User login
Tennessee’s Medicaid block grant proposal could have deep impacts
In late September, Gov. Bill Lee (R) submitted a draft proposal to the Trump administration requesting the government convert the federal share of the state’s Medicaid funding into a $7.9 billion lump sum.
Under the proposal, the federal spending cap would apply to currently eligible children and adults, patients covered based on disability, and currently eligible seniors, but not to newly eligible patients, prescription drugs, dually eligible enrollees, and reimbursement to disproportionate-share hospitals and critical access hospitals. The proposed block grant would increase on a per capita basis for new patients and would adjust annually for inflation.
Gov. Lee contends the block grant would allow for more efficiency in the state’s Medicaid program – called TennCare – and generate significant savings, which the state would split 50/50 with the federal government, according to the proposal.
“The traditional model of Medicaid financing is an outdated model of fundamentally misaligned incentives,” Gov. Lee said in the waiver request. “New models of Medicaid financing are needed that reward states for promoting value and health, not merely spending more money. Tennessee’s Medicaid block grant proposal represents a natural progression of the state’s history of nationally recognized innovation and financial management. It also ensures that TennCare members continue to receive high-quality, cost-effective care well into the future.”
Tennessee’s proposal aims to provide care to low-income recipients more efficiently, while breaking Medicaid’s “perverse dynamic” in which states seek to extract more money from the federal government, said Doug Badger, a visiting fellow in domestic policy studies at the conservative-leaning Heritage Foundation.
“The waiver would incentivize Tennessee to reduce federal spending while maintaining access to care,” Mr. Badger said in an interview. “It is an idea worth testing. Tennessee will have to convince federal officials that its proposal would maintain quality of care without increasing federal spending. If they clear those hurdles, the federal government should approve the waiver.”
However, Dan Hawkins, senior vice president for public policy and research at the nonpartisan National Association of Community Health Centers said the proposal contains some concerning omissions and extreme demands that could affect access to care. For instance, the waiver calls for complete exemption from federal Medicaid requirements, including managed care rules that ensure access and network adequacy protections, pregnancy care rules, requirements pertaining to the treatment of disabled adults and children, and early and periodic screening, diagnostic, and treatment (EPSDT) requirements. Under the waiver, the state would no longer be subject to federal oversight for its enrollment, coverage, or management decisions.
“The most shocking thing is that [Tennessee has] provided no estimate of impact on the population served, either in terms of their health or their ability to access care or the amount of money it would cost to serve them,” Mr. Hawkins said in an interview. “They are essentially saying, ‘Send [the block grant] to us and go away.’ ”
Without the federal protections, Tennessee could take any number of measures to reduce costs, adds Andy Schneider a research professor at Georgetown University and an analyst for Georgetown University Health Policy Institute’s Center for Children and Families, both in Washington. Mr. Schneider served as a senior advisor at the Centers for Medicare & Medicaid Services under the Obama administration.
“What would the state do in a world where it no longer had to follow federal rules in contracting with risk-based managed care organizations [MCOs] and in a world where it wanted to save money?” Mr. Schneider said in an interview. “Well, without the federal protections, it could reduce its payments on behalf of enrolled beneficiaries to the MCOs, it could not require the MCOs to have adequate provider networks, not require the MCOs to describe what services they were or were not providing, it could eliminate appeals protections for beneficiaries. Just let your imagination run wild.”
It is uncertain whether the waiver will be approved. Mr. Hawkins noted that CMS leadership has expressed support for the notion of a block grant. In his 2020 fiscal year federal budget for example, President Trump proposed a number of changes to Medicaid, including the potential for state Medicaid block grants or per capita caps. The Office of Management and Budget, meanwhile, is currently preparing guidance for state governments on creating block grants for Medicaid.
“On the other hand, this proposal is so outrageous I have to believe that even the federal government, even CMS, would blanch and come back requiring some additional changes in return for approving the proposal,” Mr. Hawkins said.
Mr. Schneider believes if the block grant is approved, it will likely face immediate litigation.
“The Department of Health & Human Services does not have the authority to give the state a block grant, only Congress can do that,” said Mr. Schneider who recently blogged about the proposal. “The reason for that is that, as the state makes clear in its proposal, it wants to reimagine the traditional Medicaid financing arrangement. That traditional financing arrangement has been in place since 1965. The executive branch can’t [change] it. It simply does not have the authority.”
Mr. Badger emphasized there is no shortage of speculation about the impact of fixed Medicaid allotments. What is needed now, he said, is evidence.
“A well-designed demonstration project will yield that evidence,” he said. “The statute gives the [HHS] Secretary authority to authorize demonstrations precisely to replace speculation with data. The best way to assess the impact of a fixed allotment is to test it in the real world.”
Tennessee will hold public hearings about its proposal in early October and collect feedback through Oct. 18. A final proposal is expected in November.
In late September, Gov. Bill Lee (R) submitted a draft proposal to the Trump administration requesting the government convert the federal share of the state’s Medicaid funding into a $7.9 billion lump sum.
Under the proposal, the federal spending cap would apply to currently eligible children and adults, patients covered based on disability, and currently eligible seniors, but not to newly eligible patients, prescription drugs, dually eligible enrollees, and reimbursement to disproportionate-share hospitals and critical access hospitals. The proposed block grant would increase on a per capita basis for new patients and would adjust annually for inflation.
Gov. Lee contends the block grant would allow for more efficiency in the state’s Medicaid program – called TennCare – and generate significant savings, which the state would split 50/50 with the federal government, according to the proposal.
“The traditional model of Medicaid financing is an outdated model of fundamentally misaligned incentives,” Gov. Lee said in the waiver request. “New models of Medicaid financing are needed that reward states for promoting value and health, not merely spending more money. Tennessee’s Medicaid block grant proposal represents a natural progression of the state’s history of nationally recognized innovation and financial management. It also ensures that TennCare members continue to receive high-quality, cost-effective care well into the future.”
Tennessee’s proposal aims to provide care to low-income recipients more efficiently, while breaking Medicaid’s “perverse dynamic” in which states seek to extract more money from the federal government, said Doug Badger, a visiting fellow in domestic policy studies at the conservative-leaning Heritage Foundation.
“The waiver would incentivize Tennessee to reduce federal spending while maintaining access to care,” Mr. Badger said in an interview. “It is an idea worth testing. Tennessee will have to convince federal officials that its proposal would maintain quality of care without increasing federal spending. If they clear those hurdles, the federal government should approve the waiver.”
However, Dan Hawkins, senior vice president for public policy and research at the nonpartisan National Association of Community Health Centers said the proposal contains some concerning omissions and extreme demands that could affect access to care. For instance, the waiver calls for complete exemption from federal Medicaid requirements, including managed care rules that ensure access and network adequacy protections, pregnancy care rules, requirements pertaining to the treatment of disabled adults and children, and early and periodic screening, diagnostic, and treatment (EPSDT) requirements. Under the waiver, the state would no longer be subject to federal oversight for its enrollment, coverage, or management decisions.
“The most shocking thing is that [Tennessee has] provided no estimate of impact on the population served, either in terms of their health or their ability to access care or the amount of money it would cost to serve them,” Mr. Hawkins said in an interview. “They are essentially saying, ‘Send [the block grant] to us and go away.’ ”
Without the federal protections, Tennessee could take any number of measures to reduce costs, adds Andy Schneider a research professor at Georgetown University and an analyst for Georgetown University Health Policy Institute’s Center for Children and Families, both in Washington. Mr. Schneider served as a senior advisor at the Centers for Medicare & Medicaid Services under the Obama administration.
“What would the state do in a world where it no longer had to follow federal rules in contracting with risk-based managed care organizations [MCOs] and in a world where it wanted to save money?” Mr. Schneider said in an interview. “Well, without the federal protections, it could reduce its payments on behalf of enrolled beneficiaries to the MCOs, it could not require the MCOs to have adequate provider networks, not require the MCOs to describe what services they were or were not providing, it could eliminate appeals protections for beneficiaries. Just let your imagination run wild.”
It is uncertain whether the waiver will be approved. Mr. Hawkins noted that CMS leadership has expressed support for the notion of a block grant. In his 2020 fiscal year federal budget for example, President Trump proposed a number of changes to Medicaid, including the potential for state Medicaid block grants or per capita caps. The Office of Management and Budget, meanwhile, is currently preparing guidance for state governments on creating block grants for Medicaid.
“On the other hand, this proposal is so outrageous I have to believe that even the federal government, even CMS, would blanch and come back requiring some additional changes in return for approving the proposal,” Mr. Hawkins said.
Mr. Schneider believes if the block grant is approved, it will likely face immediate litigation.
“The Department of Health & Human Services does not have the authority to give the state a block grant, only Congress can do that,” said Mr. Schneider who recently blogged about the proposal. “The reason for that is that, as the state makes clear in its proposal, it wants to reimagine the traditional Medicaid financing arrangement. That traditional financing arrangement has been in place since 1965. The executive branch can’t [change] it. It simply does not have the authority.”
Mr. Badger emphasized there is no shortage of speculation about the impact of fixed Medicaid allotments. What is needed now, he said, is evidence.
“A well-designed demonstration project will yield that evidence,” he said. “The statute gives the [HHS] Secretary authority to authorize demonstrations precisely to replace speculation with data. The best way to assess the impact of a fixed allotment is to test it in the real world.”
Tennessee will hold public hearings about its proposal in early October and collect feedback through Oct. 18. A final proposal is expected in November.
In late September, Gov. Bill Lee (R) submitted a draft proposal to the Trump administration requesting the government convert the federal share of the state’s Medicaid funding into a $7.9 billion lump sum.
Under the proposal, the federal spending cap would apply to currently eligible children and adults, patients covered based on disability, and currently eligible seniors, but not to newly eligible patients, prescription drugs, dually eligible enrollees, and reimbursement to disproportionate-share hospitals and critical access hospitals. The proposed block grant would increase on a per capita basis for new patients and would adjust annually for inflation.
Gov. Lee contends the block grant would allow for more efficiency in the state’s Medicaid program – called TennCare – and generate significant savings, which the state would split 50/50 with the federal government, according to the proposal.
“The traditional model of Medicaid financing is an outdated model of fundamentally misaligned incentives,” Gov. Lee said in the waiver request. “New models of Medicaid financing are needed that reward states for promoting value and health, not merely spending more money. Tennessee’s Medicaid block grant proposal represents a natural progression of the state’s history of nationally recognized innovation and financial management. It also ensures that TennCare members continue to receive high-quality, cost-effective care well into the future.”
Tennessee’s proposal aims to provide care to low-income recipients more efficiently, while breaking Medicaid’s “perverse dynamic” in which states seek to extract more money from the federal government, said Doug Badger, a visiting fellow in domestic policy studies at the conservative-leaning Heritage Foundation.
“The waiver would incentivize Tennessee to reduce federal spending while maintaining access to care,” Mr. Badger said in an interview. “It is an idea worth testing. Tennessee will have to convince federal officials that its proposal would maintain quality of care without increasing federal spending. If they clear those hurdles, the federal government should approve the waiver.”
However, Dan Hawkins, senior vice president for public policy and research at the nonpartisan National Association of Community Health Centers said the proposal contains some concerning omissions and extreme demands that could affect access to care. For instance, the waiver calls for complete exemption from federal Medicaid requirements, including managed care rules that ensure access and network adequacy protections, pregnancy care rules, requirements pertaining to the treatment of disabled adults and children, and early and periodic screening, diagnostic, and treatment (EPSDT) requirements. Under the waiver, the state would no longer be subject to federal oversight for its enrollment, coverage, or management decisions.
“The most shocking thing is that [Tennessee has] provided no estimate of impact on the population served, either in terms of their health or their ability to access care or the amount of money it would cost to serve them,” Mr. Hawkins said in an interview. “They are essentially saying, ‘Send [the block grant] to us and go away.’ ”
Without the federal protections, Tennessee could take any number of measures to reduce costs, adds Andy Schneider a research professor at Georgetown University and an analyst for Georgetown University Health Policy Institute’s Center for Children and Families, both in Washington. Mr. Schneider served as a senior advisor at the Centers for Medicare & Medicaid Services under the Obama administration.
“What would the state do in a world where it no longer had to follow federal rules in contracting with risk-based managed care organizations [MCOs] and in a world where it wanted to save money?” Mr. Schneider said in an interview. “Well, without the federal protections, it could reduce its payments on behalf of enrolled beneficiaries to the MCOs, it could not require the MCOs to have adequate provider networks, not require the MCOs to describe what services they were or were not providing, it could eliminate appeals protections for beneficiaries. Just let your imagination run wild.”
It is uncertain whether the waiver will be approved. Mr. Hawkins noted that CMS leadership has expressed support for the notion of a block grant. In his 2020 fiscal year federal budget for example, President Trump proposed a number of changes to Medicaid, including the potential for state Medicaid block grants or per capita caps. The Office of Management and Budget, meanwhile, is currently preparing guidance for state governments on creating block grants for Medicaid.
“On the other hand, this proposal is so outrageous I have to believe that even the federal government, even CMS, would blanch and come back requiring some additional changes in return for approving the proposal,” Mr. Hawkins said.
Mr. Schneider believes if the block grant is approved, it will likely face immediate litigation.
“The Department of Health & Human Services does not have the authority to give the state a block grant, only Congress can do that,” said Mr. Schneider who recently blogged about the proposal. “The reason for that is that, as the state makes clear in its proposal, it wants to reimagine the traditional Medicaid financing arrangement. That traditional financing arrangement has been in place since 1965. The executive branch can’t [change] it. It simply does not have the authority.”
Mr. Badger emphasized there is no shortage of speculation about the impact of fixed Medicaid allotments. What is needed now, he said, is evidence.
“A well-designed demonstration project will yield that evidence,” he said. “The statute gives the [HHS] Secretary authority to authorize demonstrations precisely to replace speculation with data. The best way to assess the impact of a fixed allotment is to test it in the real world.”
Tennessee will hold public hearings about its proposal in early October and collect feedback through Oct. 18. A final proposal is expected in November.
AAFP Congress adopts resolutions on physician privileges, medical education, employee benefits
PHILADELPHIA –

Practice enhancement
Hospital privileges were a hot topic for the reference committee on practice enhancement.
Adopted Resolution No. 304 calls on AAFP to oppose health insurance companies “privileging physicians based solely on their hospital privileges and hospital credentials.” The new rule also resolves that AAFP engage major national health insurance companies to develop methods to credential physicians that do not depend on hospital privileges.
The Congress also adopted Substitute Resolution No. 305, which calls on the AAFP to collaborate with the Joint Commission and other appropriate entities to create policy stating that hospitals remove undue barriers and restriction of privileges to hospitals and intensive care units for qualified family physicians who practice hospital medicine.
Delegates requested amendments to a resolution that called on AAFP to oppose nonphysician health care professionals making credentialing or privileging decisions regarding family physicians and that the AAFP oppose the use of nonphysician health care professionals in providing consultations requested of other physicians. The Congress could not agree on a final wording of the resolution.
Douglas J. Gruenbacher, MD, a Kansas delegate who works in a small hospital, said, “We actually credential in our hospital radiologists, orthopedists ... urologists. Do I know what they know? So we have to have many of our nonphysician providers, our nurse practitioners, help us. ... Should they be independent? No, of course not, but they do play an important part of our team.”
Douglas W. Curran, MD, a delegate from Texas, said, “I think we continue to give away stuff without taking care of ourselves. I have seen it for 4 years and, as result, we have seen this expansion of second-class care for people. ... Those are huge decisions, especially thinking about who’s going to do what in our hospitals. That includes small hospitals; I practice in a small hospital. I get all of that.”
After much debate, the Congress voted in favor of referring two proposed amendments to the board.
The Congress also adopted an amended version of Substitute Resolution No. 303, which calls on AAFP to support insurance coverage of acupuncture for pain control when ordered by a licensed physician or licensed collaborating advanced clinician on their practice team.
Education
Multipronged Substitute Resolution No. 606 – adopted by the Congress – aims to address racial inequities in medical education. Specifically, it calls on AAFP to do the following:
- Instruct the Liaison Committee for Medical Education to add race to its existing Cultural Competence and Health Care Disparities section 7.6 of Functions and Structure of a Medical School: Standards for Accreditation of Medical Education Programs Leading to the MD Degree.
- Ask the Accreditation Council for Graduate Medical Education to adopt an antiracism policy that includes corresponding curricular requirements,
- Develop and implement a policy on training in racism and implicit bias for officeholders and commission members.
- Take an active stance against racism when racist events occur within the medical community.
The Congress also adopted Resolution No. 611, which calls for the AAFP to encourage the expansion of clinical behavioral health fellowships for family medicine physicians.
The resolution received mixed testimony during the reference committee meeting, with those in favor of the resolution having cited the need for more education in behavioral health due to shortages in many communities. Opponents argued that completing the fellowship would not have added value in hospital privileging and insurance payment, because it would not lead physicians to earn a certificate of added qualification.
Delegates also passed Resolution 608, over the objections of the reference committee.
As adopted, the resolution calls for AAFP to express its concern that the American Board of Family Medicine (ABFM) Family Medicine Certification Longitudinal Assessment is the only alternative to 1-day-only certification exam, and for the AAFP to urge the ABFM to offer a longitudinal self-assessment process similar to the American Board of Obstetricians and Gynecologists self-assessment process to satisfy the cognitive component of ABFM’s continued certification requirement.
The Reference Committee on Education also referred several hotly debated resolutions back to the AAFP board of directors. No. 604 called on AAFP to support forgiving 1 year of federal medical student loans for every 2 years of full-time work in a primary care position, as well as tax credits for those working in rural or underserved areas.
Advocacy
The delegates also approved most of the recommendations of the reference committee on advocacy with little discussion.
Substitute Resolution No. 515, which was ultimately adopted with an amendment by the Congress, states that AAFP support policies that provide employees with reasonable benefits, including job security, wage replacement, and continued availability of health plan coverage in the event that leave by an employee becomes necessary for documented medical conditions, with protections for small businesses. Among the policies this resolution includes are the following:
- Medical leave for the employee, including pregnancy.
- Parental leave for the employee-parent, including leave for birth, adoption or foster care leading to adoption.
- Leave if medically appropriate to care for a member of the employee’s immediate family.
The Congress adopted several other resolutions recommended by the advocacy committee:
- Resolution No. 501, calling on AAFP to advocate for state-level adoption of the Interstate Medical Licensure Compact.
- Substitute Resolution No. 505, asking AAFP to request a National Coverage Determination for Cardiac Rehabilitation Programs to allow such programs to operate without physician supervision when an AED is immediately available, and the patient is attended by nursing staff currently trained in basic life support.
- Substitute Resolutions No. 506 and 507 to support and encourage the ability of parents to breastfeed in the workplace through its advocacy efforts, as well as promote the enforcements of current law.
- Resolution No. 508, to petition CMS, national health insurance companies, and pharmacy benefits managers to include all generic medications in a class within a health plan’s formulary and implement a system that informs the prescriber of all formulary alternatives to a medication when denying the same medication immediately upon denial, while also providing a mechanism to rapidly appeal the denial.
- Substitute Resolution No. 512, to petition the CMS to reevaluate its current policy on the time requirements for discharge summaries from hospitals and post-acute care facilities and specifically require such facilities to provide primary care physicians with discharge summaries within 7 days.
- Substitute Resolution No. 517, to unequivocally support the right of physicians to organize and bargain collectively.
- Substitute Resolution No. 519, to support legislation that decriminalizes people who are solicited for sex or sexual activities in exchange for money or goods, without supporting the legalization of the selling of sex, and advocate against legislation that decriminalizes sex-buying and third-parties who promote and/or profit from sex buying.
Some of the resolutions that incited many passionate responses during the reference committee on advocacy were not discussed during the Congress of Delegates meeting.
One of these asked the AAFP to oppose legislation of physician-patient decision making in child and adolescent gender-affirming care. Some in support of this resolution referred to this type of care as evidence-based medicine and said that legislators should be kept out of the exam room. Those opposed disagreed with classifying this type of care that way, noting that the long-term effects of some of the treatments are unknown.
During the reference committee, one opponent of the resolution, Lisa Gilbert, MD, claimed that gender-affirming care refers to blocking puberty, followed by cross-sex hormones,which would permanently sterilize the children. Dr. Gilbert, who identified herself as a member from Kansas speaking independently, added that if children have gone through puberty naturally, this would be a different discussion.
Kevin Wang, MD, an alternate delegate from Washington, who supported the resolution, noted that the rate of suicide in the transgender population is nine times that of the general population.
“I do want to emphasize that doing nothing does cause significant harm,” Dr. Wang said, The committee referred resolution No. 509 to the board for further clarification and study.
The merits of two other resolutions (No. 510 and No. 511), which called for the AAFP to no longer reject the use of “physician-assisted suicide” and “assisted suicide” and avoid the use of vague and euphemistic terms when referring to lethal medications prescribed with the intention of ending a patient’s life in statements and documents, also were heavily debated during the advocacy reference committee meeting. The committee recommended such resolutions be referred to the board for discussion.
The delegates approved the advocacy committee’s recommendations for Resolutions 509, 510, and 511.
PHILADELPHIA –
Practice enhancement
Hospital privileges were a hot topic for the reference committee on practice enhancement.
Adopted Resolution No. 304 calls on AAFP to oppose health insurance companies “privileging physicians based solely on their hospital privileges and hospital credentials.” The new rule also resolves that AAFP engage major national health insurance companies to develop methods to credential physicians that do not depend on hospital privileges.
The Congress also adopted Substitute Resolution No. 305, which calls on the AAFP to collaborate with the Joint Commission and other appropriate entities to create policy stating that hospitals remove undue barriers and restriction of privileges to hospitals and intensive care units for qualified family physicians who practice hospital medicine.
Delegates requested amendments to a resolution that called on AAFP to oppose nonphysician health care professionals making credentialing or privileging decisions regarding family physicians and that the AAFP oppose the use of nonphysician health care professionals in providing consultations requested of other physicians. The Congress could not agree on a final wording of the resolution.
Douglas J. Gruenbacher, MD, a Kansas delegate who works in a small hospital, said, “We actually credential in our hospital radiologists, orthopedists ... urologists. Do I know what they know? So we have to have many of our nonphysician providers, our nurse practitioners, help us. ... Should they be independent? No, of course not, but they do play an important part of our team.”
Douglas W. Curran, MD, a delegate from Texas, said, “I think we continue to give away stuff without taking care of ourselves. I have seen it for 4 years and, as result, we have seen this expansion of second-class care for people. ... Those are huge decisions, especially thinking about who’s going to do what in our hospitals. That includes small hospitals; I practice in a small hospital. I get all of that.”
After much debate, the Congress voted in favor of referring two proposed amendments to the board.
The Congress also adopted an amended version of Substitute Resolution No. 303, which calls on AAFP to support insurance coverage of acupuncture for pain control when ordered by a licensed physician or licensed collaborating advanced clinician on their practice team.
Education
Multipronged Substitute Resolution No. 606 – adopted by the Congress – aims to address racial inequities in medical education. Specifically, it calls on AAFP to do the following:
- Instruct the Liaison Committee for Medical Education to add race to its existing Cultural Competence and Health Care Disparities section 7.6 of Functions and Structure of a Medical School: Standards for Accreditation of Medical Education Programs Leading to the MD Degree.
- Ask the Accreditation Council for Graduate Medical Education to adopt an antiracism policy that includes corresponding curricular requirements,
- Develop and implement a policy on training in racism and implicit bias for officeholders and commission members.
- Take an active stance against racism when racist events occur within the medical community.
The Congress also adopted Resolution No. 611, which calls for the AAFP to encourage the expansion of clinical behavioral health fellowships for family medicine physicians.
The resolution received mixed testimony during the reference committee meeting, with those in favor of the resolution having cited the need for more education in behavioral health due to shortages in many communities. Opponents argued that completing the fellowship would not have added value in hospital privileging and insurance payment, because it would not lead physicians to earn a certificate of added qualification.
Delegates also passed Resolution 608, over the objections of the reference committee.
As adopted, the resolution calls for AAFP to express its concern that the American Board of Family Medicine (ABFM) Family Medicine Certification Longitudinal Assessment is the only alternative to 1-day-only certification exam, and for the AAFP to urge the ABFM to offer a longitudinal self-assessment process similar to the American Board of Obstetricians and Gynecologists self-assessment process to satisfy the cognitive component of ABFM’s continued certification requirement.
The Reference Committee on Education also referred several hotly debated resolutions back to the AAFP board of directors. No. 604 called on AAFP to support forgiving 1 year of federal medical student loans for every 2 years of full-time work in a primary care position, as well as tax credits for those working in rural or underserved areas.
Advocacy
The delegates also approved most of the recommendations of the reference committee on advocacy with little discussion.
Substitute Resolution No. 515, which was ultimately adopted with an amendment by the Congress, states that AAFP support policies that provide employees with reasonable benefits, including job security, wage replacement, and continued availability of health plan coverage in the event that leave by an employee becomes necessary for documented medical conditions, with protections for small businesses. Among the policies this resolution includes are the following:
- Medical leave for the employee, including pregnancy.
- Parental leave for the employee-parent, including leave for birth, adoption or foster care leading to adoption.
- Leave if medically appropriate to care for a member of the employee’s immediate family.
The Congress adopted several other resolutions recommended by the advocacy committee:
- Resolution No. 501, calling on AAFP to advocate for state-level adoption of the Interstate Medical Licensure Compact.
- Substitute Resolution No. 505, asking AAFP to request a National Coverage Determination for Cardiac Rehabilitation Programs to allow such programs to operate without physician supervision when an AED is immediately available, and the patient is attended by nursing staff currently trained in basic life support.
- Substitute Resolutions No. 506 and 507 to support and encourage the ability of parents to breastfeed in the workplace through its advocacy efforts, as well as promote the enforcements of current law.
- Resolution No. 508, to petition CMS, national health insurance companies, and pharmacy benefits managers to include all generic medications in a class within a health plan’s formulary and implement a system that informs the prescriber of all formulary alternatives to a medication when denying the same medication immediately upon denial, while also providing a mechanism to rapidly appeal the denial.
- Substitute Resolution No. 512, to petition the CMS to reevaluate its current policy on the time requirements for discharge summaries from hospitals and post-acute care facilities and specifically require such facilities to provide primary care physicians with discharge summaries within 7 days.
- Substitute Resolution No. 517, to unequivocally support the right of physicians to organize and bargain collectively.
- Substitute Resolution No. 519, to support legislation that decriminalizes people who are solicited for sex or sexual activities in exchange for money or goods, without supporting the legalization of the selling of sex, and advocate against legislation that decriminalizes sex-buying and third-parties who promote and/or profit from sex buying.
Some of the resolutions that incited many passionate responses during the reference committee on advocacy were not discussed during the Congress of Delegates meeting.
One of these asked the AAFP to oppose legislation of physician-patient decision making in child and adolescent gender-affirming care. Some in support of this resolution referred to this type of care as evidence-based medicine and said that legislators should be kept out of the exam room. Those opposed disagreed with classifying this type of care that way, noting that the long-term effects of some of the treatments are unknown.
During the reference committee, one opponent of the resolution, Lisa Gilbert, MD, claimed that gender-affirming care refers to blocking puberty, followed by cross-sex hormones,which would permanently sterilize the children. Dr. Gilbert, who identified herself as a member from Kansas speaking independently, added that if children have gone through puberty naturally, this would be a different discussion.
Kevin Wang, MD, an alternate delegate from Washington, who supported the resolution, noted that the rate of suicide in the transgender population is nine times that of the general population.
“I do want to emphasize that doing nothing does cause significant harm,” Dr. Wang said, The committee referred resolution No. 509 to the board for further clarification and study.
The merits of two other resolutions (No. 510 and No. 511), which called for the AAFP to no longer reject the use of “physician-assisted suicide” and “assisted suicide” and avoid the use of vague and euphemistic terms when referring to lethal medications prescribed with the intention of ending a patient’s life in statements and documents, also were heavily debated during the advocacy reference committee meeting. The committee recommended such resolutions be referred to the board for discussion.
The delegates approved the advocacy committee’s recommendations for Resolutions 509, 510, and 511.
PHILADELPHIA –
Practice enhancement
Hospital privileges were a hot topic for the reference committee on practice enhancement.
Adopted Resolution No. 304 calls on AAFP to oppose health insurance companies “privileging physicians based solely on their hospital privileges and hospital credentials.” The new rule also resolves that AAFP engage major national health insurance companies to develop methods to credential physicians that do not depend on hospital privileges.
The Congress also adopted Substitute Resolution No. 305, which calls on the AAFP to collaborate with the Joint Commission and other appropriate entities to create policy stating that hospitals remove undue barriers and restriction of privileges to hospitals and intensive care units for qualified family physicians who practice hospital medicine.
Delegates requested amendments to a resolution that called on AAFP to oppose nonphysician health care professionals making credentialing or privileging decisions regarding family physicians and that the AAFP oppose the use of nonphysician health care professionals in providing consultations requested of other physicians. The Congress could not agree on a final wording of the resolution.
Douglas J. Gruenbacher, MD, a Kansas delegate who works in a small hospital, said, “We actually credential in our hospital radiologists, orthopedists ... urologists. Do I know what they know? So we have to have many of our nonphysician providers, our nurse practitioners, help us. ... Should they be independent? No, of course not, but they do play an important part of our team.”
Douglas W. Curran, MD, a delegate from Texas, said, “I think we continue to give away stuff without taking care of ourselves. I have seen it for 4 years and, as result, we have seen this expansion of second-class care for people. ... Those are huge decisions, especially thinking about who’s going to do what in our hospitals. That includes small hospitals; I practice in a small hospital. I get all of that.”
After much debate, the Congress voted in favor of referring two proposed amendments to the board.
The Congress also adopted an amended version of Substitute Resolution No. 303, which calls on AAFP to support insurance coverage of acupuncture for pain control when ordered by a licensed physician or licensed collaborating advanced clinician on their practice team.
Education
Multipronged Substitute Resolution No. 606 – adopted by the Congress – aims to address racial inequities in medical education. Specifically, it calls on AAFP to do the following:
- Instruct the Liaison Committee for Medical Education to add race to its existing Cultural Competence and Health Care Disparities section 7.6 of Functions and Structure of a Medical School: Standards for Accreditation of Medical Education Programs Leading to the MD Degree.
- Ask the Accreditation Council for Graduate Medical Education to adopt an antiracism policy that includes corresponding curricular requirements,
- Develop and implement a policy on training in racism and implicit bias for officeholders and commission members.
- Take an active stance against racism when racist events occur within the medical community.
The Congress also adopted Resolution No. 611, which calls for the AAFP to encourage the expansion of clinical behavioral health fellowships for family medicine physicians.
The resolution received mixed testimony during the reference committee meeting, with those in favor of the resolution having cited the need for more education in behavioral health due to shortages in many communities. Opponents argued that completing the fellowship would not have added value in hospital privileging and insurance payment, because it would not lead physicians to earn a certificate of added qualification.
Delegates also passed Resolution 608, over the objections of the reference committee.
As adopted, the resolution calls for AAFP to express its concern that the American Board of Family Medicine (ABFM) Family Medicine Certification Longitudinal Assessment is the only alternative to 1-day-only certification exam, and for the AAFP to urge the ABFM to offer a longitudinal self-assessment process similar to the American Board of Obstetricians and Gynecologists self-assessment process to satisfy the cognitive component of ABFM’s continued certification requirement.
The Reference Committee on Education also referred several hotly debated resolutions back to the AAFP board of directors. No. 604 called on AAFP to support forgiving 1 year of federal medical student loans for every 2 years of full-time work in a primary care position, as well as tax credits for those working in rural or underserved areas.
Advocacy
The delegates also approved most of the recommendations of the reference committee on advocacy with little discussion.
Substitute Resolution No. 515, which was ultimately adopted with an amendment by the Congress, states that AAFP support policies that provide employees with reasonable benefits, including job security, wage replacement, and continued availability of health plan coverage in the event that leave by an employee becomes necessary for documented medical conditions, with protections for small businesses. Among the policies this resolution includes are the following:
- Medical leave for the employee, including pregnancy.
- Parental leave for the employee-parent, including leave for birth, adoption or foster care leading to adoption.
- Leave if medically appropriate to care for a member of the employee’s immediate family.
The Congress adopted several other resolutions recommended by the advocacy committee:
- Resolution No. 501, calling on AAFP to advocate for state-level adoption of the Interstate Medical Licensure Compact.
- Substitute Resolution No. 505, asking AAFP to request a National Coverage Determination for Cardiac Rehabilitation Programs to allow such programs to operate without physician supervision when an AED is immediately available, and the patient is attended by nursing staff currently trained in basic life support.
- Substitute Resolutions No. 506 and 507 to support and encourage the ability of parents to breastfeed in the workplace through its advocacy efforts, as well as promote the enforcements of current law.
- Resolution No. 508, to petition CMS, national health insurance companies, and pharmacy benefits managers to include all generic medications in a class within a health plan’s formulary and implement a system that informs the prescriber of all formulary alternatives to a medication when denying the same medication immediately upon denial, while also providing a mechanism to rapidly appeal the denial.
- Substitute Resolution No. 512, to petition the CMS to reevaluate its current policy on the time requirements for discharge summaries from hospitals and post-acute care facilities and specifically require such facilities to provide primary care physicians with discharge summaries within 7 days.
- Substitute Resolution No. 517, to unequivocally support the right of physicians to organize and bargain collectively.
- Substitute Resolution No. 519, to support legislation that decriminalizes people who are solicited for sex or sexual activities in exchange for money or goods, without supporting the legalization of the selling of sex, and advocate against legislation that decriminalizes sex-buying and third-parties who promote and/or profit from sex buying.
Some of the resolutions that incited many passionate responses during the reference committee on advocacy were not discussed during the Congress of Delegates meeting.
One of these asked the AAFP to oppose legislation of physician-patient decision making in child and adolescent gender-affirming care. Some in support of this resolution referred to this type of care as evidence-based medicine and said that legislators should be kept out of the exam room. Those opposed disagreed with classifying this type of care that way, noting that the long-term effects of some of the treatments are unknown.
During the reference committee, one opponent of the resolution, Lisa Gilbert, MD, claimed that gender-affirming care refers to blocking puberty, followed by cross-sex hormones,which would permanently sterilize the children. Dr. Gilbert, who identified herself as a member from Kansas speaking independently, added that if children have gone through puberty naturally, this would be a different discussion.
Kevin Wang, MD, an alternate delegate from Washington, who supported the resolution, noted that the rate of suicide in the transgender population is nine times that of the general population.
“I do want to emphasize that doing nothing does cause significant harm,” Dr. Wang said, The committee referred resolution No. 509 to the board for further clarification and study.
The merits of two other resolutions (No. 510 and No. 511), which called for the AAFP to no longer reject the use of “physician-assisted suicide” and “assisted suicide” and avoid the use of vague and euphemistic terms when referring to lethal medications prescribed with the intention of ending a patient’s life in statements and documents, also were heavily debated during the advocacy reference committee meeting. The committee recommended such resolutions be referred to the board for discussion.
The delegates approved the advocacy committee’s recommendations for Resolutions 509, 510, and 511.
REPORTING FROM AAFP Congress
I have seen the future
Many patients have seen their long-term physicians retire. When I ask how they like their new doctors, they say: “She’s okay, I guess. Quite efficient. Seems thorough. But it’s not the same. It’s just business. Nothing personal.”

Sometimes you have to look backward to look forward. So it’s perhaps fitting that I glimpsed the future at my last colonoscopy.
In recent years, I’ve had such procedures at a local suburban surgicenter. Easy access, plenty of parking.
The woman who checks me in is all business. She scans my insurance cards and hands me a clipboard with a medical history form. Have I ever had cancer? A hernia? Am I pregnant? I wonder whether anyone reads these.
A different young woman brings me inside, the first of many new faces. Their roles are murky.
In a curtained cubby, yet another staff person asks me to pack my clothes in a plastic bag and put on a johnny. Then an older man enters, initiating furious multitasking. A different nursing assistant asks me to confirm my name and date of birth, then inserts an intravenous line in one arm, while the old doctor hands me an anesthesia consent form to sign with the other hand. I check many answers very fast, ignore the small-print boilerplate, and sign.
I am handed two more consent forms to sign, one from each side. The staff makes no pretense of explaining them or even telling me what they are for, and I make none of reading them.
They depart, replaced by still another person, who rolls me into the next room. He confirms my name and date of birth, and which procedure I am there for. The purpose of these multiple checks is clear, along with dispiriting depersonalization. One could mitigate this with some light banter, but no one bothers. No time.
My physician – whom I actually know – enters, says hello, and exchanges pleasantries. The last guy asks me to turn onto my left side. Intravenous sedation flows into my veins. The rest is silence.
Sometime later I wake up, greeted by another staff person. She asks if I am okay and offers me water or juice and saltines. Noting her Boston Red Sox sweatshirt, I say, “Great game last night,” but she does not know what I am talking about. She cares only for football and plans to fly to Nashville, Tenn., to watch her favorites.
Curtains are closed, and I am asked to dress. Another assistant directs me to a chair, where I will await my ride home. Through I try to walk alone, she takes my arm. “We assist everyone,” she explains.
As the sedation wears off, I observe. All around me I see movement, brisk and purposeful. Staff members crisscross before me from all angles, striding from one task to another, from prep room A to cubby D, walking with or pushing patients from procedure room M to holding area 8H. No one I’ve just met recognizes me, or acknowledges having met me before.
At last, the final staff member approaches. She flashes a kind smile as she takes my arm to walk me to the door. I take this for a personal touch, until she explains that she must make sure I don’t fall and that I get into the right car. As we pass, no one in the waiting room, neither staff nor patients, takes any notice.
My wife is outside, idling in the correct car. She’s brought coffee and a chocolate croissant, which – almost – makes last night’s prep worthwhile. She confirms neither my name nor date of birth.
Altogether, I have been in and out in 90 minutes. In the car, I peruse the handout that had been given to me as I exited. Drinking my coffee, I read the postcare instructions and enjoy its full-color pictures. Seldom has my cecum looked more radiant.
In “The Checklist Manifesto,” Atul Gawande described the outcome improvement that systematized practice can achieve. Data analysis confirms the measurably superior efficacy of such a method.
As for me, I feel like output from one of today’s cataract factories: like a car just extruded from an automated wash, with a photo on its front seat of the shiny, Simonized hubcaps included with the premium service package.
Just business, though. Nothing personal.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at dermnews@mdedge.com.
Many patients have seen their long-term physicians retire. When I ask how they like their new doctors, they say: “She’s okay, I guess. Quite efficient. Seems thorough. But it’s not the same. It’s just business. Nothing personal.”
Sometimes you have to look backward to look forward. So it’s perhaps fitting that I glimpsed the future at my last colonoscopy.
In recent years, I’ve had such procedures at a local suburban surgicenter. Easy access, plenty of parking.
The woman who checks me in is all business. She scans my insurance cards and hands me a clipboard with a medical history form. Have I ever had cancer? A hernia? Am I pregnant? I wonder whether anyone reads these.
A different young woman brings me inside, the first of many new faces. Their roles are murky.
In a curtained cubby, yet another staff person asks me to pack my clothes in a plastic bag and put on a johnny. Then an older man enters, initiating furious multitasking. A different nursing assistant asks me to confirm my name and date of birth, then inserts an intravenous line in one arm, while the old doctor hands me an anesthesia consent form to sign with the other hand. I check many answers very fast, ignore the small-print boilerplate, and sign.
I am handed two more consent forms to sign, one from each side. The staff makes no pretense of explaining them or even telling me what they are for, and I make none of reading them.
They depart, replaced by still another person, who rolls me into the next room. He confirms my name and date of birth, and which procedure I am there for. The purpose of these multiple checks is clear, along with dispiriting depersonalization. One could mitigate this with some light banter, but no one bothers. No time.
My physician – whom I actually know – enters, says hello, and exchanges pleasantries. The last guy asks me to turn onto my left side. Intravenous sedation flows into my veins. The rest is silence.
Sometime later I wake up, greeted by another staff person. She asks if I am okay and offers me water or juice and saltines. Noting her Boston Red Sox sweatshirt, I say, “Great game last night,” but she does not know what I am talking about. She cares only for football and plans to fly to Nashville, Tenn., to watch her favorites.
Curtains are closed, and I am asked to dress. Another assistant directs me to a chair, where I will await my ride home. Through I try to walk alone, she takes my arm. “We assist everyone,” she explains.
As the sedation wears off, I observe. All around me I see movement, brisk and purposeful. Staff members crisscross before me from all angles, striding from one task to another, from prep room A to cubby D, walking with or pushing patients from procedure room M to holding area 8H. No one I’ve just met recognizes me, or acknowledges having met me before.
At last, the final staff member approaches. She flashes a kind smile as she takes my arm to walk me to the door. I take this for a personal touch, until she explains that she must make sure I don’t fall and that I get into the right car. As we pass, no one in the waiting room, neither staff nor patients, takes any notice.
My wife is outside, idling in the correct car. She’s brought coffee and a chocolate croissant, which – almost – makes last night’s prep worthwhile. She confirms neither my name nor date of birth.
Altogether, I have been in and out in 90 minutes. In the car, I peruse the handout that had been given to me as I exited. Drinking my coffee, I read the postcare instructions and enjoy its full-color pictures. Seldom has my cecum looked more radiant.
In “The Checklist Manifesto,” Atul Gawande described the outcome improvement that systematized practice can achieve. Data analysis confirms the measurably superior efficacy of such a method.
As for me, I feel like output from one of today’s cataract factories: like a car just extruded from an automated wash, with a photo on its front seat of the shiny, Simonized hubcaps included with the premium service package.
Just business, though. Nothing personal.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at dermnews@mdedge.com.
Many patients have seen their long-term physicians retire. When I ask how they like their new doctors, they say: “She’s okay, I guess. Quite efficient. Seems thorough. But it’s not the same. It’s just business. Nothing personal.”
Sometimes you have to look backward to look forward. So it’s perhaps fitting that I glimpsed the future at my last colonoscopy.
In recent years, I’ve had such procedures at a local suburban surgicenter. Easy access, plenty of parking.
The woman who checks me in is all business. She scans my insurance cards and hands me a clipboard with a medical history form. Have I ever had cancer? A hernia? Am I pregnant? I wonder whether anyone reads these.
A different young woman brings me inside, the first of many new faces. Their roles are murky.
In a curtained cubby, yet another staff person asks me to pack my clothes in a plastic bag and put on a johnny. Then an older man enters, initiating furious multitasking. A different nursing assistant asks me to confirm my name and date of birth, then inserts an intravenous line in one arm, while the old doctor hands me an anesthesia consent form to sign with the other hand. I check many answers very fast, ignore the small-print boilerplate, and sign.
I am handed two more consent forms to sign, one from each side. The staff makes no pretense of explaining them or even telling me what they are for, and I make none of reading them.
They depart, replaced by still another person, who rolls me into the next room. He confirms my name and date of birth, and which procedure I am there for. The purpose of these multiple checks is clear, along with dispiriting depersonalization. One could mitigate this with some light banter, but no one bothers. No time.
My physician – whom I actually know – enters, says hello, and exchanges pleasantries. The last guy asks me to turn onto my left side. Intravenous sedation flows into my veins. The rest is silence.
Sometime later I wake up, greeted by another staff person. She asks if I am okay and offers me water or juice and saltines. Noting her Boston Red Sox sweatshirt, I say, “Great game last night,” but she does not know what I am talking about. She cares only for football and plans to fly to Nashville, Tenn., to watch her favorites.
Curtains are closed, and I am asked to dress. Another assistant directs me to a chair, where I will await my ride home. Through I try to walk alone, she takes my arm. “We assist everyone,” she explains.
As the sedation wears off, I observe. All around me I see movement, brisk and purposeful. Staff members crisscross before me from all angles, striding from one task to another, from prep room A to cubby D, walking with or pushing patients from procedure room M to holding area 8H. No one I’ve just met recognizes me, or acknowledges having met me before.
At last, the final staff member approaches. She flashes a kind smile as she takes my arm to walk me to the door. I take this for a personal touch, until she explains that she must make sure I don’t fall and that I get into the right car. As we pass, no one in the waiting room, neither staff nor patients, takes any notice.
My wife is outside, idling in the correct car. She’s brought coffee and a chocolate croissant, which – almost – makes last night’s prep worthwhile. She confirms neither my name nor date of birth.
Altogether, I have been in and out in 90 minutes. In the car, I peruse the handout that had been given to me as I exited. Drinking my coffee, I read the postcare instructions and enjoy its full-color pictures. Seldom has my cecum looked more radiant.
In “The Checklist Manifesto,” Atul Gawande described the outcome improvement that systematized practice can achieve. Data analysis confirms the measurably superior efficacy of such a method.
As for me, I feel like output from one of today’s cataract factories: like a car just extruded from an automated wash, with a photo on its front seat of the shiny, Simonized hubcaps included with the premium service package.
Just business, though. Nothing personal.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at dermnews@mdedge.com.
Advancing Order Set Design
In the current health care environment, hospitals are constantly challenged to improve quality metrics and deliver better health care outcomes. One means to achieving quality improvement is through the use of order sets, groups of related orders that a health care provider (HCP) can place with either a few keystrokes or mouse clicks.1
Historically, design of order sets has largely focused on clicking checkboxes containing evidence-based practices. According to Bates and colleagues and the Institute for Safe Medication Practices, incorporating evidence-based medicine (EBM) into order sets is not by itself sufficient.2,3Execution of proper design coupled with simplicity and provider efficiency is paramount to HCP buy-in, increased likelihood of order set adherence, and to potentially better outcomes.
In this article, we outline advancements in order set design. These improvements increase provider efficiency and ease of use; incorporate human factors engineering (HFE); apply failure mode and effects analysis; and include EBM.
Methods
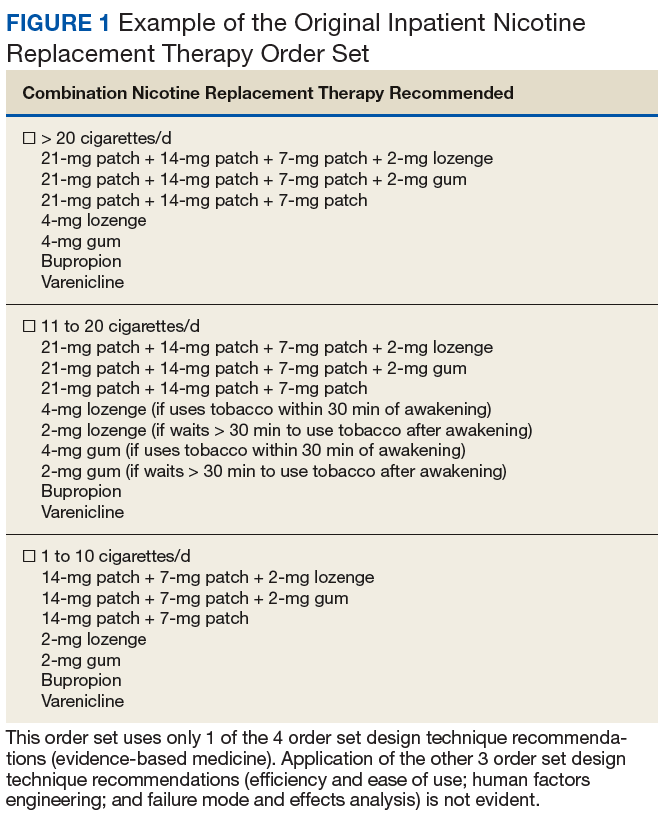
An inpatient nicotine replacement therapy (NRT) order was developed as part of a multifaceted solution to improve tobacco cessation care at the James A. Haley Veterans’ Hospital (JAHVH) in Tampa, Florida, a complexity level 1a facility. This NRT order set used the 4-step order set design framework the authors’ developed (for additional information about the NRT order set, contact the authors). We distinguish order set design technique between 2 different inpatient NRT order sets. The first order set in the comparison (Figure 1) is an inpatient NRT order set of unknown origin—it is common for US Department of Veterans Affairs (VA) medical facilities to share order sets and other resources. The second order set (Figure 2) is an inpatient NRT order set we designed using our 4-step process for comparison in this article. No institutional review board approval was required as this work met criteria for operational improvement activities exempt from ethics review.
Justin Iannello, DO, MBA, was the team leader and developer of the 4-step order set design technique. The intervention team consisted of 4 internal medicine physicians with expertise in quality improvement and patient safety: 1 certified professional in patient safety and certified as a Lean Six Sigma Black Belt; 2 physicians certified as Lean Six Sigma Black Belts; and 1 physician certified as a Lean Six Sigma Green Belt. Two inpatient clinical pharmacists and 1 quality management specialist also were involved in its development.

Development of a new NRT order set was felt to be an integral part of the tobacco cessation care delivery process. An NRT order set perceived by users as value-added required a solution that merged EBM with standardization and applied quality improvement principles. The result was an approach to order set design that focused on 4 key questions: Is the order set efficient and easy to use/navigate? Is human factors engineering incorporated? Is failure mode and effects analysis applied? Are evidence-based practices included?
Ease of Use and Navigation
Implementing an order set that is efficient and easy to use or navigate seems straightforward but can be difficult to execute. Figure 1 shows many detailed options consisting of different combinations of nicotine patches, lozenges, and gum. Also included are oral tobacco cessation options (bupropion and varenicline). Although more options may seem better, confusion about appropriate medication selection can occur.
According to Heath and Heath, too many options can result in lack of action.4 For example, Heath and Heath discuss a food store that offered 6 free samples of different jams on one day and 24 jams the following day. The customers who sampled 6 different types of jam were 10 times more likely to buy jam. The authors concluded that the more options available, the more difficulty a potential buyer has in deciding on a course of action.4
In clinical situations where a HCP is using an order set, the number of options can mean the difference between use vs avoidance if the choices are overwhelming. HCPs process layers of detail every day when creating differential diagnoses and treatment plans. While that level of detail is necessary clinically, that same level of detail included in orders sets can create challenges for HCPs.
Figure 2 advances the order set in Figure 1 by providing a simpler and cleaner design, so HCPs can more easily review and process the information. This order set design minimizes the number of options available to help users make the right decision, focusing on value for the appropriate setting and audience. In other words, order sets should not be a “one size fits all” approach.
Order sets should be tailored to the appropriate clinical setting (eg, inpatient acute care, outpatient clinic setting, etc) and HCP (eg, hospitalist, tobacco cessation specialist, etc). We are comparing NRT order sets designed for HCPs who do not routinely prescribe oral tobacco cessation products in the inpatient setting. When possible, autogenerated bundle orders should also be used according to evidence-based recommendations (such as nicotine patch tapers) for ease of use and further simplification of order sets.
Finally, usability testing known as “evaluating a product or service by testing it with representative users” helps further refine an order set.5Usability testing should be applied during all phases of order set development with end user(s) as it helps identify problems with order set design prior to implementation. By applying usability testing, the order set becomes more meaningful and valued by the user.
Human Factors Engineering
HFE is “the study of all the factors that make it easier to do the work in the right way.”6 HFE seeks to identify, align, and apply processes for people and the world within which they live and work to promote safe and efficient practices, especially in relation to the technology and physical design features in their work environment.6
The average American adult makes about 35,000 decisions per day.7 Thus, there is potential for error at any moment. Design that does not take HFE into account can be dangerous. For example, when tube feed and IV line connectors look similar and are compatible, patients may inadvertently receive food administered directly into their bloodstream.8
HFE can and should be applied to order sets. Everything from the look, feel, and verbiage of an order set affects potential outcomes. For example, consider the impact even seemingly minor modifications can have on outcomes simply by guiding users in a different way: Figure 1 provides NRT options based on cigarette use per day, whereas Figure 2 conveys pack use per day in relation to the equivalent number of cigarettes used daily. These differences may seem small; however, it helps guide users to the right choice when considering that health care providers have been historically trained on social history gathering that emphasizes packs per day and pack-years.
Failure Mode and Effects Analysis
Failure mode and effects analysis (FMEA) is “a structured way to identify and address potential problems, or failures and their resulting effects on the system or process before an adverse event occurs.”9 The benefit of an order set must be weighed against the risk during development. FMEA should be applied during order set design to assess and limit risk just as with any other clinical care process.
FMEA examines both level of risk and frequency of risk occurrence associated with a new proposed process. For example, let’s evaluate an order set designed for pain control after surgery that consists of multiple high-risk opioids along with antihistamine medications for as-needed itch relief (a non-life-threatening adverse event (AE) of opioids well known by the medical community). An interdisciplinary FMEA team consisting of subject matter experts may examine how the process should flow in step-by-step detail and then discuss the benefit of a process and risk for potential error. A FMEA team would then analyze what could go wrong with each part of the process and assign a level of risk and risk frequency for various steps in the process, and then decide that certain steps should be modified or eliminated. Perhaps after FMEA, a facility might conclude that the risk of serious complications is high when you combine opioid use with antihistamine medications. The facility could decide to remove antihistamine medications from an order set if it is determined that risks outweigh benefits. While a root cause analysis might identify the cause of an AE after order set use, these situations can be prevented with FMEA.
When applying FMEA to Figure 1, while bupropion is known as an evidence-based oral tobacco cessation option, there is the possibility that bupropion could be inadvertently prescribed from the order set in a hospitalized patient with alcohol withdrawal and withdrawal seizure history. These potentially dangerous situations can be avoided with FMEA. Thus, although bupropion may be evidence-based for NRT, decisions regarding order set design using EBM alone are insufficient.
The practitioner must consider possible unintended consequences within order sets and target treatment options to the appropriate setting and audience. Although Figure 1 may appear to be more inclusive, the interdisciplinary committee designing the inpatient NRT order set felt there was heightened risk with introducing bupropion in Figure 1 and decided the risk would be lowered by removing bupropion from the redesigned NRT order set (Figure 2). In addition to the goal of balancing availability of NRT options with acceptable risk, Figure 2 also focused on building an NRT order set most applicable to the inpatient setting.
Including Evidence-Based Practices
EBM has become a routine part of clinical decision making. Therefore, including EBM in order set design is vital. EBM for NRT has demonstrated that combination therapy is more effective than is monotherapy to help tobacco users quit. Incremental doses of NRT are recommended for patients who use tobacco more frequently.10
As shown in Figures 1 and 2, both order set designs incorporate EBM for NRT. Although the importance of implementing EBM is evident, critical factors, such as HFE and FMEA make a difference with well-designed order sets.
Results
The 4-step order set design technique was used during development of an inpatient NRT order set at the JAHVH. Results for the inpatient Joint Commission Tobacco Treatment Measures were obtained from the Veterans Health Administration quality metric reporting system known as Strategic Analytics for Improvement and Learning (SAIL). SAIL performance measure outcomes, which include the inpatient Joint Commission Tobacco Treatment Measures, are derived from chart reviews conducted by the External Peer Review Program. Outcomes demonstrated that TOB-2 and TOB-3 (2 inpatient Joint Commission Tobacco Treatment Measures) known as tob20 and tob40, respectively, within SAIL improved by more than 300% after development of an NRT order set using the 4-step order set design framework along with implementation of a multifaceted tobacco cessation care delivery system at JAHVH.
Discussion
While the overall tobacco cessation care delivery system contributed to improved outcomes with the inpatient Joint Commission Tobacco Treatment Measures at JAHVH, the NRT order set was a cornerstone of the design. Although using our order set design technique does not necessarily guarantee successful outcomes, we believe using the 4-step order set design process increases the value of order sets and has potential to improve quality outcomes.
Limitations
Although improved outcomes following implementation of our NRT order set suggest correlation, causation cannot be proven. Also while the NRT order set is believed to have helped tremendously with outcomes, the entire tobacco cessation care delivery system at JAHVH contributed to the results. In addition, the inpatient Joint Commission Tobacco Treatment Measures help improve processes for tobacco cessation care. However, we are uncertain whether the results of our improvement efforts helped patients stop tobacco use. Further studies are needed to determine impact on population health. Finally, our results were based on improvement work done at a single center. Further studies are necessary to see whether results are reproducible.
Conclusion
There was significant improvement with the inpatient Joint Commission Tobacco Treatment Measures outcomes following development of a tobacco cessation care delivery system that included design of an inpatient NRT order set using a 4-step process we developed. This 4-step structure includes emphasis on efficiency and ease of use; human factors engineering; failure mode and effects analysis; and incorporation of evidence-based medicine (Box.) Postimplementation results showed improvement of the inpatient Joint Commission Tobacco Treatment Measures by greater than 3-fold at a single hospital.

The next steps for this initiative include testing the 4-step order set design process in multiple clinical settings to determine the effectiveness of this approach in other areas of clinical care.
1. Order set. http://clinfowiki.org/wiki/index.php/Order_set. Updated October 15, 2015. Accessed August 30, 2019.
2. Bates DW, Kuperman GJ, Wang S, et al. Ten commandments for effective clinical decision support: making the practice of evidence-based medicine a reality. J Am Med Inform Assoc. 2003;10(6):523-530.
3. Institute for Safe Medication Practices. Guidelines for standard order sets. https://www.ismp.org/tools/guidelines/standardordersets.pdf. Published January 12, 2010. Accessed August 30, 2019.
4. Heath C, Heath D. Switch: How to Change Things When Change Is Hard. New York, NY: Crown Business; 2010:50-51.
5. US Department of Health and Human Services. Usability testing. https://www.usability.gov/how-to-and-tools/methods/usability-testing.html. Accessed August 30, 2019.
6. World Health Organization. What is human factors and why is it important to patient safety? www.who.int/patientsafety/education/curriculum/who_mc_topic-2.pdf. Accessed August 30, 2019.
7. Sollisch J. The cure for decision fatigue. Wall Street Journal. June 10, 2016. https://www.wsj.com/articles/the-cure-for-decision-fatigue-1465596928. Accessed August 30, 2019.
8. ECRI Institute. Implementing the ENFit initiative for preventing enteral tubing misconnections. https://www.ecri.org/components/HDJournal/Pages/ENFit-for-Preventing-Enteral-Tubing-Misconnections.aspx. Published March 29, 2017. Accessed August 30, 2019.
9. Guidance for performing failure mode and effects analysis with performance improvement projects. https://www.cms.gov/Medicare/Provider-Enrollment-and-Certification/QAPI/downloads/GuidanceForFMEA.pdf. Accessed August 30, 2019.
10. Diefanbach LJ, Smith PO, Nashelsky JM, Lindbloom E. What is the most effective nicotine replacement therapy? J Fam Pract. 2003;52(6):492-497.
In the current health care environment, hospitals are constantly challenged to improve quality metrics and deliver better health care outcomes. One means to achieving quality improvement is through the use of order sets, groups of related orders that a health care provider (HCP) can place with either a few keystrokes or mouse clicks.1
Historically, design of order sets has largely focused on clicking checkboxes containing evidence-based practices. According to Bates and colleagues and the Institute for Safe Medication Practices, incorporating evidence-based medicine (EBM) into order sets is not by itself sufficient.2,3Execution of proper design coupled with simplicity and provider efficiency is paramount to HCP buy-in, increased likelihood of order set adherence, and to potentially better outcomes.
In this article, we outline advancements in order set design. These improvements increase provider efficiency and ease of use; incorporate human factors engineering (HFE); apply failure mode and effects analysis; and include EBM.
Methods
An inpatient nicotine replacement therapy (NRT) order was developed as part of a multifaceted solution to improve tobacco cessation care at the James A. Haley Veterans’ Hospital (JAHVH) in Tampa, Florida, a complexity level 1a facility. This NRT order set used the 4-step order set design framework the authors’ developed (for additional information about the NRT order set, contact the authors). We distinguish order set design technique between 2 different inpatient NRT order sets. The first order set in the comparison (Figure 1) is an inpatient NRT order set of unknown origin—it is common for US Department of Veterans Affairs (VA) medical facilities to share order sets and other resources. The second order set (Figure 2) is an inpatient NRT order set we designed using our 4-step process for comparison in this article. No institutional review board approval was required as this work met criteria for operational improvement activities exempt from ethics review.
Justin Iannello, DO, MBA, was the team leader and developer of the 4-step order set design technique. The intervention team consisted of 4 internal medicine physicians with expertise in quality improvement and patient safety: 1 certified professional in patient safety and certified as a Lean Six Sigma Black Belt; 2 physicians certified as Lean Six Sigma Black Belts; and 1 physician certified as a Lean Six Sigma Green Belt. Two inpatient clinical pharmacists and 1 quality management specialist also were involved in its development.
Development of a new NRT order set was felt to be an integral part of the tobacco cessation care delivery process. An NRT order set perceived by users as value-added required a solution that merged EBM with standardization and applied quality improvement principles. The result was an approach to order set design that focused on 4 key questions: Is the order set efficient and easy to use/navigate? Is human factors engineering incorporated? Is failure mode and effects analysis applied? Are evidence-based practices included?
Ease of Use and Navigation
Implementing an order set that is efficient and easy to use or navigate seems straightforward but can be difficult to execute. Figure 1 shows many detailed options consisting of different combinations of nicotine patches, lozenges, and gum. Also included are oral tobacco cessation options (bupropion and varenicline). Although more options may seem better, confusion about appropriate medication selection can occur.
According to Heath and Heath, too many options can result in lack of action.4 For example, Heath and Heath discuss a food store that offered 6 free samples of different jams on one day and 24 jams the following day. The customers who sampled 6 different types of jam were 10 times more likely to buy jam. The authors concluded that the more options available, the more difficulty a potential buyer has in deciding on a course of action.4
In clinical situations where a HCP is using an order set, the number of options can mean the difference between use vs avoidance if the choices are overwhelming. HCPs process layers of detail every day when creating differential diagnoses and treatment plans. While that level of detail is necessary clinically, that same level of detail included in orders sets can create challenges for HCPs.
Figure 2 advances the order set in Figure 1 by providing a simpler and cleaner design, so HCPs can more easily review and process the information. This order set design minimizes the number of options available to help users make the right decision, focusing on value for the appropriate setting and audience. In other words, order sets should not be a “one size fits all” approach.
Order sets should be tailored to the appropriate clinical setting (eg, inpatient acute care, outpatient clinic setting, etc) and HCP (eg, hospitalist, tobacco cessation specialist, etc). We are comparing NRT order sets designed for HCPs who do not routinely prescribe oral tobacco cessation products in the inpatient setting. When possible, autogenerated bundle orders should also be used according to evidence-based recommendations (such as nicotine patch tapers) for ease of use and further simplification of order sets.
Finally, usability testing known as “evaluating a product or service by testing it with representative users” helps further refine an order set.5Usability testing should be applied during all phases of order set development with end user(s) as it helps identify problems with order set design prior to implementation. By applying usability testing, the order set becomes more meaningful and valued by the user.
Human Factors Engineering
HFE is “the study of all the factors that make it easier to do the work in the right way.”6 HFE seeks to identify, align, and apply processes for people and the world within which they live and work to promote safe and efficient practices, especially in relation to the technology and physical design features in their work environment.6
The average American adult makes about 35,000 decisions per day.7 Thus, there is potential for error at any moment. Design that does not take HFE into account can be dangerous. For example, when tube feed and IV line connectors look similar and are compatible, patients may inadvertently receive food administered directly into their bloodstream.8
HFE can and should be applied to order sets. Everything from the look, feel, and verbiage of an order set affects potential outcomes. For example, consider the impact even seemingly minor modifications can have on outcomes simply by guiding users in a different way: Figure 1 provides NRT options based on cigarette use per day, whereas Figure 2 conveys pack use per day in relation to the equivalent number of cigarettes used daily. These differences may seem small; however, it helps guide users to the right choice when considering that health care providers have been historically trained on social history gathering that emphasizes packs per day and pack-years.
Failure Mode and Effects Analysis
Failure mode and effects analysis (FMEA) is “a structured way to identify and address potential problems, or failures and their resulting effects on the system or process before an adverse event occurs.”9 The benefit of an order set must be weighed against the risk during development. FMEA should be applied during order set design to assess and limit risk just as with any other clinical care process.
FMEA examines both level of risk and frequency of risk occurrence associated with a new proposed process. For example, let’s evaluate an order set designed for pain control after surgery that consists of multiple high-risk opioids along with antihistamine medications for as-needed itch relief (a non-life-threatening adverse event (AE) of opioids well known by the medical community). An interdisciplinary FMEA team consisting of subject matter experts may examine how the process should flow in step-by-step detail and then discuss the benefit of a process and risk for potential error. A FMEA team would then analyze what could go wrong with each part of the process and assign a level of risk and risk frequency for various steps in the process, and then decide that certain steps should be modified or eliminated. Perhaps after FMEA, a facility might conclude that the risk of serious complications is high when you combine opioid use with antihistamine medications. The facility could decide to remove antihistamine medications from an order set if it is determined that risks outweigh benefits. While a root cause analysis might identify the cause of an AE after order set use, these situations can be prevented with FMEA.
When applying FMEA to Figure 1, while bupropion is known as an evidence-based oral tobacco cessation option, there is the possibility that bupropion could be inadvertently prescribed from the order set in a hospitalized patient with alcohol withdrawal and withdrawal seizure history. These potentially dangerous situations can be avoided with FMEA. Thus, although bupropion may be evidence-based for NRT, decisions regarding order set design using EBM alone are insufficient.
The practitioner must consider possible unintended consequences within order sets and target treatment options to the appropriate setting and audience. Although Figure 1 may appear to be more inclusive, the interdisciplinary committee designing the inpatient NRT order set felt there was heightened risk with introducing bupropion in Figure 1 and decided the risk would be lowered by removing bupropion from the redesigned NRT order set (Figure 2). In addition to the goal of balancing availability of NRT options with acceptable risk, Figure 2 also focused on building an NRT order set most applicable to the inpatient setting.
Including Evidence-Based Practices
EBM has become a routine part of clinical decision making. Therefore, including EBM in order set design is vital. EBM for NRT has demonstrated that combination therapy is more effective than is monotherapy to help tobacco users quit. Incremental doses of NRT are recommended for patients who use tobacco more frequently.10
As shown in Figures 1 and 2, both order set designs incorporate EBM for NRT. Although the importance of implementing EBM is evident, critical factors, such as HFE and FMEA make a difference with well-designed order sets.
Results
The 4-step order set design technique was used during development of an inpatient NRT order set at the JAHVH. Results for the inpatient Joint Commission Tobacco Treatment Measures were obtained from the Veterans Health Administration quality metric reporting system known as Strategic Analytics for Improvement and Learning (SAIL). SAIL performance measure outcomes, which include the inpatient Joint Commission Tobacco Treatment Measures, are derived from chart reviews conducted by the External Peer Review Program. Outcomes demonstrated that TOB-2 and TOB-3 (2 inpatient Joint Commission Tobacco Treatment Measures) known as tob20 and tob40, respectively, within SAIL improved by more than 300% after development of an NRT order set using the 4-step order set design framework along with implementation of a multifaceted tobacco cessation care delivery system at JAHVH.
Discussion
While the overall tobacco cessation care delivery system contributed to improved outcomes with the inpatient Joint Commission Tobacco Treatment Measures at JAHVH, the NRT order set was a cornerstone of the design. Although using our order set design technique does not necessarily guarantee successful outcomes, we believe using the 4-step order set design process increases the value of order sets and has potential to improve quality outcomes.
Limitations
Although improved outcomes following implementation of our NRT order set suggest correlation, causation cannot be proven. Also while the NRT order set is believed to have helped tremendously with outcomes, the entire tobacco cessation care delivery system at JAHVH contributed to the results. In addition, the inpatient Joint Commission Tobacco Treatment Measures help improve processes for tobacco cessation care. However, we are uncertain whether the results of our improvement efforts helped patients stop tobacco use. Further studies are needed to determine impact on population health. Finally, our results were based on improvement work done at a single center. Further studies are necessary to see whether results are reproducible.
Conclusion
There was significant improvement with the inpatient Joint Commission Tobacco Treatment Measures outcomes following development of a tobacco cessation care delivery system that included design of an inpatient NRT order set using a 4-step process we developed. This 4-step structure includes emphasis on efficiency and ease of use; human factors engineering; failure mode and effects analysis; and incorporation of evidence-based medicine (Box.) Postimplementation results showed improvement of the inpatient Joint Commission Tobacco Treatment Measures by greater than 3-fold at a single hospital.
The next steps for this initiative include testing the 4-step order set design process in multiple clinical settings to determine the effectiveness of this approach in other areas of clinical care.
In the current health care environment, hospitals are constantly challenged to improve quality metrics and deliver better health care outcomes. One means to achieving quality improvement is through the use of order sets, groups of related orders that a health care provider (HCP) can place with either a few keystrokes or mouse clicks.1
Historically, design of order sets has largely focused on clicking checkboxes containing evidence-based practices. According to Bates and colleagues and the Institute for Safe Medication Practices, incorporating evidence-based medicine (EBM) into order sets is not by itself sufficient.2,3Execution of proper design coupled with simplicity and provider efficiency is paramount to HCP buy-in, increased likelihood of order set adherence, and to potentially better outcomes.
In this article, we outline advancements in order set design. These improvements increase provider efficiency and ease of use; incorporate human factors engineering (HFE); apply failure mode and effects analysis; and include EBM.
Methods
An inpatient nicotine replacement therapy (NRT) order was developed as part of a multifaceted solution to improve tobacco cessation care at the James A. Haley Veterans’ Hospital (JAHVH) in Tampa, Florida, a complexity level 1a facility. This NRT order set used the 4-step order set design framework the authors’ developed (for additional information about the NRT order set, contact the authors). We distinguish order set design technique between 2 different inpatient NRT order sets. The first order set in the comparison (Figure 1) is an inpatient NRT order set of unknown origin—it is common for US Department of Veterans Affairs (VA) medical facilities to share order sets and other resources. The second order set (Figure 2) is an inpatient NRT order set we designed using our 4-step process for comparison in this article. No institutional review board approval was required as this work met criteria for operational improvement activities exempt from ethics review.
Justin Iannello, DO, MBA, was the team leader and developer of the 4-step order set design technique. The intervention team consisted of 4 internal medicine physicians with expertise in quality improvement and patient safety: 1 certified professional in patient safety and certified as a Lean Six Sigma Black Belt; 2 physicians certified as Lean Six Sigma Black Belts; and 1 physician certified as a Lean Six Sigma Green Belt. Two inpatient clinical pharmacists and 1 quality management specialist also were involved in its development.
Development of a new NRT order set was felt to be an integral part of the tobacco cessation care delivery process. An NRT order set perceived by users as value-added required a solution that merged EBM with standardization and applied quality improvement principles. The result was an approach to order set design that focused on 4 key questions: Is the order set efficient and easy to use/navigate? Is human factors engineering incorporated? Is failure mode and effects analysis applied? Are evidence-based practices included?
Ease of Use and Navigation
Implementing an order set that is efficient and easy to use or navigate seems straightforward but can be difficult to execute. Figure 1 shows many detailed options consisting of different combinations of nicotine patches, lozenges, and gum. Also included are oral tobacco cessation options (bupropion and varenicline). Although more options may seem better, confusion about appropriate medication selection can occur.
According to Heath and Heath, too many options can result in lack of action.4 For example, Heath and Heath discuss a food store that offered 6 free samples of different jams on one day and 24 jams the following day. The customers who sampled 6 different types of jam were 10 times more likely to buy jam. The authors concluded that the more options available, the more difficulty a potential buyer has in deciding on a course of action.4
In clinical situations where a HCP is using an order set, the number of options can mean the difference between use vs avoidance if the choices are overwhelming. HCPs process layers of detail every day when creating differential diagnoses and treatment plans. While that level of detail is necessary clinically, that same level of detail included in orders sets can create challenges for HCPs.
Figure 2 advances the order set in Figure 1 by providing a simpler and cleaner design, so HCPs can more easily review and process the information. This order set design minimizes the number of options available to help users make the right decision, focusing on value for the appropriate setting and audience. In other words, order sets should not be a “one size fits all” approach.
Order sets should be tailored to the appropriate clinical setting (eg, inpatient acute care, outpatient clinic setting, etc) and HCP (eg, hospitalist, tobacco cessation specialist, etc). We are comparing NRT order sets designed for HCPs who do not routinely prescribe oral tobacco cessation products in the inpatient setting. When possible, autogenerated bundle orders should also be used according to evidence-based recommendations (such as nicotine patch tapers) for ease of use and further simplification of order sets.
Finally, usability testing known as “evaluating a product or service by testing it with representative users” helps further refine an order set.5Usability testing should be applied during all phases of order set development with end user(s) as it helps identify problems with order set design prior to implementation. By applying usability testing, the order set becomes more meaningful and valued by the user.
Human Factors Engineering
HFE is “the study of all the factors that make it easier to do the work in the right way.”6 HFE seeks to identify, align, and apply processes for people and the world within which they live and work to promote safe and efficient practices, especially in relation to the technology and physical design features in their work environment.6
The average American adult makes about 35,000 decisions per day.7 Thus, there is potential for error at any moment. Design that does not take HFE into account can be dangerous. For example, when tube feed and IV line connectors look similar and are compatible, patients may inadvertently receive food administered directly into their bloodstream.8
HFE can and should be applied to order sets. Everything from the look, feel, and verbiage of an order set affects potential outcomes. For example, consider the impact even seemingly minor modifications can have on outcomes simply by guiding users in a different way: Figure 1 provides NRT options based on cigarette use per day, whereas Figure 2 conveys pack use per day in relation to the equivalent number of cigarettes used daily. These differences may seem small; however, it helps guide users to the right choice when considering that health care providers have been historically trained on social history gathering that emphasizes packs per day and pack-years.
Failure Mode and Effects Analysis
Failure mode and effects analysis (FMEA) is “a structured way to identify and address potential problems, or failures and their resulting effects on the system or process before an adverse event occurs.”9 The benefit of an order set must be weighed against the risk during development. FMEA should be applied during order set design to assess and limit risk just as with any other clinical care process.
FMEA examines both level of risk and frequency of risk occurrence associated with a new proposed process. For example, let’s evaluate an order set designed for pain control after surgery that consists of multiple high-risk opioids along with antihistamine medications for as-needed itch relief (a non-life-threatening adverse event (AE) of opioids well known by the medical community). An interdisciplinary FMEA team consisting of subject matter experts may examine how the process should flow in step-by-step detail and then discuss the benefit of a process and risk for potential error. A FMEA team would then analyze what could go wrong with each part of the process and assign a level of risk and risk frequency for various steps in the process, and then decide that certain steps should be modified or eliminated. Perhaps after FMEA, a facility might conclude that the risk of serious complications is high when you combine opioid use with antihistamine medications. The facility could decide to remove antihistamine medications from an order set if it is determined that risks outweigh benefits. While a root cause analysis might identify the cause of an AE after order set use, these situations can be prevented with FMEA.
When applying FMEA to Figure 1, while bupropion is known as an evidence-based oral tobacco cessation option, there is the possibility that bupropion could be inadvertently prescribed from the order set in a hospitalized patient with alcohol withdrawal and withdrawal seizure history. These potentially dangerous situations can be avoided with FMEA. Thus, although bupropion may be evidence-based for NRT, decisions regarding order set design using EBM alone are insufficient.
The practitioner must consider possible unintended consequences within order sets and target treatment options to the appropriate setting and audience. Although Figure 1 may appear to be more inclusive, the interdisciplinary committee designing the inpatient NRT order set felt there was heightened risk with introducing bupropion in Figure 1 and decided the risk would be lowered by removing bupropion from the redesigned NRT order set (Figure 2). In addition to the goal of balancing availability of NRT options with acceptable risk, Figure 2 also focused on building an NRT order set most applicable to the inpatient setting.
Including Evidence-Based Practices
EBM has become a routine part of clinical decision making. Therefore, including EBM in order set design is vital. EBM for NRT has demonstrated that combination therapy is more effective than is monotherapy to help tobacco users quit. Incremental doses of NRT are recommended for patients who use tobacco more frequently.10
As shown in Figures 1 and 2, both order set designs incorporate EBM for NRT. Although the importance of implementing EBM is evident, critical factors, such as HFE and FMEA make a difference with well-designed order sets.
Results
The 4-step order set design technique was used during development of an inpatient NRT order set at the JAHVH. Results for the inpatient Joint Commission Tobacco Treatment Measures were obtained from the Veterans Health Administration quality metric reporting system known as Strategic Analytics for Improvement and Learning (SAIL). SAIL performance measure outcomes, which include the inpatient Joint Commission Tobacco Treatment Measures, are derived from chart reviews conducted by the External Peer Review Program. Outcomes demonstrated that TOB-2 and TOB-3 (2 inpatient Joint Commission Tobacco Treatment Measures) known as tob20 and tob40, respectively, within SAIL improved by more than 300% after development of an NRT order set using the 4-step order set design framework along with implementation of a multifaceted tobacco cessation care delivery system at JAHVH.
Discussion
While the overall tobacco cessation care delivery system contributed to improved outcomes with the inpatient Joint Commission Tobacco Treatment Measures at JAHVH, the NRT order set was a cornerstone of the design. Although using our order set design technique does not necessarily guarantee successful outcomes, we believe using the 4-step order set design process increases the value of order sets and has potential to improve quality outcomes.
Limitations
Although improved outcomes following implementation of our NRT order set suggest correlation, causation cannot be proven. Also while the NRT order set is believed to have helped tremendously with outcomes, the entire tobacco cessation care delivery system at JAHVH contributed to the results. In addition, the inpatient Joint Commission Tobacco Treatment Measures help improve processes for tobacco cessation care. However, we are uncertain whether the results of our improvement efforts helped patients stop tobacco use. Further studies are needed to determine impact on population health. Finally, our results were based on improvement work done at a single center. Further studies are necessary to see whether results are reproducible.
Conclusion
There was significant improvement with the inpatient Joint Commission Tobacco Treatment Measures outcomes following development of a tobacco cessation care delivery system that included design of an inpatient NRT order set using a 4-step process we developed. This 4-step structure includes emphasis on efficiency and ease of use; human factors engineering; failure mode and effects analysis; and incorporation of evidence-based medicine (Box.) Postimplementation results showed improvement of the inpatient Joint Commission Tobacco Treatment Measures by greater than 3-fold at a single hospital.
The next steps for this initiative include testing the 4-step order set design process in multiple clinical settings to determine the effectiveness of this approach in other areas of clinical care.
1. Order set. http://clinfowiki.org/wiki/index.php/Order_set. Updated October 15, 2015. Accessed August 30, 2019.
2. Bates DW, Kuperman GJ, Wang S, et al. Ten commandments for effective clinical decision support: making the practice of evidence-based medicine a reality. J Am Med Inform Assoc. 2003;10(6):523-530.
3. Institute for Safe Medication Practices. Guidelines for standard order sets. https://www.ismp.org/tools/guidelines/standardordersets.pdf. Published January 12, 2010. Accessed August 30, 2019.
4. Heath C, Heath D. Switch: How to Change Things When Change Is Hard. New York, NY: Crown Business; 2010:50-51.
5. US Department of Health and Human Services. Usability testing. https://www.usability.gov/how-to-and-tools/methods/usability-testing.html. Accessed August 30, 2019.
6. World Health Organization. What is human factors and why is it important to patient safety? www.who.int/patientsafety/education/curriculum/who_mc_topic-2.pdf. Accessed August 30, 2019.
7. Sollisch J. The cure for decision fatigue. Wall Street Journal. June 10, 2016. https://www.wsj.com/articles/the-cure-for-decision-fatigue-1465596928. Accessed August 30, 2019.
8. ECRI Institute. Implementing the ENFit initiative for preventing enteral tubing misconnections. https://www.ecri.org/components/HDJournal/Pages/ENFit-for-Preventing-Enteral-Tubing-Misconnections.aspx. Published March 29, 2017. Accessed August 30, 2019.
9. Guidance for performing failure mode and effects analysis with performance improvement projects. https://www.cms.gov/Medicare/Provider-Enrollment-and-Certification/QAPI/downloads/GuidanceForFMEA.pdf. Accessed August 30, 2019.
10. Diefanbach LJ, Smith PO, Nashelsky JM, Lindbloom E. What is the most effective nicotine replacement therapy? J Fam Pract. 2003;52(6):492-497.
1. Order set. http://clinfowiki.org/wiki/index.php/Order_set. Updated October 15, 2015. Accessed August 30, 2019.
2. Bates DW, Kuperman GJ, Wang S, et al. Ten commandments for effective clinical decision support: making the practice of evidence-based medicine a reality. J Am Med Inform Assoc. 2003;10(6):523-530.
3. Institute for Safe Medication Practices. Guidelines for standard order sets. https://www.ismp.org/tools/guidelines/standardordersets.pdf. Published January 12, 2010. Accessed August 30, 2019.
4. Heath C, Heath D. Switch: How to Change Things When Change Is Hard. New York, NY: Crown Business; 2010:50-51.
5. US Department of Health and Human Services. Usability testing. https://www.usability.gov/how-to-and-tools/methods/usability-testing.html. Accessed August 30, 2019.
6. World Health Organization. What is human factors and why is it important to patient safety? www.who.int/patientsafety/education/curriculum/who_mc_topic-2.pdf. Accessed August 30, 2019.
7. Sollisch J. The cure for decision fatigue. Wall Street Journal. June 10, 2016. https://www.wsj.com/articles/the-cure-for-decision-fatigue-1465596928. Accessed August 30, 2019.
8. ECRI Institute. Implementing the ENFit initiative for preventing enteral tubing misconnections. https://www.ecri.org/components/HDJournal/Pages/ENFit-for-Preventing-Enteral-Tubing-Misconnections.aspx. Published March 29, 2017. Accessed August 30, 2019.
9. Guidance for performing failure mode and effects analysis with performance improvement projects. https://www.cms.gov/Medicare/Provider-Enrollment-and-Certification/QAPI/downloads/GuidanceForFMEA.pdf. Accessed August 30, 2019.
10. Diefanbach LJ, Smith PO, Nashelsky JM, Lindbloom E. What is the most effective nicotine replacement therapy? J Fam Pract. 2003;52(6):492-497.
Q&A: Drug costs and value in cancer
Skyrocketing drug costs are a key issue facing physicians, patients, and policymakers, but an even thornier problem may be determining a drug’s value.

In this Q&A, Richard L. Schilsky, MD, senior vice president and chief medical officer at the American Society of Clinical Oncology (ASCO), weighs in on the value proposition for cancer drugs and the implications for physicians.
Q: What tools exist for determining a drug’s value?
A: A number of organizations have developed tools to try to determine the value of cancer drug treatments. ASCO, the European Society for Medical Oncology (ESMO), the Institute for Clinical and Economic Review, Memorial Sloan Kettering Cancer Center, and the National Comprehensive Cancer Network have all developed tools for this purpose.
Our tool, the ASCO Value Framework, assesses the value of new cancer drug treatments based on clinical benefit, side effects, and improvements in patient symptoms or quality of life in the context of cost. While it’s hard to directly compare frameworks – given differences in methodology and the many nuances of evaluating clinical trial results – in 2018, ASCO and ESMO published a joint analysis of our value frameworks in the Journal of Clinical Oncology (2018; 37[4]:336-49).
The analysis found that the frameworks produce comparable measures of the clinical benefits of new therapies in approximately two-thirds of the more than 100 treatment comparisons that were examined. It also identified a number of factors that may contribute to the discordant scores, revealing potential ways for both of our organizations to refine our frameworks in the future.
That said, ASCO’s Value Framework is just one part of our broader, multifaceted effort to achieve high-quality, high-value care for all patients with cancer. Other efforts include ASCO’s proposed Patient-Centered Oncology Payment model, the Choosing Wisely campaign to identify low-value clinical strategies, and CancerLinQ and the Quality Oncology Practice Initiative to implement quality measurement and improvement.
Q: How can the issues around drug price and value be addressed earlier in the context of clinical trials?
A: The definition of value ultimately comes down to the price that must be paid to achieve meaningfully improved health outcomes for individual patients or the broader population of affected individuals. Optimizing the value of a new cancer drug treatment begins with an innovation to address an unmet medical need, followed by defining and achieving clinically meaningful improvements in health outcomes through well-designed and efficiently conducted clinical trials. Effectiveness research is also essential to determining how well new treatments perform compared with available alternatives and how they perform in more diverse populations than those typically included in the clinical trials used to establish efficacy.
Patient goals, preferences, and choices shape the real-world experience of a new product, and the direct and indirect costs of a treatment to patients and their families significantly affect whether it is adopted widely. Until their value is clearly established, new and costly products should be deployed judiciously and after careful consideration of the goals of treatment, available options, and the unique needs, preferences, and goals of individual patients.
More research is needed to improve how we assess the value of new cancer drug treatments. New clinical efficacy endpoints – both provider- and patient-reported ones – that accurately describe how a patient feels and functions must be developed and should reflect outcomes of value to patients other than survival, particularly in noncurative settings.
Better predictive biomarkers can transform a drug of modest efficacy in an unselected population to one of high efficacy in a biomarker-defined subgroup and thereby contribute to improving the value of a treatment.
Regulatory and policy initiatives such as adaptive licensing, value-based insurance, and indication-specific pricing that affect marketing approval, reimbursement, or price, respectively, based on treatment effectiveness, also deserve careful consideration and further research to determine their effects on aligning cost with benefit while ensuring patient access to potentially life-extending therapies and continued innovation in drug development.
Q: Aside from the policy options, what’s the role of the oncologist in discussing the value of drugs with patients when determining a treatment plan?
A: Since oncologists don’t control drug prices, our role in improving the value of cancer care involves appropriately managing how resources are used and guiding patients during discussions around the right treatment plan for their particular diagnosis, prognosis, and treatment goals.
Adopting and adhering to high-quality oncology clinical pathways is an important way to improve the quality, efficiency, and value of cancer care. High-quality oncology pathways are detailed, evidence-based treatment protocols for delivering cancer care to patients with specific disease types and stages. When properly designed and implemented, oncology pathways serve as an important tool in appropriately managing cancer care resources and improving the quality of care that patients with cancer receive, while also reducing costs.
Dr. Schilsky is the senior vice president and chief medical officer of ASCO. Formerly the chief of hematology/oncology in the department of medicine and deputy director of the University of Chicago Comprehensive Cancer Center, he is a leader in the field of clinical oncology, specializing in new drug development and the treatment of gastrointestinal cancers. Dr. Schilsky reported research funding from several pharmaceutical companies to ASCO for the Targeted Agent and Profiling Utilization Registry (TAPUR) clinical trial. He also reported travel/accommodation/expense support from Varian.
Skyrocketing drug costs are a key issue facing physicians, patients, and policymakers, but an even thornier problem may be determining a drug’s value.
In this Q&A, Richard L. Schilsky, MD, senior vice president and chief medical officer at the American Society of Clinical Oncology (ASCO), weighs in on the value proposition for cancer drugs and the implications for physicians.
Q: What tools exist for determining a drug’s value?
A: A number of organizations have developed tools to try to determine the value of cancer drug treatments. ASCO, the European Society for Medical Oncology (ESMO), the Institute for Clinical and Economic Review, Memorial Sloan Kettering Cancer Center, and the National Comprehensive Cancer Network have all developed tools for this purpose.
Our tool, the ASCO Value Framework, assesses the value of new cancer drug treatments based on clinical benefit, side effects, and improvements in patient symptoms or quality of life in the context of cost. While it’s hard to directly compare frameworks – given differences in methodology and the many nuances of evaluating clinical trial results – in 2018, ASCO and ESMO published a joint analysis of our value frameworks in the Journal of Clinical Oncology (2018; 37[4]:336-49).
The analysis found that the frameworks produce comparable measures of the clinical benefits of new therapies in approximately two-thirds of the more than 100 treatment comparisons that were examined. It also identified a number of factors that may contribute to the discordant scores, revealing potential ways for both of our organizations to refine our frameworks in the future.
That said, ASCO’s Value Framework is just one part of our broader, multifaceted effort to achieve high-quality, high-value care for all patients with cancer. Other efforts include ASCO’s proposed Patient-Centered Oncology Payment model, the Choosing Wisely campaign to identify low-value clinical strategies, and CancerLinQ and the Quality Oncology Practice Initiative to implement quality measurement and improvement.
Q: How can the issues around drug price and value be addressed earlier in the context of clinical trials?
A: The definition of value ultimately comes down to the price that must be paid to achieve meaningfully improved health outcomes for individual patients or the broader population of affected individuals. Optimizing the value of a new cancer drug treatment begins with an innovation to address an unmet medical need, followed by defining and achieving clinically meaningful improvements in health outcomes through well-designed and efficiently conducted clinical trials. Effectiveness research is also essential to determining how well new treatments perform compared with available alternatives and how they perform in more diverse populations than those typically included in the clinical trials used to establish efficacy.
Patient goals, preferences, and choices shape the real-world experience of a new product, and the direct and indirect costs of a treatment to patients and their families significantly affect whether it is adopted widely. Until their value is clearly established, new and costly products should be deployed judiciously and after careful consideration of the goals of treatment, available options, and the unique needs, preferences, and goals of individual patients.
More research is needed to improve how we assess the value of new cancer drug treatments. New clinical efficacy endpoints – both provider- and patient-reported ones – that accurately describe how a patient feels and functions must be developed and should reflect outcomes of value to patients other than survival, particularly in noncurative settings.
Better predictive biomarkers can transform a drug of modest efficacy in an unselected population to one of high efficacy in a biomarker-defined subgroup and thereby contribute to improving the value of a treatment.
Regulatory and policy initiatives such as adaptive licensing, value-based insurance, and indication-specific pricing that affect marketing approval, reimbursement, or price, respectively, based on treatment effectiveness, also deserve careful consideration and further research to determine their effects on aligning cost with benefit while ensuring patient access to potentially life-extending therapies and continued innovation in drug development.
Q: Aside from the policy options, what’s the role of the oncologist in discussing the value of drugs with patients when determining a treatment plan?
A: Since oncologists don’t control drug prices, our role in improving the value of cancer care involves appropriately managing how resources are used and guiding patients during discussions around the right treatment plan for their particular diagnosis, prognosis, and treatment goals.
Adopting and adhering to high-quality oncology clinical pathways is an important way to improve the quality, efficiency, and value of cancer care. High-quality oncology pathways are detailed, evidence-based treatment protocols for delivering cancer care to patients with specific disease types and stages. When properly designed and implemented, oncology pathways serve as an important tool in appropriately managing cancer care resources and improving the quality of care that patients with cancer receive, while also reducing costs.
Dr. Schilsky is the senior vice president and chief medical officer of ASCO. Formerly the chief of hematology/oncology in the department of medicine and deputy director of the University of Chicago Comprehensive Cancer Center, he is a leader in the field of clinical oncology, specializing in new drug development and the treatment of gastrointestinal cancers. Dr. Schilsky reported research funding from several pharmaceutical companies to ASCO for the Targeted Agent and Profiling Utilization Registry (TAPUR) clinical trial. He also reported travel/accommodation/expense support from Varian.
Skyrocketing drug costs are a key issue facing physicians, patients, and policymakers, but an even thornier problem may be determining a drug’s value.
In this Q&A, Richard L. Schilsky, MD, senior vice president and chief medical officer at the American Society of Clinical Oncology (ASCO), weighs in on the value proposition for cancer drugs and the implications for physicians.
Q: What tools exist for determining a drug’s value?
A: A number of organizations have developed tools to try to determine the value of cancer drug treatments. ASCO, the European Society for Medical Oncology (ESMO), the Institute for Clinical and Economic Review, Memorial Sloan Kettering Cancer Center, and the National Comprehensive Cancer Network have all developed tools for this purpose.
Our tool, the ASCO Value Framework, assesses the value of new cancer drug treatments based on clinical benefit, side effects, and improvements in patient symptoms or quality of life in the context of cost. While it’s hard to directly compare frameworks – given differences in methodology and the many nuances of evaluating clinical trial results – in 2018, ASCO and ESMO published a joint analysis of our value frameworks in the Journal of Clinical Oncology (2018; 37[4]:336-49).
The analysis found that the frameworks produce comparable measures of the clinical benefits of new therapies in approximately two-thirds of the more than 100 treatment comparisons that were examined. It also identified a number of factors that may contribute to the discordant scores, revealing potential ways for both of our organizations to refine our frameworks in the future.
That said, ASCO’s Value Framework is just one part of our broader, multifaceted effort to achieve high-quality, high-value care for all patients with cancer. Other efforts include ASCO’s proposed Patient-Centered Oncology Payment model, the Choosing Wisely campaign to identify low-value clinical strategies, and CancerLinQ and the Quality Oncology Practice Initiative to implement quality measurement and improvement.
Q: How can the issues around drug price and value be addressed earlier in the context of clinical trials?
A: The definition of value ultimately comes down to the price that must be paid to achieve meaningfully improved health outcomes for individual patients or the broader population of affected individuals. Optimizing the value of a new cancer drug treatment begins with an innovation to address an unmet medical need, followed by defining and achieving clinically meaningful improvements in health outcomes through well-designed and efficiently conducted clinical trials. Effectiveness research is also essential to determining how well new treatments perform compared with available alternatives and how they perform in more diverse populations than those typically included in the clinical trials used to establish efficacy.
Patient goals, preferences, and choices shape the real-world experience of a new product, and the direct and indirect costs of a treatment to patients and their families significantly affect whether it is adopted widely. Until their value is clearly established, new and costly products should be deployed judiciously and after careful consideration of the goals of treatment, available options, and the unique needs, preferences, and goals of individual patients.
More research is needed to improve how we assess the value of new cancer drug treatments. New clinical efficacy endpoints – both provider- and patient-reported ones – that accurately describe how a patient feels and functions must be developed and should reflect outcomes of value to patients other than survival, particularly in noncurative settings.
Better predictive biomarkers can transform a drug of modest efficacy in an unselected population to one of high efficacy in a biomarker-defined subgroup and thereby contribute to improving the value of a treatment.
Regulatory and policy initiatives such as adaptive licensing, value-based insurance, and indication-specific pricing that affect marketing approval, reimbursement, or price, respectively, based on treatment effectiveness, also deserve careful consideration and further research to determine their effects on aligning cost with benefit while ensuring patient access to potentially life-extending therapies and continued innovation in drug development.
Q: Aside from the policy options, what’s the role of the oncologist in discussing the value of drugs with patients when determining a treatment plan?
A: Since oncologists don’t control drug prices, our role in improving the value of cancer care involves appropriately managing how resources are used and guiding patients during discussions around the right treatment plan for their particular diagnosis, prognosis, and treatment goals.
Adopting and adhering to high-quality oncology clinical pathways is an important way to improve the quality, efficiency, and value of cancer care. High-quality oncology pathways are detailed, evidence-based treatment protocols for delivering cancer care to patients with specific disease types and stages. When properly designed and implemented, oncology pathways serve as an important tool in appropriately managing cancer care resources and improving the quality of care that patients with cancer receive, while also reducing costs.
Dr. Schilsky is the senior vice president and chief medical officer of ASCO. Formerly the chief of hematology/oncology in the department of medicine and deputy director of the University of Chicago Comprehensive Cancer Center, he is a leader in the field of clinical oncology, specializing in new drug development and the treatment of gastrointestinal cancers. Dr. Schilsky reported research funding from several pharmaceutical companies to ASCO for the Targeted Agent and Profiling Utilization Registry (TAPUR) clinical trial. He also reported travel/accommodation/expense support from Varian.
Medical boards change or consider amending mental health-related licensing questions
Delicia M. Haynes, MD, wants the Florida Board of Medicine to take another look at how its licensing applications query physicians about their mental health history. Her state’s board is one of several nationwide that has, in recent years, mulled whether the phrasing of its questions poses an unintended hurdle for physicians who need help for conditions such as depression.

The Federation of State Medical Boards (FSMB) recommends limiting such queries, if they must be asked, to questions about potential current impairment. But the Florida license application takes a more sweeping approach that may discourage physicians from seeking treatment, Dr. Haynes, founder and CEO of Family First Health Center in Daytona Beach, Fla., said in a video. She describes herself as a direct primary care physician.
In the video, which was posted on YouTube in January, 2019, Dr. Haynes discussed her own experience with having been treated for depression and then needing to report that to the state board. Florida’s license application asks if physicians have been treated within the past 5 years for a mental disorder that has impaired the ability to practice medicine. Those who have had such a condition may need to give the board an explanation providing the names of the physicians, therapists, and counselors they have seen, as well as details and dates for the institutions where they received treatment.
State medical boards play a critical role in protecting people from physicians whose current mental health conditions may risk harming patients, Dr. Haynes said. But they must balance that against the consequences of overly intrusive questioning. Florida’s current application wording may deter physicians from seeking care if they develop conditions such as depression, she said.
“It’s the fear of what’s going to happen if I have to check yes,” Dr. Haynes said in her YouTube video. Physicians will wonder how getting treatment could put their license and their employment at risk, she added.
“It’s really important that [state medical boards] are asking the questions that matter and those questions are questions that talk about impairment, and not just having a history,” Dr. Haynes said.
Members of Florida’s board of medicine last year favorably discussed making such a change during a rules/legislative committee meeting, but have not yet implemented it. The board has postponed further discussion of this topic until its December meeting, said Brad Dalton, a spokesman for the Florida Department of Health.
Dr. Haynes said in an email that she will continue to press for changes in her state’s licensing application.
There’s been a wave of reconsideration of these kinds of questions, spurred by FSMB efforts, which included the board’s offering specific advice to state medical boards about questions on licensing applications as part of a set of recommendations on addressing physician wellness and burnout last year in a report.
Boards have the option of omitting or dropping specific inquiries about mental health. But if they choose to retain this question, FSMB recommends using this phrasing: “Are you currently suffering from any condition for which you are not being appropriately treated that impairs your judgment or that would otherwise adversely affect your ability to practice medicine in a competent, ethical and professional manner? (Yes/No)” the report says.
The staff of the Medical Board of California cited FSMB’s recommendation in a January 2019 report about revising the state’s approach to asking physicians about their mental health. In May 2019, the board voted unanimously to revise its questions on physicians’ mental health, narrowing its inquiry on the licensing application to focus on current impairment.
“There are many doctors who do not seek treatment or find it threatening to seek treatment, because they are concerned that they may lose their license,” Peter Yellowlees, MBBS, MD, chief wellness officer and a professor of psychiatry at University of California, Davis, said in a public comment offered at the meeting. “This is ultimately all about patient safety.”
said Joe Knickrehm, a spokesman for the FSMB. Over the past few years, Kentucky, New Hampshire, New Mexico, North Carolina, North Dakota, Ohio, Vermont and Washington have made changes to their licensing questions, he added.
Still, state boards are not moving quickly enough to remove or revise these questions about mental health, said Katherine J. Gold, MD MSW, associate professor in the department of family medicine at University of Michigan, Ann Arbor. She has studied how these queries can deter physicians from seeking treatment for mental illness. A past treatment for postpartum depression may have no bearing on a physician’s practice, yet members of many state medical boards hesitate to alter their approach, she noted.
Groups including the American Medical Association (AMA) and the American Psychiatric Association also have pushed in recent years for changes in state rules about what a physician has to disclose about mental health, with the FSMB taking a lead in these efforts. In many cases, physicians face questions about their mental health that are beyond the limits of standards set by the Americans With Disabilities Act, according to an article published this year in the FSMB’s Journal of Medical Regulation. As of 2017, a review of questions on initial licensure applications for all 50 states and the District of Columbia showed that 32 licensing boards ask questions beyond the limits of ADA standards, the article said.
Consequences of “reporting stable and easily treatable conditions such as anxiety or depression to a state licensing board can range from a physician simply being required to submit a letter from their primary care provider documenting fitness to practice, to a request to appear before the board, to being required to undergo and pay for an examination by a board-appointed physician,” wrote Catherine M. Welcher, an AMA senior policy analyst, and coauthors of the paper, including Humayun J. Chaudhry, DO, FSMB’s CEO.
Nearly 40% of physicians surveyed about medical licensing questions (2,325 of 5,829) indicated they would be reluctant to seek formal medical care for a mental health condition because of potential repercussions, Liselotte N. Dyrbye, MD, and coauthors reported in 2017 in the Mayo Clinic Proceedings.
“You’re sort of stuck. You recognize you have a problem and you want to seek care so that you can get better, but if you do that, you might have to tell the board,” Dr. Dyrbye said in an interview. “You could be mandated potentially to have an outside psychiatric evaluation, to have your personal medical records reviewed by people other than your personal physician.”
Rising levels of burnout among physicians make it critical to remove obstacles to care, said Arthur S. Hengerer, MD, a former chair of the FSMB who has been a leader in efforts to revise medical licensing questions.

“The only thing that really brings joy to medicine are the relationships you develop with patients, the trust that comes from that, and when you can’t develop relationships because you can’t spend the time, because you are clicking and documenting, it hurts,” said Andrew Lamb, MD, who aided in successful efforts to change North Carolina’s licensing questions.
North Carolina approached this issue in steps. In 2017, it removed a question about mental health from its annual physician license renewal form. In 2018, the North Carolina Medical Board voted to drop questions about mental and physical health from physician license applications.
“It’s hoped that removing this question will encourage clinicians to get the help they need, without fear it will compromise their chances to obtain a professional license,” the board said in an annual report.
Other states are retaining these questions, but allowing physicians to check “no” if they are in an approved program for mental health treatment. The North Dakota Board of Medicine took this approach.
“Because our mission is protection of the public, we didn’t feel that we could eliminate that question,” Bonnie Storbakken, JD, executive secretary of the North Dakota Board of Medicine, said in an interview.
The North Dakota board voted in March 2018 to change its question on mental health after the FSMB spurred a national dialogue on this issue, she said. The North Dakota board opted to allow physicians to check ‘no’ on the question about current mental health and or substance issues if they had voluntarily sought assistance from the North Dakota Professional Health Program or a professional health program in another state. The approach preserves anonymity. The question as previously phrased made some physicians feel they had to report their treatment, she said.
In Alabama, physicians also have the option to check “no” on questions about whether they are undergoing mental health treatment if they are participating in a professional assistance program. Mark Jackson, executive director of the Medical Association of Alabama, said he hopes more states give physicians this option for seeking help not only with mental health issues, but with addiction as well.
“You need to get people in and get them help and not jeopardize their career in the process. Society in general has got that problem going on and physicians are not immune from that,” Mr. Jackson said. “Hopefully other states will see that we need to do something to make sure that the physician population is taken care of.”
Pamela Wible, MD, an advocate for physicians’ mental health, did her own review of state board’s licensing questions. In an assessment posted on her website, Dr. Wible, who practices family medicine, gives 13 states an “A” grade for having no mental health questions or one or two straightforward current questions about impairment that do not mention mental health. “Connecticut, Hawaii, Michigan, and New York are the most physician-friendly of all states with no mental health or impairment questions,” Dr. Wible wrote.
The Texas Medical Board earned a “C” grade in Dr. Wible’s survey. In May 2019, that board’s chairman, Sherif Zaafran, MD, wrote about how his organization had taken a new look at the licensing questions, which were last revised in 2014. While the board has not decided on any immediate changes, it is “maintaining an open dialogue to address future concerns,” Dr. Zaafran wrote in the board’s bulletin.
The Texas Medical Board’s physician licensing form, as posted on its website, includes a broadly phrased question on mental health: “Within the past five (5) years, have you been diagnosed with or treated for any: psychotic disorder, delusional disorder, mood disorder, major depression, personality disorder, or any other mental condition which impaired or does impair your behavior, judgment, or ability to function in school or work?”
Physicians who have had treatment for mental health conditions then must submit another form. They may have to provide details on diagnosis, prognosis, and medications prescribed. Compliance requirements include counseling records, contracts with impairment support groups, and records on file with law enforcement agencies and licensing agencies.
State officials are trying to figure out how best to learn of potential impairments that could pose a risk for patients, without scaring physicians away from getting the care they themselves need said Stephen ‘Brint’ Carlton, JD, executive director of the Texas Medical Board.
Delicia M. Haynes, MD, wants the Florida Board of Medicine to take another look at how its licensing applications query physicians about their mental health history. Her state’s board is one of several nationwide that has, in recent years, mulled whether the phrasing of its questions poses an unintended hurdle for physicians who need help for conditions such as depression.
The Federation of State Medical Boards (FSMB) recommends limiting such queries, if they must be asked, to questions about potential current impairment. But the Florida license application takes a more sweeping approach that may discourage physicians from seeking treatment, Dr. Haynes, founder and CEO of Family First Health Center in Daytona Beach, Fla., said in a video. She describes herself as a direct primary care physician.
In the video, which was posted on YouTube in January, 2019, Dr. Haynes discussed her own experience with having been treated for depression and then needing to report that to the state board. Florida’s license application asks if physicians have been treated within the past 5 years for a mental disorder that has impaired the ability to practice medicine. Those who have had such a condition may need to give the board an explanation providing the names of the physicians, therapists, and counselors they have seen, as well as details and dates for the institutions where they received treatment.
State medical boards play a critical role in protecting people from physicians whose current mental health conditions may risk harming patients, Dr. Haynes said. But they must balance that against the consequences of overly intrusive questioning. Florida’s current application wording may deter physicians from seeking care if they develop conditions such as depression, she said.
“It’s the fear of what’s going to happen if I have to check yes,” Dr. Haynes said in her YouTube video. Physicians will wonder how getting treatment could put their license and their employment at risk, she added.
“It’s really important that [state medical boards] are asking the questions that matter and those questions are questions that talk about impairment, and not just having a history,” Dr. Haynes said.
Members of Florida’s board of medicine last year favorably discussed making such a change during a rules/legislative committee meeting, but have not yet implemented it. The board has postponed further discussion of this topic until its December meeting, said Brad Dalton, a spokesman for the Florida Department of Health.
Dr. Haynes said in an email that she will continue to press for changes in her state’s licensing application.
There’s been a wave of reconsideration of these kinds of questions, spurred by FSMB efforts, which included the board’s offering specific advice to state medical boards about questions on licensing applications as part of a set of recommendations on addressing physician wellness and burnout last year in a report.
Boards have the option of omitting or dropping specific inquiries about mental health. But if they choose to retain this question, FSMB recommends using this phrasing: “Are you currently suffering from any condition for which you are not being appropriately treated that impairs your judgment or that would otherwise adversely affect your ability to practice medicine in a competent, ethical and professional manner? (Yes/No)” the report says.
The staff of the Medical Board of California cited FSMB’s recommendation in a January 2019 report about revising the state’s approach to asking physicians about their mental health. In May 2019, the board voted unanimously to revise its questions on physicians’ mental health, narrowing its inquiry on the licensing application to focus on current impairment.
“There are many doctors who do not seek treatment or find it threatening to seek treatment, because they are concerned that they may lose their license,” Peter Yellowlees, MBBS, MD, chief wellness officer and a professor of psychiatry at University of California, Davis, said in a public comment offered at the meeting. “This is ultimately all about patient safety.”
said Joe Knickrehm, a spokesman for the FSMB. Over the past few years, Kentucky, New Hampshire, New Mexico, North Carolina, North Dakota, Ohio, Vermont and Washington have made changes to their licensing questions, he added.
Still, state boards are not moving quickly enough to remove or revise these questions about mental health, said Katherine J. Gold, MD MSW, associate professor in the department of family medicine at University of Michigan, Ann Arbor. She has studied how these queries can deter physicians from seeking treatment for mental illness. A past treatment for postpartum depression may have no bearing on a physician’s practice, yet members of many state medical boards hesitate to alter their approach, she noted.
Groups including the American Medical Association (AMA) and the American Psychiatric Association also have pushed in recent years for changes in state rules about what a physician has to disclose about mental health, with the FSMB taking a lead in these efforts. In many cases, physicians face questions about their mental health that are beyond the limits of standards set by the Americans With Disabilities Act, according to an article published this year in the FSMB’s Journal of Medical Regulation. As of 2017, a review of questions on initial licensure applications for all 50 states and the District of Columbia showed that 32 licensing boards ask questions beyond the limits of ADA standards, the article said.
Consequences of “reporting stable and easily treatable conditions such as anxiety or depression to a state licensing board can range from a physician simply being required to submit a letter from their primary care provider documenting fitness to practice, to a request to appear before the board, to being required to undergo and pay for an examination by a board-appointed physician,” wrote Catherine M. Welcher, an AMA senior policy analyst, and coauthors of the paper, including Humayun J. Chaudhry, DO, FSMB’s CEO.
Nearly 40% of physicians surveyed about medical licensing questions (2,325 of 5,829) indicated they would be reluctant to seek formal medical care for a mental health condition because of potential repercussions, Liselotte N. Dyrbye, MD, and coauthors reported in 2017 in the Mayo Clinic Proceedings.
“You’re sort of stuck. You recognize you have a problem and you want to seek care so that you can get better, but if you do that, you might have to tell the board,” Dr. Dyrbye said in an interview. “You could be mandated potentially to have an outside psychiatric evaluation, to have your personal medical records reviewed by people other than your personal physician.”
Rising levels of burnout among physicians make it critical to remove obstacles to care, said Arthur S. Hengerer, MD, a former chair of the FSMB who has been a leader in efforts to revise medical licensing questions.
“The only thing that really brings joy to medicine are the relationships you develop with patients, the trust that comes from that, and when you can’t develop relationships because you can’t spend the time, because you are clicking and documenting, it hurts,” said Andrew Lamb, MD, who aided in successful efforts to change North Carolina’s licensing questions.
North Carolina approached this issue in steps. In 2017, it removed a question about mental health from its annual physician license renewal form. In 2018, the North Carolina Medical Board voted to drop questions about mental and physical health from physician license applications.
“It’s hoped that removing this question will encourage clinicians to get the help they need, without fear it will compromise their chances to obtain a professional license,” the board said in an annual report.
Other states are retaining these questions, but allowing physicians to check “no” if they are in an approved program for mental health treatment. The North Dakota Board of Medicine took this approach.
“Because our mission is protection of the public, we didn’t feel that we could eliminate that question,” Bonnie Storbakken, JD, executive secretary of the North Dakota Board of Medicine, said in an interview.
The North Dakota board voted in March 2018 to change its question on mental health after the FSMB spurred a national dialogue on this issue, she said. The North Dakota board opted to allow physicians to check ‘no’ on the question about current mental health and or substance issues if they had voluntarily sought assistance from the North Dakota Professional Health Program or a professional health program in another state. The approach preserves anonymity. The question as previously phrased made some physicians feel they had to report their treatment, she said.
In Alabama, physicians also have the option to check “no” on questions about whether they are undergoing mental health treatment if they are participating in a professional assistance program. Mark Jackson, executive director of the Medical Association of Alabama, said he hopes more states give physicians this option for seeking help not only with mental health issues, but with addiction as well.
“You need to get people in and get them help and not jeopardize their career in the process. Society in general has got that problem going on and physicians are not immune from that,” Mr. Jackson said. “Hopefully other states will see that we need to do something to make sure that the physician population is taken care of.”
Pamela Wible, MD, an advocate for physicians’ mental health, did her own review of state board’s licensing questions. In an assessment posted on her website, Dr. Wible, who practices family medicine, gives 13 states an “A” grade for having no mental health questions or one or two straightforward current questions about impairment that do not mention mental health. “Connecticut, Hawaii, Michigan, and New York are the most physician-friendly of all states with no mental health or impairment questions,” Dr. Wible wrote.
The Texas Medical Board earned a “C” grade in Dr. Wible’s survey. In May 2019, that board’s chairman, Sherif Zaafran, MD, wrote about how his organization had taken a new look at the licensing questions, which were last revised in 2014. While the board has not decided on any immediate changes, it is “maintaining an open dialogue to address future concerns,” Dr. Zaafran wrote in the board’s bulletin.
The Texas Medical Board’s physician licensing form, as posted on its website, includes a broadly phrased question on mental health: “Within the past five (5) years, have you been diagnosed with or treated for any: psychotic disorder, delusional disorder, mood disorder, major depression, personality disorder, or any other mental condition which impaired or does impair your behavior, judgment, or ability to function in school or work?”
Physicians who have had treatment for mental health conditions then must submit another form. They may have to provide details on diagnosis, prognosis, and medications prescribed. Compliance requirements include counseling records, contracts with impairment support groups, and records on file with law enforcement agencies and licensing agencies.
State officials are trying to figure out how best to learn of potential impairments that could pose a risk for patients, without scaring physicians away from getting the care they themselves need said Stephen ‘Brint’ Carlton, JD, executive director of the Texas Medical Board.
Delicia M. Haynes, MD, wants the Florida Board of Medicine to take another look at how its licensing applications query physicians about their mental health history. Her state’s board is one of several nationwide that has, in recent years, mulled whether the phrasing of its questions poses an unintended hurdle for physicians who need help for conditions such as depression.
The Federation of State Medical Boards (FSMB) recommends limiting such queries, if they must be asked, to questions about potential current impairment. But the Florida license application takes a more sweeping approach that may discourage physicians from seeking treatment, Dr. Haynes, founder and CEO of Family First Health Center in Daytona Beach, Fla., said in a video. She describes herself as a direct primary care physician.
In the video, which was posted on YouTube in January, 2019, Dr. Haynes discussed her own experience with having been treated for depression and then needing to report that to the state board. Florida’s license application asks if physicians have been treated within the past 5 years for a mental disorder that has impaired the ability to practice medicine. Those who have had such a condition may need to give the board an explanation providing the names of the physicians, therapists, and counselors they have seen, as well as details and dates for the institutions where they received treatment.
State medical boards play a critical role in protecting people from physicians whose current mental health conditions may risk harming patients, Dr. Haynes said. But they must balance that against the consequences of overly intrusive questioning. Florida’s current application wording may deter physicians from seeking care if they develop conditions such as depression, she said.
“It’s the fear of what’s going to happen if I have to check yes,” Dr. Haynes said in her YouTube video. Physicians will wonder how getting treatment could put their license and their employment at risk, she added.
“It’s really important that [state medical boards] are asking the questions that matter and those questions are questions that talk about impairment, and not just having a history,” Dr. Haynes said.
Members of Florida’s board of medicine last year favorably discussed making such a change during a rules/legislative committee meeting, but have not yet implemented it. The board has postponed further discussion of this topic until its December meeting, said Brad Dalton, a spokesman for the Florida Department of Health.
Dr. Haynes said in an email that she will continue to press for changes in her state’s licensing application.
There’s been a wave of reconsideration of these kinds of questions, spurred by FSMB efforts, which included the board’s offering specific advice to state medical boards about questions on licensing applications as part of a set of recommendations on addressing physician wellness and burnout last year in a report.
Boards have the option of omitting or dropping specific inquiries about mental health. But if they choose to retain this question, FSMB recommends using this phrasing: “Are you currently suffering from any condition for which you are not being appropriately treated that impairs your judgment or that would otherwise adversely affect your ability to practice medicine in a competent, ethical and professional manner? (Yes/No)” the report says.
The staff of the Medical Board of California cited FSMB’s recommendation in a January 2019 report about revising the state’s approach to asking physicians about their mental health. In May 2019, the board voted unanimously to revise its questions on physicians’ mental health, narrowing its inquiry on the licensing application to focus on current impairment.
“There are many doctors who do not seek treatment or find it threatening to seek treatment, because they are concerned that they may lose their license,” Peter Yellowlees, MBBS, MD, chief wellness officer and a professor of psychiatry at University of California, Davis, said in a public comment offered at the meeting. “This is ultimately all about patient safety.”
said Joe Knickrehm, a spokesman for the FSMB. Over the past few years, Kentucky, New Hampshire, New Mexico, North Carolina, North Dakota, Ohio, Vermont and Washington have made changes to their licensing questions, he added.
Still, state boards are not moving quickly enough to remove or revise these questions about mental health, said Katherine J. Gold, MD MSW, associate professor in the department of family medicine at University of Michigan, Ann Arbor. She has studied how these queries can deter physicians from seeking treatment for mental illness. A past treatment for postpartum depression may have no bearing on a physician’s practice, yet members of many state medical boards hesitate to alter their approach, she noted.
Groups including the American Medical Association (AMA) and the American Psychiatric Association also have pushed in recent years for changes in state rules about what a physician has to disclose about mental health, with the FSMB taking a lead in these efforts. In many cases, physicians face questions about their mental health that are beyond the limits of standards set by the Americans With Disabilities Act, according to an article published this year in the FSMB’s Journal of Medical Regulation. As of 2017, a review of questions on initial licensure applications for all 50 states and the District of Columbia showed that 32 licensing boards ask questions beyond the limits of ADA standards, the article said.
Consequences of “reporting stable and easily treatable conditions such as anxiety or depression to a state licensing board can range from a physician simply being required to submit a letter from their primary care provider documenting fitness to practice, to a request to appear before the board, to being required to undergo and pay for an examination by a board-appointed physician,” wrote Catherine M. Welcher, an AMA senior policy analyst, and coauthors of the paper, including Humayun J. Chaudhry, DO, FSMB’s CEO.
Nearly 40% of physicians surveyed about medical licensing questions (2,325 of 5,829) indicated they would be reluctant to seek formal medical care for a mental health condition because of potential repercussions, Liselotte N. Dyrbye, MD, and coauthors reported in 2017 in the Mayo Clinic Proceedings.
“You’re sort of stuck. You recognize you have a problem and you want to seek care so that you can get better, but if you do that, you might have to tell the board,” Dr. Dyrbye said in an interview. “You could be mandated potentially to have an outside psychiatric evaluation, to have your personal medical records reviewed by people other than your personal physician.”
Rising levels of burnout among physicians make it critical to remove obstacles to care, said Arthur S. Hengerer, MD, a former chair of the FSMB who has been a leader in efforts to revise medical licensing questions.
“The only thing that really brings joy to medicine are the relationships you develop with patients, the trust that comes from that, and when you can’t develop relationships because you can’t spend the time, because you are clicking and documenting, it hurts,” said Andrew Lamb, MD, who aided in successful efforts to change North Carolina’s licensing questions.
North Carolina approached this issue in steps. In 2017, it removed a question about mental health from its annual physician license renewal form. In 2018, the North Carolina Medical Board voted to drop questions about mental and physical health from physician license applications.
“It’s hoped that removing this question will encourage clinicians to get the help they need, without fear it will compromise their chances to obtain a professional license,” the board said in an annual report.
Other states are retaining these questions, but allowing physicians to check “no” if they are in an approved program for mental health treatment. The North Dakota Board of Medicine took this approach.
“Because our mission is protection of the public, we didn’t feel that we could eliminate that question,” Bonnie Storbakken, JD, executive secretary of the North Dakota Board of Medicine, said in an interview.
The North Dakota board voted in March 2018 to change its question on mental health after the FSMB spurred a national dialogue on this issue, she said. The North Dakota board opted to allow physicians to check ‘no’ on the question about current mental health and or substance issues if they had voluntarily sought assistance from the North Dakota Professional Health Program or a professional health program in another state. The approach preserves anonymity. The question as previously phrased made some physicians feel they had to report their treatment, she said.
In Alabama, physicians also have the option to check “no” on questions about whether they are undergoing mental health treatment if they are participating in a professional assistance program. Mark Jackson, executive director of the Medical Association of Alabama, said he hopes more states give physicians this option for seeking help not only with mental health issues, but with addiction as well.
“You need to get people in and get them help and not jeopardize their career in the process. Society in general has got that problem going on and physicians are not immune from that,” Mr. Jackson said. “Hopefully other states will see that we need to do something to make sure that the physician population is taken care of.”
Pamela Wible, MD, an advocate for physicians’ mental health, did her own review of state board’s licensing questions. In an assessment posted on her website, Dr. Wible, who practices family medicine, gives 13 states an “A” grade for having no mental health questions or one or two straightforward current questions about impairment that do not mention mental health. “Connecticut, Hawaii, Michigan, and New York are the most physician-friendly of all states with no mental health or impairment questions,” Dr. Wible wrote.
The Texas Medical Board earned a “C” grade in Dr. Wible’s survey. In May 2019, that board’s chairman, Sherif Zaafran, MD, wrote about how his organization had taken a new look at the licensing questions, which were last revised in 2014. While the board has not decided on any immediate changes, it is “maintaining an open dialogue to address future concerns,” Dr. Zaafran wrote in the board’s bulletin.
The Texas Medical Board’s physician licensing form, as posted on its website, includes a broadly phrased question on mental health: “Within the past five (5) years, have you been diagnosed with or treated for any: psychotic disorder, delusional disorder, mood disorder, major depression, personality disorder, or any other mental condition which impaired or does impair your behavior, judgment, or ability to function in school or work?”
Physicians who have had treatment for mental health conditions then must submit another form. They may have to provide details on diagnosis, prognosis, and medications prescribed. Compliance requirements include counseling records, contracts with impairment support groups, and records on file with law enforcement agencies and licensing agencies.
State officials are trying to figure out how best to learn of potential impairments that could pose a risk for patients, without scaring physicians away from getting the care they themselves need said Stephen ‘Brint’ Carlton, JD, executive director of the Texas Medical Board.
NICUs admitting more normal-weight newborns
Almost half of the newborns admitted to U.S. neonatal intensive care units in 2017 were of normal birth weight, according to a new report from the Dartmouth Institute for Health Policy & Clinical Practice.
The proportion of NICU admissions involving normal-weight (2,500-3,999 g) newborns increased from 42% in 2007 to 48% in 2017, investigators said in the Dartmouth Atlas of Neonatal Intensive Care. Over that same period, admissions of very-low-birth-weight (500-1,499 g) babies dropped from 16% to 13% of the total.
Those changes were part of a larger, longer-term trend. “The expansion of NICUs and beds in recent decades has been associated with changes in the newborn population receiving NICU care,” the investigators said in the report.
The number of NICU beds increased by 65% from 1995 to 2013, and the number of neonatologists rose by 75% from 1996 to 2013. “At the same time, ” they said in a written statement.
The increases in NICU and neonatologist supply, however, did not always follow the need for such care. Areas of the country with high rates of newborn prematurity, or of risk factors such as low maternal education levels or high cesarean section rates, do not have higher supplies of NICU beds or neonatologists, the researchers noted.
“We should not spare a dollar in providing the best care for newborns. But spending more doesn’t help infants if they could receive the care they need in a maternity unit or home with their mothers. It is very troubling that such a valuable and expensive health care resource is not distributed where it is needed,” said principal author David C. Goodman, MD, of the Dartmouth Institute for Health Policy & Clinical Practice in Lebanon, N.H.
Almost half of the newborns admitted to U.S. neonatal intensive care units in 2017 were of normal birth weight, according to a new report from the Dartmouth Institute for Health Policy & Clinical Practice.
The proportion of NICU admissions involving normal-weight (2,500-3,999 g) newborns increased from 42% in 2007 to 48% in 2017, investigators said in the Dartmouth Atlas of Neonatal Intensive Care. Over that same period, admissions of very-low-birth-weight (500-1,499 g) babies dropped from 16% to 13% of the total.
Those changes were part of a larger, longer-term trend. “The expansion of NICUs and beds in recent decades has been associated with changes in the newborn population receiving NICU care,” the investigators said in the report.
The number of NICU beds increased by 65% from 1995 to 2013, and the number of neonatologists rose by 75% from 1996 to 2013. “At the same time, ” they said in a written statement.
The increases in NICU and neonatologist supply, however, did not always follow the need for such care. Areas of the country with high rates of newborn prematurity, or of risk factors such as low maternal education levels or high cesarean section rates, do not have higher supplies of NICU beds or neonatologists, the researchers noted.
“We should not spare a dollar in providing the best care for newborns. But spending more doesn’t help infants if they could receive the care they need in a maternity unit or home with their mothers. It is very troubling that such a valuable and expensive health care resource is not distributed where it is needed,” said principal author David C. Goodman, MD, of the Dartmouth Institute for Health Policy & Clinical Practice in Lebanon, N.H.
Almost half of the newborns admitted to U.S. neonatal intensive care units in 2017 were of normal birth weight, according to a new report from the Dartmouth Institute for Health Policy & Clinical Practice.
The proportion of NICU admissions involving normal-weight (2,500-3,999 g) newborns increased from 42% in 2007 to 48% in 2017, investigators said in the Dartmouth Atlas of Neonatal Intensive Care. Over that same period, admissions of very-low-birth-weight (500-1,499 g) babies dropped from 16% to 13% of the total.
Those changes were part of a larger, longer-term trend. “The expansion of NICUs and beds in recent decades has been associated with changes in the newborn population receiving NICU care,” the investigators said in the report.
The number of NICU beds increased by 65% from 1995 to 2013, and the number of neonatologists rose by 75% from 1996 to 2013. “At the same time, ” they said in a written statement.
The increases in NICU and neonatologist supply, however, did not always follow the need for such care. Areas of the country with high rates of newborn prematurity, or of risk factors such as low maternal education levels or high cesarean section rates, do not have higher supplies of NICU beds or neonatologists, the researchers noted.
“We should not spare a dollar in providing the best care for newborns. But spending more doesn’t help infants if they could receive the care they need in a maternity unit or home with their mothers. It is very troubling that such a valuable and expensive health care resource is not distributed where it is needed,” said principal author David C. Goodman, MD, of the Dartmouth Institute for Health Policy & Clinical Practice in Lebanon, N.H.
Labeling of medication warnings
Question: Which one of the following statements regarding medication warnings is incorrect?
A. The drug package “insert” or “label” contains, among other things, a drug’s pharmacology, indications, contraindications, risks and warnings.
B. The Physicians’ Desk Reference (PDR) is an annually updated drug compendium, which can be admitted into evidence as a learned treatise.
C. Drug labeling is a dual responsibility of the manufacturer and the Food and Drug Administration.
D. The FDA is solely responsible for a drug’s warnings and sets the absolute standard of care regarding side effects and complications.
E. State law can impose liability for negligent failure to warn even if the FDA has not included the warning in the drug’s label.
Answer: D. In medical products liability, injured plaintiffs frequently claim a failure to warn of known risks. An example is the cardiovascular deaths caused by Vioxx, a nonsteroidal, anti-inflammatory drug that was withdrawn in 2004. Other examples alleging failure to warn are Actos-associated bladder cancer and Baycol-related rhabdomyolysis. At the time of product approval, the FDA sets out the labeling that goes with each drug, and then makes periodic changes to reflect new indications, warnings and risks. The manufacturer has the prime responsibility for submitting all updated information, especially of augmented risks that come with field experience. In 2012, for example, the FDA mandated the revision of the labeling of Lipitor and other statins to warn of the increased risk of diabetes.

The drug manufacturer stands in the unique position as having the most detailed and up-to-date data and bears a serious responsibility to submit its full findings to the FDA, including its request for label change. Litigation over failure to warn of risks frequently turns on whether the drug manufacturer knew or should have known, had failed to inform the FDA, or whether the FDA itself had declined to make the changes, e.g., because of incomplete or premature data. Notwithstanding the FDA’s overarching federal status, a plaintiff may still attempt to use state tort law to hold a manufacturer liable should the federally approved labeling be silent on the matter.
Two U.S. Supreme Court cases sought to clarify the rules under which a drug manufacturer, when sued for failure to warn, may seek protection under its FDA-approved labeling. The first case involved Diana Levine, a Vermont musician and migraine sufferer, who lost her arm after the drug Phenergan, given by intravenous push, accidentally entered an artery and caused gangrene. Although the intravenous use of Phenergan is approved by the FDA and the risk of such use is clearly stated in the drug’s package insert, the lawsuit alleged that under state law, such a warning was inadequate and should have been strengthened to prohibit this mode of administration. A Vermont jury awarded damages of $6.7 million. On appeal, Wyeth, the defendant pharmaceutical company, maintained that its warning was appropriate, as it had been approved by the federal government through the FDA. It further argued that the drug’s package insert could not be unilaterally altered or modified without running afoul of federal regulations.
In a 6-3 decision,1 the U.S. Supreme Court ruled that the manufacturer was in fact at liberty to issue a more stringent warning, and FDA approval does not bar lawsuits. The Court opined that “Federal law does not pre-empt Levine’s claim that Phenergan’s label did not contain an adequate warning about the IV-push method of administration.” Wyeth had argued that it was impossible for the company to provide additional warnings, since it was the FDA that made the sole determination of the nature and scope of a drug’s label. However, the court held that Wyeth never attempted to change the label to warn of the risk and failed to provide “clear” evidence that the FDA would have prevented it from changing its label. Without defining what constituted “clear” evidence, it rejected Wyeth’s broad assertion that unilaterally changing the Phenergan label would have violated federal law, which was based on the fundamental misunderstanding that the FDA, rather than the manufacturer, bears primary responsibility for drug labeling.
In 2019, the landmark case of Merck Sharp & Dohme Corp v. Albrecht et al.2 reached the U.S. Supreme Court. This class-action suit involved more than 500 individuals who took Fosamax, an effective anti-resorptive drug for treating osteoporosis, and suffered atypical femoral fractures between 1999 and 2010. When the FDA first approved of the manufacture and sale of Fosamax in 1995, the Fosamax label did not warn of the then-speculative risk of atypical femoral fractures. But stronger evidence connecting Fosamax to atypical fractures developed after 1995, prompting the FDA to add a warning in 2011. Merck argued that plaintiffs’ state-law failure-to-warn claims should be dismissed as preempted by federal law. It conceded that the FDA regulations would have permitted Merck to try to change the label to add a warning before 2010 but believed the FDA would have rejected that attempt. In particular, it claimed that the FDA’s rejection of Merck’s 2008 attempt to warn of a risk of “stress fractures” showed that the FDA would also have rejected any attempt by Merck to warn of the risk of atypical femoral fractures. In short, Merck was relying on the legal doctrine of “impossibility preemption,” i.e., it was impossible to comply with both state law (adequate label warning of atypical fractures) and federal law (FDA control of warning labels). The plaintiffs’ position was that Merck’s proposed warning to the FDA had minimized the seriousness of the femoral fracture risk, characterizing them only as “stress fractures.”3
The Court’s earlier Levine decision had held that a state-law failure-to-warn claim is preempted where there is “clear” evidence the FDA would not have approved a label change. In the Albrecht decision, which also sided with the plaintiffs, the court indicated that “Clear evidence is evidence that shows the court that the drug manufacturer fully informed the FDA of the justifications for the warning required by state law and that the FDA, in turn, informed the drug manufacturer that the FDA would not approve a change to the drug’s label to include that warning.” The court also held that issues relating to presumption of impossibility are law-based, and thus it remains for the judge, not the jury, to make that determination.
Issuing timely warnings regarding medical products promotes patient safety, and the law appears to place the major onus on the manufacturer. Still, striking the proper balance is important. During oral arguments in Albrecht, Associate Justice Neil Gorsuch is said to have cautioned against “ ... incentives for companies to submit weakly supported label changes to the agency, knowing that when those label changes are rejected the companies will be free of further liability.” And as pointed out in the earlier cited Johnston article: “ ... a system that creates incentives for manufacturers to over-warn physicians and patients could harm patients by listing the important warnings of adverse effects among numerous less important warnings, which may discourage physicians and patients from choosing potentially useful drugs. On the other hand, a shift of responsibility for labeling to the FDA raises questions about whether the agency, which has resources that are dwarfed by the combined resources of industry, is necessarily capable to serve in this role ...”
Finally, this issue is more complex for devices because of the Medical Device Amendments Act of 1976 (MDA), which may preempt state-based lawsuits. In a claim brought after a Medtronic catheter ruptured in a patient’s coronary artery during heart surgery, the plaintiff alleged that the device was designed, labeled, and manufactured in a manner that violated New York common law. The case was appealed to the U.S. Supreme Court. The court held that the MDA preempted petitioner’s common-law claims challenging the safety or effectiveness of a medical device marketed in a form that received premarket approval from the FDA.4 The court ruled that MDA created a scheme of federal safety oversight for medical devices while sweeping back state oversight schemes.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. This article is meant to be educational and does not constitute medical, ethical, or legal advice. For additional information, readers may contact the author at siang@hawaii.edu.
References
1. Wyeth v. Levine, 555 U.S. 2 (2009).
2. Merck, Sharp & Dohme Corp. v. Albrecht et al., 587 U. S. ____ (2019).
3. Johnston MC et al., A new Supreme Court ruling on drug liability. JAMA 2019;322(7):607-8.
4. Riegel v. Medtronic, 128 S. Ct. 999 (2008).
Question: Which one of the following statements regarding medication warnings is incorrect?
A. The drug package “insert” or “label” contains, among other things, a drug’s pharmacology, indications, contraindications, risks and warnings.
B. The Physicians’ Desk Reference (PDR) is an annually updated drug compendium, which can be admitted into evidence as a learned treatise.
C. Drug labeling is a dual responsibility of the manufacturer and the Food and Drug Administration.
D. The FDA is solely responsible for a drug’s warnings and sets the absolute standard of care regarding side effects and complications.
E. State law can impose liability for negligent failure to warn even if the FDA has not included the warning in the drug’s label.
Answer: D. In medical products liability, injured plaintiffs frequently claim a failure to warn of known risks. An example is the cardiovascular deaths caused by Vioxx, a nonsteroidal, anti-inflammatory drug that was withdrawn in 2004. Other examples alleging failure to warn are Actos-associated bladder cancer and Baycol-related rhabdomyolysis. At the time of product approval, the FDA sets out the labeling that goes with each drug, and then makes periodic changes to reflect new indications, warnings and risks. The manufacturer has the prime responsibility for submitting all updated information, especially of augmented risks that come with field experience. In 2012, for example, the FDA mandated the revision of the labeling of Lipitor and other statins to warn of the increased risk of diabetes.
The drug manufacturer stands in the unique position as having the most detailed and up-to-date data and bears a serious responsibility to submit its full findings to the FDA, including its request for label change. Litigation over failure to warn of risks frequently turns on whether the drug manufacturer knew or should have known, had failed to inform the FDA, or whether the FDA itself had declined to make the changes, e.g., because of incomplete or premature data. Notwithstanding the FDA’s overarching federal status, a plaintiff may still attempt to use state tort law to hold a manufacturer liable should the federally approved labeling be silent on the matter.
Two U.S. Supreme Court cases sought to clarify the rules under which a drug manufacturer, when sued for failure to warn, may seek protection under its FDA-approved labeling. The first case involved Diana Levine, a Vermont musician and migraine sufferer, who lost her arm after the drug Phenergan, given by intravenous push, accidentally entered an artery and caused gangrene. Although the intravenous use of Phenergan is approved by the FDA and the risk of such use is clearly stated in the drug’s package insert, the lawsuit alleged that under state law, such a warning was inadequate and should have been strengthened to prohibit this mode of administration. A Vermont jury awarded damages of $6.7 million. On appeal, Wyeth, the defendant pharmaceutical company, maintained that its warning was appropriate, as it had been approved by the federal government through the FDA. It further argued that the drug’s package insert could not be unilaterally altered or modified without running afoul of federal regulations.
In a 6-3 decision,1 the U.S. Supreme Court ruled that the manufacturer was in fact at liberty to issue a more stringent warning, and FDA approval does not bar lawsuits. The Court opined that “Federal law does not pre-empt Levine’s claim that Phenergan’s label did not contain an adequate warning about the IV-push method of administration.” Wyeth had argued that it was impossible for the company to provide additional warnings, since it was the FDA that made the sole determination of the nature and scope of a drug’s label. However, the court held that Wyeth never attempted to change the label to warn of the risk and failed to provide “clear” evidence that the FDA would have prevented it from changing its label. Without defining what constituted “clear” evidence, it rejected Wyeth’s broad assertion that unilaterally changing the Phenergan label would have violated federal law, which was based on the fundamental misunderstanding that the FDA, rather than the manufacturer, bears primary responsibility for drug labeling.
In 2019, the landmark case of Merck Sharp & Dohme Corp v. Albrecht et al.2 reached the U.S. Supreme Court. This class-action suit involved more than 500 individuals who took Fosamax, an effective anti-resorptive drug for treating osteoporosis, and suffered atypical femoral fractures between 1999 and 2010. When the FDA first approved of the manufacture and sale of Fosamax in 1995, the Fosamax label did not warn of the then-speculative risk of atypical femoral fractures. But stronger evidence connecting Fosamax to atypical fractures developed after 1995, prompting the FDA to add a warning in 2011. Merck argued that plaintiffs’ state-law failure-to-warn claims should be dismissed as preempted by federal law. It conceded that the FDA regulations would have permitted Merck to try to change the label to add a warning before 2010 but believed the FDA would have rejected that attempt. In particular, it claimed that the FDA’s rejection of Merck’s 2008 attempt to warn of a risk of “stress fractures” showed that the FDA would also have rejected any attempt by Merck to warn of the risk of atypical femoral fractures. In short, Merck was relying on the legal doctrine of “impossibility preemption,” i.e., it was impossible to comply with both state law (adequate label warning of atypical fractures) and federal law (FDA control of warning labels). The plaintiffs’ position was that Merck’s proposed warning to the FDA had minimized the seriousness of the femoral fracture risk, characterizing them only as “stress fractures.”3
The Court’s earlier Levine decision had held that a state-law failure-to-warn claim is preempted where there is “clear” evidence the FDA would not have approved a label change. In the Albrecht decision, which also sided with the plaintiffs, the court indicated that “Clear evidence is evidence that shows the court that the drug manufacturer fully informed the FDA of the justifications for the warning required by state law and that the FDA, in turn, informed the drug manufacturer that the FDA would not approve a change to the drug’s label to include that warning.” The court also held that issues relating to presumption of impossibility are law-based, and thus it remains for the judge, not the jury, to make that determination.
Issuing timely warnings regarding medical products promotes patient safety, and the law appears to place the major onus on the manufacturer. Still, striking the proper balance is important. During oral arguments in Albrecht, Associate Justice Neil Gorsuch is said to have cautioned against “ ... incentives for companies to submit weakly supported label changes to the agency, knowing that when those label changes are rejected the companies will be free of further liability.” And as pointed out in the earlier cited Johnston article: “ ... a system that creates incentives for manufacturers to over-warn physicians and patients could harm patients by listing the important warnings of adverse effects among numerous less important warnings, which may discourage physicians and patients from choosing potentially useful drugs. On the other hand, a shift of responsibility for labeling to the FDA raises questions about whether the agency, which has resources that are dwarfed by the combined resources of industry, is necessarily capable to serve in this role ...”
Finally, this issue is more complex for devices because of the Medical Device Amendments Act of 1976 (MDA), which may preempt state-based lawsuits. In a claim brought after a Medtronic catheter ruptured in a patient’s coronary artery during heart surgery, the plaintiff alleged that the device was designed, labeled, and manufactured in a manner that violated New York common law. The case was appealed to the U.S. Supreme Court. The court held that the MDA preempted petitioner’s common-law claims challenging the safety or effectiveness of a medical device marketed in a form that received premarket approval from the FDA.4 The court ruled that MDA created a scheme of federal safety oversight for medical devices while sweeping back state oversight schemes.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. This article is meant to be educational and does not constitute medical, ethical, or legal advice. For additional information, readers may contact the author at siang@hawaii.edu.
References
1. Wyeth v. Levine, 555 U.S. 2 (2009).
2. Merck, Sharp & Dohme Corp. v. Albrecht et al., 587 U. S. ____ (2019).
3. Johnston MC et al., A new Supreme Court ruling on drug liability. JAMA 2019;322(7):607-8.
4. Riegel v. Medtronic, 128 S. Ct. 999 (2008).
Question: Which one of the following statements regarding medication warnings is incorrect?
A. The drug package “insert” or “label” contains, among other things, a drug’s pharmacology, indications, contraindications, risks and warnings.
B. The Physicians’ Desk Reference (PDR) is an annually updated drug compendium, which can be admitted into evidence as a learned treatise.
C. Drug labeling is a dual responsibility of the manufacturer and the Food and Drug Administration.
D. The FDA is solely responsible for a drug’s warnings and sets the absolute standard of care regarding side effects and complications.
E. State law can impose liability for negligent failure to warn even if the FDA has not included the warning in the drug’s label.
Answer: D. In medical products liability, injured plaintiffs frequently claim a failure to warn of known risks. An example is the cardiovascular deaths caused by Vioxx, a nonsteroidal, anti-inflammatory drug that was withdrawn in 2004. Other examples alleging failure to warn are Actos-associated bladder cancer and Baycol-related rhabdomyolysis. At the time of product approval, the FDA sets out the labeling that goes with each drug, and then makes periodic changes to reflect new indications, warnings and risks. The manufacturer has the prime responsibility for submitting all updated information, especially of augmented risks that come with field experience. In 2012, for example, the FDA mandated the revision of the labeling of Lipitor and other statins to warn of the increased risk of diabetes.
The drug manufacturer stands in the unique position as having the most detailed and up-to-date data and bears a serious responsibility to submit its full findings to the FDA, including its request for label change. Litigation over failure to warn of risks frequently turns on whether the drug manufacturer knew or should have known, had failed to inform the FDA, or whether the FDA itself had declined to make the changes, e.g., because of incomplete or premature data. Notwithstanding the FDA’s overarching federal status, a plaintiff may still attempt to use state tort law to hold a manufacturer liable should the federally approved labeling be silent on the matter.
Two U.S. Supreme Court cases sought to clarify the rules under which a drug manufacturer, when sued for failure to warn, may seek protection under its FDA-approved labeling. The first case involved Diana Levine, a Vermont musician and migraine sufferer, who lost her arm after the drug Phenergan, given by intravenous push, accidentally entered an artery and caused gangrene. Although the intravenous use of Phenergan is approved by the FDA and the risk of such use is clearly stated in the drug’s package insert, the lawsuit alleged that under state law, such a warning was inadequate and should have been strengthened to prohibit this mode of administration. A Vermont jury awarded damages of $6.7 million. On appeal, Wyeth, the defendant pharmaceutical company, maintained that its warning was appropriate, as it had been approved by the federal government through the FDA. It further argued that the drug’s package insert could not be unilaterally altered or modified without running afoul of federal regulations.
In a 6-3 decision,1 the U.S. Supreme Court ruled that the manufacturer was in fact at liberty to issue a more stringent warning, and FDA approval does not bar lawsuits. The Court opined that “Federal law does not pre-empt Levine’s claim that Phenergan’s label did not contain an adequate warning about the IV-push method of administration.” Wyeth had argued that it was impossible for the company to provide additional warnings, since it was the FDA that made the sole determination of the nature and scope of a drug’s label. However, the court held that Wyeth never attempted to change the label to warn of the risk and failed to provide “clear” evidence that the FDA would have prevented it from changing its label. Without defining what constituted “clear” evidence, it rejected Wyeth’s broad assertion that unilaterally changing the Phenergan label would have violated federal law, which was based on the fundamental misunderstanding that the FDA, rather than the manufacturer, bears primary responsibility for drug labeling.
In 2019, the landmark case of Merck Sharp & Dohme Corp v. Albrecht et al.2 reached the U.S. Supreme Court. This class-action suit involved more than 500 individuals who took Fosamax, an effective anti-resorptive drug for treating osteoporosis, and suffered atypical femoral fractures between 1999 and 2010. When the FDA first approved of the manufacture and sale of Fosamax in 1995, the Fosamax label did not warn of the then-speculative risk of atypical femoral fractures. But stronger evidence connecting Fosamax to atypical fractures developed after 1995, prompting the FDA to add a warning in 2011. Merck argued that plaintiffs’ state-law failure-to-warn claims should be dismissed as preempted by federal law. It conceded that the FDA regulations would have permitted Merck to try to change the label to add a warning before 2010 but believed the FDA would have rejected that attempt. In particular, it claimed that the FDA’s rejection of Merck’s 2008 attempt to warn of a risk of “stress fractures” showed that the FDA would also have rejected any attempt by Merck to warn of the risk of atypical femoral fractures. In short, Merck was relying on the legal doctrine of “impossibility preemption,” i.e., it was impossible to comply with both state law (adequate label warning of atypical fractures) and federal law (FDA control of warning labels). The plaintiffs’ position was that Merck’s proposed warning to the FDA had minimized the seriousness of the femoral fracture risk, characterizing them only as “stress fractures.”3
The Court’s earlier Levine decision had held that a state-law failure-to-warn claim is preempted where there is “clear” evidence the FDA would not have approved a label change. In the Albrecht decision, which also sided with the plaintiffs, the court indicated that “Clear evidence is evidence that shows the court that the drug manufacturer fully informed the FDA of the justifications for the warning required by state law and that the FDA, in turn, informed the drug manufacturer that the FDA would not approve a change to the drug’s label to include that warning.” The court also held that issues relating to presumption of impossibility are law-based, and thus it remains for the judge, not the jury, to make that determination.
Issuing timely warnings regarding medical products promotes patient safety, and the law appears to place the major onus on the manufacturer. Still, striking the proper balance is important. During oral arguments in Albrecht, Associate Justice Neil Gorsuch is said to have cautioned against “ ... incentives for companies to submit weakly supported label changes to the agency, knowing that when those label changes are rejected the companies will be free of further liability.” And as pointed out in the earlier cited Johnston article: “ ... a system that creates incentives for manufacturers to over-warn physicians and patients could harm patients by listing the important warnings of adverse effects among numerous less important warnings, which may discourage physicians and patients from choosing potentially useful drugs. On the other hand, a shift of responsibility for labeling to the FDA raises questions about whether the agency, which has resources that are dwarfed by the combined resources of industry, is necessarily capable to serve in this role ...”
Finally, this issue is more complex for devices because of the Medical Device Amendments Act of 1976 (MDA), which may preempt state-based lawsuits. In a claim brought after a Medtronic catheter ruptured in a patient’s coronary artery during heart surgery, the plaintiff alleged that the device was designed, labeled, and manufactured in a manner that violated New York common law. The case was appealed to the U.S. Supreme Court. The court held that the MDA preempted petitioner’s common-law claims challenging the safety or effectiveness of a medical device marketed in a form that received premarket approval from the FDA.4 The court ruled that MDA created a scheme of federal safety oversight for medical devices while sweeping back state oversight schemes.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. This article is meant to be educational and does not constitute medical, ethical, or legal advice. For additional information, readers may contact the author at siang@hawaii.edu.
References
1. Wyeth v. Levine, 555 U.S. 2 (2009).
2. Merck, Sharp & Dohme Corp. v. Albrecht et al., 587 U. S. ____ (2019).
3. Johnston MC et al., A new Supreme Court ruling on drug liability. JAMA 2019;322(7):607-8.
4. Riegel v. Medtronic, 128 S. Ct. 999 (2008).
Judge dismisses doctors’ lawsuit against ABIM
A district court has dismissed a lawsuit levied by a group of physicians against the American Board of Internal Medicine (ABIM) over its maintenance of certification (MOC) program, calling the legal challenge “flawed.”

In a Sept. 26 decision, U.S. District Court Judge for the Eastern District of Pennsylvania Robert F. Kelly Sr. said the plaintiffs failed to demonstrate sufficient evidence for their antitrust and unjust enrichment claims against ABIM. The doctors also did not establish any showing of anticompetitive conduct by ABIM to support a monopolization claim, the judge ruled.
“We disagree with plaintiffs and find that ABIM’s initial certification and MOC products are part of a single product and do not occupy distinct markets,” Judge Kelly wrote in his decision. “Not only are we unconvinced by plaintiffs’ arguments, we find that plaintiffs’ entire framing of the ABIM certification to be flawed. In essence, plaintiffs are arguing that, in order to purchase ABIM’s initial certification, internists are forced to purchase MOC products as well. However, this is not the case. ... Nowhere in the amended complaint do plaintiffs allege that they were forced to buy MOC products in order to purchase the initial certification.”
The judge dismissed the suit, but allowed the plaintiffs 14 days to submit an amended complaint reoutlining their claims of illegal monopolization and racketeering against the board. If the amended complaint passes legal muster, the judge could revive those claims.
ABIM President Richard J. Baron, MD, expressed satisfaction that the court granted the board’s motion to dismiss the case for failure to state a valid claim.
“ABIM is pleased that the United States District Court for the Eastern District of Pennsylvania dismissed in its entirety a lawsuit that alleged physicians were harmed by the requirements for maintaining ABIM board certification,” Dr. Baron said in a statement.
C. Philip Curley, a Chicago-based attorney for the physician plaintiffs, said the case is far from over.
“The four internists who brought the lawsuit were invited to file amended claims, which is certainly being considered,” Mr. Curley said in an interview. “If necessary, all available appeals will also be pursued to the fullest. No one was under the impression that the fight to bring MOC to an end would be quick or easy.”
The original lawsuit, filed Dec. 6, 2018, in a Pennsylvania district court, claims that ABIM is charging inflated monopoly prices for maintaining certification, that the organization is forcing physicians to purchase MOC, and that ABIM is inducing employers and others to require ABIM certification. On Jan. 23 of this year the legal challenge was amended to include racketeering and unjust enrichment claims.
The four plaintiff-physicians want the court to find ABIM in violation of federal antitrust law and to bar the board from continuing its MOC process. The suit is filed as a class action on behalf of all internists and subspecialists required by ABIM to purchase MOC to maintain their ABIM certifications.
Two other lawsuits challenging MOC, one against the American Board of Psychiatry and Neurology and another against the American Board of Radiology, are ongoing. A fourth lawsuit against the American Board of Medical Specialties, the American Board of Emergency Medicine, and the American Board of Anesthesiology was filed in February.
Chicago-based cardiologist Wes Fisher, MD, and fellow physicians with the Practicing Physicians of America are funding the plaintiffs’ legal efforts through a fundraising campaign that has raised more than $300,000.
In an interview, Dr. Fisher called the legal fight against ABIM “a David versus Goliath effort” and said the battle will continue.
“The ABIM may have won this first round, but ... they have only dodged the antitrust tying claim and unjust enrichment claims,” Dr. Fisher said. “The monopoly claim and racketeering claims are still very much open. Plaintiffs have 14 days to amend their compliant.”
A district court has dismissed a lawsuit levied by a group of physicians against the American Board of Internal Medicine (ABIM) over its maintenance of certification (MOC) program, calling the legal challenge “flawed.”
In a Sept. 26 decision, U.S. District Court Judge for the Eastern District of Pennsylvania Robert F. Kelly Sr. said the plaintiffs failed to demonstrate sufficient evidence for their antitrust and unjust enrichment claims against ABIM. The doctors also did not establish any showing of anticompetitive conduct by ABIM to support a monopolization claim, the judge ruled.
“We disagree with plaintiffs and find that ABIM’s initial certification and MOC products are part of a single product and do not occupy distinct markets,” Judge Kelly wrote in his decision. “Not only are we unconvinced by plaintiffs’ arguments, we find that plaintiffs’ entire framing of the ABIM certification to be flawed. In essence, plaintiffs are arguing that, in order to purchase ABIM’s initial certification, internists are forced to purchase MOC products as well. However, this is not the case. ... Nowhere in the amended complaint do plaintiffs allege that they were forced to buy MOC products in order to purchase the initial certification.”
The judge dismissed the suit, but allowed the plaintiffs 14 days to submit an amended complaint reoutlining their claims of illegal monopolization and racketeering against the board. If the amended complaint passes legal muster, the judge could revive those claims.
ABIM President Richard J. Baron, MD, expressed satisfaction that the court granted the board’s motion to dismiss the case for failure to state a valid claim.
“ABIM is pleased that the United States District Court for the Eastern District of Pennsylvania dismissed in its entirety a lawsuit that alleged physicians were harmed by the requirements for maintaining ABIM board certification,” Dr. Baron said in a statement.
C. Philip Curley, a Chicago-based attorney for the physician plaintiffs, said the case is far from over.
“The four internists who brought the lawsuit were invited to file amended claims, which is certainly being considered,” Mr. Curley said in an interview. “If necessary, all available appeals will also be pursued to the fullest. No one was under the impression that the fight to bring MOC to an end would be quick or easy.”
The original lawsuit, filed Dec. 6, 2018, in a Pennsylvania district court, claims that ABIM is charging inflated monopoly prices for maintaining certification, that the organization is forcing physicians to purchase MOC, and that ABIM is inducing employers and others to require ABIM certification. On Jan. 23 of this year the legal challenge was amended to include racketeering and unjust enrichment claims.
The four plaintiff-physicians want the court to find ABIM in violation of federal antitrust law and to bar the board from continuing its MOC process. The suit is filed as a class action on behalf of all internists and subspecialists required by ABIM to purchase MOC to maintain their ABIM certifications.
Two other lawsuits challenging MOC, one against the American Board of Psychiatry and Neurology and another against the American Board of Radiology, are ongoing. A fourth lawsuit against the American Board of Medical Specialties, the American Board of Emergency Medicine, and the American Board of Anesthesiology was filed in February.
Chicago-based cardiologist Wes Fisher, MD, and fellow physicians with the Practicing Physicians of America are funding the plaintiffs’ legal efforts through a fundraising campaign that has raised more than $300,000.
In an interview, Dr. Fisher called the legal fight against ABIM “a David versus Goliath effort” and said the battle will continue.
“The ABIM may have won this first round, but ... they have only dodged the antitrust tying claim and unjust enrichment claims,” Dr. Fisher said. “The monopoly claim and racketeering claims are still very much open. Plaintiffs have 14 days to amend their compliant.”
A district court has dismissed a lawsuit levied by a group of physicians against the American Board of Internal Medicine (ABIM) over its maintenance of certification (MOC) program, calling the legal challenge “flawed.”
In a Sept. 26 decision, U.S. District Court Judge for the Eastern District of Pennsylvania Robert F. Kelly Sr. said the plaintiffs failed to demonstrate sufficient evidence for their antitrust and unjust enrichment claims against ABIM. The doctors also did not establish any showing of anticompetitive conduct by ABIM to support a monopolization claim, the judge ruled.
“We disagree with plaintiffs and find that ABIM’s initial certification and MOC products are part of a single product and do not occupy distinct markets,” Judge Kelly wrote in his decision. “Not only are we unconvinced by plaintiffs’ arguments, we find that plaintiffs’ entire framing of the ABIM certification to be flawed. In essence, plaintiffs are arguing that, in order to purchase ABIM’s initial certification, internists are forced to purchase MOC products as well. However, this is not the case. ... Nowhere in the amended complaint do plaintiffs allege that they were forced to buy MOC products in order to purchase the initial certification.”
The judge dismissed the suit, but allowed the plaintiffs 14 days to submit an amended complaint reoutlining their claims of illegal monopolization and racketeering against the board. If the amended complaint passes legal muster, the judge could revive those claims.
ABIM President Richard J. Baron, MD, expressed satisfaction that the court granted the board’s motion to dismiss the case for failure to state a valid claim.
“ABIM is pleased that the United States District Court for the Eastern District of Pennsylvania dismissed in its entirety a lawsuit that alleged physicians were harmed by the requirements for maintaining ABIM board certification,” Dr. Baron said in a statement.
C. Philip Curley, a Chicago-based attorney for the physician plaintiffs, said the case is far from over.
“The four internists who brought the lawsuit were invited to file amended claims, which is certainly being considered,” Mr. Curley said in an interview. “If necessary, all available appeals will also be pursued to the fullest. No one was under the impression that the fight to bring MOC to an end would be quick or easy.”
The original lawsuit, filed Dec. 6, 2018, in a Pennsylvania district court, claims that ABIM is charging inflated monopoly prices for maintaining certification, that the organization is forcing physicians to purchase MOC, and that ABIM is inducing employers and others to require ABIM certification. On Jan. 23 of this year the legal challenge was amended to include racketeering and unjust enrichment claims.
The four plaintiff-physicians want the court to find ABIM in violation of federal antitrust law and to bar the board from continuing its MOC process. The suit is filed as a class action on behalf of all internists and subspecialists required by ABIM to purchase MOC to maintain their ABIM certifications.
Two other lawsuits challenging MOC, one against the American Board of Psychiatry and Neurology and another against the American Board of Radiology, are ongoing. A fourth lawsuit against the American Board of Medical Specialties, the American Board of Emergency Medicine, and the American Board of Anesthesiology was filed in February.
Chicago-based cardiologist Wes Fisher, MD, and fellow physicians with the Practicing Physicians of America are funding the plaintiffs’ legal efforts through a fundraising campaign that has raised more than $300,000.
In an interview, Dr. Fisher called the legal fight against ABIM “a David versus Goliath effort” and said the battle will continue.
“The ABIM may have won this first round, but ... they have only dodged the antitrust tying claim and unjust enrichment claims,” Dr. Fisher said. “The monopoly claim and racketeering claims are still very much open. Plaintiffs have 14 days to amend their compliant.”
Patient-reported outcomes are here to stay
WASHINGTON – The federal official who helps oversee Medicare’s use of quality measures predicted a continued emphasis on patient-reported outcomes in the assessments of physician performance.
Reena Duseja, MD, chief medical officer for quality measurement at the Centers for Medicare & Medicaid Services, said she has seen “more emphasis” in her 2 years with the agency in collecting outcome measures, including ones reported by patients. In doing this, CMS officials are also looking to identify the core elements that willl be part of patient-reported outcomes (PROs).
“We really have to get better at standardization,” Dr. Duseja said during a policy summit sponsored by the National Comprehensive Cancer Network (NCCN). “There is room for improvement there. We’re continuing to think of ways that we can support that.”
She also said the CMS is working, in general, to try to give physicians feedback sooner on how they are faring on measurements.
“The commitment of our agency is trying to think about how we collect data in a way that shortens the cycle of measure development” and speeds the delivery of this data back to providers, Dr. Duseja said.
Her fellow panelists discussed the difficulties in designing PRO measures, including the need to account for special challenges for patients living in or near poverty. Avoiding emergency department visits and hospitalizations, for example, may be a key priority for people who are paid hourly wages, said Kashyap Patel, MD, managing partner of Carolina Blood and Cancer Care in Rock Hill, S.C. These patients will not only face the inconvenience and cost of a hospital stay, but will also lose wages from missed work. He urged policymakers to take these factors into consideration in designing quality measures, and not to forget that “there is a human being behind this.”
Ronald S. Walters, MD, associate head of the Institute for Cancer Care Innovation at the University of Texas MD Anderson Cancer Center, and chair of NCCN’s board of directors, cautioned against continued attempts to devise a “Nirvana” list of outcome measures that can be universally applied. Instead, payers may be better off with a “mix and match” approach. Certain measures may be used across the board, such as pain and quality of life metrics, while other measures could be more tailored.
Dr. Walters also called out a missed opportunity to tie PROs to Medicare payment in the area of chimeric antigen receptor (CAR) T-cell therapy.
In 2018, the CMS indicated it was considering the use of PROs in connection with CAR T-cell payment. The CMS asked its Medicare Evidence Development and Coverage Advisory Committee (MEDCAC) to consider the role of PROs in connection with payment for CAR T-cell therapy. At an August 2018 meeting, MEDCAC panelists generally expressed confidence in PROs in a series of votes about the use of this approach to quality measurement in cancer trials.
But drugmakers and physician groups raised strong objections at the MEDCAC meeting. In its national coverage decision on CAR T, issued in August 2019, the CMS said it had received many comments on PROs “ranging from support of their collection to recommendations for additional assessment tools to request to remove PRO requirements.” The CMS opted at this time to encourage participation in the Center for International Blood and Marrow Transplantation Research (CIBMTR) database “that currently collects health outcomes (and aims to collect patient reported outcomes in the future) on patients who have received CAR T-cell treatments.”
For Dr. Walters, this setback for the use of PROs in CAR T therapy payment is telling, as the treatment is known to produce serious side effects and is administered in well-controlled circumstances.
“If you can’t organize collecting patient-reported outcomes after CAR T cell, that kind of tells you the state of where we are on collecting them on everybody,” Dr. Walters said.
WASHINGTON – The federal official who helps oversee Medicare’s use of quality measures predicted a continued emphasis on patient-reported outcomes in the assessments of physician performance.
Reena Duseja, MD, chief medical officer for quality measurement at the Centers for Medicare & Medicaid Services, said she has seen “more emphasis” in her 2 years with the agency in collecting outcome measures, including ones reported by patients. In doing this, CMS officials are also looking to identify the core elements that willl be part of patient-reported outcomes (PROs).
“We really have to get better at standardization,” Dr. Duseja said during a policy summit sponsored by the National Comprehensive Cancer Network (NCCN). “There is room for improvement there. We’re continuing to think of ways that we can support that.”
She also said the CMS is working, in general, to try to give physicians feedback sooner on how they are faring on measurements.
“The commitment of our agency is trying to think about how we collect data in a way that shortens the cycle of measure development” and speeds the delivery of this data back to providers, Dr. Duseja said.
Her fellow panelists discussed the difficulties in designing PRO measures, including the need to account for special challenges for patients living in or near poverty. Avoiding emergency department visits and hospitalizations, for example, may be a key priority for people who are paid hourly wages, said Kashyap Patel, MD, managing partner of Carolina Blood and Cancer Care in Rock Hill, S.C. These patients will not only face the inconvenience and cost of a hospital stay, but will also lose wages from missed work. He urged policymakers to take these factors into consideration in designing quality measures, and not to forget that “there is a human being behind this.”
Ronald S. Walters, MD, associate head of the Institute for Cancer Care Innovation at the University of Texas MD Anderson Cancer Center, and chair of NCCN’s board of directors, cautioned against continued attempts to devise a “Nirvana” list of outcome measures that can be universally applied. Instead, payers may be better off with a “mix and match” approach. Certain measures may be used across the board, such as pain and quality of life metrics, while other measures could be more tailored.
Dr. Walters also called out a missed opportunity to tie PROs to Medicare payment in the area of chimeric antigen receptor (CAR) T-cell therapy.
In 2018, the CMS indicated it was considering the use of PROs in connection with CAR T-cell payment. The CMS asked its Medicare Evidence Development and Coverage Advisory Committee (MEDCAC) to consider the role of PROs in connection with payment for CAR T-cell therapy. At an August 2018 meeting, MEDCAC panelists generally expressed confidence in PROs in a series of votes about the use of this approach to quality measurement in cancer trials.
But drugmakers and physician groups raised strong objections at the MEDCAC meeting. In its national coverage decision on CAR T, issued in August 2019, the CMS said it had received many comments on PROs “ranging from support of their collection to recommendations for additional assessment tools to request to remove PRO requirements.” The CMS opted at this time to encourage participation in the Center for International Blood and Marrow Transplantation Research (CIBMTR) database “that currently collects health outcomes (and aims to collect patient reported outcomes in the future) on patients who have received CAR T-cell treatments.”
For Dr. Walters, this setback for the use of PROs in CAR T therapy payment is telling, as the treatment is known to produce serious side effects and is administered in well-controlled circumstances.
“If you can’t organize collecting patient-reported outcomes after CAR T cell, that kind of tells you the state of where we are on collecting them on everybody,” Dr. Walters said.
WASHINGTON – The federal official who helps oversee Medicare’s use of quality measures predicted a continued emphasis on patient-reported outcomes in the assessments of physician performance.
Reena Duseja, MD, chief medical officer for quality measurement at the Centers for Medicare & Medicaid Services, said she has seen “more emphasis” in her 2 years with the agency in collecting outcome measures, including ones reported by patients. In doing this, CMS officials are also looking to identify the core elements that willl be part of patient-reported outcomes (PROs).
“We really have to get better at standardization,” Dr. Duseja said during a policy summit sponsored by the National Comprehensive Cancer Network (NCCN). “There is room for improvement there. We’re continuing to think of ways that we can support that.”
She also said the CMS is working, in general, to try to give physicians feedback sooner on how they are faring on measurements.
“The commitment of our agency is trying to think about how we collect data in a way that shortens the cycle of measure development” and speeds the delivery of this data back to providers, Dr. Duseja said.
Her fellow panelists discussed the difficulties in designing PRO measures, including the need to account for special challenges for patients living in or near poverty. Avoiding emergency department visits and hospitalizations, for example, may be a key priority for people who are paid hourly wages, said Kashyap Patel, MD, managing partner of Carolina Blood and Cancer Care in Rock Hill, S.C. These patients will not only face the inconvenience and cost of a hospital stay, but will also lose wages from missed work. He urged policymakers to take these factors into consideration in designing quality measures, and not to forget that “there is a human being behind this.”
Ronald S. Walters, MD, associate head of the Institute for Cancer Care Innovation at the University of Texas MD Anderson Cancer Center, and chair of NCCN’s board of directors, cautioned against continued attempts to devise a “Nirvana” list of outcome measures that can be universally applied. Instead, payers may be better off with a “mix and match” approach. Certain measures may be used across the board, such as pain and quality of life metrics, while other measures could be more tailored.
Dr. Walters also called out a missed opportunity to tie PROs to Medicare payment in the area of chimeric antigen receptor (CAR) T-cell therapy.
In 2018, the CMS indicated it was considering the use of PROs in connection with CAR T-cell payment. The CMS asked its Medicare Evidence Development and Coverage Advisory Committee (MEDCAC) to consider the role of PROs in connection with payment for CAR T-cell therapy. At an August 2018 meeting, MEDCAC panelists generally expressed confidence in PROs in a series of votes about the use of this approach to quality measurement in cancer trials.
But drugmakers and physician groups raised strong objections at the MEDCAC meeting. In its national coverage decision on CAR T, issued in August 2019, the CMS said it had received many comments on PROs “ranging from support of their collection to recommendations for additional assessment tools to request to remove PRO requirements.” The CMS opted at this time to encourage participation in the Center for International Blood and Marrow Transplantation Research (CIBMTR) database “that currently collects health outcomes (and aims to collect patient reported outcomes in the future) on patients who have received CAR T-cell treatments.”
For Dr. Walters, this setback for the use of PROs in CAR T therapy payment is telling, as the treatment is known to produce serious side effects and is administered in well-controlled circumstances.
“If you can’t organize collecting patient-reported outcomes after CAR T cell, that kind of tells you the state of where we are on collecting them on everybody,” Dr. Walters said.
REPORTING FROM NCCN POLICY SUMMIT 2019