User login
A 43-year-old woman with chest pressure
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
- White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
- Hemoglobin 15.4 g/dL (12.0–16.0)
- Platelet count 122 × 109/L (150–400)
- International normalized ratio (INR) 1.1 (0.9–1.1)
- Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
- Atherosclerosis
- Protein C deficiency
- Use of oral contraceptive pills
- The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma

The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
- Yes
- No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
- Antithrombin levels
- Protein C and S levels
- Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
- Antiphospholipid antibody syndrome
- Heparin-induced thrombocytopenia
- Malignancy
- All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4

At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.

DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
- Yes
- No
- Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
- Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
- Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
- Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
- Lupus anticoagulant present
- Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
- Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
- Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
- Positron emission tomography and tumor marker levels
- A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
- True
- False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
- Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
- Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
- Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
- Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
- Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
- Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
- In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Lee KW, Lip GY. Acute coronary syndromes: Virchow’s triad revisited. Blood Coagul Fibrinolysis 2003; 14:605–625.
- Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis 1999; 42:91–138.
- Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe’s Clinical Hematology. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2004.
- Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med 2001; 135:367–373.
- Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:401S–428S.
- Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract 2003; 52:770–777.
- Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol 2001; 114:512–528.
- Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007; 110:1723–1729.
- Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci 1974; 230:262–270.
- Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res 1983; 43:3963–3968.
- Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry 1985; 24:5558–5567.
- Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res 2001; 61:795–798.
- Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost 1995; 74:1597–1603.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
- White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
- Hemoglobin 15.4 g/dL (12.0–16.0)
- Platelet count 122 × 109/L (150–400)
- International normalized ratio (INR) 1.1 (0.9–1.1)
- Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
- Atherosclerosis
- Protein C deficiency
- Use of oral contraceptive pills
- The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma
The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
- Yes
- No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
- Antithrombin levels
- Protein C and S levels
- Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
- Antiphospholipid antibody syndrome
- Heparin-induced thrombocytopenia
- Malignancy
- All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4
At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.
DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
- Yes
- No
- Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
- Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
- Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
- Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
- Lupus anticoagulant present
- Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
- Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
- Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
- Positron emission tomography and tumor marker levels
- A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
- True
- False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
- Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
- Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
- Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
- Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
- Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
- Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
- In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
- White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
- Hemoglobin 15.4 g/dL (12.0–16.0)
- Platelet count 122 × 109/L (150–400)
- International normalized ratio (INR) 1.1 (0.9–1.1)
- Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
- Atherosclerosis
- Protein C deficiency
- Use of oral contraceptive pills
- The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma
The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
- Yes
- No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
- Antithrombin levels
- Protein C and S levels
- Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
- Antiphospholipid antibody syndrome
- Heparin-induced thrombocytopenia
- Malignancy
- All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4
At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.
DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
- Yes
- No
- Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
- Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
- Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
- Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
- Lupus anticoagulant present
- Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
- Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
- Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
- Positron emission tomography and tumor marker levels
- A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
- True
- False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
- Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
- Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
- Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
- Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
- Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
- Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
- In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Lee KW, Lip GY. Acute coronary syndromes: Virchow’s triad revisited. Blood Coagul Fibrinolysis 2003; 14:605–625.
- Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis 1999; 42:91–138.
- Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe’s Clinical Hematology. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2004.
- Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med 2001; 135:367–373.
- Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:401S–428S.
- Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract 2003; 52:770–777.
- Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol 2001; 114:512–528.
- Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007; 110:1723–1729.
- Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci 1974; 230:262–270.
- Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res 1983; 43:3963–3968.
- Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry 1985; 24:5558–5567.
- Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res 2001; 61:795–798.
- Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost 1995; 74:1597–1603.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Lee KW, Lip GY. Acute coronary syndromes: Virchow’s triad revisited. Blood Coagul Fibrinolysis 2003; 14:605–625.
- Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis 1999; 42:91–138.
- Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe’s Clinical Hematology. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2004.
- Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med 2001; 135:367–373.
- Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:401S–428S.
- Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract 2003; 52:770–777.
- Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol 2001; 114:512–528.
- Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007; 110:1723–1729.
- Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci 1974; 230:262–270.
- Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res 1983; 43:3963–3968.
- Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry 1985; 24:5558–5567.
- Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res 2001; 61:795–798.
- Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost 1995; 74:1597–1603.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
A new, precise definition of acute myocardial infarction
Acute myocardial infarction (MI) portends important and substantial consequences. Angioplasty or fibrinolytic therapy to open the blocked coronary artery is proven to improve the patient’s chances of surviving without consequent morbidity or death. But the diagnosis is not always straightforward. The presentation of acute MI can vary widely, and a number of other conditions—many of them equally serious emergencies—can mimic its symptoms, electrocardiographic signs, and biomarker patterns.
In an attempt to improve the accuracy of the diagnosis of MI, a multinational task force met in 1999 under the auspices of the European Society of Cardiology and the American College of Cardiology. The goal was to develop a simple, clinically oriented definition of MI that could be widely adopted. A document was created and published simultaneously in 2000 in the European Heart Journal and the Journal of the American College of Cardiology.1 These organizations updated their paper in 2007 with a new definition of acute MI to account for advances in diagnosis and management.2
In this article we will review the new definition and how to make the diagnosis of acute MI today. Specifically, the updated definition includes:
- Subtypes of acute MI
- Imaging tests supporting the diagnosis
- Biomarker thresholds after percutaneous coronary intervention or bypass grafting.
TROPONIN: BETTER THAN CK, BUT NOT PERFECT
The original 2000 paper1 and the 2007 update2 featured the use of the cardiac biomarker troponin, which is considerably more sensitive and specific for heart damage than total creatine kinase (CK) or its isoform, CK-MB.
The new, more-sensitive biomarker-based definition of MI resulted in more cases of MI being diagnosed, and this has attracted the attention and scrutiny of many, especially population scientists and interventional cardiologists.3 This change has caused some controversy, especially when dealing with small rises in troponin following percutaneous coronary intervention.
In addition, some confusion over terminology remains. For example, the phrase “troponin leak” is often used to describe cases in which serum troponin levels rise but there is no MI. However, most experts believe that a rise and fall in troponin is due to true myocardial cell death. Troponin I and T are such large molecules that they cannot “leak” from a cardiac cell unless there has been irreparable cellular damage—that is, cell death.

Creatine kinase still has a role
In some cases, CK and CK-MB may be helpful in determining the acuity of myocardial necrosis, but their use will vary by institution. These biomarkers typically rise 2 to 4 hours after the initial event and fall within 24 to 48 hours, whereas troponin levels stay elevated for days or weeks. Thus, the presence of troponin without CK and CK-MB in the right clinical context may indicate a past MI that is no longer acute.
INFARCTION: CELL DEATH DUE TO ISCHEMIA
MI is myocardial cell death due to prolonged ischemia. Under the microscope, it can be categorized as coagulation necrosis in which ghost-like cell structures remain after hypoxic insult (typical of most MIs) or contraction band necrosis with amorphous cells that cannot contract anymore, the latter often a hallmark of excessive catecholamine damage or reperfusion injury. Apoptosis occurs in the heart but is technically not considered necrosis and is thought not to be associated with elevated troponin levels.6,7
In experiments in animals, cell death can occur as little as 20 minutes after coronary artery occlusion, although completion of infarction is thought to take 2 to 4 hours. The time to infarct completion may be longer in patients with collateral circulation or when the culprit coronary artery has intermittent (“stuttering”) occlusion. Preconditioning of myocardial cells with intermittent ischemia can also influence the timing of myocardial necrosis by protecting against cell death to some extent. Alteration in myocardial demand can influence the time required for completion of infarction either favorably or unfavorably; hence, reducing myocardial demand is beneficial in acute MI.
Three pathologic phases of MI
MI can be categorized pathologically as acute, healing, or healed.
Acute MI. In the first 6 hours after coronary artery occlusion, coagulation necrosis can be seen with no cellular infiltration. After 6 hours, polymorphonuclear leukocytes infiltrate the infarcted area, and this may continue for up to 7 days if coronary perfusion does not increase or myocardial demand does not decrease.
Healing MI is characterized by mononuclear cells and fibroblasts and the absence of polymorphonuclear leukocytes. The entire healing process takes 5 to 6 weeks and can be altered by coronary reperfusion.
Healed MI refers to scar tissue without cellular infiltration.
CLINICAL FEATURES VARY WIDELY
Sir William Osler said, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”8
Just so, patients with acute MI display a wide variety of presentations, from no symptoms (about 25%) to severe, crushing chest pain. Discomfort may occur in the upper back, neck, jaw, teeth, arms, wrist, and epigastrium. Shortness of breath, diaphoresis, nausea, vomiting, and even syncope may occur. Unlike in acute aortic dissection, the discomfort is not usually maximal at its onset: it builds up in a crescendo manner. It is not usually changed by position, but can lessen in intensity upon standing. The discomfort in the chest is deep and visceral, and typically not well localized. A pressure sensation, air hunger, or “gas buildup” can be described. The only symptom may be shortness of breath or severe diaphoresis. The symptoms can last from minutes to hours and can be relieved by sublingual nitroglycerin. Atypical or less-prominent symptoms may make the diagnosis more difficult in the elderly, patients with diabetes mellitus, and women.
The physical examination during acute MI usually finds no clear-cut distinguishing features. The patient may appear pale and diaphoretic, and the skin cool to the touch. Heart sounds are generally soft. A fourth heart sound may be audible. Blood pressure may be low, but it can vary widely. Tachycardia, particularly sinus tachycardia, and pulmonary edema are poor prognostic signs.
In view of the wide variation in presentations, the history and physical findings can raise the suspicion of acute MI, but sequential electrocardiograms and measurements of biomarkers (troponin) are always necessary.
ELECTROCARDIOGRAPHY: NECESSARY BUT NOT SUFFICIENT
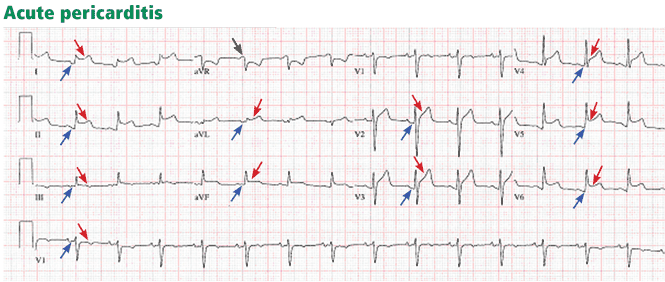

ST-elevation MI vs non-ST-elevation MI
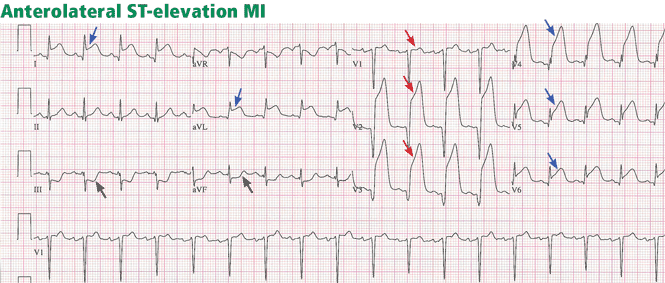
Changes in the ST segment can be very dynamic, making sequential tracings very useful. Rhythm disturbances and heart block are also more likely to be recorded when using sequential readings.
Pitfalls to electrocardiographic diagnosis
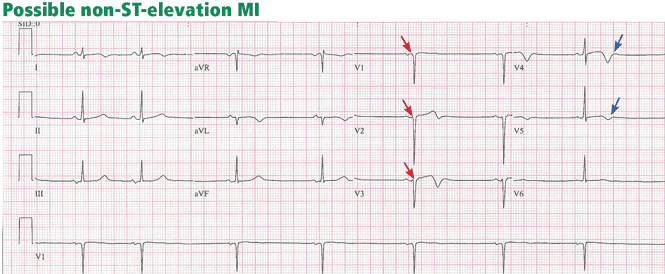
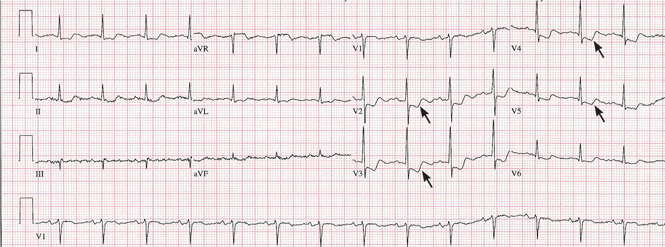
Prior MI. Q waves or QS complexes, when the Q wave is sufficiently wide (≥ 0.03 msec) or deep (≥ 1 mV), usually indicate a previous MI. However, many nuances that further raise or lower the suspicion for previous MI need to be considered. These are beyond the scope of this brief review but are available in the 2007 update.

Right ventricular infarction often requires the use of right-sided leads, which may reveal ST elevation in V4R.
ECHOCARDIOGRAPHY IF THE DIAGNOSIS IS IN DOUBT
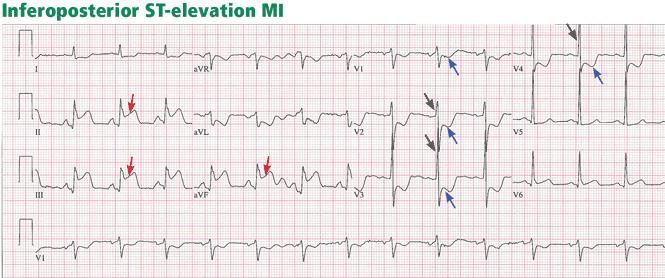
Echocardiography is an excellent way to assess wall-motion abnormalities. In the absence of any wall-motion abnormality, a large ST-elevation MI is unlikely. A large wall-motion abnormality would verify the probability of ongoing acute MI and thus would help with rapid decision-making.
Furthermore, echocardiography can help determine the likelihood that the patient has aortic dissection or pulmonary embolism, either of which can mimic acute MI but requires very different treatment.
CLINICAL CLASSIFICATION OF ACUTE MI

The new classification scheme of the different types of MI is shown in Table 4.
The new classification scheme does not include myocardial necrosis from mechanical manipulation of the heart during open heart surgery, from cardioversion, or from toxic drugs.
As clinicians are aware, it is not unusual to see elevated biomarker levels in a host of conditions unrelated to acute myocardial ischemia or MI. The new classification of acute MI is most helpful in this regard. It will likely be even more helpful in guiding treatment and management when new ultrasensitive troponin assays are widely introduced into clinical practice.
The new classification also negotiates the controversy regarding elevated biomarker levels following percutaneous coronary intervention. In brief, elevation of biomarkers is not entirely avoidable even with a successful percutaneous coronary intervention, and furthermore, there is no scientific cutoff for biomarker elevations. So, by arbitrary convention, the troponin level must rise to more than three times the 99th percentile upper reference limit to make the diagnosis of type 4a MI. A separate type 4b MI is ascribed to angiographic or autopsy-proven stent thrombosis.
The new guidelines also suggest that troponin values be more than five times the 99th percentile of the normal reference range during the first 72 hours following coronary artery bypass graft surgery (CABG) when considering a CABG-related MI (type 5). Whenever new pathologic Q waves appear in territories other than those identified before the procedure, MI should be considered, especially if associated with elevated biomarkers, new wall-motion abnormalities, or hemodynamic instability.
Thus, the diagnosis of acute MI now has widely accepted global criteria that distinguish various types of acute MI that occur under multiple circumstances. It is expected that describing the type of acute MI according to the new criteria will further enhance our understanding of the event, its proper management, and its prognosis.
- The Joint European Society of Cardiology/American College of Cardiology Committee. Myocardial infarction redefined—a consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the Redefinition of Myocardial Infarction. J Am Coll Cardiol 2000; 36:959–969.
- Thygesen K, Alpert JS, White HD, on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. Universal definition of myocardial infarction. J Am Coll Cardiol 2007; 50:2173–2188.
- Roger VL, Killian JM, Weston SA, et al. Redefinition of myocardial infarction—prospective evaluation in the community. Circulation 2006; 114:790–797.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease. J Am Coll Cardiol 2006; 48:1–11.
- French JK, White HD. Clinical implications of the new definition of myocardial infarction. Heart 2004; 90:99–106.
- James TN. The variable morphological coexistence of apoptosis and necrosis in human myocardial infarction: significance for understanding its pathogenesis, clinical course, diagnosis and prognosis. Coron Artery Dis 1998; 9:291–307.
- Sobel BE, LeWinter MM. Ingenuous interpretation of elevated blood levels of macromolecular markers of myocardial injury: a recipe for confusion. J Am Coll Cardiol 2000; 35:1355–1358.
- Osler W. Aequanimitas: With Other Addresses to Medical Students, Nurses and Practitioners of Medicine.Osler William Edition: 3, revised. Philadelphia: Blakiston’s, 1932.
- Wang F, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
Acute myocardial infarction (MI) portends important and substantial consequences. Angioplasty or fibrinolytic therapy to open the blocked coronary artery is proven to improve the patient’s chances of surviving without consequent morbidity or death. But the diagnosis is not always straightforward. The presentation of acute MI can vary widely, and a number of other conditions—many of them equally serious emergencies—can mimic its symptoms, electrocardiographic signs, and biomarker patterns.
In an attempt to improve the accuracy of the diagnosis of MI, a multinational task force met in 1999 under the auspices of the European Society of Cardiology and the American College of Cardiology. The goal was to develop a simple, clinically oriented definition of MI that could be widely adopted. A document was created and published simultaneously in 2000 in the European Heart Journal and the Journal of the American College of Cardiology.1 These organizations updated their paper in 2007 with a new definition of acute MI to account for advances in diagnosis and management.2
In this article we will review the new definition and how to make the diagnosis of acute MI today. Specifically, the updated definition includes:
- Subtypes of acute MI
- Imaging tests supporting the diagnosis
- Biomarker thresholds after percutaneous coronary intervention or bypass grafting.
TROPONIN: BETTER THAN CK, BUT NOT PERFECT
The original 2000 paper1 and the 2007 update2 featured the use of the cardiac biomarker troponin, which is considerably more sensitive and specific for heart damage than total creatine kinase (CK) or its isoform, CK-MB.
The new, more-sensitive biomarker-based definition of MI resulted in more cases of MI being diagnosed, and this has attracted the attention and scrutiny of many, especially population scientists and interventional cardiologists.3 This change has caused some controversy, especially when dealing with small rises in troponin following percutaneous coronary intervention.
In addition, some confusion over terminology remains. For example, the phrase “troponin leak” is often used to describe cases in which serum troponin levels rise but there is no MI. However, most experts believe that a rise and fall in troponin is due to true myocardial cell death. Troponin I and T are such large molecules that they cannot “leak” from a cardiac cell unless there has been irreparable cellular damage—that is, cell death.
Creatine kinase still has a role
In some cases, CK and CK-MB may be helpful in determining the acuity of myocardial necrosis, but their use will vary by institution. These biomarkers typically rise 2 to 4 hours after the initial event and fall within 24 to 48 hours, whereas troponin levels stay elevated for days or weeks. Thus, the presence of troponin without CK and CK-MB in the right clinical context may indicate a past MI that is no longer acute.
INFARCTION: CELL DEATH DUE TO ISCHEMIA
MI is myocardial cell death due to prolonged ischemia. Under the microscope, it can be categorized as coagulation necrosis in which ghost-like cell structures remain after hypoxic insult (typical of most MIs) or contraction band necrosis with amorphous cells that cannot contract anymore, the latter often a hallmark of excessive catecholamine damage or reperfusion injury. Apoptosis occurs in the heart but is technically not considered necrosis and is thought not to be associated with elevated troponin levels.6,7
In experiments in animals, cell death can occur as little as 20 minutes after coronary artery occlusion, although completion of infarction is thought to take 2 to 4 hours. The time to infarct completion may be longer in patients with collateral circulation or when the culprit coronary artery has intermittent (“stuttering”) occlusion. Preconditioning of myocardial cells with intermittent ischemia can also influence the timing of myocardial necrosis by protecting against cell death to some extent. Alteration in myocardial demand can influence the time required for completion of infarction either favorably or unfavorably; hence, reducing myocardial demand is beneficial in acute MI.
Three pathologic phases of MI
MI can be categorized pathologically as acute, healing, or healed.
Acute MI. In the first 6 hours after coronary artery occlusion, coagulation necrosis can be seen with no cellular infiltration. After 6 hours, polymorphonuclear leukocytes infiltrate the infarcted area, and this may continue for up to 7 days if coronary perfusion does not increase or myocardial demand does not decrease.
Healing MI is characterized by mononuclear cells and fibroblasts and the absence of polymorphonuclear leukocytes. The entire healing process takes 5 to 6 weeks and can be altered by coronary reperfusion.
Healed MI refers to scar tissue without cellular infiltration.
CLINICAL FEATURES VARY WIDELY
Sir William Osler said, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”8
Just so, patients with acute MI display a wide variety of presentations, from no symptoms (about 25%) to severe, crushing chest pain. Discomfort may occur in the upper back, neck, jaw, teeth, arms, wrist, and epigastrium. Shortness of breath, diaphoresis, nausea, vomiting, and even syncope may occur. Unlike in acute aortic dissection, the discomfort is not usually maximal at its onset: it builds up in a crescendo manner. It is not usually changed by position, but can lessen in intensity upon standing. The discomfort in the chest is deep and visceral, and typically not well localized. A pressure sensation, air hunger, or “gas buildup” can be described. The only symptom may be shortness of breath or severe diaphoresis. The symptoms can last from minutes to hours and can be relieved by sublingual nitroglycerin. Atypical or less-prominent symptoms may make the diagnosis more difficult in the elderly, patients with diabetes mellitus, and women.
The physical examination during acute MI usually finds no clear-cut distinguishing features. The patient may appear pale and diaphoretic, and the skin cool to the touch. Heart sounds are generally soft. A fourth heart sound may be audible. Blood pressure may be low, but it can vary widely. Tachycardia, particularly sinus tachycardia, and pulmonary edema are poor prognostic signs.
In view of the wide variation in presentations, the history and physical findings can raise the suspicion of acute MI, but sequential electrocardiograms and measurements of biomarkers (troponin) are always necessary.
ELECTROCARDIOGRAPHY: NECESSARY BUT NOT SUFFICIENT
ST-elevation MI vs non-ST-elevation MI
Changes in the ST segment can be very dynamic, making sequential tracings very useful. Rhythm disturbances and heart block are also more likely to be recorded when using sequential readings.
Pitfalls to electrocardiographic diagnosis
Prior MI. Q waves or QS complexes, when the Q wave is sufficiently wide (≥ 0.03 msec) or deep (≥ 1 mV), usually indicate a previous MI. However, many nuances that further raise or lower the suspicion for previous MI need to be considered. These are beyond the scope of this brief review but are available in the 2007 update.
Right ventricular infarction often requires the use of right-sided leads, which may reveal ST elevation in V4R.
ECHOCARDIOGRAPHY IF THE DIAGNOSIS IS IN DOUBT
Echocardiography is an excellent way to assess wall-motion abnormalities. In the absence of any wall-motion abnormality, a large ST-elevation MI is unlikely. A large wall-motion abnormality would verify the probability of ongoing acute MI and thus would help with rapid decision-making.
Furthermore, echocardiography can help determine the likelihood that the patient has aortic dissection or pulmonary embolism, either of which can mimic acute MI but requires very different treatment.
CLINICAL CLASSIFICATION OF ACUTE MI
The new classification scheme of the different types of MI is shown in Table 4.
The new classification scheme does not include myocardial necrosis from mechanical manipulation of the heart during open heart surgery, from cardioversion, or from toxic drugs.
As clinicians are aware, it is not unusual to see elevated biomarker levels in a host of conditions unrelated to acute myocardial ischemia or MI. The new classification of acute MI is most helpful in this regard. It will likely be even more helpful in guiding treatment and management when new ultrasensitive troponin assays are widely introduced into clinical practice.
The new classification also negotiates the controversy regarding elevated biomarker levels following percutaneous coronary intervention. In brief, elevation of biomarkers is not entirely avoidable even with a successful percutaneous coronary intervention, and furthermore, there is no scientific cutoff for biomarker elevations. So, by arbitrary convention, the troponin level must rise to more than three times the 99th percentile upper reference limit to make the diagnosis of type 4a MI. A separate type 4b MI is ascribed to angiographic or autopsy-proven stent thrombosis.
The new guidelines also suggest that troponin values be more than five times the 99th percentile of the normal reference range during the first 72 hours following coronary artery bypass graft surgery (CABG) when considering a CABG-related MI (type 5). Whenever new pathologic Q waves appear in territories other than those identified before the procedure, MI should be considered, especially if associated with elevated biomarkers, new wall-motion abnormalities, or hemodynamic instability.
Thus, the diagnosis of acute MI now has widely accepted global criteria that distinguish various types of acute MI that occur under multiple circumstances. It is expected that describing the type of acute MI according to the new criteria will further enhance our understanding of the event, its proper management, and its prognosis.
Acute myocardial infarction (MI) portends important and substantial consequences. Angioplasty or fibrinolytic therapy to open the blocked coronary artery is proven to improve the patient’s chances of surviving without consequent morbidity or death. But the diagnosis is not always straightforward. The presentation of acute MI can vary widely, and a number of other conditions—many of them equally serious emergencies—can mimic its symptoms, electrocardiographic signs, and biomarker patterns.
In an attempt to improve the accuracy of the diagnosis of MI, a multinational task force met in 1999 under the auspices of the European Society of Cardiology and the American College of Cardiology. The goal was to develop a simple, clinically oriented definition of MI that could be widely adopted. A document was created and published simultaneously in 2000 in the European Heart Journal and the Journal of the American College of Cardiology.1 These organizations updated their paper in 2007 with a new definition of acute MI to account for advances in diagnosis and management.2
In this article we will review the new definition and how to make the diagnosis of acute MI today. Specifically, the updated definition includes:
- Subtypes of acute MI
- Imaging tests supporting the diagnosis
- Biomarker thresholds after percutaneous coronary intervention or bypass grafting.
TROPONIN: BETTER THAN CK, BUT NOT PERFECT
The original 2000 paper1 and the 2007 update2 featured the use of the cardiac biomarker troponin, which is considerably more sensitive and specific for heart damage than total creatine kinase (CK) or its isoform, CK-MB.
The new, more-sensitive biomarker-based definition of MI resulted in more cases of MI being diagnosed, and this has attracted the attention and scrutiny of many, especially population scientists and interventional cardiologists.3 This change has caused some controversy, especially when dealing with small rises in troponin following percutaneous coronary intervention.
In addition, some confusion over terminology remains. For example, the phrase “troponin leak” is often used to describe cases in which serum troponin levels rise but there is no MI. However, most experts believe that a rise and fall in troponin is due to true myocardial cell death. Troponin I and T are such large molecules that they cannot “leak” from a cardiac cell unless there has been irreparable cellular damage—that is, cell death.
Creatine kinase still has a role
In some cases, CK and CK-MB may be helpful in determining the acuity of myocardial necrosis, but their use will vary by institution. These biomarkers typically rise 2 to 4 hours after the initial event and fall within 24 to 48 hours, whereas troponin levels stay elevated for days or weeks. Thus, the presence of troponin without CK and CK-MB in the right clinical context may indicate a past MI that is no longer acute.
INFARCTION: CELL DEATH DUE TO ISCHEMIA
MI is myocardial cell death due to prolonged ischemia. Under the microscope, it can be categorized as coagulation necrosis in which ghost-like cell structures remain after hypoxic insult (typical of most MIs) or contraction band necrosis with amorphous cells that cannot contract anymore, the latter often a hallmark of excessive catecholamine damage or reperfusion injury. Apoptosis occurs in the heart but is technically not considered necrosis and is thought not to be associated with elevated troponin levels.6,7
In experiments in animals, cell death can occur as little as 20 minutes after coronary artery occlusion, although completion of infarction is thought to take 2 to 4 hours. The time to infarct completion may be longer in patients with collateral circulation or when the culprit coronary artery has intermittent (“stuttering”) occlusion. Preconditioning of myocardial cells with intermittent ischemia can also influence the timing of myocardial necrosis by protecting against cell death to some extent. Alteration in myocardial demand can influence the time required for completion of infarction either favorably or unfavorably; hence, reducing myocardial demand is beneficial in acute MI.
Three pathologic phases of MI
MI can be categorized pathologically as acute, healing, or healed.
Acute MI. In the first 6 hours after coronary artery occlusion, coagulation necrosis can be seen with no cellular infiltration. After 6 hours, polymorphonuclear leukocytes infiltrate the infarcted area, and this may continue for up to 7 days if coronary perfusion does not increase or myocardial demand does not decrease.
Healing MI is characterized by mononuclear cells and fibroblasts and the absence of polymorphonuclear leukocytes. The entire healing process takes 5 to 6 weeks and can be altered by coronary reperfusion.
Healed MI refers to scar tissue without cellular infiltration.
CLINICAL FEATURES VARY WIDELY
Sir William Osler said, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”8
Just so, patients with acute MI display a wide variety of presentations, from no symptoms (about 25%) to severe, crushing chest pain. Discomfort may occur in the upper back, neck, jaw, teeth, arms, wrist, and epigastrium. Shortness of breath, diaphoresis, nausea, vomiting, and even syncope may occur. Unlike in acute aortic dissection, the discomfort is not usually maximal at its onset: it builds up in a crescendo manner. It is not usually changed by position, but can lessen in intensity upon standing. The discomfort in the chest is deep and visceral, and typically not well localized. A pressure sensation, air hunger, or “gas buildup” can be described. The only symptom may be shortness of breath or severe diaphoresis. The symptoms can last from minutes to hours and can be relieved by sublingual nitroglycerin. Atypical or less-prominent symptoms may make the diagnosis more difficult in the elderly, patients with diabetes mellitus, and women.
The physical examination during acute MI usually finds no clear-cut distinguishing features. The patient may appear pale and diaphoretic, and the skin cool to the touch. Heart sounds are generally soft. A fourth heart sound may be audible. Blood pressure may be low, but it can vary widely. Tachycardia, particularly sinus tachycardia, and pulmonary edema are poor prognostic signs.
In view of the wide variation in presentations, the history and physical findings can raise the suspicion of acute MI, but sequential electrocardiograms and measurements of biomarkers (troponin) are always necessary.
ELECTROCARDIOGRAPHY: NECESSARY BUT NOT SUFFICIENT
ST-elevation MI vs non-ST-elevation MI
Changes in the ST segment can be very dynamic, making sequential tracings very useful. Rhythm disturbances and heart block are also more likely to be recorded when using sequential readings.
Pitfalls to electrocardiographic diagnosis
Prior MI. Q waves or QS complexes, when the Q wave is sufficiently wide (≥ 0.03 msec) or deep (≥ 1 mV), usually indicate a previous MI. However, many nuances that further raise or lower the suspicion for previous MI need to be considered. These are beyond the scope of this brief review but are available in the 2007 update.
Right ventricular infarction often requires the use of right-sided leads, which may reveal ST elevation in V4R.
ECHOCARDIOGRAPHY IF THE DIAGNOSIS IS IN DOUBT
Echocardiography is an excellent way to assess wall-motion abnormalities. In the absence of any wall-motion abnormality, a large ST-elevation MI is unlikely. A large wall-motion abnormality would verify the probability of ongoing acute MI and thus would help with rapid decision-making.
Furthermore, echocardiography can help determine the likelihood that the patient has aortic dissection or pulmonary embolism, either of which can mimic acute MI but requires very different treatment.
CLINICAL CLASSIFICATION OF ACUTE MI
The new classification scheme of the different types of MI is shown in Table 4.
The new classification scheme does not include myocardial necrosis from mechanical manipulation of the heart during open heart surgery, from cardioversion, or from toxic drugs.
As clinicians are aware, it is not unusual to see elevated biomarker levels in a host of conditions unrelated to acute myocardial ischemia or MI. The new classification of acute MI is most helpful in this regard. It will likely be even more helpful in guiding treatment and management when new ultrasensitive troponin assays are widely introduced into clinical practice.
The new classification also negotiates the controversy regarding elevated biomarker levels following percutaneous coronary intervention. In brief, elevation of biomarkers is not entirely avoidable even with a successful percutaneous coronary intervention, and furthermore, there is no scientific cutoff for biomarker elevations. So, by arbitrary convention, the troponin level must rise to more than three times the 99th percentile upper reference limit to make the diagnosis of type 4a MI. A separate type 4b MI is ascribed to angiographic or autopsy-proven stent thrombosis.
The new guidelines also suggest that troponin values be more than five times the 99th percentile of the normal reference range during the first 72 hours following coronary artery bypass graft surgery (CABG) when considering a CABG-related MI (type 5). Whenever new pathologic Q waves appear in territories other than those identified before the procedure, MI should be considered, especially if associated with elevated biomarkers, new wall-motion abnormalities, or hemodynamic instability.
Thus, the diagnosis of acute MI now has widely accepted global criteria that distinguish various types of acute MI that occur under multiple circumstances. It is expected that describing the type of acute MI according to the new criteria will further enhance our understanding of the event, its proper management, and its prognosis.
- The Joint European Society of Cardiology/American College of Cardiology Committee. Myocardial infarction redefined—a consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the Redefinition of Myocardial Infarction. J Am Coll Cardiol 2000; 36:959–969.
- Thygesen K, Alpert JS, White HD, on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. Universal definition of myocardial infarction. J Am Coll Cardiol 2007; 50:2173–2188.
- Roger VL, Killian JM, Weston SA, et al. Redefinition of myocardial infarction—prospective evaluation in the community. Circulation 2006; 114:790–797.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease. J Am Coll Cardiol 2006; 48:1–11.
- French JK, White HD. Clinical implications of the new definition of myocardial infarction. Heart 2004; 90:99–106.
- James TN. The variable morphological coexistence of apoptosis and necrosis in human myocardial infarction: significance for understanding its pathogenesis, clinical course, diagnosis and prognosis. Coron Artery Dis 1998; 9:291–307.
- Sobel BE, LeWinter MM. Ingenuous interpretation of elevated blood levels of macromolecular markers of myocardial injury: a recipe for confusion. J Am Coll Cardiol 2000; 35:1355–1358.
- Osler W. Aequanimitas: With Other Addresses to Medical Students, Nurses and Practitioners of Medicine.Osler William Edition: 3, revised. Philadelphia: Blakiston’s, 1932.
- Wang F, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
- The Joint European Society of Cardiology/American College of Cardiology Committee. Myocardial infarction redefined—a consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the Redefinition of Myocardial Infarction. J Am Coll Cardiol 2000; 36:959–969.
- Thygesen K, Alpert JS, White HD, on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. Universal definition of myocardial infarction. J Am Coll Cardiol 2007; 50:2173–2188.
- Roger VL, Killian JM, Weston SA, et al. Redefinition of myocardial infarction—prospective evaluation in the community. Circulation 2006; 114:790–797.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease. J Am Coll Cardiol 2006; 48:1–11.
- French JK, White HD. Clinical implications of the new definition of myocardial infarction. Heart 2004; 90:99–106.
- James TN. The variable morphological coexistence of apoptosis and necrosis in human myocardial infarction: significance for understanding its pathogenesis, clinical course, diagnosis and prognosis. Coron Artery Dis 1998; 9:291–307.
- Sobel BE, LeWinter MM. Ingenuous interpretation of elevated blood levels of macromolecular markers of myocardial injury: a recipe for confusion. J Am Coll Cardiol 2000; 35:1355–1358.
- Osler W. Aequanimitas: With Other Addresses to Medical Students, Nurses and Practitioners of Medicine.Osler William Edition: 3, revised. Philadelphia: Blakiston’s, 1932.
- Wang F, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
KEY POINTS
- The clinical presentation of acute MI varies considerably from patient to patient. Therefore, one must consider the symptoms, serial electrocardiographic findings, and serial biomarker results in concert.
- Troponin I or T is now the preferred biomarker of myocardial necrosis. Still, troponin can be elevated in many conditions other than ischemic heart disease.
- Electrocardiographic signs of acute ischemia have been precisely defined, but electrocardiography can give false-positive or false-negative results in a number of conditions.
- MI is now categorized into five types depending on cause.
Grand Rounds: Man, 82, With New-Onset Headaches
An 82-year-old man presented to his primary care provider complaining of headaches for the past week. At the time of presentation, he reported persistent, nonthrobbing pain behind his right eye. Previously, he had experienced pain on the top and right side of his head.
The patient denied any recent visual changes. His last eye examination had taken place four weeks earlier. He was prescribed new eyeglasses, but he had not yet filled the prescription. He denied having symptoms of transient ischemic attack or stroke. He denied any nasal drainage, fever, or chills and reported no prior history of headaches. For the current headache, he had been taking acetaminophen intermittently and said it provided some relief.
The patient’s prior diagnoses included type 2 diabetes, hypertension, dyslipidemia, gout, metabolic syndrome, osteoarthritis, leg edema, and atrial fibrillation. His current medications were allopurinol, diltiazem, glipizide, hydrochlorothiazide, rosiglitazone, valsartan, vardenafil, and warfarin.
His most recent international normalized ratio (INR), measured five days earlier, was 3.34. Fifteen days earlier, however, his INR had been measured at 4.6.
The patient described himself as active, riding his bicycle 50 miles each week. He denied using tobacco but admitted to having “a couple of cocktails” before dinner each evening. He was a widower who lived alone. He owned an advertising company and was involved in its day-to-day operation.
On examination, the patient was alert and oriented. He had an irregularly irregular heart rate with a controlled ventricular response. Cranial nerves II through XII were intact. No papilledema was noted.
The patient was given a diagnosis of headaches of unknown etiology. He was told that he could continue using acetaminophen and was scheduled for head CT with and without contrast the following day.
CT revealed a 2.3-cm, right-sided subacute (mixed-density) subdural hematoma (SDH) with midline shift of 1.8 cm (see Figure 1). The patient’s provider was notified of the CT results, and the patient was sent directly from radiology to the emergency department. His INR was 2.7. The patient was given a partial dose of recombinant factor VIIa (rFVIIa), then emergently transferred to another facility for neurosurgical care.
Upon his arrival there, the patient was noted to be drowsy but oriented, without any focal neurologic deficits. The dose of rFVIIa was completed, and he was given 5 mg of vitamin K. He underwent an emergency craniotomy for clot evacuation. Intraoperatively, his INR was measured at 1.5, and he was given two units of fresh frozen plasma (FFP) to further reverse his coagulopathy.
Repeat head CT the following morning revealed nearly complete removal of the clot, with reexpansion of the brain (see Figure 2). The patient’s INR was 1.1. Additional doses of FFP or rFVIIa were deemed unnecessary. The patient recovered and was discharged from the hospital four days after his surgery. When he was seen at the clinic one month later, he had no neurologic deficits. Head CT was found stable with only a thin rim of residual subdural fluid noted (see Figure 3). He was followed as an outpatient with serial head CTs until all the subdural fluid completely resolved. At that time, he was allowed to restart warfarin.
Discussion
Use of anticoagulation therapy will become increasingly common as our population ages. While anticoagulants are important for preventing thromboembolic events that may result from use of mechanical heart valves, atrial fibrillation, and other conditions, their use is not without risk. The most significant and potentially lethal complication is hemorrhage.
Warfarin-Associated Hemorrhage
In patients who take warfarin, hemorrhage can occur in a variety of areas—most commonly, cerebral and gastrointestinal sites, the nose, the airways, the urinary tract, muscle, and skin.1,2 The site of hemorrhage that carries the highest risk of mortality and morbidity is cerebral.3-5 Among anticoagulated patients experiencing intracranial hemorrhage, a fourfold to fivefold increase in mortality has been reported.6 Among study patients who experienced intracranial hemorrhages while taking warfarin, only 14% were able to return to living independently.4
Excessive Anticoagulation
Recent studies have led to the conclusion that excessive anticoagulation, not anticoagulation targeting specific therapeutic levels, is associated with major bleeding events.7,8 In a review of 2,460 patients from 2000 to 2003 at Brigham and Women’s Hospital in Boston, Fanikos et al8 found that 83% of major bleeding events occurred in patients with an INR exceeding 3.0.
In addition, excessive anticoagulation has been associated with increased morbidity and mortality.5,9,10 Pieracci et al9 found that among patients who experienced a traumatic intracranial hemorrhage with an INR exceeding 3.5, the mortality rate was nearly 75%.
Intracranial Hemorrhage
Subdural hematoma is one of the most common types of intracranial hemorrhage. SDHs are classified based on radiographic findings and age. Acute SDHs are those less than three days old, subacute (mixed-density) SDHs are three to 20 days old, and chronic SDHs (CSDHs) are at least 21 days old.
Acute hemorrhages are more dense and appear white on CT, whereas CSDHs are hypodense and appear darker than the brain parenchyma. Subacute SDHs may have features of both acute SDHs and CSDHs or may appear isodense. While acute SDHs are often associated with trauma and are readily diagnosed, chronic and subacute SDHs present a greater diagnostic challenge. Clinically, subacute SDHs act like CSDHs and are treated similarly.11 For the purposes of this discussion, the case patient’s SDH will be considered a form of CSDH.
Pathophysiology of Chronic Subdural Hematomas
Chronic subdural hematomas form in a number of ways. Major causes are related to brain atrophy resulting from advanced age, alcoholism, brain injury, stroke, or other conditions.11 Atrophy of the brain causes the size of the subdural space to increase. This increased space causes the bridging veins between the cortical surface of the brain and the dura to become stretched and easily torn. As a result, seemingly minor trauma can easily lead to hemorrhage.
Over time, these small, acute hemorrhages in the subdural space may liquefy into CSDHs. Bleeding triggers an inflammatory response, and gradually, blood begins to break down, as with any bruise. Unlike most blood clots, however, blood in the subdural space is affected by fluid dynamics, fibrinolysis, and the formation of neomembranes.11,12 As a result, the blood may not be completely reabsorbed and may actually expand, causing patients to experience symptoms.
Potentially, SDHs can also be caused by subdural hygromas, low intracranial pressure, dehydration, or overdrainage of cerebrospinal fluid during lumbar puncture, spinal anesthesia, or shunting.13
Epidemiology
The annual incidence of CSDH is one to two cases per 100,000 persons. Incidence increases to seven cases per 100,000 among persons older than 70.13 The mortality rate for SDH is 31% to 36%.14,15 The mortality rate for CSDH is approximately 6%. For patients older than 60, the rate increases to 8.8%.16 Rates of morbidity (ie, severe disability or persistent vegetative state) associated with CSDHs have been reported at about 10%.16,17
Men are affected more commonly than are women (accounting for 61% to 70% of cases), and median ages between 71 and 78 have been reported.4,12,18,19
The risk factors for CSDH are listed in Table 1.4,10 SDHs frequently occur in the context of trauma, but they can occur spontaneously, especially in coagulopathic patients. Among patients with CSDHs who are taking warfarin, 45.5% to 52% deny recent experiences of trauma.4,14
Signs and Symptoms of Chronic Subdural Hematomas
The clinical onset of CSDH is insidious. Possible presenting symptoms are listed in Table 2.14,18,20,21 Frequently, the neurologic examination fails to reveal any focal deficits. Many of the symptoms are vague and nonspecific and may mimic those of other conditions that are common in the elderly, thus making diagnosis difficult. Despite clinical suspicion, the definitive diagnosis of SDH is based on CT results.
Reversing Warfarin-Induced Coagulopathy
In all patients with intracranial hemorrhages who are taking warfarin, the coagulopathy must be reversed. The agents commonly used to reverse the effects of warfarin include vitamin K, FFP, and rFVIIa.9,22-24 The choice of agents depends on the timing of intervention.
Vitamin K is commonly given to patients either intravenously or orally in combination with FFP and/or rFVIIa to promote the reversal of warfarin-induced coagulopathy. Vitamin K is seldom used alone, as its effects may not be seen for 24 hours or longer, and may not completely reverse the effects of warfarin.25
Another frequently used product is FFP. Unfortunately, FFP has been associated with complications such as fluid overload, infectious disease transmission, and anaphylaxis. Additionally, FFP too reverses coagulopathy very slowly. Boulis et al26 found that in patients given FFP with single-dose vitamin K, INR reduction averaged 0.18/hour. At this rate, it would take approximately 11 hours to correct an INR of 3.0 to the desired target of 1.0.
In contrast, rFVIIa, used off-label, has proved highly effective in rapidly reversing coagulopathy and allowing patients to safely undergo immediate surgical treatment.23,24 To its disadvantage, rFVIIa increases the risk of thromboembolism and is significantly more expensive than FFP. Compared with $105 for one unit of FFP, the cost of an 80-mcg/kg dose of rFVIIa for a patient weighing 80 kg is about $6,400.27
Factors Predicting Outcome for Subdural Hematomas
A number of factors determine post-SDH outcome. Rozzelle et al14 found that a Glasgow Coma Scale score below 7, age greater than 80, more acute hemorrhages, and hemorrhages requiring craniotomy rather than burr-hole drainage were associated with significantly higher mortality rates than when these factors were absent.
Other studies have revealed that patients with poor clinical status and larger hematomas with more midline shift are also prone to higher mortality rates.20,28 Merlicco et al29 found that younger, nonalcoholic patients without severe trauma whose hematomas were under high pressure had better chances for full recovery than other patients.
Patient Outcome
This case study illustrates the importance of patient education. The patient described here was aware of his excessive anticoagulation and told his provider that he was concerned about bleeding in the brain. Because the patient had been educated about the potential risks of warfarin therapy, he was able to alert his provider when he experienced symptoms of a possible complication. As a result, his condition was quickly diagnosed and treated, with an excellent outcome.
Conclusion
Intracranial hemorrhage is a serious and potentially life-threatening complication of warfarin therapy. CSDHs in particular are a significant cause of mortality and morbidity in older patients. The risk of death or disability increases in patients who are undergoing anticoagulation therapy. In addition, patients with an INR elevated above therapeutic levels face a significantly higher risk for major bleeding events. For this reason, it is important that anticoagulation be tightly controlled within the therapeutic range. It is equally important to educate patients and their families about anticoagulation’s potential risks and complications.
Making the diagnosis of CSDH can be difficult because its symptoms are so often nonspecific and a concomitant illness may be present. Thus, providers must maintain a low threshold for evaluating even minor patient complaints that may signal a complication of warfarin therapy. All too often, minor signs and symptoms go unrecognized, sometimes leading to devastating consequences.
Although many factors predict outcomes for CSDHs, the most important can be controlled by patients and their providers. If patients are well educated and providers listen to their patients, then early diagnosis of SDH can lead to early intervention and improved outcomes.
1. Pullicino P, Thompson JL. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med. 2003;348(3): 256-257.
2. Hurlen M, Abdelnoor M, Smith P, et al. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med. 2002;347(13):969-974.
3. DeSilvey DL. Clinical trials: advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Am J Geriatr Cardiol. 2005;14(2):98-99.
4. Fang MC, Chang Y, Hylek EM, et al. Advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Ann Intern Med. 2004;141(10):745-752.
5. Koo S, Kucher N, Nguyen PL, et al. The effect of excessive anticoagulation on mortality and morbidity in hospitalized patients with anticoagulant-related major hemorrhage. Arch Intern Med. 2004;164(14):1557-1560.
6. Mina AA, Knipfer JF, Park DY, et al. Intracranial complications of preinjury anticoagulation in trauma patients with head injury. J Trauma. 2002;53(4):668-672.
7. Pieracci FM, Eachempati SR, Shou J, et al. Degree of anticoagulation, but not warfarin use itself, predicts adverse outcomes after traumatic brain injury in elderly trauma patients. J Trauma. 2007;63(3):525-530.
8. Fanikos J, Grasso-Correnti N, Shah R, et al. Major bleeding complications in a specialized anticoagulation service. Am J Cardiol. 2005;96(4):595-598.
9. Pieracci FM, Eachempati SR, Shou J, et al. Use of long-term anticoagulation is associated with traumatic intracranial hemorrhage and subsequent mortality in elderly patients hospitalized after falls: analysis of the New York State Administrative Database. J Trauma. 2007;63(3):519-524.
10. Franko J, Kish KJ, O’Connell BG, et al. Advanced age and preinjury warfarin anticoagulation increase the risk of mortality after head trauma. J Trauma. 2006; 61(1):107-110.
11. Drapkin AJ. Chronic subdural hematoma: pathophysiological basis for treatment. Br J Neurosurg. 1991; 5(5):467-473.
12. Yamamoto H, Hirashima Y, Hamada H, et al. Independent predictors of recurrence of chronic subdural hematoma: results of multivariate analysis performed using a logistic regression model. J Neurosurg. 2003;98(6):1217-1221.
13. Iantosca MR, Simon RH. Chronic subdural hematoma in adult and elderly patients. Neurosurg Clin N Am. 2000;11(3):447-454.
14. Rozzelle CJ, Wofford JL, Branch CL. Predictors of hospital mortality in older patients with subdural hematoma. J Am Geriatr Soc. 1995;43(3):240-244.
15. Wintzen AR, Tijssen JG. Subdural hematoma and oral anticoagulant therapy. Arch Neurol. 1982;39(2): 69-72.
16. Ramachandran R, Hegde T. Chronic subdural hematomas: causes of morbidity and mortality. Surg Neurol. 2007;67(4):367-372.
17. Amirjamshidi A, Eftekhar B, Abouzari M, Rashidi A. The relationship between Glasgow coma/outcome scores and abnormal CT scan findings in chronic subdural hematoma. Clin Neurol Neurosurg. 2007;109(2): 152-157.
18. Lee JY, Ebel H, Ernestus RI, Klug N. Various surgical treatments of chronic subdural hematoma and outcome in 172 patients: is membranectomy necessary? Surg Neurol. 2004;61(6):523-527.
19. Gelabert-González M, Iglesias-Pais M, García-Allut A, Martínez-Rumbo R. Chronic subdural haematoma: surgical treatment and outcome in 1000 cases. Clin Neurol Neurosurg. 2005;107(3):223-229.
20. Mattle H, Kohler S, Huber P, et al. Anticoagulation-related intracranial extracerebral haemorrhage. J Neurol Neurosurg Psychiatry. 1989;52(7):829-837.
21. Sambasivan M. An overview of chronic subdural hematoma: experience with 2300 cases. Surg Neurol. 1997;47(5):418-422.
22. Lin J, Hanigan WC, Tarantino M, Wang J. The use of recombinant activated factor VII to reverse warfarin-induced anticoagulation in patients with hemorrhages in the central nervous system: preliminary findings. J Neurosurg. 2003;98(4):737-740.
23. Freeman WD, Brott TG, Barrett KM, et al. Recombinant factor VIIa for rapid reversal of warfarin anticoagulation in acute intracranial hemorrhage. Mayo Clin Proc. 2004;79(12):1495-1500.
24. Dager WE, King JH, Regalia RC, et al. Reversal of elevated international normalized ratios and bleeding with low-dose recombinant activated factor VII in patients receiving warfarin. Pharmacotherapy. 2006;26(8): 1091-1098.
25. Denas G, Marzot F, Offelli P, et al. Effectiveness and safety of a management protocol to correct over-anticoagulation with oral vitamin K: a retrospective study of 1,043 cases. J Thromb Thrombolysis. 2008 Mar 13; [Epub ahead of print].
26. Boulis NM, Bobek MP, Schmaier A, Hoff JT. Use of factor IX complex in warfarin-related intracranial hemorrhage. Neurosurgery. 1999;45(5):1113-1118.
27. Kissela BM, Eckman MH. Cost effectiveness of recombinant factor VIIa for treatment of intracerebral hemorrhage. BMC Neurol. 2008;8:17.
28. Ernestus RI, Beldzinski P, Lanfermann H, Klug N. Chronic subdural hematoma: surgical treatment and outcome in 104 patients. Surg Neurol. 1997;48(3): 220-225.
29. Merlicco G, Pierangeli E, di Padova PL. Chronic subdural hematomas in adults: prognostic factors: analysis of 70 cases. Neurosurg Rev. 1995;18(4):247-251.
An 82-year-old man presented to his primary care provider complaining of headaches for the past week. At the time of presentation, he reported persistent, nonthrobbing pain behind his right eye. Previously, he had experienced pain on the top and right side of his head.
The patient denied any recent visual changes. His last eye examination had taken place four weeks earlier. He was prescribed new eyeglasses, but he had not yet filled the prescription. He denied having symptoms of transient ischemic attack or stroke. He denied any nasal drainage, fever, or chills and reported no prior history of headaches. For the current headache, he had been taking acetaminophen intermittently and said it provided some relief.
The patient’s prior diagnoses included type 2 diabetes, hypertension, dyslipidemia, gout, metabolic syndrome, osteoarthritis, leg edema, and atrial fibrillation. His current medications were allopurinol, diltiazem, glipizide, hydrochlorothiazide, rosiglitazone, valsartan, vardenafil, and warfarin.
His most recent international normalized ratio (INR), measured five days earlier, was 3.34. Fifteen days earlier, however, his INR had been measured at 4.6.
The patient described himself as active, riding his bicycle 50 miles each week. He denied using tobacco but admitted to having “a couple of cocktails” before dinner each evening. He was a widower who lived alone. He owned an advertising company and was involved in its day-to-day operation.
On examination, the patient was alert and oriented. He had an irregularly irregular heart rate with a controlled ventricular response. Cranial nerves II through XII were intact. No papilledema was noted.
The patient was given a diagnosis of headaches of unknown etiology. He was told that he could continue using acetaminophen and was scheduled for head CT with and without contrast the following day.
CT revealed a 2.3-cm, right-sided subacute (mixed-density) subdural hematoma (SDH) with midline shift of 1.8 cm (see Figure 1). The patient’s provider was notified of the CT results, and the patient was sent directly from radiology to the emergency department. His INR was 2.7. The patient was given a partial dose of recombinant factor VIIa (rFVIIa), then emergently transferred to another facility for neurosurgical care.
Upon his arrival there, the patient was noted to be drowsy but oriented, without any focal neurologic deficits. The dose of rFVIIa was completed, and he was given 5 mg of vitamin K. He underwent an emergency craniotomy for clot evacuation. Intraoperatively, his INR was measured at 1.5, and he was given two units of fresh frozen plasma (FFP) to further reverse his coagulopathy.
Repeat head CT the following morning revealed nearly complete removal of the clot, with reexpansion of the brain (see Figure 2). The patient’s INR was 1.1. Additional doses of FFP or rFVIIa were deemed unnecessary. The patient recovered and was discharged from the hospital four days after his surgery. When he was seen at the clinic one month later, he had no neurologic deficits. Head CT was found stable with only a thin rim of residual subdural fluid noted (see Figure 3). He was followed as an outpatient with serial head CTs until all the subdural fluid completely resolved. At that time, he was allowed to restart warfarin.
Discussion
Use of anticoagulation therapy will become increasingly common as our population ages. While anticoagulants are important for preventing thromboembolic events that may result from use of mechanical heart valves, atrial fibrillation, and other conditions, their use is not without risk. The most significant and potentially lethal complication is hemorrhage.
Warfarin-Associated Hemorrhage
In patients who take warfarin, hemorrhage can occur in a variety of areas—most commonly, cerebral and gastrointestinal sites, the nose, the airways, the urinary tract, muscle, and skin.1,2 The site of hemorrhage that carries the highest risk of mortality and morbidity is cerebral.3-5 Among anticoagulated patients experiencing intracranial hemorrhage, a fourfold to fivefold increase in mortality has been reported.6 Among study patients who experienced intracranial hemorrhages while taking warfarin, only 14% were able to return to living independently.4
Excessive Anticoagulation
Recent studies have led to the conclusion that excessive anticoagulation, not anticoagulation targeting specific therapeutic levels, is associated with major bleeding events.7,8 In a review of 2,460 patients from 2000 to 2003 at Brigham and Women’s Hospital in Boston, Fanikos et al8 found that 83% of major bleeding events occurred in patients with an INR exceeding 3.0.
In addition, excessive anticoagulation has been associated with increased morbidity and mortality.5,9,10 Pieracci et al9 found that among patients who experienced a traumatic intracranial hemorrhage with an INR exceeding 3.5, the mortality rate was nearly 75%.
Intracranial Hemorrhage
Subdural hematoma is one of the most common types of intracranial hemorrhage. SDHs are classified based on radiographic findings and age. Acute SDHs are those less than three days old, subacute (mixed-density) SDHs are three to 20 days old, and chronic SDHs (CSDHs) are at least 21 days old.
Acute hemorrhages are more dense and appear white on CT, whereas CSDHs are hypodense and appear darker than the brain parenchyma. Subacute SDHs may have features of both acute SDHs and CSDHs or may appear isodense. While acute SDHs are often associated with trauma and are readily diagnosed, chronic and subacute SDHs present a greater diagnostic challenge. Clinically, subacute SDHs act like CSDHs and are treated similarly.11 For the purposes of this discussion, the case patient’s SDH will be considered a form of CSDH.
Pathophysiology of Chronic Subdural Hematomas
Chronic subdural hematomas form in a number of ways. Major causes are related to brain atrophy resulting from advanced age, alcoholism, brain injury, stroke, or other conditions.11 Atrophy of the brain causes the size of the subdural space to increase. This increased space causes the bridging veins between the cortical surface of the brain and the dura to become stretched and easily torn. As a result, seemingly minor trauma can easily lead to hemorrhage.
Over time, these small, acute hemorrhages in the subdural space may liquefy into CSDHs. Bleeding triggers an inflammatory response, and gradually, blood begins to break down, as with any bruise. Unlike most blood clots, however, blood in the subdural space is affected by fluid dynamics, fibrinolysis, and the formation of neomembranes.11,12 As a result, the blood may not be completely reabsorbed and may actually expand, causing patients to experience symptoms.
Potentially, SDHs can also be caused by subdural hygromas, low intracranial pressure, dehydration, or overdrainage of cerebrospinal fluid during lumbar puncture, spinal anesthesia, or shunting.13
Epidemiology
The annual incidence of CSDH is one to two cases per 100,000 persons. Incidence increases to seven cases per 100,000 among persons older than 70.13 The mortality rate for SDH is 31% to 36%.14,15 The mortality rate for CSDH is approximately 6%. For patients older than 60, the rate increases to 8.8%.16 Rates of morbidity (ie, severe disability or persistent vegetative state) associated with CSDHs have been reported at about 10%.16,17
Men are affected more commonly than are women (accounting for 61% to 70% of cases), and median ages between 71 and 78 have been reported.4,12,18,19
The risk factors for CSDH are listed in Table 1.4,10 SDHs frequently occur in the context of trauma, but they can occur spontaneously, especially in coagulopathic patients. Among patients with CSDHs who are taking warfarin, 45.5% to 52% deny recent experiences of trauma.4,14
Signs and Symptoms of Chronic Subdural Hematomas
The clinical onset of CSDH is insidious. Possible presenting symptoms are listed in Table 2.14,18,20,21 Frequently, the neurologic examination fails to reveal any focal deficits. Many of the symptoms are vague and nonspecific and may mimic those of other conditions that are common in the elderly, thus making diagnosis difficult. Despite clinical suspicion, the definitive diagnosis of SDH is based on CT results.
Reversing Warfarin-Induced Coagulopathy
In all patients with intracranial hemorrhages who are taking warfarin, the coagulopathy must be reversed. The agents commonly used to reverse the effects of warfarin include vitamin K, FFP, and rFVIIa.9,22-24 The choice of agents depends on the timing of intervention.
Vitamin K is commonly given to patients either intravenously or orally in combination with FFP and/or rFVIIa to promote the reversal of warfarin-induced coagulopathy. Vitamin K is seldom used alone, as its effects may not be seen for 24 hours or longer, and may not completely reverse the effects of warfarin.25
Another frequently used product is FFP. Unfortunately, FFP has been associated with complications such as fluid overload, infectious disease transmission, and anaphylaxis. Additionally, FFP too reverses coagulopathy very slowly. Boulis et al26 found that in patients given FFP with single-dose vitamin K, INR reduction averaged 0.18/hour. At this rate, it would take approximately 11 hours to correct an INR of 3.0 to the desired target of 1.0.
In contrast, rFVIIa, used off-label, has proved highly effective in rapidly reversing coagulopathy and allowing patients to safely undergo immediate surgical treatment.23,24 To its disadvantage, rFVIIa increases the risk of thromboembolism and is significantly more expensive than FFP. Compared with $105 for one unit of FFP, the cost of an 80-mcg/kg dose of rFVIIa for a patient weighing 80 kg is about $6,400.27
Factors Predicting Outcome for Subdural Hematomas
A number of factors determine post-SDH outcome. Rozzelle et al14 found that a Glasgow Coma Scale score below 7, age greater than 80, more acute hemorrhages, and hemorrhages requiring craniotomy rather than burr-hole drainage were associated with significantly higher mortality rates than when these factors were absent.
Other studies have revealed that patients with poor clinical status and larger hematomas with more midline shift are also prone to higher mortality rates.20,28 Merlicco et al29 found that younger, nonalcoholic patients without severe trauma whose hematomas were under high pressure had better chances for full recovery than other patients.
Patient Outcome
This case study illustrates the importance of patient education. The patient described here was aware of his excessive anticoagulation and told his provider that he was concerned about bleeding in the brain. Because the patient had been educated about the potential risks of warfarin therapy, he was able to alert his provider when he experienced symptoms of a possible complication. As a result, his condition was quickly diagnosed and treated, with an excellent outcome.
Conclusion
Intracranial hemorrhage is a serious and potentially life-threatening complication of warfarin therapy. CSDHs in particular are a significant cause of mortality and morbidity in older patients. The risk of death or disability increases in patients who are undergoing anticoagulation therapy. In addition, patients with an INR elevated above therapeutic levels face a significantly higher risk for major bleeding events. For this reason, it is important that anticoagulation be tightly controlled within the therapeutic range. It is equally important to educate patients and their families about anticoagulation’s potential risks and complications.
Making the diagnosis of CSDH can be difficult because its symptoms are so often nonspecific and a concomitant illness may be present. Thus, providers must maintain a low threshold for evaluating even minor patient complaints that may signal a complication of warfarin therapy. All too often, minor signs and symptoms go unrecognized, sometimes leading to devastating consequences.
Although many factors predict outcomes for CSDHs, the most important can be controlled by patients and their providers. If patients are well educated and providers listen to their patients, then early diagnosis of SDH can lead to early intervention and improved outcomes.
An 82-year-old man presented to his primary care provider complaining of headaches for the past week. At the time of presentation, he reported persistent, nonthrobbing pain behind his right eye. Previously, he had experienced pain on the top and right side of his head.
The patient denied any recent visual changes. His last eye examination had taken place four weeks earlier. He was prescribed new eyeglasses, but he had not yet filled the prescription. He denied having symptoms of transient ischemic attack or stroke. He denied any nasal drainage, fever, or chills and reported no prior history of headaches. For the current headache, he had been taking acetaminophen intermittently and said it provided some relief.
The patient’s prior diagnoses included type 2 diabetes, hypertension, dyslipidemia, gout, metabolic syndrome, osteoarthritis, leg edema, and atrial fibrillation. His current medications were allopurinol, diltiazem, glipizide, hydrochlorothiazide, rosiglitazone, valsartan, vardenafil, and warfarin.
His most recent international normalized ratio (INR), measured five days earlier, was 3.34. Fifteen days earlier, however, his INR had been measured at 4.6.
The patient described himself as active, riding his bicycle 50 miles each week. He denied using tobacco but admitted to having “a couple of cocktails” before dinner each evening. He was a widower who lived alone. He owned an advertising company and was involved in its day-to-day operation.
On examination, the patient was alert and oriented. He had an irregularly irregular heart rate with a controlled ventricular response. Cranial nerves II through XII were intact. No papilledema was noted.
The patient was given a diagnosis of headaches of unknown etiology. He was told that he could continue using acetaminophen and was scheduled for head CT with and without contrast the following day.
CT revealed a 2.3-cm, right-sided subacute (mixed-density) subdural hematoma (SDH) with midline shift of 1.8 cm (see Figure 1). The patient’s provider was notified of the CT results, and the patient was sent directly from radiology to the emergency department. His INR was 2.7. The patient was given a partial dose of recombinant factor VIIa (rFVIIa), then emergently transferred to another facility for neurosurgical care.
Upon his arrival there, the patient was noted to be drowsy but oriented, without any focal neurologic deficits. The dose of rFVIIa was completed, and he was given 5 mg of vitamin K. He underwent an emergency craniotomy for clot evacuation. Intraoperatively, his INR was measured at 1.5, and he was given two units of fresh frozen plasma (FFP) to further reverse his coagulopathy.
Repeat head CT the following morning revealed nearly complete removal of the clot, with reexpansion of the brain (see Figure 2). The patient’s INR was 1.1. Additional doses of FFP or rFVIIa were deemed unnecessary. The patient recovered and was discharged from the hospital four days after his surgery. When he was seen at the clinic one month later, he had no neurologic deficits. Head CT was found stable with only a thin rim of residual subdural fluid noted (see Figure 3). He was followed as an outpatient with serial head CTs until all the subdural fluid completely resolved. At that time, he was allowed to restart warfarin.
Discussion
Use of anticoagulation therapy will become increasingly common as our population ages. While anticoagulants are important for preventing thromboembolic events that may result from use of mechanical heart valves, atrial fibrillation, and other conditions, their use is not without risk. The most significant and potentially lethal complication is hemorrhage.
Warfarin-Associated Hemorrhage
In patients who take warfarin, hemorrhage can occur in a variety of areas—most commonly, cerebral and gastrointestinal sites, the nose, the airways, the urinary tract, muscle, and skin.1,2 The site of hemorrhage that carries the highest risk of mortality and morbidity is cerebral.3-5 Among anticoagulated patients experiencing intracranial hemorrhage, a fourfold to fivefold increase in mortality has been reported.6 Among study patients who experienced intracranial hemorrhages while taking warfarin, only 14% were able to return to living independently.4
Excessive Anticoagulation
Recent studies have led to the conclusion that excessive anticoagulation, not anticoagulation targeting specific therapeutic levels, is associated with major bleeding events.7,8 In a review of 2,460 patients from 2000 to 2003 at Brigham and Women’s Hospital in Boston, Fanikos et al8 found that 83% of major bleeding events occurred in patients with an INR exceeding 3.0.
In addition, excessive anticoagulation has been associated with increased morbidity and mortality.5,9,10 Pieracci et al9 found that among patients who experienced a traumatic intracranial hemorrhage with an INR exceeding 3.5, the mortality rate was nearly 75%.
Intracranial Hemorrhage
Subdural hematoma is one of the most common types of intracranial hemorrhage. SDHs are classified based on radiographic findings and age. Acute SDHs are those less than three days old, subacute (mixed-density) SDHs are three to 20 days old, and chronic SDHs (CSDHs) are at least 21 days old.
Acute hemorrhages are more dense and appear white on CT, whereas CSDHs are hypodense and appear darker than the brain parenchyma. Subacute SDHs may have features of both acute SDHs and CSDHs or may appear isodense. While acute SDHs are often associated with trauma and are readily diagnosed, chronic and subacute SDHs present a greater diagnostic challenge. Clinically, subacute SDHs act like CSDHs and are treated similarly.11 For the purposes of this discussion, the case patient’s SDH will be considered a form of CSDH.
Pathophysiology of Chronic Subdural Hematomas
Chronic subdural hematomas form in a number of ways. Major causes are related to brain atrophy resulting from advanced age, alcoholism, brain injury, stroke, or other conditions.11 Atrophy of the brain causes the size of the subdural space to increase. This increased space causes the bridging veins between the cortical surface of the brain and the dura to become stretched and easily torn. As a result, seemingly minor trauma can easily lead to hemorrhage.
Over time, these small, acute hemorrhages in the subdural space may liquefy into CSDHs. Bleeding triggers an inflammatory response, and gradually, blood begins to break down, as with any bruise. Unlike most blood clots, however, blood in the subdural space is affected by fluid dynamics, fibrinolysis, and the formation of neomembranes.11,12 As a result, the blood may not be completely reabsorbed and may actually expand, causing patients to experience symptoms.
Potentially, SDHs can also be caused by subdural hygromas, low intracranial pressure, dehydration, or overdrainage of cerebrospinal fluid during lumbar puncture, spinal anesthesia, or shunting.13
Epidemiology
The annual incidence of CSDH is one to two cases per 100,000 persons. Incidence increases to seven cases per 100,000 among persons older than 70.13 The mortality rate for SDH is 31% to 36%.14,15 The mortality rate for CSDH is approximately 6%. For patients older than 60, the rate increases to 8.8%.16 Rates of morbidity (ie, severe disability or persistent vegetative state) associated with CSDHs have been reported at about 10%.16,17
Men are affected more commonly than are women (accounting for 61% to 70% of cases), and median ages between 71 and 78 have been reported.4,12,18,19
The risk factors for CSDH are listed in Table 1.4,10 SDHs frequently occur in the context of trauma, but they can occur spontaneously, especially in coagulopathic patients. Among patients with CSDHs who are taking warfarin, 45.5% to 52% deny recent experiences of trauma.4,14
Signs and Symptoms of Chronic Subdural Hematomas
The clinical onset of CSDH is insidious. Possible presenting symptoms are listed in Table 2.14,18,20,21 Frequently, the neurologic examination fails to reveal any focal deficits. Many of the symptoms are vague and nonspecific and may mimic those of other conditions that are common in the elderly, thus making diagnosis difficult. Despite clinical suspicion, the definitive diagnosis of SDH is based on CT results.
Reversing Warfarin-Induced Coagulopathy
In all patients with intracranial hemorrhages who are taking warfarin, the coagulopathy must be reversed. The agents commonly used to reverse the effects of warfarin include vitamin K, FFP, and rFVIIa.9,22-24 The choice of agents depends on the timing of intervention.
Vitamin K is commonly given to patients either intravenously or orally in combination with FFP and/or rFVIIa to promote the reversal of warfarin-induced coagulopathy. Vitamin K is seldom used alone, as its effects may not be seen for 24 hours or longer, and may not completely reverse the effects of warfarin.25
Another frequently used product is FFP. Unfortunately, FFP has been associated with complications such as fluid overload, infectious disease transmission, and anaphylaxis. Additionally, FFP too reverses coagulopathy very slowly. Boulis et al26 found that in patients given FFP with single-dose vitamin K, INR reduction averaged 0.18/hour. At this rate, it would take approximately 11 hours to correct an INR of 3.0 to the desired target of 1.0.
In contrast, rFVIIa, used off-label, has proved highly effective in rapidly reversing coagulopathy and allowing patients to safely undergo immediate surgical treatment.23,24 To its disadvantage, rFVIIa increases the risk of thromboembolism and is significantly more expensive than FFP. Compared with $105 for one unit of FFP, the cost of an 80-mcg/kg dose of rFVIIa for a patient weighing 80 kg is about $6,400.27
Factors Predicting Outcome for Subdural Hematomas
A number of factors determine post-SDH outcome. Rozzelle et al14 found that a Glasgow Coma Scale score below 7, age greater than 80, more acute hemorrhages, and hemorrhages requiring craniotomy rather than burr-hole drainage were associated with significantly higher mortality rates than when these factors were absent.
Other studies have revealed that patients with poor clinical status and larger hematomas with more midline shift are also prone to higher mortality rates.20,28 Merlicco et al29 found that younger, nonalcoholic patients without severe trauma whose hematomas were under high pressure had better chances for full recovery than other patients.
Patient Outcome
This case study illustrates the importance of patient education. The patient described here was aware of his excessive anticoagulation and told his provider that he was concerned about bleeding in the brain. Because the patient had been educated about the potential risks of warfarin therapy, he was able to alert his provider when he experienced symptoms of a possible complication. As a result, his condition was quickly diagnosed and treated, with an excellent outcome.
Conclusion
Intracranial hemorrhage is a serious and potentially life-threatening complication of warfarin therapy. CSDHs in particular are a significant cause of mortality and morbidity in older patients. The risk of death or disability increases in patients who are undergoing anticoagulation therapy. In addition, patients with an INR elevated above therapeutic levels face a significantly higher risk for major bleeding events. For this reason, it is important that anticoagulation be tightly controlled within the therapeutic range. It is equally important to educate patients and their families about anticoagulation’s potential risks and complications.
Making the diagnosis of CSDH can be difficult because its symptoms are so often nonspecific and a concomitant illness may be present. Thus, providers must maintain a low threshold for evaluating even minor patient complaints that may signal a complication of warfarin therapy. All too often, minor signs and symptoms go unrecognized, sometimes leading to devastating consequences.
Although many factors predict outcomes for CSDHs, the most important can be controlled by patients and their providers. If patients are well educated and providers listen to their patients, then early diagnosis of SDH can lead to early intervention and improved outcomes.
1. Pullicino P, Thompson JL. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med. 2003;348(3): 256-257.
2. Hurlen M, Abdelnoor M, Smith P, et al. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med. 2002;347(13):969-974.
3. DeSilvey DL. Clinical trials: advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Am J Geriatr Cardiol. 2005;14(2):98-99.
4. Fang MC, Chang Y, Hylek EM, et al. Advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Ann Intern Med. 2004;141(10):745-752.
5. Koo S, Kucher N, Nguyen PL, et al. The effect of excessive anticoagulation on mortality and morbidity in hospitalized patients with anticoagulant-related major hemorrhage. Arch Intern Med. 2004;164(14):1557-1560.
6. Mina AA, Knipfer JF, Park DY, et al. Intracranial complications of preinjury anticoagulation in trauma patients with head injury. J Trauma. 2002;53(4):668-672.
7. Pieracci FM, Eachempati SR, Shou J, et al. Degree of anticoagulation, but not warfarin use itself, predicts adverse outcomes after traumatic brain injury in elderly trauma patients. J Trauma. 2007;63(3):525-530.
8. Fanikos J, Grasso-Correnti N, Shah R, et al. Major bleeding complications in a specialized anticoagulation service. Am J Cardiol. 2005;96(4):595-598.
9. Pieracci FM, Eachempati SR, Shou J, et al. Use of long-term anticoagulation is associated with traumatic intracranial hemorrhage and subsequent mortality in elderly patients hospitalized after falls: analysis of the New York State Administrative Database. J Trauma. 2007;63(3):519-524.
10. Franko J, Kish KJ, O’Connell BG, et al. Advanced age and preinjury warfarin anticoagulation increase the risk of mortality after head trauma. J Trauma. 2006; 61(1):107-110.
11. Drapkin AJ. Chronic subdural hematoma: pathophysiological basis for treatment. Br J Neurosurg. 1991; 5(5):467-473.
12. Yamamoto H, Hirashima Y, Hamada H, et al. Independent predictors of recurrence of chronic subdural hematoma: results of multivariate analysis performed using a logistic regression model. J Neurosurg. 2003;98(6):1217-1221.
13. Iantosca MR, Simon RH. Chronic subdural hematoma in adult and elderly patients. Neurosurg Clin N Am. 2000;11(3):447-454.
14. Rozzelle CJ, Wofford JL, Branch CL. Predictors of hospital mortality in older patients with subdural hematoma. J Am Geriatr Soc. 1995;43(3):240-244.
15. Wintzen AR, Tijssen JG. Subdural hematoma and oral anticoagulant therapy. Arch Neurol. 1982;39(2): 69-72.
16. Ramachandran R, Hegde T. Chronic subdural hematomas: causes of morbidity and mortality. Surg Neurol. 2007;67(4):367-372.
17. Amirjamshidi A, Eftekhar B, Abouzari M, Rashidi A. The relationship between Glasgow coma/outcome scores and abnormal CT scan findings in chronic subdural hematoma. Clin Neurol Neurosurg. 2007;109(2): 152-157.
18. Lee JY, Ebel H, Ernestus RI, Klug N. Various surgical treatments of chronic subdural hematoma and outcome in 172 patients: is membranectomy necessary? Surg Neurol. 2004;61(6):523-527.
19. Gelabert-González M, Iglesias-Pais M, García-Allut A, Martínez-Rumbo R. Chronic subdural haematoma: surgical treatment and outcome in 1000 cases. Clin Neurol Neurosurg. 2005;107(3):223-229.
20. Mattle H, Kohler S, Huber P, et al. Anticoagulation-related intracranial extracerebral haemorrhage. J Neurol Neurosurg Psychiatry. 1989;52(7):829-837.
21. Sambasivan M. An overview of chronic subdural hematoma: experience with 2300 cases. Surg Neurol. 1997;47(5):418-422.
22. Lin J, Hanigan WC, Tarantino M, Wang J. The use of recombinant activated factor VII to reverse warfarin-induced anticoagulation in patients with hemorrhages in the central nervous system: preliminary findings. J Neurosurg. 2003;98(4):737-740.
23. Freeman WD, Brott TG, Barrett KM, et al. Recombinant factor VIIa for rapid reversal of warfarin anticoagulation in acute intracranial hemorrhage. Mayo Clin Proc. 2004;79(12):1495-1500.
24. Dager WE, King JH, Regalia RC, et al. Reversal of elevated international normalized ratios and bleeding with low-dose recombinant activated factor VII in patients receiving warfarin. Pharmacotherapy. 2006;26(8): 1091-1098.
25. Denas G, Marzot F, Offelli P, et al. Effectiveness and safety of a management protocol to correct over-anticoagulation with oral vitamin K: a retrospective study of 1,043 cases. J Thromb Thrombolysis. 2008 Mar 13; [Epub ahead of print].
26. Boulis NM, Bobek MP, Schmaier A, Hoff JT. Use of factor IX complex in warfarin-related intracranial hemorrhage. Neurosurgery. 1999;45(5):1113-1118.
27. Kissela BM, Eckman MH. Cost effectiveness of recombinant factor VIIa for treatment of intracerebral hemorrhage. BMC Neurol. 2008;8:17.
28. Ernestus RI, Beldzinski P, Lanfermann H, Klug N. Chronic subdural hematoma: surgical treatment and outcome in 104 patients. Surg Neurol. 1997;48(3): 220-225.
29. Merlicco G, Pierangeli E, di Padova PL. Chronic subdural hematomas in adults: prognostic factors: analysis of 70 cases. Neurosurg Rev. 1995;18(4):247-251.
1. Pullicino P, Thompson JL. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med. 2003;348(3): 256-257.
2. Hurlen M, Abdelnoor M, Smith P, et al. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med. 2002;347(13):969-974.
3. DeSilvey DL. Clinical trials: advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Am J Geriatr Cardiol. 2005;14(2):98-99.
4. Fang MC, Chang Y, Hylek EM, et al. Advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Ann Intern Med. 2004;141(10):745-752.
5. Koo S, Kucher N, Nguyen PL, et al. The effect of excessive anticoagulation on mortality and morbidity in hospitalized patients with anticoagulant-related major hemorrhage. Arch Intern Med. 2004;164(14):1557-1560.
6. Mina AA, Knipfer JF, Park DY, et al. Intracranial complications of preinjury anticoagulation in trauma patients with head injury. J Trauma. 2002;53(4):668-672.
7. Pieracci FM, Eachempati SR, Shou J, et al. Degree of anticoagulation, but not warfarin use itself, predicts adverse outcomes after traumatic brain injury in elderly trauma patients. J Trauma. 2007;63(3):525-530.
8. Fanikos J, Grasso-Correnti N, Shah R, et al. Major bleeding complications in a specialized anticoagulation service. Am J Cardiol. 2005;96(4):595-598.
9. Pieracci FM, Eachempati SR, Shou J, et al. Use of long-term anticoagulation is associated with traumatic intracranial hemorrhage and subsequent mortality in elderly patients hospitalized after falls: analysis of the New York State Administrative Database. J Trauma. 2007;63(3):519-524.
10. Franko J, Kish KJ, O’Connell BG, et al. Advanced age and preinjury warfarin anticoagulation increase the risk of mortality after head trauma. J Trauma. 2006; 61(1):107-110.
11. Drapkin AJ. Chronic subdural hematoma: pathophysiological basis for treatment. Br J Neurosurg. 1991; 5(5):467-473.
12. Yamamoto H, Hirashima Y, Hamada H, et al. Independent predictors of recurrence of chronic subdural hematoma: results of multivariate analysis performed using a logistic regression model. J Neurosurg. 2003;98(6):1217-1221.
13. Iantosca MR, Simon RH. Chronic subdural hematoma in adult and elderly patients. Neurosurg Clin N Am. 2000;11(3):447-454.
14. Rozzelle CJ, Wofford JL, Branch CL. Predictors of hospital mortality in older patients with subdural hematoma. J Am Geriatr Soc. 1995;43(3):240-244.
15. Wintzen AR, Tijssen JG. Subdural hematoma and oral anticoagulant therapy. Arch Neurol. 1982;39(2): 69-72.
16. Ramachandran R, Hegde T. Chronic subdural hematomas: causes of morbidity and mortality. Surg Neurol. 2007;67(4):367-372.
17. Amirjamshidi A, Eftekhar B, Abouzari M, Rashidi A. The relationship between Glasgow coma/outcome scores and abnormal CT scan findings in chronic subdural hematoma. Clin Neurol Neurosurg. 2007;109(2): 152-157.
18. Lee JY, Ebel H, Ernestus RI, Klug N. Various surgical treatments of chronic subdural hematoma and outcome in 172 patients: is membranectomy necessary? Surg Neurol. 2004;61(6):523-527.
19. Gelabert-González M, Iglesias-Pais M, García-Allut A, Martínez-Rumbo R. Chronic subdural haematoma: surgical treatment and outcome in 1000 cases. Clin Neurol Neurosurg. 2005;107(3):223-229.
20. Mattle H, Kohler S, Huber P, et al. Anticoagulation-related intracranial extracerebral haemorrhage. J Neurol Neurosurg Psychiatry. 1989;52(7):829-837.
21. Sambasivan M. An overview of chronic subdural hematoma: experience with 2300 cases. Surg Neurol. 1997;47(5):418-422.
22. Lin J, Hanigan WC, Tarantino M, Wang J. The use of recombinant activated factor VII to reverse warfarin-induced anticoagulation in patients with hemorrhages in the central nervous system: preliminary findings. J Neurosurg. 2003;98(4):737-740.
23. Freeman WD, Brott TG, Barrett KM, et al. Recombinant factor VIIa for rapid reversal of warfarin anticoagulation in acute intracranial hemorrhage. Mayo Clin Proc. 2004;79(12):1495-1500.
24. Dager WE, King JH, Regalia RC, et al. Reversal of elevated international normalized ratios and bleeding with low-dose recombinant activated factor VII in patients receiving warfarin. Pharmacotherapy. 2006;26(8): 1091-1098.
25. Denas G, Marzot F, Offelli P, et al. Effectiveness and safety of a management protocol to correct over-anticoagulation with oral vitamin K: a retrospective study of 1,043 cases. J Thromb Thrombolysis. 2008 Mar 13; [Epub ahead of print].
26. Boulis NM, Bobek MP, Schmaier A, Hoff JT. Use of factor IX complex in warfarin-related intracranial hemorrhage. Neurosurgery. 1999;45(5):1113-1118.
27. Kissela BM, Eckman MH. Cost effectiveness of recombinant factor VIIa for treatment of intracerebral hemorrhage. BMC Neurol. 2008;8:17.
28. Ernestus RI, Beldzinski P, Lanfermann H, Klug N. Chronic subdural hematoma: surgical treatment and outcome in 104 patients. Surg Neurol. 1997;48(3): 220-225.
29. Merlicco G, Pierangeli E, di Padova PL. Chronic subdural hematomas in adults: prognostic factors: analysis of 70 cases. Neurosurg Rev. 1995;18(4):247-251.
A young pregnant woman with shortness of breath
A 21-year-old woman who is 12 weeks pregnant according to the date of her last menstrual period comes to the emergency department with shortness of breath and chest pain.
One week ago she began experiencing pre-syncope and shortness of breath on minimal exertion and then even at rest on most days. The shortness of breath worsened throughout the week, eventually limiting her daily activities to such a degree that she restricted herself to bed rest.
Her chest pain started today while she was sitting in church, without any apparent provocation. It is right-sided, sharp, and focal, and it does not radiate. At the same time, her shortness of breath was more severe than before, so she immediately came to the emergency department.
This is her third pregnancy; she has had one live birth and one abortion. Her last pregnancy was full-term, with routine prenatal care and no complications. However, so far during this pregnancy, she has had no prenatal care, she has not taken prenatal vitamins, and she has been unable to maintain adequate nutrition because of persistent emesis, which began early in her pregnancy and continues to occur as often as two or three times daily. She has lost 20 pounds over the past 12 weeks.
She says she has no close contacts who are sick, and she has had no fever, diarrhea, dysuria, urinary frequency or urgency, palpitations, swelling of the legs or feet, blurry vision, or increase in neck girth. She says she does not smoke or use alcohol or illicit substances. Her only previous surgery was laser-assisted in situ keratoplasty (LASIK) eye surgery in 1998. She is allergic to seafood only. She has not eaten at any new places recently. She is up to date with her childhood vaccinations. She has no family history of hypercoagulability or venous thrombotic events.
PHYSICAL EXAMINATION
She is breathing rapidly—as fast as 45 breaths per minute. Her temperature is 37.2°C (98.9°F), blood pressure 95/60 mm Hg, oxygen saturation 100% while on 10 L of oxygen using a nonrebreather mask, pulse 102 beats per minute, and weight 55.9 kg (123.2 pounds). She appears alert, oriented, and comfortable, with a thin body habitus. She has no jugular venous distention, neck mass, or thyromegaly. Her lungs are clear to auscultation, with no wheezes or rales. The cardiovascular examination is normal. She has a regular heart rate and rhythm, normal S1 and S2 sounds, and no rubs, clicks, or murmurs. Pulses in the extremities are normal, and she has no peripheral edema. The neurologic examination is normal.
Electrocardiography shows sinus tachycardia with first-degree atrioventricular block.
DIFFERENTIAL DIAGNOSIS
1. At this point, which is the most probable cause of her symptoms?
- Pulmonary embolism
- Peripartum cardiomyopathy
- Acute coronary syndrome
- Aortic dissection
- Expected physiologic changes of pregnancy
Pulmonary embolism would be the most probable diagnosis, given the patient’s pregnancy, shortness of breath, and tachycardia and the pleuritic quality of her chest pain.
Peripartum cardiomyopathy is also a possible cause, as it may present with profound shortness of breath and markedly decreased cardiac function. But it is much less likely in this patient because she is early in her pregnancy, and peripartum cardiomyopathy usually is seen during the last month of gestation or the first months after delivery.
Acute coronary syndrome is unlikely, given her young age and the lack of significant risk factors or a supporting history.
Aortic dissection is unlikely in view of her medical history.
Physiologic changes of pregnancy. Many pregnant women experience a sensation of not being able to catch their breath or expand their lungs fully, as the diaphragm is limited by the gravid abdomen. They also present with dyspnea, fatigue, reduced exercise capacity, peripheral edema, or volume overload.1 However, these changes tend to occur gradually and worsen over time. This patient’s degree of shortness of breath and its sudden onset do not seem like normal physiologic changes of pregnancy.
Other possible causes of dyspnea in a pregnant woman include asthma, pleural empyema, pneumonia, and severe anemia. Asthma should be considered in anyone with a history of wheezing, cough, and dyspnea. Fever and sputum production would support a diagnosis of pneumonia or empyema. In addition, maternal heart disease (eg, endocarditis, pulmonary hypertension) complicates 0.2% to 3% of pregnancies.1
CASE CONTINUED
The emergency department staff decide to evaluate the patient for heart failure and pulmonary embolism.
Bedside echocardiography reveals an ejection fraction of 55% (normal range 50%–75%), normal heart function and size, and no valvular abnormalities.
Chest radiography is normal.
Lower-extremity duplex ultrasonography is negative for deep-vein thrombosis.
The D-dimer level is 380 ng/mL (normal range < 500 ng/mL).
The medical intensive care unit is consulted about the patient’s continued tachypnea and the possible need for intubation. A ventilation-perfusion scan is performed to screen for pulmonary embolism, and it is negative.
An obstetric team performs Doppler ultrasonography at the bedside; a fetal heartbeat can be heard, thus confirming a viable pregnancy.
The patient has normal serum levels of the cardiac enzymes troponin T and creatine kinase-MB fraction, thus all but ruling out myocardial ischemia.
The patient is admitted to the hospital the next day, and a cardiology consult is obtained.
RULING OUT PULMONARY EMBOLISM
2. Has pulmonary embolism been definitively ruled out at this point?
- Yes
- No
The answer is no. The negative ventilation-perfusion scan and normal D-dimer test in this patient are not enough to rule out pulmonary embolism. The diagnosis of pulmonary embolism should be based on the clinician’s estimation of the pretest probability of pulmonary embolism (which is based on presenting signs and symptoms), as well as on a variety of tests, including spiral computed tomography (CT), ventilation-perfusion lung scanning, and serum D-dimer testing. Signs and symptoms that may guide the clinician are chest pain (present in 70% of patients with pulmonary embolism), tachypnea (70%), cough (40%), shortness of breath (25%), and tachycardia (33%).2 A history of pregnancy, malignancy, immobility, or recent surgery may also increase the pretest probability of pulmonary embolism. In many cases, one’s clinical suspicion is highly predictive and is useful in diagnosing pulmonary embolism.
The accuracy of the tests varies widely, depending on the pretest probability of pulmonary embolism. For instance, in a patient with a high pretest probability but a low-probability ventilation-perfusion scan, the true probability of pulmonary embolism is 40%, but in a patient with a low pretest probability and a low-probability scan, the probability is only 4%.
The Wells criteria can be used to calculate the pretest probability of pulmonary embolism. Given this patient’s tachycardia and clinical presentation, her pretest probability according to the Wells criteria indicates increased risk. However, because her D-dimer test, lower-extremity Doppler test, and ventilation-perfusion scan were normal, pulmonary embolism is less likely.3
However, if one’s clinical suspicion is high enough, further investigation of pulmonary embolism would proceed despite the encouraging test results.
CASE CONTINUED
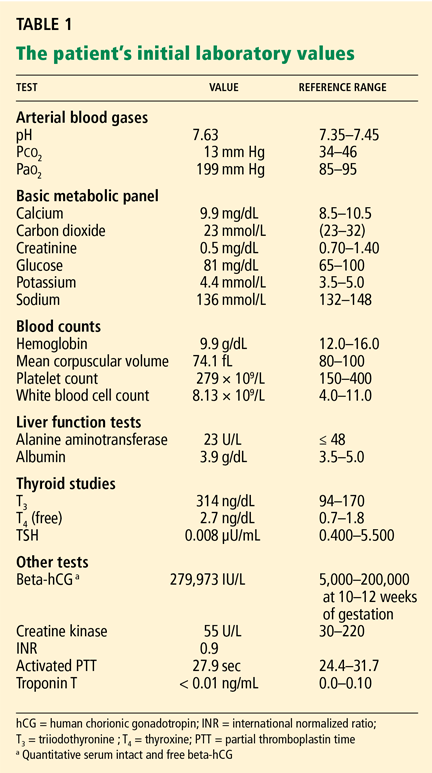
The cardiology consult team notes that her beta human chorionic gonadotropin (beta-hCG) level is much higher than would be expected at 12 weeks of pregnancy, and so they are concerned about the possibility of a molar pregnancy. In addition, her level of thyroid-stimulating hormone (TSH, or thyrotropin) is markedly low.
HYPERTHYROIDISM IN PREGNANCY
3. Which of the following would not explain this patient’s markedly low TSH level?
- Graves disease
- Molar pregnancy
- TSH-secreting pituitary adenoma
- Gestational transient thyrotoxicosis
- Twin pregnancy
Hyperthyroidism (also called thyrotoxicosis) has many causes, including but not limited to Graves disease, pituitary adenoma, struma ovarii (teratoma), hCG-secreting hydatidiform mole, and thyroid carcinoma (which is rare).4 In most of these disorders, the TSH level is low while the levels of thyroxine (T4), triiodothyronine (T3), or both are high.
Symptoms of hyperthyroidism are the effect of elevated T4 and T3 levels on the target organs themselves. Common symptoms include fever, tachycardia, tremor, stare, sweating, and lid lag. Other symptoms include nervousness, delirium, hypersensitivity to heat, flushing, palpitations, fatigue, weight loss, dyspnea, weakness, increased appetite, swelling of the legs, nausea, vomiting, diarrhea, goiter, tremor, atrial fibrillation, and cardiac failure.4 In its extreme form, called thyroid storm, thyrotoxicosis can be life-threatening. The likelihood of an impending thyroid storm can be assessed by clinical variables such as the patient’s temperature and heart rate and whether he or she has heart failure or gastrointestinal manifestations.5
Graves disease, the most common cause of hyperthyroidism in pregnancy, is due to stimulation of TSH receptors by antibodies against these receptors. Graves disease is possible in this patient, but a subsequent TSH receptor antibody test is negative.
Pituitary adenomas are one of the few causes of hyperthyroidism in which the TSH level is high, not low. Therefore, this is the correct answer.
Gestational transient thyrotoxicosis is a nonautoimmune condition that results in transient hyperthyroidism of variable severity.6 Usually, it occurs in otherwise normal pregnancies without complications, but the initial manifestation is hyper- emesis.6 It can be differentiated from Graves disease by the absence of TSH receptor antibodies and by no history of thyroid disorder.7 Common symptoms of gestational transient thyrotoxicosis include weight loss (or failure to gain weight), tachycardia, and fatigue.
The reason for the transient rise in T4 may be that beta-hCG is structurally similar to TSH (and also to luteinizing hormone and follicle-stimulating hormone), so that it has mild thyroid-stimulating effects.7 Sustained high levels of beta-hCG may in time give rise to the manifestations of thyrotoxicosis.
Molar pregnancy also can cause hyper-thyroidism via elevated levels of beta-hCG. However, twin pregnancy is more common and can produce sustained levels of beta-hCG above 100,000 IU/L. In most cases of twin pregnancy, the TSH level is decreased and the T4 level transiently elevated.6 The elevated beta-hCG and the subsequent thyrotropic manifestations are thought to be directly related, and symptoms resolve when beta-hCG levels go down.6
In most cases of hyperthyroidism in pregnancy, the acute condition can be managed by a short (≤ 2-month) course of a beta-blocker. In rare cases, propylthiouracil treatment may be required. Gestational transient thyrotoxicosis is not associated with detrimental outcomes.
Case continued
Our patient’s TSH level is low and her free T4 and T3 levels are elevated. Her high beta-hCG level may be stimulating the thyroid gland and may account for the low TSH value, as well as for her tachycardia, emesis, shortness of breath, and weight loss.
After an obstetric consult, it is determined that our patient has a viable pregnancy. However, further investigation with transvaginal ultrasonography reveals that she has two viable, single-placenta, intrauterine gestations, separated by a thin chorionic membrane.
Beta-hCG and free T4 levels are significantly higher in twin pregnancies than in single pregnancies, especially in the early stages.6 In our patient, the twin pregnancy led to the elevated beta-hCG, which eventually manifested as thyrotoxicosis, which caused the shortness of breath, hyperemesis, weight loss, tachycardia, and nausea.
Shortness of breath in patients with thyrotoxicosis is well recognized but not well explained. It may be caused by decreased lung compliance, engorged capillaries in the lung, or left ventricular failure, as well as by chest pain due to increased myocardial demand or coronary artery vasospasm.4 The dyspnea is present at rest and during exertion, and the high metabolic rate is thought to lead to an inappropriate response of the ventilatory system.3,8
WHAT TREATMENT?
4. How would you treat this patient at this point?
- No drug therapy, just supportive care
- Propranolol (Inderal)
- Levothyroxine
- Propylthiouracil
Several types of drugs are used to manage hyperthyroidism.
Antithyroid drugs such as propylthiouracil, methimazole (Northyx, Tapazole), and carbimazole block thyroid hormone synthesis by inhibiting thyroid peroxidase. Propylthiouracil also blocks peripheral conversion of T4 to T3. Side effects of these agents include abnormal sense of taste, pruritus, urticaria, agranulocytosis, and hepatotoxicity.4
Usually, hyperthyroidism is treated with propylthiouracil at the smallest effective dose. This has been proven to be safe to the fetus and mother during pregnancy.9 Propylthiouracil and the other drugs in its class cross the placenta, but propylthiouracil crosses at one-quarter the rate of the other two.9
Beta-blockers are effective in the acute phase of thyrotoxicosis against tachycardia, hypertension, and atrial fibrillation. They also decrease conversion of T4 to T3, which is an added benefit. Beta-blockers can be tapered as thyroid hormone levels decrease.
A short course of a short-acting beta-blocker would be an option for our patient and would decrease her symptoms, although she does not have the typical markedly elevated T4 or T3 levels. In the long term, a beta-blocker would present a fetal risk, but short courses can be tolerated without incident.9
Radioactive iodine 131 is used in patients with Graves disease. 131Iodine therapy is safe for most adults, but in pregnancy its use is contraindicated. Fetal thyroid tissue is thought to be present after 10 weeks of gestation and could be damaged by the use of radioactive iodine. Another warning with the use of radioactive iodine is that patients should avoid close contact with other adults for a few days after treatment, and should avoid close contact with children and pregnant women for 2 to 3 weeks after treatment because of the risk of exposure to radiation emanating from the thyroid gland.
Levothyroxine is a treatment for hypothyroidism, not hyperthyroidism.
CASE CONTINUED
Our patient is treated with propranolol and monitored for several days in the hospital, during which her symptoms markedly improve. She is discharged without complications.
TAKE-HOME POINTS
The evaluation of shortness of breath in adult patients can be difficult, given the many possible causes. It is especially challenging in pregnant patients, since normal physiologic changes of pregnancy may produce these symptoms.
In many instances, cardiomyopathy must be suspected if a pregnant patient complains of shortness of breath. However, it is not the only possible cause.
- Dobbenga-Rhodes YA, Prive AM. Assessment and evaluation of the woman with cardiac disease during pregnancy. J Perinat Neonatal Nurs 2006; 20:295–302.
- Carman TL, Deitcher SR. Advances in diagnosing and excluding pulmonary embolism: spiral CT and D-dimer measurement. Cleve Clin J Med 2002; 69:721–729.
- Wells PS, Anderson DR, Rodger M, et al. Derivation of a simple clinical model to categorize patients’ probability of pulmonary embolism: increasing the model’s utility with the SimpliRED D-dimer. Thromb Haemost 2000; 83:416–420.
- Nayak B, Burman K. Thyrotoxicosis and thyroid storm. Endocrinol Metab Clin North Am 2006; 35:663–686.
- Burch HB, Wartofsky L. Life-threatening thyrotoxicosis. Thyroid storm. Endocrinol Metab Clin North Am 1993; 22:263–277.
- Grün JP, Meuris S, De Nayer P, Glinoer D. The thyrotrophic role of human chorionic gonadotrophin (hCG) in the early stages of twin (versus single) pregnancies. Clin Endocrinol (Oxf) 1997; 46:719–725.
- Glinoer D, De Nayer P, Robyn C, Lejeune B, Kinthaert J, Meuris S. Serum levels of intact human chorionic gonadotropin (HCG) and its free alpha and beta subunits, in relation to maternal thyroid stimulation during normal pregnancy. J Endocrinol Invest 1993; 16:881–888.
- Small D, Gibbons W, Levy RD, de Lucas P, Gregory W, Cosio MG. Exertional dyspnea and ventilation in hyper-thyroidism. Chest 1992; 101:1268–1273.
- Atkins P, Cohen SB, Phillips BJ. Drug therapy for hyper-thyroidism in pregnancy: safety issues for mother and fetus. Drug Saf 2000; 23:229–244.
A 21-year-old woman who is 12 weeks pregnant according to the date of her last menstrual period comes to the emergency department with shortness of breath and chest pain.
One week ago she began experiencing pre-syncope and shortness of breath on minimal exertion and then even at rest on most days. The shortness of breath worsened throughout the week, eventually limiting her daily activities to such a degree that she restricted herself to bed rest.
Her chest pain started today while she was sitting in church, without any apparent provocation. It is right-sided, sharp, and focal, and it does not radiate. At the same time, her shortness of breath was more severe than before, so she immediately came to the emergency department.
This is her third pregnancy; she has had one live birth and one abortion. Her last pregnancy was full-term, with routine prenatal care and no complications. However, so far during this pregnancy, she has had no prenatal care, she has not taken prenatal vitamins, and she has been unable to maintain adequate nutrition because of persistent emesis, which began early in her pregnancy and continues to occur as often as two or three times daily. She has lost 20 pounds over the past 12 weeks.
She says she has no close contacts who are sick, and she has had no fever, diarrhea, dysuria, urinary frequency or urgency, palpitations, swelling of the legs or feet, blurry vision, or increase in neck girth. She says she does not smoke or use alcohol or illicit substances. Her only previous surgery was laser-assisted in situ keratoplasty (LASIK) eye surgery in 1998. She is allergic to seafood only. She has not eaten at any new places recently. She is up to date with her childhood vaccinations. She has no family history of hypercoagulability or venous thrombotic events.
PHYSICAL EXAMINATION
She is breathing rapidly—as fast as 45 breaths per minute. Her temperature is 37.2°C (98.9°F), blood pressure 95/60 mm Hg, oxygen saturation 100% while on 10 L of oxygen using a nonrebreather mask, pulse 102 beats per minute, and weight 55.9 kg (123.2 pounds). She appears alert, oriented, and comfortable, with a thin body habitus. She has no jugular venous distention, neck mass, or thyromegaly. Her lungs are clear to auscultation, with no wheezes or rales. The cardiovascular examination is normal. She has a regular heart rate and rhythm, normal S1 and S2 sounds, and no rubs, clicks, or murmurs. Pulses in the extremities are normal, and she has no peripheral edema. The neurologic examination is normal.
Electrocardiography shows sinus tachycardia with first-degree atrioventricular block.
DIFFERENTIAL DIAGNOSIS
1. At this point, which is the most probable cause of her symptoms?
- Pulmonary embolism
- Peripartum cardiomyopathy
- Acute coronary syndrome
- Aortic dissection
- Expected physiologic changes of pregnancy
Pulmonary embolism would be the most probable diagnosis, given the patient’s pregnancy, shortness of breath, and tachycardia and the pleuritic quality of her chest pain.
Peripartum cardiomyopathy is also a possible cause, as it may present with profound shortness of breath and markedly decreased cardiac function. But it is much less likely in this patient because she is early in her pregnancy, and peripartum cardiomyopathy usually is seen during the last month of gestation or the first months after delivery.
Acute coronary syndrome is unlikely, given her young age and the lack of significant risk factors or a supporting history.
Aortic dissection is unlikely in view of her medical history.
Physiologic changes of pregnancy. Many pregnant women experience a sensation of not being able to catch their breath or expand their lungs fully, as the diaphragm is limited by the gravid abdomen. They also present with dyspnea, fatigue, reduced exercise capacity, peripheral edema, or volume overload.1 However, these changes tend to occur gradually and worsen over time. This patient’s degree of shortness of breath and its sudden onset do not seem like normal physiologic changes of pregnancy.
Other possible causes of dyspnea in a pregnant woman include asthma, pleural empyema, pneumonia, and severe anemia. Asthma should be considered in anyone with a history of wheezing, cough, and dyspnea. Fever and sputum production would support a diagnosis of pneumonia or empyema. In addition, maternal heart disease (eg, endocarditis, pulmonary hypertension) complicates 0.2% to 3% of pregnancies.1
CASE CONTINUED
The emergency department staff decide to evaluate the patient for heart failure and pulmonary embolism.
Bedside echocardiography reveals an ejection fraction of 55% (normal range 50%–75%), normal heart function and size, and no valvular abnormalities.
Chest radiography is normal.
Lower-extremity duplex ultrasonography is negative for deep-vein thrombosis.
The D-dimer level is 380 ng/mL (normal range < 500 ng/mL).
The medical intensive care unit is consulted about the patient’s continued tachypnea and the possible need for intubation. A ventilation-perfusion scan is performed to screen for pulmonary embolism, and it is negative.
An obstetric team performs Doppler ultrasonography at the bedside; a fetal heartbeat can be heard, thus confirming a viable pregnancy.
The patient has normal serum levels of the cardiac enzymes troponin T and creatine kinase-MB fraction, thus all but ruling out myocardial ischemia.
The patient is admitted to the hospital the next day, and a cardiology consult is obtained.
RULING OUT PULMONARY EMBOLISM
2. Has pulmonary embolism been definitively ruled out at this point?
- Yes
- No
The answer is no. The negative ventilation-perfusion scan and normal D-dimer test in this patient are not enough to rule out pulmonary embolism. The diagnosis of pulmonary embolism should be based on the clinician’s estimation of the pretest probability of pulmonary embolism (which is based on presenting signs and symptoms), as well as on a variety of tests, including spiral computed tomography (CT), ventilation-perfusion lung scanning, and serum D-dimer testing. Signs and symptoms that may guide the clinician are chest pain (present in 70% of patients with pulmonary embolism), tachypnea (70%), cough (40%), shortness of breath (25%), and tachycardia (33%).2 A history of pregnancy, malignancy, immobility, or recent surgery may also increase the pretest probability of pulmonary embolism. In many cases, one’s clinical suspicion is highly predictive and is useful in diagnosing pulmonary embolism.
The accuracy of the tests varies widely, depending on the pretest probability of pulmonary embolism. For instance, in a patient with a high pretest probability but a low-probability ventilation-perfusion scan, the true probability of pulmonary embolism is 40%, but in a patient with a low pretest probability and a low-probability scan, the probability is only 4%.
The Wells criteria can be used to calculate the pretest probability of pulmonary embolism. Given this patient’s tachycardia and clinical presentation, her pretest probability according to the Wells criteria indicates increased risk. However, because her D-dimer test, lower-extremity Doppler test, and ventilation-perfusion scan were normal, pulmonary embolism is less likely.3
However, if one’s clinical suspicion is high enough, further investigation of pulmonary embolism would proceed despite the encouraging test results.
CASE CONTINUED
The cardiology consult team notes that her beta human chorionic gonadotropin (beta-hCG) level is much higher than would be expected at 12 weeks of pregnancy, and so they are concerned about the possibility of a molar pregnancy. In addition, her level of thyroid-stimulating hormone (TSH, or thyrotropin) is markedly low.
HYPERTHYROIDISM IN PREGNANCY
3. Which of the following would not explain this patient’s markedly low TSH level?
- Graves disease
- Molar pregnancy
- TSH-secreting pituitary adenoma
- Gestational transient thyrotoxicosis
- Twin pregnancy
Hyperthyroidism (also called thyrotoxicosis) has many causes, including but not limited to Graves disease, pituitary adenoma, struma ovarii (teratoma), hCG-secreting hydatidiform mole, and thyroid carcinoma (which is rare).4 In most of these disorders, the TSH level is low while the levels of thyroxine (T4), triiodothyronine (T3), or both are high.
Symptoms of hyperthyroidism are the effect of elevated T4 and T3 levels on the target organs themselves. Common symptoms include fever, tachycardia, tremor, stare, sweating, and lid lag. Other symptoms include nervousness, delirium, hypersensitivity to heat, flushing, palpitations, fatigue, weight loss, dyspnea, weakness, increased appetite, swelling of the legs, nausea, vomiting, diarrhea, goiter, tremor, atrial fibrillation, and cardiac failure.4 In its extreme form, called thyroid storm, thyrotoxicosis can be life-threatening. The likelihood of an impending thyroid storm can be assessed by clinical variables such as the patient’s temperature and heart rate and whether he or she has heart failure or gastrointestinal manifestations.5
Graves disease, the most common cause of hyperthyroidism in pregnancy, is due to stimulation of TSH receptors by antibodies against these receptors. Graves disease is possible in this patient, but a subsequent TSH receptor antibody test is negative.
Pituitary adenomas are one of the few causes of hyperthyroidism in which the TSH level is high, not low. Therefore, this is the correct answer.
Gestational transient thyrotoxicosis is a nonautoimmune condition that results in transient hyperthyroidism of variable severity.6 Usually, it occurs in otherwise normal pregnancies without complications, but the initial manifestation is hyper- emesis.6 It can be differentiated from Graves disease by the absence of TSH receptor antibodies and by no history of thyroid disorder.7 Common symptoms of gestational transient thyrotoxicosis include weight loss (or failure to gain weight), tachycardia, and fatigue.
The reason for the transient rise in T4 may be that beta-hCG is structurally similar to TSH (and also to luteinizing hormone and follicle-stimulating hormone), so that it has mild thyroid-stimulating effects.7 Sustained high levels of beta-hCG may in time give rise to the manifestations of thyrotoxicosis.
Molar pregnancy also can cause hyper-thyroidism via elevated levels of beta-hCG. However, twin pregnancy is more common and can produce sustained levels of beta-hCG above 100,000 IU/L. In most cases of twin pregnancy, the TSH level is decreased and the T4 level transiently elevated.6 The elevated beta-hCG and the subsequent thyrotropic manifestations are thought to be directly related, and symptoms resolve when beta-hCG levels go down.6
In most cases of hyperthyroidism in pregnancy, the acute condition can be managed by a short (≤ 2-month) course of a beta-blocker. In rare cases, propylthiouracil treatment may be required. Gestational transient thyrotoxicosis is not associated with detrimental outcomes.
Case continued
Our patient’s TSH level is low and her free T4 and T3 levels are elevated. Her high beta-hCG level may be stimulating the thyroid gland and may account for the low TSH value, as well as for her tachycardia, emesis, shortness of breath, and weight loss.
After an obstetric consult, it is determined that our patient has a viable pregnancy. However, further investigation with transvaginal ultrasonography reveals that she has two viable, single-placenta, intrauterine gestations, separated by a thin chorionic membrane.
Beta-hCG and free T4 levels are significantly higher in twin pregnancies than in single pregnancies, especially in the early stages.6 In our patient, the twin pregnancy led to the elevated beta-hCG, which eventually manifested as thyrotoxicosis, which caused the shortness of breath, hyperemesis, weight loss, tachycardia, and nausea.
Shortness of breath in patients with thyrotoxicosis is well recognized but not well explained. It may be caused by decreased lung compliance, engorged capillaries in the lung, or left ventricular failure, as well as by chest pain due to increased myocardial demand or coronary artery vasospasm.4 The dyspnea is present at rest and during exertion, and the high metabolic rate is thought to lead to an inappropriate response of the ventilatory system.3,8
WHAT TREATMENT?
4. How would you treat this patient at this point?
- No drug therapy, just supportive care
- Propranolol (Inderal)
- Levothyroxine
- Propylthiouracil
Several types of drugs are used to manage hyperthyroidism.
Antithyroid drugs such as propylthiouracil, methimazole (Northyx, Tapazole), and carbimazole block thyroid hormone synthesis by inhibiting thyroid peroxidase. Propylthiouracil also blocks peripheral conversion of T4 to T3. Side effects of these agents include abnormal sense of taste, pruritus, urticaria, agranulocytosis, and hepatotoxicity.4
Usually, hyperthyroidism is treated with propylthiouracil at the smallest effective dose. This has been proven to be safe to the fetus and mother during pregnancy.9 Propylthiouracil and the other drugs in its class cross the placenta, but propylthiouracil crosses at one-quarter the rate of the other two.9
Beta-blockers are effective in the acute phase of thyrotoxicosis against tachycardia, hypertension, and atrial fibrillation. They also decrease conversion of T4 to T3, which is an added benefit. Beta-blockers can be tapered as thyroid hormone levels decrease.
A short course of a short-acting beta-blocker would be an option for our patient and would decrease her symptoms, although she does not have the typical markedly elevated T4 or T3 levels. In the long term, a beta-blocker would present a fetal risk, but short courses can be tolerated without incident.9
Radioactive iodine 131 is used in patients with Graves disease. 131Iodine therapy is safe for most adults, but in pregnancy its use is contraindicated. Fetal thyroid tissue is thought to be present after 10 weeks of gestation and could be damaged by the use of radioactive iodine. Another warning with the use of radioactive iodine is that patients should avoid close contact with other adults for a few days after treatment, and should avoid close contact with children and pregnant women for 2 to 3 weeks after treatment because of the risk of exposure to radiation emanating from the thyroid gland.
Levothyroxine is a treatment for hypothyroidism, not hyperthyroidism.
CASE CONTINUED
Our patient is treated with propranolol and monitored for several days in the hospital, during which her symptoms markedly improve. She is discharged without complications.
TAKE-HOME POINTS
The evaluation of shortness of breath in adult patients can be difficult, given the many possible causes. It is especially challenging in pregnant patients, since normal physiologic changes of pregnancy may produce these symptoms.
In many instances, cardiomyopathy must be suspected if a pregnant patient complains of shortness of breath. However, it is not the only possible cause.
A 21-year-old woman who is 12 weeks pregnant according to the date of her last menstrual period comes to the emergency department with shortness of breath and chest pain.
One week ago she began experiencing pre-syncope and shortness of breath on minimal exertion and then even at rest on most days. The shortness of breath worsened throughout the week, eventually limiting her daily activities to such a degree that she restricted herself to bed rest.
Her chest pain started today while she was sitting in church, without any apparent provocation. It is right-sided, sharp, and focal, and it does not radiate. At the same time, her shortness of breath was more severe than before, so she immediately came to the emergency department.
This is her third pregnancy; she has had one live birth and one abortion. Her last pregnancy was full-term, with routine prenatal care and no complications. However, so far during this pregnancy, she has had no prenatal care, she has not taken prenatal vitamins, and she has been unable to maintain adequate nutrition because of persistent emesis, which began early in her pregnancy and continues to occur as often as two or three times daily. She has lost 20 pounds over the past 12 weeks.
She says she has no close contacts who are sick, and she has had no fever, diarrhea, dysuria, urinary frequency or urgency, palpitations, swelling of the legs or feet, blurry vision, or increase in neck girth. She says she does not smoke or use alcohol or illicit substances. Her only previous surgery was laser-assisted in situ keratoplasty (LASIK) eye surgery in 1998. She is allergic to seafood only. She has not eaten at any new places recently. She is up to date with her childhood vaccinations. She has no family history of hypercoagulability or venous thrombotic events.
PHYSICAL EXAMINATION
She is breathing rapidly—as fast as 45 breaths per minute. Her temperature is 37.2°C (98.9°F), blood pressure 95/60 mm Hg, oxygen saturation 100% while on 10 L of oxygen using a nonrebreather mask, pulse 102 beats per minute, and weight 55.9 kg (123.2 pounds). She appears alert, oriented, and comfortable, with a thin body habitus. She has no jugular venous distention, neck mass, or thyromegaly. Her lungs are clear to auscultation, with no wheezes or rales. The cardiovascular examination is normal. She has a regular heart rate and rhythm, normal S1 and S2 sounds, and no rubs, clicks, or murmurs. Pulses in the extremities are normal, and she has no peripheral edema. The neurologic examination is normal.
Electrocardiography shows sinus tachycardia with first-degree atrioventricular block.
DIFFERENTIAL DIAGNOSIS
1. At this point, which is the most probable cause of her symptoms?
- Pulmonary embolism
- Peripartum cardiomyopathy
- Acute coronary syndrome
- Aortic dissection
- Expected physiologic changes of pregnancy
Pulmonary embolism would be the most probable diagnosis, given the patient’s pregnancy, shortness of breath, and tachycardia and the pleuritic quality of her chest pain.
Peripartum cardiomyopathy is also a possible cause, as it may present with profound shortness of breath and markedly decreased cardiac function. But it is much less likely in this patient because she is early in her pregnancy, and peripartum cardiomyopathy usually is seen during the last month of gestation or the first months after delivery.
Acute coronary syndrome is unlikely, given her young age and the lack of significant risk factors or a supporting history.
Aortic dissection is unlikely in view of her medical history.
Physiologic changes of pregnancy. Many pregnant women experience a sensation of not being able to catch their breath or expand their lungs fully, as the diaphragm is limited by the gravid abdomen. They also present with dyspnea, fatigue, reduced exercise capacity, peripheral edema, or volume overload.1 However, these changes tend to occur gradually and worsen over time. This patient’s degree of shortness of breath and its sudden onset do not seem like normal physiologic changes of pregnancy.
Other possible causes of dyspnea in a pregnant woman include asthma, pleural empyema, pneumonia, and severe anemia. Asthma should be considered in anyone with a history of wheezing, cough, and dyspnea. Fever and sputum production would support a diagnosis of pneumonia or empyema. In addition, maternal heart disease (eg, endocarditis, pulmonary hypertension) complicates 0.2% to 3% of pregnancies.1
CASE CONTINUED
The emergency department staff decide to evaluate the patient for heart failure and pulmonary embolism.
Bedside echocardiography reveals an ejection fraction of 55% (normal range 50%–75%), normal heart function and size, and no valvular abnormalities.
Chest radiography is normal.
Lower-extremity duplex ultrasonography is negative for deep-vein thrombosis.
The D-dimer level is 380 ng/mL (normal range < 500 ng/mL).
The medical intensive care unit is consulted about the patient’s continued tachypnea and the possible need for intubation. A ventilation-perfusion scan is performed to screen for pulmonary embolism, and it is negative.
An obstetric team performs Doppler ultrasonography at the bedside; a fetal heartbeat can be heard, thus confirming a viable pregnancy.
The patient has normal serum levels of the cardiac enzymes troponin T and creatine kinase-MB fraction, thus all but ruling out myocardial ischemia.
The patient is admitted to the hospital the next day, and a cardiology consult is obtained.
RULING OUT PULMONARY EMBOLISM
2. Has pulmonary embolism been definitively ruled out at this point?
- Yes
- No
The answer is no. The negative ventilation-perfusion scan and normal D-dimer test in this patient are not enough to rule out pulmonary embolism. The diagnosis of pulmonary embolism should be based on the clinician’s estimation of the pretest probability of pulmonary embolism (which is based on presenting signs and symptoms), as well as on a variety of tests, including spiral computed tomography (CT), ventilation-perfusion lung scanning, and serum D-dimer testing. Signs and symptoms that may guide the clinician are chest pain (present in 70% of patients with pulmonary embolism), tachypnea (70%), cough (40%), shortness of breath (25%), and tachycardia (33%).2 A history of pregnancy, malignancy, immobility, or recent surgery may also increase the pretest probability of pulmonary embolism. In many cases, one’s clinical suspicion is highly predictive and is useful in diagnosing pulmonary embolism.
The accuracy of the tests varies widely, depending on the pretest probability of pulmonary embolism. For instance, in a patient with a high pretest probability but a low-probability ventilation-perfusion scan, the true probability of pulmonary embolism is 40%, but in a patient with a low pretest probability and a low-probability scan, the probability is only 4%.
The Wells criteria can be used to calculate the pretest probability of pulmonary embolism. Given this patient’s tachycardia and clinical presentation, her pretest probability according to the Wells criteria indicates increased risk. However, because her D-dimer test, lower-extremity Doppler test, and ventilation-perfusion scan were normal, pulmonary embolism is less likely.3
However, if one’s clinical suspicion is high enough, further investigation of pulmonary embolism would proceed despite the encouraging test results.
CASE CONTINUED
The cardiology consult team notes that her beta human chorionic gonadotropin (beta-hCG) level is much higher than would be expected at 12 weeks of pregnancy, and so they are concerned about the possibility of a molar pregnancy. In addition, her level of thyroid-stimulating hormone (TSH, or thyrotropin) is markedly low.
HYPERTHYROIDISM IN PREGNANCY
3. Which of the following would not explain this patient’s markedly low TSH level?
- Graves disease
- Molar pregnancy
- TSH-secreting pituitary adenoma
- Gestational transient thyrotoxicosis
- Twin pregnancy
Hyperthyroidism (also called thyrotoxicosis) has many causes, including but not limited to Graves disease, pituitary adenoma, struma ovarii (teratoma), hCG-secreting hydatidiform mole, and thyroid carcinoma (which is rare).4 In most of these disorders, the TSH level is low while the levels of thyroxine (T4), triiodothyronine (T3), or both are high.
Symptoms of hyperthyroidism are the effect of elevated T4 and T3 levels on the target organs themselves. Common symptoms include fever, tachycardia, tremor, stare, sweating, and lid lag. Other symptoms include nervousness, delirium, hypersensitivity to heat, flushing, palpitations, fatigue, weight loss, dyspnea, weakness, increased appetite, swelling of the legs, nausea, vomiting, diarrhea, goiter, tremor, atrial fibrillation, and cardiac failure.4 In its extreme form, called thyroid storm, thyrotoxicosis can be life-threatening. The likelihood of an impending thyroid storm can be assessed by clinical variables such as the patient’s temperature and heart rate and whether he or she has heart failure or gastrointestinal manifestations.5
Graves disease, the most common cause of hyperthyroidism in pregnancy, is due to stimulation of TSH receptors by antibodies against these receptors. Graves disease is possible in this patient, but a subsequent TSH receptor antibody test is negative.
Pituitary adenomas are one of the few causes of hyperthyroidism in which the TSH level is high, not low. Therefore, this is the correct answer.
Gestational transient thyrotoxicosis is a nonautoimmune condition that results in transient hyperthyroidism of variable severity.6 Usually, it occurs in otherwise normal pregnancies without complications, but the initial manifestation is hyper- emesis.6 It can be differentiated from Graves disease by the absence of TSH receptor antibodies and by no history of thyroid disorder.7 Common symptoms of gestational transient thyrotoxicosis include weight loss (or failure to gain weight), tachycardia, and fatigue.
The reason for the transient rise in T4 may be that beta-hCG is structurally similar to TSH (and also to luteinizing hormone and follicle-stimulating hormone), so that it has mild thyroid-stimulating effects.7 Sustained high levels of beta-hCG may in time give rise to the manifestations of thyrotoxicosis.
Molar pregnancy also can cause hyper-thyroidism via elevated levels of beta-hCG. However, twin pregnancy is more common and can produce sustained levels of beta-hCG above 100,000 IU/L. In most cases of twin pregnancy, the TSH level is decreased and the T4 level transiently elevated.6 The elevated beta-hCG and the subsequent thyrotropic manifestations are thought to be directly related, and symptoms resolve when beta-hCG levels go down.6
In most cases of hyperthyroidism in pregnancy, the acute condition can be managed by a short (≤ 2-month) course of a beta-blocker. In rare cases, propylthiouracil treatment may be required. Gestational transient thyrotoxicosis is not associated with detrimental outcomes.
Case continued
Our patient’s TSH level is low and her free T4 and T3 levels are elevated. Her high beta-hCG level may be stimulating the thyroid gland and may account for the low TSH value, as well as for her tachycardia, emesis, shortness of breath, and weight loss.
After an obstetric consult, it is determined that our patient has a viable pregnancy. However, further investigation with transvaginal ultrasonography reveals that she has two viable, single-placenta, intrauterine gestations, separated by a thin chorionic membrane.
Beta-hCG and free T4 levels are significantly higher in twin pregnancies than in single pregnancies, especially in the early stages.6 In our patient, the twin pregnancy led to the elevated beta-hCG, which eventually manifested as thyrotoxicosis, which caused the shortness of breath, hyperemesis, weight loss, tachycardia, and nausea.
Shortness of breath in patients with thyrotoxicosis is well recognized but not well explained. It may be caused by decreased lung compliance, engorged capillaries in the lung, or left ventricular failure, as well as by chest pain due to increased myocardial demand or coronary artery vasospasm.4 The dyspnea is present at rest and during exertion, and the high metabolic rate is thought to lead to an inappropriate response of the ventilatory system.3,8
WHAT TREATMENT?
4. How would you treat this patient at this point?
- No drug therapy, just supportive care
- Propranolol (Inderal)
- Levothyroxine
- Propylthiouracil
Several types of drugs are used to manage hyperthyroidism.
Antithyroid drugs such as propylthiouracil, methimazole (Northyx, Tapazole), and carbimazole block thyroid hormone synthesis by inhibiting thyroid peroxidase. Propylthiouracil also blocks peripheral conversion of T4 to T3. Side effects of these agents include abnormal sense of taste, pruritus, urticaria, agranulocytosis, and hepatotoxicity.4
Usually, hyperthyroidism is treated with propylthiouracil at the smallest effective dose. This has been proven to be safe to the fetus and mother during pregnancy.9 Propylthiouracil and the other drugs in its class cross the placenta, but propylthiouracil crosses at one-quarter the rate of the other two.9
Beta-blockers are effective in the acute phase of thyrotoxicosis against tachycardia, hypertension, and atrial fibrillation. They also decrease conversion of T4 to T3, which is an added benefit. Beta-blockers can be tapered as thyroid hormone levels decrease.
A short course of a short-acting beta-blocker would be an option for our patient and would decrease her symptoms, although she does not have the typical markedly elevated T4 or T3 levels. In the long term, a beta-blocker would present a fetal risk, but short courses can be tolerated without incident.9
Radioactive iodine 131 is used in patients with Graves disease. 131Iodine therapy is safe for most adults, but in pregnancy its use is contraindicated. Fetal thyroid tissue is thought to be present after 10 weeks of gestation and could be damaged by the use of radioactive iodine. Another warning with the use of radioactive iodine is that patients should avoid close contact with other adults for a few days after treatment, and should avoid close contact with children and pregnant women for 2 to 3 weeks after treatment because of the risk of exposure to radiation emanating from the thyroid gland.
Levothyroxine is a treatment for hypothyroidism, not hyperthyroidism.
CASE CONTINUED
Our patient is treated with propranolol and monitored for several days in the hospital, during which her symptoms markedly improve. She is discharged without complications.
TAKE-HOME POINTS
The evaluation of shortness of breath in adult patients can be difficult, given the many possible causes. It is especially challenging in pregnant patients, since normal physiologic changes of pregnancy may produce these symptoms.
In many instances, cardiomyopathy must be suspected if a pregnant patient complains of shortness of breath. However, it is not the only possible cause.
- Dobbenga-Rhodes YA, Prive AM. Assessment and evaluation of the woman with cardiac disease during pregnancy. J Perinat Neonatal Nurs 2006; 20:295–302.
- Carman TL, Deitcher SR. Advances in diagnosing and excluding pulmonary embolism: spiral CT and D-dimer measurement. Cleve Clin J Med 2002; 69:721–729.
- Wells PS, Anderson DR, Rodger M, et al. Derivation of a simple clinical model to categorize patients’ probability of pulmonary embolism: increasing the model’s utility with the SimpliRED D-dimer. Thromb Haemost 2000; 83:416–420.
- Nayak B, Burman K. Thyrotoxicosis and thyroid storm. Endocrinol Metab Clin North Am 2006; 35:663–686.
- Burch HB, Wartofsky L. Life-threatening thyrotoxicosis. Thyroid storm. Endocrinol Metab Clin North Am 1993; 22:263–277.
- Grün JP, Meuris S, De Nayer P, Glinoer D. The thyrotrophic role of human chorionic gonadotrophin (hCG) in the early stages of twin (versus single) pregnancies. Clin Endocrinol (Oxf) 1997; 46:719–725.
- Glinoer D, De Nayer P, Robyn C, Lejeune B, Kinthaert J, Meuris S. Serum levels of intact human chorionic gonadotropin (HCG) and its free alpha and beta subunits, in relation to maternal thyroid stimulation during normal pregnancy. J Endocrinol Invest 1993; 16:881–888.
- Small D, Gibbons W, Levy RD, de Lucas P, Gregory W, Cosio MG. Exertional dyspnea and ventilation in hyper-thyroidism. Chest 1992; 101:1268–1273.
- Atkins P, Cohen SB, Phillips BJ. Drug therapy for hyper-thyroidism in pregnancy: safety issues for mother and fetus. Drug Saf 2000; 23:229–244.
- Dobbenga-Rhodes YA, Prive AM. Assessment and evaluation of the woman with cardiac disease during pregnancy. J Perinat Neonatal Nurs 2006; 20:295–302.
- Carman TL, Deitcher SR. Advances in diagnosing and excluding pulmonary embolism: spiral CT and D-dimer measurement. Cleve Clin J Med 2002; 69:721–729.
- Wells PS, Anderson DR, Rodger M, et al. Derivation of a simple clinical model to categorize patients’ probability of pulmonary embolism: increasing the model’s utility with the SimpliRED D-dimer. Thromb Haemost 2000; 83:416–420.
- Nayak B, Burman K. Thyrotoxicosis and thyroid storm. Endocrinol Metab Clin North Am 2006; 35:663–686.
- Burch HB, Wartofsky L. Life-threatening thyrotoxicosis. Thyroid storm. Endocrinol Metab Clin North Am 1993; 22:263–277.
- Grün JP, Meuris S, De Nayer P, Glinoer D. The thyrotrophic role of human chorionic gonadotrophin (hCG) in the early stages of twin (versus single) pregnancies. Clin Endocrinol (Oxf) 1997; 46:719–725.
- Glinoer D, De Nayer P, Robyn C, Lejeune B, Kinthaert J, Meuris S. Serum levels of intact human chorionic gonadotropin (HCG) and its free alpha and beta subunits, in relation to maternal thyroid stimulation during normal pregnancy. J Endocrinol Invest 1993; 16:881–888.
- Small D, Gibbons W, Levy RD, de Lucas P, Gregory W, Cosio MG. Exertional dyspnea and ventilation in hyper-thyroidism. Chest 1992; 101:1268–1273.
- Atkins P, Cohen SB, Phillips BJ. Drug therapy for hyper-thyroidism in pregnancy: safety issues for mother and fetus. Drug Saf 2000; 23:229–244.
A judgment call
A 22-year-old African American man with sickle cell disease comes to the in his joints and chest—a presentation similar to those of his previous sickle cell crises. He is given intravenous fluids for dehydration and morphine sulfate for pain via a peripheral line, and he is admitted to the hospital.
Shortly afterward, the peripheral line becomes infiltrated. After failed attempts at peripheral cannulation, central venous cannulation via an internal jugular site is considered.
WHERE IS THE CATHETER TIP?
HAZARDS OF ABERRANT LINE PLACEMENT
Central venous catheters are commonly used to give various infusions (eg, parenteral nutrition), to draw blood, and to monitor the central venous pressure.1 The internal jugular vein is one of the preferred veins for central venous access.1,2 Normally, the anatomy of the jugular venous system and the design of the catheter facilitate proper insertion. Occasionally, however, despite proper technique, the tip of the catheter may not terminate at the desired level, resulting in aberrant placement in the internal thoracic vein, superior vena cava, azygos vein, accessory hemiazygos vein, or axillary vein.1–8
The use of chest radiographs to establish the correct placement of internal jugular and subclavian lines has been advocated and is routinely practiced.6,7 Obtaining a chest x-ray to confirm line placement is particularly important in patients with prior multiple and difficult catheterizations. The lateral view is seldom obtained when confirming central neck line placement, but when the anteroposterior view is not reassuring, it may be prudent to obtain this alternate view.
In a large retrospective analysis,9 cannulation of the azygos arch occurred in about 1.2% of insertions, and the rate was about seven times higher when the left jugular vein was used than when the right one was used. A smaller study gave the frequency of azygos arch cannulation as 0.7%.10
All procedure-related complications of central line insertion in the neck can also occur with aberrant azygos vein cannulation. These include infection, bacteremia, pneumothorax, hemothorax, arterial puncture, and various other mechanical complications. It should be noted that aberrant cannulation of the azygos arch is particularly hazardous, and that complication rates are typically higher. These complications are mainly due to the smaller vascular lumen and to the direction of blood flow in the azygos venous system.
KNOWING THE ANATOMY IS CRUCIAL
Knowledge of venous anatomy and its variants is crucial both for insertion and for ascertaining the correct placement of central venous lines.
The azygos vein has a much smaller lumen than the superior vena cava. Although the lumen size may vary significantly, the maximum diameter of the anterior arch of the azygos vein is about 6 to 7 mm,11 whereas the superior vena cava lumen is typically 1.5 to 2 cm in diameter.12 In addition, when a catheter is inserted into the superior vena cava, the direction of blood flow and the direction of the infusion are the same, but when the catheter is inserted into the azygos system, the directions of blood flow and infusion are opposite, contributing to local turbulence.
Both these factors increase the chance of puncturing the vein when the azygos arch is aberrantly cannulated for central venous access.9 Venous perforation has been reported in as many as 19% of cases in which the azygos arch was inadvertently cannulated. Venous perforation can lead to hemopericardium, hemomediastinum, and hemorrhagic pleural effusions, which can lead to significant morbidity and even death. Perforation, thrombosis, stenosis, and complete occlusion have been reported subsequent to catheter malposition in the azygos vein.13
Patients in whom the azygos vein is inadvertently cannulated may tolerate infusions and blood draws, but this does not mean that inadvertent azygos vein cannulation is acceptable, especially given the late complications of vascular perforation that can occur.9
The cannulation of the azygos vein in our patient was completely unintentional; nevertheless, the line was kept in and used for a short period for the initial rehydration and pain control and was subsequently removed without any complications.
WHEN IS CANNULATION OF THE AZYGOS VEIN AN OPTION?
In patients with previous multiple central vein cannulations, the rates of thrombosis and of fibrotic changes in these veins are high. In patients with thrombosis of both the superior vena cava and the inferior vena cava, direct insertion of a catheter into the azygos vein has been suggested as an alternate route to obtain access for dialysis.8 This approach has also been used successfully to administer total parenteral nutrition for a prolonged time in pediatric patients.14 In short, the azygos vein is never preferred for central venous access, but it can occasionally serve as an alternate route.5–9
TAKE-HOME POINTS
The radiographic assessment of an internal jugular or subclavian line may occasionally be deceptive if based solely on the anteroposterior view; confirmation may require a lateral view. We found no guidelines for using the azygos vein for central venous access. The options in cases of aberrant cannulation include leaving the line in, removing and reinserting it at the same or another site under fluoroscopy, and attempting to reposition it after changing the catheter over a guidewire.
The use of central lines found to be in an aberrant position should be driven by clinical judgment based on the urgency of the need of access, the availability or feasibility of other access options, and the intended use. The use of the azygos vein is fraught with procedural complications, as well as postprocedural complications related to vascular perforation. If the position of the catheter tip on the anteroposterior radiographic view is not satisfactory, obtaining a lateral view should be considered.
- McGee DC, Goud MK. Preventing complications of central venous catheterization. N Engl J Med. 2003; 348:1123–1133.
- Pittiruti M, Malerba M, Carriero C, Tazza L, Gui D. Which is the easiest and safest technique for central venous access? A retrospective survey of more than 5,400 cases. J Vasc Access. 2000; 1:100–107.
- Towers MJ. Preventing complications of central venous catheterization. N Engl J Med 2003; 348:2684–2686; author reply 2684–2686.
- Langston CS. The aberrant central venous catheter and its complications. Radiology. 1971; 100:55–59.
- Smith DC, Pop PM. Malposition of a total parenteral nutrition catheter in the accessory hemiazygos vein. JPEN J Parenter Enteral Nutr. 1983; 7:289–292.
- Abood GJ, Davis KA, Esposito TJ, Luchette FA, Gamelli RL. Comparison of routine chest radiograph versus clinician judgment to determine adequate central line placement in critically ill patients. J Trauma. 2007; 63:50–56.
- Gladwin MT, Slonim A, Landucci DL, Gutierrez DC, Cunnion RE. Cannulation of the internal jugular vein: is postprocedural chest radiography always necessary? Crit Care Med 1999; 27:1819–1823.
- Meranze SG, McLean GK, Stein EJ, Jordan HA. Catheter placement in the azygos system: an unusual approach to venous access. Am J Roentgenol. 1985; 144:1075–1076.
- Bankier AA, Mallek R, Wiesmayr MN, et al. Azygos arch cannulation by central venous catheters: radiographic detection of malposition and subsequent complications. J Thorac Imaging. 1997; 12:64–69.
- Langston CT. The aberrant central venous catheter and its complications. Radiology. 1971; 100:55–59.
- Heitzman ER. Radiologic appearance of the azygos vein in cardiovascular disease. Circulation. 1973; 47:628–634.
- McGowan AR, Pugatch RD. Partial obstruction of the superior vena cava. BrighamRAD. Available at: http://brighamrad.harvard.edu/Cases/bwh/hcache/58/full.html. Accessed 9/4/2008.
- Granata A, Figuera M, Castellino S, Logias F, Basile A. Azygos arch cannulation by central venous catheters for hemodialysis. J Vasc Access. 2006; 7:43–45.
- Malt RA, Kempster M. Direct azygos vein and superior vena cava cannulation for parenteral nutrition. JPEN J Parenter Enteral Nutr. 1983; 7:580–581.
A 22-year-old African American man with sickle cell disease comes to the in his joints and chest—a presentation similar to those of his previous sickle cell crises. He is given intravenous fluids for dehydration and morphine sulfate for pain via a peripheral line, and he is admitted to the hospital.
Shortly afterward, the peripheral line becomes infiltrated. After failed attempts at peripheral cannulation, central venous cannulation via an internal jugular site is considered.
WHERE IS THE CATHETER TIP?
HAZARDS OF ABERRANT LINE PLACEMENT
Central venous catheters are commonly used to give various infusions (eg, parenteral nutrition), to draw blood, and to monitor the central venous pressure.1 The internal jugular vein is one of the preferred veins for central venous access.1,2 Normally, the anatomy of the jugular venous system and the design of the catheter facilitate proper insertion. Occasionally, however, despite proper technique, the tip of the catheter may not terminate at the desired level, resulting in aberrant placement in the internal thoracic vein, superior vena cava, azygos vein, accessory hemiazygos vein, or axillary vein.1–8
The use of chest radiographs to establish the correct placement of internal jugular and subclavian lines has been advocated and is routinely practiced.6,7 Obtaining a chest x-ray to confirm line placement is particularly important in patients with prior multiple and difficult catheterizations. The lateral view is seldom obtained when confirming central neck line placement, but when the anteroposterior view is not reassuring, it may be prudent to obtain this alternate view.
In a large retrospective analysis,9 cannulation of the azygos arch occurred in about 1.2% of insertions, and the rate was about seven times higher when the left jugular vein was used than when the right one was used. A smaller study gave the frequency of azygos arch cannulation as 0.7%.10
All procedure-related complications of central line insertion in the neck can also occur with aberrant azygos vein cannulation. These include infection, bacteremia, pneumothorax, hemothorax, arterial puncture, and various other mechanical complications. It should be noted that aberrant cannulation of the azygos arch is particularly hazardous, and that complication rates are typically higher. These complications are mainly due to the smaller vascular lumen and to the direction of blood flow in the azygos venous system.
KNOWING THE ANATOMY IS CRUCIAL
Knowledge of venous anatomy and its variants is crucial both for insertion and for ascertaining the correct placement of central venous lines.
The azygos vein has a much smaller lumen than the superior vena cava. Although the lumen size may vary significantly, the maximum diameter of the anterior arch of the azygos vein is about 6 to 7 mm,11 whereas the superior vena cava lumen is typically 1.5 to 2 cm in diameter.12 In addition, when a catheter is inserted into the superior vena cava, the direction of blood flow and the direction of the infusion are the same, but when the catheter is inserted into the azygos system, the directions of blood flow and infusion are opposite, contributing to local turbulence.
Both these factors increase the chance of puncturing the vein when the azygos arch is aberrantly cannulated for central venous access.9 Venous perforation has been reported in as many as 19% of cases in which the azygos arch was inadvertently cannulated. Venous perforation can lead to hemopericardium, hemomediastinum, and hemorrhagic pleural effusions, which can lead to significant morbidity and even death. Perforation, thrombosis, stenosis, and complete occlusion have been reported subsequent to catheter malposition in the azygos vein.13
Patients in whom the azygos vein is inadvertently cannulated may tolerate infusions and blood draws, but this does not mean that inadvertent azygos vein cannulation is acceptable, especially given the late complications of vascular perforation that can occur.9
The cannulation of the azygos vein in our patient was completely unintentional; nevertheless, the line was kept in and used for a short period for the initial rehydration and pain control and was subsequently removed without any complications.
WHEN IS CANNULATION OF THE AZYGOS VEIN AN OPTION?
In patients with previous multiple central vein cannulations, the rates of thrombosis and of fibrotic changes in these veins are high. In patients with thrombosis of both the superior vena cava and the inferior vena cava, direct insertion of a catheter into the azygos vein has been suggested as an alternate route to obtain access for dialysis.8 This approach has also been used successfully to administer total parenteral nutrition for a prolonged time in pediatric patients.14 In short, the azygos vein is never preferred for central venous access, but it can occasionally serve as an alternate route.5–9
TAKE-HOME POINTS
The radiographic assessment of an internal jugular or subclavian line may occasionally be deceptive if based solely on the anteroposterior view; confirmation may require a lateral view. We found no guidelines for using the azygos vein for central venous access. The options in cases of aberrant cannulation include leaving the line in, removing and reinserting it at the same or another site under fluoroscopy, and attempting to reposition it after changing the catheter over a guidewire.
The use of central lines found to be in an aberrant position should be driven by clinical judgment based on the urgency of the need of access, the availability or feasibility of other access options, and the intended use. The use of the azygos vein is fraught with procedural complications, as well as postprocedural complications related to vascular perforation. If the position of the catheter tip on the anteroposterior radiographic view is not satisfactory, obtaining a lateral view should be considered.
A 22-year-old African American man with sickle cell disease comes to the in his joints and chest—a presentation similar to those of his previous sickle cell crises. He is given intravenous fluids for dehydration and morphine sulfate for pain via a peripheral line, and he is admitted to the hospital.
Shortly afterward, the peripheral line becomes infiltrated. After failed attempts at peripheral cannulation, central venous cannulation via an internal jugular site is considered.
WHERE IS THE CATHETER TIP?
HAZARDS OF ABERRANT LINE PLACEMENT
Central venous catheters are commonly used to give various infusions (eg, parenteral nutrition), to draw blood, and to monitor the central venous pressure.1 The internal jugular vein is one of the preferred veins for central venous access.1,2 Normally, the anatomy of the jugular venous system and the design of the catheter facilitate proper insertion. Occasionally, however, despite proper technique, the tip of the catheter may not terminate at the desired level, resulting in aberrant placement in the internal thoracic vein, superior vena cava, azygos vein, accessory hemiazygos vein, or axillary vein.1–8
The use of chest radiographs to establish the correct placement of internal jugular and subclavian lines has been advocated and is routinely practiced.6,7 Obtaining a chest x-ray to confirm line placement is particularly important in patients with prior multiple and difficult catheterizations. The lateral view is seldom obtained when confirming central neck line placement, but when the anteroposterior view is not reassuring, it may be prudent to obtain this alternate view.
In a large retrospective analysis,9 cannulation of the azygos arch occurred in about 1.2% of insertions, and the rate was about seven times higher when the left jugular vein was used than when the right one was used. A smaller study gave the frequency of azygos arch cannulation as 0.7%.10
All procedure-related complications of central line insertion in the neck can also occur with aberrant azygos vein cannulation. These include infection, bacteremia, pneumothorax, hemothorax, arterial puncture, and various other mechanical complications. It should be noted that aberrant cannulation of the azygos arch is particularly hazardous, and that complication rates are typically higher. These complications are mainly due to the smaller vascular lumen and to the direction of blood flow in the azygos venous system.
KNOWING THE ANATOMY IS CRUCIAL
Knowledge of venous anatomy and its variants is crucial both for insertion and for ascertaining the correct placement of central venous lines.
The azygos vein has a much smaller lumen than the superior vena cava. Although the lumen size may vary significantly, the maximum diameter of the anterior arch of the azygos vein is about 6 to 7 mm,11 whereas the superior vena cava lumen is typically 1.5 to 2 cm in diameter.12 In addition, when a catheter is inserted into the superior vena cava, the direction of blood flow and the direction of the infusion are the same, but when the catheter is inserted into the azygos system, the directions of blood flow and infusion are opposite, contributing to local turbulence.
Both these factors increase the chance of puncturing the vein when the azygos arch is aberrantly cannulated for central venous access.9 Venous perforation has been reported in as many as 19% of cases in which the azygos arch was inadvertently cannulated. Venous perforation can lead to hemopericardium, hemomediastinum, and hemorrhagic pleural effusions, which can lead to significant morbidity and even death. Perforation, thrombosis, stenosis, and complete occlusion have been reported subsequent to catheter malposition in the azygos vein.13
Patients in whom the azygos vein is inadvertently cannulated may tolerate infusions and blood draws, but this does not mean that inadvertent azygos vein cannulation is acceptable, especially given the late complications of vascular perforation that can occur.9
The cannulation of the azygos vein in our patient was completely unintentional; nevertheless, the line was kept in and used for a short period for the initial rehydration and pain control and was subsequently removed without any complications.
WHEN IS CANNULATION OF THE AZYGOS VEIN AN OPTION?
In patients with previous multiple central vein cannulations, the rates of thrombosis and of fibrotic changes in these veins are high. In patients with thrombosis of both the superior vena cava and the inferior vena cava, direct insertion of a catheter into the azygos vein has been suggested as an alternate route to obtain access for dialysis.8 This approach has also been used successfully to administer total parenteral nutrition for a prolonged time in pediatric patients.14 In short, the azygos vein is never preferred for central venous access, but it can occasionally serve as an alternate route.5–9
TAKE-HOME POINTS
The radiographic assessment of an internal jugular or subclavian line may occasionally be deceptive if based solely on the anteroposterior view; confirmation may require a lateral view. We found no guidelines for using the azygos vein for central venous access. The options in cases of aberrant cannulation include leaving the line in, removing and reinserting it at the same or another site under fluoroscopy, and attempting to reposition it after changing the catheter over a guidewire.
The use of central lines found to be in an aberrant position should be driven by clinical judgment based on the urgency of the need of access, the availability or feasibility of other access options, and the intended use. The use of the azygos vein is fraught with procedural complications, as well as postprocedural complications related to vascular perforation. If the position of the catheter tip on the anteroposterior radiographic view is not satisfactory, obtaining a lateral view should be considered.
- McGee DC, Goud MK. Preventing complications of central venous catheterization. N Engl J Med. 2003; 348:1123–1133.
- Pittiruti M, Malerba M, Carriero C, Tazza L, Gui D. Which is the easiest and safest technique for central venous access? A retrospective survey of more than 5,400 cases. J Vasc Access. 2000; 1:100–107.
- Towers MJ. Preventing complications of central venous catheterization. N Engl J Med 2003; 348:2684–2686; author reply 2684–2686.
- Langston CS. The aberrant central venous catheter and its complications. Radiology. 1971; 100:55–59.
- Smith DC, Pop PM. Malposition of a total parenteral nutrition catheter in the accessory hemiazygos vein. JPEN J Parenter Enteral Nutr. 1983; 7:289–292.
- Abood GJ, Davis KA, Esposito TJ, Luchette FA, Gamelli RL. Comparison of routine chest radiograph versus clinician judgment to determine adequate central line placement in critically ill patients. J Trauma. 2007; 63:50–56.
- Gladwin MT, Slonim A, Landucci DL, Gutierrez DC, Cunnion RE. Cannulation of the internal jugular vein: is postprocedural chest radiography always necessary? Crit Care Med 1999; 27:1819–1823.
- Meranze SG, McLean GK, Stein EJ, Jordan HA. Catheter placement in the azygos system: an unusual approach to venous access. Am J Roentgenol. 1985; 144:1075–1076.
- Bankier AA, Mallek R, Wiesmayr MN, et al. Azygos arch cannulation by central venous catheters: radiographic detection of malposition and subsequent complications. J Thorac Imaging. 1997; 12:64–69.
- Langston CT. The aberrant central venous catheter and its complications. Radiology. 1971; 100:55–59.
- Heitzman ER. Radiologic appearance of the azygos vein in cardiovascular disease. Circulation. 1973; 47:628–634.
- McGowan AR, Pugatch RD. Partial obstruction of the superior vena cava. BrighamRAD. Available at: http://brighamrad.harvard.edu/Cases/bwh/hcache/58/full.html. Accessed 9/4/2008.
- Granata A, Figuera M, Castellino S, Logias F, Basile A. Azygos arch cannulation by central venous catheters for hemodialysis. J Vasc Access. 2006; 7:43–45.
- Malt RA, Kempster M. Direct azygos vein and superior vena cava cannulation for parenteral nutrition. JPEN J Parenter Enteral Nutr. 1983; 7:580–581.
- McGee DC, Goud MK. Preventing complications of central venous catheterization. N Engl J Med. 2003; 348:1123–1133.
- Pittiruti M, Malerba M, Carriero C, Tazza L, Gui D. Which is the easiest and safest technique for central venous access? A retrospective survey of more than 5,400 cases. J Vasc Access. 2000; 1:100–107.
- Towers MJ. Preventing complications of central venous catheterization. N Engl J Med 2003; 348:2684–2686; author reply 2684–2686.
- Langston CS. The aberrant central venous catheter and its complications. Radiology. 1971; 100:55–59.
- Smith DC, Pop PM. Malposition of a total parenteral nutrition catheter in the accessory hemiazygos vein. JPEN J Parenter Enteral Nutr. 1983; 7:289–292.
- Abood GJ, Davis KA, Esposito TJ, Luchette FA, Gamelli RL. Comparison of routine chest radiograph versus clinician judgment to determine adequate central line placement in critically ill patients. J Trauma. 2007; 63:50–56.
- Gladwin MT, Slonim A, Landucci DL, Gutierrez DC, Cunnion RE. Cannulation of the internal jugular vein: is postprocedural chest radiography always necessary? Crit Care Med 1999; 27:1819–1823.
- Meranze SG, McLean GK, Stein EJ, Jordan HA. Catheter placement in the azygos system: an unusual approach to venous access. Am J Roentgenol. 1985; 144:1075–1076.
- Bankier AA, Mallek R, Wiesmayr MN, et al. Azygos arch cannulation by central venous catheters: radiographic detection of malposition and subsequent complications. J Thorac Imaging. 1997; 12:64–69.
- Langston CT. The aberrant central venous catheter and its complications. Radiology. 1971; 100:55–59.
- Heitzman ER. Radiologic appearance of the azygos vein in cardiovascular disease. Circulation. 1973; 47:628–634.
- McGowan AR, Pugatch RD. Partial obstruction of the superior vena cava. BrighamRAD. Available at: http://brighamrad.harvard.edu/Cases/bwh/hcache/58/full.html. Accessed 9/4/2008.
- Granata A, Figuera M, Castellino S, Logias F, Basile A. Azygos arch cannulation by central venous catheters for hemodialysis. J Vasc Access. 2006; 7:43–45.
- Malt RA, Kempster M. Direct azygos vein and superior vena cava cannulation for parenteral nutrition. JPEN J Parenter Enteral Nutr. 1983; 7:580–581.
New asthma guidelines emphasize control, regular monitoring
This review focuses on several elements in the National Asthma Education and Prevention Program’s new guidelines, the third Expert Panel Report (EPR3),1 that differ substantially from those in EPR2,2 issued in 1997 and updated in 2002.3 These differences in approach to the management of asthma described in EPR3 offer a clear potential for reducing the gap between optimal asthma care outcomes as described in guidelines and normative asthma care outcomes in the “real world.”
GREATER EMPHASIS ON CONTROL
The EPR2 guidelines2 recommended that asthma management be carried out in an algorithmic manner. Patients were classified into four severity categories: mild intermittent, mild persistent, moderate persistent, and severe persistent asthma, based on assessment of the level of symptoms (day/night), reliance on “reliever” medication, and lung function at the time of presentation. Pharmacologic management was then assigned according to each respective categorization in an evidence-based fashion.
In an ideal world, this would result in patients with asthma receiving appropriate pharmacotherapeutic agents associated with favorable asthma care outcomes, which were also advantageous from both cost- and risk-benefit standpoints. In the real world, however, this paradigm was flawed, as it relied on accurate categorization of patients in order for pharmacotherapy to be prescribed appropriately. Both providers and patients are prone to underestimate asthma severity,4,5 and for this reason many patients managed on the basis of this paradigm were undertreated.
A new paradigm, based on the assessment of asthma control, has been encouraged in the EPR3 guidelines.1
Severity and control are not synonymous
More than a decade ago, Cockroft and Swystun6 pointed out that asthma control (or lack thereof) is often used inappropriately to define asthma severity: ie, well-controlled asthma is seen as synonymous with mild asthma, and poorly controlled asthma with severe asthma.
Asthma severity can be defined as the intrinsic intensity of the disease process, while asthma control is the degree to which the manifestations of asthma are minimized. Asthma severity is clearly a determinant of asthma control, but its impact is affected by a variety of factors, including but not limited to:
- Whether appropriate medication is prescribed
- Patterns of therapeutic adherence
- The degree to which recommended measures for avoiding for clinically relevant aeroallergens are pursued.
Health care utilization, including hospitalizations and emergency department visits, correlates more closely with asthma control than with asthma severity.7–9 Indeed, a patient with severe persistent asthma who is treated appropriately with multiple “controller” medications and who takes his or her medications and avoids allergens as directed can achieve well-controlled or totally controlled asthma, and is not likely to require hospitalization or emergency department management, to miss school or work, or to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has severe persistent asthma that is well controlled.
In contrast, a patient with mild or moderate persistent asthma who does not receive appropriate instructions for avoiding allergens or taking controller medication regularly or who is poorly adherent will likely have poor asthma control. This patient is more likely to require hospitalization or emergency department management, to miss school or work, and to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has mild persistent asthma that is poorly controlled.
Assess asthma severity in the first visit, and control in subsequent visits
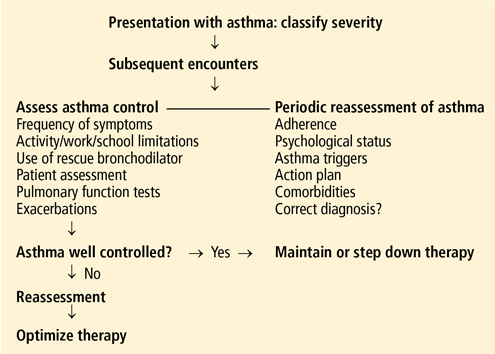
How to assess severity
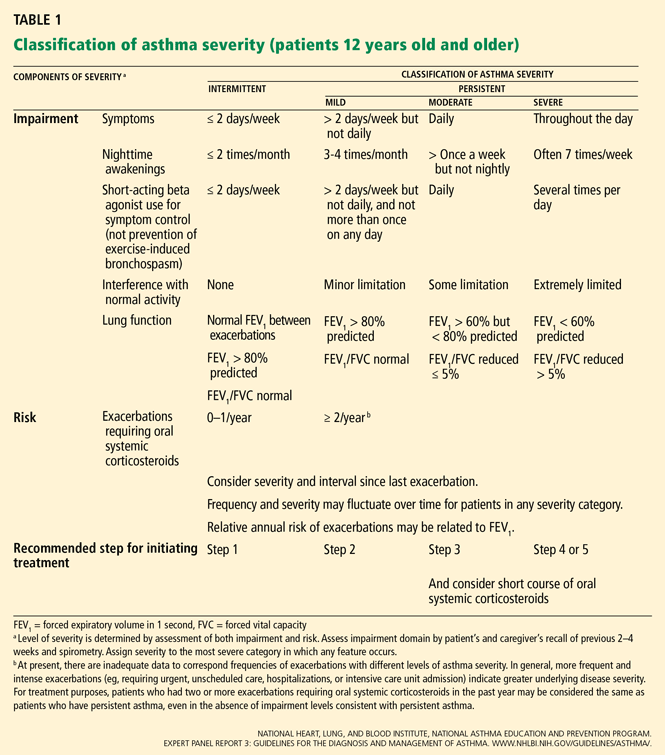
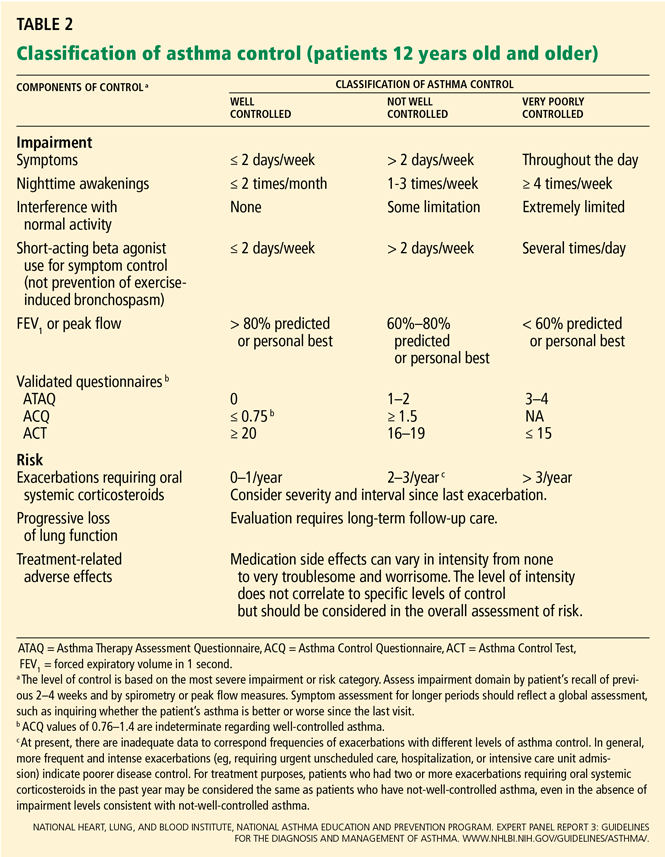
How to measure control
For all patients with asthma, regardless of severity, the goal is the same: to achieve control by reducing both impairment and risk. Asthma is classified as well controlled, not well controlled, or poorly controlled (Table 2).1
Validated tests are available to assess control
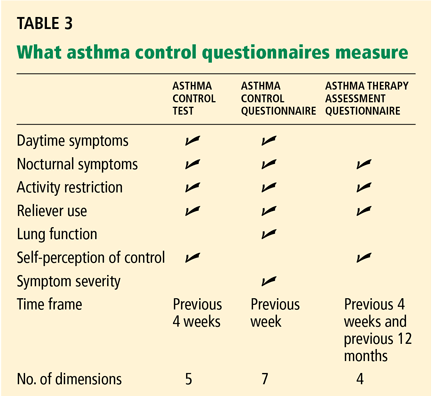
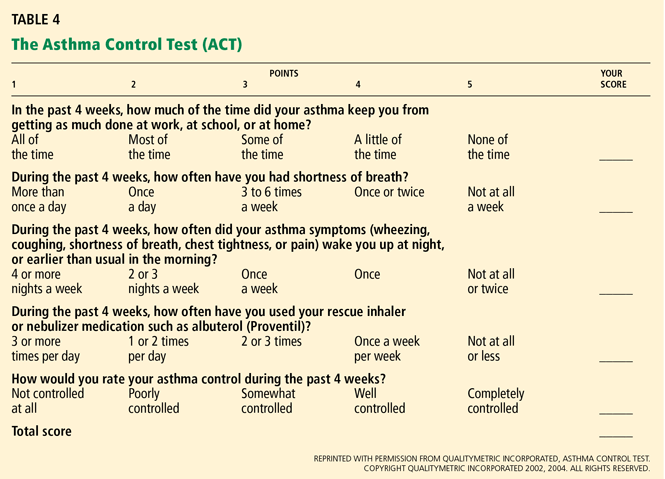
Serial testing as a quality indicator
Serial ACT scores give an objective measure of the degree to which the goals of management1 are being achieved, and in so doing can encourage optimal outcomes.14
Another use of these tests is to document whether asthma control improves over time when patients receive care from a particular physician or group. This use may become increasingly important in view of efforts underway to implement a pay-for-performance model for asthma care, in which providers will be financially rewarded for improved patient care outcomes and adherence to standards of practice based on Health Plan Employer Data and Information Set measures.15
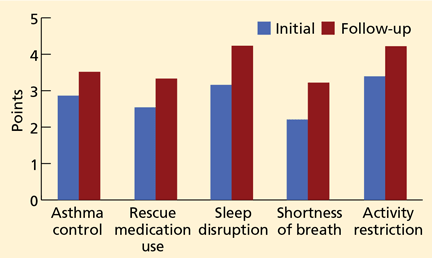
We have used the ACT in the Section of Allergy/Immunology at Cleveland Clinic for 3 years on a routine basis. All patients with asthma being seen either for the first time or as follow-up complete the ACT, which has been entered in a flow sheet in our electronic medical record, at the same time they undergo spirometry. We have shown that care in the Section of Allergy/Immunology is associated with improvement in asthma control over time, in patients who have completed serial ACT measurements at initial visits and at follow-up visits (Figure 2).
Objective measurement of lung function is also important
Serial monitoring of lung function at every patient visit with spirometry is also important, as some patients may be “poor perceivers,”16 ie, they may have little or no subjective awareness of moderate or even severe ventilatory impairment. A number of studies17,18 support the contention that symptoms and lung function are separate and independent dimensions of asthma control, and that both of them need to be assessed.
Responding to changes in control

THE STEP 3 CONTROVERSY
Salmeterol Multicenter Asthma Research Trial
In the Salmeterol Multicenter Asthma Research Trial (SMART), patients randomized to the long-acting beta agonist (LABA) salmeterol (Serevent)—particularly African Americans—had a statistically significant increase in the risk of untoward asthma care outcomes.20
SMART was launched in 1996. Patients were randomized in a double-blind fashion to receive either salmeterol 42 μg twice a day or placebo in addition to their usual asthma therapy for 28 weeks. The rate of the primary outcome (respiratory-related deaths or life-threatening experiences) was not significantly different with salmeterol than with placebo (relative risk [RR] = 1.40, 95% confidence interval [CI] 0.91–2.14). However, in 2003, the study was halted prematurely because of difficulty enrolling the targeted number of 60,000 patients, and an interim analysis that revealed significantly higher rates of secondary outcomes in subjects randomized to salmeterol. Compared with the placebo group, the salmeterol group had significantly higher rates of respiratory-related deaths (RR 2.16, 95% CI 1.06–4.41), asthma-related deaths (RR = 4.37, 95% CI = 1.25–15.34), and combined asthma-related deaths or life-threatening experiences (RR = 1.71, 95% CI 1.01–2.89). There were 13 asthma-related deaths and 37 combined asthma-related deaths or life-threatening experiences in the salmeterol group, compared with 3 and 22, respectively, in the placebo group. Of the 16 asthma deaths in the study, 13 (81%) occurred in the initial phase of SMART, when patients were recruited via print, radio, and television advertising; afterward, patients were recruited directly by investigators.
Statistically significant differences in outcomes occurred primarily in African Americans. African Americans who received salmeterol had higher rates of respiratory death or life-threatening experiences (RR = 4.10, 95% CI 1.54–10.90), the primary end point for the study, as well as higher rates of combined asthma-related deaths or life-threatening experiences (RR = 10.46, 95% CI 1.34–81.58), a secondary end point. No statistically significant differences were observed in white patients randomized to salmeterol with respect to the primary end point (RR = 1.05, 95% = 0.62–1.76); the secondary end point of combined asthma-related deaths or life-threatening experiences (RR = 1.08, 95% CI 0.55–2.14); or other end points.
Medication exposures were not tracked during the study, and allocation to inhaled corticosteroids combined with salmeterol was not randomized, so the effect of concomitant inhaled corticosteroid use cannot be determined from these data.
As a result of SMART, medications that contain either of the two LABAs, salmeterol or formoterol (Foradil), carry a black-box warning.
LABAs: Risks and benefits
Two studies21,22 have suggested that asthmatic patients who are homozygous for Arg/Arg at codon 16 of the beta-2 adrenergic receptor are predisposed to untoward asthma outcomes with regular exposure to LABAs. However, other data23–25 do not support the contention that B16 Arg/Arg patients experience adverse asthma outcomes with LABA exposure. In two recently published studies, no difference in rates of exacerbations, severe exacerbations, lung function, frequency of reliance on SABA, or nocturnal awakenings was observed in patients receiving formoterol combined with budesonide24 or salmeterol combined with fluticasone25 according to genotype. A prospective study26 also found no statistically significant difference in exacerbation rates according to beta adrenergic receptor genotype in individuals randomized to LABA monotherapy, or LABA combined with inhaled corticosteroids.
The updated EPR2 asthma guidelines,3 published in November 2002, stipulated that LABAs were the preferred controller agent to “add on” to low-dose inhaled corticosteroids for patients with moderate persistent asthma, and that the combination of low-dose inhaled corticosteroids and LABA was associated with superior outcomes: reduction of symptoms, including nocturnal awakening, increase in lung function, improvement in health-related quality of life, decreased use of “rescue” medication, and reduced rate of exacerbations and severe exacerbations, compared with higher-dose inhaled corticosteroid monotherapy. This management recommendation was categorized as level A, on the basis of data from multiple randomized, controlled, double-blinded trials.27–29 Additional evidence14,30 and data from two meta-analyses31,32 have provided further support for this recommendation, while no evidence linking LABA exposure to risk for fatal or near-fatal asthma has been found in cohort or case-control studies.33–38
Based on safety concerns, the EPR3 guidelines1 recommend that medium-dose inhaled corticosteroids be regarded as equivalent to adding LABAs to low-dose inhaled corticosteroids, and state: “the established, beneficial effects of LABA for the great majority of patients whose asthma is not well controlled with [inhaled corticosteroids] alone should be weighed against the increased risk for severe exacerbations, although uncommon, associated with daily use of LABA.”1
There is currently an honest difference of opinion39,40 among asthma specialists as to how this management recommendation for moderate persistent asthma—now depicted at “step 3” in the EPR3 guidelines (Table 4)—should be implemented. The LABA controversy was reviewed previously in the Cleveland Clinic Journal of Medicine.41
THE ROLE OF OMALIZUMAB: WEIGHING COST VS BENEFIT
The 2002 update to the EPR2 guidelines3 was issued before omalizumab (Xolair) was approved in June 2003.
Patients with severe persistent asthma are categorized in steps 5 or 6 in the EPR3 guidelines (Table 5).1 Preferred management for these patients includes inhaled corticosteroids in high doses combined with long-acting beta agonists and, for step 6 patients, oral corticosteroids.
Omalizumab was approved for management of patients with moderate or severe persistent asthma who are not achieving the goals of asthma management on inhaled corticosteroids, who exhibit a wheal-flare reaction to a perennial allergen, and whose immunoglobulin E (IgE) level is in the range of 30 to 700 IU/mL.42 Omalizumab dosing is based on the serum IgE level and on body weight.
Omalizumab, an anti-IgE monoclonal antibody
Omalizumab is a recombinant, humanized, monoclonal anti-IgE antibody that binds to IgE at the same Fc site as the high-affinity IgE receptor. Its primary mechanism of action is the binding of free IgE in the circulation, forming biologically inert, small complexes that do not activate complement and are cleared by the reticuloendothelial system.42 Its secondary mechanism of action entails a reduction in the number of high-affinity receptors on basophils, from approximately 220,000 to 8,300 receptors per cell. The latter effect was associated with a 90% reduction in histamine release from basophils in response to ex vivo challenge with dust mite allergen.43
Benefit in randomized trials
Omalizumab has been associated with statistically and clinically significant benefit in randomized, double-blind, placebo-controlled trials.44,45
Humbert et al46 randomized 419 patients whose asthma was not adequately controlled on high-dose inhaled corticosteroids and long-acting beta agonists, who were 12 to 75 years old, with reduced lung function and a history of recent asthma exacerbation, to treatment with omalizumab or placebo. Omalizumab was associated with a statistically significant reduction in the rate of asthma exacerbations and severe asthma exacerbations, as well as statistically significant improvements in asthma-related quality of life, morning peak expiratory flow rate, and asthma symptom scores.
These data support the recommendation in EPR3 to consider a trial of omalizumab in properly selected patients with severe, persistent allergic asthma.
Omalizumab is cost-beneficial in properly selected patients
The current wholesale acquisition cost of omalizumab is $532 for one 150-mg vial (David Zito, personal communication). The cost of treatment varies based on body weight and IgE level but may range from a wholesale cost of $6,388 to $38,326 per year.
However, as asthma severity increases, both direct and indirect medical expenditures increase substantially.47,48 Annual costs are approximately four times higher for severe asthma compared with mild asthma49; not only are treatment and exacerbation costs higher, but indirect costs are also disproportionately greater. Annual costs for severe asthma are significantly greater if the disease is inadequately controlled.50 For these reasons, an intervention that leads to improved outcomes for severe, poorly controlled asthma carries the potential for the greatest cost-utility for society, as it can lower direct costs by reducing the frequency and severity of exacerbations, in addition to reducing indirect medical expenditures on the basis of increased productivity and fewer days of missed work or school. The cost of omalizumab in quality-adjusted life years compares favorably with that of biologicals used in managing rheumatoid arthritis, Crohn disease, and multiple sclerosis.50
Adverse effects of omalizumab
In pivotal trials,43,44 omalizumab was associated with a substantial rate of local reactions. The rate of anaphylaxis was slightly less than 1 in 1,000, and this has been confirmed by surveillance data recorded since approval of the drug in 2003. Based on the observed risk of anaphylaxis, in July 2007, the US Food and Drug Administration added a black-box warning to the omalizumab label and stipulated that a medication guide should be provided for patients.51 The warning indicates that health care providers administering omalizumab should be prepared to manage anaphylaxis and that patients should be closely observed for an appropriate period after omalizumab administration.
The package insert also describes a numerical, but not statistically significant, increase in the rate of malignancy in patients receiving omalizumab.42 Malignancy developed in 0.5% of patients receiving omalizumab, compared with 0.2% of patients who received placebo. Because these malignancies were diagnosed over a shorter period than the time required for oncogenesis (ie, 6 months in 60% of cases), and because a heterogeneous variety of tumors was observed, there is reason to doubt these tumors were causally associated with omalizumab.
Postmarketing surveillance studies are in progress that will provide more definitive data on the potential relationship between malignancy and omalizumab exposure.
Omalizumab: Guideline recommendations
The EPR3 guidelines1 state that omalizumab is the only adjunctive therapy to demonstrate efficacy when added to high-dose inhaled corticosteroids plus long-acting beta agonists in patients with severe, persistent, allergic asthma and that evidence does not support use of the following agents, which in some cases are approved for managing other conditions and have been advocated for management of severe, refractory asthma: methotrexate, soluble interleukin (IL)-4 receptor, anti-IL-5, anti-IL-12, cyclosporine A, intravenous immune globulin, gold, troleandomycin, and colchicine. The data supporting use of macrolides were characterized as “encouraging but insufficient to support a recommendation.”
The strength of evidence for the use of omalizumab for patients in steps 5 and 6 who fulfill the criteria for its use (see above) was classified in the EPR3 guidelines1 as category B. The guidelines also say that omalizumab may be considered for adjunctive therapy in properly selected patients in step 4, as a means to avoid higher doses of inhaled corticosteroids, but that additional studies are needed to establish its utility for such patients. This recommendation was classified as category D because of the lack of published comparator trials.
ALLERGEN IMMUNOTHERAPY FOR PATIENTS WITH ASTHMA
Many patients with asthma have clinically relevant, IgE-mediated (allergic) potential to inhaled allergens.1 For patients with persistent asthma (steps 2–6 in Table 5), allergic reactions can contribute to airway inflammation, provoke symptoms, and lead to more use of medications. For this reason, identification and management of clinically relevant allergy merits consideration.52
The EPR3 guidelines1 recommend considering allergen immunotherapy for patients with mild or moderate persistent asthma (steps 2–4) who have a clinically relevant component of allergy to inhaled substances.
Changing the immune response
Allergen immunotherapy entails the incremental administration of inhalant allergens by subcutaneous injection for the purpose of inducing immune system changes in the host response. The goal of immunotherapy is to protect against allergic reactions that can be expected to occur with ongoing exposure to clinically relevant allergens.53
The immunologic changes that develop with allergen immunotherapy are complex.53,54 Successful immunotherapy results in generation of a population of CD4+/CD25+ T lymphocytes producing IL-10, transforming growth factor beta, or both. Allergen immunotherapy has been shown to block the immediate- and late-phase allergic response; to decrease recruitment of mast cells, basophils, and eosinophils on provocation or natural exposure to allergens in the skin, nose, eye, and bronchial mucosa; to blunt the seasonal rise in specific IgE; and to suppress late-phase inflammatory responses in the skin and respiratory tract. However, the efficacy of immunotherapy in relation to these immunologic changes is not completely understood.54
Many patients need skin testing
Allergen immunotherapy may be considered for patients with asthma for whom a clear relationship exists between symptoms and exposure to an allergen to which the patient is sensitive.53 Because it is often not possible to determine whether a patient is sensitive to a perennial indoor allergen (eg, dust mite) on the basis of the medical history alone,55 many patients with asthma benefit from immediate hypersensitivity skin testing to objectively assess or rule out allergy to common inhalants. In certain situations, in vitro testing may be performed, but skin testing has a higher negative predictive value and is recommended as a better screening test.56
Benefits of allergen immunotherapy
Numerous randomized, double-blind, placebo-controlled trials have shown that allergen immunotherapy is associated with benefit for reducing symptoms and medication reliance.57–63
A meta-analysis of 75 randomized, placebo-controlled studies confirmed the effectiveness of immunotherapy in asthma, with a significant reduction in asthma symptoms and medication use and with improvement in bronchial hyperreactivity.64 This meta-analysis included 36 trials of dust mite allergen, 20 of pollen, and 10 of animal dander. Immunotherapy is efficacious for pollen, mold, dust mite, cockroach, and animal allergens; however, its effectiveness is more established for dust mite, animal dander, and pollen allergens, as fewer studies have been published demonstrating efficacy using mold and cockroach allergens.53
In addition, several studies have found that children with allergic rhinitis who receive allergen immunotherapy are significantly less likely to develop asthma.65–67 Immunotherapy has also been associated with a statistically significant reduction in future sensitization to other aeroallergens.68,69
Risk of systemic reaction from allergen immunotherapy
The decision to begin allergen immunotherapy should be individualized on the basis of symptom severity, relative benefit compared with drug therapy, and whether comorbid conditions such as cardiovascular disease or beta-blocker exposure are present. These comorbid conditions are associated with heightened risk of (more serious) anaphylaxis—the major hazard of allergen immunotherapy.70 Systemic reactions during allergen immunotherapy occur at a rate of approximately 3 to 5 per 1,000 injections; for this reason, allergen immunotherapy should only be administered in a medical facility where personnel, supplies, and equipment are available to treat anaphylaxis.5
- National Heart, Lung, and Blood institute, National Asthma education and Prevention Program. Expert Panel Report 3: guidelines for the diagnosis and management of asthma. www.nhlbi.nih.gov/guidelines/asthma. Accessed 8/7/08.
- Expert Panel Report 2: Guidelines for the diagnosis and management of asthma. U.S. Department of Health and Human Services. Publication No. 97-4051; 1997.
- Expert Panel Report: Guidelines for the diagnosis and management of asthma. Update on Selected Topics—2002. J Allergy Clin Immunol 2002; 110:S141–S207.
- FitzGerald JM, Boulet LP, McIvor RA, Zimmerman S, Chapman KR. Asthma control in Canada remains suboptimal: the Reality of Asthma Control (TRAC) study. Can Respir J 2006; 13:253–259.
- Braganza S, Sharif I, Ozuah P. Documenting asthma severity: do we get it right? J Asthma 2003; 40:661–665.
- Cockcroft DW, Swystun VA. Asthma control versus asthma severity. J Allergy Clin Immunol 1996; 98:1016–1018.
- Peters SP, Jones CA, Haselkorn T, Mink DR, Valacer DJ, Weiss ST. Real-world Evaluation of Asthma Control and Treatment (REACT): findings from a national Web-based survey. J Allergy Clin Immunol. 2007; 119:1454–1461.
- Osborne ML, Vollmer WM, Pedula KL, Wilkins J, Buist AS, O’Hollaren M. Lack of correlation of symptoms with specialist-assessed long-term asthma severity. Chest 1999; 115:85–91.
- Li JT, Oppenheimer J, Bernstein IL, et al. Attaining optimal asthma control: a practice parameter. J Allergy Clin Immunol 2005; 116:S3–S11.
- Nathan RA, Sorkness C, Kosinski M, et al. Development of the Asthma Control Test: a survey for assessing asthma control. J Allergy Clin Immunol 2004; 113:59–65.
- Schatz M, Zeiger RS, Drane A, et al. Reliability and predictive validity of the Asthma Control Test administered by telephone calls using speech recognition technology. J Allergy Clin Immunol 2007; 119:336–343.
- Peters D, Chen C, Markson LE, Allen-Ramey FC, Vollmer WM. Using an asthma control questionnaire and administrative data to predict healthcare utilization. Chest 2006; 129:918–924.
- Schatz M, Sorkness C, Li JT, et al. Asthma Control Test: reliability, validity, and responsiveness in patients not previously followed by asthma specialists. J Allergy Clin Immunol 2006; 117:549–556.
- Bateman E, Boushey H, Bousquet J, et al. Can guideline-defined asthma control be achieved? Am J Respir Crit Care Med 2004; 170:836–844.
- Davies TJ, Bunn WB, Fromer L, Gelfand EW, Colice GL. A focus on the asthma HEDIS measure and its implications for clinical practice. Manag Care Interface 2006; 19:29–36.
- Rubinfeld AR, Pain MC. Perception of asthma. Lancet 1976; 1:882–884.
- Teeter J, Bleecker E. Relationship between airway obstruction and respiratory symptoms in adult asthmatics. Chest 1998; 113:272–277.
- Shingo S, Zhang J, Reiss T. Correlation of airway obstruction and patient reported endpoints in clinical studies. Eur Resp J 2001; 17:220–224.
- Juniper EF, Bousquet J, Abetz L, Bateman ED; GOAL Committee. Identifying ‘well-controlled’ and ‘not well-controlled’ asthma using the Asthma Control Questionnaire. Respir Med 2006; 100:616–621.
- Nelson H, Weiss S, Bleecker E, Yancey S, Dorinsky P. The Salmeterol Multicenter Asthma Research Trial. Chest 2006; 129:15–26.
- Wechsler M, Lehman E, Lazarus S, et al. ß-Adrenergic receptor polymorphisms and response to salmeterol. Am J Respir Crit Care Med 2006; 173:519–526.
- Palmer CNA, Lipworth BJ, Lee S, Ismail T, MacGregor DF, Mukhopadhyay S. Arginine-16 beta-2 adrenoceptor genotype predisposes to exacerbations in young asthmatics taking regular salmeterol. Thorax 2006; 61:940–944.
- Taylor DR, Drazen JM, Herbison GP, Yandava CN, Hancox RJ, Town GI. Asthma exacerbations during long term beta agonist use: influence of beta 2 adrenoceptor polymorphism. Thorax 2000; 55:762–727.
- Bleecker E, Postma D, Lawrance R, Meyers D, Ambrose H, Goldman M. Effect of ADRB2 polymorphisms on response to long-acting beta2-agonist therapy: a pharmacogenetic analysis of two randomized studies. Lancet 2007; 370:2118–2125.
- Bleecker E, Yancey S, Baitinger L, et al. Salmeterol response is not affected by beta-2 adrenergic receptor genotype in subjects with persistent asthma. J Allergy Clin Immunol 2006; 118:809–816.
- Nelson H, Bleecker E, Corren J, et al. Characterization of asthma exacerbations by Arg16Gly genotype in subjects with asthma receiving salmeterol alone or with fluticasone propionate. J Allergy Clin Immunol 2008; 121:S131.
- O’Byrne P, Barnes P, Rodriguez-Roisin R, et al. Low dose Inhaled budesonide and formoterol in mild persistent asthma. The OPTIMA Randomized Trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Greening AP, Ind PW, Northfield M, Shaw G. Added salmeterol versus higher dose corticosteroid in asthma patients with symptoms on existing inhaled corticosteroid. Lancet 1994; 344:219–224.
- Woolcock A, Lundback B, Ringdal N, Jacques LA. Comparison of addition of salmeterol to inhaled steroids with doubling of the dose of inhaled steroids. Am J Respir Crit Care Med 1996; 153:1481–1488.
- Walters EH, Walters JAE, Gibson MDP. Long-acting beta2-agonists for stable chronic asthma. Cochrane Database Syst Rev 2003; (3):CD001385. doi:10.1002/14651858.CD001385.
- Masoli M, Weatherall M, Holt S, Beasley R. Moderate dose inhaled corticosteroids plus salmeterol versus higher doses of inhaled corticosteroid in symptomatic asthma. Thorax 2005; 60:730–734.
- Sin DD, Man J, Sharpe H, Gan WQ, Man SFP. Pharmacological management to reduce exacerbations in adults with asthma. A systematic review and meta-analysis. JAMA 2004; 292:367–376.
- Mann RD, Kubota K, Pearce G, Wilton L. Salmeterol: a study by prescription event monitoring in a UK cohort of 15,407 patients. J Clin Epidemiol 1996; 49:247–250.
- Lanes S, Lanza L, Wentworth C. Risk of emergency care, hospitalization, and ICU stays for acute asthma among recipients of salmeterol. Am J Respir Crit Care Med 1998; 158:857–861.
- Meier CR, Jick H. Drug use and pulmonary death rates in increasingly symptomatic asthma patients in the UK. Thorax 1997; 52:612–617.
- Williams C, Crossland L, Finnerty J, et al. A case control study of salmeterol and near-fatal attacks of asthma. Thorax 1998; 53:7–13.
- Lanes S, Garcia Rodriguez LA, Herta C. Respiratory medications and risk of asthma death. Thorax 2002; 57:683–686.
- Anderson HR, Ayres JG, Sturdy PM, et al. Bronchodilator treatment and deaths from asthma: case control study. Br Med J 2005; 330:117–124.
- Martinez FD. Safety of long-acting beta agonists—an urgent need to clear the air. N Engl J Med 2005; 353:2637–2639.
- Nelson HS. Long-acting beta-agonists in adult asthma: evidence that these drugs are safe. Prim Care Respir J 2006; 15:271–277.
- Lang DM. The long-acting beta agonist controversy: a critical examination of the evidence. Cleve Clin J Med 2006; 73:973–992.
- Rambasek T, Lang DM, Kavuru M. Omalizumab: where does it fit in current asthma management? Cleve Clin J Med 2004; 71:251–261.
- McGlashan D, Bochner B, Adelman D, et al. Down regulation of Fc(epsilon)RI expression on human basophils during in vivo treatment of atopic patients with anti-IgE antibody. J Immunol 1997; 158:1438–1445.
- Busse W, Corren J, Lanier B, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol 2001; 108:184–190.
- Soler M, Matz J, Townley R, et al. The anti-IgE antibody omalizumab reduces asthma exacerbations and steroid requirement in allergic asthmatics. Eur Respir J 2001; 18:254–261.
- Humbert M, Beasley R, Ayres J, et al. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy 2005; 60:309–316.
- Van Ganse E, Antonicelli L, Zhang Q, et al. Asthma-related resource use and cost by GINA classification of severity in three European countries. Respir Med 2006; 100:140–147.
- Godard P, Chanez P, Siraudin L, Nicoloyannis N, Duru G. Costs of asthma are correlated with severity: a 1-yr prospective study. Eur Respir J 2002; 19:61–67.
- Cisternas MG, Blanc PH, Yen IH, et al. A comprehensive study of the direct and indirect costs of adult asthma. J Allergy Clin Immunol 2003; 111:1212–1218.
- Sullivan S, Turk F. An evaluation of the cost effectiveness of omalizumab for the treatment of severe persistent asthma. Allergy 2008; 63:670–684.
- US Food and Drug Administration. Omalizumab (marketed as Xolair) information. www.fda.gov/cder/drug/infopage/omalizumab/default.htm. Accessed August 31, 2007.
- Williams SG, Schmidt DK, Redd SC, Storms W. Key clinical activities for quality asthma care. Recommendations of the National Asthma Education and Prevention Program. MMWR Recomm Rep 2003; 52 RR-6:1–8.
- Cox L, Li J, Nelson H, Lockey R, et al. Allergy Immunotherapy: a practice parameter second update. J Allergy Clin Immunol 2007; 120:S25–S85.
- Akdis M, Akdis CA. Mechanisms of allergen-specific immunotherapy. J Allergy Clin Immunol 2007; 119:780–789.
- Murray AB, Milner RA. The accuracy of features in the clinical history for predicting atopic sensitization to airborne allergens in children. J Allergy Clin Immunol 1995; 96:588–596.
- Bernstein IL, Li JT, Bernstein DI, et al. Allergy diagnostic testing: an updated practice parameter. Ann Allergy Asthma Immunol 2008; 100 suppl 3:1S–148S.
- Walker S, Pajno GB, Lima MT, Wilson DR, Durham SR. Grass pollen immunotherapy for seasonal rhinitis and asthma: a randomized, controlled trial. J Allergy Clin Immunol 2001; 107:87–93.
- Varney VA, Edwards J, Tabbah K, Brewster H, Mavroleon G, Frew AJ. Clinical efficacy of specific immunotherapy to cat dander: a double-blind placebo-controlled trial. Clin Exp Allergy 1997; 27:860–867.
- Cantani A, Arcese G, Lucenti P, Gagliesi D, Bartolucci M. A three-year prospective study of specific immunotherapy to inhalant allergens: evidence of safety and efficacy in 300 children with allergic asthma. J Investig Allergol Clin Immunol 1997; 7:90–97.
- Hedlin G, Wille S, Browaldh L, et al. Immunotherapy in children with allergic asthma: effect on bronchial hyperreactivity and pharmacotherapy. J Allergy Clin Immunol 1999; 103:609–614.
- Arvidsson MB, Löwhagen O, Rak S. Allergen specific immunotherapy attenuates early and late phase reactions in lower airways of birch pollen asthmatic patients: a double blind placebo-controlled study. Allergy 2004; 59:74–80.
- Pichler CE, Helbling A, Pichler WJ. Three years of specific immunotherapy with house-dust-mite extracts in patients with rhinitis and asthma: significant improvement of allergen-specific parameters and of nonspecific bronchial hyperreactivity. Allergy 2001; 56:301–306.
- Mirone C, Albert F, Tosi A, et al. Efficacy and safety of subcutaneous immunotherapy with a biologically standardized extract of Ambrosia artemisiifolia pollen: a double-blind, placebo-controlled study. Clin Exp Allergy 2004; 34:1408–1414.
- Abramson MJ, Puy RM, Weiner JM. Allergen immunotherapy for asthma. Cochrane Database Syst Rev 2003; (4):CD001186.
- Jacobsen L. Preventive aspects of immunotherapy: prevention for children at risk of developing asthma. Ann Allergy Asthma Immunol 2001; 87:43–46.
- Moller C, Dreborg S, Ferdousi HA, et al. Pollen immunotherapy reduces the development of asthma in children with seasonal rhinoconjunctivitis (the PAT study). J Allergy Clin Immunol 2002; 109:251–256.
- Niggemann B, Jacobsen L, Dreborg S, et al; PAT Investigator Group. Five year follow-up on the PAT study: specific immunotherapy and long-term prevention of asthma in children. Allergy 2006: 61:855–859.
- Des Roches A, Paradis L, Menardo JL, et al. Immunotherapy with a standardized Dermatophagoides pteronyssinus extract VI: specific immunotherapy prevents the onset of new sensitizations in children. J Allergy Clin Immunol 1997; 99:450–453.
- Pajno GB, Barberio G, DeLuca F, et al. Prevention of new sensitizations in asthmatic children monosensitized to the house dust mite by specific immunotherapy: a six year follow up study. Clin Exp Allergy 2001; 31:1392–1397.
- Lang DM. Do beta blockers really enhance the risk of anaphylaxis during immunotherapy? Curr Allergy Asthma Rep 2008; 8:37–44.
This review focuses on several elements in the National Asthma Education and Prevention Program’s new guidelines, the third Expert Panel Report (EPR3),1 that differ substantially from those in EPR2,2 issued in 1997 and updated in 2002.3 These differences in approach to the management of asthma described in EPR3 offer a clear potential for reducing the gap between optimal asthma care outcomes as described in guidelines and normative asthma care outcomes in the “real world.”
GREATER EMPHASIS ON CONTROL
The EPR2 guidelines2 recommended that asthma management be carried out in an algorithmic manner. Patients were classified into four severity categories: mild intermittent, mild persistent, moderate persistent, and severe persistent asthma, based on assessment of the level of symptoms (day/night), reliance on “reliever” medication, and lung function at the time of presentation. Pharmacologic management was then assigned according to each respective categorization in an evidence-based fashion.
In an ideal world, this would result in patients with asthma receiving appropriate pharmacotherapeutic agents associated with favorable asthma care outcomes, which were also advantageous from both cost- and risk-benefit standpoints. In the real world, however, this paradigm was flawed, as it relied on accurate categorization of patients in order for pharmacotherapy to be prescribed appropriately. Both providers and patients are prone to underestimate asthma severity,4,5 and for this reason many patients managed on the basis of this paradigm were undertreated.
A new paradigm, based on the assessment of asthma control, has been encouraged in the EPR3 guidelines.1
Severity and control are not synonymous
More than a decade ago, Cockroft and Swystun6 pointed out that asthma control (or lack thereof) is often used inappropriately to define asthma severity: ie, well-controlled asthma is seen as synonymous with mild asthma, and poorly controlled asthma with severe asthma.
Asthma severity can be defined as the intrinsic intensity of the disease process, while asthma control is the degree to which the manifestations of asthma are minimized. Asthma severity is clearly a determinant of asthma control, but its impact is affected by a variety of factors, including but not limited to:
- Whether appropriate medication is prescribed
- Patterns of therapeutic adherence
- The degree to which recommended measures for avoiding for clinically relevant aeroallergens are pursued.
Health care utilization, including hospitalizations and emergency department visits, correlates more closely with asthma control than with asthma severity.7–9 Indeed, a patient with severe persistent asthma who is treated appropriately with multiple “controller” medications and who takes his or her medications and avoids allergens as directed can achieve well-controlled or totally controlled asthma, and is not likely to require hospitalization or emergency department management, to miss school or work, or to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has severe persistent asthma that is well controlled.
In contrast, a patient with mild or moderate persistent asthma who does not receive appropriate instructions for avoiding allergens or taking controller medication regularly or who is poorly adherent will likely have poor asthma control. This patient is more likely to require hospitalization or emergency department management, to miss school or work, and to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has mild persistent asthma that is poorly controlled.
Assess asthma severity in the first visit, and control in subsequent visits
How to assess severity
How to measure control
For all patients with asthma, regardless of severity, the goal is the same: to achieve control by reducing both impairment and risk. Asthma is classified as well controlled, not well controlled, or poorly controlled (Table 2).1
Validated tests are available to assess control
Serial testing as a quality indicator
Serial ACT scores give an objective measure of the degree to which the goals of management1 are being achieved, and in so doing can encourage optimal outcomes.14
Another use of these tests is to document whether asthma control improves over time when patients receive care from a particular physician or group. This use may become increasingly important in view of efforts underway to implement a pay-for-performance model for asthma care, in which providers will be financially rewarded for improved patient care outcomes and adherence to standards of practice based on Health Plan Employer Data and Information Set measures.15
We have used the ACT in the Section of Allergy/Immunology at Cleveland Clinic for 3 years on a routine basis. All patients with asthma being seen either for the first time or as follow-up complete the ACT, which has been entered in a flow sheet in our electronic medical record, at the same time they undergo spirometry. We have shown that care in the Section of Allergy/Immunology is associated with improvement in asthma control over time, in patients who have completed serial ACT measurements at initial visits and at follow-up visits (Figure 2).
Objective measurement of lung function is also important
Serial monitoring of lung function at every patient visit with spirometry is also important, as some patients may be “poor perceivers,”16 ie, they may have little or no subjective awareness of moderate or even severe ventilatory impairment. A number of studies17,18 support the contention that symptoms and lung function are separate and independent dimensions of asthma control, and that both of them need to be assessed.
Responding to changes in control
THE STEP 3 CONTROVERSY
Salmeterol Multicenter Asthma Research Trial
In the Salmeterol Multicenter Asthma Research Trial (SMART), patients randomized to the long-acting beta agonist (LABA) salmeterol (Serevent)—particularly African Americans—had a statistically significant increase in the risk of untoward asthma care outcomes.20
SMART was launched in 1996. Patients were randomized in a double-blind fashion to receive either salmeterol 42 μg twice a day or placebo in addition to their usual asthma therapy for 28 weeks. The rate of the primary outcome (respiratory-related deaths or life-threatening experiences) was not significantly different with salmeterol than with placebo (relative risk [RR] = 1.40, 95% confidence interval [CI] 0.91–2.14). However, in 2003, the study was halted prematurely because of difficulty enrolling the targeted number of 60,000 patients, and an interim analysis that revealed significantly higher rates of secondary outcomes in subjects randomized to salmeterol. Compared with the placebo group, the salmeterol group had significantly higher rates of respiratory-related deaths (RR 2.16, 95% CI 1.06–4.41), asthma-related deaths (RR = 4.37, 95% CI = 1.25–15.34), and combined asthma-related deaths or life-threatening experiences (RR = 1.71, 95% CI 1.01–2.89). There were 13 asthma-related deaths and 37 combined asthma-related deaths or life-threatening experiences in the salmeterol group, compared with 3 and 22, respectively, in the placebo group. Of the 16 asthma deaths in the study, 13 (81%) occurred in the initial phase of SMART, when patients were recruited via print, radio, and television advertising; afterward, patients were recruited directly by investigators.
Statistically significant differences in outcomes occurred primarily in African Americans. African Americans who received salmeterol had higher rates of respiratory death or life-threatening experiences (RR = 4.10, 95% CI 1.54–10.90), the primary end point for the study, as well as higher rates of combined asthma-related deaths or life-threatening experiences (RR = 10.46, 95% CI 1.34–81.58), a secondary end point. No statistically significant differences were observed in white patients randomized to salmeterol with respect to the primary end point (RR = 1.05, 95% = 0.62–1.76); the secondary end point of combined asthma-related deaths or life-threatening experiences (RR = 1.08, 95% CI 0.55–2.14); or other end points.
Medication exposures were not tracked during the study, and allocation to inhaled corticosteroids combined with salmeterol was not randomized, so the effect of concomitant inhaled corticosteroid use cannot be determined from these data.
As a result of SMART, medications that contain either of the two LABAs, salmeterol or formoterol (Foradil), carry a black-box warning.
LABAs: Risks and benefits
Two studies21,22 have suggested that asthmatic patients who are homozygous for Arg/Arg at codon 16 of the beta-2 adrenergic receptor are predisposed to untoward asthma outcomes with regular exposure to LABAs. However, other data23–25 do not support the contention that B16 Arg/Arg patients experience adverse asthma outcomes with LABA exposure. In two recently published studies, no difference in rates of exacerbations, severe exacerbations, lung function, frequency of reliance on SABA, or nocturnal awakenings was observed in patients receiving formoterol combined with budesonide24 or salmeterol combined with fluticasone25 according to genotype. A prospective study26 also found no statistically significant difference in exacerbation rates according to beta adrenergic receptor genotype in individuals randomized to LABA monotherapy, or LABA combined with inhaled corticosteroids.
The updated EPR2 asthma guidelines,3 published in November 2002, stipulated that LABAs were the preferred controller agent to “add on” to low-dose inhaled corticosteroids for patients with moderate persistent asthma, and that the combination of low-dose inhaled corticosteroids and LABA was associated with superior outcomes: reduction of symptoms, including nocturnal awakening, increase in lung function, improvement in health-related quality of life, decreased use of “rescue” medication, and reduced rate of exacerbations and severe exacerbations, compared with higher-dose inhaled corticosteroid monotherapy. This management recommendation was categorized as level A, on the basis of data from multiple randomized, controlled, double-blinded trials.27–29 Additional evidence14,30 and data from two meta-analyses31,32 have provided further support for this recommendation, while no evidence linking LABA exposure to risk for fatal or near-fatal asthma has been found in cohort or case-control studies.33–38
Based on safety concerns, the EPR3 guidelines1 recommend that medium-dose inhaled corticosteroids be regarded as equivalent to adding LABAs to low-dose inhaled corticosteroids, and state: “the established, beneficial effects of LABA for the great majority of patients whose asthma is not well controlled with [inhaled corticosteroids] alone should be weighed against the increased risk for severe exacerbations, although uncommon, associated with daily use of LABA.”1
There is currently an honest difference of opinion39,40 among asthma specialists as to how this management recommendation for moderate persistent asthma—now depicted at “step 3” in the EPR3 guidelines (Table 4)—should be implemented. The LABA controversy was reviewed previously in the Cleveland Clinic Journal of Medicine.41
THE ROLE OF OMALIZUMAB: WEIGHING COST VS BENEFIT
The 2002 update to the EPR2 guidelines3 was issued before omalizumab (Xolair) was approved in June 2003.
Patients with severe persistent asthma are categorized in steps 5 or 6 in the EPR3 guidelines (Table 5).1 Preferred management for these patients includes inhaled corticosteroids in high doses combined with long-acting beta agonists and, for step 6 patients, oral corticosteroids.
Omalizumab was approved for management of patients with moderate or severe persistent asthma who are not achieving the goals of asthma management on inhaled corticosteroids, who exhibit a wheal-flare reaction to a perennial allergen, and whose immunoglobulin E (IgE) level is in the range of 30 to 700 IU/mL.42 Omalizumab dosing is based on the serum IgE level and on body weight.
Omalizumab, an anti-IgE monoclonal antibody
Omalizumab is a recombinant, humanized, monoclonal anti-IgE antibody that binds to IgE at the same Fc site as the high-affinity IgE receptor. Its primary mechanism of action is the binding of free IgE in the circulation, forming biologically inert, small complexes that do not activate complement and are cleared by the reticuloendothelial system.42 Its secondary mechanism of action entails a reduction in the number of high-affinity receptors on basophils, from approximately 220,000 to 8,300 receptors per cell. The latter effect was associated with a 90% reduction in histamine release from basophils in response to ex vivo challenge with dust mite allergen.43
Benefit in randomized trials
Omalizumab has been associated with statistically and clinically significant benefit in randomized, double-blind, placebo-controlled trials.44,45
Humbert et al46 randomized 419 patients whose asthma was not adequately controlled on high-dose inhaled corticosteroids and long-acting beta agonists, who were 12 to 75 years old, with reduced lung function and a history of recent asthma exacerbation, to treatment with omalizumab or placebo. Omalizumab was associated with a statistically significant reduction in the rate of asthma exacerbations and severe asthma exacerbations, as well as statistically significant improvements in asthma-related quality of life, morning peak expiratory flow rate, and asthma symptom scores.
These data support the recommendation in EPR3 to consider a trial of omalizumab in properly selected patients with severe, persistent allergic asthma.
Omalizumab is cost-beneficial in properly selected patients
The current wholesale acquisition cost of omalizumab is $532 for one 150-mg vial (David Zito, personal communication). The cost of treatment varies based on body weight and IgE level but may range from a wholesale cost of $6,388 to $38,326 per year.
However, as asthma severity increases, both direct and indirect medical expenditures increase substantially.47,48 Annual costs are approximately four times higher for severe asthma compared with mild asthma49; not only are treatment and exacerbation costs higher, but indirect costs are also disproportionately greater. Annual costs for severe asthma are significantly greater if the disease is inadequately controlled.50 For these reasons, an intervention that leads to improved outcomes for severe, poorly controlled asthma carries the potential for the greatest cost-utility for society, as it can lower direct costs by reducing the frequency and severity of exacerbations, in addition to reducing indirect medical expenditures on the basis of increased productivity and fewer days of missed work or school. The cost of omalizumab in quality-adjusted life years compares favorably with that of biologicals used in managing rheumatoid arthritis, Crohn disease, and multiple sclerosis.50
Adverse effects of omalizumab
In pivotal trials,43,44 omalizumab was associated with a substantial rate of local reactions. The rate of anaphylaxis was slightly less than 1 in 1,000, and this has been confirmed by surveillance data recorded since approval of the drug in 2003. Based on the observed risk of anaphylaxis, in July 2007, the US Food and Drug Administration added a black-box warning to the omalizumab label and stipulated that a medication guide should be provided for patients.51 The warning indicates that health care providers administering omalizumab should be prepared to manage anaphylaxis and that patients should be closely observed for an appropriate period after omalizumab administration.
The package insert also describes a numerical, but not statistically significant, increase in the rate of malignancy in patients receiving omalizumab.42 Malignancy developed in 0.5% of patients receiving omalizumab, compared with 0.2% of patients who received placebo. Because these malignancies were diagnosed over a shorter period than the time required for oncogenesis (ie, 6 months in 60% of cases), and because a heterogeneous variety of tumors was observed, there is reason to doubt these tumors were causally associated with omalizumab.
Postmarketing surveillance studies are in progress that will provide more definitive data on the potential relationship between malignancy and omalizumab exposure.
Omalizumab: Guideline recommendations
The EPR3 guidelines1 state that omalizumab is the only adjunctive therapy to demonstrate efficacy when added to high-dose inhaled corticosteroids plus long-acting beta agonists in patients with severe, persistent, allergic asthma and that evidence does not support use of the following agents, which in some cases are approved for managing other conditions and have been advocated for management of severe, refractory asthma: methotrexate, soluble interleukin (IL)-4 receptor, anti-IL-5, anti-IL-12, cyclosporine A, intravenous immune globulin, gold, troleandomycin, and colchicine. The data supporting use of macrolides were characterized as “encouraging but insufficient to support a recommendation.”
The strength of evidence for the use of omalizumab for patients in steps 5 and 6 who fulfill the criteria for its use (see above) was classified in the EPR3 guidelines1 as category B. The guidelines also say that omalizumab may be considered for adjunctive therapy in properly selected patients in step 4, as a means to avoid higher doses of inhaled corticosteroids, but that additional studies are needed to establish its utility for such patients. This recommendation was classified as category D because of the lack of published comparator trials.
ALLERGEN IMMUNOTHERAPY FOR PATIENTS WITH ASTHMA
Many patients with asthma have clinically relevant, IgE-mediated (allergic) potential to inhaled allergens.1 For patients with persistent asthma (steps 2–6 in Table 5), allergic reactions can contribute to airway inflammation, provoke symptoms, and lead to more use of medications. For this reason, identification and management of clinically relevant allergy merits consideration.52
The EPR3 guidelines1 recommend considering allergen immunotherapy for patients with mild or moderate persistent asthma (steps 2–4) who have a clinically relevant component of allergy to inhaled substances.
Changing the immune response
Allergen immunotherapy entails the incremental administration of inhalant allergens by subcutaneous injection for the purpose of inducing immune system changes in the host response. The goal of immunotherapy is to protect against allergic reactions that can be expected to occur with ongoing exposure to clinically relevant allergens.53
The immunologic changes that develop with allergen immunotherapy are complex.53,54 Successful immunotherapy results in generation of a population of CD4+/CD25+ T lymphocytes producing IL-10, transforming growth factor beta, or both. Allergen immunotherapy has been shown to block the immediate- and late-phase allergic response; to decrease recruitment of mast cells, basophils, and eosinophils on provocation or natural exposure to allergens in the skin, nose, eye, and bronchial mucosa; to blunt the seasonal rise in specific IgE; and to suppress late-phase inflammatory responses in the skin and respiratory tract. However, the efficacy of immunotherapy in relation to these immunologic changes is not completely understood.54
Many patients need skin testing
Allergen immunotherapy may be considered for patients with asthma for whom a clear relationship exists between symptoms and exposure to an allergen to which the patient is sensitive.53 Because it is often not possible to determine whether a patient is sensitive to a perennial indoor allergen (eg, dust mite) on the basis of the medical history alone,55 many patients with asthma benefit from immediate hypersensitivity skin testing to objectively assess or rule out allergy to common inhalants. In certain situations, in vitro testing may be performed, but skin testing has a higher negative predictive value and is recommended as a better screening test.56
Benefits of allergen immunotherapy
Numerous randomized, double-blind, placebo-controlled trials have shown that allergen immunotherapy is associated with benefit for reducing symptoms and medication reliance.57–63
A meta-analysis of 75 randomized, placebo-controlled studies confirmed the effectiveness of immunotherapy in asthma, with a significant reduction in asthma symptoms and medication use and with improvement in bronchial hyperreactivity.64 This meta-analysis included 36 trials of dust mite allergen, 20 of pollen, and 10 of animal dander. Immunotherapy is efficacious for pollen, mold, dust mite, cockroach, and animal allergens; however, its effectiveness is more established for dust mite, animal dander, and pollen allergens, as fewer studies have been published demonstrating efficacy using mold and cockroach allergens.53
In addition, several studies have found that children with allergic rhinitis who receive allergen immunotherapy are significantly less likely to develop asthma.65–67 Immunotherapy has also been associated with a statistically significant reduction in future sensitization to other aeroallergens.68,69
Risk of systemic reaction from allergen immunotherapy
The decision to begin allergen immunotherapy should be individualized on the basis of symptom severity, relative benefit compared with drug therapy, and whether comorbid conditions such as cardiovascular disease or beta-blocker exposure are present. These comorbid conditions are associated with heightened risk of (more serious) anaphylaxis—the major hazard of allergen immunotherapy.70 Systemic reactions during allergen immunotherapy occur at a rate of approximately 3 to 5 per 1,000 injections; for this reason, allergen immunotherapy should only be administered in a medical facility where personnel, supplies, and equipment are available to treat anaphylaxis.5
This review focuses on several elements in the National Asthma Education and Prevention Program’s new guidelines, the third Expert Panel Report (EPR3),1 that differ substantially from those in EPR2,2 issued in 1997 and updated in 2002.3 These differences in approach to the management of asthma described in EPR3 offer a clear potential for reducing the gap between optimal asthma care outcomes as described in guidelines and normative asthma care outcomes in the “real world.”
GREATER EMPHASIS ON CONTROL
The EPR2 guidelines2 recommended that asthma management be carried out in an algorithmic manner. Patients were classified into four severity categories: mild intermittent, mild persistent, moderate persistent, and severe persistent asthma, based on assessment of the level of symptoms (day/night), reliance on “reliever” medication, and lung function at the time of presentation. Pharmacologic management was then assigned according to each respective categorization in an evidence-based fashion.
In an ideal world, this would result in patients with asthma receiving appropriate pharmacotherapeutic agents associated with favorable asthma care outcomes, which were also advantageous from both cost- and risk-benefit standpoints. In the real world, however, this paradigm was flawed, as it relied on accurate categorization of patients in order for pharmacotherapy to be prescribed appropriately. Both providers and patients are prone to underestimate asthma severity,4,5 and for this reason many patients managed on the basis of this paradigm were undertreated.
A new paradigm, based on the assessment of asthma control, has been encouraged in the EPR3 guidelines.1
Severity and control are not synonymous
More than a decade ago, Cockroft and Swystun6 pointed out that asthma control (or lack thereof) is often used inappropriately to define asthma severity: ie, well-controlled asthma is seen as synonymous with mild asthma, and poorly controlled asthma with severe asthma.
Asthma severity can be defined as the intrinsic intensity of the disease process, while asthma control is the degree to which the manifestations of asthma are minimized. Asthma severity is clearly a determinant of asthma control, but its impact is affected by a variety of factors, including but not limited to:
- Whether appropriate medication is prescribed
- Patterns of therapeutic adherence
- The degree to which recommended measures for avoiding for clinically relevant aeroallergens are pursued.
Health care utilization, including hospitalizations and emergency department visits, correlates more closely with asthma control than with asthma severity.7–9 Indeed, a patient with severe persistent asthma who is treated appropriately with multiple “controller” medications and who takes his or her medications and avoids allergens as directed can achieve well-controlled or totally controlled asthma, and is not likely to require hospitalization or emergency department management, to miss school or work, or to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has severe persistent asthma that is well controlled.
In contrast, a patient with mild or moderate persistent asthma who does not receive appropriate instructions for avoiding allergens or taking controller medication regularly or who is poorly adherent will likely have poor asthma control. This patient is more likely to require hospitalization or emergency department management, to miss school or work, and to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has mild persistent asthma that is poorly controlled.
Assess asthma severity in the first visit, and control in subsequent visits
How to assess severity
How to measure control
For all patients with asthma, regardless of severity, the goal is the same: to achieve control by reducing both impairment and risk. Asthma is classified as well controlled, not well controlled, or poorly controlled (Table 2).1
Validated tests are available to assess control
Serial testing as a quality indicator
Serial ACT scores give an objective measure of the degree to which the goals of management1 are being achieved, and in so doing can encourage optimal outcomes.14
Another use of these tests is to document whether asthma control improves over time when patients receive care from a particular physician or group. This use may become increasingly important in view of efforts underway to implement a pay-for-performance model for asthma care, in which providers will be financially rewarded for improved patient care outcomes and adherence to standards of practice based on Health Plan Employer Data and Information Set measures.15
We have used the ACT in the Section of Allergy/Immunology at Cleveland Clinic for 3 years on a routine basis. All patients with asthma being seen either for the first time or as follow-up complete the ACT, which has been entered in a flow sheet in our electronic medical record, at the same time they undergo spirometry. We have shown that care in the Section of Allergy/Immunology is associated with improvement in asthma control over time, in patients who have completed serial ACT measurements at initial visits and at follow-up visits (Figure 2).
Objective measurement of lung function is also important
Serial monitoring of lung function at every patient visit with spirometry is also important, as some patients may be “poor perceivers,”16 ie, they may have little or no subjective awareness of moderate or even severe ventilatory impairment. A number of studies17,18 support the contention that symptoms and lung function are separate and independent dimensions of asthma control, and that both of them need to be assessed.
Responding to changes in control
THE STEP 3 CONTROVERSY
Salmeterol Multicenter Asthma Research Trial
In the Salmeterol Multicenter Asthma Research Trial (SMART), patients randomized to the long-acting beta agonist (LABA) salmeterol (Serevent)—particularly African Americans—had a statistically significant increase in the risk of untoward asthma care outcomes.20
SMART was launched in 1996. Patients were randomized in a double-blind fashion to receive either salmeterol 42 μg twice a day or placebo in addition to their usual asthma therapy for 28 weeks. The rate of the primary outcome (respiratory-related deaths or life-threatening experiences) was not significantly different with salmeterol than with placebo (relative risk [RR] = 1.40, 95% confidence interval [CI] 0.91–2.14). However, in 2003, the study was halted prematurely because of difficulty enrolling the targeted number of 60,000 patients, and an interim analysis that revealed significantly higher rates of secondary outcomes in subjects randomized to salmeterol. Compared with the placebo group, the salmeterol group had significantly higher rates of respiratory-related deaths (RR 2.16, 95% CI 1.06–4.41), asthma-related deaths (RR = 4.37, 95% CI = 1.25–15.34), and combined asthma-related deaths or life-threatening experiences (RR = 1.71, 95% CI 1.01–2.89). There were 13 asthma-related deaths and 37 combined asthma-related deaths or life-threatening experiences in the salmeterol group, compared with 3 and 22, respectively, in the placebo group. Of the 16 asthma deaths in the study, 13 (81%) occurred in the initial phase of SMART, when patients were recruited via print, radio, and television advertising; afterward, patients were recruited directly by investigators.
Statistically significant differences in outcomes occurred primarily in African Americans. African Americans who received salmeterol had higher rates of respiratory death or life-threatening experiences (RR = 4.10, 95% CI 1.54–10.90), the primary end point for the study, as well as higher rates of combined asthma-related deaths or life-threatening experiences (RR = 10.46, 95% CI 1.34–81.58), a secondary end point. No statistically significant differences were observed in white patients randomized to salmeterol with respect to the primary end point (RR = 1.05, 95% = 0.62–1.76); the secondary end point of combined asthma-related deaths or life-threatening experiences (RR = 1.08, 95% CI 0.55–2.14); or other end points.
Medication exposures were not tracked during the study, and allocation to inhaled corticosteroids combined with salmeterol was not randomized, so the effect of concomitant inhaled corticosteroid use cannot be determined from these data.
As a result of SMART, medications that contain either of the two LABAs, salmeterol or formoterol (Foradil), carry a black-box warning.
LABAs: Risks and benefits
Two studies21,22 have suggested that asthmatic patients who are homozygous for Arg/Arg at codon 16 of the beta-2 adrenergic receptor are predisposed to untoward asthma outcomes with regular exposure to LABAs. However, other data23–25 do not support the contention that B16 Arg/Arg patients experience adverse asthma outcomes with LABA exposure. In two recently published studies, no difference in rates of exacerbations, severe exacerbations, lung function, frequency of reliance on SABA, or nocturnal awakenings was observed in patients receiving formoterol combined with budesonide24 or salmeterol combined with fluticasone25 according to genotype. A prospective study26 also found no statistically significant difference in exacerbation rates according to beta adrenergic receptor genotype in individuals randomized to LABA monotherapy, or LABA combined with inhaled corticosteroids.
The updated EPR2 asthma guidelines,3 published in November 2002, stipulated that LABAs were the preferred controller agent to “add on” to low-dose inhaled corticosteroids for patients with moderate persistent asthma, and that the combination of low-dose inhaled corticosteroids and LABA was associated with superior outcomes: reduction of symptoms, including nocturnal awakening, increase in lung function, improvement in health-related quality of life, decreased use of “rescue” medication, and reduced rate of exacerbations and severe exacerbations, compared with higher-dose inhaled corticosteroid monotherapy. This management recommendation was categorized as level A, on the basis of data from multiple randomized, controlled, double-blinded trials.27–29 Additional evidence14,30 and data from two meta-analyses31,32 have provided further support for this recommendation, while no evidence linking LABA exposure to risk for fatal or near-fatal asthma has been found in cohort or case-control studies.33–38
Based on safety concerns, the EPR3 guidelines1 recommend that medium-dose inhaled corticosteroids be regarded as equivalent to adding LABAs to low-dose inhaled corticosteroids, and state: “the established, beneficial effects of LABA for the great majority of patients whose asthma is not well controlled with [inhaled corticosteroids] alone should be weighed against the increased risk for severe exacerbations, although uncommon, associated with daily use of LABA.”1
There is currently an honest difference of opinion39,40 among asthma specialists as to how this management recommendation for moderate persistent asthma—now depicted at “step 3” in the EPR3 guidelines (Table 4)—should be implemented. The LABA controversy was reviewed previously in the Cleveland Clinic Journal of Medicine.41
THE ROLE OF OMALIZUMAB: WEIGHING COST VS BENEFIT
The 2002 update to the EPR2 guidelines3 was issued before omalizumab (Xolair) was approved in June 2003.
Patients with severe persistent asthma are categorized in steps 5 or 6 in the EPR3 guidelines (Table 5).1 Preferred management for these patients includes inhaled corticosteroids in high doses combined with long-acting beta agonists and, for step 6 patients, oral corticosteroids.
Omalizumab was approved for management of patients with moderate or severe persistent asthma who are not achieving the goals of asthma management on inhaled corticosteroids, who exhibit a wheal-flare reaction to a perennial allergen, and whose immunoglobulin E (IgE) level is in the range of 30 to 700 IU/mL.42 Omalizumab dosing is based on the serum IgE level and on body weight.
Omalizumab, an anti-IgE monoclonal antibody
Omalizumab is a recombinant, humanized, monoclonal anti-IgE antibody that binds to IgE at the same Fc site as the high-affinity IgE receptor. Its primary mechanism of action is the binding of free IgE in the circulation, forming biologically inert, small complexes that do not activate complement and are cleared by the reticuloendothelial system.42 Its secondary mechanism of action entails a reduction in the number of high-affinity receptors on basophils, from approximately 220,000 to 8,300 receptors per cell. The latter effect was associated with a 90% reduction in histamine release from basophils in response to ex vivo challenge with dust mite allergen.43
Benefit in randomized trials
Omalizumab has been associated with statistically and clinically significant benefit in randomized, double-blind, placebo-controlled trials.44,45
Humbert et al46 randomized 419 patients whose asthma was not adequately controlled on high-dose inhaled corticosteroids and long-acting beta agonists, who were 12 to 75 years old, with reduced lung function and a history of recent asthma exacerbation, to treatment with omalizumab or placebo. Omalizumab was associated with a statistically significant reduction in the rate of asthma exacerbations and severe asthma exacerbations, as well as statistically significant improvements in asthma-related quality of life, morning peak expiratory flow rate, and asthma symptom scores.
These data support the recommendation in EPR3 to consider a trial of omalizumab in properly selected patients with severe, persistent allergic asthma.
Omalizumab is cost-beneficial in properly selected patients
The current wholesale acquisition cost of omalizumab is $532 for one 150-mg vial (David Zito, personal communication). The cost of treatment varies based on body weight and IgE level but may range from a wholesale cost of $6,388 to $38,326 per year.
However, as asthma severity increases, both direct and indirect medical expenditures increase substantially.47,48 Annual costs are approximately four times higher for severe asthma compared with mild asthma49; not only are treatment and exacerbation costs higher, but indirect costs are also disproportionately greater. Annual costs for severe asthma are significantly greater if the disease is inadequately controlled.50 For these reasons, an intervention that leads to improved outcomes for severe, poorly controlled asthma carries the potential for the greatest cost-utility for society, as it can lower direct costs by reducing the frequency and severity of exacerbations, in addition to reducing indirect medical expenditures on the basis of increased productivity and fewer days of missed work or school. The cost of omalizumab in quality-adjusted life years compares favorably with that of biologicals used in managing rheumatoid arthritis, Crohn disease, and multiple sclerosis.50
Adverse effects of omalizumab
In pivotal trials,43,44 omalizumab was associated with a substantial rate of local reactions. The rate of anaphylaxis was slightly less than 1 in 1,000, and this has been confirmed by surveillance data recorded since approval of the drug in 2003. Based on the observed risk of anaphylaxis, in July 2007, the US Food and Drug Administration added a black-box warning to the omalizumab label and stipulated that a medication guide should be provided for patients.51 The warning indicates that health care providers administering omalizumab should be prepared to manage anaphylaxis and that patients should be closely observed for an appropriate period after omalizumab administration.
The package insert also describes a numerical, but not statistically significant, increase in the rate of malignancy in patients receiving omalizumab.42 Malignancy developed in 0.5% of patients receiving omalizumab, compared with 0.2% of patients who received placebo. Because these malignancies were diagnosed over a shorter period than the time required for oncogenesis (ie, 6 months in 60% of cases), and because a heterogeneous variety of tumors was observed, there is reason to doubt these tumors were causally associated with omalizumab.
Postmarketing surveillance studies are in progress that will provide more definitive data on the potential relationship between malignancy and omalizumab exposure.
Omalizumab: Guideline recommendations
The EPR3 guidelines1 state that omalizumab is the only adjunctive therapy to demonstrate efficacy when added to high-dose inhaled corticosteroids plus long-acting beta agonists in patients with severe, persistent, allergic asthma and that evidence does not support use of the following agents, which in some cases are approved for managing other conditions and have been advocated for management of severe, refractory asthma: methotrexate, soluble interleukin (IL)-4 receptor, anti-IL-5, anti-IL-12, cyclosporine A, intravenous immune globulin, gold, troleandomycin, and colchicine. The data supporting use of macrolides were characterized as “encouraging but insufficient to support a recommendation.”
The strength of evidence for the use of omalizumab for patients in steps 5 and 6 who fulfill the criteria for its use (see above) was classified in the EPR3 guidelines1 as category B. The guidelines also say that omalizumab may be considered for adjunctive therapy in properly selected patients in step 4, as a means to avoid higher doses of inhaled corticosteroids, but that additional studies are needed to establish its utility for such patients. This recommendation was classified as category D because of the lack of published comparator trials.
ALLERGEN IMMUNOTHERAPY FOR PATIENTS WITH ASTHMA
Many patients with asthma have clinically relevant, IgE-mediated (allergic) potential to inhaled allergens.1 For patients with persistent asthma (steps 2–6 in Table 5), allergic reactions can contribute to airway inflammation, provoke symptoms, and lead to more use of medications. For this reason, identification and management of clinically relevant allergy merits consideration.52
The EPR3 guidelines1 recommend considering allergen immunotherapy for patients with mild or moderate persistent asthma (steps 2–4) who have a clinically relevant component of allergy to inhaled substances.
Changing the immune response
Allergen immunotherapy entails the incremental administration of inhalant allergens by subcutaneous injection for the purpose of inducing immune system changes in the host response. The goal of immunotherapy is to protect against allergic reactions that can be expected to occur with ongoing exposure to clinically relevant allergens.53
The immunologic changes that develop with allergen immunotherapy are complex.53,54 Successful immunotherapy results in generation of a population of CD4+/CD25+ T lymphocytes producing IL-10, transforming growth factor beta, or both. Allergen immunotherapy has been shown to block the immediate- and late-phase allergic response; to decrease recruitment of mast cells, basophils, and eosinophils on provocation or natural exposure to allergens in the skin, nose, eye, and bronchial mucosa; to blunt the seasonal rise in specific IgE; and to suppress late-phase inflammatory responses in the skin and respiratory tract. However, the efficacy of immunotherapy in relation to these immunologic changes is not completely understood.54
Many patients need skin testing
Allergen immunotherapy may be considered for patients with asthma for whom a clear relationship exists between symptoms and exposure to an allergen to which the patient is sensitive.53 Because it is often not possible to determine whether a patient is sensitive to a perennial indoor allergen (eg, dust mite) on the basis of the medical history alone,55 many patients with asthma benefit from immediate hypersensitivity skin testing to objectively assess or rule out allergy to common inhalants. In certain situations, in vitro testing may be performed, but skin testing has a higher negative predictive value and is recommended as a better screening test.56
Benefits of allergen immunotherapy
Numerous randomized, double-blind, placebo-controlled trials have shown that allergen immunotherapy is associated with benefit for reducing symptoms and medication reliance.57–63
A meta-analysis of 75 randomized, placebo-controlled studies confirmed the effectiveness of immunotherapy in asthma, with a significant reduction in asthma symptoms and medication use and with improvement in bronchial hyperreactivity.64 This meta-analysis included 36 trials of dust mite allergen, 20 of pollen, and 10 of animal dander. Immunotherapy is efficacious for pollen, mold, dust mite, cockroach, and animal allergens; however, its effectiveness is more established for dust mite, animal dander, and pollen allergens, as fewer studies have been published demonstrating efficacy using mold and cockroach allergens.53
In addition, several studies have found that children with allergic rhinitis who receive allergen immunotherapy are significantly less likely to develop asthma.65–67 Immunotherapy has also been associated with a statistically significant reduction in future sensitization to other aeroallergens.68,69
Risk of systemic reaction from allergen immunotherapy
The decision to begin allergen immunotherapy should be individualized on the basis of symptom severity, relative benefit compared with drug therapy, and whether comorbid conditions such as cardiovascular disease or beta-blocker exposure are present. These comorbid conditions are associated with heightened risk of (more serious) anaphylaxis—the major hazard of allergen immunotherapy.70 Systemic reactions during allergen immunotherapy occur at a rate of approximately 3 to 5 per 1,000 injections; for this reason, allergen immunotherapy should only be administered in a medical facility where personnel, supplies, and equipment are available to treat anaphylaxis.5
- National Heart, Lung, and Blood institute, National Asthma education and Prevention Program. Expert Panel Report 3: guidelines for the diagnosis and management of asthma. www.nhlbi.nih.gov/guidelines/asthma. Accessed 8/7/08.
- Expert Panel Report 2: Guidelines for the diagnosis and management of asthma. U.S. Department of Health and Human Services. Publication No. 97-4051; 1997.
- Expert Panel Report: Guidelines for the diagnosis and management of asthma. Update on Selected Topics—2002. J Allergy Clin Immunol 2002; 110:S141–S207.
- FitzGerald JM, Boulet LP, McIvor RA, Zimmerman S, Chapman KR. Asthma control in Canada remains suboptimal: the Reality of Asthma Control (TRAC) study. Can Respir J 2006; 13:253–259.
- Braganza S, Sharif I, Ozuah P. Documenting asthma severity: do we get it right? J Asthma 2003; 40:661–665.
- Cockcroft DW, Swystun VA. Asthma control versus asthma severity. J Allergy Clin Immunol 1996; 98:1016–1018.
- Peters SP, Jones CA, Haselkorn T, Mink DR, Valacer DJ, Weiss ST. Real-world Evaluation of Asthma Control and Treatment (REACT): findings from a national Web-based survey. J Allergy Clin Immunol. 2007; 119:1454–1461.
- Osborne ML, Vollmer WM, Pedula KL, Wilkins J, Buist AS, O’Hollaren M. Lack of correlation of symptoms with specialist-assessed long-term asthma severity. Chest 1999; 115:85–91.
- Li JT, Oppenheimer J, Bernstein IL, et al. Attaining optimal asthma control: a practice parameter. J Allergy Clin Immunol 2005; 116:S3–S11.
- Nathan RA, Sorkness C, Kosinski M, et al. Development of the Asthma Control Test: a survey for assessing asthma control. J Allergy Clin Immunol 2004; 113:59–65.
- Schatz M, Zeiger RS, Drane A, et al. Reliability and predictive validity of the Asthma Control Test administered by telephone calls using speech recognition technology. J Allergy Clin Immunol 2007; 119:336–343.
- Peters D, Chen C, Markson LE, Allen-Ramey FC, Vollmer WM. Using an asthma control questionnaire and administrative data to predict healthcare utilization. Chest 2006; 129:918–924.
- Schatz M, Sorkness C, Li JT, et al. Asthma Control Test: reliability, validity, and responsiveness in patients not previously followed by asthma specialists. J Allergy Clin Immunol 2006; 117:549–556.
- Bateman E, Boushey H, Bousquet J, et al. Can guideline-defined asthma control be achieved? Am J Respir Crit Care Med 2004; 170:836–844.
- Davies TJ, Bunn WB, Fromer L, Gelfand EW, Colice GL. A focus on the asthma HEDIS measure and its implications for clinical practice. Manag Care Interface 2006; 19:29–36.
- Rubinfeld AR, Pain MC. Perception of asthma. Lancet 1976; 1:882–884.
- Teeter J, Bleecker E. Relationship between airway obstruction and respiratory symptoms in adult asthmatics. Chest 1998; 113:272–277.
- Shingo S, Zhang J, Reiss T. Correlation of airway obstruction and patient reported endpoints in clinical studies. Eur Resp J 2001; 17:220–224.
- Juniper EF, Bousquet J, Abetz L, Bateman ED; GOAL Committee. Identifying ‘well-controlled’ and ‘not well-controlled’ asthma using the Asthma Control Questionnaire. Respir Med 2006; 100:616–621.
- Nelson H, Weiss S, Bleecker E, Yancey S, Dorinsky P. The Salmeterol Multicenter Asthma Research Trial. Chest 2006; 129:15–26.
- Wechsler M, Lehman E, Lazarus S, et al. ß-Adrenergic receptor polymorphisms and response to salmeterol. Am J Respir Crit Care Med 2006; 173:519–526.
- Palmer CNA, Lipworth BJ, Lee S, Ismail T, MacGregor DF, Mukhopadhyay S. Arginine-16 beta-2 adrenoceptor genotype predisposes to exacerbations in young asthmatics taking regular salmeterol. Thorax 2006; 61:940–944.
- Taylor DR, Drazen JM, Herbison GP, Yandava CN, Hancox RJ, Town GI. Asthma exacerbations during long term beta agonist use: influence of beta 2 adrenoceptor polymorphism. Thorax 2000; 55:762–727.
- Bleecker E, Postma D, Lawrance R, Meyers D, Ambrose H, Goldman M. Effect of ADRB2 polymorphisms on response to long-acting beta2-agonist therapy: a pharmacogenetic analysis of two randomized studies. Lancet 2007; 370:2118–2125.
- Bleecker E, Yancey S, Baitinger L, et al. Salmeterol response is not affected by beta-2 adrenergic receptor genotype in subjects with persistent asthma. J Allergy Clin Immunol 2006; 118:809–816.
- Nelson H, Bleecker E, Corren J, et al. Characterization of asthma exacerbations by Arg16Gly genotype in subjects with asthma receiving salmeterol alone or with fluticasone propionate. J Allergy Clin Immunol 2008; 121:S131.
- O’Byrne P, Barnes P, Rodriguez-Roisin R, et al. Low dose Inhaled budesonide and formoterol in mild persistent asthma. The OPTIMA Randomized Trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Greening AP, Ind PW, Northfield M, Shaw G. Added salmeterol versus higher dose corticosteroid in asthma patients with symptoms on existing inhaled corticosteroid. Lancet 1994; 344:219–224.
- Woolcock A, Lundback B, Ringdal N, Jacques LA. Comparison of addition of salmeterol to inhaled steroids with doubling of the dose of inhaled steroids. Am J Respir Crit Care Med 1996; 153:1481–1488.
- Walters EH, Walters JAE, Gibson MDP. Long-acting beta2-agonists for stable chronic asthma. Cochrane Database Syst Rev 2003; (3):CD001385. doi:10.1002/14651858.CD001385.
- Masoli M, Weatherall M, Holt S, Beasley R. Moderate dose inhaled corticosteroids plus salmeterol versus higher doses of inhaled corticosteroid in symptomatic asthma. Thorax 2005; 60:730–734.
- Sin DD, Man J, Sharpe H, Gan WQ, Man SFP. Pharmacological management to reduce exacerbations in adults with asthma. A systematic review and meta-analysis. JAMA 2004; 292:367–376.
- Mann RD, Kubota K, Pearce G, Wilton L. Salmeterol: a study by prescription event monitoring in a UK cohort of 15,407 patients. J Clin Epidemiol 1996; 49:247–250.
- Lanes S, Lanza L, Wentworth C. Risk of emergency care, hospitalization, and ICU stays for acute asthma among recipients of salmeterol. Am J Respir Crit Care Med 1998; 158:857–861.
- Meier CR, Jick H. Drug use and pulmonary death rates in increasingly symptomatic asthma patients in the UK. Thorax 1997; 52:612–617.
- Williams C, Crossland L, Finnerty J, et al. A case control study of salmeterol and near-fatal attacks of asthma. Thorax 1998; 53:7–13.
- Lanes S, Garcia Rodriguez LA, Herta C. Respiratory medications and risk of asthma death. Thorax 2002; 57:683–686.
- Anderson HR, Ayres JG, Sturdy PM, et al. Bronchodilator treatment and deaths from asthma: case control study. Br Med J 2005; 330:117–124.
- Martinez FD. Safety of long-acting beta agonists—an urgent need to clear the air. N Engl J Med 2005; 353:2637–2639.
- Nelson HS. Long-acting beta-agonists in adult asthma: evidence that these drugs are safe. Prim Care Respir J 2006; 15:271–277.
- Lang DM. The long-acting beta agonist controversy: a critical examination of the evidence. Cleve Clin J Med 2006; 73:973–992.
- Rambasek T, Lang DM, Kavuru M. Omalizumab: where does it fit in current asthma management? Cleve Clin J Med 2004; 71:251–261.
- McGlashan D, Bochner B, Adelman D, et al. Down regulation of Fc(epsilon)RI expression on human basophils during in vivo treatment of atopic patients with anti-IgE antibody. J Immunol 1997; 158:1438–1445.
- Busse W, Corren J, Lanier B, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol 2001; 108:184–190.
- Soler M, Matz J, Townley R, et al. The anti-IgE antibody omalizumab reduces asthma exacerbations and steroid requirement in allergic asthmatics. Eur Respir J 2001; 18:254–261.
- Humbert M, Beasley R, Ayres J, et al. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy 2005; 60:309–316.
- Van Ganse E, Antonicelli L, Zhang Q, et al. Asthma-related resource use and cost by GINA classification of severity in three European countries. Respir Med 2006; 100:140–147.
- Godard P, Chanez P, Siraudin L, Nicoloyannis N, Duru G. Costs of asthma are correlated with severity: a 1-yr prospective study. Eur Respir J 2002; 19:61–67.
- Cisternas MG, Blanc PH, Yen IH, et al. A comprehensive study of the direct and indirect costs of adult asthma. J Allergy Clin Immunol 2003; 111:1212–1218.
- Sullivan S, Turk F. An evaluation of the cost effectiveness of omalizumab for the treatment of severe persistent asthma. Allergy 2008; 63:670–684.
- US Food and Drug Administration. Omalizumab (marketed as Xolair) information. www.fda.gov/cder/drug/infopage/omalizumab/default.htm. Accessed August 31, 2007.
- Williams SG, Schmidt DK, Redd SC, Storms W. Key clinical activities for quality asthma care. Recommendations of the National Asthma Education and Prevention Program. MMWR Recomm Rep 2003; 52 RR-6:1–8.
- Cox L, Li J, Nelson H, Lockey R, et al. Allergy Immunotherapy: a practice parameter second update. J Allergy Clin Immunol 2007; 120:S25–S85.
- Akdis M, Akdis CA. Mechanisms of allergen-specific immunotherapy. J Allergy Clin Immunol 2007; 119:780–789.
- Murray AB, Milner RA. The accuracy of features in the clinical history for predicting atopic sensitization to airborne allergens in children. J Allergy Clin Immunol 1995; 96:588–596.
- Bernstein IL, Li JT, Bernstein DI, et al. Allergy diagnostic testing: an updated practice parameter. Ann Allergy Asthma Immunol 2008; 100 suppl 3:1S–148S.
- Walker S, Pajno GB, Lima MT, Wilson DR, Durham SR. Grass pollen immunotherapy for seasonal rhinitis and asthma: a randomized, controlled trial. J Allergy Clin Immunol 2001; 107:87–93.
- Varney VA, Edwards J, Tabbah K, Brewster H, Mavroleon G, Frew AJ. Clinical efficacy of specific immunotherapy to cat dander: a double-blind placebo-controlled trial. Clin Exp Allergy 1997; 27:860–867.
- Cantani A, Arcese G, Lucenti P, Gagliesi D, Bartolucci M. A three-year prospective study of specific immunotherapy to inhalant allergens: evidence of safety and efficacy in 300 children with allergic asthma. J Investig Allergol Clin Immunol 1997; 7:90–97.
- Hedlin G, Wille S, Browaldh L, et al. Immunotherapy in children with allergic asthma: effect on bronchial hyperreactivity and pharmacotherapy. J Allergy Clin Immunol 1999; 103:609–614.
- Arvidsson MB, Löwhagen O, Rak S. Allergen specific immunotherapy attenuates early and late phase reactions in lower airways of birch pollen asthmatic patients: a double blind placebo-controlled study. Allergy 2004; 59:74–80.
- Pichler CE, Helbling A, Pichler WJ. Three years of specific immunotherapy with house-dust-mite extracts in patients with rhinitis and asthma: significant improvement of allergen-specific parameters and of nonspecific bronchial hyperreactivity. Allergy 2001; 56:301–306.
- Mirone C, Albert F, Tosi A, et al. Efficacy and safety of subcutaneous immunotherapy with a biologically standardized extract of Ambrosia artemisiifolia pollen: a double-blind, placebo-controlled study. Clin Exp Allergy 2004; 34:1408–1414.
- Abramson MJ, Puy RM, Weiner JM. Allergen immunotherapy for asthma. Cochrane Database Syst Rev 2003; (4):CD001186.
- Jacobsen L. Preventive aspects of immunotherapy: prevention for children at risk of developing asthma. Ann Allergy Asthma Immunol 2001; 87:43–46.
- Moller C, Dreborg S, Ferdousi HA, et al. Pollen immunotherapy reduces the development of asthma in children with seasonal rhinoconjunctivitis (the PAT study). J Allergy Clin Immunol 2002; 109:251–256.
- Niggemann B, Jacobsen L, Dreborg S, et al; PAT Investigator Group. Five year follow-up on the PAT study: specific immunotherapy and long-term prevention of asthma in children. Allergy 2006: 61:855–859.
- Des Roches A, Paradis L, Menardo JL, et al. Immunotherapy with a standardized Dermatophagoides pteronyssinus extract VI: specific immunotherapy prevents the onset of new sensitizations in children. J Allergy Clin Immunol 1997; 99:450–453.
- Pajno GB, Barberio G, DeLuca F, et al. Prevention of new sensitizations in asthmatic children monosensitized to the house dust mite by specific immunotherapy: a six year follow up study. Clin Exp Allergy 2001; 31:1392–1397.
- Lang DM. Do beta blockers really enhance the risk of anaphylaxis during immunotherapy? Curr Allergy Asthma Rep 2008; 8:37–44.
- National Heart, Lung, and Blood institute, National Asthma education and Prevention Program. Expert Panel Report 3: guidelines for the diagnosis and management of asthma. www.nhlbi.nih.gov/guidelines/asthma. Accessed 8/7/08.
- Expert Panel Report 2: Guidelines for the diagnosis and management of asthma. U.S. Department of Health and Human Services. Publication No. 97-4051; 1997.
- Expert Panel Report: Guidelines for the diagnosis and management of asthma. Update on Selected Topics—2002. J Allergy Clin Immunol 2002; 110:S141–S207.
- FitzGerald JM, Boulet LP, McIvor RA, Zimmerman S, Chapman KR. Asthma control in Canada remains suboptimal: the Reality of Asthma Control (TRAC) study. Can Respir J 2006; 13:253–259.
- Braganza S, Sharif I, Ozuah P. Documenting asthma severity: do we get it right? J Asthma 2003; 40:661–665.
- Cockcroft DW, Swystun VA. Asthma control versus asthma severity. J Allergy Clin Immunol 1996; 98:1016–1018.
- Peters SP, Jones CA, Haselkorn T, Mink DR, Valacer DJ, Weiss ST. Real-world Evaluation of Asthma Control and Treatment (REACT): findings from a national Web-based survey. J Allergy Clin Immunol. 2007; 119:1454–1461.
- Osborne ML, Vollmer WM, Pedula KL, Wilkins J, Buist AS, O’Hollaren M. Lack of correlation of symptoms with specialist-assessed long-term asthma severity. Chest 1999; 115:85–91.
- Li JT, Oppenheimer J, Bernstein IL, et al. Attaining optimal asthma control: a practice parameter. J Allergy Clin Immunol 2005; 116:S3–S11.
- Nathan RA, Sorkness C, Kosinski M, et al. Development of the Asthma Control Test: a survey for assessing asthma control. J Allergy Clin Immunol 2004; 113:59–65.
- Schatz M, Zeiger RS, Drane A, et al. Reliability and predictive validity of the Asthma Control Test administered by telephone calls using speech recognition technology. J Allergy Clin Immunol 2007; 119:336–343.
- Peters D, Chen C, Markson LE, Allen-Ramey FC, Vollmer WM. Using an asthma control questionnaire and administrative data to predict healthcare utilization. Chest 2006; 129:918–924.
- Schatz M, Sorkness C, Li JT, et al. Asthma Control Test: reliability, validity, and responsiveness in patients not previously followed by asthma specialists. J Allergy Clin Immunol 2006; 117:549–556.
- Bateman E, Boushey H, Bousquet J, et al. Can guideline-defined asthma control be achieved? Am J Respir Crit Care Med 2004; 170:836–844.
- Davies TJ, Bunn WB, Fromer L, Gelfand EW, Colice GL. A focus on the asthma HEDIS measure and its implications for clinical practice. Manag Care Interface 2006; 19:29–36.
- Rubinfeld AR, Pain MC. Perception of asthma. Lancet 1976; 1:882–884.
- Teeter J, Bleecker E. Relationship between airway obstruction and respiratory symptoms in adult asthmatics. Chest 1998; 113:272–277.
- Shingo S, Zhang J, Reiss T. Correlation of airway obstruction and patient reported endpoints in clinical studies. Eur Resp J 2001; 17:220–224.
- Juniper EF, Bousquet J, Abetz L, Bateman ED; GOAL Committee. Identifying ‘well-controlled’ and ‘not well-controlled’ asthma using the Asthma Control Questionnaire. Respir Med 2006; 100:616–621.
- Nelson H, Weiss S, Bleecker E, Yancey S, Dorinsky P. The Salmeterol Multicenter Asthma Research Trial. Chest 2006; 129:15–26.
- Wechsler M, Lehman E, Lazarus S, et al. ß-Adrenergic receptor polymorphisms and response to salmeterol. Am J Respir Crit Care Med 2006; 173:519–526.
- Palmer CNA, Lipworth BJ, Lee S, Ismail T, MacGregor DF, Mukhopadhyay S. Arginine-16 beta-2 adrenoceptor genotype predisposes to exacerbations in young asthmatics taking regular salmeterol. Thorax 2006; 61:940–944.
- Taylor DR, Drazen JM, Herbison GP, Yandava CN, Hancox RJ, Town GI. Asthma exacerbations during long term beta agonist use: influence of beta 2 adrenoceptor polymorphism. Thorax 2000; 55:762–727.
- Bleecker E, Postma D, Lawrance R, Meyers D, Ambrose H, Goldman M. Effect of ADRB2 polymorphisms on response to long-acting beta2-agonist therapy: a pharmacogenetic analysis of two randomized studies. Lancet 2007; 370:2118–2125.
- Bleecker E, Yancey S, Baitinger L, et al. Salmeterol response is not affected by beta-2 adrenergic receptor genotype in subjects with persistent asthma. J Allergy Clin Immunol 2006; 118:809–816.
- Nelson H, Bleecker E, Corren J, et al. Characterization of asthma exacerbations by Arg16Gly genotype in subjects with asthma receiving salmeterol alone or with fluticasone propionate. J Allergy Clin Immunol 2008; 121:S131.
- O’Byrne P, Barnes P, Rodriguez-Roisin R, et al. Low dose Inhaled budesonide and formoterol in mild persistent asthma. The OPTIMA Randomized Trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Greening AP, Ind PW, Northfield M, Shaw G. Added salmeterol versus higher dose corticosteroid in asthma patients with symptoms on existing inhaled corticosteroid. Lancet 1994; 344:219–224.
- Woolcock A, Lundback B, Ringdal N, Jacques LA. Comparison of addition of salmeterol to inhaled steroids with doubling of the dose of inhaled steroids. Am J Respir Crit Care Med 1996; 153:1481–1488.
- Walters EH, Walters JAE, Gibson MDP. Long-acting beta2-agonists for stable chronic asthma. Cochrane Database Syst Rev 2003; (3):CD001385. doi:10.1002/14651858.CD001385.
- Masoli M, Weatherall M, Holt S, Beasley R. Moderate dose inhaled corticosteroids plus salmeterol versus higher doses of inhaled corticosteroid in symptomatic asthma. Thorax 2005; 60:730–734.
- Sin DD, Man J, Sharpe H, Gan WQ, Man SFP. Pharmacological management to reduce exacerbations in adults with asthma. A systematic review and meta-analysis. JAMA 2004; 292:367–376.
- Mann RD, Kubota K, Pearce G, Wilton L. Salmeterol: a study by prescription event monitoring in a UK cohort of 15,407 patients. J Clin Epidemiol 1996; 49:247–250.
- Lanes S, Lanza L, Wentworth C. Risk of emergency care, hospitalization, and ICU stays for acute asthma among recipients of salmeterol. Am J Respir Crit Care Med 1998; 158:857–861.
- Meier CR, Jick H. Drug use and pulmonary death rates in increasingly symptomatic asthma patients in the UK. Thorax 1997; 52:612–617.
- Williams C, Crossland L, Finnerty J, et al. A case control study of salmeterol and near-fatal attacks of asthma. Thorax 1998; 53:7–13.
- Lanes S, Garcia Rodriguez LA, Herta C. Respiratory medications and risk of asthma death. Thorax 2002; 57:683–686.
- Anderson HR, Ayres JG, Sturdy PM, et al. Bronchodilator treatment and deaths from asthma: case control study. Br Med J 2005; 330:117–124.
- Martinez FD. Safety of long-acting beta agonists—an urgent need to clear the air. N Engl J Med 2005; 353:2637–2639.
- Nelson HS. Long-acting beta-agonists in adult asthma: evidence that these drugs are safe. Prim Care Respir J 2006; 15:271–277.
- Lang DM. The long-acting beta agonist controversy: a critical examination of the evidence. Cleve Clin J Med 2006; 73:973–992.
- Rambasek T, Lang DM, Kavuru M. Omalizumab: where does it fit in current asthma management? Cleve Clin J Med 2004; 71:251–261.
- McGlashan D, Bochner B, Adelman D, et al. Down regulation of Fc(epsilon)RI expression on human basophils during in vivo treatment of atopic patients with anti-IgE antibody. J Immunol 1997; 158:1438–1445.
- Busse W, Corren J, Lanier B, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol 2001; 108:184–190.
- Soler M, Matz J, Townley R, et al. The anti-IgE antibody omalizumab reduces asthma exacerbations and steroid requirement in allergic asthmatics. Eur Respir J 2001; 18:254–261.
- Humbert M, Beasley R, Ayres J, et al. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy 2005; 60:309–316.
- Van Ganse E, Antonicelli L, Zhang Q, et al. Asthma-related resource use and cost by GINA classification of severity in three European countries. Respir Med 2006; 100:140–147.
- Godard P, Chanez P, Siraudin L, Nicoloyannis N, Duru G. Costs of asthma are correlated with severity: a 1-yr prospective study. Eur Respir J 2002; 19:61–67.
- Cisternas MG, Blanc PH, Yen IH, et al. A comprehensive study of the direct and indirect costs of adult asthma. J Allergy Clin Immunol 2003; 111:1212–1218.
- Sullivan S, Turk F. An evaluation of the cost effectiveness of omalizumab for the treatment of severe persistent asthma. Allergy 2008; 63:670–684.
- US Food and Drug Administration. Omalizumab (marketed as Xolair) information. www.fda.gov/cder/drug/infopage/omalizumab/default.htm. Accessed August 31, 2007.
- Williams SG, Schmidt DK, Redd SC, Storms W. Key clinical activities for quality asthma care. Recommendations of the National Asthma Education and Prevention Program. MMWR Recomm Rep 2003; 52 RR-6:1–8.
- Cox L, Li J, Nelson H, Lockey R, et al. Allergy Immunotherapy: a practice parameter second update. J Allergy Clin Immunol 2007; 120:S25–S85.
- Akdis M, Akdis CA. Mechanisms of allergen-specific immunotherapy. J Allergy Clin Immunol 2007; 119:780–789.
- Murray AB, Milner RA. The accuracy of features in the clinical history for predicting atopic sensitization to airborne allergens in children. J Allergy Clin Immunol 1995; 96:588–596.
- Bernstein IL, Li JT, Bernstein DI, et al. Allergy diagnostic testing: an updated practice parameter. Ann Allergy Asthma Immunol 2008; 100 suppl 3:1S–148S.
- Walker S, Pajno GB, Lima MT, Wilson DR, Durham SR. Grass pollen immunotherapy for seasonal rhinitis and asthma: a randomized, controlled trial. J Allergy Clin Immunol 2001; 107:87–93.
- Varney VA, Edwards J, Tabbah K, Brewster H, Mavroleon G, Frew AJ. Clinical efficacy of specific immunotherapy to cat dander: a double-blind placebo-controlled trial. Clin Exp Allergy 1997; 27:860–867.
- Cantani A, Arcese G, Lucenti P, Gagliesi D, Bartolucci M. A three-year prospective study of specific immunotherapy to inhalant allergens: evidence of safety and efficacy in 300 children with allergic asthma. J Investig Allergol Clin Immunol 1997; 7:90–97.
- Hedlin G, Wille S, Browaldh L, et al. Immunotherapy in children with allergic asthma: effect on bronchial hyperreactivity and pharmacotherapy. J Allergy Clin Immunol 1999; 103:609–614.
- Arvidsson MB, Löwhagen O, Rak S. Allergen specific immunotherapy attenuates early and late phase reactions in lower airways of birch pollen asthmatic patients: a double blind placebo-controlled study. Allergy 2004; 59:74–80.
- Pichler CE, Helbling A, Pichler WJ. Three years of specific immunotherapy with house-dust-mite extracts in patients with rhinitis and asthma: significant improvement of allergen-specific parameters and of nonspecific bronchial hyperreactivity. Allergy 2001; 56:301–306.
- Mirone C, Albert F, Tosi A, et al. Efficacy and safety of subcutaneous immunotherapy with a biologically standardized extract of Ambrosia artemisiifolia pollen: a double-blind, placebo-controlled study. Clin Exp Allergy 2004; 34:1408–1414.
- Abramson MJ, Puy RM, Weiner JM. Allergen immunotherapy for asthma. Cochrane Database Syst Rev 2003; (4):CD001186.
- Jacobsen L. Preventive aspects of immunotherapy: prevention for children at risk of developing asthma. Ann Allergy Asthma Immunol 2001; 87:43–46.
- Moller C, Dreborg S, Ferdousi HA, et al. Pollen immunotherapy reduces the development of asthma in children with seasonal rhinoconjunctivitis (the PAT study). J Allergy Clin Immunol 2002; 109:251–256.
- Niggemann B, Jacobsen L, Dreborg S, et al; PAT Investigator Group. Five year follow-up on the PAT study: specific immunotherapy and long-term prevention of asthma in children. Allergy 2006: 61:855–859.
- Des Roches A, Paradis L, Menardo JL, et al. Immunotherapy with a standardized Dermatophagoides pteronyssinus extract VI: specific immunotherapy prevents the onset of new sensitizations in children. J Allergy Clin Immunol 1997; 99:450–453.
- Pajno GB, Barberio G, DeLuca F, et al. Prevention of new sensitizations in asthmatic children monosensitized to the house dust mite by specific immunotherapy: a six year follow up study. Clin Exp Allergy 2001; 31:1392–1397.
- Lang DM. Do beta blockers really enhance the risk of anaphylaxis during immunotherapy? Curr Allergy Asthma Rep 2008; 8:37–44.
KEY POINTS
- The EPR3 recommends that management decisions be based initially on asthma severity, and subsequently on asthma control as assessed serially by validated tests.
- Omalizumab, a monoclonal antibody against immunoglobulin E, is the only adjunctive therapy to demonstrate efficacy when added to high-dose inhaled corticosteroids plus long-acting beta agonists in patients with severe, persistent, allergic asthma.
- The EPR3 guidelines recommend consideration of allergen immunotherapy for patients with mild or moderate persistent allergic asthma.
Man, 48, With Excruciating Leg Pain
A 48-year-old black man, on hemodialysis since August 2002, presented to his primary care provider (PCP) in July 2006 with excruciating leg pain. According to the patient, the leg pain had worsened during the previous six months and was so severe that he was barely able to walk without pain. He was a full-time night security guard and reported walking three to five miles each night.
The man was undergoing hemodialysis three times per week, necessitated by nephritic range proteinuria. He had a questionable history of diabetes but a known diagnosis of hypertension. Definitive diagnosis through kidney biopsy was not obtained because of the associated risk, the patient's obesity, and his aversion to the procedure.
The patient had recently been hospitalized with shortness of breath and fluid overload. Intensive dialysis allowed a significant drop in his dialysis target weight. He was readmitted a few days later with chills, fever, cough, and shortness of breath. He was diagnosed with bilateral pulmonary emboli. The patient said his hypercoagulation work-up was negative, but he was started on warfarin before discharge.
On current presentation, he had swollen, tender legs and multiple excoriations over the calves, explained by the patient's admitted scratching. His skin was shiny and tight. He was still taking warfarin, with an international normalized ratio of 2.1. The patient denied shortness of breath, pruritus (any more than expected with renal disease), or increased fluid.
In addition to warfarin, he was taking esomeprazole 40 mg/d, extended-release metoprolol 25 mg bid, cinacalcet 90 mg/d, sevelamer 4,000 mg and lanthanum 5,000 mg before every meal, mometasone furoate as needed, hydroxyzine 25 mg every four hours as needed, miconazole powder applied to the feet as needed, and a daily prescription multivitamin complex.
Laboratory tests included normal findings (for a dialysis patient) on the complete blood count; blood urea nitrogen, 101 mg/dL (reference range, 7 to 20 mg/dL); serum creatinine, 16.6 mg/dL (0.8 to 1.4 mg/dL); Kt/V (a measure of adequacy of dialysis), 1.37 (acceptable); calcium, 9.6 mg/dL (8.2 to 10.2 mg/dL); serum phosphorus, 5.6 mg/dL (2.4 to 4.1 mg/dL); intact parathyroid hormone, 359 ng/L (10 to 65 ng/L).
The patient's PCP prescribed oxycodone for the pain and referred him to the vascular clinic for evaluation of his legs. A lower leg duplex scan with ankle/brachial indices performed on July 18 showed significant bilateral peripheral vascular disease. Subsequent magnetic resonance angiography (MRA) showed a questionable adrenal gland mass. Abdominal CT with and without contrast yielded negative results for the adrenal mass but showed a cyst in the right kidney. Although cysts are commonly found in dialysis patients, the vascular surgeon elected to evaluate the cyst with an MRI with gadolinium; the mass was found to be hemorrhagic.
Further vascular work-up continued, including MRI with gadolinium on September 26, 2006, which revealed two-vessel runoff in the right foot and three-vessel runoff in the left foot. According to the vascular consult, there was no area to bypass. The patient was sent back to his PCP. At this point, he was taking oxycodone four times per day and continuing to work full-time as a night security guard.
The patient was then sent to neurology for evaluation. By this time, the severity of his leg pain had increased 90%, with worsening swelling and persistent shininess (see figure). The neurologist was unable to obtain electromyograms due to the severity of the patient's pain and lower extremity swelling. No definitive diagnosis could be made.
About one year later, the man's attending nephrology group received copies of the work-up that the PCP sent to the dialysis center. It was apparent that neither the patient's PCP nor the vascular, radiology, or neurology consultants had seen the FDA warning released in June 20061 regarding the use of gadolinium in patients with renal disease. What had started out as a peripheral neuropathy (either renal or diabetic in etiology) was now a full-blown case of nephrogenic systemic fibrosis (NSF).
Open biopsy performed on October 29, 2007, confirmed the presence of gadolinium in the patient's epidermis. He became the first documented case of NSF in the Washington, DC area.
Discussion
In the late 1990s, several reports of an unknown sclerosing dermopathy in patients with chronic kidney disease began to emerge. In 2000, the new entity was named nephrogenic systemic fibrosis, with a disease course demonstrating systemic involvement that affected multiple organ systems and often resulted in severe joint limitations. A Web-based reporting system for this newly described disease, created by Shawn Cowper, MD, of Yale University,2 made it possible to investigate associated epidemiologic factors.
Neither gender, race, nor age appeared relevant. However, all patients had renal disease—acute, chronic, or transient—and more than 90% of patients were dialysis dependent. Factors since recognized to confirm a diagnosis of NSF are severe renal impairment (ie, glomerular filtration rate [GFR] < 30 mL/min/1.73 m2),3 CD34+ dendritic cells found on deep biopsy,4 and the following clinical manifestations:
• Skin. Burning or itching, reddened or darkened patches; possible skin swelling, hardening, and/or tightening.
• Eyes. Yellow raised spots in the whites of the eyes.
• Bones, joints, muscles. Joint stiffness; limited range of motion in the arms, hands, legs, or feet; pain deep in the hip bone or ribs; and/or muscle weakness.3
Theories abounded on the cause of NSF. While the presence of renal disease is a requirement, dialysis did not seem to be.5 Ten percent of NSF cases are patients who have never been dialyzed, and thousands of dialysis patients never develop NSF. Neither was any temporal correlation to dialysis found: While some patients developed NSF soon after starting dialysis, many had been on dialysis for years before NSF occurred. No association was found between NSF and the type of dialysis (inpatient, outpatient, hemodialysis, or peritoneal dialysis), the filter, manufacturer, dialysate, technique, or dialysis unit.2
Authors of a retrospective study involving two large tissue repositories looked for cases of NSF before 1997, but none were found.6 If dialysis was not causing NSF, and the disease did not appear to have existed before 1997, what renal toxin had been introduced in the 1990s to explain it?
One early suspicion involved erythropoietin (EPO), used to treat anemia in patients with kidney disease. Skin changes had been reported in some patients after initiation of treatment with EPO, and the NSF patients received a significantly higher mean dose of EPO than controls received.7
Ninety percent of patients with NSF had fistula reconstruction or dialysis catheter placement, but these are common in renal disease patients.8 Forty-eight percent of patients had had liver or kidney transplants, and 12% had hypercoagulable states. Most patients with NSF had never received ACE inhibitors. Were the protective antifibrogenic properties of these agents missing?
Mystery Solved
In a triumph for the Internet and its capacity to disseminate information around the world, a breakthrough came in 2006 from a small town in Austria. Grobner9 described nine patients who had received gadodiamide (Omniscan™)–enhanced MRA, five of whom developed NSF. Upon release of this report, researchers reexamined the original cases and detected a clear correlation between gadolinium and NSF. Because the contrast dose given for MRA can be as much as three times that required for routine MRI, the absence of NSF cases before 1997 suddenly made sense.
In May 2006, researchers for the Danish Medicines Agency reported 13 cases of NSF in patients injected with gadodiamide.10 Within months, 28 biopsy-proven cases were reported in St. Louis, six in Texas, and 13 at the University of Wisconsin—all involving patients exposed to gadolinium.11-13 It was apparent that NSF was iatrogenic and could be controlled.
What We Have Learned Since
In subsequent research, it has been found that more than 90% of reported cases of NSF occurred following exposure to gadodiamide—although gadodiamide accounts for only 15% of all gadolinium injections worldwide,14 and this number is decreasing as more cases are reported. The correlation between gadodiamide and NSF is so strong that its manufacturer, GE Healthcare, sent practitioners a letter in June 2006 warning of NSF as an adverse effect of gadolinium exposure.15 Two days later, the FDA issued an advisory on gadolinium-enhanced imaging procedures, recommending prompt hemodialysis after gadolinium exposure and reminding radiologists and nephrologists that gadolinium is not FDA approved for MRA.1
Although the 44% incidence rate of NSF reported by Grobner9 has never been replicated, a retrospective review of all known NSF cases affirmed that more than 90% of patients had been exposed to gadolinium.14 Two 2007 reports published in the Journal of the American Academy of Dermatology demonstrated that gadolinium was detectable in the tissues of patients with NSF.16,17
In Europe, in response to the May 2006 report from the Danish Medicines Agency,10 the European Society of Urogenital Radiology revised its guidelines with a directive that gadodiamide not be administered in any patients who had reduced kidney function or were undergoing dialysis.18 Shortly thereafter, the European Committee for Medicinal Products for Human Use issued a contraindication for gadodiamide use in patients with severe renal impairment and advised that these patients not be given gadolinium unless there was no other choice.19 A contraindication was also issued for gadodiamide use in patients with previous or anticipated liver transplantation.
The American College of Radiology guidelines published in 200720 stated that patients with any level of renal disease should not receive gadodiamide.
In March 2007, GE Healthcare published a paper on NSF, reiterating the safety of gadodiamide while acknowledging that 120 more cases had been reported to them ("usually associated with exposure at high doses").21 The FDA upholds an alert regarding use of all gadolinium-based contrast agents for patients with acute or chronic severe renal insufficiency,3 while stopping short of a ban on gadodiamide in such patients.
How Common Is NSF?
In a 2007 study conducted at the University of Wisconsin, Sadowski et al13 reported 13 cases of gadolinium-induced NSF, 11 involving patients with a GFR below 30 mL/min/1.73 m2 but two with a GFR between 30 and 60 mL/min/1.73 m2 (ie, with renal insufficiency, although the authors noted that renal insufficiency was acute in these two patients). The incidence of NSF was 4.6% among hospitalized patients with a GFR be-low 60 mL/min/1.73 m2 who underwent gadolinium-enhanced MRI at the university hospital's radiology department. A reexamination of the charts of the patients with a GFR between 30 and 60 mL/min/1.73 m2 revealed that these patients had levels below 30 mL/min/1.73 m2 when their gadolinium exposure took place.
In an outpatient population–based calculation performed by Deo et al,22 a 2.4% chance of NSF was determined for each gadolinium exposure. Incidence of NSF was calculated at 4.3 cases per 1,000 patient-years in this population, making NSF as common as contrast-induced nephropathy. Nearly 5% of patients with NSF have an exceedingly rapid and fulminant disease course that may result in death. NSF, of itself, is not a cause of death but may contribute to death by restricting effective ventilation or by restricting mobility to the point of causing an accidental fall that may be further exacerbated by fractures and clotting complications. NSF survivors may experience disabling systemic symptoms. Full recovery occurs only in patients who recover renal function, either naturally or by kidney transplantation.4
Why Is NSF More Common With Gadodiamide?
As of June 2008, five gadolinium-based contrast agents were FDA approved for use with MRI (none with MRA)3: gadobenate (MultiHance®), gadodiamide (Omniscan), gadopentetate (Magnevist®), gadoteridol (ProHance®), and gadoversetamide (Opti-MARK®). More than 90% of NSF cases are associated with gadodiamide. Because this agent is the least stable thermodynamically, it may be more likely than the others to transmetallate.14 All gadolinium chelates are excreted by the kidney, and the decreased renal clearances associated with renal impairment may expose patients to prolonged gadolinium transmetallation, allowing the agent to accumulate in bone and other tissue.
Gadoterate (Dotarem®), a cyclic gadolinium-based agent that is available in Europe but not the US, is considered more stable than other agents. It has been suggested that such agents may be safer choices for patients with decreased renal function.14,19
Strategies to Prevent NSF
In the US and Europe, only a physician who has consulted with a radiologist can write an order for gadolinium use in a patient with a GFR below 30 mL/min/1.73 m2.18,20 European guidelines do not allow use of gadodiamide in such patients.
Although the actual population-based occurrence of NSF is low, the nature of the disease calls for an effort to limit vulnerable patients' exposure to gadolinium (see box). Outside of withholding imaging procedures, the only currently known strategies to reduce the incidence of NSF are to use a more stable, nonchelating gadolinium14 and to remove the gadolinium as soon as possible.3,24
It has been recommended that patients with renal disease who are presently undergoing dialysis be dialyzed within two to three hours of gadolinium exposure, then again within 24 and 48 hours, provided it is clinically safe.20,24 This has been shown to remove 99% of the gadolinium.23
Since peritoneal dialysis clears gadolinium poorly, hemodialysis is recommended for peritoneal dialysis patients after gadolinium exposure, following the regimen outlined above.20
No consensus has been reached regarding the patient with a GFR between 30 and 60 mL/min/1.73 m2, nor for the patient with a lower GFR and no access for dialysis to be administered. Placement of a catheter for two days' dialysis incurs both surgical and renal risks for these patients.8
Patient Outcome
The only known cure for NSF is kidney transplantation, which is associated with a complete cure rate of 40%.4,25 Nevertheless, while this manuscript was in preparation, the patient presented in this case study underwent kidney transplantation. On day 8 postsurgery, he was no longer taking oxycodone, his skin condition was clearing up, and he was feeling considerably better. His health care providers hope for further regression from his disease.
Conclusion
NSF is just one example of iatrogenic conditions that can occur in any hospital, office, or clinic. Health care providers cannot be too vigilant in keeping abreast of warnings from the FDA and other agencies. In this case, several clinicians overlooked a recent, urgent public health advisory, with significant consequences.
1. US Food and Drug Administration. Public health advisory: gadolinium-containing contrast agents for magnetic resonance imaging (MRI): Omniscan, OptiMARK, Magnevist, ProHance, and MultiHance. www.fda.gov/cder/drug/advisory/gadolinium_agents.htm. Accessed July 24, 2008.
2. Cowper SE, Su L, Bhawan J, et al. Nephrogenic fibrosing dermopathy. Am J Dermatopathol. 2001;23(5):383-393.
3. US Food and Drug Administration. Information for healthcare professionals: gadolinium-based contrast agents for magnetic resonance imaging (marketed as Magnevist, MultiHance, Omniscan, OptiMARK, ProHance). Last updated June 4, 2008. www.fda.gov/cder/drug/InfoSheets/HCP/gcca_200705.htm. Accessed July 24, 2008.
4. International Center for Nephrogenic Fibrosing Dermopathy Research. www.icnfdr.org. Accessed July 24, 2008.
5. DeHoratius DM, Cowper SE. Nephrogenic systemic fibrosis: an emerging threat among renal patients. Semin Dial. 2006;19(3):191-194.
6. Galan A, Cowper SE, Bucala R. Nephrogenic systemic fibrosis (nephrogenic fibrosing dermopathy). Curr Opin Rheumatol. 2006;18(6):614-617.
7. Swaminathan S, Ahmed I, McCarthy JT, et al. Nephrogenic fibrosing dermopathy and high-dose erythropoietin therapy. Ann Intern Med. 2006;145(3):234-235.
8. Miskulin D, Gul A, Rudnick MR, Cowper SE. Nephrogenic systemic fibrosis/nephrogenic fibrosing dermopathy in advanced renal failure. www.uptodate.com/patients/content/topic.do?topicKey=dialysis/48700. Accessed July 24, 2008.
9. Grobner T. Gadolinium: a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol Dial Transplant. 2006;21(4):1104-1108.
10. Marckmann P, Skov L, Rossen K, et al. Nephrogenic systemic fibrosis: suspected causative role of gadodiamide used for contrast-enhanced magnetic resonance imaging. J Am Soc Nephrol. 2006;17(9):2359-2362.
11. Centers for Disease Control and Prevention. Nephrogenic fibrosing dermopathy associated with exposure to gadolinium-containing contrast agents—St. Louis, Missouri, 2002-2006. MMWR Morb Mortal Wkly Rep. 2007;56(7):137-141.
12. Khurana A, Runge VM, Narayanan M, et al. Nephrogenic systemic fibrosis: a review of 6 cases temporally related to gadodiamide injection (Omniscan). Invest Radiol. 2007;42(2):139-145.
13. Sadowski EA, Bennett LK, Chan MR, et al. Nephrogenic systemic fibrosis: risk factors and incidence estimation. Radiology. 2007;243(1):148-157.
14. Morcos SK. Nephrogenic systemic fibrosis following the administration of extracellular gadolinium based contrast agents: is the stability of the contrast agent molecule an important factor in the pathogenesis of this condition? Br J Radiol. 2007;80(950):73-76.
15. GE Healthcare. Omniscan safety update. http://md.gehealthcare.com/omniscan/safety/index.html. Accessed July 24, 2008.
16. Boyd AS, Zic JA, Abraham JL. Gadolinium deposition in nephrogenic fibrosing dermopathy. J Am Acad Dermatol. 2007;56(1):27-30.
17. High WA, Ayers RA, Chandler J, et al. Gadolinium is detectable within the tissue of patients with nephrogenic systemic fibrosis. J Am Acad Dermatol. 2007;56(1):21-26.
18. Thomsen H; European Society of Urogenital Radiology. European Society of Urogenital Radiology guidelines on contrast media application. Curr Opin Urol. 2007;17(1):70-76.
19. Bongartz G. Imaging in the time of NFD/NSF: do we have to change our routines concerning renal insufficiency? MAGMA. 2007;20(2):57-62.
20. Kanal E, Barkovich AJ, Bell C, et al; ACR Blue Ribbon Panel on MR Safety. ACR guidance document for safe MR practices: 2007. AJR Am J Roentgenol. 2007;188(6):1447-1474.
21. GE Healthcare Paper on Nephrogenic Systemic Fibrosis (March 2007). http://md.gehealthcare.com/omniscan/GE% 20Healthcare%20Paper%20On%20Nephrogenic%20 Systemic%20Fibrosis.pdf. Accessed July 24, 2008.
22. Deo A, Fogel M, Cowper SE. Nephrogenic systemic fibrosis: a population study examining the relationship of disease development to gadolinium exposure. Clin J Am Soc Nephrol. 2007;2(2):264-267.
23. Okada S, Katagiri K, Kumazaki T, Yokoyama H. Safety of gadolinium contrast agent in hemodialysis patients. Acta Radiol. 2001;42(3):339-341.
24. Kuo PH, Kanal E, Abu-Alfa AK, Cowper SE. Gadolinium-based MR contrast agents and nephrogenic systemic fibrosis. Radiology. 2007;242(3):647-649.
25. Cowper SE. Nephrogenic systemic fibrosis: the nosological and conceptual evolution of nephrogenic fibrosing dermopathy. Am J Kidney Dis. 2005;46(4):763-765.
A 48-year-old black man, on hemodialysis since August 2002, presented to his primary care provider (PCP) in July 2006 with excruciating leg pain. According to the patient, the leg pain had worsened during the previous six months and was so severe that he was barely able to walk without pain. He was a full-time night security guard and reported walking three to five miles each night.
The man was undergoing hemodialysis three times per week, necessitated by nephritic range proteinuria. He had a questionable history of diabetes but a known diagnosis of hypertension. Definitive diagnosis through kidney biopsy was not obtained because of the associated risk, the patient's obesity, and his aversion to the procedure.
The patient had recently been hospitalized with shortness of breath and fluid overload. Intensive dialysis allowed a significant drop in his dialysis target weight. He was readmitted a few days later with chills, fever, cough, and shortness of breath. He was diagnosed with bilateral pulmonary emboli. The patient said his hypercoagulation work-up was negative, but he was started on warfarin before discharge.
On current presentation, he had swollen, tender legs and multiple excoriations over the calves, explained by the patient's admitted scratching. His skin was shiny and tight. He was still taking warfarin, with an international normalized ratio of 2.1. The patient denied shortness of breath, pruritus (any more than expected with renal disease), or increased fluid.
In addition to warfarin, he was taking esomeprazole 40 mg/d, extended-release metoprolol 25 mg bid, cinacalcet 90 mg/d, sevelamer 4,000 mg and lanthanum 5,000 mg before every meal, mometasone furoate as needed, hydroxyzine 25 mg every four hours as needed, miconazole powder applied to the feet as needed, and a daily prescription multivitamin complex.
Laboratory tests included normal findings (for a dialysis patient) on the complete blood count; blood urea nitrogen, 101 mg/dL (reference range, 7 to 20 mg/dL); serum creatinine, 16.6 mg/dL (0.8 to 1.4 mg/dL); Kt/V (a measure of adequacy of dialysis), 1.37 (acceptable); calcium, 9.6 mg/dL (8.2 to 10.2 mg/dL); serum phosphorus, 5.6 mg/dL (2.4 to 4.1 mg/dL); intact parathyroid hormone, 359 ng/L (10 to 65 ng/L).
The patient's PCP prescribed oxycodone for the pain and referred him to the vascular clinic for evaluation of his legs. A lower leg duplex scan with ankle/brachial indices performed on July 18 showed significant bilateral peripheral vascular disease. Subsequent magnetic resonance angiography (MRA) showed a questionable adrenal gland mass. Abdominal CT with and without contrast yielded negative results for the adrenal mass but showed a cyst in the right kidney. Although cysts are commonly found in dialysis patients, the vascular surgeon elected to evaluate the cyst with an MRI with gadolinium; the mass was found to be hemorrhagic.
Further vascular work-up continued, including MRI with gadolinium on September 26, 2006, which revealed two-vessel runoff in the right foot and three-vessel runoff in the left foot. According to the vascular consult, there was no area to bypass. The patient was sent back to his PCP. At this point, he was taking oxycodone four times per day and continuing to work full-time as a night security guard.
The patient was then sent to neurology for evaluation. By this time, the severity of his leg pain had increased 90%, with worsening swelling and persistent shininess (see figure). The neurologist was unable to obtain electromyograms due to the severity of the patient's pain and lower extremity swelling. No definitive diagnosis could be made.
About one year later, the man's attending nephrology group received copies of the work-up that the PCP sent to the dialysis center. It was apparent that neither the patient's PCP nor the vascular, radiology, or neurology consultants had seen the FDA warning released in June 20061 regarding the use of gadolinium in patients with renal disease. What had started out as a peripheral neuropathy (either renal or diabetic in etiology) was now a full-blown case of nephrogenic systemic fibrosis (NSF).
Open biopsy performed on October 29, 2007, confirmed the presence of gadolinium in the patient's epidermis. He became the first documented case of NSF in the Washington, DC area.
Discussion
In the late 1990s, several reports of an unknown sclerosing dermopathy in patients with chronic kidney disease began to emerge. In 2000, the new entity was named nephrogenic systemic fibrosis, with a disease course demonstrating systemic involvement that affected multiple organ systems and often resulted in severe joint limitations. A Web-based reporting system for this newly described disease, created by Shawn Cowper, MD, of Yale University,2 made it possible to investigate associated epidemiologic factors.
Neither gender, race, nor age appeared relevant. However, all patients had renal disease—acute, chronic, or transient—and more than 90% of patients were dialysis dependent. Factors since recognized to confirm a diagnosis of NSF are severe renal impairment (ie, glomerular filtration rate [GFR] < 30 mL/min/1.73 m2),3 CD34+ dendritic cells found on deep biopsy,4 and the following clinical manifestations:
• Skin. Burning or itching, reddened or darkened patches; possible skin swelling, hardening, and/or tightening.
• Eyes. Yellow raised spots in the whites of the eyes.
• Bones, joints, muscles. Joint stiffness; limited range of motion in the arms, hands, legs, or feet; pain deep in the hip bone or ribs; and/or muscle weakness.3
Theories abounded on the cause of NSF. While the presence of renal disease is a requirement, dialysis did not seem to be.5 Ten percent of NSF cases are patients who have never been dialyzed, and thousands of dialysis patients never develop NSF. Neither was any temporal correlation to dialysis found: While some patients developed NSF soon after starting dialysis, many had been on dialysis for years before NSF occurred. No association was found between NSF and the type of dialysis (inpatient, outpatient, hemodialysis, or peritoneal dialysis), the filter, manufacturer, dialysate, technique, or dialysis unit.2
Authors of a retrospective study involving two large tissue repositories looked for cases of NSF before 1997, but none were found.6 If dialysis was not causing NSF, and the disease did not appear to have existed before 1997, what renal toxin had been introduced in the 1990s to explain it?
One early suspicion involved erythropoietin (EPO), used to treat anemia in patients with kidney disease. Skin changes had been reported in some patients after initiation of treatment with EPO, and the NSF patients received a significantly higher mean dose of EPO than controls received.7
Ninety percent of patients with NSF had fistula reconstruction or dialysis catheter placement, but these are common in renal disease patients.8 Forty-eight percent of patients had had liver or kidney transplants, and 12% had hypercoagulable states. Most patients with NSF had never received ACE inhibitors. Were the protective antifibrogenic properties of these agents missing?
Mystery Solved
In a triumph for the Internet and its capacity to disseminate information around the world, a breakthrough came in 2006 from a small town in Austria. Grobner9 described nine patients who had received gadodiamide (Omniscan™)–enhanced MRA, five of whom developed NSF. Upon release of this report, researchers reexamined the original cases and detected a clear correlation between gadolinium and NSF. Because the contrast dose given for MRA can be as much as three times that required for routine MRI, the absence of NSF cases before 1997 suddenly made sense.
In May 2006, researchers for the Danish Medicines Agency reported 13 cases of NSF in patients injected with gadodiamide.10 Within months, 28 biopsy-proven cases were reported in St. Louis, six in Texas, and 13 at the University of Wisconsin—all involving patients exposed to gadolinium.11-13 It was apparent that NSF was iatrogenic and could be controlled.
What We Have Learned Since
In subsequent research, it has been found that more than 90% of reported cases of NSF occurred following exposure to gadodiamide—although gadodiamide accounts for only 15% of all gadolinium injections worldwide,14 and this number is decreasing as more cases are reported. The correlation between gadodiamide and NSF is so strong that its manufacturer, GE Healthcare, sent practitioners a letter in June 2006 warning of NSF as an adverse effect of gadolinium exposure.15 Two days later, the FDA issued an advisory on gadolinium-enhanced imaging procedures, recommending prompt hemodialysis after gadolinium exposure and reminding radiologists and nephrologists that gadolinium is not FDA approved for MRA.1
Although the 44% incidence rate of NSF reported by Grobner9 has never been replicated, a retrospective review of all known NSF cases affirmed that more than 90% of patients had been exposed to gadolinium.14 Two 2007 reports published in the Journal of the American Academy of Dermatology demonstrated that gadolinium was detectable in the tissues of patients with NSF.16,17
In Europe, in response to the May 2006 report from the Danish Medicines Agency,10 the European Society of Urogenital Radiology revised its guidelines with a directive that gadodiamide not be administered in any patients who had reduced kidney function or were undergoing dialysis.18 Shortly thereafter, the European Committee for Medicinal Products for Human Use issued a contraindication for gadodiamide use in patients with severe renal impairment and advised that these patients not be given gadolinium unless there was no other choice.19 A contraindication was also issued for gadodiamide use in patients with previous or anticipated liver transplantation.
The American College of Radiology guidelines published in 200720 stated that patients with any level of renal disease should not receive gadodiamide.
In March 2007, GE Healthcare published a paper on NSF, reiterating the safety of gadodiamide while acknowledging that 120 more cases had been reported to them ("usually associated with exposure at high doses").21 The FDA upholds an alert regarding use of all gadolinium-based contrast agents for patients with acute or chronic severe renal insufficiency,3 while stopping short of a ban on gadodiamide in such patients.
How Common Is NSF?
In a 2007 study conducted at the University of Wisconsin, Sadowski et al13 reported 13 cases of gadolinium-induced NSF, 11 involving patients with a GFR below 30 mL/min/1.73 m2 but two with a GFR between 30 and 60 mL/min/1.73 m2 (ie, with renal insufficiency, although the authors noted that renal insufficiency was acute in these two patients). The incidence of NSF was 4.6% among hospitalized patients with a GFR be-low 60 mL/min/1.73 m2 who underwent gadolinium-enhanced MRI at the university hospital's radiology department. A reexamination of the charts of the patients with a GFR between 30 and 60 mL/min/1.73 m2 revealed that these patients had levels below 30 mL/min/1.73 m2 when their gadolinium exposure took place.
In an outpatient population–based calculation performed by Deo et al,22 a 2.4% chance of NSF was determined for each gadolinium exposure. Incidence of NSF was calculated at 4.3 cases per 1,000 patient-years in this population, making NSF as common as contrast-induced nephropathy. Nearly 5% of patients with NSF have an exceedingly rapid and fulminant disease course that may result in death. NSF, of itself, is not a cause of death but may contribute to death by restricting effective ventilation or by restricting mobility to the point of causing an accidental fall that may be further exacerbated by fractures and clotting complications. NSF survivors may experience disabling systemic symptoms. Full recovery occurs only in patients who recover renal function, either naturally or by kidney transplantation.4
Why Is NSF More Common With Gadodiamide?
As of June 2008, five gadolinium-based contrast agents were FDA approved for use with MRI (none with MRA)3: gadobenate (MultiHance®), gadodiamide (Omniscan), gadopentetate (Magnevist®), gadoteridol (ProHance®), and gadoversetamide (Opti-MARK®). More than 90% of NSF cases are associated with gadodiamide. Because this agent is the least stable thermodynamically, it may be more likely than the others to transmetallate.14 All gadolinium chelates are excreted by the kidney, and the decreased renal clearances associated with renal impairment may expose patients to prolonged gadolinium transmetallation, allowing the agent to accumulate in bone and other tissue.
Gadoterate (Dotarem®), a cyclic gadolinium-based agent that is available in Europe but not the US, is considered more stable than other agents. It has been suggested that such agents may be safer choices for patients with decreased renal function.14,19
Strategies to Prevent NSF
In the US and Europe, only a physician who has consulted with a radiologist can write an order for gadolinium use in a patient with a GFR below 30 mL/min/1.73 m2.18,20 European guidelines do not allow use of gadodiamide in such patients.
Although the actual population-based occurrence of NSF is low, the nature of the disease calls for an effort to limit vulnerable patients' exposure to gadolinium (see box). Outside of withholding imaging procedures, the only currently known strategies to reduce the incidence of NSF are to use a more stable, nonchelating gadolinium14 and to remove the gadolinium as soon as possible.3,24
It has been recommended that patients with renal disease who are presently undergoing dialysis be dialyzed within two to three hours of gadolinium exposure, then again within 24 and 48 hours, provided it is clinically safe.20,24 This has been shown to remove 99% of the gadolinium.23
Since peritoneal dialysis clears gadolinium poorly, hemodialysis is recommended for peritoneal dialysis patients after gadolinium exposure, following the regimen outlined above.20
No consensus has been reached regarding the patient with a GFR between 30 and 60 mL/min/1.73 m2, nor for the patient with a lower GFR and no access for dialysis to be administered. Placement of a catheter for two days' dialysis incurs both surgical and renal risks for these patients.8
Patient Outcome
The only known cure for NSF is kidney transplantation, which is associated with a complete cure rate of 40%.4,25 Nevertheless, while this manuscript was in preparation, the patient presented in this case study underwent kidney transplantation. On day 8 postsurgery, he was no longer taking oxycodone, his skin condition was clearing up, and he was feeling considerably better. His health care providers hope for further regression from his disease.
Conclusion
NSF is just one example of iatrogenic conditions that can occur in any hospital, office, or clinic. Health care providers cannot be too vigilant in keeping abreast of warnings from the FDA and other agencies. In this case, several clinicians overlooked a recent, urgent public health advisory, with significant consequences.
A 48-year-old black man, on hemodialysis since August 2002, presented to his primary care provider (PCP) in July 2006 with excruciating leg pain. According to the patient, the leg pain had worsened during the previous six months and was so severe that he was barely able to walk without pain. He was a full-time night security guard and reported walking three to five miles each night.
The man was undergoing hemodialysis three times per week, necessitated by nephritic range proteinuria. He had a questionable history of diabetes but a known diagnosis of hypertension. Definitive diagnosis through kidney biopsy was not obtained because of the associated risk, the patient's obesity, and his aversion to the procedure.
The patient had recently been hospitalized with shortness of breath and fluid overload. Intensive dialysis allowed a significant drop in his dialysis target weight. He was readmitted a few days later with chills, fever, cough, and shortness of breath. He was diagnosed with bilateral pulmonary emboli. The patient said his hypercoagulation work-up was negative, but he was started on warfarin before discharge.
On current presentation, he had swollen, tender legs and multiple excoriations over the calves, explained by the patient's admitted scratching. His skin was shiny and tight. He was still taking warfarin, with an international normalized ratio of 2.1. The patient denied shortness of breath, pruritus (any more than expected with renal disease), or increased fluid.
In addition to warfarin, he was taking esomeprazole 40 mg/d, extended-release metoprolol 25 mg bid, cinacalcet 90 mg/d, sevelamer 4,000 mg and lanthanum 5,000 mg before every meal, mometasone furoate as needed, hydroxyzine 25 mg every four hours as needed, miconazole powder applied to the feet as needed, and a daily prescription multivitamin complex.
Laboratory tests included normal findings (for a dialysis patient) on the complete blood count; blood urea nitrogen, 101 mg/dL (reference range, 7 to 20 mg/dL); serum creatinine, 16.6 mg/dL (0.8 to 1.4 mg/dL); Kt/V (a measure of adequacy of dialysis), 1.37 (acceptable); calcium, 9.6 mg/dL (8.2 to 10.2 mg/dL); serum phosphorus, 5.6 mg/dL (2.4 to 4.1 mg/dL); intact parathyroid hormone, 359 ng/L (10 to 65 ng/L).
The patient's PCP prescribed oxycodone for the pain and referred him to the vascular clinic for evaluation of his legs. A lower leg duplex scan with ankle/brachial indices performed on July 18 showed significant bilateral peripheral vascular disease. Subsequent magnetic resonance angiography (MRA) showed a questionable adrenal gland mass. Abdominal CT with and without contrast yielded negative results for the adrenal mass but showed a cyst in the right kidney. Although cysts are commonly found in dialysis patients, the vascular surgeon elected to evaluate the cyst with an MRI with gadolinium; the mass was found to be hemorrhagic.
Further vascular work-up continued, including MRI with gadolinium on September 26, 2006, which revealed two-vessel runoff in the right foot and three-vessel runoff in the left foot. According to the vascular consult, there was no area to bypass. The patient was sent back to his PCP. At this point, he was taking oxycodone four times per day and continuing to work full-time as a night security guard.
The patient was then sent to neurology for evaluation. By this time, the severity of his leg pain had increased 90%, with worsening swelling and persistent shininess (see figure). The neurologist was unable to obtain electromyograms due to the severity of the patient's pain and lower extremity swelling. No definitive diagnosis could be made.
About one year later, the man's attending nephrology group received copies of the work-up that the PCP sent to the dialysis center. It was apparent that neither the patient's PCP nor the vascular, radiology, or neurology consultants had seen the FDA warning released in June 20061 regarding the use of gadolinium in patients with renal disease. What had started out as a peripheral neuropathy (either renal or diabetic in etiology) was now a full-blown case of nephrogenic systemic fibrosis (NSF).
Open biopsy performed on October 29, 2007, confirmed the presence of gadolinium in the patient's epidermis. He became the first documented case of NSF in the Washington, DC area.
Discussion
In the late 1990s, several reports of an unknown sclerosing dermopathy in patients with chronic kidney disease began to emerge. In 2000, the new entity was named nephrogenic systemic fibrosis, with a disease course demonstrating systemic involvement that affected multiple organ systems and often resulted in severe joint limitations. A Web-based reporting system for this newly described disease, created by Shawn Cowper, MD, of Yale University,2 made it possible to investigate associated epidemiologic factors.
Neither gender, race, nor age appeared relevant. However, all patients had renal disease—acute, chronic, or transient—and more than 90% of patients were dialysis dependent. Factors since recognized to confirm a diagnosis of NSF are severe renal impairment (ie, glomerular filtration rate [GFR] < 30 mL/min/1.73 m2),3 CD34+ dendritic cells found on deep biopsy,4 and the following clinical manifestations:
• Skin. Burning or itching, reddened or darkened patches; possible skin swelling, hardening, and/or tightening.
• Eyes. Yellow raised spots in the whites of the eyes.
• Bones, joints, muscles. Joint stiffness; limited range of motion in the arms, hands, legs, or feet; pain deep in the hip bone or ribs; and/or muscle weakness.3
Theories abounded on the cause of NSF. While the presence of renal disease is a requirement, dialysis did not seem to be.5 Ten percent of NSF cases are patients who have never been dialyzed, and thousands of dialysis patients never develop NSF. Neither was any temporal correlation to dialysis found: While some patients developed NSF soon after starting dialysis, many had been on dialysis for years before NSF occurred. No association was found between NSF and the type of dialysis (inpatient, outpatient, hemodialysis, or peritoneal dialysis), the filter, manufacturer, dialysate, technique, or dialysis unit.2
Authors of a retrospective study involving two large tissue repositories looked for cases of NSF before 1997, but none were found.6 If dialysis was not causing NSF, and the disease did not appear to have existed before 1997, what renal toxin had been introduced in the 1990s to explain it?
One early suspicion involved erythropoietin (EPO), used to treat anemia in patients with kidney disease. Skin changes had been reported in some patients after initiation of treatment with EPO, and the NSF patients received a significantly higher mean dose of EPO than controls received.7
Ninety percent of patients with NSF had fistula reconstruction or dialysis catheter placement, but these are common in renal disease patients.8 Forty-eight percent of patients had had liver or kidney transplants, and 12% had hypercoagulable states. Most patients with NSF had never received ACE inhibitors. Were the protective antifibrogenic properties of these agents missing?
Mystery Solved
In a triumph for the Internet and its capacity to disseminate information around the world, a breakthrough came in 2006 from a small town in Austria. Grobner9 described nine patients who had received gadodiamide (Omniscan™)–enhanced MRA, five of whom developed NSF. Upon release of this report, researchers reexamined the original cases and detected a clear correlation between gadolinium and NSF. Because the contrast dose given for MRA can be as much as three times that required for routine MRI, the absence of NSF cases before 1997 suddenly made sense.
In May 2006, researchers for the Danish Medicines Agency reported 13 cases of NSF in patients injected with gadodiamide.10 Within months, 28 biopsy-proven cases were reported in St. Louis, six in Texas, and 13 at the University of Wisconsin—all involving patients exposed to gadolinium.11-13 It was apparent that NSF was iatrogenic and could be controlled.
What We Have Learned Since
In subsequent research, it has been found that more than 90% of reported cases of NSF occurred following exposure to gadodiamide—although gadodiamide accounts for only 15% of all gadolinium injections worldwide,14 and this number is decreasing as more cases are reported. The correlation between gadodiamide and NSF is so strong that its manufacturer, GE Healthcare, sent practitioners a letter in June 2006 warning of NSF as an adverse effect of gadolinium exposure.15 Two days later, the FDA issued an advisory on gadolinium-enhanced imaging procedures, recommending prompt hemodialysis after gadolinium exposure and reminding radiologists and nephrologists that gadolinium is not FDA approved for MRA.1
Although the 44% incidence rate of NSF reported by Grobner9 has never been replicated, a retrospective review of all known NSF cases affirmed that more than 90% of patients had been exposed to gadolinium.14 Two 2007 reports published in the Journal of the American Academy of Dermatology demonstrated that gadolinium was detectable in the tissues of patients with NSF.16,17
In Europe, in response to the May 2006 report from the Danish Medicines Agency,10 the European Society of Urogenital Radiology revised its guidelines with a directive that gadodiamide not be administered in any patients who had reduced kidney function or were undergoing dialysis.18 Shortly thereafter, the European Committee for Medicinal Products for Human Use issued a contraindication for gadodiamide use in patients with severe renal impairment and advised that these patients not be given gadolinium unless there was no other choice.19 A contraindication was also issued for gadodiamide use in patients with previous or anticipated liver transplantation.
The American College of Radiology guidelines published in 200720 stated that patients with any level of renal disease should not receive gadodiamide.
In March 2007, GE Healthcare published a paper on NSF, reiterating the safety of gadodiamide while acknowledging that 120 more cases had been reported to them ("usually associated with exposure at high doses").21 The FDA upholds an alert regarding use of all gadolinium-based contrast agents for patients with acute or chronic severe renal insufficiency,3 while stopping short of a ban on gadodiamide in such patients.
How Common Is NSF?
In a 2007 study conducted at the University of Wisconsin, Sadowski et al13 reported 13 cases of gadolinium-induced NSF, 11 involving patients with a GFR below 30 mL/min/1.73 m2 but two with a GFR between 30 and 60 mL/min/1.73 m2 (ie, with renal insufficiency, although the authors noted that renal insufficiency was acute in these two patients). The incidence of NSF was 4.6% among hospitalized patients with a GFR be-low 60 mL/min/1.73 m2 who underwent gadolinium-enhanced MRI at the university hospital's radiology department. A reexamination of the charts of the patients with a GFR between 30 and 60 mL/min/1.73 m2 revealed that these patients had levels below 30 mL/min/1.73 m2 when their gadolinium exposure took place.
In an outpatient population–based calculation performed by Deo et al,22 a 2.4% chance of NSF was determined for each gadolinium exposure. Incidence of NSF was calculated at 4.3 cases per 1,000 patient-years in this population, making NSF as common as contrast-induced nephropathy. Nearly 5% of patients with NSF have an exceedingly rapid and fulminant disease course that may result in death. NSF, of itself, is not a cause of death but may contribute to death by restricting effective ventilation or by restricting mobility to the point of causing an accidental fall that may be further exacerbated by fractures and clotting complications. NSF survivors may experience disabling systemic symptoms. Full recovery occurs only in patients who recover renal function, either naturally or by kidney transplantation.4
Why Is NSF More Common With Gadodiamide?
As of June 2008, five gadolinium-based contrast agents were FDA approved for use with MRI (none with MRA)3: gadobenate (MultiHance®), gadodiamide (Omniscan), gadopentetate (Magnevist®), gadoteridol (ProHance®), and gadoversetamide (Opti-MARK®). More than 90% of NSF cases are associated with gadodiamide. Because this agent is the least stable thermodynamically, it may be more likely than the others to transmetallate.14 All gadolinium chelates are excreted by the kidney, and the decreased renal clearances associated with renal impairment may expose patients to prolonged gadolinium transmetallation, allowing the agent to accumulate in bone and other tissue.
Gadoterate (Dotarem®), a cyclic gadolinium-based agent that is available in Europe but not the US, is considered more stable than other agents. It has been suggested that such agents may be safer choices for patients with decreased renal function.14,19
Strategies to Prevent NSF
In the US and Europe, only a physician who has consulted with a radiologist can write an order for gadolinium use in a patient with a GFR below 30 mL/min/1.73 m2.18,20 European guidelines do not allow use of gadodiamide in such patients.
Although the actual population-based occurrence of NSF is low, the nature of the disease calls for an effort to limit vulnerable patients' exposure to gadolinium (see box). Outside of withholding imaging procedures, the only currently known strategies to reduce the incidence of NSF are to use a more stable, nonchelating gadolinium14 and to remove the gadolinium as soon as possible.3,24
It has been recommended that patients with renal disease who are presently undergoing dialysis be dialyzed within two to three hours of gadolinium exposure, then again within 24 and 48 hours, provided it is clinically safe.20,24 This has been shown to remove 99% of the gadolinium.23
Since peritoneal dialysis clears gadolinium poorly, hemodialysis is recommended for peritoneal dialysis patients after gadolinium exposure, following the regimen outlined above.20
No consensus has been reached regarding the patient with a GFR between 30 and 60 mL/min/1.73 m2, nor for the patient with a lower GFR and no access for dialysis to be administered. Placement of a catheter for two days' dialysis incurs both surgical and renal risks for these patients.8
Patient Outcome
The only known cure for NSF is kidney transplantation, which is associated with a complete cure rate of 40%.4,25 Nevertheless, while this manuscript was in preparation, the patient presented in this case study underwent kidney transplantation. On day 8 postsurgery, he was no longer taking oxycodone, his skin condition was clearing up, and he was feeling considerably better. His health care providers hope for further regression from his disease.
Conclusion
NSF is just one example of iatrogenic conditions that can occur in any hospital, office, or clinic. Health care providers cannot be too vigilant in keeping abreast of warnings from the FDA and other agencies. In this case, several clinicians overlooked a recent, urgent public health advisory, with significant consequences.
1. US Food and Drug Administration. Public health advisory: gadolinium-containing contrast agents for magnetic resonance imaging (MRI): Omniscan, OptiMARK, Magnevist, ProHance, and MultiHance. www.fda.gov/cder/drug/advisory/gadolinium_agents.htm. Accessed July 24, 2008.
2. Cowper SE, Su L, Bhawan J, et al. Nephrogenic fibrosing dermopathy. Am J Dermatopathol. 2001;23(5):383-393.
3. US Food and Drug Administration. Information for healthcare professionals: gadolinium-based contrast agents for magnetic resonance imaging (marketed as Magnevist, MultiHance, Omniscan, OptiMARK, ProHance). Last updated June 4, 2008. www.fda.gov/cder/drug/InfoSheets/HCP/gcca_200705.htm. Accessed July 24, 2008.
4. International Center for Nephrogenic Fibrosing Dermopathy Research. www.icnfdr.org. Accessed July 24, 2008.
5. DeHoratius DM, Cowper SE. Nephrogenic systemic fibrosis: an emerging threat among renal patients. Semin Dial. 2006;19(3):191-194.
6. Galan A, Cowper SE, Bucala R. Nephrogenic systemic fibrosis (nephrogenic fibrosing dermopathy). Curr Opin Rheumatol. 2006;18(6):614-617.
7. Swaminathan S, Ahmed I, McCarthy JT, et al. Nephrogenic fibrosing dermopathy and high-dose erythropoietin therapy. Ann Intern Med. 2006;145(3):234-235.
8. Miskulin D, Gul A, Rudnick MR, Cowper SE. Nephrogenic systemic fibrosis/nephrogenic fibrosing dermopathy in advanced renal failure. www.uptodate.com/patients/content/topic.do?topicKey=dialysis/48700. Accessed July 24, 2008.
9. Grobner T. Gadolinium: a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol Dial Transplant. 2006;21(4):1104-1108.
10. Marckmann P, Skov L, Rossen K, et al. Nephrogenic systemic fibrosis: suspected causative role of gadodiamide used for contrast-enhanced magnetic resonance imaging. J Am Soc Nephrol. 2006;17(9):2359-2362.
11. Centers for Disease Control and Prevention. Nephrogenic fibrosing dermopathy associated with exposure to gadolinium-containing contrast agents—St. Louis, Missouri, 2002-2006. MMWR Morb Mortal Wkly Rep. 2007;56(7):137-141.
12. Khurana A, Runge VM, Narayanan M, et al. Nephrogenic systemic fibrosis: a review of 6 cases temporally related to gadodiamide injection (Omniscan). Invest Radiol. 2007;42(2):139-145.
13. Sadowski EA, Bennett LK, Chan MR, et al. Nephrogenic systemic fibrosis: risk factors and incidence estimation. Radiology. 2007;243(1):148-157.
14. Morcos SK. Nephrogenic systemic fibrosis following the administration of extracellular gadolinium based contrast agents: is the stability of the contrast agent molecule an important factor in the pathogenesis of this condition? Br J Radiol. 2007;80(950):73-76.
15. GE Healthcare. Omniscan safety update. http://md.gehealthcare.com/omniscan/safety/index.html. Accessed July 24, 2008.
16. Boyd AS, Zic JA, Abraham JL. Gadolinium deposition in nephrogenic fibrosing dermopathy. J Am Acad Dermatol. 2007;56(1):27-30.
17. High WA, Ayers RA, Chandler J, et al. Gadolinium is detectable within the tissue of patients with nephrogenic systemic fibrosis. J Am Acad Dermatol. 2007;56(1):21-26.
18. Thomsen H; European Society of Urogenital Radiology. European Society of Urogenital Radiology guidelines on contrast media application. Curr Opin Urol. 2007;17(1):70-76.
19. Bongartz G. Imaging in the time of NFD/NSF: do we have to change our routines concerning renal insufficiency? MAGMA. 2007;20(2):57-62.
20. Kanal E, Barkovich AJ, Bell C, et al; ACR Blue Ribbon Panel on MR Safety. ACR guidance document for safe MR practices: 2007. AJR Am J Roentgenol. 2007;188(6):1447-1474.
21. GE Healthcare Paper on Nephrogenic Systemic Fibrosis (March 2007). http://md.gehealthcare.com/omniscan/GE% 20Healthcare%20Paper%20On%20Nephrogenic%20 Systemic%20Fibrosis.pdf. Accessed July 24, 2008.
22. Deo A, Fogel M, Cowper SE. Nephrogenic systemic fibrosis: a population study examining the relationship of disease development to gadolinium exposure. Clin J Am Soc Nephrol. 2007;2(2):264-267.
23. Okada S, Katagiri K, Kumazaki T, Yokoyama H. Safety of gadolinium contrast agent in hemodialysis patients. Acta Radiol. 2001;42(3):339-341.
24. Kuo PH, Kanal E, Abu-Alfa AK, Cowper SE. Gadolinium-based MR contrast agents and nephrogenic systemic fibrosis. Radiology. 2007;242(3):647-649.
25. Cowper SE. Nephrogenic systemic fibrosis: the nosological and conceptual evolution of nephrogenic fibrosing dermopathy. Am J Kidney Dis. 2005;46(4):763-765.
1. US Food and Drug Administration. Public health advisory: gadolinium-containing contrast agents for magnetic resonance imaging (MRI): Omniscan, OptiMARK, Magnevist, ProHance, and MultiHance. www.fda.gov/cder/drug/advisory/gadolinium_agents.htm. Accessed July 24, 2008.
2. Cowper SE, Su L, Bhawan J, et al. Nephrogenic fibrosing dermopathy. Am J Dermatopathol. 2001;23(5):383-393.
3. US Food and Drug Administration. Information for healthcare professionals: gadolinium-based contrast agents for magnetic resonance imaging (marketed as Magnevist, MultiHance, Omniscan, OptiMARK, ProHance). Last updated June 4, 2008. www.fda.gov/cder/drug/InfoSheets/HCP/gcca_200705.htm. Accessed July 24, 2008.
4. International Center for Nephrogenic Fibrosing Dermopathy Research. www.icnfdr.org. Accessed July 24, 2008.
5. DeHoratius DM, Cowper SE. Nephrogenic systemic fibrosis: an emerging threat among renal patients. Semin Dial. 2006;19(3):191-194.
6. Galan A, Cowper SE, Bucala R. Nephrogenic systemic fibrosis (nephrogenic fibrosing dermopathy). Curr Opin Rheumatol. 2006;18(6):614-617.
7. Swaminathan S, Ahmed I, McCarthy JT, et al. Nephrogenic fibrosing dermopathy and high-dose erythropoietin therapy. Ann Intern Med. 2006;145(3):234-235.
8. Miskulin D, Gul A, Rudnick MR, Cowper SE. Nephrogenic systemic fibrosis/nephrogenic fibrosing dermopathy in advanced renal failure. www.uptodate.com/patients/content/topic.do?topicKey=dialysis/48700. Accessed July 24, 2008.
9. Grobner T. Gadolinium: a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol Dial Transplant. 2006;21(4):1104-1108.
10. Marckmann P, Skov L, Rossen K, et al. Nephrogenic systemic fibrosis: suspected causative role of gadodiamide used for contrast-enhanced magnetic resonance imaging. J Am Soc Nephrol. 2006;17(9):2359-2362.
11. Centers for Disease Control and Prevention. Nephrogenic fibrosing dermopathy associated with exposure to gadolinium-containing contrast agents—St. Louis, Missouri, 2002-2006. MMWR Morb Mortal Wkly Rep. 2007;56(7):137-141.
12. Khurana A, Runge VM, Narayanan M, et al. Nephrogenic systemic fibrosis: a review of 6 cases temporally related to gadodiamide injection (Omniscan). Invest Radiol. 2007;42(2):139-145.
13. Sadowski EA, Bennett LK, Chan MR, et al. Nephrogenic systemic fibrosis: risk factors and incidence estimation. Radiology. 2007;243(1):148-157.
14. Morcos SK. Nephrogenic systemic fibrosis following the administration of extracellular gadolinium based contrast agents: is the stability of the contrast agent molecule an important factor in the pathogenesis of this condition? Br J Radiol. 2007;80(950):73-76.
15. GE Healthcare. Omniscan safety update. http://md.gehealthcare.com/omniscan/safety/index.html. Accessed July 24, 2008.
16. Boyd AS, Zic JA, Abraham JL. Gadolinium deposition in nephrogenic fibrosing dermopathy. J Am Acad Dermatol. 2007;56(1):27-30.
17. High WA, Ayers RA, Chandler J, et al. Gadolinium is detectable within the tissue of patients with nephrogenic systemic fibrosis. J Am Acad Dermatol. 2007;56(1):21-26.
18. Thomsen H; European Society of Urogenital Radiology. European Society of Urogenital Radiology guidelines on contrast media application. Curr Opin Urol. 2007;17(1):70-76.
19. Bongartz G. Imaging in the time of NFD/NSF: do we have to change our routines concerning renal insufficiency? MAGMA. 2007;20(2):57-62.
20. Kanal E, Barkovich AJ, Bell C, et al; ACR Blue Ribbon Panel on MR Safety. ACR guidance document for safe MR practices: 2007. AJR Am J Roentgenol. 2007;188(6):1447-1474.
21. GE Healthcare Paper on Nephrogenic Systemic Fibrosis (March 2007). http://md.gehealthcare.com/omniscan/GE% 20Healthcare%20Paper%20On%20Nephrogenic%20 Systemic%20Fibrosis.pdf. Accessed July 24, 2008.
22. Deo A, Fogel M, Cowper SE. Nephrogenic systemic fibrosis: a population study examining the relationship of disease development to gadolinium exposure. Clin J Am Soc Nephrol. 2007;2(2):264-267.
23. Okada S, Katagiri K, Kumazaki T, Yokoyama H. Safety of gadolinium contrast agent in hemodialysis patients. Acta Radiol. 2001;42(3):339-341.
24. Kuo PH, Kanal E, Abu-Alfa AK, Cowper SE. Gadolinium-based MR contrast agents and nephrogenic systemic fibrosis. Radiology. 2007;242(3):647-649.
25. Cowper SE. Nephrogenic systemic fibrosis: the nosological and conceptual evolution of nephrogenic fibrosing dermopathy. Am J Kidney Dis. 2005;46(4):763-765.
Movement disorder emergencies in the elderly: Recognizing and treating an often-iatrogenic problem
Although we tend to think of movement disorders as chronic conditions, some of them can present as true emergencies in which failure to diagnose the condition and treat it promptly can result in significant sickness or even death.
Many cases are iatrogenic, occurring in patients with Parkinson disease or those taking antipsychotic or antidepressant medications when their regimen is started or altered. Elderly patients are particularly at risk, as they take more drugs and have less physiologic reserve.
Movement disorder emergencies in elderly patients can be difficult to diagnose and treat, since many patients are taking more than one medication: polypharmacy raises the possibility of interactions, and different drugs can cause different movement disorder syndromes. Moreover, because so many patients are at risk—for example, more than 1 million people in the United States now have Parkinson disease, and the number is growing—it is important for physicians who take care of the elderly to be more informed about these disorders, especially the presenting symptoms.
SCOPE OF THIS ARTICLE

- Disorders presenting with rigidity or stiffness
- Disorders presenting with dystonia
- Disorders presenting with hyperkinetic movements
- Disorders presenting with psychiatric disturbances.
Of these, the scenarios most likely to require emergency evaluation in the elderly are acute hypokinetic and hyperkinetic syndromes and psychiatric presentations. This article discusses movement disorder emergencies in the elderly, focusing on the more common disorders with common presentations.
DISORDERS PRESENTING WITH RIGIDITY OR STIFFNESS
Serotonin syndrome

Chief among the offenders are the selective serotonin-reuptake inhibitors (SSRIs), either alone or in combination. This syndrome occurs in 14% to 16% of patients who overdose on SSRIs.1 Examples of combinations that can lead to serotonin syndrome are an SSRI plus any of the following:
- An anxiolytic such as buspirone (BuSpar; this combination is popular for the treatment of depression and anxiety)
- A tricyclic agent such as imipramine (Tofranil)
- A serotonin and norepinephrine reuptake inhibitor such as venlafaxine (Effexor).
In addition, antiparkinson drugs such as levodopa and selegiline (Eldepryl) enhance serotonin release.
Signs and symptoms. Serotonin syndrome is characterized by:
- Severe rigidity
- Dysautonomia
- Change in mental status.
Other clinical findings include fever, gastrointestinal disturbances, and motor restlessness. Clonus is the most important finding in establishing the diagnosis.2
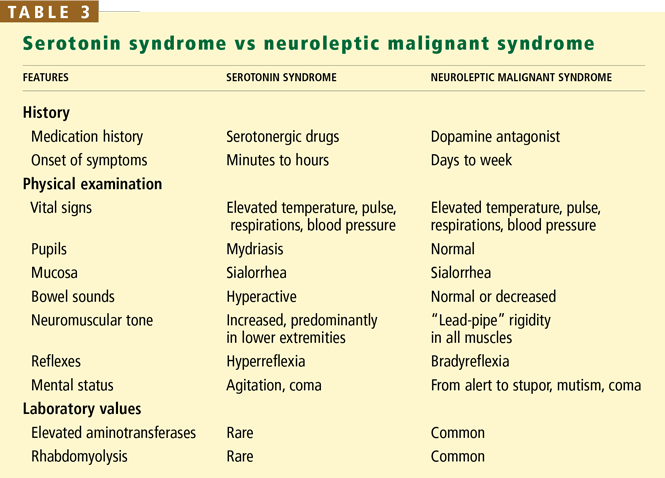
The syndrome may initially go unrecognized and can be mistaken for viral illness or anxiety.4 Manifestations range from mild to life-threatening; initially, it may present with akathisia and tremor. The symptoms progress rapidly over hours and can range from myoclonus, hyperreflexia, and seizures to severe forms of rhabdomyolysis, renal failure, and respiratory failure. The hyperreflexia and clonus seen in moderate cases may be considerably greater in the lower extremities than in the upper extremities.5
No laboratory test confirms the diagnosis, but tremor, clonus, or akathisia without additional extrapyramidal signs should lead to the diagnosis if the patient was taking a serotonergic medication.5 The onset of symptoms is usually rapid. The majority of patients present within 6 hours after initial use of the medication, an overdose, or a change in dosing.5
Treatment. The first steps are to stop the serotonergic medication and to hydrate and cool the patient to counteract the hyperpyrexic state. Benzodiazepine drugs are important in controlling agitation, regardless of its severity.5 Propranolol (Inderal) is not recommended, as it may cause hypotension and shock in patients with autonomic instability.5
Patients with moderate cases may additionally benefit from cyproheptadine (Periactin), an antihistamine that antagonizes serotonin. The initial dose is 4 to 8 mg orally, with a repeat dose after 2 hours.6 Whether to continue this treatment depends on the response after two doses.
If medications must be given parenterally, physicians can consider chlorpromazine (Thorazine) 50 to 100 mg intramuscularly.5
Vital signs should be monitored. In severe cases, intensive care may be required with immediate sedation, neuromuscular paralysis, and intubation.
In most cases, patients improve rapidly.
Comment. Serotonin syndrome can be avoided by educating physicians and by modifying prescribing practices.5 Avoiding multidrug regimens is critical to preventing serotonin syndrome. Computer-based ordering systems and personal digital assistants can help one avoid drug interactions.5
Neuroleptic malignant syndrome
This syndrome is an infrequent but potentially lethal complication associated with therapy with antipsychotic drugs such as haloperidol (Haldol) and lithium (Eskalith) and with other medications with dopamine type-2 receptor antagonist activity such as metoclopramide (Reglan) and prochlorperazine (Compazine). It has become rare since the introduction of atypical antipsychotics and now occurs in 0.2% of patients receiving atypical antipsychotics.7 Its pathogenesis is not fully understood.
This syndrome occurs mainly in young or middle-aged patients receiving doses of neuroleptics within the usual therapeutic range, but it also appears to occur in elderly patients who receive higher doses.8 Although most cases develop in the first 2 weeks of treatment, it can develop at any time during therapy.
Signs and symptoms. Four features characterize neuroleptic malignant syndrome9:
- Muscle rigidity—generalized (“lead-pipe”) muscular rigidity is accompanied by bradykinesia or akinesia.
- Autonomic dysregulation, with tachycardia, tachypnea, alterations in blood pressure, excessive sweating, and incontinence.
- Hyperthermia—fever can begin hours to days after initiating or increasing the dose of a dopamine antagonist.
- Altered sensorium, ranging from confusion to disorientation and coma.
Symptoms of neuroleptic malignant syndrome typically evolve over several days, in contrast to the rapid onset of the serotonin syndrome. Knowing the precipitating drug also helps distinguish the syndromes: dopamine antagonists produce bradykinesia, whereas serotonin agonists produce hyperkinesia.5
Laboratory abnormalities include elevated serum creatine kinase concentrations and white blood cell counts. Renal function should be assessed when renal failure and rhabdomyolysis are suspected.
Treatment involves stopping the causative medication, cooling the patient, and supporting vital functions.
In mild cases (eg, low-grade fever) benzodiazepines such as lorazepam (Ativan) can stabilize the condition. In moderate cases (eg, more significant rigidity), dopaminergic agonists such as bromocriptine (Parlodel) can be given, although there is no strong clinical evidence for their use. Bromocriptine is usually started at 2.5 mg three times a day and gradually increased in dose if tolerated.
In severe cases, muscle rigidity can be reduced with dantrolene (Dantrium), a muscle relaxant. Dantrolene is started at 1 mg/kg intravenously every 6 hours and gradually increased up to 10 mg/kg total per day.
Some patients remain rigid and febrile up to 4 weeks after the causative agent has been withdrawn. Therefore, these treatments can be continued for a few weeks. After the patient has recovered fully, if it is necessary to resume antipsychotic therapy, an atypical antipsychotic such as quetiapine (Seroquel) can be started after 2 weeks.8
Comment. Although uncommon, neuroleptic malignant syndrome is the most serious adverse effect of neuroleptic drugs, and it is potentially fatal. When neuroleptic malignant syndrome is suspected, treatment should be prompt, and the neuroleptic medication should be immediately stopped.
Parkinsonism-hyperpyrexia syndrome
Withdrawing or decreasing the dose of dopaminergic medications in patients with Parkinson disease can cause parkinsonism-hyperpyrexia syndrome, a condition that is similar to neuroleptic malignant syndrome. It can also arise after sudden withdrawal of amantadine (Symmetrel) or anticholinergics. In view of this concern, adjustments to antiparkinson drugs may need to be more gradual in some elderly patients.
Patients present with fever, rigidity, and autonomic instability and are at risk of aspiration pneumonia.
Treatment includes resuming dopaminergic therapy and giving supportive care.
Apomorphine (Apokyn), a dopaminergic agonist, was used in a 71-year-old female parkinsonian patient who developed parkinsonism-hyperpyrexia syndrome after abrupt reduction of chronic levodopa treatment.10 The symptoms resolved within 24 hours of the addition of apomorphine to her previous levodopa therapy. If the patient is taking apomorphine for the first time, the injections should be given in low doses, 0.2 mL subcutaneously. Apomorphine can induce vomiting, and if this occurs an antiemetic such as trimethobenzamide (Tigan) should be given before subsequent injections. In the elderly, caution is advised as apomorphine may cause severe orthostasis.
Methylprednisolone (Solu-Medrol) pulse therapy has been shown to shorten the duration of this syndrome in a randomized, controlled study.11
Akinetic syndrome after failure of deep brain stimulator
Deep brain stimulation involves surgical placement of a pacemaker with electrodes in specific areas of the brain. It is used to control Parkinson disease, tremor, and, less commonly, dystonia, and a number of other uses are under investigation. Continuous electrical stimulation of different nuclei in the brain has been shown to alleviate some symptoms of Parkinson disease (eg, rigidity) and to enable some patients to decrease the dose of their antiparkinson medications.
Several cases have been reported of sudden, unexpected reappearance of freezing, gait disturbance, or severe akinesia in Parkinson disease patients whose stimulators had been turned off inadvertently (eg, by a magnet in a dicating machine that was placed too close to the stimulator) and who presented to an emergency room.12
Treatment is easy if this diagnosis is considered. Checking the neurostimulator and switching it to “on” are all that is needed. Since patients and their caregivers are trained how to check and turn on the stimulator, the role of the geriatrician is simply to remind the caregiver of this possibility.
FDA warning. The US Food and Drug Administration has issued a warning against use of shortwave or microwave diathermy for patients with deep brain stimulation or other implanted leads (www.fda.gov/cdrh/safety/121902.html), stating: “There are three types of diathermy equipment used by physicians, dentists, physical therapists, chiropractors, sports therapists, and others: radio frequency (shortwave) diathermy, microwave diathermy, and ultrasound diathermy. Shortwave and microwave diathermy, in both heating and nonheating modes, can result in serious injury or death to patients with implanted devices with leads. This kind of interaction is not expected with ultrasound diathermy. Electrocautery devices are not included in this notification.” If a patient has an implanted deep brain stimulator, magnetic resonance imaging should be done only if absolutely needed and then only if the guidelines are followed.
DISORDERS PRESENTING WITH DYSTONIA
Acute dystonic reaction
Medications are a common cause of acute focal dystonia. The symptoms, which can be life-threatening, usually occur within 24 hours after taking the medication.12 The most common offenders are neuroleptic drugs and antiemetic drugs with dopamine-blocking activity (eg, metoclopramide), although in older patients, they are more likely to cause tardive dyskinesia and parkinsonism.13,14
Metoclopramide accounts for nearly one-third of all drug-induced movement disorders, and this adverse effect is a common reason for malpractice suits. The entire spectrum of drug-induced movement disorders, ranging from subtle to life-threatening, can ensue from its use; akathisia and dystonia are generally seen early in the course of metoclopramide-induced movement disorders, whereas tardive dyskinesia and parkinsonism seem to be more prevalent in long-term users.15
Treatment includes stopping the precipitating medication and reversing dystonia with anticholinergic medications such as benztropine (Cogentin). Anticholinergic therapy is given intravenously or intramuscularly followed by oral therapy for few days, as the acute dystonic reaction may recur after the effect of parenteral medication wears off.
Intravenous diphenhydramine (Benadryl), an antihistamine with additional anticholiner-gic effects, can abort dystonia in a few minutes.16
Laryngeal dystonia accompanied by multiple system atrophy
Multiple system atrophy, a Parkinson-plus syndrome, is characterized by parkinsonism (mostly with poor response to levodopa) and early onset of dysfunction of the autonomic nervous system, urinary tract, cerebellum, and corticospinal tract (hyperreflexia).17
In the course of the disease, about one-third of patients develop respiratory stridor due to abnormal movements of the vocal cords.18 Nocturnal stridor portends a poor prognosis,19 with an increased risk of sudden death. Geriatricians should be aware of these symptoms, as these patients may seek care because of hoarseness or difficulty swallowing.
Treatment. Laryngeal dystonia can be improved with continuous positive airway pressure. In some cases, tracheostomy may be needed.19
Sudden withdrawal of baclofen
Baclofen (Lioresal), a treatment for spasticity and dystonia, is delivered via a pump through a catheter into an intrathecal space. The pump needs to be refilled every 3 to 6 months. Sudden discontinuation of medication caused by a dislodged catheter tip or forgetting to refill the pump provokes withdrawal symptoms. Patients with this life-threatening syndrome can present with rigidity, fever, change in mental status, and worsening dystonic symptoms.
Treatment involves high doses of baclofen (up to 120 mg/day in divided doses).6
DISORDERS PRESENTING WITH HYPERKINETIC MOVEMENTS
Chorea, ballism (ballismus), and athetosis constitute a range of involuntary, hyperkinetic movement disorders. Chorea consists of involuntary, continuous, sudden, brief, unsustained, irregular movements that flow from one part of the body to another. Hemiballism presents as forceful flinging movements of the limbs or high-amplitude chorea that affects one side of the body.
Acute hemichorea and hemiballism
Acute hemichorea and hemiballism commonly result from infarction or hemorrhage of the basal ganglia.20 Computed tomography and especially magnetic resonance imaging can show the lesions in patients with ballism. Stroke-induced ballism is usually self-limited and resolves after a few weeks. Acute hemiballism generally evolves to hemichorea or hemiathetosis in a few days, which requires only protective measures.
Treatment. Mild cases do not need treatment but severe cases call for medical therapy. Antidopaminergics are the drugs of choice. A dopamine depletor such as reserpine (Serpasil) 0.1 mg once or twice daily or dopamine receptor blockers such as neuroleptics are considered.16 The combination of a benzodiazepine plus an antipsychotic such as olanzapine (Zyprexa) has been suggested.6
Severe parkinsonian dyskinesia
Dyskinesia is common in Parkinson disease, and patients may present to an emergency room with severe levodopa-induced dyskinesia. Dyskinesia can be exhausting if prolonged and severe. Elevated levels of creatine kinase raise the concern of rhabdomyolysis. In rare cases, the patient develops respiratory dyskinesia when respiratory muscles such as those in the diaphragm become involved.21
The risk of levodopa-induced dyskinesia increases with disease severity and higher levodopa doses. Using a dopamine agonist as initial therapy delays the onset of levodopa-induced dyskinesia in early Parkinson disease. However, Factor and Molho,21 in a case series, reported that adding dopamine agonists to the regimen was a precipitating factor; another was infection.
Treatment. A reasonable approach to treating peak-dose dyskinesia is to lower the doses of dopaminergic medications.
A mild sedative such as lorazepam, alprazolam (Xanax, Niravam), or clonazepam (Klonopin) may reduce the severity of dyski-nesia.21 These drugs are particularly useful if the dyskinesia is worse at night, and they can be used in the emergency department while waiting for the effect of the dopaminergic medications to wear off.
Amantadine ameliorates levodopa-induced peak-dose dyskinesia without worsening parkinsonian symptoms in some patients.22
Drug-induced myoclonus
Myoclonus is sudden, jerky, brief involuntary movement of the face, limbs, or trunk. Unlike tics, myoclonus cannot be controlled by the patient.
Myoclonus has various pathophysiologic mechanisms. Most myoclonic emergencies are epileptic myoclonic seizures, which are beyond the scope of this article. Often, myoclonus is caused by opiate overdose or withdrawal. It can also be a side effect of SSRIs, tricyclic anti-depressants, lithium, amantadine, and rarely, antibiotics such as imipenem (Primaxin).23
Treatment. Opiate-induced myoclonus may respond to naloxone (Narcan), whereas opiate withdrawal responds to benzodi-azepines.6
Acute akathisia
Acute akathisia occurs in susceptible patients after exposure to dopamine receptor blockers or dopamine depletors. It is characterized by subjective restless feelings accompanied by objective restless movements. The course is usually self-limited after the causative medication is discontinued.
Treatment. Symptomatic treatment may be needed in most cases for several days. Anticholinergics are effective. Additionally, vitamin B6, mianserine, propranolol, and mirtazapine (Remeron) in a low dose (15 mg/day) have been shown to be effective16,24,25
DISORDERS WITH PSYCHIATRIC PRESENTATIONS
Hallucinations and psychosis in Parkinson disease
Neuropsychiatric or behavioral complications of Parkinson disease include hallucinations, dementia, depression, psychosis, and sleep disorders.21,26 Psychosis is the leading reason for nursing home placement in advanced cases.27 Psychosis can present as hallucinations or a paranoid delusional state in association with clear sensorium.28 However, hallucinations accounted for only 3% of emergency admissions to the hospital for Parkinson disease patients in one series.29
Risk factors for hallucinations in parkinsonian patients include dementia, long-term therapy with dopaminergic drugs, long duration of disease, advanced age, anticholinergic drugs, and sleep disorders. Severe cognitive impairment or dementia is a major and independent predictive factor for visual hallucinations.30
Most hallucinations are visual; auditory, tactile, and olfactory hallucinations are rare.30
Treatment initially should be the same as in any patient with delirium. The systemic disorders that can aggravate or cause hallucinations such as electrolyte abnormalities, urinary or respiratory infection, and systemic illness should be ruled out.
The next step is to reduce or discontinue the adjunctive drugs that have the least antiparkinsonian effect and the greatest potential of inducing hallucination or psychosis. Examples of such medications include histamine-2 antagonists (eg, cimetidine [Tagamet], amantadine, selegeline, and anti-cholinergics). Selegeline can be discontinued abruptly because it has a long duration of action in the brain, but amantadine and anti-cholinergics should be tapered. Dopamine agonists can be discontinued. Levodopa can be reduced until the side effects begin to subside without significant worsening of motor symptoms.
If all the above adjustments fail, an antipsychotic medication can be considered.26 Clozapine (Clozaril) has the best result and is nearly free of extrapyramidal side effects but can cause agranulocytosis, which requires frequent blood counts. The Parkinson Study Group suggested that clozapine, at daily doses of 50 mg or less, is safe and significantly improves drug-induced psychosis without worsening parkinsonism.31 Clozapine may be impractical for elderly patients due to its side effect profile.
Quetiapine is a good alternative to cloza-pine and is less likely to worsen parkinsonian symptoms than other atypical antipsy-chotics.32 Olanzapine and risperidone (Risperdal) are reported to worsen parkinsonian symptoms.33 Not enough data have been published about the efficacy of the newer medications such as ziprasidone (Geodon) and aripiprazole (Abilify) to advocate their routine clinical use.
Rivastigmine (Exelon) was reported to improve hallucinations, sleep disturbance, and caregiver distress in addition to enhancing cognitive performance in advanced Parkinson disease in a small study.34 Burn and colleagues35 reported that rivastigmine was beneficial in patients with dementia associated with Parkinson disease, with or without hallucinations. Efficacy measures were cognitive scales, activities of daily living, behavioral symptoms, and executive and attentional functions. The differences in these measures between rivastigmine and placebo recipients tended to be larger in patients with visual hallucinations than in those without hallucinations. The study was not designed to assess the effect of treatment on psychosis or hallucination.
WHEN PATIENTS WITH MOVEMENT DISORDERS NEED SURGERY
Some of these syndromes can be prevented, especially in patients who are known to have movement disorders and are undergoing surgery.
One problem is stopping oral dopaminergic drugs before the operation. Parkinson disease patients on dopaminergic drugs can develop parkinsonism-hyperpyrexia syndrome or akinetic crisis if the drug is stopped suddenly. Restarting dopaminergic therapy and supportive measures are the main treatments. Patients who have Parkinson disease should receive their usual dose of levodopa, dopamine agonist, or amantadine up until the time of surgery and then again as soon as they awaken in the recovery room.36 That goal can be achieved more easily now that these drugs come in transdermal patches and long-acting formulas.37 Droperidol (Inapsine) and metoclopramide worsen parkinsonism and should be avoided.
Myoclonus is the most common movement disorder seen in the postoperative period. In fact, myoclonic shivering is common as patients awaken from general anesthesia.36 The anesthetic agents etomidate (Amidate) and enflurane (Ethrane) and the opioids fentanyl (Actiq, Duralgesic, Sublimaze) and meperidine (Demerol) can cause myoclonus.38
Occasionally, a patient in the recovery room suddenly develops a neurologic deficit that is inconsistent with the history and physical findings. Psychogenic movement disorders should be considered in the differential diagnosis. Reassurance and occasionally psychiatric intervention are required in these cases.36
IN THE ELDERLY, GO EASY
Polypharmacy is a huge issue in the elderly. Some of the principles in prescribing medications in the elderly can be helpful in preventing movement disorder emergencies:
- Assess the current regimen, including over-the-counter drugs, before prescribing a new drug.
- Begin with a low dose and increase as necessary. “Start low, go slow.”
- Consider the possibility that any new symptoms can be a drug side effect or due to withdrawal of a drug.
- Discuss with the patient or caregiver what kind of side effect to expect and advise him or her to report serious ones.
- Isbister GK, Bowe SJ, Dawson A, Whyte IM. Relative toxicity of selective serotonin reuptake inhibitors (SSRIs) in overdose. J Toxicol Clin Toxicol 2004; 42:277–285.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003; 96:635–642.
- Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000; 79:201–209.
- LoCurto MJ. The serotonin syndrome. Emerg Med Clin North Am 1997; 15( 3):665–675.
- Boyer E, Shannon S. The serotonin syndrome. N Engl J Med 2005; 352:1112–1120.
- Kipps CM, Fung VS, Grattan-Smith P, de Moore GM, Morris JG. Movement disorder emergencies. Mov Disord 2005; 20:322–334.
- Shalev A, Munitz H. The neuroleptic malignant syndrome: agent and host interaction. Acta Psychiatr Scand 1986; 73:337–347.
- Rosebush PI, Stewart TD, Gelenberg AJ. Twenty neuroleptic rechallenges after neuroleptic malignant syndrome in 15 patients. J Clin Psychiatry 1989; 50:295–298.
- Adityanjee , Singh S, Singh G, Ong S. Spectrum concept of neuroleptic malignant syndrome. Br J Psychiatry 1988; 153:107–111.
- Bonuccelli U, Piccini P, Corsini GU, Muratorio A. Apomorphine in malignant syndrome due to levodopa withdrawal. Ital J Neurol Sci 1992; 13:169–170.
- Sato Y, Asoh T, Metoki N, et al. Efficacy of methylprednisolone pulse therapy on neuroleptic malignant syndrome in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2004; 74:574–576.
- Hariz MI, Johansson F. Hardware failure in parkinsonian patients with chronic subthalamic nucleus stimulation is a medical emergency. Mov Disord 2001; 16:166–168.
- Pollera CF, Cognetli F, Nardi M, Mozza D. Sudden death after acute dystonic reaction to high-dose metoclopramide. Lancet 1984; 2:460–461.
- Bateman DN, Rawlins MD, Simpson JM. Extrapyramidal reactions with metoclopramide. Br Med J 1985; 291:930–932.
- Pasricha PJ, Pehlivanov N, Sugumar A, Jankovic J. Drug insight: from disturbed motility to disordered movement—a review of the clinical benefits and medicolegal risks of metoclopramide. Nat Clin Pract Gastroenterol Hepatol 2006; 3:138–148.
- Hu S, Frucht S. Emergency treatment of movement disorders. Curr Treat Options Neurol 2007; 9:103–114.
- Tousi B, Schuele SU, Subramanian T. A 46-year-old woman with rigidity and frequent falls. Cleve Clin J Med 2005; 72:57–63.
- Merlo IM, Occhini A, Pacchetti C, Alfonsi E. Not paralysis, but dystonia causes stridor in multiple system atrophy. Neurology 2002; 58:649–652.
- Silber MH, Levine S. Stridor and death in multiple system atrophy. Mov Disord 2000; 15:699–704.
- Bhatia KP, Marsden CD. The behavioural and motor consequences of focal lesions of the basal ganglia in man. Brain 1994; 117:859–876.
- Factor SA, Molho ES. Emergency department presentations of patients with Parkinson’s disease. Am J Emerg Med 2000; 18:209–215.
- Verhagen Metman L, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as a treatment for dyskinesia and motor fluctuations in Parkinson’s disease. Neurology 1998; 50:1323–1326.
- Frucht S, Eidelberg D. Imipenem-induced myoclonus. Mov Disord 1997; 12:621–622.
- Miodownik C, Lerner V, Statsenko N, et al. Vitamin B6 versus mianserin and placebo in acute neuroleptic-induced akathisia: a randomized, double-blind, controlled study. Clin Neuropharmacol 2006; 29:68–72.
- Poyurovsky M, Pashinian A, Weizman R, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry 2006; 59:1071–1077.
- Tousi B, Subramanian T. Hallucinations in Parkinson’s disease: approach and management. Clin Geriatr 2004: 12:19–24.
- Goetz CG, Stebbins GT. Risk factors for nursing home placement in advanced Parkinson’s disease. Neurology 1993; 43:2227–2229.
- Factor SA, Molho ES, Podskalny GD, Brown D. Parkinson’s disease: drug-induced psychiatric states. Adv Neurol 1995; 65:115–138.
- Woodford H, Walker R. Emergency hospital admissions in idiopathic Parkinson’s disease. Mov Disord 2005; 20:1104–1108.
- Tousi B, Frankel M. Olfactory and visual hallucinations in Parkinson’s disease. Parkinsonism Relat Disord 2004; 10:253–254.
- The Parkinson Study Group. Low-dose clozapine for the treatment of drug-induced psychosis in Parkinson’s disease. N Engl J Med 1999; 340:757–763.
- Merims D, Balas M, Pertez C, Shabtai H, Giladi N. Rater-blinded, prospective comparison: quetiapine versus clozapine for Parkinson’s disease psychosis. Clin Neuropharmacol 2006; 29:331–337.
- Goetz CG, Blasucci LM, Leurgans S, Pappert EJ. Olanzapine and clozapine: comparative effects on motor function in hallucinating PD patients. Neurology 2000; 55:789–794.
- Reading PJ, Luce AK, McKeith IG. Rivastigmine in the treatment of parkinsonian psychosis and cognitive impairment: preliminary findings from an open trial. Mov Disord 2001; 16:1171–1174.
- Burn D, Emre M, McKeith I, et al. Effects of rivastigmine in patients with and without visual hallucinations in dementia associated with Parkinson’s disease. Mov Disord 2006; 21:1899–1907.
- Frucht SJ. Movement disorder emergencies in the perioperative period. Neurol Clin 2004; 22:379–387.
- Korczyn AD, Reichmann H, Boroojerdi B, et al. Rotigotin trans-dermal system for perioperative administration. J Neural Transm 2007; 114:219–221.
- Gordon MF. Toxin and drug-induced myoclonus. Adv Neurol 2002; 89:49–76.
Although we tend to think of movement disorders as chronic conditions, some of them can present as true emergencies in which failure to diagnose the condition and treat it promptly can result in significant sickness or even death.
Many cases are iatrogenic, occurring in patients with Parkinson disease or those taking antipsychotic or antidepressant medications when their regimen is started or altered. Elderly patients are particularly at risk, as they take more drugs and have less physiologic reserve.
Movement disorder emergencies in elderly patients can be difficult to diagnose and treat, since many patients are taking more than one medication: polypharmacy raises the possibility of interactions, and different drugs can cause different movement disorder syndromes. Moreover, because so many patients are at risk—for example, more than 1 million people in the United States now have Parkinson disease, and the number is growing—it is important for physicians who take care of the elderly to be more informed about these disorders, especially the presenting symptoms.
SCOPE OF THIS ARTICLE
- Disorders presenting with rigidity or stiffness
- Disorders presenting with dystonia
- Disorders presenting with hyperkinetic movements
- Disorders presenting with psychiatric disturbances.
Of these, the scenarios most likely to require emergency evaluation in the elderly are acute hypokinetic and hyperkinetic syndromes and psychiatric presentations. This article discusses movement disorder emergencies in the elderly, focusing on the more common disorders with common presentations.
DISORDERS PRESENTING WITH RIGIDITY OR STIFFNESS
Serotonin syndrome
Chief among the offenders are the selective serotonin-reuptake inhibitors (SSRIs), either alone or in combination. This syndrome occurs in 14% to 16% of patients who overdose on SSRIs.1 Examples of combinations that can lead to serotonin syndrome are an SSRI plus any of the following:
- An anxiolytic such as buspirone (BuSpar; this combination is popular for the treatment of depression and anxiety)
- A tricyclic agent such as imipramine (Tofranil)
- A serotonin and norepinephrine reuptake inhibitor such as venlafaxine (Effexor).
In addition, antiparkinson drugs such as levodopa and selegiline (Eldepryl) enhance serotonin release.
Signs and symptoms. Serotonin syndrome is characterized by:
- Severe rigidity
- Dysautonomia
- Change in mental status.
Other clinical findings include fever, gastrointestinal disturbances, and motor restlessness. Clonus is the most important finding in establishing the diagnosis.2
The syndrome may initially go unrecognized and can be mistaken for viral illness or anxiety.4 Manifestations range from mild to life-threatening; initially, it may present with akathisia and tremor. The symptoms progress rapidly over hours and can range from myoclonus, hyperreflexia, and seizures to severe forms of rhabdomyolysis, renal failure, and respiratory failure. The hyperreflexia and clonus seen in moderate cases may be considerably greater in the lower extremities than in the upper extremities.5
No laboratory test confirms the diagnosis, but tremor, clonus, or akathisia without additional extrapyramidal signs should lead to the diagnosis if the patient was taking a serotonergic medication.5 The onset of symptoms is usually rapid. The majority of patients present within 6 hours after initial use of the medication, an overdose, or a change in dosing.5
Treatment. The first steps are to stop the serotonergic medication and to hydrate and cool the patient to counteract the hyperpyrexic state. Benzodiazepine drugs are important in controlling agitation, regardless of its severity.5 Propranolol (Inderal) is not recommended, as it may cause hypotension and shock in patients with autonomic instability.5
Patients with moderate cases may additionally benefit from cyproheptadine (Periactin), an antihistamine that antagonizes serotonin. The initial dose is 4 to 8 mg orally, with a repeat dose after 2 hours.6 Whether to continue this treatment depends on the response after two doses.
If medications must be given parenterally, physicians can consider chlorpromazine (Thorazine) 50 to 100 mg intramuscularly.5
Vital signs should be monitored. In severe cases, intensive care may be required with immediate sedation, neuromuscular paralysis, and intubation.
In most cases, patients improve rapidly.
Comment. Serotonin syndrome can be avoided by educating physicians and by modifying prescribing practices.5 Avoiding multidrug regimens is critical to preventing serotonin syndrome. Computer-based ordering systems and personal digital assistants can help one avoid drug interactions.5
Neuroleptic malignant syndrome
This syndrome is an infrequent but potentially lethal complication associated with therapy with antipsychotic drugs such as haloperidol (Haldol) and lithium (Eskalith) and with other medications with dopamine type-2 receptor antagonist activity such as metoclopramide (Reglan) and prochlorperazine (Compazine). It has become rare since the introduction of atypical antipsychotics and now occurs in 0.2% of patients receiving atypical antipsychotics.7 Its pathogenesis is not fully understood.
This syndrome occurs mainly in young or middle-aged patients receiving doses of neuroleptics within the usual therapeutic range, but it also appears to occur in elderly patients who receive higher doses.8 Although most cases develop in the first 2 weeks of treatment, it can develop at any time during therapy.
Signs and symptoms. Four features characterize neuroleptic malignant syndrome9:
- Muscle rigidity—generalized (“lead-pipe”) muscular rigidity is accompanied by bradykinesia or akinesia.
- Autonomic dysregulation, with tachycardia, tachypnea, alterations in blood pressure, excessive sweating, and incontinence.
- Hyperthermia—fever can begin hours to days after initiating or increasing the dose of a dopamine antagonist.
- Altered sensorium, ranging from confusion to disorientation and coma.
Symptoms of neuroleptic malignant syndrome typically evolve over several days, in contrast to the rapid onset of the serotonin syndrome. Knowing the precipitating drug also helps distinguish the syndromes: dopamine antagonists produce bradykinesia, whereas serotonin agonists produce hyperkinesia.5
Laboratory abnormalities include elevated serum creatine kinase concentrations and white blood cell counts. Renal function should be assessed when renal failure and rhabdomyolysis are suspected.
Treatment involves stopping the causative medication, cooling the patient, and supporting vital functions.
In mild cases (eg, low-grade fever) benzodiazepines such as lorazepam (Ativan) can stabilize the condition. In moderate cases (eg, more significant rigidity), dopaminergic agonists such as bromocriptine (Parlodel) can be given, although there is no strong clinical evidence for their use. Bromocriptine is usually started at 2.5 mg three times a day and gradually increased in dose if tolerated.
In severe cases, muscle rigidity can be reduced with dantrolene (Dantrium), a muscle relaxant. Dantrolene is started at 1 mg/kg intravenously every 6 hours and gradually increased up to 10 mg/kg total per day.
Some patients remain rigid and febrile up to 4 weeks after the causative agent has been withdrawn. Therefore, these treatments can be continued for a few weeks. After the patient has recovered fully, if it is necessary to resume antipsychotic therapy, an atypical antipsychotic such as quetiapine (Seroquel) can be started after 2 weeks.8
Comment. Although uncommon, neuroleptic malignant syndrome is the most serious adverse effect of neuroleptic drugs, and it is potentially fatal. When neuroleptic malignant syndrome is suspected, treatment should be prompt, and the neuroleptic medication should be immediately stopped.
Parkinsonism-hyperpyrexia syndrome
Withdrawing or decreasing the dose of dopaminergic medications in patients with Parkinson disease can cause parkinsonism-hyperpyrexia syndrome, a condition that is similar to neuroleptic malignant syndrome. It can also arise after sudden withdrawal of amantadine (Symmetrel) or anticholinergics. In view of this concern, adjustments to antiparkinson drugs may need to be more gradual in some elderly patients.
Patients present with fever, rigidity, and autonomic instability and are at risk of aspiration pneumonia.
Treatment includes resuming dopaminergic therapy and giving supportive care.
Apomorphine (Apokyn), a dopaminergic agonist, was used in a 71-year-old female parkinsonian patient who developed parkinsonism-hyperpyrexia syndrome after abrupt reduction of chronic levodopa treatment.10 The symptoms resolved within 24 hours of the addition of apomorphine to her previous levodopa therapy. If the patient is taking apomorphine for the first time, the injections should be given in low doses, 0.2 mL subcutaneously. Apomorphine can induce vomiting, and if this occurs an antiemetic such as trimethobenzamide (Tigan) should be given before subsequent injections. In the elderly, caution is advised as apomorphine may cause severe orthostasis.
Methylprednisolone (Solu-Medrol) pulse therapy has been shown to shorten the duration of this syndrome in a randomized, controlled study.11
Akinetic syndrome after failure of deep brain stimulator
Deep brain stimulation involves surgical placement of a pacemaker with electrodes in specific areas of the brain. It is used to control Parkinson disease, tremor, and, less commonly, dystonia, and a number of other uses are under investigation. Continuous electrical stimulation of different nuclei in the brain has been shown to alleviate some symptoms of Parkinson disease (eg, rigidity) and to enable some patients to decrease the dose of their antiparkinson medications.
Several cases have been reported of sudden, unexpected reappearance of freezing, gait disturbance, or severe akinesia in Parkinson disease patients whose stimulators had been turned off inadvertently (eg, by a magnet in a dicating machine that was placed too close to the stimulator) and who presented to an emergency room.12
Treatment is easy if this diagnosis is considered. Checking the neurostimulator and switching it to “on” are all that is needed. Since patients and their caregivers are trained how to check and turn on the stimulator, the role of the geriatrician is simply to remind the caregiver of this possibility.
FDA warning. The US Food and Drug Administration has issued a warning against use of shortwave or microwave diathermy for patients with deep brain stimulation or other implanted leads (www.fda.gov/cdrh/safety/121902.html), stating: “There are three types of diathermy equipment used by physicians, dentists, physical therapists, chiropractors, sports therapists, and others: radio frequency (shortwave) diathermy, microwave diathermy, and ultrasound diathermy. Shortwave and microwave diathermy, in both heating and nonheating modes, can result in serious injury or death to patients with implanted devices with leads. This kind of interaction is not expected with ultrasound diathermy. Electrocautery devices are not included in this notification.” If a patient has an implanted deep brain stimulator, magnetic resonance imaging should be done only if absolutely needed and then only if the guidelines are followed.
DISORDERS PRESENTING WITH DYSTONIA
Acute dystonic reaction
Medications are a common cause of acute focal dystonia. The symptoms, which can be life-threatening, usually occur within 24 hours after taking the medication.12 The most common offenders are neuroleptic drugs and antiemetic drugs with dopamine-blocking activity (eg, metoclopramide), although in older patients, they are more likely to cause tardive dyskinesia and parkinsonism.13,14
Metoclopramide accounts for nearly one-third of all drug-induced movement disorders, and this adverse effect is a common reason for malpractice suits. The entire spectrum of drug-induced movement disorders, ranging from subtle to life-threatening, can ensue from its use; akathisia and dystonia are generally seen early in the course of metoclopramide-induced movement disorders, whereas tardive dyskinesia and parkinsonism seem to be more prevalent in long-term users.15
Treatment includes stopping the precipitating medication and reversing dystonia with anticholinergic medications such as benztropine (Cogentin). Anticholinergic therapy is given intravenously or intramuscularly followed by oral therapy for few days, as the acute dystonic reaction may recur after the effect of parenteral medication wears off.
Intravenous diphenhydramine (Benadryl), an antihistamine with additional anticholiner-gic effects, can abort dystonia in a few minutes.16
Laryngeal dystonia accompanied by multiple system atrophy
Multiple system atrophy, a Parkinson-plus syndrome, is characterized by parkinsonism (mostly with poor response to levodopa) and early onset of dysfunction of the autonomic nervous system, urinary tract, cerebellum, and corticospinal tract (hyperreflexia).17
In the course of the disease, about one-third of patients develop respiratory stridor due to abnormal movements of the vocal cords.18 Nocturnal stridor portends a poor prognosis,19 with an increased risk of sudden death. Geriatricians should be aware of these symptoms, as these patients may seek care because of hoarseness or difficulty swallowing.
Treatment. Laryngeal dystonia can be improved with continuous positive airway pressure. In some cases, tracheostomy may be needed.19
Sudden withdrawal of baclofen
Baclofen (Lioresal), a treatment for spasticity and dystonia, is delivered via a pump through a catheter into an intrathecal space. The pump needs to be refilled every 3 to 6 months. Sudden discontinuation of medication caused by a dislodged catheter tip or forgetting to refill the pump provokes withdrawal symptoms. Patients with this life-threatening syndrome can present with rigidity, fever, change in mental status, and worsening dystonic symptoms.
Treatment involves high doses of baclofen (up to 120 mg/day in divided doses).6
DISORDERS PRESENTING WITH HYPERKINETIC MOVEMENTS
Chorea, ballism (ballismus), and athetosis constitute a range of involuntary, hyperkinetic movement disorders. Chorea consists of involuntary, continuous, sudden, brief, unsustained, irregular movements that flow from one part of the body to another. Hemiballism presents as forceful flinging movements of the limbs or high-amplitude chorea that affects one side of the body.
Acute hemichorea and hemiballism
Acute hemichorea and hemiballism commonly result from infarction or hemorrhage of the basal ganglia.20 Computed tomography and especially magnetic resonance imaging can show the lesions in patients with ballism. Stroke-induced ballism is usually self-limited and resolves after a few weeks. Acute hemiballism generally evolves to hemichorea or hemiathetosis in a few days, which requires only protective measures.
Treatment. Mild cases do not need treatment but severe cases call for medical therapy. Antidopaminergics are the drugs of choice. A dopamine depletor such as reserpine (Serpasil) 0.1 mg once or twice daily or dopamine receptor blockers such as neuroleptics are considered.16 The combination of a benzodiazepine plus an antipsychotic such as olanzapine (Zyprexa) has been suggested.6
Severe parkinsonian dyskinesia
Dyskinesia is common in Parkinson disease, and patients may present to an emergency room with severe levodopa-induced dyskinesia. Dyskinesia can be exhausting if prolonged and severe. Elevated levels of creatine kinase raise the concern of rhabdomyolysis. In rare cases, the patient develops respiratory dyskinesia when respiratory muscles such as those in the diaphragm become involved.21
The risk of levodopa-induced dyskinesia increases with disease severity and higher levodopa doses. Using a dopamine agonist as initial therapy delays the onset of levodopa-induced dyskinesia in early Parkinson disease. However, Factor and Molho,21 in a case series, reported that adding dopamine agonists to the regimen was a precipitating factor; another was infection.
Treatment. A reasonable approach to treating peak-dose dyskinesia is to lower the doses of dopaminergic medications.
A mild sedative such as lorazepam, alprazolam (Xanax, Niravam), or clonazepam (Klonopin) may reduce the severity of dyski-nesia.21 These drugs are particularly useful if the dyskinesia is worse at night, and they can be used in the emergency department while waiting for the effect of the dopaminergic medications to wear off.
Amantadine ameliorates levodopa-induced peak-dose dyskinesia without worsening parkinsonian symptoms in some patients.22
Drug-induced myoclonus
Myoclonus is sudden, jerky, brief involuntary movement of the face, limbs, or trunk. Unlike tics, myoclonus cannot be controlled by the patient.
Myoclonus has various pathophysiologic mechanisms. Most myoclonic emergencies are epileptic myoclonic seizures, which are beyond the scope of this article. Often, myoclonus is caused by opiate overdose or withdrawal. It can also be a side effect of SSRIs, tricyclic anti-depressants, lithium, amantadine, and rarely, antibiotics such as imipenem (Primaxin).23
Treatment. Opiate-induced myoclonus may respond to naloxone (Narcan), whereas opiate withdrawal responds to benzodi-azepines.6
Acute akathisia
Acute akathisia occurs in susceptible patients after exposure to dopamine receptor blockers or dopamine depletors. It is characterized by subjective restless feelings accompanied by objective restless movements. The course is usually self-limited after the causative medication is discontinued.
Treatment. Symptomatic treatment may be needed in most cases for several days. Anticholinergics are effective. Additionally, vitamin B6, mianserine, propranolol, and mirtazapine (Remeron) in a low dose (15 mg/day) have been shown to be effective16,24,25
DISORDERS WITH PSYCHIATRIC PRESENTATIONS
Hallucinations and psychosis in Parkinson disease
Neuropsychiatric or behavioral complications of Parkinson disease include hallucinations, dementia, depression, psychosis, and sleep disorders.21,26 Psychosis is the leading reason for nursing home placement in advanced cases.27 Psychosis can present as hallucinations or a paranoid delusional state in association with clear sensorium.28 However, hallucinations accounted for only 3% of emergency admissions to the hospital for Parkinson disease patients in one series.29
Risk factors for hallucinations in parkinsonian patients include dementia, long-term therapy with dopaminergic drugs, long duration of disease, advanced age, anticholinergic drugs, and sleep disorders. Severe cognitive impairment or dementia is a major and independent predictive factor for visual hallucinations.30
Most hallucinations are visual; auditory, tactile, and olfactory hallucinations are rare.30
Treatment initially should be the same as in any patient with delirium. The systemic disorders that can aggravate or cause hallucinations such as electrolyte abnormalities, urinary or respiratory infection, and systemic illness should be ruled out.
The next step is to reduce or discontinue the adjunctive drugs that have the least antiparkinsonian effect and the greatest potential of inducing hallucination or psychosis. Examples of such medications include histamine-2 antagonists (eg, cimetidine [Tagamet], amantadine, selegeline, and anti-cholinergics). Selegeline can be discontinued abruptly because it has a long duration of action in the brain, but amantadine and anti-cholinergics should be tapered. Dopamine agonists can be discontinued. Levodopa can be reduced until the side effects begin to subside without significant worsening of motor symptoms.
If all the above adjustments fail, an antipsychotic medication can be considered.26 Clozapine (Clozaril) has the best result and is nearly free of extrapyramidal side effects but can cause agranulocytosis, which requires frequent blood counts. The Parkinson Study Group suggested that clozapine, at daily doses of 50 mg or less, is safe and significantly improves drug-induced psychosis without worsening parkinsonism.31 Clozapine may be impractical for elderly patients due to its side effect profile.
Quetiapine is a good alternative to cloza-pine and is less likely to worsen parkinsonian symptoms than other atypical antipsy-chotics.32 Olanzapine and risperidone (Risperdal) are reported to worsen parkinsonian symptoms.33 Not enough data have been published about the efficacy of the newer medications such as ziprasidone (Geodon) and aripiprazole (Abilify) to advocate their routine clinical use.
Rivastigmine (Exelon) was reported to improve hallucinations, sleep disturbance, and caregiver distress in addition to enhancing cognitive performance in advanced Parkinson disease in a small study.34 Burn and colleagues35 reported that rivastigmine was beneficial in patients with dementia associated with Parkinson disease, with or without hallucinations. Efficacy measures were cognitive scales, activities of daily living, behavioral symptoms, and executive and attentional functions. The differences in these measures between rivastigmine and placebo recipients tended to be larger in patients with visual hallucinations than in those without hallucinations. The study was not designed to assess the effect of treatment on psychosis or hallucination.
WHEN PATIENTS WITH MOVEMENT DISORDERS NEED SURGERY
Some of these syndromes can be prevented, especially in patients who are known to have movement disorders and are undergoing surgery.
One problem is stopping oral dopaminergic drugs before the operation. Parkinson disease patients on dopaminergic drugs can develop parkinsonism-hyperpyrexia syndrome or akinetic crisis if the drug is stopped suddenly. Restarting dopaminergic therapy and supportive measures are the main treatments. Patients who have Parkinson disease should receive their usual dose of levodopa, dopamine agonist, or amantadine up until the time of surgery and then again as soon as they awaken in the recovery room.36 That goal can be achieved more easily now that these drugs come in transdermal patches and long-acting formulas.37 Droperidol (Inapsine) and metoclopramide worsen parkinsonism and should be avoided.
Myoclonus is the most common movement disorder seen in the postoperative period. In fact, myoclonic shivering is common as patients awaken from general anesthesia.36 The anesthetic agents etomidate (Amidate) and enflurane (Ethrane) and the opioids fentanyl (Actiq, Duralgesic, Sublimaze) and meperidine (Demerol) can cause myoclonus.38
Occasionally, a patient in the recovery room suddenly develops a neurologic deficit that is inconsistent with the history and physical findings. Psychogenic movement disorders should be considered in the differential diagnosis. Reassurance and occasionally psychiatric intervention are required in these cases.36
IN THE ELDERLY, GO EASY
Polypharmacy is a huge issue in the elderly. Some of the principles in prescribing medications in the elderly can be helpful in preventing movement disorder emergencies:
- Assess the current regimen, including over-the-counter drugs, before prescribing a new drug.
- Begin with a low dose and increase as necessary. “Start low, go slow.”
- Consider the possibility that any new symptoms can be a drug side effect or due to withdrawal of a drug.
- Discuss with the patient or caregiver what kind of side effect to expect and advise him or her to report serious ones.
Although we tend to think of movement disorders as chronic conditions, some of them can present as true emergencies in which failure to diagnose the condition and treat it promptly can result in significant sickness or even death.
Many cases are iatrogenic, occurring in patients with Parkinson disease or those taking antipsychotic or antidepressant medications when their regimen is started or altered. Elderly patients are particularly at risk, as they take more drugs and have less physiologic reserve.
Movement disorder emergencies in elderly patients can be difficult to diagnose and treat, since many patients are taking more than one medication: polypharmacy raises the possibility of interactions, and different drugs can cause different movement disorder syndromes. Moreover, because so many patients are at risk—for example, more than 1 million people in the United States now have Parkinson disease, and the number is growing—it is important for physicians who take care of the elderly to be more informed about these disorders, especially the presenting symptoms.
SCOPE OF THIS ARTICLE
- Disorders presenting with rigidity or stiffness
- Disorders presenting with dystonia
- Disorders presenting with hyperkinetic movements
- Disorders presenting with psychiatric disturbances.
Of these, the scenarios most likely to require emergency evaluation in the elderly are acute hypokinetic and hyperkinetic syndromes and psychiatric presentations. This article discusses movement disorder emergencies in the elderly, focusing on the more common disorders with common presentations.
DISORDERS PRESENTING WITH RIGIDITY OR STIFFNESS
Serotonin syndrome
Chief among the offenders are the selective serotonin-reuptake inhibitors (SSRIs), either alone or in combination. This syndrome occurs in 14% to 16% of patients who overdose on SSRIs.1 Examples of combinations that can lead to serotonin syndrome are an SSRI plus any of the following:
- An anxiolytic such as buspirone (BuSpar; this combination is popular for the treatment of depression and anxiety)
- A tricyclic agent such as imipramine (Tofranil)
- A serotonin and norepinephrine reuptake inhibitor such as venlafaxine (Effexor).
In addition, antiparkinson drugs such as levodopa and selegiline (Eldepryl) enhance serotonin release.
Signs and symptoms. Serotonin syndrome is characterized by:
- Severe rigidity
- Dysautonomia
- Change in mental status.
Other clinical findings include fever, gastrointestinal disturbances, and motor restlessness. Clonus is the most important finding in establishing the diagnosis.2
The syndrome may initially go unrecognized and can be mistaken for viral illness or anxiety.4 Manifestations range from mild to life-threatening; initially, it may present with akathisia and tremor. The symptoms progress rapidly over hours and can range from myoclonus, hyperreflexia, and seizures to severe forms of rhabdomyolysis, renal failure, and respiratory failure. The hyperreflexia and clonus seen in moderate cases may be considerably greater in the lower extremities than in the upper extremities.5
No laboratory test confirms the diagnosis, but tremor, clonus, or akathisia without additional extrapyramidal signs should lead to the diagnosis if the patient was taking a serotonergic medication.5 The onset of symptoms is usually rapid. The majority of patients present within 6 hours after initial use of the medication, an overdose, or a change in dosing.5
Treatment. The first steps are to stop the serotonergic medication and to hydrate and cool the patient to counteract the hyperpyrexic state. Benzodiazepine drugs are important in controlling agitation, regardless of its severity.5 Propranolol (Inderal) is not recommended, as it may cause hypotension and shock in patients with autonomic instability.5
Patients with moderate cases may additionally benefit from cyproheptadine (Periactin), an antihistamine that antagonizes serotonin. The initial dose is 4 to 8 mg orally, with a repeat dose after 2 hours.6 Whether to continue this treatment depends on the response after two doses.
If medications must be given parenterally, physicians can consider chlorpromazine (Thorazine) 50 to 100 mg intramuscularly.5
Vital signs should be monitored. In severe cases, intensive care may be required with immediate sedation, neuromuscular paralysis, and intubation.
In most cases, patients improve rapidly.
Comment. Serotonin syndrome can be avoided by educating physicians and by modifying prescribing practices.5 Avoiding multidrug regimens is critical to preventing serotonin syndrome. Computer-based ordering systems and personal digital assistants can help one avoid drug interactions.5
Neuroleptic malignant syndrome
This syndrome is an infrequent but potentially lethal complication associated with therapy with antipsychotic drugs such as haloperidol (Haldol) and lithium (Eskalith) and with other medications with dopamine type-2 receptor antagonist activity such as metoclopramide (Reglan) and prochlorperazine (Compazine). It has become rare since the introduction of atypical antipsychotics and now occurs in 0.2% of patients receiving atypical antipsychotics.7 Its pathogenesis is not fully understood.
This syndrome occurs mainly in young or middle-aged patients receiving doses of neuroleptics within the usual therapeutic range, but it also appears to occur in elderly patients who receive higher doses.8 Although most cases develop in the first 2 weeks of treatment, it can develop at any time during therapy.
Signs and symptoms. Four features characterize neuroleptic malignant syndrome9:
- Muscle rigidity—generalized (“lead-pipe”) muscular rigidity is accompanied by bradykinesia or akinesia.
- Autonomic dysregulation, with tachycardia, tachypnea, alterations in blood pressure, excessive sweating, and incontinence.
- Hyperthermia—fever can begin hours to days after initiating or increasing the dose of a dopamine antagonist.
- Altered sensorium, ranging from confusion to disorientation and coma.
Symptoms of neuroleptic malignant syndrome typically evolve over several days, in contrast to the rapid onset of the serotonin syndrome. Knowing the precipitating drug also helps distinguish the syndromes: dopamine antagonists produce bradykinesia, whereas serotonin agonists produce hyperkinesia.5
Laboratory abnormalities include elevated serum creatine kinase concentrations and white blood cell counts. Renal function should be assessed when renal failure and rhabdomyolysis are suspected.
Treatment involves stopping the causative medication, cooling the patient, and supporting vital functions.
In mild cases (eg, low-grade fever) benzodiazepines such as lorazepam (Ativan) can stabilize the condition. In moderate cases (eg, more significant rigidity), dopaminergic agonists such as bromocriptine (Parlodel) can be given, although there is no strong clinical evidence for their use. Bromocriptine is usually started at 2.5 mg three times a day and gradually increased in dose if tolerated.
In severe cases, muscle rigidity can be reduced with dantrolene (Dantrium), a muscle relaxant. Dantrolene is started at 1 mg/kg intravenously every 6 hours and gradually increased up to 10 mg/kg total per day.
Some patients remain rigid and febrile up to 4 weeks after the causative agent has been withdrawn. Therefore, these treatments can be continued for a few weeks. After the patient has recovered fully, if it is necessary to resume antipsychotic therapy, an atypical antipsychotic such as quetiapine (Seroquel) can be started after 2 weeks.8
Comment. Although uncommon, neuroleptic malignant syndrome is the most serious adverse effect of neuroleptic drugs, and it is potentially fatal. When neuroleptic malignant syndrome is suspected, treatment should be prompt, and the neuroleptic medication should be immediately stopped.
Parkinsonism-hyperpyrexia syndrome
Withdrawing or decreasing the dose of dopaminergic medications in patients with Parkinson disease can cause parkinsonism-hyperpyrexia syndrome, a condition that is similar to neuroleptic malignant syndrome. It can also arise after sudden withdrawal of amantadine (Symmetrel) or anticholinergics. In view of this concern, adjustments to antiparkinson drugs may need to be more gradual in some elderly patients.
Patients present with fever, rigidity, and autonomic instability and are at risk of aspiration pneumonia.
Treatment includes resuming dopaminergic therapy and giving supportive care.
Apomorphine (Apokyn), a dopaminergic agonist, was used in a 71-year-old female parkinsonian patient who developed parkinsonism-hyperpyrexia syndrome after abrupt reduction of chronic levodopa treatment.10 The symptoms resolved within 24 hours of the addition of apomorphine to her previous levodopa therapy. If the patient is taking apomorphine for the first time, the injections should be given in low doses, 0.2 mL subcutaneously. Apomorphine can induce vomiting, and if this occurs an antiemetic such as trimethobenzamide (Tigan) should be given before subsequent injections. In the elderly, caution is advised as apomorphine may cause severe orthostasis.
Methylprednisolone (Solu-Medrol) pulse therapy has been shown to shorten the duration of this syndrome in a randomized, controlled study.11
Akinetic syndrome after failure of deep brain stimulator
Deep brain stimulation involves surgical placement of a pacemaker with electrodes in specific areas of the brain. It is used to control Parkinson disease, tremor, and, less commonly, dystonia, and a number of other uses are under investigation. Continuous electrical stimulation of different nuclei in the brain has been shown to alleviate some symptoms of Parkinson disease (eg, rigidity) and to enable some patients to decrease the dose of their antiparkinson medications.
Several cases have been reported of sudden, unexpected reappearance of freezing, gait disturbance, or severe akinesia in Parkinson disease patients whose stimulators had been turned off inadvertently (eg, by a magnet in a dicating machine that was placed too close to the stimulator) and who presented to an emergency room.12
Treatment is easy if this diagnosis is considered. Checking the neurostimulator and switching it to “on” are all that is needed. Since patients and their caregivers are trained how to check and turn on the stimulator, the role of the geriatrician is simply to remind the caregiver of this possibility.
FDA warning. The US Food and Drug Administration has issued a warning against use of shortwave or microwave diathermy for patients with deep brain stimulation or other implanted leads (www.fda.gov/cdrh/safety/121902.html), stating: “There are three types of diathermy equipment used by physicians, dentists, physical therapists, chiropractors, sports therapists, and others: radio frequency (shortwave) diathermy, microwave diathermy, and ultrasound diathermy. Shortwave and microwave diathermy, in both heating and nonheating modes, can result in serious injury or death to patients with implanted devices with leads. This kind of interaction is not expected with ultrasound diathermy. Electrocautery devices are not included in this notification.” If a patient has an implanted deep brain stimulator, magnetic resonance imaging should be done only if absolutely needed and then only if the guidelines are followed.
DISORDERS PRESENTING WITH DYSTONIA
Acute dystonic reaction
Medications are a common cause of acute focal dystonia. The symptoms, which can be life-threatening, usually occur within 24 hours after taking the medication.12 The most common offenders are neuroleptic drugs and antiemetic drugs with dopamine-blocking activity (eg, metoclopramide), although in older patients, they are more likely to cause tardive dyskinesia and parkinsonism.13,14
Metoclopramide accounts for nearly one-third of all drug-induced movement disorders, and this adverse effect is a common reason for malpractice suits. The entire spectrum of drug-induced movement disorders, ranging from subtle to life-threatening, can ensue from its use; akathisia and dystonia are generally seen early in the course of metoclopramide-induced movement disorders, whereas tardive dyskinesia and parkinsonism seem to be more prevalent in long-term users.15
Treatment includes stopping the precipitating medication and reversing dystonia with anticholinergic medications such as benztropine (Cogentin). Anticholinergic therapy is given intravenously or intramuscularly followed by oral therapy for few days, as the acute dystonic reaction may recur after the effect of parenteral medication wears off.
Intravenous diphenhydramine (Benadryl), an antihistamine with additional anticholiner-gic effects, can abort dystonia in a few minutes.16
Laryngeal dystonia accompanied by multiple system atrophy
Multiple system atrophy, a Parkinson-plus syndrome, is characterized by parkinsonism (mostly with poor response to levodopa) and early onset of dysfunction of the autonomic nervous system, urinary tract, cerebellum, and corticospinal tract (hyperreflexia).17
In the course of the disease, about one-third of patients develop respiratory stridor due to abnormal movements of the vocal cords.18 Nocturnal stridor portends a poor prognosis,19 with an increased risk of sudden death. Geriatricians should be aware of these symptoms, as these patients may seek care because of hoarseness or difficulty swallowing.
Treatment. Laryngeal dystonia can be improved with continuous positive airway pressure. In some cases, tracheostomy may be needed.19
Sudden withdrawal of baclofen
Baclofen (Lioresal), a treatment for spasticity and dystonia, is delivered via a pump through a catheter into an intrathecal space. The pump needs to be refilled every 3 to 6 months. Sudden discontinuation of medication caused by a dislodged catheter tip or forgetting to refill the pump provokes withdrawal symptoms. Patients with this life-threatening syndrome can present with rigidity, fever, change in mental status, and worsening dystonic symptoms.
Treatment involves high doses of baclofen (up to 120 mg/day in divided doses).6
DISORDERS PRESENTING WITH HYPERKINETIC MOVEMENTS
Chorea, ballism (ballismus), and athetosis constitute a range of involuntary, hyperkinetic movement disorders. Chorea consists of involuntary, continuous, sudden, brief, unsustained, irregular movements that flow from one part of the body to another. Hemiballism presents as forceful flinging movements of the limbs or high-amplitude chorea that affects one side of the body.
Acute hemichorea and hemiballism
Acute hemichorea and hemiballism commonly result from infarction or hemorrhage of the basal ganglia.20 Computed tomography and especially magnetic resonance imaging can show the lesions in patients with ballism. Stroke-induced ballism is usually self-limited and resolves after a few weeks. Acute hemiballism generally evolves to hemichorea or hemiathetosis in a few days, which requires only protective measures.
Treatment. Mild cases do not need treatment but severe cases call for medical therapy. Antidopaminergics are the drugs of choice. A dopamine depletor such as reserpine (Serpasil) 0.1 mg once or twice daily or dopamine receptor blockers such as neuroleptics are considered.16 The combination of a benzodiazepine plus an antipsychotic such as olanzapine (Zyprexa) has been suggested.6
Severe parkinsonian dyskinesia
Dyskinesia is common in Parkinson disease, and patients may present to an emergency room with severe levodopa-induced dyskinesia. Dyskinesia can be exhausting if prolonged and severe. Elevated levels of creatine kinase raise the concern of rhabdomyolysis. In rare cases, the patient develops respiratory dyskinesia when respiratory muscles such as those in the diaphragm become involved.21
The risk of levodopa-induced dyskinesia increases with disease severity and higher levodopa doses. Using a dopamine agonist as initial therapy delays the onset of levodopa-induced dyskinesia in early Parkinson disease. However, Factor and Molho,21 in a case series, reported that adding dopamine agonists to the regimen was a precipitating factor; another was infection.
Treatment. A reasonable approach to treating peak-dose dyskinesia is to lower the doses of dopaminergic medications.
A mild sedative such as lorazepam, alprazolam (Xanax, Niravam), or clonazepam (Klonopin) may reduce the severity of dyski-nesia.21 These drugs are particularly useful if the dyskinesia is worse at night, and they can be used in the emergency department while waiting for the effect of the dopaminergic medications to wear off.
Amantadine ameliorates levodopa-induced peak-dose dyskinesia without worsening parkinsonian symptoms in some patients.22
Drug-induced myoclonus
Myoclonus is sudden, jerky, brief involuntary movement of the face, limbs, or trunk. Unlike tics, myoclonus cannot be controlled by the patient.
Myoclonus has various pathophysiologic mechanisms. Most myoclonic emergencies are epileptic myoclonic seizures, which are beyond the scope of this article. Often, myoclonus is caused by opiate overdose or withdrawal. It can also be a side effect of SSRIs, tricyclic anti-depressants, lithium, amantadine, and rarely, antibiotics such as imipenem (Primaxin).23
Treatment. Opiate-induced myoclonus may respond to naloxone (Narcan), whereas opiate withdrawal responds to benzodi-azepines.6
Acute akathisia
Acute akathisia occurs in susceptible patients after exposure to dopamine receptor blockers or dopamine depletors. It is characterized by subjective restless feelings accompanied by objective restless movements. The course is usually self-limited after the causative medication is discontinued.
Treatment. Symptomatic treatment may be needed in most cases for several days. Anticholinergics are effective. Additionally, vitamin B6, mianserine, propranolol, and mirtazapine (Remeron) in a low dose (15 mg/day) have been shown to be effective16,24,25
DISORDERS WITH PSYCHIATRIC PRESENTATIONS
Hallucinations and psychosis in Parkinson disease
Neuropsychiatric or behavioral complications of Parkinson disease include hallucinations, dementia, depression, psychosis, and sleep disorders.21,26 Psychosis is the leading reason for nursing home placement in advanced cases.27 Psychosis can present as hallucinations or a paranoid delusional state in association with clear sensorium.28 However, hallucinations accounted for only 3% of emergency admissions to the hospital for Parkinson disease patients in one series.29
Risk factors for hallucinations in parkinsonian patients include dementia, long-term therapy with dopaminergic drugs, long duration of disease, advanced age, anticholinergic drugs, and sleep disorders. Severe cognitive impairment or dementia is a major and independent predictive factor for visual hallucinations.30
Most hallucinations are visual; auditory, tactile, and olfactory hallucinations are rare.30
Treatment initially should be the same as in any patient with delirium. The systemic disorders that can aggravate or cause hallucinations such as electrolyte abnormalities, urinary or respiratory infection, and systemic illness should be ruled out.
The next step is to reduce or discontinue the adjunctive drugs that have the least antiparkinsonian effect and the greatest potential of inducing hallucination or psychosis. Examples of such medications include histamine-2 antagonists (eg, cimetidine [Tagamet], amantadine, selegeline, and anti-cholinergics). Selegeline can be discontinued abruptly because it has a long duration of action in the brain, but amantadine and anti-cholinergics should be tapered. Dopamine agonists can be discontinued. Levodopa can be reduced until the side effects begin to subside without significant worsening of motor symptoms.
If all the above adjustments fail, an antipsychotic medication can be considered.26 Clozapine (Clozaril) has the best result and is nearly free of extrapyramidal side effects but can cause agranulocytosis, which requires frequent blood counts. The Parkinson Study Group suggested that clozapine, at daily doses of 50 mg or less, is safe and significantly improves drug-induced psychosis without worsening parkinsonism.31 Clozapine may be impractical for elderly patients due to its side effect profile.
Quetiapine is a good alternative to cloza-pine and is less likely to worsen parkinsonian symptoms than other atypical antipsy-chotics.32 Olanzapine and risperidone (Risperdal) are reported to worsen parkinsonian symptoms.33 Not enough data have been published about the efficacy of the newer medications such as ziprasidone (Geodon) and aripiprazole (Abilify) to advocate their routine clinical use.
Rivastigmine (Exelon) was reported to improve hallucinations, sleep disturbance, and caregiver distress in addition to enhancing cognitive performance in advanced Parkinson disease in a small study.34 Burn and colleagues35 reported that rivastigmine was beneficial in patients with dementia associated with Parkinson disease, with or without hallucinations. Efficacy measures were cognitive scales, activities of daily living, behavioral symptoms, and executive and attentional functions. The differences in these measures between rivastigmine and placebo recipients tended to be larger in patients with visual hallucinations than in those without hallucinations. The study was not designed to assess the effect of treatment on psychosis or hallucination.
WHEN PATIENTS WITH MOVEMENT DISORDERS NEED SURGERY
Some of these syndromes can be prevented, especially in patients who are known to have movement disorders and are undergoing surgery.
One problem is stopping oral dopaminergic drugs before the operation. Parkinson disease patients on dopaminergic drugs can develop parkinsonism-hyperpyrexia syndrome or akinetic crisis if the drug is stopped suddenly. Restarting dopaminergic therapy and supportive measures are the main treatments. Patients who have Parkinson disease should receive their usual dose of levodopa, dopamine agonist, or amantadine up until the time of surgery and then again as soon as they awaken in the recovery room.36 That goal can be achieved more easily now that these drugs come in transdermal patches and long-acting formulas.37 Droperidol (Inapsine) and metoclopramide worsen parkinsonism and should be avoided.
Myoclonus is the most common movement disorder seen in the postoperative period. In fact, myoclonic shivering is common as patients awaken from general anesthesia.36 The anesthetic agents etomidate (Amidate) and enflurane (Ethrane) and the opioids fentanyl (Actiq, Duralgesic, Sublimaze) and meperidine (Demerol) can cause myoclonus.38
Occasionally, a patient in the recovery room suddenly develops a neurologic deficit that is inconsistent with the history and physical findings. Psychogenic movement disorders should be considered in the differential diagnosis. Reassurance and occasionally psychiatric intervention are required in these cases.36
IN THE ELDERLY, GO EASY
Polypharmacy is a huge issue in the elderly. Some of the principles in prescribing medications in the elderly can be helpful in preventing movement disorder emergencies:
- Assess the current regimen, including over-the-counter drugs, before prescribing a new drug.
- Begin with a low dose and increase as necessary. “Start low, go slow.”
- Consider the possibility that any new symptoms can be a drug side effect or due to withdrawal of a drug.
- Discuss with the patient or caregiver what kind of side effect to expect and advise him or her to report serious ones.
- Isbister GK, Bowe SJ, Dawson A, Whyte IM. Relative toxicity of selective serotonin reuptake inhibitors (SSRIs) in overdose. J Toxicol Clin Toxicol 2004; 42:277–285.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003; 96:635–642.
- Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000; 79:201–209.
- LoCurto MJ. The serotonin syndrome. Emerg Med Clin North Am 1997; 15( 3):665–675.
- Boyer E, Shannon S. The serotonin syndrome. N Engl J Med 2005; 352:1112–1120.
- Kipps CM, Fung VS, Grattan-Smith P, de Moore GM, Morris JG. Movement disorder emergencies. Mov Disord 2005; 20:322–334.
- Shalev A, Munitz H. The neuroleptic malignant syndrome: agent and host interaction. Acta Psychiatr Scand 1986; 73:337–347.
- Rosebush PI, Stewart TD, Gelenberg AJ. Twenty neuroleptic rechallenges after neuroleptic malignant syndrome in 15 patients. J Clin Psychiatry 1989; 50:295–298.
- Adityanjee , Singh S, Singh G, Ong S. Spectrum concept of neuroleptic malignant syndrome. Br J Psychiatry 1988; 153:107–111.
- Bonuccelli U, Piccini P, Corsini GU, Muratorio A. Apomorphine in malignant syndrome due to levodopa withdrawal. Ital J Neurol Sci 1992; 13:169–170.
- Sato Y, Asoh T, Metoki N, et al. Efficacy of methylprednisolone pulse therapy on neuroleptic malignant syndrome in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2004; 74:574–576.
- Hariz MI, Johansson F. Hardware failure in parkinsonian patients with chronic subthalamic nucleus stimulation is a medical emergency. Mov Disord 2001; 16:166–168.
- Pollera CF, Cognetli F, Nardi M, Mozza D. Sudden death after acute dystonic reaction to high-dose metoclopramide. Lancet 1984; 2:460–461.
- Bateman DN, Rawlins MD, Simpson JM. Extrapyramidal reactions with metoclopramide. Br Med J 1985; 291:930–932.
- Pasricha PJ, Pehlivanov N, Sugumar A, Jankovic J. Drug insight: from disturbed motility to disordered movement—a review of the clinical benefits and medicolegal risks of metoclopramide. Nat Clin Pract Gastroenterol Hepatol 2006; 3:138–148.
- Hu S, Frucht S. Emergency treatment of movement disorders. Curr Treat Options Neurol 2007; 9:103–114.
- Tousi B, Schuele SU, Subramanian T. A 46-year-old woman with rigidity and frequent falls. Cleve Clin J Med 2005; 72:57–63.
- Merlo IM, Occhini A, Pacchetti C, Alfonsi E. Not paralysis, but dystonia causes stridor in multiple system atrophy. Neurology 2002; 58:649–652.
- Silber MH, Levine S. Stridor and death in multiple system atrophy. Mov Disord 2000; 15:699–704.
- Bhatia KP, Marsden CD. The behavioural and motor consequences of focal lesions of the basal ganglia in man. Brain 1994; 117:859–876.
- Factor SA, Molho ES. Emergency department presentations of patients with Parkinson’s disease. Am J Emerg Med 2000; 18:209–215.
- Verhagen Metman L, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as a treatment for dyskinesia and motor fluctuations in Parkinson’s disease. Neurology 1998; 50:1323–1326.
- Frucht S, Eidelberg D. Imipenem-induced myoclonus. Mov Disord 1997; 12:621–622.
- Miodownik C, Lerner V, Statsenko N, et al. Vitamin B6 versus mianserin and placebo in acute neuroleptic-induced akathisia: a randomized, double-blind, controlled study. Clin Neuropharmacol 2006; 29:68–72.
- Poyurovsky M, Pashinian A, Weizman R, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry 2006; 59:1071–1077.
- Tousi B, Subramanian T. Hallucinations in Parkinson’s disease: approach and management. Clin Geriatr 2004: 12:19–24.
- Goetz CG, Stebbins GT. Risk factors for nursing home placement in advanced Parkinson’s disease. Neurology 1993; 43:2227–2229.
- Factor SA, Molho ES, Podskalny GD, Brown D. Parkinson’s disease: drug-induced psychiatric states. Adv Neurol 1995; 65:115–138.
- Woodford H, Walker R. Emergency hospital admissions in idiopathic Parkinson’s disease. Mov Disord 2005; 20:1104–1108.
- Tousi B, Frankel M. Olfactory and visual hallucinations in Parkinson’s disease. Parkinsonism Relat Disord 2004; 10:253–254.
- The Parkinson Study Group. Low-dose clozapine for the treatment of drug-induced psychosis in Parkinson’s disease. N Engl J Med 1999; 340:757–763.
- Merims D, Balas M, Pertez C, Shabtai H, Giladi N. Rater-blinded, prospective comparison: quetiapine versus clozapine for Parkinson’s disease psychosis. Clin Neuropharmacol 2006; 29:331–337.
- Goetz CG, Blasucci LM, Leurgans S, Pappert EJ. Olanzapine and clozapine: comparative effects on motor function in hallucinating PD patients. Neurology 2000; 55:789–794.
- Reading PJ, Luce AK, McKeith IG. Rivastigmine in the treatment of parkinsonian psychosis and cognitive impairment: preliminary findings from an open trial. Mov Disord 2001; 16:1171–1174.
- Burn D, Emre M, McKeith I, et al. Effects of rivastigmine in patients with and without visual hallucinations in dementia associated with Parkinson’s disease. Mov Disord 2006; 21:1899–1907.
- Frucht SJ. Movement disorder emergencies in the perioperative period. Neurol Clin 2004; 22:379–387.
- Korczyn AD, Reichmann H, Boroojerdi B, et al. Rotigotin trans-dermal system for perioperative administration. J Neural Transm 2007; 114:219–221.
- Gordon MF. Toxin and drug-induced myoclonus. Adv Neurol 2002; 89:49–76.
- Isbister GK, Bowe SJ, Dawson A, Whyte IM. Relative toxicity of selective serotonin reuptake inhibitors (SSRIs) in overdose. J Toxicol Clin Toxicol 2004; 42:277–285.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003; 96:635–642.
- Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000; 79:201–209.
- LoCurto MJ. The serotonin syndrome. Emerg Med Clin North Am 1997; 15( 3):665–675.
- Boyer E, Shannon S. The serotonin syndrome. N Engl J Med 2005; 352:1112–1120.
- Kipps CM, Fung VS, Grattan-Smith P, de Moore GM, Morris JG. Movement disorder emergencies. Mov Disord 2005; 20:322–334.
- Shalev A, Munitz H. The neuroleptic malignant syndrome: agent and host interaction. Acta Psychiatr Scand 1986; 73:337–347.
- Rosebush PI, Stewart TD, Gelenberg AJ. Twenty neuroleptic rechallenges after neuroleptic malignant syndrome in 15 patients. J Clin Psychiatry 1989; 50:295–298.
- Adityanjee , Singh S, Singh G, Ong S. Spectrum concept of neuroleptic malignant syndrome. Br J Psychiatry 1988; 153:107–111.
- Bonuccelli U, Piccini P, Corsini GU, Muratorio A. Apomorphine in malignant syndrome due to levodopa withdrawal. Ital J Neurol Sci 1992; 13:169–170.
- Sato Y, Asoh T, Metoki N, et al. Efficacy of methylprednisolone pulse therapy on neuroleptic malignant syndrome in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2004; 74:574–576.
- Hariz MI, Johansson F. Hardware failure in parkinsonian patients with chronic subthalamic nucleus stimulation is a medical emergency. Mov Disord 2001; 16:166–168.
- Pollera CF, Cognetli F, Nardi M, Mozza D. Sudden death after acute dystonic reaction to high-dose metoclopramide. Lancet 1984; 2:460–461.
- Bateman DN, Rawlins MD, Simpson JM. Extrapyramidal reactions with metoclopramide. Br Med J 1985; 291:930–932.
- Pasricha PJ, Pehlivanov N, Sugumar A, Jankovic J. Drug insight: from disturbed motility to disordered movement—a review of the clinical benefits and medicolegal risks of metoclopramide. Nat Clin Pract Gastroenterol Hepatol 2006; 3:138–148.
- Hu S, Frucht S. Emergency treatment of movement disorders. Curr Treat Options Neurol 2007; 9:103–114.
- Tousi B, Schuele SU, Subramanian T. A 46-year-old woman with rigidity and frequent falls. Cleve Clin J Med 2005; 72:57–63.
- Merlo IM, Occhini A, Pacchetti C, Alfonsi E. Not paralysis, but dystonia causes stridor in multiple system atrophy. Neurology 2002; 58:649–652.
- Silber MH, Levine S. Stridor and death in multiple system atrophy. Mov Disord 2000; 15:699–704.
- Bhatia KP, Marsden CD. The behavioural and motor consequences of focal lesions of the basal ganglia in man. Brain 1994; 117:859–876.
- Factor SA, Molho ES. Emergency department presentations of patients with Parkinson’s disease. Am J Emerg Med 2000; 18:209–215.
- Verhagen Metman L, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as a treatment for dyskinesia and motor fluctuations in Parkinson’s disease. Neurology 1998; 50:1323–1326.
- Frucht S, Eidelberg D. Imipenem-induced myoclonus. Mov Disord 1997; 12:621–622.
- Miodownik C, Lerner V, Statsenko N, et al. Vitamin B6 versus mianserin and placebo in acute neuroleptic-induced akathisia: a randomized, double-blind, controlled study. Clin Neuropharmacol 2006; 29:68–72.
- Poyurovsky M, Pashinian A, Weizman R, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry 2006; 59:1071–1077.
- Tousi B, Subramanian T. Hallucinations in Parkinson’s disease: approach and management. Clin Geriatr 2004: 12:19–24.
- Goetz CG, Stebbins GT. Risk factors for nursing home placement in advanced Parkinson’s disease. Neurology 1993; 43:2227–2229.
- Factor SA, Molho ES, Podskalny GD, Brown D. Parkinson’s disease: drug-induced psychiatric states. Adv Neurol 1995; 65:115–138.
- Woodford H, Walker R. Emergency hospital admissions in idiopathic Parkinson’s disease. Mov Disord 2005; 20:1104–1108.
- Tousi B, Frankel M. Olfactory and visual hallucinations in Parkinson’s disease. Parkinsonism Relat Disord 2004; 10:253–254.
- The Parkinson Study Group. Low-dose clozapine for the treatment of drug-induced psychosis in Parkinson’s disease. N Engl J Med 1999; 340:757–763.
- Merims D, Balas M, Pertez C, Shabtai H, Giladi N. Rater-blinded, prospective comparison: quetiapine versus clozapine for Parkinson’s disease psychosis. Clin Neuropharmacol 2006; 29:331–337.
- Goetz CG, Blasucci LM, Leurgans S, Pappert EJ. Olanzapine and clozapine: comparative effects on motor function in hallucinating PD patients. Neurology 2000; 55:789–794.
- Reading PJ, Luce AK, McKeith IG. Rivastigmine in the treatment of parkinsonian psychosis and cognitive impairment: preliminary findings from an open trial. Mov Disord 2001; 16:1171–1174.
- Burn D, Emre M, McKeith I, et al. Effects of rivastigmine in patients with and without visual hallucinations in dementia associated with Parkinson’s disease. Mov Disord 2006; 21:1899–1907.
- Frucht SJ. Movement disorder emergencies in the perioperative period. Neurol Clin 2004; 22:379–387.
- Korczyn AD, Reichmann H, Boroojerdi B, et al. Rotigotin trans-dermal system for perioperative administration. J Neural Transm 2007; 114:219–221.
- Gordon MF. Toxin and drug-induced myoclonus. Adv Neurol 2002; 89:49–76.
KEY POINTS
- Supportive measures must be taken immediately to maintain the functions of vital organs.
- Serotonin syndrome, which can cause rigidity or stiffness, can be prevented by avoiding multidrug regimens.
- Withdrawing or decreasing the dose of dopaminergic drugs in patients with Parkinson disease can cause parkinsonism-hyperpyrexia syndrome, a condition similar to neuroleptic malignant syndrome.
- Metoclopramide (Reglan) accounts for nearly one-third of all drug-induced movement disorders. The entire spectrum of drug-induced movement disorders, ranging from subtle to life-threatening, can ensue from its use.
- Complications of Parkinson disease include hallucinations, dementia, depression, psychosis, and sleep disorders.
Medical causes of back pain
To the Editor: In their otherwise excellent review, “Masquerade: Medical causes of back pain” (Cleve Clin J Med 2007; 74:905–913), Dr. Klineberg et al seem to confuse two distinct pathologic processes—aortic dissection and rupture of an aortic aneurysm. Parts of their description seem to fit the pathology of abdominal aortic aneurysm, with a pulsatile abdominal mass, sentinel bleeding, and rupture risk with a size over 6 cm, whereas other parts seem to correspond to aortic dissection, with severe, ripping pain and an association with Marfan syndrome. They also use the terminology “dissecting aortic aneurysm,” which again implies a single entity, when in fact the two conditions rarely occur together. The authors are not alone in their use of this misnomer: a review of the Web sites of renowned universities reveals use of the same terminology. The readers would have been better served if the authors had discussed “acute aortic dissection” and “ruptured aortic aneurysm” as two separate causes of back pain, with a note that in rare cases an aortic aneurysm can develop a dissection.
To the Editor: In their otherwise excellent review, “Masquerade: Medical causes of back pain” (Cleve Clin J Med 2007; 74:905–913), Dr. Klineberg et al seem to confuse two distinct pathologic processes—aortic dissection and rupture of an aortic aneurysm. Parts of their description seem to fit the pathology of abdominal aortic aneurysm, with a pulsatile abdominal mass, sentinel bleeding, and rupture risk with a size over 6 cm, whereas other parts seem to correspond to aortic dissection, with severe, ripping pain and an association with Marfan syndrome. They also use the terminology “dissecting aortic aneurysm,” which again implies a single entity, when in fact the two conditions rarely occur together. The authors are not alone in their use of this misnomer: a review of the Web sites of renowned universities reveals use of the same terminology. The readers would have been better served if the authors had discussed “acute aortic dissection” and “ruptured aortic aneurysm” as two separate causes of back pain, with a note that in rare cases an aortic aneurysm can develop a dissection.
To the Editor: In their otherwise excellent review, “Masquerade: Medical causes of back pain” (Cleve Clin J Med 2007; 74:905–913), Dr. Klineberg et al seem to confuse two distinct pathologic processes—aortic dissection and rupture of an aortic aneurysm. Parts of their description seem to fit the pathology of abdominal aortic aneurysm, with a pulsatile abdominal mass, sentinel bleeding, and rupture risk with a size over 6 cm, whereas other parts seem to correspond to aortic dissection, with severe, ripping pain and an association with Marfan syndrome. They also use the terminology “dissecting aortic aneurysm,” which again implies a single entity, when in fact the two conditions rarely occur together. The authors are not alone in their use of this misnomer: a review of the Web sites of renowned universities reveals use of the same terminology. The readers would have been better served if the authors had discussed “acute aortic dissection” and “ruptured aortic aneurysm” as two separate causes of back pain, with a note that in rare cases an aortic aneurysm can develop a dissection.
In reply: Medical causes of back pain
In Reply: We appreciate Dr. Hirsch’s comments and are pleased to expand the discussion of this important point.
He is correct in his assertion that dissection and aneurysm are distinct processes. But the goal of this review was to remind practitioners to consider the aorta as a possible source of pain when it occurs acutely or in an atypical manner.
A number of aortic processes can cause back pain, and aneurysm and dissection are two of them, aneurysm being more common than aortic dissection. But the pain can also be from aortic ulceration, aortitis, contained rupture of an aneurysm, and other more esoteric problems.
Aortic dissection often presents as a tearing, severe, thoracic back pain. Pain from a progressive abdominal aneurysm is more commonly referred to the lower back or flank and can be severe and unrelenting. It is rarely described as a tearing pain like that of dissection.
It is difficult on initial physical examination to distinguish aneurysm from dissection. The key to diagnosing aneurysm is to detect the pulsatile abdominal mass. A pulsatile, tender abdominal mass with hypotension and back pain is classically associated with rupture of an abdominal aortic aneurysm. The combination of back pain, a deficit in peripheral pulses, and hypertension is more often associated with dissection.
Without imaging and appropriate consultation, it is difficult for even an experienced provider to definitively diagnose these disorders. Without a bit of suspicion, even with a careful physical examination either disorder might be overlooked entirely, with disastrous effect. The purpose of our review was to remind the reader that these conditions, while uncommon or even rare, do occur and should be sought out in patients presenting with acute, atypical lumbar and thoracic back pain. As with each of the conditions discussed in this review, the decision to linger a bit over the patient’s history and then perform a basic, focused physical examination can be life-saving.
In Reply: We appreciate Dr. Hirsch’s comments and are pleased to expand the discussion of this important point.
He is correct in his assertion that dissection and aneurysm are distinct processes. But the goal of this review was to remind practitioners to consider the aorta as a possible source of pain when it occurs acutely or in an atypical manner.
A number of aortic processes can cause back pain, and aneurysm and dissection are two of them, aneurysm being more common than aortic dissection. But the pain can also be from aortic ulceration, aortitis, contained rupture of an aneurysm, and other more esoteric problems.
Aortic dissection often presents as a tearing, severe, thoracic back pain. Pain from a progressive abdominal aneurysm is more commonly referred to the lower back or flank and can be severe and unrelenting. It is rarely described as a tearing pain like that of dissection.
It is difficult on initial physical examination to distinguish aneurysm from dissection. The key to diagnosing aneurysm is to detect the pulsatile abdominal mass. A pulsatile, tender abdominal mass with hypotension and back pain is classically associated with rupture of an abdominal aortic aneurysm. The combination of back pain, a deficit in peripheral pulses, and hypertension is more often associated with dissection.
Without imaging and appropriate consultation, it is difficult for even an experienced provider to definitively diagnose these disorders. Without a bit of suspicion, even with a careful physical examination either disorder might be overlooked entirely, with disastrous effect. The purpose of our review was to remind the reader that these conditions, while uncommon or even rare, do occur and should be sought out in patients presenting with acute, atypical lumbar and thoracic back pain. As with each of the conditions discussed in this review, the decision to linger a bit over the patient’s history and then perform a basic, focused physical examination can be life-saving.
In Reply: We appreciate Dr. Hirsch’s comments and are pleased to expand the discussion of this important point.
He is correct in his assertion that dissection and aneurysm are distinct processes. But the goal of this review was to remind practitioners to consider the aorta as a possible source of pain when it occurs acutely or in an atypical manner.
A number of aortic processes can cause back pain, and aneurysm and dissection are two of them, aneurysm being more common than aortic dissection. But the pain can also be from aortic ulceration, aortitis, contained rupture of an aneurysm, and other more esoteric problems.
Aortic dissection often presents as a tearing, severe, thoracic back pain. Pain from a progressive abdominal aneurysm is more commonly referred to the lower back or flank and can be severe and unrelenting. It is rarely described as a tearing pain like that of dissection.
It is difficult on initial physical examination to distinguish aneurysm from dissection. The key to diagnosing aneurysm is to detect the pulsatile abdominal mass. A pulsatile, tender abdominal mass with hypotension and back pain is classically associated with rupture of an abdominal aortic aneurysm. The combination of back pain, a deficit in peripheral pulses, and hypertension is more often associated with dissection.
Without imaging and appropriate consultation, it is difficult for even an experienced provider to definitively diagnose these disorders. Without a bit of suspicion, even with a careful physical examination either disorder might be overlooked entirely, with disastrous effect. The purpose of our review was to remind the reader that these conditions, while uncommon or even rare, do occur and should be sought out in patients presenting with acute, atypical lumbar and thoracic back pain. As with each of the conditions discussed in this review, the decision to linger a bit over the patient’s history and then perform a basic, focused physical examination can be life-saving.