User login
Immunogenicity concerns for biosimilars so far don’t go beyond originator biologics
A key factor that impacts the efficacy and the toxicity of biologics used for rheumatic diseases is their immunogenicity, and this factor doesn’t appear to be any different for biosimilars in studies conducted so far.
In a meta-analysis of 63 studies of tumor necrosis factor (TNF) inhibitors, investigators including Daniel E. Furst, MD, professor of rheumatology at the University of Washington, Seattle, who also is affiliated with the University of California, Los Angeles, and the University of Florence, Italy, found that antidrug antibodies developed in 17% of patients (BioDrugs 2015;29:241-58).
“That doesn’t sound too bad, but does it differ by medication?” asked Dr. Furst, who spoke at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education. “For infliximab, 30% of the time, there are antidrug antibodies. So if that has a clinical effect, that’s a big deal. For certolizumab ... it’s only 6%, so there is a huge difference within the TNF inhibitors.” At the same time, antidrug antibodies developed in patients on adalimumab 23% of the time, followed by certolizumab (6%), golimumab (4%), and etanercept (2%).
One approach to circumventing the impact of antidrug antibodies on clinical response is by using immunosuppression, which in the meta-analysis had a 70% probability for decreasing antidrug antibodies. But this approach comes with a hitch, Dr. Furst said. The effect of immunosuppression on antidrug antibody positivity differs by disease. Immunosuppression had a 78% probability for decreasing antidrug antibody positivity in RA, 63% probability in inflammatory bowel disease, and 32% probability in ankylosing spondylitis. “That’s a wild mix of immunogenicity and the possibility that it’s going to affect the underlying response,” he said. The best studies in this area are of infliximab and adalimumab, which showed that antidrug antibodies in patients on infliximab reduced the probability of a clinical response by 54% and for adalimumab by 65%.
The same meta-analysis found that there were more antibodies to adalimumab than to certolizumab, golimumab, or etanercept, and also more antibodies to infliximab than to certolizumab, golimumab, or etanercept, but no difference between adalimumab and infliximab. “You’d think that maybe there is a difference between adalimumab and infliximab, but that’s not true,” Dr. Furst said. The apparent effect on the percentage of antidrug antibodies depends on what type of assay is used. For example, radioimmunoassay measures about 11% more antidrug antibodies, compared with enzyme-linked immunosorbent assay. Disease duration also matters. Each year of disease duration increases the antidrug antibodies by 1%.
What about the non-TNF inhibitors? A meta-analysis of five core trials of tocilizumab for RA found that the antidrug antibodies ranged from 1.7% at baseline to 2.3% during the respective trials (Clin Ther. 2010;32:1597-1609). “Anywhere along the way, the percentage of antidrug antibodies was about 1.5%,” said Dr. Furst, who was not involved with the study. “So as opposed to the TNF inhibitors, when you look at tocilizumab, this whole immunity question is probably a non-issue. The same is true for abatacept. With rituximab, the question is a little bit different. In one randomized, controlled trial, immunogenicity was 2.9%, while in the next it was 7.9%. In an open-label trial, it was 11.5%. So I think for rituximab we have to assume that you have some potentially important antidrug antibodies that might affect response.”
Studies have shown that antidrug antibodies do affect the pharmacokinetics of a biologic (and thereby clinical response) by producing lower trough levels of the biologic through an increase in its clearance. However, studies of infliximab and its biosimilars have not yielded any significant differences in clinical responses rates despite small differences in rates of antidrug antibodies or in rates of treatment-emergent adverse events, he said.
There has also been no difference in disease worsening in the only reported double-blind, randomized, switching study for infliximab and an infliximab biosimilar, the NOR-SWITCH study. However, the small numbers of patients in the study with certain diseases for which infliximab is indicated do not allow for conclusions to be drawn for specific diseases, Dr. Furst said.
Dr. Furst disclosed that he has received grant/research support from AbbVie, Actelion, Amgen, Bristol-Myers Squibb, Corbus, the National Institutes of Health, Novartis, and Roche/Genentech. He is also a consultant for AbbVie, Actelion, Amgen, Bristol-Myers Squibb, Cytori, Novartis, Pfizer, and Roche/Genentech. Global Academy for Medical Education and this news organization are owned by the same parent company.
A key factor that impacts the efficacy and the toxicity of biologics used for rheumatic diseases is their immunogenicity, and this factor doesn’t appear to be any different for biosimilars in studies conducted so far.
In a meta-analysis of 63 studies of tumor necrosis factor (TNF) inhibitors, investigators including Daniel E. Furst, MD, professor of rheumatology at the University of Washington, Seattle, who also is affiliated with the University of California, Los Angeles, and the University of Florence, Italy, found that antidrug antibodies developed in 17% of patients (BioDrugs 2015;29:241-58).
“That doesn’t sound too bad, but does it differ by medication?” asked Dr. Furst, who spoke at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education. “For infliximab, 30% of the time, there are antidrug antibodies. So if that has a clinical effect, that’s a big deal. For certolizumab ... it’s only 6%, so there is a huge difference within the TNF inhibitors.” At the same time, antidrug antibodies developed in patients on adalimumab 23% of the time, followed by certolizumab (6%), golimumab (4%), and etanercept (2%).
One approach to circumventing the impact of antidrug antibodies on clinical response is by using immunosuppression, which in the meta-analysis had a 70% probability for decreasing antidrug antibodies. But this approach comes with a hitch, Dr. Furst said. The effect of immunosuppression on antidrug antibody positivity differs by disease. Immunosuppression had a 78% probability for decreasing antidrug antibody positivity in RA, 63% probability in inflammatory bowel disease, and 32% probability in ankylosing spondylitis. “That’s a wild mix of immunogenicity and the possibility that it’s going to affect the underlying response,” he said. The best studies in this area are of infliximab and adalimumab, which showed that antidrug antibodies in patients on infliximab reduced the probability of a clinical response by 54% and for adalimumab by 65%.
The same meta-analysis found that there were more antibodies to adalimumab than to certolizumab, golimumab, or etanercept, and also more antibodies to infliximab than to certolizumab, golimumab, or etanercept, but no difference between adalimumab and infliximab. “You’d think that maybe there is a difference between adalimumab and infliximab, but that’s not true,” Dr. Furst said. The apparent effect on the percentage of antidrug antibodies depends on what type of assay is used. For example, radioimmunoassay measures about 11% more antidrug antibodies, compared with enzyme-linked immunosorbent assay. Disease duration also matters. Each year of disease duration increases the antidrug antibodies by 1%.
What about the non-TNF inhibitors? A meta-analysis of five core trials of tocilizumab for RA found that the antidrug antibodies ranged from 1.7% at baseline to 2.3% during the respective trials (Clin Ther. 2010;32:1597-1609). “Anywhere along the way, the percentage of antidrug antibodies was about 1.5%,” said Dr. Furst, who was not involved with the study. “So as opposed to the TNF inhibitors, when you look at tocilizumab, this whole immunity question is probably a non-issue. The same is true for abatacept. With rituximab, the question is a little bit different. In one randomized, controlled trial, immunogenicity was 2.9%, while in the next it was 7.9%. In an open-label trial, it was 11.5%. So I think for rituximab we have to assume that you have some potentially important antidrug antibodies that might affect response.”
Studies have shown that antidrug antibodies do affect the pharmacokinetics of a biologic (and thereby clinical response) by producing lower trough levels of the biologic through an increase in its clearance. However, studies of infliximab and its biosimilars have not yielded any significant differences in clinical responses rates despite small differences in rates of antidrug antibodies or in rates of treatment-emergent adverse events, he said.
There has also been no difference in disease worsening in the only reported double-blind, randomized, switching study for infliximab and an infliximab biosimilar, the NOR-SWITCH study. However, the small numbers of patients in the study with certain diseases for which infliximab is indicated do not allow for conclusions to be drawn for specific diseases, Dr. Furst said.
Dr. Furst disclosed that he has received grant/research support from AbbVie, Actelion, Amgen, Bristol-Myers Squibb, Corbus, the National Institutes of Health, Novartis, and Roche/Genentech. He is also a consultant for AbbVie, Actelion, Amgen, Bristol-Myers Squibb, Cytori, Novartis, Pfizer, and Roche/Genentech. Global Academy for Medical Education and this news organization are owned by the same parent company.
A key factor that impacts the efficacy and the toxicity of biologics used for rheumatic diseases is their immunogenicity, and this factor doesn’t appear to be any different for biosimilars in studies conducted so far.
In a meta-analysis of 63 studies of tumor necrosis factor (TNF) inhibitors, investigators including Daniel E. Furst, MD, professor of rheumatology at the University of Washington, Seattle, who also is affiliated with the University of California, Los Angeles, and the University of Florence, Italy, found that antidrug antibodies developed in 17% of patients (BioDrugs 2015;29:241-58).
“That doesn’t sound too bad, but does it differ by medication?” asked Dr. Furst, who spoke at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education. “For infliximab, 30% of the time, there are antidrug antibodies. So if that has a clinical effect, that’s a big deal. For certolizumab ... it’s only 6%, so there is a huge difference within the TNF inhibitors.” At the same time, antidrug antibodies developed in patients on adalimumab 23% of the time, followed by certolizumab (6%), golimumab (4%), and etanercept (2%).
One approach to circumventing the impact of antidrug antibodies on clinical response is by using immunosuppression, which in the meta-analysis had a 70% probability for decreasing antidrug antibodies. But this approach comes with a hitch, Dr. Furst said. The effect of immunosuppression on antidrug antibody positivity differs by disease. Immunosuppression had a 78% probability for decreasing antidrug antibody positivity in RA, 63% probability in inflammatory bowel disease, and 32% probability in ankylosing spondylitis. “That’s a wild mix of immunogenicity and the possibility that it’s going to affect the underlying response,” he said. The best studies in this area are of infliximab and adalimumab, which showed that antidrug antibodies in patients on infliximab reduced the probability of a clinical response by 54% and for adalimumab by 65%.
The same meta-analysis found that there were more antibodies to adalimumab than to certolizumab, golimumab, or etanercept, and also more antibodies to infliximab than to certolizumab, golimumab, or etanercept, but no difference between adalimumab and infliximab. “You’d think that maybe there is a difference between adalimumab and infliximab, but that’s not true,” Dr. Furst said. The apparent effect on the percentage of antidrug antibodies depends on what type of assay is used. For example, radioimmunoassay measures about 11% more antidrug antibodies, compared with enzyme-linked immunosorbent assay. Disease duration also matters. Each year of disease duration increases the antidrug antibodies by 1%.
What about the non-TNF inhibitors? A meta-analysis of five core trials of tocilizumab for RA found that the antidrug antibodies ranged from 1.7% at baseline to 2.3% during the respective trials (Clin Ther. 2010;32:1597-1609). “Anywhere along the way, the percentage of antidrug antibodies was about 1.5%,” said Dr. Furst, who was not involved with the study. “So as opposed to the TNF inhibitors, when you look at tocilizumab, this whole immunity question is probably a non-issue. The same is true for abatacept. With rituximab, the question is a little bit different. In one randomized, controlled trial, immunogenicity was 2.9%, while in the next it was 7.9%. In an open-label trial, it was 11.5%. So I think for rituximab we have to assume that you have some potentially important antidrug antibodies that might affect response.”
Studies have shown that antidrug antibodies do affect the pharmacokinetics of a biologic (and thereby clinical response) by producing lower trough levels of the biologic through an increase in its clearance. However, studies of infliximab and its biosimilars have not yielded any significant differences in clinical responses rates despite small differences in rates of antidrug antibodies or in rates of treatment-emergent adverse events, he said.
There has also been no difference in disease worsening in the only reported double-blind, randomized, switching study for infliximab and an infliximab biosimilar, the NOR-SWITCH study. However, the small numbers of patients in the study with certain diseases for which infliximab is indicated do not allow for conclusions to be drawn for specific diseases, Dr. Furst said.
Dr. Furst disclosed that he has received grant/research support from AbbVie, Actelion, Amgen, Bristol-Myers Squibb, Corbus, the National Institutes of Health, Novartis, and Roche/Genentech. He is also a consultant for AbbVie, Actelion, Amgen, Bristol-Myers Squibb, Cytori, Novartis, Pfizer, and Roche/Genentech. Global Academy for Medical Education and this news organization are owned by the same parent company.
EXPERT ANALYSIS FROM THE ANNUAL PERSPECTIVES IN RHEUMATIC DISEASES
‘Multimorbidities’ in RA make impact on treatment efficacy, disease activity
While clinicians are accustomed to managing patients with rheumatoid arthritis and comorbidities, RA patients more frequently have “multimorbidities” – the simultaneous presence of two or more chronic conditions.
“For anyone who sees patients, this is the reality of our lives,” said Jeffrey R. Curtis, MD, at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education. Some conditions are “bystanders” that don’t impact RA, some are risk factors for RA, and some can be caused by RA treatments, said Dr. Curtis, professor of medicine and William J. Koopman Professor in Rheumatology and Immunology at the University of Alabama at Birmingham. He discussed several comorbid conditions and their interactions with RA.
Obesity’s toll on disease activity
It’s long been recognized that obesity along with RA makes things worse for patients, he said: “If you have a patient who is obese, with a BMI [body mass index] over 30-35, they’re less likely clinically to achieve low disease activity or remission.”

Obesity also affects biomarkers, he said. The multibiomarker disease activity test, marketed as Vectra DA, incorporates several adipokines in its score, but given any level of RA disease, that score is roughly 5 units higher if someone is obese (Sem Arthritis Rheum. 2017 Aug 2. doi: 10.1016/j.semarthrit.2017.07.010). “The company that manufactures the test is working on an adjustment factor to optimize the role of obesity and what those adipokines are doing to better predict x-ray progression to take this influence into account,” he said. “How we measure obesity and how we’re measuring RA are probably very much affected by this issue.”
RA and the somatization comorbidity phenotype
A second common co-occurrence is what Dr. Curtis calls a somatization comorbidity phenotype (SCP), defined as RA with several other ailments such as fibromyalgia, depression, anxiety, sleep apnea, or neuropathy. “The troubling thing about this is I could cure this patient’s RA tomorrow, but they actually wouldn’t feel better in their overall health because of other things dragging down their function,” he said, noting patients are more concerned about their fatigue and pain than their swollen joint count or DAS28 score.
Dr. Curtis and his colleagues completed a recent analysis that’s in press in Arthritis Research & Therapy of about 800 RA patients in the United States starting certolizumab pegol (Cimzia) to look for how well patients with this phenotype responded to a tumor necrosis factor (TNF) inhibitor. The percentage of patients with RA plus SCP who achieved American College of Rheumatology (ACR) 20/50/70 scores were all about 10% lower than for patients with RA alone, and about 15% fewer patients achieved remission using a score of less than 2.6 on the DAS28, based on erythrocyte sedimentation rate. There was about a 10% higher withdrawal rate among the RA plus SCP group, compared with those with RA alone, mainly for lack of efficacy, and the RA plus SCP patients had three times the rate of serious infections and twice the rate of nonserious infections of typical RA patients.
Impact on choice of therapy
Clinicians have questioned whether multimorbidities should affect the choice of RA therapy, Dr. Curtis said. One of the most vexing scenarios has been in patients with a history of cancer, he noted. There have been five studies worldwide looking at RA therapy in this population. One from Sweden (Ann Rheum Dis. 2015 Dec;74[12]:2137-43), of 240 breast cancer survivors, found no difference in the occurrence or hazard ratio of recurrent breast cancer between those treated with TNF inhibitors versus those who were not. Another study coauthored by Dr. Curtis found similar results (Arthritis Rheumatol. 2016 Dec;68[10]:2403–11).
According to the ACR’s 2015 treatment guidelines, “If your RA patient has a previously treated, solid organ malignancy, there’s no restrictions on what you and I should use,” he said. “Basically, treat them like they hadn’t had a history of cancer.” The exception is for patients with previously treated nonmelanoma skin cancer or melanoma, in whom conventional synthetic DMARDs are preferred over biologics or tofacitinib (Xeljanz). In addition, patients with previously treated lymphoproliferative disorders should be given rituximab (Rituxan) – or DMARDs, abatacept (Orencia), or tocilizumab (Actemra) – over TNF inhibitors. “This is good news,” Dr. Curtis said. “It really expands the range of options for patients, now supported by data, who have a history of cancer and bad RA.”
Global Academy for Medical Education and this news organization are owned by the same parent company. Dr. Curtis receives research support or serves as a consultant for Amgen, Bristol-Myers Squibb, Corrona, Lilly, Janssen, Myriad, Pfizer, and Sanofi/Regeneron.
While clinicians are accustomed to managing patients with rheumatoid arthritis and comorbidities, RA patients more frequently have “multimorbidities” – the simultaneous presence of two or more chronic conditions.
“For anyone who sees patients, this is the reality of our lives,” said Jeffrey R. Curtis, MD, at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education. Some conditions are “bystanders” that don’t impact RA, some are risk factors for RA, and some can be caused by RA treatments, said Dr. Curtis, professor of medicine and William J. Koopman Professor in Rheumatology and Immunology at the University of Alabama at Birmingham. He discussed several comorbid conditions and their interactions with RA.
Obesity’s toll on disease activity
It’s long been recognized that obesity along with RA makes things worse for patients, he said: “If you have a patient who is obese, with a BMI [body mass index] over 30-35, they’re less likely clinically to achieve low disease activity or remission.”
Obesity also affects biomarkers, he said. The multibiomarker disease activity test, marketed as Vectra DA, incorporates several adipokines in its score, but given any level of RA disease, that score is roughly 5 units higher if someone is obese (Sem Arthritis Rheum. 2017 Aug 2. doi: 10.1016/j.semarthrit.2017.07.010). “The company that manufactures the test is working on an adjustment factor to optimize the role of obesity and what those adipokines are doing to better predict x-ray progression to take this influence into account,” he said. “How we measure obesity and how we’re measuring RA are probably very much affected by this issue.”
RA and the somatization comorbidity phenotype
A second common co-occurrence is what Dr. Curtis calls a somatization comorbidity phenotype (SCP), defined as RA with several other ailments such as fibromyalgia, depression, anxiety, sleep apnea, or neuropathy. “The troubling thing about this is I could cure this patient’s RA tomorrow, but they actually wouldn’t feel better in their overall health because of other things dragging down their function,” he said, noting patients are more concerned about their fatigue and pain than their swollen joint count or DAS28 score.
Dr. Curtis and his colleagues completed a recent analysis that’s in press in Arthritis Research & Therapy of about 800 RA patients in the United States starting certolizumab pegol (Cimzia) to look for how well patients with this phenotype responded to a tumor necrosis factor (TNF) inhibitor. The percentage of patients with RA plus SCP who achieved American College of Rheumatology (ACR) 20/50/70 scores were all about 10% lower than for patients with RA alone, and about 15% fewer patients achieved remission using a score of less than 2.6 on the DAS28, based on erythrocyte sedimentation rate. There was about a 10% higher withdrawal rate among the RA plus SCP group, compared with those with RA alone, mainly for lack of efficacy, and the RA plus SCP patients had three times the rate of serious infections and twice the rate of nonserious infections of typical RA patients.
Impact on choice of therapy
Clinicians have questioned whether multimorbidities should affect the choice of RA therapy, Dr. Curtis said. One of the most vexing scenarios has been in patients with a history of cancer, he noted. There have been five studies worldwide looking at RA therapy in this population. One from Sweden (Ann Rheum Dis. 2015 Dec;74[12]:2137-43), of 240 breast cancer survivors, found no difference in the occurrence or hazard ratio of recurrent breast cancer between those treated with TNF inhibitors versus those who were not. Another study coauthored by Dr. Curtis found similar results (Arthritis Rheumatol. 2016 Dec;68[10]:2403–11).
According to the ACR’s 2015 treatment guidelines, “If your RA patient has a previously treated, solid organ malignancy, there’s no restrictions on what you and I should use,” he said. “Basically, treat them like they hadn’t had a history of cancer.” The exception is for patients with previously treated nonmelanoma skin cancer or melanoma, in whom conventional synthetic DMARDs are preferred over biologics or tofacitinib (Xeljanz). In addition, patients with previously treated lymphoproliferative disorders should be given rituximab (Rituxan) – or DMARDs, abatacept (Orencia), or tocilizumab (Actemra) – over TNF inhibitors. “This is good news,” Dr. Curtis said. “It really expands the range of options for patients, now supported by data, who have a history of cancer and bad RA.”
Global Academy for Medical Education and this news organization are owned by the same parent company. Dr. Curtis receives research support or serves as a consultant for Amgen, Bristol-Myers Squibb, Corrona, Lilly, Janssen, Myriad, Pfizer, and Sanofi/Regeneron.
While clinicians are accustomed to managing patients with rheumatoid arthritis and comorbidities, RA patients more frequently have “multimorbidities” – the simultaneous presence of two or more chronic conditions.
“For anyone who sees patients, this is the reality of our lives,” said Jeffrey R. Curtis, MD, at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education. Some conditions are “bystanders” that don’t impact RA, some are risk factors for RA, and some can be caused by RA treatments, said Dr. Curtis, professor of medicine and William J. Koopman Professor in Rheumatology and Immunology at the University of Alabama at Birmingham. He discussed several comorbid conditions and their interactions with RA.
Obesity’s toll on disease activity
It’s long been recognized that obesity along with RA makes things worse for patients, he said: “If you have a patient who is obese, with a BMI [body mass index] over 30-35, they’re less likely clinically to achieve low disease activity or remission.”
Obesity also affects biomarkers, he said. The multibiomarker disease activity test, marketed as Vectra DA, incorporates several adipokines in its score, but given any level of RA disease, that score is roughly 5 units higher if someone is obese (Sem Arthritis Rheum. 2017 Aug 2. doi: 10.1016/j.semarthrit.2017.07.010). “The company that manufactures the test is working on an adjustment factor to optimize the role of obesity and what those adipokines are doing to better predict x-ray progression to take this influence into account,” he said. “How we measure obesity and how we’re measuring RA are probably very much affected by this issue.”
RA and the somatization comorbidity phenotype
A second common co-occurrence is what Dr. Curtis calls a somatization comorbidity phenotype (SCP), defined as RA with several other ailments such as fibromyalgia, depression, anxiety, sleep apnea, or neuropathy. “The troubling thing about this is I could cure this patient’s RA tomorrow, but they actually wouldn’t feel better in their overall health because of other things dragging down their function,” he said, noting patients are more concerned about their fatigue and pain than their swollen joint count or DAS28 score.
Dr. Curtis and his colleagues completed a recent analysis that’s in press in Arthritis Research & Therapy of about 800 RA patients in the United States starting certolizumab pegol (Cimzia) to look for how well patients with this phenotype responded to a tumor necrosis factor (TNF) inhibitor. The percentage of patients with RA plus SCP who achieved American College of Rheumatology (ACR) 20/50/70 scores were all about 10% lower than for patients with RA alone, and about 15% fewer patients achieved remission using a score of less than 2.6 on the DAS28, based on erythrocyte sedimentation rate. There was about a 10% higher withdrawal rate among the RA plus SCP group, compared with those with RA alone, mainly for lack of efficacy, and the RA plus SCP patients had three times the rate of serious infections and twice the rate of nonserious infections of typical RA patients.
Impact on choice of therapy
Clinicians have questioned whether multimorbidities should affect the choice of RA therapy, Dr. Curtis said. One of the most vexing scenarios has been in patients with a history of cancer, he noted. There have been five studies worldwide looking at RA therapy in this population. One from Sweden (Ann Rheum Dis. 2015 Dec;74[12]:2137-43), of 240 breast cancer survivors, found no difference in the occurrence or hazard ratio of recurrent breast cancer between those treated with TNF inhibitors versus those who were not. Another study coauthored by Dr. Curtis found similar results (Arthritis Rheumatol. 2016 Dec;68[10]:2403–11).
According to the ACR’s 2015 treatment guidelines, “If your RA patient has a previously treated, solid organ malignancy, there’s no restrictions on what you and I should use,” he said. “Basically, treat them like they hadn’t had a history of cancer.” The exception is for patients with previously treated nonmelanoma skin cancer or melanoma, in whom conventional synthetic DMARDs are preferred over biologics or tofacitinib (Xeljanz). In addition, patients with previously treated lymphoproliferative disorders should be given rituximab (Rituxan) – or DMARDs, abatacept (Orencia), or tocilizumab (Actemra) – over TNF inhibitors. “This is good news,” Dr. Curtis said. “It really expands the range of options for patients, now supported by data, who have a history of cancer and bad RA.”
Global Academy for Medical Education and this news organization are owned by the same parent company. Dr. Curtis receives research support or serves as a consultant for Amgen, Bristol-Myers Squibb, Corrona, Lilly, Janssen, Myriad, Pfizer, and Sanofi/Regeneron.
EXPERT ANALYSIS FROM THE ANNUAL PERSPECTIVES IN RHEUMATIC DISEASES
Rheumatoid arthritis characteristics make large contribution to cardiovascular risk
Nearly one-third of cardiovascular events in patients with rheumatoid arthritis can be attributed to their rheumatoid arthritis characteristics, such as Disease Activity Score and rheumatoid factor or anticitrullinated protein antibody positivity, research suggests.
A prospective, international cohort study published in Annals of the Rheumatic Diseases followed 5,638 patients with rheumatoid arthritis (RA) and no history of cardiovascular disease for a mean of 5.8 years to look at their risk of myocardial infarction, angina, revascularization, stroke, peripheral vascular disease, and death from cardiovascular disease.
Overall, the 10-year cumulative incidence of cardiovascular events was 20.9% in men and 11.1% in women.
Smoking and hypertension were the strongest predictors of cardiovascular disease in both men and women and had the highest population attributable risk (PAR), even after adjustment for other cardiovascular risk factors.
The PAR for triglycerides was 11.5% overall, but it was 12.6% for Disease Activity Score in 28 joints (DAS28) and 12.2% for rheumatoid factor (RF)/anticitrullinated protein antibody (ACPA) positivity. Other RA-related factors, such as erythrocyte sedimentation rate (ESR) and C-reactive protein, did not have a significant effect on cardiovascular event risk.
When combined, cardiovascular risk factors such as blood pressure, cholesterol levels, smoking, body mass index, diabetes, and family history accounted for 49% of the PAR of cardiovascular events in people with RA, and the RA characteristics explained 30.3% of the risk.
Together, the cardiovascular and RA risk factors accounted for 69.6% of the risk of cardiovascular events, and the remaining 30.4% could not be explained.
While the PAR associated with the combined cardiovascular risk factors was higher in men than in women, the contribution of all the RA characteristics combined proved to be greater in women than in men. However, neither sex difference was statistically significant.
“While the prevalence of RF/ACPA positivity and DAS28 levels was similar between the sexes, the effect sizes of RA characteristics appeared to be larger among women than men, despite lack of statistical significance,” the authors wrote.
“Moreover, higher levels of ESR in women than men may partially explain this apparent difference in PAR [and] RA disease duration was longer among women, and more women than men were receiving biological [disease-modifying antirheumatic drugs] at baseline.”
Eli Lilly, the National Institute of Arthritis and Musculoskeletal and Skin Diseases, and the Norwegian South East Health Authority supported the study. Two authors declared honoraria, fees, and grants from the pharmaceutical industry, including Eli Lilly. No other conflicts of interest were declared.
Nearly one-third of cardiovascular events in patients with rheumatoid arthritis can be attributed to their rheumatoid arthritis characteristics, such as Disease Activity Score and rheumatoid factor or anticitrullinated protein antibody positivity, research suggests.
A prospective, international cohort study published in Annals of the Rheumatic Diseases followed 5,638 patients with rheumatoid arthritis (RA) and no history of cardiovascular disease for a mean of 5.8 years to look at their risk of myocardial infarction, angina, revascularization, stroke, peripheral vascular disease, and death from cardiovascular disease.
Overall, the 10-year cumulative incidence of cardiovascular events was 20.9% in men and 11.1% in women.
Smoking and hypertension were the strongest predictors of cardiovascular disease in both men and women and had the highest population attributable risk (PAR), even after adjustment for other cardiovascular risk factors.
The PAR for triglycerides was 11.5% overall, but it was 12.6% for Disease Activity Score in 28 joints (DAS28) and 12.2% for rheumatoid factor (RF)/anticitrullinated protein antibody (ACPA) positivity. Other RA-related factors, such as erythrocyte sedimentation rate (ESR) and C-reactive protein, did not have a significant effect on cardiovascular event risk.
When combined, cardiovascular risk factors such as blood pressure, cholesterol levels, smoking, body mass index, diabetes, and family history accounted for 49% of the PAR of cardiovascular events in people with RA, and the RA characteristics explained 30.3% of the risk.
Together, the cardiovascular and RA risk factors accounted for 69.6% of the risk of cardiovascular events, and the remaining 30.4% could not be explained.
While the PAR associated with the combined cardiovascular risk factors was higher in men than in women, the contribution of all the RA characteristics combined proved to be greater in women than in men. However, neither sex difference was statistically significant.
“While the prevalence of RF/ACPA positivity and DAS28 levels was similar between the sexes, the effect sizes of RA characteristics appeared to be larger among women than men, despite lack of statistical significance,” the authors wrote.
“Moreover, higher levels of ESR in women than men may partially explain this apparent difference in PAR [and] RA disease duration was longer among women, and more women than men were receiving biological [disease-modifying antirheumatic drugs] at baseline.”
Eli Lilly, the National Institute of Arthritis and Musculoskeletal and Skin Diseases, and the Norwegian South East Health Authority supported the study. Two authors declared honoraria, fees, and grants from the pharmaceutical industry, including Eli Lilly. No other conflicts of interest were declared.
Nearly one-third of cardiovascular events in patients with rheumatoid arthritis can be attributed to their rheumatoid arthritis characteristics, such as Disease Activity Score and rheumatoid factor or anticitrullinated protein antibody positivity, research suggests.
A prospective, international cohort study published in Annals of the Rheumatic Diseases followed 5,638 patients with rheumatoid arthritis (RA) and no history of cardiovascular disease for a mean of 5.8 years to look at their risk of myocardial infarction, angina, revascularization, stroke, peripheral vascular disease, and death from cardiovascular disease.
Overall, the 10-year cumulative incidence of cardiovascular events was 20.9% in men and 11.1% in women.
Smoking and hypertension were the strongest predictors of cardiovascular disease in both men and women and had the highest population attributable risk (PAR), even after adjustment for other cardiovascular risk factors.
The PAR for triglycerides was 11.5% overall, but it was 12.6% for Disease Activity Score in 28 joints (DAS28) and 12.2% for rheumatoid factor (RF)/anticitrullinated protein antibody (ACPA) positivity. Other RA-related factors, such as erythrocyte sedimentation rate (ESR) and C-reactive protein, did not have a significant effect on cardiovascular event risk.
When combined, cardiovascular risk factors such as blood pressure, cholesterol levels, smoking, body mass index, diabetes, and family history accounted for 49% of the PAR of cardiovascular events in people with RA, and the RA characteristics explained 30.3% of the risk.
Together, the cardiovascular and RA risk factors accounted for 69.6% of the risk of cardiovascular events, and the remaining 30.4% could not be explained.
While the PAR associated with the combined cardiovascular risk factors was higher in men than in women, the contribution of all the RA characteristics combined proved to be greater in women than in men. However, neither sex difference was statistically significant.
“While the prevalence of RF/ACPA positivity and DAS28 levels was similar between the sexes, the effect sizes of RA characteristics appeared to be larger among women than men, despite lack of statistical significance,” the authors wrote.
“Moreover, higher levels of ESR in women than men may partially explain this apparent difference in PAR [and] RA disease duration was longer among women, and more women than men were receiving biological [disease-modifying antirheumatic drugs] at baseline.”
Eli Lilly, the National Institute of Arthritis and Musculoskeletal and Skin Diseases, and the Norwegian South East Health Authority supported the study. Two authors declared honoraria, fees, and grants from the pharmaceutical industry, including Eli Lilly. No other conflicts of interest were declared.
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point:
Major finding: Rheumatoid arthritis characteristics explained 30.3% of the risk of cardiovascular events in individuals with RA.
Data source: A prospective, international cohort study of 5,638 patients with RA.
Disclosures: Eli Lilly, the National Institute of Arthritis and Musculoskeletal and Skin Diseases, and the Norwegian South East Health Authority supported the study. Two authors declared honoraria, fees, and grants from the pharmaceutical industry, including Eli Lilly. No other conflicts of interest were declared.
Ultrasound’s value for arthralgia may be to rule out IA
Ultrasound evaluations to look for subclinical inflammation in joints of patients with arthralgia appear best at ruling out inflammatory arthritis (IA) 1 year in the future rather than ruling it in, according to findings from a multicenter cohort study published online in Arthritis Research and Therapy.
The imaging modality’s ability to identify those who will not go on to develop IA complemented the serologic and clinical factors that help to discriminate the individuals with arthralgia who are most at risk of the condition.
The ultimate goal of using imaging such as ultrasound in patients with arthralgia is to identify those who would benefit from starting treatment with disease-modifying antirheumatic drugs as early as possible to potentially improve outcomes, but it also could help to discriminate between the anti-citrullinated protein antibody (ACPA)-positive and seronegative individuals without clinical signs of inflammation at baseline who may progress from arthralgia to IA.
“Although ACPA positivity is a very good predictor for those patients who will develop IA within 1 year, it is still difficult to identify the exact individuals who will develop IA, because any ACPA-positive individual has an a priori chance of 50% of developing IA. In seronegative patients, the prediction of IA is even more difficult, because only 5% develop IA within the subsequent year. Imaging techniques have been shown to be able to detect synovitis before its clinical appearance and could be of help in identifying those at risk of IA,” the investigators wrote (Arthritis Res Ther. 2017;19:202. doi: 10.1186/s13075-017-1405-y).
Dr. van der Ven and her associates found that 31 (16%) of 196 patients who had arthralgia for less than 1 year in the hands, feet, or shoulders went on to develop IA after 1 year of follow-up. In this group of 196 patients at baseline, 72 (37%) had synovitis on ultrasound – defined as a greyscale grade of 2 or 3 and/or the presence of power Doppler signal (grade 1, 2, or 3) – including 32 with a positive power Doppler. A total of 18 patients were lost to follow-up during the first 6 months and another 19 were lost during months 6-12.
Rheumatologists who were unaware of ultrasound findings had to confirm soft-tissue swelling as arthritis at 1 year to classify it as incident IA. The positive predictive value of ultrasound for IA was only 26% when at least 1 joint out of 26 assessed was positive, but the negative predictive value when no joints were positive on ultrasound was 89%.
Overall, at 1 year, 15 of the 31 patients with IA had started therapy with a disease-modifying antirheumatic drug and 22 did not have a definite diagnosis; 12 had monoarthritis and 10 had polyarthritis. The remaining nine patients included four with rheumatoid arthritis, four with psoriatic arthritis, and one with spondyloarthritis.
At baseline, individuals with IA were more often older (mean age 50 vs. 44 years; P = .005), had synovitis on ultrasound (59% vs. 32%; P = .007), and had a positive power Doppler signal (31% vs. 12%; P = .012). A multivariate analysis revealed that IA at 1 year of follow-up could be independently predicted according to age (odds ratio, 1.06), morning stiffness lasting more than 30 minutes (OR, 2.80), ACPA positivity (OR, 2.35), and synovitis on ultrasound (OR, 2.65).
The investigators noted that the study’s limitations relate to requirements for patients to have at least two painful joints in hands, feet, or shoulders at baseline and two criteria related to inflammation. The possible inflammation-related criteria required for entry included morning stiffness for more than 1 hour, inability to clench a fist in the morning, pain when shaking someone’s hand, pins and needles in the fingers, difficulties wearing rings or shoes, family history of rheumatoid arthritis, and/or unexplained fatigue for less than 1 year.
Rheumatologists who enrolled patients into the cohort also may have “recruited clinically suspected patients with possibly more severe symptoms,” the investigators noted. Another potential source of bias related to the group of 38 patients who chose not to participate: It’s possible these patients had less severe symptoms than those who participated in the study.
The study was funded by an investigator-initiated grant from Pfizer. The authors declared that they have no competing interests.
Ultrasound evaluations to look for subclinical inflammation in joints of patients with arthralgia appear best at ruling out inflammatory arthritis (IA) 1 year in the future rather than ruling it in, according to findings from a multicenter cohort study published online in Arthritis Research and Therapy.
The imaging modality’s ability to identify those who will not go on to develop IA complemented the serologic and clinical factors that help to discriminate the individuals with arthralgia who are most at risk of the condition.
The ultimate goal of using imaging such as ultrasound in patients with arthralgia is to identify those who would benefit from starting treatment with disease-modifying antirheumatic drugs as early as possible to potentially improve outcomes, but it also could help to discriminate between the anti-citrullinated protein antibody (ACPA)-positive and seronegative individuals without clinical signs of inflammation at baseline who may progress from arthralgia to IA.
“Although ACPA positivity is a very good predictor for those patients who will develop IA within 1 year, it is still difficult to identify the exact individuals who will develop IA, because any ACPA-positive individual has an a priori chance of 50% of developing IA. In seronegative patients, the prediction of IA is even more difficult, because only 5% develop IA within the subsequent year. Imaging techniques have been shown to be able to detect synovitis before its clinical appearance and could be of help in identifying those at risk of IA,” the investigators wrote (Arthritis Res Ther. 2017;19:202. doi: 10.1186/s13075-017-1405-y).
Dr. van der Ven and her associates found that 31 (16%) of 196 patients who had arthralgia for less than 1 year in the hands, feet, or shoulders went on to develop IA after 1 year of follow-up. In this group of 196 patients at baseline, 72 (37%) had synovitis on ultrasound – defined as a greyscale grade of 2 or 3 and/or the presence of power Doppler signal (grade 1, 2, or 3) – including 32 with a positive power Doppler. A total of 18 patients were lost to follow-up during the first 6 months and another 19 were lost during months 6-12.
Rheumatologists who were unaware of ultrasound findings had to confirm soft-tissue swelling as arthritis at 1 year to classify it as incident IA. The positive predictive value of ultrasound for IA was only 26% when at least 1 joint out of 26 assessed was positive, but the negative predictive value when no joints were positive on ultrasound was 89%.
Overall, at 1 year, 15 of the 31 patients with IA had started therapy with a disease-modifying antirheumatic drug and 22 did not have a definite diagnosis; 12 had monoarthritis and 10 had polyarthritis. The remaining nine patients included four with rheumatoid arthritis, four with psoriatic arthritis, and one with spondyloarthritis.
At baseline, individuals with IA were more often older (mean age 50 vs. 44 years; P = .005), had synovitis on ultrasound (59% vs. 32%; P = .007), and had a positive power Doppler signal (31% vs. 12%; P = .012). A multivariate analysis revealed that IA at 1 year of follow-up could be independently predicted according to age (odds ratio, 1.06), morning stiffness lasting more than 30 minutes (OR, 2.80), ACPA positivity (OR, 2.35), and synovitis on ultrasound (OR, 2.65).
The investigators noted that the study’s limitations relate to requirements for patients to have at least two painful joints in hands, feet, or shoulders at baseline and two criteria related to inflammation. The possible inflammation-related criteria required for entry included morning stiffness for more than 1 hour, inability to clench a fist in the morning, pain when shaking someone’s hand, pins and needles in the fingers, difficulties wearing rings or shoes, family history of rheumatoid arthritis, and/or unexplained fatigue for less than 1 year.
Rheumatologists who enrolled patients into the cohort also may have “recruited clinically suspected patients with possibly more severe symptoms,” the investigators noted. Another potential source of bias related to the group of 38 patients who chose not to participate: It’s possible these patients had less severe symptoms than those who participated in the study.
The study was funded by an investigator-initiated grant from Pfizer. The authors declared that they have no competing interests.
Ultrasound evaluations to look for subclinical inflammation in joints of patients with arthralgia appear best at ruling out inflammatory arthritis (IA) 1 year in the future rather than ruling it in, according to findings from a multicenter cohort study published online in Arthritis Research and Therapy.
The imaging modality’s ability to identify those who will not go on to develop IA complemented the serologic and clinical factors that help to discriminate the individuals with arthralgia who are most at risk of the condition.
The ultimate goal of using imaging such as ultrasound in patients with arthralgia is to identify those who would benefit from starting treatment with disease-modifying antirheumatic drugs as early as possible to potentially improve outcomes, but it also could help to discriminate between the anti-citrullinated protein antibody (ACPA)-positive and seronegative individuals without clinical signs of inflammation at baseline who may progress from arthralgia to IA.
“Although ACPA positivity is a very good predictor for those patients who will develop IA within 1 year, it is still difficult to identify the exact individuals who will develop IA, because any ACPA-positive individual has an a priori chance of 50% of developing IA. In seronegative patients, the prediction of IA is even more difficult, because only 5% develop IA within the subsequent year. Imaging techniques have been shown to be able to detect synovitis before its clinical appearance and could be of help in identifying those at risk of IA,” the investigators wrote (Arthritis Res Ther. 2017;19:202. doi: 10.1186/s13075-017-1405-y).
Dr. van der Ven and her associates found that 31 (16%) of 196 patients who had arthralgia for less than 1 year in the hands, feet, or shoulders went on to develop IA after 1 year of follow-up. In this group of 196 patients at baseline, 72 (37%) had synovitis on ultrasound – defined as a greyscale grade of 2 or 3 and/or the presence of power Doppler signal (grade 1, 2, or 3) – including 32 with a positive power Doppler. A total of 18 patients were lost to follow-up during the first 6 months and another 19 were lost during months 6-12.
Rheumatologists who were unaware of ultrasound findings had to confirm soft-tissue swelling as arthritis at 1 year to classify it as incident IA. The positive predictive value of ultrasound for IA was only 26% when at least 1 joint out of 26 assessed was positive, but the negative predictive value when no joints were positive on ultrasound was 89%.
Overall, at 1 year, 15 of the 31 patients with IA had started therapy with a disease-modifying antirheumatic drug and 22 did not have a definite diagnosis; 12 had monoarthritis and 10 had polyarthritis. The remaining nine patients included four with rheumatoid arthritis, four with psoriatic arthritis, and one with spondyloarthritis.
At baseline, individuals with IA were more often older (mean age 50 vs. 44 years; P = .005), had synovitis on ultrasound (59% vs. 32%; P = .007), and had a positive power Doppler signal (31% vs. 12%; P = .012). A multivariate analysis revealed that IA at 1 year of follow-up could be independently predicted according to age (odds ratio, 1.06), morning stiffness lasting more than 30 minutes (OR, 2.80), ACPA positivity (OR, 2.35), and synovitis on ultrasound (OR, 2.65).
The investigators noted that the study’s limitations relate to requirements for patients to have at least two painful joints in hands, feet, or shoulders at baseline and two criteria related to inflammation. The possible inflammation-related criteria required for entry included morning stiffness for more than 1 hour, inability to clench a fist in the morning, pain when shaking someone’s hand, pins and needles in the fingers, difficulties wearing rings or shoes, family history of rheumatoid arthritis, and/or unexplained fatigue for less than 1 year.
Rheumatologists who enrolled patients into the cohort also may have “recruited clinically suspected patients with possibly more severe symptoms,” the investigators noted. Another potential source of bias related to the group of 38 patients who chose not to participate: It’s possible these patients had less severe symptoms than those who participated in the study.
The study was funded by an investigator-initiated grant from Pfizer. The authors declared that they have no competing interests.
FROM ARTHRITIS RESEARCH AND THERAPY
Key clinical point:
Major finding: The positive predictive value of ultrasound for IA was only 26% when at least 1 joint out of 26 assessed was positive, but the negative predictive value when no joints were positive on ultrasound was 89%.
Data source: A multicenter cohort study of 196 patients with arthralgia in at least two joints for less than 1 year.
Disclosures: The study was funded by an investigator-initiated grant from Pfizer. The authors declared that they have no competing interests.
Tocilizumab looks promising for corticosteroid refractory anti-PD-1-related adverse events
CHICAGO – Tocilizumab may be a therapeutic option for steroid-refractory immune-related adverse events that are secondary to PD-1 blockade, according to findings from a review of patients treated with nivolumab for various malignancies.
Of 87 patients who were treated with the PD-1 inhibitor between April 2015 and October 2016, 34 received tocilizumab for high-grade immune-related adverse events (irAEs) that were refractory to corticosteroids. Of those, 27 experienced clinical improvement, which was defined as documentation of symptom resolution or hospital discharge within 7 days, Aparna Hegde, MD, of East Carolina University, Greenville, N.C., reported at the Chicago Multidisciplinary Symposium in Thoracic Oncology.
The median time to discharge was 4 days, and no adverse effect on median overall survival was seen between those who received tocilizumab and those who did not (6.1 vs, 6.7 months, respectively), Dr. Hegde said.
There was, however, a trend toward inferior overall survival in patients who required more than one dose of tocilizumab, but the difference was not statistically significant (hazard ratio, 1.72), she noted.
“Immune checkpoint inhibitors are associated with an unprecedented clinical benefit in patients with lung cancer. However, they are also associated with a unique spectrum of immune-mediated adverse events. While the standard of care for initial management of these adverse events is corticosteroids, the management of steroid-refractory events is poorly defined,” she said, adding that data from randomized trials on which consensus guidelines could be based are lacking.
At East Carolina University, a significant proportion of patients treated with PD-1 inhibitors were presenting with systemic inflammatory response syndrome (SIRS)-like symptoms and immune-related organ toxicities similar to what has been described in cytokine release syndrome, Dr. Hegde said.
Such symptoms have also been reported with other types of immune therapy, such as CAR T-cell therapy and bispecific T-cell receptor–engaging antibodies in hematologic malignancies, she noted.
“In our experience, when we treated these patients with tocilizumab, which is an [anti-interleukin-6] receptor monoclonal antibody, we saw dramatic and rapid responses, not only in the SIRS symptoms, but also in other immune-related organ toxicities. Therefore, we adopted the use of tocilizumab as our standard treatment for high-grade immune-related adverse events,” she said, explaining that it has been well documented that interleukin (IL)-6 levels increase during cytokine release syndrome and are mediators of inflammation, suggesting that blocking IL-6 may treat irAEs without compromising the efficacy of immune therapy.
The current study was undertaken to look more closely at the responses to tocilizumab and to assess overall survival in those who received tocilizumab.
Study participants were being prospectively followed as part of another ongoing study looking at the relationship between systemic inflammation and cancer-related symptom burden. Most (77) were being treated for lung cancer and 10 had other types of malignancy. They received nivolumab at a dose of 3 mg/kg (or a flat dose of 240 mg after September 2016) every 2 weeks, and received tocilizumab at a dose of 4 mg/kg given over 1 hour. Those with grade 3 or 4 irAEs also received supportive care and corticosteroids. Median follow-up was 10.6 months.
C-reactive protein (CRP), a reliable surrogate marker of IL-6, was drawn at the first nivolumab infusion and before each subsequent infusion as part of the study in which the patients were enrolled, and for the current analysis was measured in relation to irAEs.
Significant reductions were seen in CRP levels after tocilizumab treatment; similar responses have been described in cytokine release syndrome, Dr. Hegde noted.
“Tocilizumab is a therapeutic option for management of immune-related adverse events in patients who are already on corticosteroids. CRP may be of clinical utility in detecting immune-related adverse events as well as monitoring the response to tocilizumab,” she said, adding that the current analysis is limited by the small patient number, single-center setting, use of tocilizumab outside of a clinical trial setting, and short follow-up.
Therefore, the findings require confirmation in multicenter randomized trials to determine “the definitive utility of tocilizumab, as well as CRP as an accompanying biomarker in the management of high-grade steroid refractory immune-related adverse events.”
Heather Wakelee, MD, an invited discussant at the symposium, commended Dr. Hegde and her colleagues for “coming up with a novel idea about how to treat [irAEs],” but also stressed the need for further study.
“This is a novel agent that has the potential ability to manage toxicity, and that’s important, because when you get beyond the steroids that we use as a first-line approach ... there’s not a whole lot else. We definitely have a clear unmet need,” said Dr. Wakelee of Stanford (Calif.) University.
However, she stressed that the approach must be evaluated in multicenter randomized trials “before we can be widely discussing this as a good thing to be doing.”
Dr. Hegde reported having no disclosures. Dr. Wakelee has been the institutional principal investigator for studies of nivolumab, tocilizumab, and other agents. She has consulted for Peregrine, ACEA, Pfizer, Helsinn, Genentech/Roche, Clovis, and Lilly, and received research/grant support from Clovis, Exelixis, AstraZeneca/Medimmune, Genentech/Roche, BMS, Gilead, Novartis, Xcovery, Pfizer, Celgene, Gilead, Pharmacyclics, and Lilly.
CHICAGO – Tocilizumab may be a therapeutic option for steroid-refractory immune-related adverse events that are secondary to PD-1 blockade, according to findings from a review of patients treated with nivolumab for various malignancies.
Of 87 patients who were treated with the PD-1 inhibitor between April 2015 and October 2016, 34 received tocilizumab for high-grade immune-related adverse events (irAEs) that were refractory to corticosteroids. Of those, 27 experienced clinical improvement, which was defined as documentation of symptom resolution or hospital discharge within 7 days, Aparna Hegde, MD, of East Carolina University, Greenville, N.C., reported at the Chicago Multidisciplinary Symposium in Thoracic Oncology.
The median time to discharge was 4 days, and no adverse effect on median overall survival was seen between those who received tocilizumab and those who did not (6.1 vs, 6.7 months, respectively), Dr. Hegde said.
There was, however, a trend toward inferior overall survival in patients who required more than one dose of tocilizumab, but the difference was not statistically significant (hazard ratio, 1.72), she noted.
“Immune checkpoint inhibitors are associated with an unprecedented clinical benefit in patients with lung cancer. However, they are also associated with a unique spectrum of immune-mediated adverse events. While the standard of care for initial management of these adverse events is corticosteroids, the management of steroid-refractory events is poorly defined,” she said, adding that data from randomized trials on which consensus guidelines could be based are lacking.
At East Carolina University, a significant proportion of patients treated with PD-1 inhibitors were presenting with systemic inflammatory response syndrome (SIRS)-like symptoms and immune-related organ toxicities similar to what has been described in cytokine release syndrome, Dr. Hegde said.
Such symptoms have also been reported with other types of immune therapy, such as CAR T-cell therapy and bispecific T-cell receptor–engaging antibodies in hematologic malignancies, she noted.
“In our experience, when we treated these patients with tocilizumab, which is an [anti-interleukin-6] receptor monoclonal antibody, we saw dramatic and rapid responses, not only in the SIRS symptoms, but also in other immune-related organ toxicities. Therefore, we adopted the use of tocilizumab as our standard treatment for high-grade immune-related adverse events,” she said, explaining that it has been well documented that interleukin (IL)-6 levels increase during cytokine release syndrome and are mediators of inflammation, suggesting that blocking IL-6 may treat irAEs without compromising the efficacy of immune therapy.
The current study was undertaken to look more closely at the responses to tocilizumab and to assess overall survival in those who received tocilizumab.
Study participants were being prospectively followed as part of another ongoing study looking at the relationship between systemic inflammation and cancer-related symptom burden. Most (77) were being treated for lung cancer and 10 had other types of malignancy. They received nivolumab at a dose of 3 mg/kg (or a flat dose of 240 mg after September 2016) every 2 weeks, and received tocilizumab at a dose of 4 mg/kg given over 1 hour. Those with grade 3 or 4 irAEs also received supportive care and corticosteroids. Median follow-up was 10.6 months.
C-reactive protein (CRP), a reliable surrogate marker of IL-6, was drawn at the first nivolumab infusion and before each subsequent infusion as part of the study in which the patients were enrolled, and for the current analysis was measured in relation to irAEs.
Significant reductions were seen in CRP levels after tocilizumab treatment; similar responses have been described in cytokine release syndrome, Dr. Hegde noted.
“Tocilizumab is a therapeutic option for management of immune-related adverse events in patients who are already on corticosteroids. CRP may be of clinical utility in detecting immune-related adverse events as well as monitoring the response to tocilizumab,” she said, adding that the current analysis is limited by the small patient number, single-center setting, use of tocilizumab outside of a clinical trial setting, and short follow-up.
Therefore, the findings require confirmation in multicenter randomized trials to determine “the definitive utility of tocilizumab, as well as CRP as an accompanying biomarker in the management of high-grade steroid refractory immune-related adverse events.”
Heather Wakelee, MD, an invited discussant at the symposium, commended Dr. Hegde and her colleagues for “coming up with a novel idea about how to treat [irAEs],” but also stressed the need for further study.
“This is a novel agent that has the potential ability to manage toxicity, and that’s important, because when you get beyond the steroids that we use as a first-line approach ... there’s not a whole lot else. We definitely have a clear unmet need,” said Dr. Wakelee of Stanford (Calif.) University.
However, she stressed that the approach must be evaluated in multicenter randomized trials “before we can be widely discussing this as a good thing to be doing.”
Dr. Hegde reported having no disclosures. Dr. Wakelee has been the institutional principal investigator for studies of nivolumab, tocilizumab, and other agents. She has consulted for Peregrine, ACEA, Pfizer, Helsinn, Genentech/Roche, Clovis, and Lilly, and received research/grant support from Clovis, Exelixis, AstraZeneca/Medimmune, Genentech/Roche, BMS, Gilead, Novartis, Xcovery, Pfizer, Celgene, Gilead, Pharmacyclics, and Lilly.
CHICAGO – Tocilizumab may be a therapeutic option for steroid-refractory immune-related adverse events that are secondary to PD-1 blockade, according to findings from a review of patients treated with nivolumab for various malignancies.
Of 87 patients who were treated with the PD-1 inhibitor between April 2015 and October 2016, 34 received tocilizumab for high-grade immune-related adverse events (irAEs) that were refractory to corticosteroids. Of those, 27 experienced clinical improvement, which was defined as documentation of symptom resolution or hospital discharge within 7 days, Aparna Hegde, MD, of East Carolina University, Greenville, N.C., reported at the Chicago Multidisciplinary Symposium in Thoracic Oncology.
The median time to discharge was 4 days, and no adverse effect on median overall survival was seen between those who received tocilizumab and those who did not (6.1 vs, 6.7 months, respectively), Dr. Hegde said.
There was, however, a trend toward inferior overall survival in patients who required more than one dose of tocilizumab, but the difference was not statistically significant (hazard ratio, 1.72), she noted.
“Immune checkpoint inhibitors are associated with an unprecedented clinical benefit in patients with lung cancer. However, they are also associated with a unique spectrum of immune-mediated adverse events. While the standard of care for initial management of these adverse events is corticosteroids, the management of steroid-refractory events is poorly defined,” she said, adding that data from randomized trials on which consensus guidelines could be based are lacking.
At East Carolina University, a significant proportion of patients treated with PD-1 inhibitors were presenting with systemic inflammatory response syndrome (SIRS)-like symptoms and immune-related organ toxicities similar to what has been described in cytokine release syndrome, Dr. Hegde said.
Such symptoms have also been reported with other types of immune therapy, such as CAR T-cell therapy and bispecific T-cell receptor–engaging antibodies in hematologic malignancies, she noted.
“In our experience, when we treated these patients with tocilizumab, which is an [anti-interleukin-6] receptor monoclonal antibody, we saw dramatic and rapid responses, not only in the SIRS symptoms, but also in other immune-related organ toxicities. Therefore, we adopted the use of tocilizumab as our standard treatment for high-grade immune-related adverse events,” she said, explaining that it has been well documented that interleukin (IL)-6 levels increase during cytokine release syndrome and are mediators of inflammation, suggesting that blocking IL-6 may treat irAEs without compromising the efficacy of immune therapy.
The current study was undertaken to look more closely at the responses to tocilizumab and to assess overall survival in those who received tocilizumab.
Study participants were being prospectively followed as part of another ongoing study looking at the relationship between systemic inflammation and cancer-related symptom burden. Most (77) were being treated for lung cancer and 10 had other types of malignancy. They received nivolumab at a dose of 3 mg/kg (or a flat dose of 240 mg after September 2016) every 2 weeks, and received tocilizumab at a dose of 4 mg/kg given over 1 hour. Those with grade 3 or 4 irAEs also received supportive care and corticosteroids. Median follow-up was 10.6 months.
C-reactive protein (CRP), a reliable surrogate marker of IL-6, was drawn at the first nivolumab infusion and before each subsequent infusion as part of the study in which the patients were enrolled, and for the current analysis was measured in relation to irAEs.
Significant reductions were seen in CRP levels after tocilizumab treatment; similar responses have been described in cytokine release syndrome, Dr. Hegde noted.
“Tocilizumab is a therapeutic option for management of immune-related adverse events in patients who are already on corticosteroids. CRP may be of clinical utility in detecting immune-related adverse events as well as monitoring the response to tocilizumab,” she said, adding that the current analysis is limited by the small patient number, single-center setting, use of tocilizumab outside of a clinical trial setting, and short follow-up.
Therefore, the findings require confirmation in multicenter randomized trials to determine “the definitive utility of tocilizumab, as well as CRP as an accompanying biomarker in the management of high-grade steroid refractory immune-related adverse events.”
Heather Wakelee, MD, an invited discussant at the symposium, commended Dr. Hegde and her colleagues for “coming up with a novel idea about how to treat [irAEs],” but also stressed the need for further study.
“This is a novel agent that has the potential ability to manage toxicity, and that’s important, because when you get beyond the steroids that we use as a first-line approach ... there’s not a whole lot else. We definitely have a clear unmet need,” said Dr. Wakelee of Stanford (Calif.) University.
However, she stressed that the approach must be evaluated in multicenter randomized trials “before we can be widely discussing this as a good thing to be doing.”
Dr. Hegde reported having no disclosures. Dr. Wakelee has been the institutional principal investigator for studies of nivolumab, tocilizumab, and other agents. She has consulted for Peregrine, ACEA, Pfizer, Helsinn, Genentech/Roche, Clovis, and Lilly, and received research/grant support from Clovis, Exelixis, AstraZeneca/Medimmune, Genentech/Roche, BMS, Gilead, Novartis, Xcovery, Pfizer, Celgene, Gilead, Pharmacyclics, and Lilly.
AT A SYMPOSIUM IN THORACIC ONCOLOGY
Key clinical point:
Major finding: Twenty-seven of 34 patients treated with tocilizumab experienced clinical improvement.
Data source: A review of 87 patients.
Disclosures: Dr. Hegde reported having no disclosures. Dr. Wakelee has been the institutional principal investigator for studies of nivolumab, tocilizumab, and other agents. She has consulted for Peregrine, ACEA, Pfizer, Helsinn, Genentech/Roche, Clovis, and Lilly, and received research/grant support from Clovis, Exelixis, AstraZeneca/Medimmune, Genentech/Roche, BMS, Gilead, Novartis, Xcovery, Pfizer, Celgene, Gilead, Pharmacyclics, and Lilly.
Adjuvant-boosted shingles vaccine earns FDA panel’s unanimous nod
A new vaccine for herpes zoster is both safe and effective in preventing herpes zoster, and in reducing the incidence of postherpetic neuralgia in older adults, according to a Food and Drug Administration advisory committee, which voted unanimously to recommend the vaccine.
The FDA generally follows the recommendations of its advisory committees.
The recombinant vaccine, dubbed HZ/su during the trial phase, showed efficacy of 97.2% against herpes zoster infection in adults aged 50 years and older, and 91.3% in adults aged 70 years and older. The effect persisted for up to the 4 years of study follow-up.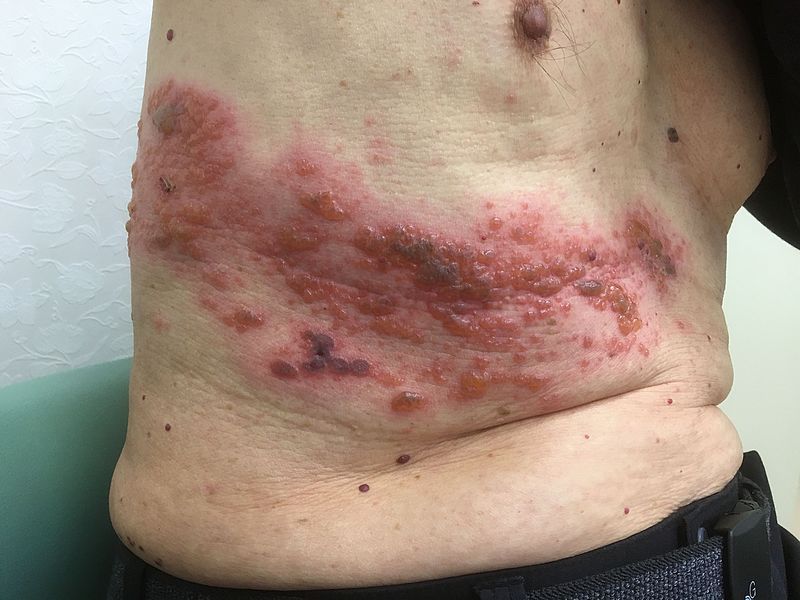
HZ/su had a generally favorable safety profile, though early constitutional symptoms and local site reactions were common, according to data presented by GlaxoSmithKline. HZ/su uses an adjuvant not found in any other U.S.-approved vaccine.
The incidence of postherpetic neuralgia, a common, persistent, and costly complication of herpes zoster, was 0.1 per 1,000 person-years in those receiving vaccine, compared with 0.9-1.2 per 1,000 person-years for those receiving placebo in the pivotal clinical trials for a median follow-up of 4 years.
In the vaccine’s pivotal clinical trials, efficacy was significantly higher than the levels seen for the only currently approved zoster live vaccine, Zostavax, especially for older populations. Zostavax’s efficacy for those aged 50-59 years is 69.8%, dropping to 18% for those aged 80 years or older.
The results of the two pivotal clinical trials were presented and analyzed by the sponsor and by FDA staff during a meeting of the Vaccines and Related Biological Products Advisory Committee of the FDA’s Center for Biologics Evaluation and Research (CBER).
During pre-vote discussions, committee members were unanimous in noting with favor the high and sustained efficacy seen for HZ/su in the trial data, especially for older populations. However, some participants wondered about the generalizability of both safety and efficacy data to all populations, given the very low trial enrollment numbers for Africans, African Americans, and individuals of Hispanic origin.
The two studies, Zoster-006 and Zoster-022, were similar in design and were conducted in parallel across 18 countries; data were able to be pooled for key efficacy and safety outcomes. Study Zoster-006 enrolled patients aged 50 years and older, while study Zoster-022 began enrollment at age 70. Patients were randomized to receive vaccine or placebo, and were followed for a median of 3.1 years for efficacy in Zoster-006 and a median of 3.9 years for Zoster-022. Safety data were obtained for a median 4.4 years for both studies.
The primary outcome measure for both studies in pooled analysis was the vaccine’s effectiveness against herpes zoster and postherpetic neuropathy in adults aged 70 and over. Safety was also assessed using pooled data.
The United States was represented by 3,934 of more than 29,000 patients enrolled globally. The remainder were primarily in Western Europe, with some sites in Australia and eastern Asia, Canada, and Latin America.
The vaccine consists of a recombinant, lyophilized truncated form of the varicella zoster virus (VZV) glycoprotein E (gE) antigen protein that, at the time of administration, is reconstituted with a novel adjuvant suspension. The antigen selection was based on the fact that gE is expressed on the surface of infected cells and is the target of both humoral and cellular immune responses in the host, said GSK’s Arnaud Didierlaurent, PhD, director and head of the adjuvant platform for GSK Vaccine’s Belgium research and development division.
The adjuvant, termed ASO1B, is not currently in use for any U.S.-approved vaccine, though it was developed more than 20 years ago, said Dr. Didierlaurent. Its combination with recombinant VZV gE was found to significantly boost the antigen’s immunogenicity during GSK’s vaccine development program. The adjuvant enhances a transient innate response in the first 3 days after administration that later helps maintain durably high levels of gE-specific antibodies and strengthens gE-specific cell-mediated immunity.
Mechanistically, the robust initial innate response is responsible for the constitutional symptoms and local site reactions seen in pooled data from the two pivotal clinical trials: 70%-85% of participants receiving HZ/su reported injection site pain, 38% of participants receiving HZ/su reported redness, and about a quarter reported swelling. By comparison, 9%-13% of those receiving placebo reported injection site pain, and about 1% reported redness and swelling.
Fatigue, headache, mild fever, myalgia, and shivering were all more common in those receiving HZ/su; both local and generalized symptoms were more common in younger recipients.
“I think this is a very good case for the first licensure of this adjuvant in the United States, because the efficacy seems pretty compelling, the disease is morbid, and there are a lot of people whose lives would be changed,” said committee member Sarah Long, MD, professor of pediatrics at Drexel University, Philadelphia.
Both the GSK and FDA presentations were in agreement that serious adverse events were in the range to be expected for an older population, and balanced across study arms. However, particular attention will be given to certain potential complications during the proposed pharmacovigilance plan.
“An imbalance toward vaccine versus placebo was observed” for gout, optic ischemic neuropathy, amyotrophic lateral sclerosis, osteonecrosis, convulsion-type reactions, and supraventricular tachycardias. “All are an adverse event of interest and will be included in planned targeted safety study,” said Dr. Didierlaurent.
Several committee members remarked on the difficulty of evaluating vaccine safety in an older population, where analysis takes place against the backdrop of more comorbidities and acute illnesses than in the younger population.
“There has been a thoughtful job both by the sponsor and by CBER in looking at complicated data,” said Melinda Wharton, MD, the director of the immunization services division of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, Atlanta.
The committee’s chair, Kathryn Edwards, MD, agreed. “I applaud the comprehensive analysis of all these safety signals. Both the sponsor and the FDA have done a wonderful job of drilling down and answering these questions,” she said. Dr. Edwards is the Sarah H. Sell and Cornelius Vanderbilt chair in pediatrics at Vanderbilt University, Nashville, Tenn.
Herpes zoster, a reactivation of the varicella virus that lies dormant in dorsal root or cranial nerve ganglia from earlier infection, is seen in about 1 million cases per year in the United States, with about 100,000 to 200,000 cases of postherpetic neuralgia occurring, said Jeffrey Cohen, MD, chief of the laboratory of infectious diseases at the National Institute of Allergy and Infectious Diseases, Bethesda, Md. The rates of herpes zoster are increasing in the United States for unknown reasons, and direct medical costs may currently exceed $1 billion annually, he said.
Each 0.5 mL dose of the HZ/su vaccine contains 50 mcg each of the recombinant VZV gE antigen and each of the two component parts of the ASO1B adjuvant. Two doses of the vaccine are administered intramuscularly 2-6 months apart. Dose-ranging studies were conducted before the pivotal clinical trials to ascertain the optimal dose of all of the vaccine components, the need for two doses, and the optimal spacing between doses.
All committee participants submitted conflict of interest statements to the FDA, and any potential conflicts were resolved before the hearing.
koakes@frontlinemedcom.com
On Twitter @karioakes
A new vaccine for herpes zoster is both safe and effective in preventing herpes zoster, and in reducing the incidence of postherpetic neuralgia in older adults, according to a Food and Drug Administration advisory committee, which voted unanimously to recommend the vaccine.
The FDA generally follows the recommendations of its advisory committees.
The recombinant vaccine, dubbed HZ/su during the trial phase, showed efficacy of 97.2% against herpes zoster infection in adults aged 50 years and older, and 91.3% in adults aged 70 years and older. The effect persisted for up to the 4 years of study follow-up.
HZ/su had a generally favorable safety profile, though early constitutional symptoms and local site reactions were common, according to data presented by GlaxoSmithKline. HZ/su uses an adjuvant not found in any other U.S.-approved vaccine.
The incidence of postherpetic neuralgia, a common, persistent, and costly complication of herpes zoster, was 0.1 per 1,000 person-years in those receiving vaccine, compared with 0.9-1.2 per 1,000 person-years for those receiving placebo in the pivotal clinical trials for a median follow-up of 4 years.
In the vaccine’s pivotal clinical trials, efficacy was significantly higher than the levels seen for the only currently approved zoster live vaccine, Zostavax, especially for older populations. Zostavax’s efficacy for those aged 50-59 years is 69.8%, dropping to 18% for those aged 80 years or older.
The results of the two pivotal clinical trials were presented and analyzed by the sponsor and by FDA staff during a meeting of the Vaccines and Related Biological Products Advisory Committee of the FDA’s Center for Biologics Evaluation and Research (CBER).
During pre-vote discussions, committee members were unanimous in noting with favor the high and sustained efficacy seen for HZ/su in the trial data, especially for older populations. However, some participants wondered about the generalizability of both safety and efficacy data to all populations, given the very low trial enrollment numbers for Africans, African Americans, and individuals of Hispanic origin.
The two studies, Zoster-006 and Zoster-022, were similar in design and were conducted in parallel across 18 countries; data were able to be pooled for key efficacy and safety outcomes. Study Zoster-006 enrolled patients aged 50 years and older, while study Zoster-022 began enrollment at age 70. Patients were randomized to receive vaccine or placebo, and were followed for a median of 3.1 years for efficacy in Zoster-006 and a median of 3.9 years for Zoster-022. Safety data were obtained for a median 4.4 years for both studies.
The primary outcome measure for both studies in pooled analysis was the vaccine’s effectiveness against herpes zoster and postherpetic neuropathy in adults aged 70 and over. Safety was also assessed using pooled data.
The United States was represented by 3,934 of more than 29,000 patients enrolled globally. The remainder were primarily in Western Europe, with some sites in Australia and eastern Asia, Canada, and Latin America.
The vaccine consists of a recombinant, lyophilized truncated form of the varicella zoster virus (VZV) glycoprotein E (gE) antigen protein that, at the time of administration, is reconstituted with a novel adjuvant suspension. The antigen selection was based on the fact that gE is expressed on the surface of infected cells and is the target of both humoral and cellular immune responses in the host, said GSK’s Arnaud Didierlaurent, PhD, director and head of the adjuvant platform for GSK Vaccine’s Belgium research and development division.
The adjuvant, termed ASO1B, is not currently in use for any U.S.-approved vaccine, though it was developed more than 20 years ago, said Dr. Didierlaurent. Its combination with recombinant VZV gE was found to significantly boost the antigen’s immunogenicity during GSK’s vaccine development program. The adjuvant enhances a transient innate response in the first 3 days after administration that later helps maintain durably high levels of gE-specific antibodies and strengthens gE-specific cell-mediated immunity.
Mechanistically, the robust initial innate response is responsible for the constitutional symptoms and local site reactions seen in pooled data from the two pivotal clinical trials: 70%-85% of participants receiving HZ/su reported injection site pain, 38% of participants receiving HZ/su reported redness, and about a quarter reported swelling. By comparison, 9%-13% of those receiving placebo reported injection site pain, and about 1% reported redness and swelling.
Fatigue, headache, mild fever, myalgia, and shivering were all more common in those receiving HZ/su; both local and generalized symptoms were more common in younger recipients.
“I think this is a very good case for the first licensure of this adjuvant in the United States, because the efficacy seems pretty compelling, the disease is morbid, and there are a lot of people whose lives would be changed,” said committee member Sarah Long, MD, professor of pediatrics at Drexel University, Philadelphia.
Both the GSK and FDA presentations were in agreement that serious adverse events were in the range to be expected for an older population, and balanced across study arms. However, particular attention will be given to certain potential complications during the proposed pharmacovigilance plan.
“An imbalance toward vaccine versus placebo was observed” for gout, optic ischemic neuropathy, amyotrophic lateral sclerosis, osteonecrosis, convulsion-type reactions, and supraventricular tachycardias. “All are an adverse event of interest and will be included in planned targeted safety study,” said Dr. Didierlaurent.
Several committee members remarked on the difficulty of evaluating vaccine safety in an older population, where analysis takes place against the backdrop of more comorbidities and acute illnesses than in the younger population.
“There has been a thoughtful job both by the sponsor and by CBER in looking at complicated data,” said Melinda Wharton, MD, the director of the immunization services division of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, Atlanta.
The committee’s chair, Kathryn Edwards, MD, agreed. “I applaud the comprehensive analysis of all these safety signals. Both the sponsor and the FDA have done a wonderful job of drilling down and answering these questions,” she said. Dr. Edwards is the Sarah H. Sell and Cornelius Vanderbilt chair in pediatrics at Vanderbilt University, Nashville, Tenn.
Herpes zoster, a reactivation of the varicella virus that lies dormant in dorsal root or cranial nerve ganglia from earlier infection, is seen in about 1 million cases per year in the United States, with about 100,000 to 200,000 cases of postherpetic neuralgia occurring, said Jeffrey Cohen, MD, chief of the laboratory of infectious diseases at the National Institute of Allergy and Infectious Diseases, Bethesda, Md. The rates of herpes zoster are increasing in the United States for unknown reasons, and direct medical costs may currently exceed $1 billion annually, he said.
Each 0.5 mL dose of the HZ/su vaccine contains 50 mcg each of the recombinant VZV gE antigen and each of the two component parts of the ASO1B adjuvant. Two doses of the vaccine are administered intramuscularly 2-6 months apart. Dose-ranging studies were conducted before the pivotal clinical trials to ascertain the optimal dose of all of the vaccine components, the need for two doses, and the optimal spacing between doses.
All committee participants submitted conflict of interest statements to the FDA, and any potential conflicts were resolved before the hearing.
koakes@frontlinemedcom.com
On Twitter @karioakes
A new vaccine for herpes zoster is both safe and effective in preventing herpes zoster, and in reducing the incidence of postherpetic neuralgia in older adults, according to a Food and Drug Administration advisory committee, which voted unanimously to recommend the vaccine.
The FDA generally follows the recommendations of its advisory committees.
The recombinant vaccine, dubbed HZ/su during the trial phase, showed efficacy of 97.2% against herpes zoster infection in adults aged 50 years and older, and 91.3% in adults aged 70 years and older. The effect persisted for up to the 4 years of study follow-up.
HZ/su had a generally favorable safety profile, though early constitutional symptoms and local site reactions were common, according to data presented by GlaxoSmithKline. HZ/su uses an adjuvant not found in any other U.S.-approved vaccine.
The incidence of postherpetic neuralgia, a common, persistent, and costly complication of herpes zoster, was 0.1 per 1,000 person-years in those receiving vaccine, compared with 0.9-1.2 per 1,000 person-years for those receiving placebo in the pivotal clinical trials for a median follow-up of 4 years.
In the vaccine’s pivotal clinical trials, efficacy was significantly higher than the levels seen for the only currently approved zoster live vaccine, Zostavax, especially for older populations. Zostavax’s efficacy for those aged 50-59 years is 69.8%, dropping to 18% for those aged 80 years or older.
The results of the two pivotal clinical trials were presented and analyzed by the sponsor and by FDA staff during a meeting of the Vaccines and Related Biological Products Advisory Committee of the FDA’s Center for Biologics Evaluation and Research (CBER).
During pre-vote discussions, committee members were unanimous in noting with favor the high and sustained efficacy seen for HZ/su in the trial data, especially for older populations. However, some participants wondered about the generalizability of both safety and efficacy data to all populations, given the very low trial enrollment numbers for Africans, African Americans, and individuals of Hispanic origin.
The two studies, Zoster-006 and Zoster-022, were similar in design and were conducted in parallel across 18 countries; data were able to be pooled for key efficacy and safety outcomes. Study Zoster-006 enrolled patients aged 50 years and older, while study Zoster-022 began enrollment at age 70. Patients were randomized to receive vaccine or placebo, and were followed for a median of 3.1 years for efficacy in Zoster-006 and a median of 3.9 years for Zoster-022. Safety data were obtained for a median 4.4 years for both studies.
The primary outcome measure for both studies in pooled analysis was the vaccine’s effectiveness against herpes zoster and postherpetic neuropathy in adults aged 70 and over. Safety was also assessed using pooled data.
The United States was represented by 3,934 of more than 29,000 patients enrolled globally. The remainder were primarily in Western Europe, with some sites in Australia and eastern Asia, Canada, and Latin America.
The vaccine consists of a recombinant, lyophilized truncated form of the varicella zoster virus (VZV) glycoprotein E (gE) antigen protein that, at the time of administration, is reconstituted with a novel adjuvant suspension. The antigen selection was based on the fact that gE is expressed on the surface of infected cells and is the target of both humoral and cellular immune responses in the host, said GSK’s Arnaud Didierlaurent, PhD, director and head of the adjuvant platform for GSK Vaccine’s Belgium research and development division.
The adjuvant, termed ASO1B, is not currently in use for any U.S.-approved vaccine, though it was developed more than 20 years ago, said Dr. Didierlaurent. Its combination with recombinant VZV gE was found to significantly boost the antigen’s immunogenicity during GSK’s vaccine development program. The adjuvant enhances a transient innate response in the first 3 days after administration that later helps maintain durably high levels of gE-specific antibodies and strengthens gE-specific cell-mediated immunity.
Mechanistically, the robust initial innate response is responsible for the constitutional symptoms and local site reactions seen in pooled data from the two pivotal clinical trials: 70%-85% of participants receiving HZ/su reported injection site pain, 38% of participants receiving HZ/su reported redness, and about a quarter reported swelling. By comparison, 9%-13% of those receiving placebo reported injection site pain, and about 1% reported redness and swelling.
Fatigue, headache, mild fever, myalgia, and shivering were all more common in those receiving HZ/su; both local and generalized symptoms were more common in younger recipients.
“I think this is a very good case for the first licensure of this adjuvant in the United States, because the efficacy seems pretty compelling, the disease is morbid, and there are a lot of people whose lives would be changed,” said committee member Sarah Long, MD, professor of pediatrics at Drexel University, Philadelphia.
Both the GSK and FDA presentations were in agreement that serious adverse events were in the range to be expected for an older population, and balanced across study arms. However, particular attention will be given to certain potential complications during the proposed pharmacovigilance plan.
“An imbalance toward vaccine versus placebo was observed” for gout, optic ischemic neuropathy, amyotrophic lateral sclerosis, osteonecrosis, convulsion-type reactions, and supraventricular tachycardias. “All are an adverse event of interest and will be included in planned targeted safety study,” said Dr. Didierlaurent.
Several committee members remarked on the difficulty of evaluating vaccine safety in an older population, where analysis takes place against the backdrop of more comorbidities and acute illnesses than in the younger population.
“There has been a thoughtful job both by the sponsor and by CBER in looking at complicated data,” said Melinda Wharton, MD, the director of the immunization services division of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, Atlanta.
The committee’s chair, Kathryn Edwards, MD, agreed. “I applaud the comprehensive analysis of all these safety signals. Both the sponsor and the FDA have done a wonderful job of drilling down and answering these questions,” she said. Dr. Edwards is the Sarah H. Sell and Cornelius Vanderbilt chair in pediatrics at Vanderbilt University, Nashville, Tenn.
Herpes zoster, a reactivation of the varicella virus that lies dormant in dorsal root or cranial nerve ganglia from earlier infection, is seen in about 1 million cases per year in the United States, with about 100,000 to 200,000 cases of postherpetic neuralgia occurring, said Jeffrey Cohen, MD, chief of the laboratory of infectious diseases at the National Institute of Allergy and Infectious Diseases, Bethesda, Md. The rates of herpes zoster are increasing in the United States for unknown reasons, and direct medical costs may currently exceed $1 billion annually, he said.
Each 0.5 mL dose of the HZ/su vaccine contains 50 mcg each of the recombinant VZV gE antigen and each of the two component parts of the ASO1B adjuvant. Two doses of the vaccine are administered intramuscularly 2-6 months apart. Dose-ranging studies were conducted before the pivotal clinical trials to ascertain the optimal dose of all of the vaccine components, the need for two doses, and the optimal spacing between doses.
All committee participants submitted conflict of interest statements to the FDA, and any potential conflicts were resolved before the hearing.
koakes@frontlinemedcom.com
On Twitter @karioakes
Worsening osteoporosis care for RA patients shows need for action
Osteoporosis care in patients with rheumatoid arthritis is suboptimal, and the relative risk of the application of appropriate osteoporosis care in both RA and osteoarthritis patients has been declining steadily over the past decade, according to findings from a large, prospective, observational study.
The study included 11,669 RA patients and 2,829 OA patients in the National Data Bank for Rheumatic Diseases who were followed from 2003 through 2014 and showed that about half of the RA patients in whom osteoporosis treatment was indicated never received an osteoporosis medication. Further, the decline in the application of appropriate osteoporosis care was apparent even after the release of the 2010 American College of Rheumatology Guideline for the Prevention and Treatment of Glucocorticoid-Induced Osteoporosis, according to the findings, which have been accepted for publication in Arthritis Care & Research (doi: 10.1002/acr.23331).
A more recent study in the general population showed that, regardless of glucocorticoid (GC) use, there was a significant decrease in the use of oral bisphosphonates from 1996 to 2012, noted Dr. Ozen of the University of Nebraska, Omaha, and Marmara University, Istanbul, Turkey.
“The evidence from all these studies suggests that, despite a slight improvement in GIOP care, with a decline in overall OP care in the early 2000s, the OP care gap has always been suboptimal and declined over the last decade significantly,” she said, adding that this is “especially more worrisome in glucocorticoid-receiving RA patients,” in whom bone loss and fracture risk are dramatically increased.
“Additionally, considering that rheumatologists do better regarding comorbidity follow-up and management than other specialists who follow patients with rheumatic diseases, the gap for osteoporosis care might be even higher than we reported,” she said.
Indeed, among physicians caring for patients with osteoporosis, there is concern that “we’re on the precipice of further increase in fracture burden,” according to Kenneth Saag, MD, a rheumatologist and professor at the University of Alabama at Birmingham.
The conclusions of the study by Dr. Ozen and her colleagues are disturbing, but “not tremendously surprising,” said Dr. Saag, who also is president of the National Osteoporosis Foundation.
The findings are consistent with those from previous studies, he explained.
For example, Daniel H. Solomon, MD, and his associates showed that only 23% of 623 RA patients, including 236 patients who were taking glucocorticoids at an index visit in 1999, underwent bone densitometry, and only 42% were prescribed a medication (other than calcium and/or vitamin D), that reduces bone loss (Arthritis Rheum. 2002;46:3136-42). They also showed that, in 2004, despite a slight improvement vs. prior years, only 48% of 193 RA patients included in a chart review had received a bone mineral density test or medication for osteoporosis, and 64% of those taking more than 5 mg of prednisone for more than 3 months were receiving osteoporosis management (Arthritis Care Res. 2006;55:873-7).
In the current study, Dr. Ozen and her colleagues looked at both RA and OA patients and found that overall, osteoporosis treatment or screening was reported in 67.4% of study subjects over a mean of 5.5 years follow-up. Of those eligible for osteoporosis treatment based on the 2010 ACR guidelines, including 48.4% of RA patients and 17.6% of OA patients, 55% reported osteoporosis medication use. Despite their increased risk of osteoporosis, particularly in the setting of glucocorticoid use, RA patients were not more likely than OA patients to undergo screening or receive treatment (hazard ratio, 1.04).
“This suboptimal and decreasing trend for osteoporosis treatment and screening in RA patients is important as the prevalence of osteoporosis leading to fractures in RA, regardless of GC use, is still high, despite aggressive management strategies and the use of [biologic disease-modifying antirheumatic agents],” they wrote.
Factors contributing to the decline
The reasons for the decline after 2008 are not fully known, but data suggest that both patient and clinician factors play a role.
Lack of knowledge, unwillingness to take additional drugs, and cost are among patient factors that may interfere with screening and treatment, and lack of experience and time, and a focus more on disease activity and other comorbid conditions may be among clinician factors, they said.
Other factors that may have affected patient and clinician willingness to pursue care include 2007 cuts in Medicare reimbursement for dual-energy x-ray absorptiometry (DXA), a 2006 warning regarding jaw osteonecrosis with bisphosphonate use, and 2007 and 2010 publications about atrial fibrillation and atypical femoral fractures associated with long-term bisphosphonate treatment, they noted.
Dr. Saag agreed that it is difficult to pinpoint exact reasons for the decline, but said the DXA reimbursement cuts have had a significant effect.
“Declining reimbursement is likely one of the major factors, if not the major factor in the decline in testing that has been observed nationally,” he said, explaining that reimbursement was below the break-even point for many physicians in freestanding medical practices, and that slight adjustments have applied only to facilities located in hospitals. As a result, the availability of DXA screening has declined.
The National Osteoporosis Foundation and other organizations such as the International Society for Clinical Densitometry are lobbying for changes, as there is a very strong link between testing and treatment.
“If we can’t get more people tested, we’re unlikely to get more people treated,” Dr. Saag said.
The availability of new agents for the treatment of osteoporosis could also lead to improvements in screening and care, he added.
In July, Amgen submitted to the Food and Drug Administration a supplemental new Biologics License Application for Prolia (denosumab) for the treatment of patients with GIOP. The application is based on a phase 3 study evaluating the safety and efficacy of Prolia, compared with risedronate, in patients receiving glucocorticoid treatment. Denosumab was also shown in a phase 3 study presented at the 2016 ACR meeting to result in greater improvement in bone mineral density vs. bisphosphonates across multiple sites, said Dr. Saag, an investigator for the trial.
“So we’re pleased about that finding, and we’re hopeful that eventually denosumab may become another treatment option,” he said, adding that having more choices leads to more personalized care for patients.
“We hope that, if it does receive approval and is utilized, that it may partially help address the gap in treatment that has been identified,” he said, adding that abaloparatide (Tymlos), which is approved for use in postmenopausal women with osteoporosis and high fracture risk, and romosozumab, currently in late-phase development, could also eventually help bridge the gap.
Recent guideline update
There is also optimism that a recent update to the 2010 guideline, published in August in Arthritis Care & Research, will help address the problem (Arthritis Care Res. 2017;69:1095-1110).
“We hope that one the most important impacts of the new 2017 ACR GIOP guideline will be increasing the awareness of GIOP care,” Dr. Ozen said, noting that changes in the new guideline will also be helpful for guiding management of certain patients not addressed in the 2010 guideline, including adults under age 40 years and children.
The new guideline also incorporates FRAX hip fracture scores into the risk-assessment recommendations .
“Lastly, the addition of other oral bisphosphonates and denosumab to the guideline has broadened the potential therapeutic options for physicians and patients,” she said.
Efforts by the ACR to make these guidelines widely known among clinicians could also help promote improved screening and treatment, said Dr. Buckley, a professor of internal medicine and pediatrics at Yale University, New Haven, Conn.
And that awareness is what is needed most, Dr. Ozen said.
“What we need in osteoporosis care of RA or GIOP patients is more awareness and education about risk-benefit ratios of antiosteoporotic medications instead of focusing on the medication type. We hope that our paper showing the worsening osteoporosis care and the new 2017 ACR GIOP guideline provide more awareness and guidance for the physicians managing these patients,” she said, adding that overcoming obstacles in osteoporosis care requires identification and attention to all potential causes for the suboptimal management of osteoporosis from both patients’ and physicians’ perspectives.
“With the improvement in management strategies of rheumatic diseases ... we anticipate improvement in life expectancy and cardiovascular outcomes in RA and inflammatory rheumatic diseases. In this regard, with aging and decreased but ongoing clinical or subclinical chronic inflammation, it is highly likely that we might be dealing with much older patients with higher osteoporosis and fracture risks. Considering the significant contribution of fractures on cost, disability, morbidity, and mortality of these diseases, rheumatologists and other specialists should work together more vigilantly to overcome the obstacles and improve osteoporosis care,” she said.
Dr. Ozen and Dr. Buckley reported having no conflicts of interest. Dr. Saag is an investigator and consultant for Amgen, Merck, and Radius Health, and is a consultant for Lilly.
Osteoporosis care in patients with rheumatoid arthritis is suboptimal, and the relative risk of the application of appropriate osteoporosis care in both RA and osteoarthritis patients has been declining steadily over the past decade, according to findings from a large, prospective, observational study.
The study included 11,669 RA patients and 2,829 OA patients in the National Data Bank for Rheumatic Diseases who were followed from 2003 through 2014 and showed that about half of the RA patients in whom osteoporosis treatment was indicated never received an osteoporosis medication. Further, the decline in the application of appropriate osteoporosis care was apparent even after the release of the 2010 American College of Rheumatology Guideline for the Prevention and Treatment of Glucocorticoid-Induced Osteoporosis, according to the findings, which have been accepted for publication in Arthritis Care & Research (doi: 10.1002/acr.23331).
A more recent study in the general population showed that, regardless of glucocorticoid (GC) use, there was a significant decrease in the use of oral bisphosphonates from 1996 to 2012, noted Dr. Ozen of the University of Nebraska, Omaha, and Marmara University, Istanbul, Turkey.
“The evidence from all these studies suggests that, despite a slight improvement in GIOP care, with a decline in overall OP care in the early 2000s, the OP care gap has always been suboptimal and declined over the last decade significantly,” she said, adding that this is “especially more worrisome in glucocorticoid-receiving RA patients,” in whom bone loss and fracture risk are dramatically increased.
“Additionally, considering that rheumatologists do better regarding comorbidity follow-up and management than other specialists who follow patients with rheumatic diseases, the gap for osteoporosis care might be even higher than we reported,” she said.
Indeed, among physicians caring for patients with osteoporosis, there is concern that “we’re on the precipice of further increase in fracture burden,” according to Kenneth Saag, MD, a rheumatologist and professor at the University of Alabama at Birmingham.
The conclusions of the study by Dr. Ozen and her colleagues are disturbing, but “not tremendously surprising,” said Dr. Saag, who also is president of the National Osteoporosis Foundation.
The findings are consistent with those from previous studies, he explained.
For example, Daniel H. Solomon, MD, and his associates showed that only 23% of 623 RA patients, including 236 patients who were taking glucocorticoids at an index visit in 1999, underwent bone densitometry, and only 42% were prescribed a medication (other than calcium and/or vitamin D), that reduces bone loss (Arthritis Rheum. 2002;46:3136-42). They also showed that, in 2004, despite a slight improvement vs. prior years, only 48% of 193 RA patients included in a chart review had received a bone mineral density test or medication for osteoporosis, and 64% of those taking more than 5 mg of prednisone for more than 3 months were receiving osteoporosis management (Arthritis Care Res. 2006;55:873-7).
In the current study, Dr. Ozen and her colleagues looked at both RA and OA patients and found that overall, osteoporosis treatment or screening was reported in 67.4% of study subjects over a mean of 5.5 years follow-up. Of those eligible for osteoporosis treatment based on the 2010 ACR guidelines, including 48.4% of RA patients and 17.6% of OA patients, 55% reported osteoporosis medication use. Despite their increased risk of osteoporosis, particularly in the setting of glucocorticoid use, RA patients were not more likely than OA patients to undergo screening or receive treatment (hazard ratio, 1.04).
“This suboptimal and decreasing trend for osteoporosis treatment and screening in RA patients is important as the prevalence of osteoporosis leading to fractures in RA, regardless of GC use, is still high, despite aggressive management strategies and the use of [biologic disease-modifying antirheumatic agents],” they wrote.
Factors contributing to the decline
The reasons for the decline after 2008 are not fully known, but data suggest that both patient and clinician factors play a role.
Lack of knowledge, unwillingness to take additional drugs, and cost are among patient factors that may interfere with screening and treatment, and lack of experience and time, and a focus more on disease activity and other comorbid conditions may be among clinician factors, they said.
Other factors that may have affected patient and clinician willingness to pursue care include 2007 cuts in Medicare reimbursement for dual-energy x-ray absorptiometry (DXA), a 2006 warning regarding jaw osteonecrosis with bisphosphonate use, and 2007 and 2010 publications about atrial fibrillation and atypical femoral fractures associated with long-term bisphosphonate treatment, they noted.
Dr. Saag agreed that it is difficult to pinpoint exact reasons for the decline, but said the DXA reimbursement cuts have had a significant effect.
“Declining reimbursement is likely one of the major factors, if not the major factor in the decline in testing that has been observed nationally,” he said, explaining that reimbursement was below the break-even point for many physicians in freestanding medical practices, and that slight adjustments have applied only to facilities located in hospitals. As a result, the availability of DXA screening has declined.
The National Osteoporosis Foundation and other organizations such as the International Society for Clinical Densitometry are lobbying for changes, as there is a very strong link between testing and treatment.
“If we can’t get more people tested, we’re unlikely to get more people treated,” Dr. Saag said.
The availability of new agents for the treatment of osteoporosis could also lead to improvements in screening and care, he added.
In July, Amgen submitted to the Food and Drug Administration a supplemental new Biologics License Application for Prolia (denosumab) for the treatment of patients with GIOP. The application is based on a phase 3 study evaluating the safety and efficacy of Prolia, compared with risedronate, in patients receiving glucocorticoid treatment. Denosumab was also shown in a phase 3 study presented at the 2016 ACR meeting to result in greater improvement in bone mineral density vs. bisphosphonates across multiple sites, said Dr. Saag, an investigator for the trial.
“So we’re pleased about that finding, and we’re hopeful that eventually denosumab may become another treatment option,” he said, adding that having more choices leads to more personalized care for patients.
“We hope that, if it does receive approval and is utilized, that it may partially help address the gap in treatment that has been identified,” he said, adding that abaloparatide (Tymlos), which is approved for use in postmenopausal women with osteoporosis and high fracture risk, and romosozumab, currently in late-phase development, could also eventually help bridge the gap.
Recent guideline update
There is also optimism that a recent update to the 2010 guideline, published in August in Arthritis Care & Research, will help address the problem (Arthritis Care Res. 2017;69:1095-1110).
“We hope that one the most important impacts of the new 2017 ACR GIOP guideline will be increasing the awareness of GIOP care,” Dr. Ozen said, noting that changes in the new guideline will also be helpful for guiding management of certain patients not addressed in the 2010 guideline, including adults under age 40 years and children.
The new guideline also incorporates FRAX hip fracture scores into the risk-assessment recommendations .
“Lastly, the addition of other oral bisphosphonates and denosumab to the guideline has broadened the potential therapeutic options for physicians and patients,” she said.
Efforts by the ACR to make these guidelines widely known among clinicians could also help promote improved screening and treatment, said Dr. Buckley, a professor of internal medicine and pediatrics at Yale University, New Haven, Conn.
And that awareness is what is needed most, Dr. Ozen said.
“What we need in osteoporosis care of RA or GIOP patients is more awareness and education about risk-benefit ratios of antiosteoporotic medications instead of focusing on the medication type. We hope that our paper showing the worsening osteoporosis care and the new 2017 ACR GIOP guideline provide more awareness and guidance for the physicians managing these patients,” she said, adding that overcoming obstacles in osteoporosis care requires identification and attention to all potential causes for the suboptimal management of osteoporosis from both patients’ and physicians’ perspectives.
“With the improvement in management strategies of rheumatic diseases ... we anticipate improvement in life expectancy and cardiovascular outcomes in RA and inflammatory rheumatic diseases. In this regard, with aging and decreased but ongoing clinical or subclinical chronic inflammation, it is highly likely that we might be dealing with much older patients with higher osteoporosis and fracture risks. Considering the significant contribution of fractures on cost, disability, morbidity, and mortality of these diseases, rheumatologists and other specialists should work together more vigilantly to overcome the obstacles and improve osteoporosis care,” she said.
Dr. Ozen and Dr. Buckley reported having no conflicts of interest. Dr. Saag is an investigator and consultant for Amgen, Merck, and Radius Health, and is a consultant for Lilly.
Osteoporosis care in patients with rheumatoid arthritis is suboptimal, and the relative risk of the application of appropriate osteoporosis care in both RA and osteoarthritis patients has been declining steadily over the past decade, according to findings from a large, prospective, observational study.
The study included 11,669 RA patients and 2,829 OA patients in the National Data Bank for Rheumatic Diseases who were followed from 2003 through 2014 and showed that about half of the RA patients in whom osteoporosis treatment was indicated never received an osteoporosis medication. Further, the decline in the application of appropriate osteoporosis care was apparent even after the release of the 2010 American College of Rheumatology Guideline for the Prevention and Treatment of Glucocorticoid-Induced Osteoporosis, according to the findings, which have been accepted for publication in Arthritis Care & Research (doi: 10.1002/acr.23331).
A more recent study in the general population showed that, regardless of glucocorticoid (GC) use, there was a significant decrease in the use of oral bisphosphonates from 1996 to 2012, noted Dr. Ozen of the University of Nebraska, Omaha, and Marmara University, Istanbul, Turkey.
“The evidence from all these studies suggests that, despite a slight improvement in GIOP care, with a decline in overall OP care in the early 2000s, the OP care gap has always been suboptimal and declined over the last decade significantly,” she said, adding that this is “especially more worrisome in glucocorticoid-receiving RA patients,” in whom bone loss and fracture risk are dramatically increased.
“Additionally, considering that rheumatologists do better regarding comorbidity follow-up and management than other specialists who follow patients with rheumatic diseases, the gap for osteoporosis care might be even higher than we reported,” she said.
Indeed, among physicians caring for patients with osteoporosis, there is concern that “we’re on the precipice of further increase in fracture burden,” according to Kenneth Saag, MD, a rheumatologist and professor at the University of Alabama at Birmingham.
The conclusions of the study by Dr. Ozen and her colleagues are disturbing, but “not tremendously surprising,” said Dr. Saag, who also is president of the National Osteoporosis Foundation.
The findings are consistent with those from previous studies, he explained.
For example, Daniel H. Solomon, MD, and his associates showed that only 23% of 623 RA patients, including 236 patients who were taking glucocorticoids at an index visit in 1999, underwent bone densitometry, and only 42% were prescribed a medication (other than calcium and/or vitamin D), that reduces bone loss (Arthritis Rheum. 2002;46:3136-42). They also showed that, in 2004, despite a slight improvement vs. prior years, only 48% of 193 RA patients included in a chart review had received a bone mineral density test or medication for osteoporosis, and 64% of those taking more than 5 mg of prednisone for more than 3 months were receiving osteoporosis management (Arthritis Care Res. 2006;55:873-7).
In the current study, Dr. Ozen and her colleagues looked at both RA and OA patients and found that overall, osteoporosis treatment or screening was reported in 67.4% of study subjects over a mean of 5.5 years follow-up. Of those eligible for osteoporosis treatment based on the 2010 ACR guidelines, including 48.4% of RA patients and 17.6% of OA patients, 55% reported osteoporosis medication use. Despite their increased risk of osteoporosis, particularly in the setting of glucocorticoid use, RA patients were not more likely than OA patients to undergo screening or receive treatment (hazard ratio, 1.04).
“This suboptimal and decreasing trend for osteoporosis treatment and screening in RA patients is important as the prevalence of osteoporosis leading to fractures in RA, regardless of GC use, is still high, despite aggressive management strategies and the use of [biologic disease-modifying antirheumatic agents],” they wrote.
Factors contributing to the decline
The reasons for the decline after 2008 are not fully known, but data suggest that both patient and clinician factors play a role.
Lack of knowledge, unwillingness to take additional drugs, and cost are among patient factors that may interfere with screening and treatment, and lack of experience and time, and a focus more on disease activity and other comorbid conditions may be among clinician factors, they said.
Other factors that may have affected patient and clinician willingness to pursue care include 2007 cuts in Medicare reimbursement for dual-energy x-ray absorptiometry (DXA), a 2006 warning regarding jaw osteonecrosis with bisphosphonate use, and 2007 and 2010 publications about atrial fibrillation and atypical femoral fractures associated with long-term bisphosphonate treatment, they noted.
Dr. Saag agreed that it is difficult to pinpoint exact reasons for the decline, but said the DXA reimbursement cuts have had a significant effect.
“Declining reimbursement is likely one of the major factors, if not the major factor in the decline in testing that has been observed nationally,” he said, explaining that reimbursement was below the break-even point for many physicians in freestanding medical practices, and that slight adjustments have applied only to facilities located in hospitals. As a result, the availability of DXA screening has declined.
The National Osteoporosis Foundation and other organizations such as the International Society for Clinical Densitometry are lobbying for changes, as there is a very strong link between testing and treatment.
“If we can’t get more people tested, we’re unlikely to get more people treated,” Dr. Saag said.
The availability of new agents for the treatment of osteoporosis could also lead to improvements in screening and care, he added.
In July, Amgen submitted to the Food and Drug Administration a supplemental new Biologics License Application for Prolia (denosumab) for the treatment of patients with GIOP. The application is based on a phase 3 study evaluating the safety and efficacy of Prolia, compared with risedronate, in patients receiving glucocorticoid treatment. Denosumab was also shown in a phase 3 study presented at the 2016 ACR meeting to result in greater improvement in bone mineral density vs. bisphosphonates across multiple sites, said Dr. Saag, an investigator for the trial.
“So we’re pleased about that finding, and we’re hopeful that eventually denosumab may become another treatment option,” he said, adding that having more choices leads to more personalized care for patients.
“We hope that, if it does receive approval and is utilized, that it may partially help address the gap in treatment that has been identified,” he said, adding that abaloparatide (Tymlos), which is approved for use in postmenopausal women with osteoporosis and high fracture risk, and romosozumab, currently in late-phase development, could also eventually help bridge the gap.
Recent guideline update
There is also optimism that a recent update to the 2010 guideline, published in August in Arthritis Care & Research, will help address the problem (Arthritis Care Res. 2017;69:1095-1110).
“We hope that one the most important impacts of the new 2017 ACR GIOP guideline will be increasing the awareness of GIOP care,” Dr. Ozen said, noting that changes in the new guideline will also be helpful for guiding management of certain patients not addressed in the 2010 guideline, including adults under age 40 years and children.
The new guideline also incorporates FRAX hip fracture scores into the risk-assessment recommendations .
“Lastly, the addition of other oral bisphosphonates and denosumab to the guideline has broadened the potential therapeutic options for physicians and patients,” she said.
Efforts by the ACR to make these guidelines widely known among clinicians could also help promote improved screening and treatment, said Dr. Buckley, a professor of internal medicine and pediatrics at Yale University, New Haven, Conn.
And that awareness is what is needed most, Dr. Ozen said.
“What we need in osteoporosis care of RA or GIOP patients is more awareness and education about risk-benefit ratios of antiosteoporotic medications instead of focusing on the medication type. We hope that our paper showing the worsening osteoporosis care and the new 2017 ACR GIOP guideline provide more awareness and guidance for the physicians managing these patients,” she said, adding that overcoming obstacles in osteoporosis care requires identification and attention to all potential causes for the suboptimal management of osteoporosis from both patients’ and physicians’ perspectives.
“With the improvement in management strategies of rheumatic diseases ... we anticipate improvement in life expectancy and cardiovascular outcomes in RA and inflammatory rheumatic diseases. In this regard, with aging and decreased but ongoing clinical or subclinical chronic inflammation, it is highly likely that we might be dealing with much older patients with higher osteoporosis and fracture risks. Considering the significant contribution of fractures on cost, disability, morbidity, and mortality of these diseases, rheumatologists and other specialists should work together more vigilantly to overcome the obstacles and improve osteoporosis care,” she said.
Dr. Ozen and Dr. Buckley reported having no conflicts of interest. Dr. Saag is an investigator and consultant for Amgen, Merck, and Radius Health, and is a consultant for Lilly.
FROM ARTHRITIS CARE & RESEARCH
New recommendations tout biosimilars for rheumatologic diseases
The available evidence is sufficient to support switching appropriate patients with rheumatologic diseases from a bio-originator agent to an approved biosimilar agent, according to new consensus-based recommendations from an international multidisciplinary task force.
“Treatment with biological agents has dramatically improved the outcome for patients with inflammatory diseases. However, the high cost of these medications has limited access for many patients,” Jonathan Kay, MD, of UMass Memorial Medical Center and the University of Massachusetts, Worcester, and his colleagues wrote on behalf of the Task Force on the Use of Biosimilars to Treat Rheumatological Diseases. Biosimilars of agents no longer protected by patent allow for increased availability at lower costs, they noted. In the European Union, the United States, Japan, and other countries, biosimilars of adalimumab, etanercept, infliximab, and rituximab have been approved, and those for which the bio-originator is no longer protected by patent have been marketed.
The task force, convened in 2016 to address the matter at an international level, included 25 experts from Europe, Japan, and the United States, including 17 rheumatologists, a rheumatologist/regulator, a dermatologist, a gastroenterologist, 2 pharmacologists, 2 patients, and a research fellow. The task force identified five overarching principles and made eight specific recommendations, based on expert opinion and an extensive literature review that yielded 29 relevant full-text papers and 20 relevant abstracts from the 2015 and 2016 American College of Rheumatology and European League Against Rheumatism annual meetings (Ann Rheum Dis. 2017 Sep 2. doi: 10.1136/annrheumdis-2017-211937).
“This statement was intended both to guide clinicians and to serve as a framework for future educational efforts,” they wrote.
The experts based all five overarching principles for the use of biosimilars on level 5, grade D evidence, indicating that they were derived mainly from expert opinion. They determined that:
- Treatment of rheumatic diseases is based on a shared decision-making process between patients and their rheumatologists.
- The contextual aspects of the health care system should be taken into consideration when treatment decisions are made.
- A biosimilar, as approved by authorities in a highly regulated area, is neither better nor worse in efficacy and is not inferior in safety to its bio-originator.
- Patients and health care providers should be informed about the nature of biosimilars, their approval process, and their safety and efficacy.
- Harmonized methods should be established to obtain reliable pharmacovigilance data, including traceability, about both biosimilars and bio-originators.
These principles represent the key issues regarding biosimilars as identified by the task force. As for the specific recommendations, the task force agreed that:
1. The availability of biosimilars must significantly lower the cost of treating an individual patient and increase access to optimal therapy for all patients with rheumatic diseases (level 5, grade D evidence).
2. Approved biosimilars can be used to treat appropriate patients in the same way as their bio-originators (level 1b, grade A evidence, indicating that the recommendation is based on an individual randomized, controlled trial and that the level 1 evidence is consistent).
3. Antidrug antibodies to biosimilars need not be measured in clinical practice as no significant differences have been detected between biosimilars and their bio-originators (level 2b, grade B evidence, indicating that the recommendation is based on an individual cohort study/low-quality randomized, controlled trial and consistent level 2 or 3 evidence).
4. Relevant preclinical and phase 1 data on a biosimilar should be available when phase 3 data are published (level 5, grade D evidence).
5. Confirmation of efficacy and safety in a single indication is sufficient for extrapolation to other diseases for which the bio-originator has been approved because biosimilars are equivalent in physiochemical, functional, and pharmacokinetic properties to the bio-originator (level 5, grade D evidence).
6. Available evidence suggests that a single switch from a bio-originator to one of its biosimilars is safe and effective; there is no reason to expect a different clinical outcome. However, patient perspectives must be considered (level 1b, grade A evidence).
7. Multiple switching between biosimilars and their bio-originators or other biosimilars should be assessed in registries (level 5, grade D evidence).
8. No switch to or among biosimilars should be initiated without the prior awareness of the patient and the treating health care provider (level 5, grade D evidence).
Differing opinions about the use of biosimilars as published by various subspecialty organizations highlight a lack of confidence among many clinicians with respect to appropriate use of the products, but that is changing amid a rapidly growing body of evidence, the task force said. The group achieved a high level of agreement about both the evaluation of biosimilars and their use to treat rheumatologic diseases, reaching 100% consensus for six of the recommendations and 91% and 96% for the other two.
“Data available as of December 2016 support the use of biosimilars by rheumatologists to encourage a fair and competitive market for biologics. Biosimilars now provide an opportunity to expand access to effective but expensive medications, increasing the number of available treatment choices and helping to control rapidly increasing drug expenditures,” they concluded.
The task force’s work was funded by an unrestricted educational grant from Amgen. Dr. Kay and his coauthors reported financial relationships with multiple pharmaceutical companies, many of which are developing biosimilars.
The available evidence is sufficient to support switching appropriate patients with rheumatologic diseases from a bio-originator agent to an approved biosimilar agent, according to new consensus-based recommendations from an international multidisciplinary task force.
“Treatment with biological agents has dramatically improved the outcome for patients with inflammatory diseases. However, the high cost of these medications has limited access for many patients,” Jonathan Kay, MD, of UMass Memorial Medical Center and the University of Massachusetts, Worcester, and his colleagues wrote on behalf of the Task Force on the Use of Biosimilars to Treat Rheumatological Diseases. Biosimilars of agents no longer protected by patent allow for increased availability at lower costs, they noted. In the European Union, the United States, Japan, and other countries, biosimilars of adalimumab, etanercept, infliximab, and rituximab have been approved, and those for which the bio-originator is no longer protected by patent have been marketed.
The task force, convened in 2016 to address the matter at an international level, included 25 experts from Europe, Japan, and the United States, including 17 rheumatologists, a rheumatologist/regulator, a dermatologist, a gastroenterologist, 2 pharmacologists, 2 patients, and a research fellow. The task force identified five overarching principles and made eight specific recommendations, based on expert opinion and an extensive literature review that yielded 29 relevant full-text papers and 20 relevant abstracts from the 2015 and 2016 American College of Rheumatology and European League Against Rheumatism annual meetings (Ann Rheum Dis. 2017 Sep 2. doi: 10.1136/annrheumdis-2017-211937).
“This statement was intended both to guide clinicians and to serve as a framework for future educational efforts,” they wrote.
The experts based all five overarching principles for the use of biosimilars on level 5, grade D evidence, indicating that they were derived mainly from expert opinion. They determined that:
- Treatment of rheumatic diseases is based on a shared decision-making process between patients and their rheumatologists.
- The contextual aspects of the health care system should be taken into consideration when treatment decisions are made.
- A biosimilar, as approved by authorities in a highly regulated area, is neither better nor worse in efficacy and is not inferior in safety to its bio-originator.
- Patients and health care providers should be informed about the nature of biosimilars, their approval process, and their safety and efficacy.
- Harmonized methods should be established to obtain reliable pharmacovigilance data, including traceability, about both biosimilars and bio-originators.
These principles represent the key issues regarding biosimilars as identified by the task force. As for the specific recommendations, the task force agreed that:
1. The availability of biosimilars must significantly lower the cost of treating an individual patient and increase access to optimal therapy for all patients with rheumatic diseases (level 5, grade D evidence).
2. Approved biosimilars can be used to treat appropriate patients in the same way as their bio-originators (level 1b, grade A evidence, indicating that the recommendation is based on an individual randomized, controlled trial and that the level 1 evidence is consistent).
3. Antidrug antibodies to biosimilars need not be measured in clinical practice as no significant differences have been detected between biosimilars and their bio-originators (level 2b, grade B evidence, indicating that the recommendation is based on an individual cohort study/low-quality randomized, controlled trial and consistent level 2 or 3 evidence).
4. Relevant preclinical and phase 1 data on a biosimilar should be available when phase 3 data are published (level 5, grade D evidence).
5. Confirmation of efficacy and safety in a single indication is sufficient for extrapolation to other diseases for which the bio-originator has been approved because biosimilars are equivalent in physiochemical, functional, and pharmacokinetic properties to the bio-originator (level 5, grade D evidence).
6. Available evidence suggests that a single switch from a bio-originator to one of its biosimilars is safe and effective; there is no reason to expect a different clinical outcome. However, patient perspectives must be considered (level 1b, grade A evidence).
7. Multiple switching between biosimilars and their bio-originators or other biosimilars should be assessed in registries (level 5, grade D evidence).
8. No switch to or among biosimilars should be initiated without the prior awareness of the patient and the treating health care provider (level 5, grade D evidence).
Differing opinions about the use of biosimilars as published by various subspecialty organizations highlight a lack of confidence among many clinicians with respect to appropriate use of the products, but that is changing amid a rapidly growing body of evidence, the task force said. The group achieved a high level of agreement about both the evaluation of biosimilars and their use to treat rheumatologic diseases, reaching 100% consensus for six of the recommendations and 91% and 96% for the other two.
“Data available as of December 2016 support the use of biosimilars by rheumatologists to encourage a fair and competitive market for biologics. Biosimilars now provide an opportunity to expand access to effective but expensive medications, increasing the number of available treatment choices and helping to control rapidly increasing drug expenditures,” they concluded.
The task force’s work was funded by an unrestricted educational grant from Amgen. Dr. Kay and his coauthors reported financial relationships with multiple pharmaceutical companies, many of which are developing biosimilars.
The available evidence is sufficient to support switching appropriate patients with rheumatologic diseases from a bio-originator agent to an approved biosimilar agent, according to new consensus-based recommendations from an international multidisciplinary task force.
“Treatment with biological agents has dramatically improved the outcome for patients with inflammatory diseases. However, the high cost of these medications has limited access for many patients,” Jonathan Kay, MD, of UMass Memorial Medical Center and the University of Massachusetts, Worcester, and his colleagues wrote on behalf of the Task Force on the Use of Biosimilars to Treat Rheumatological Diseases. Biosimilars of agents no longer protected by patent allow for increased availability at lower costs, they noted. In the European Union, the United States, Japan, and other countries, biosimilars of adalimumab, etanercept, infliximab, and rituximab have been approved, and those for which the bio-originator is no longer protected by patent have been marketed.
The task force, convened in 2016 to address the matter at an international level, included 25 experts from Europe, Japan, and the United States, including 17 rheumatologists, a rheumatologist/regulator, a dermatologist, a gastroenterologist, 2 pharmacologists, 2 patients, and a research fellow. The task force identified five overarching principles and made eight specific recommendations, based on expert opinion and an extensive literature review that yielded 29 relevant full-text papers and 20 relevant abstracts from the 2015 and 2016 American College of Rheumatology and European League Against Rheumatism annual meetings (Ann Rheum Dis. 2017 Sep 2. doi: 10.1136/annrheumdis-2017-211937).
“This statement was intended both to guide clinicians and to serve as a framework for future educational efforts,” they wrote.
The experts based all five overarching principles for the use of biosimilars on level 5, grade D evidence, indicating that they were derived mainly from expert opinion. They determined that:
- Treatment of rheumatic diseases is based on a shared decision-making process between patients and their rheumatologists.
- The contextual aspects of the health care system should be taken into consideration when treatment decisions are made.
- A biosimilar, as approved by authorities in a highly regulated area, is neither better nor worse in efficacy and is not inferior in safety to its bio-originator.
- Patients and health care providers should be informed about the nature of biosimilars, their approval process, and their safety and efficacy.
- Harmonized methods should be established to obtain reliable pharmacovigilance data, including traceability, about both biosimilars and bio-originators.
These principles represent the key issues regarding biosimilars as identified by the task force. As for the specific recommendations, the task force agreed that:
1. The availability of biosimilars must significantly lower the cost of treating an individual patient and increase access to optimal therapy for all patients with rheumatic diseases (level 5, grade D evidence).
2. Approved biosimilars can be used to treat appropriate patients in the same way as their bio-originators (level 1b, grade A evidence, indicating that the recommendation is based on an individual randomized, controlled trial and that the level 1 evidence is consistent).
3. Antidrug antibodies to biosimilars need not be measured in clinical practice as no significant differences have been detected between biosimilars and their bio-originators (level 2b, grade B evidence, indicating that the recommendation is based on an individual cohort study/low-quality randomized, controlled trial and consistent level 2 or 3 evidence).
4. Relevant preclinical and phase 1 data on a biosimilar should be available when phase 3 data are published (level 5, grade D evidence).
5. Confirmation of efficacy and safety in a single indication is sufficient for extrapolation to other diseases for which the bio-originator has been approved because biosimilars are equivalent in physiochemical, functional, and pharmacokinetic properties to the bio-originator (level 5, grade D evidence).
6. Available evidence suggests that a single switch from a bio-originator to one of its biosimilars is safe and effective; there is no reason to expect a different clinical outcome. However, patient perspectives must be considered (level 1b, grade A evidence).
7. Multiple switching between biosimilars and their bio-originators or other biosimilars should be assessed in registries (level 5, grade D evidence).
8. No switch to or among biosimilars should be initiated without the prior awareness of the patient and the treating health care provider (level 5, grade D evidence).
Differing opinions about the use of biosimilars as published by various subspecialty organizations highlight a lack of confidence among many clinicians with respect to appropriate use of the products, but that is changing amid a rapidly growing body of evidence, the task force said. The group achieved a high level of agreement about both the evaluation of biosimilars and their use to treat rheumatologic diseases, reaching 100% consensus for six of the recommendations and 91% and 96% for the other two.
“Data available as of December 2016 support the use of biosimilars by rheumatologists to encourage a fair and competitive market for biologics. Biosimilars now provide an opportunity to expand access to effective but expensive medications, increasing the number of available treatment choices and helping to control rapidly increasing drug expenditures,” they concluded.
The task force’s work was funded by an unrestricted educational grant from Amgen. Dr. Kay and his coauthors reported financial relationships with multiple pharmaceutical companies, many of which are developing biosimilars.
FROM ANNALS OF THE RHEUMATIC DISEASES
Vaccinate and consider tofacitinib monotherapy to prevent herpes zoster in RA
The results of two studies of tofacitinib treatment for rheumatoid arthritis offer evidence to support the use of the drug without concomitant conventional synthetic disease-modifying antirheumatic drugs in order to reduce the risk of risk of herpes zoster infection and the safety of starting the drug 2-3 weeks after administering live zoster vaccine.
The risk of herpes zoster infection was elevated among rheumatoid arthritis (RA) patients receiving tofacitinib (Xeljanz) with glucocorticoids, compared with those receiving tofacitinib monotherapy, according to an analysis of data from 19 phase 2, phase 3, and long-term extension studies of tofacitinib.
“Further, physicians should continue to consider shingles vaccination prior to starting tofacitinib or biologic therapy,” they wrote.
Dr. Winthrop is the first author of a separate phase 2 study that also appears in Arthritis & Rheumatology, which suggests that live zoster vaccine (LZV) is safe in RA patients who start tofacitinib 2-3 weeks after vaccination. The study also showed that varicella-zoster virus (VZV)-specific humoral and cell-mediated immune responses to LZV are similar in tofacitinib- and placebo-treated patients.
“Our study provides the first data with this vaccine in the RA setting and suggests that these patients, even while using nonbiologic DMARDs at the time of vaccination, are capable of mounting adequate immune responses to this vaccine. Further, our data suggest that the use of tofacitinib following VZV vaccination in the RA setting did not impact negatively the vaccine immunogenicity or the time course of the immune response to the vaccine,” he and his colleagues wrote (Arthritis Rheumatol. 2017 Aug 28. doi: 10.1002/art.40187).
Increased herpes zoster risk in combination therapy
In the first study, herpes zoster (HZ) was reported in 636 of 6,192 patients over a median follow-up of 3 years of tofacitinib exposure, for an incident rate (IR) of 4.0 per 100 patient-years. However, IRs varied by region, ranging from 2.4 in Eastern Europe to 8.0 and 8.4 in Japan and Korea, respectively.
Further, in phase 3 studies, the IRs varied by tofacitinib dose, background use of conventional csDMARDs, and baseline glucocorticoid use; the rates were lowest among patients on tofacitinib monotherapy at a dose of 5 mg twice daily (IR, 0.6), and highest in those on tofacitinib at 10 mg twice daily with csDMARDs and glucocorticoids (IR, 5.4), the investigators found.
Independent risk factors for HZ included age, glucocorticoid use, tofacitinib dose, and enrollment within Asia, they said.
“Shingles, or reactivation of varicella virus, is a common and potentially debilitating illness. Around one-third of the general population will develop HZ in their lifetime, and approximately 10% of these patients develop postherpetic neuralgia which can last months to years and cause significant pain and morbidity,” the investigators wrote, adding that RA patients are at 1.5- to 2-fold greater risk vs. similarly aged individuals in the general population.
RA itself and treatment with glucocorticoids are known to increase HZ risk, but recent data have suggested that Janus kinase inhibitors, such as tofacitinib, and tumor necrosis factor antagonists are also associated with a higher rate of HZ. Additionally, a theoretical risk exists with various csDMARDs, they said.
“Given the increased risk of HZ observed among patients with RA versus the general population and the risk associated with RA therapies, it is possible that risk of HZ may be further increased when such therapies are combined,” they wrote.
Indeed, the findings of the study demonstrate an increased risk of HZ with tofacitinib in combination with glucocorticoids vs. tofacitinib monotherapy.
Further research is necessary to understand why Japanese and Korean patients are at elevated risk, and to understand the mechanism for the effects of combination therapy on VZV reactivation, they concluded.
LZV immunogenicity holds up during tofacitinib treatment
In the second study, 112 patients aged 50 years and older with active RA on background methotrexate received LZV and were then randomized to receive 5 mg tofacitinib twice daily or placebo 2-3 weeks after vaccination.
At 6 weeks after vaccination, VZV-specific IgG geometric mean fold rise (GMFR) was 2.11 and 1.74 in the tofacitinib and placebo patients, respectively; at all postvaccination time points at which VZV-specific IgG levels were evaluated, there was a trend toward numerically higher GMFR in tofacitinib patients, but the differences were not statistically significant. Also, the proportion of patients developing a 1.5-fold or greater postvaccination rise in IgG levels at 6 weeks trended higher for tofacitinib (57.4% vs. 43.4% with placebo).
VZV-specific T-cell GMFR at 6 weeks increased similarly in the groups (1.50 with tofacitinib and 1.29 with placebo).
Serious adverse events occurred in three patients in the tofacitinib group (5.5%) and in none of the placebo patients.
“The three SAEs included one case each of cholangitis and bronchitis, and once case of disseminated primary varicella,” the investigators said, noting that the onset of the latter was 16 days postvaccination, 2 days after starting tofacitinib. The rash resolved with discontinuation of tofacitinib and treatment with valacyclovir for 7 days.
The findings suggest that patients with active RA develop robust immune responses to HZ vaccine, and that starting tofacitinib 2-3 weeks after vaccination has no negative impact on the established immune response.
“Importantly, while our results suggest the vaccine is safe for patients with RA with prior VZV exposure, they also indicate the potential need to either screen for prior exposure before giving this vaccine or waiting longer than 2-3 weeks before starting immunosuppression with tofacitinib,” they said, noting that the current data suggest 4 weeks might be preferable.
Alternatively, testing patients who don’t recollect a history of chickenpox to ensure prior VZV exposure prior to vaccination could also mitigate the risk, they said.
“Further research is necessary to understand the risk of this complication, as well as the long-term effectiveness of this vaccine to prevent HZ in this high-risk population,” they concluded.
Both studies were sponsored by Pfizer. Dr. Winthrop has received research grants from and served as a scientific consultant to Pfizer. Other authors also reported financial relationships with Pfizer.
At a symposium during the 2015 annual meeting of the American College of Rheumatology, William Schaffner, MD, highlighted the connection between the seriousness of an infection, and the respect one has for the solution.
Dr. Schaffner said that “if you don’t fear the infection, you won’t value the solution.” Herpes zoster (HZ) should be feared, and the solution – the live zoster vaccine – valued.
Live zoster vaccine (LZV) was approved in 2006 on the basis of a trial involving more than 38,500 adults over age 60 years, which showed a 51% HZ prevention rate (64% protection in the 60-69 year age group) and a two-thirds reduction in postherpetic neuralgia. Complications and disseminated infection were rare.
HZ vaccination should be offered regardless of a history of varicella infection or prior shingles, as HZ may recur.
There is an imperative need to know who is at risk, when and how they should be vaccinated, and what other risk reduction measures should be considered.
John J. Cush, MD, is director of clinical rheumatology at Baylor Scott & White Research Institute and professor of medicine and rheumatology at Baylor University Medical Center, both in Dallas. His comments are taken from his editorial accompanying the two studies by Winthrop et al. (Arthritis Rheumatol. 2017 Aug 28. doi: 10.1002/art.40188).
At a symposium during the 2015 annual meeting of the American College of Rheumatology, William Schaffner, MD, highlighted the connection between the seriousness of an infection, and the respect one has for the solution.
Dr. Schaffner said that “if you don’t fear the infection, you won’t value the solution.” Herpes zoster (HZ) should be feared, and the solution – the live zoster vaccine – valued.
Live zoster vaccine (LZV) was approved in 2006 on the basis of a trial involving more than 38,500 adults over age 60 years, which showed a 51% HZ prevention rate (64% protection in the 60-69 year age group) and a two-thirds reduction in postherpetic neuralgia. Complications and disseminated infection were rare.
HZ vaccination should be offered regardless of a history of varicella infection or prior shingles, as HZ may recur.
There is an imperative need to know who is at risk, when and how they should be vaccinated, and what other risk reduction measures should be considered.
John J. Cush, MD, is director of clinical rheumatology at Baylor Scott & White Research Institute and professor of medicine and rheumatology at Baylor University Medical Center, both in Dallas. His comments are taken from his editorial accompanying the two studies by Winthrop et al. (Arthritis Rheumatol. 2017 Aug 28. doi: 10.1002/art.40188).
At a symposium during the 2015 annual meeting of the American College of Rheumatology, William Schaffner, MD, highlighted the connection between the seriousness of an infection, and the respect one has for the solution.
Dr. Schaffner said that “if you don’t fear the infection, you won’t value the solution.” Herpes zoster (HZ) should be feared, and the solution – the live zoster vaccine – valued.
Live zoster vaccine (LZV) was approved in 2006 on the basis of a trial involving more than 38,500 adults over age 60 years, which showed a 51% HZ prevention rate (64% protection in the 60-69 year age group) and a two-thirds reduction in postherpetic neuralgia. Complications and disseminated infection were rare.
HZ vaccination should be offered regardless of a history of varicella infection or prior shingles, as HZ may recur.
There is an imperative need to know who is at risk, when and how they should be vaccinated, and what other risk reduction measures should be considered.
John J. Cush, MD, is director of clinical rheumatology at Baylor Scott & White Research Institute and professor of medicine and rheumatology at Baylor University Medical Center, both in Dallas. His comments are taken from his editorial accompanying the two studies by Winthrop et al. (Arthritis Rheumatol. 2017 Aug 28. doi: 10.1002/art.40188).
The results of two studies of tofacitinib treatment for rheumatoid arthritis offer evidence to support the use of the drug without concomitant conventional synthetic disease-modifying antirheumatic drugs in order to reduce the risk of risk of herpes zoster infection and the safety of starting the drug 2-3 weeks after administering live zoster vaccine.
The risk of herpes zoster infection was elevated among rheumatoid arthritis (RA) patients receiving tofacitinib (Xeljanz) with glucocorticoids, compared with those receiving tofacitinib monotherapy, according to an analysis of data from 19 phase 2, phase 3, and long-term extension studies of tofacitinib.
“Further, physicians should continue to consider shingles vaccination prior to starting tofacitinib or biologic therapy,” they wrote.
Dr. Winthrop is the first author of a separate phase 2 study that also appears in Arthritis & Rheumatology, which suggests that live zoster vaccine (LZV) is safe in RA patients who start tofacitinib 2-3 weeks after vaccination. The study also showed that varicella-zoster virus (VZV)-specific humoral and cell-mediated immune responses to LZV are similar in tofacitinib- and placebo-treated patients.
“Our study provides the first data with this vaccine in the RA setting and suggests that these patients, even while using nonbiologic DMARDs at the time of vaccination, are capable of mounting adequate immune responses to this vaccine. Further, our data suggest that the use of tofacitinib following VZV vaccination in the RA setting did not impact negatively the vaccine immunogenicity or the time course of the immune response to the vaccine,” he and his colleagues wrote (Arthritis Rheumatol. 2017 Aug 28. doi: 10.1002/art.40187).
Increased herpes zoster risk in combination therapy
In the first study, herpes zoster (HZ) was reported in 636 of 6,192 patients over a median follow-up of 3 years of tofacitinib exposure, for an incident rate (IR) of 4.0 per 100 patient-years. However, IRs varied by region, ranging from 2.4 in Eastern Europe to 8.0 and 8.4 in Japan and Korea, respectively.
Further, in phase 3 studies, the IRs varied by tofacitinib dose, background use of conventional csDMARDs, and baseline glucocorticoid use; the rates were lowest among patients on tofacitinib monotherapy at a dose of 5 mg twice daily (IR, 0.6), and highest in those on tofacitinib at 10 mg twice daily with csDMARDs and glucocorticoids (IR, 5.4), the investigators found.
Independent risk factors for HZ included age, glucocorticoid use, tofacitinib dose, and enrollment within Asia, they said.
“Shingles, or reactivation of varicella virus, is a common and potentially debilitating illness. Around one-third of the general population will develop HZ in their lifetime, and approximately 10% of these patients develop postherpetic neuralgia which can last months to years and cause significant pain and morbidity,” the investigators wrote, adding that RA patients are at 1.5- to 2-fold greater risk vs. similarly aged individuals in the general population.
RA itself and treatment with glucocorticoids are known to increase HZ risk, but recent data have suggested that Janus kinase inhibitors, such as tofacitinib, and tumor necrosis factor antagonists are also associated with a higher rate of HZ. Additionally, a theoretical risk exists with various csDMARDs, they said.
“Given the increased risk of HZ observed among patients with RA versus the general population and the risk associated with RA therapies, it is possible that risk of HZ may be further increased when such therapies are combined,” they wrote.
Indeed, the findings of the study demonstrate an increased risk of HZ with tofacitinib in combination with glucocorticoids vs. tofacitinib monotherapy.
Further research is necessary to understand why Japanese and Korean patients are at elevated risk, and to understand the mechanism for the effects of combination therapy on VZV reactivation, they concluded.
LZV immunogenicity holds up during tofacitinib treatment
In the second study, 112 patients aged 50 years and older with active RA on background methotrexate received LZV and were then randomized to receive 5 mg tofacitinib twice daily or placebo 2-3 weeks after vaccination.
At 6 weeks after vaccination, VZV-specific IgG geometric mean fold rise (GMFR) was 2.11 and 1.74 in the tofacitinib and placebo patients, respectively; at all postvaccination time points at which VZV-specific IgG levels were evaluated, there was a trend toward numerically higher GMFR in tofacitinib patients, but the differences were not statistically significant. Also, the proportion of patients developing a 1.5-fold or greater postvaccination rise in IgG levels at 6 weeks trended higher for tofacitinib (57.4% vs. 43.4% with placebo).
VZV-specific T-cell GMFR at 6 weeks increased similarly in the groups (1.50 with tofacitinib and 1.29 with placebo).
Serious adverse events occurred in three patients in the tofacitinib group (5.5%) and in none of the placebo patients.
“The three SAEs included one case each of cholangitis and bronchitis, and once case of disseminated primary varicella,” the investigators said, noting that the onset of the latter was 16 days postvaccination, 2 days after starting tofacitinib. The rash resolved with discontinuation of tofacitinib and treatment with valacyclovir for 7 days.
The findings suggest that patients with active RA develop robust immune responses to HZ vaccine, and that starting tofacitinib 2-3 weeks after vaccination has no negative impact on the established immune response.
“Importantly, while our results suggest the vaccine is safe for patients with RA with prior VZV exposure, they also indicate the potential need to either screen for prior exposure before giving this vaccine or waiting longer than 2-3 weeks before starting immunosuppression with tofacitinib,” they said, noting that the current data suggest 4 weeks might be preferable.
Alternatively, testing patients who don’t recollect a history of chickenpox to ensure prior VZV exposure prior to vaccination could also mitigate the risk, they said.
“Further research is necessary to understand the risk of this complication, as well as the long-term effectiveness of this vaccine to prevent HZ in this high-risk population,” they concluded.
Both studies were sponsored by Pfizer. Dr. Winthrop has received research grants from and served as a scientific consultant to Pfizer. Other authors also reported financial relationships with Pfizer.
The results of two studies of tofacitinib treatment for rheumatoid arthritis offer evidence to support the use of the drug without concomitant conventional synthetic disease-modifying antirheumatic drugs in order to reduce the risk of risk of herpes zoster infection and the safety of starting the drug 2-3 weeks after administering live zoster vaccine.
The risk of herpes zoster infection was elevated among rheumatoid arthritis (RA) patients receiving tofacitinib (Xeljanz) with glucocorticoids, compared with those receiving tofacitinib monotherapy, according to an analysis of data from 19 phase 2, phase 3, and long-term extension studies of tofacitinib.
“Further, physicians should continue to consider shingles vaccination prior to starting tofacitinib or biologic therapy,” they wrote.
Dr. Winthrop is the first author of a separate phase 2 study that also appears in Arthritis & Rheumatology, which suggests that live zoster vaccine (LZV) is safe in RA patients who start tofacitinib 2-3 weeks after vaccination. The study also showed that varicella-zoster virus (VZV)-specific humoral and cell-mediated immune responses to LZV are similar in tofacitinib- and placebo-treated patients.
“Our study provides the first data with this vaccine in the RA setting and suggests that these patients, even while using nonbiologic DMARDs at the time of vaccination, are capable of mounting adequate immune responses to this vaccine. Further, our data suggest that the use of tofacitinib following VZV vaccination in the RA setting did not impact negatively the vaccine immunogenicity or the time course of the immune response to the vaccine,” he and his colleagues wrote (Arthritis Rheumatol. 2017 Aug 28. doi: 10.1002/art.40187).
Increased herpes zoster risk in combination therapy
In the first study, herpes zoster (HZ) was reported in 636 of 6,192 patients over a median follow-up of 3 years of tofacitinib exposure, for an incident rate (IR) of 4.0 per 100 patient-years. However, IRs varied by region, ranging from 2.4 in Eastern Europe to 8.0 and 8.4 in Japan and Korea, respectively.
Further, in phase 3 studies, the IRs varied by tofacitinib dose, background use of conventional csDMARDs, and baseline glucocorticoid use; the rates were lowest among patients on tofacitinib monotherapy at a dose of 5 mg twice daily (IR, 0.6), and highest in those on tofacitinib at 10 mg twice daily with csDMARDs and glucocorticoids (IR, 5.4), the investigators found.
Independent risk factors for HZ included age, glucocorticoid use, tofacitinib dose, and enrollment within Asia, they said.
“Shingles, or reactivation of varicella virus, is a common and potentially debilitating illness. Around one-third of the general population will develop HZ in their lifetime, and approximately 10% of these patients develop postherpetic neuralgia which can last months to years and cause significant pain and morbidity,” the investigators wrote, adding that RA patients are at 1.5- to 2-fold greater risk vs. similarly aged individuals in the general population.
RA itself and treatment with glucocorticoids are known to increase HZ risk, but recent data have suggested that Janus kinase inhibitors, such as tofacitinib, and tumor necrosis factor antagonists are also associated with a higher rate of HZ. Additionally, a theoretical risk exists with various csDMARDs, they said.
“Given the increased risk of HZ observed among patients with RA versus the general population and the risk associated with RA therapies, it is possible that risk of HZ may be further increased when such therapies are combined,” they wrote.
Indeed, the findings of the study demonstrate an increased risk of HZ with tofacitinib in combination with glucocorticoids vs. tofacitinib monotherapy.
Further research is necessary to understand why Japanese and Korean patients are at elevated risk, and to understand the mechanism for the effects of combination therapy on VZV reactivation, they concluded.
LZV immunogenicity holds up during tofacitinib treatment
In the second study, 112 patients aged 50 years and older with active RA on background methotrexate received LZV and were then randomized to receive 5 mg tofacitinib twice daily or placebo 2-3 weeks after vaccination.
At 6 weeks after vaccination, VZV-specific IgG geometric mean fold rise (GMFR) was 2.11 and 1.74 in the tofacitinib and placebo patients, respectively; at all postvaccination time points at which VZV-specific IgG levels were evaluated, there was a trend toward numerically higher GMFR in tofacitinib patients, but the differences were not statistically significant. Also, the proportion of patients developing a 1.5-fold or greater postvaccination rise in IgG levels at 6 weeks trended higher for tofacitinib (57.4% vs. 43.4% with placebo).
VZV-specific T-cell GMFR at 6 weeks increased similarly in the groups (1.50 with tofacitinib and 1.29 with placebo).
Serious adverse events occurred in three patients in the tofacitinib group (5.5%) and in none of the placebo patients.
“The three SAEs included one case each of cholangitis and bronchitis, and once case of disseminated primary varicella,” the investigators said, noting that the onset of the latter was 16 days postvaccination, 2 days after starting tofacitinib. The rash resolved with discontinuation of tofacitinib and treatment with valacyclovir for 7 days.
The findings suggest that patients with active RA develop robust immune responses to HZ vaccine, and that starting tofacitinib 2-3 weeks after vaccination has no negative impact on the established immune response.
“Importantly, while our results suggest the vaccine is safe for patients with RA with prior VZV exposure, they also indicate the potential need to either screen for prior exposure before giving this vaccine or waiting longer than 2-3 weeks before starting immunosuppression with tofacitinib,” they said, noting that the current data suggest 4 weeks might be preferable.
Alternatively, testing patients who don’t recollect a history of chickenpox to ensure prior VZV exposure prior to vaccination could also mitigate the risk, they said.
“Further research is necessary to understand the risk of this complication, as well as the long-term effectiveness of this vaccine to prevent HZ in this high-risk population,” they concluded.
Both studies were sponsored by Pfizer. Dr. Winthrop has received research grants from and served as a scientific consultant to Pfizer. Other authors also reported financial relationships with Pfizer.
FROM ARTHRITIS & RHEUMATOLOGY
Key clinical point:
Major finding: HZ rates were lowest among patients on tofacitinib monotherapy at a dose of 5 mg twice daily (IR, 0.6).
Data source: A phase 2 trial of 112 patients, and a review of 19 studies involving 6,192 patients.
Disclosures: Both studies were sponsored by Pfizer. Dr. Winthrop has received research grants from and served as a scientific consultant to Pfizer. Other authors also reported financial relationships with Pfizer.
VIDEO: Prescription-strength ibuprofen worsens blood pressure more than other NSAIDs
BARCELONA – Prescription-strength ibuprofen has a bigger adverse effect on blood pressure than celecoxib or naproxen, a finding that suggests a likely mechanism for the worse cardiovascular event rate documented in ibuprofen-treated arthritis patients in the PRECISION trial, Frank Ruschitzka, MD, said at the annual congress of the European Society of Cardiology.
“Prescription-strength ibuprofen is under pressure – it has a high incidence of new-onset hypertension, particularly when compared to the more selective COX-2 inhibitor celecoxib. Before we did this study, many would have said it’s the other way around,” observed Dr. Ruschitzka, professor of cardiology at the University of Zurich.
He presented the results of PRECISION-ABPM (Prospective Randomized Evaluation of Celecoxib Integrated Safety Versus Ibuprofen or Naproxen Ambulatory Blood Pressure Measurement).
“These results will have impact on your daily practice when you go home,” the cardiologist promised.
PRECISION-ABPM was a prespecified double-blind, randomized, 60-center substudy of the published PRECISION trial, which included 24,081 U.S. patients who needed daily NSAIDs for arthritis and were also at increased cardiovascular risk. They were randomized to the COX-2 inhibitor celecoxib at 100-200 mg b.i.d. or the nonselective NSAIDs ibuprofen at 600-800 mg three times a day or naproxen at 375-500 mg twice daily. Participants also received a proton pump inhibitor to protect against NSAID-related GI bleeding. In the on-treatment analysis, the ibuprofen group was significantly more likely to experience cardiovascular and all-cause mortality and renal events than were those on celecoxib (N Engl J Med. 2016 Dec 29;375[26]:2519-29).
The PRECISION-ABPM substudy included 444 arthritis patients, 92% of whom had osteoarthritis. During the 4-month study, investigators amassed roughly 60,000 automated blood pressure measurements across the three study arms.
The primary outcome was change from baseline in mean 24-hour systolic blood pressure (SBP). It increased by 3.7 mm Hg in the ibuprofen group and declined by 0.3 mm Hg in the celecoxib group, while the naproxen group occupied the middle ground with a 1.6-mm Hg increase.
The nearly 4-mm Hg increase in mean 24-hour SBP at 4 months in the ibuprofen group is of sufficient magnitude to be clinically important, Dr. Ruschitzka noted. He noted that fully 23.2% of ibuprofen-treated patients who had normal baseline blood pressure developed hypertension as defined by a mean 24-hour SBP of at least 130 and/or a diastolic blood pressure of at least 80 mm Hg. In contrast, incident hypertension occurred in only 10.3% of the celecoxib group and 19% of naproxen-treated patients. Thus, the likelihood of developing hypertension was 61% less with celecoxib than ibuprofen and 51% less with celecoxib than naproxen.
Not treating chronic arthritic pain to avoid the cardiovascular risk of NSAIDs is not a legitimate option.
“Pain is a cardiovascular risk factor,” Dr. Ruschitzka emphasized. “It’s unethical not to treat it. If you don’t treat pain, the patient’s blood pressure goes up, heart rate goes up, and you’re driving patients into inactivity.”
Although he’s convinced there’s no such thing as a safe NSAID from a cardiovascular risk standpoint, the PRECISION and PRECISION-ABPM data show celecoxib is less unsafe than ibuprofen. And as for the oft-heard statement that naproxen is the safest NSAID for the heart, Dr. Ruschitzka snorted, “What an urban legend.”
Discussant Scott Solomon, MD, opined that, while PRECISION-ABPM doesn’t support the notion that conventional NSAIDs such as naproxen or ibuprofen are any safer than celecoxib, it would be wrong to conclude from the study that celecoxib doesn’t affect blood pressure and is safer than the others from a cardiovascular standpoint. That’s because the three study drugs weren’t compared in an equipotent way. Because of safety concerns, the Food and Drug Administration required that the daily dose of celecoxib be capped at the low end of the therapeutic range, while no such constraints were placed on the two nonselective NSAIDS.
“Compared to placebo, all NSAIDs likely raise blood pressure, especially in patients prone to hypertension, those with chronic kidney disease, the elderly – and this is exactly the type of patients who require NSAIDs for arthritis. Whichever NSAID is chosen, clinicians should be aware of this effect and treat hypertension according to guidelines,” said Dr. Solomon, director of noninvasive cardiology at Brigham and Women’s Hospital, Boston, and professor of medicine at Harvard Medical School.
Dr. Solomon has been a key figure in the COX-2 inhibitor controversy of the last decade. He was lead author of a 2005 review of data from clinical trials of COX-2 inhibitors for colorectal adenoma prevention, which concluded that the drugs had a cardiovascular safety issue in that setting (N Engl J Med. 2005 Mar 17;352[11]:1071-80).
“Our analysis of celecoxib concluded that a dose-dependent increase in cardiovascular events was there, was real, but notably occurred at doses which were substantially higher than what we typically use for patients with arthritis,” he said.
That report triggered a fevered reaction.
“Amid an enormous amount of hype, hyperbole, and hysteria, the safety of these agents was thrown into question, leading to the withdrawal of all but one of them from the market and a black-box warning around the one remaining agent, celecoxib,” he recalled.
Dr. Ruschitzka discussed his findings in a video interview.
PRECISION-ABPM was sponsored by Pfizer. Dr. Ruschitzka and Dr. Solomon reported having no financial conflicts of interest regarding their presentations.
BARCELONA – Prescription-strength ibuprofen has a bigger adverse effect on blood pressure than celecoxib or naproxen, a finding that suggests a likely mechanism for the worse cardiovascular event rate documented in ibuprofen-treated arthritis patients in the PRECISION trial, Frank Ruschitzka, MD, said at the annual congress of the European Society of Cardiology.
“Prescription-strength ibuprofen is under pressure – it has a high incidence of new-onset hypertension, particularly when compared to the more selective COX-2 inhibitor celecoxib. Before we did this study, many would have said it’s the other way around,” observed Dr. Ruschitzka, professor of cardiology at the University of Zurich.
He presented the results of PRECISION-ABPM (Prospective Randomized Evaluation of Celecoxib Integrated Safety Versus Ibuprofen or Naproxen Ambulatory Blood Pressure Measurement).
“These results will have impact on your daily practice when you go home,” the cardiologist promised.
PRECISION-ABPM was a prespecified double-blind, randomized, 60-center substudy of the published PRECISION trial, which included 24,081 U.S. patients who needed daily NSAIDs for arthritis and were also at increased cardiovascular risk. They were randomized to the COX-2 inhibitor celecoxib at 100-200 mg b.i.d. or the nonselective NSAIDs ibuprofen at 600-800 mg three times a day or naproxen at 375-500 mg twice daily. Participants also received a proton pump inhibitor to protect against NSAID-related GI bleeding. In the on-treatment analysis, the ibuprofen group was significantly more likely to experience cardiovascular and all-cause mortality and renal events than were those on celecoxib (N Engl J Med. 2016 Dec 29;375[26]:2519-29).
The PRECISION-ABPM substudy included 444 arthritis patients, 92% of whom had osteoarthritis. During the 4-month study, investigators amassed roughly 60,000 automated blood pressure measurements across the three study arms.
The primary outcome was change from baseline in mean 24-hour systolic blood pressure (SBP). It increased by 3.7 mm Hg in the ibuprofen group and declined by 0.3 mm Hg in the celecoxib group, while the naproxen group occupied the middle ground with a 1.6-mm Hg increase.
The nearly 4-mm Hg increase in mean 24-hour SBP at 4 months in the ibuprofen group is of sufficient magnitude to be clinically important, Dr. Ruschitzka noted. He noted that fully 23.2% of ibuprofen-treated patients who had normal baseline blood pressure developed hypertension as defined by a mean 24-hour SBP of at least 130 and/or a diastolic blood pressure of at least 80 mm Hg. In contrast, incident hypertension occurred in only 10.3% of the celecoxib group and 19% of naproxen-treated patients. Thus, the likelihood of developing hypertension was 61% less with celecoxib than ibuprofen and 51% less with celecoxib than naproxen.
Not treating chronic arthritic pain to avoid the cardiovascular risk of NSAIDs is not a legitimate option.
“Pain is a cardiovascular risk factor,” Dr. Ruschitzka emphasized. “It’s unethical not to treat it. If you don’t treat pain, the patient’s blood pressure goes up, heart rate goes up, and you’re driving patients into inactivity.”
Although he’s convinced there’s no such thing as a safe NSAID from a cardiovascular risk standpoint, the PRECISION and PRECISION-ABPM data show celecoxib is less unsafe than ibuprofen. And as for the oft-heard statement that naproxen is the safest NSAID for the heart, Dr. Ruschitzka snorted, “What an urban legend.”
Discussant Scott Solomon, MD, opined that, while PRECISION-ABPM doesn’t support the notion that conventional NSAIDs such as naproxen or ibuprofen are any safer than celecoxib, it would be wrong to conclude from the study that celecoxib doesn’t affect blood pressure and is safer than the others from a cardiovascular standpoint. That’s because the three study drugs weren’t compared in an equipotent way. Because of safety concerns, the Food and Drug Administration required that the daily dose of celecoxib be capped at the low end of the therapeutic range, while no such constraints were placed on the two nonselective NSAIDS.
“Compared to placebo, all NSAIDs likely raise blood pressure, especially in patients prone to hypertension, those with chronic kidney disease, the elderly – and this is exactly the type of patients who require NSAIDs for arthritis. Whichever NSAID is chosen, clinicians should be aware of this effect and treat hypertension according to guidelines,” said Dr. Solomon, director of noninvasive cardiology at Brigham and Women’s Hospital, Boston, and professor of medicine at Harvard Medical School.
Dr. Solomon has been a key figure in the COX-2 inhibitor controversy of the last decade. He was lead author of a 2005 review of data from clinical trials of COX-2 inhibitors for colorectal adenoma prevention, which concluded that the drugs had a cardiovascular safety issue in that setting (N Engl J Med. 2005 Mar 17;352[11]:1071-80).
“Our analysis of celecoxib concluded that a dose-dependent increase in cardiovascular events was there, was real, but notably occurred at doses which were substantially higher than what we typically use for patients with arthritis,” he said.
That report triggered a fevered reaction.
“Amid an enormous amount of hype, hyperbole, and hysteria, the safety of these agents was thrown into question, leading to the withdrawal of all but one of them from the market and a black-box warning around the one remaining agent, celecoxib,” he recalled.
Dr. Ruschitzka discussed his findings in a video interview.
PRECISION-ABPM was sponsored by Pfizer. Dr. Ruschitzka and Dr. Solomon reported having no financial conflicts of interest regarding their presentations.
BARCELONA – Prescription-strength ibuprofen has a bigger adverse effect on blood pressure than celecoxib or naproxen, a finding that suggests a likely mechanism for the worse cardiovascular event rate documented in ibuprofen-treated arthritis patients in the PRECISION trial, Frank Ruschitzka, MD, said at the annual congress of the European Society of Cardiology.
“Prescription-strength ibuprofen is under pressure – it has a high incidence of new-onset hypertension, particularly when compared to the more selective COX-2 inhibitor celecoxib. Before we did this study, many would have said it’s the other way around,” observed Dr. Ruschitzka, professor of cardiology at the University of Zurich.
He presented the results of PRECISION-ABPM (Prospective Randomized Evaluation of Celecoxib Integrated Safety Versus Ibuprofen or Naproxen Ambulatory Blood Pressure Measurement).
“These results will have impact on your daily practice when you go home,” the cardiologist promised.
PRECISION-ABPM was a prespecified double-blind, randomized, 60-center substudy of the published PRECISION trial, which included 24,081 U.S. patients who needed daily NSAIDs for arthritis and were also at increased cardiovascular risk. They were randomized to the COX-2 inhibitor celecoxib at 100-200 mg b.i.d. or the nonselective NSAIDs ibuprofen at 600-800 mg three times a day or naproxen at 375-500 mg twice daily. Participants also received a proton pump inhibitor to protect against NSAID-related GI bleeding. In the on-treatment analysis, the ibuprofen group was significantly more likely to experience cardiovascular and all-cause mortality and renal events than were those on celecoxib (N Engl J Med. 2016 Dec 29;375[26]:2519-29).
The PRECISION-ABPM substudy included 444 arthritis patients, 92% of whom had osteoarthritis. During the 4-month study, investigators amassed roughly 60,000 automated blood pressure measurements across the three study arms.
The primary outcome was change from baseline in mean 24-hour systolic blood pressure (SBP). It increased by 3.7 mm Hg in the ibuprofen group and declined by 0.3 mm Hg in the celecoxib group, while the naproxen group occupied the middle ground with a 1.6-mm Hg increase.
The nearly 4-mm Hg increase in mean 24-hour SBP at 4 months in the ibuprofen group is of sufficient magnitude to be clinically important, Dr. Ruschitzka noted. He noted that fully 23.2% of ibuprofen-treated patients who had normal baseline blood pressure developed hypertension as defined by a mean 24-hour SBP of at least 130 and/or a diastolic blood pressure of at least 80 mm Hg. In contrast, incident hypertension occurred in only 10.3% of the celecoxib group and 19% of naproxen-treated patients. Thus, the likelihood of developing hypertension was 61% less with celecoxib than ibuprofen and 51% less with celecoxib than naproxen.
Not treating chronic arthritic pain to avoid the cardiovascular risk of NSAIDs is not a legitimate option.
“Pain is a cardiovascular risk factor,” Dr. Ruschitzka emphasized. “It’s unethical not to treat it. If you don’t treat pain, the patient’s blood pressure goes up, heart rate goes up, and you’re driving patients into inactivity.”
Although he’s convinced there’s no such thing as a safe NSAID from a cardiovascular risk standpoint, the PRECISION and PRECISION-ABPM data show celecoxib is less unsafe than ibuprofen. And as for the oft-heard statement that naproxen is the safest NSAID for the heart, Dr. Ruschitzka snorted, “What an urban legend.”
Discussant Scott Solomon, MD, opined that, while PRECISION-ABPM doesn’t support the notion that conventional NSAIDs such as naproxen or ibuprofen are any safer than celecoxib, it would be wrong to conclude from the study that celecoxib doesn’t affect blood pressure and is safer than the others from a cardiovascular standpoint. That’s because the three study drugs weren’t compared in an equipotent way. Because of safety concerns, the Food and Drug Administration required that the daily dose of celecoxib be capped at the low end of the therapeutic range, while no such constraints were placed on the two nonselective NSAIDS.
“Compared to placebo, all NSAIDs likely raise blood pressure, especially in patients prone to hypertension, those with chronic kidney disease, the elderly – and this is exactly the type of patients who require NSAIDs for arthritis. Whichever NSAID is chosen, clinicians should be aware of this effect and treat hypertension according to guidelines,” said Dr. Solomon, director of noninvasive cardiology at Brigham and Women’s Hospital, Boston, and professor of medicine at Harvard Medical School.
Dr. Solomon has been a key figure in the COX-2 inhibitor controversy of the last decade. He was lead author of a 2005 review of data from clinical trials of COX-2 inhibitors for colorectal adenoma prevention, which concluded that the drugs had a cardiovascular safety issue in that setting (N Engl J Med. 2005 Mar 17;352[11]:1071-80).
“Our analysis of celecoxib concluded that a dose-dependent increase in cardiovascular events was there, was real, but notably occurred at doses which were substantially higher than what we typically use for patients with arthritis,” he said.
That report triggered a fevered reaction.
“Amid an enormous amount of hype, hyperbole, and hysteria, the safety of these agents was thrown into question, leading to the withdrawal of all but one of them from the market and a black-box warning around the one remaining agent, celecoxib,” he recalled.
Dr. Ruschitzka discussed his findings in a video interview.
PRECISION-ABPM was sponsored by Pfizer. Dr. Ruschitzka and Dr. Solomon reported having no financial conflicts of interest regarding their presentations.
AT THE ESC CONGRESS 2017
Key clinical point:
Major finding: Incident hypertension occurred within 4 months in 23.2% of arthritis patients on ibuprofen, compared with 10.3% taking celecoxib and 19% on naproxen.
Data source: This was a randomized, double-blind, multicenter, prospective trial including 444 arthritis patients at increased cardiovascular risk who underwent 4 months of ambulatory blood pressure monitoring after being assigned to prescription-strength ibuprofen, naproxen, or celecoxib.
Disclosures: The PRECISION-ABPM trial was sponsored by Pfizer. The presenter reported having no financial conflicts of interest.
