User login
Grand Rounds: Five-Day-Old Infant With Hip "Clunk"
A 5-day-old infant was referred to the pediatric orthopedic clinic for evaluation of a left hip “clunk.” She is a firstborn child, born at full term (39 weeks) via cesarean delivery secondary to breech presentation. Her weight at birth was 7 lb 6 oz. The infant was noted to have a left hip clunk during a routine physical examination by her pediatrician, who made a referral to the pediatric orthopedic clinic for possible hip dysplasia. This is the patient’s first visit to the clinic.
There is no family history of hip dysplasia or other orthopedic abnormalities. The infant is a well-appearing, alert female measuring 20.5” in length and weighing 7 lb 4 oz. Vital signs are stable with no abnormality detected. The heart is regular in rate and rhythm, and the chest is clear bilaterally.
No cutaneous abnormalities are noted. The patient is able to move all her extremities spontaneously, and her spine is straight and normal with no evidence of spinal dysraphism. Her feet are normal bilaterally, with full range of motion and no equinovarus or metatarsus adductus deformity.
The neurologic examination is also unremarkable, with normal neonatal reflexes and excellent muscle tone throughout.
Examination of the infant’s hips reveals a positive result on the Barlow test on the left side (the hip can be dislocated). There is also a positive Ortolani sign (the hip can be reduced), with asymmetric thigh skin folds noted (see Figures 1A and 1B, respectively).
Based on these positive physical examination findings, the patient was diagnosed with developmental dysplasia of the hip (DDH). Initial ultrasonography to confirm the diagnosis was not considered necessary, as the physical examination demonstrated obvious instability.1 The infant was placed in a Pavlik harness, which her parents were instructed should be worn full-time (see Figures 2A and 2B). She was scheduled for weekly follow-up visits for adjustments to the harness and serial hip examinations.
At the second follow-up visit, ultrasonography was performed, confirming the presence of dysplasia with decreased femoral head coverage and a steep socket (acetabulum). Use of the Pavlik harness was continued full-time for six weeks.
At age 6 weeks, the infant underwent a follow-up ultrasound to assess for improvement in the degree of dysplasia. The test revealed normal hips bilaterally with no evidence of DDH. Therefore, use of the Pavlik harness was discontinued. The parents were instructed to bring the child back in six months for a repeat clinical examination and an anteroposterior x-ray of the pelvis.1
Discussion
The term developmental dysplasia of the hip (DDH) has replaced the more traditional term congenital hip dislocation because DDH more accurately reflects the variable characteristics that can be seen with this condition. As DDH may not be present at birth, the term congenital is misleading. We now know that DDH may occur in utero, perinatally, or during infancy and childhood.2,3
Generally, DDH is used to describe an abnormal relationship between the femoral head and the acetabulum (see Figure 34). The term represents a wide spectrum of abnormality, as shown in the Graf classification of hips in infants: type I refers to a normal hip; type II, immature development to mild dysplasia; type III, subluxation of the femoral head; and type 4, frank dislocation with severe instability.5
Diagnosing and managing DDH correctly requires the clinician to have a thorough understanding of the normal growth and development that occurs in the hip joint. Embryologically, the joint (including the femoral head and acetabulum) develops from the same primitive mesenchymal cells.6 By 11 to 12 weeks’ gestation, the initial structures of the hip joint are fully formed; theoretically, this is the earliest time at which a dislocation can occur.2,7 DDH that develops at this stage would be called teratologic; this condition is seen most frequently in patients who have underlying neuromuscular conditions, such as myelodysplasia (spina bifida) or arthrogryposis. A typical dislocation takes place during the perinatal period in an infant who is otherwise healthy.2
Etiology
DDH occurs in about 11 of every 1,000 infants, with frank dislocations occurring in one to two infants per 10,000.8 The left hip is involved in approximately 60% of cases, the right in 20%, and both hips in about 20%. In the most common intrauterine fetal position, the left hip is lower than the right (usually abutting the mother’s sacrum) and is often in adduction. This is likely the reason that the left hip is more commonly affected by DDH.
DDH is believed to be multifactorial, with physiologic, genetic, and mechanical factors implicated in the etiology.3 The incidence of DDH varies with factors such as the patient’s age, race, and gender, the experience and training of the examiner, and the diagnostic criteria that are used.
Known risk factors for a positive newborn screening are shown in the table.9,10 It is often helpful for clinicians to remember the “4F” mnemonic associated with DDH: female, firstborn, foot first, and family history.9
There is also an increased risk for DDH in patients with other conditions that are associated with intrauterine crowding. These include congenital muscular torticollis, metatarsus adductus, and congenital dislocation of the knee.2
Physical Examination
All newborn infants should be screened for DDH as part of the initial physical examination, with ultrasonography recommended for infants deemed at high risk for DDH and for those with inconclusive results on examination.1,10,11 Providers should be aware that the newborn hip examination requires a considerable amount of practice and expertise.
A thorough medical history should always be obtained first, including gestational age, presentation (breech vs vertex), type of delivery (cesarean vs vaginal), gender, birth order, family history of DDH, ligamentous laxity, or myopathy.8
The examining clinician begins by placing the infant on a firm, flat surface. The infant should be as relaxed as possible. Next, the clinician observes both lower extremities for asymmetric thigh or buttock skin folds. Bilateral DDH can be very difficult to diagnose on the basis of this examination due to the lack of asymmetry (hips will have symmetric abnormality).
The Galeazzi sign is elicited by placing the infant supine with the hips and knees flexed to 90°.12 With the hips in neutral abduction, the provider should determine whether the knees are at the same height. Unequal knee heights—a positive result for the Galeazzi sign—suggest femoral shortening (apparent leg length discrepancy), which may be explained by a hip dislocation. If both hips are dislocated, a false-negative result will often occur, since both will appear short and there will be no discrepancy.2,12
Among physical examination techniques, the Ortolani and Barlow maneuvers are considered most reliable to detect hip instability in newborns and infants younger than 6 months2,13,14 (review Figures 1A and 1B). The Ortolani test is used to detect the sensation of the dislocated hip reducing into the acetabulum, and the Barlow test elicits the unstable hip dislocating.2 A palpable and occasionally audible clunk is considered a positive result on the Barlow test and usually indicates a diagnosis of DDH.14 High-pitched clicks or snaps frequently occur with hip range-of-motion maneuvers and during Ortolani and Barlow testing. These sounds are often attributed to snapping of the iliotibial band over the greater trochanter and do not usually signify dysplasia.15
Because DDH is a dynamic and evolving process, the physical findings on clinical examination change significantly, depending on the age of the infant or child. As an infant approaches age 3 months, limited hip abduction (especially when asymmetric) is often the most reliable physical examination finding in patients with DDH.12 After age 3 to 4 months, Ortolani and Barlow testing will often produce negative results as progressive soft tissue contractures evolve.
Once a child begins to walk, gait abnormalities (eg, a short-limbed or waddling gait pattern) may raise suspicion for a diagnosis of DDH.7 It has been recommended that evaluation for DDH be performed at each routine office examination until the child is 12 months of age.1
Treatment
The Pavlik harness is considered first-line treatment for DDH in infants younger than 6 months. The harness is a dynamic splint that allows the infant to engage in a sphere of active motion that encourages stabilization and deepening of the socket. The harness is applied with the knees flexed to about 90° and the hips in about 70° of abduction and 100° to 110° of flexion (as shown in Figures 2A and 2B).9
The duration of treatment depends on the infant’s age at presentation and the severity of DDH. Progress is judged by serial examinations and dynamic ultrasounds. The harness is worn full-time until clinical and radiographic examinations both yield normal results. After six weeks of treatment, the hips are examined out of the harness, and a repeat ultrasound is usually obtained. If findings are normal, use of the harness is ordinarily discontinued. Some patients will require harness use for a longer period in cases of delayed development of the acetabulum and/or severe laxity of the ligaments.9
The Pavlik harness is successful more than 90% of the time in newborns with DDH.8 Success rates have been reported as greatest in infants younger than 8 weeks at the time of treatment initiation, those with only one affected hip, and those with less severe disease (Graf types II or III).16
If ultrasonography shows no improvement after two to three weeks, it is usually recommended that the harness be discontinued; most orthopedic surgeons will then proceed with a closed or open reduction and spica body casting. Similarly, when the diagnosis of DDH is delayed until after ages 6 to 8 months, a closed reduction under anesthesia and placement of a spica body cast is usually the recommended treatment to maintain the hip in the reduced position.17,18 Some older children (ages 1 to 5 years) may require bracing, traction, open reduction, and/or femoral or pelvic osteotomy.17,18 It is believed that undiagnosed, untreated DDH can lead to early-onset degenerative hip disease (arthritis).1
Patient/Family Education
The Pavlik harness is most effective when a consistent support system exists to educate parents about the importance of the harness, its care and maintenance, and the consequences of failure. Close monitoring of the infant’s progress is also essential to promoting adherence. Application and removal of the harness should be demonstrated to the parent or caregiver, as well as diapering, dressing, and undressing the infant; they should then be encouraged to practice immediately in the clinic or office.
During visits for harness adjustment, the strap position should be marked with indelible ink, allowing parents to reapply the device correctly, should removal be required (eg, for bathing).9 Ten percent of parents reportedly find reapplying the harness difficult during the first weeks of use. Difficulty in dressing and carrying an infant in a harness, feet slipping out of the harness, and skin irritation have been reported by about one-third of parents.19
Treatment adherence and subsequent success with the Pavlik harness is reported greatest (95%) in patients whose parents engage in demonstrations of harness use and follow instructions precisely.19 By providing a contact name and office number and following up with a phone call a few days after the harness is first applied, clinicians can significantly decrease parents’ anxiety and increase overall compliance.9
Conclusion
Despite recent increased awareness of DDH and the importance of thorough screening programs, hip dysplasia continues to be a frequently missed diagnosis in pediatrics. It is often up to the primary care clinician to screen for, assess, and potentially diagnose DDH. Therefore, a thorough understanding of this condition can promote early detection and diagnosis, with less invasive treatment and a more favorable outcome.
A proper hip examination should be a standard component of all newborn and infant well-child examinations. If DDH is suspected, appropriate referral to a pediatric orthopedic surgeon must be made so that timely treatment can be initiated. Early use of the Pavlik harness is significantly easier than the invasive surgery and prolonged immobilization necessitated by a delayed diagnosis. Whatever the course of treatment required, it is important for clinicians to support the patient and family: training and anticipatory guidance are essential components of DDH management.
1. Karmazyn BK, Gunderman RB, Coley BD, et al; American College of Radiology. ACR appropriateness criteria on developmental dysplasia of the hip—child. J Am Coll Radiol. 2009;6(8):551-557.
2. American Academy of Pediatrics, Committee on Quality Improvement, Subcommittee on Developmental Dysplasia of the Hip. Clinical practice guideline: early detection of developmental dysplasia of the hip. Pediatrics. 2000;105(4 pt 1):896-905.
3. Mencio GA. Developmental dysplasia of the hip. In: Sponseller PD, ed. Orthopaedic Knowledge Update: Pediatrics–2. Rosemont, IL: American Academy of Orthopaedic Surgeons; 2002:161-171.
4. Children’s Hospital at Westmead. Developmental dysplasia of the hip (DDH). www.chw.edu.au/parents/factsheets/developj.htm. Accessed March 26, 2010.
5. Graf R. Classification of hip joint dysplasia by means of sonography. Arch Orthop Trauma Surg. 1984; 102:248-255.
6. Weinstein SL. Developmental hip dysplasia and dislocation. In: Morrissy RT, Weinstein SL, eds. Lovell and Winter’s Pediatric Orthopaedics. 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005:905-956.
7. Aronsson DD, Goldberg MJ, Kling TF Jr, Roy DR. Developmental dysplasia of the hip. Pediatrics. 1994; 94(2 pt 1):201-208.
8. Guille JT, Pizzutillo PD, MacEwan GD. Developmental dysplasia of the hip from birth to six months. J Am Acad Orthop Surg. 2000;8(4):232-242.
9. Hart ES, Albright MB, Rebello GN, Grottkau BE. Developmental dysplasia of the hip: nursing implications and anticipatory guidelines for parents. Orthop Nurs. 2006;25(2):100-109.
10. Dogruel H, Atalar H, Yavus OY, Sayli U. Clinical examination versus ultrasonography in detecting developmental dysplasia of the hip. Int Orthop. 2008; 32(3):415-419.
11. Mahan ST, Katz JN, Kim YJ. To screen or not to screen? A decision analysis of the utility of screening for developmental dysplasia of the hip. J Bone Joint Surg Am. 2009;91(7);1705-1719.
12. Storer SK, Skaggs DL. Developmental dysplasia of the hip. Am Fam Physician. 2006;74(8):1310-1316.
13. Ortolani M. Congenital hip dysplasia in the light of early and very early diagnosis. Clin Orthop Relat Res. 1976;119(1):6-10.
14. Barlow TG. Congenital dislocation of the hip in the newborn. Proc R Soc Med. 1966;59(11 part 1):1103-1106.
15. Bond CD, Hennrikus WL, DellaMaggiore ED. Prospective evaluation of newborn soft-tissue “clicks” with ultrasound. J Pediatr Orthop. 1997;17(2):199-201.
16. Atalar H, Sayli U, Yavuz OY, et al. Indicators of successful use of the Pavlik harness in infants with developmental dysplasia of the hip. Int Orthop. 2007; 31(2):145-150.
17. Rampal V, Sabourin M, Erdeneshoo E, et al. Closed reduction with traction for developmental dysplasia of the hip in children aged between one and five years. J Bone Joint Surg Br. 2008;90-B(7):858-863.
18. Clarke NMP, Sakthivel K. The diagnosis and management of congenital dislocation of the hip. Paediatr Child Health. 2008;18(6):268-271.
19. Hassan FA. Compliance of parents with regard to Pavlik harness treatment in developmental dysplasia of the hip. J Pediatr Orthop. 2009;18(3):111-115.
A 5-day-old infant was referred to the pediatric orthopedic clinic for evaluation of a left hip “clunk.” She is a firstborn child, born at full term (39 weeks) via cesarean delivery secondary to breech presentation. Her weight at birth was 7 lb 6 oz. The infant was noted to have a left hip clunk during a routine physical examination by her pediatrician, who made a referral to the pediatric orthopedic clinic for possible hip dysplasia. This is the patient’s first visit to the clinic.
There is no family history of hip dysplasia or other orthopedic abnormalities. The infant is a well-appearing, alert female measuring 20.5” in length and weighing 7 lb 4 oz. Vital signs are stable with no abnormality detected. The heart is regular in rate and rhythm, and the chest is clear bilaterally.
No cutaneous abnormalities are noted. The patient is able to move all her extremities spontaneously, and her spine is straight and normal with no evidence of spinal dysraphism. Her feet are normal bilaterally, with full range of motion and no equinovarus or metatarsus adductus deformity.
The neurologic examination is also unremarkable, with normal neonatal reflexes and excellent muscle tone throughout.
Examination of the infant’s hips reveals a positive result on the Barlow test on the left side (the hip can be dislocated). There is also a positive Ortolani sign (the hip can be reduced), with asymmetric thigh skin folds noted (see Figures 1A and 1B, respectively).
Based on these positive physical examination findings, the patient was diagnosed with developmental dysplasia of the hip (DDH). Initial ultrasonography to confirm the diagnosis was not considered necessary, as the physical examination demonstrated obvious instability.1 The infant was placed in a Pavlik harness, which her parents were instructed should be worn full-time (see Figures 2A and 2B). She was scheduled for weekly follow-up visits for adjustments to the harness and serial hip examinations.
At the second follow-up visit, ultrasonography was performed, confirming the presence of dysplasia with decreased femoral head coverage and a steep socket (acetabulum). Use of the Pavlik harness was continued full-time for six weeks.
At age 6 weeks, the infant underwent a follow-up ultrasound to assess for improvement in the degree of dysplasia. The test revealed normal hips bilaterally with no evidence of DDH. Therefore, use of the Pavlik harness was discontinued. The parents were instructed to bring the child back in six months for a repeat clinical examination and an anteroposterior x-ray of the pelvis.1
Discussion
The term developmental dysplasia of the hip (DDH) has replaced the more traditional term congenital hip dislocation because DDH more accurately reflects the variable characteristics that can be seen with this condition. As DDH may not be present at birth, the term congenital is misleading. We now know that DDH may occur in utero, perinatally, or during infancy and childhood.2,3
Generally, DDH is used to describe an abnormal relationship between the femoral head and the acetabulum (see Figure 34). The term represents a wide spectrum of abnormality, as shown in the Graf classification of hips in infants: type I refers to a normal hip; type II, immature development to mild dysplasia; type III, subluxation of the femoral head; and type 4, frank dislocation with severe instability.5
Diagnosing and managing DDH correctly requires the clinician to have a thorough understanding of the normal growth and development that occurs in the hip joint. Embryologically, the joint (including the femoral head and acetabulum) develops from the same primitive mesenchymal cells.6 By 11 to 12 weeks’ gestation, the initial structures of the hip joint are fully formed; theoretically, this is the earliest time at which a dislocation can occur.2,7 DDH that develops at this stage would be called teratologic; this condition is seen most frequently in patients who have underlying neuromuscular conditions, such as myelodysplasia (spina bifida) or arthrogryposis. A typical dislocation takes place during the perinatal period in an infant who is otherwise healthy.2
Etiology
DDH occurs in about 11 of every 1,000 infants, with frank dislocations occurring in one to two infants per 10,000.8 The left hip is involved in approximately 60% of cases, the right in 20%, and both hips in about 20%. In the most common intrauterine fetal position, the left hip is lower than the right (usually abutting the mother’s sacrum) and is often in adduction. This is likely the reason that the left hip is more commonly affected by DDH.
DDH is believed to be multifactorial, with physiologic, genetic, and mechanical factors implicated in the etiology.3 The incidence of DDH varies with factors such as the patient’s age, race, and gender, the experience and training of the examiner, and the diagnostic criteria that are used.
Known risk factors for a positive newborn screening are shown in the table.9,10 It is often helpful for clinicians to remember the “4F” mnemonic associated with DDH: female, firstborn, foot first, and family history.9
There is also an increased risk for DDH in patients with other conditions that are associated with intrauterine crowding. These include congenital muscular torticollis, metatarsus adductus, and congenital dislocation of the knee.2
Physical Examination
All newborn infants should be screened for DDH as part of the initial physical examination, with ultrasonography recommended for infants deemed at high risk for DDH and for those with inconclusive results on examination.1,10,11 Providers should be aware that the newborn hip examination requires a considerable amount of practice and expertise.
A thorough medical history should always be obtained first, including gestational age, presentation (breech vs vertex), type of delivery (cesarean vs vaginal), gender, birth order, family history of DDH, ligamentous laxity, or myopathy.8
The examining clinician begins by placing the infant on a firm, flat surface. The infant should be as relaxed as possible. Next, the clinician observes both lower extremities for asymmetric thigh or buttock skin folds. Bilateral DDH can be very difficult to diagnose on the basis of this examination due to the lack of asymmetry (hips will have symmetric abnormality).
The Galeazzi sign is elicited by placing the infant supine with the hips and knees flexed to 90°.12 With the hips in neutral abduction, the provider should determine whether the knees are at the same height. Unequal knee heights—a positive result for the Galeazzi sign—suggest femoral shortening (apparent leg length discrepancy), which may be explained by a hip dislocation. If both hips are dislocated, a false-negative result will often occur, since both will appear short and there will be no discrepancy.2,12
Among physical examination techniques, the Ortolani and Barlow maneuvers are considered most reliable to detect hip instability in newborns and infants younger than 6 months2,13,14 (review Figures 1A and 1B). The Ortolani test is used to detect the sensation of the dislocated hip reducing into the acetabulum, and the Barlow test elicits the unstable hip dislocating.2 A palpable and occasionally audible clunk is considered a positive result on the Barlow test and usually indicates a diagnosis of DDH.14 High-pitched clicks or snaps frequently occur with hip range-of-motion maneuvers and during Ortolani and Barlow testing. These sounds are often attributed to snapping of the iliotibial band over the greater trochanter and do not usually signify dysplasia.15
Because DDH is a dynamic and evolving process, the physical findings on clinical examination change significantly, depending on the age of the infant or child. As an infant approaches age 3 months, limited hip abduction (especially when asymmetric) is often the most reliable physical examination finding in patients with DDH.12 After age 3 to 4 months, Ortolani and Barlow testing will often produce negative results as progressive soft tissue contractures evolve.
Once a child begins to walk, gait abnormalities (eg, a short-limbed or waddling gait pattern) may raise suspicion for a diagnosis of DDH.7 It has been recommended that evaluation for DDH be performed at each routine office examination until the child is 12 months of age.1
Treatment
The Pavlik harness is considered first-line treatment for DDH in infants younger than 6 months. The harness is a dynamic splint that allows the infant to engage in a sphere of active motion that encourages stabilization and deepening of the socket. The harness is applied with the knees flexed to about 90° and the hips in about 70° of abduction and 100° to 110° of flexion (as shown in Figures 2A and 2B).9
The duration of treatment depends on the infant’s age at presentation and the severity of DDH. Progress is judged by serial examinations and dynamic ultrasounds. The harness is worn full-time until clinical and radiographic examinations both yield normal results. After six weeks of treatment, the hips are examined out of the harness, and a repeat ultrasound is usually obtained. If findings are normal, use of the harness is ordinarily discontinued. Some patients will require harness use for a longer period in cases of delayed development of the acetabulum and/or severe laxity of the ligaments.9
The Pavlik harness is successful more than 90% of the time in newborns with DDH.8 Success rates have been reported as greatest in infants younger than 8 weeks at the time of treatment initiation, those with only one affected hip, and those with less severe disease (Graf types II or III).16
If ultrasonography shows no improvement after two to three weeks, it is usually recommended that the harness be discontinued; most orthopedic surgeons will then proceed with a closed or open reduction and spica body casting. Similarly, when the diagnosis of DDH is delayed until after ages 6 to 8 months, a closed reduction under anesthesia and placement of a spica body cast is usually the recommended treatment to maintain the hip in the reduced position.17,18 Some older children (ages 1 to 5 years) may require bracing, traction, open reduction, and/or femoral or pelvic osteotomy.17,18 It is believed that undiagnosed, untreated DDH can lead to early-onset degenerative hip disease (arthritis).1
Patient/Family Education
The Pavlik harness is most effective when a consistent support system exists to educate parents about the importance of the harness, its care and maintenance, and the consequences of failure. Close monitoring of the infant’s progress is also essential to promoting adherence. Application and removal of the harness should be demonstrated to the parent or caregiver, as well as diapering, dressing, and undressing the infant; they should then be encouraged to practice immediately in the clinic or office.
During visits for harness adjustment, the strap position should be marked with indelible ink, allowing parents to reapply the device correctly, should removal be required (eg, for bathing).9 Ten percent of parents reportedly find reapplying the harness difficult during the first weeks of use. Difficulty in dressing and carrying an infant in a harness, feet slipping out of the harness, and skin irritation have been reported by about one-third of parents.19
Treatment adherence and subsequent success with the Pavlik harness is reported greatest (95%) in patients whose parents engage in demonstrations of harness use and follow instructions precisely.19 By providing a contact name and office number and following up with a phone call a few days after the harness is first applied, clinicians can significantly decrease parents’ anxiety and increase overall compliance.9
Conclusion
Despite recent increased awareness of DDH and the importance of thorough screening programs, hip dysplasia continues to be a frequently missed diagnosis in pediatrics. It is often up to the primary care clinician to screen for, assess, and potentially diagnose DDH. Therefore, a thorough understanding of this condition can promote early detection and diagnosis, with less invasive treatment and a more favorable outcome.
A proper hip examination should be a standard component of all newborn and infant well-child examinations. If DDH is suspected, appropriate referral to a pediatric orthopedic surgeon must be made so that timely treatment can be initiated. Early use of the Pavlik harness is significantly easier than the invasive surgery and prolonged immobilization necessitated by a delayed diagnosis. Whatever the course of treatment required, it is important for clinicians to support the patient and family: training and anticipatory guidance are essential components of DDH management.
A 5-day-old infant was referred to the pediatric orthopedic clinic for evaluation of a left hip “clunk.” She is a firstborn child, born at full term (39 weeks) via cesarean delivery secondary to breech presentation. Her weight at birth was 7 lb 6 oz. The infant was noted to have a left hip clunk during a routine physical examination by her pediatrician, who made a referral to the pediatric orthopedic clinic for possible hip dysplasia. This is the patient’s first visit to the clinic.
There is no family history of hip dysplasia or other orthopedic abnormalities. The infant is a well-appearing, alert female measuring 20.5” in length and weighing 7 lb 4 oz. Vital signs are stable with no abnormality detected. The heart is regular in rate and rhythm, and the chest is clear bilaterally.
No cutaneous abnormalities are noted. The patient is able to move all her extremities spontaneously, and her spine is straight and normal with no evidence of spinal dysraphism. Her feet are normal bilaterally, with full range of motion and no equinovarus or metatarsus adductus deformity.
The neurologic examination is also unremarkable, with normal neonatal reflexes and excellent muscle tone throughout.
Examination of the infant’s hips reveals a positive result on the Barlow test on the left side (the hip can be dislocated). There is also a positive Ortolani sign (the hip can be reduced), with asymmetric thigh skin folds noted (see Figures 1A and 1B, respectively).
Based on these positive physical examination findings, the patient was diagnosed with developmental dysplasia of the hip (DDH). Initial ultrasonography to confirm the diagnosis was not considered necessary, as the physical examination demonstrated obvious instability.1 The infant was placed in a Pavlik harness, which her parents were instructed should be worn full-time (see Figures 2A and 2B). She was scheduled for weekly follow-up visits for adjustments to the harness and serial hip examinations.
At the second follow-up visit, ultrasonography was performed, confirming the presence of dysplasia with decreased femoral head coverage and a steep socket (acetabulum). Use of the Pavlik harness was continued full-time for six weeks.
At age 6 weeks, the infant underwent a follow-up ultrasound to assess for improvement in the degree of dysplasia. The test revealed normal hips bilaterally with no evidence of DDH. Therefore, use of the Pavlik harness was discontinued. The parents were instructed to bring the child back in six months for a repeat clinical examination and an anteroposterior x-ray of the pelvis.1
Discussion
The term developmental dysplasia of the hip (DDH) has replaced the more traditional term congenital hip dislocation because DDH more accurately reflects the variable characteristics that can be seen with this condition. As DDH may not be present at birth, the term congenital is misleading. We now know that DDH may occur in utero, perinatally, or during infancy and childhood.2,3
Generally, DDH is used to describe an abnormal relationship between the femoral head and the acetabulum (see Figure 34). The term represents a wide spectrum of abnormality, as shown in the Graf classification of hips in infants: type I refers to a normal hip; type II, immature development to mild dysplasia; type III, subluxation of the femoral head; and type 4, frank dislocation with severe instability.5
Diagnosing and managing DDH correctly requires the clinician to have a thorough understanding of the normal growth and development that occurs in the hip joint. Embryologically, the joint (including the femoral head and acetabulum) develops from the same primitive mesenchymal cells.6 By 11 to 12 weeks’ gestation, the initial structures of the hip joint are fully formed; theoretically, this is the earliest time at which a dislocation can occur.2,7 DDH that develops at this stage would be called teratologic; this condition is seen most frequently in patients who have underlying neuromuscular conditions, such as myelodysplasia (spina bifida) or arthrogryposis. A typical dislocation takes place during the perinatal period in an infant who is otherwise healthy.2
Etiology
DDH occurs in about 11 of every 1,000 infants, with frank dislocations occurring in one to two infants per 10,000.8 The left hip is involved in approximately 60% of cases, the right in 20%, and both hips in about 20%. In the most common intrauterine fetal position, the left hip is lower than the right (usually abutting the mother’s sacrum) and is often in adduction. This is likely the reason that the left hip is more commonly affected by DDH.
DDH is believed to be multifactorial, with physiologic, genetic, and mechanical factors implicated in the etiology.3 The incidence of DDH varies with factors such as the patient’s age, race, and gender, the experience and training of the examiner, and the diagnostic criteria that are used.
Known risk factors for a positive newborn screening are shown in the table.9,10 It is often helpful for clinicians to remember the “4F” mnemonic associated with DDH: female, firstborn, foot first, and family history.9
There is also an increased risk for DDH in patients with other conditions that are associated with intrauterine crowding. These include congenital muscular torticollis, metatarsus adductus, and congenital dislocation of the knee.2
Physical Examination
All newborn infants should be screened for DDH as part of the initial physical examination, with ultrasonography recommended for infants deemed at high risk for DDH and for those with inconclusive results on examination.1,10,11 Providers should be aware that the newborn hip examination requires a considerable amount of practice and expertise.
A thorough medical history should always be obtained first, including gestational age, presentation (breech vs vertex), type of delivery (cesarean vs vaginal), gender, birth order, family history of DDH, ligamentous laxity, or myopathy.8
The examining clinician begins by placing the infant on a firm, flat surface. The infant should be as relaxed as possible. Next, the clinician observes both lower extremities for asymmetric thigh or buttock skin folds. Bilateral DDH can be very difficult to diagnose on the basis of this examination due to the lack of asymmetry (hips will have symmetric abnormality).
The Galeazzi sign is elicited by placing the infant supine with the hips and knees flexed to 90°.12 With the hips in neutral abduction, the provider should determine whether the knees are at the same height. Unequal knee heights—a positive result for the Galeazzi sign—suggest femoral shortening (apparent leg length discrepancy), which may be explained by a hip dislocation. If both hips are dislocated, a false-negative result will often occur, since both will appear short and there will be no discrepancy.2,12
Among physical examination techniques, the Ortolani and Barlow maneuvers are considered most reliable to detect hip instability in newborns and infants younger than 6 months2,13,14 (review Figures 1A and 1B). The Ortolani test is used to detect the sensation of the dislocated hip reducing into the acetabulum, and the Barlow test elicits the unstable hip dislocating.2 A palpable and occasionally audible clunk is considered a positive result on the Barlow test and usually indicates a diagnosis of DDH.14 High-pitched clicks or snaps frequently occur with hip range-of-motion maneuvers and during Ortolani and Barlow testing. These sounds are often attributed to snapping of the iliotibial band over the greater trochanter and do not usually signify dysplasia.15
Because DDH is a dynamic and evolving process, the physical findings on clinical examination change significantly, depending on the age of the infant or child. As an infant approaches age 3 months, limited hip abduction (especially when asymmetric) is often the most reliable physical examination finding in patients with DDH.12 After age 3 to 4 months, Ortolani and Barlow testing will often produce negative results as progressive soft tissue contractures evolve.
Once a child begins to walk, gait abnormalities (eg, a short-limbed or waddling gait pattern) may raise suspicion for a diagnosis of DDH.7 It has been recommended that evaluation for DDH be performed at each routine office examination until the child is 12 months of age.1
Treatment
The Pavlik harness is considered first-line treatment for DDH in infants younger than 6 months. The harness is a dynamic splint that allows the infant to engage in a sphere of active motion that encourages stabilization and deepening of the socket. The harness is applied with the knees flexed to about 90° and the hips in about 70° of abduction and 100° to 110° of flexion (as shown in Figures 2A and 2B).9
The duration of treatment depends on the infant’s age at presentation and the severity of DDH. Progress is judged by serial examinations and dynamic ultrasounds. The harness is worn full-time until clinical and radiographic examinations both yield normal results. After six weeks of treatment, the hips are examined out of the harness, and a repeat ultrasound is usually obtained. If findings are normal, use of the harness is ordinarily discontinued. Some patients will require harness use for a longer period in cases of delayed development of the acetabulum and/or severe laxity of the ligaments.9
The Pavlik harness is successful more than 90% of the time in newborns with DDH.8 Success rates have been reported as greatest in infants younger than 8 weeks at the time of treatment initiation, those with only one affected hip, and those with less severe disease (Graf types II or III).16
If ultrasonography shows no improvement after two to three weeks, it is usually recommended that the harness be discontinued; most orthopedic surgeons will then proceed with a closed or open reduction and spica body casting. Similarly, when the diagnosis of DDH is delayed until after ages 6 to 8 months, a closed reduction under anesthesia and placement of a spica body cast is usually the recommended treatment to maintain the hip in the reduced position.17,18 Some older children (ages 1 to 5 years) may require bracing, traction, open reduction, and/or femoral or pelvic osteotomy.17,18 It is believed that undiagnosed, untreated DDH can lead to early-onset degenerative hip disease (arthritis).1
Patient/Family Education
The Pavlik harness is most effective when a consistent support system exists to educate parents about the importance of the harness, its care and maintenance, and the consequences of failure. Close monitoring of the infant’s progress is also essential to promoting adherence. Application and removal of the harness should be demonstrated to the parent or caregiver, as well as diapering, dressing, and undressing the infant; they should then be encouraged to practice immediately in the clinic or office.
During visits for harness adjustment, the strap position should be marked with indelible ink, allowing parents to reapply the device correctly, should removal be required (eg, for bathing).9 Ten percent of parents reportedly find reapplying the harness difficult during the first weeks of use. Difficulty in dressing and carrying an infant in a harness, feet slipping out of the harness, and skin irritation have been reported by about one-third of parents.19
Treatment adherence and subsequent success with the Pavlik harness is reported greatest (95%) in patients whose parents engage in demonstrations of harness use and follow instructions precisely.19 By providing a contact name and office number and following up with a phone call a few days after the harness is first applied, clinicians can significantly decrease parents’ anxiety and increase overall compliance.9
Conclusion
Despite recent increased awareness of DDH and the importance of thorough screening programs, hip dysplasia continues to be a frequently missed diagnosis in pediatrics. It is often up to the primary care clinician to screen for, assess, and potentially diagnose DDH. Therefore, a thorough understanding of this condition can promote early detection and diagnosis, with less invasive treatment and a more favorable outcome.
A proper hip examination should be a standard component of all newborn and infant well-child examinations. If DDH is suspected, appropriate referral to a pediatric orthopedic surgeon must be made so that timely treatment can be initiated. Early use of the Pavlik harness is significantly easier than the invasive surgery and prolonged immobilization necessitated by a delayed diagnosis. Whatever the course of treatment required, it is important for clinicians to support the patient and family: training and anticipatory guidance are essential components of DDH management.
1. Karmazyn BK, Gunderman RB, Coley BD, et al; American College of Radiology. ACR appropriateness criteria on developmental dysplasia of the hip—child. J Am Coll Radiol. 2009;6(8):551-557.
2. American Academy of Pediatrics, Committee on Quality Improvement, Subcommittee on Developmental Dysplasia of the Hip. Clinical practice guideline: early detection of developmental dysplasia of the hip. Pediatrics. 2000;105(4 pt 1):896-905.
3. Mencio GA. Developmental dysplasia of the hip. In: Sponseller PD, ed. Orthopaedic Knowledge Update: Pediatrics–2. Rosemont, IL: American Academy of Orthopaedic Surgeons; 2002:161-171.
4. Children’s Hospital at Westmead. Developmental dysplasia of the hip (DDH). www.chw.edu.au/parents/factsheets/developj.htm. Accessed March 26, 2010.
5. Graf R. Classification of hip joint dysplasia by means of sonography. Arch Orthop Trauma Surg. 1984; 102:248-255.
6. Weinstein SL. Developmental hip dysplasia and dislocation. In: Morrissy RT, Weinstein SL, eds. Lovell and Winter’s Pediatric Orthopaedics. 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005:905-956.
7. Aronsson DD, Goldberg MJ, Kling TF Jr, Roy DR. Developmental dysplasia of the hip. Pediatrics. 1994; 94(2 pt 1):201-208.
8. Guille JT, Pizzutillo PD, MacEwan GD. Developmental dysplasia of the hip from birth to six months. J Am Acad Orthop Surg. 2000;8(4):232-242.
9. Hart ES, Albright MB, Rebello GN, Grottkau BE. Developmental dysplasia of the hip: nursing implications and anticipatory guidelines for parents. Orthop Nurs. 2006;25(2):100-109.
10. Dogruel H, Atalar H, Yavus OY, Sayli U. Clinical examination versus ultrasonography in detecting developmental dysplasia of the hip. Int Orthop. 2008; 32(3):415-419.
11. Mahan ST, Katz JN, Kim YJ. To screen or not to screen? A decision analysis of the utility of screening for developmental dysplasia of the hip. J Bone Joint Surg Am. 2009;91(7);1705-1719.
12. Storer SK, Skaggs DL. Developmental dysplasia of the hip. Am Fam Physician. 2006;74(8):1310-1316.
13. Ortolani M. Congenital hip dysplasia in the light of early and very early diagnosis. Clin Orthop Relat Res. 1976;119(1):6-10.
14. Barlow TG. Congenital dislocation of the hip in the newborn. Proc R Soc Med. 1966;59(11 part 1):1103-1106.
15. Bond CD, Hennrikus WL, DellaMaggiore ED. Prospective evaluation of newborn soft-tissue “clicks” with ultrasound. J Pediatr Orthop. 1997;17(2):199-201.
16. Atalar H, Sayli U, Yavuz OY, et al. Indicators of successful use of the Pavlik harness in infants with developmental dysplasia of the hip. Int Orthop. 2007; 31(2):145-150.
17. Rampal V, Sabourin M, Erdeneshoo E, et al. Closed reduction with traction for developmental dysplasia of the hip in children aged between one and five years. J Bone Joint Surg Br. 2008;90-B(7):858-863.
18. Clarke NMP, Sakthivel K. The diagnosis and management of congenital dislocation of the hip. Paediatr Child Health. 2008;18(6):268-271.
19. Hassan FA. Compliance of parents with regard to Pavlik harness treatment in developmental dysplasia of the hip. J Pediatr Orthop. 2009;18(3):111-115.
1. Karmazyn BK, Gunderman RB, Coley BD, et al; American College of Radiology. ACR appropriateness criteria on developmental dysplasia of the hip—child. J Am Coll Radiol. 2009;6(8):551-557.
2. American Academy of Pediatrics, Committee on Quality Improvement, Subcommittee on Developmental Dysplasia of the Hip. Clinical practice guideline: early detection of developmental dysplasia of the hip. Pediatrics. 2000;105(4 pt 1):896-905.
3. Mencio GA. Developmental dysplasia of the hip. In: Sponseller PD, ed. Orthopaedic Knowledge Update: Pediatrics–2. Rosemont, IL: American Academy of Orthopaedic Surgeons; 2002:161-171.
4. Children’s Hospital at Westmead. Developmental dysplasia of the hip (DDH). www.chw.edu.au/parents/factsheets/developj.htm. Accessed March 26, 2010.
5. Graf R. Classification of hip joint dysplasia by means of sonography. Arch Orthop Trauma Surg. 1984; 102:248-255.
6. Weinstein SL. Developmental hip dysplasia and dislocation. In: Morrissy RT, Weinstein SL, eds. Lovell and Winter’s Pediatric Orthopaedics. 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005:905-956.
7. Aronsson DD, Goldberg MJ, Kling TF Jr, Roy DR. Developmental dysplasia of the hip. Pediatrics. 1994; 94(2 pt 1):201-208.
8. Guille JT, Pizzutillo PD, MacEwan GD. Developmental dysplasia of the hip from birth to six months. J Am Acad Orthop Surg. 2000;8(4):232-242.
9. Hart ES, Albright MB, Rebello GN, Grottkau BE. Developmental dysplasia of the hip: nursing implications and anticipatory guidelines for parents. Orthop Nurs. 2006;25(2):100-109.
10. Dogruel H, Atalar H, Yavus OY, Sayli U. Clinical examination versus ultrasonography in detecting developmental dysplasia of the hip. Int Orthop. 2008; 32(3):415-419.
11. Mahan ST, Katz JN, Kim YJ. To screen or not to screen? A decision analysis of the utility of screening for developmental dysplasia of the hip. J Bone Joint Surg Am. 2009;91(7);1705-1719.
12. Storer SK, Skaggs DL. Developmental dysplasia of the hip. Am Fam Physician. 2006;74(8):1310-1316.
13. Ortolani M. Congenital hip dysplasia in the light of early and very early diagnosis. Clin Orthop Relat Res. 1976;119(1):6-10.
14. Barlow TG. Congenital dislocation of the hip in the newborn. Proc R Soc Med. 1966;59(11 part 1):1103-1106.
15. Bond CD, Hennrikus WL, DellaMaggiore ED. Prospective evaluation of newborn soft-tissue “clicks” with ultrasound. J Pediatr Orthop. 1997;17(2):199-201.
16. Atalar H, Sayli U, Yavuz OY, et al. Indicators of successful use of the Pavlik harness in infants with developmental dysplasia of the hip. Int Orthop. 2007; 31(2):145-150.
17. Rampal V, Sabourin M, Erdeneshoo E, et al. Closed reduction with traction for developmental dysplasia of the hip in children aged between one and five years. J Bone Joint Surg Br. 2008;90-B(7):858-863.
18. Clarke NMP, Sakthivel K. The diagnosis and management of congenital dislocation of the hip. Paediatr Child Health. 2008;18(6):268-271.
19. Hassan FA. Compliance of parents with regard to Pavlik harness treatment in developmental dysplasia of the hip. J Pediatr Orthop. 2009;18(3):111-115.
Managing acute upper GI bleeding, preventing recurrences
Upper gastrointestinal (GI) bleeding is common, costly, and potentially life-threatening. It must be managed promptly and appropriately to prevent adverse outcomes.
More people are admitted to the hospital for upper GI bleeding than for congestive heart failure or deep vein thrombosis. In the United States, the annual rate of hospitalization for upper GI bleeding is estimated to be 165 per 100,000—more than 300,000 hospitalizations per year, at a cost of $2.5 billion.1,2
Furthermore, despite advances in therapy, the case-fatality rate has remained unchanged at 7% to 10%.3 This may be because today’s patients are older and have more comorbidities than those in the past.4
CAUSES OF UPPER GI BLEEDING
Peptic ulcers account for about 60% of severe cases of upper GI bleeding,5 and they are the focus of this paper. Fortunately, up to 80% of bleeding ulcers stop bleeding spontaneously without any intervention.6
Gastroduodenal erosions account for about 12%.3
Varices due to cirrhosis are less common but more dangerous. Variceal bleeding accounts for a relatively small percentage (6%) of upper GI bleeding, but the mortality rate from a single episode of variceal bleeding is 30%, and 60% to 70% of patients die within 1 year, mostly of underlying liver disease.
Less frequent causes include Mallory-Weiss tears, erosive duodenitis, Dieulafoy ulcer (a type of vascular malformation), other vascular lesions, neoplasms, aortoenteric fistula, gastric antral vascular ectasia, and prolapse gastropathy.5
HEMATEMESIS AND MELENA
The most common presenting signs of acute upper GI bleeding are hematemesis (vomiting of blood), “coffee grounds” emesis, and melena (tarry black stools). About 30% of patients with bleeding ulcers present with hematemesis, 20% with melena, and 50% with both.7
Hematochezia (red blood in the stool) usually suggests a lower GI source of bleeding, since blood from an upper source turns black and tarry as it passes through the gut, producing melena. However, up to 5% of patients with bleeding ulcers have hematochezia,7 and it indicates heavy bleeding: bleeding of approximately 1,000 mL into the upper GI tract is needed to cause hematochezia, whereas only 50 to 100 mL is needed to cause melena.8,9 Hematochezia with signs and symptoms of hemodynamic compromise such as syncope, postural hypotension, tachycardia, and shock should therefore direct one’s attention to an upper GI source of bleeding.
Nonspecific features include nausea, vomiting, epigastric pain, vasovagal phenomena, and syncope.
WHAT IS THE PATIENT’S RISK?
An assessment of clinical severity is the first critical task, as it helps in planning treatment. Advanced age, multiple comorbidities, and hemodynamic instability call for aggressive treatment. Apart from this simple clinical rule, scoring systems have been developed.
The Rockall scoring system, the most widely used, gives estimates of the risks of recurrent bleeding and death. It is based on the three clinical factors mentioned above and on two endoscopic ones, awarding points for:
- Age—0 points if less than 60; 1 point if 60 to 79; or 2 points if 80 years or older
- Shock—1 point if the pulse is more than 100; 2 points if the systolic blood pressure is less than 100 mm Hg
- Comorbid illness—2 points for ischemic heart disease, congestive heart failure, or other major comorbidity; 3 points for renal failure, hepatic failure, or metastatic disease
- Endoscopic diagnosis—0 points if no lesion found or a Mallory-Weiss tear; 1 point for peptic ulcer, esophagitis, or erosive disease; 2 points for GI malignancy
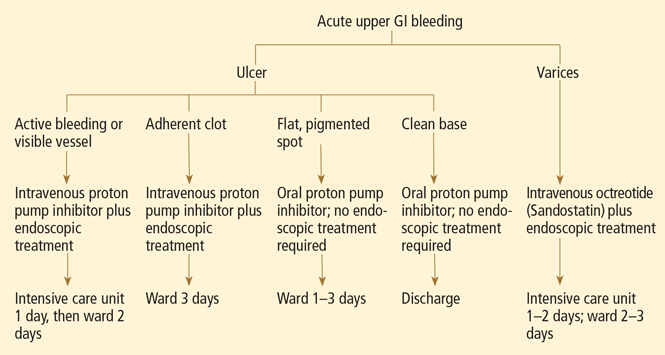
- Endoscopic stigmata or recent hemorrhage—0 points for a clean-based ulcer or flat pigmented spot; 2 points for blood in the upper GI tract, active bleeding, a nonbleeding visible vessel, or adherent clot.
The Rockall score can thus range from 0 to 11 points, with an overall score of 0, 1, or 2 associated with an excellent prognosis.10

Other systems that are used less often include the Baylor severity scale and the Acute Physiology and Chronic Health Evaluation (APACHE) II score.
Does the patient have varices?
All variceal bleeding should be considered severe, since the 1-year death rate is so high (up to 70%). Clues pointing to variceal bleeding include previous variceal bleeding, thrombocytopenia, history of liver disease, and signs of liver disease on clinical examination.
All patients suspected of having bleeding varices should be admitted to the intensive care unit for close monitoring and should be given the highest priority, even if they are hemodynamically stable.
Is the patient hemodynamically stable?
Appropriate hemodynamic assessment includes monitoring of heart rate, blood pressure, and mental status. Tachycardia at rest, hypotension, and orthostatic changes in vital signs indicate a considerable loss of blood volume. Low urine output, dry mucous membranes, and sunken neck veins are also useful signs. (Tachycardia may be blunted if the patient is taking a beta-blocker.)
If these signs of hypovolemia are present, the initial management focuses on treating shock and on improving oxygen delivery to the vital organs. This involves repletion of the intravascular volume with intravenous infusions or blood transfusions. Supplemental oxygen also is useful, especially in elderly patients with heart disease.12
Inspection of nasogastric aspirate
In the initial assessment, it is useful to insert a nasogastric tube and inspect the aspirate. If it contains bright red blood, the patient needs an urgent endoscopic evaluation and an intensive level of care13,14; if it contains coffee-grounds material, the patient needs to be admitted to the hospital and to undergo endoscopic evaluation within 24 hours.
However, a normal aspirate does not rule out upper GI bleeding. Aljebreen et al15 found that 15% of patients with upper GI bleeding and normal nasogastric aspirate still had high-risk lesions (ie, visible bleeding or nonbleeding visible vessels) on endoscopy.
ACID-SUPPRESSION HELPS ULCERS HEAL
Acid and pepsin interfere with the healing of ulcers and other nonvariceal upper GI lesions. Further, an acidic environment promotes platelet disaggregation and fibrinolysis and impairs clot formation.16 This suggests that inhibiting gastric acid secretion and raising the gastric pH to 6 or higher may stabilize clots. Moreover, pepsinogen in the stomach is converted to its active form (pepsin) if the pH is less than 4. Therefore, keeping the pH above 4 keeps pepsinogen in an inactive form.
Histamine-2 receptor antagonists
Histamine-2 receptor antagonists were the first drugs to inhibit acid secretion, reversibly blocking histamine-2 receptors on the basolateral membrane of parietal cells. However, these drugs did not prove very useful in managing upper GI bleeding in clinical trials.17,18 In their intravenous form, they often fail to keep the gastric pH at 6 or higher, due to tachyphylaxis.19 The use of this class of drugs has declined in favor of proton pump inhibitors.
Proton pump inhibitors
Proton pump inhibitors reduce both basal and stimulated acid secretion by inhibiting hydrogen-potassium adenosine triphosphatase, the proton pump of the parietal cell.
Multiple studies have shown that proton pump inhibitors raise the gastric pH and keep it high. For example, an infusion of omeprazole (Prilosec) can keep the gastric pH above 6 for 72 hours without inducing tachyphylaxis.20,21
Started after endoscopy. Randomized controlled trials have found proton pump inhibitors to be effective when given in high doses intravenously for 72 hours after successful endoscopic treatment of bleeding ulcers with high-risk endoscopic signs, such as active bleeding or nonbleeding visible vessels.22,23
A meta-analysis indicated that these drugs decrease the incidence of recurrent peptic ulcer bleeding, the need for blood transfusions, the need for surgery, and the duration of hospitalization, but not the mortality rate.24,25 These studies also illustrate the benefit of following up endoscopic treatment to stop the bleeding with an intravenous infusion of a proton pump inhibitor.
The recommended dose of omeprazole for patients with high-risk findings on endoscopy is an 80-mg bolus followed by an 8-mg/hour infusion for 72 hours. After the patient’s condition stabilizes, oral therapy can be substituted for intravenous therapy. In patients with low-risk endoscopic findings (a clean-based ulcer or flat spot), oral proton pump inhibitors in high doses are recommended.
In either case, after the initial bleeding is treated endoscopically and hemostasis is achieved, a proton pump inhibitor is recommended for 6 to 8 weeks, or longer if the patient is also positive for Helicobacter pylori or is on daily treatment with aspirin or a nonsteroidal anti-inflammatory drug (NSAID) that is not selective for cyclo-oxygenase 2 (see below).
Started before endoscopy, these drugs reduced the frequency of actively bleeding ulcers, the duration of hospitalization, and the need for endoscopic therapy in a randomized controlled trial.26 A meta-analysis found that significantly fewer patients had signs of recent bleeding on endoscopy if they received a proton pump inhibitor 24 to 48 hours before the procedure, but it did not find any significant difference in important clinical outcomes such as death, recurrent bleeding, or surgery.27 Nevertheless, we believe that intravenous proton pump inhibitor therapy should be started before endoscopy in patients with upper GI bleeding.
Somatostatin analogues
Octreotide (Sandostatin), an analogue of the hormone somatostatin, decreases splanchnic blood flow, decreases secretion of gastric acid and pepsin, and stimulates mucus production. Although it is beneficial in treating upper GI bleeding due to varices, its benefit has not been confirmed in patients with nonvariceal upper GI bleeding.
A meta-analysis revealed that outcomes were better with high-dose intravenous proton pump inhibitor therapy than with octreotide when these drugs were started after endoscopic treatment of acute peptic ulcer bleeding.28 Nevertheless, octreotide may be useful in patients with uncontrolled nonvariceal bleeding who are awaiting endoscopy, since it is relatively safe to use.
ALL PATIENTS NEED ENDOSCOPY
All patients with upper GI bleeding need an upper endoscopic examination to diagnose and assess the risk posed by the bleeding lesion and to treat the lesion, reducing the risk of recurrent bleeding.
How urgently does endoscopy need to be done?
Endoscopy within the first 24 hours of upper GI bleeding is considered the standard of care. Patients with uncontrolled or recurrent bleeding should undergo endoscopy on an urgent basis to control the bleeding and reduce the risk of death.
However, how urgently endoscopy needs to be done is often debated. A multicenter randomized controlled trial compared outcomes in patients who underwent endoscopy within 6 hours of coming to the emergency department vs within 24 hours after the initial evaluation. The study found no significant difference in outcomes between the two groups; however, the group that underwent endoscopy sooner needed fewer transfusions.29
For a better view of the stomach
Gastric lavage improves the view of the gastric fundus but has not been proven to improve outcome.30
Promotility agents such as erythromycin and metoclopramide (Reglan) are also used to empty the stomach for better visualization.31–35 Erythromycin has been shown to improve visualization, shorten the procedure time, and prevent the need for additional endoscopy attempts in two randomized controlled studies.33,34 Furthermore, a cost-effectiveness study confirmed that giving intravenous erythromycin before endoscopy for acute upper GI bleeding saved money and resulted in an increase in quality-adjusted life-years.35
Endoscopy to diagnose bleeding and assess risk
Upper endoscopy is 90% to 95% diagnostic for acute upper GI bleeding.36
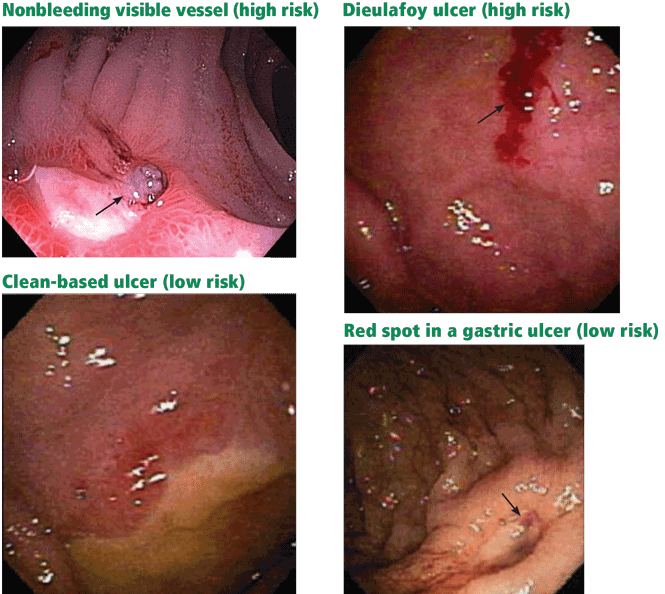
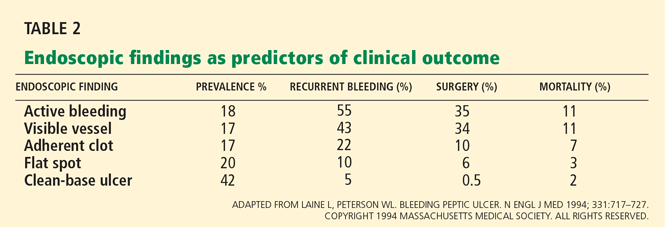
Patients at high risk (ie, older than 60 years, with severe comorbidity, or hemodynamically compromised) who have active bleeding (ie, witnessed hematemesis, red blood per nasogastric tube, or fresh blood per rectum) or a nonbleeding visible vessel should be admitted to a monitored bed or intensive care unit. Observation in a regular medical ward is appropriate for high-risk patients found to have an adherent clot. Patients with low-risk findings (eg, a clean ulcer base) are at low risk of recurrent bleeding and may be considered for early hospital discharge with appropriate outpatient follow-up.
Endoscopy to treat bleeding
About 25% of endoscopic procedures performed for upper GI bleeding include some type of treatment,39 such as injections of epinephrine, normal saline, or sclerosants; thermal cautery; argon plasma coagulation; electrocautery; or application of clips or bands. They are all equally effective, and combinations of these therapies are more effective than when they are used individually. A recent meta-analysis found dual therapy to be superior to epinephrine monotherapy in preventing recurrent bleeding, need for surgery, and death.40

How to manage adherent clots is controversial, but recent studies have revealed a significant benefit from removing them and treating the underlying lesions compared with drug therapy alone.43,45
Flat, pigmented spots and nonbleeding ulcers with a clean base do not require endoscopic treatment because the risk of recurrent bleeding is low.
Endoscopic therapy stops the bleeding in more than 90% of patients, but bleeding recurs after endoscopic therapy in 10% to 25%.46 Reversal of any severe coagulopathy with transfusions of platelets or fresh frozen plasma is essential for endoscopic hemostasis. However, coagulopathy at the time of initial bleeding and endoscopy does not appear to be associated with higher rates of recurrent bleeding following endoscopic therapy for nonvariceal upper GI bleeding.47
Patients with refractory bleeding are candidates for angiography or surgery. However, even when endoscopic hemostasis fails, endoscopy is important before angiography or surgery to pinpoint the site of bleeding and diagnose the cause.
A second endoscopic procedure is generally not recommended within 24 hours after the initial procedure.48 However, it is appropriate in cases in which clinical signs indicate recurrent bleeding or if hemostasis during the initial procedure is questionable. A meta-analysis found that routinely repeating endoscopy reduces the rate of recurrent bleeding but not the need for surgery or the risk of death.49
ALL PATIENTS SHOULD BE ADMITTED
VARICEAL BLEEDING
Variceal bleeding, a severe outcome of portal hypertension secondary to cirrhosis, carries a 6-week mortality rate of 10% to 20%.50 In view of the risk, primary prevention is indicated in patients with high-risk varices.
The mainstays of primary and secondary prevention are the nonselective beta-blockers such as nadolol (Corgard) and propranolol (Inderal). Several randomized controlled trials have shown lower rates of recurrent bleeding and death with propranolol or nadolol than with placebo.51 In doses that decrease the heart rate by 25%, beta-blockers have been shown to delay and decrease variceal hemorrhage. However, most patients require prophylactic endoscopic variceal ligation because they cannot tolerate beta-blocker therapy.
In suspected acute variceal bleeding, a somatostatin analogue should be started to decrease the portal pressure, and antibiotics should be started to reduce the risks of infection and death. Vasoactive drugs, ie, somatostatin analogues, should be started before endoscopy and continued for 5 days to reduce the chances of recurrent bleeding.52,53
Terlipressin is the only drug proven to improve the odds of survival in acute variceal bleeding. Although widely used in Europe, it has not been approved for use in the United States.
Octreotide, another option, improves hemostasis to the same extent, although it does not increase the survival rate.54,55 The recommended dose of octreotide for patients with variceal bleeding is a 50-μg intravenous bolus, followed by a 50-μg/hour infusion for 5 days.
Combining endoscopic and drug therapy improves the chances of stopping the bleeding and reduces the risk of recurrent bleeding compared with endoscopic therapy alone.56
Transjugular intrahepatic portosystemic shunting is indicated in recurrent variceal hemorrhage or in those with initial bleeding that is refractory to standard medical and endoscopic therapy. It is not the primary therapy because it doubles the risk of encephalopathy and has a high stent occlusion rate (up to 60%, lower with covered stents).
GI BLEEDING CAN CAUSE ACUTE MYOCARDIAL INFARCTION
The simultaneous presentation of acute myocardial infarction (MI) and GI hemorrhage is very serious and unfortunately common.
An acute MI occurring simultaneously with or after GI bleeding is usually precipitated by massive bleeding causing hypovolemia, hemodynamic compromise, and hypoperfusion. Conversely, the anticoagulant, antiplatelet, or thrombolytic drugs given to treat MI can precipitate GI bleeding (see below).
This distinction is important because the two scenarios have different clinical courses and prognoses. GI bleeding that precipitates an acute MI tends to be massive, whereas GI bleeding after treatment of acute MI tends to be self-limited and often resolves with reversal of underlying coagulopathy.57
Endoscopy carries a higher than average risk in patients with recent acute MI, with all-cause mortality rates as high as 1%.58 (The usual rate is 0.0004%.59) Nevertheless, endoscopy can be safely performed early on in patients with acute MI if it is done under strict monitoring in a coronary care unit.
Several studies have shown that MI patients who present with upper GI bleeding as the inciting event or patients with acute MI who are vomiting blood or who are hemodynamically unstable due to GI bleeding are significantly more likely to have a high-risk lesion and so have the greatest need for endoscopic therapy. Therefore, endoscopic intervention may be offered to MI patients at high risk who have been started on antiplatelet agents.
WARFARIN CAN PRECIPITATE BLEEDING
Acute upper GI bleeding can be a severe complication of long-term oral anticoagulation, not because the drugs cause ulcers, but rather because they exacerbate ulcers that are already present.60 Therefore, when starting warfarin (Coumadin), patients should be evaluated to determine if they have other risk factors for GI bleeding, such as ulcers.
The number of people presenting with upper GI bleeding while on warfarin therapy is increasing because of the expanding indications for long-term anticoagulation therapy, such as atrial fibrillation and deep venous thrombosis.
The risk of GI bleeding in patients who use oral anticoagulants is estimated to be 2.3 to 4.9 times higher than in nonusers.61
The goal international normalized ratio (INR) for patients on warfarin therapy is usually 2.0 to 3.0. Recent studies found that endoscopy can be safely performed in patients with acute GI bleeding whose INR is between 2.0 and 3.0.62,63 Some suggest that both the length of warfarin therapy and the INR affect the risk of bleeding.64,65
Managing patients with an INR higher than 3.0 who have an episode of GI bleeding is always a challenge. It is not uncommon to find pathologic lesions causing GI bleeding in patients who are on warfarin with a supratherapeutic INR, and thus, endoscopy is indicated. However, before endoscopy, reversal of anticoagulation should be considered.
BLEEDING IN PATIENTS ON ANTIPLATELET DRUGS
Aspirin
Aspirin decreases production of prostaglandins in the GI tract, thereby decreasing the protective and restorative properties of the gastric and duodenal mucosa and predisposing to ulcers and bleeding.
The higher the aspirin dose, the higher the risk. Aspirin doubles the risk of upper GI bleeding at daily doses of 75 mg and quadruples it at doses of 300 mg.66 Even doses as low as 10 mg can decrease gastric mucosal prostaglandin production.67 Thus, it appears that there is no risk-free dose of aspirin, and enteric-coated or buffered formulations do not appear to reduce the risk.68–70
The most important risk factor for upper GI bleeding in patients taking aspirin is a history of peptic ulcer bleeding. Approximately 15% of aspirin users who have bleeding from ulcers have recurrent bleeding within 1 year.71
As aspirin-induced GI bleeding becomes more common, health care providers often feel caught between the GI risk and the cardiovascular benefit. When considering whether to discontinue antiplatelet therapy, a cardiologist should be consulted along with a gastroenterologist to weigh the risks of GI bleeding vs thrombosis. To date, there have been no clinical trials published to suggest when antiplatelet therapy should be stopped to optimize GI and cardiovascular outcomes. An alternative is to replace aspirin with another antiplatelet drug that does not induce ulcers.
Clopidogrel
Clopidogrel (Plavix) is recommended for hospitalized patients with acute coronary syndrome who cannot tolerate the GI side effects of aspirin, according to the joint guidelines of the American College of Cardiology and the American Heart Association, with the highest level of evidence.72 This recommendation was largely based on the safety data from the CAPRIE (Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events) trial, in which the incidence of major GI bleeding was lower in the clopidogrel group (0.52%) than in the aspirin group (0.72%; P < .05).73
Aspirin plus a proton pump inhibitor
Patients who have had an episode of upper GI bleeding and who need long-term aspirin therapy should also receive a proton pump inhibitor indefinitely to prevent ulcer recurrence.
In a recent double-blind randomized controlled trial in patients with a history of aspirin-induced bleeding, the combination of low-dose aspirin plus esomeprazole (Nexium) twice a day was superior to clopidogrel by itself in terms of the rate of recurrent bleeding (0.7% vs 8.6%; P < .05).74 A similar trial showed nearly identical results: 0% upper GI bleeding in the group receiving aspirin plus esomeprazole 20 mg daily, vs 13.6% in the clopidogrel group (P = .0019).75 These studies suggest that a once-daily proton pump inhibitor combined with aspirin is a safer alternative than clopidogrel alone.
Clopidogrel plus a proton pump inhibitor
Interestingly, recent studies have shown that omeprazole decreases the antiplatelet effect of clopidogrel, possibly by inhibiting the CYP2C19 enzyme.76 However, concomitant use of pantoprazole (Protonix), lansoprazole (Prevacid), and esomeprazole did not have this effect, suggesting that although all proton pump inhibitors are metabolized to a varying degree by CYP2C19, the interaction between proton pump inhibitors and clopidogrel is not a class effect.77–79 Therefore, pantoprazole, lansoprazole, and esomeprazole may be the appropriate proton pump inhibitors to use with clopidogrel in patients who have a clear indication for the medication, consistent with current guideline recommendations.
Helicobacter pylori infection in antiplatelet drug users
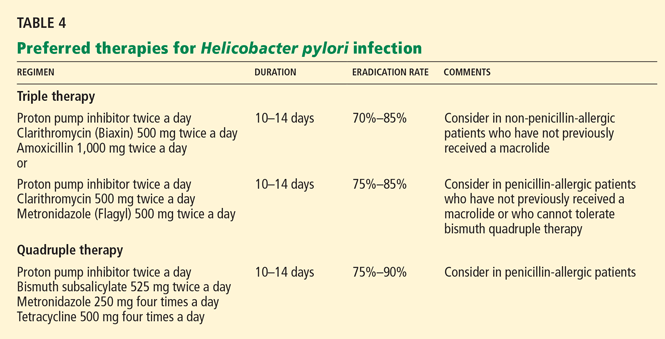
TREATMENT AND PREVENTION OF NSAID-RELATED GI INJURY
About 1 in 20 users of NSAIDs develop GI complications and ulcers of varying degrees of severity, as do one in seven NSAID users over the age of 65. In fact, NSAID use accounts for 30% of hospitalizations for upper GI bleeding and deaths from this cause.82–85 In addition, approximately 15% to 30% of NSAID users have clinically silent but endoscopically evident peptic ulcers.86
NSAIDs contribute to ulcer development by depleting prostaglandins. Thus, misoprostol (Cytotec), a synthetic prostaglandin, has been used to reduce this side effect.
In a clinical trial, misoprostol reduced the incidence of NSAID-associated GI complications by 40%.87 Furthermore, it has been shown to be better than placebo in preventing recurrent gastric ulcers in patients with a history of gastric ulcer who were receiving low-dose aspirin.88
However, misoprostol is rarely used because it can cause diarrhea and abdominal cramping. Rather, the preferred drugs for preventing and treating NSAID- and aspirin-related GI lesions are proton pump inhibitors.
Numerous clinical trials using endoscopic end points showed that proton pump inhibitors in standard doses significantly reduce the incidence of ulcers associated with the use of NSAIDs.89 Proton pump inhibitor therapy has achieved a significant reduction in relative risk of upper GI bleeding in patients who received low-dose aspirin therapy, as confirmed by epidemiologic studies.90,91 The number of NSAID-related ulcers found on endoscopy could be reduced by an estimated 90% simply by using proton pump inhibitors.92
- Lewis JD, Bilker WB, Brensinger C, Farrar JT, Strom BL. Hospitalization and mortality rates from peptic ulcer disease and GI bleeding in the 1990s: relationship to sales of non-steroidal anti-inflammatory drugs and acid suppression medications. Am J Gastroenterol 2002; 97:2540–2549.
- Viviane A, Alan BN. Estimates of costs of hospital stays for variceal and nonvariceal upper gastrointestinal bleeding in the United States. Value Health 2008; 11:1–3.
- Yavorski RT, Wong RK, Maydonovitch C, Battin LS, Furnia A, Amundson DE. Analysis of 3,294 cases of upper gastrointestinal bleeding in military medical facilities. Am J Gastroenterol 1995; 90:568–573.
- Kaplan RC, Heckbert SR, Koepsell TD, et al. Risk factors for hospitalized gastrointestinal bleeding among older persons. Cardiovascular Health Study Investigators. J Am Geriatr Soc 2001; 49:126–133.
- Longstreth GF. Epidemiology of hospitalization for acute upper gastrointestinal hemorrhage: a population-based study. Am J Gastroenterol 1995; 90:206–210.
- Laine L, Peterson WL. Bleeding peptic ulcer. N Engl J Med 1994; 331:717–727.
- Wara P, Stodkilde H. Bleeding pattern before admission as guideline for emergency endoscopy. Scand J Gastroenterol 1985; 20:72–78.
- Jensen DM, Machicado GA. Diagnosis and treatment of severe hematochezia. The role of urgent colonoscopy after purge. Gastroenterology 1988; 95:1569–1574.
- Daniel WA, Egan S. The quantity of blood required to produce a tarry stool. J Am Med Assoc 1939; 113:2232.
- Rockall TA, Logan RF, Devlin HB, Northfield TC. Risk assessment after acute upper gastrointestinal hemorrhage. Gut 1996; 38:316–321.
- Blatchford O, Murray WR, Blatchford M. A risk score to predict need for treatment for upper-gastrointestinal hemorrhage. Lancet 2000; 356:1318–1321.
- Gralnek IM, Barkun AN, Bardou M. Management of acute bleeding from a peptic ulcer. N Engl J Med 2008; 359:928–937.
- Silverstein FE, Gilbert DA, Tedesco FJ, Buenger NK, Persing J. The national ASGE survey on upper gastrointestinal bleeding II. Clinical prognostic factors. Gastrointest Endosc 1981; 27:80–93.
- Corley DA, Stefan AM, Wolf M, Cook EF, Lee TH. Early indicators of prognosis in upper gastrointestinal hemorrhage. Am J Gastroenterol 1998; 93:336–340.
- Aljebreen AM, Fallone CA, Barkun AN. Nasogastric aspirate predicts high-risk endoscopic lesions in patients with acute upper-GI bleeding. Gastrointest Endosc 2004; 59:172–178.
- Barkun AN, Cockeram AW, Plourde V, Fedorak RN. Review article: acid suppression in non-variceal acute upper gastrointestinal bleeding. Aliment Pharmacol Ther 1999; 13:1565–1584.
- Levine JE, Leontiadis JI, Sharma VK, Howden CW. Meta-analysis: the efficacy of intravenous H2-receptor antagonists in bleeding peptic ulcer. Aliment Pharmacol Ther 2002; 16:1137–1142.
- Walt RP, Cottrell J, Mann SG, Freemantle NP, Langman MJ. Continuous intravenous famotidine for hemorrhage from peptic ulcer. Lancet 1992; 340:1058–1062.
- Labenz J, Peitz U, Leusing C, Tillenburg B, Blum AL, Börsch G. Efficacy of primed infusion with high dose ranitidine and omeprazole to maintain high intragastric pH in patients with peptic ulcer bleeding: a prospective randomized controlled study. Gut 1997; 40:36–41.
- Merki HS, Wilder-Smith CH. Do continuous infusions of omeprazole and ranitidine retain their effect with prolonged dosing? Gastroenterology 1994; 106:60–64.
- Netzer P, Gaia C, Sandoz M, et al. Effect of repeated injection and continuous infusion of omeprazole and ranitidine on intragastric pH over 72 hours. Am J Gastroenterol 1999; 94:351–357.
- Lin HJ, Lo WC, Cheng YC, Perng CL. Role of intravenous omeprazole in patients with high-risk peptic ulcer bleeding after successful endoscopic epinephrine injection: a prospective randomized comparative trial. Am J Gastroenterol 2006; 101:500–505.
- Lau JY, Sung JJ, Lee KK, et al. Effects of intravenous omeprazole on recurrent bleeding after endoscopic treatment of bleeding peptic ulcers. N Engl J Med 2000; 343:310–316.
- Leontiadis GI, Sharma VK, Howden CW. Proton pump inhibitor treatment for acute peptic ulcer bleeding. Cochrane Database Syst Rev 2006;CD002094.
- Andriulli A, Annese V, Caruso N, et al. Proton-pump inhibitors and outcome of endoscopic hemostasis in bleeding peptic ulcers: a series of meta-analyses. Am J Gastroenterol 2005; 100:207–219.
- Lau JY, Leung WK, Wu JC, et al. Omeprazole before endoscopy in patients with gastrointestinal bleeding. N Engl J Med 2007; 356:1631–1640.
- Dorward S, Sreedharan A, Leontiadis GI, Howden CW, Moayyedi P, Forman D. Proton pump inhibitor treatment initiated prior to endoscopic diagnosis in upper gastrointestinal bleeding. Cochrane Database Syst Rev 2006;CD005415.
- Bardou M, Toubouti Y, Benhaberou-Brun D, Rahme E, Barkun AN. Meta-analysis: proton-pump inhibition in high-risk patients with acute peptic ulcer bleeding. Aliment Pharmacol Ther 2005; 21:677–686.
- Bjorkman DJ, Zaman A, Fennerty MB, Lieberman D, Disario JA, Guest-Warnick G. Urgent vs elective endoscopy for acute non-variceal upper-GI bleeding: an effectiveness study. Gastointest Endosc 2004; 60:1–8.
- Lee SD, Kearney DJ. A randomized controlled trial of gastric lavage prior to endoscopy for acute upper gastrointestinal bleeding. J Clin Gastroenterol 2004; 38:861–865.
- Tack J, Janssens J, Vantrappen G, et al. Effect of erythromycin on gastric motility in controls and in diabetic gastroparesis. Gastroenterology 1992; 103:72–79.
- Xynos E, Mantides A, Papageorgiou A, Fountos A, Pechlivanides G, Vassilakis JS. Erythromycin accelerates delayed gastric emptying of solids in patients after truncal vagotomy and pyloroplasty. Eur J Surg 1992; 158:407–411.
- Coffin B, Pocard M, Panis Y, et al; Groupe des endoscopistes de garde á l’AP-HP. Erythromycin improves the quality of EGD in patients with acute upper GI bleeding: a randomized controlled study. Gastrointest Endosc 2002; 56:174–179.
- Frossard JL, Spahr L, Queneau PE, et al. Erythromycin intravenous bolus infusion in acute upper gastrointestinal bleeding: a randomized, controlled, double-blind trial. Gastroenterology 2002; 123:17–23.
- Winstead NS, Wilcox CM. Erythromycin prior to endoscopy for acute upper gastrointestinal hemorrhage: a cost-effectiveness analysis. Aliment Pharmacol Ther 2007; 26:1371–1377.
- Chak A, Cooper GS, Lloyd LE, Kolz CS, Barnhart BA, Wong RC. Effectiveness of endoscopy in patients admitted to the intensive care unit with upper GI hemorrhage. Gastrointest Endosc 2001; 53:6–13.
- Lau JY, Chung SC, Leung JW, Lo KK, Yung MY, Li AK. The evolution of stigmata of hemorrhage in bleeding peptic ulcers: a sequential endoscopic study. Endoscopy 1998; 30:513–518.
- Chung IK, Kim EJ, Lee MS, et al. Endoscopic factors predisposing to rebleeding following endoscopic hemostasis in bleeding peptic ulcers. Endoscopy 2001; 33:969–975.
- Elta GH. Acute nonvariceal upper gastrointestinal hemorrhage. Curr Treat Options Gastroenterol 2002; 5:147–152.
- Marmo R, Rotondano G, Piscopo R, Bianco MA, D’Angella R, Cipolletta L. Dual therapy versus monotherapy in the endoscopic treatment of high-risk bleeding ulcers: a meta-analysis of controlled trials. Am J Gastroenterol 2007; 102:279–289.
- Kovacs TO, Jensen DM. Recent advances in the endoscopic diagnosis and therapy of upper gastrointestinal, small intestinal, and colonic bleeding. Med Clin North Am 2002; 86:1319–1356.
- Kovacs TO, Jensen DM. Endoscopic treatment of ulcer bleeding. Curr Treat Options Gastroenterol 2007; 10:143–148.
- Jensen DM, Kovacs TO, Jutabha R, et al. Randomized trial of medical or endoscopic therapy to prevent recurrent ulcer hemorrhage in patients with adherent clots. Gastroenterology 2002; 123:407–413.
- Jensen DM, Machicado GA. Endoscopic hemostasis of ulcer hemorrhage with injection, thermal, and combination methods. Techniques Gastrointest Endosc 2005; 7:124–131.
- Bleau BL, Gostout CJ, Sherman KE, et al. Recurrent bleeding from peptic ulcer associated with adherent clot: a randomized study comparing endoscopic treatment with medical therapy. Gastrointest Endosc 2002; 56:1–6.
- Lau JY, Sung JJ, Lam YH, et al. Endoscopic retreatment compared with surgery in patients with recurrent bleeding after initial endoscopic control of bleeding ulcers. N Engl J Med 1999; 340:751–756.
- Wolf AT, Wasan SK, Saltzman JR. Impact of anticoagulation on rebleeding following endoscopic therapy for nonvariceal upper gastrointestinal hemorrhage. Am J Gastroenterol 2007; 102:290–296.
- Barkun A, Bardou M, Marshall JK; Nonvariceal Upper GI Bleeding Consensus Conference Group. Consensus recommendations for managing patients with nonvariceal upper gastrointestinal bleeding. Ann Intern Med 2003; 139:843–857.
- Marmo R, Rotondano G, Bianco MA, Piscopo R, Prisco A, Cipolletta L. Outcome of endoscopic treatment for peptic ulcer bleeding: is a second look necessary? A meta-analysis. Gastrointest Endosc 2003; 57:62–67.
- Dell’Era A, deFrancis R, Iannuzzi F. Acute variceal bleeding: pharmacological treatment and primary/secondary prophylaxis. Best Pract Res Clin Gastroenterol 2008; 22:279–294.
- Jalan R, Hayes PC. UK guidelines on the management of variceal hemorrhage in cirrhotic patients. British Society of Gastroenterology. Gut 2000; 46( suppl 3–4):III1–III15.
- Bernard B, Lebrec D, Mathurin P, Opolon P, Poynard T. Beta-adrenergic antagonists in the prevention of gastrointestinal rebleeding in patients with cirrhosis: a meta-analysis. Hepatology 1997; 25:63–70.
- De Franchis R. Evolving consensus in portal hypertension. Report of the Baveno IV consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol 2005; 43:167–176.
- Levacher S, Letoumelin P, Pateron D, Blaise M, Lapandry C, Pourriat JL. Early administration of terlipressin plus glyceryl trinitrate to control active upper gastrointestinal bleeding in cirrhotic patients. Lancet 1995; 346:865–868.
- Abraldes JG, Bosch J. Somatostatin and analogues in portal hypertension. Hepatology 2002; 35:1305–1312.
- Banares R, Albillos A, Rincon D, et al. Endoscopic treatment versus endoscopic plus pharmacological treatment for acute variceal bleeding: a meta analysis. Hepatology 2002; 35:609–615.
- Cappell M. Gastrointenstinal bleeding associated with myocardial infarction. Gastroenterol Clin North Am 2000; 29:423–444.
- Lin S, Konstance R, Jollis J, Fisher DA. The utility of upper endoscopy in patients with concomitant upper gastrointestinal bleeding and acute myocardial infarction. Dig Dis Sci 2006; 51:2377–2383.
- Silvis SE, Nebel O, Rogers G, Sugawa C, Mandelstam P. Endoscopic complications. Results of the 1974 American Society for Gastrointestinal Endoscopy Survey. JAMA 1976; 235:928–930.
- Lanas A, Garcia-Rodriguez LA, Arroyo MT, et al; Investigators of the Asociación Española de Gastroenterología (AEG). Effect of antisecretory drugs and nitrates on the risk of ulcer bleeding associated with nonsteroidal anti-inflammatory drugs, antiplatelet agents, and anticoagulants. Am J Gastroenterol 2007; 102:507–515.
- Shorr RI, Ray WA, Daugherty JR, Griffin MR. Concurrent use of nonsteroidal anti-inflammatory drugs and oral anticoagulants places elderly persons at high risk for hemorrhagic peptic ulcer disease. Arch Intern Med 1993; 153:1665–1670.
- Tabibian N. Acute gastrointestinal bleeding in anticoagulated patients: a prospective evaluation. Am J Gastroenterol 1989; 84:10–12.
- Choudari CP, Rajgopal C, Palmer KR. Acute gastrointestinal hemorrhage in anticoagulated patients: diagnoses and response to endoscopic treatment. Gut 1994; 35:464–466.
- Petty GW, Brown RD, Whisnant JP, Sicks JD, O’Fallon WM, Wiebers DO. Frequency of major complications of aspirin, warfarin, and intravenous heparin for secondary stroke prevention: a population-based study. Ann Intern Med 1999; 130:14–22.
- Landefeld CS, Rosenblatt MW, Goldman L. Bleeding in outpatients treated with warfarin: relation to the prothrombin time and important remediable lesions. Am J Med 1989; 87:153–159.
- Weil J, Colin-Jones D, Langman M, et al. Prophylactic aspirin and risk of peptic ulcer bleeding. BMJ 1995; 310:827–830.
- Cryer B, Feldman M. Effects of very low dose daily, long-term aspirin therapy on gastric, duodenal, and rectal prostaglandin levels and on mucosal injury in healthy humans. Gastroenterology 1999; 117:17–25.
- De Abajo FJ, Garcia Rodriguez LA. Risk of upper gastrointestinal bleeding and perforation associated with low-dose aspirin as plain and enteric-coated formulations. BMC Clin Pharmacol 2001; 1:1.
- Kelly JP, Kaufman DW, Jurgelon JM, Sheehan J, Koff RS, Shapiro S. Risk of aspirin-associated major upper gastrointestinal bleeding with enteric coated or buffered product. Lancet 1996; 348:1413–1416.
- Garcia Rodriguez LA, Hernandez-Diaz S, De Abajo FJ. Association between aspirin and upper gastrointestinal complications: systematic review of epidemiological studies. Br J Clin Pharmacol 2001; 52:563–571.
- Wilcox CM, Ladabaum U. A patient with high risk of gastrointestinal bleeding requiring nonsteroidal anti-inflammatory drugs. Clin Gastroenterol Hepatol 2006; 4:1090–1093.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996; 348:1329–1339.
- Chan FK, Ching JY, Hung LC, et al. Clopidogrel versus aspirin and esomeprazole to prevent recurrent ulcer bleeding. N Engl J Med 2005; 352:238–244.
- Lai KC, Chu KM, Hui WM, et al. Esomeprazole with aspirin versus clopidogrel for prevention of recurrent gastrointestinal ulcer complications. Clin Gastroenterol Hepatol 2006; 4:860–865.
- Ho MP, Maddox TM, Wang L, et al. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. JAMA 2009; 301:937–944.
- Siller-Matula JM, Spiel AO, Lang IM, Kreiner G, Christ G, Jilma B. Effects of pantoprazole and esomeprazole on platelet inhibition by clopidogrel. Am Heart J 2009; 157:148.e1–e5.
- Small DS, Farid NA, Payne CD, et al. Effects of proton pump inhibitor lansoprazole on the pharmacokinetics and pharmacodynamics of prasugel and clopidogrel. J Clin Pharmacol 2008; 48:475–484.
- Ishizaki T, Horai Y. Review article: cytochrome P450 and the metabolism of proton pump inhibitors—emphasis on rabeprazole. Aliment Pharacol Ther 1999; 13 (suppl 3):27–36.
- Lanas A, Fuentes J, Benito R, Serrano P, Bajador E, Sainz R. Helicobacter pylori increases the risk of upper gastrointestinal bleeding in patients taking low-dose aspirin. Aliment Pharmacol Ther 2002; 16:779–786.
- Chan FK. NSAID-Induced peptic ulcers and Helicobacter pylori infection: implications for patient management. Drug Saf 2005; 28:287–300.
- Bombardier V, Laine L, Reicin A, et al; VIGOR Study Group. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis: VIGOR Study Group. N Eng J Med 2000; 343:1520–1528.
- Griffin MR, Ray WA, Schaffner W. Nonsteroidal anti-inflammatory drug use and death from peptic ulcer in elderly persons. Ann Intern Med 1988; 109:359–363.
- Griffin MR, Piper JM, Daugherty JR, Snowden M, Ray WA. Nonsteroidal anti-inflammatory drug use and increased risk for peptic ulcer disease in elderly persons. Ann Intern Med 1991; 114:257–263.
- Smalley WE, Ray WA, Daugherty JR, Griffin MR. Nonsteroidal anti-inflammatory drugs and the incidence of hospitalizations for peptic ulcer disease in elderly persons. Am J Epidemiol 1995; 141:539–545.
- Laine L. Approaches to nonsteroidal anti-inflammatory drug use in the high-risk patient. Gastroenterology 2001; 120:594–606.
- Silverstein FE, Graham DY, Senior JR, et al. Misoprostol reduces serious gastrointestinal complications in patients with rheumatoid arthritis receiving nonsteroidal anti-inflammatory drugs. A randomized, double blind, placebo controlled trial. Ann Intern Med 1995; 123:241–249.
- Goldstein JL, Huang B, Amer F, Christopoulos NG. Ulcer recurrence in high-risk patients receiving nonsteroidal anti-inflammatory drugs plus low dose aspirin: results of a post hoc subanalysis. Clin Ther 2004; 26:1637–1643.
- Berger JS, Stebbins A, Granger CB, et al. Initial aspirin dose and outcome among ST-elevation myocardial infarction patients treated with fibrinolytic therapy. Circulation 2008; 117:192–199.
- Lanas A, Garcia-Rodriguez LA, Arroyo MT, et al; Investigators of the Asociación Española de Gastroenterología (AEG). Effect of antisecretory drugs and nitrates on the risk of ulcer bleeding associated with nonsteroidal anti-inflammatory drugs, antiplatelet agents, and anticoagulants. Am J Gastroenterol 2007; 102:507–515.
- Chin MW, Yong G, Bulsara MK, Rankin J, Forbes GM. Predictive and protective factors associated with upper gastrointestinal bleeding after percutaneous coronary intervention: a case-control study. Am J Gastroenterol 2007; 102:2411–2416.
- Hunt RH, Bazzoli F. Should NSAID/low dose aspirin takers be tested routinely for H. Pylori infection and treated if positive? Implications for primary risk of ulcer and ulcer relapse after initial healing. Aliment Pharmacol Ther 2004; 19 (suppl 1):9–16.
Upper gastrointestinal (GI) bleeding is common, costly, and potentially life-threatening. It must be managed promptly and appropriately to prevent adverse outcomes.
More people are admitted to the hospital for upper GI bleeding than for congestive heart failure or deep vein thrombosis. In the United States, the annual rate of hospitalization for upper GI bleeding is estimated to be 165 per 100,000—more than 300,000 hospitalizations per year, at a cost of $2.5 billion.1,2
Furthermore, despite advances in therapy, the case-fatality rate has remained unchanged at 7% to 10%.3 This may be because today’s patients are older and have more comorbidities than those in the past.4
CAUSES OF UPPER GI BLEEDING
Peptic ulcers account for about 60% of severe cases of upper GI bleeding,5 and they are the focus of this paper. Fortunately, up to 80% of bleeding ulcers stop bleeding spontaneously without any intervention.6
Gastroduodenal erosions account for about 12%.3
Varices due to cirrhosis are less common but more dangerous. Variceal bleeding accounts for a relatively small percentage (6%) of upper GI bleeding, but the mortality rate from a single episode of variceal bleeding is 30%, and 60% to 70% of patients die within 1 year, mostly of underlying liver disease.
Less frequent causes include Mallory-Weiss tears, erosive duodenitis, Dieulafoy ulcer (a type of vascular malformation), other vascular lesions, neoplasms, aortoenteric fistula, gastric antral vascular ectasia, and prolapse gastropathy.5
HEMATEMESIS AND MELENA
The most common presenting signs of acute upper GI bleeding are hematemesis (vomiting of blood), “coffee grounds” emesis, and melena (tarry black stools). About 30% of patients with bleeding ulcers present with hematemesis, 20% with melena, and 50% with both.7
Hematochezia (red blood in the stool) usually suggests a lower GI source of bleeding, since blood from an upper source turns black and tarry as it passes through the gut, producing melena. However, up to 5% of patients with bleeding ulcers have hematochezia,7 and it indicates heavy bleeding: bleeding of approximately 1,000 mL into the upper GI tract is needed to cause hematochezia, whereas only 50 to 100 mL is needed to cause melena.8,9 Hematochezia with signs and symptoms of hemodynamic compromise such as syncope, postural hypotension, tachycardia, and shock should therefore direct one’s attention to an upper GI source of bleeding.
Nonspecific features include nausea, vomiting, epigastric pain, vasovagal phenomena, and syncope.
WHAT IS THE PATIENT’S RISK?
An assessment of clinical severity is the first critical task, as it helps in planning treatment. Advanced age, multiple comorbidities, and hemodynamic instability call for aggressive treatment. Apart from this simple clinical rule, scoring systems have been developed.
The Rockall scoring system, the most widely used, gives estimates of the risks of recurrent bleeding and death. It is based on the three clinical factors mentioned above and on two endoscopic ones, awarding points for:
- Age—0 points if less than 60; 1 point if 60 to 79; or 2 points if 80 years or older
- Shock—1 point if the pulse is more than 100; 2 points if the systolic blood pressure is less than 100 mm Hg
- Comorbid illness—2 points for ischemic heart disease, congestive heart failure, or other major comorbidity; 3 points for renal failure, hepatic failure, or metastatic disease
- Endoscopic diagnosis—0 points if no lesion found or a Mallory-Weiss tear; 1 point for peptic ulcer, esophagitis, or erosive disease; 2 points for GI malignancy
- Endoscopic stigmata or recent hemorrhage—0 points for a clean-based ulcer or flat pigmented spot; 2 points for blood in the upper GI tract, active bleeding, a nonbleeding visible vessel, or adherent clot.
The Rockall score can thus range from 0 to 11 points, with an overall score of 0, 1, or 2 associated with an excellent prognosis.10
Other systems that are used less often include the Baylor severity scale and the Acute Physiology and Chronic Health Evaluation (APACHE) II score.
Does the patient have varices?
All variceal bleeding should be considered severe, since the 1-year death rate is so high (up to 70%). Clues pointing to variceal bleeding include previous variceal bleeding, thrombocytopenia, history of liver disease, and signs of liver disease on clinical examination.
All patients suspected of having bleeding varices should be admitted to the intensive care unit for close monitoring and should be given the highest priority, even if they are hemodynamically stable.
Is the patient hemodynamically stable?
Appropriate hemodynamic assessment includes monitoring of heart rate, blood pressure, and mental status. Tachycardia at rest, hypotension, and orthostatic changes in vital signs indicate a considerable loss of blood volume. Low urine output, dry mucous membranes, and sunken neck veins are also useful signs. (Tachycardia may be blunted if the patient is taking a beta-blocker.)
If these signs of hypovolemia are present, the initial management focuses on treating shock and on improving oxygen delivery to the vital organs. This involves repletion of the intravascular volume with intravenous infusions or blood transfusions. Supplemental oxygen also is useful, especially in elderly patients with heart disease.12
Inspection of nasogastric aspirate
In the initial assessment, it is useful to insert a nasogastric tube and inspect the aspirate. If it contains bright red blood, the patient needs an urgent endoscopic evaluation and an intensive level of care13,14; if it contains coffee-grounds material, the patient needs to be admitted to the hospital and to undergo endoscopic evaluation within 24 hours.
However, a normal aspirate does not rule out upper GI bleeding. Aljebreen et al15 found that 15% of patients with upper GI bleeding and normal nasogastric aspirate still had high-risk lesions (ie, visible bleeding or nonbleeding visible vessels) on endoscopy.
ACID-SUPPRESSION HELPS ULCERS HEAL
Acid and pepsin interfere with the healing of ulcers and other nonvariceal upper GI lesions. Further, an acidic environment promotes platelet disaggregation and fibrinolysis and impairs clot formation.16 This suggests that inhibiting gastric acid secretion and raising the gastric pH to 6 or higher may stabilize clots. Moreover, pepsinogen in the stomach is converted to its active form (pepsin) if the pH is less than 4. Therefore, keeping the pH above 4 keeps pepsinogen in an inactive form.
Histamine-2 receptor antagonists
Histamine-2 receptor antagonists were the first drugs to inhibit acid secretion, reversibly blocking histamine-2 receptors on the basolateral membrane of parietal cells. However, these drugs did not prove very useful in managing upper GI bleeding in clinical trials.17,18 In their intravenous form, they often fail to keep the gastric pH at 6 or higher, due to tachyphylaxis.19 The use of this class of drugs has declined in favor of proton pump inhibitors.
Proton pump inhibitors
Proton pump inhibitors reduce both basal and stimulated acid secretion by inhibiting hydrogen-potassium adenosine triphosphatase, the proton pump of the parietal cell.
Multiple studies have shown that proton pump inhibitors raise the gastric pH and keep it high. For example, an infusion of omeprazole (Prilosec) can keep the gastric pH above 6 for 72 hours without inducing tachyphylaxis.20,21
Started after endoscopy. Randomized controlled trials have found proton pump inhibitors to be effective when given in high doses intravenously for 72 hours after successful endoscopic treatment of bleeding ulcers with high-risk endoscopic signs, such as active bleeding or nonbleeding visible vessels.22,23
A meta-analysis indicated that these drugs decrease the incidence of recurrent peptic ulcer bleeding, the need for blood transfusions, the need for surgery, and the duration of hospitalization, but not the mortality rate.24,25 These studies also illustrate the benefit of following up endoscopic treatment to stop the bleeding with an intravenous infusion of a proton pump inhibitor.
The recommended dose of omeprazole for patients with high-risk findings on endoscopy is an 80-mg bolus followed by an 8-mg/hour infusion for 72 hours. After the patient’s condition stabilizes, oral therapy can be substituted for intravenous therapy. In patients with low-risk endoscopic findings (a clean-based ulcer or flat spot), oral proton pump inhibitors in high doses are recommended.
In either case, after the initial bleeding is treated endoscopically and hemostasis is achieved, a proton pump inhibitor is recommended for 6 to 8 weeks, or longer if the patient is also positive for Helicobacter pylori or is on daily treatment with aspirin or a nonsteroidal anti-inflammatory drug (NSAID) that is not selective for cyclo-oxygenase 2 (see below).
Started before endoscopy, these drugs reduced the frequency of actively bleeding ulcers, the duration of hospitalization, and the need for endoscopic therapy in a randomized controlled trial.26 A meta-analysis found that significantly fewer patients had signs of recent bleeding on endoscopy if they received a proton pump inhibitor 24 to 48 hours before the procedure, but it did not find any significant difference in important clinical outcomes such as death, recurrent bleeding, or surgery.27 Nevertheless, we believe that intravenous proton pump inhibitor therapy should be started before endoscopy in patients with upper GI bleeding.
Somatostatin analogues
Octreotide (Sandostatin), an analogue of the hormone somatostatin, decreases splanchnic blood flow, decreases secretion of gastric acid and pepsin, and stimulates mucus production. Although it is beneficial in treating upper GI bleeding due to varices, its benefit has not been confirmed in patients with nonvariceal upper GI bleeding.
A meta-analysis revealed that outcomes were better with high-dose intravenous proton pump inhibitor therapy than with octreotide when these drugs were started after endoscopic treatment of acute peptic ulcer bleeding.28 Nevertheless, octreotide may be useful in patients with uncontrolled nonvariceal bleeding who are awaiting endoscopy, since it is relatively safe to use.
ALL PATIENTS NEED ENDOSCOPY
All patients with upper GI bleeding need an upper endoscopic examination to diagnose and assess the risk posed by the bleeding lesion and to treat the lesion, reducing the risk of recurrent bleeding.
How urgently does endoscopy need to be done?
Endoscopy within the first 24 hours of upper GI bleeding is considered the standard of care. Patients with uncontrolled or recurrent bleeding should undergo endoscopy on an urgent basis to control the bleeding and reduce the risk of death.
However, how urgently endoscopy needs to be done is often debated. A multicenter randomized controlled trial compared outcomes in patients who underwent endoscopy within 6 hours of coming to the emergency department vs within 24 hours after the initial evaluation. The study found no significant difference in outcomes between the two groups; however, the group that underwent endoscopy sooner needed fewer transfusions.29
For a better view of the stomach
Gastric lavage improves the view of the gastric fundus but has not been proven to improve outcome.30
Promotility agents such as erythromycin and metoclopramide (Reglan) are also used to empty the stomach for better visualization.31–35 Erythromycin has been shown to improve visualization, shorten the procedure time, and prevent the need for additional endoscopy attempts in two randomized controlled studies.33,34 Furthermore, a cost-effectiveness study confirmed that giving intravenous erythromycin before endoscopy for acute upper GI bleeding saved money and resulted in an increase in quality-adjusted life-years.35
Endoscopy to diagnose bleeding and assess risk
Upper endoscopy is 90% to 95% diagnostic for acute upper GI bleeding.36
Patients at high risk (ie, older than 60 years, with severe comorbidity, or hemodynamically compromised) who have active bleeding (ie, witnessed hematemesis, red blood per nasogastric tube, or fresh blood per rectum) or a nonbleeding visible vessel should be admitted to a monitored bed or intensive care unit. Observation in a regular medical ward is appropriate for high-risk patients found to have an adherent clot. Patients with low-risk findings (eg, a clean ulcer base) are at low risk of recurrent bleeding and may be considered for early hospital discharge with appropriate outpatient follow-up.
Endoscopy to treat bleeding
About 25% of endoscopic procedures performed for upper GI bleeding include some type of treatment,39 such as injections of epinephrine, normal saline, or sclerosants; thermal cautery; argon plasma coagulation; electrocautery; or application of clips or bands. They are all equally effective, and combinations of these therapies are more effective than when they are used individually. A recent meta-analysis found dual therapy to be superior to epinephrine monotherapy in preventing recurrent bleeding, need for surgery, and death.40
How to manage adherent clots is controversial, but recent studies have revealed a significant benefit from removing them and treating the underlying lesions compared with drug therapy alone.43,45
Flat, pigmented spots and nonbleeding ulcers with a clean base do not require endoscopic treatment because the risk of recurrent bleeding is low.
Endoscopic therapy stops the bleeding in more than 90% of patients, but bleeding recurs after endoscopic therapy in 10% to 25%.46 Reversal of any severe coagulopathy with transfusions of platelets or fresh frozen plasma is essential for endoscopic hemostasis. However, coagulopathy at the time of initial bleeding and endoscopy does not appear to be associated with higher rates of recurrent bleeding following endoscopic therapy for nonvariceal upper GI bleeding.47
Patients with refractory bleeding are candidates for angiography or surgery. However, even when endoscopic hemostasis fails, endoscopy is important before angiography or surgery to pinpoint the site of bleeding and diagnose the cause.
A second endoscopic procedure is generally not recommended within 24 hours after the initial procedure.48 However, it is appropriate in cases in which clinical signs indicate recurrent bleeding or if hemostasis during the initial procedure is questionable. A meta-analysis found that routinely repeating endoscopy reduces the rate of recurrent bleeding but not the need for surgery or the risk of death.49
ALL PATIENTS SHOULD BE ADMITTED
VARICEAL BLEEDING
Variceal bleeding, a severe outcome of portal hypertension secondary to cirrhosis, carries a 6-week mortality rate of 10% to 20%.50 In view of the risk, primary prevention is indicated in patients with high-risk varices.
The mainstays of primary and secondary prevention are the nonselective beta-blockers such as nadolol (Corgard) and propranolol (Inderal). Several randomized controlled trials have shown lower rates of recurrent bleeding and death with propranolol or nadolol than with placebo.51 In doses that decrease the heart rate by 25%, beta-blockers have been shown to delay and decrease variceal hemorrhage. However, most patients require prophylactic endoscopic variceal ligation because they cannot tolerate beta-blocker therapy.
In suspected acute variceal bleeding, a somatostatin analogue should be started to decrease the portal pressure, and antibiotics should be started to reduce the risks of infection and death. Vasoactive drugs, ie, somatostatin analogues, should be started before endoscopy and continued for 5 days to reduce the chances of recurrent bleeding.52,53
Terlipressin is the only drug proven to improve the odds of survival in acute variceal bleeding. Although widely used in Europe, it has not been approved for use in the United States.
Octreotide, another option, improves hemostasis to the same extent, although it does not increase the survival rate.54,55 The recommended dose of octreotide for patients with variceal bleeding is a 50-μg intravenous bolus, followed by a 50-μg/hour infusion for 5 days.
Combining endoscopic and drug therapy improves the chances of stopping the bleeding and reduces the risk of recurrent bleeding compared with endoscopic therapy alone.56
Transjugular intrahepatic portosystemic shunting is indicated in recurrent variceal hemorrhage or in those with initial bleeding that is refractory to standard medical and endoscopic therapy. It is not the primary therapy because it doubles the risk of encephalopathy and has a high stent occlusion rate (up to 60%, lower with covered stents).
GI BLEEDING CAN CAUSE ACUTE MYOCARDIAL INFARCTION
The simultaneous presentation of acute myocardial infarction (MI) and GI hemorrhage is very serious and unfortunately common.
An acute MI occurring simultaneously with or after GI bleeding is usually precipitated by massive bleeding causing hypovolemia, hemodynamic compromise, and hypoperfusion. Conversely, the anticoagulant, antiplatelet, or thrombolytic drugs given to treat MI can precipitate GI bleeding (see below).
This distinction is important because the two scenarios have different clinical courses and prognoses. GI bleeding that precipitates an acute MI tends to be massive, whereas GI bleeding after treatment of acute MI tends to be self-limited and often resolves with reversal of underlying coagulopathy.57
Endoscopy carries a higher than average risk in patients with recent acute MI, with all-cause mortality rates as high as 1%.58 (The usual rate is 0.0004%.59) Nevertheless, endoscopy can be safely performed early on in patients with acute MI if it is done under strict monitoring in a coronary care unit.
Several studies have shown that MI patients who present with upper GI bleeding as the inciting event or patients with acute MI who are vomiting blood or who are hemodynamically unstable due to GI bleeding are significantly more likely to have a high-risk lesion and so have the greatest need for endoscopic therapy. Therefore, endoscopic intervention may be offered to MI patients at high risk who have been started on antiplatelet agents.
WARFARIN CAN PRECIPITATE BLEEDING
Acute upper GI bleeding can be a severe complication of long-term oral anticoagulation, not because the drugs cause ulcers, but rather because they exacerbate ulcers that are already present.60 Therefore, when starting warfarin (Coumadin), patients should be evaluated to determine if they have other risk factors for GI bleeding, such as ulcers.
The number of people presenting with upper GI bleeding while on warfarin therapy is increasing because of the expanding indications for long-term anticoagulation therapy, such as atrial fibrillation and deep venous thrombosis.
The risk of GI bleeding in patients who use oral anticoagulants is estimated to be 2.3 to 4.9 times higher than in nonusers.61
The goal international normalized ratio (INR) for patients on warfarin therapy is usually 2.0 to 3.0. Recent studies found that endoscopy can be safely performed in patients with acute GI bleeding whose INR is between 2.0 and 3.0.62,63 Some suggest that both the length of warfarin therapy and the INR affect the risk of bleeding.64,65
Managing patients with an INR higher than 3.0 who have an episode of GI bleeding is always a challenge. It is not uncommon to find pathologic lesions causing GI bleeding in patients who are on warfarin with a supratherapeutic INR, and thus, endoscopy is indicated. However, before endoscopy, reversal of anticoagulation should be considered.
BLEEDING IN PATIENTS ON ANTIPLATELET DRUGS
Aspirin
Aspirin decreases production of prostaglandins in the GI tract, thereby decreasing the protective and restorative properties of the gastric and duodenal mucosa and predisposing to ulcers and bleeding.
The higher the aspirin dose, the higher the risk. Aspirin doubles the risk of upper GI bleeding at daily doses of 75 mg and quadruples it at doses of 300 mg.66 Even doses as low as 10 mg can decrease gastric mucosal prostaglandin production.67 Thus, it appears that there is no risk-free dose of aspirin, and enteric-coated or buffered formulations do not appear to reduce the risk.68–70
The most important risk factor for upper GI bleeding in patients taking aspirin is a history of peptic ulcer bleeding. Approximately 15% of aspirin users who have bleeding from ulcers have recurrent bleeding within 1 year.71
As aspirin-induced GI bleeding becomes more common, health care providers often feel caught between the GI risk and the cardiovascular benefit. When considering whether to discontinue antiplatelet therapy, a cardiologist should be consulted along with a gastroenterologist to weigh the risks of GI bleeding vs thrombosis. To date, there have been no clinical trials published to suggest when antiplatelet therapy should be stopped to optimize GI and cardiovascular outcomes. An alternative is to replace aspirin with another antiplatelet drug that does not induce ulcers.
Clopidogrel
Clopidogrel (Plavix) is recommended for hospitalized patients with acute coronary syndrome who cannot tolerate the GI side effects of aspirin, according to the joint guidelines of the American College of Cardiology and the American Heart Association, with the highest level of evidence.72 This recommendation was largely based on the safety data from the CAPRIE (Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events) trial, in which the incidence of major GI bleeding was lower in the clopidogrel group (0.52%) than in the aspirin group (0.72%; P < .05).73
Aspirin plus a proton pump inhibitor
Patients who have had an episode of upper GI bleeding and who need long-term aspirin therapy should also receive a proton pump inhibitor indefinitely to prevent ulcer recurrence.
In a recent double-blind randomized controlled trial in patients with a history of aspirin-induced bleeding, the combination of low-dose aspirin plus esomeprazole (Nexium) twice a day was superior to clopidogrel by itself in terms of the rate of recurrent bleeding (0.7% vs 8.6%; P < .05).74 A similar trial showed nearly identical results: 0% upper GI bleeding in the group receiving aspirin plus esomeprazole 20 mg daily, vs 13.6% in the clopidogrel group (P = .0019).75 These studies suggest that a once-daily proton pump inhibitor combined with aspirin is a safer alternative than clopidogrel alone.
Clopidogrel plus a proton pump inhibitor
Interestingly, recent studies have shown that omeprazole decreases the antiplatelet effect of clopidogrel, possibly by inhibiting the CYP2C19 enzyme.76 However, concomitant use of pantoprazole (Protonix), lansoprazole (Prevacid), and esomeprazole did not have this effect, suggesting that although all proton pump inhibitors are metabolized to a varying degree by CYP2C19, the interaction between proton pump inhibitors and clopidogrel is not a class effect.77–79 Therefore, pantoprazole, lansoprazole, and esomeprazole may be the appropriate proton pump inhibitors to use with clopidogrel in patients who have a clear indication for the medication, consistent with current guideline recommendations.
Helicobacter pylori infection in antiplatelet drug users
TREATMENT AND PREVENTION OF NSAID-RELATED GI INJURY
About 1 in 20 users of NSAIDs develop GI complications and ulcers of varying degrees of severity, as do one in seven NSAID users over the age of 65. In fact, NSAID use accounts for 30% of hospitalizations for upper GI bleeding and deaths from this cause.82–85 In addition, approximately 15% to 30% of NSAID users have clinically silent but endoscopically evident peptic ulcers.86
NSAIDs contribute to ulcer development by depleting prostaglandins. Thus, misoprostol (Cytotec), a synthetic prostaglandin, has been used to reduce this side effect.
In a clinical trial, misoprostol reduced the incidence of NSAID-associated GI complications by 40%.87 Furthermore, it has been shown to be better than placebo in preventing recurrent gastric ulcers in patients with a history of gastric ulcer who were receiving low-dose aspirin.88
However, misoprostol is rarely used because it can cause diarrhea and abdominal cramping. Rather, the preferred drugs for preventing and treating NSAID- and aspirin-related GI lesions are proton pump inhibitors.
Numerous clinical trials using endoscopic end points showed that proton pump inhibitors in standard doses significantly reduce the incidence of ulcers associated with the use of NSAIDs.89 Proton pump inhibitor therapy has achieved a significant reduction in relative risk of upper GI bleeding in patients who received low-dose aspirin therapy, as confirmed by epidemiologic studies.90,91 The number of NSAID-related ulcers found on endoscopy could be reduced by an estimated 90% simply by using proton pump inhibitors.92
Upper gastrointestinal (GI) bleeding is common, costly, and potentially life-threatening. It must be managed promptly and appropriately to prevent adverse outcomes.
More people are admitted to the hospital for upper GI bleeding than for congestive heart failure or deep vein thrombosis. In the United States, the annual rate of hospitalization for upper GI bleeding is estimated to be 165 per 100,000—more than 300,000 hospitalizations per year, at a cost of $2.5 billion.1,2
Furthermore, despite advances in therapy, the case-fatality rate has remained unchanged at 7% to 10%.3 This may be because today’s patients are older and have more comorbidities than those in the past.4
CAUSES OF UPPER GI BLEEDING
Peptic ulcers account for about 60% of severe cases of upper GI bleeding,5 and they are the focus of this paper. Fortunately, up to 80% of bleeding ulcers stop bleeding spontaneously without any intervention.6
Gastroduodenal erosions account for about 12%.3
Varices due to cirrhosis are less common but more dangerous. Variceal bleeding accounts for a relatively small percentage (6%) of upper GI bleeding, but the mortality rate from a single episode of variceal bleeding is 30%, and 60% to 70% of patients die within 1 year, mostly of underlying liver disease.
Less frequent causes include Mallory-Weiss tears, erosive duodenitis, Dieulafoy ulcer (a type of vascular malformation), other vascular lesions, neoplasms, aortoenteric fistula, gastric antral vascular ectasia, and prolapse gastropathy.5
HEMATEMESIS AND MELENA
The most common presenting signs of acute upper GI bleeding are hematemesis (vomiting of blood), “coffee grounds” emesis, and melena (tarry black stools). About 30% of patients with bleeding ulcers present with hematemesis, 20% with melena, and 50% with both.7
Hematochezia (red blood in the stool) usually suggests a lower GI source of bleeding, since blood from an upper source turns black and tarry as it passes through the gut, producing melena. However, up to 5% of patients with bleeding ulcers have hematochezia,7 and it indicates heavy bleeding: bleeding of approximately 1,000 mL into the upper GI tract is needed to cause hematochezia, whereas only 50 to 100 mL is needed to cause melena.8,9 Hematochezia with signs and symptoms of hemodynamic compromise such as syncope, postural hypotension, tachycardia, and shock should therefore direct one’s attention to an upper GI source of bleeding.
Nonspecific features include nausea, vomiting, epigastric pain, vasovagal phenomena, and syncope.
WHAT IS THE PATIENT’S RISK?
An assessment of clinical severity is the first critical task, as it helps in planning treatment. Advanced age, multiple comorbidities, and hemodynamic instability call for aggressive treatment. Apart from this simple clinical rule, scoring systems have been developed.
The Rockall scoring system, the most widely used, gives estimates of the risks of recurrent bleeding and death. It is based on the three clinical factors mentioned above and on two endoscopic ones, awarding points for:
- Age—0 points if less than 60; 1 point if 60 to 79; or 2 points if 80 years or older
- Shock—1 point if the pulse is more than 100; 2 points if the systolic blood pressure is less than 100 mm Hg
- Comorbid illness—2 points for ischemic heart disease, congestive heart failure, or other major comorbidity; 3 points for renal failure, hepatic failure, or metastatic disease
- Endoscopic diagnosis—0 points if no lesion found or a Mallory-Weiss tear; 1 point for peptic ulcer, esophagitis, or erosive disease; 2 points for GI malignancy
- Endoscopic stigmata or recent hemorrhage—0 points for a clean-based ulcer or flat pigmented spot; 2 points for blood in the upper GI tract, active bleeding, a nonbleeding visible vessel, or adherent clot.
The Rockall score can thus range from 0 to 11 points, with an overall score of 0, 1, or 2 associated with an excellent prognosis.10
Other systems that are used less often include the Baylor severity scale and the Acute Physiology and Chronic Health Evaluation (APACHE) II score.
Does the patient have varices?
All variceal bleeding should be considered severe, since the 1-year death rate is so high (up to 70%). Clues pointing to variceal bleeding include previous variceal bleeding, thrombocytopenia, history of liver disease, and signs of liver disease on clinical examination.
All patients suspected of having bleeding varices should be admitted to the intensive care unit for close monitoring and should be given the highest priority, even if they are hemodynamically stable.
Is the patient hemodynamically stable?
Appropriate hemodynamic assessment includes monitoring of heart rate, blood pressure, and mental status. Tachycardia at rest, hypotension, and orthostatic changes in vital signs indicate a considerable loss of blood volume. Low urine output, dry mucous membranes, and sunken neck veins are also useful signs. (Tachycardia may be blunted if the patient is taking a beta-blocker.)
If these signs of hypovolemia are present, the initial management focuses on treating shock and on improving oxygen delivery to the vital organs. This involves repletion of the intravascular volume with intravenous infusions or blood transfusions. Supplemental oxygen also is useful, especially in elderly patients with heart disease.12
Inspection of nasogastric aspirate
In the initial assessment, it is useful to insert a nasogastric tube and inspect the aspirate. If it contains bright red blood, the patient needs an urgent endoscopic evaluation and an intensive level of care13,14; if it contains coffee-grounds material, the patient needs to be admitted to the hospital and to undergo endoscopic evaluation within 24 hours.
However, a normal aspirate does not rule out upper GI bleeding. Aljebreen et al15 found that 15% of patients with upper GI bleeding and normal nasogastric aspirate still had high-risk lesions (ie, visible bleeding or nonbleeding visible vessels) on endoscopy.
ACID-SUPPRESSION HELPS ULCERS HEAL
Acid and pepsin interfere with the healing of ulcers and other nonvariceal upper GI lesions. Further, an acidic environment promotes platelet disaggregation and fibrinolysis and impairs clot formation.16 This suggests that inhibiting gastric acid secretion and raising the gastric pH to 6 or higher may stabilize clots. Moreover, pepsinogen in the stomach is converted to its active form (pepsin) if the pH is less than 4. Therefore, keeping the pH above 4 keeps pepsinogen in an inactive form.
Histamine-2 receptor antagonists
Histamine-2 receptor antagonists were the first drugs to inhibit acid secretion, reversibly blocking histamine-2 receptors on the basolateral membrane of parietal cells. However, these drugs did not prove very useful in managing upper GI bleeding in clinical trials.17,18 In their intravenous form, they often fail to keep the gastric pH at 6 or higher, due to tachyphylaxis.19 The use of this class of drugs has declined in favor of proton pump inhibitors.
Proton pump inhibitors
Proton pump inhibitors reduce both basal and stimulated acid secretion by inhibiting hydrogen-potassium adenosine triphosphatase, the proton pump of the parietal cell.
Multiple studies have shown that proton pump inhibitors raise the gastric pH and keep it high. For example, an infusion of omeprazole (Prilosec) can keep the gastric pH above 6 for 72 hours without inducing tachyphylaxis.20,21
Started after endoscopy. Randomized controlled trials have found proton pump inhibitors to be effective when given in high doses intravenously for 72 hours after successful endoscopic treatment of bleeding ulcers with high-risk endoscopic signs, such as active bleeding or nonbleeding visible vessels.22,23
A meta-analysis indicated that these drugs decrease the incidence of recurrent peptic ulcer bleeding, the need for blood transfusions, the need for surgery, and the duration of hospitalization, but not the mortality rate.24,25 These studies also illustrate the benefit of following up endoscopic treatment to stop the bleeding with an intravenous infusion of a proton pump inhibitor.
The recommended dose of omeprazole for patients with high-risk findings on endoscopy is an 80-mg bolus followed by an 8-mg/hour infusion for 72 hours. After the patient’s condition stabilizes, oral therapy can be substituted for intravenous therapy. In patients with low-risk endoscopic findings (a clean-based ulcer or flat spot), oral proton pump inhibitors in high doses are recommended.
In either case, after the initial bleeding is treated endoscopically and hemostasis is achieved, a proton pump inhibitor is recommended for 6 to 8 weeks, or longer if the patient is also positive for Helicobacter pylori or is on daily treatment with aspirin or a nonsteroidal anti-inflammatory drug (NSAID) that is not selective for cyclo-oxygenase 2 (see below).
Started before endoscopy, these drugs reduced the frequency of actively bleeding ulcers, the duration of hospitalization, and the need for endoscopic therapy in a randomized controlled trial.26 A meta-analysis found that significantly fewer patients had signs of recent bleeding on endoscopy if they received a proton pump inhibitor 24 to 48 hours before the procedure, but it did not find any significant difference in important clinical outcomes such as death, recurrent bleeding, or surgery.27 Nevertheless, we believe that intravenous proton pump inhibitor therapy should be started before endoscopy in patients with upper GI bleeding.
Somatostatin analogues
Octreotide (Sandostatin), an analogue of the hormone somatostatin, decreases splanchnic blood flow, decreases secretion of gastric acid and pepsin, and stimulates mucus production. Although it is beneficial in treating upper GI bleeding due to varices, its benefit has not been confirmed in patients with nonvariceal upper GI bleeding.
A meta-analysis revealed that outcomes were better with high-dose intravenous proton pump inhibitor therapy than with octreotide when these drugs were started after endoscopic treatment of acute peptic ulcer bleeding.28 Nevertheless, octreotide may be useful in patients with uncontrolled nonvariceal bleeding who are awaiting endoscopy, since it is relatively safe to use.
ALL PATIENTS NEED ENDOSCOPY
All patients with upper GI bleeding need an upper endoscopic examination to diagnose and assess the risk posed by the bleeding lesion and to treat the lesion, reducing the risk of recurrent bleeding.
How urgently does endoscopy need to be done?
Endoscopy within the first 24 hours of upper GI bleeding is considered the standard of care. Patients with uncontrolled or recurrent bleeding should undergo endoscopy on an urgent basis to control the bleeding and reduce the risk of death.
However, how urgently endoscopy needs to be done is often debated. A multicenter randomized controlled trial compared outcomes in patients who underwent endoscopy within 6 hours of coming to the emergency department vs within 24 hours after the initial evaluation. The study found no significant difference in outcomes between the two groups; however, the group that underwent endoscopy sooner needed fewer transfusions.29
For a better view of the stomach
Gastric lavage improves the view of the gastric fundus but has not been proven to improve outcome.30
Promotility agents such as erythromycin and metoclopramide (Reglan) are also used to empty the stomach for better visualization.31–35 Erythromycin has been shown to improve visualization, shorten the procedure time, and prevent the need for additional endoscopy attempts in two randomized controlled studies.33,34 Furthermore, a cost-effectiveness study confirmed that giving intravenous erythromycin before endoscopy for acute upper GI bleeding saved money and resulted in an increase in quality-adjusted life-years.35
Endoscopy to diagnose bleeding and assess risk
Upper endoscopy is 90% to 95% diagnostic for acute upper GI bleeding.36
Patients at high risk (ie, older than 60 years, with severe comorbidity, or hemodynamically compromised) who have active bleeding (ie, witnessed hematemesis, red blood per nasogastric tube, or fresh blood per rectum) or a nonbleeding visible vessel should be admitted to a monitored bed or intensive care unit. Observation in a regular medical ward is appropriate for high-risk patients found to have an adherent clot. Patients with low-risk findings (eg, a clean ulcer base) are at low risk of recurrent bleeding and may be considered for early hospital discharge with appropriate outpatient follow-up.
Endoscopy to treat bleeding
About 25% of endoscopic procedures performed for upper GI bleeding include some type of treatment,39 such as injections of epinephrine, normal saline, or sclerosants; thermal cautery; argon plasma coagulation; electrocautery; or application of clips or bands. They are all equally effective, and combinations of these therapies are more effective than when they are used individually. A recent meta-analysis found dual therapy to be superior to epinephrine monotherapy in preventing recurrent bleeding, need for surgery, and death.40
How to manage adherent clots is controversial, but recent studies have revealed a significant benefit from removing them and treating the underlying lesions compared with drug therapy alone.43,45
Flat, pigmented spots and nonbleeding ulcers with a clean base do not require endoscopic treatment because the risk of recurrent bleeding is low.
Endoscopic therapy stops the bleeding in more than 90% of patients, but bleeding recurs after endoscopic therapy in 10% to 25%.46 Reversal of any severe coagulopathy with transfusions of platelets or fresh frozen plasma is essential for endoscopic hemostasis. However, coagulopathy at the time of initial bleeding and endoscopy does not appear to be associated with higher rates of recurrent bleeding following endoscopic therapy for nonvariceal upper GI bleeding.47
Patients with refractory bleeding are candidates for angiography or surgery. However, even when endoscopic hemostasis fails, endoscopy is important before angiography or surgery to pinpoint the site of bleeding and diagnose the cause.
A second endoscopic procedure is generally not recommended within 24 hours after the initial procedure.48 However, it is appropriate in cases in which clinical signs indicate recurrent bleeding or if hemostasis during the initial procedure is questionable. A meta-analysis found that routinely repeating endoscopy reduces the rate of recurrent bleeding but not the need for surgery or the risk of death.49
ALL PATIENTS SHOULD BE ADMITTED
VARICEAL BLEEDING
Variceal bleeding, a severe outcome of portal hypertension secondary to cirrhosis, carries a 6-week mortality rate of 10% to 20%.50 In view of the risk, primary prevention is indicated in patients with high-risk varices.
The mainstays of primary and secondary prevention are the nonselective beta-blockers such as nadolol (Corgard) and propranolol (Inderal). Several randomized controlled trials have shown lower rates of recurrent bleeding and death with propranolol or nadolol than with placebo.51 In doses that decrease the heart rate by 25%, beta-blockers have been shown to delay and decrease variceal hemorrhage. However, most patients require prophylactic endoscopic variceal ligation because they cannot tolerate beta-blocker therapy.
In suspected acute variceal bleeding, a somatostatin analogue should be started to decrease the portal pressure, and antibiotics should be started to reduce the risks of infection and death. Vasoactive drugs, ie, somatostatin analogues, should be started before endoscopy and continued for 5 days to reduce the chances of recurrent bleeding.52,53
Terlipressin is the only drug proven to improve the odds of survival in acute variceal bleeding. Although widely used in Europe, it has not been approved for use in the United States.
Octreotide, another option, improves hemostasis to the same extent, although it does not increase the survival rate.54,55 The recommended dose of octreotide for patients with variceal bleeding is a 50-μg intravenous bolus, followed by a 50-μg/hour infusion for 5 days.
Combining endoscopic and drug therapy improves the chances of stopping the bleeding and reduces the risk of recurrent bleeding compared with endoscopic therapy alone.56
Transjugular intrahepatic portosystemic shunting is indicated in recurrent variceal hemorrhage or in those with initial bleeding that is refractory to standard medical and endoscopic therapy. It is not the primary therapy because it doubles the risk of encephalopathy and has a high stent occlusion rate (up to 60%, lower with covered stents).
GI BLEEDING CAN CAUSE ACUTE MYOCARDIAL INFARCTION
The simultaneous presentation of acute myocardial infarction (MI) and GI hemorrhage is very serious and unfortunately common.
An acute MI occurring simultaneously with or after GI bleeding is usually precipitated by massive bleeding causing hypovolemia, hemodynamic compromise, and hypoperfusion. Conversely, the anticoagulant, antiplatelet, or thrombolytic drugs given to treat MI can precipitate GI bleeding (see below).
This distinction is important because the two scenarios have different clinical courses and prognoses. GI bleeding that precipitates an acute MI tends to be massive, whereas GI bleeding after treatment of acute MI tends to be self-limited and often resolves with reversal of underlying coagulopathy.57
Endoscopy carries a higher than average risk in patients with recent acute MI, with all-cause mortality rates as high as 1%.58 (The usual rate is 0.0004%.59) Nevertheless, endoscopy can be safely performed early on in patients with acute MI if it is done under strict monitoring in a coronary care unit.
Several studies have shown that MI patients who present with upper GI bleeding as the inciting event or patients with acute MI who are vomiting blood or who are hemodynamically unstable due to GI bleeding are significantly more likely to have a high-risk lesion and so have the greatest need for endoscopic therapy. Therefore, endoscopic intervention may be offered to MI patients at high risk who have been started on antiplatelet agents.
WARFARIN CAN PRECIPITATE BLEEDING
Acute upper GI bleeding can be a severe complication of long-term oral anticoagulation, not because the drugs cause ulcers, but rather because they exacerbate ulcers that are already present.60 Therefore, when starting warfarin (Coumadin), patients should be evaluated to determine if they have other risk factors for GI bleeding, such as ulcers.
The number of people presenting with upper GI bleeding while on warfarin therapy is increasing because of the expanding indications for long-term anticoagulation therapy, such as atrial fibrillation and deep venous thrombosis.
The risk of GI bleeding in patients who use oral anticoagulants is estimated to be 2.3 to 4.9 times higher than in nonusers.61
The goal international normalized ratio (INR) for patients on warfarin therapy is usually 2.0 to 3.0. Recent studies found that endoscopy can be safely performed in patients with acute GI bleeding whose INR is between 2.0 and 3.0.62,63 Some suggest that both the length of warfarin therapy and the INR affect the risk of bleeding.64,65
Managing patients with an INR higher than 3.0 who have an episode of GI bleeding is always a challenge. It is not uncommon to find pathologic lesions causing GI bleeding in patients who are on warfarin with a supratherapeutic INR, and thus, endoscopy is indicated. However, before endoscopy, reversal of anticoagulation should be considered.
BLEEDING IN PATIENTS ON ANTIPLATELET DRUGS
Aspirin
Aspirin decreases production of prostaglandins in the GI tract, thereby decreasing the protective and restorative properties of the gastric and duodenal mucosa and predisposing to ulcers and bleeding.
The higher the aspirin dose, the higher the risk. Aspirin doubles the risk of upper GI bleeding at daily doses of 75 mg and quadruples it at doses of 300 mg.66 Even doses as low as 10 mg can decrease gastric mucosal prostaglandin production.67 Thus, it appears that there is no risk-free dose of aspirin, and enteric-coated or buffered formulations do not appear to reduce the risk.68–70
The most important risk factor for upper GI bleeding in patients taking aspirin is a history of peptic ulcer bleeding. Approximately 15% of aspirin users who have bleeding from ulcers have recurrent bleeding within 1 year.71
As aspirin-induced GI bleeding becomes more common, health care providers often feel caught between the GI risk and the cardiovascular benefit. When considering whether to discontinue antiplatelet therapy, a cardiologist should be consulted along with a gastroenterologist to weigh the risks of GI bleeding vs thrombosis. To date, there have been no clinical trials published to suggest when antiplatelet therapy should be stopped to optimize GI and cardiovascular outcomes. An alternative is to replace aspirin with another antiplatelet drug that does not induce ulcers.
Clopidogrel
Clopidogrel (Plavix) is recommended for hospitalized patients with acute coronary syndrome who cannot tolerate the GI side effects of aspirin, according to the joint guidelines of the American College of Cardiology and the American Heart Association, with the highest level of evidence.72 This recommendation was largely based on the safety data from the CAPRIE (Clopidogrel Versus Aspirin in Patients at Risk of Ischemic Events) trial, in which the incidence of major GI bleeding was lower in the clopidogrel group (0.52%) than in the aspirin group (0.72%; P < .05).73
Aspirin plus a proton pump inhibitor
Patients who have had an episode of upper GI bleeding and who need long-term aspirin therapy should also receive a proton pump inhibitor indefinitely to prevent ulcer recurrence.
In a recent double-blind randomized controlled trial in patients with a history of aspirin-induced bleeding, the combination of low-dose aspirin plus esomeprazole (Nexium) twice a day was superior to clopidogrel by itself in terms of the rate of recurrent bleeding (0.7% vs 8.6%; P < .05).74 A similar trial showed nearly identical results: 0% upper GI bleeding in the group receiving aspirin plus esomeprazole 20 mg daily, vs 13.6% in the clopidogrel group (P = .0019).75 These studies suggest that a once-daily proton pump inhibitor combined with aspirin is a safer alternative than clopidogrel alone.
Clopidogrel plus a proton pump inhibitor
Interestingly, recent studies have shown that omeprazole decreases the antiplatelet effect of clopidogrel, possibly by inhibiting the CYP2C19 enzyme.76 However, concomitant use of pantoprazole (Protonix), lansoprazole (Prevacid), and esomeprazole did not have this effect, suggesting that although all proton pump inhibitors are metabolized to a varying degree by CYP2C19, the interaction between proton pump inhibitors and clopidogrel is not a class effect.77–79 Therefore, pantoprazole, lansoprazole, and esomeprazole may be the appropriate proton pump inhibitors to use with clopidogrel in patients who have a clear indication for the medication, consistent with current guideline recommendations.
Helicobacter pylori infection in antiplatelet drug users
TREATMENT AND PREVENTION OF NSAID-RELATED GI INJURY
About 1 in 20 users of NSAIDs develop GI complications and ulcers of varying degrees of severity, as do one in seven NSAID users over the age of 65. In fact, NSAID use accounts for 30% of hospitalizations for upper GI bleeding and deaths from this cause.82–85 In addition, approximately 15% to 30% of NSAID users have clinically silent but endoscopically evident peptic ulcers.86
NSAIDs contribute to ulcer development by depleting prostaglandins. Thus, misoprostol (Cytotec), a synthetic prostaglandin, has been used to reduce this side effect.
In a clinical trial, misoprostol reduced the incidence of NSAID-associated GI complications by 40%.87 Furthermore, it has been shown to be better than placebo in preventing recurrent gastric ulcers in patients with a history of gastric ulcer who were receiving low-dose aspirin.88
However, misoprostol is rarely used because it can cause diarrhea and abdominal cramping. Rather, the preferred drugs for preventing and treating NSAID- and aspirin-related GI lesions are proton pump inhibitors.
Numerous clinical trials using endoscopic end points showed that proton pump inhibitors in standard doses significantly reduce the incidence of ulcers associated with the use of NSAIDs.89 Proton pump inhibitor therapy has achieved a significant reduction in relative risk of upper GI bleeding in patients who received low-dose aspirin therapy, as confirmed by epidemiologic studies.90,91 The number of NSAID-related ulcers found on endoscopy could be reduced by an estimated 90% simply by using proton pump inhibitors.92
- Lewis JD, Bilker WB, Brensinger C, Farrar JT, Strom BL. Hospitalization and mortality rates from peptic ulcer disease and GI bleeding in the 1990s: relationship to sales of non-steroidal anti-inflammatory drugs and acid suppression medications. Am J Gastroenterol 2002; 97:2540–2549.
- Viviane A, Alan BN. Estimates of costs of hospital stays for variceal and nonvariceal upper gastrointestinal bleeding in the United States. Value Health 2008; 11:1–3.
- Yavorski RT, Wong RK, Maydonovitch C, Battin LS, Furnia A, Amundson DE. Analysis of 3,294 cases of upper gastrointestinal bleeding in military medical facilities. Am J Gastroenterol 1995; 90:568–573.
- Kaplan RC, Heckbert SR, Koepsell TD, et al. Risk factors for hospitalized gastrointestinal bleeding among older persons. Cardiovascular Health Study Investigators. J Am Geriatr Soc 2001; 49:126–133.
- Longstreth GF. Epidemiology of hospitalization for acute upper gastrointestinal hemorrhage: a population-based study. Am J Gastroenterol 1995; 90:206–210.
- Laine L, Peterson WL. Bleeding peptic ulcer. N Engl J Med 1994; 331:717–727.
- Wara P, Stodkilde H. Bleeding pattern before admission as guideline for emergency endoscopy. Scand J Gastroenterol 1985; 20:72–78.
- Jensen DM, Machicado GA. Diagnosis and treatment of severe hematochezia. The role of urgent colonoscopy after purge. Gastroenterology 1988; 95:1569–1574.
- Daniel WA, Egan S. The quantity of blood required to produce a tarry stool. J Am Med Assoc 1939; 113:2232.
- Rockall TA, Logan RF, Devlin HB, Northfield TC. Risk assessment after acute upper gastrointestinal hemorrhage. Gut 1996; 38:316–321.
- Blatchford O, Murray WR, Blatchford M. A risk score to predict need for treatment for upper-gastrointestinal hemorrhage. Lancet 2000; 356:1318–1321.
- Gralnek IM, Barkun AN, Bardou M. Management of acute bleeding from a peptic ulcer. N Engl J Med 2008; 359:928–937.
- Silverstein FE, Gilbert DA, Tedesco FJ, Buenger NK, Persing J. The national ASGE survey on upper gastrointestinal bleeding II. Clinical prognostic factors. Gastrointest Endosc 1981; 27:80–93.
- Corley DA, Stefan AM, Wolf M, Cook EF, Lee TH. Early indicators of prognosis in upper gastrointestinal hemorrhage. Am J Gastroenterol 1998; 93:336–340.
- Aljebreen AM, Fallone CA, Barkun AN. Nasogastric aspirate predicts high-risk endoscopic lesions in patients with acute upper-GI bleeding. Gastrointest Endosc 2004; 59:172–178.
- Barkun AN, Cockeram AW, Plourde V, Fedorak RN. Review article: acid suppression in non-variceal acute upper gastrointestinal bleeding. Aliment Pharmacol Ther 1999; 13:1565–1584.
- Levine JE, Leontiadis JI, Sharma VK, Howden CW. Meta-analysis: the efficacy of intravenous H2-receptor antagonists in bleeding peptic ulcer. Aliment Pharmacol Ther 2002; 16:1137–1142.
- Walt RP, Cottrell J, Mann SG, Freemantle NP, Langman MJ. Continuous intravenous famotidine for hemorrhage from peptic ulcer. Lancet 1992; 340:1058–1062.
- Labenz J, Peitz U, Leusing C, Tillenburg B, Blum AL, Börsch G. Efficacy of primed infusion with high dose ranitidine and omeprazole to maintain high intragastric pH in patients with peptic ulcer bleeding: a prospective randomized controlled study. Gut 1997; 40:36–41.
- Merki HS, Wilder-Smith CH. Do continuous infusions of omeprazole and ranitidine retain their effect with prolonged dosing? Gastroenterology 1994; 106:60–64.
- Netzer P, Gaia C, Sandoz M, et al. Effect of repeated injection and continuous infusion of omeprazole and ranitidine on intragastric pH over 72 hours. Am J Gastroenterol 1999; 94:351–357.
- Lin HJ, Lo WC, Cheng YC, Perng CL. Role of intravenous omeprazole in patients with high-risk peptic ulcer bleeding after successful endoscopic epinephrine injection: a prospective randomized comparative trial. Am J Gastroenterol 2006; 101:500–505.
- Lau JY, Sung JJ, Lee KK, et al. Effects of intravenous omeprazole on recurrent bleeding after endoscopic treatment of bleeding peptic ulcers. N Engl J Med 2000; 343:310–316.
- Leontiadis GI, Sharma VK, Howden CW. Proton pump inhibitor treatment for acute peptic ulcer bleeding. Cochrane Database Syst Rev 2006;CD002094.
- Andriulli A, Annese V, Caruso N, et al. Proton-pump inhibitors and outcome of endoscopic hemostasis in bleeding peptic ulcers: a series of meta-analyses. Am J Gastroenterol 2005; 100:207–219.
- Lau JY, Leung WK, Wu JC, et al. Omeprazole before endoscopy in patients with gastrointestinal bleeding. N Engl J Med 2007; 356:1631–1640.
- Dorward S, Sreedharan A, Leontiadis GI, Howden CW, Moayyedi P, Forman D. Proton pump inhibitor treatment initiated prior to endoscopic diagnosis in upper gastrointestinal bleeding. Cochrane Database Syst Rev 2006;CD005415.
- Bardou M, Toubouti Y, Benhaberou-Brun D, Rahme E, Barkun AN. Meta-analysis: proton-pump inhibition in high-risk patients with acute peptic ulcer bleeding. Aliment Pharmacol Ther 2005; 21:677–686.
- Bjorkman DJ, Zaman A, Fennerty MB, Lieberman D, Disario JA, Guest-Warnick G. Urgent vs elective endoscopy for acute non-variceal upper-GI bleeding: an effectiveness study. Gastointest Endosc 2004; 60:1–8.
- Lee SD, Kearney DJ. A randomized controlled trial of gastric lavage prior to endoscopy for acute upper gastrointestinal bleeding. J Clin Gastroenterol 2004; 38:861–865.
- Tack J, Janssens J, Vantrappen G, et al. Effect of erythromycin on gastric motility in controls and in diabetic gastroparesis. Gastroenterology 1992; 103:72–79.
- Xynos E, Mantides A, Papageorgiou A, Fountos A, Pechlivanides G, Vassilakis JS. Erythromycin accelerates delayed gastric emptying of solids in patients after truncal vagotomy and pyloroplasty. Eur J Surg 1992; 158:407–411.
- Coffin B, Pocard M, Panis Y, et al; Groupe des endoscopistes de garde á l’AP-HP. Erythromycin improves the quality of EGD in patients with acute upper GI bleeding: a randomized controlled study. Gastrointest Endosc 2002; 56:174–179.
- Frossard JL, Spahr L, Queneau PE, et al. Erythromycin intravenous bolus infusion in acute upper gastrointestinal bleeding: a randomized, controlled, double-blind trial. Gastroenterology 2002; 123:17–23.
- Winstead NS, Wilcox CM. Erythromycin prior to endoscopy for acute upper gastrointestinal hemorrhage: a cost-effectiveness analysis. Aliment Pharmacol Ther 2007; 26:1371–1377.
- Chak A, Cooper GS, Lloyd LE, Kolz CS, Barnhart BA, Wong RC. Effectiveness of endoscopy in patients admitted to the intensive care unit with upper GI hemorrhage. Gastrointest Endosc 2001; 53:6–13.
- Lau JY, Chung SC, Leung JW, Lo KK, Yung MY, Li AK. The evolution of stigmata of hemorrhage in bleeding peptic ulcers: a sequential endoscopic study. Endoscopy 1998; 30:513–518.
- Chung IK, Kim EJ, Lee MS, et al. Endoscopic factors predisposing to rebleeding following endoscopic hemostasis in bleeding peptic ulcers. Endoscopy 2001; 33:969–975.
- Elta GH. Acute nonvariceal upper gastrointestinal hemorrhage. Curr Treat Options Gastroenterol 2002; 5:147–152.
- Marmo R, Rotondano G, Piscopo R, Bianco MA, D’Angella R, Cipolletta L. Dual therapy versus monotherapy in the endoscopic treatment of high-risk bleeding ulcers: a meta-analysis of controlled trials. Am J Gastroenterol 2007; 102:279–289.
- Kovacs TO, Jensen DM. Recent advances in the endoscopic diagnosis and therapy of upper gastrointestinal, small intestinal, and colonic bleeding. Med Clin North Am 2002; 86:1319–1356.
- Kovacs TO, Jensen DM. Endoscopic treatment of ulcer bleeding. Curr Treat Options Gastroenterol 2007; 10:143–148.
- Jensen DM, Kovacs TO, Jutabha R, et al. Randomized trial of medical or endoscopic therapy to prevent recurrent ulcer hemorrhage in patients with adherent clots. Gastroenterology 2002; 123:407–413.
- Jensen DM, Machicado GA. Endoscopic hemostasis of ulcer hemorrhage with injection, thermal, and combination methods. Techniques Gastrointest Endosc 2005; 7:124–131.
- Bleau BL, Gostout CJ, Sherman KE, et al. Recurrent bleeding from peptic ulcer associated with adherent clot: a randomized study comparing endoscopic treatment with medical therapy. Gastrointest Endosc 2002; 56:1–6.
- Lau JY, Sung JJ, Lam YH, et al. Endoscopic retreatment compared with surgery in patients with recurrent bleeding after initial endoscopic control of bleeding ulcers. N Engl J Med 1999; 340:751–756.
- Wolf AT, Wasan SK, Saltzman JR. Impact of anticoagulation on rebleeding following endoscopic therapy for nonvariceal upper gastrointestinal hemorrhage. Am J Gastroenterol 2007; 102:290–296.
- Barkun A, Bardou M, Marshall JK; Nonvariceal Upper GI Bleeding Consensus Conference Group. Consensus recommendations for managing patients with nonvariceal upper gastrointestinal bleeding. Ann Intern Med 2003; 139:843–857.
- Marmo R, Rotondano G, Bianco MA, Piscopo R, Prisco A, Cipolletta L. Outcome of endoscopic treatment for peptic ulcer bleeding: is a second look necessary? A meta-analysis. Gastrointest Endosc 2003; 57:62–67.
- Dell’Era A, deFrancis R, Iannuzzi F. Acute variceal bleeding: pharmacological treatment and primary/secondary prophylaxis. Best Pract Res Clin Gastroenterol 2008; 22:279–294.
- Jalan R, Hayes PC. UK guidelines on the management of variceal hemorrhage in cirrhotic patients. British Society of Gastroenterology. Gut 2000; 46( suppl 3–4):III1–III15.
- Bernard B, Lebrec D, Mathurin P, Opolon P, Poynard T. Beta-adrenergic antagonists in the prevention of gastrointestinal rebleeding in patients with cirrhosis: a meta-analysis. Hepatology 1997; 25:63–70.
- De Franchis R. Evolving consensus in portal hypertension. Report of the Baveno IV consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol 2005; 43:167–176.
- Levacher S, Letoumelin P, Pateron D, Blaise M, Lapandry C, Pourriat JL. Early administration of terlipressin plus glyceryl trinitrate to control active upper gastrointestinal bleeding in cirrhotic patients. Lancet 1995; 346:865–868.
- Abraldes JG, Bosch J. Somatostatin and analogues in portal hypertension. Hepatology 2002; 35:1305–1312.
- Banares R, Albillos A, Rincon D, et al. Endoscopic treatment versus endoscopic plus pharmacological treatment for acute variceal bleeding: a meta analysis. Hepatology 2002; 35:609–615.
- Cappell M. Gastrointenstinal bleeding associated with myocardial infarction. Gastroenterol Clin North Am 2000; 29:423–444.
- Lin S, Konstance R, Jollis J, Fisher DA. The utility of upper endoscopy in patients with concomitant upper gastrointestinal bleeding and acute myocardial infarction. Dig Dis Sci 2006; 51:2377–2383.
- Silvis SE, Nebel O, Rogers G, Sugawa C, Mandelstam P. Endoscopic complications. Results of the 1974 American Society for Gastrointestinal Endoscopy Survey. JAMA 1976; 235:928–930.
- Lanas A, Garcia-Rodriguez LA, Arroyo MT, et al; Investigators of the Asociación Española de Gastroenterología (AEG). Effect of antisecretory drugs and nitrates on the risk of ulcer bleeding associated with nonsteroidal anti-inflammatory drugs, antiplatelet agents, and anticoagulants. Am J Gastroenterol 2007; 102:507–515.
- Shorr RI, Ray WA, Daugherty JR, Griffin MR. Concurrent use of nonsteroidal anti-inflammatory drugs and oral anticoagulants places elderly persons at high risk for hemorrhagic peptic ulcer disease. Arch Intern Med 1993; 153:1665–1670.
- Tabibian N. Acute gastrointestinal bleeding in anticoagulated patients: a prospective evaluation. Am J Gastroenterol 1989; 84:10–12.
- Choudari CP, Rajgopal C, Palmer KR. Acute gastrointestinal hemorrhage in anticoagulated patients: diagnoses and response to endoscopic treatment. Gut 1994; 35:464–466.
- Petty GW, Brown RD, Whisnant JP, Sicks JD, O’Fallon WM, Wiebers DO. Frequency of major complications of aspirin, warfarin, and intravenous heparin for secondary stroke prevention: a population-based study. Ann Intern Med 1999; 130:14–22.
- Landefeld CS, Rosenblatt MW, Goldman L. Bleeding in outpatients treated with warfarin: relation to the prothrombin time and important remediable lesions. Am J Med 1989; 87:153–159.
- Weil J, Colin-Jones D, Langman M, et al. Prophylactic aspirin and risk of peptic ulcer bleeding. BMJ 1995; 310:827–830.
- Cryer B, Feldman M. Effects of very low dose daily, long-term aspirin therapy on gastric, duodenal, and rectal prostaglandin levels and on mucosal injury in healthy humans. Gastroenterology 1999; 117:17–25.
- De Abajo FJ, Garcia Rodriguez LA. Risk of upper gastrointestinal bleeding and perforation associated with low-dose aspirin as plain and enteric-coated formulations. BMC Clin Pharmacol 2001; 1:1.
- Kelly JP, Kaufman DW, Jurgelon JM, Sheehan J, Koff RS, Shapiro S. Risk of aspirin-associated major upper gastrointestinal bleeding with enteric coated or buffered product. Lancet 1996; 348:1413–1416.
- Garcia Rodriguez LA, Hernandez-Diaz S, De Abajo FJ. Association between aspirin and upper gastrointestinal complications: systematic review of epidemiological studies. Br J Clin Pharmacol 2001; 52:563–571.
- Wilcox CM, Ladabaum U. A patient with high risk of gastrointestinal bleeding requiring nonsteroidal anti-inflammatory drugs. Clin Gastroenterol Hepatol 2006; 4:1090–1093.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996; 348:1329–1339.
- Chan FK, Ching JY, Hung LC, et al. Clopidogrel versus aspirin and esomeprazole to prevent recurrent ulcer bleeding. N Engl J Med 2005; 352:238–244.
- Lai KC, Chu KM, Hui WM, et al. Esomeprazole with aspirin versus clopidogrel for prevention of recurrent gastrointestinal ulcer complications. Clin Gastroenterol Hepatol 2006; 4:860–865.
- Ho MP, Maddox TM, Wang L, et al. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. JAMA 2009; 301:937–944.
- Siller-Matula JM, Spiel AO, Lang IM, Kreiner G, Christ G, Jilma B. Effects of pantoprazole and esomeprazole on platelet inhibition by clopidogrel. Am Heart J 2009; 157:148.e1–e5.
- Small DS, Farid NA, Payne CD, et al. Effects of proton pump inhibitor lansoprazole on the pharmacokinetics and pharmacodynamics of prasugel and clopidogrel. J Clin Pharmacol 2008; 48:475–484.
- Ishizaki T, Horai Y. Review article: cytochrome P450 and the metabolism of proton pump inhibitors—emphasis on rabeprazole. Aliment Pharacol Ther 1999; 13 (suppl 3):27–36.
- Lanas A, Fuentes J, Benito R, Serrano P, Bajador E, Sainz R. Helicobacter pylori increases the risk of upper gastrointestinal bleeding in patients taking low-dose aspirin. Aliment Pharmacol Ther 2002; 16:779–786.
- Chan FK. NSAID-Induced peptic ulcers and Helicobacter pylori infection: implications for patient management. Drug Saf 2005; 28:287–300.
- Bombardier V, Laine L, Reicin A, et al; VIGOR Study Group. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis: VIGOR Study Group. N Eng J Med 2000; 343:1520–1528.
- Griffin MR, Ray WA, Schaffner W. Nonsteroidal anti-inflammatory drug use and death from peptic ulcer in elderly persons. Ann Intern Med 1988; 109:359–363.
- Griffin MR, Piper JM, Daugherty JR, Snowden M, Ray WA. Nonsteroidal anti-inflammatory drug use and increased risk for peptic ulcer disease in elderly persons. Ann Intern Med 1991; 114:257–263.
- Smalley WE, Ray WA, Daugherty JR, Griffin MR. Nonsteroidal anti-inflammatory drugs and the incidence of hospitalizations for peptic ulcer disease in elderly persons. Am J Epidemiol 1995; 141:539–545.
- Laine L. Approaches to nonsteroidal anti-inflammatory drug use in the high-risk patient. Gastroenterology 2001; 120:594–606.
- Silverstein FE, Graham DY, Senior JR, et al. Misoprostol reduces serious gastrointestinal complications in patients with rheumatoid arthritis receiving nonsteroidal anti-inflammatory drugs. A randomized, double blind, placebo controlled trial. Ann Intern Med 1995; 123:241–249.
- Goldstein JL, Huang B, Amer F, Christopoulos NG. Ulcer recurrence in high-risk patients receiving nonsteroidal anti-inflammatory drugs plus low dose aspirin: results of a post hoc subanalysis. Clin Ther 2004; 26:1637–1643.
- Berger JS, Stebbins A, Granger CB, et al. Initial aspirin dose and outcome among ST-elevation myocardial infarction patients treated with fibrinolytic therapy. Circulation 2008; 117:192–199.
- Lanas A, Garcia-Rodriguez LA, Arroyo MT, et al; Investigators of the Asociación Española de Gastroenterología (AEG). Effect of antisecretory drugs and nitrates on the risk of ulcer bleeding associated with nonsteroidal anti-inflammatory drugs, antiplatelet agents, and anticoagulants. Am J Gastroenterol 2007; 102:507–515.
- Chin MW, Yong G, Bulsara MK, Rankin J, Forbes GM. Predictive and protective factors associated with upper gastrointestinal bleeding after percutaneous coronary intervention: a case-control study. Am J Gastroenterol 2007; 102:2411–2416.
- Hunt RH, Bazzoli F. Should NSAID/low dose aspirin takers be tested routinely for H. Pylori infection and treated if positive? Implications for primary risk of ulcer and ulcer relapse after initial healing. Aliment Pharmacol Ther 2004; 19 (suppl 1):9–16.
- Lewis JD, Bilker WB, Brensinger C, Farrar JT, Strom BL. Hospitalization and mortality rates from peptic ulcer disease and GI bleeding in the 1990s: relationship to sales of non-steroidal anti-inflammatory drugs and acid suppression medications. Am J Gastroenterol 2002; 97:2540–2549.
- Viviane A, Alan BN. Estimates of costs of hospital stays for variceal and nonvariceal upper gastrointestinal bleeding in the United States. Value Health 2008; 11:1–3.
- Yavorski RT, Wong RK, Maydonovitch C, Battin LS, Furnia A, Amundson DE. Analysis of 3,294 cases of upper gastrointestinal bleeding in military medical facilities. Am J Gastroenterol 1995; 90:568–573.
- Kaplan RC, Heckbert SR, Koepsell TD, et al. Risk factors for hospitalized gastrointestinal bleeding among older persons. Cardiovascular Health Study Investigators. J Am Geriatr Soc 2001; 49:126–133.
- Longstreth GF. Epidemiology of hospitalization for acute upper gastrointestinal hemorrhage: a population-based study. Am J Gastroenterol 1995; 90:206–210.
- Laine L, Peterson WL. Bleeding peptic ulcer. N Engl J Med 1994; 331:717–727.
- Wara P, Stodkilde H. Bleeding pattern before admission as guideline for emergency endoscopy. Scand J Gastroenterol 1985; 20:72–78.
- Jensen DM, Machicado GA. Diagnosis and treatment of severe hematochezia. The role of urgent colonoscopy after purge. Gastroenterology 1988; 95:1569–1574.
- Daniel WA, Egan S. The quantity of blood required to produce a tarry stool. J Am Med Assoc 1939; 113:2232.
- Rockall TA, Logan RF, Devlin HB, Northfield TC. Risk assessment after acute upper gastrointestinal hemorrhage. Gut 1996; 38:316–321.
- Blatchford O, Murray WR, Blatchford M. A risk score to predict need for treatment for upper-gastrointestinal hemorrhage. Lancet 2000; 356:1318–1321.
- Gralnek IM, Barkun AN, Bardou M. Management of acute bleeding from a peptic ulcer. N Engl J Med 2008; 359:928–937.
- Silverstein FE, Gilbert DA, Tedesco FJ, Buenger NK, Persing J. The national ASGE survey on upper gastrointestinal bleeding II. Clinical prognostic factors. Gastrointest Endosc 1981; 27:80–93.
- Corley DA, Stefan AM, Wolf M, Cook EF, Lee TH. Early indicators of prognosis in upper gastrointestinal hemorrhage. Am J Gastroenterol 1998; 93:336–340.
- Aljebreen AM, Fallone CA, Barkun AN. Nasogastric aspirate predicts high-risk endoscopic lesions in patients with acute upper-GI bleeding. Gastrointest Endosc 2004; 59:172–178.
- Barkun AN, Cockeram AW, Plourde V, Fedorak RN. Review article: acid suppression in non-variceal acute upper gastrointestinal bleeding. Aliment Pharmacol Ther 1999; 13:1565–1584.
- Levine JE, Leontiadis JI, Sharma VK, Howden CW. Meta-analysis: the efficacy of intravenous H2-receptor antagonists in bleeding peptic ulcer. Aliment Pharmacol Ther 2002; 16:1137–1142.
- Walt RP, Cottrell J, Mann SG, Freemantle NP, Langman MJ. Continuous intravenous famotidine for hemorrhage from peptic ulcer. Lancet 1992; 340:1058–1062.
- Labenz J, Peitz U, Leusing C, Tillenburg B, Blum AL, Börsch G. Efficacy of primed infusion with high dose ranitidine and omeprazole to maintain high intragastric pH in patients with peptic ulcer bleeding: a prospective randomized controlled study. Gut 1997; 40:36–41.
- Merki HS, Wilder-Smith CH. Do continuous infusions of omeprazole and ranitidine retain their effect with prolonged dosing? Gastroenterology 1994; 106:60–64.
- Netzer P, Gaia C, Sandoz M, et al. Effect of repeated injection and continuous infusion of omeprazole and ranitidine on intragastric pH over 72 hours. Am J Gastroenterol 1999; 94:351–357.
- Lin HJ, Lo WC, Cheng YC, Perng CL. Role of intravenous omeprazole in patients with high-risk peptic ulcer bleeding after successful endoscopic epinephrine injection: a prospective randomized comparative trial. Am J Gastroenterol 2006; 101:500–505.
- Lau JY, Sung JJ, Lee KK, et al. Effects of intravenous omeprazole on recurrent bleeding after endoscopic treatment of bleeding peptic ulcers. N Engl J Med 2000; 343:310–316.
- Leontiadis GI, Sharma VK, Howden CW. Proton pump inhibitor treatment for acute peptic ulcer bleeding. Cochrane Database Syst Rev 2006;CD002094.
- Andriulli A, Annese V, Caruso N, et al. Proton-pump inhibitors and outcome of endoscopic hemostasis in bleeding peptic ulcers: a series of meta-analyses. Am J Gastroenterol 2005; 100:207–219.
- Lau JY, Leung WK, Wu JC, et al. Omeprazole before endoscopy in patients with gastrointestinal bleeding. N Engl J Med 2007; 356:1631–1640.
- Dorward S, Sreedharan A, Leontiadis GI, Howden CW, Moayyedi P, Forman D. Proton pump inhibitor treatment initiated prior to endoscopic diagnosis in upper gastrointestinal bleeding. Cochrane Database Syst Rev 2006;CD005415.
- Bardou M, Toubouti Y, Benhaberou-Brun D, Rahme E, Barkun AN. Meta-analysis: proton-pump inhibition in high-risk patients with acute peptic ulcer bleeding. Aliment Pharmacol Ther 2005; 21:677–686.
- Bjorkman DJ, Zaman A, Fennerty MB, Lieberman D, Disario JA, Guest-Warnick G. Urgent vs elective endoscopy for acute non-variceal upper-GI bleeding: an effectiveness study. Gastointest Endosc 2004; 60:1–8.
- Lee SD, Kearney DJ. A randomized controlled trial of gastric lavage prior to endoscopy for acute upper gastrointestinal bleeding. J Clin Gastroenterol 2004; 38:861–865.
- Tack J, Janssens J, Vantrappen G, et al. Effect of erythromycin on gastric motility in controls and in diabetic gastroparesis. Gastroenterology 1992; 103:72–79.
- Xynos E, Mantides A, Papageorgiou A, Fountos A, Pechlivanides G, Vassilakis JS. Erythromycin accelerates delayed gastric emptying of solids in patients after truncal vagotomy and pyloroplasty. Eur J Surg 1992; 158:407–411.
- Coffin B, Pocard M, Panis Y, et al; Groupe des endoscopistes de garde á l’AP-HP. Erythromycin improves the quality of EGD in patients with acute upper GI bleeding: a randomized controlled study. Gastrointest Endosc 2002; 56:174–179.
- Frossard JL, Spahr L, Queneau PE, et al. Erythromycin intravenous bolus infusion in acute upper gastrointestinal bleeding: a randomized, controlled, double-blind trial. Gastroenterology 2002; 123:17–23.
- Winstead NS, Wilcox CM. Erythromycin prior to endoscopy for acute upper gastrointestinal hemorrhage: a cost-effectiveness analysis. Aliment Pharmacol Ther 2007; 26:1371–1377.
- Chak A, Cooper GS, Lloyd LE, Kolz CS, Barnhart BA, Wong RC. Effectiveness of endoscopy in patients admitted to the intensive care unit with upper GI hemorrhage. Gastrointest Endosc 2001; 53:6–13.
- Lau JY, Chung SC, Leung JW, Lo KK, Yung MY, Li AK. The evolution of stigmata of hemorrhage in bleeding peptic ulcers: a sequential endoscopic study. Endoscopy 1998; 30:513–518.
- Chung IK, Kim EJ, Lee MS, et al. Endoscopic factors predisposing to rebleeding following endoscopic hemostasis in bleeding peptic ulcers. Endoscopy 2001; 33:969–975.
- Elta GH. Acute nonvariceal upper gastrointestinal hemorrhage. Curr Treat Options Gastroenterol 2002; 5:147–152.
- Marmo R, Rotondano G, Piscopo R, Bianco MA, D’Angella R, Cipolletta L. Dual therapy versus monotherapy in the endoscopic treatment of high-risk bleeding ulcers: a meta-analysis of controlled trials. Am J Gastroenterol 2007; 102:279–289.
- Kovacs TO, Jensen DM. Recent advances in the endoscopic diagnosis and therapy of upper gastrointestinal, small intestinal, and colonic bleeding. Med Clin North Am 2002; 86:1319–1356.
- Kovacs TO, Jensen DM. Endoscopic treatment of ulcer bleeding. Curr Treat Options Gastroenterol 2007; 10:143–148.
- Jensen DM, Kovacs TO, Jutabha R, et al. Randomized trial of medical or endoscopic therapy to prevent recurrent ulcer hemorrhage in patients with adherent clots. Gastroenterology 2002; 123:407–413.
- Jensen DM, Machicado GA. Endoscopic hemostasis of ulcer hemorrhage with injection, thermal, and combination methods. Techniques Gastrointest Endosc 2005; 7:124–131.
- Bleau BL, Gostout CJ, Sherman KE, et al. Recurrent bleeding from peptic ulcer associated with adherent clot: a randomized study comparing endoscopic treatment with medical therapy. Gastrointest Endosc 2002; 56:1–6.
- Lau JY, Sung JJ, Lam YH, et al. Endoscopic retreatment compared with surgery in patients with recurrent bleeding after initial endoscopic control of bleeding ulcers. N Engl J Med 1999; 340:751–756.
- Wolf AT, Wasan SK, Saltzman JR. Impact of anticoagulation on rebleeding following endoscopic therapy for nonvariceal upper gastrointestinal hemorrhage. Am J Gastroenterol 2007; 102:290–296.
- Barkun A, Bardou M, Marshall JK; Nonvariceal Upper GI Bleeding Consensus Conference Group. Consensus recommendations for managing patients with nonvariceal upper gastrointestinal bleeding. Ann Intern Med 2003; 139:843–857.
- Marmo R, Rotondano G, Bianco MA, Piscopo R, Prisco A, Cipolletta L. Outcome of endoscopic treatment for peptic ulcer bleeding: is a second look necessary? A meta-analysis. Gastrointest Endosc 2003; 57:62–67.
- Dell’Era A, deFrancis R, Iannuzzi F. Acute variceal bleeding: pharmacological treatment and primary/secondary prophylaxis. Best Pract Res Clin Gastroenterol 2008; 22:279–294.
- Jalan R, Hayes PC. UK guidelines on the management of variceal hemorrhage in cirrhotic patients. British Society of Gastroenterology. Gut 2000; 46( suppl 3–4):III1–III15.
- Bernard B, Lebrec D, Mathurin P, Opolon P, Poynard T. Beta-adrenergic antagonists in the prevention of gastrointestinal rebleeding in patients with cirrhosis: a meta-analysis. Hepatology 1997; 25:63–70.
- De Franchis R. Evolving consensus in portal hypertension. Report of the Baveno IV consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol 2005; 43:167–176.
- Levacher S, Letoumelin P, Pateron D, Blaise M, Lapandry C, Pourriat JL. Early administration of terlipressin plus glyceryl trinitrate to control active upper gastrointestinal bleeding in cirrhotic patients. Lancet 1995; 346:865–868.
- Abraldes JG, Bosch J. Somatostatin and analogues in portal hypertension. Hepatology 2002; 35:1305–1312.
- Banares R, Albillos A, Rincon D, et al. Endoscopic treatment versus endoscopic plus pharmacological treatment for acute variceal bleeding: a meta analysis. Hepatology 2002; 35:609–615.
- Cappell M. Gastrointenstinal bleeding associated with myocardial infarction. Gastroenterol Clin North Am 2000; 29:423–444.
- Lin S, Konstance R, Jollis J, Fisher DA. The utility of upper endoscopy in patients with concomitant upper gastrointestinal bleeding and acute myocardial infarction. Dig Dis Sci 2006; 51:2377–2383.
- Silvis SE, Nebel O, Rogers G, Sugawa C, Mandelstam P. Endoscopic complications. Results of the 1974 American Society for Gastrointestinal Endoscopy Survey. JAMA 1976; 235:928–930.
- Lanas A, Garcia-Rodriguez LA, Arroyo MT, et al; Investigators of the Asociación Española de Gastroenterología (AEG). Effect of antisecretory drugs and nitrates on the risk of ulcer bleeding associated with nonsteroidal anti-inflammatory drugs, antiplatelet agents, and anticoagulants. Am J Gastroenterol 2007; 102:507–515.
- Shorr RI, Ray WA, Daugherty JR, Griffin MR. Concurrent use of nonsteroidal anti-inflammatory drugs and oral anticoagulants places elderly persons at high risk for hemorrhagic peptic ulcer disease. Arch Intern Med 1993; 153:1665–1670.
- Tabibian N. Acute gastrointestinal bleeding in anticoagulated patients: a prospective evaluation. Am J Gastroenterol 1989; 84:10–12.
- Choudari CP, Rajgopal C, Palmer KR. Acute gastrointestinal hemorrhage in anticoagulated patients: diagnoses and response to endoscopic treatment. Gut 1994; 35:464–466.
- Petty GW, Brown RD, Whisnant JP, Sicks JD, O’Fallon WM, Wiebers DO. Frequency of major complications of aspirin, warfarin, and intravenous heparin for secondary stroke prevention: a population-based study. Ann Intern Med 1999; 130:14–22.
- Landefeld CS, Rosenblatt MW, Goldman L. Bleeding in outpatients treated with warfarin: relation to the prothrombin time and important remediable lesions. Am J Med 1989; 87:153–159.
- Weil J, Colin-Jones D, Langman M, et al. Prophylactic aspirin and risk of peptic ulcer bleeding. BMJ 1995; 310:827–830.
- Cryer B, Feldman M. Effects of very low dose daily, long-term aspirin therapy on gastric, duodenal, and rectal prostaglandin levels and on mucosal injury in healthy humans. Gastroenterology 1999; 117:17–25.
- De Abajo FJ, Garcia Rodriguez LA. Risk of upper gastrointestinal bleeding and perforation associated with low-dose aspirin as plain and enteric-coated formulations. BMC Clin Pharmacol 2001; 1:1.
- Kelly JP, Kaufman DW, Jurgelon JM, Sheehan J, Koff RS, Shapiro S. Risk of aspirin-associated major upper gastrointestinal bleeding with enteric coated or buffered product. Lancet 1996; 348:1413–1416.
- Garcia Rodriguez LA, Hernandez-Diaz S, De Abajo FJ. Association between aspirin and upper gastrointestinal complications: systematic review of epidemiological studies. Br J Clin Pharmacol 2001; 52:563–571.
- Wilcox CM, Ladabaum U. A patient with high risk of gastrointestinal bleeding requiring nonsteroidal anti-inflammatory drugs. Clin Gastroenterol Hepatol 2006; 4:1090–1093.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996; 348:1329–1339.
- Chan FK, Ching JY, Hung LC, et al. Clopidogrel versus aspirin and esomeprazole to prevent recurrent ulcer bleeding. N Engl J Med 2005; 352:238–244.
- Lai KC, Chu KM, Hui WM, et al. Esomeprazole with aspirin versus clopidogrel for prevention of recurrent gastrointestinal ulcer complications. Clin Gastroenterol Hepatol 2006; 4:860–865.
- Ho MP, Maddox TM, Wang L, et al. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. JAMA 2009; 301:937–944.
- Siller-Matula JM, Spiel AO, Lang IM, Kreiner G, Christ G, Jilma B. Effects of pantoprazole and esomeprazole on platelet inhibition by clopidogrel. Am Heart J 2009; 157:148.e1–e5.
- Small DS, Farid NA, Payne CD, et al. Effects of proton pump inhibitor lansoprazole on the pharmacokinetics and pharmacodynamics of prasugel and clopidogrel. J Clin Pharmacol 2008; 48:475–484.
- Ishizaki T, Horai Y. Review article: cytochrome P450 and the metabolism of proton pump inhibitors—emphasis on rabeprazole. Aliment Pharacol Ther 1999; 13 (suppl 3):27–36.
- Lanas A, Fuentes J, Benito R, Serrano P, Bajador E, Sainz R. Helicobacter pylori increases the risk of upper gastrointestinal bleeding in patients taking low-dose aspirin. Aliment Pharmacol Ther 2002; 16:779–786.
- Chan FK. NSAID-Induced peptic ulcers and Helicobacter pylori infection: implications for patient management. Drug Saf 2005; 28:287–300.
- Bombardier V, Laine L, Reicin A, et al; VIGOR Study Group. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis: VIGOR Study Group. N Eng J Med 2000; 343:1520–1528.
- Griffin MR, Ray WA, Schaffner W. Nonsteroidal anti-inflammatory drug use and death from peptic ulcer in elderly persons. Ann Intern Med 1988; 109:359–363.
- Griffin MR, Piper JM, Daugherty JR, Snowden M, Ray WA. Nonsteroidal anti-inflammatory drug use and increased risk for peptic ulcer disease in elderly persons. Ann Intern Med 1991; 114:257–263.
- Smalley WE, Ray WA, Daugherty JR, Griffin MR. Nonsteroidal anti-inflammatory drugs and the incidence of hospitalizations for peptic ulcer disease in elderly persons. Am J Epidemiol 1995; 141:539–545.
- Laine L. Approaches to nonsteroidal anti-inflammatory drug use in the high-risk patient. Gastroenterology 2001; 120:594–606.
- Silverstein FE, Graham DY, Senior JR, et al. Misoprostol reduces serious gastrointestinal complications in patients with rheumatoid arthritis receiving nonsteroidal anti-inflammatory drugs. A randomized, double blind, placebo controlled trial. Ann Intern Med 1995; 123:241–249.
- Goldstein JL, Huang B, Amer F, Christopoulos NG. Ulcer recurrence in high-risk patients receiving nonsteroidal anti-inflammatory drugs plus low dose aspirin: results of a post hoc subanalysis. Clin Ther 2004; 26:1637–1643.
- Berger JS, Stebbins A, Granger CB, et al. Initial aspirin dose and outcome among ST-elevation myocardial infarction patients treated with fibrinolytic therapy. Circulation 2008; 117:192–199.
- Lanas A, Garcia-Rodriguez LA, Arroyo MT, et al; Investigators of the Asociación Española de Gastroenterología (AEG). Effect of antisecretory drugs and nitrates on the risk of ulcer bleeding associated with nonsteroidal anti-inflammatory drugs, antiplatelet agents, and anticoagulants. Am J Gastroenterol 2007; 102:507–515.
- Chin MW, Yong G, Bulsara MK, Rankin J, Forbes GM. Predictive and protective factors associated with upper gastrointestinal bleeding after percutaneous coronary intervention: a case-control study. Am J Gastroenterol 2007; 102:2411–2416.
- Hunt RH, Bazzoli F. Should NSAID/low dose aspirin takers be tested routinely for H. Pylori infection and treated if positive? Implications for primary risk of ulcer and ulcer relapse after initial healing. Aliment Pharmacol Ther 2004; 19 (suppl 1):9–16.
KEY POINTS
- The first priority is to ensure that the patient is hemodynamically stable, which often requires admission to the intensive care unit for monitoring and fluid resuscitation.
- Peptic ulcers account for most cases of upper GI bleeding, but bleeding from varices has a much higher case-fatality rate and always demands aggressive treatment.
- Patients with ulcer disease should be tested and treated for Helicobacter pylori infection.
- Patients with a history of bleeding ulcers who need long-term treatment with aspirin or a nonsteroidal anti-inflammatory drug should also be prescribed a proton pump inhibitor.
Controversies in non-ST-elevation acute coronary syndromes and percutaneous coronary interventions
Despite all the attention paid to ST-segment-elevation myocardial infarction (MI), in terms of sheer numbers, non-ST-elevation MI and unstable angina are where the action is. Acute coronary syndromes account for 2.43 million hospital discharges per year. Of these, 0.46 million are for ST-elevation MI and 1.97 million are for non-ST-elevation MI and unstable angina.1,2
A number of recent studies have begun to answer some of the pressing questions about treating these types of acute coronary syndromes. In this article, I update the reader on these studies, along with recent findings regarding stenting and antiplatelet agents. As you will see, they are all interconnected.
TO CATHETERIZE IS BETTER THAN NOT TO CATHETERIZE
In the 1990s, a topic of debate was whether patients presenting with unstable angina or non-ST-elevation MI should routinely undergo catheterization or whether they would do just as well with a conservative approach, ie, undergoing catheterization only if they developed recurrent, spontaneous, or stress-induced ischemia. Now, the data are reasonably clear and favor an aggressive strategy.3
Mehta et al4 performed a meta-analysis of seven randomized controlled trials (N = 9,212 patients) of aggressive vs conservative angiography and revascularization for non-ST-elevation MI or unstable angina. The results favored the aggressive strategy. At 17 months of follow-up, death or MI had occurred in 7.4% of patients who received the aggressive therapy compared with 11.0% of those who received the conservative therapy, for an odds ratio of 0.82 (P = .001).
The CRUSADE (Can Rapid Risk Stratification of Unstable Angina Patients Suppress Adverse Outcomes With Early Implemention of the ACC/AHA Guidelines?) Quality Improvement Initiative5 analyzed data from a registry of 17,926 patients with non-ST-elevation acute coronary syndrome who were at high risk because of positive cardiac markers or ischemic electrocardiographic changes. Overall, 2.0% of patients who received early invasive care (catheterization within the first 48 hours) died in the hospital compared with 6.2% of those who got no early invasive care, for an adjusted odds ratio of 0.63 (95% confidence interval [CI] 0.52–0.77).
The investigators also stratified the patients into those at low, medium, and high risk, using the criteria of the PURSUIT (Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin [eptifibatide] Therapy) risk score. There were fewer deaths with early invasive therapy in each risk group, and the risk reduction was greatest in the high-risk group.5
Bavry et al6 performed an updated meta-analysis of randomized trials. At a mean follow-up of 24 months, the relative risk of death from any cause was 0.75 in patients who received early invasive therapy.
In another meta-analysis, O’Donoghue et al7 found that the odds ratio of death, MI, or rehospitalization with acute coronary syndromes was 0.73 (95% CI 0.55–0.98) in men who received invasive vs conservative therapy; in women it was 0.81 (95% CI 0.65–1.01). In women, the benefit was statistically significant in those who had elevations of creatine kinase MB or troponin but not in those who did not, though the benefit in men appeared to be less dependent on the presence of biomarker abnormalities.
MUST ANGIOGRAPHY BE DONE IN THE FIRST 24 HOURS?
Although a number of trials showed that a routine invasive strategy leads to better outcomes than a conservative strategy, until recently we had no information as to whether the catheterization needed to be done early (eg, within the first 24 hours) or if it could be delayed a day or two while the patient received medical therapy.
Mehta et al8 conducted a trial to find out: the Timing of Intervention in Acute Coronary Syndrome (TIMACS) trial. Patients were included if they had unstable angina or non-ST-elevation MI, presented to a hospital within 24 hours of the onset of symptoms, and had two of three high-risk features: age 60 years or older, elevated cardiac biomarkers, or electrocardiographic findings compatible with ischemia. All received standard medical therapy, and 3,031 were randomly assigned to undergo angiography either within 24 hours after randomization or 36 or more hours after randomization.
At 6 months, the primary outcome of death, new MI, or stroke had occurred in 9.6% of the patients in the early-intervention group and in 11.3% of those in the delayed-intervention group, but the difference was not statistically significant. However, the difference in the rate of a secondary end point, death, MI, or refractory ischemia, was statistically significant: 9.5% vs 12.9%, P = .003, owing mainly to less refractory ischemia with early intervention.
The patients were also stratified into two groups by baseline risk. The rate of the primary outcome was significantly lower with early intervention in high-risk patients, but not in those at intermediate or low risk. Thus, early intervention may be beneficial in patients at high risk, such as those with ongoing chest pain, but not necessarily in those at low risk.
LEAVE NO LESION BEHIND?
Coronary artery disease often affects more than one segment. Until recently, it was not known whether we should stent all stenotic segments in patients presenting with non-ST-elevation MI or unstable angina, or only the “culprit lesion.”
Shishehbor et al9 examined data from a Cleveland Clinic registry of 1,240 patients with acute coronary syndrome and multivessel coronary artery disease who underwent bare-metal stenting. The median follow-up was 2.3 years. Using a propensity model to match patients in the two groups with similar baseline characteristics, they found that the rate of repeat revascularization was less with multivessel intervention than with culprit-only stenting, as was the rate of the combined end point of death, MI, or revascularization, but not that of all-cause mortality or the composite of death or MI.
BARE-METAL VS DRUG-ELUTING STENTS: BALANCING THE RISKS AND BENEFITS
After a patient receives a stent, two bad things can happen: the artery can close up again either gradually, in a process called restenosis, or suddenly, via thrombosis.
Drug-eluting stents were invented to solve the problem of restenosis, and they work very well. Stone et al10 pooled the data from four double-blind trials of sirolimus (Rapamune) stents and five double-blind trials of paclitaxel (Taxol) stents and found that, at 4 years, the rates of target-lesion revascularization (for restenosis) were 7.8% with sirolimus stents vs 23.6% with bare-metal stents (P < .001), and 10.1% with paclitaxel stents vs 20.0% with bare-metal stents (P < .001).
Thrombosis was much less common in these studies, occurring in 1.2% of the sirolimus stent groups vs 0.6% of the bare-metal stent groups (P = .20), and in 1.3% of the paclitaxel stent groups vs 0.9% of the bare-metal stent groups (P = .30).10
However, drug-eluting stents appear to increase the risk of thrombosis later on, ie, after 1 year. Bavry et al,11 in a meta-analysis, calculated that when stent thrombosis occurred, the median time after implantation was 15.5 months with sirolimus stents vs 4 months with bare-metal stents (P = .0052), and 18 months with paclitaxel stents vs 3.5 months with bare-metal stents (P = .04). The absolute risk of very late stent thrombosis after 1 year was very low, with five events per 1,000 patients with drug-eluting stents vs no events with bare-metal stents (P = .02). Nevertheless, this finding has practical implications. How long must patients continue dual antiplatelet therapy? And what if a patient needs surgery a year later?
Restenosis is not always so gradual
Although stent thrombosis is serious and often fatal, bare-metal stent restenosis is not always benign either, despite the classic view that stent restenosis is a gradual process that results in exertional angina. Reviewing 1,186 cases of bare-metal stent restenosis in 984 patients at Cleveland Clinic, Chen et al12 reported that 9.5% of cases presented as acute MI (2.2% as ST-elevation MI and 7.3% as non-ST-elevation MI), and 26.4% as unstable angina requiring hospitalization.
A Mayo Clinic study13 corroborated these findings. The 10-year incidence of clinical bare-metal stent restenosis was 18.1%, and the incidence of MI was 2.1%. The 10-year rate of bare-metal stent thrombosis was 2%. Off-label use, primarily in saphenous vein grafts, increased the incidence; other correlates were prior MI, peripheral arterial disease, and ulcerated lesions.
Furthermore, bare-metal stent thrombosis can also occur later. We saw a case that occurred 13 years after the procedure, 3 days after the patient stopped taking aspirin because he was experiencing flu-like symptoms, ran out of aspirin, and felt too sick to go out and buy more. The presentation was with ST-elevation MI. The patient recovered after treatment with intracoronary abciximab (ReoPro), percutaneous thrombectomy, balloon angioplasty, and, eventually, bypass surgery.14
No difference in risk of death with drug-eluting vs bare-metal stents
Even though drug-eluting stents pose a slightly higher risk of thrombosis than bare-metal stents, the risk of death is no higher.15
I believe the reason is that there are competing risks, and that the higher risk of thrombosis with first-generation drug-eluting stents and the higher risk of restenosis with bare-metal stents essentially cancel each other out. For most patients, there is an absolute benefit with drug-eluting stents, which reduce the need for revascularization with no effect in terms of either increasing or decreasing the risk of MI or death. Second-generation drug-eluting stents may have advantages in reducing rates of death or MI compared with first-generation drug-eluting stents, though this remains to be proven conclusively.
The right revascularization for the right patient
Bavry and I16 developed an algorithm for deciding on revascularization, posing a series of questions:
- Does the patient need any form of revascularization?
- Is he or she at higher risk of both stent thrombosis and restenosis, as in patients with diabetes, diffuse multivessel disease with bifurcation lesions, or chronic total occlusions? If so, coronary artery bypass grafting remains an excellent option.
- Does he or she have a low risk of restenosis, as in patients without diabetes with focal lesions in large vessels? If so, one could consider a bare-metal stent, which would probably be more cost-effective than a drug-eluting stent in this situation.
- Does the patient have relative contraindications to drug-eluting stents? Examples are a history of noncompliance with medical therapy, financial issues such as lack of insurance that would make buying clopidogrel (Plavix) a problem, long-term anticoagulation, or anticipated need for surgery in the next few years.
If a drug-eluting stent is used, certain measures can help ensure that it is used optimally. It should often be placed under high pressure with a noncompliant balloon so that it achieves contact with the artery wall all around. One should consider intravascular ultrasonographic guidance to make sure the stent is well opposed if it is in a very calcified lesion. Dual antiplatelet therapy with clopidogrel and aspirin should be given for at least 1 year, and if there is no bleeding, perhaps longer, pending further data.16
LEAVE NO PLATELET ACTIVATED?
Platelets have several types of receptors that, when bound by their respective ligands, lead to platelet activation and aggregation and, ultimately, thrombus formation. Antagonists to some of these receptors are available or are being developed.17
For long-term therapy, blocking the process “upstream,” ie, preventing platelet activation, is better than blocking it “downstream,” ie, preventing aggregation. For example, clopidogrel, ticlopipine (Ticlid), and prasugrel (Effient) have active metabolites that bind to a subtype of the adenosine diphosphate receptor and prevent platelet activation, whereas the glycoprotein IIb/IIIa inhibitors such as abciximab work downstream, binding to a different receptor and preventing aggregation.18
Dual therapy for 1 year is the standard of care after acute coronary syndromes
The evidence for using dual antiplatelet therapy (ie, aspirin plus clopidogrel) in patients with acute coronary syndromes without ST-elevation is very well established.
The Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) trial,19 published in 2001, found a 20% relative risk reduction and a 2% absolute risk reduction in the incidence of MI, stroke, or cardiovascular death in patients randomly assigned to receive clopidogrel plus aspirin for 1 year vs aspirin alone for 1 year (P < .001). In the subgroup of patients who underwent percutaneous coronary intervention, the relative risk reduction in the incidence of MI or cardiovascular death at 1 year of follow-up was 31% (P = .002).20
As a result of these findings, the cardiology society guidelines21 recommend a year of dual antiplatelet therapy after acute coronary syndromes, regardless of whether the patient is treated medically, percutaneously, or surgically.
But what happens after clopidogrel is withdrawn? Ho et al22 retrospectively analyzed data from Veterans Affairs hospitals and found a spike in the incidence of death or MI in the first 90 days after stopping clopidogrel treatment. This was true in medically treated patients as well as in those treated with percutaneous coronary interventions, in those with or without diabetes mellitus, in those who received a drug-eluting stent or a bare-metal stent, and in those treated longer than 9 months.
The investigators concluded that there might be a “clopidogrel rebound effect.” However, I believe that a true rebound effect, such as after withdrawal of heparin or warfarin, is biologically unlikely with clopidogrel, since clopidogrel irreversibly binds to its receptor for the 7- to 10-day life span of the platelet. Rather, I believe the phenomenon must be due to withdrawal of protection in patients at risk.
In stable patients, dual therapy is not as beneficial
Would dual antiplatelet therapy with clopidogrel and aspirin also benefit patients at risk of atherothrombotic events but without acute coronary syndromes?
The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial23 included 15,603 patients with either clinically evident but stable cardiovascular disease or multiple risk factors for athero-thrombosis. They were randomly assigned to receive either clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin. At a median of 28 months, the groups did not differ significantly in the rate of MI, stroke, or death from cardiovascular causes.
However, the subgroup of patients who had documented prior MI, ischemic stroke, or symptomatic peripheral arterial disease did appear to derive significant benefit from dual therapy.24 In this subgroup, the rate of MI, stroke, or cardiovascular death at a median follow-up of 27.6 months was 8.8% with placebo plus aspirin compared with 7.3% with clopidogrel plus aspirin, for a hazard ratio of 0.83 (95% CI 0.72–0.96, P = .01). Unstented patients with stable coronary artery disease but without prior MI derived no benefit.
Bleeding and thrombosis: The Scylla and Charybdis of antiplatelet therapy
However, with dual antiplatelet therapy, we steer between the Scylla of bleeding and the Charybdis of thrombosis.25
In the CHARISMA subgroup who had prior MI, ischemic stroke, or symptomatic peripheral arterial disease, the incidence of moderate or severe bleeding was higher with dual therapy than with aspirin alone, but the rates converged after about 1 year of treatment.24 Further, there was no difference in fatal bleeding or intracranial bleeding, although the rate of moderate bleeding (defined as the need for transfusion) was higher with dual therapy (2.0% vs 1.3%, P = .004).
I believe the data indicate that if a patient can tolerate dual antiplatelet therapy for 9 to 12 months without any bleeding issues, he or she is unlikely to have a major bleeding episode if dual therapy is continued beyond this time.
About half of bleeding events in patients on chronic antiplatelet therapy are gastrointestinal. To address this risk, in 2008 an expert committee from the American College of Cardiology, American College of Gastroenterology, and American Heart Association issued a consensus document26 in which they recommended assessing gastrointestinal risk factors in patients on antiplatelet therapy, such as history of ulcers (and testing for and treating Helicobacter pylori infection if present), history of gastrointestinal bleeding, concomitant anticoagulant therapy, and dual antiplatelet therapy. If any of these were present, the committee recommended considering a proton pump inhibitor. The committee also recommended a proton pump inhibitor for patients on antiplatelet therapy who have more than one of the following: age 60 years or more, corticosteroid use, or dyspepsia or gastroesophageal reflux symptoms.
Some ex vivo platelet studies and observational analyses have suggested that there might be an adverse interaction between clopidogrel and proton pump inhibitors due to a blunting of clopidogrel’s antiplatelet effect. A large randomized clinical trial was designed and launched to determine if a single-pill combination of the proton pump inhibitor omeprazole (Prilosec) and clopidogrel would be safer than clopidogrel alone when added to aspirin. Called COGENT-1 (Clopidogrel and the Optimization of GI Events Trial), it was halted early in 2009 when it lost its funding. However, preliminary data did not show an adverse interaction between clopidogrel and omeprazole.
What is the right dose of aspirin?
Steinhubl et al27 performed a post hoc observational analysis of data from the CHARISMA trial. Their findings suggested that higher doses of aspirin are not more effective than lower doses for chronic therapy. Furthermore, in the group receiving clopidogrel plus aspirin, the incidence of severe or life-threatening bleeding was significantly greater with aspirin doses higher than 100 mg than with doses lower than 100 mg, 2.6% vs 1.7%, P = .040.
A randomized, controlled trial called Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent Events/Optimal Antiplatelet Strategy for Interventions (CURRENT/OASIS 7)28 recently reported that higher-dose aspirin (ie, 325 mg) may be better than lower dose aspirin (ie, 81 mg) in patients with acute coronary syndromes undergoing percutaneous coronary intervention and receiving clopidogrel. During this 30-day study, there was no increase in overall bleeding with the higher dose of aspirin, though gastrointestinal bleeding was slightly increased.29 In a factorial design, the second part of this trial found that a higher-dose clopidogrel regimen reduced stent thrombosis.29
Should nonresponders get higher doses of clopidogrel?
In vitro, response to clopidogrel shows a normal bell-shaped distribution.30 In theory, therefore, patients who are hyperresponders may be at higher risk of bleeding, and those who are hyporesponders may be at risk of ischemic events.
A clinical trial is under way to examine whether hyporesponders should get higher doses. Called GRAVITAS (Gauging Responsiveness With a VerifyNow Assay Impact on Thrombosis and Safety), it will use a point-of-care platelet assay and then allocate patients to receive either standard therapy or double the dose of clopidogrel. The primary end point will be the rate of cardiovascular death, nonfatal MI, or stent thrombosis at 6 months.
Is prasugrel better than clopidogrel?
Prasugrel (Effient) is a new drug of the same class as clopidogrel, ie, a thienopyridine, with its active metabolite binding to the same platelet receptor as clopidogrel and inhibiting platelet aggregation more rapidly, more consistently, and to a greater extent than clopidogrel. Prasugrel was recently approved by the Food and Drug Administration. But is it better?31
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel–Thrombolysis in Myocardial Infarction (TRITON-TIMI 38) compared prasugrel and clopidogrel in 13,608 patients with moderate- to high-risk acute coronary syndromes who were scheduled to undergo percutaneous coronary intervention.32
Overall, prasugrel was better. At 15 months, the incidence of the primary end point (death from cardiovascular causes, nonfatal MI, or nonfatal stroke) was significantly lower with prasugrel therapy than with clopidogrel in the entire cohort (9.9% vs 12.1%, hazard ratio 0.81, 95% CI 0.73–0.90, P < .001), in the subgroup with ST-segment elevation MI, and in the subgroup with unstable angina or non-ST-elevation MI.
However, there was a price to pay. The rate of major bleeding was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% CI 1.03–1.68, P = .03). Assessing the balance between the risk and the benefit, the investigators identified three subgroups who did not derive a net clinical benefit from prasugrel: patients who had had a previous stroke or transient ischemic attack (this group actually had a net harm from prasugrel), patients 75 years of age or older, and patients weighing less than 60 kg (132 pounds).
More work is needed to determine which patients are best served by standard-dose clopidogrel, higher doses of clopidogrel, platelet-assay-guided dosing of clopidogrel, or prasugrel.24
Short-acting, potent intravenous platelet blockade with an agent such as cangrelor is theoretically appealing, but further research is necessary.33,34 Ticagrelor, a reversible adenosine diphosphate receptor antagonist, provides yet another potential option in antiplatelet therapy for acute coronary syndromes. In the recent PLATO trial (Study of Platelet Inhibition and Patient Outcomes), compared with clopidogrel, ticagrelor reduced the risk of ischemic events, including death.35,36 Here, too, there was more major bleeding (unrelated to coronary artery bypass grafting) with ticagrelor.
Thus, clinical assessment of an individual patient’s ischemic and bleeding risks will continue to be critical as therapeutic strategies evolve.
- Wiviott SD, Morrow DA, Giugliano RP, et al. Performance of the Thrombolysis In Myocardial Infarction risk index for early acute coronary syndrome in the National Registry of Myocardial Infarction: a simple risk index predicts mortality in both ST and non-ST elevation myocardial infarction [abstract]. J Am Coll Cardiol 2003; 43( suppl 2):365A–366A.
- Thom T, Haase N, Rosamond W, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2006; 113:e85–e151. Errata in Circulation 2006; 113:e696 and Circulation 2006 114:e630.
- Bhatt DL. To cath or not to cath. That is no longer the question. JAMA 2005; 293:2935–2937.
- Mehta SR, Cannon CP, Fox KA, et al. Routine vs selective invasive strategies in patients with acute coronary syndromes: a collaborative meta-analysis of randomized trials. JAMA 2005; 293:2908–2917.
- Bhatt DL, Roe MT, Peterson ED, et al; for the CRUSADE Investigators. Utilization of early invasive management strategies for high-risk patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE Quality Improvement Initiative. JAMA 2004; 292:2096–2104.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- O’Donoghue MO, Boden WE, Braunwald E, et al. Early invasive vs conservative treatment strategies in women and men with unstable angina and non-ST segment elevation myocardial infarction: a meta-analysis. JAMA 2008; 300:71–80.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Shishehbor MH, Lauer MS, Singh IM, et al. In unstable angina or non-ST-segment acute coronary syndrome, should patients with multivessel coronary artery disease undergo multivessel or culpritonly stenting? J Am Coll Cardiol 2007; 49:849–854.
- Stone GW, Moses JW, Ellis SG, et al. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. N Engl J Med 2007; 356:998–1008.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Chen MS, John JM, Chew DP, Lee DS, Ellis SG, Bhatt DL. Bare metal stent restenosis is not a benign clinical entity. Am Heart J 2006; 151:1260–1264.
- Doyle B, Rihal CS, O’Sullivan CJ, et al. Outcomes of stent thrombosis and restenosis during extended follow-up of patients treated with bare-metal coronary stents. Circulation 2007; 116:2391–2398.
- Sarkees ML, Bavry AA, Galla JM, Bhatt DL. Bare metal stent thrombosis 13 years after implantation. Cardiovasc Revasc Med 2009; 10:58–91.
- Bavry AA, Bhatt DL. Appropriate use of drug-eluting stents: balancing the reduction in restenosis with the concern of late thrombosis. Lancet 2008; 371:2134–2143.
- Bavry AA, Bhatt DL. Drug-eluting stents: dual antiplatelet therapy for every survivor? Circulation 2007; 116:696–699.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov 2003; 2:15–28.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502. Errata in N Engl J Med 2001; 345:1506 and N Engl J Med 2001; 345:1716.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- Anderson JL, Adams CD, Antman EM, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction); american College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons; American Association of Cardiovascular and Pulmonary Rehabilitation; Society for Academic Emergency Medicine. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol 2007; 50:e1–e157.
- Ho PM, Peterson ED, Wang L, et al. Incidence of death and acute myocardial infarction associated with stopping clopidogrel after acute coronary syndrome. JAMA 2008; 299:532–539. Erratum in JAMA 2008; 299:2390.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL. Intensifying platelet inhibition—navigating between Scylla and Charybdis. N Engl J Med 2007; 357:2078–2081.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2008; 118:1894–1909.
- Steinhubl SR, Bhatt DL, Brennan DM, et al; CHARISMA Investigators. Aspirin to prevent cardiovascular disease: the association of aspirin dose and clopidogrel with thrombosis and bleeding. Ann Intern Med 2009; 150:379–386.
- Mehta SR, Bassand JP, Chrolavicius S, et al; CURRENT-OASIS 7 Steering Committee. Design and rationale of CURRENT-OASIS 7: a randomized, 2 x 2 factorial trial evaluating optimal dosing strategies for clopidogrel and aspirin in patients with ST and non-ST-elevation acute coronary syndromes managed with an early invasive strategy. Am Heart J 2008; 156:1080–1088.
- Mehta SR, Van de Werf F. A randomized comparison of a clopidogrel high loading and maintenance dose regimen versus standard dose and high versus low dose aspirin in 25,000 patients with acute coronary syndromes: results of the CURRENT OASIS 7 trial. Paper presented at the European Society of Cardiology Congress; August 30, 2009; Barcelona, Spain. Also available online at www.Escardio.org/congresses/esc-2009/congress-reports. Accessed December 12, 2009.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AT, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
- Bhatt DL. Prasugrel in clinical practice [perspective]. N Engl J Med 2009; 361:940–942.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Bhatt DL, Lincoff AM, Gibson CM, et al; for the CHAMPION PLATFORM Investigators. Intravenous platelet blockade with cangrelor during PCI. N Engl J Med 2009 Nov 15(epub ahead of print).
- Harrington RA, Stone GW, McNulty S, et al. Platelet inhibition with cangrelor in patient sundergoing PCI. N Engl J Med 2009 Nov 17(epub ahead of print).
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Bhatt DL. Ticagrelor in ACS—what does PLATO teach us? Nat Rev Cardiol 2009; 6:737–738.
Despite all the attention paid to ST-segment-elevation myocardial infarction (MI), in terms of sheer numbers, non-ST-elevation MI and unstable angina are where the action is. Acute coronary syndromes account for 2.43 million hospital discharges per year. Of these, 0.46 million are for ST-elevation MI and 1.97 million are for non-ST-elevation MI and unstable angina.1,2
A number of recent studies have begun to answer some of the pressing questions about treating these types of acute coronary syndromes. In this article, I update the reader on these studies, along with recent findings regarding stenting and antiplatelet agents. As you will see, they are all interconnected.
TO CATHETERIZE IS BETTER THAN NOT TO CATHETERIZE
In the 1990s, a topic of debate was whether patients presenting with unstable angina or non-ST-elevation MI should routinely undergo catheterization or whether they would do just as well with a conservative approach, ie, undergoing catheterization only if they developed recurrent, spontaneous, or stress-induced ischemia. Now, the data are reasonably clear and favor an aggressive strategy.3
Mehta et al4 performed a meta-analysis of seven randomized controlled trials (N = 9,212 patients) of aggressive vs conservative angiography and revascularization for non-ST-elevation MI or unstable angina. The results favored the aggressive strategy. At 17 months of follow-up, death or MI had occurred in 7.4% of patients who received the aggressive therapy compared with 11.0% of those who received the conservative therapy, for an odds ratio of 0.82 (P = .001).
The CRUSADE (Can Rapid Risk Stratification of Unstable Angina Patients Suppress Adverse Outcomes With Early Implemention of the ACC/AHA Guidelines?) Quality Improvement Initiative5 analyzed data from a registry of 17,926 patients with non-ST-elevation acute coronary syndrome who were at high risk because of positive cardiac markers or ischemic electrocardiographic changes. Overall, 2.0% of patients who received early invasive care (catheterization within the first 48 hours) died in the hospital compared with 6.2% of those who got no early invasive care, for an adjusted odds ratio of 0.63 (95% confidence interval [CI] 0.52–0.77).
The investigators also stratified the patients into those at low, medium, and high risk, using the criteria of the PURSUIT (Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin [eptifibatide] Therapy) risk score. There were fewer deaths with early invasive therapy in each risk group, and the risk reduction was greatest in the high-risk group.5
Bavry et al6 performed an updated meta-analysis of randomized trials. At a mean follow-up of 24 months, the relative risk of death from any cause was 0.75 in patients who received early invasive therapy.
In another meta-analysis, O’Donoghue et al7 found that the odds ratio of death, MI, or rehospitalization with acute coronary syndromes was 0.73 (95% CI 0.55–0.98) in men who received invasive vs conservative therapy; in women it was 0.81 (95% CI 0.65–1.01). In women, the benefit was statistically significant in those who had elevations of creatine kinase MB or troponin but not in those who did not, though the benefit in men appeared to be less dependent on the presence of biomarker abnormalities.
MUST ANGIOGRAPHY BE DONE IN THE FIRST 24 HOURS?
Although a number of trials showed that a routine invasive strategy leads to better outcomes than a conservative strategy, until recently we had no information as to whether the catheterization needed to be done early (eg, within the first 24 hours) or if it could be delayed a day or two while the patient received medical therapy.
Mehta et al8 conducted a trial to find out: the Timing of Intervention in Acute Coronary Syndrome (TIMACS) trial. Patients were included if they had unstable angina or non-ST-elevation MI, presented to a hospital within 24 hours of the onset of symptoms, and had two of three high-risk features: age 60 years or older, elevated cardiac biomarkers, or electrocardiographic findings compatible with ischemia. All received standard medical therapy, and 3,031 were randomly assigned to undergo angiography either within 24 hours after randomization or 36 or more hours after randomization.
At 6 months, the primary outcome of death, new MI, or stroke had occurred in 9.6% of the patients in the early-intervention group and in 11.3% of those in the delayed-intervention group, but the difference was not statistically significant. However, the difference in the rate of a secondary end point, death, MI, or refractory ischemia, was statistically significant: 9.5% vs 12.9%, P = .003, owing mainly to less refractory ischemia with early intervention.
The patients were also stratified into two groups by baseline risk. The rate of the primary outcome was significantly lower with early intervention in high-risk patients, but not in those at intermediate or low risk. Thus, early intervention may be beneficial in patients at high risk, such as those with ongoing chest pain, but not necessarily in those at low risk.
LEAVE NO LESION BEHIND?
Coronary artery disease often affects more than one segment. Until recently, it was not known whether we should stent all stenotic segments in patients presenting with non-ST-elevation MI or unstable angina, or only the “culprit lesion.”
Shishehbor et al9 examined data from a Cleveland Clinic registry of 1,240 patients with acute coronary syndrome and multivessel coronary artery disease who underwent bare-metal stenting. The median follow-up was 2.3 years. Using a propensity model to match patients in the two groups with similar baseline characteristics, they found that the rate of repeat revascularization was less with multivessel intervention than with culprit-only stenting, as was the rate of the combined end point of death, MI, or revascularization, but not that of all-cause mortality or the composite of death or MI.
BARE-METAL VS DRUG-ELUTING STENTS: BALANCING THE RISKS AND BENEFITS
After a patient receives a stent, two bad things can happen: the artery can close up again either gradually, in a process called restenosis, or suddenly, via thrombosis.
Drug-eluting stents were invented to solve the problem of restenosis, and they work very well. Stone et al10 pooled the data from four double-blind trials of sirolimus (Rapamune) stents and five double-blind trials of paclitaxel (Taxol) stents and found that, at 4 years, the rates of target-lesion revascularization (for restenosis) were 7.8% with sirolimus stents vs 23.6% with bare-metal stents (P < .001), and 10.1% with paclitaxel stents vs 20.0% with bare-metal stents (P < .001).
Thrombosis was much less common in these studies, occurring in 1.2% of the sirolimus stent groups vs 0.6% of the bare-metal stent groups (P = .20), and in 1.3% of the paclitaxel stent groups vs 0.9% of the bare-metal stent groups (P = .30).10
However, drug-eluting stents appear to increase the risk of thrombosis later on, ie, after 1 year. Bavry et al,11 in a meta-analysis, calculated that when stent thrombosis occurred, the median time after implantation was 15.5 months with sirolimus stents vs 4 months with bare-metal stents (P = .0052), and 18 months with paclitaxel stents vs 3.5 months with bare-metal stents (P = .04). The absolute risk of very late stent thrombosis after 1 year was very low, with five events per 1,000 patients with drug-eluting stents vs no events with bare-metal stents (P = .02). Nevertheless, this finding has practical implications. How long must patients continue dual antiplatelet therapy? And what if a patient needs surgery a year later?
Restenosis is not always so gradual
Although stent thrombosis is serious and often fatal, bare-metal stent restenosis is not always benign either, despite the classic view that stent restenosis is a gradual process that results in exertional angina. Reviewing 1,186 cases of bare-metal stent restenosis in 984 patients at Cleveland Clinic, Chen et al12 reported that 9.5% of cases presented as acute MI (2.2% as ST-elevation MI and 7.3% as non-ST-elevation MI), and 26.4% as unstable angina requiring hospitalization.
A Mayo Clinic study13 corroborated these findings. The 10-year incidence of clinical bare-metal stent restenosis was 18.1%, and the incidence of MI was 2.1%. The 10-year rate of bare-metal stent thrombosis was 2%. Off-label use, primarily in saphenous vein grafts, increased the incidence; other correlates were prior MI, peripheral arterial disease, and ulcerated lesions.
Furthermore, bare-metal stent thrombosis can also occur later. We saw a case that occurred 13 years after the procedure, 3 days after the patient stopped taking aspirin because he was experiencing flu-like symptoms, ran out of aspirin, and felt too sick to go out and buy more. The presentation was with ST-elevation MI. The patient recovered after treatment with intracoronary abciximab (ReoPro), percutaneous thrombectomy, balloon angioplasty, and, eventually, bypass surgery.14
No difference in risk of death with drug-eluting vs bare-metal stents
Even though drug-eluting stents pose a slightly higher risk of thrombosis than bare-metal stents, the risk of death is no higher.15
I believe the reason is that there are competing risks, and that the higher risk of thrombosis with first-generation drug-eluting stents and the higher risk of restenosis with bare-metal stents essentially cancel each other out. For most patients, there is an absolute benefit with drug-eluting stents, which reduce the need for revascularization with no effect in terms of either increasing or decreasing the risk of MI or death. Second-generation drug-eluting stents may have advantages in reducing rates of death or MI compared with first-generation drug-eluting stents, though this remains to be proven conclusively.
The right revascularization for the right patient
Bavry and I16 developed an algorithm for deciding on revascularization, posing a series of questions:
- Does the patient need any form of revascularization?
- Is he or she at higher risk of both stent thrombosis and restenosis, as in patients with diabetes, diffuse multivessel disease with bifurcation lesions, or chronic total occlusions? If so, coronary artery bypass grafting remains an excellent option.
- Does he or she have a low risk of restenosis, as in patients without diabetes with focal lesions in large vessels? If so, one could consider a bare-metal stent, which would probably be more cost-effective than a drug-eluting stent in this situation.
- Does the patient have relative contraindications to drug-eluting stents? Examples are a history of noncompliance with medical therapy, financial issues such as lack of insurance that would make buying clopidogrel (Plavix) a problem, long-term anticoagulation, or anticipated need for surgery in the next few years.
If a drug-eluting stent is used, certain measures can help ensure that it is used optimally. It should often be placed under high pressure with a noncompliant balloon so that it achieves contact with the artery wall all around. One should consider intravascular ultrasonographic guidance to make sure the stent is well opposed if it is in a very calcified lesion. Dual antiplatelet therapy with clopidogrel and aspirin should be given for at least 1 year, and if there is no bleeding, perhaps longer, pending further data.16
LEAVE NO PLATELET ACTIVATED?
Platelets have several types of receptors that, when bound by their respective ligands, lead to platelet activation and aggregation and, ultimately, thrombus formation. Antagonists to some of these receptors are available or are being developed.17
For long-term therapy, blocking the process “upstream,” ie, preventing platelet activation, is better than blocking it “downstream,” ie, preventing aggregation. For example, clopidogrel, ticlopipine (Ticlid), and prasugrel (Effient) have active metabolites that bind to a subtype of the adenosine diphosphate receptor and prevent platelet activation, whereas the glycoprotein IIb/IIIa inhibitors such as abciximab work downstream, binding to a different receptor and preventing aggregation.18
Dual therapy for 1 year is the standard of care after acute coronary syndromes
The evidence for using dual antiplatelet therapy (ie, aspirin plus clopidogrel) in patients with acute coronary syndromes without ST-elevation is very well established.
The Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) trial,19 published in 2001, found a 20% relative risk reduction and a 2% absolute risk reduction in the incidence of MI, stroke, or cardiovascular death in patients randomly assigned to receive clopidogrel plus aspirin for 1 year vs aspirin alone for 1 year (P < .001). In the subgroup of patients who underwent percutaneous coronary intervention, the relative risk reduction in the incidence of MI or cardiovascular death at 1 year of follow-up was 31% (P = .002).20
As a result of these findings, the cardiology society guidelines21 recommend a year of dual antiplatelet therapy after acute coronary syndromes, regardless of whether the patient is treated medically, percutaneously, or surgically.
But what happens after clopidogrel is withdrawn? Ho et al22 retrospectively analyzed data from Veterans Affairs hospitals and found a spike in the incidence of death or MI in the first 90 days after stopping clopidogrel treatment. This was true in medically treated patients as well as in those treated with percutaneous coronary interventions, in those with or without diabetes mellitus, in those who received a drug-eluting stent or a bare-metal stent, and in those treated longer than 9 months.
The investigators concluded that there might be a “clopidogrel rebound effect.” However, I believe that a true rebound effect, such as after withdrawal of heparin or warfarin, is biologically unlikely with clopidogrel, since clopidogrel irreversibly binds to its receptor for the 7- to 10-day life span of the platelet. Rather, I believe the phenomenon must be due to withdrawal of protection in patients at risk.
In stable patients, dual therapy is not as beneficial
Would dual antiplatelet therapy with clopidogrel and aspirin also benefit patients at risk of atherothrombotic events but without acute coronary syndromes?
The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial23 included 15,603 patients with either clinically evident but stable cardiovascular disease or multiple risk factors for athero-thrombosis. They were randomly assigned to receive either clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin. At a median of 28 months, the groups did not differ significantly in the rate of MI, stroke, or death from cardiovascular causes.
However, the subgroup of patients who had documented prior MI, ischemic stroke, or symptomatic peripheral arterial disease did appear to derive significant benefit from dual therapy.24 In this subgroup, the rate of MI, stroke, or cardiovascular death at a median follow-up of 27.6 months was 8.8% with placebo plus aspirin compared with 7.3% with clopidogrel plus aspirin, for a hazard ratio of 0.83 (95% CI 0.72–0.96, P = .01). Unstented patients with stable coronary artery disease but without prior MI derived no benefit.
Bleeding and thrombosis: The Scylla and Charybdis of antiplatelet therapy
However, with dual antiplatelet therapy, we steer between the Scylla of bleeding and the Charybdis of thrombosis.25
In the CHARISMA subgroup who had prior MI, ischemic stroke, or symptomatic peripheral arterial disease, the incidence of moderate or severe bleeding was higher with dual therapy than with aspirin alone, but the rates converged after about 1 year of treatment.24 Further, there was no difference in fatal bleeding or intracranial bleeding, although the rate of moderate bleeding (defined as the need for transfusion) was higher with dual therapy (2.0% vs 1.3%, P = .004).
I believe the data indicate that if a patient can tolerate dual antiplatelet therapy for 9 to 12 months without any bleeding issues, he or she is unlikely to have a major bleeding episode if dual therapy is continued beyond this time.
About half of bleeding events in patients on chronic antiplatelet therapy are gastrointestinal. To address this risk, in 2008 an expert committee from the American College of Cardiology, American College of Gastroenterology, and American Heart Association issued a consensus document26 in which they recommended assessing gastrointestinal risk factors in patients on antiplatelet therapy, such as history of ulcers (and testing for and treating Helicobacter pylori infection if present), history of gastrointestinal bleeding, concomitant anticoagulant therapy, and dual antiplatelet therapy. If any of these were present, the committee recommended considering a proton pump inhibitor. The committee also recommended a proton pump inhibitor for patients on antiplatelet therapy who have more than one of the following: age 60 years or more, corticosteroid use, or dyspepsia or gastroesophageal reflux symptoms.
Some ex vivo platelet studies and observational analyses have suggested that there might be an adverse interaction between clopidogrel and proton pump inhibitors due to a blunting of clopidogrel’s antiplatelet effect. A large randomized clinical trial was designed and launched to determine if a single-pill combination of the proton pump inhibitor omeprazole (Prilosec) and clopidogrel would be safer than clopidogrel alone when added to aspirin. Called COGENT-1 (Clopidogrel and the Optimization of GI Events Trial), it was halted early in 2009 when it lost its funding. However, preliminary data did not show an adverse interaction between clopidogrel and omeprazole.
What is the right dose of aspirin?
Steinhubl et al27 performed a post hoc observational analysis of data from the CHARISMA trial. Their findings suggested that higher doses of aspirin are not more effective than lower doses for chronic therapy. Furthermore, in the group receiving clopidogrel plus aspirin, the incidence of severe or life-threatening bleeding was significantly greater with aspirin doses higher than 100 mg than with doses lower than 100 mg, 2.6% vs 1.7%, P = .040.
A randomized, controlled trial called Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent Events/Optimal Antiplatelet Strategy for Interventions (CURRENT/OASIS 7)28 recently reported that higher-dose aspirin (ie, 325 mg) may be better than lower dose aspirin (ie, 81 mg) in patients with acute coronary syndromes undergoing percutaneous coronary intervention and receiving clopidogrel. During this 30-day study, there was no increase in overall bleeding with the higher dose of aspirin, though gastrointestinal bleeding was slightly increased.29 In a factorial design, the second part of this trial found that a higher-dose clopidogrel regimen reduced stent thrombosis.29
Should nonresponders get higher doses of clopidogrel?
In vitro, response to clopidogrel shows a normal bell-shaped distribution.30 In theory, therefore, patients who are hyperresponders may be at higher risk of bleeding, and those who are hyporesponders may be at risk of ischemic events.
A clinical trial is under way to examine whether hyporesponders should get higher doses. Called GRAVITAS (Gauging Responsiveness With a VerifyNow Assay Impact on Thrombosis and Safety), it will use a point-of-care platelet assay and then allocate patients to receive either standard therapy or double the dose of clopidogrel. The primary end point will be the rate of cardiovascular death, nonfatal MI, or stent thrombosis at 6 months.
Is prasugrel better than clopidogrel?
Prasugrel (Effient) is a new drug of the same class as clopidogrel, ie, a thienopyridine, with its active metabolite binding to the same platelet receptor as clopidogrel and inhibiting platelet aggregation more rapidly, more consistently, and to a greater extent than clopidogrel. Prasugrel was recently approved by the Food and Drug Administration. But is it better?31
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel–Thrombolysis in Myocardial Infarction (TRITON-TIMI 38) compared prasugrel and clopidogrel in 13,608 patients with moderate- to high-risk acute coronary syndromes who were scheduled to undergo percutaneous coronary intervention.32
Overall, prasugrel was better. At 15 months, the incidence of the primary end point (death from cardiovascular causes, nonfatal MI, or nonfatal stroke) was significantly lower with prasugrel therapy than with clopidogrel in the entire cohort (9.9% vs 12.1%, hazard ratio 0.81, 95% CI 0.73–0.90, P < .001), in the subgroup with ST-segment elevation MI, and in the subgroup with unstable angina or non-ST-elevation MI.
However, there was a price to pay. The rate of major bleeding was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% CI 1.03–1.68, P = .03). Assessing the balance between the risk and the benefit, the investigators identified three subgroups who did not derive a net clinical benefit from prasugrel: patients who had had a previous stroke or transient ischemic attack (this group actually had a net harm from prasugrel), patients 75 years of age or older, and patients weighing less than 60 kg (132 pounds).
More work is needed to determine which patients are best served by standard-dose clopidogrel, higher doses of clopidogrel, platelet-assay-guided dosing of clopidogrel, or prasugrel.24
Short-acting, potent intravenous platelet blockade with an agent such as cangrelor is theoretically appealing, but further research is necessary.33,34 Ticagrelor, a reversible adenosine diphosphate receptor antagonist, provides yet another potential option in antiplatelet therapy for acute coronary syndromes. In the recent PLATO trial (Study of Platelet Inhibition and Patient Outcomes), compared with clopidogrel, ticagrelor reduced the risk of ischemic events, including death.35,36 Here, too, there was more major bleeding (unrelated to coronary artery bypass grafting) with ticagrelor.
Thus, clinical assessment of an individual patient’s ischemic and bleeding risks will continue to be critical as therapeutic strategies evolve.
Despite all the attention paid to ST-segment-elevation myocardial infarction (MI), in terms of sheer numbers, non-ST-elevation MI and unstable angina are where the action is. Acute coronary syndromes account for 2.43 million hospital discharges per year. Of these, 0.46 million are for ST-elevation MI and 1.97 million are for non-ST-elevation MI and unstable angina.1,2
A number of recent studies have begun to answer some of the pressing questions about treating these types of acute coronary syndromes. In this article, I update the reader on these studies, along with recent findings regarding stenting and antiplatelet agents. As you will see, they are all interconnected.
TO CATHETERIZE IS BETTER THAN NOT TO CATHETERIZE
In the 1990s, a topic of debate was whether patients presenting with unstable angina or non-ST-elevation MI should routinely undergo catheterization or whether they would do just as well with a conservative approach, ie, undergoing catheterization only if they developed recurrent, spontaneous, or stress-induced ischemia. Now, the data are reasonably clear and favor an aggressive strategy.3
Mehta et al4 performed a meta-analysis of seven randomized controlled trials (N = 9,212 patients) of aggressive vs conservative angiography and revascularization for non-ST-elevation MI or unstable angina. The results favored the aggressive strategy. At 17 months of follow-up, death or MI had occurred in 7.4% of patients who received the aggressive therapy compared with 11.0% of those who received the conservative therapy, for an odds ratio of 0.82 (P = .001).
The CRUSADE (Can Rapid Risk Stratification of Unstable Angina Patients Suppress Adverse Outcomes With Early Implemention of the ACC/AHA Guidelines?) Quality Improvement Initiative5 analyzed data from a registry of 17,926 patients with non-ST-elevation acute coronary syndrome who were at high risk because of positive cardiac markers or ischemic electrocardiographic changes. Overall, 2.0% of patients who received early invasive care (catheterization within the first 48 hours) died in the hospital compared with 6.2% of those who got no early invasive care, for an adjusted odds ratio of 0.63 (95% confidence interval [CI] 0.52–0.77).
The investigators also stratified the patients into those at low, medium, and high risk, using the criteria of the PURSUIT (Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin [eptifibatide] Therapy) risk score. There were fewer deaths with early invasive therapy in each risk group, and the risk reduction was greatest in the high-risk group.5
Bavry et al6 performed an updated meta-analysis of randomized trials. At a mean follow-up of 24 months, the relative risk of death from any cause was 0.75 in patients who received early invasive therapy.
In another meta-analysis, O’Donoghue et al7 found that the odds ratio of death, MI, or rehospitalization with acute coronary syndromes was 0.73 (95% CI 0.55–0.98) in men who received invasive vs conservative therapy; in women it was 0.81 (95% CI 0.65–1.01). In women, the benefit was statistically significant in those who had elevations of creatine kinase MB or troponin but not in those who did not, though the benefit in men appeared to be less dependent on the presence of biomarker abnormalities.
MUST ANGIOGRAPHY BE DONE IN THE FIRST 24 HOURS?
Although a number of trials showed that a routine invasive strategy leads to better outcomes than a conservative strategy, until recently we had no information as to whether the catheterization needed to be done early (eg, within the first 24 hours) or if it could be delayed a day or two while the patient received medical therapy.
Mehta et al8 conducted a trial to find out: the Timing of Intervention in Acute Coronary Syndrome (TIMACS) trial. Patients were included if they had unstable angina or non-ST-elevation MI, presented to a hospital within 24 hours of the onset of symptoms, and had two of three high-risk features: age 60 years or older, elevated cardiac biomarkers, or electrocardiographic findings compatible with ischemia. All received standard medical therapy, and 3,031 were randomly assigned to undergo angiography either within 24 hours after randomization or 36 or more hours after randomization.
At 6 months, the primary outcome of death, new MI, or stroke had occurred in 9.6% of the patients in the early-intervention group and in 11.3% of those in the delayed-intervention group, but the difference was not statistically significant. However, the difference in the rate of a secondary end point, death, MI, or refractory ischemia, was statistically significant: 9.5% vs 12.9%, P = .003, owing mainly to less refractory ischemia with early intervention.
The patients were also stratified into two groups by baseline risk. The rate of the primary outcome was significantly lower with early intervention in high-risk patients, but not in those at intermediate or low risk. Thus, early intervention may be beneficial in patients at high risk, such as those with ongoing chest pain, but not necessarily in those at low risk.
LEAVE NO LESION BEHIND?
Coronary artery disease often affects more than one segment. Until recently, it was not known whether we should stent all stenotic segments in patients presenting with non-ST-elevation MI or unstable angina, or only the “culprit lesion.”
Shishehbor et al9 examined data from a Cleveland Clinic registry of 1,240 patients with acute coronary syndrome and multivessel coronary artery disease who underwent bare-metal stenting. The median follow-up was 2.3 years. Using a propensity model to match patients in the two groups with similar baseline characteristics, they found that the rate of repeat revascularization was less with multivessel intervention than with culprit-only stenting, as was the rate of the combined end point of death, MI, or revascularization, but not that of all-cause mortality or the composite of death or MI.
BARE-METAL VS DRUG-ELUTING STENTS: BALANCING THE RISKS AND BENEFITS
After a patient receives a stent, two bad things can happen: the artery can close up again either gradually, in a process called restenosis, or suddenly, via thrombosis.
Drug-eluting stents were invented to solve the problem of restenosis, and they work very well. Stone et al10 pooled the data from four double-blind trials of sirolimus (Rapamune) stents and five double-blind trials of paclitaxel (Taxol) stents and found that, at 4 years, the rates of target-lesion revascularization (for restenosis) were 7.8% with sirolimus stents vs 23.6% with bare-metal stents (P < .001), and 10.1% with paclitaxel stents vs 20.0% with bare-metal stents (P < .001).
Thrombosis was much less common in these studies, occurring in 1.2% of the sirolimus stent groups vs 0.6% of the bare-metal stent groups (P = .20), and in 1.3% of the paclitaxel stent groups vs 0.9% of the bare-metal stent groups (P = .30).10
However, drug-eluting stents appear to increase the risk of thrombosis later on, ie, after 1 year. Bavry et al,11 in a meta-analysis, calculated that when stent thrombosis occurred, the median time after implantation was 15.5 months with sirolimus stents vs 4 months with bare-metal stents (P = .0052), and 18 months with paclitaxel stents vs 3.5 months with bare-metal stents (P = .04). The absolute risk of very late stent thrombosis after 1 year was very low, with five events per 1,000 patients with drug-eluting stents vs no events with bare-metal stents (P = .02). Nevertheless, this finding has practical implications. How long must patients continue dual antiplatelet therapy? And what if a patient needs surgery a year later?
Restenosis is not always so gradual
Although stent thrombosis is serious and often fatal, bare-metal stent restenosis is not always benign either, despite the classic view that stent restenosis is a gradual process that results in exertional angina. Reviewing 1,186 cases of bare-metal stent restenosis in 984 patients at Cleveland Clinic, Chen et al12 reported that 9.5% of cases presented as acute MI (2.2% as ST-elevation MI and 7.3% as non-ST-elevation MI), and 26.4% as unstable angina requiring hospitalization.
A Mayo Clinic study13 corroborated these findings. The 10-year incidence of clinical bare-metal stent restenosis was 18.1%, and the incidence of MI was 2.1%. The 10-year rate of bare-metal stent thrombosis was 2%. Off-label use, primarily in saphenous vein grafts, increased the incidence; other correlates were prior MI, peripheral arterial disease, and ulcerated lesions.
Furthermore, bare-metal stent thrombosis can also occur later. We saw a case that occurred 13 years after the procedure, 3 days after the patient stopped taking aspirin because he was experiencing flu-like symptoms, ran out of aspirin, and felt too sick to go out and buy more. The presentation was with ST-elevation MI. The patient recovered after treatment with intracoronary abciximab (ReoPro), percutaneous thrombectomy, balloon angioplasty, and, eventually, bypass surgery.14
No difference in risk of death with drug-eluting vs bare-metal stents
Even though drug-eluting stents pose a slightly higher risk of thrombosis than bare-metal stents, the risk of death is no higher.15
I believe the reason is that there are competing risks, and that the higher risk of thrombosis with first-generation drug-eluting stents and the higher risk of restenosis with bare-metal stents essentially cancel each other out. For most patients, there is an absolute benefit with drug-eluting stents, which reduce the need for revascularization with no effect in terms of either increasing or decreasing the risk of MI or death. Second-generation drug-eluting stents may have advantages in reducing rates of death or MI compared with first-generation drug-eluting stents, though this remains to be proven conclusively.
The right revascularization for the right patient
Bavry and I16 developed an algorithm for deciding on revascularization, posing a series of questions:
- Does the patient need any form of revascularization?
- Is he or she at higher risk of both stent thrombosis and restenosis, as in patients with diabetes, diffuse multivessel disease with bifurcation lesions, or chronic total occlusions? If so, coronary artery bypass grafting remains an excellent option.
- Does he or she have a low risk of restenosis, as in patients without diabetes with focal lesions in large vessels? If so, one could consider a bare-metal stent, which would probably be more cost-effective than a drug-eluting stent in this situation.
- Does the patient have relative contraindications to drug-eluting stents? Examples are a history of noncompliance with medical therapy, financial issues such as lack of insurance that would make buying clopidogrel (Plavix) a problem, long-term anticoagulation, or anticipated need for surgery in the next few years.
If a drug-eluting stent is used, certain measures can help ensure that it is used optimally. It should often be placed under high pressure with a noncompliant balloon so that it achieves contact with the artery wall all around. One should consider intravascular ultrasonographic guidance to make sure the stent is well opposed if it is in a very calcified lesion. Dual antiplatelet therapy with clopidogrel and aspirin should be given for at least 1 year, and if there is no bleeding, perhaps longer, pending further data.16
LEAVE NO PLATELET ACTIVATED?
Platelets have several types of receptors that, when bound by their respective ligands, lead to platelet activation and aggregation and, ultimately, thrombus formation. Antagonists to some of these receptors are available or are being developed.17
For long-term therapy, blocking the process “upstream,” ie, preventing platelet activation, is better than blocking it “downstream,” ie, preventing aggregation. For example, clopidogrel, ticlopipine (Ticlid), and prasugrel (Effient) have active metabolites that bind to a subtype of the adenosine diphosphate receptor and prevent platelet activation, whereas the glycoprotein IIb/IIIa inhibitors such as abciximab work downstream, binding to a different receptor and preventing aggregation.18
Dual therapy for 1 year is the standard of care after acute coronary syndromes
The evidence for using dual antiplatelet therapy (ie, aspirin plus clopidogrel) in patients with acute coronary syndromes without ST-elevation is very well established.
The Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) trial,19 published in 2001, found a 20% relative risk reduction and a 2% absolute risk reduction in the incidence of MI, stroke, or cardiovascular death in patients randomly assigned to receive clopidogrel plus aspirin for 1 year vs aspirin alone for 1 year (P < .001). In the subgroup of patients who underwent percutaneous coronary intervention, the relative risk reduction in the incidence of MI or cardiovascular death at 1 year of follow-up was 31% (P = .002).20
As a result of these findings, the cardiology society guidelines21 recommend a year of dual antiplatelet therapy after acute coronary syndromes, regardless of whether the patient is treated medically, percutaneously, or surgically.
But what happens after clopidogrel is withdrawn? Ho et al22 retrospectively analyzed data from Veterans Affairs hospitals and found a spike in the incidence of death or MI in the first 90 days after stopping clopidogrel treatment. This was true in medically treated patients as well as in those treated with percutaneous coronary interventions, in those with or without diabetes mellitus, in those who received a drug-eluting stent or a bare-metal stent, and in those treated longer than 9 months.
The investigators concluded that there might be a “clopidogrel rebound effect.” However, I believe that a true rebound effect, such as after withdrawal of heparin or warfarin, is biologically unlikely with clopidogrel, since clopidogrel irreversibly binds to its receptor for the 7- to 10-day life span of the platelet. Rather, I believe the phenomenon must be due to withdrawal of protection in patients at risk.
In stable patients, dual therapy is not as beneficial
Would dual antiplatelet therapy with clopidogrel and aspirin also benefit patients at risk of atherothrombotic events but without acute coronary syndromes?
The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial23 included 15,603 patients with either clinically evident but stable cardiovascular disease or multiple risk factors for athero-thrombosis. They were randomly assigned to receive either clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin. At a median of 28 months, the groups did not differ significantly in the rate of MI, stroke, or death from cardiovascular causes.
However, the subgroup of patients who had documented prior MI, ischemic stroke, or symptomatic peripheral arterial disease did appear to derive significant benefit from dual therapy.24 In this subgroup, the rate of MI, stroke, or cardiovascular death at a median follow-up of 27.6 months was 8.8% with placebo plus aspirin compared with 7.3% with clopidogrel plus aspirin, for a hazard ratio of 0.83 (95% CI 0.72–0.96, P = .01). Unstented patients with stable coronary artery disease but without prior MI derived no benefit.
Bleeding and thrombosis: The Scylla and Charybdis of antiplatelet therapy
However, with dual antiplatelet therapy, we steer between the Scylla of bleeding and the Charybdis of thrombosis.25
In the CHARISMA subgroup who had prior MI, ischemic stroke, or symptomatic peripheral arterial disease, the incidence of moderate or severe bleeding was higher with dual therapy than with aspirin alone, but the rates converged after about 1 year of treatment.24 Further, there was no difference in fatal bleeding or intracranial bleeding, although the rate of moderate bleeding (defined as the need for transfusion) was higher with dual therapy (2.0% vs 1.3%, P = .004).
I believe the data indicate that if a patient can tolerate dual antiplatelet therapy for 9 to 12 months without any bleeding issues, he or she is unlikely to have a major bleeding episode if dual therapy is continued beyond this time.
About half of bleeding events in patients on chronic antiplatelet therapy are gastrointestinal. To address this risk, in 2008 an expert committee from the American College of Cardiology, American College of Gastroenterology, and American Heart Association issued a consensus document26 in which they recommended assessing gastrointestinal risk factors in patients on antiplatelet therapy, such as history of ulcers (and testing for and treating Helicobacter pylori infection if present), history of gastrointestinal bleeding, concomitant anticoagulant therapy, and dual antiplatelet therapy. If any of these were present, the committee recommended considering a proton pump inhibitor. The committee also recommended a proton pump inhibitor for patients on antiplatelet therapy who have more than one of the following: age 60 years or more, corticosteroid use, or dyspepsia or gastroesophageal reflux symptoms.
Some ex vivo platelet studies and observational analyses have suggested that there might be an adverse interaction between clopidogrel and proton pump inhibitors due to a blunting of clopidogrel’s antiplatelet effect. A large randomized clinical trial was designed and launched to determine if a single-pill combination of the proton pump inhibitor omeprazole (Prilosec) and clopidogrel would be safer than clopidogrel alone when added to aspirin. Called COGENT-1 (Clopidogrel and the Optimization of GI Events Trial), it was halted early in 2009 when it lost its funding. However, preliminary data did not show an adverse interaction between clopidogrel and omeprazole.
What is the right dose of aspirin?
Steinhubl et al27 performed a post hoc observational analysis of data from the CHARISMA trial. Their findings suggested that higher doses of aspirin are not more effective than lower doses for chronic therapy. Furthermore, in the group receiving clopidogrel plus aspirin, the incidence of severe or life-threatening bleeding was significantly greater with aspirin doses higher than 100 mg than with doses lower than 100 mg, 2.6% vs 1.7%, P = .040.
A randomized, controlled trial called Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent Events/Optimal Antiplatelet Strategy for Interventions (CURRENT/OASIS 7)28 recently reported that higher-dose aspirin (ie, 325 mg) may be better than lower dose aspirin (ie, 81 mg) in patients with acute coronary syndromes undergoing percutaneous coronary intervention and receiving clopidogrel. During this 30-day study, there was no increase in overall bleeding with the higher dose of aspirin, though gastrointestinal bleeding was slightly increased.29 In a factorial design, the second part of this trial found that a higher-dose clopidogrel regimen reduced stent thrombosis.29
Should nonresponders get higher doses of clopidogrel?
In vitro, response to clopidogrel shows a normal bell-shaped distribution.30 In theory, therefore, patients who are hyperresponders may be at higher risk of bleeding, and those who are hyporesponders may be at risk of ischemic events.
A clinical trial is under way to examine whether hyporesponders should get higher doses. Called GRAVITAS (Gauging Responsiveness With a VerifyNow Assay Impact on Thrombosis and Safety), it will use a point-of-care platelet assay and then allocate patients to receive either standard therapy or double the dose of clopidogrel. The primary end point will be the rate of cardiovascular death, nonfatal MI, or stent thrombosis at 6 months.
Is prasugrel better than clopidogrel?
Prasugrel (Effient) is a new drug of the same class as clopidogrel, ie, a thienopyridine, with its active metabolite binding to the same platelet receptor as clopidogrel and inhibiting platelet aggregation more rapidly, more consistently, and to a greater extent than clopidogrel. Prasugrel was recently approved by the Food and Drug Administration. But is it better?31
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel–Thrombolysis in Myocardial Infarction (TRITON-TIMI 38) compared prasugrel and clopidogrel in 13,608 patients with moderate- to high-risk acute coronary syndromes who were scheduled to undergo percutaneous coronary intervention.32
Overall, prasugrel was better. At 15 months, the incidence of the primary end point (death from cardiovascular causes, nonfatal MI, or nonfatal stroke) was significantly lower with prasugrel therapy than with clopidogrel in the entire cohort (9.9% vs 12.1%, hazard ratio 0.81, 95% CI 0.73–0.90, P < .001), in the subgroup with ST-segment elevation MI, and in the subgroup with unstable angina or non-ST-elevation MI.
However, there was a price to pay. The rate of major bleeding was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% CI 1.03–1.68, P = .03). Assessing the balance between the risk and the benefit, the investigators identified three subgroups who did not derive a net clinical benefit from prasugrel: patients who had had a previous stroke or transient ischemic attack (this group actually had a net harm from prasugrel), patients 75 years of age or older, and patients weighing less than 60 kg (132 pounds).
More work is needed to determine which patients are best served by standard-dose clopidogrel, higher doses of clopidogrel, platelet-assay-guided dosing of clopidogrel, or prasugrel.24
Short-acting, potent intravenous platelet blockade with an agent such as cangrelor is theoretically appealing, but further research is necessary.33,34 Ticagrelor, a reversible adenosine diphosphate receptor antagonist, provides yet another potential option in antiplatelet therapy for acute coronary syndromes. In the recent PLATO trial (Study of Platelet Inhibition and Patient Outcomes), compared with clopidogrel, ticagrelor reduced the risk of ischemic events, including death.35,36 Here, too, there was more major bleeding (unrelated to coronary artery bypass grafting) with ticagrelor.
Thus, clinical assessment of an individual patient’s ischemic and bleeding risks will continue to be critical as therapeutic strategies evolve.
- Wiviott SD, Morrow DA, Giugliano RP, et al. Performance of the Thrombolysis In Myocardial Infarction risk index for early acute coronary syndrome in the National Registry of Myocardial Infarction: a simple risk index predicts mortality in both ST and non-ST elevation myocardial infarction [abstract]. J Am Coll Cardiol 2003; 43( suppl 2):365A–366A.
- Thom T, Haase N, Rosamond W, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2006; 113:e85–e151. Errata in Circulation 2006; 113:e696 and Circulation 2006 114:e630.
- Bhatt DL. To cath or not to cath. That is no longer the question. JAMA 2005; 293:2935–2937.
- Mehta SR, Cannon CP, Fox KA, et al. Routine vs selective invasive strategies in patients with acute coronary syndromes: a collaborative meta-analysis of randomized trials. JAMA 2005; 293:2908–2917.
- Bhatt DL, Roe MT, Peterson ED, et al; for the CRUSADE Investigators. Utilization of early invasive management strategies for high-risk patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE Quality Improvement Initiative. JAMA 2004; 292:2096–2104.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- O’Donoghue MO, Boden WE, Braunwald E, et al. Early invasive vs conservative treatment strategies in women and men with unstable angina and non-ST segment elevation myocardial infarction: a meta-analysis. JAMA 2008; 300:71–80.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Shishehbor MH, Lauer MS, Singh IM, et al. In unstable angina or non-ST-segment acute coronary syndrome, should patients with multivessel coronary artery disease undergo multivessel or culpritonly stenting? J Am Coll Cardiol 2007; 49:849–854.
- Stone GW, Moses JW, Ellis SG, et al. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. N Engl J Med 2007; 356:998–1008.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Chen MS, John JM, Chew DP, Lee DS, Ellis SG, Bhatt DL. Bare metal stent restenosis is not a benign clinical entity. Am Heart J 2006; 151:1260–1264.
- Doyle B, Rihal CS, O’Sullivan CJ, et al. Outcomes of stent thrombosis and restenosis during extended follow-up of patients treated with bare-metal coronary stents. Circulation 2007; 116:2391–2398.
- Sarkees ML, Bavry AA, Galla JM, Bhatt DL. Bare metal stent thrombosis 13 years after implantation. Cardiovasc Revasc Med 2009; 10:58–91.
- Bavry AA, Bhatt DL. Appropriate use of drug-eluting stents: balancing the reduction in restenosis with the concern of late thrombosis. Lancet 2008; 371:2134–2143.
- Bavry AA, Bhatt DL. Drug-eluting stents: dual antiplatelet therapy for every survivor? Circulation 2007; 116:696–699.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov 2003; 2:15–28.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502. Errata in N Engl J Med 2001; 345:1506 and N Engl J Med 2001; 345:1716.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- Anderson JL, Adams CD, Antman EM, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction); american College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons; American Association of Cardiovascular and Pulmonary Rehabilitation; Society for Academic Emergency Medicine. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol 2007; 50:e1–e157.
- Ho PM, Peterson ED, Wang L, et al. Incidence of death and acute myocardial infarction associated with stopping clopidogrel after acute coronary syndrome. JAMA 2008; 299:532–539. Erratum in JAMA 2008; 299:2390.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL. Intensifying platelet inhibition—navigating between Scylla and Charybdis. N Engl J Med 2007; 357:2078–2081.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2008; 118:1894–1909.
- Steinhubl SR, Bhatt DL, Brennan DM, et al; CHARISMA Investigators. Aspirin to prevent cardiovascular disease: the association of aspirin dose and clopidogrel with thrombosis and bleeding. Ann Intern Med 2009; 150:379–386.
- Mehta SR, Bassand JP, Chrolavicius S, et al; CURRENT-OASIS 7 Steering Committee. Design and rationale of CURRENT-OASIS 7: a randomized, 2 x 2 factorial trial evaluating optimal dosing strategies for clopidogrel and aspirin in patients with ST and non-ST-elevation acute coronary syndromes managed with an early invasive strategy. Am Heart J 2008; 156:1080–1088.
- Mehta SR, Van de Werf F. A randomized comparison of a clopidogrel high loading and maintenance dose regimen versus standard dose and high versus low dose aspirin in 25,000 patients with acute coronary syndromes: results of the CURRENT OASIS 7 trial. Paper presented at the European Society of Cardiology Congress; August 30, 2009; Barcelona, Spain. Also available online at www.Escardio.org/congresses/esc-2009/congress-reports. Accessed December 12, 2009.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AT, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
- Bhatt DL. Prasugrel in clinical practice [perspective]. N Engl J Med 2009; 361:940–942.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Bhatt DL, Lincoff AM, Gibson CM, et al; for the CHAMPION PLATFORM Investigators. Intravenous platelet blockade with cangrelor during PCI. N Engl J Med 2009 Nov 15(epub ahead of print).
- Harrington RA, Stone GW, McNulty S, et al. Platelet inhibition with cangrelor in patient sundergoing PCI. N Engl J Med 2009 Nov 17(epub ahead of print).
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Bhatt DL. Ticagrelor in ACS—what does PLATO teach us? Nat Rev Cardiol 2009; 6:737–738.
- Wiviott SD, Morrow DA, Giugliano RP, et al. Performance of the Thrombolysis In Myocardial Infarction risk index for early acute coronary syndrome in the National Registry of Myocardial Infarction: a simple risk index predicts mortality in both ST and non-ST elevation myocardial infarction [abstract]. J Am Coll Cardiol 2003; 43( suppl 2):365A–366A.
- Thom T, Haase N, Rosamond W, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2006; 113:e85–e151. Errata in Circulation 2006; 113:e696 and Circulation 2006 114:e630.
- Bhatt DL. To cath or not to cath. That is no longer the question. JAMA 2005; 293:2935–2937.
- Mehta SR, Cannon CP, Fox KA, et al. Routine vs selective invasive strategies in patients with acute coronary syndromes: a collaborative meta-analysis of randomized trials. JAMA 2005; 293:2908–2917.
- Bhatt DL, Roe MT, Peterson ED, et al; for the CRUSADE Investigators. Utilization of early invasive management strategies for high-risk patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE Quality Improvement Initiative. JAMA 2004; 292:2096–2104.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- O’Donoghue MO, Boden WE, Braunwald E, et al. Early invasive vs conservative treatment strategies in women and men with unstable angina and non-ST segment elevation myocardial infarction: a meta-analysis. JAMA 2008; 300:71–80.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Shishehbor MH, Lauer MS, Singh IM, et al. In unstable angina or non-ST-segment acute coronary syndrome, should patients with multivessel coronary artery disease undergo multivessel or culpritonly stenting? J Am Coll Cardiol 2007; 49:849–854.
- Stone GW, Moses JW, Ellis SG, et al. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. N Engl J Med 2007; 356:998–1008.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Chen MS, John JM, Chew DP, Lee DS, Ellis SG, Bhatt DL. Bare metal stent restenosis is not a benign clinical entity. Am Heart J 2006; 151:1260–1264.
- Doyle B, Rihal CS, O’Sullivan CJ, et al. Outcomes of stent thrombosis and restenosis during extended follow-up of patients treated with bare-metal coronary stents. Circulation 2007; 116:2391–2398.
- Sarkees ML, Bavry AA, Galla JM, Bhatt DL. Bare metal stent thrombosis 13 years after implantation. Cardiovasc Revasc Med 2009; 10:58–91.
- Bavry AA, Bhatt DL. Appropriate use of drug-eluting stents: balancing the reduction in restenosis with the concern of late thrombosis. Lancet 2008; 371:2134–2143.
- Bavry AA, Bhatt DL. Drug-eluting stents: dual antiplatelet therapy for every survivor? Circulation 2007; 116:696–699.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov 2003; 2:15–28.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502. Errata in N Engl J Med 2001; 345:1506 and N Engl J Med 2001; 345:1716.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- Anderson JL, Adams CD, Antman EM, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction); american College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons; American Association of Cardiovascular and Pulmonary Rehabilitation; Society for Academic Emergency Medicine. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol 2007; 50:e1–e157.
- Ho PM, Peterson ED, Wang L, et al. Incidence of death and acute myocardial infarction associated with stopping clopidogrel after acute coronary syndrome. JAMA 2008; 299:532–539. Erratum in JAMA 2008; 299:2390.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL. Intensifying platelet inhibition—navigating between Scylla and Charybdis. N Engl J Med 2007; 357:2078–2081.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2008; 118:1894–1909.
- Steinhubl SR, Bhatt DL, Brennan DM, et al; CHARISMA Investigators. Aspirin to prevent cardiovascular disease: the association of aspirin dose and clopidogrel with thrombosis and bleeding. Ann Intern Med 2009; 150:379–386.
- Mehta SR, Bassand JP, Chrolavicius S, et al; CURRENT-OASIS 7 Steering Committee. Design and rationale of CURRENT-OASIS 7: a randomized, 2 x 2 factorial trial evaluating optimal dosing strategies for clopidogrel and aspirin in patients with ST and non-ST-elevation acute coronary syndromes managed with an early invasive strategy. Am Heart J 2008; 156:1080–1088.
- Mehta SR, Van de Werf F. A randomized comparison of a clopidogrel high loading and maintenance dose regimen versus standard dose and high versus low dose aspirin in 25,000 patients with acute coronary syndromes: results of the CURRENT OASIS 7 trial. Paper presented at the European Society of Cardiology Congress; August 30, 2009; Barcelona, Spain. Also available online at www.Escardio.org/congresses/esc-2009/congress-reports. Accessed December 12, 2009.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AT, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
- Bhatt DL. Prasugrel in clinical practice [perspective]. N Engl J Med 2009; 361:940–942.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Bhatt DL, Lincoff AM, Gibson CM, et al; for the CHAMPION PLATFORM Investigators. Intravenous platelet blockade with cangrelor during PCI. N Engl J Med 2009 Nov 15(epub ahead of print).
- Harrington RA, Stone GW, McNulty S, et al. Platelet inhibition with cangrelor in patient sundergoing PCI. N Engl J Med 2009 Nov 17(epub ahead of print).
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Bhatt DL. Ticagrelor in ACS—what does PLATO teach us? Nat Rev Cardiol 2009; 6:737–738.
KEY POINTS
- The data favor an aggressive strategy of routine catheterization, rather than a conservative strategy of catheterization only if a patient develops recurrent, spontaneous, or stress-induced ischemia.
- Early percutaneous intervention (within 24 hours) may be beneficial in patients at higher risk, but not necessarily in those at lower risk.
- Drug-eluting stents appear safe, assuming dual antiplatelet therapy is used. It is unclear how long this therapy needs to be continued.
- The choice of revascularization strategy—bypass surgery, bare-metal stent, or drug-eluting stent—should be individualized based on the risk of restenosis, thrombosis, and other factors.
Grand Rounds: Girl, 6, With Rapid Heart Rate
A 6-year-old girl was brought by her parents to the emergency department (ED) with an elevated heart rate. According to the parents, the girl was carrying her younger sister when they both fell, landing on their buttocks. The child told them that her heart was beating fast, and the parents said she appeared to be on the verge of fainting.
They stated that their daughter was healthy and active; they denied previous episodes of shortness of breath, headache, weakness, tachycardia, syncope, or fatigue with exercise. Her caffeine intake, they claimed, was limited to one small cup of soda they allowed her each week.
Initial evaluation in the ED revealed an anxious child with tachycardia and shortness of breath. She presented with a temperature of 98.3°F (36.8°C); pulse, 210 beats/min; respirations, 33 breaths/min; blood pressure, 100/72 mm Hg; weight, 78 lb; height, 45 in; and BMI, 27.1. ECG revealed a heart rate exceeding 210 beats/min, and a pediatric cardiologist made a diagnosis of supraventricular tachycardia (SVT).
The pediatric cardiologist prescribed an adenosine IV drip, which successfully stabilized the child’s heart to sinus rhythm. After three hours in the ED, the patient was discharged with a stable heart rate of 100 beats/min. (It is well known that heart rate regulation changes significantly during development; this is most obvious in higher basal rates in infants and children, compared with adults.1)
The parents were advised to administer atenolol 12.5 mg (one tablet) twice daily and to make a follow-up appointment with a pediatric electrophysiologist. (Although atenolol is not currently FDA approved for this use, a multicenter prospective randomized controlled trial comparing digoxin with beta-blockers for the treatment of SVT in children is presently under way.2)
At that appointment, the pediatric electrophysiologist provided information to the parents regarding the therapeutic options for SVT. The parents continued to administer atenolol to the child, as was deemed necessary until any accessory electrical pathway could be identified and, if so, an ablation procedure could be performed. They were uncertain how to proceed so long as their daughter experienced no recurrent episodes of SVT while receiving pharmacologic therapy.
However, six months after the initial episode, the child (then age 7) presented to the ED once again with recurrent SVT. The pediatric cardiologist ordered an adenosine IV drip, which resulted in successful conversion to sinus rhythm. The parents were instructed to increase the child’s atenolol dosage to 25 mg twice a day.
Six months later, after extensive research and consultation, the parents agreed to an ablation procedure in order to prevent further episodes of SVT. Upon their informed consent, the child was sent to a cardiac catheterization laboratory for an electrophysiology study (EPS), which confirmed the presence of an accessory pathway, as well as the diagnosis of atrioventricular reciprocating tachycardia (AVRT). The procedure was followed by radiofrequency catheter ablation to correct the 7-year-old patient’s accessory pathway–mediated reentry tachycardia.
Discussion
SVT, also known as paroxysmal supraventricular tachycardia (PSVT), is one of the most common symptomatic pediatric arrhythmias, affecting between one in 25,000 and one in 250 children.3 It is defined as rapid heart rhythm (140 to 240 beats/min) that is caused by the presence of additional electrical connections and/or congenital muscle fibers between the atrium and the ventricle or within the atrioventricular (AV) node that did not, for unknown reasons, separate completely during development.4 SVT can be triggered by physical or psychological stress automaticity.3
Approximately 50% of children with SVT present with a first episode before age 1. SVT usually occurs in early childhood, between ages 6 and 9.4 Almost 90% of pediatric patients with SVT are diagnosed with a reentry mechanism.3 The symptoms experienced may be resolved pharmacologically or by means of an invasive therapy. Serious sequelae associated with SVT include heart failure and cardiac arrest.
For children with rare and mildly symptomatic episodes in whom SVT is easily terminated, the SVT may not warrant treatment. However, it may be advisable to offer medical therapy or transcatheter ablation as therapeutic options for children with episodes that are difficult to terminate, occur frequently, or occur during participation in athletics.4
Pathophysiology
SVT generally presents as one of three types: AVRT, which is also known as Wolff-Parkinson-White syndrome; atrioventricular nodal reentry tachycardia (AVNRT); and automatic tachycardia (AT).
AVRT, the most common type of SVT, comprises about 90% of pediatric cases. It is defined by the presence of one or more accessory conduction pathways that are anatomically separated from the normal cardiac conduction system.5 AVRT may be orthodromic (that is, the arrhythmia circuit proceeds down the AV node and retrograde up the accessory conduction pathway) or antedromic (ie, proceeding down the accessory pathway and up the AV node5; see figure.6,7)
AVNRT, considered the second most common type of SVT in children, accounts for about 10% of pediatric cases. AVNRT is caused by an interaction between the two types of pathways within the AV node—one with a fast conduction time and a short refractory period, and the other with a slow conduction time and a long refractory period. AVNRT occurs when the antegrade conduction block in the fast pathway results in conduction over the slow pathway and back up the fast pathway, forming a microreentrant circuit.5
AT is the result of rapid depolarization from an automatic focus originating within the atria but outside the sinus node.3
Patient Presentation and History
The typical presentation of AVRT in children of school age includes palpitations, chest pain or tightness, dizziness, anxiety, decrease in exercise tolerance, easy fatigability, and/or shortness of breath.3 Onset is described as abrupt, while termination of SVT is described as slower because the catecholamine levels are typically elevated.4
The frequency and duration of SVT can vary greatly, from a few minutes to a few hours; it can occur as regularly as daily or as uncommonly as once or twice per year.4 Additionally, SVT symptoms can go unrecognized until a cardiac dysfunction develops. As for the patient in the case study, no apparent factor in her history was identified that may have induced SVT.
The differential diagnosis for SVT is broad, including sinus tachycardia, multifocal atrial tachycardia, and SVT with aberrancy.8 Additional considerations include stress, anxiety, hyperthyroidism, electrolyte abnormalities, and dehydration—any of which can present with a tachycardia response.4 Furthermore, clinicians are often unlikely to diagnose a child with any cardiac problem because chest pain is more commonly a presenting symptom of a pulmonary or musculoskeletal condition than a cardiac problem.3
Physical Examination
SVT can be diagnosed based on medical history and physical examination. During the physical examination, providers will assess the patient’s blood pressure and pulse, auscultate heart and lung sounds, assess the veins in the patient’s neck for different types of pulsations, and conduct cardiac maneuvers, including the Valsalva maneuver and carotid sinus massage.9,10
Laboratory Work-up and Diagnosis
Three specific tests help clinicians monitor and evaluate a patient’s conduction system. ECG is important to assess the heart rhythm both at baseline and when symptoms are occurring, if possible.3 Ambulatory ECG (ie, Holter monitoring, event recorders) record the patient’s heart rhythm on a continuous basis.
An EPS, which is performed to classify the mechanism of SVT, is conducted by inserting one or more electrocatheters into the heart by way of the femoral vein or other peripheral vessel.3 Pacing and sensing electrodes at the ends of the catheters record local intracardiac electrical activity and timing information, providing a detailed analysis of the heart’s electrical activity. The EPS is critical to determine the presence of one or more extra electrical pathways within the heart and to localize it by region.3,11 An ablation procedure may follow.
Management Options
SVT can be treated pharmacologically or nonpharmacologically. First-line pharmacologic options include certain beta-blockers (including atenolol and propranolol), digoxin, and calcium channel blockers. Second-line pharmacologic treatments include amiodarone, flecainide, and sotalol,4 all of which are contraindicated in children younger than 1 year because of these patients’ hemodynamic dependency on the heart and inability to generate stroke volume.3 Pharmacologic treatment of SVT is associated with a 68% success rate in children4 (see Table 14).
For children in whom pharmacologic treatment is ineffective, an ablation procedure may be performed. Radiofrequency catheter ablation is currently considered first-line therapy for AVRT and AVNRT.12 In this invasive procedure, intracardiac electrical mapping is performed and the initiating focus of the arrhythmia or the accessory electrical pathway that has been identified within the heart is destroyed by radiofrequency energy, delivered by electrocatheter. Ablations performed during the acute phase of SVT have a 95% success rate.3,13
Cryoablation is a relatively new treatment in which liquid nitrous oxide is used to cool the catheter to subfreezing temperatures, enabling it to destroy the myocardial tissue by freezing.3,14 The advantage of cryoablation is the option of reversible cooling, which allows the electrophysiologist to test the area first, confirming the accuracy of the apparent location accessory pathway.15
Noninvasive, nonpharmacologic interventions that increase the refractoriness of the AV node may be successful in terminating the tachyarrhythmia during episodes of SVT (see Table 23,9,13,16). They are used to terminate and diagnose tachydysrhythmias, increase parasympathetic tone, and slow conduction through the AV node.3
Patient Education
It is very important for health care providers to relieve parents’ and caregivers’ stress, anxiety, and uncertainty by educating them about the child’s cardiac condition of SVT. Information to convey include an understanding of what SVT is, what may cause it, what triggers the patient should avoid, what treatments are available and appropriate (including the maneuvers shown in Table 2), and what outcomes may be expected. An excellent patient/family education handout from the Children’s Hospitals and Clinics of Minnesota17 is available at www.childrensmn.org/Manuals/PFS/Condill/018303.pdf.
Follow-Up
Primary care providers must emphasize the importance of monitoring the patient’s progress, based on the severity of his or her SVT symptoms. The provider may choose to monitor the patient for a few weeks or a few months, assessing the frequency of arrhythmia recurrence and the heart rate, to adjust or substitute medications based on repeat ECG or Holter evaluations, and to plan further therapy, should the condition worsen.5
The Case Patient
One month after undergoing radiofrequency catheter ablation, the child presented to the pediatric cardiologist for follow-up. Since the procedure, she had been without any symptoms referable to the cardiovascular system. She denied experiencing any fast heart rate, palpitations, chest pain, shortness of breath, or dizziness. ECG demonstrated normal sinus rhythm.
Two years after undergoing radiofrequency ablation, the child is functioning at a normal activity level with no recurrence of SVT episodes.
Conclusion
The purpose of this case study is to improve primary care providers’ understanding of SVT in children and to convey the importance of identifying the condition in a timely manner and referring patients to a pediatric cardiologist or electrophysiologist. For most children affected by SVT, a regimen of pharmacologic and/or nonpharmacologic treatment—supported by detailed education for their parents and caregivers—can allow them to live a healthy, normal life.
1. Kudielka BM, Buske-Kirschbaum A, Hellhammer DH, Kirschbaum C. Differential heart rate reactivity and recovery after psychosocial stress (TSST) in healthy children, younger adults, and elderly adults: the impact of age and gender. Int J Behav Med. 2004;11(2):116-121.
2. Multicenter Study of Antiarrhythmic Medications for Treatment of Infants With Supraventricular Tachycardia. www.clinicaltrials.gov/ct2/results?term=NCT00390546. Accessed January 26, 2010.
3. Schlechte EA, Boramanand N, Funk M. Supraventricular tachycardia in the pediatric primary care setting: age-related presentation, diagnosis, and management. J Pediatr Health Care. 2008;22(5): 289-299.
4. Salerno JC, Seslar SP. Supraventricular tachycardia. Arch Pediatr Adolesc Med. 2009;163(3): 268-274.
5. Doniger SJ, Sharieff GQ. Pediatric dysrhythmias. Pediatr Clin North Am. 2006;53(1):85-105, vi.
6. Mavroudis C, Deal BJ, Backer CL, Tsao S. Arrhythmia surgery in patients with and without congenital heart disease. Ann Thorac Surg. 2008;86(3):857-868.
7. Wang PJ, Estes NAM III. Supraventricular tachycardia. Circulation. 2002;106(25):e206-e208.
8. Buttaro TM, Trybulski J, Bailey PP, Sandberg-Cook J. Primary Care: A Collaborative Practice. 3rd ed. St. Louis, MO: Mosby Elsevier; 2008.
9. Wen ZC, Chen SA, Tai CT, et al. Electrophysiological mechanisms and determinants of vagal maneuvers for termination of paroxysmal supraventricular tachycardia. Circulation.1998;98(24):2716-2723.
10. Julian MR. Treatment of paroxysmal supraventricular tachycardia using instrument-assisted manipulation of the fourth rib: a 6-year case study. J Manipulative Physiol Ther. 2008;31(5):389-391.
11. Calkins H, Kumar VKA, Francis J. Radiofrequency catheter ablation of supraventricular tachycardia. Indian Pacing Electrophysiol J. 2002;2(2):45-49.
12. Nakagawa H, Jackman WM. Catheter ablation of paroxysmal supraventricular tachycardia. Circulation. 2007;116(21):2465-2478.
13. Kugler JD, Danford DA, Houston K, Felix G; Pediatric Radiofrequency Ablation Registry of the Pediatric Electrophysiology Society. Pediatric radiofrequency catheter ablation registry success, fluoroscopy time, and complication rate for supraventricular tachycardia: comparison of early and recent eras. J Cardiovasc Electrophysiol. 2002;13(4):336-341.
14. Chun TU, Van Hare GF. Advances in the approach to treatment of supraventricular tachycardia in the pediatric population. Curr Cardiol Rep. 2004; 6(5):322-326.
15. Friedman PL, Dubuc M, Green MS, et al. Catheter cryoablation of supraventricular tachycardia: results of the multicenter prospective “frosty” trial. Heart Rhythm. 2004;1(2):129-138.
16. Bosen DM. Atrio-ventricular nodal reentry tachycardia in children. Dimens Crit Care Nurs. 2002; 21(4):134-139.
17. Children’s Hospitals and Clinics of Minnesota. Patient and family education: supraventricular tachycardia (2009). www.childrensmn.org/Manuals/PFS/Condill/018303.pdf. Accessed January 26, 2010.
A 6-year-old girl was brought by her parents to the emergency department (ED) with an elevated heart rate. According to the parents, the girl was carrying her younger sister when they both fell, landing on their buttocks. The child told them that her heart was beating fast, and the parents said she appeared to be on the verge of fainting.
They stated that their daughter was healthy and active; they denied previous episodes of shortness of breath, headache, weakness, tachycardia, syncope, or fatigue with exercise. Her caffeine intake, they claimed, was limited to one small cup of soda they allowed her each week.
Initial evaluation in the ED revealed an anxious child with tachycardia and shortness of breath. She presented with a temperature of 98.3°F (36.8°C); pulse, 210 beats/min; respirations, 33 breaths/min; blood pressure, 100/72 mm Hg; weight, 78 lb; height, 45 in; and BMI, 27.1. ECG revealed a heart rate exceeding 210 beats/min, and a pediatric cardiologist made a diagnosis of supraventricular tachycardia (SVT).
The pediatric cardiologist prescribed an adenosine IV drip, which successfully stabilized the child’s heart to sinus rhythm. After three hours in the ED, the patient was discharged with a stable heart rate of 100 beats/min. (It is well known that heart rate regulation changes significantly during development; this is most obvious in higher basal rates in infants and children, compared with adults.1)
The parents were advised to administer atenolol 12.5 mg (one tablet) twice daily and to make a follow-up appointment with a pediatric electrophysiologist. (Although atenolol is not currently FDA approved for this use, a multicenter prospective randomized controlled trial comparing digoxin with beta-blockers for the treatment of SVT in children is presently under way.2)
At that appointment, the pediatric electrophysiologist provided information to the parents regarding the therapeutic options for SVT. The parents continued to administer atenolol to the child, as was deemed necessary until any accessory electrical pathway could be identified and, if so, an ablation procedure could be performed. They were uncertain how to proceed so long as their daughter experienced no recurrent episodes of SVT while receiving pharmacologic therapy.
However, six months after the initial episode, the child (then age 7) presented to the ED once again with recurrent SVT. The pediatric cardiologist ordered an adenosine IV drip, which resulted in successful conversion to sinus rhythm. The parents were instructed to increase the child’s atenolol dosage to 25 mg twice a day.
Six months later, after extensive research and consultation, the parents agreed to an ablation procedure in order to prevent further episodes of SVT. Upon their informed consent, the child was sent to a cardiac catheterization laboratory for an electrophysiology study (EPS), which confirmed the presence of an accessory pathway, as well as the diagnosis of atrioventricular reciprocating tachycardia (AVRT). The procedure was followed by radiofrequency catheter ablation to correct the 7-year-old patient’s accessory pathway–mediated reentry tachycardia.
Discussion
SVT, also known as paroxysmal supraventricular tachycardia (PSVT), is one of the most common symptomatic pediatric arrhythmias, affecting between one in 25,000 and one in 250 children.3 It is defined as rapid heart rhythm (140 to 240 beats/min) that is caused by the presence of additional electrical connections and/or congenital muscle fibers between the atrium and the ventricle or within the atrioventricular (AV) node that did not, for unknown reasons, separate completely during development.4 SVT can be triggered by physical or psychological stress automaticity.3
Approximately 50% of children with SVT present with a first episode before age 1. SVT usually occurs in early childhood, between ages 6 and 9.4 Almost 90% of pediatric patients with SVT are diagnosed with a reentry mechanism.3 The symptoms experienced may be resolved pharmacologically or by means of an invasive therapy. Serious sequelae associated with SVT include heart failure and cardiac arrest.
For children with rare and mildly symptomatic episodes in whom SVT is easily terminated, the SVT may not warrant treatment. However, it may be advisable to offer medical therapy or transcatheter ablation as therapeutic options for children with episodes that are difficult to terminate, occur frequently, or occur during participation in athletics.4
Pathophysiology
SVT generally presents as one of three types: AVRT, which is also known as Wolff-Parkinson-White syndrome; atrioventricular nodal reentry tachycardia (AVNRT); and automatic tachycardia (AT).
AVRT, the most common type of SVT, comprises about 90% of pediatric cases. It is defined by the presence of one or more accessory conduction pathways that are anatomically separated from the normal cardiac conduction system.5 AVRT may be orthodromic (that is, the arrhythmia circuit proceeds down the AV node and retrograde up the accessory conduction pathway) or antedromic (ie, proceeding down the accessory pathway and up the AV node5; see figure.6,7)
AVNRT, considered the second most common type of SVT in children, accounts for about 10% of pediatric cases. AVNRT is caused by an interaction between the two types of pathways within the AV node—one with a fast conduction time and a short refractory period, and the other with a slow conduction time and a long refractory period. AVNRT occurs when the antegrade conduction block in the fast pathway results in conduction over the slow pathway and back up the fast pathway, forming a microreentrant circuit.5
AT is the result of rapid depolarization from an automatic focus originating within the atria but outside the sinus node.3
Patient Presentation and History
The typical presentation of AVRT in children of school age includes palpitations, chest pain or tightness, dizziness, anxiety, decrease in exercise tolerance, easy fatigability, and/or shortness of breath.3 Onset is described as abrupt, while termination of SVT is described as slower because the catecholamine levels are typically elevated.4
The frequency and duration of SVT can vary greatly, from a few minutes to a few hours; it can occur as regularly as daily or as uncommonly as once or twice per year.4 Additionally, SVT symptoms can go unrecognized until a cardiac dysfunction develops. As for the patient in the case study, no apparent factor in her history was identified that may have induced SVT.
The differential diagnosis for SVT is broad, including sinus tachycardia, multifocal atrial tachycardia, and SVT with aberrancy.8 Additional considerations include stress, anxiety, hyperthyroidism, electrolyte abnormalities, and dehydration—any of which can present with a tachycardia response.4 Furthermore, clinicians are often unlikely to diagnose a child with any cardiac problem because chest pain is more commonly a presenting symptom of a pulmonary or musculoskeletal condition than a cardiac problem.3
Physical Examination
SVT can be diagnosed based on medical history and physical examination. During the physical examination, providers will assess the patient’s blood pressure and pulse, auscultate heart and lung sounds, assess the veins in the patient’s neck for different types of pulsations, and conduct cardiac maneuvers, including the Valsalva maneuver and carotid sinus massage.9,10
Laboratory Work-up and Diagnosis
Three specific tests help clinicians monitor and evaluate a patient’s conduction system. ECG is important to assess the heart rhythm both at baseline and when symptoms are occurring, if possible.3 Ambulatory ECG (ie, Holter monitoring, event recorders) record the patient’s heart rhythm on a continuous basis.
An EPS, which is performed to classify the mechanism of SVT, is conducted by inserting one or more electrocatheters into the heart by way of the femoral vein or other peripheral vessel.3 Pacing and sensing electrodes at the ends of the catheters record local intracardiac electrical activity and timing information, providing a detailed analysis of the heart’s electrical activity. The EPS is critical to determine the presence of one or more extra electrical pathways within the heart and to localize it by region.3,11 An ablation procedure may follow.
Management Options
SVT can be treated pharmacologically or nonpharmacologically. First-line pharmacologic options include certain beta-blockers (including atenolol and propranolol), digoxin, and calcium channel blockers. Second-line pharmacologic treatments include amiodarone, flecainide, and sotalol,4 all of which are contraindicated in children younger than 1 year because of these patients’ hemodynamic dependency on the heart and inability to generate stroke volume.3 Pharmacologic treatment of SVT is associated with a 68% success rate in children4 (see Table 14).
For children in whom pharmacologic treatment is ineffective, an ablation procedure may be performed. Radiofrequency catheter ablation is currently considered first-line therapy for AVRT and AVNRT.12 In this invasive procedure, intracardiac electrical mapping is performed and the initiating focus of the arrhythmia or the accessory electrical pathway that has been identified within the heart is destroyed by radiofrequency energy, delivered by electrocatheter. Ablations performed during the acute phase of SVT have a 95% success rate.3,13
Cryoablation is a relatively new treatment in which liquid nitrous oxide is used to cool the catheter to subfreezing temperatures, enabling it to destroy the myocardial tissue by freezing.3,14 The advantage of cryoablation is the option of reversible cooling, which allows the electrophysiologist to test the area first, confirming the accuracy of the apparent location accessory pathway.15
Noninvasive, nonpharmacologic interventions that increase the refractoriness of the AV node may be successful in terminating the tachyarrhythmia during episodes of SVT (see Table 23,9,13,16). They are used to terminate and diagnose tachydysrhythmias, increase parasympathetic tone, and slow conduction through the AV node.3
Patient Education
It is very important for health care providers to relieve parents’ and caregivers’ stress, anxiety, and uncertainty by educating them about the child’s cardiac condition of SVT. Information to convey include an understanding of what SVT is, what may cause it, what triggers the patient should avoid, what treatments are available and appropriate (including the maneuvers shown in Table 2), and what outcomes may be expected. An excellent patient/family education handout from the Children’s Hospitals and Clinics of Minnesota17 is available at www.childrensmn.org/Manuals/PFS/Condill/018303.pdf.
Follow-Up
Primary care providers must emphasize the importance of monitoring the patient’s progress, based on the severity of his or her SVT symptoms. The provider may choose to monitor the patient for a few weeks or a few months, assessing the frequency of arrhythmia recurrence and the heart rate, to adjust or substitute medications based on repeat ECG or Holter evaluations, and to plan further therapy, should the condition worsen.5
The Case Patient
One month after undergoing radiofrequency catheter ablation, the child presented to the pediatric cardiologist for follow-up. Since the procedure, she had been without any symptoms referable to the cardiovascular system. She denied experiencing any fast heart rate, palpitations, chest pain, shortness of breath, or dizziness. ECG demonstrated normal sinus rhythm.
Two years after undergoing radiofrequency ablation, the child is functioning at a normal activity level with no recurrence of SVT episodes.
Conclusion
The purpose of this case study is to improve primary care providers’ understanding of SVT in children and to convey the importance of identifying the condition in a timely manner and referring patients to a pediatric cardiologist or electrophysiologist. For most children affected by SVT, a regimen of pharmacologic and/or nonpharmacologic treatment—supported by detailed education for their parents and caregivers—can allow them to live a healthy, normal life.
A 6-year-old girl was brought by her parents to the emergency department (ED) with an elevated heart rate. According to the parents, the girl was carrying her younger sister when they both fell, landing on their buttocks. The child told them that her heart was beating fast, and the parents said she appeared to be on the verge of fainting.
They stated that their daughter was healthy and active; they denied previous episodes of shortness of breath, headache, weakness, tachycardia, syncope, or fatigue with exercise. Her caffeine intake, they claimed, was limited to one small cup of soda they allowed her each week.
Initial evaluation in the ED revealed an anxious child with tachycardia and shortness of breath. She presented with a temperature of 98.3°F (36.8°C); pulse, 210 beats/min; respirations, 33 breaths/min; blood pressure, 100/72 mm Hg; weight, 78 lb; height, 45 in; and BMI, 27.1. ECG revealed a heart rate exceeding 210 beats/min, and a pediatric cardiologist made a diagnosis of supraventricular tachycardia (SVT).
The pediatric cardiologist prescribed an adenosine IV drip, which successfully stabilized the child’s heart to sinus rhythm. After three hours in the ED, the patient was discharged with a stable heart rate of 100 beats/min. (It is well known that heart rate regulation changes significantly during development; this is most obvious in higher basal rates in infants and children, compared with adults.1)
The parents were advised to administer atenolol 12.5 mg (one tablet) twice daily and to make a follow-up appointment with a pediatric electrophysiologist. (Although atenolol is not currently FDA approved for this use, a multicenter prospective randomized controlled trial comparing digoxin with beta-blockers for the treatment of SVT in children is presently under way.2)
At that appointment, the pediatric electrophysiologist provided information to the parents regarding the therapeutic options for SVT. The parents continued to administer atenolol to the child, as was deemed necessary until any accessory electrical pathway could be identified and, if so, an ablation procedure could be performed. They were uncertain how to proceed so long as their daughter experienced no recurrent episodes of SVT while receiving pharmacologic therapy.
However, six months after the initial episode, the child (then age 7) presented to the ED once again with recurrent SVT. The pediatric cardiologist ordered an adenosine IV drip, which resulted in successful conversion to sinus rhythm. The parents were instructed to increase the child’s atenolol dosage to 25 mg twice a day.
Six months later, after extensive research and consultation, the parents agreed to an ablation procedure in order to prevent further episodes of SVT. Upon their informed consent, the child was sent to a cardiac catheterization laboratory for an electrophysiology study (EPS), which confirmed the presence of an accessory pathway, as well as the diagnosis of atrioventricular reciprocating tachycardia (AVRT). The procedure was followed by radiofrequency catheter ablation to correct the 7-year-old patient’s accessory pathway–mediated reentry tachycardia.
Discussion
SVT, also known as paroxysmal supraventricular tachycardia (PSVT), is one of the most common symptomatic pediatric arrhythmias, affecting between one in 25,000 and one in 250 children.3 It is defined as rapid heart rhythm (140 to 240 beats/min) that is caused by the presence of additional electrical connections and/or congenital muscle fibers between the atrium and the ventricle or within the atrioventricular (AV) node that did not, for unknown reasons, separate completely during development.4 SVT can be triggered by physical or psychological stress automaticity.3
Approximately 50% of children with SVT present with a first episode before age 1. SVT usually occurs in early childhood, between ages 6 and 9.4 Almost 90% of pediatric patients with SVT are diagnosed with a reentry mechanism.3 The symptoms experienced may be resolved pharmacologically or by means of an invasive therapy. Serious sequelae associated with SVT include heart failure and cardiac arrest.
For children with rare and mildly symptomatic episodes in whom SVT is easily terminated, the SVT may not warrant treatment. However, it may be advisable to offer medical therapy or transcatheter ablation as therapeutic options for children with episodes that are difficult to terminate, occur frequently, or occur during participation in athletics.4
Pathophysiology
SVT generally presents as one of three types: AVRT, which is also known as Wolff-Parkinson-White syndrome; atrioventricular nodal reentry tachycardia (AVNRT); and automatic tachycardia (AT).
AVRT, the most common type of SVT, comprises about 90% of pediatric cases. It is defined by the presence of one or more accessory conduction pathways that are anatomically separated from the normal cardiac conduction system.5 AVRT may be orthodromic (that is, the arrhythmia circuit proceeds down the AV node and retrograde up the accessory conduction pathway) or antedromic (ie, proceeding down the accessory pathway and up the AV node5; see figure.6,7)
AVNRT, considered the second most common type of SVT in children, accounts for about 10% of pediatric cases. AVNRT is caused by an interaction between the two types of pathways within the AV node—one with a fast conduction time and a short refractory period, and the other with a slow conduction time and a long refractory period. AVNRT occurs when the antegrade conduction block in the fast pathway results in conduction over the slow pathway and back up the fast pathway, forming a microreentrant circuit.5
AT is the result of rapid depolarization from an automatic focus originating within the atria but outside the sinus node.3
Patient Presentation and History
The typical presentation of AVRT in children of school age includes palpitations, chest pain or tightness, dizziness, anxiety, decrease in exercise tolerance, easy fatigability, and/or shortness of breath.3 Onset is described as abrupt, while termination of SVT is described as slower because the catecholamine levels are typically elevated.4
The frequency and duration of SVT can vary greatly, from a few minutes to a few hours; it can occur as regularly as daily or as uncommonly as once or twice per year.4 Additionally, SVT symptoms can go unrecognized until a cardiac dysfunction develops. As for the patient in the case study, no apparent factor in her history was identified that may have induced SVT.
The differential diagnosis for SVT is broad, including sinus tachycardia, multifocal atrial tachycardia, and SVT with aberrancy.8 Additional considerations include stress, anxiety, hyperthyroidism, electrolyte abnormalities, and dehydration—any of which can present with a tachycardia response.4 Furthermore, clinicians are often unlikely to diagnose a child with any cardiac problem because chest pain is more commonly a presenting symptom of a pulmonary or musculoskeletal condition than a cardiac problem.3
Physical Examination
SVT can be diagnosed based on medical history and physical examination. During the physical examination, providers will assess the patient’s blood pressure and pulse, auscultate heart and lung sounds, assess the veins in the patient’s neck for different types of pulsations, and conduct cardiac maneuvers, including the Valsalva maneuver and carotid sinus massage.9,10
Laboratory Work-up and Diagnosis
Three specific tests help clinicians monitor and evaluate a patient’s conduction system. ECG is important to assess the heart rhythm both at baseline and when symptoms are occurring, if possible.3 Ambulatory ECG (ie, Holter monitoring, event recorders) record the patient’s heart rhythm on a continuous basis.
An EPS, which is performed to classify the mechanism of SVT, is conducted by inserting one or more electrocatheters into the heart by way of the femoral vein or other peripheral vessel.3 Pacing and sensing electrodes at the ends of the catheters record local intracardiac electrical activity and timing information, providing a detailed analysis of the heart’s electrical activity. The EPS is critical to determine the presence of one or more extra electrical pathways within the heart and to localize it by region.3,11 An ablation procedure may follow.
Management Options
SVT can be treated pharmacologically or nonpharmacologically. First-line pharmacologic options include certain beta-blockers (including atenolol and propranolol), digoxin, and calcium channel blockers. Second-line pharmacologic treatments include amiodarone, flecainide, and sotalol,4 all of which are contraindicated in children younger than 1 year because of these patients’ hemodynamic dependency on the heart and inability to generate stroke volume.3 Pharmacologic treatment of SVT is associated with a 68% success rate in children4 (see Table 14).
For children in whom pharmacologic treatment is ineffective, an ablation procedure may be performed. Radiofrequency catheter ablation is currently considered first-line therapy for AVRT and AVNRT.12 In this invasive procedure, intracardiac electrical mapping is performed and the initiating focus of the arrhythmia or the accessory electrical pathway that has been identified within the heart is destroyed by radiofrequency energy, delivered by electrocatheter. Ablations performed during the acute phase of SVT have a 95% success rate.3,13
Cryoablation is a relatively new treatment in which liquid nitrous oxide is used to cool the catheter to subfreezing temperatures, enabling it to destroy the myocardial tissue by freezing.3,14 The advantage of cryoablation is the option of reversible cooling, which allows the electrophysiologist to test the area first, confirming the accuracy of the apparent location accessory pathway.15
Noninvasive, nonpharmacologic interventions that increase the refractoriness of the AV node may be successful in terminating the tachyarrhythmia during episodes of SVT (see Table 23,9,13,16). They are used to terminate and diagnose tachydysrhythmias, increase parasympathetic tone, and slow conduction through the AV node.3
Patient Education
It is very important for health care providers to relieve parents’ and caregivers’ stress, anxiety, and uncertainty by educating them about the child’s cardiac condition of SVT. Information to convey include an understanding of what SVT is, what may cause it, what triggers the patient should avoid, what treatments are available and appropriate (including the maneuvers shown in Table 2), and what outcomes may be expected. An excellent patient/family education handout from the Children’s Hospitals and Clinics of Minnesota17 is available at www.childrensmn.org/Manuals/PFS/Condill/018303.pdf.
Follow-Up
Primary care providers must emphasize the importance of monitoring the patient’s progress, based on the severity of his or her SVT symptoms. The provider may choose to monitor the patient for a few weeks or a few months, assessing the frequency of arrhythmia recurrence and the heart rate, to adjust or substitute medications based on repeat ECG or Holter evaluations, and to plan further therapy, should the condition worsen.5
The Case Patient
One month after undergoing radiofrequency catheter ablation, the child presented to the pediatric cardiologist for follow-up. Since the procedure, she had been without any symptoms referable to the cardiovascular system. She denied experiencing any fast heart rate, palpitations, chest pain, shortness of breath, or dizziness. ECG demonstrated normal sinus rhythm.
Two years after undergoing radiofrequency ablation, the child is functioning at a normal activity level with no recurrence of SVT episodes.
Conclusion
The purpose of this case study is to improve primary care providers’ understanding of SVT in children and to convey the importance of identifying the condition in a timely manner and referring patients to a pediatric cardiologist or electrophysiologist. For most children affected by SVT, a regimen of pharmacologic and/or nonpharmacologic treatment—supported by detailed education for their parents and caregivers—can allow them to live a healthy, normal life.
1. Kudielka BM, Buske-Kirschbaum A, Hellhammer DH, Kirschbaum C. Differential heart rate reactivity and recovery after psychosocial stress (TSST) in healthy children, younger adults, and elderly adults: the impact of age and gender. Int J Behav Med. 2004;11(2):116-121.
2. Multicenter Study of Antiarrhythmic Medications for Treatment of Infants With Supraventricular Tachycardia. www.clinicaltrials.gov/ct2/results?term=NCT00390546. Accessed January 26, 2010.
3. Schlechte EA, Boramanand N, Funk M. Supraventricular tachycardia in the pediatric primary care setting: age-related presentation, diagnosis, and management. J Pediatr Health Care. 2008;22(5): 289-299.
4. Salerno JC, Seslar SP. Supraventricular tachycardia. Arch Pediatr Adolesc Med. 2009;163(3): 268-274.
5. Doniger SJ, Sharieff GQ. Pediatric dysrhythmias. Pediatr Clin North Am. 2006;53(1):85-105, vi.
6. Mavroudis C, Deal BJ, Backer CL, Tsao S. Arrhythmia surgery in patients with and without congenital heart disease. Ann Thorac Surg. 2008;86(3):857-868.
7. Wang PJ, Estes NAM III. Supraventricular tachycardia. Circulation. 2002;106(25):e206-e208.
8. Buttaro TM, Trybulski J, Bailey PP, Sandberg-Cook J. Primary Care: A Collaborative Practice. 3rd ed. St. Louis, MO: Mosby Elsevier; 2008.
9. Wen ZC, Chen SA, Tai CT, et al. Electrophysiological mechanisms and determinants of vagal maneuvers for termination of paroxysmal supraventricular tachycardia. Circulation.1998;98(24):2716-2723.
10. Julian MR. Treatment of paroxysmal supraventricular tachycardia using instrument-assisted manipulation of the fourth rib: a 6-year case study. J Manipulative Physiol Ther. 2008;31(5):389-391.
11. Calkins H, Kumar VKA, Francis J. Radiofrequency catheter ablation of supraventricular tachycardia. Indian Pacing Electrophysiol J. 2002;2(2):45-49.
12. Nakagawa H, Jackman WM. Catheter ablation of paroxysmal supraventricular tachycardia. Circulation. 2007;116(21):2465-2478.
13. Kugler JD, Danford DA, Houston K, Felix G; Pediatric Radiofrequency Ablation Registry of the Pediatric Electrophysiology Society. Pediatric radiofrequency catheter ablation registry success, fluoroscopy time, and complication rate for supraventricular tachycardia: comparison of early and recent eras. J Cardiovasc Electrophysiol. 2002;13(4):336-341.
14. Chun TU, Van Hare GF. Advances in the approach to treatment of supraventricular tachycardia in the pediatric population. Curr Cardiol Rep. 2004; 6(5):322-326.
15. Friedman PL, Dubuc M, Green MS, et al. Catheter cryoablation of supraventricular tachycardia: results of the multicenter prospective “frosty” trial. Heart Rhythm. 2004;1(2):129-138.
16. Bosen DM. Atrio-ventricular nodal reentry tachycardia in children. Dimens Crit Care Nurs. 2002; 21(4):134-139.
17. Children’s Hospitals and Clinics of Minnesota. Patient and family education: supraventricular tachycardia (2009). www.childrensmn.org/Manuals/PFS/Condill/018303.pdf. Accessed January 26, 2010.
1. Kudielka BM, Buske-Kirschbaum A, Hellhammer DH, Kirschbaum C. Differential heart rate reactivity and recovery after psychosocial stress (TSST) in healthy children, younger adults, and elderly adults: the impact of age and gender. Int J Behav Med. 2004;11(2):116-121.
2. Multicenter Study of Antiarrhythmic Medications for Treatment of Infants With Supraventricular Tachycardia. www.clinicaltrials.gov/ct2/results?term=NCT00390546. Accessed January 26, 2010.
3. Schlechte EA, Boramanand N, Funk M. Supraventricular tachycardia in the pediatric primary care setting: age-related presentation, diagnosis, and management. J Pediatr Health Care. 2008;22(5): 289-299.
4. Salerno JC, Seslar SP. Supraventricular tachycardia. Arch Pediatr Adolesc Med. 2009;163(3): 268-274.
5. Doniger SJ, Sharieff GQ. Pediatric dysrhythmias. Pediatr Clin North Am. 2006;53(1):85-105, vi.
6. Mavroudis C, Deal BJ, Backer CL, Tsao S. Arrhythmia surgery in patients with and without congenital heart disease. Ann Thorac Surg. 2008;86(3):857-868.
7. Wang PJ, Estes NAM III. Supraventricular tachycardia. Circulation. 2002;106(25):e206-e208.
8. Buttaro TM, Trybulski J, Bailey PP, Sandberg-Cook J. Primary Care: A Collaborative Practice. 3rd ed. St. Louis, MO: Mosby Elsevier; 2008.
9. Wen ZC, Chen SA, Tai CT, et al. Electrophysiological mechanisms and determinants of vagal maneuvers for termination of paroxysmal supraventricular tachycardia. Circulation.1998;98(24):2716-2723.
10. Julian MR. Treatment of paroxysmal supraventricular tachycardia using instrument-assisted manipulation of the fourth rib: a 6-year case study. J Manipulative Physiol Ther. 2008;31(5):389-391.
11. Calkins H, Kumar VKA, Francis J. Radiofrequency catheter ablation of supraventricular tachycardia. Indian Pacing Electrophysiol J. 2002;2(2):45-49.
12. Nakagawa H, Jackman WM. Catheter ablation of paroxysmal supraventricular tachycardia. Circulation. 2007;116(21):2465-2478.
13. Kugler JD, Danford DA, Houston K, Felix G; Pediatric Radiofrequency Ablation Registry of the Pediatric Electrophysiology Society. Pediatric radiofrequency catheter ablation registry success, fluoroscopy time, and complication rate for supraventricular tachycardia: comparison of early and recent eras. J Cardiovasc Electrophysiol. 2002;13(4):336-341.
14. Chun TU, Van Hare GF. Advances in the approach to treatment of supraventricular tachycardia in the pediatric population. Curr Cardiol Rep. 2004; 6(5):322-326.
15. Friedman PL, Dubuc M, Green MS, et al. Catheter cryoablation of supraventricular tachycardia: results of the multicenter prospective “frosty” trial. Heart Rhythm. 2004;1(2):129-138.
16. Bosen DM. Atrio-ventricular nodal reentry tachycardia in children. Dimens Crit Care Nurs. 2002; 21(4):134-139.
17. Children’s Hospitals and Clinics of Minnesota. Patient and family education: supraventricular tachycardia (2009). www.childrensmn.org/Manuals/PFS/Condill/018303.pdf. Accessed January 26, 2010.
Grand Rounds: Woman, 39, With Leg Weakness After Exercise Class
A 39-year-old woman presented to the emergency department (ED) with a chief complaint of muscle aches and pain. She stated that three days earlier, she had begun exercising in a 45-minute “spinning” class (ie, riding a stationary bicycle with a weighted front wheel). The patient had not engaged in any aerobic exercise for at least six months before the spinning class. She mentioned that much older participants in the class were outperforming her, but she did not feel the need to keep up with them.
After dismounting, the woman said, she experienced weakness in her legs and had great difficulty ambulating. She went home, took 400 mg of ibuprofen, and went to bed. She awoke with pain and swelling in both thighs and continued to take ibuprofen, in addition to applying a topical mentholated preparation to her thighs. She took an Epsom salts bath two days later.
On the morning of the third day after the spinning class, she voided black urine and presented to the ED.
The patient had no significant medical history. Surgical history was limited to removal of a ganglion cyst on her wrist. She denied any history of seizure disorder, thyroid disease, hepatitis, heart disease, or hyperlipidemia.
The patient had been taking ibuprofen as needed since the spinning class. She was taking no other medications. She denied any allergies to drugs or food.
The patient admitted to smoking one pack of cigarettes per week and to occasional alcohol consumption but denied use of illicit drugs. She was employed as an executive officer for a large business association.
On physical examination, the patient’s vital signs were blood pressure, 134/73 mm/Hg; pulse, 86 beats/min; and respirations, 16 breaths/min. She was afebrile, alert, and oriented. Her sclera were nonicteric. Her neck was supple with no anterior cervical lymphadenopathy. There was no thyroid enlargement, her lungs were clear to auscultation, and her heart sounds were regular. There was no peripheral edema, and dorsalis pedis pulses were present bilaterally. Her thighs appeared swollen but were not tender to palpation.
The patient’s history, combined with an extremely high level of serum creatine phosphokinase (CPK; ie, 123,800 U/L [reference range, 45 to 260 U/L1]), confirmed a diagnosis of rhabdomyolysis. She was admitted for close observation. The patient’s urinalysis revealed 2 to 5 red blood cells and 6 to 10 white blood cells per high-power field. Moderate occult blood was detected, with no casts or protein noted. A urine myoglobin test was not performed.
The patient underwent IV hydration with dextrose 5% in water and three ampules of sodium bicarbonate after being given a 2.0-L saline bolus. Ibuprofen was discontinued. IV hydration with bicarbonate solution was continued until the patient’s CPK level declined significantly. She underwent daily laboratory testing (see Table 1). Her renal function remained stable, and she was discharged on hospital day 7.
DISCUSSION
Rhabdomyolysis is a clinical condition defined as muscle necrosis resulting from the release of intracellular skeletal muscle components (including myoglobin, CPK, potassium, phosphorus, and aldolase) into the extracellular compartment.1-3 The condition was first described during the bombing of London in World War II, with high incidence of crush injuries, shock, and associated kidney damage.4 The preponderance of such injuries during a 1988 earthquake in Armenia led the International Society of Nephrology to form its Renal Disaster Relief Task Force, which has provided support at numerous other disaster scenes since then.5
Rhabdomyolysis has been identified with a variety of pathologic events: those that cause muscle trauma, those associated with muscle use or overuse, and other etiologies involving genetic, metabolic, infectious, or pharmaceutical factors.1 Many of the reported causes of rhabdomyolysis are listed in Table 2.1,2
For patients with muscle trauma, the etiology of rhabdomyolysis is clear, but for those with other disease states, diagnosis may be more elusive. Patients who present with rhabdomyolysis after excessive exercise, for example, may have underlying metabolic disorders that predispose them to exertional rhabdomyolysis, such as chronic hypokalemia resulting from primary hyperaldosteronism.1 Others may have a muscle enzyme deficiency, as in McArdle’s syndrome or carnitine deficiency.6
Alterations in blood chemistries can also contribute to development of rhabdomyolysis, even when more obvious etiologies for muscle necrosis are evident. Hypokalemia interferes with the vasodilation that normally occurs during exercise to increase muscle blood flow.1,7,8 Continued exercise can lead to muscle necrosis, raising a concern for athletes who take diuretics.1 Hypophosphatemia leads to a state of muscle necrosis; this is of particular concern for alcoholic patients who receive hyperalimentation without repletion of phosphates.9
Diagnosis
Patients with rhabdomyolysis usually present with myalgias, darkened urine (red, brown, or black), and a clinical scenario that corroborates the diagnosis (ie, history of trauma, excessive exercise, use of an offending medication).1 Some patients may have minimal to absent symptoms or symptoms that occur only after exercise.3
A careful history is key. While traumatic causes are obvious, it is important to ask a patient with rhabdomyolysis after exertion about previous history of excessive weakness during or immediately after exercise, excessive cramping, or discoloration of urine after exercise. The family history may point to a genetic abnormality. A thorough understanding of the patient’s use of medications, including OTC agents, is also important. Rhabdomyolysis has been reported in patients who use herbal remedies, including those taken to facilitate weight loss or to improve lipid profiles.10,11
For patients suspected of having rhabdomyolysis, a serum CPK level should be obtained; results exceeding normal values by five times confirm the diagnosis.3 Measurements for potassium, phosphorus, and calcium are also important to determine, as is renal function. A high level of serum aldolase (an enzyme that breaks down glucose in muscle tissue) can also support a diagnosis of rhabdomyolysis.1,12 Urinalysis and urine myoglobin testing are also warranted, although a negative urine myoglobin test result does not rule out rhabdomyolysis in the presence of an elevated CPK level. Myoglobin is cleared rapidly by the kidneys, whereas serum CPK levels change slowly.1
Any patient who presents with acute rhabdomyolysis and low to normal values for potassium or phosphate should be evaluated further for hypokalemia and hypophosphatemia as contributing or etiologic factors. Hypocalcemia may occur in the early course of rhabdomyolysis as calcium salt is deposited in muscle tissue. Patients recovering from rhabdomyolysis may experience rebound hypercalcemia as the damaged muscle releases the deposited calcium.7,13
In most cases of rhabdomyolysis, only laboratory values are needed to make the diagnosis and follow the course of the episode.1 However, when the etiology appears to involve metabolic deficiencies or genetic etiologies, it may become necessary to order additional diagnostic tests. These may include tests for thyroid function, a carnitine level to screen for glycogen storage diseases, and toxin screening (eg, for illicit drugs, such as cocaine).2,6
Treatment and Management
Effective treatment of rhabdomyolysis relies on recognizing the underlying disorder.1 For patients with muscle trauma (eg, crush injury) or muscle overuse, the mainstay of treatment is aggressive fluid resuscitation and prevention of acute injury to the kidneys.13 As for patients with an injury induced by a pharmaceutical agent or a toxin, removal of the offending agent is required, followed by hydration and prevention of renal damage. Supportive care during an infectious illness is also essential.14
Additionally, treatment must address the complications inherent with rhabdomyolysis.1 In addition to CPK, potassium, phosphorus, and myoglobin are also released from skeletal muscle tissue. Hyperkalemia can be fatal, and potassium levels must be monitored closely to avert this condition.7,8,13 During an episode of rhabdomyolysis, normal levels of both potassium and phosphorus should raise the clinician’s suspicion for underlying hypokalemia and hypophosphatemia—conditions that may have contributed to the episode of rhabdomyolysis. Hypocalcemia may also develop.13
Released myoglobin may cause acute kidney injury, as is the case in 33% to 50% of patients with rhabdomyolysis.3 In early studies, it was determined that alkalinizing the urine with IV isotonic bicarbonate might thwart onset of acute kidney injury.1,2,15 Time is critical, and even on the battlefield or at the scene of a recent disaster, most attempts at resuscitation are begun immediately. IV access may be problematic, but administration of oral bicarbonate solutions has also proven effective.15 Close follow-up of the serum urea and creatinine levels and measurement of the urine pH during alkalinization is warranted throughout the course of the episode.
Unfortunately, some patients respond poorly to these conservative measures, and the released myoglobin can cause renal tubular blockage and necrosis, resulting in acute kidney injury.1 Renal replacement therapy may be required.16 However, most episodes of dialysis-dependent acute renal injury do subside with time.
For patients with less elusive causes of rhabdomyolysis, treatment will hinge on a workup of the possible etiologies and follow-up treatment to target the apparent cause. For example, carnitine may be administered to patients with carnitine deficiency, and hypokalemic patients may be given potassium.1,6,7 These patients will also need counseling before they consider engaging in an exercise program.
Patient’s Outcome
The case patient presented with exertional rhabdomyolysis; improper hydration, severe deconditioning, and a relatively low serum potassium level may all have contributed to the muscle necrosis she experienced. She was given IV alkaline solutions and did not develop acute kidney injury. She was discharged from the hospital and at the time of this writing was awaiting outpatient follow-up.
It should be interesting to see whether the case patient experiences any further episodes of severe weakness after engaging in exercise. Her low-normal potassium level (reference range, 3.5 to 5.3 mmol/L17) warrants further follow-up, as does her mildly elevated thyroid-stimulating hormone level (reference range, 0.5 to 4.7 mcIU/mL17).
CONCLUSION
Patients with rhabdomyolysis may present with muscle aches, darkened urine, and/or weakness; an elevated CPK level confirms the diagnosis. Management is mainly conservative, with IV hydration accompanied by alkalinizing the urine and correcting any metabolic abnormalities, such as potassium deficiencies. For the few patients who experience severe acute kidney injury, renal replacement therapy may be necessary.
While most causes of rhabdomyolysis have obvious clinical scenarios, such as a crush injury, a search for muscle enzyme deficiencies, disorders of potassium homeostasis, and thyroid abnormalities is also warranted in patients who present with exertional rhabdomyolysis.
1. Huerta-Alardín AL, Varon J, Marik PE. Bench-to-bedside review: rhabdomyolysis—an overview for clinicians. Crit Care. 2005;9(2):158-169.
2. Warren JD, Blumbergs PC, Thompson PD. Rhabdomyolysis: a review. Muscle Nerve. 2002;25(3):332-347.
3. Lima RSA, da Silva GB Jr, Liborio AB, Daher ED. Acute kidney injury due to rhabdomyolysis. Saudi J Kidney Dis Transpl. 2008;19(5):721-729.
4. Bywaters EG, Beall D. Crush injuries with impairment of renal function [reprinted from BMJ, 1941]. J Am Soc Nephrol. 1998;9(2):322-332.
5. Vanholder R, Van Biesen W, Lameire N, Sever MS; International Society of Nephrology/Renal Disaster Relief Task Force. The role of the International Society of Nephrology/Renal Disaster Relief Task Force in the rescue of renal disaster victims. Contrib Nephrol. 2007;156:325-332.
6. Toledo R, López V, Martín G, et al. Rhabdomyolysis due to enzyme deficiency in muscles. Nefrología. 2009;29(1):77-80.
7. Agrawal S, Agrawal V, Taneja A. Hypokalemia causing rhabdomyolysis resulting in life-threatening hyperkalemia. Pediatr Nephrol. 2006;221(2): 289-291.
8. Knochel JP, Schlein EM. On the mechanism of rhabdomyolysis in potassium depletion. J Clin Invest. 1972:51(7):1750-1758.
9. Knochel JP. Hypophosphatemia and rhabdomyolysis. Am J Med. 1992;92(5):455-457.
10. Mansi IA, Huang J. Rhabdomyolysis in response to weight-loss herbal medicine. Am J Med Sci. 2004; 327(6): 356-357.
11. Heber D, Yip I, Ashley JM, et al. Cholesterol-lowering effects of a proprietary Chinese red-yeast-rice dietary supplement. Am J Clin Nutr. 1999;69(2): 231-236.
12. Hooda AK, Narula AS. Exertional rhabdomyolysis causing acute renal failure. Med J Armed Forces India. 2005;61(4):395-396.
13. Chatzizisis YS, Misirli G, Hatzitolios AI, Giannoglou GD. The syndrome of rhabdomyolysis: complications and treatment. Eur J Intern Med. 2008;19(8): 568-574.
14. Blanco JR, Zabalza M, Salcedo J, et al. Rhabdomyolysis of infectious and noninfectious causes. South Med J. 2002;95(5):542-544.
15. Ron D, Taitelman U, Michaelson M, et al. Prevention of acute renal failure in traumatic rhabdomyolysis. Arch Intern Med. 1984;144(2):277-280.
16. Soni SS, Nagarik AP, Adikey GK, Raman A. Using continuous renal replacement therapy to manage patients of shock and acute renal failure. J Emerg Trauma Shock. 2009;2(1):19-22.
17. Normal laboratory values. In: Beers MH, Berkow R, eds. The Merck Manual of Diagnosis and Therapy. 17th ed. Whitehouse Station, NJ: Merck Research Laboratories; 1999:2526-2546.
A 39-year-old woman presented to the emergency department (ED) with a chief complaint of muscle aches and pain. She stated that three days earlier, she had begun exercising in a 45-minute “spinning” class (ie, riding a stationary bicycle with a weighted front wheel). The patient had not engaged in any aerobic exercise for at least six months before the spinning class. She mentioned that much older participants in the class were outperforming her, but she did not feel the need to keep up with them.
After dismounting, the woman said, she experienced weakness in her legs and had great difficulty ambulating. She went home, took 400 mg of ibuprofen, and went to bed. She awoke with pain and swelling in both thighs and continued to take ibuprofen, in addition to applying a topical mentholated preparation to her thighs. She took an Epsom salts bath two days later.
On the morning of the third day after the spinning class, she voided black urine and presented to the ED.
The patient had no significant medical history. Surgical history was limited to removal of a ganglion cyst on her wrist. She denied any history of seizure disorder, thyroid disease, hepatitis, heart disease, or hyperlipidemia.
The patient had been taking ibuprofen as needed since the spinning class. She was taking no other medications. She denied any allergies to drugs or food.
The patient admitted to smoking one pack of cigarettes per week and to occasional alcohol consumption but denied use of illicit drugs. She was employed as an executive officer for a large business association.
On physical examination, the patient’s vital signs were blood pressure, 134/73 mm/Hg; pulse, 86 beats/min; and respirations, 16 breaths/min. She was afebrile, alert, and oriented. Her sclera were nonicteric. Her neck was supple with no anterior cervical lymphadenopathy. There was no thyroid enlargement, her lungs were clear to auscultation, and her heart sounds were regular. There was no peripheral edema, and dorsalis pedis pulses were present bilaterally. Her thighs appeared swollen but were not tender to palpation.
The patient’s history, combined with an extremely high level of serum creatine phosphokinase (CPK; ie, 123,800 U/L [reference range, 45 to 260 U/L1]), confirmed a diagnosis of rhabdomyolysis. She was admitted for close observation. The patient’s urinalysis revealed 2 to 5 red blood cells and 6 to 10 white blood cells per high-power field. Moderate occult blood was detected, with no casts or protein noted. A urine myoglobin test was not performed.
The patient underwent IV hydration with dextrose 5% in water and three ampules of sodium bicarbonate after being given a 2.0-L saline bolus. Ibuprofen was discontinued. IV hydration with bicarbonate solution was continued until the patient’s CPK level declined significantly. She underwent daily laboratory testing (see Table 1). Her renal function remained stable, and she was discharged on hospital day 7.
DISCUSSION
Rhabdomyolysis is a clinical condition defined as muscle necrosis resulting from the release of intracellular skeletal muscle components (including myoglobin, CPK, potassium, phosphorus, and aldolase) into the extracellular compartment.1-3 The condition was first described during the bombing of London in World War II, with high incidence of crush injuries, shock, and associated kidney damage.4 The preponderance of such injuries during a 1988 earthquake in Armenia led the International Society of Nephrology to form its Renal Disaster Relief Task Force, which has provided support at numerous other disaster scenes since then.5
Rhabdomyolysis has been identified with a variety of pathologic events: those that cause muscle trauma, those associated with muscle use or overuse, and other etiologies involving genetic, metabolic, infectious, or pharmaceutical factors.1 Many of the reported causes of rhabdomyolysis are listed in Table 2.1,2
For patients with muscle trauma, the etiology of rhabdomyolysis is clear, but for those with other disease states, diagnosis may be more elusive. Patients who present with rhabdomyolysis after excessive exercise, for example, may have underlying metabolic disorders that predispose them to exertional rhabdomyolysis, such as chronic hypokalemia resulting from primary hyperaldosteronism.1 Others may have a muscle enzyme deficiency, as in McArdle’s syndrome or carnitine deficiency.6
Alterations in blood chemistries can also contribute to development of rhabdomyolysis, even when more obvious etiologies for muscle necrosis are evident. Hypokalemia interferes with the vasodilation that normally occurs during exercise to increase muscle blood flow.1,7,8 Continued exercise can lead to muscle necrosis, raising a concern for athletes who take diuretics.1 Hypophosphatemia leads to a state of muscle necrosis; this is of particular concern for alcoholic patients who receive hyperalimentation without repletion of phosphates.9
Diagnosis
Patients with rhabdomyolysis usually present with myalgias, darkened urine (red, brown, or black), and a clinical scenario that corroborates the diagnosis (ie, history of trauma, excessive exercise, use of an offending medication).1 Some patients may have minimal to absent symptoms or symptoms that occur only after exercise.3
A careful history is key. While traumatic causes are obvious, it is important to ask a patient with rhabdomyolysis after exertion about previous history of excessive weakness during or immediately after exercise, excessive cramping, or discoloration of urine after exercise. The family history may point to a genetic abnormality. A thorough understanding of the patient’s use of medications, including OTC agents, is also important. Rhabdomyolysis has been reported in patients who use herbal remedies, including those taken to facilitate weight loss or to improve lipid profiles.10,11
For patients suspected of having rhabdomyolysis, a serum CPK level should be obtained; results exceeding normal values by five times confirm the diagnosis.3 Measurements for potassium, phosphorus, and calcium are also important to determine, as is renal function. A high level of serum aldolase (an enzyme that breaks down glucose in muscle tissue) can also support a diagnosis of rhabdomyolysis.1,12 Urinalysis and urine myoglobin testing are also warranted, although a negative urine myoglobin test result does not rule out rhabdomyolysis in the presence of an elevated CPK level. Myoglobin is cleared rapidly by the kidneys, whereas serum CPK levels change slowly.1
Any patient who presents with acute rhabdomyolysis and low to normal values for potassium or phosphate should be evaluated further for hypokalemia and hypophosphatemia as contributing or etiologic factors. Hypocalcemia may occur in the early course of rhabdomyolysis as calcium salt is deposited in muscle tissue. Patients recovering from rhabdomyolysis may experience rebound hypercalcemia as the damaged muscle releases the deposited calcium.7,13
In most cases of rhabdomyolysis, only laboratory values are needed to make the diagnosis and follow the course of the episode.1 However, when the etiology appears to involve metabolic deficiencies or genetic etiologies, it may become necessary to order additional diagnostic tests. These may include tests for thyroid function, a carnitine level to screen for glycogen storage diseases, and toxin screening (eg, for illicit drugs, such as cocaine).2,6
Treatment and Management
Effective treatment of rhabdomyolysis relies on recognizing the underlying disorder.1 For patients with muscle trauma (eg, crush injury) or muscle overuse, the mainstay of treatment is aggressive fluid resuscitation and prevention of acute injury to the kidneys.13 As for patients with an injury induced by a pharmaceutical agent or a toxin, removal of the offending agent is required, followed by hydration and prevention of renal damage. Supportive care during an infectious illness is also essential.14
Additionally, treatment must address the complications inherent with rhabdomyolysis.1 In addition to CPK, potassium, phosphorus, and myoglobin are also released from skeletal muscle tissue. Hyperkalemia can be fatal, and potassium levels must be monitored closely to avert this condition.7,8,13 During an episode of rhabdomyolysis, normal levels of both potassium and phosphorus should raise the clinician’s suspicion for underlying hypokalemia and hypophosphatemia—conditions that may have contributed to the episode of rhabdomyolysis. Hypocalcemia may also develop.13
Released myoglobin may cause acute kidney injury, as is the case in 33% to 50% of patients with rhabdomyolysis.3 In early studies, it was determined that alkalinizing the urine with IV isotonic bicarbonate might thwart onset of acute kidney injury.1,2,15 Time is critical, and even on the battlefield or at the scene of a recent disaster, most attempts at resuscitation are begun immediately. IV access may be problematic, but administration of oral bicarbonate solutions has also proven effective.15 Close follow-up of the serum urea and creatinine levels and measurement of the urine pH during alkalinization is warranted throughout the course of the episode.
Unfortunately, some patients respond poorly to these conservative measures, and the released myoglobin can cause renal tubular blockage and necrosis, resulting in acute kidney injury.1 Renal replacement therapy may be required.16 However, most episodes of dialysis-dependent acute renal injury do subside with time.
For patients with less elusive causes of rhabdomyolysis, treatment will hinge on a workup of the possible etiologies and follow-up treatment to target the apparent cause. For example, carnitine may be administered to patients with carnitine deficiency, and hypokalemic patients may be given potassium.1,6,7 These patients will also need counseling before they consider engaging in an exercise program.
Patient’s Outcome
The case patient presented with exertional rhabdomyolysis; improper hydration, severe deconditioning, and a relatively low serum potassium level may all have contributed to the muscle necrosis she experienced. She was given IV alkaline solutions and did not develop acute kidney injury. She was discharged from the hospital and at the time of this writing was awaiting outpatient follow-up.
It should be interesting to see whether the case patient experiences any further episodes of severe weakness after engaging in exercise. Her low-normal potassium level (reference range, 3.5 to 5.3 mmol/L17) warrants further follow-up, as does her mildly elevated thyroid-stimulating hormone level (reference range, 0.5 to 4.7 mcIU/mL17).
CONCLUSION
Patients with rhabdomyolysis may present with muscle aches, darkened urine, and/or weakness; an elevated CPK level confirms the diagnosis. Management is mainly conservative, with IV hydration accompanied by alkalinizing the urine and correcting any metabolic abnormalities, such as potassium deficiencies. For the few patients who experience severe acute kidney injury, renal replacement therapy may be necessary.
While most causes of rhabdomyolysis have obvious clinical scenarios, such as a crush injury, a search for muscle enzyme deficiencies, disorders of potassium homeostasis, and thyroid abnormalities is also warranted in patients who present with exertional rhabdomyolysis.
A 39-year-old woman presented to the emergency department (ED) with a chief complaint of muscle aches and pain. She stated that three days earlier, she had begun exercising in a 45-minute “spinning” class (ie, riding a stationary bicycle with a weighted front wheel). The patient had not engaged in any aerobic exercise for at least six months before the spinning class. She mentioned that much older participants in the class were outperforming her, but she did not feel the need to keep up with them.
After dismounting, the woman said, she experienced weakness in her legs and had great difficulty ambulating. She went home, took 400 mg of ibuprofen, and went to bed. She awoke with pain and swelling in both thighs and continued to take ibuprofen, in addition to applying a topical mentholated preparation to her thighs. She took an Epsom salts bath two days later.
On the morning of the third day after the spinning class, she voided black urine and presented to the ED.
The patient had no significant medical history. Surgical history was limited to removal of a ganglion cyst on her wrist. She denied any history of seizure disorder, thyroid disease, hepatitis, heart disease, or hyperlipidemia.
The patient had been taking ibuprofen as needed since the spinning class. She was taking no other medications. She denied any allergies to drugs or food.
The patient admitted to smoking one pack of cigarettes per week and to occasional alcohol consumption but denied use of illicit drugs. She was employed as an executive officer for a large business association.
On physical examination, the patient’s vital signs were blood pressure, 134/73 mm/Hg; pulse, 86 beats/min; and respirations, 16 breaths/min. She was afebrile, alert, and oriented. Her sclera were nonicteric. Her neck was supple with no anterior cervical lymphadenopathy. There was no thyroid enlargement, her lungs were clear to auscultation, and her heart sounds were regular. There was no peripheral edema, and dorsalis pedis pulses were present bilaterally. Her thighs appeared swollen but were not tender to palpation.
The patient’s history, combined with an extremely high level of serum creatine phosphokinase (CPK; ie, 123,800 U/L [reference range, 45 to 260 U/L1]), confirmed a diagnosis of rhabdomyolysis. She was admitted for close observation. The patient’s urinalysis revealed 2 to 5 red blood cells and 6 to 10 white blood cells per high-power field. Moderate occult blood was detected, with no casts or protein noted. A urine myoglobin test was not performed.
The patient underwent IV hydration with dextrose 5% in water and three ampules of sodium bicarbonate after being given a 2.0-L saline bolus. Ibuprofen was discontinued. IV hydration with bicarbonate solution was continued until the patient’s CPK level declined significantly. She underwent daily laboratory testing (see Table 1). Her renal function remained stable, and she was discharged on hospital day 7.
DISCUSSION
Rhabdomyolysis is a clinical condition defined as muscle necrosis resulting from the release of intracellular skeletal muscle components (including myoglobin, CPK, potassium, phosphorus, and aldolase) into the extracellular compartment.1-3 The condition was first described during the bombing of London in World War II, with high incidence of crush injuries, shock, and associated kidney damage.4 The preponderance of such injuries during a 1988 earthquake in Armenia led the International Society of Nephrology to form its Renal Disaster Relief Task Force, which has provided support at numerous other disaster scenes since then.5
Rhabdomyolysis has been identified with a variety of pathologic events: those that cause muscle trauma, those associated with muscle use or overuse, and other etiologies involving genetic, metabolic, infectious, or pharmaceutical factors.1 Many of the reported causes of rhabdomyolysis are listed in Table 2.1,2
For patients with muscle trauma, the etiology of rhabdomyolysis is clear, but for those with other disease states, diagnosis may be more elusive. Patients who present with rhabdomyolysis after excessive exercise, for example, may have underlying metabolic disorders that predispose them to exertional rhabdomyolysis, such as chronic hypokalemia resulting from primary hyperaldosteronism.1 Others may have a muscle enzyme deficiency, as in McArdle’s syndrome or carnitine deficiency.6
Alterations in blood chemistries can also contribute to development of rhabdomyolysis, even when more obvious etiologies for muscle necrosis are evident. Hypokalemia interferes with the vasodilation that normally occurs during exercise to increase muscle blood flow.1,7,8 Continued exercise can lead to muscle necrosis, raising a concern for athletes who take diuretics.1 Hypophosphatemia leads to a state of muscle necrosis; this is of particular concern for alcoholic patients who receive hyperalimentation without repletion of phosphates.9
Diagnosis
Patients with rhabdomyolysis usually present with myalgias, darkened urine (red, brown, or black), and a clinical scenario that corroborates the diagnosis (ie, history of trauma, excessive exercise, use of an offending medication).1 Some patients may have minimal to absent symptoms or symptoms that occur only after exercise.3
A careful history is key. While traumatic causes are obvious, it is important to ask a patient with rhabdomyolysis after exertion about previous history of excessive weakness during or immediately after exercise, excessive cramping, or discoloration of urine after exercise. The family history may point to a genetic abnormality. A thorough understanding of the patient’s use of medications, including OTC agents, is also important. Rhabdomyolysis has been reported in patients who use herbal remedies, including those taken to facilitate weight loss or to improve lipid profiles.10,11
For patients suspected of having rhabdomyolysis, a serum CPK level should be obtained; results exceeding normal values by five times confirm the diagnosis.3 Measurements for potassium, phosphorus, and calcium are also important to determine, as is renal function. A high level of serum aldolase (an enzyme that breaks down glucose in muscle tissue) can also support a diagnosis of rhabdomyolysis.1,12 Urinalysis and urine myoglobin testing are also warranted, although a negative urine myoglobin test result does not rule out rhabdomyolysis in the presence of an elevated CPK level. Myoglobin is cleared rapidly by the kidneys, whereas serum CPK levels change slowly.1
Any patient who presents with acute rhabdomyolysis and low to normal values for potassium or phosphate should be evaluated further for hypokalemia and hypophosphatemia as contributing or etiologic factors. Hypocalcemia may occur in the early course of rhabdomyolysis as calcium salt is deposited in muscle tissue. Patients recovering from rhabdomyolysis may experience rebound hypercalcemia as the damaged muscle releases the deposited calcium.7,13
In most cases of rhabdomyolysis, only laboratory values are needed to make the diagnosis and follow the course of the episode.1 However, when the etiology appears to involve metabolic deficiencies or genetic etiologies, it may become necessary to order additional diagnostic tests. These may include tests for thyroid function, a carnitine level to screen for glycogen storage diseases, and toxin screening (eg, for illicit drugs, such as cocaine).2,6
Treatment and Management
Effective treatment of rhabdomyolysis relies on recognizing the underlying disorder.1 For patients with muscle trauma (eg, crush injury) or muscle overuse, the mainstay of treatment is aggressive fluid resuscitation and prevention of acute injury to the kidneys.13 As for patients with an injury induced by a pharmaceutical agent or a toxin, removal of the offending agent is required, followed by hydration and prevention of renal damage. Supportive care during an infectious illness is also essential.14
Additionally, treatment must address the complications inherent with rhabdomyolysis.1 In addition to CPK, potassium, phosphorus, and myoglobin are also released from skeletal muscle tissue. Hyperkalemia can be fatal, and potassium levels must be monitored closely to avert this condition.7,8,13 During an episode of rhabdomyolysis, normal levels of both potassium and phosphorus should raise the clinician’s suspicion for underlying hypokalemia and hypophosphatemia—conditions that may have contributed to the episode of rhabdomyolysis. Hypocalcemia may also develop.13
Released myoglobin may cause acute kidney injury, as is the case in 33% to 50% of patients with rhabdomyolysis.3 In early studies, it was determined that alkalinizing the urine with IV isotonic bicarbonate might thwart onset of acute kidney injury.1,2,15 Time is critical, and even on the battlefield or at the scene of a recent disaster, most attempts at resuscitation are begun immediately. IV access may be problematic, but administration of oral bicarbonate solutions has also proven effective.15 Close follow-up of the serum urea and creatinine levels and measurement of the urine pH during alkalinization is warranted throughout the course of the episode.
Unfortunately, some patients respond poorly to these conservative measures, and the released myoglobin can cause renal tubular blockage and necrosis, resulting in acute kidney injury.1 Renal replacement therapy may be required.16 However, most episodes of dialysis-dependent acute renal injury do subside with time.
For patients with less elusive causes of rhabdomyolysis, treatment will hinge on a workup of the possible etiologies and follow-up treatment to target the apparent cause. For example, carnitine may be administered to patients with carnitine deficiency, and hypokalemic patients may be given potassium.1,6,7 These patients will also need counseling before they consider engaging in an exercise program.
Patient’s Outcome
The case patient presented with exertional rhabdomyolysis; improper hydration, severe deconditioning, and a relatively low serum potassium level may all have contributed to the muscle necrosis she experienced. She was given IV alkaline solutions and did not develop acute kidney injury. She was discharged from the hospital and at the time of this writing was awaiting outpatient follow-up.
It should be interesting to see whether the case patient experiences any further episodes of severe weakness after engaging in exercise. Her low-normal potassium level (reference range, 3.5 to 5.3 mmol/L17) warrants further follow-up, as does her mildly elevated thyroid-stimulating hormone level (reference range, 0.5 to 4.7 mcIU/mL17).
CONCLUSION
Patients with rhabdomyolysis may present with muscle aches, darkened urine, and/or weakness; an elevated CPK level confirms the diagnosis. Management is mainly conservative, with IV hydration accompanied by alkalinizing the urine and correcting any metabolic abnormalities, such as potassium deficiencies. For the few patients who experience severe acute kidney injury, renal replacement therapy may be necessary.
While most causes of rhabdomyolysis have obvious clinical scenarios, such as a crush injury, a search for muscle enzyme deficiencies, disorders of potassium homeostasis, and thyroid abnormalities is also warranted in patients who present with exertional rhabdomyolysis.
1. Huerta-Alardín AL, Varon J, Marik PE. Bench-to-bedside review: rhabdomyolysis—an overview for clinicians. Crit Care. 2005;9(2):158-169.
2. Warren JD, Blumbergs PC, Thompson PD. Rhabdomyolysis: a review. Muscle Nerve. 2002;25(3):332-347.
3. Lima RSA, da Silva GB Jr, Liborio AB, Daher ED. Acute kidney injury due to rhabdomyolysis. Saudi J Kidney Dis Transpl. 2008;19(5):721-729.
4. Bywaters EG, Beall D. Crush injuries with impairment of renal function [reprinted from BMJ, 1941]. J Am Soc Nephrol. 1998;9(2):322-332.
5. Vanholder R, Van Biesen W, Lameire N, Sever MS; International Society of Nephrology/Renal Disaster Relief Task Force. The role of the International Society of Nephrology/Renal Disaster Relief Task Force in the rescue of renal disaster victims. Contrib Nephrol. 2007;156:325-332.
6. Toledo R, López V, Martín G, et al. Rhabdomyolysis due to enzyme deficiency in muscles. Nefrología. 2009;29(1):77-80.
7. Agrawal S, Agrawal V, Taneja A. Hypokalemia causing rhabdomyolysis resulting in life-threatening hyperkalemia. Pediatr Nephrol. 2006;221(2): 289-291.
8. Knochel JP, Schlein EM. On the mechanism of rhabdomyolysis in potassium depletion. J Clin Invest. 1972:51(7):1750-1758.
9. Knochel JP. Hypophosphatemia and rhabdomyolysis. Am J Med. 1992;92(5):455-457.
10. Mansi IA, Huang J. Rhabdomyolysis in response to weight-loss herbal medicine. Am J Med Sci. 2004; 327(6): 356-357.
11. Heber D, Yip I, Ashley JM, et al. Cholesterol-lowering effects of a proprietary Chinese red-yeast-rice dietary supplement. Am J Clin Nutr. 1999;69(2): 231-236.
12. Hooda AK, Narula AS. Exertional rhabdomyolysis causing acute renal failure. Med J Armed Forces India. 2005;61(4):395-396.
13. Chatzizisis YS, Misirli G, Hatzitolios AI, Giannoglou GD. The syndrome of rhabdomyolysis: complications and treatment. Eur J Intern Med. 2008;19(8): 568-574.
14. Blanco JR, Zabalza M, Salcedo J, et al. Rhabdomyolysis of infectious and noninfectious causes. South Med J. 2002;95(5):542-544.
15. Ron D, Taitelman U, Michaelson M, et al. Prevention of acute renal failure in traumatic rhabdomyolysis. Arch Intern Med. 1984;144(2):277-280.
16. Soni SS, Nagarik AP, Adikey GK, Raman A. Using continuous renal replacement therapy to manage patients of shock and acute renal failure. J Emerg Trauma Shock. 2009;2(1):19-22.
17. Normal laboratory values. In: Beers MH, Berkow R, eds. The Merck Manual of Diagnosis and Therapy. 17th ed. Whitehouse Station, NJ: Merck Research Laboratories; 1999:2526-2546.
1. Huerta-Alardín AL, Varon J, Marik PE. Bench-to-bedside review: rhabdomyolysis—an overview for clinicians. Crit Care. 2005;9(2):158-169.
2. Warren JD, Blumbergs PC, Thompson PD. Rhabdomyolysis: a review. Muscle Nerve. 2002;25(3):332-347.
3. Lima RSA, da Silva GB Jr, Liborio AB, Daher ED. Acute kidney injury due to rhabdomyolysis. Saudi J Kidney Dis Transpl. 2008;19(5):721-729.
4. Bywaters EG, Beall D. Crush injuries with impairment of renal function [reprinted from BMJ, 1941]. J Am Soc Nephrol. 1998;9(2):322-332.
5. Vanholder R, Van Biesen W, Lameire N, Sever MS; International Society of Nephrology/Renal Disaster Relief Task Force. The role of the International Society of Nephrology/Renal Disaster Relief Task Force in the rescue of renal disaster victims. Contrib Nephrol. 2007;156:325-332.
6. Toledo R, López V, Martín G, et al. Rhabdomyolysis due to enzyme deficiency in muscles. Nefrología. 2009;29(1):77-80.
7. Agrawal S, Agrawal V, Taneja A. Hypokalemia causing rhabdomyolysis resulting in life-threatening hyperkalemia. Pediatr Nephrol. 2006;221(2): 289-291.
8. Knochel JP, Schlein EM. On the mechanism of rhabdomyolysis in potassium depletion. J Clin Invest. 1972:51(7):1750-1758.
9. Knochel JP. Hypophosphatemia and rhabdomyolysis. Am J Med. 1992;92(5):455-457.
10. Mansi IA, Huang J. Rhabdomyolysis in response to weight-loss herbal medicine. Am J Med Sci. 2004; 327(6): 356-357.
11. Heber D, Yip I, Ashley JM, et al. Cholesterol-lowering effects of a proprietary Chinese red-yeast-rice dietary supplement. Am J Clin Nutr. 1999;69(2): 231-236.
12. Hooda AK, Narula AS. Exertional rhabdomyolysis causing acute renal failure. Med J Armed Forces India. 2005;61(4):395-396.
13. Chatzizisis YS, Misirli G, Hatzitolios AI, Giannoglou GD. The syndrome of rhabdomyolysis: complications and treatment. Eur J Intern Med. 2008;19(8): 568-574.
14. Blanco JR, Zabalza M, Salcedo J, et al. Rhabdomyolysis of infectious and noninfectious causes. South Med J. 2002;95(5):542-544.
15. Ron D, Taitelman U, Michaelson M, et al. Prevention of acute renal failure in traumatic rhabdomyolysis. Arch Intern Med. 1984;144(2):277-280.
16. Soni SS, Nagarik AP, Adikey GK, Raman A. Using continuous renal replacement therapy to manage patients of shock and acute renal failure. J Emerg Trauma Shock. 2009;2(1):19-22.
17. Normal laboratory values. In: Beers MH, Berkow R, eds. The Merck Manual of Diagnosis and Therapy. 17th ed. Whitehouse Station, NJ: Merck Research Laboratories; 1999:2526-2546.
Grand Rounds: Man, 29, With Apparent Throat Obstruction
A 29-year-old man presented to the emergency department (ED) with a chief complaint of food stuck in his throat. He reported that he had swallowed a piece of chicken and felt it get stuck. Drinking water to help it go down was unsuccessful.
The patient’s history was positive for childhood asthma and nine years of solid food dysphagia. There was no history of a caustic chemical ingestion or of drug-induced esophagitis. He denied having dyspepsia, heartburn, or chest pain. He was not taking any medications and had no allergies.
When his dysphagia symptoms began nine years ago, he was diagnosed with acid reflux disease, confirmed by an upper gastrointestinal (GI) tract x-ray. Since that time, he reported having to swallow liquid after every bite of food and said he suffered from severe anxiety over fear of choking.
Evaluation in the ED consisted of endoscopic examination by a gastroenterologist. In addition to dislodging a food bolus, the endoscope revealed a narrowed, ringed esophagus with mucosal changes throughout the length of the esophagus (see Figures 1 through 3). Esophageal biopsies were taken, and the esophagus was dilated successfully with a 40-Fr Maloney dilator. The endoscopist detected too much resistance to pass a larger dilator.
Biopsy results revealed eosinophilic esophagitis. The patient was given oral fluticasone propionate. At one-month follow-up, he reported feeling much better. Upper endoscopy revealed some improvement, and the gastroenterologist was able to pass both a 46- and a 48-Fr Maloney dilator with only mild resistance. (The largest Maloney dilator, a 60-Fr dilator, should easily pass through a normal esophagus, according to T. L. Sack, MD, oral communication, June 2009.)
Discussion
Eosinophilic esophagitis (EE) involves the infiltration of the esophageal mucosa with eosinophils, causing edema, inflammation, and eventually, thickening and stenotic changes of the esophageal mucosa.1
The normal esophageal mucosa contains lymphocytes, mast cells, and dendritic cells, which protect the esophagus from invading toxins and microorganisms. Eosinophils are not usually present, but when they are, they can have toxic effects on the esophageal mucosa.2 EE is associated with solid food dysphagia, a direct result of damage to the esophageal mucosa, and other causes that are not clearly understood.
Research findings suggest that symptoms of dysphagia may be caused by degranulating eosinophils and mast cells, which have an antagonistic effect on the muscarinic receptors and cause smooth muscle to contract.3,4 The proposed triggering mechanism of EE is an immunoglobulin E (IgE) immune–mediated response to an allergen.2 Based on results from IgE radioallergosorbent testing (RAST), aeroallergens are more likely than food to act as triggers.5
EE in the Adult Patient
Traditionally, EE has been a condition seen in the pediatric population, with symptoms of nausea, vomiting, and failure to thrive; however, it is becoming increasingly recognized among adults. The typical patient is a man in his 20s or 30s (although cases of EE have been reported among women and older adults) with acute and recurrent solid food dysphagia, with or without food impaction.4
Often the patient reports a history of environmental or food allergies, asthma, rhinitis, or eczema.2,4-6 Researchers have reported the presence of allergic symptoms in at least 50% of patients diagnosed with EE,2 and many patients experience exacerbations associated with seasonal changes.7
GERD may coexist with EE; however, no relationship has been identified between the two.8 EE should be considered in patients with gastrointestinal symptoms that persist despite at least four weeks’ treatment with a proton pump inhibitor (PPI).2
Dysphagia: Differential Diagnosis
Adult patients with esophageal dysphagia usually report the feeling of food getting stuck when they try to swallow.9 Dysphagia may result from a mechanical obstruction or a neuromuscular/motility condition. Patients with mechanical obstructions usually have difficulty swallowing solids, while those with motility disorders tend to have difficulty with both liquids and solids.1,9
Mechanical obstructions may include carcinomas (intrinsic and extrinsic), strictures, or Schatzki rings (small thin mucosal rings of unknown etiology located at the gastroesophageal junction).1,9 Progressive dysphagia to solids over a short period of time is often indicative of esophageal carcinoma. GERD, pill-induced trauma, previous ingestion of a caustic chemical, and radiation are common causes of esophageal stricture formation. For a list of medications that are particularly caustic to the esophageal mucosa, see the table.9,10
Neuromuscular manifestations of dysphagia include achalasia, diffuse esophageal spasm, nutcracker esophagus, and scleroderma.1 These are usually associated with progressive difficulty in swallowing.9
Evaluating the Patient
A thorough patient history can often reveal potential causes of dysphagia and eliminate others. This should include current medications, chronic medical conditions and details regarding their onset and duration, and symptoms associated with dysphagia.9
Physical examination should include palpation of the thyroid because of the potential for a thyroid mass to cause extrinsic compression of the esophagus, palpation of the abdomen for masses or organomegaly, and a complete neurologic evaluation.9
Laboratory tests should be ordered based on the information obtained from the history and physical. Testing may include thyroid studies to eliminate hypothyroid or hyperthyroid causes of dysphagia, and complete blood count (CBC) with differential to rule out inflammatory or infectious processes.9 While eosinophilia may be present in the differential, it is not a universally accepted marker for establishing the diagnosis of EE.2,5 Stools should be checked for occult blood, because a positive finding may suggest esophageal carcinoma.9
Diagnosis
In the primary care setting, a barium esophagram may be used during the initial workup to evaluate the anatomic structures of the esophagus and to differentiate between a mechanical obstruction and a neuromuscular disorder.1,9 This noninvasive test requires the patient to swallow a radiopaque liquid as x-rays are taken.
The gold standard for diagnosing EE, however, is upper endoscopy with biopsy of the esophageal mucosa.6 Endoscopic findings that indicate EE are atypical of GERD; they may include a narrowed, small-caliber esophagus, concentric mucosal rings, proximal stenosis, linear ulcerations, atrophic changes, and white papules associated with eosinophilic microabscesses.6
Although there is no consensus regarding the number of eosinophils that should be present for an accurate diagnosis of EE, microscopic interpretation of the biopsy from both the proximal and the distal esophageal epithelia5 usually shows 15 or more eosinophils per high-power field.2,11 It has been suggested that mucosal biopsies be taken along the entire length of the esophagus, as eosinophilic infiltration may extend from the proximal to the distal esophagus.2
GERD and trauma induced by medication use may also be associated with esophageal eosinophilic infiltration5; however, eosinophils are usually present only in the distal esophageal mucosa3 and are not as abundant as in EE.7 If endoscopy reveals persistent eosinophilia despite four to eight weeks’ treatment with a PPI, the diagnosis of EE is confirmed.2
Treatment
Treatment for EE is still under investigation. Research has examined the association between EE and food allergies or aeroallergens.4 Evaluation by an allergist using skin prick tests or RAST is recommended in the adult patient to help determine the source of the underlying inflammation.5,7 Eliminating any identified allergen should help alleviate symptoms.4
For patients in whom no source of inflammation can be identified, treatment with 1.0 to 2.0 mg/kg/d of oral prednisone for acute exacerbations has been shown to significantly improve symptoms and histology12; however, because of the associated risk for adverse systemic effects, long-term use is not recommended.
In many patients, the inhaled corticosteroid fluticasone has also proved successful in reducing EE—associated inflammation.6 Current evidence supports adult dosing between 880 and 1,760 mcg per day for six to eight weeks, administered with a metered-dose inhaler and no spacer. Fluticasone should be sprayed directly into the mouth and swallowed, after which the patient should take nothing by mouth for 30 minutes.13 Prolonged fluticasone use has been associated with esophageal candidiasis.2 There are currently no recommendations regarding its use as maintenance therapy.
Montelukast, a leukotriene receptor antagonist, has also been shown in some studies to reduce the inflammatory process11; however, one study team recently found it to have no therapeutic effect.13
PPIs may be effective for improving EE symptoms even in the absence of GERD because of the reduced gastric acid production,7 but they do not usually improve EE’s histologic features.3
Use of esophageal dilation in patients with EE is controversial because of an associated risk for perforation.14 If this intervention is to be performed, the patient should be treated in advance with oral corticosteroids to reduce esophageal inflammation.15,16 In addition, the endoscopist should start with small-sized dilators and carefully proceed to larger sizes.11 Critics of esophageal dilation argue that the procedure is only a temporary solution and does nothing for the underlying condition.4,8
Regarding endoscopic surveillance, an interval of at least four weeks between interventions is recommended.13
Role of the Primary Care Clinician
Undiagnosed EE can cause the patient discomfort, frustration, and anxiety, as seen in the case study. Many patients with undiagnosed EE have been exposed to unnecessary medical therapy and antireflux surgery.3 Without proper diagnosis and treatment, EE may worsen, causing complications associated with chronic inflammation (ie, esophageal fibrosis and strictures).2,6
The long-term prognosis of EE is unknown at this time.8 The disease is usually chronic, with periods of remission and exacerbation. With an understanding of EE and appropriate therapies, the primary care practitioner can team with the gastroenterologist to provide effective disease management through endoscopic surveillance and intervention for acute exacerbations. Guidelines recommend that patients be closely followed with regular office visits to reassess symptoms, compliance with therapy, and adverse effects, with the goal of preventing complications associated with EE.13
Conclusion
To effectively evaluate the patient who presents with dysphagia, the primary care provider should have a working knowledge of EE, as well as an understanding of the key elements in the history and physical examination to help ensure an accurate diagnosis. This will facilitate timely referral to a gastroenterologist for endoscopic evaluation, when indicated.
1. McQuaid KR. Gastrointestinal disorders. In: McPhee S, Papadakis M. CURRENT Medical Diagnosis & Treatment 2009. New York: McGraw-Hill: 2009:487-581.
2. Nurko S, Furuta GT. Eosinophilic esophagitis (2006). GI Motility Online. www.nature.com/gimo/contents/pt1/full/gimo49.html. Accessed July 27, 2009.
3. Parfitt JR, Gregor JC, Suskin NG, et al. Eosinophilic esophagitis in adults: distinguishing features from gastroesophageal reflux disease: a study of 41 patients. Mod Pathol. 2006;19(1):90-96.
4. Swoger JM, Weiler CR, Arora AS. Eosinophilic esophagitis: is it all allergies? Mayo Clin Proc. 2007;82(12):1541-1549.
5. Conus S, Simon HU. General laboratory diagnostics of eosinophilic GI diseases. Best Pract Res Clin Gastroenterol. 2008;22(3):441-453.
6. Remedios M, Campbell C, Jones DM, Kerlin P. Eosinophilic esophagitis in adults: clinical, endoscopic, histologic findings, and response to treatment with fluticasone propionate. Gastrointest Endosc. 2006;63(1):3-12.
7. Rothenberg ME. Eosinophilic gastrointestinal disorders (EGID). J Allergy Clin Immunol. 2004; 113(1):11-28.
8. Lucendo AJ, Carrion G, Navarro M, et al. Eosinophilic esophagitis in adults: an emerging disease. Dig Dis Sci. 2004;49(11-12):1884-1888.
9. Spieker MR. Evaluating dysphagia. Am Fam Physician. 2000;61(12):3639-3648.
10. Boyce HW. Drug-induced esophageal damage: diseases of medical progress. Gastrointest Endosc. 1998;47:547-550.
11. Potter JW, Saeian K, Staff D, et al. Eosinophilic esophagitis in adults: an emerging problem with unique esophageal features. Gastrointest Endosc. 2004;59(3):355-361.
12. Schaefer ET, Fitzgerald JF, Molleston JP, et al. Comparison of oral prednisone and topical fluticasone in the treatment of eosinophilic esophagitis: a randomized trial in children. Clin Gastroenterol Hepatol. 2008;6(2):165-173.
13. Furuta GT, Liacouras CA, Collins MH, et al; First International Gastrointestinal Eosinophil Research Symposium (FIGERS) Subcommittees. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendation for diagnosis and treatment. Gastroenterology. 2007;133(4): 1342-1363.
14. Straumann A, Rossi L, Simon HU, et al. Fragility of the esophageal mucosa: a pathognomonic endoscopic sign of primary eosinophilic esophagitis. Gastrointest Endosc. 2003;57(3):407-412.
15. Hawari R, Pasricha PJ. Images in clinical medicine: eosinophilic esophagitis. N Engl J Med. 2007; 356(20):e20.
16. Leclercq P, Marting A, Gast P. Eosinophilic esophagitis. N Engl J Med. 2007;357(14):1446.
A 29-year-old man presented to the emergency department (ED) with a chief complaint of food stuck in his throat. He reported that he had swallowed a piece of chicken and felt it get stuck. Drinking water to help it go down was unsuccessful.
The patient’s history was positive for childhood asthma and nine years of solid food dysphagia. There was no history of a caustic chemical ingestion or of drug-induced esophagitis. He denied having dyspepsia, heartburn, or chest pain. He was not taking any medications and had no allergies.
When his dysphagia symptoms began nine years ago, he was diagnosed with acid reflux disease, confirmed by an upper gastrointestinal (GI) tract x-ray. Since that time, he reported having to swallow liquid after every bite of food and said he suffered from severe anxiety over fear of choking.
Evaluation in the ED consisted of endoscopic examination by a gastroenterologist. In addition to dislodging a food bolus, the endoscope revealed a narrowed, ringed esophagus with mucosal changes throughout the length of the esophagus (see Figures 1 through 3). Esophageal biopsies were taken, and the esophagus was dilated successfully with a 40-Fr Maloney dilator. The endoscopist detected too much resistance to pass a larger dilator.
Biopsy results revealed eosinophilic esophagitis. The patient was given oral fluticasone propionate. At one-month follow-up, he reported feeling much better. Upper endoscopy revealed some improvement, and the gastroenterologist was able to pass both a 46- and a 48-Fr Maloney dilator with only mild resistance. (The largest Maloney dilator, a 60-Fr dilator, should easily pass through a normal esophagus, according to T. L. Sack, MD, oral communication, June 2009.)
Discussion
Eosinophilic esophagitis (EE) involves the infiltration of the esophageal mucosa with eosinophils, causing edema, inflammation, and eventually, thickening and stenotic changes of the esophageal mucosa.1
The normal esophageal mucosa contains lymphocytes, mast cells, and dendritic cells, which protect the esophagus from invading toxins and microorganisms. Eosinophils are not usually present, but when they are, they can have toxic effects on the esophageal mucosa.2 EE is associated with solid food dysphagia, a direct result of damage to the esophageal mucosa, and other causes that are not clearly understood.
Research findings suggest that symptoms of dysphagia may be caused by degranulating eosinophils and mast cells, which have an antagonistic effect on the muscarinic receptors and cause smooth muscle to contract.3,4 The proposed triggering mechanism of EE is an immunoglobulin E (IgE) immune–mediated response to an allergen.2 Based on results from IgE radioallergosorbent testing (RAST), aeroallergens are more likely than food to act as triggers.5
EE in the Adult Patient
Traditionally, EE has been a condition seen in the pediatric population, with symptoms of nausea, vomiting, and failure to thrive; however, it is becoming increasingly recognized among adults. The typical patient is a man in his 20s or 30s (although cases of EE have been reported among women and older adults) with acute and recurrent solid food dysphagia, with or without food impaction.4
Often the patient reports a history of environmental or food allergies, asthma, rhinitis, or eczema.2,4-6 Researchers have reported the presence of allergic symptoms in at least 50% of patients diagnosed with EE,2 and many patients experience exacerbations associated with seasonal changes.7
GERD may coexist with EE; however, no relationship has been identified between the two.8 EE should be considered in patients with gastrointestinal symptoms that persist despite at least four weeks’ treatment with a proton pump inhibitor (PPI).2
Dysphagia: Differential Diagnosis
Adult patients with esophageal dysphagia usually report the feeling of food getting stuck when they try to swallow.9 Dysphagia may result from a mechanical obstruction or a neuromuscular/motility condition. Patients with mechanical obstructions usually have difficulty swallowing solids, while those with motility disorders tend to have difficulty with both liquids and solids.1,9
Mechanical obstructions may include carcinomas (intrinsic and extrinsic), strictures, or Schatzki rings (small thin mucosal rings of unknown etiology located at the gastroesophageal junction).1,9 Progressive dysphagia to solids over a short period of time is often indicative of esophageal carcinoma. GERD, pill-induced trauma, previous ingestion of a caustic chemical, and radiation are common causes of esophageal stricture formation. For a list of medications that are particularly caustic to the esophageal mucosa, see the table.9,10
Neuromuscular manifestations of dysphagia include achalasia, diffuse esophageal spasm, nutcracker esophagus, and scleroderma.1 These are usually associated with progressive difficulty in swallowing.9
Evaluating the Patient
A thorough patient history can often reveal potential causes of dysphagia and eliminate others. This should include current medications, chronic medical conditions and details regarding their onset and duration, and symptoms associated with dysphagia.9
Physical examination should include palpation of the thyroid because of the potential for a thyroid mass to cause extrinsic compression of the esophagus, palpation of the abdomen for masses or organomegaly, and a complete neurologic evaluation.9
Laboratory tests should be ordered based on the information obtained from the history and physical. Testing may include thyroid studies to eliminate hypothyroid or hyperthyroid causes of dysphagia, and complete blood count (CBC) with differential to rule out inflammatory or infectious processes.9 While eosinophilia may be present in the differential, it is not a universally accepted marker for establishing the diagnosis of EE.2,5 Stools should be checked for occult blood, because a positive finding may suggest esophageal carcinoma.9
Diagnosis
In the primary care setting, a barium esophagram may be used during the initial workup to evaluate the anatomic structures of the esophagus and to differentiate between a mechanical obstruction and a neuromuscular disorder.1,9 This noninvasive test requires the patient to swallow a radiopaque liquid as x-rays are taken.
The gold standard for diagnosing EE, however, is upper endoscopy with biopsy of the esophageal mucosa.6 Endoscopic findings that indicate EE are atypical of GERD; they may include a narrowed, small-caliber esophagus, concentric mucosal rings, proximal stenosis, linear ulcerations, atrophic changes, and white papules associated with eosinophilic microabscesses.6
Although there is no consensus regarding the number of eosinophils that should be present for an accurate diagnosis of EE, microscopic interpretation of the biopsy from both the proximal and the distal esophageal epithelia5 usually shows 15 or more eosinophils per high-power field.2,11 It has been suggested that mucosal biopsies be taken along the entire length of the esophagus, as eosinophilic infiltration may extend from the proximal to the distal esophagus.2
GERD and trauma induced by medication use may also be associated with esophageal eosinophilic infiltration5; however, eosinophils are usually present only in the distal esophageal mucosa3 and are not as abundant as in EE.7 If endoscopy reveals persistent eosinophilia despite four to eight weeks’ treatment with a PPI, the diagnosis of EE is confirmed.2
Treatment
Treatment for EE is still under investigation. Research has examined the association between EE and food allergies or aeroallergens.4 Evaluation by an allergist using skin prick tests or RAST is recommended in the adult patient to help determine the source of the underlying inflammation.5,7 Eliminating any identified allergen should help alleviate symptoms.4
For patients in whom no source of inflammation can be identified, treatment with 1.0 to 2.0 mg/kg/d of oral prednisone for acute exacerbations has been shown to significantly improve symptoms and histology12; however, because of the associated risk for adverse systemic effects, long-term use is not recommended.
In many patients, the inhaled corticosteroid fluticasone has also proved successful in reducing EE—associated inflammation.6 Current evidence supports adult dosing between 880 and 1,760 mcg per day for six to eight weeks, administered with a metered-dose inhaler and no spacer. Fluticasone should be sprayed directly into the mouth and swallowed, after which the patient should take nothing by mouth for 30 minutes.13 Prolonged fluticasone use has been associated with esophageal candidiasis.2 There are currently no recommendations regarding its use as maintenance therapy.
Montelukast, a leukotriene receptor antagonist, has also been shown in some studies to reduce the inflammatory process11; however, one study team recently found it to have no therapeutic effect.13
PPIs may be effective for improving EE symptoms even in the absence of GERD because of the reduced gastric acid production,7 but they do not usually improve EE’s histologic features.3
Use of esophageal dilation in patients with EE is controversial because of an associated risk for perforation.14 If this intervention is to be performed, the patient should be treated in advance with oral corticosteroids to reduce esophageal inflammation.15,16 In addition, the endoscopist should start with small-sized dilators and carefully proceed to larger sizes.11 Critics of esophageal dilation argue that the procedure is only a temporary solution and does nothing for the underlying condition.4,8
Regarding endoscopic surveillance, an interval of at least four weeks between interventions is recommended.13
Role of the Primary Care Clinician
Undiagnosed EE can cause the patient discomfort, frustration, and anxiety, as seen in the case study. Many patients with undiagnosed EE have been exposed to unnecessary medical therapy and antireflux surgery.3 Without proper diagnosis and treatment, EE may worsen, causing complications associated with chronic inflammation (ie, esophageal fibrosis and strictures).2,6
The long-term prognosis of EE is unknown at this time.8 The disease is usually chronic, with periods of remission and exacerbation. With an understanding of EE and appropriate therapies, the primary care practitioner can team with the gastroenterologist to provide effective disease management through endoscopic surveillance and intervention for acute exacerbations. Guidelines recommend that patients be closely followed with regular office visits to reassess symptoms, compliance with therapy, and adverse effects, with the goal of preventing complications associated with EE.13
Conclusion
To effectively evaluate the patient who presents with dysphagia, the primary care provider should have a working knowledge of EE, as well as an understanding of the key elements in the history and physical examination to help ensure an accurate diagnosis. This will facilitate timely referral to a gastroenterologist for endoscopic evaluation, when indicated.
A 29-year-old man presented to the emergency department (ED) with a chief complaint of food stuck in his throat. He reported that he had swallowed a piece of chicken and felt it get stuck. Drinking water to help it go down was unsuccessful.
The patient’s history was positive for childhood asthma and nine years of solid food dysphagia. There was no history of a caustic chemical ingestion or of drug-induced esophagitis. He denied having dyspepsia, heartburn, or chest pain. He was not taking any medications and had no allergies.
When his dysphagia symptoms began nine years ago, he was diagnosed with acid reflux disease, confirmed by an upper gastrointestinal (GI) tract x-ray. Since that time, he reported having to swallow liquid after every bite of food and said he suffered from severe anxiety over fear of choking.
Evaluation in the ED consisted of endoscopic examination by a gastroenterologist. In addition to dislodging a food bolus, the endoscope revealed a narrowed, ringed esophagus with mucosal changes throughout the length of the esophagus (see Figures 1 through 3). Esophageal biopsies were taken, and the esophagus was dilated successfully with a 40-Fr Maloney dilator. The endoscopist detected too much resistance to pass a larger dilator.
Biopsy results revealed eosinophilic esophagitis. The patient was given oral fluticasone propionate. At one-month follow-up, he reported feeling much better. Upper endoscopy revealed some improvement, and the gastroenterologist was able to pass both a 46- and a 48-Fr Maloney dilator with only mild resistance. (The largest Maloney dilator, a 60-Fr dilator, should easily pass through a normal esophagus, according to T. L. Sack, MD, oral communication, June 2009.)
Discussion
Eosinophilic esophagitis (EE) involves the infiltration of the esophageal mucosa with eosinophils, causing edema, inflammation, and eventually, thickening and stenotic changes of the esophageal mucosa.1
The normal esophageal mucosa contains lymphocytes, mast cells, and dendritic cells, which protect the esophagus from invading toxins and microorganisms. Eosinophils are not usually present, but when they are, they can have toxic effects on the esophageal mucosa.2 EE is associated with solid food dysphagia, a direct result of damage to the esophageal mucosa, and other causes that are not clearly understood.
Research findings suggest that symptoms of dysphagia may be caused by degranulating eosinophils and mast cells, which have an antagonistic effect on the muscarinic receptors and cause smooth muscle to contract.3,4 The proposed triggering mechanism of EE is an immunoglobulin E (IgE) immune–mediated response to an allergen.2 Based on results from IgE radioallergosorbent testing (RAST), aeroallergens are more likely than food to act as triggers.5
EE in the Adult Patient
Traditionally, EE has been a condition seen in the pediatric population, with symptoms of nausea, vomiting, and failure to thrive; however, it is becoming increasingly recognized among adults. The typical patient is a man in his 20s or 30s (although cases of EE have been reported among women and older adults) with acute and recurrent solid food dysphagia, with or without food impaction.4
Often the patient reports a history of environmental or food allergies, asthma, rhinitis, or eczema.2,4-6 Researchers have reported the presence of allergic symptoms in at least 50% of patients diagnosed with EE,2 and many patients experience exacerbations associated with seasonal changes.7
GERD may coexist with EE; however, no relationship has been identified between the two.8 EE should be considered in patients with gastrointestinal symptoms that persist despite at least four weeks’ treatment with a proton pump inhibitor (PPI).2
Dysphagia: Differential Diagnosis
Adult patients with esophageal dysphagia usually report the feeling of food getting stuck when they try to swallow.9 Dysphagia may result from a mechanical obstruction or a neuromuscular/motility condition. Patients with mechanical obstructions usually have difficulty swallowing solids, while those with motility disorders tend to have difficulty with both liquids and solids.1,9
Mechanical obstructions may include carcinomas (intrinsic and extrinsic), strictures, or Schatzki rings (small thin mucosal rings of unknown etiology located at the gastroesophageal junction).1,9 Progressive dysphagia to solids over a short period of time is often indicative of esophageal carcinoma. GERD, pill-induced trauma, previous ingestion of a caustic chemical, and radiation are common causes of esophageal stricture formation. For a list of medications that are particularly caustic to the esophageal mucosa, see the table.9,10
Neuromuscular manifestations of dysphagia include achalasia, diffuse esophageal spasm, nutcracker esophagus, and scleroderma.1 These are usually associated with progressive difficulty in swallowing.9
Evaluating the Patient
A thorough patient history can often reveal potential causes of dysphagia and eliminate others. This should include current medications, chronic medical conditions and details regarding their onset and duration, and symptoms associated with dysphagia.9
Physical examination should include palpation of the thyroid because of the potential for a thyroid mass to cause extrinsic compression of the esophagus, palpation of the abdomen for masses or organomegaly, and a complete neurologic evaluation.9
Laboratory tests should be ordered based on the information obtained from the history and physical. Testing may include thyroid studies to eliminate hypothyroid or hyperthyroid causes of dysphagia, and complete blood count (CBC) with differential to rule out inflammatory or infectious processes.9 While eosinophilia may be present in the differential, it is not a universally accepted marker for establishing the diagnosis of EE.2,5 Stools should be checked for occult blood, because a positive finding may suggest esophageal carcinoma.9
Diagnosis
In the primary care setting, a barium esophagram may be used during the initial workup to evaluate the anatomic structures of the esophagus and to differentiate between a mechanical obstruction and a neuromuscular disorder.1,9 This noninvasive test requires the patient to swallow a radiopaque liquid as x-rays are taken.
The gold standard for diagnosing EE, however, is upper endoscopy with biopsy of the esophageal mucosa.6 Endoscopic findings that indicate EE are atypical of GERD; they may include a narrowed, small-caliber esophagus, concentric mucosal rings, proximal stenosis, linear ulcerations, atrophic changes, and white papules associated with eosinophilic microabscesses.6
Although there is no consensus regarding the number of eosinophils that should be present for an accurate diagnosis of EE, microscopic interpretation of the biopsy from both the proximal and the distal esophageal epithelia5 usually shows 15 or more eosinophils per high-power field.2,11 It has been suggested that mucosal biopsies be taken along the entire length of the esophagus, as eosinophilic infiltration may extend from the proximal to the distal esophagus.2
GERD and trauma induced by medication use may also be associated with esophageal eosinophilic infiltration5; however, eosinophils are usually present only in the distal esophageal mucosa3 and are not as abundant as in EE.7 If endoscopy reveals persistent eosinophilia despite four to eight weeks’ treatment with a PPI, the diagnosis of EE is confirmed.2
Treatment
Treatment for EE is still under investigation. Research has examined the association between EE and food allergies or aeroallergens.4 Evaluation by an allergist using skin prick tests or RAST is recommended in the adult patient to help determine the source of the underlying inflammation.5,7 Eliminating any identified allergen should help alleviate symptoms.4
For patients in whom no source of inflammation can be identified, treatment with 1.0 to 2.0 mg/kg/d of oral prednisone for acute exacerbations has been shown to significantly improve symptoms and histology12; however, because of the associated risk for adverse systemic effects, long-term use is not recommended.
In many patients, the inhaled corticosteroid fluticasone has also proved successful in reducing EE—associated inflammation.6 Current evidence supports adult dosing between 880 and 1,760 mcg per day for six to eight weeks, administered with a metered-dose inhaler and no spacer. Fluticasone should be sprayed directly into the mouth and swallowed, after which the patient should take nothing by mouth for 30 minutes.13 Prolonged fluticasone use has been associated with esophageal candidiasis.2 There are currently no recommendations regarding its use as maintenance therapy.
Montelukast, a leukotriene receptor antagonist, has also been shown in some studies to reduce the inflammatory process11; however, one study team recently found it to have no therapeutic effect.13
PPIs may be effective for improving EE symptoms even in the absence of GERD because of the reduced gastric acid production,7 but they do not usually improve EE’s histologic features.3
Use of esophageal dilation in patients with EE is controversial because of an associated risk for perforation.14 If this intervention is to be performed, the patient should be treated in advance with oral corticosteroids to reduce esophageal inflammation.15,16 In addition, the endoscopist should start with small-sized dilators and carefully proceed to larger sizes.11 Critics of esophageal dilation argue that the procedure is only a temporary solution and does nothing for the underlying condition.4,8
Regarding endoscopic surveillance, an interval of at least four weeks between interventions is recommended.13
Role of the Primary Care Clinician
Undiagnosed EE can cause the patient discomfort, frustration, and anxiety, as seen in the case study. Many patients with undiagnosed EE have been exposed to unnecessary medical therapy and antireflux surgery.3 Without proper diagnosis and treatment, EE may worsen, causing complications associated with chronic inflammation (ie, esophageal fibrosis and strictures).2,6
The long-term prognosis of EE is unknown at this time.8 The disease is usually chronic, with periods of remission and exacerbation. With an understanding of EE and appropriate therapies, the primary care practitioner can team with the gastroenterologist to provide effective disease management through endoscopic surveillance and intervention for acute exacerbations. Guidelines recommend that patients be closely followed with regular office visits to reassess symptoms, compliance with therapy, and adverse effects, with the goal of preventing complications associated with EE.13
Conclusion
To effectively evaluate the patient who presents with dysphagia, the primary care provider should have a working knowledge of EE, as well as an understanding of the key elements in the history and physical examination to help ensure an accurate diagnosis. This will facilitate timely referral to a gastroenterologist for endoscopic evaluation, when indicated.
1. McQuaid KR. Gastrointestinal disorders. In: McPhee S, Papadakis M. CURRENT Medical Diagnosis & Treatment 2009. New York: McGraw-Hill: 2009:487-581.
2. Nurko S, Furuta GT. Eosinophilic esophagitis (2006). GI Motility Online. www.nature.com/gimo/contents/pt1/full/gimo49.html. Accessed July 27, 2009.
3. Parfitt JR, Gregor JC, Suskin NG, et al. Eosinophilic esophagitis in adults: distinguishing features from gastroesophageal reflux disease: a study of 41 patients. Mod Pathol. 2006;19(1):90-96.
4. Swoger JM, Weiler CR, Arora AS. Eosinophilic esophagitis: is it all allergies? Mayo Clin Proc. 2007;82(12):1541-1549.
5. Conus S, Simon HU. General laboratory diagnostics of eosinophilic GI diseases. Best Pract Res Clin Gastroenterol. 2008;22(3):441-453.
6. Remedios M, Campbell C, Jones DM, Kerlin P. Eosinophilic esophagitis in adults: clinical, endoscopic, histologic findings, and response to treatment with fluticasone propionate. Gastrointest Endosc. 2006;63(1):3-12.
7. Rothenberg ME. Eosinophilic gastrointestinal disorders (EGID). J Allergy Clin Immunol. 2004; 113(1):11-28.
8. Lucendo AJ, Carrion G, Navarro M, et al. Eosinophilic esophagitis in adults: an emerging disease. Dig Dis Sci. 2004;49(11-12):1884-1888.
9. Spieker MR. Evaluating dysphagia. Am Fam Physician. 2000;61(12):3639-3648.
10. Boyce HW. Drug-induced esophageal damage: diseases of medical progress. Gastrointest Endosc. 1998;47:547-550.
11. Potter JW, Saeian K, Staff D, et al. Eosinophilic esophagitis in adults: an emerging problem with unique esophageal features. Gastrointest Endosc. 2004;59(3):355-361.
12. Schaefer ET, Fitzgerald JF, Molleston JP, et al. Comparison of oral prednisone and topical fluticasone in the treatment of eosinophilic esophagitis: a randomized trial in children. Clin Gastroenterol Hepatol. 2008;6(2):165-173.
13. Furuta GT, Liacouras CA, Collins MH, et al; First International Gastrointestinal Eosinophil Research Symposium (FIGERS) Subcommittees. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendation for diagnosis and treatment. Gastroenterology. 2007;133(4): 1342-1363.
14. Straumann A, Rossi L, Simon HU, et al. Fragility of the esophageal mucosa: a pathognomonic endoscopic sign of primary eosinophilic esophagitis. Gastrointest Endosc. 2003;57(3):407-412.
15. Hawari R, Pasricha PJ. Images in clinical medicine: eosinophilic esophagitis. N Engl J Med. 2007; 356(20):e20.
16. Leclercq P, Marting A, Gast P. Eosinophilic esophagitis. N Engl J Med. 2007;357(14):1446.
1. McQuaid KR. Gastrointestinal disorders. In: McPhee S, Papadakis M. CURRENT Medical Diagnosis & Treatment 2009. New York: McGraw-Hill: 2009:487-581.
2. Nurko S, Furuta GT. Eosinophilic esophagitis (2006). GI Motility Online. www.nature.com/gimo/contents/pt1/full/gimo49.html. Accessed July 27, 2009.
3. Parfitt JR, Gregor JC, Suskin NG, et al. Eosinophilic esophagitis in adults: distinguishing features from gastroesophageal reflux disease: a study of 41 patients. Mod Pathol. 2006;19(1):90-96.
4. Swoger JM, Weiler CR, Arora AS. Eosinophilic esophagitis: is it all allergies? Mayo Clin Proc. 2007;82(12):1541-1549.
5. Conus S, Simon HU. General laboratory diagnostics of eosinophilic GI diseases. Best Pract Res Clin Gastroenterol. 2008;22(3):441-453.
6. Remedios M, Campbell C, Jones DM, Kerlin P. Eosinophilic esophagitis in adults: clinical, endoscopic, histologic findings, and response to treatment with fluticasone propionate. Gastrointest Endosc. 2006;63(1):3-12.
7. Rothenberg ME. Eosinophilic gastrointestinal disorders (EGID). J Allergy Clin Immunol. 2004; 113(1):11-28.
8. Lucendo AJ, Carrion G, Navarro M, et al. Eosinophilic esophagitis in adults: an emerging disease. Dig Dis Sci. 2004;49(11-12):1884-1888.
9. Spieker MR. Evaluating dysphagia. Am Fam Physician. 2000;61(12):3639-3648.
10. Boyce HW. Drug-induced esophageal damage: diseases of medical progress. Gastrointest Endosc. 1998;47:547-550.
11. Potter JW, Saeian K, Staff D, et al. Eosinophilic esophagitis in adults: an emerging problem with unique esophageal features. Gastrointest Endosc. 2004;59(3):355-361.
12. Schaefer ET, Fitzgerald JF, Molleston JP, et al. Comparison of oral prednisone and topical fluticasone in the treatment of eosinophilic esophagitis: a randomized trial in children. Clin Gastroenterol Hepatol. 2008;6(2):165-173.
13. Furuta GT, Liacouras CA, Collins MH, et al; First International Gastrointestinal Eosinophil Research Symposium (FIGERS) Subcommittees. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendation for diagnosis and treatment. Gastroenterology. 2007;133(4): 1342-1363.
14. Straumann A, Rossi L, Simon HU, et al. Fragility of the esophageal mucosa: a pathognomonic endoscopic sign of primary eosinophilic esophagitis. Gastrointest Endosc. 2003;57(3):407-412.
15. Hawari R, Pasricha PJ. Images in clinical medicine: eosinophilic esophagitis. N Engl J Med. 2007; 356(20):e20.
16. Leclercq P, Marting A, Gast P. Eosinophilic esophagitis. N Engl J Med. 2007;357(14):1446.
Grand Rounds: Man, 60, With Abdominal Pain
A 60-year-old white man with a history of hyperlipidemia, hypertension, and anxiety presented with complaints of abdominal pain, localized to an area left of the umbilicus. He described the pain as constant and rated it 6 on a scale of 1 to 10. He said the pain had been present for longer than three weeks.
The man said he had been seen by another health care provider shortly after the pain began, but he did not think the provider took his complaint seriously. At that visit, antacids were prescribed, blood work was ordered, and the man was told to return if there was no improvement. He felt that because he was being treated for anxiety, the provider believed he was just imagining the pain.
At the current visit, the review of systems revealed additional complaints of shakiness and nausea without vomiting, with other findings unremarkable. The persistent pain did not seem related to eating, and the patient had no history of any surgeries that might help explain his current complaints. He had smoked a pack of cigarettes daily for 40 years and had a history of heavy alcohol use, although he denied having consumed any alcohol during the previous five years.
His prescribed medications included gemfibrozil 600 mg per day, hydrochlorothiazide 25 mg each morning, and diazepam 5 mg twice daily, with an OTC antacid.
The patient’s recent laboratory results were normal; they included a complete blood count, comprehensive metabolic panel, liver enzyme levels, and a serum amylase level. The patient weighed 280 lb and his height was 5’10”; his BMI was 40. His temperature was 97.7°F, with a regular heart rate of 88 beats/min; blood pressure, 140/90 mm Hg; and respiratory rate, 18 breaths/min.
The patient did not appear to be in acute distress. A bruit was heard in the indicated area of pain. No mass was palpated, and the width of his aorta could not be determined because of his obesity. His physical exam was otherwise normal.
Abdominal ultrasonography (US) revealed a 5.5-cm abdominal aortic aneurysm (AAA), and the man was referred for immediate surgery. The aneurysm was repaired in an open abdominal procedure with a polyester prosthetic graft. The surgery was successful.
Discussion
AAA is a permanent bulging area of the aorta that exceeds 3.0 cm in diameter (see Figure 1). It is a potentially life-threatening condition due to the possibility of rupture. Often an aneurysm is asymptomatic until it ruptures, making this a difficult illness to diagnose.1
Each year, an estimated 10,000 deaths result from a ruptured AAA, making this condition the 14th leading cause of death in the United States.2,3 Incidence of AAA appears to have increased over the past two decades. Causes for this may include the aging of the US population, an increase in the number of smokers, and a trend toward diets that are higher in fat.
Prognosis among patients with AAA can be improved with increased awareness of the disease among health care providers, earlier detection of AAAs at risk for rupture, and timely, effective interventions.
Symptomatology
In about one-third of patients with a ruptured AAA, a clinical triad of symptoms is present: abdominal and/or back pain, a pulsatile abdominal mass, and hypotension.4,5 In these cases, according to the American College of Cardiology/American Heart Association (ACC/AHA),4 immediate surgical evaluation is indicated.
Prior to the rupture of an AAA, the patient may feel a pulsing sensation in the abdomen or may experience no symptoms at all. Some patients report vague complaints, such as back, flank, groin, or abdominal pain. Syncope may be the chief complaint as the aneurysm expands, so it is important for primary care providers to be alert to progressive symptoms, including this signal that an aneurysm may exist and may be expanding.6
Pain may also be abrupt and severe in the lower abdomen and back, including tenderness in the area over the aneurysm. Shock can develop rapidly and symptoms such as cyanosis, mottling, altered mental status, tachycardia, and hypotension may be present.1,4
Since symptoms may be vague, the differential diagnosis can be broad (see Table 14,7,8), necessitating a detailed patient history and a careful physical examination. In an elderly patient, low back pain should be evaluated for AAA.9 In addition, acute abdominal pain in a patient older than 50 should be presumed to be a ruptured AAA.8
Risk Factors
A clinician should be familiar with the risk factors for AAA so that diagnosis can be made before a rupture occurs. Male gender and age greater than 65 are important risk factors for AAA, but one of the most important environmental risks is cigarette smoking.9,10 Current smokers are more than seven times more likely than nonsmokers to have an aneurysm.10 Atherosclerosis, which weakens the wall of the aorta, is also believed to contribute to the risk for AAA.11
Other contributing factors include hypertension, chronic obstructive pulmonary disease, hyperlipidemia, and family history. Chronic infection, inflammatory illnesses, and connective tissue disorders (eg, Marfan syndrome) can also increase the risk for aneurysm. Less frequent causes of AAA are trauma and infectious diseases, such as syphilis.1,12
In 85% of patients with femoral aneurysms, AAA has been found to coexist, as it has in 62% of patients with popliteal aneurysms. Patients previously diagnosed with these conditions should be screened for AAA.4,13,14
Diagnosis
An abdominal bruit or a pulsating mass may be found on palpation, but the sensitivity for detection of AAA is related to its size. An aneurysm greater than 5.0 cm has an 82% chance of detection by palpation.15 To assess for the presence of an abdominal aneurysm, the examiner should press the midline between the xiphoid and umbilicus bimanually, firmly but gently.12 There is no evidence to suggest that palpating the abdomen can cause an aneurysm to rupture.
The most useful tests for diagnosis of AAA are US, CT, and MRI.6 US is the simplest and least costly of these diagnostic procedures; it is noninvasive and has a sensitivity of 95% and specificity of nearly 100%. Bedside US can provide a rapid diagnosis in an unstable patient.16
CT is nearly 100% effective in diagnosing AAA and is usually used to help decide on appropriate treatment, as it can determine the size and shape of the aneurysm.17 However, CT should not be used for unstable patients.
MRI is useful in diagnosing AAA, but it is expensive, and inappropriate for unstable patients. Currently, conventional aortography is rarely used for preoperative assessment but may still be used for placement of endovascular devices or in patients with renal complications.1,12
Screening Recommendations
The US Preventive Services Task Force (USPSTF) recommends that all men ages 65 to 74 who have a lifelong history of smoking at least 100 cigarettes should be screened for AAA with abdominal US.3,18 Screening is not recommended for those younger than 65 who have never smoked, but this decision must be individualized to the patient, with other risk factors considered.
The ACC/AHA4 advises that men whose parents or siblings have a history of AAA and who are older than 60 should undergo physical examination and screening US for AAA. In addition, patients with a small AAA should receive US surveillance until the aneurysm reaches 5.5 cm in diameter; survival has not been shown to improve if an AAA is repaired before it reaches this size.1,2,19 In consideration of increased comorbidities and decreased life expectancy, screening is not recommended for men older than 75, but this too should be determined individually.3
Screening for women is not recommended by the USPSTF.3,18 The document states that the prevalence of large AAAs in women is low and that screening may lead to an increased number of unnecessary surgeries with associated morbidity and mortality. Clinical judgment must be used in making this decision, however, as several studies have shown that women have an AAA rupture rate that is three times higher than that in men; they also have an increased in-hospital mortality rate when rupture does occur. Thus, women are less likely to experience AAA but have a worse prognosis when AAA does develop.20-22
Management
The size of an AAA is the most important predictor of rupture. According to the ACC/AHA,4 the associated risk for rupture is about 20% for aneurysms that measure 5.0 cm in diameter, 40% for those measuring at least 6.0 cm, and at least 50% for aneurysms exceeding 7.0 cm.4,23,24 Regarding surveillance of known aneurysms, it is recommended that a patient with an aneurysm smaller than 3.0 cm in diameter requires no further testing. If an AAA measures 3.0 to 4.0 cm, US should be performed yearly; if it is 4.0 to 4.9 cm, US should be performed every six months.4,25
If an identified AAA is larger than 4.5 cm, or if any segment of the aorta is more than 1.5 times the diameter of an adjacent section, referral to a vascular surgeon for further evaluation is indicated. The vascular surgeon should be consulted immediately regarding a symptomatic patient with an AAA, or one with an aneurysm that measures 5.5 cm or larger, as the risk for rupture is high.4,26
Preventing rupture of an AAA is the primary aim in management. Beta-blockers may be used to reduce systolic hypertension in cardiac patients, thus slowing the rate of expansion in those with aortic aneurysms. Patients with a known AAA should undergo frequent monitoring for blood pressure and lipid levels and be advised to stop smoking. Smoking cessation interventions such as behavior modification, nicotine replacement, or bupropion should be offered.27,28
There is evidence that statin use may reduce the size of aneurysms, even in patients without hypercholesterolemia, possibly due to statins’ anti-inflammatory properties.22,29 ACE inhibitors may also be beneficial in reducing AAA growth and in lowering blood pressure. Antiplatelet medications are important in general cardiovascular risk reduction in the patient with AAA. Aspirin is the drug of choice.27,29
Surgical Repair
AAAs are usually repaired by one of two types of surgery: endovascular repair (EVR) or open surgery. Open surgical repair, the more traditional method, involves an incision into the abdomen from the breastbone to below the navel. The weakened area is replaced with a graft made of synthetic material. Open repair of an intact AAA, performed under general anesthesia, takes from three to six hours, and the patient must be hospitalized for five to eight days.30
In EVR, the patient is given epidural anesthesia and an incision is made in the right groin, allowing a synthetic stent graft to be threaded by way of a catheter through the femoral artery to repair the lesion (see Figure 2). EVR generally takes two to five hours, followed by a two- to five-day hospital stay. EVR is usually recommended for patients who are at high risk for complications from open operations because of severe cardiopulmonary disease or other risk factors, such as advanced age, morbid obesity, or a history of multiple abdominal operations.1,2,4,19
Prognosis
Patients with a ruptured AAA have a survival rate of less than 50%, with most deaths occurring before surgical repair has been attempted.3,31 In patients with kidney failure resulting from AAA (whether ruptured or unruptured, an AAA can disrupt renal blood flow), the chance for survival is poor. By contrast, the risk for death during surgical graft repair of an AAA is only about 2% to 8%.1,12
In a systematic review, EVR was associated with a lower 30-day mortality rate compared with open surgical repair (1.6% vs 4.7%, respectively), but this reduction did not persist over two years’ follow-up; neither did EVR improve overall survival or quality of life, compared with open surgery.1 Additionally, EVR requires periodic imaging throughout the patient’s life, which is associated with more reinterventions.1,19
Patient Education
Clinicians should encourage all patients to stop smoking, follow a low-cholesterol diet, control hypertension, and exercise regularly to lower the risk for AAAs. Screening recommendations should be explained to patients at risk, as should the signs and symptoms of an aneurysm. These patients should be instructed to call their health care provider immediately if they suspect a problem.
Conclusion
The incidence of AAA is increasing, and primary care providers must be prepared to act promptly in any case of suspected AAA to ensure a safe outcome. For aneurysms measuring greater than 5.5 cm in diameter, open or endovascular surgical repair should be considered. Patients with smaller aneurysms or contraindications for surgery should receive careful medical management and education to reduce the risks of AAA expansion leading to possible rupture.
1. Wilt TJ, Lederle FA, MacDonald R, et al; Agency for Healthcare Research and Quality. Comparison of Endovascular and Open Surgical Repairs for Abdominal Aortic Aneurysm. Rockville, MD: Agency for Healthcare Research and Quality; 2006. AHRQ publication 06-E107. Evidence Report/Technology Assessment 144. www.ahrq.gov/CLINIC/tp/aaareptp.htm. Accessed June 23, 2009.
2. Birkmeyer JD, Upchurch GR Jr. Evidence-based screening and management of abdominal aortic aneurysm. Ann Intern Med. 2007;146(10):749-750.
3. Fleming C, Whitlock EP, Beil TL, Lederle FA. Screening for abdominal aortic aneurysm: a best-evidence systematic review for the US Preventive Services Task Force. Ann Intern Med. 2005;142(3):203-211.
4. Hirsch AT, Haskal ZJ, Hertzer NR, et al. ACC/AHA guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): executive summary a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease) endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. J Am Coll Cardiol. 2006;47(6):1239-1312.
5. Kiell CS, Ernst CB. Advances in management of abdominal aortic aneurysm. Adv Surg. 1993;26:73–98.
6. O’Connor RE. Aneurysm, abdominal. http://emedicine.medscape.com/article/756735-overview. Accessed June 23, 2009.
7. Lederle FA, Parenti CM, Chute EP. Ruptured abdominal aortic aneurysm: the internist as diagnostician. Am J Med. 1994;96:163-167.
8. Cartwright SL, Knudson MP. Evaluation of acute abdominal pain in adults. Am Fam Physician. 2008;77(7): 971-978.
9. Lyon C, Clark DC. Diagnosis of acute abdominal pain in older patients. Am Fam Physician. 2006;74(9):1537-1544.
10. Wilmink TB, Quick CR, Day NE. The association between cigarette smoking and abdominal aortic aneurysms. J Vasc Surg. 1999;30(6):1099-1105.
11. Palazzuoli P, Gallotta M, Guerrieri G, et al. Prevalence of risk factors, coronary and systemic atherosclerosis in abdominal aortic aneurysm: comparison with high cardiovascular risk population. Vasc Health Risk Manag. 2008;4(4):877-883.
12. Sakalihasan N, Limet R, Defawe OD. Abdominal aortic aneurysm. Lancet. 2005;365(9470):1577-1589.
13. Graham LM, Zelenock GB, Whitehouse WM Jr, et al. Clinical significance of arteriosclerotic femoral artery aneurysms. Arch Surg. 1980;115(4):502–507.
14. Whitehouse WM Jr, Wakefield TW, Graham LM, et al. Limb-threatening potential of arteriosclerotic popliteal artery aneurysms. Surgery. 1983;93(5):694–699.
15. Fink HA, Lederle FA, Roth CS, et al. The accuracy of physical examination to detect abdominal aortic aneurysm. Arch Intern Med. 2000;160:833-836.
16. Bentz S, Jones J. Accuracy of emergency department ultrasound scanning in detecting abdominal aortic aneurysm. Emerg Med J. 2006;23(10):803-804.
17. Kvilekval KH, Best IM, Mason RA, et al. The value of computed tomography in the management of symptomatic abdominal aortic aneurysm. J Vasc Surg. 1990;12(1):28-33.
18. US Preventive Services Task Force. Screening for abdominal aortic aneurysm: recommendation statement. Ann Intern Med. 2005;142(3):198-202.
19. Lederle FA, Kane RL, MacDonald R, Wilt TJ. Systematic review: repair of unruptured abdominal aortic aneurysm. Ann Intern Med. 2007;146(10):735-741.
20. McPhee JT, Hill JS, Elami MH. The impact of gender on presentation, therapy, and mortality of abdominal aortic aneurysm in the United States, 2001-2004. J Vasc Surg. 2007;45(5):891-899.
21. Mofidi R, Goldie VJ, Kelman J, et al. Influence of sex on expansion rate of abdominal aortic aneurysms. Br J Surg. 2007;94(3):310-314.
22. Norman PE, Powell JT. Abdominal aortic aneurysm: the prognosis in women is worse than in men. Circulation. 2007;115(22):2865-2869.
23. Englund R, Hudson P, Hanel K, Stanton A. Expansion rates of small abdominal aortic aneurysms. Aust N Z J Surg. 1998;68(1):21–24.
24. Conway KP, Byrne J, Townsend M, Lane IF. Prognosis of patients turned down for conventional abdominal aortic aneurysm repair in the endovascular and sonographic era: Szilagyi revisited? J Vasc Surg. 2001;33(4):752–757.
25. Cook TA, Galland RB. A prospective study to define the optimum rescreening interval for small abdominal aortic aneurysm. Cardiovasc Surg. 1996;4(4):441–444.
26. Kent KC, Zwolak RM, Jaff MR, et al; Society for Vascular Surgery; American Association of Vascular Surgery; Society for Vascular Medicine and Biology. Screening for abdominal aortic aneurysm: a consensus statement. J Vasc Surg. 2004;39(1):267-269.
27. Golledge J, Powell JT. Medical management of abdominal aortic aneurysm. Eur J Vasc Endovasc Surg. 2007;4(3):267-273.
28. Sule S, Aronow WS. Management of abdominal aortic aneurysms. Compr Ther. 2009;35(1):3-8.
29. Powell JT. Non-operative or medical management of abdominal aortic aneurysm. Scand J Surg. 2008;97(2): 121-124.
30. Huber TS, Wang JG, Derrow AE, et al. Experience in the United States with intact abdominal aortic aneurysm repair. J Vasc Surg. 2001;33(2):304-310.
31. Adam DJ, Mohan IV, Stuart WP, et al. Community and hospital outcome from ruptured abdominal aortic aneurysm within the catchment area of a regional vascular surgical service. J Vasc Surg. 1999;30(5):922-928.
A 60-year-old white man with a history of hyperlipidemia, hypertension, and anxiety presented with complaints of abdominal pain, localized to an area left of the umbilicus. He described the pain as constant and rated it 6 on a scale of 1 to 10. He said the pain had been present for longer than three weeks.
The man said he had been seen by another health care provider shortly after the pain began, but he did not think the provider took his complaint seriously. At that visit, antacids were prescribed, blood work was ordered, and the man was told to return if there was no improvement. He felt that because he was being treated for anxiety, the provider believed he was just imagining the pain.
At the current visit, the review of systems revealed additional complaints of shakiness and nausea without vomiting, with other findings unremarkable. The persistent pain did not seem related to eating, and the patient had no history of any surgeries that might help explain his current complaints. He had smoked a pack of cigarettes daily for 40 years and had a history of heavy alcohol use, although he denied having consumed any alcohol during the previous five years.
His prescribed medications included gemfibrozil 600 mg per day, hydrochlorothiazide 25 mg each morning, and diazepam 5 mg twice daily, with an OTC antacid.
The patient’s recent laboratory results were normal; they included a complete blood count, comprehensive metabolic panel, liver enzyme levels, and a serum amylase level. The patient weighed 280 lb and his height was 5’10”; his BMI was 40. His temperature was 97.7°F, with a regular heart rate of 88 beats/min; blood pressure, 140/90 mm Hg; and respiratory rate, 18 breaths/min.
The patient did not appear to be in acute distress. A bruit was heard in the indicated area of pain. No mass was palpated, and the width of his aorta could not be determined because of his obesity. His physical exam was otherwise normal.
Abdominal ultrasonography (US) revealed a 5.5-cm abdominal aortic aneurysm (AAA), and the man was referred for immediate surgery. The aneurysm was repaired in an open abdominal procedure with a polyester prosthetic graft. The surgery was successful.
Discussion
AAA is a permanent bulging area of the aorta that exceeds 3.0 cm in diameter (see Figure 1). It is a potentially life-threatening condition due to the possibility of rupture. Often an aneurysm is asymptomatic until it ruptures, making this a difficult illness to diagnose.1
Each year, an estimated 10,000 deaths result from a ruptured AAA, making this condition the 14th leading cause of death in the United States.2,3 Incidence of AAA appears to have increased over the past two decades. Causes for this may include the aging of the US population, an increase in the number of smokers, and a trend toward diets that are higher in fat.
Prognosis among patients with AAA can be improved with increased awareness of the disease among health care providers, earlier detection of AAAs at risk for rupture, and timely, effective interventions.
Symptomatology
In about one-third of patients with a ruptured AAA, a clinical triad of symptoms is present: abdominal and/or back pain, a pulsatile abdominal mass, and hypotension.4,5 In these cases, according to the American College of Cardiology/American Heart Association (ACC/AHA),4 immediate surgical evaluation is indicated.
Prior to the rupture of an AAA, the patient may feel a pulsing sensation in the abdomen or may experience no symptoms at all. Some patients report vague complaints, such as back, flank, groin, or abdominal pain. Syncope may be the chief complaint as the aneurysm expands, so it is important for primary care providers to be alert to progressive symptoms, including this signal that an aneurysm may exist and may be expanding.6
Pain may also be abrupt and severe in the lower abdomen and back, including tenderness in the area over the aneurysm. Shock can develop rapidly and symptoms such as cyanosis, mottling, altered mental status, tachycardia, and hypotension may be present.1,4
Since symptoms may be vague, the differential diagnosis can be broad (see Table 14,7,8), necessitating a detailed patient history and a careful physical examination. In an elderly patient, low back pain should be evaluated for AAA.9 In addition, acute abdominal pain in a patient older than 50 should be presumed to be a ruptured AAA.8
Risk Factors
A clinician should be familiar with the risk factors for AAA so that diagnosis can be made before a rupture occurs. Male gender and age greater than 65 are important risk factors for AAA, but one of the most important environmental risks is cigarette smoking.9,10 Current smokers are more than seven times more likely than nonsmokers to have an aneurysm.10 Atherosclerosis, which weakens the wall of the aorta, is also believed to contribute to the risk for AAA.11
Other contributing factors include hypertension, chronic obstructive pulmonary disease, hyperlipidemia, and family history. Chronic infection, inflammatory illnesses, and connective tissue disorders (eg, Marfan syndrome) can also increase the risk for aneurysm. Less frequent causes of AAA are trauma and infectious diseases, such as syphilis.1,12
In 85% of patients with femoral aneurysms, AAA has been found to coexist, as it has in 62% of patients with popliteal aneurysms. Patients previously diagnosed with these conditions should be screened for AAA.4,13,14
Diagnosis
An abdominal bruit or a pulsating mass may be found on palpation, but the sensitivity for detection of AAA is related to its size. An aneurysm greater than 5.0 cm has an 82% chance of detection by palpation.15 To assess for the presence of an abdominal aneurysm, the examiner should press the midline between the xiphoid and umbilicus bimanually, firmly but gently.12 There is no evidence to suggest that palpating the abdomen can cause an aneurysm to rupture.
The most useful tests for diagnosis of AAA are US, CT, and MRI.6 US is the simplest and least costly of these diagnostic procedures; it is noninvasive and has a sensitivity of 95% and specificity of nearly 100%. Bedside US can provide a rapid diagnosis in an unstable patient.16
CT is nearly 100% effective in diagnosing AAA and is usually used to help decide on appropriate treatment, as it can determine the size and shape of the aneurysm.17 However, CT should not be used for unstable patients.
MRI is useful in diagnosing AAA, but it is expensive, and inappropriate for unstable patients. Currently, conventional aortography is rarely used for preoperative assessment but may still be used for placement of endovascular devices or in patients with renal complications.1,12
Screening Recommendations
The US Preventive Services Task Force (USPSTF) recommends that all men ages 65 to 74 who have a lifelong history of smoking at least 100 cigarettes should be screened for AAA with abdominal US.3,18 Screening is not recommended for those younger than 65 who have never smoked, but this decision must be individualized to the patient, with other risk factors considered.
The ACC/AHA4 advises that men whose parents or siblings have a history of AAA and who are older than 60 should undergo physical examination and screening US for AAA. In addition, patients with a small AAA should receive US surveillance until the aneurysm reaches 5.5 cm in diameter; survival has not been shown to improve if an AAA is repaired before it reaches this size.1,2,19 In consideration of increased comorbidities and decreased life expectancy, screening is not recommended for men older than 75, but this too should be determined individually.3
Screening for women is not recommended by the USPSTF.3,18 The document states that the prevalence of large AAAs in women is low and that screening may lead to an increased number of unnecessary surgeries with associated morbidity and mortality. Clinical judgment must be used in making this decision, however, as several studies have shown that women have an AAA rupture rate that is three times higher than that in men; they also have an increased in-hospital mortality rate when rupture does occur. Thus, women are less likely to experience AAA but have a worse prognosis when AAA does develop.20-22
Management
The size of an AAA is the most important predictor of rupture. According to the ACC/AHA,4 the associated risk for rupture is about 20% for aneurysms that measure 5.0 cm in diameter, 40% for those measuring at least 6.0 cm, and at least 50% for aneurysms exceeding 7.0 cm.4,23,24 Regarding surveillance of known aneurysms, it is recommended that a patient with an aneurysm smaller than 3.0 cm in diameter requires no further testing. If an AAA measures 3.0 to 4.0 cm, US should be performed yearly; if it is 4.0 to 4.9 cm, US should be performed every six months.4,25
If an identified AAA is larger than 4.5 cm, or if any segment of the aorta is more than 1.5 times the diameter of an adjacent section, referral to a vascular surgeon for further evaluation is indicated. The vascular surgeon should be consulted immediately regarding a symptomatic patient with an AAA, or one with an aneurysm that measures 5.5 cm or larger, as the risk for rupture is high.4,26
Preventing rupture of an AAA is the primary aim in management. Beta-blockers may be used to reduce systolic hypertension in cardiac patients, thus slowing the rate of expansion in those with aortic aneurysms. Patients with a known AAA should undergo frequent monitoring for blood pressure and lipid levels and be advised to stop smoking. Smoking cessation interventions such as behavior modification, nicotine replacement, or bupropion should be offered.27,28
There is evidence that statin use may reduce the size of aneurysms, even in patients without hypercholesterolemia, possibly due to statins’ anti-inflammatory properties.22,29 ACE inhibitors may also be beneficial in reducing AAA growth and in lowering blood pressure. Antiplatelet medications are important in general cardiovascular risk reduction in the patient with AAA. Aspirin is the drug of choice.27,29
Surgical Repair
AAAs are usually repaired by one of two types of surgery: endovascular repair (EVR) or open surgery. Open surgical repair, the more traditional method, involves an incision into the abdomen from the breastbone to below the navel. The weakened area is replaced with a graft made of synthetic material. Open repair of an intact AAA, performed under general anesthesia, takes from three to six hours, and the patient must be hospitalized for five to eight days.30
In EVR, the patient is given epidural anesthesia and an incision is made in the right groin, allowing a synthetic stent graft to be threaded by way of a catheter through the femoral artery to repair the lesion (see Figure 2). EVR generally takes two to five hours, followed by a two- to five-day hospital stay. EVR is usually recommended for patients who are at high risk for complications from open operations because of severe cardiopulmonary disease or other risk factors, such as advanced age, morbid obesity, or a history of multiple abdominal operations.1,2,4,19
Prognosis
Patients with a ruptured AAA have a survival rate of less than 50%, with most deaths occurring before surgical repair has been attempted.3,31 In patients with kidney failure resulting from AAA (whether ruptured or unruptured, an AAA can disrupt renal blood flow), the chance for survival is poor. By contrast, the risk for death during surgical graft repair of an AAA is only about 2% to 8%.1,12
In a systematic review, EVR was associated with a lower 30-day mortality rate compared with open surgical repair (1.6% vs 4.7%, respectively), but this reduction did not persist over two years’ follow-up; neither did EVR improve overall survival or quality of life, compared with open surgery.1 Additionally, EVR requires periodic imaging throughout the patient’s life, which is associated with more reinterventions.1,19
Patient Education
Clinicians should encourage all patients to stop smoking, follow a low-cholesterol diet, control hypertension, and exercise regularly to lower the risk for AAAs. Screening recommendations should be explained to patients at risk, as should the signs and symptoms of an aneurysm. These patients should be instructed to call their health care provider immediately if they suspect a problem.
Conclusion
The incidence of AAA is increasing, and primary care providers must be prepared to act promptly in any case of suspected AAA to ensure a safe outcome. For aneurysms measuring greater than 5.5 cm in diameter, open or endovascular surgical repair should be considered. Patients with smaller aneurysms or contraindications for surgery should receive careful medical management and education to reduce the risks of AAA expansion leading to possible rupture.
A 60-year-old white man with a history of hyperlipidemia, hypertension, and anxiety presented with complaints of abdominal pain, localized to an area left of the umbilicus. He described the pain as constant and rated it 6 on a scale of 1 to 10. He said the pain had been present for longer than three weeks.
The man said he had been seen by another health care provider shortly after the pain began, but he did not think the provider took his complaint seriously. At that visit, antacids were prescribed, blood work was ordered, and the man was told to return if there was no improvement. He felt that because he was being treated for anxiety, the provider believed he was just imagining the pain.
At the current visit, the review of systems revealed additional complaints of shakiness and nausea without vomiting, with other findings unremarkable. The persistent pain did not seem related to eating, and the patient had no history of any surgeries that might help explain his current complaints. He had smoked a pack of cigarettes daily for 40 years and had a history of heavy alcohol use, although he denied having consumed any alcohol during the previous five years.
His prescribed medications included gemfibrozil 600 mg per day, hydrochlorothiazide 25 mg each morning, and diazepam 5 mg twice daily, with an OTC antacid.
The patient’s recent laboratory results were normal; they included a complete blood count, comprehensive metabolic panel, liver enzyme levels, and a serum amylase level. The patient weighed 280 lb and his height was 5’10”; his BMI was 40. His temperature was 97.7°F, with a regular heart rate of 88 beats/min; blood pressure, 140/90 mm Hg; and respiratory rate, 18 breaths/min.
The patient did not appear to be in acute distress. A bruit was heard in the indicated area of pain. No mass was palpated, and the width of his aorta could not be determined because of his obesity. His physical exam was otherwise normal.
Abdominal ultrasonography (US) revealed a 5.5-cm abdominal aortic aneurysm (AAA), and the man was referred for immediate surgery. The aneurysm was repaired in an open abdominal procedure with a polyester prosthetic graft. The surgery was successful.
Discussion
AAA is a permanent bulging area of the aorta that exceeds 3.0 cm in diameter (see Figure 1). It is a potentially life-threatening condition due to the possibility of rupture. Often an aneurysm is asymptomatic until it ruptures, making this a difficult illness to diagnose.1
Each year, an estimated 10,000 deaths result from a ruptured AAA, making this condition the 14th leading cause of death in the United States.2,3 Incidence of AAA appears to have increased over the past two decades. Causes for this may include the aging of the US population, an increase in the number of smokers, and a trend toward diets that are higher in fat.
Prognosis among patients with AAA can be improved with increased awareness of the disease among health care providers, earlier detection of AAAs at risk for rupture, and timely, effective interventions.
Symptomatology
In about one-third of patients with a ruptured AAA, a clinical triad of symptoms is present: abdominal and/or back pain, a pulsatile abdominal mass, and hypotension.4,5 In these cases, according to the American College of Cardiology/American Heart Association (ACC/AHA),4 immediate surgical evaluation is indicated.
Prior to the rupture of an AAA, the patient may feel a pulsing sensation in the abdomen or may experience no symptoms at all. Some patients report vague complaints, such as back, flank, groin, or abdominal pain. Syncope may be the chief complaint as the aneurysm expands, so it is important for primary care providers to be alert to progressive symptoms, including this signal that an aneurysm may exist and may be expanding.6
Pain may also be abrupt and severe in the lower abdomen and back, including tenderness in the area over the aneurysm. Shock can develop rapidly and symptoms such as cyanosis, mottling, altered mental status, tachycardia, and hypotension may be present.1,4
Since symptoms may be vague, the differential diagnosis can be broad (see Table 14,7,8), necessitating a detailed patient history and a careful physical examination. In an elderly patient, low back pain should be evaluated for AAA.9 In addition, acute abdominal pain in a patient older than 50 should be presumed to be a ruptured AAA.8
Risk Factors
A clinician should be familiar with the risk factors for AAA so that diagnosis can be made before a rupture occurs. Male gender and age greater than 65 are important risk factors for AAA, but one of the most important environmental risks is cigarette smoking.9,10 Current smokers are more than seven times more likely than nonsmokers to have an aneurysm.10 Atherosclerosis, which weakens the wall of the aorta, is also believed to contribute to the risk for AAA.11
Other contributing factors include hypertension, chronic obstructive pulmonary disease, hyperlipidemia, and family history. Chronic infection, inflammatory illnesses, and connective tissue disorders (eg, Marfan syndrome) can also increase the risk for aneurysm. Less frequent causes of AAA are trauma and infectious diseases, such as syphilis.1,12
In 85% of patients with femoral aneurysms, AAA has been found to coexist, as it has in 62% of patients with popliteal aneurysms. Patients previously diagnosed with these conditions should be screened for AAA.4,13,14
Diagnosis
An abdominal bruit or a pulsating mass may be found on palpation, but the sensitivity for detection of AAA is related to its size. An aneurysm greater than 5.0 cm has an 82% chance of detection by palpation.15 To assess for the presence of an abdominal aneurysm, the examiner should press the midline between the xiphoid and umbilicus bimanually, firmly but gently.12 There is no evidence to suggest that palpating the abdomen can cause an aneurysm to rupture.
The most useful tests for diagnosis of AAA are US, CT, and MRI.6 US is the simplest and least costly of these diagnostic procedures; it is noninvasive and has a sensitivity of 95% and specificity of nearly 100%. Bedside US can provide a rapid diagnosis in an unstable patient.16
CT is nearly 100% effective in diagnosing AAA and is usually used to help decide on appropriate treatment, as it can determine the size and shape of the aneurysm.17 However, CT should not be used for unstable patients.
MRI is useful in diagnosing AAA, but it is expensive, and inappropriate for unstable patients. Currently, conventional aortography is rarely used for preoperative assessment but may still be used for placement of endovascular devices or in patients with renal complications.1,12
Screening Recommendations
The US Preventive Services Task Force (USPSTF) recommends that all men ages 65 to 74 who have a lifelong history of smoking at least 100 cigarettes should be screened for AAA with abdominal US.3,18 Screening is not recommended for those younger than 65 who have never smoked, but this decision must be individualized to the patient, with other risk factors considered.
The ACC/AHA4 advises that men whose parents or siblings have a history of AAA and who are older than 60 should undergo physical examination and screening US for AAA. In addition, patients with a small AAA should receive US surveillance until the aneurysm reaches 5.5 cm in diameter; survival has not been shown to improve if an AAA is repaired before it reaches this size.1,2,19 In consideration of increased comorbidities and decreased life expectancy, screening is not recommended for men older than 75, but this too should be determined individually.3
Screening for women is not recommended by the USPSTF.3,18 The document states that the prevalence of large AAAs in women is low and that screening may lead to an increased number of unnecessary surgeries with associated morbidity and mortality. Clinical judgment must be used in making this decision, however, as several studies have shown that women have an AAA rupture rate that is three times higher than that in men; they also have an increased in-hospital mortality rate when rupture does occur. Thus, women are less likely to experience AAA but have a worse prognosis when AAA does develop.20-22
Management
The size of an AAA is the most important predictor of rupture. According to the ACC/AHA,4 the associated risk for rupture is about 20% for aneurysms that measure 5.0 cm in diameter, 40% for those measuring at least 6.0 cm, and at least 50% for aneurysms exceeding 7.0 cm.4,23,24 Regarding surveillance of known aneurysms, it is recommended that a patient with an aneurysm smaller than 3.0 cm in diameter requires no further testing. If an AAA measures 3.0 to 4.0 cm, US should be performed yearly; if it is 4.0 to 4.9 cm, US should be performed every six months.4,25
If an identified AAA is larger than 4.5 cm, or if any segment of the aorta is more than 1.5 times the diameter of an adjacent section, referral to a vascular surgeon for further evaluation is indicated. The vascular surgeon should be consulted immediately regarding a symptomatic patient with an AAA, or one with an aneurysm that measures 5.5 cm or larger, as the risk for rupture is high.4,26
Preventing rupture of an AAA is the primary aim in management. Beta-blockers may be used to reduce systolic hypertension in cardiac patients, thus slowing the rate of expansion in those with aortic aneurysms. Patients with a known AAA should undergo frequent monitoring for blood pressure and lipid levels and be advised to stop smoking. Smoking cessation interventions such as behavior modification, nicotine replacement, or bupropion should be offered.27,28
There is evidence that statin use may reduce the size of aneurysms, even in patients without hypercholesterolemia, possibly due to statins’ anti-inflammatory properties.22,29 ACE inhibitors may also be beneficial in reducing AAA growth and in lowering blood pressure. Antiplatelet medications are important in general cardiovascular risk reduction in the patient with AAA. Aspirin is the drug of choice.27,29
Surgical Repair
AAAs are usually repaired by one of two types of surgery: endovascular repair (EVR) or open surgery. Open surgical repair, the more traditional method, involves an incision into the abdomen from the breastbone to below the navel. The weakened area is replaced with a graft made of synthetic material. Open repair of an intact AAA, performed under general anesthesia, takes from three to six hours, and the patient must be hospitalized for five to eight days.30
In EVR, the patient is given epidural anesthesia and an incision is made in the right groin, allowing a synthetic stent graft to be threaded by way of a catheter through the femoral artery to repair the lesion (see Figure 2). EVR generally takes two to five hours, followed by a two- to five-day hospital stay. EVR is usually recommended for patients who are at high risk for complications from open operations because of severe cardiopulmonary disease or other risk factors, such as advanced age, morbid obesity, or a history of multiple abdominal operations.1,2,4,19
Prognosis
Patients with a ruptured AAA have a survival rate of less than 50%, with most deaths occurring before surgical repair has been attempted.3,31 In patients with kidney failure resulting from AAA (whether ruptured or unruptured, an AAA can disrupt renal blood flow), the chance for survival is poor. By contrast, the risk for death during surgical graft repair of an AAA is only about 2% to 8%.1,12
In a systematic review, EVR was associated with a lower 30-day mortality rate compared with open surgical repair (1.6% vs 4.7%, respectively), but this reduction did not persist over two years’ follow-up; neither did EVR improve overall survival or quality of life, compared with open surgery.1 Additionally, EVR requires periodic imaging throughout the patient’s life, which is associated with more reinterventions.1,19
Patient Education
Clinicians should encourage all patients to stop smoking, follow a low-cholesterol diet, control hypertension, and exercise regularly to lower the risk for AAAs. Screening recommendations should be explained to patients at risk, as should the signs and symptoms of an aneurysm. These patients should be instructed to call their health care provider immediately if they suspect a problem.
Conclusion
The incidence of AAA is increasing, and primary care providers must be prepared to act promptly in any case of suspected AAA to ensure a safe outcome. For aneurysms measuring greater than 5.5 cm in diameter, open or endovascular surgical repair should be considered. Patients with smaller aneurysms or contraindications for surgery should receive careful medical management and education to reduce the risks of AAA expansion leading to possible rupture.
1. Wilt TJ, Lederle FA, MacDonald R, et al; Agency for Healthcare Research and Quality. Comparison of Endovascular and Open Surgical Repairs for Abdominal Aortic Aneurysm. Rockville, MD: Agency for Healthcare Research and Quality; 2006. AHRQ publication 06-E107. Evidence Report/Technology Assessment 144. www.ahrq.gov/CLINIC/tp/aaareptp.htm. Accessed June 23, 2009.
2. Birkmeyer JD, Upchurch GR Jr. Evidence-based screening and management of abdominal aortic aneurysm. Ann Intern Med. 2007;146(10):749-750.
3. Fleming C, Whitlock EP, Beil TL, Lederle FA. Screening for abdominal aortic aneurysm: a best-evidence systematic review for the US Preventive Services Task Force. Ann Intern Med. 2005;142(3):203-211.
4. Hirsch AT, Haskal ZJ, Hertzer NR, et al. ACC/AHA guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): executive summary a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease) endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. J Am Coll Cardiol. 2006;47(6):1239-1312.
5. Kiell CS, Ernst CB. Advances in management of abdominal aortic aneurysm. Adv Surg. 1993;26:73–98.
6. O’Connor RE. Aneurysm, abdominal. http://emedicine.medscape.com/article/756735-overview. Accessed June 23, 2009.
7. Lederle FA, Parenti CM, Chute EP. Ruptured abdominal aortic aneurysm: the internist as diagnostician. Am J Med. 1994;96:163-167.
8. Cartwright SL, Knudson MP. Evaluation of acute abdominal pain in adults. Am Fam Physician. 2008;77(7): 971-978.
9. Lyon C, Clark DC. Diagnosis of acute abdominal pain in older patients. Am Fam Physician. 2006;74(9):1537-1544.
10. Wilmink TB, Quick CR, Day NE. The association between cigarette smoking and abdominal aortic aneurysms. J Vasc Surg. 1999;30(6):1099-1105.
11. Palazzuoli P, Gallotta M, Guerrieri G, et al. Prevalence of risk factors, coronary and systemic atherosclerosis in abdominal aortic aneurysm: comparison with high cardiovascular risk population. Vasc Health Risk Manag. 2008;4(4):877-883.
12. Sakalihasan N, Limet R, Defawe OD. Abdominal aortic aneurysm. Lancet. 2005;365(9470):1577-1589.
13. Graham LM, Zelenock GB, Whitehouse WM Jr, et al. Clinical significance of arteriosclerotic femoral artery aneurysms. Arch Surg. 1980;115(4):502–507.
14. Whitehouse WM Jr, Wakefield TW, Graham LM, et al. Limb-threatening potential of arteriosclerotic popliteal artery aneurysms. Surgery. 1983;93(5):694–699.
15. Fink HA, Lederle FA, Roth CS, et al. The accuracy of physical examination to detect abdominal aortic aneurysm. Arch Intern Med. 2000;160:833-836.
16. Bentz S, Jones J. Accuracy of emergency department ultrasound scanning in detecting abdominal aortic aneurysm. Emerg Med J. 2006;23(10):803-804.
17. Kvilekval KH, Best IM, Mason RA, et al. The value of computed tomography in the management of symptomatic abdominal aortic aneurysm. J Vasc Surg. 1990;12(1):28-33.
18. US Preventive Services Task Force. Screening for abdominal aortic aneurysm: recommendation statement. Ann Intern Med. 2005;142(3):198-202.
19. Lederle FA, Kane RL, MacDonald R, Wilt TJ. Systematic review: repair of unruptured abdominal aortic aneurysm. Ann Intern Med. 2007;146(10):735-741.
20. McPhee JT, Hill JS, Elami MH. The impact of gender on presentation, therapy, and mortality of abdominal aortic aneurysm in the United States, 2001-2004. J Vasc Surg. 2007;45(5):891-899.
21. Mofidi R, Goldie VJ, Kelman J, et al. Influence of sex on expansion rate of abdominal aortic aneurysms. Br J Surg. 2007;94(3):310-314.
22. Norman PE, Powell JT. Abdominal aortic aneurysm: the prognosis in women is worse than in men. Circulation. 2007;115(22):2865-2869.
23. Englund R, Hudson P, Hanel K, Stanton A. Expansion rates of small abdominal aortic aneurysms. Aust N Z J Surg. 1998;68(1):21–24.
24. Conway KP, Byrne J, Townsend M, Lane IF. Prognosis of patients turned down for conventional abdominal aortic aneurysm repair in the endovascular and sonographic era: Szilagyi revisited? J Vasc Surg. 2001;33(4):752–757.
25. Cook TA, Galland RB. A prospective study to define the optimum rescreening interval for small abdominal aortic aneurysm. Cardiovasc Surg. 1996;4(4):441–444.
26. Kent KC, Zwolak RM, Jaff MR, et al; Society for Vascular Surgery; American Association of Vascular Surgery; Society for Vascular Medicine and Biology. Screening for abdominal aortic aneurysm: a consensus statement. J Vasc Surg. 2004;39(1):267-269.
27. Golledge J, Powell JT. Medical management of abdominal aortic aneurysm. Eur J Vasc Endovasc Surg. 2007;4(3):267-273.
28. Sule S, Aronow WS. Management of abdominal aortic aneurysms. Compr Ther. 2009;35(1):3-8.
29. Powell JT. Non-operative or medical management of abdominal aortic aneurysm. Scand J Surg. 2008;97(2): 121-124.
30. Huber TS, Wang JG, Derrow AE, et al. Experience in the United States with intact abdominal aortic aneurysm repair. J Vasc Surg. 2001;33(2):304-310.
31. Adam DJ, Mohan IV, Stuart WP, et al. Community and hospital outcome from ruptured abdominal aortic aneurysm within the catchment area of a regional vascular surgical service. J Vasc Surg. 1999;30(5):922-928.
1. Wilt TJ, Lederle FA, MacDonald R, et al; Agency for Healthcare Research and Quality. Comparison of Endovascular and Open Surgical Repairs for Abdominal Aortic Aneurysm. Rockville, MD: Agency for Healthcare Research and Quality; 2006. AHRQ publication 06-E107. Evidence Report/Technology Assessment 144. www.ahrq.gov/CLINIC/tp/aaareptp.htm. Accessed June 23, 2009.
2. Birkmeyer JD, Upchurch GR Jr. Evidence-based screening and management of abdominal aortic aneurysm. Ann Intern Med. 2007;146(10):749-750.
3. Fleming C, Whitlock EP, Beil TL, Lederle FA. Screening for abdominal aortic aneurysm: a best-evidence systematic review for the US Preventive Services Task Force. Ann Intern Med. 2005;142(3):203-211.
4. Hirsch AT, Haskal ZJ, Hertzer NR, et al. ACC/AHA guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): executive summary a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease) endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. J Am Coll Cardiol. 2006;47(6):1239-1312.
5. Kiell CS, Ernst CB. Advances in management of abdominal aortic aneurysm. Adv Surg. 1993;26:73–98.
6. O’Connor RE. Aneurysm, abdominal. http://emedicine.medscape.com/article/756735-overview. Accessed June 23, 2009.
7. Lederle FA, Parenti CM, Chute EP. Ruptured abdominal aortic aneurysm: the internist as diagnostician. Am J Med. 1994;96:163-167.
8. Cartwright SL, Knudson MP. Evaluation of acute abdominal pain in adults. Am Fam Physician. 2008;77(7): 971-978.
9. Lyon C, Clark DC. Diagnosis of acute abdominal pain in older patients. Am Fam Physician. 2006;74(9):1537-1544.
10. Wilmink TB, Quick CR, Day NE. The association between cigarette smoking and abdominal aortic aneurysms. J Vasc Surg. 1999;30(6):1099-1105.
11. Palazzuoli P, Gallotta M, Guerrieri G, et al. Prevalence of risk factors, coronary and systemic atherosclerosis in abdominal aortic aneurysm: comparison with high cardiovascular risk population. Vasc Health Risk Manag. 2008;4(4):877-883.
12. Sakalihasan N, Limet R, Defawe OD. Abdominal aortic aneurysm. Lancet. 2005;365(9470):1577-1589.
13. Graham LM, Zelenock GB, Whitehouse WM Jr, et al. Clinical significance of arteriosclerotic femoral artery aneurysms. Arch Surg. 1980;115(4):502–507.
14. Whitehouse WM Jr, Wakefield TW, Graham LM, et al. Limb-threatening potential of arteriosclerotic popliteal artery aneurysms. Surgery. 1983;93(5):694–699.
15. Fink HA, Lederle FA, Roth CS, et al. The accuracy of physical examination to detect abdominal aortic aneurysm. Arch Intern Med. 2000;160:833-836.
16. Bentz S, Jones J. Accuracy of emergency department ultrasound scanning in detecting abdominal aortic aneurysm. Emerg Med J. 2006;23(10):803-804.
17. Kvilekval KH, Best IM, Mason RA, et al. The value of computed tomography in the management of symptomatic abdominal aortic aneurysm. J Vasc Surg. 1990;12(1):28-33.
18. US Preventive Services Task Force. Screening for abdominal aortic aneurysm: recommendation statement. Ann Intern Med. 2005;142(3):198-202.
19. Lederle FA, Kane RL, MacDonald R, Wilt TJ. Systematic review: repair of unruptured abdominal aortic aneurysm. Ann Intern Med. 2007;146(10):735-741.
20. McPhee JT, Hill JS, Elami MH. The impact of gender on presentation, therapy, and mortality of abdominal aortic aneurysm in the United States, 2001-2004. J Vasc Surg. 2007;45(5):891-899.
21. Mofidi R, Goldie VJ, Kelman J, et al. Influence of sex on expansion rate of abdominal aortic aneurysms. Br J Surg. 2007;94(3):310-314.
22. Norman PE, Powell JT. Abdominal aortic aneurysm: the prognosis in women is worse than in men. Circulation. 2007;115(22):2865-2869.
23. Englund R, Hudson P, Hanel K, Stanton A. Expansion rates of small abdominal aortic aneurysms. Aust N Z J Surg. 1998;68(1):21–24.
24. Conway KP, Byrne J, Townsend M, Lane IF. Prognosis of patients turned down for conventional abdominal aortic aneurysm repair in the endovascular and sonographic era: Szilagyi revisited? J Vasc Surg. 2001;33(4):752–757.
25. Cook TA, Galland RB. A prospective study to define the optimum rescreening interval for small abdominal aortic aneurysm. Cardiovasc Surg. 1996;4(4):441–444.
26. Kent KC, Zwolak RM, Jaff MR, et al; Society for Vascular Surgery; American Association of Vascular Surgery; Society for Vascular Medicine and Biology. Screening for abdominal aortic aneurysm: a consensus statement. J Vasc Surg. 2004;39(1):267-269.
27. Golledge J, Powell JT. Medical management of abdominal aortic aneurysm. Eur J Vasc Endovasc Surg. 2007;4(3):267-273.
28. Sule S, Aronow WS. Management of abdominal aortic aneurysms. Compr Ther. 2009;35(1):3-8.
29. Powell JT. Non-operative or medical management of abdominal aortic aneurysm. Scand J Surg. 2008;97(2): 121-124.
30. Huber TS, Wang JG, Derrow AE, et al. Experience in the United States with intact abdominal aortic aneurysm repair. J Vasc Surg. 2001;33(2):304-310.
31. Adam DJ, Mohan IV, Stuart WP, et al. Community and hospital outcome from ruptured abdominal aortic aneurysm within the catchment area of a regional vascular surgical service. J Vasc Surg. 1999;30(5):922-928.
Grand Rounds: Boy, 10, With Knee Pain
A 10-year-old boy first complained of right knee pain two months prior to presentation. There was no traumatic event to explain the pain and no prior viral or bacterial illness. Radiographs taken earlier at another facility were initially pronounced normal. One month later, repeat x-rays showed a possible hairline fracture, and MRI was ordered. MRI documented a destructive lesion in the right distal femur with a soft-tissue mass that was worrisome for primary bone malignancy.
The boy was placed on weight-bearing restrictions and was given a wheelchair. Unfortunately, he fell from the wheelchair and sustained a pathologic fracture through the lesion (see Figure 1). He was transported to the hospital and admitted. A biopsy was performed with a closed reduction, as the fracture was maligned. The patient was placed in a long leg cast with a pelvic band.
His history was previously unremarkable. He was taking no medications and had experienced no recent illnesses. His surgical/medical history was positive for a tonsillectomy at an early age and a fracture of the right proximal femur at age 2. On examination, he was noted to be talkative with his family but guarded during conversations with staff.
His physical exam was positive for pain at the right distal femur and knee with palpation; otherwise, all other systems were unremarkable. The patient was in too much pain to range the knee and had been placed in a long posterior leg splint (prior to surgery and application of the cast). Distally, his right lower extremity motor and sensory function were intact.
The patient’s vital signs were within normal limits, and results from his blood chemistries and alkaline phosphatase and C-reactive protein levels were unremarkable. Findings on the complete blood cell count were slightly abnormal: Hemoglobin was 11 g and the hematocrit, 33% (both within normal limits); however, in the differential there was an elevation in segmented neutrophils (72%, compared with a reference range of 31% to 61%), with Döhle bodies present—possibly signifying acute and/or chronic systemic infection or malignancy. The lymphocyte count represented 11% of the total white blood cell count (range, 28% to 48%), and platelets were 82 x 103/mL (normal range, 150 to 350 x 103/mL). The patient’s erythrocyte sedimentation rate was 44 mm/h (normal range, 0 to 20).
Result from pathology were positive for osteosarcoma, telangiectatic type. The patient underwent a nuclear medicine bone scan that showed no metastases, and chest CT was negative for pulmonary lesions as well. After a psychology consult, the boy was gently told about his condition.
Treatment then proceeded, including surgical placement of a double-lumen chest catheter for delivery of neoadjuvant and adjuvant chemotherapy. Doxorubicin, cisplatin, and methotrexate were used because the boy was enrolled in an international cooperative trial through the Children’s Oncology Group for treatment of localized osteosarcoma.
Discussion
Osteosarcoma (OS) is the most common primary bone malignancy.1,2 Approximately 5% of all pediatric patients with tumors present with this diagnosis, and about 400 new cases are diagnosed in the United States each year.1 Most osteosarcomas develop in the bones of the lower extremities and in the humerus, affecting males more often than females.1-3 This kind of malignancy is frequently seen during the adolescent growth spurt, but it can affect patients of any age.1,2 Patients usually present with pain or functional limitation in gait or daily activities or both.1-3
The telangiectatic subtype of OS is a rare, aggressive variant that represents 2% to 12% of all cases of OS.4-6 Telangiectatic OS (TOS) is characterized by multiple aneurysmally dilated, blood-filled cavities with high-grade sarcomatous cells seen in the peripheral rim and septae.3,7,8 This process can cause the lesion to resemble an aneurysmal bone cyst, explaining why some cases of TOS are misdiagnosed—with delayed time to treatment and increased morbidity and mortality.3,5 Generally, TOS patients are more likely than other OS patients to have tumors of femoral location, larger lesions, and normal alkaline phosphatase values. Many have pathologic fractures on presentation.7
The medical literature chronicles a long debate regarding the difference in mortality between patients with OS and those with TOS. It was once believed that patients with TOS were at higher risk for recurrence (especially those with a pathologic fracture) and mortality. However, in recent studies examining newer neoadjuvant and adjuvant chemotherapies, mortality rates for the two conditions are similar and certainly lower than they were many years ago.7,8 In one study, a better histologic response was reported to neoadjuvant chemotherapy in patients with TOS than with OS.7
Diagnosis
The first diagnostic tool used for patients with suspected OS or TOS is a plain radiographic film. A TOS lesion is lytic, with no areas of sclerosis, and almost always involves the long bones. It is poorly defined, destroying the cortex with formation of periosteal bone and invading the soft tissue. An initial pattern of parallel striations is highly suggestive of TOS.5
MRI and CT often reveal thick nodular tissue in a largely hemorrhagic and/or necrotic osseous lesion, with an associated soft-tissue mass that allows distinction from an aneurysmal bone cyst.3 Next, patients generally undergo a nuclear medicine bone scan and CT of the chest to observe for signs of metastases. Chest CT is commonly repeated on a regular basis during and after treatment.9
Pathologic evaluation, the final step to diagnosis, is very important, especially in the effort to differentiate TOS from an aneurysmal bone cyst. The typical gross findings for a TOS tumor include a dominant cystic cavity–like architecture, with a pushing peripheral margin that frequently expands and erodes the adjacent cortex and extends into the surrounding tissue. There is usually no area of intramural bone tissue.
Microscopically, the cystic areas contain clots and fragments of tumor that are often lined with a layer of neoplasm. The blood-filled telangiectatic spaces form in these areas. The spaces are irregularly shaped and typically traversed by septae composed in part of neoplastic cells. Osteoid formation through these cells can appear as a fine, ice-like material between tumor cells.4,7
Treatment
The main goals of treatment are to limit the anatomical extent of the disease, decrease the possibility of recurrence, and restore the highest possible level of function.2 Initial treatment of any OS or TOS consists of aggressive, immediate chemotherapy prior to and after any surgical intervention.1 (Chemotherapy will not be discussed in further detail here.) Surgical treatments for patients younger than 14 include amputation (above the lesion with wide margins), an expanding prosthesis, or rotationplasty. The location and extent of the tumor, the patient’s age, and his or her desired lifestyle will all have an impact on the choice of surgery.10
Historic data demonstrate that patients who undergo amputation alone almost always develop metastatic disease.1 Other data show that only 10% of patients with OS have been cured by chemotherapy alone. Yet when medical treatment is combined with surgical treatment, the overall expected cure rate can be as high as 65%.2
Discussing amputation with a young patient and the family can be emotionally difficult. If functional levels are to be restored, above-knee amputation (AKA) is the least favored surgical method. Compared with healthy individuals, patients who undergo AKA will walk 43% less quickly and will expend much more energy. These patients frequently have an inefficient gait and, given their limited reserve, they may lose the ability to walk altogether.2
Reconstructive surgical options include limb-salvage procedures; since the late 1980s, these have become the standard of care for OS at all sites.11 One such option includes removal of the lesion (eg, a distal femoral or proximal tibial lesion) with acceptable margins and replacement of the lost bone with an allograft or with a metallic prosthesis and knee joint (called arthroplasty). This endoprosthesis expands as the child grows (by way of a minor surgical procedure or a magnetic spring) so there is no apparent discrepancy between limb lengths, and the patient’s appearance is as normal and socially acceptable as possible.1,2
Because the case patient developed a pathologic fracture through his TOS tumor, he was not a candidate for endoprosthesis. His options were AKA or rotationplasty.
This procedure was first described in 195012 for treatment of proximal focal femoral deficiency. It is considered an alternative for skeletally immature individuals for whom the goal is to preserve function.
When AKA is indicated, the lower limb can be salvaged to allow functioning similar to that of a patient with a below-knee amputation (BKA). During rotationplasty, all but the most proximal aspect of the femur is resected. The tibia is externally rotated on the axis of the neurovascular bundle, then an arthrodesis of the proximal portion of the femur and the tibial plateau is performed (see Figure 2).
The end result is an extremity with the appearance, dimensions, and functional potential of a BKA. The ankle is rotated 180° so that it can serve as the new knee joint, and the attached foot, now pointing in the opposite direction, acts as the residual limb for fitting a prosthesis.2 This procedure is favored in patients with an extensive soft-tissue mass, intra-articular extension of the tumor, and/or pathologic fractures. It can also help prevent phantom pain.13
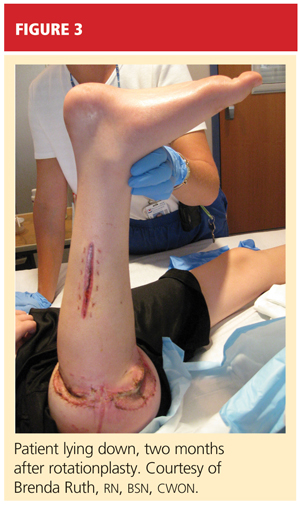
The Case Patient
After psychological evaluation of the patient and extensive family discussion, he underwent successful rotationplasty. The day after his surgery, however, he developed compartment syndrome and was required to undergo fasciotomies of the calf and proximal thigh. His wounds were treated, a skin graft was performed to close the proximal thigh wound, and his calf wounds were sutured closed (see Figures 3 and 4). His hip range of motion is excellent, and his ankle range of motion continues to improve with physical therapy.
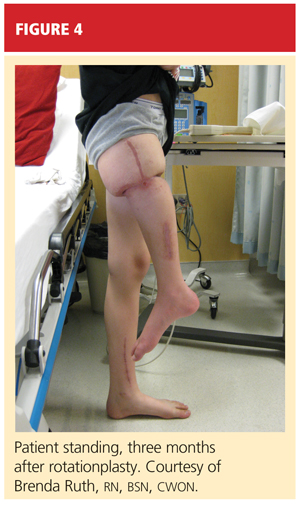
At this writing, the patient was scheduled for his first prosthetic fitting, and he had nearly completed his chemotherapy. His outlook is very promising.
Conclusion
TOS is a rare, aggressive subtype of OS but the most common primary malignant bone tumor of childhood. In the past, outcomes in patients treated with surgery alone were poor. With the advent of chemotherapy and the combination of medical and surgical treatment, TOS-associated mortality has continued to decline. There is no significant difference in outcomes among the available surgical options, but limb-salvage surgical procedures usually offer patients much better function and quality of life. The most important consideration is early diagnosis followed by immediate treatment.
1. Siegel HJ, Pressey JG. Current concepts on the surgical and medical management of osteosarcoma. Expert Rev Anticancer Ther. 2008;8(8):1257-1269.
2. Marulanda GA, Henderson ER, Johnson DA, et al. Orthopedic surgery options for the treatment of primary osteosarcoma. Cancer Control. 2008;15(1):13-20.
3. Murphey MD, wan Jaovisidha S, Temple HT, et al. Telangiectatic osteosarcoma: radiologic-pathologic comparison. Radiology. 2003;229(2):545-553.
4. Mervak TR, Unni KK, Pritchard DJ, McLeod RA. Telangiectatic osteosarcoma. Clin Orthop Relat Res. 1991 Sep;270:135-139.
5. Vanel D, Tcheng S, Contesso G, et al. The radiological appearances of telangiectatic osteosarcoma: a study of 14 cases. Skeletal Radiol. 1987;16(3):196-200.
6. Ferrari S, Smeland S, Mercuri M, et al. Neoadjuvant chemotherapy with high-dose ifosfamide, high-dose methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of the extremity: a joint study by the Italian and Scandinavian Sarcoma Groups. J Clin Oncol. 2005;23(34):8845-8852.
7. Bacci G, Ferrari S, Ruggieri P, et al. Telangiectatic osteosarcoma of the extremity: neoadjuvant chemotherapy in 24 cases. Acta Orthop Scand. 2001;72(2):167-172.
8. Weiss A, Khoury JD, Hoffer FA, et al. Telangiectatic osteosarcoma: the St. Jude Children’s Research Hospital’s experience. Cancer. 2007;109(8):1627-1637.
9. Agarwal M, Anchan C, Shah M, et al. Limb salvage surgery for osteosarcoma: effective low-cost treatment. Clin Orthop Relat Res. 2007;459:82-91.
10. Bacci G, Ferrari S, Lari S, et al. Osteosarcoma of the limb: amputation or limb salvage in patients treated by neoadjuvant chemotherapy. J Bone Joint Surg Br. 2002;84(1):88-92.
11. Simon MA, Aschliman MA, Thomas N, Mankin HJ. Limb-salvage treatment versus amputation for osteosarcoma of the distal end of the femur. J Bone Joint Surg Am. 1986;68(9):1331-1337.
12. Van Nes CP. Rotation-plasty for congenital defects of the femur: making use of the shortened limb to control the knee joint of a prosthesis. J Bone Joint Surg. 1950;32B:12-16.
13. Sawamura C, Hornicek FJ, Gebhardt MC. Complications and risk factors for failure of rotationplasty: review of 25 patients. Clin Orthop Relat Res. 2008;466(6):1302-1308.
A 10-year-old boy first complained of right knee pain two months prior to presentation. There was no traumatic event to explain the pain and no prior viral or bacterial illness. Radiographs taken earlier at another facility were initially pronounced normal. One month later, repeat x-rays showed a possible hairline fracture, and MRI was ordered. MRI documented a destructive lesion in the right distal femur with a soft-tissue mass that was worrisome for primary bone malignancy.
The boy was placed on weight-bearing restrictions and was given a wheelchair. Unfortunately, he fell from the wheelchair and sustained a pathologic fracture through the lesion (see Figure 1). He was transported to the hospital and admitted. A biopsy was performed with a closed reduction, as the fracture was maligned. The patient was placed in a long leg cast with a pelvic band.
His history was previously unremarkable. He was taking no medications and had experienced no recent illnesses. His surgical/medical history was positive for a tonsillectomy at an early age and a fracture of the right proximal femur at age 2. On examination, he was noted to be talkative with his family but guarded during conversations with staff.
His physical exam was positive for pain at the right distal femur and knee with palpation; otherwise, all other systems were unremarkable. The patient was in too much pain to range the knee and had been placed in a long posterior leg splint (prior to surgery and application of the cast). Distally, his right lower extremity motor and sensory function were intact.
The patient’s vital signs were within normal limits, and results from his blood chemistries and alkaline phosphatase and C-reactive protein levels were unremarkable. Findings on the complete blood cell count were slightly abnormal: Hemoglobin was 11 g and the hematocrit, 33% (both within normal limits); however, in the differential there was an elevation in segmented neutrophils (72%, compared with a reference range of 31% to 61%), with Döhle bodies present—possibly signifying acute and/or chronic systemic infection or malignancy. The lymphocyte count represented 11% of the total white blood cell count (range, 28% to 48%), and platelets were 82 x 103/mL (normal range, 150 to 350 x 103/mL). The patient’s erythrocyte sedimentation rate was 44 mm/h (normal range, 0 to 20).
Result from pathology were positive for osteosarcoma, telangiectatic type. The patient underwent a nuclear medicine bone scan that showed no metastases, and chest CT was negative for pulmonary lesions as well. After a psychology consult, the boy was gently told about his condition.
Treatment then proceeded, including surgical placement of a double-lumen chest catheter for delivery of neoadjuvant and adjuvant chemotherapy. Doxorubicin, cisplatin, and methotrexate were used because the boy was enrolled in an international cooperative trial through the Children’s Oncology Group for treatment of localized osteosarcoma.
Discussion
Osteosarcoma (OS) is the most common primary bone malignancy.1,2 Approximately 5% of all pediatric patients with tumors present with this diagnosis, and about 400 new cases are diagnosed in the United States each year.1 Most osteosarcomas develop in the bones of the lower extremities and in the humerus, affecting males more often than females.1-3 This kind of malignancy is frequently seen during the adolescent growth spurt, but it can affect patients of any age.1,2 Patients usually present with pain or functional limitation in gait or daily activities or both.1-3
The telangiectatic subtype of OS is a rare, aggressive variant that represents 2% to 12% of all cases of OS.4-6 Telangiectatic OS (TOS) is characterized by multiple aneurysmally dilated, blood-filled cavities with high-grade sarcomatous cells seen in the peripheral rim and septae.3,7,8 This process can cause the lesion to resemble an aneurysmal bone cyst, explaining why some cases of TOS are misdiagnosed—with delayed time to treatment and increased morbidity and mortality.3,5 Generally, TOS patients are more likely than other OS patients to have tumors of femoral location, larger lesions, and normal alkaline phosphatase values. Many have pathologic fractures on presentation.7
The medical literature chronicles a long debate regarding the difference in mortality between patients with OS and those with TOS. It was once believed that patients with TOS were at higher risk for recurrence (especially those with a pathologic fracture) and mortality. However, in recent studies examining newer neoadjuvant and adjuvant chemotherapies, mortality rates for the two conditions are similar and certainly lower than they were many years ago.7,8 In one study, a better histologic response was reported to neoadjuvant chemotherapy in patients with TOS than with OS.7
Diagnosis
The first diagnostic tool used for patients with suspected OS or TOS is a plain radiographic film. A TOS lesion is lytic, with no areas of sclerosis, and almost always involves the long bones. It is poorly defined, destroying the cortex with formation of periosteal bone and invading the soft tissue. An initial pattern of parallel striations is highly suggestive of TOS.5
MRI and CT often reveal thick nodular tissue in a largely hemorrhagic and/or necrotic osseous lesion, with an associated soft-tissue mass that allows distinction from an aneurysmal bone cyst.3 Next, patients generally undergo a nuclear medicine bone scan and CT of the chest to observe for signs of metastases. Chest CT is commonly repeated on a regular basis during and after treatment.9
Pathologic evaluation, the final step to diagnosis, is very important, especially in the effort to differentiate TOS from an aneurysmal bone cyst. The typical gross findings for a TOS tumor include a dominant cystic cavity–like architecture, with a pushing peripheral margin that frequently expands and erodes the adjacent cortex and extends into the surrounding tissue. There is usually no area of intramural bone tissue.
Microscopically, the cystic areas contain clots and fragments of tumor that are often lined with a layer of neoplasm. The blood-filled telangiectatic spaces form in these areas. The spaces are irregularly shaped and typically traversed by septae composed in part of neoplastic cells. Osteoid formation through these cells can appear as a fine, ice-like material between tumor cells.4,7
Treatment
The main goals of treatment are to limit the anatomical extent of the disease, decrease the possibility of recurrence, and restore the highest possible level of function.2 Initial treatment of any OS or TOS consists of aggressive, immediate chemotherapy prior to and after any surgical intervention.1 (Chemotherapy will not be discussed in further detail here.) Surgical treatments for patients younger than 14 include amputation (above the lesion with wide margins), an expanding prosthesis, or rotationplasty. The location and extent of the tumor, the patient’s age, and his or her desired lifestyle will all have an impact on the choice of surgery.10
Historic data demonstrate that patients who undergo amputation alone almost always develop metastatic disease.1 Other data show that only 10% of patients with OS have been cured by chemotherapy alone. Yet when medical treatment is combined with surgical treatment, the overall expected cure rate can be as high as 65%.2
Discussing amputation with a young patient and the family can be emotionally difficult. If functional levels are to be restored, above-knee amputation (AKA) is the least favored surgical method. Compared with healthy individuals, patients who undergo AKA will walk 43% less quickly and will expend much more energy. These patients frequently have an inefficient gait and, given their limited reserve, they may lose the ability to walk altogether.2
Reconstructive surgical options include limb-salvage procedures; since the late 1980s, these have become the standard of care for OS at all sites.11 One such option includes removal of the lesion (eg, a distal femoral or proximal tibial lesion) with acceptable margins and replacement of the lost bone with an allograft or with a metallic prosthesis and knee joint (called arthroplasty). This endoprosthesis expands as the child grows (by way of a minor surgical procedure or a magnetic spring) so there is no apparent discrepancy between limb lengths, and the patient’s appearance is as normal and socially acceptable as possible.1,2
Because the case patient developed a pathologic fracture through his TOS tumor, he was not a candidate for endoprosthesis. His options were AKA or rotationplasty.
This procedure was first described in 195012 for treatment of proximal focal femoral deficiency. It is considered an alternative for skeletally immature individuals for whom the goal is to preserve function.
When AKA is indicated, the lower limb can be salvaged to allow functioning similar to that of a patient with a below-knee amputation (BKA). During rotationplasty, all but the most proximal aspect of the femur is resected. The tibia is externally rotated on the axis of the neurovascular bundle, then an arthrodesis of the proximal portion of the femur and the tibial plateau is performed (see Figure 2).
The end result is an extremity with the appearance, dimensions, and functional potential of a BKA. The ankle is rotated 180° so that it can serve as the new knee joint, and the attached foot, now pointing in the opposite direction, acts as the residual limb for fitting a prosthesis.2 This procedure is favored in patients with an extensive soft-tissue mass, intra-articular extension of the tumor, and/or pathologic fractures. It can also help prevent phantom pain.13
The Case Patient
After psychological evaluation of the patient and extensive family discussion, he underwent successful rotationplasty. The day after his surgery, however, he developed compartment syndrome and was required to undergo fasciotomies of the calf and proximal thigh. His wounds were treated, a skin graft was performed to close the proximal thigh wound, and his calf wounds were sutured closed (see Figures 3 and 4). His hip range of motion is excellent, and his ankle range of motion continues to improve with physical therapy.
At this writing, the patient was scheduled for his first prosthetic fitting, and he had nearly completed his chemotherapy. His outlook is very promising.
Conclusion
TOS is a rare, aggressive subtype of OS but the most common primary malignant bone tumor of childhood. In the past, outcomes in patients treated with surgery alone were poor. With the advent of chemotherapy and the combination of medical and surgical treatment, TOS-associated mortality has continued to decline. There is no significant difference in outcomes among the available surgical options, but limb-salvage surgical procedures usually offer patients much better function and quality of life. The most important consideration is early diagnosis followed by immediate treatment.
A 10-year-old boy first complained of right knee pain two months prior to presentation. There was no traumatic event to explain the pain and no prior viral or bacterial illness. Radiographs taken earlier at another facility were initially pronounced normal. One month later, repeat x-rays showed a possible hairline fracture, and MRI was ordered. MRI documented a destructive lesion in the right distal femur with a soft-tissue mass that was worrisome for primary bone malignancy.
The boy was placed on weight-bearing restrictions and was given a wheelchair. Unfortunately, he fell from the wheelchair and sustained a pathologic fracture through the lesion (see Figure 1). He was transported to the hospital and admitted. A biopsy was performed with a closed reduction, as the fracture was maligned. The patient was placed in a long leg cast with a pelvic band.
His history was previously unremarkable. He was taking no medications and had experienced no recent illnesses. His surgical/medical history was positive for a tonsillectomy at an early age and a fracture of the right proximal femur at age 2. On examination, he was noted to be talkative with his family but guarded during conversations with staff.
His physical exam was positive for pain at the right distal femur and knee with palpation; otherwise, all other systems were unremarkable. The patient was in too much pain to range the knee and had been placed in a long posterior leg splint (prior to surgery and application of the cast). Distally, his right lower extremity motor and sensory function were intact.
The patient’s vital signs were within normal limits, and results from his blood chemistries and alkaline phosphatase and C-reactive protein levels were unremarkable. Findings on the complete blood cell count were slightly abnormal: Hemoglobin was 11 g and the hematocrit, 33% (both within normal limits); however, in the differential there was an elevation in segmented neutrophils (72%, compared with a reference range of 31% to 61%), with Döhle bodies present—possibly signifying acute and/or chronic systemic infection or malignancy. The lymphocyte count represented 11% of the total white blood cell count (range, 28% to 48%), and platelets were 82 x 103/mL (normal range, 150 to 350 x 103/mL). The patient’s erythrocyte sedimentation rate was 44 mm/h (normal range, 0 to 20).
Result from pathology were positive for osteosarcoma, telangiectatic type. The patient underwent a nuclear medicine bone scan that showed no metastases, and chest CT was negative for pulmonary lesions as well. After a psychology consult, the boy was gently told about his condition.
Treatment then proceeded, including surgical placement of a double-lumen chest catheter for delivery of neoadjuvant and adjuvant chemotherapy. Doxorubicin, cisplatin, and methotrexate were used because the boy was enrolled in an international cooperative trial through the Children’s Oncology Group for treatment of localized osteosarcoma.
Discussion
Osteosarcoma (OS) is the most common primary bone malignancy.1,2 Approximately 5% of all pediatric patients with tumors present with this diagnosis, and about 400 new cases are diagnosed in the United States each year.1 Most osteosarcomas develop in the bones of the lower extremities and in the humerus, affecting males more often than females.1-3 This kind of malignancy is frequently seen during the adolescent growth spurt, but it can affect patients of any age.1,2 Patients usually present with pain or functional limitation in gait or daily activities or both.1-3
The telangiectatic subtype of OS is a rare, aggressive variant that represents 2% to 12% of all cases of OS.4-6 Telangiectatic OS (TOS) is characterized by multiple aneurysmally dilated, blood-filled cavities with high-grade sarcomatous cells seen in the peripheral rim and septae.3,7,8 This process can cause the lesion to resemble an aneurysmal bone cyst, explaining why some cases of TOS are misdiagnosed—with delayed time to treatment and increased morbidity and mortality.3,5 Generally, TOS patients are more likely than other OS patients to have tumors of femoral location, larger lesions, and normal alkaline phosphatase values. Many have pathologic fractures on presentation.7
The medical literature chronicles a long debate regarding the difference in mortality between patients with OS and those with TOS. It was once believed that patients with TOS were at higher risk for recurrence (especially those with a pathologic fracture) and mortality. However, in recent studies examining newer neoadjuvant and adjuvant chemotherapies, mortality rates for the two conditions are similar and certainly lower than they were many years ago.7,8 In one study, a better histologic response was reported to neoadjuvant chemotherapy in patients with TOS than with OS.7
Diagnosis
The first diagnostic tool used for patients with suspected OS or TOS is a plain radiographic film. A TOS lesion is lytic, with no areas of sclerosis, and almost always involves the long bones. It is poorly defined, destroying the cortex with formation of periosteal bone and invading the soft tissue. An initial pattern of parallel striations is highly suggestive of TOS.5
MRI and CT often reveal thick nodular tissue in a largely hemorrhagic and/or necrotic osseous lesion, with an associated soft-tissue mass that allows distinction from an aneurysmal bone cyst.3 Next, patients generally undergo a nuclear medicine bone scan and CT of the chest to observe for signs of metastases. Chest CT is commonly repeated on a regular basis during and after treatment.9
Pathologic evaluation, the final step to diagnosis, is very important, especially in the effort to differentiate TOS from an aneurysmal bone cyst. The typical gross findings for a TOS tumor include a dominant cystic cavity–like architecture, with a pushing peripheral margin that frequently expands and erodes the adjacent cortex and extends into the surrounding tissue. There is usually no area of intramural bone tissue.
Microscopically, the cystic areas contain clots and fragments of tumor that are often lined with a layer of neoplasm. The blood-filled telangiectatic spaces form in these areas. The spaces are irregularly shaped and typically traversed by septae composed in part of neoplastic cells. Osteoid formation through these cells can appear as a fine, ice-like material between tumor cells.4,7
Treatment
The main goals of treatment are to limit the anatomical extent of the disease, decrease the possibility of recurrence, and restore the highest possible level of function.2 Initial treatment of any OS or TOS consists of aggressive, immediate chemotherapy prior to and after any surgical intervention.1 (Chemotherapy will not be discussed in further detail here.) Surgical treatments for patients younger than 14 include amputation (above the lesion with wide margins), an expanding prosthesis, or rotationplasty. The location and extent of the tumor, the patient’s age, and his or her desired lifestyle will all have an impact on the choice of surgery.10
Historic data demonstrate that patients who undergo amputation alone almost always develop metastatic disease.1 Other data show that only 10% of patients with OS have been cured by chemotherapy alone. Yet when medical treatment is combined with surgical treatment, the overall expected cure rate can be as high as 65%.2
Discussing amputation with a young patient and the family can be emotionally difficult. If functional levels are to be restored, above-knee amputation (AKA) is the least favored surgical method. Compared with healthy individuals, patients who undergo AKA will walk 43% less quickly and will expend much more energy. These patients frequently have an inefficient gait and, given their limited reserve, they may lose the ability to walk altogether.2
Reconstructive surgical options include limb-salvage procedures; since the late 1980s, these have become the standard of care for OS at all sites.11 One such option includes removal of the lesion (eg, a distal femoral or proximal tibial lesion) with acceptable margins and replacement of the lost bone with an allograft or with a metallic prosthesis and knee joint (called arthroplasty). This endoprosthesis expands as the child grows (by way of a minor surgical procedure or a magnetic spring) so there is no apparent discrepancy between limb lengths, and the patient’s appearance is as normal and socially acceptable as possible.1,2
Because the case patient developed a pathologic fracture through his TOS tumor, he was not a candidate for endoprosthesis. His options were AKA or rotationplasty.
This procedure was first described in 195012 for treatment of proximal focal femoral deficiency. It is considered an alternative for skeletally immature individuals for whom the goal is to preserve function.
When AKA is indicated, the lower limb can be salvaged to allow functioning similar to that of a patient with a below-knee amputation (BKA). During rotationplasty, all but the most proximal aspect of the femur is resected. The tibia is externally rotated on the axis of the neurovascular bundle, then an arthrodesis of the proximal portion of the femur and the tibial plateau is performed (see Figure 2).
The end result is an extremity with the appearance, dimensions, and functional potential of a BKA. The ankle is rotated 180° so that it can serve as the new knee joint, and the attached foot, now pointing in the opposite direction, acts as the residual limb for fitting a prosthesis.2 This procedure is favored in patients with an extensive soft-tissue mass, intra-articular extension of the tumor, and/or pathologic fractures. It can also help prevent phantom pain.13
The Case Patient
After psychological evaluation of the patient and extensive family discussion, he underwent successful rotationplasty. The day after his surgery, however, he developed compartment syndrome and was required to undergo fasciotomies of the calf and proximal thigh. His wounds were treated, a skin graft was performed to close the proximal thigh wound, and his calf wounds were sutured closed (see Figures 3 and 4). His hip range of motion is excellent, and his ankle range of motion continues to improve with physical therapy.
At this writing, the patient was scheduled for his first prosthetic fitting, and he had nearly completed his chemotherapy. His outlook is very promising.
Conclusion
TOS is a rare, aggressive subtype of OS but the most common primary malignant bone tumor of childhood. In the past, outcomes in patients treated with surgery alone were poor. With the advent of chemotherapy and the combination of medical and surgical treatment, TOS-associated mortality has continued to decline. There is no significant difference in outcomes among the available surgical options, but limb-salvage surgical procedures usually offer patients much better function and quality of life. The most important consideration is early diagnosis followed by immediate treatment.
1. Siegel HJ, Pressey JG. Current concepts on the surgical and medical management of osteosarcoma. Expert Rev Anticancer Ther. 2008;8(8):1257-1269.
2. Marulanda GA, Henderson ER, Johnson DA, et al. Orthopedic surgery options for the treatment of primary osteosarcoma. Cancer Control. 2008;15(1):13-20.
3. Murphey MD, wan Jaovisidha S, Temple HT, et al. Telangiectatic osteosarcoma: radiologic-pathologic comparison. Radiology. 2003;229(2):545-553.
4. Mervak TR, Unni KK, Pritchard DJ, McLeod RA. Telangiectatic osteosarcoma. Clin Orthop Relat Res. 1991 Sep;270:135-139.
5. Vanel D, Tcheng S, Contesso G, et al. The radiological appearances of telangiectatic osteosarcoma: a study of 14 cases. Skeletal Radiol. 1987;16(3):196-200.
6. Ferrari S, Smeland S, Mercuri M, et al. Neoadjuvant chemotherapy with high-dose ifosfamide, high-dose methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of the extremity: a joint study by the Italian and Scandinavian Sarcoma Groups. J Clin Oncol. 2005;23(34):8845-8852.
7. Bacci G, Ferrari S, Ruggieri P, et al. Telangiectatic osteosarcoma of the extremity: neoadjuvant chemotherapy in 24 cases. Acta Orthop Scand. 2001;72(2):167-172.
8. Weiss A, Khoury JD, Hoffer FA, et al. Telangiectatic osteosarcoma: the St. Jude Children’s Research Hospital’s experience. Cancer. 2007;109(8):1627-1637.
9. Agarwal M, Anchan C, Shah M, et al. Limb salvage surgery for osteosarcoma: effective low-cost treatment. Clin Orthop Relat Res. 2007;459:82-91.
10. Bacci G, Ferrari S, Lari S, et al. Osteosarcoma of the limb: amputation or limb salvage in patients treated by neoadjuvant chemotherapy. J Bone Joint Surg Br. 2002;84(1):88-92.
11. Simon MA, Aschliman MA, Thomas N, Mankin HJ. Limb-salvage treatment versus amputation for osteosarcoma of the distal end of the femur. J Bone Joint Surg Am. 1986;68(9):1331-1337.
12. Van Nes CP. Rotation-plasty for congenital defects of the femur: making use of the shortened limb to control the knee joint of a prosthesis. J Bone Joint Surg. 1950;32B:12-16.
13. Sawamura C, Hornicek FJ, Gebhardt MC. Complications and risk factors for failure of rotationplasty: review of 25 patients. Clin Orthop Relat Res. 2008;466(6):1302-1308.
1. Siegel HJ, Pressey JG. Current concepts on the surgical and medical management of osteosarcoma. Expert Rev Anticancer Ther. 2008;8(8):1257-1269.
2. Marulanda GA, Henderson ER, Johnson DA, et al. Orthopedic surgery options for the treatment of primary osteosarcoma. Cancer Control. 2008;15(1):13-20.
3. Murphey MD, wan Jaovisidha S, Temple HT, et al. Telangiectatic osteosarcoma: radiologic-pathologic comparison. Radiology. 2003;229(2):545-553.
4. Mervak TR, Unni KK, Pritchard DJ, McLeod RA. Telangiectatic osteosarcoma. Clin Orthop Relat Res. 1991 Sep;270:135-139.
5. Vanel D, Tcheng S, Contesso G, et al. The radiological appearances of telangiectatic osteosarcoma: a study of 14 cases. Skeletal Radiol. 1987;16(3):196-200.
6. Ferrari S, Smeland S, Mercuri M, et al. Neoadjuvant chemotherapy with high-dose ifosfamide, high-dose methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of the extremity: a joint study by the Italian and Scandinavian Sarcoma Groups. J Clin Oncol. 2005;23(34):8845-8852.
7. Bacci G, Ferrari S, Ruggieri P, et al. Telangiectatic osteosarcoma of the extremity: neoadjuvant chemotherapy in 24 cases. Acta Orthop Scand. 2001;72(2):167-172.
8. Weiss A, Khoury JD, Hoffer FA, et al. Telangiectatic osteosarcoma: the St. Jude Children’s Research Hospital’s experience. Cancer. 2007;109(8):1627-1637.
9. Agarwal M, Anchan C, Shah M, et al. Limb salvage surgery for osteosarcoma: effective low-cost treatment. Clin Orthop Relat Res. 2007;459:82-91.
10. Bacci G, Ferrari S, Lari S, et al. Osteosarcoma of the limb: amputation or limb salvage in patients treated by neoadjuvant chemotherapy. J Bone Joint Surg Br. 2002;84(1):88-92.
11. Simon MA, Aschliman MA, Thomas N, Mankin HJ. Limb-salvage treatment versus amputation for osteosarcoma of the distal end of the femur. J Bone Joint Surg Am. 1986;68(9):1331-1337.
12. Van Nes CP. Rotation-plasty for congenital defects of the femur: making use of the shortened limb to control the knee joint of a prosthesis. J Bone Joint Surg. 1950;32B:12-16.
13. Sawamura C, Hornicek FJ, Gebhardt MC. Complications and risk factors for failure of rotationplasty: review of 25 patients. Clin Orthop Relat Res. 2008;466(6):1302-1308.
When and how to image a suspected broken rib
A 70-year-old man falls in his bathroom and subsequently presents to an urgent care clinic. Among his complaints is right-sided chest pain. On physical examination he has point tenderness over the lateral right thorax with some superficial swelling and bruising. The chest is normal on auscultation.
Should this patient undergo imaging to determine if he has a rib fracture? And which imaging study would be appropriate?
This article outlines the use of various imaging tests in the evaluation of suspected rib fractures and recommends an approach to management. This article does not address fractures in children.
MANY CAUSES OF RIB FRACTURES
Trauma, the most common cause of rib fractures, includes penetrating injuries and blunt injury to the chest wall. Between 10% and 66% of traumatic injuries result in rib fractures. 1 Traumatic injury can result from motor vehicle accidents, assault, sports, cardiopulmonary resuscitation, physical abuse (“nonaccidental” trauma), and, rarely, severe paroxysms of coughing.2
Cancer can cause pathologic fractures of the rib.
Stress fractures of the ribs are more likely to occur in high-level athletes whose activity involves repetitive musculoskeletal loading, although they can also occur in people with repetitive coughing paroxysms.3 Sports and activities that result in stress fractures include rowing, pitching or throwing, basketball, weight-lifting, ballet, golf, gymnastics, and swimming.4
WHICH RIB IS BROKEN?
The fourth through 10th ribs are the most often fractured. Fractures of the first through the third ribs can be associated with underlying nerve and vascular injuries, and fractures of the 10th through 12th ribs are associated with damage to abdominal organs,5 most commonly the liver, spleen, kidneys, and diaphragm.3
Fractures of the costal cartilage can occur by any of the mechanisms described above. The true incidence of costal cartilage fractures is not known because plain radiography, the traditional method of evaluation, does not reliably detect them.
WHY CONFIRM A RIB FRACTURE?
For many rib fractures without associated injury, a radiographic diagnosis has little impact on patient management, which consists mainly of pain control. But knowing whether a patient has a broken rib can often be important.
To detect associated injury. The rate of associated injury in patients with rib fractures is high.6 Potentially severe complications include:
- Pneumothorax
- Hemothorax
- Pulmonary contusion
- Flail chest
- Pneumonia
- Vascular and nerve damage (especially with trauma to the upper chest or the first through third ribs)
- Abdominal organ injury (particularly with trauma to the lower thorax or lower ribs).
The absence of a rib fracture does not preclude these conditions, however.
To prevent complications. Even in the absence of associated injuries, radiographic confirmation of a rib fracture can help prevent complications such as atelectasis and is particularly important in patients with comorbidities such as chronic obstructive pulmonary disease, cardiac disease, hepatic disease, renal disease, dementia, and coagulopathy.1
To document the injury. Radiographic documentation of a rib fracture may be required for medical-legal issues in cases of assault, motor vehicle accident, occupational injury, and abuse.
To help manage pain. Confirmation of rib fracture can facilitate pain management, particularly in patients with undiagnosed fractures with long-standing refractory pain. For example, conservative pain control with nonsteroidal anti-inflammatory drugs may be sufficient for a soft-tissue injury but may not be enough for a rib fracture. Intravenous narcotics or nerve blocks might be preferable.3,7 Controlling pain helps limit the incidence of associated complications.
To count how many ribs are broken. The more ribs broken, the greater the likelihood of illness and death in certain populations, such as the elderly. One study8 found that patients over age 45 with more than four broken ribs are at a significantly higher risk of prolonged stay in the intensive care unit, prolonged ventilator support, and prolonged overall hospital stay.
Knowing the number of ribs fractured may also influence other treatment decisions, such as whether to transfer the patient to a trauma center: a study showed that the more ribs broken, the greater the death rate, and that more than three rib fractures may indicate the need to transfer to a trauma center.6
HOW TO DIAGNOSE A BROKEN RIB
Signs and symptoms are unreliable but important
Clinical symptoms do not reliably tell us if a rib is broken.9,10 Nevertheless, the history and physical examination can uncover possible complications or associated injuries,10,11 such as flail chest, pneumothorax, or vascular injury.
Classic clinical signs and symptoms of rib fracture include point tenderness, focally referred pain with general chest compression, splinting, bony crepitus, and ecchymosis.9 A history of a motor vehicle accident (especially on a motorcycle) or other injury due to rapid deceleration, a fall from higher than 20 feet, a gunshot wound, assault, or a crushing injury would indicate a greater risk of complications.
Signs of complications may include decreased oxygen saturation, decreased or absent breath sounds, dullness or hyperresonance to percussion, tracheal deviation, hypotension, arrythmia, subcutaneous emphysema, neck vein distension, neck hematoma, a focal neurologic deficit below the clavicles or in the upper extremities, and flail chest.11 Flail chest results from multiple fractures in the same rib, so that a segment of chest wall does not contribute to breathing.
Further research is needed into the correlation of clinical symptoms with rib fractures. Much of the evidence that clinical symptoms correlate poorly with fractures comes from studies that used plain radiography to detect the fractures. However, ultrasonography and computed tomography (CT) can detect fractures that plain radiography cannot, and studies using these newer imaging tests may reveal a better correlation between clinical symptoms and rib fracture than previously thought.6
Chest radiography may miss 50% of rib fractures, but is still useful
Plain radiography of the chest with or without oblique views and optimized by the technologist for bony detail (“bone technique”) has historically been the imaging test of choice. However, it may miss up to 50% of fractures.10 Furthermore, it is not sensitive for costal cartilage3 or stress fractures.
Despite these limitations, plain radiography is vitally important in diagnosing complications and associated injuries such as a pneumothorax, hemothorax, pulmonary contusion, pneumomediastinum, or pneumoperitoneum. Also, a widened mediastinum could indicate aortic injury.
Currently, a standard chest x-ray is often the initial study of choice in the evaluation of chest pain and in cases of minor blunt trauma. If rib fractures are suspected clinically, a rib series can be of benefit. A rib series consists of a marker placed over the region of interest, oblique views, and optimization of the radiograph by the technologist to highlight bony detail. The decision to image a rib fracture in the absence of other underlying abnormalities or associated injuries depends on the clinical scenario.
Computed tomography provides more detail
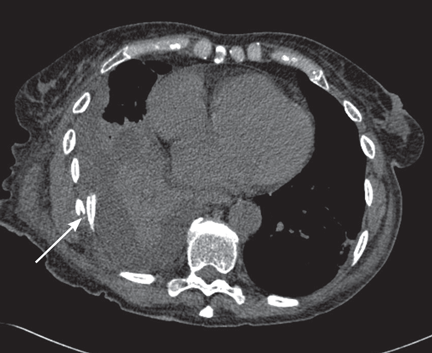
While CT appears to be the best imaging test for evaluating for rib fractures and associated injuries, it is relatively costly, is time-consuming, is not always available, and exposes the patient to a significant amount of radiation.
Also, while CT plays a vital role in major and penetrating trauma of the chest or abdomen, its use in other situations is more limited. Again, the issue of clinical impact of a diagnosis of rib fracture comes into play, and in this setting CT competes with plain radiography and ultrasonography, which are less costly and involve less or no radiation exposure.
Ultrasonography has advantages but is not widely used
Ultrasonography can be used to look for broken ribs and costal cartilage fractures. Associated injuries such as pneumothorax, hemothorax, and abdominal organ injury can also be evaluated. Studies have found it to be much more sensitive than plain radiography in detecting rib fractures,3,15 whereas other studies have suggested it is only equally sensitive or slightly better.7 It also has the advantage of not using radiation.
Because of a number of disadvantages, ultrasonography is rarely used in the evaluation of rib fracture. It is time-consuming and more costly than plain radiography. It is often not readily available. It can be painful, making it impractical for trauma patients. Its results depend greatly on the skill of the technician, and it is unable to adequately assess certain portions of the thorax (eg, the first rib under the clavicle, and the upper ribs under the scapula).7,15 Although able to detect some associated injuries, ultrasonography is not as sensitive and comprehensive as plain radiography and CT. Its role is therefore limited to situations in which the diagnosis of a rib fracture alone, in an accessible rib, is important.
Bone scan: Sensitive but not specific
Technetium Tc 99m methylene diphosphonate bone scanning can be used to look for bone pathology, including rib fractures. Bone scans are sensitive but not specific, and abnormal uptake generates an extensive differential diagnosis.16 Single-photon emission CT, or SPECT, can help localize the abnormality. 4 Because a hot spot on a bone scan can represent a number of conditions besides rib fractures, including cancer, focal sclerosis, and focal osteosclerosis, bone scanning is not routinely used for evaluating rib fractures, although it is very sensitive for stress fractures.
Occasionally, in a patient undergoing a bone scan as part of a workup for cancer, a scan shows a lesion that might be a rib fracture. In this case, one should correlate the results with those of plain radiography or CT.16
Magnetic resonance imaging: no role yet in rib fracture evaluation
MRI is not considered appropriate for evaluating rib fractures. It may be useful if there is concern about soft-tissue or vascular abnormalities. Beyond this, further research is needed to elucidate its role in rib fracture.
THE CHOICE OF TEST DEPENDS ON THE SITUATION
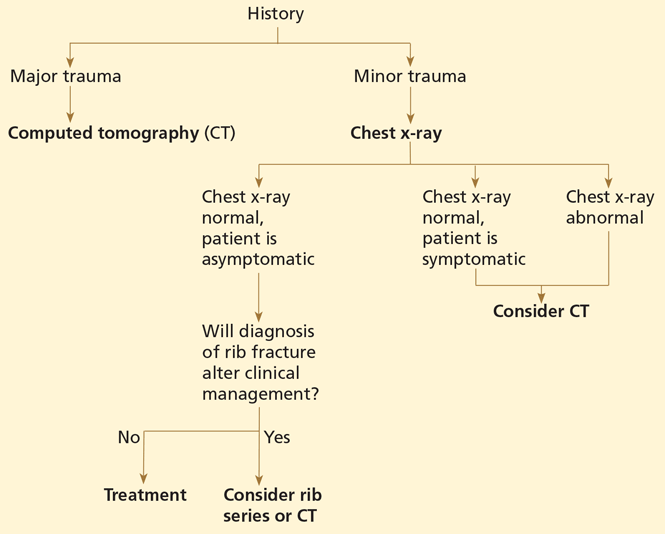
In patients with penetrating or major chest or abdominal trauma, CT is the study of choice. It provides the most information about associated injuries, and it accurately detects rib fractures. This helps target treatment of associated injuries, and helps identify patients at higher risk, such as those with significant vascular, pulmonary, or abdominal injuries and those with a greater number of fractures. An unstable, critically injured patient would not be a candidate for CT because of the risk of transport to the scanner; chest radiography would have to suffice in these cases.
In cases of minor blunt trauma when there is little suspicion of associated injuries or complications, plain radiography is likely sufficient. If there is suspicion of a rib fracture alone and confirmation is of clinical importance (eg, in the elderly or those with long-standing refractory pain, or when certain pain management treatments are being considered), then oblique radiographic views, bone technique, and marker placement over the concerning region are recommended. The role of ultrasonography in this setting is still up for debate.
In cases of suspected rib fracture with longstanding pain refractory to conservative pain management, plain radiography with oblique views, bone technique, and marker placement is useful. If the radiograph is negative or if there is a high suspicion of cartilage fracture, CT or ultrasonography may be of benefit only if the diagnosis will alter clinical management.
If stress fracture is suspected, a nuclear bone scan may be helpful to first detect an abnormality, and CT may then be used for correlation if needed.
CASE CONCLUDED: LIVING WITH UNCERTAINTY
As for the 70-year-old man presented at the beginning of this article, the first question is whether we suspect an associated injury on the basis of clinical features. If we had clinical findings suspicious for pneumothorax or hemothorax, plain radiography of the chest would be indicated. Since the patient was not involved in major trauma, a CT scan is not indicated as the first study.
Our patient has clinical findings suggesting a rib fracture without associated injury. In this setting, routine posteroanterior and lateral chest radiography would be useful to rule out major associated injuries and, perhaps, to find a rib fracture. If the chest film is normal and rib fracture is still suspected, we must decide whether the diagnosis would alter our clinical management. Our patient would likely be treated the same regardless of whether or not he has a fracture; therefore, we would prescribe pain management.
Chest radiography was performed to rule out associated injuries, especially since the patient was elderly, but the chest x-ray did not reveal anything. On follow-up approximately 1 month later, he appeared improved, with less pain and tenderness. This may be due to healing of a rib fracture or healing of his soft-tissue injury. We will never know whether he truly had a fracture, but it is irrelevant to his care.
- Bergeron E, Lavoie A, Clas D, et al. Elderly trauma patients with rib fractures are at greater risk of death and pneumonia. J Trauma 2003; 54:478–485.
- Lederer W, Mair D, Rabl W, Baubin M. Frequency of rib and sternum fractures associated with out-of-hospital cardiopulmonary resuscitation is underestimated by conventional chest x-ray. Resuscitation 2004; 60:157–162.
- Kara M, Dikmen E, Erdal HH, Simsir I, Kara SA. Disclosure of unnoticed rib fractures with the use of ultrasonography in minor blunt chest trauma. Eur J Cardiothorac Surg 2003; 24:608–613.
- Connolly LP, Connolly SA. Rib stress fractures. Clin Nucl Med 2004; 29:614–616.
- Bansidhar BJ, Lagares-Garcia JA, Miller SL. Clinical rib fractures: are follow-up chest x-rays a waste of resources? Am Surg 2002; 68:449–453.
- Stawicki SP, Grossman MD, Hoey BA, Miller DL, Reed JF. Rib fractures in the elderly: a marker of injury severity. J Am Geriatr Soc 2004; 52:805–808.
- Hurley ME, Keye GD, Hamilton S. Is ultrasound really helpful in the detection of rib fractures? Injury 2004; 35:562–566.
- Holcomb JB, McMullin NR, Kozar RA, Lygas MH, Moore FA. Morbidity from rib fractures increases after age 45. J Am Coll Surg 2003; 196:549–555.
- Deluca SA, Rhea JT, O’Malley TO. Radiographic evaluation of rib fractures. AJR Am J Roentgenol 1982; 138:91–92.
- Dubinsky I, Low A. Non-life threatening blunt chest trauma: appropriate investigation and treatment. Am J Emerg Med 1997; 15:240–243.
- Sears BW, Luchette FA, Esposito TJ, et al. Old fashion clinical judgment in the era of protocols: is mandatory chest x-ray necessary in injured patients? J Trauma 2005; 59:324–332.
- Traub M, Stevenson M, McEvoy S, et al. The use of chest computed tomography versus chest x-ray in patients with major blunt trauma. Injury 2007; 38:43–47.
- Trupka A, Waydhas C, Hallfeldt KK, Nast-Kolb D, Pfeifer KJ, Schweiberer L. Value of thoracic computed tomography in the first assessment of severely injured patients with blunt chest trauma: results of a prospective study. J Trauma 1997; 43:405–412.
- Malghem J, Vande Berg B, Lecouvet F, Maldague B. Costal cartilage fractures as revealed on CT and sonography. AJR Am J Roentgenol 2001; 176:429–432.
- Griffith JF, Rainer TH, Ching AS, Law KL, Cocks RA, Metreweli C. Sonography compared with radiography in revealing acute rib fracture. AJR Am J Roentgenol 1999; 173:1603–1609.
- Niitsu M, Takeda T. Solitary hot spots in the ribs on bone scan: value of thin-section reformatted computed tomography to exclude radiography negative fractures. J Comput Assist Tomogr 2003; 27:469–474.
A 70-year-old man falls in his bathroom and subsequently presents to an urgent care clinic. Among his complaints is right-sided chest pain. On physical examination he has point tenderness over the lateral right thorax with some superficial swelling and bruising. The chest is normal on auscultation.
Should this patient undergo imaging to determine if he has a rib fracture? And which imaging study would be appropriate?
This article outlines the use of various imaging tests in the evaluation of suspected rib fractures and recommends an approach to management. This article does not address fractures in children.
MANY CAUSES OF RIB FRACTURES
Trauma, the most common cause of rib fractures, includes penetrating injuries and blunt injury to the chest wall. Between 10% and 66% of traumatic injuries result in rib fractures. 1 Traumatic injury can result from motor vehicle accidents, assault, sports, cardiopulmonary resuscitation, physical abuse (“nonaccidental” trauma), and, rarely, severe paroxysms of coughing.2
Cancer can cause pathologic fractures of the rib.
Stress fractures of the ribs are more likely to occur in high-level athletes whose activity involves repetitive musculoskeletal loading, although they can also occur in people with repetitive coughing paroxysms.3 Sports and activities that result in stress fractures include rowing, pitching or throwing, basketball, weight-lifting, ballet, golf, gymnastics, and swimming.4
WHICH RIB IS BROKEN?
The fourth through 10th ribs are the most often fractured. Fractures of the first through the third ribs can be associated with underlying nerve and vascular injuries, and fractures of the 10th through 12th ribs are associated with damage to abdominal organs,5 most commonly the liver, spleen, kidneys, and diaphragm.3
Fractures of the costal cartilage can occur by any of the mechanisms described above. The true incidence of costal cartilage fractures is not known because plain radiography, the traditional method of evaluation, does not reliably detect them.
WHY CONFIRM A RIB FRACTURE?
For many rib fractures without associated injury, a radiographic diagnosis has little impact on patient management, which consists mainly of pain control. But knowing whether a patient has a broken rib can often be important.
To detect associated injury. The rate of associated injury in patients with rib fractures is high.6 Potentially severe complications include:
- Pneumothorax
- Hemothorax
- Pulmonary contusion
- Flail chest
- Pneumonia
- Vascular and nerve damage (especially with trauma to the upper chest or the first through third ribs)
- Abdominal organ injury (particularly with trauma to the lower thorax or lower ribs).
The absence of a rib fracture does not preclude these conditions, however.
To prevent complications. Even in the absence of associated injuries, radiographic confirmation of a rib fracture can help prevent complications such as atelectasis and is particularly important in patients with comorbidities such as chronic obstructive pulmonary disease, cardiac disease, hepatic disease, renal disease, dementia, and coagulopathy.1
To document the injury. Radiographic documentation of a rib fracture may be required for medical-legal issues in cases of assault, motor vehicle accident, occupational injury, and abuse.
To help manage pain. Confirmation of rib fracture can facilitate pain management, particularly in patients with undiagnosed fractures with long-standing refractory pain. For example, conservative pain control with nonsteroidal anti-inflammatory drugs may be sufficient for a soft-tissue injury but may not be enough for a rib fracture. Intravenous narcotics or nerve blocks might be preferable.3,7 Controlling pain helps limit the incidence of associated complications.
To count how many ribs are broken. The more ribs broken, the greater the likelihood of illness and death in certain populations, such as the elderly. One study8 found that patients over age 45 with more than four broken ribs are at a significantly higher risk of prolonged stay in the intensive care unit, prolonged ventilator support, and prolonged overall hospital stay.
Knowing the number of ribs fractured may also influence other treatment decisions, such as whether to transfer the patient to a trauma center: a study showed that the more ribs broken, the greater the death rate, and that more than three rib fractures may indicate the need to transfer to a trauma center.6
HOW TO DIAGNOSE A BROKEN RIB
Signs and symptoms are unreliable but important
Clinical symptoms do not reliably tell us if a rib is broken.9,10 Nevertheless, the history and physical examination can uncover possible complications or associated injuries,10,11 such as flail chest, pneumothorax, or vascular injury.
Classic clinical signs and symptoms of rib fracture include point tenderness, focally referred pain with general chest compression, splinting, bony crepitus, and ecchymosis.9 A history of a motor vehicle accident (especially on a motorcycle) or other injury due to rapid deceleration, a fall from higher than 20 feet, a gunshot wound, assault, or a crushing injury would indicate a greater risk of complications.
Signs of complications may include decreased oxygen saturation, decreased or absent breath sounds, dullness or hyperresonance to percussion, tracheal deviation, hypotension, arrythmia, subcutaneous emphysema, neck vein distension, neck hematoma, a focal neurologic deficit below the clavicles or in the upper extremities, and flail chest.11 Flail chest results from multiple fractures in the same rib, so that a segment of chest wall does not contribute to breathing.
Further research is needed into the correlation of clinical symptoms with rib fractures. Much of the evidence that clinical symptoms correlate poorly with fractures comes from studies that used plain radiography to detect the fractures. However, ultrasonography and computed tomography (CT) can detect fractures that plain radiography cannot, and studies using these newer imaging tests may reveal a better correlation between clinical symptoms and rib fracture than previously thought.6
Chest radiography may miss 50% of rib fractures, but is still useful
Plain radiography of the chest with or without oblique views and optimized by the technologist for bony detail (“bone technique”) has historically been the imaging test of choice. However, it may miss up to 50% of fractures.10 Furthermore, it is not sensitive for costal cartilage3 or stress fractures.
Despite these limitations, plain radiography is vitally important in diagnosing complications and associated injuries such as a pneumothorax, hemothorax, pulmonary contusion, pneumomediastinum, or pneumoperitoneum. Also, a widened mediastinum could indicate aortic injury.
Currently, a standard chest x-ray is often the initial study of choice in the evaluation of chest pain and in cases of minor blunt trauma. If rib fractures are suspected clinically, a rib series can be of benefit. A rib series consists of a marker placed over the region of interest, oblique views, and optimization of the radiograph by the technologist to highlight bony detail. The decision to image a rib fracture in the absence of other underlying abnormalities or associated injuries depends on the clinical scenario.
Computed tomography provides more detail
While CT appears to be the best imaging test for evaluating for rib fractures and associated injuries, it is relatively costly, is time-consuming, is not always available, and exposes the patient to a significant amount of radiation.
Also, while CT plays a vital role in major and penetrating trauma of the chest or abdomen, its use in other situations is more limited. Again, the issue of clinical impact of a diagnosis of rib fracture comes into play, and in this setting CT competes with plain radiography and ultrasonography, which are less costly and involve less or no radiation exposure.
Ultrasonography has advantages but is not widely used
Ultrasonography can be used to look for broken ribs and costal cartilage fractures. Associated injuries such as pneumothorax, hemothorax, and abdominal organ injury can also be evaluated. Studies have found it to be much more sensitive than plain radiography in detecting rib fractures,3,15 whereas other studies have suggested it is only equally sensitive or slightly better.7 It also has the advantage of not using radiation.
Because of a number of disadvantages, ultrasonography is rarely used in the evaluation of rib fracture. It is time-consuming and more costly than plain radiography. It is often not readily available. It can be painful, making it impractical for trauma patients. Its results depend greatly on the skill of the technician, and it is unable to adequately assess certain portions of the thorax (eg, the first rib under the clavicle, and the upper ribs under the scapula).7,15 Although able to detect some associated injuries, ultrasonography is not as sensitive and comprehensive as plain radiography and CT. Its role is therefore limited to situations in which the diagnosis of a rib fracture alone, in an accessible rib, is important.
Bone scan: Sensitive but not specific
Technetium Tc 99m methylene diphosphonate bone scanning can be used to look for bone pathology, including rib fractures. Bone scans are sensitive but not specific, and abnormal uptake generates an extensive differential diagnosis.16 Single-photon emission CT, or SPECT, can help localize the abnormality. 4 Because a hot spot on a bone scan can represent a number of conditions besides rib fractures, including cancer, focal sclerosis, and focal osteosclerosis, bone scanning is not routinely used for evaluating rib fractures, although it is very sensitive for stress fractures.
Occasionally, in a patient undergoing a bone scan as part of a workup for cancer, a scan shows a lesion that might be a rib fracture. In this case, one should correlate the results with those of plain radiography or CT.16
Magnetic resonance imaging: no role yet in rib fracture evaluation
MRI is not considered appropriate for evaluating rib fractures. It may be useful if there is concern about soft-tissue or vascular abnormalities. Beyond this, further research is needed to elucidate its role in rib fracture.
THE CHOICE OF TEST DEPENDS ON THE SITUATION
In patients with penetrating or major chest or abdominal trauma, CT is the study of choice. It provides the most information about associated injuries, and it accurately detects rib fractures. This helps target treatment of associated injuries, and helps identify patients at higher risk, such as those with significant vascular, pulmonary, or abdominal injuries and those with a greater number of fractures. An unstable, critically injured patient would not be a candidate for CT because of the risk of transport to the scanner; chest radiography would have to suffice in these cases.
In cases of minor blunt trauma when there is little suspicion of associated injuries or complications, plain radiography is likely sufficient. If there is suspicion of a rib fracture alone and confirmation is of clinical importance (eg, in the elderly or those with long-standing refractory pain, or when certain pain management treatments are being considered), then oblique radiographic views, bone technique, and marker placement over the concerning region are recommended. The role of ultrasonography in this setting is still up for debate.
In cases of suspected rib fracture with longstanding pain refractory to conservative pain management, plain radiography with oblique views, bone technique, and marker placement is useful. If the radiograph is negative or if there is a high suspicion of cartilage fracture, CT or ultrasonography may be of benefit only if the diagnosis will alter clinical management.
If stress fracture is suspected, a nuclear bone scan may be helpful to first detect an abnormality, and CT may then be used for correlation if needed.
CASE CONCLUDED: LIVING WITH UNCERTAINTY
As for the 70-year-old man presented at the beginning of this article, the first question is whether we suspect an associated injury on the basis of clinical features. If we had clinical findings suspicious for pneumothorax or hemothorax, plain radiography of the chest would be indicated. Since the patient was not involved in major trauma, a CT scan is not indicated as the first study.
Our patient has clinical findings suggesting a rib fracture without associated injury. In this setting, routine posteroanterior and lateral chest radiography would be useful to rule out major associated injuries and, perhaps, to find a rib fracture. If the chest film is normal and rib fracture is still suspected, we must decide whether the diagnosis would alter our clinical management. Our patient would likely be treated the same regardless of whether or not he has a fracture; therefore, we would prescribe pain management.
Chest radiography was performed to rule out associated injuries, especially since the patient was elderly, but the chest x-ray did not reveal anything. On follow-up approximately 1 month later, he appeared improved, with less pain and tenderness. This may be due to healing of a rib fracture or healing of his soft-tissue injury. We will never know whether he truly had a fracture, but it is irrelevant to his care.
A 70-year-old man falls in his bathroom and subsequently presents to an urgent care clinic. Among his complaints is right-sided chest pain. On physical examination he has point tenderness over the lateral right thorax with some superficial swelling and bruising. The chest is normal on auscultation.
Should this patient undergo imaging to determine if he has a rib fracture? And which imaging study would be appropriate?
This article outlines the use of various imaging tests in the evaluation of suspected rib fractures and recommends an approach to management. This article does not address fractures in children.
MANY CAUSES OF RIB FRACTURES
Trauma, the most common cause of rib fractures, includes penetrating injuries and blunt injury to the chest wall. Between 10% and 66% of traumatic injuries result in rib fractures. 1 Traumatic injury can result from motor vehicle accidents, assault, sports, cardiopulmonary resuscitation, physical abuse (“nonaccidental” trauma), and, rarely, severe paroxysms of coughing.2
Cancer can cause pathologic fractures of the rib.
Stress fractures of the ribs are more likely to occur in high-level athletes whose activity involves repetitive musculoskeletal loading, although they can also occur in people with repetitive coughing paroxysms.3 Sports and activities that result in stress fractures include rowing, pitching or throwing, basketball, weight-lifting, ballet, golf, gymnastics, and swimming.4
WHICH RIB IS BROKEN?
The fourth through 10th ribs are the most often fractured. Fractures of the first through the third ribs can be associated with underlying nerve and vascular injuries, and fractures of the 10th through 12th ribs are associated with damage to abdominal organs,5 most commonly the liver, spleen, kidneys, and diaphragm.3
Fractures of the costal cartilage can occur by any of the mechanisms described above. The true incidence of costal cartilage fractures is not known because plain radiography, the traditional method of evaluation, does not reliably detect them.
WHY CONFIRM A RIB FRACTURE?
For many rib fractures without associated injury, a radiographic diagnosis has little impact on patient management, which consists mainly of pain control. But knowing whether a patient has a broken rib can often be important.
To detect associated injury. The rate of associated injury in patients with rib fractures is high.6 Potentially severe complications include:
- Pneumothorax
- Hemothorax
- Pulmonary contusion
- Flail chest
- Pneumonia
- Vascular and nerve damage (especially with trauma to the upper chest or the first through third ribs)
- Abdominal organ injury (particularly with trauma to the lower thorax or lower ribs).
The absence of a rib fracture does not preclude these conditions, however.
To prevent complications. Even in the absence of associated injuries, radiographic confirmation of a rib fracture can help prevent complications such as atelectasis and is particularly important in patients with comorbidities such as chronic obstructive pulmonary disease, cardiac disease, hepatic disease, renal disease, dementia, and coagulopathy.1
To document the injury. Radiographic documentation of a rib fracture may be required for medical-legal issues in cases of assault, motor vehicle accident, occupational injury, and abuse.
To help manage pain. Confirmation of rib fracture can facilitate pain management, particularly in patients with undiagnosed fractures with long-standing refractory pain. For example, conservative pain control with nonsteroidal anti-inflammatory drugs may be sufficient for a soft-tissue injury but may not be enough for a rib fracture. Intravenous narcotics or nerve blocks might be preferable.3,7 Controlling pain helps limit the incidence of associated complications.
To count how many ribs are broken. The more ribs broken, the greater the likelihood of illness and death in certain populations, such as the elderly. One study8 found that patients over age 45 with more than four broken ribs are at a significantly higher risk of prolonged stay in the intensive care unit, prolonged ventilator support, and prolonged overall hospital stay.
Knowing the number of ribs fractured may also influence other treatment decisions, such as whether to transfer the patient to a trauma center: a study showed that the more ribs broken, the greater the death rate, and that more than three rib fractures may indicate the need to transfer to a trauma center.6
HOW TO DIAGNOSE A BROKEN RIB
Signs and symptoms are unreliable but important
Clinical symptoms do not reliably tell us if a rib is broken.9,10 Nevertheless, the history and physical examination can uncover possible complications or associated injuries,10,11 such as flail chest, pneumothorax, or vascular injury.
Classic clinical signs and symptoms of rib fracture include point tenderness, focally referred pain with general chest compression, splinting, bony crepitus, and ecchymosis.9 A history of a motor vehicle accident (especially on a motorcycle) or other injury due to rapid deceleration, a fall from higher than 20 feet, a gunshot wound, assault, or a crushing injury would indicate a greater risk of complications.
Signs of complications may include decreased oxygen saturation, decreased or absent breath sounds, dullness or hyperresonance to percussion, tracheal deviation, hypotension, arrythmia, subcutaneous emphysema, neck vein distension, neck hematoma, a focal neurologic deficit below the clavicles or in the upper extremities, and flail chest.11 Flail chest results from multiple fractures in the same rib, so that a segment of chest wall does not contribute to breathing.
Further research is needed into the correlation of clinical symptoms with rib fractures. Much of the evidence that clinical symptoms correlate poorly with fractures comes from studies that used plain radiography to detect the fractures. However, ultrasonography and computed tomography (CT) can detect fractures that plain radiography cannot, and studies using these newer imaging tests may reveal a better correlation between clinical symptoms and rib fracture than previously thought.6
Chest radiography may miss 50% of rib fractures, but is still useful
Plain radiography of the chest with or without oblique views and optimized by the technologist for bony detail (“bone technique”) has historically been the imaging test of choice. However, it may miss up to 50% of fractures.10 Furthermore, it is not sensitive for costal cartilage3 or stress fractures.
Despite these limitations, plain radiography is vitally important in diagnosing complications and associated injuries such as a pneumothorax, hemothorax, pulmonary contusion, pneumomediastinum, or pneumoperitoneum. Also, a widened mediastinum could indicate aortic injury.
Currently, a standard chest x-ray is often the initial study of choice in the evaluation of chest pain and in cases of minor blunt trauma. If rib fractures are suspected clinically, a rib series can be of benefit. A rib series consists of a marker placed over the region of interest, oblique views, and optimization of the radiograph by the technologist to highlight bony detail. The decision to image a rib fracture in the absence of other underlying abnormalities or associated injuries depends on the clinical scenario.
Computed tomography provides more detail
While CT appears to be the best imaging test for evaluating for rib fractures and associated injuries, it is relatively costly, is time-consuming, is not always available, and exposes the patient to a significant amount of radiation.
Also, while CT plays a vital role in major and penetrating trauma of the chest or abdomen, its use in other situations is more limited. Again, the issue of clinical impact of a diagnosis of rib fracture comes into play, and in this setting CT competes with plain radiography and ultrasonography, which are less costly and involve less or no radiation exposure.
Ultrasonography has advantages but is not widely used
Ultrasonography can be used to look for broken ribs and costal cartilage fractures. Associated injuries such as pneumothorax, hemothorax, and abdominal organ injury can also be evaluated. Studies have found it to be much more sensitive than plain radiography in detecting rib fractures,3,15 whereas other studies have suggested it is only equally sensitive or slightly better.7 It also has the advantage of not using radiation.
Because of a number of disadvantages, ultrasonography is rarely used in the evaluation of rib fracture. It is time-consuming and more costly than plain radiography. It is often not readily available. It can be painful, making it impractical for trauma patients. Its results depend greatly on the skill of the technician, and it is unable to adequately assess certain portions of the thorax (eg, the first rib under the clavicle, and the upper ribs under the scapula).7,15 Although able to detect some associated injuries, ultrasonography is not as sensitive and comprehensive as plain radiography and CT. Its role is therefore limited to situations in which the diagnosis of a rib fracture alone, in an accessible rib, is important.
Bone scan: Sensitive but not specific
Technetium Tc 99m methylene diphosphonate bone scanning can be used to look for bone pathology, including rib fractures. Bone scans are sensitive but not specific, and abnormal uptake generates an extensive differential diagnosis.16 Single-photon emission CT, or SPECT, can help localize the abnormality. 4 Because a hot spot on a bone scan can represent a number of conditions besides rib fractures, including cancer, focal sclerosis, and focal osteosclerosis, bone scanning is not routinely used for evaluating rib fractures, although it is very sensitive for stress fractures.
Occasionally, in a patient undergoing a bone scan as part of a workup for cancer, a scan shows a lesion that might be a rib fracture. In this case, one should correlate the results with those of plain radiography or CT.16
Magnetic resonance imaging: no role yet in rib fracture evaluation
MRI is not considered appropriate for evaluating rib fractures. It may be useful if there is concern about soft-tissue or vascular abnormalities. Beyond this, further research is needed to elucidate its role in rib fracture.
THE CHOICE OF TEST DEPENDS ON THE SITUATION
In patients with penetrating or major chest or abdominal trauma, CT is the study of choice. It provides the most information about associated injuries, and it accurately detects rib fractures. This helps target treatment of associated injuries, and helps identify patients at higher risk, such as those with significant vascular, pulmonary, or abdominal injuries and those with a greater number of fractures. An unstable, critically injured patient would not be a candidate for CT because of the risk of transport to the scanner; chest radiography would have to suffice in these cases.
In cases of minor blunt trauma when there is little suspicion of associated injuries or complications, plain radiography is likely sufficient. If there is suspicion of a rib fracture alone and confirmation is of clinical importance (eg, in the elderly or those with long-standing refractory pain, or when certain pain management treatments are being considered), then oblique radiographic views, bone technique, and marker placement over the concerning region are recommended. The role of ultrasonography in this setting is still up for debate.
In cases of suspected rib fracture with longstanding pain refractory to conservative pain management, plain radiography with oblique views, bone technique, and marker placement is useful. If the radiograph is negative or if there is a high suspicion of cartilage fracture, CT or ultrasonography may be of benefit only if the diagnosis will alter clinical management.
If stress fracture is suspected, a nuclear bone scan may be helpful to first detect an abnormality, and CT may then be used for correlation if needed.
CASE CONCLUDED: LIVING WITH UNCERTAINTY
As for the 70-year-old man presented at the beginning of this article, the first question is whether we suspect an associated injury on the basis of clinical features. If we had clinical findings suspicious for pneumothorax or hemothorax, plain radiography of the chest would be indicated. Since the patient was not involved in major trauma, a CT scan is not indicated as the first study.
Our patient has clinical findings suggesting a rib fracture without associated injury. In this setting, routine posteroanterior and lateral chest radiography would be useful to rule out major associated injuries and, perhaps, to find a rib fracture. If the chest film is normal and rib fracture is still suspected, we must decide whether the diagnosis would alter our clinical management. Our patient would likely be treated the same regardless of whether or not he has a fracture; therefore, we would prescribe pain management.
Chest radiography was performed to rule out associated injuries, especially since the patient was elderly, but the chest x-ray did not reveal anything. On follow-up approximately 1 month later, he appeared improved, with less pain and tenderness. This may be due to healing of a rib fracture or healing of his soft-tissue injury. We will never know whether he truly had a fracture, but it is irrelevant to his care.
- Bergeron E, Lavoie A, Clas D, et al. Elderly trauma patients with rib fractures are at greater risk of death and pneumonia. J Trauma 2003; 54:478–485.
- Lederer W, Mair D, Rabl W, Baubin M. Frequency of rib and sternum fractures associated with out-of-hospital cardiopulmonary resuscitation is underestimated by conventional chest x-ray. Resuscitation 2004; 60:157–162.
- Kara M, Dikmen E, Erdal HH, Simsir I, Kara SA. Disclosure of unnoticed rib fractures with the use of ultrasonography in minor blunt chest trauma. Eur J Cardiothorac Surg 2003; 24:608–613.
- Connolly LP, Connolly SA. Rib stress fractures. Clin Nucl Med 2004; 29:614–616.
- Bansidhar BJ, Lagares-Garcia JA, Miller SL. Clinical rib fractures: are follow-up chest x-rays a waste of resources? Am Surg 2002; 68:449–453.
- Stawicki SP, Grossman MD, Hoey BA, Miller DL, Reed JF. Rib fractures in the elderly: a marker of injury severity. J Am Geriatr Soc 2004; 52:805–808.
- Hurley ME, Keye GD, Hamilton S. Is ultrasound really helpful in the detection of rib fractures? Injury 2004; 35:562–566.
- Holcomb JB, McMullin NR, Kozar RA, Lygas MH, Moore FA. Morbidity from rib fractures increases after age 45. J Am Coll Surg 2003; 196:549–555.
- Deluca SA, Rhea JT, O’Malley TO. Radiographic evaluation of rib fractures. AJR Am J Roentgenol 1982; 138:91–92.
- Dubinsky I, Low A. Non-life threatening blunt chest trauma: appropriate investigation and treatment. Am J Emerg Med 1997; 15:240–243.
- Sears BW, Luchette FA, Esposito TJ, et al. Old fashion clinical judgment in the era of protocols: is mandatory chest x-ray necessary in injured patients? J Trauma 2005; 59:324–332.
- Traub M, Stevenson M, McEvoy S, et al. The use of chest computed tomography versus chest x-ray in patients with major blunt trauma. Injury 2007; 38:43–47.
- Trupka A, Waydhas C, Hallfeldt KK, Nast-Kolb D, Pfeifer KJ, Schweiberer L. Value of thoracic computed tomography in the first assessment of severely injured patients with blunt chest trauma: results of a prospective study. J Trauma 1997; 43:405–412.
- Malghem J, Vande Berg B, Lecouvet F, Maldague B. Costal cartilage fractures as revealed on CT and sonography. AJR Am J Roentgenol 2001; 176:429–432.
- Griffith JF, Rainer TH, Ching AS, Law KL, Cocks RA, Metreweli C. Sonography compared with radiography in revealing acute rib fracture. AJR Am J Roentgenol 1999; 173:1603–1609.
- Niitsu M, Takeda T. Solitary hot spots in the ribs on bone scan: value of thin-section reformatted computed tomography to exclude radiography negative fractures. J Comput Assist Tomogr 2003; 27:469–474.
- Bergeron E, Lavoie A, Clas D, et al. Elderly trauma patients with rib fractures are at greater risk of death and pneumonia. J Trauma 2003; 54:478–485.
- Lederer W, Mair D, Rabl W, Baubin M. Frequency of rib and sternum fractures associated with out-of-hospital cardiopulmonary resuscitation is underestimated by conventional chest x-ray. Resuscitation 2004; 60:157–162.
- Kara M, Dikmen E, Erdal HH, Simsir I, Kara SA. Disclosure of unnoticed rib fractures with the use of ultrasonography in minor blunt chest trauma. Eur J Cardiothorac Surg 2003; 24:608–613.
- Connolly LP, Connolly SA. Rib stress fractures. Clin Nucl Med 2004; 29:614–616.
- Bansidhar BJ, Lagares-Garcia JA, Miller SL. Clinical rib fractures: are follow-up chest x-rays a waste of resources? Am Surg 2002; 68:449–453.
- Stawicki SP, Grossman MD, Hoey BA, Miller DL, Reed JF. Rib fractures in the elderly: a marker of injury severity. J Am Geriatr Soc 2004; 52:805–808.
- Hurley ME, Keye GD, Hamilton S. Is ultrasound really helpful in the detection of rib fractures? Injury 2004; 35:562–566.
- Holcomb JB, McMullin NR, Kozar RA, Lygas MH, Moore FA. Morbidity from rib fractures increases after age 45. J Am Coll Surg 2003; 196:549–555.
- Deluca SA, Rhea JT, O’Malley TO. Radiographic evaluation of rib fractures. AJR Am J Roentgenol 1982; 138:91–92.
- Dubinsky I, Low A. Non-life threatening blunt chest trauma: appropriate investigation and treatment. Am J Emerg Med 1997; 15:240–243.
- Sears BW, Luchette FA, Esposito TJ, et al. Old fashion clinical judgment in the era of protocols: is mandatory chest x-ray necessary in injured patients? J Trauma 2005; 59:324–332.
- Traub M, Stevenson M, McEvoy S, et al. The use of chest computed tomography versus chest x-ray in patients with major blunt trauma. Injury 2007; 38:43–47.
- Trupka A, Waydhas C, Hallfeldt KK, Nast-Kolb D, Pfeifer KJ, Schweiberer L. Value of thoracic computed tomography in the first assessment of severely injured patients with blunt chest trauma: results of a prospective study. J Trauma 1997; 43:405–412.
- Malghem J, Vande Berg B, Lecouvet F, Maldague B. Costal cartilage fractures as revealed on CT and sonography. AJR Am J Roentgenol 2001; 176:429–432.
- Griffith JF, Rainer TH, Ching AS, Law KL, Cocks RA, Metreweli C. Sonography compared with radiography in revealing acute rib fracture. AJR Am J Roentgenol 1999; 173:1603–1609.
- Niitsu M, Takeda T. Solitary hot spots in the ribs on bone scan: value of thin-section reformatted computed tomography to exclude radiography negative fractures. J Comput Assist Tomogr 2003; 27:469–474.
KEY POINTS
- Knowing the number of ribs fractured may influence treatment decisions, such as whether to transfer a patient to a trauma center.
- Classic clinical signs and symptoms of rib fracture include point tenderness, focally referred pain with general chest compression, splinting, bony crepitus, and ecchymosis.
- In a patient with minor blunt trauma, when there is little suspicion of associated injury or complication, plain radiography is likely sufficient.
- Computed tomography is the imaging study of choice in patients with penetrating or major chest or abdominal trauma.
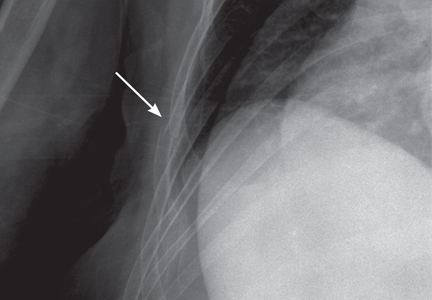
A 37-year-old man with chest pain, ECG changes, and elevated cardiac enzymes
A 37-year-old African American man presents to the emergency department with chest pain and dyspnea, which began suddenly 30 minutes ago. The pain is severe, pressure-like, nonradiating, and pleuritic.
His heart rate is 88 beats per minute, blood pressure 135/72 mm Hg, respiratory rate 12 per minute, and oral temperature 38.5°C (101.3°F). His oxygen saturation by pulse oximetry is 99% while breathing room air. He is given sublingual nitroglycerin, but this does not alleviate his pain.
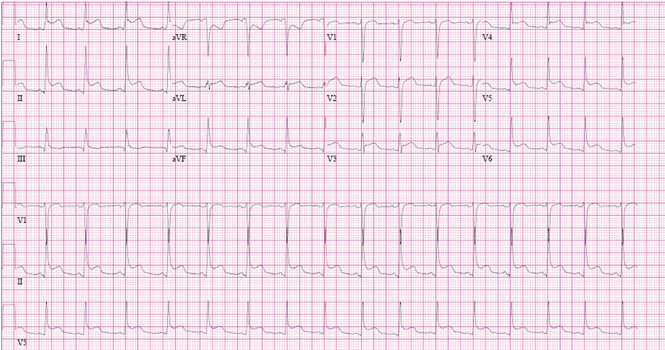
While blood samples are being drawn, we learn more about his history. He has hypertension, for which he takes amlodipine (Norvasc), and gastroesophageal reflux under control with esomeprazole (Nexium). He says he does not have hyperlipidemia, diabetes, or coronary artery disease and his surgical history is unremarkable. He says he does not smoke, rarely drinks, and does not use any drugs. No one in his family has had premature coronary artery disease.
He says he has had similar symptoms in the past few months, which resulted in two emergency room visits. Electrocardiograms at those times were unremarkable, and a stress test was negative for ischemia.
A computed tomographic (CT) scan of the chest was also obtained during one of those visits. The scan was negative for a pulmonary embolus but incidentally showed liver hemangiomas.
The patient’s initial laboratory results are shown in Table 1.
WHAT IS THE CAUSE OF HIS CHEST PAIN?
1. Which is the most likely cause of this patient’s chest pain?
- Acute myocardial infarction
- Acute pericarditis
- Myocarditis
- Pulmonary embolism
- Aortic dissection
- Pneumonia
Acute myocardial infarction. This is a young man with chest pain, ST-segment elevation, and elevated cardiac enzymes. Acute myocardial infarction should always be included in the differential diagnosis of such a patient, as recognizing it early and making an effort to rapidly restore blood flow to the myocardium can greatly improve the clinical outcome. However, particular features in his electrocardiogram and the duration and nature of his chest pain suggest another diagnosis.
Acute pericarditis causes pleuritic chest pain with diffuse ST-segment elevation, and its electrocardiographic changes may be difficult to distinguish from those of ischemia. The features in our patient’s electrocardiogram that point to pericarditis are1:
- ST-segment elevation that is concave upward, occurring in all leads except aVR
- T waves concordant with ST-segment deviation
- PR-segment depression, sparing V1 and aVR
- PR-segment elevation and ST depression in aVR.
Pleuritic chest pain is the most common symptom in acute pericarditis. A prodrome of fever, myalgia, and malaise is also common, especially in younger patients.2 On physical examination, a pericardial friction rub is pathognomonic.
Our patient has most if not all of the classic features of acute pericarditis. Elevated cardiac enzymes, which this patient has, are not a classic feature of pericarditis and are generally considered a marker of cardiac ischemia. However, because the myocardium is adjacent to the pericardium, the acute inflammatory process of acute pericarditis may also result in myocardial injury, resulting in release of creatine kinase-MB.3
An increase in cardiac troponin is also frequently observed in acute pericarditis, reflecting biochemical evidence of inflammatory myocardial cell damage.4 Furthermore, cardiac troponin can be elevated in several other medical conditions,5 such as ischemic heart disease, congestive heart failure, myocarditis, pulmonary embolism, severe pulmonary hypertension, significant left ventricular hypertrophy, renal failure, sepsis, critical illness, and subarachnoid hemorrhage. Therefore, cardiac enzymes are not good markers to distinguish between acute myocardial infarction and acute pericarditis. However, echocardiography is an effective way to help differentiate pericarditis from myocardial ischemia in the setting of elevated troponins and electrocardiographic changes, by determining if wall-motion abnormalities are present or absent.
Hence, the diagnosis of acute pericarditis should take into account the combination of the clinical picture, electrocardiographic findings, and laboratory values. Overreliance on any of these in isolation can lead to misdiagnosis.
Pulmonary embolism is another common cause of acute-onset pleuritic chest pain and dyspnea. Electrocardiographic changes can include ST-segment elevation, and cardiac enzymes can be elevated, although this is uncommon.
Myocarditis is commonly due to infections, collagen vascular diseases, or medications. Hallmarks of this disease are elevated cardiac enzymes and myocardial damage that results in reduction in heart function.
Aortic dissection typically causes a sharp, tearing chest pain that radiates to the back. This diagnosis is unlikely in this patient.
Pneumonia. Although our patient did not have a cough and no crackles were heard on lung examination to suggest pneumonia, his fever, pleuritic chest pain, and leukocytosis with a left shift warrant a workup for it. A parapneumonic effusion could manifest with fevers and pleuritic chest pain. However, the acuity of the symptoms and the characteristic electrocardiographic changes and elevated cardiac enzymes are better explained by the other diagnoses, notably acute pericarditis.
ACUTE PERICARDITIS: WHAT IS THE CAUSE?
2. Which is the most common cause of acute pericarditis?
- Idiopathic
- Neoplasm
- Autoimmune
- Tuberculosis
Most (approximately 80%) of cases of acute pericarditis are idiopathic.6,7 In a study in 100 patients with acute pericarditis,6 a specific cause was identified in only 22. The most common identified cause was neoplasm, which was present in seven patients: four with lung cancer and one each with breast carcinoma, cystic duct adenocarcinoma, and cardiac angiosarcoma.

CASE CONTINUES: PERICARDIAL EFFUSION
The patient is admitted to the hospital for additional workup. His fever, myalgia, and chest pain persist, though the pain is less intense than before.
A chest roentgenogram and transthoracic echocardiogram are ordered and blood cultures are drawn.
The roentgenogram shows marked cardiomegaly, bilateral small pleural effusions, and minimal atelectatic changes in the lungs.
Echocardiography reveals a normal ejection fraction (60%) and a moderate-sized pericardial effusion without evidence of tamponade.
3. Which is the most common cause of pericardial effusion?
- Idiopathic
- Infection
- Malignancy
- Collagen vascular disease
Pericardial effusion is relatively common after acute pericarditis but also has many other possible causes. In a study of 204 patients with pericardial effusion,8 48% of cases were labeled as idiopathic. Of the remaining 52%, the most common specific diagnoses were infection (16%) and cancer (15%). Collagen vascular disease accounted for 8% of the cases and included systemic lupus erythematosus, rheumatoid arthritis, and scleroderma.
Although small pericardial effusions are common in pericarditis, larger pericardial effusions or failure to respond to therapy necessitates additional workup.2
In our patient, an extensive workup is initiated to look for bacterial, viral, fungal, and autoimmune causes of pericardial effusion, but the results of the workup are negative.
TREATING ACUTE PERICARDITIS
4. Which is the most appropriate treatment for acute pericarditis?
- Steroids
- A nonsteroidal anti-inflammatory drug (NSAID) or aspirin
- Opioids
- Colchicine
- Colchicine plus an NSAID or aspirin
An NSAID or aspirin is the basis of treatment for acute pericarditis and is very effective in relieving symptoms. Aspirin 2–4 g daily, indomethacin (Indocin) 75–225 mg daily, or ibuprofen (Motrin) 1,600–3,200 mg daily are prescribed most often; ibuprofen is preferred because it has a lower incidence of adverse effects than the others.9
Colchicine is recommended in addition to aspirin or NSAIDs for the treatment of acute pericarditis. Although in the past colchicine was reserved for recurrent pericarditis, the Colchicine for Acute Pericarditis (COPE) trial10 found it to be beneficial for first episodes of pericarditis as well.10 In this study, patients were randomized to receive conventional treatment with aspirin 800 mg every 6 or 8 hours or aspirin at the same dose combined with colchicine 0.5 to 1.0 mg daily. Colchicine showed significant benefit over conventional therapy, resulting in reduced rates of recurrence.
CASE CONTINUES: HEPATIC LESIONS ON MRI
Although aspirin and colchicine were started at the time of admission, our patient’s symptoms fail to improve. A suspicion remains that a neoplastic disorder could be the underlying cause of the presentation and could explain his chronic malaise, pericardial disease, and fever. In view of the liver hemangiomas reported previously on CT, we decide to evaluate the liver further with magnetic resonance imaging (MRI).
Since our patient’s symptoms have improved significantly during the past few days and his fever has resolved, biopsy is scheduled on an outpatient basis. Biopsy with ultrasonographic guidance is performed a week later and yields a pathologic diagnosis of hemangioma. The improvement, however, is short-lived, and his pain and dyspnea recur after 2 months. A follow-up echocardiogram is ordered.
A remarkable echocardiographic finding
The original echocardiogram that was performed a little over 2-1/2 months ago is re-reviewed. It very subtly suggests a complexity to the pericardial effusion in the area of the current mass, apparent only when the two studies are directly compared. Clearly, there has been interval development of a mass easily detectable by echocardiography. Although a small mass may have been obscured by the pericardial effusion in the original echocardiogram, the development of a mass of this size in such a short time suggests a rapidly growing tumor.
CARDIAC TUMORS
5. Which is the most common primary cardiac tumor?
- Myxoma
- Papillary fibroelastoma
- Sarcoma
- Lymphoma
Primary cardiac tumors are rare, with an incidence on autopsy series ranging between 0.0017% and 0.33%,11,12 making them far less common than metastases to the heart.
Myxomas are benign cardiac tumors and are the most common primary cardiac neoplasm. Approximately 80% of myxomas originate in the left atrium, typically presenting with one or more of the triad of intracardiac obstruction, systemic embolization, and constitutional symptoms.14
Cardiac papillary fibroelastomas, the second most common cardiac tumors, are benign and predominantly affect the cardiac valves.15
Only one-fourth of all cardiac tumors are malignant. Nearly all of these malignant tumors are sarcomas, with angiosarcoma being the most common morphologic type, accounting for 30% of primary cardiac sarcomas.13
Primary cardiac lymphomas are extremely rare and account for only 1.3% of all primary cardiac tumors.16
A DIAGNOSIS IS MADE
CARDIAC ANGIOSARCOMA
Cardiac angiosarcoma, the most common malignant primary cardiac tumor, has a predilection for the right atrium.13 These tumors tend to occur between the third and fifth decade of life and are three times more common in men than in women. Cardiac sarcomas proliferate rapidly and commonly extend into the pericardial space, causing pericardial effusion in up to one-fourth of patients.
Surgical resection is the treatment of choice, but due to the location and extent of involvement, complete resection is often difficult. Also, distant metastases are present at the time of diagnosis in 80% of cases, precluding a surgical cure.17 Adjuvant chemotherapy, radiotherapy, and even heart transplantation do not substantially improve the survival of these patients.18–20 Because no effective treatment is available, the prognosis is dismal, with a median survival of 6 to 12 months.
Our patient is discharged home to follow up with an oncologist and initiate chemotherapy.
Acknowledgment: We thank Lisa M. Yerian, MD, for interpreting the biopsy specimens described in this article.
- Ariyarajah V, Spodick DH. Acute pericarditis: diagnostic cues and common electrocardiographic manifestations. Cardiol Rev 2007; 15:24–30.
- LeWinter MM, Kabbani S. Pericardial disease. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1757–1781.
- Karjalainen J, Heikkila J. “Acute pericarditis”: myocardial enzyme release as evidence for myocarditis”. Am Heart J 1986; 111:546–552.
- Brandt RR, Filzmaier K, Hanrath P. Circulating cardiac troponin I in acute pericarditis. Am J Cardiol 2001; 87:1326–1328.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease: the present and the future. J Am Coll Cardiol 2006; 48:1–11.
- Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol 1995; 75:378–382.
- Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation 2007; 115:2739–2744.
- Levy PY, Corey R, Berger P, et al. Etiologic diagnosis of 204 pericardial effusions. Medicine (Baltimore) 2003; 82:385–391.
- Lange RA, Hillis LD. Clinical practice. Acute pericarditis. N Engl J Med 2004; 351:2195–2202.
- Imazio M, Bobbio M, Cecchi E, et al. Colchicine in addition to conventional therapy for acute pericarditis: Results of the COlchicine for acute PEricarditis (COPE) trial. Circulation 2005; 112:2012–2016.
- Heath D. Pathology of cardiac tumors. Am J Cardiol 1968; 21:315–327.
- Wold LE, Lie JT. Cardiac myxomas: a clinicopathologic profile. Am J Pathol 1980; 101:219–240.
- Sabatine MS, Colucci WS, Schoen FS. Primary tumors of the heart. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1741–1757.
- Gowda RM, Khan IA, Nair CK, Mehta NJ, Vasavada BC, Sacchi TJ. Cardiac papillary fibroelastoma: a comprehensive analysis of 725 cases. Am Heart J 2003; 146:404–410.
- Glancy DL, Morales JB, Roberts WC. Angiosarcoma of the heart. Am J Cardiol 1968; 21:413–419.
- Ceresoli GL, Ferreri AJ, Bucci E, Ripa C, Ponzoni M, Villa E. Primary cardiac lymphoma in immunocompetent patients: diagnostic and therapeutic management. Cancer 1997; 80:1497–1506.
- Bear PA, Moodie DS. Malignant primary cardiac tumors. The Cleveland Clinic experience, 1956 to 1986. Chest 1987; 92:860–862.
- Llombart–Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer 1998; 78:1624–1628.
- Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DA. Primary cardiac sarcomas. Ann Thorac Surg 1991; 51:906–910.
- Conklin LD, Reardon MJ. Autotransplantation of the heart for primary cardiac malignancy: development and surgical technique. Tex Heart Inst J 2002; 29:105–108.
A 37-year-old African American man presents to the emergency department with chest pain and dyspnea, which began suddenly 30 minutes ago. The pain is severe, pressure-like, nonradiating, and pleuritic.
His heart rate is 88 beats per minute, blood pressure 135/72 mm Hg, respiratory rate 12 per minute, and oral temperature 38.5°C (101.3°F). His oxygen saturation by pulse oximetry is 99% while breathing room air. He is given sublingual nitroglycerin, but this does not alleviate his pain.
While blood samples are being drawn, we learn more about his history. He has hypertension, for which he takes amlodipine (Norvasc), and gastroesophageal reflux under control with esomeprazole (Nexium). He says he does not have hyperlipidemia, diabetes, or coronary artery disease and his surgical history is unremarkable. He says he does not smoke, rarely drinks, and does not use any drugs. No one in his family has had premature coronary artery disease.
He says he has had similar symptoms in the past few months, which resulted in two emergency room visits. Electrocardiograms at those times were unremarkable, and a stress test was negative for ischemia.
A computed tomographic (CT) scan of the chest was also obtained during one of those visits. The scan was negative for a pulmonary embolus but incidentally showed liver hemangiomas.
The patient’s initial laboratory results are shown in Table 1.
WHAT IS THE CAUSE OF HIS CHEST PAIN?
1. Which is the most likely cause of this patient’s chest pain?
- Acute myocardial infarction
- Acute pericarditis
- Myocarditis
- Pulmonary embolism
- Aortic dissection
- Pneumonia
Acute myocardial infarction. This is a young man with chest pain, ST-segment elevation, and elevated cardiac enzymes. Acute myocardial infarction should always be included in the differential diagnosis of such a patient, as recognizing it early and making an effort to rapidly restore blood flow to the myocardium can greatly improve the clinical outcome. However, particular features in his electrocardiogram and the duration and nature of his chest pain suggest another diagnosis.
Acute pericarditis causes pleuritic chest pain with diffuse ST-segment elevation, and its electrocardiographic changes may be difficult to distinguish from those of ischemia. The features in our patient’s electrocardiogram that point to pericarditis are1:
- ST-segment elevation that is concave upward, occurring in all leads except aVR
- T waves concordant with ST-segment deviation
- PR-segment depression, sparing V1 and aVR
- PR-segment elevation and ST depression in aVR.
Pleuritic chest pain is the most common symptom in acute pericarditis. A prodrome of fever, myalgia, and malaise is also common, especially in younger patients.2 On physical examination, a pericardial friction rub is pathognomonic.
Our patient has most if not all of the classic features of acute pericarditis. Elevated cardiac enzymes, which this patient has, are not a classic feature of pericarditis and are generally considered a marker of cardiac ischemia. However, because the myocardium is adjacent to the pericardium, the acute inflammatory process of acute pericarditis may also result in myocardial injury, resulting in release of creatine kinase-MB.3
An increase in cardiac troponin is also frequently observed in acute pericarditis, reflecting biochemical evidence of inflammatory myocardial cell damage.4 Furthermore, cardiac troponin can be elevated in several other medical conditions,5 such as ischemic heart disease, congestive heart failure, myocarditis, pulmonary embolism, severe pulmonary hypertension, significant left ventricular hypertrophy, renal failure, sepsis, critical illness, and subarachnoid hemorrhage. Therefore, cardiac enzymes are not good markers to distinguish between acute myocardial infarction and acute pericarditis. However, echocardiography is an effective way to help differentiate pericarditis from myocardial ischemia in the setting of elevated troponins and electrocardiographic changes, by determining if wall-motion abnormalities are present or absent.
Hence, the diagnosis of acute pericarditis should take into account the combination of the clinical picture, electrocardiographic findings, and laboratory values. Overreliance on any of these in isolation can lead to misdiagnosis.
Pulmonary embolism is another common cause of acute-onset pleuritic chest pain and dyspnea. Electrocardiographic changes can include ST-segment elevation, and cardiac enzymes can be elevated, although this is uncommon.
Myocarditis is commonly due to infections, collagen vascular diseases, or medications. Hallmarks of this disease are elevated cardiac enzymes and myocardial damage that results in reduction in heart function.
Aortic dissection typically causes a sharp, tearing chest pain that radiates to the back. This diagnosis is unlikely in this patient.
Pneumonia. Although our patient did not have a cough and no crackles were heard on lung examination to suggest pneumonia, his fever, pleuritic chest pain, and leukocytosis with a left shift warrant a workup for it. A parapneumonic effusion could manifest with fevers and pleuritic chest pain. However, the acuity of the symptoms and the characteristic electrocardiographic changes and elevated cardiac enzymes are better explained by the other diagnoses, notably acute pericarditis.
ACUTE PERICARDITIS: WHAT IS THE CAUSE?
2. Which is the most common cause of acute pericarditis?
- Idiopathic
- Neoplasm
- Autoimmune
- Tuberculosis
Most (approximately 80%) of cases of acute pericarditis are idiopathic.6,7 In a study in 100 patients with acute pericarditis,6 a specific cause was identified in only 22. The most common identified cause was neoplasm, which was present in seven patients: four with lung cancer and one each with breast carcinoma, cystic duct adenocarcinoma, and cardiac angiosarcoma.
CASE CONTINUES: PERICARDIAL EFFUSION
The patient is admitted to the hospital for additional workup. His fever, myalgia, and chest pain persist, though the pain is less intense than before.
A chest roentgenogram and transthoracic echocardiogram are ordered and blood cultures are drawn.
The roentgenogram shows marked cardiomegaly, bilateral small pleural effusions, and minimal atelectatic changes in the lungs.
Echocardiography reveals a normal ejection fraction (60%) and a moderate-sized pericardial effusion without evidence of tamponade.
3. Which is the most common cause of pericardial effusion?
- Idiopathic
- Infection
- Malignancy
- Collagen vascular disease
Pericardial effusion is relatively common after acute pericarditis but also has many other possible causes. In a study of 204 patients with pericardial effusion,8 48% of cases were labeled as idiopathic. Of the remaining 52%, the most common specific diagnoses were infection (16%) and cancer (15%). Collagen vascular disease accounted for 8% of the cases and included systemic lupus erythematosus, rheumatoid arthritis, and scleroderma.
Although small pericardial effusions are common in pericarditis, larger pericardial effusions or failure to respond to therapy necessitates additional workup.2
In our patient, an extensive workup is initiated to look for bacterial, viral, fungal, and autoimmune causes of pericardial effusion, but the results of the workup are negative.
TREATING ACUTE PERICARDITIS
4. Which is the most appropriate treatment for acute pericarditis?
- Steroids
- A nonsteroidal anti-inflammatory drug (NSAID) or aspirin
- Opioids
- Colchicine
- Colchicine plus an NSAID or aspirin
An NSAID or aspirin is the basis of treatment for acute pericarditis and is very effective in relieving symptoms. Aspirin 2–4 g daily, indomethacin (Indocin) 75–225 mg daily, or ibuprofen (Motrin) 1,600–3,200 mg daily are prescribed most often; ibuprofen is preferred because it has a lower incidence of adverse effects than the others.9
Colchicine is recommended in addition to aspirin or NSAIDs for the treatment of acute pericarditis. Although in the past colchicine was reserved for recurrent pericarditis, the Colchicine for Acute Pericarditis (COPE) trial10 found it to be beneficial for first episodes of pericarditis as well.10 In this study, patients were randomized to receive conventional treatment with aspirin 800 mg every 6 or 8 hours or aspirin at the same dose combined with colchicine 0.5 to 1.0 mg daily. Colchicine showed significant benefit over conventional therapy, resulting in reduced rates of recurrence.
CASE CONTINUES: HEPATIC LESIONS ON MRI
Although aspirin and colchicine were started at the time of admission, our patient’s symptoms fail to improve. A suspicion remains that a neoplastic disorder could be the underlying cause of the presentation and could explain his chronic malaise, pericardial disease, and fever. In view of the liver hemangiomas reported previously on CT, we decide to evaluate the liver further with magnetic resonance imaging (MRI).
Since our patient’s symptoms have improved significantly during the past few days and his fever has resolved, biopsy is scheduled on an outpatient basis. Biopsy with ultrasonographic guidance is performed a week later and yields a pathologic diagnosis of hemangioma. The improvement, however, is short-lived, and his pain and dyspnea recur after 2 months. A follow-up echocardiogram is ordered.
A remarkable echocardiographic finding
The original echocardiogram that was performed a little over 2-1/2 months ago is re-reviewed. It very subtly suggests a complexity to the pericardial effusion in the area of the current mass, apparent only when the two studies are directly compared. Clearly, there has been interval development of a mass easily detectable by echocardiography. Although a small mass may have been obscured by the pericardial effusion in the original echocardiogram, the development of a mass of this size in such a short time suggests a rapidly growing tumor.
CARDIAC TUMORS
5. Which is the most common primary cardiac tumor?
- Myxoma
- Papillary fibroelastoma
- Sarcoma
- Lymphoma
Primary cardiac tumors are rare, with an incidence on autopsy series ranging between 0.0017% and 0.33%,11,12 making them far less common than metastases to the heart.
Myxomas are benign cardiac tumors and are the most common primary cardiac neoplasm. Approximately 80% of myxomas originate in the left atrium, typically presenting with one or more of the triad of intracardiac obstruction, systemic embolization, and constitutional symptoms.14
Cardiac papillary fibroelastomas, the second most common cardiac tumors, are benign and predominantly affect the cardiac valves.15
Only one-fourth of all cardiac tumors are malignant. Nearly all of these malignant tumors are sarcomas, with angiosarcoma being the most common morphologic type, accounting for 30% of primary cardiac sarcomas.13
Primary cardiac lymphomas are extremely rare and account for only 1.3% of all primary cardiac tumors.16
A DIAGNOSIS IS MADE
CARDIAC ANGIOSARCOMA
Cardiac angiosarcoma, the most common malignant primary cardiac tumor, has a predilection for the right atrium.13 These tumors tend to occur between the third and fifth decade of life and are three times more common in men than in women. Cardiac sarcomas proliferate rapidly and commonly extend into the pericardial space, causing pericardial effusion in up to one-fourth of patients.
Surgical resection is the treatment of choice, but due to the location and extent of involvement, complete resection is often difficult. Also, distant metastases are present at the time of diagnosis in 80% of cases, precluding a surgical cure.17 Adjuvant chemotherapy, radiotherapy, and even heart transplantation do not substantially improve the survival of these patients.18–20 Because no effective treatment is available, the prognosis is dismal, with a median survival of 6 to 12 months.
Our patient is discharged home to follow up with an oncologist and initiate chemotherapy.
Acknowledgment: We thank Lisa M. Yerian, MD, for interpreting the biopsy specimens described in this article.
A 37-year-old African American man presents to the emergency department with chest pain and dyspnea, which began suddenly 30 minutes ago. The pain is severe, pressure-like, nonradiating, and pleuritic.
His heart rate is 88 beats per minute, blood pressure 135/72 mm Hg, respiratory rate 12 per minute, and oral temperature 38.5°C (101.3°F). His oxygen saturation by pulse oximetry is 99% while breathing room air. He is given sublingual nitroglycerin, but this does not alleviate his pain.
While blood samples are being drawn, we learn more about his history. He has hypertension, for which he takes amlodipine (Norvasc), and gastroesophageal reflux under control with esomeprazole (Nexium). He says he does not have hyperlipidemia, diabetes, or coronary artery disease and his surgical history is unremarkable. He says he does not smoke, rarely drinks, and does not use any drugs. No one in his family has had premature coronary artery disease.
He says he has had similar symptoms in the past few months, which resulted in two emergency room visits. Electrocardiograms at those times were unremarkable, and a stress test was negative for ischemia.
A computed tomographic (CT) scan of the chest was also obtained during one of those visits. The scan was negative for a pulmonary embolus but incidentally showed liver hemangiomas.
The patient’s initial laboratory results are shown in Table 1.
WHAT IS THE CAUSE OF HIS CHEST PAIN?
1. Which is the most likely cause of this patient’s chest pain?
- Acute myocardial infarction
- Acute pericarditis
- Myocarditis
- Pulmonary embolism
- Aortic dissection
- Pneumonia
Acute myocardial infarction. This is a young man with chest pain, ST-segment elevation, and elevated cardiac enzymes. Acute myocardial infarction should always be included in the differential diagnosis of such a patient, as recognizing it early and making an effort to rapidly restore blood flow to the myocardium can greatly improve the clinical outcome. However, particular features in his electrocardiogram and the duration and nature of his chest pain suggest another diagnosis.
Acute pericarditis causes pleuritic chest pain with diffuse ST-segment elevation, and its electrocardiographic changes may be difficult to distinguish from those of ischemia. The features in our patient’s electrocardiogram that point to pericarditis are1:
- ST-segment elevation that is concave upward, occurring in all leads except aVR
- T waves concordant with ST-segment deviation
- PR-segment depression, sparing V1 and aVR
- PR-segment elevation and ST depression in aVR.
Pleuritic chest pain is the most common symptom in acute pericarditis. A prodrome of fever, myalgia, and malaise is also common, especially in younger patients.2 On physical examination, a pericardial friction rub is pathognomonic.
Our patient has most if not all of the classic features of acute pericarditis. Elevated cardiac enzymes, which this patient has, are not a classic feature of pericarditis and are generally considered a marker of cardiac ischemia. However, because the myocardium is adjacent to the pericardium, the acute inflammatory process of acute pericarditis may also result in myocardial injury, resulting in release of creatine kinase-MB.3
An increase in cardiac troponin is also frequently observed in acute pericarditis, reflecting biochemical evidence of inflammatory myocardial cell damage.4 Furthermore, cardiac troponin can be elevated in several other medical conditions,5 such as ischemic heart disease, congestive heart failure, myocarditis, pulmonary embolism, severe pulmonary hypertension, significant left ventricular hypertrophy, renal failure, sepsis, critical illness, and subarachnoid hemorrhage. Therefore, cardiac enzymes are not good markers to distinguish between acute myocardial infarction and acute pericarditis. However, echocardiography is an effective way to help differentiate pericarditis from myocardial ischemia in the setting of elevated troponins and electrocardiographic changes, by determining if wall-motion abnormalities are present or absent.
Hence, the diagnosis of acute pericarditis should take into account the combination of the clinical picture, electrocardiographic findings, and laboratory values. Overreliance on any of these in isolation can lead to misdiagnosis.
Pulmonary embolism is another common cause of acute-onset pleuritic chest pain and dyspnea. Electrocardiographic changes can include ST-segment elevation, and cardiac enzymes can be elevated, although this is uncommon.
Myocarditis is commonly due to infections, collagen vascular diseases, or medications. Hallmarks of this disease are elevated cardiac enzymes and myocardial damage that results in reduction in heart function.
Aortic dissection typically causes a sharp, tearing chest pain that radiates to the back. This diagnosis is unlikely in this patient.
Pneumonia. Although our patient did not have a cough and no crackles were heard on lung examination to suggest pneumonia, his fever, pleuritic chest pain, and leukocytosis with a left shift warrant a workup for it. A parapneumonic effusion could manifest with fevers and pleuritic chest pain. However, the acuity of the symptoms and the characteristic electrocardiographic changes and elevated cardiac enzymes are better explained by the other diagnoses, notably acute pericarditis.
ACUTE PERICARDITIS: WHAT IS THE CAUSE?
2. Which is the most common cause of acute pericarditis?
- Idiopathic
- Neoplasm
- Autoimmune
- Tuberculosis
Most (approximately 80%) of cases of acute pericarditis are idiopathic.6,7 In a study in 100 patients with acute pericarditis,6 a specific cause was identified in only 22. The most common identified cause was neoplasm, which was present in seven patients: four with lung cancer and one each with breast carcinoma, cystic duct adenocarcinoma, and cardiac angiosarcoma.
CASE CONTINUES: PERICARDIAL EFFUSION
The patient is admitted to the hospital for additional workup. His fever, myalgia, and chest pain persist, though the pain is less intense than before.
A chest roentgenogram and transthoracic echocardiogram are ordered and blood cultures are drawn.
The roentgenogram shows marked cardiomegaly, bilateral small pleural effusions, and minimal atelectatic changes in the lungs.
Echocardiography reveals a normal ejection fraction (60%) and a moderate-sized pericardial effusion without evidence of tamponade.
3. Which is the most common cause of pericardial effusion?
- Idiopathic
- Infection
- Malignancy
- Collagen vascular disease
Pericardial effusion is relatively common after acute pericarditis but also has many other possible causes. In a study of 204 patients with pericardial effusion,8 48% of cases were labeled as idiopathic. Of the remaining 52%, the most common specific diagnoses were infection (16%) and cancer (15%). Collagen vascular disease accounted for 8% of the cases and included systemic lupus erythematosus, rheumatoid arthritis, and scleroderma.
Although small pericardial effusions are common in pericarditis, larger pericardial effusions or failure to respond to therapy necessitates additional workup.2
In our patient, an extensive workup is initiated to look for bacterial, viral, fungal, and autoimmune causes of pericardial effusion, but the results of the workup are negative.
TREATING ACUTE PERICARDITIS
4. Which is the most appropriate treatment for acute pericarditis?
- Steroids
- A nonsteroidal anti-inflammatory drug (NSAID) or aspirin
- Opioids
- Colchicine
- Colchicine plus an NSAID or aspirin
An NSAID or aspirin is the basis of treatment for acute pericarditis and is very effective in relieving symptoms. Aspirin 2–4 g daily, indomethacin (Indocin) 75–225 mg daily, or ibuprofen (Motrin) 1,600–3,200 mg daily are prescribed most often; ibuprofen is preferred because it has a lower incidence of adverse effects than the others.9
Colchicine is recommended in addition to aspirin or NSAIDs for the treatment of acute pericarditis. Although in the past colchicine was reserved for recurrent pericarditis, the Colchicine for Acute Pericarditis (COPE) trial10 found it to be beneficial for first episodes of pericarditis as well.10 In this study, patients were randomized to receive conventional treatment with aspirin 800 mg every 6 or 8 hours or aspirin at the same dose combined with colchicine 0.5 to 1.0 mg daily. Colchicine showed significant benefit over conventional therapy, resulting in reduced rates of recurrence.
CASE CONTINUES: HEPATIC LESIONS ON MRI
Although aspirin and colchicine were started at the time of admission, our patient’s symptoms fail to improve. A suspicion remains that a neoplastic disorder could be the underlying cause of the presentation and could explain his chronic malaise, pericardial disease, and fever. In view of the liver hemangiomas reported previously on CT, we decide to evaluate the liver further with magnetic resonance imaging (MRI).
Since our patient’s symptoms have improved significantly during the past few days and his fever has resolved, biopsy is scheduled on an outpatient basis. Biopsy with ultrasonographic guidance is performed a week later and yields a pathologic diagnosis of hemangioma. The improvement, however, is short-lived, and his pain and dyspnea recur after 2 months. A follow-up echocardiogram is ordered.
A remarkable echocardiographic finding
The original echocardiogram that was performed a little over 2-1/2 months ago is re-reviewed. It very subtly suggests a complexity to the pericardial effusion in the area of the current mass, apparent only when the two studies are directly compared. Clearly, there has been interval development of a mass easily detectable by echocardiography. Although a small mass may have been obscured by the pericardial effusion in the original echocardiogram, the development of a mass of this size in such a short time suggests a rapidly growing tumor.
CARDIAC TUMORS
5. Which is the most common primary cardiac tumor?
- Myxoma
- Papillary fibroelastoma
- Sarcoma
- Lymphoma
Primary cardiac tumors are rare, with an incidence on autopsy series ranging between 0.0017% and 0.33%,11,12 making them far less common than metastases to the heart.
Myxomas are benign cardiac tumors and are the most common primary cardiac neoplasm. Approximately 80% of myxomas originate in the left atrium, typically presenting with one or more of the triad of intracardiac obstruction, systemic embolization, and constitutional symptoms.14
Cardiac papillary fibroelastomas, the second most common cardiac tumors, are benign and predominantly affect the cardiac valves.15
Only one-fourth of all cardiac tumors are malignant. Nearly all of these malignant tumors are sarcomas, with angiosarcoma being the most common morphologic type, accounting for 30% of primary cardiac sarcomas.13
Primary cardiac lymphomas are extremely rare and account for only 1.3% of all primary cardiac tumors.16
A DIAGNOSIS IS MADE
CARDIAC ANGIOSARCOMA
Cardiac angiosarcoma, the most common malignant primary cardiac tumor, has a predilection for the right atrium.13 These tumors tend to occur between the third and fifth decade of life and are three times more common in men than in women. Cardiac sarcomas proliferate rapidly and commonly extend into the pericardial space, causing pericardial effusion in up to one-fourth of patients.
Surgical resection is the treatment of choice, but due to the location and extent of involvement, complete resection is often difficult. Also, distant metastases are present at the time of diagnosis in 80% of cases, precluding a surgical cure.17 Adjuvant chemotherapy, radiotherapy, and even heart transplantation do not substantially improve the survival of these patients.18–20 Because no effective treatment is available, the prognosis is dismal, with a median survival of 6 to 12 months.
Our patient is discharged home to follow up with an oncologist and initiate chemotherapy.
Acknowledgment: We thank Lisa M. Yerian, MD, for interpreting the biopsy specimens described in this article.
- Ariyarajah V, Spodick DH. Acute pericarditis: diagnostic cues and common electrocardiographic manifestations. Cardiol Rev 2007; 15:24–30.
- LeWinter MM, Kabbani S. Pericardial disease. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1757–1781.
- Karjalainen J, Heikkila J. “Acute pericarditis”: myocardial enzyme release as evidence for myocarditis”. Am Heart J 1986; 111:546–552.
- Brandt RR, Filzmaier K, Hanrath P. Circulating cardiac troponin I in acute pericarditis. Am J Cardiol 2001; 87:1326–1328.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease: the present and the future. J Am Coll Cardiol 2006; 48:1–11.
- Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol 1995; 75:378–382.
- Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation 2007; 115:2739–2744.
- Levy PY, Corey R, Berger P, et al. Etiologic diagnosis of 204 pericardial effusions. Medicine (Baltimore) 2003; 82:385–391.
- Lange RA, Hillis LD. Clinical practice. Acute pericarditis. N Engl J Med 2004; 351:2195–2202.
- Imazio M, Bobbio M, Cecchi E, et al. Colchicine in addition to conventional therapy for acute pericarditis: Results of the COlchicine for acute PEricarditis (COPE) trial. Circulation 2005; 112:2012–2016.
- Heath D. Pathology of cardiac tumors. Am J Cardiol 1968; 21:315–327.
- Wold LE, Lie JT. Cardiac myxomas: a clinicopathologic profile. Am J Pathol 1980; 101:219–240.
- Sabatine MS, Colucci WS, Schoen FS. Primary tumors of the heart. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1741–1757.
- Gowda RM, Khan IA, Nair CK, Mehta NJ, Vasavada BC, Sacchi TJ. Cardiac papillary fibroelastoma: a comprehensive analysis of 725 cases. Am Heart J 2003; 146:404–410.
- Glancy DL, Morales JB, Roberts WC. Angiosarcoma of the heart. Am J Cardiol 1968; 21:413–419.
- Ceresoli GL, Ferreri AJ, Bucci E, Ripa C, Ponzoni M, Villa E. Primary cardiac lymphoma in immunocompetent patients: diagnostic and therapeutic management. Cancer 1997; 80:1497–1506.
- Bear PA, Moodie DS. Malignant primary cardiac tumors. The Cleveland Clinic experience, 1956 to 1986. Chest 1987; 92:860–862.
- Llombart–Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer 1998; 78:1624–1628.
- Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DA. Primary cardiac sarcomas. Ann Thorac Surg 1991; 51:906–910.
- Conklin LD, Reardon MJ. Autotransplantation of the heart for primary cardiac malignancy: development and surgical technique. Tex Heart Inst J 2002; 29:105–108.
- Ariyarajah V, Spodick DH. Acute pericarditis: diagnostic cues and common electrocardiographic manifestations. Cardiol Rev 2007; 15:24–30.
- LeWinter MM, Kabbani S. Pericardial disease. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1757–1781.
- Karjalainen J, Heikkila J. “Acute pericarditis”: myocardial enzyme release as evidence for myocarditis”. Am Heart J 1986; 111:546–552.
- Brandt RR, Filzmaier K, Hanrath P. Circulating cardiac troponin I in acute pericarditis. Am J Cardiol 2001; 87:1326–1328.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease: the present and the future. J Am Coll Cardiol 2006; 48:1–11.
- Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol 1995; 75:378–382.
- Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation 2007; 115:2739–2744.
- Levy PY, Corey R, Berger P, et al. Etiologic diagnosis of 204 pericardial effusions. Medicine (Baltimore) 2003; 82:385–391.
- Lange RA, Hillis LD. Clinical practice. Acute pericarditis. N Engl J Med 2004; 351:2195–2202.
- Imazio M, Bobbio M, Cecchi E, et al. Colchicine in addition to conventional therapy for acute pericarditis: Results of the COlchicine for acute PEricarditis (COPE) trial. Circulation 2005; 112:2012–2016.
- Heath D. Pathology of cardiac tumors. Am J Cardiol 1968; 21:315–327.
- Wold LE, Lie JT. Cardiac myxomas: a clinicopathologic profile. Am J Pathol 1980; 101:219–240.
- Sabatine MS, Colucci WS, Schoen FS. Primary tumors of the heart. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1741–1757.
- Gowda RM, Khan IA, Nair CK, Mehta NJ, Vasavada BC, Sacchi TJ. Cardiac papillary fibroelastoma: a comprehensive analysis of 725 cases. Am Heart J 2003; 146:404–410.
- Glancy DL, Morales JB, Roberts WC. Angiosarcoma of the heart. Am J Cardiol 1968; 21:413–419.
- Ceresoli GL, Ferreri AJ, Bucci E, Ripa C, Ponzoni M, Villa E. Primary cardiac lymphoma in immunocompetent patients: diagnostic and therapeutic management. Cancer 1997; 80:1497–1506.
- Bear PA, Moodie DS. Malignant primary cardiac tumors. The Cleveland Clinic experience, 1956 to 1986. Chest 1987; 92:860–862.
- Llombart–Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer 1998; 78:1624–1628.
- Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DA. Primary cardiac sarcomas. Ann Thorac Surg 1991; 51:906–910.
- Conklin LD, Reardon MJ. Autotransplantation of the heart for primary cardiac malignancy: development and surgical technique. Tex Heart Inst J 2002; 29:105–108.