User login
Expanding treatment options for diverse neuroendocrine tumors
Neuroendocrine tumors (NETs) are an extremely diverse group of cancers that have steadily increased in incidence in recent years. They can prove challenging to treat but, as we discuss here, a steady evolution in our understanding of NETs has significantly expanded the scope of therapeutic options.
A unique tumor type
NETs arise from neuroendocrine cells – cells with features of both nerve and endocrine cells that have important physiological functions, including the production and release of hormones. These tumors were first recognized by a German pathologist in the mid-1800s and were initially referred to as carcinoids in reference to their carcinoma-like appearance but lack of other malignant features.1
Unlike other solid tumors, which are associated with a particular primary location, NETs can arise anywhere in the body where neuroendocrine cells are found. They are also unique in their ability to oversecrete bioactive substances that regulate bodily functions, which results in an associated clinical syndrome, known as carcinoid syndrome, in up to 35% of patients.2,3
Although they are considered to be a relatively rare type of tumor, the incidence of NETs has been increasing in recent years. According to data from the Surveillance, Epidemiology and End Results (SEER) program, the age-adjusted incidence of NETs increased more than two-and-a-half fold during 1973-2004 and the rise is predicted to continue at an accelerated rate.4
Historically, NETs have been thought of as relatively benign because of their slow-growing nature, but it is now widely appreciated that they often metastasize. Furthermore, many patients are not symptomatic at first, so around half of all cases are not diagnosed until they have reached this more aggressive stage.4
The challenge of NET diversity
The most common type of NETs are those that arise in the gastrointestinal tract (GI-NET), representing more than 65% of cases, and for which the “carcinoid” terminology often is still applied. GI-NETs most frequently arise in the small intestine (41.8%), rectum (27.4%), and stomach (8.7%).4,5
About a quarter of NETs originate in the bronchopulmonary system, including the lungs and the thymus. Thymic NETs are particularly aggressive and are associated with a poor prognosis. Pancreatic NETs (pNETs) make up the next largest group, although they represent less than 1% of total NETs. Compared with the most common type of pancreatic cancer, pancreatic ductal adenocarcinoma, they have a more favorable prognosis. pNETs are often grouped together with GI-NETs and referred to as gastroenteropancreatic NETs (GEP-NETs).3-5 Other rarer types of NET include Merkel cell carcinoma (a type of skin cancer) and medullary thyroid cancers.
The classification network
NETs are classified according to the anatomic site from which they originate, as well as their histology, grade, and stage. Another important consideration is their level of hormone secretion. “Functional” and “nonfunctional” NETs both produce hormones, but only the former cause related symptoms.3,4,6
Functionality plays a particularly important role in the subclassification of GEP-NETs. Functional pNETs, for instance, are further divided according to the clinical syndromes associated with the hormones they produce, as insulinomas, glucagonomas, gastrinomas, somatostatinomas, and VIPomas (producing vasoactive intestinal peptide).7,8
In 2010, the World Health Organization developed a classification system for GEP-NETs that categorized these tumors as well differentiated (grade 1 or 2, depending on their rate of proliferation) and poorly differentiated (grade 3).9 The WHO classification of bronchopulmonary NETs, published in 2015, is slightly different; broken down into 3 subgroups, typical carcinoid, atypical carcinoid (corresponding to grade 1 and 2 GEP-NETs), and large and small-cell NETs (equivalent to grade 3 GEP-NETs).10
Although NETs develop from the same cell type, they in fact comprise a spectrum of diseases that vary extensively in their underlying biology, histology, and clinical behavior. Both the diversity and unique nature of NETs have become increasingly evident in recent years with the application of next-generation sequencing technologies to this tumor type. In general, NETs seem to be more genetically stable than other tumor types from the same primary location, and have fewer somatic mutations. The classic tumor suppressors and oncogenes that drive other tumor types are not common in NETs.6,11
The diversity of NETs presents a diagnostic and therapeutic challenge and, until recently, there was a paucity of effective treatment options. In the past decade, an evolution in our understanding of the molecular mechanisms underlying these tumors has altered the treatment landscape for well-differentiated tumors as an expanding array of targeted therapies with proven efficacy have become available (Table 1).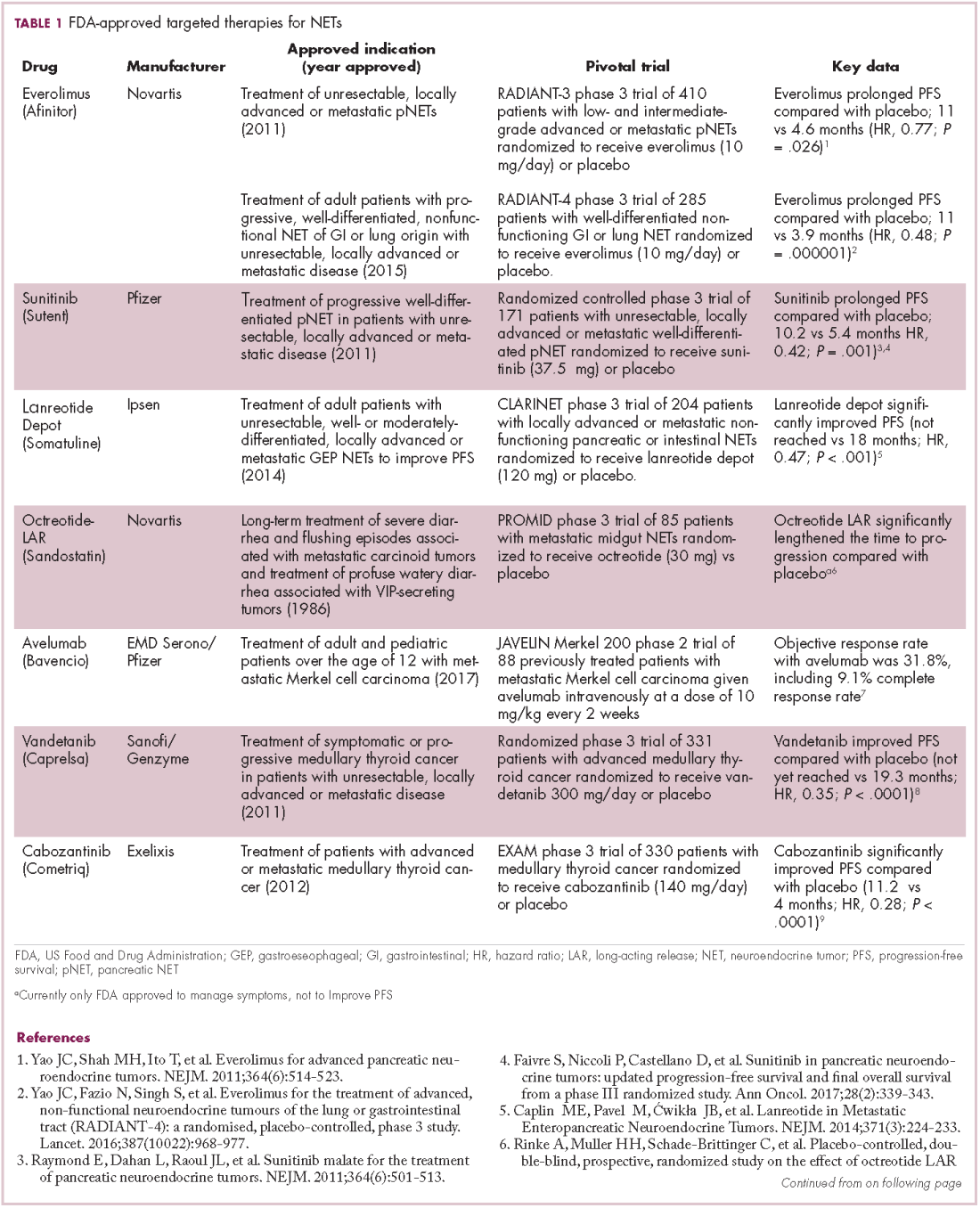

Their poorly differentiated counterparts, on the other hand, continue to present a significant unmet need.
Somatostatin analogs lead the charge
The fact that many NETs overexpress hormone receptors presents a significant therapeutic opportunity, and among the most successful targets to date are the somatostatin receptors (SSTRs). There are 5 main SSTRs that each bind to somatostatin with different effects on cell signaling and expression that varies according to the type of NET.
More stable synthetic analogs of the somatostatin hormone (somatostatin analogs [SSAs]), which has a very short half-life in the circulation, have been developed that act as SSTR agonists. Two long-acting SSAs, octreotide (Sandostatin LAR Depot) and lanreotide (Somatuline Depot), which bind SSTR2 and SSTR5, have been approved by the United States Food and Drug Administration (FDA), but were primarily used for the alleviation of the symptoms associated with NETs resulting from carcinoid syndrome.
In recent years, evidence has begun to emerge that SSAs also have an anti-tumor effect, which is thought to be both direct and indirect in nature. Direct effects result from the interaction between the SSA and SSTRs expressed on tumor cells, blocking the protumor cellular effects of SSTR signaling that are poorly understood but thought to involve the mitogen-activated protein kinase (MAPK) pathway. Indirect effects are fortuitous side effects mediated through off-target effects, such as the suppression of other cellular activities of SSTRs and the other growth factors that they bind to, which can impact processes such as angiogenesis and immune modulation.7,12
Several clinical trials have been designed to test the anti-tumor effects of NETs, including the PROMID trial of octreotide and the CLARINET trial of lanreotide, the latter leading to the 2014 approval of lanreotide for the improvement of progression-free survival (PFS) in patients with advanced GI- and pNETs.
The randomized phase 3 study compared lanreotide 120 mg with placebo in 204 patients with locally advanced or metastatic nonfunctioning pancreatic or intestinal NETs. Lanreotide treatment resulted in a significant improvement in PFS (Not yet reached vs 18 months for placebo; hazard ratio [HR], 0.47; P < .001).13
Meanwhile, the PROMID trial compared octreotide 30 mg with placebo in 85 patients with advanced midgut NETs and demonstrated that octreotide increased time to progression (TTP; 14.3 months vs 6 months for placebo; P = .000072) with no significant difference in side effects.14
Pasireotide is a second-generation SSA with improved binding affinity to SSTR1, 3, and 5. Despite its improved specificity, pasireotide has not proved more effective than other SSAs and its development for the treatment of NETs has been discontinued.
Coupling radioisotopes to SSAs provides another promising therapeutic option for NETs, known as peptide receptor radionuclide therapy, or PRRT, which uses SSAs to deliver therapeutic radiation directly to the tumor cells. Several variations have been studied with different radioactive isotopes, but most promising is lutetium-177 (177Lu). A 177Lu-labelled octreotide (177Lu-Dotatate) recently demonstrated significant efficacy in the phase 3 NETTER-1 clinical trial in patients with advanced stage NETs of the small bowel. The trial randomly assigned 229 patients who were progressing on an SSA to either 177Lu-Dotatate or high-dose octreotide LAR (long-acting release). There was a significant increase in PFS in the 177Lu-Dotatate arm (Not yet reached vs 8.4 months; P < .0001). There was also a trend toward improved overall survival (OS), and longer follow-up is eagerly anticipated for confirmation. 177Lu-Dotatate has been granted priority review by the FDA, and a decision on its approval is expected in the next few months.11,15-17
Molecularly and immune-targeted therapies continue to take aim
The mammalian target of rapamycin, or mTOR, is a serine/threonine kinase that sits at the confluence of a number of different upstream signaling pathways and mediates key cellular processes including cell proliferation and survival (Figure 1).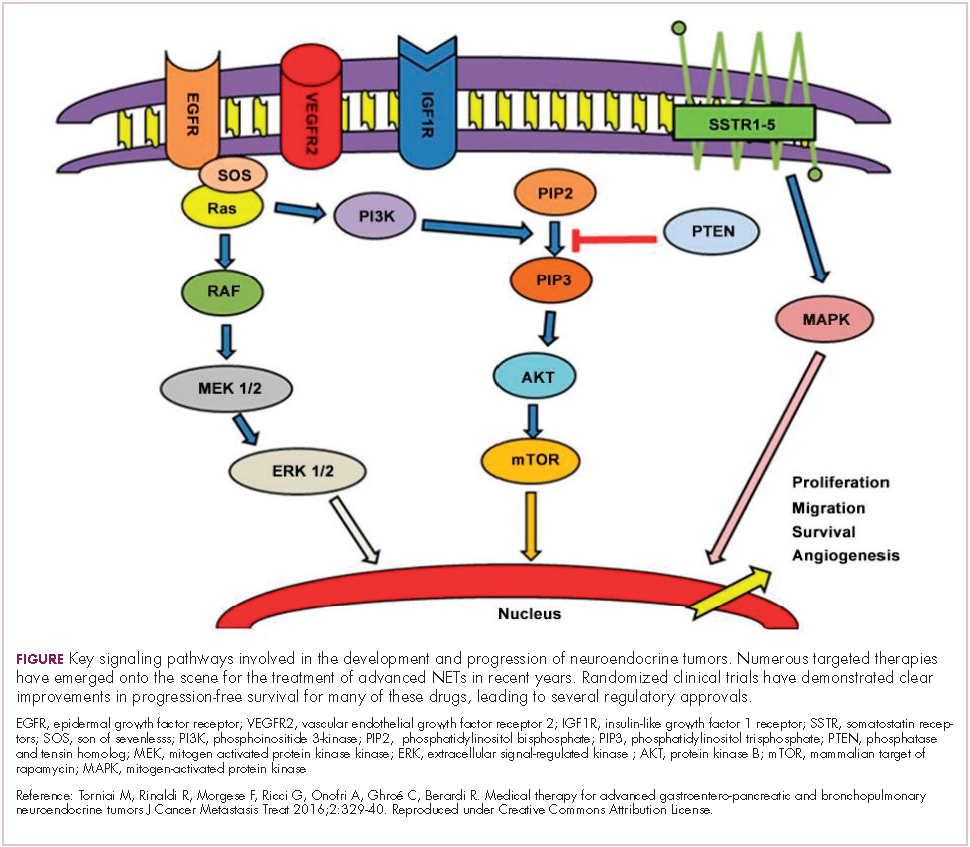
Alterations in nearly all members of the mTOR pathway, including upstream activators and downstream effectors, have been observed in NETs, in both sporadic disease and the genetic syndromes associated with the development of NETs.18
The involvement of the mTOR pathway in the pathogenesis of NETs first came into focus in pNETs and the mTOR inhibitor, everolimus (Afinitor) has been extensively studied in this indication, culminating in its regulatory approval in 2011. In the pivotal trial (RADIANT-3), everolimus monotherapy was compared with placebo in 410 patients with low- and intermediate-grade pNETs. There was a statistically significant improvement in PFS from 4.6 months to 11 months (HR, 0.77; P = .026).19 The final OS analysis for this trial also revealed a benefit of more than 6 months in the everolimus arm, although this was not statistically significant, which the study authors attribute to the high rate of crossover from the placebo arm after progression.20
More recently, the results of the RADIANT-4 trial, in which everolimus was compare with placebo in patients with advanced, well-differentiated, nonfunctioning NETs of the GI tract and lung, led to a new approved indication for the mTOR inhibitor and the first approved targeted therapy for advanced lung NETs. In the overall study population (n = 285), everolimus prolonged PFS by more than 7 months (11 months vs 3.9 months for placebo; HR, 0.48; P = .000001), corresponding to a 52% reduction in the risk of disease progression or death.21,22
Everolimus continues to be evaluated, with a particular focus on combination therapy to overcome the resistance that commonly occurs after treatment with molecularly targeted drugs (Table 2). For example, preclinical studies suggested that mTOR inhibitors and SSAs may have synergistic activity owing to combined inhibition of the mTOR and insulin-like growth factor pathways. In a phase 1 study, the combination of pasireotide and everolimus was found to be safe and to have preliminary anti-tumor activity. However, the subsequent phase 2 COOPERATE-2 study failed to show improved PFS.23,24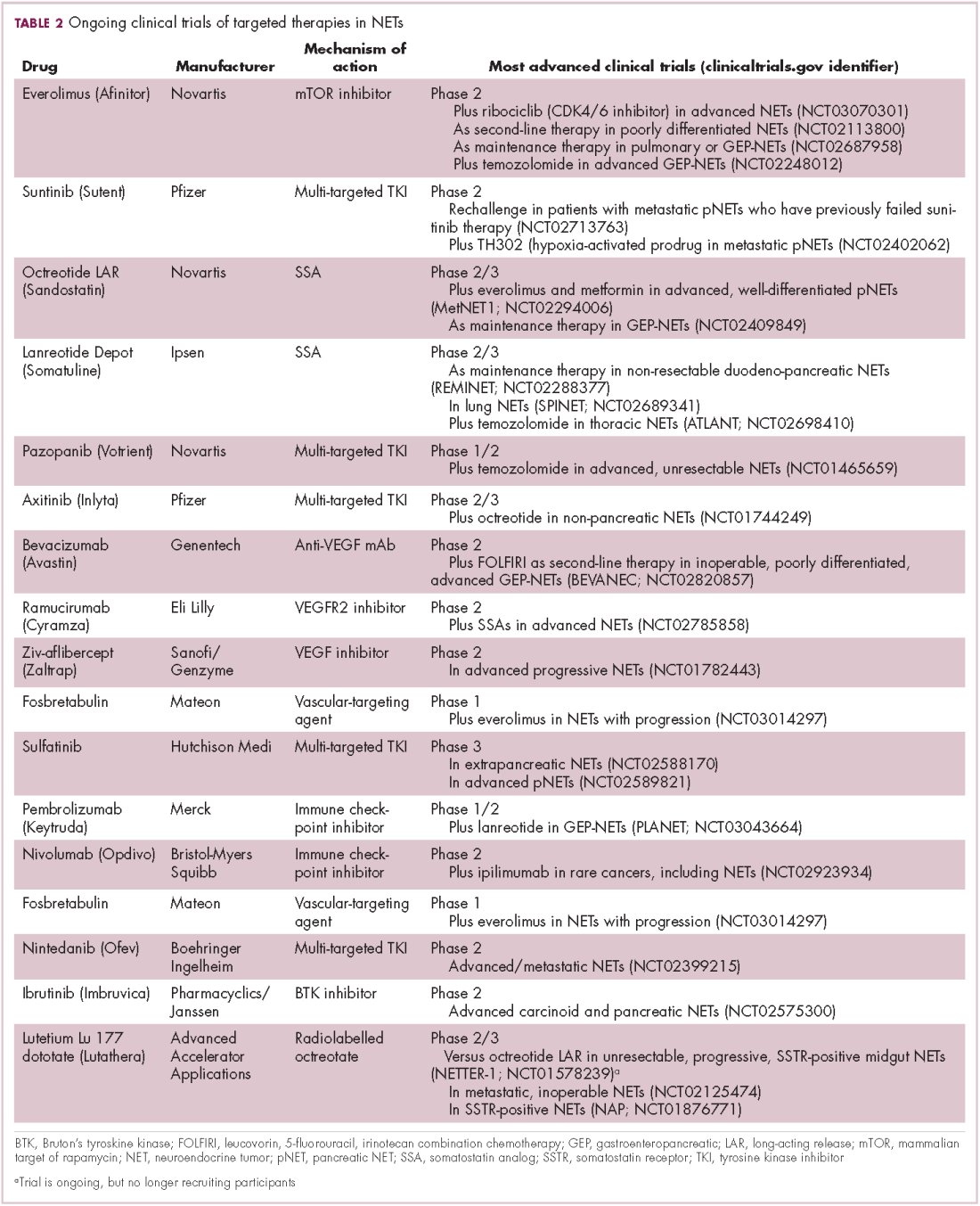
The observation that NETs are highly vascularized and frequently express vascular endothelial growth factor (VEGF) and its receptor (VEGFR), which play a key role in coordinating angiogenesis, led to the pursuit of anti-angiogenic therapies in NETs. Both the anti-VEGF monoclonal antibody bevacizumab and small molecule tyrosine kinase inhibitors that include among their targets VEGFRs and other receptors involved in angiogenesis, such as platelet-derived growth factor receptor, have been tested.
Sunitinib was approved for the treatment of pNETs in 2011, making it a banner year for this tumor type. Approval was granted on the basis of significantly improved PFS in the sunitinib arm of a phase 3 randomized trial, but long-term follow-up suggested that sunitinib also improved OS by 10 months. Like everolimus, the OS benefit was not statistically significant, and again this was thought to be the result of extensive crossover.
Two other multikinase inhibitors have received regulatory approval for a much rarer form of NET, medullary thyroid cancer. Vandetanib and cabozantinib were approved for this indication in 2011 and 2012, respectively. Early in 2017, the results of a single-arm phase 2 trial of cabozantinib suggested that this drug may also have significant activity in other types of NET. In patients with advanced carcinoid and pNETs who received cabozantinib at 60 mg/day orally, partial responses were observed in 15% of patients and the median PFS was 21.8 months in the pNET cohort and >30 months in the carcinoid tumor cohort.25 Confirmatory phase 3 trials are planned but not currently underway.
Sulfatinib is a novel kinase inhibitor that targets the VEGFRs and fibroblast growth factor receptor 1. It has recently shown significant promise in the treatment of patients with advanced NETs. According to data presented at this year’s annual conference of the European Neuroendocrine Tumor Society in Barcelona, sulfatinib demonstrated an overall response rate of 17.1% in pancreatic NETs and 15% in extra-pancreatic NETs, with an overall disease control rate of 91.4%, and was well tolerated.26 Based on these and other promising phase 1 and 2 data, 2 phase 3 trials are ongoing.
Meanwhile, earlier this year, Mateon Therapeutics presented data from a phase 2 trial of a different kind of anti-angiogenic drug in patients with GI- or pNETs. Fosbretabulin is a vascular disrupting agent that targets the existing tumor vasculature rather than preventing the formation of new blood vessels. They do this via a number of different mechanisms, in the case of fosbretabulin it specifically targets endothelial cells and inhibits the assembly of microtubules and, hence, blocks mitosis. In 18 patients, fosbretabulin treatment resulted in 1 partial response and 7 patients who had stable disease; more than half of the patients reported improved quality of life.27 Fosbretabulin continues to be studied in NETs in combination with everolimus.
Finally, researchers are beginning to make a foray into the immunotherapy field that has revolutionized the treatment of many other tumor types. The immune checkpoint inhibitors nivolumab and pembrolizumab are being evaluated in ongoing phase 1 and 2 trials, while avelumab (Bavencio) was very recently approved by the FDA for the treatment of Merkel cell carcinoma.28,29
1. Pinchot SN, Holen K, Sippel RS, Chen H. Carcinoid tumors. Oncologist. 2008;13(12):1255-1269.
2. Rorstad O. Prognostic indicators for carcinoid neuroendocrine tumors of the gastrointestinal tract. J Surg Oncol. 2005;89(3):151-160.
3. The NET Alliance. Characterizing a challenging cancer. http://www.thenetalliance.com/hcp/facts-about-net/characterization/. Publishing date not provided. Accessed October 18, 2017.
4. Yao JC, Hassan M, Phan A, et al. One hundred years after ‘carcinoid’: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063-3072.
5. Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97(4):934-959.
6. Spada F, Valente M. Review of recent advances in medical treatment for neuroendocrine neoplasms: somatostatin analogs and chemotherapy. J Cancer Metastasis Treat. 2016;2(8):313-320.
7. Kelgiorgi D, Dervenis C. Pancreatic neuroendocrine tumors: the basics, the gray zone, and the target. F1000Research. 2017;6:663.
8. Viudez A, De Jesus-Acosta A, Carvalho FL, Vera R, Martin-Algarra S, Ramirez N. Pancreatic neuroendocrine tumors: Challenges in an underestimated disease. Crit Rev Oncol Hematol. 2016;101:193-206.
9. World Health Organization, International Agency for Research on Cancer. Bosman FT, Carneiro F, Hruban RH, Theise ND (eds). WHO classification of tumours of the digestive system. 2010, 4th ed (vol 3).
10. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors. J Thorac Oncol. 2015;10(9):1243-1260.
11. Lee A, Chan DL, Wong MH, et al. Systematic review of the role of targeted therapy in metastatic neuroendocrine tumors. Neuroendocrinology. 2017;104(3):209-222.
12. Theodoropoulou M, Stalla GK. Somatostatin receptors: from signaling to clinical practice. Front Neuroendocrinol. 2013;34(3):228-252.
13. Caplin ME, Pavel M, Cwikła JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224-233.
14. Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656-4663.
15. Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125-135.
16. Falconi M, Partelli S. Neuroendocrine tumours in 2016: defining rules for increasingly personalized treatments. Nat Rev Clin Oncol. 2017;14(2):80-82.
17. Hutchinson L. Targeted therapies: widening the treatment NET. Nat Rev Clin Oncol. 2017;14(1):2-3.
18. Cingarlini S, Bonomi M, Corbo V, Scarpa A, Tortora G. Profiling mTOR pathway in neuroendocrine tumors. Target Oncol. 2012;7(3):183-188.
19. Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514-523.
20. Yao JC, Pavel M, Lombard-Bohas C, et al. Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: overall survival and circulating biomarkers from the randomized, phase III RADIANT-3 study. J Clin Oncol. http://ascopubs.org/ doi/abs/10.1200/JCO.2016.68.0702?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dpubmed. September 12, 2016. E-pub ahead of print.
21. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968-977.
22. Gajate P, Martínez-Sáez O, Alonso-Gordoa T, Grande E. Emerging use of everolimus in the treatment of neuroendocrine tumors. Cancer Manage Res. 2017;9:215-224.
23. Chan JA, Ryan DP, Zhu AX, et al. Phase I study of pasireotide (SOM 230) and everolimus (RAD001) in advanced neuroendocrine tumors. Endocr Relat Cancer. 2012;19(5):615-623.
24. Kulke MH, Ruszniewski P, Van Cutsem E, et al. A randomized, open-label, phase 2 study of everolimus in combination with pasireotide LAR or everolimus alone in advanced, well-dierentiated, progressive pancreatic neuroendocrine tumors: COOPERATE-2 trial. Ann Oncol. 2017;28(6):1309-1315.
25. Chan JA, Faris JE, Murphy JE, et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J Clin Oncol. 2017;35(4 suppl):228-228.
26. Xu J, Li J, Bai CM, et al. An open-label phase Ib/II study of sulfatinib in patients with advanced neuroendocrine tumors (NCT02267967). Paper presented at the 14th Annual European Neuroendocrine Tumor Society Conference for the Diagnosis and Treatment of Neuroendocrine Tumor Disease; March 8-10, 2017, Barcelona, Spain.
27. Libutti SK, Anthony LB, Chaplin DJ, Sosa JA. A phase II study of combretastatin A4-phosphate (CA4P) in the treatment of well-differentiated, low- to intermediate-grade, unresectable, recurrent, or metastatic pancreatic, or GI neuroendocrine tumors/carcinoid (GI-NETs/PNETs) with elevated biomarkers. J Clin Oncol. 2017;35(4 suppl):432-432.
28. Cordes LM, Gulley JL. Avelumab for the treatment of metastatic Merkel cell carcinoma. Drugs Today (Barc). 2017;53(7):377-383.
29. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374-1385.
Neuroendocrine tumors (NETs) are an extremely diverse group of cancers that have steadily increased in incidence in recent years. They can prove challenging to treat but, as we discuss here, a steady evolution in our understanding of NETs has significantly expanded the scope of therapeutic options.
A unique tumor type
NETs arise from neuroendocrine cells – cells with features of both nerve and endocrine cells that have important physiological functions, including the production and release of hormones. These tumors were first recognized by a German pathologist in the mid-1800s and were initially referred to as carcinoids in reference to their carcinoma-like appearance but lack of other malignant features.1
Unlike other solid tumors, which are associated with a particular primary location, NETs can arise anywhere in the body where neuroendocrine cells are found. They are also unique in their ability to oversecrete bioactive substances that regulate bodily functions, which results in an associated clinical syndrome, known as carcinoid syndrome, in up to 35% of patients.2,3
Although they are considered to be a relatively rare type of tumor, the incidence of NETs has been increasing in recent years. According to data from the Surveillance, Epidemiology and End Results (SEER) program, the age-adjusted incidence of NETs increased more than two-and-a-half fold during 1973-2004 and the rise is predicted to continue at an accelerated rate.4
Historically, NETs have been thought of as relatively benign because of their slow-growing nature, but it is now widely appreciated that they often metastasize. Furthermore, many patients are not symptomatic at first, so around half of all cases are not diagnosed until they have reached this more aggressive stage.4
The challenge of NET diversity
The most common type of NETs are those that arise in the gastrointestinal tract (GI-NET), representing more than 65% of cases, and for which the “carcinoid” terminology often is still applied. GI-NETs most frequently arise in the small intestine (41.8%), rectum (27.4%), and stomach (8.7%).4,5
About a quarter of NETs originate in the bronchopulmonary system, including the lungs and the thymus. Thymic NETs are particularly aggressive and are associated with a poor prognosis. Pancreatic NETs (pNETs) make up the next largest group, although they represent less than 1% of total NETs. Compared with the most common type of pancreatic cancer, pancreatic ductal adenocarcinoma, they have a more favorable prognosis. pNETs are often grouped together with GI-NETs and referred to as gastroenteropancreatic NETs (GEP-NETs).3-5 Other rarer types of NET include Merkel cell carcinoma (a type of skin cancer) and medullary thyroid cancers.
The classification network
NETs are classified according to the anatomic site from which they originate, as well as their histology, grade, and stage. Another important consideration is their level of hormone secretion. “Functional” and “nonfunctional” NETs both produce hormones, but only the former cause related symptoms.3,4,6
Functionality plays a particularly important role in the subclassification of GEP-NETs. Functional pNETs, for instance, are further divided according to the clinical syndromes associated with the hormones they produce, as insulinomas, glucagonomas, gastrinomas, somatostatinomas, and VIPomas (producing vasoactive intestinal peptide).7,8
In 2010, the World Health Organization developed a classification system for GEP-NETs that categorized these tumors as well differentiated (grade 1 or 2, depending on their rate of proliferation) and poorly differentiated (grade 3).9 The WHO classification of bronchopulmonary NETs, published in 2015, is slightly different; broken down into 3 subgroups, typical carcinoid, atypical carcinoid (corresponding to grade 1 and 2 GEP-NETs), and large and small-cell NETs (equivalent to grade 3 GEP-NETs).10
Although NETs develop from the same cell type, they in fact comprise a spectrum of diseases that vary extensively in their underlying biology, histology, and clinical behavior. Both the diversity and unique nature of NETs have become increasingly evident in recent years with the application of next-generation sequencing technologies to this tumor type. In general, NETs seem to be more genetically stable than other tumor types from the same primary location, and have fewer somatic mutations. The classic tumor suppressors and oncogenes that drive other tumor types are not common in NETs.6,11
The diversity of NETs presents a diagnostic and therapeutic challenge and, until recently, there was a paucity of effective treatment options. In the past decade, an evolution in our understanding of the molecular mechanisms underlying these tumors has altered the treatment landscape for well-differentiated tumors as an expanding array of targeted therapies with proven efficacy have become available (Table 1).
Their poorly differentiated counterparts, on the other hand, continue to present a significant unmet need.
Somatostatin analogs lead the charge
The fact that many NETs overexpress hormone receptors presents a significant therapeutic opportunity, and among the most successful targets to date are the somatostatin receptors (SSTRs). There are 5 main SSTRs that each bind to somatostatin with different effects on cell signaling and expression that varies according to the type of NET.
More stable synthetic analogs of the somatostatin hormone (somatostatin analogs [SSAs]), which has a very short half-life in the circulation, have been developed that act as SSTR agonists. Two long-acting SSAs, octreotide (Sandostatin LAR Depot) and lanreotide (Somatuline Depot), which bind SSTR2 and SSTR5, have been approved by the United States Food and Drug Administration (FDA), but were primarily used for the alleviation of the symptoms associated with NETs resulting from carcinoid syndrome.
In recent years, evidence has begun to emerge that SSAs also have an anti-tumor effect, which is thought to be both direct and indirect in nature. Direct effects result from the interaction between the SSA and SSTRs expressed on tumor cells, blocking the protumor cellular effects of SSTR signaling that are poorly understood but thought to involve the mitogen-activated protein kinase (MAPK) pathway. Indirect effects are fortuitous side effects mediated through off-target effects, such as the suppression of other cellular activities of SSTRs and the other growth factors that they bind to, which can impact processes such as angiogenesis and immune modulation.7,12
Several clinical trials have been designed to test the anti-tumor effects of NETs, including the PROMID trial of octreotide and the CLARINET trial of lanreotide, the latter leading to the 2014 approval of lanreotide for the improvement of progression-free survival (PFS) in patients with advanced GI- and pNETs.
The randomized phase 3 study compared lanreotide 120 mg with placebo in 204 patients with locally advanced or metastatic nonfunctioning pancreatic or intestinal NETs. Lanreotide treatment resulted in a significant improvement in PFS (Not yet reached vs 18 months for placebo; hazard ratio [HR], 0.47; P < .001).13
Meanwhile, the PROMID trial compared octreotide 30 mg with placebo in 85 patients with advanced midgut NETs and demonstrated that octreotide increased time to progression (TTP; 14.3 months vs 6 months for placebo; P = .000072) with no significant difference in side effects.14
Pasireotide is a second-generation SSA with improved binding affinity to SSTR1, 3, and 5. Despite its improved specificity, pasireotide has not proved more effective than other SSAs and its development for the treatment of NETs has been discontinued.
Coupling radioisotopes to SSAs provides another promising therapeutic option for NETs, known as peptide receptor radionuclide therapy, or PRRT, which uses SSAs to deliver therapeutic radiation directly to the tumor cells. Several variations have been studied with different radioactive isotopes, but most promising is lutetium-177 (177Lu). A 177Lu-labelled octreotide (177Lu-Dotatate) recently demonstrated significant efficacy in the phase 3 NETTER-1 clinical trial in patients with advanced stage NETs of the small bowel. The trial randomly assigned 229 patients who were progressing on an SSA to either 177Lu-Dotatate or high-dose octreotide LAR (long-acting release). There was a significant increase in PFS in the 177Lu-Dotatate arm (Not yet reached vs 8.4 months; P < .0001). There was also a trend toward improved overall survival (OS), and longer follow-up is eagerly anticipated for confirmation. 177Lu-Dotatate has been granted priority review by the FDA, and a decision on its approval is expected in the next few months.11,15-17
Molecularly and immune-targeted therapies continue to take aim
The mammalian target of rapamycin, or mTOR, is a serine/threonine kinase that sits at the confluence of a number of different upstream signaling pathways and mediates key cellular processes including cell proliferation and survival (Figure 1).
Alterations in nearly all members of the mTOR pathway, including upstream activators and downstream effectors, have been observed in NETs, in both sporadic disease and the genetic syndromes associated with the development of NETs.18
The involvement of the mTOR pathway in the pathogenesis of NETs first came into focus in pNETs and the mTOR inhibitor, everolimus (Afinitor) has been extensively studied in this indication, culminating in its regulatory approval in 2011. In the pivotal trial (RADIANT-3), everolimus monotherapy was compared with placebo in 410 patients with low- and intermediate-grade pNETs. There was a statistically significant improvement in PFS from 4.6 months to 11 months (HR, 0.77; P = .026).19 The final OS analysis for this trial also revealed a benefit of more than 6 months in the everolimus arm, although this was not statistically significant, which the study authors attribute to the high rate of crossover from the placebo arm after progression.20
More recently, the results of the RADIANT-4 trial, in which everolimus was compare with placebo in patients with advanced, well-differentiated, nonfunctioning NETs of the GI tract and lung, led to a new approved indication for the mTOR inhibitor and the first approved targeted therapy for advanced lung NETs. In the overall study population (n = 285), everolimus prolonged PFS by more than 7 months (11 months vs 3.9 months for placebo; HR, 0.48; P = .000001), corresponding to a 52% reduction in the risk of disease progression or death.21,22
Everolimus continues to be evaluated, with a particular focus on combination therapy to overcome the resistance that commonly occurs after treatment with molecularly targeted drugs (Table 2). For example, preclinical studies suggested that mTOR inhibitors and SSAs may have synergistic activity owing to combined inhibition of the mTOR and insulin-like growth factor pathways. In a phase 1 study, the combination of pasireotide and everolimus was found to be safe and to have preliminary anti-tumor activity. However, the subsequent phase 2 COOPERATE-2 study failed to show improved PFS.23,24
The observation that NETs are highly vascularized and frequently express vascular endothelial growth factor (VEGF) and its receptor (VEGFR), which play a key role in coordinating angiogenesis, led to the pursuit of anti-angiogenic therapies in NETs. Both the anti-VEGF monoclonal antibody bevacizumab and small molecule tyrosine kinase inhibitors that include among their targets VEGFRs and other receptors involved in angiogenesis, such as platelet-derived growth factor receptor, have been tested.
Sunitinib was approved for the treatment of pNETs in 2011, making it a banner year for this tumor type. Approval was granted on the basis of significantly improved PFS in the sunitinib arm of a phase 3 randomized trial, but long-term follow-up suggested that sunitinib also improved OS by 10 months. Like everolimus, the OS benefit was not statistically significant, and again this was thought to be the result of extensive crossover.
Two other multikinase inhibitors have received regulatory approval for a much rarer form of NET, medullary thyroid cancer. Vandetanib and cabozantinib were approved for this indication in 2011 and 2012, respectively. Early in 2017, the results of a single-arm phase 2 trial of cabozantinib suggested that this drug may also have significant activity in other types of NET. In patients with advanced carcinoid and pNETs who received cabozantinib at 60 mg/day orally, partial responses were observed in 15% of patients and the median PFS was 21.8 months in the pNET cohort and >30 months in the carcinoid tumor cohort.25 Confirmatory phase 3 trials are planned but not currently underway.
Sulfatinib is a novel kinase inhibitor that targets the VEGFRs and fibroblast growth factor receptor 1. It has recently shown significant promise in the treatment of patients with advanced NETs. According to data presented at this year’s annual conference of the European Neuroendocrine Tumor Society in Barcelona, sulfatinib demonstrated an overall response rate of 17.1% in pancreatic NETs and 15% in extra-pancreatic NETs, with an overall disease control rate of 91.4%, and was well tolerated.26 Based on these and other promising phase 1 and 2 data, 2 phase 3 trials are ongoing.
Meanwhile, earlier this year, Mateon Therapeutics presented data from a phase 2 trial of a different kind of anti-angiogenic drug in patients with GI- or pNETs. Fosbretabulin is a vascular disrupting agent that targets the existing tumor vasculature rather than preventing the formation of new blood vessels. They do this via a number of different mechanisms, in the case of fosbretabulin it specifically targets endothelial cells and inhibits the assembly of microtubules and, hence, blocks mitosis. In 18 patients, fosbretabulin treatment resulted in 1 partial response and 7 patients who had stable disease; more than half of the patients reported improved quality of life.27 Fosbretabulin continues to be studied in NETs in combination with everolimus.
Finally, researchers are beginning to make a foray into the immunotherapy field that has revolutionized the treatment of many other tumor types. The immune checkpoint inhibitors nivolumab and pembrolizumab are being evaluated in ongoing phase 1 and 2 trials, while avelumab (Bavencio) was very recently approved by the FDA for the treatment of Merkel cell carcinoma.28,29
Neuroendocrine tumors (NETs) are an extremely diverse group of cancers that have steadily increased in incidence in recent years. They can prove challenging to treat but, as we discuss here, a steady evolution in our understanding of NETs has significantly expanded the scope of therapeutic options.
A unique tumor type
NETs arise from neuroendocrine cells – cells with features of both nerve and endocrine cells that have important physiological functions, including the production and release of hormones. These tumors were first recognized by a German pathologist in the mid-1800s and were initially referred to as carcinoids in reference to their carcinoma-like appearance but lack of other malignant features.1
Unlike other solid tumors, which are associated with a particular primary location, NETs can arise anywhere in the body where neuroendocrine cells are found. They are also unique in their ability to oversecrete bioactive substances that regulate bodily functions, which results in an associated clinical syndrome, known as carcinoid syndrome, in up to 35% of patients.2,3
Although they are considered to be a relatively rare type of tumor, the incidence of NETs has been increasing in recent years. According to data from the Surveillance, Epidemiology and End Results (SEER) program, the age-adjusted incidence of NETs increased more than two-and-a-half fold during 1973-2004 and the rise is predicted to continue at an accelerated rate.4
Historically, NETs have been thought of as relatively benign because of their slow-growing nature, but it is now widely appreciated that they often metastasize. Furthermore, many patients are not symptomatic at first, so around half of all cases are not diagnosed until they have reached this more aggressive stage.4
The challenge of NET diversity
The most common type of NETs are those that arise in the gastrointestinal tract (GI-NET), representing more than 65% of cases, and for which the “carcinoid” terminology often is still applied. GI-NETs most frequently arise in the small intestine (41.8%), rectum (27.4%), and stomach (8.7%).4,5
About a quarter of NETs originate in the bronchopulmonary system, including the lungs and the thymus. Thymic NETs are particularly aggressive and are associated with a poor prognosis. Pancreatic NETs (pNETs) make up the next largest group, although they represent less than 1% of total NETs. Compared with the most common type of pancreatic cancer, pancreatic ductal adenocarcinoma, they have a more favorable prognosis. pNETs are often grouped together with GI-NETs and referred to as gastroenteropancreatic NETs (GEP-NETs).3-5 Other rarer types of NET include Merkel cell carcinoma (a type of skin cancer) and medullary thyroid cancers.
The classification network
NETs are classified according to the anatomic site from which they originate, as well as their histology, grade, and stage. Another important consideration is their level of hormone secretion. “Functional” and “nonfunctional” NETs both produce hormones, but only the former cause related symptoms.3,4,6
Functionality plays a particularly important role in the subclassification of GEP-NETs. Functional pNETs, for instance, are further divided according to the clinical syndromes associated with the hormones they produce, as insulinomas, glucagonomas, gastrinomas, somatostatinomas, and VIPomas (producing vasoactive intestinal peptide).7,8
In 2010, the World Health Organization developed a classification system for GEP-NETs that categorized these tumors as well differentiated (grade 1 or 2, depending on their rate of proliferation) and poorly differentiated (grade 3).9 The WHO classification of bronchopulmonary NETs, published in 2015, is slightly different; broken down into 3 subgroups, typical carcinoid, atypical carcinoid (corresponding to grade 1 and 2 GEP-NETs), and large and small-cell NETs (equivalent to grade 3 GEP-NETs).10
Although NETs develop from the same cell type, they in fact comprise a spectrum of diseases that vary extensively in their underlying biology, histology, and clinical behavior. Both the diversity and unique nature of NETs have become increasingly evident in recent years with the application of next-generation sequencing technologies to this tumor type. In general, NETs seem to be more genetically stable than other tumor types from the same primary location, and have fewer somatic mutations. The classic tumor suppressors and oncogenes that drive other tumor types are not common in NETs.6,11
The diversity of NETs presents a diagnostic and therapeutic challenge and, until recently, there was a paucity of effective treatment options. In the past decade, an evolution in our understanding of the molecular mechanisms underlying these tumors has altered the treatment landscape for well-differentiated tumors as an expanding array of targeted therapies with proven efficacy have become available (Table 1).
Their poorly differentiated counterparts, on the other hand, continue to present a significant unmet need.
Somatostatin analogs lead the charge
The fact that many NETs overexpress hormone receptors presents a significant therapeutic opportunity, and among the most successful targets to date are the somatostatin receptors (SSTRs). There are 5 main SSTRs that each bind to somatostatin with different effects on cell signaling and expression that varies according to the type of NET.
More stable synthetic analogs of the somatostatin hormone (somatostatin analogs [SSAs]), which has a very short half-life in the circulation, have been developed that act as SSTR agonists. Two long-acting SSAs, octreotide (Sandostatin LAR Depot) and lanreotide (Somatuline Depot), which bind SSTR2 and SSTR5, have been approved by the United States Food and Drug Administration (FDA), but were primarily used for the alleviation of the symptoms associated with NETs resulting from carcinoid syndrome.
In recent years, evidence has begun to emerge that SSAs also have an anti-tumor effect, which is thought to be both direct and indirect in nature. Direct effects result from the interaction between the SSA and SSTRs expressed on tumor cells, blocking the protumor cellular effects of SSTR signaling that are poorly understood but thought to involve the mitogen-activated protein kinase (MAPK) pathway. Indirect effects are fortuitous side effects mediated through off-target effects, such as the suppression of other cellular activities of SSTRs and the other growth factors that they bind to, which can impact processes such as angiogenesis and immune modulation.7,12
Several clinical trials have been designed to test the anti-tumor effects of NETs, including the PROMID trial of octreotide and the CLARINET trial of lanreotide, the latter leading to the 2014 approval of lanreotide for the improvement of progression-free survival (PFS) in patients with advanced GI- and pNETs.
The randomized phase 3 study compared lanreotide 120 mg with placebo in 204 patients with locally advanced or metastatic nonfunctioning pancreatic or intestinal NETs. Lanreotide treatment resulted in a significant improvement in PFS (Not yet reached vs 18 months for placebo; hazard ratio [HR], 0.47; P < .001).13
Meanwhile, the PROMID trial compared octreotide 30 mg with placebo in 85 patients with advanced midgut NETs and demonstrated that octreotide increased time to progression (TTP; 14.3 months vs 6 months for placebo; P = .000072) with no significant difference in side effects.14
Pasireotide is a second-generation SSA with improved binding affinity to SSTR1, 3, and 5. Despite its improved specificity, pasireotide has not proved more effective than other SSAs and its development for the treatment of NETs has been discontinued.
Coupling radioisotopes to SSAs provides another promising therapeutic option for NETs, known as peptide receptor radionuclide therapy, or PRRT, which uses SSAs to deliver therapeutic radiation directly to the tumor cells. Several variations have been studied with different radioactive isotopes, but most promising is lutetium-177 (177Lu). A 177Lu-labelled octreotide (177Lu-Dotatate) recently demonstrated significant efficacy in the phase 3 NETTER-1 clinical trial in patients with advanced stage NETs of the small bowel. The trial randomly assigned 229 patients who were progressing on an SSA to either 177Lu-Dotatate or high-dose octreotide LAR (long-acting release). There was a significant increase in PFS in the 177Lu-Dotatate arm (Not yet reached vs 8.4 months; P < .0001). There was also a trend toward improved overall survival (OS), and longer follow-up is eagerly anticipated for confirmation. 177Lu-Dotatate has been granted priority review by the FDA, and a decision on its approval is expected in the next few months.11,15-17
Molecularly and immune-targeted therapies continue to take aim
The mammalian target of rapamycin, or mTOR, is a serine/threonine kinase that sits at the confluence of a number of different upstream signaling pathways and mediates key cellular processes including cell proliferation and survival (Figure 1).
Alterations in nearly all members of the mTOR pathway, including upstream activators and downstream effectors, have been observed in NETs, in both sporadic disease and the genetic syndromes associated with the development of NETs.18
The involvement of the mTOR pathway in the pathogenesis of NETs first came into focus in pNETs and the mTOR inhibitor, everolimus (Afinitor) has been extensively studied in this indication, culminating in its regulatory approval in 2011. In the pivotal trial (RADIANT-3), everolimus monotherapy was compared with placebo in 410 patients with low- and intermediate-grade pNETs. There was a statistically significant improvement in PFS from 4.6 months to 11 months (HR, 0.77; P = .026).19 The final OS analysis for this trial also revealed a benefit of more than 6 months in the everolimus arm, although this was not statistically significant, which the study authors attribute to the high rate of crossover from the placebo arm after progression.20
More recently, the results of the RADIANT-4 trial, in which everolimus was compare with placebo in patients with advanced, well-differentiated, nonfunctioning NETs of the GI tract and lung, led to a new approved indication for the mTOR inhibitor and the first approved targeted therapy for advanced lung NETs. In the overall study population (n = 285), everolimus prolonged PFS by more than 7 months (11 months vs 3.9 months for placebo; HR, 0.48; P = .000001), corresponding to a 52% reduction in the risk of disease progression or death.21,22
Everolimus continues to be evaluated, with a particular focus on combination therapy to overcome the resistance that commonly occurs after treatment with molecularly targeted drugs (Table 2). For example, preclinical studies suggested that mTOR inhibitors and SSAs may have synergistic activity owing to combined inhibition of the mTOR and insulin-like growth factor pathways. In a phase 1 study, the combination of pasireotide and everolimus was found to be safe and to have preliminary anti-tumor activity. However, the subsequent phase 2 COOPERATE-2 study failed to show improved PFS.23,24
The observation that NETs are highly vascularized and frequently express vascular endothelial growth factor (VEGF) and its receptor (VEGFR), which play a key role in coordinating angiogenesis, led to the pursuit of anti-angiogenic therapies in NETs. Both the anti-VEGF monoclonal antibody bevacizumab and small molecule tyrosine kinase inhibitors that include among their targets VEGFRs and other receptors involved in angiogenesis, such as platelet-derived growth factor receptor, have been tested.
Sunitinib was approved for the treatment of pNETs in 2011, making it a banner year for this tumor type. Approval was granted on the basis of significantly improved PFS in the sunitinib arm of a phase 3 randomized trial, but long-term follow-up suggested that sunitinib also improved OS by 10 months. Like everolimus, the OS benefit was not statistically significant, and again this was thought to be the result of extensive crossover.
Two other multikinase inhibitors have received regulatory approval for a much rarer form of NET, medullary thyroid cancer. Vandetanib and cabozantinib were approved for this indication in 2011 and 2012, respectively. Early in 2017, the results of a single-arm phase 2 trial of cabozantinib suggested that this drug may also have significant activity in other types of NET. In patients with advanced carcinoid and pNETs who received cabozantinib at 60 mg/day orally, partial responses were observed in 15% of patients and the median PFS was 21.8 months in the pNET cohort and >30 months in the carcinoid tumor cohort.25 Confirmatory phase 3 trials are planned but not currently underway.
Sulfatinib is a novel kinase inhibitor that targets the VEGFRs and fibroblast growth factor receptor 1. It has recently shown significant promise in the treatment of patients with advanced NETs. According to data presented at this year’s annual conference of the European Neuroendocrine Tumor Society in Barcelona, sulfatinib demonstrated an overall response rate of 17.1% in pancreatic NETs and 15% in extra-pancreatic NETs, with an overall disease control rate of 91.4%, and was well tolerated.26 Based on these and other promising phase 1 and 2 data, 2 phase 3 trials are ongoing.
Meanwhile, earlier this year, Mateon Therapeutics presented data from a phase 2 trial of a different kind of anti-angiogenic drug in patients with GI- or pNETs. Fosbretabulin is a vascular disrupting agent that targets the existing tumor vasculature rather than preventing the formation of new blood vessels. They do this via a number of different mechanisms, in the case of fosbretabulin it specifically targets endothelial cells and inhibits the assembly of microtubules and, hence, blocks mitosis. In 18 patients, fosbretabulin treatment resulted in 1 partial response and 7 patients who had stable disease; more than half of the patients reported improved quality of life.27 Fosbretabulin continues to be studied in NETs in combination with everolimus.
Finally, researchers are beginning to make a foray into the immunotherapy field that has revolutionized the treatment of many other tumor types. The immune checkpoint inhibitors nivolumab and pembrolizumab are being evaluated in ongoing phase 1 and 2 trials, while avelumab (Bavencio) was very recently approved by the FDA for the treatment of Merkel cell carcinoma.28,29
1. Pinchot SN, Holen K, Sippel RS, Chen H. Carcinoid tumors. Oncologist. 2008;13(12):1255-1269.
2. Rorstad O. Prognostic indicators for carcinoid neuroendocrine tumors of the gastrointestinal tract. J Surg Oncol. 2005;89(3):151-160.
3. The NET Alliance. Characterizing a challenging cancer. http://www.thenetalliance.com/hcp/facts-about-net/characterization/. Publishing date not provided. Accessed October 18, 2017.
4. Yao JC, Hassan M, Phan A, et al. One hundred years after ‘carcinoid’: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063-3072.
5. Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97(4):934-959.
6. Spada F, Valente M. Review of recent advances in medical treatment for neuroendocrine neoplasms: somatostatin analogs and chemotherapy. J Cancer Metastasis Treat. 2016;2(8):313-320.
7. Kelgiorgi D, Dervenis C. Pancreatic neuroendocrine tumors: the basics, the gray zone, and the target. F1000Research. 2017;6:663.
8. Viudez A, De Jesus-Acosta A, Carvalho FL, Vera R, Martin-Algarra S, Ramirez N. Pancreatic neuroendocrine tumors: Challenges in an underestimated disease. Crit Rev Oncol Hematol. 2016;101:193-206.
9. World Health Organization, International Agency for Research on Cancer. Bosman FT, Carneiro F, Hruban RH, Theise ND (eds). WHO classification of tumours of the digestive system. 2010, 4th ed (vol 3).
10. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors. J Thorac Oncol. 2015;10(9):1243-1260.
11. Lee A, Chan DL, Wong MH, et al. Systematic review of the role of targeted therapy in metastatic neuroendocrine tumors. Neuroendocrinology. 2017;104(3):209-222.
12. Theodoropoulou M, Stalla GK. Somatostatin receptors: from signaling to clinical practice. Front Neuroendocrinol. 2013;34(3):228-252.
13. Caplin ME, Pavel M, Cwikła JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224-233.
14. Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656-4663.
15. Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125-135.
16. Falconi M, Partelli S. Neuroendocrine tumours in 2016: defining rules for increasingly personalized treatments. Nat Rev Clin Oncol. 2017;14(2):80-82.
17. Hutchinson L. Targeted therapies: widening the treatment NET. Nat Rev Clin Oncol. 2017;14(1):2-3.
18. Cingarlini S, Bonomi M, Corbo V, Scarpa A, Tortora G. Profiling mTOR pathway in neuroendocrine tumors. Target Oncol. 2012;7(3):183-188.
19. Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514-523.
20. Yao JC, Pavel M, Lombard-Bohas C, et al. Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: overall survival and circulating biomarkers from the randomized, phase III RADIANT-3 study. J Clin Oncol. http://ascopubs.org/ doi/abs/10.1200/JCO.2016.68.0702?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dpubmed. September 12, 2016. E-pub ahead of print.
21. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968-977.
22. Gajate P, Martínez-Sáez O, Alonso-Gordoa T, Grande E. Emerging use of everolimus in the treatment of neuroendocrine tumors. Cancer Manage Res. 2017;9:215-224.
23. Chan JA, Ryan DP, Zhu AX, et al. Phase I study of pasireotide (SOM 230) and everolimus (RAD001) in advanced neuroendocrine tumors. Endocr Relat Cancer. 2012;19(5):615-623.
24. Kulke MH, Ruszniewski P, Van Cutsem E, et al. A randomized, open-label, phase 2 study of everolimus in combination with pasireotide LAR or everolimus alone in advanced, well-dierentiated, progressive pancreatic neuroendocrine tumors: COOPERATE-2 trial. Ann Oncol. 2017;28(6):1309-1315.
25. Chan JA, Faris JE, Murphy JE, et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J Clin Oncol. 2017;35(4 suppl):228-228.
26. Xu J, Li J, Bai CM, et al. An open-label phase Ib/II study of sulfatinib in patients with advanced neuroendocrine tumors (NCT02267967). Paper presented at the 14th Annual European Neuroendocrine Tumor Society Conference for the Diagnosis and Treatment of Neuroendocrine Tumor Disease; March 8-10, 2017, Barcelona, Spain.
27. Libutti SK, Anthony LB, Chaplin DJ, Sosa JA. A phase II study of combretastatin A4-phosphate (CA4P) in the treatment of well-differentiated, low- to intermediate-grade, unresectable, recurrent, or metastatic pancreatic, or GI neuroendocrine tumors/carcinoid (GI-NETs/PNETs) with elevated biomarkers. J Clin Oncol. 2017;35(4 suppl):432-432.
28. Cordes LM, Gulley JL. Avelumab for the treatment of metastatic Merkel cell carcinoma. Drugs Today (Barc). 2017;53(7):377-383.
29. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374-1385.
1. Pinchot SN, Holen K, Sippel RS, Chen H. Carcinoid tumors. Oncologist. 2008;13(12):1255-1269.
2. Rorstad O. Prognostic indicators for carcinoid neuroendocrine tumors of the gastrointestinal tract. J Surg Oncol. 2005;89(3):151-160.
3. The NET Alliance. Characterizing a challenging cancer. http://www.thenetalliance.com/hcp/facts-about-net/characterization/. Publishing date not provided. Accessed October 18, 2017.
4. Yao JC, Hassan M, Phan A, et al. One hundred years after ‘carcinoid’: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063-3072.
5. Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97(4):934-959.
6. Spada F, Valente M. Review of recent advances in medical treatment for neuroendocrine neoplasms: somatostatin analogs and chemotherapy. J Cancer Metastasis Treat. 2016;2(8):313-320.
7. Kelgiorgi D, Dervenis C. Pancreatic neuroendocrine tumors: the basics, the gray zone, and the target. F1000Research. 2017;6:663.
8. Viudez A, De Jesus-Acosta A, Carvalho FL, Vera R, Martin-Algarra S, Ramirez N. Pancreatic neuroendocrine tumors: Challenges in an underestimated disease. Crit Rev Oncol Hematol. 2016;101:193-206.
9. World Health Organization, International Agency for Research on Cancer. Bosman FT, Carneiro F, Hruban RH, Theise ND (eds). WHO classification of tumours of the digestive system. 2010, 4th ed (vol 3).
10. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors. J Thorac Oncol. 2015;10(9):1243-1260.
11. Lee A, Chan DL, Wong MH, et al. Systematic review of the role of targeted therapy in metastatic neuroendocrine tumors. Neuroendocrinology. 2017;104(3):209-222.
12. Theodoropoulou M, Stalla GK. Somatostatin receptors: from signaling to clinical practice. Front Neuroendocrinol. 2013;34(3):228-252.
13. Caplin ME, Pavel M, Cwikła JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224-233.
14. Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656-4663.
15. Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125-135.
16. Falconi M, Partelli S. Neuroendocrine tumours in 2016: defining rules for increasingly personalized treatments. Nat Rev Clin Oncol. 2017;14(2):80-82.
17. Hutchinson L. Targeted therapies: widening the treatment NET. Nat Rev Clin Oncol. 2017;14(1):2-3.
18. Cingarlini S, Bonomi M, Corbo V, Scarpa A, Tortora G. Profiling mTOR pathway in neuroendocrine tumors. Target Oncol. 2012;7(3):183-188.
19. Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514-523.
20. Yao JC, Pavel M, Lombard-Bohas C, et al. Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: overall survival and circulating biomarkers from the randomized, phase III RADIANT-3 study. J Clin Oncol. http://ascopubs.org/ doi/abs/10.1200/JCO.2016.68.0702?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dpubmed. September 12, 2016. E-pub ahead of print.
21. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968-977.
22. Gajate P, Martínez-Sáez O, Alonso-Gordoa T, Grande E. Emerging use of everolimus in the treatment of neuroendocrine tumors. Cancer Manage Res. 2017;9:215-224.
23. Chan JA, Ryan DP, Zhu AX, et al. Phase I study of pasireotide (SOM 230) and everolimus (RAD001) in advanced neuroendocrine tumors. Endocr Relat Cancer. 2012;19(5):615-623.
24. Kulke MH, Ruszniewski P, Van Cutsem E, et al. A randomized, open-label, phase 2 study of everolimus in combination with pasireotide LAR or everolimus alone in advanced, well-dierentiated, progressive pancreatic neuroendocrine tumors: COOPERATE-2 trial. Ann Oncol. 2017;28(6):1309-1315.
25. Chan JA, Faris JE, Murphy JE, et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J Clin Oncol. 2017;35(4 suppl):228-228.
26. Xu J, Li J, Bai CM, et al. An open-label phase Ib/II study of sulfatinib in patients with advanced neuroendocrine tumors (NCT02267967). Paper presented at the 14th Annual European Neuroendocrine Tumor Society Conference for the Diagnosis and Treatment of Neuroendocrine Tumor Disease; March 8-10, 2017, Barcelona, Spain.
27. Libutti SK, Anthony LB, Chaplin DJ, Sosa JA. A phase II study of combretastatin A4-phosphate (CA4P) in the treatment of well-differentiated, low- to intermediate-grade, unresectable, recurrent, or metastatic pancreatic, or GI neuroendocrine tumors/carcinoid (GI-NETs/PNETs) with elevated biomarkers. J Clin Oncol. 2017;35(4 suppl):432-432.
28. Cordes LM, Gulley JL. Avelumab for the treatment of metastatic Merkel cell carcinoma. Drugs Today (Barc). 2017;53(7):377-383.
29. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374-1385.
Brigatinib approval yields additional treatment options for crizotinib-resistant, ALK-positive NSCLC patients
The accelerated approval by the United States Food and Drug Administration (FDA) of the anaplastic lymphoma kinase (ALK) inhibitor brigatinib, marked the fourth approved drug in this class.1 The most recent approval expands the available treatment options for patients with metastatic ALK-positive non–small-cell lung cancer (NSCLC) whose disease is no longer responding to the first-line ALK inhibitor crizotinib. The FDA based its decision on the results of the phase 2 ALTA trial, in which a significant proportion of patients experienced tumor shrinkage.2
The pivotal trial was a noncomparative, 2-arm, open-label, multicenter study that was carried out during June 2014-September 2015 at 71 centers across 18 countries. Eligible patients were 18 years or older, with locally advanced or metastatic ALK-positive NSCLC, disease progression while taking crizotinib, at least 1 measurable lesion, adequate organ and hematologic function, and Eastern Cooperative Oncology Group (ECOG) performance status of ≤2 (range, 0-5, where 0 means the patient is fully active, and 2, ambulatory and capable of all self-care but not able to carry out any work activities).
Patients were excluded from the trial if they had received previous ALK inhibitor therapy, other than crizotinib, or had received crizotinib within 3 days of the first dose of brigatinib, or they had received chemotherapy, radiation therapy, or investigational drugs within 14 days or monoclonal antibody therapy within 30 days of the first dose of the study drug. Anyone with a history or the presence of pulmonary interstitial disease or drug-related pneumonitis or symptomatic central nervous system (CNS) metastases that were neurologically unstable or required an increasing dose of corticosteroids was also ineligible.
A total of 222 patients were randomized to receive one of two brigatinib doses, either 90 mg daily or 180 mg daily after a 7-day lead-in at 90 mg (the latter to help mitigate pulmonary adverse events observed in previous studies). Randomization was stratified according to baseline brain metastases (present or absent) and best investigator-assessed response to crizotinib (complete response [CR] or partial response [PR] vs other or unknown)
Chest and abdomen imaging by computed-tomography (CT) or magnetic resonance imaging (MRI) with contrast were performed to assess disease at screening and every 8 weeks through cycle 15, and then every 12 weeks until disease progression. Contrast-enhanced brain MRI was carried out at screening and repeated after baseline for the 68% of patients who had CNS metastases at the time of enrollment.
The primary endpoint was confirmed investigator-assessed objective response rate (ORR) per Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), and secondary endpoints included CNS response, duration of response (DoR), progression-free and overall survival (PFS and OS, respectively). ORRs for the 90-mg and 180-mg doses were 48% and 53%, respectively. Responses occurred quickly and were durable in both arms; after a median follow-up of 8 months, median DoR was 13.8 months for both doses. Among the patients with brain metastases, the intracranial response rates for the two doses were 42% and 67%, respectively, notable because of the poor ability of crizotinib to penetrate the blood-brain barrier.
Other secondary outcomes also favored the 180-mg dose. Investigator-assessed PFS for the 90-mg and 180-mg doses were 9.2 months and 12.9 months, respectively, and estimated 1-year OS was 71% and 80%, respectively, the latter representing a nonstatistically significant 43% reduction in the risk of death with the 180 mg dose. There were 4 confirmed CRs in the 180-mg arm and 1 in the 90-mg arm.
The safety of brigatinib was evaluated in 219 patients who received at least 1 dose of brigatinib. Treatment was discontinued in 8% of patients in the 180-mg arm and 3% in the 90-mg arm because of adverse events (AEs). The most common AEs were nausea, diarrhea, fatigue, cough, and headache, and visual disturbances also occurred. The most common serious AEs were pneumonia and interstitial lung disease/pneumonitis.
The prescribing information details warnings and precautions about these and other potential toxicities, including hypertension, bradycardia, creatine phosphokinase (CPK) and pancreatic enzyme elevation, and hyperglycemia.3 Patients should be monitored for new or worsening respiratory symptoms, especially during the first week of initiating brigatinib treatment; blood pressure should be controlled before treatment initiation and monitored after 2 weeks and at least monthly thereafter; heart rate and blood pressure should be monitored frequently; patients should be advised to report any visual symptoms, or any unexplained muscle pain, tenderness or weakness; CPK, lipase, and amylase levels should be monitored during treatment, and fasting glucose tested before starting treatment and periodically thereafter.
Brigatinib should be withheld in any patient with new or worsening respiratory symptoms, for grade 3 hypertension despite optimal antihypertensive therapy, for symptomatic bradycardia, for patients with new or worsening visual symptoms of grade 2 or above, for grade 3 or 4 CPK or pancreatic enzyme elevation, or if adequate hyperglycemia control cannot be achieved. Treatment should be permanently discontinued for grade 3 or 4 or recurrent interstitial lung disease/pneumonitis, grade 4 or recurrent grade 3 hypertension, life-threatening bradycardia, and grade 4 visual disturbance.
Based on its mechanism of action, brigatinib can cause fetal harm and patients of reproductive potential should be advised of the risks and necessary precautions. Brigatinib is marketed as Alunbrig. It was discovered by Ariad Pharmaceuticals Inc, which was acquired by Takeda in February 2017.
1. United States Food and Drug Administration. Brigatinib. US FDA Web site. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm555841.htm. Last updated April 28, 2017. Accessed July 15, 2017
2. Kim D-W, Tiseo M, Ahn M-J, Reckamp KL, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non–small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol. 2017;35(22):2490-2498.
3. Alunbrig (brigatinib) tablets, for oral use. Prescribing information. Ariad Pharmaceuticals Inc. https://www.alunbrig.com/assets/pi.pdf. Posted April 2017. Accessed July 15, 2017.
The accelerated approval by the United States Food and Drug Administration (FDA) of the anaplastic lymphoma kinase (ALK) inhibitor brigatinib, marked the fourth approved drug in this class.1 The most recent approval expands the available treatment options for patients with metastatic ALK-positive non–small-cell lung cancer (NSCLC) whose disease is no longer responding to the first-line ALK inhibitor crizotinib. The FDA based its decision on the results of the phase 2 ALTA trial, in which a significant proportion of patients experienced tumor shrinkage.2
The pivotal trial was a noncomparative, 2-arm, open-label, multicenter study that was carried out during June 2014-September 2015 at 71 centers across 18 countries. Eligible patients were 18 years or older, with locally advanced or metastatic ALK-positive NSCLC, disease progression while taking crizotinib, at least 1 measurable lesion, adequate organ and hematologic function, and Eastern Cooperative Oncology Group (ECOG) performance status of ≤2 (range, 0-5, where 0 means the patient is fully active, and 2, ambulatory and capable of all self-care but not able to carry out any work activities).
Patients were excluded from the trial if they had received previous ALK inhibitor therapy, other than crizotinib, or had received crizotinib within 3 days of the first dose of brigatinib, or they had received chemotherapy, radiation therapy, or investigational drugs within 14 days or monoclonal antibody therapy within 30 days of the first dose of the study drug. Anyone with a history or the presence of pulmonary interstitial disease or drug-related pneumonitis or symptomatic central nervous system (CNS) metastases that were neurologically unstable or required an increasing dose of corticosteroids was also ineligible.
A total of 222 patients were randomized to receive one of two brigatinib doses, either 90 mg daily or 180 mg daily after a 7-day lead-in at 90 mg (the latter to help mitigate pulmonary adverse events observed in previous studies). Randomization was stratified according to baseline brain metastases (present or absent) and best investigator-assessed response to crizotinib (complete response [CR] or partial response [PR] vs other or unknown)
Chest and abdomen imaging by computed-tomography (CT) or magnetic resonance imaging (MRI) with contrast were performed to assess disease at screening and every 8 weeks through cycle 15, and then every 12 weeks until disease progression. Contrast-enhanced brain MRI was carried out at screening and repeated after baseline for the 68% of patients who had CNS metastases at the time of enrollment.
The primary endpoint was confirmed investigator-assessed objective response rate (ORR) per Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), and secondary endpoints included CNS response, duration of response (DoR), progression-free and overall survival (PFS and OS, respectively). ORRs for the 90-mg and 180-mg doses were 48% and 53%, respectively. Responses occurred quickly and were durable in both arms; after a median follow-up of 8 months, median DoR was 13.8 months for both doses. Among the patients with brain metastases, the intracranial response rates for the two doses were 42% and 67%, respectively, notable because of the poor ability of crizotinib to penetrate the blood-brain barrier.
Other secondary outcomes also favored the 180-mg dose. Investigator-assessed PFS for the 90-mg and 180-mg doses were 9.2 months and 12.9 months, respectively, and estimated 1-year OS was 71% and 80%, respectively, the latter representing a nonstatistically significant 43% reduction in the risk of death with the 180 mg dose. There were 4 confirmed CRs in the 180-mg arm and 1 in the 90-mg arm.
The safety of brigatinib was evaluated in 219 patients who received at least 1 dose of brigatinib. Treatment was discontinued in 8% of patients in the 180-mg arm and 3% in the 90-mg arm because of adverse events (AEs). The most common AEs were nausea, diarrhea, fatigue, cough, and headache, and visual disturbances also occurred. The most common serious AEs were pneumonia and interstitial lung disease/pneumonitis.
The prescribing information details warnings and precautions about these and other potential toxicities, including hypertension, bradycardia, creatine phosphokinase (CPK) and pancreatic enzyme elevation, and hyperglycemia.3 Patients should be monitored for new or worsening respiratory symptoms, especially during the first week of initiating brigatinib treatment; blood pressure should be controlled before treatment initiation and monitored after 2 weeks and at least monthly thereafter; heart rate and blood pressure should be monitored frequently; patients should be advised to report any visual symptoms, or any unexplained muscle pain, tenderness or weakness; CPK, lipase, and amylase levels should be monitored during treatment, and fasting glucose tested before starting treatment and periodically thereafter.
Brigatinib should be withheld in any patient with new or worsening respiratory symptoms, for grade 3 hypertension despite optimal antihypertensive therapy, for symptomatic bradycardia, for patients with new or worsening visual symptoms of grade 2 or above, for grade 3 or 4 CPK or pancreatic enzyme elevation, or if adequate hyperglycemia control cannot be achieved. Treatment should be permanently discontinued for grade 3 or 4 or recurrent interstitial lung disease/pneumonitis, grade 4 or recurrent grade 3 hypertension, life-threatening bradycardia, and grade 4 visual disturbance.
Based on its mechanism of action, brigatinib can cause fetal harm and patients of reproductive potential should be advised of the risks and necessary precautions. Brigatinib is marketed as Alunbrig. It was discovered by Ariad Pharmaceuticals Inc, which was acquired by Takeda in February 2017.
The accelerated approval by the United States Food and Drug Administration (FDA) of the anaplastic lymphoma kinase (ALK) inhibitor brigatinib, marked the fourth approved drug in this class.1 The most recent approval expands the available treatment options for patients with metastatic ALK-positive non–small-cell lung cancer (NSCLC) whose disease is no longer responding to the first-line ALK inhibitor crizotinib. The FDA based its decision on the results of the phase 2 ALTA trial, in which a significant proportion of patients experienced tumor shrinkage.2
The pivotal trial was a noncomparative, 2-arm, open-label, multicenter study that was carried out during June 2014-September 2015 at 71 centers across 18 countries. Eligible patients were 18 years or older, with locally advanced or metastatic ALK-positive NSCLC, disease progression while taking crizotinib, at least 1 measurable lesion, adequate organ and hematologic function, and Eastern Cooperative Oncology Group (ECOG) performance status of ≤2 (range, 0-5, where 0 means the patient is fully active, and 2, ambulatory and capable of all self-care but not able to carry out any work activities).
Patients were excluded from the trial if they had received previous ALK inhibitor therapy, other than crizotinib, or had received crizotinib within 3 days of the first dose of brigatinib, or they had received chemotherapy, radiation therapy, or investigational drugs within 14 days or monoclonal antibody therapy within 30 days of the first dose of the study drug. Anyone with a history or the presence of pulmonary interstitial disease or drug-related pneumonitis or symptomatic central nervous system (CNS) metastases that were neurologically unstable or required an increasing dose of corticosteroids was also ineligible.
A total of 222 patients were randomized to receive one of two brigatinib doses, either 90 mg daily or 180 mg daily after a 7-day lead-in at 90 mg (the latter to help mitigate pulmonary adverse events observed in previous studies). Randomization was stratified according to baseline brain metastases (present or absent) and best investigator-assessed response to crizotinib (complete response [CR] or partial response [PR] vs other or unknown)
Chest and abdomen imaging by computed-tomography (CT) or magnetic resonance imaging (MRI) with contrast were performed to assess disease at screening and every 8 weeks through cycle 15, and then every 12 weeks until disease progression. Contrast-enhanced brain MRI was carried out at screening and repeated after baseline for the 68% of patients who had CNS metastases at the time of enrollment.
The primary endpoint was confirmed investigator-assessed objective response rate (ORR) per Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), and secondary endpoints included CNS response, duration of response (DoR), progression-free and overall survival (PFS and OS, respectively). ORRs for the 90-mg and 180-mg doses were 48% and 53%, respectively. Responses occurred quickly and were durable in both arms; after a median follow-up of 8 months, median DoR was 13.8 months for both doses. Among the patients with brain metastases, the intracranial response rates for the two doses were 42% and 67%, respectively, notable because of the poor ability of crizotinib to penetrate the blood-brain barrier.
Other secondary outcomes also favored the 180-mg dose. Investigator-assessed PFS for the 90-mg and 180-mg doses were 9.2 months and 12.9 months, respectively, and estimated 1-year OS was 71% and 80%, respectively, the latter representing a nonstatistically significant 43% reduction in the risk of death with the 180 mg dose. There were 4 confirmed CRs in the 180-mg arm and 1 in the 90-mg arm.
The safety of brigatinib was evaluated in 219 patients who received at least 1 dose of brigatinib. Treatment was discontinued in 8% of patients in the 180-mg arm and 3% in the 90-mg arm because of adverse events (AEs). The most common AEs were nausea, diarrhea, fatigue, cough, and headache, and visual disturbances also occurred. The most common serious AEs were pneumonia and interstitial lung disease/pneumonitis.
The prescribing information details warnings and precautions about these and other potential toxicities, including hypertension, bradycardia, creatine phosphokinase (CPK) and pancreatic enzyme elevation, and hyperglycemia.3 Patients should be monitored for new or worsening respiratory symptoms, especially during the first week of initiating brigatinib treatment; blood pressure should be controlled before treatment initiation and monitored after 2 weeks and at least monthly thereafter; heart rate and blood pressure should be monitored frequently; patients should be advised to report any visual symptoms, or any unexplained muscle pain, tenderness or weakness; CPK, lipase, and amylase levels should be monitored during treatment, and fasting glucose tested before starting treatment and periodically thereafter.
Brigatinib should be withheld in any patient with new or worsening respiratory symptoms, for grade 3 hypertension despite optimal antihypertensive therapy, for symptomatic bradycardia, for patients with new or worsening visual symptoms of grade 2 or above, for grade 3 or 4 CPK or pancreatic enzyme elevation, or if adequate hyperglycemia control cannot be achieved. Treatment should be permanently discontinued for grade 3 or 4 or recurrent interstitial lung disease/pneumonitis, grade 4 or recurrent grade 3 hypertension, life-threatening bradycardia, and grade 4 visual disturbance.
Based on its mechanism of action, brigatinib can cause fetal harm and patients of reproductive potential should be advised of the risks and necessary precautions. Brigatinib is marketed as Alunbrig. It was discovered by Ariad Pharmaceuticals Inc, which was acquired by Takeda in February 2017.
1. United States Food and Drug Administration. Brigatinib. US FDA Web site. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm555841.htm. Last updated April 28, 2017. Accessed July 15, 2017
2. Kim D-W, Tiseo M, Ahn M-J, Reckamp KL, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non–small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol. 2017;35(22):2490-2498.
3. Alunbrig (brigatinib) tablets, for oral use. Prescribing information. Ariad Pharmaceuticals Inc. https://www.alunbrig.com/assets/pi.pdf. Posted April 2017. Accessed July 15, 2017.
1. United States Food and Drug Administration. Brigatinib. US FDA Web site. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm555841.htm. Last updated April 28, 2017. Accessed July 15, 2017
2. Kim D-W, Tiseo M, Ahn M-J, Reckamp KL, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non–small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol. 2017;35(22):2490-2498.
3. Alunbrig (brigatinib) tablets, for oral use. Prescribing information. Ariad Pharmaceuticals Inc. https://www.alunbrig.com/assets/pi.pdf. Posted April 2017. Accessed July 15, 2017.
Ultrathin bronchoscopy plus radial EBUS unreliable at making diagnoses
TORONTO – Ultrathin bronchoscopy plus radial endobronchial ultrasound is not a great method for determining whether a suspicious lesion is cancerous or benign, suggests new research.
In this study of patients with CT-detected solid lung lesions, the researchers were able to make a diagnosis for only 49% of those whose nodules were evaluated using ultrathin bronchoscopy plus radial endobronchial ultrasound (EBUS).
“When you do CT-guided biopsies of lung lesions, the [diagnostic] yield is about 94%. So do the math” by comparing it to the roughly 50% yield from ultrathin bronchoscopy plus radial EBUS to decide whether the latter procedure is worth doing, she noted.
The study Dr. Tanner and her associates designed compared the diagnostic yield of ultrathin bronchoscopy plus radial EBUS with standard bronchoscopy and fluoroscopy in patients with CT-detected solid lung lesions 1.5-5.0 cm in size. It ran at five U.S. centers and randomized 221 patients: 85 evaluable patients were tested using the standard methods, and 112 evaluable patients were tested using ultrathin bronchoscopy plus radial EBUS. Patients averaged 65-68 years of age and were divided evenly between women and men. Their lesions averaged slightly more than 3 cm. The ultrathin device had a 4 mm wide diameter and had a 2 mm working channel.
The diagnostic yield was 38% among patients who underwent standard bronchoscopy and fluoroscopy, and 49% among those biopsied using ultrathin bronchoscopy and radial EBUS, Dr. Tanner reported. The between-group difference in yield fell short of being statistically significant.
Forty-six of the 53 patients who were not diagnosable using standard bronchoscopy and fluoroscopy crossed over to the investigational method, which produced a diagnosis for an additional seven patients (15% of the biopsied crossover patients).
The results showed that standard bronchoscopy plus fluoroscopy is “very poor” for distinguishing cancerous and benign pulmonary lesions, Dr. Tanner concluded. The yield from ultrathin bronchoscopy plus radial EBUS in her study was similar to the diagnostic yields reported in prior studies of guided bronchoscopy, even when also using radial EBUS, she added.
Given the limitations of ultrathin bronchoscopy plus radial EBUS, Dr. Tanner suggested that the best scenario for using this diagnostic method would be in patients who need a linear EBUS procedure for mediastinal lymph node staging. Such staging often requires a biopsy of the primary tumor to make a cancer diagnosis, and in such cases, “while you’re in the neighborhood, you could do bronchoscopy with an ultrathin scope,” she suggested.
The potential also exists to augment the diagnostic yield of ultrathin bronchoscopy by applying a navigational software platform and needle biopsy, two methods not included in the study, Dr. Tanner noted. “More studies should be done using this combination,” she said.
The study was funded by Olympus. Dr. Tanner has been a consultant to and has received research funding from Olympus. She has also been a consultant to Cook Medical, Integrated Diagnostics, Oncocyte, Veracyte, and Veran Medical Technologies, and she has also received research funding from Cook, Integrated Diagnostics, Oncocyte, Oncimmune, and Veracyte.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
Although bronchoscopic tools are safe and accurate to evaluate both central and peripheral lung lesions, the diagnostic yield of the different available techniques is variable. In this study, a diagnostic yield of only 49% was achieved when ultrathin bronchoscopy with radial EBUS was performed for diagnosis of solid nodules. This yield is not much better than that obtained from conventional bronchoscopy with fluoroscopic guidance and much lower than the diagnostic yield from transthoracic needle biopsy. While there is no doubt that the advances in minimally invasive technologies for diagnosing lung nodules and diagnosing and staging lung cancer have revolutionized clinical practice, pulmonologists and thoracic surgeons need to recognize not only the utility but also the limitations of the available diagnostic procedures (as well as the cost). These technologies are complimentary and multidisciplinary discussions should facilitate selection of the best procedure for each individual case.
Although bronchoscopic tools are safe and accurate to evaluate both central and peripheral lung lesions, the diagnostic yield of the different available techniques is variable. In this study, a diagnostic yield of only 49% was achieved when ultrathin bronchoscopy with radial EBUS was performed for diagnosis of solid nodules. This yield is not much better than that obtained from conventional bronchoscopy with fluoroscopic guidance and much lower than the diagnostic yield from transthoracic needle biopsy. While there is no doubt that the advances in minimally invasive technologies for diagnosing lung nodules and diagnosing and staging lung cancer have revolutionized clinical practice, pulmonologists and thoracic surgeons need to recognize not only the utility but also the limitations of the available diagnostic procedures (as well as the cost). These technologies are complimentary and multidisciplinary discussions should facilitate selection of the best procedure for each individual case.
Although bronchoscopic tools are safe and accurate to evaluate both central and peripheral lung lesions, the diagnostic yield of the different available techniques is variable. In this study, a diagnostic yield of only 49% was achieved when ultrathin bronchoscopy with radial EBUS was performed for diagnosis of solid nodules. This yield is not much better than that obtained from conventional bronchoscopy with fluoroscopic guidance and much lower than the diagnostic yield from transthoracic needle biopsy. While there is no doubt that the advances in minimally invasive technologies for diagnosing lung nodules and diagnosing and staging lung cancer have revolutionized clinical practice, pulmonologists and thoracic surgeons need to recognize not only the utility but also the limitations of the available diagnostic procedures (as well as the cost). These technologies are complimentary and multidisciplinary discussions should facilitate selection of the best procedure for each individual case.
TORONTO – Ultrathin bronchoscopy plus radial endobronchial ultrasound is not a great method for determining whether a suspicious lesion is cancerous or benign, suggests new research.
In this study of patients with CT-detected solid lung lesions, the researchers were able to make a diagnosis for only 49% of those whose nodules were evaluated using ultrathin bronchoscopy plus radial endobronchial ultrasound (EBUS).
“When you do CT-guided biopsies of lung lesions, the [diagnostic] yield is about 94%. So do the math” by comparing it to the roughly 50% yield from ultrathin bronchoscopy plus radial EBUS to decide whether the latter procedure is worth doing, she noted.
The study Dr. Tanner and her associates designed compared the diagnostic yield of ultrathin bronchoscopy plus radial EBUS with standard bronchoscopy and fluoroscopy in patients with CT-detected solid lung lesions 1.5-5.0 cm in size. It ran at five U.S. centers and randomized 221 patients: 85 evaluable patients were tested using the standard methods, and 112 evaluable patients were tested using ultrathin bronchoscopy plus radial EBUS. Patients averaged 65-68 years of age and were divided evenly between women and men. Their lesions averaged slightly more than 3 cm. The ultrathin device had a 4 mm wide diameter and had a 2 mm working channel.
The diagnostic yield was 38% among patients who underwent standard bronchoscopy and fluoroscopy, and 49% among those biopsied using ultrathin bronchoscopy and radial EBUS, Dr. Tanner reported. The between-group difference in yield fell short of being statistically significant.
Forty-six of the 53 patients who were not diagnosable using standard bronchoscopy and fluoroscopy crossed over to the investigational method, which produced a diagnosis for an additional seven patients (15% of the biopsied crossover patients).
The results showed that standard bronchoscopy plus fluoroscopy is “very poor” for distinguishing cancerous and benign pulmonary lesions, Dr. Tanner concluded. The yield from ultrathin bronchoscopy plus radial EBUS in her study was similar to the diagnostic yields reported in prior studies of guided bronchoscopy, even when also using radial EBUS, she added.
Given the limitations of ultrathin bronchoscopy plus radial EBUS, Dr. Tanner suggested that the best scenario for using this diagnostic method would be in patients who need a linear EBUS procedure for mediastinal lymph node staging. Such staging often requires a biopsy of the primary tumor to make a cancer diagnosis, and in such cases, “while you’re in the neighborhood, you could do bronchoscopy with an ultrathin scope,” she suggested.
The potential also exists to augment the diagnostic yield of ultrathin bronchoscopy by applying a navigational software platform and needle biopsy, two methods not included in the study, Dr. Tanner noted. “More studies should be done using this combination,” she said.
The study was funded by Olympus. Dr. Tanner has been a consultant to and has received research funding from Olympus. She has also been a consultant to Cook Medical, Integrated Diagnostics, Oncocyte, Veracyte, and Veran Medical Technologies, and she has also received research funding from Cook, Integrated Diagnostics, Oncocyte, Oncimmune, and Veracyte.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
TORONTO – Ultrathin bronchoscopy plus radial endobronchial ultrasound is not a great method for determining whether a suspicious lesion is cancerous or benign, suggests new research.
In this study of patients with CT-detected solid lung lesions, the researchers were able to make a diagnosis for only 49% of those whose nodules were evaluated using ultrathin bronchoscopy plus radial endobronchial ultrasound (EBUS).
“When you do CT-guided biopsies of lung lesions, the [diagnostic] yield is about 94%. So do the math” by comparing it to the roughly 50% yield from ultrathin bronchoscopy plus radial EBUS to decide whether the latter procedure is worth doing, she noted.
The study Dr. Tanner and her associates designed compared the diagnostic yield of ultrathin bronchoscopy plus radial EBUS with standard bronchoscopy and fluoroscopy in patients with CT-detected solid lung lesions 1.5-5.0 cm in size. It ran at five U.S. centers and randomized 221 patients: 85 evaluable patients were tested using the standard methods, and 112 evaluable patients were tested using ultrathin bronchoscopy plus radial EBUS. Patients averaged 65-68 years of age and were divided evenly between women and men. Their lesions averaged slightly more than 3 cm. The ultrathin device had a 4 mm wide diameter and had a 2 mm working channel.
The diagnostic yield was 38% among patients who underwent standard bronchoscopy and fluoroscopy, and 49% among those biopsied using ultrathin bronchoscopy and radial EBUS, Dr. Tanner reported. The between-group difference in yield fell short of being statistically significant.
Forty-six of the 53 patients who were not diagnosable using standard bronchoscopy and fluoroscopy crossed over to the investigational method, which produced a diagnosis for an additional seven patients (15% of the biopsied crossover patients).
The results showed that standard bronchoscopy plus fluoroscopy is “very poor” for distinguishing cancerous and benign pulmonary lesions, Dr. Tanner concluded. The yield from ultrathin bronchoscopy plus radial EBUS in her study was similar to the diagnostic yields reported in prior studies of guided bronchoscopy, even when also using radial EBUS, she added.
Given the limitations of ultrathin bronchoscopy plus radial EBUS, Dr. Tanner suggested that the best scenario for using this diagnostic method would be in patients who need a linear EBUS procedure for mediastinal lymph node staging. Such staging often requires a biopsy of the primary tumor to make a cancer diagnosis, and in such cases, “while you’re in the neighborhood, you could do bronchoscopy with an ultrathin scope,” she suggested.
The potential also exists to augment the diagnostic yield of ultrathin bronchoscopy by applying a navigational software platform and needle biopsy, two methods not included in the study, Dr. Tanner noted. “More studies should be done using this combination,” she said.
The study was funded by Olympus. Dr. Tanner has been a consultant to and has received research funding from Olympus. She has also been a consultant to Cook Medical, Integrated Diagnostics, Oncocyte, Veracyte, and Veran Medical Technologies, and she has also received research funding from Cook, Integrated Diagnostics, Oncocyte, Oncimmune, and Veracyte.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
AT CHEST 2017
Key clinical point:
Major finding: The diagnostic yield using ultrathin bronchoscopy with radial EBUS was 49%, while standard bronchoscopy had a 38% yield.
Data source: Multicenter, randomized study with 221 total patients and 197 evaluable patients.
Disclosures: The study was funded by Olympus. Dr. Tanner has been a consultant to and has received research funding from Olympus. She has also been a consultant to Cook Medical, Integrated Diagnostics, Oncocyte, Veracyte, and Veran Medical Technologies, and she has also received research funding from Cook, Integrated Diagnostics, Oncocyte, Oncimmune, and Veracyte.
First-in-class glutaminase inhibitor combats anti-PD-1/PD-L1 resistance
NATIONAL HARBOR, MD. – Combination treatment with the first-in-class glutaminase inhibitor CB-839 and nivolumab is well-tolerated and shows clinical activity in patients with advanced melanoma, renal cell carcinoma, or non-small cell lung cancer, including anti-PD-1/PD-L1 refractory patients, according to initial results from a phase 1/2 study.
Responses in melanoma patients who were progressing on nivolumab at study entry and who were refractory to multiple prior immunotherapy regimens are particularly notable, as they highlight the potential for CB-839, when added to nivolumab (Opdivo), to help overcome resistance to anti-PD-L1 therapy, Funda Meric‐Bernstam, MD, reported at the annual meeting of the Society for Immunotherapy of Cancer.
CB‐839 is highly selective and targets tumor glutamine metabolism, said Dr. Meric-Bernstam of the University of Texas MD Anderson Cancer Center, Houston.
Competition between tumor cells and immune cells for nutrients such as glutamine in the tumor microenvironment can create a metabolic checkpoint that induces local immune suppression. CB‐839 inhibits tumor glutamine consumption, thereby increasing glutamine availability to support T‐cell activity, she explained, noting that in preclinical models, CB‐839 increased intra‐tumoral glutamine and enhanced antitumor activity of PD‐1/PD‐L1 inhibitors.
In the phase 1 dose escalation study, she and her colleagues evaluated the safety and efficacy of CB-839 in combination with the PD‐1 inhibitor nivolumab in patients with melanoma, non-small cell lung cancer (NSCLC), or renal cell carcinoma (RCC). Phase 2 expansion cohorts include a melanoma rescue cohort of patients progressing on anti-PD-L1 therapy at study entry (22 patients), an NSCLC and RCC rescue cohort of patients who were progressing on anti-PD-L1 therapy at study entry or who had stable disease for 6 months or longer without a response (11 NSCLC and 11 RCC), an RCC cohort of patients with prior immunotherapy exposure and no response (10 patients), and an RCC cohort of patents who had no prior immunotherapy exposure (28 patients).
During dose escalation, patients received oral CB‐839 at 600 mg or 800 mg twice daily in combination with standard‐dose nivolumab. In the ongoing phase 2 expansion study, which continues to enroll, patients are receiving 800 mg of CB-839 twice daily with standard‐dose nivolumab, Dr. Meric-Bernstam said.
Patients in each of the cohorts were high risk and/or had intermediate or poor prognostic status at study entry. For example, 50% of patients in the melanoma rescue cohort had liver metastases, 77% had other visceral metastases, and 18% had brain metastases, and the majority of patients in the lung cancer/RCC cohort had visceral metastases. Most had progressive disease as their best response on their last line of immunotherapy.
Of 16 response-evaluable melanoma patients, 1 experienced a complete response, 2 had partial responses, and 4 had stable disease.
“So overall in this patient population that was progressing on a PD-1/PD-L1 inhibitor at enrollment, 19% had an objective response. The disease control rate in this group was 44%,” she said.
In evaluable patients in the lung cancer rescue cohort (6 patients), RCC rescue cohort (8 patients), and RCC prior exposure cohort (7 patients), disease control rates ranged from 57% to 75%, and in the immunotherapy-naive RCC cohort (19 patients), the partial response rate was 21%, and 53% had stable disease, so the overall disease control rate was 74%. Half of the patients in that group remain on study, she noted.
A closer look at the melanoma rescue cohort showed dramatic and rapid responses in two patients who each achieved a partial response in about 8 weeks with response durations of 3.7 months and 5.4 months, respectively. Additionally, pre-treatment biopsies in this cohort showed an elevated T-cell inflamed signature associated with clinical benefit from the addition of CB-839, and in one patient who had both a pretreatment and on-treatment biopsy that was evaluable, the latter showed an increase in T-cell inflamed signature and T-cell effector genes.
In all cohorts, the combination therapy was generally well tolerated. A maximum tolerated dose was not reached. Dose-limiting toxicity – a grade 3 alanine aminotransferase (ALT) increase – occurred in one patient on the 800-mg dose. The most common grade 3 or greater adverse events were fatigue, nausea, photophobia, rash, and elevated ALT, she said, noting that two patients discontinued for treatment-related adverse events (one for a grade 3 rash and one for grade 2 pneumonitis).
“Overall there appeared to be no apparent increase in immune-related adverse events, either in rate or severity, compared with [nivolumab] monotherapy,” she said.
The combination of CB-839 and nivolumab was well tolerated, and in some patients – as seen in the melanoma cohort – adding CB-839 to checkpoint blockade can overcome checkpoint blockade resistance, Dr. Meric-Bernstam concluded, noting that the disease control rates seen in the majority of lung cancer and RCC patients who were progressing on checkpoint blockade is encouraging, as is the objective response rate seen thus far in the RCC therapy-naive patients, and the stable and deep responses seen in the melanoma rescue cohort.
“Based on our encouraging signal in the melanoma rescue cohort, this [cohort] has been expanded,” she said.
Calithera Biosciences sponsored the study. Bristol-Myers Squibb provided nivolumab for the study. Dr. Meric-Bernstam has received grant or research support from Calithera Biosciences and many other companies. She also reported being a paid consultant for several companies and serving on an advisory committee or review panel, or as a board member for multiple companies.
NATIONAL HARBOR, MD. – Combination treatment with the first-in-class glutaminase inhibitor CB-839 and nivolumab is well-tolerated and shows clinical activity in patients with advanced melanoma, renal cell carcinoma, or non-small cell lung cancer, including anti-PD-1/PD-L1 refractory patients, according to initial results from a phase 1/2 study.
Responses in melanoma patients who were progressing on nivolumab at study entry and who were refractory to multiple prior immunotherapy regimens are particularly notable, as they highlight the potential for CB-839, when added to nivolumab (Opdivo), to help overcome resistance to anti-PD-L1 therapy, Funda Meric‐Bernstam, MD, reported at the annual meeting of the Society for Immunotherapy of Cancer.
CB‐839 is highly selective and targets tumor glutamine metabolism, said Dr. Meric-Bernstam of the University of Texas MD Anderson Cancer Center, Houston.
Competition between tumor cells and immune cells for nutrients such as glutamine in the tumor microenvironment can create a metabolic checkpoint that induces local immune suppression. CB‐839 inhibits tumor glutamine consumption, thereby increasing glutamine availability to support T‐cell activity, she explained, noting that in preclinical models, CB‐839 increased intra‐tumoral glutamine and enhanced antitumor activity of PD‐1/PD‐L1 inhibitors.
In the phase 1 dose escalation study, she and her colleagues evaluated the safety and efficacy of CB-839 in combination with the PD‐1 inhibitor nivolumab in patients with melanoma, non-small cell lung cancer (NSCLC), or renal cell carcinoma (RCC). Phase 2 expansion cohorts include a melanoma rescue cohort of patients progressing on anti-PD-L1 therapy at study entry (22 patients), an NSCLC and RCC rescue cohort of patients who were progressing on anti-PD-L1 therapy at study entry or who had stable disease for 6 months or longer without a response (11 NSCLC and 11 RCC), an RCC cohort of patients with prior immunotherapy exposure and no response (10 patients), and an RCC cohort of patents who had no prior immunotherapy exposure (28 patients).
During dose escalation, patients received oral CB‐839 at 600 mg or 800 mg twice daily in combination with standard‐dose nivolumab. In the ongoing phase 2 expansion study, which continues to enroll, patients are receiving 800 mg of CB-839 twice daily with standard‐dose nivolumab, Dr. Meric-Bernstam said.
Patients in each of the cohorts were high risk and/or had intermediate or poor prognostic status at study entry. For example, 50% of patients in the melanoma rescue cohort had liver metastases, 77% had other visceral metastases, and 18% had brain metastases, and the majority of patients in the lung cancer/RCC cohort had visceral metastases. Most had progressive disease as their best response on their last line of immunotherapy.
Of 16 response-evaluable melanoma patients, 1 experienced a complete response, 2 had partial responses, and 4 had stable disease.
“So overall in this patient population that was progressing on a PD-1/PD-L1 inhibitor at enrollment, 19% had an objective response. The disease control rate in this group was 44%,” she said.
In evaluable patients in the lung cancer rescue cohort (6 patients), RCC rescue cohort (8 patients), and RCC prior exposure cohort (7 patients), disease control rates ranged from 57% to 75%, and in the immunotherapy-naive RCC cohort (19 patients), the partial response rate was 21%, and 53% had stable disease, so the overall disease control rate was 74%. Half of the patients in that group remain on study, she noted.
A closer look at the melanoma rescue cohort showed dramatic and rapid responses in two patients who each achieved a partial response in about 8 weeks with response durations of 3.7 months and 5.4 months, respectively. Additionally, pre-treatment biopsies in this cohort showed an elevated T-cell inflamed signature associated with clinical benefit from the addition of CB-839, and in one patient who had both a pretreatment and on-treatment biopsy that was evaluable, the latter showed an increase in T-cell inflamed signature and T-cell effector genes.
In all cohorts, the combination therapy was generally well tolerated. A maximum tolerated dose was not reached. Dose-limiting toxicity – a grade 3 alanine aminotransferase (ALT) increase – occurred in one patient on the 800-mg dose. The most common grade 3 or greater adverse events were fatigue, nausea, photophobia, rash, and elevated ALT, she said, noting that two patients discontinued for treatment-related adverse events (one for a grade 3 rash and one for grade 2 pneumonitis).
“Overall there appeared to be no apparent increase in immune-related adverse events, either in rate or severity, compared with [nivolumab] monotherapy,” she said.
The combination of CB-839 and nivolumab was well tolerated, and in some patients – as seen in the melanoma cohort – adding CB-839 to checkpoint blockade can overcome checkpoint blockade resistance, Dr. Meric-Bernstam concluded, noting that the disease control rates seen in the majority of lung cancer and RCC patients who were progressing on checkpoint blockade is encouraging, as is the objective response rate seen thus far in the RCC therapy-naive patients, and the stable and deep responses seen in the melanoma rescue cohort.
“Based on our encouraging signal in the melanoma rescue cohort, this [cohort] has been expanded,” she said.
Calithera Biosciences sponsored the study. Bristol-Myers Squibb provided nivolumab for the study. Dr. Meric-Bernstam has received grant or research support from Calithera Biosciences and many other companies. She also reported being a paid consultant for several companies and serving on an advisory committee or review panel, or as a board member for multiple companies.
NATIONAL HARBOR, MD. – Combination treatment with the first-in-class glutaminase inhibitor CB-839 and nivolumab is well-tolerated and shows clinical activity in patients with advanced melanoma, renal cell carcinoma, or non-small cell lung cancer, including anti-PD-1/PD-L1 refractory patients, according to initial results from a phase 1/2 study.
Responses in melanoma patients who were progressing on nivolumab at study entry and who were refractory to multiple prior immunotherapy regimens are particularly notable, as they highlight the potential for CB-839, when added to nivolumab (Opdivo), to help overcome resistance to anti-PD-L1 therapy, Funda Meric‐Bernstam, MD, reported at the annual meeting of the Society for Immunotherapy of Cancer.
CB‐839 is highly selective and targets tumor glutamine metabolism, said Dr. Meric-Bernstam of the University of Texas MD Anderson Cancer Center, Houston.
Competition between tumor cells and immune cells for nutrients such as glutamine in the tumor microenvironment can create a metabolic checkpoint that induces local immune suppression. CB‐839 inhibits tumor glutamine consumption, thereby increasing glutamine availability to support T‐cell activity, she explained, noting that in preclinical models, CB‐839 increased intra‐tumoral glutamine and enhanced antitumor activity of PD‐1/PD‐L1 inhibitors.
In the phase 1 dose escalation study, she and her colleagues evaluated the safety and efficacy of CB-839 in combination with the PD‐1 inhibitor nivolumab in patients with melanoma, non-small cell lung cancer (NSCLC), or renal cell carcinoma (RCC). Phase 2 expansion cohorts include a melanoma rescue cohort of patients progressing on anti-PD-L1 therapy at study entry (22 patients), an NSCLC and RCC rescue cohort of patients who were progressing on anti-PD-L1 therapy at study entry or who had stable disease for 6 months or longer without a response (11 NSCLC and 11 RCC), an RCC cohort of patients with prior immunotherapy exposure and no response (10 patients), and an RCC cohort of patents who had no prior immunotherapy exposure (28 patients).
During dose escalation, patients received oral CB‐839 at 600 mg or 800 mg twice daily in combination with standard‐dose nivolumab. In the ongoing phase 2 expansion study, which continues to enroll, patients are receiving 800 mg of CB-839 twice daily with standard‐dose nivolumab, Dr. Meric-Bernstam said.
Patients in each of the cohorts were high risk and/or had intermediate or poor prognostic status at study entry. For example, 50% of patients in the melanoma rescue cohort had liver metastases, 77% had other visceral metastases, and 18% had brain metastases, and the majority of patients in the lung cancer/RCC cohort had visceral metastases. Most had progressive disease as their best response on their last line of immunotherapy.
Of 16 response-evaluable melanoma patients, 1 experienced a complete response, 2 had partial responses, and 4 had stable disease.
“So overall in this patient population that was progressing on a PD-1/PD-L1 inhibitor at enrollment, 19% had an objective response. The disease control rate in this group was 44%,” she said.
In evaluable patients in the lung cancer rescue cohort (6 patients), RCC rescue cohort (8 patients), and RCC prior exposure cohort (7 patients), disease control rates ranged from 57% to 75%, and in the immunotherapy-naive RCC cohort (19 patients), the partial response rate was 21%, and 53% had stable disease, so the overall disease control rate was 74%. Half of the patients in that group remain on study, she noted.
A closer look at the melanoma rescue cohort showed dramatic and rapid responses in two patients who each achieved a partial response in about 8 weeks with response durations of 3.7 months and 5.4 months, respectively. Additionally, pre-treatment biopsies in this cohort showed an elevated T-cell inflamed signature associated with clinical benefit from the addition of CB-839, and in one patient who had both a pretreatment and on-treatment biopsy that was evaluable, the latter showed an increase in T-cell inflamed signature and T-cell effector genes.
In all cohorts, the combination therapy was generally well tolerated. A maximum tolerated dose was not reached. Dose-limiting toxicity – a grade 3 alanine aminotransferase (ALT) increase – occurred in one patient on the 800-mg dose. The most common grade 3 or greater adverse events were fatigue, nausea, photophobia, rash, and elevated ALT, she said, noting that two patients discontinued for treatment-related adverse events (one for a grade 3 rash and one for grade 2 pneumonitis).
“Overall there appeared to be no apparent increase in immune-related adverse events, either in rate or severity, compared with [nivolumab] monotherapy,” she said.
The combination of CB-839 and nivolumab was well tolerated, and in some patients – as seen in the melanoma cohort – adding CB-839 to checkpoint blockade can overcome checkpoint blockade resistance, Dr. Meric-Bernstam concluded, noting that the disease control rates seen in the majority of lung cancer and RCC patients who were progressing on checkpoint blockade is encouraging, as is the objective response rate seen thus far in the RCC therapy-naive patients, and the stable and deep responses seen in the melanoma rescue cohort.
“Based on our encouraging signal in the melanoma rescue cohort, this [cohort] has been expanded,” she said.
Calithera Biosciences sponsored the study. Bristol-Myers Squibb provided nivolumab for the study. Dr. Meric-Bernstam has received grant or research support from Calithera Biosciences and many other companies. She also reported being a paid consultant for several companies and serving on an advisory committee or review panel, or as a board member for multiple companies.
AT SITC 2017
Key clinical point:
Major finding: The objective response rate in advanced melanoma patients refractory to anti-PD-1/PD-L1 therapy was 19%.
Data source: A phase 1/2 study of 82 patients.
Disclosures: Calithera Biosciences sponsored the study. Bristol-Myers Squibb provided nivolumab for the study. Dr. Meric-Bernstam has received grant or research support from Calithera Biosciences and many other companies. She also reported being a paid consultant for several companies and serving on an advisory committee or review panel or as a board member for multiple companies.
ENCORE 601 study: Entinostat shows promise in NSCLC
NATIONAL HARBOR, MD. – The oral, class I selective histone deacetylase (HDAC) inhibitor entinostat given in combination with pembrolizumab demonstrated antitumor activity and acceptable safety in patients with non–small cell lung cancer in the phase 1b/2 ENCORE 601 study.
Entinostat, which has been shown in preclinical models to enhance suppressor cells in the tumor microenvironment, was evaluated in ENCORE 601 as a treatment for non–small cell lung cancer (NSCLC), melanoma, and colorectal cancer. Previously reported phase 1 results showed that an oral dose of 5 mg weekly plus 200 mg of pembrolizumab given intravenously every 3 weeks deserved further exploration for these indications, according to Leena Gandhi, MD, who reported phase 2, stage 1 results from the lung cancer arm of the Simon two-stage study at the annual meeting of the Society for Immunotherapy of Cancer.
Treatment at that dose was studied in both anti-PD-L1–naive patients with advanced NSCLC, and in NSCLC patients who progressed on anti-PD-L1 treatment, said Dr. Gandhi of New York University Langone Medical Center.
The primary objective of stage 1 was objective response rate, and criteria for advancement were 4 or more responses out of 17 evaluable anti-PD-L1–naive patients (cohort 1), and at least 3 responses out of 31 patients who progressed on anti-PD-L1 therapy (cohort 2).
Both cohorts met the endpoint, with 4 of 17 evaluable cohort 1 patients (24%) achieving a partial response, and 3 of 31 evaluable cohort 2 patients (10%) achieving a partial response.
In cohort 1, two responses were confirmed and two were unconfirmed. One of the unconfirmed patients had malignant pericardial effusion, but remains on study with continued clinical benefit, Dr. Gandhi said, noting that three patients remain on study in all.
“The other notable thing I’d like to point out here … is that the majority of these were patients who did not have high levels of expression of PD-L1,” she said.
In cohort 2 patients, two responses were confirmed and one was unconfirmed. Three patients remain on study.
“In both of these cohorts there are a couple of patients who’ve had quite durable responses,” she said.
The best response to prior anti-PD-1therapy in the cohort 2 patients who had a response was stable disease (two patients). The response to prior therapy was unknown in one patient, she noted.
“All of them had clear regressions, after that initial PD-1 therapy, with this combination,” she said, noting that two had “essentially negative PD-L1 expression, and none had high levels of expression.”
Treatment was associated with grade 3/4 adverse events deemed drug related in 31% of patients; the most common of these events, occurring in at least 10% of patients in cohort 1, were hypophosphatemia and neutropenia, and in cohort 2 were fatigue, anemia, anorexia, and pneumonitis; 13% of patients discontinued treatment due to an adverse event, Dr. Gandhi said.
Of note, there were reductions in circulating myeloid derived suppressor cells in both cohorts following treatment.
Based on the responses seen in this first stage of the study, cohort 2 has advanced to stage 2 and has completed enrollment. Additional patients have not been enrolled in cohort 1, but that is still under consideration, she said.
Dr. Gandhi reported having no disclosures.
NATIONAL HARBOR, MD. – The oral, class I selective histone deacetylase (HDAC) inhibitor entinostat given in combination with pembrolizumab demonstrated antitumor activity and acceptable safety in patients with non–small cell lung cancer in the phase 1b/2 ENCORE 601 study.
Entinostat, which has been shown in preclinical models to enhance suppressor cells in the tumor microenvironment, was evaluated in ENCORE 601 as a treatment for non–small cell lung cancer (NSCLC), melanoma, and colorectal cancer. Previously reported phase 1 results showed that an oral dose of 5 mg weekly plus 200 mg of pembrolizumab given intravenously every 3 weeks deserved further exploration for these indications, according to Leena Gandhi, MD, who reported phase 2, stage 1 results from the lung cancer arm of the Simon two-stage study at the annual meeting of the Society for Immunotherapy of Cancer.
Treatment at that dose was studied in both anti-PD-L1–naive patients with advanced NSCLC, and in NSCLC patients who progressed on anti-PD-L1 treatment, said Dr. Gandhi of New York University Langone Medical Center.
The primary objective of stage 1 was objective response rate, and criteria for advancement were 4 or more responses out of 17 evaluable anti-PD-L1–naive patients (cohort 1), and at least 3 responses out of 31 patients who progressed on anti-PD-L1 therapy (cohort 2).
Both cohorts met the endpoint, with 4 of 17 evaluable cohort 1 patients (24%) achieving a partial response, and 3 of 31 evaluable cohort 2 patients (10%) achieving a partial response.
In cohort 1, two responses were confirmed and two were unconfirmed. One of the unconfirmed patients had malignant pericardial effusion, but remains on study with continued clinical benefit, Dr. Gandhi said, noting that three patients remain on study in all.
“The other notable thing I’d like to point out here … is that the majority of these were patients who did not have high levels of expression of PD-L1,” she said.
In cohort 2 patients, two responses were confirmed and one was unconfirmed. Three patients remain on study.
“In both of these cohorts there are a couple of patients who’ve had quite durable responses,” she said.
The best response to prior anti-PD-1therapy in the cohort 2 patients who had a response was stable disease (two patients). The response to prior therapy was unknown in one patient, she noted.
“All of them had clear regressions, after that initial PD-1 therapy, with this combination,” she said, noting that two had “essentially negative PD-L1 expression, and none had high levels of expression.”
Treatment was associated with grade 3/4 adverse events deemed drug related in 31% of patients; the most common of these events, occurring in at least 10% of patients in cohort 1, were hypophosphatemia and neutropenia, and in cohort 2 were fatigue, anemia, anorexia, and pneumonitis; 13% of patients discontinued treatment due to an adverse event, Dr. Gandhi said.
Of note, there were reductions in circulating myeloid derived suppressor cells in both cohorts following treatment.
Based on the responses seen in this first stage of the study, cohort 2 has advanced to stage 2 and has completed enrollment. Additional patients have not been enrolled in cohort 1, but that is still under consideration, she said.
Dr. Gandhi reported having no disclosures.
NATIONAL HARBOR, MD. – The oral, class I selective histone deacetylase (HDAC) inhibitor entinostat given in combination with pembrolizumab demonstrated antitumor activity and acceptable safety in patients with non–small cell lung cancer in the phase 1b/2 ENCORE 601 study.
Entinostat, which has been shown in preclinical models to enhance suppressor cells in the tumor microenvironment, was evaluated in ENCORE 601 as a treatment for non–small cell lung cancer (NSCLC), melanoma, and colorectal cancer. Previously reported phase 1 results showed that an oral dose of 5 mg weekly plus 200 mg of pembrolizumab given intravenously every 3 weeks deserved further exploration for these indications, according to Leena Gandhi, MD, who reported phase 2, stage 1 results from the lung cancer arm of the Simon two-stage study at the annual meeting of the Society for Immunotherapy of Cancer.
Treatment at that dose was studied in both anti-PD-L1–naive patients with advanced NSCLC, and in NSCLC patients who progressed on anti-PD-L1 treatment, said Dr. Gandhi of New York University Langone Medical Center.
The primary objective of stage 1 was objective response rate, and criteria for advancement were 4 or more responses out of 17 evaluable anti-PD-L1–naive patients (cohort 1), and at least 3 responses out of 31 patients who progressed on anti-PD-L1 therapy (cohort 2).
Both cohorts met the endpoint, with 4 of 17 evaluable cohort 1 patients (24%) achieving a partial response, and 3 of 31 evaluable cohort 2 patients (10%) achieving a partial response.
In cohort 1, two responses were confirmed and two were unconfirmed. One of the unconfirmed patients had malignant pericardial effusion, but remains on study with continued clinical benefit, Dr. Gandhi said, noting that three patients remain on study in all.
“The other notable thing I’d like to point out here … is that the majority of these were patients who did not have high levels of expression of PD-L1,” she said.
In cohort 2 patients, two responses were confirmed and one was unconfirmed. Three patients remain on study.
“In both of these cohorts there are a couple of patients who’ve had quite durable responses,” she said.
The best response to prior anti-PD-1therapy in the cohort 2 patients who had a response was stable disease (two patients). The response to prior therapy was unknown in one patient, she noted.
“All of them had clear regressions, after that initial PD-1 therapy, with this combination,” she said, noting that two had “essentially negative PD-L1 expression, and none had high levels of expression.”
Treatment was associated with grade 3/4 adverse events deemed drug related in 31% of patients; the most common of these events, occurring in at least 10% of patients in cohort 1, were hypophosphatemia and neutropenia, and in cohort 2 were fatigue, anemia, anorexia, and pneumonitis; 13% of patients discontinued treatment due to an adverse event, Dr. Gandhi said.
Of note, there were reductions in circulating myeloid derived suppressor cells in both cohorts following treatment.
Based on the responses seen in this first stage of the study, cohort 2 has advanced to stage 2 and has completed enrollment. Additional patients have not been enrolled in cohort 1, but that is still under consideration, she said.
Dr. Gandhi reported having no disclosures.
AT SITC 2017
Key clinical point:
Major finding: Partial responses were seen in 24% of cohort 1 patients and 10% of cohort 2 patients.
Data source: Stage 1 of a phase 2 Simon two-stage study (48 evaluable patients).
Disclosures: Dr. Gandhi reported having no disclosures.
Breakthrough cancer gene assay approved, CMS proposes coverage
The Food and Drug Administration approved a new genetic sequencing test that detects mutations across 324 genes in tumor biopsy specimens with an accuracy of 94.6%.
The FoundationOne CDx (F1CDx) test from Foundation Medicine “can identify which patients with any of five tumor types” – non–small-cell lung cancer, melanoma, breast cancer, colorectal cancer, or ovarian cancer – “may benefit from 15 different FDA-approved targeted treatment options,” as well as clinical trial eligibility, “with one test report, avoiding duplicative biopsies,” the agency said in a statement.
On the same day as the approval, the Centers for Medicare & Medicaid Services proposed nationwide coverage for Medicare beneficiaries with recurrent or metastatic disease. CMS is accepting public comments on the proposal for 30 days. The cost of the test is $5,800.
F1CDx went through the FDA and CMS Parallel Review Program, in which the agencies review medical devices concurrently to help reduce the time between approval and Medicare coverage.
F1CDx reads the order of nucleotides on DNA isolated from biopsy specimens to detect a range of genetic anomalies, including base substitutions, insertion and deletion alterations, copy number alterations, and select gene rearrangements, as well as genomic signatures including microsatellite instability and tumor mutational burden. Clinical performance was established by comparing the F1CDx to previously approved tests.
The Food and Drug Administration approved a new genetic sequencing test that detects mutations across 324 genes in tumor biopsy specimens with an accuracy of 94.6%.
The FoundationOne CDx (F1CDx) test from Foundation Medicine “can identify which patients with any of five tumor types” – non–small-cell lung cancer, melanoma, breast cancer, colorectal cancer, or ovarian cancer – “may benefit from 15 different FDA-approved targeted treatment options,” as well as clinical trial eligibility, “with one test report, avoiding duplicative biopsies,” the agency said in a statement.
On the same day as the approval, the Centers for Medicare & Medicaid Services proposed nationwide coverage for Medicare beneficiaries with recurrent or metastatic disease. CMS is accepting public comments on the proposal for 30 days. The cost of the test is $5,800.
F1CDx went through the FDA and CMS Parallel Review Program, in which the agencies review medical devices concurrently to help reduce the time between approval and Medicare coverage.
F1CDx reads the order of nucleotides on DNA isolated from biopsy specimens to detect a range of genetic anomalies, including base substitutions, insertion and deletion alterations, copy number alterations, and select gene rearrangements, as well as genomic signatures including microsatellite instability and tumor mutational burden. Clinical performance was established by comparing the F1CDx to previously approved tests.
The Food and Drug Administration approved a new genetic sequencing test that detects mutations across 324 genes in tumor biopsy specimens with an accuracy of 94.6%.
The FoundationOne CDx (F1CDx) test from Foundation Medicine “can identify which patients with any of five tumor types” – non–small-cell lung cancer, melanoma, breast cancer, colorectal cancer, or ovarian cancer – “may benefit from 15 different FDA-approved targeted treatment options,” as well as clinical trial eligibility, “with one test report, avoiding duplicative biopsies,” the agency said in a statement.
On the same day as the approval, the Centers for Medicare & Medicaid Services proposed nationwide coverage for Medicare beneficiaries with recurrent or metastatic disease. CMS is accepting public comments on the proposal for 30 days. The cost of the test is $5,800.
F1CDx went through the FDA and CMS Parallel Review Program, in which the agencies review medical devices concurrently to help reduce the time between approval and Medicare coverage.
F1CDx reads the order of nucleotides on DNA isolated from biopsy specimens to detect a range of genetic anomalies, including base substitutions, insertion and deletion alterations, copy number alterations, and select gene rearrangements, as well as genomic signatures including microsatellite instability and tumor mutational burden. Clinical performance was established by comparing the F1CDx to previously approved tests.
PD-L1-targeting drug atezolizumab nabs approval for non-small cell lung cancer
The approval last fall by the US Food and Drug Administration (FDA) of the immune checkpoint inhibitor atezolizumab for the treatment of metastatic non–small cell lung cancer (NSCLC) marked the second approved indication for the drug in a single year. Atezolizumab, which targets the programmed cell death protein-ligand 1 (PD-L1), has a unique mechanism of action compared with other approved immune checkpoint inhibitors and had been previously approved for the treatment of patients with advanced urothelial carcinoma. The current approval was based on 2 international, randomized, open-label trials, involving more than 1,000 patients, in which atezolizumab outperformed the chemotherapeutic drug docetaxel.
The POPLAR trial1 was a phase 2 study performed at 61 academic centers and community oncology practices across 13 countries in Europe and North America and enrolled 287 patients, while the phase 3 OAK trial2 was carried out at 193 centers in 31 countries and enrolled 850 patients.
Eligibility criteria for the 2 studies were similar; patients were 18 years or older, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, had measurable disease as per Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), and adequate hematologic and end-organ function. Patients with asymptomatic or treated central nervous system (CNS) metastases were also eligible.
Patients were excluded from both studies if they had a history of autoimmune disease, pneumonitis or chronic viral diseases, or had received previous treatment with docetaxel, CD137 agonists, anti-CTLA4, anti-PD-L1 or anti-PD-1 therapeutics.
Patients were randomized 1:1 to receive atezolizumab, administered intravenously at a fixed dose of 1,200 mg, or a 75 mg/m2 dose of docetaxel, every 3 weeks on day 1 of each 3-week cycle. Randomization was stratified according to PD-L1 expression, number of previous chemotherapy regimens, and histology (squamous vs nonsquamous). Tumors were assessed at baseline and every 6 weeks for 36 weeks, and every 9 weeks thereafter until progression or, in patients who continued beyond progression, until discontinuation. PD-L1 expression was assessed prospectively on tumor cells and tumor-infiltrating immune cells, using the FDA-approved companion diagnostic, Ventana SP142 PD-L1 immunohistochemistry assay.
The primary endpoint in both studies was overall survival (OS), which was significantly improved in the atezolizumab treatment arm. In the POPLAR trial, over a minimum follow-up of 13 months, the median OS was 12.6 months in atezolizumab-treated patients, compared with 9.7 months in docetaxel-treated patients (hazard ratio [HR], 0.73; P = .04) and in the OAK trial, over a median follow-up of 21 months, the median OS was 15.7 months and 10.3 months, respectively.
The main difference between the 2 trials was that the OS benefit in the POPLAR trial reached statistical significance only in patients with the highest levels of PD-L1 expression, whereas in the OAK study, the OS was significantly improved regardless of PD-L1 expression status, although the greatest benefit was derived in patients with higher levels of PD-L1 expression (20.5 months and 8.9 months, respectively). The OS benefit was observed across all other prespecified subgroups, except in patients with EGFR mutation-positivity in the OAK trial. Progression-free survival and overall response rates were similar in the 2 treatment arms in both studies.
An additional 375 patients were enrolled in the OAK trial and included in the safety analyses. In both studies, the safety profile of atezolizumab was similar to that observed in previous studies of this drug. The most commonly observed adverse events (AEs) with atezolizumab treatment were fatigue, decreased appetite, dyspnea, cough, nausea, musculoskeletal pain, and constipation. Despite longer treatment duration, there were fewer incidences of grade 3/4 AEs with atezolizumab compared with docetaxel (37% vs 54%, respectively, in the OAK trial), a lower rate of discontinuation with atezolizumab treatment, and no deaths related to the study drug. The most common grade 3/4 AEs included dyspnea, pneumonia, hypoxia, hyponatremia, fatigue, and anemia. Clinically significant immune-related AEs included pneumonitis, hepatitis, colitis, and thyroid disease.
Atezolizumab is marketed as Tecentriq by Genentech and the prescribing information details warnings and precautions about immune-related AEs; pneumonitis, colitis, endocrinopathies (eg, thyroid disorders, adrenal insufficiency, and diabetes mellitus), and others, as well as infections, infusion-related reactions, and embryofetal toxicity.3
Atezolizumab treatment should be withheld for grade 2 pneumonitis; aspartate aminotransferase (AST) or alanine aminotransferase (ALT) levels of >3-5 times the upper limit of normal (ULN) or total bilirubin levels of >1.5 and up to 3 times the ULN; grade 2/3 diarrhea or colitis, adrenal insufficiency, hypothyroidism, or grade 3/4 hyperglycemia; grade 2 ocular inflammatory toxicity; grade 2/3 pancreatitis or grade 3/4 increases in amylase or lipase levels; grade 3/4 infection; grade 2 infusion-related reactions; and grade 3 rash. Treatment can then be resumed in patients whose AEs recover to grade 0 or 1.
Treatment should be permanently discontinued in the event of grade 3/4 pneumonitis; AST/ALT levels of >5 times the ULN or bilirubin levels of >3 times the ULN; grade 4 diarrhea or colitis; myasthenic syndrome/myasthenia gravis, Guillain-Barre syndrome or meningoencephalitis (all grades); grade 3/4 ocular inflammatory toxicity; grade 4 or recurrent (any grade) pancreatitis; grade 3/4 infusion-related reactions; or grade 4 rash. Patients should also be advised of the risk of fetal harm.
The approval last fall by the US Food and Drug Administration (FDA) of the immune checkpoint inhibitor atezolizumab for the treatment of metastatic non–small cell lung cancer (NSCLC) marked the second approved indication for the drug in a single year. Atezolizumab, which targets the programmed cell death protein-ligand 1 (PD-L1), has a unique mechanism of action compared with other approved immune checkpoint inhibitors and had been previously approved for the treatment of patients with advanced urothelial carcinoma. The current approval was based on 2 international, randomized, open-label trials, involving more than 1,000 patients, in which atezolizumab outperformed the chemotherapeutic drug docetaxel.
The POPLAR trial1 was a phase 2 study performed at 61 academic centers and community oncology practices across 13 countries in Europe and North America and enrolled 287 patients, while the phase 3 OAK trial2 was carried out at 193 centers in 31 countries and enrolled 850 patients.
Eligibility criteria for the 2 studies were similar; patients were 18 years or older, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, had measurable disease as per Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), and adequate hematologic and end-organ function. Patients with asymptomatic or treated central nervous system (CNS) metastases were also eligible.
Patients were excluded from both studies if they had a history of autoimmune disease, pneumonitis or chronic viral diseases, or had received previous treatment with docetaxel, CD137 agonists, anti-CTLA4, anti-PD-L1 or anti-PD-1 therapeutics.
Patients were randomized 1:1 to receive atezolizumab, administered intravenously at a fixed dose of 1,200 mg, or a 75 mg/m2 dose of docetaxel, every 3 weeks on day 1 of each 3-week cycle. Randomization was stratified according to PD-L1 expression, number of previous chemotherapy regimens, and histology (squamous vs nonsquamous). Tumors were assessed at baseline and every 6 weeks for 36 weeks, and every 9 weeks thereafter until progression or, in patients who continued beyond progression, until discontinuation. PD-L1 expression was assessed prospectively on tumor cells and tumor-infiltrating immune cells, using the FDA-approved companion diagnostic, Ventana SP142 PD-L1 immunohistochemistry assay.
The primary endpoint in both studies was overall survival (OS), which was significantly improved in the atezolizumab treatment arm. In the POPLAR trial, over a minimum follow-up of 13 months, the median OS was 12.6 months in atezolizumab-treated patients, compared with 9.7 months in docetaxel-treated patients (hazard ratio [HR], 0.73; P = .04) and in the OAK trial, over a median follow-up of 21 months, the median OS was 15.7 months and 10.3 months, respectively.
The main difference between the 2 trials was that the OS benefit in the POPLAR trial reached statistical significance only in patients with the highest levels of PD-L1 expression, whereas in the OAK study, the OS was significantly improved regardless of PD-L1 expression status, although the greatest benefit was derived in patients with higher levels of PD-L1 expression (20.5 months and 8.9 months, respectively). The OS benefit was observed across all other prespecified subgroups, except in patients with EGFR mutation-positivity in the OAK trial. Progression-free survival and overall response rates were similar in the 2 treatment arms in both studies.
An additional 375 patients were enrolled in the OAK trial and included in the safety analyses. In both studies, the safety profile of atezolizumab was similar to that observed in previous studies of this drug. The most commonly observed adverse events (AEs) with atezolizumab treatment were fatigue, decreased appetite, dyspnea, cough, nausea, musculoskeletal pain, and constipation. Despite longer treatment duration, there were fewer incidences of grade 3/4 AEs with atezolizumab compared with docetaxel (37% vs 54%, respectively, in the OAK trial), a lower rate of discontinuation with atezolizumab treatment, and no deaths related to the study drug. The most common grade 3/4 AEs included dyspnea, pneumonia, hypoxia, hyponatremia, fatigue, and anemia. Clinically significant immune-related AEs included pneumonitis, hepatitis, colitis, and thyroid disease.
Atezolizumab is marketed as Tecentriq by Genentech and the prescribing information details warnings and precautions about immune-related AEs; pneumonitis, colitis, endocrinopathies (eg, thyroid disorders, adrenal insufficiency, and diabetes mellitus), and others, as well as infections, infusion-related reactions, and embryofetal toxicity.3
Atezolizumab treatment should be withheld for grade 2 pneumonitis; aspartate aminotransferase (AST) or alanine aminotransferase (ALT) levels of >3-5 times the upper limit of normal (ULN) or total bilirubin levels of >1.5 and up to 3 times the ULN; grade 2/3 diarrhea or colitis, adrenal insufficiency, hypothyroidism, or grade 3/4 hyperglycemia; grade 2 ocular inflammatory toxicity; grade 2/3 pancreatitis or grade 3/4 increases in amylase or lipase levels; grade 3/4 infection; grade 2 infusion-related reactions; and grade 3 rash. Treatment can then be resumed in patients whose AEs recover to grade 0 or 1.
Treatment should be permanently discontinued in the event of grade 3/4 pneumonitis; AST/ALT levels of >5 times the ULN or bilirubin levels of >3 times the ULN; grade 4 diarrhea or colitis; myasthenic syndrome/myasthenia gravis, Guillain-Barre syndrome or meningoencephalitis (all grades); grade 3/4 ocular inflammatory toxicity; grade 4 or recurrent (any grade) pancreatitis; grade 3/4 infusion-related reactions; or grade 4 rash. Patients should also be advised of the risk of fetal harm.
The approval last fall by the US Food and Drug Administration (FDA) of the immune checkpoint inhibitor atezolizumab for the treatment of metastatic non–small cell lung cancer (NSCLC) marked the second approved indication for the drug in a single year. Atezolizumab, which targets the programmed cell death protein-ligand 1 (PD-L1), has a unique mechanism of action compared with other approved immune checkpoint inhibitors and had been previously approved for the treatment of patients with advanced urothelial carcinoma. The current approval was based on 2 international, randomized, open-label trials, involving more than 1,000 patients, in which atezolizumab outperformed the chemotherapeutic drug docetaxel.
The POPLAR trial1 was a phase 2 study performed at 61 academic centers and community oncology practices across 13 countries in Europe and North America and enrolled 287 patients, while the phase 3 OAK trial2 was carried out at 193 centers in 31 countries and enrolled 850 patients.
Eligibility criteria for the 2 studies were similar; patients were 18 years or older, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, had measurable disease as per Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), and adequate hematologic and end-organ function. Patients with asymptomatic or treated central nervous system (CNS) metastases were also eligible.
Patients were excluded from both studies if they had a history of autoimmune disease, pneumonitis or chronic viral diseases, or had received previous treatment with docetaxel, CD137 agonists, anti-CTLA4, anti-PD-L1 or anti-PD-1 therapeutics.
Patients were randomized 1:1 to receive atezolizumab, administered intravenously at a fixed dose of 1,200 mg, or a 75 mg/m2 dose of docetaxel, every 3 weeks on day 1 of each 3-week cycle. Randomization was stratified according to PD-L1 expression, number of previous chemotherapy regimens, and histology (squamous vs nonsquamous). Tumors were assessed at baseline and every 6 weeks for 36 weeks, and every 9 weeks thereafter until progression or, in patients who continued beyond progression, until discontinuation. PD-L1 expression was assessed prospectively on tumor cells and tumor-infiltrating immune cells, using the FDA-approved companion diagnostic, Ventana SP142 PD-L1 immunohistochemistry assay.
The primary endpoint in both studies was overall survival (OS), which was significantly improved in the atezolizumab treatment arm. In the POPLAR trial, over a minimum follow-up of 13 months, the median OS was 12.6 months in atezolizumab-treated patients, compared with 9.7 months in docetaxel-treated patients (hazard ratio [HR], 0.73; P = .04) and in the OAK trial, over a median follow-up of 21 months, the median OS was 15.7 months and 10.3 months, respectively.
The main difference between the 2 trials was that the OS benefit in the POPLAR trial reached statistical significance only in patients with the highest levels of PD-L1 expression, whereas in the OAK study, the OS was significantly improved regardless of PD-L1 expression status, although the greatest benefit was derived in patients with higher levels of PD-L1 expression (20.5 months and 8.9 months, respectively). The OS benefit was observed across all other prespecified subgroups, except in patients with EGFR mutation-positivity in the OAK trial. Progression-free survival and overall response rates were similar in the 2 treatment arms in both studies.
An additional 375 patients were enrolled in the OAK trial and included in the safety analyses. In both studies, the safety profile of atezolizumab was similar to that observed in previous studies of this drug. The most commonly observed adverse events (AEs) with atezolizumab treatment were fatigue, decreased appetite, dyspnea, cough, nausea, musculoskeletal pain, and constipation. Despite longer treatment duration, there were fewer incidences of grade 3/4 AEs with atezolizumab compared with docetaxel (37% vs 54%, respectively, in the OAK trial), a lower rate of discontinuation with atezolizumab treatment, and no deaths related to the study drug. The most common grade 3/4 AEs included dyspnea, pneumonia, hypoxia, hyponatremia, fatigue, and anemia. Clinically significant immune-related AEs included pneumonitis, hepatitis, colitis, and thyroid disease.
Atezolizumab is marketed as Tecentriq by Genentech and the prescribing information details warnings and precautions about immune-related AEs; pneumonitis, colitis, endocrinopathies (eg, thyroid disorders, adrenal insufficiency, and diabetes mellitus), and others, as well as infections, infusion-related reactions, and embryofetal toxicity.3
Atezolizumab treatment should be withheld for grade 2 pneumonitis; aspartate aminotransferase (AST) or alanine aminotransferase (ALT) levels of >3-5 times the upper limit of normal (ULN) or total bilirubin levels of >1.5 and up to 3 times the ULN; grade 2/3 diarrhea or colitis, adrenal insufficiency, hypothyroidism, or grade 3/4 hyperglycemia; grade 2 ocular inflammatory toxicity; grade 2/3 pancreatitis or grade 3/4 increases in amylase or lipase levels; grade 3/4 infection; grade 2 infusion-related reactions; and grade 3 rash. Treatment can then be resumed in patients whose AEs recover to grade 0 or 1.
Treatment should be permanently discontinued in the event of grade 3/4 pneumonitis; AST/ALT levels of >5 times the ULN or bilirubin levels of >3 times the ULN; grade 4 diarrhea or colitis; myasthenic syndrome/myasthenia gravis, Guillain-Barre syndrome or meningoencephalitis (all grades); grade 3/4 ocular inflammatory toxicity; grade 4 or recurrent (any grade) pancreatitis; grade 3/4 infusion-related reactions; or grade 4 rash. Patients should also be advised of the risk of fetal harm.
Defining quality in lung cancer surgery
Implementing quality initiatives and creating reporting mechanisms for lung cancer patients can lead to better outcomes, including overall survival. While barriers exist – namely the conflicting perspectives of providers, payers, hospitals, and patients – thoracic oncologic surgeons should seize the opportunity to establish robust quality and value metrics for lung cancer programs, said Whitney S. Brandt, MD, and her coauthors in an expert opinion in the Journal of Thoracic and Cardiovascular Surgery (2017;154:1397-403).
Dr. Brandt, a surgeon at Memorial Sloan Kettering Cancer Center in New York, and her coauthors examined the key elements of quality and value initiatives, categorizing them into preoperative, intraoperative, and postoperative components and primarily focusing on early stage lung cancer. The National Institutes of Health/National Cancer Center provided a grant for the authors’ work.
The preoperative evaluation should at least include CT imaging of the tumor and, for smokers, smoking cessation, said Dr. Brandt and her coauthors. All candidates for pulmonary lung resection should have spirometry and diffusion capacity tests; furthermore, both predicted postoperative forced expiratory volume in 1 second and diffusing capacity of the lungs for CO should be calculated. “Patients with a predicted postoperative value less than 40% for either measurement should be considered high risk for lobectomy and should be offered either sublobar resection or nonsurgical therapy,” they recommended.
Dr. Brandt and her colleagues also clarified preoperative management of patients with cardiac disease. Only patients with significant cardiac disease risk factors need to undergo cardiac testing before lung surgery, and patients with stable cardiac disease do not require revascularization beforehand.
For preoperative staging, the most comprehensive clinical guidelines come from the National Comprehensive Cancer Network, they stated. The guidelines recommend that all patients with a small cell lung cancer or stage II to IV non–small cell lung cancer (NSCLC) receive a brain MRI or – if that’s not available – a head CT with contrast to assess for brain metastasis.
Intraoperative quality measures take into account the surgical approach, including cost, resection and margins, and lymph node evaluation. With regard to surgical approach, trials have shown traditional video-assisted surgery (VATS) lobectomy results in shorter hospital stays and thereby lower costs, as well as fewer complications and deaths, than thoracotomy, said Dr. Brandt and her coauthors. But that cost advantage has not yet carried over to robotic-assisted VATS. That said, “robotic-assisted VATS remains a relatively new technology, and with time and increased robotic platform competition, costs will likely decrease.”
Dr. Brandt and her coauthors also noted that clinical trials support resection margins of 2 cm in patients having surgery for NSCLC and that adequate lymph node evaluation is a critical component of a lung cancer quality initiative. “Regardless of whether lymph nodes are sampled or dissected, we believe that systematic acquisition of mediastinal nodal tissue based on nodal station(s) is a useful quality metric, and, therefore, we recommend each program adopt a preferred approach and track adherence,” they said.
As for postoperative quality metrics, the most obvious are morbidity and mortality. “A quality program should track 30-day or in-hospital mortality, as well as 90-day mortality, following lung cancer resection.” Such metrics can serve as “starting points” for quality improvement initiatives. Length of stay has also emerged as an important metric because it is a surrogate of other metrics, such as patient comorbidities, age, and socioeconomic status. “Length-of-stay metrics likely need to be risk-stratified on the basis of these and other variables to be meaningful to a practicing surgeon,” Dr. Brandt and her coauthors said, adding that: “Studying the effectiveness of enhanced recovery after surgery programs in thoracic surgical oncology poses an opportunity for a well-designed trial.”
Two other key quality metrics for lung cancer programs that need further development were pointed out in the paper: hospital readmissions and tracking of adjuvant therapies. “Programmatic oncologic quality metrics to track appropriate and inappropriate referrals for adjuvant therapy and the number of patients who complete such therapy are important,” they said.
Another step programs should take: Participating in a national or regional database, as recommended by the Society of Thoracic Surgeons, and taking advantage of the “clear benefits to benchmarking your program to others.”
Dr. Brandt and her coauthors reported having no financial disclosures. The National Institutes of Health/National Cancer Center provided grant support.
Whitney S. Brandt, MD, and her coauthors pointed out the difficulty of finding a comprehensive quality metric because of the multitude of contributing indicators, said Alessandro Brunelli, MD, of St. James University Hospital in Leeds, England, in his invited commentary (J Thorac Cardiovasc Surg. 2017;154:1404-5). But he added that two nonclinical indicators needed further consideration: patient perspectives and costs.
“Satisfaction with care depends on multiple subjective factors and is affected by different socioeconomic and cultural backgrounds,” Dr. Brunelli said. “There have been very few attempts to use patient satisfaction scales as a measure of quality in our specialty.” Residual quality of life after surgery is another key measure of patient perspective. “Long-term survival in fact cannot be assessed in isolation and without taking into consideration the actual quality of life of the cancer survivors,” he said. That information would help inform surgical decision-making.
To be meaningful as a quality metric, cost requires clinical risk adjustment, Dr. Brunelli wrote, and surgeons should take the lead here “to prevent misleading evaluations by third parties.” He added, “There have been few studies reporting on financial risk models in our specialty, and more research is needed in this field.”
Dr. Brunelli reported having no financial disclosures.
Whitney S. Brandt, MD, and her coauthors pointed out the difficulty of finding a comprehensive quality metric because of the multitude of contributing indicators, said Alessandro Brunelli, MD, of St. James University Hospital in Leeds, England, in his invited commentary (J Thorac Cardiovasc Surg. 2017;154:1404-5). But he added that two nonclinical indicators needed further consideration: patient perspectives and costs.
“Satisfaction with care depends on multiple subjective factors and is affected by different socioeconomic and cultural backgrounds,” Dr. Brunelli said. “There have been very few attempts to use patient satisfaction scales as a measure of quality in our specialty.” Residual quality of life after surgery is another key measure of patient perspective. “Long-term survival in fact cannot be assessed in isolation and without taking into consideration the actual quality of life of the cancer survivors,” he said. That information would help inform surgical decision-making.
To be meaningful as a quality metric, cost requires clinical risk adjustment, Dr. Brunelli wrote, and surgeons should take the lead here “to prevent misleading evaluations by third parties.” He added, “There have been few studies reporting on financial risk models in our specialty, and more research is needed in this field.”
Dr. Brunelli reported having no financial disclosures.
Whitney S. Brandt, MD, and her coauthors pointed out the difficulty of finding a comprehensive quality metric because of the multitude of contributing indicators, said Alessandro Brunelli, MD, of St. James University Hospital in Leeds, England, in his invited commentary (J Thorac Cardiovasc Surg. 2017;154:1404-5). But he added that two nonclinical indicators needed further consideration: patient perspectives and costs.
“Satisfaction with care depends on multiple subjective factors and is affected by different socioeconomic and cultural backgrounds,” Dr. Brunelli said. “There have been very few attempts to use patient satisfaction scales as a measure of quality in our specialty.” Residual quality of life after surgery is another key measure of patient perspective. “Long-term survival in fact cannot be assessed in isolation and without taking into consideration the actual quality of life of the cancer survivors,” he said. That information would help inform surgical decision-making.
To be meaningful as a quality metric, cost requires clinical risk adjustment, Dr. Brunelli wrote, and surgeons should take the lead here “to prevent misleading evaluations by third parties.” He added, “There have been few studies reporting on financial risk models in our specialty, and more research is needed in this field.”
Dr. Brunelli reported having no financial disclosures.
Implementing quality initiatives and creating reporting mechanisms for lung cancer patients can lead to better outcomes, including overall survival. While barriers exist – namely the conflicting perspectives of providers, payers, hospitals, and patients – thoracic oncologic surgeons should seize the opportunity to establish robust quality and value metrics for lung cancer programs, said Whitney S. Brandt, MD, and her coauthors in an expert opinion in the Journal of Thoracic and Cardiovascular Surgery (2017;154:1397-403).
Dr. Brandt, a surgeon at Memorial Sloan Kettering Cancer Center in New York, and her coauthors examined the key elements of quality and value initiatives, categorizing them into preoperative, intraoperative, and postoperative components and primarily focusing on early stage lung cancer. The National Institutes of Health/National Cancer Center provided a grant for the authors’ work.
The preoperative evaluation should at least include CT imaging of the tumor and, for smokers, smoking cessation, said Dr. Brandt and her coauthors. All candidates for pulmonary lung resection should have spirometry and diffusion capacity tests; furthermore, both predicted postoperative forced expiratory volume in 1 second and diffusing capacity of the lungs for CO should be calculated. “Patients with a predicted postoperative value less than 40% for either measurement should be considered high risk for lobectomy and should be offered either sublobar resection or nonsurgical therapy,” they recommended.
Dr. Brandt and her colleagues also clarified preoperative management of patients with cardiac disease. Only patients with significant cardiac disease risk factors need to undergo cardiac testing before lung surgery, and patients with stable cardiac disease do not require revascularization beforehand.
For preoperative staging, the most comprehensive clinical guidelines come from the National Comprehensive Cancer Network, they stated. The guidelines recommend that all patients with a small cell lung cancer or stage II to IV non–small cell lung cancer (NSCLC) receive a brain MRI or – if that’s not available – a head CT with contrast to assess for brain metastasis.
Intraoperative quality measures take into account the surgical approach, including cost, resection and margins, and lymph node evaluation. With regard to surgical approach, trials have shown traditional video-assisted surgery (VATS) lobectomy results in shorter hospital stays and thereby lower costs, as well as fewer complications and deaths, than thoracotomy, said Dr. Brandt and her coauthors. But that cost advantage has not yet carried over to robotic-assisted VATS. That said, “robotic-assisted VATS remains a relatively new technology, and with time and increased robotic platform competition, costs will likely decrease.”
Dr. Brandt and her coauthors also noted that clinical trials support resection margins of 2 cm in patients having surgery for NSCLC and that adequate lymph node evaluation is a critical component of a lung cancer quality initiative. “Regardless of whether lymph nodes are sampled or dissected, we believe that systematic acquisition of mediastinal nodal tissue based on nodal station(s) is a useful quality metric, and, therefore, we recommend each program adopt a preferred approach and track adherence,” they said.
As for postoperative quality metrics, the most obvious are morbidity and mortality. “A quality program should track 30-day or in-hospital mortality, as well as 90-day mortality, following lung cancer resection.” Such metrics can serve as “starting points” for quality improvement initiatives. Length of stay has also emerged as an important metric because it is a surrogate of other metrics, such as patient comorbidities, age, and socioeconomic status. “Length-of-stay metrics likely need to be risk-stratified on the basis of these and other variables to be meaningful to a practicing surgeon,” Dr. Brandt and her coauthors said, adding that: “Studying the effectiveness of enhanced recovery after surgery programs in thoracic surgical oncology poses an opportunity for a well-designed trial.”
Two other key quality metrics for lung cancer programs that need further development were pointed out in the paper: hospital readmissions and tracking of adjuvant therapies. “Programmatic oncologic quality metrics to track appropriate and inappropriate referrals for adjuvant therapy and the number of patients who complete such therapy are important,” they said.
Another step programs should take: Participating in a national or regional database, as recommended by the Society of Thoracic Surgeons, and taking advantage of the “clear benefits to benchmarking your program to others.”
Dr. Brandt and her coauthors reported having no financial disclosures. The National Institutes of Health/National Cancer Center provided grant support.
Implementing quality initiatives and creating reporting mechanisms for lung cancer patients can lead to better outcomes, including overall survival. While barriers exist – namely the conflicting perspectives of providers, payers, hospitals, and patients – thoracic oncologic surgeons should seize the opportunity to establish robust quality and value metrics for lung cancer programs, said Whitney S. Brandt, MD, and her coauthors in an expert opinion in the Journal of Thoracic and Cardiovascular Surgery (2017;154:1397-403).
Dr. Brandt, a surgeon at Memorial Sloan Kettering Cancer Center in New York, and her coauthors examined the key elements of quality and value initiatives, categorizing them into preoperative, intraoperative, and postoperative components and primarily focusing on early stage lung cancer. The National Institutes of Health/National Cancer Center provided a grant for the authors’ work.
The preoperative evaluation should at least include CT imaging of the tumor and, for smokers, smoking cessation, said Dr. Brandt and her coauthors. All candidates for pulmonary lung resection should have spirometry and diffusion capacity tests; furthermore, both predicted postoperative forced expiratory volume in 1 second and diffusing capacity of the lungs for CO should be calculated. “Patients with a predicted postoperative value less than 40% for either measurement should be considered high risk for lobectomy and should be offered either sublobar resection or nonsurgical therapy,” they recommended.
Dr. Brandt and her colleagues also clarified preoperative management of patients with cardiac disease. Only patients with significant cardiac disease risk factors need to undergo cardiac testing before lung surgery, and patients with stable cardiac disease do not require revascularization beforehand.
For preoperative staging, the most comprehensive clinical guidelines come from the National Comprehensive Cancer Network, they stated. The guidelines recommend that all patients with a small cell lung cancer or stage II to IV non–small cell lung cancer (NSCLC) receive a brain MRI or – if that’s not available – a head CT with contrast to assess for brain metastasis.
Intraoperative quality measures take into account the surgical approach, including cost, resection and margins, and lymph node evaluation. With regard to surgical approach, trials have shown traditional video-assisted surgery (VATS) lobectomy results in shorter hospital stays and thereby lower costs, as well as fewer complications and deaths, than thoracotomy, said Dr. Brandt and her coauthors. But that cost advantage has not yet carried over to robotic-assisted VATS. That said, “robotic-assisted VATS remains a relatively new technology, and with time and increased robotic platform competition, costs will likely decrease.”
Dr. Brandt and her coauthors also noted that clinical trials support resection margins of 2 cm in patients having surgery for NSCLC and that adequate lymph node evaluation is a critical component of a lung cancer quality initiative. “Regardless of whether lymph nodes are sampled or dissected, we believe that systematic acquisition of mediastinal nodal tissue based on nodal station(s) is a useful quality metric, and, therefore, we recommend each program adopt a preferred approach and track adherence,” they said.
As for postoperative quality metrics, the most obvious are morbidity and mortality. “A quality program should track 30-day or in-hospital mortality, as well as 90-day mortality, following lung cancer resection.” Such metrics can serve as “starting points” for quality improvement initiatives. Length of stay has also emerged as an important metric because it is a surrogate of other metrics, such as patient comorbidities, age, and socioeconomic status. “Length-of-stay metrics likely need to be risk-stratified on the basis of these and other variables to be meaningful to a practicing surgeon,” Dr. Brandt and her coauthors said, adding that: “Studying the effectiveness of enhanced recovery after surgery programs in thoracic surgical oncology poses an opportunity for a well-designed trial.”
Two other key quality metrics for lung cancer programs that need further development were pointed out in the paper: hospital readmissions and tracking of adjuvant therapies. “Programmatic oncologic quality metrics to track appropriate and inappropriate referrals for adjuvant therapy and the number of patients who complete such therapy are important,” they said.
Another step programs should take: Participating in a national or regional database, as recommended by the Society of Thoracic Surgeons, and taking advantage of the “clear benefits to benchmarking your program to others.”
Dr. Brandt and her coauthors reported having no financial disclosures. The National Institutes of Health/National Cancer Center provided grant support.
FROM THE JOURNAL OF THORACIC AND CARDIOVASCULAR SURGERY
Key clinical point: Quality and value initiatives in lung cancer surgery are complex and multifaceted.
Major finding: Expert opinion identifies quality and value strategies for the preoperative, intraoperative, and postoperative stages.
Data source: Review of elements of quality and value for lung cancer surgery, including the Donabedian classification of structure, process and outcomes.
Disclosures: Dr. Brandt and co-authors reported having no financial disclosures. The National Institutes of Health/National Cancer Center provided grant support.
Cancer patients with TKI-induced hypothyroidism had better survival rates
VICTORIA, B.C. – When it comes to the adverse effects of tyrosine kinase inhibitors (TKIs), hypothyroidism appears to have a bright side, according to a retrospective cohort study among patients with nonthyroid cancers.
While taking one of these targeted agents, roughly a quarter of patients became overtly hypothyroid, an adverse effect that appears to be due in part to immune destruction. Risk was higher for women and earlier in therapy.
Relative to counterparts who remained euthyroid, overtly hypothyroid patients were 44% less likely to die after other factors were taken into account.
Hypothyroidism may reflect changes in immune activation, Dr. Angell proposed. “Additional studies may be helpful, both prospectively looking at the clinical importance of this finding [of survival benefit], and also potentially mechanistically, to understand the relationship between hypothyroidism and survival in these patients.”
“This is an innovative study that looked at an interesting clinical question,” observed session cochair Angela M. Leung, MD, of the University of California, Los Angeles, and an endocrinologist at both UCLA and the VA Greater Los Angeles Healthcare System.
Thyroid dysfunction is a well-known, common side effect of TKI therapy, Dr. Angell noted. “The possible mechanisms that have been suggested for this are direct toxicity on the thyroid gland, destructive thyroiditis, increased thyroid hormone clearance, and vascular endothelial growth factor (VEGF) inhibition, among others.”
Some previous research has suggested a possible survival benefit of TKI-induced hypothyroidism. But “there are limitations in our understanding of hypothyroidism in this setting, including the timing of onset, what risk factors there may be, and the effect of additional clinical variables on the survival effect seen,” Dr. Angell pointed out.
He and his coinvestigators studied 538 adult patients with nonthyroid cancers (mostly stage III or IV) who received a first TKI during 2000-2013 and were followed up through 2017. They excluded those who had preexisting thyroid disease or were on thyroid-related medications.
During TKI therapy, 26.7% of patients developed overt hypothyroidism, and another 13.2% developed subclinical hypothyroidism.
“For a given drug, patients were less likely to develop hypothyroidism when they were given it subsequent to another TKI, as opposed to it being the initial TKI,” Dr. Angell reported. But median time to onset of hypothyroidism was about 2.5 months, regardless.
Cumulative months of all TKI exposure during cancer treatment were not significantly associated with development of hypothyroidism.
In a multivariate analysis, patients were significantly more likely to develop hypothyroidism if they were female (odds ratio, 1.99) and significantly less likely if they had a longer total time on treatment (OR, 0.98) or received a non-TKI VEGF inhibitor (OR, 0.43). Age, race, and cumulative TKI exposure did not influence the outcome.
In a second multivariate analysis, patients’ risk of death was significantly lower if they developed overt hypothyroidism (hazard ratio, 0.56; P less than .0001), but not if they developed subclinical hypothyroidism (HR, 0.79; P = .1655).
Treatment of hypothyroidism did not appear to influence survival, according to Dr. Angell. However, “there wasn’t a specific decision on who was treated, how they were treated, [or] when they were treated,” he said. “So, it is difficult within this cohort to look specifically at which cutoff would be ideal” for initiating treatment.
Similarly, thyroid function testing was not standardized in this retrospectively identified cohort, so it was not possible to determine how long patients were hypothyroid and whether that had an impact, according to Dr. Angell.
Dr. Angell had no relevant conflicts of interest.
VICTORIA, B.C. – When it comes to the adverse effects of tyrosine kinase inhibitors (TKIs), hypothyroidism appears to have a bright side, according to a retrospective cohort study among patients with nonthyroid cancers.
While taking one of these targeted agents, roughly a quarter of patients became overtly hypothyroid, an adverse effect that appears to be due in part to immune destruction. Risk was higher for women and earlier in therapy.
Relative to counterparts who remained euthyroid, overtly hypothyroid patients were 44% less likely to die after other factors were taken into account.
Hypothyroidism may reflect changes in immune activation, Dr. Angell proposed. “Additional studies may be helpful, both prospectively looking at the clinical importance of this finding [of survival benefit], and also potentially mechanistically, to understand the relationship between hypothyroidism and survival in these patients.”
“This is an innovative study that looked at an interesting clinical question,” observed session cochair Angela M. Leung, MD, of the University of California, Los Angeles, and an endocrinologist at both UCLA and the VA Greater Los Angeles Healthcare System.
Thyroid dysfunction is a well-known, common side effect of TKI therapy, Dr. Angell noted. “The possible mechanisms that have been suggested for this are direct toxicity on the thyroid gland, destructive thyroiditis, increased thyroid hormone clearance, and vascular endothelial growth factor (VEGF) inhibition, among others.”
Some previous research has suggested a possible survival benefit of TKI-induced hypothyroidism. But “there are limitations in our understanding of hypothyroidism in this setting, including the timing of onset, what risk factors there may be, and the effect of additional clinical variables on the survival effect seen,” Dr. Angell pointed out.
He and his coinvestigators studied 538 adult patients with nonthyroid cancers (mostly stage III or IV) who received a first TKI during 2000-2013 and were followed up through 2017. They excluded those who had preexisting thyroid disease or were on thyroid-related medications.
During TKI therapy, 26.7% of patients developed overt hypothyroidism, and another 13.2% developed subclinical hypothyroidism.
“For a given drug, patients were less likely to develop hypothyroidism when they were given it subsequent to another TKI, as opposed to it being the initial TKI,” Dr. Angell reported. But median time to onset of hypothyroidism was about 2.5 months, regardless.
Cumulative months of all TKI exposure during cancer treatment were not significantly associated with development of hypothyroidism.
In a multivariate analysis, patients were significantly more likely to develop hypothyroidism if they were female (odds ratio, 1.99) and significantly less likely if they had a longer total time on treatment (OR, 0.98) or received a non-TKI VEGF inhibitor (OR, 0.43). Age, race, and cumulative TKI exposure did not influence the outcome.
In a second multivariate analysis, patients’ risk of death was significantly lower if they developed overt hypothyroidism (hazard ratio, 0.56; P less than .0001), but not if they developed subclinical hypothyroidism (HR, 0.79; P = .1655).
Treatment of hypothyroidism did not appear to influence survival, according to Dr. Angell. However, “there wasn’t a specific decision on who was treated, how they were treated, [or] when they were treated,” he said. “So, it is difficult within this cohort to look specifically at which cutoff would be ideal” for initiating treatment.
Similarly, thyroid function testing was not standardized in this retrospectively identified cohort, so it was not possible to determine how long patients were hypothyroid and whether that had an impact, according to Dr. Angell.
Dr. Angell had no relevant conflicts of interest.
VICTORIA, B.C. – When it comes to the adverse effects of tyrosine kinase inhibitors (TKIs), hypothyroidism appears to have a bright side, according to a retrospective cohort study among patients with nonthyroid cancers.
While taking one of these targeted agents, roughly a quarter of patients became overtly hypothyroid, an adverse effect that appears to be due in part to immune destruction. Risk was higher for women and earlier in therapy.
Relative to counterparts who remained euthyroid, overtly hypothyroid patients were 44% less likely to die after other factors were taken into account.
Hypothyroidism may reflect changes in immune activation, Dr. Angell proposed. “Additional studies may be helpful, both prospectively looking at the clinical importance of this finding [of survival benefit], and also potentially mechanistically, to understand the relationship between hypothyroidism and survival in these patients.”
“This is an innovative study that looked at an interesting clinical question,” observed session cochair Angela M. Leung, MD, of the University of California, Los Angeles, and an endocrinologist at both UCLA and the VA Greater Los Angeles Healthcare System.
Thyroid dysfunction is a well-known, common side effect of TKI therapy, Dr. Angell noted. “The possible mechanisms that have been suggested for this are direct toxicity on the thyroid gland, destructive thyroiditis, increased thyroid hormone clearance, and vascular endothelial growth factor (VEGF) inhibition, among others.”
Some previous research has suggested a possible survival benefit of TKI-induced hypothyroidism. But “there are limitations in our understanding of hypothyroidism in this setting, including the timing of onset, what risk factors there may be, and the effect of additional clinical variables on the survival effect seen,” Dr. Angell pointed out.
He and his coinvestigators studied 538 adult patients with nonthyroid cancers (mostly stage III or IV) who received a first TKI during 2000-2013 and were followed up through 2017. They excluded those who had preexisting thyroid disease or were on thyroid-related medications.
During TKI therapy, 26.7% of patients developed overt hypothyroidism, and another 13.2% developed subclinical hypothyroidism.
“For a given drug, patients were less likely to develop hypothyroidism when they were given it subsequent to another TKI, as opposed to it being the initial TKI,” Dr. Angell reported. But median time to onset of hypothyroidism was about 2.5 months, regardless.
Cumulative months of all TKI exposure during cancer treatment were not significantly associated with development of hypothyroidism.
In a multivariate analysis, patients were significantly more likely to develop hypothyroidism if they were female (odds ratio, 1.99) and significantly less likely if they had a longer total time on treatment (OR, 0.98) or received a non-TKI VEGF inhibitor (OR, 0.43). Age, race, and cumulative TKI exposure did not influence the outcome.
In a second multivariate analysis, patients’ risk of death was significantly lower if they developed overt hypothyroidism (hazard ratio, 0.56; P less than .0001), but not if they developed subclinical hypothyroidism (HR, 0.79; P = .1655).
Treatment of hypothyroidism did not appear to influence survival, according to Dr. Angell. However, “there wasn’t a specific decision on who was treated, how they were treated, [or] when they were treated,” he said. “So, it is difficult within this cohort to look specifically at which cutoff would be ideal” for initiating treatment.
Similarly, thyroid function testing was not standardized in this retrospectively identified cohort, so it was not possible to determine how long patients were hypothyroid and whether that had an impact, according to Dr. Angell.
Dr. Angell had no relevant conflicts of interest.
AT ATA 2017
Key clinical point:
Major finding: Relative to peers who remained euthyroid, patients who developed overt hypothyroidism had a reduced risk of death (HR, 0.56; P less than .0001).
Data source: A retrospective cohort study of 538 adult patients with mainly advanced nonthyroid cancers treated with a tyrosine kinase inhibitor.
Disclosures: Dr. Angell had no relevant conflicts of interest.
Gut bacteria influenced response to checkpoint inhibitors
The gut microbome may influence responses to immune checkpoint inhibitors, based on results from two studies, and one of the investigators is now gearing up for the next step - evaluating in a clinical trial whether altering the microflora will actually improve responses.
In the first study, investigators carried out a series of experiments using fecal microbiome samples from patients with metastatic melanoma embarking on therapy with a PD-1 (programmed cell death protein 1) inhibitor.
“In melanoma patients, there were differential signals in the gut microbiome of responders versus nonresponders, and I think the clincher was when we transplanted fecal samples from responders to nonresponders in germ-free mice, essentially reconstituting the microbiome and showing that it equally affected the systemic immunity and antitumor immunity when we implanted tumors, as well as response to checkpoint blockade,” lead author Jennifer A. Wargo, MD, MMSc, of the University of Texas MD Anderson Cancer Center in Houston, said in an interview.
Dr. Wargo and her colleagues first collected buccal and fecal microbiome samples from 112 patients with metastatic melanoma before they began therapy with a PD-1 inhibitor. After performing taxonomic profiling on all samples, they found that there was a clustering effect by response status in the gut microbiome, but not the oral microbiome, and because changes in the oral microbiome did not appear to be related to treatment response, they focused on the gut.
When Dr. Wargo and her colleagues studied the posttherapy microbiomes of 43 patients (30 responders and 13 nonresponders) according to Response Evaluation Criteria in Solid Tumors (RECIST 1.1), they found that the responders had a significantly higher degree of alpha diversity, a measure of species diversity within a specific environment, compared with nonresponders (P less than .01). In addition, responders had a relative abundance of Ruminococcaceae, commonly occurring gut microbes that break down complex carbohydrates, the investigators reported (Science. 2017 Nov. 2. doi: 10.1126/science.aan4236).
They found that patients whose microbiomes were diverse in general, and in particular were enriched with Faecalibacterium and Clostridiales species, were more likely to respond to immunotherapy with a PD-1 inhibitor and have a longer duration of progression-free survival. In contrast, patients whose microbiomes were more enriched with Bacteroidales species were more likely to be nonresponders.
To get a better understanding of the mechanisms whereby gut bacteria may influence response to PD-1 inhibitors, they performed metagenomic analysis on samples from 14 responders and 11 nonresponders, and found that responders had micro-organisms predominantly associated with anabolic functions that may support host immunity, whereas nonresponders had microbiomes where catabolic functions were more common.
The investigators next performed immune profiling, and found that both systemic immunity and local immunity in the tumor microenvironment in responders were associated with the aforementioned favorable gut microbiome.
The researchers then transplanted feces from the human donors into germ-free mice and then injected tumor cells into the mice, and found that tumor growth was significantly reduced, and response to PD-1 inhibition was significantly enhanced, in mice who received feces from responders.
“An obvious next step is to run a clinical trial to test the hypothesis that by modulating the microbiome, you can actually enhance responses to therapy,” Dr. Wargo said. Details of the clinical trial are still being worked out, but will likely involve fecal transfers and other mechanisms for modulating the microbiome in hopes of improving responses to PD-1 inhibitors.
“It’s going to be a very biomarker-heavy trial,” she said. “We’re going to look, certainly, for changes in the microbiome, and will also do a lot of profiling in the blood, the tumor, and in the microbiome to see if there are changes that occur by modulating that microbiome. Then of course we’ll look for differences in response rates in patients as well.”
Bacteria also affect epithelial cancers
In a separate study, also published in Science, investigators led by Bertrand Routy, MD, of the Gustave Roussy Cancer Institute in Villejuif, France, reported that patients with non–small cell lung cancer and urothelial carcinoma who had previously used systemic antibiotics had reduced survival when treated with a PD-1 inhibitor, compared with patients who had never taken antibiotics (Science. 2017 Nov. 2 doi: 10.1126/science.aan3706).
Analysis of the gut microbiome in these patients showed that higher levels of Akkermansia muciniphila were associated with the best clinical outcomes, with the species detectable in the microbiome of 69% of patients who had partial responses to anti–PD-1 therapy, and in 58% of those with stable disease. In contrast, the bacterium was detectable in only 34% of patients who experienced disease progression.
As in the experiments by Dr. Wargo and her associates, when the French investigators first treated mice with antibiotics and then gave them oral supplements containing the bacteria, the supplements restored response to PD-1 blockade,
“We conclude from the study that the gut microbiome markedly influences the outcome of PD-1 blockade in mice and patients,” Dr. Routy and his associates wrote.
They acknowledged that the mechanism whereby a common organism such as Akkermansia muciniphila might have an immunomodulatory effect is still unknown,
“Irrespective of these remaining questions, our findings suggest that the microbiome governs the cancer-immune set point of cancer-bearing individuals and offer[s] novel avenues for manipulating the gut ecosystem to circumvent primary resistance to [immune checkpoint inhibitors],” they wrote.
The study by Dr. Wargo and her colleagues was supported by contributions to the University of Texas MD Anderson Melanoma Moon Shots program. Dr. Wargo is supported by the Binational Science Foundation, Melanoma Research Alliance, Stand Up to Cancer, and the MDACC Melanoma Moon Shots Program. The work by Dr. Routy and his associates was supported by the Goustave Roussy Cancer Institute and McGill University. Coauthors were supported by the National Cancer Institute of France and other agencies and philanthropies.
The gut microbome may influence responses to immune checkpoint inhibitors, based on results from two studies, and one of the investigators is now gearing up for the next step - evaluating in a clinical trial whether altering the microflora will actually improve responses.
In the first study, investigators carried out a series of experiments using fecal microbiome samples from patients with metastatic melanoma embarking on therapy with a PD-1 (programmed cell death protein 1) inhibitor.
“In melanoma patients, there were differential signals in the gut microbiome of responders versus nonresponders, and I think the clincher was when we transplanted fecal samples from responders to nonresponders in germ-free mice, essentially reconstituting the microbiome and showing that it equally affected the systemic immunity and antitumor immunity when we implanted tumors, as well as response to checkpoint blockade,” lead author Jennifer A. Wargo, MD, MMSc, of the University of Texas MD Anderson Cancer Center in Houston, said in an interview.
Dr. Wargo and her colleagues first collected buccal and fecal microbiome samples from 112 patients with metastatic melanoma before they began therapy with a PD-1 inhibitor. After performing taxonomic profiling on all samples, they found that there was a clustering effect by response status in the gut microbiome, but not the oral microbiome, and because changes in the oral microbiome did not appear to be related to treatment response, they focused on the gut.
When Dr. Wargo and her colleagues studied the posttherapy microbiomes of 43 patients (30 responders and 13 nonresponders) according to Response Evaluation Criteria in Solid Tumors (RECIST 1.1), they found that the responders had a significantly higher degree of alpha diversity, a measure of species diversity within a specific environment, compared with nonresponders (P less than .01). In addition, responders had a relative abundance of Ruminococcaceae, commonly occurring gut microbes that break down complex carbohydrates, the investigators reported (Science. 2017 Nov. 2. doi: 10.1126/science.aan4236).
They found that patients whose microbiomes were diverse in general, and in particular were enriched with Faecalibacterium and Clostridiales species, were more likely to respond to immunotherapy with a PD-1 inhibitor and have a longer duration of progression-free survival. In contrast, patients whose microbiomes were more enriched with Bacteroidales species were more likely to be nonresponders.
To get a better understanding of the mechanisms whereby gut bacteria may influence response to PD-1 inhibitors, they performed metagenomic analysis on samples from 14 responders and 11 nonresponders, and found that responders had micro-organisms predominantly associated with anabolic functions that may support host immunity, whereas nonresponders had microbiomes where catabolic functions were more common.
The investigators next performed immune profiling, and found that both systemic immunity and local immunity in the tumor microenvironment in responders were associated with the aforementioned favorable gut microbiome.
The researchers then transplanted feces from the human donors into germ-free mice and then injected tumor cells into the mice, and found that tumor growth was significantly reduced, and response to PD-1 inhibition was significantly enhanced, in mice who received feces from responders.
“An obvious next step is to run a clinical trial to test the hypothesis that by modulating the microbiome, you can actually enhance responses to therapy,” Dr. Wargo said. Details of the clinical trial are still being worked out, but will likely involve fecal transfers and other mechanisms for modulating the microbiome in hopes of improving responses to PD-1 inhibitors.
“It’s going to be a very biomarker-heavy trial,” she said. “We’re going to look, certainly, for changes in the microbiome, and will also do a lot of profiling in the blood, the tumor, and in the microbiome to see if there are changes that occur by modulating that microbiome. Then of course we’ll look for differences in response rates in patients as well.”
Bacteria also affect epithelial cancers
In a separate study, also published in Science, investigators led by Bertrand Routy, MD, of the Gustave Roussy Cancer Institute in Villejuif, France, reported that patients with non–small cell lung cancer and urothelial carcinoma who had previously used systemic antibiotics had reduced survival when treated with a PD-1 inhibitor, compared with patients who had never taken antibiotics (Science. 2017 Nov. 2 doi: 10.1126/science.aan3706).
Analysis of the gut microbiome in these patients showed that higher levels of Akkermansia muciniphila were associated with the best clinical outcomes, with the species detectable in the microbiome of 69% of patients who had partial responses to anti–PD-1 therapy, and in 58% of those with stable disease. In contrast, the bacterium was detectable in only 34% of patients who experienced disease progression.
As in the experiments by Dr. Wargo and her associates, when the French investigators first treated mice with antibiotics and then gave them oral supplements containing the bacteria, the supplements restored response to PD-1 blockade,
“We conclude from the study that the gut microbiome markedly influences the outcome of PD-1 blockade in mice and patients,” Dr. Routy and his associates wrote.
They acknowledged that the mechanism whereby a common organism such as Akkermansia muciniphila might have an immunomodulatory effect is still unknown,
“Irrespective of these remaining questions, our findings suggest that the microbiome governs the cancer-immune set point of cancer-bearing individuals and offer[s] novel avenues for manipulating the gut ecosystem to circumvent primary resistance to [immune checkpoint inhibitors],” they wrote.
The study by Dr. Wargo and her colleagues was supported by contributions to the University of Texas MD Anderson Melanoma Moon Shots program. Dr. Wargo is supported by the Binational Science Foundation, Melanoma Research Alliance, Stand Up to Cancer, and the MDACC Melanoma Moon Shots Program. The work by Dr. Routy and his associates was supported by the Goustave Roussy Cancer Institute and McGill University. Coauthors were supported by the National Cancer Institute of France and other agencies and philanthropies.
The gut microbome may influence responses to immune checkpoint inhibitors, based on results from two studies, and one of the investigators is now gearing up for the next step - evaluating in a clinical trial whether altering the microflora will actually improve responses.
In the first study, investigators carried out a series of experiments using fecal microbiome samples from patients with metastatic melanoma embarking on therapy with a PD-1 (programmed cell death protein 1) inhibitor.
“In melanoma patients, there were differential signals in the gut microbiome of responders versus nonresponders, and I think the clincher was when we transplanted fecal samples from responders to nonresponders in germ-free mice, essentially reconstituting the microbiome and showing that it equally affected the systemic immunity and antitumor immunity when we implanted tumors, as well as response to checkpoint blockade,” lead author Jennifer A. Wargo, MD, MMSc, of the University of Texas MD Anderson Cancer Center in Houston, said in an interview.
Dr. Wargo and her colleagues first collected buccal and fecal microbiome samples from 112 patients with metastatic melanoma before they began therapy with a PD-1 inhibitor. After performing taxonomic profiling on all samples, they found that there was a clustering effect by response status in the gut microbiome, but not the oral microbiome, and because changes in the oral microbiome did not appear to be related to treatment response, they focused on the gut.
When Dr. Wargo and her colleagues studied the posttherapy microbiomes of 43 patients (30 responders and 13 nonresponders) according to Response Evaluation Criteria in Solid Tumors (RECIST 1.1), they found that the responders had a significantly higher degree of alpha diversity, a measure of species diversity within a specific environment, compared with nonresponders (P less than .01). In addition, responders had a relative abundance of Ruminococcaceae, commonly occurring gut microbes that break down complex carbohydrates, the investigators reported (Science. 2017 Nov. 2. doi: 10.1126/science.aan4236).
They found that patients whose microbiomes were diverse in general, and in particular were enriched with Faecalibacterium and Clostridiales species, were more likely to respond to immunotherapy with a PD-1 inhibitor and have a longer duration of progression-free survival. In contrast, patients whose microbiomes were more enriched with Bacteroidales species were more likely to be nonresponders.
To get a better understanding of the mechanisms whereby gut bacteria may influence response to PD-1 inhibitors, they performed metagenomic analysis on samples from 14 responders and 11 nonresponders, and found that responders had micro-organisms predominantly associated with anabolic functions that may support host immunity, whereas nonresponders had microbiomes where catabolic functions were more common.
The investigators next performed immune profiling, and found that both systemic immunity and local immunity in the tumor microenvironment in responders were associated with the aforementioned favorable gut microbiome.
The researchers then transplanted feces from the human donors into germ-free mice and then injected tumor cells into the mice, and found that tumor growth was significantly reduced, and response to PD-1 inhibition was significantly enhanced, in mice who received feces from responders.
“An obvious next step is to run a clinical trial to test the hypothesis that by modulating the microbiome, you can actually enhance responses to therapy,” Dr. Wargo said. Details of the clinical trial are still being worked out, but will likely involve fecal transfers and other mechanisms for modulating the microbiome in hopes of improving responses to PD-1 inhibitors.
“It’s going to be a very biomarker-heavy trial,” she said. “We’re going to look, certainly, for changes in the microbiome, and will also do a lot of profiling in the blood, the tumor, and in the microbiome to see if there are changes that occur by modulating that microbiome. Then of course we’ll look for differences in response rates in patients as well.”
Bacteria also affect epithelial cancers
In a separate study, also published in Science, investigators led by Bertrand Routy, MD, of the Gustave Roussy Cancer Institute in Villejuif, France, reported that patients with non–small cell lung cancer and urothelial carcinoma who had previously used systemic antibiotics had reduced survival when treated with a PD-1 inhibitor, compared with patients who had never taken antibiotics (Science. 2017 Nov. 2 doi: 10.1126/science.aan3706).
Analysis of the gut microbiome in these patients showed that higher levels of Akkermansia muciniphila were associated with the best clinical outcomes, with the species detectable in the microbiome of 69% of patients who had partial responses to anti–PD-1 therapy, and in 58% of those with stable disease. In contrast, the bacterium was detectable in only 34% of patients who experienced disease progression.
As in the experiments by Dr. Wargo and her associates, when the French investigators first treated mice with antibiotics and then gave them oral supplements containing the bacteria, the supplements restored response to PD-1 blockade,
“We conclude from the study that the gut microbiome markedly influences the outcome of PD-1 blockade in mice and patients,” Dr. Routy and his associates wrote.
They acknowledged that the mechanism whereby a common organism such as Akkermansia muciniphila might have an immunomodulatory effect is still unknown,
“Irrespective of these remaining questions, our findings suggest that the microbiome governs the cancer-immune set point of cancer-bearing individuals and offer[s] novel avenues for manipulating the gut ecosystem to circumvent primary resistance to [immune checkpoint inhibitors],” they wrote.
The study by Dr. Wargo and her colleagues was supported by contributions to the University of Texas MD Anderson Melanoma Moon Shots program. Dr. Wargo is supported by the Binational Science Foundation, Melanoma Research Alliance, Stand Up to Cancer, and the MDACC Melanoma Moon Shots Program. The work by Dr. Routy and his associates was supported by the Goustave Roussy Cancer Institute and McGill University. Coauthors were supported by the National Cancer Institute of France and other agencies and philanthropies.
FROM SCIENCE
Key clinical point: Modulating the gut microbome may improve responses to immune checkpoint inhibitors in patients with advanced melanoma, non–small cell lung cancer, and urothelial carcinoma.
Major finding: Responders to a checkpoint inhibitor had a significantly higher degree of alpha diversity, a measure of species diversity within a specific environment, compared with nonresponders (P less than .01).
Data source: A series of studies using microbiome samples from cancer patients receiving immune checkpoint inhibitors.
Disclosures: The study by Dr. Wargo and her colleagues was supported by contributions to the University of Texas MD Anderson Melanoma Moon Shots Program. Dr. Wargo is supported by the Binational Science Foundation, Melanoma Research Alliance, Stand Up to Cancer, and the MDACC Melanoma Moon Shots Program. The work by Dr. Routy and his colleagues was supported by the Goustave Roussy Cancer Institute and McGill University. Coauthors were supported by the National Cancer Institute of France and other agencies and philanthropies.