User login
Association of Atrial Fibrillation and/or Flutter With Adverse Cardiac Outcomes and Mortality in Patients With Wolff-Parkinson-White Syndrome
Wolff-Parkinson-White (WPW) syndrome is characterized by the presence of ≥ 1 accessory pathways and the development of both recurrent paroxysmal atrial fibrillation (AF) and supraventricular tachycardia that can lead to further malignant arrhythmias resulting in sudden cardiac death (SCD).1-7 Historically, incidental, ventricular pre-excitation on electrocardiogram has conferred a relatively low SCD risk in adults; however, newer WPW syndrome data suggest the endpoint may not be as benign as previously thought.7 The current literature has defined atrioventricular reentrant tachycardia triggering AF, rather than symptoms, as an independent risk factor for malignant arrhythmias. Still, long-term data detailing the association of AF with serious cardiac events and death in patients with WPW syndrome are still limited.1-7
While previous guidelines for the treatment of WPW syndrome only recommended routine electrophysiology testing (EPT) with liberal catheter ablation for symptomatic individuals, the 2015 American College of Cardiology/American Heart Association/Heart Rhythm Society guidelines now suggest its potential benefit for risk stratification in the asymptomatic population.8-12 Given the limited existing data, more long-term studies are needed to corroborate the latest EPT recommendations before routinely applying them in practice. Furthermore, since concomitant AF can lead to adverse cardiac outcomes in patients with WPW syndrome, additional data evaluating this association are also necessary. In this study, we aimed to determine the impact of atrial fibrillation and/or flutter (AF/AFL) on adverse cardiac outcomes and mortality in patients with WPW syndrome.
METHODS
This study used data from the Military Health System (MHS) Database Repository. The MHS is one of the largest health care systems in the country and includes information on about 10 million active duty and retired military service members and their families (51% male; 49% female).13,14 Data were fully anonymized and complied in accordance with federal and state laws, including the Health Insurance Portability and Accountability Act of 1996. The Naval Medical Center Portsmouth Institutional Review Board approved this study.
Study Design
This retrospective, observational cohort study identified MHS patients with WPW syndrome from January 1, 2014, to December 31, 2019. Patients were included if they had ≥ 2 International Classification of Diseases, Ninth Revision (ICD-9) or International Classification of Diseases, Tenth Revision (ICD-10) diagnosis codes for WPW syndrome (ICD-9, 426.7; ICD-10, I45.6) on separate dates; were aged ≥ 18 years at index date; and had ≥ 1 year of continuous eligibility prior to the index date (enrollment gaps ≤ 30 days were considered continuous). Patients were then divided into 2 subgroups by the presence or absence of AF/AFL using diagnostic codes. Patients were excluded if they had evidence of an implantable cardioverter-defibrillator, permanent pacemaker or were missing age or sex data. Patients were followed from index date until the first occurrence of the outcome of interest, MHS disenrollment, or the end of the study period.
Cardiac composite outcomes comprised of sudden cardiac arrest (SCA), ventricular fibrillation (VF), ventricular tachycardia and death, as well as death specifically, were the outcomes of interest and assessed after index date using ICD-9 and ICD-10 codes. Death was defined as all-cause mortality. Time to event was calculated based on the date of the initial component from the composite outcome and date of death specifically for mortality. Those not experiencing an outcome were followed until MHS disenrollment or the end of the study period.
Various patient characteristics were assessed at index including age, sex, military sponsor (the patient’s active or retired duty member through which their dependent receives TRICARE benefits) rank and branch, geographic region, type of US Department of Defense beneficiary, and index year. Clinical characteristics were assessed over a 1-year baseline period prior to index date and included the number of cardiologist and clinical visits for WPW syndrome, Charlson Comorbidity Index (CCI) scores calculated from diagnostic codes outlined in the Quan coding method, and preindex time.15 Comorbidities were assessed at baseline and defined as having ≥ 1 ICD-9 or ICD-10 code for a corresponding condition within 1 year prior to index.
Statistical Analysis
Baseline characteristics were assessed and descriptive statistics for categorical and continuous variables were presented accordingly. To assess bivariate association with exposure, χ2 tests were used to compare categorical variables, while t tests were used to compare continuous variables by exposure status. Incidence proportions and rates were reported for each outcome of interest. Kaplan-Meier curves were constructed to assess the bivariate association between exposure and study outcomes. Cox proportional hazard modeling was performed to estimate the association between AF/AFL and time to each of the outcomes. Multiple models were designed to assess cardiac and metabolic covariates, in addition to baseline characteristics. This included a base model adjusted for age, sex, military sponsor rank and branch, geographic region, and duty status.
Additional models adjusted for cardiac and metabolic confounders and CCI score. A comprehensive model included the base, cardiac, and metabolic covariates. Multicollinearity between covariates was assessed. Variables with a variance inflation factor > 4 or a tolerance level < 0.1 were added to the models. Cox proportional hazard models were used to estimate the unadjusted and adjusted hazard ratios (HRs) and 95% CIs of the association between AF/AFL and the study outcomes. Data were analyzed using SAS, version 9.4 for Windows.
RESULTS

From 2014 through 2019, 35,539 patients with WPW syndrome were identified in the MHS, 5291 had AF/AFL (14.9%); 19,961 were female (56.2%), the mean (SD) age was 62.9 (18.0) years, and 11,742 were aged ≥ 75 years (33.0%) (Table 1).

There were 4121 (11.6%), 322 (0.9%), and 848 (2.4%) patients with AF, AFL, and both arrhythmias, respectively. The mean (SD) number of cardiology visits was 3.9 (3.0). The mean (SD) baseline CCI score for the AF/AFL subgroup was 5.9 (3.5) vs 3.7 (2.2) for the non-AF/AFL subgroup (P < .001). The most prevalent comorbid conditions were hypertension, hyperlipidemia, chronic obstructive pulmonary disease, and diabetes (P < .001) (Figure 1).
Composite Outcomes

In the overall cohort, during a mean (SD) follow-up time of 3.4 (2.0) years comprising 119,682 total person-years, the components of the composite outcome occurred 6506 times with an incidence rate of 5.44 per 100 person-years. Ventricular tachycardia was the most common event, occurring 3281 times with an incidence rate of 2.74 per 100 person-years. SCA and VF occurred 841 and 135 times with incidence rates of 0.70 and 0.11 per 100 person-years, respectively. Death was the initial event 2249 times with an incidence rate of 1.88 per 100 person-years. Figure 2 shows the Kaplan-Meier curve of cardiac composite outcome by AF/AFL status.

The subgroup with AF/AFL comprised 17,412 total person-years and 1424 cardiac composite incidences compared with 102,270 person years and 5082 incidences in the no AF/AFL group (Table 2). Comparing AF/AFL vs no AF/AFL incidence rates were 8.18 vs 4.97 per 100 person-years, respectively (P < .001). SCA and VF occurred 233 and 38 times and respectively had incidence rates of 1.34 and 0.22 per 100 person-years in the AF/AFL group vs 0.59 and 0.09 per 100 person-years in the no AF/AFL group (P < .001). There were 549 deaths and a 3.15 per 100 person-years incidence rate in the AF/AFL group vs 1700 deaths and a 1.66 incidence rate in the no AF/AFL group (P < .001).

The HR for the composite outcome in the base model was 1.33 (95% CI, 1.26-1.42, P < .001) (Table 3). The association between AF/AFL and the composite outcome remained significant after adjusting for additional metabolic and cardiac covariates. The HRs for the metabolic and cardiac models were 1.30 (95% CI, 1.23-1.38, P < .001) and 1.11 (95% CI, 1.05-1.18, P < .001), respectively. After adjusting for the full model, the HR was 1.12 (95% CI, 1.05-1.19, P < .001).
Mortality

Over the 6-year study period, there was a lower survival probability for patients with AF/AFL. In the overall cohort, during a mean (SD) follow-up time of 3.7 (1.9) years comprising 129,391 total person-years, there were 3130 (8.8%) deaths and an incidence rate of 2.42 per 100 person-years. Death occurred 786 times with a 4.09 incidence rate per 100 person-years in the AF/AFL vs 2344 deaths and a 2.13 incidence rate per 100 person-years in the no AF/AFL group (P < .001). In the non-AF/AFL subgroup, death occurred 2344 times during a mean (SD) follow-up of 3.7 (1.9) years comprising 110,151 total person-years. Figure 3 shows the Kaplan-Meier curve of mortality by AF/AFL status.

After adjusting for the base, metabolic and cardiac covariates, the HRs for mortality were 1.45 (95% CI, 1.33-1.57, P < .001), 1.40 (95% CI, 1.29-1.51, P < .001) and 1.15 (95% CI, 1.06-1.25, P = .001), respectively (Table 4). The HR after adjusting for the full model was 1.16 (95% CI, 1.07-1.26, P < .001).
DISCUSSION
In this large retrospective cohort study, patients with WPW syndrome and comorbid AF/AFL had a significantly higher association with the cardiac composite outcome and death during a 3-year follow-up period when compared with patients without AF/AFL. After adjusting for confounding variables, the AF/AFL subgroup maintained a 12% and 16% higher association with the composite outcome and mortality, respectively. There was minimal difference in confounding effects between demographic data and metabolic profiles, suggesting one may serve as a proxy for the other.
To our knowledge, this is the largest WPW syndrome cohort study evaluating cardiac outcomes and mortality to date. Although previous research has shown the relatively low and mostly anecdotal SCD incidence within this population,our results demonstrate a higher association of adverse cardiac outcomes and death in an AF/AFL subgroup.16-18 Notably, in this study the AF/AFL cohort was older and had higher CCI scores than their counterparts (P < .001), thus inferring an inherently greater degree of morbidity and 10-year mortality risk. Our study is also unique in that the mean patient age was significantly older than previously reported (63 vs 27 years), which may suggest a longer living history of both ventricular pre-excitation and the comorbidities outlined in Figure 1.19 Given these age discrepancies, it is possible that our overall study population was still relatively low risk and that not all reported deaths were necessarily related to WPW syndrome. Despite these assumptions, when comparing the WPW syndrome subgroups, we still found the AF/AFL cohort maintained a statistically significant higher association with the 2 study outcomes, even after adjusting for the greater presence of comorbidities. This suggests that the presence of AF/AFL may still portend a worse prognosis in patients with WPW syndrome.
Although the association of AF and development of VF in patients with WPW syndrome—due to rapid conduction over the accessory pathway(s)—was first reported > 40 years ago, there has still been few large, long-term data studies exploring mortality in this cohort.19-25 Furthermore, even though the current literature attributes the development of AF with the electrophysiologic properties of the accessory pathway, as well as intrinsic atrial architecture and muscle vulnerability, there is still equivocal consensus regarding EPT screening and ablation indications for asymptomatic patients with WPW syndrome.26-28 Notably, Pappone and colleagues demonstrated the potential benefit of liberal ablation indications for asymptomatic patients, arguing that the intrinsic electrophysiologic properties of the accessory pathway—ie, short accessory-pathway antegrade effective refractory period, inducibility of atrioventricular reentrant tachycardia triggering AF, and multiple accessory pathway—rather than symptoms, are independent predictors of developing malignant arrhythmia.1-5
These findings contradict those reported by Obeyesekere and colleagues, who concluded that the low SCD incidence rates in patients with WPW syndrome precluded routine invasive screening.19,28 They argued that Pappone and colleagues used malignant arrhythmia as a surrogate marker for death, and that the positive predictive value of a short accessory-pathway antegrade effective refractory period for developing malignant arrhythmia was lower than reported (15% vs 82%, respectively) and that its negative predictive value was 100%.1,19,28 Given these conflicting recommendations, we hope our data elucidates the higher association of adverse outcomes and support considerations for more intensive EPT indications in patients with WPW syndrome.
While our study does not report SCD incidence, it does provide robust and reliable mortality data that suggests a greater association of death within an AF/AFL subgroup. Our findings would support more liberal EPT recommendations in patients with WPW syndrome.1-5,8,9 In this study, the SCA incidence rate was more than double the rate in the AF/AFL cohort (P < .001) and is commonly the initial presenting event in WPW syndrome.9 Even though the reported SCD incidence rate is low in WPW syndrome, our data demonstrated an increased association of death within the AF/AFL cohort. Physicians should consider early risk stratification and ablation to prevent potential recurrent malignant arrhythmia leading to death.1-5,8,9,12,19,20
Limitations
As a retrospective study and without access to the National Death Index, we were unable to determine the exact cause or events leading to death and instead utilized all-cause mortality data. Subsequently, our observations may only demonstrate association, rather than causality, between AF/AFL and death in patients with WPW syndrome. Additionally, we could not distinguish between AF and AFL as the arrhythmia leading to death. However, since overall survivability was the outcome of interest, our adjusted HR models were still able to demonstrate the increased association of the composite outcome and death within an AF/AFL cohort.
Although a large cohort was analyzed, due to the constraints of utilizing diagnostic codes to determine study outcomes, we could not distinguish between symptomatic and asymptomatic patients, nor how they were managed prior to the outcome event. However, as recent literature demonstrates, updated predictors of malignant arrhythmia and decisions for early EPT are similar for both symptomatic and asymptomatic patients and should be driven by the intrinsic electrophysiologic properties of the accessory pathway, rather than symptomatology;thus, our inability to discern this should have negligible consequence in determining when to perform risk stratification and ablation.1
MHS eligible patients have direct access to care; the generalizability of our data may not necessarily correspond to a community population with lower socioeconomic status (we did adjust for military sponsor rank which has been used as a proxy), reduced access to care, or uninsured individuals. However, the prevalence of WPW syndrome within our cohort was comparable to the general population, 0.4% vs 0.1%-0.3%, respectively.13,14,19 Similarly, the incidence of AF within our population was comparable to the general population, 15% vs 16%-26%, respectively.23 These similar data points suggest our results may apply beyond MHS patients.
CONCLUSIONS
Patients with WPW syndrome and AF/AFL have a higher association with adverse cardiac outcomes and death. Despite previously reported low SCD incidence rates in this population, our study demonstrates the increased association of mortality in an AF/AFL cohort. The limitations of utilizing all-cause mortality data necessitate further investigation into the etiology behind the deaths in our study population. Since ventricular pre-excitation can predispose patients to AF and potentially lead to malignant arrhythmia and SCD, understanding the cause of mortality will allow physicians to determine the appropriate monitoring and intervention strategies to improve outcomes in this population. Our results suggest consideration for more aggressive EPT screening and ablation recommendations in patients with WPW syndrome may be warranted.
1. Pappone C, Vicedomini G, Manguso F, et al. The natural history of WPW syndrome. Eur Heart J Suppl. 2015; 17 (Supplement A):A8-A11.doi:10.1093/eurheartj/suv004
2. Pappone C, Vicedomini G, Manguso F, et al. Risk of malignant arrhythmias in initially symptomatic patients with Wolff-Parkinson-White syndrome: results of a prospective long-term electrophysiological follow-up study. Circulation. 2012;125(5):661-668. doi:10.1161/CIRCULATIONAHA.111.065722
3. Pappone C, Santinelli V, Rosanio S, et al. Usefulness of invasive electrophysiologic testing to stratify the risk of arrhythmic events in asymptomatic patients with Wolff-Parkinson-White pattern: results from a large prospective long-term follow-up study. J Am Coll Cardiol. 2003;41(2):239-244. doi:10.1016/s0735-1097(02)02706-7
4. Pappone C, Vicedomini G, Manguso F, et al. Wolff-Parkinson-White syndrome in the era of catheter ablation: insights from a registry study of 2169 patients. Circulation. 2014;130(10):811-819. doi:10.1161/CIRCULATIONAHA.114.011154
5. Pappone C, Santinelli V, Manguso F, et al. A randomized study of prophylactic catheter ablation in asymptomatic patients with the Wolff-Parkinson-White syndrome. N Engl J Med. 2003;349(19):1803-1811. doi:10.1056/NEJMoa035345
6. Santinelli V, Radinovic A, Manguso F, et al. Asymptomatic ventricular preexcitation: a long-term prospective follow-up study of 293 adult patients. Circ Arrhythm Electrophysiol. 2009;2(2):102-107. doi:10.1161/CIRCEP.108.827550
7. Santinelli V, Radinovic A, Manguso F, et al. The natural history of asymptomatic ventricular pre-excitation a long-term prospective follow-up study of 184 asymptomatic children. J Am Coll Cardiol. 2009;53(3):275-280. doi:10.1016/j.jacc.2008.09.037
8. Al-Khatib SM, Arshad A, Balk EM, et al. Risk Stratification for Arrhythmic Events in Patients With Asymptomatic Pre-Excitation: A Systematic Review for the 2015 ACC/AHA/HRS Guideline for the Management of Adult Patients With Supraventricular Tachycardia: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2016;67(13):1624-1638. doi:10.1016/j.jacc.2015.09.018
9. Blomström-Lundqvist C, Scheinman MM, Aliot EM, et al. ACC/AHA/ESC guidelines for the management of patients with supraventricular arrhythmias--executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Supraventricular Arrhythmias). Circulation. 2003;108(15):1871-1909.doi:10.1161/01.CIR.0000091380.04100.84
10. Pediatric and Congenital Electrophysiology Society (PACES); Heart Rhythm Society (HRS); American College of Cardiology Foundation (ACCF); PACES/HRS expert consensus statement on the management of the asymptomatic young patient with a Wolff-Parkinson-White (WPW, ventricular preexcitation) electrocardiographic pattern: developed in partnership between the Pediatric and Congenital Electrophysiology Society (PACES) and the Heart Rhythm Society (HRS). Endorsed by the governing bodies of PACES, HRS, the American College of Cardiology Foundation (ACCF), the American Heart Association (AHA), the American Academy of Pediatrics (AAP), and the Canadian Heart Rhythm Society (CHRS). Heart Rhythm. 2012;9(6):1006-1024. doi:10.1016/j.hrthm.2012.03.050
11. Cohen M, Triedman J. Guidelines for management of asymptomatic ventricular pre-excitation: brave new world or Pandora’s box?. Circ Arrhythm Electrophysiol. 2014;7(2):187-189. doi:10.1161/CIRCEP.114.001528
12. Svendsen JH, Dagres N, Dobreanu D, et al. Current strategy for treatment of patients with Wolff-Parkinson-White syndrome and asymptomatic preexcitation in Europe: European Heart Rhythm Association survey. Europace. 2013;15(5):750-753. doi:10.1093/europace/eut094
13. Gimbel RW, Pangaro L, Barbour G. America’s “undiscovered” laboratory for health services research. Med Care. 2010;48(8):751-756. doi:10.1097/MLR.0b013e3181e35be8
14. Dorrance KA, Ramchandani S, Neil N, Fisher H. Leveraging the military health system as a laboratory for health care reform. Mil Med. 2013;178(2):142-145. doi:10.7205/milmed-d-12-00168
15. Quan H, Sundararajan V, Halfon P, et al. Coding algorithms for defining comorbidities in ICD-9-CM and ICD-10 administrative data. Med Care. 2005;43(11):1130-1139. doi:10.1097/01.mlr.0000182534.19832.83
16. Finocchiaro G, Papadakis M, Behr ER, Sharma S, Sheppard M. Sudden Cardiac Death in Pre-Excitation and Wolff-Parkinson-White: Demographic and Clinical Features. J Am Coll Cardiol. 2017;69(12):1644-1645. doi:10.1016/j.jacc.2017.01.023
17. Munger TM, Packer DL, Hammill SC, et al. A population study of the natural history of Wolff-Parkinson-White syndrome in Olmsted County, Minnesota, 1953-1989. Circulation. 1993;87(3):866-873. doi:10.1161/01.cir.87.3.866
18. Fitzsimmons PJ, McWhirter PD, Peterson DW, Kruyer WB. The natural history of Wolff-Parkinson-White syndrome in 228 military aviators: a long-term follow-up of 22 years. Am Heart J. 2001;142(3):530-536. doi:10.1067/mhj.2001.117779
19. Obeyesekere MN, Leong-Sit P, Massel D, et al. Risk of arrhythmia and sudden death in patients with asymptomatic preexcitation: a meta-analysis. Circulation. 2012;125(19):2308-2315. doi:10.1161/CIRCULATIONAHA.111.055350
20. Waspe LE, Brodman R, Kim SG, Fisher JD. Susceptibility to atrial fibrillation and ventricular tachyarrhythmia in the Wolff-Parkinson-White syndrome: role of the accessory pathway. Am Heart J. 1986;112(6):1141-1152. doi:10.1016/0002-8703(86)90342-x
21. Pietersen AH, Andersen ED, Sandøe E. Atrial fibrillation in the Wolff-Parkinson-White syndrome. Am J Cardiol. 1992;70(5):38A-43A. doi:10.1016/0002-9149(92)91076-g
22. Della Bella P, Brugada P, Talajic M, et al. Atrial fibrillation in patients with an accessory pathway: importance of the conduction properties of the accessory pathway. J Am Coll Cardiol. 1991;17(6):1352-1356. doi:10.1016/s0735-1097(10)80146-9
23. Fujimura O, Klein GJ, Yee R, Sharma AD. Mode of onset of atrial fibrillation in the Wolff-Parkinson-White syndrome: how important is the accessory pathway?. J Am Coll Cardiol. 1990;15(5):1082-1086. doi:10.1016/0735-1097(90)90244-j
24. Montoya PT, Brugada P, Smeets J, et al. Ventricular fibrillation in the Wolff-Parkinson-White syndrome. Eur Heart J. 1991;12(2):144-150. doi:10.1093/oxfordjournals.eurheartj.a059860
25. Klein GJ, Bashore TM, Sellers TD, Pritchett EL, Smith WM, Gallagher JJ. Ventricular fibrillation in the Wolff-Parkinson-White syndrome. N Engl J Med. 1979;301(20):1080-1085. doi:10.1056/NEJM197911153012003
26. Centurion OA. Atrial Fibrillation in the Wolff-Parkinson-White Syndrome. J Atr Fibrillation. 2011;4(1):287. Published 2011 May 4. doi:10.4022/jafib.287
27. Song C, Guo Y, Zheng X, et al. Prognostic Significance and Risk of Atrial Fibrillation of Wolff-Parkinson-White Syndrome in Patients With Hypertrophic Cardiomyopathy. Am J Cardiol. 2018;122(9):1546-1550. doi:10.1016/j.amjcard.2018.07.021
28. Obeyesekere M, Gula LJ, Skanes AC, Leong-Sit P, Klein GJ. Risk of sudden death in Wolff-Parkinson-White syndrome: how high is the risk?. Circulation. 2012;125(5):659-660. doi:10.1161/CIRCULATIONAHA.111.085159
Wolff-Parkinson-White (WPW) syndrome is characterized by the presence of ≥ 1 accessory pathways and the development of both recurrent paroxysmal atrial fibrillation (AF) and supraventricular tachycardia that can lead to further malignant arrhythmias resulting in sudden cardiac death (SCD).1-7 Historically, incidental, ventricular pre-excitation on electrocardiogram has conferred a relatively low SCD risk in adults; however, newer WPW syndrome data suggest the endpoint may not be as benign as previously thought.7 The current literature has defined atrioventricular reentrant tachycardia triggering AF, rather than symptoms, as an independent risk factor for malignant arrhythmias. Still, long-term data detailing the association of AF with serious cardiac events and death in patients with WPW syndrome are still limited.1-7
While previous guidelines for the treatment of WPW syndrome only recommended routine electrophysiology testing (EPT) with liberal catheter ablation for symptomatic individuals, the 2015 American College of Cardiology/American Heart Association/Heart Rhythm Society guidelines now suggest its potential benefit for risk stratification in the asymptomatic population.8-12 Given the limited existing data, more long-term studies are needed to corroborate the latest EPT recommendations before routinely applying them in practice. Furthermore, since concomitant AF can lead to adverse cardiac outcomes in patients with WPW syndrome, additional data evaluating this association are also necessary. In this study, we aimed to determine the impact of atrial fibrillation and/or flutter (AF/AFL) on adverse cardiac outcomes and mortality in patients with WPW syndrome.
METHODS
This study used data from the Military Health System (MHS) Database Repository. The MHS is one of the largest health care systems in the country and includes information on about 10 million active duty and retired military service members and their families (51% male; 49% female).13,14 Data were fully anonymized and complied in accordance with federal and state laws, including the Health Insurance Portability and Accountability Act of 1996. The Naval Medical Center Portsmouth Institutional Review Board approved this study.
Study Design
This retrospective, observational cohort study identified MHS patients with WPW syndrome from January 1, 2014, to December 31, 2019. Patients were included if they had ≥ 2 International Classification of Diseases, Ninth Revision (ICD-9) or International Classification of Diseases, Tenth Revision (ICD-10) diagnosis codes for WPW syndrome (ICD-9, 426.7; ICD-10, I45.6) on separate dates; were aged ≥ 18 years at index date; and had ≥ 1 year of continuous eligibility prior to the index date (enrollment gaps ≤ 30 days were considered continuous). Patients were then divided into 2 subgroups by the presence or absence of AF/AFL using diagnostic codes. Patients were excluded if they had evidence of an implantable cardioverter-defibrillator, permanent pacemaker or were missing age or sex data. Patients were followed from index date until the first occurrence of the outcome of interest, MHS disenrollment, or the end of the study period.
Cardiac composite outcomes comprised of sudden cardiac arrest (SCA), ventricular fibrillation (VF), ventricular tachycardia and death, as well as death specifically, were the outcomes of interest and assessed after index date using ICD-9 and ICD-10 codes. Death was defined as all-cause mortality. Time to event was calculated based on the date of the initial component from the composite outcome and date of death specifically for mortality. Those not experiencing an outcome were followed until MHS disenrollment or the end of the study period.
Various patient characteristics were assessed at index including age, sex, military sponsor (the patient’s active or retired duty member through which their dependent receives TRICARE benefits) rank and branch, geographic region, type of US Department of Defense beneficiary, and index year. Clinical characteristics were assessed over a 1-year baseline period prior to index date and included the number of cardiologist and clinical visits for WPW syndrome, Charlson Comorbidity Index (CCI) scores calculated from diagnostic codes outlined in the Quan coding method, and preindex time.15 Comorbidities were assessed at baseline and defined as having ≥ 1 ICD-9 or ICD-10 code for a corresponding condition within 1 year prior to index.
Statistical Analysis
Baseline characteristics were assessed and descriptive statistics for categorical and continuous variables were presented accordingly. To assess bivariate association with exposure, χ2 tests were used to compare categorical variables, while t tests were used to compare continuous variables by exposure status. Incidence proportions and rates were reported for each outcome of interest. Kaplan-Meier curves were constructed to assess the bivariate association between exposure and study outcomes. Cox proportional hazard modeling was performed to estimate the association between AF/AFL and time to each of the outcomes. Multiple models were designed to assess cardiac and metabolic covariates, in addition to baseline characteristics. This included a base model adjusted for age, sex, military sponsor rank and branch, geographic region, and duty status.
Additional models adjusted for cardiac and metabolic confounders and CCI score. A comprehensive model included the base, cardiac, and metabolic covariates. Multicollinearity between covariates was assessed. Variables with a variance inflation factor > 4 or a tolerance level < 0.1 were added to the models. Cox proportional hazard models were used to estimate the unadjusted and adjusted hazard ratios (HRs) and 95% CIs of the association between AF/AFL and the study outcomes. Data were analyzed using SAS, version 9.4 for Windows.
RESULTS
From 2014 through 2019, 35,539 patients with WPW syndrome were identified in the MHS, 5291 had AF/AFL (14.9%); 19,961 were female (56.2%), the mean (SD) age was 62.9 (18.0) years, and 11,742 were aged ≥ 75 years (33.0%) (Table 1).
There were 4121 (11.6%), 322 (0.9%), and 848 (2.4%) patients with AF, AFL, and both arrhythmias, respectively. The mean (SD) number of cardiology visits was 3.9 (3.0). The mean (SD) baseline CCI score for the AF/AFL subgroup was 5.9 (3.5) vs 3.7 (2.2) for the non-AF/AFL subgroup (P < .001). The most prevalent comorbid conditions were hypertension, hyperlipidemia, chronic obstructive pulmonary disease, and diabetes (P < .001) (Figure 1).
Composite Outcomes
In the overall cohort, during a mean (SD) follow-up time of 3.4 (2.0) years comprising 119,682 total person-years, the components of the composite outcome occurred 6506 times with an incidence rate of 5.44 per 100 person-years. Ventricular tachycardia was the most common event, occurring 3281 times with an incidence rate of 2.74 per 100 person-years. SCA and VF occurred 841 and 135 times with incidence rates of 0.70 and 0.11 per 100 person-years, respectively. Death was the initial event 2249 times with an incidence rate of 1.88 per 100 person-years. Figure 2 shows the Kaplan-Meier curve of cardiac composite outcome by AF/AFL status.
The subgroup with AF/AFL comprised 17,412 total person-years and 1424 cardiac composite incidences compared with 102,270 person years and 5082 incidences in the no AF/AFL group (Table 2). Comparing AF/AFL vs no AF/AFL incidence rates were 8.18 vs 4.97 per 100 person-years, respectively (P < .001). SCA and VF occurred 233 and 38 times and respectively had incidence rates of 1.34 and 0.22 per 100 person-years in the AF/AFL group vs 0.59 and 0.09 per 100 person-years in the no AF/AFL group (P < .001). There were 549 deaths and a 3.15 per 100 person-years incidence rate in the AF/AFL group vs 1700 deaths and a 1.66 incidence rate in the no AF/AFL group (P < .001).
The HR for the composite outcome in the base model was 1.33 (95% CI, 1.26-1.42, P < .001) (Table 3). The association between AF/AFL and the composite outcome remained significant after adjusting for additional metabolic and cardiac covariates. The HRs for the metabolic and cardiac models were 1.30 (95% CI, 1.23-1.38, P < .001) and 1.11 (95% CI, 1.05-1.18, P < .001), respectively. After adjusting for the full model, the HR was 1.12 (95% CI, 1.05-1.19, P < .001).
Mortality
Over the 6-year study period, there was a lower survival probability for patients with AF/AFL. In the overall cohort, during a mean (SD) follow-up time of 3.7 (1.9) years comprising 129,391 total person-years, there were 3130 (8.8%) deaths and an incidence rate of 2.42 per 100 person-years. Death occurred 786 times with a 4.09 incidence rate per 100 person-years in the AF/AFL vs 2344 deaths and a 2.13 incidence rate per 100 person-years in the no AF/AFL group (P < .001). In the non-AF/AFL subgroup, death occurred 2344 times during a mean (SD) follow-up of 3.7 (1.9) years comprising 110,151 total person-years. Figure 3 shows the Kaplan-Meier curve of mortality by AF/AFL status.
After adjusting for the base, metabolic and cardiac covariates, the HRs for mortality were 1.45 (95% CI, 1.33-1.57, P < .001), 1.40 (95% CI, 1.29-1.51, P < .001) and 1.15 (95% CI, 1.06-1.25, P = .001), respectively (Table 4). The HR after adjusting for the full model was 1.16 (95% CI, 1.07-1.26, P < .001).
DISCUSSION
In this large retrospective cohort study, patients with WPW syndrome and comorbid AF/AFL had a significantly higher association with the cardiac composite outcome and death during a 3-year follow-up period when compared with patients without AF/AFL. After adjusting for confounding variables, the AF/AFL subgroup maintained a 12% and 16% higher association with the composite outcome and mortality, respectively. There was minimal difference in confounding effects between demographic data and metabolic profiles, suggesting one may serve as a proxy for the other.
To our knowledge, this is the largest WPW syndrome cohort study evaluating cardiac outcomes and mortality to date. Although previous research has shown the relatively low and mostly anecdotal SCD incidence within this population,our results demonstrate a higher association of adverse cardiac outcomes and death in an AF/AFL subgroup.16-18 Notably, in this study the AF/AFL cohort was older and had higher CCI scores than their counterparts (P < .001), thus inferring an inherently greater degree of morbidity and 10-year mortality risk. Our study is also unique in that the mean patient age was significantly older than previously reported (63 vs 27 years), which may suggest a longer living history of both ventricular pre-excitation and the comorbidities outlined in Figure 1.19 Given these age discrepancies, it is possible that our overall study population was still relatively low risk and that not all reported deaths were necessarily related to WPW syndrome. Despite these assumptions, when comparing the WPW syndrome subgroups, we still found the AF/AFL cohort maintained a statistically significant higher association with the 2 study outcomes, even after adjusting for the greater presence of comorbidities. This suggests that the presence of AF/AFL may still portend a worse prognosis in patients with WPW syndrome.
Although the association of AF and development of VF in patients with WPW syndrome—due to rapid conduction over the accessory pathway(s)—was first reported > 40 years ago, there has still been few large, long-term data studies exploring mortality in this cohort.19-25 Furthermore, even though the current literature attributes the development of AF with the electrophysiologic properties of the accessory pathway, as well as intrinsic atrial architecture and muscle vulnerability, there is still equivocal consensus regarding EPT screening and ablation indications for asymptomatic patients with WPW syndrome.26-28 Notably, Pappone and colleagues demonstrated the potential benefit of liberal ablation indications for asymptomatic patients, arguing that the intrinsic electrophysiologic properties of the accessory pathway—ie, short accessory-pathway antegrade effective refractory period, inducibility of atrioventricular reentrant tachycardia triggering AF, and multiple accessory pathway—rather than symptoms, are independent predictors of developing malignant arrhythmia.1-5
These findings contradict those reported by Obeyesekere and colleagues, who concluded that the low SCD incidence rates in patients with WPW syndrome precluded routine invasive screening.19,28 They argued that Pappone and colleagues used malignant arrhythmia as a surrogate marker for death, and that the positive predictive value of a short accessory-pathway antegrade effective refractory period for developing malignant arrhythmia was lower than reported (15% vs 82%, respectively) and that its negative predictive value was 100%.1,19,28 Given these conflicting recommendations, we hope our data elucidates the higher association of adverse outcomes and support considerations for more intensive EPT indications in patients with WPW syndrome.
While our study does not report SCD incidence, it does provide robust and reliable mortality data that suggests a greater association of death within an AF/AFL subgroup. Our findings would support more liberal EPT recommendations in patients with WPW syndrome.1-5,8,9 In this study, the SCA incidence rate was more than double the rate in the AF/AFL cohort (P < .001) and is commonly the initial presenting event in WPW syndrome.9 Even though the reported SCD incidence rate is low in WPW syndrome, our data demonstrated an increased association of death within the AF/AFL cohort. Physicians should consider early risk stratification and ablation to prevent potential recurrent malignant arrhythmia leading to death.1-5,8,9,12,19,20
Limitations
As a retrospective study and without access to the National Death Index, we were unable to determine the exact cause or events leading to death and instead utilized all-cause mortality data. Subsequently, our observations may only demonstrate association, rather than causality, between AF/AFL and death in patients with WPW syndrome. Additionally, we could not distinguish between AF and AFL as the arrhythmia leading to death. However, since overall survivability was the outcome of interest, our adjusted HR models were still able to demonstrate the increased association of the composite outcome and death within an AF/AFL cohort.
Although a large cohort was analyzed, due to the constraints of utilizing diagnostic codes to determine study outcomes, we could not distinguish between symptomatic and asymptomatic patients, nor how they were managed prior to the outcome event. However, as recent literature demonstrates, updated predictors of malignant arrhythmia and decisions for early EPT are similar for both symptomatic and asymptomatic patients and should be driven by the intrinsic electrophysiologic properties of the accessory pathway, rather than symptomatology;thus, our inability to discern this should have negligible consequence in determining when to perform risk stratification and ablation.1
MHS eligible patients have direct access to care; the generalizability of our data may not necessarily correspond to a community population with lower socioeconomic status (we did adjust for military sponsor rank which has been used as a proxy), reduced access to care, or uninsured individuals. However, the prevalence of WPW syndrome within our cohort was comparable to the general population, 0.4% vs 0.1%-0.3%, respectively.13,14,19 Similarly, the incidence of AF within our population was comparable to the general population, 15% vs 16%-26%, respectively.23 These similar data points suggest our results may apply beyond MHS patients.
CONCLUSIONS
Patients with WPW syndrome and AF/AFL have a higher association with adverse cardiac outcomes and death. Despite previously reported low SCD incidence rates in this population, our study demonstrates the increased association of mortality in an AF/AFL cohort. The limitations of utilizing all-cause mortality data necessitate further investigation into the etiology behind the deaths in our study population. Since ventricular pre-excitation can predispose patients to AF and potentially lead to malignant arrhythmia and SCD, understanding the cause of mortality will allow physicians to determine the appropriate monitoring and intervention strategies to improve outcomes in this population. Our results suggest consideration for more aggressive EPT screening and ablation recommendations in patients with WPW syndrome may be warranted.
Wolff-Parkinson-White (WPW) syndrome is characterized by the presence of ≥ 1 accessory pathways and the development of both recurrent paroxysmal atrial fibrillation (AF) and supraventricular tachycardia that can lead to further malignant arrhythmias resulting in sudden cardiac death (SCD).1-7 Historically, incidental, ventricular pre-excitation on electrocardiogram has conferred a relatively low SCD risk in adults; however, newer WPW syndrome data suggest the endpoint may not be as benign as previously thought.7 The current literature has defined atrioventricular reentrant tachycardia triggering AF, rather than symptoms, as an independent risk factor for malignant arrhythmias. Still, long-term data detailing the association of AF with serious cardiac events and death in patients with WPW syndrome are still limited.1-7
While previous guidelines for the treatment of WPW syndrome only recommended routine electrophysiology testing (EPT) with liberal catheter ablation for symptomatic individuals, the 2015 American College of Cardiology/American Heart Association/Heart Rhythm Society guidelines now suggest its potential benefit for risk stratification in the asymptomatic population.8-12 Given the limited existing data, more long-term studies are needed to corroborate the latest EPT recommendations before routinely applying them in practice. Furthermore, since concomitant AF can lead to adverse cardiac outcomes in patients with WPW syndrome, additional data evaluating this association are also necessary. In this study, we aimed to determine the impact of atrial fibrillation and/or flutter (AF/AFL) on adverse cardiac outcomes and mortality in patients with WPW syndrome.
METHODS
This study used data from the Military Health System (MHS) Database Repository. The MHS is one of the largest health care systems in the country and includes information on about 10 million active duty and retired military service members and their families (51% male; 49% female).13,14 Data were fully anonymized and complied in accordance with federal and state laws, including the Health Insurance Portability and Accountability Act of 1996. The Naval Medical Center Portsmouth Institutional Review Board approved this study.
Study Design
This retrospective, observational cohort study identified MHS patients with WPW syndrome from January 1, 2014, to December 31, 2019. Patients were included if they had ≥ 2 International Classification of Diseases, Ninth Revision (ICD-9) or International Classification of Diseases, Tenth Revision (ICD-10) diagnosis codes for WPW syndrome (ICD-9, 426.7; ICD-10, I45.6) on separate dates; were aged ≥ 18 years at index date; and had ≥ 1 year of continuous eligibility prior to the index date (enrollment gaps ≤ 30 days were considered continuous). Patients were then divided into 2 subgroups by the presence or absence of AF/AFL using diagnostic codes. Patients were excluded if they had evidence of an implantable cardioverter-defibrillator, permanent pacemaker or were missing age or sex data. Patients were followed from index date until the first occurrence of the outcome of interest, MHS disenrollment, or the end of the study period.
Cardiac composite outcomes comprised of sudden cardiac arrest (SCA), ventricular fibrillation (VF), ventricular tachycardia and death, as well as death specifically, were the outcomes of interest and assessed after index date using ICD-9 and ICD-10 codes. Death was defined as all-cause mortality. Time to event was calculated based on the date of the initial component from the composite outcome and date of death specifically for mortality. Those not experiencing an outcome were followed until MHS disenrollment or the end of the study period.
Various patient characteristics were assessed at index including age, sex, military sponsor (the patient’s active or retired duty member through which their dependent receives TRICARE benefits) rank and branch, geographic region, type of US Department of Defense beneficiary, and index year. Clinical characteristics were assessed over a 1-year baseline period prior to index date and included the number of cardiologist and clinical visits for WPW syndrome, Charlson Comorbidity Index (CCI) scores calculated from diagnostic codes outlined in the Quan coding method, and preindex time.15 Comorbidities were assessed at baseline and defined as having ≥ 1 ICD-9 or ICD-10 code for a corresponding condition within 1 year prior to index.
Statistical Analysis
Baseline characteristics were assessed and descriptive statistics for categorical and continuous variables were presented accordingly. To assess bivariate association with exposure, χ2 tests were used to compare categorical variables, while t tests were used to compare continuous variables by exposure status. Incidence proportions and rates were reported for each outcome of interest. Kaplan-Meier curves were constructed to assess the bivariate association between exposure and study outcomes. Cox proportional hazard modeling was performed to estimate the association between AF/AFL and time to each of the outcomes. Multiple models were designed to assess cardiac and metabolic covariates, in addition to baseline characteristics. This included a base model adjusted for age, sex, military sponsor rank and branch, geographic region, and duty status.
Additional models adjusted for cardiac and metabolic confounders and CCI score. A comprehensive model included the base, cardiac, and metabolic covariates. Multicollinearity between covariates was assessed. Variables with a variance inflation factor > 4 or a tolerance level < 0.1 were added to the models. Cox proportional hazard models were used to estimate the unadjusted and adjusted hazard ratios (HRs) and 95% CIs of the association between AF/AFL and the study outcomes. Data were analyzed using SAS, version 9.4 for Windows.
RESULTS
From 2014 through 2019, 35,539 patients with WPW syndrome were identified in the MHS, 5291 had AF/AFL (14.9%); 19,961 were female (56.2%), the mean (SD) age was 62.9 (18.0) years, and 11,742 were aged ≥ 75 years (33.0%) (Table 1).
There were 4121 (11.6%), 322 (0.9%), and 848 (2.4%) patients with AF, AFL, and both arrhythmias, respectively. The mean (SD) number of cardiology visits was 3.9 (3.0). The mean (SD) baseline CCI score for the AF/AFL subgroup was 5.9 (3.5) vs 3.7 (2.2) for the non-AF/AFL subgroup (P < .001). The most prevalent comorbid conditions were hypertension, hyperlipidemia, chronic obstructive pulmonary disease, and diabetes (P < .001) (Figure 1).
Composite Outcomes
In the overall cohort, during a mean (SD) follow-up time of 3.4 (2.0) years comprising 119,682 total person-years, the components of the composite outcome occurred 6506 times with an incidence rate of 5.44 per 100 person-years. Ventricular tachycardia was the most common event, occurring 3281 times with an incidence rate of 2.74 per 100 person-years. SCA and VF occurred 841 and 135 times with incidence rates of 0.70 and 0.11 per 100 person-years, respectively. Death was the initial event 2249 times with an incidence rate of 1.88 per 100 person-years. Figure 2 shows the Kaplan-Meier curve of cardiac composite outcome by AF/AFL status.
The subgroup with AF/AFL comprised 17,412 total person-years and 1424 cardiac composite incidences compared with 102,270 person years and 5082 incidences in the no AF/AFL group (Table 2). Comparing AF/AFL vs no AF/AFL incidence rates were 8.18 vs 4.97 per 100 person-years, respectively (P < .001). SCA and VF occurred 233 and 38 times and respectively had incidence rates of 1.34 and 0.22 per 100 person-years in the AF/AFL group vs 0.59 and 0.09 per 100 person-years in the no AF/AFL group (P < .001). There were 549 deaths and a 3.15 per 100 person-years incidence rate in the AF/AFL group vs 1700 deaths and a 1.66 incidence rate in the no AF/AFL group (P < .001).
The HR for the composite outcome in the base model was 1.33 (95% CI, 1.26-1.42, P < .001) (Table 3). The association between AF/AFL and the composite outcome remained significant after adjusting for additional metabolic and cardiac covariates. The HRs for the metabolic and cardiac models were 1.30 (95% CI, 1.23-1.38, P < .001) and 1.11 (95% CI, 1.05-1.18, P < .001), respectively. After adjusting for the full model, the HR was 1.12 (95% CI, 1.05-1.19, P < .001).
Mortality
Over the 6-year study period, there was a lower survival probability for patients with AF/AFL. In the overall cohort, during a mean (SD) follow-up time of 3.7 (1.9) years comprising 129,391 total person-years, there were 3130 (8.8%) deaths and an incidence rate of 2.42 per 100 person-years. Death occurred 786 times with a 4.09 incidence rate per 100 person-years in the AF/AFL vs 2344 deaths and a 2.13 incidence rate per 100 person-years in the no AF/AFL group (P < .001). In the non-AF/AFL subgroup, death occurred 2344 times during a mean (SD) follow-up of 3.7 (1.9) years comprising 110,151 total person-years. Figure 3 shows the Kaplan-Meier curve of mortality by AF/AFL status.
After adjusting for the base, metabolic and cardiac covariates, the HRs for mortality were 1.45 (95% CI, 1.33-1.57, P < .001), 1.40 (95% CI, 1.29-1.51, P < .001) and 1.15 (95% CI, 1.06-1.25, P = .001), respectively (Table 4). The HR after adjusting for the full model was 1.16 (95% CI, 1.07-1.26, P < .001).
DISCUSSION
In this large retrospective cohort study, patients with WPW syndrome and comorbid AF/AFL had a significantly higher association with the cardiac composite outcome and death during a 3-year follow-up period when compared with patients without AF/AFL. After adjusting for confounding variables, the AF/AFL subgroup maintained a 12% and 16% higher association with the composite outcome and mortality, respectively. There was minimal difference in confounding effects between demographic data and metabolic profiles, suggesting one may serve as a proxy for the other.
To our knowledge, this is the largest WPW syndrome cohort study evaluating cardiac outcomes and mortality to date. Although previous research has shown the relatively low and mostly anecdotal SCD incidence within this population,our results demonstrate a higher association of adverse cardiac outcomes and death in an AF/AFL subgroup.16-18 Notably, in this study the AF/AFL cohort was older and had higher CCI scores than their counterparts (P < .001), thus inferring an inherently greater degree of morbidity and 10-year mortality risk. Our study is also unique in that the mean patient age was significantly older than previously reported (63 vs 27 years), which may suggest a longer living history of both ventricular pre-excitation and the comorbidities outlined in Figure 1.19 Given these age discrepancies, it is possible that our overall study population was still relatively low risk and that not all reported deaths were necessarily related to WPW syndrome. Despite these assumptions, when comparing the WPW syndrome subgroups, we still found the AF/AFL cohort maintained a statistically significant higher association with the 2 study outcomes, even after adjusting for the greater presence of comorbidities. This suggests that the presence of AF/AFL may still portend a worse prognosis in patients with WPW syndrome.
Although the association of AF and development of VF in patients with WPW syndrome—due to rapid conduction over the accessory pathway(s)—was first reported > 40 years ago, there has still been few large, long-term data studies exploring mortality in this cohort.19-25 Furthermore, even though the current literature attributes the development of AF with the electrophysiologic properties of the accessory pathway, as well as intrinsic atrial architecture and muscle vulnerability, there is still equivocal consensus regarding EPT screening and ablation indications for asymptomatic patients with WPW syndrome.26-28 Notably, Pappone and colleagues demonstrated the potential benefit of liberal ablation indications for asymptomatic patients, arguing that the intrinsic electrophysiologic properties of the accessory pathway—ie, short accessory-pathway antegrade effective refractory period, inducibility of atrioventricular reentrant tachycardia triggering AF, and multiple accessory pathway—rather than symptoms, are independent predictors of developing malignant arrhythmia.1-5
These findings contradict those reported by Obeyesekere and colleagues, who concluded that the low SCD incidence rates in patients with WPW syndrome precluded routine invasive screening.19,28 They argued that Pappone and colleagues used malignant arrhythmia as a surrogate marker for death, and that the positive predictive value of a short accessory-pathway antegrade effective refractory period for developing malignant arrhythmia was lower than reported (15% vs 82%, respectively) and that its negative predictive value was 100%.1,19,28 Given these conflicting recommendations, we hope our data elucidates the higher association of adverse outcomes and support considerations for more intensive EPT indications in patients with WPW syndrome.
While our study does not report SCD incidence, it does provide robust and reliable mortality data that suggests a greater association of death within an AF/AFL subgroup. Our findings would support more liberal EPT recommendations in patients with WPW syndrome.1-5,8,9 In this study, the SCA incidence rate was more than double the rate in the AF/AFL cohort (P < .001) and is commonly the initial presenting event in WPW syndrome.9 Even though the reported SCD incidence rate is low in WPW syndrome, our data demonstrated an increased association of death within the AF/AFL cohort. Physicians should consider early risk stratification and ablation to prevent potential recurrent malignant arrhythmia leading to death.1-5,8,9,12,19,20
Limitations
As a retrospective study and without access to the National Death Index, we were unable to determine the exact cause or events leading to death and instead utilized all-cause mortality data. Subsequently, our observations may only demonstrate association, rather than causality, between AF/AFL and death in patients with WPW syndrome. Additionally, we could not distinguish between AF and AFL as the arrhythmia leading to death. However, since overall survivability was the outcome of interest, our adjusted HR models were still able to demonstrate the increased association of the composite outcome and death within an AF/AFL cohort.
Although a large cohort was analyzed, due to the constraints of utilizing diagnostic codes to determine study outcomes, we could not distinguish between symptomatic and asymptomatic patients, nor how they were managed prior to the outcome event. However, as recent literature demonstrates, updated predictors of malignant arrhythmia and decisions for early EPT are similar for both symptomatic and asymptomatic patients and should be driven by the intrinsic electrophysiologic properties of the accessory pathway, rather than symptomatology;thus, our inability to discern this should have negligible consequence in determining when to perform risk stratification and ablation.1
MHS eligible patients have direct access to care; the generalizability of our data may not necessarily correspond to a community population with lower socioeconomic status (we did adjust for military sponsor rank which has been used as a proxy), reduced access to care, or uninsured individuals. However, the prevalence of WPW syndrome within our cohort was comparable to the general population, 0.4% vs 0.1%-0.3%, respectively.13,14,19 Similarly, the incidence of AF within our population was comparable to the general population, 15% vs 16%-26%, respectively.23 These similar data points suggest our results may apply beyond MHS patients.
CONCLUSIONS
Patients with WPW syndrome and AF/AFL have a higher association with adverse cardiac outcomes and death. Despite previously reported low SCD incidence rates in this population, our study demonstrates the increased association of mortality in an AF/AFL cohort. The limitations of utilizing all-cause mortality data necessitate further investigation into the etiology behind the deaths in our study population. Since ventricular pre-excitation can predispose patients to AF and potentially lead to malignant arrhythmia and SCD, understanding the cause of mortality will allow physicians to determine the appropriate monitoring and intervention strategies to improve outcomes in this population. Our results suggest consideration for more aggressive EPT screening and ablation recommendations in patients with WPW syndrome may be warranted.
1. Pappone C, Vicedomini G, Manguso F, et al. The natural history of WPW syndrome. Eur Heart J Suppl. 2015; 17 (Supplement A):A8-A11.doi:10.1093/eurheartj/suv004
2. Pappone C, Vicedomini G, Manguso F, et al. Risk of malignant arrhythmias in initially symptomatic patients with Wolff-Parkinson-White syndrome: results of a prospective long-term electrophysiological follow-up study. Circulation. 2012;125(5):661-668. doi:10.1161/CIRCULATIONAHA.111.065722
3. Pappone C, Santinelli V, Rosanio S, et al. Usefulness of invasive electrophysiologic testing to stratify the risk of arrhythmic events in asymptomatic patients with Wolff-Parkinson-White pattern: results from a large prospective long-term follow-up study. J Am Coll Cardiol. 2003;41(2):239-244. doi:10.1016/s0735-1097(02)02706-7
4. Pappone C, Vicedomini G, Manguso F, et al. Wolff-Parkinson-White syndrome in the era of catheter ablation: insights from a registry study of 2169 patients. Circulation. 2014;130(10):811-819. doi:10.1161/CIRCULATIONAHA.114.011154
5. Pappone C, Santinelli V, Manguso F, et al. A randomized study of prophylactic catheter ablation in asymptomatic patients with the Wolff-Parkinson-White syndrome. N Engl J Med. 2003;349(19):1803-1811. doi:10.1056/NEJMoa035345
6. Santinelli V, Radinovic A, Manguso F, et al. Asymptomatic ventricular preexcitation: a long-term prospective follow-up study of 293 adult patients. Circ Arrhythm Electrophysiol. 2009;2(2):102-107. doi:10.1161/CIRCEP.108.827550
7. Santinelli V, Radinovic A, Manguso F, et al. The natural history of asymptomatic ventricular pre-excitation a long-term prospective follow-up study of 184 asymptomatic children. J Am Coll Cardiol. 2009;53(3):275-280. doi:10.1016/j.jacc.2008.09.037
8. Al-Khatib SM, Arshad A, Balk EM, et al. Risk Stratification for Arrhythmic Events in Patients With Asymptomatic Pre-Excitation: A Systematic Review for the 2015 ACC/AHA/HRS Guideline for the Management of Adult Patients With Supraventricular Tachycardia: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2016;67(13):1624-1638. doi:10.1016/j.jacc.2015.09.018
9. Blomström-Lundqvist C, Scheinman MM, Aliot EM, et al. ACC/AHA/ESC guidelines for the management of patients with supraventricular arrhythmias--executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Supraventricular Arrhythmias). Circulation. 2003;108(15):1871-1909.doi:10.1161/01.CIR.0000091380.04100.84
10. Pediatric and Congenital Electrophysiology Society (PACES); Heart Rhythm Society (HRS); American College of Cardiology Foundation (ACCF); PACES/HRS expert consensus statement on the management of the asymptomatic young patient with a Wolff-Parkinson-White (WPW, ventricular preexcitation) electrocardiographic pattern: developed in partnership between the Pediatric and Congenital Electrophysiology Society (PACES) and the Heart Rhythm Society (HRS). Endorsed by the governing bodies of PACES, HRS, the American College of Cardiology Foundation (ACCF), the American Heart Association (AHA), the American Academy of Pediatrics (AAP), and the Canadian Heart Rhythm Society (CHRS). Heart Rhythm. 2012;9(6):1006-1024. doi:10.1016/j.hrthm.2012.03.050
11. Cohen M, Triedman J. Guidelines for management of asymptomatic ventricular pre-excitation: brave new world or Pandora’s box?. Circ Arrhythm Electrophysiol. 2014;7(2):187-189. doi:10.1161/CIRCEP.114.001528
12. Svendsen JH, Dagres N, Dobreanu D, et al. Current strategy for treatment of patients with Wolff-Parkinson-White syndrome and asymptomatic preexcitation in Europe: European Heart Rhythm Association survey. Europace. 2013;15(5):750-753. doi:10.1093/europace/eut094
13. Gimbel RW, Pangaro L, Barbour G. America’s “undiscovered” laboratory for health services research. Med Care. 2010;48(8):751-756. doi:10.1097/MLR.0b013e3181e35be8
14. Dorrance KA, Ramchandani S, Neil N, Fisher H. Leveraging the military health system as a laboratory for health care reform. Mil Med. 2013;178(2):142-145. doi:10.7205/milmed-d-12-00168
15. Quan H, Sundararajan V, Halfon P, et al. Coding algorithms for defining comorbidities in ICD-9-CM and ICD-10 administrative data. Med Care. 2005;43(11):1130-1139. doi:10.1097/01.mlr.0000182534.19832.83
16. Finocchiaro G, Papadakis M, Behr ER, Sharma S, Sheppard M. Sudden Cardiac Death in Pre-Excitation and Wolff-Parkinson-White: Demographic and Clinical Features. J Am Coll Cardiol. 2017;69(12):1644-1645. doi:10.1016/j.jacc.2017.01.023
17. Munger TM, Packer DL, Hammill SC, et al. A population study of the natural history of Wolff-Parkinson-White syndrome in Olmsted County, Minnesota, 1953-1989. Circulation. 1993;87(3):866-873. doi:10.1161/01.cir.87.3.866
18. Fitzsimmons PJ, McWhirter PD, Peterson DW, Kruyer WB. The natural history of Wolff-Parkinson-White syndrome in 228 military aviators: a long-term follow-up of 22 years. Am Heart J. 2001;142(3):530-536. doi:10.1067/mhj.2001.117779
19. Obeyesekere MN, Leong-Sit P, Massel D, et al. Risk of arrhythmia and sudden death in patients with asymptomatic preexcitation: a meta-analysis. Circulation. 2012;125(19):2308-2315. doi:10.1161/CIRCULATIONAHA.111.055350
20. Waspe LE, Brodman R, Kim SG, Fisher JD. Susceptibility to atrial fibrillation and ventricular tachyarrhythmia in the Wolff-Parkinson-White syndrome: role of the accessory pathway. Am Heart J. 1986;112(6):1141-1152. doi:10.1016/0002-8703(86)90342-x
21. Pietersen AH, Andersen ED, Sandøe E. Atrial fibrillation in the Wolff-Parkinson-White syndrome. Am J Cardiol. 1992;70(5):38A-43A. doi:10.1016/0002-9149(92)91076-g
22. Della Bella P, Brugada P, Talajic M, et al. Atrial fibrillation in patients with an accessory pathway: importance of the conduction properties of the accessory pathway. J Am Coll Cardiol. 1991;17(6):1352-1356. doi:10.1016/s0735-1097(10)80146-9
23. Fujimura O, Klein GJ, Yee R, Sharma AD. Mode of onset of atrial fibrillation in the Wolff-Parkinson-White syndrome: how important is the accessory pathway?. J Am Coll Cardiol. 1990;15(5):1082-1086. doi:10.1016/0735-1097(90)90244-j
24. Montoya PT, Brugada P, Smeets J, et al. Ventricular fibrillation in the Wolff-Parkinson-White syndrome. Eur Heart J. 1991;12(2):144-150. doi:10.1093/oxfordjournals.eurheartj.a059860
25. Klein GJ, Bashore TM, Sellers TD, Pritchett EL, Smith WM, Gallagher JJ. Ventricular fibrillation in the Wolff-Parkinson-White syndrome. N Engl J Med. 1979;301(20):1080-1085. doi:10.1056/NEJM197911153012003
26. Centurion OA. Atrial Fibrillation in the Wolff-Parkinson-White Syndrome. J Atr Fibrillation. 2011;4(1):287. Published 2011 May 4. doi:10.4022/jafib.287
27. Song C, Guo Y, Zheng X, et al. Prognostic Significance and Risk of Atrial Fibrillation of Wolff-Parkinson-White Syndrome in Patients With Hypertrophic Cardiomyopathy. Am J Cardiol. 2018;122(9):1546-1550. doi:10.1016/j.amjcard.2018.07.021
28. Obeyesekere M, Gula LJ, Skanes AC, Leong-Sit P, Klein GJ. Risk of sudden death in Wolff-Parkinson-White syndrome: how high is the risk?. Circulation. 2012;125(5):659-660. doi:10.1161/CIRCULATIONAHA.111.085159
1. Pappone C, Vicedomini G, Manguso F, et al. The natural history of WPW syndrome. Eur Heart J Suppl. 2015; 17 (Supplement A):A8-A11.doi:10.1093/eurheartj/suv004
2. Pappone C, Vicedomini G, Manguso F, et al. Risk of malignant arrhythmias in initially symptomatic patients with Wolff-Parkinson-White syndrome: results of a prospective long-term electrophysiological follow-up study. Circulation. 2012;125(5):661-668. doi:10.1161/CIRCULATIONAHA.111.065722
3. Pappone C, Santinelli V, Rosanio S, et al. Usefulness of invasive electrophysiologic testing to stratify the risk of arrhythmic events in asymptomatic patients with Wolff-Parkinson-White pattern: results from a large prospective long-term follow-up study. J Am Coll Cardiol. 2003;41(2):239-244. doi:10.1016/s0735-1097(02)02706-7
4. Pappone C, Vicedomini G, Manguso F, et al. Wolff-Parkinson-White syndrome in the era of catheter ablation: insights from a registry study of 2169 patients. Circulation. 2014;130(10):811-819. doi:10.1161/CIRCULATIONAHA.114.011154
5. Pappone C, Santinelli V, Manguso F, et al. A randomized study of prophylactic catheter ablation in asymptomatic patients with the Wolff-Parkinson-White syndrome. N Engl J Med. 2003;349(19):1803-1811. doi:10.1056/NEJMoa035345
6. Santinelli V, Radinovic A, Manguso F, et al. Asymptomatic ventricular preexcitation: a long-term prospective follow-up study of 293 adult patients. Circ Arrhythm Electrophysiol. 2009;2(2):102-107. doi:10.1161/CIRCEP.108.827550
7. Santinelli V, Radinovic A, Manguso F, et al. The natural history of asymptomatic ventricular pre-excitation a long-term prospective follow-up study of 184 asymptomatic children. J Am Coll Cardiol. 2009;53(3):275-280. doi:10.1016/j.jacc.2008.09.037
8. Al-Khatib SM, Arshad A, Balk EM, et al. Risk Stratification for Arrhythmic Events in Patients With Asymptomatic Pre-Excitation: A Systematic Review for the 2015 ACC/AHA/HRS Guideline for the Management of Adult Patients With Supraventricular Tachycardia: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2016;67(13):1624-1638. doi:10.1016/j.jacc.2015.09.018
9. Blomström-Lundqvist C, Scheinman MM, Aliot EM, et al. ACC/AHA/ESC guidelines for the management of patients with supraventricular arrhythmias--executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Supraventricular Arrhythmias). Circulation. 2003;108(15):1871-1909.doi:10.1161/01.CIR.0000091380.04100.84
10. Pediatric and Congenital Electrophysiology Society (PACES); Heart Rhythm Society (HRS); American College of Cardiology Foundation (ACCF); PACES/HRS expert consensus statement on the management of the asymptomatic young patient with a Wolff-Parkinson-White (WPW, ventricular preexcitation) electrocardiographic pattern: developed in partnership between the Pediatric and Congenital Electrophysiology Society (PACES) and the Heart Rhythm Society (HRS). Endorsed by the governing bodies of PACES, HRS, the American College of Cardiology Foundation (ACCF), the American Heart Association (AHA), the American Academy of Pediatrics (AAP), and the Canadian Heart Rhythm Society (CHRS). Heart Rhythm. 2012;9(6):1006-1024. doi:10.1016/j.hrthm.2012.03.050
11. Cohen M, Triedman J. Guidelines for management of asymptomatic ventricular pre-excitation: brave new world or Pandora’s box?. Circ Arrhythm Electrophysiol. 2014;7(2):187-189. doi:10.1161/CIRCEP.114.001528
12. Svendsen JH, Dagres N, Dobreanu D, et al. Current strategy for treatment of patients with Wolff-Parkinson-White syndrome and asymptomatic preexcitation in Europe: European Heart Rhythm Association survey. Europace. 2013;15(5):750-753. doi:10.1093/europace/eut094
13. Gimbel RW, Pangaro L, Barbour G. America’s “undiscovered” laboratory for health services research. Med Care. 2010;48(8):751-756. doi:10.1097/MLR.0b013e3181e35be8
14. Dorrance KA, Ramchandani S, Neil N, Fisher H. Leveraging the military health system as a laboratory for health care reform. Mil Med. 2013;178(2):142-145. doi:10.7205/milmed-d-12-00168
15. Quan H, Sundararajan V, Halfon P, et al. Coding algorithms for defining comorbidities in ICD-9-CM and ICD-10 administrative data. Med Care. 2005;43(11):1130-1139. doi:10.1097/01.mlr.0000182534.19832.83
16. Finocchiaro G, Papadakis M, Behr ER, Sharma S, Sheppard M. Sudden Cardiac Death in Pre-Excitation and Wolff-Parkinson-White: Demographic and Clinical Features. J Am Coll Cardiol. 2017;69(12):1644-1645. doi:10.1016/j.jacc.2017.01.023
17. Munger TM, Packer DL, Hammill SC, et al. A population study of the natural history of Wolff-Parkinson-White syndrome in Olmsted County, Minnesota, 1953-1989. Circulation. 1993;87(3):866-873. doi:10.1161/01.cir.87.3.866
18. Fitzsimmons PJ, McWhirter PD, Peterson DW, Kruyer WB. The natural history of Wolff-Parkinson-White syndrome in 228 military aviators: a long-term follow-up of 22 years. Am Heart J. 2001;142(3):530-536. doi:10.1067/mhj.2001.117779
19. Obeyesekere MN, Leong-Sit P, Massel D, et al. Risk of arrhythmia and sudden death in patients with asymptomatic preexcitation: a meta-analysis. Circulation. 2012;125(19):2308-2315. doi:10.1161/CIRCULATIONAHA.111.055350
20. Waspe LE, Brodman R, Kim SG, Fisher JD. Susceptibility to atrial fibrillation and ventricular tachyarrhythmia in the Wolff-Parkinson-White syndrome: role of the accessory pathway. Am Heart J. 1986;112(6):1141-1152. doi:10.1016/0002-8703(86)90342-x
21. Pietersen AH, Andersen ED, Sandøe E. Atrial fibrillation in the Wolff-Parkinson-White syndrome. Am J Cardiol. 1992;70(5):38A-43A. doi:10.1016/0002-9149(92)91076-g
22. Della Bella P, Brugada P, Talajic M, et al. Atrial fibrillation in patients with an accessory pathway: importance of the conduction properties of the accessory pathway. J Am Coll Cardiol. 1991;17(6):1352-1356. doi:10.1016/s0735-1097(10)80146-9
23. Fujimura O, Klein GJ, Yee R, Sharma AD. Mode of onset of atrial fibrillation in the Wolff-Parkinson-White syndrome: how important is the accessory pathway?. J Am Coll Cardiol. 1990;15(5):1082-1086. doi:10.1016/0735-1097(90)90244-j
24. Montoya PT, Brugada P, Smeets J, et al. Ventricular fibrillation in the Wolff-Parkinson-White syndrome. Eur Heart J. 1991;12(2):144-150. doi:10.1093/oxfordjournals.eurheartj.a059860
25. Klein GJ, Bashore TM, Sellers TD, Pritchett EL, Smith WM, Gallagher JJ. Ventricular fibrillation in the Wolff-Parkinson-White syndrome. N Engl J Med. 1979;301(20):1080-1085. doi:10.1056/NEJM197911153012003
26. Centurion OA. Atrial Fibrillation in the Wolff-Parkinson-White Syndrome. J Atr Fibrillation. 2011;4(1):287. Published 2011 May 4. doi:10.4022/jafib.287
27. Song C, Guo Y, Zheng X, et al. Prognostic Significance and Risk of Atrial Fibrillation of Wolff-Parkinson-White Syndrome in Patients With Hypertrophic Cardiomyopathy. Am J Cardiol. 2018;122(9):1546-1550. doi:10.1016/j.amjcard.2018.07.021
28. Obeyesekere M, Gula LJ, Skanes AC, Leong-Sit P, Klein GJ. Risk of sudden death in Wolff-Parkinson-White syndrome: how high is the risk?. Circulation. 2012;125(5):659-660. doi:10.1161/CIRCULATIONAHA.111.085159
Nasal Cannula Dislodgement During Sleep in Veterans Receiving Long-term Oxygen Therapy for Hypoxemic Chronic Respiratory Failure
The prevalence of chronic obstructive pulmonary disease (COPD) among male US veterans is higher than in the general population.1 Veterans with COPD have higher rates of comorbidities and increased respiratory-related and all-cause health care use, including the use of long-term oxygen therapy (LTOT).2-5 It has been well established that LTOT reduces all-cause mortality in patients with COPD and
Delivery of domiciliary LTOT entails placing a nasal cannula into both nostrils and loosely securing it around both ears throughout the wake-sleep cycle. Several veterans with hypoxemic CRF due to COPD at the Jesse Brown Veterans Affairs Medical Center (JBVAMC) in Chicago, Illinois, who were receiving LTOT reported nasal cannula dislodgement (NCD) while they slept. However, the clinical significance and impact of these repeated episodes on respiratory-related health care utilization, such as frequent COPD exacerbations with hospitalization, were not recognized.
The purpose of this study was to determine whether veterans with hypoxemic CRF due to COPD and receiving 24-hour LTOT at JBVAMC were experiencing NCD during sleep and, if so, its impact on
METHODS
We reviewed electronic health records (EHRs) of veterans with hypoxemic CRF from COPD who received 24-hour LTOT administered through nasal cannula and were followed
Pertinent patient demographics, clinical and physiologic variables, and hospitalizations with length of JBVAMC stay for each physician-diagnosed COPD exacerbation in the preceding year from the date last seen in the clinic were abstracted from EHRs. Overall hospital cost, defined as a veteran overnight stay in either the medical intensive care unit (MICU) or a general acute medicine bed in a US Department of Veterans Affairs (VA) facility, was calculated for each hospitalization for physician-diagnosed COPD exacerbation using VA Managerial Cost Accounting System National Cost Extracts for inpatient encounters.15 We then contacted each veteran by telephone and asked whether they had experienced NCD and, if so, its weekly frequency ranging from once to nightly.
Data Analysis
Data were reported as mean (SD) where appropriate. The t test and Fisher exact test were used as indicated. P < .05 was considered statistically significant. The study protocol
RESULTS

During the study period,

Of the 75 patients, 66 (88%) responded to the telephone survey and 22 patients (33%) reported weekly episodes of NCD while they slept (median, 4 dislodgments per week). (Table 1). Eight patients (36%) reported nightly NCDs (Figure). All 66 respondents were male and 14 of 22 in the NCD group as well as 21 of 44 in the no NCD group were Black veterans. The mean age was similar in both groups: 71 years in the NCD group and 72 years in the no NCD group. There were no statistically significant differences in demographics, including prevalence of obstructive sleep apnea (OSA), supplemental oxygen flow rate, and duration of LTOT, or in pulmonary function test results between patients who did and did not experience NCD while sleeping (Table 2).


Ten of 22 patients (45%) with NCD and 9 of 44 patients (20%) without NCD were hospitalized at the JBVAMC for ≥ 1 COPD exacerbation in the preceding year that was diagnosed by a physician (P = .045). Three of 22 patients (14%) with NCD and no patients in the no NCD group were admitted to the MICU. No patients required intubation and mechanical ventilation during hospitalization, and no patients died. Overall hospital costs were 25% ($64,342) higher in NCD group compared with the no NCD group and were attributed to the MICU admissions in the NCD group (Table 3). Nine veterans did not respond to repeated telephone calls. One physician-diagnosed COPD exacerbation requiring hospitalization was documented in the nonresponder group; the patient was hospitalized for 2 days. One veteran died before being contacted.
DISCUSSION
There are 3 new findings in this study.
Nocturnal arterial oxygen desaturation in patients with COPD without evidence of OSA may contribute to the frequency of exacerbations.16 Although the mechanism(s) underlying this phenomenon is uncertain, we posit that prolonged nocturnal airway wall hypoxia could amplify underlying chronic inflammation through local generation of reactive oxygen species, thereby predisposing patients to exacerbations. Frequent COPD exacerbations promote disease progression and health status decline and are associated with increased mortality.11,13 Moreover, hospitalization of patients with COPD is the largest contributor to the annual direct cost of COPD per patient.10,12 The higher hospitalization rate observed in the NCD group in our study suggests that interruption of supplemental oxygen delivery while asleep may be a risk factor for COPD exacerbation. Alternatively, an independent factor or factors may have contributed to both NCD during sleep and COPD exacerbation in these patients or an impending exacerbation resulted in sleep disturbances that led to NCD. Additional research is warranted on veterans with hypoxemic CRF from COPD who are receiving LTOT and report frequent NCD during sleep that may support or refute these hypotheses.
To the best of our knowledge, NCD during sleep has not been previously reported in patients
Limitations
This was a small, single-site study, comprised entirely of male patients who are predominantly Black veterans. The telephone interviews with veterans self-reporting NCD during their sleep are prone to recall bias. In addition, the validity and reproducibility of NCD during sleep were not addressed in this study. Missing data from 9 nonresponders may have introduced a nonresponse bias in data analysis and interpretation. The overall hospital cost for a COPD exacerbation at JBVAMC was derived from VA data; US Centers for Medicare & Medicaid Services or commercial carrier data may be different.15,21 Lastly, access to LTOT for veterans with hypoxemic CRF from COPD is regulated and supervised at VA medical facilities.14 This process may be different for patients outside the VA. Taken together, it is difficult to generalize our initial observations to non-VA patients with hypoxemic CRF from COPD who are receiving LTOT. We suggest a large, prospective study of veterans be conducted to determine the prevalence of NCD during sleep and its relationship with COPD exacerbations in veterans receiving LTOT with hypoxemic CRF due to COPD.
CONCLUSIONS
Acknowledgments
We thank Yolanda Davis, RRT, and George Adam for their assistance with this project.
1. Boersma P, Cohen RA, Zelaya CE, Moy E. Multiple chronic conditions among veterans and nonveterans: United States, 2015-2018. Natl Health Stat Report. 2021;(153):1-13. doi:10.15620/cdc:101659
2. Sharafkhaneh A, Petersen NJ, Yu H-J, Dalal AA, Johnson ML, Hanania NA. Burden of COPD in a government health care system: a retrospective observational study using data from the US Veterans Affairs population. Int J Chron Obstruct Pulmon Dis. 2010;5:125-132. doi:10.2147/copd.s8047
3. LaBedz SL, Krishnan JA, Chung Y-C, et al. Chronic obstructive pulmonary disease outcomes at Veterans Affairs versus non-Veterans Affairs hospitals. Chronic Obstr Pulm Dis. 2021;8(3):306-313. doi:10.15326/jcopdf.2021.0201
4. Darnell K, Dwivedi AK, Weng Z, Panos RJ. Disproportionate utilization of healthcare resources among veterans with COPD: a retrospective analysis of factors associated with COPD healthcare cost. Cost Eff Resour Alloc. 2013;11:13. doi:10.1186/1478-7547-11-13
5. Bamonti PM, Robinson SA, Wan ES, Moy ML. Improving physiological, physical, and psychological health outcomes: a narrative review in US Veterans with COPD. Int J Chron Obstruct Pulmon Dis. 2022;17:1269-1283. doi:10.2147/COPD.S339323
6. Cranston JM, Crockett AJ, Moss JR, Alpers JH. Domiciliary oxygen for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2005;2005(4):CD001744. doi:10.1002/14651858.CD001744.pub2
7. Lacasse Y, Tan AM, Maltais F, Krishnan JA. Home oxygen in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2018;197(10):1254-1264. doi:10.1164/rccm.201802-0382CI
8. Jacobs SS, Krishnan JA, Lederer DJ, et al. Home oxygen therapy for adults with chronic lung disease. An official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2020;202(10):e121-e141. doi:10.1164/rccm.202009-3608ST
9. AARC. AARC clinical practice guideline. Oxygen therapy in the home or alternate site health care facility--2007 revision & update. Respir Care. 2007;52(8):1063-1068.
10. Foo J, Landis SH, Maskell J, et al. Continuing to confront COPD international patient survey: economic impact of COPD in 12 countries. PLoS One. 2016;11(4):e0152618. doi:10.1371/journal.pone.0152618
11. Rothnie KJ, Müllerová H, Smeeth L, Quint JK. Natural history of chronic obstructive pulmonary disease exacerbations in a general practice-based population with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2018;198(4):464-471. doi:10.1164/rccm.201710-2029OC
12. Stanford RH, Engel-Nitz NM, Bancroft T, Essoi B. The identification and cost of acute chronic obstructive pulmonary disease exacerbations in a United States population healthcare claims database. COPD. 2020;17(5):499-508. doi:10.1080/15412555.2020.1817357
13. Hurst JR, Han MK, Singh B, et al. Prognostic risk factors for moderate-to-severe exacerbations in patients with chronic obstructive pulmonary disease: a systematic literature review. Respir Res. 2022;23(1):213. doi:10.1186/s12931-022-02123-5
14. US Department of Veterans Affairs, Veterans Health Administration. Home oxygen program. VHA Directive 1173.13(1). Published August 5, 2020. Accessed February 28, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=8947
15. Phibbs CS, Barnett PG, Fan A, Harden C, King SS, Scott JY. Research guide to decision support system national cost extracts. Health Economics Resource Center of Health Service R&D Services, US Department of Veterans Affairs. September 2010. Accessed February 14, 2024. https://www.herc.research.va.gov/files/book_621.pdf
16. Agusti A, Hedner J, Marin JM, Barbé F, Cazzola M, Rennard S. Night-time symptoms: a forgotten dimension of COPD. Eur Respir Rev. 2011;20(121):183-194. doi:10.1183/09059180.00004311
17. Croxton TL, Bailey WC. Long-term oxygen treatment in chronic obstructive pulmonary disease: recommendations for future research: an NHLBI workshop report. Am J Respir Crit Care Med. 2006;174(4):373-378. doi:10.1164/rccm.200507-1161WS
18. Melani AS, Sestini P, Rottoli P. Home oxygen therapy: re-thinking the role of devices. Expert Rev Clin Pharmacol. 2018;11(3):279-289. doi:10.1080/17512433.2018.1421457
19. Sculley JA, Corbridge SJ, Prieto-Centurion V, et al. Home oxygen therapy for patients with COPD: time for a reboot. Respir Care. 2019;64(12):1574-1585. doi:10.4187/respcare.07135
20. Jacobs SS, Lindell KO, Collins EG, et al. Patient perceptions of the adequacy of supplemental oxygen therapy. Results of the American Thoracic Society Nursing Assembly Oxygen Working Group Survey. Ann Am Thorac Soc. 2018;15:24-32. doi:10.1513/AnnalsATS.201703-209OC
21. US Centers for Medicare & Medicaid Services. Home use of oxygen. Publication number 100-3. January 3, 2023. Accessed February 14, 2024. https://www.cms.gov/medicare-coverage-database/view/ncd.aspx?NCDId=169
The prevalence of chronic obstructive pulmonary disease (COPD) among male US veterans is higher than in the general population.1 Veterans with COPD have higher rates of comorbidities and increased respiratory-related and all-cause health care use, including the use of long-term oxygen therapy (LTOT).2-5 It has been well established that LTOT reduces all-cause mortality in patients with COPD and
Delivery of domiciliary LTOT entails placing a nasal cannula into both nostrils and loosely securing it around both ears throughout the wake-sleep cycle. Several veterans with hypoxemic CRF due to COPD at the Jesse Brown Veterans Affairs Medical Center (JBVAMC) in Chicago, Illinois, who were receiving LTOT reported nasal cannula dislodgement (NCD) while they slept. However, the clinical significance and impact of these repeated episodes on respiratory-related health care utilization, such as frequent COPD exacerbations with hospitalization, were not recognized.
The purpose of this study was to determine whether veterans with hypoxemic CRF due to COPD and receiving 24-hour LTOT at JBVAMC were experiencing NCD during sleep and, if so, its impact on
METHODS
We reviewed electronic health records (EHRs) of veterans with hypoxemic CRF from COPD who received 24-hour LTOT administered through nasal cannula and were followed
Pertinent patient demographics, clinical and physiologic variables, and hospitalizations with length of JBVAMC stay for each physician-diagnosed COPD exacerbation in the preceding year from the date last seen in the clinic were abstracted from EHRs. Overall hospital cost, defined as a veteran overnight stay in either the medical intensive care unit (MICU) or a general acute medicine bed in a US Department of Veterans Affairs (VA) facility, was calculated for each hospitalization for physician-diagnosed COPD exacerbation using VA Managerial Cost Accounting System National Cost Extracts for inpatient encounters.15 We then contacted each veteran by telephone and asked whether they had experienced NCD and, if so, its weekly frequency ranging from once to nightly.
Data Analysis
Data were reported as mean (SD) where appropriate. The t test and Fisher exact test were used as indicated. P < .05 was considered statistically significant. The study protocol
RESULTS
During the study period,
Of the 75 patients, 66 (88%) responded to the telephone survey and 22 patients (33%) reported weekly episodes of NCD while they slept (median, 4 dislodgments per week). (Table 1). Eight patients (36%) reported nightly NCDs (Figure). All 66 respondents were male and 14 of 22 in the NCD group as well as 21 of 44 in the no NCD group were Black veterans. The mean age was similar in both groups: 71 years in the NCD group and 72 years in the no NCD group. There were no statistically significant differences in demographics, including prevalence of obstructive sleep apnea (OSA), supplemental oxygen flow rate, and duration of LTOT, or in pulmonary function test results between patients who did and did not experience NCD while sleeping (Table 2).
Ten of 22 patients (45%) with NCD and 9 of 44 patients (20%) without NCD were hospitalized at the JBVAMC for ≥ 1 COPD exacerbation in the preceding year that was diagnosed by a physician (P = .045). Three of 22 patients (14%) with NCD and no patients in the no NCD group were admitted to the MICU. No patients required intubation and mechanical ventilation during hospitalization, and no patients died. Overall hospital costs were 25% ($64,342) higher in NCD group compared with the no NCD group and were attributed to the MICU admissions in the NCD group (Table 3). Nine veterans did not respond to repeated telephone calls. One physician-diagnosed COPD exacerbation requiring hospitalization was documented in the nonresponder group; the patient was hospitalized for 2 days. One veteran died before being contacted.
DISCUSSION
There are 3 new findings in this study.
Nocturnal arterial oxygen desaturation in patients with COPD without evidence of OSA may contribute to the frequency of exacerbations.16 Although the mechanism(s) underlying this phenomenon is uncertain, we posit that prolonged nocturnal airway wall hypoxia could amplify underlying chronic inflammation through local generation of reactive oxygen species, thereby predisposing patients to exacerbations. Frequent COPD exacerbations promote disease progression and health status decline and are associated with increased mortality.11,13 Moreover, hospitalization of patients with COPD is the largest contributor to the annual direct cost of COPD per patient.10,12 The higher hospitalization rate observed in the NCD group in our study suggests that interruption of supplemental oxygen delivery while asleep may be a risk factor for COPD exacerbation. Alternatively, an independent factor or factors may have contributed to both NCD during sleep and COPD exacerbation in these patients or an impending exacerbation resulted in sleep disturbances that led to NCD. Additional research is warranted on veterans with hypoxemic CRF from COPD who are receiving LTOT and report frequent NCD during sleep that may support or refute these hypotheses.
To the best of our knowledge, NCD during sleep has not been previously reported in patients
Limitations
This was a small, single-site study, comprised entirely of male patients who are predominantly Black veterans. The telephone interviews with veterans self-reporting NCD during their sleep are prone to recall bias. In addition, the validity and reproducibility of NCD during sleep were not addressed in this study. Missing data from 9 nonresponders may have introduced a nonresponse bias in data analysis and interpretation. The overall hospital cost for a COPD exacerbation at JBVAMC was derived from VA data; US Centers for Medicare & Medicaid Services or commercial carrier data may be different.15,21 Lastly, access to LTOT for veterans with hypoxemic CRF from COPD is regulated and supervised at VA medical facilities.14 This process may be different for patients outside the VA. Taken together, it is difficult to generalize our initial observations to non-VA patients with hypoxemic CRF from COPD who are receiving LTOT. We suggest a large, prospective study of veterans be conducted to determine the prevalence of NCD during sleep and its relationship with COPD exacerbations in veterans receiving LTOT with hypoxemic CRF due to COPD.
CONCLUSIONS
Acknowledgments
We thank Yolanda Davis, RRT, and George Adam for their assistance with this project.
The prevalence of chronic obstructive pulmonary disease (COPD) among male US veterans is higher than in the general population.1 Veterans with COPD have higher rates of comorbidities and increased respiratory-related and all-cause health care use, including the use of long-term oxygen therapy (LTOT).2-5 It has been well established that LTOT reduces all-cause mortality in patients with COPD and
Delivery of domiciliary LTOT entails placing a nasal cannula into both nostrils and loosely securing it around both ears throughout the wake-sleep cycle. Several veterans with hypoxemic CRF due to COPD at the Jesse Brown Veterans Affairs Medical Center (JBVAMC) in Chicago, Illinois, who were receiving LTOT reported nasal cannula dislodgement (NCD) while they slept. However, the clinical significance and impact of these repeated episodes on respiratory-related health care utilization, such as frequent COPD exacerbations with hospitalization, were not recognized.
The purpose of this study was to determine whether veterans with hypoxemic CRF due to COPD and receiving 24-hour LTOT at JBVAMC were experiencing NCD during sleep and, if so, its impact on
METHODS
We reviewed electronic health records (EHRs) of veterans with hypoxemic CRF from COPD who received 24-hour LTOT administered through nasal cannula and were followed
Pertinent patient demographics, clinical and physiologic variables, and hospitalizations with length of JBVAMC stay for each physician-diagnosed COPD exacerbation in the preceding year from the date last seen in the clinic were abstracted from EHRs. Overall hospital cost, defined as a veteran overnight stay in either the medical intensive care unit (MICU) or a general acute medicine bed in a US Department of Veterans Affairs (VA) facility, was calculated for each hospitalization for physician-diagnosed COPD exacerbation using VA Managerial Cost Accounting System National Cost Extracts for inpatient encounters.15 We then contacted each veteran by telephone and asked whether they had experienced NCD and, if so, its weekly frequency ranging from once to nightly.
Data Analysis
Data were reported as mean (SD) where appropriate. The t test and Fisher exact test were used as indicated. P < .05 was considered statistically significant. The study protocol
RESULTS
During the study period,
Of the 75 patients, 66 (88%) responded to the telephone survey and 22 patients (33%) reported weekly episodes of NCD while they slept (median, 4 dislodgments per week). (Table 1). Eight patients (36%) reported nightly NCDs (Figure). All 66 respondents were male and 14 of 22 in the NCD group as well as 21 of 44 in the no NCD group were Black veterans. The mean age was similar in both groups: 71 years in the NCD group and 72 years in the no NCD group. There were no statistically significant differences in demographics, including prevalence of obstructive sleep apnea (OSA), supplemental oxygen flow rate, and duration of LTOT, or in pulmonary function test results between patients who did and did not experience NCD while sleeping (Table 2).
Ten of 22 patients (45%) with NCD and 9 of 44 patients (20%) without NCD were hospitalized at the JBVAMC for ≥ 1 COPD exacerbation in the preceding year that was diagnosed by a physician (P = .045). Three of 22 patients (14%) with NCD and no patients in the no NCD group were admitted to the MICU. No patients required intubation and mechanical ventilation during hospitalization, and no patients died. Overall hospital costs were 25% ($64,342) higher in NCD group compared with the no NCD group and were attributed to the MICU admissions in the NCD group (Table 3). Nine veterans did not respond to repeated telephone calls. One physician-diagnosed COPD exacerbation requiring hospitalization was documented in the nonresponder group; the patient was hospitalized for 2 days. One veteran died before being contacted.
DISCUSSION
There are 3 new findings in this study.
Nocturnal arterial oxygen desaturation in patients with COPD without evidence of OSA may contribute to the frequency of exacerbations.16 Although the mechanism(s) underlying this phenomenon is uncertain, we posit that prolonged nocturnal airway wall hypoxia could amplify underlying chronic inflammation through local generation of reactive oxygen species, thereby predisposing patients to exacerbations. Frequent COPD exacerbations promote disease progression and health status decline and are associated with increased mortality.11,13 Moreover, hospitalization of patients with COPD is the largest contributor to the annual direct cost of COPD per patient.10,12 The higher hospitalization rate observed in the NCD group in our study suggests that interruption of supplemental oxygen delivery while asleep may be a risk factor for COPD exacerbation. Alternatively, an independent factor or factors may have contributed to both NCD during sleep and COPD exacerbation in these patients or an impending exacerbation resulted in sleep disturbances that led to NCD. Additional research is warranted on veterans with hypoxemic CRF from COPD who are receiving LTOT and report frequent NCD during sleep that may support or refute these hypotheses.
To the best of our knowledge, NCD during sleep has not been previously reported in patients
Limitations
This was a small, single-site study, comprised entirely of male patients who are predominantly Black veterans. The telephone interviews with veterans self-reporting NCD during their sleep are prone to recall bias. In addition, the validity and reproducibility of NCD during sleep were not addressed in this study. Missing data from 9 nonresponders may have introduced a nonresponse bias in data analysis and interpretation. The overall hospital cost for a COPD exacerbation at JBVAMC was derived from VA data; US Centers for Medicare & Medicaid Services or commercial carrier data may be different.15,21 Lastly, access to LTOT for veterans with hypoxemic CRF from COPD is regulated and supervised at VA medical facilities.14 This process may be different for patients outside the VA. Taken together, it is difficult to generalize our initial observations to non-VA patients with hypoxemic CRF from COPD who are receiving LTOT. We suggest a large, prospective study of veterans be conducted to determine the prevalence of NCD during sleep and its relationship with COPD exacerbations in veterans receiving LTOT with hypoxemic CRF due to COPD.
CONCLUSIONS
Acknowledgments
We thank Yolanda Davis, RRT, and George Adam for their assistance with this project.
1. Boersma P, Cohen RA, Zelaya CE, Moy E. Multiple chronic conditions among veterans and nonveterans: United States, 2015-2018. Natl Health Stat Report. 2021;(153):1-13. doi:10.15620/cdc:101659
2. Sharafkhaneh A, Petersen NJ, Yu H-J, Dalal AA, Johnson ML, Hanania NA. Burden of COPD in a government health care system: a retrospective observational study using data from the US Veterans Affairs population. Int J Chron Obstruct Pulmon Dis. 2010;5:125-132. doi:10.2147/copd.s8047
3. LaBedz SL, Krishnan JA, Chung Y-C, et al. Chronic obstructive pulmonary disease outcomes at Veterans Affairs versus non-Veterans Affairs hospitals. Chronic Obstr Pulm Dis. 2021;8(3):306-313. doi:10.15326/jcopdf.2021.0201
4. Darnell K, Dwivedi AK, Weng Z, Panos RJ. Disproportionate utilization of healthcare resources among veterans with COPD: a retrospective analysis of factors associated with COPD healthcare cost. Cost Eff Resour Alloc. 2013;11:13. doi:10.1186/1478-7547-11-13
5. Bamonti PM, Robinson SA, Wan ES, Moy ML. Improving physiological, physical, and psychological health outcomes: a narrative review in US Veterans with COPD. Int J Chron Obstruct Pulmon Dis. 2022;17:1269-1283. doi:10.2147/COPD.S339323
6. Cranston JM, Crockett AJ, Moss JR, Alpers JH. Domiciliary oxygen for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2005;2005(4):CD001744. doi:10.1002/14651858.CD001744.pub2
7. Lacasse Y, Tan AM, Maltais F, Krishnan JA. Home oxygen in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2018;197(10):1254-1264. doi:10.1164/rccm.201802-0382CI
8. Jacobs SS, Krishnan JA, Lederer DJ, et al. Home oxygen therapy for adults with chronic lung disease. An official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2020;202(10):e121-e141. doi:10.1164/rccm.202009-3608ST
9. AARC. AARC clinical practice guideline. Oxygen therapy in the home or alternate site health care facility--2007 revision & update. Respir Care. 2007;52(8):1063-1068.
10. Foo J, Landis SH, Maskell J, et al. Continuing to confront COPD international patient survey: economic impact of COPD in 12 countries. PLoS One. 2016;11(4):e0152618. doi:10.1371/journal.pone.0152618
11. Rothnie KJ, Müllerová H, Smeeth L, Quint JK. Natural history of chronic obstructive pulmonary disease exacerbations in a general practice-based population with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2018;198(4):464-471. doi:10.1164/rccm.201710-2029OC
12. Stanford RH, Engel-Nitz NM, Bancroft T, Essoi B. The identification and cost of acute chronic obstructive pulmonary disease exacerbations in a United States population healthcare claims database. COPD. 2020;17(5):499-508. doi:10.1080/15412555.2020.1817357
13. Hurst JR, Han MK, Singh B, et al. Prognostic risk factors for moderate-to-severe exacerbations in patients with chronic obstructive pulmonary disease: a systematic literature review. Respir Res. 2022;23(1):213. doi:10.1186/s12931-022-02123-5
14. US Department of Veterans Affairs, Veterans Health Administration. Home oxygen program. VHA Directive 1173.13(1). Published August 5, 2020. Accessed February 28, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=8947
15. Phibbs CS, Barnett PG, Fan A, Harden C, King SS, Scott JY. Research guide to decision support system national cost extracts. Health Economics Resource Center of Health Service R&D Services, US Department of Veterans Affairs. September 2010. Accessed February 14, 2024. https://www.herc.research.va.gov/files/book_621.pdf
16. Agusti A, Hedner J, Marin JM, Barbé F, Cazzola M, Rennard S. Night-time symptoms: a forgotten dimension of COPD. Eur Respir Rev. 2011;20(121):183-194. doi:10.1183/09059180.00004311
17. Croxton TL, Bailey WC. Long-term oxygen treatment in chronic obstructive pulmonary disease: recommendations for future research: an NHLBI workshop report. Am J Respir Crit Care Med. 2006;174(4):373-378. doi:10.1164/rccm.200507-1161WS
18. Melani AS, Sestini P, Rottoli P. Home oxygen therapy: re-thinking the role of devices. Expert Rev Clin Pharmacol. 2018;11(3):279-289. doi:10.1080/17512433.2018.1421457
19. Sculley JA, Corbridge SJ, Prieto-Centurion V, et al. Home oxygen therapy for patients with COPD: time for a reboot. Respir Care. 2019;64(12):1574-1585. doi:10.4187/respcare.07135
20. Jacobs SS, Lindell KO, Collins EG, et al. Patient perceptions of the adequacy of supplemental oxygen therapy. Results of the American Thoracic Society Nursing Assembly Oxygen Working Group Survey. Ann Am Thorac Soc. 2018;15:24-32. doi:10.1513/AnnalsATS.201703-209OC
21. US Centers for Medicare & Medicaid Services. Home use of oxygen. Publication number 100-3. January 3, 2023. Accessed February 14, 2024. https://www.cms.gov/medicare-coverage-database/view/ncd.aspx?NCDId=169
1. Boersma P, Cohen RA, Zelaya CE, Moy E. Multiple chronic conditions among veterans and nonveterans: United States, 2015-2018. Natl Health Stat Report. 2021;(153):1-13. doi:10.15620/cdc:101659
2. Sharafkhaneh A, Petersen NJ, Yu H-J, Dalal AA, Johnson ML, Hanania NA. Burden of COPD in a government health care system: a retrospective observational study using data from the US Veterans Affairs population. Int J Chron Obstruct Pulmon Dis. 2010;5:125-132. doi:10.2147/copd.s8047
3. LaBedz SL, Krishnan JA, Chung Y-C, et al. Chronic obstructive pulmonary disease outcomes at Veterans Affairs versus non-Veterans Affairs hospitals. Chronic Obstr Pulm Dis. 2021;8(3):306-313. doi:10.15326/jcopdf.2021.0201
4. Darnell K, Dwivedi AK, Weng Z, Panos RJ. Disproportionate utilization of healthcare resources among veterans with COPD: a retrospective analysis of factors associated with COPD healthcare cost. Cost Eff Resour Alloc. 2013;11:13. doi:10.1186/1478-7547-11-13
5. Bamonti PM, Robinson SA, Wan ES, Moy ML. Improving physiological, physical, and psychological health outcomes: a narrative review in US Veterans with COPD. Int J Chron Obstruct Pulmon Dis. 2022;17:1269-1283. doi:10.2147/COPD.S339323
6. Cranston JM, Crockett AJ, Moss JR, Alpers JH. Domiciliary oxygen for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2005;2005(4):CD001744. doi:10.1002/14651858.CD001744.pub2
7. Lacasse Y, Tan AM, Maltais F, Krishnan JA. Home oxygen in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2018;197(10):1254-1264. doi:10.1164/rccm.201802-0382CI
8. Jacobs SS, Krishnan JA, Lederer DJ, et al. Home oxygen therapy for adults with chronic lung disease. An official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2020;202(10):e121-e141. doi:10.1164/rccm.202009-3608ST
9. AARC. AARC clinical practice guideline. Oxygen therapy in the home or alternate site health care facility--2007 revision & update. Respir Care. 2007;52(8):1063-1068.
10. Foo J, Landis SH, Maskell J, et al. Continuing to confront COPD international patient survey: economic impact of COPD in 12 countries. PLoS One. 2016;11(4):e0152618. doi:10.1371/journal.pone.0152618
11. Rothnie KJ, Müllerová H, Smeeth L, Quint JK. Natural history of chronic obstructive pulmonary disease exacerbations in a general practice-based population with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2018;198(4):464-471. doi:10.1164/rccm.201710-2029OC
12. Stanford RH, Engel-Nitz NM, Bancroft T, Essoi B. The identification and cost of acute chronic obstructive pulmonary disease exacerbations in a United States population healthcare claims database. COPD. 2020;17(5):499-508. doi:10.1080/15412555.2020.1817357
13. Hurst JR, Han MK, Singh B, et al. Prognostic risk factors for moderate-to-severe exacerbations in patients with chronic obstructive pulmonary disease: a systematic literature review. Respir Res. 2022;23(1):213. doi:10.1186/s12931-022-02123-5
14. US Department of Veterans Affairs, Veterans Health Administration. Home oxygen program. VHA Directive 1173.13(1). Published August 5, 2020. Accessed February 28, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=8947
15. Phibbs CS, Barnett PG, Fan A, Harden C, King SS, Scott JY. Research guide to decision support system national cost extracts. Health Economics Resource Center of Health Service R&D Services, US Department of Veterans Affairs. September 2010. Accessed February 14, 2024. https://www.herc.research.va.gov/files/book_621.pdf
16. Agusti A, Hedner J, Marin JM, Barbé F, Cazzola M, Rennard S. Night-time symptoms: a forgotten dimension of COPD. Eur Respir Rev. 2011;20(121):183-194. doi:10.1183/09059180.00004311
17. Croxton TL, Bailey WC. Long-term oxygen treatment in chronic obstructive pulmonary disease: recommendations for future research: an NHLBI workshop report. Am J Respir Crit Care Med. 2006;174(4):373-378. doi:10.1164/rccm.200507-1161WS
18. Melani AS, Sestini P, Rottoli P. Home oxygen therapy: re-thinking the role of devices. Expert Rev Clin Pharmacol. 2018;11(3):279-289. doi:10.1080/17512433.2018.1421457
19. Sculley JA, Corbridge SJ, Prieto-Centurion V, et al. Home oxygen therapy for patients with COPD: time for a reboot. Respir Care. 2019;64(12):1574-1585. doi:10.4187/respcare.07135
20. Jacobs SS, Lindell KO, Collins EG, et al. Patient perceptions of the adequacy of supplemental oxygen therapy. Results of the American Thoracic Society Nursing Assembly Oxygen Working Group Survey. Ann Am Thorac Soc. 2018;15:24-32. doi:10.1513/AnnalsATS.201703-209OC
21. US Centers for Medicare & Medicaid Services. Home use of oxygen. Publication number 100-3. January 3, 2023. Accessed February 14, 2024. https://www.cms.gov/medicare-coverage-database/view/ncd.aspx?NCDId=169
Comorbidities of Migraine
Dx Across the Skin Color Spectrum: Longitudinal Melanonychia
![]()
Longitudinal melanonychia (LM) is a pigmented linear band—brown, black, or gray—spanning the length of the nail plate due to the presence of excess melanin, which may be attributed to a benign or malignant process and may warrant further investigation.1,2 The majority of patients who present with LM are diagnosed with melanocytic activation of the nail matrix due to their inherent darker skin tone or various triggers including trauma, infection, and medications. Longitudinal melanonychia secondary to melanocytic activation often occurs spontaneously in patients with skin of color.3 Less commonly, LM is caused by a nail matrix nevus or lentigo; however, LM may arise secondary to subungual melanoma, a more dangerous cause.
A thorough clinical history including duration, recent changes in LM manifestation, nail trauma, or infection is helpful in evaluating patients with LM; however, a history of nail trauma can be misleading, as nail changes attributed to the trauma may in fact be melanoma. Irregularly spaced vertical lines of pigmentation ranging from brown to black with variations in spacing and width are characteristic of subungual melanoma.4 Nail dystrophy, granular hyperpigmentation, and Hutchinson sign (extension of pigmentation to the nail folds) also are worrisome features.5 In recent years, dermoscopy has become an important tool in the clinical examination of LM, with the development of criteria based on color and pattern recognition.5,6 Dermoscopy can be useful in screening potential candidates for biopsy. Although clinical examination and dermoscopy are essential to evaluating LM, the gold-standard diagnostic test when malignancy is suspected is a nail matrix biopsy.1,2,6,7
Epidemiology
It is not unusual for patients with darker skin tones to develop LM due to melanocytic activation of multiple nails with age. This finding can be seen in approximately 80% of African American individuals, 30% of Japanese individuals, and 50% of Hispanic individuals.2 It has even been reported that approximately 100% of Black patients older than 50 years will have evidence of LM.3
In a retrospective analysis, children presenting with LM tend to have a higher prevalence of nail matrix nevi compared to adults (56.1% [60/106] vs 34.3% [23/66]; P =.005).8 Involvement of a single digit in children is most likely indicative of a nevus; however, when an adult presents with LM in a single digit, suspicion for subungual melanoma should be raised.2,3,9
Two separate single-center retrospective studies showed the prevalence of subungual melanoma in patients presenting with melanonychia in Asia. Jin et al10 reported subungual melanoma in 6.2% (17/275) of Korean patients presenting with melanonychia at a general dermatology clinic from 2002 to 2014. Lyu et al8 studied LM in 172 Chinese patients in a dermatology clinic from 2018 to 2021 and reported 9% (6/66) of adults (aged ≥ 18 years) with subungual melanoma, with no reported cases in childhood (aged < 18 years).
Although the prevalence of subungual melanoma in patients with LM is low, it is an important diagnosis that should not be missed. In confirmed cases of subungual melanoma, two-thirds of lesions manifested as LM.3,10,11 Thus, LM arising in an adult in a single digit is more concerning for malignancy.2,3,7,9
Individuals of African and Asian descent as well as American Indian individuals are at highest risk for subungual melanoma with a poor prognosis compared to other types of melanoma, largely due to diagnosis at an advanced stage of disease.3,9 In a retrospective study of 25 patients with surgically treated subungual melanoma, the mean recurrence-free survival was 33.6 months. The recurrence-free survival was 66% at 1 year and 40% at 3 years, and the overall survival rate was 37% at 3 years.12
Key clinical features in individuals with darker skin tones
• In patients with darker skin tones, LM tends to occur on multiple nails as a result of melanocytic activation.2,13
• Several longitudinal bands may be noted on the same nail and the pigmentation of the bands may vary. With age, these longitudinal bands typically increase in number and width.13
• Pseudo-Hutchinson sign may be present due to ethnic melanosis of the proximal nail fold.13,14
• Dermoscopic findings of LM in patients with skin of color include wider bands (P = .0125), lower band brightness (P < .032), and higher frequency of changing appearance of bands (P = .0071).15
Worth noting
When patients present with LM, thorough examination of the nail plate, periungual skin, and distal pulp of all digits on all extremities with adequate lighting is important.2 Dermoscopy is useful, and a gel interface helps for examining the nail plates.7
Clinicians should be encouraged to biopsy or immediately refer patients with concerning nail unit lesions. Cases of LM most likely are benign, but if some doubt exists, the lesions should be biopsied or tracked closely with clinical and dermoscopic images, with a biopsy if changes occur.16 In conjunction with evaluation by a qualified clinician, patients also should be encouraged to take photographs, as the evolution of nail changes is a critical part of clinical decision-making on the need for a biopsy or referral.
Health disparity highlight
Despite the disproportionately high mortality rates from subungual melanoma in Black and Hispanic populations,3,9 studies often do not adequately represent these populations. Although subungual melanoma is rare, a delay in the diagnosis contributes to high morbidity and mortality rates.
1. Tosti A, Piraccini BM, de Farias DC. Dealing with melanonychia. Semin Cutan Med Surg. 2009;28:49-54. doi:10.1016/j.sder.2008.12.004
2. Piraccini BM, Dika E, Fanti PA. Tips for diagnosis and treatment of nail pigmentation with practical algorithm. Dermatol Clin. 2015;33:185-195. doi:10.1016/j.det.2014.12.002
3. Halteh P, Scher R, Artis A, et al. Assessment of patient knowledge of longitudinal melanonychia: a survey study of patients in outpatient clinics. Skin Appendage Disord. 2016;2:156-161. doi:10.1159/000452673
4. Singal A, Bisherwal K. Melanonychia: etiology, diagnosis, and treatment. Indian Dermatol J Online. 2020;11:1-11. doi:10.4103/idoj.IDOJ_167_19
5. Benati E, Ribero S, Longo C, et al. Clinical and dermoscopic clues to differentiate pigmented nail bands: an International Dermoscopy Society study. J Eur Acad Dermatol Venereol. 2017;31:732-736. doi:10.1111/jdv.13991
6. Sawada M, Yokota K, Matsumoto T, et al. Proposed classification of longitudinal melanonychia based on clinical and dermoscopic criteria. Int J Dermatol. 2014;53:581-585. doi:10.1111/ijd.12001
7. Starace M, Alessandrini A, Brandi N, et al. Use of nail dermoscopy in the management of melanonychia. Dermatol Pract Concept. 2019;9:38-43. doi:10.5826/dpc.0901a10
8. Lyu A, Hou Y, Wang Q. Retrospective analysis of longitudinal melanonychia: a Chinese experience. Front Pediatr. 2023;10:1065758. doi:10.3389/fped.2022.1065758
9. Williams NM, Obayomi AO, Diaz-Perez, JA, et al. Monodactylous longitudinal melanonychia: a sign of Bowen’s disease in skin of color. Skin Appendage Disord. 2021;7:306-310. doi:10.1159/000514221
10. Jin H, Kim JM, Kim GW, et al. Diagnostic criteria for and clinical review of melanonychia in Korean patients. J Am Acad Dermatol. 2016;74,1121-1127. doi:10.1016/j.jaad.2015.12.039
11. Halteh P, Scher R, Artis A, et al. A survey-based study of management of longitudinal melanonychia amongst attending and resident dermatologists. J Am Acad Dermatol. 2017;76:994-996. doi:10.1016/j.jaad.2016.11.053
12. LaRocca CJ, Lai L, Nelson RA, et al. Subungual melanoma: a single institution experience. Med Sci (Basel). 2021;9:57. doi:10.3390/medsci9030057
13. Baran LR, Ruben BS, Kechijian P, et al. Non‐melanoma Hutchinson’s sign: a reappraisal of this important, remarkable melanoma simulant. J Eur Acad Dermatol Venereol. 2018;32:495-501. doi:10.1111/jdv.14715
14. Sladden MJ, Mortimer NJ, Osborne JE. Longitudinal melanonychia and pseudo‐Hutchinson sign associated with amlodipine. Br J Dermatol. 2005;153:219-220. doi:10.1111/j.13652133.2005.06668.x
15. Lee DK, Chang MJ, Desai AD, et al. Clinical and dermoscopic findings of benign longitudinal melanonychia due to melanocytic activation differ by skin type and predict likelihood of nail matrix biopsy. J Am Acad Dermatol. 2022;87:792-799. doi:10.1016/j.jaad.2022.06.1165
16. Hogue L, Harvey VM. Basal cell carcinoma, squamous cell carcinoma, and cutaneous melanoma in skin of color patients. Dermatol Clin. 2019;37:519-526. doi:10.1016/j.det.2019.05.009
![]()
Longitudinal melanonychia (LM) is a pigmented linear band—brown, black, or gray—spanning the length of the nail plate due to the presence of excess melanin, which may be attributed to a benign or malignant process and may warrant further investigation.1,2 The majority of patients who present with LM are diagnosed with melanocytic activation of the nail matrix due to their inherent darker skin tone or various triggers including trauma, infection, and medications. Longitudinal melanonychia secondary to melanocytic activation often occurs spontaneously in patients with skin of color.3 Less commonly, LM is caused by a nail matrix nevus or lentigo; however, LM may arise secondary to subungual melanoma, a more dangerous cause.
A thorough clinical history including duration, recent changes in LM manifestation, nail trauma, or infection is helpful in evaluating patients with LM; however, a history of nail trauma can be misleading, as nail changes attributed to the trauma may in fact be melanoma. Irregularly spaced vertical lines of pigmentation ranging from brown to black with variations in spacing and width are characteristic of subungual melanoma.4 Nail dystrophy, granular hyperpigmentation, and Hutchinson sign (extension of pigmentation to the nail folds) also are worrisome features.5 In recent years, dermoscopy has become an important tool in the clinical examination of LM, with the development of criteria based on color and pattern recognition.5,6 Dermoscopy can be useful in screening potential candidates for biopsy. Although clinical examination and dermoscopy are essential to evaluating LM, the gold-standard diagnostic test when malignancy is suspected is a nail matrix biopsy.1,2,6,7
Epidemiology
It is not unusual for patients with darker skin tones to develop LM due to melanocytic activation of multiple nails with age. This finding can be seen in approximately 80% of African American individuals, 30% of Japanese individuals, and 50% of Hispanic individuals.2 It has even been reported that approximately 100% of Black patients older than 50 years will have evidence of LM.3
In a retrospective analysis, children presenting with LM tend to have a higher prevalence of nail matrix nevi compared to adults (56.1% [60/106] vs 34.3% [23/66]; P =.005).8 Involvement of a single digit in children is most likely indicative of a nevus; however, when an adult presents with LM in a single digit, suspicion for subungual melanoma should be raised.2,3,9
Two separate single-center retrospective studies showed the prevalence of subungual melanoma in patients presenting with melanonychia in Asia. Jin et al10 reported subungual melanoma in 6.2% (17/275) of Korean patients presenting with melanonychia at a general dermatology clinic from 2002 to 2014. Lyu et al8 studied LM in 172 Chinese patients in a dermatology clinic from 2018 to 2021 and reported 9% (6/66) of adults (aged ≥ 18 years) with subungual melanoma, with no reported cases in childhood (aged < 18 years).
Although the prevalence of subungual melanoma in patients with LM is low, it is an important diagnosis that should not be missed. In confirmed cases of subungual melanoma, two-thirds of lesions manifested as LM.3,10,11 Thus, LM arising in an adult in a single digit is more concerning for malignancy.2,3,7,9
Individuals of African and Asian descent as well as American Indian individuals are at highest risk for subungual melanoma with a poor prognosis compared to other types of melanoma, largely due to diagnosis at an advanced stage of disease.3,9 In a retrospective study of 25 patients with surgically treated subungual melanoma, the mean recurrence-free survival was 33.6 months. The recurrence-free survival was 66% at 1 year and 40% at 3 years, and the overall survival rate was 37% at 3 years.12
Key clinical features in individuals with darker skin tones
• In patients with darker skin tones, LM tends to occur on multiple nails as a result of melanocytic activation.2,13
• Several longitudinal bands may be noted on the same nail and the pigmentation of the bands may vary. With age, these longitudinal bands typically increase in number and width.13
• Pseudo-Hutchinson sign may be present due to ethnic melanosis of the proximal nail fold.13,14
• Dermoscopic findings of LM in patients with skin of color include wider bands (P = .0125), lower band brightness (P < .032), and higher frequency of changing appearance of bands (P = .0071).15
Worth noting
When patients present with LM, thorough examination of the nail plate, periungual skin, and distal pulp of all digits on all extremities with adequate lighting is important.2 Dermoscopy is useful, and a gel interface helps for examining the nail plates.7
Clinicians should be encouraged to biopsy or immediately refer patients with concerning nail unit lesions. Cases of LM most likely are benign, but if some doubt exists, the lesions should be biopsied or tracked closely with clinical and dermoscopic images, with a biopsy if changes occur.16 In conjunction with evaluation by a qualified clinician, patients also should be encouraged to take photographs, as the evolution of nail changes is a critical part of clinical decision-making on the need for a biopsy or referral.
Health disparity highlight
Despite the disproportionately high mortality rates from subungual melanoma in Black and Hispanic populations,3,9 studies often do not adequately represent these populations. Although subungual melanoma is rare, a delay in the diagnosis contributes to high morbidity and mortality rates.
![]()
Longitudinal melanonychia (LM) is a pigmented linear band—brown, black, or gray—spanning the length of the nail plate due to the presence of excess melanin, which may be attributed to a benign or malignant process and may warrant further investigation.1,2 The majority of patients who present with LM are diagnosed with melanocytic activation of the nail matrix due to their inherent darker skin tone or various triggers including trauma, infection, and medications. Longitudinal melanonychia secondary to melanocytic activation often occurs spontaneously in patients with skin of color.3 Less commonly, LM is caused by a nail matrix nevus or lentigo; however, LM may arise secondary to subungual melanoma, a more dangerous cause.
A thorough clinical history including duration, recent changes in LM manifestation, nail trauma, or infection is helpful in evaluating patients with LM; however, a history of nail trauma can be misleading, as nail changes attributed to the trauma may in fact be melanoma. Irregularly spaced vertical lines of pigmentation ranging from brown to black with variations in spacing and width are characteristic of subungual melanoma.4 Nail dystrophy, granular hyperpigmentation, and Hutchinson sign (extension of pigmentation to the nail folds) also are worrisome features.5 In recent years, dermoscopy has become an important tool in the clinical examination of LM, with the development of criteria based on color and pattern recognition.5,6 Dermoscopy can be useful in screening potential candidates for biopsy. Although clinical examination and dermoscopy are essential to evaluating LM, the gold-standard diagnostic test when malignancy is suspected is a nail matrix biopsy.1,2,6,7
Epidemiology
It is not unusual for patients with darker skin tones to develop LM due to melanocytic activation of multiple nails with age. This finding can be seen in approximately 80% of African American individuals, 30% of Japanese individuals, and 50% of Hispanic individuals.2 It has even been reported that approximately 100% of Black patients older than 50 years will have evidence of LM.3
In a retrospective analysis, children presenting with LM tend to have a higher prevalence of nail matrix nevi compared to adults (56.1% [60/106] vs 34.3% [23/66]; P =.005).8 Involvement of a single digit in children is most likely indicative of a nevus; however, when an adult presents with LM in a single digit, suspicion for subungual melanoma should be raised.2,3,9
Two separate single-center retrospective studies showed the prevalence of subungual melanoma in patients presenting with melanonychia in Asia. Jin et al10 reported subungual melanoma in 6.2% (17/275) of Korean patients presenting with melanonychia at a general dermatology clinic from 2002 to 2014. Lyu et al8 studied LM in 172 Chinese patients in a dermatology clinic from 2018 to 2021 and reported 9% (6/66) of adults (aged ≥ 18 years) with subungual melanoma, with no reported cases in childhood (aged < 18 years).
Although the prevalence of subungual melanoma in patients with LM is low, it is an important diagnosis that should not be missed. In confirmed cases of subungual melanoma, two-thirds of lesions manifested as LM.3,10,11 Thus, LM arising in an adult in a single digit is more concerning for malignancy.2,3,7,9
Individuals of African and Asian descent as well as American Indian individuals are at highest risk for subungual melanoma with a poor prognosis compared to other types of melanoma, largely due to diagnosis at an advanced stage of disease.3,9 In a retrospective study of 25 patients with surgically treated subungual melanoma, the mean recurrence-free survival was 33.6 months. The recurrence-free survival was 66% at 1 year and 40% at 3 years, and the overall survival rate was 37% at 3 years.12
Key clinical features in individuals with darker skin tones
• In patients with darker skin tones, LM tends to occur on multiple nails as a result of melanocytic activation.2,13
• Several longitudinal bands may be noted on the same nail and the pigmentation of the bands may vary. With age, these longitudinal bands typically increase in number and width.13
• Pseudo-Hutchinson sign may be present due to ethnic melanosis of the proximal nail fold.13,14
• Dermoscopic findings of LM in patients with skin of color include wider bands (P = .0125), lower band brightness (P < .032), and higher frequency of changing appearance of bands (P = .0071).15
Worth noting
When patients present with LM, thorough examination of the nail plate, periungual skin, and distal pulp of all digits on all extremities with adequate lighting is important.2 Dermoscopy is useful, and a gel interface helps for examining the nail plates.7
Clinicians should be encouraged to biopsy or immediately refer patients with concerning nail unit lesions. Cases of LM most likely are benign, but if some doubt exists, the lesions should be biopsied or tracked closely with clinical and dermoscopic images, with a biopsy if changes occur.16 In conjunction with evaluation by a qualified clinician, patients also should be encouraged to take photographs, as the evolution of nail changes is a critical part of clinical decision-making on the need for a biopsy or referral.
Health disparity highlight
Despite the disproportionately high mortality rates from subungual melanoma in Black and Hispanic populations,3,9 studies often do not adequately represent these populations. Although subungual melanoma is rare, a delay in the diagnosis contributes to high morbidity and mortality rates.
1. Tosti A, Piraccini BM, de Farias DC. Dealing with melanonychia. Semin Cutan Med Surg. 2009;28:49-54. doi:10.1016/j.sder.2008.12.004
2. Piraccini BM, Dika E, Fanti PA. Tips for diagnosis and treatment of nail pigmentation with practical algorithm. Dermatol Clin. 2015;33:185-195. doi:10.1016/j.det.2014.12.002
3. Halteh P, Scher R, Artis A, et al. Assessment of patient knowledge of longitudinal melanonychia: a survey study of patients in outpatient clinics. Skin Appendage Disord. 2016;2:156-161. doi:10.1159/000452673
4. Singal A, Bisherwal K. Melanonychia: etiology, diagnosis, and treatment. Indian Dermatol J Online. 2020;11:1-11. doi:10.4103/idoj.IDOJ_167_19
5. Benati E, Ribero S, Longo C, et al. Clinical and dermoscopic clues to differentiate pigmented nail bands: an International Dermoscopy Society study. J Eur Acad Dermatol Venereol. 2017;31:732-736. doi:10.1111/jdv.13991
6. Sawada M, Yokota K, Matsumoto T, et al. Proposed classification of longitudinal melanonychia based on clinical and dermoscopic criteria. Int J Dermatol. 2014;53:581-585. doi:10.1111/ijd.12001
7. Starace M, Alessandrini A, Brandi N, et al. Use of nail dermoscopy in the management of melanonychia. Dermatol Pract Concept. 2019;9:38-43. doi:10.5826/dpc.0901a10
8. Lyu A, Hou Y, Wang Q. Retrospective analysis of longitudinal melanonychia: a Chinese experience. Front Pediatr. 2023;10:1065758. doi:10.3389/fped.2022.1065758
9. Williams NM, Obayomi AO, Diaz-Perez, JA, et al. Monodactylous longitudinal melanonychia: a sign of Bowen’s disease in skin of color. Skin Appendage Disord. 2021;7:306-310. doi:10.1159/000514221
10. Jin H, Kim JM, Kim GW, et al. Diagnostic criteria for and clinical review of melanonychia in Korean patients. J Am Acad Dermatol. 2016;74,1121-1127. doi:10.1016/j.jaad.2015.12.039
11. Halteh P, Scher R, Artis A, et al. A survey-based study of management of longitudinal melanonychia amongst attending and resident dermatologists. J Am Acad Dermatol. 2017;76:994-996. doi:10.1016/j.jaad.2016.11.053
12. LaRocca CJ, Lai L, Nelson RA, et al. Subungual melanoma: a single institution experience. Med Sci (Basel). 2021;9:57. doi:10.3390/medsci9030057
13. Baran LR, Ruben BS, Kechijian P, et al. Non‐melanoma Hutchinson’s sign: a reappraisal of this important, remarkable melanoma simulant. J Eur Acad Dermatol Venereol. 2018;32:495-501. doi:10.1111/jdv.14715
14. Sladden MJ, Mortimer NJ, Osborne JE. Longitudinal melanonychia and pseudo‐Hutchinson sign associated with amlodipine. Br J Dermatol. 2005;153:219-220. doi:10.1111/j.13652133.2005.06668.x
15. Lee DK, Chang MJ, Desai AD, et al. Clinical and dermoscopic findings of benign longitudinal melanonychia due to melanocytic activation differ by skin type and predict likelihood of nail matrix biopsy. J Am Acad Dermatol. 2022;87:792-799. doi:10.1016/j.jaad.2022.06.1165
16. Hogue L, Harvey VM. Basal cell carcinoma, squamous cell carcinoma, and cutaneous melanoma in skin of color patients. Dermatol Clin. 2019;37:519-526. doi:10.1016/j.det.2019.05.009
1. Tosti A, Piraccini BM, de Farias DC. Dealing with melanonychia. Semin Cutan Med Surg. 2009;28:49-54. doi:10.1016/j.sder.2008.12.004
2. Piraccini BM, Dika E, Fanti PA. Tips for diagnosis and treatment of nail pigmentation with practical algorithm. Dermatol Clin. 2015;33:185-195. doi:10.1016/j.det.2014.12.002
3. Halteh P, Scher R, Artis A, et al. Assessment of patient knowledge of longitudinal melanonychia: a survey study of patients in outpatient clinics. Skin Appendage Disord. 2016;2:156-161. doi:10.1159/000452673
4. Singal A, Bisherwal K. Melanonychia: etiology, diagnosis, and treatment. Indian Dermatol J Online. 2020;11:1-11. doi:10.4103/idoj.IDOJ_167_19
5. Benati E, Ribero S, Longo C, et al. Clinical and dermoscopic clues to differentiate pigmented nail bands: an International Dermoscopy Society study. J Eur Acad Dermatol Venereol. 2017;31:732-736. doi:10.1111/jdv.13991
6. Sawada M, Yokota K, Matsumoto T, et al. Proposed classification of longitudinal melanonychia based on clinical and dermoscopic criteria. Int J Dermatol. 2014;53:581-585. doi:10.1111/ijd.12001
7. Starace M, Alessandrini A, Brandi N, et al. Use of nail dermoscopy in the management of melanonychia. Dermatol Pract Concept. 2019;9:38-43. doi:10.5826/dpc.0901a10
8. Lyu A, Hou Y, Wang Q. Retrospective analysis of longitudinal melanonychia: a Chinese experience. Front Pediatr. 2023;10:1065758. doi:10.3389/fped.2022.1065758
9. Williams NM, Obayomi AO, Diaz-Perez, JA, et al. Monodactylous longitudinal melanonychia: a sign of Bowen’s disease in skin of color. Skin Appendage Disord. 2021;7:306-310. doi:10.1159/000514221
10. Jin H, Kim JM, Kim GW, et al. Diagnostic criteria for and clinical review of melanonychia in Korean patients. J Am Acad Dermatol. 2016;74,1121-1127. doi:10.1016/j.jaad.2015.12.039
11. Halteh P, Scher R, Artis A, et al. A survey-based study of management of longitudinal melanonychia amongst attending and resident dermatologists. J Am Acad Dermatol. 2017;76:994-996. doi:10.1016/j.jaad.2016.11.053
12. LaRocca CJ, Lai L, Nelson RA, et al. Subungual melanoma: a single institution experience. Med Sci (Basel). 2021;9:57. doi:10.3390/medsci9030057
13. Baran LR, Ruben BS, Kechijian P, et al. Non‐melanoma Hutchinson’s sign: a reappraisal of this important, remarkable melanoma simulant. J Eur Acad Dermatol Venereol. 2018;32:495-501. doi:10.1111/jdv.14715
14. Sladden MJ, Mortimer NJ, Osborne JE. Longitudinal melanonychia and pseudo‐Hutchinson sign associated with amlodipine. Br J Dermatol. 2005;153:219-220. doi:10.1111/j.13652133.2005.06668.x
15. Lee DK, Chang MJ, Desai AD, et al. Clinical and dermoscopic findings of benign longitudinal melanonychia due to melanocytic activation differ by skin type and predict likelihood of nail matrix biopsy. J Am Acad Dermatol. 2022;87:792-799. doi:10.1016/j.jaad.2022.06.1165
16. Hogue L, Harvey VM. Basal cell carcinoma, squamous cell carcinoma, and cutaneous melanoma in skin of color patients. Dermatol Clin. 2019;37:519-526. doi:10.1016/j.det.2019.05.009
Graduate Medical Education Financing in the US Department of Veterans Affairs

The US Department of Veterans Affairs (VA) has partnered with academic medical centers and programs since 1946 to provide clinical training for physician residents. Ranking second in federal graduate medical education (GME) funding to the Centers for Medicare and Medicaid Services (CMS), the $850 million VA GME budget annually reimburses > 250 GME-sponsoring institutions (affiliates) of 8000 GME programs for the clinical training of 49,000 individual residents rotating through > 11,000 full-time equivalent (FTE) positions.1 The VA also distributes $1.6 billion to VA facilities to offset the costs of conducting health professions education (HPE) (eg, facility infrastructure, salary support for VA instructors and preceptors, education office administration, and instructional equipment).2 The VA financial and educational contributions account for payment of 11% of resident positions nationally and allow academic medical centers to be less reliant on CMS GME funding.3,4 The VA contributions also provide opportunities for GME expansion,1,5,6 educational innovations,5,7 interprofessional and team-based care,8,9 and quality and safety training.10,11 The Table provides a comparison of CMS and VA GME reimbursability based on activity.
GME financing is complex, particularly the formulaic approach used by CMS, the details of which are often obscured in federal regulations. Due to this complexity and the $16 billion CMS GME budget, academic publications have focused on CMS GME financing while not fully explaining the VA GME policies and processes.4,12-14 By comparison, the VA GME financing model is relatively straightforward and governed by different statues and VA regulations, yet sharing some of the same principles as CMS regulations. Given the challenges in CMS reimbursement to fully support the cost of resident education, as well as the educational opportunities at the VA, the VA designs its reimbursement model to assure that affiliates receive appropriate payments.4,12,15 To ensure the continued success of VA GME partnerships, knowledge of VA GME financing has become increasingly important for designated institutional officers (DIOs) and residency program directors, particularly in light of recent investigations into oversight of the VA’s reimbursement to academic affiliates.
VA AUTHORITY
While the VA’s primary mission is “to provide a complete hospital medical service for the medical care and treatment of veterans,”early VA leaders recognized the importance of affiliating with the nation’s academic institutions.19 In 1946, the VA Policy Memorandum Number 2 established a partnership between the VA and the academic medical community.20 Additional legislation authorized specific agreements with academic affiliates for the central administration of salary and benefits for residents rotating at VA facilities. This process, known as disbursement, is an alternative payroll mechanism whereby the VA reimburses the academic affiliate for resident salary and benefits and the affiliate acts as the disbursing agent, issuing paychecks to residents.21,22
Resident FUNDING
By policy, with rare exceptions, the VA does not sponsor residency programs due to the challenges of providing an appropriate patient mix of age, sex, and medical conditions to meet accreditation standards.4 Nearly all VA reimbursements are for residents in affiliate-sponsored programs, while just 1% pays for residents in legacy, VA-sponsored residency programs at 2 VA facilities. The VA budget for resident (including fellows) salary and benefits is managed by the VA Office of Academic Affiliations (OAA), the national VA office responsible for oversight, policy, and funding of VA HPE programs.
Resident Salaries and Benefits
VA funding of resident salary and benefits are analogous with CMS direct GME (DGME), which is designed to cover resident salary and benefits costs.4,14,23 CMS DGME payments depend on a hospital’s volume of CMS inpatients and are based on a statutory formula, which uses the hospital’s resident FTE positions, the per-resident amount, and Medicare’s share of inpatient beds (Medicare patient load) to determine payments.12 The per-resident amount is set by statute, varies geographically, and is calculated by dividing the hospital’s allowable costs of GME (percentage of CMS inpatient days) divided by the number of residents.12,24
By comparison, the VA GME payment reimburses for each FTE based on the salary and benefits rate set by the academic affiliate. Reimbursement is calculated based on resident time spent at the VA multiplied by a daily salary rate. The daily salary rate is determined by dividing the resident’s total compensation (salary and benefits) by the number of calendar days in an academic year. Resident time spent at the VA facility is determined by obtaining rotation schedules provided by the academic affiliate and verifying resident clinical and educational activity during scheduled rotations.
Indirect Medical Education Funding
In addition to resident salary and benefits, funds to offset the cost of conducting HPE are provided to VA facilities. These funds are intended to improve and maintain necessary infrastructure for all HPE programs not just GME, including education office administration needs, teaching costs (ie, a portion of VA preceptors salary), and instructional equipment.

The Veterans Equitable Resource Allocation (VERA) is a national budgeting process for VA medical facilities that funds facility operational needs such as staff salary and benefits, infrastructure, and equipment.2 The education portion of the VERA, the VERA Education Support Component (VESC), is not managed by the OAA, but rather is distributed through the VERA model to the general budget of VA facilities hosting HPE (Figure). VESC funding in the VA budget is based on labor mapping of physician time spent in education; other labor mapping categories include clinical care, research, and administration. VA facility VESC funding is calculated based on the number of paid health profession trainees (HPTs) from all professions, apportioned according to the number of FTEs for physician residents and VA-paid HPTs in other disciplines. In fiscal year 2024, VA facilities received $115,812 for each physician resident FTE position and $84,906 for each VA-paid, non-GME FTE position.
The VESC is like CMS's indirect GME funding, termed Indirect Medical Education (IME), an additional payment for each Medicare patient discharged reflecting teaching hospitals’ higher patient care costs relative to nonteaching hospitals. Described elsewhere, IME is calculated using a resident-to-bed ratio and a multiplier, which is set by statute.4,25 While IME can be used for reimbursement for some resident clinical and educational activities(eg, research), VA VESC funds cannot be used for such activities and are part of the general facility budget and appropriated per the discretion of the medical facility director.
ESTABLISHING GME PARTNERSHIPS
An affiliation agreement establishes the administrative and legal requirements for educational relationships with academic affiliates and includes standards for conducting HPE, responsibilities for accreditation standards, program leadership, faculty, resources, supervision, academic policies, and procedures. The VA uses standardized affiliation agreement templates that have been vetted with accrediting bodies and the VA Office of General Counsel.
A disbursement agreement authorizes the VA to reimburse affiliates for resident salary and benefits for VA clinical and educational activities. The disbursement agreement details the fiscal arrangements (eg, payment in advance vs arrears, salary, and benefit rates, leave) for the reimbursement payments. Veterans Health Administration (VHA) Directive 1400.05 provides the policy and procedures for calculating reimbursement for HPT educational activities.26
The VA facility designated education officer (DEO) oversees all HPE programs and coordinates the affiliation and disbursement agreement processes.27 The DEO, affiliate DIO, residency program director, and VA residency site director determine the physician resident FTE positions assigned to a VA facility based on educational objectives and availability of educational resources at the VA facility, such as patient care opportunities, faculty supervisors, space, and equipment. The VA facility requests for resident FTE positions are submitted to the OAA by the facility DEO.
Once GME FTE positions are approved by the OAA, VA facilities work with their academic affiliate to submit the physician resident salary and benefit rate. Affiliate DIOs attest to the accuracy of the salary rate schedule and the local DEO submits the budget request to the OAA. Upon approval, the funds are transferred to the VA facility each fiscal year, which begins October 1. DEOs report quarterly to the OAA both budget needs and excesses based on variations in the approved FTEs due to additional VA rotations, physician resident attrition, or reassignment.
Resident Position Allocation
VA GME financing provides flexibility through periodic needs assessments and expansion initiatives. In August and December, DEOs collaborate with an academic affiliate to submit reports to the OAA confirming their projected GME needs for the next academic year. Additional positions requests are reviewed by the OAA; funding depends on budget and the educational justification. The OAA periodically issues GME expansion requests for proposal, which typically arise from legislation to address specific VA workforce needs. The VA facility DEO and affiliate GME leaders collaborate to apply for additional positions. For example, a VA GME expansion under the Veterans Access, Choice, and Accountability Act of 2014 added 1500 GME positions in 8 years for critically needed specialties and in rural and underserved areas.5 The Maintaining Internal Systems and Strengthening Outside Networks (MISSION) Act of 2018 authorized a pilot program for VA to fund residents at non-VA facilities with priority for Indian Health Services, Tribes and Tribal Organizations, Federally Qualified Health Centers, and US Department of Defense facilities to provide access to veterans in underserved areas.6
The VA GME financing system has flexibility to meet local needs for additional resident positions and to address broader VA workforce gaps through targeted expansion. Generally, CMS does not fund positions to address workforce needs, place residents in specific geographic areas, or require the training of certain types of residents.4 However, the Consolidated Appropriations Act of 2021 has provided the opportunity to address rural workforce needs.28
Reimbursement
The VA provides reimbursement for clinical and educational activities performed in VA facilities for the benefit of veterans as well as research, didactics, meetings and conferences, annual and sick leave, and orientation. The VA also may provide reimbursement for educational activities that occur off VA grounds (eg, the VA proportional share of a residency program’s didactic sessions). The VA does not reimburse for affiliate clinical duties or administrative costs, although a national policy allows VA facilities to reimburse affiliates for some GME overhead costs.29
CMS similarly reimburses for residency training time spent in patient care activities as well as orientation activities, didactics, leave, and, in some cases, research.4,30,31 CMS makes payments to hospitals, which may include sponsoring institutions and Medicare-eligible participating training sites.4,30,31 For both the VA and CMS, residents may not be counted twice for reimbursement by 2 federal agencies; in other words, a resident may not count for > 1 FTE.4,30-32
GME Oversight
VA GME funding came under significant scrutiny. At a 2016 House Veterans Affairs Committee hearing, Representative Phil Roe, MD (R-Tennessee), noted that no process existed at many VA facilities for “determining trainee presence” and that many VA medical centers had “difficulty tracking resident rotations”16 A VA Office of the Inspector General investigation recommended that the VA implement policies and procedures to improve oversight to “ensure residents are fully participating in educational activities” and that the VA is “paying the correct amount” to the affiliate.17 A 2020 General Accountability Office report outlined unclear policy guidance, incomplete tracking of resident activities, and improper fiscal processes for reimbursement and reconciliation of affiliate invoices.18

In response, the OAA created an oversight and compliance unit, revised VHA Directive 1400.05 (the policy for disbursement), and improved resident tracking procedures.26 The standard operating procedure that accompanied VHA Directive 1400.05 provides detailed information for the DEO and VA facility staff for tracking resident clinical and educational activities. FTE counts are essential to both VA and CMS for accurate reimbursement. The eAppendix and the Table provide a guide to reimbursable activities in the VA for the calculation of reimbursement, with a comparison to CMS.33,34 The OAA in cooperation with other VA staff and officers periodically conducts audits to assess compliance with disbursement policy and affiliate reimbursement accuracy.
In the VA, resident activities are captured on the VA Educational Activity Record, a standardized spreadsheet to track activities and calculate reimbursement. Each VA facility hosting resident physicians manually records resident activity by the half-day. This process is labor intensive, involving both VA and affiliate staff to accurately reconcile payments. To address the workload demands, the OAA is developing an online tool that will automate aspects of the tracking process. Also, to ensure adequate staffing, the OAA is in the process of implementing an office optimization project, providing standardized position descriptions, an organizational chart, and staffing levels for DEO offices in VA facilities.
Conclusions
This report describes the key policies and principles of VA GME financing, highlighting the essential similarities and differences between VA and CMS. Neither the VA nor CMS regulations allow for reimbursement for > 1 FTE position per resident, a principle that underpins the assignment of resident rotations and federal funding for GME and are similar with respect to reimbursement for patient care activities, didactics, research, orientation, and scholarly activity. While reimbursable activities in the VA require physical presence and care of veteran patients, CMS also limits reimbursement to resident activities in the hospital and approved other settings if the hospital is paying for resident salary and benefits in these settings. The VA provides some flexibility for offsite activities including didactics and, in specific circumstances, remote care of veteran patients (eg, teleradiology).
The VA and CMS use different GME financing models. For example, the CMS calculations for resident FTEs are complex, whereas VA calculations reimburse the salary and benefits as set by the academic affiliate. The VA process accounts for local variation in salary rates, whereas the per-resident amount set by CMS varies regionally and does not fully account for differences in the cost of living.24 Because all patients in VA facilities are veterans, VA calculations for reimbursement do not involve ratios of beds like the CMS calculations to determine a proportional share of reimbursement. The VA GME expansion tends to be more directed to VA health workforce needs than CMS, specifying the types of programs and geographic locations to address these needs.
The VA regularly reevaluates how affiliates are reimbursed for VA resident activity, balancing compliance with VA policies and the workload for VA and its affiliates. The VA obtains input from key stakeholders including DEOs, DIOs, and professional organizations such as the Association of American Medical Colleges and the Accreditation Council for Graduate Medical Education.35,36
Looking ahead, the VA is developing an online tool to improve the accuracy of affiliate reimbursement. The VA will also implement a standardized staffing model, organizational structure, and position descriptions for DEO offices. These initiatives will help reduce the burden of tracking and verifying resident activity and continue to support the 77-year partnership between VA and its affiliated institutions.
1. Klink KA, Albanese AP, Bope ET, Sanders KM. Veterans Affairs graduate medical education expansion addresses US physician workforce needs. Acad Med. 2022;97(8):1144-1150. doi:10.1097/ACM.0000000000004545
2. Andrus CH, Johnson K, Pierce E, Romito PJ, Hartel P, Berrios‐Guccione S, Best W. Finance modeling in the delivery of medical care in tertiary‐care hospitals in the Department of Veterans Affairs. J Surg Res. 2001;96(2):152-157. doi:10.1006/jsre.1999.5728
3. Petrakis IL, Kozal M. Academic medical centers and the U.S. Department of Veterans Affairs: a 75-year partnership influences medical education, scientific discovery, and clinical care. Acad Med. 2022;97(8):1110-1113. doi:10.1097/ACM.0000000000004734
4. Heisler EJ, Mendez BH, Mitchell A, Panangala SV, Villagrana MA. Federal support for graduate medical education: an overview (R44376). Congressional Research Service report R44376; version 11. Updated December 27, 2018. Accessed March 2, 2024. https://crsreports.congress.gov/product/pdf/R/R44376/11
5. Chang BK, Brannen JL. The Veterans Access, Choice, and Accountability Act of 2014: examining graduate medical education enhancement in the Department of Veterans Affairs. Acad Med. 2015;90(9):1196-1198. doi:10.1097/ACM.0000000000000795
6. Albanese AP, Bope ET, Sanders KM, Bowman M. The VA MISSION Act of 2018: a potential game changer for rural GME expansion and veteran health care. J Rural Health. 2020;36(1):133-136. doi:10.1111/jrh.12360
7. Lypson ML, Roberts LW. Valuing the partnership between the Veterans Health Administration and academic medicine. Acad Med. 2022;97(8):1091-1093. doi:10.1097/ACM.0000000000004748
8. Harada ND, Traylor L, Rugen KW, et al. Interprofessional transformation of clinical education: the first six years of the Veterans Affairs Centers of Excellence in Primary Care Education. J Interprof Care. 2023;37(suppl 1):S86-S94. doi:10.1080/13561820.2018.1433642

9. Harada ND, Rajashekara S, Sansgiry S, et al. Developing interprofessional primary care teams: alumni evaluation of the Department of Veterans Affairs Centers of Excellence in Primary Care Education Program. J Med Educ Curric Dev. 2019;6:2382120519875455. doi:10.1177/2382120519875455
10. Splaine ME, Ogrinc G, Gilman SC, et al. The Department of Veterans Affairs National Quality Scholars Fellowship Program: experience from 10 years of training quality scholars. Acad Med. 2009;84(12):1741-1748. doi:10.1097/ACM.0b013e3181bfdcef
11. Watts BV, Paull DE, Williams LC, Neily J, Hemphill RR, Brannen JL. Department of Veterans Affairs chief resident in quality and patient safety program: a model to spread change. Am J Med Qual. 2016;31(6):598-600. doi:10.1177/1062860616643403
12. He K, Whang E, Kristo G. Graduate medical education funding mechanisms, challenges, and solutions: a narrative review. Am J Surg. 2021;221(1):65-71. doi:10.1016/j.amjsurg.2020.06.007
13. Villagrana M. Medicare graduate medical education payments: an overview. Congressional Research Service report IF10960. Updated September 29, 2022. Accessed March 2, 2024. https://crsreports.congress.gov/product/pdf/IF/IF10960
14. Committee on the Governance and Financing of Graduate Medical Education; Board on Health Care Services; Institute of Medicine. Graduate Medical Education That Meets the Nation’s Health Needs. Eden J, Berwick DM, Wilensky GR, eds. Washington, DC: National Academies Press; 2014. doi:10.17226/18754
15. Physician workforce: caps on Medicare-funded graduate medical education at teaching hospitals. Report to congressional requesters. GAO-21-391. May 21, 2021. Accessed March 1, 2024. https://www.gao.gov/assets/gao-21-391.pdf
16. VA and Academic Affiliates: Who Benefits? Hearing Before the Subcommittee on Oversight and Investigations of the Committee on Veterans’ Affairs, 114th Cong, 2nd Sess (2016). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/CHRG-115hhrg29685/html/CHRG-115hhrg29685.htm
17. US Department of Veterans Affairs, Office of Inspector General (OIG). Veterans Health Administration. Review of resident and part-time physician time and attendance at the Oklahoma City VA Health Care System. OIG report 17-00253-93. March 28, 2018. Accessed March 1, 2024. https://www.oversight.gov/sites/default/files/oig-reports/VAOIG-17-00253-93.pdf
18. VA health care: actions needed to improve oversight of graduate medical education reimbursement. Report to the ranking member, Committee on Veterans’ Affairs, House of Representatives. GAO-20-553. July 2020. Accessed March 1, 2024. https://www.gao.gov/assets/710/708275.pdf
19. Functions of Veterans Health Administration: in general, 38 USC §7301 (2022). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/USCODE-2022-title38/pdf/USCODE-2022-title38-partV-chap73-subchapI-sec7301.pdf
20. US Department of Veterans Affairs. Policy memorandum no. 2, policy in association of veterans’ hospitals with medical schools. January 30, 1946.
21. Veterans Health Care Expansion Act of 1973, Public Law 93-82. August 2, 1973. Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/STATUTE-87/pdf/STATUTE-87-Pg179.pdf
22. Residencies and internships, 38 USC § 7406 (2022). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/USCODE-2022-title38/pdf/USCODE-2022-title38-partV-chap74-subchapI-sec7406.pdf
23. Direct graduate medical education (DGME). Centers for Medicaid and Medicare Services. Updated December 5, 2023. Accessed March 1, 2024. https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/AcuteInpatientPPS/DGME
24. Drezdzon MK, Cowley NJ, Sweeney DP, et al. Going for broke: the impact of cost of living on surgery resident stipend value. Ann Surg. 2023;278(6):1053-1059. doi:10.1097/SLA.0000000000005923
25. Special treatment: hospitals that incur indirect costs for graduate medical education programs, 42 CFR § 412.105 (2023). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/CFR-2023-title42-vol2/pdf/CFR-2023-title42-vol2-sec412-105.pdf
26. US Department of Veterans Affairs, Veterans Health Administration. VHA Directive 1400.05, Disbursement agreements for health professions trainees appointed under 38 U.S.C. § 7406. June 2, 2021. Accessed March 1, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=9293
27. Harada ND, Sanders KM, Bowman MA. Health systems education leadership: learning from the VA designated education officer role. Fed Pract. 2022;39(6):266-273. doi:10.12788/fp.0278
28. Schleiter Hitchell K, Johnson L. CMS finalizes rules for distribution of 1000 new Medicare-funded residency positions and changes to rural training track programs. J Grad Med Educ. 2022;14(2):245-249. doi:10.4300/JGME-D-22-00193.1
29. US Department of Veterans Affairs, Veterans Health Administration. VHA Directive 1400.10, Educational cost contracts for health professions education. September 25, 2023. Accessed March 1, 2024. https://www.va.gov/VHAPUBLICATIONS/ViewPublication.asp?pub_ID=11480
30. Direct GME payments: general requirements, 42 CFR § 413.75 (2023). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/CFR-2023-title42-vol2/pdf/CFR-2023-title42-vol2-sec413-75.pdf
31. Direct GME payments: determination of the total number of FTE residents, 42 CFR § 413.78 (2023). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/CFR-2023-title42-vol2/pdf/CFR-2023-title42-vol2-sec413-78.pdf
32. US Department of Health and Human Services, Centers for Medicare and Medicaid Services. Medicare financial management manual, chapter 8. Contractor procedures for provider audits. Accessed March 1, 2024. https://www.cms.gov/regulations-and-guidance/guidance/manuals/downloads/fin106c08.pdf
33. US Department of Health and Human Services, Office of Inspector General. CMS did not always ensure hospitals complied with Medicare reimbursement requirements for graduate medical education. OIG report A-02-17-01017. November 2018. Accessed March 1, 2024. https://oig.hhs.gov/oas/reports/region2/21701017.pdf
34. US Department of Health and Human Services, Centers for Medicare and Medicaid Services. Interns and Residents Information System (IRIS) XML format. Publication 100-20. Transmittal 11418. Change request 12724. May 19, 2022. Accessed March 1, 2024. https://www.hhs.gov/guidance/sites/default/files/hhs-guidance-documents/R11418OTN.pdf
35. Birnbaum AD, Byrne J, on behalf of the VA Office of Academic Affiliations. VHA Updates: Disbursement Policy and Education Cost Contracts. Presented at: American Association of Medical Colleges Webinar; June 2021. Accessed March 1, 2024. https://vimeo.com/644415670
36. Byrne JM, on behalf of the VA Office of Academic Affiliations. Disbursement procedures update for AY 23-24. Accessed March 1, 2024. https://www.va.gov/oaa/Videos/AffiliatePresentationDisbursementandEARsAY23-24.pptx
The US Department of Veterans Affairs (VA) has partnered with academic medical centers and programs since 1946 to provide clinical training for physician residents. Ranking second in federal graduate medical education (GME) funding to the Centers for Medicare and Medicaid Services (CMS), the $850 million VA GME budget annually reimburses > 250 GME-sponsoring institutions (affiliates) of 8000 GME programs for the clinical training of 49,000 individual residents rotating through > 11,000 full-time equivalent (FTE) positions.1 The VA also distributes $1.6 billion to VA facilities to offset the costs of conducting health professions education (HPE) (eg, facility infrastructure, salary support for VA instructors and preceptors, education office administration, and instructional equipment).2 The VA financial and educational contributions account for payment of 11% of resident positions nationally and allow academic medical centers to be less reliant on CMS GME funding.3,4 The VA contributions also provide opportunities for GME expansion,1,5,6 educational innovations,5,7 interprofessional and team-based care,8,9 and quality and safety training.10,11 The Table provides a comparison of CMS and VA GME reimbursability based on activity.
GME financing is complex, particularly the formulaic approach used by CMS, the details of which are often obscured in federal regulations. Due to this complexity and the $16 billion CMS GME budget, academic publications have focused on CMS GME financing while not fully explaining the VA GME policies and processes.4,12-14 By comparison, the VA GME financing model is relatively straightforward and governed by different statues and VA regulations, yet sharing some of the same principles as CMS regulations. Given the challenges in CMS reimbursement to fully support the cost of resident education, as well as the educational opportunities at the VA, the VA designs its reimbursement model to assure that affiliates receive appropriate payments.4,12,15 To ensure the continued success of VA GME partnerships, knowledge of VA GME financing has become increasingly important for designated institutional officers (DIOs) and residency program directors, particularly in light of recent investigations into oversight of the VA’s reimbursement to academic affiliates.
VA AUTHORITY
While the VA’s primary mission is “to provide a complete hospital medical service for the medical care and treatment of veterans,”early VA leaders recognized the importance of affiliating with the nation’s academic institutions.19 In 1946, the VA Policy Memorandum Number 2 established a partnership between the VA and the academic medical community.20 Additional legislation authorized specific agreements with academic affiliates for the central administration of salary and benefits for residents rotating at VA facilities. This process, known as disbursement, is an alternative payroll mechanism whereby the VA reimburses the academic affiliate for resident salary and benefits and the affiliate acts as the disbursing agent, issuing paychecks to residents.21,22
Resident FUNDING
By policy, with rare exceptions, the VA does not sponsor residency programs due to the challenges of providing an appropriate patient mix of age, sex, and medical conditions to meet accreditation standards.4 Nearly all VA reimbursements are for residents in affiliate-sponsored programs, while just 1% pays for residents in legacy, VA-sponsored residency programs at 2 VA facilities. The VA budget for resident (including fellows) salary and benefits is managed by the VA Office of Academic Affiliations (OAA), the national VA office responsible for oversight, policy, and funding of VA HPE programs.
Resident Salaries and Benefits
VA funding of resident salary and benefits are analogous with CMS direct GME (DGME), which is designed to cover resident salary and benefits costs.4,14,23 CMS DGME payments depend on a hospital’s volume of CMS inpatients and are based on a statutory formula, which uses the hospital’s resident FTE positions, the per-resident amount, and Medicare’s share of inpatient beds (Medicare patient load) to determine payments.12 The per-resident amount is set by statute, varies geographically, and is calculated by dividing the hospital’s allowable costs of GME (percentage of CMS inpatient days) divided by the number of residents.12,24
By comparison, the VA GME payment reimburses for each FTE based on the salary and benefits rate set by the academic affiliate. Reimbursement is calculated based on resident time spent at the VA multiplied by a daily salary rate. The daily salary rate is determined by dividing the resident’s total compensation (salary and benefits) by the number of calendar days in an academic year. Resident time spent at the VA facility is determined by obtaining rotation schedules provided by the academic affiliate and verifying resident clinical and educational activity during scheduled rotations.
Indirect Medical Education Funding
In addition to resident salary and benefits, funds to offset the cost of conducting HPE are provided to VA facilities. These funds are intended to improve and maintain necessary infrastructure for all HPE programs not just GME, including education office administration needs, teaching costs (ie, a portion of VA preceptors salary), and instructional equipment.
The Veterans Equitable Resource Allocation (VERA) is a national budgeting process for VA medical facilities that funds facility operational needs such as staff salary and benefits, infrastructure, and equipment.2 The education portion of the VERA, the VERA Education Support Component (VESC), is not managed by the OAA, but rather is distributed through the VERA model to the general budget of VA facilities hosting HPE (Figure). VESC funding in the VA budget is based on labor mapping of physician time spent in education; other labor mapping categories include clinical care, research, and administration. VA facility VESC funding is calculated based on the number of paid health profession trainees (HPTs) from all professions, apportioned according to the number of FTEs for physician residents and VA-paid HPTs in other disciplines. In fiscal year 2024, VA facilities received $115,812 for each physician resident FTE position and $84,906 for each VA-paid, non-GME FTE position.
The VESC is like CMS's indirect GME funding, termed Indirect Medical Education (IME), an additional payment for each Medicare patient discharged reflecting teaching hospitals’ higher patient care costs relative to nonteaching hospitals. Described elsewhere, IME is calculated using a resident-to-bed ratio and a multiplier, which is set by statute.4,25 While IME can be used for reimbursement for some resident clinical and educational activities(eg, research), VA VESC funds cannot be used for such activities and are part of the general facility budget and appropriated per the discretion of the medical facility director.
ESTABLISHING GME PARTNERSHIPS
An affiliation agreement establishes the administrative and legal requirements for educational relationships with academic affiliates and includes standards for conducting HPE, responsibilities for accreditation standards, program leadership, faculty, resources, supervision, academic policies, and procedures. The VA uses standardized affiliation agreement templates that have been vetted with accrediting bodies and the VA Office of General Counsel.
A disbursement agreement authorizes the VA to reimburse affiliates for resident salary and benefits for VA clinical and educational activities. The disbursement agreement details the fiscal arrangements (eg, payment in advance vs arrears, salary, and benefit rates, leave) for the reimbursement payments. Veterans Health Administration (VHA) Directive 1400.05 provides the policy and procedures for calculating reimbursement for HPT educational activities.26
The VA facility designated education officer (DEO) oversees all HPE programs and coordinates the affiliation and disbursement agreement processes.27 The DEO, affiliate DIO, residency program director, and VA residency site director determine the physician resident FTE positions assigned to a VA facility based on educational objectives and availability of educational resources at the VA facility, such as patient care opportunities, faculty supervisors, space, and equipment. The VA facility requests for resident FTE positions are submitted to the OAA by the facility DEO.
Once GME FTE positions are approved by the OAA, VA facilities work with their academic affiliate to submit the physician resident salary and benefit rate. Affiliate DIOs attest to the accuracy of the salary rate schedule and the local DEO submits the budget request to the OAA. Upon approval, the funds are transferred to the VA facility each fiscal year, which begins October 1. DEOs report quarterly to the OAA both budget needs and excesses based on variations in the approved FTEs due to additional VA rotations, physician resident attrition, or reassignment.
Resident Position Allocation
VA GME financing provides flexibility through periodic needs assessments and expansion initiatives. In August and December, DEOs collaborate with an academic affiliate to submit reports to the OAA confirming their projected GME needs for the next academic year. Additional positions requests are reviewed by the OAA; funding depends on budget and the educational justification. The OAA periodically issues GME expansion requests for proposal, which typically arise from legislation to address specific VA workforce needs. The VA facility DEO and affiliate GME leaders collaborate to apply for additional positions. For example, a VA GME expansion under the Veterans Access, Choice, and Accountability Act of 2014 added 1500 GME positions in 8 years for critically needed specialties and in rural and underserved areas.5 The Maintaining Internal Systems and Strengthening Outside Networks (MISSION) Act of 2018 authorized a pilot program for VA to fund residents at non-VA facilities with priority for Indian Health Services, Tribes and Tribal Organizations, Federally Qualified Health Centers, and US Department of Defense facilities to provide access to veterans in underserved areas.6
The VA GME financing system has flexibility to meet local needs for additional resident positions and to address broader VA workforce gaps through targeted expansion. Generally, CMS does not fund positions to address workforce needs, place residents in specific geographic areas, or require the training of certain types of residents.4 However, the Consolidated Appropriations Act of 2021 has provided the opportunity to address rural workforce needs.28
Reimbursement
The VA provides reimbursement for clinical and educational activities performed in VA facilities for the benefit of veterans as well as research, didactics, meetings and conferences, annual and sick leave, and orientation. The VA also may provide reimbursement for educational activities that occur off VA grounds (eg, the VA proportional share of a residency program’s didactic sessions). The VA does not reimburse for affiliate clinical duties or administrative costs, although a national policy allows VA facilities to reimburse affiliates for some GME overhead costs.29
CMS similarly reimburses for residency training time spent in patient care activities as well as orientation activities, didactics, leave, and, in some cases, research.4,30,31 CMS makes payments to hospitals, which may include sponsoring institutions and Medicare-eligible participating training sites.4,30,31 For both the VA and CMS, residents may not be counted twice for reimbursement by 2 federal agencies; in other words, a resident may not count for > 1 FTE.4,30-32
GME Oversight
VA GME funding came under significant scrutiny. At a 2016 House Veterans Affairs Committee hearing, Representative Phil Roe, MD (R-Tennessee), noted that no process existed at many VA facilities for “determining trainee presence” and that many VA medical centers had “difficulty tracking resident rotations”16 A VA Office of the Inspector General investigation recommended that the VA implement policies and procedures to improve oversight to “ensure residents are fully participating in educational activities” and that the VA is “paying the correct amount” to the affiliate.17 A 2020 General Accountability Office report outlined unclear policy guidance, incomplete tracking of resident activities, and improper fiscal processes for reimbursement and reconciliation of affiliate invoices.18
In response, the OAA created an oversight and compliance unit, revised VHA Directive 1400.05 (the policy for disbursement), and improved resident tracking procedures.26 The standard operating procedure that accompanied VHA Directive 1400.05 provides detailed information for the DEO and VA facility staff for tracking resident clinical and educational activities. FTE counts are essential to both VA and CMS for accurate reimbursement. The eAppendix and the Table provide a guide to reimbursable activities in the VA for the calculation of reimbursement, with a comparison to CMS.33,34 The OAA in cooperation with other VA staff and officers periodically conducts audits to assess compliance with disbursement policy and affiliate reimbursement accuracy.
In the VA, resident activities are captured on the VA Educational Activity Record, a standardized spreadsheet to track activities and calculate reimbursement. Each VA facility hosting resident physicians manually records resident activity by the half-day. This process is labor intensive, involving both VA and affiliate staff to accurately reconcile payments. To address the workload demands, the OAA is developing an online tool that will automate aspects of the tracking process. Also, to ensure adequate staffing, the OAA is in the process of implementing an office optimization project, providing standardized position descriptions, an organizational chart, and staffing levels for DEO offices in VA facilities.
Conclusions
This report describes the key policies and principles of VA GME financing, highlighting the essential similarities and differences between VA and CMS. Neither the VA nor CMS regulations allow for reimbursement for > 1 FTE position per resident, a principle that underpins the assignment of resident rotations and federal funding for GME and are similar with respect to reimbursement for patient care activities, didactics, research, orientation, and scholarly activity. While reimbursable activities in the VA require physical presence and care of veteran patients, CMS also limits reimbursement to resident activities in the hospital and approved other settings if the hospital is paying for resident salary and benefits in these settings. The VA provides some flexibility for offsite activities including didactics and, in specific circumstances, remote care of veteran patients (eg, teleradiology).
The VA and CMS use different GME financing models. For example, the CMS calculations for resident FTEs are complex, whereas VA calculations reimburse the salary and benefits as set by the academic affiliate. The VA process accounts for local variation in salary rates, whereas the per-resident amount set by CMS varies regionally and does not fully account for differences in the cost of living.24 Because all patients in VA facilities are veterans, VA calculations for reimbursement do not involve ratios of beds like the CMS calculations to determine a proportional share of reimbursement. The VA GME expansion tends to be more directed to VA health workforce needs than CMS, specifying the types of programs and geographic locations to address these needs.
The VA regularly reevaluates how affiliates are reimbursed for VA resident activity, balancing compliance with VA policies and the workload for VA and its affiliates. The VA obtains input from key stakeholders including DEOs, DIOs, and professional organizations such as the Association of American Medical Colleges and the Accreditation Council for Graduate Medical Education.35,36
Looking ahead, the VA is developing an online tool to improve the accuracy of affiliate reimbursement. The VA will also implement a standardized staffing model, organizational structure, and position descriptions for DEO offices. These initiatives will help reduce the burden of tracking and verifying resident activity and continue to support the 77-year partnership between VA and its affiliated institutions.
The US Department of Veterans Affairs (VA) has partnered with academic medical centers and programs since 1946 to provide clinical training for physician residents. Ranking second in federal graduate medical education (GME) funding to the Centers for Medicare and Medicaid Services (CMS), the $850 million VA GME budget annually reimburses > 250 GME-sponsoring institutions (affiliates) of 8000 GME programs for the clinical training of 49,000 individual residents rotating through > 11,000 full-time equivalent (FTE) positions.1 The VA also distributes $1.6 billion to VA facilities to offset the costs of conducting health professions education (HPE) (eg, facility infrastructure, salary support for VA instructors and preceptors, education office administration, and instructional equipment).2 The VA financial and educational contributions account for payment of 11% of resident positions nationally and allow academic medical centers to be less reliant on CMS GME funding.3,4 The VA contributions also provide opportunities for GME expansion,1,5,6 educational innovations,5,7 interprofessional and team-based care,8,9 and quality and safety training.10,11 The Table provides a comparison of CMS and VA GME reimbursability based on activity.
GME financing is complex, particularly the formulaic approach used by CMS, the details of which are often obscured in federal regulations. Due to this complexity and the $16 billion CMS GME budget, academic publications have focused on CMS GME financing while not fully explaining the VA GME policies and processes.4,12-14 By comparison, the VA GME financing model is relatively straightforward and governed by different statues and VA regulations, yet sharing some of the same principles as CMS regulations. Given the challenges in CMS reimbursement to fully support the cost of resident education, as well as the educational opportunities at the VA, the VA designs its reimbursement model to assure that affiliates receive appropriate payments.4,12,15 To ensure the continued success of VA GME partnerships, knowledge of VA GME financing has become increasingly important for designated institutional officers (DIOs) and residency program directors, particularly in light of recent investigations into oversight of the VA’s reimbursement to academic affiliates.
VA AUTHORITY
While the VA’s primary mission is “to provide a complete hospital medical service for the medical care and treatment of veterans,”early VA leaders recognized the importance of affiliating with the nation’s academic institutions.19 In 1946, the VA Policy Memorandum Number 2 established a partnership between the VA and the academic medical community.20 Additional legislation authorized specific agreements with academic affiliates for the central administration of salary and benefits for residents rotating at VA facilities. This process, known as disbursement, is an alternative payroll mechanism whereby the VA reimburses the academic affiliate for resident salary and benefits and the affiliate acts as the disbursing agent, issuing paychecks to residents.21,22
Resident FUNDING
By policy, with rare exceptions, the VA does not sponsor residency programs due to the challenges of providing an appropriate patient mix of age, sex, and medical conditions to meet accreditation standards.4 Nearly all VA reimbursements are for residents in affiliate-sponsored programs, while just 1% pays for residents in legacy, VA-sponsored residency programs at 2 VA facilities. The VA budget for resident (including fellows) salary and benefits is managed by the VA Office of Academic Affiliations (OAA), the national VA office responsible for oversight, policy, and funding of VA HPE programs.
Resident Salaries and Benefits
VA funding of resident salary and benefits are analogous with CMS direct GME (DGME), which is designed to cover resident salary and benefits costs.4,14,23 CMS DGME payments depend on a hospital’s volume of CMS inpatients and are based on a statutory formula, which uses the hospital’s resident FTE positions, the per-resident amount, and Medicare’s share of inpatient beds (Medicare patient load) to determine payments.12 The per-resident amount is set by statute, varies geographically, and is calculated by dividing the hospital’s allowable costs of GME (percentage of CMS inpatient days) divided by the number of residents.12,24
By comparison, the VA GME payment reimburses for each FTE based on the salary and benefits rate set by the academic affiliate. Reimbursement is calculated based on resident time spent at the VA multiplied by a daily salary rate. The daily salary rate is determined by dividing the resident’s total compensation (salary and benefits) by the number of calendar days in an academic year. Resident time spent at the VA facility is determined by obtaining rotation schedules provided by the academic affiliate and verifying resident clinical and educational activity during scheduled rotations.
Indirect Medical Education Funding
In addition to resident salary and benefits, funds to offset the cost of conducting HPE are provided to VA facilities. These funds are intended to improve and maintain necessary infrastructure for all HPE programs not just GME, including education office administration needs, teaching costs (ie, a portion of VA preceptors salary), and instructional equipment.
The Veterans Equitable Resource Allocation (VERA) is a national budgeting process for VA medical facilities that funds facility operational needs such as staff salary and benefits, infrastructure, and equipment.2 The education portion of the VERA, the VERA Education Support Component (VESC), is not managed by the OAA, but rather is distributed through the VERA model to the general budget of VA facilities hosting HPE (Figure). VESC funding in the VA budget is based on labor mapping of physician time spent in education; other labor mapping categories include clinical care, research, and administration. VA facility VESC funding is calculated based on the number of paid health profession trainees (HPTs) from all professions, apportioned according to the number of FTEs for physician residents and VA-paid HPTs in other disciplines. In fiscal year 2024, VA facilities received $115,812 for each physician resident FTE position and $84,906 for each VA-paid, non-GME FTE position.
The VESC is like CMS's indirect GME funding, termed Indirect Medical Education (IME), an additional payment for each Medicare patient discharged reflecting teaching hospitals’ higher patient care costs relative to nonteaching hospitals. Described elsewhere, IME is calculated using a resident-to-bed ratio and a multiplier, which is set by statute.4,25 While IME can be used for reimbursement for some resident clinical and educational activities(eg, research), VA VESC funds cannot be used for such activities and are part of the general facility budget and appropriated per the discretion of the medical facility director.
ESTABLISHING GME PARTNERSHIPS
An affiliation agreement establishes the administrative and legal requirements for educational relationships with academic affiliates and includes standards for conducting HPE, responsibilities for accreditation standards, program leadership, faculty, resources, supervision, academic policies, and procedures. The VA uses standardized affiliation agreement templates that have been vetted with accrediting bodies and the VA Office of General Counsel.
A disbursement agreement authorizes the VA to reimburse affiliates for resident salary and benefits for VA clinical and educational activities. The disbursement agreement details the fiscal arrangements (eg, payment in advance vs arrears, salary, and benefit rates, leave) for the reimbursement payments. Veterans Health Administration (VHA) Directive 1400.05 provides the policy and procedures for calculating reimbursement for HPT educational activities.26
The VA facility designated education officer (DEO) oversees all HPE programs and coordinates the affiliation and disbursement agreement processes.27 The DEO, affiliate DIO, residency program director, and VA residency site director determine the physician resident FTE positions assigned to a VA facility based on educational objectives and availability of educational resources at the VA facility, such as patient care opportunities, faculty supervisors, space, and equipment. The VA facility requests for resident FTE positions are submitted to the OAA by the facility DEO.
Once GME FTE positions are approved by the OAA, VA facilities work with their academic affiliate to submit the physician resident salary and benefit rate. Affiliate DIOs attest to the accuracy of the salary rate schedule and the local DEO submits the budget request to the OAA. Upon approval, the funds are transferred to the VA facility each fiscal year, which begins October 1. DEOs report quarterly to the OAA both budget needs and excesses based on variations in the approved FTEs due to additional VA rotations, physician resident attrition, or reassignment.
Resident Position Allocation
VA GME financing provides flexibility through periodic needs assessments and expansion initiatives. In August and December, DEOs collaborate with an academic affiliate to submit reports to the OAA confirming their projected GME needs for the next academic year. Additional positions requests are reviewed by the OAA; funding depends on budget and the educational justification. The OAA periodically issues GME expansion requests for proposal, which typically arise from legislation to address specific VA workforce needs. The VA facility DEO and affiliate GME leaders collaborate to apply for additional positions. For example, a VA GME expansion under the Veterans Access, Choice, and Accountability Act of 2014 added 1500 GME positions in 8 years for critically needed specialties and in rural and underserved areas.5 The Maintaining Internal Systems and Strengthening Outside Networks (MISSION) Act of 2018 authorized a pilot program for VA to fund residents at non-VA facilities with priority for Indian Health Services, Tribes and Tribal Organizations, Federally Qualified Health Centers, and US Department of Defense facilities to provide access to veterans in underserved areas.6
The VA GME financing system has flexibility to meet local needs for additional resident positions and to address broader VA workforce gaps through targeted expansion. Generally, CMS does not fund positions to address workforce needs, place residents in specific geographic areas, or require the training of certain types of residents.4 However, the Consolidated Appropriations Act of 2021 has provided the opportunity to address rural workforce needs.28
Reimbursement
The VA provides reimbursement for clinical and educational activities performed in VA facilities for the benefit of veterans as well as research, didactics, meetings and conferences, annual and sick leave, and orientation. The VA also may provide reimbursement for educational activities that occur off VA grounds (eg, the VA proportional share of a residency program’s didactic sessions). The VA does not reimburse for affiliate clinical duties or administrative costs, although a national policy allows VA facilities to reimburse affiliates for some GME overhead costs.29
CMS similarly reimburses for residency training time spent in patient care activities as well as orientation activities, didactics, leave, and, in some cases, research.4,30,31 CMS makes payments to hospitals, which may include sponsoring institutions and Medicare-eligible participating training sites.4,30,31 For both the VA and CMS, residents may not be counted twice for reimbursement by 2 federal agencies; in other words, a resident may not count for > 1 FTE.4,30-32
GME Oversight
VA GME funding came under significant scrutiny. At a 2016 House Veterans Affairs Committee hearing, Representative Phil Roe, MD (R-Tennessee), noted that no process existed at many VA facilities for “determining trainee presence” and that many VA medical centers had “difficulty tracking resident rotations”16 A VA Office of the Inspector General investigation recommended that the VA implement policies and procedures to improve oversight to “ensure residents are fully participating in educational activities” and that the VA is “paying the correct amount” to the affiliate.17 A 2020 General Accountability Office report outlined unclear policy guidance, incomplete tracking of resident activities, and improper fiscal processes for reimbursement and reconciliation of affiliate invoices.18
In response, the OAA created an oversight and compliance unit, revised VHA Directive 1400.05 (the policy for disbursement), and improved resident tracking procedures.26 The standard operating procedure that accompanied VHA Directive 1400.05 provides detailed information for the DEO and VA facility staff for tracking resident clinical and educational activities. FTE counts are essential to both VA and CMS for accurate reimbursement. The eAppendix and the Table provide a guide to reimbursable activities in the VA for the calculation of reimbursement, with a comparison to CMS.33,34 The OAA in cooperation with other VA staff and officers periodically conducts audits to assess compliance with disbursement policy and affiliate reimbursement accuracy.
In the VA, resident activities are captured on the VA Educational Activity Record, a standardized spreadsheet to track activities and calculate reimbursement. Each VA facility hosting resident physicians manually records resident activity by the half-day. This process is labor intensive, involving both VA and affiliate staff to accurately reconcile payments. To address the workload demands, the OAA is developing an online tool that will automate aspects of the tracking process. Also, to ensure adequate staffing, the OAA is in the process of implementing an office optimization project, providing standardized position descriptions, an organizational chart, and staffing levels for DEO offices in VA facilities.
Conclusions
This report describes the key policies and principles of VA GME financing, highlighting the essential similarities and differences between VA and CMS. Neither the VA nor CMS regulations allow for reimbursement for > 1 FTE position per resident, a principle that underpins the assignment of resident rotations and federal funding for GME and are similar with respect to reimbursement for patient care activities, didactics, research, orientation, and scholarly activity. While reimbursable activities in the VA require physical presence and care of veteran patients, CMS also limits reimbursement to resident activities in the hospital and approved other settings if the hospital is paying for resident salary and benefits in these settings. The VA provides some flexibility for offsite activities including didactics and, in specific circumstances, remote care of veteran patients (eg, teleradiology).
The VA and CMS use different GME financing models. For example, the CMS calculations for resident FTEs are complex, whereas VA calculations reimburse the salary and benefits as set by the academic affiliate. The VA process accounts for local variation in salary rates, whereas the per-resident amount set by CMS varies regionally and does not fully account for differences in the cost of living.24 Because all patients in VA facilities are veterans, VA calculations for reimbursement do not involve ratios of beds like the CMS calculations to determine a proportional share of reimbursement. The VA GME expansion tends to be more directed to VA health workforce needs than CMS, specifying the types of programs and geographic locations to address these needs.
The VA regularly reevaluates how affiliates are reimbursed for VA resident activity, balancing compliance with VA policies and the workload for VA and its affiliates. The VA obtains input from key stakeholders including DEOs, DIOs, and professional organizations such as the Association of American Medical Colleges and the Accreditation Council for Graduate Medical Education.35,36
Looking ahead, the VA is developing an online tool to improve the accuracy of affiliate reimbursement. The VA will also implement a standardized staffing model, organizational structure, and position descriptions for DEO offices. These initiatives will help reduce the burden of tracking and verifying resident activity and continue to support the 77-year partnership between VA and its affiliated institutions.
1. Klink KA, Albanese AP, Bope ET, Sanders KM. Veterans Affairs graduate medical education expansion addresses US physician workforce needs. Acad Med. 2022;97(8):1144-1150. doi:10.1097/ACM.0000000000004545
2. Andrus CH, Johnson K, Pierce E, Romito PJ, Hartel P, Berrios‐Guccione S, Best W. Finance modeling in the delivery of medical care in tertiary‐care hospitals in the Department of Veterans Affairs. J Surg Res. 2001;96(2):152-157. doi:10.1006/jsre.1999.5728
3. Petrakis IL, Kozal M. Academic medical centers and the U.S. Department of Veterans Affairs: a 75-year partnership influences medical education, scientific discovery, and clinical care. Acad Med. 2022;97(8):1110-1113. doi:10.1097/ACM.0000000000004734
4. Heisler EJ, Mendez BH, Mitchell A, Panangala SV, Villagrana MA. Federal support for graduate medical education: an overview (R44376). Congressional Research Service report R44376; version 11. Updated December 27, 2018. Accessed March 2, 2024. https://crsreports.congress.gov/product/pdf/R/R44376/11
5. Chang BK, Brannen JL. The Veterans Access, Choice, and Accountability Act of 2014: examining graduate medical education enhancement in the Department of Veterans Affairs. Acad Med. 2015;90(9):1196-1198. doi:10.1097/ACM.0000000000000795
6. Albanese AP, Bope ET, Sanders KM, Bowman M. The VA MISSION Act of 2018: a potential game changer for rural GME expansion and veteran health care. J Rural Health. 2020;36(1):133-136. doi:10.1111/jrh.12360
7. Lypson ML, Roberts LW. Valuing the partnership between the Veterans Health Administration and academic medicine. Acad Med. 2022;97(8):1091-1093. doi:10.1097/ACM.0000000000004748
8. Harada ND, Traylor L, Rugen KW, et al. Interprofessional transformation of clinical education: the first six years of the Veterans Affairs Centers of Excellence in Primary Care Education. J Interprof Care. 2023;37(suppl 1):S86-S94. doi:10.1080/13561820.2018.1433642
9. Harada ND, Rajashekara S, Sansgiry S, et al. Developing interprofessional primary care teams: alumni evaluation of the Department of Veterans Affairs Centers of Excellence in Primary Care Education Program. J Med Educ Curric Dev. 2019;6:2382120519875455. doi:10.1177/2382120519875455
10. Splaine ME, Ogrinc G, Gilman SC, et al. The Department of Veterans Affairs National Quality Scholars Fellowship Program: experience from 10 years of training quality scholars. Acad Med. 2009;84(12):1741-1748. doi:10.1097/ACM.0b013e3181bfdcef
11. Watts BV, Paull DE, Williams LC, Neily J, Hemphill RR, Brannen JL. Department of Veterans Affairs chief resident in quality and patient safety program: a model to spread change. Am J Med Qual. 2016;31(6):598-600. doi:10.1177/1062860616643403
12. He K, Whang E, Kristo G. Graduate medical education funding mechanisms, challenges, and solutions: a narrative review. Am J Surg. 2021;221(1):65-71. doi:10.1016/j.amjsurg.2020.06.007
13. Villagrana M. Medicare graduate medical education payments: an overview. Congressional Research Service report IF10960. Updated September 29, 2022. Accessed March 2, 2024. https://crsreports.congress.gov/product/pdf/IF/IF10960
14. Committee on the Governance and Financing of Graduate Medical Education; Board on Health Care Services; Institute of Medicine. Graduate Medical Education That Meets the Nation’s Health Needs. Eden J, Berwick DM, Wilensky GR, eds. Washington, DC: National Academies Press; 2014. doi:10.17226/18754
15. Physician workforce: caps on Medicare-funded graduate medical education at teaching hospitals. Report to congressional requesters. GAO-21-391. May 21, 2021. Accessed March 1, 2024. https://www.gao.gov/assets/gao-21-391.pdf
16. VA and Academic Affiliates: Who Benefits? Hearing Before the Subcommittee on Oversight and Investigations of the Committee on Veterans’ Affairs, 114th Cong, 2nd Sess (2016). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/CHRG-115hhrg29685/html/CHRG-115hhrg29685.htm
17. US Department of Veterans Affairs, Office of Inspector General (OIG). Veterans Health Administration. Review of resident and part-time physician time and attendance at the Oklahoma City VA Health Care System. OIG report 17-00253-93. March 28, 2018. Accessed March 1, 2024. https://www.oversight.gov/sites/default/files/oig-reports/VAOIG-17-00253-93.pdf
18. VA health care: actions needed to improve oversight of graduate medical education reimbursement. Report to the ranking member, Committee on Veterans’ Affairs, House of Representatives. GAO-20-553. July 2020. Accessed March 1, 2024. https://www.gao.gov/assets/710/708275.pdf
19. Functions of Veterans Health Administration: in general, 38 USC §7301 (2022). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/USCODE-2022-title38/pdf/USCODE-2022-title38-partV-chap73-subchapI-sec7301.pdf
20. US Department of Veterans Affairs. Policy memorandum no. 2, policy in association of veterans’ hospitals with medical schools. January 30, 1946.
21. Veterans Health Care Expansion Act of 1973, Public Law 93-82. August 2, 1973. Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/STATUTE-87/pdf/STATUTE-87-Pg179.pdf
22. Residencies and internships, 38 USC § 7406 (2022). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/USCODE-2022-title38/pdf/USCODE-2022-title38-partV-chap74-subchapI-sec7406.pdf
23. Direct graduate medical education (DGME). Centers for Medicaid and Medicare Services. Updated December 5, 2023. Accessed March 1, 2024. https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/AcuteInpatientPPS/DGME
24. Drezdzon MK, Cowley NJ, Sweeney DP, et al. Going for broke: the impact of cost of living on surgery resident stipend value. Ann Surg. 2023;278(6):1053-1059. doi:10.1097/SLA.0000000000005923
25. Special treatment: hospitals that incur indirect costs for graduate medical education programs, 42 CFR § 412.105 (2023). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/CFR-2023-title42-vol2/pdf/CFR-2023-title42-vol2-sec412-105.pdf
26. US Department of Veterans Affairs, Veterans Health Administration. VHA Directive 1400.05, Disbursement agreements for health professions trainees appointed under 38 U.S.C. § 7406. June 2, 2021. Accessed March 1, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=9293
27. Harada ND, Sanders KM, Bowman MA. Health systems education leadership: learning from the VA designated education officer role. Fed Pract. 2022;39(6):266-273. doi:10.12788/fp.0278
28. Schleiter Hitchell K, Johnson L. CMS finalizes rules for distribution of 1000 new Medicare-funded residency positions and changes to rural training track programs. J Grad Med Educ. 2022;14(2):245-249. doi:10.4300/JGME-D-22-00193.1
29. US Department of Veterans Affairs, Veterans Health Administration. VHA Directive 1400.10, Educational cost contracts for health professions education. September 25, 2023. Accessed March 1, 2024. https://www.va.gov/VHAPUBLICATIONS/ViewPublication.asp?pub_ID=11480
30. Direct GME payments: general requirements, 42 CFR § 413.75 (2023). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/CFR-2023-title42-vol2/pdf/CFR-2023-title42-vol2-sec413-75.pdf
31. Direct GME payments: determination of the total number of FTE residents, 42 CFR § 413.78 (2023). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/CFR-2023-title42-vol2/pdf/CFR-2023-title42-vol2-sec413-78.pdf
32. US Department of Health and Human Services, Centers for Medicare and Medicaid Services. Medicare financial management manual, chapter 8. Contractor procedures for provider audits. Accessed March 1, 2024. https://www.cms.gov/regulations-and-guidance/guidance/manuals/downloads/fin106c08.pdf
33. US Department of Health and Human Services, Office of Inspector General. CMS did not always ensure hospitals complied with Medicare reimbursement requirements for graduate medical education. OIG report A-02-17-01017. November 2018. Accessed March 1, 2024. https://oig.hhs.gov/oas/reports/region2/21701017.pdf
34. US Department of Health and Human Services, Centers for Medicare and Medicaid Services. Interns and Residents Information System (IRIS) XML format. Publication 100-20. Transmittal 11418. Change request 12724. May 19, 2022. Accessed March 1, 2024. https://www.hhs.gov/guidance/sites/default/files/hhs-guidance-documents/R11418OTN.pdf
35. Birnbaum AD, Byrne J, on behalf of the VA Office of Academic Affiliations. VHA Updates: Disbursement Policy and Education Cost Contracts. Presented at: American Association of Medical Colleges Webinar; June 2021. Accessed March 1, 2024. https://vimeo.com/644415670
36. Byrne JM, on behalf of the VA Office of Academic Affiliations. Disbursement procedures update for AY 23-24. Accessed March 1, 2024. https://www.va.gov/oaa/Videos/AffiliatePresentationDisbursementandEARsAY23-24.pptx
1. Klink KA, Albanese AP, Bope ET, Sanders KM. Veterans Affairs graduate medical education expansion addresses US physician workforce needs. Acad Med. 2022;97(8):1144-1150. doi:10.1097/ACM.0000000000004545
2. Andrus CH, Johnson K, Pierce E, Romito PJ, Hartel P, Berrios‐Guccione S, Best W. Finance modeling in the delivery of medical care in tertiary‐care hospitals in the Department of Veterans Affairs. J Surg Res. 2001;96(2):152-157. doi:10.1006/jsre.1999.5728
3. Petrakis IL, Kozal M. Academic medical centers and the U.S. Department of Veterans Affairs: a 75-year partnership influences medical education, scientific discovery, and clinical care. Acad Med. 2022;97(8):1110-1113. doi:10.1097/ACM.0000000000004734
4. Heisler EJ, Mendez BH, Mitchell A, Panangala SV, Villagrana MA. Federal support for graduate medical education: an overview (R44376). Congressional Research Service report R44376; version 11. Updated December 27, 2018. Accessed March 2, 2024. https://crsreports.congress.gov/product/pdf/R/R44376/11
5. Chang BK, Brannen JL. The Veterans Access, Choice, and Accountability Act of 2014: examining graduate medical education enhancement in the Department of Veterans Affairs. Acad Med. 2015;90(9):1196-1198. doi:10.1097/ACM.0000000000000795
6. Albanese AP, Bope ET, Sanders KM, Bowman M. The VA MISSION Act of 2018: a potential game changer for rural GME expansion and veteran health care. J Rural Health. 2020;36(1):133-136. doi:10.1111/jrh.12360
7. Lypson ML, Roberts LW. Valuing the partnership between the Veterans Health Administration and academic medicine. Acad Med. 2022;97(8):1091-1093. doi:10.1097/ACM.0000000000004748
8. Harada ND, Traylor L, Rugen KW, et al. Interprofessional transformation of clinical education: the first six years of the Veterans Affairs Centers of Excellence in Primary Care Education. J Interprof Care. 2023;37(suppl 1):S86-S94. doi:10.1080/13561820.2018.1433642
9. Harada ND, Rajashekara S, Sansgiry S, et al. Developing interprofessional primary care teams: alumni evaluation of the Department of Veterans Affairs Centers of Excellence in Primary Care Education Program. J Med Educ Curric Dev. 2019;6:2382120519875455. doi:10.1177/2382120519875455
10. Splaine ME, Ogrinc G, Gilman SC, et al. The Department of Veterans Affairs National Quality Scholars Fellowship Program: experience from 10 years of training quality scholars. Acad Med. 2009;84(12):1741-1748. doi:10.1097/ACM.0b013e3181bfdcef
11. Watts BV, Paull DE, Williams LC, Neily J, Hemphill RR, Brannen JL. Department of Veterans Affairs chief resident in quality and patient safety program: a model to spread change. Am J Med Qual. 2016;31(6):598-600. doi:10.1177/1062860616643403
12. He K, Whang E, Kristo G. Graduate medical education funding mechanisms, challenges, and solutions: a narrative review. Am J Surg. 2021;221(1):65-71. doi:10.1016/j.amjsurg.2020.06.007
13. Villagrana M. Medicare graduate medical education payments: an overview. Congressional Research Service report IF10960. Updated September 29, 2022. Accessed March 2, 2024. https://crsreports.congress.gov/product/pdf/IF/IF10960
14. Committee on the Governance and Financing of Graduate Medical Education; Board on Health Care Services; Institute of Medicine. Graduate Medical Education That Meets the Nation’s Health Needs. Eden J, Berwick DM, Wilensky GR, eds. Washington, DC: National Academies Press; 2014. doi:10.17226/18754
15. Physician workforce: caps on Medicare-funded graduate medical education at teaching hospitals. Report to congressional requesters. GAO-21-391. May 21, 2021. Accessed March 1, 2024. https://www.gao.gov/assets/gao-21-391.pdf
16. VA and Academic Affiliates: Who Benefits? Hearing Before the Subcommittee on Oversight and Investigations of the Committee on Veterans’ Affairs, 114th Cong, 2nd Sess (2016). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/CHRG-115hhrg29685/html/CHRG-115hhrg29685.htm
17. US Department of Veterans Affairs, Office of Inspector General (OIG). Veterans Health Administration. Review of resident and part-time physician time and attendance at the Oklahoma City VA Health Care System. OIG report 17-00253-93. March 28, 2018. Accessed March 1, 2024. https://www.oversight.gov/sites/default/files/oig-reports/VAOIG-17-00253-93.pdf
18. VA health care: actions needed to improve oversight of graduate medical education reimbursement. Report to the ranking member, Committee on Veterans’ Affairs, House of Representatives. GAO-20-553. July 2020. Accessed March 1, 2024. https://www.gao.gov/assets/710/708275.pdf
19. Functions of Veterans Health Administration: in general, 38 USC §7301 (2022). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/USCODE-2022-title38/pdf/USCODE-2022-title38-partV-chap73-subchapI-sec7301.pdf
20. US Department of Veterans Affairs. Policy memorandum no. 2, policy in association of veterans’ hospitals with medical schools. January 30, 1946.
21. Veterans Health Care Expansion Act of 1973, Public Law 93-82. August 2, 1973. Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/STATUTE-87/pdf/STATUTE-87-Pg179.pdf
22. Residencies and internships, 38 USC § 7406 (2022). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/USCODE-2022-title38/pdf/USCODE-2022-title38-partV-chap74-subchapI-sec7406.pdf
23. Direct graduate medical education (DGME). Centers for Medicaid and Medicare Services. Updated December 5, 2023. Accessed March 1, 2024. https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/AcuteInpatientPPS/DGME
24. Drezdzon MK, Cowley NJ, Sweeney DP, et al. Going for broke: the impact of cost of living on surgery resident stipend value. Ann Surg. 2023;278(6):1053-1059. doi:10.1097/SLA.0000000000005923
25. Special treatment: hospitals that incur indirect costs for graduate medical education programs, 42 CFR § 412.105 (2023). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/CFR-2023-title42-vol2/pdf/CFR-2023-title42-vol2-sec412-105.pdf
26. US Department of Veterans Affairs, Veterans Health Administration. VHA Directive 1400.05, Disbursement agreements for health professions trainees appointed under 38 U.S.C. § 7406. June 2, 2021. Accessed March 1, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=9293
27. Harada ND, Sanders KM, Bowman MA. Health systems education leadership: learning from the VA designated education officer role. Fed Pract. 2022;39(6):266-273. doi:10.12788/fp.0278
28. Schleiter Hitchell K, Johnson L. CMS finalizes rules for distribution of 1000 new Medicare-funded residency positions and changes to rural training track programs. J Grad Med Educ. 2022;14(2):245-249. doi:10.4300/JGME-D-22-00193.1
29. US Department of Veterans Affairs, Veterans Health Administration. VHA Directive 1400.10, Educational cost contracts for health professions education. September 25, 2023. Accessed March 1, 2024. https://www.va.gov/VHAPUBLICATIONS/ViewPublication.asp?pub_ID=11480
30. Direct GME payments: general requirements, 42 CFR § 413.75 (2023). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/CFR-2023-title42-vol2/pdf/CFR-2023-title42-vol2-sec413-75.pdf
31. Direct GME payments: determination of the total number of FTE residents, 42 CFR § 413.78 (2023). Accessed March 1, 2024. https://www.govinfo.gov/content/pkg/CFR-2023-title42-vol2/pdf/CFR-2023-title42-vol2-sec413-78.pdf
32. US Department of Health and Human Services, Centers for Medicare and Medicaid Services. Medicare financial management manual, chapter 8. Contractor procedures for provider audits. Accessed March 1, 2024. https://www.cms.gov/regulations-and-guidance/guidance/manuals/downloads/fin106c08.pdf
33. US Department of Health and Human Services, Office of Inspector General. CMS did not always ensure hospitals complied with Medicare reimbursement requirements for graduate medical education. OIG report A-02-17-01017. November 2018. Accessed March 1, 2024. https://oig.hhs.gov/oas/reports/region2/21701017.pdf
34. US Department of Health and Human Services, Centers for Medicare and Medicaid Services. Interns and Residents Information System (IRIS) XML format. Publication 100-20. Transmittal 11418. Change request 12724. May 19, 2022. Accessed March 1, 2024. https://www.hhs.gov/guidance/sites/default/files/hhs-guidance-documents/R11418OTN.pdf
35. Birnbaum AD, Byrne J, on behalf of the VA Office of Academic Affiliations. VHA Updates: Disbursement Policy and Education Cost Contracts. Presented at: American Association of Medical Colleges Webinar; June 2021. Accessed March 1, 2024. https://vimeo.com/644415670
36. Byrne JM, on behalf of the VA Office of Academic Affiliations. Disbursement procedures update for AY 23-24. Accessed March 1, 2024. https://www.va.gov/oaa/Videos/AffiliatePresentationDisbursementandEARsAY23-24.pptx
The Future of Polycythemia Vera


There are several new therapies on the horizon for polycythemia vera. What is the potential impact of these treatments coming to market?
Dr. Richard: There are a number of emerging therapies for polycythemia vera (PV), such as PTG-300, idasanutlin, and givinostat. PTG-300, or rusfertide, is a hepcidin mimetic that works by regulating iron metabolism and potentially controlling erythropoiesis, limiting the need for phlebotomy. Idasanutlin, a selective MDM2 inhibitor, targets p53 activity. Even though this drug is early in its development, everyone who treats patients with cancer has been hoping for a drug that works through p53. If it is effective here, who knows where else it could be effective across various other conditions.
Givinostat is well along the development pathway in advanced trials. This drug shows promise in modulating gene expression and reducing the inflammation and fibrosis associated with PV, potentially improving patient outcomes and quality of life. Everyone is hopeful that givinostat could show some effect on disease control and potentially an effect on the myeloproliferative clone. However, rigorous clinical trials and further research are necessary to validate their efficacy, safety profiles, and long-term impacts on patients with PV.
Now, with the approval of peginterferon, the next step is going to be to see how effective it will be and what the adverse events might be. I think we will be getting more data as it starts to be used more. My prediction is that there will be a slow uptake, largely because many older physicians such as myself remember the significant side effects from interferon in the past. Despite being an FDA-approved treatment, it remains an emerging therapy, particularly in the United States. Its adoption and efficacy will become clearer as time progresses.
Another promising drug early in its development is bomedemstat, which functions through a different mechanism as a deacetylase. While the potential effect of histone deacetylase drugs on patient treatment outcomes remains uncertain this year, there might be significant data—either positive or negative—that accelerate the progress of these drugs in their developmental trajectory.
We know that ruxolitinib can be used effectively for patients once they fail hydroxyurea. And now there has been the development of other JAK2 inhibitors that are approved for myelofibrosis. I am not quite sure how they can be evaluated in PV, since we are talking about relatively small numbers of patients, but they do seem to have some slight differences that may be significant and could be used in this space.
Those are the main therapies that I will have my eye on this year.
What is the potential significance of an accelerated dosing schedule for BESREMi (ropeginterferon-alfa-2b-njft), which is being investigated in the ECLIPSE PV phase 3b clinical trial?
Dr. Richard: The potential significance of an accelerated dosing schedule for BESREMi, as investigated in the ECLIPSE PV phase 3b clinical trial, lies in its capacity to enhance treatment efficacy and outcomes for patients with PV. I am incredibly pleased that it is being done as a trial, partly because a lot of people assume that once a phase 3 study is complete and a drug receives FDA approval, everything is finished and done, and we will move on to the next thing. I really appreciate it when phase 3b or 4 studies are performed, and the data get collected and published.
This study is going to follow a group of patients closely for adverse events and for the JAK2 signal. By administering BESREMi at an accelerated pace, researchers can evaluate its ability to better control hematocrit levels and symptoms associated with PV. In addition, an accelerated dosing schedule could potentially offer patients more efficient symptom management and disease control, leading to improved quality of life and reduced complications associated with PV. I believe that findings from this trial could thus pave the way for optimized treatment strategies and better outcomes for individuals living with PV.
What should future trials focus on to help improve prognosis and survival for patients with PV?
Dr. Richard: We are starting to move increasingly into finding better therapies for patients with PV, and I’ll add in essential thrombocytosis, which are based on informed prognostication. I would love to see studies that just pull out the patients at the highest risk, where the survival is down around 5 years—those are small numbers of patients. To conduct a study like that is exceedingly difficult to do. We are seeing increased consortiums of myeloproliferative neoplasm physicians. Europe has always been particularly good at this. The United States is getting better at it, so it is possible that a trial like that could be pulled together, where centers put in 1 or 2 patients at a time.
Future trials aimed at improving prognosis and survival for PV should prioritize several critical areas. First, there is a need for comprehensive studies to better understand the molecular mechanisms underlying PV pathogenesis, including the JAK2 mutation and its downstream effects. Exploring new therapeutic implications and improve long-term outcomes. Additionally, identifying reliable biomarkers for disease progression and treatment response can facilitate early intervention and personalized treatment approaches. Finally, trials should focus on assessing the impact of treatment on quality of life and addressing the unique needs of patients with PV to optimize overall prognosis and survival.
I have always held hope that the Veterans Administration could serve as a platform for conducting some of these studies, given that we possess the largest healthcare system in the country. Whether we participate in larger studies or conduct our research internally, this is something I have long envisioned.
There are several new therapies on the horizon for polycythemia vera. What is the potential impact of these treatments coming to market?
Dr. Richard: There are a number of emerging therapies for polycythemia vera (PV), such as PTG-300, idasanutlin, and givinostat. PTG-300, or rusfertide, is a hepcidin mimetic that works by regulating iron metabolism and potentially controlling erythropoiesis, limiting the need for phlebotomy. Idasanutlin, a selective MDM2 inhibitor, targets p53 activity. Even though this drug is early in its development, everyone who treats patients with cancer has been hoping for a drug that works through p53. If it is effective here, who knows where else it could be effective across various other conditions.
Givinostat is well along the development pathway in advanced trials. This drug shows promise in modulating gene expression and reducing the inflammation and fibrosis associated with PV, potentially improving patient outcomes and quality of life. Everyone is hopeful that givinostat could show some effect on disease control and potentially an effect on the myeloproliferative clone. However, rigorous clinical trials and further research are necessary to validate their efficacy, safety profiles, and long-term impacts on patients with PV.
Now, with the approval of peginterferon, the next step is going to be to see how effective it will be and what the adverse events might be. I think we will be getting more data as it starts to be used more. My prediction is that there will be a slow uptake, largely because many older physicians such as myself remember the significant side effects from interferon in the past. Despite being an FDA-approved treatment, it remains an emerging therapy, particularly in the United States. Its adoption and efficacy will become clearer as time progresses.
Another promising drug early in its development is bomedemstat, which functions through a different mechanism as a deacetylase. While the potential effect of histone deacetylase drugs on patient treatment outcomes remains uncertain this year, there might be significant data—either positive or negative—that accelerate the progress of these drugs in their developmental trajectory.
We know that ruxolitinib can be used effectively for patients once they fail hydroxyurea. And now there has been the development of other JAK2 inhibitors that are approved for myelofibrosis. I am not quite sure how they can be evaluated in PV, since we are talking about relatively small numbers of patients, but they do seem to have some slight differences that may be significant and could be used in this space.
Those are the main therapies that I will have my eye on this year.
What is the potential significance of an accelerated dosing schedule for BESREMi (ropeginterferon-alfa-2b-njft), which is being investigated in the ECLIPSE PV phase 3b clinical trial?
Dr. Richard: The potential significance of an accelerated dosing schedule for BESREMi, as investigated in the ECLIPSE PV phase 3b clinical trial, lies in its capacity to enhance treatment efficacy and outcomes for patients with PV. I am incredibly pleased that it is being done as a trial, partly because a lot of people assume that once a phase 3 study is complete and a drug receives FDA approval, everything is finished and done, and we will move on to the next thing. I really appreciate it when phase 3b or 4 studies are performed, and the data get collected and published.
This study is going to follow a group of patients closely for adverse events and for the JAK2 signal. By administering BESREMi at an accelerated pace, researchers can evaluate its ability to better control hematocrit levels and symptoms associated with PV. In addition, an accelerated dosing schedule could potentially offer patients more efficient symptom management and disease control, leading to improved quality of life and reduced complications associated with PV. I believe that findings from this trial could thus pave the way for optimized treatment strategies and better outcomes for individuals living with PV.
What should future trials focus on to help improve prognosis and survival for patients with PV?
Dr. Richard: We are starting to move increasingly into finding better therapies for patients with PV, and I’ll add in essential thrombocytosis, which are based on informed prognostication. I would love to see studies that just pull out the patients at the highest risk, where the survival is down around 5 years—those are small numbers of patients. To conduct a study like that is exceedingly difficult to do. We are seeing increased consortiums of myeloproliferative neoplasm physicians. Europe has always been particularly good at this. The United States is getting better at it, so it is possible that a trial like that could be pulled together, where centers put in 1 or 2 patients at a time.
Future trials aimed at improving prognosis and survival for PV should prioritize several critical areas. First, there is a need for comprehensive studies to better understand the molecular mechanisms underlying PV pathogenesis, including the JAK2 mutation and its downstream effects. Exploring new therapeutic implications and improve long-term outcomes. Additionally, identifying reliable biomarkers for disease progression and treatment response can facilitate early intervention and personalized treatment approaches. Finally, trials should focus on assessing the impact of treatment on quality of life and addressing the unique needs of patients with PV to optimize overall prognosis and survival.
I have always held hope that the Veterans Administration could serve as a platform for conducting some of these studies, given that we possess the largest healthcare system in the country. Whether we participate in larger studies or conduct our research internally, this is something I have long envisioned.
There are several new therapies on the horizon for polycythemia vera. What is the potential impact of these treatments coming to market?
Dr. Richard: There are a number of emerging therapies for polycythemia vera (PV), such as PTG-300, idasanutlin, and givinostat. PTG-300, or rusfertide, is a hepcidin mimetic that works by regulating iron metabolism and potentially controlling erythropoiesis, limiting the need for phlebotomy. Idasanutlin, a selective MDM2 inhibitor, targets p53 activity. Even though this drug is early in its development, everyone who treats patients with cancer has been hoping for a drug that works through p53. If it is effective here, who knows where else it could be effective across various other conditions.
Givinostat is well along the development pathway in advanced trials. This drug shows promise in modulating gene expression and reducing the inflammation and fibrosis associated with PV, potentially improving patient outcomes and quality of life. Everyone is hopeful that givinostat could show some effect on disease control and potentially an effect on the myeloproliferative clone. However, rigorous clinical trials and further research are necessary to validate their efficacy, safety profiles, and long-term impacts on patients with PV.
Now, with the approval of peginterferon, the next step is going to be to see how effective it will be and what the adverse events might be. I think we will be getting more data as it starts to be used more. My prediction is that there will be a slow uptake, largely because many older physicians such as myself remember the significant side effects from interferon in the past. Despite being an FDA-approved treatment, it remains an emerging therapy, particularly in the United States. Its adoption and efficacy will become clearer as time progresses.
Another promising drug early in its development is bomedemstat, which functions through a different mechanism as a deacetylase. While the potential effect of histone deacetylase drugs on patient treatment outcomes remains uncertain this year, there might be significant data—either positive or negative—that accelerate the progress of these drugs in their developmental trajectory.
We know that ruxolitinib can be used effectively for patients once they fail hydroxyurea. And now there has been the development of other JAK2 inhibitors that are approved for myelofibrosis. I am not quite sure how they can be evaluated in PV, since we are talking about relatively small numbers of patients, but they do seem to have some slight differences that may be significant and could be used in this space.
Those are the main therapies that I will have my eye on this year.
What is the potential significance of an accelerated dosing schedule for BESREMi (ropeginterferon-alfa-2b-njft), which is being investigated in the ECLIPSE PV phase 3b clinical trial?
Dr. Richard: The potential significance of an accelerated dosing schedule for BESREMi, as investigated in the ECLIPSE PV phase 3b clinical trial, lies in its capacity to enhance treatment efficacy and outcomes for patients with PV. I am incredibly pleased that it is being done as a trial, partly because a lot of people assume that once a phase 3 study is complete and a drug receives FDA approval, everything is finished and done, and we will move on to the next thing. I really appreciate it when phase 3b or 4 studies are performed, and the data get collected and published.
This study is going to follow a group of patients closely for adverse events and for the JAK2 signal. By administering BESREMi at an accelerated pace, researchers can evaluate its ability to better control hematocrit levels and symptoms associated with PV. In addition, an accelerated dosing schedule could potentially offer patients more efficient symptom management and disease control, leading to improved quality of life and reduced complications associated with PV. I believe that findings from this trial could thus pave the way for optimized treatment strategies and better outcomes for individuals living with PV.
What should future trials focus on to help improve prognosis and survival for patients with PV?
Dr. Richard: We are starting to move increasingly into finding better therapies for patients with PV, and I’ll add in essential thrombocytosis, which are based on informed prognostication. I would love to see studies that just pull out the patients at the highest risk, where the survival is down around 5 years—those are small numbers of patients. To conduct a study like that is exceedingly difficult to do. We are seeing increased consortiums of myeloproliferative neoplasm physicians. Europe has always been particularly good at this. The United States is getting better at it, so it is possible that a trial like that could be pulled together, where centers put in 1 or 2 patients at a time.
Future trials aimed at improving prognosis and survival for PV should prioritize several critical areas. First, there is a need for comprehensive studies to better understand the molecular mechanisms underlying PV pathogenesis, including the JAK2 mutation and its downstream effects. Exploring new therapeutic implications and improve long-term outcomes. Additionally, identifying reliable biomarkers for disease progression and treatment response can facilitate early intervention and personalized treatment approaches. Finally, trials should focus on assessing the impact of treatment on quality of life and addressing the unique needs of patients with PV to optimize overall prognosis and survival.
I have always held hope that the Veterans Administration could serve as a platform for conducting some of these studies, given that we possess the largest healthcare system in the country. Whether we participate in larger studies or conduct our research internally, this is something I have long envisioned.
Tender Dermal Nodule on the Temple
The Diagnosis: Lymphoepithelioma-like Carcinoma
Lymphoepithelioma-like carcinoma (LELC) is a rare, poorly differentiated, primary cutaneous neoplasm that occurs on sun-exposed skin, particularly on the head and neck of elderly individuals. It often manifests as an asymptomatic, slow-growing, flesh-colored or erythematous dermal nodule, though ulceration and tenderness have been reported.1 Histopathologically, these neoplasms often are poorly circumscribed and can infiltrate surrounding subcutaneous and soft tissue. As a biphasic tumor, LELC is characterized by islands, nests, or trabeculae of epithelioid cells within the mid dermis surrounded by a dense lymphocytic infiltrate with plasma cells (Figure 1).1 The epithelial component rarely communicates with the overlying epidermis and is composed of atypical polygonal cells with eosinophilic cytoplasm, vesicular nuclei, prominent nucleoli, and frequent mitosis.2 These epithelial nests can be highlighted by pancytokeratin AE1/AE3 or other epithelial differentiation markers (eg, CAM 5.2, CK5/6, epithelial membrane antigen, high-molecular-weight cytokeratin), while the surrounding lymphocytic infiltrate consists of an admixture of T cells and B cells. Lymphoepithelioma-like carcinomas also can demonstrate sebaceous, eccrine, or follicular differentiations.3 The epithelial nests of LELC also are positive for p63 and epithelial membrane antigen.2
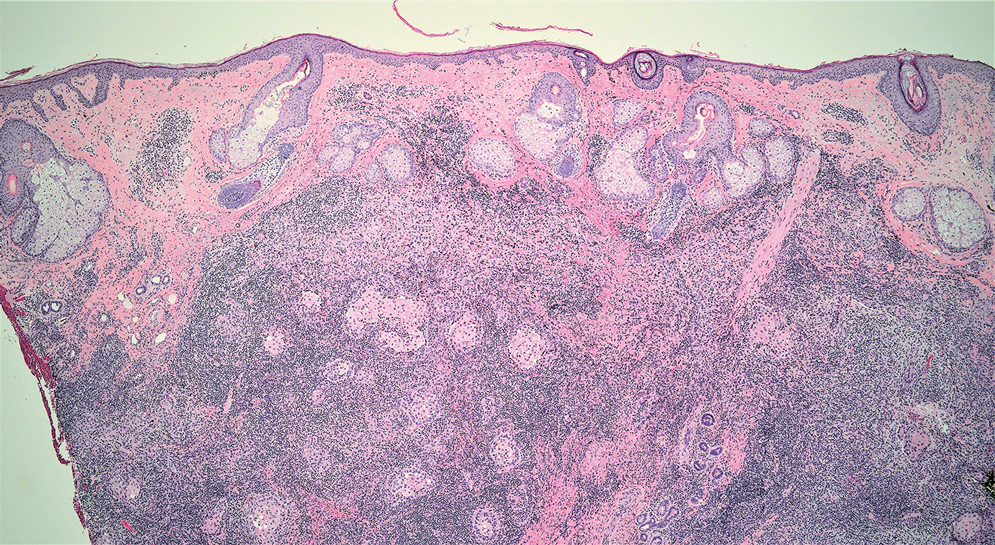
The usual treatment of LELC is wide local excision or Mohs micrographic surgery.1 Despite the poorly differentiated morphology of the tumor, LELC has a generally good prognosis with low metastatic potential and few reports of local recurrence after incomplete excision.3 Patients who are not candidates for surgery as well as recalcitrant cases are managed with radiotherapy.1
Cutaneous lymphadenoma (CL) is a benign adnexal neoplasm that manifests as a small, solitary, fleshcolored nodule usually in the head and neck region.4 Histologically, CL consists of well-circumscribed epithelial nests within the dermis that are peripherally outlined by palisading basaloid cells and filled with clear to eosinophilic epithelioid cells (Figure 2).5 The fibrotic tumor stroma often is infiltrated by numerous intralobular dendritic cells and lymphocytes that occasionally can be arranged in germinal center–like nodules.4 The lymphoepithelial nature of CL can be challenging to distinguish morphologically from LELC, and immunohistochemistry stains may be required. In CL, both the basaloid and epithelioid cells stain positive for pancytokeratin AE1/ AE3, but the peripheral palisaded basaloid cells also stain positive for BerEP4. Additionally, the fibrotic stroma can be highlighted by CD34 and the intralobular dendritic cells by S-100.4
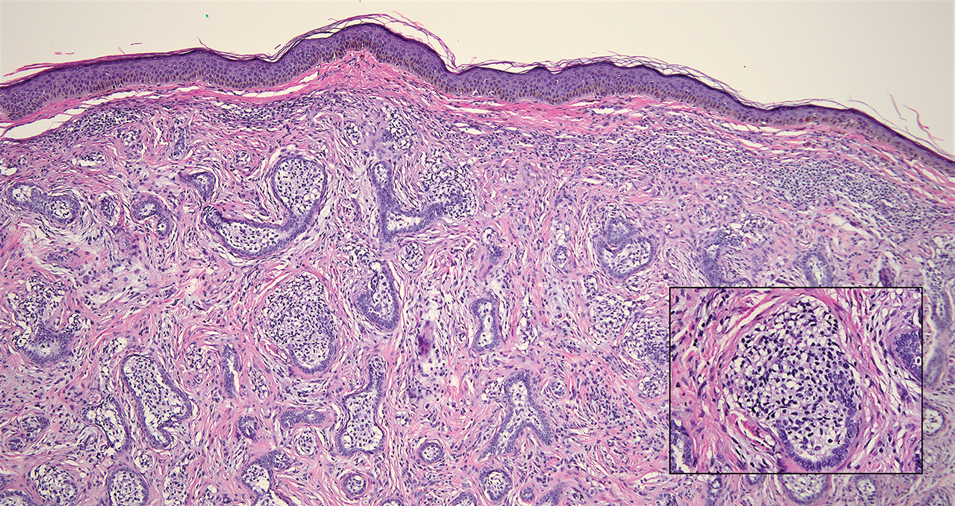
Nasopharyngeal carcinoma (NPC), formerly known as lymphoepithelioma, refers to carcinoma arising within the epithelium of the nasopharynx.6 Endemic to China, NPC manifests as an enlarging nasopharyngeal mass, causing clinical symptoms such as nasal obstruction and epistaxis.7 Histologically, nonkeratinizing NPC exhibits a biphasic morphology consisting of epithelioid neoplastic cells and background lymphocytic infiltrates (Figure 3). The epithelial component consists of round to oval neoplastic cells with amphophilic to eosinophilic cytoplasm, vesicular nuclei, and prominent nucleoli.6 Nasopharyngeal carcinoma is associated strongly with the Epstein-Barr virus while LELC is not; thus, Epstein- Barr encoding region in situ hybridization can reliably distinguish these entities. Metastatic NPC is rare but has been reported; therefore, it is highly recommended to perform an otolaryngologic examination in addition to testing for Epstein-Barr virus reactivity as part of a complete evaluation.8
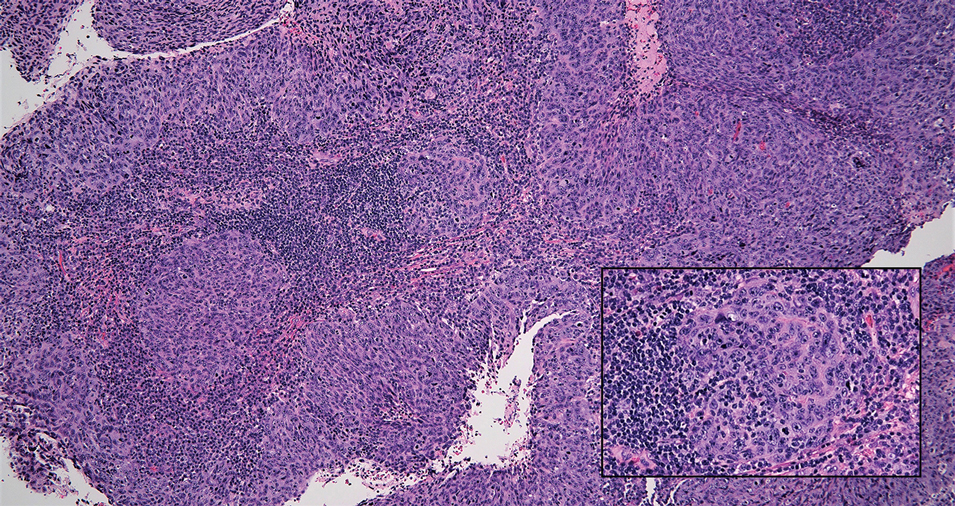
Cutaneous squamous cell carcinoma (SCC) is a common epidermal malignancy with multiple subtypes and variable morphology. The clinical presentation of SCC is similar to LELC—an enlarging hyperkeratotic papule or nodule on sun-exposed skin that often is ulcerated and tender.9 Histologically, poorly differentiated nonkeratinizing SCC can form nests and trabeculae of epithelioid cells that are stained by epithelial differentiation markers, resembling the epithelioid nests of LELC. Distinguishing between LELC and poorly differentiated SCC with robust inflammatory infiltrate can be challenging (Figure 4). In fact, some experts support LELC as an SCC variant rather than a separate entity.9 However, in contrast to LELC, the dermal nests of SCC usually maintain an epidermal connection and often are associated with an overlying area of SCC in situ or welldifferentiated SCC.3
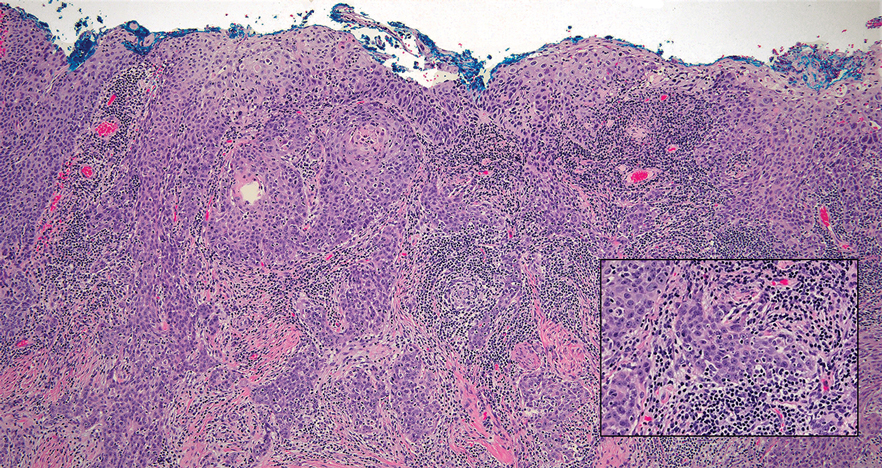
Mycosis fungoides (MF) is a primary cutaneous T-cell lymphoma. It is the most common type of cutaneous lymphoma, accounting for almost 50% of all reported cases.10 Classic MF has an indolent course and progresses through several clinical stages. Patches and plaques characterize early stages; lymphadenopathy indicates progression to later stages in which erythroderma may develop with coalescence of patches, plaques, and tumors; and MF present in blood or lymph nodes characterizes the late stage. Each stage of MF is different histologically—from a superficial lichenoid infiltrate with exocytosis of malignant T cells in the patch stage, to more robust epidermotropism and dermal infiltrate in the plaque stage, and finally a dense dermal infiltrate in the late stage.11 The rare syringotropic variant of MF clinically manifests as solitary or multiple erythematous lesions, often with overlying alopecia. Syringotropic MF uniquely exhibits folliculotropism and syringotropism along with syringometaplasia on histologic evaluation (Figure 5).12 The syringometaplasia can be difficult to distinguish from the epithelial nests of LELC, particularly with the lymphocytic background. Immunohistochemical panels for T-cell markers can highlight aberrant T cells in syringotropic MF through their usual loss of CD5 and CD7, in comparison to normal T cells in LELC.11 An elevated CD4:CD8 ratio of 4:1 and molecular analysis for T-cell receptor gene clonal rearrangements also can support the diagnosis of MF.12
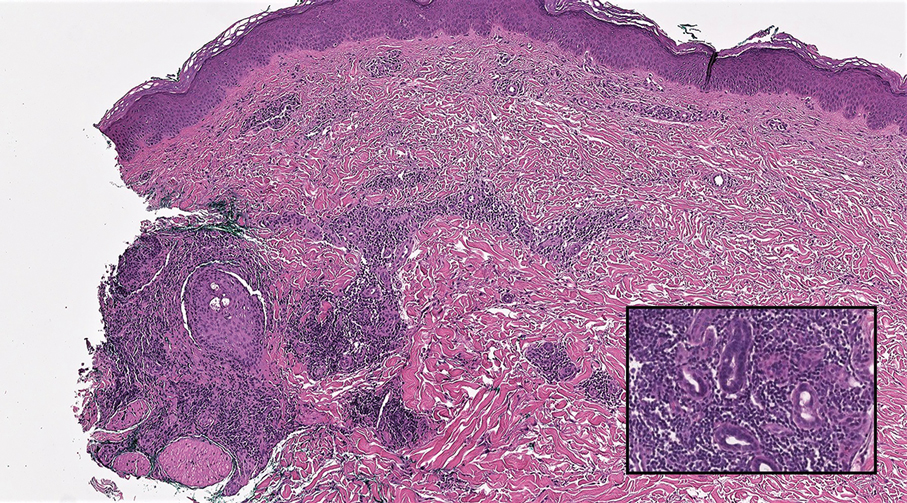
- Morteza Abedi S, Salama S, Alowami S. Lymphoepithelioma-like carcinoma of the skin: case report and approach to surgical pathology sign out. Rare Tumors. 2013;5:E47.
- Fisher JC, White RM, Hurd DS. Lymphoepithelioma-like carcinoma of the skin: a case of one patient presenting with two primary cutaneous neoplasms. J Am Osteopath Coll Dermatol. 2015;33:40-41.
- Welch PQ, Williams SB, Foss RD, et al. Lymphoepithelioma-like carcinoma of head and neck skin: a systematic analysis of 11 cases and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:78-86.
- Yu R, Salama S, Alowami S. Cutaneous lymphadenoma: a rare case and brief review of a diagnostic pitfall. Rare Tumors. 2014;6:5358.
- Monteagudo C, Fúnez R, Sánchez-Sendra B, et al. Cutaneous lymphadenoma is a distinct trichoblastoma-like lymphoepithelial tumor with diffuse androgen receptor immunoreactivity, Notch1 ligand in Reed-Sternberg-like Cells, and common EGFR somatic mutations. Am J Surg Pathol. 2021;45:1382-1390.
- Stelow EB, Wenig BM. Update from the 4th edition of the World Health Organization classification of head and neck tumours: nasopharynx. Head Neck Pathol. 2017;11:16-22.
- Almomani MH, Zulfiqar H, Nagalli S. Nasopharyngeal carcinoma (NPC, lymphoepithelioma). StatPearls Publishing; 2022.
- Lassen CB, Lock-Andersen J. Lymphoepithelioma-like carcinoma of the skin: a case with perineural invasion. Plast Reconstr Surg Glob Open. 2014;2:E252.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer Staging Guidelines, Prognostic Factors, and Histopathologic Variants. Adv Anat Pathol. 2017;24:171-194.
- Pileri A, Facchetti F, Rütten A, et al. Syringotropic mycosis fungoides: a rare variant of the disease with peculiar clinicopathologic features. Am J Surg Pathol. 2011;35:100-109.
- Ryu HJ, Kim SI, Jang HO, et al. Evaluation of the International Society for Cutaneous Lymphoma Algorithm for the Diagnosis of Early Mycosis Fungoides [published October 15, 2021]. Cells. 2021;10:2758. doi:10.3390/cells10102758
- Lehmer LM, Amber KT, de Feraudy SM. Syringotropic mycosis fungoides: a rare form of cutaneous T-cell lymphoma enabling a histopathologic “sigh of relief.” Am J Dermatopathol. 2017;39:920-923.
The Diagnosis: Lymphoepithelioma-like Carcinoma
Lymphoepithelioma-like carcinoma (LELC) is a rare, poorly differentiated, primary cutaneous neoplasm that occurs on sun-exposed skin, particularly on the head and neck of elderly individuals. It often manifests as an asymptomatic, slow-growing, flesh-colored or erythematous dermal nodule, though ulceration and tenderness have been reported.1 Histopathologically, these neoplasms often are poorly circumscribed and can infiltrate surrounding subcutaneous and soft tissue. As a biphasic tumor, LELC is characterized by islands, nests, or trabeculae of epithelioid cells within the mid dermis surrounded by a dense lymphocytic infiltrate with plasma cells (Figure 1).1 The epithelial component rarely communicates with the overlying epidermis and is composed of atypical polygonal cells with eosinophilic cytoplasm, vesicular nuclei, prominent nucleoli, and frequent mitosis.2 These epithelial nests can be highlighted by pancytokeratin AE1/AE3 or other epithelial differentiation markers (eg, CAM 5.2, CK5/6, epithelial membrane antigen, high-molecular-weight cytokeratin), while the surrounding lymphocytic infiltrate consists of an admixture of T cells and B cells. Lymphoepithelioma-like carcinomas also can demonstrate sebaceous, eccrine, or follicular differentiations.3 The epithelial nests of LELC also are positive for p63 and epithelial membrane antigen.2
The usual treatment of LELC is wide local excision or Mohs micrographic surgery.1 Despite the poorly differentiated morphology of the tumor, LELC has a generally good prognosis with low metastatic potential and few reports of local recurrence after incomplete excision.3 Patients who are not candidates for surgery as well as recalcitrant cases are managed with radiotherapy.1
Cutaneous lymphadenoma (CL) is a benign adnexal neoplasm that manifests as a small, solitary, fleshcolored nodule usually in the head and neck region.4 Histologically, CL consists of well-circumscribed epithelial nests within the dermis that are peripherally outlined by palisading basaloid cells and filled with clear to eosinophilic epithelioid cells (Figure 2).5 The fibrotic tumor stroma often is infiltrated by numerous intralobular dendritic cells and lymphocytes that occasionally can be arranged in germinal center–like nodules.4 The lymphoepithelial nature of CL can be challenging to distinguish morphologically from LELC, and immunohistochemistry stains may be required. In CL, both the basaloid and epithelioid cells stain positive for pancytokeratin AE1/ AE3, but the peripheral palisaded basaloid cells also stain positive for BerEP4. Additionally, the fibrotic stroma can be highlighted by CD34 and the intralobular dendritic cells by S-100.4
Nasopharyngeal carcinoma (NPC), formerly known as lymphoepithelioma, refers to carcinoma arising within the epithelium of the nasopharynx.6 Endemic to China, NPC manifests as an enlarging nasopharyngeal mass, causing clinical symptoms such as nasal obstruction and epistaxis.7 Histologically, nonkeratinizing NPC exhibits a biphasic morphology consisting of epithelioid neoplastic cells and background lymphocytic infiltrates (Figure 3). The epithelial component consists of round to oval neoplastic cells with amphophilic to eosinophilic cytoplasm, vesicular nuclei, and prominent nucleoli.6 Nasopharyngeal carcinoma is associated strongly with the Epstein-Barr virus while LELC is not; thus, Epstein- Barr encoding region in situ hybridization can reliably distinguish these entities. Metastatic NPC is rare but has been reported; therefore, it is highly recommended to perform an otolaryngologic examination in addition to testing for Epstein-Barr virus reactivity as part of a complete evaluation.8
Cutaneous squamous cell carcinoma (SCC) is a common epidermal malignancy with multiple subtypes and variable morphology. The clinical presentation of SCC is similar to LELC—an enlarging hyperkeratotic papule or nodule on sun-exposed skin that often is ulcerated and tender.9 Histologically, poorly differentiated nonkeratinizing SCC can form nests and trabeculae of epithelioid cells that are stained by epithelial differentiation markers, resembling the epithelioid nests of LELC. Distinguishing between LELC and poorly differentiated SCC with robust inflammatory infiltrate can be challenging (Figure 4). In fact, some experts support LELC as an SCC variant rather than a separate entity.9 However, in contrast to LELC, the dermal nests of SCC usually maintain an epidermal connection and often are associated with an overlying area of SCC in situ or welldifferentiated SCC.3
Mycosis fungoides (MF) is a primary cutaneous T-cell lymphoma. It is the most common type of cutaneous lymphoma, accounting for almost 50% of all reported cases.10 Classic MF has an indolent course and progresses through several clinical stages. Patches and plaques characterize early stages; lymphadenopathy indicates progression to later stages in which erythroderma may develop with coalescence of patches, plaques, and tumors; and MF present in blood or lymph nodes characterizes the late stage. Each stage of MF is different histologically—from a superficial lichenoid infiltrate with exocytosis of malignant T cells in the patch stage, to more robust epidermotropism and dermal infiltrate in the plaque stage, and finally a dense dermal infiltrate in the late stage.11 The rare syringotropic variant of MF clinically manifests as solitary or multiple erythematous lesions, often with overlying alopecia. Syringotropic MF uniquely exhibits folliculotropism and syringotropism along with syringometaplasia on histologic evaluation (Figure 5).12 The syringometaplasia can be difficult to distinguish from the epithelial nests of LELC, particularly with the lymphocytic background. Immunohistochemical panels for T-cell markers can highlight aberrant T cells in syringotropic MF through their usual loss of CD5 and CD7, in comparison to normal T cells in LELC.11 An elevated CD4:CD8 ratio of 4:1 and molecular analysis for T-cell receptor gene clonal rearrangements also can support the diagnosis of MF.12
The Diagnosis: Lymphoepithelioma-like Carcinoma
Lymphoepithelioma-like carcinoma (LELC) is a rare, poorly differentiated, primary cutaneous neoplasm that occurs on sun-exposed skin, particularly on the head and neck of elderly individuals. It often manifests as an asymptomatic, slow-growing, flesh-colored or erythematous dermal nodule, though ulceration and tenderness have been reported.1 Histopathologically, these neoplasms often are poorly circumscribed and can infiltrate surrounding subcutaneous and soft tissue. As a biphasic tumor, LELC is characterized by islands, nests, or trabeculae of epithelioid cells within the mid dermis surrounded by a dense lymphocytic infiltrate with plasma cells (Figure 1).1 The epithelial component rarely communicates with the overlying epidermis and is composed of atypical polygonal cells with eosinophilic cytoplasm, vesicular nuclei, prominent nucleoli, and frequent mitosis.2 These epithelial nests can be highlighted by pancytokeratin AE1/AE3 or other epithelial differentiation markers (eg, CAM 5.2, CK5/6, epithelial membrane antigen, high-molecular-weight cytokeratin), while the surrounding lymphocytic infiltrate consists of an admixture of T cells and B cells. Lymphoepithelioma-like carcinomas also can demonstrate sebaceous, eccrine, or follicular differentiations.3 The epithelial nests of LELC also are positive for p63 and epithelial membrane antigen.2
The usual treatment of LELC is wide local excision or Mohs micrographic surgery.1 Despite the poorly differentiated morphology of the tumor, LELC has a generally good prognosis with low metastatic potential and few reports of local recurrence after incomplete excision.3 Patients who are not candidates for surgery as well as recalcitrant cases are managed with radiotherapy.1
Cutaneous lymphadenoma (CL) is a benign adnexal neoplasm that manifests as a small, solitary, fleshcolored nodule usually in the head and neck region.4 Histologically, CL consists of well-circumscribed epithelial nests within the dermis that are peripherally outlined by palisading basaloid cells and filled with clear to eosinophilic epithelioid cells (Figure 2).5 The fibrotic tumor stroma often is infiltrated by numerous intralobular dendritic cells and lymphocytes that occasionally can be arranged in germinal center–like nodules.4 The lymphoepithelial nature of CL can be challenging to distinguish morphologically from LELC, and immunohistochemistry stains may be required. In CL, both the basaloid and epithelioid cells stain positive for pancytokeratin AE1/ AE3, but the peripheral palisaded basaloid cells also stain positive for BerEP4. Additionally, the fibrotic stroma can be highlighted by CD34 and the intralobular dendritic cells by S-100.4
Nasopharyngeal carcinoma (NPC), formerly known as lymphoepithelioma, refers to carcinoma arising within the epithelium of the nasopharynx.6 Endemic to China, NPC manifests as an enlarging nasopharyngeal mass, causing clinical symptoms such as nasal obstruction and epistaxis.7 Histologically, nonkeratinizing NPC exhibits a biphasic morphology consisting of epithelioid neoplastic cells and background lymphocytic infiltrates (Figure 3). The epithelial component consists of round to oval neoplastic cells with amphophilic to eosinophilic cytoplasm, vesicular nuclei, and prominent nucleoli.6 Nasopharyngeal carcinoma is associated strongly with the Epstein-Barr virus while LELC is not; thus, Epstein- Barr encoding region in situ hybridization can reliably distinguish these entities. Metastatic NPC is rare but has been reported; therefore, it is highly recommended to perform an otolaryngologic examination in addition to testing for Epstein-Barr virus reactivity as part of a complete evaluation.8
Cutaneous squamous cell carcinoma (SCC) is a common epidermal malignancy with multiple subtypes and variable morphology. The clinical presentation of SCC is similar to LELC—an enlarging hyperkeratotic papule or nodule on sun-exposed skin that often is ulcerated and tender.9 Histologically, poorly differentiated nonkeratinizing SCC can form nests and trabeculae of epithelioid cells that are stained by epithelial differentiation markers, resembling the epithelioid nests of LELC. Distinguishing between LELC and poorly differentiated SCC with robust inflammatory infiltrate can be challenging (Figure 4). In fact, some experts support LELC as an SCC variant rather than a separate entity.9 However, in contrast to LELC, the dermal nests of SCC usually maintain an epidermal connection and often are associated with an overlying area of SCC in situ or welldifferentiated SCC.3
Mycosis fungoides (MF) is a primary cutaneous T-cell lymphoma. It is the most common type of cutaneous lymphoma, accounting for almost 50% of all reported cases.10 Classic MF has an indolent course and progresses through several clinical stages. Patches and plaques characterize early stages; lymphadenopathy indicates progression to later stages in which erythroderma may develop with coalescence of patches, plaques, and tumors; and MF present in blood or lymph nodes characterizes the late stage. Each stage of MF is different histologically—from a superficial lichenoid infiltrate with exocytosis of malignant T cells in the patch stage, to more robust epidermotropism and dermal infiltrate in the plaque stage, and finally a dense dermal infiltrate in the late stage.11 The rare syringotropic variant of MF clinically manifests as solitary or multiple erythematous lesions, often with overlying alopecia. Syringotropic MF uniquely exhibits folliculotropism and syringotropism along with syringometaplasia on histologic evaluation (Figure 5).12 The syringometaplasia can be difficult to distinguish from the epithelial nests of LELC, particularly with the lymphocytic background. Immunohistochemical panels for T-cell markers can highlight aberrant T cells in syringotropic MF through their usual loss of CD5 and CD7, in comparison to normal T cells in LELC.11 An elevated CD4:CD8 ratio of 4:1 and molecular analysis for T-cell receptor gene clonal rearrangements also can support the diagnosis of MF.12
- Morteza Abedi S, Salama S, Alowami S. Lymphoepithelioma-like carcinoma of the skin: case report and approach to surgical pathology sign out. Rare Tumors. 2013;5:E47.
- Fisher JC, White RM, Hurd DS. Lymphoepithelioma-like carcinoma of the skin: a case of one patient presenting with two primary cutaneous neoplasms. J Am Osteopath Coll Dermatol. 2015;33:40-41.
- Welch PQ, Williams SB, Foss RD, et al. Lymphoepithelioma-like carcinoma of head and neck skin: a systematic analysis of 11 cases and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:78-86.
- Yu R, Salama S, Alowami S. Cutaneous lymphadenoma: a rare case and brief review of a diagnostic pitfall. Rare Tumors. 2014;6:5358.
- Monteagudo C, Fúnez R, Sánchez-Sendra B, et al. Cutaneous lymphadenoma is a distinct trichoblastoma-like lymphoepithelial tumor with diffuse androgen receptor immunoreactivity, Notch1 ligand in Reed-Sternberg-like Cells, and common EGFR somatic mutations. Am J Surg Pathol. 2021;45:1382-1390.
- Stelow EB, Wenig BM. Update from the 4th edition of the World Health Organization classification of head and neck tumours: nasopharynx. Head Neck Pathol. 2017;11:16-22.
- Almomani MH, Zulfiqar H, Nagalli S. Nasopharyngeal carcinoma (NPC, lymphoepithelioma). StatPearls Publishing; 2022.
- Lassen CB, Lock-Andersen J. Lymphoepithelioma-like carcinoma of the skin: a case with perineural invasion. Plast Reconstr Surg Glob Open. 2014;2:E252.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer Staging Guidelines, Prognostic Factors, and Histopathologic Variants. Adv Anat Pathol. 2017;24:171-194.
- Pileri A, Facchetti F, Rütten A, et al. Syringotropic mycosis fungoides: a rare variant of the disease with peculiar clinicopathologic features. Am J Surg Pathol. 2011;35:100-109.
- Ryu HJ, Kim SI, Jang HO, et al. Evaluation of the International Society for Cutaneous Lymphoma Algorithm for the Diagnosis of Early Mycosis Fungoides [published October 15, 2021]. Cells. 2021;10:2758. doi:10.3390/cells10102758
- Lehmer LM, Amber KT, de Feraudy SM. Syringotropic mycosis fungoides: a rare form of cutaneous T-cell lymphoma enabling a histopathologic “sigh of relief.” Am J Dermatopathol. 2017;39:920-923.
- Morteza Abedi S, Salama S, Alowami S. Lymphoepithelioma-like carcinoma of the skin: case report and approach to surgical pathology sign out. Rare Tumors. 2013;5:E47.
- Fisher JC, White RM, Hurd DS. Lymphoepithelioma-like carcinoma of the skin: a case of one patient presenting with two primary cutaneous neoplasms. J Am Osteopath Coll Dermatol. 2015;33:40-41.
- Welch PQ, Williams SB, Foss RD, et al. Lymphoepithelioma-like carcinoma of head and neck skin: a systematic analysis of 11 cases and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:78-86.
- Yu R, Salama S, Alowami S. Cutaneous lymphadenoma: a rare case and brief review of a diagnostic pitfall. Rare Tumors. 2014;6:5358.
- Monteagudo C, Fúnez R, Sánchez-Sendra B, et al. Cutaneous lymphadenoma is a distinct trichoblastoma-like lymphoepithelial tumor with diffuse androgen receptor immunoreactivity, Notch1 ligand in Reed-Sternberg-like Cells, and common EGFR somatic mutations. Am J Surg Pathol. 2021;45:1382-1390.
- Stelow EB, Wenig BM. Update from the 4th edition of the World Health Organization classification of head and neck tumours: nasopharynx. Head Neck Pathol. 2017;11:16-22.
- Almomani MH, Zulfiqar H, Nagalli S. Nasopharyngeal carcinoma (NPC, lymphoepithelioma). StatPearls Publishing; 2022.
- Lassen CB, Lock-Andersen J. Lymphoepithelioma-like carcinoma of the skin: a case with perineural invasion. Plast Reconstr Surg Glob Open. 2014;2:E252.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer Staging Guidelines, Prognostic Factors, and Histopathologic Variants. Adv Anat Pathol. 2017;24:171-194.
- Pileri A, Facchetti F, Rütten A, et al. Syringotropic mycosis fungoides: a rare variant of the disease with peculiar clinicopathologic features. Am J Surg Pathol. 2011;35:100-109.
- Ryu HJ, Kim SI, Jang HO, et al. Evaluation of the International Society for Cutaneous Lymphoma Algorithm for the Diagnosis of Early Mycosis Fungoides [published October 15, 2021]. Cells. 2021;10:2758. doi:10.3390/cells10102758
- Lehmer LM, Amber KT, de Feraudy SM. Syringotropic mycosis fungoides: a rare form of cutaneous T-cell lymphoma enabling a histopathologic “sigh of relief.” Am J Dermatopathol. 2017;39:920-923.
A 77-year-old man presented with a 1.2-cm dermal nodule on the left temple of 1 year’s duration. The lesion had become tender and darker in color. An excision was performed and submitted for histologic examination. Additional immunohistochemistry staining for Epstein-Barr virus was negative.
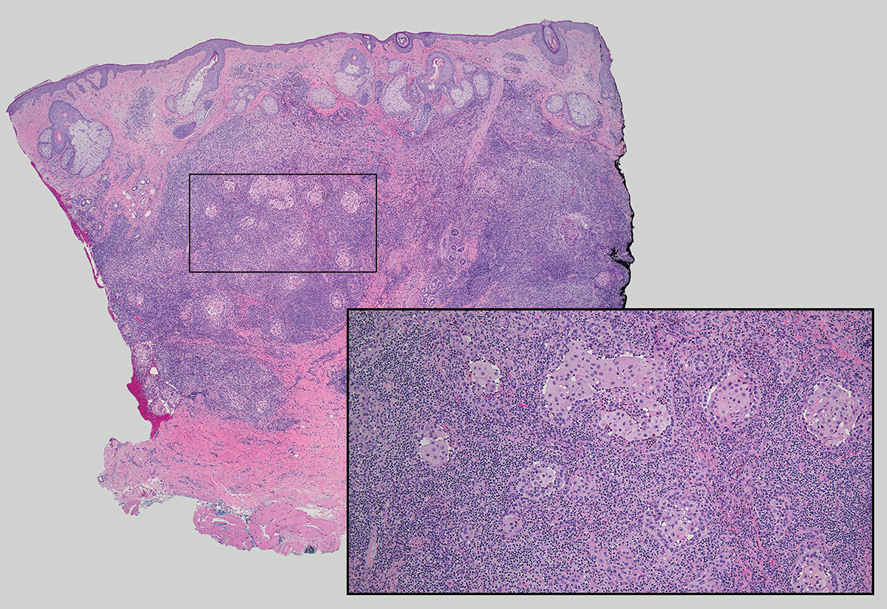
Obesity in Children
Botanical Briefs: Fig Phytophotodermatitis (Ficus carica)
Plant Parts and Nomenclature
Ficus carica (common fig) is a deciduous shrub or small tree with smooth gray bark that can grow up to 10 m in height (Figure 1). It is characterized by many spreading branches, but the trunk rarely grows beyond a diameter of 7 in. Its hairy leaves are coarse on the upper side and soft underneath with 3 to 7 deep lobes that can extend up to 25 cm in length or width; the leaves grow individually, alternating along the sides of the branches. Fig trees often can be seen adorning yards, gardens, and parks, especially in tropical and subtropical climates. Ficus carica should not be confused with Ficus benjamina (weeping fig), a common ornamental tree that also is used to provide shade in hot climates, though both can cause phototoxic skin eruptions.
.")
The common fig tree originated in the Mediterranean and western Asia1 and has been cultivated by humans since the second and third millennia

Ficus carica is a member of the Moraceae family (derived from the Latin name for the mulberry tree), which includes 53 genera and approximately 1400 species, of which about 850 belong to the genus Ficus (the Latin name for a fig tree). The term carica likely comes from the Latin word carricare (to load) to describe a tree loaded with figs. Family members include trees, shrubs, lianas, and herbs that usually contain laticifers with a milky latex.
Traditional Uses
For centuries, components of the fig tree have been used in herbal teas and pastes to treat ailments ranging from sore throats to diarrhea, though there is no evidence to support their efficacy.4 Ancient Indians and Egyptians used plants such as the common fig tree containing furocoumarins to induce hyperpigmentation in vitiligo.5
Phototoxic Components
The leaves and sap of the common fig tree contain psoralens, which are members of the furocoumarin group of chemical compounds and are the source of its phototoxicity. The fruit does not contain psoralens.6-9 The tree also produces proteolytic enzymes such as protease, amylase, ficin, triterpenoids, and lipodiastase that enhance its phototoxic effects.8 Exposure to UV light between 320 and 400 nm following contact with these phototoxic components triggers a reaction in the skin over the course of 1 to 3 days.5 The psoralens bind in epidermal cells, cross-link the DNA, and cause cell-membrane destruction, leading to edema and necrosis.10 The delay in symptoms may be attributed to the time needed to synthesize acute-phase reaction proteins such as tumor necrosis factor α and IL-1.11 In spring and summer months, an increased concentration of psoralens in the leaves and sap contribute to an increased incidence of phytophotodermatitis.9 Humidity and sweat also increase the percutaneous absorption of psoralens.12,13
Allergens
Fig trees produce a latex protein that can cause cross-reactive hypersensitivity reactions in those allergic to F benjamina latex and rubber latex.6 The latex proteins in fig trees can act as airborne respiratory allergens. Ingestion of figs can produce anaphylactic reactions in those sensitized to rubber latex and F benjamina latex.7 Other plant families associated with phototoxic reactions include Rutaceae (lemon, lime, bitter orange), Apiaceae (formerly Umbelliferae)(carrot, parsnip, parsley, dill, celery, hogweed), and Fabaceae (prairie turnip).
Cutaneous Manifestations
Most cases of fig phytophotodermatitis begin with burning, pain, and/or itching within hours of sunlight exposure in areas of the skin that encountered components of the fig tree, often in a linear pattern. The affected areas become erythematous and edematous with formation of bullae and unilocular vesicles over the course of 1 to 3 days.12,14,15 Lesions may extend beyond the region of contact with the fig tree as they spread across the skin due to sweat or friction, and pain may linger even after the lesions resolve.12,13,16 Adults who handle fig trees (eg, pruning) are susceptible to phototoxic reactions, especially those using chain saws or other mechanisms that result in spray exposure, as the photosensitizing sap permeates the wood and bark of the entire tree.17 Similarly, children who handle fig leaves or sap during outdoor play can develop bullous eruptions. Severe cases have resulted in hospital admission after prolonged exposure.16 Additionally, irritant dermatitis may arise from contact with the trichomes or “hairs” on various parts of the plant.

Patients who use natural remedies containing components of the fig tree without the supervision of a medical provider put themselves at risk for unsafe or unwanted adverse effects, such as phytophotodermatitis.12,15,16,18 An entire family presented with burns after they applied fig leaf extract to the skin prior to tanning outside in the sun.19 A 42-year-old woman acquired a severe burn covering 81% of the body surface after topically applying fig leaf tea to the skin as a tanning agent.20 A subset of patients ingesting or applying fig tree components for conditions such as vitiligo, dermatitis, onychomycosis, and motor retardation developed similar cutaneous reactions.13,14,21,22 Lesions resembling finger marks can raise concerns for potential abuse or neglect in children.22
The differential diagnosis for fig phytophotodermatitis includes sunburn, chemical burns, drug-related photosensitivity, infectious lesions (eg, herpes simplex, bullous impetigo, Lyme disease, superficial lymphangitis), connective tissue disease (eg, systemic lupus erythematosus), contact dermatitis, and nonaccidental trauma.12,15,18 Compared to sunburn, phytophotodermatitis tends to increase in severity over days following exposure and heals with dramatic hyperpigmentation, which also prompts visits to dermatology.12
Treatment
Treatment of fig phytophotodermatitis chiefly is symptomatic, including analgesia, appropriate wound care, and infection prophylaxis. Topical and systemic corticosteroids may aid in the resolution of moderate to severe reactions.15,23,24 Even severe injuries over small areas or mild injuries to a high percentage of the total body surface area may require treatment in a burn unit. Patients should be encouraged to use mineral-based sunscreens on the affected areas to reduce the risk for hyperpigmentation. Individuals who regularly handle fig trees should use contact barriers including gloves and protective clothing (eg, long-sleeved shirts, long pants).
- Ikegami H, Nogata H, Hirashima K, et al. Analysis of genetic diversity among European and Asian fig varieties (Ficus carica L.) using ISSR, RAPD, and SSR markers. Genetic Resources and Crop Evolution. 2009;56:201-209.
- Zohary D, Spiegel-Roy P. Beginnings of fruit growing in the Old World. Science. 1975;187:319-327.
- Young R. Young’s Analytical Concordance. Thomas Nelson; 1982.
- Duke JA. Handbook of Medicinal Herbs. CRC Press; 2002.
- Pathak MA, Fitzpatrick TB. Bioassay of natural and synthetic furocoumarins (psoralens). J Invest Dermatol. 1959;32:509-518.
- Focke M, Hemmer W, Wöhrl S, et al. Cross-reactivity between Ficus benjamina latex and fig fruit in patients with clinical fig allergy. Clin Exp Allergy. 2003;33:971-977.
- Hemmer W, Focke M, Götz M, et al. Sensitization to Ficus benjamina: relationship to natural rubber latex allergy and identification of foods implicated in the Ficus-fruit syndrome. Clin Exp Allergy. 2004;34:1251-1258.
- Bonamonte D, Foti C, Lionetti N, et al. Photoallergic contact dermatitis to 8-methoxypsoralen in Ficus carica. Contact Dermatitis. 2010;62:343-348.
- Zaynoun ST, Aftimos BG, Abi Ali L, et al. Ficus carica; isolation and quantification of the photoactive components. Contact Dermatitis. 1984;11:21-25.
- Tessman JW, Isaacs ST, Hearst JE. Photochemistry of the furan-side 8-methoxypsoralen-thymidine monoadduct inside the DNA helix. conversion to diadduct and to pyrone-side monoadduct. Biochemistry. 1985;24:1669-1676.
- Geary P. Burns related to the use of psoralens as a tanning agent. Burns. 1996;22:636-637.
- Redgrave N, Solomon J. Severe phytophotodermatitis from fig sap: a little known phenomenon. BMJ Case Rep. 2021;14:E238745.
- Ozdamar E, Ozbek S, Akin S. An unusual cause of burn injury: fig leaf decoction used as a remedy for a dermatitis of unknown etiology. J Burn Care Rehabil. 2003;24:229-233; discussion 228.
- Berakha GJ, Lefkovits G. Psoralen phototherapy and phototoxicity. Ann Plast Surg. 1985;14:458-461.
- Papazoglou A, Mantadakis E. Fig tree leaves phytophotodermatitis. J Pediatr. 2021;239:244-245.
- Imen MS, Ahmadabadi A, Tavousi SH, et al. The curious cases of burn by fig tree leaves. Indian J Dermatol. 2019;64:71-73.
- Rouaiguia-Bouakkaz S, Amira-Guebailia H, Rivière C, et al. Identification and quantification of furanocoumarins in stem bark and wood of eight Algerian varieties of Ficus carica by RP-HPLC-DAD and RP-HPLC-DAD-MS. Nat Prod Commun. 2013;8:485-486.
- Oliveira AA, Morais J, Pires O, et al. Fig tree induced phytophotodermatitis. BMJ Case Rep. 2020;13:E233392.
- Bassioukas K, Stergiopoulou C, Hatzis J. Erythrodermic phytophotodermatitis after application of aqueous fig-leaf extract as an artificial suntan promoter and sunbathing. Contact Dermatitis. 2004;51:94-95.
- Sforza M, Andjelkov K, Zaccheddu R. Severe burn on 81% of body surface after sun tanning. Ulus Travma Acil Cerrahi Derg. 2013;19:383-384.
- Son JH, Jin H, You HS, et al. Five cases of phytophotodermatitis caused by fig leaves and relevant literature review. Ann Dermatol. 2017;29:86-90.
- Abali AE, Aka M, Aydogan C, et al. Burns or phytophotodermatitis, abuse or neglect: confusing aspects of skin lesions caused by the superstitious use of fig leaves. J Burn Care Res. 2012;33:E309-E312.
- Picard C, Morice C, Moreau A, et al. Phytophotodermatitis in children: a difficult diagnosis mimicking other dermatitis. 2017;5:1-3.
- Enjolras O, Soupre V, Picard A. Uncommon benign infantile vascular tumors. Adv Dermatol. 2008;24:105-124.
Plant Parts and Nomenclature
Ficus carica (common fig) is a deciduous shrub or small tree with smooth gray bark that can grow up to 10 m in height (Figure 1). It is characterized by many spreading branches, but the trunk rarely grows beyond a diameter of 7 in. Its hairy leaves are coarse on the upper side and soft underneath with 3 to 7 deep lobes that can extend up to 25 cm in length or width; the leaves grow individually, alternating along the sides of the branches. Fig trees often can be seen adorning yards, gardens, and parks, especially in tropical and subtropical climates. Ficus carica should not be confused with Ficus benjamina (weeping fig), a common ornamental tree that also is used to provide shade in hot climates, though both can cause phototoxic skin eruptions.
The common fig tree originated in the Mediterranean and western Asia1 and has been cultivated by humans since the second and third millennia
Ficus carica is a member of the Moraceae family (derived from the Latin name for the mulberry tree), which includes 53 genera and approximately 1400 species, of which about 850 belong to the genus Ficus (the Latin name for a fig tree). The term carica likely comes from the Latin word carricare (to load) to describe a tree loaded with figs. Family members include trees, shrubs, lianas, and herbs that usually contain laticifers with a milky latex.
Traditional Uses
For centuries, components of the fig tree have been used in herbal teas and pastes to treat ailments ranging from sore throats to diarrhea, though there is no evidence to support their efficacy.4 Ancient Indians and Egyptians used plants such as the common fig tree containing furocoumarins to induce hyperpigmentation in vitiligo.5
Phototoxic Components
The leaves and sap of the common fig tree contain psoralens, which are members of the furocoumarin group of chemical compounds and are the source of its phototoxicity. The fruit does not contain psoralens.6-9 The tree also produces proteolytic enzymes such as protease, amylase, ficin, triterpenoids, and lipodiastase that enhance its phototoxic effects.8 Exposure to UV light between 320 and 400 nm following contact with these phototoxic components triggers a reaction in the skin over the course of 1 to 3 days.5 The psoralens bind in epidermal cells, cross-link the DNA, and cause cell-membrane destruction, leading to edema and necrosis.10 The delay in symptoms may be attributed to the time needed to synthesize acute-phase reaction proteins such as tumor necrosis factor α and IL-1.11 In spring and summer months, an increased concentration of psoralens in the leaves and sap contribute to an increased incidence of phytophotodermatitis.9 Humidity and sweat also increase the percutaneous absorption of psoralens.12,13
Allergens
Fig trees produce a latex protein that can cause cross-reactive hypersensitivity reactions in those allergic to F benjamina latex and rubber latex.6 The latex proteins in fig trees can act as airborne respiratory allergens. Ingestion of figs can produce anaphylactic reactions in those sensitized to rubber latex and F benjamina latex.7 Other plant families associated with phototoxic reactions include Rutaceae (lemon, lime, bitter orange), Apiaceae (formerly Umbelliferae)(carrot, parsnip, parsley, dill, celery, hogweed), and Fabaceae (prairie turnip).
Cutaneous Manifestations
Most cases of fig phytophotodermatitis begin with burning, pain, and/or itching within hours of sunlight exposure in areas of the skin that encountered components of the fig tree, often in a linear pattern. The affected areas become erythematous and edematous with formation of bullae and unilocular vesicles over the course of 1 to 3 days.12,14,15 Lesions may extend beyond the region of contact with the fig tree as they spread across the skin due to sweat or friction, and pain may linger even after the lesions resolve.12,13,16 Adults who handle fig trees (eg, pruning) are susceptible to phototoxic reactions, especially those using chain saws or other mechanisms that result in spray exposure, as the photosensitizing sap permeates the wood and bark of the entire tree.17 Similarly, children who handle fig leaves or sap during outdoor play can develop bullous eruptions. Severe cases have resulted in hospital admission after prolonged exposure.16 Additionally, irritant dermatitis may arise from contact with the trichomes or “hairs” on various parts of the plant.
Patients who use natural remedies containing components of the fig tree without the supervision of a medical provider put themselves at risk for unsafe or unwanted adverse effects, such as phytophotodermatitis.12,15,16,18 An entire family presented with burns after they applied fig leaf extract to the skin prior to tanning outside in the sun.19 A 42-year-old woman acquired a severe burn covering 81% of the body surface after topically applying fig leaf tea to the skin as a tanning agent.20 A subset of patients ingesting or applying fig tree components for conditions such as vitiligo, dermatitis, onychomycosis, and motor retardation developed similar cutaneous reactions.13,14,21,22 Lesions resembling finger marks can raise concerns for potential abuse or neglect in children.22
The differential diagnosis for fig phytophotodermatitis includes sunburn, chemical burns, drug-related photosensitivity, infectious lesions (eg, herpes simplex, bullous impetigo, Lyme disease, superficial lymphangitis), connective tissue disease (eg, systemic lupus erythematosus), contact dermatitis, and nonaccidental trauma.12,15,18 Compared to sunburn, phytophotodermatitis tends to increase in severity over days following exposure and heals with dramatic hyperpigmentation, which also prompts visits to dermatology.12
Treatment
Treatment of fig phytophotodermatitis chiefly is symptomatic, including analgesia, appropriate wound care, and infection prophylaxis. Topical and systemic corticosteroids may aid in the resolution of moderate to severe reactions.15,23,24 Even severe injuries over small areas or mild injuries to a high percentage of the total body surface area may require treatment in a burn unit. Patients should be encouraged to use mineral-based sunscreens on the affected areas to reduce the risk for hyperpigmentation. Individuals who regularly handle fig trees should use contact barriers including gloves and protective clothing (eg, long-sleeved shirts, long pants).
Plant Parts and Nomenclature
Ficus carica (common fig) is a deciduous shrub or small tree with smooth gray bark that can grow up to 10 m in height (Figure 1). It is characterized by many spreading branches, but the trunk rarely grows beyond a diameter of 7 in. Its hairy leaves are coarse on the upper side and soft underneath with 3 to 7 deep lobes that can extend up to 25 cm in length or width; the leaves grow individually, alternating along the sides of the branches. Fig trees often can be seen adorning yards, gardens, and parks, especially in tropical and subtropical climates. Ficus carica should not be confused with Ficus benjamina (weeping fig), a common ornamental tree that also is used to provide shade in hot climates, though both can cause phototoxic skin eruptions.
The common fig tree originated in the Mediterranean and western Asia1 and has been cultivated by humans since the second and third millennia
Ficus carica is a member of the Moraceae family (derived from the Latin name for the mulberry tree), which includes 53 genera and approximately 1400 species, of which about 850 belong to the genus Ficus (the Latin name for a fig tree). The term carica likely comes from the Latin word carricare (to load) to describe a tree loaded with figs. Family members include trees, shrubs, lianas, and herbs that usually contain laticifers with a milky latex.
Traditional Uses
For centuries, components of the fig tree have been used in herbal teas and pastes to treat ailments ranging from sore throats to diarrhea, though there is no evidence to support their efficacy.4 Ancient Indians and Egyptians used plants such as the common fig tree containing furocoumarins to induce hyperpigmentation in vitiligo.5
Phototoxic Components
The leaves and sap of the common fig tree contain psoralens, which are members of the furocoumarin group of chemical compounds and are the source of its phototoxicity. The fruit does not contain psoralens.6-9 The tree also produces proteolytic enzymes such as protease, amylase, ficin, triterpenoids, and lipodiastase that enhance its phototoxic effects.8 Exposure to UV light between 320 and 400 nm following contact with these phototoxic components triggers a reaction in the skin over the course of 1 to 3 days.5 The psoralens bind in epidermal cells, cross-link the DNA, and cause cell-membrane destruction, leading to edema and necrosis.10 The delay in symptoms may be attributed to the time needed to synthesize acute-phase reaction proteins such as tumor necrosis factor α and IL-1.11 In spring and summer months, an increased concentration of psoralens in the leaves and sap contribute to an increased incidence of phytophotodermatitis.9 Humidity and sweat also increase the percutaneous absorption of psoralens.12,13
Allergens
Fig trees produce a latex protein that can cause cross-reactive hypersensitivity reactions in those allergic to F benjamina latex and rubber latex.6 The latex proteins in fig trees can act as airborne respiratory allergens. Ingestion of figs can produce anaphylactic reactions in those sensitized to rubber latex and F benjamina latex.7 Other plant families associated with phototoxic reactions include Rutaceae (lemon, lime, bitter orange), Apiaceae (formerly Umbelliferae)(carrot, parsnip, parsley, dill, celery, hogweed), and Fabaceae (prairie turnip).
Cutaneous Manifestations
Most cases of fig phytophotodermatitis begin with burning, pain, and/or itching within hours of sunlight exposure in areas of the skin that encountered components of the fig tree, often in a linear pattern. The affected areas become erythematous and edematous with formation of bullae and unilocular vesicles over the course of 1 to 3 days.12,14,15 Lesions may extend beyond the region of contact with the fig tree as they spread across the skin due to sweat or friction, and pain may linger even after the lesions resolve.12,13,16 Adults who handle fig trees (eg, pruning) are susceptible to phototoxic reactions, especially those using chain saws or other mechanisms that result in spray exposure, as the photosensitizing sap permeates the wood and bark of the entire tree.17 Similarly, children who handle fig leaves or sap during outdoor play can develop bullous eruptions. Severe cases have resulted in hospital admission after prolonged exposure.16 Additionally, irritant dermatitis may arise from contact with the trichomes or “hairs” on various parts of the plant.
Patients who use natural remedies containing components of the fig tree without the supervision of a medical provider put themselves at risk for unsafe or unwanted adverse effects, such as phytophotodermatitis.12,15,16,18 An entire family presented with burns after they applied fig leaf extract to the skin prior to tanning outside in the sun.19 A 42-year-old woman acquired a severe burn covering 81% of the body surface after topically applying fig leaf tea to the skin as a tanning agent.20 A subset of patients ingesting or applying fig tree components for conditions such as vitiligo, dermatitis, onychomycosis, and motor retardation developed similar cutaneous reactions.13,14,21,22 Lesions resembling finger marks can raise concerns for potential abuse or neglect in children.22
The differential diagnosis for fig phytophotodermatitis includes sunburn, chemical burns, drug-related photosensitivity, infectious lesions (eg, herpes simplex, bullous impetigo, Lyme disease, superficial lymphangitis), connective tissue disease (eg, systemic lupus erythematosus), contact dermatitis, and nonaccidental trauma.12,15,18 Compared to sunburn, phytophotodermatitis tends to increase in severity over days following exposure and heals with dramatic hyperpigmentation, which also prompts visits to dermatology.12
Treatment
Treatment of fig phytophotodermatitis chiefly is symptomatic, including analgesia, appropriate wound care, and infection prophylaxis. Topical and systemic corticosteroids may aid in the resolution of moderate to severe reactions.15,23,24 Even severe injuries over small areas or mild injuries to a high percentage of the total body surface area may require treatment in a burn unit. Patients should be encouraged to use mineral-based sunscreens on the affected areas to reduce the risk for hyperpigmentation. Individuals who regularly handle fig trees should use contact barriers including gloves and protective clothing (eg, long-sleeved shirts, long pants).
- Ikegami H, Nogata H, Hirashima K, et al. Analysis of genetic diversity among European and Asian fig varieties (Ficus carica L.) using ISSR, RAPD, and SSR markers. Genetic Resources and Crop Evolution. 2009;56:201-209.
- Zohary D, Spiegel-Roy P. Beginnings of fruit growing in the Old World. Science. 1975;187:319-327.
- Young R. Young’s Analytical Concordance. Thomas Nelson; 1982.
- Duke JA. Handbook of Medicinal Herbs. CRC Press; 2002.
- Pathak MA, Fitzpatrick TB. Bioassay of natural and synthetic furocoumarins (psoralens). J Invest Dermatol. 1959;32:509-518.
- Focke M, Hemmer W, Wöhrl S, et al. Cross-reactivity between Ficus benjamina latex and fig fruit in patients with clinical fig allergy. Clin Exp Allergy. 2003;33:971-977.
- Hemmer W, Focke M, Götz M, et al. Sensitization to Ficus benjamina: relationship to natural rubber latex allergy and identification of foods implicated in the Ficus-fruit syndrome. Clin Exp Allergy. 2004;34:1251-1258.
- Bonamonte D, Foti C, Lionetti N, et al. Photoallergic contact dermatitis to 8-methoxypsoralen in Ficus carica. Contact Dermatitis. 2010;62:343-348.
- Zaynoun ST, Aftimos BG, Abi Ali L, et al. Ficus carica; isolation and quantification of the photoactive components. Contact Dermatitis. 1984;11:21-25.
- Tessman JW, Isaacs ST, Hearst JE. Photochemistry of the furan-side 8-methoxypsoralen-thymidine monoadduct inside the DNA helix. conversion to diadduct and to pyrone-side monoadduct. Biochemistry. 1985;24:1669-1676.
- Geary P. Burns related to the use of psoralens as a tanning agent. Burns. 1996;22:636-637.
- Redgrave N, Solomon J. Severe phytophotodermatitis from fig sap: a little known phenomenon. BMJ Case Rep. 2021;14:E238745.
- Ozdamar E, Ozbek S, Akin S. An unusual cause of burn injury: fig leaf decoction used as a remedy for a dermatitis of unknown etiology. J Burn Care Rehabil. 2003;24:229-233; discussion 228.
- Berakha GJ, Lefkovits G. Psoralen phototherapy and phototoxicity. Ann Plast Surg. 1985;14:458-461.
- Papazoglou A, Mantadakis E. Fig tree leaves phytophotodermatitis. J Pediatr. 2021;239:244-245.
- Imen MS, Ahmadabadi A, Tavousi SH, et al. The curious cases of burn by fig tree leaves. Indian J Dermatol. 2019;64:71-73.
- Rouaiguia-Bouakkaz S, Amira-Guebailia H, Rivière C, et al. Identification and quantification of furanocoumarins in stem bark and wood of eight Algerian varieties of Ficus carica by RP-HPLC-DAD and RP-HPLC-DAD-MS. Nat Prod Commun. 2013;8:485-486.
- Oliveira AA, Morais J, Pires O, et al. Fig tree induced phytophotodermatitis. BMJ Case Rep. 2020;13:E233392.
- Bassioukas K, Stergiopoulou C, Hatzis J. Erythrodermic phytophotodermatitis after application of aqueous fig-leaf extract as an artificial suntan promoter and sunbathing. Contact Dermatitis. 2004;51:94-95.
- Sforza M, Andjelkov K, Zaccheddu R. Severe burn on 81% of body surface after sun tanning. Ulus Travma Acil Cerrahi Derg. 2013;19:383-384.
- Son JH, Jin H, You HS, et al. Five cases of phytophotodermatitis caused by fig leaves and relevant literature review. Ann Dermatol. 2017;29:86-90.
- Abali AE, Aka M, Aydogan C, et al. Burns or phytophotodermatitis, abuse or neglect: confusing aspects of skin lesions caused by the superstitious use of fig leaves. J Burn Care Res. 2012;33:E309-E312.
- Picard C, Morice C, Moreau A, et al. Phytophotodermatitis in children: a difficult diagnosis mimicking other dermatitis. 2017;5:1-3.
- Enjolras O, Soupre V, Picard A. Uncommon benign infantile vascular tumors. Adv Dermatol. 2008;24:105-124.
- Ikegami H, Nogata H, Hirashima K, et al. Analysis of genetic diversity among European and Asian fig varieties (Ficus carica L.) using ISSR, RAPD, and SSR markers. Genetic Resources and Crop Evolution. 2009;56:201-209.
- Zohary D, Spiegel-Roy P. Beginnings of fruit growing in the Old World. Science. 1975;187:319-327.
- Young R. Young’s Analytical Concordance. Thomas Nelson; 1982.
- Duke JA. Handbook of Medicinal Herbs. CRC Press; 2002.
- Pathak MA, Fitzpatrick TB. Bioassay of natural and synthetic furocoumarins (psoralens). J Invest Dermatol. 1959;32:509-518.
- Focke M, Hemmer W, Wöhrl S, et al. Cross-reactivity between Ficus benjamina latex and fig fruit in patients with clinical fig allergy. Clin Exp Allergy. 2003;33:971-977.
- Hemmer W, Focke M, Götz M, et al. Sensitization to Ficus benjamina: relationship to natural rubber latex allergy and identification of foods implicated in the Ficus-fruit syndrome. Clin Exp Allergy. 2004;34:1251-1258.
- Bonamonte D, Foti C, Lionetti N, et al. Photoallergic contact dermatitis to 8-methoxypsoralen in Ficus carica. Contact Dermatitis. 2010;62:343-348.
- Zaynoun ST, Aftimos BG, Abi Ali L, et al. Ficus carica; isolation and quantification of the photoactive components. Contact Dermatitis. 1984;11:21-25.
- Tessman JW, Isaacs ST, Hearst JE. Photochemistry of the furan-side 8-methoxypsoralen-thymidine monoadduct inside the DNA helix. conversion to diadduct and to pyrone-side monoadduct. Biochemistry. 1985;24:1669-1676.
- Geary P. Burns related to the use of psoralens as a tanning agent. Burns. 1996;22:636-637.
- Redgrave N, Solomon J. Severe phytophotodermatitis from fig sap: a little known phenomenon. BMJ Case Rep. 2021;14:E238745.
- Ozdamar E, Ozbek S, Akin S. An unusual cause of burn injury: fig leaf decoction used as a remedy for a dermatitis of unknown etiology. J Burn Care Rehabil. 2003;24:229-233; discussion 228.
- Berakha GJ, Lefkovits G. Psoralen phototherapy and phototoxicity. Ann Plast Surg. 1985;14:458-461.
- Papazoglou A, Mantadakis E. Fig tree leaves phytophotodermatitis. J Pediatr. 2021;239:244-245.
- Imen MS, Ahmadabadi A, Tavousi SH, et al. The curious cases of burn by fig tree leaves. Indian J Dermatol. 2019;64:71-73.
- Rouaiguia-Bouakkaz S, Amira-Guebailia H, Rivière C, et al. Identification and quantification of furanocoumarins in stem bark and wood of eight Algerian varieties of Ficus carica by RP-HPLC-DAD and RP-HPLC-DAD-MS. Nat Prod Commun. 2013;8:485-486.
- Oliveira AA, Morais J, Pires O, et al. Fig tree induced phytophotodermatitis. BMJ Case Rep. 2020;13:E233392.
- Bassioukas K, Stergiopoulou C, Hatzis J. Erythrodermic phytophotodermatitis after application of aqueous fig-leaf extract as an artificial suntan promoter and sunbathing. Contact Dermatitis. 2004;51:94-95.
- Sforza M, Andjelkov K, Zaccheddu R. Severe burn on 81% of body surface after sun tanning. Ulus Travma Acil Cerrahi Derg. 2013;19:383-384.
- Son JH, Jin H, You HS, et al. Five cases of phytophotodermatitis caused by fig leaves and relevant literature review. Ann Dermatol. 2017;29:86-90.
- Abali AE, Aka M, Aydogan C, et al. Burns or phytophotodermatitis, abuse or neglect: confusing aspects of skin lesions caused by the superstitious use of fig leaves. J Burn Care Res. 2012;33:E309-E312.
- Picard C, Morice C, Moreau A, et al. Phytophotodermatitis in children: a difficult diagnosis mimicking other dermatitis. 2017;5:1-3.
- Enjolras O, Soupre V, Picard A. Uncommon benign infantile vascular tumors. Adv Dermatol. 2008;24:105-124.
Practice Points
- Exposure to the components of the common fig tree (Ficus carica) can induce phytophotodermatitis.
- Notable postinflammatory hyperpigmentation typically occurs in the healing stage of fig phytophotodermatitis.

Micronutrient Deficiencies in Patients With Inflammatory Bowel Disease
In 2023, ESPEN (the European Society for Clinical Nutrition and Metabolism) published consensus recommendations highlighting the importance of regular monitoring and treatment of nutrient deficiencies in patients with inflammatory bowel disease (IBD) for improved prognosis, mortality, and quality of life.1 Suboptimal nutrition in patients with IBD predominantly results from inflammation of the gastrointestinal (GI) tract leading to malabsorption; however, medications commonly used to manage IBD also can contribute to malnutrition.2,3 Additionally, patients may develop nausea and food avoidance due to medication or the disease itself, leading to nutritional withdrawal and eventual deficiency.4 Even with the development of diets focused on balancing nutritional needs and decreasing inflammation,5 offsetting this aversion to food can be difficult to overcome.2
Cutaneous manifestations of IBD are multifaceted and can be secondary to the disease, reactive to or associated with IBD, or effects from nutritional deficiencies. The most common vitamin and nutrient deficiencies in patients with IBD include iron; zinc; calcium; vitamin D; and vitamins B6 (pyridoxine), B9 (folic acid), and B12.6 Malnutrition may manifest with cutaneous disease, and dermatologists can be the first to identify and assess for nutritional deficiencies. In this article, we review the mechanisms of these micronutrient depletions in the context of IBD, their subsequent dermatologic manifestations (Table), and treatment and monitoring guidelines for each deficiency.

Iron
A systematic review conducted from 2007 to 2012 in European patients with IBD (N=2192) found the overall prevalence of anemia in this population to be 24% (95% CI, 18%-31%), with 57% of patients with anemia experiencing iron deficiency.7 Anemia is observed more commonly in patients hospitalized with IBD and is common in patients with both Crohn disease and ulcerative colitis.8
Pathophysiology—Iron is critically important in oxygen transportation throughout the body as a major component of hemoglobin. Physiologically, the low pH of the duodenum and proximal jejunum allows divalent metal transporter 1 to transfer dietary Fe3+ into enterocytes, where it is reduced to the transportable Fe2+.9,10 Distribution of Fe2+ ions from enterocytes relies on ferroportin, an iron-transporting protein, which is heavily regulated by the protein hepcidin.11 Hepcidin, a known acute phase reactant, will increase in the setting of active IBD, causing a depletion of ferroportin and an inability of the body to utilize the stored iron in enterocytes.12 This poor utilization of iron stores combined with blood loss caused by inflammation in the GI tract is the proposed primary mechanism of iron-deficiency anemia observed in patients with IBD.13
Cutaneous Manifestations—From a dermatologic perspective, iron-deficiency anemia can manifest with a wide range of symptoms including glossitis, koilonychia, xerosis and/or pruritus, and brittle hair or hair loss.14,15 Although the underlying pathophysiology of these cutaneous manifestations is not fully understood, there are several theories assessing the mechanisms behind the skin findings of iron deficiency.
Atrophic glossitis has been observed in many patients with iron deficiency and is thought to manifest due to low iron concentrations in the blood, thereby decreasing oxygen delivery to the papillae of the dorsal tongue with resultant atrophy.16,17 Similarly, decreased oxygen delivery to the nail bed capillaries may cause deformities in the nail called koilonychia (or “spoon nails”).18 Iron is a key co-factor in collagen lysyl hydroxylase that promotes collagen binding; iron deficiency may lead to disruptions in the epidermal barrier that can cause pruritus and xerosis.19 An observational study of 200 healthy patients with a primary concern of pruritus found a correlation between low serum ferritin and a higher degree of pruritus (r=−0.768; P<.00001).20
Evidence for iron’s role in hair growth comes from a mouse model study with a mutation in the serine protease TMPRSS6—a protein that regulates hepcidin and iron absorption—which caused an increase in hepcidin production and subsequent systemic iron deficiency. Mice at 4 weeks of age were devoid of all body hair but had substantial regrowth after initiation of a 2-week iron-rich diet, which suggests a connection between iron repletion and hair growth in mice with iron deficiency.21 Additionally, a meta-analysis analyzing the comorbidities of patients with alopecia areata found them to have higher odds (odds ratio [OR]=2.78; 95% CI, 1.23-6.29) of iron-deficiency anemia but no association with IBD (OR=1.48; 95% CI, 0.32-6.82).22
Diagnosis and Monitoring—The American Gastroenterological Association recommends a complete blood cell count (CBC), serum ferritin, transferrin saturation (TfS), and C-reactive protein (CRP) as standard evaluations for iron deficiency in patients with IBD. Patients with active IBD should be screened every 3 months,and patients with inactive disease should be screened every 6 to 12 months.23
Although ferritin and TfS often are used as markers for iron status in healthy individuals, they are positive and negative acute phase reactants, respectively. Using them to assess iron status in patients with IBD may inaccurately represent iron status in the setting of inflammation from the disease.24 The European Crohn’s and Colitis Organisation (ECCO) produced guidelines to define iron deficiency as a TfS less than 20% or a ferritin level less than 30 µg/L in patients without evidence of active IBD and a ferritin level less than 100 µg/L for patients with active inflammation.25
A 2020 multicenter observational study of 202 patients with diagnosed IBD found that the ECCO guideline of ferritin less than 30 µg/L had an area under the receiver operating characteristic (AUROC) curve of 0.69, a sensitivity of 0.43, and a specificity of 0.95 in their population.26 In a sensitivity analysis stratifying patients by CRP level (<10 or ≥10 mg/L), the authors found that for patients with ulcerative colitis and a CRP less than 10 mg/L, a cut-off value of ferritin less than 65 µg/L (AUROC=0.78) had a sensitivity of 0.78 and specificity of 0.76, and a TfS value of less than 16% (AUROC=0.88) had a sensitivity of 0.79 and a specificity of 0.9. In patients with a CRP of 10 mg/L or greater, a cut-off value of ferritin 80 µg/L (AUROC=0.76) had a sensitivity of 0.75 and a specificity of 0.82, and a TfS value of less than 11% (AUROC=0.69) had a sensitivity of 0.79 and a specificity of 0.88. There were no ferritin cut-off values associated with good diagnostic performance (defined as both sensitivity and specificity >0.70) for iron deficiency in patients with Crohn disease.26
The authors recommended using an alternative iron measurement such as soluble transferrin receptor (sTfR)/log ferritin ratio (TfR-F) that is not influenced by active inflammation and has a good correlation with ferritin values (TfR-F: r=0.66; P<.001).26 However, both sTfR and TfR-F have high costs and intermethod variability as well as differences in their reference ranges depending on which laboratory performs the analysis, limiting the accessibility and practicality of easily obtaining these tests.27 Although there may be inaccuracies for standard ferritin or TfS under ECCO guidelines, proposed alternatives have their own limitations, which may make ferritin and TfS the most reasonable evaluations of iron status as long as disease activity status at the time of testing is taken into consideration.
Treatment—Treatment of underlying iron deficiency in patients with IBD requires reversing the cause of the deficiency and supplementing iron. In patients with IBD, the options to supplement iron may be limited by active disease, making oral intake less effective. Oral iron supplementation also is associated with notable GI adverse effects that may be exacerbated in patients with IBD. A systematic review of 43 randomized controlled trials (RCTs) evaluating GI adverse effects (eg, nausea, abdominal pain, diarrhea, constipation, and black or tarry stools) of oral ferrous sulfate compared with placebo or intravenous (IV) iron supplementation in healthy nonanemic individuals found a significant increase in GI adverse effects with oral supplementation (placebo: OR=2.32; P<.0001; IV: OR=3.05; P<.0001).28
Therefore, IV iron repletion may be necessary in patients with IBD and may require numerous infusions depending on the formulation of iron. In an RCT conducted in 2011, patients with iron-deficiency anemia with quiescent or mild to moderate IBD were treated with either IV iron sulfate or ferric carboxymaltose.29 With a primary end point of hemoglobin response greater than 2 g/dL, the authors found that 150 of 240 patients responded to ferric carboxymaltose vs 118 of 235 treated with iron sulfate (P=.004). The dosing for ferric carboxymaltose was 1 to 3 infusions of 500 to 1000 mg of iron and for iron sulfate up to 11 infusions of 200 mg of iron.29
Zinc
A systematic review of zinc deficiency in patients with IBD identified 7 studies including 2413 patients and revealed those with Crohn disease had a higher prevalence of zinc deficiency compared with patients with ulcerative colitis (54% vs 41%).30
Pathophysiology—Zinc serves as a catalytic cofactor for enzymatic activity within proteins and immune cells.31 The homeostasis of zinc is tightly regulated within the brush border of the small intestine by zinc transporters ZIP4 and ZIP1 from the lumen of enterocytes into the bloodstream.32 Inflammation in the small intestine due to Crohn disease can result in zinc malabsorption.
Ranaldi et al33 exposed intestinal cells and zinc-depleted intestinal cells to tumor necrosis factor α media to simulate an inflammatory environment. They measured transepithelial electrical resistance as a surrogate for transmembrane permeability and found that zinc-depleted cells had a statistically significantly higher transepithelial electrical resistance percentage (60% reduction after 4 hours; P<1.10–6) when exposed to tumor necrosis factor α signaling compared with normal intestinal cells. They concluded that zinc deficiency can increase intestinal permeability in the presence of inflammation, creating a cycle of further nutrient malabsorption and inflammation exacerbating IBD symptoms.33
Cutaneous Manifestations—After absorption in the small intestine, approximately 5% of zinc resides in the skin, with the highest concentration in the stratum spinosum.34 A cell study found that keratinocytes in zinc-deficient environments had higher rates of apoptosis compared with cells in normal media. The authors proposed that this higher rate of apoptosis and the resulting inflammation could be a mechanism for developing the desquamative or eczematous scaly plaques that are common cutaneous manifestations of zinc deficiency.35
Other cutaneous findings may include angular cheilitis, stomatitis, glossitis, paronychia, onychodystrophy, generalized alopecia, and delayed wound healing.36 The histopathology of these skin lesions is characterized by granular layer loss, epidermal pallor, confluent parakeratosis, spongiosis, dyskeratosis, and psoriasiform hyperplasia.37
Diagnosis and Monitoring—Assessing serum zinc levels is challenging, as they may decrease during states of inflammation.38 A mouse model study showed a 3.1-fold increase (P<.001) in ZIP14 expression in wild-type mice compared with an IL-6 -/- knock-down model after IL-6 exposure. The authors concluded that the upregulation of ZIP14 in the liver due to inflammatory cytokine upregulation decreases zinc availability in serum.39 Additionally, serum zinc can overestimate the level of deficiency in IBD because approximately 75% of serum zinc is bound to albumin, which decreases in the setting of inflammation.40-42
Alternatively, alkaline phosphatase (AP), a zinc-dependent metalloenzyme, may be a better evaluator of zinc status during periods of inflammation. A study in rats evaluated zinc through serum zinc levels and AP levels after a period of induced stress to mimic a short-term inflammatory state.43 The researchers found that total body stores of zinc were unaffected throughout the experiment; only serum zinc declined throughout the experiment duration while AP did not. Because approximately 75% of serum zinc is bound to serum albumin,42 the researchers concluded the induced inflammatory state depleted serum albumin and redistributed zinc to the liver, causing the observed serum zinc changes, while total body zinc levels and AP were largely unaffected in comparison.43 Comorbid conditions such as liver or bone disease can increase AP levels, which limits the utility of AP as a surrogate for zinc in patients with comorbidities.44 However, even in the context of active IBD, serum zinc still is currently considered the best biomarker to evaluate zinc status.45
Treatment—The recommended dose for zinc supplementation is 20 to 40 mg daily with higher doses (>50 mg/d) for patients with malabsorptive syndromes such as IBD.46 It can be administered orally or parenterally. Although rare, zinc replacement therapy may be associated with diarrhea, nausea, vomiting, mild headaches, and fatigue.46 Additional considerations should be taken when repleting other micronutrients with zinc, as calcium and folate can inhibit zinc reabsorption, while zinc itself can inhibit iron and copper reabsorption.47
Vitamin D and Calcium
Low vitamin D levels (<50 nmol/L) and hypocalcemia (<8.8 mg/dL) are common in patients with IBD.48,49
Pathophysiology—Vitamin D levels are maintained via 2 mechanisms. The first mechanism is through the skin, as keratinocytes produce 7-dehydrocholesterol after exposure to UV light, which is converted into previtamin D3 and then thermally isomerizes into vitamin D3. This vitamin D3 is then transported to the liver on vitamin D–binding protein.50 The second mechanism is through oral vitamin D3 that is absorbed through vitamin D receptors in intestinal epithelium and transported to the liver, where it is hydroxylated into 25-hydroxyvitamin D (25[OH]D), then to the kidneys for hydroxylation to 1,25(OH)2D for redistribution throughout the body.50 This activated form of vitamin D regulates calcium absorption in the intestine, and optimal vitamin D levels are necessary to absorb calcium efficiently.51 Inflammation from IBD within the small intestine can downregulate vitamin D receptors, causing malabsorption and decreased serum vitamin D.52
Vitamin D signaling also is vital to maintaining the tight junctions and adherens junctions of the intestinal epithelium. Weakening the permeability of the epithelium further exacerbates malabsorption and subsequent vitamin D deficiency.52 A meta-analysis of 27 studies including 8316 patients with IBD showed low vitamin D levels were associated with increased odds of disease activity (OR=1.53; 95% CI, 1.32-1.77), mucosal inflammation (OR=1.25; 95% CI, 1.06-1.47), and future clinical relapse (OR=1.23; 95% CI, 1.03-1.47) in patients with Crohn disease. The authors concluded that low levels of vitamin D could be used as a potential biomarker of inflammatory status in Crohn disease.53
Vitamin D and calcium are further implicated in maintaining skeletal health,47 while vitamin D specifically helps maintain intestinal homeostasis54 and immune system modulation in the skin.55
Cutaneous Manifestations—Vitamin D is thought to play crucial roles in skin differentiation and proliferation, cutaneous innate immunity, hair follicle cycling, photoprotection, and wound healing.56 Vitamin D deficiency has been observed in a large range of cutaneous diseases including skin cancer, psoriasis, vitiligo, bullous pemphigoid, atopic dermatitis, and various types of alopecia.56-59 It is unclear whether vitamin D deficiency facilitates these disease processes or is merely the consequence of a disrupted cutaneous surface with the inability to complete the first step in vitamin D processing. A 2014 meta-analysis of 290 prospective cohort studies and 172 randomized trials concluded that 25(OH)D deficiency was associated with ill health and did not find causal evidence for any specific disease, dermatologic or otherwise.60 Calcium deficiency may cause epidermal changes including dry skin, coarse hair, and brittle nails.61
Diagnosis and Monitoring—The ECCO guidelines recommend obtaining serum 25(OH)D levels every 3 months in patients with IBD.62 Levels less than 75 nmol/L are considered deficient, and a value less than 30 nmol/L increases the risk for osteomalacia and nutritional rickets, constituting severe vitamin D deficiency.63-65
An observational study of 325 patients with IBD showed a statistically significant negative correlation between serum vitamin D and fecal calprotectin (r=−0.19; P<.001), a stool-based marker for gut inflammation, supporting vitamin D as a potential biomarker in IBD.66
Evaluation of calcium can be done through serum levels in patients with IBD.67 Patients with IBD are at risk for hypoalbuminemia; therefore, consideration should be taken to ensure calcium levels are corrected, as approximately 50% of calcium is bound to albumin or other ions in the body,68 which can be done by adjusting the calcium concentration by 0.02 mmol/L for every 1 g/L of albumin above or below 40 g/L. In the most critically ill patients, a direct ionized calcium blood level should be used instead because the previously mentioned correction calculations are inaccurate when albumin is critically low.69
Treatment—The ECCO guidelines recommend calcium and vitamin D repletion of 500 to 1000 mg and 800 to 1000 U, respectively, in patients with IBD on systemic corticosteroids to prevent the negative effects of bone loss.62 Calcium repletion in patients with IBD who are not on systemic steroids are the same as for the general population.65
Vitamin D repletion also may help decrease IBD activity. In a prospective study, 10,000 IU/d of vitamin D in 10 patients with IBD—adjusted over 12 weeks to a target of 100 to 125 nmol/L of serum 25(OH)D—showed a significant reduction in clinical Crohn activity (P=.019) over the study period.70 In contrast, 2000 IU/d for 3 months in an RCT of 27 patients with Crohn disease found significantly lower CRP (P=.019) and significantly higher self-reported quality of life (P=.037) but nonsignificant decreases in Crohn activity (P=.082) in patients with 25(OH)D levels of 75 nmol/L or higher compared with those with 25(OH)D levels less than 75 nmol/L.71
These discrepancies illustrate the need for expanded clinical trials to elucidate the optimal vitamin D dosing for patients with IBD. Ultimately, assessing vitamin D and calcium status and considering repletion in patients with IBD, especially those with comorbid dermatologic diseases such as poor wound healing, psoriasis, or atopic dermatitis, is important.
Vitamin B6 (Pyridoxine)
Pathophysiology—Pyridoxine is an important coenzyme for many functions including amino acid transamination, fatty acid metabolism, and conversion of tryptophan to niacin. It is absorbed in the jejunum and ileum and subsequently transported to the liver for rephosphorylation and release into its active form.36 An observational study assessing the nutritional status of patients with IBD found that only 5.7% of 105 patients with food records had inadequate dietary intake of pyridoxine, but 29% of all patients with IBD had subnormal pyridoxine levels.72 Additionally, they found no significant difference in the prevalence of subnormal pyridoxine levels in patients with active IBD vs IBD in remission. The authors suggested that the subnormal pyridoxine levels in patients with IBD likely were multifactorial and resulted from malabsorption due to active disease, inflammation, and inadequate intake.72
Cutaneous Manifestations—Cutaneous findings associated with pyridoxine deficiency include periorificial and perineal dermatitis,73 angular stomatitis, and cheilitis with associated burning, redness, and tongue edema.36 Additionally, pyridoxine is involved in the conversion of tryptophan to niacin, and its deficiency may manifest with pellagralike findings.74
Because pyridoxine is critical to protein metabolism, its deficiency may disrupt key cellular structures that rely on protein concentrations to maintain structural integrity. One such structure in the skin that heavily relies on protein concentrations is the ground substance of the extracellular matrix—the amorphous gelatinous spaces that occupy the areas between the extracellular matrix, which consists of cross-linked glycosaminoglycans and proteins.75 Without protein, ground substance increases in viscosity and can disrupt the epidermal barrier, leading to increased transepidermal water loss and ultimately inflammation.76 Although this theory has yet to be validated fully, this is a potential mechanistic explanation for the inflammation in dermal papillae that leads to dermatitis observed in pyridoxine deficiency.
Diagnosis and Monitoring—Direct biomarkers of pyridoxine status are in serum, plasma, erythrocytes, and urine, with the most common measurement in plasma as pyridoxal 5′-phosphate (PLP).77 Plasma PLP concentrations lower than 20 nmol/L are suggestive of deficiency.78 Plasma PLP has shown inverse relationships with acute phase inflammatory markers CRP79 and AP,78 thereby raising concerns for its validity to assess pyridoxine status in patients with symptomatic IBD.80
Alternative evaluations of pyridoxine include tryptophan and methionine loading tests,36 which are measured via urinary excretion and require normal kidney function to be accurate. They should be considered in IBD if necessary, but routine testing, even in patients with symptomatic IBD, is not recommended in the ECCO guidelines. Additional considerations should be taken in patients with altered nutrient requirements such as those who have undergone bowel resection due to highly active disease or those who receive parenteral nutritional supplementation.81
Treatment—Recommendations for oral pyridoxine supplementation range from 25 to 600 mg daily,82 with symptoms typically improving on 100 mg daily.36 Pyridoxine supplementation may have additional benefits for patients with IBD and potentially modulate disease severity. An IL-10 knockout mouse supplemented with pyridoxine had an approximately 60% reduction (P<.05) in inflammation compared to mice deficient in pyridoxine.83 The authors suggest that PLP-dependent enzymes can inhibit further proinflammatory signaling and T-cell migration that can exacerbate IBD. Ultimately, more data is needed before determining the efficacy of pyridoxine supplementation for active IBD.
Vitamin B12 and Vitamin B9 (Folic Acid)
Pathophysiology—Vitamin B12 is reabsorbed in the terminal ileum, the distal portion of the small intestine. The American Gastroenterological Association recommends that patients with a history of extensive ileal disease or prior ileal surgery, which is the case for many patients with Crohn disease, be monitored for vitamin B12 deficiency.23 Monitoring and rapid supplementation of vitamin B12 can prevent pernicious anemia and irreversible neurologic damage that may result from deficiency.84
Folic acid is primarily absorbed in the duodenum and jejunum of the small intestine. A meta-analysis performed in 2017 assessed studies observing folic acid and vitamin B12 levels in 1086 patients with IBD compared with 1484 healthy controls and found an average difference in serum folate concentration of 0.46 nmol/L (P<.001).84 Interestingly, this study did not find a significant difference in serum vitamin B12 levels between patients with IBD and healthy controls, highlighting the mechanism of vitamin B12 deficiency in IBD because only patients with terminal ileal involvement are at risk for malabsorption and subsequent deficiency.
Cutaneous Manifestations—Both vitamin B12 and folic acid deficiency can manifest as cheilitis, glossitis, and/or generalized hyperpigmentation that is accentuated in the flexural areas, palms, soles, and oral cavity.85,86 Systemic symptoms of patients with vitamin B12 and folic acid deficiency include megaloblastic anemia, pallor, and fatigue. A potential mechanism for the hyperpigmentation observed from vitamin B12 deficiency came from an electron microscope study that showed an increased concentration of melanosomes in a patient with deficiency.87
Diagnosis and Monitoring—In patients with suspected vitamin B12 and/or folic acid deficiency, initial evaluation should include a CBC with peripheral smear and serum vitamin B12 and folate levels. In cases for which the diagnosis still is unclear after initial testing,
Treatment—According to the Centers for Disease Control and Prevention, supplementation of vitamin B12 can be done orally with 1000 µg daily in patients with deficiency. In patients with active IBD, oral reabsorption of vitamin B12 can be less effective, making subcutaneous or intramuscular administration (1000 µg/wk for 8 weeks, then monthly for life) better options.89
Patients with IBD managed with methotrexate should be screened carefully for folate deficiency. Methotrexate is a folate analog that sometimes is used for the treatment of IBD. Reversible competitive inhibition of dihydrofolate reductase can precipitate a systemic folic acid decrease.91 Typically, oral folic acid (1 to 5 mg/d) is sufficient to treat folate deficiency, with the ESPEN recommending 5 mg once weekly 24 to 72 hours after methotrexate treatment or 1 mg daily for 5 days per week in patients with IBD.1 Alternative formulations—IV, subcutaneous, or intramuscular—are available for patients who cannot tolerate oral intake.92
Final Thoughts
Dermatologists can be the first to observe the cutaneous manifestations of micronutrient deficiencies. Although the symptoms of each micronutrient deficiency discussed may overlap, attention to small clinical clues in patients with IBD can improve patient outcomes and quality of life. For example, koilonychia with glossitis and xerosis likely is due to iron deficiency, while zinc deficiency should be suspected in patients with scaly eczematous plaques in skin folds. A high level of suspicion for micronutrient deficiencies in patients with IBD should be followed by a complete patient history, review of systems, and thorough clinical examination. A thorough laboratory evaluation can pinpoint nutritional deficiencies in patients with IBD, keeping in mind that specific biomarkers such as ferritin and serum zinc also act as acute phase reactants and should be interpreted in this context. Co-management with gastroenterologists should be a priority in patients with IBD, as gaining control of inflammatory disease is crucial for the prevention of recurrent vitamin and micronutrient deficiencies in addition to long-term health in this population.
- Bischoff SC, Bager P, Escher J, et al. ESPEN guideline on clinical nutrition in inflammatory bowel disease. Clin Nutr. 2023;42:352-379. doi:10.1016/j.clnu.2022.12.004
- Gerasimidis K, McGrogan P, Edwards CA. The aetiology and impact of malnutrition in paediatric inflammator y bowel disease. J Hum Nutr Diet. 2011;24:313-326. doi:10.1111/j.1365-277X.2011.01171.x
- Mentella MC, Scaldaferri F, Pizzoferrato M, et al. Nutrition, IBD and gut microbiota: a review. Nutrients. 2020;12:944. doi:10.3390/nu12040944
- Bonsack O, Caron B, Baumann C, et al. Food avoidance and fasting in patients with inflammatory bowel disease: experience from the Nancy IBD nutrition clinic. United European Gastroenterol J. 2023;11:361-370. doi:10.1002/ueg2.1238521
- Campmans-Kuijpers MJE, Dijkstra G. Food and food groups in inflammatory bowel disease (IBD): the design of the Groningen Anti-Inflammatory Diet (GrAID). Nutrients. 2021;13:1067. doi:10.3390/nu13041067
- Hwang C, Issokson K, Giguere-Rich C, et al. Development and pilot testing of the inflammatory bowel disease nutrition care pathway. Clin Gastroenterol Hepatol. 2020;18:2645-2649.e4. doi:10.1016/j.cgh.2020.06.039
- Filmann N, Rey J, Schneeweiss S, et al. Prevalence of anemia in inflammatory bowel diseases in European countries: a systematic review and individual patient data meta-analysis. Inflamm Bowel Dis. 2014;20:936-945. doi:10.1097/01.MIB.0000442728.74340.fd
- Stein J, Hartmann F, Dignass AU. Diagnosis and management of iron deficiency anemia in patients with IBD. Nat Rev Gastroenterol Hepatol. 2010;7:599-610. doi:10.1038/nrgastro.2010.151
- Ems T, St Lucia K, Huecker MR. Biochemistry, iron absorption. StatPearls [Internet]. Updated April 17, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK448204/
- Evstatiev R, Gasche C. Iron sensing and signalling. Gut. 2012;61:933-952. doi:10.1136/gut.2010.214312
- Przybyszewska J, Zekanowska E. The role of hepcidin, ferroportin, HCP1, and DMT1 protein in iron absorption in the human digestive tract. Prz Gastroenterol. 2014;9:208-213. doi:10.5114/pg.2014.45102
- Weiss G, Gasche C. Pathogenesis and treatment of anemia in inflammatory bowel disease. Haematologica. 2010;95:175-178. doi:10.3324/haematol.2009.017046
- Kaitha S, Bashir M, Ali T. Iron deficiency anemia in inflammatory bowel disease. World J Gastrointest Pathophysiol. 2015;6:62-72. doi:10.4291/wjgp.v6.i3.62
- Moiz B. Spoon nails: still seen in today’s world. Clin Case Rep. 2018;6:547-548. doi:10.1002/ccr3.1404
- St Pierre SA, Vercellotti GM, Donovan JC, et al. Iron deficiency and diffuse nonscarring scalp alopecia in women: more pieces to the puzzle. J Am Acad Dermatol. 2010;63:1070-1076. doi:10.1016/j.jaad.2009.05.054
- Chiang CP, Yu-Fong Chang J, Wang YP, et al. Anemia, hematinic deficiencies, hyperhomocysteinemia, and serum gastric parietal cell antibody positivity in atrophic glossitis patients with or without microcytosis. J Formos Med Assoc. 2019;118:1401-1407. doi:10.1016/j.jfma.2019.06.004
- Chiang CP, Chang JY, Wang YP, et al. Atrophic glossitis: Etiology, serum autoantibodies, anemia, hematinic deficiencies, hyperhomocysteinemia, and management. J Formos Med Assoc. 2020;119:774-780. doi:10.1016/j.jfma.2019.04.015
- Walker J, Baran R, Vélez N, et al. Koilonychia: an update on pathophysiology, differential diagnosis and clinical relevance. J Eur Acad Dermatol Venereol. 2016;30:1985-1991. doi:10.1111/jdv.13610
- Guo HF, Tsai CL, Terajima M, et al. Pro-metastatic collagen lysyl hydroxylase dimer assemblies stabilized by Fe2+-binding. Nat Commun. 2018;9:512. doi:10.1038/s41467-018-02859-z
- Saini S, Jain AK, Agarwal S, et al. Iron deficiency and pruritus: a cross-sectional analysis to assess its association and relationship. Indian J Dermatol. 2021;66:705. doi:10.4103/ijd.ijd_326_21
- Du X, She E, Gelbart T, et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science. 2008;320:1088-1092. doi:10.1126/science.1157121
- Lee S, Lee H, Lee CH, et al. Comorbidities in alopecia areata: a systematic review and meta-analysis. J Am Acad Dermatol. 2019;80:466-477.e16. doi:10.1016/j.jaad.2018.07.013
- Hashash JG, Elkins J, Lewis JD, et al. AGA Clinical Practice Update on diet and nutritional therapies in patients with inflammatory bowel disease: expert review [published online January 23, 2024]. Gastroenterology. doi:10.1053/j.gastro.2023.11.303
- Choudhuri S, Chowdhury IH, Saha A, et al. Acute monocyte pro- inflammatory response predicts higher positive to negative acute phase reactants ratio and severe hemostatic derangement in dengue fever. Cytokine. 2021;146:155644. doi:10.1016/j.cyto.2021.155644
- Dignass AU, Gasche C, Bettenworth D, et al; European Crohn’s and Colitis Organisation. European consensus on the diagnosis and management of iron deficiency and anaemia in inflammatory bowel diseases. J Crohn’s Colitis. 2015;9:211-222. doi:10.1093/ecco-jcc/jju009
- Daude S, Remen T, Chateau T, et al. Comparative accuracy of ferritin, transferrin saturation and soluble transferrin receptor for the diagnosis of iron deficiency in inflammatory bowel disease. Aliment Pharmacol Ther. 2020;51:1087-1095. doi:10.1111/apt.15739
- Pfeiffer CM, Looker AC. Laboratory methodologies for indicators of iron status: strengths, limitations, and analytical challenges. Am J Clin Nutr. 2017;106(suppl 6):1606S-1614S. doi:10.3945/ajcn.117.155887
- Tolkien Z, Stecher L, Mander AP, et al. Ferrous sulfate supplementation causes significant gastrointestinal side-effects in adults: a systematic review and meta-analysis. PLoS One. 2015;10:e0117383. doi:10.1371/journal.pone.0117383
- Evstatiev R, Marteau P, Iqbal T, et al. FERGIcor, a randomized controlled trial on ferric carboxymaltose for iron deficiency anemia in inflammatory bowel disease. Gastroenterology. 2011;141:846-853.e8532. doi:10.1053/j.gastro.2011.06.005
- Zupo R, Sila A, Castellana F, et al. Prevalence of zinc deficiency in inflammatory bowel disease: a systematic review and meta-analysis. Nutrients. 2022;14:4052. doi:10.3390/nu14194052
- Thompson MW. Regulation of zinc-dependent enzymes by metal carrier proteins. Biometals. 2022;35:187-213. doi:10.1007/s10534-022-00373-w
- Maares M, Haase H. A guide to human zinc absorption: general overview and recent advances of in vitro intestinal models. Nutrients. 2020;12:762. doi:10.3390/nu12030762
- Ranaldi G, Ferruzza S, Canali R, et al. Intracellular zinc is required for intestinal cell survival signals triggered by the inflammatory cytokine TNFα. J Nutr Biochem. 2013;24:967-976. doi:10.1016/j.jnutbio.2012.06.020
- Ogawa Y, Kawamura T, Shimada S. Zinc and skin biology. Arch Biochem Biophys. 2016;611:113-119. doi:10.1016/j.abb.2016.06.003
- Wilson D, Varigos G, Ackland ML. Apoptosis may underlie the pathology of zinc-deficient skin. Immunol Cell Biol. 2006;84:28-37. doi:10.1111/j.1440-1711.2005.01391.x
- Jen M, Yan AC. Syndromes associated with nutritional deficiency and excess. Clin Dermatol. 2010;28:669-685. doi:10.1016/j.clindermatol.2010.03.029
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Gammoh NZ, Rink L. Zinc in infection and inflammation. Nutrients. 2017;9:624. doi:10.3390/nu9060624
- Liuzzi JP, Lichten LA, Rivera S, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to the hypozincemia of the acute-phase response. Proc Natl Acad Sci U S A. 2005;102:6843-6848. doi:10.1073/pnas.0502257102
- Vermeire S, Van Assche G, Rutgeerts P. Laboratory markers in IBD: useful, magic, or unnecessary toys?. Gut. 2006;55:426-431. doi:10.1136/gut.2005.069476
- Morisaku M, Ito K, Ogiso A, et al. Correlation between serum albumin and serum zinc in malignant lymphoma. Fujita Med J. 2022;8:59-64. doi:10.20407/fmj.2021-006
- Falchuk KH. Effect of acute disease and ACTH on serum zinc proteins. N Engl J Med. 1977:296:1129-1134.
- Naber TH, Baadenhuysen H, Jansen JB, et al. Serum alkaline phosphatase activity during zinc deficiency and long-term inflammatory stress. Clin Chim Acta. 1996;249:109-127. doi:10.1016/0009-8981(96)06281-x
- Lowe D, Sanvictores T, Zubair M, et al. Alkaline phosphatase. StatPearls [Internet]. Updated October 29, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK459201/
- Krebs NF. Update on zinc deficiency and excess in clinical pediatric practice. Ann Nutr Metab. 2013;62 suppl 1:19-29. doi:10.1159/000348261
- Maxfield L, Shukla S, Crane JS. Zinc deficiency. StatPearls [Internet]. Updated June 28, 2023. Accessed March 25, 2024. https://www.ncbi.nlm.nih.gov/books/NBK493231/
- Ghishan FK, Kiela PR. Vitamins and minerals in inflammatory bowel disease. Gastroenterol Clin North Am. 2017;46:797-808. doi:10.1016/j.gtc.2017.08.011
- Caviezel D, Maissen S, Niess JH, et al. High prevalence of vitamin D deficiency among patients with inflammatory bowel disease. Inflamm Intest Dis. 2018;2:200-210. doi:10.1159/000489010
- Jasielska M, Grzybowska-Chlebowczyk U. Hypocalcemia and vitamin D deficiency in children with inflammatory bowel diseases and lactose intolerance. Nutrients. 2021;13:2583. doi:10.3390/nu13082583
- Vernia F, Valvano M, Longo S, et al. Vitamin D in inflammatory bowel diseases. Mechanisms of action and therapeutic implications. Nutrients. 2022;14:269. doi:10.3390/nu14020269
- Khazai N, Judd SE, Tangpricha V. Calcium and vitamin D: skeletal and extraskeletal health. Curr Rheumatol Rep. 2008;10:110-117. doi:10.1007/s11926-008-0020-y
- Domazetovic V, Iantomasi T, Bonanomi AG, et al. Vitamin D regulates claudin-2 and claudin-4 expression in active ulcerative colitis by p-Stat-6 and Smad-7 signaling. Int J Colorectal Dis. 2020;35:1231-1242. doi:10.1007/s00384-020-03576-0
- Gubatan J, Chou ND, Nielsen OH, et al. Systematic review with meta-analysis: association of vitamin D status with clinical outcomes in adult patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2019;50:1146-1158. doi:10.1111/apt.15506
- Fakhoury HMA, Kvietys PR, AlKattan W, et al. Vitamin D and intestinal homeostasis: barrier, microbiota, and immune modulation. J Steroid Biochem Mol Biol. 2020;200:105663. doi:10.1016/j.jsbmb.2020.105663
- Liu PT, Stenger S, Li H, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770-1773. doi:10.1126/science.1123933
- Mostafa WZ, Hegazy RA. Vitamin D and the skin: focus on a complex relationship: a review. J Adv Res. 2015;6:793-804. doi:10.1016/j.jare.2014.01.011
- Searing DA, Leung DY. Vitamin D in atopic dermatitis, asthma and allergic diseases. Immunol Allergy Clin North Am. 2010;30:397-409.
- Lee YH, Song GG. Association between circulating 25-hydroxyvitamin D levels and psoriasis, and correlation with disease severity: a meta-analysis. Clin Exp Dermatol. 2018;43:529-535.
- Adorini L, Penna G. Control of autoimmune diseases by the vitamin D endocrine system. Nat Clin Pract Rheumatol. 2008;4:404-412.
- Autier P, Boniol M, Pizot C, et al. Vitamin D status and ill health: a systematic review. Lancet Diabetes Endocrinol. 2014;2:76-89. doi:10.1016/S2213-8587(13)70165-7
- Schafer AL, Shoback DM. Hypocalcemia: diagnosis and treatment. In: Feingold KR, Anawalt B, Blackman MR, et al, eds. Endotext [Internet]. Updated January 3, 2016. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK279022/
- Magro F, Gionchetti P, Eliakim R, et al. Third European Evidence-based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 1: Definitions, diagnosis, extra-intestinal manifestations, pregnancy, cancer surveillance, surgery, and ileo-anal pouch disorders. J Crohns Colitis. 2017;11:649-670. doi:10.1093/ecco-jcc/jjx008
- Amrein K, Scherkl M, Hoffmann M, et al. Vitamin D deficiency 2.0: an update on the current status worldwide. Eur J Clin Nutr. 2020;74:1498-1513. doi:10.1038/s41430-020-0558-y
- Munns CF, Shaw N, Kiely M, et al. Global consensus recommendations on prevention and management of nutritional rickets. J Clin Endocrinol Metab. 2016;101:394-415. doi:10.1210/jc.2015-2175
- Institute of Medicine (US) Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Ross AC, Taylor CL, Yaktine AL, Del Valle HB, eds. Dietary Reference Intakes for Calcium and Vitamin D. National Academies Press (US); 2011.
- Yeaman F, Nguyen A, Abasszade J, et al. Assessing vitamin D as a biomarker in inflammatory bowel disease. JGH Open. 2023;7:953-958. doi:10.1002/jgh3.13010
- Vernia P, Loizos P, Di Giuseppantonio I, et al S. Dietary calcium intake in patients with inflammatory bowel disease. J Crohns Colitis. 2014;8:312-317. doi:10.1016/j.crohns.2013.09.008
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ. 2008;336:1298-1302. doi:10.1136/bmj.39582.589433.BE
- Kenny CM, Murphy CE, Boyce DS, et al. Things we do for no reason™: calculating a “corrected calcium” level. J Hosp Med. 2021;16:499-501. doi:10.12788/jhm.3619
- Garg M, Rosella O, Rosella G, et al. Evaluation of a 12-week targeted vitamin D supplementation regimen in patients with active inflammatory bowel disease. Clin Nutr. 2018;37:1375-1382. doi:10.1016/j.clnu.2017.06.011
- Raftery T, Martineau AR, Greiller CL, et al. Effects of vitamin D supplementation on intestinal permeability, cathelicidin and disease markers in Crohn’s disease: results from a randomised double-blind placebo-controlled study. United European Gastroenterol J. 2015;3:294-302. doi:10.1177/2050640615572176
- Vagianos K, Bector S, McConnell J, et al. Nutrition assessment of patients with inflammatory bowel disease. JPEN J Parenter Enteral Nutr. 2007;31:311-319. doi:10.1177/0148607107031004311
- Barthelemy H, Chouvet B, Cambazard F. Skin and mucosal manifestations in vitamin deficiency. J Am Acad Dermatol. 1986;15:1263-1274. doi:10.1016/s0190-9622(86)70301-0
- Galimberti F, Mesinkovska NA. Skin findings associated with nutritional deficiencies. Cleve Clin J Med. 2016;83:731-739. doi:10.3949/ccjm.83a.15061
- Elgharably N, Al Abadie M, Al Abadie M, et al. Vitamin B group levels and supplementations in dermatology. Dermatol Reports. 2022;15:9511. doi:10.4081/dr.2022.9511
- Hołubiec P, Leon´czyk M, Staszewski F, et al. Pathophysiology and clinical management of pellagra—a review. Folia Med Cracov. 2021;61:125-137. doi:10.24425/fmc.2021.138956
- Ink SL, Henderson LM. Vitamin B6 metabolism. Annu Rev Nutr. 1984;4:455-470. doi:10.1146/annurev.nu.04.070184.002323
- Brown MJ, Ameer MA, Daley SF, et al. Vitamin B6 deficiency. StatPearls [Internet]. Updated August 8, 2023. Accessed March 25, 2024. https://www.ncbi.nlm.nih.gov/books/NBK470579/.
- Vasilaki AT, McMillan DC, Kinsella J, et al. Relation between pyridoxal and pyridoxal phosphate concentrations in plasma, red cells, and white cells in patients with critical illness. Am J Clin Nutr. 2008;88:140-146. doi:10.1093/ajcn/88.1.140
- Chiang EP, Bagley PJ, Selhub J, et al. Abnormal vitamin B(6) status is associated with severity of symptoms in patients with rheumatoid arthritis. Am J Med. 2003;114:283-287. doi:10.1016/s0002-9343(02)01528-0
- Maaser C, Sturm A, Vavricka SR, et al. ECCO-ESGAR guideline for diagnostic assessment in IBD. Part 1: initial diagnosis, monitoring of known IBD, detection of complications. J Crohns Colitis. 2019;13:144-164. doi:10.1093/ecco-jcc/jjy113
- Spinneker A, Sola R, Lemmen V, et al. Vitamin B6 status, deficiency and its consequences—an overview. Nutr Hosp. 2007;22:7-24.
- Selhub J, Byun A, Liu Z, et al. Dietary vitamin B6 intake modulates colonic inflammation in the IL10-/- model of inflammatory bowel disease. J Nutr Biochem. 2013;24:2138-2143. doi:10.1016/j.jnutbio.2013.08.005
- Pan Y, Liu Y, Guo H, et al. Associations between folate and vitamin B12 levels and inflammatory bowel disease: a meta-analysis. Nutrients. 2017;9:382. doi:10.3390/nu9040382
- Brescoll J, Daveluy S. A review of vitamin B12 in dermatology. Am J Clin Dermatol. 2015;16:27-33. doi:10.1007/s40257-014-0107-3
- DiBaise M, Tarleton SM. Hair, nails, and skin: differentiating cutaneous manifestations of micronutrient deficiency. Nutr Clin Pract. 2019;34:490-503. doi:10.1002/ncp.10321
- Mori K, Ando I, Kukita A. Generalized hyperpigmentation of the skin due to vitamin B12 deficiency. J Dermatol. 2001;28:282-285. doi:10.1111/j.1346-8138.2001.tb00134.x
- Green R. Indicators for assessing folate and vitamin B-12 status and for monitoring the efficacy of intervention strategies. Am J Clin Nutr. 2011;94:666S-672S. doi:10.3945/ajcn.110.009613
- NIH Office of Dietary Supplements. Vitamin B12: fact sheet for health professionals. Updated February 27, 2024. Accessed March 19, 2024. https://ods.od.nih.gov/factsheets/VitaminB12-HealthProfessional/
- NIH Office of Dietary Supplements. Folate: fact sheet for health professionals. Updated November 20, 2023. Accessed March 19, 2024. https://ods.od.nih.gov/factsheets/Folate-HealthProfessional/.
- Saibeni S, Bollani S, Losco A, et al. The use of methotrexate for treatment of inflammatory bowel disease in clinical practice. Dig Liver Dis. 2012;44:123-127. doi:10.1016/j.dld.2011.09.015
- Khan KM, Jialal I. Folic acid deficiency. StatPearls [Internet]. Updated June 26, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK535377/
In 2023, ESPEN (the European Society for Clinical Nutrition and Metabolism) published consensus recommendations highlighting the importance of regular monitoring and treatment of nutrient deficiencies in patients with inflammatory bowel disease (IBD) for improved prognosis, mortality, and quality of life.1 Suboptimal nutrition in patients with IBD predominantly results from inflammation of the gastrointestinal (GI) tract leading to malabsorption; however, medications commonly used to manage IBD also can contribute to malnutrition.2,3 Additionally, patients may develop nausea and food avoidance due to medication or the disease itself, leading to nutritional withdrawal and eventual deficiency.4 Even with the development of diets focused on balancing nutritional needs and decreasing inflammation,5 offsetting this aversion to food can be difficult to overcome.2
Cutaneous manifestations of IBD are multifaceted and can be secondary to the disease, reactive to or associated with IBD, or effects from nutritional deficiencies. The most common vitamin and nutrient deficiencies in patients with IBD include iron; zinc; calcium; vitamin D; and vitamins B6 (pyridoxine), B9 (folic acid), and B12.6 Malnutrition may manifest with cutaneous disease, and dermatologists can be the first to identify and assess for nutritional deficiencies. In this article, we review the mechanisms of these micronutrient depletions in the context of IBD, their subsequent dermatologic manifestations (Table), and treatment and monitoring guidelines for each deficiency.
Iron
A systematic review conducted from 2007 to 2012 in European patients with IBD (N=2192) found the overall prevalence of anemia in this population to be 24% (95% CI, 18%-31%), with 57% of patients with anemia experiencing iron deficiency.7 Anemia is observed more commonly in patients hospitalized with IBD and is common in patients with both Crohn disease and ulcerative colitis.8
Pathophysiology—Iron is critically important in oxygen transportation throughout the body as a major component of hemoglobin. Physiologically, the low pH of the duodenum and proximal jejunum allows divalent metal transporter 1 to transfer dietary Fe3+ into enterocytes, where it is reduced to the transportable Fe2+.9,10 Distribution of Fe2+ ions from enterocytes relies on ferroportin, an iron-transporting protein, which is heavily regulated by the protein hepcidin.11 Hepcidin, a known acute phase reactant, will increase in the setting of active IBD, causing a depletion of ferroportin and an inability of the body to utilize the stored iron in enterocytes.12 This poor utilization of iron stores combined with blood loss caused by inflammation in the GI tract is the proposed primary mechanism of iron-deficiency anemia observed in patients with IBD.13
Cutaneous Manifestations—From a dermatologic perspective, iron-deficiency anemia can manifest with a wide range of symptoms including glossitis, koilonychia, xerosis and/or pruritus, and brittle hair or hair loss.14,15 Although the underlying pathophysiology of these cutaneous manifestations is not fully understood, there are several theories assessing the mechanisms behind the skin findings of iron deficiency.
Atrophic glossitis has been observed in many patients with iron deficiency and is thought to manifest due to low iron concentrations in the blood, thereby decreasing oxygen delivery to the papillae of the dorsal tongue with resultant atrophy.16,17 Similarly, decreased oxygen delivery to the nail bed capillaries may cause deformities in the nail called koilonychia (or “spoon nails”).18 Iron is a key co-factor in collagen lysyl hydroxylase that promotes collagen binding; iron deficiency may lead to disruptions in the epidermal barrier that can cause pruritus and xerosis.19 An observational study of 200 healthy patients with a primary concern of pruritus found a correlation between low serum ferritin and a higher degree of pruritus (r=−0.768; P<.00001).20
Evidence for iron’s role in hair growth comes from a mouse model study with a mutation in the serine protease TMPRSS6—a protein that regulates hepcidin and iron absorption—which caused an increase in hepcidin production and subsequent systemic iron deficiency. Mice at 4 weeks of age were devoid of all body hair but had substantial regrowth after initiation of a 2-week iron-rich diet, which suggests a connection between iron repletion and hair growth in mice with iron deficiency.21 Additionally, a meta-analysis analyzing the comorbidities of patients with alopecia areata found them to have higher odds (odds ratio [OR]=2.78; 95% CI, 1.23-6.29) of iron-deficiency anemia but no association with IBD (OR=1.48; 95% CI, 0.32-6.82).22
Diagnosis and Monitoring—The American Gastroenterological Association recommends a complete blood cell count (CBC), serum ferritin, transferrin saturation (TfS), and C-reactive protein (CRP) as standard evaluations for iron deficiency in patients with IBD. Patients with active IBD should be screened every 3 months,and patients with inactive disease should be screened every 6 to 12 months.23
Although ferritin and TfS often are used as markers for iron status in healthy individuals, they are positive and negative acute phase reactants, respectively. Using them to assess iron status in patients with IBD may inaccurately represent iron status in the setting of inflammation from the disease.24 The European Crohn’s and Colitis Organisation (ECCO) produced guidelines to define iron deficiency as a TfS less than 20% or a ferritin level less than 30 µg/L in patients without evidence of active IBD and a ferritin level less than 100 µg/L for patients with active inflammation.25
A 2020 multicenter observational study of 202 patients with diagnosed IBD found that the ECCO guideline of ferritin less than 30 µg/L had an area under the receiver operating characteristic (AUROC) curve of 0.69, a sensitivity of 0.43, and a specificity of 0.95 in their population.26 In a sensitivity analysis stratifying patients by CRP level (<10 or ≥10 mg/L), the authors found that for patients with ulcerative colitis and a CRP less than 10 mg/L, a cut-off value of ferritin less than 65 µg/L (AUROC=0.78) had a sensitivity of 0.78 and specificity of 0.76, and a TfS value of less than 16% (AUROC=0.88) had a sensitivity of 0.79 and a specificity of 0.9. In patients with a CRP of 10 mg/L or greater, a cut-off value of ferritin 80 µg/L (AUROC=0.76) had a sensitivity of 0.75 and a specificity of 0.82, and a TfS value of less than 11% (AUROC=0.69) had a sensitivity of 0.79 and a specificity of 0.88. There were no ferritin cut-off values associated with good diagnostic performance (defined as both sensitivity and specificity >0.70) for iron deficiency in patients with Crohn disease.26
The authors recommended using an alternative iron measurement such as soluble transferrin receptor (sTfR)/log ferritin ratio (TfR-F) that is not influenced by active inflammation and has a good correlation with ferritin values (TfR-F: r=0.66; P<.001).26 However, both sTfR and TfR-F have high costs and intermethod variability as well as differences in their reference ranges depending on which laboratory performs the analysis, limiting the accessibility and practicality of easily obtaining these tests.27 Although there may be inaccuracies for standard ferritin or TfS under ECCO guidelines, proposed alternatives have their own limitations, which may make ferritin and TfS the most reasonable evaluations of iron status as long as disease activity status at the time of testing is taken into consideration.
Treatment—Treatment of underlying iron deficiency in patients with IBD requires reversing the cause of the deficiency and supplementing iron. In patients with IBD, the options to supplement iron may be limited by active disease, making oral intake less effective. Oral iron supplementation also is associated with notable GI adverse effects that may be exacerbated in patients with IBD. A systematic review of 43 randomized controlled trials (RCTs) evaluating GI adverse effects (eg, nausea, abdominal pain, diarrhea, constipation, and black or tarry stools) of oral ferrous sulfate compared with placebo or intravenous (IV) iron supplementation in healthy nonanemic individuals found a significant increase in GI adverse effects with oral supplementation (placebo: OR=2.32; P<.0001; IV: OR=3.05; P<.0001).28
Therefore, IV iron repletion may be necessary in patients with IBD and may require numerous infusions depending on the formulation of iron. In an RCT conducted in 2011, patients with iron-deficiency anemia with quiescent or mild to moderate IBD were treated with either IV iron sulfate or ferric carboxymaltose.29 With a primary end point of hemoglobin response greater than 2 g/dL, the authors found that 150 of 240 patients responded to ferric carboxymaltose vs 118 of 235 treated with iron sulfate (P=.004). The dosing for ferric carboxymaltose was 1 to 3 infusions of 500 to 1000 mg of iron and for iron sulfate up to 11 infusions of 200 mg of iron.29
Zinc
A systematic review of zinc deficiency in patients with IBD identified 7 studies including 2413 patients and revealed those with Crohn disease had a higher prevalence of zinc deficiency compared with patients with ulcerative colitis (54% vs 41%).30
Pathophysiology—Zinc serves as a catalytic cofactor for enzymatic activity within proteins and immune cells.31 The homeostasis of zinc is tightly regulated within the brush border of the small intestine by zinc transporters ZIP4 and ZIP1 from the lumen of enterocytes into the bloodstream.32 Inflammation in the small intestine due to Crohn disease can result in zinc malabsorption.
Ranaldi et al33 exposed intestinal cells and zinc-depleted intestinal cells to tumor necrosis factor α media to simulate an inflammatory environment. They measured transepithelial electrical resistance as a surrogate for transmembrane permeability and found that zinc-depleted cells had a statistically significantly higher transepithelial electrical resistance percentage (60% reduction after 4 hours; P<1.10–6) when exposed to tumor necrosis factor α signaling compared with normal intestinal cells. They concluded that zinc deficiency can increase intestinal permeability in the presence of inflammation, creating a cycle of further nutrient malabsorption and inflammation exacerbating IBD symptoms.33
Cutaneous Manifestations—After absorption in the small intestine, approximately 5% of zinc resides in the skin, with the highest concentration in the stratum spinosum.34 A cell study found that keratinocytes in zinc-deficient environments had higher rates of apoptosis compared with cells in normal media. The authors proposed that this higher rate of apoptosis and the resulting inflammation could be a mechanism for developing the desquamative or eczematous scaly plaques that are common cutaneous manifestations of zinc deficiency.35
Other cutaneous findings may include angular cheilitis, stomatitis, glossitis, paronychia, onychodystrophy, generalized alopecia, and delayed wound healing.36 The histopathology of these skin lesions is characterized by granular layer loss, epidermal pallor, confluent parakeratosis, spongiosis, dyskeratosis, and psoriasiform hyperplasia.37
Diagnosis and Monitoring—Assessing serum zinc levels is challenging, as they may decrease during states of inflammation.38 A mouse model study showed a 3.1-fold increase (P<.001) in ZIP14 expression in wild-type mice compared with an IL-6 -/- knock-down model after IL-6 exposure. The authors concluded that the upregulation of ZIP14 in the liver due to inflammatory cytokine upregulation decreases zinc availability in serum.39 Additionally, serum zinc can overestimate the level of deficiency in IBD because approximately 75% of serum zinc is bound to albumin, which decreases in the setting of inflammation.40-42
Alternatively, alkaline phosphatase (AP), a zinc-dependent metalloenzyme, may be a better evaluator of zinc status during periods of inflammation. A study in rats evaluated zinc through serum zinc levels and AP levels after a period of induced stress to mimic a short-term inflammatory state.43 The researchers found that total body stores of zinc were unaffected throughout the experiment; only serum zinc declined throughout the experiment duration while AP did not. Because approximately 75% of serum zinc is bound to serum albumin,42 the researchers concluded the induced inflammatory state depleted serum albumin and redistributed zinc to the liver, causing the observed serum zinc changes, while total body zinc levels and AP were largely unaffected in comparison.43 Comorbid conditions such as liver or bone disease can increase AP levels, which limits the utility of AP as a surrogate for zinc in patients with comorbidities.44 However, even in the context of active IBD, serum zinc still is currently considered the best biomarker to evaluate zinc status.45
Treatment—The recommended dose for zinc supplementation is 20 to 40 mg daily with higher doses (>50 mg/d) for patients with malabsorptive syndromes such as IBD.46 It can be administered orally or parenterally. Although rare, zinc replacement therapy may be associated with diarrhea, nausea, vomiting, mild headaches, and fatigue.46 Additional considerations should be taken when repleting other micronutrients with zinc, as calcium and folate can inhibit zinc reabsorption, while zinc itself can inhibit iron and copper reabsorption.47
Vitamin D and Calcium
Low vitamin D levels (<50 nmol/L) and hypocalcemia (<8.8 mg/dL) are common in patients with IBD.48,49
Pathophysiology—Vitamin D levels are maintained via 2 mechanisms. The first mechanism is through the skin, as keratinocytes produce 7-dehydrocholesterol after exposure to UV light, which is converted into previtamin D3 and then thermally isomerizes into vitamin D3. This vitamin D3 is then transported to the liver on vitamin D–binding protein.50 The second mechanism is through oral vitamin D3 that is absorbed through vitamin D receptors in intestinal epithelium and transported to the liver, where it is hydroxylated into 25-hydroxyvitamin D (25[OH]D), then to the kidneys for hydroxylation to 1,25(OH)2D for redistribution throughout the body.50 This activated form of vitamin D regulates calcium absorption in the intestine, and optimal vitamin D levels are necessary to absorb calcium efficiently.51 Inflammation from IBD within the small intestine can downregulate vitamin D receptors, causing malabsorption and decreased serum vitamin D.52
Vitamin D signaling also is vital to maintaining the tight junctions and adherens junctions of the intestinal epithelium. Weakening the permeability of the epithelium further exacerbates malabsorption and subsequent vitamin D deficiency.52 A meta-analysis of 27 studies including 8316 patients with IBD showed low vitamin D levels were associated with increased odds of disease activity (OR=1.53; 95% CI, 1.32-1.77), mucosal inflammation (OR=1.25; 95% CI, 1.06-1.47), and future clinical relapse (OR=1.23; 95% CI, 1.03-1.47) in patients with Crohn disease. The authors concluded that low levels of vitamin D could be used as a potential biomarker of inflammatory status in Crohn disease.53
Vitamin D and calcium are further implicated in maintaining skeletal health,47 while vitamin D specifically helps maintain intestinal homeostasis54 and immune system modulation in the skin.55
Cutaneous Manifestations—Vitamin D is thought to play crucial roles in skin differentiation and proliferation, cutaneous innate immunity, hair follicle cycling, photoprotection, and wound healing.56 Vitamin D deficiency has been observed in a large range of cutaneous diseases including skin cancer, psoriasis, vitiligo, bullous pemphigoid, atopic dermatitis, and various types of alopecia.56-59 It is unclear whether vitamin D deficiency facilitates these disease processes or is merely the consequence of a disrupted cutaneous surface with the inability to complete the first step in vitamin D processing. A 2014 meta-analysis of 290 prospective cohort studies and 172 randomized trials concluded that 25(OH)D deficiency was associated with ill health and did not find causal evidence for any specific disease, dermatologic or otherwise.60 Calcium deficiency may cause epidermal changes including dry skin, coarse hair, and brittle nails.61
Diagnosis and Monitoring—The ECCO guidelines recommend obtaining serum 25(OH)D levels every 3 months in patients with IBD.62 Levels less than 75 nmol/L are considered deficient, and a value less than 30 nmol/L increases the risk for osteomalacia and nutritional rickets, constituting severe vitamin D deficiency.63-65
An observational study of 325 patients with IBD showed a statistically significant negative correlation between serum vitamin D and fecal calprotectin (r=−0.19; P<.001), a stool-based marker for gut inflammation, supporting vitamin D as a potential biomarker in IBD.66
Evaluation of calcium can be done through serum levels in patients with IBD.67 Patients with IBD are at risk for hypoalbuminemia; therefore, consideration should be taken to ensure calcium levels are corrected, as approximately 50% of calcium is bound to albumin or other ions in the body,68 which can be done by adjusting the calcium concentration by 0.02 mmol/L for every 1 g/L of albumin above or below 40 g/L. In the most critically ill patients, a direct ionized calcium blood level should be used instead because the previously mentioned correction calculations are inaccurate when albumin is critically low.69
Treatment—The ECCO guidelines recommend calcium and vitamin D repletion of 500 to 1000 mg and 800 to 1000 U, respectively, in patients with IBD on systemic corticosteroids to prevent the negative effects of bone loss.62 Calcium repletion in patients with IBD who are not on systemic steroids are the same as for the general population.65
Vitamin D repletion also may help decrease IBD activity. In a prospective study, 10,000 IU/d of vitamin D in 10 patients with IBD—adjusted over 12 weeks to a target of 100 to 125 nmol/L of serum 25(OH)D—showed a significant reduction in clinical Crohn activity (P=.019) over the study period.70 In contrast, 2000 IU/d for 3 months in an RCT of 27 patients with Crohn disease found significantly lower CRP (P=.019) and significantly higher self-reported quality of life (P=.037) but nonsignificant decreases in Crohn activity (P=.082) in patients with 25(OH)D levels of 75 nmol/L or higher compared with those with 25(OH)D levels less than 75 nmol/L.71
These discrepancies illustrate the need for expanded clinical trials to elucidate the optimal vitamin D dosing for patients with IBD. Ultimately, assessing vitamin D and calcium status and considering repletion in patients with IBD, especially those with comorbid dermatologic diseases such as poor wound healing, psoriasis, or atopic dermatitis, is important.
Vitamin B6 (Pyridoxine)
Pathophysiology—Pyridoxine is an important coenzyme for many functions including amino acid transamination, fatty acid metabolism, and conversion of tryptophan to niacin. It is absorbed in the jejunum and ileum and subsequently transported to the liver for rephosphorylation and release into its active form.36 An observational study assessing the nutritional status of patients with IBD found that only 5.7% of 105 patients with food records had inadequate dietary intake of pyridoxine, but 29% of all patients with IBD had subnormal pyridoxine levels.72 Additionally, they found no significant difference in the prevalence of subnormal pyridoxine levels in patients with active IBD vs IBD in remission. The authors suggested that the subnormal pyridoxine levels in patients with IBD likely were multifactorial and resulted from malabsorption due to active disease, inflammation, and inadequate intake.72
Cutaneous Manifestations—Cutaneous findings associated with pyridoxine deficiency include periorificial and perineal dermatitis,73 angular stomatitis, and cheilitis with associated burning, redness, and tongue edema.36 Additionally, pyridoxine is involved in the conversion of tryptophan to niacin, and its deficiency may manifest with pellagralike findings.74
Because pyridoxine is critical to protein metabolism, its deficiency may disrupt key cellular structures that rely on protein concentrations to maintain structural integrity. One such structure in the skin that heavily relies on protein concentrations is the ground substance of the extracellular matrix—the amorphous gelatinous spaces that occupy the areas between the extracellular matrix, which consists of cross-linked glycosaminoglycans and proteins.75 Without protein, ground substance increases in viscosity and can disrupt the epidermal barrier, leading to increased transepidermal water loss and ultimately inflammation.76 Although this theory has yet to be validated fully, this is a potential mechanistic explanation for the inflammation in dermal papillae that leads to dermatitis observed in pyridoxine deficiency.
Diagnosis and Monitoring—Direct biomarkers of pyridoxine status are in serum, plasma, erythrocytes, and urine, with the most common measurement in plasma as pyridoxal 5′-phosphate (PLP).77 Plasma PLP concentrations lower than 20 nmol/L are suggestive of deficiency.78 Plasma PLP has shown inverse relationships with acute phase inflammatory markers CRP79 and AP,78 thereby raising concerns for its validity to assess pyridoxine status in patients with symptomatic IBD.80
Alternative evaluations of pyridoxine include tryptophan and methionine loading tests,36 which are measured via urinary excretion and require normal kidney function to be accurate. They should be considered in IBD if necessary, but routine testing, even in patients with symptomatic IBD, is not recommended in the ECCO guidelines. Additional considerations should be taken in patients with altered nutrient requirements such as those who have undergone bowel resection due to highly active disease or those who receive parenteral nutritional supplementation.81
Treatment—Recommendations for oral pyridoxine supplementation range from 25 to 600 mg daily,82 with symptoms typically improving on 100 mg daily.36 Pyridoxine supplementation may have additional benefits for patients with IBD and potentially modulate disease severity. An IL-10 knockout mouse supplemented with pyridoxine had an approximately 60% reduction (P<.05) in inflammation compared to mice deficient in pyridoxine.83 The authors suggest that PLP-dependent enzymes can inhibit further proinflammatory signaling and T-cell migration that can exacerbate IBD. Ultimately, more data is needed before determining the efficacy of pyridoxine supplementation for active IBD.
Vitamin B12 and Vitamin B9 (Folic Acid)
Pathophysiology—Vitamin B12 is reabsorbed in the terminal ileum, the distal portion of the small intestine. The American Gastroenterological Association recommends that patients with a history of extensive ileal disease or prior ileal surgery, which is the case for many patients with Crohn disease, be monitored for vitamin B12 deficiency.23 Monitoring and rapid supplementation of vitamin B12 can prevent pernicious anemia and irreversible neurologic damage that may result from deficiency.84
Folic acid is primarily absorbed in the duodenum and jejunum of the small intestine. A meta-analysis performed in 2017 assessed studies observing folic acid and vitamin B12 levels in 1086 patients with IBD compared with 1484 healthy controls and found an average difference in serum folate concentration of 0.46 nmol/L (P<.001).84 Interestingly, this study did not find a significant difference in serum vitamin B12 levels between patients with IBD and healthy controls, highlighting the mechanism of vitamin B12 deficiency in IBD because only patients with terminal ileal involvement are at risk for malabsorption and subsequent deficiency.
Cutaneous Manifestations—Both vitamin B12 and folic acid deficiency can manifest as cheilitis, glossitis, and/or generalized hyperpigmentation that is accentuated in the flexural areas, palms, soles, and oral cavity.85,86 Systemic symptoms of patients with vitamin B12 and folic acid deficiency include megaloblastic anemia, pallor, and fatigue. A potential mechanism for the hyperpigmentation observed from vitamin B12 deficiency came from an electron microscope study that showed an increased concentration of melanosomes in a patient with deficiency.87
Diagnosis and Monitoring—In patients with suspected vitamin B12 and/or folic acid deficiency, initial evaluation should include a CBC with peripheral smear and serum vitamin B12 and folate levels. In cases for which the diagnosis still is unclear after initial testing,
Treatment—According to the Centers for Disease Control and Prevention, supplementation of vitamin B12 can be done orally with 1000 µg daily in patients with deficiency. In patients with active IBD, oral reabsorption of vitamin B12 can be less effective, making subcutaneous or intramuscular administration (1000 µg/wk for 8 weeks, then monthly for life) better options.89
Patients with IBD managed with methotrexate should be screened carefully for folate deficiency. Methotrexate is a folate analog that sometimes is used for the treatment of IBD. Reversible competitive inhibition of dihydrofolate reductase can precipitate a systemic folic acid decrease.91 Typically, oral folic acid (1 to 5 mg/d) is sufficient to treat folate deficiency, with the ESPEN recommending 5 mg once weekly 24 to 72 hours after methotrexate treatment or 1 mg daily for 5 days per week in patients with IBD.1 Alternative formulations—IV, subcutaneous, or intramuscular—are available for patients who cannot tolerate oral intake.92
Final Thoughts
Dermatologists can be the first to observe the cutaneous manifestations of micronutrient deficiencies. Although the symptoms of each micronutrient deficiency discussed may overlap, attention to small clinical clues in patients with IBD can improve patient outcomes and quality of life. For example, koilonychia with glossitis and xerosis likely is due to iron deficiency, while zinc deficiency should be suspected in patients with scaly eczematous plaques in skin folds. A high level of suspicion for micronutrient deficiencies in patients with IBD should be followed by a complete patient history, review of systems, and thorough clinical examination. A thorough laboratory evaluation can pinpoint nutritional deficiencies in patients with IBD, keeping in mind that specific biomarkers such as ferritin and serum zinc also act as acute phase reactants and should be interpreted in this context. Co-management with gastroenterologists should be a priority in patients with IBD, as gaining control of inflammatory disease is crucial for the prevention of recurrent vitamin and micronutrient deficiencies in addition to long-term health in this population.
In 2023, ESPEN (the European Society for Clinical Nutrition and Metabolism) published consensus recommendations highlighting the importance of regular monitoring and treatment of nutrient deficiencies in patients with inflammatory bowel disease (IBD) for improved prognosis, mortality, and quality of life.1 Suboptimal nutrition in patients with IBD predominantly results from inflammation of the gastrointestinal (GI) tract leading to malabsorption; however, medications commonly used to manage IBD also can contribute to malnutrition.2,3 Additionally, patients may develop nausea and food avoidance due to medication or the disease itself, leading to nutritional withdrawal and eventual deficiency.4 Even with the development of diets focused on balancing nutritional needs and decreasing inflammation,5 offsetting this aversion to food can be difficult to overcome.2
Cutaneous manifestations of IBD are multifaceted and can be secondary to the disease, reactive to or associated with IBD, or effects from nutritional deficiencies. The most common vitamin and nutrient deficiencies in patients with IBD include iron; zinc; calcium; vitamin D; and vitamins B6 (pyridoxine), B9 (folic acid), and B12.6 Malnutrition may manifest with cutaneous disease, and dermatologists can be the first to identify and assess for nutritional deficiencies. In this article, we review the mechanisms of these micronutrient depletions in the context of IBD, their subsequent dermatologic manifestations (Table), and treatment and monitoring guidelines for each deficiency.
Iron
A systematic review conducted from 2007 to 2012 in European patients with IBD (N=2192) found the overall prevalence of anemia in this population to be 24% (95% CI, 18%-31%), with 57% of patients with anemia experiencing iron deficiency.7 Anemia is observed more commonly in patients hospitalized with IBD and is common in patients with both Crohn disease and ulcerative colitis.8
Pathophysiology—Iron is critically important in oxygen transportation throughout the body as a major component of hemoglobin. Physiologically, the low pH of the duodenum and proximal jejunum allows divalent metal transporter 1 to transfer dietary Fe3+ into enterocytes, where it is reduced to the transportable Fe2+.9,10 Distribution of Fe2+ ions from enterocytes relies on ferroportin, an iron-transporting protein, which is heavily regulated by the protein hepcidin.11 Hepcidin, a known acute phase reactant, will increase in the setting of active IBD, causing a depletion of ferroportin and an inability of the body to utilize the stored iron in enterocytes.12 This poor utilization of iron stores combined with blood loss caused by inflammation in the GI tract is the proposed primary mechanism of iron-deficiency anemia observed in patients with IBD.13
Cutaneous Manifestations—From a dermatologic perspective, iron-deficiency anemia can manifest with a wide range of symptoms including glossitis, koilonychia, xerosis and/or pruritus, and brittle hair or hair loss.14,15 Although the underlying pathophysiology of these cutaneous manifestations is not fully understood, there are several theories assessing the mechanisms behind the skin findings of iron deficiency.
Atrophic glossitis has been observed in many patients with iron deficiency and is thought to manifest due to low iron concentrations in the blood, thereby decreasing oxygen delivery to the papillae of the dorsal tongue with resultant atrophy.16,17 Similarly, decreased oxygen delivery to the nail bed capillaries may cause deformities in the nail called koilonychia (or “spoon nails”).18 Iron is a key co-factor in collagen lysyl hydroxylase that promotes collagen binding; iron deficiency may lead to disruptions in the epidermal barrier that can cause pruritus and xerosis.19 An observational study of 200 healthy patients with a primary concern of pruritus found a correlation between low serum ferritin and a higher degree of pruritus (r=−0.768; P<.00001).20
Evidence for iron’s role in hair growth comes from a mouse model study with a mutation in the serine protease TMPRSS6—a protein that regulates hepcidin and iron absorption—which caused an increase in hepcidin production and subsequent systemic iron deficiency. Mice at 4 weeks of age were devoid of all body hair but had substantial regrowth after initiation of a 2-week iron-rich diet, which suggests a connection between iron repletion and hair growth in mice with iron deficiency.21 Additionally, a meta-analysis analyzing the comorbidities of patients with alopecia areata found them to have higher odds (odds ratio [OR]=2.78; 95% CI, 1.23-6.29) of iron-deficiency anemia but no association with IBD (OR=1.48; 95% CI, 0.32-6.82).22
Diagnosis and Monitoring—The American Gastroenterological Association recommends a complete blood cell count (CBC), serum ferritin, transferrin saturation (TfS), and C-reactive protein (CRP) as standard evaluations for iron deficiency in patients with IBD. Patients with active IBD should be screened every 3 months,and patients with inactive disease should be screened every 6 to 12 months.23
Although ferritin and TfS often are used as markers for iron status in healthy individuals, they are positive and negative acute phase reactants, respectively. Using them to assess iron status in patients with IBD may inaccurately represent iron status in the setting of inflammation from the disease.24 The European Crohn’s and Colitis Organisation (ECCO) produced guidelines to define iron deficiency as a TfS less than 20% or a ferritin level less than 30 µg/L in patients without evidence of active IBD and a ferritin level less than 100 µg/L for patients with active inflammation.25
A 2020 multicenter observational study of 202 patients with diagnosed IBD found that the ECCO guideline of ferritin less than 30 µg/L had an area under the receiver operating characteristic (AUROC) curve of 0.69, a sensitivity of 0.43, and a specificity of 0.95 in their population.26 In a sensitivity analysis stratifying patients by CRP level (<10 or ≥10 mg/L), the authors found that for patients with ulcerative colitis and a CRP less than 10 mg/L, a cut-off value of ferritin less than 65 µg/L (AUROC=0.78) had a sensitivity of 0.78 and specificity of 0.76, and a TfS value of less than 16% (AUROC=0.88) had a sensitivity of 0.79 and a specificity of 0.9. In patients with a CRP of 10 mg/L or greater, a cut-off value of ferritin 80 µg/L (AUROC=0.76) had a sensitivity of 0.75 and a specificity of 0.82, and a TfS value of less than 11% (AUROC=0.69) had a sensitivity of 0.79 and a specificity of 0.88. There were no ferritin cut-off values associated with good diagnostic performance (defined as both sensitivity and specificity >0.70) for iron deficiency in patients with Crohn disease.26
The authors recommended using an alternative iron measurement such as soluble transferrin receptor (sTfR)/log ferritin ratio (TfR-F) that is not influenced by active inflammation and has a good correlation with ferritin values (TfR-F: r=0.66; P<.001).26 However, both sTfR and TfR-F have high costs and intermethod variability as well as differences in their reference ranges depending on which laboratory performs the analysis, limiting the accessibility and practicality of easily obtaining these tests.27 Although there may be inaccuracies for standard ferritin or TfS under ECCO guidelines, proposed alternatives have their own limitations, which may make ferritin and TfS the most reasonable evaluations of iron status as long as disease activity status at the time of testing is taken into consideration.
Treatment—Treatment of underlying iron deficiency in patients with IBD requires reversing the cause of the deficiency and supplementing iron. In patients with IBD, the options to supplement iron may be limited by active disease, making oral intake less effective. Oral iron supplementation also is associated with notable GI adverse effects that may be exacerbated in patients with IBD. A systematic review of 43 randomized controlled trials (RCTs) evaluating GI adverse effects (eg, nausea, abdominal pain, diarrhea, constipation, and black or tarry stools) of oral ferrous sulfate compared with placebo or intravenous (IV) iron supplementation in healthy nonanemic individuals found a significant increase in GI adverse effects with oral supplementation (placebo: OR=2.32; P<.0001; IV: OR=3.05; P<.0001).28
Therefore, IV iron repletion may be necessary in patients with IBD and may require numerous infusions depending on the formulation of iron. In an RCT conducted in 2011, patients with iron-deficiency anemia with quiescent or mild to moderate IBD were treated with either IV iron sulfate or ferric carboxymaltose.29 With a primary end point of hemoglobin response greater than 2 g/dL, the authors found that 150 of 240 patients responded to ferric carboxymaltose vs 118 of 235 treated with iron sulfate (P=.004). The dosing for ferric carboxymaltose was 1 to 3 infusions of 500 to 1000 mg of iron and for iron sulfate up to 11 infusions of 200 mg of iron.29
Zinc
A systematic review of zinc deficiency in patients with IBD identified 7 studies including 2413 patients and revealed those with Crohn disease had a higher prevalence of zinc deficiency compared with patients with ulcerative colitis (54% vs 41%).30
Pathophysiology—Zinc serves as a catalytic cofactor for enzymatic activity within proteins and immune cells.31 The homeostasis of zinc is tightly regulated within the brush border of the small intestine by zinc transporters ZIP4 and ZIP1 from the lumen of enterocytes into the bloodstream.32 Inflammation in the small intestine due to Crohn disease can result in zinc malabsorption.
Ranaldi et al33 exposed intestinal cells and zinc-depleted intestinal cells to tumor necrosis factor α media to simulate an inflammatory environment. They measured transepithelial electrical resistance as a surrogate for transmembrane permeability and found that zinc-depleted cells had a statistically significantly higher transepithelial electrical resistance percentage (60% reduction after 4 hours; P<1.10–6) when exposed to tumor necrosis factor α signaling compared with normal intestinal cells. They concluded that zinc deficiency can increase intestinal permeability in the presence of inflammation, creating a cycle of further nutrient malabsorption and inflammation exacerbating IBD symptoms.33
Cutaneous Manifestations—After absorption in the small intestine, approximately 5% of zinc resides in the skin, with the highest concentration in the stratum spinosum.34 A cell study found that keratinocytes in zinc-deficient environments had higher rates of apoptosis compared with cells in normal media. The authors proposed that this higher rate of apoptosis and the resulting inflammation could be a mechanism for developing the desquamative or eczematous scaly plaques that are common cutaneous manifestations of zinc deficiency.35
Other cutaneous findings may include angular cheilitis, stomatitis, glossitis, paronychia, onychodystrophy, generalized alopecia, and delayed wound healing.36 The histopathology of these skin lesions is characterized by granular layer loss, epidermal pallor, confluent parakeratosis, spongiosis, dyskeratosis, and psoriasiform hyperplasia.37
Diagnosis and Monitoring—Assessing serum zinc levels is challenging, as they may decrease during states of inflammation.38 A mouse model study showed a 3.1-fold increase (P<.001) in ZIP14 expression in wild-type mice compared with an IL-6 -/- knock-down model after IL-6 exposure. The authors concluded that the upregulation of ZIP14 in the liver due to inflammatory cytokine upregulation decreases zinc availability in serum.39 Additionally, serum zinc can overestimate the level of deficiency in IBD because approximately 75% of serum zinc is bound to albumin, which decreases in the setting of inflammation.40-42
Alternatively, alkaline phosphatase (AP), a zinc-dependent metalloenzyme, may be a better evaluator of zinc status during periods of inflammation. A study in rats evaluated zinc through serum zinc levels and AP levels after a period of induced stress to mimic a short-term inflammatory state.43 The researchers found that total body stores of zinc were unaffected throughout the experiment; only serum zinc declined throughout the experiment duration while AP did not. Because approximately 75% of serum zinc is bound to serum albumin,42 the researchers concluded the induced inflammatory state depleted serum albumin and redistributed zinc to the liver, causing the observed serum zinc changes, while total body zinc levels and AP were largely unaffected in comparison.43 Comorbid conditions such as liver or bone disease can increase AP levels, which limits the utility of AP as a surrogate for zinc in patients with comorbidities.44 However, even in the context of active IBD, serum zinc still is currently considered the best biomarker to evaluate zinc status.45
Treatment—The recommended dose for zinc supplementation is 20 to 40 mg daily with higher doses (>50 mg/d) for patients with malabsorptive syndromes such as IBD.46 It can be administered orally or parenterally. Although rare, zinc replacement therapy may be associated with diarrhea, nausea, vomiting, mild headaches, and fatigue.46 Additional considerations should be taken when repleting other micronutrients with zinc, as calcium and folate can inhibit zinc reabsorption, while zinc itself can inhibit iron and copper reabsorption.47
Vitamin D and Calcium
Low vitamin D levels (<50 nmol/L) and hypocalcemia (<8.8 mg/dL) are common in patients with IBD.48,49
Pathophysiology—Vitamin D levels are maintained via 2 mechanisms. The first mechanism is through the skin, as keratinocytes produce 7-dehydrocholesterol after exposure to UV light, which is converted into previtamin D3 and then thermally isomerizes into vitamin D3. This vitamin D3 is then transported to the liver on vitamin D–binding protein.50 The second mechanism is through oral vitamin D3 that is absorbed through vitamin D receptors in intestinal epithelium and transported to the liver, where it is hydroxylated into 25-hydroxyvitamin D (25[OH]D), then to the kidneys for hydroxylation to 1,25(OH)2D for redistribution throughout the body.50 This activated form of vitamin D regulates calcium absorption in the intestine, and optimal vitamin D levels are necessary to absorb calcium efficiently.51 Inflammation from IBD within the small intestine can downregulate vitamin D receptors, causing malabsorption and decreased serum vitamin D.52
Vitamin D signaling also is vital to maintaining the tight junctions and adherens junctions of the intestinal epithelium. Weakening the permeability of the epithelium further exacerbates malabsorption and subsequent vitamin D deficiency.52 A meta-analysis of 27 studies including 8316 patients with IBD showed low vitamin D levels were associated with increased odds of disease activity (OR=1.53; 95% CI, 1.32-1.77), mucosal inflammation (OR=1.25; 95% CI, 1.06-1.47), and future clinical relapse (OR=1.23; 95% CI, 1.03-1.47) in patients with Crohn disease. The authors concluded that low levels of vitamin D could be used as a potential biomarker of inflammatory status in Crohn disease.53
Vitamin D and calcium are further implicated in maintaining skeletal health,47 while vitamin D specifically helps maintain intestinal homeostasis54 and immune system modulation in the skin.55
Cutaneous Manifestations—Vitamin D is thought to play crucial roles in skin differentiation and proliferation, cutaneous innate immunity, hair follicle cycling, photoprotection, and wound healing.56 Vitamin D deficiency has been observed in a large range of cutaneous diseases including skin cancer, psoriasis, vitiligo, bullous pemphigoid, atopic dermatitis, and various types of alopecia.56-59 It is unclear whether vitamin D deficiency facilitates these disease processes or is merely the consequence of a disrupted cutaneous surface with the inability to complete the first step in vitamin D processing. A 2014 meta-analysis of 290 prospective cohort studies and 172 randomized trials concluded that 25(OH)D deficiency was associated with ill health and did not find causal evidence for any specific disease, dermatologic or otherwise.60 Calcium deficiency may cause epidermal changes including dry skin, coarse hair, and brittle nails.61
Diagnosis and Monitoring—The ECCO guidelines recommend obtaining serum 25(OH)D levels every 3 months in patients with IBD.62 Levels less than 75 nmol/L are considered deficient, and a value less than 30 nmol/L increases the risk for osteomalacia and nutritional rickets, constituting severe vitamin D deficiency.63-65
An observational study of 325 patients with IBD showed a statistically significant negative correlation between serum vitamin D and fecal calprotectin (r=−0.19; P<.001), a stool-based marker for gut inflammation, supporting vitamin D as a potential biomarker in IBD.66
Evaluation of calcium can be done through serum levels in patients with IBD.67 Patients with IBD are at risk for hypoalbuminemia; therefore, consideration should be taken to ensure calcium levels are corrected, as approximately 50% of calcium is bound to albumin or other ions in the body,68 which can be done by adjusting the calcium concentration by 0.02 mmol/L for every 1 g/L of albumin above or below 40 g/L. In the most critically ill patients, a direct ionized calcium blood level should be used instead because the previously mentioned correction calculations are inaccurate when albumin is critically low.69
Treatment—The ECCO guidelines recommend calcium and vitamin D repletion of 500 to 1000 mg and 800 to 1000 U, respectively, in patients with IBD on systemic corticosteroids to prevent the negative effects of bone loss.62 Calcium repletion in patients with IBD who are not on systemic steroids are the same as for the general population.65
Vitamin D repletion also may help decrease IBD activity. In a prospective study, 10,000 IU/d of vitamin D in 10 patients with IBD—adjusted over 12 weeks to a target of 100 to 125 nmol/L of serum 25(OH)D—showed a significant reduction in clinical Crohn activity (P=.019) over the study period.70 In contrast, 2000 IU/d for 3 months in an RCT of 27 patients with Crohn disease found significantly lower CRP (P=.019) and significantly higher self-reported quality of life (P=.037) but nonsignificant decreases in Crohn activity (P=.082) in patients with 25(OH)D levels of 75 nmol/L or higher compared with those with 25(OH)D levels less than 75 nmol/L.71
These discrepancies illustrate the need for expanded clinical trials to elucidate the optimal vitamin D dosing for patients with IBD. Ultimately, assessing vitamin D and calcium status and considering repletion in patients with IBD, especially those with comorbid dermatologic diseases such as poor wound healing, psoriasis, or atopic dermatitis, is important.
Vitamin B6 (Pyridoxine)
Pathophysiology—Pyridoxine is an important coenzyme for many functions including amino acid transamination, fatty acid metabolism, and conversion of tryptophan to niacin. It is absorbed in the jejunum and ileum and subsequently transported to the liver for rephosphorylation and release into its active form.36 An observational study assessing the nutritional status of patients with IBD found that only 5.7% of 105 patients with food records had inadequate dietary intake of pyridoxine, but 29% of all patients with IBD had subnormal pyridoxine levels.72 Additionally, they found no significant difference in the prevalence of subnormal pyridoxine levels in patients with active IBD vs IBD in remission. The authors suggested that the subnormal pyridoxine levels in patients with IBD likely were multifactorial and resulted from malabsorption due to active disease, inflammation, and inadequate intake.72
Cutaneous Manifestations—Cutaneous findings associated with pyridoxine deficiency include periorificial and perineal dermatitis,73 angular stomatitis, and cheilitis with associated burning, redness, and tongue edema.36 Additionally, pyridoxine is involved in the conversion of tryptophan to niacin, and its deficiency may manifest with pellagralike findings.74
Because pyridoxine is critical to protein metabolism, its deficiency may disrupt key cellular structures that rely on protein concentrations to maintain structural integrity. One such structure in the skin that heavily relies on protein concentrations is the ground substance of the extracellular matrix—the amorphous gelatinous spaces that occupy the areas between the extracellular matrix, which consists of cross-linked glycosaminoglycans and proteins.75 Without protein, ground substance increases in viscosity and can disrupt the epidermal barrier, leading to increased transepidermal water loss and ultimately inflammation.76 Although this theory has yet to be validated fully, this is a potential mechanistic explanation for the inflammation in dermal papillae that leads to dermatitis observed in pyridoxine deficiency.
Diagnosis and Monitoring—Direct biomarkers of pyridoxine status are in serum, plasma, erythrocytes, and urine, with the most common measurement in plasma as pyridoxal 5′-phosphate (PLP).77 Plasma PLP concentrations lower than 20 nmol/L are suggestive of deficiency.78 Plasma PLP has shown inverse relationships with acute phase inflammatory markers CRP79 and AP,78 thereby raising concerns for its validity to assess pyridoxine status in patients with symptomatic IBD.80
Alternative evaluations of pyridoxine include tryptophan and methionine loading tests,36 which are measured via urinary excretion and require normal kidney function to be accurate. They should be considered in IBD if necessary, but routine testing, even in patients with symptomatic IBD, is not recommended in the ECCO guidelines. Additional considerations should be taken in patients with altered nutrient requirements such as those who have undergone bowel resection due to highly active disease or those who receive parenteral nutritional supplementation.81
Treatment—Recommendations for oral pyridoxine supplementation range from 25 to 600 mg daily,82 with symptoms typically improving on 100 mg daily.36 Pyridoxine supplementation may have additional benefits for patients with IBD and potentially modulate disease severity. An IL-10 knockout mouse supplemented with pyridoxine had an approximately 60% reduction (P<.05) in inflammation compared to mice deficient in pyridoxine.83 The authors suggest that PLP-dependent enzymes can inhibit further proinflammatory signaling and T-cell migration that can exacerbate IBD. Ultimately, more data is needed before determining the efficacy of pyridoxine supplementation for active IBD.
Vitamin B12 and Vitamin B9 (Folic Acid)
Pathophysiology—Vitamin B12 is reabsorbed in the terminal ileum, the distal portion of the small intestine. The American Gastroenterological Association recommends that patients with a history of extensive ileal disease or prior ileal surgery, which is the case for many patients with Crohn disease, be monitored for vitamin B12 deficiency.23 Monitoring and rapid supplementation of vitamin B12 can prevent pernicious anemia and irreversible neurologic damage that may result from deficiency.84
Folic acid is primarily absorbed in the duodenum and jejunum of the small intestine. A meta-analysis performed in 2017 assessed studies observing folic acid and vitamin B12 levels in 1086 patients with IBD compared with 1484 healthy controls and found an average difference in serum folate concentration of 0.46 nmol/L (P<.001).84 Interestingly, this study did not find a significant difference in serum vitamin B12 levels between patients with IBD and healthy controls, highlighting the mechanism of vitamin B12 deficiency in IBD because only patients with terminal ileal involvement are at risk for malabsorption and subsequent deficiency.
Cutaneous Manifestations—Both vitamin B12 and folic acid deficiency can manifest as cheilitis, glossitis, and/or generalized hyperpigmentation that is accentuated in the flexural areas, palms, soles, and oral cavity.85,86 Systemic symptoms of patients with vitamin B12 and folic acid deficiency include megaloblastic anemia, pallor, and fatigue. A potential mechanism for the hyperpigmentation observed from vitamin B12 deficiency came from an electron microscope study that showed an increased concentration of melanosomes in a patient with deficiency.87
Diagnosis and Monitoring—In patients with suspected vitamin B12 and/or folic acid deficiency, initial evaluation should include a CBC with peripheral smear and serum vitamin B12 and folate levels. In cases for which the diagnosis still is unclear after initial testing,
Treatment—According to the Centers for Disease Control and Prevention, supplementation of vitamin B12 can be done orally with 1000 µg daily in patients with deficiency. In patients with active IBD, oral reabsorption of vitamin B12 can be less effective, making subcutaneous or intramuscular administration (1000 µg/wk for 8 weeks, then monthly for life) better options.89
Patients with IBD managed with methotrexate should be screened carefully for folate deficiency. Methotrexate is a folate analog that sometimes is used for the treatment of IBD. Reversible competitive inhibition of dihydrofolate reductase can precipitate a systemic folic acid decrease.91 Typically, oral folic acid (1 to 5 mg/d) is sufficient to treat folate deficiency, with the ESPEN recommending 5 mg once weekly 24 to 72 hours after methotrexate treatment or 1 mg daily for 5 days per week in patients with IBD.1 Alternative formulations—IV, subcutaneous, or intramuscular—are available for patients who cannot tolerate oral intake.92
Final Thoughts
Dermatologists can be the first to observe the cutaneous manifestations of micronutrient deficiencies. Although the symptoms of each micronutrient deficiency discussed may overlap, attention to small clinical clues in patients with IBD can improve patient outcomes and quality of life. For example, koilonychia with glossitis and xerosis likely is due to iron deficiency, while zinc deficiency should be suspected in patients with scaly eczematous plaques in skin folds. A high level of suspicion for micronutrient deficiencies in patients with IBD should be followed by a complete patient history, review of systems, and thorough clinical examination. A thorough laboratory evaluation can pinpoint nutritional deficiencies in patients with IBD, keeping in mind that specific biomarkers such as ferritin and serum zinc also act as acute phase reactants and should be interpreted in this context. Co-management with gastroenterologists should be a priority in patients with IBD, as gaining control of inflammatory disease is crucial for the prevention of recurrent vitamin and micronutrient deficiencies in addition to long-term health in this population.
- Bischoff SC, Bager P, Escher J, et al. ESPEN guideline on clinical nutrition in inflammatory bowel disease. Clin Nutr. 2023;42:352-379. doi:10.1016/j.clnu.2022.12.004
- Gerasimidis K, McGrogan P, Edwards CA. The aetiology and impact of malnutrition in paediatric inflammator y bowel disease. J Hum Nutr Diet. 2011;24:313-326. doi:10.1111/j.1365-277X.2011.01171.x
- Mentella MC, Scaldaferri F, Pizzoferrato M, et al. Nutrition, IBD and gut microbiota: a review. Nutrients. 2020;12:944. doi:10.3390/nu12040944
- Bonsack O, Caron B, Baumann C, et al. Food avoidance and fasting in patients with inflammatory bowel disease: experience from the Nancy IBD nutrition clinic. United European Gastroenterol J. 2023;11:361-370. doi:10.1002/ueg2.1238521
- Campmans-Kuijpers MJE, Dijkstra G. Food and food groups in inflammatory bowel disease (IBD): the design of the Groningen Anti-Inflammatory Diet (GrAID). Nutrients. 2021;13:1067. doi:10.3390/nu13041067
- Hwang C, Issokson K, Giguere-Rich C, et al. Development and pilot testing of the inflammatory bowel disease nutrition care pathway. Clin Gastroenterol Hepatol. 2020;18:2645-2649.e4. doi:10.1016/j.cgh.2020.06.039
- Filmann N, Rey J, Schneeweiss S, et al. Prevalence of anemia in inflammatory bowel diseases in European countries: a systematic review and individual patient data meta-analysis. Inflamm Bowel Dis. 2014;20:936-945. doi:10.1097/01.MIB.0000442728.74340.fd
- Stein J, Hartmann F, Dignass AU. Diagnosis and management of iron deficiency anemia in patients with IBD. Nat Rev Gastroenterol Hepatol. 2010;7:599-610. doi:10.1038/nrgastro.2010.151
- Ems T, St Lucia K, Huecker MR. Biochemistry, iron absorption. StatPearls [Internet]. Updated April 17, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK448204/
- Evstatiev R, Gasche C. Iron sensing and signalling. Gut. 2012;61:933-952. doi:10.1136/gut.2010.214312
- Przybyszewska J, Zekanowska E. The role of hepcidin, ferroportin, HCP1, and DMT1 protein in iron absorption in the human digestive tract. Prz Gastroenterol. 2014;9:208-213. doi:10.5114/pg.2014.45102
- Weiss G, Gasche C. Pathogenesis and treatment of anemia in inflammatory bowel disease. Haematologica. 2010;95:175-178. doi:10.3324/haematol.2009.017046
- Kaitha S, Bashir M, Ali T. Iron deficiency anemia in inflammatory bowel disease. World J Gastrointest Pathophysiol. 2015;6:62-72. doi:10.4291/wjgp.v6.i3.62
- Moiz B. Spoon nails: still seen in today’s world. Clin Case Rep. 2018;6:547-548. doi:10.1002/ccr3.1404
- St Pierre SA, Vercellotti GM, Donovan JC, et al. Iron deficiency and diffuse nonscarring scalp alopecia in women: more pieces to the puzzle. J Am Acad Dermatol. 2010;63:1070-1076. doi:10.1016/j.jaad.2009.05.054
- Chiang CP, Yu-Fong Chang J, Wang YP, et al. Anemia, hematinic deficiencies, hyperhomocysteinemia, and serum gastric parietal cell antibody positivity in atrophic glossitis patients with or without microcytosis. J Formos Med Assoc. 2019;118:1401-1407. doi:10.1016/j.jfma.2019.06.004
- Chiang CP, Chang JY, Wang YP, et al. Atrophic glossitis: Etiology, serum autoantibodies, anemia, hematinic deficiencies, hyperhomocysteinemia, and management. J Formos Med Assoc. 2020;119:774-780. doi:10.1016/j.jfma.2019.04.015
- Walker J, Baran R, Vélez N, et al. Koilonychia: an update on pathophysiology, differential diagnosis and clinical relevance. J Eur Acad Dermatol Venereol. 2016;30:1985-1991. doi:10.1111/jdv.13610
- Guo HF, Tsai CL, Terajima M, et al. Pro-metastatic collagen lysyl hydroxylase dimer assemblies stabilized by Fe2+-binding. Nat Commun. 2018;9:512. doi:10.1038/s41467-018-02859-z
- Saini S, Jain AK, Agarwal S, et al. Iron deficiency and pruritus: a cross-sectional analysis to assess its association and relationship. Indian J Dermatol. 2021;66:705. doi:10.4103/ijd.ijd_326_21
- Du X, She E, Gelbart T, et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science. 2008;320:1088-1092. doi:10.1126/science.1157121
- Lee S, Lee H, Lee CH, et al. Comorbidities in alopecia areata: a systematic review and meta-analysis. J Am Acad Dermatol. 2019;80:466-477.e16. doi:10.1016/j.jaad.2018.07.013
- Hashash JG, Elkins J, Lewis JD, et al. AGA Clinical Practice Update on diet and nutritional therapies in patients with inflammatory bowel disease: expert review [published online January 23, 2024]. Gastroenterology. doi:10.1053/j.gastro.2023.11.303
- Choudhuri S, Chowdhury IH, Saha A, et al. Acute monocyte pro- inflammatory response predicts higher positive to negative acute phase reactants ratio and severe hemostatic derangement in dengue fever. Cytokine. 2021;146:155644. doi:10.1016/j.cyto.2021.155644
- Dignass AU, Gasche C, Bettenworth D, et al; European Crohn’s and Colitis Organisation. European consensus on the diagnosis and management of iron deficiency and anaemia in inflammatory bowel diseases. J Crohn’s Colitis. 2015;9:211-222. doi:10.1093/ecco-jcc/jju009
- Daude S, Remen T, Chateau T, et al. Comparative accuracy of ferritin, transferrin saturation and soluble transferrin receptor for the diagnosis of iron deficiency in inflammatory bowel disease. Aliment Pharmacol Ther. 2020;51:1087-1095. doi:10.1111/apt.15739
- Pfeiffer CM, Looker AC. Laboratory methodologies for indicators of iron status: strengths, limitations, and analytical challenges. Am J Clin Nutr. 2017;106(suppl 6):1606S-1614S. doi:10.3945/ajcn.117.155887
- Tolkien Z, Stecher L, Mander AP, et al. Ferrous sulfate supplementation causes significant gastrointestinal side-effects in adults: a systematic review and meta-analysis. PLoS One. 2015;10:e0117383. doi:10.1371/journal.pone.0117383
- Evstatiev R, Marteau P, Iqbal T, et al. FERGIcor, a randomized controlled trial on ferric carboxymaltose for iron deficiency anemia in inflammatory bowel disease. Gastroenterology. 2011;141:846-853.e8532. doi:10.1053/j.gastro.2011.06.005
- Zupo R, Sila A, Castellana F, et al. Prevalence of zinc deficiency in inflammatory bowel disease: a systematic review and meta-analysis. Nutrients. 2022;14:4052. doi:10.3390/nu14194052
- Thompson MW. Regulation of zinc-dependent enzymes by metal carrier proteins. Biometals. 2022;35:187-213. doi:10.1007/s10534-022-00373-w
- Maares M, Haase H. A guide to human zinc absorption: general overview and recent advances of in vitro intestinal models. Nutrients. 2020;12:762. doi:10.3390/nu12030762
- Ranaldi G, Ferruzza S, Canali R, et al. Intracellular zinc is required for intestinal cell survival signals triggered by the inflammatory cytokine TNFα. J Nutr Biochem. 2013;24:967-976. doi:10.1016/j.jnutbio.2012.06.020
- Ogawa Y, Kawamura T, Shimada S. Zinc and skin biology. Arch Biochem Biophys. 2016;611:113-119. doi:10.1016/j.abb.2016.06.003
- Wilson D, Varigos G, Ackland ML. Apoptosis may underlie the pathology of zinc-deficient skin. Immunol Cell Biol. 2006;84:28-37. doi:10.1111/j.1440-1711.2005.01391.x
- Jen M, Yan AC. Syndromes associated with nutritional deficiency and excess. Clin Dermatol. 2010;28:669-685. doi:10.1016/j.clindermatol.2010.03.029
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Gammoh NZ, Rink L. Zinc in infection and inflammation. Nutrients. 2017;9:624. doi:10.3390/nu9060624
- Liuzzi JP, Lichten LA, Rivera S, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to the hypozincemia of the acute-phase response. Proc Natl Acad Sci U S A. 2005;102:6843-6848. doi:10.1073/pnas.0502257102
- Vermeire S, Van Assche G, Rutgeerts P. Laboratory markers in IBD: useful, magic, or unnecessary toys?. Gut. 2006;55:426-431. doi:10.1136/gut.2005.069476
- Morisaku M, Ito K, Ogiso A, et al. Correlation between serum albumin and serum zinc in malignant lymphoma. Fujita Med J. 2022;8:59-64. doi:10.20407/fmj.2021-006
- Falchuk KH. Effect of acute disease and ACTH on serum zinc proteins. N Engl J Med. 1977:296:1129-1134.
- Naber TH, Baadenhuysen H, Jansen JB, et al. Serum alkaline phosphatase activity during zinc deficiency and long-term inflammatory stress. Clin Chim Acta. 1996;249:109-127. doi:10.1016/0009-8981(96)06281-x
- Lowe D, Sanvictores T, Zubair M, et al. Alkaline phosphatase. StatPearls [Internet]. Updated October 29, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK459201/
- Krebs NF. Update on zinc deficiency and excess in clinical pediatric practice. Ann Nutr Metab. 2013;62 suppl 1:19-29. doi:10.1159/000348261
- Maxfield L, Shukla S, Crane JS. Zinc deficiency. StatPearls [Internet]. Updated June 28, 2023. Accessed March 25, 2024. https://www.ncbi.nlm.nih.gov/books/NBK493231/
- Ghishan FK, Kiela PR. Vitamins and minerals in inflammatory bowel disease. Gastroenterol Clin North Am. 2017;46:797-808. doi:10.1016/j.gtc.2017.08.011
- Caviezel D, Maissen S, Niess JH, et al. High prevalence of vitamin D deficiency among patients with inflammatory bowel disease. Inflamm Intest Dis. 2018;2:200-210. doi:10.1159/000489010
- Jasielska M, Grzybowska-Chlebowczyk U. Hypocalcemia and vitamin D deficiency in children with inflammatory bowel diseases and lactose intolerance. Nutrients. 2021;13:2583. doi:10.3390/nu13082583
- Vernia F, Valvano M, Longo S, et al. Vitamin D in inflammatory bowel diseases. Mechanisms of action and therapeutic implications. Nutrients. 2022;14:269. doi:10.3390/nu14020269
- Khazai N, Judd SE, Tangpricha V. Calcium and vitamin D: skeletal and extraskeletal health. Curr Rheumatol Rep. 2008;10:110-117. doi:10.1007/s11926-008-0020-y
- Domazetovic V, Iantomasi T, Bonanomi AG, et al. Vitamin D regulates claudin-2 and claudin-4 expression in active ulcerative colitis by p-Stat-6 and Smad-7 signaling. Int J Colorectal Dis. 2020;35:1231-1242. doi:10.1007/s00384-020-03576-0
- Gubatan J, Chou ND, Nielsen OH, et al. Systematic review with meta-analysis: association of vitamin D status with clinical outcomes in adult patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2019;50:1146-1158. doi:10.1111/apt.15506
- Fakhoury HMA, Kvietys PR, AlKattan W, et al. Vitamin D and intestinal homeostasis: barrier, microbiota, and immune modulation. J Steroid Biochem Mol Biol. 2020;200:105663. doi:10.1016/j.jsbmb.2020.105663
- Liu PT, Stenger S, Li H, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770-1773. doi:10.1126/science.1123933
- Mostafa WZ, Hegazy RA. Vitamin D and the skin: focus on a complex relationship: a review. J Adv Res. 2015;6:793-804. doi:10.1016/j.jare.2014.01.011
- Searing DA, Leung DY. Vitamin D in atopic dermatitis, asthma and allergic diseases. Immunol Allergy Clin North Am. 2010;30:397-409.
- Lee YH, Song GG. Association between circulating 25-hydroxyvitamin D levels and psoriasis, and correlation with disease severity: a meta-analysis. Clin Exp Dermatol. 2018;43:529-535.
- Adorini L, Penna G. Control of autoimmune diseases by the vitamin D endocrine system. Nat Clin Pract Rheumatol. 2008;4:404-412.
- Autier P, Boniol M, Pizot C, et al. Vitamin D status and ill health: a systematic review. Lancet Diabetes Endocrinol. 2014;2:76-89. doi:10.1016/S2213-8587(13)70165-7
- Schafer AL, Shoback DM. Hypocalcemia: diagnosis and treatment. In: Feingold KR, Anawalt B, Blackman MR, et al, eds. Endotext [Internet]. Updated January 3, 2016. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK279022/
- Magro F, Gionchetti P, Eliakim R, et al. Third European Evidence-based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 1: Definitions, diagnosis, extra-intestinal manifestations, pregnancy, cancer surveillance, surgery, and ileo-anal pouch disorders. J Crohns Colitis. 2017;11:649-670. doi:10.1093/ecco-jcc/jjx008
- Amrein K, Scherkl M, Hoffmann M, et al. Vitamin D deficiency 2.0: an update on the current status worldwide. Eur J Clin Nutr. 2020;74:1498-1513. doi:10.1038/s41430-020-0558-y
- Munns CF, Shaw N, Kiely M, et al. Global consensus recommendations on prevention and management of nutritional rickets. J Clin Endocrinol Metab. 2016;101:394-415. doi:10.1210/jc.2015-2175
- Institute of Medicine (US) Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Ross AC, Taylor CL, Yaktine AL, Del Valle HB, eds. Dietary Reference Intakes for Calcium and Vitamin D. National Academies Press (US); 2011.
- Yeaman F, Nguyen A, Abasszade J, et al. Assessing vitamin D as a biomarker in inflammatory bowel disease. JGH Open. 2023;7:953-958. doi:10.1002/jgh3.13010
- Vernia P, Loizos P, Di Giuseppantonio I, et al S. Dietary calcium intake in patients with inflammatory bowel disease. J Crohns Colitis. 2014;8:312-317. doi:10.1016/j.crohns.2013.09.008
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ. 2008;336:1298-1302. doi:10.1136/bmj.39582.589433.BE
- Kenny CM, Murphy CE, Boyce DS, et al. Things we do for no reason™: calculating a “corrected calcium” level. J Hosp Med. 2021;16:499-501. doi:10.12788/jhm.3619
- Garg M, Rosella O, Rosella G, et al. Evaluation of a 12-week targeted vitamin D supplementation regimen in patients with active inflammatory bowel disease. Clin Nutr. 2018;37:1375-1382. doi:10.1016/j.clnu.2017.06.011
- Raftery T, Martineau AR, Greiller CL, et al. Effects of vitamin D supplementation on intestinal permeability, cathelicidin and disease markers in Crohn’s disease: results from a randomised double-blind placebo-controlled study. United European Gastroenterol J. 2015;3:294-302. doi:10.1177/2050640615572176
- Vagianos K, Bector S, McConnell J, et al. Nutrition assessment of patients with inflammatory bowel disease. JPEN J Parenter Enteral Nutr. 2007;31:311-319. doi:10.1177/0148607107031004311
- Barthelemy H, Chouvet B, Cambazard F. Skin and mucosal manifestations in vitamin deficiency. J Am Acad Dermatol. 1986;15:1263-1274. doi:10.1016/s0190-9622(86)70301-0
- Galimberti F, Mesinkovska NA. Skin findings associated with nutritional deficiencies. Cleve Clin J Med. 2016;83:731-739. doi:10.3949/ccjm.83a.15061
- Elgharably N, Al Abadie M, Al Abadie M, et al. Vitamin B group levels and supplementations in dermatology. Dermatol Reports. 2022;15:9511. doi:10.4081/dr.2022.9511
- Hołubiec P, Leon´czyk M, Staszewski F, et al. Pathophysiology and clinical management of pellagra—a review. Folia Med Cracov. 2021;61:125-137. doi:10.24425/fmc.2021.138956
- Ink SL, Henderson LM. Vitamin B6 metabolism. Annu Rev Nutr. 1984;4:455-470. doi:10.1146/annurev.nu.04.070184.002323
- Brown MJ, Ameer MA, Daley SF, et al. Vitamin B6 deficiency. StatPearls [Internet]. Updated August 8, 2023. Accessed March 25, 2024. https://www.ncbi.nlm.nih.gov/books/NBK470579/.
- Vasilaki AT, McMillan DC, Kinsella J, et al. Relation between pyridoxal and pyridoxal phosphate concentrations in plasma, red cells, and white cells in patients with critical illness. Am J Clin Nutr. 2008;88:140-146. doi:10.1093/ajcn/88.1.140
- Chiang EP, Bagley PJ, Selhub J, et al. Abnormal vitamin B(6) status is associated with severity of symptoms in patients with rheumatoid arthritis. Am J Med. 2003;114:283-287. doi:10.1016/s0002-9343(02)01528-0
- Maaser C, Sturm A, Vavricka SR, et al. ECCO-ESGAR guideline for diagnostic assessment in IBD. Part 1: initial diagnosis, monitoring of known IBD, detection of complications. J Crohns Colitis. 2019;13:144-164. doi:10.1093/ecco-jcc/jjy113
- Spinneker A, Sola R, Lemmen V, et al. Vitamin B6 status, deficiency and its consequences—an overview. Nutr Hosp. 2007;22:7-24.
- Selhub J, Byun A, Liu Z, et al. Dietary vitamin B6 intake modulates colonic inflammation in the IL10-/- model of inflammatory bowel disease. J Nutr Biochem. 2013;24:2138-2143. doi:10.1016/j.jnutbio.2013.08.005
- Pan Y, Liu Y, Guo H, et al. Associations between folate and vitamin B12 levels and inflammatory bowel disease: a meta-analysis. Nutrients. 2017;9:382. doi:10.3390/nu9040382
- Brescoll J, Daveluy S. A review of vitamin B12 in dermatology. Am J Clin Dermatol. 2015;16:27-33. doi:10.1007/s40257-014-0107-3
- DiBaise M, Tarleton SM. Hair, nails, and skin: differentiating cutaneous manifestations of micronutrient deficiency. Nutr Clin Pract. 2019;34:490-503. doi:10.1002/ncp.10321
- Mori K, Ando I, Kukita A. Generalized hyperpigmentation of the skin due to vitamin B12 deficiency. J Dermatol. 2001;28:282-285. doi:10.1111/j.1346-8138.2001.tb00134.x
- Green R. Indicators for assessing folate and vitamin B-12 status and for monitoring the efficacy of intervention strategies. Am J Clin Nutr. 2011;94:666S-672S. doi:10.3945/ajcn.110.009613
- NIH Office of Dietary Supplements. Vitamin B12: fact sheet for health professionals. Updated February 27, 2024. Accessed March 19, 2024. https://ods.od.nih.gov/factsheets/VitaminB12-HealthProfessional/
- NIH Office of Dietary Supplements. Folate: fact sheet for health professionals. Updated November 20, 2023. Accessed March 19, 2024. https://ods.od.nih.gov/factsheets/Folate-HealthProfessional/.
- Saibeni S, Bollani S, Losco A, et al. The use of methotrexate for treatment of inflammatory bowel disease in clinical practice. Dig Liver Dis. 2012;44:123-127. doi:10.1016/j.dld.2011.09.015
- Khan KM, Jialal I. Folic acid deficiency. StatPearls [Internet]. Updated June 26, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK535377/
- Bischoff SC, Bager P, Escher J, et al. ESPEN guideline on clinical nutrition in inflammatory bowel disease. Clin Nutr. 2023;42:352-379. doi:10.1016/j.clnu.2022.12.004
- Gerasimidis K, McGrogan P, Edwards CA. The aetiology and impact of malnutrition in paediatric inflammator y bowel disease. J Hum Nutr Diet. 2011;24:313-326. doi:10.1111/j.1365-277X.2011.01171.x
- Mentella MC, Scaldaferri F, Pizzoferrato M, et al. Nutrition, IBD and gut microbiota: a review. Nutrients. 2020;12:944. doi:10.3390/nu12040944
- Bonsack O, Caron B, Baumann C, et al. Food avoidance and fasting in patients with inflammatory bowel disease: experience from the Nancy IBD nutrition clinic. United European Gastroenterol J. 2023;11:361-370. doi:10.1002/ueg2.1238521
- Campmans-Kuijpers MJE, Dijkstra G. Food and food groups in inflammatory bowel disease (IBD): the design of the Groningen Anti-Inflammatory Diet (GrAID). Nutrients. 2021;13:1067. doi:10.3390/nu13041067
- Hwang C, Issokson K, Giguere-Rich C, et al. Development and pilot testing of the inflammatory bowel disease nutrition care pathway. Clin Gastroenterol Hepatol. 2020;18:2645-2649.e4. doi:10.1016/j.cgh.2020.06.039
- Filmann N, Rey J, Schneeweiss S, et al. Prevalence of anemia in inflammatory bowel diseases in European countries: a systematic review and individual patient data meta-analysis. Inflamm Bowel Dis. 2014;20:936-945. doi:10.1097/01.MIB.0000442728.74340.fd
- Stein J, Hartmann F, Dignass AU. Diagnosis and management of iron deficiency anemia in patients with IBD. Nat Rev Gastroenterol Hepatol. 2010;7:599-610. doi:10.1038/nrgastro.2010.151
- Ems T, St Lucia K, Huecker MR. Biochemistry, iron absorption. StatPearls [Internet]. Updated April 17, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK448204/
- Evstatiev R, Gasche C. Iron sensing and signalling. Gut. 2012;61:933-952. doi:10.1136/gut.2010.214312
- Przybyszewska J, Zekanowska E. The role of hepcidin, ferroportin, HCP1, and DMT1 protein in iron absorption in the human digestive tract. Prz Gastroenterol. 2014;9:208-213. doi:10.5114/pg.2014.45102
- Weiss G, Gasche C. Pathogenesis and treatment of anemia in inflammatory bowel disease. Haematologica. 2010;95:175-178. doi:10.3324/haematol.2009.017046
- Kaitha S, Bashir M, Ali T. Iron deficiency anemia in inflammatory bowel disease. World J Gastrointest Pathophysiol. 2015;6:62-72. doi:10.4291/wjgp.v6.i3.62
- Moiz B. Spoon nails: still seen in today’s world. Clin Case Rep. 2018;6:547-548. doi:10.1002/ccr3.1404
- St Pierre SA, Vercellotti GM, Donovan JC, et al. Iron deficiency and diffuse nonscarring scalp alopecia in women: more pieces to the puzzle. J Am Acad Dermatol. 2010;63:1070-1076. doi:10.1016/j.jaad.2009.05.054
- Chiang CP, Yu-Fong Chang J, Wang YP, et al. Anemia, hematinic deficiencies, hyperhomocysteinemia, and serum gastric parietal cell antibody positivity in atrophic glossitis patients with or without microcytosis. J Formos Med Assoc. 2019;118:1401-1407. doi:10.1016/j.jfma.2019.06.004
- Chiang CP, Chang JY, Wang YP, et al. Atrophic glossitis: Etiology, serum autoantibodies, anemia, hematinic deficiencies, hyperhomocysteinemia, and management. J Formos Med Assoc. 2020;119:774-780. doi:10.1016/j.jfma.2019.04.015
- Walker J, Baran R, Vélez N, et al. Koilonychia: an update on pathophysiology, differential diagnosis and clinical relevance. J Eur Acad Dermatol Venereol. 2016;30:1985-1991. doi:10.1111/jdv.13610
- Guo HF, Tsai CL, Terajima M, et al. Pro-metastatic collagen lysyl hydroxylase dimer assemblies stabilized by Fe2+-binding. Nat Commun. 2018;9:512. doi:10.1038/s41467-018-02859-z
- Saini S, Jain AK, Agarwal S, et al. Iron deficiency and pruritus: a cross-sectional analysis to assess its association and relationship. Indian J Dermatol. 2021;66:705. doi:10.4103/ijd.ijd_326_21
- Du X, She E, Gelbart T, et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science. 2008;320:1088-1092. doi:10.1126/science.1157121
- Lee S, Lee H, Lee CH, et al. Comorbidities in alopecia areata: a systematic review and meta-analysis. J Am Acad Dermatol. 2019;80:466-477.e16. doi:10.1016/j.jaad.2018.07.013
- Hashash JG, Elkins J, Lewis JD, et al. AGA Clinical Practice Update on diet and nutritional therapies in patients with inflammatory bowel disease: expert review [published online January 23, 2024]. Gastroenterology. doi:10.1053/j.gastro.2023.11.303
- Choudhuri S, Chowdhury IH, Saha A, et al. Acute monocyte pro- inflammatory response predicts higher positive to negative acute phase reactants ratio and severe hemostatic derangement in dengue fever. Cytokine. 2021;146:155644. doi:10.1016/j.cyto.2021.155644
- Dignass AU, Gasche C, Bettenworth D, et al; European Crohn’s and Colitis Organisation. European consensus on the diagnosis and management of iron deficiency and anaemia in inflammatory bowel diseases. J Crohn’s Colitis. 2015;9:211-222. doi:10.1093/ecco-jcc/jju009
- Daude S, Remen T, Chateau T, et al. Comparative accuracy of ferritin, transferrin saturation and soluble transferrin receptor for the diagnosis of iron deficiency in inflammatory bowel disease. Aliment Pharmacol Ther. 2020;51:1087-1095. doi:10.1111/apt.15739
- Pfeiffer CM, Looker AC. Laboratory methodologies for indicators of iron status: strengths, limitations, and analytical challenges. Am J Clin Nutr. 2017;106(suppl 6):1606S-1614S. doi:10.3945/ajcn.117.155887
- Tolkien Z, Stecher L, Mander AP, et al. Ferrous sulfate supplementation causes significant gastrointestinal side-effects in adults: a systematic review and meta-analysis. PLoS One. 2015;10:e0117383. doi:10.1371/journal.pone.0117383
- Evstatiev R, Marteau P, Iqbal T, et al. FERGIcor, a randomized controlled trial on ferric carboxymaltose for iron deficiency anemia in inflammatory bowel disease. Gastroenterology. 2011;141:846-853.e8532. doi:10.1053/j.gastro.2011.06.005
- Zupo R, Sila A, Castellana F, et al. Prevalence of zinc deficiency in inflammatory bowel disease: a systematic review and meta-analysis. Nutrients. 2022;14:4052. doi:10.3390/nu14194052
- Thompson MW. Regulation of zinc-dependent enzymes by metal carrier proteins. Biometals. 2022;35:187-213. doi:10.1007/s10534-022-00373-w
- Maares M, Haase H. A guide to human zinc absorption: general overview and recent advances of in vitro intestinal models. Nutrients. 2020;12:762. doi:10.3390/nu12030762
- Ranaldi G, Ferruzza S, Canali R, et al. Intracellular zinc is required for intestinal cell survival signals triggered by the inflammatory cytokine TNFα. J Nutr Biochem. 2013;24:967-976. doi:10.1016/j.jnutbio.2012.06.020
- Ogawa Y, Kawamura T, Shimada S. Zinc and skin biology. Arch Biochem Biophys. 2016;611:113-119. doi:10.1016/j.abb.2016.06.003
- Wilson D, Varigos G, Ackland ML. Apoptosis may underlie the pathology of zinc-deficient skin. Immunol Cell Biol. 2006;84:28-37. doi:10.1111/j.1440-1711.2005.01391.x
- Jen M, Yan AC. Syndromes associated with nutritional deficiency and excess. Clin Dermatol. 2010;28:669-685. doi:10.1016/j.clindermatol.2010.03.029
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Gammoh NZ, Rink L. Zinc in infection and inflammation. Nutrients. 2017;9:624. doi:10.3390/nu9060624
- Liuzzi JP, Lichten LA, Rivera S, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to the hypozincemia of the acute-phase response. Proc Natl Acad Sci U S A. 2005;102:6843-6848. doi:10.1073/pnas.0502257102
- Vermeire S, Van Assche G, Rutgeerts P. Laboratory markers in IBD: useful, magic, or unnecessary toys?. Gut. 2006;55:426-431. doi:10.1136/gut.2005.069476
- Morisaku M, Ito K, Ogiso A, et al. Correlation between serum albumin and serum zinc in malignant lymphoma. Fujita Med J. 2022;8:59-64. doi:10.20407/fmj.2021-006
- Falchuk KH. Effect of acute disease and ACTH on serum zinc proteins. N Engl J Med. 1977:296:1129-1134.
- Naber TH, Baadenhuysen H, Jansen JB, et al. Serum alkaline phosphatase activity during zinc deficiency and long-term inflammatory stress. Clin Chim Acta. 1996;249:109-127. doi:10.1016/0009-8981(96)06281-x
- Lowe D, Sanvictores T, Zubair M, et al. Alkaline phosphatase. StatPearls [Internet]. Updated October 29, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK459201/
- Krebs NF. Update on zinc deficiency and excess in clinical pediatric practice. Ann Nutr Metab. 2013;62 suppl 1:19-29. doi:10.1159/000348261
- Maxfield L, Shukla S, Crane JS. Zinc deficiency. StatPearls [Internet]. Updated June 28, 2023. Accessed March 25, 2024. https://www.ncbi.nlm.nih.gov/books/NBK493231/
- Ghishan FK, Kiela PR. Vitamins and minerals in inflammatory bowel disease. Gastroenterol Clin North Am. 2017;46:797-808. doi:10.1016/j.gtc.2017.08.011
- Caviezel D, Maissen S, Niess JH, et al. High prevalence of vitamin D deficiency among patients with inflammatory bowel disease. Inflamm Intest Dis. 2018;2:200-210. doi:10.1159/000489010
- Jasielska M, Grzybowska-Chlebowczyk U. Hypocalcemia and vitamin D deficiency in children with inflammatory bowel diseases and lactose intolerance. Nutrients. 2021;13:2583. doi:10.3390/nu13082583
- Vernia F, Valvano M, Longo S, et al. Vitamin D in inflammatory bowel diseases. Mechanisms of action and therapeutic implications. Nutrients. 2022;14:269. doi:10.3390/nu14020269
- Khazai N, Judd SE, Tangpricha V. Calcium and vitamin D: skeletal and extraskeletal health. Curr Rheumatol Rep. 2008;10:110-117. doi:10.1007/s11926-008-0020-y
- Domazetovic V, Iantomasi T, Bonanomi AG, et al. Vitamin D regulates claudin-2 and claudin-4 expression in active ulcerative colitis by p-Stat-6 and Smad-7 signaling. Int J Colorectal Dis. 2020;35:1231-1242. doi:10.1007/s00384-020-03576-0
- Gubatan J, Chou ND, Nielsen OH, et al. Systematic review with meta-analysis: association of vitamin D status with clinical outcomes in adult patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2019;50:1146-1158. doi:10.1111/apt.15506
- Fakhoury HMA, Kvietys PR, AlKattan W, et al. Vitamin D and intestinal homeostasis: barrier, microbiota, and immune modulation. J Steroid Biochem Mol Biol. 2020;200:105663. doi:10.1016/j.jsbmb.2020.105663
- Liu PT, Stenger S, Li H, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770-1773. doi:10.1126/science.1123933
- Mostafa WZ, Hegazy RA. Vitamin D and the skin: focus on a complex relationship: a review. J Adv Res. 2015;6:793-804. doi:10.1016/j.jare.2014.01.011
- Searing DA, Leung DY. Vitamin D in atopic dermatitis, asthma and allergic diseases. Immunol Allergy Clin North Am. 2010;30:397-409.
- Lee YH, Song GG. Association between circulating 25-hydroxyvitamin D levels and psoriasis, and correlation with disease severity: a meta-analysis. Clin Exp Dermatol. 2018;43:529-535.
- Adorini L, Penna G. Control of autoimmune diseases by the vitamin D endocrine system. Nat Clin Pract Rheumatol. 2008;4:404-412.
- Autier P, Boniol M, Pizot C, et al. Vitamin D status and ill health: a systematic review. Lancet Diabetes Endocrinol. 2014;2:76-89. doi:10.1016/S2213-8587(13)70165-7
- Schafer AL, Shoback DM. Hypocalcemia: diagnosis and treatment. In: Feingold KR, Anawalt B, Blackman MR, et al, eds. Endotext [Internet]. Updated January 3, 2016. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK279022/
- Magro F, Gionchetti P, Eliakim R, et al. Third European Evidence-based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 1: Definitions, diagnosis, extra-intestinal manifestations, pregnancy, cancer surveillance, surgery, and ileo-anal pouch disorders. J Crohns Colitis. 2017;11:649-670. doi:10.1093/ecco-jcc/jjx008
- Amrein K, Scherkl M, Hoffmann M, et al. Vitamin D deficiency 2.0: an update on the current status worldwide. Eur J Clin Nutr. 2020;74:1498-1513. doi:10.1038/s41430-020-0558-y
- Munns CF, Shaw N, Kiely M, et al. Global consensus recommendations on prevention and management of nutritional rickets. J Clin Endocrinol Metab. 2016;101:394-415. doi:10.1210/jc.2015-2175
- Institute of Medicine (US) Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Ross AC, Taylor CL, Yaktine AL, Del Valle HB, eds. Dietary Reference Intakes for Calcium and Vitamin D. National Academies Press (US); 2011.
- Yeaman F, Nguyen A, Abasszade J, et al. Assessing vitamin D as a biomarker in inflammatory bowel disease. JGH Open. 2023;7:953-958. doi:10.1002/jgh3.13010
- Vernia P, Loizos P, Di Giuseppantonio I, et al S. Dietary calcium intake in patients with inflammatory bowel disease. J Crohns Colitis. 2014;8:312-317. doi:10.1016/j.crohns.2013.09.008
- Cooper MS, Gittoes NJ. Diagnosis and management of hypocalcaemia. BMJ. 2008;336:1298-1302. doi:10.1136/bmj.39582.589433.BE
- Kenny CM, Murphy CE, Boyce DS, et al. Things we do for no reason™: calculating a “corrected calcium” level. J Hosp Med. 2021;16:499-501. doi:10.12788/jhm.3619
- Garg M, Rosella O, Rosella G, et al. Evaluation of a 12-week targeted vitamin D supplementation regimen in patients with active inflammatory bowel disease. Clin Nutr. 2018;37:1375-1382. doi:10.1016/j.clnu.2017.06.011
- Raftery T, Martineau AR, Greiller CL, et al. Effects of vitamin D supplementation on intestinal permeability, cathelicidin and disease markers in Crohn’s disease: results from a randomised double-blind placebo-controlled study. United European Gastroenterol J. 2015;3:294-302. doi:10.1177/2050640615572176
- Vagianos K, Bector S, McConnell J, et al. Nutrition assessment of patients with inflammatory bowel disease. JPEN J Parenter Enteral Nutr. 2007;31:311-319. doi:10.1177/0148607107031004311
- Barthelemy H, Chouvet B, Cambazard F. Skin and mucosal manifestations in vitamin deficiency. J Am Acad Dermatol. 1986;15:1263-1274. doi:10.1016/s0190-9622(86)70301-0
- Galimberti F, Mesinkovska NA. Skin findings associated with nutritional deficiencies. Cleve Clin J Med. 2016;83:731-739. doi:10.3949/ccjm.83a.15061
- Elgharably N, Al Abadie M, Al Abadie M, et al. Vitamin B group levels and supplementations in dermatology. Dermatol Reports. 2022;15:9511. doi:10.4081/dr.2022.9511
- Hołubiec P, Leon´czyk M, Staszewski F, et al. Pathophysiology and clinical management of pellagra—a review. Folia Med Cracov. 2021;61:125-137. doi:10.24425/fmc.2021.138956
- Ink SL, Henderson LM. Vitamin B6 metabolism. Annu Rev Nutr. 1984;4:455-470. doi:10.1146/annurev.nu.04.070184.002323
- Brown MJ, Ameer MA, Daley SF, et al. Vitamin B6 deficiency. StatPearls [Internet]. Updated August 8, 2023. Accessed March 25, 2024. https://www.ncbi.nlm.nih.gov/books/NBK470579/.
- Vasilaki AT, McMillan DC, Kinsella J, et al. Relation between pyridoxal and pyridoxal phosphate concentrations in plasma, red cells, and white cells in patients with critical illness. Am J Clin Nutr. 2008;88:140-146. doi:10.1093/ajcn/88.1.140
- Chiang EP, Bagley PJ, Selhub J, et al. Abnormal vitamin B(6) status is associated with severity of symptoms in patients with rheumatoid arthritis. Am J Med. 2003;114:283-287. doi:10.1016/s0002-9343(02)01528-0
- Maaser C, Sturm A, Vavricka SR, et al. ECCO-ESGAR guideline for diagnostic assessment in IBD. Part 1: initial diagnosis, monitoring of known IBD, detection of complications. J Crohns Colitis. 2019;13:144-164. doi:10.1093/ecco-jcc/jjy113
- Spinneker A, Sola R, Lemmen V, et al. Vitamin B6 status, deficiency and its consequences—an overview. Nutr Hosp. 2007;22:7-24.
- Selhub J, Byun A, Liu Z, et al. Dietary vitamin B6 intake modulates colonic inflammation in the IL10-/- model of inflammatory bowel disease. J Nutr Biochem. 2013;24:2138-2143. doi:10.1016/j.jnutbio.2013.08.005
- Pan Y, Liu Y, Guo H, et al. Associations between folate and vitamin B12 levels and inflammatory bowel disease: a meta-analysis. Nutrients. 2017;9:382. doi:10.3390/nu9040382
- Brescoll J, Daveluy S. A review of vitamin B12 in dermatology. Am J Clin Dermatol. 2015;16:27-33. doi:10.1007/s40257-014-0107-3
- DiBaise M, Tarleton SM. Hair, nails, and skin: differentiating cutaneous manifestations of micronutrient deficiency. Nutr Clin Pract. 2019;34:490-503. doi:10.1002/ncp.10321
- Mori K, Ando I, Kukita A. Generalized hyperpigmentation of the skin due to vitamin B12 deficiency. J Dermatol. 2001;28:282-285. doi:10.1111/j.1346-8138.2001.tb00134.x
- Green R. Indicators for assessing folate and vitamin B-12 status and for monitoring the efficacy of intervention strategies. Am J Clin Nutr. 2011;94:666S-672S. doi:10.3945/ajcn.110.009613
- NIH Office of Dietary Supplements. Vitamin B12: fact sheet for health professionals. Updated February 27, 2024. Accessed March 19, 2024. https://ods.od.nih.gov/factsheets/VitaminB12-HealthProfessional/
- NIH Office of Dietary Supplements. Folate: fact sheet for health professionals. Updated November 20, 2023. Accessed March 19, 2024. https://ods.od.nih.gov/factsheets/Folate-HealthProfessional/.
- Saibeni S, Bollani S, Losco A, et al. The use of methotrexate for treatment of inflammatory bowel disease in clinical practice. Dig Liver Dis. 2012;44:123-127. doi:10.1016/j.dld.2011.09.015
- Khan KM, Jialal I. Folic acid deficiency. StatPearls [Internet]. Updated June 26, 2023. Accessed March 19, 2024. https://www.ncbi.nlm.nih.gov/books/NBK535377/
Practice Points
- Patients with inflammatory bowel disease (IBD) are at increased risk for vitamin and nutrient deficiencies that may be identified first through cutaneous manifestations.
- Because active inflammation in IBD may skew routine laboratory values used for screening of micronutrient deficiencies, be cautious when interpreting these values.
- Patients taking systemic therapies for IBD such as corticosteroids and methotrexate are at higher risk for nutritional deficiencies.