Usery JB. Acute ascending muscle weakness secondary to medication-induced hyperkalemia. Case Rep Med. 2014;2014:789529. doi:10.1155/2014/789529
- Naik KR, Saroja AO, Khanpet MS. Reversible electrophysiological abnormalities in acute secondary hyperkalemic paralysis. Ann Indian Acad Neurol. 2012;15:339-343. doi:10.4103/0972-2327.104354
- Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol. 2008;3:324-330. doi:10.2215/CJN.04611007
- Larivée NL, Michaud JB, More KM, Wilson JA, Tennankore KK. Hyperkalemia: prevalence, predictors and emerging treatments. Cardiol Ther. 2023;12:35-63. doi:10.1007/s40119-022-00289-z
- Shingarev R, Allon M. A physiologic-based approach to the treatment of acute hyperkalemia. Am J Kidney Dis. 2010;56:578-584. doi:10.1053/j.ajkd.2010.03.014
- Parham WA, Mehdirad AA, Biermann KM, Fredman CS. Hyperkalemia revisited. Tex Heart Inst J. 2006;33:40-47.
- Ng KE, Lee CS. Updated treatment options in the management of hyperkalemia. U.S. Pharmacist. February 16, 2017. Accessed October 1, 2025. www.uspharmacist.com/article/updated-treatment-options-in-the-management-of-hyperkalemia
- Quick G, Bastani B. Prolonged asystolic hyperkalemic cardiac arrest with no neurologic sequelae. Ann Emerg Med. 1994;24:305-311. doi:10.1016/s0196-0644(94)70144-x 18.
- Allon M, Dunlay R, Copkney C. Nebulized albuterol for acute hyperkalemia in patients on hemodialysis. Ann Intern Med. 1989;110:426-429. doi:10.7326/0003-4819-110-6-42619.
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024;105(4 suppl):S117-S314. doi:10.1016/j.kint.2023.10.018
- Youn JH, McDonough AA. Recent advances in understanding integrative control of potassium homeostasis. Annu Rev Physiol. 2009;71:381-401. doi:10.1146/annurev.physiol.010908.163241 2.
- Simon LV, Hashmi MF, Farrell MW. Hyperkalemia. In: StatPearls. StatPearls Publishing; September 4, 2023. Accessed October 22, 2025.
- Mu F, Betts KA, Woolley JM, et al. Prevalence and economic burden of hyperkalemia in the United States Medicare population. Curr Med Res Opin. 2020;36:1333-1341. doi:10.1080/03007995.2020.1775072
- Loutradis C, Tolika P, Skodra A, et al. Prevalence of hyperkalemia in diabetic and non-diabetic patients with chronic kidney disease: a nested case-control study. Am J Nephrol. 2015;42:351-360. doi:10.1159/000442393
- Grodzinsky A, Goyal A, Gosch K, et al. Prevalence and prognosis of hyperkalemia in patients with acute myocardial infarction. Am J Med. 2016;129:858-865. doi:10.1016/j.amjmed.2016.03.008
- Hunter RW, Bailey MA. Hyperkalemia: pathophysiology, risk factors and consequences. Nephrol Dial Transplant. 2019;34(suppl 3):iii2-iii11. doi:10.1093/ndt/gfz206
- Luo J, Brunelli SM, Jensen DE, Yang A. Association between serum potassium and outcomes in patients with reduced kidney function. Clin J Am Soc Nephrol. 2016;11:90-100. doi:10.2215/CJN.01730215
- Montford JR, Linas S. How dangerous is hyperkalemia? J Am Soc Nephrol. 2017;28:3155-3165. doi:10.1681/ASN.2016121344
- Mattu A, Brady WJ, Robinson DA. Electrocardiographic manifestations of hyperkalemia. Am J Emerg Med. 2000;18:721-729. doi:10.1053/ajem.2000.7344
- Kimmons LA, Usery JB. Acute ascending muscle weakness secondary to medication-induced hyperkalemia. Case Rep Med. 2014;2014:789529. doi:10.1155/2014/789529
- Naik KR, Saroja AO, Khanpet MS. Reversible electrophysiological abnormalities in acute secondary hyperkalemic paralysis. Ann Indian Acad Neurol. 2012;15:339-343. doi:10.4103/0972-2327.104354
- Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol. 2008;3:324-330. doi:10.2215/CJN.04611007
- Larivée NL, Michaud JB, More KM, Wilson JA, Tennankore KK. Hyperkalemia: prevalence, predictors and emerging treatments. Cardiol Ther. 2023;12:35-63. doi:10.1007/s40119-022-00289-z
- Shingarev R, Allon M. A physiologic-based approach to the treatment of acute hyperkalemia. Am J Kidney Dis. 2010;56:578-584. doi:10.1053/j.ajkd.2010.03.014
- Parham WA, Mehdirad AA, Biermann KM, Fredman CS. Hyperkalemia revisited. Tex Heart Inst J. 2006;33:40-47.
- Ng KE, Lee CS. Updated treatment options in the management of hyperkalemia. U.S. Pharmacist. February 16, 2017. Accessed October 1, 2025. www.uspharmacist.com/article/updated-treatment-options-in-the-management-of-hyperkalemia
- Quick G, Bastani B. Prolonged asystolic hyperkalemic cardiac arrest with no neurologic sequelae. Ann Emerg Med. 1994;24:305-311. doi:10.1016/s0196-0644(94)70144-x 18.
- Allon M, Dunlay R, Copkney C. Nebulized albuterol for acute hyperkalemia in patients on hemodialysis. Ann Intern Med. 1989;110:426-429. doi:10.7326/0003-4819-110-6-42619.
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024;105(4 suppl):S117-S314. doi:10.1016/j.kint.2023.10.018
Following the Hyperkalemia Trail: A Case Report of ECG Changes and Treatment Responses
Following the Hyperkalemia Trail: A Case Report of ECG Changes and Treatment Responses

Gadolinium Intermediate Elimination and Persistent Symptoms After Magnetic Resonance Imaging Contrast Agent Exposure
Gadolinium Intermediate Elimination and Persistent Symptoms After Magnetic Resonance Imaging Contrast Agent Exposure
Magnetic resonance image (MRI) contrast agents can induce profound complications, including gadolinium encephalopathy, kidney injury, gadolinium-associated plaques, and progressive systemic fibrosis, which can be fatal.1-10 About 50% of MRIs use gadolinium-based contrast (Gd3+), a toxic rare earth metal ion that enhances imaging but requires binding with pharmaceutical ligands to reduce toxicity and promote renal elimination (Figure 1). Despite these measures, Gd3+ can persist in the body, including the brain.11,12 Wastewater treatment fails to remove these agents, making Gd3+ a growing pollutant in water and the food chain.13-15 Because Gd3+ is a rare earth metal ion in the milieu intérieur, there is an urgent need to study its biological and long-term effects (Appendix 1).
Case Presentation
A 65-year-old Vietnam-era veteran presented to nephrology at the Raymond G. Murphy Veterans Affairs Medical Center (RGMVAMC) in Albuquerque, New Mexico, for evaluation of gadolinium-induced symptoms. His medical history included metabolic syndrome, hypertension, hyperlipidemia, hypogonadism, cervical spondylosis, and an elevated prostate-specific antigen, previously assessed with a contrast-enhanced MRI in 2019 (Gadobenic acid, 19 mL). Surgical history included cervical fusion and ankle hardware.
The patient had a scheduled MRI 25 days earlier, following an elevated prostate specific antigen test result, prompting urologic surveillance and concern for malignancy. In preparation for the contrast-enhanced MRI, his right arm was cannulated with a line primed with gadobenic acid contrast. Though the technician stated the infusion had not started, the patient’s symptoms began shortly after entry into the scanner, before any programmed pulse sequences. The patient experienced claustrophobia, diaphoresis, palpitations, xerostomia, dysgeusia, shortness of breath, and a sensation of heat in his groin, chest, “kidneys,” and lower back. The MRI was terminated prematurely in response to the patient’s acute symptomatology. The patient continued experiencing new symptoms intermittently during the following week, including lightheadedness, headaches, right clavicular pain, raspy voice, edema, and a sense of doom.
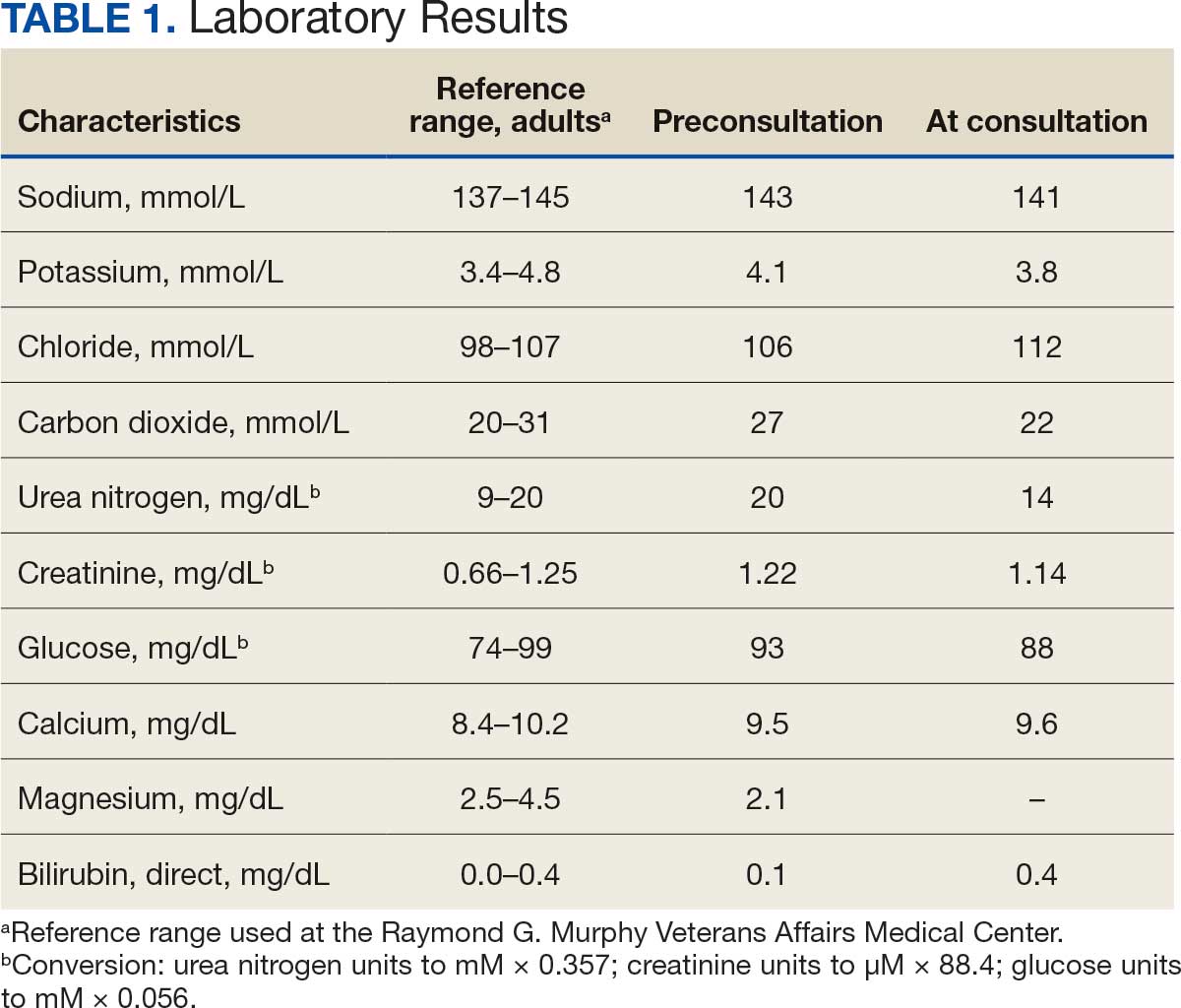
The patient presented to the RGMVAMC emergency department (ED) 8 days after the MRI with worsening symptoms and was hospitalized for 10 days. During this time, he was referred to nephrology for outpatient evaluation. While awaiting his nephrology appointment, the patient presented to the RGMVAMC ED 20 days after the initial episode with ongoing symptoms. “I thought I was dying,” he said. Laboratory results and a 12-lead electrocardiogram showed a finely static background, wide P waves (> 80 ms) with notching in lead II, sinusoidal P waves in V1, R transition in V2, RR’ in V2, ST flat in lead III, and sinus bradycardia (Table 1 and Appendix 2).
The patient’s medical and surgical histories were reviewed at the nephrology evaluation 25 days following the MRI. He reported that household water was sourced from a well and that he filtered his drinking water with a reverse osmosis system. He served in the US Army for 10 years as an engineer specializing in mechanical systems, power generation, and vehicles. Following Army retirement, the patient served in the US Air Force Reserves for 15 years, working as a crew chief in pneudraulics. The patient reported stopping tobacco use 1 year before and also reported regular use of a broad array of prescription medications and dietary supplements, including dexamethasone (4 mg twice daily), fluticasone nasal spray (50 mcg per nostril, twice daily), ibuprofen (400 mg twice daily, as needed), loratadine (10 mg daily), aspirin (81 mg daily), and metoprolol succinate (50 mg nightly). In addition, he reported consistent use of cholecalciferol (3000 IU daily), another supplemental vitamin D preparation, chelated magnesium glycinate (3 tablets daily for bone issues), turmeric (1 tablet daily), a multivitamin (Living Green Liquid Gel, daily), and a mega-B complex.
Physical examination revealed a well-nourished, tall man with hypertension (145/87 mmHg) and bilateral lower extremity edema. Oral examination showed poor dentition, including missing molars (#1-3, #14-16, #17-19, #30-31), with the anterior teeth replaced by bridges supported by dental implants. The review of systems was otherwise unremarkable, with nocturia noted before the consultation.
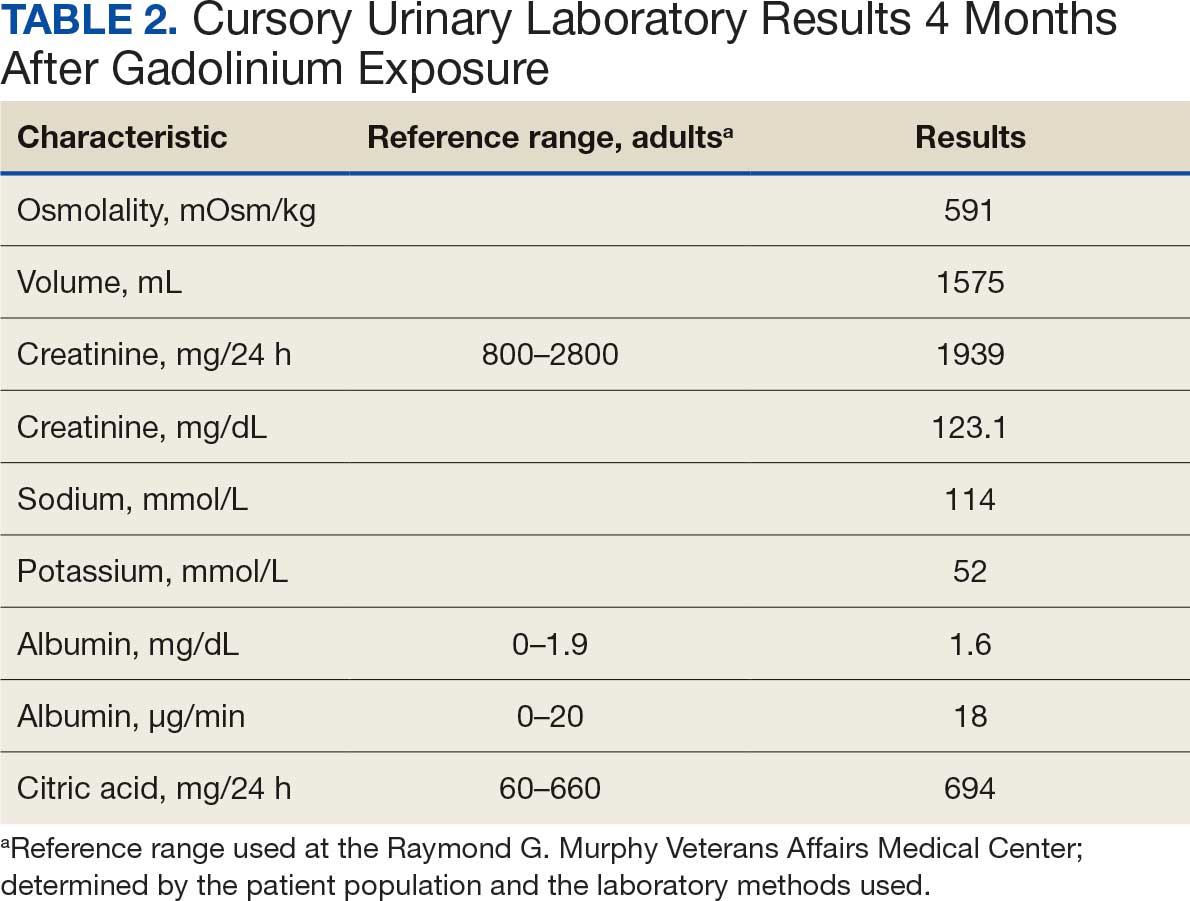
Serum and urine gadolinium testing, (Mayo Clinic Laboratories) revealed gadolinium levels of 0.3 mcg/24 h in the urine and 0.1 ng/mL in the serum. Nonzero values indicated detectable gadolinium, suggesting retention. The patient had a prior gadolinium exposure during a 2019 MRI (about 1340 days before) and suspected a repeat exposure on day 0, although the MRI technician stated that no contrast was administered. Given his elevated vitamin D levels, the patient was advised to minimize dietary supplements, particularly vitamin D, to avoid confounding symptoms. The plan included monitoring symptoms and a follow-up evaluation with repeat laboratory tests on day 116.
At the nephrology follow-up 4 months postexposure, the patient's symptoms had primarily abated, with a marked reduction in the previously noted metallic dysgeusia. Physical examination remained consistent with prior findings. He was afebrile (97.7 °F) with a blood pressure of 111/72 mmHg, a pulse of 63 beats per minute, and an oxygen saturation of 98% on ambient air. Laboratory analysis revealed serum and urine gadolinium levels below detectable thresholds (< 0.1 ng/mL and < 0.1 mcg/24 h). A 24-hour creatinine clearance, calculated from a urine volume of 1300 mL, measured at an optimal 106 mL/min, indicating preserved renal function (Tables 2 and 3). Of note, his 24-hour oxalate was above the reference range, with a urine pH below the reference range and a high supersaturation index for calcium oxalate.
Discussion
Use of enhanced MRI has increased in the Veterans Health Administration (Figure 2). A growing range of indications for enhanced procedures (eg, cardiac MRI) has contributed to this rise. The market has grown with new gadolinium-based contrast agents, such as gadopiclenol. However, reliance on untested assumptions about the safety of newer agents and need for robust clinical trials pose potential risks to patient safety.
Without prospective evidence, the American College of Radiology (ACR) classifies gadolinium-based contrast agents into 3 groups: Group 1, associated with the highest number of nephrogenic systemic fibrosis cases; Group 2, linked to few, if any, unconfounded cases; and Group 3, where data on nephrogenic systemic fibrosis risk have been limited. As of April 2024, the ACR reclassified Group 3 agents (Ablavar/Vasovist/Angiomark and Primovist/Eovist) into Group 2. Curiously, Vueway and Elucirem were approved in late 2022 and should clearly be categorized as Group 3 (Table 4).There were 19 cases of nephrogenic systemic fibrosis or similar manifestations, 8 of which were unconfounded by other factors. These patients had been exposed to gadobutrol, often combined with other agents. Gadobutrol—like other Group 2 agents—has been associated with nephrogenic systemic fibrosis.16,17 Despite US Food and Drug Administration (FDA) documentation of rising reports, many clinicians remain unaware that nephrogenic systemic fibrosis is increasingly linked to Group 2 agents classified by the ACR.18 While declines in reported cases of nephrogenic systemic fibrosis may suggest reduced incidence, this trend may reflect diminished clinical vigilance and underreporting, particularly given emerging evidence implicating even Group 2 gadolinium-based contrast agents in delayed and underrecognized presentations. This information has yet to permeate the medical community, particularly among nephrologists. Considering these cases, revisiting the ACR guidelines may be prudent.
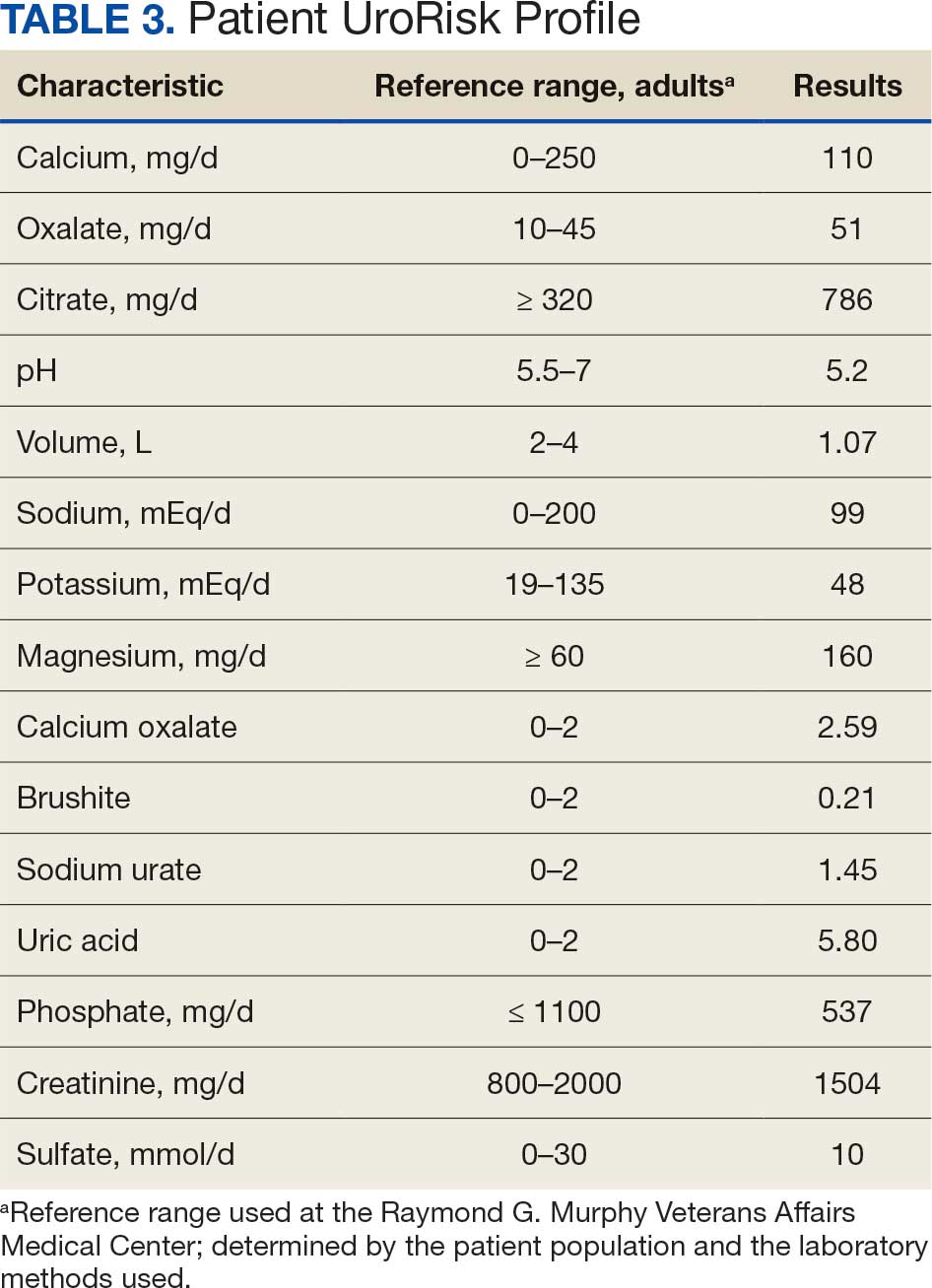
To address this growing concern, clinicians must adopt stricter vigilance and actively pursue updated information to mitigate patient risks tied to these contrast agents.
There exists an illusion of knowledge in disregarding the confounded exposures of MRI contrast agents. Ten distinct brands of contrast agents have been approved for clinical use. With repeated imaging, patients are often exposed to varying formulations of gadolinium-based agents. Yet investigators commonly discard these data points when assessing risk. By doing so, they assume—without evidence—that some formulations are inherently less likely to provoke adverse effects (AEs) than others. This untested presumption becomes perilous, especially given the limited understanding of the mechanisms underlying gadolinium-induced pathologies. As Aldous Huxley warned, “Facts do not cease to exist because they are ignored.”19
Gadolinium Persistence
Contrary to expectations, gadolinium persists in the body far longer than initially presumed. Symptoms associated with gadolinium exposure (SAGE) encapsulate the chronic, often enigmatic maladies tied to MRI contrast agents.20 The prolonged retention of this rare earth metal offers a compelling hypothesis for the etiology of SAGE. It has been hypothesized that Lewis base-rich metabolites increase susceptibility to gadolinium-based contrast agent complications.21
The blood and urine concentration elimination curves of gadolinium are exponential and categorized as fast, intermediate, and long-term.1 For urinary elimination, the function of the curves is exponential. The quantity of gadolinium in the urine at a time (t) after exposure (D[Gd](t)) is equal to the product of the amount of gadolinium in the sample (urine or blood) at the end of the fast elimination period (D[Gd](t0)) and the exponential decay with k being a rate constant.
To the authors’ knowledge, we are the only research team currently investigating the rate constant for the intermediate- and long-term phase gadolinium elimination. The Retention and Toxicity of Gadolinium-based Contrast Agents study was approved by the University of New Mexico Health Sciences Center Institutional Review Board on May 27, 2020 (IRB ID 19-660). The data for the patient in this case were compared with preliminary results for patients with exposure-to-measurement intervals < 100 days.
The patient in this case presented with detectable gadolinium levels in urine and serum shortly after an attempted contrast-enhanced MRI procedure (Figure 3). The presence of detectable gadolinium levels in the patient’s urine and serum suggests a likely exposure to a contrast agent about 27 days before his consultation. While the technician reported that no contrast was administered during the attempted MRI, it remains possible that a small amount was introduced during cannulation, potentially triggering the patient’s symptoms. Linear modeling of semilogarithmic plots for participants exposed to contrast agents within 100 days (urine: P = 1.8 × 10ˉ8, adjusted r² = 0.62; blood: P = .005, adjusted r² = 0.21) provided clearance rates (k values) for urine and blood. Extrapolating from these models to the presumed exposure date, the intercepts estimate that the patient received between 0.5% and 8% of a standard contrast dose.

MRI contrast agents can cause skin disease. Systemic fibrosis is considered one of the most severe AEs. Skin pathophysiology involving myeloid cells is driven by elevated levels of monocyte chemoattractant protein-1, which recruits circulating fibroblasts via the C-C chemokine receptor 2.22,23 This occurs alongside activation of NADPH oxidase Nox4.4,24,25 Intracellular gadolinium-rich nanoparticles likely serve as catalysts for this reactive cascade.2,18,22,26,27 These particles assemble around intracellular lipid droplets and ferrule them in spiculated rare earth-rich shells that compromise cellular architecture.2,18,21,22,26,27 Frequently sequestered within endosomal compartments, they disrupt vesicular integrity and threaten cellular homeostasis. Interference with degradative systems such as the endolysosomal axis perturbs energy-recycling pathways—an insidious disturbance, particularly in cells with high metabolic demand. Skin-related symptoms are among the most frequently reported AEs, according to the FDA AE reporting system.18
Studies indicate repeated exposure to MRI contrast agents can lead to permanent gadolinium retention in the brain and other vital organs. Intravenous (IV) contrast agents cross the blood-brain barrier rapidly, while intrathecal administration has been linked to significant and lasting neurologic effects.18
Gadolinium is chemically bound to pharmaceutical ligands to enhance renal clearance and reduce toxicity. However, available data from human samples suggest potential ligand exchanges with undefined physiologic substances. This exchange may facilitate gadolinium precipitation and accumulation within cells into spiculated nanoparticles. Transmission electron microscopy reveals the formation of unilamellar bodies associated with mitochondriopathy and cellular damage, particularly in renal proximal tubules.2,18,22,26,27 It is proposed that intracellular nanoparticle formation represents a key mechanism driving the systemic symptoms observed in patients.1,2,18, 22,26,27
Any hypothesis based on free soluble gadolinium—or concept derived from it—should be discarded. The high affinity of pharmaceutical ligands for gadolinium suggests that the cationic rare earth metal remains predominantly in a ligand-bound, soluble form. It is hypothesized that gadolinium undergoes ligand exchange with physiologic substances, directly leading to nanoparticle formation. Current data demonstrate gadolinium precipitation according to the Le Chatelier’s principle. Since precipitated gadolinium does not readily re-equilibrate with pharmaceutical ligands, repeated administration of different contrast agent brands may contribute to nanoparticle growth.26
Meanwhile, a growing number of patients are turning to chelation therapy, a largely untested treatment. The premise of chelation therapy is rooted in several unproven assumptions.18,21 First, it assumes that clinically significant amounts of gadolinium persist in compartments such as the extracellular space, where they can be effectively chelated and cleared. Second, it presumes that free gadolinium is the primary driver of chronic symptoms, an assertion that remains scientifically unsubstantiated. Finally, chelation proponents overlook the potential harm caused by depleting essential physiological metals during the process, assuming without evidence that the scant removal of gadolinium outweighs the risk of physiological mineral depletion.
These assumptions underpin an unproven remedy that demands critical scrutiny. Recent findings reveal that gadolinium deposits in the skin and kidney often take the form of intracellular nanoparticles, directly challenging the foundation of chelation therapy. Chelation advocates must demonstrate that these intracellular gadolinium deposits neither trigger cellular toxicity nor initiate a cytokine cascade. Chelation supporters must prove that the systemic response to these foreign particles is unrelated to the symptoms reported by patients. Until then, the validity of chelation therapy remains highly questionable.
The causality of the symptoms, mainly whether IV gadolinium was administered, was examined. The null hypothesis stated that the patient was not exposed to gadolinium. However, this hypothesis was contradicted by the detection of gadolinium in the serum and urine 27 days after the potential exposure.
Two plausible explanations exist for the nonzero gadolinium levels detected in the serum and urine. The first possibility is that minute quantities of gadolinium were introduced during cannulation, with the amount being sufficient to persist in measurable concentrations 27 days postexposure. The second possibility is that the gadolinium originated from an MRI contrast agent administered 4 years earlier. In this scenario, gadolinium stored in organ reservoirs such as bone, liver, or kidneys may have been mobilized into the extracellular fluid compartment due to the administration of high-dose steroids 20 days after the recent contrast-enhanced MRI procedure attempt. Coyte et al reported elevated gadolinium levels in the serum, cord blood, breast milk, and placenta of pregnant women with prior exposure to MRI contrast agents.28 These findings suggest that gadolinium, stored in organs such as bone may be remobilized by variables affecting bone remodeling (eg, high-dose steroids).
Significantly, the patient exhibited elevated urinary oxalate levels. Previous research has found that oxalic acid reacts rapidly with MRI contrast agents, forming digadolinium trioxalate. While the gadolinium-rich nanoparticles identified in tissues such as the skin and kidney (including the human kidney) are amorphous, these in vitro findings establish a proof-of-concept: the intracellular environment facilitates gadolinium dissociation from pharmaceutical chelates.
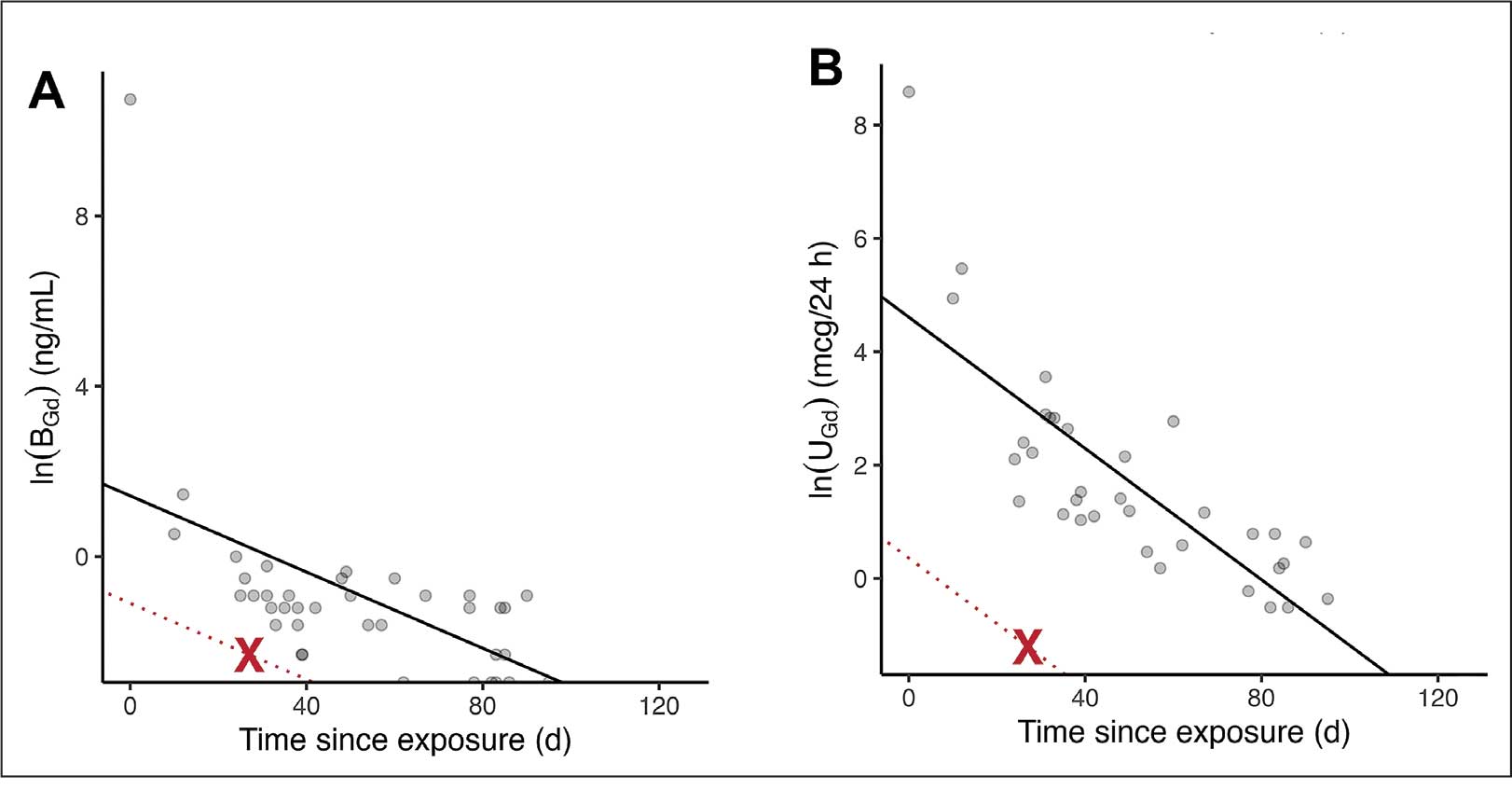
Furthermore, in vitro experiments show that proteins and lysosomal pH promote this dissociation, underscoring how human metabolic conditions—particularly oxalic acid concentration—may drive intracellular gadolinium deposition.
Patient Perspective
“They put something into my body that they cannot get out.” This stark realization underpins the patient’s profound concern about gadolinium-based contrast agents and their potential long-term effects. Reflecting on his experience, the patient expressed deep fears about the unknown future impacts: “I’m concerned about my kidneys, I’m concerned about my heart, and I’m concerned about my brain. I don’t know how this stuff is going to affect me in the future.”
He drew an unsettling parallel between gadolinium and heavy metals: “Heavy metal is poison. The body does not produce this kind of stuff on its own.” His reaction to the procedure left a lasting impression, prompting him to question the logic of using a substance that cannot be purged: “Why would you put something into someone’s body that you cannot extract? Nobody—nobody—should experience what I went through.”
The patient emphasized the lack of clear research on long-term outcomes, which compounds his anxiety: “If there was research that said, ‘Well, this is only going to affect these organs for this long,’ OK, I might be able to accept that. But there is no research like that. Nobody can tell me what’s going to happen in 5 years.”
Strengths and Limitations
A significant strength of this approach is the ability to track gadolinium elimination and symptom resolution over time, supported by unique access to intermediate and long-term clearance data from our ongoing research protocol. The investigators were equipped to back-extrapolate the exposure, which provided a rare opportunity to correlate gadolinium levels with clinical outcomes. The primary limitation is the lack of a defined clinical case definition for gadolinium toxicity and limited mechanistic understanding of SAGE, which hinders diagnosis and management.
Metabolites, proteins, and lipids rich in Lewis bases could initiate this process as substrates for intracellular gadolinium sedimentation. Future studies should investigate whether metabolic conditions such as oxalate burden or altered parathyroid hormone levels modulate gadolinium compartmentalization and tissue retention. If gadolinium-rich nanoparticle formation and accumulation disrupt cellular equilibrium, it underscores an urgent need to understand the implications of long-term gadolinium retention. The research team continues to gather evidence that the gadolinium cation remains chelated from the moment MRI contrast agents are administered through to the formation of intracellular nanoparticles. Retained gadolinium nanoparticles may act as a nidus, triggering cellular signaling cascades that lead to multisymptomatic illnesses. Intracellular and insoluble retained gadolinium challenges proponents of untested chelation therapies.
Conclusions
This case highlights emerging clinical and ethical concerns surrounding gadolinium-based contrast agent use. Clinicians may benefit from considering gadolinium retention as a contributor to persistent, unexplained symptoms—particularly in patients with recent imaging exposure. As contrast use continues to rise within federal health systems, regulatory and administrative stakeholders would do well to re-examine current safety frameworks. Informed consent should reflect what is known: gadolinium can remain in the body long after administration, potentially indefinitely. The long-term consequences of cumulative exposure remain poorly defined, but the presence of a lanthanide element in human tissue warrants greater attention from researchers and regulators alike. Interest in alternative imaging modalities and long-term safety monitoring would mark progress toward more transparent, accountable care.
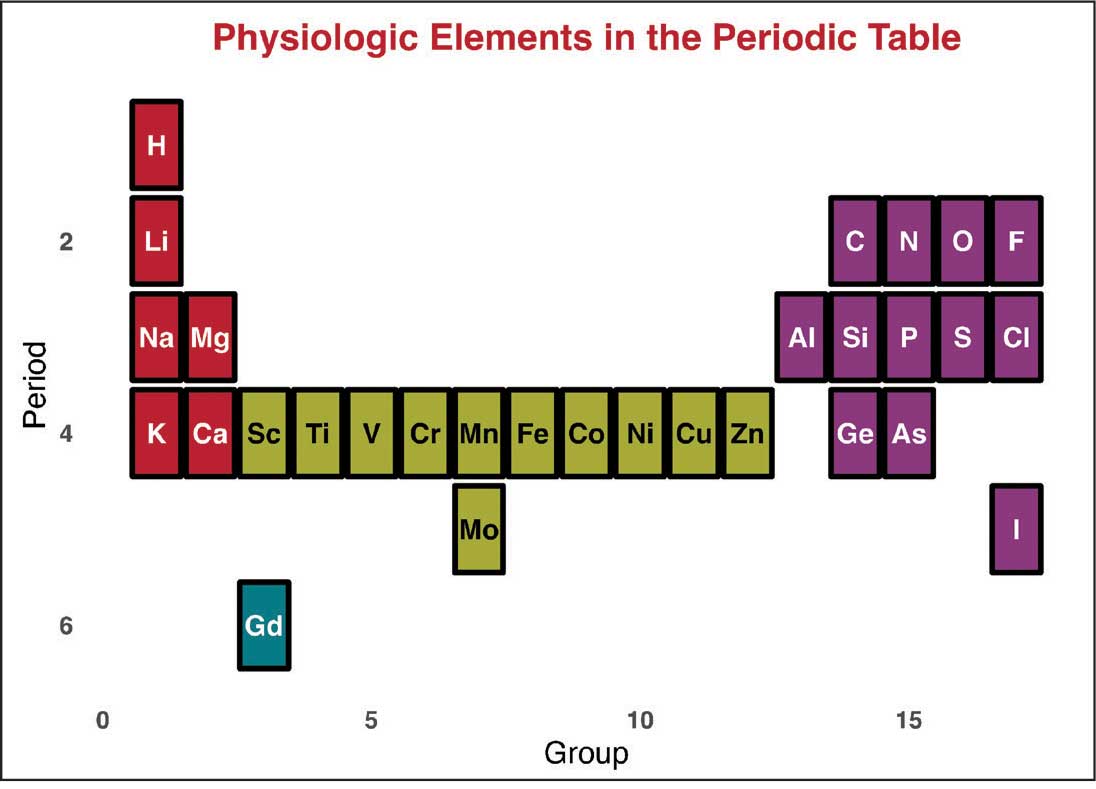
Jackson DB, MacIntyre T, Duarte-Miramontes V, et al. Gadolinium deposition disease: a case report and the prevalence of enhanced MRI procedures within the Veterans Health Administration. Fed Pract. 2022;39:218-225. doi:10.12788/fp.0258
Do C, DeAguero J, Brearley A, et al. Gadolinium-based contrast agent use, their safety, and practice evolution. Kidney360. 2020;1:561-568.doi:10.34067/kid.0000272019
Leyba K, Wagner B. Gadolinium-based contrast agents: why nephrologists need to be concerned. Curr Opin Nephrol Hypertens. 2019;28:154-162. doi:10.1097/MNH.0000000000000475
Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;311:F1-F11. doi:10.1152/ajprenal.00166.2016
Maramattom BV, Manno EM, Wijdicks EF, et al. Gadolinium encephalopathy in a patient with renal failure. Neurology. 2005;64:1276-1278.doi:10.1212/01.WNL.0000156805.45547.6E
Sam AD II, Morasch MD, Collins J, et al. Safety of gadolinium contrast angiography in patients with chronic renal insufficiency. J Vasc Surg. 2003;38:313-318. doi:10.1016/s0741-5214(03)00315-x
Schenker MP, Solomon JA, Roberts DA. Gadolinium arteriography complicated by acute pancreatitis and acute renal failure. J Vasc Interv Radiol. 2001;12:393. doi:10.1016/s1051-0443(07)61925-3
Gemery J, Idelson B, Reid S, et al. Acute renal failure after arteriography with a gadolinium-based contrast agent. AJR Am J Roentgenol. 1998;171:1277-1278. doi:10.2214/ajr.171.5.9798860
Akgun H, Gonlusen G, Cartwright J Jr, et al. Are gadolinium-based contrast media nephrotoxic? A renal biopsy study. Arch Pathol Lab Med. 2006;130:1354-1357. doi:10.5858/2006-130-1354-AGCMNA
Gathings RM, Reddy R, Santa Cruz D, et al. Gadolinium-associated plaques: a new, distinctive clinical entity. JAMA Dermatol. 2015;151:316-319. doi:10.1001/jamadermatol.2014.2660
McDonald RJ, McDonald JS, Kallmes DF, et al. Gadolinium deposition in human brain tissues after contrast-enhanced MR imaging in adult patients without intracranial abnormalities. Radiology. 2017;285(2):546-554. doi:10.1148/radiol.2017161595
Kanda T, Ishii K, Kawaguchi H, et al. High signal intensity in the dentate nucleus and globus pallidus on unenhanced T1-weighted MR images: relationship with increasing cumulative dose of a gadolinium-based contrast material. Radiology. 2014;270(3):834-841. doi:10.1148/radiol.13131669
Schmidt K, Bau M, Merschel G, et al. Anthropogenic gadolinium in tap water and in tap water-based beverages from fast-food franchises in six major cities in Germany. Sci Total Environ. 2019;687:1401-1408. doi:10.1016/j.scitotenv.2019.07.075
Kulaksız S, Bau M. Anthropogenic gadolinium as a microcontaminant in tap water used as drinking water in urban areas and megacities. Appl Geochem. 2011;26:1877-1885.
Brunjes R, Hofmann T. Anthropogenic gadolinium in freshwater and drinking water systems. Water Res. 2020;182:115966. doi:10.1016/j.watres.2020.115966
Endrikat J, Gutberlet M, Hoffmann KT, et al. Clinical safety of gadobutrol: review of over 25 years of use exceeding 100 million administrations. Invest Radiol. 2024;59(9):605-613. doi:10.1097/RLI.0000000000001072
Elmholdt TR, Jørgensen B, Ramsing M, et al. Two cases of nephrogenic systemic fibrosis after exposure to the macrocyclic compound gadobutrol. NDT Plus. 2010;3(3):285-287. doi:10.1093/ndtplus/sfq028
Cunningham A, Kirk M, Hong E, et al. The safety of magnetic resonance imaging contrast agents. Front Toxicol. 2024;6:1376587. doi:10.3389/ftox.2024.1376587
Huxley A. Complete Essays. Volume II, 1926-1929. Chicago; 2000:227.
McDonald RJ, Weinreb JC, Davenport MS. Symptoms associated with gadolinium exposure (SAGE): a suggested term. Radiology. 2022;302(2):270-273. doi:10.1148/radiol.2021211349
Henderson IM, Benevidez AD, Mowry CD, et al. Precipitation of gadolinium from magnetic resonance imaging contrast agents may be the Brass tacks of toxicity. Magn Reson Imaging. 2025;119:110383. doi:10.1016/j.mri.2025.110383
Do C, Drel V, Tan C, et al. Nephrogenic systemic fibrosis is mediated by myeloid C-C chemokine receptor 2. J Invest Dermatol. 2019;139(10):2134-2143. doi:10.1016/j.jid.2019.03.1145
Drel VR, Tan C, Barnes JL, et al. Centrality of bone marrow in the severity of gadolinium-based contrast-induced systemic fibrosis. FASEB J. 2016;30(9):3026-3038. doi:10.1096/fj.201500188R
Bruno F, DeAguero J, Do C, et al. Overlapping roles of NADPH oxidase 4 for diabetic and gadolinium-based contrast agent-induced systemic fibrosis. Am J Physiol Renal Physiol. 2021;320(4):F617-F627. doi:10.1152/ajprenal.00456.2020
Wagner B, Tan C, Barnes JL, et al. Nephrogenic systemic fibrosis: evidence for oxidative stress and bone marrow-derived fibrocytes in skin, liver, and heart lesions using a 5/6 nephrectomy rodent model. Am J Pathol. 2012;181(6):1941-1952. doi:10.1016/j.ajpath.2012.08.026
DeAguero J, Howard T, Kusewitt D, et al. The onset of rare earth metallosis begins with renal gadolinium-rich nanoparticles from magnetic resonance imaging contrast agent exposure. Sci Rep. 2023;13(1):2025. doi:10.1038/s41598-023-28666-1
Do C, Ford B, Lee DY, et al. Gadolinium-based contrast agents: Stimulators of myeloid-induced renal fibrosis and major metabolic disruptors. Toxicol Appl Pharmacol. 2019;375:32-45. doi:10.1016/j.taap.2019.05.009
Coyte RM, Darrah T, Olesik J, et al. Gadolinium during human pregnancy following administration of gadolinium chelate before pregnancy. Birth Defects Res. 2023;115(14):1264-1273. doi:10.1002/bdr2.2209
Magnetic resonance image (MRI) contrast agents can induce profound complications, including gadolinium encephalopathy, kidney injury, gadolinium-associated plaques, and progressive systemic fibrosis, which can be fatal.1-10 About 50% of MRIs use gadolinium-based contrast (Gd3+), a toxic rare earth metal ion that enhances imaging but requires binding with pharmaceutical ligands to reduce toxicity and promote renal elimination (Figure 1). Despite these measures, Gd3+ can persist in the body, including the brain.11,12 Wastewater treatment fails to remove these agents, making Gd3+ a growing pollutant in water and the food chain.13-15 Because Gd3+ is a rare earth metal ion in the milieu intérieur, there is an urgent need to study its biological and long-term effects (Appendix 1).
Case Presentation
A 65-year-old Vietnam-era veteran presented to nephrology at the Raymond G. Murphy Veterans Affairs Medical Center (RGMVAMC) in Albuquerque, New Mexico, for evaluation of gadolinium-induced symptoms. His medical history included metabolic syndrome, hypertension, hyperlipidemia, hypogonadism, cervical spondylosis, and an elevated prostate-specific antigen, previously assessed with a contrast-enhanced MRI in 2019 (Gadobenic acid, 19 mL). Surgical history included cervical fusion and ankle hardware.
The patient had a scheduled MRI 25 days earlier, following an elevated prostate specific antigen test result, prompting urologic surveillance and concern for malignancy. In preparation for the contrast-enhanced MRI, his right arm was cannulated with a line primed with gadobenic acid contrast. Though the technician stated the infusion had not started, the patient’s symptoms began shortly after entry into the scanner, before any programmed pulse sequences. The patient experienced claustrophobia, diaphoresis, palpitations, xerostomia, dysgeusia, shortness of breath, and a sensation of heat in his groin, chest, “kidneys,” and lower back. The MRI was terminated prematurely in response to the patient’s acute symptomatology. The patient continued experiencing new symptoms intermittently during the following week, including lightheadedness, headaches, right clavicular pain, raspy voice, edema, and a sense of doom.
The patient presented to the RGMVAMC emergency department (ED) 8 days after the MRI with worsening symptoms and was hospitalized for 10 days. During this time, he was referred to nephrology for outpatient evaluation. While awaiting his nephrology appointment, the patient presented to the RGMVAMC ED 20 days after the initial episode with ongoing symptoms. “I thought I was dying,” he said. Laboratory results and a 12-lead electrocardiogram showed a finely static background, wide P waves (> 80 ms) with notching in lead II, sinusoidal P waves in V1, R transition in V2, RR’ in V2, ST flat in lead III, and sinus bradycardia (Table 1 and Appendix 2).
The patient’s medical and surgical histories were reviewed at the nephrology evaluation 25 days following the MRI. He reported that household water was sourced from a well and that he filtered his drinking water with a reverse osmosis system. He served in the US Army for 10 years as an engineer specializing in mechanical systems, power generation, and vehicles. Following Army retirement, the patient served in the US Air Force Reserves for 15 years, working as a crew chief in pneudraulics. The patient reported stopping tobacco use 1 year before and also reported regular use of a broad array of prescription medications and dietary supplements, including dexamethasone (4 mg twice daily), fluticasone nasal spray (50 mcg per nostril, twice daily), ibuprofen (400 mg twice daily, as needed), loratadine (10 mg daily), aspirin (81 mg daily), and metoprolol succinate (50 mg nightly). In addition, he reported consistent use of cholecalciferol (3000 IU daily), another supplemental vitamin D preparation, chelated magnesium glycinate (3 tablets daily for bone issues), turmeric (1 tablet daily), a multivitamin (Living Green Liquid Gel, daily), and a mega-B complex.
Physical examination revealed a well-nourished, tall man with hypertension (145/87 mmHg) and bilateral lower extremity edema. Oral examination showed poor dentition, including missing molars (#1-3, #14-16, #17-19, #30-31), with the anterior teeth replaced by bridges supported by dental implants. The review of systems was otherwise unremarkable, with nocturia noted before the consultation.
Serum and urine gadolinium testing, (Mayo Clinic Laboratories) revealed gadolinium levels of 0.3 mcg/24 h in the urine and 0.1 ng/mL in the serum. Nonzero values indicated detectable gadolinium, suggesting retention. The patient had a prior gadolinium exposure during a 2019 MRI (about 1340 days before) and suspected a repeat exposure on day 0, although the MRI technician stated that no contrast was administered. Given his elevated vitamin D levels, the patient was advised to minimize dietary supplements, particularly vitamin D, to avoid confounding symptoms. The plan included monitoring symptoms and a follow-up evaluation with repeat laboratory tests on day 116.
At the nephrology follow-up 4 months postexposure, the patient's symptoms had primarily abated, with a marked reduction in the previously noted metallic dysgeusia. Physical examination remained consistent with prior findings. He was afebrile (97.7 °F) with a blood pressure of 111/72 mmHg, a pulse of 63 beats per minute, and an oxygen saturation of 98% on ambient air. Laboratory analysis revealed serum and urine gadolinium levels below detectable thresholds (< 0.1 ng/mL and < 0.1 mcg/24 h). A 24-hour creatinine clearance, calculated from a urine volume of 1300 mL, measured at an optimal 106 mL/min, indicating preserved renal function (Tables 2 and 3). Of note, his 24-hour oxalate was above the reference range, with a urine pH below the reference range and a high supersaturation index for calcium oxalate.
Discussion
Use of enhanced MRI has increased in the Veterans Health Administration (Figure 2). A growing range of indications for enhanced procedures (eg, cardiac MRI) has contributed to this rise. The market has grown with new gadolinium-based contrast agents, such as gadopiclenol. However, reliance on untested assumptions about the safety of newer agents and need for robust clinical trials pose potential risks to patient safety.
Without prospective evidence, the American College of Radiology (ACR) classifies gadolinium-based contrast agents into 3 groups: Group 1, associated with the highest number of nephrogenic systemic fibrosis cases; Group 2, linked to few, if any, unconfounded cases; and Group 3, where data on nephrogenic systemic fibrosis risk have been limited. As of April 2024, the ACR reclassified Group 3 agents (Ablavar/Vasovist/Angiomark and Primovist/Eovist) into Group 2. Curiously, Vueway and Elucirem were approved in late 2022 and should clearly be categorized as Group 3 (Table 4).There were 19 cases of nephrogenic systemic fibrosis or similar manifestations, 8 of which were unconfounded by other factors. These patients had been exposed to gadobutrol, often combined with other agents. Gadobutrol—like other Group 2 agents—has been associated with nephrogenic systemic fibrosis.16,17 Despite US Food and Drug Administration (FDA) documentation of rising reports, many clinicians remain unaware that nephrogenic systemic fibrosis is increasingly linked to Group 2 agents classified by the ACR.18 While declines in reported cases of nephrogenic systemic fibrosis may suggest reduced incidence, this trend may reflect diminished clinical vigilance and underreporting, particularly given emerging evidence implicating even Group 2 gadolinium-based contrast agents in delayed and underrecognized presentations. This information has yet to permeate the medical community, particularly among nephrologists. Considering these cases, revisiting the ACR guidelines may be prudent.
To address this growing concern, clinicians must adopt stricter vigilance and actively pursue updated information to mitigate patient risks tied to these contrast agents.
There exists an illusion of knowledge in disregarding the confounded exposures of MRI contrast agents. Ten distinct brands of contrast agents have been approved for clinical use. With repeated imaging, patients are often exposed to varying formulations of gadolinium-based agents. Yet investigators commonly discard these data points when assessing risk. By doing so, they assume—without evidence—that some formulations are inherently less likely to provoke adverse effects (AEs) than others. This untested presumption becomes perilous, especially given the limited understanding of the mechanisms underlying gadolinium-induced pathologies. As Aldous Huxley warned, “Facts do not cease to exist because they are ignored.”19
Gadolinium Persistence
Contrary to expectations, gadolinium persists in the body far longer than initially presumed. Symptoms associated with gadolinium exposure (SAGE) encapsulate the chronic, often enigmatic maladies tied to MRI contrast agents.20 The prolonged retention of this rare earth metal offers a compelling hypothesis for the etiology of SAGE. It has been hypothesized that Lewis base-rich metabolites increase susceptibility to gadolinium-based contrast agent complications.21
The blood and urine concentration elimination curves of gadolinium are exponential and categorized as fast, intermediate, and long-term.1 For urinary elimination, the function of the curves is exponential. The quantity of gadolinium in the urine at a time (t) after exposure (D[Gd](t)) is equal to the product of the amount of gadolinium in the sample (urine or blood) at the end of the fast elimination period (D[Gd](t0)) and the exponential decay with k being a rate constant.
To the authors’ knowledge, we are the only research team currently investigating the rate constant for the intermediate- and long-term phase gadolinium elimination. The Retention and Toxicity of Gadolinium-based Contrast Agents study was approved by the University of New Mexico Health Sciences Center Institutional Review Board on May 27, 2020 (IRB ID 19-660). The data for the patient in this case were compared with preliminary results for patients with exposure-to-measurement intervals < 100 days.
The patient in this case presented with detectable gadolinium levels in urine and serum shortly after an attempted contrast-enhanced MRI procedure (Figure 3). The presence of detectable gadolinium levels in the patient’s urine and serum suggests a likely exposure to a contrast agent about 27 days before his consultation. While the technician reported that no contrast was administered during the attempted MRI, it remains possible that a small amount was introduced during cannulation, potentially triggering the patient’s symptoms. Linear modeling of semilogarithmic plots for participants exposed to contrast agents within 100 days (urine: P = 1.8 × 10ˉ8, adjusted r² = 0.62; blood: P = .005, adjusted r² = 0.21) provided clearance rates (k values) for urine and blood. Extrapolating from these models to the presumed exposure date, the intercepts estimate that the patient received between 0.5% and 8% of a standard contrast dose.
MRI contrast agents can cause skin disease. Systemic fibrosis is considered one of the most severe AEs. Skin pathophysiology involving myeloid cells is driven by elevated levels of monocyte chemoattractant protein-1, which recruits circulating fibroblasts via the C-C chemokine receptor 2.22,23 This occurs alongside activation of NADPH oxidase Nox4.4,24,25 Intracellular gadolinium-rich nanoparticles likely serve as catalysts for this reactive cascade.2,18,22,26,27 These particles assemble around intracellular lipid droplets and ferrule them in spiculated rare earth-rich shells that compromise cellular architecture.2,18,21,22,26,27 Frequently sequestered within endosomal compartments, they disrupt vesicular integrity and threaten cellular homeostasis. Interference with degradative systems such as the endolysosomal axis perturbs energy-recycling pathways—an insidious disturbance, particularly in cells with high metabolic demand. Skin-related symptoms are among the most frequently reported AEs, according to the FDA AE reporting system.18
Studies indicate repeated exposure to MRI contrast agents can lead to permanent gadolinium retention in the brain and other vital organs. Intravenous (IV) contrast agents cross the blood-brain barrier rapidly, while intrathecal administration has been linked to significant and lasting neurologic effects.18
Gadolinium is chemically bound to pharmaceutical ligands to enhance renal clearance and reduce toxicity. However, available data from human samples suggest potential ligand exchanges with undefined physiologic substances. This exchange may facilitate gadolinium precipitation and accumulation within cells into spiculated nanoparticles. Transmission electron microscopy reveals the formation of unilamellar bodies associated with mitochondriopathy and cellular damage, particularly in renal proximal tubules.2,18,22,26,27 It is proposed that intracellular nanoparticle formation represents a key mechanism driving the systemic symptoms observed in patients.1,2,18, 22,26,27
Any hypothesis based on free soluble gadolinium—or concept derived from it—should be discarded. The high affinity of pharmaceutical ligands for gadolinium suggests that the cationic rare earth metal remains predominantly in a ligand-bound, soluble form. It is hypothesized that gadolinium undergoes ligand exchange with physiologic substances, directly leading to nanoparticle formation. Current data demonstrate gadolinium precipitation according to the Le Chatelier’s principle. Since precipitated gadolinium does not readily re-equilibrate with pharmaceutical ligands, repeated administration of different contrast agent brands may contribute to nanoparticle growth.26
Meanwhile, a growing number of patients are turning to chelation therapy, a largely untested treatment. The premise of chelation therapy is rooted in several unproven assumptions.18,21 First, it assumes that clinically significant amounts of gadolinium persist in compartments such as the extracellular space, where they can be effectively chelated and cleared. Second, it presumes that free gadolinium is the primary driver of chronic symptoms, an assertion that remains scientifically unsubstantiated. Finally, chelation proponents overlook the potential harm caused by depleting essential physiological metals during the process, assuming without evidence that the scant removal of gadolinium outweighs the risk of physiological mineral depletion.
These assumptions underpin an unproven remedy that demands critical scrutiny. Recent findings reveal that gadolinium deposits in the skin and kidney often take the form of intracellular nanoparticles, directly challenging the foundation of chelation therapy. Chelation advocates must demonstrate that these intracellular gadolinium deposits neither trigger cellular toxicity nor initiate a cytokine cascade. Chelation supporters must prove that the systemic response to these foreign particles is unrelated to the symptoms reported by patients. Until then, the validity of chelation therapy remains highly questionable.
The causality of the symptoms, mainly whether IV gadolinium was administered, was examined. The null hypothesis stated that the patient was not exposed to gadolinium. However, this hypothesis was contradicted by the detection of gadolinium in the serum and urine 27 days after the potential exposure.
Two plausible explanations exist for the nonzero gadolinium levels detected in the serum and urine. The first possibility is that minute quantities of gadolinium were introduced during cannulation, with the amount being sufficient to persist in measurable concentrations 27 days postexposure. The second possibility is that the gadolinium originated from an MRI contrast agent administered 4 years earlier. In this scenario, gadolinium stored in organ reservoirs such as bone, liver, or kidneys may have been mobilized into the extracellular fluid compartment due to the administration of high-dose steroids 20 days after the recent contrast-enhanced MRI procedure attempt. Coyte et al reported elevated gadolinium levels in the serum, cord blood, breast milk, and placenta of pregnant women with prior exposure to MRI contrast agents.28 These findings suggest that gadolinium, stored in organs such as bone may be remobilized by variables affecting bone remodeling (eg, high-dose steroids).
Significantly, the patient exhibited elevated urinary oxalate levels. Previous research has found that oxalic acid reacts rapidly with MRI contrast agents, forming digadolinium trioxalate. While the gadolinium-rich nanoparticles identified in tissues such as the skin and kidney (including the human kidney) are amorphous, these in vitro findings establish a proof-of-concept: the intracellular environment facilitates gadolinium dissociation from pharmaceutical chelates.
Furthermore, in vitro experiments show that proteins and lysosomal pH promote this dissociation, underscoring how human metabolic conditions—particularly oxalic acid concentration—may drive intracellular gadolinium deposition.
Patient Perspective
“They put something into my body that they cannot get out.” This stark realization underpins the patient’s profound concern about gadolinium-based contrast agents and their potential long-term effects. Reflecting on his experience, the patient expressed deep fears about the unknown future impacts: “I’m concerned about my kidneys, I’m concerned about my heart, and I’m concerned about my brain. I don’t know how this stuff is going to affect me in the future.”
He drew an unsettling parallel between gadolinium and heavy metals: “Heavy metal is poison. The body does not produce this kind of stuff on its own.” His reaction to the procedure left a lasting impression, prompting him to question the logic of using a substance that cannot be purged: “Why would you put something into someone’s body that you cannot extract? Nobody—nobody—should experience what I went through.”
The patient emphasized the lack of clear research on long-term outcomes, which compounds his anxiety: “If there was research that said, ‘Well, this is only going to affect these organs for this long,’ OK, I might be able to accept that. But there is no research like that. Nobody can tell me what’s going to happen in 5 years.”
Strengths and Limitations
A significant strength of this approach is the ability to track gadolinium elimination and symptom resolution over time, supported by unique access to intermediate and long-term clearance data from our ongoing research protocol. The investigators were equipped to back-extrapolate the exposure, which provided a rare opportunity to correlate gadolinium levels with clinical outcomes. The primary limitation is the lack of a defined clinical case definition for gadolinium toxicity and limited mechanistic understanding of SAGE, which hinders diagnosis and management.
Metabolites, proteins, and lipids rich in Lewis bases could initiate this process as substrates for intracellular gadolinium sedimentation. Future studies should investigate whether metabolic conditions such as oxalate burden or altered parathyroid hormone levels modulate gadolinium compartmentalization and tissue retention. If gadolinium-rich nanoparticle formation and accumulation disrupt cellular equilibrium, it underscores an urgent need to understand the implications of long-term gadolinium retention. The research team continues to gather evidence that the gadolinium cation remains chelated from the moment MRI contrast agents are administered through to the formation of intracellular nanoparticles. Retained gadolinium nanoparticles may act as a nidus, triggering cellular signaling cascades that lead to multisymptomatic illnesses. Intracellular and insoluble retained gadolinium challenges proponents of untested chelation therapies.
Conclusions
This case highlights emerging clinical and ethical concerns surrounding gadolinium-based contrast agent use. Clinicians may benefit from considering gadolinium retention as a contributor to persistent, unexplained symptoms—particularly in patients with recent imaging exposure. As contrast use continues to rise within federal health systems, regulatory and administrative stakeholders would do well to re-examine current safety frameworks. Informed consent should reflect what is known: gadolinium can remain in the body long after administration, potentially indefinitely. The long-term consequences of cumulative exposure remain poorly defined, but the presence of a lanthanide element in human tissue warrants greater attention from researchers and regulators alike. Interest in alternative imaging modalities and long-term safety monitoring would mark progress toward more transparent, accountable care.
Magnetic resonance image (MRI) contrast agents can induce profound complications, including gadolinium encephalopathy, kidney injury, gadolinium-associated plaques, and progressive systemic fibrosis, which can be fatal.1-10 About 50% of MRIs use gadolinium-based contrast (Gd3+), a toxic rare earth metal ion that enhances imaging but requires binding with pharmaceutical ligands to reduce toxicity and promote renal elimination (Figure 1). Despite these measures, Gd3+ can persist in the body, including the brain.11,12 Wastewater treatment fails to remove these agents, making Gd3+ a growing pollutant in water and the food chain.13-15 Because Gd3+ is a rare earth metal ion in the milieu intérieur, there is an urgent need to study its biological and long-term effects (Appendix 1).
Case Presentation
A 65-year-old Vietnam-era veteran presented to nephrology at the Raymond G. Murphy Veterans Affairs Medical Center (RGMVAMC) in Albuquerque, New Mexico, for evaluation of gadolinium-induced symptoms. His medical history included metabolic syndrome, hypertension, hyperlipidemia, hypogonadism, cervical spondylosis, and an elevated prostate-specific antigen, previously assessed with a contrast-enhanced MRI in 2019 (Gadobenic acid, 19 mL). Surgical history included cervical fusion and ankle hardware.
The patient had a scheduled MRI 25 days earlier, following an elevated prostate specific antigen test result, prompting urologic surveillance and concern for malignancy. In preparation for the contrast-enhanced MRI, his right arm was cannulated with a line primed with gadobenic acid contrast. Though the technician stated the infusion had not started, the patient’s symptoms began shortly after entry into the scanner, before any programmed pulse sequences. The patient experienced claustrophobia, diaphoresis, palpitations, xerostomia, dysgeusia, shortness of breath, and a sensation of heat in his groin, chest, “kidneys,” and lower back. The MRI was terminated prematurely in response to the patient’s acute symptomatology. The patient continued experiencing new symptoms intermittently during the following week, including lightheadedness, headaches, right clavicular pain, raspy voice, edema, and a sense of doom.
The patient presented to the RGMVAMC emergency department (ED) 8 days after the MRI with worsening symptoms and was hospitalized for 10 days. During this time, he was referred to nephrology for outpatient evaluation. While awaiting his nephrology appointment, the patient presented to the RGMVAMC ED 20 days after the initial episode with ongoing symptoms. “I thought I was dying,” he said. Laboratory results and a 12-lead electrocardiogram showed a finely static background, wide P waves (> 80 ms) with notching in lead II, sinusoidal P waves in V1, R transition in V2, RR’ in V2, ST flat in lead III, and sinus bradycardia (Table 1 and Appendix 2).
The patient’s medical and surgical histories were reviewed at the nephrology evaluation 25 days following the MRI. He reported that household water was sourced from a well and that he filtered his drinking water with a reverse osmosis system. He served in the US Army for 10 years as an engineer specializing in mechanical systems, power generation, and vehicles. Following Army retirement, the patient served in the US Air Force Reserves for 15 years, working as a crew chief in pneudraulics. The patient reported stopping tobacco use 1 year before and also reported regular use of a broad array of prescription medications and dietary supplements, including dexamethasone (4 mg twice daily), fluticasone nasal spray (50 mcg per nostril, twice daily), ibuprofen (400 mg twice daily, as needed), loratadine (10 mg daily), aspirin (81 mg daily), and metoprolol succinate (50 mg nightly). In addition, he reported consistent use of cholecalciferol (3000 IU daily), another supplemental vitamin D preparation, chelated magnesium glycinate (3 tablets daily for bone issues), turmeric (1 tablet daily), a multivitamin (Living Green Liquid Gel, daily), and a mega-B complex.
Physical examination revealed a well-nourished, tall man with hypertension (145/87 mmHg) and bilateral lower extremity edema. Oral examination showed poor dentition, including missing molars (#1-3, #14-16, #17-19, #30-31), with the anterior teeth replaced by bridges supported by dental implants. The review of systems was otherwise unremarkable, with nocturia noted before the consultation.
Serum and urine gadolinium testing, (Mayo Clinic Laboratories) revealed gadolinium levels of 0.3 mcg/24 h in the urine and 0.1 ng/mL in the serum. Nonzero values indicated detectable gadolinium, suggesting retention. The patient had a prior gadolinium exposure during a 2019 MRI (about 1340 days before) and suspected a repeat exposure on day 0, although the MRI technician stated that no contrast was administered. Given his elevated vitamin D levels, the patient was advised to minimize dietary supplements, particularly vitamin D, to avoid confounding symptoms. The plan included monitoring symptoms and a follow-up evaluation with repeat laboratory tests on day 116.
At the nephrology follow-up 4 months postexposure, the patient's symptoms had primarily abated, with a marked reduction in the previously noted metallic dysgeusia. Physical examination remained consistent with prior findings. He was afebrile (97.7 °F) with a blood pressure of 111/72 mmHg, a pulse of 63 beats per minute, and an oxygen saturation of 98% on ambient air. Laboratory analysis revealed serum and urine gadolinium levels below detectable thresholds (< 0.1 ng/mL and < 0.1 mcg/24 h). A 24-hour creatinine clearance, calculated from a urine volume of 1300 mL, measured at an optimal 106 mL/min, indicating preserved renal function (Tables 2 and 3). Of note, his 24-hour oxalate was above the reference range, with a urine pH below the reference range and a high supersaturation index for calcium oxalate.
Discussion
Use of enhanced MRI has increased in the Veterans Health Administration (Figure 2). A growing range of indications for enhanced procedures (eg, cardiac MRI) has contributed to this rise. The market has grown with new gadolinium-based contrast agents, such as gadopiclenol. However, reliance on untested assumptions about the safety of newer agents and need for robust clinical trials pose potential risks to patient safety.
Without prospective evidence, the American College of Radiology (ACR) classifies gadolinium-based contrast agents into 3 groups: Group 1, associated with the highest number of nephrogenic systemic fibrosis cases; Group 2, linked to few, if any, unconfounded cases; and Group 3, where data on nephrogenic systemic fibrosis risk have been limited. As of April 2024, the ACR reclassified Group 3 agents (Ablavar/Vasovist/Angiomark and Primovist/Eovist) into Group 2. Curiously, Vueway and Elucirem were approved in late 2022 and should clearly be categorized as Group 3 (Table 4).There were 19 cases of nephrogenic systemic fibrosis or similar manifestations, 8 of which were unconfounded by other factors. These patients had been exposed to gadobutrol, often combined with other agents. Gadobutrol—like other Group 2 agents—has been associated with nephrogenic systemic fibrosis.16,17 Despite US Food and Drug Administration (FDA) documentation of rising reports, many clinicians remain unaware that nephrogenic systemic fibrosis is increasingly linked to Group 2 agents classified by the ACR.18 While declines in reported cases of nephrogenic systemic fibrosis may suggest reduced incidence, this trend may reflect diminished clinical vigilance and underreporting, particularly given emerging evidence implicating even Group 2 gadolinium-based contrast agents in delayed and underrecognized presentations. This information has yet to permeate the medical community, particularly among nephrologists. Considering these cases, revisiting the ACR guidelines may be prudent.
To address this growing concern, clinicians must adopt stricter vigilance and actively pursue updated information to mitigate patient risks tied to these contrast agents.
There exists an illusion of knowledge in disregarding the confounded exposures of MRI contrast agents. Ten distinct brands of contrast agents have been approved for clinical use. With repeated imaging, patients are often exposed to varying formulations of gadolinium-based agents. Yet investigators commonly discard these data points when assessing risk. By doing so, they assume—without evidence—that some formulations are inherently less likely to provoke adverse effects (AEs) than others. This untested presumption becomes perilous, especially given the limited understanding of the mechanisms underlying gadolinium-induced pathologies. As Aldous Huxley warned, “Facts do not cease to exist because they are ignored.”19
Gadolinium Persistence
Contrary to expectations, gadolinium persists in the body far longer than initially presumed. Symptoms associated with gadolinium exposure (SAGE) encapsulate the chronic, often enigmatic maladies tied to MRI contrast agents.20 The prolonged retention of this rare earth metal offers a compelling hypothesis for the etiology of SAGE. It has been hypothesized that Lewis base-rich metabolites increase susceptibility to gadolinium-based contrast agent complications.21
The blood and urine concentration elimination curves of gadolinium are exponential and categorized as fast, intermediate, and long-term.1 For urinary elimination, the function of the curves is exponential. The quantity of gadolinium in the urine at a time (t) after exposure (D[Gd](t)) is equal to the product of the amount of gadolinium in the sample (urine or blood) at the end of the fast elimination period (D[Gd](t0)) and the exponential decay with k being a rate constant.
To the authors’ knowledge, we are the only research team currently investigating the rate constant for the intermediate- and long-term phase gadolinium elimination. The Retention and Toxicity of Gadolinium-based Contrast Agents study was approved by the University of New Mexico Health Sciences Center Institutional Review Board on May 27, 2020 (IRB ID 19-660). The data for the patient in this case were compared with preliminary results for patients with exposure-to-measurement intervals < 100 days.
The patient in this case presented with detectable gadolinium levels in urine and serum shortly after an attempted contrast-enhanced MRI procedure (Figure 3). The presence of detectable gadolinium levels in the patient’s urine and serum suggests a likely exposure to a contrast agent about 27 days before his consultation. While the technician reported that no contrast was administered during the attempted MRI, it remains possible that a small amount was introduced during cannulation, potentially triggering the patient’s symptoms. Linear modeling of semilogarithmic plots for participants exposed to contrast agents within 100 days (urine: P = 1.8 × 10ˉ8, adjusted r² = 0.62; blood: P = .005, adjusted r² = 0.21) provided clearance rates (k values) for urine and blood. Extrapolating from these models to the presumed exposure date, the intercepts estimate that the patient received between 0.5% and 8% of a standard contrast dose.
MRI contrast agents can cause skin disease. Systemic fibrosis is considered one of the most severe AEs. Skin pathophysiology involving myeloid cells is driven by elevated levels of monocyte chemoattractant protein-1, which recruits circulating fibroblasts via the C-C chemokine receptor 2.22,23 This occurs alongside activation of NADPH oxidase Nox4.4,24,25 Intracellular gadolinium-rich nanoparticles likely serve as catalysts for this reactive cascade.2,18,22,26,27 These particles assemble around intracellular lipid droplets and ferrule them in spiculated rare earth-rich shells that compromise cellular architecture.2,18,21,22,26,27 Frequently sequestered within endosomal compartments, they disrupt vesicular integrity and threaten cellular homeostasis. Interference with degradative systems such as the endolysosomal axis perturbs energy-recycling pathways—an insidious disturbance, particularly in cells with high metabolic demand. Skin-related symptoms are among the most frequently reported AEs, according to the FDA AE reporting system.18
Studies indicate repeated exposure to MRI contrast agents can lead to permanent gadolinium retention in the brain and other vital organs. Intravenous (IV) contrast agents cross the blood-brain barrier rapidly, while intrathecal administration has been linked to significant and lasting neurologic effects.18
Gadolinium is chemically bound to pharmaceutical ligands to enhance renal clearance and reduce toxicity. However, available data from human samples suggest potential ligand exchanges with undefined physiologic substances. This exchange may facilitate gadolinium precipitation and accumulation within cells into spiculated nanoparticles. Transmission electron microscopy reveals the formation of unilamellar bodies associated with mitochondriopathy and cellular damage, particularly in renal proximal tubules.2,18,22,26,27 It is proposed that intracellular nanoparticle formation represents a key mechanism driving the systemic symptoms observed in patients.1,2,18, 22,26,27
Any hypothesis based on free soluble gadolinium—or concept derived from it—should be discarded. The high affinity of pharmaceutical ligands for gadolinium suggests that the cationic rare earth metal remains predominantly in a ligand-bound, soluble form. It is hypothesized that gadolinium undergoes ligand exchange with physiologic substances, directly leading to nanoparticle formation. Current data demonstrate gadolinium precipitation according to the Le Chatelier’s principle. Since precipitated gadolinium does not readily re-equilibrate with pharmaceutical ligands, repeated administration of different contrast agent brands may contribute to nanoparticle growth.26
Meanwhile, a growing number of patients are turning to chelation therapy, a largely untested treatment. The premise of chelation therapy is rooted in several unproven assumptions.18,21 First, it assumes that clinically significant amounts of gadolinium persist in compartments such as the extracellular space, where they can be effectively chelated and cleared. Second, it presumes that free gadolinium is the primary driver of chronic symptoms, an assertion that remains scientifically unsubstantiated. Finally, chelation proponents overlook the potential harm caused by depleting essential physiological metals during the process, assuming without evidence that the scant removal of gadolinium outweighs the risk of physiological mineral depletion.
These assumptions underpin an unproven remedy that demands critical scrutiny. Recent findings reveal that gadolinium deposits in the skin and kidney often take the form of intracellular nanoparticles, directly challenging the foundation of chelation therapy. Chelation advocates must demonstrate that these intracellular gadolinium deposits neither trigger cellular toxicity nor initiate a cytokine cascade. Chelation supporters must prove that the systemic response to these foreign particles is unrelated to the symptoms reported by patients. Until then, the validity of chelation therapy remains highly questionable.
The causality of the symptoms, mainly whether IV gadolinium was administered, was examined. The null hypothesis stated that the patient was not exposed to gadolinium. However, this hypothesis was contradicted by the detection of gadolinium in the serum and urine 27 days after the potential exposure.
Two plausible explanations exist for the nonzero gadolinium levels detected in the serum and urine. The first possibility is that minute quantities of gadolinium were introduced during cannulation, with the amount being sufficient to persist in measurable concentrations 27 days postexposure. The second possibility is that the gadolinium originated from an MRI contrast agent administered 4 years earlier. In this scenario, gadolinium stored in organ reservoirs such as bone, liver, or kidneys may have been mobilized into the extracellular fluid compartment due to the administration of high-dose steroids 20 days after the recent contrast-enhanced MRI procedure attempt. Coyte et al reported elevated gadolinium levels in the serum, cord blood, breast milk, and placenta of pregnant women with prior exposure to MRI contrast agents.28 These findings suggest that gadolinium, stored in organs such as bone may be remobilized by variables affecting bone remodeling (eg, high-dose steroids).
Significantly, the patient exhibited elevated urinary oxalate levels. Previous research has found that oxalic acid reacts rapidly with MRI contrast agents, forming digadolinium trioxalate. While the gadolinium-rich nanoparticles identified in tissues such as the skin and kidney (including the human kidney) are amorphous, these in vitro findings establish a proof-of-concept: the intracellular environment facilitates gadolinium dissociation from pharmaceutical chelates.
Furthermore, in vitro experiments show that proteins and lysosomal pH promote this dissociation, underscoring how human metabolic conditions—particularly oxalic acid concentration—may drive intracellular gadolinium deposition.
Patient Perspective
“They put something into my body that they cannot get out.” This stark realization underpins the patient’s profound concern about gadolinium-based contrast agents and their potential long-term effects. Reflecting on his experience, the patient expressed deep fears about the unknown future impacts: “I’m concerned about my kidneys, I’m concerned about my heart, and I’m concerned about my brain. I don’t know how this stuff is going to affect me in the future.”
He drew an unsettling parallel between gadolinium and heavy metals: “Heavy metal is poison. The body does not produce this kind of stuff on its own.” His reaction to the procedure left a lasting impression, prompting him to question the logic of using a substance that cannot be purged: “Why would you put something into someone’s body that you cannot extract? Nobody—nobody—should experience what I went through.”
The patient emphasized the lack of clear research on long-term outcomes, which compounds his anxiety: “If there was research that said, ‘Well, this is only going to affect these organs for this long,’ OK, I might be able to accept that. But there is no research like that. Nobody can tell me what’s going to happen in 5 years.”
Strengths and Limitations
A significant strength of this approach is the ability to track gadolinium elimination and symptom resolution over time, supported by unique access to intermediate and long-term clearance data from our ongoing research protocol. The investigators were equipped to back-extrapolate the exposure, which provided a rare opportunity to correlate gadolinium levels with clinical outcomes. The primary limitation is the lack of a defined clinical case definition for gadolinium toxicity and limited mechanistic understanding of SAGE, which hinders diagnosis and management.
Metabolites, proteins, and lipids rich in Lewis bases could initiate this process as substrates for intracellular gadolinium sedimentation. Future studies should investigate whether metabolic conditions such as oxalate burden or altered parathyroid hormone levels modulate gadolinium compartmentalization and tissue retention. If gadolinium-rich nanoparticle formation and accumulation disrupt cellular equilibrium, it underscores an urgent need to understand the implications of long-term gadolinium retention. The research team continues to gather evidence that the gadolinium cation remains chelated from the moment MRI contrast agents are administered through to the formation of intracellular nanoparticles. Retained gadolinium nanoparticles may act as a nidus, triggering cellular signaling cascades that lead to multisymptomatic illnesses. Intracellular and insoluble retained gadolinium challenges proponents of untested chelation therapies.
Conclusions
This case highlights emerging clinical and ethical concerns surrounding gadolinium-based contrast agent use. Clinicians may benefit from considering gadolinium retention as a contributor to persistent, unexplained symptoms—particularly in patients with recent imaging exposure. As contrast use continues to rise within federal health systems, regulatory and administrative stakeholders would do well to re-examine current safety frameworks. Informed consent should reflect what is known: gadolinium can remain in the body long after administration, potentially indefinitely. The long-term consequences of cumulative exposure remain poorly defined, but the presence of a lanthanide element in human tissue warrants greater attention from researchers and regulators alike. Interest in alternative imaging modalities and long-term safety monitoring would mark progress toward more transparent, accountable care.
Jackson DB, MacIntyre T, Duarte-Miramontes V, et al. Gadolinium deposition disease: a case report and the prevalence of enhanced MRI procedures within the Veterans Health Administration. Fed Pract. 2022;39:218-225. doi:10.12788/fp.0258
Do C, DeAguero J, Brearley A, et al. Gadolinium-based contrast agent use, their safety, and practice evolution. Kidney360. 2020;1:561-568.doi:10.34067/kid.0000272019
Leyba K, Wagner B. Gadolinium-based contrast agents: why nephrologists need to be concerned. Curr Opin Nephrol Hypertens. 2019;28:154-162. doi:10.1097/MNH.0000000000000475
Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;311:F1-F11. doi:10.1152/ajprenal.00166.2016
Maramattom BV, Manno EM, Wijdicks EF, et al. Gadolinium encephalopathy in a patient with renal failure. Neurology. 2005;64:1276-1278.doi:10.1212/01.WNL.0000156805.45547.6E
Sam AD II, Morasch MD, Collins J, et al. Safety of gadolinium contrast angiography in patients with chronic renal insufficiency. J Vasc Surg. 2003;38:313-318. doi:10.1016/s0741-5214(03)00315-x
Schenker MP, Solomon JA, Roberts DA. Gadolinium arteriography complicated by acute pancreatitis and acute renal failure. J Vasc Interv Radiol. 2001;12:393. doi:10.1016/s1051-0443(07)61925-3
Gemery J, Idelson B, Reid S, et al. Acute renal failure after arteriography with a gadolinium-based contrast agent. AJR Am J Roentgenol. 1998;171:1277-1278. doi:10.2214/ajr.171.5.9798860
Akgun H, Gonlusen G, Cartwright J Jr, et al. Are gadolinium-based contrast media nephrotoxic? A renal biopsy study. Arch Pathol Lab Med. 2006;130:1354-1357. doi:10.5858/2006-130-1354-AGCMNA
Gathings RM, Reddy R, Santa Cruz D, et al. Gadolinium-associated plaques: a new, distinctive clinical entity. JAMA Dermatol. 2015;151:316-319. doi:10.1001/jamadermatol.2014.2660
McDonald RJ, McDonald JS, Kallmes DF, et al. Gadolinium deposition in human brain tissues after contrast-enhanced MR imaging in adult patients without intracranial abnormalities. Radiology. 2017;285(2):546-554. doi:10.1148/radiol.2017161595
Kanda T, Ishii K, Kawaguchi H, et al. High signal intensity in the dentate nucleus and globus pallidus on unenhanced T1-weighted MR images: relationship with increasing cumulative dose of a gadolinium-based contrast material. Radiology. 2014;270(3):834-841. doi:10.1148/radiol.13131669
Schmidt K, Bau M, Merschel G, et al. Anthropogenic gadolinium in tap water and in tap water-based beverages from fast-food franchises in six major cities in Germany. Sci Total Environ. 2019;687:1401-1408. doi:10.1016/j.scitotenv.2019.07.075
Kulaksız S, Bau M. Anthropogenic gadolinium as a microcontaminant in tap water used as drinking water in urban areas and megacities. Appl Geochem. 2011;26:1877-1885.
Brunjes R, Hofmann T. Anthropogenic gadolinium in freshwater and drinking water systems. Water Res. 2020;182:115966. doi:10.1016/j.watres.2020.115966
Endrikat J, Gutberlet M, Hoffmann KT, et al. Clinical safety of gadobutrol: review of over 25 years of use exceeding 100 million administrations. Invest Radiol. 2024;59(9):605-613. doi:10.1097/RLI.0000000000001072
Elmholdt TR, Jørgensen B, Ramsing M, et al. Two cases of nephrogenic systemic fibrosis after exposure to the macrocyclic compound gadobutrol. NDT Plus. 2010;3(3):285-287. doi:10.1093/ndtplus/sfq028
Cunningham A, Kirk M, Hong E, et al. The safety of magnetic resonance imaging contrast agents. Front Toxicol. 2024;6:1376587. doi:10.3389/ftox.2024.1376587
Huxley A. Complete Essays. Volume II, 1926-1929. Chicago; 2000:227.
McDonald RJ, Weinreb JC, Davenport MS. Symptoms associated with gadolinium exposure (SAGE): a suggested term. Radiology. 2022;302(2):270-273. doi:10.1148/radiol.2021211349
Henderson IM, Benevidez AD, Mowry CD, et al. Precipitation of gadolinium from magnetic resonance imaging contrast agents may be the Brass tacks of toxicity. Magn Reson Imaging. 2025;119:110383. doi:10.1016/j.mri.2025.110383
Do C, Drel V, Tan C, et al. Nephrogenic systemic fibrosis is mediated by myeloid C-C chemokine receptor 2. J Invest Dermatol. 2019;139(10):2134-2143. doi:10.1016/j.jid.2019.03.1145
Drel VR, Tan C, Barnes JL, et al. Centrality of bone marrow in the severity of gadolinium-based contrast-induced systemic fibrosis. FASEB J. 2016;30(9):3026-3038. doi:10.1096/fj.201500188R
Bruno F, DeAguero J, Do C, et al. Overlapping roles of NADPH oxidase 4 for diabetic and gadolinium-based contrast agent-induced systemic fibrosis. Am J Physiol Renal Physiol. 2021;320(4):F617-F627. doi:10.1152/ajprenal.00456.2020
Wagner B, Tan C, Barnes JL, et al. Nephrogenic systemic fibrosis: evidence for oxidative stress and bone marrow-derived fibrocytes in skin, liver, and heart lesions using a 5/6 nephrectomy rodent model. Am J Pathol. 2012;181(6):1941-1952. doi:10.1016/j.ajpath.2012.08.026
DeAguero J, Howard T, Kusewitt D, et al. The onset of rare earth metallosis begins with renal gadolinium-rich nanoparticles from magnetic resonance imaging contrast agent exposure. Sci Rep. 2023;13(1):2025. doi:10.1038/s41598-023-28666-1
Do C, Ford B, Lee DY, et al. Gadolinium-based contrast agents: Stimulators of myeloid-induced renal fibrosis and major metabolic disruptors. Toxicol Appl Pharmacol. 2019;375:32-45. doi:10.1016/j.taap.2019.05.009
Coyte RM, Darrah T, Olesik J, et al. Gadolinium during human pregnancy following administration of gadolinium chelate before pregnancy. Birth Defects Res. 2023;115(14):1264-1273. doi:10.1002/bdr2.2209
Jackson DB, MacIntyre T, Duarte-Miramontes V, et al. Gadolinium deposition disease: a case report and the prevalence of enhanced MRI procedures within the Veterans Health Administration. Fed Pract. 2022;39:218-225. doi:10.12788/fp.0258
Do C, DeAguero J, Brearley A, et al. Gadolinium-based contrast agent use, their safety, and practice evolution. Kidney360. 2020;1:561-568.doi:10.34067/kid.0000272019
Leyba K, Wagner B. Gadolinium-based contrast agents: why nephrologists need to be concerned. Curr Opin Nephrol Hypertens. 2019;28:154-162. doi:10.1097/MNH.0000000000000475
Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;311:F1-F11. doi:10.1152/ajprenal.00166.2016
Maramattom BV, Manno EM, Wijdicks EF, et al. Gadolinium encephalopathy in a patient with renal failure. Neurology. 2005;64:1276-1278.doi:10.1212/01.WNL.0000156805.45547.6E
Sam AD II, Morasch MD, Collins J, et al. Safety of gadolinium contrast angiography in patients with chronic renal insufficiency. J Vasc Surg. 2003;38:313-318. doi:10.1016/s0741-5214(03)00315-x
Schenker MP, Solomon JA, Roberts DA. Gadolinium arteriography complicated by acute pancreatitis and acute renal failure. J Vasc Interv Radiol. 2001;12:393. doi:10.1016/s1051-0443(07)61925-3
Gemery J, Idelson B, Reid S, et al. Acute renal failure after arteriography with a gadolinium-based contrast agent. AJR Am J Roentgenol. 1998;171:1277-1278. doi:10.2214/ajr.171.5.9798860
Akgun H, Gonlusen G, Cartwright J Jr, et al. Are gadolinium-based contrast media nephrotoxic? A renal biopsy study. Arch Pathol Lab Med. 2006;130:1354-1357. doi:10.5858/2006-130-1354-AGCMNA
Gathings RM, Reddy R, Santa Cruz D, et al. Gadolinium-associated plaques: a new, distinctive clinical entity. JAMA Dermatol. 2015;151:316-319. doi:10.1001/jamadermatol.2014.2660
McDonald RJ, McDonald JS, Kallmes DF, et al. Gadolinium deposition in human brain tissues after contrast-enhanced MR imaging in adult patients without intracranial abnormalities. Radiology. 2017;285(2):546-554. doi:10.1148/radiol.2017161595
Kanda T, Ishii K, Kawaguchi H, et al. High signal intensity in the dentate nucleus and globus pallidus on unenhanced T1-weighted MR images: relationship with increasing cumulative dose of a gadolinium-based contrast material. Radiology. 2014;270(3):834-841. doi:10.1148/radiol.13131669
Schmidt K, Bau M, Merschel G, et al. Anthropogenic gadolinium in tap water and in tap water-based beverages from fast-food franchises in six major cities in Germany. Sci Total Environ. 2019;687:1401-1408. doi:10.1016/j.scitotenv.2019.07.075
Kulaksız S, Bau M. Anthropogenic gadolinium as a microcontaminant in tap water used as drinking water in urban areas and megacities. Appl Geochem. 2011;26:1877-1885.
Brunjes R, Hofmann T. Anthropogenic gadolinium in freshwater and drinking water systems. Water Res. 2020;182:115966. doi:10.1016/j.watres.2020.115966
Endrikat J, Gutberlet M, Hoffmann KT, et al. Clinical safety of gadobutrol: review of over 25 years of use exceeding 100 million administrations. Invest Radiol. 2024;59(9):605-613. doi:10.1097/RLI.0000000000001072
Elmholdt TR, Jørgensen B, Ramsing M, et al. Two cases of nephrogenic systemic fibrosis after exposure to the macrocyclic compound gadobutrol. NDT Plus. 2010;3(3):285-287. doi:10.1093/ndtplus/sfq028
Cunningham A, Kirk M, Hong E, et al. The safety of magnetic resonance imaging contrast agents. Front Toxicol. 2024;6:1376587. doi:10.3389/ftox.2024.1376587
Huxley A. Complete Essays. Volume II, 1926-1929. Chicago; 2000:227.
McDonald RJ, Weinreb JC, Davenport MS. Symptoms associated with gadolinium exposure (SAGE): a suggested term. Radiology. 2022;302(2):270-273. doi:10.1148/radiol.2021211349
Henderson IM, Benevidez AD, Mowry CD, et al. Precipitation of gadolinium from magnetic resonance imaging contrast agents may be the Brass tacks of toxicity. Magn Reson Imaging. 2025;119:110383. doi:10.1016/j.mri.2025.110383
Do C, Drel V, Tan C, et al. Nephrogenic systemic fibrosis is mediated by myeloid C-C chemokine receptor 2. J Invest Dermatol. 2019;139(10):2134-2143. doi:10.1016/j.jid.2019.03.1145
Drel VR, Tan C, Barnes JL, et al. Centrality of bone marrow in the severity of gadolinium-based contrast-induced systemic fibrosis. FASEB J. 2016;30(9):3026-3038. doi:10.1096/fj.201500188R
Bruno F, DeAguero J, Do C, et al. Overlapping roles of NADPH oxidase 4 for diabetic and gadolinium-based contrast agent-induced systemic fibrosis. Am J Physiol Renal Physiol. 2021;320(4):F617-F627. doi:10.1152/ajprenal.00456.2020
Wagner B, Tan C, Barnes JL, et al. Nephrogenic systemic fibrosis: evidence for oxidative stress and bone marrow-derived fibrocytes in skin, liver, and heart lesions using a 5/6 nephrectomy rodent model. Am J Pathol. 2012;181(6):1941-1952. doi:10.1016/j.ajpath.2012.08.026
DeAguero J, Howard T, Kusewitt D, et al. The onset of rare earth metallosis begins with renal gadolinium-rich nanoparticles from magnetic resonance imaging contrast agent exposure. Sci Rep. 2023;13(1):2025. doi:10.1038/s41598-023-28666-1
Do C, Ford B, Lee DY, et al. Gadolinium-based contrast agents: Stimulators of myeloid-induced renal fibrosis and major metabolic disruptors. Toxicol Appl Pharmacol. 2019;375:32-45. doi:10.1016/j.taap.2019.05.009
Coyte RM, Darrah T, Olesik J, et al. Gadolinium during human pregnancy following administration of gadolinium chelate before pregnancy. Birth Defects Res. 2023;115(14):1264-1273. doi:10.1002/bdr2.2209
Gadolinium Intermediate Elimination and Persistent Symptoms After Magnetic Resonance Imaging Contrast Agent Exposure
Gadolinium Intermediate Elimination and Persistent Symptoms After Magnetic Resonance Imaging Contrast Agent Exposure
Atrophic Areas on the Axillary and Anogenital Anatomy
Atrophic Areas on the Axillary and Anogenital Anatomy
Discussion
A diagnosis of lichen sclerosus (LS) was made based on clinical and dermoscopic features, followed by confirmation with histology. The patient’s presentation included typical signs and symptoms of LS: itching, burning, intermittent bleeding, perianal hemorrhage, fusion of the clitoral head, and fissures. Other presentations can include dyspareunia, erosions, and excoriations; however, these symptoms and signs were not reported or seen in this patient.
LS typically affects the anogenital region and has 2 peak incidences: in preadolescent teens and during the fifth to sixth decade of life.1 This patient presented with a case of extragenital LS, which is less common than the classic presentation of LS that affects the genitals. This variant’s epidemiology differs, as it is less common in children and more common in postmenopausal women.2 Extragenital LS presents as white, atrophic plaques with a predilection for sites including the trunk, breasts, upper arms, and sites of physical trauma, with symptoms of dryness and pruritus. Over time, the papules can coalesce and form ivory, scar-like papules or plaques with a wrinkled surface. In advanced stages, telangiectasia or follicular plugging can be present, along with flattening of the dermal-epidermal junction. This flat interface is fragile and can result in bullae that may become hemorrhagic.
Cutaneous squamous cell carcinoma (SCC) may infrequently arise from LS, similar to other chronic inflammatory dermatoses.3 Lichen planus is typically not associated with an increased risk of SCC, except in the oral and hypertrophic variants. However, LS may be considered a premalignant process, and many vulvar SCC cases are noted to have adjacent LS lesions.3
Autoimmune and genetic factors contribute to the pathogenesis of LS. Extracellular matrix protein 1 (ECM1) binds molecules of the basement membrane zone and dermis, contributing to the structure and integrity of skin. Autoantibodies against ECM1 and other antigens of the basement membrane zone, including BP180 and BP320, were found in LS.2 HLA-DQ7 major histocompatibility complex class II antigens have been associated with LS.1
On histologic examination, the epidermis of LS is atrophic with hyperkeratosis. The dermis shows homogenization and sclerosis of superficial collagen with a band-like lymphocytic infiltrate below the sclerosis. The basal layer is thickened, showing basal cell vacuolization and hydropic degeneration.4
First-line treatment for genital and extragenital variants of LS is high-potency topical steroids for 3 months or until the skin texture and color resolve (ie, clobetasol 0.05% cream or ointment). The second-line treatment is a topical calcineurin inhibitor. These treatments are used for management. They are not cures for LS, as relapse is possible after the initial treatment course is completed. Adverse effects of high potency topical steroids are skin burning, skin atrophy, and fragility, telangiectasia. The adverse effects of topical calcineurin inhibitors are stinging and burning on application.
Other Diagnostic Considerations
Inverse psoriasis (IP) is a variant of psoriasis that presents as erythematous, well-demarcated plaques with minimal scale in intertriginous areas and flexural surfaces. Localized dermatophyte, candidal, or bacterial infections can trigger IP.5 It occurs in about 3% to 7% of patients with plaque psoriasis and is thought to form due to koebnerization via mechanical friction of flexural zones.6 The patient described in this case did not have IP because IP would be more likely to present as a well-demarcated erythematous plaque rather than a patch.
Histologically, IP shows regular psoriasiform acanthosis and hypogranulosis of the epidermis, Munro microabscess, spongiform pustules of Kogoj, dilated tortuous dermal vessels, and thinning of the suprapapillary plates.5
Lichen planus pigmentosus-inversus (LPPI) is also known as lichen planus pigmentosus—intertriginous variant. This variant of lichen planus pigmentosus presents as multiple gray to dark brown macules and patches with poorly defined borders in a linear distribution limited to intertriginous areas, flexural surfaces, or following the lines of Blaschko.7 About 20% of cases present with frontal fibrosing alopecia. It is most common in individuals with intermediate and darker skin pigmentation, has a higher prevalence in females, and typically occurs within the third and fifth decades of life. Friction is a common trigger of LPPI.7 A diagnosis of LPPI is incorrect because the lesions would present as gray to dark brown macules, as opposed to the shiny white atrophic thin papules with surrounding pink and purple patches seen in this case.
Histologically, while both LS and LPPI share band-like lymphocytic infiltrate and basal cell vacuolization, findings in the dermis differ. LPPI shows melanophages and prominent melanin incontinence, while LS shows homogenization and sclerosis of superficial collagen.1,8 LPPI also shows absence of compensatory keratinocyte proliferation.
Morphea is an inflammatory disease that affects the dermis and subcutaneous fat, resulting in sclerosis that appears scarlike. Its prevalence increases with age and has a 4:1 prevalence in females, with the plaque type being the most common variant. 9 The typical presentation of plaque-type morphea is an insidious onset of asymptomatic, slightly elevated, erythematous or violaceous, slightly edematous plaques with centrifugal expansion. The center of the plaque may become sclerotic and indurated, acquiring a shiny white color with a peripheral “lilac” ring. Trunk and upper extremity involvement is common. Morphea is associated with increased antisingle-stranded DNA, antitopoisomerase IIa, antiphospholipid, antifibrillin-1, and antihistone antibodies. Triggers of morphea are believed to be localized insults to the skin, including mechanical trauma, injections, vaccinations, and irradiation.9 This answer is incorrect because the patient’s lesions were pruritic and had genital involvement, which are not typical of morphea. Morphea can be differentiated with based on symptoms (lack of pruritus, pain, burning), morphology of lesions (induration versus atrophy), dermoscopy (fibrotic beams with less scale and hemorrhage vs keratotic follicular plugs), and histopathology (depth of inflammation in superficial and deep dermis).
Histology of morphea can differ based on the stage, whether the lesion is sampled in the inflammatory margin or central sclerosis, and the depth of affected skin. At the inflammatory margin, vascular changes, including endothelial swelling and edema, are present, as well as CD4+ T cells, eosinophils, plasma cells, and mast cells surrounding smaller blood vessels. In late stages, the inflammatory infiltrate is no longer present, the epidermis appears regular, and there is a flattened dermal-epidermal junction. Distinct features include homogenous collagen bundles that replace many dermal structures, with atrophic eccrine glands that appear “trapped” in the thickened dermis, and homogenized and hyalinized subcutis.9
Mycosis fungoides (MF) is the most common type of cutaneous T-cell lymphoma and presents as annular, erythematous or hypopigmented patches and plaques with fine scale and tumors on the buttocks and sun-protected areas of the limbs and trunk. Lesions can appear with prominent poikiloderma or atrophic or lichenified skin.10 It is most common in males of African descent aged 50 to 55 years. The etiology is largely unknown but believed to be multifactorial. This answer is incorrect because the lesions in this patient appeared more atrophic, were less well demarcated, and lacked the scale that would be present in MF.
On histology, both LS and MF show band-like lymphocytic infiltrate, however MF lacks the homogenization and sclerosis of superficial collagen that is present in the dermis of LS. Also, MF demonstrates epidermotropism of atypical lymphocytes forming Pautrier microabscess.10
Primary Care Role
Primary care physicians can diagnose and treat LS. Referral to dermatology is not mandatory. Note that topical steroids can be used daily for up to 12 weeks. In LS, early treatment is associated with improved outcomes and minimizes the risk of irreversible skin changes.11 Follow-up during the treatment period is recommended to monitor subjective and objective response to treatment. Follow-up after the initial treatment is recommended since LS is typically chronic, can relapse, and SCC can infrequently arise from LS lesions.11
- Tran DA, Tan X, Macri CJ, Goldstein AT, Fu SW. Lichen sclerosus: an autoimmunopathogenic and genomic enigma with emerging genetic and immune targets. Int J Biol Sci. 2019;15:1429-1439. doi:10.7150/ijbs.34613
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389/fmed.2023.1106318
- Kuraitis D, Murina A. Squamous cell carcinoma arising in chronic inflammatory dermatoses. Cutis. 2024;113:29-34. doi:10.12788/cutis.0914
- Gaertner E, Elstein W. Lichen planus pigmentosus-inversus: case report and review of an unusual entity. Dermatol Online J. 2012;18:11.
- Micali G, Verzì AE, Giuffrida G, et al. Inverse psoriasis: from diagnosis to current treatment options. Clin Cosmet Investig Dermatol. 2019;12:953-959. doi:10.2147/CCID.S189000
- Syed ZU, Khachemoune A. Inverse psoriasis: case presentation and review. Am J Clin Dermatol. 2011;12:143-146. doi:10.2165/11532060-000000000-00000
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514. doi:10.1111/ijd.13806
- Vinay K, Kumar S, Bishnoi A, et al. A clinico-demographic study of 344 patients with lichen planus pigmentosus seen in a tertiary care center in India over an 8-year period. Int J Dermatol. 2020;59:245-252. doi:10.1111/ijd.14540
- Papara C, De Luca DA, Bieber K, et al. Morphea: the 2023 update. Front Med (Lausanne). 2023;10:1108623. doi:10.3389/fmed.2023.1108623
- Zinzani PL, Ferreri AJ, Cerroni L. Mycosis fungoides. Cri t Rev Oncol Hematol. 2008;65:172-182. doi:10.1016/j.critrevonc.2007.08.004
- Lee A, Bradford J, Fischer G. Long-term management of adult vulvar lichen sclerosus: a prospective cohort study of 507 women. JAMA Dermatol. 2015;151(10):1061-1067. doi:10.1001/jamadermatol.2015.0643
Discussion
A diagnosis of lichen sclerosus (LS) was made based on clinical and dermoscopic features, followed by confirmation with histology. The patient’s presentation included typical signs and symptoms of LS: itching, burning, intermittent bleeding, perianal hemorrhage, fusion of the clitoral head, and fissures. Other presentations can include dyspareunia, erosions, and excoriations; however, these symptoms and signs were not reported or seen in this patient.
LS typically affects the anogenital region and has 2 peak incidences: in preadolescent teens and during the fifth to sixth decade of life.1 This patient presented with a case of extragenital LS, which is less common than the classic presentation of LS that affects the genitals. This variant’s epidemiology differs, as it is less common in children and more common in postmenopausal women.2 Extragenital LS presents as white, atrophic plaques with a predilection for sites including the trunk, breasts, upper arms, and sites of physical trauma, with symptoms of dryness and pruritus. Over time, the papules can coalesce and form ivory, scar-like papules or plaques with a wrinkled surface. In advanced stages, telangiectasia or follicular plugging can be present, along with flattening of the dermal-epidermal junction. This flat interface is fragile and can result in bullae that may become hemorrhagic.
Cutaneous squamous cell carcinoma (SCC) may infrequently arise from LS, similar to other chronic inflammatory dermatoses.3 Lichen planus is typically not associated with an increased risk of SCC, except in the oral and hypertrophic variants. However, LS may be considered a premalignant process, and many vulvar SCC cases are noted to have adjacent LS lesions.3
Autoimmune and genetic factors contribute to the pathogenesis of LS. Extracellular matrix protein 1 (ECM1) binds molecules of the basement membrane zone and dermis, contributing to the structure and integrity of skin. Autoantibodies against ECM1 and other antigens of the basement membrane zone, including BP180 and BP320, were found in LS.2 HLA-DQ7 major histocompatibility complex class II antigens have been associated with LS.1
On histologic examination, the epidermis of LS is atrophic with hyperkeratosis. The dermis shows homogenization and sclerosis of superficial collagen with a band-like lymphocytic infiltrate below the sclerosis. The basal layer is thickened, showing basal cell vacuolization and hydropic degeneration.4
First-line treatment for genital and extragenital variants of LS is high-potency topical steroids for 3 months or until the skin texture and color resolve (ie, clobetasol 0.05% cream or ointment). The second-line treatment is a topical calcineurin inhibitor. These treatments are used for management. They are not cures for LS, as relapse is possible after the initial treatment course is completed. Adverse effects of high potency topical steroids are skin burning, skin atrophy, and fragility, telangiectasia. The adverse effects of topical calcineurin inhibitors are stinging and burning on application.
Other Diagnostic Considerations
Inverse psoriasis (IP) is a variant of psoriasis that presents as erythematous, well-demarcated plaques with minimal scale in intertriginous areas and flexural surfaces. Localized dermatophyte, candidal, or bacterial infections can trigger IP.5 It occurs in about 3% to 7% of patients with plaque psoriasis and is thought to form due to koebnerization via mechanical friction of flexural zones.6 The patient described in this case did not have IP because IP would be more likely to present as a well-demarcated erythematous plaque rather than a patch.
Histologically, IP shows regular psoriasiform acanthosis and hypogranulosis of the epidermis, Munro microabscess, spongiform pustules of Kogoj, dilated tortuous dermal vessels, and thinning of the suprapapillary plates.5
Lichen planus pigmentosus-inversus (LPPI) is also known as lichen planus pigmentosus—intertriginous variant. This variant of lichen planus pigmentosus presents as multiple gray to dark brown macules and patches with poorly defined borders in a linear distribution limited to intertriginous areas, flexural surfaces, or following the lines of Blaschko.7 About 20% of cases present with frontal fibrosing alopecia. It is most common in individuals with intermediate and darker skin pigmentation, has a higher prevalence in females, and typically occurs within the third and fifth decades of life. Friction is a common trigger of LPPI.7 A diagnosis of LPPI is incorrect because the lesions would present as gray to dark brown macules, as opposed to the shiny white atrophic thin papules with surrounding pink and purple patches seen in this case.
Histologically, while both LS and LPPI share band-like lymphocytic infiltrate and basal cell vacuolization, findings in the dermis differ. LPPI shows melanophages and prominent melanin incontinence, while LS shows homogenization and sclerosis of superficial collagen.1,8 LPPI also shows absence of compensatory keratinocyte proliferation.
Morphea is an inflammatory disease that affects the dermis and subcutaneous fat, resulting in sclerosis that appears scarlike. Its prevalence increases with age and has a 4:1 prevalence in females, with the plaque type being the most common variant. 9 The typical presentation of plaque-type morphea is an insidious onset of asymptomatic, slightly elevated, erythematous or violaceous, slightly edematous plaques with centrifugal expansion. The center of the plaque may become sclerotic and indurated, acquiring a shiny white color with a peripheral “lilac” ring. Trunk and upper extremity involvement is common. Morphea is associated with increased antisingle-stranded DNA, antitopoisomerase IIa, antiphospholipid, antifibrillin-1, and antihistone antibodies. Triggers of morphea are believed to be localized insults to the skin, including mechanical trauma, injections, vaccinations, and irradiation.9 This answer is incorrect because the patient’s lesions were pruritic and had genital involvement, which are not typical of morphea. Morphea can be differentiated with based on symptoms (lack of pruritus, pain, burning), morphology of lesions (induration versus atrophy), dermoscopy (fibrotic beams with less scale and hemorrhage vs keratotic follicular plugs), and histopathology (depth of inflammation in superficial and deep dermis).
Histology of morphea can differ based on the stage, whether the lesion is sampled in the inflammatory margin or central sclerosis, and the depth of affected skin. At the inflammatory margin, vascular changes, including endothelial swelling and edema, are present, as well as CD4+ T cells, eosinophils, plasma cells, and mast cells surrounding smaller blood vessels. In late stages, the inflammatory infiltrate is no longer present, the epidermis appears regular, and there is a flattened dermal-epidermal junction. Distinct features include homogenous collagen bundles that replace many dermal structures, with atrophic eccrine glands that appear “trapped” in the thickened dermis, and homogenized and hyalinized subcutis.9
Mycosis fungoides (MF) is the most common type of cutaneous T-cell lymphoma and presents as annular, erythematous or hypopigmented patches and plaques with fine scale and tumors on the buttocks and sun-protected areas of the limbs and trunk. Lesions can appear with prominent poikiloderma or atrophic or lichenified skin.10 It is most common in males of African descent aged 50 to 55 years. The etiology is largely unknown but believed to be multifactorial. This answer is incorrect because the lesions in this patient appeared more atrophic, were less well demarcated, and lacked the scale that would be present in MF.
On histology, both LS and MF show band-like lymphocytic infiltrate, however MF lacks the homogenization and sclerosis of superficial collagen that is present in the dermis of LS. Also, MF demonstrates epidermotropism of atypical lymphocytes forming Pautrier microabscess.10
Primary Care Role
Primary care physicians can diagnose and treat LS. Referral to dermatology is not mandatory. Note that topical steroids can be used daily for up to 12 weeks. In LS, early treatment is associated with improved outcomes and minimizes the risk of irreversible skin changes.11 Follow-up during the treatment period is recommended to monitor subjective and objective response to treatment. Follow-up after the initial treatment is recommended since LS is typically chronic, can relapse, and SCC can infrequently arise from LS lesions.11
Discussion
A diagnosis of lichen sclerosus (LS) was made based on clinical and dermoscopic features, followed by confirmation with histology. The patient’s presentation included typical signs and symptoms of LS: itching, burning, intermittent bleeding, perianal hemorrhage, fusion of the clitoral head, and fissures. Other presentations can include dyspareunia, erosions, and excoriations; however, these symptoms and signs were not reported or seen in this patient.
LS typically affects the anogenital region and has 2 peak incidences: in preadolescent teens and during the fifth to sixth decade of life.1 This patient presented with a case of extragenital LS, which is less common than the classic presentation of LS that affects the genitals. This variant’s epidemiology differs, as it is less common in children and more common in postmenopausal women.2 Extragenital LS presents as white, atrophic plaques with a predilection for sites including the trunk, breasts, upper arms, and sites of physical trauma, with symptoms of dryness and pruritus. Over time, the papules can coalesce and form ivory, scar-like papules or plaques with a wrinkled surface. In advanced stages, telangiectasia or follicular plugging can be present, along with flattening of the dermal-epidermal junction. This flat interface is fragile and can result in bullae that may become hemorrhagic.
Cutaneous squamous cell carcinoma (SCC) may infrequently arise from LS, similar to other chronic inflammatory dermatoses.3 Lichen planus is typically not associated with an increased risk of SCC, except in the oral and hypertrophic variants. However, LS may be considered a premalignant process, and many vulvar SCC cases are noted to have adjacent LS lesions.3
Autoimmune and genetic factors contribute to the pathogenesis of LS. Extracellular matrix protein 1 (ECM1) binds molecules of the basement membrane zone and dermis, contributing to the structure and integrity of skin. Autoantibodies against ECM1 and other antigens of the basement membrane zone, including BP180 and BP320, were found in LS.2 HLA-DQ7 major histocompatibility complex class II antigens have been associated with LS.1
On histologic examination, the epidermis of LS is atrophic with hyperkeratosis. The dermis shows homogenization and sclerosis of superficial collagen with a band-like lymphocytic infiltrate below the sclerosis. The basal layer is thickened, showing basal cell vacuolization and hydropic degeneration.4
First-line treatment for genital and extragenital variants of LS is high-potency topical steroids for 3 months or until the skin texture and color resolve (ie, clobetasol 0.05% cream or ointment). The second-line treatment is a topical calcineurin inhibitor. These treatments are used for management. They are not cures for LS, as relapse is possible after the initial treatment course is completed. Adverse effects of high potency topical steroids are skin burning, skin atrophy, and fragility, telangiectasia. The adverse effects of topical calcineurin inhibitors are stinging and burning on application.
Other Diagnostic Considerations
Inverse psoriasis (IP) is a variant of psoriasis that presents as erythematous, well-demarcated plaques with minimal scale in intertriginous areas and flexural surfaces. Localized dermatophyte, candidal, or bacterial infections can trigger IP.5 It occurs in about 3% to 7% of patients with plaque psoriasis and is thought to form due to koebnerization via mechanical friction of flexural zones.6 The patient described in this case did not have IP because IP would be more likely to present as a well-demarcated erythematous plaque rather than a patch.
Histologically, IP shows regular psoriasiform acanthosis and hypogranulosis of the epidermis, Munro microabscess, spongiform pustules of Kogoj, dilated tortuous dermal vessels, and thinning of the suprapapillary plates.5
Lichen planus pigmentosus-inversus (LPPI) is also known as lichen planus pigmentosus—intertriginous variant. This variant of lichen planus pigmentosus presents as multiple gray to dark brown macules and patches with poorly defined borders in a linear distribution limited to intertriginous areas, flexural surfaces, or following the lines of Blaschko.7 About 20% of cases present with frontal fibrosing alopecia. It is most common in individuals with intermediate and darker skin pigmentation, has a higher prevalence in females, and typically occurs within the third and fifth decades of life. Friction is a common trigger of LPPI.7 A diagnosis of LPPI is incorrect because the lesions would present as gray to dark brown macules, as opposed to the shiny white atrophic thin papules with surrounding pink and purple patches seen in this case.
Histologically, while both LS and LPPI share band-like lymphocytic infiltrate and basal cell vacuolization, findings in the dermis differ. LPPI shows melanophages and prominent melanin incontinence, while LS shows homogenization and sclerosis of superficial collagen.1,8 LPPI also shows absence of compensatory keratinocyte proliferation.
Morphea is an inflammatory disease that affects the dermis and subcutaneous fat, resulting in sclerosis that appears scarlike. Its prevalence increases with age and has a 4:1 prevalence in females, with the plaque type being the most common variant. 9 The typical presentation of plaque-type morphea is an insidious onset of asymptomatic, slightly elevated, erythematous or violaceous, slightly edematous plaques with centrifugal expansion. The center of the plaque may become sclerotic and indurated, acquiring a shiny white color with a peripheral “lilac” ring. Trunk and upper extremity involvement is common. Morphea is associated with increased antisingle-stranded DNA, antitopoisomerase IIa, antiphospholipid, antifibrillin-1, and antihistone antibodies. Triggers of morphea are believed to be localized insults to the skin, including mechanical trauma, injections, vaccinations, and irradiation.9 This answer is incorrect because the patient’s lesions were pruritic and had genital involvement, which are not typical of morphea. Morphea can be differentiated with based on symptoms (lack of pruritus, pain, burning), morphology of lesions (induration versus atrophy), dermoscopy (fibrotic beams with less scale and hemorrhage vs keratotic follicular plugs), and histopathology (depth of inflammation in superficial and deep dermis).
Histology of morphea can differ based on the stage, whether the lesion is sampled in the inflammatory margin or central sclerosis, and the depth of affected skin. At the inflammatory margin, vascular changes, including endothelial swelling and edema, are present, as well as CD4+ T cells, eosinophils, plasma cells, and mast cells surrounding smaller blood vessels. In late stages, the inflammatory infiltrate is no longer present, the epidermis appears regular, and there is a flattened dermal-epidermal junction. Distinct features include homogenous collagen bundles that replace many dermal structures, with atrophic eccrine glands that appear “trapped” in the thickened dermis, and homogenized and hyalinized subcutis.9
Mycosis fungoides (MF) is the most common type of cutaneous T-cell lymphoma and presents as annular, erythematous or hypopigmented patches and plaques with fine scale and tumors on the buttocks and sun-protected areas of the limbs and trunk. Lesions can appear with prominent poikiloderma or atrophic or lichenified skin.10 It is most common in males of African descent aged 50 to 55 years. The etiology is largely unknown but believed to be multifactorial. This answer is incorrect because the lesions in this patient appeared more atrophic, were less well demarcated, and lacked the scale that would be present in MF.
On histology, both LS and MF show band-like lymphocytic infiltrate, however MF lacks the homogenization and sclerosis of superficial collagen that is present in the dermis of LS. Also, MF demonstrates epidermotropism of atypical lymphocytes forming Pautrier microabscess.10
Primary Care Role
Primary care physicians can diagnose and treat LS. Referral to dermatology is not mandatory. Note that topical steroids can be used daily for up to 12 weeks. In LS, early treatment is associated with improved outcomes and minimizes the risk of irreversible skin changes.11 Follow-up during the treatment period is recommended to monitor subjective and objective response to treatment. Follow-up after the initial treatment is recommended since LS is typically chronic, can relapse, and SCC can infrequently arise from LS lesions.11
- Tran DA, Tan X, Macri CJ, Goldstein AT, Fu SW. Lichen sclerosus: an autoimmunopathogenic and genomic enigma with emerging genetic and immune targets. Int J Biol Sci. 2019;15:1429-1439. doi:10.7150/ijbs.34613
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389/fmed.2023.1106318
- Kuraitis D, Murina A. Squamous cell carcinoma arising in chronic inflammatory dermatoses. Cutis. 2024;113:29-34. doi:10.12788/cutis.0914
- Gaertner E, Elstein W. Lichen planus pigmentosus-inversus: case report and review of an unusual entity. Dermatol Online J. 2012;18:11.
- Micali G, Verzì AE, Giuffrida G, et al. Inverse psoriasis: from diagnosis to current treatment options. Clin Cosmet Investig Dermatol. 2019;12:953-959. doi:10.2147/CCID.S189000
- Syed ZU, Khachemoune A. Inverse psoriasis: case presentation and review. Am J Clin Dermatol. 2011;12:143-146. doi:10.2165/11532060-000000000-00000
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514. doi:10.1111/ijd.13806
- Vinay K, Kumar S, Bishnoi A, et al. A clinico-demographic study of 344 patients with lichen planus pigmentosus seen in a tertiary care center in India over an 8-year period. Int J Dermatol. 2020;59:245-252. doi:10.1111/ijd.14540
- Papara C, De Luca DA, Bieber K, et al. Morphea: the 2023 update. Front Med (Lausanne). 2023;10:1108623. doi:10.3389/fmed.2023.1108623
- Zinzani PL, Ferreri AJ, Cerroni L. Mycosis fungoides. Cri t Rev Oncol Hematol. 2008;65:172-182. doi:10.1016/j.critrevonc.2007.08.004
- Lee A, Bradford J, Fischer G. Long-term management of adult vulvar lichen sclerosus: a prospective cohort study of 507 women. JAMA Dermatol. 2015;151(10):1061-1067. doi:10.1001/jamadermatol.2015.0643
- Tran DA, Tan X, Macri CJ, Goldstein AT, Fu SW. Lichen sclerosus: an autoimmunopathogenic and genomic enigma with emerging genetic and immune targets. Int J Biol Sci. 2019;15:1429-1439. doi:10.7150/ijbs.34613
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389/fmed.2023.1106318
- Kuraitis D, Murina A. Squamous cell carcinoma arising in chronic inflammatory dermatoses. Cutis. 2024;113:29-34. doi:10.12788/cutis.0914
- Gaertner E, Elstein W. Lichen planus pigmentosus-inversus: case report and review of an unusual entity. Dermatol Online J. 2012;18:11.
- Micali G, Verzì AE, Giuffrida G, et al. Inverse psoriasis: from diagnosis to current treatment options. Clin Cosmet Investig Dermatol. 2019;12:953-959. doi:10.2147/CCID.S189000
- Syed ZU, Khachemoune A. Inverse psoriasis: case presentation and review. Am J Clin Dermatol. 2011;12:143-146. doi:10.2165/11532060-000000000-00000
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514. doi:10.1111/ijd.13806
- Vinay K, Kumar S, Bishnoi A, et al. A clinico-demographic study of 344 patients with lichen planus pigmentosus seen in a tertiary care center in India over an 8-year period. Int J Dermatol. 2020;59:245-252. doi:10.1111/ijd.14540
- Papara C, De Luca DA, Bieber K, et al. Morphea: the 2023 update. Front Med (Lausanne). 2023;10:1108623. doi:10.3389/fmed.2023.1108623
- Zinzani PL, Ferreri AJ, Cerroni L. Mycosis fungoides. Cri t Rev Oncol Hematol. 2008;65:172-182. doi:10.1016/j.critrevonc.2007.08.004
- Lee A, Bradford J, Fischer G. Long-term management of adult vulvar lichen sclerosus: a prospective cohort study of 507 women. JAMA Dermatol. 2015;151(10):1061-1067. doi:10.1001/jamadermatol.2015.0643
Atrophic Areas on the Axillary and Anogenital Anatomy
Atrophic Areas on the Axillary and Anogenital Anatomy
A 62-year-old woman presented for a fullbody skin examination and was found to have a rash in her axillae and inframammary regions. The rash was intermittently pruritic, and the patient felt that the inframammary rash had started from contact with brassiere underwires. She had no oral lesions but noted intermittent burning and itching of the vaginal folds and intermittent bleeding near her anus. Physical examination revealed confluent, shiny, white, atrophic, thin papules with surrounding pink and purple patches on bilateral axillae, bilateral inframammary folds, bilateral inner thighs, and on the clitoral hood and labia minora. There was also an hourglass-shaped erythematous patch involving the vagina and anus. A small fissure was noted perianally, and small hemorrhage was noted on the clitoral head, with fusion of the clitoral head and superior labia minora (Figures 1 and 2).
lesion from punch biopsy of the patient’s left axilla.
sclerosus plaque showing bright white grouped dots
on a pink background with follicular plugging and linear
branching vessels.
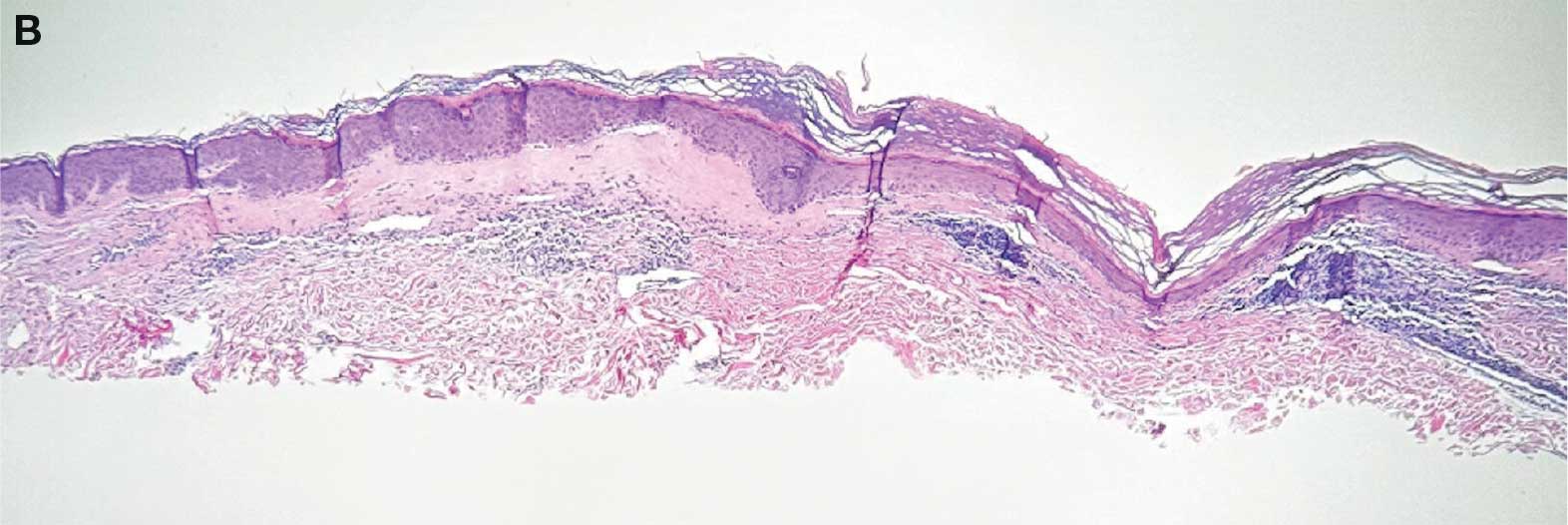
showing a compact corneal layer with a pale papillary
dermis and an underlying lymphocytic infiltrate. These
findings give the “red, white, and blue” appearance.
Low power 20× magnification.
nsbp;
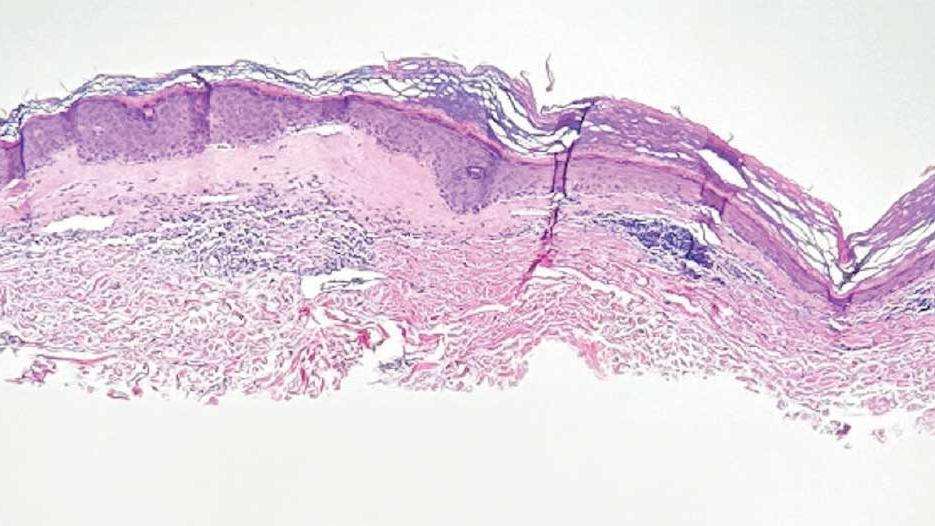
A Case Report of Unanticipated Difficult Intubation Due to Posterior Tracheal Angulation
A Case Report of Unanticipated Difficult Intubation Due to Posterior Tracheal Angulation
Tracheal deviation mostly occurs from mechanical compression of the trachea, and can be caused by a variety of clinical conditions, including trauma,¹ pharyngeal abscess,² neck hematoma,³ thyroid enlargement,4 and kyphoscoliosis.5 These conditions often result in lateral tracheal deviation, which can be associated with tracheal compression and reduction in tracheal caliber.
Anterior-posterior (A-P) tracheal deviation has rarely been reported. Kyphoscoliosis, scarring after a tracheostomy, or innominate vein compression are probable causes of A-P tracheal deviation and can be associated with tracheal narrowing and vascular fistula formation. This report describes a case of difficult endotracheal tube (ETT) advancement secondary to unexpected acute posterior tracheal deviation encountered during cardiopulmonary resuscitation (CPR). A waiver of patient consent was obtained from the Human Research Protection Program at the US Department of Veterans Affairs (VA) Puget Sound Health Care System.
Case Presentation
A 50-year-old male with a history of chronic cerebral venous sinus thrombosis and taking enoxaparin, presented to the emergency department for recurrent headaches. He experienced sudden cardiac arrest, and CPR in the form of chest compression and bag mask ventilation was immediately initiated. With the patient's head in an extended position and using a video laryngoscope, a Cormack–Lehane grade 1 view of the glottic opening was obtained and the trachea was intubated with an 8 mm (internal diameter) polyvinyl chloride ETT. Tracheal intubation was confirmed by utilizing continuous EtCO2 monitoring. The ETT was secured at 22 cm measured at the teeth.
After about 40 minutes of CPR, spontaneous circulation restarted and a portable A-P chest X-ray with the head in a neutral position indicated the ETT tip was at the level of the first rib (Figure 1). This finding, along with a persistent air leak, prompted blind advancement of the ETT to 26 cm at the teeth, but resistance to advancement was noted. A subsequent chest computed tomography (CT) with the head in a neutral position revealed the ETT remained inappropriately positioned with the tip measured 8.2 cm above the carina (Figure 2A). Concurrently, a sagittal CT view demonstrated significant posterior deviation of the mid and lower trachea. This deviation was determined to be the most likely cause of the difficulty encountered in advancing the ETT. No masses or lesions contributing to the acute tracheal angulation could be identified. Comparing CT imaging from 2 months prior, the trachea was of normal caliber and ordinarily aligned with the vertebral column (Figure 2B).
With the patient in Fowler position with the head midline, a flexible fiber-optic bronchoscopy was performed. Acute, almost 90-degree tracheal angulation was encountered and navigated by retroflexion of the flexible bronchoscope. Once the posterior tracheal wall was encountered, retroflexion was relaxed and the carina was visualized. The bronchoscope tip was placed near the carina, and the ETT was advanced over the fiber-optic bronchoscope to terminate 3 cm above the carina. A subsequent chest X-ray confirmed appropriate ETT position (Figure 3).
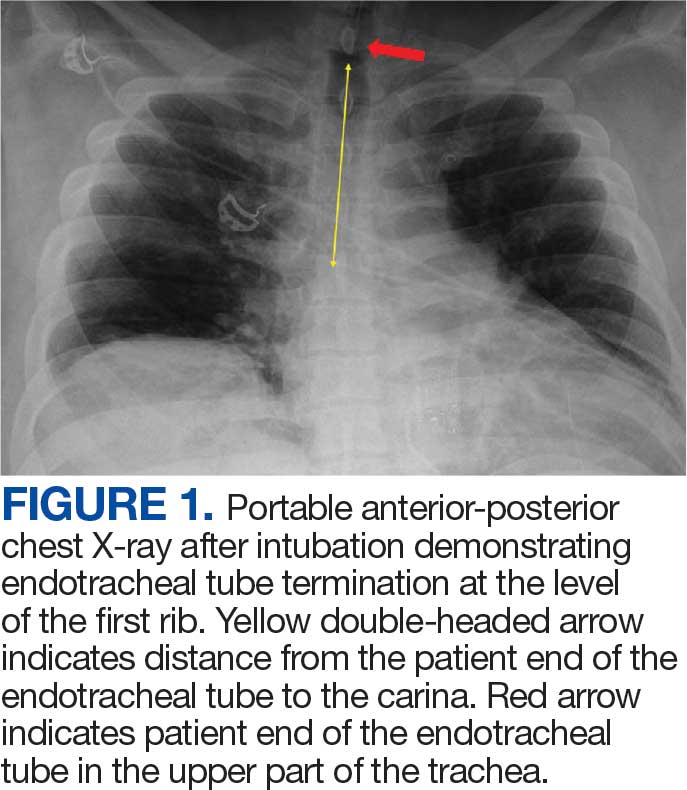
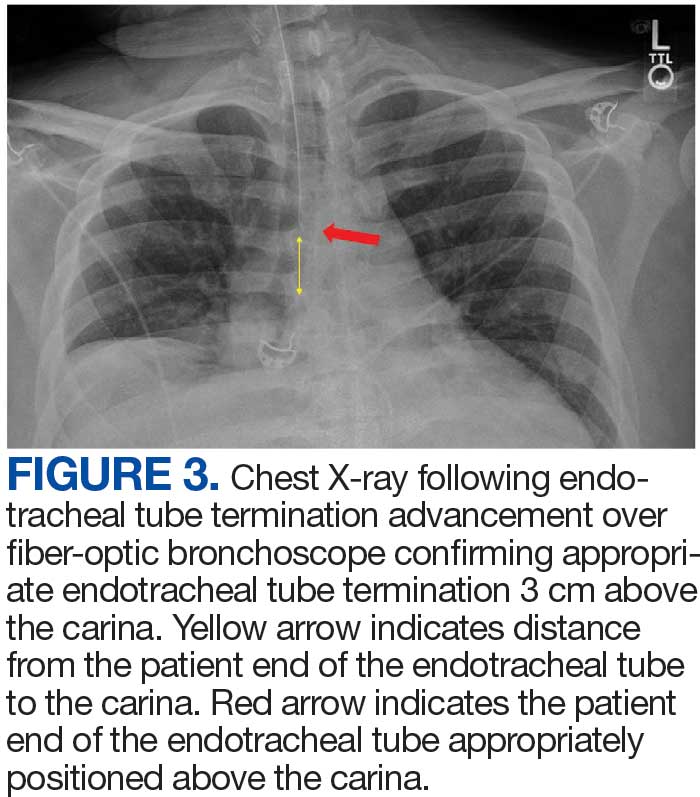
Discussion
Tracheal deviation in the A-P dimension resulting in difficult tracheal intubation has rarely been reported. Previous reports have described anatomical lesions contributing to similar tracheal deviation, such as retro-tracheal thyroid tissue, pronounced cervical lordosis, and severe kyphoscoliosis with destructive cervical fusion.5-8 In a study of the anatomical correlation of double lumen tube placement while using positron emission tomography CT, Cameron et al evaluated the size and angulation of the glottis and proximal trachea using calibrated CT measurements and an online digital protractor and note nearly perfect alignment of the pharynx and glottis.9 However, the trachea turned posteriorly relative to the glottis, resulting in an overall posterior angle of the proximal trachea compared to the glottis of 30.4 to 50.1 degrees, with no sex differences. The need to maneuver similar proximal tracheal angulation during endotracheal intubation has been reported as a cause of difficult intubation.10
In this case, the posterior angulation was not encountered in the proximal trachea but rather in the more distal trachea. The extreme A-P tracheal deviation was not associated with any identifiable masses or lesions. A CT performed 2 months prior demonstrated normal tracheal anatomy, and there was no interval history of neck trauma or tracheal obstruction suggestive of a likely cause for this deviation. This change in the patient’s tracheal anatomy was only discovered after CPR had been performed and as part of the workup for cardiac arrest. Iatrogenic injuries are known to occur during CPR. Common CPR-related airway injuries include tracheal mucosal injury from traumatic intubation and bony injuries to the chest wall from compressions.11 Laryngeal cartilage damage from intubation may also occur, but tracheal displacement following CPR has not been previously reported.11
This case of tracheal deviation is unlikely to be related to patient positioning, as the A-P deviation persisted in 3 separate head and neck alignments. First, during indirect laryngoscopy, performed in a standard sniffing position. Second, during the CT, performed in the supine position, with no head support. The acute A-P deviation seen in Figure 2 was clearly noted in this position. Lastly, flexible fiber-optic bronchoscopy was performed in a semiupright position with the head supported on a pillow. A-P deviation was encountered and navigated in this position during flexible fiber-optic guided ETT repositioning.
Using magnetic resonance imaging, alterations in the alignment of pharyngeal and tracheal axes have been described with changes in neck positioning; however, tracheal deviation has not been described with changes in head and neck alignment.12 Although the clinical presentation in this case was consistent with prior reports, we were unable to identify any previously reported anatomic cause for the tracheal deviation.5,6,8 Initial glottic visualization with a video laryngoscope was unremarkable, but resistance to sufficient ETT advancement past the vocal cords and a persistent air leak due to cuff herniation through the glottic opening was noticeable. The ETT was maneuvered to an appropriate position in the trachea using a flexible fiber-optic bronchoscope. The acute angulation of the trachea that was appreciated on bronchoscopy did not result in kinking of the ETT both initially and after in-situ thermosoftening of the polyvinyl chloride tube.13 Previously reported instances of A-P tracheal deviation have outlined the necessity of using alternative techniques to establish a patent airway, including the use of a laryngeal mask airway and a cuffless ETT with saline-soaked gauze packing.5,8 In 1 reported case, awake fiber-optic intubation was performed when difficult tracheal intubation was anticipated due to known A-P tracheal deviation.6
Failure of ETT advancement can be due to obstruction from the arytenoids and at the level of the vocal cords.14 When the ETT has been visualized to have traversed the vocal cords, tracheal A-P deviation should be considered as a cause of difficult ETT advancement. If an adequate endotracheal airway cannot be established, prompt consideration should be given to placement of a supraglottic airway. Early fiber-optic bronchoscopy should be used to establish the diagnosis and assist with proper ETT positioning.
Conclusions
This case illustrates the rare occurrence of A-P tracheal deviation leading to difficult intubation during CPR. The findings underscore the importance of considering A-P deviation as a potential cause of airway complications in emergency settings, especially in patients with previously normal tracheal anatomy. The successful use of flexible fiber-optic bronchoscopy in this case provides a valuable technique for addressing acute tracheal angulation. This report contributes to the limited literature on A-P tracheal deviation and serves as a reminder for clinicians to maintain a high index of suspicion for unusual airway challenges during critical interventions.
Creasy JD, Chiles C, Routh WD, et al. Overview of traumatic injury of the thoracic aorta. Radiogr Rev Publ Radiol Soc N Am Inc. 1997;17:27-45. doi:10.1148/radiographics.17.1.9017797
Yee AM, Christensen DN, Waterbrook AL, et al. Parapharyngeal abscess with tracheal deviation. Intern Emerg Med. 2017;12:1077-1078.doi:10.1007/s11739-017-1634-8
Querney J, Singh SI, Sebbag I. Tracheal deviation with phrenic nerve palsy after brachial plexus block. Anaesth Rep. 2021;9:41-43. doi:10.1002/anr3.12100
Geissler B, Wagner T, Dorn R, et al. Extensive sterile abscess in an invasive fibrous thyroiditis (Riedel’s thyroiditis) caused by an occlusive vasculitis. J Endocrinol Invest. 2001;24:111-115. doi:10.1007/BF03343824
Kim HJ, Choi YS, Park SH, et al. Difficult endotracheal intubation secondary to tracheal deviation and stenosis in a patient with severe kyphoscoliosis: a case report. Korean J Anesthesiol. 2016;69:386-389. doi:10.4097/kjae.2016.69.4.386
Crabb IJ. Anterior deviation of the trachea. Anaesthesia. 2001;56:284-286.doi:10.1046/j.1365-2044.2001.01918-17.x
De Cassai A, Boscolo A, Rose K, et al. Predictive parameters of difficult intubation in thyroid surgery: a meta-analysis. Minerva Anestesiol. 2020;86:317-326. doi:10.23736/S0375-9393.19.14127-2
Davies R. Difficult tracheal intubation secondary to a tracheal diverticulum and a 90 degree deviation in the trachea. Anaesthesia. 2000;55:923-925. doi:10.1046/j.1365-2044.2000.01664-18.x
Cameron RB, Peacock WJ, Chang XG, et al. Double lumen endobronchial tube intubation: lessons learned from anatomy. BMC Anesthesiol. 2024;24:150. doi:10.1186/s12871-024-02517-6
Walls RM, Samuels-Kalow M, Perkins A. A new maneuver for endotracheal tube insertion during difficult GlideScope intubation. J Emerg Med. 2010;39:86-88. doi:10.1016/j.jemermed.2009.11.005
Buschmann CT, Tsokos M. Frequent and rare complications of resuscitation attempts. Intensive Care Med. 2009;35:397-404. doi:10.1007/s00134-008-1255-9
Greenland KB, Edwards MJ, Hutton NJ, et al. Changes in airway configuration with different head and neck positions using magnetic resonance imaging of normal airways: a new concept with possible clinical applications. Br J Anaesth. 2010;105:683-690. doi:10.1093/bja/aeq239
Takasugi Y, Futagawa K, Umeda T, et al. Thermophysical Properties of Thermosoftening Nasotracheal Tubes. Anesth Prog. 2018;65:100-105. doi:10.2344/anpr-65-02-06
Phelan MP. Use of the endotracheal bougie introducer for difficult intubations. Am J Emerg Med. 2004;22:479-482. doi:10.1016/j.ajem.2004.07.017
Tracheal deviation mostly occurs from mechanical compression of the trachea, and can be caused by a variety of clinical conditions, including trauma,¹ pharyngeal abscess,² neck hematoma,³ thyroid enlargement,4 and kyphoscoliosis.5 These conditions often result in lateral tracheal deviation, which can be associated with tracheal compression and reduction in tracheal caliber.
Anterior-posterior (A-P) tracheal deviation has rarely been reported. Kyphoscoliosis, scarring after a tracheostomy, or innominate vein compression are probable causes of A-P tracheal deviation and can be associated with tracheal narrowing and vascular fistula formation. This report describes a case of difficult endotracheal tube (ETT) advancement secondary to unexpected acute posterior tracheal deviation encountered during cardiopulmonary resuscitation (CPR). A waiver of patient consent was obtained from the Human Research Protection Program at the US Department of Veterans Affairs (VA) Puget Sound Health Care System.
Case Presentation
A 50-year-old male with a history of chronic cerebral venous sinus thrombosis and taking enoxaparin, presented to the emergency department for recurrent headaches. He experienced sudden cardiac arrest, and CPR in the form of chest compression and bag mask ventilation was immediately initiated. With the patient's head in an extended position and using a video laryngoscope, a Cormack–Lehane grade 1 view of the glottic opening was obtained and the trachea was intubated with an 8 mm (internal diameter) polyvinyl chloride ETT. Tracheal intubation was confirmed by utilizing continuous EtCO2 monitoring. The ETT was secured at 22 cm measured at the teeth.
After about 40 minutes of CPR, spontaneous circulation restarted and a portable A-P chest X-ray with the head in a neutral position indicated the ETT tip was at the level of the first rib (Figure 1). This finding, along with a persistent air leak, prompted blind advancement of the ETT to 26 cm at the teeth, but resistance to advancement was noted. A subsequent chest computed tomography (CT) with the head in a neutral position revealed the ETT remained inappropriately positioned with the tip measured 8.2 cm above the carina (Figure 2A). Concurrently, a sagittal CT view demonstrated significant posterior deviation of the mid and lower trachea. This deviation was determined to be the most likely cause of the difficulty encountered in advancing the ETT. No masses or lesions contributing to the acute tracheal angulation could be identified. Comparing CT imaging from 2 months prior, the trachea was of normal caliber and ordinarily aligned with the vertebral column (Figure 2B).
With the patient in Fowler position with the head midline, a flexible fiber-optic bronchoscopy was performed. Acute, almost 90-degree tracheal angulation was encountered and navigated by retroflexion of the flexible bronchoscope. Once the posterior tracheal wall was encountered, retroflexion was relaxed and the carina was visualized. The bronchoscope tip was placed near the carina, and the ETT was advanced over the fiber-optic bronchoscope to terminate 3 cm above the carina. A subsequent chest X-ray confirmed appropriate ETT position (Figure 3).
Discussion
Tracheal deviation in the A-P dimension resulting in difficult tracheal intubation has rarely been reported. Previous reports have described anatomical lesions contributing to similar tracheal deviation, such as retro-tracheal thyroid tissue, pronounced cervical lordosis, and severe kyphoscoliosis with destructive cervical fusion.5-8 In a study of the anatomical correlation of double lumen tube placement while using positron emission tomography CT, Cameron et al evaluated the size and angulation of the glottis and proximal trachea using calibrated CT measurements and an online digital protractor and note nearly perfect alignment of the pharynx and glottis.9 However, the trachea turned posteriorly relative to the glottis, resulting in an overall posterior angle of the proximal trachea compared to the glottis of 30.4 to 50.1 degrees, with no sex differences. The need to maneuver similar proximal tracheal angulation during endotracheal intubation has been reported as a cause of difficult intubation.10
In this case, the posterior angulation was not encountered in the proximal trachea but rather in the more distal trachea. The extreme A-P tracheal deviation was not associated with any identifiable masses or lesions. A CT performed 2 months prior demonstrated normal tracheal anatomy, and there was no interval history of neck trauma or tracheal obstruction suggestive of a likely cause for this deviation. This change in the patient’s tracheal anatomy was only discovered after CPR had been performed and as part of the workup for cardiac arrest. Iatrogenic injuries are known to occur during CPR. Common CPR-related airway injuries include tracheal mucosal injury from traumatic intubation and bony injuries to the chest wall from compressions.11 Laryngeal cartilage damage from intubation may also occur, but tracheal displacement following CPR has not been previously reported.11
This case of tracheal deviation is unlikely to be related to patient positioning, as the A-P deviation persisted in 3 separate head and neck alignments. First, during indirect laryngoscopy, performed in a standard sniffing position. Second, during the CT, performed in the supine position, with no head support. The acute A-P deviation seen in Figure 2 was clearly noted in this position. Lastly, flexible fiber-optic bronchoscopy was performed in a semiupright position with the head supported on a pillow. A-P deviation was encountered and navigated in this position during flexible fiber-optic guided ETT repositioning.
Using magnetic resonance imaging, alterations in the alignment of pharyngeal and tracheal axes have been described with changes in neck positioning; however, tracheal deviation has not been described with changes in head and neck alignment.12 Although the clinical presentation in this case was consistent with prior reports, we were unable to identify any previously reported anatomic cause for the tracheal deviation.5,6,8 Initial glottic visualization with a video laryngoscope was unremarkable, but resistance to sufficient ETT advancement past the vocal cords and a persistent air leak due to cuff herniation through the glottic opening was noticeable. The ETT was maneuvered to an appropriate position in the trachea using a flexible fiber-optic bronchoscope. The acute angulation of the trachea that was appreciated on bronchoscopy did not result in kinking of the ETT both initially and after in-situ thermosoftening of the polyvinyl chloride tube.13 Previously reported instances of A-P tracheal deviation have outlined the necessity of using alternative techniques to establish a patent airway, including the use of a laryngeal mask airway and a cuffless ETT with saline-soaked gauze packing.5,8 In 1 reported case, awake fiber-optic intubation was performed when difficult tracheal intubation was anticipated due to known A-P tracheal deviation.6
Failure of ETT advancement can be due to obstruction from the arytenoids and at the level of the vocal cords.14 When the ETT has been visualized to have traversed the vocal cords, tracheal A-P deviation should be considered as a cause of difficult ETT advancement. If an adequate endotracheal airway cannot be established, prompt consideration should be given to placement of a supraglottic airway. Early fiber-optic bronchoscopy should be used to establish the diagnosis and assist with proper ETT positioning.
Conclusions
This case illustrates the rare occurrence of A-P tracheal deviation leading to difficult intubation during CPR. The findings underscore the importance of considering A-P deviation as a potential cause of airway complications in emergency settings, especially in patients with previously normal tracheal anatomy. The successful use of flexible fiber-optic bronchoscopy in this case provides a valuable technique for addressing acute tracheal angulation. This report contributes to the limited literature on A-P tracheal deviation and serves as a reminder for clinicians to maintain a high index of suspicion for unusual airway challenges during critical interventions.
Tracheal deviation mostly occurs from mechanical compression of the trachea, and can be caused by a variety of clinical conditions, including trauma,¹ pharyngeal abscess,² neck hematoma,³ thyroid enlargement,4 and kyphoscoliosis.5 These conditions often result in lateral tracheal deviation, which can be associated with tracheal compression and reduction in tracheal caliber.
Anterior-posterior (A-P) tracheal deviation has rarely been reported. Kyphoscoliosis, scarring after a tracheostomy, or innominate vein compression are probable causes of A-P tracheal deviation and can be associated with tracheal narrowing and vascular fistula formation. This report describes a case of difficult endotracheal tube (ETT) advancement secondary to unexpected acute posterior tracheal deviation encountered during cardiopulmonary resuscitation (CPR). A waiver of patient consent was obtained from the Human Research Protection Program at the US Department of Veterans Affairs (VA) Puget Sound Health Care System.
Case Presentation
A 50-year-old male with a history of chronic cerebral venous sinus thrombosis and taking enoxaparin, presented to the emergency department for recurrent headaches. He experienced sudden cardiac arrest, and CPR in the form of chest compression and bag mask ventilation was immediately initiated. With the patient's head in an extended position and using a video laryngoscope, a Cormack–Lehane grade 1 view of the glottic opening was obtained and the trachea was intubated with an 8 mm (internal diameter) polyvinyl chloride ETT. Tracheal intubation was confirmed by utilizing continuous EtCO2 monitoring. The ETT was secured at 22 cm measured at the teeth.
After about 40 minutes of CPR, spontaneous circulation restarted and a portable A-P chest X-ray with the head in a neutral position indicated the ETT tip was at the level of the first rib (Figure 1). This finding, along with a persistent air leak, prompted blind advancement of the ETT to 26 cm at the teeth, but resistance to advancement was noted. A subsequent chest computed tomography (CT) with the head in a neutral position revealed the ETT remained inappropriately positioned with the tip measured 8.2 cm above the carina (Figure 2A). Concurrently, a sagittal CT view demonstrated significant posterior deviation of the mid and lower trachea. This deviation was determined to be the most likely cause of the difficulty encountered in advancing the ETT. No masses or lesions contributing to the acute tracheal angulation could be identified. Comparing CT imaging from 2 months prior, the trachea was of normal caliber and ordinarily aligned with the vertebral column (Figure 2B).
With the patient in Fowler position with the head midline, a flexible fiber-optic bronchoscopy was performed. Acute, almost 90-degree tracheal angulation was encountered and navigated by retroflexion of the flexible bronchoscope. Once the posterior tracheal wall was encountered, retroflexion was relaxed and the carina was visualized. The bronchoscope tip was placed near the carina, and the ETT was advanced over the fiber-optic bronchoscope to terminate 3 cm above the carina. A subsequent chest X-ray confirmed appropriate ETT position (Figure 3).
Discussion
Tracheal deviation in the A-P dimension resulting in difficult tracheal intubation has rarely been reported. Previous reports have described anatomical lesions contributing to similar tracheal deviation, such as retro-tracheal thyroid tissue, pronounced cervical lordosis, and severe kyphoscoliosis with destructive cervical fusion.5-8 In a study of the anatomical correlation of double lumen tube placement while using positron emission tomography CT, Cameron et al evaluated the size and angulation of the glottis and proximal trachea using calibrated CT measurements and an online digital protractor and note nearly perfect alignment of the pharynx and glottis.9 However, the trachea turned posteriorly relative to the glottis, resulting in an overall posterior angle of the proximal trachea compared to the glottis of 30.4 to 50.1 degrees, with no sex differences. The need to maneuver similar proximal tracheal angulation during endotracheal intubation has been reported as a cause of difficult intubation.10
In this case, the posterior angulation was not encountered in the proximal trachea but rather in the more distal trachea. The extreme A-P tracheal deviation was not associated with any identifiable masses or lesions. A CT performed 2 months prior demonstrated normal tracheal anatomy, and there was no interval history of neck trauma or tracheal obstruction suggestive of a likely cause for this deviation. This change in the patient’s tracheal anatomy was only discovered after CPR had been performed and as part of the workup for cardiac arrest. Iatrogenic injuries are known to occur during CPR. Common CPR-related airway injuries include tracheal mucosal injury from traumatic intubation and bony injuries to the chest wall from compressions.11 Laryngeal cartilage damage from intubation may also occur, but tracheal displacement following CPR has not been previously reported.11
This case of tracheal deviation is unlikely to be related to patient positioning, as the A-P deviation persisted in 3 separate head and neck alignments. First, during indirect laryngoscopy, performed in a standard sniffing position. Second, during the CT, performed in the supine position, with no head support. The acute A-P deviation seen in Figure 2 was clearly noted in this position. Lastly, flexible fiber-optic bronchoscopy was performed in a semiupright position with the head supported on a pillow. A-P deviation was encountered and navigated in this position during flexible fiber-optic guided ETT repositioning.
Using magnetic resonance imaging, alterations in the alignment of pharyngeal and tracheal axes have been described with changes in neck positioning; however, tracheal deviation has not been described with changes in head and neck alignment.12 Although the clinical presentation in this case was consistent with prior reports, we were unable to identify any previously reported anatomic cause for the tracheal deviation.5,6,8 Initial glottic visualization with a video laryngoscope was unremarkable, but resistance to sufficient ETT advancement past the vocal cords and a persistent air leak due to cuff herniation through the glottic opening was noticeable. The ETT was maneuvered to an appropriate position in the trachea using a flexible fiber-optic bronchoscope. The acute angulation of the trachea that was appreciated on bronchoscopy did not result in kinking of the ETT both initially and after in-situ thermosoftening of the polyvinyl chloride tube.13 Previously reported instances of A-P tracheal deviation have outlined the necessity of using alternative techniques to establish a patent airway, including the use of a laryngeal mask airway and a cuffless ETT with saline-soaked gauze packing.5,8 In 1 reported case, awake fiber-optic intubation was performed when difficult tracheal intubation was anticipated due to known A-P tracheal deviation.6
Failure of ETT advancement can be due to obstruction from the arytenoids and at the level of the vocal cords.14 When the ETT has been visualized to have traversed the vocal cords, tracheal A-P deviation should be considered as a cause of difficult ETT advancement. If an adequate endotracheal airway cannot be established, prompt consideration should be given to placement of a supraglottic airway. Early fiber-optic bronchoscopy should be used to establish the diagnosis and assist with proper ETT positioning.
Conclusions
This case illustrates the rare occurrence of A-P tracheal deviation leading to difficult intubation during CPR. The findings underscore the importance of considering A-P deviation as a potential cause of airway complications in emergency settings, especially in patients with previously normal tracheal anatomy. The successful use of flexible fiber-optic bronchoscopy in this case provides a valuable technique for addressing acute tracheal angulation. This report contributes to the limited literature on A-P tracheal deviation and serves as a reminder for clinicians to maintain a high index of suspicion for unusual airway challenges during critical interventions.
Creasy JD, Chiles C, Routh WD, et al. Overview of traumatic injury of the thoracic aorta. Radiogr Rev Publ Radiol Soc N Am Inc. 1997;17:27-45. doi:10.1148/radiographics.17.1.9017797
Yee AM, Christensen DN, Waterbrook AL, et al. Parapharyngeal abscess with tracheal deviation. Intern Emerg Med. 2017;12:1077-1078.doi:10.1007/s11739-017-1634-8
Querney J, Singh SI, Sebbag I. Tracheal deviation with phrenic nerve palsy after brachial plexus block. Anaesth Rep. 2021;9:41-43. doi:10.1002/anr3.12100
Geissler B, Wagner T, Dorn R, et al. Extensive sterile abscess in an invasive fibrous thyroiditis (Riedel’s thyroiditis) caused by an occlusive vasculitis. J Endocrinol Invest. 2001;24:111-115. doi:10.1007/BF03343824
Kim HJ, Choi YS, Park SH, et al. Difficult endotracheal intubation secondary to tracheal deviation and stenosis in a patient with severe kyphoscoliosis: a case report. Korean J Anesthesiol. 2016;69:386-389. doi:10.4097/kjae.2016.69.4.386
Crabb IJ. Anterior deviation of the trachea. Anaesthesia. 2001;56:284-286.doi:10.1046/j.1365-2044.2001.01918-17.x
De Cassai A, Boscolo A, Rose K, et al. Predictive parameters of difficult intubation in thyroid surgery: a meta-analysis. Minerva Anestesiol. 2020;86:317-326. doi:10.23736/S0375-9393.19.14127-2
Davies R. Difficult tracheal intubation secondary to a tracheal diverticulum and a 90 degree deviation in the trachea. Anaesthesia. 2000;55:923-925. doi:10.1046/j.1365-2044.2000.01664-18.x
Cameron RB, Peacock WJ, Chang XG, et al. Double lumen endobronchial tube intubation: lessons learned from anatomy. BMC Anesthesiol. 2024;24:150. doi:10.1186/s12871-024-02517-6
Walls RM, Samuels-Kalow M, Perkins A. A new maneuver for endotracheal tube insertion during difficult GlideScope intubation. J Emerg Med. 2010;39:86-88. doi:10.1016/j.jemermed.2009.11.005
Buschmann CT, Tsokos M. Frequent and rare complications of resuscitation attempts. Intensive Care Med. 2009;35:397-404. doi:10.1007/s00134-008-1255-9
Greenland KB, Edwards MJ, Hutton NJ, et al. Changes in airway configuration with different head and neck positions using magnetic resonance imaging of normal airways: a new concept with possible clinical applications. Br J Anaesth. 2010;105:683-690. doi:10.1093/bja/aeq239
Takasugi Y, Futagawa K, Umeda T, et al. Thermophysical Properties of Thermosoftening Nasotracheal Tubes. Anesth Prog. 2018;65:100-105. doi:10.2344/anpr-65-02-06
Phelan MP. Use of the endotracheal bougie introducer for difficult intubations. Am J Emerg Med. 2004;22:479-482. doi:10.1016/j.ajem.2004.07.017
Creasy JD, Chiles C, Routh WD, et al. Overview of traumatic injury of the thoracic aorta. Radiogr Rev Publ Radiol Soc N Am Inc. 1997;17:27-45. doi:10.1148/radiographics.17.1.9017797
Yee AM, Christensen DN, Waterbrook AL, et al. Parapharyngeal abscess with tracheal deviation. Intern Emerg Med. 2017;12:1077-1078.doi:10.1007/s11739-017-1634-8
Querney J, Singh SI, Sebbag I. Tracheal deviation with phrenic nerve palsy after brachial plexus block. Anaesth Rep. 2021;9:41-43. doi:10.1002/anr3.12100
Geissler B, Wagner T, Dorn R, et al. Extensive sterile abscess in an invasive fibrous thyroiditis (Riedel’s thyroiditis) caused by an occlusive vasculitis. J Endocrinol Invest. 2001;24:111-115. doi:10.1007/BF03343824
Kim HJ, Choi YS, Park SH, et al. Difficult endotracheal intubation secondary to tracheal deviation and stenosis in a patient with severe kyphoscoliosis: a case report. Korean J Anesthesiol. 2016;69:386-389. doi:10.4097/kjae.2016.69.4.386
Crabb IJ. Anterior deviation of the trachea. Anaesthesia. 2001;56:284-286.doi:10.1046/j.1365-2044.2001.01918-17.x
De Cassai A, Boscolo A, Rose K, et al. Predictive parameters of difficult intubation in thyroid surgery: a meta-analysis. Minerva Anestesiol. 2020;86:317-326. doi:10.23736/S0375-9393.19.14127-2
Davies R. Difficult tracheal intubation secondary to a tracheal diverticulum and a 90 degree deviation in the trachea. Anaesthesia. 2000;55:923-925. doi:10.1046/j.1365-2044.2000.01664-18.x
Cameron RB, Peacock WJ, Chang XG, et al. Double lumen endobronchial tube intubation: lessons learned from anatomy. BMC Anesthesiol. 2024;24:150. doi:10.1186/s12871-024-02517-6
Walls RM, Samuels-Kalow M, Perkins A. A new maneuver for endotracheal tube insertion during difficult GlideScope intubation. J Emerg Med. 2010;39:86-88. doi:10.1016/j.jemermed.2009.11.005
Buschmann CT, Tsokos M. Frequent and rare complications of resuscitation attempts. Intensive Care Med. 2009;35:397-404. doi:10.1007/s00134-008-1255-9
Greenland KB, Edwards MJ, Hutton NJ, et al. Changes in airway configuration with different head and neck positions using magnetic resonance imaging of normal airways: a new concept with possible clinical applications. Br J Anaesth. 2010;105:683-690. doi:10.1093/bja/aeq239
Takasugi Y, Futagawa K, Umeda T, et al. Thermophysical Properties of Thermosoftening Nasotracheal Tubes. Anesth Prog. 2018;65:100-105. doi:10.2344/anpr-65-02-06
Phelan MP. Use of the endotracheal bougie introducer for difficult intubations. Am J Emerg Med. 2004;22:479-482. doi:10.1016/j.ajem.2004.07.017
A Case Report of Unanticipated Difficult Intubation Due to Posterior Tracheal Angulation
A Case Report of Unanticipated Difficult Intubation Due to Posterior Tracheal Angulation
Destructive Facial Granuloma Following Self-Treatment With Vitamin E Oil and an At-Home Microneedling Device
Destructive Facial Granuloma Following Self-Treatment With Vitamin E Oil and an At-Home Microneedling Device
Topical application or injection of cosmeceuticals in conjunction with procedures such as facial microneedling (MN) has been associated with local and systemic complications.1
Although at-home options may be more accessible and affordable for patients, they also increase the risk for improper use and subsequent infection. Additionally, the use of cosmeceuticals such as vitamin E oil in conjunction with MN to enhance the effects of the procedure can lead to further complications. We report the case of a 44-year-old woman who developed a necrotic ulcer on the chin following self-treatment with vitamin E oil and an at-home MN device. While MN has been reported to be relatively safe when performed by board-certified dermatologists, clinicians should be vigilant in correlating clinical history and recent cosmetic procedures with the histologic findings for timely diagnosis and treatment of unusual lesions on the face.
Case Report
A 44-year-old woman presented to the emergency department with a progressively enlarging, necrotic, ulcerative lesion on the midline chin of 4 months’ duration. The patient reported that the lesion started as redness that developed into a painful oozing ulcer following application of vitamin E oil in conjunction with an at-home MN device (Figure 1). She purchased the vitamin E oil and MN device online and performed the procedure herself, applying the vitamin E oil to her whole face before, during, and after using the MN device, which contained 0.25-mm titanium needles. She denied undergoing any other recent cosmetic procedures.
The lesion initially was treated by the patient’s primary care physician with oral doxycycline for 6 weeks, followed by oral cephalexin and clindamycin for 2 weeks. Although the redness stabilized, the lesion continued to enlarge, which prompted her initial visit to our hospital 1 month after seeing her primary care physician. During this visit, the patient was given penicillin, and the ulcer was debrided and biopsied; however, no clinical improvement was seen.
A biopsy during her initial emergency department visit and a repeat biopsy after 1 month showed similar findings of diffuse lymphohistiocytic and eosinophilic inflammation in the dermis (Figure 2) with poorly defined granulomas and multinucleated giant cells containing nonpolarizable exogenous material (Figure 3). Similar detached exogenous materials also were identified adjacent to the tissue. Diffuse re-epithelialization was seen, featuring pseudoepitheliomatous hyperplasia in association with the inflammatory process and granulation tissue (Figures 3 and 4). A higher-power view of the dermis showed foci of sclerosing lipogranuloma (Figure 4). Periodic acid–Schiff, Grocott methenamine silver, acid-fast bacilli, Fite, and Wright-Giemsa stains all were negative for microorganisms, and pancytokeratin staining was negative for carcinoma. These findings supported the diagnosis of a foreign body granulomatous reaction to an exogenous material—in this case, the vitamin E oil. Subsequent treatment with intralesional triamcinolone 10 mg/mL injection over 18 months resulted in progressive and drastic improvement of the lesion (Figure 5). A scar excision was performed, which further improved the lesion’s cosmetic appearance.
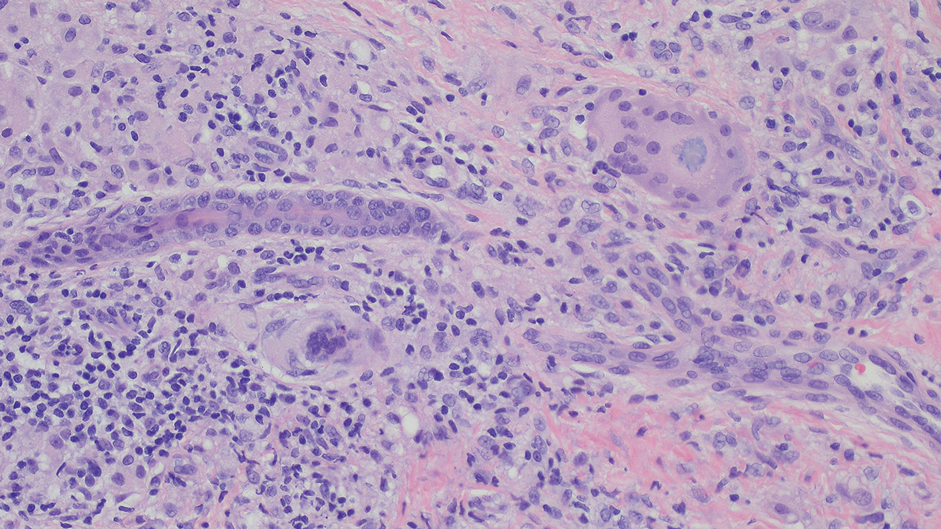
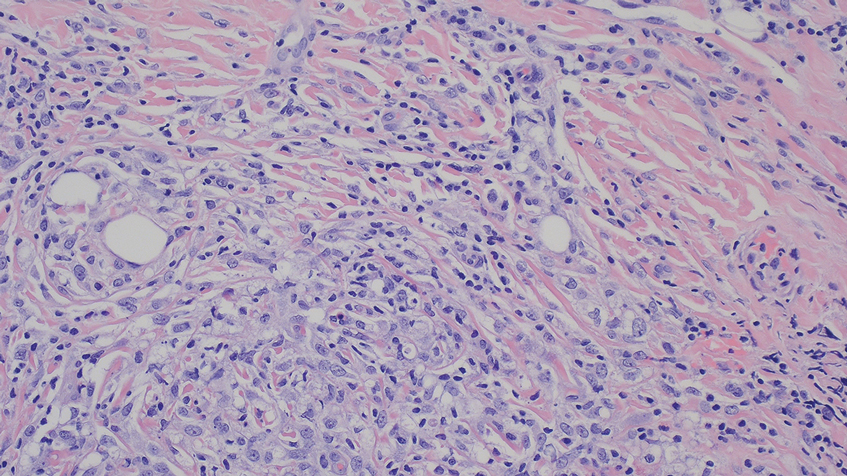
Comment
Application of various topical cosmeceuticals before, during, or after MN to enhance the effects of the procedure can introduce particles into the dermis, resulting in local or systemic hypersensitivity reactions. The associated adverse events can be divided into 2 main categories: adverse reactions related to the topical product or to the materials of the MN device itself.
A study showed that topical application of vitamin E oil to wounds on the skin does not improve the cosmetic appearance of scars.3 Instead, it is associated with a high incidence of contact dermatitis. A similar case of vitamin E injection, although without the concurrent use of an MN device, complicated by a facial lipogranuloma has been described.4 Sclerodermoid reaction, subcutaneous nodules, persistent edema, and ulceration at the site of vitamin E injection also have been described following the injection.5,6 Because vitamin E is a lipid-soluble vitamin, its absorption in the human body is dependent on the presence of lipid or oil-like substances. The reactions mentioned above are associated with the vitamin E oil, which acts as a helper vehicle for lipid-soluble vitamins to be absorbed.7 Other ingredients in topical vitamin E oil include a combination of D-alpha-tocopherol, D-alpha-tocopheryl acetate, D-alpha-tocopheryl succinate, or mixed tocopherols.8 These ester conjugate forms of vitamin E also may play a role in its immunogenic properties and
Hyaluronic acid is a relatively safe and commonly used topical treatment that acts as a lubricant during MN procedures to help the needles glide across the skin and prevent dragging. It also can be applied after the procedure for hydration purposes. Other common alternatives include peptides, ceramides, and epidermal growth factors. Topical products to avoid before, during, and 48 hours after undergoing MN include retinoids, vitamin C, vitamin E, exfoliants, serums that contain acids (eg, alpha hydroxy acids, beta hydroxy acids, glycolic acid, and lactic acid), serums that contain fragrance, and oil-based serums because they are associated with similar adverse effects.8-10 A granulomatous reaction after an MN procedure also has been reported with the use of vitamin C serum.11
The
Most MN devices are made of nickel and various other metals. Cases of contact dermatitis and delayed-type hypersensitivity granulomatous reaction with systemic symptoms have been reported after MN procedures due to the material of the MN device.1,13,14
Conclusion
Microneedling is a minimally invasive procedure that causes nominal damage to the epidermis and superficial papillary dermis, stimulating a wound-healing cascade for collagen production.15,16 Although not approved by the US Food and Drug Administration, MN performed at dermatology offices sometimes can be used in conjunction with topical products to enhance their absorption; however, while vitamin E is known for its antioxidant properties and potential skin benefits, the lipid substance acting as the vehicle is not absorbable by the skin and may cause a granulomatous reaction as the body attempts to encapsulate and digest the foreign substance.10,17 Although rarely reported, the use of topical vitamins with MN—through intradermal injection or combined with MN—can be associated with severe complications, including local, sometimes systemic, and life-threatening complications. Clinicians should be vigilant in order to correlate clinical background and history of recent cosmetic procedures with the histologic findings for prompt diagnosis and timely treatment.
- Soltani-Arabshahi R, Wong JW, Duffy KL, et al. Facial allergic granulomatous reaction and systemic hypersensitivity associated with microneedle therapy for skin rejuvenation. JAMA Dermatol. 2014;150:68-72. doi:10.1001/jamadermatol.2013.6955
- Microneedling market. The Brainy Insights. Published January, 2023. Accessed September 9, 2023. https://www.thebrainyinsights.com/report/microneedling-market-13269
- Baumann LS, Spencer J. The effects of topical vitamin E on the cosmetic appearance of scars. Dermatol Surg. 1999;25:311-315. doi:10.1046/j.1524-4725.1999.08223.x
- Abtahi-Naeini B, Rastegarnasab F, Saffaei A. Liquid vitamin E injection for cosmetic facial rejuvenation: a disaster report of lipogranuloma. J Cosmet Dermatol. 2022;21:5549-5554. doi:10.1111/jocd.15294
- Kamouna B, Litov I, Bardarov E, et al. Granuloma formation after oil-soluble vitamin D injection for lip augmentation - case report. J Eur Acad Dermatol Venereol. 2016;30:1435-1436. doi:10.1111/jdv.13277
- Kamouna B, Darlenski R, Kazandjieva J, et al. Complications of injected vitamin E as a filler for lip augmentation: case series and therapeutic approach. Dermatol Ther. 2015;28:94-97. doi:10.1111/dth.12203
- Kosari P, Alikhan A, Sockolov M, et al. Vitamin E and allergic contact dermatitis. Dermatitis. 2010;21:148-153
- Thiele JJ, Ekanayake-Mudiyanselage S. Vitamin E in human skin: organ-specific physiology and considerations for its use in dermatology. Mol Aspects Med. 2007;28:646-667. doi:10.1016/j.mam.2007.06.001
- Spataro EA, Dierks K, Carniol PJ. Microneedling-associated procedures to enhance facial rejuvenation. Facial Plast Surg Clin North Am. 2022;30:389-397. doi:10.1016/j.fsc.2022.03.012
- Setterfield L. The Concise Guide to Dermal Needling. Acacia Dermacare; 2017.
- Handal M, Kyriakides K, Cohen J, et al. Sarcoidal granulomatous reaction to microneedling with vitamin C serum. JAAD Case Rep. 2023;36:67-69. doi:10.1016/j.jdcr.2023.04.015
- Microneedling devices. U.S. Food and Drug Administration. Published 2020. Accessed September 9, 2025. https://www.fda.gov/medical-devices/aesthetic-cosmetic-devices/microneedling-devices#risks
- Gowda A, Healey B, Ezaldein H, et al. A systematic review examining the potential adverse effects of microneedling. J Clin Aesthet Dermatol. 2021;14:45-54.
- Hou A, Cohen B, Haimovic A, et al. Microneedling: a comprehensive review. Dermatol Surg. 2017;43:321-339. doi:10.1097/DSS.0000000000000924
- Hogan S, Velez MW, Ibrahim O. Microneedling: a new approach for treating textural abnormalities and scars. Semin Cutan Med Surg. 2017;36:155-163. doi:10.12788/j.sder.2017.042
- Schmitt L, Marquardt Y, Amann P, et al. Comprehensive molecular characterization of microneedling therapy in a human three-dimensional skin model. PLoS One. 2018;13:e0204318. doi:10.1371/journal.pone.0204318
- Friedmann DP, Mehta E, Verma KK, et al. Granulomatous reactions from microneedling: a systematic review of the literature. Dermatol Surg. 2025;51:263-266. doi:10.1097/DSS.0000000000004450
Topical application or injection of cosmeceuticals in conjunction with procedures such as facial microneedling (MN) has been associated with local and systemic complications.1
Although at-home options may be more accessible and affordable for patients, they also increase the risk for improper use and subsequent infection. Additionally, the use of cosmeceuticals such as vitamin E oil in conjunction with MN to enhance the effects of the procedure can lead to further complications. We report the case of a 44-year-old woman who developed a necrotic ulcer on the chin following self-treatment with vitamin E oil and an at-home MN device. While MN has been reported to be relatively safe when performed by board-certified dermatologists, clinicians should be vigilant in correlating clinical history and recent cosmetic procedures with the histologic findings for timely diagnosis and treatment of unusual lesions on the face.
Case Report
A 44-year-old woman presented to the emergency department with a progressively enlarging, necrotic, ulcerative lesion on the midline chin of 4 months’ duration. The patient reported that the lesion started as redness that developed into a painful oozing ulcer following application of vitamin E oil in conjunction with an at-home MN device (Figure 1). She purchased the vitamin E oil and MN device online and performed the procedure herself, applying the vitamin E oil to her whole face before, during, and after using the MN device, which contained 0.25-mm titanium needles. She denied undergoing any other recent cosmetic procedures.
The lesion initially was treated by the patient’s primary care physician with oral doxycycline for 6 weeks, followed by oral cephalexin and clindamycin for 2 weeks. Although the redness stabilized, the lesion continued to enlarge, which prompted her initial visit to our hospital 1 month after seeing her primary care physician. During this visit, the patient was given penicillin, and the ulcer was debrided and biopsied; however, no clinical improvement was seen.
A biopsy during her initial emergency department visit and a repeat biopsy after 1 month showed similar findings of diffuse lymphohistiocytic and eosinophilic inflammation in the dermis (Figure 2) with poorly defined granulomas and multinucleated giant cells containing nonpolarizable exogenous material (Figure 3). Similar detached exogenous materials also were identified adjacent to the tissue. Diffuse re-epithelialization was seen, featuring pseudoepitheliomatous hyperplasia in association with the inflammatory process and granulation tissue (Figures 3 and 4). A higher-power view of the dermis showed foci of sclerosing lipogranuloma (Figure 4). Periodic acid–Schiff, Grocott methenamine silver, acid-fast bacilli, Fite, and Wright-Giemsa stains all were negative for microorganisms, and pancytokeratin staining was negative for carcinoma. These findings supported the diagnosis of a foreign body granulomatous reaction to an exogenous material—in this case, the vitamin E oil. Subsequent treatment with intralesional triamcinolone 10 mg/mL injection over 18 months resulted in progressive and drastic improvement of the lesion (Figure 5). A scar excision was performed, which further improved the lesion’s cosmetic appearance.
Comment
Application of various topical cosmeceuticals before, during, or after MN to enhance the effects of the procedure can introduce particles into the dermis, resulting in local or systemic hypersensitivity reactions. The associated adverse events can be divided into 2 main categories: adverse reactions related to the topical product or to the materials of the MN device itself.
A study showed that topical application of vitamin E oil to wounds on the skin does not improve the cosmetic appearance of scars.3 Instead, it is associated with a high incidence of contact dermatitis. A similar case of vitamin E injection, although without the concurrent use of an MN device, complicated by a facial lipogranuloma has been described.4 Sclerodermoid reaction, subcutaneous nodules, persistent edema, and ulceration at the site of vitamin E injection also have been described following the injection.5,6 Because vitamin E is a lipid-soluble vitamin, its absorption in the human body is dependent on the presence of lipid or oil-like substances. The reactions mentioned above are associated with the vitamin E oil, which acts as a helper vehicle for lipid-soluble vitamins to be absorbed.7 Other ingredients in topical vitamin E oil include a combination of D-alpha-tocopherol, D-alpha-tocopheryl acetate, D-alpha-tocopheryl succinate, or mixed tocopherols.8 These ester conjugate forms of vitamin E also may play a role in its immunogenic properties and
Hyaluronic acid is a relatively safe and commonly used topical treatment that acts as a lubricant during MN procedures to help the needles glide across the skin and prevent dragging. It also can be applied after the procedure for hydration purposes. Other common alternatives include peptides, ceramides, and epidermal growth factors. Topical products to avoid before, during, and 48 hours after undergoing MN include retinoids, vitamin C, vitamin E, exfoliants, serums that contain acids (eg, alpha hydroxy acids, beta hydroxy acids, glycolic acid, and lactic acid), serums that contain fragrance, and oil-based serums because they are associated with similar adverse effects.8-10 A granulomatous reaction after an MN procedure also has been reported with the use of vitamin C serum.11
The
Most MN devices are made of nickel and various other metals. Cases of contact dermatitis and delayed-type hypersensitivity granulomatous reaction with systemic symptoms have been reported after MN procedures due to the material of the MN device.1,13,14
Conclusion
Microneedling is a minimally invasive procedure that causes nominal damage to the epidermis and superficial papillary dermis, stimulating a wound-healing cascade for collagen production.15,16 Although not approved by the US Food and Drug Administration, MN performed at dermatology offices sometimes can be used in conjunction with topical products to enhance their absorption; however, while vitamin E is known for its antioxidant properties and potential skin benefits, the lipid substance acting as the vehicle is not absorbable by the skin and may cause a granulomatous reaction as the body attempts to encapsulate and digest the foreign substance.10,17 Although rarely reported, the use of topical vitamins with MN—through intradermal injection or combined with MN—can be associated with severe complications, including local, sometimes systemic, and life-threatening complications. Clinicians should be vigilant in order to correlate clinical background and history of recent cosmetic procedures with the histologic findings for prompt diagnosis and timely treatment.
Topical application or injection of cosmeceuticals in conjunction with procedures such as facial microneedling (MN) has been associated with local and systemic complications.1
Although at-home options may be more accessible and affordable for patients, they also increase the risk for improper use and subsequent infection. Additionally, the use of cosmeceuticals such as vitamin E oil in conjunction with MN to enhance the effects of the procedure can lead to further complications. We report the case of a 44-year-old woman who developed a necrotic ulcer on the chin following self-treatment with vitamin E oil and an at-home MN device. While MN has been reported to be relatively safe when performed by board-certified dermatologists, clinicians should be vigilant in correlating clinical history and recent cosmetic procedures with the histologic findings for timely diagnosis and treatment of unusual lesions on the face.
Case Report
A 44-year-old woman presented to the emergency department with a progressively enlarging, necrotic, ulcerative lesion on the midline chin of 4 months’ duration. The patient reported that the lesion started as redness that developed into a painful oozing ulcer following application of vitamin E oil in conjunction with an at-home MN device (Figure 1). She purchased the vitamin E oil and MN device online and performed the procedure herself, applying the vitamin E oil to her whole face before, during, and after using the MN device, which contained 0.25-mm titanium needles. She denied undergoing any other recent cosmetic procedures.
The lesion initially was treated by the patient’s primary care physician with oral doxycycline for 6 weeks, followed by oral cephalexin and clindamycin for 2 weeks. Although the redness stabilized, the lesion continued to enlarge, which prompted her initial visit to our hospital 1 month after seeing her primary care physician. During this visit, the patient was given penicillin, and the ulcer was debrided and biopsied; however, no clinical improvement was seen.
A biopsy during her initial emergency department visit and a repeat biopsy after 1 month showed similar findings of diffuse lymphohistiocytic and eosinophilic inflammation in the dermis (Figure 2) with poorly defined granulomas and multinucleated giant cells containing nonpolarizable exogenous material (Figure 3). Similar detached exogenous materials also were identified adjacent to the tissue. Diffuse re-epithelialization was seen, featuring pseudoepitheliomatous hyperplasia in association with the inflammatory process and granulation tissue (Figures 3 and 4). A higher-power view of the dermis showed foci of sclerosing lipogranuloma (Figure 4). Periodic acid–Schiff, Grocott methenamine silver, acid-fast bacilli, Fite, and Wright-Giemsa stains all were negative for microorganisms, and pancytokeratin staining was negative for carcinoma. These findings supported the diagnosis of a foreign body granulomatous reaction to an exogenous material—in this case, the vitamin E oil. Subsequent treatment with intralesional triamcinolone 10 mg/mL injection over 18 months resulted in progressive and drastic improvement of the lesion (Figure 5). A scar excision was performed, which further improved the lesion’s cosmetic appearance.
Comment
Application of various topical cosmeceuticals before, during, or after MN to enhance the effects of the procedure can introduce particles into the dermis, resulting in local or systemic hypersensitivity reactions. The associated adverse events can be divided into 2 main categories: adverse reactions related to the topical product or to the materials of the MN device itself.
A study showed that topical application of vitamin E oil to wounds on the skin does not improve the cosmetic appearance of scars.3 Instead, it is associated with a high incidence of contact dermatitis. A similar case of vitamin E injection, although without the concurrent use of an MN device, complicated by a facial lipogranuloma has been described.4 Sclerodermoid reaction, subcutaneous nodules, persistent edema, and ulceration at the site of vitamin E injection also have been described following the injection.5,6 Because vitamin E is a lipid-soluble vitamin, its absorption in the human body is dependent on the presence of lipid or oil-like substances. The reactions mentioned above are associated with the vitamin E oil, which acts as a helper vehicle for lipid-soluble vitamins to be absorbed.7 Other ingredients in topical vitamin E oil include a combination of D-alpha-tocopherol, D-alpha-tocopheryl acetate, D-alpha-tocopheryl succinate, or mixed tocopherols.8 These ester conjugate forms of vitamin E also may play a role in its immunogenic properties and
Hyaluronic acid is a relatively safe and commonly used topical treatment that acts as a lubricant during MN procedures to help the needles glide across the skin and prevent dragging. It also can be applied after the procedure for hydration purposes. Other common alternatives include peptides, ceramides, and epidermal growth factors. Topical products to avoid before, during, and 48 hours after undergoing MN include retinoids, vitamin C, vitamin E, exfoliants, serums that contain acids (eg, alpha hydroxy acids, beta hydroxy acids, glycolic acid, and lactic acid), serums that contain fragrance, and oil-based serums because they are associated with similar adverse effects.8-10 A granulomatous reaction after an MN procedure also has been reported with the use of vitamin C serum.11
The
Most MN devices are made of nickel and various other metals. Cases of contact dermatitis and delayed-type hypersensitivity granulomatous reaction with systemic symptoms have been reported after MN procedures due to the material of the MN device.1,13,14
Conclusion
Microneedling is a minimally invasive procedure that causes nominal damage to the epidermis and superficial papillary dermis, stimulating a wound-healing cascade for collagen production.15,16 Although not approved by the US Food and Drug Administration, MN performed at dermatology offices sometimes can be used in conjunction with topical products to enhance their absorption; however, while vitamin E is known for its antioxidant properties and potential skin benefits, the lipid substance acting as the vehicle is not absorbable by the skin and may cause a granulomatous reaction as the body attempts to encapsulate and digest the foreign substance.10,17 Although rarely reported, the use of topical vitamins with MN—through intradermal injection or combined with MN—can be associated with severe complications, including local, sometimes systemic, and life-threatening complications. Clinicians should be vigilant in order to correlate clinical background and history of recent cosmetic procedures with the histologic findings for prompt diagnosis and timely treatment.
- Soltani-Arabshahi R, Wong JW, Duffy KL, et al. Facial allergic granulomatous reaction and systemic hypersensitivity associated with microneedle therapy for skin rejuvenation. JAMA Dermatol. 2014;150:68-72. doi:10.1001/jamadermatol.2013.6955
- Microneedling market. The Brainy Insights. Published January, 2023. Accessed September 9, 2023. https://www.thebrainyinsights.com/report/microneedling-market-13269
- Baumann LS, Spencer J. The effects of topical vitamin E on the cosmetic appearance of scars. Dermatol Surg. 1999;25:311-315. doi:10.1046/j.1524-4725.1999.08223.x
- Abtahi-Naeini B, Rastegarnasab F, Saffaei A. Liquid vitamin E injection for cosmetic facial rejuvenation: a disaster report of lipogranuloma. J Cosmet Dermatol. 2022;21:5549-5554. doi:10.1111/jocd.15294
- Kamouna B, Litov I, Bardarov E, et al. Granuloma formation after oil-soluble vitamin D injection for lip augmentation - case report. J Eur Acad Dermatol Venereol. 2016;30:1435-1436. doi:10.1111/jdv.13277
- Kamouna B, Darlenski R, Kazandjieva J, et al. Complications of injected vitamin E as a filler for lip augmentation: case series and therapeutic approach. Dermatol Ther. 2015;28:94-97. doi:10.1111/dth.12203
- Kosari P, Alikhan A, Sockolov M, et al. Vitamin E and allergic contact dermatitis. Dermatitis. 2010;21:148-153
- Thiele JJ, Ekanayake-Mudiyanselage S. Vitamin E in human skin: organ-specific physiology and considerations for its use in dermatology. Mol Aspects Med. 2007;28:646-667. doi:10.1016/j.mam.2007.06.001
- Spataro EA, Dierks K, Carniol PJ. Microneedling-associated procedures to enhance facial rejuvenation. Facial Plast Surg Clin North Am. 2022;30:389-397. doi:10.1016/j.fsc.2022.03.012
- Setterfield L. The Concise Guide to Dermal Needling. Acacia Dermacare; 2017.
- Handal M, Kyriakides K, Cohen J, et al. Sarcoidal granulomatous reaction to microneedling with vitamin C serum. JAAD Case Rep. 2023;36:67-69. doi:10.1016/j.jdcr.2023.04.015
- Microneedling devices. U.S. Food and Drug Administration. Published 2020. Accessed September 9, 2025. https://www.fda.gov/medical-devices/aesthetic-cosmetic-devices/microneedling-devices#risks
- Gowda A, Healey B, Ezaldein H, et al. A systematic review examining the potential adverse effects of microneedling. J Clin Aesthet Dermatol. 2021;14:45-54.
- Hou A, Cohen B, Haimovic A, et al. Microneedling: a comprehensive review. Dermatol Surg. 2017;43:321-339. doi:10.1097/DSS.0000000000000924
- Hogan S, Velez MW, Ibrahim O. Microneedling: a new approach for treating textural abnormalities and scars. Semin Cutan Med Surg. 2017;36:155-163. doi:10.12788/j.sder.2017.042
- Schmitt L, Marquardt Y, Amann P, et al. Comprehensive molecular characterization of microneedling therapy in a human three-dimensional skin model. PLoS One. 2018;13:e0204318. doi:10.1371/journal.pone.0204318
- Friedmann DP, Mehta E, Verma KK, et al. Granulomatous reactions from microneedling: a systematic review of the literature. Dermatol Surg. 2025;51:263-266. doi:10.1097/DSS.0000000000004450
- Soltani-Arabshahi R, Wong JW, Duffy KL, et al. Facial allergic granulomatous reaction and systemic hypersensitivity associated with microneedle therapy for skin rejuvenation. JAMA Dermatol. 2014;150:68-72. doi:10.1001/jamadermatol.2013.6955
- Microneedling market. The Brainy Insights. Published January, 2023. Accessed September 9, 2023. https://www.thebrainyinsights.com/report/microneedling-market-13269
- Baumann LS, Spencer J. The effects of topical vitamin E on the cosmetic appearance of scars. Dermatol Surg. 1999;25:311-315. doi:10.1046/j.1524-4725.1999.08223.x
- Abtahi-Naeini B, Rastegarnasab F, Saffaei A. Liquid vitamin E injection for cosmetic facial rejuvenation: a disaster report of lipogranuloma. J Cosmet Dermatol. 2022;21:5549-5554. doi:10.1111/jocd.15294
- Kamouna B, Litov I, Bardarov E, et al. Granuloma formation after oil-soluble vitamin D injection for lip augmentation - case report. J Eur Acad Dermatol Venereol. 2016;30:1435-1436. doi:10.1111/jdv.13277
- Kamouna B, Darlenski R, Kazandjieva J, et al. Complications of injected vitamin E as a filler for lip augmentation: case series and therapeutic approach. Dermatol Ther. 2015;28:94-97. doi:10.1111/dth.12203
- Kosari P, Alikhan A, Sockolov M, et al. Vitamin E and allergic contact dermatitis. Dermatitis. 2010;21:148-153
- Thiele JJ, Ekanayake-Mudiyanselage S. Vitamin E in human skin: organ-specific physiology and considerations for its use in dermatology. Mol Aspects Med. 2007;28:646-667. doi:10.1016/j.mam.2007.06.001
- Spataro EA, Dierks K, Carniol PJ. Microneedling-associated procedures to enhance facial rejuvenation. Facial Plast Surg Clin North Am. 2022;30:389-397. doi:10.1016/j.fsc.2022.03.012
- Setterfield L. The Concise Guide to Dermal Needling. Acacia Dermacare; 2017.
- Handal M, Kyriakides K, Cohen J, et al. Sarcoidal granulomatous reaction to microneedling with vitamin C serum. JAAD Case Rep. 2023;36:67-69. doi:10.1016/j.jdcr.2023.04.015
- Microneedling devices. U.S. Food and Drug Administration. Published 2020. Accessed September 9, 2025. https://www.fda.gov/medical-devices/aesthetic-cosmetic-devices/microneedling-devices#risks
- Gowda A, Healey B, Ezaldein H, et al. A systematic review examining the potential adverse effects of microneedling. J Clin Aesthet Dermatol. 2021;14:45-54.
- Hou A, Cohen B, Haimovic A, et al. Microneedling: a comprehensive review. Dermatol Surg. 2017;43:321-339. doi:10.1097/DSS.0000000000000924
- Hogan S, Velez MW, Ibrahim O. Microneedling: a new approach for treating textural abnormalities and scars. Semin Cutan Med Surg. 2017;36:155-163. doi:10.12788/j.sder.2017.042
- Schmitt L, Marquardt Y, Amann P, et al. Comprehensive molecular characterization of microneedling therapy in a human three-dimensional skin model. PLoS One. 2018;13:e0204318. doi:10.1371/journal.pone.0204318
- Friedmann DP, Mehta E, Verma KK, et al. Granulomatous reactions from microneedling: a systematic review of the literature. Dermatol Surg. 2025;51:263-266. doi:10.1097/DSS.0000000000004450
Destructive Facial Granuloma Following Self-Treatment With Vitamin E Oil and an At-Home Microneedling Device
Destructive Facial Granuloma Following Self-Treatment With Vitamin E Oil and an At-Home Microneedling Device
Practice Points
- Severe complications can potentially arise from at-home microneedling procedures when combined with cosmeceuticals such as vitamin E oil.
- Clinicopathologic correlation with cosmetic procedures is imperative to prompt diagnosis and treatment of these skin reactions.
- Microneedling procedures should be performed under the supervision of a board-certified dermatologist to avoid complications, and clinicians should inquire specifically about skin care routines and cosmetic procedures when patients present with unusual lesions on the face.
