User login
Woman, 78, With Dyspnea, Dry Cough, and Fatigue
A 78-year-old woman presented to the emergency department (ED) complaining of shortness of breath, a dry nonproductive cough, fatigue, hypoxia, and general malaise lasting for several months and worsening over a two-week period. She denied having fever, chills, hemoptysis, weight loss, headache, rashes, or joint pain. She reported sweats, decrease in appetite, wheezing, cough without sputum production, and slight swelling of the legs. The patient complained of chest pain upon admission, but it resolved quickly.
The patient, a retired widow with five grown children, denied recent surgery or exposure to sick people, had not travelled, and reported no changes in her home environment. She claimed to have no pets but admitted to currently smoking about four cigarettes a day; she had previously smoked, on average, three packs of cigarettes per day for 60 years. She denied using alcohol or drugs, including intravenous agents.
The patient’s medical history was significant for paroxysmal atrial fibrillation. She had also been diagnosed with chronic obstructive pulmonary disease (COPD), transient ischemic attack, patent foramen ovale, hyperlipidemia, seizure disorder, and hypothyroidism. She had no known HIV risk factors and had had no exposure to asbestos or tuberculosis.
The patient’s current medications included amiodarone (200 mg/d) for four years; valproic acid (500 mg/d); aspirin (325 mg/d); levothyroxine (50 g/d); rosuvastatin (10 mg/d); daily warfarin, dosed according to the international normalized ratio (INR); and budesonide/formoterol (160/4.5 mg, one puff bid). She denied having any drug allergies.
Physical examination in the ED revealed a pulse of 63 beats/min; blood pressure, 108/50 mm Hg; and respiratory rate, 16 to 20 breaths/min. The patient’s O2 saturation was 84% on room air; 82% to 84% on 4 L to 6 L of supplemental oxygen; 87% to 92% with a venturi mask; and 95% on biphasic positive airway pressure (BiPAP) device. She was afebrile with hypoxia and able to speak in full sentences. Crackles were detected in the upper lung fields, best heard anteriorly, as well as a few scattered wheezes and rhonchi. Her heart sounds were normal with a regular rhythm; her extremities exhibited trace edema bilaterally. The remainder of the physical exam was normal.
The patient’s laboratory values included a normal white blood cell (WBC) count, elevated lactic acid dehydrogenase (LDH) at 448 IU/L (reference range, 84 to 246 IU/L), and no eosinophils. The erythrocyte sedimentation rate (ESR) was not measured on admission. Blood analysis of her N-terminal pro-brain natriuretic peptide (NT-proBNP) was 4,877 pg/mL; for women older than 75, a level higher than 1,800 pg/mL is abnormal.
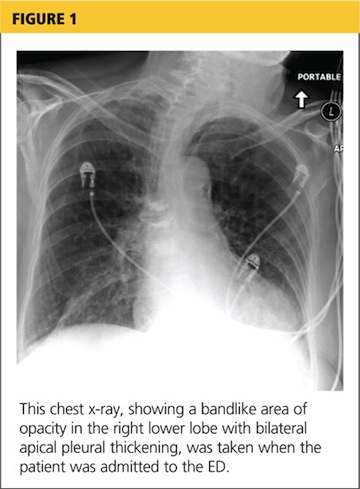
A chest x-ray was performed on admission, showing hyperinflation of the lungs with mild coarsening of the lung markings. A bandlike area of opacity in the right lower lobe with bilateral apical pleural thickening was noted (see Figure 1). Noncontrast CT of the chest revealed diffuse upper lobe ground glass opacities in both lungs, extending into the right middle lobe and lingula as well the superior segments of the lower lobes, with areas of emphysema and septal thickening. Numerous nodules, some of which appeared cavitary, were apparent in the lower lobes.
A two-dimensional echocardiogram demonstrated normal left ventricular size and systolic function, mild tricuspid regurgitation without evidence of pulmonary hypertension, and mild left atrial enlargement.
The patient was admitted to the cardiac unit for evaluation. While there, she received one dose of methylprednisolone (125 mg IV), three doses of ipratropium bromide/albuterol, one dose of ceftriaxone (1 g IV), and one dose of azithromycin (500 mg po). In the absence of significant leg edema and an elevation of jugular venous distention with a normal two-dimensional echocardiogram, heart failure was ruled out. The chest pains reported on initial presentation were ultimately felt to be noncardiac in nature.
After the patient was transferred to the medical floor with an initial diagnosis of exacerbation of her COPD, she was treated with antibiotics, nebulizers, and corticosteroids. She continued to experience episodes of O2 desaturation while on 4 L to 6 L of oxygen via nasal cannula and on a venturi mask. She was then placed on a BiPAP device, set to 12/5, and 50% Fio2 (fraction of inspired oxygen), which improved her oxygenation.
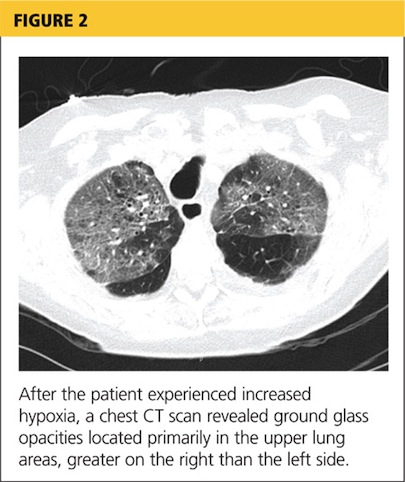
Her hypoxia prompted further radiographic studies. The resulting chest CT scan showed ground glass opacities located primarily in the upper lung areas, greater on the right than on the left side (see Figure 2). The radiologist suggested that the hypoxia was caused by an infection, but because the patient’s presenting symptoms were chronic in nature, drug-induced causes were considered as well. Amiodarone was discontinued.
Cardiology was consulted and agreed that stopping amiodarone was acceptable since the patient was in sinus rhythm at the time. The patient continued to take antibiotics and prednisone. Her symptoms slowly improved during hospitalization, and she required less oxygen. Based on the patient’s presentation, physical exam findings, imaging studies, and laboratory findings, amiodarone-induced pulmonary toxicity (APT) was diagnosed.
She was discharged home on supplemental oxygen at 4 L via cannula, a tapering dosage of prednisone, and metered-dose inhalers for fluticasone/salmeterol and tiotropium bromide. She also had outpatient appointments scheduled, one with the pulmonologist to follow up on her imaging studies and to manage the prednisone taper and the other with the cardiologist to manage her atrial fibrillation.
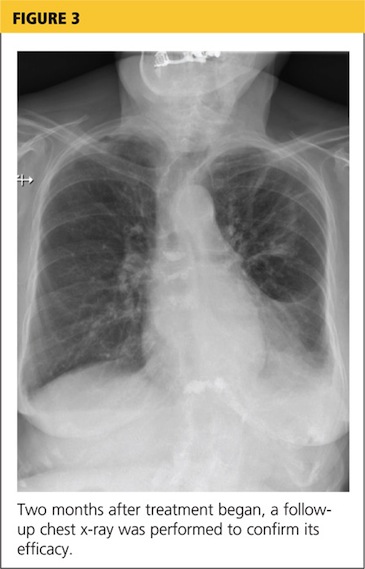
At pulmonology two months later, she had a chest x-ray (see Figure 3) and pulmonary function tests (PFTs). The patient reported feeling progressively better in the past month. Her dyspnea on exertion had improved, and she did not require supplemental oxygen anymore. She stopped smoking cigarettes.
The patient continued to use fluticasone/salmeterol but stopped tiotropium bromide. On physical exam, her O2 saturation was 95% on room air, heart rhythm and rate were regular, and her lungs revealed very minimal crackles at the right base but were otherwise clear.
The plan specified continuing the prednisone taper. The patient was asked to call the office if she had any worsening shortness of breath, cough, and sputum production. She was also encouraged to continue refraining from smoking cigarettes. This patient had done very well, with near complete resolution of symptoms and a clear chest x-ray.
Continue reading for discussion...
DISCUSSION
Amiodarone, a highly effective antiarrhythmic drug, is FDA approved for suppressing ventricular fibrillation and ventricular tachycardia. It is also used off-label as a second- or third-line choice for atrial fibrillation.1
Standard of care requires that, prior to starting amiodarone therapy, patients have a baseline chest x-ray and PFTs with diffusing capacity performed. Thereafter, the patient should be monitored with annual chest x-rays, with one performed promptly if new symptoms develop. Serial PFTs have not offered any benefit for monitoring, but a decrease of more than 15% in total lung capacity or more than 20% in diffusing capacity from baseline is consistent with APT.2
Adverse effects, both cardiac and noncardiac, are common with amiodarone therapy. They include proarrhythmias, bradycardia, and heart block, as well as thyroid and liver dysfunctions; dermatologic conditions such as blue-gray discoloration of the skin and photosensitivity; neurologic effects such as ataxia, paresthesias, and tremor; ocular problems, including corneal microdeposits; gastrointestinal problems such as nausea, anorexia, and constipation; and lung problems such as pulmonary toxicity, pleural effusion, and pleural thickening.3-6 Of these, pulmonary toxicity is the most severe and life threatening.7
APT, also known as amiodarone pneumonitis and amiodarone lung, typically manifests from a few months to a year and a half after treatment is commenced.6 APT can occur even after the drug is discontinued, because amiodarone has a very long elimination half-life of approximately 15 to 45 days and a tendency to concentrate in organs with high blood perfusion and in adipose tissues.8 Patients taking 400 mg/d for two months or longer or 200 mg/d for more than two years are considered at higher risk for APT.9 The severity of disease appears to correlate with the cumulative dose and length of treatment.10
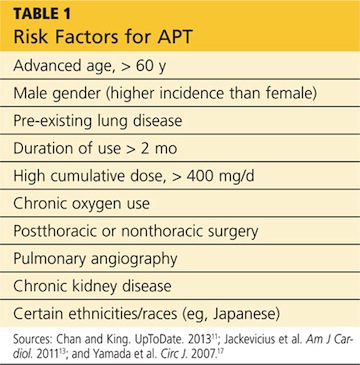
Numerous risk factors for pulmonary toxicity have been reported, including high drug dosage, pre-existing lung disease, patient age, and prior surgery (see Table 1).11 According to an analysis of a database of 237 patients, only age and duration of amiodarone therapy were significant risk factors for APT.9 Its incidence is not precisely known; reported rates range from 1% to 17%.6,12,13
Presentation with such nonspecific symptoms as shortness of breath, nonproductive cough, fatigue, hypoxia, and general malaise is typical for many pulmonary and cardiac illnesses (see Table 2), making APT difficult to diagnose.14 Occasionally, rapid onset with progression to pneumonitis and respiratory failure masquerades as acute respiratory distress syndrome (ARDS).15
Notable, however, is that APT can manifest with nonproductive cough and dyspnea in 50% to 75% of cases. In addition, presenting symptoms will include fever (33% to 50% of cases) with associated malaise, fatigue, chest pain, and weight loss. In patients with APT, the physical exam usually reveals bilateral crackles on inspiration, but diffuse rales may be heard as well.11
Laboratory studies are not very helpful in diagnosing APT. Patients may present with nonspecific elevated WBCs without eosinophilia and an elevated LDH level.11 An elevated ESR may be detected before symptoms of APT manifest and can be present at the time of diagnosis.6
Imaging studies are far more helpful and specific in diagnosing APT. The typical chest x-ray shows bilateral patchy diffuse infiltrates.12 CT of the chest is usually more revealing, demonstrating ground glass opacities in the periphery and subpleural thickening, especially where infiltrates are denser. This thickening may result in pleuritic chest pain.6
The right upper lobe is more often affected in these cases than the left lung.6 Numerous pulmonary nodules in the upper lobes are found rarely and can be confused with lung cancer. These nodules are likely the result of an accumulation of the drug in areas of previous inflammation; a lung mass should prompt the addition of APT in the differential.2,16
APT is a diagnosis of exclusion, requiring clinical suspicion, drug history, imaging, and consideration of the differential. The presence of three or more clinical factors supports a diagnosis of APT (see Table 3).11

Once APT is recognized, the first action is to have the patient stop taking amiodarone, followed by the administration of corticosteroids (eg, prednisone 40 to 60 mg/d11) for four to 12 months.17 Patients, especially those with underlying lung disease, will typically require temporary oxygen supplementation until hypoxia resolves. Even after the drug has been discontinued, some patients experience worsening symptoms before they see improvement simply because the drug can persist in lung tissue for up to a year following cessation of therapy.6
If APT is diagnosed early, the prognosis is favorable. In one study, a significant number of APT patients stabilized or improved after withdrawal of the drug, regardless of concurrent treatment with corticosteroids.18 Follow-up studies, both imaging and PFT, indicate complete clearing of lung opacities in the majority of patients treated for APT.19 Radiologic improvement may be seen six months after cessation of amiodarone.20 Patients who develop ARDS tend to do poorly and have a mortality rate of approximately 50%.11
Continue reading for the conclusion...
CONCLUSION
Among patients who are taking long-term or high-dose amiodarone, particularly those older than 60, new-onset nonproductive cough and dyspnea signal the need for pulmonary and cardiac work-up. Once the diagnosis of APT is made, treatment is straightforward: Withdraw the amiodarone, and initiate corticosteroid therapy.
REFERENCES
1. Fuster V, Rydén LE, Asinger RW, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; European Society of Cardiology Committee for Practice Guidelines and Policy Conferences (Committee to Develop Guidelines for the Management of Patients With Atrial Fibrillation); North American Society of Pacing and Electrophysiology. ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation: executive summary. Circulation. 2001; 104(17):2118-2150.
2. Jarand J, Lee A, Leigh R. Amiodaronoma: an unusual form of amiodarone-induced pulmonary toxicity. CMAJ. 2007;176(10):1411-1413.
3. Connolly S. Evidence-based analysis of amiodarone efficacy and safety. Circulation. 1999;100:2025-2034.
4. Amiodarone Trials Meta-Analysis Investigators. Effect of prophylactic amiodarone on mortality after acute myocardial infarction and in congestive heart failure: meta-analysis of individual data from 6500 patients in randomised trials. Lancet. 1997;350(9089):1417-1424.
5. Pollak PT. Clinical organ toxicity of antiarrhythmic compounds: ocular and pulmonary manifestations. Am J Cardiol. 1999;84(9A):37R-45R.
6. Camus P, Martin W, Rosenow E. Amiodarone pulmonary toxicity. Clin Chest Med. 2004;25(1):65-75.
7. Rady MY, Ryan T, Starr NJ. Preoperative therapy with amiodarone and the incidence of acute organ dysfunction after cardiac surgery. Anesth Analg. 1997;85(3):489-497.
8. Canada A, Lesko L, Haffajee C, et al. Amiodarone for tachyarrhythmias: kinetics, and efficacy. Drug Intell Clin Pharm. 1983;17(2):100-104.
9. Ernawati DK, Stafford L, Hughes JD. Amiodarone-induced pulmonary toxicity. Br J Clin Pharmacol. 2008;66(1):82-87.
10. Liu FL, Cohen RD, Downar E, et al. Amiodarone pulmonary toxicity: functional and ultrastructural evaluation. Thorax. 1986;41(2):100-105.
11. Chan E, King TE. Amiodarone pulmonary toxicity. UpToDate. 2013. www.uptodate.com/contents/amiodarone-pulmonary-toxicity. Accessed January 17, 2014.
12. Wolkove N, Baltzan M. Amiodarone pulmonary toxicity. Can Respir J. 2009;16(2):43-48.
13. Jackevicius CA, Tom A, Essebag V, et al. Population-level incidence and risk factors for pulmonary toxicity associated with amiodarone. Am J Cardiol. 2011;108:705-710.
14. Jessurun G, Crijns H. Amiodarone pulmonary toxicity [editorial]. BMJ. 1997;314(7081):619-620.
15. Nacca N, Castigliano B, Yuhico L, et al. Severe amiodarone induced pulmonary toxicity. J Thorac Dis. 2012;4(6):667-670.
16. Arnon R, Raz I, Chajek-Shaul T, et al. Amiodarone pulmonary toxicity presenting as a solitary lung mass. Chest. 1988;93(2):425-427.
17. Yamada Y, Shiga T, Matsuda N, et al. Incidence and predictors of pulmonary toxicity in Japanese patients receiving low-dose amiodarone. Circ J. 2007;71(10):1610-1616.
18. Coudert B, Bailly F, Lombard JN, et al. Amiodarone pneumonitis: bronchoalveolar lavage findings in 15 patients and review of the literature. Chest. 1992;102(4):1005-1012.
19. Vernhet H, Bousquet C, Durand G, et al. Reversible amiodarone-induced lung disease: HRCT findings. Eur Radiol. 2001;11(9):1697-1703.
20. Olson LK, Forrest JV, Friedman PJ, et al. Pneumonitis after amiodarone therapy. Radiology. 1984;150(2):327-330.
A 78-year-old woman presented to the emergency department (ED) complaining of shortness of breath, a dry nonproductive cough, fatigue, hypoxia, and general malaise lasting for several months and worsening over a two-week period. She denied having fever, chills, hemoptysis, weight loss, headache, rashes, or joint pain. She reported sweats, decrease in appetite, wheezing, cough without sputum production, and slight swelling of the legs. The patient complained of chest pain upon admission, but it resolved quickly.
The patient, a retired widow with five grown children, denied recent surgery or exposure to sick people, had not travelled, and reported no changes in her home environment. She claimed to have no pets but admitted to currently smoking about four cigarettes a day; she had previously smoked, on average, three packs of cigarettes per day for 60 years. She denied using alcohol or drugs, including intravenous agents.
The patient’s medical history was significant for paroxysmal atrial fibrillation. She had also been diagnosed with chronic obstructive pulmonary disease (COPD), transient ischemic attack, patent foramen ovale, hyperlipidemia, seizure disorder, and hypothyroidism. She had no known HIV risk factors and had had no exposure to asbestos or tuberculosis.
The patient’s current medications included amiodarone (200 mg/d) for four years; valproic acid (500 mg/d); aspirin (325 mg/d); levothyroxine (50 g/d); rosuvastatin (10 mg/d); daily warfarin, dosed according to the international normalized ratio (INR); and budesonide/formoterol (160/4.5 mg, one puff bid). She denied having any drug allergies.
Physical examination in the ED revealed a pulse of 63 beats/min; blood pressure, 108/50 mm Hg; and respiratory rate, 16 to 20 breaths/min. The patient’s O2 saturation was 84% on room air; 82% to 84% on 4 L to 6 L of supplemental oxygen; 87% to 92% with a venturi mask; and 95% on biphasic positive airway pressure (BiPAP) device. She was afebrile with hypoxia and able to speak in full sentences. Crackles were detected in the upper lung fields, best heard anteriorly, as well as a few scattered wheezes and rhonchi. Her heart sounds were normal with a regular rhythm; her extremities exhibited trace edema bilaterally. The remainder of the physical exam was normal.
The patient’s laboratory values included a normal white blood cell (WBC) count, elevated lactic acid dehydrogenase (LDH) at 448 IU/L (reference range, 84 to 246 IU/L), and no eosinophils. The erythrocyte sedimentation rate (ESR) was not measured on admission. Blood analysis of her N-terminal pro-brain natriuretic peptide (NT-proBNP) was 4,877 pg/mL; for women older than 75, a level higher than 1,800 pg/mL is abnormal.
A chest x-ray was performed on admission, showing hyperinflation of the lungs with mild coarsening of the lung markings. A bandlike area of opacity in the right lower lobe with bilateral apical pleural thickening was noted (see Figure 1). Noncontrast CT of the chest revealed diffuse upper lobe ground glass opacities in both lungs, extending into the right middle lobe and lingula as well the superior segments of the lower lobes, with areas of emphysema and septal thickening. Numerous nodules, some of which appeared cavitary, were apparent in the lower lobes.
A two-dimensional echocardiogram demonstrated normal left ventricular size and systolic function, mild tricuspid regurgitation without evidence of pulmonary hypertension, and mild left atrial enlargement.
The patient was admitted to the cardiac unit for evaluation. While there, she received one dose of methylprednisolone (125 mg IV), three doses of ipratropium bromide/albuterol, one dose of ceftriaxone (1 g IV), and one dose of azithromycin (500 mg po). In the absence of significant leg edema and an elevation of jugular venous distention with a normal two-dimensional echocardiogram, heart failure was ruled out. The chest pains reported on initial presentation were ultimately felt to be noncardiac in nature.
After the patient was transferred to the medical floor with an initial diagnosis of exacerbation of her COPD, she was treated with antibiotics, nebulizers, and corticosteroids. She continued to experience episodes of O2 desaturation while on 4 L to 6 L of oxygen via nasal cannula and on a venturi mask. She was then placed on a BiPAP device, set to 12/5, and 50% Fio2 (fraction of inspired oxygen), which improved her oxygenation.
Her hypoxia prompted further radiographic studies. The resulting chest CT scan showed ground glass opacities located primarily in the upper lung areas, greater on the right than on the left side (see Figure 2). The radiologist suggested that the hypoxia was caused by an infection, but because the patient’s presenting symptoms were chronic in nature, drug-induced causes were considered as well. Amiodarone was discontinued.
Cardiology was consulted and agreed that stopping amiodarone was acceptable since the patient was in sinus rhythm at the time. The patient continued to take antibiotics and prednisone. Her symptoms slowly improved during hospitalization, and she required less oxygen. Based on the patient’s presentation, physical exam findings, imaging studies, and laboratory findings, amiodarone-induced pulmonary toxicity (APT) was diagnosed.
She was discharged home on supplemental oxygen at 4 L via cannula, a tapering dosage of prednisone, and metered-dose inhalers for fluticasone/salmeterol and tiotropium bromide. She also had outpatient appointments scheduled, one with the pulmonologist to follow up on her imaging studies and to manage the prednisone taper and the other with the cardiologist to manage her atrial fibrillation.
At pulmonology two months later, she had a chest x-ray (see Figure 3) and pulmonary function tests (PFTs). The patient reported feeling progressively better in the past month. Her dyspnea on exertion had improved, and she did not require supplemental oxygen anymore. She stopped smoking cigarettes.
The patient continued to use fluticasone/salmeterol but stopped tiotropium bromide. On physical exam, her O2 saturation was 95% on room air, heart rhythm and rate were regular, and her lungs revealed very minimal crackles at the right base but were otherwise clear.
The plan specified continuing the prednisone taper. The patient was asked to call the office if she had any worsening shortness of breath, cough, and sputum production. She was also encouraged to continue refraining from smoking cigarettes. This patient had done very well, with near complete resolution of symptoms and a clear chest x-ray.
Continue reading for discussion...
DISCUSSION
Amiodarone, a highly effective antiarrhythmic drug, is FDA approved for suppressing ventricular fibrillation and ventricular tachycardia. It is also used off-label as a second- or third-line choice for atrial fibrillation.1
Standard of care requires that, prior to starting amiodarone therapy, patients have a baseline chest x-ray and PFTs with diffusing capacity performed. Thereafter, the patient should be monitored with annual chest x-rays, with one performed promptly if new symptoms develop. Serial PFTs have not offered any benefit for monitoring, but a decrease of more than 15% in total lung capacity or more than 20% in diffusing capacity from baseline is consistent with APT.2
Adverse effects, both cardiac and noncardiac, are common with amiodarone therapy. They include proarrhythmias, bradycardia, and heart block, as well as thyroid and liver dysfunctions; dermatologic conditions such as blue-gray discoloration of the skin and photosensitivity; neurologic effects such as ataxia, paresthesias, and tremor; ocular problems, including corneal microdeposits; gastrointestinal problems such as nausea, anorexia, and constipation; and lung problems such as pulmonary toxicity, pleural effusion, and pleural thickening.3-6 Of these, pulmonary toxicity is the most severe and life threatening.7
APT, also known as amiodarone pneumonitis and amiodarone lung, typically manifests from a few months to a year and a half after treatment is commenced.6 APT can occur even after the drug is discontinued, because amiodarone has a very long elimination half-life of approximately 15 to 45 days and a tendency to concentrate in organs with high blood perfusion and in adipose tissues.8 Patients taking 400 mg/d for two months or longer or 200 mg/d for more than two years are considered at higher risk for APT.9 The severity of disease appears to correlate with the cumulative dose and length of treatment.10
Numerous risk factors for pulmonary toxicity have been reported, including high drug dosage, pre-existing lung disease, patient age, and prior surgery (see Table 1).11 According to an analysis of a database of 237 patients, only age and duration of amiodarone therapy were significant risk factors for APT.9 Its incidence is not precisely known; reported rates range from 1% to 17%.6,12,13
Presentation with such nonspecific symptoms as shortness of breath, nonproductive cough, fatigue, hypoxia, and general malaise is typical for many pulmonary and cardiac illnesses (see Table 2), making APT difficult to diagnose.14 Occasionally, rapid onset with progression to pneumonitis and respiratory failure masquerades as acute respiratory distress syndrome (ARDS).15
Notable, however, is that APT can manifest with nonproductive cough and dyspnea in 50% to 75% of cases. In addition, presenting symptoms will include fever (33% to 50% of cases) with associated malaise, fatigue, chest pain, and weight loss. In patients with APT, the physical exam usually reveals bilateral crackles on inspiration, but diffuse rales may be heard as well.11
Laboratory studies are not very helpful in diagnosing APT. Patients may present with nonspecific elevated WBCs without eosinophilia and an elevated LDH level.11 An elevated ESR may be detected before symptoms of APT manifest and can be present at the time of diagnosis.6
Imaging studies are far more helpful and specific in diagnosing APT. The typical chest x-ray shows bilateral patchy diffuse infiltrates.12 CT of the chest is usually more revealing, demonstrating ground glass opacities in the periphery and subpleural thickening, especially where infiltrates are denser. This thickening may result in pleuritic chest pain.6
The right upper lobe is more often affected in these cases than the left lung.6 Numerous pulmonary nodules in the upper lobes are found rarely and can be confused with lung cancer. These nodules are likely the result of an accumulation of the drug in areas of previous inflammation; a lung mass should prompt the addition of APT in the differential.2,16
APT is a diagnosis of exclusion, requiring clinical suspicion, drug history, imaging, and consideration of the differential. The presence of three or more clinical factors supports a diagnosis of APT (see Table 3).11
Once APT is recognized, the first action is to have the patient stop taking amiodarone, followed by the administration of corticosteroids (eg, prednisone 40 to 60 mg/d11) for four to 12 months.17 Patients, especially those with underlying lung disease, will typically require temporary oxygen supplementation until hypoxia resolves. Even after the drug has been discontinued, some patients experience worsening symptoms before they see improvement simply because the drug can persist in lung tissue for up to a year following cessation of therapy.6
If APT is diagnosed early, the prognosis is favorable. In one study, a significant number of APT patients stabilized or improved after withdrawal of the drug, regardless of concurrent treatment with corticosteroids.18 Follow-up studies, both imaging and PFT, indicate complete clearing of lung opacities in the majority of patients treated for APT.19 Radiologic improvement may be seen six months after cessation of amiodarone.20 Patients who develop ARDS tend to do poorly and have a mortality rate of approximately 50%.11
Continue reading for the conclusion...
CONCLUSION
Among patients who are taking long-term or high-dose amiodarone, particularly those older than 60, new-onset nonproductive cough and dyspnea signal the need for pulmonary and cardiac work-up. Once the diagnosis of APT is made, treatment is straightforward: Withdraw the amiodarone, and initiate corticosteroid therapy.
REFERENCES
1. Fuster V, Rydén LE, Asinger RW, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; European Society of Cardiology Committee for Practice Guidelines and Policy Conferences (Committee to Develop Guidelines for the Management of Patients With Atrial Fibrillation); North American Society of Pacing and Electrophysiology. ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation: executive summary. Circulation. 2001; 104(17):2118-2150.
2. Jarand J, Lee A, Leigh R. Amiodaronoma: an unusual form of amiodarone-induced pulmonary toxicity. CMAJ. 2007;176(10):1411-1413.
3. Connolly S. Evidence-based analysis of amiodarone efficacy and safety. Circulation. 1999;100:2025-2034.
4. Amiodarone Trials Meta-Analysis Investigators. Effect of prophylactic amiodarone on mortality after acute myocardial infarction and in congestive heart failure: meta-analysis of individual data from 6500 patients in randomised trials. Lancet. 1997;350(9089):1417-1424.
5. Pollak PT. Clinical organ toxicity of antiarrhythmic compounds: ocular and pulmonary manifestations. Am J Cardiol. 1999;84(9A):37R-45R.
6. Camus P, Martin W, Rosenow E. Amiodarone pulmonary toxicity. Clin Chest Med. 2004;25(1):65-75.
7. Rady MY, Ryan T, Starr NJ. Preoperative therapy with amiodarone and the incidence of acute organ dysfunction after cardiac surgery. Anesth Analg. 1997;85(3):489-497.
8. Canada A, Lesko L, Haffajee C, et al. Amiodarone for tachyarrhythmias: kinetics, and efficacy. Drug Intell Clin Pharm. 1983;17(2):100-104.
9. Ernawati DK, Stafford L, Hughes JD. Amiodarone-induced pulmonary toxicity. Br J Clin Pharmacol. 2008;66(1):82-87.
10. Liu FL, Cohen RD, Downar E, et al. Amiodarone pulmonary toxicity: functional and ultrastructural evaluation. Thorax. 1986;41(2):100-105.
11. Chan E, King TE. Amiodarone pulmonary toxicity. UpToDate. 2013. www.uptodate.com/contents/amiodarone-pulmonary-toxicity. Accessed January 17, 2014.
12. Wolkove N, Baltzan M. Amiodarone pulmonary toxicity. Can Respir J. 2009;16(2):43-48.
13. Jackevicius CA, Tom A, Essebag V, et al. Population-level incidence and risk factors for pulmonary toxicity associated with amiodarone. Am J Cardiol. 2011;108:705-710.
14. Jessurun G, Crijns H. Amiodarone pulmonary toxicity [editorial]. BMJ. 1997;314(7081):619-620.
15. Nacca N, Castigliano B, Yuhico L, et al. Severe amiodarone induced pulmonary toxicity. J Thorac Dis. 2012;4(6):667-670.
16. Arnon R, Raz I, Chajek-Shaul T, et al. Amiodarone pulmonary toxicity presenting as a solitary lung mass. Chest. 1988;93(2):425-427.
17. Yamada Y, Shiga T, Matsuda N, et al. Incidence and predictors of pulmonary toxicity in Japanese patients receiving low-dose amiodarone. Circ J. 2007;71(10):1610-1616.
18. Coudert B, Bailly F, Lombard JN, et al. Amiodarone pneumonitis: bronchoalveolar lavage findings in 15 patients and review of the literature. Chest. 1992;102(4):1005-1012.
19. Vernhet H, Bousquet C, Durand G, et al. Reversible amiodarone-induced lung disease: HRCT findings. Eur Radiol. 2001;11(9):1697-1703.
20. Olson LK, Forrest JV, Friedman PJ, et al. Pneumonitis after amiodarone therapy. Radiology. 1984;150(2):327-330.
A 78-year-old woman presented to the emergency department (ED) complaining of shortness of breath, a dry nonproductive cough, fatigue, hypoxia, and general malaise lasting for several months and worsening over a two-week period. She denied having fever, chills, hemoptysis, weight loss, headache, rashes, or joint pain. She reported sweats, decrease in appetite, wheezing, cough without sputum production, and slight swelling of the legs. The patient complained of chest pain upon admission, but it resolved quickly.
The patient, a retired widow with five grown children, denied recent surgery or exposure to sick people, had not travelled, and reported no changes in her home environment. She claimed to have no pets but admitted to currently smoking about four cigarettes a day; she had previously smoked, on average, three packs of cigarettes per day for 60 years. She denied using alcohol or drugs, including intravenous agents.
The patient’s medical history was significant for paroxysmal atrial fibrillation. She had also been diagnosed with chronic obstructive pulmonary disease (COPD), transient ischemic attack, patent foramen ovale, hyperlipidemia, seizure disorder, and hypothyroidism. She had no known HIV risk factors and had had no exposure to asbestos or tuberculosis.
The patient’s current medications included amiodarone (200 mg/d) for four years; valproic acid (500 mg/d); aspirin (325 mg/d); levothyroxine (50 g/d); rosuvastatin (10 mg/d); daily warfarin, dosed according to the international normalized ratio (INR); and budesonide/formoterol (160/4.5 mg, one puff bid). She denied having any drug allergies.
Physical examination in the ED revealed a pulse of 63 beats/min; blood pressure, 108/50 mm Hg; and respiratory rate, 16 to 20 breaths/min. The patient’s O2 saturation was 84% on room air; 82% to 84% on 4 L to 6 L of supplemental oxygen; 87% to 92% with a venturi mask; and 95% on biphasic positive airway pressure (BiPAP) device. She was afebrile with hypoxia and able to speak in full sentences. Crackles were detected in the upper lung fields, best heard anteriorly, as well as a few scattered wheezes and rhonchi. Her heart sounds were normal with a regular rhythm; her extremities exhibited trace edema bilaterally. The remainder of the physical exam was normal.
The patient’s laboratory values included a normal white blood cell (WBC) count, elevated lactic acid dehydrogenase (LDH) at 448 IU/L (reference range, 84 to 246 IU/L), and no eosinophils. The erythrocyte sedimentation rate (ESR) was not measured on admission. Blood analysis of her N-terminal pro-brain natriuretic peptide (NT-proBNP) was 4,877 pg/mL; for women older than 75, a level higher than 1,800 pg/mL is abnormal.
A chest x-ray was performed on admission, showing hyperinflation of the lungs with mild coarsening of the lung markings. A bandlike area of opacity in the right lower lobe with bilateral apical pleural thickening was noted (see Figure 1). Noncontrast CT of the chest revealed diffuse upper lobe ground glass opacities in both lungs, extending into the right middle lobe and lingula as well the superior segments of the lower lobes, with areas of emphysema and septal thickening. Numerous nodules, some of which appeared cavitary, were apparent in the lower lobes.
A two-dimensional echocardiogram demonstrated normal left ventricular size and systolic function, mild tricuspid regurgitation without evidence of pulmonary hypertension, and mild left atrial enlargement.
The patient was admitted to the cardiac unit for evaluation. While there, she received one dose of methylprednisolone (125 mg IV), three doses of ipratropium bromide/albuterol, one dose of ceftriaxone (1 g IV), and one dose of azithromycin (500 mg po). In the absence of significant leg edema and an elevation of jugular venous distention with a normal two-dimensional echocardiogram, heart failure was ruled out. The chest pains reported on initial presentation were ultimately felt to be noncardiac in nature.
After the patient was transferred to the medical floor with an initial diagnosis of exacerbation of her COPD, she was treated with antibiotics, nebulizers, and corticosteroids. She continued to experience episodes of O2 desaturation while on 4 L to 6 L of oxygen via nasal cannula and on a venturi mask. She was then placed on a BiPAP device, set to 12/5, and 50% Fio2 (fraction of inspired oxygen), which improved her oxygenation.
Her hypoxia prompted further radiographic studies. The resulting chest CT scan showed ground glass opacities located primarily in the upper lung areas, greater on the right than on the left side (see Figure 2). The radiologist suggested that the hypoxia was caused by an infection, but because the patient’s presenting symptoms were chronic in nature, drug-induced causes were considered as well. Amiodarone was discontinued.
Cardiology was consulted and agreed that stopping amiodarone was acceptable since the patient was in sinus rhythm at the time. The patient continued to take antibiotics and prednisone. Her symptoms slowly improved during hospitalization, and she required less oxygen. Based on the patient’s presentation, physical exam findings, imaging studies, and laboratory findings, amiodarone-induced pulmonary toxicity (APT) was diagnosed.
She was discharged home on supplemental oxygen at 4 L via cannula, a tapering dosage of prednisone, and metered-dose inhalers for fluticasone/salmeterol and tiotropium bromide. She also had outpatient appointments scheduled, one with the pulmonologist to follow up on her imaging studies and to manage the prednisone taper and the other with the cardiologist to manage her atrial fibrillation.
At pulmonology two months later, she had a chest x-ray (see Figure 3) and pulmonary function tests (PFTs). The patient reported feeling progressively better in the past month. Her dyspnea on exertion had improved, and she did not require supplemental oxygen anymore. She stopped smoking cigarettes.
The patient continued to use fluticasone/salmeterol but stopped tiotropium bromide. On physical exam, her O2 saturation was 95% on room air, heart rhythm and rate were regular, and her lungs revealed very minimal crackles at the right base but were otherwise clear.
The plan specified continuing the prednisone taper. The patient was asked to call the office if she had any worsening shortness of breath, cough, and sputum production. She was also encouraged to continue refraining from smoking cigarettes. This patient had done very well, with near complete resolution of symptoms and a clear chest x-ray.
Continue reading for discussion...
DISCUSSION
Amiodarone, a highly effective antiarrhythmic drug, is FDA approved for suppressing ventricular fibrillation and ventricular tachycardia. It is also used off-label as a second- or third-line choice for atrial fibrillation.1
Standard of care requires that, prior to starting amiodarone therapy, patients have a baseline chest x-ray and PFTs with diffusing capacity performed. Thereafter, the patient should be monitored with annual chest x-rays, with one performed promptly if new symptoms develop. Serial PFTs have not offered any benefit for monitoring, but a decrease of more than 15% in total lung capacity or more than 20% in diffusing capacity from baseline is consistent with APT.2
Adverse effects, both cardiac and noncardiac, are common with amiodarone therapy. They include proarrhythmias, bradycardia, and heart block, as well as thyroid and liver dysfunctions; dermatologic conditions such as blue-gray discoloration of the skin and photosensitivity; neurologic effects such as ataxia, paresthesias, and tremor; ocular problems, including corneal microdeposits; gastrointestinal problems such as nausea, anorexia, and constipation; and lung problems such as pulmonary toxicity, pleural effusion, and pleural thickening.3-6 Of these, pulmonary toxicity is the most severe and life threatening.7
APT, also known as amiodarone pneumonitis and amiodarone lung, typically manifests from a few months to a year and a half after treatment is commenced.6 APT can occur even after the drug is discontinued, because amiodarone has a very long elimination half-life of approximately 15 to 45 days and a tendency to concentrate in organs with high blood perfusion and in adipose tissues.8 Patients taking 400 mg/d for two months or longer or 200 mg/d for more than two years are considered at higher risk for APT.9 The severity of disease appears to correlate with the cumulative dose and length of treatment.10
Numerous risk factors for pulmonary toxicity have been reported, including high drug dosage, pre-existing lung disease, patient age, and prior surgery (see Table 1).11 According to an analysis of a database of 237 patients, only age and duration of amiodarone therapy were significant risk factors for APT.9 Its incidence is not precisely known; reported rates range from 1% to 17%.6,12,13
Presentation with such nonspecific symptoms as shortness of breath, nonproductive cough, fatigue, hypoxia, and general malaise is typical for many pulmonary and cardiac illnesses (see Table 2), making APT difficult to diagnose.14 Occasionally, rapid onset with progression to pneumonitis and respiratory failure masquerades as acute respiratory distress syndrome (ARDS).15
Notable, however, is that APT can manifest with nonproductive cough and dyspnea in 50% to 75% of cases. In addition, presenting symptoms will include fever (33% to 50% of cases) with associated malaise, fatigue, chest pain, and weight loss. In patients with APT, the physical exam usually reveals bilateral crackles on inspiration, but diffuse rales may be heard as well.11
Laboratory studies are not very helpful in diagnosing APT. Patients may present with nonspecific elevated WBCs without eosinophilia and an elevated LDH level.11 An elevated ESR may be detected before symptoms of APT manifest and can be present at the time of diagnosis.6
Imaging studies are far more helpful and specific in diagnosing APT. The typical chest x-ray shows bilateral patchy diffuse infiltrates.12 CT of the chest is usually more revealing, demonstrating ground glass opacities in the periphery and subpleural thickening, especially where infiltrates are denser. This thickening may result in pleuritic chest pain.6
The right upper lobe is more often affected in these cases than the left lung.6 Numerous pulmonary nodules in the upper lobes are found rarely and can be confused with lung cancer. These nodules are likely the result of an accumulation of the drug in areas of previous inflammation; a lung mass should prompt the addition of APT in the differential.2,16
APT is a diagnosis of exclusion, requiring clinical suspicion, drug history, imaging, and consideration of the differential. The presence of three or more clinical factors supports a diagnosis of APT (see Table 3).11
Once APT is recognized, the first action is to have the patient stop taking amiodarone, followed by the administration of corticosteroids (eg, prednisone 40 to 60 mg/d11) for four to 12 months.17 Patients, especially those with underlying lung disease, will typically require temporary oxygen supplementation until hypoxia resolves. Even after the drug has been discontinued, some patients experience worsening symptoms before they see improvement simply because the drug can persist in lung tissue for up to a year following cessation of therapy.6
If APT is diagnosed early, the prognosis is favorable. In one study, a significant number of APT patients stabilized or improved after withdrawal of the drug, regardless of concurrent treatment with corticosteroids.18 Follow-up studies, both imaging and PFT, indicate complete clearing of lung opacities in the majority of patients treated for APT.19 Radiologic improvement may be seen six months after cessation of amiodarone.20 Patients who develop ARDS tend to do poorly and have a mortality rate of approximately 50%.11
Continue reading for the conclusion...
CONCLUSION
Among patients who are taking long-term or high-dose amiodarone, particularly those older than 60, new-onset nonproductive cough and dyspnea signal the need for pulmonary and cardiac work-up. Once the diagnosis of APT is made, treatment is straightforward: Withdraw the amiodarone, and initiate corticosteroid therapy.
REFERENCES
1. Fuster V, Rydén LE, Asinger RW, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; European Society of Cardiology Committee for Practice Guidelines and Policy Conferences (Committee to Develop Guidelines for the Management of Patients With Atrial Fibrillation); North American Society of Pacing and Electrophysiology. ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation: executive summary. Circulation. 2001; 104(17):2118-2150.
2. Jarand J, Lee A, Leigh R. Amiodaronoma: an unusual form of amiodarone-induced pulmonary toxicity. CMAJ. 2007;176(10):1411-1413.
3. Connolly S. Evidence-based analysis of amiodarone efficacy and safety. Circulation. 1999;100:2025-2034.
4. Amiodarone Trials Meta-Analysis Investigators. Effect of prophylactic amiodarone on mortality after acute myocardial infarction and in congestive heart failure: meta-analysis of individual data from 6500 patients in randomised trials. Lancet. 1997;350(9089):1417-1424.
5. Pollak PT. Clinical organ toxicity of antiarrhythmic compounds: ocular and pulmonary manifestations. Am J Cardiol. 1999;84(9A):37R-45R.
6. Camus P, Martin W, Rosenow E. Amiodarone pulmonary toxicity. Clin Chest Med. 2004;25(1):65-75.
7. Rady MY, Ryan T, Starr NJ. Preoperative therapy with amiodarone and the incidence of acute organ dysfunction after cardiac surgery. Anesth Analg. 1997;85(3):489-497.
8. Canada A, Lesko L, Haffajee C, et al. Amiodarone for tachyarrhythmias: kinetics, and efficacy. Drug Intell Clin Pharm. 1983;17(2):100-104.
9. Ernawati DK, Stafford L, Hughes JD. Amiodarone-induced pulmonary toxicity. Br J Clin Pharmacol. 2008;66(1):82-87.
10. Liu FL, Cohen RD, Downar E, et al. Amiodarone pulmonary toxicity: functional and ultrastructural evaluation. Thorax. 1986;41(2):100-105.
11. Chan E, King TE. Amiodarone pulmonary toxicity. UpToDate. 2013. www.uptodate.com/contents/amiodarone-pulmonary-toxicity. Accessed January 17, 2014.
12. Wolkove N, Baltzan M. Amiodarone pulmonary toxicity. Can Respir J. 2009;16(2):43-48.
13. Jackevicius CA, Tom A, Essebag V, et al. Population-level incidence and risk factors for pulmonary toxicity associated with amiodarone. Am J Cardiol. 2011;108:705-710.
14. Jessurun G, Crijns H. Amiodarone pulmonary toxicity [editorial]. BMJ. 1997;314(7081):619-620.
15. Nacca N, Castigliano B, Yuhico L, et al. Severe amiodarone induced pulmonary toxicity. J Thorac Dis. 2012;4(6):667-670.
16. Arnon R, Raz I, Chajek-Shaul T, et al. Amiodarone pulmonary toxicity presenting as a solitary lung mass. Chest. 1988;93(2):425-427.
17. Yamada Y, Shiga T, Matsuda N, et al. Incidence and predictors of pulmonary toxicity in Japanese patients receiving low-dose amiodarone. Circ J. 2007;71(10):1610-1616.
18. Coudert B, Bailly F, Lombard JN, et al. Amiodarone pneumonitis: bronchoalveolar lavage findings in 15 patients and review of the literature. Chest. 1992;102(4):1005-1012.
19. Vernhet H, Bousquet C, Durand G, et al. Reversible amiodarone-induced lung disease: HRCT findings. Eur Radiol. 2001;11(9):1697-1703.
20. Olson LK, Forrest JV, Friedman PJ, et al. Pneumonitis after amiodarone therapy. Radiology. 1984;150(2):327-330.
Man, 45, With Greasy Rash and Deformed Nails
A 45-year-old man presented to the dermatology office complaining of a pruritic rash on his neck, chest, abdomen, and upper back. The rash had been present since the patient was 20, intermittently flaring and causing severe pruritus. For the past two weeks, it had become increasingly bothersome.
The patient described the rash as “greasy” brown plaques diffusely scattered on his body. The rash on his neck was the most bothersome, and the patient felt an uncontrollable need to scratch that area.
Since it first developed 25 years ago, he had used OTC hydrocortisone cream as needed to treat the rash. Although effective for past flares, the cream provided only minimal relief during the current episode.
The patient’s medical history included brittle nails with a worsening of nail quality in recent years. The family history revealed that the patient’s father and sister were affected by the same type of rash, which developed in adolescence for each of them, as well as brittle nails.
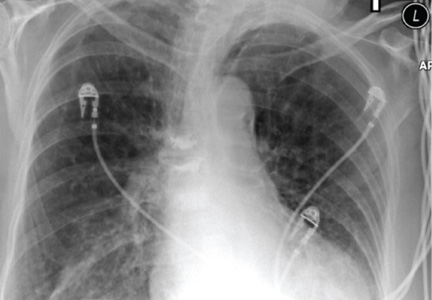
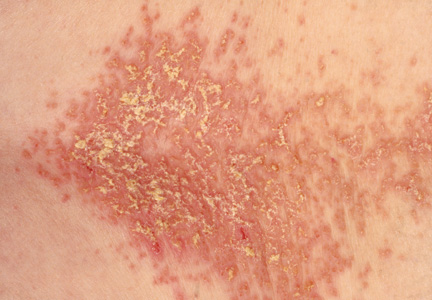
On physical examination, the skin was warm and moist to the touch. Flat, slightly elevated, greasy brown papules were scattered on the chest, abdomen, and upper back, with mild surrounding erythema (see Figure 1). Excoriated lesions were noted on the anterior surface of the neck, with pinpoint bleeding resulting from constant irritation. The patient’s fingernails were deformed, with longitudinal ridges and v-shaped notching of the free margin. The remainder of the physical exam was unremarkable, and review of systems was negative.
This patient’s symptoms could result from a variety of causes. Seborrheic dermatitis is a common skin condition that presents with brown plaques similar to those on the patient’s trunk. Another possible diagnosis is Grover’s disease, a rare disorder also known as transient acantholytic dermatosis, in which keratotic plaques appear on the torso and are thought to occur from trauma to sun-damaged skin. An additional consideration is Hailey-Hailey disease, a rare genetic disorder also known as benign familial pemphigus, which is characterized by red-brown plaques located predominantly on flexure surfaces.1 Skin biopsy should be performed for a definitive diagnosis.
Given the family history of a similar rash occurring in first-degree relatives and the distinct physical exam findings, the most likely diagnosis for this patient is keratosis follicularis, also known as Darier disease (DD) or Darier-White disease.
DISCUSSION
Named after Ferdinand-Jean Darier, who discovered this rare genodermatosis, DD is a rare genetic skin disorder caused by mutations of the ATP2A2 gene, located on the long arm of chromosome 12 at position 24,11.1,2 The mutation disrupts the encoding of the enzyme sarco/endoplasmic reticulum calcium-ATPase 2 (SERCA2). This enzyme is important in the transport of calcium ions across the cell membrane, and insufficient amounts lead to a defect in intracellular calcium signaling.2,3
This genetic mutation is inherited as an autosomal dominant trait with complete penetrance. DD affects men and women equally, with progressive skin signs of interfamilial and intrafamilial variability.4 Skin manifestations occur from late childhood to early adulthood and are typical during adolescence.4 Acute flare-ups can be triggered by heat, perspiration, sunlight, ultraviolet B exposure, stress, or certain medications (in particular, lithium).2 DD is not contagious.2
CLINICAL PRESENTATION
The characteristics of DD include yellow or brown, rough, firm papules that are frequently crusted. The papules often appear in seborrheic areas of the body, such as the chest, back, ears, nasolabial fold, forehead, scalp, and groin.4 The severity of expression varies from mild, with few lesions, to severe, in which the entire body is covered with disfiguring, macerated plaques emitting a strong odor. On biopsy, the histopathologic findings are typical of dyskeratosis and acantholysis.4
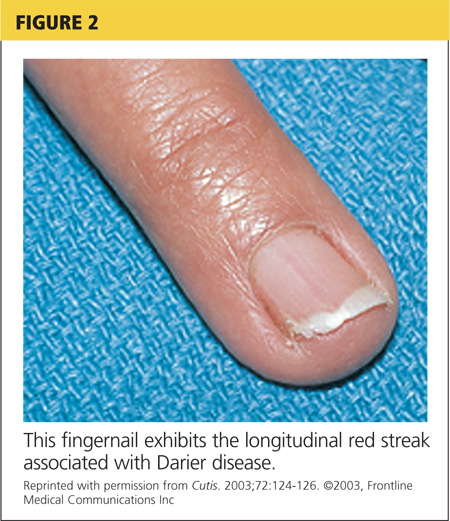
Fingernails (and occasionally toenails) display broad, white or red, somewhat translucent, longitudinal bands accompanied by v-shaped notching1,4,5 (see Figure 2). Such nail changes are diagnostic and occur in 92% to 95% of patients with DD.6 They may, in fact, occur in the absence of cutaneous disease. All nails may be affected, but usually only two to three are involved.6
Although uncommon in DD, white, umbilicated, or cobblestone plaques may be found on intraoral mucous membranes (ie, tongue, buccal mucosa, palate, epiglottis, pharyngeal wall, and esophagus); due to confluence, papules may mimic leukoplakia.7 Lesions may also appear on the vulva or rectum.1,5 In severe cases, the salivary glands can become blocked, and the gums can hypertrophy.5
Since epidermal and brain tissue both derive from ectoderm, pathologic processes that affect one organ system may also affect the other.8 Indeed, among patients with DD, neuropsychiatric problems—including epilepsy, learning difficulties, and schizoaffective disorder—are commonly reported.1 To confirm an association between DD and ATP2A2 mutations, Jacobsen and colleagues performed an analysis of 19 unrelated DD patients with neuropsychiatric phenotypes. They discovered evidence to support the gene’s pleiotropic effects in the brain and hypothesized that mutations in the enzyme SERCA2 correlate with these phenotypes, most specifically for mood disorders.9
TREATMENT AND MANAGEMENT
Although no cure is currently available for DD, both short- and long-term treatment options are available; the choice should be based on the severity of an individual patient’s signs and symptoms. For mild cases, topical therapy, such as general emollients, corticosteroid ointments, and high sun protection factor sunscreen, is sufficient.1
For moderate cases, topical retinoids, including tretinoin cream, adapalene gel or cream, and tazarotene gel, may be necessary.4 Keratolytics, including salicylic acid in propylene glycol gel, may be used to regulate hyperkeratosis.4 Celecoxib, a COX-2 inhibitor, is another option that may restore the down regulation of SERCA2. This can prevent progression of the disease.10
Long-term management includes use of oral retinoid therapy (eg, acitretin), which might reduce the frequency of inflammatory flares.1 Systemic adverse effects from long-term use of oral retinoids are cause for concern, however. Close monitoring along with patient education can limit the occurrence of complications.11
If DD is uncontrolled with medication, dermabrasion and erbium:YAG laser ablation have been used to successfully treat chronic cases.12 Although these treatment options may remove existing lesions, it is important to inform patients that the disease has not been cured, that remission is difficult to attain, and that lesions may recur.
Because viral, bacterial, and fungal superinfections are common and may exacerbate the disease, be sure to check for signs of infection while examining the patient.4 Patients should be advised to avoid hot environments, and if that is not possible, to dress in cool cotton clothing to allow for proper ventilation and avoid the build-up of perspiration. Excessive perspiration along with poor hygiene can contribute to the formation of infections as well as trigger a flare-up. If an infection develops, patients should consult a health care provider.
Keeping the skin well moisturized can alleviate the constant pruritus that many patients experience. Daily sunscreen use is essential to avoid skin irritation caused by the sun, which can trigger an acute flare-up. Patients should be advised to avoid the long-term use of corticosteroid ointment. They should also contact their health care provider before using OTC treatments such as Burow’s solution.
CONCLUSION
A thorough history and physical exam are crucial in the diagnosis of DD. In this particular case, inquiry into family history was the key to proper diagnosis. That information, paired with a thorough physical exam, led to the correct diagnosis of this rare genetic skin disorder. A skin biopsy provided definitive confirmation.
This patient had a mild-to-moderate manifestation of DD. He was prescribed retinoid therapy, and routine follow-up visits were recommended to monitor the efficacy of medical therapy and to screen for secondary infections or neuropsychiatric disorders.
This case illustrates the importance of taking a full history and performing an in-depth physical exam when a patient presents with an unfamiliar complaint. Being thorough reduces the risk of missing a crucial element that can guide the diagnostic process.
REFERENCES
1. Creamer D, Barker J, Kerdel FA. Papular and papulosquamous dermatoses. In: Acute Adult Dermatology: Diagnosis and Management (A Colour Handbook). London, UK: Manson Publishing Ltd; 2011:48.
2. Kelly EB. Darier disease (DAR). In: Encyclopedia of Human Genetics and Disease. Santa Barbara, CA: ABC-CLIO; 2013:186-187.
3. Klausegger A, Laimer M, Bauer JW. Darier disease. [In German.] Hautarzt. 2013;64:22-25.
4. Ringpfeil F. Dermatologic disorders. In: NORD Guide to Rare Disorders. Philadelphia, PA: Lippincott Williams & Wilkins; 2003:101.
5. Disorders of keratinization. In: Ostler HB, Maibach HI, Hoke AW, Schwab IR, eds. Diseases of the Eye and Skin: A Color Atlas. Philadelphia, PA: Lippincott Williams & Wilkins; 2004:23-34.
6. Baran R, de Berker D, Holzberg M, Thomas L, eds. Baran & Dawber’s Diseases of the Nails and their Management. 4th ed. West Sussex, UK: John Wiley & Sons, Ltd; 2012:295-296.
7. Thiagarajan MK, Narasimhan M, Sankarasubramanian A. Darier disease with oral and esophageal involvement: a case report. Indian J Dent Res. 2011;22:843-846.
8. Medansky RS, Woloshin AA. Darier’s disease: an evaluation of its neuropsychiatric component. Arch Dermatol. 1961;84:482-484.
9. Jacobsen NJ, Lyons I, Hoogendoorn B, et al. ATP2A2 mutations in Darier’s disease and their relationship to neuropsychiatric phenotypes. Hum Mol Genet. 1999;8:1631-1636.
10. Kamijo M, Nishiyama C, Takagi A, et al. Cyclooxygenase-2 inhibition restores ultraviolet B-induced downregulation of ATP2A2/SERCA2 in keratinocytes: possible therapeutic approach of cyclooxygenase-2 inhibition for treatment of Darier disease. Br J Dermatol. 2012;166: 1017-1022.
11. Brecher AR, Orlow SJ. Oral retinoid therapy for dermatologic conditions in children and adolescents. J Am Acad Dermatol. 2003;49:171-182.
12. Beier C, Kaufmann R. Efficacy of erbium:YAG laser ablation in Darier disease and Hailey-Hailey disease. Arch Dermatol. 1999;35:423-427.
A 45-year-old man presented to the dermatology office complaining of a pruritic rash on his neck, chest, abdomen, and upper back. The rash had been present since the patient was 20, intermittently flaring and causing severe pruritus. For the past two weeks, it had become increasingly bothersome.
The patient described the rash as “greasy” brown plaques diffusely scattered on his body. The rash on his neck was the most bothersome, and the patient felt an uncontrollable need to scratch that area.
Since it first developed 25 years ago, he had used OTC hydrocortisone cream as needed to treat the rash. Although effective for past flares, the cream provided only minimal relief during the current episode.
The patient’s medical history included brittle nails with a worsening of nail quality in recent years. The family history revealed that the patient’s father and sister were affected by the same type of rash, which developed in adolescence for each of them, as well as brittle nails.
On physical examination, the skin was warm and moist to the touch. Flat, slightly elevated, greasy brown papules were scattered on the chest, abdomen, and upper back, with mild surrounding erythema (see Figure 1). Excoriated lesions were noted on the anterior surface of the neck, with pinpoint bleeding resulting from constant irritation. The patient’s fingernails were deformed, with longitudinal ridges and v-shaped notching of the free margin. The remainder of the physical exam was unremarkable, and review of systems was negative.
This patient’s symptoms could result from a variety of causes. Seborrheic dermatitis is a common skin condition that presents with brown plaques similar to those on the patient’s trunk. Another possible diagnosis is Grover’s disease, a rare disorder also known as transient acantholytic dermatosis, in which keratotic plaques appear on the torso and are thought to occur from trauma to sun-damaged skin. An additional consideration is Hailey-Hailey disease, a rare genetic disorder also known as benign familial pemphigus, which is characterized by red-brown plaques located predominantly on flexure surfaces.1 Skin biopsy should be performed for a definitive diagnosis.
Given the family history of a similar rash occurring in first-degree relatives and the distinct physical exam findings, the most likely diagnosis for this patient is keratosis follicularis, also known as Darier disease (DD) or Darier-White disease.
DISCUSSION
Named after Ferdinand-Jean Darier, who discovered this rare genodermatosis, DD is a rare genetic skin disorder caused by mutations of the ATP2A2 gene, located on the long arm of chromosome 12 at position 24,11.1,2 The mutation disrupts the encoding of the enzyme sarco/endoplasmic reticulum calcium-ATPase 2 (SERCA2). This enzyme is important in the transport of calcium ions across the cell membrane, and insufficient amounts lead to a defect in intracellular calcium signaling.2,3
This genetic mutation is inherited as an autosomal dominant trait with complete penetrance. DD affects men and women equally, with progressive skin signs of interfamilial and intrafamilial variability.4 Skin manifestations occur from late childhood to early adulthood and are typical during adolescence.4 Acute flare-ups can be triggered by heat, perspiration, sunlight, ultraviolet B exposure, stress, or certain medications (in particular, lithium).2 DD is not contagious.2
CLINICAL PRESENTATION
The characteristics of DD include yellow or brown, rough, firm papules that are frequently crusted. The papules often appear in seborrheic areas of the body, such as the chest, back, ears, nasolabial fold, forehead, scalp, and groin.4 The severity of expression varies from mild, with few lesions, to severe, in which the entire body is covered with disfiguring, macerated plaques emitting a strong odor. On biopsy, the histopathologic findings are typical of dyskeratosis and acantholysis.4
Fingernails (and occasionally toenails) display broad, white or red, somewhat translucent, longitudinal bands accompanied by v-shaped notching1,4,5 (see Figure 2). Such nail changes are diagnostic and occur in 92% to 95% of patients with DD.6 They may, in fact, occur in the absence of cutaneous disease. All nails may be affected, but usually only two to three are involved.6
Although uncommon in DD, white, umbilicated, or cobblestone plaques may be found on intraoral mucous membranes (ie, tongue, buccal mucosa, palate, epiglottis, pharyngeal wall, and esophagus); due to confluence, papules may mimic leukoplakia.7 Lesions may also appear on the vulva or rectum.1,5 In severe cases, the salivary glands can become blocked, and the gums can hypertrophy.5
Since epidermal and brain tissue both derive from ectoderm, pathologic processes that affect one organ system may also affect the other.8 Indeed, among patients with DD, neuropsychiatric problems—including epilepsy, learning difficulties, and schizoaffective disorder—are commonly reported.1 To confirm an association between DD and ATP2A2 mutations, Jacobsen and colleagues performed an analysis of 19 unrelated DD patients with neuropsychiatric phenotypes. They discovered evidence to support the gene’s pleiotropic effects in the brain and hypothesized that mutations in the enzyme SERCA2 correlate with these phenotypes, most specifically for mood disorders.9
TREATMENT AND MANAGEMENT
Although no cure is currently available for DD, both short- and long-term treatment options are available; the choice should be based on the severity of an individual patient’s signs and symptoms. For mild cases, topical therapy, such as general emollients, corticosteroid ointments, and high sun protection factor sunscreen, is sufficient.1
For moderate cases, topical retinoids, including tretinoin cream, adapalene gel or cream, and tazarotene gel, may be necessary.4 Keratolytics, including salicylic acid in propylene glycol gel, may be used to regulate hyperkeratosis.4 Celecoxib, a COX-2 inhibitor, is another option that may restore the down regulation of SERCA2. This can prevent progression of the disease.10
Long-term management includes use of oral retinoid therapy (eg, acitretin), which might reduce the frequency of inflammatory flares.1 Systemic adverse effects from long-term use of oral retinoids are cause for concern, however. Close monitoring along with patient education can limit the occurrence of complications.11
If DD is uncontrolled with medication, dermabrasion and erbium:YAG laser ablation have been used to successfully treat chronic cases.12 Although these treatment options may remove existing lesions, it is important to inform patients that the disease has not been cured, that remission is difficult to attain, and that lesions may recur.
Because viral, bacterial, and fungal superinfections are common and may exacerbate the disease, be sure to check for signs of infection while examining the patient.4 Patients should be advised to avoid hot environments, and if that is not possible, to dress in cool cotton clothing to allow for proper ventilation and avoid the build-up of perspiration. Excessive perspiration along with poor hygiene can contribute to the formation of infections as well as trigger a flare-up. If an infection develops, patients should consult a health care provider.
Keeping the skin well moisturized can alleviate the constant pruritus that many patients experience. Daily sunscreen use is essential to avoid skin irritation caused by the sun, which can trigger an acute flare-up. Patients should be advised to avoid the long-term use of corticosteroid ointment. They should also contact their health care provider before using OTC treatments such as Burow’s solution.
CONCLUSION
A thorough history and physical exam are crucial in the diagnosis of DD. In this particular case, inquiry into family history was the key to proper diagnosis. That information, paired with a thorough physical exam, led to the correct diagnosis of this rare genetic skin disorder. A skin biopsy provided definitive confirmation.
This patient had a mild-to-moderate manifestation of DD. He was prescribed retinoid therapy, and routine follow-up visits were recommended to monitor the efficacy of medical therapy and to screen for secondary infections or neuropsychiatric disorders.
This case illustrates the importance of taking a full history and performing an in-depth physical exam when a patient presents with an unfamiliar complaint. Being thorough reduces the risk of missing a crucial element that can guide the diagnostic process.
REFERENCES
1. Creamer D, Barker J, Kerdel FA. Papular and papulosquamous dermatoses. In: Acute Adult Dermatology: Diagnosis and Management (A Colour Handbook). London, UK: Manson Publishing Ltd; 2011:48.
2. Kelly EB. Darier disease (DAR). In: Encyclopedia of Human Genetics and Disease. Santa Barbara, CA: ABC-CLIO; 2013:186-187.
3. Klausegger A, Laimer M, Bauer JW. Darier disease. [In German.] Hautarzt. 2013;64:22-25.
4. Ringpfeil F. Dermatologic disorders. In: NORD Guide to Rare Disorders. Philadelphia, PA: Lippincott Williams & Wilkins; 2003:101.
5. Disorders of keratinization. In: Ostler HB, Maibach HI, Hoke AW, Schwab IR, eds. Diseases of the Eye and Skin: A Color Atlas. Philadelphia, PA: Lippincott Williams & Wilkins; 2004:23-34.
6. Baran R, de Berker D, Holzberg M, Thomas L, eds. Baran & Dawber’s Diseases of the Nails and their Management. 4th ed. West Sussex, UK: John Wiley & Sons, Ltd; 2012:295-296.
7. Thiagarajan MK, Narasimhan M, Sankarasubramanian A. Darier disease with oral and esophageal involvement: a case report. Indian J Dent Res. 2011;22:843-846.
8. Medansky RS, Woloshin AA. Darier’s disease: an evaluation of its neuropsychiatric component. Arch Dermatol. 1961;84:482-484.
9. Jacobsen NJ, Lyons I, Hoogendoorn B, et al. ATP2A2 mutations in Darier’s disease and their relationship to neuropsychiatric phenotypes. Hum Mol Genet. 1999;8:1631-1636.
10. Kamijo M, Nishiyama C, Takagi A, et al. Cyclooxygenase-2 inhibition restores ultraviolet B-induced downregulation of ATP2A2/SERCA2 in keratinocytes: possible therapeutic approach of cyclooxygenase-2 inhibition for treatment of Darier disease. Br J Dermatol. 2012;166: 1017-1022.
11. Brecher AR, Orlow SJ. Oral retinoid therapy for dermatologic conditions in children and adolescents. J Am Acad Dermatol. 2003;49:171-182.
12. Beier C, Kaufmann R. Efficacy of erbium:YAG laser ablation in Darier disease and Hailey-Hailey disease. Arch Dermatol. 1999;35:423-427.
A 45-year-old man presented to the dermatology office complaining of a pruritic rash on his neck, chest, abdomen, and upper back. The rash had been present since the patient was 20, intermittently flaring and causing severe pruritus. For the past two weeks, it had become increasingly bothersome.
The patient described the rash as “greasy” brown plaques diffusely scattered on his body. The rash on his neck was the most bothersome, and the patient felt an uncontrollable need to scratch that area.
Since it first developed 25 years ago, he had used OTC hydrocortisone cream as needed to treat the rash. Although effective for past flares, the cream provided only minimal relief during the current episode.
The patient’s medical history included brittle nails with a worsening of nail quality in recent years. The family history revealed that the patient’s father and sister were affected by the same type of rash, which developed in adolescence for each of them, as well as brittle nails.
On physical examination, the skin was warm and moist to the touch. Flat, slightly elevated, greasy brown papules were scattered on the chest, abdomen, and upper back, with mild surrounding erythema (see Figure 1). Excoriated lesions were noted on the anterior surface of the neck, with pinpoint bleeding resulting from constant irritation. The patient’s fingernails were deformed, with longitudinal ridges and v-shaped notching of the free margin. The remainder of the physical exam was unremarkable, and review of systems was negative.
This patient’s symptoms could result from a variety of causes. Seborrheic dermatitis is a common skin condition that presents with brown plaques similar to those on the patient’s trunk. Another possible diagnosis is Grover’s disease, a rare disorder also known as transient acantholytic dermatosis, in which keratotic plaques appear on the torso and are thought to occur from trauma to sun-damaged skin. An additional consideration is Hailey-Hailey disease, a rare genetic disorder also known as benign familial pemphigus, which is characterized by red-brown plaques located predominantly on flexure surfaces.1 Skin biopsy should be performed for a definitive diagnosis.
Given the family history of a similar rash occurring in first-degree relatives and the distinct physical exam findings, the most likely diagnosis for this patient is keratosis follicularis, also known as Darier disease (DD) or Darier-White disease.
DISCUSSION
Named after Ferdinand-Jean Darier, who discovered this rare genodermatosis, DD is a rare genetic skin disorder caused by mutations of the ATP2A2 gene, located on the long arm of chromosome 12 at position 24,11.1,2 The mutation disrupts the encoding of the enzyme sarco/endoplasmic reticulum calcium-ATPase 2 (SERCA2). This enzyme is important in the transport of calcium ions across the cell membrane, and insufficient amounts lead to a defect in intracellular calcium signaling.2,3
This genetic mutation is inherited as an autosomal dominant trait with complete penetrance. DD affects men and women equally, with progressive skin signs of interfamilial and intrafamilial variability.4 Skin manifestations occur from late childhood to early adulthood and are typical during adolescence.4 Acute flare-ups can be triggered by heat, perspiration, sunlight, ultraviolet B exposure, stress, or certain medications (in particular, lithium).2 DD is not contagious.2
CLINICAL PRESENTATION
The characteristics of DD include yellow or brown, rough, firm papules that are frequently crusted. The papules often appear in seborrheic areas of the body, such as the chest, back, ears, nasolabial fold, forehead, scalp, and groin.4 The severity of expression varies from mild, with few lesions, to severe, in which the entire body is covered with disfiguring, macerated plaques emitting a strong odor. On biopsy, the histopathologic findings are typical of dyskeratosis and acantholysis.4
Fingernails (and occasionally toenails) display broad, white or red, somewhat translucent, longitudinal bands accompanied by v-shaped notching1,4,5 (see Figure 2). Such nail changes are diagnostic and occur in 92% to 95% of patients with DD.6 They may, in fact, occur in the absence of cutaneous disease. All nails may be affected, but usually only two to three are involved.6
Although uncommon in DD, white, umbilicated, or cobblestone plaques may be found on intraoral mucous membranes (ie, tongue, buccal mucosa, palate, epiglottis, pharyngeal wall, and esophagus); due to confluence, papules may mimic leukoplakia.7 Lesions may also appear on the vulva or rectum.1,5 In severe cases, the salivary glands can become blocked, and the gums can hypertrophy.5
Since epidermal and brain tissue both derive from ectoderm, pathologic processes that affect one organ system may also affect the other.8 Indeed, among patients with DD, neuropsychiatric problems—including epilepsy, learning difficulties, and schizoaffective disorder—are commonly reported.1 To confirm an association between DD and ATP2A2 mutations, Jacobsen and colleagues performed an analysis of 19 unrelated DD patients with neuropsychiatric phenotypes. They discovered evidence to support the gene’s pleiotropic effects in the brain and hypothesized that mutations in the enzyme SERCA2 correlate with these phenotypes, most specifically for mood disorders.9
TREATMENT AND MANAGEMENT
Although no cure is currently available for DD, both short- and long-term treatment options are available; the choice should be based on the severity of an individual patient’s signs and symptoms. For mild cases, topical therapy, such as general emollients, corticosteroid ointments, and high sun protection factor sunscreen, is sufficient.1
For moderate cases, topical retinoids, including tretinoin cream, adapalene gel or cream, and tazarotene gel, may be necessary.4 Keratolytics, including salicylic acid in propylene glycol gel, may be used to regulate hyperkeratosis.4 Celecoxib, a COX-2 inhibitor, is another option that may restore the down regulation of SERCA2. This can prevent progression of the disease.10
Long-term management includes use of oral retinoid therapy (eg, acitretin), which might reduce the frequency of inflammatory flares.1 Systemic adverse effects from long-term use of oral retinoids are cause for concern, however. Close monitoring along with patient education can limit the occurrence of complications.11
If DD is uncontrolled with medication, dermabrasion and erbium:YAG laser ablation have been used to successfully treat chronic cases.12 Although these treatment options may remove existing lesions, it is important to inform patients that the disease has not been cured, that remission is difficult to attain, and that lesions may recur.
Because viral, bacterial, and fungal superinfections are common and may exacerbate the disease, be sure to check for signs of infection while examining the patient.4 Patients should be advised to avoid hot environments, and if that is not possible, to dress in cool cotton clothing to allow for proper ventilation and avoid the build-up of perspiration. Excessive perspiration along with poor hygiene can contribute to the formation of infections as well as trigger a flare-up. If an infection develops, patients should consult a health care provider.
Keeping the skin well moisturized can alleviate the constant pruritus that many patients experience. Daily sunscreen use is essential to avoid skin irritation caused by the sun, which can trigger an acute flare-up. Patients should be advised to avoid the long-term use of corticosteroid ointment. They should also contact their health care provider before using OTC treatments such as Burow’s solution.
CONCLUSION
A thorough history and physical exam are crucial in the diagnosis of DD. In this particular case, inquiry into family history was the key to proper diagnosis. That information, paired with a thorough physical exam, led to the correct diagnosis of this rare genetic skin disorder. A skin biopsy provided definitive confirmation.
This patient had a mild-to-moderate manifestation of DD. He was prescribed retinoid therapy, and routine follow-up visits were recommended to monitor the efficacy of medical therapy and to screen for secondary infections or neuropsychiatric disorders.
This case illustrates the importance of taking a full history and performing an in-depth physical exam when a patient presents with an unfamiliar complaint. Being thorough reduces the risk of missing a crucial element that can guide the diagnostic process.
REFERENCES
1. Creamer D, Barker J, Kerdel FA. Papular and papulosquamous dermatoses. In: Acute Adult Dermatology: Diagnosis and Management (A Colour Handbook). London, UK: Manson Publishing Ltd; 2011:48.
2. Kelly EB. Darier disease (DAR). In: Encyclopedia of Human Genetics and Disease. Santa Barbara, CA: ABC-CLIO; 2013:186-187.
3. Klausegger A, Laimer M, Bauer JW. Darier disease. [In German.] Hautarzt. 2013;64:22-25.
4. Ringpfeil F. Dermatologic disorders. In: NORD Guide to Rare Disorders. Philadelphia, PA: Lippincott Williams & Wilkins; 2003:101.
5. Disorders of keratinization. In: Ostler HB, Maibach HI, Hoke AW, Schwab IR, eds. Diseases of the Eye and Skin: A Color Atlas. Philadelphia, PA: Lippincott Williams & Wilkins; 2004:23-34.
6. Baran R, de Berker D, Holzberg M, Thomas L, eds. Baran & Dawber’s Diseases of the Nails and their Management. 4th ed. West Sussex, UK: John Wiley & Sons, Ltd; 2012:295-296.
7. Thiagarajan MK, Narasimhan M, Sankarasubramanian A. Darier disease with oral and esophageal involvement: a case report. Indian J Dent Res. 2011;22:843-846.
8. Medansky RS, Woloshin AA. Darier’s disease: an evaluation of its neuropsychiatric component. Arch Dermatol. 1961;84:482-484.
9. Jacobsen NJ, Lyons I, Hoogendoorn B, et al. ATP2A2 mutations in Darier’s disease and their relationship to neuropsychiatric phenotypes. Hum Mol Genet. 1999;8:1631-1636.
10. Kamijo M, Nishiyama C, Takagi A, et al. Cyclooxygenase-2 inhibition restores ultraviolet B-induced downregulation of ATP2A2/SERCA2 in keratinocytes: possible therapeutic approach of cyclooxygenase-2 inhibition for treatment of Darier disease. Br J Dermatol. 2012;166: 1017-1022.
11. Brecher AR, Orlow SJ. Oral retinoid therapy for dermatologic conditions in children and adolescents. J Am Acad Dermatol. 2003;49:171-182.
12. Beier C, Kaufmann R. Efficacy of erbium:YAG laser ablation in Darier disease and Hailey-Hailey disease. Arch Dermatol. 1999;35:423-427.
Man, 55, With Mild Chest Discomfort
A 55-year-old white man with controlled hypertension and hypercholesterolemia awoke with mild chest discomfort that he believed was mild gastroesophageal reflux. He denied radiation of pain to the shoulders, arms, back, or neck; dyspnea; palpitations; diaphoresis; nausea/vomiting; cough; or fever, during the first 30 hours of discomfort. There was no change in discomfort with deep breath, palpation of the chest, or administration of antacids. Minimal, short-lived improvement was noted with belching.
The patient had no trouble sleeping in the prone position and did not notice an increase in discomfort or unusual difficulty during his daily vigorous 30-minute aerobic workout. In fact, his symptoms seemed to improve or disappear during exercise. The patient denied any recent illness or exposure to sick people, had not traveled outside the United States, and had not been exposed to radiation of the chest wall. At the end of the second day of discomfort, the patient noted irregular palpitations with mild shortness of breath and was transported to the hospital for evaluation. He denied being a cigarette smoker or illicit drug user.
The patient had no history of MI or diabetes. The patient’s father had an MI in his 80s, and two uncles died suddenly in their 50s of “massive heart attacks.” His mother, who had died of sepsis of uncertain etiology approximately 10 days earlier, also had hypertension and hypercholesterolemia but no history of coronary artery disease (CAD). Both of the patient’s adult daughters had been diagnosed with celiac disease in the preceding three years. His elder daughter had also been diagnosed with type 1 diabetes within the past two years.
On examination, the patient was afebrile, with a blood pressure of 143/87 mm Hg; pulse, 53 beats/min; and respiratory rate, 17 breaths/min. The patient’s weight was 204 lb and his height, 75 in (BMI, 25.5). The patient was in no apparent distress. Head, eyes, ears, nose, and throat were unremarkable. There was no significant jugular venous distention. The carotid pulses were full, and no bruits were appreciated. S1 and S2 sounds were within normal limits. No murmurs or S3 or S4 gallops were appreciated. The chest was clear on auscultation. Results of the abdominal exam were negative, no edema was noted in the extremities, and pulses were symmetrical.
ECG demonstrated subtle ST-segment elevation in leads I and aVL with a prominent R wave in lead V1. This pattern was interpreted as consistent with an acute inferolateral MI. A baseline ECG, previously obtained by the patient’s internist, had been interpreted as normal.
Peak troponin level was 55 ng/mL (normal, < 0.03 ng/mL); total creatine kinase (CK), 807 U/L (reference range, 20 to 259 U/L); and mass CK-MB fraction, 44 ng/mL (0.1 to 6.6 ng/mL). Total cholesterol was 105 mg/dL, with both LDL- and HDL-cholesterol fractions at 46 mg/dL. A complete blood count without differential revealed a total white blood cell count of 53,000/µL. Hemoglobin and hematocrit were both low (12.3 g/dL and 34.5%, respectively). All indices were within normal limits, as was the platelet count. Glucose, blood urea nitrogen, creatinine, potassium chloride bicarbonate, and calcium were all within normal limits. The sodium level was slightly low (132 mEq/L). Emergency catheterization revealed an ejection fraction of 45% (reference range, 55% to 70%), with mild-to-moderate diffuse hypokinesis but normal coronary arteries.
The patient was diagnosed with myocarditis, likely of viral origin.
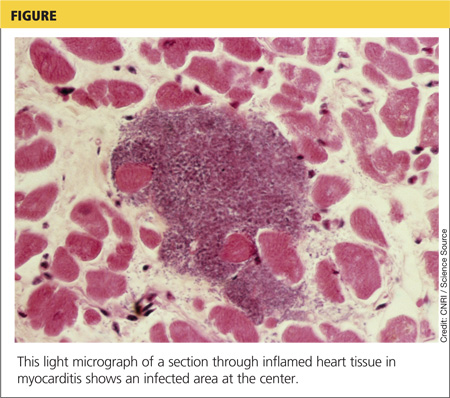
DISCUSSION
Although the incidence of myocarditis in the US is difficult to assess, autopsy reports implicate it in 8.6% to 12% of cases of sudden cardiac death in young adults,1,2 and a large prospective series implicated myocarditis in 9% of cases of dilated cardiomyopathy.3 Myocarditis is considered to be at one extreme of a spectrum of perimyocardial processes that result in inflammation of the myocardium (see figure), pericardium, or both.4
The underlying pathology involves an acute injury to the myocyte. This activates the innate and humoral immune systems, resulting in severe inflammation. The immune reaction eventually subsides, and the myocardium recovers. In certain patients, however, myocardial inflammation persists, resulting in ongoing myocyte damage, relentless symptomatic heart failure, or even death.5
Although a variety of diagnostic criteria have been developed and employed, the diagnosis of myocarditis is often one of exclusion. First proposed in 1986, the Dallas criteria—a histopathologic classification for myocarditis diagnosis—are based on endomyocardial biopsy, with inflammatory cellular infiltrate (with or without associated myocyte necrosis) visible on conventionally stained myocardial tissue sections.5 However, this method poses significant practical limitations, including low sensitivity (43% to 64%) and complication and death rates of 6% and 0.4%, respectively.5,6
An empiric diagnosis of myocarditis is often based on a combination of clinical findings including altered ECG, increase in myocardial enzymes, and lack of significant CAD.6 The recommended diagnostic cardiac magnetic resonance (CMR) imaging criteria for clinically suspected myocardial inflammation (ie, the Lake Louise Criteria) include at least two of the following:7
• Regional or global myocardial signal intensity increase in T2-weighted images.
• Increased global myocardial early gadolinium enhancement ratio between myocardium and skeletal muscle in gadolinium-enhanced T1-weighted images.
• At least one focal lesion with nonischemic regional distribution in inversion recovery–prepared gadolinium-enhanced T1-weighted images (“late gadolinium enhancement”).
Because of its reported high sensitivity and specificity (100% and 90%, respectively), CMR was used in the case patient to confirm the diagnosis of myocarditis.8 Specifically, CMR with contrast demonstrated normal left ventricular cavity size and mild reduction in overall left ventricular systolic function, with a visually estimated left ventricular ejection fraction of 45% to 50%. Regional hypokinesis of the mid-inferior wall and apical inferior septum was noted. Delayed contrast imaging demonstrated extensive non-CAD scarring and fibrosis, involving the basal anterior wall, basal inferior wall, and basal and midlateral wall in a pattern consistent with acute myocarditis.
Just as there is variability in the specific criteria by which the diagnosis of myocarditis can be made, the array of clinical findings with which it can manifest range from fatigue and other nonspecific symptoms to fulminant congestive heart failure and sudden death.6 Often, but not always, a viral prodrome precedes the onset of “cardiac symptoms” (eg, chest pain, dyspnea, palpitations, or syncope).5 This patient’s multiple risk factors for CAD and a suggestive, albeit atypical, history of chest discomfort, palpitations, and shortness of breath helped to focus the clinicians’ evaluation on the heart.
Potential Causes
Once a diagnosis of myocarditis is rendered, the next challenge is distinguishing its specific source from a plethora of potential etiologies, including infection, toxic exposure, or hypersensitivity/autoimmune reaction. Viral infections (mostly herpes, parvovirus, and cytomegalovirus) are thought to cause most cases of myocarditis in developed countries.5,9
Viral myocarditis results when viruses enter cardiac myocytes and incite a cytotoxic effect with activation of the immune response, including expression of interferon , natural killer cells, and release of nitric oxide. The majority of patients recover, but some develop an adaptive immune response, which further causes cardiac damage. In this response, antibodies to viral and to some cardiac proteins are produced, and effector T lymphocytes proliferate. Viral genome or inflammatory mechanisms may persist, contributing to ventricular dysfunction leading to heart failure and arrhythmias.10
Celiac disease is a chronic gastroenterologic disease caused by an immune response to a gluten protein. Damage to the brush border of the small intestine results in an inability to absorb fat, protein, vitamins, and minerals. Intermittent diarrhea, abdominal pain, and bloating are most commonly reported, but celiac disease may also manifest less obviously with iron deficiency anemia, joint pain, muscle cramps, osteoporosis, and neuropathy.11 Iron deficiency anemia that is refractory to iron replacement may offer insight into diagnosing myocarditis due to celiac disease.12 Although studies have found that more than 4% of patients with myocarditis also had celiac disease, none had the classic GI symptoms of celiac disease.12
Takotsubo cardiomyopathy is a transient left ventricular apical ballooning syndrome of unknown etiology. (For more information, see Fasolino T. Takotsubo cardiomyopathy: a clinical overview). Patients who have experienced emotional or physiologic stress and postmenopausal women appear to be at greatest risk. The clinical symptoms mimic MI, including chest pain with ST-segment elevation in the precordial leads on ECG13 and minor elevation of the cardiac enzyme and biomarker levels.14 However, patients experiencing this stress cardiomyopathy lack evidence of atherosclerotic CAD.15 An echocardiogram or CMR imaging reveals characteristic wall motion hypokinesis, akinesis, or dyskinesis of the left ventricular apex and mid-ventricle that help to differentiate it from other forms of myocarditis.15,16 Patient prognosis is favorable, with 95% of patients experiencing a full recovery; left ventricular dysfunction usually begins to improve in a few weeks.13,14
Sarcoidosis is a systemic disease resulting in noncaseating granulomas in multiple organs.17 Initial presentation typically includes bilateral hilar adenopathy, pulmonary reticular opacities, and/or skin, joint, or eye lesions.18 Patients with cardiac sarcoidosis most commonly present with conduction disturbances and ventricular arrhythmias.17 Although frequently absent, clinical symptoms may include palpitations, syncope, dizziness, or chest pain and clinical heart failure.17,18 It is difficult to distinguish cardiac sarcoidosis from other forms of myocarditis unless signs of systemic sarcoidosis are evident. A patient with suspected cardiac sarcoidosis should have an ECG to detect subclinical conduction abnormalities.17 The patient should wear a Holter monitor for 24 hours to screen for cardiac involvement, and echocardiography should be performed to define cardiac abnormalities.19
Giant-cell myocarditis (GCM) is a rare, rapidly progressive, and frequently fatal myocardial disease. Based on endomyocardial or surgical biopsy, GCM is histologically defined by multinucleated giant cells, a lymphocytic inflammatory infiltrate, and myocyte necrosis. It is often found in association with various immune-related systemic disorders.20 Patients present with heart failure, ventricular arrhythmias, and atrioventricular block that fails to improve with standard therapy.21
Treatment and Management
The typical management of acute myocarditis includes supportive care for left ventricular dysfunction and arrhythmia control.22 Many of the standard heart failure therapies—β-blockers, ACE inhibitors, angiotensin receptor blockers, and aldosterone antagonists—are efficacious; several, at least in animal models, appear to exert anti-inflammatory as well as the standard cardiovascular effects.23
Caution is advised regarding the selection of specific therapies. For example, in one study, metoprolol produced deleterious effects in acute murine Coxsackie virus myocarditis; inflammation, necrosis, and mortality significantly increased in the treatment group, compared with the placebo group.23
Information on the effects of particular therapies for specific etiologies of myocarditis are limited, but some evidence supports immunosuppressive and immune-modulating therapies for chronic, virus-negative inflammatory cardiomyopathy. Immunosuppressive therapy is also beneficial for acute GCM and sarcoidosis.23 For patients with myocarditis associated with celiac disease, a gluten-free diet alone or in combination with immunosuppressive agents can significantly improve clinical outcomes.12
OUTCOME FOR THE CASE PATIENT
Because the patient was already taking a statin and an ACE inhibitor for hypercholesterolemia and hypertension, respectively, as well as one baby aspirin per day, only a β-blocker was added to his discharge medication regimen.
Three months after hospital discharge, the patient underwent repeat CMR imaging. The ejection fraction had markedly improved to the 55%-to-60% range, although extensive midmyocardial-to-epicardial scarring in a multifocal pattern, primarily involving the basilar anterior and anterolateral wall, was still present, as was a small focus of an active (albeit healing) process in the inferior wall. Clinically, the patient was doing reasonably well and was vigorously exercising daily without dizziness, syncope, chest discomfort, or shortness of breath.
However, within several weeks of discharge, the patient reported having one two-hour episode of frequent palpitations at rest. Since that episode, palpitations have occurred infrequently. A 48-hour Holter monitor was ordered to better evaluate the palpitations and showed only rare premature ventricular contractions and isolated premature atrial contractions; no complex ectopy was noted. A follow-up stress echocardiogram was scheduled for 12 months, assuming the patient was free of clinical signs and symptoms of heart failure and arrhythmias at that time.
CONCLUSION
Myocarditis can manifest with a broad spectrum of signs and symptoms that may make its identification difficult, especially if a cardiac source is not initially considered in the differential diagnosis. However, for patients who present with elevated biomarkers and normal coronary artery anatomy, the identification of myocarditis is relatively easy; the difficulty in this circumstance relates to the identification of the specific etiology of the myocarditis.
The long-term prognosis for myocarditis is frequently good and the treatment straightforward, using medications that are modeled after standard heart failure therapy. However, depending on the etiology, specific treatment may be advisable—or required—in order to improve outcomes.
References
1. Fabre A, Sheppard MN. Sudden adult death syndrome and other nonischaemic causes of sudden cardiac death: a UK experience. Heart. 2006;92:316-320.
2. Doolan A, Semsarian C, Langlois N. Causes of sudden cardiac death in young Australians. Med J Aust. 2004;180:110-112.
3. Felker GM, Hu W, Hare JM, Hruban RH, et al. The spectrum of dilated cardiomyopathy: the Johns Hopkins experience with 1,278 patients. Medicine (Baltimore). 1999;78:270-283.
4. Leitman M, Tyomkin V, Peleg E, et al. Left ventricular function in acute inflammatory peri-myocardial diseases—new insights and long-term follow-up. Cardiovasc Ultrasound. 2012;10:42.
5. Blauwet LA, Cooper LT. Myocarditis. Prog Cardiovasc Dis. 2010;52:274-288.
6. Testani JM, Kolansky DM, Litt H, Gerstenfeld EP. Focal myocarditis mimicking acute ST-elevation myocardial infarction: diagnosis using cardiac magnetic resonance imaging. Tex Heart Inst J. 2006;33:256-259.
7. Friedrich MG, Sechtem U, Schulz-Menger J, et al. Cardiovascular magnetic resonance in myocarditis: a JACC white paper. J Am Coll Cardiol. 2009;53: 1475-1487.
8. Olimulder MA, van Es J, Galjee MA. The importance of cardiac MRI as a diagnostic tool in viral myocarditis-induced cardiomyopathy. Neth Heart J. 2009;17:481-486.
9. Mavrogeni S, Bratis K, Markussis V, et al. The diagnostic role of cardiac magnetic resonance imaging in detecting myocardial inflammation in systemic lupus erythematosus. Differentiation from viral myocarditis. Lupus. 2013;22:34-43.
10. Schultz JC, Hilliard AA, Cooper LT, Rihal CS. Diagnosis and treatment of viral myocarditis. Mayo Clin Proc. 2009;84:1001-1009.
11. Schuppan D, Dieterich W. Pathogenesis, epidemiology, and clinical manifestations of celiac disease in adults (2013). www.uptodate.com/contents/pathogenesis-epidemiology-and-clinical-manifestations-of-celiac-disease-in-adults. Accessed November 14, 2013.
12. Frustaci A, Cuoco L, Chimenti C, et al. Celiac disease associated with autoimmune myocarditis. Circulation. 2002;105:2611-2618.
13. Thakar S, Chandra P, Hollander G, Lichstein E. Electrocardiographic changes in Takotsubo cardiomyopathy. Pacing Clin Electrophysiol. 2011;34:
1278-1282.
14. Fefer P, Chelvanathan A, Dick A, et al. Takotsubo cardiomyopathy and left ventricular outflow tract obstruction. J Interv Cardiol. 2009;22:444-452.
15. Stensaeth KH, Fossum E, Hoffmann P, et al. Takotsubo cardiomyopathy in acute coronary syndrome; clinical features and contribution of cardiac magnetic resonance during the acute and convalescent phase. Scand Cardiovasc J. 2011;45:77-85.
16. Omerovic E. How to think about stress-induced cardiomyopathy?—Think “out of the box”! Scand Cardiovasc J. 2011;45:67-71.
17. McKenna WJ. Cardiac sarcoidosis (2013). www.uptodate.com/contents/cardiac-sarcoidosis. Accessed November 14, 2013.
18. King TE Jr. Clinical manifestations and diagnosis of sarcoidosis (2013). www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-sarcoidosis. Accessed November 14, 2013.
19. Bussinguer M, Danielian A, Sharma O. Cardiac sarcoidosis: diagnosis and management. Curr Treat Options Cardiovasc Med. 2012;14:652-664.
20. Cooper LT Jr, Berry GJ, Shabetai R; Multicenter Giant Cell Myocarditis Study Group Investigators. Idiopathic giant-cell myocarditis—natural history and treatment. N Engl J Med. 1997;336(26):1860-1866.
21. Kandolin R, Lehtonen J, Salmenkivi K, et al. Diagnosis, treatment, and outcome of giant-cell myocarditis in the era of combined immunosuppression. Circ Heart Fail. 2013;6:15-22.
22. Htwe TH, Khardori NM. Cardiac emergencies: infective endocarditis, pericarditis, and myocarditis. Med Clin North Am. 2012;96:1149-1169.
23. Kindermann I, Barth C, Mahfoud F, et al. Update on myocarditis. J Am Coll Cardiol. 2012;59:779-792.
A 55-year-old white man with controlled hypertension and hypercholesterolemia awoke with mild chest discomfort that he believed was mild gastroesophageal reflux. He denied radiation of pain to the shoulders, arms, back, or neck; dyspnea; palpitations; diaphoresis; nausea/vomiting; cough; or fever, during the first 30 hours of discomfort. There was no change in discomfort with deep breath, palpation of the chest, or administration of antacids. Minimal, short-lived improvement was noted with belching.
The patient had no trouble sleeping in the prone position and did not notice an increase in discomfort or unusual difficulty during his daily vigorous 30-minute aerobic workout. In fact, his symptoms seemed to improve or disappear during exercise. The patient denied any recent illness or exposure to sick people, had not traveled outside the United States, and had not been exposed to radiation of the chest wall. At the end of the second day of discomfort, the patient noted irregular palpitations with mild shortness of breath and was transported to the hospital for evaluation. He denied being a cigarette smoker or illicit drug user.
The patient had no history of MI or diabetes. The patient’s father had an MI in his 80s, and two uncles died suddenly in their 50s of “massive heart attacks.” His mother, who had died of sepsis of uncertain etiology approximately 10 days earlier, also had hypertension and hypercholesterolemia but no history of coronary artery disease (CAD). Both of the patient’s adult daughters had been diagnosed with celiac disease in the preceding three years. His elder daughter had also been diagnosed with type 1 diabetes within the past two years.
On examination, the patient was afebrile, with a blood pressure of 143/87 mm Hg; pulse, 53 beats/min; and respiratory rate, 17 breaths/min. The patient’s weight was 204 lb and his height, 75 in (BMI, 25.5). The patient was in no apparent distress. Head, eyes, ears, nose, and throat were unremarkable. There was no significant jugular venous distention. The carotid pulses were full, and no bruits were appreciated. S1 and S2 sounds were within normal limits. No murmurs or S3 or S4 gallops were appreciated. The chest was clear on auscultation. Results of the abdominal exam were negative, no edema was noted in the extremities, and pulses were symmetrical.
ECG demonstrated subtle ST-segment elevation in leads I and aVL with a prominent R wave in lead V1. This pattern was interpreted as consistent with an acute inferolateral MI. A baseline ECG, previously obtained by the patient’s internist, had been interpreted as normal.
Peak troponin level was 55 ng/mL (normal, < 0.03 ng/mL); total creatine kinase (CK), 807 U/L (reference range, 20 to 259 U/L); and mass CK-MB fraction, 44 ng/mL (0.1 to 6.6 ng/mL). Total cholesterol was 105 mg/dL, with both LDL- and HDL-cholesterol fractions at 46 mg/dL. A complete blood count without differential revealed a total white blood cell count of 53,000/µL. Hemoglobin and hematocrit were both low (12.3 g/dL and 34.5%, respectively). All indices were within normal limits, as was the platelet count. Glucose, blood urea nitrogen, creatinine, potassium chloride bicarbonate, and calcium were all within normal limits. The sodium level was slightly low (132 mEq/L). Emergency catheterization revealed an ejection fraction of 45% (reference range, 55% to 70%), with mild-to-moderate diffuse hypokinesis but normal coronary arteries.
The patient was diagnosed with myocarditis, likely of viral origin.
DISCUSSION
Although the incidence of myocarditis in the US is difficult to assess, autopsy reports implicate it in 8.6% to 12% of cases of sudden cardiac death in young adults,1,2 and a large prospective series implicated myocarditis in 9% of cases of dilated cardiomyopathy.3 Myocarditis is considered to be at one extreme of a spectrum of perimyocardial processes that result in inflammation of the myocardium (see figure), pericardium, or both.4
The underlying pathology involves an acute injury to the myocyte. This activates the innate and humoral immune systems, resulting in severe inflammation. The immune reaction eventually subsides, and the myocardium recovers. In certain patients, however, myocardial inflammation persists, resulting in ongoing myocyte damage, relentless symptomatic heart failure, or even death.5
Although a variety of diagnostic criteria have been developed and employed, the diagnosis of myocarditis is often one of exclusion. First proposed in 1986, the Dallas criteria—a histopathologic classification for myocarditis diagnosis—are based on endomyocardial biopsy, with inflammatory cellular infiltrate (with or without associated myocyte necrosis) visible on conventionally stained myocardial tissue sections.5 However, this method poses significant practical limitations, including low sensitivity (43% to 64%) and complication and death rates of 6% and 0.4%, respectively.5,6
An empiric diagnosis of myocarditis is often based on a combination of clinical findings including altered ECG, increase in myocardial enzymes, and lack of significant CAD.6 The recommended diagnostic cardiac magnetic resonance (CMR) imaging criteria for clinically suspected myocardial inflammation (ie, the Lake Louise Criteria) include at least two of the following:7
• Regional or global myocardial signal intensity increase in T2-weighted images.
• Increased global myocardial early gadolinium enhancement ratio between myocardium and skeletal muscle in gadolinium-enhanced T1-weighted images.
• At least one focal lesion with nonischemic regional distribution in inversion recovery–prepared gadolinium-enhanced T1-weighted images (“late gadolinium enhancement”).
Because of its reported high sensitivity and specificity (100% and 90%, respectively), CMR was used in the case patient to confirm the diagnosis of myocarditis.8 Specifically, CMR with contrast demonstrated normal left ventricular cavity size and mild reduction in overall left ventricular systolic function, with a visually estimated left ventricular ejection fraction of 45% to 50%. Regional hypokinesis of the mid-inferior wall and apical inferior septum was noted. Delayed contrast imaging demonstrated extensive non-CAD scarring and fibrosis, involving the basal anterior wall, basal inferior wall, and basal and midlateral wall in a pattern consistent with acute myocarditis.
Just as there is variability in the specific criteria by which the diagnosis of myocarditis can be made, the array of clinical findings with which it can manifest range from fatigue and other nonspecific symptoms to fulminant congestive heart failure and sudden death.6 Often, but not always, a viral prodrome precedes the onset of “cardiac symptoms” (eg, chest pain, dyspnea, palpitations, or syncope).5 This patient’s multiple risk factors for CAD and a suggestive, albeit atypical, history of chest discomfort, palpitations, and shortness of breath helped to focus the clinicians’ evaluation on the heart.
Potential Causes
Once a diagnosis of myocarditis is rendered, the next challenge is distinguishing its specific source from a plethora of potential etiologies, including infection, toxic exposure, or hypersensitivity/autoimmune reaction. Viral infections (mostly herpes, parvovirus, and cytomegalovirus) are thought to cause most cases of myocarditis in developed countries.5,9
Viral myocarditis results when viruses enter cardiac myocytes and incite a cytotoxic effect with activation of the immune response, including expression of interferon , natural killer cells, and release of nitric oxide. The majority of patients recover, but some develop an adaptive immune response, which further causes cardiac damage. In this response, antibodies to viral and to some cardiac proteins are produced, and effector T lymphocytes proliferate. Viral genome or inflammatory mechanisms may persist, contributing to ventricular dysfunction leading to heart failure and arrhythmias.10
Celiac disease is a chronic gastroenterologic disease caused by an immune response to a gluten protein. Damage to the brush border of the small intestine results in an inability to absorb fat, protein, vitamins, and minerals. Intermittent diarrhea, abdominal pain, and bloating are most commonly reported, but celiac disease may also manifest less obviously with iron deficiency anemia, joint pain, muscle cramps, osteoporosis, and neuropathy.11 Iron deficiency anemia that is refractory to iron replacement may offer insight into diagnosing myocarditis due to celiac disease.12 Although studies have found that more than 4% of patients with myocarditis also had celiac disease, none had the classic GI symptoms of celiac disease.12
Takotsubo cardiomyopathy is a transient left ventricular apical ballooning syndrome of unknown etiology. (For more information, see Fasolino T. Takotsubo cardiomyopathy: a clinical overview). Patients who have experienced emotional or physiologic stress and postmenopausal women appear to be at greatest risk. The clinical symptoms mimic MI, including chest pain with ST-segment elevation in the precordial leads on ECG13 and minor elevation of the cardiac enzyme and biomarker levels.14 However, patients experiencing this stress cardiomyopathy lack evidence of atherosclerotic CAD.15 An echocardiogram or CMR imaging reveals characteristic wall motion hypokinesis, akinesis, or dyskinesis of the left ventricular apex and mid-ventricle that help to differentiate it from other forms of myocarditis.15,16 Patient prognosis is favorable, with 95% of patients experiencing a full recovery; left ventricular dysfunction usually begins to improve in a few weeks.13,14
Sarcoidosis is a systemic disease resulting in noncaseating granulomas in multiple organs.17 Initial presentation typically includes bilateral hilar adenopathy, pulmonary reticular opacities, and/or skin, joint, or eye lesions.18 Patients with cardiac sarcoidosis most commonly present with conduction disturbances and ventricular arrhythmias.17 Although frequently absent, clinical symptoms may include palpitations, syncope, dizziness, or chest pain and clinical heart failure.17,18 It is difficult to distinguish cardiac sarcoidosis from other forms of myocarditis unless signs of systemic sarcoidosis are evident. A patient with suspected cardiac sarcoidosis should have an ECG to detect subclinical conduction abnormalities.17 The patient should wear a Holter monitor for 24 hours to screen for cardiac involvement, and echocardiography should be performed to define cardiac abnormalities.19
Giant-cell myocarditis (GCM) is a rare, rapidly progressive, and frequently fatal myocardial disease. Based on endomyocardial or surgical biopsy, GCM is histologically defined by multinucleated giant cells, a lymphocytic inflammatory infiltrate, and myocyte necrosis. It is often found in association with various immune-related systemic disorders.20 Patients present with heart failure, ventricular arrhythmias, and atrioventricular block that fails to improve with standard therapy.21
Treatment and Management
The typical management of acute myocarditis includes supportive care for left ventricular dysfunction and arrhythmia control.22 Many of the standard heart failure therapies—β-blockers, ACE inhibitors, angiotensin receptor blockers, and aldosterone antagonists—are efficacious; several, at least in animal models, appear to exert anti-inflammatory as well as the standard cardiovascular effects.23
Caution is advised regarding the selection of specific therapies. For example, in one study, metoprolol produced deleterious effects in acute murine Coxsackie virus myocarditis; inflammation, necrosis, and mortality significantly increased in the treatment group, compared with the placebo group.23
Information on the effects of particular therapies for specific etiologies of myocarditis are limited, but some evidence supports immunosuppressive and immune-modulating therapies for chronic, virus-negative inflammatory cardiomyopathy. Immunosuppressive therapy is also beneficial for acute GCM and sarcoidosis.23 For patients with myocarditis associated with celiac disease, a gluten-free diet alone or in combination with immunosuppressive agents can significantly improve clinical outcomes.12
OUTCOME FOR THE CASE PATIENT
Because the patient was already taking a statin and an ACE inhibitor for hypercholesterolemia and hypertension, respectively, as well as one baby aspirin per day, only a β-blocker was added to his discharge medication regimen.
Three months after hospital discharge, the patient underwent repeat CMR imaging. The ejection fraction had markedly improved to the 55%-to-60% range, although extensive midmyocardial-to-epicardial scarring in a multifocal pattern, primarily involving the basilar anterior and anterolateral wall, was still present, as was a small focus of an active (albeit healing) process in the inferior wall. Clinically, the patient was doing reasonably well and was vigorously exercising daily without dizziness, syncope, chest discomfort, or shortness of breath.
However, within several weeks of discharge, the patient reported having one two-hour episode of frequent palpitations at rest. Since that episode, palpitations have occurred infrequently. A 48-hour Holter monitor was ordered to better evaluate the palpitations and showed only rare premature ventricular contractions and isolated premature atrial contractions; no complex ectopy was noted. A follow-up stress echocardiogram was scheduled for 12 months, assuming the patient was free of clinical signs and symptoms of heart failure and arrhythmias at that time.
CONCLUSION
Myocarditis can manifest with a broad spectrum of signs and symptoms that may make its identification difficult, especially if a cardiac source is not initially considered in the differential diagnosis. However, for patients who present with elevated biomarkers and normal coronary artery anatomy, the identification of myocarditis is relatively easy; the difficulty in this circumstance relates to the identification of the specific etiology of the myocarditis.
The long-term prognosis for myocarditis is frequently good and the treatment straightforward, using medications that are modeled after standard heart failure therapy. However, depending on the etiology, specific treatment may be advisable—or required—in order to improve outcomes.
References
1. Fabre A, Sheppard MN. Sudden adult death syndrome and other nonischaemic causes of sudden cardiac death: a UK experience. Heart. 2006;92:316-320.
2. Doolan A, Semsarian C, Langlois N. Causes of sudden cardiac death in young Australians. Med J Aust. 2004;180:110-112.
3. Felker GM, Hu W, Hare JM, Hruban RH, et al. The spectrum of dilated cardiomyopathy: the Johns Hopkins experience with 1,278 patients. Medicine (Baltimore). 1999;78:270-283.
4. Leitman M, Tyomkin V, Peleg E, et al. Left ventricular function in acute inflammatory peri-myocardial diseases—new insights and long-term follow-up. Cardiovasc Ultrasound. 2012;10:42.
5. Blauwet LA, Cooper LT. Myocarditis. Prog Cardiovasc Dis. 2010;52:274-288.
6. Testani JM, Kolansky DM, Litt H, Gerstenfeld EP. Focal myocarditis mimicking acute ST-elevation myocardial infarction: diagnosis using cardiac magnetic resonance imaging. Tex Heart Inst J. 2006;33:256-259.
7. Friedrich MG, Sechtem U, Schulz-Menger J, et al. Cardiovascular magnetic resonance in myocarditis: a JACC white paper. J Am Coll Cardiol. 2009;53: 1475-1487.
8. Olimulder MA, van Es J, Galjee MA. The importance of cardiac MRI as a diagnostic tool in viral myocarditis-induced cardiomyopathy. Neth Heart J. 2009;17:481-486.
9. Mavrogeni S, Bratis K, Markussis V, et al. The diagnostic role of cardiac magnetic resonance imaging in detecting myocardial inflammation in systemic lupus erythematosus. Differentiation from viral myocarditis. Lupus. 2013;22:34-43.
10. Schultz JC, Hilliard AA, Cooper LT, Rihal CS. Diagnosis and treatment of viral myocarditis. Mayo Clin Proc. 2009;84:1001-1009.
11. Schuppan D, Dieterich W. Pathogenesis, epidemiology, and clinical manifestations of celiac disease in adults (2013). www.uptodate.com/contents/pathogenesis-epidemiology-and-clinical-manifestations-of-celiac-disease-in-adults. Accessed November 14, 2013.
12. Frustaci A, Cuoco L, Chimenti C, et al. Celiac disease associated with autoimmune myocarditis. Circulation. 2002;105:2611-2618.
13. Thakar S, Chandra P, Hollander G, Lichstein E. Electrocardiographic changes in Takotsubo cardiomyopathy. Pacing Clin Electrophysiol. 2011;34:
1278-1282.
14. Fefer P, Chelvanathan A, Dick A, et al. Takotsubo cardiomyopathy and left ventricular outflow tract obstruction. J Interv Cardiol. 2009;22:444-452.
15. Stensaeth KH, Fossum E, Hoffmann P, et al. Takotsubo cardiomyopathy in acute coronary syndrome; clinical features and contribution of cardiac magnetic resonance during the acute and convalescent phase. Scand Cardiovasc J. 2011;45:77-85.
16. Omerovic E. How to think about stress-induced cardiomyopathy?—Think “out of the box”! Scand Cardiovasc J. 2011;45:67-71.
17. McKenna WJ. Cardiac sarcoidosis (2013). www.uptodate.com/contents/cardiac-sarcoidosis. Accessed November 14, 2013.
18. King TE Jr. Clinical manifestations and diagnosis of sarcoidosis (2013). www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-sarcoidosis. Accessed November 14, 2013.
19. Bussinguer M, Danielian A, Sharma O. Cardiac sarcoidosis: diagnosis and management. Curr Treat Options Cardiovasc Med. 2012;14:652-664.
20. Cooper LT Jr, Berry GJ, Shabetai R; Multicenter Giant Cell Myocarditis Study Group Investigators. Idiopathic giant-cell myocarditis—natural history and treatment. N Engl J Med. 1997;336(26):1860-1866.
21. Kandolin R, Lehtonen J, Salmenkivi K, et al. Diagnosis, treatment, and outcome of giant-cell myocarditis in the era of combined immunosuppression. Circ Heart Fail. 2013;6:15-22.
22. Htwe TH, Khardori NM. Cardiac emergencies: infective endocarditis, pericarditis, and myocarditis. Med Clin North Am. 2012;96:1149-1169.
23. Kindermann I, Barth C, Mahfoud F, et al. Update on myocarditis. J Am Coll Cardiol. 2012;59:779-792.
A 55-year-old white man with controlled hypertension and hypercholesterolemia awoke with mild chest discomfort that he believed was mild gastroesophageal reflux. He denied radiation of pain to the shoulders, arms, back, or neck; dyspnea; palpitations; diaphoresis; nausea/vomiting; cough; or fever, during the first 30 hours of discomfort. There was no change in discomfort with deep breath, palpation of the chest, or administration of antacids. Minimal, short-lived improvement was noted with belching.
The patient had no trouble sleeping in the prone position and did not notice an increase in discomfort or unusual difficulty during his daily vigorous 30-minute aerobic workout. In fact, his symptoms seemed to improve or disappear during exercise. The patient denied any recent illness or exposure to sick people, had not traveled outside the United States, and had not been exposed to radiation of the chest wall. At the end of the second day of discomfort, the patient noted irregular palpitations with mild shortness of breath and was transported to the hospital for evaluation. He denied being a cigarette smoker or illicit drug user.
The patient had no history of MI or diabetes. The patient’s father had an MI in his 80s, and two uncles died suddenly in their 50s of “massive heart attacks.” His mother, who had died of sepsis of uncertain etiology approximately 10 days earlier, also had hypertension and hypercholesterolemia but no history of coronary artery disease (CAD). Both of the patient’s adult daughters had been diagnosed with celiac disease in the preceding three years. His elder daughter had also been diagnosed with type 1 diabetes within the past two years.
On examination, the patient was afebrile, with a blood pressure of 143/87 mm Hg; pulse, 53 beats/min; and respiratory rate, 17 breaths/min. The patient’s weight was 204 lb and his height, 75 in (BMI, 25.5). The patient was in no apparent distress. Head, eyes, ears, nose, and throat were unremarkable. There was no significant jugular venous distention. The carotid pulses were full, and no bruits were appreciated. S1 and S2 sounds were within normal limits. No murmurs or S3 or S4 gallops were appreciated. The chest was clear on auscultation. Results of the abdominal exam were negative, no edema was noted in the extremities, and pulses were symmetrical.
ECG demonstrated subtle ST-segment elevation in leads I and aVL with a prominent R wave in lead V1. This pattern was interpreted as consistent with an acute inferolateral MI. A baseline ECG, previously obtained by the patient’s internist, had been interpreted as normal.
Peak troponin level was 55 ng/mL (normal, < 0.03 ng/mL); total creatine kinase (CK), 807 U/L (reference range, 20 to 259 U/L); and mass CK-MB fraction, 44 ng/mL (0.1 to 6.6 ng/mL). Total cholesterol was 105 mg/dL, with both LDL- and HDL-cholesterol fractions at 46 mg/dL. A complete blood count without differential revealed a total white blood cell count of 53,000/µL. Hemoglobin and hematocrit were both low (12.3 g/dL and 34.5%, respectively). All indices were within normal limits, as was the platelet count. Glucose, blood urea nitrogen, creatinine, potassium chloride bicarbonate, and calcium were all within normal limits. The sodium level was slightly low (132 mEq/L). Emergency catheterization revealed an ejection fraction of 45% (reference range, 55% to 70%), with mild-to-moderate diffuse hypokinesis but normal coronary arteries.
The patient was diagnosed with myocarditis, likely of viral origin.
DISCUSSION
Although the incidence of myocarditis in the US is difficult to assess, autopsy reports implicate it in 8.6% to 12% of cases of sudden cardiac death in young adults,1,2 and a large prospective series implicated myocarditis in 9% of cases of dilated cardiomyopathy.3 Myocarditis is considered to be at one extreme of a spectrum of perimyocardial processes that result in inflammation of the myocardium (see figure), pericardium, or both.4
The underlying pathology involves an acute injury to the myocyte. This activates the innate and humoral immune systems, resulting in severe inflammation. The immune reaction eventually subsides, and the myocardium recovers. In certain patients, however, myocardial inflammation persists, resulting in ongoing myocyte damage, relentless symptomatic heart failure, or even death.5
Although a variety of diagnostic criteria have been developed and employed, the diagnosis of myocarditis is often one of exclusion. First proposed in 1986, the Dallas criteria—a histopathologic classification for myocarditis diagnosis—are based on endomyocardial biopsy, with inflammatory cellular infiltrate (with or without associated myocyte necrosis) visible on conventionally stained myocardial tissue sections.5 However, this method poses significant practical limitations, including low sensitivity (43% to 64%) and complication and death rates of 6% and 0.4%, respectively.5,6
An empiric diagnosis of myocarditis is often based on a combination of clinical findings including altered ECG, increase in myocardial enzymes, and lack of significant CAD.6 The recommended diagnostic cardiac magnetic resonance (CMR) imaging criteria for clinically suspected myocardial inflammation (ie, the Lake Louise Criteria) include at least two of the following:7
• Regional or global myocardial signal intensity increase in T2-weighted images.
• Increased global myocardial early gadolinium enhancement ratio between myocardium and skeletal muscle in gadolinium-enhanced T1-weighted images.
• At least one focal lesion with nonischemic regional distribution in inversion recovery–prepared gadolinium-enhanced T1-weighted images (“late gadolinium enhancement”).
Because of its reported high sensitivity and specificity (100% and 90%, respectively), CMR was used in the case patient to confirm the diagnosis of myocarditis.8 Specifically, CMR with contrast demonstrated normal left ventricular cavity size and mild reduction in overall left ventricular systolic function, with a visually estimated left ventricular ejection fraction of 45% to 50%. Regional hypokinesis of the mid-inferior wall and apical inferior septum was noted. Delayed contrast imaging demonstrated extensive non-CAD scarring and fibrosis, involving the basal anterior wall, basal inferior wall, and basal and midlateral wall in a pattern consistent with acute myocarditis.
Just as there is variability in the specific criteria by which the diagnosis of myocarditis can be made, the array of clinical findings with which it can manifest range from fatigue and other nonspecific symptoms to fulminant congestive heart failure and sudden death.6 Often, but not always, a viral prodrome precedes the onset of “cardiac symptoms” (eg, chest pain, dyspnea, palpitations, or syncope).5 This patient’s multiple risk factors for CAD and a suggestive, albeit atypical, history of chest discomfort, palpitations, and shortness of breath helped to focus the clinicians’ evaluation on the heart.
Potential Causes
Once a diagnosis of myocarditis is rendered, the next challenge is distinguishing its specific source from a plethora of potential etiologies, including infection, toxic exposure, or hypersensitivity/autoimmune reaction. Viral infections (mostly herpes, parvovirus, and cytomegalovirus) are thought to cause most cases of myocarditis in developed countries.5,9
Viral myocarditis results when viruses enter cardiac myocytes and incite a cytotoxic effect with activation of the immune response, including expression of interferon , natural killer cells, and release of nitric oxide. The majority of patients recover, but some develop an adaptive immune response, which further causes cardiac damage. In this response, antibodies to viral and to some cardiac proteins are produced, and effector T lymphocytes proliferate. Viral genome or inflammatory mechanisms may persist, contributing to ventricular dysfunction leading to heart failure and arrhythmias.10
Celiac disease is a chronic gastroenterologic disease caused by an immune response to a gluten protein. Damage to the brush border of the small intestine results in an inability to absorb fat, protein, vitamins, and minerals. Intermittent diarrhea, abdominal pain, and bloating are most commonly reported, but celiac disease may also manifest less obviously with iron deficiency anemia, joint pain, muscle cramps, osteoporosis, and neuropathy.11 Iron deficiency anemia that is refractory to iron replacement may offer insight into diagnosing myocarditis due to celiac disease.12 Although studies have found that more than 4% of patients with myocarditis also had celiac disease, none had the classic GI symptoms of celiac disease.12
Takotsubo cardiomyopathy is a transient left ventricular apical ballooning syndrome of unknown etiology. (For more information, see Fasolino T. Takotsubo cardiomyopathy: a clinical overview). Patients who have experienced emotional or physiologic stress and postmenopausal women appear to be at greatest risk. The clinical symptoms mimic MI, including chest pain with ST-segment elevation in the precordial leads on ECG13 and minor elevation of the cardiac enzyme and biomarker levels.14 However, patients experiencing this stress cardiomyopathy lack evidence of atherosclerotic CAD.15 An echocardiogram or CMR imaging reveals characteristic wall motion hypokinesis, akinesis, or dyskinesis of the left ventricular apex and mid-ventricle that help to differentiate it from other forms of myocarditis.15,16 Patient prognosis is favorable, with 95% of patients experiencing a full recovery; left ventricular dysfunction usually begins to improve in a few weeks.13,14
Sarcoidosis is a systemic disease resulting in noncaseating granulomas in multiple organs.17 Initial presentation typically includes bilateral hilar adenopathy, pulmonary reticular opacities, and/or skin, joint, or eye lesions.18 Patients with cardiac sarcoidosis most commonly present with conduction disturbances and ventricular arrhythmias.17 Although frequently absent, clinical symptoms may include palpitations, syncope, dizziness, or chest pain and clinical heart failure.17,18 It is difficult to distinguish cardiac sarcoidosis from other forms of myocarditis unless signs of systemic sarcoidosis are evident. A patient with suspected cardiac sarcoidosis should have an ECG to detect subclinical conduction abnormalities.17 The patient should wear a Holter monitor for 24 hours to screen for cardiac involvement, and echocardiography should be performed to define cardiac abnormalities.19
Giant-cell myocarditis (GCM) is a rare, rapidly progressive, and frequently fatal myocardial disease. Based on endomyocardial or surgical biopsy, GCM is histologically defined by multinucleated giant cells, a lymphocytic inflammatory infiltrate, and myocyte necrosis. It is often found in association with various immune-related systemic disorders.20 Patients present with heart failure, ventricular arrhythmias, and atrioventricular block that fails to improve with standard therapy.21
Treatment and Management
The typical management of acute myocarditis includes supportive care for left ventricular dysfunction and arrhythmia control.22 Many of the standard heart failure therapies—β-blockers, ACE inhibitors, angiotensin receptor blockers, and aldosterone antagonists—are efficacious; several, at least in animal models, appear to exert anti-inflammatory as well as the standard cardiovascular effects.23
Caution is advised regarding the selection of specific therapies. For example, in one study, metoprolol produced deleterious effects in acute murine Coxsackie virus myocarditis; inflammation, necrosis, and mortality significantly increased in the treatment group, compared with the placebo group.23
Information on the effects of particular therapies for specific etiologies of myocarditis are limited, but some evidence supports immunosuppressive and immune-modulating therapies for chronic, virus-negative inflammatory cardiomyopathy. Immunosuppressive therapy is also beneficial for acute GCM and sarcoidosis.23 For patients with myocarditis associated with celiac disease, a gluten-free diet alone or in combination with immunosuppressive agents can significantly improve clinical outcomes.12
OUTCOME FOR THE CASE PATIENT
Because the patient was already taking a statin and an ACE inhibitor for hypercholesterolemia and hypertension, respectively, as well as one baby aspirin per day, only a β-blocker was added to his discharge medication regimen.
Three months after hospital discharge, the patient underwent repeat CMR imaging. The ejection fraction had markedly improved to the 55%-to-60% range, although extensive midmyocardial-to-epicardial scarring in a multifocal pattern, primarily involving the basilar anterior and anterolateral wall, was still present, as was a small focus of an active (albeit healing) process in the inferior wall. Clinically, the patient was doing reasonably well and was vigorously exercising daily without dizziness, syncope, chest discomfort, or shortness of breath.
However, within several weeks of discharge, the patient reported having one two-hour episode of frequent palpitations at rest. Since that episode, palpitations have occurred infrequently. A 48-hour Holter monitor was ordered to better evaluate the palpitations and showed only rare premature ventricular contractions and isolated premature atrial contractions; no complex ectopy was noted. A follow-up stress echocardiogram was scheduled for 12 months, assuming the patient was free of clinical signs and symptoms of heart failure and arrhythmias at that time.
CONCLUSION
Myocarditis can manifest with a broad spectrum of signs and symptoms that may make its identification difficult, especially if a cardiac source is not initially considered in the differential diagnosis. However, for patients who present with elevated biomarkers and normal coronary artery anatomy, the identification of myocarditis is relatively easy; the difficulty in this circumstance relates to the identification of the specific etiology of the myocarditis.
The long-term prognosis for myocarditis is frequently good and the treatment straightforward, using medications that are modeled after standard heart failure therapy. However, depending on the etiology, specific treatment may be advisable—or required—in order to improve outcomes.
References
1. Fabre A, Sheppard MN. Sudden adult death syndrome and other nonischaemic causes of sudden cardiac death: a UK experience. Heart. 2006;92:316-320.
2. Doolan A, Semsarian C, Langlois N. Causes of sudden cardiac death in young Australians. Med J Aust. 2004;180:110-112.
3. Felker GM, Hu W, Hare JM, Hruban RH, et al. The spectrum of dilated cardiomyopathy: the Johns Hopkins experience with 1,278 patients. Medicine (Baltimore). 1999;78:270-283.
4. Leitman M, Tyomkin V, Peleg E, et al. Left ventricular function in acute inflammatory peri-myocardial diseases—new insights and long-term follow-up. Cardiovasc Ultrasound. 2012;10:42.
5. Blauwet LA, Cooper LT. Myocarditis. Prog Cardiovasc Dis. 2010;52:274-288.
6. Testani JM, Kolansky DM, Litt H, Gerstenfeld EP. Focal myocarditis mimicking acute ST-elevation myocardial infarction: diagnosis using cardiac magnetic resonance imaging. Tex Heart Inst J. 2006;33:256-259.
7. Friedrich MG, Sechtem U, Schulz-Menger J, et al. Cardiovascular magnetic resonance in myocarditis: a JACC white paper. J Am Coll Cardiol. 2009;53: 1475-1487.
8. Olimulder MA, van Es J, Galjee MA. The importance of cardiac MRI as a diagnostic tool in viral myocarditis-induced cardiomyopathy. Neth Heart J. 2009;17:481-486.
9. Mavrogeni S, Bratis K, Markussis V, et al. The diagnostic role of cardiac magnetic resonance imaging in detecting myocardial inflammation in systemic lupus erythematosus. Differentiation from viral myocarditis. Lupus. 2013;22:34-43.
10. Schultz JC, Hilliard AA, Cooper LT, Rihal CS. Diagnosis and treatment of viral myocarditis. Mayo Clin Proc. 2009;84:1001-1009.
11. Schuppan D, Dieterich W. Pathogenesis, epidemiology, and clinical manifestations of celiac disease in adults (2013). www.uptodate.com/contents/pathogenesis-epidemiology-and-clinical-manifestations-of-celiac-disease-in-adults. Accessed November 14, 2013.
12. Frustaci A, Cuoco L, Chimenti C, et al. Celiac disease associated with autoimmune myocarditis. Circulation. 2002;105:2611-2618.
13. Thakar S, Chandra P, Hollander G, Lichstein E. Electrocardiographic changes in Takotsubo cardiomyopathy. Pacing Clin Electrophysiol. 2011;34:
1278-1282.
14. Fefer P, Chelvanathan A, Dick A, et al. Takotsubo cardiomyopathy and left ventricular outflow tract obstruction. J Interv Cardiol. 2009;22:444-452.
15. Stensaeth KH, Fossum E, Hoffmann P, et al. Takotsubo cardiomyopathy in acute coronary syndrome; clinical features and contribution of cardiac magnetic resonance during the acute and convalescent phase. Scand Cardiovasc J. 2011;45:77-85.
16. Omerovic E. How to think about stress-induced cardiomyopathy?—Think “out of the box”! Scand Cardiovasc J. 2011;45:67-71.
17. McKenna WJ. Cardiac sarcoidosis (2013). www.uptodate.com/contents/cardiac-sarcoidosis. Accessed November 14, 2013.
18. King TE Jr. Clinical manifestations and diagnosis of sarcoidosis (2013). www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-sarcoidosis. Accessed November 14, 2013.
19. Bussinguer M, Danielian A, Sharma O. Cardiac sarcoidosis: diagnosis and management. Curr Treat Options Cardiovasc Med. 2012;14:652-664.
20. Cooper LT Jr, Berry GJ, Shabetai R; Multicenter Giant Cell Myocarditis Study Group Investigators. Idiopathic giant-cell myocarditis—natural history and treatment. N Engl J Med. 1997;336(26):1860-1866.
21. Kandolin R, Lehtonen J, Salmenkivi K, et al. Diagnosis, treatment, and outcome of giant-cell myocarditis in the era of combined immunosuppression. Circ Heart Fail. 2013;6:15-22.
22. Htwe TH, Khardori NM. Cardiac emergencies: infective endocarditis, pericarditis, and myocarditis. Med Clin North Am. 2012;96:1149-1169.
23. Kindermann I, Barth C, Mahfoud F, et al. Update on myocarditis. J Am Coll Cardiol. 2012;59:779-792.
Woman, 45, With Red, Scaly Nipple
A 45-year-old woman noticed some redness and scaling around her right nipple. She applied peroxide and OTC antibiotic ointment for approximately seven months with mixed results. She sought medical attention when pain developed in the breast, along with some bloody discharge from the nipple (see Figure 1). Around that time, she also noticed three small nodules in the upper outer portion of the breast.
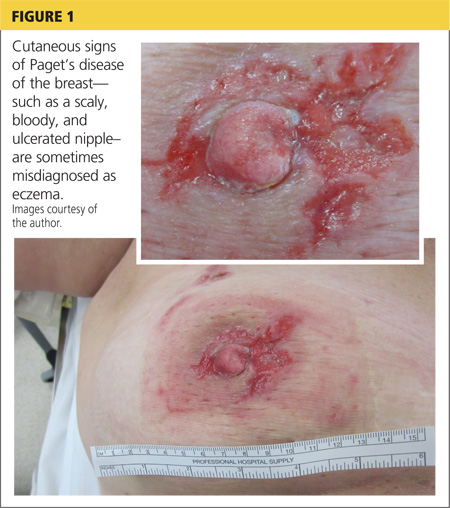
A mammogram and ultrasound revealed a 1.7 × 2.0–cm spiculated mass in the axillary tail, as well as two smaller breast lesions. A PET/CT scan ordered subsequently revealed intense uptake in the periareolar region and a suspicious axillary node. By then, the biopsy results had confirmed invasive ductal carcinoma, later determined to be Paget’s disease of the breast (PDB).
The patient’s previous medical history was significant for cystic breasts (never biopsied), chronic back pain, anxiety, and obesity. She was perimenopausal with irregular periods, the last one about 10 months ago. Her obstetric history included two pregnancies resulting in live births and no history of abortion; her menarche occurred at age 14 and her first pregnancy at 27. Family history was significant for leukemia in her maternal grandmother and niece. She did not use tobacco, alcohol, or illicit drugs. She lived at home with her husband and two daughters, who were all very supportive.
The patient elected to undergo a right modified radical mastectomy (MRM) and prophylactic left total mastectomy. MRM was performed on the right breast because sentinel lymph node identification was unsuccessful. This may have been due to involvement of the right subareolar plexus. Five of eight lymph nodes later tested positive for malignancy. The surgery was completed by placement of bilateral tissue expanders for eventual breast reconstruction.
Chemotherapy was started six weeks after surgery and included 15 weeks (five cycles) of docetaxel, carboplatin, and trastuzumab (a combination known as TCH), followed by 51 weeks (17 cycles) of trastuzumab, along with daily tamoxifen. The TCH regimen was followed by four weekly cycles of external beam radiation therapy (EBRT). Adverse effects of treatment have included chest wall dermatitis, right upper extremity lymphedema, nausea/vomiting, dyspnea, peripheral neuropathy, alopecia, and fatigue.
Discussion
Nearly 150 years ago, James Paget recognized a connection between skin changes around the nipple and deeper lesions in the breast.1 The disease that Paget identified is defined as the presence of intraepithelial adenocarcinoma cells (ie, Paget’s cells) within the epidermis of the nipple, with or without an underlying carcinoma.
An underlying breast cancer is present 85% to 95% of the time but is palpable in only approximately 50% of cases (see Figure 2). However, 25% of the time there is neither a palpable mass nor a mammographic abnormality. In these cases particularly, timely diagnosis depends on recognition of suspicious nipple changes, followed by a prompt and thorough diagnostic workup. Unfortunately, the accurate diagnosis of Paget’s disease still takes an average of several months.2
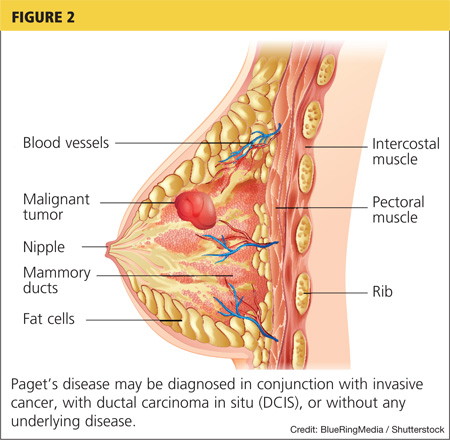
Paget’s disease is rare; it represents only 1% to 3% of new cases of female breast cancer, or about 2,250 cases a year.2-4 (The number of Paget’s disease cases per year was calculated by the author, based on the reported incidence of all breast cancers.) It is even more rare among men. For both genders, the peak age for this disease is between 50 and 60.2
Paget’s disease is an important entity for primary care PAs and NPs because it presents an opportunity to make a timely and life-changing diagnosis, and because it provides an elegant model for understanding current diagnostic and therapeutic approaches to breast cancer.
Clinical Presentation and Pathophysiology
The hallmark of PDB is scaly, vesicular, or ulcerated changes that begin in the nipple and spread to the areola. These changes are most often unilateral and may occur with pruritus, burning pain, and/or oozing from the nipple.5 This presentation is often mistaken for common skin conditions, such as eczema. Like eczema, changes in PDB may improve spontaneously and fluctuate over time, which is confusing for both the patient and clinician. A clinical pearl is that eczema is more likely to spread from the areola to the nipple, and will usually respond to topical corticosteroids. By contrast, changes in PDB tend to spread from the nipple to the areola, and corticosteroids do not provide a sustained response. Of note, Paget’s lesions may heal spontaneously even as the underlying malignancy progresses.6
PDB is unique because the underlying lesion and skin changes are not just coincidental. The cutaneous changes and the malignancy that lies beneath have a causal, not merely co-occurring, relationship. Paget himself believed that the nipple changes were both a precursor, and a promoter, of the underlying cancer.1 This transformation theory states that normal nipple epidermis turns into Paget’s cells spontaneously, before there is any underlying disease. This theory is supported by the fact that, occasionally (though rarely), no underlying breast cancer is ever found. Also, the concomitant tumor may be some distance (> 2 cm) from the nipple-areolar complex (NAC), suggesting a synchronous but causally unrelated lesion.6-8
Modern immunochemistry has turned PDB inside out. Today, PDB is believed to begin within the breast and then to spread “upward” to the NAC, called the epidermotrophic theory. This theory is supported by the fact that Paget’s cells share several molecular markers with their respective parenchymal tumors. Some researchers now propose that there is a single Paget’s progenitor cell with a motility factor that allows it to traverse the ductal system, resulting in nipple and skin changes that have come to be recognized as PDB.6-8
The invasive cancers that are associated with PDB are most likely to be both estrogen- and progesterone-receptor–negative and of a high histologic grade.3,7 Estrogen- and progesterone-sensitive tumors respond to hormonal manipulation therapy. Tumors that are receptor-negative and that have a more aggressive grade are more difficult to treat.
Differential Diagnosis
PDB may be confused with the early stages of inflammatory breast cancer (IBC), an aggressive malignant disease (see Table 1). Both conditions may present with erythema and skin thickening and may be mistaken for mastitis. However, IBC spreads rapidly through the entire breast, and clinical features may include tenderness, a feeling of heat or heaviness, breast enlargement, and significant lymphadenopathy. Current recommendations call for a biopsy of any area of breast inflammation that does not respond to antibiotics within seven days.9
PDB is not the only cutaneous manifestation of breast cancer. Others include carcinoma erysipeloides (inflammatory changes that resemble cellulitis), carcinoma telangiectaticum (vascularized plaques), and/or inflammatory papules or nodules appearing on the breast, back, neck, or scalp. Each of these non–Paget’s conditions involves lymphatic (versus ductal) spread and signifies advanced malignancy.10
Diagnosis and Staging
After biopsy of the nipple lesion(s), diagnosis proceeds to the assessment of the breast itself and ultimately to cancer staging. PDB may occur (in order of incidence):
• In conjunction with an invasive cancer
• With underlying ductal carcinoma in situ (DCIS)
• Without any underlying disease.7
Mammography is used to determine the extent and location of the underlying lesion(s), which is more likely to be peripheral and/or multicentric. However, in some cases, there are no mammographic changes, which is now recognized as an indication for performing a breast MRI.11 Once the lesion is located, direct or image-guided biopsy confirms whether it is invasive cancer or DCIS. Palpable masses that occur with PDB are usually invasive and signal advanced disease.2,6,12,13 Sentinel lymph node biopsy (SLNB), which is usually performed at the time of surgery, plays a critical role in cancer staging and treatment planning. SLNB reliably diagnoses axillary metastasis in approximately 98% of patients.14
Like other breast cancers, PDB is also categorized by the expression of molecular markers, including HER2 (human epidermal growth factor receptor 2). Cancer cells in which HER2 gene is overexpressed tend to proliferate more rapidly than others. HER-status can also provide a clue as to which chemotherapy agents are likely to be most effective.2
Treatment and Management
The primary treatment for breast cancer is surgery, which serves both diagnostic and therapeutic purposes. To be effective, surgical treatment of PDB requires excision of the NAC, also called central lumpectomy. This may be sufficient treatment in those rare cases in which the disease is confined to the NAC.11,12
For underlying tumors, partial mastectomy is an option when the tumor is small (< 2 cm) and located close enough to the NAC to achieve negative margins, while leaving a cosmetically acceptable breast. Partial mastectomy is usually followed by whole breast irradiation. A few centers offer intraoperative radiation therapy (IORT)—performed before the surgeon closes the incision—for patients who wish to avoid or limit the duration of postoperative radiation treatment.15-17
Complete mastectomy (including excision of the NAC) should be considered when:
• The distance between the NAC and the underlying tumor is significant
• Multicentric disease and/or diffuse calcifications exist
• Achieving negative margins would remove too much tissue to leave a cosmetically acceptable breast.
Evaluation of the axillary nodes is the same in PDB as with other breast cancers. Patients with disease localized to the NAC and no underlying carcinoma may choose to forego lymph node biopsy. The same is true for patients who have PDB with a single underlying DCIS. However, lymph node biopsy is always recommended in cases of multicentric DCIS or invasive disease, or if a mastectomy is planned.18,19
Sentinel node biopsy results determine whether the mastectomy should be simple (excision of the breast alone) or modified radical (breast and axillary nodes). Today, complete radical mastectomy (excision of the breast, axillary nodes, and pectoral muscle) is reserved for cases in which disease invades the chest wall.18,19
The use of adjuvant (postoperative) therapy in patients with DCIS (whether or not related to PDB) is still debated. For patients with invasive cancers, both radiation therapy and chemotherapy are usually indicated. The decision to use neoadjuvant (preoperative) chemotherapy is made on a case-by-case basis. All decisions are based on the nature of the underlying cancer, regardless of whether the diagnosis is PDB.
Because PDB is categorized as invasive in at least 85% of cases, and because all invasive breast cancers carry about twice the risk for newly diagnosed contralateral disease, systematic follow-up is extremely important for patients with PDB. A clinical exam and updated history should be performed every four to six months during the first two years and at least annually after that. Screening recommendations, including a yearly mammogram, remain the same for asymptomatic patients. Patients with new or recurring symptoms—because they are at high risk for cancer recurrence—or who are undergoing treatment may have additional testing, including assessing for tumor markers, ultrasound, or MRI.2
PDB is treated with the same chemotherapy regimens as other breast cancers. In the early stages, chemotherapy reduces the risk for recurrence. In advanced breast cancer, the goal of chemotherapy is to reduce tumor size and achieve local control.
Prognosis
Patients with negative lymph node biopsy results have survival rates of 85% and 79% at five and 10 years, respectively. Patients with positive node results face survival rates of 32% at five years and 28% at 10 years. As with other cancers, anything that contributes to disease progression (including delayed diagnosis or treatment) decreases the patient’s survival rate.2,3 The overall prognosis for PDB is based on the nature of the underlying breast cancer, including its stage and other predictive factors—not on the fact that it is PDB.
Patient Outcome
Nearing the end of her treatment with trastuzumab, the patient became concerned about new-onset vaginal and left pelvic pain, along with some lower back discomfort. She mentioned these symptoms to her oncologist immediately. A transvaginal ultrasound could not rule out an ovarian neoplasm.
The patient elected to undergo total abdominal hysterectomy and bilateral salpingo-oophorectomy (TAH/BSO). This option allowed for removal of a mass discovered during the procedure, minimized the risk for subsequent endometrial cancer, and reduced the chance of recurrence of the patient’s estrogen/progesterone receptor–positive breast cancer. The mass itself turned out to be a benign pedunculated fibroid tumor.
The patient was relieved and continues to recover well. A follow-up PET/CT scan is scheduled for three months from now.
Conclusion
PDB is a complex disease that challenges our current understanding of breast cancer and its diagnosis and treatment. It depends uniquely upon ductal (versus blood or lymphatic) spread. Little did Paget and his contemporaries realize they had opened up such a porthole into modern histology. Nor did they appreciate the fact that they had identified an insidious breast cancer that declares itself through the skin.
Today, it is understood that by the time nipple changes of PDB appear, an underlying breast cancer most likely exists. In at least 25% of cases, there is neither a palpable mass nor a positive mammogram finding. For this reason, clinicians must maintain a high level of clinical suspicion and a low threshold for biopsy when there are skin changes at the nipple. This is especially true because the underlying lesions are more likely to be invasive cancers.
Surgical treatment will often mean complete mastectomy, whether simple, modified radical, or radical. This choice will be driven by the extent and location of the underlying disease. There is a role for partial mastectomy followed by radiation therapy in those rare cases in which PDB is confined to the NAC with no underlying tumor. Partial mastectomy is also a consideration when the underlying tumor is small and/or located close to the NAC. Patients with PDB may consider whole-breast or NAC reconstruction once radiation therapy and/or chemotherapy are completed.
PDB remains a poignant reminder for all clinicians of the importance of a thorough clinical exam and a well-focused history in all patients at risk for breast cancer. Moreover, it is an enduring example of the fact that common symptoms sometimes do signify something uncommon and potentially life- changing.
References
1. Paget J. On disease of the mammary areola preceding cancer of the mammary gland. In: Paget S, ed. Selected Essays and Addresses by Sir James Paget. London: Longmans, Green and Co.; 1902:145-148.
2. Sabel MS, Weaver DL. Paget disease of the breast. In: UpToDate. Chagpar AE, Hayes DF, Pierce LJ, eds. www.uptodate.com/contents/paget-disease-of-the-breast. Updated November 27, 2012. Accessed September 9, 2013.
3. Ortiz-Pagan S, Cunto-Amesty G, Narayan S. Effect of Paget’s disease on survival in breast cancer. Arch Surg. 2001;146:1267-1270.
4. American Cancer Society. Cancer facts & figures 2012. www.cancer.org/research/cancerfactsfigures/cancerfactsfigures/cancer-facts-figures-2012. Accessed September 9, 2013.
5. Ashikari R, Park K, Huvos AG, Urban JA. Paget’s disease of the breast. Cancer. 1970;3:680-685.
6. Sakorafas GH, Blanchard K, Sarr MG, Farley DR. Paget’s disease of the breast. Cancer Treatment Rev. 2001;27:9-18.
7. Chen C-Y, Sun L-M, Anderson BO. Paget disease of the breast: changing patterns of incidence, clinical presentation, and treatment in the U.S. Cancer. 2006;107:1448-1458.
8. Paone JF, Baker R. Pathogenesis and treatment of Paget’s disease of the breast. Cancer. 1981;48:825-829.
9. Nelson JA, Patel D, Mancuso P. Inflammatory breast cancer. ADVANCE for NPs and PAs. 2011;2(10):25-28.
10. Ngan V. Skin metastasis. DermNet NZ. New Zealand Dermatological Society. http://dermnetnz.org/lesions/metastasis.html. Accessed September 9, 2013.
11. Amano G, Yajima M, Moroboshi Y, et al. MRI accurately depicts underlying DCIS in a patient with Paget’s disease of the breast without palpable mass and mammography findings. Jpn J Clin Oncol. 2005;35:149-153.
12. Burrell HC, Evans AJ. Radiological assessment of the breast: what the surgical oncologist needs to know. Eur J Surg Oncol. 2001;27:689-691.
13. Muttarak M, Siriya B, Kongmebhol P. Paget’s disease of the breast: clinical, imaging and pathologic findings: a review of 16 patients. Biomed Imaging Interv J. 2001;7:e16, 1-7.
14. Laronga C, Hasson D, Hoover S, et al. Paget’s disease in the era of sentinel lymph node biopsy. Am J Surg. 2006;192:481-483.
15. Pezzi CM, Kukora JS, Audet IM. Breast conservation surgery using nipple-areolar resection for central breast cancers. Arch Surg. 2004;139:32-37.
16. Polgar C, Zsolt O, Tibor K, Janos F. Breast-conserving therapy for Paget disease of the nipple. Cancer. 2002;94:1904-1905.
17. Marshall JK, Griffith KA, Haffty BG, Solin LJ. Conservative management of Paget disease of the breast with radiotherapy. Cancer. 2003;97:2142-2149.
18. Vasquez B, Rousseau D, Hurd TC. Surgical management of breast cancer. Sem Oncol. 2007;34:234-240.
19. Mamounas EP. Continuing evolution in breast cancer surgical management. J Clin Oncol. 2005;23:1603-1606.
20. Nicholson BT, Harvey JA, Cohen MA. Nipple-areolar complex: normal anatomy and benign and malignant processes. Radiographics. 2009;29:509-523.
A 45-year-old woman noticed some redness and scaling around her right nipple. She applied peroxide and OTC antibiotic ointment for approximately seven months with mixed results. She sought medical attention when pain developed in the breast, along with some bloody discharge from the nipple (see Figure 1). Around that time, she also noticed three small nodules in the upper outer portion of the breast.
A mammogram and ultrasound revealed a 1.7 × 2.0–cm spiculated mass in the axillary tail, as well as two smaller breast lesions. A PET/CT scan ordered subsequently revealed intense uptake in the periareolar region and a suspicious axillary node. By then, the biopsy results had confirmed invasive ductal carcinoma, later determined to be Paget’s disease of the breast (PDB).
The patient’s previous medical history was significant for cystic breasts (never biopsied), chronic back pain, anxiety, and obesity. She was perimenopausal with irregular periods, the last one about 10 months ago. Her obstetric history included two pregnancies resulting in live births and no history of abortion; her menarche occurred at age 14 and her first pregnancy at 27. Family history was significant for leukemia in her maternal grandmother and niece. She did not use tobacco, alcohol, or illicit drugs. She lived at home with her husband and two daughters, who were all very supportive.
The patient elected to undergo a right modified radical mastectomy (MRM) and prophylactic left total mastectomy. MRM was performed on the right breast because sentinel lymph node identification was unsuccessful. This may have been due to involvement of the right subareolar plexus. Five of eight lymph nodes later tested positive for malignancy. The surgery was completed by placement of bilateral tissue expanders for eventual breast reconstruction.
Chemotherapy was started six weeks after surgery and included 15 weeks (five cycles) of docetaxel, carboplatin, and trastuzumab (a combination known as TCH), followed by 51 weeks (17 cycles) of trastuzumab, along with daily tamoxifen. The TCH regimen was followed by four weekly cycles of external beam radiation therapy (EBRT). Adverse effects of treatment have included chest wall dermatitis, right upper extremity lymphedema, nausea/vomiting, dyspnea, peripheral neuropathy, alopecia, and fatigue.
Discussion
Nearly 150 years ago, James Paget recognized a connection between skin changes around the nipple and deeper lesions in the breast.1 The disease that Paget identified is defined as the presence of intraepithelial adenocarcinoma cells (ie, Paget’s cells) within the epidermis of the nipple, with or without an underlying carcinoma.
An underlying breast cancer is present 85% to 95% of the time but is palpable in only approximately 50% of cases (see Figure 2). However, 25% of the time there is neither a palpable mass nor a mammographic abnormality. In these cases particularly, timely diagnosis depends on recognition of suspicious nipple changes, followed by a prompt and thorough diagnostic workup. Unfortunately, the accurate diagnosis of Paget’s disease still takes an average of several months.2
Paget’s disease is rare; it represents only 1% to 3% of new cases of female breast cancer, or about 2,250 cases a year.2-4 (The number of Paget’s disease cases per year was calculated by the author, based on the reported incidence of all breast cancers.) It is even more rare among men. For both genders, the peak age for this disease is between 50 and 60.2
Paget’s disease is an important entity for primary care PAs and NPs because it presents an opportunity to make a timely and life-changing diagnosis, and because it provides an elegant model for understanding current diagnostic and therapeutic approaches to breast cancer.
Clinical Presentation and Pathophysiology
The hallmark of PDB is scaly, vesicular, or ulcerated changes that begin in the nipple and spread to the areola. These changes are most often unilateral and may occur with pruritus, burning pain, and/or oozing from the nipple.5 This presentation is often mistaken for common skin conditions, such as eczema. Like eczema, changes in PDB may improve spontaneously and fluctuate over time, which is confusing for both the patient and clinician. A clinical pearl is that eczema is more likely to spread from the areola to the nipple, and will usually respond to topical corticosteroids. By contrast, changes in PDB tend to spread from the nipple to the areola, and corticosteroids do not provide a sustained response. Of note, Paget’s lesions may heal spontaneously even as the underlying malignancy progresses.6
PDB is unique because the underlying lesion and skin changes are not just coincidental. The cutaneous changes and the malignancy that lies beneath have a causal, not merely co-occurring, relationship. Paget himself believed that the nipple changes were both a precursor, and a promoter, of the underlying cancer.1 This transformation theory states that normal nipple epidermis turns into Paget’s cells spontaneously, before there is any underlying disease. This theory is supported by the fact that, occasionally (though rarely), no underlying breast cancer is ever found. Also, the concomitant tumor may be some distance (> 2 cm) from the nipple-areolar complex (NAC), suggesting a synchronous but causally unrelated lesion.6-8
Modern immunochemistry has turned PDB inside out. Today, PDB is believed to begin within the breast and then to spread “upward” to the NAC, called the epidermotrophic theory. This theory is supported by the fact that Paget’s cells share several molecular markers with their respective parenchymal tumors. Some researchers now propose that there is a single Paget’s progenitor cell with a motility factor that allows it to traverse the ductal system, resulting in nipple and skin changes that have come to be recognized as PDB.6-8
The invasive cancers that are associated with PDB are most likely to be both estrogen- and progesterone-receptor–negative and of a high histologic grade.3,7 Estrogen- and progesterone-sensitive tumors respond to hormonal manipulation therapy. Tumors that are receptor-negative and that have a more aggressive grade are more difficult to treat.
Differential Diagnosis
PDB may be confused with the early stages of inflammatory breast cancer (IBC), an aggressive malignant disease (see Table 1). Both conditions may present with erythema and skin thickening and may be mistaken for mastitis. However, IBC spreads rapidly through the entire breast, and clinical features may include tenderness, a feeling of heat or heaviness, breast enlargement, and significant lymphadenopathy. Current recommendations call for a biopsy of any area of breast inflammation that does not respond to antibiotics within seven days.9
PDB is not the only cutaneous manifestation of breast cancer. Others include carcinoma erysipeloides (inflammatory changes that resemble cellulitis), carcinoma telangiectaticum (vascularized plaques), and/or inflammatory papules or nodules appearing on the breast, back, neck, or scalp. Each of these non–Paget’s conditions involves lymphatic (versus ductal) spread and signifies advanced malignancy.10
Diagnosis and Staging
After biopsy of the nipple lesion(s), diagnosis proceeds to the assessment of the breast itself and ultimately to cancer staging. PDB may occur (in order of incidence):
• In conjunction with an invasive cancer
• With underlying ductal carcinoma in situ (DCIS)
• Without any underlying disease.7
Mammography is used to determine the extent and location of the underlying lesion(s), which is more likely to be peripheral and/or multicentric. However, in some cases, there are no mammographic changes, which is now recognized as an indication for performing a breast MRI.11 Once the lesion is located, direct or image-guided biopsy confirms whether it is invasive cancer or DCIS. Palpable masses that occur with PDB are usually invasive and signal advanced disease.2,6,12,13 Sentinel lymph node biopsy (SLNB), which is usually performed at the time of surgery, plays a critical role in cancer staging and treatment planning. SLNB reliably diagnoses axillary metastasis in approximately 98% of patients.14
Like other breast cancers, PDB is also categorized by the expression of molecular markers, including HER2 (human epidermal growth factor receptor 2). Cancer cells in which HER2 gene is overexpressed tend to proliferate more rapidly than others. HER-status can also provide a clue as to which chemotherapy agents are likely to be most effective.2
Treatment and Management
The primary treatment for breast cancer is surgery, which serves both diagnostic and therapeutic purposes. To be effective, surgical treatment of PDB requires excision of the NAC, also called central lumpectomy. This may be sufficient treatment in those rare cases in which the disease is confined to the NAC.11,12
For underlying tumors, partial mastectomy is an option when the tumor is small (< 2 cm) and located close enough to the NAC to achieve negative margins, while leaving a cosmetically acceptable breast. Partial mastectomy is usually followed by whole breast irradiation. A few centers offer intraoperative radiation therapy (IORT)—performed before the surgeon closes the incision—for patients who wish to avoid or limit the duration of postoperative radiation treatment.15-17
Complete mastectomy (including excision of the NAC) should be considered when:
• The distance between the NAC and the underlying tumor is significant
• Multicentric disease and/or diffuse calcifications exist
• Achieving negative margins would remove too much tissue to leave a cosmetically acceptable breast.
Evaluation of the axillary nodes is the same in PDB as with other breast cancers. Patients with disease localized to the NAC and no underlying carcinoma may choose to forego lymph node biopsy. The same is true for patients who have PDB with a single underlying DCIS. However, lymph node biopsy is always recommended in cases of multicentric DCIS or invasive disease, or if a mastectomy is planned.18,19
Sentinel node biopsy results determine whether the mastectomy should be simple (excision of the breast alone) or modified radical (breast and axillary nodes). Today, complete radical mastectomy (excision of the breast, axillary nodes, and pectoral muscle) is reserved for cases in which disease invades the chest wall.18,19
The use of adjuvant (postoperative) therapy in patients with DCIS (whether or not related to PDB) is still debated. For patients with invasive cancers, both radiation therapy and chemotherapy are usually indicated. The decision to use neoadjuvant (preoperative) chemotherapy is made on a case-by-case basis. All decisions are based on the nature of the underlying cancer, regardless of whether the diagnosis is PDB.
Because PDB is categorized as invasive in at least 85% of cases, and because all invasive breast cancers carry about twice the risk for newly diagnosed contralateral disease, systematic follow-up is extremely important for patients with PDB. A clinical exam and updated history should be performed every four to six months during the first two years and at least annually after that. Screening recommendations, including a yearly mammogram, remain the same for asymptomatic patients. Patients with new or recurring symptoms—because they are at high risk for cancer recurrence—or who are undergoing treatment may have additional testing, including assessing for tumor markers, ultrasound, or MRI.2
PDB is treated with the same chemotherapy regimens as other breast cancers. In the early stages, chemotherapy reduces the risk for recurrence. In advanced breast cancer, the goal of chemotherapy is to reduce tumor size and achieve local control.
Prognosis
Patients with negative lymph node biopsy results have survival rates of 85% and 79% at five and 10 years, respectively. Patients with positive node results face survival rates of 32% at five years and 28% at 10 years. As with other cancers, anything that contributes to disease progression (including delayed diagnosis or treatment) decreases the patient’s survival rate.2,3 The overall prognosis for PDB is based on the nature of the underlying breast cancer, including its stage and other predictive factors—not on the fact that it is PDB.
Patient Outcome
Nearing the end of her treatment with trastuzumab, the patient became concerned about new-onset vaginal and left pelvic pain, along with some lower back discomfort. She mentioned these symptoms to her oncologist immediately. A transvaginal ultrasound could not rule out an ovarian neoplasm.
The patient elected to undergo total abdominal hysterectomy and bilateral salpingo-oophorectomy (TAH/BSO). This option allowed for removal of a mass discovered during the procedure, minimized the risk for subsequent endometrial cancer, and reduced the chance of recurrence of the patient’s estrogen/progesterone receptor–positive breast cancer. The mass itself turned out to be a benign pedunculated fibroid tumor.
The patient was relieved and continues to recover well. A follow-up PET/CT scan is scheduled for three months from now.
Conclusion
PDB is a complex disease that challenges our current understanding of breast cancer and its diagnosis and treatment. It depends uniquely upon ductal (versus blood or lymphatic) spread. Little did Paget and his contemporaries realize they had opened up such a porthole into modern histology. Nor did they appreciate the fact that they had identified an insidious breast cancer that declares itself through the skin.
Today, it is understood that by the time nipple changes of PDB appear, an underlying breast cancer most likely exists. In at least 25% of cases, there is neither a palpable mass nor a positive mammogram finding. For this reason, clinicians must maintain a high level of clinical suspicion and a low threshold for biopsy when there are skin changes at the nipple. This is especially true because the underlying lesions are more likely to be invasive cancers.
Surgical treatment will often mean complete mastectomy, whether simple, modified radical, or radical. This choice will be driven by the extent and location of the underlying disease. There is a role for partial mastectomy followed by radiation therapy in those rare cases in which PDB is confined to the NAC with no underlying tumor. Partial mastectomy is also a consideration when the underlying tumor is small and/or located close to the NAC. Patients with PDB may consider whole-breast or NAC reconstruction once radiation therapy and/or chemotherapy are completed.
PDB remains a poignant reminder for all clinicians of the importance of a thorough clinical exam and a well-focused history in all patients at risk for breast cancer. Moreover, it is an enduring example of the fact that common symptoms sometimes do signify something uncommon and potentially life- changing.
References
1. Paget J. On disease of the mammary areola preceding cancer of the mammary gland. In: Paget S, ed. Selected Essays and Addresses by Sir James Paget. London: Longmans, Green and Co.; 1902:145-148.
2. Sabel MS, Weaver DL. Paget disease of the breast. In: UpToDate. Chagpar AE, Hayes DF, Pierce LJ, eds. www.uptodate.com/contents/paget-disease-of-the-breast. Updated November 27, 2012. Accessed September 9, 2013.
3. Ortiz-Pagan S, Cunto-Amesty G, Narayan S. Effect of Paget’s disease on survival in breast cancer. Arch Surg. 2001;146:1267-1270.
4. American Cancer Society. Cancer facts & figures 2012. www.cancer.org/research/cancerfactsfigures/cancerfactsfigures/cancer-facts-figures-2012. Accessed September 9, 2013.
5. Ashikari R, Park K, Huvos AG, Urban JA. Paget’s disease of the breast. Cancer. 1970;3:680-685.
6. Sakorafas GH, Blanchard K, Sarr MG, Farley DR. Paget’s disease of the breast. Cancer Treatment Rev. 2001;27:9-18.
7. Chen C-Y, Sun L-M, Anderson BO. Paget disease of the breast: changing patterns of incidence, clinical presentation, and treatment in the U.S. Cancer. 2006;107:1448-1458.
8. Paone JF, Baker R. Pathogenesis and treatment of Paget’s disease of the breast. Cancer. 1981;48:825-829.
9. Nelson JA, Patel D, Mancuso P. Inflammatory breast cancer. ADVANCE for NPs and PAs. 2011;2(10):25-28.
10. Ngan V. Skin metastasis. DermNet NZ. New Zealand Dermatological Society. http://dermnetnz.org/lesions/metastasis.html. Accessed September 9, 2013.
11. Amano G, Yajima M, Moroboshi Y, et al. MRI accurately depicts underlying DCIS in a patient with Paget’s disease of the breast without palpable mass and mammography findings. Jpn J Clin Oncol. 2005;35:149-153.
12. Burrell HC, Evans AJ. Radiological assessment of the breast: what the surgical oncologist needs to know. Eur J Surg Oncol. 2001;27:689-691.
13. Muttarak M, Siriya B, Kongmebhol P. Paget’s disease of the breast: clinical, imaging and pathologic findings: a review of 16 patients. Biomed Imaging Interv J. 2001;7:e16, 1-7.
14. Laronga C, Hasson D, Hoover S, et al. Paget’s disease in the era of sentinel lymph node biopsy. Am J Surg. 2006;192:481-483.
15. Pezzi CM, Kukora JS, Audet IM. Breast conservation surgery using nipple-areolar resection for central breast cancers. Arch Surg. 2004;139:32-37.
16. Polgar C, Zsolt O, Tibor K, Janos F. Breast-conserving therapy for Paget disease of the nipple. Cancer. 2002;94:1904-1905.
17. Marshall JK, Griffith KA, Haffty BG, Solin LJ. Conservative management of Paget disease of the breast with radiotherapy. Cancer. 2003;97:2142-2149.
18. Vasquez B, Rousseau D, Hurd TC. Surgical management of breast cancer. Sem Oncol. 2007;34:234-240.
19. Mamounas EP. Continuing evolution in breast cancer surgical management. J Clin Oncol. 2005;23:1603-1606.
20. Nicholson BT, Harvey JA, Cohen MA. Nipple-areolar complex: normal anatomy and benign and malignant processes. Radiographics. 2009;29:509-523.
A 45-year-old woman noticed some redness and scaling around her right nipple. She applied peroxide and OTC antibiotic ointment for approximately seven months with mixed results. She sought medical attention when pain developed in the breast, along with some bloody discharge from the nipple (see Figure 1). Around that time, she also noticed three small nodules in the upper outer portion of the breast.
A mammogram and ultrasound revealed a 1.7 × 2.0–cm spiculated mass in the axillary tail, as well as two smaller breast lesions. A PET/CT scan ordered subsequently revealed intense uptake in the periareolar region and a suspicious axillary node. By then, the biopsy results had confirmed invasive ductal carcinoma, later determined to be Paget’s disease of the breast (PDB).
The patient’s previous medical history was significant for cystic breasts (never biopsied), chronic back pain, anxiety, and obesity. She was perimenopausal with irregular periods, the last one about 10 months ago. Her obstetric history included two pregnancies resulting in live births and no history of abortion; her menarche occurred at age 14 and her first pregnancy at 27. Family history was significant for leukemia in her maternal grandmother and niece. She did not use tobacco, alcohol, or illicit drugs. She lived at home with her husband and two daughters, who were all very supportive.
The patient elected to undergo a right modified radical mastectomy (MRM) and prophylactic left total mastectomy. MRM was performed on the right breast because sentinel lymph node identification was unsuccessful. This may have been due to involvement of the right subareolar plexus. Five of eight lymph nodes later tested positive for malignancy. The surgery was completed by placement of bilateral tissue expanders for eventual breast reconstruction.
Chemotherapy was started six weeks after surgery and included 15 weeks (five cycles) of docetaxel, carboplatin, and trastuzumab (a combination known as TCH), followed by 51 weeks (17 cycles) of trastuzumab, along with daily tamoxifen. The TCH regimen was followed by four weekly cycles of external beam radiation therapy (EBRT). Adverse effects of treatment have included chest wall dermatitis, right upper extremity lymphedema, nausea/vomiting, dyspnea, peripheral neuropathy, alopecia, and fatigue.
Discussion
Nearly 150 years ago, James Paget recognized a connection between skin changes around the nipple and deeper lesions in the breast.1 The disease that Paget identified is defined as the presence of intraepithelial adenocarcinoma cells (ie, Paget’s cells) within the epidermis of the nipple, with or without an underlying carcinoma.
An underlying breast cancer is present 85% to 95% of the time but is palpable in only approximately 50% of cases (see Figure 2). However, 25% of the time there is neither a palpable mass nor a mammographic abnormality. In these cases particularly, timely diagnosis depends on recognition of suspicious nipple changes, followed by a prompt and thorough diagnostic workup. Unfortunately, the accurate diagnosis of Paget’s disease still takes an average of several months.2
Paget’s disease is rare; it represents only 1% to 3% of new cases of female breast cancer, or about 2,250 cases a year.2-4 (The number of Paget’s disease cases per year was calculated by the author, based on the reported incidence of all breast cancers.) It is even more rare among men. For both genders, the peak age for this disease is between 50 and 60.2
Paget’s disease is an important entity for primary care PAs and NPs because it presents an opportunity to make a timely and life-changing diagnosis, and because it provides an elegant model for understanding current diagnostic and therapeutic approaches to breast cancer.
Clinical Presentation and Pathophysiology
The hallmark of PDB is scaly, vesicular, or ulcerated changes that begin in the nipple and spread to the areola. These changes are most often unilateral and may occur with pruritus, burning pain, and/or oozing from the nipple.5 This presentation is often mistaken for common skin conditions, such as eczema. Like eczema, changes in PDB may improve spontaneously and fluctuate over time, which is confusing for both the patient and clinician. A clinical pearl is that eczema is more likely to spread from the areola to the nipple, and will usually respond to topical corticosteroids. By contrast, changes in PDB tend to spread from the nipple to the areola, and corticosteroids do not provide a sustained response. Of note, Paget’s lesions may heal spontaneously even as the underlying malignancy progresses.6
PDB is unique because the underlying lesion and skin changes are not just coincidental. The cutaneous changes and the malignancy that lies beneath have a causal, not merely co-occurring, relationship. Paget himself believed that the nipple changes were both a precursor, and a promoter, of the underlying cancer.1 This transformation theory states that normal nipple epidermis turns into Paget’s cells spontaneously, before there is any underlying disease. This theory is supported by the fact that, occasionally (though rarely), no underlying breast cancer is ever found. Also, the concomitant tumor may be some distance (> 2 cm) from the nipple-areolar complex (NAC), suggesting a synchronous but causally unrelated lesion.6-8
Modern immunochemistry has turned PDB inside out. Today, PDB is believed to begin within the breast and then to spread “upward” to the NAC, called the epidermotrophic theory. This theory is supported by the fact that Paget’s cells share several molecular markers with their respective parenchymal tumors. Some researchers now propose that there is a single Paget’s progenitor cell with a motility factor that allows it to traverse the ductal system, resulting in nipple and skin changes that have come to be recognized as PDB.6-8
The invasive cancers that are associated with PDB are most likely to be both estrogen- and progesterone-receptor–negative and of a high histologic grade.3,7 Estrogen- and progesterone-sensitive tumors respond to hormonal manipulation therapy. Tumors that are receptor-negative and that have a more aggressive grade are more difficult to treat.
Differential Diagnosis
PDB may be confused with the early stages of inflammatory breast cancer (IBC), an aggressive malignant disease (see Table 1). Both conditions may present with erythema and skin thickening and may be mistaken for mastitis. However, IBC spreads rapidly through the entire breast, and clinical features may include tenderness, a feeling of heat or heaviness, breast enlargement, and significant lymphadenopathy. Current recommendations call for a biopsy of any area of breast inflammation that does not respond to antibiotics within seven days.9
PDB is not the only cutaneous manifestation of breast cancer. Others include carcinoma erysipeloides (inflammatory changes that resemble cellulitis), carcinoma telangiectaticum (vascularized plaques), and/or inflammatory papules or nodules appearing on the breast, back, neck, or scalp. Each of these non–Paget’s conditions involves lymphatic (versus ductal) spread and signifies advanced malignancy.10
Diagnosis and Staging
After biopsy of the nipple lesion(s), diagnosis proceeds to the assessment of the breast itself and ultimately to cancer staging. PDB may occur (in order of incidence):
• In conjunction with an invasive cancer
• With underlying ductal carcinoma in situ (DCIS)
• Without any underlying disease.7
Mammography is used to determine the extent and location of the underlying lesion(s), which is more likely to be peripheral and/or multicentric. However, in some cases, there are no mammographic changes, which is now recognized as an indication for performing a breast MRI.11 Once the lesion is located, direct or image-guided biopsy confirms whether it is invasive cancer or DCIS. Palpable masses that occur with PDB are usually invasive and signal advanced disease.2,6,12,13 Sentinel lymph node biopsy (SLNB), which is usually performed at the time of surgery, plays a critical role in cancer staging and treatment planning. SLNB reliably diagnoses axillary metastasis in approximately 98% of patients.14
Like other breast cancers, PDB is also categorized by the expression of molecular markers, including HER2 (human epidermal growth factor receptor 2). Cancer cells in which HER2 gene is overexpressed tend to proliferate more rapidly than others. HER-status can also provide a clue as to which chemotherapy agents are likely to be most effective.2
Treatment and Management
The primary treatment for breast cancer is surgery, which serves both diagnostic and therapeutic purposes. To be effective, surgical treatment of PDB requires excision of the NAC, also called central lumpectomy. This may be sufficient treatment in those rare cases in which the disease is confined to the NAC.11,12
For underlying tumors, partial mastectomy is an option when the tumor is small (< 2 cm) and located close enough to the NAC to achieve negative margins, while leaving a cosmetically acceptable breast. Partial mastectomy is usually followed by whole breast irradiation. A few centers offer intraoperative radiation therapy (IORT)—performed before the surgeon closes the incision—for patients who wish to avoid or limit the duration of postoperative radiation treatment.15-17
Complete mastectomy (including excision of the NAC) should be considered when:
• The distance between the NAC and the underlying tumor is significant
• Multicentric disease and/or diffuse calcifications exist
• Achieving negative margins would remove too much tissue to leave a cosmetically acceptable breast.
Evaluation of the axillary nodes is the same in PDB as with other breast cancers. Patients with disease localized to the NAC and no underlying carcinoma may choose to forego lymph node biopsy. The same is true for patients who have PDB with a single underlying DCIS. However, lymph node biopsy is always recommended in cases of multicentric DCIS or invasive disease, or if a mastectomy is planned.18,19
Sentinel node biopsy results determine whether the mastectomy should be simple (excision of the breast alone) or modified radical (breast and axillary nodes). Today, complete radical mastectomy (excision of the breast, axillary nodes, and pectoral muscle) is reserved for cases in which disease invades the chest wall.18,19
The use of adjuvant (postoperative) therapy in patients with DCIS (whether or not related to PDB) is still debated. For patients with invasive cancers, both radiation therapy and chemotherapy are usually indicated. The decision to use neoadjuvant (preoperative) chemotherapy is made on a case-by-case basis. All decisions are based on the nature of the underlying cancer, regardless of whether the diagnosis is PDB.
Because PDB is categorized as invasive in at least 85% of cases, and because all invasive breast cancers carry about twice the risk for newly diagnosed contralateral disease, systematic follow-up is extremely important for patients with PDB. A clinical exam and updated history should be performed every four to six months during the first two years and at least annually after that. Screening recommendations, including a yearly mammogram, remain the same for asymptomatic patients. Patients with new or recurring symptoms—because they are at high risk for cancer recurrence—or who are undergoing treatment may have additional testing, including assessing for tumor markers, ultrasound, or MRI.2
PDB is treated with the same chemotherapy regimens as other breast cancers. In the early stages, chemotherapy reduces the risk for recurrence. In advanced breast cancer, the goal of chemotherapy is to reduce tumor size and achieve local control.
Prognosis
Patients with negative lymph node biopsy results have survival rates of 85% and 79% at five and 10 years, respectively. Patients with positive node results face survival rates of 32% at five years and 28% at 10 years. As with other cancers, anything that contributes to disease progression (including delayed diagnosis or treatment) decreases the patient’s survival rate.2,3 The overall prognosis for PDB is based on the nature of the underlying breast cancer, including its stage and other predictive factors—not on the fact that it is PDB.
Patient Outcome
Nearing the end of her treatment with trastuzumab, the patient became concerned about new-onset vaginal and left pelvic pain, along with some lower back discomfort. She mentioned these symptoms to her oncologist immediately. A transvaginal ultrasound could not rule out an ovarian neoplasm.
The patient elected to undergo total abdominal hysterectomy and bilateral salpingo-oophorectomy (TAH/BSO). This option allowed for removal of a mass discovered during the procedure, minimized the risk for subsequent endometrial cancer, and reduced the chance of recurrence of the patient’s estrogen/progesterone receptor–positive breast cancer. The mass itself turned out to be a benign pedunculated fibroid tumor.
The patient was relieved and continues to recover well. A follow-up PET/CT scan is scheduled for three months from now.
Conclusion
PDB is a complex disease that challenges our current understanding of breast cancer and its diagnosis and treatment. It depends uniquely upon ductal (versus blood or lymphatic) spread. Little did Paget and his contemporaries realize they had opened up such a porthole into modern histology. Nor did they appreciate the fact that they had identified an insidious breast cancer that declares itself through the skin.
Today, it is understood that by the time nipple changes of PDB appear, an underlying breast cancer most likely exists. In at least 25% of cases, there is neither a palpable mass nor a positive mammogram finding. For this reason, clinicians must maintain a high level of clinical suspicion and a low threshold for biopsy when there are skin changes at the nipple. This is especially true because the underlying lesions are more likely to be invasive cancers.
Surgical treatment will often mean complete mastectomy, whether simple, modified radical, or radical. This choice will be driven by the extent and location of the underlying disease. There is a role for partial mastectomy followed by radiation therapy in those rare cases in which PDB is confined to the NAC with no underlying tumor. Partial mastectomy is also a consideration when the underlying tumor is small and/or located close to the NAC. Patients with PDB may consider whole-breast or NAC reconstruction once radiation therapy and/or chemotherapy are completed.
PDB remains a poignant reminder for all clinicians of the importance of a thorough clinical exam and a well-focused history in all patients at risk for breast cancer. Moreover, it is an enduring example of the fact that common symptoms sometimes do signify something uncommon and potentially life- changing.
References
1. Paget J. On disease of the mammary areola preceding cancer of the mammary gland. In: Paget S, ed. Selected Essays and Addresses by Sir James Paget. London: Longmans, Green and Co.; 1902:145-148.
2. Sabel MS, Weaver DL. Paget disease of the breast. In: UpToDate. Chagpar AE, Hayes DF, Pierce LJ, eds. www.uptodate.com/contents/paget-disease-of-the-breast. Updated November 27, 2012. Accessed September 9, 2013.
3. Ortiz-Pagan S, Cunto-Amesty G, Narayan S. Effect of Paget’s disease on survival in breast cancer. Arch Surg. 2001;146:1267-1270.
4. American Cancer Society. Cancer facts & figures 2012. www.cancer.org/research/cancerfactsfigures/cancerfactsfigures/cancer-facts-figures-2012. Accessed September 9, 2013.
5. Ashikari R, Park K, Huvos AG, Urban JA. Paget’s disease of the breast. Cancer. 1970;3:680-685.
6. Sakorafas GH, Blanchard K, Sarr MG, Farley DR. Paget’s disease of the breast. Cancer Treatment Rev. 2001;27:9-18.
7. Chen C-Y, Sun L-M, Anderson BO. Paget disease of the breast: changing patterns of incidence, clinical presentation, and treatment in the U.S. Cancer. 2006;107:1448-1458.
8. Paone JF, Baker R. Pathogenesis and treatment of Paget’s disease of the breast. Cancer. 1981;48:825-829.
9. Nelson JA, Patel D, Mancuso P. Inflammatory breast cancer. ADVANCE for NPs and PAs. 2011;2(10):25-28.
10. Ngan V. Skin metastasis. DermNet NZ. New Zealand Dermatological Society. http://dermnetnz.org/lesions/metastasis.html. Accessed September 9, 2013.
11. Amano G, Yajima M, Moroboshi Y, et al. MRI accurately depicts underlying DCIS in a patient with Paget’s disease of the breast without palpable mass and mammography findings. Jpn J Clin Oncol. 2005;35:149-153.
12. Burrell HC, Evans AJ. Radiological assessment of the breast: what the surgical oncologist needs to know. Eur J Surg Oncol. 2001;27:689-691.
13. Muttarak M, Siriya B, Kongmebhol P. Paget’s disease of the breast: clinical, imaging and pathologic findings: a review of 16 patients. Biomed Imaging Interv J. 2001;7:e16, 1-7.
14. Laronga C, Hasson D, Hoover S, et al. Paget’s disease in the era of sentinel lymph node biopsy. Am J Surg. 2006;192:481-483.
15. Pezzi CM, Kukora JS, Audet IM. Breast conservation surgery using nipple-areolar resection for central breast cancers. Arch Surg. 2004;139:32-37.
16. Polgar C, Zsolt O, Tibor K, Janos F. Breast-conserving therapy for Paget disease of the nipple. Cancer. 2002;94:1904-1905.
17. Marshall JK, Griffith KA, Haffty BG, Solin LJ. Conservative management of Paget disease of the breast with radiotherapy. Cancer. 2003;97:2142-2149.
18. Vasquez B, Rousseau D, Hurd TC. Surgical management of breast cancer. Sem Oncol. 2007;34:234-240.
19. Mamounas EP. Continuing evolution in breast cancer surgical management. J Clin Oncol. 2005;23:1603-1606.
20. Nicholson BT, Harvey JA, Cohen MA. Nipple-areolar complex: normal anatomy and benign and malignant processes. Radiographics. 2009;29:509-523.
Girl, 13, With a Bump on Her Leg
A girl, age 13 years, 4 months, presented to her primary care provider’s office for a well visit. Among her concerns, she mentioned a “bump” she had had on her right leg “for the past six months, maybe longer.” The area felt irritated when touched or when the patient “ran too much.” She had seen no change in the bump since she first noticed it. The patient knew of no trauma or other preceding factors. She denied any fever or warmth, redness, or ecchymosis to the area.
Medical history was unremarkable except for familial short stature and myopia. The patient was the fifth of eight children born to nonconsanguinous parents. She denied any surgical history or hospitalizations and was premenarcheal. She was up to date on all age-appropriate vaccines, with her meningococcal vaccine administered at that visit.
The patient’s blood pressure was 99/58 mm Hg with an apical pulse rate of 82 beats/min. Her growth parameters were following her curve. Her height was 55” (0.3 percentile); weight, 81 lb (7.5 percentile); and BMI, 18.8 (48.6 percentile).
The physical exam was normal with the exception of the musculoskeletal exam. Examination of the lower extremities revealed a palpable, 4 cm x 5 cm lesion at the right distal medial thigh just proximal to the knee. The lesion could not be visualized but on palpation was tender and firm. There was some question as to whether the lesion itself or inflamed soft tissue overlying the lesion was mobile. No overlying warmth, induration, erythema, or ecchymosis was noted.
Passive and active range of motion was intact at the hip and knee. No lesions to the upper extremities were evident, and no scoliosis was seen.
Blood work was done to rule out certain diagnoses. Results from a complete blood count with differential, lactate dehydrogenase (LDH), parathyroid hormone, lipid profiles, thyroid function, and a comprehensive metabolic profile were unremarkable. A low level of vitamin D 25-OH was detected: 21.7 ng/mL (normal range, 32 to 100 ng/mL).
Distal femur x-rays with posteroanterior, lateral, and oblique views were ordered. The imaging revealed a 3 cm x 3 cm lesion projecting from the “distal, somewhat medial” femur, which was diagnosed as a benign femoral osteochondroma. Significant inflammation to the surrounding soft-tissue structures was observed. A questionable old fracture of the osteochondroma was noted. The remaining bony structures and joints appeared normal.
An ultrasound of the lesion was also ordered to investigate soft-tissue swelling. This revealed a hypoechoic collar around the distal end of the osteochondroma, which could represent a fluid collection, hematoma from trauma, or bursitis. The soft tissues were deemed normal.
Because of the extent of inflammation, the radiologist recommended MRI without contrast to rule out bursitis or trauma to the osteochondroma.
DISCUSSION
Osteochondromas, which may be present in up to 3% of the general population, are the most common benign bone tumors.1-3 An osteochondroma is a cartilage-capped bony projection that arises on the external surface of the bone; it contains a marrow cavity that is continuous with the underlying bone.2,4 The majority of osteochondromas are solitary, accounting for perhaps 85% to 90% of all such lesions, and they are typically nonhereditary; the remaining 10% to 15% of osteochondromas are hereditary multiple osteochondromas or exostoses1,2 (see “Definition of Multiple Exostoses Syndrome”2,5,6,7).

Most lesions are painless and slow growing, and they usually occur in children and adolescents.2 They typically stop growing at skeletal maturity with the closure of the growth plates.3,8,9 There is no predilection for males or females in single lesions.2
Solitary osteochondromas typically appear in the lower extremities and at long tubular bone metaphyses,1-3,10 especially on the femur, humerus, tibia, spine, and hip. Any part of the skeleton can be affected, but 30% of lesions occur on the femur and 40% at either the proximal metaphysis of the tibia or the distal metaphysis of the femur.2,11
Most osteochondromas are asymptomatic and are found incidentally.1,3 However, some patients present with local pain as a result of irritation to adjacent structures, limitation of joint motion, growth disturbance, or fracture of the pedicle.3,4,9,11,12 A very small proportion of patients (no more than 1%) with solitary osteochondromas experience malignant transformation.2,3,6,7 No particular blood work is recommended for patients with solitary osteochondromas.2
Differential Diagnosis
In addition to osteochondromas, several other lesions should be considered in the patient with musculoskeletal lesions (see Table 15,6,13-19).
Cartilaginous tumors. Chondrosarcomas are malignant cartilaginous tumors.20-22 They commonly affect long bones, including the humerus and femur, and some flat bones, such as the pelvic bones.13,22 They are most commonly seen in adults, and have no predisposition by gender.13
Chondrosarcomas can be primary (ie, arising de novo) or secondary (developing on preexisting benign cartilaginous neoplasms, including osteochondromas). The majority of chondrosarcomas are slow growing, and they rarely metastasize. It is difficult to differentiate between a benign lesion (such as an osteochondroma) and a chondrosarcoma by either histology or radiology. However, reliable predictors for malignancy include size exceeding 5 cm and location in the axial skeleton.20
Bone tumors.Osteosarcomas are the most common malignant bone tumors in children and adolescents, with 400 to 560 US patients in this age-group diagnosed each year.14-16 Osteosarcomas are uncommon in children younger than 10; their incidence peaks during the early teenage years (median peak age, 16), then declines rapidly among older patients. They are more common in males than females.15
Osteosarcomas commonly develop during periods of rapid bone turnover, such as the adolescent growth spurt. Common sites include the distal femur, proximal humerus, and proximal tibia,15,16 particularly near the knee.13 Usually, osteosarcomas present with nonspecific symptoms, including strain-related pain of several months’ duration, which may disrupt sleep.16 Laboratory findings in affected patients may include elevations in LDH, alkaline phosphatase, and/or ESR.15,23
Physical exam reveals a visible or palpable mass in the affected area, along with decreased joint motion; localized warmth or erythema may also be present. Late signs of osteosarcoma include weight loss, general malaise, and fever. First-line imaging for the patient with a suspected osteosarcoma is x-ray, which will show ill-defined borders, osteoblastic and/or osteolytic features, and an associated soft tissue mass. Advanced imaging, such as MRI, is warranted.16
Ewing sarcoma, the second most common bone tumor in children and adolescents, is an aggressive form of childhood cancer.14,18 Approximately 25% of all Ewing sarcomas arise in soft tissues rather than bones.18 They are more common in whites than in other ethnic groups and have a slight male predominance.13,18 The median age at diagnosis is 15.13 The most common presenting symptoms are tumor related, such as pain or a noticeable mass. While x-rays are usually ordered first, MRI is preferred.18
Soft tissue tumors and masses.Rhabdomyosarcomas are malignancies that account for more than half of the soft tissue sarcomas in children and adolescents. Less than one-fifth of cases occur in the extremities, and most occur in children younger than 10. These lesions have a slight male predominance and are more common in whites than in other patients.14,17,24
Approximately 6% of childhood soft tissue tumors are adipose tissue tumors, which may be benign (eg, lipomas) or malignant (eg, liposarcomas). Lipomas in children account for nearly 4% of all soft tissue tumors and can be classified as superficial (which are often diagnosed clinically) or deep (frequently requiring imaging).25
Lymphomaaccounts for 7% of cancers in US children and adolescents and more than 25% of newly diagnosed cancers in patients between 15 and 19, making it the most common malignancy in adolescents and the third most common in children.26,27 Non-Hodgkin lymphoma is the fourth leading type of malignancy in US adolescents.27 Rarely, lymphomas present with primary event soft tissue involvement.28
Myositis ossificans (MO) is a rare benign disorder involving formation of heterotrophic bone in skeletal muscles and soft tissues.29 Though possible in patients of any age, MO is most commonly seen in adolescents and young adults. Often the result of soft tissue injury (in which case it is referred to as myositis ossificans circumscripta or traumatic), MO develops in areas that are exposed to trauma, such as the anterior thighs or arms. Lesions can be diagnosed via plain x-ray or CT, although MRI and ultrasound can also be useful evaluation tools.17,29,30
Because MO circumscripta typically presents as a painful soft tissue mass, it can be mistaken for a soft tissue sarcoma or an osteosarcoma; radiologic evaluation is required to make the proper diagnosis. Less common forms of MO are myositis ossificans progressiva and myositis ossificans without a history of trauma.29
Ollier diseaseis a rare, nonfamilial disorder characterized by multiple enchondromas (or enchondromatoses), which are distributed asymmetrically with areas of dysplastic cartilage. Enchondromas are benign cartilage tumors that frequently affect long tubular bones along the metaphyses in proximity to the growth plate. The enchondromas result in significant growth abnormalities. About one in 1 million people are diagnosed yearly.5,19 (Similarly, Maffucci syndrome is represented by multiple enchondromas in association with hemangiomas.5)
Ollier disease typically manifests during childhood5 with bone swelling, local pain, and palpable bony masses, which are often associated with bone deformities.19 Patients generally present with an asymmetric shortening of one extremity and the appearance of palpable bony masses on their fingers or toes, which may or may not be associated with pathologic fractures.5,19 In 20% to 50% of patients with Ollier disease, enchondromas are at risk for malignant transformation into chondrosarcomas.5
Vascular malformations. Certain abnormalities of vascular development cause birthmarks and abnormalities of varying degree in underlying tissues.31 They are usually present at birth and grow proportionally to the child’s growth.25,31 However, they can also be seen in later childhood and adolescence.
Radiologic Investigation
Plain radiography of the affected area is the first-line radiologic study to be performed.13 While most osteochondromas can be diagnosed by plain x-ray, cross-sectional imaging via CT or MRI is recommended in lesions with certain characteristics, such as a broad stalk or location in the axial skeleton. Because MRI involves no radiation exposure, it is a particularly good diagnostic tool for children.32
Ultrasound is a good imaging method for evaluating for complications of osteochondromas, including bursa formation or vascular compromise.32
Treatment and Management
Although usually asymptomatic, osteochondroma can trigger some significant symptoms. Osteochondromas are at risk for fracture and can cause body deformities, mechanical joint problems, weakness of the affected limb, numbness, vascular compression, aneurysm, arterial thrombosis, venous thrombosis, pain, acute ischemia, and nerve compression. Clinical signs of malignant transformation include pain, swelling, and increased lesion size.2
Surgical excision is recommended but should be delayed until after the patient has reached skeletal maturity in order to decrease the risk for recurrence.33
Patient Education and Follow-up
In addition to explaining appropriate pain management (eg, NSAIDs), it is especially important for the pediatric NP or PA to encourage the patient with a solitary osteochondroma to follow up with the pediatric orthopedic surgeon. Reasons include the need to monitor growth of the lesion (which is likely to continue in a patient who has not yet reached skeletal maturity) and assess for associated functional or joint problems. Patients should also be advised to seek the specialist’s attention if such problems develop or if pain increases.
Generally, the pediatric clinician should be sufficiently informed to answer questions about this condition from the patient or family. Any follow-up laboratory work recommended by the specialist can also be performed by the pediatric NP or PA.
OUTCOME FOR THE CASE PATIENT
MRI without contrast, as recommended by the radiologist to rule out a bursa or trauma to the osteochondroma, was considered an important part of the follow-up plan. As the patient had not yet reached skeletal maturity, she was referred to a pediatric orthopedic surgeon for possible excision of the lesion, due to its size and the pain associated with running or other exertion.
CONCLUSION
Solitary osteochondromas are the most common benign bone tumors. Although they are generally asymptomatic, pain and other symptoms can arise as a result of irritation to the adjacent structures. In this case, the patient’s chief complaint was an irritating “bump” that she had had on her right leg for at least six months.
Generally, follow-up monitoring of the osteochondroma and orthopedic follow-up care are warranted, at least until the patient reaches skeletal maturity. At that point, surgical excision of the lesion is recommended.
REFERENCES
1. Florez B, Mönckeberg J, Castillo G, Beguiristain J. Solitary osteochondroma long-term follow-up. J Pediatr Orthop B. 2008;17:91-94.
2. Kitsoulis P, Galani V, Stefanaki K, et al. Osteochondromas: review of the clinical, radiological and pathological features. In Vivo. 2008;22:633-646.
3. Ramos-Pascua LR, Sánchez-Herráez S, Alonso-Barrio JA, Alonso-León A. Solitary proximal end of femur osteochondroma: an indication and result of the en bloc resection without hip luxation [in Spanish]. Rev Esp Cir Ortop Traumatol. 2012;56:24-31.
4. Payne WT, Merrell G. Benign bony and soft tissue tumors of the hand. J Hand Surg. 2010;35:1901-1910.
5. Pannier S, Legeai-Mallet L. Hereditary multiple exostoses and enchondromatosis. Best Pract Res Clin Rheumatol. 2008;22:45-54.
6. Bovée JV. Multiple osteochondromas. Orphanet J Rare Dis. 2008;3(3).
7. Staals EL, Bacchini P, Mercuri M, Bertoni F. Dedifferentiated chondrosarcomas arising in preexisting osteochondromas. J Bone Joint Surg Am. 2007;89:987-993.
8. Singh R, Jain M, Siwach R, et al. Large para-articular osteochondroma of the knee joint: a case report. Acta Orthop Traumatol Turc. 2012;46:139-143.
9. Lee JY, Lee S, Joo KB, et al. Intraarticular osteochondroma of shoulder: a case report. Clin Imaging. 2013;37:379-381.
10. Kim Y-C, Ahn JH, Lee JW. Osteochondroma of the distal tibia complicated by a tibialis posterior tendon tear. J Foot Ankle Surg. 2012;51: 660-663.
11. Allagui M, Amara K, Aloui I, et al. Historical giant near-circumferential osteochondroma of the proximal humerus. J Shoulder Elbow Surg. 2010;19:e12-e15.
12. Li M, Luettringhaus T, Walker KR, Cole PA. Operative treatment of femoral neck osteochondroma through a digastric approach in a pediatric patient: a case report and review of the literature. J Pediatr Orthop B. 2012;21:230-234.
13. Hogendoorn PC, Athanasou N, Bielack S, et al; ESMO/EUROBONET Working Group. Bone sarcomas: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21 suppl 5:v204-v213.
14. Arndt CAS, Rose PS, Folpe AL, Laack NN. Common musculoskeletal tumors of childhood and adolescence. Mayo Clin Proc. 2012;87:475-487.
15. Ta HT, Dass CR, Choong PF, Dunstan DE. Osteosarcoma treatment: state of the art. Cancer Metastasis Rev. 2009;28:247-263.
16. Messerschmitt PJ, Garcia RM, Abdul-Karim FW, et al. Osteosarcoma. J Am Acad Orthop Surg. 2009;17:515-527.
17. Laffan EE, Ngan B-Y, Navarro OM. Pediatric soft-tissue tumors and pseudotumors: MR imaging features with pathologic correlation: Part 2. Tumors of fibroblastic/myofibroblastic, so-called fibrohistiocytic, muscular, lymphomatous, neurogenic, hair matrix, and uncertain origin. Radiographics. 2009;29:e36.
18. Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncol. 2010;11(2):184.
19. D’Angelo L, Massimi L, Narducci A, Di Rocco C. Ollier disease. Childs Nerv Syst. 2009;25:647-653.
20. Gelderblom H, Hogendoorn PC, Dijkstra SD, et al. The clinical approach towards chondrosarcoma. Oncologist. 2008;13:320-329.
21. Nosratzehi T, Pakfetrat A. Chondrosarcoma. Zahedan J Res Med Sci. 2013;15:64-64.
22. Prado FO, Nishimoto IN, Perez DE, et al. Head and neck chondrosarcoma: analysis of 16 cases. Br J Oral Maxillofacial Surg. 2009;47:555-557.
23. Kim HJ, Chalmers PN, Morris CD. Pediatric osteogenic sarcoma. Curr Opin Pediatr. 2010;22:61-66.
24. Sultan I, Qaddoumi I, Yaser S, et al. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol. 2009;27:3391-3397.
25. Navarro OM, Laffan EE, Ngan B-Y. Pediatric soft-tissue tumors and pseudo-tumors: MR imaging features with pathologic correlation: Part 1. Imaging approach, pseudotumors, vascular lesions, and adipocytic tumors. Radiographics. 2009;29:887-906.
26. Gross TG, Termuhlen AM. Pediatric non-Hodgkin lymphoma. Curr Hematol Malig Rep. 2008;3:167-173.
27. Hochberg J, Waxman IM, Kelly KM, et al. Adolescent non-Hodgkin lymphoma and Hodgkin lymphoma: state of the science. Br J Haematol. 2009;144:24-40.
28. Derenzini E, Casadei B, Pellegrini C, et al. Non-Hodgkin lymphomas presenting as soft tissue masses: A single center experience and meta-analysis of the published series. Clin Lymphoma Myeloma Leuk. 2012 Dec 12. [Epub ahead of print]
29. Micheli A, Trapani S, Brizzi I, et al. Myositis ossificans circumscripta: a paediatric case and review of the literature. Eur J Pediatr. 2009;168:523-529.
30. McKenzie G, Raby N, Ritchie D. Non-neoplastic soft-tissue masses. Br J Radiol. 2009;82:775-785.
31. Buckmiller LM, Richter GT, Suen JY. Diagnosis and management of hemangiomas and vascular malformations of the head and neck. Oral Dis. 2010;16:405-418.
32. Khanna G, Bennett DL. Pediatric bone lesions: beyond the plain radiographic evaluation. Semin Roentgenol. 2012;47:90-99.
33. Rijal L, Nepal P, Baral S, et al. Solitary diaphyseal exostosis of femur, how common is it? Eur J Orthop Surg Traumatol. 2011;21:363-365.
A girl, age 13 years, 4 months, presented to her primary care provider’s office for a well visit. Among her concerns, she mentioned a “bump” she had had on her right leg “for the past six months, maybe longer.” The area felt irritated when touched or when the patient “ran too much.” She had seen no change in the bump since she first noticed it. The patient knew of no trauma or other preceding factors. She denied any fever or warmth, redness, or ecchymosis to the area.
Medical history was unremarkable except for familial short stature and myopia. The patient was the fifth of eight children born to nonconsanguinous parents. She denied any surgical history or hospitalizations and was premenarcheal. She was up to date on all age-appropriate vaccines, with her meningococcal vaccine administered at that visit.
The patient’s blood pressure was 99/58 mm Hg with an apical pulse rate of 82 beats/min. Her growth parameters were following her curve. Her height was 55” (0.3 percentile); weight, 81 lb (7.5 percentile); and BMI, 18.8 (48.6 percentile).
The physical exam was normal with the exception of the musculoskeletal exam. Examination of the lower extremities revealed a palpable, 4 cm x 5 cm lesion at the right distal medial thigh just proximal to the knee. The lesion could not be visualized but on palpation was tender and firm. There was some question as to whether the lesion itself or inflamed soft tissue overlying the lesion was mobile. No overlying warmth, induration, erythema, or ecchymosis was noted.
Passive and active range of motion was intact at the hip and knee. No lesions to the upper extremities were evident, and no scoliosis was seen.
Blood work was done to rule out certain diagnoses. Results from a complete blood count with differential, lactate dehydrogenase (LDH), parathyroid hormone, lipid profiles, thyroid function, and a comprehensive metabolic profile were unremarkable. A low level of vitamin D 25-OH was detected: 21.7 ng/mL (normal range, 32 to 100 ng/mL).
Distal femur x-rays with posteroanterior, lateral, and oblique views were ordered. The imaging revealed a 3 cm x 3 cm lesion projecting from the “distal, somewhat medial” femur, which was diagnosed as a benign femoral osteochondroma. Significant inflammation to the surrounding soft-tissue structures was observed. A questionable old fracture of the osteochondroma was noted. The remaining bony structures and joints appeared normal.
An ultrasound of the lesion was also ordered to investigate soft-tissue swelling. This revealed a hypoechoic collar around the distal end of the osteochondroma, which could represent a fluid collection, hematoma from trauma, or bursitis. The soft tissues were deemed normal.
Because of the extent of inflammation, the radiologist recommended MRI without contrast to rule out bursitis or trauma to the osteochondroma.
DISCUSSION
Osteochondromas, which may be present in up to 3% of the general population, are the most common benign bone tumors.1-3 An osteochondroma is a cartilage-capped bony projection that arises on the external surface of the bone; it contains a marrow cavity that is continuous with the underlying bone.2,4 The majority of osteochondromas are solitary, accounting for perhaps 85% to 90% of all such lesions, and they are typically nonhereditary; the remaining 10% to 15% of osteochondromas are hereditary multiple osteochondromas or exostoses1,2 (see “Definition of Multiple Exostoses Syndrome”2,5,6,7).
Most lesions are painless and slow growing, and they usually occur in children and adolescents.2 They typically stop growing at skeletal maturity with the closure of the growth plates.3,8,9 There is no predilection for males or females in single lesions.2
Solitary osteochondromas typically appear in the lower extremities and at long tubular bone metaphyses,1-3,10 especially on the femur, humerus, tibia, spine, and hip. Any part of the skeleton can be affected, but 30% of lesions occur on the femur and 40% at either the proximal metaphysis of the tibia or the distal metaphysis of the femur.2,11
Most osteochondromas are asymptomatic and are found incidentally.1,3 However, some patients present with local pain as a result of irritation to adjacent structures, limitation of joint motion, growth disturbance, or fracture of the pedicle.3,4,9,11,12 A very small proportion of patients (no more than 1%) with solitary osteochondromas experience malignant transformation.2,3,6,7 No particular blood work is recommended for patients with solitary osteochondromas.2
Differential Diagnosis
In addition to osteochondromas, several other lesions should be considered in the patient with musculoskeletal lesions (see Table 15,6,13-19).
Cartilaginous tumors. Chondrosarcomas are malignant cartilaginous tumors.20-22 They commonly affect long bones, including the humerus and femur, and some flat bones, such as the pelvic bones.13,22 They are most commonly seen in adults, and have no predisposition by gender.13
Chondrosarcomas can be primary (ie, arising de novo) or secondary (developing on preexisting benign cartilaginous neoplasms, including osteochondromas). The majority of chondrosarcomas are slow growing, and they rarely metastasize. It is difficult to differentiate between a benign lesion (such as an osteochondroma) and a chondrosarcoma by either histology or radiology. However, reliable predictors for malignancy include size exceeding 5 cm and location in the axial skeleton.20
Bone tumors.Osteosarcomas are the most common malignant bone tumors in children and adolescents, with 400 to 560 US patients in this age-group diagnosed each year.14-16 Osteosarcomas are uncommon in children younger than 10; their incidence peaks during the early teenage years (median peak age, 16), then declines rapidly among older patients. They are more common in males than females.15
Osteosarcomas commonly develop during periods of rapid bone turnover, such as the adolescent growth spurt. Common sites include the distal femur, proximal humerus, and proximal tibia,15,16 particularly near the knee.13 Usually, osteosarcomas present with nonspecific symptoms, including strain-related pain of several months’ duration, which may disrupt sleep.16 Laboratory findings in affected patients may include elevations in LDH, alkaline phosphatase, and/or ESR.15,23
Physical exam reveals a visible or palpable mass in the affected area, along with decreased joint motion; localized warmth or erythema may also be present. Late signs of osteosarcoma include weight loss, general malaise, and fever. First-line imaging for the patient with a suspected osteosarcoma is x-ray, which will show ill-defined borders, osteoblastic and/or osteolytic features, and an associated soft tissue mass. Advanced imaging, such as MRI, is warranted.16
Ewing sarcoma, the second most common bone tumor in children and adolescents, is an aggressive form of childhood cancer.14,18 Approximately 25% of all Ewing sarcomas arise in soft tissues rather than bones.18 They are more common in whites than in other ethnic groups and have a slight male predominance.13,18 The median age at diagnosis is 15.13 The most common presenting symptoms are tumor related, such as pain or a noticeable mass. While x-rays are usually ordered first, MRI is preferred.18
Soft tissue tumors and masses.Rhabdomyosarcomas are malignancies that account for more than half of the soft tissue sarcomas in children and adolescents. Less than one-fifth of cases occur in the extremities, and most occur in children younger than 10. These lesions have a slight male predominance and are more common in whites than in other patients.14,17,24
Approximately 6% of childhood soft tissue tumors are adipose tissue tumors, which may be benign (eg, lipomas) or malignant (eg, liposarcomas). Lipomas in children account for nearly 4% of all soft tissue tumors and can be classified as superficial (which are often diagnosed clinically) or deep (frequently requiring imaging).25
Lymphomaaccounts for 7% of cancers in US children and adolescents and more than 25% of newly diagnosed cancers in patients between 15 and 19, making it the most common malignancy in adolescents and the third most common in children.26,27 Non-Hodgkin lymphoma is the fourth leading type of malignancy in US adolescents.27 Rarely, lymphomas present with primary event soft tissue involvement.28
Myositis ossificans (MO) is a rare benign disorder involving formation of heterotrophic bone in skeletal muscles and soft tissues.29 Though possible in patients of any age, MO is most commonly seen in adolescents and young adults. Often the result of soft tissue injury (in which case it is referred to as myositis ossificans circumscripta or traumatic), MO develops in areas that are exposed to trauma, such as the anterior thighs or arms. Lesions can be diagnosed via plain x-ray or CT, although MRI and ultrasound can also be useful evaluation tools.17,29,30
Because MO circumscripta typically presents as a painful soft tissue mass, it can be mistaken for a soft tissue sarcoma or an osteosarcoma; radiologic evaluation is required to make the proper diagnosis. Less common forms of MO are myositis ossificans progressiva and myositis ossificans without a history of trauma.29
Ollier diseaseis a rare, nonfamilial disorder characterized by multiple enchondromas (or enchondromatoses), which are distributed asymmetrically with areas of dysplastic cartilage. Enchondromas are benign cartilage tumors that frequently affect long tubular bones along the metaphyses in proximity to the growth plate. The enchondromas result in significant growth abnormalities. About one in 1 million people are diagnosed yearly.5,19 (Similarly, Maffucci syndrome is represented by multiple enchondromas in association with hemangiomas.5)
Ollier disease typically manifests during childhood5 with bone swelling, local pain, and palpable bony masses, which are often associated with bone deformities.19 Patients generally present with an asymmetric shortening of one extremity and the appearance of palpable bony masses on their fingers or toes, which may or may not be associated with pathologic fractures.5,19 In 20% to 50% of patients with Ollier disease, enchondromas are at risk for malignant transformation into chondrosarcomas.5
Vascular malformations. Certain abnormalities of vascular development cause birthmarks and abnormalities of varying degree in underlying tissues.31 They are usually present at birth and grow proportionally to the child’s growth.25,31 However, they can also be seen in later childhood and adolescence.
Radiologic Investigation
Plain radiography of the affected area is the first-line radiologic study to be performed.13 While most osteochondromas can be diagnosed by plain x-ray, cross-sectional imaging via CT or MRI is recommended in lesions with certain characteristics, such as a broad stalk or location in the axial skeleton. Because MRI involves no radiation exposure, it is a particularly good diagnostic tool for children.32
Ultrasound is a good imaging method for evaluating for complications of osteochondromas, including bursa formation or vascular compromise.32
Treatment and Management
Although usually asymptomatic, osteochondroma can trigger some significant symptoms. Osteochondromas are at risk for fracture and can cause body deformities, mechanical joint problems, weakness of the affected limb, numbness, vascular compression, aneurysm, arterial thrombosis, venous thrombosis, pain, acute ischemia, and nerve compression. Clinical signs of malignant transformation include pain, swelling, and increased lesion size.2
Surgical excision is recommended but should be delayed until after the patient has reached skeletal maturity in order to decrease the risk for recurrence.33
Patient Education and Follow-up
In addition to explaining appropriate pain management (eg, NSAIDs), it is especially important for the pediatric NP or PA to encourage the patient with a solitary osteochondroma to follow up with the pediatric orthopedic surgeon. Reasons include the need to monitor growth of the lesion (which is likely to continue in a patient who has not yet reached skeletal maturity) and assess for associated functional or joint problems. Patients should also be advised to seek the specialist’s attention if such problems develop or if pain increases.
Generally, the pediatric clinician should be sufficiently informed to answer questions about this condition from the patient or family. Any follow-up laboratory work recommended by the specialist can also be performed by the pediatric NP or PA.
OUTCOME FOR THE CASE PATIENT
MRI without contrast, as recommended by the radiologist to rule out a bursa or trauma to the osteochondroma, was considered an important part of the follow-up plan. As the patient had not yet reached skeletal maturity, she was referred to a pediatric orthopedic surgeon for possible excision of the lesion, due to its size and the pain associated with running or other exertion.
CONCLUSION
Solitary osteochondromas are the most common benign bone tumors. Although they are generally asymptomatic, pain and other symptoms can arise as a result of irritation to the adjacent structures. In this case, the patient’s chief complaint was an irritating “bump” that she had had on her right leg for at least six months.
Generally, follow-up monitoring of the osteochondroma and orthopedic follow-up care are warranted, at least until the patient reaches skeletal maturity. At that point, surgical excision of the lesion is recommended.
REFERENCES
1. Florez B, Mönckeberg J, Castillo G, Beguiristain J. Solitary osteochondroma long-term follow-up. J Pediatr Orthop B. 2008;17:91-94.
2. Kitsoulis P, Galani V, Stefanaki K, et al. Osteochondromas: review of the clinical, radiological and pathological features. In Vivo. 2008;22:633-646.
3. Ramos-Pascua LR, Sánchez-Herráez S, Alonso-Barrio JA, Alonso-León A. Solitary proximal end of femur osteochondroma: an indication and result of the en bloc resection without hip luxation [in Spanish]. Rev Esp Cir Ortop Traumatol. 2012;56:24-31.
4. Payne WT, Merrell G. Benign bony and soft tissue tumors of the hand. J Hand Surg. 2010;35:1901-1910.
5. Pannier S, Legeai-Mallet L. Hereditary multiple exostoses and enchondromatosis. Best Pract Res Clin Rheumatol. 2008;22:45-54.
6. Bovée JV. Multiple osteochondromas. Orphanet J Rare Dis. 2008;3(3).
7. Staals EL, Bacchini P, Mercuri M, Bertoni F. Dedifferentiated chondrosarcomas arising in preexisting osteochondromas. J Bone Joint Surg Am. 2007;89:987-993.
8. Singh R, Jain M, Siwach R, et al. Large para-articular osteochondroma of the knee joint: a case report. Acta Orthop Traumatol Turc. 2012;46:139-143.
9. Lee JY, Lee S, Joo KB, et al. Intraarticular osteochondroma of shoulder: a case report. Clin Imaging. 2013;37:379-381.
10. Kim Y-C, Ahn JH, Lee JW. Osteochondroma of the distal tibia complicated by a tibialis posterior tendon tear. J Foot Ankle Surg. 2012;51: 660-663.
11. Allagui M, Amara K, Aloui I, et al. Historical giant near-circumferential osteochondroma of the proximal humerus. J Shoulder Elbow Surg. 2010;19:e12-e15.
12. Li M, Luettringhaus T, Walker KR, Cole PA. Operative treatment of femoral neck osteochondroma through a digastric approach in a pediatric patient: a case report and review of the literature. J Pediatr Orthop B. 2012;21:230-234.
13. Hogendoorn PC, Athanasou N, Bielack S, et al; ESMO/EUROBONET Working Group. Bone sarcomas: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21 suppl 5:v204-v213.
14. Arndt CAS, Rose PS, Folpe AL, Laack NN. Common musculoskeletal tumors of childhood and adolescence. Mayo Clin Proc. 2012;87:475-487.
15. Ta HT, Dass CR, Choong PF, Dunstan DE. Osteosarcoma treatment: state of the art. Cancer Metastasis Rev. 2009;28:247-263.
16. Messerschmitt PJ, Garcia RM, Abdul-Karim FW, et al. Osteosarcoma. J Am Acad Orthop Surg. 2009;17:515-527.
17. Laffan EE, Ngan B-Y, Navarro OM. Pediatric soft-tissue tumors and pseudotumors: MR imaging features with pathologic correlation: Part 2. Tumors of fibroblastic/myofibroblastic, so-called fibrohistiocytic, muscular, lymphomatous, neurogenic, hair matrix, and uncertain origin. Radiographics. 2009;29:e36.
18. Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncol. 2010;11(2):184.
19. D’Angelo L, Massimi L, Narducci A, Di Rocco C. Ollier disease. Childs Nerv Syst. 2009;25:647-653.
20. Gelderblom H, Hogendoorn PC, Dijkstra SD, et al. The clinical approach towards chondrosarcoma. Oncologist. 2008;13:320-329.
21. Nosratzehi T, Pakfetrat A. Chondrosarcoma. Zahedan J Res Med Sci. 2013;15:64-64.
22. Prado FO, Nishimoto IN, Perez DE, et al. Head and neck chondrosarcoma: analysis of 16 cases. Br J Oral Maxillofacial Surg. 2009;47:555-557.
23. Kim HJ, Chalmers PN, Morris CD. Pediatric osteogenic sarcoma. Curr Opin Pediatr. 2010;22:61-66.
24. Sultan I, Qaddoumi I, Yaser S, et al. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol. 2009;27:3391-3397.
25. Navarro OM, Laffan EE, Ngan B-Y. Pediatric soft-tissue tumors and pseudo-tumors: MR imaging features with pathologic correlation: Part 1. Imaging approach, pseudotumors, vascular lesions, and adipocytic tumors. Radiographics. 2009;29:887-906.
26. Gross TG, Termuhlen AM. Pediatric non-Hodgkin lymphoma. Curr Hematol Malig Rep. 2008;3:167-173.
27. Hochberg J, Waxman IM, Kelly KM, et al. Adolescent non-Hodgkin lymphoma and Hodgkin lymphoma: state of the science. Br J Haematol. 2009;144:24-40.
28. Derenzini E, Casadei B, Pellegrini C, et al. Non-Hodgkin lymphomas presenting as soft tissue masses: A single center experience and meta-analysis of the published series. Clin Lymphoma Myeloma Leuk. 2012 Dec 12. [Epub ahead of print]
29. Micheli A, Trapani S, Brizzi I, et al. Myositis ossificans circumscripta: a paediatric case and review of the literature. Eur J Pediatr. 2009;168:523-529.
30. McKenzie G, Raby N, Ritchie D. Non-neoplastic soft-tissue masses. Br J Radiol. 2009;82:775-785.
31. Buckmiller LM, Richter GT, Suen JY. Diagnosis and management of hemangiomas and vascular malformations of the head and neck. Oral Dis. 2010;16:405-418.
32. Khanna G, Bennett DL. Pediatric bone lesions: beyond the plain radiographic evaluation. Semin Roentgenol. 2012;47:90-99.
33. Rijal L, Nepal P, Baral S, et al. Solitary diaphyseal exostosis of femur, how common is it? Eur J Orthop Surg Traumatol. 2011;21:363-365.
A girl, age 13 years, 4 months, presented to her primary care provider’s office for a well visit. Among her concerns, she mentioned a “bump” she had had on her right leg “for the past six months, maybe longer.” The area felt irritated when touched or when the patient “ran too much.” She had seen no change in the bump since she first noticed it. The patient knew of no trauma or other preceding factors. She denied any fever or warmth, redness, or ecchymosis to the area.
Medical history was unremarkable except for familial short stature and myopia. The patient was the fifth of eight children born to nonconsanguinous parents. She denied any surgical history or hospitalizations and was premenarcheal. She was up to date on all age-appropriate vaccines, with her meningococcal vaccine administered at that visit.
The patient’s blood pressure was 99/58 mm Hg with an apical pulse rate of 82 beats/min. Her growth parameters were following her curve. Her height was 55” (0.3 percentile); weight, 81 lb (7.5 percentile); and BMI, 18.8 (48.6 percentile).
The physical exam was normal with the exception of the musculoskeletal exam. Examination of the lower extremities revealed a palpable, 4 cm x 5 cm lesion at the right distal medial thigh just proximal to the knee. The lesion could not be visualized but on palpation was tender and firm. There was some question as to whether the lesion itself or inflamed soft tissue overlying the lesion was mobile. No overlying warmth, induration, erythema, or ecchymosis was noted.
Passive and active range of motion was intact at the hip and knee. No lesions to the upper extremities were evident, and no scoliosis was seen.
Blood work was done to rule out certain diagnoses. Results from a complete blood count with differential, lactate dehydrogenase (LDH), parathyroid hormone, lipid profiles, thyroid function, and a comprehensive metabolic profile were unremarkable. A low level of vitamin D 25-OH was detected: 21.7 ng/mL (normal range, 32 to 100 ng/mL).
Distal femur x-rays with posteroanterior, lateral, and oblique views were ordered. The imaging revealed a 3 cm x 3 cm lesion projecting from the “distal, somewhat medial” femur, which was diagnosed as a benign femoral osteochondroma. Significant inflammation to the surrounding soft-tissue structures was observed. A questionable old fracture of the osteochondroma was noted. The remaining bony structures and joints appeared normal.
An ultrasound of the lesion was also ordered to investigate soft-tissue swelling. This revealed a hypoechoic collar around the distal end of the osteochondroma, which could represent a fluid collection, hematoma from trauma, or bursitis. The soft tissues were deemed normal.
Because of the extent of inflammation, the radiologist recommended MRI without contrast to rule out bursitis or trauma to the osteochondroma.
DISCUSSION
Osteochondromas, which may be present in up to 3% of the general population, are the most common benign bone tumors.1-3 An osteochondroma is a cartilage-capped bony projection that arises on the external surface of the bone; it contains a marrow cavity that is continuous with the underlying bone.2,4 The majority of osteochondromas are solitary, accounting for perhaps 85% to 90% of all such lesions, and they are typically nonhereditary; the remaining 10% to 15% of osteochondromas are hereditary multiple osteochondromas or exostoses1,2 (see “Definition of Multiple Exostoses Syndrome”2,5,6,7).
Most lesions are painless and slow growing, and they usually occur in children and adolescents.2 They typically stop growing at skeletal maturity with the closure of the growth plates.3,8,9 There is no predilection for males or females in single lesions.2
Solitary osteochondromas typically appear in the lower extremities and at long tubular bone metaphyses,1-3,10 especially on the femur, humerus, tibia, spine, and hip. Any part of the skeleton can be affected, but 30% of lesions occur on the femur and 40% at either the proximal metaphysis of the tibia or the distal metaphysis of the femur.2,11
Most osteochondromas are asymptomatic and are found incidentally.1,3 However, some patients present with local pain as a result of irritation to adjacent structures, limitation of joint motion, growth disturbance, or fracture of the pedicle.3,4,9,11,12 A very small proportion of patients (no more than 1%) with solitary osteochondromas experience malignant transformation.2,3,6,7 No particular blood work is recommended for patients with solitary osteochondromas.2
Differential Diagnosis
In addition to osteochondromas, several other lesions should be considered in the patient with musculoskeletal lesions (see Table 15,6,13-19).
Cartilaginous tumors. Chondrosarcomas are malignant cartilaginous tumors.20-22 They commonly affect long bones, including the humerus and femur, and some flat bones, such as the pelvic bones.13,22 They are most commonly seen in adults, and have no predisposition by gender.13
Chondrosarcomas can be primary (ie, arising de novo) or secondary (developing on preexisting benign cartilaginous neoplasms, including osteochondromas). The majority of chondrosarcomas are slow growing, and they rarely metastasize. It is difficult to differentiate between a benign lesion (such as an osteochondroma) and a chondrosarcoma by either histology or radiology. However, reliable predictors for malignancy include size exceeding 5 cm and location in the axial skeleton.20
Bone tumors.Osteosarcomas are the most common malignant bone tumors in children and adolescents, with 400 to 560 US patients in this age-group diagnosed each year.14-16 Osteosarcomas are uncommon in children younger than 10; their incidence peaks during the early teenage years (median peak age, 16), then declines rapidly among older patients. They are more common in males than females.15
Osteosarcomas commonly develop during periods of rapid bone turnover, such as the adolescent growth spurt. Common sites include the distal femur, proximal humerus, and proximal tibia,15,16 particularly near the knee.13 Usually, osteosarcomas present with nonspecific symptoms, including strain-related pain of several months’ duration, which may disrupt sleep.16 Laboratory findings in affected patients may include elevations in LDH, alkaline phosphatase, and/or ESR.15,23
Physical exam reveals a visible or palpable mass in the affected area, along with decreased joint motion; localized warmth or erythema may also be present. Late signs of osteosarcoma include weight loss, general malaise, and fever. First-line imaging for the patient with a suspected osteosarcoma is x-ray, which will show ill-defined borders, osteoblastic and/or osteolytic features, and an associated soft tissue mass. Advanced imaging, such as MRI, is warranted.16
Ewing sarcoma, the second most common bone tumor in children and adolescents, is an aggressive form of childhood cancer.14,18 Approximately 25% of all Ewing sarcomas arise in soft tissues rather than bones.18 They are more common in whites than in other ethnic groups and have a slight male predominance.13,18 The median age at diagnosis is 15.13 The most common presenting symptoms are tumor related, such as pain or a noticeable mass. While x-rays are usually ordered first, MRI is preferred.18
Soft tissue tumors and masses.Rhabdomyosarcomas are malignancies that account for more than half of the soft tissue sarcomas in children and adolescents. Less than one-fifth of cases occur in the extremities, and most occur in children younger than 10. These lesions have a slight male predominance and are more common in whites than in other patients.14,17,24
Approximately 6% of childhood soft tissue tumors are adipose tissue tumors, which may be benign (eg, lipomas) or malignant (eg, liposarcomas). Lipomas in children account for nearly 4% of all soft tissue tumors and can be classified as superficial (which are often diagnosed clinically) or deep (frequently requiring imaging).25
Lymphomaaccounts for 7% of cancers in US children and adolescents and more than 25% of newly diagnosed cancers in patients between 15 and 19, making it the most common malignancy in adolescents and the third most common in children.26,27 Non-Hodgkin lymphoma is the fourth leading type of malignancy in US adolescents.27 Rarely, lymphomas present with primary event soft tissue involvement.28
Myositis ossificans (MO) is a rare benign disorder involving formation of heterotrophic bone in skeletal muscles and soft tissues.29 Though possible in patients of any age, MO is most commonly seen in adolescents and young adults. Often the result of soft tissue injury (in which case it is referred to as myositis ossificans circumscripta or traumatic), MO develops in areas that are exposed to trauma, such as the anterior thighs or arms. Lesions can be diagnosed via plain x-ray or CT, although MRI and ultrasound can also be useful evaluation tools.17,29,30
Because MO circumscripta typically presents as a painful soft tissue mass, it can be mistaken for a soft tissue sarcoma or an osteosarcoma; radiologic evaluation is required to make the proper diagnosis. Less common forms of MO are myositis ossificans progressiva and myositis ossificans without a history of trauma.29
Ollier diseaseis a rare, nonfamilial disorder characterized by multiple enchondromas (or enchondromatoses), which are distributed asymmetrically with areas of dysplastic cartilage. Enchondromas are benign cartilage tumors that frequently affect long tubular bones along the metaphyses in proximity to the growth plate. The enchondromas result in significant growth abnormalities. About one in 1 million people are diagnosed yearly.5,19 (Similarly, Maffucci syndrome is represented by multiple enchondromas in association with hemangiomas.5)
Ollier disease typically manifests during childhood5 with bone swelling, local pain, and palpable bony masses, which are often associated with bone deformities.19 Patients generally present with an asymmetric shortening of one extremity and the appearance of palpable bony masses on their fingers or toes, which may or may not be associated with pathologic fractures.5,19 In 20% to 50% of patients with Ollier disease, enchondromas are at risk for malignant transformation into chondrosarcomas.5
Vascular malformations. Certain abnormalities of vascular development cause birthmarks and abnormalities of varying degree in underlying tissues.31 They are usually present at birth and grow proportionally to the child’s growth.25,31 However, they can also be seen in later childhood and adolescence.
Radiologic Investigation
Plain radiography of the affected area is the first-line radiologic study to be performed.13 While most osteochondromas can be diagnosed by plain x-ray, cross-sectional imaging via CT or MRI is recommended in lesions with certain characteristics, such as a broad stalk or location in the axial skeleton. Because MRI involves no radiation exposure, it is a particularly good diagnostic tool for children.32
Ultrasound is a good imaging method for evaluating for complications of osteochondromas, including bursa formation or vascular compromise.32
Treatment and Management
Although usually asymptomatic, osteochondroma can trigger some significant symptoms. Osteochondromas are at risk for fracture and can cause body deformities, mechanical joint problems, weakness of the affected limb, numbness, vascular compression, aneurysm, arterial thrombosis, venous thrombosis, pain, acute ischemia, and nerve compression. Clinical signs of malignant transformation include pain, swelling, and increased lesion size.2
Surgical excision is recommended but should be delayed until after the patient has reached skeletal maturity in order to decrease the risk for recurrence.33
Patient Education and Follow-up
In addition to explaining appropriate pain management (eg, NSAIDs), it is especially important for the pediatric NP or PA to encourage the patient with a solitary osteochondroma to follow up with the pediatric orthopedic surgeon. Reasons include the need to monitor growth of the lesion (which is likely to continue in a patient who has not yet reached skeletal maturity) and assess for associated functional or joint problems. Patients should also be advised to seek the specialist’s attention if such problems develop or if pain increases.
Generally, the pediatric clinician should be sufficiently informed to answer questions about this condition from the patient or family. Any follow-up laboratory work recommended by the specialist can also be performed by the pediatric NP or PA.
OUTCOME FOR THE CASE PATIENT
MRI without contrast, as recommended by the radiologist to rule out a bursa or trauma to the osteochondroma, was considered an important part of the follow-up plan. As the patient had not yet reached skeletal maturity, she was referred to a pediatric orthopedic surgeon for possible excision of the lesion, due to its size and the pain associated with running or other exertion.
CONCLUSION
Solitary osteochondromas are the most common benign bone tumors. Although they are generally asymptomatic, pain and other symptoms can arise as a result of irritation to the adjacent structures. In this case, the patient’s chief complaint was an irritating “bump” that she had had on her right leg for at least six months.
Generally, follow-up monitoring of the osteochondroma and orthopedic follow-up care are warranted, at least until the patient reaches skeletal maturity. At that point, surgical excision of the lesion is recommended.
REFERENCES
1. Florez B, Mönckeberg J, Castillo G, Beguiristain J. Solitary osteochondroma long-term follow-up. J Pediatr Orthop B. 2008;17:91-94.
2. Kitsoulis P, Galani V, Stefanaki K, et al. Osteochondromas: review of the clinical, radiological and pathological features. In Vivo. 2008;22:633-646.
3. Ramos-Pascua LR, Sánchez-Herráez S, Alonso-Barrio JA, Alonso-León A. Solitary proximal end of femur osteochondroma: an indication and result of the en bloc resection without hip luxation [in Spanish]. Rev Esp Cir Ortop Traumatol. 2012;56:24-31.
4. Payne WT, Merrell G. Benign bony and soft tissue tumors of the hand. J Hand Surg. 2010;35:1901-1910.
5. Pannier S, Legeai-Mallet L. Hereditary multiple exostoses and enchondromatosis. Best Pract Res Clin Rheumatol. 2008;22:45-54.
6. Bovée JV. Multiple osteochondromas. Orphanet J Rare Dis. 2008;3(3).
7. Staals EL, Bacchini P, Mercuri M, Bertoni F. Dedifferentiated chondrosarcomas arising in preexisting osteochondromas. J Bone Joint Surg Am. 2007;89:987-993.
8. Singh R, Jain M, Siwach R, et al. Large para-articular osteochondroma of the knee joint: a case report. Acta Orthop Traumatol Turc. 2012;46:139-143.
9. Lee JY, Lee S, Joo KB, et al. Intraarticular osteochondroma of shoulder: a case report. Clin Imaging. 2013;37:379-381.
10. Kim Y-C, Ahn JH, Lee JW. Osteochondroma of the distal tibia complicated by a tibialis posterior tendon tear. J Foot Ankle Surg. 2012;51: 660-663.
11. Allagui M, Amara K, Aloui I, et al. Historical giant near-circumferential osteochondroma of the proximal humerus. J Shoulder Elbow Surg. 2010;19:e12-e15.
12. Li M, Luettringhaus T, Walker KR, Cole PA. Operative treatment of femoral neck osteochondroma through a digastric approach in a pediatric patient: a case report and review of the literature. J Pediatr Orthop B. 2012;21:230-234.
13. Hogendoorn PC, Athanasou N, Bielack S, et al; ESMO/EUROBONET Working Group. Bone sarcomas: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21 suppl 5:v204-v213.
14. Arndt CAS, Rose PS, Folpe AL, Laack NN. Common musculoskeletal tumors of childhood and adolescence. Mayo Clin Proc. 2012;87:475-487.
15. Ta HT, Dass CR, Choong PF, Dunstan DE. Osteosarcoma treatment: state of the art. Cancer Metastasis Rev. 2009;28:247-263.
16. Messerschmitt PJ, Garcia RM, Abdul-Karim FW, et al. Osteosarcoma. J Am Acad Orthop Surg. 2009;17:515-527.
17. Laffan EE, Ngan B-Y, Navarro OM. Pediatric soft-tissue tumors and pseudotumors: MR imaging features with pathologic correlation: Part 2. Tumors of fibroblastic/myofibroblastic, so-called fibrohistiocytic, muscular, lymphomatous, neurogenic, hair matrix, and uncertain origin. Radiographics. 2009;29:e36.
18. Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncol. 2010;11(2):184.
19. D’Angelo L, Massimi L, Narducci A, Di Rocco C. Ollier disease. Childs Nerv Syst. 2009;25:647-653.
20. Gelderblom H, Hogendoorn PC, Dijkstra SD, et al. The clinical approach towards chondrosarcoma. Oncologist. 2008;13:320-329.
21. Nosratzehi T, Pakfetrat A. Chondrosarcoma. Zahedan J Res Med Sci. 2013;15:64-64.
22. Prado FO, Nishimoto IN, Perez DE, et al. Head and neck chondrosarcoma: analysis of 16 cases. Br J Oral Maxillofacial Surg. 2009;47:555-557.
23. Kim HJ, Chalmers PN, Morris CD. Pediatric osteogenic sarcoma. Curr Opin Pediatr. 2010;22:61-66.
24. Sultan I, Qaddoumi I, Yaser S, et al. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol. 2009;27:3391-3397.
25. Navarro OM, Laffan EE, Ngan B-Y. Pediatric soft-tissue tumors and pseudo-tumors: MR imaging features with pathologic correlation: Part 1. Imaging approach, pseudotumors, vascular lesions, and adipocytic tumors. Radiographics. 2009;29:887-906.
26. Gross TG, Termuhlen AM. Pediatric non-Hodgkin lymphoma. Curr Hematol Malig Rep. 2008;3:167-173.
27. Hochberg J, Waxman IM, Kelly KM, et al. Adolescent non-Hodgkin lymphoma and Hodgkin lymphoma: state of the science. Br J Haematol. 2009;144:24-40.
28. Derenzini E, Casadei B, Pellegrini C, et al. Non-Hodgkin lymphomas presenting as soft tissue masses: A single center experience and meta-analysis of the published series. Clin Lymphoma Myeloma Leuk. 2012 Dec 12. [Epub ahead of print]
29. Micheli A, Trapani S, Brizzi I, et al. Myositis ossificans circumscripta: a paediatric case and review of the literature. Eur J Pediatr. 2009;168:523-529.
30. McKenzie G, Raby N, Ritchie D. Non-neoplastic soft-tissue masses. Br J Radiol. 2009;82:775-785.
31. Buckmiller LM, Richter GT, Suen JY. Diagnosis and management of hemangiomas and vascular malformations of the head and neck. Oral Dis. 2010;16:405-418.
32. Khanna G, Bennett DL. Pediatric bone lesions: beyond the plain radiographic evaluation. Semin Roentgenol. 2012;47:90-99.
33. Rijal L, Nepal P, Baral S, et al. Solitary diaphyseal exostosis of femur, how common is it? Eur J Orthop Surg Traumatol. 2011;21:363-365.
Man, 57, With Dyspnea After Chiropractic Manipulation
A 57-year-old man presented to the emergency department (ED) with a two-day history of worsening shortness of breath, light-headedness, and back pain. The patient, who had a history of ankylosing spondylitis, had been receiving weekly therapy from a chiropractor for about 10 years. One week before presenting to the ED, he had begun to undergo daily manipulations under anesthesia (MUA)—an aggressive chiropractic procedure that is administered while the patient is under monitored, procedural sedation. After the second day of treatment, the patient began to experience worsening back pain and progressive light-headedness and shortness of breath.
At a follow-up visit with his chiropractor, he was found to have decreased O2 saturation and was directed to go to the hospital for evaluation. On arrival at the ED, the patient was awake and alert. He had intact motor strength in all extremities, no sensory abnormalities, intact symmetric reflexes, and no bladder or bowel dysfunction, with a negative Babinski sign. His O2 saturation was 92% on 5 L of oxygen. An absence of breath sounds was noted on the left side.
Chest x-ray (see Figure 1) was performed, which demonstrated complete opacification of the left hemithorax, consistent with a large pleural effusion or hemothorax. CT scan of the thoracic spine showed diffuse ankylosis. A complex oblique coronal and transversely oriented fracture with 7 mm of displacement was identified, beginning at the right anterior inferior lateral margin of the T8 vertebral body and extending centrally and inferiorly to the left and right into the T9 vertebral body. The fracture continued through the right T9-10 neural foramen and what was probably the right fused T9-10 facet joint. The fracture exited through the left superior and lateral margin of the T10 vertebral body and the left T10-11 neural foramen (see Figures 2, 3, and 4).
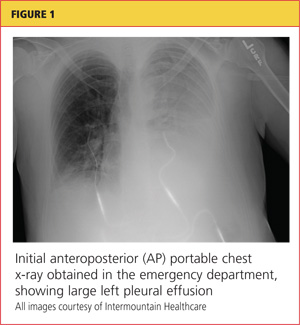
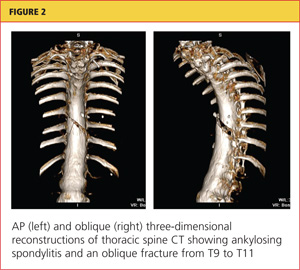
A chest tube was inserted in the ED, and 1,600 mL of old blood was immediately drained. The patient was admitted to the ICU on the trauma service. He was taken to surgery for open reduction and internal fixation of his unstable thoracic spine fracture on day 3 of hospitalization, after his pulmonary condition stabilized. Pedicle screws were placed from T7 through T12 during the spinal fusion. Good reduction of the fracture was observed following the spine surgery (see Figures 5 and 6). At the conclusion of surgery, an epidural catheter was placed in the thoracic spine to administer pain control.
After the spine portion of the procedure, the patient was repositioned and underwent video-assisted thoracoscopic surgery of the left hemithorax for evacuation of retained hemothorax. The patient tolerated the procedure well and was taken to the ICU for recovery.
On postoperative day 2, the patient complained of chest pain and experienced hypoxemia with activity. CT angiography of the chest demonstrated bilateral segmental and subsegmental pulmonary emboli. The epidural catheter was discontinued. Six hours later, a heparin drip was started, and the patient was transitioned to therapeutic enoxaparin and warfarin. When methicillin-sensitive Staphylococcus aureus (MSSA) was detected in his hemothorax fluid, he was treated with a course of nafcillin.
The patient was discharged to home on postoperative day 12. He has remained neurologically intact and has returned to his former work activities. He is not taking narcotic pain medications.
Discussion
Chiropractic care is a popular alternative health care modality in the United States. Researchers for the 2007 National Health Interview Study1 reported an annual use of chiropractic manipulation of 8.6%, while the Medical Expenditure Panel Survey2 data yielded an estimate of 12.6 million adults using chiropractic manipulation in 2006—translating to a prevalence of 5.6%. Despite the popularity of chiropractic medicine, few well-designed studies have been conducted to support its use.3,4 Because of its designation as an alternative therapy, however, chiropractic manipulation has not been subjected to rigorous efficacy and safety evaluations.5
Given the inconsistency of the evidence to support chiropractic manipulation, the practice's safety profile is a concern. The risks associated with spinal manipulation are generally described in case reports and small series. Most serious adverse events described in the literature are cerebrovascular in nature and tend to occur after cervical manipulation.6,7 Fractures after spine manipulation are exceedingly rare, and published literature on this topic consists of a few isolated case reports, with all fractures occurring in the cervical spine in patients with an underlying pathologic condition.8-10
In 2009, Gouveia et al5 reviewed the published literature regarding all adverse events resulting from chiropractic manipulation. The authors found one randomized controlled trial, two case-control studies, six prospective studies, 12 surveys, three retrospective studies, and 100 case reports. The spectrum of complications identified ranged from benign and transient, such as local discomfort, to far more serious: stroke, myelopathy, radiculopathy, subdural hematoma, spinal fluid leakage, cauda equina syndrome, herniated disc, diaphragmatic palsy, and vertebral fractures. The authors were unable to perform a true meta-analysis because of the heterogeneity of the data, but they concluded that complications associated with chiropractic procedures are "frequent."5
Manipulations Under Anesthesia
MUA is a procedure that combines chiropractic adjustments and manipulations with general anesthesia or procedural sedation.11 The theory behind this strategy is that the anesthesia or sedation reduces pain and muscle spasm that may hinder the manipulation, allowing the practitioner to more effectively break up joint adhesions and reduce segmental dysfunction than if the patient had not undergone anesthesia.11
MUA is generally indicated in patients who have not responded to a 4- to 8-week trial of traditional manipulation therapy.12 It is also considered in patients who have "painful and restricting muscular guarding [that] interferes with the performance of spinal adjustments, mobilizations, and soft tissue release techniques."13
In the chiropractic literature, between 3% and 10% of patients are estimated to be candidates for MUA.12,14 It is not completely clear, however, what diagnoses are most likely to be treated successfully with this technique. Contraindications to MUA are generally the same as those for manipulation in conscious patients. A published list of contraindications from the Committee for Manipulation under Anesthesia (2003)15 included malignancy with bony metastasis, tuberculosis of the bone, recent fracture, acute arthritis, acute gout, diabetic neuropathy, syphilitic articular lesions, excessive spinal osteoporosis, disk fragmentation, direct nerve root impingement, and evidence of cord or caudal compression by tumor, ankylosis, or other space-occupying lesions.
MUA generally begins with deep procedural sedation, managed by an anesthesiologist. Once an adequate level of sedation is achieved, the manipulations are performed. Both high- and low-velocity thrusts are used, but it is recommended that the force exerted should be much less, and the manipulations performed with more caution, than in patients who are not anesthetized.12
For the thoracic spine, the patient is manipulated in the supine position with the arms crossed over the chest. The practitioner places one hand in a fist under the spine with the other hand on the patient's crossed arms, then delivers an anterior-to-posterior thrust. This is repeated until all affected segments have been treated.11,12
Literature to support the use of MUA for various indications is largely anecdotal. The largest published series13 is of 177 patients with chronic spinal pain who each underwent three MUA sessions followed by four to six weeks of traditional manipulations. The authors found that pain, as measured by visual analog scale, was reduced by 62% in patients with cervical spine pain, and by 60% in patients with lumbar pain. No adverse events were reported in the study.
Kohlbeck and Haldeman12 reviewed the reported complications of MUA across all published literature. They found that in 17 published papers, the overall complication rate was 0.7%, mainly represented by transitory increased pain. No spinal fractures were reported.
This case demonstrates a rare but serious complication of chiropractic MUA. It is unclear exactly what mechanism of injury led to an unstable thoracic spine fracture with massive hemothorax, and the precise cause will probably never be known. The clinicians who treated the case patient find it curious that the reported rate of adverse events following this procedure is so low, but they suspect an element of reporting bias in the chiropractic literature.
Conclusion
Iatrogenic injury after chiropractic manipulation is uncommon, but it can be devastating. Few serious complications of chiropractic MUA have been reported, but the literature is lacking in well-designed research studies. Despite the dearth of clinical trials to support its safety and efficacy, use of MUA has continued in the chiropractic community. This case demonstrates that serious adverse outcomes can occur, and more rigorous studies are needed to delineate the true benefits and risks of this set of chiropractic procedures.
References
1. Barnes PM, Bloom B, Nahin RL. Complementary and alternative medicine use among adults and children: United States, 2007. Natl Health Stat Report. 2008;12:1-9.
2. Davis MA, Sirovich BE, Weeks WB. Utilization and expenditures on chiropractic care in the United States from 1997 to 2006. Health Serv Res. 2009;45:748-761.
3. Canadian Chiropractic Association; Canadian Federation of Chiropractic Regulatory Boards; Clinical Practice Guidelines Development Initiative; Guidelines Development Committee. Chiropractic clinical practice guideline: evidence-based treatment of adult neck pain not due to whiplash. J Can Chiropr Assoc. 2005;49:417-421.
4.
Hurwitz EL, Aker PD, Adams AH, et al. Manipulation and mobilization of the cervical spine: a systematic review of the literature. Spine (Phila Pa 1976). 1996;21:1746-1760.
5.Gouveia LO, Castanho P, Ferreira JJ. Safety of chiropractic interventions: a systematic review. Spine (Phila Pa 1976). 2009;34:E405-E413.
6. Di Fabio RP. Manipulation of the cervical spine: risks and benefits. Phys Ther. 1999;79:50-65.
7. Nadareishvili Z, Norris JW. Stroke from traumatic arterial dissection. Lancet. 1999;354:159-160.
8. Austin RT. Pathological vertebral fractures after spinal manipulation. Br Med J (Clin Res Ed). 1985;291:1114-1115.
9. Ea HK, Weber AJ, Yon F, Lioté F. Osteoporotic fracture of the dens revealed by cervical manipulation. Joint Bone Spine. 2004;71:246-250.
10. Schmitz A, Lutterbey G, von Engelhardt L, et al. Pathological cervical fracture after spinal manipulation in a pregnant patient. J Manipulative Physiol Ther. 2005;28:633-636.
11. Cremata E, Collins S, Clauson W, et al. Manipulation under anesthesia: a report of four cases. J Manipulative Physiol Ther. 2005;28:526-533.
12. Kohlbeck FJ, Haldeman S. Medication-assisted spinal manipulation. Spine J. 2002;2:288-302.
13. West DT, Mathews RS, Miller MR, Kent GM. Effective management of spinal pain in one hundred seventy-seven patients evaluated for manipulation under anesthesia. J Manipulative Physiol Ther. 1999;22:299-308.
14. Morey LW Jr. Osteopathic manipulation under general anesthesia. J Am Osteopath Assoc. 1973;73:116-127.
15. Tain L, Gunderson C, Cremata E, et al; Committee for Manipulation Under Anesthesia. Recommendations to the Industrial Medical Council Work Group of California for manipulation under anesthesia use for injured workers. Sacramento, CA: Industrial Medical Council; 2003.
A 57-year-old man presented to the emergency department (ED) with a two-day history of worsening shortness of breath, light-headedness, and back pain. The patient, who had a history of ankylosing spondylitis, had been receiving weekly therapy from a chiropractor for about 10 years. One week before presenting to the ED, he had begun to undergo daily manipulations under anesthesia (MUA)—an aggressive chiropractic procedure that is administered while the patient is under monitored, procedural sedation. After the second day of treatment, the patient began to experience worsening back pain and progressive light-headedness and shortness of breath.
At a follow-up visit with his chiropractor, he was found to have decreased O2 saturation and was directed to go to the hospital for evaluation. On arrival at the ED, the patient was awake and alert. He had intact motor strength in all extremities, no sensory abnormalities, intact symmetric reflexes, and no bladder or bowel dysfunction, with a negative Babinski sign. His O2 saturation was 92% on 5 L of oxygen. An absence of breath sounds was noted on the left side.
Chest x-ray (see Figure 1) was performed, which demonstrated complete opacification of the left hemithorax, consistent with a large pleural effusion or hemothorax. CT scan of the thoracic spine showed diffuse ankylosis. A complex oblique coronal and transversely oriented fracture with 7 mm of displacement was identified, beginning at the right anterior inferior lateral margin of the T8 vertebral body and extending centrally and inferiorly to the left and right into the T9 vertebral body. The fracture continued through the right T9-10 neural foramen and what was probably the right fused T9-10 facet joint. The fracture exited through the left superior and lateral margin of the T10 vertebral body and the left T10-11 neural foramen (see Figures 2, 3, and 4).
A chest tube was inserted in the ED, and 1,600 mL of old blood was immediately drained. The patient was admitted to the ICU on the trauma service. He was taken to surgery for open reduction and internal fixation of his unstable thoracic spine fracture on day 3 of hospitalization, after his pulmonary condition stabilized. Pedicle screws were placed from T7 through T12 during the spinal fusion. Good reduction of the fracture was observed following the spine surgery (see Figures 5 and 6). At the conclusion of surgery, an epidural catheter was placed in the thoracic spine to administer pain control.
After the spine portion of the procedure, the patient was repositioned and underwent video-assisted thoracoscopic surgery of the left hemithorax for evacuation of retained hemothorax. The patient tolerated the procedure well and was taken to the ICU for recovery.
On postoperative day 2, the patient complained of chest pain and experienced hypoxemia with activity. CT angiography of the chest demonstrated bilateral segmental and subsegmental pulmonary emboli. The epidural catheter was discontinued. Six hours later, a heparin drip was started, and the patient was transitioned to therapeutic enoxaparin and warfarin. When methicillin-sensitive Staphylococcus aureus (MSSA) was detected in his hemothorax fluid, he was treated with a course of nafcillin.
The patient was discharged to home on postoperative day 12. He has remained neurologically intact and has returned to his former work activities. He is not taking narcotic pain medications.
Discussion
Chiropractic care is a popular alternative health care modality in the United States. Researchers for the 2007 National Health Interview Study1 reported an annual use of chiropractic manipulation of 8.6%, while the Medical Expenditure Panel Survey2 data yielded an estimate of 12.6 million adults using chiropractic manipulation in 2006—translating to a prevalence of 5.6%. Despite the popularity of chiropractic medicine, few well-designed studies have been conducted to support its use.3,4 Because of its designation as an alternative therapy, however, chiropractic manipulation has not been subjected to rigorous efficacy and safety evaluations.5
Given the inconsistency of the evidence to support chiropractic manipulation, the practice's safety profile is a concern. The risks associated with spinal manipulation are generally described in case reports and small series. Most serious adverse events described in the literature are cerebrovascular in nature and tend to occur after cervical manipulation.6,7 Fractures after spine manipulation are exceedingly rare, and published literature on this topic consists of a few isolated case reports, with all fractures occurring in the cervical spine in patients with an underlying pathologic condition.8-10
In 2009, Gouveia et al5 reviewed the published literature regarding all adverse events resulting from chiropractic manipulation. The authors found one randomized controlled trial, two case-control studies, six prospective studies, 12 surveys, three retrospective studies, and 100 case reports. The spectrum of complications identified ranged from benign and transient, such as local discomfort, to far more serious: stroke, myelopathy, radiculopathy, subdural hematoma, spinal fluid leakage, cauda equina syndrome, herniated disc, diaphragmatic palsy, and vertebral fractures. The authors were unable to perform a true meta-analysis because of the heterogeneity of the data, but they concluded that complications associated with chiropractic procedures are "frequent."5
Manipulations Under Anesthesia
MUA is a procedure that combines chiropractic adjustments and manipulations with general anesthesia or procedural sedation.11 The theory behind this strategy is that the anesthesia or sedation reduces pain and muscle spasm that may hinder the manipulation, allowing the practitioner to more effectively break up joint adhesions and reduce segmental dysfunction than if the patient had not undergone anesthesia.11
MUA is generally indicated in patients who have not responded to a 4- to 8-week trial of traditional manipulation therapy.12 It is also considered in patients who have "painful and restricting muscular guarding [that] interferes with the performance of spinal adjustments, mobilizations, and soft tissue release techniques."13
In the chiropractic literature, between 3% and 10% of patients are estimated to be candidates for MUA.12,14 It is not completely clear, however, what diagnoses are most likely to be treated successfully with this technique. Contraindications to MUA are generally the same as those for manipulation in conscious patients. A published list of contraindications from the Committee for Manipulation under Anesthesia (2003)15 included malignancy with bony metastasis, tuberculosis of the bone, recent fracture, acute arthritis, acute gout, diabetic neuropathy, syphilitic articular lesions, excessive spinal osteoporosis, disk fragmentation, direct nerve root impingement, and evidence of cord or caudal compression by tumor, ankylosis, or other space-occupying lesions.
MUA generally begins with deep procedural sedation, managed by an anesthesiologist. Once an adequate level of sedation is achieved, the manipulations are performed. Both high- and low-velocity thrusts are used, but it is recommended that the force exerted should be much less, and the manipulations performed with more caution, than in patients who are not anesthetized.12
For the thoracic spine, the patient is manipulated in the supine position with the arms crossed over the chest. The practitioner places one hand in a fist under the spine with the other hand on the patient's crossed arms, then delivers an anterior-to-posterior thrust. This is repeated until all affected segments have been treated.11,12
Literature to support the use of MUA for various indications is largely anecdotal. The largest published series13 is of 177 patients with chronic spinal pain who each underwent three MUA sessions followed by four to six weeks of traditional manipulations. The authors found that pain, as measured by visual analog scale, was reduced by 62% in patients with cervical spine pain, and by 60% in patients with lumbar pain. No adverse events were reported in the study.
Kohlbeck and Haldeman12 reviewed the reported complications of MUA across all published literature. They found that in 17 published papers, the overall complication rate was 0.7%, mainly represented by transitory increased pain. No spinal fractures were reported.
This case demonstrates a rare but serious complication of chiropractic MUA. It is unclear exactly what mechanism of injury led to an unstable thoracic spine fracture with massive hemothorax, and the precise cause will probably never be known. The clinicians who treated the case patient find it curious that the reported rate of adverse events following this procedure is so low, but they suspect an element of reporting bias in the chiropractic literature.
Conclusion
Iatrogenic injury after chiropractic manipulation is uncommon, but it can be devastating. Few serious complications of chiropractic MUA have been reported, but the literature is lacking in well-designed research studies. Despite the dearth of clinical trials to support its safety and efficacy, use of MUA has continued in the chiropractic community. This case demonstrates that serious adverse outcomes can occur, and more rigorous studies are needed to delineate the true benefits and risks of this set of chiropractic procedures.
References
1. Barnes PM, Bloom B, Nahin RL. Complementary and alternative medicine use among adults and children: United States, 2007. Natl Health Stat Report. 2008;12:1-9.
2. Davis MA, Sirovich BE, Weeks WB. Utilization and expenditures on chiropractic care in the United States from 1997 to 2006. Health Serv Res. 2009;45:748-761.
3. Canadian Chiropractic Association; Canadian Federation of Chiropractic Regulatory Boards; Clinical Practice Guidelines Development Initiative; Guidelines Development Committee. Chiropractic clinical practice guideline: evidence-based treatment of adult neck pain not due to whiplash. J Can Chiropr Assoc. 2005;49:417-421.
4.
Hurwitz EL, Aker PD, Adams AH, et al. Manipulation and mobilization of the cervical spine: a systematic review of the literature. Spine (Phila Pa 1976). 1996;21:1746-1760.
5.Gouveia LO, Castanho P, Ferreira JJ. Safety of chiropractic interventions: a systematic review. Spine (Phila Pa 1976). 2009;34:E405-E413.
6. Di Fabio RP. Manipulation of the cervical spine: risks and benefits. Phys Ther. 1999;79:50-65.
7. Nadareishvili Z, Norris JW. Stroke from traumatic arterial dissection. Lancet. 1999;354:159-160.
8. Austin RT. Pathological vertebral fractures after spinal manipulation. Br Med J (Clin Res Ed). 1985;291:1114-1115.
9. Ea HK, Weber AJ, Yon F, Lioté F. Osteoporotic fracture of the dens revealed by cervical manipulation. Joint Bone Spine. 2004;71:246-250.
10. Schmitz A, Lutterbey G, von Engelhardt L, et al. Pathological cervical fracture after spinal manipulation in a pregnant patient. J Manipulative Physiol Ther. 2005;28:633-636.
11. Cremata E, Collins S, Clauson W, et al. Manipulation under anesthesia: a report of four cases. J Manipulative Physiol Ther. 2005;28:526-533.
12. Kohlbeck FJ, Haldeman S. Medication-assisted spinal manipulation. Spine J. 2002;2:288-302.
13. West DT, Mathews RS, Miller MR, Kent GM. Effective management of spinal pain in one hundred seventy-seven patients evaluated for manipulation under anesthesia. J Manipulative Physiol Ther. 1999;22:299-308.
14. Morey LW Jr. Osteopathic manipulation under general anesthesia. J Am Osteopath Assoc. 1973;73:116-127.
15. Tain L, Gunderson C, Cremata E, et al; Committee for Manipulation Under Anesthesia. Recommendations to the Industrial Medical Council Work Group of California for manipulation under anesthesia use for injured workers. Sacramento, CA: Industrial Medical Council; 2003.
A 57-year-old man presented to the emergency department (ED) with a two-day history of worsening shortness of breath, light-headedness, and back pain. The patient, who had a history of ankylosing spondylitis, had been receiving weekly therapy from a chiropractor for about 10 years. One week before presenting to the ED, he had begun to undergo daily manipulations under anesthesia (MUA)—an aggressive chiropractic procedure that is administered while the patient is under monitored, procedural sedation. After the second day of treatment, the patient began to experience worsening back pain and progressive light-headedness and shortness of breath.
At a follow-up visit with his chiropractor, he was found to have decreased O2 saturation and was directed to go to the hospital for evaluation. On arrival at the ED, the patient was awake and alert. He had intact motor strength in all extremities, no sensory abnormalities, intact symmetric reflexes, and no bladder or bowel dysfunction, with a negative Babinski sign. His O2 saturation was 92% on 5 L of oxygen. An absence of breath sounds was noted on the left side.
Chest x-ray (see Figure 1) was performed, which demonstrated complete opacification of the left hemithorax, consistent with a large pleural effusion or hemothorax. CT scan of the thoracic spine showed diffuse ankylosis. A complex oblique coronal and transversely oriented fracture with 7 mm of displacement was identified, beginning at the right anterior inferior lateral margin of the T8 vertebral body and extending centrally and inferiorly to the left and right into the T9 vertebral body. The fracture continued through the right T9-10 neural foramen and what was probably the right fused T9-10 facet joint. The fracture exited through the left superior and lateral margin of the T10 vertebral body and the left T10-11 neural foramen (see Figures 2, 3, and 4).
A chest tube was inserted in the ED, and 1,600 mL of old blood was immediately drained. The patient was admitted to the ICU on the trauma service. He was taken to surgery for open reduction and internal fixation of his unstable thoracic spine fracture on day 3 of hospitalization, after his pulmonary condition stabilized. Pedicle screws were placed from T7 through T12 during the spinal fusion. Good reduction of the fracture was observed following the spine surgery (see Figures 5 and 6). At the conclusion of surgery, an epidural catheter was placed in the thoracic spine to administer pain control.
After the spine portion of the procedure, the patient was repositioned and underwent video-assisted thoracoscopic surgery of the left hemithorax for evacuation of retained hemothorax. The patient tolerated the procedure well and was taken to the ICU for recovery.
On postoperative day 2, the patient complained of chest pain and experienced hypoxemia with activity. CT angiography of the chest demonstrated bilateral segmental and subsegmental pulmonary emboli. The epidural catheter was discontinued. Six hours later, a heparin drip was started, and the patient was transitioned to therapeutic enoxaparin and warfarin. When methicillin-sensitive Staphylococcus aureus (MSSA) was detected in his hemothorax fluid, he was treated with a course of nafcillin.
The patient was discharged to home on postoperative day 12. He has remained neurologically intact and has returned to his former work activities. He is not taking narcotic pain medications.
Discussion
Chiropractic care is a popular alternative health care modality in the United States. Researchers for the 2007 National Health Interview Study1 reported an annual use of chiropractic manipulation of 8.6%, while the Medical Expenditure Panel Survey2 data yielded an estimate of 12.6 million adults using chiropractic manipulation in 2006—translating to a prevalence of 5.6%. Despite the popularity of chiropractic medicine, few well-designed studies have been conducted to support its use.3,4 Because of its designation as an alternative therapy, however, chiropractic manipulation has not been subjected to rigorous efficacy and safety evaluations.5
Given the inconsistency of the evidence to support chiropractic manipulation, the practice's safety profile is a concern. The risks associated with spinal manipulation are generally described in case reports and small series. Most serious adverse events described in the literature are cerebrovascular in nature and tend to occur after cervical manipulation.6,7 Fractures after spine manipulation are exceedingly rare, and published literature on this topic consists of a few isolated case reports, with all fractures occurring in the cervical spine in patients with an underlying pathologic condition.8-10
In 2009, Gouveia et al5 reviewed the published literature regarding all adverse events resulting from chiropractic manipulation. The authors found one randomized controlled trial, two case-control studies, six prospective studies, 12 surveys, three retrospective studies, and 100 case reports. The spectrum of complications identified ranged from benign and transient, such as local discomfort, to far more serious: stroke, myelopathy, radiculopathy, subdural hematoma, spinal fluid leakage, cauda equina syndrome, herniated disc, diaphragmatic palsy, and vertebral fractures. The authors were unable to perform a true meta-analysis because of the heterogeneity of the data, but they concluded that complications associated with chiropractic procedures are "frequent."5
Manipulations Under Anesthesia
MUA is a procedure that combines chiropractic adjustments and manipulations with general anesthesia or procedural sedation.11 The theory behind this strategy is that the anesthesia or sedation reduces pain and muscle spasm that may hinder the manipulation, allowing the practitioner to more effectively break up joint adhesions and reduce segmental dysfunction than if the patient had not undergone anesthesia.11
MUA is generally indicated in patients who have not responded to a 4- to 8-week trial of traditional manipulation therapy.12 It is also considered in patients who have "painful and restricting muscular guarding [that] interferes with the performance of spinal adjustments, mobilizations, and soft tissue release techniques."13
In the chiropractic literature, between 3% and 10% of patients are estimated to be candidates for MUA.12,14 It is not completely clear, however, what diagnoses are most likely to be treated successfully with this technique. Contraindications to MUA are generally the same as those for manipulation in conscious patients. A published list of contraindications from the Committee for Manipulation under Anesthesia (2003)15 included malignancy with bony metastasis, tuberculosis of the bone, recent fracture, acute arthritis, acute gout, diabetic neuropathy, syphilitic articular lesions, excessive spinal osteoporosis, disk fragmentation, direct nerve root impingement, and evidence of cord or caudal compression by tumor, ankylosis, or other space-occupying lesions.
MUA generally begins with deep procedural sedation, managed by an anesthesiologist. Once an adequate level of sedation is achieved, the manipulations are performed. Both high- and low-velocity thrusts are used, but it is recommended that the force exerted should be much less, and the manipulations performed with more caution, than in patients who are not anesthetized.12
For the thoracic spine, the patient is manipulated in the supine position with the arms crossed over the chest. The practitioner places one hand in a fist under the spine with the other hand on the patient's crossed arms, then delivers an anterior-to-posterior thrust. This is repeated until all affected segments have been treated.11,12
Literature to support the use of MUA for various indications is largely anecdotal. The largest published series13 is of 177 patients with chronic spinal pain who each underwent three MUA sessions followed by four to six weeks of traditional manipulations. The authors found that pain, as measured by visual analog scale, was reduced by 62% in patients with cervical spine pain, and by 60% in patients with lumbar pain. No adverse events were reported in the study.
Kohlbeck and Haldeman12 reviewed the reported complications of MUA across all published literature. They found that in 17 published papers, the overall complication rate was 0.7%, mainly represented by transitory increased pain. No spinal fractures were reported.
This case demonstrates a rare but serious complication of chiropractic MUA. It is unclear exactly what mechanism of injury led to an unstable thoracic spine fracture with massive hemothorax, and the precise cause will probably never be known. The clinicians who treated the case patient find it curious that the reported rate of adverse events following this procedure is so low, but they suspect an element of reporting bias in the chiropractic literature.
Conclusion
Iatrogenic injury after chiropractic manipulation is uncommon, but it can be devastating. Few serious complications of chiropractic MUA have been reported, but the literature is lacking in well-designed research studies. Despite the dearth of clinical trials to support its safety and efficacy, use of MUA has continued in the chiropractic community. This case demonstrates that serious adverse outcomes can occur, and more rigorous studies are needed to delineate the true benefits and risks of this set of chiropractic procedures.
References
1. Barnes PM, Bloom B, Nahin RL. Complementary and alternative medicine use among adults and children: United States, 2007. Natl Health Stat Report. 2008;12:1-9.
2. Davis MA, Sirovich BE, Weeks WB. Utilization and expenditures on chiropractic care in the United States from 1997 to 2006. Health Serv Res. 2009;45:748-761.
3. Canadian Chiropractic Association; Canadian Federation of Chiropractic Regulatory Boards; Clinical Practice Guidelines Development Initiative; Guidelines Development Committee. Chiropractic clinical practice guideline: evidence-based treatment of adult neck pain not due to whiplash. J Can Chiropr Assoc. 2005;49:417-421.
4.
Hurwitz EL, Aker PD, Adams AH, et al. Manipulation and mobilization of the cervical spine: a systematic review of the literature. Spine (Phila Pa 1976). 1996;21:1746-1760.
5.Gouveia LO, Castanho P, Ferreira JJ. Safety of chiropractic interventions: a systematic review. Spine (Phila Pa 1976). 2009;34:E405-E413.
6. Di Fabio RP. Manipulation of the cervical spine: risks and benefits. Phys Ther. 1999;79:50-65.
7. Nadareishvili Z, Norris JW. Stroke from traumatic arterial dissection. Lancet. 1999;354:159-160.
8. Austin RT. Pathological vertebral fractures after spinal manipulation. Br Med J (Clin Res Ed). 1985;291:1114-1115.
9. Ea HK, Weber AJ, Yon F, Lioté F. Osteoporotic fracture of the dens revealed by cervical manipulation. Joint Bone Spine. 2004;71:246-250.
10. Schmitz A, Lutterbey G, von Engelhardt L, et al. Pathological cervical fracture after spinal manipulation in a pregnant patient. J Manipulative Physiol Ther. 2005;28:633-636.
11. Cremata E, Collins S, Clauson W, et al. Manipulation under anesthesia: a report of four cases. J Manipulative Physiol Ther. 2005;28:526-533.
12. Kohlbeck FJ, Haldeman S. Medication-assisted spinal manipulation. Spine J. 2002;2:288-302.
13. West DT, Mathews RS, Miller MR, Kent GM. Effective management of spinal pain in one hundred seventy-seven patients evaluated for manipulation under anesthesia. J Manipulative Physiol Ther. 1999;22:299-308.
14. Morey LW Jr. Osteopathic manipulation under general anesthesia. J Am Osteopath Assoc. 1973;73:116-127.
15. Tain L, Gunderson C, Cremata E, et al; Committee for Manipulation Under Anesthesia. Recommendations to the Industrial Medical Council Work Group of California for manipulation under anesthesia use for injured workers. Sacramento, CA: Industrial Medical Council; 2003.
Pregnant Woman, 39, With Hypertension and New-Onset Proteinuria
A 39-year-old black woman, gravida 1, para 0, with an intrauterine pregnancy of 34 weeks and three days (according to last menstrual period and nine-week ultrasound) presented to her Ob-Gyn office for a routine prenatal visit. She was found to have an elevated blood pressure with new onset of 2+ proteinuria. The patient was sent to the labor and delivery unit at the adjoining hospital for serial blood pressure readings, laboratory work, and fetal monitoring.
The patient’s previous medical history was limited to sinusitis. She was taking no prescription medications, and her only listed allergy was to pineapple. Initial lab studies revealed elevations in liver enzymes, lactate dehydrogenase (LDH), uric acid, and serum creatinine, as well as thrombocytopenia (see Table 11-5). She also had a critically low blood glucose level, which conflicted with a normal follow-up reading.

At this point, the patient was thought to have HELLP syndrome6 (ie, hemolysis, elevated liver enzymes, low platelet count), or possibly acute fatty liver of pregnancy (AFLP).2,4,7-11 Additional labs were drawn immediately to confirm or rule out AFLP. These included repeat serum glucose (following a second reading with normal results), a serum ammonia level, prothrombin time (PT), and partial thromboplastin time (PTT). The most reliable values to distinguish AFLP from HELLP are profound hypoglycemia (found in 94% of women with AFLP12) and an elevated serum ammonia level.4
Given the serious nature of either diagnosis, immediate delivery of the infant was deemed necessary. Because the patient’s cervix was not found favorable for induction, she underwent low-transverse cesarean delivery without complications. She was noted to have essentially normal anatomy with the exception of a small subserosal fibroid posteriorly. Meconium-stained amniotic fluid was present. A male infant was delivered, weighing 5 lb with 1-minute and 5-minute Apgar scores of 8 and 9, respectively.
Postoperatively, the patient remained in the recovery area, where she received intensive monitoring. She experienced fluctuations in blood glucose, ranging from 33 to 144 mg/dL; she was started on 5% dextrose in lactated Ringer’s solution and treated with IV dextrose 50 g. While the patient was in surgical recovery, results from the second set of labs, drawn before surgery, were returned; findings included an elevated ammonia level and an abnormal coagulation panel, including PT of 25.3 sec, PTT of 48.4 sec, and a fibrinogen level of 116 mg/dL, confirming the suspected diagnosis of AFLP.
Magnesium sulfate, which had been started immediately postop, was discontinued on confirmation of the diagnosis of AFLP. The patient was initially somnolent as a result of general anesthesia but gradually returned to a fully normal sensorium by early morning on postop day 1. Postoperatively, the patient’s hemoglobin was found to be low (8.6 g/dL; reference range, 13.5 to 18.5 g/dL), so she was transfused with two units of packed red blood cells (PRBCs) and given fresh frozen plasma (FFP) to correct this coagulopathy. The patient’s platelets were also low at 82,000/mm3 (reference range, 140,000 to 340,000/mm3).
On postop day 1, the patient’s serum creatinine rose to 4.2 mg/dL and her total bilirubin increased to 14.4 mg/dL (reference ranges, 0.6 to 1.2 mg/dL and < 1.0 mg/dL, respectively). Given the multiple systems affected by AFLP and the need for intensive supportive care, the patient was transferred to the ICU.
On her arrival at the ICU, the patient’s vital signs were initially stable, and she was alert and oriented. However, within the next few hours, she became hypotensive and encephalopathic. She required aggressive fluid resuscitation and multiple transfusions of PRBCs and FFP due to persistent anemia and coagulopathy. Her vital signs were stabilized, but she continued to need blood transfusions.
Postop day 2, the patient became less responsive and was soon unable to follow commands or speak clearly. Her breathing remained stable with just 3 L of oxygen by nasal cannula, but in order to prevent aspiration and in consideration of a postoperative ileus, it was necessary to place a nasogastric tube with low intermittent suction. This produced a bloody return, but no intervention other than close monitoring and transfusion was performed at that time.
Abdominal ultrasound showed ascites and mild left-sided hydronephrosis with no gallstones. The common bile duct measured 3 mm in diameter.
Although liver biopsy is considered the gold standard for a confirmed diagnosis of AFLP,13,14 this procedure was contraindicated by the patient’s coagulopathy. Concern was also expressed by one consultant that the patient might have thrombotic thrombocytopenic purpura (TTP) in addition to AFLP. TTP can manifest with similar findings, such as anemia, thrombocytopenia, neurologic symptoms, and renal abnormalities, but usually fever is involved, and the patient was afebrile. A catheter was placed for hemodialysis and therapeutic plasma exchange (TPE). Given that TTP-associated mortality is significantly decreased by use of TPE,15 this intervention was deemed prudent. The patient underwent TPE on three consecutive days, postop days 2 through 4.
The patient’s mental status began to improve, and by postop day 6, she was able to follow commands and engage in brief conversations. By postop day 9, she had returned almost completely to her baseline mental status.
The patient’s liver function test results and total bilirubin, ammonia, and creatinine levels all improved over the first few postoperative days but began to rise again by day 6. In response to worsening renal and hepatic functioning, the decision was made on postop day 9 to transfer the patient to a hospital with liver transplantation capabilities, should this procedure become necessary.
Discussion
AFLP is a rare condition specific to pregnancy, affecting 1/7,000 to 1/20,000 pregnancies. Due to the low incidence of this disease, randomized controlled trials to study it are not possible. Instead, clinicians must learn either from individual case studies or from retrospective syntheses of cases reported over time.1,2,7 Fortunately, the wealth of information gleaned over the past 30 years has significantly reduced AFLP-associated maternal and fetal mortality and morbidity rates. In the 1980s, maternal and fetal mortality rates as high as 85% were reported.3 Worldwide, maternal mortality associated with AFLP has decreased significantly to 7% to 18%, whereas the fetal mortality rate has fallen to between 9% and 23%.1,16,17
Common trends among women who have developed AFLP include nulliparity, multiparity, and advanced maternal age. One retrospective study of 57 women who had developed AFLP revealed that 35 cases (61%) involved first-time pregnancies. It also showed that 10 (18%) of the women had twins, and 14 (25%) were older than 35.2 In another study of 35 cases of AFLP, 40% of the women were nulliparous, and 11.4% were multiparous, including one triplet gestation.12 In a third, smaller study, 80% of women affected by AFLP were multiparous.10 Currently, there is no known evidence linking any maternal behavior to development of AFLP.
Presentation
Women who present with AFLP often experience vague, nonspecific symptoms, leading to misdiagnosis or delayed diagnosis. Objective measurements, including physical exam findings, laboratory studies, and other diagnostic tests, will help with a diagnosis. The most frequent initial symptoms are nausea and vomiting (in 70% of patients) and abdominal pain (50% to 80%), epigastric or right upper-quadrant.3 Other common symptoms include fatigue, malaise, anorexia, weight gain, polyuria, and polydipsia.2,3,9,18,19
Because the presenting symptoms in AFLP can be vague, clinicians should complete a thorough physical exam to differentiate accurately among conditions associated with pregnancy. Physical signs present in women with AFLP can include jaundice, ascites, edema, confusion, abdominal tenderness, and fever. More severe cases can present with multisystem involvement, including acute renal failure, gastrointestinal bleeding, pancreatitis, coagulopathy, and hepatic encephalopathy.3,4,9,18
Diagnostic Tests
Relevant laboratory tests include a complete blood count (CBC), liver studies, chemistry, coagulation studies, and urinalysis (see Table 1). Viral causes should be ruled out by way of a hepatitis panel.3 In AFLP, the CBC may show elevated white blood cells, decreased hemoglobin and hematocrit, and decreased platelets. Liver studies show elevated hepatic aminotransferase, bilirubin, LDH, and ammonia levels. Chemistry results show elevated blood urea nitrogen and creatinine, and decreased blood glucose. Coagulation factors are affected, and prolonged PTT, decreased fibrinogen, and proteinurea may also be found.9
Though invasive and not often necessary4,13 (and not possible for the case patient), the definitive diagnostic test for AFLP is liver biopsy.13,14 Biopsy reveals a microvesicular fatty infiltration of the hepatocytes as minute fat droplets surrounding a centrally located nucleus. These fatty infiltrates stain with oil red O, specific for fat. Inflammation is present in 50% of cases. There may also be a picture similar to cholestasis with bile thrombi or deposits within the hepatocytes.20
Due to the risk for hemorrhage and the critical status of women with AFLP, biopsy is often not possible. Ultrasonography may show increased echogenicity; CT may show decreased or diffuse attenuation in the liver. These imaging studies, though possibly helpful in severe cases, often yield false-negative results.3,20
In the absence of another explanation for the patient’s symptoms, the Swansea criteria are used for diagnosis of AFLP.1 Six or more of the following criteria must be present to confirm this diagnosis: vomiting, abdominal pain, polydipsia or polyuria, encephalopathy, leukocytosis, elevated bilirubin, elevated liver enzymes, elevated ammonia, hypoglycemia, renal dysfunction, coagulopathy, elevated uric acid, ascites on ultrasound, and microvesicular steatosis on liver biopsy.1,2,5
Pathophysiology
Normal functions of the liver include metabolism, protein synthesis, and manufacturing of blood coagulation proteins. These functions are disturbed in the presence of AFLP. Thus, women with this disease experience signs and symptoms related directly to the dysfunction of these processes.20-22
Disturbances in the hepatocytes due to excess fatty acids impair the liver’s ability to convert unconjugated bilirubin into conjugated bilirubin, causing plasma levels of unconjugated bilirubin to rise. This increase in bilirubin explains the jaundiced appearance of women with AFLP. AFLP is often thought to occur in conjunction with preeclampsia in many, but not all, patients. Thrombocytopenia in these patients is felt to be secondary to peripheral vascular consumption. Conjugated bilirubin levels may also be increased due to decreased flow of conjugated bilirubin into the common bile duct.21
Another liver function that is disrupted is that of glycogen storage and conversion to glucose, and the liver’s ability to convert nutrients into glycogen is also impaired. Decreased storage of glycogen, along with the liver’s inability to break down previously stored glycogen, causes a decrease in serum glucose levels. Women with AFLP often require treatment with IV dextrose in response to marked hypoglycemia.16,21,23
The liver dysfunction associated with AFLP reduces adequate production of clotting factors and coagulation proteins. Thrombocytopenia, elevated clotting times, and bleeding are all problems seen in AFLP. Mild to moderate elevations in serum aminotransferases and elevated LDH also occur in patients with AFLP.23,24
Genetic Factor
There is little known about the etiology of AFLP, although recent data point to a genetic component that was found in as many as 62% of mothers in one study and in 25% of infants in another study.20-22 Fatty acid oxidation (FAO) is one of the processes of hepatic mitochondria, a process that relies on several enzymes. When FAO is interrupted, fatty acids are deposited in the liver cells, as seen in histologic studies of AFLP.25,26 The common thread in women with this disease is a mutation in one of the enzymes needed for FAO. This enzyme is the long-chain 3-hydroxyacyl-CoA dehydrogenase. Deficiencies in this enzyme are common in mothers with AFLP and their infants.3,16,20,23,27
Differential Diagnosis
Several complications of pregnancy that involve the liver may, on presentation, mimic AFLP.16,20,23,24,28 The most common are hyperemesis gravidarum and intrahepatic cholestasis of pregnancy23 (see Table 216,20,23,24,28); others are preeclampsia/eclampsia and HELLP syndrome. It is important to distinguish between the signs and symptoms associated with each of these disorders in order to provide the most effective treatment. Hepatitis serologies are important in the differential diagnosis when liver enzyme levels are exceptionally high.4,16,22,28
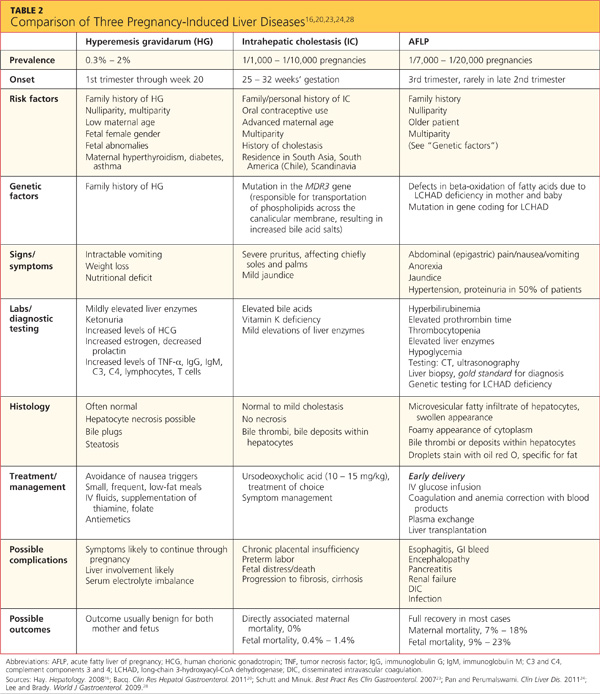
Treatment
The most effective treatment for AFLP is delivery of the infant; often, this alone causes the signs and symptoms of AFLP to resolve.8,21,27,29 In two of three cases in a small study by Aso et al,8 early delivery of the fetus led to complete resolution of symptoms and return to normal liver function. One of these patients was sent home four days after delivery; the other, 14 days later. Other patients may require more invasive treatment and support.8
Management in the ICU is often required to provide appropriate supportive care to the mother after delivery. Acute respiratory distress syndrome, pancreatitis, hemorrhage, encephalopathy, renal failure, and continual liver failure are among the severe complications associated with AFLP.4,8,10 Many women require intubation, dialysis, fluid resuscitation, blood product transfusion, and vasopressor therapy.3,8,11 Prophylactic antibiotics, IV steroids, and glucose may all be required in the supportive care and recovery of a mother with AFLP.3,8,11
TPE has also been useful in instances of severe complications.1,3,6 In one retrospective study, Martin et al1 recommended administration of TPE in patients with AFLP under the following circumstances:
(1) Deteriorating central nervous system abnormalities, such as sensorium changes or coma;
(2) Persistent coagulopathy requiring continued and aggressive blood product support with plasma, red cells, and/or cryoprecipitate;
(3) Advanced renal dysfunction that compromised fluid management;
(4) Progressive cardiopulmonary compromise; and/or
(5) Ongoing fluid management concerns, including significant ascites, edema, anuria/oliguria, and/or fluid overload.1
In rare cases, liver transplantation is needed in patients with AFLP. Westbrook et al18 reviewed 54 cases of liver disease in pregnancy in one UK hospital between 1997 and 2008. Of these, six patients with encephalopathy or elevated lactate were listed for liver transplant, including just one with a diagnosis of AFLP. This woman never actually underwent transplant but recovered in response to medical management alone.18 According to data reported in June 2011 by the Organ Procurement and Transplantation Network,30 liver transplantation was needed in only three US patients with AFLP between 2000 and 2011. Further retrospective studies on outcomes from transplant versus medical management should be considered to guide future decision making involving this invasive therapy.
The Case Patient
This 39-year-old patient presented during a routine prenatal visit with proteinuria and hypertension, possibly indicative of preeclampsia. Because of the serious nature of this potential diagnosis in pregnancy, she was admitted for monitoring and further testing. Although the diagnosis of AFLP was not confirmed until later, the patient’s preliminary lab studies showed elevated liver enzymes and low platelet counts, signifying the need for prompt intervention and delivery of the infant. At this point, the patient met criteria for HELLP syndrome, but AFLP was suspected after the initial finding of profound hypoglycemia led to further testing.
As an older mother experiencing pregnancy for the first time, this patient fit the profile for AFLP. She initially responded well after delivery of her infant but continued to experience complications. On the days that the patient was treated with TPE, her total bilirubin and liver enzymes were at their lowest. Perhaps this treatment should be considered in more cases of AFLP.
The patient was transferred to a hospital with liver transplantation capabilities, but she ultimately recovered without undergoing transplant.
Conclusion
For the primary obstetric care provider, being aware of the possible complications associated with pregnancy is important. Though uncommon, AFLP is a serious complication that should be ruled out in women who present with vague symptoms such as nausea, vomiting, and abdominal pain in the third trimester of pregnancy. The reduction in AFLP-associated morbidity and mortality during the past 20 years is a direct result of increased early recognition and therapeutic delivery.
Referral to a maternal fetal medicine specialist, gastroenterologist, hematologist, and/or nephrologist may be necessary and appropriate in the management of a woman with AFLP. Further study is indicated for use of TPE in more severe cases of AFLP, particularly in women affected by persistent thrombocytopenia and anemia.
The author would like to thank C. Leanne Browning, MD, obstetrics/gynecology, for her invaluable guidance and advice on this project.
References
1. Martin JN Jr, Briery CM, Rose CH, et al. Postpartum plasma exchange as adjunctive therapy for severe acute fatty liver of pregnancy. J Clin Apher. 2009;23(4):138-143.
2. Knight M, Nelson-Piercy C, Kurinczuk JJ; UK Obstetric Surveillance System. A prospective national study of acute fatty liver of pregnancy in the UK. Gut. 2008;57(7):951-956.
3. Barsoom MJ, Tierney BJ. Acute fatty liver of pregnancy (2011). http://emedicine.medscape.com/article/1562425-overview. Accessed January 21, 2013.
4. Ko HH, Yoshida E. Acute fatty liver of pregnancy. Can J Gastroenterol. 2006;20(1):25-30.
5. Rathi U, Bapat M, Rathi P, Abraham P. Effect of liver disease on maternal and fetal outcome: a prospective study. Indian J Gastroenterol. 2007;26(2):59-63.
6. Myers L. Postpartum plasma exchange in a woman with suspected thrombotic thrombocytopenic purpura (TTP) vs hemolysis, elevated liver enzymes, and low platelet syndrome (HELLP): a case study. Nephrol Nurs J. 2010;37(4):399-402.
7. Vigil-de Gracia P. Acute fatty liver and HELLP syndrome: two distinct pregnancy disorders. Int J Gynaecol Obstet. 2001;73(3):215-220.
8. Aso K, Hojo S, Yumoto Y, et al. Three cases of acute fatty liver of pregnancy: postpartum clinical course depends on interval between onset of symptoms and termination of pregnancy. J Matern Fetal Neonatal Med. 2010;23(9):1047-1049.
9. Wei Q, Zhang L, Liu X. Clinical diagnosis and treatment of acute fatty liver of pregnancy: a literature review and 11 new cases. J Obstet Gynaecol Res. 2010;36(4):751-756.
10. Barber MA, Eguiluz I, Martin A, et al. Acute fatty liver of pregnancy: analysis of five consecutive cases from a tertiary centre. J Obstet Gynaecol. 2010;30(3):241-243.
11. Ajayi AO, Alao MO. Case report: acute fatty liver of pregnancy in a 30-year-old Nigerian primigravida. Niger J Clin Pract. 2008;11(4):389-391.
12. Vigíl-de Gracia P, Montufar-Rueda C. Acute fatty liver of pregnancy: diagnosis, treatment, and outcome based on 35 consecutive cases. J Matern Fetal Neonatal Med. 2011;24(9):1143-1146.
13. Dey M, Reema K. Acute fatty liver of pregnancy. N Am J Med Sci. 2012;4(11):611-612.
14. Castro MA, Goodwin TM, Shaw KJ, et al. Disseminated intravascular coagulation and antithrombin III depression in acute fatty liver of pregnancy. Am J Obstet Gynecol. 1996;174(1 pt 1):211-216.
15. Altuntas F, Aydogdu I, Kabukcu S, et al. Therapeutic plasma exchange for the treatment of thrombotic thrombocytopenic purpura: a retrospective multicenter study. Transfus Apher Sci. 2007;36(1):57-67.
16. Hay JE. Liver disease in pregnancy. Hepatology. 2008;47(3):1067-1076.
17. Wand S, Waeschle RM, Von Ahsen N, et al. Acute fatty liver failure due to acute fatty liver of pregnancy. Minerva Anesthesiol. 2012;78(4):503-506.
18. Westbrook RH, Yeoman AD, Joshi D, et al. Outcomes of severe pregnancy-related liver disease: refining the role of transplantation. Am J Transplant. 2010;10(11):2520-2526.
19. Fesenmeier MF, Coppage KH, Lambers DS, et al. Acute fatty liver of pregnancy in 3 tertiary care centers. Am J Obstet Gynecol. 2005;192(5):1416-1419.
20. Bacq Y. Liver diseases unique to pregnancy: a 2010 update. Clin Res Hepatol Gastroenterol. 2011;35(3):182-193.
21. Huether SE. Alterations of digestive function. In: McCance KL, Huether SE, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 6th ed. St. Louis, MO: Mosby; 2009:1452-1515.
22. Huether SE. Structure and function of the digestive system. In: McCance KL, Huether SE, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 6th ed. St. Louis, MO: Mosby; 2009:1420-1451.
23. Schutt VA, Minuk GY. Liver diseases unique to pregnancy. Best Pract Res Clin Gastroenterol. 2007;21(5):771-792.
24. Pan C, Perumalswami PV. Pregnancy-related liver diseases. Clin Liver Dis. 2011;15(1):199-208.
25. Ibdah JA. Acute fatty liver of pregnancy: an update on pathogenesis and clinical implications. World J Gastroenterol. 2006;12(46):7397-7404.
26. Browning MF, Levy HL, Wilkins-Haug LE, et al. Fetal fatty acid oxidation defects and maternal liver disease in pregnancy. Obstet Gynecol. 2006;107(1):115-120.
27. Dekker RR, Schutte JM, Stekelenburg J, et al. Maternal mortality and severe maternal morbidity from acute fatty liver of pregnancy in the Netherlands. Eur J Obstet Gynecol Reprod Biol. 2011;157(1):27-31.
28. Lee NM, Brady CW. Liver disease in pregnancy. World J Gastroenterol. 2009;15(8):897-906.
29. Vora KS, Shah VR, Parikh GP. Acute fatty liver of pregnancy: a case report of an uncommon disease. Indian J Crit Care Med. 2009;13(1):34-36.
30. Organ Procurement and Transplantation Network, Scientific Registry of Transplant Recipients. OPTN/SRTR 2011 Annual Data Report: Liver. http://srtr.transplant.hrsa.gov/annual_reports/2011/pdf/03_%20liver_12.pdf. Accessed January 18, 2013.
A 39-year-old black woman, gravida 1, para 0, with an intrauterine pregnancy of 34 weeks and three days (according to last menstrual period and nine-week ultrasound) presented to her Ob-Gyn office for a routine prenatal visit. She was found to have an elevated blood pressure with new onset of 2+ proteinuria. The patient was sent to the labor and delivery unit at the adjoining hospital for serial blood pressure readings, laboratory work, and fetal monitoring.
The patient’s previous medical history was limited to sinusitis. She was taking no prescription medications, and her only listed allergy was to pineapple. Initial lab studies revealed elevations in liver enzymes, lactate dehydrogenase (LDH), uric acid, and serum creatinine, as well as thrombocytopenia (see Table 11-5). She also had a critically low blood glucose level, which conflicted with a normal follow-up reading.
At this point, the patient was thought to have HELLP syndrome6 (ie, hemolysis, elevated liver enzymes, low platelet count), or possibly acute fatty liver of pregnancy (AFLP).2,4,7-11 Additional labs were drawn immediately to confirm or rule out AFLP. These included repeat serum glucose (following a second reading with normal results), a serum ammonia level, prothrombin time (PT), and partial thromboplastin time (PTT). The most reliable values to distinguish AFLP from HELLP are profound hypoglycemia (found in 94% of women with AFLP12) and an elevated serum ammonia level.4
Given the serious nature of either diagnosis, immediate delivery of the infant was deemed necessary. Because the patient’s cervix was not found favorable for induction, she underwent low-transverse cesarean delivery without complications. She was noted to have essentially normal anatomy with the exception of a small subserosal fibroid posteriorly. Meconium-stained amniotic fluid was present. A male infant was delivered, weighing 5 lb with 1-minute and 5-minute Apgar scores of 8 and 9, respectively.
Postoperatively, the patient remained in the recovery area, where she received intensive monitoring. She experienced fluctuations in blood glucose, ranging from 33 to 144 mg/dL; she was started on 5% dextrose in lactated Ringer’s solution and treated with IV dextrose 50 g. While the patient was in surgical recovery, results from the second set of labs, drawn before surgery, were returned; findings included an elevated ammonia level and an abnormal coagulation panel, including PT of 25.3 sec, PTT of 48.4 sec, and a fibrinogen level of 116 mg/dL, confirming the suspected diagnosis of AFLP.
Magnesium sulfate, which had been started immediately postop, was discontinued on confirmation of the diagnosis of AFLP. The patient was initially somnolent as a result of general anesthesia but gradually returned to a fully normal sensorium by early morning on postop day 1. Postoperatively, the patient’s hemoglobin was found to be low (8.6 g/dL; reference range, 13.5 to 18.5 g/dL), so she was transfused with two units of packed red blood cells (PRBCs) and given fresh frozen plasma (FFP) to correct this coagulopathy. The patient’s platelets were also low at 82,000/mm3 (reference range, 140,000 to 340,000/mm3).
On postop day 1, the patient’s serum creatinine rose to 4.2 mg/dL and her total bilirubin increased to 14.4 mg/dL (reference ranges, 0.6 to 1.2 mg/dL and < 1.0 mg/dL, respectively). Given the multiple systems affected by AFLP and the need for intensive supportive care, the patient was transferred to the ICU.
On her arrival at the ICU, the patient’s vital signs were initially stable, and she was alert and oriented. However, within the next few hours, she became hypotensive and encephalopathic. She required aggressive fluid resuscitation and multiple transfusions of PRBCs and FFP due to persistent anemia and coagulopathy. Her vital signs were stabilized, but she continued to need blood transfusions.
Postop day 2, the patient became less responsive and was soon unable to follow commands or speak clearly. Her breathing remained stable with just 3 L of oxygen by nasal cannula, but in order to prevent aspiration and in consideration of a postoperative ileus, it was necessary to place a nasogastric tube with low intermittent suction. This produced a bloody return, but no intervention other than close monitoring and transfusion was performed at that time.
Abdominal ultrasound showed ascites and mild left-sided hydronephrosis with no gallstones. The common bile duct measured 3 mm in diameter.
Although liver biopsy is considered the gold standard for a confirmed diagnosis of AFLP,13,14 this procedure was contraindicated by the patient’s coagulopathy. Concern was also expressed by one consultant that the patient might have thrombotic thrombocytopenic purpura (TTP) in addition to AFLP. TTP can manifest with similar findings, such as anemia, thrombocytopenia, neurologic symptoms, and renal abnormalities, but usually fever is involved, and the patient was afebrile. A catheter was placed for hemodialysis and therapeutic plasma exchange (TPE). Given that TTP-associated mortality is significantly decreased by use of TPE,15 this intervention was deemed prudent. The patient underwent TPE on three consecutive days, postop days 2 through 4.
The patient’s mental status began to improve, and by postop day 6, she was able to follow commands and engage in brief conversations. By postop day 9, she had returned almost completely to her baseline mental status.
The patient’s liver function test results and total bilirubin, ammonia, and creatinine levels all improved over the first few postoperative days but began to rise again by day 6. In response to worsening renal and hepatic functioning, the decision was made on postop day 9 to transfer the patient to a hospital with liver transplantation capabilities, should this procedure become necessary.
Discussion
AFLP is a rare condition specific to pregnancy, affecting 1/7,000 to 1/20,000 pregnancies. Due to the low incidence of this disease, randomized controlled trials to study it are not possible. Instead, clinicians must learn either from individual case studies or from retrospective syntheses of cases reported over time.1,2,7 Fortunately, the wealth of information gleaned over the past 30 years has significantly reduced AFLP-associated maternal and fetal mortality and morbidity rates. In the 1980s, maternal and fetal mortality rates as high as 85% were reported.3 Worldwide, maternal mortality associated with AFLP has decreased significantly to 7% to 18%, whereas the fetal mortality rate has fallen to between 9% and 23%.1,16,17
Common trends among women who have developed AFLP include nulliparity, multiparity, and advanced maternal age. One retrospective study of 57 women who had developed AFLP revealed that 35 cases (61%) involved first-time pregnancies. It also showed that 10 (18%) of the women had twins, and 14 (25%) were older than 35.2 In another study of 35 cases of AFLP, 40% of the women were nulliparous, and 11.4% were multiparous, including one triplet gestation.12 In a third, smaller study, 80% of women affected by AFLP were multiparous.10 Currently, there is no known evidence linking any maternal behavior to development of AFLP.
Presentation
Women who present with AFLP often experience vague, nonspecific symptoms, leading to misdiagnosis or delayed diagnosis. Objective measurements, including physical exam findings, laboratory studies, and other diagnostic tests, will help with a diagnosis. The most frequent initial symptoms are nausea and vomiting (in 70% of patients) and abdominal pain (50% to 80%), epigastric or right upper-quadrant.3 Other common symptoms include fatigue, malaise, anorexia, weight gain, polyuria, and polydipsia.2,3,9,18,19
Because the presenting symptoms in AFLP can be vague, clinicians should complete a thorough physical exam to differentiate accurately among conditions associated with pregnancy. Physical signs present in women with AFLP can include jaundice, ascites, edema, confusion, abdominal tenderness, and fever. More severe cases can present with multisystem involvement, including acute renal failure, gastrointestinal bleeding, pancreatitis, coagulopathy, and hepatic encephalopathy.3,4,9,18
Diagnostic Tests
Relevant laboratory tests include a complete blood count (CBC), liver studies, chemistry, coagulation studies, and urinalysis (see Table 1). Viral causes should be ruled out by way of a hepatitis panel.3 In AFLP, the CBC may show elevated white blood cells, decreased hemoglobin and hematocrit, and decreased platelets. Liver studies show elevated hepatic aminotransferase, bilirubin, LDH, and ammonia levels. Chemistry results show elevated blood urea nitrogen and creatinine, and decreased blood glucose. Coagulation factors are affected, and prolonged PTT, decreased fibrinogen, and proteinurea may also be found.9
Though invasive and not often necessary4,13 (and not possible for the case patient), the definitive diagnostic test for AFLP is liver biopsy.13,14 Biopsy reveals a microvesicular fatty infiltration of the hepatocytes as minute fat droplets surrounding a centrally located nucleus. These fatty infiltrates stain with oil red O, specific for fat. Inflammation is present in 50% of cases. There may also be a picture similar to cholestasis with bile thrombi or deposits within the hepatocytes.20
Due to the risk for hemorrhage and the critical status of women with AFLP, biopsy is often not possible. Ultrasonography may show increased echogenicity; CT may show decreased or diffuse attenuation in the liver. These imaging studies, though possibly helpful in severe cases, often yield false-negative results.3,20
In the absence of another explanation for the patient’s symptoms, the Swansea criteria are used for diagnosis of AFLP.1 Six or more of the following criteria must be present to confirm this diagnosis: vomiting, abdominal pain, polydipsia or polyuria, encephalopathy, leukocytosis, elevated bilirubin, elevated liver enzymes, elevated ammonia, hypoglycemia, renal dysfunction, coagulopathy, elevated uric acid, ascites on ultrasound, and microvesicular steatosis on liver biopsy.1,2,5
Pathophysiology
Normal functions of the liver include metabolism, protein synthesis, and manufacturing of blood coagulation proteins. These functions are disturbed in the presence of AFLP. Thus, women with this disease experience signs and symptoms related directly to the dysfunction of these processes.20-22
Disturbances in the hepatocytes due to excess fatty acids impair the liver’s ability to convert unconjugated bilirubin into conjugated bilirubin, causing plasma levels of unconjugated bilirubin to rise. This increase in bilirubin explains the jaundiced appearance of women with AFLP. AFLP is often thought to occur in conjunction with preeclampsia in many, but not all, patients. Thrombocytopenia in these patients is felt to be secondary to peripheral vascular consumption. Conjugated bilirubin levels may also be increased due to decreased flow of conjugated bilirubin into the common bile duct.21
Another liver function that is disrupted is that of glycogen storage and conversion to glucose, and the liver’s ability to convert nutrients into glycogen is also impaired. Decreased storage of glycogen, along with the liver’s inability to break down previously stored glycogen, causes a decrease in serum glucose levels. Women with AFLP often require treatment with IV dextrose in response to marked hypoglycemia.16,21,23
The liver dysfunction associated with AFLP reduces adequate production of clotting factors and coagulation proteins. Thrombocytopenia, elevated clotting times, and bleeding are all problems seen in AFLP. Mild to moderate elevations in serum aminotransferases and elevated LDH also occur in patients with AFLP.23,24
Genetic Factor
There is little known about the etiology of AFLP, although recent data point to a genetic component that was found in as many as 62% of mothers in one study and in 25% of infants in another study.20-22 Fatty acid oxidation (FAO) is one of the processes of hepatic mitochondria, a process that relies on several enzymes. When FAO is interrupted, fatty acids are deposited in the liver cells, as seen in histologic studies of AFLP.25,26 The common thread in women with this disease is a mutation in one of the enzymes needed for FAO. This enzyme is the long-chain 3-hydroxyacyl-CoA dehydrogenase. Deficiencies in this enzyme are common in mothers with AFLP and their infants.3,16,20,23,27
Differential Diagnosis
Several complications of pregnancy that involve the liver may, on presentation, mimic AFLP.16,20,23,24,28 The most common are hyperemesis gravidarum and intrahepatic cholestasis of pregnancy23 (see Table 216,20,23,24,28); others are preeclampsia/eclampsia and HELLP syndrome. It is important to distinguish between the signs and symptoms associated with each of these disorders in order to provide the most effective treatment. Hepatitis serologies are important in the differential diagnosis when liver enzyme levels are exceptionally high.4,16,22,28
Treatment
The most effective treatment for AFLP is delivery of the infant; often, this alone causes the signs and symptoms of AFLP to resolve.8,21,27,29 In two of three cases in a small study by Aso et al,8 early delivery of the fetus led to complete resolution of symptoms and return to normal liver function. One of these patients was sent home four days after delivery; the other, 14 days later. Other patients may require more invasive treatment and support.8
Management in the ICU is often required to provide appropriate supportive care to the mother after delivery. Acute respiratory distress syndrome, pancreatitis, hemorrhage, encephalopathy, renal failure, and continual liver failure are among the severe complications associated with AFLP.4,8,10 Many women require intubation, dialysis, fluid resuscitation, blood product transfusion, and vasopressor therapy.3,8,11 Prophylactic antibiotics, IV steroids, and glucose may all be required in the supportive care and recovery of a mother with AFLP.3,8,11
TPE has also been useful in instances of severe complications.1,3,6 In one retrospective study, Martin et al1 recommended administration of TPE in patients with AFLP under the following circumstances:
(1) Deteriorating central nervous system abnormalities, such as sensorium changes or coma;
(2) Persistent coagulopathy requiring continued and aggressive blood product support with plasma, red cells, and/or cryoprecipitate;
(3) Advanced renal dysfunction that compromised fluid management;
(4) Progressive cardiopulmonary compromise; and/or
(5) Ongoing fluid management concerns, including significant ascites, edema, anuria/oliguria, and/or fluid overload.1
In rare cases, liver transplantation is needed in patients with AFLP. Westbrook et al18 reviewed 54 cases of liver disease in pregnancy in one UK hospital between 1997 and 2008. Of these, six patients with encephalopathy or elevated lactate were listed for liver transplant, including just one with a diagnosis of AFLP. This woman never actually underwent transplant but recovered in response to medical management alone.18 According to data reported in June 2011 by the Organ Procurement and Transplantation Network,30 liver transplantation was needed in only three US patients with AFLP between 2000 and 2011. Further retrospective studies on outcomes from transplant versus medical management should be considered to guide future decision making involving this invasive therapy.
The Case Patient
This 39-year-old patient presented during a routine prenatal visit with proteinuria and hypertension, possibly indicative of preeclampsia. Because of the serious nature of this potential diagnosis in pregnancy, she was admitted for monitoring and further testing. Although the diagnosis of AFLP was not confirmed until later, the patient’s preliminary lab studies showed elevated liver enzymes and low platelet counts, signifying the need for prompt intervention and delivery of the infant. At this point, the patient met criteria for HELLP syndrome, but AFLP was suspected after the initial finding of profound hypoglycemia led to further testing.
As an older mother experiencing pregnancy for the first time, this patient fit the profile for AFLP. She initially responded well after delivery of her infant but continued to experience complications. On the days that the patient was treated with TPE, her total bilirubin and liver enzymes were at their lowest. Perhaps this treatment should be considered in more cases of AFLP.
The patient was transferred to a hospital with liver transplantation capabilities, but she ultimately recovered without undergoing transplant.
Conclusion
For the primary obstetric care provider, being aware of the possible complications associated with pregnancy is important. Though uncommon, AFLP is a serious complication that should be ruled out in women who present with vague symptoms such as nausea, vomiting, and abdominal pain in the third trimester of pregnancy. The reduction in AFLP-associated morbidity and mortality during the past 20 years is a direct result of increased early recognition and therapeutic delivery.
Referral to a maternal fetal medicine specialist, gastroenterologist, hematologist, and/or nephrologist may be necessary and appropriate in the management of a woman with AFLP. Further study is indicated for use of TPE in more severe cases of AFLP, particularly in women affected by persistent thrombocytopenia and anemia.
The author would like to thank C. Leanne Browning, MD, obstetrics/gynecology, for her invaluable guidance and advice on this project.
References
1. Martin JN Jr, Briery CM, Rose CH, et al. Postpartum plasma exchange as adjunctive therapy for severe acute fatty liver of pregnancy. J Clin Apher. 2009;23(4):138-143.
2. Knight M, Nelson-Piercy C, Kurinczuk JJ; UK Obstetric Surveillance System. A prospective national study of acute fatty liver of pregnancy in the UK. Gut. 2008;57(7):951-956.
3. Barsoom MJ, Tierney BJ. Acute fatty liver of pregnancy (2011). http://emedicine.medscape.com/article/1562425-overview. Accessed January 21, 2013.
4. Ko HH, Yoshida E. Acute fatty liver of pregnancy. Can J Gastroenterol. 2006;20(1):25-30.
5. Rathi U, Bapat M, Rathi P, Abraham P. Effect of liver disease on maternal and fetal outcome: a prospective study. Indian J Gastroenterol. 2007;26(2):59-63.
6. Myers L. Postpartum plasma exchange in a woman with suspected thrombotic thrombocytopenic purpura (TTP) vs hemolysis, elevated liver enzymes, and low platelet syndrome (HELLP): a case study. Nephrol Nurs J. 2010;37(4):399-402.
7. Vigil-de Gracia P. Acute fatty liver and HELLP syndrome: two distinct pregnancy disorders. Int J Gynaecol Obstet. 2001;73(3):215-220.
8. Aso K, Hojo S, Yumoto Y, et al. Three cases of acute fatty liver of pregnancy: postpartum clinical course depends on interval between onset of symptoms and termination of pregnancy. J Matern Fetal Neonatal Med. 2010;23(9):1047-1049.
9. Wei Q, Zhang L, Liu X. Clinical diagnosis and treatment of acute fatty liver of pregnancy: a literature review and 11 new cases. J Obstet Gynaecol Res. 2010;36(4):751-756.
10. Barber MA, Eguiluz I, Martin A, et al. Acute fatty liver of pregnancy: analysis of five consecutive cases from a tertiary centre. J Obstet Gynaecol. 2010;30(3):241-243.
11. Ajayi AO, Alao MO. Case report: acute fatty liver of pregnancy in a 30-year-old Nigerian primigravida. Niger J Clin Pract. 2008;11(4):389-391.
12. Vigíl-de Gracia P, Montufar-Rueda C. Acute fatty liver of pregnancy: diagnosis, treatment, and outcome based on 35 consecutive cases. J Matern Fetal Neonatal Med. 2011;24(9):1143-1146.
13. Dey M, Reema K. Acute fatty liver of pregnancy. N Am J Med Sci. 2012;4(11):611-612.
14. Castro MA, Goodwin TM, Shaw KJ, et al. Disseminated intravascular coagulation and antithrombin III depression in acute fatty liver of pregnancy. Am J Obstet Gynecol. 1996;174(1 pt 1):211-216.
15. Altuntas F, Aydogdu I, Kabukcu S, et al. Therapeutic plasma exchange for the treatment of thrombotic thrombocytopenic purpura: a retrospective multicenter study. Transfus Apher Sci. 2007;36(1):57-67.
16. Hay JE. Liver disease in pregnancy. Hepatology. 2008;47(3):1067-1076.
17. Wand S, Waeschle RM, Von Ahsen N, et al. Acute fatty liver failure due to acute fatty liver of pregnancy. Minerva Anesthesiol. 2012;78(4):503-506.
18. Westbrook RH, Yeoman AD, Joshi D, et al. Outcomes of severe pregnancy-related liver disease: refining the role of transplantation. Am J Transplant. 2010;10(11):2520-2526.
19. Fesenmeier MF, Coppage KH, Lambers DS, et al. Acute fatty liver of pregnancy in 3 tertiary care centers. Am J Obstet Gynecol. 2005;192(5):1416-1419.
20. Bacq Y. Liver diseases unique to pregnancy: a 2010 update. Clin Res Hepatol Gastroenterol. 2011;35(3):182-193.
21. Huether SE. Alterations of digestive function. In: McCance KL, Huether SE, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 6th ed. St. Louis, MO: Mosby; 2009:1452-1515.
22. Huether SE. Structure and function of the digestive system. In: McCance KL, Huether SE, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 6th ed. St. Louis, MO: Mosby; 2009:1420-1451.
23. Schutt VA, Minuk GY. Liver diseases unique to pregnancy. Best Pract Res Clin Gastroenterol. 2007;21(5):771-792.
24. Pan C, Perumalswami PV. Pregnancy-related liver diseases. Clin Liver Dis. 2011;15(1):199-208.
25. Ibdah JA. Acute fatty liver of pregnancy: an update on pathogenesis and clinical implications. World J Gastroenterol. 2006;12(46):7397-7404.
26. Browning MF, Levy HL, Wilkins-Haug LE, et al. Fetal fatty acid oxidation defects and maternal liver disease in pregnancy. Obstet Gynecol. 2006;107(1):115-120.
27. Dekker RR, Schutte JM, Stekelenburg J, et al. Maternal mortality and severe maternal morbidity from acute fatty liver of pregnancy in the Netherlands. Eur J Obstet Gynecol Reprod Biol. 2011;157(1):27-31.
28. Lee NM, Brady CW. Liver disease in pregnancy. World J Gastroenterol. 2009;15(8):897-906.
29. Vora KS, Shah VR, Parikh GP. Acute fatty liver of pregnancy: a case report of an uncommon disease. Indian J Crit Care Med. 2009;13(1):34-36.
30. Organ Procurement and Transplantation Network, Scientific Registry of Transplant Recipients. OPTN/SRTR 2011 Annual Data Report: Liver. http://srtr.transplant.hrsa.gov/annual_reports/2011/pdf/03_%20liver_12.pdf. Accessed January 18, 2013.
A 39-year-old black woman, gravida 1, para 0, with an intrauterine pregnancy of 34 weeks and three days (according to last menstrual period and nine-week ultrasound) presented to her Ob-Gyn office for a routine prenatal visit. She was found to have an elevated blood pressure with new onset of 2+ proteinuria. The patient was sent to the labor and delivery unit at the adjoining hospital for serial blood pressure readings, laboratory work, and fetal monitoring.
The patient’s previous medical history was limited to sinusitis. She was taking no prescription medications, and her only listed allergy was to pineapple. Initial lab studies revealed elevations in liver enzymes, lactate dehydrogenase (LDH), uric acid, and serum creatinine, as well as thrombocytopenia (see Table 11-5). She also had a critically low blood glucose level, which conflicted with a normal follow-up reading.
At this point, the patient was thought to have HELLP syndrome6 (ie, hemolysis, elevated liver enzymes, low platelet count), or possibly acute fatty liver of pregnancy (AFLP).2,4,7-11 Additional labs were drawn immediately to confirm or rule out AFLP. These included repeat serum glucose (following a second reading with normal results), a serum ammonia level, prothrombin time (PT), and partial thromboplastin time (PTT). The most reliable values to distinguish AFLP from HELLP are profound hypoglycemia (found in 94% of women with AFLP12) and an elevated serum ammonia level.4
Given the serious nature of either diagnosis, immediate delivery of the infant was deemed necessary. Because the patient’s cervix was not found favorable for induction, she underwent low-transverse cesarean delivery without complications. She was noted to have essentially normal anatomy with the exception of a small subserosal fibroid posteriorly. Meconium-stained amniotic fluid was present. A male infant was delivered, weighing 5 lb with 1-minute and 5-minute Apgar scores of 8 and 9, respectively.
Postoperatively, the patient remained in the recovery area, where she received intensive monitoring. She experienced fluctuations in blood glucose, ranging from 33 to 144 mg/dL; she was started on 5% dextrose in lactated Ringer’s solution and treated with IV dextrose 50 g. While the patient was in surgical recovery, results from the second set of labs, drawn before surgery, were returned; findings included an elevated ammonia level and an abnormal coagulation panel, including PT of 25.3 sec, PTT of 48.4 sec, and a fibrinogen level of 116 mg/dL, confirming the suspected diagnosis of AFLP.
Magnesium sulfate, which had been started immediately postop, was discontinued on confirmation of the diagnosis of AFLP. The patient was initially somnolent as a result of general anesthesia but gradually returned to a fully normal sensorium by early morning on postop day 1. Postoperatively, the patient’s hemoglobin was found to be low (8.6 g/dL; reference range, 13.5 to 18.5 g/dL), so she was transfused with two units of packed red blood cells (PRBCs) and given fresh frozen plasma (FFP) to correct this coagulopathy. The patient’s platelets were also low at 82,000/mm3 (reference range, 140,000 to 340,000/mm3).
On postop day 1, the patient’s serum creatinine rose to 4.2 mg/dL and her total bilirubin increased to 14.4 mg/dL (reference ranges, 0.6 to 1.2 mg/dL and < 1.0 mg/dL, respectively). Given the multiple systems affected by AFLP and the need for intensive supportive care, the patient was transferred to the ICU.
On her arrival at the ICU, the patient’s vital signs were initially stable, and she was alert and oriented. However, within the next few hours, she became hypotensive and encephalopathic. She required aggressive fluid resuscitation and multiple transfusions of PRBCs and FFP due to persistent anemia and coagulopathy. Her vital signs were stabilized, but she continued to need blood transfusions.
Postop day 2, the patient became less responsive and was soon unable to follow commands or speak clearly. Her breathing remained stable with just 3 L of oxygen by nasal cannula, but in order to prevent aspiration and in consideration of a postoperative ileus, it was necessary to place a nasogastric tube with low intermittent suction. This produced a bloody return, but no intervention other than close monitoring and transfusion was performed at that time.
Abdominal ultrasound showed ascites and mild left-sided hydronephrosis with no gallstones. The common bile duct measured 3 mm in diameter.
Although liver biopsy is considered the gold standard for a confirmed diagnosis of AFLP,13,14 this procedure was contraindicated by the patient’s coagulopathy. Concern was also expressed by one consultant that the patient might have thrombotic thrombocytopenic purpura (TTP) in addition to AFLP. TTP can manifest with similar findings, such as anemia, thrombocytopenia, neurologic symptoms, and renal abnormalities, but usually fever is involved, and the patient was afebrile. A catheter was placed for hemodialysis and therapeutic plasma exchange (TPE). Given that TTP-associated mortality is significantly decreased by use of TPE,15 this intervention was deemed prudent. The patient underwent TPE on three consecutive days, postop days 2 through 4.
The patient’s mental status began to improve, and by postop day 6, she was able to follow commands and engage in brief conversations. By postop day 9, she had returned almost completely to her baseline mental status.
The patient’s liver function test results and total bilirubin, ammonia, and creatinine levels all improved over the first few postoperative days but began to rise again by day 6. In response to worsening renal and hepatic functioning, the decision was made on postop day 9 to transfer the patient to a hospital with liver transplantation capabilities, should this procedure become necessary.
Discussion
AFLP is a rare condition specific to pregnancy, affecting 1/7,000 to 1/20,000 pregnancies. Due to the low incidence of this disease, randomized controlled trials to study it are not possible. Instead, clinicians must learn either from individual case studies or from retrospective syntheses of cases reported over time.1,2,7 Fortunately, the wealth of information gleaned over the past 30 years has significantly reduced AFLP-associated maternal and fetal mortality and morbidity rates. In the 1980s, maternal and fetal mortality rates as high as 85% were reported.3 Worldwide, maternal mortality associated with AFLP has decreased significantly to 7% to 18%, whereas the fetal mortality rate has fallen to between 9% and 23%.1,16,17
Common trends among women who have developed AFLP include nulliparity, multiparity, and advanced maternal age. One retrospective study of 57 women who had developed AFLP revealed that 35 cases (61%) involved first-time pregnancies. It also showed that 10 (18%) of the women had twins, and 14 (25%) were older than 35.2 In another study of 35 cases of AFLP, 40% of the women were nulliparous, and 11.4% were multiparous, including one triplet gestation.12 In a third, smaller study, 80% of women affected by AFLP were multiparous.10 Currently, there is no known evidence linking any maternal behavior to development of AFLP.
Presentation
Women who present with AFLP often experience vague, nonspecific symptoms, leading to misdiagnosis or delayed diagnosis. Objective measurements, including physical exam findings, laboratory studies, and other diagnostic tests, will help with a diagnosis. The most frequent initial symptoms are nausea and vomiting (in 70% of patients) and abdominal pain (50% to 80%), epigastric or right upper-quadrant.3 Other common symptoms include fatigue, malaise, anorexia, weight gain, polyuria, and polydipsia.2,3,9,18,19
Because the presenting symptoms in AFLP can be vague, clinicians should complete a thorough physical exam to differentiate accurately among conditions associated with pregnancy. Physical signs present in women with AFLP can include jaundice, ascites, edema, confusion, abdominal tenderness, and fever. More severe cases can present with multisystem involvement, including acute renal failure, gastrointestinal bleeding, pancreatitis, coagulopathy, and hepatic encephalopathy.3,4,9,18
Diagnostic Tests
Relevant laboratory tests include a complete blood count (CBC), liver studies, chemistry, coagulation studies, and urinalysis (see Table 1). Viral causes should be ruled out by way of a hepatitis panel.3 In AFLP, the CBC may show elevated white blood cells, decreased hemoglobin and hematocrit, and decreased platelets. Liver studies show elevated hepatic aminotransferase, bilirubin, LDH, and ammonia levels. Chemistry results show elevated blood urea nitrogen and creatinine, and decreased blood glucose. Coagulation factors are affected, and prolonged PTT, decreased fibrinogen, and proteinurea may also be found.9
Though invasive and not often necessary4,13 (and not possible for the case patient), the definitive diagnostic test for AFLP is liver biopsy.13,14 Biopsy reveals a microvesicular fatty infiltration of the hepatocytes as minute fat droplets surrounding a centrally located nucleus. These fatty infiltrates stain with oil red O, specific for fat. Inflammation is present in 50% of cases. There may also be a picture similar to cholestasis with bile thrombi or deposits within the hepatocytes.20
Due to the risk for hemorrhage and the critical status of women with AFLP, biopsy is often not possible. Ultrasonography may show increased echogenicity; CT may show decreased or diffuse attenuation in the liver. These imaging studies, though possibly helpful in severe cases, often yield false-negative results.3,20
In the absence of another explanation for the patient’s symptoms, the Swansea criteria are used for diagnosis of AFLP.1 Six or more of the following criteria must be present to confirm this diagnosis: vomiting, abdominal pain, polydipsia or polyuria, encephalopathy, leukocytosis, elevated bilirubin, elevated liver enzymes, elevated ammonia, hypoglycemia, renal dysfunction, coagulopathy, elevated uric acid, ascites on ultrasound, and microvesicular steatosis on liver biopsy.1,2,5
Pathophysiology
Normal functions of the liver include metabolism, protein synthesis, and manufacturing of blood coagulation proteins. These functions are disturbed in the presence of AFLP. Thus, women with this disease experience signs and symptoms related directly to the dysfunction of these processes.20-22
Disturbances in the hepatocytes due to excess fatty acids impair the liver’s ability to convert unconjugated bilirubin into conjugated bilirubin, causing plasma levels of unconjugated bilirubin to rise. This increase in bilirubin explains the jaundiced appearance of women with AFLP. AFLP is often thought to occur in conjunction with preeclampsia in many, but not all, patients. Thrombocytopenia in these patients is felt to be secondary to peripheral vascular consumption. Conjugated bilirubin levels may also be increased due to decreased flow of conjugated bilirubin into the common bile duct.21
Another liver function that is disrupted is that of glycogen storage and conversion to glucose, and the liver’s ability to convert nutrients into glycogen is also impaired. Decreased storage of glycogen, along with the liver’s inability to break down previously stored glycogen, causes a decrease in serum glucose levels. Women with AFLP often require treatment with IV dextrose in response to marked hypoglycemia.16,21,23
The liver dysfunction associated with AFLP reduces adequate production of clotting factors and coagulation proteins. Thrombocytopenia, elevated clotting times, and bleeding are all problems seen in AFLP. Mild to moderate elevations in serum aminotransferases and elevated LDH also occur in patients with AFLP.23,24
Genetic Factor
There is little known about the etiology of AFLP, although recent data point to a genetic component that was found in as many as 62% of mothers in one study and in 25% of infants in another study.20-22 Fatty acid oxidation (FAO) is one of the processes of hepatic mitochondria, a process that relies on several enzymes. When FAO is interrupted, fatty acids are deposited in the liver cells, as seen in histologic studies of AFLP.25,26 The common thread in women with this disease is a mutation in one of the enzymes needed for FAO. This enzyme is the long-chain 3-hydroxyacyl-CoA dehydrogenase. Deficiencies in this enzyme are common in mothers with AFLP and their infants.3,16,20,23,27
Differential Diagnosis
Several complications of pregnancy that involve the liver may, on presentation, mimic AFLP.16,20,23,24,28 The most common are hyperemesis gravidarum and intrahepatic cholestasis of pregnancy23 (see Table 216,20,23,24,28); others are preeclampsia/eclampsia and HELLP syndrome. It is important to distinguish between the signs and symptoms associated with each of these disorders in order to provide the most effective treatment. Hepatitis serologies are important in the differential diagnosis when liver enzyme levels are exceptionally high.4,16,22,28
Treatment
The most effective treatment for AFLP is delivery of the infant; often, this alone causes the signs and symptoms of AFLP to resolve.8,21,27,29 In two of three cases in a small study by Aso et al,8 early delivery of the fetus led to complete resolution of symptoms and return to normal liver function. One of these patients was sent home four days after delivery; the other, 14 days later. Other patients may require more invasive treatment and support.8
Management in the ICU is often required to provide appropriate supportive care to the mother after delivery. Acute respiratory distress syndrome, pancreatitis, hemorrhage, encephalopathy, renal failure, and continual liver failure are among the severe complications associated with AFLP.4,8,10 Many women require intubation, dialysis, fluid resuscitation, blood product transfusion, and vasopressor therapy.3,8,11 Prophylactic antibiotics, IV steroids, and glucose may all be required in the supportive care and recovery of a mother with AFLP.3,8,11
TPE has also been useful in instances of severe complications.1,3,6 In one retrospective study, Martin et al1 recommended administration of TPE in patients with AFLP under the following circumstances:
(1) Deteriorating central nervous system abnormalities, such as sensorium changes or coma;
(2) Persistent coagulopathy requiring continued and aggressive blood product support with plasma, red cells, and/or cryoprecipitate;
(3) Advanced renal dysfunction that compromised fluid management;
(4) Progressive cardiopulmonary compromise; and/or
(5) Ongoing fluid management concerns, including significant ascites, edema, anuria/oliguria, and/or fluid overload.1
In rare cases, liver transplantation is needed in patients with AFLP. Westbrook et al18 reviewed 54 cases of liver disease in pregnancy in one UK hospital between 1997 and 2008. Of these, six patients with encephalopathy or elevated lactate were listed for liver transplant, including just one with a diagnosis of AFLP. This woman never actually underwent transplant but recovered in response to medical management alone.18 According to data reported in June 2011 by the Organ Procurement and Transplantation Network,30 liver transplantation was needed in only three US patients with AFLP between 2000 and 2011. Further retrospective studies on outcomes from transplant versus medical management should be considered to guide future decision making involving this invasive therapy.
The Case Patient
This 39-year-old patient presented during a routine prenatal visit with proteinuria and hypertension, possibly indicative of preeclampsia. Because of the serious nature of this potential diagnosis in pregnancy, she was admitted for monitoring and further testing. Although the diagnosis of AFLP was not confirmed until later, the patient’s preliminary lab studies showed elevated liver enzymes and low platelet counts, signifying the need for prompt intervention and delivery of the infant. At this point, the patient met criteria for HELLP syndrome, but AFLP was suspected after the initial finding of profound hypoglycemia led to further testing.
As an older mother experiencing pregnancy for the first time, this patient fit the profile for AFLP. She initially responded well after delivery of her infant but continued to experience complications. On the days that the patient was treated with TPE, her total bilirubin and liver enzymes were at their lowest. Perhaps this treatment should be considered in more cases of AFLP.
The patient was transferred to a hospital with liver transplantation capabilities, but she ultimately recovered without undergoing transplant.
Conclusion
For the primary obstetric care provider, being aware of the possible complications associated with pregnancy is important. Though uncommon, AFLP is a serious complication that should be ruled out in women who present with vague symptoms such as nausea, vomiting, and abdominal pain in the third trimester of pregnancy. The reduction in AFLP-associated morbidity and mortality during the past 20 years is a direct result of increased early recognition and therapeutic delivery.
Referral to a maternal fetal medicine specialist, gastroenterologist, hematologist, and/or nephrologist may be necessary and appropriate in the management of a woman with AFLP. Further study is indicated for use of TPE in more severe cases of AFLP, particularly in women affected by persistent thrombocytopenia and anemia.
The author would like to thank C. Leanne Browning, MD, obstetrics/gynecology, for her invaluable guidance and advice on this project.
References
1. Martin JN Jr, Briery CM, Rose CH, et al. Postpartum plasma exchange as adjunctive therapy for severe acute fatty liver of pregnancy. J Clin Apher. 2009;23(4):138-143.
2. Knight M, Nelson-Piercy C, Kurinczuk JJ; UK Obstetric Surveillance System. A prospective national study of acute fatty liver of pregnancy in the UK. Gut. 2008;57(7):951-956.
3. Barsoom MJ, Tierney BJ. Acute fatty liver of pregnancy (2011). http://emedicine.medscape.com/article/1562425-overview. Accessed January 21, 2013.
4. Ko HH, Yoshida E. Acute fatty liver of pregnancy. Can J Gastroenterol. 2006;20(1):25-30.
5. Rathi U, Bapat M, Rathi P, Abraham P. Effect of liver disease on maternal and fetal outcome: a prospective study. Indian J Gastroenterol. 2007;26(2):59-63.
6. Myers L. Postpartum plasma exchange in a woman with suspected thrombotic thrombocytopenic purpura (TTP) vs hemolysis, elevated liver enzymes, and low platelet syndrome (HELLP): a case study. Nephrol Nurs J. 2010;37(4):399-402.
7. Vigil-de Gracia P. Acute fatty liver and HELLP syndrome: two distinct pregnancy disorders. Int J Gynaecol Obstet. 2001;73(3):215-220.
8. Aso K, Hojo S, Yumoto Y, et al. Three cases of acute fatty liver of pregnancy: postpartum clinical course depends on interval between onset of symptoms and termination of pregnancy. J Matern Fetal Neonatal Med. 2010;23(9):1047-1049.
9. Wei Q, Zhang L, Liu X. Clinical diagnosis and treatment of acute fatty liver of pregnancy: a literature review and 11 new cases. J Obstet Gynaecol Res. 2010;36(4):751-756.
10. Barber MA, Eguiluz I, Martin A, et al. Acute fatty liver of pregnancy: analysis of five consecutive cases from a tertiary centre. J Obstet Gynaecol. 2010;30(3):241-243.
11. Ajayi AO, Alao MO. Case report: acute fatty liver of pregnancy in a 30-year-old Nigerian primigravida. Niger J Clin Pract. 2008;11(4):389-391.
12. Vigíl-de Gracia P, Montufar-Rueda C. Acute fatty liver of pregnancy: diagnosis, treatment, and outcome based on 35 consecutive cases. J Matern Fetal Neonatal Med. 2011;24(9):1143-1146.
13. Dey M, Reema K. Acute fatty liver of pregnancy. N Am J Med Sci. 2012;4(11):611-612.
14. Castro MA, Goodwin TM, Shaw KJ, et al. Disseminated intravascular coagulation and antithrombin III depression in acute fatty liver of pregnancy. Am J Obstet Gynecol. 1996;174(1 pt 1):211-216.
15. Altuntas F, Aydogdu I, Kabukcu S, et al. Therapeutic plasma exchange for the treatment of thrombotic thrombocytopenic purpura: a retrospective multicenter study. Transfus Apher Sci. 2007;36(1):57-67.
16. Hay JE. Liver disease in pregnancy. Hepatology. 2008;47(3):1067-1076.
17. Wand S, Waeschle RM, Von Ahsen N, et al. Acute fatty liver failure due to acute fatty liver of pregnancy. Minerva Anesthesiol. 2012;78(4):503-506.
18. Westbrook RH, Yeoman AD, Joshi D, et al. Outcomes of severe pregnancy-related liver disease: refining the role of transplantation. Am J Transplant. 2010;10(11):2520-2526.
19. Fesenmeier MF, Coppage KH, Lambers DS, et al. Acute fatty liver of pregnancy in 3 tertiary care centers. Am J Obstet Gynecol. 2005;192(5):1416-1419.
20. Bacq Y. Liver diseases unique to pregnancy: a 2010 update. Clin Res Hepatol Gastroenterol. 2011;35(3):182-193.
21. Huether SE. Alterations of digestive function. In: McCance KL, Huether SE, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 6th ed. St. Louis, MO: Mosby; 2009:1452-1515.
22. Huether SE. Structure and function of the digestive system. In: McCance KL, Huether SE, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 6th ed. St. Louis, MO: Mosby; 2009:1420-1451.
23. Schutt VA, Minuk GY. Liver diseases unique to pregnancy. Best Pract Res Clin Gastroenterol. 2007;21(5):771-792.
24. Pan C, Perumalswami PV. Pregnancy-related liver diseases. Clin Liver Dis. 2011;15(1):199-208.
25. Ibdah JA. Acute fatty liver of pregnancy: an update on pathogenesis and clinical implications. World J Gastroenterol. 2006;12(46):7397-7404.
26. Browning MF, Levy HL, Wilkins-Haug LE, et al. Fetal fatty acid oxidation defects and maternal liver disease in pregnancy. Obstet Gynecol. 2006;107(1):115-120.
27. Dekker RR, Schutte JM, Stekelenburg J, et al. Maternal mortality and severe maternal morbidity from acute fatty liver of pregnancy in the Netherlands. Eur J Obstet Gynecol Reprod Biol. 2011;157(1):27-31.
28. Lee NM, Brady CW. Liver disease in pregnancy. World J Gastroenterol. 2009;15(8):897-906.
29. Vora KS, Shah VR, Parikh GP. Acute fatty liver of pregnancy: a case report of an uncommon disease. Indian J Crit Care Med. 2009;13(1):34-36.
30. Organ Procurement and Transplantation Network, Scientific Registry of Transplant Recipients. OPTN/SRTR 2011 Annual Data Report: Liver. http://srtr.transplant.hrsa.gov/annual_reports/2011/pdf/03_%20liver_12.pdf. Accessed January 18, 2013.
Man, 56, With Wrist Pain After a Fall
A white man, age 56, presented to his primary care clinician with wrist pain and swelling. Two days earlier, he had fallen from a step stool and landed on his right wrist. He treated the pain by resting, elevating his arm, applying ice, and taking ibuprofen 800 mg tid. He said he had lost strength in his hand and arm and was experiencing numbness and tingling in his right hand and fingers.
The patient’s medical history included hypertension, type 2 diabetes mellitus, morbid obesity, obstructive sleep apnea, asthma, carpel tunnel syndrome, and peripheral neuropathy. His surgical history was significant for duodenal switch gastric bypass surgery, performed eight years earlier, and his weight at the time of presentation was 200 lb; before his gastric bypass, he weighed 385 lb. Since the surgery, his hypertension, diabetes, asthma, and sleep apnea had all resolved. Table 1 shows a list of medications he was taking at the time of presentation.
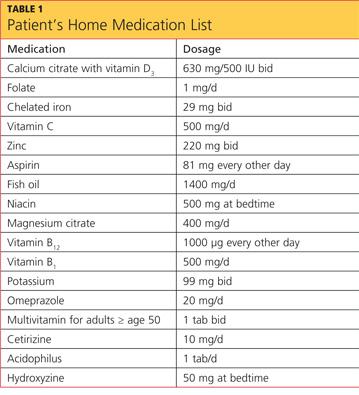
The patient, a registered nurse, had been married for 30 years and had one child. He had quit smoking 15 years earlier, with a 43–pack-year smoking history. He reported social drinking but denied any recreational drug use. He was unaware of having any allergies to food or medication.
His vital signs on presentation were blood pressure, 110/75 mm Hg; heart rate, 53 beats/min; respiration, 18 breaths/min; O2 saturation, 97% on room air; and temperature, 97.5°F.
Physical exam revealed that the patient’s right wrist was ecchymotic and swollen with +1 pitting edema. The skin was warm and dry to the touch. Decreased range of motion was noted in the right wrist, compared with the left. Pain with point tenderness was noted at the right lateral wrist. Pulses were +3 with capillary refill of less than 3 seconds. The rest of the exam was unremarkable.
The differential diagnosis included fracture secondary to the fall, osteoporosis, osteopenia, osteomalacia, Paget’s disease, tumor, infection, and sprain or strain of the wrist. A wrist x-ray was ordered, as were the following baseline labs: complete blood count with differential (CBC), vitamin B12 and folate levels, blood chemistry, lipid profile, liver profile, total vitamin D, and sensitive thyroid-stimulating hormone. Test results are shown in Table 2.
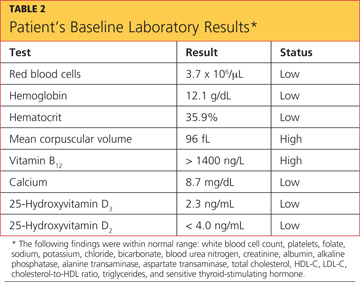
X-ray of the wrist showed fracture only, making it possible to rule out Paget’s disease (ie, no patchy white areas noted in the bone) and tumor (no masses seen) as the immediate cause of fracture. Normal body temperature and normal white blood cell count eliminated the possibility of infection.
Because the patient was only 56 and had a history of bariatric surgery, further testing was pursued to investigate a cause for the weakened bone. Bone mineral density (BMD) testing revealed the following results:
• The lumbar spine in frontal projection measured 0.968 g/cm2 with a T-score of –2.2 and a Z-score of –2.2.
• Total BMD of the left hip was 0.863 g/cm2 with a T-score of –1.7 and a Z-score of –1.4.
• Total BMD of the left femoral neck was 0.863 g/cm2 with a T-score of 1.7 and a Z-score of –1.1.
These findings suggested osteopenia1,2 (not osteoporosis) in all sites, with a 12% decrease of BMD in the spine (suggesting increased risk for spinal fracture) and a 16.3% decrease of BMD in the hip since the patient’s most recent bone scan five years earlier (radiologist’s report). Other abnormal findings were elevated parathyroid hormone (PTH) serum, 95.7 pg/mL (reference range, 10 to 65 pg/mL); low total calcium serum, 8.7 mg/dL (reference range, 8.9 to 10.2 mg/dL), and low 25-hydroxyvitamin D total, 12.3 ng/mL (reference range, 25 to 80 ng/mL).
A 2010 clinical practice guideline from the Endocrine Society3 specifies that after malabsorptive surgery, vitamin D and calcium supplementation should be adjusted by a qualified medical professional, based on serum markers and measures of bone density. An endocrinologist who was consulted at the patient’s initial visit prescribed the following medications: vitamin D2, 50,000 U/wk PO; combined calcium citrate (vitamin D3) 500 IU with calcium 630 mg, 1 tab bid; and calcitriol 0.5 μg bid.
The patient’s final diagnosis was osteomalacia secondary to gastric bypass surgery. (See “Making the Diagnosis of Osteomalacia.”4-6)

DISCUSSION
According to 2008 data from the World Health Organization (WHO),7 1.4 billion persons older than 20 worldwide were overweight, and 200 million men and 300 million women were considered obese—meaning that one in every 10 adults worldwide is overweight or obese. In 2010, the WHO reports, 40 million children younger than 5 worldwide were considered overweight.7 Health care providers need to be prepared to care for the increasing number of patients who will undergo bariatric surgeries to treat obesity and its related comorbidities.8
Postoperative follow-up for the malabsorption deficiencies related to bariatric procedures should be performed every six months, including obtaining levels of alkaline phosphatase and others previously discussed. In addition, the Endocrine Society guideline3 recommends measuring levels of vitamin B12, albumin, pre-albumin, iron, and ferritin, and obtaining a CBC, a liver profile, glucose reading, creatinine measurement, and a metabolic profile at one month and two months after surgery, then every six months until two years after surgery, then annually if findings are stable.
Furthermore, the Endocrine Society3 recommends obtaining zinc levels every six months for the first year, then annually. An annual vitamin A level is optional.9 Yearly bone density testing is recommended until the patient’s BMD is deemed stable.3
Additionally, Koch and Finelli10 recommend performing the following labs postoperatively: hemoglobin A1C every three months; copper, magnesium, whole blood thiamine, vitamin B12, and a 24-hour urinary calcium every six months for the first three years, then once a year if findings remain stable.
Use of alcohol should be discouraged among patients who have undergone bariatric surgery, as its use alters micronutrient requirements and metabolism. Alcohol consumption may also contribute to dumping syndrome (ie, rapid gastric emptying).11
Any patient with a history of malabsorptive bypass surgery who complains of neurologic, visual, or skin disorders, anemia, or edema may require a further workup to rule out other absorptive deficiencies. These include vitamins A, E, and B12, zinc, folate, thiamine, niacin, selenium, and ferritin.10
Osteomalacia
Metabolic bone diseases can result from genetics, dietary factors, medication use, surgery, or hormonal irregularities. They alter the normal biochemical reactions in bone structure.
The three most common forms of metabolic bone disease are osteoporosis, osteopenia, and osteomalacia. The WHO diagnostic classifications and associated T-scores for bone mineral density1,2 indicate a T-score above –1.0 as normal. A score between –1.0 and –2.5 is indicative of osteopenia, and a score below –2.5 indicates osteoporosis. A T-score below –2.5 in the patient with a history of fragility fracture indicates severe osteoporosis.1,2
In osteomalacia, bone volume remains unchanged, but mineralization of osteoid in the mature compact and spongy bone is either delayed or inadequate. The remolding cycle continues unchanged in the formation of osteoid, but mineral calcification and deposition do not occur.3-5
Osteomalacia is normally considered a rare disorder, but it may become more common as increasing numbers of patients undergo gastric bypass operations.12,13 Primary care practitioners should monitor for this condition in such patients before serious bone loss or other problems develop.9,13,14
Vitamin D deficiency (see “Vitamin D Metabolism,”4,15-19 below), whether or not the result of gastric bypass surgery, is a major risk factor for osteomalacia. Disorders of the small bowel, the hepatobiliary system, and the pancreas are all common causes of vitamin D deficiency. Liver disease interferes with the metabolism of vitamin D. Diseases of the pancreas may cause a deficiency of bile salts, which are vital for the intestinal absorption of vitamin D.17

Restriction and Malabsorption
The case patient had undergone a gastric bypass (duodenal switch), in which a large portion of the stomach is removed and a large part of the small bowel rerouted—with both parts of the procedure causing malabsorption.11 It is in the small bowel that absorption of vitamin D and calcium takes place.
The duodenal switch gastric bypass surgery causes both restriction and malabsorption. Though similar to a biliopancreatic diversion, the duodenal switch preserves the distal stomach and the pylorus20 by way of a sleeve gastrectomy that is performed to reduce the gastric reservoir; the common channel length after revision is 100 cm, not 50 cm (as in conventional biliopancreatic diversion).13 The sleeve gastrectomy involves removal of parietal cells, reducing production of hydrochloric acid (which is necessary to break down food), and hindering the absorption of certain nutrients, including the fat-soluble vitamins, vitamin B12, and iron.12 Patients who take H2-blockers or proton pump inhibitors experience an additional decrease in the production and availability of HCl and may have an increased risk for fracture.14,20,21
In addition to its biliopancreatic diversion component, the duodenal switch diverts a large portion of the small bowel, with food restricted from moving through it. Vitamin D and protein are normally absorbed at the jejunum and ileum, but only when bile salts are present; after a duodenal switch, bile and pancreatic enzymes are not introduced into the small intestines until 75 to 100 cm before they reach the large intestine. Thus, absorption of vitamin D, protein, calcium, and other nutrients is impaired.20,22
Since phosphorus and magnesium are also absorbed at the sites of the duodenum and jejunum, malabsorption of these nutrients may occur in a patient who has undergone a duodenal switch. Although vitamin B12 is absorbed at the site of the distal ileum, it also requires gastric acid to free it from the food. Zinc absorption, which normally occurs at the site of the jejunum, may be impaired after duodenal switch surgery, and calcium supplementation, though essential, may further reduce zinc absorption.9 Iron absorption requires HCl, facilitated by the presence of vitamin C. Use of H2-blockers and proton pump inhibitors may impair iron metabolism, resulting in anemia.20
In a randomized controlled trial, Aasheim et al23 compared the effects of Roux-en-Y gastric bypass with those of duodenal switch gastric bypass on patients’ vitamin metabolism. The researchers concluded that patients who undergo a duodenal switch are at greater risk for vitamin A and D deficiencies in the first year after surgery; and for thiamine deficiency in the months following surgery as a result of malabsorption, compared with patients who undergo Roux-en-Y gastric bypass.20,23
Patient Management
The case patient’s care necessitated consultations with endocrinology, dermatology, and gastroenterology (GI). Table 3 (below) shows the laboratory findings and the medication changes prompted by the patient’s physical exam and lab results. Table 4 lists the findings from other lab studies ordered throughout the patient’s course of treatment.
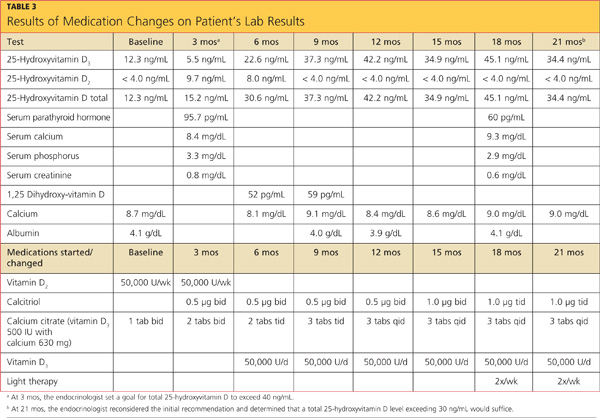
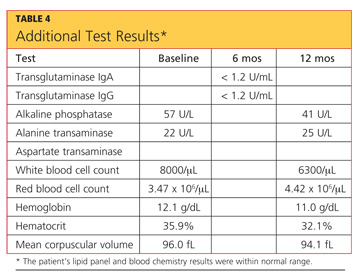
The endocrinologist was consulted at the first sign of osteopenia, and a workup was soon initiated, followed by treatment. GI was consulted six months after the beginning of treatment, when the patient began to complain of reflux while sleeping and frequent diarrhea throughout the day.
Results of esophagogastroduodenoscopy with biopsy ruled out celiac disease and mucosal ulceration, but a small hiatal hernia that was detected (< 3 cm) was determined to be an aggravating factor for the patient’s reflux. The patient was instructed in lifestyle modifications for hiatal hernia, including the need to remain upright one to two hours after eating before going to sleep to prevent aspiration. The patient was instructed to avoid taking iron and calcium within two hours of each other and to limit his alcohol intake. He was also educated in precautions against falls.
Dermatology was consulted nine months into treatment so that light therapy could be initiated, allowing the patient to take advantage of the body’s natural pathway to manufacture vitamin D3.
CONCLUSION
For post–bariatric surgery patients, primary care practitioners are in a position to coordinate care recommendations from multiple specialists, including those in nutrition, to determine the best course of action.
This case illustrates complications of bariatric surgery (malabsorption of key vitamins and minerals, wrist fracture, osteopenia, osteomalacia) that require diagnosis and treatment. The specialists and the primary care practitioner, along with the patient, had to weigh the risks and benefits of continued proton pump inhibitor use, as such medications can increase the risk for fracture. They also addressed the patient’s anemia and remained attentive to his preventive health care needs.
REFERENCES
1. Brusin JH. Update on bone densitometry. Radiol Technol. 2009;81(2):153BD-170BD.
2. Wilson CR. Essentials of bone densitometry for the medical physicist. Presented at: The American Association of Physicists in Medicine 2003 Annual Meeting; July 22-26, 2003; San Diego, CA.
3. Heber D, Greenway FL, Kaplan LM. et al. Endocrine and nutritional management of the post-bariatric surgery patient: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95(11):4825-4843.
4. Osteomalacia: step-by-step diagnostic approach (2011). http://bestpractice.bmj.com/best-practice/monograph/517/diagnosis/step-by-step.html. Accessed December 18, 2012.
5. Gifre L, Peris P, Monegal A, et al. Osteomalacia revisited : a report on 28 cases. Clin Rheumatol. 2011;30(5):639-645.
6. Bingham CT, Fitzpatrick LA. Noninvasive testing in the diagnosis of osteomalacia. Am J Med. 1993;95(5):519-523.
7. World Health Organization. Obesity and overweight (May 2012). Fact Sheet No 311. www.who.int/mediacentre/factsheets/fs311/en/index.html. Accessed December 18, 2012.
8. Tanner BD, Allen JW. Complications of bariatric surgery: implications for the covering physician. Am Surg. 2009;75(2):103-112.
9. Soleymani T, Tejavanija S, Morgan S. Obesity, bariatric surgery, and bone. Curr Opin Rheumatol. 2011;23(4):396-405.
10. Koch TR, Finelli FC. Postoperative metabolic and nutritional complications of bariatric surgery. Gastroenterol Clin North Am. 2010;39(1):109-124.
11. Manchester S, Roye GD. Bariatric surgery: an overview for dietetics professionals. Nutr Today. 2011;46(6):264-275.
12. Bal BS, Finelli FC, Shope TR, Koch TR. Nutritional deficiencies after bariatric surgery. Nat Rev Endocrinol. 2012;8(9):544-546.
13. Iannelli A, Schneck AS, Dahman M, et al. Two-step laparoscopic duodenal switch for superobesity: a feasibility study. Surg Endosc. 2009;23(10):2385-2389.
14. Lalmohamed A, de Vries F, Bazelier MT, et al. Risk of fracture after bariatric surgery in the United Kingdom: population based, retrospective cohort study. BMJ. 2012;345:e5085.
15. Holrick MF. Vitamin D: important for prevention of osteoporosis, cardiovascular heart disease, type 1 diabetes, autoimmune diseases, and some cancers. South Med J. 2005;98 (10):1024-1027.
16. Kalro BN. Vitamin D and the skeleton. Alt Ther Womens Health. 2009;2(4):25-32.
17. Crowther-Radulewicz CL, McCance KL. Alterations of musculoskeletal function. In: McCance KL, Huether SE, Brashers VL, Rote NS, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 6th ed. Maryland Heights, MO: Mosby Elsevier; 2010:1568-1617.
18. Huether SE. Structure and function of the renal and urologic systems. In: McCance KL, Huether SE, Brashers VL, Rote NS, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 6th ed. Maryland Heights, MO: Mosby Elsevier; 2010:1344-1364.
19. Bhan A, Rao AD, Rao DS. Osteomalacia as a result of vitamin D deficiency. Endocrinol Metab Clin North Am. 2010;39(2):321-331.
20. Decker GA, Swain JM, Crowell MD. Gastrointestinal and nutritional complications after bariatric surgery. Am J Gastroenterol. 2007;102(11):2571-2580.
21. Targownik LE, Lix LM, Metge C, et al. Use of proton pump inhibitors and risk of osteoporosis-related fractures. CMAJ. 2008;179(4):319-326.
22. Ybarra J, Sánchez-Hernández J, Pérez A. Hypovitaminosis D and morbid obesity. Nurs Clin North Am. 2007;42(1):19-27.
23. Aasheim ET, Björkman S, Søvik TT, et al. Vitamin status after bariatric surgery: a randomized study of gastric bypass and duodenal switch. Am J Clin Nutr. 2009;90(1):15-22.
A white man, age 56, presented to his primary care clinician with wrist pain and swelling. Two days earlier, he had fallen from a step stool and landed on his right wrist. He treated the pain by resting, elevating his arm, applying ice, and taking ibuprofen 800 mg tid. He said he had lost strength in his hand and arm and was experiencing numbness and tingling in his right hand and fingers.
The patient’s medical history included hypertension, type 2 diabetes mellitus, morbid obesity, obstructive sleep apnea, asthma, carpel tunnel syndrome, and peripheral neuropathy. His surgical history was significant for duodenal switch gastric bypass surgery, performed eight years earlier, and his weight at the time of presentation was 200 lb; before his gastric bypass, he weighed 385 lb. Since the surgery, his hypertension, diabetes, asthma, and sleep apnea had all resolved. Table 1 shows a list of medications he was taking at the time of presentation.
The patient, a registered nurse, had been married for 30 years and had one child. He had quit smoking 15 years earlier, with a 43–pack-year smoking history. He reported social drinking but denied any recreational drug use. He was unaware of having any allergies to food or medication.
His vital signs on presentation were blood pressure, 110/75 mm Hg; heart rate, 53 beats/min; respiration, 18 breaths/min; O2 saturation, 97% on room air; and temperature, 97.5°F.
Physical exam revealed that the patient’s right wrist was ecchymotic and swollen with +1 pitting edema. The skin was warm and dry to the touch. Decreased range of motion was noted in the right wrist, compared with the left. Pain with point tenderness was noted at the right lateral wrist. Pulses were +3 with capillary refill of less than 3 seconds. The rest of the exam was unremarkable.
The differential diagnosis included fracture secondary to the fall, osteoporosis, osteopenia, osteomalacia, Paget’s disease, tumor, infection, and sprain or strain of the wrist. A wrist x-ray was ordered, as were the following baseline labs: complete blood count with differential (CBC), vitamin B12 and folate levels, blood chemistry, lipid profile, liver profile, total vitamin D, and sensitive thyroid-stimulating hormone. Test results are shown in Table 2.
X-ray of the wrist showed fracture only, making it possible to rule out Paget’s disease (ie, no patchy white areas noted in the bone) and tumor (no masses seen) as the immediate cause of fracture. Normal body temperature and normal white blood cell count eliminated the possibility of infection.
Because the patient was only 56 and had a history of bariatric surgery, further testing was pursued to investigate a cause for the weakened bone. Bone mineral density (BMD) testing revealed the following results:
• The lumbar spine in frontal projection measured 0.968 g/cm2 with a T-score of –2.2 and a Z-score of –2.2.
• Total BMD of the left hip was 0.863 g/cm2 with a T-score of –1.7 and a Z-score of –1.4.
• Total BMD of the left femoral neck was 0.863 g/cm2 with a T-score of 1.7 and a Z-score of –1.1.
These findings suggested osteopenia1,2 (not osteoporosis) in all sites, with a 12% decrease of BMD in the spine (suggesting increased risk for spinal fracture) and a 16.3% decrease of BMD in the hip since the patient’s most recent bone scan five years earlier (radiologist’s report). Other abnormal findings were elevated parathyroid hormone (PTH) serum, 95.7 pg/mL (reference range, 10 to 65 pg/mL); low total calcium serum, 8.7 mg/dL (reference range, 8.9 to 10.2 mg/dL), and low 25-hydroxyvitamin D total, 12.3 ng/mL (reference range, 25 to 80 ng/mL).
A 2010 clinical practice guideline from the Endocrine Society3 specifies that after malabsorptive surgery, vitamin D and calcium supplementation should be adjusted by a qualified medical professional, based on serum markers and measures of bone density. An endocrinologist who was consulted at the patient’s initial visit prescribed the following medications: vitamin D2, 50,000 U/wk PO; combined calcium citrate (vitamin D3) 500 IU with calcium 630 mg, 1 tab bid; and calcitriol 0.5 μg bid.
The patient’s final diagnosis was osteomalacia secondary to gastric bypass surgery. (See “Making the Diagnosis of Osteomalacia.”4-6)
DISCUSSION
According to 2008 data from the World Health Organization (WHO),7 1.4 billion persons older than 20 worldwide were overweight, and 200 million men and 300 million women were considered obese—meaning that one in every 10 adults worldwide is overweight or obese. In 2010, the WHO reports, 40 million children younger than 5 worldwide were considered overweight.7 Health care providers need to be prepared to care for the increasing number of patients who will undergo bariatric surgeries to treat obesity and its related comorbidities.8
Postoperative follow-up for the malabsorption deficiencies related to bariatric procedures should be performed every six months, including obtaining levels of alkaline phosphatase and others previously discussed. In addition, the Endocrine Society guideline3 recommends measuring levels of vitamin B12, albumin, pre-albumin, iron, and ferritin, and obtaining a CBC, a liver profile, glucose reading, creatinine measurement, and a metabolic profile at one month and two months after surgery, then every six months until two years after surgery, then annually if findings are stable.
Furthermore, the Endocrine Society3 recommends obtaining zinc levels every six months for the first year, then annually. An annual vitamin A level is optional.9 Yearly bone density testing is recommended until the patient’s BMD is deemed stable.3
Additionally, Koch and Finelli10 recommend performing the following labs postoperatively: hemoglobin A1C every three months; copper, magnesium, whole blood thiamine, vitamin B12, and a 24-hour urinary calcium every six months for the first three years, then once a year if findings remain stable.
Use of alcohol should be discouraged among patients who have undergone bariatric surgery, as its use alters micronutrient requirements and metabolism. Alcohol consumption may also contribute to dumping syndrome (ie, rapid gastric emptying).11
Any patient with a history of malabsorptive bypass surgery who complains of neurologic, visual, or skin disorders, anemia, or edema may require a further workup to rule out other absorptive deficiencies. These include vitamins A, E, and B12, zinc, folate, thiamine, niacin, selenium, and ferritin.10
Osteomalacia
Metabolic bone diseases can result from genetics, dietary factors, medication use, surgery, or hormonal irregularities. They alter the normal biochemical reactions in bone structure.
The three most common forms of metabolic bone disease are osteoporosis, osteopenia, and osteomalacia. The WHO diagnostic classifications and associated T-scores for bone mineral density1,2 indicate a T-score above –1.0 as normal. A score between –1.0 and –2.5 is indicative of osteopenia, and a score below –2.5 indicates osteoporosis. A T-score below –2.5 in the patient with a history of fragility fracture indicates severe osteoporosis.1,2
In osteomalacia, bone volume remains unchanged, but mineralization of osteoid in the mature compact and spongy bone is either delayed or inadequate. The remolding cycle continues unchanged in the formation of osteoid, but mineral calcification and deposition do not occur.3-5
Osteomalacia is normally considered a rare disorder, but it may become more common as increasing numbers of patients undergo gastric bypass operations.12,13 Primary care practitioners should monitor for this condition in such patients before serious bone loss or other problems develop.9,13,14
Vitamin D deficiency (see “Vitamin D Metabolism,”4,15-19 below), whether or not the result of gastric bypass surgery, is a major risk factor for osteomalacia. Disorders of the small bowel, the hepatobiliary system, and the pancreas are all common causes of vitamin D deficiency. Liver disease interferes with the metabolism of vitamin D. Diseases of the pancreas may cause a deficiency of bile salts, which are vital for the intestinal absorption of vitamin D.17
Restriction and Malabsorption
The case patient had undergone a gastric bypass (duodenal switch), in which a large portion of the stomach is removed and a large part of the small bowel rerouted—with both parts of the procedure causing malabsorption.11 It is in the small bowel that absorption of vitamin D and calcium takes place.
The duodenal switch gastric bypass surgery causes both restriction and malabsorption. Though similar to a biliopancreatic diversion, the duodenal switch preserves the distal stomach and the pylorus20 by way of a sleeve gastrectomy that is performed to reduce the gastric reservoir; the common channel length after revision is 100 cm, not 50 cm (as in conventional biliopancreatic diversion).13 The sleeve gastrectomy involves removal of parietal cells, reducing production of hydrochloric acid (which is necessary to break down food), and hindering the absorption of certain nutrients, including the fat-soluble vitamins, vitamin B12, and iron.12 Patients who take H2-blockers or proton pump inhibitors experience an additional decrease in the production and availability of HCl and may have an increased risk for fracture.14,20,21
In addition to its biliopancreatic diversion component, the duodenal switch diverts a large portion of the small bowel, with food restricted from moving through it. Vitamin D and protein are normally absorbed at the jejunum and ileum, but only when bile salts are present; after a duodenal switch, bile and pancreatic enzymes are not introduced into the small intestines until 75 to 100 cm before they reach the large intestine. Thus, absorption of vitamin D, protein, calcium, and other nutrients is impaired.20,22
Since phosphorus and magnesium are also absorbed at the sites of the duodenum and jejunum, malabsorption of these nutrients may occur in a patient who has undergone a duodenal switch. Although vitamin B12 is absorbed at the site of the distal ileum, it also requires gastric acid to free it from the food. Zinc absorption, which normally occurs at the site of the jejunum, may be impaired after duodenal switch surgery, and calcium supplementation, though essential, may further reduce zinc absorption.9 Iron absorption requires HCl, facilitated by the presence of vitamin C. Use of H2-blockers and proton pump inhibitors may impair iron metabolism, resulting in anemia.20
In a randomized controlled trial, Aasheim et al23 compared the effects of Roux-en-Y gastric bypass with those of duodenal switch gastric bypass on patients’ vitamin metabolism. The researchers concluded that patients who undergo a duodenal switch are at greater risk for vitamin A and D deficiencies in the first year after surgery; and for thiamine deficiency in the months following surgery as a result of malabsorption, compared with patients who undergo Roux-en-Y gastric bypass.20,23
Patient Management
The case patient’s care necessitated consultations with endocrinology, dermatology, and gastroenterology (GI). Table 3 (below) shows the laboratory findings and the medication changes prompted by the patient’s physical exam and lab results. Table 4 lists the findings from other lab studies ordered throughout the patient’s course of treatment.
The endocrinologist was consulted at the first sign of osteopenia, and a workup was soon initiated, followed by treatment. GI was consulted six months after the beginning of treatment, when the patient began to complain of reflux while sleeping and frequent diarrhea throughout the day.
Results of esophagogastroduodenoscopy with biopsy ruled out celiac disease and mucosal ulceration, but a small hiatal hernia that was detected (< 3 cm) was determined to be an aggravating factor for the patient’s reflux. The patient was instructed in lifestyle modifications for hiatal hernia, including the need to remain upright one to two hours after eating before going to sleep to prevent aspiration. The patient was instructed to avoid taking iron and calcium within two hours of each other and to limit his alcohol intake. He was also educated in precautions against falls.
Dermatology was consulted nine months into treatment so that light therapy could be initiated, allowing the patient to take advantage of the body’s natural pathway to manufacture vitamin D3.
CONCLUSION
For post–bariatric surgery patients, primary care practitioners are in a position to coordinate care recommendations from multiple specialists, including those in nutrition, to determine the best course of action.
This case illustrates complications of bariatric surgery (malabsorption of key vitamins and minerals, wrist fracture, osteopenia, osteomalacia) that require diagnosis and treatment. The specialists and the primary care practitioner, along with the patient, had to weigh the risks and benefits of continued proton pump inhibitor use, as such medications can increase the risk for fracture. They also addressed the patient’s anemia and remained attentive to his preventive health care needs.
REFERENCES
1. Brusin JH. Update on bone densitometry. Radiol Technol. 2009;81(2):153BD-170BD.
2. Wilson CR. Essentials of bone densitometry for the medical physicist. Presented at: The American Association of Physicists in Medicine 2003 Annual Meeting; July 22-26, 2003; San Diego, CA.
3. Heber D, Greenway FL, Kaplan LM. et al. Endocrine and nutritional management of the post-bariatric surgery patient: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95(11):4825-4843.
4. Osteomalacia: step-by-step diagnostic approach (2011). http://bestpractice.bmj.com/best-practice/monograph/517/diagnosis/step-by-step.html. Accessed December 18, 2012.
5. Gifre L, Peris P, Monegal A, et al. Osteomalacia revisited : a report on 28 cases. Clin Rheumatol. 2011;30(5):639-645.
6. Bingham CT, Fitzpatrick LA. Noninvasive testing in the diagnosis of osteomalacia. Am J Med. 1993;95(5):519-523.
7. World Health Organization. Obesity and overweight (May 2012). Fact Sheet No 311. www.who.int/mediacentre/factsheets/fs311/en/index.html. Accessed December 18, 2012.
8. Tanner BD, Allen JW. Complications of bariatric surgery: implications for the covering physician. Am Surg. 2009;75(2):103-112.
9. Soleymani T, Tejavanija S, Morgan S. Obesity, bariatric surgery, and bone. Curr Opin Rheumatol. 2011;23(4):396-405.
10. Koch TR, Finelli FC. Postoperative metabolic and nutritional complications of bariatric surgery. Gastroenterol Clin North Am. 2010;39(1):109-124.
11. Manchester S, Roye GD. Bariatric surgery: an overview for dietetics professionals. Nutr Today. 2011;46(6):264-275.
12. Bal BS, Finelli FC, Shope TR, Koch TR. Nutritional deficiencies after bariatric surgery. Nat Rev Endocrinol. 2012;8(9):544-546.
13. Iannelli A, Schneck AS, Dahman M, et al. Two-step laparoscopic duodenal switch for superobesity: a feasibility study. Surg Endosc. 2009;23(10):2385-2389.
14. Lalmohamed A, de Vries F, Bazelier MT, et al. Risk of fracture after bariatric surgery in the United Kingdom: population based, retrospective cohort study. BMJ. 2012;345:e5085.
15. Holrick MF. Vitamin D: important for prevention of osteoporosis, cardiovascular heart disease, type 1 diabetes, autoimmune diseases, and some cancers. South Med J. 2005;98 (10):1024-1027.
16. Kalro BN. Vitamin D and the skeleton. Alt Ther Womens Health. 2009;2(4):25-32.
17. Crowther-Radulewicz CL, McCance KL. Alterations of musculoskeletal function. In: McCance KL, Huether SE, Brashers VL, Rote NS, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 6th ed. Maryland Heights, MO: Mosby Elsevier; 2010:1568-1617.
18. Huether SE. Structure and function of the renal and urologic systems. In: McCance KL, Huether SE, Brashers VL, Rote NS, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 6th ed. Maryland Heights, MO: Mosby Elsevier; 2010:1344-1364.
19. Bhan A, Rao AD, Rao DS. Osteomalacia as a result of vitamin D deficiency. Endocrinol Metab Clin North Am. 2010;39(2):321-331.
20. Decker GA, Swain JM, Crowell MD. Gastrointestinal and nutritional complications after bariatric surgery. Am J Gastroenterol. 2007;102(11):2571-2580.
21. Targownik LE, Lix LM, Metge C, et al. Use of proton pump inhibitors and risk of osteoporosis-related fractures. CMAJ. 2008;179(4):319-326.
22. Ybarra J, Sánchez-Hernández J, Pérez A. Hypovitaminosis D and morbid obesity. Nurs Clin North Am. 2007;42(1):19-27.
23. Aasheim ET, Björkman S, Søvik TT, et al. Vitamin status after bariatric surgery: a randomized study of gastric bypass and duodenal switch. Am J Clin Nutr. 2009;90(1):15-22.
A white man, age 56, presented to his primary care clinician with wrist pain and swelling. Two days earlier, he had fallen from a step stool and landed on his right wrist. He treated the pain by resting, elevating his arm, applying ice, and taking ibuprofen 800 mg tid. He said he had lost strength in his hand and arm and was experiencing numbness and tingling in his right hand and fingers.
The patient’s medical history included hypertension, type 2 diabetes mellitus, morbid obesity, obstructive sleep apnea, asthma, carpel tunnel syndrome, and peripheral neuropathy. His surgical history was significant for duodenal switch gastric bypass surgery, performed eight years earlier, and his weight at the time of presentation was 200 lb; before his gastric bypass, he weighed 385 lb. Since the surgery, his hypertension, diabetes, asthma, and sleep apnea had all resolved. Table 1 shows a list of medications he was taking at the time of presentation.
The patient, a registered nurse, had been married for 30 years and had one child. He had quit smoking 15 years earlier, with a 43–pack-year smoking history. He reported social drinking but denied any recreational drug use. He was unaware of having any allergies to food or medication.
His vital signs on presentation were blood pressure, 110/75 mm Hg; heart rate, 53 beats/min; respiration, 18 breaths/min; O2 saturation, 97% on room air; and temperature, 97.5°F.
Physical exam revealed that the patient’s right wrist was ecchymotic and swollen with +1 pitting edema. The skin was warm and dry to the touch. Decreased range of motion was noted in the right wrist, compared with the left. Pain with point tenderness was noted at the right lateral wrist. Pulses were +3 with capillary refill of less than 3 seconds. The rest of the exam was unremarkable.
The differential diagnosis included fracture secondary to the fall, osteoporosis, osteopenia, osteomalacia, Paget’s disease, tumor, infection, and sprain or strain of the wrist. A wrist x-ray was ordered, as were the following baseline labs: complete blood count with differential (CBC), vitamin B12 and folate levels, blood chemistry, lipid profile, liver profile, total vitamin D, and sensitive thyroid-stimulating hormone. Test results are shown in Table 2.
X-ray of the wrist showed fracture only, making it possible to rule out Paget’s disease (ie, no patchy white areas noted in the bone) and tumor (no masses seen) as the immediate cause of fracture. Normal body temperature and normal white blood cell count eliminated the possibility of infection.
Because the patient was only 56 and had a history of bariatric surgery, further testing was pursued to investigate a cause for the weakened bone. Bone mineral density (BMD) testing revealed the following results:
• The lumbar spine in frontal projection measured 0.968 g/cm2 with a T-score of –2.2 and a Z-score of –2.2.
• Total BMD of the left hip was 0.863 g/cm2 with a T-score of –1.7 and a Z-score of –1.4.
• Total BMD of the left femoral neck was 0.863 g/cm2 with a T-score of 1.7 and a Z-score of –1.1.
These findings suggested osteopenia1,2 (not osteoporosis) in all sites, with a 12% decrease of BMD in the spine (suggesting increased risk for spinal fracture) and a 16.3% decrease of BMD in the hip since the patient’s most recent bone scan five years earlier (radiologist’s report). Other abnormal findings were elevated parathyroid hormone (PTH) serum, 95.7 pg/mL (reference range, 10 to 65 pg/mL); low total calcium serum, 8.7 mg/dL (reference range, 8.9 to 10.2 mg/dL), and low 25-hydroxyvitamin D total, 12.3 ng/mL (reference range, 25 to 80 ng/mL).
A 2010 clinical practice guideline from the Endocrine Society3 specifies that after malabsorptive surgery, vitamin D and calcium supplementation should be adjusted by a qualified medical professional, based on serum markers and measures of bone density. An endocrinologist who was consulted at the patient’s initial visit prescribed the following medications: vitamin D2, 50,000 U/wk PO; combined calcium citrate (vitamin D3) 500 IU with calcium 630 mg, 1 tab bid; and calcitriol 0.5 μg bid.
The patient’s final diagnosis was osteomalacia secondary to gastric bypass surgery. (See “Making the Diagnosis of Osteomalacia.”4-6)
DISCUSSION
According to 2008 data from the World Health Organization (WHO),7 1.4 billion persons older than 20 worldwide were overweight, and 200 million men and 300 million women were considered obese—meaning that one in every 10 adults worldwide is overweight or obese. In 2010, the WHO reports, 40 million children younger than 5 worldwide were considered overweight.7 Health care providers need to be prepared to care for the increasing number of patients who will undergo bariatric surgeries to treat obesity and its related comorbidities.8
Postoperative follow-up for the malabsorption deficiencies related to bariatric procedures should be performed every six months, including obtaining levels of alkaline phosphatase and others previously discussed. In addition, the Endocrine Society guideline3 recommends measuring levels of vitamin B12, albumin, pre-albumin, iron, and ferritin, and obtaining a CBC, a liver profile, glucose reading, creatinine measurement, and a metabolic profile at one month and two months after surgery, then every six months until two years after surgery, then annually if findings are stable.
Furthermore, the Endocrine Society3 recommends obtaining zinc levels every six months for the first year, then annually. An annual vitamin A level is optional.9 Yearly bone density testing is recommended until the patient’s BMD is deemed stable.3
Additionally, Koch and Finelli10 recommend performing the following labs postoperatively: hemoglobin A1C every three months; copper, magnesium, whole blood thiamine, vitamin B12, and a 24-hour urinary calcium every six months for the first three years, then once a year if findings remain stable.
Use of alcohol should be discouraged among patients who have undergone bariatric surgery, as its use alters micronutrient requirements and metabolism. Alcohol consumption may also contribute to dumping syndrome (ie, rapid gastric emptying).11
Any patient with a history of malabsorptive bypass surgery who complains of neurologic, visual, or skin disorders, anemia, or edema may require a further workup to rule out other absorptive deficiencies. These include vitamins A, E, and B12, zinc, folate, thiamine, niacin, selenium, and ferritin.10
Osteomalacia
Metabolic bone diseases can result from genetics, dietary factors, medication use, surgery, or hormonal irregularities. They alter the normal biochemical reactions in bone structure.
The three most common forms of metabolic bone disease are osteoporosis, osteopenia, and osteomalacia. The WHO diagnostic classifications and associated T-scores for bone mineral density1,2 indicate a T-score above –1.0 as normal. A score between –1.0 and –2.5 is indicative of osteopenia, and a score below –2.5 indicates osteoporosis. A T-score below –2.5 in the patient with a history of fragility fracture indicates severe osteoporosis.1,2
In osteomalacia, bone volume remains unchanged, but mineralization of osteoid in the mature compact and spongy bone is either delayed or inadequate. The remolding cycle continues unchanged in the formation of osteoid, but mineral calcification and deposition do not occur.3-5
Osteomalacia is normally considered a rare disorder, but it may become more common as increasing numbers of patients undergo gastric bypass operations.12,13 Primary care practitioners should monitor for this condition in such patients before serious bone loss or other problems develop.9,13,14
Vitamin D deficiency (see “Vitamin D Metabolism,”4,15-19 below), whether or not the result of gastric bypass surgery, is a major risk factor for osteomalacia. Disorders of the small bowel, the hepatobiliary system, and the pancreas are all common causes of vitamin D deficiency. Liver disease interferes with the metabolism of vitamin D. Diseases of the pancreas may cause a deficiency of bile salts, which are vital for the intestinal absorption of vitamin D.17
Restriction and Malabsorption
The case patient had undergone a gastric bypass (duodenal switch), in which a large portion of the stomach is removed and a large part of the small bowel rerouted—with both parts of the procedure causing malabsorption.11 It is in the small bowel that absorption of vitamin D and calcium takes place.
The duodenal switch gastric bypass surgery causes both restriction and malabsorption. Though similar to a biliopancreatic diversion, the duodenal switch preserves the distal stomach and the pylorus20 by way of a sleeve gastrectomy that is performed to reduce the gastric reservoir; the common channel length after revision is 100 cm, not 50 cm (as in conventional biliopancreatic diversion).13 The sleeve gastrectomy involves removal of parietal cells, reducing production of hydrochloric acid (which is necessary to break down food), and hindering the absorption of certain nutrients, including the fat-soluble vitamins, vitamin B12, and iron.12 Patients who take H2-blockers or proton pump inhibitors experience an additional decrease in the production and availability of HCl and may have an increased risk for fracture.14,20,21
In addition to its biliopancreatic diversion component, the duodenal switch diverts a large portion of the small bowel, with food restricted from moving through it. Vitamin D and protein are normally absorbed at the jejunum and ileum, but only when bile salts are present; after a duodenal switch, bile and pancreatic enzymes are not introduced into the small intestines until 75 to 100 cm before they reach the large intestine. Thus, absorption of vitamin D, protein, calcium, and other nutrients is impaired.20,22
Since phosphorus and magnesium are also absorbed at the sites of the duodenum and jejunum, malabsorption of these nutrients may occur in a patient who has undergone a duodenal switch. Although vitamin B12 is absorbed at the site of the distal ileum, it also requires gastric acid to free it from the food. Zinc absorption, which normally occurs at the site of the jejunum, may be impaired after duodenal switch surgery, and calcium supplementation, though essential, may further reduce zinc absorption.9 Iron absorption requires HCl, facilitated by the presence of vitamin C. Use of H2-blockers and proton pump inhibitors may impair iron metabolism, resulting in anemia.20
In a randomized controlled trial, Aasheim et al23 compared the effects of Roux-en-Y gastric bypass with those of duodenal switch gastric bypass on patients’ vitamin metabolism. The researchers concluded that patients who undergo a duodenal switch are at greater risk for vitamin A and D deficiencies in the first year after surgery; and for thiamine deficiency in the months following surgery as a result of malabsorption, compared with patients who undergo Roux-en-Y gastric bypass.20,23
Patient Management
The case patient’s care necessitated consultations with endocrinology, dermatology, and gastroenterology (GI). Table 3 (below) shows the laboratory findings and the medication changes prompted by the patient’s physical exam and lab results. Table 4 lists the findings from other lab studies ordered throughout the patient’s course of treatment.
The endocrinologist was consulted at the first sign of osteopenia, and a workup was soon initiated, followed by treatment. GI was consulted six months after the beginning of treatment, when the patient began to complain of reflux while sleeping and frequent diarrhea throughout the day.
Results of esophagogastroduodenoscopy with biopsy ruled out celiac disease and mucosal ulceration, but a small hiatal hernia that was detected (< 3 cm) was determined to be an aggravating factor for the patient’s reflux. The patient was instructed in lifestyle modifications for hiatal hernia, including the need to remain upright one to two hours after eating before going to sleep to prevent aspiration. The patient was instructed to avoid taking iron and calcium within two hours of each other and to limit his alcohol intake. He was also educated in precautions against falls.
Dermatology was consulted nine months into treatment so that light therapy could be initiated, allowing the patient to take advantage of the body’s natural pathway to manufacture vitamin D3.
CONCLUSION
For post–bariatric surgery patients, primary care practitioners are in a position to coordinate care recommendations from multiple specialists, including those in nutrition, to determine the best course of action.
This case illustrates complications of bariatric surgery (malabsorption of key vitamins and minerals, wrist fracture, osteopenia, osteomalacia) that require diagnosis and treatment. The specialists and the primary care practitioner, along with the patient, had to weigh the risks and benefits of continued proton pump inhibitor use, as such medications can increase the risk for fracture. They also addressed the patient’s anemia and remained attentive to his preventive health care needs.
REFERENCES
1. Brusin JH. Update on bone densitometry. Radiol Technol. 2009;81(2):153BD-170BD.
2. Wilson CR. Essentials of bone densitometry for the medical physicist. Presented at: The American Association of Physicists in Medicine 2003 Annual Meeting; July 22-26, 2003; San Diego, CA.
3. Heber D, Greenway FL, Kaplan LM. et al. Endocrine and nutritional management of the post-bariatric surgery patient: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95(11):4825-4843.
4. Osteomalacia: step-by-step diagnostic approach (2011). http://bestpractice.bmj.com/best-practice/monograph/517/diagnosis/step-by-step.html. Accessed December 18, 2012.
5. Gifre L, Peris P, Monegal A, et al. Osteomalacia revisited : a report on 28 cases. Clin Rheumatol. 2011;30(5):639-645.
6. Bingham CT, Fitzpatrick LA. Noninvasive testing in the diagnosis of osteomalacia. Am J Med. 1993;95(5):519-523.
7. World Health Organization. Obesity and overweight (May 2012). Fact Sheet No 311. www.who.int/mediacentre/factsheets/fs311/en/index.html. Accessed December 18, 2012.
8. Tanner BD, Allen JW. Complications of bariatric surgery: implications for the covering physician. Am Surg. 2009;75(2):103-112.
9. Soleymani T, Tejavanija S, Morgan S. Obesity, bariatric surgery, and bone. Curr Opin Rheumatol. 2011;23(4):396-405.
10. Koch TR, Finelli FC. Postoperative metabolic and nutritional complications of bariatric surgery. Gastroenterol Clin North Am. 2010;39(1):109-124.
11. Manchester S, Roye GD. Bariatric surgery: an overview for dietetics professionals. Nutr Today. 2011;46(6):264-275.
12. Bal BS, Finelli FC, Shope TR, Koch TR. Nutritional deficiencies after bariatric surgery. Nat Rev Endocrinol. 2012;8(9):544-546.
13. Iannelli A, Schneck AS, Dahman M, et al. Two-step laparoscopic duodenal switch for superobesity: a feasibility study. Surg Endosc. 2009;23(10):2385-2389.
14. Lalmohamed A, de Vries F, Bazelier MT, et al. Risk of fracture after bariatric surgery in the United Kingdom: population based, retrospective cohort study. BMJ. 2012;345:e5085.
15. Holrick MF. Vitamin D: important for prevention of osteoporosis, cardiovascular heart disease, type 1 diabetes, autoimmune diseases, and some cancers. South Med J. 2005;98 (10):1024-1027.
16. Kalro BN. Vitamin D and the skeleton. Alt Ther Womens Health. 2009;2(4):25-32.
17. Crowther-Radulewicz CL, McCance KL. Alterations of musculoskeletal function. In: McCance KL, Huether SE, Brashers VL, Rote NS, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 6th ed. Maryland Heights, MO: Mosby Elsevier; 2010:1568-1617.
18. Huether SE. Structure and function of the renal and urologic systems. In: McCance KL, Huether SE, Brashers VL, Rote NS, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 6th ed. Maryland Heights, MO: Mosby Elsevier; 2010:1344-1364.
19. Bhan A, Rao AD, Rao DS. Osteomalacia as a result of vitamin D deficiency. Endocrinol Metab Clin North Am. 2010;39(2):321-331.
20. Decker GA, Swain JM, Crowell MD. Gastrointestinal and nutritional complications after bariatric surgery. Am J Gastroenterol. 2007;102(11):2571-2580.
21. Targownik LE, Lix LM, Metge C, et al. Use of proton pump inhibitors and risk of osteoporosis-related fractures. CMAJ. 2008;179(4):319-326.
22. Ybarra J, Sánchez-Hernández J, Pérez A. Hypovitaminosis D and morbid obesity. Nurs Clin North Am. 2007;42(1):19-27.
23. Aasheim ET, Björkman S, Søvik TT, et al. Vitamin status after bariatric surgery: a randomized study of gastric bypass and duodenal switch. Am J Clin Nutr. 2009;90(1):15-22.
Grand Rounds: Woman, 38, With Pulseless Electrical Activity
On an autumn day, a 38-year-old woman with a history of asthma presented to the emergency department (ED) with the chief complaint of shortness of breath (SOB). The patient described her SOB as sudden in onset and not relieved by use of her albuterol inhaler; hence the ED visit.
She denied any chest pain, palpitations, dizziness, orthopnea, upper respiratory tract infection, cough, wheezing, fever or chills, headache, vision changes, body aches, sick contacts, or pets at home. She said she uses her albuterol inhaler as needed, and that she had used it that day for the first time in “a few months.” She denied any history of intubation or steroid use. Additionally, she had not been seen by a primary care provider in years.
The woman, a native of Ghana, had been living in the United States for many years. She denied any recent travel or exposure to toxic chemicals; any use of tobacco, alcohol, or illicit drugs; or any history of sexually transmitted disease.
The patient was afebrile (temperature, 98.6°F), with a respiratory rate of 20 breaths/min; blood pressure, 144/69 mm Hg; and ventricular rate, 125 beats/min. On physical examination, her extraocular movements were intact; pupils were equal, round, reactive to light and accommodation; and sclera were nonicteric. The patient’s head was normocephalic and atraumatic, and the neck was supple with normal range of motion and no jugular venous distension or lymphadenopathy. Her mucous membranes were moist with no pharyngeal erythema or exudates. Cardiovascular examination, including ECG, revealed tachycardia but no murmurs or gallops.
While being evaluated in the ED, the patient became tachypneic and began to experience respiratory distress. She was intubated for airway protection, at which time she developed pulseless electrical activity (PEA), with 30 beats/min. She responded to atropine and epinephrine injections. A repeat ECG showed sinus tachycardia and right atrial enlargement with right-axis deviation. Chest x-ray (see Figure 1) showed no consolidation, pleural effusion, or pneumothorax.
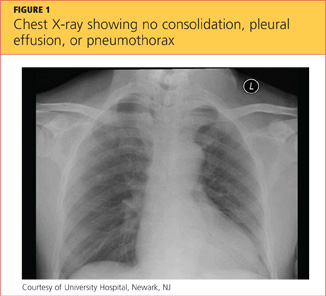
Results from the patient’s lab work are shown in the table, above. Negative results were reported for a urine pregnancy test.

Since there was no clear etiology for the patient’s PEA, she underwent pan-culturing, with the following tests ordered: HIV antibody testing, immunovirology for influenza A and B viruses, and urine toxicology. Doppler ultrasound of the bilateral lower extremities was also ordered, in addition to chest CT and transthoracic and transesophageal echocardiography (TTE and TEE, respectively). The patient was intubated and transferred to the medical ICU for further management.
The differential diagnosis included cardiac tamponade, acute MI, acute pulmonary embolus (PE), tension pneumothorax, hypovolemia, and asthma exacerbated by viral or bacterial infection.1,2 Although the case patient presented with PEA, she did not have the presenting signs of cardiac tamponade known as Beck’s triad: hypotension, jugular venous distension, and muffled heart sounds.3 TTE showed an ejection fraction of 65% and grade 2 diastolic dysfunction but no pericardial effusions (which accumulate rapidly in the patient with cardiac tamponade, resulting from fluid buildup in the pericardial layers),4 and TEE showed no atrial thrombi (which can masquerade as cardiac tamponade5). The patient had no signs of trauma and denied any history of malignancy (both potential causes of cardiac tamponade). Chest x-ray showed normal heart size and no pneumothorax, consolidations, or pleural effusions.4,6-8 Thus, the diagnosis of cardiac tamponade was ruled out.
Common presenting symptoms of acute MI include sudden-onset chest pain, SOB, palpitations, dizziness, nausea, and/or vomiting. Women may experience less dramatic symptoms—often little more than SOB and fatigue.9 According to a 2000 consensus document from a joint European Society of Cardiology/American College of Cardiology committee10 in which MI was redefined, the diagnosis of MI relies on a rise in cardiac troponin levels, typical MI symptoms, and changes in ECG showing pathological Q waves or ST elevation or depression. The case patient’s troponin I level was less than 0.02 ng/mL, and ECG did not reveal Q waves or ST-T wave changes; additionally, since the patient had no chest pain, palpitations, diaphoresis, nausea, or vomiting, acute MI was ruled out.
Blood clots capable of blocking the pulmonary artery usually originate in the deep veins of the lower extremities.11 Three main factors, called Virchow’s triad, are known to contribute to these deep vein thromboses (DVTs): venous stasis, endothelial injury, and a hypercoagulability state.12,13 The patient had denied any trauma, recent travel, history of malignancy, or use of tobacco or oral contraceptives, and the result of her urine pregnancy test was negative. Even though the patient presented with tachypnea and acute SOB, with ECG showing right-axis deviation and tachycardia (common presenting signs and symptoms for PE), her chest CT showed no evidence of PE (see Figure 2); additionally, Doppler ultrasound of the bilateral lower extremities revealed no DVTs. Thus, PE was also excluded.
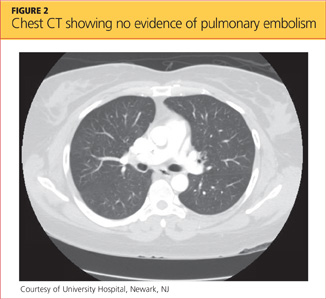
Tension pneumothorax was also ruled out, as chest x-ray showed neither mediastinal shift nor tracheal deviation, and the patient had denied any trauma. Laboratory analyses did not indicate hyponatremia, and the patient’s hemoglobin and hematocrit were satisfactory. She was tachycardic on admission, but her blood pressure was stable. As the patient denied any use of vasodilators or diuretics, hypovolemia was ruled out.
Patients experiencing asthma exacerbation can present with acute SOB, which usually resolves following use of IV steroids, nebulizer therapy, and inhaler treatments. Despite being administered IV methylprednisolone and magnesium sulfate in the ED, the patient experienced PEA and respiratory distress and required intubation for airway protection.
The HIV test was nonreactive, and blood and urine cultures did not show any growth. Results of tests for Legionella urinary antigen and Streptococcus pneumoniae antigen were negative. Sputum culture showed normal flora. Immunovirology testing, however, was positive for both influenza A and B antigens.
Chest X-ray showed no acute pulmonary pathology, nor did chest CT show any central, interlobar, or segmental embolism or mediastinal lymphadenopathy. It was determined that the patient’s acute SOB might represent asthma exacerbation secondary to influenza viral infection. Her PEA was attributed to possible acute pericarditis secondary to concomitant influenza A and B viral infection.
DISCUSSION
Currently, the CDC recognizes three types of influenza virus: A, B, and C.14 Only influenza A viruses are further classified into subtypes, based on the presence of surface proteins called hemagglutinin (HA) or neuraminidase (NA) glycoproteins. Humans can be infected by influenza A subtypes H1N1 and H3N2.14 Influenza B viruses, found mostly in humans, are associated with significant morbidity and mortality.
Influenza A and B viruses are further classified into strains that change with each flu season—thus, the need to update vaccinations against influenza A and B each year. No vaccination exists against influenza C virus, which is known to cause only mild illness in humans.15
In patients with asthma (as in the case patient), chronic bronchitis, or emphysema, infection with the influenza virus can manifest with SOB, in addition to the more common symptoms of fever, sore throat, headache, rhinorrhea, chills, muscle aches, and general discomfort.16 Patients with coronary artery disease, congestive heart failure (CHF), and/or a history of smoking may experience more severe symptoms and increased risk for influenza-associated mortality than do other patients.17,18
Rare cardiac complications of influenza infections are myocarditis and benign acute pericarditis; myocarditis can progress to CHF and death.19,20 A case of acute myopericarditis was reported by Proby et al21 in a patient with acute influenza A infection who developed pericardial effusions, myositis, tamponade, and pleurisy. That patient recovered after pericardiocentesis and administration of inotropic drugs.
In the literature, a few cases of acute pericarditis have been reported in association with administration of the influenza vaccination.22,23
In the case patient, the diagnosis of influenza A and B was made following testing of nasal and nasopharyngeal swabs with an immunochromatographic assay that uses highly sensitive monoclonal antibodies to detect influenza A and B nucleoprotein antigens.24,25
According to reports in the literature, two-thirds of cases of acute pericarditis are caused by infection, most commonly viral infection (including influenza virus, adenovirus, enterovirus, cytomegalovirus, hepatitis B virus, and herpes simplex virus).26,27 Other etiologies for acute pericarditis are autoimmune (accounting for less than 10% of cases) and neoplastic conditions (5% to 7% of cases).26
PATIENT OUTCOME
Consultation with an infectious disease specialist was obtained. The patient was placed under droplet isolation precautions and was started on a nebulizer, IV steroid treatments, and oseltamivir 75 mg by mouth every 12 hours. She was transferred to a medical floor, where she completed a five-day course of oseltamivir.
As a result of timely intervention, the patient was discharged in stable condition on a therapeutic regimen that included albuterol, fluticasone, and salmeterol inhalation, in addition to tapered-dose steroids. She was advised to follow up with her primary care provider and at the pulmonary clinic.
CONCLUSION
To our knowledge, this is the first reported case of acute pericarditis in a patient with concomitant acute infections with influenza A and B. According to conclusions reached in recent literature, further research is needed to explain the pathophysiology of influenza viral infections, associated cardiovascular morbidity and mortality, and the degree to which these can be prevented by influenza vaccination.1,28 Also to be pursued through research is a better understanding of the morbidity and mortality associated with influenza viruses, especially in children and in adults affected by asthma, cardiac disease, and/or obesity.
REFERENCES
1. Finelli L, Chaves SS. Influenza and acute myocardial infarction. J Infect Dis. 2011;203(12):
1701-1704.
2. Steiger HV, Rimbach K, Müller E, Breitkreutz R. Focused emergency echocardiography: lifesaving tool for a 14-year-old girl suffering out-of-hospital pulseless electrical activity arrest because of cardiac tamponade. Eur J Emerg Med. 2009;16(2): 103-105.
3. Goodman A, Perera P, Mailhot T, Mandavia D. The role of bedside ultrasound in the diagnosis of pericardial effusion and cardiac tamponade.
J Emerg Trauma Shock. 2012;5(1):72-75.
4. Restrepo CS, Lemos DF, Lemos JA, et al. Imaging findings in cardiac tamponade with emphasis on CT. Radiographics. 2007;27(6):1595-1610.
5. Papanagnou D, Stone MB. Massive right atrial thrombus masquerading as cardiac tamponade. Acad Emerg Med. 2010;17(2):E11.
6. Saito Y, Donohue A, Attai S, et al. The syndrome of cardiac tamponade with “small” pericardial effusion. Echocardiography. 2008;25(3): 321-327.
7. Lin E, Boire A, Hemmige V, et al. Cardiac tamponade mimicking tuberculous pericarditis as the initial presentation of chronic lymphocytic leukemia in a 58-year-old woman: a case report. J Med Case Rep. 2010;4:246.
8. Meniconi A, Attenhofer Jost CH, Jenni R. How to survive myocardial rupture after myocardial infarction. Heart. 2000;84(5):552.
9. Kosuge M, Kimura K, Ishikawa T, et al. Differences between men and women in terms of clinical features of ST-segment elevation acute myocardial infarction. Circ J. 2006;70(3):222-226.
10. Alpert JS, Thygesen K, Antman E, Bassand JP. Myocardial infarction redefined: a consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J Am Coll Cardiol. 2000;36(3):959-969.
11. Goldhaber SZ. Deep venous thrombosis and pulmonary thromboembolism. In: Fauci AS, Braunwald E, Kasper DL, et al. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill Medical; 2008:1651–1657.
12. Brooks EG, Trotman W, Wadsworth MP, et al. Valves of the deep venous system: an overlooked risk factor. Blood. 2009;114(6):1276-1279.
13. Kyrle PA, Eichinger S. Is Virchow’s triad complete? Blood. 2009;114(6):1138-1139.
14. CDC. Seasonal influenza (flu): types of influenza viruses (2012). www.cdc.gov/flu/about/viruses/types.htm. Accessed October 24, 2012.
15. CDC. Seasonal influenza (flu)(2012). www.cdc .gov/flu. Accessed October 24, 2012.
16. Eccles R. Understanding the symptoms of the common cold and influenza. Lancet Infect Dis. 2005;5(11):718-725.
17. Angelo SJ, Marshall PS, Chrissoheris MP, Chaves AM. Clinical characteristics associated with poor outcome in patients acutely infected with Influenza A. Conn Med. 2004;68(4):199-205.
18. Murin S, Bilello K. Respiratory tract infections: another reason not to smoke. Cleve Clin J Med. 2005;72(10):916-920.
19. Ray CG, Icenogle TB, Minnich LL, et al. The use of intravenous ribavirin to treat influenza virus–associated acute myocarditis. J Infect Dis. 1989; 159(5):829-836.
20. Fairley CK, Ryan M, Wall PG, Weinberg J. The organism reported to cause infective myocarditis and pericarditis in England and Wales. J Infect. 1996;32(3):223-225.
21. Proby CM, Hackett D, Gupta S, Cox TM. Acute myopericarditis in influenza A infection. Q J Med. 1986;60(233):887-892.
22. Streifler JJ, Dux S, Garty M, Rosenfeld JB. Recurrent pericarditis: a rare complication of influenza vaccination. Br Med J (Clin Res Ed). 1981; 283(6290):526-527.
23. Desson JF, Leprévost M, Vabret F, Davy A. Acute benign pericarditis after anti-influenza vaccination [in French]. Presse Med. 1997;26 (9):415.
24. BinaxNOW® Influenza A&B Test Kit (product instructions). www.diagnosticsdirect2u.com/images/PDF/Binax%20Now%20416-022%20PPI .pdf. Accessed October 24, 2012.
25. 510(k) Substantial Equivalence Determination Decision Summary [BinaxNow® Influenza A & B Test] (2009). www.accessdata.fda.gov/cdrh_docs/reviews/K062109.pdf. Accessed October 24, 2012.
26. Imazio M, Spodick DH, Brucato A, et al. Controversial issues in the management of pericardial diseases. Circulation. 2010;121(7):916-928.
27. Maisch B, Seferovic PM, Ristic AD, et al; Task Force on the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology. Guidelines on the diagnosis and management of pericardial diseases: executive summary. Eur Heart J. 2004;25(7):587-610.
28. McCullers JA, Hayden FG. Fatal influenza B infections: time to reexamine influenza research priorities. J Infect Dis. 2012;205(6):870-872.
On an autumn day, a 38-year-old woman with a history of asthma presented to the emergency department (ED) with the chief complaint of shortness of breath (SOB). The patient described her SOB as sudden in onset and not relieved by use of her albuterol inhaler; hence the ED visit.
She denied any chest pain, palpitations, dizziness, orthopnea, upper respiratory tract infection, cough, wheezing, fever or chills, headache, vision changes, body aches, sick contacts, or pets at home. She said she uses her albuterol inhaler as needed, and that she had used it that day for the first time in “a few months.” She denied any history of intubation or steroid use. Additionally, she had not been seen by a primary care provider in years.
The woman, a native of Ghana, had been living in the United States for many years. She denied any recent travel or exposure to toxic chemicals; any use of tobacco, alcohol, or illicit drugs; or any history of sexually transmitted disease.
The patient was afebrile (temperature, 98.6°F), with a respiratory rate of 20 breaths/min; blood pressure, 144/69 mm Hg; and ventricular rate, 125 beats/min. On physical examination, her extraocular movements were intact; pupils were equal, round, reactive to light and accommodation; and sclera were nonicteric. The patient’s head was normocephalic and atraumatic, and the neck was supple with normal range of motion and no jugular venous distension or lymphadenopathy. Her mucous membranes were moist with no pharyngeal erythema or exudates. Cardiovascular examination, including ECG, revealed tachycardia but no murmurs or gallops.
While being evaluated in the ED, the patient became tachypneic and began to experience respiratory distress. She was intubated for airway protection, at which time she developed pulseless electrical activity (PEA), with 30 beats/min. She responded to atropine and epinephrine injections. A repeat ECG showed sinus tachycardia and right atrial enlargement with right-axis deviation. Chest x-ray (see Figure 1) showed no consolidation, pleural effusion, or pneumothorax.
Results from the patient’s lab work are shown in the table, above. Negative results were reported for a urine pregnancy test.
Since there was no clear etiology for the patient’s PEA, she underwent pan-culturing, with the following tests ordered: HIV antibody testing, immunovirology for influenza A and B viruses, and urine toxicology. Doppler ultrasound of the bilateral lower extremities was also ordered, in addition to chest CT and transthoracic and transesophageal echocardiography (TTE and TEE, respectively). The patient was intubated and transferred to the medical ICU for further management.
The differential diagnosis included cardiac tamponade, acute MI, acute pulmonary embolus (PE), tension pneumothorax, hypovolemia, and asthma exacerbated by viral or bacterial infection.1,2 Although the case patient presented with PEA, she did not have the presenting signs of cardiac tamponade known as Beck’s triad: hypotension, jugular venous distension, and muffled heart sounds.3 TTE showed an ejection fraction of 65% and grade 2 diastolic dysfunction but no pericardial effusions (which accumulate rapidly in the patient with cardiac tamponade, resulting from fluid buildup in the pericardial layers),4 and TEE showed no atrial thrombi (which can masquerade as cardiac tamponade5). The patient had no signs of trauma and denied any history of malignancy (both potential causes of cardiac tamponade). Chest x-ray showed normal heart size and no pneumothorax, consolidations, or pleural effusions.4,6-8 Thus, the diagnosis of cardiac tamponade was ruled out.
Common presenting symptoms of acute MI include sudden-onset chest pain, SOB, palpitations, dizziness, nausea, and/or vomiting. Women may experience less dramatic symptoms—often little more than SOB and fatigue.9 According to a 2000 consensus document from a joint European Society of Cardiology/American College of Cardiology committee10 in which MI was redefined, the diagnosis of MI relies on a rise in cardiac troponin levels, typical MI symptoms, and changes in ECG showing pathological Q waves or ST elevation or depression. The case patient’s troponin I level was less than 0.02 ng/mL, and ECG did not reveal Q waves or ST-T wave changes; additionally, since the patient had no chest pain, palpitations, diaphoresis, nausea, or vomiting, acute MI was ruled out.
Blood clots capable of blocking the pulmonary artery usually originate in the deep veins of the lower extremities.11 Three main factors, called Virchow’s triad, are known to contribute to these deep vein thromboses (DVTs): venous stasis, endothelial injury, and a hypercoagulability state.12,13 The patient had denied any trauma, recent travel, history of malignancy, or use of tobacco or oral contraceptives, and the result of her urine pregnancy test was negative. Even though the patient presented with tachypnea and acute SOB, with ECG showing right-axis deviation and tachycardia (common presenting signs and symptoms for PE), her chest CT showed no evidence of PE (see Figure 2); additionally, Doppler ultrasound of the bilateral lower extremities revealed no DVTs. Thus, PE was also excluded.
Tension pneumothorax was also ruled out, as chest x-ray showed neither mediastinal shift nor tracheal deviation, and the patient had denied any trauma. Laboratory analyses did not indicate hyponatremia, and the patient’s hemoglobin and hematocrit were satisfactory. She was tachycardic on admission, but her blood pressure was stable. As the patient denied any use of vasodilators or diuretics, hypovolemia was ruled out.
Patients experiencing asthma exacerbation can present with acute SOB, which usually resolves following use of IV steroids, nebulizer therapy, and inhaler treatments. Despite being administered IV methylprednisolone and magnesium sulfate in the ED, the patient experienced PEA and respiratory distress and required intubation for airway protection.
The HIV test was nonreactive, and blood and urine cultures did not show any growth. Results of tests for Legionella urinary antigen and Streptococcus pneumoniae antigen were negative. Sputum culture showed normal flora. Immunovirology testing, however, was positive for both influenza A and B antigens.
Chest X-ray showed no acute pulmonary pathology, nor did chest CT show any central, interlobar, or segmental embolism or mediastinal lymphadenopathy. It was determined that the patient’s acute SOB might represent asthma exacerbation secondary to influenza viral infection. Her PEA was attributed to possible acute pericarditis secondary to concomitant influenza A and B viral infection.
DISCUSSION
Currently, the CDC recognizes three types of influenza virus: A, B, and C.14 Only influenza A viruses are further classified into subtypes, based on the presence of surface proteins called hemagglutinin (HA) or neuraminidase (NA) glycoproteins. Humans can be infected by influenza A subtypes H1N1 and H3N2.14 Influenza B viruses, found mostly in humans, are associated with significant morbidity and mortality.
Influenza A and B viruses are further classified into strains that change with each flu season—thus, the need to update vaccinations against influenza A and B each year. No vaccination exists against influenza C virus, which is known to cause only mild illness in humans.15
In patients with asthma (as in the case patient), chronic bronchitis, or emphysema, infection with the influenza virus can manifest with SOB, in addition to the more common symptoms of fever, sore throat, headache, rhinorrhea, chills, muscle aches, and general discomfort.16 Patients with coronary artery disease, congestive heart failure (CHF), and/or a history of smoking may experience more severe symptoms and increased risk for influenza-associated mortality than do other patients.17,18
Rare cardiac complications of influenza infections are myocarditis and benign acute pericarditis; myocarditis can progress to CHF and death.19,20 A case of acute myopericarditis was reported by Proby et al21 in a patient with acute influenza A infection who developed pericardial effusions, myositis, tamponade, and pleurisy. That patient recovered after pericardiocentesis and administration of inotropic drugs.
In the literature, a few cases of acute pericarditis have been reported in association with administration of the influenza vaccination.22,23
In the case patient, the diagnosis of influenza A and B was made following testing of nasal and nasopharyngeal swabs with an immunochromatographic assay that uses highly sensitive monoclonal antibodies to detect influenza A and B nucleoprotein antigens.24,25
According to reports in the literature, two-thirds of cases of acute pericarditis are caused by infection, most commonly viral infection (including influenza virus, adenovirus, enterovirus, cytomegalovirus, hepatitis B virus, and herpes simplex virus).26,27 Other etiologies for acute pericarditis are autoimmune (accounting for less than 10% of cases) and neoplastic conditions (5% to 7% of cases).26
PATIENT OUTCOME
Consultation with an infectious disease specialist was obtained. The patient was placed under droplet isolation precautions and was started on a nebulizer, IV steroid treatments, and oseltamivir 75 mg by mouth every 12 hours. She was transferred to a medical floor, where she completed a five-day course of oseltamivir.
As a result of timely intervention, the patient was discharged in stable condition on a therapeutic regimen that included albuterol, fluticasone, and salmeterol inhalation, in addition to tapered-dose steroids. She was advised to follow up with her primary care provider and at the pulmonary clinic.
CONCLUSION
To our knowledge, this is the first reported case of acute pericarditis in a patient with concomitant acute infections with influenza A and B. According to conclusions reached in recent literature, further research is needed to explain the pathophysiology of influenza viral infections, associated cardiovascular morbidity and mortality, and the degree to which these can be prevented by influenza vaccination.1,28 Also to be pursued through research is a better understanding of the morbidity and mortality associated with influenza viruses, especially in children and in adults affected by asthma, cardiac disease, and/or obesity.
REFERENCES
1. Finelli L, Chaves SS. Influenza and acute myocardial infarction. J Infect Dis. 2011;203(12):
1701-1704.
2. Steiger HV, Rimbach K, Müller E, Breitkreutz R. Focused emergency echocardiography: lifesaving tool for a 14-year-old girl suffering out-of-hospital pulseless electrical activity arrest because of cardiac tamponade. Eur J Emerg Med. 2009;16(2): 103-105.
3. Goodman A, Perera P, Mailhot T, Mandavia D. The role of bedside ultrasound in the diagnosis of pericardial effusion and cardiac tamponade.
J Emerg Trauma Shock. 2012;5(1):72-75.
4. Restrepo CS, Lemos DF, Lemos JA, et al. Imaging findings in cardiac tamponade with emphasis on CT. Radiographics. 2007;27(6):1595-1610.
5. Papanagnou D, Stone MB. Massive right atrial thrombus masquerading as cardiac tamponade. Acad Emerg Med. 2010;17(2):E11.
6. Saito Y, Donohue A, Attai S, et al. The syndrome of cardiac tamponade with “small” pericardial effusion. Echocardiography. 2008;25(3): 321-327.
7. Lin E, Boire A, Hemmige V, et al. Cardiac tamponade mimicking tuberculous pericarditis as the initial presentation of chronic lymphocytic leukemia in a 58-year-old woman: a case report. J Med Case Rep. 2010;4:246.
8. Meniconi A, Attenhofer Jost CH, Jenni R. How to survive myocardial rupture after myocardial infarction. Heart. 2000;84(5):552.
9. Kosuge M, Kimura K, Ishikawa T, et al. Differences between men and women in terms of clinical features of ST-segment elevation acute myocardial infarction. Circ J. 2006;70(3):222-226.
10. Alpert JS, Thygesen K, Antman E, Bassand JP. Myocardial infarction redefined: a consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J Am Coll Cardiol. 2000;36(3):959-969.
11. Goldhaber SZ. Deep venous thrombosis and pulmonary thromboembolism. In: Fauci AS, Braunwald E, Kasper DL, et al. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill Medical; 2008:1651–1657.
12. Brooks EG, Trotman W, Wadsworth MP, et al. Valves of the deep venous system: an overlooked risk factor. Blood. 2009;114(6):1276-1279.
13. Kyrle PA, Eichinger S. Is Virchow’s triad complete? Blood. 2009;114(6):1138-1139.
14. CDC. Seasonal influenza (flu): types of influenza viruses (2012). www.cdc.gov/flu/about/viruses/types.htm. Accessed October 24, 2012.
15. CDC. Seasonal influenza (flu)(2012). www.cdc .gov/flu. Accessed October 24, 2012.
16. Eccles R. Understanding the symptoms of the common cold and influenza. Lancet Infect Dis. 2005;5(11):718-725.
17. Angelo SJ, Marshall PS, Chrissoheris MP, Chaves AM. Clinical characteristics associated with poor outcome in patients acutely infected with Influenza A. Conn Med. 2004;68(4):199-205.
18. Murin S, Bilello K. Respiratory tract infections: another reason not to smoke. Cleve Clin J Med. 2005;72(10):916-920.
19. Ray CG, Icenogle TB, Minnich LL, et al. The use of intravenous ribavirin to treat influenza virus–associated acute myocarditis. J Infect Dis. 1989; 159(5):829-836.
20. Fairley CK, Ryan M, Wall PG, Weinberg J. The organism reported to cause infective myocarditis and pericarditis in England and Wales. J Infect. 1996;32(3):223-225.
21. Proby CM, Hackett D, Gupta S, Cox TM. Acute myopericarditis in influenza A infection. Q J Med. 1986;60(233):887-892.
22. Streifler JJ, Dux S, Garty M, Rosenfeld JB. Recurrent pericarditis: a rare complication of influenza vaccination. Br Med J (Clin Res Ed). 1981; 283(6290):526-527.
23. Desson JF, Leprévost M, Vabret F, Davy A. Acute benign pericarditis after anti-influenza vaccination [in French]. Presse Med. 1997;26 (9):415.
24. BinaxNOW® Influenza A&B Test Kit (product instructions). www.diagnosticsdirect2u.com/images/PDF/Binax%20Now%20416-022%20PPI .pdf. Accessed October 24, 2012.
25. 510(k) Substantial Equivalence Determination Decision Summary [BinaxNow® Influenza A & B Test] (2009). www.accessdata.fda.gov/cdrh_docs/reviews/K062109.pdf. Accessed October 24, 2012.
26. Imazio M, Spodick DH, Brucato A, et al. Controversial issues in the management of pericardial diseases. Circulation. 2010;121(7):916-928.
27. Maisch B, Seferovic PM, Ristic AD, et al; Task Force on the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology. Guidelines on the diagnosis and management of pericardial diseases: executive summary. Eur Heart J. 2004;25(7):587-610.
28. McCullers JA, Hayden FG. Fatal influenza B infections: time to reexamine influenza research priorities. J Infect Dis. 2012;205(6):870-872.
On an autumn day, a 38-year-old woman with a history of asthma presented to the emergency department (ED) with the chief complaint of shortness of breath (SOB). The patient described her SOB as sudden in onset and not relieved by use of her albuterol inhaler; hence the ED visit.
She denied any chest pain, palpitations, dizziness, orthopnea, upper respiratory tract infection, cough, wheezing, fever or chills, headache, vision changes, body aches, sick contacts, or pets at home. She said she uses her albuterol inhaler as needed, and that she had used it that day for the first time in “a few months.” She denied any history of intubation or steroid use. Additionally, she had not been seen by a primary care provider in years.
The woman, a native of Ghana, had been living in the United States for many years. She denied any recent travel or exposure to toxic chemicals; any use of tobacco, alcohol, or illicit drugs; or any history of sexually transmitted disease.
The patient was afebrile (temperature, 98.6°F), with a respiratory rate of 20 breaths/min; blood pressure, 144/69 mm Hg; and ventricular rate, 125 beats/min. On physical examination, her extraocular movements were intact; pupils were equal, round, reactive to light and accommodation; and sclera were nonicteric. The patient’s head was normocephalic and atraumatic, and the neck was supple with normal range of motion and no jugular venous distension or lymphadenopathy. Her mucous membranes were moist with no pharyngeal erythema or exudates. Cardiovascular examination, including ECG, revealed tachycardia but no murmurs or gallops.
While being evaluated in the ED, the patient became tachypneic and began to experience respiratory distress. She was intubated for airway protection, at which time she developed pulseless electrical activity (PEA), with 30 beats/min. She responded to atropine and epinephrine injections. A repeat ECG showed sinus tachycardia and right atrial enlargement with right-axis deviation. Chest x-ray (see Figure 1) showed no consolidation, pleural effusion, or pneumothorax.
Results from the patient’s lab work are shown in the table, above. Negative results were reported for a urine pregnancy test.
Since there was no clear etiology for the patient’s PEA, she underwent pan-culturing, with the following tests ordered: HIV antibody testing, immunovirology for influenza A and B viruses, and urine toxicology. Doppler ultrasound of the bilateral lower extremities was also ordered, in addition to chest CT and transthoracic and transesophageal echocardiography (TTE and TEE, respectively). The patient was intubated and transferred to the medical ICU for further management.
The differential diagnosis included cardiac tamponade, acute MI, acute pulmonary embolus (PE), tension pneumothorax, hypovolemia, and asthma exacerbated by viral or bacterial infection.1,2 Although the case patient presented with PEA, she did not have the presenting signs of cardiac tamponade known as Beck’s triad: hypotension, jugular venous distension, and muffled heart sounds.3 TTE showed an ejection fraction of 65% and grade 2 diastolic dysfunction but no pericardial effusions (which accumulate rapidly in the patient with cardiac tamponade, resulting from fluid buildup in the pericardial layers),4 and TEE showed no atrial thrombi (which can masquerade as cardiac tamponade5). The patient had no signs of trauma and denied any history of malignancy (both potential causes of cardiac tamponade). Chest x-ray showed normal heart size and no pneumothorax, consolidations, or pleural effusions.4,6-8 Thus, the diagnosis of cardiac tamponade was ruled out.
Common presenting symptoms of acute MI include sudden-onset chest pain, SOB, palpitations, dizziness, nausea, and/or vomiting. Women may experience less dramatic symptoms—often little more than SOB and fatigue.9 According to a 2000 consensus document from a joint European Society of Cardiology/American College of Cardiology committee10 in which MI was redefined, the diagnosis of MI relies on a rise in cardiac troponin levels, typical MI symptoms, and changes in ECG showing pathological Q waves or ST elevation or depression. The case patient’s troponin I level was less than 0.02 ng/mL, and ECG did not reveal Q waves or ST-T wave changes; additionally, since the patient had no chest pain, palpitations, diaphoresis, nausea, or vomiting, acute MI was ruled out.
Blood clots capable of blocking the pulmonary artery usually originate in the deep veins of the lower extremities.11 Three main factors, called Virchow’s triad, are known to contribute to these deep vein thromboses (DVTs): venous stasis, endothelial injury, and a hypercoagulability state.12,13 The patient had denied any trauma, recent travel, history of malignancy, or use of tobacco or oral contraceptives, and the result of her urine pregnancy test was negative. Even though the patient presented with tachypnea and acute SOB, with ECG showing right-axis deviation and tachycardia (common presenting signs and symptoms for PE), her chest CT showed no evidence of PE (see Figure 2); additionally, Doppler ultrasound of the bilateral lower extremities revealed no DVTs. Thus, PE was also excluded.
Tension pneumothorax was also ruled out, as chest x-ray showed neither mediastinal shift nor tracheal deviation, and the patient had denied any trauma. Laboratory analyses did not indicate hyponatremia, and the patient’s hemoglobin and hematocrit were satisfactory. She was tachycardic on admission, but her blood pressure was stable. As the patient denied any use of vasodilators or diuretics, hypovolemia was ruled out.
Patients experiencing asthma exacerbation can present with acute SOB, which usually resolves following use of IV steroids, nebulizer therapy, and inhaler treatments. Despite being administered IV methylprednisolone and magnesium sulfate in the ED, the patient experienced PEA and respiratory distress and required intubation for airway protection.
The HIV test was nonreactive, and blood and urine cultures did not show any growth. Results of tests for Legionella urinary antigen and Streptococcus pneumoniae antigen were negative. Sputum culture showed normal flora. Immunovirology testing, however, was positive for both influenza A and B antigens.
Chest X-ray showed no acute pulmonary pathology, nor did chest CT show any central, interlobar, or segmental embolism or mediastinal lymphadenopathy. It was determined that the patient’s acute SOB might represent asthma exacerbation secondary to influenza viral infection. Her PEA was attributed to possible acute pericarditis secondary to concomitant influenza A and B viral infection.
DISCUSSION
Currently, the CDC recognizes three types of influenza virus: A, B, and C.14 Only influenza A viruses are further classified into subtypes, based on the presence of surface proteins called hemagglutinin (HA) or neuraminidase (NA) glycoproteins. Humans can be infected by influenza A subtypes H1N1 and H3N2.14 Influenza B viruses, found mostly in humans, are associated with significant morbidity and mortality.
Influenza A and B viruses are further classified into strains that change with each flu season—thus, the need to update vaccinations against influenza A and B each year. No vaccination exists against influenza C virus, which is known to cause only mild illness in humans.15
In patients with asthma (as in the case patient), chronic bronchitis, or emphysema, infection with the influenza virus can manifest with SOB, in addition to the more common symptoms of fever, sore throat, headache, rhinorrhea, chills, muscle aches, and general discomfort.16 Patients with coronary artery disease, congestive heart failure (CHF), and/or a history of smoking may experience more severe symptoms and increased risk for influenza-associated mortality than do other patients.17,18
Rare cardiac complications of influenza infections are myocarditis and benign acute pericarditis; myocarditis can progress to CHF and death.19,20 A case of acute myopericarditis was reported by Proby et al21 in a patient with acute influenza A infection who developed pericardial effusions, myositis, tamponade, and pleurisy. That patient recovered after pericardiocentesis and administration of inotropic drugs.
In the literature, a few cases of acute pericarditis have been reported in association with administration of the influenza vaccination.22,23
In the case patient, the diagnosis of influenza A and B was made following testing of nasal and nasopharyngeal swabs with an immunochromatographic assay that uses highly sensitive monoclonal antibodies to detect influenza A and B nucleoprotein antigens.24,25
According to reports in the literature, two-thirds of cases of acute pericarditis are caused by infection, most commonly viral infection (including influenza virus, adenovirus, enterovirus, cytomegalovirus, hepatitis B virus, and herpes simplex virus).26,27 Other etiologies for acute pericarditis are autoimmune (accounting for less than 10% of cases) and neoplastic conditions (5% to 7% of cases).26
PATIENT OUTCOME
Consultation with an infectious disease specialist was obtained. The patient was placed under droplet isolation precautions and was started on a nebulizer, IV steroid treatments, and oseltamivir 75 mg by mouth every 12 hours. She was transferred to a medical floor, where she completed a five-day course of oseltamivir.
As a result of timely intervention, the patient was discharged in stable condition on a therapeutic regimen that included albuterol, fluticasone, and salmeterol inhalation, in addition to tapered-dose steroids. She was advised to follow up with her primary care provider and at the pulmonary clinic.
CONCLUSION
To our knowledge, this is the first reported case of acute pericarditis in a patient with concomitant acute infections with influenza A and B. According to conclusions reached in recent literature, further research is needed to explain the pathophysiology of influenza viral infections, associated cardiovascular morbidity and mortality, and the degree to which these can be prevented by influenza vaccination.1,28 Also to be pursued through research is a better understanding of the morbidity and mortality associated with influenza viruses, especially in children and in adults affected by asthma, cardiac disease, and/or obesity.
REFERENCES
1. Finelli L, Chaves SS. Influenza and acute myocardial infarction. J Infect Dis. 2011;203(12):
1701-1704.
2. Steiger HV, Rimbach K, Müller E, Breitkreutz R. Focused emergency echocardiography: lifesaving tool for a 14-year-old girl suffering out-of-hospital pulseless electrical activity arrest because of cardiac tamponade. Eur J Emerg Med. 2009;16(2): 103-105.
3. Goodman A, Perera P, Mailhot T, Mandavia D. The role of bedside ultrasound in the diagnosis of pericardial effusion and cardiac tamponade.
J Emerg Trauma Shock. 2012;5(1):72-75.
4. Restrepo CS, Lemos DF, Lemos JA, et al. Imaging findings in cardiac tamponade with emphasis on CT. Radiographics. 2007;27(6):1595-1610.
5. Papanagnou D, Stone MB. Massive right atrial thrombus masquerading as cardiac tamponade. Acad Emerg Med. 2010;17(2):E11.
6. Saito Y, Donohue A, Attai S, et al. The syndrome of cardiac tamponade with “small” pericardial effusion. Echocardiography. 2008;25(3): 321-327.
7. Lin E, Boire A, Hemmige V, et al. Cardiac tamponade mimicking tuberculous pericarditis as the initial presentation of chronic lymphocytic leukemia in a 58-year-old woman: a case report. J Med Case Rep. 2010;4:246.
8. Meniconi A, Attenhofer Jost CH, Jenni R. How to survive myocardial rupture after myocardial infarction. Heart. 2000;84(5):552.
9. Kosuge M, Kimura K, Ishikawa T, et al. Differences between men and women in terms of clinical features of ST-segment elevation acute myocardial infarction. Circ J. 2006;70(3):222-226.
10. Alpert JS, Thygesen K, Antman E, Bassand JP. Myocardial infarction redefined: a consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J Am Coll Cardiol. 2000;36(3):959-969.
11. Goldhaber SZ. Deep venous thrombosis and pulmonary thromboembolism. In: Fauci AS, Braunwald E, Kasper DL, et al. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill Medical; 2008:1651–1657.
12. Brooks EG, Trotman W, Wadsworth MP, et al. Valves of the deep venous system: an overlooked risk factor. Blood. 2009;114(6):1276-1279.
13. Kyrle PA, Eichinger S. Is Virchow’s triad complete? Blood. 2009;114(6):1138-1139.
14. CDC. Seasonal influenza (flu): types of influenza viruses (2012). www.cdc.gov/flu/about/viruses/types.htm. Accessed October 24, 2012.
15. CDC. Seasonal influenza (flu)(2012). www.cdc .gov/flu. Accessed October 24, 2012.
16. Eccles R. Understanding the symptoms of the common cold and influenza. Lancet Infect Dis. 2005;5(11):718-725.
17. Angelo SJ, Marshall PS, Chrissoheris MP, Chaves AM. Clinical characteristics associated with poor outcome in patients acutely infected with Influenza A. Conn Med. 2004;68(4):199-205.
18. Murin S, Bilello K. Respiratory tract infections: another reason not to smoke. Cleve Clin J Med. 2005;72(10):916-920.
19. Ray CG, Icenogle TB, Minnich LL, et al. The use of intravenous ribavirin to treat influenza virus–associated acute myocarditis. J Infect Dis. 1989; 159(5):829-836.
20. Fairley CK, Ryan M, Wall PG, Weinberg J. The organism reported to cause infective myocarditis and pericarditis in England and Wales. J Infect. 1996;32(3):223-225.
21. Proby CM, Hackett D, Gupta S, Cox TM. Acute myopericarditis in influenza A infection. Q J Med. 1986;60(233):887-892.
22. Streifler JJ, Dux S, Garty M, Rosenfeld JB. Recurrent pericarditis: a rare complication of influenza vaccination. Br Med J (Clin Res Ed). 1981; 283(6290):526-527.
23. Desson JF, Leprévost M, Vabret F, Davy A. Acute benign pericarditis after anti-influenza vaccination [in French]. Presse Med. 1997;26 (9):415.
24. BinaxNOW® Influenza A&B Test Kit (product instructions). www.diagnosticsdirect2u.com/images/PDF/Binax%20Now%20416-022%20PPI .pdf. Accessed October 24, 2012.
25. 510(k) Substantial Equivalence Determination Decision Summary [BinaxNow® Influenza A & B Test] (2009). www.accessdata.fda.gov/cdrh_docs/reviews/K062109.pdf. Accessed October 24, 2012.
26. Imazio M, Spodick DH, Brucato A, et al. Controversial issues in the management of pericardial diseases. Circulation. 2010;121(7):916-928.
27. Maisch B, Seferovic PM, Ristic AD, et al; Task Force on the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology. Guidelines on the diagnosis and management of pericardial diseases: executive summary. Eur Heart J. 2004;25(7):587-610.
28. McCullers JA, Hayden FG. Fatal influenza B infections: time to reexamine influenza research priorities. J Infect Dis. 2012;205(6):870-872.
Grand Rounds: Man, 65, With Heart Failure Symptoms
A black man, age 65, with no known history of cardiopulmonary disease presented with acute-onset exertional dyspnea and lower extremity edema. He also reported an episode of syncope, as well as occasional dizziness and abdominal bloating. He said he experienced exertional dyspnea while doing a routine step aerobic exercise. His exercise regimen included distance walking, yoga, and aerobics four to five days per week.
The patient’s medical history was remarkable for a single episode of a bleeding ulcer in previous years, low back pain, shoulder pain, and a septic arthritic hip. His social history was negative for use of tobacco, alcohol, or illegal drugs. He was married and had two biological daughters with fairly unremarkable medical histories. The patient had earned a master’s degree, worked full-time in the insurance business, and was an avid worldwide traveler. He reported diminished quality of life as a result of his acute-onset heart failure symptoms, which reduced his ability to exercise routinely, work full-time, or travel.
The patient’s sudden experience of exertional dyspnea prompted him to visit his primary care provider, who ordered an ECG that demonstrated low voltage patterns and a first-degree atrioventricular (AV) block. Subsequent stress echocardiography showed generalized thickening of the left ventricular myocardium. Posterior wall thickness measured 1.7 cm (normal range, 0.6 to 1.1 cm), septal thickness measured 1.9 cm (normal, 0.6 to 1.1 cm), and ejection fraction was 65%. The stress echocardiogram also showed a speckling pattern (brightly scattered spots) on the myocardium.
Although stress echocardiography results were negative for ischemic disease, the patient did experience dyspnea during the exam. He underwent cardiac catheterization, which indicated normal coronary arteries.
Additional diagnostic studies included cardiac MRI with and without contrast, which showed nulling of the heart muscle and delayed patchy hyperenhancement; this suggested myocardial tissue abnormality as result of amyloid fibril deposition.1 Both pulmonary and tricuspid aortic valves were normal, with no evidence of stenosis. No regional wall motion abnormalities were noted.
Laboratory findings during the work-up were lipid panel, unremarkable; complete blood count (CBC), mild anemia and leukopenia; and urinalysis, positive for proteinuria. Brain natriuretic peptide (BNP) was measured at 686 pg/mL (normal, 0.0 to 100 pg/mL), indicating moderate heart failure. A peripheral blood smear was negative for monoclonal plasma cells.
The patient’s physical exam was unremarkable except for 2+ pedal edema bilaterally. In consideration of normal coronary arteries on cardiac catheterization, the patient’s heart failure symptoms, and stress echocardiography abnormalities, a heart biopsy was ordered. An endomyocardial biopsy with Congo Red stain demonstrated an apple-green birefringent pattern viewed under high-definition polarized light microscope, which was consistent with amyloid deposition.2
The patient was given a diagnosis of primary amyloidosis by his local cardiologist despite negative findings on the peripheral blood smear for monoclonal plasma cells (which are typically found in primary amyloidosis).3 He presented to an institution well-known for its expertise in amyloidosis, for a second opinion. There, the diagnosis was negated, based on reevaluation of the patient’s previous heart specimen through immunohistochemical studies. These studies were positive for serum amyloid P, which is suggestive of transthyretin (TTR) or familial amyloidosis.4 Genetic testing revealed a familial amyloidosis DNA sequence analysis with the Val122Ile variant (ie, isoleucine for valine at position 1225). With the correct diagnosis confirmed, the patient was referred to another highly regarded institution to begin a work-up for cardiac transplantation. Meanwhile, he was cautiously treated with the loop diuretic furosemide to manage his shortness of breath and peripheral edema.
Fifteen months later (13 weeks after being listed for transplant), the patient underwent successful cardiac transplantation.
On pathologic review of the patient’s extricated heart, the myocardium was found to be grossly thickened (see figure, above) and weighed 540 g; the average adult heart weighs 300 to 350 g, depending on the patient’s size.6 Congo Red staining showed extensive amyloid deposits with infiltration throughout the myocardium.
Ninety percent of the amyloid deposits were interstitial, 5% were in the vessels, and 5% were noted in a nodular pattern. The left ventricular cavity showed dilated and thickened walls. Intramural and extramural blood vessels were infiltrated with amyloid as well.
Six months after transplantation, the patient underwent diagnostic testing to assess the function and structure of his new heart. Cardiac catheterization was negative for coronary artery disease. Thirteen months posttransplantation, endomyocardial biopsy with Congo Red stain was negative for amyloid deposition or organ rejection.
About 24 months posttransplantation, the patient was taking tacrolimus, pravastatin, pantoprazole, dapsone, propanolol, colchicine, and donepezil. Stress echocardiography demonstrated normal right and left ventricular systolic function; no wall-motion abnormalities or left ventricular hypertrophy were detected, and the right atrium was of normal size. There was abnormal structural enlargement of the left atrium at the site of anastamosis—a common finding in cardiac transplant patients. The aortic, tricuspid, and mitral valves were all normal.
At that time, it was decided not to repeat endomyocardial biopsy because of normal results on molecular expression testing (a noninvasive technique called AlloMap®7-9), which is performed to assess for heart transplant rejection. The patient’s lipid panel remained within normal limits. CBC indicated persistent anemia and leukopenia. Urine protein and BNP test results were not available.
Since undergoing cardiac transplantation, the patient has resumed his normal routine activities, including some type of exercise five days per week. He said his diet is maintained in moderation. He denied shortness of breath, chest pain, dizziness, or edema. He has returned to full-time employment and has vacationed in Croatia, Italy, and Central America.
DISCUSSION
Familial amyloidosis is an autosomal dominant disease characterized by the production of mutated proteins, most commonly ATTR. Presence of the ATTR Val 122Ile allele has been reported in 3.9% of all black Americans, and in one study, 23% of black Americans diagnosed with cardiac amyloidosis at autopsy were heterozygous for this variant allele.10-12 ATTR Val122Ile usually manifests in the fifth
or sixth decade of life with its characteristic presentation of infiltrative/restrictive cardiomyopathy,13 resulting in heart failure and sometimes peripheral neuropathy.10,11,14
Pathophysiology
In patients with ATTR Val122Ile, cardiomyopathy results from the deposition of mutant protein fibrils in the cardiac muscle, leading to restricted heart wall motion10,15 and stiffening of the cardiac ventricles, with subsequent disruption of the diastolic filling properties of the cardiac muscle.16 Fluid overload and heart failure follow.3,10,17 The atrium of the heart dilates, and the walls of the ventricles become thickened and fibrous.18 Liepnieks and Benson6 reported that the cadaver heart of one Val122Ile patient infiltrated with amyloid protein fibrils weighed 725 g—more than double the weight of an average adult heart.
In patients with cardiac amyloidosis, ECG can detect arrhythmia, and echocardiography shows cardiac enlargement; however, as in the case patient, cardiac catheterization shows normal coronary arteries.15,16,19,20 Thus, previously healthy patients who present with heart failure and negative results on cardiac catheterization should undergo further work-up for cardiac amyloidosis.19
Amyloidosis affects all populations globally.10 In systemic amyloidosis, amyloid releases into the plasma, infiltrating and impairing multiple organs. Poor survival has been reported in patients with heart failure symptoms resulting from amyloid deposition.20,21
Types of Amyloidosis
Primary amyloidosis, the most common of the three amyloidosis types, can be systemic or localized.22 It occurs when protein fibrils, developed from immunoglobin light chains or monoclonal plasma cells and measuring 7 to 10 nm in diameter, adhere to the heart, kidneys, peripheral nerves, eyes, and other organs.5,11,20,23,24 Known for its relation to multiple myeloma,19 primary amyloidosis is associated with a poor prognosis.3,10
Secondary amyloidosis results from a chronic inflammatory disorder, such as rheumatoid arthritis or ankylosing spondylitis—conditions that trigger the production of amyloid proteins.3,10 This type has also been associated with substance abuse and AIDS.23
Familial or hereditary amyloidosis, according to Benson,10 is a group of diseases, each resulting from mutation in a specific protein. In the United States, the most common type of familial amyloidosis is ATTR.11 More than 100 mutant types of ATTR proteins have been identified, each involving a specific nationality or group of nationalities.10,11,23
Mutant ATTR amyloid, when deposited in specific organs, leads to their dysfunction and ultimate failure.6 ATTR may affect the cardiac, gastric, renal, ophthalmic, or nervous system. Depending on the ATTR variant, the resulting clinical features are age- and time-dependent, with onset most common between the third and fifth decade of life.10
Prevalence
The prevalence of ATTR Val 122Ile amyloidosis is reportedly high in West Africa, and in the US African-American population (3.9%).4,11,14,25,26 In a study conducted at a county hospital in Indianapolis, 3% of black newborns were found positive for ATTR Val122Ile through DNA sampling of umbilical cord blood.25 These statistics are of concern, as ATTR amyloidosis could be a significant health concern in a patient population that is already medically underserved.
Yamashita et al25 estimate that 1.35 million Americans of African-American descent may be affected by ATTR Val122Ile and vulnerable to restrictive cardiomyopathy–related heart failure and death. At the very least, this disorder can impair quality of life, especially in the presence of other comorbid conditions.
Clinical Presentation
The presence of exertional syncope at presentation is ominous, as it may be a marker of severe restrictive cardiomyopathy, postural hypotension due to excessive diuresis or autonomic neuropathy, ventricular arrhythmias from localized hypoperfusion, and rarely from cardiac tamponade due to pericardial involvement.27 Despite widespread involvement of the conduction system in specimens at autopsy, high-grade IV block is unusual.3
Diagnostic Studies
ECG. Both ECG and Holter monitoring can detect the arrhythmias and conduction disturbances (eg, first-degree AV block, low voltage patterns) associated with cardiac amyloidosis. Patients often experience syncopal and near-syncopal episodes as a result of conduction disturbances.28 Patients with conditions such as cardiac amyloidosis who present with severe heart failure are at high risk for sudden death secondary to conduction disturbances. Many have benefited from implanted defibrillators.29
Echocardiography. In patients with ATTR Val122Ile cardiac amyloidosis, echocardiography reveals thickened ventricular walls (ie, measuring ≥ 15 mm; normal, ≤ 11 mm).19 Amyloid-restrictive cardiomyopathy is associated with a marked dissociation between short- and long-axis systolic function, in cases in which left ventricular ejection fraction is normal.30
Echocardiography may demonstrate the characteristic specular or granular sparkling appearance that signifies advanced disease.15,21 Only a minority have this pattern in the myocardium, however, and changes in echocardiographic technology have made this finding less noticeable.30
MRI. Among more recently used diagnostic studies, cardiac magnetic resonance (CMR) has been reported to demonstrate late gadolinium enhancement (LGE) in perhaps 80% of patients with familial amyloidosis and cardiac involvement (as determined through biopsy and Congo Red stain). LGE-CMR shows darkening of the cardiac tissue, a common occurrence in amyloidosis.31
LGE is associated with increased thickness of both left and right ventricles, lower ECG voltage patterns, elevated BNP, and elevated troponin T.31,32 Globally, LGE is associated with the worst prognosis in patients with cardiac amyloidosis. Use of LGE-CMR testing can help facilitate early detection of cardiac amyloidosis in patients who may be vulnerable to cardiac damage.31
Cardiac catheterization. In patients with cardiac amyloidosis, cardiac catheterization usually shows normal coronary arteries.3
Diagnosis
Early diagnosis of ATTR cardiac amyloidosis is crucial to the patient’s survival; it should be ruled out in any African-American patient with unexplained heart failure and echocardiography showing increased wall thickness with a nondilated left ventricular cavity. Additional clues include significant proteinuria, hepatomegaly disproportionate to the degree of heart failure, or corresponding neuropathy. Known family history of the disease, along with variant type, allows for a prompt and correct diagnosis.10
It has been reported that most clinicians who encounter heart failure, particularly in black patients, do not consider amyloidosis in the differential diagnosis, because of the high prevalence of hypertension and congestive heart failure in this population.10,15,19 As a result, amyloidosis often goes undiagnosed.19
Findings of enlarged and thickened cardiac walls on echocardiography but normal coronary arteries on cardiac catheterization should alert the treating clinician to further work-up for cardiac amyloidosis.19 In such a patient, according to Kristen et al,21 endomyocardial biopsy with Congo Red staining is the gold standard for diagnosis of amyloidosis.
In ATTR Val122Ile familial amyloidosis, it is unclear whether patients who are homozygous for the disease present with symptoms at earlier onset with more progressive illness or die sooner than those who are heterozygous.33 Nevertheless, once the diagnosis is confirmed, it is important to determine the patient’s specific variant type by DNA testing so that appropriate treatment can be initiated and the patient’s prognosis evaluated.5,10,13
Treatment
For familial amyloidosis in general, some researchers advocate liver transplantation to remove the source of mutant amyloid protein and stop all deposition of amyloid fibrils; this procedure can be followed later by transplantation of other affected organs (including the heart).5,23 Maurer et al34 have reported improved one-year survival rates among patients with ATTR amyloidosis who underwent both cardiac and liver transplantation: 75%, versus 23% in patients who did not receive transplanted organs.
Management of cardiac amyloidosis usually requires a twofold approach: treating associated congestive heart failure, and preventing further deposition of amyloid.24 In the case patient (as in most patients with ATTR amyloidosis), heart transplantation was deemed the only life-sustaining treatment option.11,19,33
Pharmacotherapeutic options are limited for patients with ATTR Val122Ile familial amyloidosis. Conventional heart failure agents (eg, ACE inhibitors, angiotensin receptor blockers, digoxin, β-blockers, calcium channel blockers) can exacerbate heart failure symptoms, leading to a potentially life-threatening arrhythmia.3,11,19,24,35 Amyloid fibrils bind to digitalis, increasing susceptibility to digitalis toxicity; and to nifedipine, causing hemodynamic deterioration. Verapamil should be avoided, as it may induce severe left ventricular dysfunction. ACE inhibitors often provoke profound hypotension in primary amyloidosis.24,35
Diuretics, too (eg, furosemide, as was prescribed for the case patient), must be used with caution.3 These agents have been used to treat fluid overload and the resulting peripheral edema and shortness of breath found in ATTR Val122Ile patients who experience heart failure.36 According to Dubrey et al,5 cautious use of diuretics is necessary for management of heart failure symptoms in these patients.
Because the risk for intracardiac thrombus is high, anticoagulation (using agents other than β-blockers or calcium channel blockers) should be implemented unless compelling risks are involved.11,24 Amiodarone is relatively well tolerated for ventricular tachydysrhythmias and in atrial fibrillation if the goal is maintaining sinus rhythm.37
Regarding heart transplantation in patients with familial amyloidosis, Jacob et al33 hypothesize that since mutant amyloid protein is synthesized by the liver, it would take approximately 50 years for a transplanted heart to become affected by amyloid deposition. In a 59-year-old Afro-Caribbean man with familial amyloidosis who underwent cardiac transplantation, Hamour et al11 reported that the donor heart remained amyloid-free three years posttransplantation, as demonstrated by serial cardiac biopsy.
On the Horizon
Clinical trials are now under way to examine pharmacotherapeutic options for patients with ATTR amyloidosis. Now being examined in clinical trials, for example, is Fx-1006A, a drug that stabilizes ATTR and prevents the misfolding of the amyloid protein fibril, in turn preventing it from binding to the target organ.38 Similarly, ALN-TTR, a drug believed to prevent disease manifestation and possibly facilitate disease regression, is being investigated in early human trials.39
Additionally, the use of genetic testing is recommended in at-risk individuals to identify the TTR gene. Affected patients may benefit from prophylactic medical management, which would halt amyloidogenesis of TTR—and possibly treat the condition as well.35 Pharmacotherapeutic agents like diflunisal, an NSAID, antagonize the aggregation of TTR protein and hinder formation of the amyloid fibrils.40
CONCLUSION
ATTR Val122Ile familial amyloidosis is a rare disorder that causes abnormal synthesis of amyloid protein in the liver, which then infiltrates the cardiac structure, leading to restrictive cardiomyopathy and progressive heart failure. Patients who present with symptoms of heart failure, cardiac enlargement on echocardiography, and a finding of granular speckling patterns, though not specific on echocardiography, should prompt the health care provider to refer the patient to a cardiologist familiar with cardiac amyloidosis for further work-up.
Diagnosed patients must undergo genetic testing to determine the specific variant type so that prompt treatment can be initiated. In patients with ATTR Val122Ile familial amyloidosis, the treatment of choice is cardiac transplantation. Although the mutant amyloid protein continues to be synthesized in the liver, the donor heart is unlikely to become affected by this substance for many years. Appropriately treated patients can maintain good quality of life, free of heart failure.
REFERENCES
1. Lim RP, Srichai MB, Lee VS. Non-ischemic causes of delayed myocardial hyperenhancement on MRI. AJR Am J Roentgenol. 2007;188 (6):1675-1681.
2. Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid. 2010;17(3-4):101-104.
3. Kendall H. Cardiac amyloidosis. Crit Care Nurse. 2010;30(2):16-23.
4. Eriksson M, Büttener J, Todorov T, et al. Prevalence of germline mutations in the TTR gene in a consecutive series of surgical pathology specimens with AATR amyloid. Am J Surg Pathol. 2009;33(1):58-65.
5. Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97(1):75-84.
6. Liepnieks JJ, Benson MD. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid. 2007;14(4):277-282.
7. XDx Expression Diagnostics. Allomap®: molecular expression testing (2004). www.allomap.com. Accessed May 14, 2012.
8. Mandras SA, Crespo J, Patel HM. Innovative application of immunologic principles in heart transplantation. Ochsner J. 2010;10(4):231-235.
9. Yamani MH, Taylor DO, Rodriguez R, et al. Transplant vasculopathy is associated with increased AlloMap gene expression score. J Heart Lung Transplant. 2007;26(4):403-406.
10. Benson MD. The hereditary amyloidoses. Best Pract Res Clin Rheumatol. 2003;17(6):909-927.
11. Hamour IM, Lachmann HJ, Goodman HJ, et al. Heart transplantation for homozygous familial transthyretin (TTR) V122I cardiac amyloidosis. Am J Transplant. 2008;8(5):1056-1059.
12. Jacobson DR, Pastore RD, Yaghoubian R, et al. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336(7):466-473.
13. Falk RH, Dubrey SW. Amyloid heart disease. Prog Cardiovasc Dis. 2010;52(4):347-361.
14. Askanas V, Engel WK, McFerrin J, Vattemi G. Transthyretin Val122Ile, accumulated Abeta, and inclusion-body myositis aspects in cultured muscle. Neurology. 2003;61(2):257-260.
15. Hamidi Asl K, Nakamura M, Yamashita T, Benson MD. Cardiac amyloidoses associated with the transthyretin lle122 mutation in a Caucasian family. Amyloid. 2001;8(4):263-269.
16. Mogensen J, Arbustini E. Restrictive cardiomyopathy. Curr Opin Cardiol. 2009;24(3): 214-220.
17. Nihoyannopoulos P, Dawson D. Restrictive cardiomyopathies. Eur J Echocardiogr. 2009;10 (8):iii23-iii33.
18. Bruce J. Getting to the heart of cardiomyopathies. Nursing. 2005;35(8):44-47.
19. Falk RH. The neglected entity of familial cardiac amyloidosis in African Americans. Ethn Dis. 2002;12(1):141-143.
20. Piper C, Butz T, Farr M, et al. How to diagnose cardiac amyloidosis early: impact of ECG, tissue Doppler echocardiography, and myocardial biopsy. Amyloid. 2010;17(1):1-9.
21. Kristen AV, Meyer FJ, Perz JB, et al. Risk stratification in cardiac amyloidosis: novel approaches. Transplantation. 2005;80(1 suppl):S151-S155.
22. Westermark P, Benson MD, Buxbaum JN, et al. A primer of amyloid nomenclature. Amyloid. 2007;14(3):179-183.
23. Picken MM. New insights into systemic amyloidosis: the importance of diagnosis of specific type. Curr Opin Nephrol Hypertens. 2007; 16(3):196-203.
24. Falk RH. Cardiac amyloidosis: a treatable disease, often overlooked. Circulation. 2011;124(9):1079-1085.
25. Yamashita T, Asl KH, Yazaki M, Benson MD. A prospective evaluation of the transthyretin Ile 122 allele frequency in an African-American population. Amyloid. 2005;12(2):127-130.
26. Benson MD, Yazaki M, Magy N. Laboratory assessment of transthyretin amyloidosis. Clin Chem Lab Med. 2002;40(12):1262-1265.
27. Chamarthi B, Dubrey SW, Cha K, et al. Features and prognosis of exertional syncope in light-chain associated AL cardiac amyloidosis. Am J Cardiol. 1997;80(9):1242-1245.
28. Correia MJ, Coutinho CA, Conceiçao I, et al. Role of heart rate variability in the assessment of autonomic dysfunction in type I familial amyloidotic polyneuropathy. Folia Cardiol. 2005;12(suppl C):459-462.
29. Kadish A, Mehra M. Heart failure devices: Implantable cardioverter-defibrillators and biventricular pacing therapy. Circulation. 2005; 111(24):3327-3335.
30. Rahman JE, Helou EF, Gelzer-Bell R, et al. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol. 2004;43(3):410-415.
31. Syed IS, Glockner JF, Feng D, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging. 2010;3(2):155-164.
32. Fealey ME, Edwards WD, Buadi FK, et al. Echocardiographic features of cardiac amyloidosis presenting as endomyocardial disease in a 54-year-old male. J Cardiol. 2009;54(1):162-166.
33. Jacob EK, Edwards WD, Zucker M, et al. Homozygous transthyretin mutation in an African American male. J Mol Diagn. 2007; 9(1):127-131.
34. Maurer MS, Raina A, Hesdorffer C, et al. Cardiac transplantation using extended-donor criteria organs for systemic amyloidosis complicated by heart failure. Transplantation. 2007;83(5):539-545.
35. Buxbaum J, Alexander A, Koziol J, et al. Significance of the amyloidogenic transthyretin Val 122 Ile allele in African-Americans in the Arteriosclerosis Risk in Communities (ARIC) and Cardiovascular Health (CHS) Studies. Am Heart J. 2010;159(5):864-870.
36. Rose BD, Colucci WS. Use of diuretics in heart failure (2010). www.uptodate.com/con tents/use-of-diuretics-in-patients-with-heart-failure. Accessed May 14, 2012.
37. Trappe H-J. Treating critical supraventricular and ventricular arrhythmias. J Emerg Trauma Shock. 2010;3(2):143-152.
38. Sekijima Y, Kelly JW, Ikeda S. Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Curr Pharm Des. 2008;14(30):3219-3230.
39. Alnylam Pharmaceuticals. TTR Amyloidosis: ALN-TTR (2011). www.alnylam.com/Programs-and-Pipeline/Programs/index.php. Accessed May 14, 2012.
40. Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogues stabilize the native state of transthyretin: potent inhibition of amyloidogenesis. J Med Chem. 2004;47(2):355-374.
A black man, age 65, with no known history of cardiopulmonary disease presented with acute-onset exertional dyspnea and lower extremity edema. He also reported an episode of syncope, as well as occasional dizziness and abdominal bloating. He said he experienced exertional dyspnea while doing a routine step aerobic exercise. His exercise regimen included distance walking, yoga, and aerobics four to five days per week.
The patient’s medical history was remarkable for a single episode of a bleeding ulcer in previous years, low back pain, shoulder pain, and a septic arthritic hip. His social history was negative for use of tobacco, alcohol, or illegal drugs. He was married and had two biological daughters with fairly unremarkable medical histories. The patient had earned a master’s degree, worked full-time in the insurance business, and was an avid worldwide traveler. He reported diminished quality of life as a result of his acute-onset heart failure symptoms, which reduced his ability to exercise routinely, work full-time, or travel.
The patient’s sudden experience of exertional dyspnea prompted him to visit his primary care provider, who ordered an ECG that demonstrated low voltage patterns and a first-degree atrioventricular (AV) block. Subsequent stress echocardiography showed generalized thickening of the left ventricular myocardium. Posterior wall thickness measured 1.7 cm (normal range, 0.6 to 1.1 cm), septal thickness measured 1.9 cm (normal, 0.6 to 1.1 cm), and ejection fraction was 65%. The stress echocardiogram also showed a speckling pattern (brightly scattered spots) on the myocardium.
Although stress echocardiography results were negative for ischemic disease, the patient did experience dyspnea during the exam. He underwent cardiac catheterization, which indicated normal coronary arteries.
Additional diagnostic studies included cardiac MRI with and without contrast, which showed nulling of the heart muscle and delayed patchy hyperenhancement; this suggested myocardial tissue abnormality as result of amyloid fibril deposition.1 Both pulmonary and tricuspid aortic valves were normal, with no evidence of stenosis. No regional wall motion abnormalities were noted.
Laboratory findings during the work-up were lipid panel, unremarkable; complete blood count (CBC), mild anemia and leukopenia; and urinalysis, positive for proteinuria. Brain natriuretic peptide (BNP) was measured at 686 pg/mL (normal, 0.0 to 100 pg/mL), indicating moderate heart failure. A peripheral blood smear was negative for monoclonal plasma cells.
The patient’s physical exam was unremarkable except for 2+ pedal edema bilaterally. In consideration of normal coronary arteries on cardiac catheterization, the patient’s heart failure symptoms, and stress echocardiography abnormalities, a heart biopsy was ordered. An endomyocardial biopsy with Congo Red stain demonstrated an apple-green birefringent pattern viewed under high-definition polarized light microscope, which was consistent with amyloid deposition.2
The patient was given a diagnosis of primary amyloidosis by his local cardiologist despite negative findings on the peripheral blood smear for monoclonal plasma cells (which are typically found in primary amyloidosis).3 He presented to an institution well-known for its expertise in amyloidosis, for a second opinion. There, the diagnosis was negated, based on reevaluation of the patient’s previous heart specimen through immunohistochemical studies. These studies were positive for serum amyloid P, which is suggestive of transthyretin (TTR) or familial amyloidosis.4 Genetic testing revealed a familial amyloidosis DNA sequence analysis with the Val122Ile variant (ie, isoleucine for valine at position 1225). With the correct diagnosis confirmed, the patient was referred to another highly regarded institution to begin a work-up for cardiac transplantation. Meanwhile, he was cautiously treated with the loop diuretic furosemide to manage his shortness of breath and peripheral edema.
Fifteen months later (13 weeks after being listed for transplant), the patient underwent successful cardiac transplantation.
On pathologic review of the patient’s extricated heart, the myocardium was found to be grossly thickened (see figure, above) and weighed 540 g; the average adult heart weighs 300 to 350 g, depending on the patient’s size.6 Congo Red staining showed extensive amyloid deposits with infiltration throughout the myocardium.
Ninety percent of the amyloid deposits were interstitial, 5% were in the vessels, and 5% were noted in a nodular pattern. The left ventricular cavity showed dilated and thickened walls. Intramural and extramural blood vessels were infiltrated with amyloid as well.
Six months after transplantation, the patient underwent diagnostic testing to assess the function and structure of his new heart. Cardiac catheterization was negative for coronary artery disease. Thirteen months posttransplantation, endomyocardial biopsy with Congo Red stain was negative for amyloid deposition or organ rejection.
About 24 months posttransplantation, the patient was taking tacrolimus, pravastatin, pantoprazole, dapsone, propanolol, colchicine, and donepezil. Stress echocardiography demonstrated normal right and left ventricular systolic function; no wall-motion abnormalities or left ventricular hypertrophy were detected, and the right atrium was of normal size. There was abnormal structural enlargement of the left atrium at the site of anastamosis—a common finding in cardiac transplant patients. The aortic, tricuspid, and mitral valves were all normal.
At that time, it was decided not to repeat endomyocardial biopsy because of normal results on molecular expression testing (a noninvasive technique called AlloMap®7-9), which is performed to assess for heart transplant rejection. The patient’s lipid panel remained within normal limits. CBC indicated persistent anemia and leukopenia. Urine protein and BNP test results were not available.
Since undergoing cardiac transplantation, the patient has resumed his normal routine activities, including some type of exercise five days per week. He said his diet is maintained in moderation. He denied shortness of breath, chest pain, dizziness, or edema. He has returned to full-time employment and has vacationed in Croatia, Italy, and Central America.
DISCUSSION
Familial amyloidosis is an autosomal dominant disease characterized by the production of mutated proteins, most commonly ATTR. Presence of the ATTR Val 122Ile allele has been reported in 3.9% of all black Americans, and in one study, 23% of black Americans diagnosed with cardiac amyloidosis at autopsy were heterozygous for this variant allele.10-12 ATTR Val122Ile usually manifests in the fifth
or sixth decade of life with its characteristic presentation of infiltrative/restrictive cardiomyopathy,13 resulting in heart failure and sometimes peripheral neuropathy.10,11,14
Pathophysiology
In patients with ATTR Val122Ile, cardiomyopathy results from the deposition of mutant protein fibrils in the cardiac muscle, leading to restricted heart wall motion10,15 and stiffening of the cardiac ventricles, with subsequent disruption of the diastolic filling properties of the cardiac muscle.16 Fluid overload and heart failure follow.3,10,17 The atrium of the heart dilates, and the walls of the ventricles become thickened and fibrous.18 Liepnieks and Benson6 reported that the cadaver heart of one Val122Ile patient infiltrated with amyloid protein fibrils weighed 725 g—more than double the weight of an average adult heart.
In patients with cardiac amyloidosis, ECG can detect arrhythmia, and echocardiography shows cardiac enlargement; however, as in the case patient, cardiac catheterization shows normal coronary arteries.15,16,19,20 Thus, previously healthy patients who present with heart failure and negative results on cardiac catheterization should undergo further work-up for cardiac amyloidosis.19
Amyloidosis affects all populations globally.10 In systemic amyloidosis, amyloid releases into the plasma, infiltrating and impairing multiple organs. Poor survival has been reported in patients with heart failure symptoms resulting from amyloid deposition.20,21
Types of Amyloidosis
Primary amyloidosis, the most common of the three amyloidosis types, can be systemic or localized.22 It occurs when protein fibrils, developed from immunoglobin light chains or monoclonal plasma cells and measuring 7 to 10 nm in diameter, adhere to the heart, kidneys, peripheral nerves, eyes, and other organs.5,11,20,23,24 Known for its relation to multiple myeloma,19 primary amyloidosis is associated with a poor prognosis.3,10
Secondary amyloidosis results from a chronic inflammatory disorder, such as rheumatoid arthritis or ankylosing spondylitis—conditions that trigger the production of amyloid proteins.3,10 This type has also been associated with substance abuse and AIDS.23
Familial or hereditary amyloidosis, according to Benson,10 is a group of diseases, each resulting from mutation in a specific protein. In the United States, the most common type of familial amyloidosis is ATTR.11 More than 100 mutant types of ATTR proteins have been identified, each involving a specific nationality or group of nationalities.10,11,23
Mutant ATTR amyloid, when deposited in specific organs, leads to their dysfunction and ultimate failure.6 ATTR may affect the cardiac, gastric, renal, ophthalmic, or nervous system. Depending on the ATTR variant, the resulting clinical features are age- and time-dependent, with onset most common between the third and fifth decade of life.10
Prevalence
The prevalence of ATTR Val 122Ile amyloidosis is reportedly high in West Africa, and in the US African-American population (3.9%).4,11,14,25,26 In a study conducted at a county hospital in Indianapolis, 3% of black newborns were found positive for ATTR Val122Ile through DNA sampling of umbilical cord blood.25 These statistics are of concern, as ATTR amyloidosis could be a significant health concern in a patient population that is already medically underserved.
Yamashita et al25 estimate that 1.35 million Americans of African-American descent may be affected by ATTR Val122Ile and vulnerable to restrictive cardiomyopathy–related heart failure and death. At the very least, this disorder can impair quality of life, especially in the presence of other comorbid conditions.
Clinical Presentation
The presence of exertional syncope at presentation is ominous, as it may be a marker of severe restrictive cardiomyopathy, postural hypotension due to excessive diuresis or autonomic neuropathy, ventricular arrhythmias from localized hypoperfusion, and rarely from cardiac tamponade due to pericardial involvement.27 Despite widespread involvement of the conduction system in specimens at autopsy, high-grade IV block is unusual.3
Diagnostic Studies
ECG. Both ECG and Holter monitoring can detect the arrhythmias and conduction disturbances (eg, first-degree AV block, low voltage patterns) associated with cardiac amyloidosis. Patients often experience syncopal and near-syncopal episodes as a result of conduction disturbances.28 Patients with conditions such as cardiac amyloidosis who present with severe heart failure are at high risk for sudden death secondary to conduction disturbances. Many have benefited from implanted defibrillators.29
Echocardiography. In patients with ATTR Val122Ile cardiac amyloidosis, echocardiography reveals thickened ventricular walls (ie, measuring ≥ 15 mm; normal, ≤ 11 mm).19 Amyloid-restrictive cardiomyopathy is associated with a marked dissociation between short- and long-axis systolic function, in cases in which left ventricular ejection fraction is normal.30
Echocardiography may demonstrate the characteristic specular or granular sparkling appearance that signifies advanced disease.15,21 Only a minority have this pattern in the myocardium, however, and changes in echocardiographic technology have made this finding less noticeable.30
MRI. Among more recently used diagnostic studies, cardiac magnetic resonance (CMR) has been reported to demonstrate late gadolinium enhancement (LGE) in perhaps 80% of patients with familial amyloidosis and cardiac involvement (as determined through biopsy and Congo Red stain). LGE-CMR shows darkening of the cardiac tissue, a common occurrence in amyloidosis.31
LGE is associated with increased thickness of both left and right ventricles, lower ECG voltage patterns, elevated BNP, and elevated troponin T.31,32 Globally, LGE is associated with the worst prognosis in patients with cardiac amyloidosis. Use of LGE-CMR testing can help facilitate early detection of cardiac amyloidosis in patients who may be vulnerable to cardiac damage.31
Cardiac catheterization. In patients with cardiac amyloidosis, cardiac catheterization usually shows normal coronary arteries.3
Diagnosis
Early diagnosis of ATTR cardiac amyloidosis is crucial to the patient’s survival; it should be ruled out in any African-American patient with unexplained heart failure and echocardiography showing increased wall thickness with a nondilated left ventricular cavity. Additional clues include significant proteinuria, hepatomegaly disproportionate to the degree of heart failure, or corresponding neuropathy. Known family history of the disease, along with variant type, allows for a prompt and correct diagnosis.10
It has been reported that most clinicians who encounter heart failure, particularly in black patients, do not consider amyloidosis in the differential diagnosis, because of the high prevalence of hypertension and congestive heart failure in this population.10,15,19 As a result, amyloidosis often goes undiagnosed.19
Findings of enlarged and thickened cardiac walls on echocardiography but normal coronary arteries on cardiac catheterization should alert the treating clinician to further work-up for cardiac amyloidosis.19 In such a patient, according to Kristen et al,21 endomyocardial biopsy with Congo Red staining is the gold standard for diagnosis of amyloidosis.
In ATTR Val122Ile familial amyloidosis, it is unclear whether patients who are homozygous for the disease present with symptoms at earlier onset with more progressive illness or die sooner than those who are heterozygous.33 Nevertheless, once the diagnosis is confirmed, it is important to determine the patient’s specific variant type by DNA testing so that appropriate treatment can be initiated and the patient’s prognosis evaluated.5,10,13
Treatment
For familial amyloidosis in general, some researchers advocate liver transplantation to remove the source of mutant amyloid protein and stop all deposition of amyloid fibrils; this procedure can be followed later by transplantation of other affected organs (including the heart).5,23 Maurer et al34 have reported improved one-year survival rates among patients with ATTR amyloidosis who underwent both cardiac and liver transplantation: 75%, versus 23% in patients who did not receive transplanted organs.
Management of cardiac amyloidosis usually requires a twofold approach: treating associated congestive heart failure, and preventing further deposition of amyloid.24 In the case patient (as in most patients with ATTR amyloidosis), heart transplantation was deemed the only life-sustaining treatment option.11,19,33
Pharmacotherapeutic options are limited for patients with ATTR Val122Ile familial amyloidosis. Conventional heart failure agents (eg, ACE inhibitors, angiotensin receptor blockers, digoxin, β-blockers, calcium channel blockers) can exacerbate heart failure symptoms, leading to a potentially life-threatening arrhythmia.3,11,19,24,35 Amyloid fibrils bind to digitalis, increasing susceptibility to digitalis toxicity; and to nifedipine, causing hemodynamic deterioration. Verapamil should be avoided, as it may induce severe left ventricular dysfunction. ACE inhibitors often provoke profound hypotension in primary amyloidosis.24,35
Diuretics, too (eg, furosemide, as was prescribed for the case patient), must be used with caution.3 These agents have been used to treat fluid overload and the resulting peripheral edema and shortness of breath found in ATTR Val122Ile patients who experience heart failure.36 According to Dubrey et al,5 cautious use of diuretics is necessary for management of heart failure symptoms in these patients.
Because the risk for intracardiac thrombus is high, anticoagulation (using agents other than β-blockers or calcium channel blockers) should be implemented unless compelling risks are involved.11,24 Amiodarone is relatively well tolerated for ventricular tachydysrhythmias and in atrial fibrillation if the goal is maintaining sinus rhythm.37
Regarding heart transplantation in patients with familial amyloidosis, Jacob et al33 hypothesize that since mutant amyloid protein is synthesized by the liver, it would take approximately 50 years for a transplanted heart to become affected by amyloid deposition. In a 59-year-old Afro-Caribbean man with familial amyloidosis who underwent cardiac transplantation, Hamour et al11 reported that the donor heart remained amyloid-free three years posttransplantation, as demonstrated by serial cardiac biopsy.
On the Horizon
Clinical trials are now under way to examine pharmacotherapeutic options for patients with ATTR amyloidosis. Now being examined in clinical trials, for example, is Fx-1006A, a drug that stabilizes ATTR and prevents the misfolding of the amyloid protein fibril, in turn preventing it from binding to the target organ.38 Similarly, ALN-TTR, a drug believed to prevent disease manifestation and possibly facilitate disease regression, is being investigated in early human trials.39
Additionally, the use of genetic testing is recommended in at-risk individuals to identify the TTR gene. Affected patients may benefit from prophylactic medical management, which would halt amyloidogenesis of TTR—and possibly treat the condition as well.35 Pharmacotherapeutic agents like diflunisal, an NSAID, antagonize the aggregation of TTR protein and hinder formation of the amyloid fibrils.40
CONCLUSION
ATTR Val122Ile familial amyloidosis is a rare disorder that causes abnormal synthesis of amyloid protein in the liver, which then infiltrates the cardiac structure, leading to restrictive cardiomyopathy and progressive heart failure. Patients who present with symptoms of heart failure, cardiac enlargement on echocardiography, and a finding of granular speckling patterns, though not specific on echocardiography, should prompt the health care provider to refer the patient to a cardiologist familiar with cardiac amyloidosis for further work-up.
Diagnosed patients must undergo genetic testing to determine the specific variant type so that prompt treatment can be initiated. In patients with ATTR Val122Ile familial amyloidosis, the treatment of choice is cardiac transplantation. Although the mutant amyloid protein continues to be synthesized in the liver, the donor heart is unlikely to become affected by this substance for many years. Appropriately treated patients can maintain good quality of life, free of heart failure.
REFERENCES
1. Lim RP, Srichai MB, Lee VS. Non-ischemic causes of delayed myocardial hyperenhancement on MRI. AJR Am J Roentgenol. 2007;188 (6):1675-1681.
2. Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid. 2010;17(3-4):101-104.
3. Kendall H. Cardiac amyloidosis. Crit Care Nurse. 2010;30(2):16-23.
4. Eriksson M, Büttener J, Todorov T, et al. Prevalence of germline mutations in the TTR gene in a consecutive series of surgical pathology specimens with AATR amyloid. Am J Surg Pathol. 2009;33(1):58-65.
5. Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97(1):75-84.
6. Liepnieks JJ, Benson MD. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid. 2007;14(4):277-282.
7. XDx Expression Diagnostics. Allomap®: molecular expression testing (2004). www.allomap.com. Accessed May 14, 2012.
8. Mandras SA, Crespo J, Patel HM. Innovative application of immunologic principles in heart transplantation. Ochsner J. 2010;10(4):231-235.
9. Yamani MH, Taylor DO, Rodriguez R, et al. Transplant vasculopathy is associated with increased AlloMap gene expression score. J Heart Lung Transplant. 2007;26(4):403-406.
10. Benson MD. The hereditary amyloidoses. Best Pract Res Clin Rheumatol. 2003;17(6):909-927.
11. Hamour IM, Lachmann HJ, Goodman HJ, et al. Heart transplantation for homozygous familial transthyretin (TTR) V122I cardiac amyloidosis. Am J Transplant. 2008;8(5):1056-1059.
12. Jacobson DR, Pastore RD, Yaghoubian R, et al. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336(7):466-473.
13. Falk RH, Dubrey SW. Amyloid heart disease. Prog Cardiovasc Dis. 2010;52(4):347-361.
14. Askanas V, Engel WK, McFerrin J, Vattemi G. Transthyretin Val122Ile, accumulated Abeta, and inclusion-body myositis aspects in cultured muscle. Neurology. 2003;61(2):257-260.
15. Hamidi Asl K, Nakamura M, Yamashita T, Benson MD. Cardiac amyloidoses associated with the transthyretin lle122 mutation in a Caucasian family. Amyloid. 2001;8(4):263-269.
16. Mogensen J, Arbustini E. Restrictive cardiomyopathy. Curr Opin Cardiol. 2009;24(3): 214-220.
17. Nihoyannopoulos P, Dawson D. Restrictive cardiomyopathies. Eur J Echocardiogr. 2009;10 (8):iii23-iii33.
18. Bruce J. Getting to the heart of cardiomyopathies. Nursing. 2005;35(8):44-47.
19. Falk RH. The neglected entity of familial cardiac amyloidosis in African Americans. Ethn Dis. 2002;12(1):141-143.
20. Piper C, Butz T, Farr M, et al. How to diagnose cardiac amyloidosis early: impact of ECG, tissue Doppler echocardiography, and myocardial biopsy. Amyloid. 2010;17(1):1-9.
21. Kristen AV, Meyer FJ, Perz JB, et al. Risk stratification in cardiac amyloidosis: novel approaches. Transplantation. 2005;80(1 suppl):S151-S155.
22. Westermark P, Benson MD, Buxbaum JN, et al. A primer of amyloid nomenclature. Amyloid. 2007;14(3):179-183.
23. Picken MM. New insights into systemic amyloidosis: the importance of diagnosis of specific type. Curr Opin Nephrol Hypertens. 2007; 16(3):196-203.
24. Falk RH. Cardiac amyloidosis: a treatable disease, often overlooked. Circulation. 2011;124(9):1079-1085.
25. Yamashita T, Asl KH, Yazaki M, Benson MD. A prospective evaluation of the transthyretin Ile 122 allele frequency in an African-American population. Amyloid. 2005;12(2):127-130.
26. Benson MD, Yazaki M, Magy N. Laboratory assessment of transthyretin amyloidosis. Clin Chem Lab Med. 2002;40(12):1262-1265.
27. Chamarthi B, Dubrey SW, Cha K, et al. Features and prognosis of exertional syncope in light-chain associated AL cardiac amyloidosis. Am J Cardiol. 1997;80(9):1242-1245.
28. Correia MJ, Coutinho CA, Conceiçao I, et al. Role of heart rate variability in the assessment of autonomic dysfunction in type I familial amyloidotic polyneuropathy. Folia Cardiol. 2005;12(suppl C):459-462.
29. Kadish A, Mehra M. Heart failure devices: Implantable cardioverter-defibrillators and biventricular pacing therapy. Circulation. 2005; 111(24):3327-3335.
30. Rahman JE, Helou EF, Gelzer-Bell R, et al. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol. 2004;43(3):410-415.
31. Syed IS, Glockner JF, Feng D, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging. 2010;3(2):155-164.
32. Fealey ME, Edwards WD, Buadi FK, et al. Echocardiographic features of cardiac amyloidosis presenting as endomyocardial disease in a 54-year-old male. J Cardiol. 2009;54(1):162-166.
33. Jacob EK, Edwards WD, Zucker M, et al. Homozygous transthyretin mutation in an African American male. J Mol Diagn. 2007; 9(1):127-131.
34. Maurer MS, Raina A, Hesdorffer C, et al. Cardiac transplantation using extended-donor criteria organs for systemic amyloidosis complicated by heart failure. Transplantation. 2007;83(5):539-545.
35. Buxbaum J, Alexander A, Koziol J, et al. Significance of the amyloidogenic transthyretin Val 122 Ile allele in African-Americans in the Arteriosclerosis Risk in Communities (ARIC) and Cardiovascular Health (CHS) Studies. Am Heart J. 2010;159(5):864-870.
36. Rose BD, Colucci WS. Use of diuretics in heart failure (2010). www.uptodate.com/con tents/use-of-diuretics-in-patients-with-heart-failure. Accessed May 14, 2012.
37. Trappe H-J. Treating critical supraventricular and ventricular arrhythmias. J Emerg Trauma Shock. 2010;3(2):143-152.
38. Sekijima Y, Kelly JW, Ikeda S. Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Curr Pharm Des. 2008;14(30):3219-3230.
39. Alnylam Pharmaceuticals. TTR Amyloidosis: ALN-TTR (2011). www.alnylam.com/Programs-and-Pipeline/Programs/index.php. Accessed May 14, 2012.
40. Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogues stabilize the native state of transthyretin: potent inhibition of amyloidogenesis. J Med Chem. 2004;47(2):355-374.
A black man, age 65, with no known history of cardiopulmonary disease presented with acute-onset exertional dyspnea and lower extremity edema. He also reported an episode of syncope, as well as occasional dizziness and abdominal bloating. He said he experienced exertional dyspnea while doing a routine step aerobic exercise. His exercise regimen included distance walking, yoga, and aerobics four to five days per week.
The patient’s medical history was remarkable for a single episode of a bleeding ulcer in previous years, low back pain, shoulder pain, and a septic arthritic hip. His social history was negative for use of tobacco, alcohol, or illegal drugs. He was married and had two biological daughters with fairly unremarkable medical histories. The patient had earned a master’s degree, worked full-time in the insurance business, and was an avid worldwide traveler. He reported diminished quality of life as a result of his acute-onset heart failure symptoms, which reduced his ability to exercise routinely, work full-time, or travel.
The patient’s sudden experience of exertional dyspnea prompted him to visit his primary care provider, who ordered an ECG that demonstrated low voltage patterns and a first-degree atrioventricular (AV) block. Subsequent stress echocardiography showed generalized thickening of the left ventricular myocardium. Posterior wall thickness measured 1.7 cm (normal range, 0.6 to 1.1 cm), septal thickness measured 1.9 cm (normal, 0.6 to 1.1 cm), and ejection fraction was 65%. The stress echocardiogram also showed a speckling pattern (brightly scattered spots) on the myocardium.
Although stress echocardiography results were negative for ischemic disease, the patient did experience dyspnea during the exam. He underwent cardiac catheterization, which indicated normal coronary arteries.
Additional diagnostic studies included cardiac MRI with and without contrast, which showed nulling of the heart muscle and delayed patchy hyperenhancement; this suggested myocardial tissue abnormality as result of amyloid fibril deposition.1 Both pulmonary and tricuspid aortic valves were normal, with no evidence of stenosis. No regional wall motion abnormalities were noted.
Laboratory findings during the work-up were lipid panel, unremarkable; complete blood count (CBC), mild anemia and leukopenia; and urinalysis, positive for proteinuria. Brain natriuretic peptide (BNP) was measured at 686 pg/mL (normal, 0.0 to 100 pg/mL), indicating moderate heart failure. A peripheral blood smear was negative for monoclonal plasma cells.
The patient’s physical exam was unremarkable except for 2+ pedal edema bilaterally. In consideration of normal coronary arteries on cardiac catheterization, the patient’s heart failure symptoms, and stress echocardiography abnormalities, a heart biopsy was ordered. An endomyocardial biopsy with Congo Red stain demonstrated an apple-green birefringent pattern viewed under high-definition polarized light microscope, which was consistent with amyloid deposition.2
The patient was given a diagnosis of primary amyloidosis by his local cardiologist despite negative findings on the peripheral blood smear for monoclonal plasma cells (which are typically found in primary amyloidosis).3 He presented to an institution well-known for its expertise in amyloidosis, for a second opinion. There, the diagnosis was negated, based on reevaluation of the patient’s previous heart specimen through immunohistochemical studies. These studies were positive for serum amyloid P, which is suggestive of transthyretin (TTR) or familial amyloidosis.4 Genetic testing revealed a familial amyloidosis DNA sequence analysis with the Val122Ile variant (ie, isoleucine for valine at position 1225). With the correct diagnosis confirmed, the patient was referred to another highly regarded institution to begin a work-up for cardiac transplantation. Meanwhile, he was cautiously treated with the loop diuretic furosemide to manage his shortness of breath and peripheral edema.
Fifteen months later (13 weeks after being listed for transplant), the patient underwent successful cardiac transplantation.
On pathologic review of the patient’s extricated heart, the myocardium was found to be grossly thickened (see figure, above) and weighed 540 g; the average adult heart weighs 300 to 350 g, depending on the patient’s size.6 Congo Red staining showed extensive amyloid deposits with infiltration throughout the myocardium.
Ninety percent of the amyloid deposits were interstitial, 5% were in the vessels, and 5% were noted in a nodular pattern. The left ventricular cavity showed dilated and thickened walls. Intramural and extramural blood vessels were infiltrated with amyloid as well.
Six months after transplantation, the patient underwent diagnostic testing to assess the function and structure of his new heart. Cardiac catheterization was negative for coronary artery disease. Thirteen months posttransplantation, endomyocardial biopsy with Congo Red stain was negative for amyloid deposition or organ rejection.
About 24 months posttransplantation, the patient was taking tacrolimus, pravastatin, pantoprazole, dapsone, propanolol, colchicine, and donepezil. Stress echocardiography demonstrated normal right and left ventricular systolic function; no wall-motion abnormalities or left ventricular hypertrophy were detected, and the right atrium was of normal size. There was abnormal structural enlargement of the left atrium at the site of anastamosis—a common finding in cardiac transplant patients. The aortic, tricuspid, and mitral valves were all normal.
At that time, it was decided not to repeat endomyocardial biopsy because of normal results on molecular expression testing (a noninvasive technique called AlloMap®7-9), which is performed to assess for heart transplant rejection. The patient’s lipid panel remained within normal limits. CBC indicated persistent anemia and leukopenia. Urine protein and BNP test results were not available.
Since undergoing cardiac transplantation, the patient has resumed his normal routine activities, including some type of exercise five days per week. He said his diet is maintained in moderation. He denied shortness of breath, chest pain, dizziness, or edema. He has returned to full-time employment and has vacationed in Croatia, Italy, and Central America.
DISCUSSION
Familial amyloidosis is an autosomal dominant disease characterized by the production of mutated proteins, most commonly ATTR. Presence of the ATTR Val 122Ile allele has been reported in 3.9% of all black Americans, and in one study, 23% of black Americans diagnosed with cardiac amyloidosis at autopsy were heterozygous for this variant allele.10-12 ATTR Val122Ile usually manifests in the fifth
or sixth decade of life with its characteristic presentation of infiltrative/restrictive cardiomyopathy,13 resulting in heart failure and sometimes peripheral neuropathy.10,11,14
Pathophysiology
In patients with ATTR Val122Ile, cardiomyopathy results from the deposition of mutant protein fibrils in the cardiac muscle, leading to restricted heart wall motion10,15 and stiffening of the cardiac ventricles, with subsequent disruption of the diastolic filling properties of the cardiac muscle.16 Fluid overload and heart failure follow.3,10,17 The atrium of the heart dilates, and the walls of the ventricles become thickened and fibrous.18 Liepnieks and Benson6 reported that the cadaver heart of one Val122Ile patient infiltrated with amyloid protein fibrils weighed 725 g—more than double the weight of an average adult heart.
In patients with cardiac amyloidosis, ECG can detect arrhythmia, and echocardiography shows cardiac enlargement; however, as in the case patient, cardiac catheterization shows normal coronary arteries.15,16,19,20 Thus, previously healthy patients who present with heart failure and negative results on cardiac catheterization should undergo further work-up for cardiac amyloidosis.19
Amyloidosis affects all populations globally.10 In systemic amyloidosis, amyloid releases into the plasma, infiltrating and impairing multiple organs. Poor survival has been reported in patients with heart failure symptoms resulting from amyloid deposition.20,21
Types of Amyloidosis
Primary amyloidosis, the most common of the three amyloidosis types, can be systemic or localized.22 It occurs when protein fibrils, developed from immunoglobin light chains or monoclonal plasma cells and measuring 7 to 10 nm in diameter, adhere to the heart, kidneys, peripheral nerves, eyes, and other organs.5,11,20,23,24 Known for its relation to multiple myeloma,19 primary amyloidosis is associated with a poor prognosis.3,10
Secondary amyloidosis results from a chronic inflammatory disorder, such as rheumatoid arthritis or ankylosing spondylitis—conditions that trigger the production of amyloid proteins.3,10 This type has also been associated with substance abuse and AIDS.23
Familial or hereditary amyloidosis, according to Benson,10 is a group of diseases, each resulting from mutation in a specific protein. In the United States, the most common type of familial amyloidosis is ATTR.11 More than 100 mutant types of ATTR proteins have been identified, each involving a specific nationality or group of nationalities.10,11,23
Mutant ATTR amyloid, when deposited in specific organs, leads to their dysfunction and ultimate failure.6 ATTR may affect the cardiac, gastric, renal, ophthalmic, or nervous system. Depending on the ATTR variant, the resulting clinical features are age- and time-dependent, with onset most common between the third and fifth decade of life.10
Prevalence
The prevalence of ATTR Val 122Ile amyloidosis is reportedly high in West Africa, and in the US African-American population (3.9%).4,11,14,25,26 In a study conducted at a county hospital in Indianapolis, 3% of black newborns were found positive for ATTR Val122Ile through DNA sampling of umbilical cord blood.25 These statistics are of concern, as ATTR amyloidosis could be a significant health concern in a patient population that is already medically underserved.
Yamashita et al25 estimate that 1.35 million Americans of African-American descent may be affected by ATTR Val122Ile and vulnerable to restrictive cardiomyopathy–related heart failure and death. At the very least, this disorder can impair quality of life, especially in the presence of other comorbid conditions.
Clinical Presentation
The presence of exertional syncope at presentation is ominous, as it may be a marker of severe restrictive cardiomyopathy, postural hypotension due to excessive diuresis or autonomic neuropathy, ventricular arrhythmias from localized hypoperfusion, and rarely from cardiac tamponade due to pericardial involvement.27 Despite widespread involvement of the conduction system in specimens at autopsy, high-grade IV block is unusual.3
Diagnostic Studies
ECG. Both ECG and Holter monitoring can detect the arrhythmias and conduction disturbances (eg, first-degree AV block, low voltage patterns) associated with cardiac amyloidosis. Patients often experience syncopal and near-syncopal episodes as a result of conduction disturbances.28 Patients with conditions such as cardiac amyloidosis who present with severe heart failure are at high risk for sudden death secondary to conduction disturbances. Many have benefited from implanted defibrillators.29
Echocardiography. In patients with ATTR Val122Ile cardiac amyloidosis, echocardiography reveals thickened ventricular walls (ie, measuring ≥ 15 mm; normal, ≤ 11 mm).19 Amyloid-restrictive cardiomyopathy is associated with a marked dissociation between short- and long-axis systolic function, in cases in which left ventricular ejection fraction is normal.30
Echocardiography may demonstrate the characteristic specular or granular sparkling appearance that signifies advanced disease.15,21 Only a minority have this pattern in the myocardium, however, and changes in echocardiographic technology have made this finding less noticeable.30
MRI. Among more recently used diagnostic studies, cardiac magnetic resonance (CMR) has been reported to demonstrate late gadolinium enhancement (LGE) in perhaps 80% of patients with familial amyloidosis and cardiac involvement (as determined through biopsy and Congo Red stain). LGE-CMR shows darkening of the cardiac tissue, a common occurrence in amyloidosis.31
LGE is associated with increased thickness of both left and right ventricles, lower ECG voltage patterns, elevated BNP, and elevated troponin T.31,32 Globally, LGE is associated with the worst prognosis in patients with cardiac amyloidosis. Use of LGE-CMR testing can help facilitate early detection of cardiac amyloidosis in patients who may be vulnerable to cardiac damage.31
Cardiac catheterization. In patients with cardiac amyloidosis, cardiac catheterization usually shows normal coronary arteries.3
Diagnosis
Early diagnosis of ATTR cardiac amyloidosis is crucial to the patient’s survival; it should be ruled out in any African-American patient with unexplained heart failure and echocardiography showing increased wall thickness with a nondilated left ventricular cavity. Additional clues include significant proteinuria, hepatomegaly disproportionate to the degree of heart failure, or corresponding neuropathy. Known family history of the disease, along with variant type, allows for a prompt and correct diagnosis.10
It has been reported that most clinicians who encounter heart failure, particularly in black patients, do not consider amyloidosis in the differential diagnosis, because of the high prevalence of hypertension and congestive heart failure in this population.10,15,19 As a result, amyloidosis often goes undiagnosed.19
Findings of enlarged and thickened cardiac walls on echocardiography but normal coronary arteries on cardiac catheterization should alert the treating clinician to further work-up for cardiac amyloidosis.19 In such a patient, according to Kristen et al,21 endomyocardial biopsy with Congo Red staining is the gold standard for diagnosis of amyloidosis.
In ATTR Val122Ile familial amyloidosis, it is unclear whether patients who are homozygous for the disease present with symptoms at earlier onset with more progressive illness or die sooner than those who are heterozygous.33 Nevertheless, once the diagnosis is confirmed, it is important to determine the patient’s specific variant type by DNA testing so that appropriate treatment can be initiated and the patient’s prognosis evaluated.5,10,13
Treatment
For familial amyloidosis in general, some researchers advocate liver transplantation to remove the source of mutant amyloid protein and stop all deposition of amyloid fibrils; this procedure can be followed later by transplantation of other affected organs (including the heart).5,23 Maurer et al34 have reported improved one-year survival rates among patients with ATTR amyloidosis who underwent both cardiac and liver transplantation: 75%, versus 23% in patients who did not receive transplanted organs.
Management of cardiac amyloidosis usually requires a twofold approach: treating associated congestive heart failure, and preventing further deposition of amyloid.24 In the case patient (as in most patients with ATTR amyloidosis), heart transplantation was deemed the only life-sustaining treatment option.11,19,33
Pharmacotherapeutic options are limited for patients with ATTR Val122Ile familial amyloidosis. Conventional heart failure agents (eg, ACE inhibitors, angiotensin receptor blockers, digoxin, β-blockers, calcium channel blockers) can exacerbate heart failure symptoms, leading to a potentially life-threatening arrhythmia.3,11,19,24,35 Amyloid fibrils bind to digitalis, increasing susceptibility to digitalis toxicity; and to nifedipine, causing hemodynamic deterioration. Verapamil should be avoided, as it may induce severe left ventricular dysfunction. ACE inhibitors often provoke profound hypotension in primary amyloidosis.24,35
Diuretics, too (eg, furosemide, as was prescribed for the case patient), must be used with caution.3 These agents have been used to treat fluid overload and the resulting peripheral edema and shortness of breath found in ATTR Val122Ile patients who experience heart failure.36 According to Dubrey et al,5 cautious use of diuretics is necessary for management of heart failure symptoms in these patients.
Because the risk for intracardiac thrombus is high, anticoagulation (using agents other than β-blockers or calcium channel blockers) should be implemented unless compelling risks are involved.11,24 Amiodarone is relatively well tolerated for ventricular tachydysrhythmias and in atrial fibrillation if the goal is maintaining sinus rhythm.37
Regarding heart transplantation in patients with familial amyloidosis, Jacob et al33 hypothesize that since mutant amyloid protein is synthesized by the liver, it would take approximately 50 years for a transplanted heart to become affected by amyloid deposition. In a 59-year-old Afro-Caribbean man with familial amyloidosis who underwent cardiac transplantation, Hamour et al11 reported that the donor heart remained amyloid-free three years posttransplantation, as demonstrated by serial cardiac biopsy.
On the Horizon
Clinical trials are now under way to examine pharmacotherapeutic options for patients with ATTR amyloidosis. Now being examined in clinical trials, for example, is Fx-1006A, a drug that stabilizes ATTR and prevents the misfolding of the amyloid protein fibril, in turn preventing it from binding to the target organ.38 Similarly, ALN-TTR, a drug believed to prevent disease manifestation and possibly facilitate disease regression, is being investigated in early human trials.39
Additionally, the use of genetic testing is recommended in at-risk individuals to identify the TTR gene. Affected patients may benefit from prophylactic medical management, which would halt amyloidogenesis of TTR—and possibly treat the condition as well.35 Pharmacotherapeutic agents like diflunisal, an NSAID, antagonize the aggregation of TTR protein and hinder formation of the amyloid fibrils.40
CONCLUSION
ATTR Val122Ile familial amyloidosis is a rare disorder that causes abnormal synthesis of amyloid protein in the liver, which then infiltrates the cardiac structure, leading to restrictive cardiomyopathy and progressive heart failure. Patients who present with symptoms of heart failure, cardiac enlargement on echocardiography, and a finding of granular speckling patterns, though not specific on echocardiography, should prompt the health care provider to refer the patient to a cardiologist familiar with cardiac amyloidosis for further work-up.
Diagnosed patients must undergo genetic testing to determine the specific variant type so that prompt treatment can be initiated. In patients with ATTR Val122Ile familial amyloidosis, the treatment of choice is cardiac transplantation. Although the mutant amyloid protein continues to be synthesized in the liver, the donor heart is unlikely to become affected by this substance for many years. Appropriately treated patients can maintain good quality of life, free of heart failure.
REFERENCES
1. Lim RP, Srichai MB, Lee VS. Non-ischemic causes of delayed myocardial hyperenhancement on MRI. AJR Am J Roentgenol. 2007;188 (6):1675-1681.
2. Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid. 2010;17(3-4):101-104.
3. Kendall H. Cardiac amyloidosis. Crit Care Nurse. 2010;30(2):16-23.
4. Eriksson M, Büttener J, Todorov T, et al. Prevalence of germline mutations in the TTR gene in a consecutive series of surgical pathology specimens with AATR amyloid. Am J Surg Pathol. 2009;33(1):58-65.
5. Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97(1):75-84.
6. Liepnieks JJ, Benson MD. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid. 2007;14(4):277-282.
7. XDx Expression Diagnostics. Allomap®: molecular expression testing (2004). www.allomap.com. Accessed May 14, 2012.
8. Mandras SA, Crespo J, Patel HM. Innovative application of immunologic principles in heart transplantation. Ochsner J. 2010;10(4):231-235.
9. Yamani MH, Taylor DO, Rodriguez R, et al. Transplant vasculopathy is associated with increased AlloMap gene expression score. J Heart Lung Transplant. 2007;26(4):403-406.
10. Benson MD. The hereditary amyloidoses. Best Pract Res Clin Rheumatol. 2003;17(6):909-927.
11. Hamour IM, Lachmann HJ, Goodman HJ, et al. Heart transplantation for homozygous familial transthyretin (TTR) V122I cardiac amyloidosis. Am J Transplant. 2008;8(5):1056-1059.
12. Jacobson DR, Pastore RD, Yaghoubian R, et al. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336(7):466-473.
13. Falk RH, Dubrey SW. Amyloid heart disease. Prog Cardiovasc Dis. 2010;52(4):347-361.
14. Askanas V, Engel WK, McFerrin J, Vattemi G. Transthyretin Val122Ile, accumulated Abeta, and inclusion-body myositis aspects in cultured muscle. Neurology. 2003;61(2):257-260.
15. Hamidi Asl K, Nakamura M, Yamashita T, Benson MD. Cardiac amyloidoses associated with the transthyretin lle122 mutation in a Caucasian family. Amyloid. 2001;8(4):263-269.
16. Mogensen J, Arbustini E. Restrictive cardiomyopathy. Curr Opin Cardiol. 2009;24(3): 214-220.
17. Nihoyannopoulos P, Dawson D. Restrictive cardiomyopathies. Eur J Echocardiogr. 2009;10 (8):iii23-iii33.
18. Bruce J. Getting to the heart of cardiomyopathies. Nursing. 2005;35(8):44-47.
19. Falk RH. The neglected entity of familial cardiac amyloidosis in African Americans. Ethn Dis. 2002;12(1):141-143.
20. Piper C, Butz T, Farr M, et al. How to diagnose cardiac amyloidosis early: impact of ECG, tissue Doppler echocardiography, and myocardial biopsy. Amyloid. 2010;17(1):1-9.
21. Kristen AV, Meyer FJ, Perz JB, et al. Risk stratification in cardiac amyloidosis: novel approaches. Transplantation. 2005;80(1 suppl):S151-S155.
22. Westermark P, Benson MD, Buxbaum JN, et al. A primer of amyloid nomenclature. Amyloid. 2007;14(3):179-183.
23. Picken MM. New insights into systemic amyloidosis: the importance of diagnosis of specific type. Curr Opin Nephrol Hypertens. 2007; 16(3):196-203.
24. Falk RH. Cardiac amyloidosis: a treatable disease, often overlooked. Circulation. 2011;124(9):1079-1085.
25. Yamashita T, Asl KH, Yazaki M, Benson MD. A prospective evaluation of the transthyretin Ile 122 allele frequency in an African-American population. Amyloid. 2005;12(2):127-130.
26. Benson MD, Yazaki M, Magy N. Laboratory assessment of transthyretin amyloidosis. Clin Chem Lab Med. 2002;40(12):1262-1265.
27. Chamarthi B, Dubrey SW, Cha K, et al. Features and prognosis of exertional syncope in light-chain associated AL cardiac amyloidosis. Am J Cardiol. 1997;80(9):1242-1245.
28. Correia MJ, Coutinho CA, Conceiçao I, et al. Role of heart rate variability in the assessment of autonomic dysfunction in type I familial amyloidotic polyneuropathy. Folia Cardiol. 2005;12(suppl C):459-462.
29. Kadish A, Mehra M. Heart failure devices: Implantable cardioverter-defibrillators and biventricular pacing therapy. Circulation. 2005; 111(24):3327-3335.
30. Rahman JE, Helou EF, Gelzer-Bell R, et al. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol. 2004;43(3):410-415.
31. Syed IS, Glockner JF, Feng D, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging. 2010;3(2):155-164.
32. Fealey ME, Edwards WD, Buadi FK, et al. Echocardiographic features of cardiac amyloidosis presenting as endomyocardial disease in a 54-year-old male. J Cardiol. 2009;54(1):162-166.
33. Jacob EK, Edwards WD, Zucker M, et al. Homozygous transthyretin mutation in an African American male. J Mol Diagn. 2007; 9(1):127-131.
34. Maurer MS, Raina A, Hesdorffer C, et al. Cardiac transplantation using extended-donor criteria organs for systemic amyloidosis complicated by heart failure. Transplantation. 2007;83(5):539-545.
35. Buxbaum J, Alexander A, Koziol J, et al. Significance of the amyloidogenic transthyretin Val 122 Ile allele in African-Americans in the Arteriosclerosis Risk in Communities (ARIC) and Cardiovascular Health (CHS) Studies. Am Heart J. 2010;159(5):864-870.
36. Rose BD, Colucci WS. Use of diuretics in heart failure (2010). www.uptodate.com/con tents/use-of-diuretics-in-patients-with-heart-failure. Accessed May 14, 2012.
37. Trappe H-J. Treating critical supraventricular and ventricular arrhythmias. J Emerg Trauma Shock. 2010;3(2):143-152.
38. Sekijima Y, Kelly JW, Ikeda S. Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Curr Pharm Des. 2008;14(30):3219-3230.
39. Alnylam Pharmaceuticals. TTR Amyloidosis: ALN-TTR (2011). www.alnylam.com/Programs-and-Pipeline/Programs/index.php. Accessed May 14, 2012.
40. Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogues stabilize the native state of transthyretin: potent inhibition of amyloidogenesis. J Med Chem. 2004;47(2):355-374.