User login
Grand Rounds: Man, 62, With New-Onset Atrial Fibrillation
A 62-year-old black nursing home resident was transported to the hospital emergency department with fever of 102°F, new-onset atrial fibrillation (A-fib), and dementia. His medical history was significant for hypertension and multiple strokes.
His inpatient work-up for A-fib and dementia revealed a thyroid-stimulating hormone (TSH) level below 0.005 µIU/mL (normal range, 0.3 to 3.0 µIU/mL). Results of thyroid function testing (TFT) revealed a triiodothyronine (T3) level within normal range but a free thyroxine (T4) level of 2.9 ng/dL (normal range, 0.7 to 1.5 ng/dL) and a total T4 of 17.8 µg/dL (normal, 4.5 to 12.0 µg/dL). The abnormal TSH and T4 levels were considered suggestive of a thyrotoxic state, warranting an endocrinology consult. Cardiology was consulted regarding new-onset A-fib.
During history taking, the patient denied any shortness of breath, cough, palpitations, heat intolerance, anxiety, tremors, insomnia, dysphagia, diarrhea, dysuria, weight loss, or recent ingestion of iodine-containing medications or supplements.
On examination, the patient was febrile, with a blood pressure of 106/71 mm Hg; pulse, 74 beats/min; respiratory rate, 20 breaths/min; and O2 saturation, 98% to 99% on room air. ECG showed a normal sinus rhythm and a ventricular rate of 64 beats/min.
The patient's weight was 58.9 kg, and his height, 63" (BMI, 22.8). The patient had no skin changes, and his mucous membranes were slightly moist. The patient's head was atraumatic and normocephalic. His extraocular movements were intact, and his pupils were equal, round, and reactive to light, with nonicteric sclera. There was no proptosis or ophthalmoplegia. The patient's neck was supple, with no jugular venous distension, tracheal deviation, or thyromegaly.
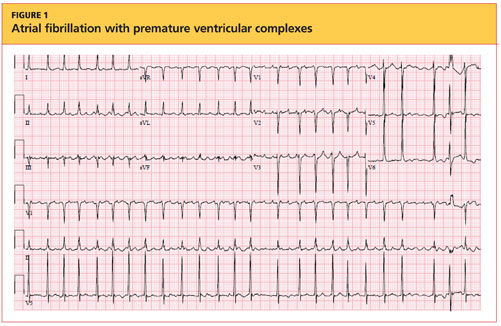
The cardiovascular exam revealed an irregular heartbeat, and repeat ECG showed A-fib with a ventricular rate of 151 beats/min (see Figure 1). The patient's chest was clear, with no wheezing or rhonchi. The abdomen was soft and slightly obese, and bowel sounds were present. The neurologic examination revealed no hyperreflexia. The patient's mental status was altered at times and he was alert, awake, and oriented to others. His speech was slightly slow, and some left-sided weakness was noted.
As recommended during the endocrinology consult, the patient underwent an I-123 sodium iodide thyroid scan, which showed faint uptake at the base of the neck, slightly to the left of midline; and a 24-hour radioactive iodide uptake (RAIU), which measured 2.8% (normal range, 8% to 35%).
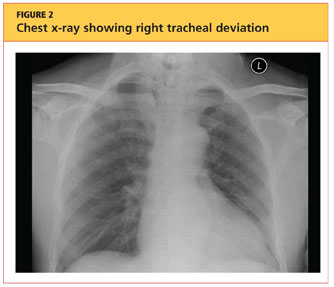
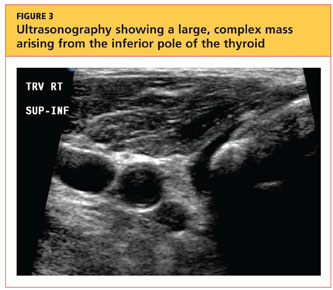
The patient's chest X-ray showed a right tracheal deviation not previously noted on physical examination (see Figure 2); the possible cause of a thyroid mass was considered. Subsequent ultrasonography of the thyroid revealed generally normal dimensions and parenchymal echogenicity. However, a large complex mass was detected, arising from the inferior pole of the thyroid and displacing the trachea toward the right (see Figure 3). According to the radiologist's notes, the mass contained both solid and cystic elements, scattered calcifications, and foci of flow on color Doppler. It measured about 6 cm in the largest (transverse) dimension. A 2.0-mm nodule was noted in the isthmus, slightly to the right of midline, consistent with multinodular goiter.
Following the cardiology consult, a diltiazem drip was initiated, but the patient was later optimized on flecainide for rhythm control and metoprolol for rate control. He was also initially anticoagulated using a heparin drip and bridged to warfarin, with target international normalized ratio (INR) between 2.0 and 3.0. Echocardiography revealed normal systolic function with ejection fraction of 55%, left ventricular hypertrophy, pulmonary artery systolic pressure of 35 mm Hg, and no pericardial effusions or valvular disease.
Regarding the patient's unexplained fever, results of chest imaging were negative for signs of pneumonia or atelectasis, which might have suggested a pulmonary cause. Urinalysis results were normal. Complete blood count showed no leukocytosis. The patient's fever subsided within 48 hours.
The differential diagnosis included Graves' disease, toxic multinodular goiter, Jod-Basedow syndrome, and subacute thyroiditis.
Graves' disease, an autoimmune disease with an unknown trigger, is the most common cause of hyperthyroidism. In affected patients, the thyroid gland overproduces thyroid hormones, leading to thyrotoxicosis. Thyrotoxicosis can result in multiple clinical signs and symptoms, including Graves' ophthalmopathy, pretibial myxedema, and goiter; TFT results typically include elevated T3 and T4 and low TSH.1-5 In the case patient (who had no history of thyroid disease, nor clinical signs or symptoms of Graves' disease), low uptake of iodine on thyroid scan precluded this diagnosis.
Toxic multinodular goiter, the second most common cause of hyperthyroidism, can be responsible for A-fib, tachycardia, and congestive heart failure.6,7 Iodine deficiency causes enlargement of the thyroid gland, where numerous nodules can develop, as seen in the case patient. These nodules can function independently, sometimes producing excess thyroid hormone; this leads to hyperplasia of the thyroid gland, resulting in a nontoxic multinodular goiter. From this goiter, a toxic multinodular goiter can emerge insidiously. However, in this condition, RAIU typically exceeds 30%; in the case patient, low 24-hour RAIU (2.8%) and the absence of functioning nodules on scanning made it possible to rule out this diagnosis.
Jod-Basedow syndrome refers to hyperthyroidism that develops as a result of administration of iodide, either as a dietary supplement or as IV contrast medium, or as an adverse effect of the antiarrhythmic drug amiodarone. This phenomenon is usually seen in a patient with endemic goiter.8-11 The relatively limited nature of the case patient's goiter and absence of a precipitating exposure to iodine made this diagnosis highly unlikely.
Subacute thyroiditis is a condition to which the patient's abnormal TFT results could reasonably be attributed. The patient had a substernal multinodular goiter that could not be palpated on physical examination, but it was visualized in the extended lower neck during thyroid scintigraphy.3 RAIU was minimal—a typical finding in this disorder,6 as TSH is suppressed by leakage of the excessive amounts of thyroid hormone. A tentative diagnosis of subacute thyroiditis was made.
As subacute thyroiditis is a self-limiting disorder, the patient was not started on any medications for hyperthyroidism but was advised to follow up with his primary care provider or an endocrinologist for repeat TFT and for fine-needle aspiration biopsy of the large thyroid nodule (a complex mass, containing cystic elements and calcifications, with a potential for malignancy) to rule out thyroid cancer.
Repeat ECG before discharge showed normal sinus rhythm with a ventricular rate of 74 beats/min. The patient was alert, awake, and oriented at discharge. He was continued on flecainide, metoprolol, and warfarin and advised to follow up with his primary care provider regarding his target INR.
DISCUSSION
The incidence of subacute thyroiditis, according to findings reported in 2003 from the Rochester Epidemiology Project in Olmsted County, Minnesota,12 is 12.1 cases per 100,000/year, with a higher incidence in women than men. It is most common in young adults and decreases with advancing age. Coxsackie virus, adenovirus, mumps, echovirus, influenza, and Epstein-Barr virus have been implicated in the disorder.12,13
Subacute thyroiditis is associated with a triphasic clinical course of hyperthyroidism, then hypothyroidism, then a return to normal thyroid function—as was seen in the case patient. Onset of subacute thyroiditis has been associated with recent viral infection, which may serve as a precipitant. The cause of this patient's high fever was never identified; thus, the etiology may have been viral.
The initial high thyroid hormone levels result from inflammation of thyroid tissue and release of preformed thyroid hormone into the circulation.6 At this point, TSH is suppressed and patients have very low RAIU, as was true in the case patient.
The condition is self-limiting and does not require treatment in the majority of patients, as TFT results return to normal levels within about two months.6 Patients can appear extremely ill due to thyrotoxicosis from subacute thyroiditis, but this usually lasts no longer than six to eight weeks.3 Subacute thyroiditis can be associated with atrial arrhythmia or heart failure.14,15
PATIENT OUTCOME
New-onset A-fib was attributed to the patient's thyrotoxicosis, which in turn was caused by subacute thyroiditis. He had a multinodular goiter, although he had not received any iodine supplements or IV contrast. As in most cases of subacute thyroiditis, no precipitating event was identified. However, given this patient's residence in a nursing facility and presentation with a high fever with no identifiable cause, a viral etiology for his subacute thyroiditis is possible.6
The patient's dementia may have been secondary to acute thyrotoxicosis, as his mental state improved during the hospital stay. His vitamin B12, folate, and A1C levels were within normal range. CT of the head showed multiple chronic infarcts and cerebral atrophy, and MRI of the brain indicated microvascular ischemic disease.
The patient was readmitted one month later for an episode of near-syncope (which, it was concluded, was a vasovagal episode). At that time, his TSH was found normal at 1.350 µIU/mL. Flecainide and metoprolol were discontinued; he was started on diltiazem for continued rate and rhythm control (as recommended by cardiology) and continued on warfarin.
CONCLUSION
In this case, subacute thyroiditis was most likely caused by a viral infection that led to destruction of the normal thyroid follicles and release of their preformed thyroid hormone into the circulation; this in turn led to sudden-onset A-fib. The diagnosis of subacute thyroiditis was suggested based on the abnormalities seen in this patient's TFT results, coupled with the suppressed RAIU—a typical finding in this disease.
Because subacute thyroiditis is a self-limiting condition, there is no role for antithyroid medication. Instead, treatment should be focused on relieving the patient's symptoms, such as ß-blockade or calcium channel blockers for tachycardia and corticosteroids or NSAIDs for neck pain.
REFERENCES
1. Weetman AP. Graves' disease. N Engl J Med. 2000;343(17):1236-1248.
2. Delgado Hurtado JJ, Pineda M. Images in medicine: Graves' disease. N Engl J Med. 2011; 364(20):1955.
3. Al-Sharif AA, Abujbara MA, Chiacchio S, et al. Contribution of radioiodine uptake measurement and thyroid scintigraphy to the differential diagnosis of thyrotoxicosis. Hell J Nucl Med. 2010;13(2):132-137.
4. Buccelletti F, Carroccia A, Marsiliani D, et al. Utility of routine thyroid-stimulating hormone determination in new-onset atrial fibrillation in the ED. Am J Emerg Med. 2011;29(9):1158-1162.
5. Ross DS. Radioiodine therapy for hyperthyroidism. N Engl J Med. 2011;364(6):542-550.
6. Bahn RS, Burch HB, Cooper DS, et al. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Endocr Pract. 2011;17(3):456-520.
7. Erickson D, Gharib H, Li H, van Heerden JA. Treatment of patients with toxic multinodular goiter. Thyroid. 1998;8(4):277-282.
8. Basaria S, Cooper DS. Amiodarone and the thyroid. Am J Med. 2005;118(7):706-714.
9. Bogazzi F, Bartalena L, Martino E. Approach to the patient with amiodarone-induced thyrotoxicosis. J Clin Endocrinol Metab. 2010;95(6):2529-2535.
10. El-Shirbiny AM, Stavrou SS, Dnistrian A, et al. Jod-Basedow syndrome following oral iodine and radioiodinated-antibody administration. J Nucl Med. 1997;38(11):1816-1817.
11. Stanbury JB, Ermans AE, Bourdoux P, et al. Iodine-induced hyperthyroidism: occurrence and epidemiology. Thyroid. 1998;8(1):83-100.
12. Fatourechi V, Aniszewski JP, Fatourechi GZ, et al. Clinical features and outcome of subacute thyroiditis in an incidence cohort: Olmsted County, Minnesota, study. J Clin Endocrinol Metab. 2003;88(5):2100-2105.
13. Golden SH, Robinson KA, Saldanha I, et al. Clinical review: prevalence and incidence of endocrine and metabolic disorders in the United States: a comprehensive review. J Clin Endocrinol Metab. 2009;94(6):1853-1878.
14. Volpé R. The management of subacute (DeQuervain's) thyroiditis. Thyroid. 1993;3(3):253-255.
15. Lee SL. Subacute thyroiditis (2009). http://emedicine.medscape.com/article/125648-overview. Accessed April 17, 2012.
A 62-year-old black nursing home resident was transported to the hospital emergency department with fever of 102°F, new-onset atrial fibrillation (A-fib), and dementia. His medical history was significant for hypertension and multiple strokes.
His inpatient work-up for A-fib and dementia revealed a thyroid-stimulating hormone (TSH) level below 0.005 µIU/mL (normal range, 0.3 to 3.0 µIU/mL). Results of thyroid function testing (TFT) revealed a triiodothyronine (T3) level within normal range but a free thyroxine (T4) level of 2.9 ng/dL (normal range, 0.7 to 1.5 ng/dL) and a total T4 of 17.8 µg/dL (normal, 4.5 to 12.0 µg/dL). The abnormal TSH and T4 levels were considered suggestive of a thyrotoxic state, warranting an endocrinology consult. Cardiology was consulted regarding new-onset A-fib.
During history taking, the patient denied any shortness of breath, cough, palpitations, heat intolerance, anxiety, tremors, insomnia, dysphagia, diarrhea, dysuria, weight loss, or recent ingestion of iodine-containing medications or supplements.
On examination, the patient was febrile, with a blood pressure of 106/71 mm Hg; pulse, 74 beats/min; respiratory rate, 20 breaths/min; and O2 saturation, 98% to 99% on room air. ECG showed a normal sinus rhythm and a ventricular rate of 64 beats/min.
The patient's weight was 58.9 kg, and his height, 63" (BMI, 22.8). The patient had no skin changes, and his mucous membranes were slightly moist. The patient's head was atraumatic and normocephalic. His extraocular movements were intact, and his pupils were equal, round, and reactive to light, with nonicteric sclera. There was no proptosis or ophthalmoplegia. The patient's neck was supple, with no jugular venous distension, tracheal deviation, or thyromegaly.
The cardiovascular exam revealed an irregular heartbeat, and repeat ECG showed A-fib with a ventricular rate of 151 beats/min (see Figure 1). The patient's chest was clear, with no wheezing or rhonchi. The abdomen was soft and slightly obese, and bowel sounds were present. The neurologic examination revealed no hyperreflexia. The patient's mental status was altered at times and he was alert, awake, and oriented to others. His speech was slightly slow, and some left-sided weakness was noted.
As recommended during the endocrinology consult, the patient underwent an I-123 sodium iodide thyroid scan, which showed faint uptake at the base of the neck, slightly to the left of midline; and a 24-hour radioactive iodide uptake (RAIU), which measured 2.8% (normal range, 8% to 35%).
The patient's chest X-ray showed a right tracheal deviation not previously noted on physical examination (see Figure 2); the possible cause of a thyroid mass was considered. Subsequent ultrasonography of the thyroid revealed generally normal dimensions and parenchymal echogenicity. However, a large complex mass was detected, arising from the inferior pole of the thyroid and displacing the trachea toward the right (see Figure 3). According to the radiologist's notes, the mass contained both solid and cystic elements, scattered calcifications, and foci of flow on color Doppler. It measured about 6 cm in the largest (transverse) dimension. A 2.0-mm nodule was noted in the isthmus, slightly to the right of midline, consistent with multinodular goiter.
Following the cardiology consult, a diltiazem drip was initiated, but the patient was later optimized on flecainide for rhythm control and metoprolol for rate control. He was also initially anticoagulated using a heparin drip and bridged to warfarin, with target international normalized ratio (INR) between 2.0 and 3.0. Echocardiography revealed normal systolic function with ejection fraction of 55%, left ventricular hypertrophy, pulmonary artery systolic pressure of 35 mm Hg, and no pericardial effusions or valvular disease.
Regarding the patient's unexplained fever, results of chest imaging were negative for signs of pneumonia or atelectasis, which might have suggested a pulmonary cause. Urinalysis results were normal. Complete blood count showed no leukocytosis. The patient's fever subsided within 48 hours.
The differential diagnosis included Graves' disease, toxic multinodular goiter, Jod-Basedow syndrome, and subacute thyroiditis.
Graves' disease, an autoimmune disease with an unknown trigger, is the most common cause of hyperthyroidism. In affected patients, the thyroid gland overproduces thyroid hormones, leading to thyrotoxicosis. Thyrotoxicosis can result in multiple clinical signs and symptoms, including Graves' ophthalmopathy, pretibial myxedema, and goiter; TFT results typically include elevated T3 and T4 and low TSH.1-5 In the case patient (who had no history of thyroid disease, nor clinical signs or symptoms of Graves' disease), low uptake of iodine on thyroid scan precluded this diagnosis.
Toxic multinodular goiter, the second most common cause of hyperthyroidism, can be responsible for A-fib, tachycardia, and congestive heart failure.6,7 Iodine deficiency causes enlargement of the thyroid gland, where numerous nodules can develop, as seen in the case patient. These nodules can function independently, sometimes producing excess thyroid hormone; this leads to hyperplasia of the thyroid gland, resulting in a nontoxic multinodular goiter. From this goiter, a toxic multinodular goiter can emerge insidiously. However, in this condition, RAIU typically exceeds 30%; in the case patient, low 24-hour RAIU (2.8%) and the absence of functioning nodules on scanning made it possible to rule out this diagnosis.
Jod-Basedow syndrome refers to hyperthyroidism that develops as a result of administration of iodide, either as a dietary supplement or as IV contrast medium, or as an adverse effect of the antiarrhythmic drug amiodarone. This phenomenon is usually seen in a patient with endemic goiter.8-11 The relatively limited nature of the case patient's goiter and absence of a precipitating exposure to iodine made this diagnosis highly unlikely.
Subacute thyroiditis is a condition to which the patient's abnormal TFT results could reasonably be attributed. The patient had a substernal multinodular goiter that could not be palpated on physical examination, but it was visualized in the extended lower neck during thyroid scintigraphy.3 RAIU was minimal—a typical finding in this disorder,6 as TSH is suppressed by leakage of the excessive amounts of thyroid hormone. A tentative diagnosis of subacute thyroiditis was made.
As subacute thyroiditis is a self-limiting disorder, the patient was not started on any medications for hyperthyroidism but was advised to follow up with his primary care provider or an endocrinologist for repeat TFT and for fine-needle aspiration biopsy of the large thyroid nodule (a complex mass, containing cystic elements and calcifications, with a potential for malignancy) to rule out thyroid cancer.
Repeat ECG before discharge showed normal sinus rhythm with a ventricular rate of 74 beats/min. The patient was alert, awake, and oriented at discharge. He was continued on flecainide, metoprolol, and warfarin and advised to follow up with his primary care provider regarding his target INR.
DISCUSSION
The incidence of subacute thyroiditis, according to findings reported in 2003 from the Rochester Epidemiology Project in Olmsted County, Minnesota,12 is 12.1 cases per 100,000/year, with a higher incidence in women than men. It is most common in young adults and decreases with advancing age. Coxsackie virus, adenovirus, mumps, echovirus, influenza, and Epstein-Barr virus have been implicated in the disorder.12,13
Subacute thyroiditis is associated with a triphasic clinical course of hyperthyroidism, then hypothyroidism, then a return to normal thyroid function—as was seen in the case patient. Onset of subacute thyroiditis has been associated with recent viral infection, which may serve as a precipitant. The cause of this patient's high fever was never identified; thus, the etiology may have been viral.
The initial high thyroid hormone levels result from inflammation of thyroid tissue and release of preformed thyroid hormone into the circulation.6 At this point, TSH is suppressed and patients have very low RAIU, as was true in the case patient.
The condition is self-limiting and does not require treatment in the majority of patients, as TFT results return to normal levels within about two months.6 Patients can appear extremely ill due to thyrotoxicosis from subacute thyroiditis, but this usually lasts no longer than six to eight weeks.3 Subacute thyroiditis can be associated with atrial arrhythmia or heart failure.14,15
PATIENT OUTCOME
New-onset A-fib was attributed to the patient's thyrotoxicosis, which in turn was caused by subacute thyroiditis. He had a multinodular goiter, although he had not received any iodine supplements or IV contrast. As in most cases of subacute thyroiditis, no precipitating event was identified. However, given this patient's residence in a nursing facility and presentation with a high fever with no identifiable cause, a viral etiology for his subacute thyroiditis is possible.6
The patient's dementia may have been secondary to acute thyrotoxicosis, as his mental state improved during the hospital stay. His vitamin B12, folate, and A1C levels were within normal range. CT of the head showed multiple chronic infarcts and cerebral atrophy, and MRI of the brain indicated microvascular ischemic disease.
The patient was readmitted one month later for an episode of near-syncope (which, it was concluded, was a vasovagal episode). At that time, his TSH was found normal at 1.350 µIU/mL. Flecainide and metoprolol were discontinued; he was started on diltiazem for continued rate and rhythm control (as recommended by cardiology) and continued on warfarin.
CONCLUSION
In this case, subacute thyroiditis was most likely caused by a viral infection that led to destruction of the normal thyroid follicles and release of their preformed thyroid hormone into the circulation; this in turn led to sudden-onset A-fib. The diagnosis of subacute thyroiditis was suggested based on the abnormalities seen in this patient's TFT results, coupled with the suppressed RAIU—a typical finding in this disease.
Because subacute thyroiditis is a self-limiting condition, there is no role for antithyroid medication. Instead, treatment should be focused on relieving the patient's symptoms, such as ß-blockade or calcium channel blockers for tachycardia and corticosteroids or NSAIDs for neck pain.
REFERENCES
1. Weetman AP. Graves' disease. N Engl J Med. 2000;343(17):1236-1248.
2. Delgado Hurtado JJ, Pineda M. Images in medicine: Graves' disease. N Engl J Med. 2011; 364(20):1955.
3. Al-Sharif AA, Abujbara MA, Chiacchio S, et al. Contribution of radioiodine uptake measurement and thyroid scintigraphy to the differential diagnosis of thyrotoxicosis. Hell J Nucl Med. 2010;13(2):132-137.
4. Buccelletti F, Carroccia A, Marsiliani D, et al. Utility of routine thyroid-stimulating hormone determination in new-onset atrial fibrillation in the ED. Am J Emerg Med. 2011;29(9):1158-1162.
5. Ross DS. Radioiodine therapy for hyperthyroidism. N Engl J Med. 2011;364(6):542-550.
6. Bahn RS, Burch HB, Cooper DS, et al. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Endocr Pract. 2011;17(3):456-520.
7. Erickson D, Gharib H, Li H, van Heerden JA. Treatment of patients with toxic multinodular goiter. Thyroid. 1998;8(4):277-282.
8. Basaria S, Cooper DS. Amiodarone and the thyroid. Am J Med. 2005;118(7):706-714.
9. Bogazzi F, Bartalena L, Martino E. Approach to the patient with amiodarone-induced thyrotoxicosis. J Clin Endocrinol Metab. 2010;95(6):2529-2535.
10. El-Shirbiny AM, Stavrou SS, Dnistrian A, et al. Jod-Basedow syndrome following oral iodine and radioiodinated-antibody administration. J Nucl Med. 1997;38(11):1816-1817.
11. Stanbury JB, Ermans AE, Bourdoux P, et al. Iodine-induced hyperthyroidism: occurrence and epidemiology. Thyroid. 1998;8(1):83-100.
12. Fatourechi V, Aniszewski JP, Fatourechi GZ, et al. Clinical features and outcome of subacute thyroiditis in an incidence cohort: Olmsted County, Minnesota, study. J Clin Endocrinol Metab. 2003;88(5):2100-2105.
13. Golden SH, Robinson KA, Saldanha I, et al. Clinical review: prevalence and incidence of endocrine and metabolic disorders in the United States: a comprehensive review. J Clin Endocrinol Metab. 2009;94(6):1853-1878.
14. Volpé R. The management of subacute (DeQuervain's) thyroiditis. Thyroid. 1993;3(3):253-255.
15. Lee SL. Subacute thyroiditis (2009). http://emedicine.medscape.com/article/125648-overview. Accessed April 17, 2012.
A 62-year-old black nursing home resident was transported to the hospital emergency department with fever of 102°F, new-onset atrial fibrillation (A-fib), and dementia. His medical history was significant for hypertension and multiple strokes.
His inpatient work-up for A-fib and dementia revealed a thyroid-stimulating hormone (TSH) level below 0.005 µIU/mL (normal range, 0.3 to 3.0 µIU/mL). Results of thyroid function testing (TFT) revealed a triiodothyronine (T3) level within normal range but a free thyroxine (T4) level of 2.9 ng/dL (normal range, 0.7 to 1.5 ng/dL) and a total T4 of 17.8 µg/dL (normal, 4.5 to 12.0 µg/dL). The abnormal TSH and T4 levels were considered suggestive of a thyrotoxic state, warranting an endocrinology consult. Cardiology was consulted regarding new-onset A-fib.
During history taking, the patient denied any shortness of breath, cough, palpitations, heat intolerance, anxiety, tremors, insomnia, dysphagia, diarrhea, dysuria, weight loss, or recent ingestion of iodine-containing medications or supplements.
On examination, the patient was febrile, with a blood pressure of 106/71 mm Hg; pulse, 74 beats/min; respiratory rate, 20 breaths/min; and O2 saturation, 98% to 99% on room air. ECG showed a normal sinus rhythm and a ventricular rate of 64 beats/min.
The patient's weight was 58.9 kg, and his height, 63" (BMI, 22.8). The patient had no skin changes, and his mucous membranes were slightly moist. The patient's head was atraumatic and normocephalic. His extraocular movements were intact, and his pupils were equal, round, and reactive to light, with nonicteric sclera. There was no proptosis or ophthalmoplegia. The patient's neck was supple, with no jugular venous distension, tracheal deviation, or thyromegaly.
The cardiovascular exam revealed an irregular heartbeat, and repeat ECG showed A-fib with a ventricular rate of 151 beats/min (see Figure 1). The patient's chest was clear, with no wheezing or rhonchi. The abdomen was soft and slightly obese, and bowel sounds were present. The neurologic examination revealed no hyperreflexia. The patient's mental status was altered at times and he was alert, awake, and oriented to others. His speech was slightly slow, and some left-sided weakness was noted.
As recommended during the endocrinology consult, the patient underwent an I-123 sodium iodide thyroid scan, which showed faint uptake at the base of the neck, slightly to the left of midline; and a 24-hour radioactive iodide uptake (RAIU), which measured 2.8% (normal range, 8% to 35%).
The patient's chest X-ray showed a right tracheal deviation not previously noted on physical examination (see Figure 2); the possible cause of a thyroid mass was considered. Subsequent ultrasonography of the thyroid revealed generally normal dimensions and parenchymal echogenicity. However, a large complex mass was detected, arising from the inferior pole of the thyroid and displacing the trachea toward the right (see Figure 3). According to the radiologist's notes, the mass contained both solid and cystic elements, scattered calcifications, and foci of flow on color Doppler. It measured about 6 cm in the largest (transverse) dimension. A 2.0-mm nodule was noted in the isthmus, slightly to the right of midline, consistent with multinodular goiter.
Following the cardiology consult, a diltiazem drip was initiated, but the patient was later optimized on flecainide for rhythm control and metoprolol for rate control. He was also initially anticoagulated using a heparin drip and bridged to warfarin, with target international normalized ratio (INR) between 2.0 and 3.0. Echocardiography revealed normal systolic function with ejection fraction of 55%, left ventricular hypertrophy, pulmonary artery systolic pressure of 35 mm Hg, and no pericardial effusions or valvular disease.
Regarding the patient's unexplained fever, results of chest imaging were negative for signs of pneumonia or atelectasis, which might have suggested a pulmonary cause. Urinalysis results were normal. Complete blood count showed no leukocytosis. The patient's fever subsided within 48 hours.
The differential diagnosis included Graves' disease, toxic multinodular goiter, Jod-Basedow syndrome, and subacute thyroiditis.
Graves' disease, an autoimmune disease with an unknown trigger, is the most common cause of hyperthyroidism. In affected patients, the thyroid gland overproduces thyroid hormones, leading to thyrotoxicosis. Thyrotoxicosis can result in multiple clinical signs and symptoms, including Graves' ophthalmopathy, pretibial myxedema, and goiter; TFT results typically include elevated T3 and T4 and low TSH.1-5 In the case patient (who had no history of thyroid disease, nor clinical signs or symptoms of Graves' disease), low uptake of iodine on thyroid scan precluded this diagnosis.
Toxic multinodular goiter, the second most common cause of hyperthyroidism, can be responsible for A-fib, tachycardia, and congestive heart failure.6,7 Iodine deficiency causes enlargement of the thyroid gland, where numerous nodules can develop, as seen in the case patient. These nodules can function independently, sometimes producing excess thyroid hormone; this leads to hyperplasia of the thyroid gland, resulting in a nontoxic multinodular goiter. From this goiter, a toxic multinodular goiter can emerge insidiously. However, in this condition, RAIU typically exceeds 30%; in the case patient, low 24-hour RAIU (2.8%) and the absence of functioning nodules on scanning made it possible to rule out this diagnosis.
Jod-Basedow syndrome refers to hyperthyroidism that develops as a result of administration of iodide, either as a dietary supplement or as IV contrast medium, or as an adverse effect of the antiarrhythmic drug amiodarone. This phenomenon is usually seen in a patient with endemic goiter.8-11 The relatively limited nature of the case patient's goiter and absence of a precipitating exposure to iodine made this diagnosis highly unlikely.
Subacute thyroiditis is a condition to which the patient's abnormal TFT results could reasonably be attributed. The patient had a substernal multinodular goiter that could not be palpated on physical examination, but it was visualized in the extended lower neck during thyroid scintigraphy.3 RAIU was minimal—a typical finding in this disorder,6 as TSH is suppressed by leakage of the excessive amounts of thyroid hormone. A tentative diagnosis of subacute thyroiditis was made.
As subacute thyroiditis is a self-limiting disorder, the patient was not started on any medications for hyperthyroidism but was advised to follow up with his primary care provider or an endocrinologist for repeat TFT and for fine-needle aspiration biopsy of the large thyroid nodule (a complex mass, containing cystic elements and calcifications, with a potential for malignancy) to rule out thyroid cancer.
Repeat ECG before discharge showed normal sinus rhythm with a ventricular rate of 74 beats/min. The patient was alert, awake, and oriented at discharge. He was continued on flecainide, metoprolol, and warfarin and advised to follow up with his primary care provider regarding his target INR.
DISCUSSION
The incidence of subacute thyroiditis, according to findings reported in 2003 from the Rochester Epidemiology Project in Olmsted County, Minnesota,12 is 12.1 cases per 100,000/year, with a higher incidence in women than men. It is most common in young adults and decreases with advancing age. Coxsackie virus, adenovirus, mumps, echovirus, influenza, and Epstein-Barr virus have been implicated in the disorder.12,13
Subacute thyroiditis is associated with a triphasic clinical course of hyperthyroidism, then hypothyroidism, then a return to normal thyroid function—as was seen in the case patient. Onset of subacute thyroiditis has been associated with recent viral infection, which may serve as a precipitant. The cause of this patient's high fever was never identified; thus, the etiology may have been viral.
The initial high thyroid hormone levels result from inflammation of thyroid tissue and release of preformed thyroid hormone into the circulation.6 At this point, TSH is suppressed and patients have very low RAIU, as was true in the case patient.
The condition is self-limiting and does not require treatment in the majority of patients, as TFT results return to normal levels within about two months.6 Patients can appear extremely ill due to thyrotoxicosis from subacute thyroiditis, but this usually lasts no longer than six to eight weeks.3 Subacute thyroiditis can be associated with atrial arrhythmia or heart failure.14,15
PATIENT OUTCOME
New-onset A-fib was attributed to the patient's thyrotoxicosis, which in turn was caused by subacute thyroiditis. He had a multinodular goiter, although he had not received any iodine supplements or IV contrast. As in most cases of subacute thyroiditis, no precipitating event was identified. However, given this patient's residence in a nursing facility and presentation with a high fever with no identifiable cause, a viral etiology for his subacute thyroiditis is possible.6
The patient's dementia may have been secondary to acute thyrotoxicosis, as his mental state improved during the hospital stay. His vitamin B12, folate, and A1C levels were within normal range. CT of the head showed multiple chronic infarcts and cerebral atrophy, and MRI of the brain indicated microvascular ischemic disease.
The patient was readmitted one month later for an episode of near-syncope (which, it was concluded, was a vasovagal episode). At that time, his TSH was found normal at 1.350 µIU/mL. Flecainide and metoprolol were discontinued; he was started on diltiazem for continued rate and rhythm control (as recommended by cardiology) and continued on warfarin.
CONCLUSION
In this case, subacute thyroiditis was most likely caused by a viral infection that led to destruction of the normal thyroid follicles and release of their preformed thyroid hormone into the circulation; this in turn led to sudden-onset A-fib. The diagnosis of subacute thyroiditis was suggested based on the abnormalities seen in this patient's TFT results, coupled with the suppressed RAIU—a typical finding in this disease.
Because subacute thyroiditis is a self-limiting condition, there is no role for antithyroid medication. Instead, treatment should be focused on relieving the patient's symptoms, such as ß-blockade or calcium channel blockers for tachycardia and corticosteroids or NSAIDs for neck pain.
REFERENCES
1. Weetman AP. Graves' disease. N Engl J Med. 2000;343(17):1236-1248.
2. Delgado Hurtado JJ, Pineda M. Images in medicine: Graves' disease. N Engl J Med. 2011; 364(20):1955.
3. Al-Sharif AA, Abujbara MA, Chiacchio S, et al. Contribution of radioiodine uptake measurement and thyroid scintigraphy to the differential diagnosis of thyrotoxicosis. Hell J Nucl Med. 2010;13(2):132-137.
4. Buccelletti F, Carroccia A, Marsiliani D, et al. Utility of routine thyroid-stimulating hormone determination in new-onset atrial fibrillation in the ED. Am J Emerg Med. 2011;29(9):1158-1162.
5. Ross DS. Radioiodine therapy for hyperthyroidism. N Engl J Med. 2011;364(6):542-550.
6. Bahn RS, Burch HB, Cooper DS, et al. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Endocr Pract. 2011;17(3):456-520.
7. Erickson D, Gharib H, Li H, van Heerden JA. Treatment of patients with toxic multinodular goiter. Thyroid. 1998;8(4):277-282.
8. Basaria S, Cooper DS. Amiodarone and the thyroid. Am J Med. 2005;118(7):706-714.
9. Bogazzi F, Bartalena L, Martino E. Approach to the patient with amiodarone-induced thyrotoxicosis. J Clin Endocrinol Metab. 2010;95(6):2529-2535.
10. El-Shirbiny AM, Stavrou SS, Dnistrian A, et al. Jod-Basedow syndrome following oral iodine and radioiodinated-antibody administration. J Nucl Med. 1997;38(11):1816-1817.
11. Stanbury JB, Ermans AE, Bourdoux P, et al. Iodine-induced hyperthyroidism: occurrence and epidemiology. Thyroid. 1998;8(1):83-100.
12. Fatourechi V, Aniszewski JP, Fatourechi GZ, et al. Clinical features and outcome of subacute thyroiditis in an incidence cohort: Olmsted County, Minnesota, study. J Clin Endocrinol Metab. 2003;88(5):2100-2105.
13. Golden SH, Robinson KA, Saldanha I, et al. Clinical review: prevalence and incidence of endocrine and metabolic disorders in the United States: a comprehensive review. J Clin Endocrinol Metab. 2009;94(6):1853-1878.
14. Volpé R. The management of subacute (DeQuervain's) thyroiditis. Thyroid. 1993;3(3):253-255.
15. Lee SL. Subacute thyroiditis (2009). http://emedicine.medscape.com/article/125648-overview. Accessed April 17, 2012.
Grand Rounds: Man, 30, With Traumatic Finger Amputations
A 30-year-old man sustained traumatic amputations of three of his left fingers while at work. A heavy object fell when a supporting chain snapped; although he moved quickly, three of his left distal fingers were caught under the object. He was flown to a hospital for definitive hand care.
During the preadmission history and physical, it was noted that the patient had mild right knee pain in addition to his finger injuries. He had experienced no head injury and no loss of consciousness or other complaints. He did not remember injuring his leg, although he said it might have been struck by the falling object; all he could remember was the injury to his fingers.
On physical exam, the only abnormality other than the man’s traumatic finger amputations was mild right knee edema and a small bruised area medially. Initially, he complained of mild pain on palpation and moderate pain with passive range of motion, but range of motion was intact. His pain was worse at the proximal, medial tibial area, and he had mild lateral mid-calf tenderness though no bruising. Distally, his right lower extremity motor and sensory function were intact, and he had no open wounds or skin breakdown. He had 2+ dorsalis pedis pulse and 1+ posterior tibial pulse. The toes were pink and warm with brisk capillary refill. All compartments were soft and compressible.
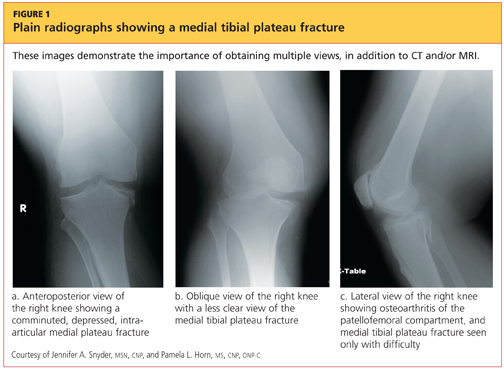
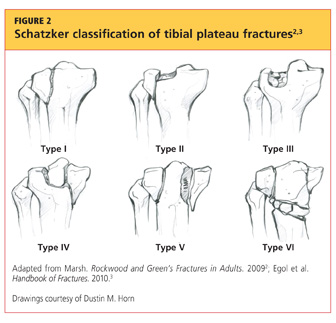
Upon review of his plain radiographs (three views of the right knee), the patient was noted to have a severely comminuted medial tibial plateau fracture that extended to the midline in the region of the tibial spine, with mild depression of the fracture fragments measuring about 6 mm (see Figures 1a, 1b, and 1c). This would translate into a Schatzker IV classification type1 fracture (see Figure 22,3).
The man was admitted and underwent emergent surgery on his injured left fingers that night. Further diagnostic knee testing was performed, including CT and MRI (see Figures 3 and 4). Three days after admission, the patient underwent open reduction and internal fixation (plating) of the right medial, proximal tibia (see Figure 5). He has done very well since without issue.
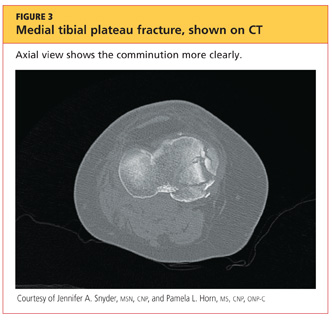
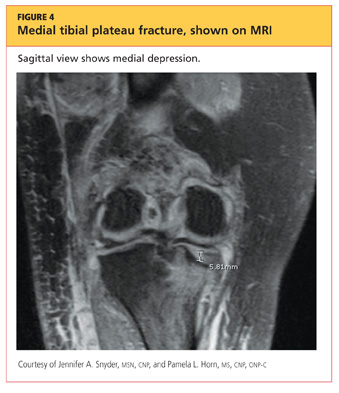
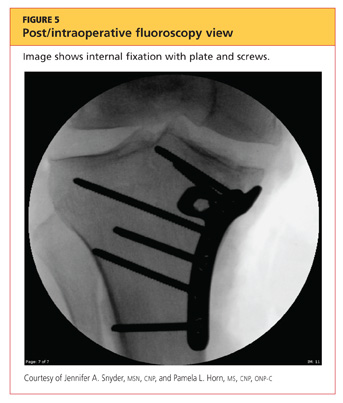
DISCUSSION
Fractures of the tibial plateau occur along the articular, or joint, surface of the proximal tibia. The plateau consists of lateral and medial condylar surfaces. These concave structures function as an articulation point for the cartilaginous menisci and the femoral condyles.4 The medial plateau and condyle are stronger than those of the lateral side, and therefore are less commonly fractured. An elevated intercondylar eminence divides the lateral and medial plateaus, providing an attachment site for the cruciate ligaments.3
The Schatzker classification system1 is most commonly used to describe the types of tibial plateau fractures (as seen in Figure 22,3). Schatzker et al1 divided these injuries into six categories, according to the impact of increased energy exerted onto the bone; the rising classification numbers indicate an increase in complexity and severity and usually a worsening prognosis.
The type I fracture represents a split fracture of the lateral plateau. Typically, a fracture of this type has depression or displacement measuring less than 4 mm.
Type II tibial plateau fractures, the most common Schatzker injury, are lateral plateau fractures with depression noted at the split. Not always evident on plain radiographs, this depression can often be overlooked, and the injury mistaken for a type I fracture. The depression is measured vertically from the lower edge of the medial plateau to the lowest depression point of the lateral plateau.5
Type III fractures, the least common among the Schatzker injuries, are described as pure depression fractures of the lateral plateau. These fractures do not have an appreciable “split” along the plateau and are usually found in older patients with osteopenia.2
The Schatzker type IV injury is a medial fracture with displacement or depression to a portion of the plateau. The fracture may be split or comminuted and may originate in the intercondylar area.
Type V fractures, also known as “bicondylar fractures,” affect both the lateral and medial plateau. An inverted “Y” pattern is frequently seen, and there may be additional involvement of the intercondylar eminence. Type V fractures differ from type VI injuries in that there is no disturbance of the metaphyseal-diaphyseal connection. Thus, type VI fractures also include a transverse component that separates the condyles (metaphysis) of the bone from the shaft (diaphysis). Wide variation is seen among type VI fractures.5
Assessment and Diagnosis
Originally termed “fender fractures” due to their frequent association with automobile injuries, fractures of the tibial plateau account for 1% of all fractures and 8% of fractures in elderly patients.6 Tibial plateau fractures occur when varus or valgus force is combined with axial loading. The fracture itself occurs when the femoral condyle is driven into the lateral or medial plateau. Bicondylar injuries occur when rigorous axial force is sustained in a fully extended knee.
Injuries may also include those of the ligaments or menisci, resulting in joint instability. Patients may present with generalized knee pain or difficulty bearing weight after sustaining injuries, such as being struck in a motor vehicle accident, being tackled, or falling from some height.4
Evaluation of a patient with a suspected tibial plateau fracture begins with a detailed history and thorough physical examination. Details regarding the mechanism of injury help to predict the pattern of the fracture and may indicate whether a more focused neurovascular exam is warranted. Low-energy injuries (often seen with Schatzker types I to III) or twisting injuries yield low suspicion for neurovascular injury or compartment syndrome. However, high-energy injuries (seen often with Schatzker types IV through VI) have a greater likelihood of resulting in complicated injuries that must be urgently or emergently treated.5
The popliteal artery is bound posteriorly and distally to the tibial plateau, and the peroneal nerve is located laterally and positioned around the fibular head. It is essential to assess for the popliteal pulse, as well as lateral lower-extremity sensation and the patient’s ability to dorsiflex. Along with motor and neurovascular injuries, presentation with a painful, strikingly swollen knee and difficulty bearing weight may indicate a hemarthrosis. Soft tissue injuries over the knee resulting from direct trauma may require a saline arthrogram to rule out communication into the joint. Furthermore, a thorough ligamentous exam of the knee is helpful in determining the extent of the injuries.3
Compartment syndrome is a serious, emergent complication that can occur with tibial plateau fractures, especially those sustained during high-energy trauma.7 The health care provider must perform serial exams of the lower extremity to assess for classic signs of compartment syndrome. Are the compartments tense or noncompressible? Does the patient have pain with passive stretch or with range of motion of the lower extremity? Is there pallor or paresthesia to the affected limb? Is the pulse weak or absent? Presence of any of the aforementioned symptoms should prompt a high suspicion for compartment syndrome, and the patient must be sent to an emergency department for urgent evaluation.5
Treatment/Rehabilitation
For Schatzker types I through III, intervention focuses on the articular cartilage examination and repair. Type IV injuries often include corresponding damage to the popliteal artery and/or peroneal nerve, and types V and VI often have such overlying soft tissue damage that temporary placement of an external fixation device is required before definitive surgical intervention can be performed.8
However, it should be noted that conservative versus surgical treatment is often debated among surgeons for treatment of Schatzker fractures. The management of a tibial plateau fracture depends on the physical demands and health of the patient, the severity of the fracture, the stability of the joint, and the surgeon’s skill set and preferences.4 Operative intervention is generally indicated for fractures with depressions greater than 2 mm (although some surgeons allow up to 1 cm of depression), fractures with joint instability, or open fractures. Injuries with concern for vascular injury or compartment syndrome are also treated both operatively and emergently. Postoperatively, patients will remain non–weight-bearing for eight to 12 weeks after surgery, and in the interim, depending on the surgeon’s preference, may or may not engage in active or passive range of motion of the knee.
Advocates of open reduction and internal fixation (ORIF) argue that this method allows for the fracture reduction and anatomic alignment to be directly examined, but they also acknowledge that this approach compromises a great deal of soft tissue surrounding the proximal tibia.9,10
In order to reduce soft tissue damage, some surgeons favor external fixation. Initial use of this surgical technique results in minimal soft tissue swelling and allows early range of motion. While the external fixation device is in place, there is a risk for pin site infection, and proper site care must be provided.6,11
Generally, the treatment of tibial plateau fractures is considered successful when the fracture reduction is sustained, the patient’s functional capacity and axial loading are restored, and the articular surface is reconstructed. As a rule, nonoperative treatment is reserved for tibial plateau fractures that are minimally depressed or nondisplaced, or for patients with advanced osteoporosis. Under these circumstances, after a non–weight-bearing period of four to eight weeks, patients will begin to perform protected and partial weight bearing using a hinged knee brace.2 Early active range of motion, along with isometric exercises to strengthen the quadriceps, is recommended.
Whether surgical or conservative treatment is chosen, complications of tibial plateau fractures include knee stiffness, wound breakdown and infection, malunion or nonunion, vascular or neurologic injury, prominent or painful hardware, or avascular necrosis of fragmented bone pieces.4
CONCLUSION
The primary care practitioner must never overlook patients’ complaints of knee pain, especially after varus or valgus stress injuries or axial loading injuries to the knee. The patient may be able to ambulate; however, ordering a radiograph is an easy method for evaluation and for ruling out tibial plateau injuries. If there is any question regarding the presence of fracture with plain radiographs and/or the clinical exam warrants it, CT is an appropriate second diagnostic intervention.
Should a tibial plateau fracture present in a primary care or urgent care setting, thorough examination of neurovascular status and risk for compartment syndrome must be done urgently, followed by a referral to an orthopedic surgeon or emergency department.
REFERENCES
1. Schatzker J, McBroom R, Bruce D. The tibial plateau fracture: the Toronto experience, 1968–1975. Clin Orthop Relat Res. 1979;(138): 94-104.
2. Marsh JL. Tibial plateau fractures. In: Bucholz RW, Court-Brown CM, Heckman HD, Tornetta P. Rockwood and Green’s Fractures in Adults. 7th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:1780-1831.
3. Egol K, Koval KJ, Zuckerman JD. Tibial plateau. In: Egol K, Koval KJ, Zuckerman JD. Handbook of Fractures. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2010:455-463.
4. Fenton PP, Porter KK. Tibial plateau fractures: a review. Trauma. 2011;13(3):181-187.
5. Markhardt BK, Gross JM, Monu JU. Schatzker classification of tibial plateau fractures: use of CT and MR imaging improves assessment. Radiographics. 2009;29(2):585-597.
6. Lewis C. Does the mode of fixation of tibial plateau fractures, i.e. external fixation versus internal fixation, influence the time to union? A systematic review of the literature. Eur J Orthopaed Surg Traumatol. 2008;18(5):365-370.
7. Weinlein J, Schmidt A. Acute compartment syndrome in tibial plateau fractures—beware! J Knee Surg. 2010;31(1):9-16.
8. te Stroet MA, Holla M, Biert J, van Kampen A. The value of CT scan compared to plain radiographs for the classification and treatment plan in tibial plateau fractures. Emerg Radiol. 2011;18(4):279-283.
9. Musahl V, Tarkin I, Kobbe P, et al. New trends and techniques in open reduction and internal fixation of fractures of the tibial plateau. J Bone Joint Surg Br. 2009;91(4):426-433.
10. Toro-Arbelaez JB, Gardner MJ, Shindle MK, et al. Open reduction and internal fixation of intraarticular tibial plateau nonunions. Injury. 2007;38(3):378-383.
11. Marsh JL, Smith ST, Do TT. External fixation and limited internal fixation for complex fractures of the tibial plateau. J Bone Joint Surg Am. 1995;77(5):661-673.
A 30-year-old man sustained traumatic amputations of three of his left fingers while at work. A heavy object fell when a supporting chain snapped; although he moved quickly, three of his left distal fingers were caught under the object. He was flown to a hospital for definitive hand care.
During the preadmission history and physical, it was noted that the patient had mild right knee pain in addition to his finger injuries. He had experienced no head injury and no loss of consciousness or other complaints. He did not remember injuring his leg, although he said it might have been struck by the falling object; all he could remember was the injury to his fingers.
On physical exam, the only abnormality other than the man’s traumatic finger amputations was mild right knee edema and a small bruised area medially. Initially, he complained of mild pain on palpation and moderate pain with passive range of motion, but range of motion was intact. His pain was worse at the proximal, medial tibial area, and he had mild lateral mid-calf tenderness though no bruising. Distally, his right lower extremity motor and sensory function were intact, and he had no open wounds or skin breakdown. He had 2+ dorsalis pedis pulse and 1+ posterior tibial pulse. The toes were pink and warm with brisk capillary refill. All compartments were soft and compressible.
Upon review of his plain radiographs (three views of the right knee), the patient was noted to have a severely comminuted medial tibial plateau fracture that extended to the midline in the region of the tibial spine, with mild depression of the fracture fragments measuring about 6 mm (see Figures 1a, 1b, and 1c). This would translate into a Schatzker IV classification type1 fracture (see Figure 22,3).
The man was admitted and underwent emergent surgery on his injured left fingers that night. Further diagnostic knee testing was performed, including CT and MRI (see Figures 3 and 4). Three days after admission, the patient underwent open reduction and internal fixation (plating) of the right medial, proximal tibia (see Figure 5). He has done very well since without issue.
DISCUSSION
Fractures of the tibial plateau occur along the articular, or joint, surface of the proximal tibia. The plateau consists of lateral and medial condylar surfaces. These concave structures function as an articulation point for the cartilaginous menisci and the femoral condyles.4 The medial plateau and condyle are stronger than those of the lateral side, and therefore are less commonly fractured. An elevated intercondylar eminence divides the lateral and medial plateaus, providing an attachment site for the cruciate ligaments.3
The Schatzker classification system1 is most commonly used to describe the types of tibial plateau fractures (as seen in Figure 22,3). Schatzker et al1 divided these injuries into six categories, according to the impact of increased energy exerted onto the bone; the rising classification numbers indicate an increase in complexity and severity and usually a worsening prognosis.
The type I fracture represents a split fracture of the lateral plateau. Typically, a fracture of this type has depression or displacement measuring less than 4 mm.
Type II tibial plateau fractures, the most common Schatzker injury, are lateral plateau fractures with depression noted at the split. Not always evident on plain radiographs, this depression can often be overlooked, and the injury mistaken for a type I fracture. The depression is measured vertically from the lower edge of the medial plateau to the lowest depression point of the lateral plateau.5
Type III fractures, the least common among the Schatzker injuries, are described as pure depression fractures of the lateral plateau. These fractures do not have an appreciable “split” along the plateau and are usually found in older patients with osteopenia.2
The Schatzker type IV injury is a medial fracture with displacement or depression to a portion of the plateau. The fracture may be split or comminuted and may originate in the intercondylar area.
Type V fractures, also known as “bicondylar fractures,” affect both the lateral and medial plateau. An inverted “Y” pattern is frequently seen, and there may be additional involvement of the intercondylar eminence. Type V fractures differ from type VI injuries in that there is no disturbance of the metaphyseal-diaphyseal connection. Thus, type VI fractures also include a transverse component that separates the condyles (metaphysis) of the bone from the shaft (diaphysis). Wide variation is seen among type VI fractures.5
Assessment and Diagnosis
Originally termed “fender fractures” due to their frequent association with automobile injuries, fractures of the tibial plateau account for 1% of all fractures and 8% of fractures in elderly patients.6 Tibial plateau fractures occur when varus or valgus force is combined with axial loading. The fracture itself occurs when the femoral condyle is driven into the lateral or medial plateau. Bicondylar injuries occur when rigorous axial force is sustained in a fully extended knee.
Injuries may also include those of the ligaments or menisci, resulting in joint instability. Patients may present with generalized knee pain or difficulty bearing weight after sustaining injuries, such as being struck in a motor vehicle accident, being tackled, or falling from some height.4
Evaluation of a patient with a suspected tibial plateau fracture begins with a detailed history and thorough physical examination. Details regarding the mechanism of injury help to predict the pattern of the fracture and may indicate whether a more focused neurovascular exam is warranted. Low-energy injuries (often seen with Schatzker types I to III) or twisting injuries yield low suspicion for neurovascular injury or compartment syndrome. However, high-energy injuries (seen often with Schatzker types IV through VI) have a greater likelihood of resulting in complicated injuries that must be urgently or emergently treated.5
The popliteal artery is bound posteriorly and distally to the tibial plateau, and the peroneal nerve is located laterally and positioned around the fibular head. It is essential to assess for the popliteal pulse, as well as lateral lower-extremity sensation and the patient’s ability to dorsiflex. Along with motor and neurovascular injuries, presentation with a painful, strikingly swollen knee and difficulty bearing weight may indicate a hemarthrosis. Soft tissue injuries over the knee resulting from direct trauma may require a saline arthrogram to rule out communication into the joint. Furthermore, a thorough ligamentous exam of the knee is helpful in determining the extent of the injuries.3
Compartment syndrome is a serious, emergent complication that can occur with tibial plateau fractures, especially those sustained during high-energy trauma.7 The health care provider must perform serial exams of the lower extremity to assess for classic signs of compartment syndrome. Are the compartments tense or noncompressible? Does the patient have pain with passive stretch or with range of motion of the lower extremity? Is there pallor or paresthesia to the affected limb? Is the pulse weak or absent? Presence of any of the aforementioned symptoms should prompt a high suspicion for compartment syndrome, and the patient must be sent to an emergency department for urgent evaluation.5
Treatment/Rehabilitation
For Schatzker types I through III, intervention focuses on the articular cartilage examination and repair. Type IV injuries often include corresponding damage to the popliteal artery and/or peroneal nerve, and types V and VI often have such overlying soft tissue damage that temporary placement of an external fixation device is required before definitive surgical intervention can be performed.8
However, it should be noted that conservative versus surgical treatment is often debated among surgeons for treatment of Schatzker fractures. The management of a tibial plateau fracture depends on the physical demands and health of the patient, the severity of the fracture, the stability of the joint, and the surgeon’s skill set and preferences.4 Operative intervention is generally indicated for fractures with depressions greater than 2 mm (although some surgeons allow up to 1 cm of depression), fractures with joint instability, or open fractures. Injuries with concern for vascular injury or compartment syndrome are also treated both operatively and emergently. Postoperatively, patients will remain non–weight-bearing for eight to 12 weeks after surgery, and in the interim, depending on the surgeon’s preference, may or may not engage in active or passive range of motion of the knee.
Advocates of open reduction and internal fixation (ORIF) argue that this method allows for the fracture reduction and anatomic alignment to be directly examined, but they also acknowledge that this approach compromises a great deal of soft tissue surrounding the proximal tibia.9,10
In order to reduce soft tissue damage, some surgeons favor external fixation. Initial use of this surgical technique results in minimal soft tissue swelling and allows early range of motion. While the external fixation device is in place, there is a risk for pin site infection, and proper site care must be provided.6,11
Generally, the treatment of tibial plateau fractures is considered successful when the fracture reduction is sustained, the patient’s functional capacity and axial loading are restored, and the articular surface is reconstructed. As a rule, nonoperative treatment is reserved for tibial plateau fractures that are minimally depressed or nondisplaced, or for patients with advanced osteoporosis. Under these circumstances, after a non–weight-bearing period of four to eight weeks, patients will begin to perform protected and partial weight bearing using a hinged knee brace.2 Early active range of motion, along with isometric exercises to strengthen the quadriceps, is recommended.
Whether surgical or conservative treatment is chosen, complications of tibial plateau fractures include knee stiffness, wound breakdown and infection, malunion or nonunion, vascular or neurologic injury, prominent or painful hardware, or avascular necrosis of fragmented bone pieces.4
CONCLUSION
The primary care practitioner must never overlook patients’ complaints of knee pain, especially after varus or valgus stress injuries or axial loading injuries to the knee. The patient may be able to ambulate; however, ordering a radiograph is an easy method for evaluation and for ruling out tibial plateau injuries. If there is any question regarding the presence of fracture with plain radiographs and/or the clinical exam warrants it, CT is an appropriate second diagnostic intervention.
Should a tibial plateau fracture present in a primary care or urgent care setting, thorough examination of neurovascular status and risk for compartment syndrome must be done urgently, followed by a referral to an orthopedic surgeon or emergency department.
REFERENCES
1. Schatzker J, McBroom R, Bruce D. The tibial plateau fracture: the Toronto experience, 1968–1975. Clin Orthop Relat Res. 1979;(138): 94-104.
2. Marsh JL. Tibial plateau fractures. In: Bucholz RW, Court-Brown CM, Heckman HD, Tornetta P. Rockwood and Green’s Fractures in Adults. 7th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:1780-1831.
3. Egol K, Koval KJ, Zuckerman JD. Tibial plateau. In: Egol K, Koval KJ, Zuckerman JD. Handbook of Fractures. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2010:455-463.
4. Fenton PP, Porter KK. Tibial plateau fractures: a review. Trauma. 2011;13(3):181-187.
5. Markhardt BK, Gross JM, Monu JU. Schatzker classification of tibial plateau fractures: use of CT and MR imaging improves assessment. Radiographics. 2009;29(2):585-597.
6. Lewis C. Does the mode of fixation of tibial plateau fractures, i.e. external fixation versus internal fixation, influence the time to union? A systematic review of the literature. Eur J Orthopaed Surg Traumatol. 2008;18(5):365-370.
7. Weinlein J, Schmidt A. Acute compartment syndrome in tibial plateau fractures—beware! J Knee Surg. 2010;31(1):9-16.
8. te Stroet MA, Holla M, Biert J, van Kampen A. The value of CT scan compared to plain radiographs for the classification and treatment plan in tibial plateau fractures. Emerg Radiol. 2011;18(4):279-283.
9. Musahl V, Tarkin I, Kobbe P, et al. New trends and techniques in open reduction and internal fixation of fractures of the tibial plateau. J Bone Joint Surg Br. 2009;91(4):426-433.
10. Toro-Arbelaez JB, Gardner MJ, Shindle MK, et al. Open reduction and internal fixation of intraarticular tibial plateau nonunions. Injury. 2007;38(3):378-383.
11. Marsh JL, Smith ST, Do TT. External fixation and limited internal fixation for complex fractures of the tibial plateau. J Bone Joint Surg Am. 1995;77(5):661-673.
A 30-year-old man sustained traumatic amputations of three of his left fingers while at work. A heavy object fell when a supporting chain snapped; although he moved quickly, three of his left distal fingers were caught under the object. He was flown to a hospital for definitive hand care.
During the preadmission history and physical, it was noted that the patient had mild right knee pain in addition to his finger injuries. He had experienced no head injury and no loss of consciousness or other complaints. He did not remember injuring his leg, although he said it might have been struck by the falling object; all he could remember was the injury to his fingers.
On physical exam, the only abnormality other than the man’s traumatic finger amputations was mild right knee edema and a small bruised area medially. Initially, he complained of mild pain on palpation and moderate pain with passive range of motion, but range of motion was intact. His pain was worse at the proximal, medial tibial area, and he had mild lateral mid-calf tenderness though no bruising. Distally, his right lower extremity motor and sensory function were intact, and he had no open wounds or skin breakdown. He had 2+ dorsalis pedis pulse and 1+ posterior tibial pulse. The toes were pink and warm with brisk capillary refill. All compartments were soft and compressible.
Upon review of his plain radiographs (three views of the right knee), the patient was noted to have a severely comminuted medial tibial plateau fracture that extended to the midline in the region of the tibial spine, with mild depression of the fracture fragments measuring about 6 mm (see Figures 1a, 1b, and 1c). This would translate into a Schatzker IV classification type1 fracture (see Figure 22,3).
The man was admitted and underwent emergent surgery on his injured left fingers that night. Further diagnostic knee testing was performed, including CT and MRI (see Figures 3 and 4). Three days after admission, the patient underwent open reduction and internal fixation (plating) of the right medial, proximal tibia (see Figure 5). He has done very well since without issue.
DISCUSSION
Fractures of the tibial plateau occur along the articular, or joint, surface of the proximal tibia. The plateau consists of lateral and medial condylar surfaces. These concave structures function as an articulation point for the cartilaginous menisci and the femoral condyles.4 The medial plateau and condyle are stronger than those of the lateral side, and therefore are less commonly fractured. An elevated intercondylar eminence divides the lateral and medial plateaus, providing an attachment site for the cruciate ligaments.3
The Schatzker classification system1 is most commonly used to describe the types of tibial plateau fractures (as seen in Figure 22,3). Schatzker et al1 divided these injuries into six categories, according to the impact of increased energy exerted onto the bone; the rising classification numbers indicate an increase in complexity and severity and usually a worsening prognosis.
The type I fracture represents a split fracture of the lateral plateau. Typically, a fracture of this type has depression or displacement measuring less than 4 mm.
Type II tibial plateau fractures, the most common Schatzker injury, are lateral plateau fractures with depression noted at the split. Not always evident on plain radiographs, this depression can often be overlooked, and the injury mistaken for a type I fracture. The depression is measured vertically from the lower edge of the medial plateau to the lowest depression point of the lateral plateau.5
Type III fractures, the least common among the Schatzker injuries, are described as pure depression fractures of the lateral plateau. These fractures do not have an appreciable “split” along the plateau and are usually found in older patients with osteopenia.2
The Schatzker type IV injury is a medial fracture with displacement or depression to a portion of the plateau. The fracture may be split or comminuted and may originate in the intercondylar area.
Type V fractures, also known as “bicondylar fractures,” affect both the lateral and medial plateau. An inverted “Y” pattern is frequently seen, and there may be additional involvement of the intercondylar eminence. Type V fractures differ from type VI injuries in that there is no disturbance of the metaphyseal-diaphyseal connection. Thus, type VI fractures also include a transverse component that separates the condyles (metaphysis) of the bone from the shaft (diaphysis). Wide variation is seen among type VI fractures.5
Assessment and Diagnosis
Originally termed “fender fractures” due to their frequent association with automobile injuries, fractures of the tibial plateau account for 1% of all fractures and 8% of fractures in elderly patients.6 Tibial plateau fractures occur when varus or valgus force is combined with axial loading. The fracture itself occurs when the femoral condyle is driven into the lateral or medial plateau. Bicondylar injuries occur when rigorous axial force is sustained in a fully extended knee.
Injuries may also include those of the ligaments or menisci, resulting in joint instability. Patients may present with generalized knee pain or difficulty bearing weight after sustaining injuries, such as being struck in a motor vehicle accident, being tackled, or falling from some height.4
Evaluation of a patient with a suspected tibial plateau fracture begins with a detailed history and thorough physical examination. Details regarding the mechanism of injury help to predict the pattern of the fracture and may indicate whether a more focused neurovascular exam is warranted. Low-energy injuries (often seen with Schatzker types I to III) or twisting injuries yield low suspicion for neurovascular injury or compartment syndrome. However, high-energy injuries (seen often with Schatzker types IV through VI) have a greater likelihood of resulting in complicated injuries that must be urgently or emergently treated.5
The popliteal artery is bound posteriorly and distally to the tibial plateau, and the peroneal nerve is located laterally and positioned around the fibular head. It is essential to assess for the popliteal pulse, as well as lateral lower-extremity sensation and the patient’s ability to dorsiflex. Along with motor and neurovascular injuries, presentation with a painful, strikingly swollen knee and difficulty bearing weight may indicate a hemarthrosis. Soft tissue injuries over the knee resulting from direct trauma may require a saline arthrogram to rule out communication into the joint. Furthermore, a thorough ligamentous exam of the knee is helpful in determining the extent of the injuries.3
Compartment syndrome is a serious, emergent complication that can occur with tibial plateau fractures, especially those sustained during high-energy trauma.7 The health care provider must perform serial exams of the lower extremity to assess for classic signs of compartment syndrome. Are the compartments tense or noncompressible? Does the patient have pain with passive stretch or with range of motion of the lower extremity? Is there pallor or paresthesia to the affected limb? Is the pulse weak or absent? Presence of any of the aforementioned symptoms should prompt a high suspicion for compartment syndrome, and the patient must be sent to an emergency department for urgent evaluation.5
Treatment/Rehabilitation
For Schatzker types I through III, intervention focuses on the articular cartilage examination and repair. Type IV injuries often include corresponding damage to the popliteal artery and/or peroneal nerve, and types V and VI often have such overlying soft tissue damage that temporary placement of an external fixation device is required before definitive surgical intervention can be performed.8
However, it should be noted that conservative versus surgical treatment is often debated among surgeons for treatment of Schatzker fractures. The management of a tibial plateau fracture depends on the physical demands and health of the patient, the severity of the fracture, the stability of the joint, and the surgeon’s skill set and preferences.4 Operative intervention is generally indicated for fractures with depressions greater than 2 mm (although some surgeons allow up to 1 cm of depression), fractures with joint instability, or open fractures. Injuries with concern for vascular injury or compartment syndrome are also treated both operatively and emergently. Postoperatively, patients will remain non–weight-bearing for eight to 12 weeks after surgery, and in the interim, depending on the surgeon’s preference, may or may not engage in active or passive range of motion of the knee.
Advocates of open reduction and internal fixation (ORIF) argue that this method allows for the fracture reduction and anatomic alignment to be directly examined, but they also acknowledge that this approach compromises a great deal of soft tissue surrounding the proximal tibia.9,10
In order to reduce soft tissue damage, some surgeons favor external fixation. Initial use of this surgical technique results in minimal soft tissue swelling and allows early range of motion. While the external fixation device is in place, there is a risk for pin site infection, and proper site care must be provided.6,11
Generally, the treatment of tibial plateau fractures is considered successful when the fracture reduction is sustained, the patient’s functional capacity and axial loading are restored, and the articular surface is reconstructed. As a rule, nonoperative treatment is reserved for tibial plateau fractures that are minimally depressed or nondisplaced, or for patients with advanced osteoporosis. Under these circumstances, after a non–weight-bearing period of four to eight weeks, patients will begin to perform protected and partial weight bearing using a hinged knee brace.2 Early active range of motion, along with isometric exercises to strengthen the quadriceps, is recommended.
Whether surgical or conservative treatment is chosen, complications of tibial plateau fractures include knee stiffness, wound breakdown and infection, malunion or nonunion, vascular or neurologic injury, prominent or painful hardware, or avascular necrosis of fragmented bone pieces.4
CONCLUSION
The primary care practitioner must never overlook patients’ complaints of knee pain, especially after varus or valgus stress injuries or axial loading injuries to the knee. The patient may be able to ambulate; however, ordering a radiograph is an easy method for evaluation and for ruling out tibial plateau injuries. If there is any question regarding the presence of fracture with plain radiographs and/or the clinical exam warrants it, CT is an appropriate second diagnostic intervention.
Should a tibial plateau fracture present in a primary care or urgent care setting, thorough examination of neurovascular status and risk for compartment syndrome must be done urgently, followed by a referral to an orthopedic surgeon or emergency department.
REFERENCES
1. Schatzker J, McBroom R, Bruce D. The tibial plateau fracture: the Toronto experience, 1968–1975. Clin Orthop Relat Res. 1979;(138): 94-104.
2. Marsh JL. Tibial plateau fractures. In: Bucholz RW, Court-Brown CM, Heckman HD, Tornetta P. Rockwood and Green’s Fractures in Adults. 7th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:1780-1831.
3. Egol K, Koval KJ, Zuckerman JD. Tibial plateau. In: Egol K, Koval KJ, Zuckerman JD. Handbook of Fractures. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2010:455-463.
4. Fenton PP, Porter KK. Tibial plateau fractures: a review. Trauma. 2011;13(3):181-187.
5. Markhardt BK, Gross JM, Monu JU. Schatzker classification of tibial plateau fractures: use of CT and MR imaging improves assessment. Radiographics. 2009;29(2):585-597.
6. Lewis C. Does the mode of fixation of tibial plateau fractures, i.e. external fixation versus internal fixation, influence the time to union? A systematic review of the literature. Eur J Orthopaed Surg Traumatol. 2008;18(5):365-370.
7. Weinlein J, Schmidt A. Acute compartment syndrome in tibial plateau fractures—beware! J Knee Surg. 2010;31(1):9-16.
8. te Stroet MA, Holla M, Biert J, van Kampen A. The value of CT scan compared to plain radiographs for the classification and treatment plan in tibial plateau fractures. Emerg Radiol. 2011;18(4):279-283.
9. Musahl V, Tarkin I, Kobbe P, et al. New trends and techniques in open reduction and internal fixation of fractures of the tibial plateau. J Bone Joint Surg Br. 2009;91(4):426-433.
10. Toro-Arbelaez JB, Gardner MJ, Shindle MK, et al. Open reduction and internal fixation of intraarticular tibial plateau nonunions. Injury. 2007;38(3):378-383.
11. Marsh JL, Smith ST, Do TT. External fixation and limited internal fixation for complex fractures of the tibial plateau. J Bone Joint Surg Am. 1995;77(5):661-673.
Grand Rounds: Man, 61, With Painful Oral Ulcerations
A 61-year-old man, who had recently emigrated from the Ukraine, presented to his primary care provider with a chief complaint of painful oral lesions and weight loss. The patient described the gradual onset of a severe sore throat and mouth pain three months earlier. Originally, he attributed his symptoms to an upper respiratory infection but became concerned when his symptoms did not resolve.
He reported that the pain had worsened over time and that he was now barely able to swallow solid food or tolerate acidic beverages due to considerable discomfort. His son, who accompanied him to the appointment, had also noted weight loss.
The patient denied any concomitant symptoms, including fever, cough, night sweats, fatigue, lymphadenopathy, abdominal pain, diarrhea, melena, or concomitant rash. His medical history was remarkable only for stage 1 hypertension, which had been well controlled on hydrochlorothiazide 12.5 mg/d for the previous three years. However, the patient had received only minimal preventive health care while living in the Ukraine. His family history was unknown.
One week earlier, the patient had seen a dentist complaining of mouth pain, and was referred to an oral medicine specialist; this specialist, in turn, referred the patient to a primary care nurse practitioner for lab work to confirm the suspected diagnosis of pemphigus vulgaris.
On physical examination, the patient appeared older than his stated age. He was a thin, mildly ill–appearing man, afebrile and normotensive, with heart rate and respirations within normal limits. However, intraoral examination revealed multiple oropharyngeal ulcerations of varying size on a base of erythematous and swollen mucosa on the inside of the man’s cheek and palatal and buccal mucosa (see Figure 1). On his upper back, two round, crusted blisters were noted in isolation (Figure 2). The remaining findings in the physical examination were unremarkable.
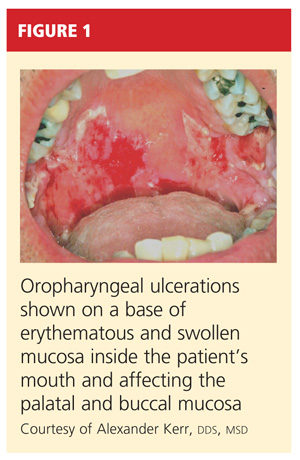
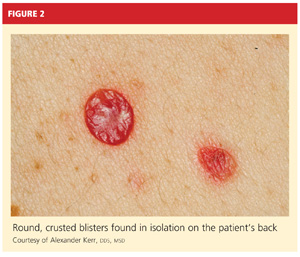
Based on the patient’s physical exam findings and clinical guideline recommendations regarding chronic oral ulcerations of unknown etiology,1,2 the patient was scheduled for a cytologic smear to be performed by oral medicine, followed by a gingival biopsy for a direct immunofluorescence test and routine histopathology.3 Unfortunately, due to extensive involvement and concern for possible mucosal shredding, an oral biopsy was not deemed possible.
However, the oral medicine specialist, because he strongly suspected pemphigus vulgaris, recommended testing for circulating autoantibodies against the antigens desmogleins 1 and/or 3 in the epidermis, which are responsible for cellular adhesion. (A positive test result supports, but does not confirm, a diagnosis of pemphigus vulgaris.4)
Additionally, baseline labs were performed for signs of systemic illness, including infection, anemia, and liver and kidney disease. Frequent monitoring was conducted for steroid-induced symptoms of elevated blood sugars; the primary care provider was responsible for monitoring the patient for weight gain and steroid-induced psychosis. The patient was referred to gastroenterology for a colonoscopy to ensure that his weight loss and anorexia were not the result of gastrointestinal malignancy. However, the patient declined this test.
DISCUSSION
Painful oral lesions can have numerous etiologies of varying severity and complexity, including herpes simplex virus infection, aphthae, lichen planus, erythema multiforme, squamous cell and other oral carcinomas, primary HIV infection, lupus, and pemphigus. Differentiating among these conditions requires a careful medical history and complete physical exam.5
Pemphigus vulgaris (PV) is the most common variant of pemphigus, a group of chronic autoimmune diseases that cause blistering and ulceration of the mucous membranes and the skin.6 From the Greek pemphix (bubble), PV is more common in people of Ashkenazi Jewish or Mediterranean descent,6,7 usually occurs in middle-aged and older persons, and occurs about 1.5 times more commonly in women than men.5,7 Until the introduction of systemic steroids, pemphigus was often a fatal disease. Significant mortality still exists, mainly as a result of infection or adverse reactions to medication therapy.5
In patients with PV, flaccid bullae are formed on the skin in a process called acantholysis, in which epidermal cells lose their ability to adhere to one another. This results in rapidly expanding, thin-walled blisters on the oral mucosa, scalp, face, axillae, and groin. The blisters burst easily, leaving irregularly shaped, painful ulcerations.4 Painful oral mucosal membrane erosions are the first presenting sign of PV and often the only sign for an average of five months before other skin lesions develop.3 These lesions are noninfectious.
To make a definitive diagnosis of PV, clinical lesions must be present, with a confirmation of histologic findings, acantholysis on biopsy, and a confirmation of autoantibodies present in tissue and/or serum.4 (For proposed detailed diagnostic criteria, see table4,8.)
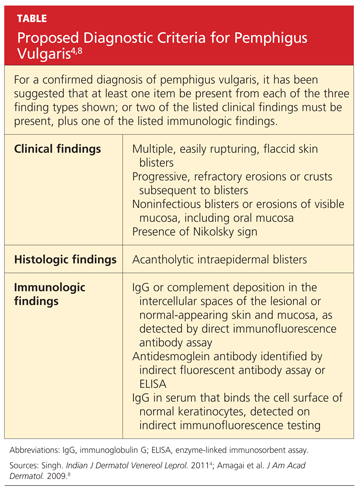
Initial misdiagnoses, which often lead to delayed or incorrect treatment, usually include aphthous stomatitis, gingivostomatitis, erythema multiforme, erosive lichen planus, herpes simplex virus, and/or oral candidiasis.3
Common Differentials
Herpes simplex virus. Affecting between 15% and 45% of the population, herpes simplex virus (HSV) infection, also known as cold sores, is the most common cause of recurrent oral ulcers.9 HSV is transmitted through direct contact with lesions or via viral shedding. Primary infection, which may occur with flu-like symptoms, causes the sudden onset of multiple clustered vesicles on an erythematous base that quickly ulcerate and crust. Recurrent infections tend to be less severe and are accompanied by minimal systemic symptoms.10
Diagnosis is usually made through history and physical exam. However, diagnostic tests, including Tzanck smears, biopsy, polymerase chain reaction (PCR) assay, and/or viral isolation in culture, are sometimes used to confirm a suspected case.10
Oral lichen planus (OLP). This is a common, chronic, mucocutaneous inflammatory disease of unknown etiology that affects skin and mucous membranes of the mouth, including the buccal mucosa, tongue, and/or gums. These lesions are noninfectious and are an immunologically mediated disease. Stress, anxiety, genetic predisposition, NSAID use, antihypertensive medications (eg, captopril, enalapril, propranolol; considered an oral lichenoid drug reaction), and altered cell-mediated immune response have been considered possible causative factors.11,12 Recent reports suggest an association between hepatitis C virus and OLP.13
Affecting about 4% of the general population, and more predominate in perimenopausal women, OLP lesions appear as white, lacey patches; red, swollen tissues; or open sores, most commonly on the inside of the mouth bilaterally. Patients will present with complaints of burning, roughness, or pain in the mouth, dry mouth, sensitivity to hot or spicy foods, and difficulty swallowing if the throat is involved. Diagnosis is based on history and physical examination and often a confirmatory biopsy. Topical high-potency corticosteroids are generally first-line therapy, with systemic medications such as oral prednisone used to treat severe cases.14,15
Oral candidiasis. Up to 80% of healthy individuals carry Candida albicans in their mouths16; this pathogen accounts for about half of all cases of oral candidiasis (oral thrush). Oral infections occur only with an underlying predisposing condition in the host. Oral thrush presents as creamy white lesions on the oral mucosa; a diagnostic feature is that the plaques can be removed to reveal an erythematous base.16,17
In the chronic form of candidiasis, the mucosal surface is bright red and smooth. When the tongue is involved, it may appear dry, fissured, or cracked. Patients may report a dry mouth, burning pain, and difficulty eating. Infection can be confirmed with periodic acid-Schiff staining of a smear to detect candidal hyphae.9
Use of antifungal creams and lozenges, as well as improved oral hygiene, will often lead to resolution of symptoms.9 Management of any associated underlying conditions, such as diabetes, asthma requiring long-term use of steroid inhalers, or infection with HIV/AIDS, is essential.18
Oral aphthae. Recurrent aphthous ulcers (commonly called canker sores; also referred to as recurrent aphthous stomatitis [RAS]) are a common oral condition. Etiology is unknown and most likely multifactorial, with a strong genetic tendency and multiple predisposing factors, including trauma, stress, food allergies, hormones, and smoking.19 Certain chronic illnesses, including celiac disease, inflammatory bowel disease (IBD), HIV, and neutropenia may also predispose patients to RAS or RAS-like syndromes.
Aphthous ulcers are classified as minor or major. Minor aphthae, which account for 90% of RAS cases, present as single or multiple, small, oval or round ulcers with an erythematous halo on the buccal or labial mucosa or tongue.19 The ulcers last 7 to 10 days and heal spontaneously without scarring.
Diagnosis, based on history and clinical presentation, may include evaluation for systemic causes of oral ulcers. Treatment for both minor and major apthae is palliative, with mainstays including topical corticosteroids, mouth rinses, and, in severe cases, thalidomide, although randomized controlled trials have not shown this agent to be of benefit.9
Treatment for Pemphigus Vulgaris
The outcome goal for management of pemphigus is to achieve and maintain remission. This includes the epithelialization of all skin and mucosal lesions, prevention of relapse, minimization of adverse treatment effects, and successful withdrawal of therapeutic medications.20
The response to treatment varies greatly among patients, as the optimal therapeutic regimen for pemphigus is unknown.20 Systemic glucocorticoids are considered the gold standard of treatment and management, but their use has been associated with several adverse effects, including weight gain and elevated blood sugar levels. Recently, the combination of IV immune globulin and biological therapies (eg, rituximab) that target specific molecules in the inflammatory process have been demonstrated as effective in cases of refractory pemphigus.21,22
PATIENT MANAGEMENT AND OUTCOME
Several referrals were made, including dermatology, for its familiarity with autoimmune diseases of the skin. There, the patient was fully examined and found to have a small truncal lesion compatible with PV. He was referred to an otolaryngologist for a nasal endoscopy to determine the extent of the lesions. They were found to extend far beyond his oral cavity into his esophagus.
Based on a positive enzyme-linked immunosorbent assay (ELISA) for PV antibodies, a cytologic smear with acantholytic cells, and a classic clinical presentation of PV, the patient was started on prednisone 80 mg/d with azathioprine 50 mg/d for the first 14 days.23,24 He responded quickly to these oral medications and underwent a confirmatory oral biopsy within a few weeks.
After several months, the patient was slowly titrated down to lower maintenance doses of prednisone and azathioprine. Now in remission, he continues to receive collaborative management from oral medicine, dermatology, and a nurse practitioner–managed primary care practice. Health care maintenance has included appropriate vaccination and discussion regarding prostate cancer screening, per 2010 guidelines from the US Preventive Services Task Force.25
CONCLUSION
Since the differential diagnosis for pemphigus vulgaris is extensive and the diagnostic criteria are exacting, many affected patients are undiagnosed or misdiagnosed, with a resulting delay in effective treatment. It is important for the primary care clinician to undertake a frequent review of common oral infections, particularly those with similar presentations.
The authors extend their thanks to Alexander Kerr, DDS, MSD, Clinical Associate Professor, Department of Oral and Maxillary Pathology, Radiology and Medicine, New York University College of Dentistry, for the images included in this article and for Dr. Kerr’s clinical expertise and partnership.
REFERENCES
1. Sciubba JJ. Oral mucosal diseases in the office setting. Part II. Oral lichen planus, pemphigus vulgaris, and mucosal pemphigoid. Gen Dent. 2007;55(5):464-476.
2. Muñoz-Corcuera M, Esparza-Gómez G, González-Moles MA, Bascones-Martínez A. Oral ulcers: clinical aspects. A tool for dermatologists. Part II. Chronic ulcers. Clin Exp Dermatol. 2009; 34(4):456-461.
3. Dagistan S, Goregen M, Miloglu O, Cakur B. Oral pemphigus vulgaris: a case report with review of the literature. J Oral Sci. 2008;50(3):359-362.
4. Singh S. Evidence-based treatments for pemphigus vulgaris, pemphigus foliaceus and bullous pemphigoid: a systematic review. Indian J Dermatol Venereol Leprol. 2011;77(4):456-469.
5. Ohta M, Osawa S, Endo H, et al. Pemphigus vulgaris confined to the gingiva: a case report. Int J Dent. 2011;2011:207153. Epub 2011 May 11.
6. Mignona MD, Fortuna G, Leuci S. Oral pemphigus. Minerva Stomatol. 2009;58(10):501-518.
7. Mimouni D, Bar H, Gdalevich M, et al. Pemphigus: analysis of epidemiological factors in 155 patients. J Eur Acad Dermatol Venereol. 2008; 22(10):1232-1235.
8. Amagai M, Ikeda S, Shimizu H, et al. A randomized double-blind trial of intravenous immunoglobulin for pemphigus. J Am Acad Dermatol. 2009;60(4):595-603.
9. Gonsalves WC, Chi AC, Neville BW. Common oral lesions: Part I. Superficial mucosal lesions. Am Fam Physician. 2007;75(4):501-507.
10. Fatahzadeh M, Schwartz R. Human herpes simplex virus infections: epidemiology, pathogenesis, symptomatology, diagnosis, and management. J Am Acad Dermatol. 2007;57(5):737-763.
11. Sugerman PB, Savage NW. Oral lichen planus: causes, diagnosis and management. Aust Dent J. 2002;47(4):290-297.
12. Kaomongkolgit R. Oral lichenoid drug reaction associated with antihypertensive and hypoglycemic drugs. J Drugs Dermatol. 2010;9(1):73-75.
13. Petti S, Rabiei M, De Luca M, Scully C. The magnitude of the association between hepatitis C virus infection and oral lichen planus: meta-analysis and case control study. Odontology. 2011;99(2):168-178.
14. Usatine RP, Tinitigan M. Diagnosis and treatment of lichen planus. Am Fam Physician. 2011;84(1): 53-60.
15. Thongprasom K, Carrozzo M, Furness S, Lodi G. Interventions for treating oral lichen planus. Cochrane Database Syst Rev. 2011 Jul 6; (7):CD001168.
16. Giannini PJ, Shetty KV. Diagnosis and management of oral candidiasis. Otolaryngol Clin North Am. 2011;44(1):231-240, vii.
17. Lynch DP. Oral candidiasis. History, classification, and clinical presentation. Oral Surg Oral Med Oral Pathol. 1994;78(2):189-193.
18. Williams D, Lewis M. Pathogenesis and treatment of oral candidosis. J Oral Microbiol. 2011 Jan 28;3. doi: 10.3402/jom.v3i0.5771.
19. Scully C, Challacombe SJ. Pemphigus vulgaris: update on etiopathogenesis, oral manifestations, and management. Crit Rev Oral Biol Med. 2002;13(5):397-408.
20. Martin LK, Werth V, Villanueva E, Murrell DF. A systematic review of randomized controlled trials for pemphigus vulgaris and pemphigus foliaceus. J Am Acad Dermatol. 2011;64(5):903-908.
21. Joly P, Mouquet H, Roujeau JC, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357(6):545-552.
22. Diaz LA. Rituximab and pemphigus: a therapeutic advance. N Engl J Med. 2007;357(6):605-607.
23. Anstey AV, Wakelin S, Reynolds NJ. Guidelines for prescribing azathioprine in dermatology. Br J Dermatol. 2004;151(6):1123-1132.
24. Chams-Davatchi C, Daneshpazhooh M. Prednisolone dosage in pemphigus vulgaris. J Am Acad Dermatol. 2005;53(3):547.
25. Agency for Healthcare Research and Quality. Guide to Clinical Preventive Services, 2010-2011: recommendations of the US Preventive Services Task Force. AHRQ Publication No. 10-05145, September 2010. www.ahrq.gov/clinic/pocketgd1011/pocketgd1011.pdf. Accessed January 23, 2012.
A 61-year-old man, who had recently emigrated from the Ukraine, presented to his primary care provider with a chief complaint of painful oral lesions and weight loss. The patient described the gradual onset of a severe sore throat and mouth pain three months earlier. Originally, he attributed his symptoms to an upper respiratory infection but became concerned when his symptoms did not resolve.
He reported that the pain had worsened over time and that he was now barely able to swallow solid food or tolerate acidic beverages due to considerable discomfort. His son, who accompanied him to the appointment, had also noted weight loss.
The patient denied any concomitant symptoms, including fever, cough, night sweats, fatigue, lymphadenopathy, abdominal pain, diarrhea, melena, or concomitant rash. His medical history was remarkable only for stage 1 hypertension, which had been well controlled on hydrochlorothiazide 12.5 mg/d for the previous three years. However, the patient had received only minimal preventive health care while living in the Ukraine. His family history was unknown.
One week earlier, the patient had seen a dentist complaining of mouth pain, and was referred to an oral medicine specialist; this specialist, in turn, referred the patient to a primary care nurse practitioner for lab work to confirm the suspected diagnosis of pemphigus vulgaris.
On physical examination, the patient appeared older than his stated age. He was a thin, mildly ill–appearing man, afebrile and normotensive, with heart rate and respirations within normal limits. However, intraoral examination revealed multiple oropharyngeal ulcerations of varying size on a base of erythematous and swollen mucosa on the inside of the man’s cheek and palatal and buccal mucosa (see Figure 1). On his upper back, two round, crusted blisters were noted in isolation (Figure 2). The remaining findings in the physical examination were unremarkable.
Based on the patient’s physical exam findings and clinical guideline recommendations regarding chronic oral ulcerations of unknown etiology,1,2 the patient was scheduled for a cytologic smear to be performed by oral medicine, followed by a gingival biopsy for a direct immunofluorescence test and routine histopathology.3 Unfortunately, due to extensive involvement and concern for possible mucosal shredding, an oral biopsy was not deemed possible.
However, the oral medicine specialist, because he strongly suspected pemphigus vulgaris, recommended testing for circulating autoantibodies against the antigens desmogleins 1 and/or 3 in the epidermis, which are responsible for cellular adhesion. (A positive test result supports, but does not confirm, a diagnosis of pemphigus vulgaris.4)
Additionally, baseline labs were performed for signs of systemic illness, including infection, anemia, and liver and kidney disease. Frequent monitoring was conducted for steroid-induced symptoms of elevated blood sugars; the primary care provider was responsible for monitoring the patient for weight gain and steroid-induced psychosis. The patient was referred to gastroenterology for a colonoscopy to ensure that his weight loss and anorexia were not the result of gastrointestinal malignancy. However, the patient declined this test.
DISCUSSION
Painful oral lesions can have numerous etiologies of varying severity and complexity, including herpes simplex virus infection, aphthae, lichen planus, erythema multiforme, squamous cell and other oral carcinomas, primary HIV infection, lupus, and pemphigus. Differentiating among these conditions requires a careful medical history and complete physical exam.5
Pemphigus vulgaris (PV) is the most common variant of pemphigus, a group of chronic autoimmune diseases that cause blistering and ulceration of the mucous membranes and the skin.6 From the Greek pemphix (bubble), PV is more common in people of Ashkenazi Jewish or Mediterranean descent,6,7 usually occurs in middle-aged and older persons, and occurs about 1.5 times more commonly in women than men.5,7 Until the introduction of systemic steroids, pemphigus was often a fatal disease. Significant mortality still exists, mainly as a result of infection or adverse reactions to medication therapy.5
In patients with PV, flaccid bullae are formed on the skin in a process called acantholysis, in which epidermal cells lose their ability to adhere to one another. This results in rapidly expanding, thin-walled blisters on the oral mucosa, scalp, face, axillae, and groin. The blisters burst easily, leaving irregularly shaped, painful ulcerations.4 Painful oral mucosal membrane erosions are the first presenting sign of PV and often the only sign for an average of five months before other skin lesions develop.3 These lesions are noninfectious.
To make a definitive diagnosis of PV, clinical lesions must be present, with a confirmation of histologic findings, acantholysis on biopsy, and a confirmation of autoantibodies present in tissue and/or serum.4 (For proposed detailed diagnostic criteria, see table4,8.)
Initial misdiagnoses, which often lead to delayed or incorrect treatment, usually include aphthous stomatitis, gingivostomatitis, erythema multiforme, erosive lichen planus, herpes simplex virus, and/or oral candidiasis.3
Common Differentials
Herpes simplex virus. Affecting between 15% and 45% of the population, herpes simplex virus (HSV) infection, also known as cold sores, is the most common cause of recurrent oral ulcers.9 HSV is transmitted through direct contact with lesions or via viral shedding. Primary infection, which may occur with flu-like symptoms, causes the sudden onset of multiple clustered vesicles on an erythematous base that quickly ulcerate and crust. Recurrent infections tend to be less severe and are accompanied by minimal systemic symptoms.10
Diagnosis is usually made through history and physical exam. However, diagnostic tests, including Tzanck smears, biopsy, polymerase chain reaction (PCR) assay, and/or viral isolation in culture, are sometimes used to confirm a suspected case.10
Oral lichen planus (OLP). This is a common, chronic, mucocutaneous inflammatory disease of unknown etiology that affects skin and mucous membranes of the mouth, including the buccal mucosa, tongue, and/or gums. These lesions are noninfectious and are an immunologically mediated disease. Stress, anxiety, genetic predisposition, NSAID use, antihypertensive medications (eg, captopril, enalapril, propranolol; considered an oral lichenoid drug reaction), and altered cell-mediated immune response have been considered possible causative factors.11,12 Recent reports suggest an association between hepatitis C virus and OLP.13
Affecting about 4% of the general population, and more predominate in perimenopausal women, OLP lesions appear as white, lacey patches; red, swollen tissues; or open sores, most commonly on the inside of the mouth bilaterally. Patients will present with complaints of burning, roughness, or pain in the mouth, dry mouth, sensitivity to hot or spicy foods, and difficulty swallowing if the throat is involved. Diagnosis is based on history and physical examination and often a confirmatory biopsy. Topical high-potency corticosteroids are generally first-line therapy, with systemic medications such as oral prednisone used to treat severe cases.14,15
Oral candidiasis. Up to 80% of healthy individuals carry Candida albicans in their mouths16; this pathogen accounts for about half of all cases of oral candidiasis (oral thrush). Oral infections occur only with an underlying predisposing condition in the host. Oral thrush presents as creamy white lesions on the oral mucosa; a diagnostic feature is that the plaques can be removed to reveal an erythematous base.16,17
In the chronic form of candidiasis, the mucosal surface is bright red and smooth. When the tongue is involved, it may appear dry, fissured, or cracked. Patients may report a dry mouth, burning pain, and difficulty eating. Infection can be confirmed with periodic acid-Schiff staining of a smear to detect candidal hyphae.9
Use of antifungal creams and lozenges, as well as improved oral hygiene, will often lead to resolution of symptoms.9 Management of any associated underlying conditions, such as diabetes, asthma requiring long-term use of steroid inhalers, or infection with HIV/AIDS, is essential.18
Oral aphthae. Recurrent aphthous ulcers (commonly called canker sores; also referred to as recurrent aphthous stomatitis [RAS]) are a common oral condition. Etiology is unknown and most likely multifactorial, with a strong genetic tendency and multiple predisposing factors, including trauma, stress, food allergies, hormones, and smoking.19 Certain chronic illnesses, including celiac disease, inflammatory bowel disease (IBD), HIV, and neutropenia may also predispose patients to RAS or RAS-like syndromes.
Aphthous ulcers are classified as minor or major. Minor aphthae, which account for 90% of RAS cases, present as single or multiple, small, oval or round ulcers with an erythematous halo on the buccal or labial mucosa or tongue.19 The ulcers last 7 to 10 days and heal spontaneously without scarring.
Diagnosis, based on history and clinical presentation, may include evaluation for systemic causes of oral ulcers. Treatment for both minor and major apthae is palliative, with mainstays including topical corticosteroids, mouth rinses, and, in severe cases, thalidomide, although randomized controlled trials have not shown this agent to be of benefit.9
Treatment for Pemphigus Vulgaris
The outcome goal for management of pemphigus is to achieve and maintain remission. This includes the epithelialization of all skin and mucosal lesions, prevention of relapse, minimization of adverse treatment effects, and successful withdrawal of therapeutic medications.20
The response to treatment varies greatly among patients, as the optimal therapeutic regimen for pemphigus is unknown.20 Systemic glucocorticoids are considered the gold standard of treatment and management, but their use has been associated with several adverse effects, including weight gain and elevated blood sugar levels. Recently, the combination of IV immune globulin and biological therapies (eg, rituximab) that target specific molecules in the inflammatory process have been demonstrated as effective in cases of refractory pemphigus.21,22
PATIENT MANAGEMENT AND OUTCOME
Several referrals were made, including dermatology, for its familiarity with autoimmune diseases of the skin. There, the patient was fully examined and found to have a small truncal lesion compatible with PV. He was referred to an otolaryngologist for a nasal endoscopy to determine the extent of the lesions. They were found to extend far beyond his oral cavity into his esophagus.
Based on a positive enzyme-linked immunosorbent assay (ELISA) for PV antibodies, a cytologic smear with acantholytic cells, and a classic clinical presentation of PV, the patient was started on prednisone 80 mg/d with azathioprine 50 mg/d for the first 14 days.23,24 He responded quickly to these oral medications and underwent a confirmatory oral biopsy within a few weeks.
After several months, the patient was slowly titrated down to lower maintenance doses of prednisone and azathioprine. Now in remission, he continues to receive collaborative management from oral medicine, dermatology, and a nurse practitioner–managed primary care practice. Health care maintenance has included appropriate vaccination and discussion regarding prostate cancer screening, per 2010 guidelines from the US Preventive Services Task Force.25
CONCLUSION
Since the differential diagnosis for pemphigus vulgaris is extensive and the diagnostic criteria are exacting, many affected patients are undiagnosed or misdiagnosed, with a resulting delay in effective treatment. It is important for the primary care clinician to undertake a frequent review of common oral infections, particularly those with similar presentations.
The authors extend their thanks to Alexander Kerr, DDS, MSD, Clinical Associate Professor, Department of Oral and Maxillary Pathology, Radiology and Medicine, New York University College of Dentistry, for the images included in this article and for Dr. Kerr’s clinical expertise and partnership.
REFERENCES
1. Sciubba JJ. Oral mucosal diseases in the office setting. Part II. Oral lichen planus, pemphigus vulgaris, and mucosal pemphigoid. Gen Dent. 2007;55(5):464-476.
2. Muñoz-Corcuera M, Esparza-Gómez G, González-Moles MA, Bascones-Martínez A. Oral ulcers: clinical aspects. A tool for dermatologists. Part II. Chronic ulcers. Clin Exp Dermatol. 2009; 34(4):456-461.
3. Dagistan S, Goregen M, Miloglu O, Cakur B. Oral pemphigus vulgaris: a case report with review of the literature. J Oral Sci. 2008;50(3):359-362.
4. Singh S. Evidence-based treatments for pemphigus vulgaris, pemphigus foliaceus and bullous pemphigoid: a systematic review. Indian J Dermatol Venereol Leprol. 2011;77(4):456-469.
5. Ohta M, Osawa S, Endo H, et al. Pemphigus vulgaris confined to the gingiva: a case report. Int J Dent. 2011;2011:207153. Epub 2011 May 11.
6. Mignona MD, Fortuna G, Leuci S. Oral pemphigus. Minerva Stomatol. 2009;58(10):501-518.
7. Mimouni D, Bar H, Gdalevich M, et al. Pemphigus: analysis of epidemiological factors in 155 patients. J Eur Acad Dermatol Venereol. 2008; 22(10):1232-1235.
8. Amagai M, Ikeda S, Shimizu H, et al. A randomized double-blind trial of intravenous immunoglobulin for pemphigus. J Am Acad Dermatol. 2009;60(4):595-603.
9. Gonsalves WC, Chi AC, Neville BW. Common oral lesions: Part I. Superficial mucosal lesions. Am Fam Physician. 2007;75(4):501-507.
10. Fatahzadeh M, Schwartz R. Human herpes simplex virus infections: epidemiology, pathogenesis, symptomatology, diagnosis, and management. J Am Acad Dermatol. 2007;57(5):737-763.
11. Sugerman PB, Savage NW. Oral lichen planus: causes, diagnosis and management. Aust Dent J. 2002;47(4):290-297.
12. Kaomongkolgit R. Oral lichenoid drug reaction associated with antihypertensive and hypoglycemic drugs. J Drugs Dermatol. 2010;9(1):73-75.
13. Petti S, Rabiei M, De Luca M, Scully C. The magnitude of the association between hepatitis C virus infection and oral lichen planus: meta-analysis and case control study. Odontology. 2011;99(2):168-178.
14. Usatine RP, Tinitigan M. Diagnosis and treatment of lichen planus. Am Fam Physician. 2011;84(1): 53-60.
15. Thongprasom K, Carrozzo M, Furness S, Lodi G. Interventions for treating oral lichen planus. Cochrane Database Syst Rev. 2011 Jul 6; (7):CD001168.
16. Giannini PJ, Shetty KV. Diagnosis and management of oral candidiasis. Otolaryngol Clin North Am. 2011;44(1):231-240, vii.
17. Lynch DP. Oral candidiasis. History, classification, and clinical presentation. Oral Surg Oral Med Oral Pathol. 1994;78(2):189-193.
18. Williams D, Lewis M. Pathogenesis and treatment of oral candidosis. J Oral Microbiol. 2011 Jan 28;3. doi: 10.3402/jom.v3i0.5771.
19. Scully C, Challacombe SJ. Pemphigus vulgaris: update on etiopathogenesis, oral manifestations, and management. Crit Rev Oral Biol Med. 2002;13(5):397-408.
20. Martin LK, Werth V, Villanueva E, Murrell DF. A systematic review of randomized controlled trials for pemphigus vulgaris and pemphigus foliaceus. J Am Acad Dermatol. 2011;64(5):903-908.
21. Joly P, Mouquet H, Roujeau JC, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357(6):545-552.
22. Diaz LA. Rituximab and pemphigus: a therapeutic advance. N Engl J Med. 2007;357(6):605-607.
23. Anstey AV, Wakelin S, Reynolds NJ. Guidelines for prescribing azathioprine in dermatology. Br J Dermatol. 2004;151(6):1123-1132.
24. Chams-Davatchi C, Daneshpazhooh M. Prednisolone dosage in pemphigus vulgaris. J Am Acad Dermatol. 2005;53(3):547.
25. Agency for Healthcare Research and Quality. Guide to Clinical Preventive Services, 2010-2011: recommendations of the US Preventive Services Task Force. AHRQ Publication No. 10-05145, September 2010. www.ahrq.gov/clinic/pocketgd1011/pocketgd1011.pdf. Accessed January 23, 2012.
A 61-year-old man, who had recently emigrated from the Ukraine, presented to his primary care provider with a chief complaint of painful oral lesions and weight loss. The patient described the gradual onset of a severe sore throat and mouth pain three months earlier. Originally, he attributed his symptoms to an upper respiratory infection but became concerned when his symptoms did not resolve.
He reported that the pain had worsened over time and that he was now barely able to swallow solid food or tolerate acidic beverages due to considerable discomfort. His son, who accompanied him to the appointment, had also noted weight loss.
The patient denied any concomitant symptoms, including fever, cough, night sweats, fatigue, lymphadenopathy, abdominal pain, diarrhea, melena, or concomitant rash. His medical history was remarkable only for stage 1 hypertension, which had been well controlled on hydrochlorothiazide 12.5 mg/d for the previous three years. However, the patient had received only minimal preventive health care while living in the Ukraine. His family history was unknown.
One week earlier, the patient had seen a dentist complaining of mouth pain, and was referred to an oral medicine specialist; this specialist, in turn, referred the patient to a primary care nurse practitioner for lab work to confirm the suspected diagnosis of pemphigus vulgaris.
On physical examination, the patient appeared older than his stated age. He was a thin, mildly ill–appearing man, afebrile and normotensive, with heart rate and respirations within normal limits. However, intraoral examination revealed multiple oropharyngeal ulcerations of varying size on a base of erythematous and swollen mucosa on the inside of the man’s cheek and palatal and buccal mucosa (see Figure 1). On his upper back, two round, crusted blisters were noted in isolation (Figure 2). The remaining findings in the physical examination were unremarkable.
Based on the patient’s physical exam findings and clinical guideline recommendations regarding chronic oral ulcerations of unknown etiology,1,2 the patient was scheduled for a cytologic smear to be performed by oral medicine, followed by a gingival biopsy for a direct immunofluorescence test and routine histopathology.3 Unfortunately, due to extensive involvement and concern for possible mucosal shredding, an oral biopsy was not deemed possible.
However, the oral medicine specialist, because he strongly suspected pemphigus vulgaris, recommended testing for circulating autoantibodies against the antigens desmogleins 1 and/or 3 in the epidermis, which are responsible for cellular adhesion. (A positive test result supports, but does not confirm, a diagnosis of pemphigus vulgaris.4)
Additionally, baseline labs were performed for signs of systemic illness, including infection, anemia, and liver and kidney disease. Frequent monitoring was conducted for steroid-induced symptoms of elevated blood sugars; the primary care provider was responsible for monitoring the patient for weight gain and steroid-induced psychosis. The patient was referred to gastroenterology for a colonoscopy to ensure that his weight loss and anorexia were not the result of gastrointestinal malignancy. However, the patient declined this test.
DISCUSSION
Painful oral lesions can have numerous etiologies of varying severity and complexity, including herpes simplex virus infection, aphthae, lichen planus, erythema multiforme, squamous cell and other oral carcinomas, primary HIV infection, lupus, and pemphigus. Differentiating among these conditions requires a careful medical history and complete physical exam.5
Pemphigus vulgaris (PV) is the most common variant of pemphigus, a group of chronic autoimmune diseases that cause blistering and ulceration of the mucous membranes and the skin.6 From the Greek pemphix (bubble), PV is more common in people of Ashkenazi Jewish or Mediterranean descent,6,7 usually occurs in middle-aged and older persons, and occurs about 1.5 times more commonly in women than men.5,7 Until the introduction of systemic steroids, pemphigus was often a fatal disease. Significant mortality still exists, mainly as a result of infection or adverse reactions to medication therapy.5
In patients with PV, flaccid bullae are formed on the skin in a process called acantholysis, in which epidermal cells lose their ability to adhere to one another. This results in rapidly expanding, thin-walled blisters on the oral mucosa, scalp, face, axillae, and groin. The blisters burst easily, leaving irregularly shaped, painful ulcerations.4 Painful oral mucosal membrane erosions are the first presenting sign of PV and often the only sign for an average of five months before other skin lesions develop.3 These lesions are noninfectious.
To make a definitive diagnosis of PV, clinical lesions must be present, with a confirmation of histologic findings, acantholysis on biopsy, and a confirmation of autoantibodies present in tissue and/or serum.4 (For proposed detailed diagnostic criteria, see table4,8.)
Initial misdiagnoses, which often lead to delayed or incorrect treatment, usually include aphthous stomatitis, gingivostomatitis, erythema multiforme, erosive lichen planus, herpes simplex virus, and/or oral candidiasis.3
Common Differentials
Herpes simplex virus. Affecting between 15% and 45% of the population, herpes simplex virus (HSV) infection, also known as cold sores, is the most common cause of recurrent oral ulcers.9 HSV is transmitted through direct contact with lesions or via viral shedding. Primary infection, which may occur with flu-like symptoms, causes the sudden onset of multiple clustered vesicles on an erythematous base that quickly ulcerate and crust. Recurrent infections tend to be less severe and are accompanied by minimal systemic symptoms.10
Diagnosis is usually made through history and physical exam. However, diagnostic tests, including Tzanck smears, biopsy, polymerase chain reaction (PCR) assay, and/or viral isolation in culture, are sometimes used to confirm a suspected case.10
Oral lichen planus (OLP). This is a common, chronic, mucocutaneous inflammatory disease of unknown etiology that affects skin and mucous membranes of the mouth, including the buccal mucosa, tongue, and/or gums. These lesions are noninfectious and are an immunologically mediated disease. Stress, anxiety, genetic predisposition, NSAID use, antihypertensive medications (eg, captopril, enalapril, propranolol; considered an oral lichenoid drug reaction), and altered cell-mediated immune response have been considered possible causative factors.11,12 Recent reports suggest an association between hepatitis C virus and OLP.13
Affecting about 4% of the general population, and more predominate in perimenopausal women, OLP lesions appear as white, lacey patches; red, swollen tissues; or open sores, most commonly on the inside of the mouth bilaterally. Patients will present with complaints of burning, roughness, or pain in the mouth, dry mouth, sensitivity to hot or spicy foods, and difficulty swallowing if the throat is involved. Diagnosis is based on history and physical examination and often a confirmatory biopsy. Topical high-potency corticosteroids are generally first-line therapy, with systemic medications such as oral prednisone used to treat severe cases.14,15
Oral candidiasis. Up to 80% of healthy individuals carry Candida albicans in their mouths16; this pathogen accounts for about half of all cases of oral candidiasis (oral thrush). Oral infections occur only with an underlying predisposing condition in the host. Oral thrush presents as creamy white lesions on the oral mucosa; a diagnostic feature is that the plaques can be removed to reveal an erythematous base.16,17
In the chronic form of candidiasis, the mucosal surface is bright red and smooth. When the tongue is involved, it may appear dry, fissured, or cracked. Patients may report a dry mouth, burning pain, and difficulty eating. Infection can be confirmed with periodic acid-Schiff staining of a smear to detect candidal hyphae.9
Use of antifungal creams and lozenges, as well as improved oral hygiene, will often lead to resolution of symptoms.9 Management of any associated underlying conditions, such as diabetes, asthma requiring long-term use of steroid inhalers, or infection with HIV/AIDS, is essential.18
Oral aphthae. Recurrent aphthous ulcers (commonly called canker sores; also referred to as recurrent aphthous stomatitis [RAS]) are a common oral condition. Etiology is unknown and most likely multifactorial, with a strong genetic tendency and multiple predisposing factors, including trauma, stress, food allergies, hormones, and smoking.19 Certain chronic illnesses, including celiac disease, inflammatory bowel disease (IBD), HIV, and neutropenia may also predispose patients to RAS or RAS-like syndromes.
Aphthous ulcers are classified as minor or major. Minor aphthae, which account for 90% of RAS cases, present as single or multiple, small, oval or round ulcers with an erythematous halo on the buccal or labial mucosa or tongue.19 The ulcers last 7 to 10 days and heal spontaneously without scarring.
Diagnosis, based on history and clinical presentation, may include evaluation for systemic causes of oral ulcers. Treatment for both minor and major apthae is palliative, with mainstays including topical corticosteroids, mouth rinses, and, in severe cases, thalidomide, although randomized controlled trials have not shown this agent to be of benefit.9
Treatment for Pemphigus Vulgaris
The outcome goal for management of pemphigus is to achieve and maintain remission. This includes the epithelialization of all skin and mucosal lesions, prevention of relapse, minimization of adverse treatment effects, and successful withdrawal of therapeutic medications.20
The response to treatment varies greatly among patients, as the optimal therapeutic regimen for pemphigus is unknown.20 Systemic glucocorticoids are considered the gold standard of treatment and management, but their use has been associated with several adverse effects, including weight gain and elevated blood sugar levels. Recently, the combination of IV immune globulin and biological therapies (eg, rituximab) that target specific molecules in the inflammatory process have been demonstrated as effective in cases of refractory pemphigus.21,22
PATIENT MANAGEMENT AND OUTCOME
Several referrals were made, including dermatology, for its familiarity with autoimmune diseases of the skin. There, the patient was fully examined and found to have a small truncal lesion compatible with PV. He was referred to an otolaryngologist for a nasal endoscopy to determine the extent of the lesions. They were found to extend far beyond his oral cavity into his esophagus.
Based on a positive enzyme-linked immunosorbent assay (ELISA) for PV antibodies, a cytologic smear with acantholytic cells, and a classic clinical presentation of PV, the patient was started on prednisone 80 mg/d with azathioprine 50 mg/d for the first 14 days.23,24 He responded quickly to these oral medications and underwent a confirmatory oral biopsy within a few weeks.
After several months, the patient was slowly titrated down to lower maintenance doses of prednisone and azathioprine. Now in remission, he continues to receive collaborative management from oral medicine, dermatology, and a nurse practitioner–managed primary care practice. Health care maintenance has included appropriate vaccination and discussion regarding prostate cancer screening, per 2010 guidelines from the US Preventive Services Task Force.25
CONCLUSION
Since the differential diagnosis for pemphigus vulgaris is extensive and the diagnostic criteria are exacting, many affected patients are undiagnosed or misdiagnosed, with a resulting delay in effective treatment. It is important for the primary care clinician to undertake a frequent review of common oral infections, particularly those with similar presentations.
The authors extend their thanks to Alexander Kerr, DDS, MSD, Clinical Associate Professor, Department of Oral and Maxillary Pathology, Radiology and Medicine, New York University College of Dentistry, for the images included in this article and for Dr. Kerr’s clinical expertise and partnership.
REFERENCES
1. Sciubba JJ. Oral mucosal diseases in the office setting. Part II. Oral lichen planus, pemphigus vulgaris, and mucosal pemphigoid. Gen Dent. 2007;55(5):464-476.
2. Muñoz-Corcuera M, Esparza-Gómez G, González-Moles MA, Bascones-Martínez A. Oral ulcers: clinical aspects. A tool for dermatologists. Part II. Chronic ulcers. Clin Exp Dermatol. 2009; 34(4):456-461.
3. Dagistan S, Goregen M, Miloglu O, Cakur B. Oral pemphigus vulgaris: a case report with review of the literature. J Oral Sci. 2008;50(3):359-362.
4. Singh S. Evidence-based treatments for pemphigus vulgaris, pemphigus foliaceus and bullous pemphigoid: a systematic review. Indian J Dermatol Venereol Leprol. 2011;77(4):456-469.
5. Ohta M, Osawa S, Endo H, et al. Pemphigus vulgaris confined to the gingiva: a case report. Int J Dent. 2011;2011:207153. Epub 2011 May 11.
6. Mignona MD, Fortuna G, Leuci S. Oral pemphigus. Minerva Stomatol. 2009;58(10):501-518.
7. Mimouni D, Bar H, Gdalevich M, et al. Pemphigus: analysis of epidemiological factors in 155 patients. J Eur Acad Dermatol Venereol. 2008; 22(10):1232-1235.
8. Amagai M, Ikeda S, Shimizu H, et al. A randomized double-blind trial of intravenous immunoglobulin for pemphigus. J Am Acad Dermatol. 2009;60(4):595-603.
9. Gonsalves WC, Chi AC, Neville BW. Common oral lesions: Part I. Superficial mucosal lesions. Am Fam Physician. 2007;75(4):501-507.
10. Fatahzadeh M, Schwartz R. Human herpes simplex virus infections: epidemiology, pathogenesis, symptomatology, diagnosis, and management. J Am Acad Dermatol. 2007;57(5):737-763.
11. Sugerman PB, Savage NW. Oral lichen planus: causes, diagnosis and management. Aust Dent J. 2002;47(4):290-297.
12. Kaomongkolgit R. Oral lichenoid drug reaction associated with antihypertensive and hypoglycemic drugs. J Drugs Dermatol. 2010;9(1):73-75.
13. Petti S, Rabiei M, De Luca M, Scully C. The magnitude of the association between hepatitis C virus infection and oral lichen planus: meta-analysis and case control study. Odontology. 2011;99(2):168-178.
14. Usatine RP, Tinitigan M. Diagnosis and treatment of lichen planus. Am Fam Physician. 2011;84(1): 53-60.
15. Thongprasom K, Carrozzo M, Furness S, Lodi G. Interventions for treating oral lichen planus. Cochrane Database Syst Rev. 2011 Jul 6; (7):CD001168.
16. Giannini PJ, Shetty KV. Diagnosis and management of oral candidiasis. Otolaryngol Clin North Am. 2011;44(1):231-240, vii.
17. Lynch DP. Oral candidiasis. History, classification, and clinical presentation. Oral Surg Oral Med Oral Pathol. 1994;78(2):189-193.
18. Williams D, Lewis M. Pathogenesis and treatment of oral candidosis. J Oral Microbiol. 2011 Jan 28;3. doi: 10.3402/jom.v3i0.5771.
19. Scully C, Challacombe SJ. Pemphigus vulgaris: update on etiopathogenesis, oral manifestations, and management. Crit Rev Oral Biol Med. 2002;13(5):397-408.
20. Martin LK, Werth V, Villanueva E, Murrell DF. A systematic review of randomized controlled trials for pemphigus vulgaris and pemphigus foliaceus. J Am Acad Dermatol. 2011;64(5):903-908.
21. Joly P, Mouquet H, Roujeau JC, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357(6):545-552.
22. Diaz LA. Rituximab and pemphigus: a therapeutic advance. N Engl J Med. 2007;357(6):605-607.
23. Anstey AV, Wakelin S, Reynolds NJ. Guidelines for prescribing azathioprine in dermatology. Br J Dermatol. 2004;151(6):1123-1132.
24. Chams-Davatchi C, Daneshpazhooh M. Prednisolone dosage in pemphigus vulgaris. J Am Acad Dermatol. 2005;53(3):547.
25. Agency for Healthcare Research and Quality. Guide to Clinical Preventive Services, 2010-2011: recommendations of the US Preventive Services Task Force. AHRQ Publication No. 10-05145, September 2010. www.ahrq.gov/clinic/pocketgd1011/pocketgd1011.pdf. Accessed January 23, 2012.
Grand Rounds: Woman, 29, With Persistent Migraine
A 29-year-old woman with a history of frequent migraines presented to her primary care provider for a refill of medication. For the past two years she had been taking rizatriptan 10 mg, but with little relief. She stated that she had continued to experience discrete migraines several days per month, often clustered around menses. The severity of the headaches had negatively affected her work attendance, productivity, and social interactions. She wondered if she should be taking a different kind of medication.
The patient had been diagnosed with migraines at age 12, just prior to menarche. She described her headache as a unilateral, sharp throbbing pain associated with increased sensitivity to light and sound as well as nausea. She denied any history of head trauma. She had no allergies, and the only other medications she was taking at the time were an oral contraceptive (ethinyl estradiol/norgestimate 0.035 mg/0.18 mg with an oral triphasic 21/7 treatment cycle) and fluoxetine 20 mg for depression.
The patient worked daytime hours as a sales representative. She considered herself active, exercised regularly, ate a balanced diet, and slept well. She consumed no more than two to four alcoholic drinks per month and denied the use of herbals, dietary supplements, tobacco, or illegal drugs.
The patient stated that her mother had frequent headaches but had never sought a medical explanation or treatment. She was unaware of any other family history of headaches, and there was no family history of cardiovascular disease. Her sister had been diagnosed with a prolactinoma at age 25. At age 26, the patient had undergone a pituitary protocol MRI of the head with and without contrast, with negative results.
On examination, the patient was alert and oriented with normal vital signs. Her pupils were equal and reactive to light, and no papilledema was evident on fundoscopic examination. The cranial nerves were grossly intact and no other neurologic deficits were appreciated. No carotid bruits were present on cardiovascular exam.
Based on the patient’s history and physical exam, she met the International Classification of Headache Disorders (ICHD-II)1 diagnostic criteria for migraine without aura (1.1). When asked to recall the onset and frequency of attacks she had had in the previous four weeks, she noted that they regularly occurred during her menstrual cycle.
She was subsequently asked to begin a diary to record her headache characteristics, severity, and duration, with days of menstruation noted. The Migraine Disability Assessment (MIDAS) questionnaire2 (see Tables 1 and 22) was performed to measure the migraine attacks’ impact on the patient’s life; her score indicated that the headaches were causing her severe disability.
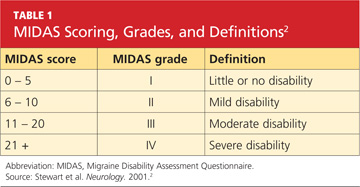
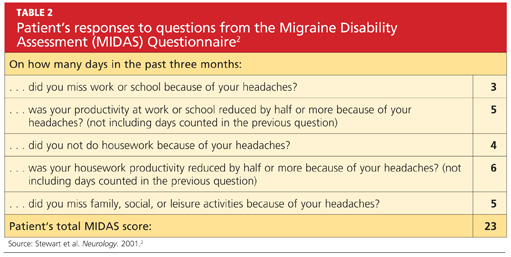
The patient’s abortive migraine medication was changed from rizatriptan 10 mg to the combination sumatriptan/naproxen sodium 85 mg/500 mg. She was instructed to take the initial dose as soon as she noticed signs of an impending migraine and to repeat the dose in two hours if symptoms persisted. The possibility of starting a preventive medication was discussed, but the patient wanted to evaluate her response to the combination triptan/NSAID before considering migraine prophylaxis.
Three months later, the patient returned for follow-up, including a review of her headache diary. She stated that the frequency and intensity of attacks had not decreased; acute treatment with sumatriptan/naproxen sodium made her headaches more bearable but did not ameliorate symptoms. The patient had recorded a detailed account of each migraine which, based on the ICHD-II criteria,1 demonstrated a pattern of headache occurrences consistent with menstrually related migraine. She reported a total of 18 headaches in the previous three months, 12 of which had occurred within the five-day perimenstrual period (see Figure 1).
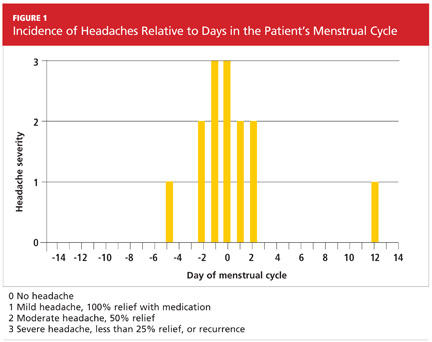
Based on this information and the fact that the patient’s headaches were resistant to previous treatments, it was decided to alter the approach to her migraine management once more. In an effort to limit estrogen fluctuations during her menstrual cycle, her oral contraceptive was changed from ethinyl estradiol/norgestimate to a 12-week placebo-free monophasic regimen of ethinyl estradiol/levonorgestrel 20 mg/90 mcg. For intermittent prophylaxis, she was instructed to take frovatriptan 2.5 mg twice daily, beginning two days prior to the start of menses and continuing through the last day of her cycle. For acute treatment of breakthrough migraines, she was prescribed sumatriptan 20-mg nasal spray to take at the first sign of migraine symptoms and instructed to repeat the dose if the pain persisted or returned.
The patient continued to track her headaches in the diary and was seen in the office after three months of following the revised menstrual migraine management plan. She reported fewer migraines associated with her menstrual cycle and noted that they were less severe and shorter in duration. When she repeated the MIDAS test, her score was reduced from 23 to 10. In the subsequent nine months she has reported a consistent decrease in migraine prevalence and now rarely needs the abortive therapy.
DISCUSSION
Migraine, though commonly encountered in clinical practice, is a complex disorder. For women, migraine headaches have been recognized by the World Health Organization as the 12th leading cause of “life lived with a disabling condition.”3 Pure menstrual migraine and menstrually related migraine will be the focus of discussion here.
Etiology
Menstrually related migraine (comparable to pure menstrual migraine, although the latter is distinguished by occurring only during the perimenstrual period1) is recognized as a distinct type of migraine associated with perimenstrual hormone fluctuations.4 Of women who experience migraine, 42% to 61% can associate their attacks with the perimenstrual period5; this is defined as two days before to three days after the start of menstruation.
It has also been determined that women are more likely to have migraine attacks during the late luteal and early follicular phases (when there is a natural drop in estrogen levels) than in other phases (when estrogen levels are higher).6 Despite clinical evidence to support this estrogen withdrawal theory, the pathophysiology is not completely understood. It is possible that affected women are more sensitive than other women to the decrease in estrogen levels that occurs with menstruation.7
History and Physical Findings of Menstrual Migraines
Almost every woman with perimenstrual migraines reports an absence of aura.7 In the evaluation of headache, the same criteria for migraine without aura pertain to the classifications of pure menstrual migraine (PMM) or menstrually related migraine (MRM).1 Correlation of migraine attacks to the onset of menses is the key finding in the patient history to differentiate menstrual migraine from migraine without aura in women.8 Furthermore, perimenstrual migraines are often of longer duration and more difficult to treat than migraines not associated with hormone fluctuations.9
In order to distinguish between PMM and MRM, it is important to understand that pure menstrual migraine attacks take place exclusively in the five-day perimenstrual window and at no other times of the cycle. The criteria for MRM allow for attacks at other times of the cycle.1
In addition to causing physical pain, menstrual migraines can impact work performance, household activities, and personal relationships. The MIDAS questionnaire is a disability assessment tool that can reveal to the practitioner how migraines have affected the patient’s life over the previous three months.10 This is a useful method to identify patients with disabling migraines, determine their need for treatment, and monitor treatment efficacy.
Diagnosis
Menstrual migraine is a clinical diagnosis made by findings from the patient’s history. The International Headache Society has established specific diagnostic criteria in the ICHD-II for both PMM and MRM.1 An accurate and detailed migraine history is invaluable for the diagnosis of menstrual migraine. Although a formal questionnaire can serve as a good screening tool, it relies on the patient’s ability to recall specific times and dates with accuracy.11 Recall bias can be misleading in any attempt to confirm a diagnosis. The patient’s conscientious use of a daily headache diary or calendar (see Figure 2, for example) can lead to a precise record of the characteristics and timing of migraines, overcoming these obstacles.
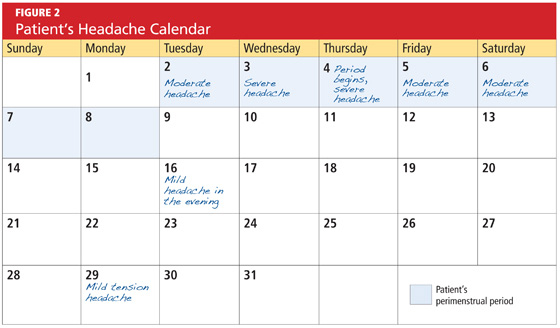
Brain imaging is necessary if the patient’s symptoms suggest a critical etiology that requires immediate diagnosis and management. Red flags include sudden onset of a severe headache, a headache characterized as “the worst headache of the patient’s life,” a change in headache pattern, altered mental status, an abnormal neurologic examination, or fever with neck stiffness.12
Treatment Options for Menstrual Migraine
There is no FDA-approved treatment specific for menstrual migraines; however, medications used for management of nonmenstrual migraines are also those most commonly prescribed for women with menstrual migraine headaches.13 Because these headaches are frequently more severe and of longer duration than nonmenstrual migraine headaches, a combination of intermittent preventive therapy, hormone manipulation, and acute treatment strategies is often necessary.4
Acute therapy is aimed to treat migraine pain quickly and effectively with minimal adverse effects or need for additional medication. Triptans have been the mainstay of menstrual migraine treatment and have been proven effective for both acute attacks and prevention.4 Sumatriptan has a rapid onset of action and may be given orally as a 50- or 100-mg tablet, as a 6-mg subcutaneous injection, or as a 20-mg nasal spray.14
Abortive therapies are most effective when taken at the first sign of an attack. Patients can repeat the dose in two hours if the headache persists or recurs, to a maximum of two doses in 24 hours.15 Rizatriptan is another triptan used for acute treatment of menstrual migraine headaches. Its initial 10-mg dose can be repeated every two hours, to a maximum of 30 mg per 24 hours. NSAIDs, such as naproxen sodium, have also been recommended in acute migraine attacks. They seem to work synergistically with triptans, inhibiting prostaglandin synthesis and blocking neurogenic inflammation.15
Clinical study results have demonstrated superior pain relief and decreased migraine recurrence when a triptan and NSAID are used in combination, compared with use of either medication alone.4 A single-tablet formulation of sumatriptan 85 mg and naproxen sodium 500 mg may be considered for initial therapy in hard-to-treat patients.14
Preventive therapy should be considered when responsiveness to acute treatment is inadequate.4 Nonhormonal intermittent prophylactic treatment is recommended two days prior to the beginning of menses, continuing for five days.16 Longer-acting triptans, such as frovatriptan 2.5 mg and naratriptan 1.0 mg, dosed twice daily, have been demonstrated as effective in clinical trials when used during the perimenstrual period.17,18
The advantage of short-term therapy over daily prophylaxis is the potential to avoid adverse effects seen with continuous exposure to the drug.3 However, successful therapy relies on consistency in menstruation, and therefore may not be ideal for women with irregular cycles or those with coexisting nonmenstrual migraines.16 Estrogen-based therapy is an option for these women and for those who have failed nonhormonal methods.19
The goal of hormone prophylaxis is to prevent or reduce the physiologic decline in estradiol that occurs in the late luteal phase.4 Clinical studies have been conducted using various hormonal strategies to maintain steady estradiol levels, all of which decreased migraine prevalence.19 Estrogen fluctuations can be minimized by eliminating the placebo week in traditional estrogen/progestin oral contraceptives to achieve an extended-cycle regimen, resembling that of the 12-week ethinyl estradiol/levonorgestrel formulation.19
Continuous use of combined oral contraceptives is also an option for relief of menstrual migraine. When cyclic or extended-cycle regimens allow for menses, supplemental estrogen (10- to 20-mg ethinyl estradiol) is recommended during the hormone-free week.14
CONCLUSION
Proper diagnosis of menstrual migraines, using screening tools and the MIDAS questionnaire, can help practitioners provide the most effective migraine management for their patients. The most important step toward a good prognosis is acknowledging menstrual migraine as a unique headache disorder and formulating a precise diagnosis in order to identify individually tailored treatment options. With proper identification and integrated acute and prophylactic treatment, women with menstrual migraines are able to lead a healthier, more satisfying life.
REFERENCES
1. International Headache Society. The International Classification of Headache Disorders. 2nd ed. Cephalalgia. 2004;24(suppl 1):1-160.
2. Stewart WF, Lipton RB, Dowson AJ, Sawyer J. Development and testing of the Migraine Disability Assessment (MIDAS) Questionnaire to assess headache-related disability. Neurology. 2001;56(6 suppl 1):S20-S28.
3. MacGregor EA. Perimenstrual headaches: unmet needs. Curr Pain Headache Rep. 2008;12(6):468-474.
4. Mannix LK. Menstrual-related pain conditions: dysmenorrhea and migraine. J Womens Health (Larchmt). 2008;17(5):879-891.
5. Martin VT. New theories in the pathogenesis of menstrual migraine. Curr Pain Headache Rep. 2008;12(6):453-462.
6. MacGregor EA. Migraine headache in perimenopausal and menopausal women. Curr Pain Headache Rep. 2009;13(5):399-403.
7. Martin VT, Wernke S, Mandell K, et al. Symptoms of premenstrual syndrome and their association with migraine headache. Headache. 2006; 46(1):125-137.
8. Martin VT, Behbehani M. Ovarian hormones and migraine headache: understanding mechanisms and pathogenesis—part 2. Headache. 2006;46(3):365-386.
9. Granella F, Sances G, Allais G, et al. Characteristics of menstrual and nonmenstrual attacks in women with menstrually related migraine referred to headache centres. Cephalalgia. 2004;24(9):707-716.
10. Hutchinson SL, Silberstein SD. Menstrual migraine: case studies of women with estrogen-related headaches. Headache. 2008;48 suppl 3:S131-S141.
11. Tepper SJ, Zatochill M, Szeto M, et al. Development of a simple menstrual migraine screening tool for obstetric and gynecology clinics: the Menstrual Migraine Assessment Tool. Headache. 2008; 48(10):1419-1425.
12. Marcus DA. Focus on primary care diagnosis and management of headache in women. Obstet Gynecol Surv. 1999;54(6):395-402.
13. Tepper SJ. Tailoring management strategies for the patient with menstrual migraine: focus on prevention and treatment. Headache. 2006;46(suppl 2):S61-S68.
14. Lay CL, Payne R. Recognition and treatment of menstrual migraine. Neurologist. 2007;13(4):197-204.
15. Henry KA, Cohen CI. Perimenstrual headache: treatment options. Curr Pain Headache Rep. 2009;13(1):82-88.
16. Calhoun AH. Estrogen-associated migraine. www.uptodate.com/contents/estrogen-associated-migraine. Accessed May 4, 2011.
17. Silberstein SD, Elkind AH, Schreiber C, et al. A randomized trial of frovatriptan for the intermittent prevention of menstrual migraine. Neurology. 2004;63:261-269.
18. Mannix LK, Savani N, Landy S, et al. Efficacy and tolerability of naratriptan for short-term prevention of menstrually related migraine: data from two randomized, double-blind, placebo-controlled studies. Headache. 2007;47(7):1037-1049.
19. Calhoun AH, Hutchinson S. Hormonal therapies for menstrual migraine. Curr Pain Headache Rep. 2009;13(5):381-385.
A 29-year-old woman with a history of frequent migraines presented to her primary care provider for a refill of medication. For the past two years she had been taking rizatriptan 10 mg, but with little relief. She stated that she had continued to experience discrete migraines several days per month, often clustered around menses. The severity of the headaches had negatively affected her work attendance, productivity, and social interactions. She wondered if she should be taking a different kind of medication.
The patient had been diagnosed with migraines at age 12, just prior to menarche. She described her headache as a unilateral, sharp throbbing pain associated with increased sensitivity to light and sound as well as nausea. She denied any history of head trauma. She had no allergies, and the only other medications she was taking at the time were an oral contraceptive (ethinyl estradiol/norgestimate 0.035 mg/0.18 mg with an oral triphasic 21/7 treatment cycle) and fluoxetine 20 mg for depression.
The patient worked daytime hours as a sales representative. She considered herself active, exercised regularly, ate a balanced diet, and slept well. She consumed no more than two to four alcoholic drinks per month and denied the use of herbals, dietary supplements, tobacco, or illegal drugs.
The patient stated that her mother had frequent headaches but had never sought a medical explanation or treatment. She was unaware of any other family history of headaches, and there was no family history of cardiovascular disease. Her sister had been diagnosed with a prolactinoma at age 25. At age 26, the patient had undergone a pituitary protocol MRI of the head with and without contrast, with negative results.
On examination, the patient was alert and oriented with normal vital signs. Her pupils were equal and reactive to light, and no papilledema was evident on fundoscopic examination. The cranial nerves were grossly intact and no other neurologic deficits were appreciated. No carotid bruits were present on cardiovascular exam.
Based on the patient’s history and physical exam, she met the International Classification of Headache Disorders (ICHD-II)1 diagnostic criteria for migraine without aura (1.1). When asked to recall the onset and frequency of attacks she had had in the previous four weeks, she noted that they regularly occurred during her menstrual cycle.
She was subsequently asked to begin a diary to record her headache characteristics, severity, and duration, with days of menstruation noted. The Migraine Disability Assessment (MIDAS) questionnaire2 (see Tables 1 and 22) was performed to measure the migraine attacks’ impact on the patient’s life; her score indicated that the headaches were causing her severe disability.
The patient’s abortive migraine medication was changed from rizatriptan 10 mg to the combination sumatriptan/naproxen sodium 85 mg/500 mg. She was instructed to take the initial dose as soon as she noticed signs of an impending migraine and to repeat the dose in two hours if symptoms persisted. The possibility of starting a preventive medication was discussed, but the patient wanted to evaluate her response to the combination triptan/NSAID before considering migraine prophylaxis.
Three months later, the patient returned for follow-up, including a review of her headache diary. She stated that the frequency and intensity of attacks had not decreased; acute treatment with sumatriptan/naproxen sodium made her headaches more bearable but did not ameliorate symptoms. The patient had recorded a detailed account of each migraine which, based on the ICHD-II criteria,1 demonstrated a pattern of headache occurrences consistent with menstrually related migraine. She reported a total of 18 headaches in the previous three months, 12 of which had occurred within the five-day perimenstrual period (see Figure 1).
Based on this information and the fact that the patient’s headaches were resistant to previous treatments, it was decided to alter the approach to her migraine management once more. In an effort to limit estrogen fluctuations during her menstrual cycle, her oral contraceptive was changed from ethinyl estradiol/norgestimate to a 12-week placebo-free monophasic regimen of ethinyl estradiol/levonorgestrel 20 mg/90 mcg. For intermittent prophylaxis, she was instructed to take frovatriptan 2.5 mg twice daily, beginning two days prior to the start of menses and continuing through the last day of her cycle. For acute treatment of breakthrough migraines, she was prescribed sumatriptan 20-mg nasal spray to take at the first sign of migraine symptoms and instructed to repeat the dose if the pain persisted or returned.
The patient continued to track her headaches in the diary and was seen in the office after three months of following the revised menstrual migraine management plan. She reported fewer migraines associated with her menstrual cycle and noted that they were less severe and shorter in duration. When she repeated the MIDAS test, her score was reduced from 23 to 10. In the subsequent nine months she has reported a consistent decrease in migraine prevalence and now rarely needs the abortive therapy.
DISCUSSION
Migraine, though commonly encountered in clinical practice, is a complex disorder. For women, migraine headaches have been recognized by the World Health Organization as the 12th leading cause of “life lived with a disabling condition.”3 Pure menstrual migraine and menstrually related migraine will be the focus of discussion here.
Etiology
Menstrually related migraine (comparable to pure menstrual migraine, although the latter is distinguished by occurring only during the perimenstrual period1) is recognized as a distinct type of migraine associated with perimenstrual hormone fluctuations.4 Of women who experience migraine, 42% to 61% can associate their attacks with the perimenstrual period5; this is defined as two days before to three days after the start of menstruation.
It has also been determined that women are more likely to have migraine attacks during the late luteal and early follicular phases (when there is a natural drop in estrogen levels) than in other phases (when estrogen levels are higher).6 Despite clinical evidence to support this estrogen withdrawal theory, the pathophysiology is not completely understood. It is possible that affected women are more sensitive than other women to the decrease in estrogen levels that occurs with menstruation.7
History and Physical Findings of Menstrual Migraines
Almost every woman with perimenstrual migraines reports an absence of aura.7 In the evaluation of headache, the same criteria for migraine without aura pertain to the classifications of pure menstrual migraine (PMM) or menstrually related migraine (MRM).1 Correlation of migraine attacks to the onset of menses is the key finding in the patient history to differentiate menstrual migraine from migraine without aura in women.8 Furthermore, perimenstrual migraines are often of longer duration and more difficult to treat than migraines not associated with hormone fluctuations.9
In order to distinguish between PMM and MRM, it is important to understand that pure menstrual migraine attacks take place exclusively in the five-day perimenstrual window and at no other times of the cycle. The criteria for MRM allow for attacks at other times of the cycle.1
In addition to causing physical pain, menstrual migraines can impact work performance, household activities, and personal relationships. The MIDAS questionnaire is a disability assessment tool that can reveal to the practitioner how migraines have affected the patient’s life over the previous three months.10 This is a useful method to identify patients with disabling migraines, determine their need for treatment, and monitor treatment efficacy.
Diagnosis
Menstrual migraine is a clinical diagnosis made by findings from the patient’s history. The International Headache Society has established specific diagnostic criteria in the ICHD-II for both PMM and MRM.1 An accurate and detailed migraine history is invaluable for the diagnosis of menstrual migraine. Although a formal questionnaire can serve as a good screening tool, it relies on the patient’s ability to recall specific times and dates with accuracy.11 Recall bias can be misleading in any attempt to confirm a diagnosis. The patient’s conscientious use of a daily headache diary or calendar (see Figure 2, for example) can lead to a precise record of the characteristics and timing of migraines, overcoming these obstacles.
Brain imaging is necessary if the patient’s symptoms suggest a critical etiology that requires immediate diagnosis and management. Red flags include sudden onset of a severe headache, a headache characterized as “the worst headache of the patient’s life,” a change in headache pattern, altered mental status, an abnormal neurologic examination, or fever with neck stiffness.12
Treatment Options for Menstrual Migraine
There is no FDA-approved treatment specific for menstrual migraines; however, medications used for management of nonmenstrual migraines are also those most commonly prescribed for women with menstrual migraine headaches.13 Because these headaches are frequently more severe and of longer duration than nonmenstrual migraine headaches, a combination of intermittent preventive therapy, hormone manipulation, and acute treatment strategies is often necessary.4
Acute therapy is aimed to treat migraine pain quickly and effectively with minimal adverse effects or need for additional medication. Triptans have been the mainstay of menstrual migraine treatment and have been proven effective for both acute attacks and prevention.4 Sumatriptan has a rapid onset of action and may be given orally as a 50- or 100-mg tablet, as a 6-mg subcutaneous injection, or as a 20-mg nasal spray.14
Abortive therapies are most effective when taken at the first sign of an attack. Patients can repeat the dose in two hours if the headache persists or recurs, to a maximum of two doses in 24 hours.15 Rizatriptan is another triptan used for acute treatment of menstrual migraine headaches. Its initial 10-mg dose can be repeated every two hours, to a maximum of 30 mg per 24 hours. NSAIDs, such as naproxen sodium, have also been recommended in acute migraine attacks. They seem to work synergistically with triptans, inhibiting prostaglandin synthesis and blocking neurogenic inflammation.15
Clinical study results have demonstrated superior pain relief and decreased migraine recurrence when a triptan and NSAID are used in combination, compared with use of either medication alone.4 A single-tablet formulation of sumatriptan 85 mg and naproxen sodium 500 mg may be considered for initial therapy in hard-to-treat patients.14
Preventive therapy should be considered when responsiveness to acute treatment is inadequate.4 Nonhormonal intermittent prophylactic treatment is recommended two days prior to the beginning of menses, continuing for five days.16 Longer-acting triptans, such as frovatriptan 2.5 mg and naratriptan 1.0 mg, dosed twice daily, have been demonstrated as effective in clinical trials when used during the perimenstrual period.17,18
The advantage of short-term therapy over daily prophylaxis is the potential to avoid adverse effects seen with continuous exposure to the drug.3 However, successful therapy relies on consistency in menstruation, and therefore may not be ideal for women with irregular cycles or those with coexisting nonmenstrual migraines.16 Estrogen-based therapy is an option for these women and for those who have failed nonhormonal methods.19
The goal of hormone prophylaxis is to prevent or reduce the physiologic decline in estradiol that occurs in the late luteal phase.4 Clinical studies have been conducted using various hormonal strategies to maintain steady estradiol levels, all of which decreased migraine prevalence.19 Estrogen fluctuations can be minimized by eliminating the placebo week in traditional estrogen/progestin oral contraceptives to achieve an extended-cycle regimen, resembling that of the 12-week ethinyl estradiol/levonorgestrel formulation.19
Continuous use of combined oral contraceptives is also an option for relief of menstrual migraine. When cyclic or extended-cycle regimens allow for menses, supplemental estrogen (10- to 20-mg ethinyl estradiol) is recommended during the hormone-free week.14
CONCLUSION
Proper diagnosis of menstrual migraines, using screening tools and the MIDAS questionnaire, can help practitioners provide the most effective migraine management for their patients. The most important step toward a good prognosis is acknowledging menstrual migraine as a unique headache disorder and formulating a precise diagnosis in order to identify individually tailored treatment options. With proper identification and integrated acute and prophylactic treatment, women with menstrual migraines are able to lead a healthier, more satisfying life.
REFERENCES
1. International Headache Society. The International Classification of Headache Disorders. 2nd ed. Cephalalgia. 2004;24(suppl 1):1-160.
2. Stewart WF, Lipton RB, Dowson AJ, Sawyer J. Development and testing of the Migraine Disability Assessment (MIDAS) Questionnaire to assess headache-related disability. Neurology. 2001;56(6 suppl 1):S20-S28.
3. MacGregor EA. Perimenstrual headaches: unmet needs. Curr Pain Headache Rep. 2008;12(6):468-474.
4. Mannix LK. Menstrual-related pain conditions: dysmenorrhea and migraine. J Womens Health (Larchmt). 2008;17(5):879-891.
5. Martin VT. New theories in the pathogenesis of menstrual migraine. Curr Pain Headache Rep. 2008;12(6):453-462.
6. MacGregor EA. Migraine headache in perimenopausal and menopausal women. Curr Pain Headache Rep. 2009;13(5):399-403.
7. Martin VT, Wernke S, Mandell K, et al. Symptoms of premenstrual syndrome and their association with migraine headache. Headache. 2006; 46(1):125-137.
8. Martin VT, Behbehani M. Ovarian hormones and migraine headache: understanding mechanisms and pathogenesis—part 2. Headache. 2006;46(3):365-386.
9. Granella F, Sances G, Allais G, et al. Characteristics of menstrual and nonmenstrual attacks in women with menstrually related migraine referred to headache centres. Cephalalgia. 2004;24(9):707-716.
10. Hutchinson SL, Silberstein SD. Menstrual migraine: case studies of women with estrogen-related headaches. Headache. 2008;48 suppl 3:S131-S141.
11. Tepper SJ, Zatochill M, Szeto M, et al. Development of a simple menstrual migraine screening tool for obstetric and gynecology clinics: the Menstrual Migraine Assessment Tool. Headache. 2008; 48(10):1419-1425.
12. Marcus DA. Focus on primary care diagnosis and management of headache in women. Obstet Gynecol Surv. 1999;54(6):395-402.
13. Tepper SJ. Tailoring management strategies for the patient with menstrual migraine: focus on prevention and treatment. Headache. 2006;46(suppl 2):S61-S68.
14. Lay CL, Payne R. Recognition and treatment of menstrual migraine. Neurologist. 2007;13(4):197-204.
15. Henry KA, Cohen CI. Perimenstrual headache: treatment options. Curr Pain Headache Rep. 2009;13(1):82-88.
16. Calhoun AH. Estrogen-associated migraine. www.uptodate.com/contents/estrogen-associated-migraine. Accessed May 4, 2011.
17. Silberstein SD, Elkind AH, Schreiber C, et al. A randomized trial of frovatriptan for the intermittent prevention of menstrual migraine. Neurology. 2004;63:261-269.
18. Mannix LK, Savani N, Landy S, et al. Efficacy and tolerability of naratriptan for short-term prevention of menstrually related migraine: data from two randomized, double-blind, placebo-controlled studies. Headache. 2007;47(7):1037-1049.
19. Calhoun AH, Hutchinson S. Hormonal therapies for menstrual migraine. Curr Pain Headache Rep. 2009;13(5):381-385.
A 29-year-old woman with a history of frequent migraines presented to her primary care provider for a refill of medication. For the past two years she had been taking rizatriptan 10 mg, but with little relief. She stated that she had continued to experience discrete migraines several days per month, often clustered around menses. The severity of the headaches had negatively affected her work attendance, productivity, and social interactions. She wondered if she should be taking a different kind of medication.
The patient had been diagnosed with migraines at age 12, just prior to menarche. She described her headache as a unilateral, sharp throbbing pain associated with increased sensitivity to light and sound as well as nausea. She denied any history of head trauma. She had no allergies, and the only other medications she was taking at the time were an oral contraceptive (ethinyl estradiol/norgestimate 0.035 mg/0.18 mg with an oral triphasic 21/7 treatment cycle) and fluoxetine 20 mg for depression.
The patient worked daytime hours as a sales representative. She considered herself active, exercised regularly, ate a balanced diet, and slept well. She consumed no more than two to four alcoholic drinks per month and denied the use of herbals, dietary supplements, tobacco, or illegal drugs.
The patient stated that her mother had frequent headaches but had never sought a medical explanation or treatment. She was unaware of any other family history of headaches, and there was no family history of cardiovascular disease. Her sister had been diagnosed with a prolactinoma at age 25. At age 26, the patient had undergone a pituitary protocol MRI of the head with and without contrast, with negative results.
On examination, the patient was alert and oriented with normal vital signs. Her pupils were equal and reactive to light, and no papilledema was evident on fundoscopic examination. The cranial nerves were grossly intact and no other neurologic deficits were appreciated. No carotid bruits were present on cardiovascular exam.
Based on the patient’s history and physical exam, she met the International Classification of Headache Disorders (ICHD-II)1 diagnostic criteria for migraine without aura (1.1). When asked to recall the onset and frequency of attacks she had had in the previous four weeks, she noted that they regularly occurred during her menstrual cycle.
She was subsequently asked to begin a diary to record her headache characteristics, severity, and duration, with days of menstruation noted. The Migraine Disability Assessment (MIDAS) questionnaire2 (see Tables 1 and 22) was performed to measure the migraine attacks’ impact on the patient’s life; her score indicated that the headaches were causing her severe disability.
The patient’s abortive migraine medication was changed from rizatriptan 10 mg to the combination sumatriptan/naproxen sodium 85 mg/500 mg. She was instructed to take the initial dose as soon as she noticed signs of an impending migraine and to repeat the dose in two hours if symptoms persisted. The possibility of starting a preventive medication was discussed, but the patient wanted to evaluate her response to the combination triptan/NSAID before considering migraine prophylaxis.
Three months later, the patient returned for follow-up, including a review of her headache diary. She stated that the frequency and intensity of attacks had not decreased; acute treatment with sumatriptan/naproxen sodium made her headaches more bearable but did not ameliorate symptoms. The patient had recorded a detailed account of each migraine which, based on the ICHD-II criteria,1 demonstrated a pattern of headache occurrences consistent with menstrually related migraine. She reported a total of 18 headaches in the previous three months, 12 of which had occurred within the five-day perimenstrual period (see Figure 1).
Based on this information and the fact that the patient’s headaches were resistant to previous treatments, it was decided to alter the approach to her migraine management once more. In an effort to limit estrogen fluctuations during her menstrual cycle, her oral contraceptive was changed from ethinyl estradiol/norgestimate to a 12-week placebo-free monophasic regimen of ethinyl estradiol/levonorgestrel 20 mg/90 mcg. For intermittent prophylaxis, she was instructed to take frovatriptan 2.5 mg twice daily, beginning two days prior to the start of menses and continuing through the last day of her cycle. For acute treatment of breakthrough migraines, she was prescribed sumatriptan 20-mg nasal spray to take at the first sign of migraine symptoms and instructed to repeat the dose if the pain persisted or returned.
The patient continued to track her headaches in the diary and was seen in the office after three months of following the revised menstrual migraine management plan. She reported fewer migraines associated with her menstrual cycle and noted that they were less severe and shorter in duration. When she repeated the MIDAS test, her score was reduced from 23 to 10. In the subsequent nine months she has reported a consistent decrease in migraine prevalence and now rarely needs the abortive therapy.
DISCUSSION
Migraine, though commonly encountered in clinical practice, is a complex disorder. For women, migraine headaches have been recognized by the World Health Organization as the 12th leading cause of “life lived with a disabling condition.”3 Pure menstrual migraine and menstrually related migraine will be the focus of discussion here.
Etiology
Menstrually related migraine (comparable to pure menstrual migraine, although the latter is distinguished by occurring only during the perimenstrual period1) is recognized as a distinct type of migraine associated with perimenstrual hormone fluctuations.4 Of women who experience migraine, 42% to 61% can associate their attacks with the perimenstrual period5; this is defined as two days before to three days after the start of menstruation.
It has also been determined that women are more likely to have migraine attacks during the late luteal and early follicular phases (when there is a natural drop in estrogen levels) than in other phases (when estrogen levels are higher).6 Despite clinical evidence to support this estrogen withdrawal theory, the pathophysiology is not completely understood. It is possible that affected women are more sensitive than other women to the decrease in estrogen levels that occurs with menstruation.7
History and Physical Findings of Menstrual Migraines
Almost every woman with perimenstrual migraines reports an absence of aura.7 In the evaluation of headache, the same criteria for migraine without aura pertain to the classifications of pure menstrual migraine (PMM) or menstrually related migraine (MRM).1 Correlation of migraine attacks to the onset of menses is the key finding in the patient history to differentiate menstrual migraine from migraine without aura in women.8 Furthermore, perimenstrual migraines are often of longer duration and more difficult to treat than migraines not associated with hormone fluctuations.9
In order to distinguish between PMM and MRM, it is important to understand that pure menstrual migraine attacks take place exclusively in the five-day perimenstrual window and at no other times of the cycle. The criteria for MRM allow for attacks at other times of the cycle.1
In addition to causing physical pain, menstrual migraines can impact work performance, household activities, and personal relationships. The MIDAS questionnaire is a disability assessment tool that can reveal to the practitioner how migraines have affected the patient’s life over the previous three months.10 This is a useful method to identify patients with disabling migraines, determine their need for treatment, and monitor treatment efficacy.
Diagnosis
Menstrual migraine is a clinical diagnosis made by findings from the patient’s history. The International Headache Society has established specific diagnostic criteria in the ICHD-II for both PMM and MRM.1 An accurate and detailed migraine history is invaluable for the diagnosis of menstrual migraine. Although a formal questionnaire can serve as a good screening tool, it relies on the patient’s ability to recall specific times and dates with accuracy.11 Recall bias can be misleading in any attempt to confirm a diagnosis. The patient’s conscientious use of a daily headache diary or calendar (see Figure 2, for example) can lead to a precise record of the characteristics and timing of migraines, overcoming these obstacles.
Brain imaging is necessary if the patient’s symptoms suggest a critical etiology that requires immediate diagnosis and management. Red flags include sudden onset of a severe headache, a headache characterized as “the worst headache of the patient’s life,” a change in headache pattern, altered mental status, an abnormal neurologic examination, or fever with neck stiffness.12
Treatment Options for Menstrual Migraine
There is no FDA-approved treatment specific for menstrual migraines; however, medications used for management of nonmenstrual migraines are also those most commonly prescribed for women with menstrual migraine headaches.13 Because these headaches are frequently more severe and of longer duration than nonmenstrual migraine headaches, a combination of intermittent preventive therapy, hormone manipulation, and acute treatment strategies is often necessary.4
Acute therapy is aimed to treat migraine pain quickly and effectively with minimal adverse effects or need for additional medication. Triptans have been the mainstay of menstrual migraine treatment and have been proven effective for both acute attacks and prevention.4 Sumatriptan has a rapid onset of action and may be given orally as a 50- or 100-mg tablet, as a 6-mg subcutaneous injection, or as a 20-mg nasal spray.14
Abortive therapies are most effective when taken at the first sign of an attack. Patients can repeat the dose in two hours if the headache persists or recurs, to a maximum of two doses in 24 hours.15 Rizatriptan is another triptan used for acute treatment of menstrual migraine headaches. Its initial 10-mg dose can be repeated every two hours, to a maximum of 30 mg per 24 hours. NSAIDs, such as naproxen sodium, have also been recommended in acute migraine attacks. They seem to work synergistically with triptans, inhibiting prostaglandin synthesis and blocking neurogenic inflammation.15
Clinical study results have demonstrated superior pain relief and decreased migraine recurrence when a triptan and NSAID are used in combination, compared with use of either medication alone.4 A single-tablet formulation of sumatriptan 85 mg and naproxen sodium 500 mg may be considered for initial therapy in hard-to-treat patients.14
Preventive therapy should be considered when responsiveness to acute treatment is inadequate.4 Nonhormonal intermittent prophylactic treatment is recommended two days prior to the beginning of menses, continuing for five days.16 Longer-acting triptans, such as frovatriptan 2.5 mg and naratriptan 1.0 mg, dosed twice daily, have been demonstrated as effective in clinical trials when used during the perimenstrual period.17,18
The advantage of short-term therapy over daily prophylaxis is the potential to avoid adverse effects seen with continuous exposure to the drug.3 However, successful therapy relies on consistency in menstruation, and therefore may not be ideal for women with irregular cycles or those with coexisting nonmenstrual migraines.16 Estrogen-based therapy is an option for these women and for those who have failed nonhormonal methods.19
The goal of hormone prophylaxis is to prevent or reduce the physiologic decline in estradiol that occurs in the late luteal phase.4 Clinical studies have been conducted using various hormonal strategies to maintain steady estradiol levels, all of which decreased migraine prevalence.19 Estrogen fluctuations can be minimized by eliminating the placebo week in traditional estrogen/progestin oral contraceptives to achieve an extended-cycle regimen, resembling that of the 12-week ethinyl estradiol/levonorgestrel formulation.19
Continuous use of combined oral contraceptives is also an option for relief of menstrual migraine. When cyclic or extended-cycle regimens allow for menses, supplemental estrogen (10- to 20-mg ethinyl estradiol) is recommended during the hormone-free week.14
CONCLUSION
Proper diagnosis of menstrual migraines, using screening tools and the MIDAS questionnaire, can help practitioners provide the most effective migraine management for their patients. The most important step toward a good prognosis is acknowledging menstrual migraine as a unique headache disorder and formulating a precise diagnosis in order to identify individually tailored treatment options. With proper identification and integrated acute and prophylactic treatment, women with menstrual migraines are able to lead a healthier, more satisfying life.
REFERENCES
1. International Headache Society. The International Classification of Headache Disorders. 2nd ed. Cephalalgia. 2004;24(suppl 1):1-160.
2. Stewart WF, Lipton RB, Dowson AJ, Sawyer J. Development and testing of the Migraine Disability Assessment (MIDAS) Questionnaire to assess headache-related disability. Neurology. 2001;56(6 suppl 1):S20-S28.
3. MacGregor EA. Perimenstrual headaches: unmet needs. Curr Pain Headache Rep. 2008;12(6):468-474.
4. Mannix LK. Menstrual-related pain conditions: dysmenorrhea and migraine. J Womens Health (Larchmt). 2008;17(5):879-891.
5. Martin VT. New theories in the pathogenesis of menstrual migraine. Curr Pain Headache Rep. 2008;12(6):453-462.
6. MacGregor EA. Migraine headache in perimenopausal and menopausal women. Curr Pain Headache Rep. 2009;13(5):399-403.
7. Martin VT, Wernke S, Mandell K, et al. Symptoms of premenstrual syndrome and their association with migraine headache. Headache. 2006; 46(1):125-137.
8. Martin VT, Behbehani M. Ovarian hormones and migraine headache: understanding mechanisms and pathogenesis—part 2. Headache. 2006;46(3):365-386.
9. Granella F, Sances G, Allais G, et al. Characteristics of menstrual and nonmenstrual attacks in women with menstrually related migraine referred to headache centres. Cephalalgia. 2004;24(9):707-716.
10. Hutchinson SL, Silberstein SD. Menstrual migraine: case studies of women with estrogen-related headaches. Headache. 2008;48 suppl 3:S131-S141.
11. Tepper SJ, Zatochill M, Szeto M, et al. Development of a simple menstrual migraine screening tool for obstetric and gynecology clinics: the Menstrual Migraine Assessment Tool. Headache. 2008; 48(10):1419-1425.
12. Marcus DA. Focus on primary care diagnosis and management of headache in women. Obstet Gynecol Surv. 1999;54(6):395-402.
13. Tepper SJ. Tailoring management strategies for the patient with menstrual migraine: focus on prevention and treatment. Headache. 2006;46(suppl 2):S61-S68.
14. Lay CL, Payne R. Recognition and treatment of menstrual migraine. Neurologist. 2007;13(4):197-204.
15. Henry KA, Cohen CI. Perimenstrual headache: treatment options. Curr Pain Headache Rep. 2009;13(1):82-88.
16. Calhoun AH. Estrogen-associated migraine. www.uptodate.com/contents/estrogen-associated-migraine. Accessed May 4, 2011.
17. Silberstein SD, Elkind AH, Schreiber C, et al. A randomized trial of frovatriptan for the intermittent prevention of menstrual migraine. Neurology. 2004;63:261-269.
18. Mannix LK, Savani N, Landy S, et al. Efficacy and tolerability of naratriptan for short-term prevention of menstrually related migraine: data from two randomized, double-blind, placebo-controlled studies. Headache. 2007;47(7):1037-1049.
19. Calhoun AH, Hutchinson S. Hormonal therapies for menstrual migraine. Curr Pain Headache Rep. 2009;13(5):381-385.
Grand Rounds: Pregnant Woman, 33, With Leg Pain and Numbness
A 33-year-old woman in her 32nd week of pregnancy (gravida 3, para 2) presented to the emergency department (ED) with a five-day history of weakness and ascending numbness below the right knee. She related a two-week history of right-sided low back pain that radiated to the right buttock and was associated with severe right lower extremity (RLE) pain, most prominent in the posterolateral aspect of the right calf. She denied perianal numbness, incontinence, or other changes in bowel or bladder function. She also denied left lower extremity involvement or trauma.
The patient had had one uneventful pregnancy to date. Her medical history included hypothyroidism, treated with levothyroxine; and anxiety, for which she was taking sertraline. She denied any history of allergies, alcohol consumption, smoking, or illicit drug use. She had been evaluated twice and received reassurance in the two weeks before her presentation to the ED. She was admitted to the obstetric service secondary to pain, and a stat MRI rather than x-ray was ordered by obstetrics. An orthopedic consult was ordered. A spine surgeon happened to be on call.
Examination revealed that the patient walked plantigrade, with her right foot slightly externally rotated. She was unable to dorsiflex or plantarflex her right foot. She was unable to heel- or toe-walk on the right side, possessed 0 out of 5 strength at the right extensor hallucis longus and 2 to 3 out of 5 at the right tibialis anterior and gastroc soleus complex. She complained of pain with right leg elevation exceeding 30° and had very limited sensation to light touch in the right L5 and S1 dermatomes. Deep tendon reflex was absent at the right ankle. The patient refused a rectal exam or post-void evaluation.
The initial diagnosis considered by the ED clinician was sciatica, with a differential diagnosis that included pelvic pain of pregnancy, lumbar sprain strain, sciatica, lumbar disk, herniated nucleus pulposus with radiculopathy, and cauda equina syndrome. Trauma was considered and ruled out, as were malignancies; inflammatory, infectious, or degenerative conditions; or other compressive processes.1
Lumbar MRI demonstrated a very large, right-sided disk herniation at L5-S1 with an extruded fragment that was severely compressing the thecal sac and the right S1 nerve root, causing severe right foraminal stenosis at the level of L5-S1. Degenerative changes were noted at L4-5 with disk dessication and no lesions seen.
The patient was diagnosed with cauda equina syndrome, which was felt to be causing severe RLE weakness and ascending numbness. The options of observation, analgesia, physical therapy, and epidural injections were discussed with the patient; however, surgery was strongly recommended due to her profound weakness and the severity of pain she was experiencing, in addition to the size of the disk herniation. She opted for surgery.
The patient was given epidural anesthesia at the L3-4 level, with a catheter left in place during the procedure. A test dose of lidocaine (1.5 cc) with epinephrine was injected to ensure proper placement, and bupivacaine 0.5% was given in increments of 5.0 cc three times during the case. Propofol was administered for sedation, and a 2.0-mg dose of a long-acting morphine was given to the patient before removal of the epidural catheter. Fetal monitoring was performed by obstetrics throughout the procedure.
A laminotomy, partial facetectomy, and diskectomy were performed at L5-S1 with excision of a free fragment. Surgical pathology described the disk as fibrocartilaginous tissue measuring 3.5 cm x 1.4 cm x 0.6 cm.
DISCUSSION
Although nearly half of pregnant women experience low back pain, cauda equina syndrome (CES), a complication of lumbar disk herniation, is extremely rare in the gravid patient.2 In a decade-long review of 48,760 consecutive deliveries, LaBan et al3 identified symptomatic lumbar herniated nucleus pulposus in only five patients (approximately one in 10,000 pregnancies). In pregnant women who do experience CES, symptoms most commonly develop between the fifth and seventh month of pregnancy.4 According to Small et al,5 “The major pitfall in diagnosis is not including CES in the back pain differential.”
True CES presents as a triad of symptoms: lower extremity weakness, altered sensation in the skin of the buttocks and upper posterior thighs (saddle anesthesia), and dysfunction or paralysis of the bowel and bladder. However, few patients present with all of the classic symptoms,6 and patients with CES are often dismissed by several clinicians in their search for relief before presenting to a subspecialist. Kostuik et al7 consider “unilateral sciatica with motor and sensory disturbance” a more common presentation of CES; also indicative of this condition, they report, is “urinary dysfunction combined with motor and sensory loss in the presence of a disc lesion.”
The polypeptide relaxin, which is secreted by the corpus luteum to promote joint laxity in late pregnancy, has been associated with low back pain and pelvic pain of pregnancy; it has also been suggested as a possible contributing cause of CES during pregnancy.8,9 Additionally, increased lumbar lordosis with positional and postural stress may cause direct pressure by the gravid uterus on nerve roots. The great vessels may also be compressed by the uterus, resulting in ischemia of the neural element and back pain that radiates to the legs.10 Many cases of lumbar disk prolapse occur during the first and second trimesters. The most clinically incapacitated patients have been found to have the highest levels of relaxin.9
The Diagnosis
Early diagnosis of CES, through proper physical examination and radiologic studies, is paramount. A rectal examination should be performed to assess for sphincter tone (which may be diminished in 80% of patients) and to assess for perineal sensation.5 Catheterization yielding a postvoid residual urine greater than 100/200 cc is reported to have a specificity and sensitivity of 90% or greater for CES. Small et al5 recommend a straight leg raise maneuver to assess for radiculopathy.
Various studies in the literature support the use of MRI in the gravid patient to confirm the diagnosis of CES and to identify the degree and level of disk protrusion.2-4,11
Treatment
CES requires urgent surgical decompression.11 Early recognition of CES attributable to lumbar disk prolapse, report O’Laoire et al,12 is essential to prevent irreversible sphincter paralysis. They liken the condition’s urgency to that of extradural hematoma in a head injury.
Disk surgery during pregnancy—preferably a team effort, with obstetrics performing perioperative fetal monitoring—has been deemed a safe management method.2,4 Spinal or general anesthesia during nonobstetric surgery is generally considered safe for both mother and fetus.13,14 Adequate oxygenation without risk for hyperventilation is considered essential.15
PATIENT OUTCOME
In the immediate postoperative period, the patient continued to complain of RLE pain, which abated significantly by the time she was discharged. When she was seen in follow-up four days later, she was able to heel- and toe-walk on the right side, and her strength had improved to 3 or 4 out of 5 at the RLE. She continued to experience diminished sensation to the plantar aspect of the right foot, which persisted at the one-month follow up. At that visit, the patient also reported occasional pain in the right buttock. Physical therapy was started to strengthen the RLE.
By three months postsurgery, the patient had undergone uneventful vaginal delivery. She had an entirely benign exam with 5 out of 5 strength at the RLE and no neurologic deficits. She was cleared to return to light weightlifting with good technique and lumbar support but was told to refrain from running until the sixth month postsurgery.
CONCLUSION
Although the case patient did not have a “true” (ie, typical) presentation of CES, her symptoms warranted a full workup and treatment to prevent possible long-term sequelae. Medical practitioners should be familiar with the triad presentation of CES. They must differentiate lower back pain of muscular origin from lumbar disk herniation and be able to appreciate the degree of symptom severity reported by the gravid patient. A thorough history and physical assessment must be performed in every such case. When in doubt, the clinician must err on the side of caution, referring the patient for MRI and consulting with a specialist.
REFERENCES
1. Johnston RA. The management of acute spinal cord compression. J Neurol Neurosurg Psychiatr. 1993;56(10):1046-1054.
2. Brown MD, Levi AD. Surgery for lumbar disc herniation during pregnancy. Spine (Phila PA 1976). 2001;26(5):440-443.
3. LaBan MM, Perrin JCS, Latimer FR. Pregnancy and the herniated lumbar disc. Arch Phys Med Rehabil. 1983;64(7):319-321.
4. LaBan MM, Rapp NS, Van Oeyen P, Meerschaert JR. The lumbar herniated disk of pregnancy: a report of six cases identified by magnetic resonance imaging. Arch Phys Med Rehabil. 1995;76(5):476-479.
5. Small SA, Perron AD, Brady WJ. Orthopedic pitfalls: cauda equina syndrome. Am J Emerg Med. 2005;23(2):159-163.
6. Tay EC, Chacha PB. Midline prolapse of a lumbar intervertebral disc with compression of the cauda equina. J Bone Joint Surg. 1979;61(1):43-46.
7. Kostuik JP, Harrington I, Alexander D, et al. Cauda equina syndrome and lumbar disc herniation. J Bone Joint Surg Am. 1986;68(3):386-391.
8. Russell R, Reynolds F. Back pain, pregnancy, and childbirth. BMJ. 1997;314(7087):1062-1063.
9. MacLennan AH, Nicholson R, Green RC, Bath M. Serum relaxin and pelvic pain of pregnancy. Lancet. 1986;2(8501):243-245.
10. Ashkan K, Casey AT, Powell M, Crockard HA. Back pain during pregnancy and after childbirth: an unusual cause not to miss. J R Soc Med. 1998;91(2):88-90.
11. Busse JW, Bhandari M, Schnittker JB, et al. Delayed presentation of cauda equina syndrome secondary to lumbar disc herniation: functional outcomes and health-related quality of life. CJEM. 2001;3(4):285-291.
12. O’Laoire SA, Crockard HA, Thomas DG. Prognosis for sphincter recovery after operation for cauda equina compression owing to lumbar disc prolapse. Br Med J (Clin Res Ed). 1981;282(6279):1852-1854.
13. Kuczkowski KM. The safety of anaesthetics in pregnant women. Expert Opin Drug Saf. 2006; 5(2):251-264.
14. Kuczkowski KM. Nonobstetric surgery during pregnancy: what are the risks of anesthesia? Obstet Gynecol Surv. 2004;59(1):52-56.
15. Birnbach DJ, Browne IM. Anesthesia for obstetrics. In: Miller RD, Eriksson LI, Fleisher LA, et al. Miller’s Anesthesia. Philadelphia, PA: Churchill Livingston, Elsevier Health Science; 2010: 2203-2240.
A 33-year-old woman in her 32nd week of pregnancy (gravida 3, para 2) presented to the emergency department (ED) with a five-day history of weakness and ascending numbness below the right knee. She related a two-week history of right-sided low back pain that radiated to the right buttock and was associated with severe right lower extremity (RLE) pain, most prominent in the posterolateral aspect of the right calf. She denied perianal numbness, incontinence, or other changes in bowel or bladder function. She also denied left lower extremity involvement or trauma.
The patient had had one uneventful pregnancy to date. Her medical history included hypothyroidism, treated with levothyroxine; and anxiety, for which she was taking sertraline. She denied any history of allergies, alcohol consumption, smoking, or illicit drug use. She had been evaluated twice and received reassurance in the two weeks before her presentation to the ED. She was admitted to the obstetric service secondary to pain, and a stat MRI rather than x-ray was ordered by obstetrics. An orthopedic consult was ordered. A spine surgeon happened to be on call.
Examination revealed that the patient walked plantigrade, with her right foot slightly externally rotated. She was unable to dorsiflex or plantarflex her right foot. She was unable to heel- or toe-walk on the right side, possessed 0 out of 5 strength at the right extensor hallucis longus and 2 to 3 out of 5 at the right tibialis anterior and gastroc soleus complex. She complained of pain with right leg elevation exceeding 30° and had very limited sensation to light touch in the right L5 and S1 dermatomes. Deep tendon reflex was absent at the right ankle. The patient refused a rectal exam or post-void evaluation.
The initial diagnosis considered by the ED clinician was sciatica, with a differential diagnosis that included pelvic pain of pregnancy, lumbar sprain strain, sciatica, lumbar disk, herniated nucleus pulposus with radiculopathy, and cauda equina syndrome. Trauma was considered and ruled out, as were malignancies; inflammatory, infectious, or degenerative conditions; or other compressive processes.1
Lumbar MRI demonstrated a very large, right-sided disk herniation at L5-S1 with an extruded fragment that was severely compressing the thecal sac and the right S1 nerve root, causing severe right foraminal stenosis at the level of L5-S1. Degenerative changes were noted at L4-5 with disk dessication and no lesions seen.
The patient was diagnosed with cauda equina syndrome, which was felt to be causing severe RLE weakness and ascending numbness. The options of observation, analgesia, physical therapy, and epidural injections were discussed with the patient; however, surgery was strongly recommended due to her profound weakness and the severity of pain she was experiencing, in addition to the size of the disk herniation. She opted for surgery.
The patient was given epidural anesthesia at the L3-4 level, with a catheter left in place during the procedure. A test dose of lidocaine (1.5 cc) with epinephrine was injected to ensure proper placement, and bupivacaine 0.5% was given in increments of 5.0 cc three times during the case. Propofol was administered for sedation, and a 2.0-mg dose of a long-acting morphine was given to the patient before removal of the epidural catheter. Fetal monitoring was performed by obstetrics throughout the procedure.
A laminotomy, partial facetectomy, and diskectomy were performed at L5-S1 with excision of a free fragment. Surgical pathology described the disk as fibrocartilaginous tissue measuring 3.5 cm x 1.4 cm x 0.6 cm.
DISCUSSION
Although nearly half of pregnant women experience low back pain, cauda equina syndrome (CES), a complication of lumbar disk herniation, is extremely rare in the gravid patient.2 In a decade-long review of 48,760 consecutive deliveries, LaBan et al3 identified symptomatic lumbar herniated nucleus pulposus in only five patients (approximately one in 10,000 pregnancies). In pregnant women who do experience CES, symptoms most commonly develop between the fifth and seventh month of pregnancy.4 According to Small et al,5 “The major pitfall in diagnosis is not including CES in the back pain differential.”
True CES presents as a triad of symptoms: lower extremity weakness, altered sensation in the skin of the buttocks and upper posterior thighs (saddle anesthesia), and dysfunction or paralysis of the bowel and bladder. However, few patients present with all of the classic symptoms,6 and patients with CES are often dismissed by several clinicians in their search for relief before presenting to a subspecialist. Kostuik et al7 consider “unilateral sciatica with motor and sensory disturbance” a more common presentation of CES; also indicative of this condition, they report, is “urinary dysfunction combined with motor and sensory loss in the presence of a disc lesion.”
The polypeptide relaxin, which is secreted by the corpus luteum to promote joint laxity in late pregnancy, has been associated with low back pain and pelvic pain of pregnancy; it has also been suggested as a possible contributing cause of CES during pregnancy.8,9 Additionally, increased lumbar lordosis with positional and postural stress may cause direct pressure by the gravid uterus on nerve roots. The great vessels may also be compressed by the uterus, resulting in ischemia of the neural element and back pain that radiates to the legs.10 Many cases of lumbar disk prolapse occur during the first and second trimesters. The most clinically incapacitated patients have been found to have the highest levels of relaxin.9
The Diagnosis
Early diagnosis of CES, through proper physical examination and radiologic studies, is paramount. A rectal examination should be performed to assess for sphincter tone (which may be diminished in 80% of patients) and to assess for perineal sensation.5 Catheterization yielding a postvoid residual urine greater than 100/200 cc is reported to have a specificity and sensitivity of 90% or greater for CES. Small et al5 recommend a straight leg raise maneuver to assess for radiculopathy.
Various studies in the literature support the use of MRI in the gravid patient to confirm the diagnosis of CES and to identify the degree and level of disk protrusion.2-4,11
Treatment
CES requires urgent surgical decompression.11 Early recognition of CES attributable to lumbar disk prolapse, report O’Laoire et al,12 is essential to prevent irreversible sphincter paralysis. They liken the condition’s urgency to that of extradural hematoma in a head injury.
Disk surgery during pregnancy—preferably a team effort, with obstetrics performing perioperative fetal monitoring—has been deemed a safe management method.2,4 Spinal or general anesthesia during nonobstetric surgery is generally considered safe for both mother and fetus.13,14 Adequate oxygenation without risk for hyperventilation is considered essential.15
PATIENT OUTCOME
In the immediate postoperative period, the patient continued to complain of RLE pain, which abated significantly by the time she was discharged. When she was seen in follow-up four days later, she was able to heel- and toe-walk on the right side, and her strength had improved to 3 or 4 out of 5 at the RLE. She continued to experience diminished sensation to the plantar aspect of the right foot, which persisted at the one-month follow up. At that visit, the patient also reported occasional pain in the right buttock. Physical therapy was started to strengthen the RLE.
By three months postsurgery, the patient had undergone uneventful vaginal delivery. She had an entirely benign exam with 5 out of 5 strength at the RLE and no neurologic deficits. She was cleared to return to light weightlifting with good technique and lumbar support but was told to refrain from running until the sixth month postsurgery.
CONCLUSION
Although the case patient did not have a “true” (ie, typical) presentation of CES, her symptoms warranted a full workup and treatment to prevent possible long-term sequelae. Medical practitioners should be familiar with the triad presentation of CES. They must differentiate lower back pain of muscular origin from lumbar disk herniation and be able to appreciate the degree of symptom severity reported by the gravid patient. A thorough history and physical assessment must be performed in every such case. When in doubt, the clinician must err on the side of caution, referring the patient for MRI and consulting with a specialist.
REFERENCES
1. Johnston RA. The management of acute spinal cord compression. J Neurol Neurosurg Psychiatr. 1993;56(10):1046-1054.
2. Brown MD, Levi AD. Surgery for lumbar disc herniation during pregnancy. Spine (Phila PA 1976). 2001;26(5):440-443.
3. LaBan MM, Perrin JCS, Latimer FR. Pregnancy and the herniated lumbar disc. Arch Phys Med Rehabil. 1983;64(7):319-321.
4. LaBan MM, Rapp NS, Van Oeyen P, Meerschaert JR. The lumbar herniated disk of pregnancy: a report of six cases identified by magnetic resonance imaging. Arch Phys Med Rehabil. 1995;76(5):476-479.
5. Small SA, Perron AD, Brady WJ. Orthopedic pitfalls: cauda equina syndrome. Am J Emerg Med. 2005;23(2):159-163.
6. Tay EC, Chacha PB. Midline prolapse of a lumbar intervertebral disc with compression of the cauda equina. J Bone Joint Surg. 1979;61(1):43-46.
7. Kostuik JP, Harrington I, Alexander D, et al. Cauda equina syndrome and lumbar disc herniation. J Bone Joint Surg Am. 1986;68(3):386-391.
8. Russell R, Reynolds F. Back pain, pregnancy, and childbirth. BMJ. 1997;314(7087):1062-1063.
9. MacLennan AH, Nicholson R, Green RC, Bath M. Serum relaxin and pelvic pain of pregnancy. Lancet. 1986;2(8501):243-245.
10. Ashkan K, Casey AT, Powell M, Crockard HA. Back pain during pregnancy and after childbirth: an unusual cause not to miss. J R Soc Med. 1998;91(2):88-90.
11. Busse JW, Bhandari M, Schnittker JB, et al. Delayed presentation of cauda equina syndrome secondary to lumbar disc herniation: functional outcomes and health-related quality of life. CJEM. 2001;3(4):285-291.
12. O’Laoire SA, Crockard HA, Thomas DG. Prognosis for sphincter recovery after operation for cauda equina compression owing to lumbar disc prolapse. Br Med J (Clin Res Ed). 1981;282(6279):1852-1854.
13. Kuczkowski KM. The safety of anaesthetics in pregnant women. Expert Opin Drug Saf. 2006; 5(2):251-264.
14. Kuczkowski KM. Nonobstetric surgery during pregnancy: what are the risks of anesthesia? Obstet Gynecol Surv. 2004;59(1):52-56.
15. Birnbach DJ, Browne IM. Anesthesia for obstetrics. In: Miller RD, Eriksson LI, Fleisher LA, et al. Miller’s Anesthesia. Philadelphia, PA: Churchill Livingston, Elsevier Health Science; 2010: 2203-2240.
A 33-year-old woman in her 32nd week of pregnancy (gravida 3, para 2) presented to the emergency department (ED) with a five-day history of weakness and ascending numbness below the right knee. She related a two-week history of right-sided low back pain that radiated to the right buttock and was associated with severe right lower extremity (RLE) pain, most prominent in the posterolateral aspect of the right calf. She denied perianal numbness, incontinence, or other changes in bowel or bladder function. She also denied left lower extremity involvement or trauma.
The patient had had one uneventful pregnancy to date. Her medical history included hypothyroidism, treated with levothyroxine; and anxiety, for which she was taking sertraline. She denied any history of allergies, alcohol consumption, smoking, or illicit drug use. She had been evaluated twice and received reassurance in the two weeks before her presentation to the ED. She was admitted to the obstetric service secondary to pain, and a stat MRI rather than x-ray was ordered by obstetrics. An orthopedic consult was ordered. A spine surgeon happened to be on call.
Examination revealed that the patient walked plantigrade, with her right foot slightly externally rotated. She was unable to dorsiflex or plantarflex her right foot. She was unable to heel- or toe-walk on the right side, possessed 0 out of 5 strength at the right extensor hallucis longus and 2 to 3 out of 5 at the right tibialis anterior and gastroc soleus complex. She complained of pain with right leg elevation exceeding 30° and had very limited sensation to light touch in the right L5 and S1 dermatomes. Deep tendon reflex was absent at the right ankle. The patient refused a rectal exam or post-void evaluation.
The initial diagnosis considered by the ED clinician was sciatica, with a differential diagnosis that included pelvic pain of pregnancy, lumbar sprain strain, sciatica, lumbar disk, herniated nucleus pulposus with radiculopathy, and cauda equina syndrome. Trauma was considered and ruled out, as were malignancies; inflammatory, infectious, or degenerative conditions; or other compressive processes.1
Lumbar MRI demonstrated a very large, right-sided disk herniation at L5-S1 with an extruded fragment that was severely compressing the thecal sac and the right S1 nerve root, causing severe right foraminal stenosis at the level of L5-S1. Degenerative changes were noted at L4-5 with disk dessication and no lesions seen.
The patient was diagnosed with cauda equina syndrome, which was felt to be causing severe RLE weakness and ascending numbness. The options of observation, analgesia, physical therapy, and epidural injections were discussed with the patient; however, surgery was strongly recommended due to her profound weakness and the severity of pain she was experiencing, in addition to the size of the disk herniation. She opted for surgery.
The patient was given epidural anesthesia at the L3-4 level, with a catheter left in place during the procedure. A test dose of lidocaine (1.5 cc) with epinephrine was injected to ensure proper placement, and bupivacaine 0.5% was given in increments of 5.0 cc three times during the case. Propofol was administered for sedation, and a 2.0-mg dose of a long-acting morphine was given to the patient before removal of the epidural catheter. Fetal monitoring was performed by obstetrics throughout the procedure.
A laminotomy, partial facetectomy, and diskectomy were performed at L5-S1 with excision of a free fragment. Surgical pathology described the disk as fibrocartilaginous tissue measuring 3.5 cm x 1.4 cm x 0.6 cm.
DISCUSSION
Although nearly half of pregnant women experience low back pain, cauda equina syndrome (CES), a complication of lumbar disk herniation, is extremely rare in the gravid patient.2 In a decade-long review of 48,760 consecutive deliveries, LaBan et al3 identified symptomatic lumbar herniated nucleus pulposus in only five patients (approximately one in 10,000 pregnancies). In pregnant women who do experience CES, symptoms most commonly develop between the fifth and seventh month of pregnancy.4 According to Small et al,5 “The major pitfall in diagnosis is not including CES in the back pain differential.”
True CES presents as a triad of symptoms: lower extremity weakness, altered sensation in the skin of the buttocks and upper posterior thighs (saddle anesthesia), and dysfunction or paralysis of the bowel and bladder. However, few patients present with all of the classic symptoms,6 and patients with CES are often dismissed by several clinicians in their search for relief before presenting to a subspecialist. Kostuik et al7 consider “unilateral sciatica with motor and sensory disturbance” a more common presentation of CES; also indicative of this condition, they report, is “urinary dysfunction combined with motor and sensory loss in the presence of a disc lesion.”
The polypeptide relaxin, which is secreted by the corpus luteum to promote joint laxity in late pregnancy, has been associated with low back pain and pelvic pain of pregnancy; it has also been suggested as a possible contributing cause of CES during pregnancy.8,9 Additionally, increased lumbar lordosis with positional and postural stress may cause direct pressure by the gravid uterus on nerve roots. The great vessels may also be compressed by the uterus, resulting in ischemia of the neural element and back pain that radiates to the legs.10 Many cases of lumbar disk prolapse occur during the first and second trimesters. The most clinically incapacitated patients have been found to have the highest levels of relaxin.9
The Diagnosis
Early diagnosis of CES, through proper physical examination and radiologic studies, is paramount. A rectal examination should be performed to assess for sphincter tone (which may be diminished in 80% of patients) and to assess for perineal sensation.5 Catheterization yielding a postvoid residual urine greater than 100/200 cc is reported to have a specificity and sensitivity of 90% or greater for CES. Small et al5 recommend a straight leg raise maneuver to assess for radiculopathy.
Various studies in the literature support the use of MRI in the gravid patient to confirm the diagnosis of CES and to identify the degree and level of disk protrusion.2-4,11
Treatment
CES requires urgent surgical decompression.11 Early recognition of CES attributable to lumbar disk prolapse, report O’Laoire et al,12 is essential to prevent irreversible sphincter paralysis. They liken the condition’s urgency to that of extradural hematoma in a head injury.
Disk surgery during pregnancy—preferably a team effort, with obstetrics performing perioperative fetal monitoring—has been deemed a safe management method.2,4 Spinal or general anesthesia during nonobstetric surgery is generally considered safe for both mother and fetus.13,14 Adequate oxygenation without risk for hyperventilation is considered essential.15
PATIENT OUTCOME
In the immediate postoperative period, the patient continued to complain of RLE pain, which abated significantly by the time she was discharged. When she was seen in follow-up four days later, she was able to heel- and toe-walk on the right side, and her strength had improved to 3 or 4 out of 5 at the RLE. She continued to experience diminished sensation to the plantar aspect of the right foot, which persisted at the one-month follow up. At that visit, the patient also reported occasional pain in the right buttock. Physical therapy was started to strengthen the RLE.
By three months postsurgery, the patient had undergone uneventful vaginal delivery. She had an entirely benign exam with 5 out of 5 strength at the RLE and no neurologic deficits. She was cleared to return to light weightlifting with good technique and lumbar support but was told to refrain from running until the sixth month postsurgery.
CONCLUSION
Although the case patient did not have a “true” (ie, typical) presentation of CES, her symptoms warranted a full workup and treatment to prevent possible long-term sequelae. Medical practitioners should be familiar with the triad presentation of CES. They must differentiate lower back pain of muscular origin from lumbar disk herniation and be able to appreciate the degree of symptom severity reported by the gravid patient. A thorough history and physical assessment must be performed in every such case. When in doubt, the clinician must err on the side of caution, referring the patient for MRI and consulting with a specialist.
REFERENCES
1. Johnston RA. The management of acute spinal cord compression. J Neurol Neurosurg Psychiatr. 1993;56(10):1046-1054.
2. Brown MD, Levi AD. Surgery for lumbar disc herniation during pregnancy. Spine (Phila PA 1976). 2001;26(5):440-443.
3. LaBan MM, Perrin JCS, Latimer FR. Pregnancy and the herniated lumbar disc. Arch Phys Med Rehabil. 1983;64(7):319-321.
4. LaBan MM, Rapp NS, Van Oeyen P, Meerschaert JR. The lumbar herniated disk of pregnancy: a report of six cases identified by magnetic resonance imaging. Arch Phys Med Rehabil. 1995;76(5):476-479.
5. Small SA, Perron AD, Brady WJ. Orthopedic pitfalls: cauda equina syndrome. Am J Emerg Med. 2005;23(2):159-163.
6. Tay EC, Chacha PB. Midline prolapse of a lumbar intervertebral disc with compression of the cauda equina. J Bone Joint Surg. 1979;61(1):43-46.
7. Kostuik JP, Harrington I, Alexander D, et al. Cauda equina syndrome and lumbar disc herniation. J Bone Joint Surg Am. 1986;68(3):386-391.
8. Russell R, Reynolds F. Back pain, pregnancy, and childbirth. BMJ. 1997;314(7087):1062-1063.
9. MacLennan AH, Nicholson R, Green RC, Bath M. Serum relaxin and pelvic pain of pregnancy. Lancet. 1986;2(8501):243-245.
10. Ashkan K, Casey AT, Powell M, Crockard HA. Back pain during pregnancy and after childbirth: an unusual cause not to miss. J R Soc Med. 1998;91(2):88-90.
11. Busse JW, Bhandari M, Schnittker JB, et al. Delayed presentation of cauda equina syndrome secondary to lumbar disc herniation: functional outcomes and health-related quality of life. CJEM. 2001;3(4):285-291.
12. O’Laoire SA, Crockard HA, Thomas DG. Prognosis for sphincter recovery after operation for cauda equina compression owing to lumbar disc prolapse. Br Med J (Clin Res Ed). 1981;282(6279):1852-1854.
13. Kuczkowski KM. The safety of anaesthetics in pregnant women. Expert Opin Drug Saf. 2006; 5(2):251-264.
14. Kuczkowski KM. Nonobstetric surgery during pregnancy: what are the risks of anesthesia? Obstet Gynecol Surv. 2004;59(1):52-56.
15. Birnbach DJ, Browne IM. Anesthesia for obstetrics. In: Miller RD, Eriksson LI, Fleisher LA, et al. Miller’s Anesthesia. Philadelphia, PA: Churchill Livingston, Elsevier Health Science; 2010: 2203-2240.
Grand Rounds: Man, 46, With a Curious Ear Pain
A 46-year-old man presented to a hospital emergency department (ED) with a four-day history of right ear pain. He described the pain as a constant, dull, burning pain radiating to the neck and face, associated with a feeling of congestion. The patient also stated that the right side of his face had felt numb for about one day.
Three days earlier, the man had been seen by his primary health care provider, who told him that his ear looked normal and free of infection. The day before his current presentation to the ED, however, he noticed what he described as an “acne-like” rash on his ear lobe. Shortly before coming to the ED, the patient also developed numbness over his right upper lip, which he likened to the effects of procaine during a dental visit. He reported drooling from the right side of his mouth while drinking water and difficulty blinking his right eye.
He denied any tinnitus, fever, headache, or change in hearing. A review of symptoms was positive only for mild dizziness during the previous two to three days.
The patient was a well-appearing white man. He was alert and oriented to identity, time, and place. His skin was warm, dry, and intact. The examiner noticed a small area of erythematous rash with vesicles on the man’s right ear lobe. The external auditory canals appeared within normal limits, with no erythema or edema, and were nontender bilaterally. The tympanic membranes were normal bilaterally, without bulging or discernible fluid levels.
The ocular exam was normal with no visual acuity changes and no fluorescein uptake; external ocular movements were intact. A slight droop was noted in the right eyelid, but there was no droop on the contralateral side of his face. When asked to puff up his cheeks, the patient found it difficult to do so on the right side of his mouth without releasing air from his lips.
The remainder of the cranial nerves were intact. Muscle strength was 5/5 in all extremities and equal bilaterally. The man’s gait was within normal limits, and the remaining findings in the physical exam were normal.
The initial diagnosis considered in the differential was otitis externa, because it is a common explanation for ear pain in patients who present to the ED.1,2 Also, in otitis, pain is characteristically present in the affected ear, and erythema is often found in the external auditory canal.3 However, this diagnosis was deemed unlikely because otitis externa would not explain the neurologic findings or the vesicular rash.1
Bell’s palsy was next in the differential, as it was considered consistent with the patient’s unilateral neurologic deficits.4 In addition to weakness or palsy of the facial nerve, many patients with Bell’s palsy complain of mastoid pain, which can be confused with a complaint of ear pain.5 However, patients with Bell’s palsy have no rash, and this diagnosis was considered unlikely.
The painful, burning rash on the patient’s face was characteristic of herpes zoster (shingles), which was next in the differential. Infrequently, shingles can also cause weakness in the nerve it affects. In the case patient, weakness that was evident in the affected nerve resembled that seen in Bell’s palsy. This combination of symptoms is referred to as Ramsay Hunt syndrome—which in this case was decided to be the correct diagnosis.
DISCUSSION
Ramsay Hunt syndrome (RHS, also known as geniculate herpes5,6) is caused by the varicella-zoster virus, most commonly known as the cause of chickenpox. In the United States, RHS is believed to affect only about one in 1,500 persons, although 20% to 30% of persons experience herpes zoster infection at some time.7
Soon after a chickenpox infection subsides, the virus spreads along the sensory nerve fibers of the peripheral and cranial nerves. The virus then becomes dormant in the dorsal root ganglion, where in some patients it later reactivates in the form of shingles.8
In RHS, the ganglia of cranial nerve VII (CN VII, the facial nerve, which innervates the facial muscles) are infected; for this reason, the condition is also referred to as zoster oticus.9 Because of the involvement and weakening of the facial nerve, the presentation of RHS often resembles that of Bell’s palsy or facial nerve palsy.
While most cases of Bell’s palsy are idiopathic,10,11 RHS can usually be attributed to viral infection—most commonly, infection with herpes simplex virus type 1 (HSV-1).12 RHS can be differentiated from Bell’s palsy by the presence of a rash on the ipsilateral side. The rash appears in the form of inflamed vesicles on an erythematous base and may be present around the ear (see figure), the eardrum, the hard and soft palate, or the tongue.6 When the rash is painful, it is often described as a burning pain. Loss of taste may occur in the anterior portion of the tongue.9,12
Unlike shingles, which usually manifests as a sensory neuropathy, RHS is distinguished by motor neuropathy.7 The patient usually reports weakness in the facial muscles on one side, leading to difficulty drinking water or puffing out the cheek and to drooling on one side of the face. A complaint of dryness in the ipsilateral eye may result from weakness or an inability to close the eyelid.
It is important to note that as in Bell’s palsy, RHS can be differentiated from stroke by the patient’s inability to wrinkle the forehead. The motor muscles of the forehead are innervated by both sides of the brain; in the case of stroke, only one side of the brain is affected, and movement of the forehead remains possible on the contralateral side. In facial nerve palsy, the nerve itself is affected; thus, no movement of the forehead is possible.13 Other common complaints in patients with facial nerve palsy include vertigo, hearing loss, and changes in facial sensation.
RHS was first described in 1907 as herpes zoster associated with Bell’s palsy by the neurologist J. Ramsay Hunt, for whom the condition is named.9,14 RHS is more common in men than women. It occurs most commonly in adults and is rare in children younger than 6.13,15
Diagnosis
In most cases, a diagnosis of RHS is made on a clinical basis.1 However, a polymerase chain reaction (PCR) assay can be performed on samples of tear fluid or submandibular saliva to detect the zoster virus.16,17 PCR can also be performed using exudates from the geniculate zone of the ear (a small area in the center of the auricle6,14), which is more sensitive than tears or blood.18,19 Findings from a complete blood count and the erythrocyte sedimentation rate can be used to differentiate between infectious and inflammatory causes.13
Head CT or MRI can be obtained to rule out any structural lesions. In one study, Kim et al20 examined MRI changes in patients with either Bell’s palsy or RHS. In both conditions, researchers were able to identify swelling of the labyrinthine segment of the facial nerve on temporal MRI scans.20 Although CT has not been shown to have any prognostic or diagnostic application, it can occasionally be used if decompression of the facial nerve is warranted.11
Treatment
Data used to support the use of corticosteroids for treatment of Bell’s palsy10,21,22 have been extrapolated to justify their use for treatment of RHS,23 and prednisolone is the most common choice.10 Steroids reduce the associated inflammation, resulting in decreased pain and neurologic symptoms. A daily dose for one to two weeks, followed by a slow taper, is the preferred prescribing method.10
The addition of acyclovir has been recommended to inhibit viral DNA replication9,23 (valacyclovir and famciclovir have also been mentioned12,18). If started within three days of symptom onset, acyclovir can help reduce pain and hasten resolution of symptoms.
In a large retrospective study, it was demonstrated that patients treated with prednisone at 1.0 mg/kg/d for five days, followed by a 10-day taper, combined with acyclovir, showed long-term improvement that was statistically significant.23 Complete facial recovery was reported in only 52% of patients, however. Risk factors for a poor prognosis include hypertension, diabetes mellitus, and advancing age.7
Artificial tears are also prescribed to keep the affected eye from becoming irritated and dry. The patient can be instructed to tape the eye closed at night.10
Early diagnosis and treatment (ie, within three days of symptom onset, and preferably with a combination of acyclovir and steroids23) is an important factor in a good prognosis.7,23 Because RHS-affected patients have only about a 50% chance of full recovery,23 proper follow-up care is extremely important. Follow-up visits are recommended at two weeks, six weeks, and three months.13 For optimal outcomes in patients with this neurologic diagnosis, referral to a neurologist is recommended for ongoing management. This practitioner is likely to detect subtle changes in patient presentation and can perform follow-up testing as needed.
THE CASE PATIENT
One week after the patient’s visit to the ED, he was contacted by hospital staff for a standard satisfaction and quality control survey. The patient (who had been treated with steroids and acyclovir, ibuprofen, and artificial tears) reported almost complete resolution of his pain; any mild pain, he said, was easily tolerated or could be resolved with OTC medication. He reported only minimal persistent facial weakness, stating that he was able to eat, drink, and speak normally.
The patient had not been seen by any health care provider for follow-up, but he agreed to make an appointment as soon as possible.
REFERENCES
1. Kim D, Bhimani M. Ramsay Hunt syndrome presenting as simple otitis externa. CJEM. 2008;10(3):247-250.
2. Agius AM, Pickles JM, Burch KL. A prospective study of otitis externa. Clin Otolaryngol. 1992;17(2):150-154.
3. Rosenfeld RM, Brown L, Cannon CR, et al; American Academy of Otolaryngology—Head and Neck Surgery Foundation. Clinical practice guideline: acute otitis externa. Otolaryngol Head Neck Surg. 2006;134(4 suppl):S4-S23.
4. Holland J, Bernstein J. Bell’s palsy. Clin Evid (Online). 2011 Mar 7;2011.pii:1204.
5. Jacewicz M. Bell’s palsy (2007). www.merckmanuals.com/professional/sec16/ch219/ch219i.html. Accessed May 26, 2011.
6. Harrison K. Discussion: the Ramsay Hunt Syndrome. Proc Royal Soc Med. 1953;47(371):11-24.
7. Bhupal HK. Ramsay Hunt syndrome presenting in primary care. Practitioner. 2010;254(1727):33-35.
8. Aizawa H, Ohtani F, Furuta Y, et al. Variable patterns of varicella-zoster virus reactivation in Ramsay Hunt syndrome. J Med Virol. 2004;74(2):355-360.
9. Gondivkar S, Parikh V, Parikh R. Herpes zoster oticus: a rare clinical entity. Contemp Clin Dent. 2010;1(2):127-129.
10. Sullivan FM, Swan IRC, Donnan PT, et al. Early treatment with prednisolone or acyclovir in Bell’s palsy. N Engl J Med. 2007;357(16):1598-1607.
11. Gilden DH. Bell’s palsy. N Engl J Med. 2004;351(13):1323-1331.
12. Diaz GA, Rakita RM, Koelle DM. A case of Ramsay Hunt–like syndrome caused by herpes simplex virus type 2. Clin Infect Dis. 2005;40(10):1545-1547.
13. Miravalle AA. Ramsay Hunt syndrome. http://emedicine.medscape.com/article/1166804-over iew. Accessed July 22, 2011.
14. Hunt JR. On herpetic inflammation of the geniculate ganglion: a new syndrome and its complications. J Nerv Ment Dis. 1907;34:73-96.
15. Sandoval CC, Núñez FA, Lizama CM, et al. Ramsay Hunt syndrome in children: four cases and review [in Spanish]. Rev Chilena Infectol. 2008; 25(6):458-464.
16. Murakami S, Nakashiro Y, Mizobuchi M, et al. Varicella-zoster virus distribution in Ramsay Hunt syndrome revealed by polymerase chain reaction. Acta Otolaryngol. 1998;118(2):145-149.
17. Hiroshige K, Ikeda M, Hondo R. Detection of varicella zoster virus DNA in tear fluid and saliva of patients with Ramsay Hunt syndrome. Otol Neurol. 2002;23(4):602-607.
18. Sweeney CJ, Gilden DH. Ramsay Hunt syndrome. J Neurol Neurosurg Psychiatr. 2001;71(2):148-154.
19. Murakami S, Honda N, Mizobuchi M, et al. Rapid diagnosis of varicella zoster virus infection in acute facial palsy. Neurology. 1998;51(4):1202-1205.
20. Kim IS, Shin SH, Kim J, et al. Correlation between MRI and operative findings in Bell’s palsy and Ramsay Hunt syndrome. Yonsei Med J. 2007;48(6):963-968.
21. Engström M, Berg T, Stjernquist-Desatnik A, et al. Prednisolone and valaciclovir in Bell’s palsy: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 2008;7(11):993-1000.
22. Hato N, Yamada H, Kohno H, et al. Valacyclovir and prednisolone treatment for Bell’s palsy: a multicenter, randomized, placebo-controlled study. Otol Neurotol. 2007;28(3):408-413.
23. Murakami S, Hato N, Horiuchi J, et al. Treatment of Ramsay Hunt syndrome with acyclovir-prednisone: significance of early diagnosis and treatment. Ann Neurol. 1997;41(3):353-357.
A 46-year-old man presented to a hospital emergency department (ED) with a four-day history of right ear pain. He described the pain as a constant, dull, burning pain radiating to the neck and face, associated with a feeling of congestion. The patient also stated that the right side of his face had felt numb for about one day.
Three days earlier, the man had been seen by his primary health care provider, who told him that his ear looked normal and free of infection. The day before his current presentation to the ED, however, he noticed what he described as an “acne-like” rash on his ear lobe. Shortly before coming to the ED, the patient also developed numbness over his right upper lip, which he likened to the effects of procaine during a dental visit. He reported drooling from the right side of his mouth while drinking water and difficulty blinking his right eye.
He denied any tinnitus, fever, headache, or change in hearing. A review of symptoms was positive only for mild dizziness during the previous two to three days.
The patient was a well-appearing white man. He was alert and oriented to identity, time, and place. His skin was warm, dry, and intact. The examiner noticed a small area of erythematous rash with vesicles on the man’s right ear lobe. The external auditory canals appeared within normal limits, with no erythema or edema, and were nontender bilaterally. The tympanic membranes were normal bilaterally, without bulging or discernible fluid levels.
The ocular exam was normal with no visual acuity changes and no fluorescein uptake; external ocular movements were intact. A slight droop was noted in the right eyelid, but there was no droop on the contralateral side of his face. When asked to puff up his cheeks, the patient found it difficult to do so on the right side of his mouth without releasing air from his lips.
The remainder of the cranial nerves were intact. Muscle strength was 5/5 in all extremities and equal bilaterally. The man’s gait was within normal limits, and the remaining findings in the physical exam were normal.
The initial diagnosis considered in the differential was otitis externa, because it is a common explanation for ear pain in patients who present to the ED.1,2 Also, in otitis, pain is characteristically present in the affected ear, and erythema is often found in the external auditory canal.3 However, this diagnosis was deemed unlikely because otitis externa would not explain the neurologic findings or the vesicular rash.1
Bell’s palsy was next in the differential, as it was considered consistent with the patient’s unilateral neurologic deficits.4 In addition to weakness or palsy of the facial nerve, many patients with Bell’s palsy complain of mastoid pain, which can be confused with a complaint of ear pain.5 However, patients with Bell’s palsy have no rash, and this diagnosis was considered unlikely.
The painful, burning rash on the patient’s face was characteristic of herpes zoster (shingles), which was next in the differential. Infrequently, shingles can also cause weakness in the nerve it affects. In the case patient, weakness that was evident in the affected nerve resembled that seen in Bell’s palsy. This combination of symptoms is referred to as Ramsay Hunt syndrome—which in this case was decided to be the correct diagnosis.
DISCUSSION
Ramsay Hunt syndrome (RHS, also known as geniculate herpes5,6) is caused by the varicella-zoster virus, most commonly known as the cause of chickenpox. In the United States, RHS is believed to affect only about one in 1,500 persons, although 20% to 30% of persons experience herpes zoster infection at some time.7
Soon after a chickenpox infection subsides, the virus spreads along the sensory nerve fibers of the peripheral and cranial nerves. The virus then becomes dormant in the dorsal root ganglion, where in some patients it later reactivates in the form of shingles.8
In RHS, the ganglia of cranial nerve VII (CN VII, the facial nerve, which innervates the facial muscles) are infected; for this reason, the condition is also referred to as zoster oticus.9 Because of the involvement and weakening of the facial nerve, the presentation of RHS often resembles that of Bell’s palsy or facial nerve palsy.
While most cases of Bell’s palsy are idiopathic,10,11 RHS can usually be attributed to viral infection—most commonly, infection with herpes simplex virus type 1 (HSV-1).12 RHS can be differentiated from Bell’s palsy by the presence of a rash on the ipsilateral side. The rash appears in the form of inflamed vesicles on an erythematous base and may be present around the ear (see figure), the eardrum, the hard and soft palate, or the tongue.6 When the rash is painful, it is often described as a burning pain. Loss of taste may occur in the anterior portion of the tongue.9,12
Unlike shingles, which usually manifests as a sensory neuropathy, RHS is distinguished by motor neuropathy.7 The patient usually reports weakness in the facial muscles on one side, leading to difficulty drinking water or puffing out the cheek and to drooling on one side of the face. A complaint of dryness in the ipsilateral eye may result from weakness or an inability to close the eyelid.
It is important to note that as in Bell’s palsy, RHS can be differentiated from stroke by the patient’s inability to wrinkle the forehead. The motor muscles of the forehead are innervated by both sides of the brain; in the case of stroke, only one side of the brain is affected, and movement of the forehead remains possible on the contralateral side. In facial nerve palsy, the nerve itself is affected; thus, no movement of the forehead is possible.13 Other common complaints in patients with facial nerve palsy include vertigo, hearing loss, and changes in facial sensation.
RHS was first described in 1907 as herpes zoster associated with Bell’s palsy by the neurologist J. Ramsay Hunt, for whom the condition is named.9,14 RHS is more common in men than women. It occurs most commonly in adults and is rare in children younger than 6.13,15
Diagnosis
In most cases, a diagnosis of RHS is made on a clinical basis.1 However, a polymerase chain reaction (PCR) assay can be performed on samples of tear fluid or submandibular saliva to detect the zoster virus.16,17 PCR can also be performed using exudates from the geniculate zone of the ear (a small area in the center of the auricle6,14), which is more sensitive than tears or blood.18,19 Findings from a complete blood count and the erythrocyte sedimentation rate can be used to differentiate between infectious and inflammatory causes.13
Head CT or MRI can be obtained to rule out any structural lesions. In one study, Kim et al20 examined MRI changes in patients with either Bell’s palsy or RHS. In both conditions, researchers were able to identify swelling of the labyrinthine segment of the facial nerve on temporal MRI scans.20 Although CT has not been shown to have any prognostic or diagnostic application, it can occasionally be used if decompression of the facial nerve is warranted.11
Treatment
Data used to support the use of corticosteroids for treatment of Bell’s palsy10,21,22 have been extrapolated to justify their use for treatment of RHS,23 and prednisolone is the most common choice.10 Steroids reduce the associated inflammation, resulting in decreased pain and neurologic symptoms. A daily dose for one to two weeks, followed by a slow taper, is the preferred prescribing method.10
The addition of acyclovir has been recommended to inhibit viral DNA replication9,23 (valacyclovir and famciclovir have also been mentioned12,18). If started within three days of symptom onset, acyclovir can help reduce pain and hasten resolution of symptoms.
In a large retrospective study, it was demonstrated that patients treated with prednisone at 1.0 mg/kg/d for five days, followed by a 10-day taper, combined with acyclovir, showed long-term improvement that was statistically significant.23 Complete facial recovery was reported in only 52% of patients, however. Risk factors for a poor prognosis include hypertension, diabetes mellitus, and advancing age.7
Artificial tears are also prescribed to keep the affected eye from becoming irritated and dry. The patient can be instructed to tape the eye closed at night.10
Early diagnosis and treatment (ie, within three days of symptom onset, and preferably with a combination of acyclovir and steroids23) is an important factor in a good prognosis.7,23 Because RHS-affected patients have only about a 50% chance of full recovery,23 proper follow-up care is extremely important. Follow-up visits are recommended at two weeks, six weeks, and three months.13 For optimal outcomes in patients with this neurologic diagnosis, referral to a neurologist is recommended for ongoing management. This practitioner is likely to detect subtle changes in patient presentation and can perform follow-up testing as needed.
THE CASE PATIENT
One week after the patient’s visit to the ED, he was contacted by hospital staff for a standard satisfaction and quality control survey. The patient (who had been treated with steroids and acyclovir, ibuprofen, and artificial tears) reported almost complete resolution of his pain; any mild pain, he said, was easily tolerated or could be resolved with OTC medication. He reported only minimal persistent facial weakness, stating that he was able to eat, drink, and speak normally.
The patient had not been seen by any health care provider for follow-up, but he agreed to make an appointment as soon as possible.
REFERENCES
1. Kim D, Bhimani M. Ramsay Hunt syndrome presenting as simple otitis externa. CJEM. 2008;10(3):247-250.
2. Agius AM, Pickles JM, Burch KL. A prospective study of otitis externa. Clin Otolaryngol. 1992;17(2):150-154.
3. Rosenfeld RM, Brown L, Cannon CR, et al; American Academy of Otolaryngology—Head and Neck Surgery Foundation. Clinical practice guideline: acute otitis externa. Otolaryngol Head Neck Surg. 2006;134(4 suppl):S4-S23.
4. Holland J, Bernstein J. Bell’s palsy. Clin Evid (Online). 2011 Mar 7;2011.pii:1204.
5. Jacewicz M. Bell’s palsy (2007). www.merckmanuals.com/professional/sec16/ch219/ch219i.html. Accessed May 26, 2011.
6. Harrison K. Discussion: the Ramsay Hunt Syndrome. Proc Royal Soc Med. 1953;47(371):11-24.
7. Bhupal HK. Ramsay Hunt syndrome presenting in primary care. Practitioner. 2010;254(1727):33-35.
8. Aizawa H, Ohtani F, Furuta Y, et al. Variable patterns of varicella-zoster virus reactivation in Ramsay Hunt syndrome. J Med Virol. 2004;74(2):355-360.
9. Gondivkar S, Parikh V, Parikh R. Herpes zoster oticus: a rare clinical entity. Contemp Clin Dent. 2010;1(2):127-129.
10. Sullivan FM, Swan IRC, Donnan PT, et al. Early treatment with prednisolone or acyclovir in Bell’s palsy. N Engl J Med. 2007;357(16):1598-1607.
11. Gilden DH. Bell’s palsy. N Engl J Med. 2004;351(13):1323-1331.
12. Diaz GA, Rakita RM, Koelle DM. A case of Ramsay Hunt–like syndrome caused by herpes simplex virus type 2. Clin Infect Dis. 2005;40(10):1545-1547.
13. Miravalle AA. Ramsay Hunt syndrome. http://emedicine.medscape.com/article/1166804-over iew. Accessed July 22, 2011.
14. Hunt JR. On herpetic inflammation of the geniculate ganglion: a new syndrome and its complications. J Nerv Ment Dis. 1907;34:73-96.
15. Sandoval CC, Núñez FA, Lizama CM, et al. Ramsay Hunt syndrome in children: four cases and review [in Spanish]. Rev Chilena Infectol. 2008; 25(6):458-464.
16. Murakami S, Nakashiro Y, Mizobuchi M, et al. Varicella-zoster virus distribution in Ramsay Hunt syndrome revealed by polymerase chain reaction. Acta Otolaryngol. 1998;118(2):145-149.
17. Hiroshige K, Ikeda M, Hondo R. Detection of varicella zoster virus DNA in tear fluid and saliva of patients with Ramsay Hunt syndrome. Otol Neurol. 2002;23(4):602-607.
18. Sweeney CJ, Gilden DH. Ramsay Hunt syndrome. J Neurol Neurosurg Psychiatr. 2001;71(2):148-154.
19. Murakami S, Honda N, Mizobuchi M, et al. Rapid diagnosis of varicella zoster virus infection in acute facial palsy. Neurology. 1998;51(4):1202-1205.
20. Kim IS, Shin SH, Kim J, et al. Correlation between MRI and operative findings in Bell’s palsy and Ramsay Hunt syndrome. Yonsei Med J. 2007;48(6):963-968.
21. Engström M, Berg T, Stjernquist-Desatnik A, et al. Prednisolone and valaciclovir in Bell’s palsy: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 2008;7(11):993-1000.
22. Hato N, Yamada H, Kohno H, et al. Valacyclovir and prednisolone treatment for Bell’s palsy: a multicenter, randomized, placebo-controlled study. Otol Neurotol. 2007;28(3):408-413.
23. Murakami S, Hato N, Horiuchi J, et al. Treatment of Ramsay Hunt syndrome with acyclovir-prednisone: significance of early diagnosis and treatment. Ann Neurol. 1997;41(3):353-357.
A 46-year-old man presented to a hospital emergency department (ED) with a four-day history of right ear pain. He described the pain as a constant, dull, burning pain radiating to the neck and face, associated with a feeling of congestion. The patient also stated that the right side of his face had felt numb for about one day.
Three days earlier, the man had been seen by his primary health care provider, who told him that his ear looked normal and free of infection. The day before his current presentation to the ED, however, he noticed what he described as an “acne-like” rash on his ear lobe. Shortly before coming to the ED, the patient also developed numbness over his right upper lip, which he likened to the effects of procaine during a dental visit. He reported drooling from the right side of his mouth while drinking water and difficulty blinking his right eye.
He denied any tinnitus, fever, headache, or change in hearing. A review of symptoms was positive only for mild dizziness during the previous two to three days.
The patient was a well-appearing white man. He was alert and oriented to identity, time, and place. His skin was warm, dry, and intact. The examiner noticed a small area of erythematous rash with vesicles on the man’s right ear lobe. The external auditory canals appeared within normal limits, with no erythema or edema, and were nontender bilaterally. The tympanic membranes were normal bilaterally, without bulging or discernible fluid levels.
The ocular exam was normal with no visual acuity changes and no fluorescein uptake; external ocular movements were intact. A slight droop was noted in the right eyelid, but there was no droop on the contralateral side of his face. When asked to puff up his cheeks, the patient found it difficult to do so on the right side of his mouth without releasing air from his lips.
The remainder of the cranial nerves were intact. Muscle strength was 5/5 in all extremities and equal bilaterally. The man’s gait was within normal limits, and the remaining findings in the physical exam were normal.
The initial diagnosis considered in the differential was otitis externa, because it is a common explanation for ear pain in patients who present to the ED.1,2 Also, in otitis, pain is characteristically present in the affected ear, and erythema is often found in the external auditory canal.3 However, this diagnosis was deemed unlikely because otitis externa would not explain the neurologic findings or the vesicular rash.1
Bell’s palsy was next in the differential, as it was considered consistent with the patient’s unilateral neurologic deficits.4 In addition to weakness or palsy of the facial nerve, many patients with Bell’s palsy complain of mastoid pain, which can be confused with a complaint of ear pain.5 However, patients with Bell’s palsy have no rash, and this diagnosis was considered unlikely.
The painful, burning rash on the patient’s face was characteristic of herpes zoster (shingles), which was next in the differential. Infrequently, shingles can also cause weakness in the nerve it affects. In the case patient, weakness that was evident in the affected nerve resembled that seen in Bell’s palsy. This combination of symptoms is referred to as Ramsay Hunt syndrome—which in this case was decided to be the correct diagnosis.
DISCUSSION
Ramsay Hunt syndrome (RHS, also known as geniculate herpes5,6) is caused by the varicella-zoster virus, most commonly known as the cause of chickenpox. In the United States, RHS is believed to affect only about one in 1,500 persons, although 20% to 30% of persons experience herpes zoster infection at some time.7
Soon after a chickenpox infection subsides, the virus spreads along the sensory nerve fibers of the peripheral and cranial nerves. The virus then becomes dormant in the dorsal root ganglion, where in some patients it later reactivates in the form of shingles.8
In RHS, the ganglia of cranial nerve VII (CN VII, the facial nerve, which innervates the facial muscles) are infected; for this reason, the condition is also referred to as zoster oticus.9 Because of the involvement and weakening of the facial nerve, the presentation of RHS often resembles that of Bell’s palsy or facial nerve palsy.
While most cases of Bell’s palsy are idiopathic,10,11 RHS can usually be attributed to viral infection—most commonly, infection with herpes simplex virus type 1 (HSV-1).12 RHS can be differentiated from Bell’s palsy by the presence of a rash on the ipsilateral side. The rash appears in the form of inflamed vesicles on an erythematous base and may be present around the ear (see figure), the eardrum, the hard and soft palate, or the tongue.6 When the rash is painful, it is often described as a burning pain. Loss of taste may occur in the anterior portion of the tongue.9,12
Unlike shingles, which usually manifests as a sensory neuropathy, RHS is distinguished by motor neuropathy.7 The patient usually reports weakness in the facial muscles on one side, leading to difficulty drinking water or puffing out the cheek and to drooling on one side of the face. A complaint of dryness in the ipsilateral eye may result from weakness or an inability to close the eyelid.
It is important to note that as in Bell’s palsy, RHS can be differentiated from stroke by the patient’s inability to wrinkle the forehead. The motor muscles of the forehead are innervated by both sides of the brain; in the case of stroke, only one side of the brain is affected, and movement of the forehead remains possible on the contralateral side. In facial nerve palsy, the nerve itself is affected; thus, no movement of the forehead is possible.13 Other common complaints in patients with facial nerve palsy include vertigo, hearing loss, and changes in facial sensation.
RHS was first described in 1907 as herpes zoster associated with Bell’s palsy by the neurologist J. Ramsay Hunt, for whom the condition is named.9,14 RHS is more common in men than women. It occurs most commonly in adults and is rare in children younger than 6.13,15
Diagnosis
In most cases, a diagnosis of RHS is made on a clinical basis.1 However, a polymerase chain reaction (PCR) assay can be performed on samples of tear fluid or submandibular saliva to detect the zoster virus.16,17 PCR can also be performed using exudates from the geniculate zone of the ear (a small area in the center of the auricle6,14), which is more sensitive than tears or blood.18,19 Findings from a complete blood count and the erythrocyte sedimentation rate can be used to differentiate between infectious and inflammatory causes.13
Head CT or MRI can be obtained to rule out any structural lesions. In one study, Kim et al20 examined MRI changes in patients with either Bell’s palsy or RHS. In both conditions, researchers were able to identify swelling of the labyrinthine segment of the facial nerve on temporal MRI scans.20 Although CT has not been shown to have any prognostic or diagnostic application, it can occasionally be used if decompression of the facial nerve is warranted.11
Treatment
Data used to support the use of corticosteroids for treatment of Bell’s palsy10,21,22 have been extrapolated to justify their use for treatment of RHS,23 and prednisolone is the most common choice.10 Steroids reduce the associated inflammation, resulting in decreased pain and neurologic symptoms. A daily dose for one to two weeks, followed by a slow taper, is the preferred prescribing method.10
The addition of acyclovir has been recommended to inhibit viral DNA replication9,23 (valacyclovir and famciclovir have also been mentioned12,18). If started within three days of symptom onset, acyclovir can help reduce pain and hasten resolution of symptoms.
In a large retrospective study, it was demonstrated that patients treated with prednisone at 1.0 mg/kg/d for five days, followed by a 10-day taper, combined with acyclovir, showed long-term improvement that was statistically significant.23 Complete facial recovery was reported in only 52% of patients, however. Risk factors for a poor prognosis include hypertension, diabetes mellitus, and advancing age.7
Artificial tears are also prescribed to keep the affected eye from becoming irritated and dry. The patient can be instructed to tape the eye closed at night.10
Early diagnosis and treatment (ie, within three days of symptom onset, and preferably with a combination of acyclovir and steroids23) is an important factor in a good prognosis.7,23 Because RHS-affected patients have only about a 50% chance of full recovery,23 proper follow-up care is extremely important. Follow-up visits are recommended at two weeks, six weeks, and three months.13 For optimal outcomes in patients with this neurologic diagnosis, referral to a neurologist is recommended for ongoing management. This practitioner is likely to detect subtle changes in patient presentation and can perform follow-up testing as needed.
THE CASE PATIENT
One week after the patient’s visit to the ED, he was contacted by hospital staff for a standard satisfaction and quality control survey. The patient (who had been treated with steroids and acyclovir, ibuprofen, and artificial tears) reported almost complete resolution of his pain; any mild pain, he said, was easily tolerated or could be resolved with OTC medication. He reported only minimal persistent facial weakness, stating that he was able to eat, drink, and speak normally.
The patient had not been seen by any health care provider for follow-up, but he agreed to make an appointment as soon as possible.
REFERENCES
1. Kim D, Bhimani M. Ramsay Hunt syndrome presenting as simple otitis externa. CJEM. 2008;10(3):247-250.
2. Agius AM, Pickles JM, Burch KL. A prospective study of otitis externa. Clin Otolaryngol. 1992;17(2):150-154.
3. Rosenfeld RM, Brown L, Cannon CR, et al; American Academy of Otolaryngology—Head and Neck Surgery Foundation. Clinical practice guideline: acute otitis externa. Otolaryngol Head Neck Surg. 2006;134(4 suppl):S4-S23.
4. Holland J, Bernstein J. Bell’s palsy. Clin Evid (Online). 2011 Mar 7;2011.pii:1204.
5. Jacewicz M. Bell’s palsy (2007). www.merckmanuals.com/professional/sec16/ch219/ch219i.html. Accessed May 26, 2011.
6. Harrison K. Discussion: the Ramsay Hunt Syndrome. Proc Royal Soc Med. 1953;47(371):11-24.
7. Bhupal HK. Ramsay Hunt syndrome presenting in primary care. Practitioner. 2010;254(1727):33-35.
8. Aizawa H, Ohtani F, Furuta Y, et al. Variable patterns of varicella-zoster virus reactivation in Ramsay Hunt syndrome. J Med Virol. 2004;74(2):355-360.
9. Gondivkar S, Parikh V, Parikh R. Herpes zoster oticus: a rare clinical entity. Contemp Clin Dent. 2010;1(2):127-129.
10. Sullivan FM, Swan IRC, Donnan PT, et al. Early treatment with prednisolone or acyclovir in Bell’s palsy. N Engl J Med. 2007;357(16):1598-1607.
11. Gilden DH. Bell’s palsy. N Engl J Med. 2004;351(13):1323-1331.
12. Diaz GA, Rakita RM, Koelle DM. A case of Ramsay Hunt–like syndrome caused by herpes simplex virus type 2. Clin Infect Dis. 2005;40(10):1545-1547.
13. Miravalle AA. Ramsay Hunt syndrome. http://emedicine.medscape.com/article/1166804-over iew. Accessed July 22, 2011.
14. Hunt JR. On herpetic inflammation of the geniculate ganglion: a new syndrome and its complications. J Nerv Ment Dis. 1907;34:73-96.
15. Sandoval CC, Núñez FA, Lizama CM, et al. Ramsay Hunt syndrome in children: four cases and review [in Spanish]. Rev Chilena Infectol. 2008; 25(6):458-464.
16. Murakami S, Nakashiro Y, Mizobuchi M, et al. Varicella-zoster virus distribution in Ramsay Hunt syndrome revealed by polymerase chain reaction. Acta Otolaryngol. 1998;118(2):145-149.
17. Hiroshige K, Ikeda M, Hondo R. Detection of varicella zoster virus DNA in tear fluid and saliva of patients with Ramsay Hunt syndrome. Otol Neurol. 2002;23(4):602-607.
18. Sweeney CJ, Gilden DH. Ramsay Hunt syndrome. J Neurol Neurosurg Psychiatr. 2001;71(2):148-154.
19. Murakami S, Honda N, Mizobuchi M, et al. Rapid diagnosis of varicella zoster virus infection in acute facial palsy. Neurology. 1998;51(4):1202-1205.
20. Kim IS, Shin SH, Kim J, et al. Correlation between MRI and operative findings in Bell’s palsy and Ramsay Hunt syndrome. Yonsei Med J. 2007;48(6):963-968.
21. Engström M, Berg T, Stjernquist-Desatnik A, et al. Prednisolone and valaciclovir in Bell’s palsy: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 2008;7(11):993-1000.
22. Hato N, Yamada H, Kohno H, et al. Valacyclovir and prednisolone treatment for Bell’s palsy: a multicenter, randomized, placebo-controlled study. Otol Neurotol. 2007;28(3):408-413.
23. Murakami S, Hato N, Horiuchi J, et al. Treatment of Ramsay Hunt syndrome with acyclovir-prednisone: significance of early diagnosis and treatment. Ann Neurol. 1997;41(3):353-357.
Grand Rounds: Woman, 20, With Difficulty Walking
A 20-year-old woman presented to her primary care clinic with a chief complaint of lower leg weakness and difficulty walking. The weakness she described had been worsening over the previous four days, with progressively worsening tingling and numbness of her toes bilaterally.
The day before the patient presented, she noticed numbness and paresthesia in both calves. At the time of her presentation to the clinic, she complained of low back ache, paresthesia of both hands, numbness bilaterally to her groin, difficulty sitting upright, ataxia, and a numb, thick-feeling tongue. She denied fever, neck stiffness, shortness of breath, headache, or visual changes.
The patient stated that 10 days earlier, she had developed an upper respiratory infection for which she was seen at the clinic and treated with a seven-day course of amoxicillin/clavulanate 875/125 mg twice daily. She said that she had recovered completely.
A review of the patient’s systems revealed proximal muscle weakness bilaterally (2/5) and loss of touch-pressure in the lower extremities. She was experiencing paresthesia of the hands and mild weakness bilaterally (4/5). She also walked with an ataxic gait and had reduced deep tendon reflexes in the lower limbs. All cranial nerves were intact, and her vital signs were stable.
The woman’s medical history was positive only for asthma. Her family history included ischemic stroke in the maternal grandfather and brain tumor in the paternal grandfather. Social history was positive for alcohol intake (ranging from four to 12 beers per week). The patient said she had never smoked or used illicit drugs. She was an unmarried college student, living in a dorm on campus. She participated in track at school.
The patient was admitted to the hospital telemetry step-down unit, and a neurology consultation was requested. Tests were ordered, among them MRI of the head and spine and comprehensive blood work, to rule out neurologic, infectious, or metabolic causes of the patient’s weakness; urinalysis was also obtained. These tests all yielded negative results.
A lumbar puncture performed the following day revealed a cerebrospinal fluid (CSF) protein level of 570 mg/L (normal range, 150 to 450 mg/L). Leukocytes numbered 2 cells/mm3 (normal count, 0 to 10 cells/mm3).
Based on the patient’s presentation, history, and symptoms, a neurologist made a diagnosis of Guillain-Barré syndrome. It was decided that no electromyographic (EMG) study was required to rule out other disease processes (eg, spinal cord disease, multiple sclerosis, tumors).
The patient underwent a five-dose course of immunomodulatory therapy with IV immunoglobulin (IVIG). In the step-down unit, she experienced one incident of sinus bradycardia (ie, resting heart rate between 40 and 50 beats/min). Her blood pressure remained stable, as did her respiratory status, according to peak expiratory flow measured frequently at her bedside.
Physical therapy was initiated, consisting of passive and active range of motion, crossovers with the patient’s feet, and stair training. This was done in response to a complaint of ankle weakness, and it helped to strengthen weakened muscles and improve alignment while the patient was bedridden and in a weakened, fatigued state. Additionally, the patient was given enoxaparin, wore antiembolic hose, and used sequential compression devices while in bed. As a result of these measures, she never experienced a pulmonary embolus or deep vein thrombosis (DVT) as a result of being immobilized.
By the seventh day of hospitalization, the patient had stable vital signs and improved lower limb strength, and numbness was resolving in her hands and lower extremities. She was discharged to home, with physical therapy to resume on an outpatient basis.
Discussion
Guillain-Barré syndrome (GBS), an acute immune-mediated paralytic disorder,1 manifests in the form of weakness and diminished reflexes. Affecting the peripheral nerves, GBS is characterized by progressive symmetrical ascending weakness with varying degrees of sensory complaints.2,3
GBS occurs worldwide, and incidence is estimated between 1.1 and 1.8 cases per 100,000 persons.4 In the United States, GBS can be found in all age-groups, with peak incidence noted in elderly persons and young adults.5,6 Even with treatment, 3% to 10% of patients are reported to die of this illness, and 20% cannot walk six months after symptom onset.7 In one prospective population-based study of patients with confirmed GBS, 6% of patients died within 30 days of symptom onset, often as a result of respiratory complications.8
GBS is a postinfectious disorder, with cases developing several days or weeks after a viral or bacterial illness—most commonly, an upper respiratory infection or diarrhea (see Table 19-13). The most common trigger of GBS is infection with the bacterial microorganism Campylobacter jejuni (occurring in 15% to 40% of patients with GBS),9,14 a pathogen that can produce demyelination-causing antibodies. Other responsible pathogens include cytomegalovirus and Epstein-Barr virus.9 In a process called molecular mimicry, the immune system is unable to distinguish the amino acid of an infectious organism from the proteinaceous content of the peripheral nerve.15 Subsequently, the immune system attacks and destroys the myelin sheath.
An example of this is the apparent cross-reaction of the ganglioside GM1 with C jejuni lipopolysaccharide antigens.14,15 The resulting effect is immunologic damage to the peripheral nervous system. The flaccid paralysis that occurs in patients with GBS is thought to be caused by lymphocytic infiltration and complement activation of the spinal roots and peripheral nerves, where macrophages strip the myelin.5,15,16
Stages and Variants
Three stages characterize the course of GBS. The acute phase, which lasts one to four weeks, begins with onset of symptoms and persists until the associated neurologic deterioration has ceased. During the second phase, the plateau period, symptoms persist with no further deterioration; this stage can last several days to several weeks or months. The final phase, the recovery period, can last from four months to two years after symptom onset.15,17,18
The clinical course of GBS is highly variable and in many cases difficult to predict. Certain factors have been associated with a poor outcome: advancing age, previous presence of diarrhea, need for mechanical ventilation, an extended plateau phase, and a lower patient score on the Erasmus GBS Outcome Scale,19 when measured two weeks after GBS onset.8,20 This score can help predict the patient’s chance of independent walking after six months.15,19
Although the classic presenting symptom of GBS is symmetric ascending weakness, several disease variants have been identified, with differing symptoms and degrees of recovery. These variants also differ in terms of the muscle groups affected; in some, visual defects may be present at onset. GBS variants include21:
• Acute motor axonal neuropathy (AMAN)1,22
• Acute inflammatory demyelinating polyneuropathy (AIDP)1
• Pharyngeal-cervical-brachial variant23
• Purely sensory variant24
• Miller-Fisher syndrome, which manifests with ophthalmoplegia, in addition to ataxia and areflexia25
• Axonal form.5,21
AMAN and AIDP are the most common subtypes of GBS.1
Symptoms, Signs, and Disease Manifestations
Limb weakness, the classic presenting symptom of GBS, is both symmetrical and ascending. Weakness can develop acutely and progress over days to weeks.2,15 Hughes and Cornblath26 also note pain, numbness, and paresthesias among the initial symptoms of GBS. Others include sensory changes, cranial nerve involvement, various autonomic changes, and respiratory or oropharyngeal weakness. Reflexes, particularly the tendon reflexes, may be diminished or absent.15,18,21 In many cases, sensory changes (ie, pain) may precede the onset of weakness, often making diagnosis difficult.15
Cranial nerves most commonly affected are V, VI, VII, X, XI, and XII, with manifestations that include dysphagia, dysarthria, diplopia, limitation to eye movements, and facial droop and weakness. Usually facial and oropharyngeal weakness occur after the extremities and trunk are affected. Blindness may occur if demyelination of the optic nerve occurs; this is seen in Miller-Fisher syndrome.10,15,25,27
In GBS, many patients report pain, which can present as bilateral sciatica or as throbbing or aching in the large muscles of the upper legs, flanks, or back.28 This pain, which results from the demyelination of the sensory nerve fibers, can be severe.10
Patients with GBS may experience manifestations of autonomic nervous system dysfunction—for example, arrhythmias, hypotension or hypertension, urinary retention, cardiomyopathy, and paralytic ileus.10,20 Dysautonomia often impedes patients’ progress in inpatient rehabilitation. Patients may have persistent problems involving postural hypotension, hypertension, excessive sympathetic outflow, or bladder and bowel dysfunction.29
Blood pressure fluctuations, often attributed to changes in catecholamine levels and disturbances in the baroreceptor reflex pathway, are common and are considered characteristic of GBS. Transient or persistent hypotension is caused by the dysregulation of the parasympathetic and sympathetic systems, with subsequent alterations in venomotor tone.3 Additionally, an increased sensitivity to catecholamine can lead to cardiovascular disturbances, resulting in denervation hypersensitivity and impairment of the carotid sinus reflex.
Arrhythmias occur in perhaps half of patients with GBS. The most common is sustained sinus tachycardia, which usually requires no treatment. Bradycardia leading to atrioventricular blocks and asystole is believed to result from afferent baroreceptor reflex failure. Treatment may be required—either administration of atropine or insertion of a pacemaker, depending on the severity of the arrhythmia.3,10
Myocardial involvement can range from asymptomatic mycocarditis to neurogenic stunned myocardium and heart failure. Patients with ECG abnormalities should undergo two-dimensional echocardiographic studies and other testing to explore cardiac involvement. Acute coronary syndromes, including ST-segment elevation MI, have been reported, in some cases associated with IVIG treatment. In one patient, coronary spasm was reported, with clean coronary arteries found on cardiac catheterization.3
Patients with GBS are at risk for compromised neuromuscular respiratory function; demyelination of the nerves that innervate the intercostal muscles and the diaphragm can result in respiratory failure. Key clinical indicators of respiratory muscle fatigue include tachypnea, diaphoresis, and asynchronous movements of the abdomen and chest;10 other symptoms relevant to respiratory or oropharyngeal weakness include slurred speech, dyspnea (with or without exertion), difficulty swallowing, and inability to cough.2,10 Serial respiratory function testing is advisable to detect patients at risk for respiratory failure.30
Diagnosis
Guillain-Barré is a syndrome diagnosed by a collection of symptoms (see Table 22,21,31), including subacute developing paralysis, symmetrical bilateral weakness beginning at onset, and diminishing to absent reflexes.21,31 Other causes for rapidly developing weaknesses should be ruled out (see Table 310,21,26,31). Lumbar puncture typically shows increased protein levels with a normal white cell count; however, neither this test nor electrophysiologic evaluation offers significant value for diagnosis of GBS.21,26,31
During the acute phase of GBS (within three weeks of onset), there is found an elevation of CSF protein (> 550 mg/L) without an elevation in white blood cells. This phenomenon, called albuminocytologic dissociation, reflects inflammation of the nerve roots and is considered the hallmark of GBS.2
MRI can also facilitate the diagnosis of GBS; it demonstrates anterior and posterior intrathecal spinal nerve roots and cauda equina.32 In patients with GBS, evidence supporting breakdown of the blood–nerve barrier can be seen in abnormal gadolinium enhancement of the intrathecal nerve roots on MRI.33
When electrophysiologic studies are performed, they typically reveal slowing nerve conduction, prolonged distal latencies, and partial motor conduction block.34 The characteristic finding of early demyelination is conduction block, a reduction in the amplitude of the muscle action potential after stimulation of the distal, as opposed to the proximal, nerve.28 Nerve conduction studies may help in the diagnosis and classification of GBS—and, to a limited extent, formulation of a prognosis. Such alternative diagnoses as myositis and myasthenia gravis may be excluded by neurophysiology.26 Early in GBS, neurophysiologic abnormalities may be very mild or occasionally normal; test results may not correlate with clinical disability.35,36
The clinician cannot depend on clinical features alone to predict respiratory decline.31 Frequent evaluations of respiratory effort, by measurement of maximal inspiratory pressures and vital capacity, should be performed at the bedside to monitor diaphragmatic strength. Respiratory ventilation should be initiated if the patient becomes hypoxic or experiences a rapid decline in vital capacity (ie, below 60% of predicted value).10 Mechanical ventilation is more likely to be required in patients with a negative inspiratory force of less than 30 cm H2O.31
Treatment
Guillain-Barré syndrome has an acute onset and progression. Patients quickly become nonambulatory and may require total ventilation due to paralysis. Therapeutic options are IVIG or plasmapheresis (plasma exchange).37-40 Corticosteroids do not appear to benefit patients with GBS.41,42
Several mechanisms appear to contribute to the effectiveness of immunoglobulin.38,39 Infused IVIG interferes with antigen presentation, inhibits antibody production, neutralizes pathologic autoantibodies, and modulates other immunologic events involved in the pathogenesis of autoimmune neuromuscular diseases, including GBS.43 Adverse reactions, which are usually minor, include headache, fever, chills, myalgia, and malaise. In rare instances, anaphylaxis or renal failure may occur.15,44
In plasmapheresis, blood is removed from the body and dialyzed, with circulating antibodies and immunoglobulins removed from the plasma; fresh frozen plasma, albumin, or saline is administered. This treatment, performed via central venous catheter, should be initiated as soon as possible after onset of symptoms but can be implemented as late as 30 days after GBS onset. Plasmapheresis requires personnel trained in dialysis, which may not be performed in all hospitals. Possible adverse events include infection and hemorrhage. Laboratory values must be monitored for hypokalemia and hypocalcemia.45,46
Supportive Care
Patients with GBS require intensive care and very close monitoring for complications of respiratory difficulty and autonomic dysfunction. Individualized programs should be initiated for patients in the acute phase of GBS, aimed at the prevention of contractures and skin breakdown.10 Exercise programs, as conducted with the case patient, should also help relieve the fatigue syndromes that accompany GBS.
Immobilization associated with bed rest incurs a risk for pulmonary emboli and DVT; this has been found true during the first 12 weeks after symptom onset in patients with GBS who remain immobile.47 The use of antiembolic hose and sequential compression devices can help reduce the risk for thrombotic events.10 Use of enoxaparin or heparin is recommended for nonambulating patients until they are able to walk, with Gaber et al47 specifying the use of low-molecular-weight heparin to reduce, but not eliminate, the risk for DVT.
The pain associated with GBS can be severe. Narcotic analgesics may be administered with careful monitoring of autonomic denervation. Long-term management of neuropathic pain may require adjuvant therapy, such as tricyclic antidepressants, gabapentin, or tramadol hydrochloride.10 According to Pandey et al,48 gabapentin alone may suffice for pain control in GBS, with minimal adverse effects. In certain rehabilitation facilities, tricyclic antidepressants, capsaicin, and transcutaneous nerve stimulation have been reported effective; during the early stages of treatment, until these treatments reach their full effect, pain medications such as tramadol or narcotics can provide temporary relief.29
More than one-half of patients with GBS in the acute phase can develop ileus. Constipation can also occur as a result of pain medication use, prolonged bed rest, and poor intake. Auscultation of bowel sounds and abdominal assessment should be performed daily to monitor for ileus. Hughes et al10 do not recommend the use of promotility drugs in patients with dysautonomia.
After hospital discharge, easy fatigability can affect work and social activities. With continued physical therapy, occupational therapy, and monitoring, however, patients with GBS can expect to return to an optimal level of functioning. Speed of recovery varies with these patients from a few months to several years, depending on such factors as age and the extent to which axonal degeneration has occurred.6,49
The Case Patient
For several weeks after discharge, the case patient continued to experience fatigue, low back pain, and general muscle pain. With her family’s support, she continued to receive outpatient physical therapy, and within one month she had regained her ankle strength. She was soon able to resume her classes, despite some lingering fatigue.
Conclusion
Guillain-Barré syndrome is a potentially life-threatening disease whose symptoms health care providers need to recognize quickly to provide prompt treatment. Supportive care for both patient and family is of key importance for maximum rehabilitation and return to the previous lifestyle. The clinical course of GBS is highly variable and difficult to predict. The patient’s outcome depends on several factors, including age and severity of illness. GBS patients can experience long-term psychosocial effects.
References
1. Magira EE, Papaioakim M, Nachamkin I, et al. Differential distribution of HLA-DQ beta/DR beta epitopes in the two forms of Guillain-Barré syndrome, acute motor axonal neuropathy and acute inflammatory demyelinating polyneuropathy (AIDP): identification of DQ beta epitopes associated with susceptibility to and protection from AIDP. J Immunol. 2003;170(6):3074-3080.
2. Tremblay ME, Closon A, D’Anjou G, Bussières JF. Guillain-Barré syndrome following H1N1 immunization in a pediatric patient. Ann Pharmacother. 2010;44(7-8):1330-1333.
3. Mukerji S, Aloka F, Farooq MU, et al. Cardiovascular complications of the Guillain-Barré syndrome. Am J Cardiol. 2009;104(10):1452-1455.
4. McGrogan A, Madle GC, Seaman HE, de Vries CS. The epidemiology of Guillain-Barré syndrome worldwide: a systematic literature review. Neuroepidemiology. 2009;32(2):150-163.
5. Haber P, Sejvar J, Mikaeloff Y, DeStefano F. Vaccines and Guillain-Barré syndrome. Drug Saf. 2009; 32(4):309-323.
6. van Doorn PA. What’s new in Guillain-Barré syndrome in 2007-2008? J Periph Nerv Syst. 2009;14(2):72-74.
7. van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain-Barré syndrome. Lancet Neurol. 2008;7(10):939-950.
8. Chiò A, Cocito D, Leone M, et al; Piemonte and alle d’Aosta Register for Guillain-Barré Syndrome. Guillain-Barré syndrome: a prospective, population-based incidence and outcome survey. Neurology. 2003; 60(7):1146-1150.
9. Hadden RD, Karch H, Hartung HP, et al. Preceding infections, immune factors, and outcome in Guillain-Barré syndrome. Neurology. 2001;56(6):758-765.
10. Hughes RA, Wijdicks EF, Benson E, et al. Supportive care for patients with Guillain-Barré syndrome. Arch Neurol. 2005;62(8):1194-1198.
11. Aluka KJ, Turner PL, Fullum TM. Guillain-Barré syndrome and postbariatric surgery polyneuropathies. JSLS. 2009;13(2):250-253.
12. Brannagan TH 3rd, Zhou Y. HIV-associated Guillain-Barré syndrome. J Neurol Sci. 2003;208(1-2):39-42.
13. Lin WC, Lee PI, Lu CY, et al. Mycoplasma pneumoniae encephalitis in childhood. J Microbiol Immunol Infect. 2002;35(3):173-178.
14. Sivadon-Tardy V, Orlikowski D, Porcher R, et al. Detection of Campylobacter jejuni by culture and real-time PCR in a French cohort of patients with Guillain-Barre syndrome. J Clin Microbiol. 2010;48 (6):2278-2281.
15. van Doorn PA, Kuitwaard K, Walgaard C, et al. IVIG treatment and prognosis in Guillain-Barré syndrome. J Clin Immunol. 2010;30 suppl 1:S74-S78.
16. Kaida K, Kusunoki S. Guillan-Barré syndrome: update on immunobiology and treatment. Expert Rev Neurother. 2009;9(9):1307-1319.
17. Forsberg A, Press R, Einarsson U, et al. Disability and health-related quality of life in Guillain-Barré syndrome during the first two years after onset: a prospective study. Clin Rehabil. 2005;19(8):900-909.
18. Criteria for diagnosis of Guillain-Barré syndrome. Ann Neurol. 1978;3(6):565-566.
19. van Koningsveld R, Steyerberg EW, Hughes RA, et al. A clinical progostic scoring system for Guillain-Barré syndrome. Lancet Neurol. 2007;6(7):589-594.
20. Koeppen S, Kraywinkel K, Wessendorf TE, et al. Long-term outcome of Guillain-Barré syndrome. Neurocrit Care. 2006;5(3)235-242.
21. Sheridan JM, Smith D. Atypical Guillain-Barré in the emergency department. West J Emerg Med. 2010;11(1):80-82.
22. Ogawara K, Kuwabara S, Koga M, et al. Anti-GM1b IgG antibody is associated with acute motor axonal neuropathy and Campylobacter jejuni infection. J Neurol Sci. 2003;210(1-2):41-45.
23. Nagashima T, Koga M, Odaka M, et al. Continuous spectrum of pharyngeal-cervical-brachial variant of Guillain-Barré syndrome. Arch Neurol. 2007;64(10):1519-1523.
24. Oh SJ, LaGanke C, Claussen GC. Sensory Guillain-Barré syndrome. Neurology. 2001;56(1):82-86.
25. Aráranyi Z, Kovács T, Sipos I, Bereczki D. Miller Fisher syndrome: brief overview and update with a focus on electrophysiological findings. Eur J Neurol. 2011 Jun 1. [Epub ahead of print]
26. Hughes RA, Cornblath, DR. Guillain-Barré syndrome. Lancet. 2005;366(9497):1653-1666.
27. Snyder LA, Rismondo V, Miller NR. The Fisher variant of Guillain-Barré syndrome (Fisher syndrome). J Neuroophthalmol. 2009;29(4):312-324.
28. Ropper AH. The Guillain-Barré syndrome. N Engl J Med.1992;326(17):1130-1136.
29. Meythaler JM. Rehabilitation of Guillain-Barré syndrome. Arch Phys Med Rehabil.1997;78(8):872-879.
30. Sharshar T, Chevret S, Bourdain F, et al; French Cooperative Group on Plasma Exchange in Guillain-Barré syndrome. Early predictors of mechanical ventilation in Guillain-Barré syndrome. Crit Care Med. 2003; 31(1):278-283.
31. McGillicuddy DC, Walker O, Shapiro NI, et al. Guillain-Barré syndrome in the emergency department. Ann Emerg Med. 2006;47(4):390-393.
32. Yikilmaz A, Doganay S, Gumus H, et al. Magnetic resonance imaging of childhood Guillain-Barré syndrome. Childs Nerv Syst. 2010;26(8):1103-1108.
33. Gonzalez-Quevedo A, Carriera RF, O’Farrill ZL, et al. An appraisal of blood-cerebrospinal fluid barrier dysfunction during the course of Guillain-Barré syndrome. Neurol India. 2009;57(3):288-294.
34. Abai S, Kim SB, Kim JP, Lim YJ. Guillan-Barré syndrome combined with acute cervical myelopathy. J Korean Neurosurg Soc. 2010;48(3):298-300.
35. Uncini A, Yuki N. Electrophysiologic and immunopathologic correlates in Guillain-Barré syndrome subtypes. Expert Rev Neurother. 2009;9(6):869-884.
36. Hadden RD, Hughes RA. Management of inflammatory neuropathies. J Neurol Neurosurg Psychiatry. 2003;74 suppl 2:ii9-ii14.
37. Raphaël JC, Chevret S, Hughes RA, Annane D. Plasma exchange for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2002;(2):CD001798.
38. Hughes RA, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2010 Jun 16; (6):CD002063.
39. Human immunoglobulin and the Guillain-Barré syndrome: new indication. An alternative to plasmapheresis. Prescrire Int. 2000;9(49):142-143.
40. van der Meché FG, Schmitz PI; Dutch Guillain-Barré Study Group. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. N Engl J Med. 1992;327(17):1123-1129.
41. Hughes RA, Swan AV, van Doorn PA. Corticosteroids for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2010 Feb 16;(2):CD001446.
42. Hahn AF. Guillain-Barré syndrome. Lancet. 1998; 352(9128):635-641.
43. Dalakas MC. Intravenous immunoglobulin in autoimmune neuromuscular diseases. JAMA. 2004;291(19):2367-2375.
44. Kuitwaard K, de Gelder J, Tio-Gillen AP, et al. Pharmacokenetics of intravenous immunoglobulin and outcome in Guillain-Barré syndrome. Ann Neurol. 2009;66(5):597-603.
45. Atkinson SB, Carr RL, Maybee P, Haynes D. The challenges of managing and treating Guillain-Barré syndrome during the acute phase. Dimens Crit Care Nurs. 2006;25(6):256-263.
46. van Doorn PA. Treatment of Guillain-Barré syndrome and CIDP. J Periph Nerv Syst. 2005;10(2):113-127.
47. Gaber TA, Kirker SGB, Jenner JR. Current practice of prophylactic anticoagulation in Guillain-Barré syndrome. Clin Rehabil. 2002;16(2):190-193.
48. Pandey CK, Bose N, Garg G, et al. Gabapentin for the treatment of pain in Guillain-Barré syndrome: a double-blinded, placebo-controlled, crossover study. Anesth Analg. 2002;95(6):1719-1723.
49. de Vries JM, Hagemans ML, Bussmann JB, et al. Fatigue in neuromuscular disorders: focus on Guillain-Barré syndrome and Pompe disease. Cell Mol Life Sci. 2010;67(5):701-713.
A 20-year-old woman presented to her primary care clinic with a chief complaint of lower leg weakness and difficulty walking. The weakness she described had been worsening over the previous four days, with progressively worsening tingling and numbness of her toes bilaterally.
The day before the patient presented, she noticed numbness and paresthesia in both calves. At the time of her presentation to the clinic, she complained of low back ache, paresthesia of both hands, numbness bilaterally to her groin, difficulty sitting upright, ataxia, and a numb, thick-feeling tongue. She denied fever, neck stiffness, shortness of breath, headache, or visual changes.
The patient stated that 10 days earlier, she had developed an upper respiratory infection for which she was seen at the clinic and treated with a seven-day course of amoxicillin/clavulanate 875/125 mg twice daily. She said that she had recovered completely.
A review of the patient’s systems revealed proximal muscle weakness bilaterally (2/5) and loss of touch-pressure in the lower extremities. She was experiencing paresthesia of the hands and mild weakness bilaterally (4/5). She also walked with an ataxic gait and had reduced deep tendon reflexes in the lower limbs. All cranial nerves were intact, and her vital signs were stable.
The woman’s medical history was positive only for asthma. Her family history included ischemic stroke in the maternal grandfather and brain tumor in the paternal grandfather. Social history was positive for alcohol intake (ranging from four to 12 beers per week). The patient said she had never smoked or used illicit drugs. She was an unmarried college student, living in a dorm on campus. She participated in track at school.
The patient was admitted to the hospital telemetry step-down unit, and a neurology consultation was requested. Tests were ordered, among them MRI of the head and spine and comprehensive blood work, to rule out neurologic, infectious, or metabolic causes of the patient’s weakness; urinalysis was also obtained. These tests all yielded negative results.
A lumbar puncture performed the following day revealed a cerebrospinal fluid (CSF) protein level of 570 mg/L (normal range, 150 to 450 mg/L). Leukocytes numbered 2 cells/mm3 (normal count, 0 to 10 cells/mm3).
Based on the patient’s presentation, history, and symptoms, a neurologist made a diagnosis of Guillain-Barré syndrome. It was decided that no electromyographic (EMG) study was required to rule out other disease processes (eg, spinal cord disease, multiple sclerosis, tumors).
The patient underwent a five-dose course of immunomodulatory therapy with IV immunoglobulin (IVIG). In the step-down unit, she experienced one incident of sinus bradycardia (ie, resting heart rate between 40 and 50 beats/min). Her blood pressure remained stable, as did her respiratory status, according to peak expiratory flow measured frequently at her bedside.
Physical therapy was initiated, consisting of passive and active range of motion, crossovers with the patient’s feet, and stair training. This was done in response to a complaint of ankle weakness, and it helped to strengthen weakened muscles and improve alignment while the patient was bedridden and in a weakened, fatigued state. Additionally, the patient was given enoxaparin, wore antiembolic hose, and used sequential compression devices while in bed. As a result of these measures, she never experienced a pulmonary embolus or deep vein thrombosis (DVT) as a result of being immobilized.
By the seventh day of hospitalization, the patient had stable vital signs and improved lower limb strength, and numbness was resolving in her hands and lower extremities. She was discharged to home, with physical therapy to resume on an outpatient basis.
Discussion
Guillain-Barré syndrome (GBS), an acute immune-mediated paralytic disorder,1 manifests in the form of weakness and diminished reflexes. Affecting the peripheral nerves, GBS is characterized by progressive symmetrical ascending weakness with varying degrees of sensory complaints.2,3
GBS occurs worldwide, and incidence is estimated between 1.1 and 1.8 cases per 100,000 persons.4 In the United States, GBS can be found in all age-groups, with peak incidence noted in elderly persons and young adults.5,6 Even with treatment, 3% to 10% of patients are reported to die of this illness, and 20% cannot walk six months after symptom onset.7 In one prospective population-based study of patients with confirmed GBS, 6% of patients died within 30 days of symptom onset, often as a result of respiratory complications.8
GBS is a postinfectious disorder, with cases developing several days or weeks after a viral or bacterial illness—most commonly, an upper respiratory infection or diarrhea (see Table 19-13). The most common trigger of GBS is infection with the bacterial microorganism Campylobacter jejuni (occurring in 15% to 40% of patients with GBS),9,14 a pathogen that can produce demyelination-causing antibodies. Other responsible pathogens include cytomegalovirus and Epstein-Barr virus.9 In a process called molecular mimicry, the immune system is unable to distinguish the amino acid of an infectious organism from the proteinaceous content of the peripheral nerve.15 Subsequently, the immune system attacks and destroys the myelin sheath.
An example of this is the apparent cross-reaction of the ganglioside GM1 with C jejuni lipopolysaccharide antigens.14,15 The resulting effect is immunologic damage to the peripheral nervous system. The flaccid paralysis that occurs in patients with GBS is thought to be caused by lymphocytic infiltration and complement activation of the spinal roots and peripheral nerves, where macrophages strip the myelin.5,15,16
Stages and Variants
Three stages characterize the course of GBS. The acute phase, which lasts one to four weeks, begins with onset of symptoms and persists until the associated neurologic deterioration has ceased. During the second phase, the plateau period, symptoms persist with no further deterioration; this stage can last several days to several weeks or months. The final phase, the recovery period, can last from four months to two years after symptom onset.15,17,18
The clinical course of GBS is highly variable and in many cases difficult to predict. Certain factors have been associated with a poor outcome: advancing age, previous presence of diarrhea, need for mechanical ventilation, an extended plateau phase, and a lower patient score on the Erasmus GBS Outcome Scale,19 when measured two weeks after GBS onset.8,20 This score can help predict the patient’s chance of independent walking after six months.15,19
Although the classic presenting symptom of GBS is symmetric ascending weakness, several disease variants have been identified, with differing symptoms and degrees of recovery. These variants also differ in terms of the muscle groups affected; in some, visual defects may be present at onset. GBS variants include21:
• Acute motor axonal neuropathy (AMAN)1,22
• Acute inflammatory demyelinating polyneuropathy (AIDP)1
• Pharyngeal-cervical-brachial variant23
• Purely sensory variant24
• Miller-Fisher syndrome, which manifests with ophthalmoplegia, in addition to ataxia and areflexia25
• Axonal form.5,21
AMAN and AIDP are the most common subtypes of GBS.1
Symptoms, Signs, and Disease Manifestations
Limb weakness, the classic presenting symptom of GBS, is both symmetrical and ascending. Weakness can develop acutely and progress over days to weeks.2,15 Hughes and Cornblath26 also note pain, numbness, and paresthesias among the initial symptoms of GBS. Others include sensory changes, cranial nerve involvement, various autonomic changes, and respiratory or oropharyngeal weakness. Reflexes, particularly the tendon reflexes, may be diminished or absent.15,18,21 In many cases, sensory changes (ie, pain) may precede the onset of weakness, often making diagnosis difficult.15
Cranial nerves most commonly affected are V, VI, VII, X, XI, and XII, with manifestations that include dysphagia, dysarthria, diplopia, limitation to eye movements, and facial droop and weakness. Usually facial and oropharyngeal weakness occur after the extremities and trunk are affected. Blindness may occur if demyelination of the optic nerve occurs; this is seen in Miller-Fisher syndrome.10,15,25,27
In GBS, many patients report pain, which can present as bilateral sciatica or as throbbing or aching in the large muscles of the upper legs, flanks, or back.28 This pain, which results from the demyelination of the sensory nerve fibers, can be severe.10
Patients with GBS may experience manifestations of autonomic nervous system dysfunction—for example, arrhythmias, hypotension or hypertension, urinary retention, cardiomyopathy, and paralytic ileus.10,20 Dysautonomia often impedes patients’ progress in inpatient rehabilitation. Patients may have persistent problems involving postural hypotension, hypertension, excessive sympathetic outflow, or bladder and bowel dysfunction.29
Blood pressure fluctuations, often attributed to changes in catecholamine levels and disturbances in the baroreceptor reflex pathway, are common and are considered characteristic of GBS. Transient or persistent hypotension is caused by the dysregulation of the parasympathetic and sympathetic systems, with subsequent alterations in venomotor tone.3 Additionally, an increased sensitivity to catecholamine can lead to cardiovascular disturbances, resulting in denervation hypersensitivity and impairment of the carotid sinus reflex.
Arrhythmias occur in perhaps half of patients with GBS. The most common is sustained sinus tachycardia, which usually requires no treatment. Bradycardia leading to atrioventricular blocks and asystole is believed to result from afferent baroreceptor reflex failure. Treatment may be required—either administration of atropine or insertion of a pacemaker, depending on the severity of the arrhythmia.3,10
Myocardial involvement can range from asymptomatic mycocarditis to neurogenic stunned myocardium and heart failure. Patients with ECG abnormalities should undergo two-dimensional echocardiographic studies and other testing to explore cardiac involvement. Acute coronary syndromes, including ST-segment elevation MI, have been reported, in some cases associated with IVIG treatment. In one patient, coronary spasm was reported, with clean coronary arteries found on cardiac catheterization.3
Patients with GBS are at risk for compromised neuromuscular respiratory function; demyelination of the nerves that innervate the intercostal muscles and the diaphragm can result in respiratory failure. Key clinical indicators of respiratory muscle fatigue include tachypnea, diaphoresis, and asynchronous movements of the abdomen and chest;10 other symptoms relevant to respiratory or oropharyngeal weakness include slurred speech, dyspnea (with or without exertion), difficulty swallowing, and inability to cough.2,10 Serial respiratory function testing is advisable to detect patients at risk for respiratory failure.30
Diagnosis
Guillain-Barré is a syndrome diagnosed by a collection of symptoms (see Table 22,21,31), including subacute developing paralysis, symmetrical bilateral weakness beginning at onset, and diminishing to absent reflexes.21,31 Other causes for rapidly developing weaknesses should be ruled out (see Table 310,21,26,31). Lumbar puncture typically shows increased protein levels with a normal white cell count; however, neither this test nor electrophysiologic evaluation offers significant value for diagnosis of GBS.21,26,31
During the acute phase of GBS (within three weeks of onset), there is found an elevation of CSF protein (> 550 mg/L) without an elevation in white blood cells. This phenomenon, called albuminocytologic dissociation, reflects inflammation of the nerve roots and is considered the hallmark of GBS.2
MRI can also facilitate the diagnosis of GBS; it demonstrates anterior and posterior intrathecal spinal nerve roots and cauda equina.32 In patients with GBS, evidence supporting breakdown of the blood–nerve barrier can be seen in abnormal gadolinium enhancement of the intrathecal nerve roots on MRI.33
When electrophysiologic studies are performed, they typically reveal slowing nerve conduction, prolonged distal latencies, and partial motor conduction block.34 The characteristic finding of early demyelination is conduction block, a reduction in the amplitude of the muscle action potential after stimulation of the distal, as opposed to the proximal, nerve.28 Nerve conduction studies may help in the diagnosis and classification of GBS—and, to a limited extent, formulation of a prognosis. Such alternative diagnoses as myositis and myasthenia gravis may be excluded by neurophysiology.26 Early in GBS, neurophysiologic abnormalities may be very mild or occasionally normal; test results may not correlate with clinical disability.35,36
The clinician cannot depend on clinical features alone to predict respiratory decline.31 Frequent evaluations of respiratory effort, by measurement of maximal inspiratory pressures and vital capacity, should be performed at the bedside to monitor diaphragmatic strength. Respiratory ventilation should be initiated if the patient becomes hypoxic or experiences a rapid decline in vital capacity (ie, below 60% of predicted value).10 Mechanical ventilation is more likely to be required in patients with a negative inspiratory force of less than 30 cm H2O.31
Treatment
Guillain-Barré syndrome has an acute onset and progression. Patients quickly become nonambulatory and may require total ventilation due to paralysis. Therapeutic options are IVIG or plasmapheresis (plasma exchange).37-40 Corticosteroids do not appear to benefit patients with GBS.41,42
Several mechanisms appear to contribute to the effectiveness of immunoglobulin.38,39 Infused IVIG interferes with antigen presentation, inhibits antibody production, neutralizes pathologic autoantibodies, and modulates other immunologic events involved in the pathogenesis of autoimmune neuromuscular diseases, including GBS.43 Adverse reactions, which are usually minor, include headache, fever, chills, myalgia, and malaise. In rare instances, anaphylaxis or renal failure may occur.15,44
In plasmapheresis, blood is removed from the body and dialyzed, with circulating antibodies and immunoglobulins removed from the plasma; fresh frozen plasma, albumin, or saline is administered. This treatment, performed via central venous catheter, should be initiated as soon as possible after onset of symptoms but can be implemented as late as 30 days after GBS onset. Plasmapheresis requires personnel trained in dialysis, which may not be performed in all hospitals. Possible adverse events include infection and hemorrhage. Laboratory values must be monitored for hypokalemia and hypocalcemia.45,46
Supportive Care
Patients with GBS require intensive care and very close monitoring for complications of respiratory difficulty and autonomic dysfunction. Individualized programs should be initiated for patients in the acute phase of GBS, aimed at the prevention of contractures and skin breakdown.10 Exercise programs, as conducted with the case patient, should also help relieve the fatigue syndromes that accompany GBS.
Immobilization associated with bed rest incurs a risk for pulmonary emboli and DVT; this has been found true during the first 12 weeks after symptom onset in patients with GBS who remain immobile.47 The use of antiembolic hose and sequential compression devices can help reduce the risk for thrombotic events.10 Use of enoxaparin or heparin is recommended for nonambulating patients until they are able to walk, with Gaber et al47 specifying the use of low-molecular-weight heparin to reduce, but not eliminate, the risk for DVT.
The pain associated with GBS can be severe. Narcotic analgesics may be administered with careful monitoring of autonomic denervation. Long-term management of neuropathic pain may require adjuvant therapy, such as tricyclic antidepressants, gabapentin, or tramadol hydrochloride.10 According to Pandey et al,48 gabapentin alone may suffice for pain control in GBS, with minimal adverse effects. In certain rehabilitation facilities, tricyclic antidepressants, capsaicin, and transcutaneous nerve stimulation have been reported effective; during the early stages of treatment, until these treatments reach their full effect, pain medications such as tramadol or narcotics can provide temporary relief.29
More than one-half of patients with GBS in the acute phase can develop ileus. Constipation can also occur as a result of pain medication use, prolonged bed rest, and poor intake. Auscultation of bowel sounds and abdominal assessment should be performed daily to monitor for ileus. Hughes et al10 do not recommend the use of promotility drugs in patients with dysautonomia.
After hospital discharge, easy fatigability can affect work and social activities. With continued physical therapy, occupational therapy, and monitoring, however, patients with GBS can expect to return to an optimal level of functioning. Speed of recovery varies with these patients from a few months to several years, depending on such factors as age and the extent to which axonal degeneration has occurred.6,49
The Case Patient
For several weeks after discharge, the case patient continued to experience fatigue, low back pain, and general muscle pain. With her family’s support, she continued to receive outpatient physical therapy, and within one month she had regained her ankle strength. She was soon able to resume her classes, despite some lingering fatigue.
Conclusion
Guillain-Barré syndrome is a potentially life-threatening disease whose symptoms health care providers need to recognize quickly to provide prompt treatment. Supportive care for both patient and family is of key importance for maximum rehabilitation and return to the previous lifestyle. The clinical course of GBS is highly variable and difficult to predict. The patient’s outcome depends on several factors, including age and severity of illness. GBS patients can experience long-term psychosocial effects.
References
1. Magira EE, Papaioakim M, Nachamkin I, et al. Differential distribution of HLA-DQ beta/DR beta epitopes in the two forms of Guillain-Barré syndrome, acute motor axonal neuropathy and acute inflammatory demyelinating polyneuropathy (AIDP): identification of DQ beta epitopes associated with susceptibility to and protection from AIDP. J Immunol. 2003;170(6):3074-3080.
2. Tremblay ME, Closon A, D’Anjou G, Bussières JF. Guillain-Barré syndrome following H1N1 immunization in a pediatric patient. Ann Pharmacother. 2010;44(7-8):1330-1333.
3. Mukerji S, Aloka F, Farooq MU, et al. Cardiovascular complications of the Guillain-Barré syndrome. Am J Cardiol. 2009;104(10):1452-1455.
4. McGrogan A, Madle GC, Seaman HE, de Vries CS. The epidemiology of Guillain-Barré syndrome worldwide: a systematic literature review. Neuroepidemiology. 2009;32(2):150-163.
5. Haber P, Sejvar J, Mikaeloff Y, DeStefano F. Vaccines and Guillain-Barré syndrome. Drug Saf. 2009; 32(4):309-323.
6. van Doorn PA. What’s new in Guillain-Barré syndrome in 2007-2008? J Periph Nerv Syst. 2009;14(2):72-74.
7. van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain-Barré syndrome. Lancet Neurol. 2008;7(10):939-950.
8. Chiò A, Cocito D, Leone M, et al; Piemonte and alle d’Aosta Register for Guillain-Barré Syndrome. Guillain-Barré syndrome: a prospective, population-based incidence and outcome survey. Neurology. 2003; 60(7):1146-1150.
9. Hadden RD, Karch H, Hartung HP, et al. Preceding infections, immune factors, and outcome in Guillain-Barré syndrome. Neurology. 2001;56(6):758-765.
10. Hughes RA, Wijdicks EF, Benson E, et al. Supportive care for patients with Guillain-Barré syndrome. Arch Neurol. 2005;62(8):1194-1198.
11. Aluka KJ, Turner PL, Fullum TM. Guillain-Barré syndrome and postbariatric surgery polyneuropathies. JSLS. 2009;13(2):250-253.
12. Brannagan TH 3rd, Zhou Y. HIV-associated Guillain-Barré syndrome. J Neurol Sci. 2003;208(1-2):39-42.
13. Lin WC, Lee PI, Lu CY, et al. Mycoplasma pneumoniae encephalitis in childhood. J Microbiol Immunol Infect. 2002;35(3):173-178.
14. Sivadon-Tardy V, Orlikowski D, Porcher R, et al. Detection of Campylobacter jejuni by culture and real-time PCR in a French cohort of patients with Guillain-Barre syndrome. J Clin Microbiol. 2010;48 (6):2278-2281.
15. van Doorn PA, Kuitwaard K, Walgaard C, et al. IVIG treatment and prognosis in Guillain-Barré syndrome. J Clin Immunol. 2010;30 suppl 1:S74-S78.
16. Kaida K, Kusunoki S. Guillan-Barré syndrome: update on immunobiology and treatment. Expert Rev Neurother. 2009;9(9):1307-1319.
17. Forsberg A, Press R, Einarsson U, et al. Disability and health-related quality of life in Guillain-Barré syndrome during the first two years after onset: a prospective study. Clin Rehabil. 2005;19(8):900-909.
18. Criteria for diagnosis of Guillain-Barré syndrome. Ann Neurol. 1978;3(6):565-566.
19. van Koningsveld R, Steyerberg EW, Hughes RA, et al. A clinical progostic scoring system for Guillain-Barré syndrome. Lancet Neurol. 2007;6(7):589-594.
20. Koeppen S, Kraywinkel K, Wessendorf TE, et al. Long-term outcome of Guillain-Barré syndrome. Neurocrit Care. 2006;5(3)235-242.
21. Sheridan JM, Smith D. Atypical Guillain-Barré in the emergency department. West J Emerg Med. 2010;11(1):80-82.
22. Ogawara K, Kuwabara S, Koga M, et al. Anti-GM1b IgG antibody is associated with acute motor axonal neuropathy and Campylobacter jejuni infection. J Neurol Sci. 2003;210(1-2):41-45.
23. Nagashima T, Koga M, Odaka M, et al. Continuous spectrum of pharyngeal-cervical-brachial variant of Guillain-Barré syndrome. Arch Neurol. 2007;64(10):1519-1523.
24. Oh SJ, LaGanke C, Claussen GC. Sensory Guillain-Barré syndrome. Neurology. 2001;56(1):82-86.
25. Aráranyi Z, Kovács T, Sipos I, Bereczki D. Miller Fisher syndrome: brief overview and update with a focus on electrophysiological findings. Eur J Neurol. 2011 Jun 1. [Epub ahead of print]
26. Hughes RA, Cornblath, DR. Guillain-Barré syndrome. Lancet. 2005;366(9497):1653-1666.
27. Snyder LA, Rismondo V, Miller NR. The Fisher variant of Guillain-Barré syndrome (Fisher syndrome). J Neuroophthalmol. 2009;29(4):312-324.
28. Ropper AH. The Guillain-Barré syndrome. N Engl J Med.1992;326(17):1130-1136.
29. Meythaler JM. Rehabilitation of Guillain-Barré syndrome. Arch Phys Med Rehabil.1997;78(8):872-879.
30. Sharshar T, Chevret S, Bourdain F, et al; French Cooperative Group on Plasma Exchange in Guillain-Barré syndrome. Early predictors of mechanical ventilation in Guillain-Barré syndrome. Crit Care Med. 2003; 31(1):278-283.
31. McGillicuddy DC, Walker O, Shapiro NI, et al. Guillain-Barré syndrome in the emergency department. Ann Emerg Med. 2006;47(4):390-393.
32. Yikilmaz A, Doganay S, Gumus H, et al. Magnetic resonance imaging of childhood Guillain-Barré syndrome. Childs Nerv Syst. 2010;26(8):1103-1108.
33. Gonzalez-Quevedo A, Carriera RF, O’Farrill ZL, et al. An appraisal of blood-cerebrospinal fluid barrier dysfunction during the course of Guillain-Barré syndrome. Neurol India. 2009;57(3):288-294.
34. Abai S, Kim SB, Kim JP, Lim YJ. Guillan-Barré syndrome combined with acute cervical myelopathy. J Korean Neurosurg Soc. 2010;48(3):298-300.
35. Uncini A, Yuki N. Electrophysiologic and immunopathologic correlates in Guillain-Barré syndrome subtypes. Expert Rev Neurother. 2009;9(6):869-884.
36. Hadden RD, Hughes RA. Management of inflammatory neuropathies. J Neurol Neurosurg Psychiatry. 2003;74 suppl 2:ii9-ii14.
37. Raphaël JC, Chevret S, Hughes RA, Annane D. Plasma exchange for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2002;(2):CD001798.
38. Hughes RA, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2010 Jun 16; (6):CD002063.
39. Human immunoglobulin and the Guillain-Barré syndrome: new indication. An alternative to plasmapheresis. Prescrire Int. 2000;9(49):142-143.
40. van der Meché FG, Schmitz PI; Dutch Guillain-Barré Study Group. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. N Engl J Med. 1992;327(17):1123-1129.
41. Hughes RA, Swan AV, van Doorn PA. Corticosteroids for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2010 Feb 16;(2):CD001446.
42. Hahn AF. Guillain-Barré syndrome. Lancet. 1998; 352(9128):635-641.
43. Dalakas MC. Intravenous immunoglobulin in autoimmune neuromuscular diseases. JAMA. 2004;291(19):2367-2375.
44. Kuitwaard K, de Gelder J, Tio-Gillen AP, et al. Pharmacokenetics of intravenous immunoglobulin and outcome in Guillain-Barré syndrome. Ann Neurol. 2009;66(5):597-603.
45. Atkinson SB, Carr RL, Maybee P, Haynes D. The challenges of managing and treating Guillain-Barré syndrome during the acute phase. Dimens Crit Care Nurs. 2006;25(6):256-263.
46. van Doorn PA. Treatment of Guillain-Barré syndrome and CIDP. J Periph Nerv Syst. 2005;10(2):113-127.
47. Gaber TA, Kirker SGB, Jenner JR. Current practice of prophylactic anticoagulation in Guillain-Barré syndrome. Clin Rehabil. 2002;16(2):190-193.
48. Pandey CK, Bose N, Garg G, et al. Gabapentin for the treatment of pain in Guillain-Barré syndrome: a double-blinded, placebo-controlled, crossover study. Anesth Analg. 2002;95(6):1719-1723.
49. de Vries JM, Hagemans ML, Bussmann JB, et al. Fatigue in neuromuscular disorders: focus on Guillain-Barré syndrome and Pompe disease. Cell Mol Life Sci. 2010;67(5):701-713.
A 20-year-old woman presented to her primary care clinic with a chief complaint of lower leg weakness and difficulty walking. The weakness she described had been worsening over the previous four days, with progressively worsening tingling and numbness of her toes bilaterally.
The day before the patient presented, she noticed numbness and paresthesia in both calves. At the time of her presentation to the clinic, she complained of low back ache, paresthesia of both hands, numbness bilaterally to her groin, difficulty sitting upright, ataxia, and a numb, thick-feeling tongue. She denied fever, neck stiffness, shortness of breath, headache, or visual changes.
The patient stated that 10 days earlier, she had developed an upper respiratory infection for which she was seen at the clinic and treated with a seven-day course of amoxicillin/clavulanate 875/125 mg twice daily. She said that she had recovered completely.
A review of the patient’s systems revealed proximal muscle weakness bilaterally (2/5) and loss of touch-pressure in the lower extremities. She was experiencing paresthesia of the hands and mild weakness bilaterally (4/5). She also walked with an ataxic gait and had reduced deep tendon reflexes in the lower limbs. All cranial nerves were intact, and her vital signs were stable.
The woman’s medical history was positive only for asthma. Her family history included ischemic stroke in the maternal grandfather and brain tumor in the paternal grandfather. Social history was positive for alcohol intake (ranging from four to 12 beers per week). The patient said she had never smoked or used illicit drugs. She was an unmarried college student, living in a dorm on campus. She participated in track at school.
The patient was admitted to the hospital telemetry step-down unit, and a neurology consultation was requested. Tests were ordered, among them MRI of the head and spine and comprehensive blood work, to rule out neurologic, infectious, or metabolic causes of the patient’s weakness; urinalysis was also obtained. These tests all yielded negative results.
A lumbar puncture performed the following day revealed a cerebrospinal fluid (CSF) protein level of 570 mg/L (normal range, 150 to 450 mg/L). Leukocytes numbered 2 cells/mm3 (normal count, 0 to 10 cells/mm3).
Based on the patient’s presentation, history, and symptoms, a neurologist made a diagnosis of Guillain-Barré syndrome. It was decided that no electromyographic (EMG) study was required to rule out other disease processes (eg, spinal cord disease, multiple sclerosis, tumors).
The patient underwent a five-dose course of immunomodulatory therapy with IV immunoglobulin (IVIG). In the step-down unit, she experienced one incident of sinus bradycardia (ie, resting heart rate between 40 and 50 beats/min). Her blood pressure remained stable, as did her respiratory status, according to peak expiratory flow measured frequently at her bedside.
Physical therapy was initiated, consisting of passive and active range of motion, crossovers with the patient’s feet, and stair training. This was done in response to a complaint of ankle weakness, and it helped to strengthen weakened muscles and improve alignment while the patient was bedridden and in a weakened, fatigued state. Additionally, the patient was given enoxaparin, wore antiembolic hose, and used sequential compression devices while in bed. As a result of these measures, she never experienced a pulmonary embolus or deep vein thrombosis (DVT) as a result of being immobilized.
By the seventh day of hospitalization, the patient had stable vital signs and improved lower limb strength, and numbness was resolving in her hands and lower extremities. She was discharged to home, with physical therapy to resume on an outpatient basis.
Discussion
Guillain-Barré syndrome (GBS), an acute immune-mediated paralytic disorder,1 manifests in the form of weakness and diminished reflexes. Affecting the peripheral nerves, GBS is characterized by progressive symmetrical ascending weakness with varying degrees of sensory complaints.2,3
GBS occurs worldwide, and incidence is estimated between 1.1 and 1.8 cases per 100,000 persons.4 In the United States, GBS can be found in all age-groups, with peak incidence noted in elderly persons and young adults.5,6 Even with treatment, 3% to 10% of patients are reported to die of this illness, and 20% cannot walk six months after symptom onset.7 In one prospective population-based study of patients with confirmed GBS, 6% of patients died within 30 days of symptom onset, often as a result of respiratory complications.8
GBS is a postinfectious disorder, with cases developing several days or weeks after a viral or bacterial illness—most commonly, an upper respiratory infection or diarrhea (see Table 19-13). The most common trigger of GBS is infection with the bacterial microorganism Campylobacter jejuni (occurring in 15% to 40% of patients with GBS),9,14 a pathogen that can produce demyelination-causing antibodies. Other responsible pathogens include cytomegalovirus and Epstein-Barr virus.9 In a process called molecular mimicry, the immune system is unable to distinguish the amino acid of an infectious organism from the proteinaceous content of the peripheral nerve.15 Subsequently, the immune system attacks and destroys the myelin sheath.
An example of this is the apparent cross-reaction of the ganglioside GM1 with C jejuni lipopolysaccharide antigens.14,15 The resulting effect is immunologic damage to the peripheral nervous system. The flaccid paralysis that occurs in patients with GBS is thought to be caused by lymphocytic infiltration and complement activation of the spinal roots and peripheral nerves, where macrophages strip the myelin.5,15,16
Stages and Variants
Three stages characterize the course of GBS. The acute phase, which lasts one to four weeks, begins with onset of symptoms and persists until the associated neurologic deterioration has ceased. During the second phase, the plateau period, symptoms persist with no further deterioration; this stage can last several days to several weeks or months. The final phase, the recovery period, can last from four months to two years after symptom onset.15,17,18
The clinical course of GBS is highly variable and in many cases difficult to predict. Certain factors have been associated with a poor outcome: advancing age, previous presence of diarrhea, need for mechanical ventilation, an extended plateau phase, and a lower patient score on the Erasmus GBS Outcome Scale,19 when measured two weeks after GBS onset.8,20 This score can help predict the patient’s chance of independent walking after six months.15,19
Although the classic presenting symptom of GBS is symmetric ascending weakness, several disease variants have been identified, with differing symptoms and degrees of recovery. These variants also differ in terms of the muscle groups affected; in some, visual defects may be present at onset. GBS variants include21:
• Acute motor axonal neuropathy (AMAN)1,22
• Acute inflammatory demyelinating polyneuropathy (AIDP)1
• Pharyngeal-cervical-brachial variant23
• Purely sensory variant24
• Miller-Fisher syndrome, which manifests with ophthalmoplegia, in addition to ataxia and areflexia25
• Axonal form.5,21
AMAN and AIDP are the most common subtypes of GBS.1
Symptoms, Signs, and Disease Manifestations
Limb weakness, the classic presenting symptom of GBS, is both symmetrical and ascending. Weakness can develop acutely and progress over days to weeks.2,15 Hughes and Cornblath26 also note pain, numbness, and paresthesias among the initial symptoms of GBS. Others include sensory changes, cranial nerve involvement, various autonomic changes, and respiratory or oropharyngeal weakness. Reflexes, particularly the tendon reflexes, may be diminished or absent.15,18,21 In many cases, sensory changes (ie, pain) may precede the onset of weakness, often making diagnosis difficult.15
Cranial nerves most commonly affected are V, VI, VII, X, XI, and XII, with manifestations that include dysphagia, dysarthria, diplopia, limitation to eye movements, and facial droop and weakness. Usually facial and oropharyngeal weakness occur after the extremities and trunk are affected. Blindness may occur if demyelination of the optic nerve occurs; this is seen in Miller-Fisher syndrome.10,15,25,27
In GBS, many patients report pain, which can present as bilateral sciatica or as throbbing or aching in the large muscles of the upper legs, flanks, or back.28 This pain, which results from the demyelination of the sensory nerve fibers, can be severe.10
Patients with GBS may experience manifestations of autonomic nervous system dysfunction—for example, arrhythmias, hypotension or hypertension, urinary retention, cardiomyopathy, and paralytic ileus.10,20 Dysautonomia often impedes patients’ progress in inpatient rehabilitation. Patients may have persistent problems involving postural hypotension, hypertension, excessive sympathetic outflow, or bladder and bowel dysfunction.29
Blood pressure fluctuations, often attributed to changes in catecholamine levels and disturbances in the baroreceptor reflex pathway, are common and are considered characteristic of GBS. Transient or persistent hypotension is caused by the dysregulation of the parasympathetic and sympathetic systems, with subsequent alterations in venomotor tone.3 Additionally, an increased sensitivity to catecholamine can lead to cardiovascular disturbances, resulting in denervation hypersensitivity and impairment of the carotid sinus reflex.
Arrhythmias occur in perhaps half of patients with GBS. The most common is sustained sinus tachycardia, which usually requires no treatment. Bradycardia leading to atrioventricular blocks and asystole is believed to result from afferent baroreceptor reflex failure. Treatment may be required—either administration of atropine or insertion of a pacemaker, depending on the severity of the arrhythmia.3,10
Myocardial involvement can range from asymptomatic mycocarditis to neurogenic stunned myocardium and heart failure. Patients with ECG abnormalities should undergo two-dimensional echocardiographic studies and other testing to explore cardiac involvement. Acute coronary syndromes, including ST-segment elevation MI, have been reported, in some cases associated with IVIG treatment. In one patient, coronary spasm was reported, with clean coronary arteries found on cardiac catheterization.3
Patients with GBS are at risk for compromised neuromuscular respiratory function; demyelination of the nerves that innervate the intercostal muscles and the diaphragm can result in respiratory failure. Key clinical indicators of respiratory muscle fatigue include tachypnea, diaphoresis, and asynchronous movements of the abdomen and chest;10 other symptoms relevant to respiratory or oropharyngeal weakness include slurred speech, dyspnea (with or without exertion), difficulty swallowing, and inability to cough.2,10 Serial respiratory function testing is advisable to detect patients at risk for respiratory failure.30
Diagnosis
Guillain-Barré is a syndrome diagnosed by a collection of symptoms (see Table 22,21,31), including subacute developing paralysis, symmetrical bilateral weakness beginning at onset, and diminishing to absent reflexes.21,31 Other causes for rapidly developing weaknesses should be ruled out (see Table 310,21,26,31). Lumbar puncture typically shows increased protein levels with a normal white cell count; however, neither this test nor electrophysiologic evaluation offers significant value for diagnosis of GBS.21,26,31
During the acute phase of GBS (within three weeks of onset), there is found an elevation of CSF protein (> 550 mg/L) without an elevation in white blood cells. This phenomenon, called albuminocytologic dissociation, reflects inflammation of the nerve roots and is considered the hallmark of GBS.2
MRI can also facilitate the diagnosis of GBS; it demonstrates anterior and posterior intrathecal spinal nerve roots and cauda equina.32 In patients with GBS, evidence supporting breakdown of the blood–nerve barrier can be seen in abnormal gadolinium enhancement of the intrathecal nerve roots on MRI.33
When electrophysiologic studies are performed, they typically reveal slowing nerve conduction, prolonged distal latencies, and partial motor conduction block.34 The characteristic finding of early demyelination is conduction block, a reduction in the amplitude of the muscle action potential after stimulation of the distal, as opposed to the proximal, nerve.28 Nerve conduction studies may help in the diagnosis and classification of GBS—and, to a limited extent, formulation of a prognosis. Such alternative diagnoses as myositis and myasthenia gravis may be excluded by neurophysiology.26 Early in GBS, neurophysiologic abnormalities may be very mild or occasionally normal; test results may not correlate with clinical disability.35,36
The clinician cannot depend on clinical features alone to predict respiratory decline.31 Frequent evaluations of respiratory effort, by measurement of maximal inspiratory pressures and vital capacity, should be performed at the bedside to monitor diaphragmatic strength. Respiratory ventilation should be initiated if the patient becomes hypoxic or experiences a rapid decline in vital capacity (ie, below 60% of predicted value).10 Mechanical ventilation is more likely to be required in patients with a negative inspiratory force of less than 30 cm H2O.31
Treatment
Guillain-Barré syndrome has an acute onset and progression. Patients quickly become nonambulatory and may require total ventilation due to paralysis. Therapeutic options are IVIG or plasmapheresis (plasma exchange).37-40 Corticosteroids do not appear to benefit patients with GBS.41,42
Several mechanisms appear to contribute to the effectiveness of immunoglobulin.38,39 Infused IVIG interferes with antigen presentation, inhibits antibody production, neutralizes pathologic autoantibodies, and modulates other immunologic events involved in the pathogenesis of autoimmune neuromuscular diseases, including GBS.43 Adverse reactions, which are usually minor, include headache, fever, chills, myalgia, and malaise. In rare instances, anaphylaxis or renal failure may occur.15,44
In plasmapheresis, blood is removed from the body and dialyzed, with circulating antibodies and immunoglobulins removed from the plasma; fresh frozen plasma, albumin, or saline is administered. This treatment, performed via central venous catheter, should be initiated as soon as possible after onset of symptoms but can be implemented as late as 30 days after GBS onset. Plasmapheresis requires personnel trained in dialysis, which may not be performed in all hospitals. Possible adverse events include infection and hemorrhage. Laboratory values must be monitored for hypokalemia and hypocalcemia.45,46
Supportive Care
Patients with GBS require intensive care and very close monitoring for complications of respiratory difficulty and autonomic dysfunction. Individualized programs should be initiated for patients in the acute phase of GBS, aimed at the prevention of contractures and skin breakdown.10 Exercise programs, as conducted with the case patient, should also help relieve the fatigue syndromes that accompany GBS.
Immobilization associated with bed rest incurs a risk for pulmonary emboli and DVT; this has been found true during the first 12 weeks after symptom onset in patients with GBS who remain immobile.47 The use of antiembolic hose and sequential compression devices can help reduce the risk for thrombotic events.10 Use of enoxaparin or heparin is recommended for nonambulating patients until they are able to walk, with Gaber et al47 specifying the use of low-molecular-weight heparin to reduce, but not eliminate, the risk for DVT.
The pain associated with GBS can be severe. Narcotic analgesics may be administered with careful monitoring of autonomic denervation. Long-term management of neuropathic pain may require adjuvant therapy, such as tricyclic antidepressants, gabapentin, or tramadol hydrochloride.10 According to Pandey et al,48 gabapentin alone may suffice for pain control in GBS, with minimal adverse effects. In certain rehabilitation facilities, tricyclic antidepressants, capsaicin, and transcutaneous nerve stimulation have been reported effective; during the early stages of treatment, until these treatments reach their full effect, pain medications such as tramadol or narcotics can provide temporary relief.29
More than one-half of patients with GBS in the acute phase can develop ileus. Constipation can also occur as a result of pain medication use, prolonged bed rest, and poor intake. Auscultation of bowel sounds and abdominal assessment should be performed daily to monitor for ileus. Hughes et al10 do not recommend the use of promotility drugs in patients with dysautonomia.
After hospital discharge, easy fatigability can affect work and social activities. With continued physical therapy, occupational therapy, and monitoring, however, patients with GBS can expect to return to an optimal level of functioning. Speed of recovery varies with these patients from a few months to several years, depending on such factors as age and the extent to which axonal degeneration has occurred.6,49
The Case Patient
For several weeks after discharge, the case patient continued to experience fatigue, low back pain, and general muscle pain. With her family’s support, she continued to receive outpatient physical therapy, and within one month she had regained her ankle strength. She was soon able to resume her classes, despite some lingering fatigue.
Conclusion
Guillain-Barré syndrome is a potentially life-threatening disease whose symptoms health care providers need to recognize quickly to provide prompt treatment. Supportive care for both patient and family is of key importance for maximum rehabilitation and return to the previous lifestyle. The clinical course of GBS is highly variable and difficult to predict. The patient’s outcome depends on several factors, including age and severity of illness. GBS patients can experience long-term psychosocial effects.
References
1. Magira EE, Papaioakim M, Nachamkin I, et al. Differential distribution of HLA-DQ beta/DR beta epitopes in the two forms of Guillain-Barré syndrome, acute motor axonal neuropathy and acute inflammatory demyelinating polyneuropathy (AIDP): identification of DQ beta epitopes associated with susceptibility to and protection from AIDP. J Immunol. 2003;170(6):3074-3080.
2. Tremblay ME, Closon A, D’Anjou G, Bussières JF. Guillain-Barré syndrome following H1N1 immunization in a pediatric patient. Ann Pharmacother. 2010;44(7-8):1330-1333.
3. Mukerji S, Aloka F, Farooq MU, et al. Cardiovascular complications of the Guillain-Barré syndrome. Am J Cardiol. 2009;104(10):1452-1455.
4. McGrogan A, Madle GC, Seaman HE, de Vries CS. The epidemiology of Guillain-Barré syndrome worldwide: a systematic literature review. Neuroepidemiology. 2009;32(2):150-163.
5. Haber P, Sejvar J, Mikaeloff Y, DeStefano F. Vaccines and Guillain-Barré syndrome. Drug Saf. 2009; 32(4):309-323.
6. van Doorn PA. What’s new in Guillain-Barré syndrome in 2007-2008? J Periph Nerv Syst. 2009;14(2):72-74.
7. van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain-Barré syndrome. Lancet Neurol. 2008;7(10):939-950.
8. Chiò A, Cocito D, Leone M, et al; Piemonte and alle d’Aosta Register for Guillain-Barré Syndrome. Guillain-Barré syndrome: a prospective, population-based incidence and outcome survey. Neurology. 2003; 60(7):1146-1150.
9. Hadden RD, Karch H, Hartung HP, et al. Preceding infections, immune factors, and outcome in Guillain-Barré syndrome. Neurology. 2001;56(6):758-765.
10. Hughes RA, Wijdicks EF, Benson E, et al. Supportive care for patients with Guillain-Barré syndrome. Arch Neurol. 2005;62(8):1194-1198.
11. Aluka KJ, Turner PL, Fullum TM. Guillain-Barré syndrome and postbariatric surgery polyneuropathies. JSLS. 2009;13(2):250-253.
12. Brannagan TH 3rd, Zhou Y. HIV-associated Guillain-Barré syndrome. J Neurol Sci. 2003;208(1-2):39-42.
13. Lin WC, Lee PI, Lu CY, et al. Mycoplasma pneumoniae encephalitis in childhood. J Microbiol Immunol Infect. 2002;35(3):173-178.
14. Sivadon-Tardy V, Orlikowski D, Porcher R, et al. Detection of Campylobacter jejuni by culture and real-time PCR in a French cohort of patients with Guillain-Barre syndrome. J Clin Microbiol. 2010;48 (6):2278-2281.
15. van Doorn PA, Kuitwaard K, Walgaard C, et al. IVIG treatment and prognosis in Guillain-Barré syndrome. J Clin Immunol. 2010;30 suppl 1:S74-S78.
16. Kaida K, Kusunoki S. Guillan-Barré syndrome: update on immunobiology and treatment. Expert Rev Neurother. 2009;9(9):1307-1319.
17. Forsberg A, Press R, Einarsson U, et al. Disability and health-related quality of life in Guillain-Barré syndrome during the first two years after onset: a prospective study. Clin Rehabil. 2005;19(8):900-909.
18. Criteria for diagnosis of Guillain-Barré syndrome. Ann Neurol. 1978;3(6):565-566.
19. van Koningsveld R, Steyerberg EW, Hughes RA, et al. A clinical progostic scoring system for Guillain-Barré syndrome. Lancet Neurol. 2007;6(7):589-594.
20. Koeppen S, Kraywinkel K, Wessendorf TE, et al. Long-term outcome of Guillain-Barré syndrome. Neurocrit Care. 2006;5(3)235-242.
21. Sheridan JM, Smith D. Atypical Guillain-Barré in the emergency department. West J Emerg Med. 2010;11(1):80-82.
22. Ogawara K, Kuwabara S, Koga M, et al. Anti-GM1b IgG antibody is associated with acute motor axonal neuropathy and Campylobacter jejuni infection. J Neurol Sci. 2003;210(1-2):41-45.
23. Nagashima T, Koga M, Odaka M, et al. Continuous spectrum of pharyngeal-cervical-brachial variant of Guillain-Barré syndrome. Arch Neurol. 2007;64(10):1519-1523.
24. Oh SJ, LaGanke C, Claussen GC. Sensory Guillain-Barré syndrome. Neurology. 2001;56(1):82-86.
25. Aráranyi Z, Kovács T, Sipos I, Bereczki D. Miller Fisher syndrome: brief overview and update with a focus on electrophysiological findings. Eur J Neurol. 2011 Jun 1. [Epub ahead of print]
26. Hughes RA, Cornblath, DR. Guillain-Barré syndrome. Lancet. 2005;366(9497):1653-1666.
27. Snyder LA, Rismondo V, Miller NR. The Fisher variant of Guillain-Barré syndrome (Fisher syndrome). J Neuroophthalmol. 2009;29(4):312-324.
28. Ropper AH. The Guillain-Barré syndrome. N Engl J Med.1992;326(17):1130-1136.
29. Meythaler JM. Rehabilitation of Guillain-Barré syndrome. Arch Phys Med Rehabil.1997;78(8):872-879.
30. Sharshar T, Chevret S, Bourdain F, et al; French Cooperative Group on Plasma Exchange in Guillain-Barré syndrome. Early predictors of mechanical ventilation in Guillain-Barré syndrome. Crit Care Med. 2003; 31(1):278-283.
31. McGillicuddy DC, Walker O, Shapiro NI, et al. Guillain-Barré syndrome in the emergency department. Ann Emerg Med. 2006;47(4):390-393.
32. Yikilmaz A, Doganay S, Gumus H, et al. Magnetic resonance imaging of childhood Guillain-Barré syndrome. Childs Nerv Syst. 2010;26(8):1103-1108.
33. Gonzalez-Quevedo A, Carriera RF, O’Farrill ZL, et al. An appraisal of blood-cerebrospinal fluid barrier dysfunction during the course of Guillain-Barré syndrome. Neurol India. 2009;57(3):288-294.
34. Abai S, Kim SB, Kim JP, Lim YJ. Guillan-Barré syndrome combined with acute cervical myelopathy. J Korean Neurosurg Soc. 2010;48(3):298-300.
35. Uncini A, Yuki N. Electrophysiologic and immunopathologic correlates in Guillain-Barré syndrome subtypes. Expert Rev Neurother. 2009;9(6):869-884.
36. Hadden RD, Hughes RA. Management of inflammatory neuropathies. J Neurol Neurosurg Psychiatry. 2003;74 suppl 2:ii9-ii14.
37. Raphaël JC, Chevret S, Hughes RA, Annane D. Plasma exchange for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2002;(2):CD001798.
38. Hughes RA, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2010 Jun 16; (6):CD002063.
39. Human immunoglobulin and the Guillain-Barré syndrome: new indication. An alternative to plasmapheresis. Prescrire Int. 2000;9(49):142-143.
40. van der Meché FG, Schmitz PI; Dutch Guillain-Barré Study Group. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. N Engl J Med. 1992;327(17):1123-1129.
41. Hughes RA, Swan AV, van Doorn PA. Corticosteroids for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2010 Feb 16;(2):CD001446.
42. Hahn AF. Guillain-Barré syndrome. Lancet. 1998; 352(9128):635-641.
43. Dalakas MC. Intravenous immunoglobulin in autoimmune neuromuscular diseases. JAMA. 2004;291(19):2367-2375.
44. Kuitwaard K, de Gelder J, Tio-Gillen AP, et al. Pharmacokenetics of intravenous immunoglobulin and outcome in Guillain-Barré syndrome. Ann Neurol. 2009;66(5):597-603.
45. Atkinson SB, Carr RL, Maybee P, Haynes D. The challenges of managing and treating Guillain-Barré syndrome during the acute phase. Dimens Crit Care Nurs. 2006;25(6):256-263.
46. van Doorn PA. Treatment of Guillain-Barré syndrome and CIDP. J Periph Nerv Syst. 2005;10(2):113-127.
47. Gaber TA, Kirker SGB, Jenner JR. Current practice of prophylactic anticoagulation in Guillain-Barré syndrome. Clin Rehabil. 2002;16(2):190-193.
48. Pandey CK, Bose N, Garg G, et al. Gabapentin for the treatment of pain in Guillain-Barré syndrome: a double-blinded, placebo-controlled, crossover study. Anesth Analg. 2002;95(6):1719-1723.
49. de Vries JM, Hagemans ML, Bussmann JB, et al. Fatigue in neuromuscular disorders: focus on Guillain-Barré syndrome and Pompe disease. Cell Mol Life Sci. 2010;67(5):701-713.
Grand Rounds: Woman, 22, With Dizziness and Headache
A 22-year-old student was brought in to a college student health center in a wheelchair by campus safety personnel. She appeared drowsy and was crying softly. She complained of a severe headache and said she was “tired of going through this all the time.” The woman said she had seen spots and become dizzy, then had gotten “the worst headache of my life” while sitting in class. She rated the headache pain at 8 on a 10-point scale and also complained of nausea and photophobia.
The history revealed dizziness that made her “feel as if I’m tipping over” and similar headaches during the previous year. The patient said she had seen “a few doctors” for her symptoms, but that they “could never find anything.” The headaches usually occurred on the left side of her head, lasted hours to days, and were only partially relieved with acetaminophen. The patient could not remember whether she had eaten breakfast and was unsure of what day it was. She described herself as frustrated and began to weep again.
She was currently under the care of a psychologist but seemed uncertain why; she said that she was sexually active and used condoms. She had undergone an appendectomy at age 12. She denied taking any medications besides acetaminophen. She denied smoking or drug use, history of migraine headaches, vision or hearing changes, facial weakness, depression, or anxiety. Her family history included a grandfather with diabetes and hypertension and an uncle with heart disease. The family history was negative for migraine or psychiatric illness.
Because of the patient’s weakness, she was assisted onto the examination table by a nurse. Physical exam revealed a pale, slightly sweaty, overweight, tearful young woman who was slow to respond. Her blood pressure was measured at 134/104 mm Hg; pulse, 100 beats/min; respirations, 14 breaths/min; and temperature, 97.0ºF. Point-of-care testing of blood glucose was 91 mg/dL, and hemoglobin was measured at 12.3 g/dL. The ophthalmologic exam was positive for photophobia and revealed slightly disconjugate gaze with horizontal nystagmus during testing of cranial nerves (CN) III, IV, and VI. The otoscopic exam revealed a slightly injected right tympanic membrane, and there were no apparent hearing deficits.
The neurologic exam showed patellar and brachial deep tendon reflexes equal, grips weak and equal, and the pupillary response intact. The patient was able to stand without assistance, although her gait was slightly unsteady. Because the patient was of college age, the clinician ruled out meningitis by negative Kernig’s and Brudzinski’s signs and absence of fever. Subarachnoid hemorrhage was also a concern when the patient mentioned the “worst headache of my life,” indicating the need for emergent imaging.
The patient’s presentation, it was felt, warranted a 911 call. The emergency medical team arrived, and its members began to question the patient. Discrepancies in the patient’s history during the paramedics’ reexamination led them to question whether an emergency department (ED) visit was necessary, but at the clinician’s insistence, they agreed to transport the student to the ED.
The following day, the student health center clinician was contacted by a member of the hospital ED staff with an update on the patient’s status. Shortly after her arrival at the hospital, she underwent MRI and was diagnosed with a vestibular schwannoma. She had surgery that same evening, during which the surgeon removed most of the tumor. Although the ED staff was not at liberty to provide more complete information, they did inform the clinician that the patient would require radiation for the remainder of the tumor.
DISCUSSION
Vestibular schwannoma is also known as acoustic schwannoma, acoustic neuroma, acoustic neurinoma, or vestibular neurilemmoma. These tumors arise from perineural elements of Schwann cells, which commonly form and lead to myelination in the vestibular area of CN VIII1 (see figure). They occur with equal frequency on the superior and inferior branches of the vestibular nerve and originate only rarely at the cochlear portion of the eighth cranial nerve. Vestibular schwannomas represent approximately 8% to 10% of brain tumors and 80% to 90% of tumors in the cerebellopontine angle in adults.2 Tumors are distributed evenly across genders, but the majority of diagnosed patients are white.3
Most likely because of improvements in diagnostic technology, the incidence of vestibular schwannoma has increased over the past 30 years. One British research team predicts that one in 1,000 persons will receive a diagnosis of vestibular schwannoma in their lifetime.4 These tumors are most commonly diagnosed in people ages 30 to 60, with a median age of 55.5
A relationship has been demonstrated between neurofibromatosis type 2 (NF2), an autosomal-dominant disease, and the development of vestibular schwannomas.6,7 NF2 has a birth prevalence of one in about 25,000 persons,4,8 and those who inherit the responsible gene inevitably develop vestibular schwannomas.9 Patients with a confirmed diagnosis of vestibular schwannoma should be screened by a geneticist for the NF2 gene; although the tumors are benign, they can cause compression of the vestibular nerve, leading to deafness and balance disorders.10 Schwannomas of the spinal nerves can also occur in persons with NF2.11 Compression of the spinal nerves in these patients can lead to significant morbidity and a shortened average life span.10
NF2 is diagnosed using the following criteria:
1) Bilateral vestibular schwannomas
2) Diagnosis of a family member with either NF2 or unilateral vestibular schwannoma, and
3) Juvenile posterior subscapular lens opacities.9,12,13
Because schwannomas grow slowly, the vestibular system can adapt to the slow destruction of CN VIII. For this reason, patients typically present with unilateral deafness or hearing impairment rather than dizziness.11 Many patients also present with tinnitus and/or vertigo.14,15
Some vestibular tumors remain stable or even regress; others progress, in some cases causing life-threatening complications.16 An extremely rare complication of a vestibular schwannoma was reported in one patient: an intratumoral hemorrhage that led to acute neurologic deterioration and death.17
Since the case patient underwent immediate surgical intervention, it appears she was experiencing significant involvement and it was likely anticipated that without surgical intervention, clinical progression would occur. Her young age could be considered a risk factor for a faster-growing neuroma.18
Clinical Presentation and Diagnosis
Primary care clinicians commonly see patients with complaints of dizziness, lightheadedness, faintness, or a sensation of spinning or tilting. Vestibular schwannoma should be considered in the differential diagnosis of the patient who presents with these complaints, as well as tinnitus or hearing loss.9 The patient with vestibular schwannoma may also have a history of headache, unsteady gait, facial pain, and numbness.19 A partial differential diagnosis is listed in the table20,21). The astute clinician will systematically rule out many of these conditions, since certain other features that may be present (eg, rapid onset, vomiting, fever) do not typically occur in the patient with vestibular schwannoma.
Because the symptoms typically associated with vestibular schwannoma are likely to occur bilaterally in patients with other conditions, unilateral symptoms should alert the clinician to investigate further. The patterns and growth rates of vestibular schwannomas are highly variable and currently unpredictable18 (according to Fortnum et al,14 at least 50% of tumors do not grow within several years after diagnosis); thus, no clear predictors of tumor growth have been identified to assist in the evaluation of an affected patient,16 although faster tumor growth rates have been reported in young patients, and Baser et al18 have called for additional research involving younger persons with vestibular schwannomas.
Standard testing is audiometry followed by MRI, which is considered the most effective means to confirm a diagnosis of vestibular schwannoma.5,14,22
Treatment for Vestibular Schwannoma
Treatment, whether with surgery or radiation, is associated with significant morbidity and possibly decreased quality of life.16 Therefore, distinguishing patients whose tumors will grow and pose a threat to them from those whose tumors are likely to remain stable is central to appropriate management.23
Treatment modalities are considered based on tumor size, growth, presence or absence of tinnitus, and the patient’s preferences and life expectancy.23 In most cases, decision making is complex and should be customized to meet the patient’s individual circumstances. Patients with similar clinical scenarios have been reported to opt for different treatment choices.24
Four treatment options are currently available for patients with vestibular schwannoma:
Serial observation with periodic MRI studies. Since vestibular schwannomas are benign and slow-growing, conservative management can be a reasonable option, particularly if the patient is elderly, the tumor is small, and/or little hearing loss has taken place. However, use of observation is associated with a risk for progressive and permanent hearing loss.2 Between 15% and 50% of patients who opt for serial observation will undergo subsequent surgical intervention, particularly in cases involving worsening tinnitus, balance problems, or hearing loss.23-25
Chemotherapy. Agents including bevacizumab (a humanized monoclonal antibody against vascular endothelial growth factor)8,26,27 and erlotinib (an epidermal growth factor receptor inhibitor) may delay progression or even facilitate regression of vestibular schwannomas.28 Hearing improvement has also been reported in patients with NF2 who were treated with bevacizumab8; research is ongoing.26
Fractionated radiotherapy. Hearing may be preserved in 60% to 95% of patients, depending on levels of dosing to the cochlea, but 3% to 7% of patients will need further treatment.29-31 Radiation treatment is a likely choice in patients with tumors measuring 2.0 cm or less. Larger tumors are considered a surgical disease, and directed radiotherapy may be administered postoperatively (as in the case patient) for residual portions of the tumor.16
Microsurgery. Compared with other treatment modalities, the emphasis of microsurgery is on removing tumors (particularly larger tumors) rather than controlling their growth.29 The three common approaches are retrosigmoid, middle fossa, or translabyrinthine.32-34 Preservation of hearing is reportedly better following retrosigmoid or middle fossa microsurgery, compared with a translabyrinthine procedure (because in the latter, the tumor cannot be exposed without damage to the inner ear).32,35
With any such surgery, risks include cranial nerve damage, leakage of cerebrospinal fluid, and infection.29,32 Postsurgically, about half of patients report frequent headaches, which are persistent in about half of these cases.36-38 Another concern is preservation of the facial nerves, with a risk for temporary facial weakness or dysfunction.3,24,39 Less than 2% of patients who undergo microsurgery require additional treatment.29
Stereotactic radiosurgery. These procedures, which are performed using the Gamma Knife,® the CyberKnife, or the linear accelerator,29,40,41 are considered appropriate for patients with smaller tumors and those who are not candidates for conventional surgery.1 Trigeminal neuropathy, injury to the facial nerves, and hydrocephaly are reported complications of Gamma Knife radiosurgery, but improvements in these technologies are ongoing.1,2,40
Patient Outcomes
The outcome in a patient with vestibular schwannoma depends on the treatment administered, but prolonged follow-up is typically necessary. For patients being managed through observation, annual brain scans are recommended for 10 years, with subsequent scans every three to five years if no tumor growth is seen. For patients who have had surgery, annual brain scans are advised for the successive eight to 10 years, with decreasing frequency if no tumor remains. In patients who undergo radiation, annual scans are recommended for 10 years, then every two years if no tumor growth is detected.36
Psychosocial experiences vary widely among patients who have undergone treatment for vestibular schwannomas. Some are unable to perform necessary or recreational activities, and others must retire early from work.42 Others, however, have minimal disruption in their lives and enjoy a good quality of life. The most difficult consequence of vestibular schwannoma and its treatment, according to patients, is the associated hearing loss.8,19
THE CASE PATIENT
The 22-year-old patient in this case had an atypical presentation of vestibular schwannoma. Although she did present with vertigo, she also complained of headache, nausea, and photophobia—which are rarely reported in investigations of these tumors. She was also younger than the typical patient and did not report tinnitus.
The case patient reportedly underwent surgery and subsequent radiation to treat the remaining portion of her tumor. She suspended her attendance at the college and, as of this writing, has not re-enrolled. She was lost to follow-up.
CONCLUSION
For the primary care provider, diagnostic challenges require great clinical acumen. Vertigo, headache, hearing loss, and tinnitus are all symptoms seen in the primary care setting; when they occur together, the clinician should be alerted to investigate further. A high level of suspicion is appropriate when a patient complains of longstanding auditory symptoms, with or without headache. Unilateral hearing loss is a common symptom in patients with vestibular schwannomas, although some may present with facial weakness or pain, imbalance, and/or vertigo.
In addition to the history and physical exam, experts recommend that audiometry and MRI be considered, particularly if hearing loss is unilateral. Genetic screening for NF2 should be performed if vestibular schwannoma is found on MRI. Referral to a neurologist, a neurosurgeon, or an otolaryngologist is appropriate.
REFERENCES
1. Arthurs BJ, Lamoreaux WT, Giddings NA, et al. Gamma Knife radiosurgery for vestibular schwannoma: case report and review of the literature. World J Surg Oncol. 2009 Dec 18;7:100.
2. Mohammed TA, Ahuja MS, Ju SS, Thomas J. Normal pressure hydrocephalus after Gamma Knife radiosurgery for vestibular schwannoma. J Postgrad Med. 2010;56(3):213-215.
3. Gal TJ, Shinn J, Huang B. Current epidemiology and management trends in acoustic neuroma. Otolaryngol Head Neck Surg. 2010;142(5):677-681.
4. Evans DG, Moran A, King A, et al. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005;26(1):93-97.
5. Haynes D. Acoustic neuroma diagnosis and treatment options. Hearing Health. 2009;25(3):32. www.drf.org/magazine/36/Summer+2009+Issue/article/272. Accessed May 16, 2011.
6. Sobel RA. Vestibular (acoustic) schwannomas: histologic features in neurofibromatosis 2 and in unilateral cases. J Neuropathol Exp Neurol. 1993;52(2):106-113.
7. Evans DG, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84(304):603-618.
8. Plotkin SR, Stemmer-Rachamimov AO, Barker FG 2nd, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361(4):358-367.
9. Evans DGR, Sainio M, Baser E. Neurofibromatosis type 2. J Med Genet. 2000:37(11):897-904.
10. Gusella JF, Ramesh V, MacCollin M, Jacoby LB. Neurofibromatosis 2: loss of Merlin’s protective spell. Curr Opin Genet Dev. 1996;6(1):87-92.
11. Sagar SM, Israel MA. Ch 374. Primary and metastatic tumors of the nervous system. In: Kasper DL, Braunwald E, Fauci AS, et al. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill Companies, Inc; 2008:2601-2610.
12. Evans DGR. Neurofibromatosis 2 [bilateral acoustic neurofibromatosis, central neurofibromatosis, NF2, neurofibromatosis type II]. Genet Med. 2009;11(9):599-610.
13. Arya R, Sahu JK, Kabra M. Neurofibromatosis type II (Wishart type). J Pediatr Neurol. 2009;7(3): 333-335.
14. Fortnum H, O’Neill C, Taylor R, et al. The role of magnetic resonance imaging in the identification of suspected acoustic neuroma: a systematic review of clinical and cost effectiveness and natural history. Health Technol Assess. 2009;13(18):iii-iv, ix-xi, 1-154.
15. Forton GE, Cremers CW, Offeciers EE. Acoustic neuroma ingrowth in the cochlear nerve: does it influence the clinical presentation? Ann Otol Rhinol Laryngol. 2004;113(7):582-586.
16. Nikolopoulos TP, Fortnum H, O’Donoghue G, Baguley D. Acoustic neuroma growth: a systematic review of the evidence. Otol Neurotol. 2010;31(3):478-485.
17. Yates CW, Weinberg M, Packer MJ, Jacob A. Fatal case of tumor-associated hemorrhage in a large vestibular schwannoma. Ann Otol Rhinol Laryngol. 2010;119(6):402-405.
18. Baser ME, Mautner VF, Parry DM, Evans DGR. Methodological issues in longitudinal studies; vestibular schwannoma growth rates in neurofibromatosis 2. J Med Genet. 2005;42(12):903-906.
19. Brooker J, Burney S, Fletcher J, Dally M. A qualitative exploration of quality of life among individuals diagnosed with an acoustic neuroma. Br J Health Psychol. 2009;14(pt 3):563-578.
20. Strupp M, Brandt T. Diagnosis and treatment of vertigo and dizziness. Dtsch Arzetbl Int. 2008;105(10):173-180.
21. Kerber KA. Dizziness and vertigo. In: Andreoli TE, Griggs RC, Benjamin I , Wing EJ, eds. Andreoli and Carpenter’s Cecil Essentials of Medicine. 8th ed. Philadelphia, PA: Elsevier Inc; 2010:1104-1105.
22. Gimsing S. Vestibular schwannoma: when to look for it? J Laryngol Otol. 2010;124(3):258-264.
23. Agrawal Y, Clark JH, Limb CJ, et al. Predictors of vestibular schwannoma growth and clinical implications. Otol Neurotol. 2010;31(5):807-812.
24. Cheung SW, Aranda D, Driscoll CLW, Parsa AT. Mapping clinical outcomes expectations to treatment decisions: an application to vestibular schwannoma management. Otol Neurotol. 2010;31(2):284-293.
25. Myrseth E, Pedersen PH, Møller P, Lund-Johansen M. Treatment of vestibular schwannomas: why, when and how? Acta Neurochir (Wien). 2007;149(7):647-660.
26. Sidney Kimmel Comprehensive Cancer Center, Massachusetts General Hospital, National Cancer Institute. Bevacizumab for symptomatic vestibular schwannoma in neurofibromatosis type 2 (NF2). http://clinicaltrials.gov/ct2/show/NCT01207687. Accessed May 16, 2011.
27. Mautner VF, Nguyen R, Kutta H, et al. Bevacizumab induces regression of vestibular schwannomas in patients with neurofibromatosis type 2. Neuro Oncol. 2010;12(1):14-18.
28. Plotkin SR, Halpin C, McKenna MJ, et al. Erlotinib for progressive vestibular schwannoma in neurofibromatosis 2 patients. Otol Neurotol. 2010;31(7):1135-1143.
29. Arthurs BJ, Fairbanks RK, Demakas JJ, et al. A review of treatment modalities for vestibular schwannoma. Neurosurg Rev. 2011 Feb 9; [Epub ahead of print].
30. Andrews DW, Werner-Wasik M, Den RB, et al. Toward dose optimization for fractionated stereotactic radiotherapy for acoustic neuromas: comparison of two dose cohorts. Int J Radiat Oncol Biol Phys. 2009;74(2):419-426.
31. Thomas C, Di Maio S, Ma R, et al. Hearing preservation following fractionated stereotactic radiotherapy for vestibular schwannomas: prognostic implications of cochlear dose. J Neurosurg. 2007;107(5):917-926.
32. Samii M, Gerganov V, Samii A. Improved preservation of hearing and facial nerve function in vestibular schwannoma surgery via the retrosigmoid approach in a series of 200 patients. J Neurosurg. 2006;105(4):527-535.
33. Shiobara R, Ohira T, Inoue Y, et al. Extended middle cranial fossa approach for vestibular schwannoma: technical note and surgical results of 896 operations. Prog Neurol Surg. 2008;21:65-72.
34. Schmerber S, Palombi O, Boubagra K, et al. Long-term control of vestibular schwannoma after a translabyrinthine complete removal. Neurosurgery. 2005;57(4):693-698.
35. Phillips DJ, Kobylarz EJ, De Peralta ET, et al. Predictive factors of hearing preservation after surgical resection of small vestibular schwannomas. Otol Neurotol. 2010;31(9):1463-1468.
36. Park JK, Black MP, Vernick DM, Ramakrishna N. Vestibular schwannoma (acoustic neuroma) (2010). www.uptodate.com/contents/vestibular-schwannoma-acoustic-neuroma. Accessed May 16, 2011.
37. Schankin CJ, Gall C, Straube A. Headache syndromes after acoustic neuroma surgery and their implications for quality of life. Cephalalgia. 2009;29(7):760-761.
38. Ryzenman JM, Pensak ML, Tew JM Jr. Headache: a quality of life analysis in a cohort of 1,657 patients undergoing acoustic neuroma surgery: results from the Acoustic Neuroma Association. Laryngoscope. 2005;115(4):703-711.
39. Sriskandan N, Connor SE. The role of radiology in the diagnosis and management of vestibular schwannoma. Clin Radiol. 2010;66(4):357-365.
40. Yang I, Sughrue ME, Han SJ, et al. Facial nerve preservation after vestibular schwannoma Gamma Knife surgery. J Neurooncol. 2009;93(1): 41-48.
41. Unger F, Dominikus K, Haselsberger K. Stereotactic radiosurgery and fractionated stereotactic radiotherapy of acoustic neuromas [in German]. HNO. 2011;59(1):31-37.
42. Tos T, Caye-Thomasen P, Stangerup SE, et al. Long-term socio-economic impact of vestibular schwannoma for patients under observation and after surgery. J Laryngol Otol. 2003;117(12):955-964.
A 22-year-old student was brought in to a college student health center in a wheelchair by campus safety personnel. She appeared drowsy and was crying softly. She complained of a severe headache and said she was “tired of going through this all the time.” The woman said she had seen spots and become dizzy, then had gotten “the worst headache of my life” while sitting in class. She rated the headache pain at 8 on a 10-point scale and also complained of nausea and photophobia.
The history revealed dizziness that made her “feel as if I’m tipping over” and similar headaches during the previous year. The patient said she had seen “a few doctors” for her symptoms, but that they “could never find anything.” The headaches usually occurred on the left side of her head, lasted hours to days, and were only partially relieved with acetaminophen. The patient could not remember whether she had eaten breakfast and was unsure of what day it was. She described herself as frustrated and began to weep again.
She was currently under the care of a psychologist but seemed uncertain why; she said that she was sexually active and used condoms. She had undergone an appendectomy at age 12. She denied taking any medications besides acetaminophen. She denied smoking or drug use, history of migraine headaches, vision or hearing changes, facial weakness, depression, or anxiety. Her family history included a grandfather with diabetes and hypertension and an uncle with heart disease. The family history was negative for migraine or psychiatric illness.
Because of the patient’s weakness, she was assisted onto the examination table by a nurse. Physical exam revealed a pale, slightly sweaty, overweight, tearful young woman who was slow to respond. Her blood pressure was measured at 134/104 mm Hg; pulse, 100 beats/min; respirations, 14 breaths/min; and temperature, 97.0ºF. Point-of-care testing of blood glucose was 91 mg/dL, and hemoglobin was measured at 12.3 g/dL. The ophthalmologic exam was positive for photophobia and revealed slightly disconjugate gaze with horizontal nystagmus during testing of cranial nerves (CN) III, IV, and VI. The otoscopic exam revealed a slightly injected right tympanic membrane, and there were no apparent hearing deficits.
The neurologic exam showed patellar and brachial deep tendon reflexes equal, grips weak and equal, and the pupillary response intact. The patient was able to stand without assistance, although her gait was slightly unsteady. Because the patient was of college age, the clinician ruled out meningitis by negative Kernig’s and Brudzinski’s signs and absence of fever. Subarachnoid hemorrhage was also a concern when the patient mentioned the “worst headache of my life,” indicating the need for emergent imaging.
The patient’s presentation, it was felt, warranted a 911 call. The emergency medical team arrived, and its members began to question the patient. Discrepancies in the patient’s history during the paramedics’ reexamination led them to question whether an emergency department (ED) visit was necessary, but at the clinician’s insistence, they agreed to transport the student to the ED.
The following day, the student health center clinician was contacted by a member of the hospital ED staff with an update on the patient’s status. Shortly after her arrival at the hospital, she underwent MRI and was diagnosed with a vestibular schwannoma. She had surgery that same evening, during which the surgeon removed most of the tumor. Although the ED staff was not at liberty to provide more complete information, they did inform the clinician that the patient would require radiation for the remainder of the tumor.
DISCUSSION
Vestibular schwannoma is also known as acoustic schwannoma, acoustic neuroma, acoustic neurinoma, or vestibular neurilemmoma. These tumors arise from perineural elements of Schwann cells, which commonly form and lead to myelination in the vestibular area of CN VIII1 (see figure). They occur with equal frequency on the superior and inferior branches of the vestibular nerve and originate only rarely at the cochlear portion of the eighth cranial nerve. Vestibular schwannomas represent approximately 8% to 10% of brain tumors and 80% to 90% of tumors in the cerebellopontine angle in adults.2 Tumors are distributed evenly across genders, but the majority of diagnosed patients are white.3
Most likely because of improvements in diagnostic technology, the incidence of vestibular schwannoma has increased over the past 30 years. One British research team predicts that one in 1,000 persons will receive a diagnosis of vestibular schwannoma in their lifetime.4 These tumors are most commonly diagnosed in people ages 30 to 60, with a median age of 55.5
A relationship has been demonstrated between neurofibromatosis type 2 (NF2), an autosomal-dominant disease, and the development of vestibular schwannomas.6,7 NF2 has a birth prevalence of one in about 25,000 persons,4,8 and those who inherit the responsible gene inevitably develop vestibular schwannomas.9 Patients with a confirmed diagnosis of vestibular schwannoma should be screened by a geneticist for the NF2 gene; although the tumors are benign, they can cause compression of the vestibular nerve, leading to deafness and balance disorders.10 Schwannomas of the spinal nerves can also occur in persons with NF2.11 Compression of the spinal nerves in these patients can lead to significant morbidity and a shortened average life span.10
NF2 is diagnosed using the following criteria:
1) Bilateral vestibular schwannomas
2) Diagnosis of a family member with either NF2 or unilateral vestibular schwannoma, and
3) Juvenile posterior subscapular lens opacities.9,12,13
Because schwannomas grow slowly, the vestibular system can adapt to the slow destruction of CN VIII. For this reason, patients typically present with unilateral deafness or hearing impairment rather than dizziness.11 Many patients also present with tinnitus and/or vertigo.14,15
Some vestibular tumors remain stable or even regress; others progress, in some cases causing life-threatening complications.16 An extremely rare complication of a vestibular schwannoma was reported in one patient: an intratumoral hemorrhage that led to acute neurologic deterioration and death.17
Since the case patient underwent immediate surgical intervention, it appears she was experiencing significant involvement and it was likely anticipated that without surgical intervention, clinical progression would occur. Her young age could be considered a risk factor for a faster-growing neuroma.18
Clinical Presentation and Diagnosis
Primary care clinicians commonly see patients with complaints of dizziness, lightheadedness, faintness, or a sensation of spinning or tilting. Vestibular schwannoma should be considered in the differential diagnosis of the patient who presents with these complaints, as well as tinnitus or hearing loss.9 The patient with vestibular schwannoma may also have a history of headache, unsteady gait, facial pain, and numbness.19 A partial differential diagnosis is listed in the table20,21). The astute clinician will systematically rule out many of these conditions, since certain other features that may be present (eg, rapid onset, vomiting, fever) do not typically occur in the patient with vestibular schwannoma.
Because the symptoms typically associated with vestibular schwannoma are likely to occur bilaterally in patients with other conditions, unilateral symptoms should alert the clinician to investigate further. The patterns and growth rates of vestibular schwannomas are highly variable and currently unpredictable18 (according to Fortnum et al,14 at least 50% of tumors do not grow within several years after diagnosis); thus, no clear predictors of tumor growth have been identified to assist in the evaluation of an affected patient,16 although faster tumor growth rates have been reported in young patients, and Baser et al18 have called for additional research involving younger persons with vestibular schwannomas.
Standard testing is audiometry followed by MRI, which is considered the most effective means to confirm a diagnosis of vestibular schwannoma.5,14,22
Treatment for Vestibular Schwannoma
Treatment, whether with surgery or radiation, is associated with significant morbidity and possibly decreased quality of life.16 Therefore, distinguishing patients whose tumors will grow and pose a threat to them from those whose tumors are likely to remain stable is central to appropriate management.23
Treatment modalities are considered based on tumor size, growth, presence or absence of tinnitus, and the patient’s preferences and life expectancy.23 In most cases, decision making is complex and should be customized to meet the patient’s individual circumstances. Patients with similar clinical scenarios have been reported to opt for different treatment choices.24
Four treatment options are currently available for patients with vestibular schwannoma:
Serial observation with periodic MRI studies. Since vestibular schwannomas are benign and slow-growing, conservative management can be a reasonable option, particularly if the patient is elderly, the tumor is small, and/or little hearing loss has taken place. However, use of observation is associated with a risk for progressive and permanent hearing loss.2 Between 15% and 50% of patients who opt for serial observation will undergo subsequent surgical intervention, particularly in cases involving worsening tinnitus, balance problems, or hearing loss.23-25
Chemotherapy. Agents including bevacizumab (a humanized monoclonal antibody against vascular endothelial growth factor)8,26,27 and erlotinib (an epidermal growth factor receptor inhibitor) may delay progression or even facilitate regression of vestibular schwannomas.28 Hearing improvement has also been reported in patients with NF2 who were treated with bevacizumab8; research is ongoing.26
Fractionated radiotherapy. Hearing may be preserved in 60% to 95% of patients, depending on levels of dosing to the cochlea, but 3% to 7% of patients will need further treatment.29-31 Radiation treatment is a likely choice in patients with tumors measuring 2.0 cm or less. Larger tumors are considered a surgical disease, and directed radiotherapy may be administered postoperatively (as in the case patient) for residual portions of the tumor.16
Microsurgery. Compared with other treatment modalities, the emphasis of microsurgery is on removing tumors (particularly larger tumors) rather than controlling their growth.29 The three common approaches are retrosigmoid, middle fossa, or translabyrinthine.32-34 Preservation of hearing is reportedly better following retrosigmoid or middle fossa microsurgery, compared with a translabyrinthine procedure (because in the latter, the tumor cannot be exposed without damage to the inner ear).32,35
With any such surgery, risks include cranial nerve damage, leakage of cerebrospinal fluid, and infection.29,32 Postsurgically, about half of patients report frequent headaches, which are persistent in about half of these cases.36-38 Another concern is preservation of the facial nerves, with a risk for temporary facial weakness or dysfunction.3,24,39 Less than 2% of patients who undergo microsurgery require additional treatment.29
Stereotactic radiosurgery. These procedures, which are performed using the Gamma Knife,® the CyberKnife, or the linear accelerator,29,40,41 are considered appropriate for patients with smaller tumors and those who are not candidates for conventional surgery.1 Trigeminal neuropathy, injury to the facial nerves, and hydrocephaly are reported complications of Gamma Knife radiosurgery, but improvements in these technologies are ongoing.1,2,40
Patient Outcomes
The outcome in a patient with vestibular schwannoma depends on the treatment administered, but prolonged follow-up is typically necessary. For patients being managed through observation, annual brain scans are recommended for 10 years, with subsequent scans every three to five years if no tumor growth is seen. For patients who have had surgery, annual brain scans are advised for the successive eight to 10 years, with decreasing frequency if no tumor remains. In patients who undergo radiation, annual scans are recommended for 10 years, then every two years if no tumor growth is detected.36
Psychosocial experiences vary widely among patients who have undergone treatment for vestibular schwannomas. Some are unable to perform necessary or recreational activities, and others must retire early from work.42 Others, however, have minimal disruption in their lives and enjoy a good quality of life. The most difficult consequence of vestibular schwannoma and its treatment, according to patients, is the associated hearing loss.8,19
THE CASE PATIENT
The 22-year-old patient in this case had an atypical presentation of vestibular schwannoma. Although she did present with vertigo, she also complained of headache, nausea, and photophobia—which are rarely reported in investigations of these tumors. She was also younger than the typical patient and did not report tinnitus.
The case patient reportedly underwent surgery and subsequent radiation to treat the remaining portion of her tumor. She suspended her attendance at the college and, as of this writing, has not re-enrolled. She was lost to follow-up.
CONCLUSION
For the primary care provider, diagnostic challenges require great clinical acumen. Vertigo, headache, hearing loss, and tinnitus are all symptoms seen in the primary care setting; when they occur together, the clinician should be alerted to investigate further. A high level of suspicion is appropriate when a patient complains of longstanding auditory symptoms, with or without headache. Unilateral hearing loss is a common symptom in patients with vestibular schwannomas, although some may present with facial weakness or pain, imbalance, and/or vertigo.
In addition to the history and physical exam, experts recommend that audiometry and MRI be considered, particularly if hearing loss is unilateral. Genetic screening for NF2 should be performed if vestibular schwannoma is found on MRI. Referral to a neurologist, a neurosurgeon, or an otolaryngologist is appropriate.
REFERENCES
1. Arthurs BJ, Lamoreaux WT, Giddings NA, et al. Gamma Knife radiosurgery for vestibular schwannoma: case report and review of the literature. World J Surg Oncol. 2009 Dec 18;7:100.
2. Mohammed TA, Ahuja MS, Ju SS, Thomas J. Normal pressure hydrocephalus after Gamma Knife radiosurgery for vestibular schwannoma. J Postgrad Med. 2010;56(3):213-215.
3. Gal TJ, Shinn J, Huang B. Current epidemiology and management trends in acoustic neuroma. Otolaryngol Head Neck Surg. 2010;142(5):677-681.
4. Evans DG, Moran A, King A, et al. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005;26(1):93-97.
5. Haynes D. Acoustic neuroma diagnosis and treatment options. Hearing Health. 2009;25(3):32. www.drf.org/magazine/36/Summer+2009+Issue/article/272. Accessed May 16, 2011.
6. Sobel RA. Vestibular (acoustic) schwannomas: histologic features in neurofibromatosis 2 and in unilateral cases. J Neuropathol Exp Neurol. 1993;52(2):106-113.
7. Evans DG, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84(304):603-618.
8. Plotkin SR, Stemmer-Rachamimov AO, Barker FG 2nd, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361(4):358-367.
9. Evans DGR, Sainio M, Baser E. Neurofibromatosis type 2. J Med Genet. 2000:37(11):897-904.
10. Gusella JF, Ramesh V, MacCollin M, Jacoby LB. Neurofibromatosis 2: loss of Merlin’s protective spell. Curr Opin Genet Dev. 1996;6(1):87-92.
11. Sagar SM, Israel MA. Ch 374. Primary and metastatic tumors of the nervous system. In: Kasper DL, Braunwald E, Fauci AS, et al. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill Companies, Inc; 2008:2601-2610.
12. Evans DGR. Neurofibromatosis 2 [bilateral acoustic neurofibromatosis, central neurofibromatosis, NF2, neurofibromatosis type II]. Genet Med. 2009;11(9):599-610.
13. Arya R, Sahu JK, Kabra M. Neurofibromatosis type II (Wishart type). J Pediatr Neurol. 2009;7(3): 333-335.
14. Fortnum H, O’Neill C, Taylor R, et al. The role of magnetic resonance imaging in the identification of suspected acoustic neuroma: a systematic review of clinical and cost effectiveness and natural history. Health Technol Assess. 2009;13(18):iii-iv, ix-xi, 1-154.
15. Forton GE, Cremers CW, Offeciers EE. Acoustic neuroma ingrowth in the cochlear nerve: does it influence the clinical presentation? Ann Otol Rhinol Laryngol. 2004;113(7):582-586.
16. Nikolopoulos TP, Fortnum H, O’Donoghue G, Baguley D. Acoustic neuroma growth: a systematic review of the evidence. Otol Neurotol. 2010;31(3):478-485.
17. Yates CW, Weinberg M, Packer MJ, Jacob A. Fatal case of tumor-associated hemorrhage in a large vestibular schwannoma. Ann Otol Rhinol Laryngol. 2010;119(6):402-405.
18. Baser ME, Mautner VF, Parry DM, Evans DGR. Methodological issues in longitudinal studies; vestibular schwannoma growth rates in neurofibromatosis 2. J Med Genet. 2005;42(12):903-906.
19. Brooker J, Burney S, Fletcher J, Dally M. A qualitative exploration of quality of life among individuals diagnosed with an acoustic neuroma. Br J Health Psychol. 2009;14(pt 3):563-578.
20. Strupp M, Brandt T. Diagnosis and treatment of vertigo and dizziness. Dtsch Arzetbl Int. 2008;105(10):173-180.
21. Kerber KA. Dizziness and vertigo. In: Andreoli TE, Griggs RC, Benjamin I , Wing EJ, eds. Andreoli and Carpenter’s Cecil Essentials of Medicine. 8th ed. Philadelphia, PA: Elsevier Inc; 2010:1104-1105.
22. Gimsing S. Vestibular schwannoma: when to look for it? J Laryngol Otol. 2010;124(3):258-264.
23. Agrawal Y, Clark JH, Limb CJ, et al. Predictors of vestibular schwannoma growth and clinical implications. Otol Neurotol. 2010;31(5):807-812.
24. Cheung SW, Aranda D, Driscoll CLW, Parsa AT. Mapping clinical outcomes expectations to treatment decisions: an application to vestibular schwannoma management. Otol Neurotol. 2010;31(2):284-293.
25. Myrseth E, Pedersen PH, Møller P, Lund-Johansen M. Treatment of vestibular schwannomas: why, when and how? Acta Neurochir (Wien). 2007;149(7):647-660.
26. Sidney Kimmel Comprehensive Cancer Center, Massachusetts General Hospital, National Cancer Institute. Bevacizumab for symptomatic vestibular schwannoma in neurofibromatosis type 2 (NF2). http://clinicaltrials.gov/ct2/show/NCT01207687. Accessed May 16, 2011.
27. Mautner VF, Nguyen R, Kutta H, et al. Bevacizumab induces regression of vestibular schwannomas in patients with neurofibromatosis type 2. Neuro Oncol. 2010;12(1):14-18.
28. Plotkin SR, Halpin C, McKenna MJ, et al. Erlotinib for progressive vestibular schwannoma in neurofibromatosis 2 patients. Otol Neurotol. 2010;31(7):1135-1143.
29. Arthurs BJ, Fairbanks RK, Demakas JJ, et al. A review of treatment modalities for vestibular schwannoma. Neurosurg Rev. 2011 Feb 9; [Epub ahead of print].
30. Andrews DW, Werner-Wasik M, Den RB, et al. Toward dose optimization for fractionated stereotactic radiotherapy for acoustic neuromas: comparison of two dose cohorts. Int J Radiat Oncol Biol Phys. 2009;74(2):419-426.
31. Thomas C, Di Maio S, Ma R, et al. Hearing preservation following fractionated stereotactic radiotherapy for vestibular schwannomas: prognostic implications of cochlear dose. J Neurosurg. 2007;107(5):917-926.
32. Samii M, Gerganov V, Samii A. Improved preservation of hearing and facial nerve function in vestibular schwannoma surgery via the retrosigmoid approach in a series of 200 patients. J Neurosurg. 2006;105(4):527-535.
33. Shiobara R, Ohira T, Inoue Y, et al. Extended middle cranial fossa approach for vestibular schwannoma: technical note and surgical results of 896 operations. Prog Neurol Surg. 2008;21:65-72.
34. Schmerber S, Palombi O, Boubagra K, et al. Long-term control of vestibular schwannoma after a translabyrinthine complete removal. Neurosurgery. 2005;57(4):693-698.
35. Phillips DJ, Kobylarz EJ, De Peralta ET, et al. Predictive factors of hearing preservation after surgical resection of small vestibular schwannomas. Otol Neurotol. 2010;31(9):1463-1468.
36. Park JK, Black MP, Vernick DM, Ramakrishna N. Vestibular schwannoma (acoustic neuroma) (2010). www.uptodate.com/contents/vestibular-schwannoma-acoustic-neuroma. Accessed May 16, 2011.
37. Schankin CJ, Gall C, Straube A. Headache syndromes after acoustic neuroma surgery and their implications for quality of life. Cephalalgia. 2009;29(7):760-761.
38. Ryzenman JM, Pensak ML, Tew JM Jr. Headache: a quality of life analysis in a cohort of 1,657 patients undergoing acoustic neuroma surgery: results from the Acoustic Neuroma Association. Laryngoscope. 2005;115(4):703-711.
39. Sriskandan N, Connor SE. The role of radiology in the diagnosis and management of vestibular schwannoma. Clin Radiol. 2010;66(4):357-365.
40. Yang I, Sughrue ME, Han SJ, et al. Facial nerve preservation after vestibular schwannoma Gamma Knife surgery. J Neurooncol. 2009;93(1): 41-48.
41. Unger F, Dominikus K, Haselsberger K. Stereotactic radiosurgery and fractionated stereotactic radiotherapy of acoustic neuromas [in German]. HNO. 2011;59(1):31-37.
42. Tos T, Caye-Thomasen P, Stangerup SE, et al. Long-term socio-economic impact of vestibular schwannoma for patients under observation and after surgery. J Laryngol Otol. 2003;117(12):955-964.
A 22-year-old student was brought in to a college student health center in a wheelchair by campus safety personnel. She appeared drowsy and was crying softly. She complained of a severe headache and said she was “tired of going through this all the time.” The woman said she had seen spots and become dizzy, then had gotten “the worst headache of my life” while sitting in class. She rated the headache pain at 8 on a 10-point scale and also complained of nausea and photophobia.
The history revealed dizziness that made her “feel as if I’m tipping over” and similar headaches during the previous year. The patient said she had seen “a few doctors” for her symptoms, but that they “could never find anything.” The headaches usually occurred on the left side of her head, lasted hours to days, and were only partially relieved with acetaminophen. The patient could not remember whether she had eaten breakfast and was unsure of what day it was. She described herself as frustrated and began to weep again.
She was currently under the care of a psychologist but seemed uncertain why; she said that she was sexually active and used condoms. She had undergone an appendectomy at age 12. She denied taking any medications besides acetaminophen. She denied smoking or drug use, history of migraine headaches, vision or hearing changes, facial weakness, depression, or anxiety. Her family history included a grandfather with diabetes and hypertension and an uncle with heart disease. The family history was negative for migraine or psychiatric illness.
Because of the patient’s weakness, she was assisted onto the examination table by a nurse. Physical exam revealed a pale, slightly sweaty, overweight, tearful young woman who was slow to respond. Her blood pressure was measured at 134/104 mm Hg; pulse, 100 beats/min; respirations, 14 breaths/min; and temperature, 97.0ºF. Point-of-care testing of blood glucose was 91 mg/dL, and hemoglobin was measured at 12.3 g/dL. The ophthalmologic exam was positive for photophobia and revealed slightly disconjugate gaze with horizontal nystagmus during testing of cranial nerves (CN) III, IV, and VI. The otoscopic exam revealed a slightly injected right tympanic membrane, and there were no apparent hearing deficits.
The neurologic exam showed patellar and brachial deep tendon reflexes equal, grips weak and equal, and the pupillary response intact. The patient was able to stand without assistance, although her gait was slightly unsteady. Because the patient was of college age, the clinician ruled out meningitis by negative Kernig’s and Brudzinski’s signs and absence of fever. Subarachnoid hemorrhage was also a concern when the patient mentioned the “worst headache of my life,” indicating the need for emergent imaging.
The patient’s presentation, it was felt, warranted a 911 call. The emergency medical team arrived, and its members began to question the patient. Discrepancies in the patient’s history during the paramedics’ reexamination led them to question whether an emergency department (ED) visit was necessary, but at the clinician’s insistence, they agreed to transport the student to the ED.
The following day, the student health center clinician was contacted by a member of the hospital ED staff with an update on the patient’s status. Shortly after her arrival at the hospital, she underwent MRI and was diagnosed with a vestibular schwannoma. She had surgery that same evening, during which the surgeon removed most of the tumor. Although the ED staff was not at liberty to provide more complete information, they did inform the clinician that the patient would require radiation for the remainder of the tumor.
DISCUSSION
Vestibular schwannoma is also known as acoustic schwannoma, acoustic neuroma, acoustic neurinoma, or vestibular neurilemmoma. These tumors arise from perineural elements of Schwann cells, which commonly form and lead to myelination in the vestibular area of CN VIII1 (see figure). They occur with equal frequency on the superior and inferior branches of the vestibular nerve and originate only rarely at the cochlear portion of the eighth cranial nerve. Vestibular schwannomas represent approximately 8% to 10% of brain tumors and 80% to 90% of tumors in the cerebellopontine angle in adults.2 Tumors are distributed evenly across genders, but the majority of diagnosed patients are white.3
Most likely because of improvements in diagnostic technology, the incidence of vestibular schwannoma has increased over the past 30 years. One British research team predicts that one in 1,000 persons will receive a diagnosis of vestibular schwannoma in their lifetime.4 These tumors are most commonly diagnosed in people ages 30 to 60, with a median age of 55.5
A relationship has been demonstrated between neurofibromatosis type 2 (NF2), an autosomal-dominant disease, and the development of vestibular schwannomas.6,7 NF2 has a birth prevalence of one in about 25,000 persons,4,8 and those who inherit the responsible gene inevitably develop vestibular schwannomas.9 Patients with a confirmed diagnosis of vestibular schwannoma should be screened by a geneticist for the NF2 gene; although the tumors are benign, they can cause compression of the vestibular nerve, leading to deafness and balance disorders.10 Schwannomas of the spinal nerves can also occur in persons with NF2.11 Compression of the spinal nerves in these patients can lead to significant morbidity and a shortened average life span.10
NF2 is diagnosed using the following criteria:
1) Bilateral vestibular schwannomas
2) Diagnosis of a family member with either NF2 or unilateral vestibular schwannoma, and
3) Juvenile posterior subscapular lens opacities.9,12,13
Because schwannomas grow slowly, the vestibular system can adapt to the slow destruction of CN VIII. For this reason, patients typically present with unilateral deafness or hearing impairment rather than dizziness.11 Many patients also present with tinnitus and/or vertigo.14,15
Some vestibular tumors remain stable or even regress; others progress, in some cases causing life-threatening complications.16 An extremely rare complication of a vestibular schwannoma was reported in one patient: an intratumoral hemorrhage that led to acute neurologic deterioration and death.17
Since the case patient underwent immediate surgical intervention, it appears she was experiencing significant involvement and it was likely anticipated that without surgical intervention, clinical progression would occur. Her young age could be considered a risk factor for a faster-growing neuroma.18
Clinical Presentation and Diagnosis
Primary care clinicians commonly see patients with complaints of dizziness, lightheadedness, faintness, or a sensation of spinning or tilting. Vestibular schwannoma should be considered in the differential diagnosis of the patient who presents with these complaints, as well as tinnitus or hearing loss.9 The patient with vestibular schwannoma may also have a history of headache, unsteady gait, facial pain, and numbness.19 A partial differential diagnosis is listed in the table20,21). The astute clinician will systematically rule out many of these conditions, since certain other features that may be present (eg, rapid onset, vomiting, fever) do not typically occur in the patient with vestibular schwannoma.
Because the symptoms typically associated with vestibular schwannoma are likely to occur bilaterally in patients with other conditions, unilateral symptoms should alert the clinician to investigate further. The patterns and growth rates of vestibular schwannomas are highly variable and currently unpredictable18 (according to Fortnum et al,14 at least 50% of tumors do not grow within several years after diagnosis); thus, no clear predictors of tumor growth have been identified to assist in the evaluation of an affected patient,16 although faster tumor growth rates have been reported in young patients, and Baser et al18 have called for additional research involving younger persons with vestibular schwannomas.
Standard testing is audiometry followed by MRI, which is considered the most effective means to confirm a diagnosis of vestibular schwannoma.5,14,22
Treatment for Vestibular Schwannoma
Treatment, whether with surgery or radiation, is associated with significant morbidity and possibly decreased quality of life.16 Therefore, distinguishing patients whose tumors will grow and pose a threat to them from those whose tumors are likely to remain stable is central to appropriate management.23
Treatment modalities are considered based on tumor size, growth, presence or absence of tinnitus, and the patient’s preferences and life expectancy.23 In most cases, decision making is complex and should be customized to meet the patient’s individual circumstances. Patients with similar clinical scenarios have been reported to opt for different treatment choices.24
Four treatment options are currently available for patients with vestibular schwannoma:
Serial observation with periodic MRI studies. Since vestibular schwannomas are benign and slow-growing, conservative management can be a reasonable option, particularly if the patient is elderly, the tumor is small, and/or little hearing loss has taken place. However, use of observation is associated with a risk for progressive and permanent hearing loss.2 Between 15% and 50% of patients who opt for serial observation will undergo subsequent surgical intervention, particularly in cases involving worsening tinnitus, balance problems, or hearing loss.23-25
Chemotherapy. Agents including bevacizumab (a humanized monoclonal antibody against vascular endothelial growth factor)8,26,27 and erlotinib (an epidermal growth factor receptor inhibitor) may delay progression or even facilitate regression of vestibular schwannomas.28 Hearing improvement has also been reported in patients with NF2 who were treated with bevacizumab8; research is ongoing.26
Fractionated radiotherapy. Hearing may be preserved in 60% to 95% of patients, depending on levels of dosing to the cochlea, but 3% to 7% of patients will need further treatment.29-31 Radiation treatment is a likely choice in patients with tumors measuring 2.0 cm or less. Larger tumors are considered a surgical disease, and directed radiotherapy may be administered postoperatively (as in the case patient) for residual portions of the tumor.16
Microsurgery. Compared with other treatment modalities, the emphasis of microsurgery is on removing tumors (particularly larger tumors) rather than controlling their growth.29 The three common approaches are retrosigmoid, middle fossa, or translabyrinthine.32-34 Preservation of hearing is reportedly better following retrosigmoid or middle fossa microsurgery, compared with a translabyrinthine procedure (because in the latter, the tumor cannot be exposed without damage to the inner ear).32,35
With any such surgery, risks include cranial nerve damage, leakage of cerebrospinal fluid, and infection.29,32 Postsurgically, about half of patients report frequent headaches, which are persistent in about half of these cases.36-38 Another concern is preservation of the facial nerves, with a risk for temporary facial weakness or dysfunction.3,24,39 Less than 2% of patients who undergo microsurgery require additional treatment.29
Stereotactic radiosurgery. These procedures, which are performed using the Gamma Knife,® the CyberKnife, or the linear accelerator,29,40,41 are considered appropriate for patients with smaller tumors and those who are not candidates for conventional surgery.1 Trigeminal neuropathy, injury to the facial nerves, and hydrocephaly are reported complications of Gamma Knife radiosurgery, but improvements in these technologies are ongoing.1,2,40
Patient Outcomes
The outcome in a patient with vestibular schwannoma depends on the treatment administered, but prolonged follow-up is typically necessary. For patients being managed through observation, annual brain scans are recommended for 10 years, with subsequent scans every three to five years if no tumor growth is seen. For patients who have had surgery, annual brain scans are advised for the successive eight to 10 years, with decreasing frequency if no tumor remains. In patients who undergo radiation, annual scans are recommended for 10 years, then every two years if no tumor growth is detected.36
Psychosocial experiences vary widely among patients who have undergone treatment for vestibular schwannomas. Some are unable to perform necessary or recreational activities, and others must retire early from work.42 Others, however, have minimal disruption in their lives and enjoy a good quality of life. The most difficult consequence of vestibular schwannoma and its treatment, according to patients, is the associated hearing loss.8,19
THE CASE PATIENT
The 22-year-old patient in this case had an atypical presentation of vestibular schwannoma. Although she did present with vertigo, she also complained of headache, nausea, and photophobia—which are rarely reported in investigations of these tumors. She was also younger than the typical patient and did not report tinnitus.
The case patient reportedly underwent surgery and subsequent radiation to treat the remaining portion of her tumor. She suspended her attendance at the college and, as of this writing, has not re-enrolled. She was lost to follow-up.
CONCLUSION
For the primary care provider, diagnostic challenges require great clinical acumen. Vertigo, headache, hearing loss, and tinnitus are all symptoms seen in the primary care setting; when they occur together, the clinician should be alerted to investigate further. A high level of suspicion is appropriate when a patient complains of longstanding auditory symptoms, with or without headache. Unilateral hearing loss is a common symptom in patients with vestibular schwannomas, although some may present with facial weakness or pain, imbalance, and/or vertigo.
In addition to the history and physical exam, experts recommend that audiometry and MRI be considered, particularly if hearing loss is unilateral. Genetic screening for NF2 should be performed if vestibular schwannoma is found on MRI. Referral to a neurologist, a neurosurgeon, or an otolaryngologist is appropriate.
REFERENCES
1. Arthurs BJ, Lamoreaux WT, Giddings NA, et al. Gamma Knife radiosurgery for vestibular schwannoma: case report and review of the literature. World J Surg Oncol. 2009 Dec 18;7:100.
2. Mohammed TA, Ahuja MS, Ju SS, Thomas J. Normal pressure hydrocephalus after Gamma Knife radiosurgery for vestibular schwannoma. J Postgrad Med. 2010;56(3):213-215.
3. Gal TJ, Shinn J, Huang B. Current epidemiology and management trends in acoustic neuroma. Otolaryngol Head Neck Surg. 2010;142(5):677-681.
4. Evans DG, Moran A, King A, et al. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005;26(1):93-97.
5. Haynes D. Acoustic neuroma diagnosis and treatment options. Hearing Health. 2009;25(3):32. www.drf.org/magazine/36/Summer+2009+Issue/article/272. Accessed May 16, 2011.
6. Sobel RA. Vestibular (acoustic) schwannomas: histologic features in neurofibromatosis 2 and in unilateral cases. J Neuropathol Exp Neurol. 1993;52(2):106-113.
7. Evans DG, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84(304):603-618.
8. Plotkin SR, Stemmer-Rachamimov AO, Barker FG 2nd, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361(4):358-367.
9. Evans DGR, Sainio M, Baser E. Neurofibromatosis type 2. J Med Genet. 2000:37(11):897-904.
10. Gusella JF, Ramesh V, MacCollin M, Jacoby LB. Neurofibromatosis 2: loss of Merlin’s protective spell. Curr Opin Genet Dev. 1996;6(1):87-92.
11. Sagar SM, Israel MA. Ch 374. Primary and metastatic tumors of the nervous system. In: Kasper DL, Braunwald E, Fauci AS, et al. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill Companies, Inc; 2008:2601-2610.
12. Evans DGR. Neurofibromatosis 2 [bilateral acoustic neurofibromatosis, central neurofibromatosis, NF2, neurofibromatosis type II]. Genet Med. 2009;11(9):599-610.
13. Arya R, Sahu JK, Kabra M. Neurofibromatosis type II (Wishart type). J Pediatr Neurol. 2009;7(3): 333-335.
14. Fortnum H, O’Neill C, Taylor R, et al. The role of magnetic resonance imaging in the identification of suspected acoustic neuroma: a systematic review of clinical and cost effectiveness and natural history. Health Technol Assess. 2009;13(18):iii-iv, ix-xi, 1-154.
15. Forton GE, Cremers CW, Offeciers EE. Acoustic neuroma ingrowth in the cochlear nerve: does it influence the clinical presentation? Ann Otol Rhinol Laryngol. 2004;113(7):582-586.
16. Nikolopoulos TP, Fortnum H, O’Donoghue G, Baguley D. Acoustic neuroma growth: a systematic review of the evidence. Otol Neurotol. 2010;31(3):478-485.
17. Yates CW, Weinberg M, Packer MJ, Jacob A. Fatal case of tumor-associated hemorrhage in a large vestibular schwannoma. Ann Otol Rhinol Laryngol. 2010;119(6):402-405.
18. Baser ME, Mautner VF, Parry DM, Evans DGR. Methodological issues in longitudinal studies; vestibular schwannoma growth rates in neurofibromatosis 2. J Med Genet. 2005;42(12):903-906.
19. Brooker J, Burney S, Fletcher J, Dally M. A qualitative exploration of quality of life among individuals diagnosed with an acoustic neuroma. Br J Health Psychol. 2009;14(pt 3):563-578.
20. Strupp M, Brandt T. Diagnosis and treatment of vertigo and dizziness. Dtsch Arzetbl Int. 2008;105(10):173-180.
21. Kerber KA. Dizziness and vertigo. In: Andreoli TE, Griggs RC, Benjamin I , Wing EJ, eds. Andreoli and Carpenter’s Cecil Essentials of Medicine. 8th ed. Philadelphia, PA: Elsevier Inc; 2010:1104-1105.
22. Gimsing S. Vestibular schwannoma: when to look for it? J Laryngol Otol. 2010;124(3):258-264.
23. Agrawal Y, Clark JH, Limb CJ, et al. Predictors of vestibular schwannoma growth and clinical implications. Otol Neurotol. 2010;31(5):807-812.
24. Cheung SW, Aranda D, Driscoll CLW, Parsa AT. Mapping clinical outcomes expectations to treatment decisions: an application to vestibular schwannoma management. Otol Neurotol. 2010;31(2):284-293.
25. Myrseth E, Pedersen PH, Møller P, Lund-Johansen M. Treatment of vestibular schwannomas: why, when and how? Acta Neurochir (Wien). 2007;149(7):647-660.
26. Sidney Kimmel Comprehensive Cancer Center, Massachusetts General Hospital, National Cancer Institute. Bevacizumab for symptomatic vestibular schwannoma in neurofibromatosis type 2 (NF2). http://clinicaltrials.gov/ct2/show/NCT01207687. Accessed May 16, 2011.
27. Mautner VF, Nguyen R, Kutta H, et al. Bevacizumab induces regression of vestibular schwannomas in patients with neurofibromatosis type 2. Neuro Oncol. 2010;12(1):14-18.
28. Plotkin SR, Halpin C, McKenna MJ, et al. Erlotinib for progressive vestibular schwannoma in neurofibromatosis 2 patients. Otol Neurotol. 2010;31(7):1135-1143.
29. Arthurs BJ, Fairbanks RK, Demakas JJ, et al. A review of treatment modalities for vestibular schwannoma. Neurosurg Rev. 2011 Feb 9; [Epub ahead of print].
30. Andrews DW, Werner-Wasik M, Den RB, et al. Toward dose optimization for fractionated stereotactic radiotherapy for acoustic neuromas: comparison of two dose cohorts. Int J Radiat Oncol Biol Phys. 2009;74(2):419-426.
31. Thomas C, Di Maio S, Ma R, et al. Hearing preservation following fractionated stereotactic radiotherapy for vestibular schwannomas: prognostic implications of cochlear dose. J Neurosurg. 2007;107(5):917-926.
32. Samii M, Gerganov V, Samii A. Improved preservation of hearing and facial nerve function in vestibular schwannoma surgery via the retrosigmoid approach in a series of 200 patients. J Neurosurg. 2006;105(4):527-535.
33. Shiobara R, Ohira T, Inoue Y, et al. Extended middle cranial fossa approach for vestibular schwannoma: technical note and surgical results of 896 operations. Prog Neurol Surg. 2008;21:65-72.
34. Schmerber S, Palombi O, Boubagra K, et al. Long-term control of vestibular schwannoma after a translabyrinthine complete removal. Neurosurgery. 2005;57(4):693-698.
35. Phillips DJ, Kobylarz EJ, De Peralta ET, et al. Predictive factors of hearing preservation after surgical resection of small vestibular schwannomas. Otol Neurotol. 2010;31(9):1463-1468.
36. Park JK, Black MP, Vernick DM, Ramakrishna N. Vestibular schwannoma (acoustic neuroma) (2010). www.uptodate.com/contents/vestibular-schwannoma-acoustic-neuroma. Accessed May 16, 2011.
37. Schankin CJ, Gall C, Straube A. Headache syndromes after acoustic neuroma surgery and their implications for quality of life. Cephalalgia. 2009;29(7):760-761.
38. Ryzenman JM, Pensak ML, Tew JM Jr. Headache: a quality of life analysis in a cohort of 1,657 patients undergoing acoustic neuroma surgery: results from the Acoustic Neuroma Association. Laryngoscope. 2005;115(4):703-711.
39. Sriskandan N, Connor SE. The role of radiology in the diagnosis and management of vestibular schwannoma. Clin Radiol. 2010;66(4):357-365.
40. Yang I, Sughrue ME, Han SJ, et al. Facial nerve preservation after vestibular schwannoma Gamma Knife surgery. J Neurooncol. 2009;93(1): 41-48.
41. Unger F, Dominikus K, Haselsberger K. Stereotactic radiosurgery and fractionated stereotactic radiotherapy of acoustic neuromas [in German]. HNO. 2011;59(1):31-37.
42. Tos T, Caye-Thomasen P, Stangerup SE, et al. Long-term socio-economic impact of vestibular schwannoma for patients under observation and after surgery. J Laryngol Otol. 2003;117(12):955-964.
Grand Rounds: Woman, 49, With Dyspnea and Chest Tightness
A 49-year-old woman presented to urgent care with complaints of worsening dyspnea for the previous two days. She reported that her symptoms had begun gradually; at the time of her presentation, however, she was also experiencing chest tightness, occasional wheezing, and a nonproductive cough. She had experienced similar symptoms in the past and obtained good results by using her albuterol inhaler. During the current episode, however, she had not had the usual response to inhaler treatment.
The patient’s medical history was positive for environmental allergies, asthma, and GERD. Two weeks earlier, she had undergone dilatation and curettage (D&C) for dysfunctional bleeding, with no associated complications.
In the social history, the patient reported drinking four to six caffeine beverages daily and consuming alcohol moderately (two to four glasses of wine per week). She was following no formal dietary regimen. The patient denied current or past history of tobacco use and had not traveled recently. She had no family history of coronary vascular disease.
Her medications included albuterol and desloratadine as needed, pantoprazole 40 mg/d, and drospirenone/ethinyl estradiol. The patient said she used her albuterol inhaler four to six times per month but more often in the summer and fall. Nighttime awakenings due to asthma symptoms occurred no more than twice per month. She denied prior history of acute asthma exacerbations requiring oral systemic corticosteroids. The patient stated that since her D&C, she had been using ibuprofen almost daily for mild abdominal cramping.
A review of systems was positive for mild fatigue since her D&C. The patient denied fever, chills, headache, sore throat, or cough. She did complain of daily nasal congestion but with no unusual drainage. The patient denied orthopnea, chest pain, palpitations, or peripheral edema, as well as nausea, vomiting, diarrhea, constipation, hematochezia, or melena. She admitted to daily heartburn for the previous two weeks that was relieved somewhat with pantoprazole. She had not experienced urinary frequency or urgency, dysuria, or hematuria. She also denied rash, pruritus, weakness, paresthesias, joint pain, or swelling.
Physical examination revealed an alert, oriented female who appeared slightly anxious but was in no acute distress. Specific findings were pulse, 110 beats/min; blood pressure, 138/88 mm Hg; respirations, 24 breaths/min; temperature, 97.7°F; O2 saturation, 92% on room air. Her height measured 5’2” and weight, 150 lb (BMI, 27.43).
Her conjunctiva were slightly injected, and the tympanic membranes were intact bilaterally with a light reflex; the septum was midline. The mucosa was pale, boggy, and moist with clear drainage and no inflammation. The nasopharynx had no erythema, and the tonsils appeared normal, although a cobblestone appearance was noted in the posterior pharynx. The neck was supple with no adenopathy.
The patient’s heart rate, 110 beats/min, was regular with no murmurs, rubs, or gallops. In the lungs, a prolonged expiratory phase was noted, with diffuse wheezing on chest auscultation bilaterally. Neither retractions nor use of accessory muscles with breathing was observed. The abdomen was soft, rounded, and nontender with no organomegaly. Bowel sounds were evident in all four quadrants. The patient’s skin was free of suspicious lesions or rashes. Her extremities were without edema, and no calf tenderness was noted; Homans’ sign was negative. Superficial varicosities were noted bilaterally.
The top differential diagnosis included:
• Acute asthma (risk factors: history of uncontrolled asthma, as evidenced by frequent use of albuterol)
• Acute anemia (risk factors: history of dysfunctional uterine bleeding, recent D&C)
• Pulmonary embolism (risk factors: recent surgery, recent start of oral contraceptive use).
Additional diagnoses to be considered less likely included:
• Acute coronary syndrome/MI (possible causes of chest tightness, dyspnea, dyspepsia; but no chest pain, diaphoresis, or nausea)
• Acute respiratory distress (history of tachycardia, possible dyspnea; but no diaphoresis, cyanosis, retractions, accessory muscle use, or lung crackles)
• Pneumonia (risk factors: recent surgery, possible cause of nonproductive cough; but no evidence of fever, chills, rales, or pleuritic chest pain).
Diagnostic testing included a 12-lead ECG to evaluate the patient for cardiac arrhythmia or injury; on it, tachycardia was noted, with a regular rate of 106 beats/min. The patient’s chest x-ray yielded normal results.
Laboratory testing included a complete blood count to screen for anemia and infection. Results included a white blood cell count of 8,200/mL (normal range, 4,500 to 11,000/mL); hematocrit, 38.2% (normal range for women, 36.1% to 44.3%); hemoglobin, 13.1 g/dL (normal for women, 12.1 to 15.1 g/dL). A comprehensive metabolic panel was performed to assess electrolyte levels and kidney and liver function; findings were normal. Results of a D-dimer assay, which was obtained to exclude pulmonary embolism,1 were normal at 0.5 mg/L (range, 0.4 to 1.4 mg/L).
In the case of heightened suspicion for MI, the patient would have been transferred to the emergency department (ED) for evaluation, including serial cardiac troponin levels; elevated troponin levels are deemed the standard criterion to define and diagnose MI in a consensus document from the European Society of Cardiology and the American College of Cardiology.2 (Troponin-T and troponin-I are more tissue-specific than the MB fraction of creatine kinase [CK-MB] in detecting MI; positive troponin levels are considered virtually diagnostic of MI.2 Typically, cardiac troponin levels are measured two to three times over a 12- to 16-hour period.)
Peak expiratory flow (PEF), which was measured to evaluate the patient’s respiratory status, was 150 L/min (compared with personal best for a patient of her height and age, approximately 460 L/min). She was given 2.5 mg/3 mL of inhaled albuterol over 15 minutes. Her PEF increased to 350 L/min. O2 saturation improved to 96% on room air, pulse to 104 beats/min, and respirations 20 breaths/min; her blood pressure reading was now 140/90 mm Hg. A prolonged expiratory phase persisted in the lungs, but diffuse wheezing decreased by 40% on chest auscultation.
A second albuterol treatment was administered 20 minutes later, and the patient’s PEF increased to 380 L/min and O2 saturation to 99%. The lungs presently cleared with no further wheezing noted.
In addition, the patient was given a GI cocktail (ie, liquid antacid combined with an anticholinergic agent and viscous lidocaine). Within 10 minutes, her chest tightness was relieved 100%. Her blood pressure was then measured at 135/84 mm Hg; respirations, 18 breaths/min; and pulse rate, 96 beats/min.
According to the National Asthma Education and Prevention Program (NAEPP) 2007 Guidelines for the Diagnosis and Management of Asthma, Expert Panel Report 3 (EPR-3),3 the patient was classified as having intermittent, not-well-controlled asthma with an acute exacerbation. In addition, she was given a diagnosis of uncontrolled GERD.
DISCUSSION
Asthma Incidence and Risk Factors
Asthma affects approximately 300 million people worldwide and remains a global respiratory concern.4 In the United States, this chronic health condition has a prevalence of 8% to 10%. It is estimated that 5% to 10% of asthmatic patients have severe disease that does not respond typically to therapeutic interventions.5
Asthma involves bronchial hyperresponsiveness, airflow obstruction, and underlying inflammation. Acute episodes of asthma, arising from bronchospasm, usually manifest with progressively worsening cough, shortness of breath, chest tightness and wheezing (asthma’s hallmark symptoms), or a combination of symptoms.3
Symptoms of asthma or exacerbations of reactive airway disease vary from patient to patient. In addition to the hallmark symptoms noted, subacute or acute episodes of asthma exacerbation are characterized by decreases in expiratory airflow that can be documented by objective measurements of lung function, such as PEF or spirometry; these measures of airflow indicate the severity of an exacerbation more reliably than does perceived symptom severity.3 The EPR-3 panelists recommend determining asthma severity using a combination of objective criteria and clinical symptoms,3 although few clinicians use the objective criteria.6
Estimates of the prevalence of GERD among patients with asthma have varied from 34% to 89%.7-9 Patients with GERD are 1.97 times more likely than patients without GERD to have asthma10; silent gastroesophageal reflux has been identified in 24% to 62% of patients with asthma, and early studies suggest that treatment for GERD may improve asthma control in patients with severe or difficult-to-control asthma.8,11,12
The exact link between the two conditions is unclear. However, possible explanations why GERD and asthma coincide are that acid flow causes injury to the lining of the throat, airways, and lungs, making inhalation difficult and often causing a persistent cough; or that when acid enters the esophagus, a nerve reflex is triggered that causes the airways to narrow in order to prevent the acid from entering; this can explain dyspnea.8,9
Economic Burden
Asthma is costly to treat, and because there is no cure, the expense is ongoing. According to a 2011 report,13 the average annual direct cost of care (eg, medications, hospital admissions, nonemergency office visits) for one asthma patient between 2002 and 2007 was $3,259. In 2007, the most current data available, the total cost of asthma in the US was $56 billion, with productivity losses due to mortality accounting for $2.1 billion and morbidity-related losses estimated at $3.8 billion.13 The economic consequences of asthma are substantial and can place a considerable burden on affected individuals, their families, the health care system, and society as a whole.3
Current Standard of Care
Based on the scientific literature and the opinions expressed by the NAEPP in the EPR-3,3 clinicians are advised to consider the following general principles and goals for managing asthma: early treatment, special attention to patients at high risk for asthma-related death, and special attention to infants.3 The guidelines emphasize the importance of a clinician/patient partnership to facilitate the asthma patient’s self-management.
Early treatment is a particularly important component for management of asthma exacerbations. Important elements of early treatment include a written asthma action plan, combined with enhanced awareness of the early indicators of an exacerbation (ie, worsening PEF).3,14 It is believed that if patients are able to monitor their respiratory condition and follow a plan of care based on their PEF and/or signs and symptoms of asthma, they are more likely to achieve optimal management of their disease.15
Written Asthma Action Plan. The EPR-33 recommends that health care providers supply all asthmatic patients with a written asthma action plan that will define and support the patient’s efforts at self-management. Written asthma action plans are particularly beneficial for patients with moderate to severe persistent asthma, poorly controlled asthma, or a history of severe exacerbations.3,14
The written asthma action plan should include instructions for daily management of asthma and ways to recognize and treat worsening asthma, including adjustments to medication dosing. Plans may be based on PEF and/or symptoms. Asthma action plans should be discussed and reevaluated at follow-up visits.3 A sample asthma action plan can be found at www.health.state.ny.us/diseases/asthma/pdf/4850.pdf.16
Peak Expiratory Flow (PEF). The EPR-33 recommends PEF monitoring in all asthma patients, regardless of the severity of their exacerbations.17 PEF-based plans are especially useful for the patient who has difficulty perceiving early signs and symptoms of worsening asthma.3,18 A PEF-based plan instructs the patient to use quick-relief medications if symptoms occur or if PEF drops below 80% of the patient’s personal or predicted best. (Measured personal best is the patient’s highest PEF in the previous two weeks of good asthma control,3,19 whereas predicted best is calculated based on findings from a 1983 study by Knudson et al.3,20)
A PEF between 50% and 79% requires the patient to carefully monitor his or her response to the quick-relief medication and, based on that response, consider whether to contact a health care provider. When PEF falls below 50%, a provider’s immediate intervention is usually recommended.3
In the urgent care or ED setting, according to EPR-3 recommendations,3 the PEF or forced expiratory volume in 1 second (FEV1) is used to indicate the following:
• ≥ 70% predicted PEF or FEV1: goal for discharge
• 40% to 69% predicted PEF or FEV1: incomplete response to treatment, frequent need for treatment in the ED
• 3
Treatment and Management
Asthma management interventions that target the treatment of active disease and predisposing triggers are designed to reduce the severity and/or duration of morbidity associated with asthma—principally, to prevent symptoms and exacerbations (see Table 13).
When patients are discharged following an asthma exacerbation, their medications should include an oral corticosteroid burst and a short-acting b2-agonist (SABA); the clinician should also consider prescribing an inhaled corticosteroid (ICS).3
It is no longer recommended that ICS dosing be doubled in place of an oral steroid burst.3,21 The addition of a leukotriene receptor antagonist (LTRA) may also be considered.3,22
Patients should be given an action plan, and follow-up with a primary care provider should be scheduled within a few days—or even the following day, depending on the severity of the patient’s condition. The importance of follow-up with a primary care provider, a pulmonologist, or an asthma/allergy specialist should be emphasized.3,23
For patients who have difficulty recognizing their symptoms, a peak flow meter may be useful. This device is also recommended for patients with moderate to severe asthma or a history of numerous severe exacerbations.3 Additionally, spacers should always be used with metered dose inhalers (MDIs), because they make it easier for medication to reach the lungs and reduce the amount deposited in the mouth and throat, where it can lead to irritation. At each office visit, use of the peak flow meter and inhaler technique should be observed, and the action plan reevaluated and changed if necessary.3,14
Additional components of patient education include instruction in controlling environmental factors: avoiding environmental tobacco smoke, exposure to insect allergens, and molds. It is also important to stress controlling comorbid conditions that influence asthma, such as allergies or GERD. Patients with symptoms of GERD should be advised to take the steps shown in Table 2.8,24
Clinical Implications
Assessment of the severity of an asthma exacerbation is an essential component of ambulatory asthma care. Underclassification of asthma severity has been associated with increased morbidity and mortality,6 and the NAEPP guidelines recommend that clinicians assess and document asthma severity at each clinic visit.3,25 Patients who receive care based on evidence-based practice guidelines have been shown to experience 28% better outcomes.26
PATIENT OUTCOME
The case patient was discharged on an oral corticosteroid burst and a low-dose ICS. She was instructed how and when to use her SABA and given a prescription for a spacer; use of a peak flow meter was initiated with an estimated personal best goal of 460 L/min. The patient was given a written asthma action plan to help her recognize early signs and symptoms of worsening asthma and was advised to use quick-relief medications if she experienced symptoms or if her PEF dropped below 80% of her predicted best.
The patient’s clinician emphasized the importance of controlling any asthma-triggering environmental factors and reviewed nonpharmacologic interventions to control GERD. The patient was advised to resume desloratadine 5 mg/d and pantoprazole 40 mg/d. She was also instructed to schedule an appointment with her primary care provider within 48 hours and to return to urgent care or the ED with any further exacerbation of respiratory symptoms not controlled by her SABA.
CONCLUSION
Asthma morbidity is a nationally recognized, major public health problem. Given the sharp rise in health care costs and limited resources, health care providers must factor in the comparative effectiveness, comparative cost, and cost-effectiveness of both new and existing health care interventions when making treatment decisions.
Many asthmatic patients face the challenges of health care access and quality. By promoting their self-care and awareness, clinicians can help asthmatic patients achieve better symptom management and use the health care system less often.
REFERENCES
1. Stein PD, Hull RD, Patel KC, et al. D-Dimer for the exclusion of acute venous thrombosis and pulmonary embolism. Ann Intern Med. 2004;140(8):589-602.
2. Alpert JS, Thygesen K, Antman E, Bassand JP. Myocardial infarction redefined: a consensus document of The Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J Am Coll Cardiol. 2000;36(3):959-969.
3. National Asthma Education and Prevention Program, National Heart, Lung, and Blood Institute. Expert Panel Report 3: Guidelines for the Diagnosis and Management of Asthma. Bethesda, MD: National Heart, Lung, and Blood Institute; 2007. US Department of Health and Human Services publication NIH 07-4051.
4. Lougheed DM. Variability in asthma: symptom perception, care, and outcomes. Can J Physiol Pharmacol. 2007;85(1):149-154.
5. Higgins JC. The ‘crashing asthmatic.’ Am Fam Physician. 2003;67(5):997-1004.
6. Cowen MK, Wakefield DB, Cloutier MM. Classifying asthma severity: objective versus subjective measures. J Asthma. 2007;44(9):711-715.
7. Takenaka R, Matsuno O, Kitajima K, et al. The use of frequency scale for the symptoms of GERD in assessment of gastro-oesophageal reflux symptoms in asthma. Allergol Immunopathol (Madr). 2010;38(1):20-24.
8. Harding SM, Barnes PJ, Hollingsworth H. Gastroesophageal reflux and asthma (2010). www.uptodate.com/contents/gastroesophageal-reflux-and-asthma. Accessed April 5, 2011.
9. Havemann BD, Henderson CA, El-Serag HB. The association between gastro-oesophageal reflux disease and asthma: a systematic review. Gut. 2007;56(12):1654-1664.
10. Tsai MC, Lin HL, Lin CC, et al. Increased risk of concurrent asthma among patients with gastroesophageal reflux disease: a nationwide population-based study. Eur J Gastroenterol Hepatol. 2010;22(10):1169-1173.
11. Harding SM, Richter JE, Guzzo MR, et al. Asthma and gastroesophageal reflux: acid suppressive therapy improves asthma outcome. Am J Med. 1996;100(4):395-405.
12. Gibson PG, Henry RL, Coughlan JL. Gastro-oesophageal reflux treatment for asthma in adults and children. Cochrane Database Syst Rev. 2003;(2):CD001496.
13. Barnett SB, Nurmagambetov TA. Costs of asthma in the United States: 2002-2007. J Allergy Clin Immunol. 2011;127(1):145-152.
14. Walders N, Kercsmar C, Schluchter M, et al. An interdisciplinary intervention for undertreated pediatric asthma. Chest. 2006;129(2):292-299.
15. Morrow R, Fletcher J, Mulvihill M, Park H. The asthma dialogues: a model of interactive education for skills. J Contin Educ Health Prof. 2007;27(1): 49-58.
16. State of New York, Department of Health. Asthma action plan. www.health.state.ny.us/diseases/asthma/pdf/4850.pdf. Accessed April 11, 2011.
17. Picken HA, Greenfield S, Teres D, et al. Effects of local standards on the implementation of national guidelines for asthma: primary care agreement with national asthma guidelines. J Gen Intern Med. 1998;13(10):659-663.
18. Hardie GE, Gold WM, Janson S, et al. Understanding how asthmatics perceive symptom distress during a methacholine challenge. J Asthma. 2002;39(7):611-618.
19. Reddel HK, Marks GB, Jenkins CR. When can personal best peak flow be determined for asthma action plans? Thorax. 2004;59(11):922-924.
20. Knudson RJ, Lebowitz MD, Holberg CJ, Burrows B. Changes in the normal maximal expiratory flow-volume curve with growth and aging. Am Rev Respir Dis. 1983;127(6):725-734.
21. Ind PW, Dal Negro R, Colman NC, et al. Addition of salmeterol to fluticasone propionate treatment in moderate-to-severe asthma. Respir Med. 2003;97(5):555-562.
22. Price DB, Hernandez D, Magyar P, et al; Clinical Outcomes with Montelukast as a Partner Agent to Corticosteroid Therapy (COMPACT) International Study Group. Randomised controlled trial of montelukast plus inhaled budesonide versus double dose inhaled budesonide in adult patients with asthma. Thorax. 2003;58(3):211-216.
23. Schatz M, Zeiger RS, Mosen D, et al. Improved asthma outcomes from allergy specialist care: a population-based cross-sectional analysis. J Allergy Clin Immunol. 2005;116(6):1307-1313.
24. Hampel H, Abraham NS, El-Serag HB. Meta-analysis: obesity and the risk for gastroesophageal reflux disease and its complications. Ann Intern Med. 2005;143(3):199-211.
25. Cabana MD, Bruckman D, Meister K, et al. Documentation of asthma severity in pediatric outpatient clinics. Clin Pediatr (Phila). 2003;42(2):121-125.
26. Heater BS, Becker AM, Olson RK. Nursing interventions and patient outcomes: a meta-analysis of studies. Nurs Res. 1988;37(5):303-307.
A 49-year-old woman presented to urgent care with complaints of worsening dyspnea for the previous two days. She reported that her symptoms had begun gradually; at the time of her presentation, however, she was also experiencing chest tightness, occasional wheezing, and a nonproductive cough. She had experienced similar symptoms in the past and obtained good results by using her albuterol inhaler. During the current episode, however, she had not had the usual response to inhaler treatment.
The patient’s medical history was positive for environmental allergies, asthma, and GERD. Two weeks earlier, she had undergone dilatation and curettage (D&C) for dysfunctional bleeding, with no associated complications.
In the social history, the patient reported drinking four to six caffeine beverages daily and consuming alcohol moderately (two to four glasses of wine per week). She was following no formal dietary regimen. The patient denied current or past history of tobacco use and had not traveled recently. She had no family history of coronary vascular disease.
Her medications included albuterol and desloratadine as needed, pantoprazole 40 mg/d, and drospirenone/ethinyl estradiol. The patient said she used her albuterol inhaler four to six times per month but more often in the summer and fall. Nighttime awakenings due to asthma symptoms occurred no more than twice per month. She denied prior history of acute asthma exacerbations requiring oral systemic corticosteroids. The patient stated that since her D&C, she had been using ibuprofen almost daily for mild abdominal cramping.
A review of systems was positive for mild fatigue since her D&C. The patient denied fever, chills, headache, sore throat, or cough. She did complain of daily nasal congestion but with no unusual drainage. The patient denied orthopnea, chest pain, palpitations, or peripheral edema, as well as nausea, vomiting, diarrhea, constipation, hematochezia, or melena. She admitted to daily heartburn for the previous two weeks that was relieved somewhat with pantoprazole. She had not experienced urinary frequency or urgency, dysuria, or hematuria. She also denied rash, pruritus, weakness, paresthesias, joint pain, or swelling.
Physical examination revealed an alert, oriented female who appeared slightly anxious but was in no acute distress. Specific findings were pulse, 110 beats/min; blood pressure, 138/88 mm Hg; respirations, 24 breaths/min; temperature, 97.7°F; O2 saturation, 92% on room air. Her height measured 5’2” and weight, 150 lb (BMI, 27.43).
Her conjunctiva were slightly injected, and the tympanic membranes were intact bilaterally with a light reflex; the septum was midline. The mucosa was pale, boggy, and moist with clear drainage and no inflammation. The nasopharynx had no erythema, and the tonsils appeared normal, although a cobblestone appearance was noted in the posterior pharynx. The neck was supple with no adenopathy.
The patient’s heart rate, 110 beats/min, was regular with no murmurs, rubs, or gallops. In the lungs, a prolonged expiratory phase was noted, with diffuse wheezing on chest auscultation bilaterally. Neither retractions nor use of accessory muscles with breathing was observed. The abdomen was soft, rounded, and nontender with no organomegaly. Bowel sounds were evident in all four quadrants. The patient’s skin was free of suspicious lesions or rashes. Her extremities were without edema, and no calf tenderness was noted; Homans’ sign was negative. Superficial varicosities were noted bilaterally.
The top differential diagnosis included:
• Acute asthma (risk factors: history of uncontrolled asthma, as evidenced by frequent use of albuterol)
• Acute anemia (risk factors: history of dysfunctional uterine bleeding, recent D&C)
• Pulmonary embolism (risk factors: recent surgery, recent start of oral contraceptive use).
Additional diagnoses to be considered less likely included:
• Acute coronary syndrome/MI (possible causes of chest tightness, dyspnea, dyspepsia; but no chest pain, diaphoresis, or nausea)
• Acute respiratory distress (history of tachycardia, possible dyspnea; but no diaphoresis, cyanosis, retractions, accessory muscle use, or lung crackles)
• Pneumonia (risk factors: recent surgery, possible cause of nonproductive cough; but no evidence of fever, chills, rales, or pleuritic chest pain).
Diagnostic testing included a 12-lead ECG to evaluate the patient for cardiac arrhythmia or injury; on it, tachycardia was noted, with a regular rate of 106 beats/min. The patient’s chest x-ray yielded normal results.
Laboratory testing included a complete blood count to screen for anemia and infection. Results included a white blood cell count of 8,200/mL (normal range, 4,500 to 11,000/mL); hematocrit, 38.2% (normal range for women, 36.1% to 44.3%); hemoglobin, 13.1 g/dL (normal for women, 12.1 to 15.1 g/dL). A comprehensive metabolic panel was performed to assess electrolyte levels and kidney and liver function; findings were normal. Results of a D-dimer assay, which was obtained to exclude pulmonary embolism,1 were normal at 0.5 mg/L (range, 0.4 to 1.4 mg/L).
In the case of heightened suspicion for MI, the patient would have been transferred to the emergency department (ED) for evaluation, including serial cardiac troponin levels; elevated troponin levels are deemed the standard criterion to define and diagnose MI in a consensus document from the European Society of Cardiology and the American College of Cardiology.2 (Troponin-T and troponin-I are more tissue-specific than the MB fraction of creatine kinase [CK-MB] in detecting MI; positive troponin levels are considered virtually diagnostic of MI.2 Typically, cardiac troponin levels are measured two to three times over a 12- to 16-hour period.)
Peak expiratory flow (PEF), which was measured to evaluate the patient’s respiratory status, was 150 L/min (compared with personal best for a patient of her height and age, approximately 460 L/min). She was given 2.5 mg/3 mL of inhaled albuterol over 15 minutes. Her PEF increased to 350 L/min. O2 saturation improved to 96% on room air, pulse to 104 beats/min, and respirations 20 breaths/min; her blood pressure reading was now 140/90 mm Hg. A prolonged expiratory phase persisted in the lungs, but diffuse wheezing decreased by 40% on chest auscultation.
A second albuterol treatment was administered 20 minutes later, and the patient’s PEF increased to 380 L/min and O2 saturation to 99%. The lungs presently cleared with no further wheezing noted.
In addition, the patient was given a GI cocktail (ie, liquid antacid combined with an anticholinergic agent and viscous lidocaine). Within 10 minutes, her chest tightness was relieved 100%. Her blood pressure was then measured at 135/84 mm Hg; respirations, 18 breaths/min; and pulse rate, 96 beats/min.
According to the National Asthma Education and Prevention Program (NAEPP) 2007 Guidelines for the Diagnosis and Management of Asthma, Expert Panel Report 3 (EPR-3),3 the patient was classified as having intermittent, not-well-controlled asthma with an acute exacerbation. In addition, she was given a diagnosis of uncontrolled GERD.
DISCUSSION
Asthma Incidence and Risk Factors
Asthma affects approximately 300 million people worldwide and remains a global respiratory concern.4 In the United States, this chronic health condition has a prevalence of 8% to 10%. It is estimated that 5% to 10% of asthmatic patients have severe disease that does not respond typically to therapeutic interventions.5
Asthma involves bronchial hyperresponsiveness, airflow obstruction, and underlying inflammation. Acute episodes of asthma, arising from bronchospasm, usually manifest with progressively worsening cough, shortness of breath, chest tightness and wheezing (asthma’s hallmark symptoms), or a combination of symptoms.3
Symptoms of asthma or exacerbations of reactive airway disease vary from patient to patient. In addition to the hallmark symptoms noted, subacute or acute episodes of asthma exacerbation are characterized by decreases in expiratory airflow that can be documented by objective measurements of lung function, such as PEF or spirometry; these measures of airflow indicate the severity of an exacerbation more reliably than does perceived symptom severity.3 The EPR-3 panelists recommend determining asthma severity using a combination of objective criteria and clinical symptoms,3 although few clinicians use the objective criteria.6
Estimates of the prevalence of GERD among patients with asthma have varied from 34% to 89%.7-9 Patients with GERD are 1.97 times more likely than patients without GERD to have asthma10; silent gastroesophageal reflux has been identified in 24% to 62% of patients with asthma, and early studies suggest that treatment for GERD may improve asthma control in patients with severe or difficult-to-control asthma.8,11,12
The exact link between the two conditions is unclear. However, possible explanations why GERD and asthma coincide are that acid flow causes injury to the lining of the throat, airways, and lungs, making inhalation difficult and often causing a persistent cough; or that when acid enters the esophagus, a nerve reflex is triggered that causes the airways to narrow in order to prevent the acid from entering; this can explain dyspnea.8,9
Economic Burden
Asthma is costly to treat, and because there is no cure, the expense is ongoing. According to a 2011 report,13 the average annual direct cost of care (eg, medications, hospital admissions, nonemergency office visits) for one asthma patient between 2002 and 2007 was $3,259. In 2007, the most current data available, the total cost of asthma in the US was $56 billion, with productivity losses due to mortality accounting for $2.1 billion and morbidity-related losses estimated at $3.8 billion.13 The economic consequences of asthma are substantial and can place a considerable burden on affected individuals, their families, the health care system, and society as a whole.3
Current Standard of Care
Based on the scientific literature and the opinions expressed by the NAEPP in the EPR-3,3 clinicians are advised to consider the following general principles and goals for managing asthma: early treatment, special attention to patients at high risk for asthma-related death, and special attention to infants.3 The guidelines emphasize the importance of a clinician/patient partnership to facilitate the asthma patient’s self-management.
Early treatment is a particularly important component for management of asthma exacerbations. Important elements of early treatment include a written asthma action plan, combined with enhanced awareness of the early indicators of an exacerbation (ie, worsening PEF).3,14 It is believed that if patients are able to monitor their respiratory condition and follow a plan of care based on their PEF and/or signs and symptoms of asthma, they are more likely to achieve optimal management of their disease.15
Written Asthma Action Plan. The EPR-33 recommends that health care providers supply all asthmatic patients with a written asthma action plan that will define and support the patient’s efforts at self-management. Written asthma action plans are particularly beneficial for patients with moderate to severe persistent asthma, poorly controlled asthma, or a history of severe exacerbations.3,14
The written asthma action plan should include instructions for daily management of asthma and ways to recognize and treat worsening asthma, including adjustments to medication dosing. Plans may be based on PEF and/or symptoms. Asthma action plans should be discussed and reevaluated at follow-up visits.3 A sample asthma action plan can be found at www.health.state.ny.us/diseases/asthma/pdf/4850.pdf.16
Peak Expiratory Flow (PEF). The EPR-33 recommends PEF monitoring in all asthma patients, regardless of the severity of their exacerbations.17 PEF-based plans are especially useful for the patient who has difficulty perceiving early signs and symptoms of worsening asthma.3,18 A PEF-based plan instructs the patient to use quick-relief medications if symptoms occur or if PEF drops below 80% of the patient’s personal or predicted best. (Measured personal best is the patient’s highest PEF in the previous two weeks of good asthma control,3,19 whereas predicted best is calculated based on findings from a 1983 study by Knudson et al.3,20)
A PEF between 50% and 79% requires the patient to carefully monitor his or her response to the quick-relief medication and, based on that response, consider whether to contact a health care provider. When PEF falls below 50%, a provider’s immediate intervention is usually recommended.3
In the urgent care or ED setting, according to EPR-3 recommendations,3 the PEF or forced expiratory volume in 1 second (FEV1) is used to indicate the following:
• ≥ 70% predicted PEF or FEV1: goal for discharge
• 40% to 69% predicted PEF or FEV1: incomplete response to treatment, frequent need for treatment in the ED
• 3
Treatment and Management
Asthma management interventions that target the treatment of active disease and predisposing triggers are designed to reduce the severity and/or duration of morbidity associated with asthma—principally, to prevent symptoms and exacerbations (see Table 13).
When patients are discharged following an asthma exacerbation, their medications should include an oral corticosteroid burst and a short-acting b2-agonist (SABA); the clinician should also consider prescribing an inhaled corticosteroid (ICS).3
It is no longer recommended that ICS dosing be doubled in place of an oral steroid burst.3,21 The addition of a leukotriene receptor antagonist (LTRA) may also be considered.3,22
Patients should be given an action plan, and follow-up with a primary care provider should be scheduled within a few days—or even the following day, depending on the severity of the patient’s condition. The importance of follow-up with a primary care provider, a pulmonologist, or an asthma/allergy specialist should be emphasized.3,23
For patients who have difficulty recognizing their symptoms, a peak flow meter may be useful. This device is also recommended for patients with moderate to severe asthma or a history of numerous severe exacerbations.3 Additionally, spacers should always be used with metered dose inhalers (MDIs), because they make it easier for medication to reach the lungs and reduce the amount deposited in the mouth and throat, where it can lead to irritation. At each office visit, use of the peak flow meter and inhaler technique should be observed, and the action plan reevaluated and changed if necessary.3,14
Additional components of patient education include instruction in controlling environmental factors: avoiding environmental tobacco smoke, exposure to insect allergens, and molds. It is also important to stress controlling comorbid conditions that influence asthma, such as allergies or GERD. Patients with symptoms of GERD should be advised to take the steps shown in Table 2.8,24
Clinical Implications
Assessment of the severity of an asthma exacerbation is an essential component of ambulatory asthma care. Underclassification of asthma severity has been associated with increased morbidity and mortality,6 and the NAEPP guidelines recommend that clinicians assess and document asthma severity at each clinic visit.3,25 Patients who receive care based on evidence-based practice guidelines have been shown to experience 28% better outcomes.26
PATIENT OUTCOME
The case patient was discharged on an oral corticosteroid burst and a low-dose ICS. She was instructed how and when to use her SABA and given a prescription for a spacer; use of a peak flow meter was initiated with an estimated personal best goal of 460 L/min. The patient was given a written asthma action plan to help her recognize early signs and symptoms of worsening asthma and was advised to use quick-relief medications if she experienced symptoms or if her PEF dropped below 80% of her predicted best.
The patient’s clinician emphasized the importance of controlling any asthma-triggering environmental factors and reviewed nonpharmacologic interventions to control GERD. The patient was advised to resume desloratadine 5 mg/d and pantoprazole 40 mg/d. She was also instructed to schedule an appointment with her primary care provider within 48 hours and to return to urgent care or the ED with any further exacerbation of respiratory symptoms not controlled by her SABA.
CONCLUSION
Asthma morbidity is a nationally recognized, major public health problem. Given the sharp rise in health care costs and limited resources, health care providers must factor in the comparative effectiveness, comparative cost, and cost-effectiveness of both new and existing health care interventions when making treatment decisions.
Many asthmatic patients face the challenges of health care access and quality. By promoting their self-care and awareness, clinicians can help asthmatic patients achieve better symptom management and use the health care system less often.
REFERENCES
1. Stein PD, Hull RD, Patel KC, et al. D-Dimer for the exclusion of acute venous thrombosis and pulmonary embolism. Ann Intern Med. 2004;140(8):589-602.
2. Alpert JS, Thygesen K, Antman E, Bassand JP. Myocardial infarction redefined: a consensus document of The Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J Am Coll Cardiol. 2000;36(3):959-969.
3. National Asthma Education and Prevention Program, National Heart, Lung, and Blood Institute. Expert Panel Report 3: Guidelines for the Diagnosis and Management of Asthma. Bethesda, MD: National Heart, Lung, and Blood Institute; 2007. US Department of Health and Human Services publication NIH 07-4051.
4. Lougheed DM. Variability in asthma: symptom perception, care, and outcomes. Can J Physiol Pharmacol. 2007;85(1):149-154.
5. Higgins JC. The ‘crashing asthmatic.’ Am Fam Physician. 2003;67(5):997-1004.
6. Cowen MK, Wakefield DB, Cloutier MM. Classifying asthma severity: objective versus subjective measures. J Asthma. 2007;44(9):711-715.
7. Takenaka R, Matsuno O, Kitajima K, et al. The use of frequency scale for the symptoms of GERD in assessment of gastro-oesophageal reflux symptoms in asthma. Allergol Immunopathol (Madr). 2010;38(1):20-24.
8. Harding SM, Barnes PJ, Hollingsworth H. Gastroesophageal reflux and asthma (2010). www.uptodate.com/contents/gastroesophageal-reflux-and-asthma. Accessed April 5, 2011.
9. Havemann BD, Henderson CA, El-Serag HB. The association between gastro-oesophageal reflux disease and asthma: a systematic review. Gut. 2007;56(12):1654-1664.
10. Tsai MC, Lin HL, Lin CC, et al. Increased risk of concurrent asthma among patients with gastroesophageal reflux disease: a nationwide population-based study. Eur J Gastroenterol Hepatol. 2010;22(10):1169-1173.
11. Harding SM, Richter JE, Guzzo MR, et al. Asthma and gastroesophageal reflux: acid suppressive therapy improves asthma outcome. Am J Med. 1996;100(4):395-405.
12. Gibson PG, Henry RL, Coughlan JL. Gastro-oesophageal reflux treatment for asthma in adults and children. Cochrane Database Syst Rev. 2003;(2):CD001496.
13. Barnett SB, Nurmagambetov TA. Costs of asthma in the United States: 2002-2007. J Allergy Clin Immunol. 2011;127(1):145-152.
14. Walders N, Kercsmar C, Schluchter M, et al. An interdisciplinary intervention for undertreated pediatric asthma. Chest. 2006;129(2):292-299.
15. Morrow R, Fletcher J, Mulvihill M, Park H. The asthma dialogues: a model of interactive education for skills. J Contin Educ Health Prof. 2007;27(1): 49-58.
16. State of New York, Department of Health. Asthma action plan. www.health.state.ny.us/diseases/asthma/pdf/4850.pdf. Accessed April 11, 2011.
17. Picken HA, Greenfield S, Teres D, et al. Effects of local standards on the implementation of national guidelines for asthma: primary care agreement with national asthma guidelines. J Gen Intern Med. 1998;13(10):659-663.
18. Hardie GE, Gold WM, Janson S, et al. Understanding how asthmatics perceive symptom distress during a methacholine challenge. J Asthma. 2002;39(7):611-618.
19. Reddel HK, Marks GB, Jenkins CR. When can personal best peak flow be determined for asthma action plans? Thorax. 2004;59(11):922-924.
20. Knudson RJ, Lebowitz MD, Holberg CJ, Burrows B. Changes in the normal maximal expiratory flow-volume curve with growth and aging. Am Rev Respir Dis. 1983;127(6):725-734.
21. Ind PW, Dal Negro R, Colman NC, et al. Addition of salmeterol to fluticasone propionate treatment in moderate-to-severe asthma. Respir Med. 2003;97(5):555-562.
22. Price DB, Hernandez D, Magyar P, et al; Clinical Outcomes with Montelukast as a Partner Agent to Corticosteroid Therapy (COMPACT) International Study Group. Randomised controlled trial of montelukast plus inhaled budesonide versus double dose inhaled budesonide in adult patients with asthma. Thorax. 2003;58(3):211-216.
23. Schatz M, Zeiger RS, Mosen D, et al. Improved asthma outcomes from allergy specialist care: a population-based cross-sectional analysis. J Allergy Clin Immunol. 2005;116(6):1307-1313.
24. Hampel H, Abraham NS, El-Serag HB. Meta-analysis: obesity and the risk for gastroesophageal reflux disease and its complications. Ann Intern Med. 2005;143(3):199-211.
25. Cabana MD, Bruckman D, Meister K, et al. Documentation of asthma severity in pediatric outpatient clinics. Clin Pediatr (Phila). 2003;42(2):121-125.
26. Heater BS, Becker AM, Olson RK. Nursing interventions and patient outcomes: a meta-analysis of studies. Nurs Res. 1988;37(5):303-307.
A 49-year-old woman presented to urgent care with complaints of worsening dyspnea for the previous two days. She reported that her symptoms had begun gradually; at the time of her presentation, however, she was also experiencing chest tightness, occasional wheezing, and a nonproductive cough. She had experienced similar symptoms in the past and obtained good results by using her albuterol inhaler. During the current episode, however, she had not had the usual response to inhaler treatment.
The patient’s medical history was positive for environmental allergies, asthma, and GERD. Two weeks earlier, she had undergone dilatation and curettage (D&C) for dysfunctional bleeding, with no associated complications.
In the social history, the patient reported drinking four to six caffeine beverages daily and consuming alcohol moderately (two to four glasses of wine per week). She was following no formal dietary regimen. The patient denied current or past history of tobacco use and had not traveled recently. She had no family history of coronary vascular disease.
Her medications included albuterol and desloratadine as needed, pantoprazole 40 mg/d, and drospirenone/ethinyl estradiol. The patient said she used her albuterol inhaler four to six times per month but more often in the summer and fall. Nighttime awakenings due to asthma symptoms occurred no more than twice per month. She denied prior history of acute asthma exacerbations requiring oral systemic corticosteroids. The patient stated that since her D&C, she had been using ibuprofen almost daily for mild abdominal cramping.
A review of systems was positive for mild fatigue since her D&C. The patient denied fever, chills, headache, sore throat, or cough. She did complain of daily nasal congestion but with no unusual drainage. The patient denied orthopnea, chest pain, palpitations, or peripheral edema, as well as nausea, vomiting, diarrhea, constipation, hematochezia, or melena. She admitted to daily heartburn for the previous two weeks that was relieved somewhat with pantoprazole. She had not experienced urinary frequency or urgency, dysuria, or hematuria. She also denied rash, pruritus, weakness, paresthesias, joint pain, or swelling.
Physical examination revealed an alert, oriented female who appeared slightly anxious but was in no acute distress. Specific findings were pulse, 110 beats/min; blood pressure, 138/88 mm Hg; respirations, 24 breaths/min; temperature, 97.7°F; O2 saturation, 92% on room air. Her height measured 5’2” and weight, 150 lb (BMI, 27.43).
Her conjunctiva were slightly injected, and the tympanic membranes were intact bilaterally with a light reflex; the septum was midline. The mucosa was pale, boggy, and moist with clear drainage and no inflammation. The nasopharynx had no erythema, and the tonsils appeared normal, although a cobblestone appearance was noted in the posterior pharynx. The neck was supple with no adenopathy.
The patient’s heart rate, 110 beats/min, was regular with no murmurs, rubs, or gallops. In the lungs, a prolonged expiratory phase was noted, with diffuse wheezing on chest auscultation bilaterally. Neither retractions nor use of accessory muscles with breathing was observed. The abdomen was soft, rounded, and nontender with no organomegaly. Bowel sounds were evident in all four quadrants. The patient’s skin was free of suspicious lesions or rashes. Her extremities were without edema, and no calf tenderness was noted; Homans’ sign was negative. Superficial varicosities were noted bilaterally.
The top differential diagnosis included:
• Acute asthma (risk factors: history of uncontrolled asthma, as evidenced by frequent use of albuterol)
• Acute anemia (risk factors: history of dysfunctional uterine bleeding, recent D&C)
• Pulmonary embolism (risk factors: recent surgery, recent start of oral contraceptive use).
Additional diagnoses to be considered less likely included:
• Acute coronary syndrome/MI (possible causes of chest tightness, dyspnea, dyspepsia; but no chest pain, diaphoresis, or nausea)
• Acute respiratory distress (history of tachycardia, possible dyspnea; but no diaphoresis, cyanosis, retractions, accessory muscle use, or lung crackles)
• Pneumonia (risk factors: recent surgery, possible cause of nonproductive cough; but no evidence of fever, chills, rales, or pleuritic chest pain).
Diagnostic testing included a 12-lead ECG to evaluate the patient for cardiac arrhythmia or injury; on it, tachycardia was noted, with a regular rate of 106 beats/min. The patient’s chest x-ray yielded normal results.
Laboratory testing included a complete blood count to screen for anemia and infection. Results included a white blood cell count of 8,200/mL (normal range, 4,500 to 11,000/mL); hematocrit, 38.2% (normal range for women, 36.1% to 44.3%); hemoglobin, 13.1 g/dL (normal for women, 12.1 to 15.1 g/dL). A comprehensive metabolic panel was performed to assess electrolyte levels and kidney and liver function; findings were normal. Results of a D-dimer assay, which was obtained to exclude pulmonary embolism,1 were normal at 0.5 mg/L (range, 0.4 to 1.4 mg/L).
In the case of heightened suspicion for MI, the patient would have been transferred to the emergency department (ED) for evaluation, including serial cardiac troponin levels; elevated troponin levels are deemed the standard criterion to define and diagnose MI in a consensus document from the European Society of Cardiology and the American College of Cardiology.2 (Troponin-T and troponin-I are more tissue-specific than the MB fraction of creatine kinase [CK-MB] in detecting MI; positive troponin levels are considered virtually diagnostic of MI.2 Typically, cardiac troponin levels are measured two to three times over a 12- to 16-hour period.)
Peak expiratory flow (PEF), which was measured to evaluate the patient’s respiratory status, was 150 L/min (compared with personal best for a patient of her height and age, approximately 460 L/min). She was given 2.5 mg/3 mL of inhaled albuterol over 15 minutes. Her PEF increased to 350 L/min. O2 saturation improved to 96% on room air, pulse to 104 beats/min, and respirations 20 breaths/min; her blood pressure reading was now 140/90 mm Hg. A prolonged expiratory phase persisted in the lungs, but diffuse wheezing decreased by 40% on chest auscultation.
A second albuterol treatment was administered 20 minutes later, and the patient’s PEF increased to 380 L/min and O2 saturation to 99%. The lungs presently cleared with no further wheezing noted.
In addition, the patient was given a GI cocktail (ie, liquid antacid combined with an anticholinergic agent and viscous lidocaine). Within 10 minutes, her chest tightness was relieved 100%. Her blood pressure was then measured at 135/84 mm Hg; respirations, 18 breaths/min; and pulse rate, 96 beats/min.
According to the National Asthma Education and Prevention Program (NAEPP) 2007 Guidelines for the Diagnosis and Management of Asthma, Expert Panel Report 3 (EPR-3),3 the patient was classified as having intermittent, not-well-controlled asthma with an acute exacerbation. In addition, she was given a diagnosis of uncontrolled GERD.
DISCUSSION
Asthma Incidence and Risk Factors
Asthma affects approximately 300 million people worldwide and remains a global respiratory concern.4 In the United States, this chronic health condition has a prevalence of 8% to 10%. It is estimated that 5% to 10% of asthmatic patients have severe disease that does not respond typically to therapeutic interventions.5
Asthma involves bronchial hyperresponsiveness, airflow obstruction, and underlying inflammation. Acute episodes of asthma, arising from bronchospasm, usually manifest with progressively worsening cough, shortness of breath, chest tightness and wheezing (asthma’s hallmark symptoms), or a combination of symptoms.3
Symptoms of asthma or exacerbations of reactive airway disease vary from patient to patient. In addition to the hallmark symptoms noted, subacute or acute episodes of asthma exacerbation are characterized by decreases in expiratory airflow that can be documented by objective measurements of lung function, such as PEF or spirometry; these measures of airflow indicate the severity of an exacerbation more reliably than does perceived symptom severity.3 The EPR-3 panelists recommend determining asthma severity using a combination of objective criteria and clinical symptoms,3 although few clinicians use the objective criteria.6
Estimates of the prevalence of GERD among patients with asthma have varied from 34% to 89%.7-9 Patients with GERD are 1.97 times more likely than patients without GERD to have asthma10; silent gastroesophageal reflux has been identified in 24% to 62% of patients with asthma, and early studies suggest that treatment for GERD may improve asthma control in patients with severe or difficult-to-control asthma.8,11,12
The exact link between the two conditions is unclear. However, possible explanations why GERD and asthma coincide are that acid flow causes injury to the lining of the throat, airways, and lungs, making inhalation difficult and often causing a persistent cough; or that when acid enters the esophagus, a nerve reflex is triggered that causes the airways to narrow in order to prevent the acid from entering; this can explain dyspnea.8,9
Economic Burden
Asthma is costly to treat, and because there is no cure, the expense is ongoing. According to a 2011 report,13 the average annual direct cost of care (eg, medications, hospital admissions, nonemergency office visits) for one asthma patient between 2002 and 2007 was $3,259. In 2007, the most current data available, the total cost of asthma in the US was $56 billion, with productivity losses due to mortality accounting for $2.1 billion and morbidity-related losses estimated at $3.8 billion.13 The economic consequences of asthma are substantial and can place a considerable burden on affected individuals, their families, the health care system, and society as a whole.3
Current Standard of Care
Based on the scientific literature and the opinions expressed by the NAEPP in the EPR-3,3 clinicians are advised to consider the following general principles and goals for managing asthma: early treatment, special attention to patients at high risk for asthma-related death, and special attention to infants.3 The guidelines emphasize the importance of a clinician/patient partnership to facilitate the asthma patient’s self-management.
Early treatment is a particularly important component for management of asthma exacerbations. Important elements of early treatment include a written asthma action plan, combined with enhanced awareness of the early indicators of an exacerbation (ie, worsening PEF).3,14 It is believed that if patients are able to monitor their respiratory condition and follow a plan of care based on their PEF and/or signs and symptoms of asthma, they are more likely to achieve optimal management of their disease.15
Written Asthma Action Plan. The EPR-33 recommends that health care providers supply all asthmatic patients with a written asthma action plan that will define and support the patient’s efforts at self-management. Written asthma action plans are particularly beneficial for patients with moderate to severe persistent asthma, poorly controlled asthma, or a history of severe exacerbations.3,14
The written asthma action plan should include instructions for daily management of asthma and ways to recognize and treat worsening asthma, including adjustments to medication dosing. Plans may be based on PEF and/or symptoms. Asthma action plans should be discussed and reevaluated at follow-up visits.3 A sample asthma action plan can be found at www.health.state.ny.us/diseases/asthma/pdf/4850.pdf.16
Peak Expiratory Flow (PEF). The EPR-33 recommends PEF monitoring in all asthma patients, regardless of the severity of their exacerbations.17 PEF-based plans are especially useful for the patient who has difficulty perceiving early signs and symptoms of worsening asthma.3,18 A PEF-based plan instructs the patient to use quick-relief medications if symptoms occur or if PEF drops below 80% of the patient’s personal or predicted best. (Measured personal best is the patient’s highest PEF in the previous two weeks of good asthma control,3,19 whereas predicted best is calculated based on findings from a 1983 study by Knudson et al.3,20)
A PEF between 50% and 79% requires the patient to carefully monitor his or her response to the quick-relief medication and, based on that response, consider whether to contact a health care provider. When PEF falls below 50%, a provider’s immediate intervention is usually recommended.3
In the urgent care or ED setting, according to EPR-3 recommendations,3 the PEF or forced expiratory volume in 1 second (FEV1) is used to indicate the following:
• ≥ 70% predicted PEF or FEV1: goal for discharge
• 40% to 69% predicted PEF or FEV1: incomplete response to treatment, frequent need for treatment in the ED
• 3
Treatment and Management
Asthma management interventions that target the treatment of active disease and predisposing triggers are designed to reduce the severity and/or duration of morbidity associated with asthma—principally, to prevent symptoms and exacerbations (see Table 13).
When patients are discharged following an asthma exacerbation, their medications should include an oral corticosteroid burst and a short-acting b2-agonist (SABA); the clinician should also consider prescribing an inhaled corticosteroid (ICS).3
It is no longer recommended that ICS dosing be doubled in place of an oral steroid burst.3,21 The addition of a leukotriene receptor antagonist (LTRA) may also be considered.3,22
Patients should be given an action plan, and follow-up with a primary care provider should be scheduled within a few days—or even the following day, depending on the severity of the patient’s condition. The importance of follow-up with a primary care provider, a pulmonologist, or an asthma/allergy specialist should be emphasized.3,23
For patients who have difficulty recognizing their symptoms, a peak flow meter may be useful. This device is also recommended for patients with moderate to severe asthma or a history of numerous severe exacerbations.3 Additionally, spacers should always be used with metered dose inhalers (MDIs), because they make it easier for medication to reach the lungs and reduce the amount deposited in the mouth and throat, where it can lead to irritation. At each office visit, use of the peak flow meter and inhaler technique should be observed, and the action plan reevaluated and changed if necessary.3,14
Additional components of patient education include instruction in controlling environmental factors: avoiding environmental tobacco smoke, exposure to insect allergens, and molds. It is also important to stress controlling comorbid conditions that influence asthma, such as allergies or GERD. Patients with symptoms of GERD should be advised to take the steps shown in Table 2.8,24
Clinical Implications
Assessment of the severity of an asthma exacerbation is an essential component of ambulatory asthma care. Underclassification of asthma severity has been associated with increased morbidity and mortality,6 and the NAEPP guidelines recommend that clinicians assess and document asthma severity at each clinic visit.3,25 Patients who receive care based on evidence-based practice guidelines have been shown to experience 28% better outcomes.26
PATIENT OUTCOME
The case patient was discharged on an oral corticosteroid burst and a low-dose ICS. She was instructed how and when to use her SABA and given a prescription for a spacer; use of a peak flow meter was initiated with an estimated personal best goal of 460 L/min. The patient was given a written asthma action plan to help her recognize early signs and symptoms of worsening asthma and was advised to use quick-relief medications if she experienced symptoms or if her PEF dropped below 80% of her predicted best.
The patient’s clinician emphasized the importance of controlling any asthma-triggering environmental factors and reviewed nonpharmacologic interventions to control GERD. The patient was advised to resume desloratadine 5 mg/d and pantoprazole 40 mg/d. She was also instructed to schedule an appointment with her primary care provider within 48 hours and to return to urgent care or the ED with any further exacerbation of respiratory symptoms not controlled by her SABA.
CONCLUSION
Asthma morbidity is a nationally recognized, major public health problem. Given the sharp rise in health care costs and limited resources, health care providers must factor in the comparative effectiveness, comparative cost, and cost-effectiveness of both new and existing health care interventions when making treatment decisions.
Many asthmatic patients face the challenges of health care access and quality. By promoting their self-care and awareness, clinicians can help asthmatic patients achieve better symptom management and use the health care system less often.
REFERENCES
1. Stein PD, Hull RD, Patel KC, et al. D-Dimer for the exclusion of acute venous thrombosis and pulmonary embolism. Ann Intern Med. 2004;140(8):589-602.
2. Alpert JS, Thygesen K, Antman E, Bassand JP. Myocardial infarction redefined: a consensus document of The Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J Am Coll Cardiol. 2000;36(3):959-969.
3. National Asthma Education and Prevention Program, National Heart, Lung, and Blood Institute. Expert Panel Report 3: Guidelines for the Diagnosis and Management of Asthma. Bethesda, MD: National Heart, Lung, and Blood Institute; 2007. US Department of Health and Human Services publication NIH 07-4051.
4. Lougheed DM. Variability in asthma: symptom perception, care, and outcomes. Can J Physiol Pharmacol. 2007;85(1):149-154.
5. Higgins JC. The ‘crashing asthmatic.’ Am Fam Physician. 2003;67(5):997-1004.
6. Cowen MK, Wakefield DB, Cloutier MM. Classifying asthma severity: objective versus subjective measures. J Asthma. 2007;44(9):711-715.
7. Takenaka R, Matsuno O, Kitajima K, et al. The use of frequency scale for the symptoms of GERD in assessment of gastro-oesophageal reflux symptoms in asthma. Allergol Immunopathol (Madr). 2010;38(1):20-24.
8. Harding SM, Barnes PJ, Hollingsworth H. Gastroesophageal reflux and asthma (2010). www.uptodate.com/contents/gastroesophageal-reflux-and-asthma. Accessed April 5, 2011.
9. Havemann BD, Henderson CA, El-Serag HB. The association between gastro-oesophageal reflux disease and asthma: a systematic review. Gut. 2007;56(12):1654-1664.
10. Tsai MC, Lin HL, Lin CC, et al. Increased risk of concurrent asthma among patients with gastroesophageal reflux disease: a nationwide population-based study. Eur J Gastroenterol Hepatol. 2010;22(10):1169-1173.
11. Harding SM, Richter JE, Guzzo MR, et al. Asthma and gastroesophageal reflux: acid suppressive therapy improves asthma outcome. Am J Med. 1996;100(4):395-405.
12. Gibson PG, Henry RL, Coughlan JL. Gastro-oesophageal reflux treatment for asthma in adults and children. Cochrane Database Syst Rev. 2003;(2):CD001496.
13. Barnett SB, Nurmagambetov TA. Costs of asthma in the United States: 2002-2007. J Allergy Clin Immunol. 2011;127(1):145-152.
14. Walders N, Kercsmar C, Schluchter M, et al. An interdisciplinary intervention for undertreated pediatric asthma. Chest. 2006;129(2):292-299.
15. Morrow R, Fletcher J, Mulvihill M, Park H. The asthma dialogues: a model of interactive education for skills. J Contin Educ Health Prof. 2007;27(1): 49-58.
16. State of New York, Department of Health. Asthma action plan. www.health.state.ny.us/diseases/asthma/pdf/4850.pdf. Accessed April 11, 2011.
17. Picken HA, Greenfield S, Teres D, et al. Effects of local standards on the implementation of national guidelines for asthma: primary care agreement with national asthma guidelines. J Gen Intern Med. 1998;13(10):659-663.
18. Hardie GE, Gold WM, Janson S, et al. Understanding how asthmatics perceive symptom distress during a methacholine challenge. J Asthma. 2002;39(7):611-618.
19. Reddel HK, Marks GB, Jenkins CR. When can personal best peak flow be determined for asthma action plans? Thorax. 2004;59(11):922-924.
20. Knudson RJ, Lebowitz MD, Holberg CJ, Burrows B. Changes in the normal maximal expiratory flow-volume curve with growth and aging. Am Rev Respir Dis. 1983;127(6):725-734.
21. Ind PW, Dal Negro R, Colman NC, et al. Addition of salmeterol to fluticasone propionate treatment in moderate-to-severe asthma. Respir Med. 2003;97(5):555-562.
22. Price DB, Hernandez D, Magyar P, et al; Clinical Outcomes with Montelukast as a Partner Agent to Corticosteroid Therapy (COMPACT) International Study Group. Randomised controlled trial of montelukast plus inhaled budesonide versus double dose inhaled budesonide in adult patients with asthma. Thorax. 2003;58(3):211-216.
23. Schatz M, Zeiger RS, Mosen D, et al. Improved asthma outcomes from allergy specialist care: a population-based cross-sectional analysis. J Allergy Clin Immunol. 2005;116(6):1307-1313.
24. Hampel H, Abraham NS, El-Serag HB. Meta-analysis: obesity and the risk for gastroesophageal reflux disease and its complications. Ann Intern Med. 2005;143(3):199-211.
25. Cabana MD, Bruckman D, Meister K, et al. Documentation of asthma severity in pediatric outpatient clinics. Clin Pediatr (Phila). 2003;42(2):121-125.
26. Heater BS, Becker AM, Olson RK. Nursing interventions and patient outcomes: a meta-analysis of studies. Nurs Res. 1988;37(5):303-307.
Man, 54, With Delusions and Seizures
A 54-year-old African-American man was brought by police officers to the emergency department (ED) after he called 911 several times to report seeing a Rottweiler looking into his second-story window. At the scene, the police were unable to confirm his story, thought the man seemed intoxicated, and brought him to the ED for evaluation.
The patient reported that he had been drinking the previous evening but denied current intoxication or illicit drug use. He denied experiencing symptoms of alcohol withdrawal.
Regarding his medical history, the patient admitted to having had seizures, including two episodes that he said required hospitalization. He described these episodes as right-hand “tingling” (paresthesias), accompanied by right-facial numbness and aphasia. The patient said his physician had instructed him to take “a few phenytoin pills” whenever these episodes occurred. He reported that the medication usually helped resolve his symptoms. He said he had taken phenytoin shortly before his current presentation.
According to friends of the patient who were questioned, he had had noticeable memory problems during the previous six to eight months. They said that he often told the same joke, day after day. His speech had become increasingly slurred, even when he was not drinking.
Once the patient’s medical records were retrieved, it was revealed that he had been hospitalized twice for witnessed grand mal seizures about six months before his current admission; he had been drinking alcohol prior to both episodes. He underwent electroencephalography (EEG) during one of these hospitalizations, with results reported as normal. On both occasions, the patient was discharged with phenytoin and was instructed to follow up with his primary care provider and neurologist.
The patient, who reported working in customer service, had no known allergies. He claimed to drink one or two 40-ounce beers twice per week and admitted to occasional cocaine use. Of significance in his family history was a fatal MI in his mother. Although the patient denied any history of rashes or lesions, his current delirium made it impossible to obtain a reliable sexual history; a friend who was questioned, however, described the patient as promiscuous.
On initial physical examination, the man was afebrile, tachycardic, and somewhat combative with the ED staff. He was fully oriented to self but only partially to place and time.
His right pupil was 3+ and his left pupil was 2+, with neither reactive to light. He spoke with tangential speech and his gait was unsteady, but no other significant abnormalities were noted. A full assessment revealed no rashes or other lesions.
Significant laboratory findings included a low level of phenytoin, a negative blood alcohol level, presence of cocaine on urine drug screening, and normal levels of thyroid-stimulating hormone (TSH), vitamin B12, and folate. The patient’s serum VDRL (venereal disease research laboratory) titer was positive at 1:256.
Electroencephalography showed diffuse slowing, and brain CT performed in the ED showed atrophy that was mild but appropriate for a person of the patient’s age, with no evidence of a cerebrovascular accident (CVA). Aneurysm was ruled out by CT angiography of the brain. MRI revealed persistent increased signal in the subarachnoid space.
The patient was admitted with an initial diagnosis of paranoid delusional psychosis and monitored for alcohol withdrawal. He was given lorazepam as needed for agitation. Consultations were arranged with the psychiatry service regarding his delusions, and with neurology to determine whether to continue phenytoin.
The patient showed little response during the next several days. Based on positive results on serum VDRL with high titer, the presence of Argyll-Robertson pupils on exam, and his history of dementia-like symptoms, a lumbar puncture was performed to rule out neurosyphilis. In the patient’s cerebral spinal fluid (CSF) analysis, the first tube was clear and colorless, with 72 cells (28% neutrophils, 59% lymphocytes); glucose, 64 mg/dL; and total protein, 117 mg/dL. The fourth tube had 34 cells (17% neutrophils, 65% lymphocytes) and a positive VDRL titer at 1:128. Results from a serum syphilis immunoglobulin G (IgG) test were positive, and HIV antibody testing was nonreactive, confirming the diagnosis of neurosyphilis.
The hospital’s infectious disease (ID) team recommended treatment with IV penicillin for 14 days. Once this was completed, the patient was discharged with instructions to follow up at the ID clinic in three months for a repeat CSF VDRL titer to monitor for resolution of the disease. His prescription for phenytoin was discontinued.
At the time of discharge, it was noted that the patient showed no evidence of having regained cognitive function. He was deemed by the psychiatry service to lack decision-making capacity—a likely sequelae of untreated neurosyphilis of unknown duration.
He did return to the ID clinic six months after his discharge. At that visit, a VDRL serum titer was drawn with a result of 1:64, a decrease from 1:128. His syphilis IgG remained positive, however.
Discussion
Definition and Epidemiology
Syphilis is commonly known as a sexually transmitted disease with primary, secondary, and tertiary (early and late latent) stages.1 Neurosyphilis is defined as a manifestation of the inflammatory response to invasion over decades by the Treponema pallidum spirochete in the CSF as a result of untreated primary and/or secondary syphilis.2 About one in 10 patients with untreated syphilis will experience neurologic involvement.3,4 Before 2005, neurosyphilis was required to be reported as a specific stage of syphilis (ie, a manifestation of tertiary syphilis4), but now should be reported as syphilis with neurologic manifestations.5
A reportable infectious disease, syphilis was widespread until the advent of penicillin. According to CDC statistics,6 the number of reported cases of primary and secondary syphilis has declined steadily since 1943. In the late 1970s and early 1980s, the number of tertiary cases also began to plateau, likely as a result of earlier diagnosis and more widespread use of penicillin. Recent case reports suggest greater prevalence of syphilis among men than women and increased incidence among men who have sex with men.7
Pathogenesis
Syphilis is most commonly spread by sexual contact or contact with an infected primary lesion (chancre). Less likely routes of transmission are placental passage or blood transfusion. Infectivity is greatest in the early disease stages.8
Primary syphilis is marked by transmission of the spirochete, ending with development of secondary syphilis (usually two to 12 weeks after transmission). A chancre commonly develops but is often missed by patients because it is painless and can heal spontaneously.7 The chancre is also often confused with two other sources of genital lesions, herpes simplex (genital herpes) and Haemophilus ducreyi (chancroid). In two-thirds of cases of untreated primary syphilis, the infection clears spontaneously, but in the remaining one-third, the disease progresses.8
Secondary syphilis, with or without presence of a chancre, manifests with constitutional symptoms, including lymphadenopathy, fever, headache, and malaise. Patients in this disease phase may also present with a generalized, nonpruritic, macular to maculopapular or pustular rash. The rash can affect the skin of the trunk, the proximal extremities, and the palms and soles. Ocular involvement may occur, especially in patients who are coinfected with HIV.8 In either primary or secondary syphilis, infection can invade the central nervous system.1
During latent syphilis, patients show serologic conversion without overt symptoms. Early latent syphilis is defined as infection within the previous year, as demonstrated by conversion from negative to positive testing, or an increase in titers within the previous year. Any case occurring after one year is defined as late or unknown latent syphilis.8
Tertiary syphilis is marked by complications resulting from untreated syphilis; affected patients commonly experience central nervous system and cardiovascular involvement. Gummatous disease is seen in 15% of patients.1
The early stages of neurosyphilis may be asymptomatic, acute meningeal, and meningovascular.1,4,8,9 Only 5% of patients with early neurosyphilis are symptomatic, with the added potential for cranial neuritis or ocular involvement.1 The late stages of neurosyphilis are detailed in the table.1,4,8
Diagnosis
A diagnosis of syphilis is made by testing blood samples or scrapings from a lesion. In patients with suspected syphilis, rapid plasma reagin (RPR) testing or a VDRL titer is commonly ordered. When results are positive, a serum treponemal test is recommended to confirm a diagnosis of syphilis. Options include the fluorescent treponemal antibody absorption test (FTA-ABS) and the microhemagglutinin assay for antibody to T pallidum (MHA-TP).5
If neurologic symptoms are present, a CSF sample should be obtained, followed by the same testing. A confirmed diagnosis of neurosyphilis is defined by the CDC as syphilis at any stage that meets laboratory criteria for neurosyphilis5; these include increased CSF protein or an elevated CSF leukocyte count with no other known cause, and clinical signs or symptoms without other known causes.7
Treatment
Treatment of syphilis generally consists of penicillin, administered intramuscularly (IM) or IV, depending on the stage. According to 2006 guidelines from the CDC,10,11 treatment for adults with primary and secondary syphilis is a single dose of IM penicillin G, 2.4 million units. If neurosyphilis is suspected, recommended treatment is IV penicillin G, 18 to 24 million units per day divided into six doses (ie, 3 to 4 million units every four hours) or continuous pump infusion for 10 to 14 days.10-12 Follow-up is recommended by monitoring CSF titers to ensure clearance of infection; retreatment may be required if CSF abnormalities persist after two years.11
Patients with a penicillin allergy should undergo desensitization, as penicillin is the preferred agent; the potential exists for cross-reactivity with ceftriaxone, a possible alternative for patients with neurosyphilis.11 All patients diagnosed with syphilis should also be tested for HIV and other sexually transmitted diseases.10-12
The prognosis of patients treated for neurosyphilis is generally good if the condition is diagnosed and treated early. In patients with cerebral atrophy, frontal lesions, dementia, or tabes dorsalis, the potential for recovery decreases.2,13,14
Teaching Points
There are several teaching points to take away from this case:
• Remember to rule out a CVA in any patient who presents with numbness, paresthesias, or slurred speech. In this case, a brain CT and CT angiography of the brain were both obtained in the ED before the patient was admitted. They both yielded negative results; because the patient’s history was consistent with alcohol and drug use and he had a history of seizures, he was monitored closely for signs of withdrawal or further seizure.
• Phenytoin is an antiepileptic agent whose use requires proper patient education and drug level monitoring. Appropriate follow-up must be ensured before phenytoin therapy is begun, as toxicity can result in nystagmus, ataxia, slurred speech, decreased coordination, mental confusion, and possibly death.15,16
• For patients with a suspected acute change in mental status, a workup is required and should be tailored appropriately, based on findings. This should include, but not be limited to, a thorough history and physical exam, CT of the brain (to rule out an acute brain injury17), and, if warranted, MRI of the brain. Also, a urine drug screen and alcohol level, a complete blood count, a TSH level (to evaluate for altered thyroid function that may explain mental status changes), comprehensive panel, RPR testing and/or a VDRL titer should be obtained, depending on the facility’s protocol18,19; at some facilities, a treponemal test, rather than VDRL, is being obtained at the outset.20 Levels of vitamin B12 (as part of the dementia workup), folate, thiamine, and ammonia (in patients with suspected liver disease) can also be obtained in patients with change in mental status.18,19 Urinalysis should not be overlooked to check for a urinary tract infection, especially in elderly patients.21
• If primary syphilis is suspected, treatment must be undertaken.20
Conclusion
Despite the decline seen since the 1940s in cases of primary and secondary syphilis, and the effectiveness of penicillin in treating the infection early, patients with late-stage syphilis, including those with neurosyphilis, may still present to the emergency care, urgent care, or primary care setting. Immediate treatment with penicillin is recommended to achieve an optimal prognosis for the affected patient.
1. Golden MR, Marra CM, Holmes KK. Update on syphilis: resurgence of an old problem. JAMA. 2003;290(11):1510-1514.
2. Simon RP. Chapter 20. Neurosyphilis. In: Klausner JD, Hook EW III, eds. Current Diagnosis & Treatment of Sexually Transmitted Diseases. USA: The McGraw-Hill Companies; 2007:130-137.
3. Sanchez FM, Zisselman MH. Treatment of psychiatric symptoms associated with neurosyphilis. Psychosomatics. 2007;48:440-445.
4. Marra CM. Neurosyphilis. Curr Neurol Neurosci Rep. 2004;4(6):435-440.
5. CDC. Sexually transmitted diseases surveillance, 2007: STD surveillance case definitions. www.cdc.gov/std/stats07/app-casedef.htm. Accessed March 23, 2011.
6. CDC. 2008 Sexually Transmitted Diseases Surveillance: Table 1. Cases of sexually transmitted diseases reported by state health departments and rates per 100,000 population: United States, 1941-2008. www.cdc.gov/std/stats08/tables/1.htm. Accessed March 23, 2011.
7. CDC. Sexually transmitted diseases (STDs): Syphilis: CDC fact sheet. www.cdc.gov/std/syphilis/STDfact-syphilis.htm. Accessed March 23, 2011.
8. Tramont EC. Chapter 238. Treponema pallidum (syphilis). In: Mandell GL, Bennett JE, Dolin R, eds. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia: Elsevier Churchill Livingstone; 2009.
9. Ghanem KG. Neurosyphilis: a historical perspective and review. CNS Neurosci Ther. 2010; 16(5):e157-e168.
10. Workowski KA, Berman SM; CDC. Sexually transmitted diseases treatment guidelines, 2006. MMWR Recomm Rep. 2006;55(RR-11):1-94.
11. CDC. Sexually transmitted diseases: treatment guidelines 2006. www.cdc.gov/std/treatment/2006/genital-ulcers.htm#genulc6. Accessed March 29, 2011.
12. Drugs for sexually transmitted infections. Treatment Guidelines from the Medical Letter. 2010;95:95a. http://secure.medicalletter.org. Accessed March 23, 2011.
13. Russouw HG, Roberts MC, Emsley RA, et al. Psychiatric manifestations and magnetic resonance imaging in HIV-negative neurosyphilis. Biol Psychiatry. 1997;41(4):467-473.
14. Hooshmand H, Escobar MR, Kopf SW. Neurosyphylis: a study of 241 patients. JAMA. 1972;219 (6):726-729.
15. Miller CA, Joyce DM. Toxicity, phenytoin. http://emedicine.medscape.com/article/816447-overview. Accessed March 23, 2011.
16. Earnest MP, Marx JA, Drury LR. Complications of intravenous phenytoin for acute treatment of seizures: recommendations for usage. JAMA. 1983; 246(6):762-765.
17. Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol. 2008;64(1): 97-108.
18. Mechem CC. Chapter 143. Altered mental status and coma. In: Ma J, Cline DM, Tintinalli JE, et al, eds. Emergency Medicine Manual, 6e. www.access emergencymedicine.com/content.aspx?aID=2020. Accessed March 23, 2011.
19. Knopman DS, DeKosky ST, Cummings JL, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: diagnosis of dementia (an evidence-based review). Neurology. 2001;56(9):1143-1153.
20. CDC. Syphilis testing algorithms using treponemal tests for initial screening—four laboratories, New York City, 2005-2006. MMWR Morb Mortal Wkly Rep. 2008;57(32):872-875.
21. Anderson CA, Filley CM. Chapter 33. Behavioral presentations of medical and neurologic disorders. In: Jacobson JL, Jacobson AM, eds. Psychiatric Secrets. 2nd ed. St. Louis, MO: Hanley & Belfus; 2001.
A 54-year-old African-American man was brought by police officers to the emergency department (ED) after he called 911 several times to report seeing a Rottweiler looking into his second-story window. At the scene, the police were unable to confirm his story, thought the man seemed intoxicated, and brought him to the ED for evaluation.
The patient reported that he had been drinking the previous evening but denied current intoxication or illicit drug use. He denied experiencing symptoms of alcohol withdrawal.
Regarding his medical history, the patient admitted to having had seizures, including two episodes that he said required hospitalization. He described these episodes as right-hand “tingling” (paresthesias), accompanied by right-facial numbness and aphasia. The patient said his physician had instructed him to take “a few phenytoin pills” whenever these episodes occurred. He reported that the medication usually helped resolve his symptoms. He said he had taken phenytoin shortly before his current presentation.
According to friends of the patient who were questioned, he had had noticeable memory problems during the previous six to eight months. They said that he often told the same joke, day after day. His speech had become increasingly slurred, even when he was not drinking.
Once the patient’s medical records were retrieved, it was revealed that he had been hospitalized twice for witnessed grand mal seizures about six months before his current admission; he had been drinking alcohol prior to both episodes. He underwent electroencephalography (EEG) during one of these hospitalizations, with results reported as normal. On both occasions, the patient was discharged with phenytoin and was instructed to follow up with his primary care provider and neurologist.
The patient, who reported working in customer service, had no known allergies. He claimed to drink one or two 40-ounce beers twice per week and admitted to occasional cocaine use. Of significance in his family history was a fatal MI in his mother. Although the patient denied any history of rashes or lesions, his current delirium made it impossible to obtain a reliable sexual history; a friend who was questioned, however, described the patient as promiscuous.
On initial physical examination, the man was afebrile, tachycardic, and somewhat combative with the ED staff. He was fully oriented to self but only partially to place and time.
His right pupil was 3+ and his left pupil was 2+, with neither reactive to light. He spoke with tangential speech and his gait was unsteady, but no other significant abnormalities were noted. A full assessment revealed no rashes or other lesions.
Significant laboratory findings included a low level of phenytoin, a negative blood alcohol level, presence of cocaine on urine drug screening, and normal levels of thyroid-stimulating hormone (TSH), vitamin B12, and folate. The patient’s serum VDRL (venereal disease research laboratory) titer was positive at 1:256.
Electroencephalography showed diffuse slowing, and brain CT performed in the ED showed atrophy that was mild but appropriate for a person of the patient’s age, with no evidence of a cerebrovascular accident (CVA). Aneurysm was ruled out by CT angiography of the brain. MRI revealed persistent increased signal in the subarachnoid space.
The patient was admitted with an initial diagnosis of paranoid delusional psychosis and monitored for alcohol withdrawal. He was given lorazepam as needed for agitation. Consultations were arranged with the psychiatry service regarding his delusions, and with neurology to determine whether to continue phenytoin.
The patient showed little response during the next several days. Based on positive results on serum VDRL with high titer, the presence of Argyll-Robertson pupils on exam, and his history of dementia-like symptoms, a lumbar puncture was performed to rule out neurosyphilis. In the patient’s cerebral spinal fluid (CSF) analysis, the first tube was clear and colorless, with 72 cells (28% neutrophils, 59% lymphocytes); glucose, 64 mg/dL; and total protein, 117 mg/dL. The fourth tube had 34 cells (17% neutrophils, 65% lymphocytes) and a positive VDRL titer at 1:128. Results from a serum syphilis immunoglobulin G (IgG) test were positive, and HIV antibody testing was nonreactive, confirming the diagnosis of neurosyphilis.
The hospital’s infectious disease (ID) team recommended treatment with IV penicillin for 14 days. Once this was completed, the patient was discharged with instructions to follow up at the ID clinic in three months for a repeat CSF VDRL titer to monitor for resolution of the disease. His prescription for phenytoin was discontinued.
At the time of discharge, it was noted that the patient showed no evidence of having regained cognitive function. He was deemed by the psychiatry service to lack decision-making capacity—a likely sequelae of untreated neurosyphilis of unknown duration.
He did return to the ID clinic six months after his discharge. At that visit, a VDRL serum titer was drawn with a result of 1:64, a decrease from 1:128. His syphilis IgG remained positive, however.
Discussion
Definition and Epidemiology
Syphilis is commonly known as a sexually transmitted disease with primary, secondary, and tertiary (early and late latent) stages.1 Neurosyphilis is defined as a manifestation of the inflammatory response to invasion over decades by the Treponema pallidum spirochete in the CSF as a result of untreated primary and/or secondary syphilis.2 About one in 10 patients with untreated syphilis will experience neurologic involvement.3,4 Before 2005, neurosyphilis was required to be reported as a specific stage of syphilis (ie, a manifestation of tertiary syphilis4), but now should be reported as syphilis with neurologic manifestations.5
A reportable infectious disease, syphilis was widespread until the advent of penicillin. According to CDC statistics,6 the number of reported cases of primary and secondary syphilis has declined steadily since 1943. In the late 1970s and early 1980s, the number of tertiary cases also began to plateau, likely as a result of earlier diagnosis and more widespread use of penicillin. Recent case reports suggest greater prevalence of syphilis among men than women and increased incidence among men who have sex with men.7
Pathogenesis
Syphilis is most commonly spread by sexual contact or contact with an infected primary lesion (chancre). Less likely routes of transmission are placental passage or blood transfusion. Infectivity is greatest in the early disease stages.8
Primary syphilis is marked by transmission of the spirochete, ending with development of secondary syphilis (usually two to 12 weeks after transmission). A chancre commonly develops but is often missed by patients because it is painless and can heal spontaneously.7 The chancre is also often confused with two other sources of genital lesions, herpes simplex (genital herpes) and Haemophilus ducreyi (chancroid). In two-thirds of cases of untreated primary syphilis, the infection clears spontaneously, but in the remaining one-third, the disease progresses.8
Secondary syphilis, with or without presence of a chancre, manifests with constitutional symptoms, including lymphadenopathy, fever, headache, and malaise. Patients in this disease phase may also present with a generalized, nonpruritic, macular to maculopapular or pustular rash. The rash can affect the skin of the trunk, the proximal extremities, and the palms and soles. Ocular involvement may occur, especially in patients who are coinfected with HIV.8 In either primary or secondary syphilis, infection can invade the central nervous system.1
During latent syphilis, patients show serologic conversion without overt symptoms. Early latent syphilis is defined as infection within the previous year, as demonstrated by conversion from negative to positive testing, or an increase in titers within the previous year. Any case occurring after one year is defined as late or unknown latent syphilis.8
Tertiary syphilis is marked by complications resulting from untreated syphilis; affected patients commonly experience central nervous system and cardiovascular involvement. Gummatous disease is seen in 15% of patients.1
The early stages of neurosyphilis may be asymptomatic, acute meningeal, and meningovascular.1,4,8,9 Only 5% of patients with early neurosyphilis are symptomatic, with the added potential for cranial neuritis or ocular involvement.1 The late stages of neurosyphilis are detailed in the table.1,4,8
Diagnosis
A diagnosis of syphilis is made by testing blood samples or scrapings from a lesion. In patients with suspected syphilis, rapid plasma reagin (RPR) testing or a VDRL titer is commonly ordered. When results are positive, a serum treponemal test is recommended to confirm a diagnosis of syphilis. Options include the fluorescent treponemal antibody absorption test (FTA-ABS) and the microhemagglutinin assay for antibody to T pallidum (MHA-TP).5
If neurologic symptoms are present, a CSF sample should be obtained, followed by the same testing. A confirmed diagnosis of neurosyphilis is defined by the CDC as syphilis at any stage that meets laboratory criteria for neurosyphilis5; these include increased CSF protein or an elevated CSF leukocyte count with no other known cause, and clinical signs or symptoms without other known causes.7
Treatment
Treatment of syphilis generally consists of penicillin, administered intramuscularly (IM) or IV, depending on the stage. According to 2006 guidelines from the CDC,10,11 treatment for adults with primary and secondary syphilis is a single dose of IM penicillin G, 2.4 million units. If neurosyphilis is suspected, recommended treatment is IV penicillin G, 18 to 24 million units per day divided into six doses (ie, 3 to 4 million units every four hours) or continuous pump infusion for 10 to 14 days.10-12 Follow-up is recommended by monitoring CSF titers to ensure clearance of infection; retreatment may be required if CSF abnormalities persist after two years.11
Patients with a penicillin allergy should undergo desensitization, as penicillin is the preferred agent; the potential exists for cross-reactivity with ceftriaxone, a possible alternative for patients with neurosyphilis.11 All patients diagnosed with syphilis should also be tested for HIV and other sexually transmitted diseases.10-12
The prognosis of patients treated for neurosyphilis is generally good if the condition is diagnosed and treated early. In patients with cerebral atrophy, frontal lesions, dementia, or tabes dorsalis, the potential for recovery decreases.2,13,14
Teaching Points
There are several teaching points to take away from this case:
• Remember to rule out a CVA in any patient who presents with numbness, paresthesias, or slurred speech. In this case, a brain CT and CT angiography of the brain were both obtained in the ED before the patient was admitted. They both yielded negative results; because the patient’s history was consistent with alcohol and drug use and he had a history of seizures, he was monitored closely for signs of withdrawal or further seizure.
• Phenytoin is an antiepileptic agent whose use requires proper patient education and drug level monitoring. Appropriate follow-up must be ensured before phenytoin therapy is begun, as toxicity can result in nystagmus, ataxia, slurred speech, decreased coordination, mental confusion, and possibly death.15,16
• For patients with a suspected acute change in mental status, a workup is required and should be tailored appropriately, based on findings. This should include, but not be limited to, a thorough history and physical exam, CT of the brain (to rule out an acute brain injury17), and, if warranted, MRI of the brain. Also, a urine drug screen and alcohol level, a complete blood count, a TSH level (to evaluate for altered thyroid function that may explain mental status changes), comprehensive panel, RPR testing and/or a VDRL titer should be obtained, depending on the facility’s protocol18,19; at some facilities, a treponemal test, rather than VDRL, is being obtained at the outset.20 Levels of vitamin B12 (as part of the dementia workup), folate, thiamine, and ammonia (in patients with suspected liver disease) can also be obtained in patients with change in mental status.18,19 Urinalysis should not be overlooked to check for a urinary tract infection, especially in elderly patients.21
• If primary syphilis is suspected, treatment must be undertaken.20
Conclusion
Despite the decline seen since the 1940s in cases of primary and secondary syphilis, and the effectiveness of penicillin in treating the infection early, patients with late-stage syphilis, including those with neurosyphilis, may still present to the emergency care, urgent care, or primary care setting. Immediate treatment with penicillin is recommended to achieve an optimal prognosis for the affected patient.
A 54-year-old African-American man was brought by police officers to the emergency department (ED) after he called 911 several times to report seeing a Rottweiler looking into his second-story window. At the scene, the police were unable to confirm his story, thought the man seemed intoxicated, and brought him to the ED for evaluation.
The patient reported that he had been drinking the previous evening but denied current intoxication or illicit drug use. He denied experiencing symptoms of alcohol withdrawal.
Regarding his medical history, the patient admitted to having had seizures, including two episodes that he said required hospitalization. He described these episodes as right-hand “tingling” (paresthesias), accompanied by right-facial numbness and aphasia. The patient said his physician had instructed him to take “a few phenytoin pills” whenever these episodes occurred. He reported that the medication usually helped resolve his symptoms. He said he had taken phenytoin shortly before his current presentation.
According to friends of the patient who were questioned, he had had noticeable memory problems during the previous six to eight months. They said that he often told the same joke, day after day. His speech had become increasingly slurred, even when he was not drinking.
Once the patient’s medical records were retrieved, it was revealed that he had been hospitalized twice for witnessed grand mal seizures about six months before his current admission; he had been drinking alcohol prior to both episodes. He underwent electroencephalography (EEG) during one of these hospitalizations, with results reported as normal. On both occasions, the patient was discharged with phenytoin and was instructed to follow up with his primary care provider and neurologist.
The patient, who reported working in customer service, had no known allergies. He claimed to drink one or two 40-ounce beers twice per week and admitted to occasional cocaine use. Of significance in his family history was a fatal MI in his mother. Although the patient denied any history of rashes or lesions, his current delirium made it impossible to obtain a reliable sexual history; a friend who was questioned, however, described the patient as promiscuous.
On initial physical examination, the man was afebrile, tachycardic, and somewhat combative with the ED staff. He was fully oriented to self but only partially to place and time.
His right pupil was 3+ and his left pupil was 2+, with neither reactive to light. He spoke with tangential speech and his gait was unsteady, but no other significant abnormalities were noted. A full assessment revealed no rashes or other lesions.
Significant laboratory findings included a low level of phenytoin, a negative blood alcohol level, presence of cocaine on urine drug screening, and normal levels of thyroid-stimulating hormone (TSH), vitamin B12, and folate. The patient’s serum VDRL (venereal disease research laboratory) titer was positive at 1:256.
Electroencephalography showed diffuse slowing, and brain CT performed in the ED showed atrophy that was mild but appropriate for a person of the patient’s age, with no evidence of a cerebrovascular accident (CVA). Aneurysm was ruled out by CT angiography of the brain. MRI revealed persistent increased signal in the subarachnoid space.
The patient was admitted with an initial diagnosis of paranoid delusional psychosis and monitored for alcohol withdrawal. He was given lorazepam as needed for agitation. Consultations were arranged with the psychiatry service regarding his delusions, and with neurology to determine whether to continue phenytoin.
The patient showed little response during the next several days. Based on positive results on serum VDRL with high titer, the presence of Argyll-Robertson pupils on exam, and his history of dementia-like symptoms, a lumbar puncture was performed to rule out neurosyphilis. In the patient’s cerebral spinal fluid (CSF) analysis, the first tube was clear and colorless, with 72 cells (28% neutrophils, 59% lymphocytes); glucose, 64 mg/dL; and total protein, 117 mg/dL. The fourth tube had 34 cells (17% neutrophils, 65% lymphocytes) and a positive VDRL titer at 1:128. Results from a serum syphilis immunoglobulin G (IgG) test were positive, and HIV antibody testing was nonreactive, confirming the diagnosis of neurosyphilis.
The hospital’s infectious disease (ID) team recommended treatment with IV penicillin for 14 days. Once this was completed, the patient was discharged with instructions to follow up at the ID clinic in three months for a repeat CSF VDRL titer to monitor for resolution of the disease. His prescription for phenytoin was discontinued.
At the time of discharge, it was noted that the patient showed no evidence of having regained cognitive function. He was deemed by the psychiatry service to lack decision-making capacity—a likely sequelae of untreated neurosyphilis of unknown duration.
He did return to the ID clinic six months after his discharge. At that visit, a VDRL serum titer was drawn with a result of 1:64, a decrease from 1:128. His syphilis IgG remained positive, however.
Discussion
Definition and Epidemiology
Syphilis is commonly known as a sexually transmitted disease with primary, secondary, and tertiary (early and late latent) stages.1 Neurosyphilis is defined as a manifestation of the inflammatory response to invasion over decades by the Treponema pallidum spirochete in the CSF as a result of untreated primary and/or secondary syphilis.2 About one in 10 patients with untreated syphilis will experience neurologic involvement.3,4 Before 2005, neurosyphilis was required to be reported as a specific stage of syphilis (ie, a manifestation of tertiary syphilis4), but now should be reported as syphilis with neurologic manifestations.5
A reportable infectious disease, syphilis was widespread until the advent of penicillin. According to CDC statistics,6 the number of reported cases of primary and secondary syphilis has declined steadily since 1943. In the late 1970s and early 1980s, the number of tertiary cases also began to plateau, likely as a result of earlier diagnosis and more widespread use of penicillin. Recent case reports suggest greater prevalence of syphilis among men than women and increased incidence among men who have sex with men.7
Pathogenesis
Syphilis is most commonly spread by sexual contact or contact with an infected primary lesion (chancre). Less likely routes of transmission are placental passage or blood transfusion. Infectivity is greatest in the early disease stages.8
Primary syphilis is marked by transmission of the spirochete, ending with development of secondary syphilis (usually two to 12 weeks after transmission). A chancre commonly develops but is often missed by patients because it is painless and can heal spontaneously.7 The chancre is also often confused with two other sources of genital lesions, herpes simplex (genital herpes) and Haemophilus ducreyi (chancroid). In two-thirds of cases of untreated primary syphilis, the infection clears spontaneously, but in the remaining one-third, the disease progresses.8
Secondary syphilis, with or without presence of a chancre, manifests with constitutional symptoms, including lymphadenopathy, fever, headache, and malaise. Patients in this disease phase may also present with a generalized, nonpruritic, macular to maculopapular or pustular rash. The rash can affect the skin of the trunk, the proximal extremities, and the palms and soles. Ocular involvement may occur, especially in patients who are coinfected with HIV.8 In either primary or secondary syphilis, infection can invade the central nervous system.1
During latent syphilis, patients show serologic conversion without overt symptoms. Early latent syphilis is defined as infection within the previous year, as demonstrated by conversion from negative to positive testing, or an increase in titers within the previous year. Any case occurring after one year is defined as late or unknown latent syphilis.8
Tertiary syphilis is marked by complications resulting from untreated syphilis; affected patients commonly experience central nervous system and cardiovascular involvement. Gummatous disease is seen in 15% of patients.1
The early stages of neurosyphilis may be asymptomatic, acute meningeal, and meningovascular.1,4,8,9 Only 5% of patients with early neurosyphilis are symptomatic, with the added potential for cranial neuritis or ocular involvement.1 The late stages of neurosyphilis are detailed in the table.1,4,8
Diagnosis
A diagnosis of syphilis is made by testing blood samples or scrapings from a lesion. In patients with suspected syphilis, rapid plasma reagin (RPR) testing or a VDRL titer is commonly ordered. When results are positive, a serum treponemal test is recommended to confirm a diagnosis of syphilis. Options include the fluorescent treponemal antibody absorption test (FTA-ABS) and the microhemagglutinin assay for antibody to T pallidum (MHA-TP).5
If neurologic symptoms are present, a CSF sample should be obtained, followed by the same testing. A confirmed diagnosis of neurosyphilis is defined by the CDC as syphilis at any stage that meets laboratory criteria for neurosyphilis5; these include increased CSF protein or an elevated CSF leukocyte count with no other known cause, and clinical signs or symptoms without other known causes.7
Treatment
Treatment of syphilis generally consists of penicillin, administered intramuscularly (IM) or IV, depending on the stage. According to 2006 guidelines from the CDC,10,11 treatment for adults with primary and secondary syphilis is a single dose of IM penicillin G, 2.4 million units. If neurosyphilis is suspected, recommended treatment is IV penicillin G, 18 to 24 million units per day divided into six doses (ie, 3 to 4 million units every four hours) or continuous pump infusion for 10 to 14 days.10-12 Follow-up is recommended by monitoring CSF titers to ensure clearance of infection; retreatment may be required if CSF abnormalities persist after two years.11
Patients with a penicillin allergy should undergo desensitization, as penicillin is the preferred agent; the potential exists for cross-reactivity with ceftriaxone, a possible alternative for patients with neurosyphilis.11 All patients diagnosed with syphilis should also be tested for HIV and other sexually transmitted diseases.10-12
The prognosis of patients treated for neurosyphilis is generally good if the condition is diagnosed and treated early. In patients with cerebral atrophy, frontal lesions, dementia, or tabes dorsalis, the potential for recovery decreases.2,13,14
Teaching Points
There are several teaching points to take away from this case:
• Remember to rule out a CVA in any patient who presents with numbness, paresthesias, or slurred speech. In this case, a brain CT and CT angiography of the brain were both obtained in the ED before the patient was admitted. They both yielded negative results; because the patient’s history was consistent with alcohol and drug use and he had a history of seizures, he was monitored closely for signs of withdrawal or further seizure.
• Phenytoin is an antiepileptic agent whose use requires proper patient education and drug level monitoring. Appropriate follow-up must be ensured before phenytoin therapy is begun, as toxicity can result in nystagmus, ataxia, slurred speech, decreased coordination, mental confusion, and possibly death.15,16
• For patients with a suspected acute change in mental status, a workup is required and should be tailored appropriately, based on findings. This should include, but not be limited to, a thorough history and physical exam, CT of the brain (to rule out an acute brain injury17), and, if warranted, MRI of the brain. Also, a urine drug screen and alcohol level, a complete blood count, a TSH level (to evaluate for altered thyroid function that may explain mental status changes), comprehensive panel, RPR testing and/or a VDRL titer should be obtained, depending on the facility’s protocol18,19; at some facilities, a treponemal test, rather than VDRL, is being obtained at the outset.20 Levels of vitamin B12 (as part of the dementia workup), folate, thiamine, and ammonia (in patients with suspected liver disease) can also be obtained in patients with change in mental status.18,19 Urinalysis should not be overlooked to check for a urinary tract infection, especially in elderly patients.21
• If primary syphilis is suspected, treatment must be undertaken.20
Conclusion
Despite the decline seen since the 1940s in cases of primary and secondary syphilis, and the effectiveness of penicillin in treating the infection early, patients with late-stage syphilis, including those with neurosyphilis, may still present to the emergency care, urgent care, or primary care setting. Immediate treatment with penicillin is recommended to achieve an optimal prognosis for the affected patient.
1. Golden MR, Marra CM, Holmes KK. Update on syphilis: resurgence of an old problem. JAMA. 2003;290(11):1510-1514.
2. Simon RP. Chapter 20. Neurosyphilis. In: Klausner JD, Hook EW III, eds. Current Diagnosis & Treatment of Sexually Transmitted Diseases. USA: The McGraw-Hill Companies; 2007:130-137.
3. Sanchez FM, Zisselman MH. Treatment of psychiatric symptoms associated with neurosyphilis. Psychosomatics. 2007;48:440-445.
4. Marra CM. Neurosyphilis. Curr Neurol Neurosci Rep. 2004;4(6):435-440.
5. CDC. Sexually transmitted diseases surveillance, 2007: STD surveillance case definitions. www.cdc.gov/std/stats07/app-casedef.htm. Accessed March 23, 2011.
6. CDC. 2008 Sexually Transmitted Diseases Surveillance: Table 1. Cases of sexually transmitted diseases reported by state health departments and rates per 100,000 population: United States, 1941-2008. www.cdc.gov/std/stats08/tables/1.htm. Accessed March 23, 2011.
7. CDC. Sexually transmitted diseases (STDs): Syphilis: CDC fact sheet. www.cdc.gov/std/syphilis/STDfact-syphilis.htm. Accessed March 23, 2011.
8. Tramont EC. Chapter 238. Treponema pallidum (syphilis). In: Mandell GL, Bennett JE, Dolin R, eds. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia: Elsevier Churchill Livingstone; 2009.
9. Ghanem KG. Neurosyphilis: a historical perspective and review. CNS Neurosci Ther. 2010; 16(5):e157-e168.
10. Workowski KA, Berman SM; CDC. Sexually transmitted diseases treatment guidelines, 2006. MMWR Recomm Rep. 2006;55(RR-11):1-94.
11. CDC. Sexually transmitted diseases: treatment guidelines 2006. www.cdc.gov/std/treatment/2006/genital-ulcers.htm#genulc6. Accessed March 29, 2011.
12. Drugs for sexually transmitted infections. Treatment Guidelines from the Medical Letter. 2010;95:95a. http://secure.medicalletter.org. Accessed March 23, 2011.
13. Russouw HG, Roberts MC, Emsley RA, et al. Psychiatric manifestations and magnetic resonance imaging in HIV-negative neurosyphilis. Biol Psychiatry. 1997;41(4):467-473.
14. Hooshmand H, Escobar MR, Kopf SW. Neurosyphylis: a study of 241 patients. JAMA. 1972;219 (6):726-729.
15. Miller CA, Joyce DM. Toxicity, phenytoin. http://emedicine.medscape.com/article/816447-overview. Accessed March 23, 2011.
16. Earnest MP, Marx JA, Drury LR. Complications of intravenous phenytoin for acute treatment of seizures: recommendations for usage. JAMA. 1983; 246(6):762-765.
17. Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol. 2008;64(1): 97-108.
18. Mechem CC. Chapter 143. Altered mental status and coma. In: Ma J, Cline DM, Tintinalli JE, et al, eds. Emergency Medicine Manual, 6e. www.access emergencymedicine.com/content.aspx?aID=2020. Accessed March 23, 2011.
19. Knopman DS, DeKosky ST, Cummings JL, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: diagnosis of dementia (an evidence-based review). Neurology. 2001;56(9):1143-1153.
20. CDC. Syphilis testing algorithms using treponemal tests for initial screening—four laboratories, New York City, 2005-2006. MMWR Morb Mortal Wkly Rep. 2008;57(32):872-875.
21. Anderson CA, Filley CM. Chapter 33. Behavioral presentations of medical and neurologic disorders. In: Jacobson JL, Jacobson AM, eds. Psychiatric Secrets. 2nd ed. St. Louis, MO: Hanley & Belfus; 2001.
1. Golden MR, Marra CM, Holmes KK. Update on syphilis: resurgence of an old problem. JAMA. 2003;290(11):1510-1514.
2. Simon RP. Chapter 20. Neurosyphilis. In: Klausner JD, Hook EW III, eds. Current Diagnosis & Treatment of Sexually Transmitted Diseases. USA: The McGraw-Hill Companies; 2007:130-137.
3. Sanchez FM, Zisselman MH. Treatment of psychiatric symptoms associated with neurosyphilis. Psychosomatics. 2007;48:440-445.
4. Marra CM. Neurosyphilis. Curr Neurol Neurosci Rep. 2004;4(6):435-440.
5. CDC. Sexually transmitted diseases surveillance, 2007: STD surveillance case definitions. www.cdc.gov/std/stats07/app-casedef.htm. Accessed March 23, 2011.
6. CDC. 2008 Sexually Transmitted Diseases Surveillance: Table 1. Cases of sexually transmitted diseases reported by state health departments and rates per 100,000 population: United States, 1941-2008. www.cdc.gov/std/stats08/tables/1.htm. Accessed March 23, 2011.
7. CDC. Sexually transmitted diseases (STDs): Syphilis: CDC fact sheet. www.cdc.gov/std/syphilis/STDfact-syphilis.htm. Accessed March 23, 2011.
8. Tramont EC. Chapter 238. Treponema pallidum (syphilis). In: Mandell GL, Bennett JE, Dolin R, eds. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia: Elsevier Churchill Livingstone; 2009.
9. Ghanem KG. Neurosyphilis: a historical perspective and review. CNS Neurosci Ther. 2010; 16(5):e157-e168.
10. Workowski KA, Berman SM; CDC. Sexually transmitted diseases treatment guidelines, 2006. MMWR Recomm Rep. 2006;55(RR-11):1-94.
11. CDC. Sexually transmitted diseases: treatment guidelines 2006. www.cdc.gov/std/treatment/2006/genital-ulcers.htm#genulc6. Accessed March 29, 2011.
12. Drugs for sexually transmitted infections. Treatment Guidelines from the Medical Letter. 2010;95:95a. http://secure.medicalletter.org. Accessed March 23, 2011.
13. Russouw HG, Roberts MC, Emsley RA, et al. Psychiatric manifestations and magnetic resonance imaging in HIV-negative neurosyphilis. Biol Psychiatry. 1997;41(4):467-473.
14. Hooshmand H, Escobar MR, Kopf SW. Neurosyphylis: a study of 241 patients. JAMA. 1972;219 (6):726-729.
15. Miller CA, Joyce DM. Toxicity, phenytoin. http://emedicine.medscape.com/article/816447-overview. Accessed March 23, 2011.
16. Earnest MP, Marx JA, Drury LR. Complications of intravenous phenytoin for acute treatment of seizures: recommendations for usage. JAMA. 1983; 246(6):762-765.
17. Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol. 2008;64(1): 97-108.
18. Mechem CC. Chapter 143. Altered mental status and coma. In: Ma J, Cline DM, Tintinalli JE, et al, eds. Emergency Medicine Manual, 6e. www.access emergencymedicine.com/content.aspx?aID=2020. Accessed March 23, 2011.
19. Knopman DS, DeKosky ST, Cummings JL, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: diagnosis of dementia (an evidence-based review). Neurology. 2001;56(9):1143-1153.
20. CDC. Syphilis testing algorithms using treponemal tests for initial screening—four laboratories, New York City, 2005-2006. MMWR Morb Mortal Wkly Rep. 2008;57(32):872-875.
21. Anderson CA, Filley CM. Chapter 33. Behavioral presentations of medical and neurologic disorders. In: Jacobson JL, Jacobson AM, eds. Psychiatric Secrets. 2nd ed. St. Louis, MO: Hanley & Belfus; 2001.