User login
Herpes zoster risk increased with some psoriasis, psoriatic arthritis treatments
All individuals with psoriasis or psoriatic arthritis aged over 50 years should receive the recombinant herpes zoster vaccine, according to a systematic review and consensus recommendations from the National Psoriasis Foundation.
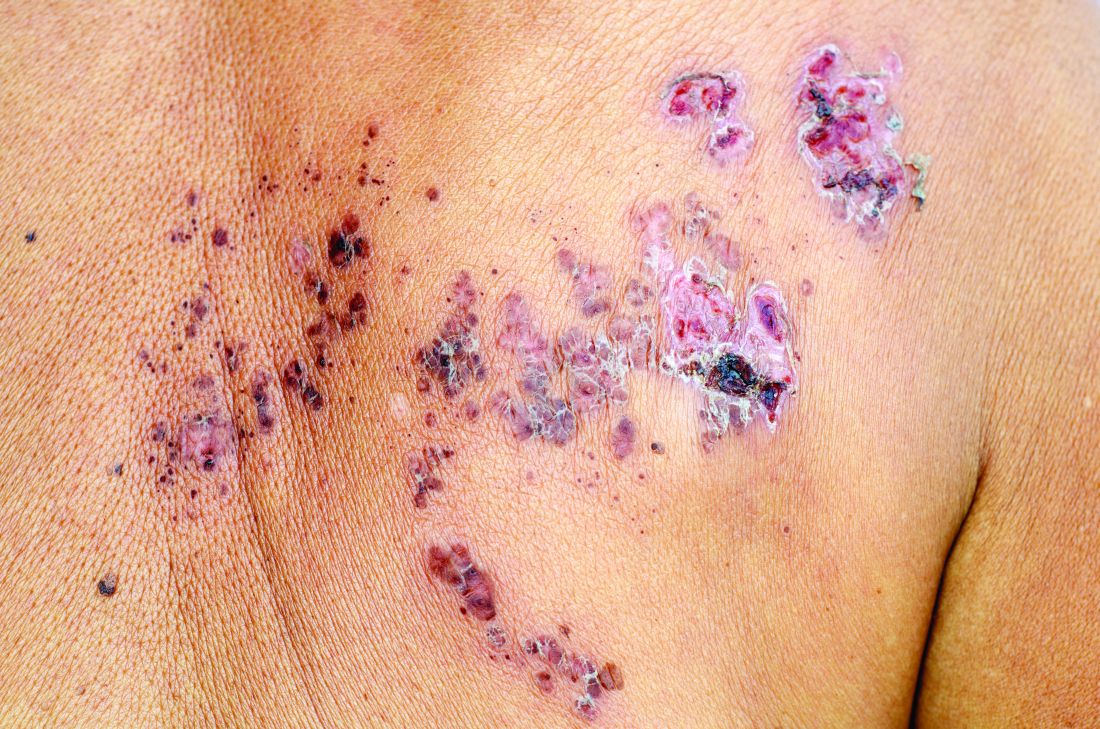
Emily Baumrin, MD, of Brigham and Women’s Hospital, Boston, and her coauthors reviewed 41 studies of herpes zoster in people with psoriasis or psoriatic arthritis according to treatment modality. Their report is in the Journal of the American Academy of Dermatology.
Overall, psoriasis was associated with an increased rate of herpes zoster when compared with the general population: 13.3 cases per 1,000 patient-years for psoriasis and 15.9 for psoriatic arthritis, compared with 8.5 in healthy controls after adjustment for age, sex, and systemic medications. Most of this increased incidence was seen in patients with more severe disease: Those with mild disease who were not receiving systemic therapy had a risk similar to that of healthy controls.
However, one study suggested much of the increased risk of herpes zoster in psoriasis was accounted for by immunosuppressive therapy; when those patients were excluded, there was an 8% increase in risk.
The authors found that people whose psoriasis was treated with tofacitinib (Xeljanz) had a two- to threefold increased risk of herpes zoster, compared with those treated with tumor necrosis factor (TNF) inhibitors or conventional synthetic disease-modifying antirheumatic drugs (DMARDs).
Corticosteroids – either alone or in combination with DMARDs – were also associated with significant increases in the risk of herpes zoster. Patients treated with TNF inhibitor monotherapy had a risk of herpes zoster similar to that of those treated with conventional synthetic DMARDs or no synthetic therapy.
On the question of immunization, the authors pointed to guidelines recommending use of the live attenuated zoster vaccine (Zostavax) in immunocompetent patients or those on low-dose immunosuppression, although they noted that the vaccine is currently contraindicated for patients on biologic DMARDs.
They also examined the evidence for the use of the recently-released non-live recombinant herpes zoster vaccine (Shingrix) in immunocompromised patients, which found no evidence of vaccine-related serious adverse events in individuals with HIV and low CD4 cell counts and in autologous hematopoietic stem cell transplant recipients.
Given this, they recommended that the recombinant vaccine be administered to all patients aged over 50 years with psoriasis or psoriatic arthritis, and to those aged under 50 years who were being treated with tofacitinib, systemic corticosteroids, or combination systemic therapy.
There were insufficient data to draw conclusions about the impact of treatment with the interleukin-12/23 blocker ustekinumab (Stelara) on herpes zoster risk, but the authors noted that there was a trend toward an increased risk. They found no increase in the risk of herpes zoster with interleukin-17 inhibitors (ixekizumab [Taltz], secukinumab [Cosentyx], and brodalumab [Siliq]) and interleukin-23 (p19 subunit) inhibitors (guselkumab [Tremfya], tildrakizumab [Ilumya], and risankizumab) but noted an absence of long-term safety data for these drugs.
Four authors declared advisory, consultancy, or speaker positions with the pharmaceutical sector.
SOURCE: Baumrin E et al. J Am Acad Dermatol. 2019 March 15. doi: 10.1016/j.jaad.2019.03.017.
All individuals with psoriasis or psoriatic arthritis aged over 50 years should receive the recombinant herpes zoster vaccine, according to a systematic review and consensus recommendations from the National Psoriasis Foundation.
Emily Baumrin, MD, of Brigham and Women’s Hospital, Boston, and her coauthors reviewed 41 studies of herpes zoster in people with psoriasis or psoriatic arthritis according to treatment modality. Their report is in the Journal of the American Academy of Dermatology.
Overall, psoriasis was associated with an increased rate of herpes zoster when compared with the general population: 13.3 cases per 1,000 patient-years for psoriasis and 15.9 for psoriatic arthritis, compared with 8.5 in healthy controls after adjustment for age, sex, and systemic medications. Most of this increased incidence was seen in patients with more severe disease: Those with mild disease who were not receiving systemic therapy had a risk similar to that of healthy controls.
However, one study suggested much of the increased risk of herpes zoster in psoriasis was accounted for by immunosuppressive therapy; when those patients were excluded, there was an 8% increase in risk.
The authors found that people whose psoriasis was treated with tofacitinib (Xeljanz) had a two- to threefold increased risk of herpes zoster, compared with those treated with tumor necrosis factor (TNF) inhibitors or conventional synthetic disease-modifying antirheumatic drugs (DMARDs).
Corticosteroids – either alone or in combination with DMARDs – were also associated with significant increases in the risk of herpes zoster. Patients treated with TNF inhibitor monotherapy had a risk of herpes zoster similar to that of those treated with conventional synthetic DMARDs or no synthetic therapy.
On the question of immunization, the authors pointed to guidelines recommending use of the live attenuated zoster vaccine (Zostavax) in immunocompetent patients or those on low-dose immunosuppression, although they noted that the vaccine is currently contraindicated for patients on biologic DMARDs.
They also examined the evidence for the use of the recently-released non-live recombinant herpes zoster vaccine (Shingrix) in immunocompromised patients, which found no evidence of vaccine-related serious adverse events in individuals with HIV and low CD4 cell counts and in autologous hematopoietic stem cell transplant recipients.
Given this, they recommended that the recombinant vaccine be administered to all patients aged over 50 years with psoriasis or psoriatic arthritis, and to those aged under 50 years who were being treated with tofacitinib, systemic corticosteroids, or combination systemic therapy.
There were insufficient data to draw conclusions about the impact of treatment with the interleukin-12/23 blocker ustekinumab (Stelara) on herpes zoster risk, but the authors noted that there was a trend toward an increased risk. They found no increase in the risk of herpes zoster with interleukin-17 inhibitors (ixekizumab [Taltz], secukinumab [Cosentyx], and brodalumab [Siliq]) and interleukin-23 (p19 subunit) inhibitors (guselkumab [Tremfya], tildrakizumab [Ilumya], and risankizumab) but noted an absence of long-term safety data for these drugs.
Four authors declared advisory, consultancy, or speaker positions with the pharmaceutical sector.
SOURCE: Baumrin E et al. J Am Acad Dermatol. 2019 March 15. doi: 10.1016/j.jaad.2019.03.017.
All individuals with psoriasis or psoriatic arthritis aged over 50 years should receive the recombinant herpes zoster vaccine, according to a systematic review and consensus recommendations from the National Psoriasis Foundation.
Emily Baumrin, MD, of Brigham and Women’s Hospital, Boston, and her coauthors reviewed 41 studies of herpes zoster in people with psoriasis or psoriatic arthritis according to treatment modality. Their report is in the Journal of the American Academy of Dermatology.
Overall, psoriasis was associated with an increased rate of herpes zoster when compared with the general population: 13.3 cases per 1,000 patient-years for psoriasis and 15.9 for psoriatic arthritis, compared with 8.5 in healthy controls after adjustment for age, sex, and systemic medications. Most of this increased incidence was seen in patients with more severe disease: Those with mild disease who were not receiving systemic therapy had a risk similar to that of healthy controls.
However, one study suggested much of the increased risk of herpes zoster in psoriasis was accounted for by immunosuppressive therapy; when those patients were excluded, there was an 8% increase in risk.
The authors found that people whose psoriasis was treated with tofacitinib (Xeljanz) had a two- to threefold increased risk of herpes zoster, compared with those treated with tumor necrosis factor (TNF) inhibitors or conventional synthetic disease-modifying antirheumatic drugs (DMARDs).
Corticosteroids – either alone or in combination with DMARDs – were also associated with significant increases in the risk of herpes zoster. Patients treated with TNF inhibitor monotherapy had a risk of herpes zoster similar to that of those treated with conventional synthetic DMARDs or no synthetic therapy.
On the question of immunization, the authors pointed to guidelines recommending use of the live attenuated zoster vaccine (Zostavax) in immunocompetent patients or those on low-dose immunosuppression, although they noted that the vaccine is currently contraindicated for patients on biologic DMARDs.
They also examined the evidence for the use of the recently-released non-live recombinant herpes zoster vaccine (Shingrix) in immunocompromised patients, which found no evidence of vaccine-related serious adverse events in individuals with HIV and low CD4 cell counts and in autologous hematopoietic stem cell transplant recipients.
Given this, they recommended that the recombinant vaccine be administered to all patients aged over 50 years with psoriasis or psoriatic arthritis, and to those aged under 50 years who were being treated with tofacitinib, systemic corticosteroids, or combination systemic therapy.
There were insufficient data to draw conclusions about the impact of treatment with the interleukin-12/23 blocker ustekinumab (Stelara) on herpes zoster risk, but the authors noted that there was a trend toward an increased risk. They found no increase in the risk of herpes zoster with interleukin-17 inhibitors (ixekizumab [Taltz], secukinumab [Cosentyx], and brodalumab [Siliq]) and interleukin-23 (p19 subunit) inhibitors (guselkumab [Tremfya], tildrakizumab [Ilumya], and risankizumab) but noted an absence of long-term safety data for these drugs.
Four authors declared advisory, consultancy, or speaker positions with the pharmaceutical sector.
SOURCE: Baumrin E et al. J Am Acad Dermatol. 2019 March 15. doi: 10.1016/j.jaad.2019.03.017.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
No biological benefits from alcohol seen in rheumatoid arthritis
, according to a study published online March 20 in Arthritis Care & Research.

Joshua F. Baker, MD, of the University of Pennsylvania, Philadelphia, and his coauthors wrote that previous studies had suggested a link between moderate alcohol consumption and lower disease activity, better quality of life, and better functional status in people with rheumatoid arthritis. This link may tempt clinicians “to encourage moderate alcohol consumption among patients with RA,” the researchers wrote, and so it prompted them to examine the relationship more closely.
The researchers studied 16,762 individuals with rheumatoid arthritis in the National Databank for Rheumatic Diseases who had been asked about alcohol use and disease activity in a series of semiannual surveys, providing a total of 121,280 observations, at which 53% reported using alcohol.
Across the observations taken from the semiannual surveys, a total of 8.2% reported discontinuing alcohol consumption from one survey to the next, and 8.4% of abstainers reported initiating alcohol use. Importantly, individuals with high disease activity had a significantly shorter time to discontinuation of alcohol, and those with a moderate or high Patient Activity Scale-II (PAS-II) score were 36% more likely to stop alcohol consumption, compared with individuals who had a low PAS-II score.
Individuals who were older or obese or had more comorbidities or greater work disability were all independently more likely to discontinue alcohol use, while those less likely to give up alcohol tended to be white, male, and have higher physical and mental quality of life, higher educational level, and greater household income.
Participants with moderate or high PAS-II scores were also less likely to start consuming alcohol in comparison to those with low scores.
“Overall, these observations suggest that patients with RA are substantially less likely to use alcohol when their disease activity is high and their health and quality of life are poor,” the authors wrote. “This study also found that active drinking, recent discontinuation of drinking, and recent initiation of drinking were not associated with disease activity or death in this population when considering the reasons for the changes in behavior.”
They said this offered a different explanation for the previously observed association between alcohol use and lower disease activity by showing an effect of reverse causality rather than any biologically protective effect of alcohol.
While the study also found a strong link between discontinuation of alcohol use and increased subsequent mortality, they suggested this was also likely a function of disease activity and disability, rather than the effect of giving up alcohol.
The study was funded by grants to several authors from the Department of Veterans Affairs, the National Institutes of Health, and the Rheumatology Research Foundation. Dr. Baker reported receiving consulting fees from Bristol-Myers Squibb outside of the current work.
SOURCE: Baker J et al. Arthritis Care Res. 2019 Mar 20. doi: 10.1002/acr.23847.
, according to a study published online March 20 in Arthritis Care & Research.
Joshua F. Baker, MD, of the University of Pennsylvania, Philadelphia, and his coauthors wrote that previous studies had suggested a link between moderate alcohol consumption and lower disease activity, better quality of life, and better functional status in people with rheumatoid arthritis. This link may tempt clinicians “to encourage moderate alcohol consumption among patients with RA,” the researchers wrote, and so it prompted them to examine the relationship more closely.
The researchers studied 16,762 individuals with rheumatoid arthritis in the National Databank for Rheumatic Diseases who had been asked about alcohol use and disease activity in a series of semiannual surveys, providing a total of 121,280 observations, at which 53% reported using alcohol.
Across the observations taken from the semiannual surveys, a total of 8.2% reported discontinuing alcohol consumption from one survey to the next, and 8.4% of abstainers reported initiating alcohol use. Importantly, individuals with high disease activity had a significantly shorter time to discontinuation of alcohol, and those with a moderate or high Patient Activity Scale-II (PAS-II) score were 36% more likely to stop alcohol consumption, compared with individuals who had a low PAS-II score.
Individuals who were older or obese or had more comorbidities or greater work disability were all independently more likely to discontinue alcohol use, while those less likely to give up alcohol tended to be white, male, and have higher physical and mental quality of life, higher educational level, and greater household income.
Participants with moderate or high PAS-II scores were also less likely to start consuming alcohol in comparison to those with low scores.
“Overall, these observations suggest that patients with RA are substantially less likely to use alcohol when their disease activity is high and their health and quality of life are poor,” the authors wrote. “This study also found that active drinking, recent discontinuation of drinking, and recent initiation of drinking were not associated with disease activity or death in this population when considering the reasons for the changes in behavior.”
They said this offered a different explanation for the previously observed association between alcohol use and lower disease activity by showing an effect of reverse causality rather than any biologically protective effect of alcohol.
While the study also found a strong link between discontinuation of alcohol use and increased subsequent mortality, they suggested this was also likely a function of disease activity and disability, rather than the effect of giving up alcohol.
The study was funded by grants to several authors from the Department of Veterans Affairs, the National Institutes of Health, and the Rheumatology Research Foundation. Dr. Baker reported receiving consulting fees from Bristol-Myers Squibb outside of the current work.
SOURCE: Baker J et al. Arthritis Care Res. 2019 Mar 20. doi: 10.1002/acr.23847.
, according to a study published online March 20 in Arthritis Care & Research.
Joshua F. Baker, MD, of the University of Pennsylvania, Philadelphia, and his coauthors wrote that previous studies had suggested a link between moderate alcohol consumption and lower disease activity, better quality of life, and better functional status in people with rheumatoid arthritis. This link may tempt clinicians “to encourage moderate alcohol consumption among patients with RA,” the researchers wrote, and so it prompted them to examine the relationship more closely.
The researchers studied 16,762 individuals with rheumatoid arthritis in the National Databank for Rheumatic Diseases who had been asked about alcohol use and disease activity in a series of semiannual surveys, providing a total of 121,280 observations, at which 53% reported using alcohol.
Across the observations taken from the semiannual surveys, a total of 8.2% reported discontinuing alcohol consumption from one survey to the next, and 8.4% of abstainers reported initiating alcohol use. Importantly, individuals with high disease activity had a significantly shorter time to discontinuation of alcohol, and those with a moderate or high Patient Activity Scale-II (PAS-II) score were 36% more likely to stop alcohol consumption, compared with individuals who had a low PAS-II score.
Individuals who were older or obese or had more comorbidities or greater work disability were all independently more likely to discontinue alcohol use, while those less likely to give up alcohol tended to be white, male, and have higher physical and mental quality of life, higher educational level, and greater household income.
Participants with moderate or high PAS-II scores were also less likely to start consuming alcohol in comparison to those with low scores.
“Overall, these observations suggest that patients with RA are substantially less likely to use alcohol when their disease activity is high and their health and quality of life are poor,” the authors wrote. “This study also found that active drinking, recent discontinuation of drinking, and recent initiation of drinking were not associated with disease activity or death in this population when considering the reasons for the changes in behavior.”
They said this offered a different explanation for the previously observed association between alcohol use and lower disease activity by showing an effect of reverse causality rather than any biologically protective effect of alcohol.
While the study also found a strong link between discontinuation of alcohol use and increased subsequent mortality, they suggested this was also likely a function of disease activity and disability, rather than the effect of giving up alcohol.
The study was funded by grants to several authors from the Department of Veterans Affairs, the National Institutes of Health, and the Rheumatology Research Foundation. Dr. Baker reported receiving consulting fees from Bristol-Myers Squibb outside of the current work.
SOURCE: Baker J et al. Arthritis Care Res. 2019 Mar 20. doi: 10.1002/acr.23847.
FROM ARTHRITIS CARE & RESEARCH
Disease burden in OA worse than RA 6 months post presentation
Patients with osteoarthritis (OA) have RAPID3 scores at their initial visit (16.0) similar to patients with rheumatoid arthritis (RA) and either prior use of disease-modifying antirheumatic drugs (DMARDs) or no exposure to DMARDs (15.6 and 15.5, respectively). After 6 months of treatment, the RAPID3 (Routine Assessment of Patient Index Data 3) score fell by just 1.7 points for patients with OA, compared with 5.7 points in RA patients naive to DMARDs and 4.3 points in those with prior DMARD exposure. These findings were published March 20 in Arthritis & Rheumatology (doi: 10.1002/art.40869).
We reported this story at the 2018 World Congress on Osteoarthritis before it was published in the journal. Read the story at the link above.
Patients with osteoarthritis (OA) have RAPID3 scores at their initial visit (16.0) similar to patients with rheumatoid arthritis (RA) and either prior use of disease-modifying antirheumatic drugs (DMARDs) or no exposure to DMARDs (15.6 and 15.5, respectively). After 6 months of treatment, the RAPID3 (Routine Assessment of Patient Index Data 3) score fell by just 1.7 points for patients with OA, compared with 5.7 points in RA patients naive to DMARDs and 4.3 points in those with prior DMARD exposure. These findings were published March 20 in Arthritis & Rheumatology (doi: 10.1002/art.40869).
We reported this story at the 2018 World Congress on Osteoarthritis before it was published in the journal. Read the story at the link above.
Patients with osteoarthritis (OA) have RAPID3 scores at their initial visit (16.0) similar to patients with rheumatoid arthritis (RA) and either prior use of disease-modifying antirheumatic drugs (DMARDs) or no exposure to DMARDs (15.6 and 15.5, respectively). After 6 months of treatment, the RAPID3 (Routine Assessment of Patient Index Data 3) score fell by just 1.7 points for patients with OA, compared with 5.7 points in RA patients naive to DMARDs and 4.3 points in those with prior DMARD exposure. These findings were published March 20 in Arthritis & Rheumatology (doi: 10.1002/art.40869).
We reported this story at the 2018 World Congress on Osteoarthritis before it was published in the journal. Read the story at the link above.
FROM ARTHRITIS & RHEUMATOLOGY
Concomitant methotrexate boosts pegloticase efficacy in gout patients
MAUI, HAWAII – , Orrin M. Troum, MD, said at the 2019 Rheumatology Winter Clinical Symposium.

He cited what he considers to be a practice-changing, prospective, observational, proof-of-concept study presented by John Botson, MD, at the 2018 annual meeting of the American College of Rheumatology.
Dr. Botson, a rheumatologist at Orthopedic Physicians Alaska, in Anchorage, reported on nine patients with refractory tophaceous gout placed on an 8-mg infusion of pegloticase every 2 weeks as third-line therapy. But 1 month beforehand he put them on oral methotrexate at 15 mg once weekly along with folic acid at 1 mg/day in an effort to prevent the development of treatment-limiting anti-pegloticase antibodies. It’s the same strategy rheumatologists often use when patients with rheumatoid arthritis on a tumor necrosis factor inhibitor begin to develop anti-drug antibodies.
At the time of the ACR meeting, all nine patients had received at least nine infusions, and six had received at least 12 infusions over the course of 6 months. The response rate was 100%, defined as more than 80% of serum uric acid levels being below 6.0 mg/dL. All patients stayed on methotrexate with no dose adjustment. And there were no infusion reactions. In contrast, the response rate in the randomized trials of pegloticase was only 42%, and 26% of pegloticase recipients experienced infusion reactions within 6 months.
“Although this is not [Food and Drug Administration] approved, it makes a lot of sense. From my standpoint, this is something that I’m doing now for my patients starting on pegloticase if there’s no contraindication to using methotrexate,” said Dr. Troum, a rheumatologist at the University of Southern California in Los Angeles.
“I’ve been doing this, too. This really did change my practice,” added his fellow panelist Alvin F. Wells, MD, PhD, director of the Rheumatology and Immunotherapy Center in Franklin, Wisc.
When they asked for a show of hands, only a handful of audience members indicated they are now using methotrexate in conjunction with pegloticase in their tophaceous gout patients.
Dr. Wells said his sole reservation about the practice involves using methotrexate in patients with an elevated creatinine level. What about using azathioprine or corticosteroids instead? he asked.
Dr. Troum replied that he monitors those patients carefully but sticks with the methotrexate because it’s only for a few months, which is the time frame in which patients are especially vulnerable to experiencing loss of response to pegloticase due to development of anti-drug antibodies.
Dr. Botson, who was in the Maui audience, rose to give a study update. With additional follow-up, he said, there has still been no signal of loss of response to pegloticase coadministered with methotrexate.
“A lot of us are starting to feel like immunosuppression, whether it’s with methotrexate or something else, is standard of care now,” according to the rheumatologist.
As to prescribing methotrexate in gout patients with renal insufficiency, he continued, he and his colleagues have given the matter quite a bit of thought.
“You’re talking about using methotrexate for 6 months in most of these cases. A lot of the patients who have really bad tophaceous gout already have renal insufficiency, and in the short term we haven’t really seen any problems with that. We work closely with a nephrologist on those cases. And a lot of nephrologists swear – although I don’t think the data are there – that they actually improve their renal function when we start to treat their tophaceous gout,” Dr. Botson said.
Dr. Troum and Dr. Wells reported serving as consultants to and on speakers bureaus for numerous pharmaceutical companies.
MAUI, HAWAII – , Orrin M. Troum, MD, said at the 2019 Rheumatology Winter Clinical Symposium.
He cited what he considers to be a practice-changing, prospective, observational, proof-of-concept study presented by John Botson, MD, at the 2018 annual meeting of the American College of Rheumatology.
Dr. Botson, a rheumatologist at Orthopedic Physicians Alaska, in Anchorage, reported on nine patients with refractory tophaceous gout placed on an 8-mg infusion of pegloticase every 2 weeks as third-line therapy. But 1 month beforehand he put them on oral methotrexate at 15 mg once weekly along with folic acid at 1 mg/day in an effort to prevent the development of treatment-limiting anti-pegloticase antibodies. It’s the same strategy rheumatologists often use when patients with rheumatoid arthritis on a tumor necrosis factor inhibitor begin to develop anti-drug antibodies.
At the time of the ACR meeting, all nine patients had received at least nine infusions, and six had received at least 12 infusions over the course of 6 months. The response rate was 100%, defined as more than 80% of serum uric acid levels being below 6.0 mg/dL. All patients stayed on methotrexate with no dose adjustment. And there were no infusion reactions. In contrast, the response rate in the randomized trials of pegloticase was only 42%, and 26% of pegloticase recipients experienced infusion reactions within 6 months.
“Although this is not [Food and Drug Administration] approved, it makes a lot of sense. From my standpoint, this is something that I’m doing now for my patients starting on pegloticase if there’s no contraindication to using methotrexate,” said Dr. Troum, a rheumatologist at the University of Southern California in Los Angeles.
“I’ve been doing this, too. This really did change my practice,” added his fellow panelist Alvin F. Wells, MD, PhD, director of the Rheumatology and Immunotherapy Center in Franklin, Wisc.
When they asked for a show of hands, only a handful of audience members indicated they are now using methotrexate in conjunction with pegloticase in their tophaceous gout patients.
Dr. Wells said his sole reservation about the practice involves using methotrexate in patients with an elevated creatinine level. What about using azathioprine or corticosteroids instead? he asked.
Dr. Troum replied that he monitors those patients carefully but sticks with the methotrexate because it’s only for a few months, which is the time frame in which patients are especially vulnerable to experiencing loss of response to pegloticase due to development of anti-drug antibodies.
Dr. Botson, who was in the Maui audience, rose to give a study update. With additional follow-up, he said, there has still been no signal of loss of response to pegloticase coadministered with methotrexate.
“A lot of us are starting to feel like immunosuppression, whether it’s with methotrexate or something else, is standard of care now,” according to the rheumatologist.
As to prescribing methotrexate in gout patients with renal insufficiency, he continued, he and his colleagues have given the matter quite a bit of thought.
“You’re talking about using methotrexate for 6 months in most of these cases. A lot of the patients who have really bad tophaceous gout already have renal insufficiency, and in the short term we haven’t really seen any problems with that. We work closely with a nephrologist on those cases. And a lot of nephrologists swear – although I don’t think the data are there – that they actually improve their renal function when we start to treat their tophaceous gout,” Dr. Botson said.
Dr. Troum and Dr. Wells reported serving as consultants to and on speakers bureaus for numerous pharmaceutical companies.
MAUI, HAWAII – , Orrin M. Troum, MD, said at the 2019 Rheumatology Winter Clinical Symposium.
He cited what he considers to be a practice-changing, prospective, observational, proof-of-concept study presented by John Botson, MD, at the 2018 annual meeting of the American College of Rheumatology.
Dr. Botson, a rheumatologist at Orthopedic Physicians Alaska, in Anchorage, reported on nine patients with refractory tophaceous gout placed on an 8-mg infusion of pegloticase every 2 weeks as third-line therapy. But 1 month beforehand he put them on oral methotrexate at 15 mg once weekly along with folic acid at 1 mg/day in an effort to prevent the development of treatment-limiting anti-pegloticase antibodies. It’s the same strategy rheumatologists often use when patients with rheumatoid arthritis on a tumor necrosis factor inhibitor begin to develop anti-drug antibodies.
At the time of the ACR meeting, all nine patients had received at least nine infusions, and six had received at least 12 infusions over the course of 6 months. The response rate was 100%, defined as more than 80% of serum uric acid levels being below 6.0 mg/dL. All patients stayed on methotrexate with no dose adjustment. And there were no infusion reactions. In contrast, the response rate in the randomized trials of pegloticase was only 42%, and 26% of pegloticase recipients experienced infusion reactions within 6 months.
“Although this is not [Food and Drug Administration] approved, it makes a lot of sense. From my standpoint, this is something that I’m doing now for my patients starting on pegloticase if there’s no contraindication to using methotrexate,” said Dr. Troum, a rheumatologist at the University of Southern California in Los Angeles.
“I’ve been doing this, too. This really did change my practice,” added his fellow panelist Alvin F. Wells, MD, PhD, director of the Rheumatology and Immunotherapy Center in Franklin, Wisc.
When they asked for a show of hands, only a handful of audience members indicated they are now using methotrexate in conjunction with pegloticase in their tophaceous gout patients.
Dr. Wells said his sole reservation about the practice involves using methotrexate in patients with an elevated creatinine level. What about using azathioprine or corticosteroids instead? he asked.
Dr. Troum replied that he monitors those patients carefully but sticks with the methotrexate because it’s only for a few months, which is the time frame in which patients are especially vulnerable to experiencing loss of response to pegloticase due to development of anti-drug antibodies.
Dr. Botson, who was in the Maui audience, rose to give a study update. With additional follow-up, he said, there has still been no signal of loss of response to pegloticase coadministered with methotrexate.
“A lot of us are starting to feel like immunosuppression, whether it’s with methotrexate or something else, is standard of care now,” according to the rheumatologist.
As to prescribing methotrexate in gout patients with renal insufficiency, he continued, he and his colleagues have given the matter quite a bit of thought.
“You’re talking about using methotrexate for 6 months in most of these cases. A lot of the patients who have really bad tophaceous gout already have renal insufficiency, and in the short term we haven’t really seen any problems with that. We work closely with a nephrologist on those cases. And a lot of nephrologists swear – although I don’t think the data are there – that they actually improve their renal function when we start to treat their tophaceous gout,” Dr. Botson said.
Dr. Troum and Dr. Wells reported serving as consultants to and on speakers bureaus for numerous pharmaceutical companies.
REPORTING FROM RWCS 2019
Patients at risk of RA may already have abnormal aortic stiffness
according to a study of potential RA patients who underwent cardiac MRI.
“To our knowledge, this is the first study showing subclinical increase in aortic stiffness in at-risk individuals for RA, with values numerically close to those seen in early, treatment-naive RA,” wrote Graham Fent, MBChB, of the University of Leeds (England) and his associates. The study was published in Annals of the Rheumatic Diseases.
Hypothesizing that patients with no systemic inflammation but circulating anti–cyclic citrullinated peptide (CCP) antibodies may already have cardiovascular concerns, Dr. Fent and his colleagues recruited 18 individuals at risk of developing RA and 30 healthy controls. The groups were matched for age and gender and then underwent multiparametric 3.0 Tesla cardiac MRI with late gadolinium enhancement. The at-risk individuals were classified as being at either low (n = 10) or high (n = 8) risk of RA. Over 12 months, five of the at-risk patients progressed to RA.
According to the cardiac MRI findings, aortic distensibility was lower – and thus arterial stiffness was greater – in the at-risk group (3.6 x 10–3 per mm Hg) versus the healthy controls (4.9 x 10–3 per mm Hg). The difference was even more distinct in the high-risk group (3.1 x 10–3 per mm Hg), compared with the low-risk group (4.2 x 10–3 per mm Hg). The group who eventually progressed to RA also showed lower levels of distensibility (3.2 x 10–3 per mm Hg).
The coauthors acknowledged that the major limitation of their study was a lack of control groups. However, they noted that such a pronounced level of aortic stiffness in the high-risk and RA groups should be seen as “implying a particular role of CCP antibodies.”
The study was supported by the U.K. National Institute for Health Research. One author reported being funded by a National Institute for Health Research grant; another reported being funded by a British Heart Foundation Personal Chair.
SOURCE: Fent G et al. Ann Rheum Dis. 2019 Mar 9. doi: 10.1136/annrheumdis-2018-214975.
according to a study of potential RA patients who underwent cardiac MRI.
“To our knowledge, this is the first study showing subclinical increase in aortic stiffness in at-risk individuals for RA, with values numerically close to those seen in early, treatment-naive RA,” wrote Graham Fent, MBChB, of the University of Leeds (England) and his associates. The study was published in Annals of the Rheumatic Diseases.
Hypothesizing that patients with no systemic inflammation but circulating anti–cyclic citrullinated peptide (CCP) antibodies may already have cardiovascular concerns, Dr. Fent and his colleagues recruited 18 individuals at risk of developing RA and 30 healthy controls. The groups were matched for age and gender and then underwent multiparametric 3.0 Tesla cardiac MRI with late gadolinium enhancement. The at-risk individuals were classified as being at either low (n = 10) or high (n = 8) risk of RA. Over 12 months, five of the at-risk patients progressed to RA.
According to the cardiac MRI findings, aortic distensibility was lower – and thus arterial stiffness was greater – in the at-risk group (3.6 x 10–3 per mm Hg) versus the healthy controls (4.9 x 10–3 per mm Hg). The difference was even more distinct in the high-risk group (3.1 x 10–3 per mm Hg), compared with the low-risk group (4.2 x 10–3 per mm Hg). The group who eventually progressed to RA also showed lower levels of distensibility (3.2 x 10–3 per mm Hg).
The coauthors acknowledged that the major limitation of their study was a lack of control groups. However, they noted that such a pronounced level of aortic stiffness in the high-risk and RA groups should be seen as “implying a particular role of CCP antibodies.”
The study was supported by the U.K. National Institute for Health Research. One author reported being funded by a National Institute for Health Research grant; another reported being funded by a British Heart Foundation Personal Chair.
SOURCE: Fent G et al. Ann Rheum Dis. 2019 Mar 9. doi: 10.1136/annrheumdis-2018-214975.
according to a study of potential RA patients who underwent cardiac MRI.
“To our knowledge, this is the first study showing subclinical increase in aortic stiffness in at-risk individuals for RA, with values numerically close to those seen in early, treatment-naive RA,” wrote Graham Fent, MBChB, of the University of Leeds (England) and his associates. The study was published in Annals of the Rheumatic Diseases.
Hypothesizing that patients with no systemic inflammation but circulating anti–cyclic citrullinated peptide (CCP) antibodies may already have cardiovascular concerns, Dr. Fent and his colleagues recruited 18 individuals at risk of developing RA and 30 healthy controls. The groups were matched for age and gender and then underwent multiparametric 3.0 Tesla cardiac MRI with late gadolinium enhancement. The at-risk individuals were classified as being at either low (n = 10) or high (n = 8) risk of RA. Over 12 months, five of the at-risk patients progressed to RA.
According to the cardiac MRI findings, aortic distensibility was lower – and thus arterial stiffness was greater – in the at-risk group (3.6 x 10–3 per mm Hg) versus the healthy controls (4.9 x 10–3 per mm Hg). The difference was even more distinct in the high-risk group (3.1 x 10–3 per mm Hg), compared with the low-risk group (4.2 x 10–3 per mm Hg). The group who eventually progressed to RA also showed lower levels of distensibility (3.2 x 10–3 per mm Hg).
The coauthors acknowledged that the major limitation of their study was a lack of control groups. However, they noted that such a pronounced level of aortic stiffness in the high-risk and RA groups should be seen as “implying a particular role of CCP antibodies.”
The study was supported by the U.K. National Institute for Health Research. One author reported being funded by a National Institute for Health Research grant; another reported being funded by a British Heart Foundation Personal Chair.
SOURCE: Fent G et al. Ann Rheum Dis. 2019 Mar 9. doi: 10.1136/annrheumdis-2018-214975.
FROM ANNALS OF THE RHEUMATIC DISEASES
Socioeconomic status affects scleroderma severity in African Americans
according to findings from an analysis of single-center cohort data over a 10-year period.

Indeed, among patients in the cohort of 402 scleroderma patients at MedStar Georgetown University Hospital in Washington, lower household income was predictive of higher mortality during follow-up, independent of race, according to first author Duncan F. Moore, MD, and his colleagues at the hospital.
Previous studies have demonstrated increased risk for scleroderma in African American patients, who also are more likely than non–African Americans to be diagnosed at a younger age and to have conditions including more diffuse cutaneous disease, more severe restrictive lung disease, more cardiac and renal involvement, and increased mortality, the authors wrote in Arthritis Care & Research.
“We did clearly show that African Americans have worse outcomes and severe pulmonary involvement, but I was surprised that there still was a major contribution of socioeconomic status affecting outcomes for all patients, even though only 10% of our patients were indigent and on medical assistance,” Virginia Steen, MD, senior author of the study and professor of rheumatology at Georgetown University, said in an interview. “I still feel strongly that there are likely genetic issues as to why African Americans have such severe disease. We are eager to learn more from the GRASP [Genome Research in African American Scleroderma Patients] study, which is specifically looking at the genetic issues in African American scleroderma patients,” she said.
Of the 402 scleroderma patients at MedStar Georgetown who were seen during 2006-2016, 202 were African American. A total of 186 African American and 184 non–African American patients in the study met the 2013 American College of Rheumatology/European League Against Rheumatism criteria for systemic sclerosis (SSc). Demographics including gender (87% female) and age (mean of 48 years) were similar between the groups.
Overall, the African American patients showed more severe lung disease, more pulmonary hypertension, and more severe cardiac involvement than did non–African American patients, and autoantibodies were significantly different between the groups.
During follow-up, mortality proved much higher among African Americans at 21%, compared with 11% in non–African Americans (P = .005). However, the unadjusted hazard ratio for death declined from 2.061 (P = .006) to a nonsignificant 1.256 after adjustment for socioeconomic variables.
All socioeconomic measures showed significant differences between the groups. African Americans were more likely to be single and disabled at the initial study visit and to have Medicaid, but they were less likely to be a homemaker, have private insurance, or have a college degree. African Americans’ $74,000 median household income (based on ZIP code) was also a statistically significant $23,000 less than non–African American patients. But the researchers noted that “for every additional $10,000 of household income, independent of race, the hazard of death during follow-up declined by 15.5%.”
Notable differences in antibodies appeared between the groups, with more African American patients having isolated nucleolar ANA, anti-U1RNP antibody, or other positive antinuclear antibodies without SSc-specific antibodies. African American patients also were less likely to have anticentromere or anti-RNA polymerase III antibodies.
The study findings were limited by several factors, including possible bias in the matching process and the use of only index values for socioeconomic variables, the researchers noted.
Regardless of relative socioeconomic and genetic influences, “it is clear that African Americans with scleroderma merit more intensive efforts to facilitate timely diagnosis and access to continued evaluation and suppressive treatment, particularly with respect to cardiopulmonary involvement,” they wrote.
Next steps for research, according to Dr. Steen, include studying clinical subsets of African American patients to try to identify factors to predict outcomes, including the nucleolar pattern ANA, overlap with lupus, history of hypertension, and the relationship with renal crisis.
“We are also looking at whether the African American patients are less responsive to mycophenolate than the non–African American patients. We definitely need to find ways to be more aggressive at identifying and treating African American patients early in their disease,” she added.
The researchers had no financial conflicts to disclose. Dr. Steen serves on the MDedge Rheumatology Editorial Advisory Board.
SOURCE: Moore DF et al. Arthritis Care Res. 2019 March 1. doi: 10.1002/acr.23861.
“Not only do patients who manifest the diffuse cutaneous subset of disease experience a more severe course, but so do affected persons of African American race,” Nadia D. Morgan, MBBS, and Allan C. Gelber, MD, wrote in an accompanying editorial. The effects of socioeconomic status should not be overlooked based on the current study, in which the inclusion of socioeconomic factors eliminated the significance of association between race and mortality among scleroderma patients, they wrote.

“Overall, and in the context of these published reports which underscore the disproportionate and adverse impact of scleroderma among African Americans, and in light of the ongoing efforts of the GRASP study, the current paper by Moore et al. emphasizes the importance of socioeconomic status, and of socioeconomic determinants of health, to account for differences in clinically relevant outcomes,” they wrote.
Dr. Gelber is affiliated with the division of rheumatology at Johns Hopkins University, Baltimore. Dr. Morgan, who was also with Johns Hopkins, died before publication of the editorial. They made no conflict of interest disclosures.
“Not only do patients who manifest the diffuse cutaneous subset of disease experience a more severe course, but so do affected persons of African American race,” Nadia D. Morgan, MBBS, and Allan C. Gelber, MD, wrote in an accompanying editorial. The effects of socioeconomic status should not be overlooked based on the current study, in which the inclusion of socioeconomic factors eliminated the significance of association between race and mortality among scleroderma patients, they wrote.
“Overall, and in the context of these published reports which underscore the disproportionate and adverse impact of scleroderma among African Americans, and in light of the ongoing efforts of the GRASP study, the current paper by Moore et al. emphasizes the importance of socioeconomic status, and of socioeconomic determinants of health, to account for differences in clinically relevant outcomes,” they wrote.
Dr. Gelber is affiliated with the division of rheumatology at Johns Hopkins University, Baltimore. Dr. Morgan, who was also with Johns Hopkins, died before publication of the editorial. They made no conflict of interest disclosures.
“Not only do patients who manifest the diffuse cutaneous subset of disease experience a more severe course, but so do affected persons of African American race,” Nadia D. Morgan, MBBS, and Allan C. Gelber, MD, wrote in an accompanying editorial. The effects of socioeconomic status should not be overlooked based on the current study, in which the inclusion of socioeconomic factors eliminated the significance of association between race and mortality among scleroderma patients, they wrote.
“Overall, and in the context of these published reports which underscore the disproportionate and adverse impact of scleroderma among African Americans, and in light of the ongoing efforts of the GRASP study, the current paper by Moore et al. emphasizes the importance of socioeconomic status, and of socioeconomic determinants of health, to account for differences in clinically relevant outcomes,” they wrote.
Dr. Gelber is affiliated with the division of rheumatology at Johns Hopkins University, Baltimore. Dr. Morgan, who was also with Johns Hopkins, died before publication of the editorial. They made no conflict of interest disclosures.
according to findings from an analysis of single-center cohort data over a 10-year period.
Indeed, among patients in the cohort of 402 scleroderma patients at MedStar Georgetown University Hospital in Washington, lower household income was predictive of higher mortality during follow-up, independent of race, according to first author Duncan F. Moore, MD, and his colleagues at the hospital.
Previous studies have demonstrated increased risk for scleroderma in African American patients, who also are more likely than non–African Americans to be diagnosed at a younger age and to have conditions including more diffuse cutaneous disease, more severe restrictive lung disease, more cardiac and renal involvement, and increased mortality, the authors wrote in Arthritis Care & Research.
“We did clearly show that African Americans have worse outcomes and severe pulmonary involvement, but I was surprised that there still was a major contribution of socioeconomic status affecting outcomes for all patients, even though only 10% of our patients were indigent and on medical assistance,” Virginia Steen, MD, senior author of the study and professor of rheumatology at Georgetown University, said in an interview. “I still feel strongly that there are likely genetic issues as to why African Americans have such severe disease. We are eager to learn more from the GRASP [Genome Research in African American Scleroderma Patients] study, which is specifically looking at the genetic issues in African American scleroderma patients,” she said.
Of the 402 scleroderma patients at MedStar Georgetown who were seen during 2006-2016, 202 were African American. A total of 186 African American and 184 non–African American patients in the study met the 2013 American College of Rheumatology/European League Against Rheumatism criteria for systemic sclerosis (SSc). Demographics including gender (87% female) and age (mean of 48 years) were similar between the groups.
Overall, the African American patients showed more severe lung disease, more pulmonary hypertension, and more severe cardiac involvement than did non–African American patients, and autoantibodies were significantly different between the groups.
During follow-up, mortality proved much higher among African Americans at 21%, compared with 11% in non–African Americans (P = .005). However, the unadjusted hazard ratio for death declined from 2.061 (P = .006) to a nonsignificant 1.256 after adjustment for socioeconomic variables.
All socioeconomic measures showed significant differences between the groups. African Americans were more likely to be single and disabled at the initial study visit and to have Medicaid, but they were less likely to be a homemaker, have private insurance, or have a college degree. African Americans’ $74,000 median household income (based on ZIP code) was also a statistically significant $23,000 less than non–African American patients. But the researchers noted that “for every additional $10,000 of household income, independent of race, the hazard of death during follow-up declined by 15.5%.”
Notable differences in antibodies appeared between the groups, with more African American patients having isolated nucleolar ANA, anti-U1RNP antibody, or other positive antinuclear antibodies without SSc-specific antibodies. African American patients also were less likely to have anticentromere or anti-RNA polymerase III antibodies.
The study findings were limited by several factors, including possible bias in the matching process and the use of only index values for socioeconomic variables, the researchers noted.
Regardless of relative socioeconomic and genetic influences, “it is clear that African Americans with scleroderma merit more intensive efforts to facilitate timely diagnosis and access to continued evaluation and suppressive treatment, particularly with respect to cardiopulmonary involvement,” they wrote.
Next steps for research, according to Dr. Steen, include studying clinical subsets of African American patients to try to identify factors to predict outcomes, including the nucleolar pattern ANA, overlap with lupus, history of hypertension, and the relationship with renal crisis.
“We are also looking at whether the African American patients are less responsive to mycophenolate than the non–African American patients. We definitely need to find ways to be more aggressive at identifying and treating African American patients early in their disease,” she added.
The researchers had no financial conflicts to disclose. Dr. Steen serves on the MDedge Rheumatology Editorial Advisory Board.
SOURCE: Moore DF et al. Arthritis Care Res. 2019 March 1. doi: 10.1002/acr.23861.
according to findings from an analysis of single-center cohort data over a 10-year period.
Indeed, among patients in the cohort of 402 scleroderma patients at MedStar Georgetown University Hospital in Washington, lower household income was predictive of higher mortality during follow-up, independent of race, according to first author Duncan F. Moore, MD, and his colleagues at the hospital.
Previous studies have demonstrated increased risk for scleroderma in African American patients, who also are more likely than non–African Americans to be diagnosed at a younger age and to have conditions including more diffuse cutaneous disease, more severe restrictive lung disease, more cardiac and renal involvement, and increased mortality, the authors wrote in Arthritis Care & Research.
“We did clearly show that African Americans have worse outcomes and severe pulmonary involvement, but I was surprised that there still was a major contribution of socioeconomic status affecting outcomes for all patients, even though only 10% of our patients were indigent and on medical assistance,” Virginia Steen, MD, senior author of the study and professor of rheumatology at Georgetown University, said in an interview. “I still feel strongly that there are likely genetic issues as to why African Americans have such severe disease. We are eager to learn more from the GRASP [Genome Research in African American Scleroderma Patients] study, which is specifically looking at the genetic issues in African American scleroderma patients,” she said.
Of the 402 scleroderma patients at MedStar Georgetown who were seen during 2006-2016, 202 were African American. A total of 186 African American and 184 non–African American patients in the study met the 2013 American College of Rheumatology/European League Against Rheumatism criteria for systemic sclerosis (SSc). Demographics including gender (87% female) and age (mean of 48 years) were similar between the groups.
Overall, the African American patients showed more severe lung disease, more pulmonary hypertension, and more severe cardiac involvement than did non–African American patients, and autoantibodies were significantly different between the groups.
During follow-up, mortality proved much higher among African Americans at 21%, compared with 11% in non–African Americans (P = .005). However, the unadjusted hazard ratio for death declined from 2.061 (P = .006) to a nonsignificant 1.256 after adjustment for socioeconomic variables.
All socioeconomic measures showed significant differences between the groups. African Americans were more likely to be single and disabled at the initial study visit and to have Medicaid, but they were less likely to be a homemaker, have private insurance, or have a college degree. African Americans’ $74,000 median household income (based on ZIP code) was also a statistically significant $23,000 less than non–African American patients. But the researchers noted that “for every additional $10,000 of household income, independent of race, the hazard of death during follow-up declined by 15.5%.”
Notable differences in antibodies appeared between the groups, with more African American patients having isolated nucleolar ANA, anti-U1RNP antibody, or other positive antinuclear antibodies without SSc-specific antibodies. African American patients also were less likely to have anticentromere or anti-RNA polymerase III antibodies.
The study findings were limited by several factors, including possible bias in the matching process and the use of only index values for socioeconomic variables, the researchers noted.
Regardless of relative socioeconomic and genetic influences, “it is clear that African Americans with scleroderma merit more intensive efforts to facilitate timely diagnosis and access to continued evaluation and suppressive treatment, particularly with respect to cardiopulmonary involvement,” they wrote.
Next steps for research, according to Dr. Steen, include studying clinical subsets of African American patients to try to identify factors to predict outcomes, including the nucleolar pattern ANA, overlap with lupus, history of hypertension, and the relationship with renal crisis.
“We are also looking at whether the African American patients are less responsive to mycophenolate than the non–African American patients. We definitely need to find ways to be more aggressive at identifying and treating African American patients early in their disease,” she added.
The researchers had no financial conflicts to disclose. Dr. Steen serves on the MDedge Rheumatology Editorial Advisory Board.
SOURCE: Moore DF et al. Arthritis Care Res. 2019 March 1. doi: 10.1002/acr.23861.
FROM ARTHRITIS CARE & RESEARCH
Resistant hypertension hits SLE patients hard
at a tertiary care center.

A patient with resistant hypertension either has blood pressure remaining above 140/90 mm Hg while taking three antihypertensive medications or requires the use of four or more antihypertensives to attain blood pressure control. Resistant hypertension, which was more likely to occur among blacks and patients with lower renal function, hypercholesterolemia, and increased inflammatory markers, increased the risk of death nearly threefold (hazard ratio, 2.91; P = .0005) when compared with those who didn’t have this condition.
The results of this analysis were published March 15 in Arthritis Care & Research (doi: 10.1002/acr.23880). We covered this study at the 2018 annual meeting of the American College of Rheumatology in Chicago before it was published in the journal. Read our previous story at the link above.
at a tertiary care center.
A patient with resistant hypertension either has blood pressure remaining above 140/90 mm Hg while taking three antihypertensive medications or requires the use of four or more antihypertensives to attain blood pressure control. Resistant hypertension, which was more likely to occur among blacks and patients with lower renal function, hypercholesterolemia, and increased inflammatory markers, increased the risk of death nearly threefold (hazard ratio, 2.91; P = .0005) when compared with those who didn’t have this condition.
The results of this analysis were published March 15 in Arthritis Care & Research (doi: 10.1002/acr.23880). We covered this study at the 2018 annual meeting of the American College of Rheumatology in Chicago before it was published in the journal. Read our previous story at the link above.
at a tertiary care center.
A patient with resistant hypertension either has blood pressure remaining above 140/90 mm Hg while taking three antihypertensive medications or requires the use of four or more antihypertensives to attain blood pressure control. Resistant hypertension, which was more likely to occur among blacks and patients with lower renal function, hypercholesterolemia, and increased inflammatory markers, increased the risk of death nearly threefold (hazard ratio, 2.91; P = .0005) when compared with those who didn’t have this condition.
The results of this analysis were published March 15 in Arthritis Care & Research (doi: 10.1002/acr.23880). We covered this study at the 2018 annual meeting of the American College of Rheumatology in Chicago before it was published in the journal. Read our previous story at the link above.
FROM ARTHRITIS CARE & RESEARCH
Recent trials advance axial spondyloarthritis therapy
MAUI, HAWAII – Arguably the most exciting therapeutic development in axial spondyloarthritis in the past year was the demonstrated efficacy and safety of the investigational oral selective Janus kinase 1 inhibitor filgotinib in the setting of active ankylosing spondylitis, speakers agreed at the 2019 Rheumatology Winter Clinical Symposium.

“This is big, big news,” commented symposium director Arthur Kavanaugh, MD. “This is going to be a big deal.”
Other recent clinical trials of note in axial spondyloarthritis (SpA) highlighted by Dr. Kavanaugh and Eric Ruderman, MD, professor of medicine at Northwestern University, Chicago, included a positive phase 3 study of certolizumab pegol (Cimzia) in nonradiographic SpA, two positive phase 3 trials of the interleukin-17A antagonist ixekizumab (Taltz) in radiographic SpA, a positive phase 2b trial of the dual IL-17A/F antagonist bimekizumab, and publication of three surprisingly negative phase 3 trials of the IL-12/23 inhibitor ustekinumab (Stelara).
Filgotinib
TORTUGA was a phase 2b, double-blind, multicenter trial of 116 European patients with active ankylosing spondylitis nonresponsive to NSAIDs who were randomized to oral filgotinib at 200 mg once daily or placebo for 12 weeks. Filgotinib reduced the Ankylosing Spondylitis Disease Activity Score (ASDAS) by a mean of 1.47 points from baseline, a significantly better result for the primary outcome than the 0.57-point decrease in controls (Lancet. 2018 Dec 1;392[10162]:2378-87).
Dr. Ruderman was also favorably impressed with the oral Janus kinase 1 (JAK1) inhibitor’s performance on the secondary outcome measures, including a mean 2.41-point reduction from baseline on the Bath Ankylosing Spondylitis Disease Activity Index, compared with a 1.44-point decrease in controls, with the difference being significant from week 8 onward. The filgotinib group also did significantly better on validated measures of physical function, spinal mobility, physical function, quality of life, peripheral arthritis, fatigue, and spinal and sacroiliac joint inflammation as assessed by MRI.
One patient in the filgotinib group, a smoker, developed pneumonia and another experienced deep venous thrombosis.
The study results are an exciting development because SpA treatments with new mechanisms of action are sorely needed. NSAIDs are considered first-line pharmacotherapy at present, with various tumor necrosis factor (TNF) inhibitors as well as the IL-17 inhibitor secukinumab (Cosentyx) the only approved biologic alternatives.
“This is the most impressive data I’ve seen that JAK inhibitors are effective in ankylosing spondyloarthritis,” commented Paul Emery, MD, professor of rheumatology and director of the University of Leeds (England) Musculoskeletal Biomedical Research Center.
TORTUGA was the first positive phase 2 trial of a selective JAK1 inhibitor in SpA. However, Dr. Kavanaugh noted that while a phase 2 trial of tofacitinib (Xeljanz) failed to meet its “very convoluted” primary endpoint, the JAK1/3 inhibitor was positive for key secondary endpoints, including favorable MRI changes. And a phase 3 trial of tofacitinib in SpA is underway.
A key remaining question pending the outcome of definitive phase 3 trials is whether specificity of JAK enzyme inhibition matters or if a class effect is at work, according to Dr. Kavanaugh, professor of medicine at the University of California, San Diego.
Certolizumab pegol
The TNF inhibitor is already approved for ankylosing spondylitis, among other indications, but it has now demonstrated efficacy and safety in nonradiographic SpA in a phase 3 trial structured with guidance from the Food and Drug Administration.
“It seems likely that UCB will get the indication for this,” according to Dr. Ruderman.
This 317-patient trial was remarkable in that it entailed a full 52 weeks of double-blind therapy with certolizumab at the standard dose of 200 mg every 2 weeks or placebo. The ASDAS Major Improvement rate, defined as at least a 2-point improvement from baseline, was 47% in the active treatment arm, compared with 7% on placebo. The Assessment in Ankylosing Spondylitis International Society 40% (ASAS 40) response rate, a more patient-reported outcome measure, was 57% in the certolizumab group and 16% in controls in this trial, which was recently published (Arthritis Rheumatol. 2019 March 8. doi: 10.1002/art.40866). All participants had to have baseline MRI evidence of sacroiliac joint inflammation and/or an elevated C-reactive protein.
By way of background, Dr. Ruderman explained that the FDA required 52 weeks of double-blind, placebo-controlled therapy because the agency’s advisory committee had formerly expressed reservations about considering an expanded indication for TNF inhibitors in nonradiographic as opposed to radiographic SpA.
“They were very concerned that approval could result in patients with mechanical back pain or fibromyalgia being treated with biologics. And they weren’t sure nonradiographic SpA was a discrete entity. They wondered if it remits on its own,” according to the rheumatologist.
Dr. Ruderman is curious to see how the FDA is going to handle this situation in light of the positive phase 3 certolizumab results. Will the agency require other companies that market TNF inhibitors to mount a similarly rigorous 52-week, double-blind, placebo-controlled trial in order to obtain an expanded indication? That would seem to pose ethical issues now. Or will the companies be able to gain an expanded indication by retrospective analysis of outcomes in patients with nonradiographic SpA in their existing trials databases? Stay tuned.
Ixekizumab
The COAST-V trial was a phase 3, randomized, double-blind, active- and placebo-controlled trial of 341 patients with radiographic SpA who hadn’t previously been treated with a biologic disease-modifying antirheumatic drug. They were assigned to 80 mg of ixekizumab every 2 weeks, 80 mg every 4 weeks, adalimumab (Humira) at 40 mg every 2 weeks, or placebo. The primary endpoint – an ASAS 40 response at week 16 – was achieved in 52% of patients on ixekizumab every 2 weeks, 48% of those who received ixekizumab every 4 weeks, 36% on adalimumab, and 18% on placebo (Lancet. 2018 Dec 8;392[10163]:2441-51).
In contrast, the phase 3 COAST-W trial included 316 patients with radiographic SpA who were inadequate responders or intolerant to one or more prior anti-TNF agents. The 16-week ASAS 40 response rate was 30.6% with ixekizumab every 2 weeks, similar at 25.4% with ixekizumab every 4 weeks, and 12.5% with placebo (Arthritis Rheumatol. 2018 Oct 20. doi: 10.1002/art.40753).
While the COAST-V trial convincingly showed both ixekizumab and adalimumab were more effective than placebo, the patient numbers were way too small to draw any conclusions about the relative efficacy of the two biologics, according to Dr. Ruderman.
“I don’t think these results are surprising,” Dr. Kavanaugh commented. “It would have been surprising if ixekizumab was ineffective, given that secukinumab works. But it’s nice to have the proof.”
Bimekizumab
This investigational dual IL-17A/F inhibitor demonstrated efficacy and safety for SpA in a 297-patient, phase 2b trial presented at the 2018 European Congress of Rheumatology.
“It’s effective, but it doesn’t look like it’s particularly more effective than either of the existing IL-17A inhibitors. We’ll see going forward if there truly is an advantage here to the additional inhibition of IL-17F in this population. I will say that the preclinical and laboratory data on the potential advantages of IL-17F inhibition are mostly in the psoriasis/psoriatic arthritis space. It’s not clear in ankylosing spondyloarthritis specifically whether we should expect to see a difference,” Dr. Ruderman said.
Ustekinumab
The IL-12/23 inhibitor proved no better than placebo in patients with SpA in three separate phase 3, randomized trials recently published as a single summary article (Arthritis Rheumatol. 2019 Feb;71[2]:258-70).
“Ustekinumab was effective in an earlier open study, so I think everybody was surprised by this,” Dr. Kavanaugh said.
He reported serving as a consultant to and/or receiving research funding from a dozen pharmaceutical companies. Dr. Ruderman reported financial relationships with eight companies.
MAUI, HAWAII – Arguably the most exciting therapeutic development in axial spondyloarthritis in the past year was the demonstrated efficacy and safety of the investigational oral selective Janus kinase 1 inhibitor filgotinib in the setting of active ankylosing spondylitis, speakers agreed at the 2019 Rheumatology Winter Clinical Symposium.
“This is big, big news,” commented symposium director Arthur Kavanaugh, MD. “This is going to be a big deal.”
Other recent clinical trials of note in axial spondyloarthritis (SpA) highlighted by Dr. Kavanaugh and Eric Ruderman, MD, professor of medicine at Northwestern University, Chicago, included a positive phase 3 study of certolizumab pegol (Cimzia) in nonradiographic SpA, two positive phase 3 trials of the interleukin-17A antagonist ixekizumab (Taltz) in radiographic SpA, a positive phase 2b trial of the dual IL-17A/F antagonist bimekizumab, and publication of three surprisingly negative phase 3 trials of the IL-12/23 inhibitor ustekinumab (Stelara).
Filgotinib
TORTUGA was a phase 2b, double-blind, multicenter trial of 116 European patients with active ankylosing spondylitis nonresponsive to NSAIDs who were randomized to oral filgotinib at 200 mg once daily or placebo for 12 weeks. Filgotinib reduced the Ankylosing Spondylitis Disease Activity Score (ASDAS) by a mean of 1.47 points from baseline, a significantly better result for the primary outcome than the 0.57-point decrease in controls (Lancet. 2018 Dec 1;392[10162]:2378-87).
Dr. Ruderman was also favorably impressed with the oral Janus kinase 1 (JAK1) inhibitor’s performance on the secondary outcome measures, including a mean 2.41-point reduction from baseline on the Bath Ankylosing Spondylitis Disease Activity Index, compared with a 1.44-point decrease in controls, with the difference being significant from week 8 onward. The filgotinib group also did significantly better on validated measures of physical function, spinal mobility, physical function, quality of life, peripheral arthritis, fatigue, and spinal and sacroiliac joint inflammation as assessed by MRI.
One patient in the filgotinib group, a smoker, developed pneumonia and another experienced deep venous thrombosis.
The study results are an exciting development because SpA treatments with new mechanisms of action are sorely needed. NSAIDs are considered first-line pharmacotherapy at present, with various tumor necrosis factor (TNF) inhibitors as well as the IL-17 inhibitor secukinumab (Cosentyx) the only approved biologic alternatives.
“This is the most impressive data I’ve seen that JAK inhibitors are effective in ankylosing spondyloarthritis,” commented Paul Emery, MD, professor of rheumatology and director of the University of Leeds (England) Musculoskeletal Biomedical Research Center.
TORTUGA was the first positive phase 2 trial of a selective JAK1 inhibitor in SpA. However, Dr. Kavanaugh noted that while a phase 2 trial of tofacitinib (Xeljanz) failed to meet its “very convoluted” primary endpoint, the JAK1/3 inhibitor was positive for key secondary endpoints, including favorable MRI changes. And a phase 3 trial of tofacitinib in SpA is underway.
A key remaining question pending the outcome of definitive phase 3 trials is whether specificity of JAK enzyme inhibition matters or if a class effect is at work, according to Dr. Kavanaugh, professor of medicine at the University of California, San Diego.
Certolizumab pegol
The TNF inhibitor is already approved for ankylosing spondylitis, among other indications, but it has now demonstrated efficacy and safety in nonradiographic SpA in a phase 3 trial structured with guidance from the Food and Drug Administration.
“It seems likely that UCB will get the indication for this,” according to Dr. Ruderman.
This 317-patient trial was remarkable in that it entailed a full 52 weeks of double-blind therapy with certolizumab at the standard dose of 200 mg every 2 weeks or placebo. The ASDAS Major Improvement rate, defined as at least a 2-point improvement from baseline, was 47% in the active treatment arm, compared with 7% on placebo. The Assessment in Ankylosing Spondylitis International Society 40% (ASAS 40) response rate, a more patient-reported outcome measure, was 57% in the certolizumab group and 16% in controls in this trial, which was recently published (Arthritis Rheumatol. 2019 March 8. doi: 10.1002/art.40866). All participants had to have baseline MRI evidence of sacroiliac joint inflammation and/or an elevated C-reactive protein.
By way of background, Dr. Ruderman explained that the FDA required 52 weeks of double-blind, placebo-controlled therapy because the agency’s advisory committee had formerly expressed reservations about considering an expanded indication for TNF inhibitors in nonradiographic as opposed to radiographic SpA.
“They were very concerned that approval could result in patients with mechanical back pain or fibromyalgia being treated with biologics. And they weren’t sure nonradiographic SpA was a discrete entity. They wondered if it remits on its own,” according to the rheumatologist.
Dr. Ruderman is curious to see how the FDA is going to handle this situation in light of the positive phase 3 certolizumab results. Will the agency require other companies that market TNF inhibitors to mount a similarly rigorous 52-week, double-blind, placebo-controlled trial in order to obtain an expanded indication? That would seem to pose ethical issues now. Or will the companies be able to gain an expanded indication by retrospective analysis of outcomes in patients with nonradiographic SpA in their existing trials databases? Stay tuned.
Ixekizumab
The COAST-V trial was a phase 3, randomized, double-blind, active- and placebo-controlled trial of 341 patients with radiographic SpA who hadn’t previously been treated with a biologic disease-modifying antirheumatic drug. They were assigned to 80 mg of ixekizumab every 2 weeks, 80 mg every 4 weeks, adalimumab (Humira) at 40 mg every 2 weeks, or placebo. The primary endpoint – an ASAS 40 response at week 16 – was achieved in 52% of patients on ixekizumab every 2 weeks, 48% of those who received ixekizumab every 4 weeks, 36% on adalimumab, and 18% on placebo (Lancet. 2018 Dec 8;392[10163]:2441-51).
In contrast, the phase 3 COAST-W trial included 316 patients with radiographic SpA who were inadequate responders or intolerant to one or more prior anti-TNF agents. The 16-week ASAS 40 response rate was 30.6% with ixekizumab every 2 weeks, similar at 25.4% with ixekizumab every 4 weeks, and 12.5% with placebo (Arthritis Rheumatol. 2018 Oct 20. doi: 10.1002/art.40753).
While the COAST-V trial convincingly showed both ixekizumab and adalimumab were more effective than placebo, the patient numbers were way too small to draw any conclusions about the relative efficacy of the two biologics, according to Dr. Ruderman.
“I don’t think these results are surprising,” Dr. Kavanaugh commented. “It would have been surprising if ixekizumab was ineffective, given that secukinumab works. But it’s nice to have the proof.”
Bimekizumab
This investigational dual IL-17A/F inhibitor demonstrated efficacy and safety for SpA in a 297-patient, phase 2b trial presented at the 2018 European Congress of Rheumatology.
“It’s effective, but it doesn’t look like it’s particularly more effective than either of the existing IL-17A inhibitors. We’ll see going forward if there truly is an advantage here to the additional inhibition of IL-17F in this population. I will say that the preclinical and laboratory data on the potential advantages of IL-17F inhibition are mostly in the psoriasis/psoriatic arthritis space. It’s not clear in ankylosing spondyloarthritis specifically whether we should expect to see a difference,” Dr. Ruderman said.
Ustekinumab
The IL-12/23 inhibitor proved no better than placebo in patients with SpA in three separate phase 3, randomized trials recently published as a single summary article (Arthritis Rheumatol. 2019 Feb;71[2]:258-70).
“Ustekinumab was effective in an earlier open study, so I think everybody was surprised by this,” Dr. Kavanaugh said.
He reported serving as a consultant to and/or receiving research funding from a dozen pharmaceutical companies. Dr. Ruderman reported financial relationships with eight companies.
MAUI, HAWAII – Arguably the most exciting therapeutic development in axial spondyloarthritis in the past year was the demonstrated efficacy and safety of the investigational oral selective Janus kinase 1 inhibitor filgotinib in the setting of active ankylosing spondylitis, speakers agreed at the 2019 Rheumatology Winter Clinical Symposium.
“This is big, big news,” commented symposium director Arthur Kavanaugh, MD. “This is going to be a big deal.”
Other recent clinical trials of note in axial spondyloarthritis (SpA) highlighted by Dr. Kavanaugh and Eric Ruderman, MD, professor of medicine at Northwestern University, Chicago, included a positive phase 3 study of certolizumab pegol (Cimzia) in nonradiographic SpA, two positive phase 3 trials of the interleukin-17A antagonist ixekizumab (Taltz) in radiographic SpA, a positive phase 2b trial of the dual IL-17A/F antagonist bimekizumab, and publication of three surprisingly negative phase 3 trials of the IL-12/23 inhibitor ustekinumab (Stelara).
Filgotinib
TORTUGA was a phase 2b, double-blind, multicenter trial of 116 European patients with active ankylosing spondylitis nonresponsive to NSAIDs who were randomized to oral filgotinib at 200 mg once daily or placebo for 12 weeks. Filgotinib reduced the Ankylosing Spondylitis Disease Activity Score (ASDAS) by a mean of 1.47 points from baseline, a significantly better result for the primary outcome than the 0.57-point decrease in controls (Lancet. 2018 Dec 1;392[10162]:2378-87).
Dr. Ruderman was also favorably impressed with the oral Janus kinase 1 (JAK1) inhibitor’s performance on the secondary outcome measures, including a mean 2.41-point reduction from baseline on the Bath Ankylosing Spondylitis Disease Activity Index, compared with a 1.44-point decrease in controls, with the difference being significant from week 8 onward. The filgotinib group also did significantly better on validated measures of physical function, spinal mobility, physical function, quality of life, peripheral arthritis, fatigue, and spinal and sacroiliac joint inflammation as assessed by MRI.
One patient in the filgotinib group, a smoker, developed pneumonia and another experienced deep venous thrombosis.
The study results are an exciting development because SpA treatments with new mechanisms of action are sorely needed. NSAIDs are considered first-line pharmacotherapy at present, with various tumor necrosis factor (TNF) inhibitors as well as the IL-17 inhibitor secukinumab (Cosentyx) the only approved biologic alternatives.
“This is the most impressive data I’ve seen that JAK inhibitors are effective in ankylosing spondyloarthritis,” commented Paul Emery, MD, professor of rheumatology and director of the University of Leeds (England) Musculoskeletal Biomedical Research Center.
TORTUGA was the first positive phase 2 trial of a selective JAK1 inhibitor in SpA. However, Dr. Kavanaugh noted that while a phase 2 trial of tofacitinib (Xeljanz) failed to meet its “very convoluted” primary endpoint, the JAK1/3 inhibitor was positive for key secondary endpoints, including favorable MRI changes. And a phase 3 trial of tofacitinib in SpA is underway.
A key remaining question pending the outcome of definitive phase 3 trials is whether specificity of JAK enzyme inhibition matters or if a class effect is at work, according to Dr. Kavanaugh, professor of medicine at the University of California, San Diego.
Certolizumab pegol
The TNF inhibitor is already approved for ankylosing spondylitis, among other indications, but it has now demonstrated efficacy and safety in nonradiographic SpA in a phase 3 trial structured with guidance from the Food and Drug Administration.
“It seems likely that UCB will get the indication for this,” according to Dr. Ruderman.
This 317-patient trial was remarkable in that it entailed a full 52 weeks of double-blind therapy with certolizumab at the standard dose of 200 mg every 2 weeks or placebo. The ASDAS Major Improvement rate, defined as at least a 2-point improvement from baseline, was 47% in the active treatment arm, compared with 7% on placebo. The Assessment in Ankylosing Spondylitis International Society 40% (ASAS 40) response rate, a more patient-reported outcome measure, was 57% in the certolizumab group and 16% in controls in this trial, which was recently published (Arthritis Rheumatol. 2019 March 8. doi: 10.1002/art.40866). All participants had to have baseline MRI evidence of sacroiliac joint inflammation and/or an elevated C-reactive protein.
By way of background, Dr. Ruderman explained that the FDA required 52 weeks of double-blind, placebo-controlled therapy because the agency’s advisory committee had formerly expressed reservations about considering an expanded indication for TNF inhibitors in nonradiographic as opposed to radiographic SpA.
“They were very concerned that approval could result in patients with mechanical back pain or fibromyalgia being treated with biologics. And they weren’t sure nonradiographic SpA was a discrete entity. They wondered if it remits on its own,” according to the rheumatologist.
Dr. Ruderman is curious to see how the FDA is going to handle this situation in light of the positive phase 3 certolizumab results. Will the agency require other companies that market TNF inhibitors to mount a similarly rigorous 52-week, double-blind, placebo-controlled trial in order to obtain an expanded indication? That would seem to pose ethical issues now. Or will the companies be able to gain an expanded indication by retrospective analysis of outcomes in patients with nonradiographic SpA in their existing trials databases? Stay tuned.
Ixekizumab
The COAST-V trial was a phase 3, randomized, double-blind, active- and placebo-controlled trial of 341 patients with radiographic SpA who hadn’t previously been treated with a biologic disease-modifying antirheumatic drug. They were assigned to 80 mg of ixekizumab every 2 weeks, 80 mg every 4 weeks, adalimumab (Humira) at 40 mg every 2 weeks, or placebo. The primary endpoint – an ASAS 40 response at week 16 – was achieved in 52% of patients on ixekizumab every 2 weeks, 48% of those who received ixekizumab every 4 weeks, 36% on adalimumab, and 18% on placebo (Lancet. 2018 Dec 8;392[10163]:2441-51).
In contrast, the phase 3 COAST-W trial included 316 patients with radiographic SpA who were inadequate responders or intolerant to one or more prior anti-TNF agents. The 16-week ASAS 40 response rate was 30.6% with ixekizumab every 2 weeks, similar at 25.4% with ixekizumab every 4 weeks, and 12.5% with placebo (Arthritis Rheumatol. 2018 Oct 20. doi: 10.1002/art.40753).
While the COAST-V trial convincingly showed both ixekizumab and adalimumab were more effective than placebo, the patient numbers were way too small to draw any conclusions about the relative efficacy of the two biologics, according to Dr. Ruderman.
“I don’t think these results are surprising,” Dr. Kavanaugh commented. “It would have been surprising if ixekizumab was ineffective, given that secukinumab works. But it’s nice to have the proof.”
Bimekizumab
This investigational dual IL-17A/F inhibitor demonstrated efficacy and safety for SpA in a 297-patient, phase 2b trial presented at the 2018 European Congress of Rheumatology.
“It’s effective, but it doesn’t look like it’s particularly more effective than either of the existing IL-17A inhibitors. We’ll see going forward if there truly is an advantage here to the additional inhibition of IL-17F in this population. I will say that the preclinical and laboratory data on the potential advantages of IL-17F inhibition are mostly in the psoriasis/psoriatic arthritis space. It’s not clear in ankylosing spondyloarthritis specifically whether we should expect to see a difference,” Dr. Ruderman said.
Ustekinumab
The IL-12/23 inhibitor proved no better than placebo in patients with SpA in three separate phase 3, randomized trials recently published as a single summary article (Arthritis Rheumatol. 2019 Feb;71[2]:258-70).
“Ustekinumab was effective in an earlier open study, so I think everybody was surprised by this,” Dr. Kavanaugh said.
He reported serving as a consultant to and/or receiving research funding from a dozen pharmaceutical companies. Dr. Ruderman reported financial relationships with eight companies.
REPORTING FROM RWCS 2019
Possible mortality risk seen with tramadol in osteoarthritis
Tramadol appears to be associated with higher mortality risk among older patients with osteoarthritis when compared against common NSAIDs, according to findings from a study published online March 12 in JAMA.

The findings from the retrospective cohort study are worth noting despite their susceptibility to confounding by indication because “tramadol is a weak opioid agonist and has been considered a potential alternative to NSAIDs and traditional opioids because of its assumed relatively lower risk of serious cardiovascular and gastrointestinal adverse effects than NSAIDs, as well as a lower risk of addiction and respiratory depression compared with other opioids,” wrote Chao Zeng, MD, PhD, of Xiangya Hospital of Central South University, Changsha, China, and his coauthors.
The investigators analyzed data from a combined total of 88,902 individuals aged 50 years and older with knee, hip, or hand osteoarthritis who were seen during 2000-2015 and had visits recorded in the United Kingdom’s The Health Improvement Network (THIN) electronic medical records database. Participants were matched on sociodemographic and lifestyle factors, as well as osteoarthritis duration, comorbidities, other prescriptions, and health care utilization prior to the index date of the study.
Over 1 year of follow-up, researchers saw a 71% higher risk of all-cause mortality in patients taking tramadol than that in seen in those taking naproxen, 88% higher than in those taking diclofenac, 70% higher than in those taking celecoxib, and about twice as high as in patients taking etoricoxib.
However, there was no significant difference in risk of all-cause mortality between tramadol and codeine, the researchers found.
The authors suggested that tramadol may have adverse effects on the neurologic system by inhibiting central serotonin and norepinephrine uptake, which could potentially lead to serotonin syndrome. They also speculated that it could increase the risk of postoperative delirium, cause fatal poisoning or respiratory depression if taken in conjunction with alcohol or other drugs, or increase the risk of hypoglycemia, hyponatremia, fractures, or falls.
The numbers of deaths from cardiovascular, gastrointestinal, infection, cancer, and respiratory diseases were all higher in the tramadol group, compared with patients taking NSAIDs, but the differences were not statistically significant because of the relatively small number of deaths, the authors said.
Overall, 44,451 patients were taking tramadol, 12,397 were taking naproxen, 6,512 were taking diclofenac, 5,674 were taking celecoxib, 2,946 were taking etoricoxib, and 16,922 were taking codeine.
Patients in the tramadol cohort were generally older, with higher body mass index, a longer duration of osteoarthritis, and had a higher prevalence of comorbidities, higher health care utilization, and more prescriptions of other medications.
The authors noted that, while the patients from each medication cohort were matched on propensity score, the results were still susceptible to confounding by indication and should be interpreted with caution.
The study was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the National Natural Science Foundation of China. One author declared funding from the National Institute on Drug Abuse during the conduct of the study and grants from Optum Labs outside the study. No other conflicts of interest were declared.
SOURCE: Zeng C et al. JAMA. 2019;321:969-82.
Tramadol appears to be associated with higher mortality risk among older patients with osteoarthritis when compared against common NSAIDs, according to findings from a study published online March 12 in JAMA.
The findings from the retrospective cohort study are worth noting despite their susceptibility to confounding by indication because “tramadol is a weak opioid agonist and has been considered a potential alternative to NSAIDs and traditional opioids because of its assumed relatively lower risk of serious cardiovascular and gastrointestinal adverse effects than NSAIDs, as well as a lower risk of addiction and respiratory depression compared with other opioids,” wrote Chao Zeng, MD, PhD, of Xiangya Hospital of Central South University, Changsha, China, and his coauthors.
The investigators analyzed data from a combined total of 88,902 individuals aged 50 years and older with knee, hip, or hand osteoarthritis who were seen during 2000-2015 and had visits recorded in the United Kingdom’s The Health Improvement Network (THIN) electronic medical records database. Participants were matched on sociodemographic and lifestyle factors, as well as osteoarthritis duration, comorbidities, other prescriptions, and health care utilization prior to the index date of the study.
Over 1 year of follow-up, researchers saw a 71% higher risk of all-cause mortality in patients taking tramadol than that in seen in those taking naproxen, 88% higher than in those taking diclofenac, 70% higher than in those taking celecoxib, and about twice as high as in patients taking etoricoxib.
However, there was no significant difference in risk of all-cause mortality between tramadol and codeine, the researchers found.
The authors suggested that tramadol may have adverse effects on the neurologic system by inhibiting central serotonin and norepinephrine uptake, which could potentially lead to serotonin syndrome. They also speculated that it could increase the risk of postoperative delirium, cause fatal poisoning or respiratory depression if taken in conjunction with alcohol or other drugs, or increase the risk of hypoglycemia, hyponatremia, fractures, or falls.
The numbers of deaths from cardiovascular, gastrointestinal, infection, cancer, and respiratory diseases were all higher in the tramadol group, compared with patients taking NSAIDs, but the differences were not statistically significant because of the relatively small number of deaths, the authors said.
Overall, 44,451 patients were taking tramadol, 12,397 were taking naproxen, 6,512 were taking diclofenac, 5,674 were taking celecoxib, 2,946 were taking etoricoxib, and 16,922 were taking codeine.
Patients in the tramadol cohort were generally older, with higher body mass index, a longer duration of osteoarthritis, and had a higher prevalence of comorbidities, higher health care utilization, and more prescriptions of other medications.
The authors noted that, while the patients from each medication cohort were matched on propensity score, the results were still susceptible to confounding by indication and should be interpreted with caution.
The study was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the National Natural Science Foundation of China. One author declared funding from the National Institute on Drug Abuse during the conduct of the study and grants from Optum Labs outside the study. No other conflicts of interest were declared.
SOURCE: Zeng C et al. JAMA. 2019;321:969-82.
Tramadol appears to be associated with higher mortality risk among older patients with osteoarthritis when compared against common NSAIDs, according to findings from a study published online March 12 in JAMA.
The findings from the retrospective cohort study are worth noting despite their susceptibility to confounding by indication because “tramadol is a weak opioid agonist and has been considered a potential alternative to NSAIDs and traditional opioids because of its assumed relatively lower risk of serious cardiovascular and gastrointestinal adverse effects than NSAIDs, as well as a lower risk of addiction and respiratory depression compared with other opioids,” wrote Chao Zeng, MD, PhD, of Xiangya Hospital of Central South University, Changsha, China, and his coauthors.
The investigators analyzed data from a combined total of 88,902 individuals aged 50 years and older with knee, hip, or hand osteoarthritis who were seen during 2000-2015 and had visits recorded in the United Kingdom’s The Health Improvement Network (THIN) electronic medical records database. Participants were matched on sociodemographic and lifestyle factors, as well as osteoarthritis duration, comorbidities, other prescriptions, and health care utilization prior to the index date of the study.
Over 1 year of follow-up, researchers saw a 71% higher risk of all-cause mortality in patients taking tramadol than that in seen in those taking naproxen, 88% higher than in those taking diclofenac, 70% higher than in those taking celecoxib, and about twice as high as in patients taking etoricoxib.
However, there was no significant difference in risk of all-cause mortality between tramadol and codeine, the researchers found.
The authors suggested that tramadol may have adverse effects on the neurologic system by inhibiting central serotonin and norepinephrine uptake, which could potentially lead to serotonin syndrome. They also speculated that it could increase the risk of postoperative delirium, cause fatal poisoning or respiratory depression if taken in conjunction with alcohol or other drugs, or increase the risk of hypoglycemia, hyponatremia, fractures, or falls.
The numbers of deaths from cardiovascular, gastrointestinal, infection, cancer, and respiratory diseases were all higher in the tramadol group, compared with patients taking NSAIDs, but the differences were not statistically significant because of the relatively small number of deaths, the authors said.
Overall, 44,451 patients were taking tramadol, 12,397 were taking naproxen, 6,512 were taking diclofenac, 5,674 were taking celecoxib, 2,946 were taking etoricoxib, and 16,922 were taking codeine.
Patients in the tramadol cohort were generally older, with higher body mass index, a longer duration of osteoarthritis, and had a higher prevalence of comorbidities, higher health care utilization, and more prescriptions of other medications.
The authors noted that, while the patients from each medication cohort were matched on propensity score, the results were still susceptible to confounding by indication and should be interpreted with caution.
The study was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the National Natural Science Foundation of China. One author declared funding from the National Institute on Drug Abuse during the conduct of the study and grants from Optum Labs outside the study. No other conflicts of interest were declared.
SOURCE: Zeng C et al. JAMA. 2019;321:969-82.
FROM JAMA
Juvenile idiopathic arthritis: Old disease, new tactics
Juvenile idiopathic arthritis (JIA) is a clinically heterogeneous group of arthritides that are characterized by onset before 16 years of age and defined in part as lasting ≥6 weeks.1 Significantly, the etiology of JIA is unknown, making it a diagnosis of exclusion.2
The most common autoimmune condition of childhood, JIA has a prevalence of 3.8 to 400 affected children for every 100,000 people.3,4 As the leading cause of musculoskeletal disability in children,5 and comprising 7 categories of disease, JIA must be managed with appropriate initial and ongoing intervention.
The amalgam of care that a JIA patient requires—medical, social, physical, psychological—calls for a primary care physician’s expert ability to collaborate and coordinate with medical specialists and subspecialists, including rheumatology, ophthalmology, social work, physical and occupational therapy, and psychology. The goal? As this article describes, the goal is to provide prompt diagnosis, suitable and effective intervention, and continuity of care. (JIA is a lifelong disease, in many cases.)
How JIA is classifiedfor diagnosis and treatment
JIA comprises 7 categories, or classes.6 The scheme devised by the International League of Associations for Rheumatology (ILAR), now widely accepted, classifies JIA on the basis of clinical and biochemical markers that aid detection and treatment of the disorder, as well as research. (See “How efforts to classify JIA have caused confusion.”7-10) The ILAR classes (TABLE11) are:
- enthesitis-related arthritis (ERA)
- extended oligo-articular JIA (eoJIA), which involves ≤4 joints
- juvenile psoriatic arthritis (jPsA)
- rheumatoid factor (RF)-positive polyarticular JIA (RF+ pJIA)
- RF-negative polyarticular JIA (RF– pJIA)
- systemic-onset JIA (sJIA)
- undifferentiated JIA, which, generally, involves ≥4 joints.

SIDEBAR
How efforts to classiy JIA have caused confusion7-10
Various classifications of juvenile arthritis have been proposed and used over the past 3 decades. First was the American College of Rheumatology’s 1972 criteria for juvenile rheumatoid arthritis7; next came the European League against Rheumatism (EULAR) criteria for juvenile chronic arthritis, developed in 1977.8 Being contemporaneous, the 2 classifications led to a complicated, dichotomous definition of JIA among clinicians and researchers.
As a result of this disarray, the 1997 Durban, South Africa, meeting of the Pediatric Standing Committee of the International League of Associations for Rheumatology (ILAR)9 proposed that juvenile idiopathic arthritis be adopted as the umbrella term for the misunderstood terms juvenile rheumatoid arthritis and juvenile chronic arthritis. The intent of including “idiopathic” in the term was to acknowledge that the cause of these diseases was (and is still) unknown.
The novel classification proposed by the Pediatric Standing Committee was followed, in 2001, by an ILAR task force meeting in Edmonton, Alberta, Canada, on the classification of childhood arthritis. The outcome was a recommendation to add exclusion and inclusion criteria, to make all classes of JIA mutually exclusive.10 Most recently, as discussed in the body of this article, updated ILAR guidelines on JIA classification emphasize 1) heterogeneity among the 7 disease subtypes and 2) the fact that overlapping and exclusive features exist from class to class.
Updated guidelines regarding the 7 ILAR classes of JIA emphasize heterogeneity among disease subtypes, with overlapping and exclusive features noted from class to class.11
Extended oligo-articular JIA (27%-56%), pJIA (13%-35%), sJIA (4%-17%), and ERA,(3%-11%) are the most common JIA subtypes,12 with age of onset and sex predilection differing according to JIA class.11 The disease occurs more often in girls than in boys,11 and the predisposition is higher among Whites and Asians. The incidence of JIA (all classes taken together, for every 100,000 people) is: in Japan, 10 to 15 cases13; in Turkey, 64 cases14; in Norway, 65 cases15; and in the United States and Canada, taken together, 10 to 15 cases.16
What causes JIA?
The etiology of JIA remains unclear. It is known that the disease involves inflammation of the synovium and destruction of hard and soft tissues in joints.17 It has been postulated, therefore, that a combination of genetic, environmental, and immunogenic mechanisms might be responsible for JIA.
Continue to: For example, there is an increased...
For example, there is an increased frequency of autoimmune diseases among JIA patients.18 There are also reports documenting an increased rate of infection, including with enteric pathogens, parvovirus B,19 rubella, mumps, hepatitis B, Epstein-Barr virus, mycoplasma, and chlamydia.19 Stress and trauma have also been implicated.12
The T-lymphocyte percentage is increased in the synovial fluid of JIA patients, although that percentage varies from subtype to subtype.20 This elevation results in an increase in the number of macrophages, which are induced by secreted cytokines to produce interleukin (IL)-1, IL-6, and tumor necrosis factor alpha (TNF-a). This activity of cellular immunity leads to joint destruction.21
Clinical features
The most common signs and symptoms of JIA are arthralgias (39%), arthritis (25%), fever (18%), limping (9%), rash (8%), abdominal pain (1.3%), and uveitis (1.3%).15 Forty percent of JIA patients are reported to have temporomandibular joint involvement at some point in their life; mandibular asymmetry secondary to condylar resorption and remodeling17 is the most common presenting complaint—not arthralgia or pain, as would be expected.
Most JIA patients (52%) first present to the emergency department; another 42% present to the office of a general medical practitioner.15 On average, 3 visits to a physician, over the course of approximately 3 months, are made before a definitive diagnosis (usually by a pediatric rheumatologist) is made.15
Pertinent questions to ask a patient who has a confirmed diagnosis of JIA include the nature, severity, and duration of morning stiffness and pain, as well as any encumbering factors to regular functioning at home or school.22 Different scoring charts can be used to determine the extent of pain and disability, including the Juvenile Arthritis Disease Activity Score (JADAS)23 and the clinical JADAS (cJADAS),24 which measure minimal disease activity25 and clinically inactive disease26 cutoffs.
Continue to: Macrophage-activating syndrome increases risk of morbidity, mortality
Macrophage-activating syndrome increases risk of morbidity, mortality
An overactivation and expansion of T lymphocytes and macrophagic histiocytes with hemophagocytic activity, macrophage-activating syndrome (MAS) occurs in approximately 10% of JIA patients,27 increasing their risk of morbidity and mortality. The syndrome, which typically presents as fever, seizures, hypotension, purpura, hepatitis, splenomegaly, and occasionally, multisystem organ failure, is seen in 30% to 40% of sJIA patients; approximately 11% of them experience sudden death as a consequence.28
The clinical setting of MAS includes presenting symptoms of fever and a salmon-pink macular rash (FIGURE). For many sJIA patients with MAS, the diagnosis is made when laboratory results show hyperferritinemia, thrombocytopenia, anemia, leukopenia, coagulopathy, and elevated levels of C-reactive protein and D-dimer.27

Different classes, different features
The following clinical profiles have been documented in different classes of JIA:
Systemic JIA presents with intermittent fever of at least 2 weeks’ duration, arthritis, and occasionally, a rash.
Extended oligo-articular JIA involves pain, in a mono-articular lower-extremity joint, that can develop suddenly or insidiously, and is characterized by early-morning stiffness and uveitis (especially in early-onset, antinuclear antibody-positive JIA patients).
Continue to: Poly-articular JIA
Poly-articular JIA patients present with mild fever, weight loss, and anemia.
Enthesis-related arthritis patients have findings of enthesopathy; asymmetric arthritis of the lower extremities, particularly the Achilles tendon29; and recurrent acute, symptomatic iridocyclitis.30
Juvenile psoriatic arthritis can involve any joint but is readily differentiated from pJIA by involvement of distal interphalangeal joints and psoriatic skin and nail changes.29
Investigations
Imaging
Radiography is still the most widely used imaging tool for making the diagnosis of JIA. Plain films demonstrate structural joint damage and disturbances of growth and maturation in bones. Radiography has poor sensitivity for detecting acute synovitis and limited utility in visualizing erosion changes early in the course of disease, however, which has led to increased use of ultrasonography (US) and contrast-enhanced magnetic resonance imaging (MRI) to diagnose JIA.30
Contrast-enhanced MRI is superior to US for detecting early inflammation and monitoring subsequent joint disease. Of course, MRI is more expensive than US, and less widely available. Other imaging options are computed tomography and positron emission tomography, but these scans are not as sensitive as contrast-enhanced MRI and have the disadvantage of radiation exposure (in the former) and cost (in the latter).
Continue to: Laboratory testing
Laboratory testing
No diagnostic tests for JIA exist. Assays of acute-phase reactants, including C-reactive protein, the erythrocyte sedimentation rate, and serum amyloid-A proteins, can be utilized to demonstrate inflammation but not to confirm the diagnosis. For some classes of JIA, various tests, including rheumatoid factor, antinuclear antibody, human leukocyte antigen B-27, and cyclic citrullated peptide antibodies, can be used to confirm a specific class but, again, are not recommended for confirming JIA.6
The complete blood count, blood cultures, and tests of uric acid and lactate dehydrogenase can be ordered during treatment to monitor for complications, such as malignancy, infection, MAS, and sepsis.
Treatment is based on disease class
Nonsteroidal anti-inflammatory drugs (NSAIDs) and intra-articular steroids are used in all JIA classes, as an adjunct to class-specific treatment, or as induction agents.31 These therapies, although they alleviate acute signs and symptoms, such as pain, inflammation, swelling and joint contractures, are not useful for long-term treatment of JIA because they do not halt disease progression.
Systemic steroids can be utilized in exceptional cases, including chronic uveitis with arthritis or in patients with destructive arthritis and poor prognostic features, including cyclic citrullated peptide antibodies, positive RF, erosions, and joint-space narrowing.32
Other drugs. Options include traditional disease-modifying anti-rheumatic drugs (csDMARDs), such as methotrexate and leflunomide; biologic agents, such as TNF-a inhibitors (eg, etanercept, adalimumab, and infliximab); and anti-IL monoclonal antibody drugs (eg, the IL-6 inhibitor tocilizumab and IL-1 inhibitors anakinra, and canakinumab).31 Indications by class include:
- csDMARDs as first-line therapy in persistent eoJIA and pJIA;
- TNF-Symbolα inhibitors for refractory eoJIA and for pJIA episodes31;
- tocilizumab, recommended for sJIA patients who have persistent systemic signs; and
- anakinra and canakinumab for refractory SJIA patients.32
Continue to: Failure
Failure
When treatment of JIA fails with a given drug, options include increasing the dosage; switching to another agent in the same drug class; switching to a different class; and combining an NSAID with a csDMARD or a biologic agent.32 In class-specific JIA cases, a change in a drug regimen is warranted on the basis of the evidence-based historical clinical response rate.32
What is the prognosis?
Treatment of JIA with novel agents, such as biologics, has opened up the possibility that JIA patients can live not just with suppressed symptoms but immunologically inactive disease. This is the result of better understanding of the pathogenesis of JIA and the mechanism of action of targeted drugs, and identification of biomarkers that are helpful in predicting prognosis, adverse effects, and response to treatment.
JIA is often a lifelong disease; one-third of patients continue to exhibit symptoms into adulthood.4 If their disease is properly managed, however, these patients do not develop typical features of rheumatoid arthritis, including hand, limb, and spine deformities. Last, patients with JIA who have only intermittent disease tend to do better over the long term than those whose disease is continual.32
The mortality rate of JIA has dropped: from 1% to 4% in the mid-1970s to 0.3% to 1% today4—an improvement in life expectancy that is echoed in enhanced quality of life for patients. According to the 4-level Steinbrocker functional classification scale33 (used to rate the extent of physical disability), 15% of JIA patients were Class III (limited to few or no activities of the patient’s usual occupation) or Class IV (bedridden with little or no self-care) in the period from 1976 to 1994—a percentage that had declined to 5% by 2002.34
The family physician plays pivotal role in JIA care
For the family physician, appropriate initial intervention in the management of JIA is imperative. This includes ordering imaging (whether plain films or MRI), laboratory tests as described earlier (although not to make the diagnosis), and the use of NSAIDs, intra-articular steroids, and other induction agents. Once the diagnosis is made, and a drug regimen is put in place, you will need to monitor for adverse effects. This monitoring will need to occur when a patient is escalated to csDMARDs, biological agents, or systemic steroids; is maintained on an NSAID; or is placed on a combination regimen.
Continue to: Before beginning therapy with a biologic agent...
Before beginning therapy with a biologic agent, it’s important to screen for hepatitis B, hepatitis C, human immunodeficiency virus infection, tuberculosis, and fungal infection (eg, Histoplasma capsulatum, Coccidioides immitis32). Be sure to make a timely referral to the ophthalmology service for a bi-annual eye exam and, in the event that surgery is necessary, conduct a preoperative evaluation, with the knowledge of how long before surgery a biologic agent must be withheld (duration varies by drug).32
CORRESPONDENCE
Tobe Momah, MD, Department of Family Medicine, Clinical Science Building, 4th Floor, University of Mississippi Medical Center, 2500 North State Street, Jackson, MS 39216; tmomah@umc.edu.
1. Adriano LS, de França Fonteles MM, de Fátima Menezes Azevedo M, et al. Medication adherence in patients with juvenile idiopathic arthritis. Rev Bras Reumatol Engl Ed. 2017;57:23-29.
2. Akioka S. A better understanding of juvenile idiopathic arthritis with classification criteria. Nihon Rinsho Meneki Gakkai Kaishi. 2016;39:513-521.
3. Thierry S, Fautrel B, Lemelle I, Guillemin F. Prevalence and incidence of juvenile idiopathic arthritis: a systematic review. Joint Bone Spine. 2014;81:112-117.
4. Petty RE, Laxer RM, Lindsley CB, et al. Pediatric Rheumatology. Philadelphia, PA: Elsevier; 2016:188-201.e6.
5. Scott C, Brice N. Juvenile idiopathic arthritis–an update on its diagnosis and management. S Afr Med J. 2015;105:1077.
6. Giancane G, Consolaro A, Lanni S, et al. Juvenile idiopathic arthritis: diagnosis and treatment. Rheumatol Ther. 2016;3:187-207.
7. Criteria for the classification of juvenile rheumatoid arthritis. Bull Rheum Dis. 1972;23:712-719.
8. Wood PHN: Special meeting on nomenclature and classification of arthritis in children. In: Munthe E, ed. The Care of Rheumatic Children. Basel, Switzerland: EULAR Publishers; 1978:47-50.
9. Petty RE, Southwood TR, Baum J, et al. Revision of the proposed classification criteria for juvenile idiopathic arthritis: Durban, 1997. J Rheumatol. 1998;25:1991-1994.
10. Petty RE, Southwood TR, Manners P, et al; International League of Associations for Rheumatology. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31:390-392.
11. Basra HAS, Humphries PD. Juvenile idiopathic arthritis: what is the utility of ultrasound? Br J Radiol. 2017;90:20160920.
12. Weiss J, Ilowite NT. Juvenile idiopathic arthritis. Pediatr Clin North Am. 2005;52:413-442, vi.
13. Fujikawa S, Okuni M. A nationwide surveillance study of rheumatic diseases among Japanese children. Acta Pediatric Jpn. 1997:39:242-244.
14. Ozen S, Karaaslan Y, Ozdemir O, et al. Prevalence of juvenile chronic arthritis and familial Mediterranean fever in Turkey: a field study. J Rheumatol. 1998;25:2445-2449.
15. Aoust L, Rossi-Semerano L, Koné-PauL I, et al. Time to diagnosis in juvenile idiopathic arthritis: a French perspective. Orphanet J Rare Dis. 2017;12:43.
16. Moe N, Rygg M. Epidemiology of juvenile chronic arthritis in northern Norway; a ten-year retrospective study. Clin Exp Rheumatol. 1998;16:99-101.
17. Abramowicz S, Kim S, Prahalad S, et al. Juvenile arthritis: current concepts in terminology, etiopathogenesis, diagnosis, and management. Int J Oral Maxillofac Surg. 2016;45:801-812.
18. Prahalad S, Shear ES, Thompson SD, et al. Increased prevalence of familial autoimmunity in simplex and multiplex families with juvenile rheumatoid arthritis. Arthritis Rheum. 2002;46:1851-1856.
19. Gonzalez B, Larrañaga C, León O, et al. Parvovirus B19 may have a role in the pathogenesis of juvenile idiopathic arthritis. J Rheumatol. 2007;34:1336-1340.
20. Prakken B, Albani S, Martini A. Juvenile idiopathic arthritis. Lancet. 2011;377:2138-2149.
21. Zhou J, Ding Y, Zhang Y, et al. CD3+CD56+ natural killer T cell activity in children with different forms of juvenile idiopathic arthritis and the influence of etanercept treatment on polyarticular subgroup. Clin Immunol. 2016;176:1-11.
22. Shoop-Worrall SJW, Verstappen SMM, Baildam E, et al. How common is clinically inactive disease in a prospective cohort of patients with juvenile idiopathic arthritis? The importance of definition. Ann Rheum Dis. 2017;0:1-8.
23. Nordal EB, Zak M, Berntson L, et al. Juvenile Arthritis Disease Activity Score (JADAS) based on CRP; validity and predictive ability in a Nordic population-based setting. Pediatr Rheumatol Online J. 2011;9(suppl 1):155.
24. Swart JF, Dijkhuizen EHP, Wulffraat NM, et al. Clinical Juvenile Arthritis Disease Activity Score proves to be a useful tool in treat-to-target therapy in juvenile idiopathic arthritis. Ann Rheum Dis. 2018;77:336-342.
25. Horneff G, Klein A, Ganser G, et al. Protocols on classification, monitoring and therapy in children’s rheumatology (PRO-KIND): results of the working group polyarticular juvenile idiopathic arthritis. Pediatr Rheumatol Online J. 2017;15:78.
26. Shoop-Worrall SJW, Verstappen SMM, McDonagh JE, et al. Long‐term outcomes following achievement of clinically inactive disease in juvenile idiopathic arthritis. Arthritis Rheumatol. 2018;70:1519-1529.
27. Ahn SS, Yoo BW, Jung SM, et al. In-hospital mortality in febrile lupus patients based on 2016 EULAR/ACR/PRINTO classification criteria for macrophage activation syndrome. Sem Arthritis Rheum. 2017;.47:216-221.
28. Yokota S, Mori M, Imagawa T, et al. Proposal for juvenile idiopathic arthritis guidance on diagnosis and treatment for primary care pediatricians and nonpediatric rheumatologists (2007). Mod Rheumatol. 2007;17:353-363.
29. Barut K, Adrovic A, Şahin S, et al. Juvenile idiopathic arthritis. Balkan Med J. 2017;34:90-101.
30. Colebatch-Bourn AN, Edwards CJ, et al. EULAR-PReS points to consider for the use of imaging in the diagnosis and management of juvenile idiopathic arthritis in clinical practice. Ann Rheum Dis. 2015;74:1946-1957.
31. Blazina Š, Markelj G, AvramoviČ MZ, et al. Management of juvenile idiopathic arthritis: a clinical guide. Pediatr Drugs. 2016;18:397-412.
32. Santos MJ, Conde M, Mourão AF, et al. 2016 update of the Portuguese recommendations for the use of biologic therapies in children and adolescents with juvenile idiopathic arthritis. Acta Rheumatol Port. 2016;41:194-212.
33. Steinbrocker 0, Traeger CH, Batterman RC. Therapeutic criteria in rheumatoid arthritis. JAMA. 1949;140:659-662.
34. Oen K, Malleson PN, Cabral D, et al. Disease course and outcome of juvenile rheumatoid arthritis in a multicenter cohort. J Rheumatol. 2002;29:1989-1999.
Juvenile idiopathic arthritis (JIA) is a clinically heterogeneous group of arthritides that are characterized by onset before 16 years of age and defined in part as lasting ≥6 weeks.1 Significantly, the etiology of JIA is unknown, making it a diagnosis of exclusion.2
The most common autoimmune condition of childhood, JIA has a prevalence of 3.8 to 400 affected children for every 100,000 people.3,4 As the leading cause of musculoskeletal disability in children,5 and comprising 7 categories of disease, JIA must be managed with appropriate initial and ongoing intervention.
The amalgam of care that a JIA patient requires—medical, social, physical, psychological—calls for a primary care physician’s expert ability to collaborate and coordinate with medical specialists and subspecialists, including rheumatology, ophthalmology, social work, physical and occupational therapy, and psychology. The goal? As this article describes, the goal is to provide prompt diagnosis, suitable and effective intervention, and continuity of care. (JIA is a lifelong disease, in many cases.)
How JIA is classifiedfor diagnosis and treatment
JIA comprises 7 categories, or classes.6 The scheme devised by the International League of Associations for Rheumatology (ILAR), now widely accepted, classifies JIA on the basis of clinical and biochemical markers that aid detection and treatment of the disorder, as well as research. (See “How efforts to classify JIA have caused confusion.”7-10) The ILAR classes (TABLE11) are:
- enthesitis-related arthritis (ERA)
- extended oligo-articular JIA (eoJIA), which involves ≤4 joints
- juvenile psoriatic arthritis (jPsA)
- rheumatoid factor (RF)-positive polyarticular JIA (RF+ pJIA)
- RF-negative polyarticular JIA (RF– pJIA)
- systemic-onset JIA (sJIA)
- undifferentiated JIA, which, generally, involves ≥4 joints.
SIDEBAR
How efforts to classiy JIA have caused confusion7-10
Various classifications of juvenile arthritis have been proposed and used over the past 3 decades. First was the American College of Rheumatology’s 1972 criteria for juvenile rheumatoid arthritis7; next came the European League against Rheumatism (EULAR) criteria for juvenile chronic arthritis, developed in 1977.8 Being contemporaneous, the 2 classifications led to a complicated, dichotomous definition of JIA among clinicians and researchers.
As a result of this disarray, the 1997 Durban, South Africa, meeting of the Pediatric Standing Committee of the International League of Associations for Rheumatology (ILAR)9 proposed that juvenile idiopathic arthritis be adopted as the umbrella term for the misunderstood terms juvenile rheumatoid arthritis and juvenile chronic arthritis. The intent of including “idiopathic” in the term was to acknowledge that the cause of these diseases was (and is still) unknown.
The novel classification proposed by the Pediatric Standing Committee was followed, in 2001, by an ILAR task force meeting in Edmonton, Alberta, Canada, on the classification of childhood arthritis. The outcome was a recommendation to add exclusion and inclusion criteria, to make all classes of JIA mutually exclusive.10 Most recently, as discussed in the body of this article, updated ILAR guidelines on JIA classification emphasize 1) heterogeneity among the 7 disease subtypes and 2) the fact that overlapping and exclusive features exist from class to class.
Updated guidelines regarding the 7 ILAR classes of JIA emphasize heterogeneity among disease subtypes, with overlapping and exclusive features noted from class to class.11
Extended oligo-articular JIA (27%-56%), pJIA (13%-35%), sJIA (4%-17%), and ERA,(3%-11%) are the most common JIA subtypes,12 with age of onset and sex predilection differing according to JIA class.11 The disease occurs more often in girls than in boys,11 and the predisposition is higher among Whites and Asians. The incidence of JIA (all classes taken together, for every 100,000 people) is: in Japan, 10 to 15 cases13; in Turkey, 64 cases14; in Norway, 65 cases15; and in the United States and Canada, taken together, 10 to 15 cases.16
What causes JIA?
The etiology of JIA remains unclear. It is known that the disease involves inflammation of the synovium and destruction of hard and soft tissues in joints.17 It has been postulated, therefore, that a combination of genetic, environmental, and immunogenic mechanisms might be responsible for JIA.
Continue to: For example, there is an increased...
For example, there is an increased frequency of autoimmune diseases among JIA patients.18 There are also reports documenting an increased rate of infection, including with enteric pathogens, parvovirus B,19 rubella, mumps, hepatitis B, Epstein-Barr virus, mycoplasma, and chlamydia.19 Stress and trauma have also been implicated.12
The T-lymphocyte percentage is increased in the synovial fluid of JIA patients, although that percentage varies from subtype to subtype.20 This elevation results in an increase in the number of macrophages, which are induced by secreted cytokines to produce interleukin (IL)-1, IL-6, and tumor necrosis factor alpha (TNF-a). This activity of cellular immunity leads to joint destruction.21
Clinical features
The most common signs and symptoms of JIA are arthralgias (39%), arthritis (25%), fever (18%), limping (9%), rash (8%), abdominal pain (1.3%), and uveitis (1.3%).15 Forty percent of JIA patients are reported to have temporomandibular joint involvement at some point in their life; mandibular asymmetry secondary to condylar resorption and remodeling17 is the most common presenting complaint—not arthralgia or pain, as would be expected.
Most JIA patients (52%) first present to the emergency department; another 42% present to the office of a general medical practitioner.15 On average, 3 visits to a physician, over the course of approximately 3 months, are made before a definitive diagnosis (usually by a pediatric rheumatologist) is made.15
Pertinent questions to ask a patient who has a confirmed diagnosis of JIA include the nature, severity, and duration of morning stiffness and pain, as well as any encumbering factors to regular functioning at home or school.22 Different scoring charts can be used to determine the extent of pain and disability, including the Juvenile Arthritis Disease Activity Score (JADAS)23 and the clinical JADAS (cJADAS),24 which measure minimal disease activity25 and clinically inactive disease26 cutoffs.
Continue to: Macrophage-activating syndrome increases risk of morbidity, mortality
Macrophage-activating syndrome increases risk of morbidity, mortality
An overactivation and expansion of T lymphocytes and macrophagic histiocytes with hemophagocytic activity, macrophage-activating syndrome (MAS) occurs in approximately 10% of JIA patients,27 increasing their risk of morbidity and mortality. The syndrome, which typically presents as fever, seizures, hypotension, purpura, hepatitis, splenomegaly, and occasionally, multisystem organ failure, is seen in 30% to 40% of sJIA patients; approximately 11% of them experience sudden death as a consequence.28
The clinical setting of MAS includes presenting symptoms of fever and a salmon-pink macular rash (FIGURE). For many sJIA patients with MAS, the diagnosis is made when laboratory results show hyperferritinemia, thrombocytopenia, anemia, leukopenia, coagulopathy, and elevated levels of C-reactive protein and D-dimer.27
Different classes, different features
The following clinical profiles have been documented in different classes of JIA:
Systemic JIA presents with intermittent fever of at least 2 weeks’ duration, arthritis, and occasionally, a rash.
Extended oligo-articular JIA involves pain, in a mono-articular lower-extremity joint, that can develop suddenly or insidiously, and is characterized by early-morning stiffness and uveitis (especially in early-onset, antinuclear antibody-positive JIA patients).
Continue to: Poly-articular JIA
Poly-articular JIA patients present with mild fever, weight loss, and anemia.
Enthesis-related arthritis patients have findings of enthesopathy; asymmetric arthritis of the lower extremities, particularly the Achilles tendon29; and recurrent acute, symptomatic iridocyclitis.30
Juvenile psoriatic arthritis can involve any joint but is readily differentiated from pJIA by involvement of distal interphalangeal joints and psoriatic skin and nail changes.29
Investigations
Imaging
Radiography is still the most widely used imaging tool for making the diagnosis of JIA. Plain films demonstrate structural joint damage and disturbances of growth and maturation in bones. Radiography has poor sensitivity for detecting acute synovitis and limited utility in visualizing erosion changes early in the course of disease, however, which has led to increased use of ultrasonography (US) and contrast-enhanced magnetic resonance imaging (MRI) to diagnose JIA.30
Contrast-enhanced MRI is superior to US for detecting early inflammation and monitoring subsequent joint disease. Of course, MRI is more expensive than US, and less widely available. Other imaging options are computed tomography and positron emission tomography, but these scans are not as sensitive as contrast-enhanced MRI and have the disadvantage of radiation exposure (in the former) and cost (in the latter).
Continue to: Laboratory testing
Laboratory testing
No diagnostic tests for JIA exist. Assays of acute-phase reactants, including C-reactive protein, the erythrocyte sedimentation rate, and serum amyloid-A proteins, can be utilized to demonstrate inflammation but not to confirm the diagnosis. For some classes of JIA, various tests, including rheumatoid factor, antinuclear antibody, human leukocyte antigen B-27, and cyclic citrullated peptide antibodies, can be used to confirm a specific class but, again, are not recommended for confirming JIA.6
The complete blood count, blood cultures, and tests of uric acid and lactate dehydrogenase can be ordered during treatment to monitor for complications, such as malignancy, infection, MAS, and sepsis.
Treatment is based on disease class
Nonsteroidal anti-inflammatory drugs (NSAIDs) and intra-articular steroids are used in all JIA classes, as an adjunct to class-specific treatment, or as induction agents.31 These therapies, although they alleviate acute signs and symptoms, such as pain, inflammation, swelling and joint contractures, are not useful for long-term treatment of JIA because they do not halt disease progression.
Systemic steroids can be utilized in exceptional cases, including chronic uveitis with arthritis or in patients with destructive arthritis and poor prognostic features, including cyclic citrullated peptide antibodies, positive RF, erosions, and joint-space narrowing.32
Other drugs. Options include traditional disease-modifying anti-rheumatic drugs (csDMARDs), such as methotrexate and leflunomide; biologic agents, such as TNF-a inhibitors (eg, etanercept, adalimumab, and infliximab); and anti-IL monoclonal antibody drugs (eg, the IL-6 inhibitor tocilizumab and IL-1 inhibitors anakinra, and canakinumab).31 Indications by class include:
- csDMARDs as first-line therapy in persistent eoJIA and pJIA;
- TNF-Symbolα inhibitors for refractory eoJIA and for pJIA episodes31;
- tocilizumab, recommended for sJIA patients who have persistent systemic signs; and
- anakinra and canakinumab for refractory SJIA patients.32
Continue to: Failure
Failure
When treatment of JIA fails with a given drug, options include increasing the dosage; switching to another agent in the same drug class; switching to a different class; and combining an NSAID with a csDMARD or a biologic agent.32 In class-specific JIA cases, a change in a drug regimen is warranted on the basis of the evidence-based historical clinical response rate.32
What is the prognosis?
Treatment of JIA with novel agents, such as biologics, has opened up the possibility that JIA patients can live not just with suppressed symptoms but immunologically inactive disease. This is the result of better understanding of the pathogenesis of JIA and the mechanism of action of targeted drugs, and identification of biomarkers that are helpful in predicting prognosis, adverse effects, and response to treatment.
JIA is often a lifelong disease; one-third of patients continue to exhibit symptoms into adulthood.4 If their disease is properly managed, however, these patients do not develop typical features of rheumatoid arthritis, including hand, limb, and spine deformities. Last, patients with JIA who have only intermittent disease tend to do better over the long term than those whose disease is continual.32
The mortality rate of JIA has dropped: from 1% to 4% in the mid-1970s to 0.3% to 1% today4—an improvement in life expectancy that is echoed in enhanced quality of life for patients. According to the 4-level Steinbrocker functional classification scale33 (used to rate the extent of physical disability), 15% of JIA patients were Class III (limited to few or no activities of the patient’s usual occupation) or Class IV (bedridden with little or no self-care) in the period from 1976 to 1994—a percentage that had declined to 5% by 2002.34
The family physician plays pivotal role in JIA care
For the family physician, appropriate initial intervention in the management of JIA is imperative. This includes ordering imaging (whether plain films or MRI), laboratory tests as described earlier (although not to make the diagnosis), and the use of NSAIDs, intra-articular steroids, and other induction agents. Once the diagnosis is made, and a drug regimen is put in place, you will need to monitor for adverse effects. This monitoring will need to occur when a patient is escalated to csDMARDs, biological agents, or systemic steroids; is maintained on an NSAID; or is placed on a combination regimen.
Continue to: Before beginning therapy with a biologic agent...
Before beginning therapy with a biologic agent, it’s important to screen for hepatitis B, hepatitis C, human immunodeficiency virus infection, tuberculosis, and fungal infection (eg, Histoplasma capsulatum, Coccidioides immitis32). Be sure to make a timely referral to the ophthalmology service for a bi-annual eye exam and, in the event that surgery is necessary, conduct a preoperative evaluation, with the knowledge of how long before surgery a biologic agent must be withheld (duration varies by drug).32
CORRESPONDENCE
Tobe Momah, MD, Department of Family Medicine, Clinical Science Building, 4th Floor, University of Mississippi Medical Center, 2500 North State Street, Jackson, MS 39216; tmomah@umc.edu.
Juvenile idiopathic arthritis (JIA) is a clinically heterogeneous group of arthritides that are characterized by onset before 16 years of age and defined in part as lasting ≥6 weeks.1 Significantly, the etiology of JIA is unknown, making it a diagnosis of exclusion.2
The most common autoimmune condition of childhood, JIA has a prevalence of 3.8 to 400 affected children for every 100,000 people.3,4 As the leading cause of musculoskeletal disability in children,5 and comprising 7 categories of disease, JIA must be managed with appropriate initial and ongoing intervention.
The amalgam of care that a JIA patient requires—medical, social, physical, psychological—calls for a primary care physician’s expert ability to collaborate and coordinate with medical specialists and subspecialists, including rheumatology, ophthalmology, social work, physical and occupational therapy, and psychology. The goal? As this article describes, the goal is to provide prompt diagnosis, suitable and effective intervention, and continuity of care. (JIA is a lifelong disease, in many cases.)
How JIA is classifiedfor diagnosis and treatment
JIA comprises 7 categories, or classes.6 The scheme devised by the International League of Associations for Rheumatology (ILAR), now widely accepted, classifies JIA on the basis of clinical and biochemical markers that aid detection and treatment of the disorder, as well as research. (See “How efforts to classify JIA have caused confusion.”7-10) The ILAR classes (TABLE11) are:
- enthesitis-related arthritis (ERA)
- extended oligo-articular JIA (eoJIA), which involves ≤4 joints
- juvenile psoriatic arthritis (jPsA)
- rheumatoid factor (RF)-positive polyarticular JIA (RF+ pJIA)
- RF-negative polyarticular JIA (RF– pJIA)
- systemic-onset JIA (sJIA)
- undifferentiated JIA, which, generally, involves ≥4 joints.
SIDEBAR
How efforts to classiy JIA have caused confusion7-10
Various classifications of juvenile arthritis have been proposed and used over the past 3 decades. First was the American College of Rheumatology’s 1972 criteria for juvenile rheumatoid arthritis7; next came the European League against Rheumatism (EULAR) criteria for juvenile chronic arthritis, developed in 1977.8 Being contemporaneous, the 2 classifications led to a complicated, dichotomous definition of JIA among clinicians and researchers.
As a result of this disarray, the 1997 Durban, South Africa, meeting of the Pediatric Standing Committee of the International League of Associations for Rheumatology (ILAR)9 proposed that juvenile idiopathic arthritis be adopted as the umbrella term for the misunderstood terms juvenile rheumatoid arthritis and juvenile chronic arthritis. The intent of including “idiopathic” in the term was to acknowledge that the cause of these diseases was (and is still) unknown.
The novel classification proposed by the Pediatric Standing Committee was followed, in 2001, by an ILAR task force meeting in Edmonton, Alberta, Canada, on the classification of childhood arthritis. The outcome was a recommendation to add exclusion and inclusion criteria, to make all classes of JIA mutually exclusive.10 Most recently, as discussed in the body of this article, updated ILAR guidelines on JIA classification emphasize 1) heterogeneity among the 7 disease subtypes and 2) the fact that overlapping and exclusive features exist from class to class.
Updated guidelines regarding the 7 ILAR classes of JIA emphasize heterogeneity among disease subtypes, with overlapping and exclusive features noted from class to class.11
Extended oligo-articular JIA (27%-56%), pJIA (13%-35%), sJIA (4%-17%), and ERA,(3%-11%) are the most common JIA subtypes,12 with age of onset and sex predilection differing according to JIA class.11 The disease occurs more often in girls than in boys,11 and the predisposition is higher among Whites and Asians. The incidence of JIA (all classes taken together, for every 100,000 people) is: in Japan, 10 to 15 cases13; in Turkey, 64 cases14; in Norway, 65 cases15; and in the United States and Canada, taken together, 10 to 15 cases.16
What causes JIA?
The etiology of JIA remains unclear. It is known that the disease involves inflammation of the synovium and destruction of hard and soft tissues in joints.17 It has been postulated, therefore, that a combination of genetic, environmental, and immunogenic mechanisms might be responsible for JIA.
Continue to: For example, there is an increased...
For example, there is an increased frequency of autoimmune diseases among JIA patients.18 There are also reports documenting an increased rate of infection, including with enteric pathogens, parvovirus B,19 rubella, mumps, hepatitis B, Epstein-Barr virus, mycoplasma, and chlamydia.19 Stress and trauma have also been implicated.12
The T-lymphocyte percentage is increased in the synovial fluid of JIA patients, although that percentage varies from subtype to subtype.20 This elevation results in an increase in the number of macrophages, which are induced by secreted cytokines to produce interleukin (IL)-1, IL-6, and tumor necrosis factor alpha (TNF-a). This activity of cellular immunity leads to joint destruction.21
Clinical features
The most common signs and symptoms of JIA are arthralgias (39%), arthritis (25%), fever (18%), limping (9%), rash (8%), abdominal pain (1.3%), and uveitis (1.3%).15 Forty percent of JIA patients are reported to have temporomandibular joint involvement at some point in their life; mandibular asymmetry secondary to condylar resorption and remodeling17 is the most common presenting complaint—not arthralgia or pain, as would be expected.
Most JIA patients (52%) first present to the emergency department; another 42% present to the office of a general medical practitioner.15 On average, 3 visits to a physician, over the course of approximately 3 months, are made before a definitive diagnosis (usually by a pediatric rheumatologist) is made.15
Pertinent questions to ask a patient who has a confirmed diagnosis of JIA include the nature, severity, and duration of morning stiffness and pain, as well as any encumbering factors to regular functioning at home or school.22 Different scoring charts can be used to determine the extent of pain and disability, including the Juvenile Arthritis Disease Activity Score (JADAS)23 and the clinical JADAS (cJADAS),24 which measure minimal disease activity25 and clinically inactive disease26 cutoffs.
Continue to: Macrophage-activating syndrome increases risk of morbidity, mortality
Macrophage-activating syndrome increases risk of morbidity, mortality
An overactivation and expansion of T lymphocytes and macrophagic histiocytes with hemophagocytic activity, macrophage-activating syndrome (MAS) occurs in approximately 10% of JIA patients,27 increasing their risk of morbidity and mortality. The syndrome, which typically presents as fever, seizures, hypotension, purpura, hepatitis, splenomegaly, and occasionally, multisystem organ failure, is seen in 30% to 40% of sJIA patients; approximately 11% of them experience sudden death as a consequence.28
The clinical setting of MAS includes presenting symptoms of fever and a salmon-pink macular rash (FIGURE). For many sJIA patients with MAS, the diagnosis is made when laboratory results show hyperferritinemia, thrombocytopenia, anemia, leukopenia, coagulopathy, and elevated levels of C-reactive protein and D-dimer.27
Different classes, different features
The following clinical profiles have been documented in different classes of JIA:
Systemic JIA presents with intermittent fever of at least 2 weeks’ duration, arthritis, and occasionally, a rash.
Extended oligo-articular JIA involves pain, in a mono-articular lower-extremity joint, that can develop suddenly or insidiously, and is characterized by early-morning stiffness and uveitis (especially in early-onset, antinuclear antibody-positive JIA patients).
Continue to: Poly-articular JIA
Poly-articular JIA patients present with mild fever, weight loss, and anemia.
Enthesis-related arthritis patients have findings of enthesopathy; asymmetric arthritis of the lower extremities, particularly the Achilles tendon29; and recurrent acute, symptomatic iridocyclitis.30
Juvenile psoriatic arthritis can involve any joint but is readily differentiated from pJIA by involvement of distal interphalangeal joints and psoriatic skin and nail changes.29
Investigations
Imaging
Radiography is still the most widely used imaging tool for making the diagnosis of JIA. Plain films demonstrate structural joint damage and disturbances of growth and maturation in bones. Radiography has poor sensitivity for detecting acute synovitis and limited utility in visualizing erosion changes early in the course of disease, however, which has led to increased use of ultrasonography (US) and contrast-enhanced magnetic resonance imaging (MRI) to diagnose JIA.30
Contrast-enhanced MRI is superior to US for detecting early inflammation and monitoring subsequent joint disease. Of course, MRI is more expensive than US, and less widely available. Other imaging options are computed tomography and positron emission tomography, but these scans are not as sensitive as contrast-enhanced MRI and have the disadvantage of radiation exposure (in the former) and cost (in the latter).
Continue to: Laboratory testing
Laboratory testing
No diagnostic tests for JIA exist. Assays of acute-phase reactants, including C-reactive protein, the erythrocyte sedimentation rate, and serum amyloid-A proteins, can be utilized to demonstrate inflammation but not to confirm the diagnosis. For some classes of JIA, various tests, including rheumatoid factor, antinuclear antibody, human leukocyte antigen B-27, and cyclic citrullated peptide antibodies, can be used to confirm a specific class but, again, are not recommended for confirming JIA.6
The complete blood count, blood cultures, and tests of uric acid and lactate dehydrogenase can be ordered during treatment to monitor for complications, such as malignancy, infection, MAS, and sepsis.
Treatment is based on disease class
Nonsteroidal anti-inflammatory drugs (NSAIDs) and intra-articular steroids are used in all JIA classes, as an adjunct to class-specific treatment, or as induction agents.31 These therapies, although they alleviate acute signs and symptoms, such as pain, inflammation, swelling and joint contractures, are not useful for long-term treatment of JIA because they do not halt disease progression.
Systemic steroids can be utilized in exceptional cases, including chronic uveitis with arthritis or in patients with destructive arthritis and poor prognostic features, including cyclic citrullated peptide antibodies, positive RF, erosions, and joint-space narrowing.32
Other drugs. Options include traditional disease-modifying anti-rheumatic drugs (csDMARDs), such as methotrexate and leflunomide; biologic agents, such as TNF-a inhibitors (eg, etanercept, adalimumab, and infliximab); and anti-IL monoclonal antibody drugs (eg, the IL-6 inhibitor tocilizumab and IL-1 inhibitors anakinra, and canakinumab).31 Indications by class include:
- csDMARDs as first-line therapy in persistent eoJIA and pJIA;
- TNF-Symbolα inhibitors for refractory eoJIA and for pJIA episodes31;
- tocilizumab, recommended for sJIA patients who have persistent systemic signs; and
- anakinra and canakinumab for refractory SJIA patients.32
Continue to: Failure
Failure
When treatment of JIA fails with a given drug, options include increasing the dosage; switching to another agent in the same drug class; switching to a different class; and combining an NSAID with a csDMARD or a biologic agent.32 In class-specific JIA cases, a change in a drug regimen is warranted on the basis of the evidence-based historical clinical response rate.32
What is the prognosis?
Treatment of JIA with novel agents, such as biologics, has opened up the possibility that JIA patients can live not just with suppressed symptoms but immunologically inactive disease. This is the result of better understanding of the pathogenesis of JIA and the mechanism of action of targeted drugs, and identification of biomarkers that are helpful in predicting prognosis, adverse effects, and response to treatment.
JIA is often a lifelong disease; one-third of patients continue to exhibit symptoms into adulthood.4 If their disease is properly managed, however, these patients do not develop typical features of rheumatoid arthritis, including hand, limb, and spine deformities. Last, patients with JIA who have only intermittent disease tend to do better over the long term than those whose disease is continual.32
The mortality rate of JIA has dropped: from 1% to 4% in the mid-1970s to 0.3% to 1% today4—an improvement in life expectancy that is echoed in enhanced quality of life for patients. According to the 4-level Steinbrocker functional classification scale33 (used to rate the extent of physical disability), 15% of JIA patients were Class III (limited to few or no activities of the patient’s usual occupation) or Class IV (bedridden with little or no self-care) in the period from 1976 to 1994—a percentage that had declined to 5% by 2002.34
The family physician plays pivotal role in JIA care
For the family physician, appropriate initial intervention in the management of JIA is imperative. This includes ordering imaging (whether plain films or MRI), laboratory tests as described earlier (although not to make the diagnosis), and the use of NSAIDs, intra-articular steroids, and other induction agents. Once the diagnosis is made, and a drug regimen is put in place, you will need to monitor for adverse effects. This monitoring will need to occur when a patient is escalated to csDMARDs, biological agents, or systemic steroids; is maintained on an NSAID; or is placed on a combination regimen.
Continue to: Before beginning therapy with a biologic agent...
Before beginning therapy with a biologic agent, it’s important to screen for hepatitis B, hepatitis C, human immunodeficiency virus infection, tuberculosis, and fungal infection (eg, Histoplasma capsulatum, Coccidioides immitis32). Be sure to make a timely referral to the ophthalmology service for a bi-annual eye exam and, in the event that surgery is necessary, conduct a preoperative evaluation, with the knowledge of how long before surgery a biologic agent must be withheld (duration varies by drug).32
CORRESPONDENCE
Tobe Momah, MD, Department of Family Medicine, Clinical Science Building, 4th Floor, University of Mississippi Medical Center, 2500 North State Street, Jackson, MS 39216; tmomah@umc.edu.
1. Adriano LS, de França Fonteles MM, de Fátima Menezes Azevedo M, et al. Medication adherence in patients with juvenile idiopathic arthritis. Rev Bras Reumatol Engl Ed. 2017;57:23-29.
2. Akioka S. A better understanding of juvenile idiopathic arthritis with classification criteria. Nihon Rinsho Meneki Gakkai Kaishi. 2016;39:513-521.
3. Thierry S, Fautrel B, Lemelle I, Guillemin F. Prevalence and incidence of juvenile idiopathic arthritis: a systematic review. Joint Bone Spine. 2014;81:112-117.
4. Petty RE, Laxer RM, Lindsley CB, et al. Pediatric Rheumatology. Philadelphia, PA: Elsevier; 2016:188-201.e6.
5. Scott C, Brice N. Juvenile idiopathic arthritis–an update on its diagnosis and management. S Afr Med J. 2015;105:1077.
6. Giancane G, Consolaro A, Lanni S, et al. Juvenile idiopathic arthritis: diagnosis and treatment. Rheumatol Ther. 2016;3:187-207.
7. Criteria for the classification of juvenile rheumatoid arthritis. Bull Rheum Dis. 1972;23:712-719.
8. Wood PHN: Special meeting on nomenclature and classification of arthritis in children. In: Munthe E, ed. The Care of Rheumatic Children. Basel, Switzerland: EULAR Publishers; 1978:47-50.
9. Petty RE, Southwood TR, Baum J, et al. Revision of the proposed classification criteria for juvenile idiopathic arthritis: Durban, 1997. J Rheumatol. 1998;25:1991-1994.
10. Petty RE, Southwood TR, Manners P, et al; International League of Associations for Rheumatology. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31:390-392.
11. Basra HAS, Humphries PD. Juvenile idiopathic arthritis: what is the utility of ultrasound? Br J Radiol. 2017;90:20160920.
12. Weiss J, Ilowite NT. Juvenile idiopathic arthritis. Pediatr Clin North Am. 2005;52:413-442, vi.
13. Fujikawa S, Okuni M. A nationwide surveillance study of rheumatic diseases among Japanese children. Acta Pediatric Jpn. 1997:39:242-244.
14. Ozen S, Karaaslan Y, Ozdemir O, et al. Prevalence of juvenile chronic arthritis and familial Mediterranean fever in Turkey: a field study. J Rheumatol. 1998;25:2445-2449.
15. Aoust L, Rossi-Semerano L, Koné-PauL I, et al. Time to diagnosis in juvenile idiopathic arthritis: a French perspective. Orphanet J Rare Dis. 2017;12:43.
16. Moe N, Rygg M. Epidemiology of juvenile chronic arthritis in northern Norway; a ten-year retrospective study. Clin Exp Rheumatol. 1998;16:99-101.
17. Abramowicz S, Kim S, Prahalad S, et al. Juvenile arthritis: current concepts in terminology, etiopathogenesis, diagnosis, and management. Int J Oral Maxillofac Surg. 2016;45:801-812.
18. Prahalad S, Shear ES, Thompson SD, et al. Increased prevalence of familial autoimmunity in simplex and multiplex families with juvenile rheumatoid arthritis. Arthritis Rheum. 2002;46:1851-1856.
19. Gonzalez B, Larrañaga C, León O, et al. Parvovirus B19 may have a role in the pathogenesis of juvenile idiopathic arthritis. J Rheumatol. 2007;34:1336-1340.
20. Prakken B, Albani S, Martini A. Juvenile idiopathic arthritis. Lancet. 2011;377:2138-2149.
21. Zhou J, Ding Y, Zhang Y, et al. CD3+CD56+ natural killer T cell activity in children with different forms of juvenile idiopathic arthritis and the influence of etanercept treatment on polyarticular subgroup. Clin Immunol. 2016;176:1-11.
22. Shoop-Worrall SJW, Verstappen SMM, Baildam E, et al. How common is clinically inactive disease in a prospective cohort of patients with juvenile idiopathic arthritis? The importance of definition. Ann Rheum Dis. 2017;0:1-8.
23. Nordal EB, Zak M, Berntson L, et al. Juvenile Arthritis Disease Activity Score (JADAS) based on CRP; validity and predictive ability in a Nordic population-based setting. Pediatr Rheumatol Online J. 2011;9(suppl 1):155.
24. Swart JF, Dijkhuizen EHP, Wulffraat NM, et al. Clinical Juvenile Arthritis Disease Activity Score proves to be a useful tool in treat-to-target therapy in juvenile idiopathic arthritis. Ann Rheum Dis. 2018;77:336-342.
25. Horneff G, Klein A, Ganser G, et al. Protocols on classification, monitoring and therapy in children’s rheumatology (PRO-KIND): results of the working group polyarticular juvenile idiopathic arthritis. Pediatr Rheumatol Online J. 2017;15:78.
26. Shoop-Worrall SJW, Verstappen SMM, McDonagh JE, et al. Long‐term outcomes following achievement of clinically inactive disease in juvenile idiopathic arthritis. Arthritis Rheumatol. 2018;70:1519-1529.
27. Ahn SS, Yoo BW, Jung SM, et al. In-hospital mortality in febrile lupus patients based on 2016 EULAR/ACR/PRINTO classification criteria for macrophage activation syndrome. Sem Arthritis Rheum. 2017;.47:216-221.
28. Yokota S, Mori M, Imagawa T, et al. Proposal for juvenile idiopathic arthritis guidance on diagnosis and treatment for primary care pediatricians and nonpediatric rheumatologists (2007). Mod Rheumatol. 2007;17:353-363.
29. Barut K, Adrovic A, Şahin S, et al. Juvenile idiopathic arthritis. Balkan Med J. 2017;34:90-101.
30. Colebatch-Bourn AN, Edwards CJ, et al. EULAR-PReS points to consider for the use of imaging in the diagnosis and management of juvenile idiopathic arthritis in clinical practice. Ann Rheum Dis. 2015;74:1946-1957.
31. Blazina Š, Markelj G, AvramoviČ MZ, et al. Management of juvenile idiopathic arthritis: a clinical guide. Pediatr Drugs. 2016;18:397-412.
32. Santos MJ, Conde M, Mourão AF, et al. 2016 update of the Portuguese recommendations for the use of biologic therapies in children and adolescents with juvenile idiopathic arthritis. Acta Rheumatol Port. 2016;41:194-212.
33. Steinbrocker 0, Traeger CH, Batterman RC. Therapeutic criteria in rheumatoid arthritis. JAMA. 1949;140:659-662.
34. Oen K, Malleson PN, Cabral D, et al. Disease course and outcome of juvenile rheumatoid arthritis in a multicenter cohort. J Rheumatol. 2002;29:1989-1999.
1. Adriano LS, de França Fonteles MM, de Fátima Menezes Azevedo M, et al. Medication adherence in patients with juvenile idiopathic arthritis. Rev Bras Reumatol Engl Ed. 2017;57:23-29.
2. Akioka S. A better understanding of juvenile idiopathic arthritis with classification criteria. Nihon Rinsho Meneki Gakkai Kaishi. 2016;39:513-521.
3. Thierry S, Fautrel B, Lemelle I, Guillemin F. Prevalence and incidence of juvenile idiopathic arthritis: a systematic review. Joint Bone Spine. 2014;81:112-117.
4. Petty RE, Laxer RM, Lindsley CB, et al. Pediatric Rheumatology. Philadelphia, PA: Elsevier; 2016:188-201.e6.
5. Scott C, Brice N. Juvenile idiopathic arthritis–an update on its diagnosis and management. S Afr Med J. 2015;105:1077.
6. Giancane G, Consolaro A, Lanni S, et al. Juvenile idiopathic arthritis: diagnosis and treatment. Rheumatol Ther. 2016;3:187-207.
7. Criteria for the classification of juvenile rheumatoid arthritis. Bull Rheum Dis. 1972;23:712-719.
8. Wood PHN: Special meeting on nomenclature and classification of arthritis in children. In: Munthe E, ed. The Care of Rheumatic Children. Basel, Switzerland: EULAR Publishers; 1978:47-50.
9. Petty RE, Southwood TR, Baum J, et al. Revision of the proposed classification criteria for juvenile idiopathic arthritis: Durban, 1997. J Rheumatol. 1998;25:1991-1994.
10. Petty RE, Southwood TR, Manners P, et al; International League of Associations for Rheumatology. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31:390-392.
11. Basra HAS, Humphries PD. Juvenile idiopathic arthritis: what is the utility of ultrasound? Br J Radiol. 2017;90:20160920.
12. Weiss J, Ilowite NT. Juvenile idiopathic arthritis. Pediatr Clin North Am. 2005;52:413-442, vi.
13. Fujikawa S, Okuni M. A nationwide surveillance study of rheumatic diseases among Japanese children. Acta Pediatric Jpn. 1997:39:242-244.
14. Ozen S, Karaaslan Y, Ozdemir O, et al. Prevalence of juvenile chronic arthritis and familial Mediterranean fever in Turkey: a field study. J Rheumatol. 1998;25:2445-2449.
15. Aoust L, Rossi-Semerano L, Koné-PauL I, et al. Time to diagnosis in juvenile idiopathic arthritis: a French perspective. Orphanet J Rare Dis. 2017;12:43.
16. Moe N, Rygg M. Epidemiology of juvenile chronic arthritis in northern Norway; a ten-year retrospective study. Clin Exp Rheumatol. 1998;16:99-101.
17. Abramowicz S, Kim S, Prahalad S, et al. Juvenile arthritis: current concepts in terminology, etiopathogenesis, diagnosis, and management. Int J Oral Maxillofac Surg. 2016;45:801-812.
18. Prahalad S, Shear ES, Thompson SD, et al. Increased prevalence of familial autoimmunity in simplex and multiplex families with juvenile rheumatoid arthritis. Arthritis Rheum. 2002;46:1851-1856.
19. Gonzalez B, Larrañaga C, León O, et al. Parvovirus B19 may have a role in the pathogenesis of juvenile idiopathic arthritis. J Rheumatol. 2007;34:1336-1340.
20. Prakken B, Albani S, Martini A. Juvenile idiopathic arthritis. Lancet. 2011;377:2138-2149.
21. Zhou J, Ding Y, Zhang Y, et al. CD3+CD56+ natural killer T cell activity in children with different forms of juvenile idiopathic arthritis and the influence of etanercept treatment on polyarticular subgroup. Clin Immunol. 2016;176:1-11.
22. Shoop-Worrall SJW, Verstappen SMM, Baildam E, et al. How common is clinically inactive disease in a prospective cohort of patients with juvenile idiopathic arthritis? The importance of definition. Ann Rheum Dis. 2017;0:1-8.
23. Nordal EB, Zak M, Berntson L, et al. Juvenile Arthritis Disease Activity Score (JADAS) based on CRP; validity and predictive ability in a Nordic population-based setting. Pediatr Rheumatol Online J. 2011;9(suppl 1):155.
24. Swart JF, Dijkhuizen EHP, Wulffraat NM, et al. Clinical Juvenile Arthritis Disease Activity Score proves to be a useful tool in treat-to-target therapy in juvenile idiopathic arthritis. Ann Rheum Dis. 2018;77:336-342.
25. Horneff G, Klein A, Ganser G, et al. Protocols on classification, monitoring and therapy in children’s rheumatology (PRO-KIND): results of the working group polyarticular juvenile idiopathic arthritis. Pediatr Rheumatol Online J. 2017;15:78.
26. Shoop-Worrall SJW, Verstappen SMM, McDonagh JE, et al. Long‐term outcomes following achievement of clinically inactive disease in juvenile idiopathic arthritis. Arthritis Rheumatol. 2018;70:1519-1529.
27. Ahn SS, Yoo BW, Jung SM, et al. In-hospital mortality in febrile lupus patients based on 2016 EULAR/ACR/PRINTO classification criteria for macrophage activation syndrome. Sem Arthritis Rheum. 2017;.47:216-221.
28. Yokota S, Mori M, Imagawa T, et al. Proposal for juvenile idiopathic arthritis guidance on diagnosis and treatment for primary care pediatricians and nonpediatric rheumatologists (2007). Mod Rheumatol. 2007;17:353-363.
29. Barut K, Adrovic A, Şahin S, et al. Juvenile idiopathic arthritis. Balkan Med J. 2017;34:90-101.
30. Colebatch-Bourn AN, Edwards CJ, et al. EULAR-PReS points to consider for the use of imaging in the diagnosis and management of juvenile idiopathic arthritis in clinical practice. Ann Rheum Dis. 2015;74:1946-1957.
31. Blazina Š, Markelj G, AvramoviČ MZ, et al. Management of juvenile idiopathic arthritis: a clinical guide. Pediatr Drugs. 2016;18:397-412.
32. Santos MJ, Conde M, Mourão AF, et al. 2016 update of the Portuguese recommendations for the use of biologic therapies in children and adolescents with juvenile idiopathic arthritis. Acta Rheumatol Port. 2016;41:194-212.
33. Steinbrocker 0, Traeger CH, Batterman RC. Therapeutic criteria in rheumatoid arthritis. JAMA. 1949;140:659-662.
34. Oen K, Malleson PN, Cabral D, et al. Disease course and outcome of juvenile rheumatoid arthritis in a multicenter cohort. J Rheumatol. 2002;29:1989-1999.
PRACTICE RECOMMENDATIONS
› Pair the findings of your clinical exam with the results of imaging and laboratory testing to make the diagnosis of juvenile idiopathic arthritis (JIA), as it is a diagnosis of exclusion. B
› Individualize treatment based on where the patient falls in the JIA disease spectrum to increase the likelihood that medical therapy will be effective. A
› Consider treating diagnosed JIA with an available biologic agent, which can provide a long asymptomatic period. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
