User login
Distance learning may cause convergence insufficiency
NEW ORLEANS – The increased use of digital screens for school during the COVID-19 pandemic may be causing convergence insufficiency in children, researchers say.
Although the long-term implications for current schoolchildren are not clear, convergence insufficiency sometimes persists for a lifetime, said Kammi Gunton, MD, interim chief of pediatric ophthalmology and strabismus at Wills Eye Hospital, Philadelphia.
“It’s important, if we use digital technology for education, that we are aware that it might contribute to increased eye symptoms in children,” Dr. Gunton told this news organization.
Dr. Gunton’s colleague, Jordan Hamburger, an MD candidate at Sidney Kimmel Medical College, Philadelphia, presented the finding at the American Academy of Ophthalmology 2021 Annual Meeting.
Convergence insufficiency is an impairment of binocularity. Symptoms include headaches while reading, words that seem to move around the page, blurriness, diplopia, and eye fatigue. It can be treated with exercise, prism glasses, or, rarely, surgery.
“We have some kids who improve with either time or maturity, then we have other patients who suffer from it for their entire lives,” Dr. Gunton said.
Previous research has linked the use of digital screens to convergence insufficiency, so when many schools shifted to distance learning for the pandemic, Dr. Gunton and her colleagues wanted to see whether it would have this effect on the students’ eyes.
They surveyed 110 healthy schoolchildren and adolescent students regarding eye symptoms before and after a day of virtual school. The mean age of the participants was 14 years (range, 10-17 years). The participants spent an average of 6.96 hours per day in virtual school. Forty-one percent also attended school in person part time. These students filled out the survey on days when they were in virtual school.
The participants answered questions on the Convergence Insufficiency Symptom Survey (CISS). The survey consists of 15 questions about eye complaints. On each question, the students rated symptoms from 0 to 4, with 4 indicating a severe symptom.
The average sum of the CISS scores rose from 5.17 before school to 9.82 after school, a statistically significant change (P < .001). Sixty-one percent of the participants reported an increase in convergence insufficiency symptoms.
Seventeen percent scored a total of at least 16, which is the threshold score considered suggestive of convergence insufficiency.
The researchers also found that, on average, the more hours each student spent in virtual school, the higher their CISS scores.
This makes sense, because reading requires convergence, Dr. Gunton said. The same problem might occur in traditional school if the students were looking at books all day instead of focusing on objects at various distances in their classrooms, such as the teacher or the whiteboard. “So, in the past, if you read a book, maybe you wouldn’t read for several hours, but now we’re asking children during virtual learning to stay on a device with the camera on,” she said.
Previous research has shown that people blink less when reading or using electronic devices, probably because of their increased concentration. This might explain symptoms such as burning and itching. Fifty-three percent of the students reported an increase in asthenopia symptoms.
The researchers would have liked to have compared the students in virtual school to a matched group of students in traditional school. However, almost all students were enrolled in virtual school when the study was conducted, making such a control difficult.
Although previous research has related virtual learning to myopia, as reported by this news organization, this study did not investigate myopia, and the researchers do not believe that convergence insufficiency causes myopia or vice versa.
Parents can help prevent convergence insufficiency during school by reminding their children to take breaks, Dr. Gunton said. She recommends the 20/20/20 rule: After 20 minutes of work that involves looking at objects nearby, students should take a 20-second break and look at something 20 feet away.
“I also think the take-home message is for parents to ask students if they’re having symptoms,” she said, “and if they hear complaints while kids are on the computers, to have them see an eye doctor and have an evaluation.”
Stephen Lipsky, MD, who wasn’t involved in the study, said he is seeing more cases of eye strain at Children’s Healthcare of Atlanta, where he is a consulting ophthalmologist.
“The study is very valuable in that it shines a light on the fact that these children do have symptoms, such as asthenopia or convergence insufficiency,” he told this news organization. “But I’m optimistic that with a return to more traditional learning, we will return the more traditional incidence of these problems.”
Dr. Gunton and Dr. Lipsky have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – The increased use of digital screens for school during the COVID-19 pandemic may be causing convergence insufficiency in children, researchers say.
Although the long-term implications for current schoolchildren are not clear, convergence insufficiency sometimes persists for a lifetime, said Kammi Gunton, MD, interim chief of pediatric ophthalmology and strabismus at Wills Eye Hospital, Philadelphia.
“It’s important, if we use digital technology for education, that we are aware that it might contribute to increased eye symptoms in children,” Dr. Gunton told this news organization.
Dr. Gunton’s colleague, Jordan Hamburger, an MD candidate at Sidney Kimmel Medical College, Philadelphia, presented the finding at the American Academy of Ophthalmology 2021 Annual Meeting.
Convergence insufficiency is an impairment of binocularity. Symptoms include headaches while reading, words that seem to move around the page, blurriness, diplopia, and eye fatigue. It can be treated with exercise, prism glasses, or, rarely, surgery.
“We have some kids who improve with either time or maturity, then we have other patients who suffer from it for their entire lives,” Dr. Gunton said.
Previous research has linked the use of digital screens to convergence insufficiency, so when many schools shifted to distance learning for the pandemic, Dr. Gunton and her colleagues wanted to see whether it would have this effect on the students’ eyes.
They surveyed 110 healthy schoolchildren and adolescent students regarding eye symptoms before and after a day of virtual school. The mean age of the participants was 14 years (range, 10-17 years). The participants spent an average of 6.96 hours per day in virtual school. Forty-one percent also attended school in person part time. These students filled out the survey on days when they were in virtual school.
The participants answered questions on the Convergence Insufficiency Symptom Survey (CISS). The survey consists of 15 questions about eye complaints. On each question, the students rated symptoms from 0 to 4, with 4 indicating a severe symptom.
The average sum of the CISS scores rose from 5.17 before school to 9.82 after school, a statistically significant change (P < .001). Sixty-one percent of the participants reported an increase in convergence insufficiency symptoms.
Seventeen percent scored a total of at least 16, which is the threshold score considered suggestive of convergence insufficiency.
The researchers also found that, on average, the more hours each student spent in virtual school, the higher their CISS scores.
This makes sense, because reading requires convergence, Dr. Gunton said. The same problem might occur in traditional school if the students were looking at books all day instead of focusing on objects at various distances in their classrooms, such as the teacher or the whiteboard. “So, in the past, if you read a book, maybe you wouldn’t read for several hours, but now we’re asking children during virtual learning to stay on a device with the camera on,” she said.
Previous research has shown that people blink less when reading or using electronic devices, probably because of their increased concentration. This might explain symptoms such as burning and itching. Fifty-three percent of the students reported an increase in asthenopia symptoms.
The researchers would have liked to have compared the students in virtual school to a matched group of students in traditional school. However, almost all students were enrolled in virtual school when the study was conducted, making such a control difficult.
Although previous research has related virtual learning to myopia, as reported by this news organization, this study did not investigate myopia, and the researchers do not believe that convergence insufficiency causes myopia or vice versa.
Parents can help prevent convergence insufficiency during school by reminding their children to take breaks, Dr. Gunton said. She recommends the 20/20/20 rule: After 20 minutes of work that involves looking at objects nearby, students should take a 20-second break and look at something 20 feet away.
“I also think the take-home message is for parents to ask students if they’re having symptoms,” she said, “and if they hear complaints while kids are on the computers, to have them see an eye doctor and have an evaluation.”
Stephen Lipsky, MD, who wasn’t involved in the study, said he is seeing more cases of eye strain at Children’s Healthcare of Atlanta, where he is a consulting ophthalmologist.
“The study is very valuable in that it shines a light on the fact that these children do have symptoms, such as asthenopia or convergence insufficiency,” he told this news organization. “But I’m optimistic that with a return to more traditional learning, we will return the more traditional incidence of these problems.”
Dr. Gunton and Dr. Lipsky have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – The increased use of digital screens for school during the COVID-19 pandemic may be causing convergence insufficiency in children, researchers say.
Although the long-term implications for current schoolchildren are not clear, convergence insufficiency sometimes persists for a lifetime, said Kammi Gunton, MD, interim chief of pediatric ophthalmology and strabismus at Wills Eye Hospital, Philadelphia.
“It’s important, if we use digital technology for education, that we are aware that it might contribute to increased eye symptoms in children,” Dr. Gunton told this news organization.
Dr. Gunton’s colleague, Jordan Hamburger, an MD candidate at Sidney Kimmel Medical College, Philadelphia, presented the finding at the American Academy of Ophthalmology 2021 Annual Meeting.
Convergence insufficiency is an impairment of binocularity. Symptoms include headaches while reading, words that seem to move around the page, blurriness, diplopia, and eye fatigue. It can be treated with exercise, prism glasses, or, rarely, surgery.
“We have some kids who improve with either time or maturity, then we have other patients who suffer from it for their entire lives,” Dr. Gunton said.
Previous research has linked the use of digital screens to convergence insufficiency, so when many schools shifted to distance learning for the pandemic, Dr. Gunton and her colleagues wanted to see whether it would have this effect on the students’ eyes.
They surveyed 110 healthy schoolchildren and adolescent students regarding eye symptoms before and after a day of virtual school. The mean age of the participants was 14 years (range, 10-17 years). The participants spent an average of 6.96 hours per day in virtual school. Forty-one percent also attended school in person part time. These students filled out the survey on days when they were in virtual school.
The participants answered questions on the Convergence Insufficiency Symptom Survey (CISS). The survey consists of 15 questions about eye complaints. On each question, the students rated symptoms from 0 to 4, with 4 indicating a severe symptom.
The average sum of the CISS scores rose from 5.17 before school to 9.82 after school, a statistically significant change (P < .001). Sixty-one percent of the participants reported an increase in convergence insufficiency symptoms.
Seventeen percent scored a total of at least 16, which is the threshold score considered suggestive of convergence insufficiency.
The researchers also found that, on average, the more hours each student spent in virtual school, the higher their CISS scores.
This makes sense, because reading requires convergence, Dr. Gunton said. The same problem might occur in traditional school if the students were looking at books all day instead of focusing on objects at various distances in their classrooms, such as the teacher or the whiteboard. “So, in the past, if you read a book, maybe you wouldn’t read for several hours, but now we’re asking children during virtual learning to stay on a device with the camera on,” she said.
Previous research has shown that people blink less when reading or using electronic devices, probably because of their increased concentration. This might explain symptoms such as burning and itching. Fifty-three percent of the students reported an increase in asthenopia symptoms.
The researchers would have liked to have compared the students in virtual school to a matched group of students in traditional school. However, almost all students were enrolled in virtual school when the study was conducted, making such a control difficult.
Although previous research has related virtual learning to myopia, as reported by this news organization, this study did not investigate myopia, and the researchers do not believe that convergence insufficiency causes myopia or vice versa.
Parents can help prevent convergence insufficiency during school by reminding their children to take breaks, Dr. Gunton said. She recommends the 20/20/20 rule: After 20 minutes of work that involves looking at objects nearby, students should take a 20-second break and look at something 20 feet away.
“I also think the take-home message is for parents to ask students if they’re having symptoms,” she said, “and if they hear complaints while kids are on the computers, to have them see an eye doctor and have an evaluation.”
Stephen Lipsky, MD, who wasn’t involved in the study, said he is seeing more cases of eye strain at Children’s Healthcare of Atlanta, where he is a consulting ophthalmologist.
“The study is very valuable in that it shines a light on the fact that these children do have symptoms, such as asthenopia or convergence insufficiency,” he told this news organization. “But I’m optimistic that with a return to more traditional learning, we will return the more traditional incidence of these problems.”
Dr. Gunton and Dr. Lipsky have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM AAO 2021
People of color missing in inflammatory bowel disease trials
LAS VEGAS – Clinical trials of treatments for inflammatory bowel disease (IBD) have disproportionately enrolled White people, researchers say.

These skewed demographics could result in researchers overlooking differences in how the disease and its treatments might affect other racial and ethnic groups, said Jellyana Peraza, MD, a chief resident at Albert Einstein College of Medicine, New York.
“The only way we can determine that therapies work differently in different populations is by including those populations in these clinical trials,” she said in an interview. “We think that diversity should be present, and that will answer some questions about the pathogenesis of the disease in general.”
Dr. Peraza presented the findings at the annual meeting of the American College of Gastroenterology.
Previous studies have found that, in trials of other conditions, such as cancer and cardiovascular disease, White people have been disproportionately represented. However, little research has been conducted regarding race and ethnicity in IBD trials.
To fill that gap, Dr. Peraza and colleagues analyzed data from completed trials through the U.S. National Library of Medicine’s registry, ClinicalTrials.gov, for the period from 2000 to 2020.
They found 22 trials conducted exclusively in the United States and 56 conducted in other countries that reported the race or ethnicity of participants; 54 trials did not include this information.
With regard to the prevalence of IBD in White people and Asian people, these populations were overrepresented in U.S. clinical trials. All other groups were underrepresented.
The researchers calculated the odds ratio of being included in an IBD clinical trial for each group. Compared with White people, all the other groups were less likely to be included except for Asian people, who were 85% more likely to be included. These ORs were all statistically significant (P < .03) except for Hispanic people (OR, 0.81; 95% confidence interval, 0.65-1.01; P = .06).
It’s not clear why Asian people are overrepresented, Dr. Peraza said. “Honestly, that was kind of surprising for us. We initially thought that could be related to where these studies were conducted, for example, if some of them were conducted on the West Coast, where maybe more Asian communities are located. However, we didn’t find any specific association between location and Asian representation.”
IBD is more prevalent among White people, although its prevalence is increasing among other groups, Dr. Peraza said. However, that is not reflected in the trials. In an analysis of data in 5-year increments, the researchers found that the participation of White and Hispanic people in IBD trials had not changed much, whereas the participation of Black people had declined, and the participation of Asian and Native American people had increased.
On the basis of work of other researchers, Dr. Peraza said that people of color are as willing to participate in trials as White people. “There is not so much a mistrust as a lack of education and a lack of access to the tertiary centers or the centers where these studies are conducted,” she said.
Clinical trial investigators should recruit more participants from community centers, and health care practitioners should talk about the trials with people in underrepresented groups, she said. “They should have the conversation with their patients about how these clinical trials can benefit the evolution of their diseases.”
One research center that is working hard to diversify its IBD trials is the Ohio State University IBD Center, Columbus, said Anita Afzali, MD, its medical director.
“We have a great team that works actively on the recruitment of all patients,” she said in an interview. “Oftentimes, it just takes a little bit of education and spending time with the patient on discussing what the options are for them.”
Some research indicates that Black people with IBD are more likely to have fistulizing disease, Dr. Afzali said. “However, it doesn’t come so much of their differences in phenotype that we’re seeing but more so the differences in access to care and getting the appropriate therapy in a timely way.”
Dr. Peraza and Dr. Afzali disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
LAS VEGAS – Clinical trials of treatments for inflammatory bowel disease (IBD) have disproportionately enrolled White people, researchers say.
These skewed demographics could result in researchers overlooking differences in how the disease and its treatments might affect other racial and ethnic groups, said Jellyana Peraza, MD, a chief resident at Albert Einstein College of Medicine, New York.
“The only way we can determine that therapies work differently in different populations is by including those populations in these clinical trials,” she said in an interview. “We think that diversity should be present, and that will answer some questions about the pathogenesis of the disease in general.”
Dr. Peraza presented the findings at the annual meeting of the American College of Gastroenterology.
Previous studies have found that, in trials of other conditions, such as cancer and cardiovascular disease, White people have been disproportionately represented. However, little research has been conducted regarding race and ethnicity in IBD trials.
To fill that gap, Dr. Peraza and colleagues analyzed data from completed trials through the U.S. National Library of Medicine’s registry, ClinicalTrials.gov, for the period from 2000 to 2020.
They found 22 trials conducted exclusively in the United States and 56 conducted in other countries that reported the race or ethnicity of participants; 54 trials did not include this information.
With regard to the prevalence of IBD in White people and Asian people, these populations were overrepresented in U.S. clinical trials. All other groups were underrepresented.
The researchers calculated the odds ratio of being included in an IBD clinical trial for each group. Compared with White people, all the other groups were less likely to be included except for Asian people, who were 85% more likely to be included. These ORs were all statistically significant (P < .03) except for Hispanic people (OR, 0.81; 95% confidence interval, 0.65-1.01; P = .06).
It’s not clear why Asian people are overrepresented, Dr. Peraza said. “Honestly, that was kind of surprising for us. We initially thought that could be related to where these studies were conducted, for example, if some of them were conducted on the West Coast, where maybe more Asian communities are located. However, we didn’t find any specific association between location and Asian representation.”
IBD is more prevalent among White people, although its prevalence is increasing among other groups, Dr. Peraza said. However, that is not reflected in the trials. In an analysis of data in 5-year increments, the researchers found that the participation of White and Hispanic people in IBD trials had not changed much, whereas the participation of Black people had declined, and the participation of Asian and Native American people had increased.
On the basis of work of other researchers, Dr. Peraza said that people of color are as willing to participate in trials as White people. “There is not so much a mistrust as a lack of education and a lack of access to the tertiary centers or the centers where these studies are conducted,” she said.
Clinical trial investigators should recruit more participants from community centers, and health care practitioners should talk about the trials with people in underrepresented groups, she said. “They should have the conversation with their patients about how these clinical trials can benefit the evolution of their diseases.”
One research center that is working hard to diversify its IBD trials is the Ohio State University IBD Center, Columbus, said Anita Afzali, MD, its medical director.
“We have a great team that works actively on the recruitment of all patients,” she said in an interview. “Oftentimes, it just takes a little bit of education and spending time with the patient on discussing what the options are for them.”
Some research indicates that Black people with IBD are more likely to have fistulizing disease, Dr. Afzali said. “However, it doesn’t come so much of their differences in phenotype that we’re seeing but more so the differences in access to care and getting the appropriate therapy in a timely way.”
Dr. Peraza and Dr. Afzali disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
LAS VEGAS – Clinical trials of treatments for inflammatory bowel disease (IBD) have disproportionately enrolled White people, researchers say.
These skewed demographics could result in researchers overlooking differences in how the disease and its treatments might affect other racial and ethnic groups, said Jellyana Peraza, MD, a chief resident at Albert Einstein College of Medicine, New York.
“The only way we can determine that therapies work differently in different populations is by including those populations in these clinical trials,” she said in an interview. “We think that diversity should be present, and that will answer some questions about the pathogenesis of the disease in general.”
Dr. Peraza presented the findings at the annual meeting of the American College of Gastroenterology.
Previous studies have found that, in trials of other conditions, such as cancer and cardiovascular disease, White people have been disproportionately represented. However, little research has been conducted regarding race and ethnicity in IBD trials.
To fill that gap, Dr. Peraza and colleagues analyzed data from completed trials through the U.S. National Library of Medicine’s registry, ClinicalTrials.gov, for the period from 2000 to 2020.
They found 22 trials conducted exclusively in the United States and 56 conducted in other countries that reported the race or ethnicity of participants; 54 trials did not include this information.
With regard to the prevalence of IBD in White people and Asian people, these populations were overrepresented in U.S. clinical trials. All other groups were underrepresented.
The researchers calculated the odds ratio of being included in an IBD clinical trial for each group. Compared with White people, all the other groups were less likely to be included except for Asian people, who were 85% more likely to be included. These ORs were all statistically significant (P < .03) except for Hispanic people (OR, 0.81; 95% confidence interval, 0.65-1.01; P = .06).
It’s not clear why Asian people are overrepresented, Dr. Peraza said. “Honestly, that was kind of surprising for us. We initially thought that could be related to where these studies were conducted, for example, if some of them were conducted on the West Coast, where maybe more Asian communities are located. However, we didn’t find any specific association between location and Asian representation.”
IBD is more prevalent among White people, although its prevalence is increasing among other groups, Dr. Peraza said. However, that is not reflected in the trials. In an analysis of data in 5-year increments, the researchers found that the participation of White and Hispanic people in IBD trials had not changed much, whereas the participation of Black people had declined, and the participation of Asian and Native American people had increased.
On the basis of work of other researchers, Dr. Peraza said that people of color are as willing to participate in trials as White people. “There is not so much a mistrust as a lack of education and a lack of access to the tertiary centers or the centers where these studies are conducted,” she said.
Clinical trial investigators should recruit more participants from community centers, and health care practitioners should talk about the trials with people in underrepresented groups, she said. “They should have the conversation with their patients about how these clinical trials can benefit the evolution of their diseases.”
One research center that is working hard to diversify its IBD trials is the Ohio State University IBD Center, Columbus, said Anita Afzali, MD, its medical director.
“We have a great team that works actively on the recruitment of all patients,” she said in an interview. “Oftentimes, it just takes a little bit of education and spending time with the patient on discussing what the options are for them.”
Some research indicates that Black people with IBD are more likely to have fistulizing disease, Dr. Afzali said. “However, it doesn’t come so much of their differences in phenotype that we’re seeing but more so the differences in access to care and getting the appropriate therapy in a timely way.”
Dr. Peraza and Dr. Afzali disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AT ACG 2021

Risankizumab has early and lasting benefits in Crohn’s disease
LAS VEGAS – Risankizumab (Skyrizi, AbbVie) provides early and lasting benefits for patients with Crohn’s disease, phase 3 trials indicate.
Based on these and other recent findings, the drug could be used as a first-line treatment and even displace ustekinumab (Stelara, Janssen), which itself was approved by the Food and Drug Administration for Crohn’s disease in 2016, according to David Rubin, MD, the Joseph B. Kirsner Professor of Medicine at the University of Chicago.
“The drug works fast,” Dr. Rubin said in an interview. “If you start this therapy in patients with moderate to severe Crohn’s disease, they’re likely to feel better within the first few weeks.”
Dr. Rubin presented the findings on the drug’s early onset at the annual meeting of the American College of Gastroenterology. A related trial presented at the meeting showed the drug continuing to perform well up to 52 weeks.
Advances in immunomodulation have allowed drug companies to feed multiple new therapies into the pipeline for Crohn’s disease and related conditions in recent years, giving hope to the many patients who have not been able to benefit from older classes of drugs, such as biologics.
A humanized immunoglobulin G1 (IgG1) monoclonal antibody, risankizumab blocks interleukin (IL) 23 by binding to its p19 subunit. IL-23 is a cytokine implicated in several chronic immune disorders, including Crohn’s disease and psoriasis. Researchers hope that risankizumab will prove more selective, with a better safety profile, than previous drugs in its class. The FDA approved risankizumab in April 2019 for the treatment of moderate to severe plaque psoriasis.
MOTIVATE and ADVANCE studies
The two induction trials for Crohn’s disease enrolled slightly different populations.
The MOTIVATE study enrolled patients who had responded inadequately or were intolerant to biologic therapy. In this trial, the investigators assigned 205 patients to 1,200 mg of risankizumab, 206 patients to 600 mg of risankizumab, and 207 patients to placebo.
The ADVANCE study enrolled patients who had responded inadequately or could not tolerate either biologic or conventional therapy. In this trial, investigators randomly assigned 372 patients to 1,200 mg of risankizumab, 373 patients to 600 mg of risankizumab, and 186 patients to placebo.
In both trials, intravenous injections were given at weeks 0, 4, and 8.
The researchers defined a Crohn’s Disease Activity Index (CDAI) clinical remission as a score less than 150. They defined a Stool Frequency and Abdominal Pain Score (SF/APS) clinical remission as a soft stool frequency of no more than 2.8, and an abdominal pain score of no more than 1 and not worse than baseline.
A CDAI clinical response was at least a 100-point decrease from baseline. The SF/APS enhanced clinical response was at least a 60% decrease in average daily stool frequency or at least a 35% decrease in average daily abdominal pain, with both not worse than baseline.
At 4 weeks, the researchers found that the percentage of patients who achieved CDAI clinical remission in both risankizumab groups of both studies was greater than in the placebo group. The difference was statistically significant (P ≤ .01 in ADVANCE and P ≤ .05 in MOTIVATE), and it continued to grow at 8 weeks and 12 weeks.
By 12 weeks in the ADVANCE trial, according to a press release from AbbVie, 45% of patients on the 600-mg dose of risankizumab and 42% on the 1,200-mg dose of risankizumab had achieved CDAI clinical remission, compared with 25% of those on placebo, which was statistically significant (P < .001). For the MOTIVATE trial, the results were significantly better for patients in the risankizumab groups than for those in the placebo group.
In both trials, the treated groups continued to improve faster than the placebo groups through 12 weeks. Improvements in SF/APS enhanced clinical response largely paralleled those for CDAI clinical remission.
“It did show very good results,” session moderator Jonathan Leighton, MD, professor of medicine and chair of the division of gastroenterology at Mayo Clinic in Phoenix, Ariz., said in an interview with Medscape Medical News. “But basically, it’s so early that we don’t have all the data.” In particular, he would have liked to see whether patients responded to the drug before week 4.
FORTIFY study
In FORTIFY, the maintenance trial that followed, the researchers rerandomized those patients who had responded to risankizumab into three groups. Two groups received subcutaneous injections of risankizumab, with 179 patients getting 360 mg and another 179 patients getting 180 mg. The placebo group included the remaining 184 patients.
At week 52, 40.9% of patients in the placebo group were in clinical remission, compared with 52.2% in the 360-mg group and 55.4% in the 180-mg group, which was statistically significant (P = .005 for 360 mg, and P = .003 for 180 mg.)
“It showed us that [risankizumab] could achieve deep remission, which means patients achieving remission endoscopically in combination with clinical remission,” the presenter, Marla Dubinsky, MD, professor of pediatrics and medicine in the division of pediatric gastroenterology at Icahn School of Medicine at Mount Sinai in New York, said in an interview.
Over the 52 weeks, deep remission and endoscopic remission rates increased in the 360-mg group, held steady in the 180-mg group, and decreased in the placebo group. Mean fecal calprotectin and C-reactive protein levels decreased in the risankizumab groups and increased in the placebo group.
There were more total treatment-emergent adverse events per 100 patient-years in the placebo group (339.7) than in the 360-mg group (269.3) or the 180-mg group (283.5). The same difference between groups was true of severe treatment-emergent adverse events. Serious events and events leading to discontinuation were similar in the three groups.
Dr. Leighton reports financial relationships to Olympus and Pfizer. Dr. Rubin reports financial relationships to AbbVie, AltruBio, Allergan, Arena Pharmaceuticals, Athos Therapeutics, Bellatrix, Boehringer Ingelheim, Bristol Myers Squibb, Celgene/Syneos, Connect Biopharma, GalenPharma/Atlantica, Genentech/Roche, Gilead, InDex Pharmaceuticals, Ironwood, Iterative Scopes, Janssen, Lilly, Materia Prima Farmaceutica, Pfizer, Prometheus Biosciences, Reistone, Takeda, and TECHLAB. Dr. Dubinsky reports financial relationships to all or most of the companies making drugs for inflammatory bowel disease. The studies were funded by AbbVie.
A version of this article first appeared on Medscape.com.
LAS VEGAS – Risankizumab (Skyrizi, AbbVie) provides early and lasting benefits for patients with Crohn’s disease, phase 3 trials indicate.
Based on these and other recent findings, the drug could be used as a first-line treatment and even displace ustekinumab (Stelara, Janssen), which itself was approved by the Food and Drug Administration for Crohn’s disease in 2016, according to David Rubin, MD, the Joseph B. Kirsner Professor of Medicine at the University of Chicago.
“The drug works fast,” Dr. Rubin said in an interview. “If you start this therapy in patients with moderate to severe Crohn’s disease, they’re likely to feel better within the first few weeks.”
Dr. Rubin presented the findings on the drug’s early onset at the annual meeting of the American College of Gastroenterology. A related trial presented at the meeting showed the drug continuing to perform well up to 52 weeks.
Advances in immunomodulation have allowed drug companies to feed multiple new therapies into the pipeline for Crohn’s disease and related conditions in recent years, giving hope to the many patients who have not been able to benefit from older classes of drugs, such as biologics.
A humanized immunoglobulin G1 (IgG1) monoclonal antibody, risankizumab blocks interleukin (IL) 23 by binding to its p19 subunit. IL-23 is a cytokine implicated in several chronic immune disorders, including Crohn’s disease and psoriasis. Researchers hope that risankizumab will prove more selective, with a better safety profile, than previous drugs in its class. The FDA approved risankizumab in April 2019 for the treatment of moderate to severe plaque psoriasis.
MOTIVATE and ADVANCE studies
The two induction trials for Crohn’s disease enrolled slightly different populations.
The MOTIVATE study enrolled patients who had responded inadequately or were intolerant to biologic therapy. In this trial, the investigators assigned 205 patients to 1,200 mg of risankizumab, 206 patients to 600 mg of risankizumab, and 207 patients to placebo.
The ADVANCE study enrolled patients who had responded inadequately or could not tolerate either biologic or conventional therapy. In this trial, investigators randomly assigned 372 patients to 1,200 mg of risankizumab, 373 patients to 600 mg of risankizumab, and 186 patients to placebo.
In both trials, intravenous injections were given at weeks 0, 4, and 8.
The researchers defined a Crohn’s Disease Activity Index (CDAI) clinical remission as a score less than 150. They defined a Stool Frequency and Abdominal Pain Score (SF/APS) clinical remission as a soft stool frequency of no more than 2.8, and an abdominal pain score of no more than 1 and not worse than baseline.
A CDAI clinical response was at least a 100-point decrease from baseline. The SF/APS enhanced clinical response was at least a 60% decrease in average daily stool frequency or at least a 35% decrease in average daily abdominal pain, with both not worse than baseline.
At 4 weeks, the researchers found that the percentage of patients who achieved CDAI clinical remission in both risankizumab groups of both studies was greater than in the placebo group. The difference was statistically significant (P ≤ .01 in ADVANCE and P ≤ .05 in MOTIVATE), and it continued to grow at 8 weeks and 12 weeks.
By 12 weeks in the ADVANCE trial, according to a press release from AbbVie, 45% of patients on the 600-mg dose of risankizumab and 42% on the 1,200-mg dose of risankizumab had achieved CDAI clinical remission, compared with 25% of those on placebo, which was statistically significant (P < .001). For the MOTIVATE trial, the results were significantly better for patients in the risankizumab groups than for those in the placebo group.
In both trials, the treated groups continued to improve faster than the placebo groups through 12 weeks. Improvements in SF/APS enhanced clinical response largely paralleled those for CDAI clinical remission.
“It did show very good results,” session moderator Jonathan Leighton, MD, professor of medicine and chair of the division of gastroenterology at Mayo Clinic in Phoenix, Ariz., said in an interview with Medscape Medical News. “But basically, it’s so early that we don’t have all the data.” In particular, he would have liked to see whether patients responded to the drug before week 4.
FORTIFY study
In FORTIFY, the maintenance trial that followed, the researchers rerandomized those patients who had responded to risankizumab into three groups. Two groups received subcutaneous injections of risankizumab, with 179 patients getting 360 mg and another 179 patients getting 180 mg. The placebo group included the remaining 184 patients.
At week 52, 40.9% of patients in the placebo group were in clinical remission, compared with 52.2% in the 360-mg group and 55.4% in the 180-mg group, which was statistically significant (P = .005 for 360 mg, and P = .003 for 180 mg.)
“It showed us that [risankizumab] could achieve deep remission, which means patients achieving remission endoscopically in combination with clinical remission,” the presenter, Marla Dubinsky, MD, professor of pediatrics and medicine in the division of pediatric gastroenterology at Icahn School of Medicine at Mount Sinai in New York, said in an interview.
Over the 52 weeks, deep remission and endoscopic remission rates increased in the 360-mg group, held steady in the 180-mg group, and decreased in the placebo group. Mean fecal calprotectin and C-reactive protein levels decreased in the risankizumab groups and increased in the placebo group.
There were more total treatment-emergent adverse events per 100 patient-years in the placebo group (339.7) than in the 360-mg group (269.3) or the 180-mg group (283.5). The same difference between groups was true of severe treatment-emergent adverse events. Serious events and events leading to discontinuation were similar in the three groups.
Dr. Leighton reports financial relationships to Olympus and Pfizer. Dr. Rubin reports financial relationships to AbbVie, AltruBio, Allergan, Arena Pharmaceuticals, Athos Therapeutics, Bellatrix, Boehringer Ingelheim, Bristol Myers Squibb, Celgene/Syneos, Connect Biopharma, GalenPharma/Atlantica, Genentech/Roche, Gilead, InDex Pharmaceuticals, Ironwood, Iterative Scopes, Janssen, Lilly, Materia Prima Farmaceutica, Pfizer, Prometheus Biosciences, Reistone, Takeda, and TECHLAB. Dr. Dubinsky reports financial relationships to all or most of the companies making drugs for inflammatory bowel disease. The studies were funded by AbbVie.
A version of this article first appeared on Medscape.com.
LAS VEGAS – Risankizumab (Skyrizi, AbbVie) provides early and lasting benefits for patients with Crohn’s disease, phase 3 trials indicate.
Based on these and other recent findings, the drug could be used as a first-line treatment and even displace ustekinumab (Stelara, Janssen), which itself was approved by the Food and Drug Administration for Crohn’s disease in 2016, according to David Rubin, MD, the Joseph B. Kirsner Professor of Medicine at the University of Chicago.
“The drug works fast,” Dr. Rubin said in an interview. “If you start this therapy in patients with moderate to severe Crohn’s disease, they’re likely to feel better within the first few weeks.”
Dr. Rubin presented the findings on the drug’s early onset at the annual meeting of the American College of Gastroenterology. A related trial presented at the meeting showed the drug continuing to perform well up to 52 weeks.
Advances in immunomodulation have allowed drug companies to feed multiple new therapies into the pipeline for Crohn’s disease and related conditions in recent years, giving hope to the many patients who have not been able to benefit from older classes of drugs, such as biologics.
A humanized immunoglobulin G1 (IgG1) monoclonal antibody, risankizumab blocks interleukin (IL) 23 by binding to its p19 subunit. IL-23 is a cytokine implicated in several chronic immune disorders, including Crohn’s disease and psoriasis. Researchers hope that risankizumab will prove more selective, with a better safety profile, than previous drugs in its class. The FDA approved risankizumab in April 2019 for the treatment of moderate to severe plaque psoriasis.
MOTIVATE and ADVANCE studies
The two induction trials for Crohn’s disease enrolled slightly different populations.
The MOTIVATE study enrolled patients who had responded inadequately or were intolerant to biologic therapy. In this trial, the investigators assigned 205 patients to 1,200 mg of risankizumab, 206 patients to 600 mg of risankizumab, and 207 patients to placebo.
The ADVANCE study enrolled patients who had responded inadequately or could not tolerate either biologic or conventional therapy. In this trial, investigators randomly assigned 372 patients to 1,200 mg of risankizumab, 373 patients to 600 mg of risankizumab, and 186 patients to placebo.
In both trials, intravenous injections were given at weeks 0, 4, and 8.
The researchers defined a Crohn’s Disease Activity Index (CDAI) clinical remission as a score less than 150. They defined a Stool Frequency and Abdominal Pain Score (SF/APS) clinical remission as a soft stool frequency of no more than 2.8, and an abdominal pain score of no more than 1 and not worse than baseline.
A CDAI clinical response was at least a 100-point decrease from baseline. The SF/APS enhanced clinical response was at least a 60% decrease in average daily stool frequency or at least a 35% decrease in average daily abdominal pain, with both not worse than baseline.
At 4 weeks, the researchers found that the percentage of patients who achieved CDAI clinical remission in both risankizumab groups of both studies was greater than in the placebo group. The difference was statistically significant (P ≤ .01 in ADVANCE and P ≤ .05 in MOTIVATE), and it continued to grow at 8 weeks and 12 weeks.
By 12 weeks in the ADVANCE trial, according to a press release from AbbVie, 45% of patients on the 600-mg dose of risankizumab and 42% on the 1,200-mg dose of risankizumab had achieved CDAI clinical remission, compared with 25% of those on placebo, which was statistically significant (P < .001). For the MOTIVATE trial, the results were significantly better for patients in the risankizumab groups than for those in the placebo group.
In both trials, the treated groups continued to improve faster than the placebo groups through 12 weeks. Improvements in SF/APS enhanced clinical response largely paralleled those for CDAI clinical remission.
“It did show very good results,” session moderator Jonathan Leighton, MD, professor of medicine and chair of the division of gastroenterology at Mayo Clinic in Phoenix, Ariz., said in an interview with Medscape Medical News. “But basically, it’s so early that we don’t have all the data.” In particular, he would have liked to see whether patients responded to the drug before week 4.
FORTIFY study
In FORTIFY, the maintenance trial that followed, the researchers rerandomized those patients who had responded to risankizumab into three groups. Two groups received subcutaneous injections of risankizumab, with 179 patients getting 360 mg and another 179 patients getting 180 mg. The placebo group included the remaining 184 patients.
At week 52, 40.9% of patients in the placebo group were in clinical remission, compared with 52.2% in the 360-mg group and 55.4% in the 180-mg group, which was statistically significant (P = .005 for 360 mg, and P = .003 for 180 mg.)
“It showed us that [risankizumab] could achieve deep remission, which means patients achieving remission endoscopically in combination with clinical remission,” the presenter, Marla Dubinsky, MD, professor of pediatrics and medicine in the division of pediatric gastroenterology at Icahn School of Medicine at Mount Sinai in New York, said in an interview.
Over the 52 weeks, deep remission and endoscopic remission rates increased in the 360-mg group, held steady in the 180-mg group, and decreased in the placebo group. Mean fecal calprotectin and C-reactive protein levels decreased in the risankizumab groups and increased in the placebo group.
There were more total treatment-emergent adverse events per 100 patient-years in the placebo group (339.7) than in the 360-mg group (269.3) or the 180-mg group (283.5). The same difference between groups was true of severe treatment-emergent adverse events. Serious events and events leading to discontinuation were similar in the three groups.
Dr. Leighton reports financial relationships to Olympus and Pfizer. Dr. Rubin reports financial relationships to AbbVie, AltruBio, Allergan, Arena Pharmaceuticals, Athos Therapeutics, Bellatrix, Boehringer Ingelheim, Bristol Myers Squibb, Celgene/Syneos, Connect Biopharma, GalenPharma/Atlantica, Genentech/Roche, Gilead, InDex Pharmaceuticals, Ironwood, Iterative Scopes, Janssen, Lilly, Materia Prima Farmaceutica, Pfizer, Prometheus Biosciences, Reistone, Takeda, and TECHLAB. Dr. Dubinsky reports financial relationships to all or most of the companies making drugs for inflammatory bowel disease. The studies were funded by AbbVie.
A version of this article first appeared on Medscape.com.
AT ACG 2021
Obesity interventions tied to colon cancer risk reduction
LAS VEGAS – People with obesity may be able to reduce their risk of colorectal cancer with weight loss surgery or medication, researchers say.
“We need to have conversations with our patients in the clinic and educate them that they have these resources available,” said Aakash Desai, MD, a hospitalist at MetroHealth Medical Center, Cleveland, in an interview with this news organization.
Dr. Desai and colleagues found that sleeve gastrectomy and four medications were associated with a reduced risk of colorectal cancer but Roux-en-Y gastrojejunostomy and orlistat were not.
Coauthor Zryan Shwani, MD, a gastroenterology fellow at Sibley Memorial Hospital, Washington, D.C., presented the findings here at the American College of Gastroenterology (ACG) 2021 Annual Scientific Meeting.
Working with an underserved population with high rates of obesity in northeastern Ohio, the researchers wondered how surgery and medication could affect these patients.
They analyzed data from the IBM Explorys clinical database, which compiles and standardizes data from electronic medical records on about 74 million patients from more than 300 U.S. hospitals. Consistent with previous studies, they determined that patients with obesity in the database were 2.5 times more likely than people with a healthy weight to be diagnosed with colorectal cancer (odds ratio, 2.48; 95% CI, 2.45-2.51).
Zeroing in on people who had weight loss interventions, they included adults aged 18-75 years who had undergone either Roux-en-Y gastrojejunostomy or sleeve gastrectomy, or had taken the medications liraglutide, orlistat, phentermine/topiramate, bupropion/naltrexone, or lorcaserin.
They excluded patients with Lynch syndrome, intestinal polyposis syndrome, a family history of gastrointestinal malignancy, inflammatory bowel disease, or tobacco or alcohol abuse. Patients who had taken one of the weight loss medications and also had type 2 diabetes were excluded. They did not include patients who had undergone gastric banding because it has become less popular.
For the weight loss medication group, they found 117,730 patients who met their criteria. For the surgery group, 43,050 patients met the criteria.
In analyzing the colorectal cancer rates, they included only diagnoses of malignant neoplasms made 2 years after the interventions.
They compared these patients to a control group of 52,540 people matched in age, with a body mass index (BMI) greater than 30 kg/m2 who did not undergo weight loss surgery or take weight loss medication.
Among the 9,370 patients who underwent Roux-en-Y gastrojejunostomy, 50 were diagnosed with colorectal cancer and 400 had benign polyps. Their rate of colorectal cancer was not statistically different from people who didn’t have surgery (OR, 1.09; 95% CI, 0.82-1.43). The rate of benign polyps after Roux-en-Y gastrojejunostomy was greater (OR, 1.72; 95% CI, 1.55-1.90).
On the other hand, among the 33,680 patients who underwent sleeve gastrectomy, 50 were diagnosed with colorectal cancer, a lower rate than in the population who didn’t have surgery (OR, 0.30; 95% CI, 0.22-0.39). Their risk of benign polyps was also reduced (OR, 0.45; 95% CI, 0.40-0.50).
All of the medications were significantly associated with a lower risk of colorectal cancer, except orlistat (OR, 0.94; 95% CI, 0.72-1.25).
The finding on Roux-en-Y gastrojejunostomy agreed with studies from England and Nordic countries showing double the risk of colorectal cancer in those patients but conflicted with a French study showing decreased risk, Dr. Shwani said.
While the study doesn’t establish a reason why Roux-en-Y gastrojejunostomy was less beneficial, other researchers have associated the procedure with biomarkers of inflammation, Dr. Shwani said. “It’s inconsistent, and I don’t think we have a clear answer why.”
As a retrospective analysis, the study could not establish a cause-and-effect relationship between surgery or medication and cancer, or adjust for such factors as diet, exercise, or genes, he acknowledged.
Colorectal cancer is just one outcome to consider when deciding whether to undergo weight loss surgery or take weight loss drugs, said session moderator Mohammad Yaghoobi, MD, an associate professor of medicine at McMaster University, Hamilton, Ont.
“The most important outcome that should be investigated is the survival of the patients after obesity surgery,” he told this news organization. “The second would be the quality of life of those patients. Colon cancer is preventable if you are having regular colonoscopies.”
Other studies have not shown much difference between patients who have weight loss surgery and those who don’t, he added.
The study was funded by Merck. Dr. Desai and Dr. Shwani have reported receiving grant funding from Merck. Dr. Yaghoobi has reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
LAS VEGAS – People with obesity may be able to reduce their risk of colorectal cancer with weight loss surgery or medication, researchers say.
“We need to have conversations with our patients in the clinic and educate them that they have these resources available,” said Aakash Desai, MD, a hospitalist at MetroHealth Medical Center, Cleveland, in an interview with this news organization.
Dr. Desai and colleagues found that sleeve gastrectomy and four medications were associated with a reduced risk of colorectal cancer but Roux-en-Y gastrojejunostomy and orlistat were not.
Coauthor Zryan Shwani, MD, a gastroenterology fellow at Sibley Memorial Hospital, Washington, D.C., presented the findings here at the American College of Gastroenterology (ACG) 2021 Annual Scientific Meeting.
Working with an underserved population with high rates of obesity in northeastern Ohio, the researchers wondered how surgery and medication could affect these patients.
They analyzed data from the IBM Explorys clinical database, which compiles and standardizes data from electronic medical records on about 74 million patients from more than 300 U.S. hospitals. Consistent with previous studies, they determined that patients with obesity in the database were 2.5 times more likely than people with a healthy weight to be diagnosed with colorectal cancer (odds ratio, 2.48; 95% CI, 2.45-2.51).
Zeroing in on people who had weight loss interventions, they included adults aged 18-75 years who had undergone either Roux-en-Y gastrojejunostomy or sleeve gastrectomy, or had taken the medications liraglutide, orlistat, phentermine/topiramate, bupropion/naltrexone, or lorcaserin.
They excluded patients with Lynch syndrome, intestinal polyposis syndrome, a family history of gastrointestinal malignancy, inflammatory bowel disease, or tobacco or alcohol abuse. Patients who had taken one of the weight loss medications and also had type 2 diabetes were excluded. They did not include patients who had undergone gastric banding because it has become less popular.
For the weight loss medication group, they found 117,730 patients who met their criteria. For the surgery group, 43,050 patients met the criteria.
In analyzing the colorectal cancer rates, they included only diagnoses of malignant neoplasms made 2 years after the interventions.
They compared these patients to a control group of 52,540 people matched in age, with a body mass index (BMI) greater than 30 kg/m2 who did not undergo weight loss surgery or take weight loss medication.
Among the 9,370 patients who underwent Roux-en-Y gastrojejunostomy, 50 were diagnosed with colorectal cancer and 400 had benign polyps. Their rate of colorectal cancer was not statistically different from people who didn’t have surgery (OR, 1.09; 95% CI, 0.82-1.43). The rate of benign polyps after Roux-en-Y gastrojejunostomy was greater (OR, 1.72; 95% CI, 1.55-1.90).
On the other hand, among the 33,680 patients who underwent sleeve gastrectomy, 50 were diagnosed with colorectal cancer, a lower rate than in the population who didn’t have surgery (OR, 0.30; 95% CI, 0.22-0.39). Their risk of benign polyps was also reduced (OR, 0.45; 95% CI, 0.40-0.50).
All of the medications were significantly associated with a lower risk of colorectal cancer, except orlistat (OR, 0.94; 95% CI, 0.72-1.25).
The finding on Roux-en-Y gastrojejunostomy agreed with studies from England and Nordic countries showing double the risk of colorectal cancer in those patients but conflicted with a French study showing decreased risk, Dr. Shwani said.
While the study doesn’t establish a reason why Roux-en-Y gastrojejunostomy was less beneficial, other researchers have associated the procedure with biomarkers of inflammation, Dr. Shwani said. “It’s inconsistent, and I don’t think we have a clear answer why.”
As a retrospective analysis, the study could not establish a cause-and-effect relationship between surgery or medication and cancer, or adjust for such factors as diet, exercise, or genes, he acknowledged.
Colorectal cancer is just one outcome to consider when deciding whether to undergo weight loss surgery or take weight loss drugs, said session moderator Mohammad Yaghoobi, MD, an associate professor of medicine at McMaster University, Hamilton, Ont.
“The most important outcome that should be investigated is the survival of the patients after obesity surgery,” he told this news organization. “The second would be the quality of life of those patients. Colon cancer is preventable if you are having regular colonoscopies.”
Other studies have not shown much difference between patients who have weight loss surgery and those who don’t, he added.
The study was funded by Merck. Dr. Desai and Dr. Shwani have reported receiving grant funding from Merck. Dr. Yaghoobi has reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
LAS VEGAS – People with obesity may be able to reduce their risk of colorectal cancer with weight loss surgery or medication, researchers say.
“We need to have conversations with our patients in the clinic and educate them that they have these resources available,” said Aakash Desai, MD, a hospitalist at MetroHealth Medical Center, Cleveland, in an interview with this news organization.
Dr. Desai and colleagues found that sleeve gastrectomy and four medications were associated with a reduced risk of colorectal cancer but Roux-en-Y gastrojejunostomy and orlistat were not.
Coauthor Zryan Shwani, MD, a gastroenterology fellow at Sibley Memorial Hospital, Washington, D.C., presented the findings here at the American College of Gastroenterology (ACG) 2021 Annual Scientific Meeting.
Working with an underserved population with high rates of obesity in northeastern Ohio, the researchers wondered how surgery and medication could affect these patients.
They analyzed data from the IBM Explorys clinical database, which compiles and standardizes data from electronic medical records on about 74 million patients from more than 300 U.S. hospitals. Consistent with previous studies, they determined that patients with obesity in the database were 2.5 times more likely than people with a healthy weight to be diagnosed with colorectal cancer (odds ratio, 2.48; 95% CI, 2.45-2.51).
Zeroing in on people who had weight loss interventions, they included adults aged 18-75 years who had undergone either Roux-en-Y gastrojejunostomy or sleeve gastrectomy, or had taken the medications liraglutide, orlistat, phentermine/topiramate, bupropion/naltrexone, or lorcaserin.
They excluded patients with Lynch syndrome, intestinal polyposis syndrome, a family history of gastrointestinal malignancy, inflammatory bowel disease, or tobacco or alcohol abuse. Patients who had taken one of the weight loss medications and also had type 2 diabetes were excluded. They did not include patients who had undergone gastric banding because it has become less popular.
For the weight loss medication group, they found 117,730 patients who met their criteria. For the surgery group, 43,050 patients met the criteria.
In analyzing the colorectal cancer rates, they included only diagnoses of malignant neoplasms made 2 years after the interventions.
They compared these patients to a control group of 52,540 people matched in age, with a body mass index (BMI) greater than 30 kg/m2 who did not undergo weight loss surgery or take weight loss medication.
Among the 9,370 patients who underwent Roux-en-Y gastrojejunostomy, 50 were diagnosed with colorectal cancer and 400 had benign polyps. Their rate of colorectal cancer was not statistically different from people who didn’t have surgery (OR, 1.09; 95% CI, 0.82-1.43). The rate of benign polyps after Roux-en-Y gastrojejunostomy was greater (OR, 1.72; 95% CI, 1.55-1.90).
On the other hand, among the 33,680 patients who underwent sleeve gastrectomy, 50 were diagnosed with colorectal cancer, a lower rate than in the population who didn’t have surgery (OR, 0.30; 95% CI, 0.22-0.39). Their risk of benign polyps was also reduced (OR, 0.45; 95% CI, 0.40-0.50).
All of the medications were significantly associated with a lower risk of colorectal cancer, except orlistat (OR, 0.94; 95% CI, 0.72-1.25).
The finding on Roux-en-Y gastrojejunostomy agreed with studies from England and Nordic countries showing double the risk of colorectal cancer in those patients but conflicted with a French study showing decreased risk, Dr. Shwani said.
While the study doesn’t establish a reason why Roux-en-Y gastrojejunostomy was less beneficial, other researchers have associated the procedure with biomarkers of inflammation, Dr. Shwani said. “It’s inconsistent, and I don’t think we have a clear answer why.”
As a retrospective analysis, the study could not establish a cause-and-effect relationship between surgery or medication and cancer, or adjust for such factors as diet, exercise, or genes, he acknowledged.
Colorectal cancer is just one outcome to consider when deciding whether to undergo weight loss surgery or take weight loss drugs, said session moderator Mohammad Yaghoobi, MD, an associate professor of medicine at McMaster University, Hamilton, Ont.
“The most important outcome that should be investigated is the survival of the patients after obesity surgery,” he told this news organization. “The second would be the quality of life of those patients. Colon cancer is preventable if you are having regular colonoscopies.”
Other studies have not shown much difference between patients who have weight loss surgery and those who don’t, he added.
The study was funded by Merck. Dr. Desai and Dr. Shwani have reported receiving grant funding from Merck. Dr. Yaghoobi has reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AT ACG 2021
Clinicians may overprescribe clarithromycin for H. pylori
Clinicians are prescribing clarithromycin at high rates for Helicobacter pylori infections, despite increasing resistance to this antibiotic, researchers say.
In an analysis of 1 million U.S. prescriptions for H. pylori infections, 80% contained clarithromycin, said Carol Rockett, PharmD, associate vice president of RedHill Biopharma in Raleigh, N.C.
Dr. Rockett presented the findings at the annual meeting of the American College of Gastroenterology.
“Multiple talks [at the meeting] have suggested that the use of clarithromycin in H. pylori is obsolete,” she told this news organization. “Clarithromycin is particularly ineffective in people with a genetic variant that causes rapid metabolism.”
According to the 2017 ACG clinical guideline for treating H. pylori, patients diagnosed with this infection should be asked about their previous antibiotic exposure prior to treatment.
Additionally, clinicians should prescribe clarithromycin triple therapy with a proton pump inhibitor (PPI) and amoxicillin or metronidazole as a first-line treatment only in “regions where H. pylori clarithromycin resistance is known to be less than 15%” and in patients with no previous history of macrolide exposure.
The guideline puts bismuth quadruple therapy, consisting of a PPI, bismuth, tetracycline, and a nitroimidazole, at the top of its list of six alternative first-line therapies. However, three of the six alternatives include clarithromycin.
ERADICATE Hp and ERADICATE Hp2
To understand how U.S. physicians are treating patients with H. pylori, Dr. Rockett’s colleagues analyzed data from two phase 3 clinical trials of RedHill’s RHB-105 (Talicia): ERADICATE Hp and ERADICATE Hp2.
RHB-105 is an all-in‐one combination of omeprazole (40 mg), amoxicillin (1,000 mg), and rifabutin (50 mg) that the Food and Drug Administration approved for treatment of H pylori in 2019.
The researchers followed 38 subjects from ERADICATE Hp who remained positive for H. pylori after the study’s completion. A total of 33 had received a placebo in that trial, while the other 5 had received RHB-105.
The researchers obtained data on 31 of these patients. The overall cure rate was 61.3%. Of the 31 patients, 27 received a regimen including clarithromycin. Their cure rate was 59.3%.
Turning to ERADICATE Hp2, the researchers obtained data on 94 patients whose H. pylori infections persisted after the trial. Of those, 67 had received an active comparator (amoxicillin 250 mg and omeprazole 10 mg) and 27 had received RHB-105.
The overall cure rate was 56.2%. For the 48 subjects who received therapies including clarithromycin, the cure rate was 60.4%. For the 22 subjects who received a bismuth-based quadruple regimen, the cure rate was 45.4%.
In another analysis, the researchers crunched 12 months of numbers from IQVIA PharMetrics Plus medical and prescription claim database of over 1 million prescriptions for H. pylori. They found that 80% of the prescriptions made by gastroenterologists were for regimens containing clarithromycin. That proportion increased to 84% for physician assistants and internists, 85% for nurse practitioners, 86% for family practitioners, and 89% for general practitioners.
Finally, the researchers also analyzed patients for CYP2C19 gene status. They tested 65 subjects who received RHB-105 in ERADICATE Hp and all 445 subjects in ERADICATE Hp2. They found that 58.5% in ERADICATE Hp and 48.6% in ERADICATE Hp2 were normal metabolizers.
In 20 normal metabolizers who received clarithromycin, the drug eradicated the infection in 16 (80%). Out of 11 rapid metabolizers, clarithromycin eradicated the bacterium in 2 (18.2%). The difference was statistically significant (P = .0017).
“With clarithromycin, you can see that the efficacy is reduced in those patients who are rapid metabolizers,” Dr. Rockett said. “We didn’t see that with rifabutin [one of the drugs in RHB-105].”
Jared Magee, DO, MPH, a gastroenterology fellow at the Walter Reed National Military Medical Center in Bethesda, Md., said in treating H. pylori infections, he checks the patients’ medical records to see what antibiotics they have received in the past and generally begins treatment with the bismuth quadruple therapy.
“There is education needed to get the data out there that clarithromycin-based therapies may not be the right choice for patients,” he said. “There is a subset who will do well with it, but I think where we’re at now, with the frequency of macrolide prescriptions for other conditions, that clarithromycin is going to be a difficult therapy for a lot of people.”
Clinicians who are not gastroenterologists may not be aware of the guideline promulgated by the ACG, he pointed out.
Dr. Rockett is an employee of RedHill Biopharma. Dr. Magee has disclosed no relevant financial relationships. The study was funded by RedHill Biopharma.
A version of this article first appeared on Medscape.com.
Clinicians are prescribing clarithromycin at high rates for Helicobacter pylori infections, despite increasing resistance to this antibiotic, researchers say.
In an analysis of 1 million U.S. prescriptions for H. pylori infections, 80% contained clarithromycin, said Carol Rockett, PharmD, associate vice president of RedHill Biopharma in Raleigh, N.C.
Dr. Rockett presented the findings at the annual meeting of the American College of Gastroenterology.
“Multiple talks [at the meeting] have suggested that the use of clarithromycin in H. pylori is obsolete,” she told this news organization. “Clarithromycin is particularly ineffective in people with a genetic variant that causes rapid metabolism.”
According to the 2017 ACG clinical guideline for treating H. pylori, patients diagnosed with this infection should be asked about their previous antibiotic exposure prior to treatment.
Additionally, clinicians should prescribe clarithromycin triple therapy with a proton pump inhibitor (PPI) and amoxicillin or metronidazole as a first-line treatment only in “regions where H. pylori clarithromycin resistance is known to be less than 15%” and in patients with no previous history of macrolide exposure.
The guideline puts bismuth quadruple therapy, consisting of a PPI, bismuth, tetracycline, and a nitroimidazole, at the top of its list of six alternative first-line therapies. However, three of the six alternatives include clarithromycin.
ERADICATE Hp and ERADICATE Hp2
To understand how U.S. physicians are treating patients with H. pylori, Dr. Rockett’s colleagues analyzed data from two phase 3 clinical trials of RedHill’s RHB-105 (Talicia): ERADICATE Hp and ERADICATE Hp2.
RHB-105 is an all-in‐one combination of omeprazole (40 mg), amoxicillin (1,000 mg), and rifabutin (50 mg) that the Food and Drug Administration approved for treatment of H pylori in 2019.
The researchers followed 38 subjects from ERADICATE Hp who remained positive for H. pylori after the study’s completion. A total of 33 had received a placebo in that trial, while the other 5 had received RHB-105.
The researchers obtained data on 31 of these patients. The overall cure rate was 61.3%. Of the 31 patients, 27 received a regimen including clarithromycin. Their cure rate was 59.3%.
Turning to ERADICATE Hp2, the researchers obtained data on 94 patients whose H. pylori infections persisted after the trial. Of those, 67 had received an active comparator (amoxicillin 250 mg and omeprazole 10 mg) and 27 had received RHB-105.
The overall cure rate was 56.2%. For the 48 subjects who received therapies including clarithromycin, the cure rate was 60.4%. For the 22 subjects who received a bismuth-based quadruple regimen, the cure rate was 45.4%.
In another analysis, the researchers crunched 12 months of numbers from IQVIA PharMetrics Plus medical and prescription claim database of over 1 million prescriptions for H. pylori. They found that 80% of the prescriptions made by gastroenterologists were for regimens containing clarithromycin. That proportion increased to 84% for physician assistants and internists, 85% for nurse practitioners, 86% for family practitioners, and 89% for general practitioners.
Finally, the researchers also analyzed patients for CYP2C19 gene status. They tested 65 subjects who received RHB-105 in ERADICATE Hp and all 445 subjects in ERADICATE Hp2. They found that 58.5% in ERADICATE Hp and 48.6% in ERADICATE Hp2 were normal metabolizers.
In 20 normal metabolizers who received clarithromycin, the drug eradicated the infection in 16 (80%). Out of 11 rapid metabolizers, clarithromycin eradicated the bacterium in 2 (18.2%). The difference was statistically significant (P = .0017).
“With clarithromycin, you can see that the efficacy is reduced in those patients who are rapid metabolizers,” Dr. Rockett said. “We didn’t see that with rifabutin [one of the drugs in RHB-105].”
Jared Magee, DO, MPH, a gastroenterology fellow at the Walter Reed National Military Medical Center in Bethesda, Md., said in treating H. pylori infections, he checks the patients’ medical records to see what antibiotics they have received in the past and generally begins treatment with the bismuth quadruple therapy.
“There is education needed to get the data out there that clarithromycin-based therapies may not be the right choice for patients,” he said. “There is a subset who will do well with it, but I think where we’re at now, with the frequency of macrolide prescriptions for other conditions, that clarithromycin is going to be a difficult therapy for a lot of people.”
Clinicians who are not gastroenterologists may not be aware of the guideline promulgated by the ACG, he pointed out.
Dr. Rockett is an employee of RedHill Biopharma. Dr. Magee has disclosed no relevant financial relationships. The study was funded by RedHill Biopharma.
A version of this article first appeared on Medscape.com.
Clinicians are prescribing clarithromycin at high rates for Helicobacter pylori infections, despite increasing resistance to this antibiotic, researchers say.
In an analysis of 1 million U.S. prescriptions for H. pylori infections, 80% contained clarithromycin, said Carol Rockett, PharmD, associate vice president of RedHill Biopharma in Raleigh, N.C.
Dr. Rockett presented the findings at the annual meeting of the American College of Gastroenterology.
“Multiple talks [at the meeting] have suggested that the use of clarithromycin in H. pylori is obsolete,” she told this news organization. “Clarithromycin is particularly ineffective in people with a genetic variant that causes rapid metabolism.”
According to the 2017 ACG clinical guideline for treating H. pylori, patients diagnosed with this infection should be asked about their previous antibiotic exposure prior to treatment.
Additionally, clinicians should prescribe clarithromycin triple therapy with a proton pump inhibitor (PPI) and amoxicillin or metronidazole as a first-line treatment only in “regions where H. pylori clarithromycin resistance is known to be less than 15%” and in patients with no previous history of macrolide exposure.
The guideline puts bismuth quadruple therapy, consisting of a PPI, bismuth, tetracycline, and a nitroimidazole, at the top of its list of six alternative first-line therapies. However, three of the six alternatives include clarithromycin.
ERADICATE Hp and ERADICATE Hp2
To understand how U.S. physicians are treating patients with H. pylori, Dr. Rockett’s colleagues analyzed data from two phase 3 clinical trials of RedHill’s RHB-105 (Talicia): ERADICATE Hp and ERADICATE Hp2.
RHB-105 is an all-in‐one combination of omeprazole (40 mg), amoxicillin (1,000 mg), and rifabutin (50 mg) that the Food and Drug Administration approved for treatment of H pylori in 2019.
The researchers followed 38 subjects from ERADICATE Hp who remained positive for H. pylori after the study’s completion. A total of 33 had received a placebo in that trial, while the other 5 had received RHB-105.
The researchers obtained data on 31 of these patients. The overall cure rate was 61.3%. Of the 31 patients, 27 received a regimen including clarithromycin. Their cure rate was 59.3%.
Turning to ERADICATE Hp2, the researchers obtained data on 94 patients whose H. pylori infections persisted after the trial. Of those, 67 had received an active comparator (amoxicillin 250 mg and omeprazole 10 mg) and 27 had received RHB-105.
The overall cure rate was 56.2%. For the 48 subjects who received therapies including clarithromycin, the cure rate was 60.4%. For the 22 subjects who received a bismuth-based quadruple regimen, the cure rate was 45.4%.
In another analysis, the researchers crunched 12 months of numbers from IQVIA PharMetrics Plus medical and prescription claim database of over 1 million prescriptions for H. pylori. They found that 80% of the prescriptions made by gastroenterologists were for regimens containing clarithromycin. That proportion increased to 84% for physician assistants and internists, 85% for nurse practitioners, 86% for family practitioners, and 89% for general practitioners.
Finally, the researchers also analyzed patients for CYP2C19 gene status. They tested 65 subjects who received RHB-105 in ERADICATE Hp and all 445 subjects in ERADICATE Hp2. They found that 58.5% in ERADICATE Hp and 48.6% in ERADICATE Hp2 were normal metabolizers.
In 20 normal metabolizers who received clarithromycin, the drug eradicated the infection in 16 (80%). Out of 11 rapid metabolizers, clarithromycin eradicated the bacterium in 2 (18.2%). The difference was statistically significant (P = .0017).
“With clarithromycin, you can see that the efficacy is reduced in those patients who are rapid metabolizers,” Dr. Rockett said. “We didn’t see that with rifabutin [one of the drugs in RHB-105].”
Jared Magee, DO, MPH, a gastroenterology fellow at the Walter Reed National Military Medical Center in Bethesda, Md., said in treating H. pylori infections, he checks the patients’ medical records to see what antibiotics they have received in the past and generally begins treatment with the bismuth quadruple therapy.
“There is education needed to get the data out there that clarithromycin-based therapies may not be the right choice for patients,” he said. “There is a subset who will do well with it, but I think where we’re at now, with the frequency of macrolide prescriptions for other conditions, that clarithromycin is going to be a difficult therapy for a lot of people.”
Clinicians who are not gastroenterologists may not be aware of the guideline promulgated by the ACG, he pointed out.
Dr. Rockett is an employee of RedHill Biopharma. Dr. Magee has disclosed no relevant financial relationships. The study was funded by RedHill Biopharma.
A version of this article first appeared on Medscape.com.
Upadacitinib shows potential for ulcerative colitis
LAS VEGAS – An oral Janus kinase 1 inhibitor upadacitinib (Rinvoq, AbbVie) showed high efficacy and good safety as a treatment for ulcerative colitis in a phase 3 trial.
The finding could provide some reassurance after the Food and Drug Administration recently warned of an increased risk of cancer and heart disease associated with medications in the same class as upadacitinib.
“Serious adverse events were numerically lower in patients on upadacitinib, and discontinuations from the study due to adverse events were also lower” than in patients taking a placebo, said Edward Loftus, MD, a gastroenterologist at the Mayo Clinic in Rochester, Minn.
Dr. Loftus presented the findings from the U-ACCOMPLISH study at the annual meeting of the American College of Gastroenterology.
Although other medications are approved for the treatment of ulcerative colitis, including biologics, many patients do not respond. In 2019, tofacitinib (Xeljanz) became the first JAK inhibitor approved for this condition. It works by blocking the JAK1 and JAK3 inflammation pathways, and at high concentrations, it also blocks the tyrosine kinase 2 and JAK2 pathways.
However, adverse events seen in clinical trials of tofacitinib include pneumonia, herpes zoster, anal abscess, and Clostridioides difficile infections. And, as reported by this news organization in September, the FDA required its manufacturer, Pfizer, to add a boxed warning that includes information about the risks of stroke, cancer, blood clots, and death.
Upadacitinib may be more selective and reversible because it preferentially blocks JAK1 or JAK1/3. In August 2019, it received FDA approval at a dose of 15 mg for adult patients with moderately to severely active RA who have had an inadequate response or intolerance to methotrexate.
But the FDA applied the same warnings to upadacitinib – and to a third related drug, baricitinib (Olumiant) – that it required for tofacitinib, even though they are not as well studied.
The FDA also limited approved uses of these three medications to patients who have not responded well to tumor necrosis factor blockers to ensure their benefits outweigh their risks.
A well-tolerated treatment
U-ACCOMPLISH is one of two phase 3 trials induction trials completed on upadacitinib.
Investigators randomized 522 people with moderately to severely active ulcerative colitis, defined as Adapted Mayo Score 5-9 with a centrally read endoscopic score of 2-3. Of those patients, the intent to treat population included 341 in the upadacitinib group (45 mg once daily) and 174 in the placebo group.
The baseline demographics and disease characteristics were similar between groups. More than two-thirds of patients in both groups were White, and more than two-thirds were men. In the upadacitinib group, 50.7% had responded inadequately to biologic treatments, compared with 51.1% in the placebo group.
After 8 weeks, a significantly higher proportion of patients receiving upadacitinib achieved clinical remission as defined by the adapted Mayo Score (stool frequency subscore ≤1 and not greater than baseline, rectal bleeding subscore of 0, and Mayo endoscopic subscore ≤1).
“In terms of the efficacy, I think it’s very, very promising,” said Derrick Eichele, MD, an assistant professor of gastroenterology-hepatology at the University of Nebraska Medical Center in Omaha, who was not involved in the trial.
The efficacy data were similar to those reported for tofacitinib in clinical trials, he said in an interview. “But I think again, what we’re waiting to see is how is this going to be positioned in relation to tofacitinib in terms of safety profile.”
More patients in the upadacitinib group reported adverse events, including those deemed related to the drug. However, the proportion that were severe, serious, or led to discontinuation was higher in the placebo group. No one in the study died, and no one in the upadacitinib group had an adjudicated major adverse cardiovascular event, tuberculosis, or malignancy.
The most common adverse events were acne, blood creatine phosphokinase elevation, and anemia, which were all more common in the upadacitinib group, and headache and worsening of ulcerative colitis, which were more common in the placebo group.
Among adverse events of special interest, anemia, neutropenia, hepatic disorder, lymphopenia, serious infection, and opportunistic infection were more common in the upadacitinib group than in the placebo group. The four opportunistic infections in the upadacitinib group included two cases of herpes zoster.
In reviewing the poster presented at this meeting, the cases of neutropenia and hepatic disorder in the upadacitinib group stood out for Dr. Eichele. But he said it’s hard to pass judgment based on this amount of data. He is looking forward to a peer-reviewed publication. “I’ll be interested to see what it shows in terms of the details.”
Phase 3 trials of upadacitinib are underway in atopic dermatitis, RA, psoriatic arthritis, axial spondyloarthritis, Crohn’s disease, giant cell arteritis, and Takayasu arteritis as well as ulcerative colitis.
In a 52-week maintenance trial, according to a press release, malignancies (excluding nonmelanoma skin cancer) included one event among 148 people taking a 15-mg dose of upadacitinib 15, two events among 154 people taking a 30-mg dose of upadacitinib, and one event among 149 people in the placebo group.
Two cases of pulmonary embolism were reported in the 15-mg group and two cases of deep vein thrombosis were reported in the 30-mg group, compared with one event of ovarian vein thrombosis in the placebo group. One adjudicated major cardiovascular event each were reported in the upadacitinib 30-mg group and the placebo group. No one died.
The study was funded by AbbVie. Dr. Loftus reported that he is a consultant for AbbVie as well as multiple other gastroenterology drug companies. Dr. Eichele disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
LAS VEGAS – An oral Janus kinase 1 inhibitor upadacitinib (Rinvoq, AbbVie) showed high efficacy and good safety as a treatment for ulcerative colitis in a phase 3 trial.
The finding could provide some reassurance after the Food and Drug Administration recently warned of an increased risk of cancer and heart disease associated with medications in the same class as upadacitinib.
“Serious adverse events were numerically lower in patients on upadacitinib, and discontinuations from the study due to adverse events were also lower” than in patients taking a placebo, said Edward Loftus, MD, a gastroenterologist at the Mayo Clinic in Rochester, Minn.
Dr. Loftus presented the findings from the U-ACCOMPLISH study at the annual meeting of the American College of Gastroenterology.
Although other medications are approved for the treatment of ulcerative colitis, including biologics, many patients do not respond. In 2019, tofacitinib (Xeljanz) became the first JAK inhibitor approved for this condition. It works by blocking the JAK1 and JAK3 inflammation pathways, and at high concentrations, it also blocks the tyrosine kinase 2 and JAK2 pathways.
However, adverse events seen in clinical trials of tofacitinib include pneumonia, herpes zoster, anal abscess, and Clostridioides difficile infections. And, as reported by this news organization in September, the FDA required its manufacturer, Pfizer, to add a boxed warning that includes information about the risks of stroke, cancer, blood clots, and death.
Upadacitinib may be more selective and reversible because it preferentially blocks JAK1 or JAK1/3. In August 2019, it received FDA approval at a dose of 15 mg for adult patients with moderately to severely active RA who have had an inadequate response or intolerance to methotrexate.
But the FDA applied the same warnings to upadacitinib – and to a third related drug, baricitinib (Olumiant) – that it required for tofacitinib, even though they are not as well studied.
The FDA also limited approved uses of these three medications to patients who have not responded well to tumor necrosis factor blockers to ensure their benefits outweigh their risks.
A well-tolerated treatment
U-ACCOMPLISH is one of two phase 3 trials induction trials completed on upadacitinib.
Investigators randomized 522 people with moderately to severely active ulcerative colitis, defined as Adapted Mayo Score 5-9 with a centrally read endoscopic score of 2-3. Of those patients, the intent to treat population included 341 in the upadacitinib group (45 mg once daily) and 174 in the placebo group.
The baseline demographics and disease characteristics were similar between groups. More than two-thirds of patients in both groups were White, and more than two-thirds were men. In the upadacitinib group, 50.7% had responded inadequately to biologic treatments, compared with 51.1% in the placebo group.
After 8 weeks, a significantly higher proportion of patients receiving upadacitinib achieved clinical remission as defined by the adapted Mayo Score (stool frequency subscore ≤1 and not greater than baseline, rectal bleeding subscore of 0, and Mayo endoscopic subscore ≤1).
“In terms of the efficacy, I think it’s very, very promising,” said Derrick Eichele, MD, an assistant professor of gastroenterology-hepatology at the University of Nebraska Medical Center in Omaha, who was not involved in the trial.
The efficacy data were similar to those reported for tofacitinib in clinical trials, he said in an interview. “But I think again, what we’re waiting to see is how is this going to be positioned in relation to tofacitinib in terms of safety profile.”
More patients in the upadacitinib group reported adverse events, including those deemed related to the drug. However, the proportion that were severe, serious, or led to discontinuation was higher in the placebo group. No one in the study died, and no one in the upadacitinib group had an adjudicated major adverse cardiovascular event, tuberculosis, or malignancy.
The most common adverse events were acne, blood creatine phosphokinase elevation, and anemia, which were all more common in the upadacitinib group, and headache and worsening of ulcerative colitis, which were more common in the placebo group.
Among adverse events of special interest, anemia, neutropenia, hepatic disorder, lymphopenia, serious infection, and opportunistic infection were more common in the upadacitinib group than in the placebo group. The four opportunistic infections in the upadacitinib group included two cases of herpes zoster.
In reviewing the poster presented at this meeting, the cases of neutropenia and hepatic disorder in the upadacitinib group stood out for Dr. Eichele. But he said it’s hard to pass judgment based on this amount of data. He is looking forward to a peer-reviewed publication. “I’ll be interested to see what it shows in terms of the details.”
Phase 3 trials of upadacitinib are underway in atopic dermatitis, RA, psoriatic arthritis, axial spondyloarthritis, Crohn’s disease, giant cell arteritis, and Takayasu arteritis as well as ulcerative colitis.
In a 52-week maintenance trial, according to a press release, malignancies (excluding nonmelanoma skin cancer) included one event among 148 people taking a 15-mg dose of upadacitinib 15, two events among 154 people taking a 30-mg dose of upadacitinib, and one event among 149 people in the placebo group.
Two cases of pulmonary embolism were reported in the 15-mg group and two cases of deep vein thrombosis were reported in the 30-mg group, compared with one event of ovarian vein thrombosis in the placebo group. One adjudicated major cardiovascular event each were reported in the upadacitinib 30-mg group and the placebo group. No one died.
The study was funded by AbbVie. Dr. Loftus reported that he is a consultant for AbbVie as well as multiple other gastroenterology drug companies. Dr. Eichele disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
LAS VEGAS – An oral Janus kinase 1 inhibitor upadacitinib (Rinvoq, AbbVie) showed high efficacy and good safety as a treatment for ulcerative colitis in a phase 3 trial.
The finding could provide some reassurance after the Food and Drug Administration recently warned of an increased risk of cancer and heart disease associated with medications in the same class as upadacitinib.
“Serious adverse events were numerically lower in patients on upadacitinib, and discontinuations from the study due to adverse events were also lower” than in patients taking a placebo, said Edward Loftus, MD, a gastroenterologist at the Mayo Clinic in Rochester, Minn.
Dr. Loftus presented the findings from the U-ACCOMPLISH study at the annual meeting of the American College of Gastroenterology.
Although other medications are approved for the treatment of ulcerative colitis, including biologics, many patients do not respond. In 2019, tofacitinib (Xeljanz) became the first JAK inhibitor approved for this condition. It works by blocking the JAK1 and JAK3 inflammation pathways, and at high concentrations, it also blocks the tyrosine kinase 2 and JAK2 pathways.
However, adverse events seen in clinical trials of tofacitinib include pneumonia, herpes zoster, anal abscess, and Clostridioides difficile infections. And, as reported by this news organization in September, the FDA required its manufacturer, Pfizer, to add a boxed warning that includes information about the risks of stroke, cancer, blood clots, and death.
Upadacitinib may be more selective and reversible because it preferentially blocks JAK1 or JAK1/3. In August 2019, it received FDA approval at a dose of 15 mg for adult patients with moderately to severely active RA who have had an inadequate response or intolerance to methotrexate.
But the FDA applied the same warnings to upadacitinib – and to a third related drug, baricitinib (Olumiant) – that it required for tofacitinib, even though they are not as well studied.
The FDA also limited approved uses of these three medications to patients who have not responded well to tumor necrosis factor blockers to ensure their benefits outweigh their risks.
A well-tolerated treatment
U-ACCOMPLISH is one of two phase 3 trials induction trials completed on upadacitinib.
Investigators randomized 522 people with moderately to severely active ulcerative colitis, defined as Adapted Mayo Score 5-9 with a centrally read endoscopic score of 2-3. Of those patients, the intent to treat population included 341 in the upadacitinib group (45 mg once daily) and 174 in the placebo group.
The baseline demographics and disease characteristics were similar between groups. More than two-thirds of patients in both groups were White, and more than two-thirds were men. In the upadacitinib group, 50.7% had responded inadequately to biologic treatments, compared with 51.1% in the placebo group.
After 8 weeks, a significantly higher proportion of patients receiving upadacitinib achieved clinical remission as defined by the adapted Mayo Score (stool frequency subscore ≤1 and not greater than baseline, rectal bleeding subscore of 0, and Mayo endoscopic subscore ≤1).
“In terms of the efficacy, I think it’s very, very promising,” said Derrick Eichele, MD, an assistant professor of gastroenterology-hepatology at the University of Nebraska Medical Center in Omaha, who was not involved in the trial.
The efficacy data were similar to those reported for tofacitinib in clinical trials, he said in an interview. “But I think again, what we’re waiting to see is how is this going to be positioned in relation to tofacitinib in terms of safety profile.”
More patients in the upadacitinib group reported adverse events, including those deemed related to the drug. However, the proportion that were severe, serious, or led to discontinuation was higher in the placebo group. No one in the study died, and no one in the upadacitinib group had an adjudicated major adverse cardiovascular event, tuberculosis, or malignancy.
The most common adverse events were acne, blood creatine phosphokinase elevation, and anemia, which were all more common in the upadacitinib group, and headache and worsening of ulcerative colitis, which were more common in the placebo group.
Among adverse events of special interest, anemia, neutropenia, hepatic disorder, lymphopenia, serious infection, and opportunistic infection were more common in the upadacitinib group than in the placebo group. The four opportunistic infections in the upadacitinib group included two cases of herpes zoster.
In reviewing the poster presented at this meeting, the cases of neutropenia and hepatic disorder in the upadacitinib group stood out for Dr. Eichele. But he said it’s hard to pass judgment based on this amount of data. He is looking forward to a peer-reviewed publication. “I’ll be interested to see what it shows in terms of the details.”
Phase 3 trials of upadacitinib are underway in atopic dermatitis, RA, psoriatic arthritis, axial spondyloarthritis, Crohn’s disease, giant cell arteritis, and Takayasu arteritis as well as ulcerative colitis.
In a 52-week maintenance trial, according to a press release, malignancies (excluding nonmelanoma skin cancer) included one event among 148 people taking a 15-mg dose of upadacitinib 15, two events among 154 people taking a 30-mg dose of upadacitinib, and one event among 149 people in the placebo group.
Two cases of pulmonary embolism were reported in the 15-mg group and two cases of deep vein thrombosis were reported in the 30-mg group, compared with one event of ovarian vein thrombosis in the placebo group. One adjudicated major cardiovascular event each were reported in the upadacitinib 30-mg group and the placebo group. No one died.
The study was funded by AbbVie. Dr. Loftus reported that he is a consultant for AbbVie as well as multiple other gastroenterology drug companies. Dr. Eichele disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AT ACG 2021
Balloon-enhanced colonoscopy finds more adenomas
LAS VEGAS – The G-EYE colonoscope facilitates detection of adenomas more so than does the Endocuff Vision, researchers say.
In the first head-to-head comparison of two mechanical enhancement colonoscopy devices, “the G-EYE demonstrated a meaningful increase in adenoma detection rate [ADR] over Endocuff, particularly for advanced adenomas,” said Seth Gross, MD, a professor of medicine at New York University.
Previous studies have shown that mechanical enhancements are more effective than optical enhancements, Dr. Gross said. “To take it a step further, when you look at mechanical devices, especially these two, in past studies, the G-EYE has been sort of the leader in adenoma detection,” he told this news organization.
But until now, no studies had compared them head to head, said Dr. Gross, who presented the finding here at the American College of Gastroenterology (ACG) 2021 Annual Scientific Meeting.
The two devices work differently. The Endocuff Vision fits onto the colonoscope tip. During withdrawal, it expands radially, and its arms flatten the folds within the colon. The G-EYE balloon is deflated at insertion, then is inflated at the cecum, smoothing the colon wall while centering the colonoscopic view.
To compare the two, Dr. Gross and colleagues randomly assigned 363 patients to undergo colonoscopy with G-EYE and 364 patients to undergo colonoscopy with Endocuff Vision. The two groups were similar in demographics.
Withdrawal times were >6 minutes in both groups. The researchers detected adenomas in a higher percentage of patients with the G-EYE than with the Endocuff Vision. The same was true for advanced adenomas.
When using the G-EYE, the researchers also found more adenomas per patient, more sessile serrated adenomas per patient, more large adenomas per patient, and more right colon adenomas per patient.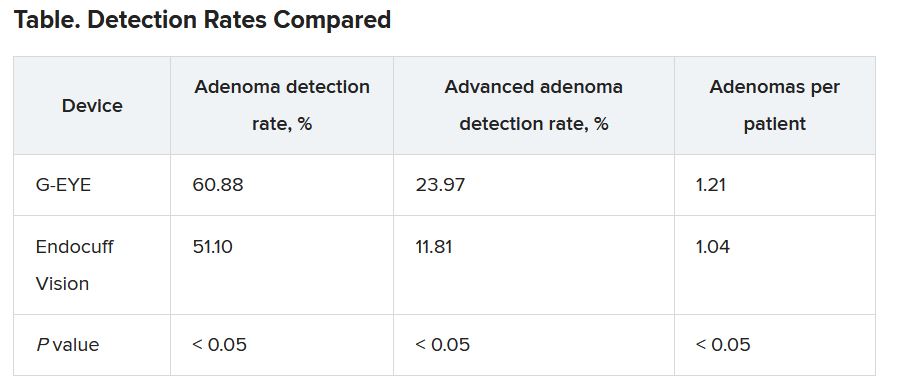
The benchmark for ADR is only 25%, Dr. Gross said, suggesting that both devices are a worthwhile improvement over standard colonoscopes. “It supports the past literature that a mechanical enhancement is something that should be considered during colonoscopy,” he said.
Costs differ as well. The G-EYE requires a permanent modification to the bending rubber of the colonoscope, so the cost is up front. The Endocuff Vision utilizes a single-use cap that is placed on the tip, so costs are spread over time.
The G-EYE gained U.S. Food and Drug Administration clearance in May 2020. In 2016, the Endocuff (an earlier version of the Endocuff Vision) became the first mechanical device the use of which the FDA acknowledged improved ADRs.
Dr. Gross said that it would be interesting to see whether the mechanical devices and artificial intelligence enhancements could complement each other so as to yield even higher detection rates.
Session moderator Brooks Cash, MD, a professor of medicine at the University of Texas Health Science Center, Houston, said the difference in detection rates made an impressive case for the G-EYE.
“I wouldn’t say I’m convinced,” Dr. Cash said in an interview. “I’d like to see more data. But I think that the plurality of the evidence that they presented and the size of the study were certainly compelling.”
He added that he’d like to see evidence that adding the balloon to a colonoscope doesn’t complicate the cleaning of the device.
Dr. Gross has a financial relationship with Olympus, the maker of the Endocuff Vision. Dr. Cash reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
LAS VEGAS – The G-EYE colonoscope facilitates detection of adenomas more so than does the Endocuff Vision, researchers say.
In the first head-to-head comparison of two mechanical enhancement colonoscopy devices, “the G-EYE demonstrated a meaningful increase in adenoma detection rate [ADR] over Endocuff, particularly for advanced adenomas,” said Seth Gross, MD, a professor of medicine at New York University.
Previous studies have shown that mechanical enhancements are more effective than optical enhancements, Dr. Gross said. “To take it a step further, when you look at mechanical devices, especially these two, in past studies, the G-EYE has been sort of the leader in adenoma detection,” he told this news organization.
But until now, no studies had compared them head to head, said Dr. Gross, who presented the finding here at the American College of Gastroenterology (ACG) 2021 Annual Scientific Meeting.
The two devices work differently. The Endocuff Vision fits onto the colonoscope tip. During withdrawal, it expands radially, and its arms flatten the folds within the colon. The G-EYE balloon is deflated at insertion, then is inflated at the cecum, smoothing the colon wall while centering the colonoscopic view.
To compare the two, Dr. Gross and colleagues randomly assigned 363 patients to undergo colonoscopy with G-EYE and 364 patients to undergo colonoscopy with Endocuff Vision. The two groups were similar in demographics.
Withdrawal times were >6 minutes in both groups. The researchers detected adenomas in a higher percentage of patients with the G-EYE than with the Endocuff Vision. The same was true for advanced adenomas.
When using the G-EYE, the researchers also found more adenomas per patient, more sessile serrated adenomas per patient, more large adenomas per patient, and more right colon adenomas per patient.
The benchmark for ADR is only 25%, Dr. Gross said, suggesting that both devices are a worthwhile improvement over standard colonoscopes. “It supports the past literature that a mechanical enhancement is something that should be considered during colonoscopy,” he said.
Costs differ as well. The G-EYE requires a permanent modification to the bending rubber of the colonoscope, so the cost is up front. The Endocuff Vision utilizes a single-use cap that is placed on the tip, so costs are spread over time.
The G-EYE gained U.S. Food and Drug Administration clearance in May 2020. In 2016, the Endocuff (an earlier version of the Endocuff Vision) became the first mechanical device the use of which the FDA acknowledged improved ADRs.
Dr. Gross said that it would be interesting to see whether the mechanical devices and artificial intelligence enhancements could complement each other so as to yield even higher detection rates.
Session moderator Brooks Cash, MD, a professor of medicine at the University of Texas Health Science Center, Houston, said the difference in detection rates made an impressive case for the G-EYE.
“I wouldn’t say I’m convinced,” Dr. Cash said in an interview. “I’d like to see more data. But I think that the plurality of the evidence that they presented and the size of the study were certainly compelling.”
He added that he’d like to see evidence that adding the balloon to a colonoscope doesn’t complicate the cleaning of the device.
Dr. Gross has a financial relationship with Olympus, the maker of the Endocuff Vision. Dr. Cash reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
LAS VEGAS – The G-EYE colonoscope facilitates detection of adenomas more so than does the Endocuff Vision, researchers say.
In the first head-to-head comparison of two mechanical enhancement colonoscopy devices, “the G-EYE demonstrated a meaningful increase in adenoma detection rate [ADR] over Endocuff, particularly for advanced adenomas,” said Seth Gross, MD, a professor of medicine at New York University.
Previous studies have shown that mechanical enhancements are more effective than optical enhancements, Dr. Gross said. “To take it a step further, when you look at mechanical devices, especially these two, in past studies, the G-EYE has been sort of the leader in adenoma detection,” he told this news organization.
But until now, no studies had compared them head to head, said Dr. Gross, who presented the finding here at the American College of Gastroenterology (ACG) 2021 Annual Scientific Meeting.
The two devices work differently. The Endocuff Vision fits onto the colonoscope tip. During withdrawal, it expands radially, and its arms flatten the folds within the colon. The G-EYE balloon is deflated at insertion, then is inflated at the cecum, smoothing the colon wall while centering the colonoscopic view.
To compare the two, Dr. Gross and colleagues randomly assigned 363 patients to undergo colonoscopy with G-EYE and 364 patients to undergo colonoscopy with Endocuff Vision. The two groups were similar in demographics.
Withdrawal times were >6 minutes in both groups. The researchers detected adenomas in a higher percentage of patients with the G-EYE than with the Endocuff Vision. The same was true for advanced adenomas.
When using the G-EYE, the researchers also found more adenomas per patient, more sessile serrated adenomas per patient, more large adenomas per patient, and more right colon adenomas per patient.
The benchmark for ADR is only 25%, Dr. Gross said, suggesting that both devices are a worthwhile improvement over standard colonoscopes. “It supports the past literature that a mechanical enhancement is something that should be considered during colonoscopy,” he said.
Costs differ as well. The G-EYE requires a permanent modification to the bending rubber of the colonoscope, so the cost is up front. The Endocuff Vision utilizes a single-use cap that is placed on the tip, so costs are spread over time.
The G-EYE gained U.S. Food and Drug Administration clearance in May 2020. In 2016, the Endocuff (an earlier version of the Endocuff Vision) became the first mechanical device the use of which the FDA acknowledged improved ADRs.
Dr. Gross said that it would be interesting to see whether the mechanical devices and artificial intelligence enhancements could complement each other so as to yield even higher detection rates.
Session moderator Brooks Cash, MD, a professor of medicine at the University of Texas Health Science Center, Houston, said the difference in detection rates made an impressive case for the G-EYE.
“I wouldn’t say I’m convinced,” Dr. Cash said in an interview. “I’d like to see more data. But I think that the plurality of the evidence that they presented and the size of the study were certainly compelling.”
He added that he’d like to see evidence that adding the balloon to a colonoscope doesn’t complicate the cleaning of the device.
Dr. Gross has a financial relationship with Olympus, the maker of the Endocuff Vision. Dr. Cash reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AT ACG 2021
A pill for C. difficile works by increasing microbiome diversity
CP101, under development by Finch Therapeutics, proved more effective than a placebo in preventing recurrent infections for up to 24 weeks.
The CP101 capsules contain a powder of freeze-dried human stools from screened donors. They restore natural diversity that has been disrupted by antibiotics, said Jessica Allegretti, MD, MPH a gastroenterologist at Brigham and Women’s Hospital in Boston.
The treatment offers an alternative to fecal microbiota transplant, which can effectively treat antibiotic-resistant C. difficile infections but is difficult to standardize and administer – and doesn’t have full approval from the U.S. Food and Drug Administration, she added.
“I think this marks a moment in this space where we’re going to have better, safer, and more available options for patients,” she said in an interview. “It’s exciting.”
Dr. Allegretti is an author on three presentations of results from PRISM3, a phase 2 trial of CP101. They will be presented this week at the annual meeting of the American College of Gastroenterology. These results extend out to 24 weeks, whereas the 8-week results of this trial were presented a year ago at the same meeting.
Study details
The study enrolled 198 people who received antibiotics for recurrent C. difficile infections. Some patients had two or more recurrences, while others had only one recurrence but were 65 years of age or older.
“That was a unique aspect of this study, to see the effect of bringing a therapy like CP101 earlier in the treatment paradigm,” said Dr. Allegretti. “You can imagine for an older, frail, or more fragile patient that you would want to get rid of this [infection] earlier.”
After waiting 2-6 days for the antibiotics to wash out, the researchers randomly assigned 102 of these patients to take the CP101 pills orally and 96 to take placebo pills, both without bowel preparation.
The two groups were not significantly different in age, gender, comorbidities, the number of C. difficile recurrences, or the type of test used to diagnose the infection (PCR-based vs. toxin EIA-based).
After 8 weeks, 74.5% of those given the CP101 pills had not had a recurrence, compared with 61.5% of those given the placebo. The difference was just barely statistically significant (P = .0488).
Sixteen weeks later, the effect endured, with 73.5% of the CP101 group and 59.4% of the placebo group still free of recurrence. The statistical significance of the difference improved slightly (P = .0347).
Drug-related emergent adverse events were similar between the two groups: 16.3% for the CP101 group vs. 19.2% for the placebo group. These were mostly gastrointestinal symptoms, and none were serious.
Some of the patients received vancomycin as a first-line treatment for C. difficile infections, and the researchers wondered if the washout period was not sufficient to purge that antibiotic, leaving enough to interfere with the effectiveness of CP101.
Therefore, they separately analyzed 40 patients treated with fidaxomicin, which they expected to wash out more quickly. Among these patients, 81% who received CP101 were free of recurrences, at 8 weeks and 24 weeks. This compared with 42.1% of those who received the placebo, at both time points. This difference was more statistically significant (P = .0211).
Understanding how it works
To understand better how CP101 achieves its effects, the researchers collected stool samples from the patients and counted the number of different kinds organisms in each sample.
At baseline, the patients had about the same number, but after a week the diversity was greater in the patients treated with CP101, and that difference had increased at week 8. The researchers also found much less diversity of organisms in the stools of those patients who had recurrences of C. difficile infection.
The diversity of microbes in the successfully treated patients appeared to have been introduced by CP101. Dr. Allegretti and colleagues measured the number of organisms in the stool samples that came from CP101. They found that 96% of patients colonized by the CP101 organisms had avoided recurrence of the C. difficile infections, compared with 54.2% of those patients not colonized by these microbes.
“We now have some microbiome-based markers that show us as early as week 1 that the patient is going to be cured or not,” Dr. Allegretti said.
Based on these results, Finch plans to launch a phase 3 trial soon, she said.
The data on colonization is interesting because it has not been found with fecal microbiota transplants, said Purna Kashyap, MBBS, codirector of the Microbiome Program at the Mayo Clinic College of Medicine in Rochester, Minn., who was not involved in the study.
But to better interpret the data, it would be helpful to know more about how the placebo and CP101 groups compared at baseline with regard to medications, immunosuppression, and antibiotics used to treat the C. difficile infections, Dr. Kashyap said. He was struck by the lower cure rate in the portion of the placebo group treated with fidaxomicin.
“Overall, I think these are exciting observations based on the data but require careful review of the entire data to make sense of [them], which will happen when it goes through peer review,” he told this news organization in an email.
Several other standardized microbiota restoration products are under development, including at least two other capsules. In contrast to CP101, which is made up of whole stool, VE303 (Vedanta Biosciences) is a “rationally defined bacterial consortium,” and SER-109 (Seres Therapeutics) is a “consortium of highly purified Firmicutes spores.” VE303 has completed a phase 2 trial, and SER-109 has completed a phase 3 trial.
Dr. Allegretti is a consultant for Finch Therapeutics, which funded the trial. Dr. Kashyap has disclosed no relevant financial relationships.
Help your patients understand their C. difficile diagnosis by sharing patient education from the AGA GI Patient Center: www.gastro.org/Cdiff.
A version of this article first appeared on Medscape.com.
CP101, under development by Finch Therapeutics, proved more effective than a placebo in preventing recurrent infections for up to 24 weeks.
The CP101 capsules contain a powder of freeze-dried human stools from screened donors. They restore natural diversity that has been disrupted by antibiotics, said Jessica Allegretti, MD, MPH a gastroenterologist at Brigham and Women’s Hospital in Boston.
The treatment offers an alternative to fecal microbiota transplant, which can effectively treat antibiotic-resistant C. difficile infections but is difficult to standardize and administer – and doesn’t have full approval from the U.S. Food and Drug Administration, she added.
“I think this marks a moment in this space where we’re going to have better, safer, and more available options for patients,” she said in an interview. “It’s exciting.”
Dr. Allegretti is an author on three presentations of results from PRISM3, a phase 2 trial of CP101. They will be presented this week at the annual meeting of the American College of Gastroenterology. These results extend out to 24 weeks, whereas the 8-week results of this trial were presented a year ago at the same meeting.
Study details
The study enrolled 198 people who received antibiotics for recurrent C. difficile infections. Some patients had two or more recurrences, while others had only one recurrence but were 65 years of age or older.
“That was a unique aspect of this study, to see the effect of bringing a therapy like CP101 earlier in the treatment paradigm,” said Dr. Allegretti. “You can imagine for an older, frail, or more fragile patient that you would want to get rid of this [infection] earlier.”
After waiting 2-6 days for the antibiotics to wash out, the researchers randomly assigned 102 of these patients to take the CP101 pills orally and 96 to take placebo pills, both without bowel preparation.
The two groups were not significantly different in age, gender, comorbidities, the number of C. difficile recurrences, or the type of test used to diagnose the infection (PCR-based vs. toxin EIA-based).
After 8 weeks, 74.5% of those given the CP101 pills had not had a recurrence, compared with 61.5% of those given the placebo. The difference was just barely statistically significant (P = .0488).
Sixteen weeks later, the effect endured, with 73.5% of the CP101 group and 59.4% of the placebo group still free of recurrence. The statistical significance of the difference improved slightly (P = .0347).
Drug-related emergent adverse events were similar between the two groups: 16.3% for the CP101 group vs. 19.2% for the placebo group. These were mostly gastrointestinal symptoms, and none were serious.
Some of the patients received vancomycin as a first-line treatment for C. difficile infections, and the researchers wondered if the washout period was not sufficient to purge that antibiotic, leaving enough to interfere with the effectiveness of CP101.
Therefore, they separately analyzed 40 patients treated with fidaxomicin, which they expected to wash out more quickly. Among these patients, 81% who received CP101 were free of recurrences, at 8 weeks and 24 weeks. This compared with 42.1% of those who received the placebo, at both time points. This difference was more statistically significant (P = .0211).
Understanding how it works
To understand better how CP101 achieves its effects, the researchers collected stool samples from the patients and counted the number of different kinds organisms in each sample.
At baseline, the patients had about the same number, but after a week the diversity was greater in the patients treated with CP101, and that difference had increased at week 8. The researchers also found much less diversity of organisms in the stools of those patients who had recurrences of C. difficile infection.
The diversity of microbes in the successfully treated patients appeared to have been introduced by CP101. Dr. Allegretti and colleagues measured the number of organisms in the stool samples that came from CP101. They found that 96% of patients colonized by the CP101 organisms had avoided recurrence of the C. difficile infections, compared with 54.2% of those patients not colonized by these microbes.
“We now have some microbiome-based markers that show us as early as week 1 that the patient is going to be cured or not,” Dr. Allegretti said.
Based on these results, Finch plans to launch a phase 3 trial soon, she said.
The data on colonization is interesting because it has not been found with fecal microbiota transplants, said Purna Kashyap, MBBS, codirector of the Microbiome Program at the Mayo Clinic College of Medicine in Rochester, Minn., who was not involved in the study.
But to better interpret the data, it would be helpful to know more about how the placebo and CP101 groups compared at baseline with regard to medications, immunosuppression, and antibiotics used to treat the C. difficile infections, Dr. Kashyap said. He was struck by the lower cure rate in the portion of the placebo group treated with fidaxomicin.
“Overall, I think these are exciting observations based on the data but require careful review of the entire data to make sense of [them], which will happen when it goes through peer review,” he told this news organization in an email.
Several other standardized microbiota restoration products are under development, including at least two other capsules. In contrast to CP101, which is made up of whole stool, VE303 (Vedanta Biosciences) is a “rationally defined bacterial consortium,” and SER-109 (Seres Therapeutics) is a “consortium of highly purified Firmicutes spores.” VE303 has completed a phase 2 trial, and SER-109 has completed a phase 3 trial.
Dr. Allegretti is a consultant for Finch Therapeutics, which funded the trial. Dr. Kashyap has disclosed no relevant financial relationships.
Help your patients understand their C. difficile diagnosis by sharing patient education from the AGA GI Patient Center: www.gastro.org/Cdiff.
A version of this article first appeared on Medscape.com.
CP101, under development by Finch Therapeutics, proved more effective than a placebo in preventing recurrent infections for up to 24 weeks.
The CP101 capsules contain a powder of freeze-dried human stools from screened donors. They restore natural diversity that has been disrupted by antibiotics, said Jessica Allegretti, MD, MPH a gastroenterologist at Brigham and Women’s Hospital in Boston.
The treatment offers an alternative to fecal microbiota transplant, which can effectively treat antibiotic-resistant C. difficile infections but is difficult to standardize and administer – and doesn’t have full approval from the U.S. Food and Drug Administration, she added.
“I think this marks a moment in this space where we’re going to have better, safer, and more available options for patients,” she said in an interview. “It’s exciting.”
Dr. Allegretti is an author on three presentations of results from PRISM3, a phase 2 trial of CP101. They will be presented this week at the annual meeting of the American College of Gastroenterology. These results extend out to 24 weeks, whereas the 8-week results of this trial were presented a year ago at the same meeting.
Study details
The study enrolled 198 people who received antibiotics for recurrent C. difficile infections. Some patients had two or more recurrences, while others had only one recurrence but were 65 years of age or older.
“That was a unique aspect of this study, to see the effect of bringing a therapy like CP101 earlier in the treatment paradigm,” said Dr. Allegretti. “You can imagine for an older, frail, or more fragile patient that you would want to get rid of this [infection] earlier.”
After waiting 2-6 days for the antibiotics to wash out, the researchers randomly assigned 102 of these patients to take the CP101 pills orally and 96 to take placebo pills, both without bowel preparation.
The two groups were not significantly different in age, gender, comorbidities, the number of C. difficile recurrences, or the type of test used to diagnose the infection (PCR-based vs. toxin EIA-based).
After 8 weeks, 74.5% of those given the CP101 pills had not had a recurrence, compared with 61.5% of those given the placebo. The difference was just barely statistically significant (P = .0488).
Sixteen weeks later, the effect endured, with 73.5% of the CP101 group and 59.4% of the placebo group still free of recurrence. The statistical significance of the difference improved slightly (P = .0347).
Drug-related emergent adverse events were similar between the two groups: 16.3% for the CP101 group vs. 19.2% for the placebo group. These were mostly gastrointestinal symptoms, and none were serious.
Some of the patients received vancomycin as a first-line treatment for C. difficile infections, and the researchers wondered if the washout period was not sufficient to purge that antibiotic, leaving enough to interfere with the effectiveness of CP101.
Therefore, they separately analyzed 40 patients treated with fidaxomicin, which they expected to wash out more quickly. Among these patients, 81% who received CP101 were free of recurrences, at 8 weeks and 24 weeks. This compared with 42.1% of those who received the placebo, at both time points. This difference was more statistically significant (P = .0211).
Understanding how it works
To understand better how CP101 achieves its effects, the researchers collected stool samples from the patients and counted the number of different kinds organisms in each sample.
At baseline, the patients had about the same number, but after a week the diversity was greater in the patients treated with CP101, and that difference had increased at week 8. The researchers also found much less diversity of organisms in the stools of those patients who had recurrences of C. difficile infection.
The diversity of microbes in the successfully treated patients appeared to have been introduced by CP101. Dr. Allegretti and colleagues measured the number of organisms in the stool samples that came from CP101. They found that 96% of patients colonized by the CP101 organisms had avoided recurrence of the C. difficile infections, compared with 54.2% of those patients not colonized by these microbes.
“We now have some microbiome-based markers that show us as early as week 1 that the patient is going to be cured or not,” Dr. Allegretti said.
Based on these results, Finch plans to launch a phase 3 trial soon, she said.
The data on colonization is interesting because it has not been found with fecal microbiota transplants, said Purna Kashyap, MBBS, codirector of the Microbiome Program at the Mayo Clinic College of Medicine in Rochester, Minn., who was not involved in the study.
But to better interpret the data, it would be helpful to know more about how the placebo and CP101 groups compared at baseline with regard to medications, immunosuppression, and antibiotics used to treat the C. difficile infections, Dr. Kashyap said. He was struck by the lower cure rate in the portion of the placebo group treated with fidaxomicin.
“Overall, I think these are exciting observations based on the data but require careful review of the entire data to make sense of [them], which will happen when it goes through peer review,” he told this news organization in an email.
Several other standardized microbiota restoration products are under development, including at least two other capsules. In contrast to CP101, which is made up of whole stool, VE303 (Vedanta Biosciences) is a “rationally defined bacterial consortium,” and SER-109 (Seres Therapeutics) is a “consortium of highly purified Firmicutes spores.” VE303 has completed a phase 2 trial, and SER-109 has completed a phase 3 trial.
Dr. Allegretti is a consultant for Finch Therapeutics, which funded the trial. Dr. Kashyap has disclosed no relevant financial relationships.
Help your patients understand their C. difficile diagnosis by sharing patient education from the AGA GI Patient Center: www.gastro.org/Cdiff.
A version of this article first appeared on Medscape.com.
FROM ACG 2021
A pill for C. difficile works by increasing microbiome diversity
CP101, under development by Finch Therapeutics, proved more effective than a placebo in preventing recurrent infections for up to 24 weeks.
The CP101 capsules contain a powder of freeze-dried human stools from screened donors. They restore natural diversity that has been disrupted by antibiotics, said Jessica Allegretti, MD, MPH a gastroenterologist at Brigham and Women’s Hospital in Boston.
The treatment offers an alternative to fecal microbiota transplant, which can effectively treat antibiotic-resistant C. difficile infections but is difficult to standardize and administer – and doesn’t have full approval from the U.S. Food and Drug Administration, she added.
“I think this marks a moment in this space where we’re going to have better, safer, and more available options for patients,” she said in an interview. “It’s exciting.”
Dr. Allegretti is an author on three presentations of results from PRISM3, a phase 2 trial of CP101. They will be presented this week at the annual meeting of the American College of Gastroenterology. These results extend out to 24 weeks, whereas the 8-week results of this trial were presented a year ago at the same meeting.
Study details
The study enrolled 198 people who received antibiotics for recurrent C. difficile infections. Some patients had two or more recurrences, while others had only one recurrence but were 65 years of age or older.
“That was a unique aspect of this study, to see the effect of bringing a therapy like CP101 earlier in the treatment paradigm,” said Dr. Allegretti. “You can imagine for an older, frail, or more fragile patient that you would want to get rid of this [infection] earlier.”
After waiting 2-6 days for the antibiotics to wash out, the researchers randomly assigned 102 of these patients to take the CP101 pills orally and 96 to take placebo pills, both without bowel preparation.
The two groups were not significantly different in age, gender, comorbidities, the number of C. difficile recurrences, or the type of test used to diagnose the infection (PCR-based vs. toxin EIA-based).
After 8 weeks, 74.5% of those given the CP101 pills had not had a recurrence, compared with 61.5% of those given the placebo. The difference was just barely statistically significant (P = .0488).
Sixteen weeks later, the effect endured, with 73.5% of the CP101 group and 59.4% of the placebo group still free of recurrence. The statistical significance of the difference improved slightly (P = .0347).
Drug-related emergent adverse events were similar between the two groups: 16.3% for the CP101 group vs. 19.2% for the placebo group. These were mostly gastrointestinal symptoms, and none were serious.
Some of the patients received vancomycin as a first-line treatment for C. difficile infections, and the researchers wondered if the washout period was not sufficient to purge that antibiotic, leaving enough to interfere with the effectiveness of CP101.
Therefore, they separately analyzed 40 patients treated with fidaxomicin, which they expected to wash out more quickly. Among these patients, 81% who received CP101 were free of recurrences, at 8 weeks and 24 weeks. This compared with 42.1% of those who received the placebo, at both time points. This difference was more statistically significant (P = .0211).
Understanding how it works
To understand better how CP101 achieves its effects, the researchers collected stool samples from the patients and counted the number of different kinds organisms in each sample.
At baseline, the patients had about the same number, but after a week the diversity was greater in the patients treated with CP101, and that difference had increased at week 8. The researchers also found much less diversity of organisms in the stools of those patients who had recurrences of C. difficile infection.
The diversity of microbes in the successfully treated patients appeared to have been introduced by CP101. Dr. Allegretti and colleagues measured the number of organisms in the stool samples that came from CP101. They found that 96% of patients colonized by the CP101 organisms had avoided recurrence of the C. difficile infections, compared with 54.2% of those patients not colonized by these microbes.
“We now have some microbiome-based markers that show us as early as week 1 that the patient is going to be cured or not,” Dr. Allegretti said.
Based on these results, Finch plans to launch a phase 3 trial soon, she said.
The data on colonization is interesting because it has not been found with fecal microbiota transplants, said Purna Kashyap, MBBS, codirector of the Microbiome Program at the Mayo Clinic College of Medicine in Rochester, Minn., who was not involved in the study.
But to better interpret the data, it would be helpful to know more about how the placebo and CP101 groups compared at baseline with regard to medications, immunosuppression, and antibiotics used to treat the C. difficile infections, Dr. Kashyap said. He was struck by the lower cure rate in the portion of the placebo group treated with fidaxomicin.
“Overall, I think these are exciting observations based on the data but require careful review of the entire data to make sense of [them], which will happen when it goes through peer review,” he told this news organization in an email.
Several other standardized microbiota restoration products are under development, including at least two other capsules. In contrast to CP101, which is made up of whole stool, VE303 (Vedanta Biosciences) is a “rationally defined bacterial consortium,” and SER-109 (Seres Therapeutics) is a “consortium of highly purified Firmicutes spores.” VE303 has completed a phase 2 trial, and SER-109 has completed a phase 3 trial.
Dr. Allegretti is a consultant for Finch Therapeutics, which funded the trial. Dr. Kashyap has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
CP101, under development by Finch Therapeutics, proved more effective than a placebo in preventing recurrent infections for up to 24 weeks.
The CP101 capsules contain a powder of freeze-dried human stools from screened donors. They restore natural diversity that has been disrupted by antibiotics, said Jessica Allegretti, MD, MPH a gastroenterologist at Brigham and Women’s Hospital in Boston.
The treatment offers an alternative to fecal microbiota transplant, which can effectively treat antibiotic-resistant C. difficile infections but is difficult to standardize and administer – and doesn’t have full approval from the U.S. Food and Drug Administration, she added.
“I think this marks a moment in this space where we’re going to have better, safer, and more available options for patients,” she said in an interview. “It’s exciting.”
Dr. Allegretti is an author on three presentations of results from PRISM3, a phase 2 trial of CP101. They will be presented this week at the annual meeting of the American College of Gastroenterology. These results extend out to 24 weeks, whereas the 8-week results of this trial were presented a year ago at the same meeting.
Study details
The study enrolled 198 people who received antibiotics for recurrent C. difficile infections. Some patients had two or more recurrences, while others had only one recurrence but were 65 years of age or older.
“That was a unique aspect of this study, to see the effect of bringing a therapy like CP101 earlier in the treatment paradigm,” said Dr. Allegretti. “You can imagine for an older, frail, or more fragile patient that you would want to get rid of this [infection] earlier.”
After waiting 2-6 days for the antibiotics to wash out, the researchers randomly assigned 102 of these patients to take the CP101 pills orally and 96 to take placebo pills, both without bowel preparation.
The two groups were not significantly different in age, gender, comorbidities, the number of C. difficile recurrences, or the type of test used to diagnose the infection (PCR-based vs. toxin EIA-based).
After 8 weeks, 74.5% of those given the CP101 pills had not had a recurrence, compared with 61.5% of those given the placebo. The difference was just barely statistically significant (P = .0488).
Sixteen weeks later, the effect endured, with 73.5% of the CP101 group and 59.4% of the placebo group still free of recurrence. The statistical significance of the difference improved slightly (P = .0347).
Drug-related emergent adverse events were similar between the two groups: 16.3% for the CP101 group vs. 19.2% for the placebo group. These were mostly gastrointestinal symptoms, and none were serious.
Some of the patients received vancomycin as a first-line treatment for C. difficile infections, and the researchers wondered if the washout period was not sufficient to purge that antibiotic, leaving enough to interfere with the effectiveness of CP101.
Therefore, they separately analyzed 40 patients treated with fidaxomicin, which they expected to wash out more quickly. Among these patients, 81% who received CP101 were free of recurrences, at 8 weeks and 24 weeks. This compared with 42.1% of those who received the placebo, at both time points. This difference was more statistically significant (P = .0211).
Understanding how it works
To understand better how CP101 achieves its effects, the researchers collected stool samples from the patients and counted the number of different kinds organisms in each sample.
At baseline, the patients had about the same number, but after a week the diversity was greater in the patients treated with CP101, and that difference had increased at week 8. The researchers also found much less diversity of organisms in the stools of those patients who had recurrences of C. difficile infection.
The diversity of microbes in the successfully treated patients appeared to have been introduced by CP101. Dr. Allegretti and colleagues measured the number of organisms in the stool samples that came from CP101. They found that 96% of patients colonized by the CP101 organisms had avoided recurrence of the C. difficile infections, compared with 54.2% of those patients not colonized by these microbes.
“We now have some microbiome-based markers that show us as early as week 1 that the patient is going to be cured or not,” Dr. Allegretti said.
Based on these results, Finch plans to launch a phase 3 trial soon, she said.
The data on colonization is interesting because it has not been found with fecal microbiota transplants, said Purna Kashyap, MBBS, codirector of the Microbiome Program at the Mayo Clinic College of Medicine in Rochester, Minn., who was not involved in the study.
But to better interpret the data, it would be helpful to know more about how the placebo and CP101 groups compared at baseline with regard to medications, immunosuppression, and antibiotics used to treat the C. difficile infections, Dr. Kashyap said. He was struck by the lower cure rate in the portion of the placebo group treated with fidaxomicin.
“Overall, I think these are exciting observations based on the data but require careful review of the entire data to make sense of [them], which will happen when it goes through peer review,” he told this news organization in an email.
Several other standardized microbiota restoration products are under development, including at least two other capsules. In contrast to CP101, which is made up of whole stool, VE303 (Vedanta Biosciences) is a “rationally defined bacterial consortium,” and SER-109 (Seres Therapeutics) is a “consortium of highly purified Firmicutes spores.” VE303 has completed a phase 2 trial, and SER-109 has completed a phase 3 trial.
Dr. Allegretti is a consultant for Finch Therapeutics, which funded the trial. Dr. Kashyap has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
CP101, under development by Finch Therapeutics, proved more effective than a placebo in preventing recurrent infections for up to 24 weeks.
The CP101 capsules contain a powder of freeze-dried human stools from screened donors. They restore natural diversity that has been disrupted by antibiotics, said Jessica Allegretti, MD, MPH a gastroenterologist at Brigham and Women’s Hospital in Boston.
The treatment offers an alternative to fecal microbiota transplant, which can effectively treat antibiotic-resistant C. difficile infections but is difficult to standardize and administer – and doesn’t have full approval from the U.S. Food and Drug Administration, she added.
“I think this marks a moment in this space where we’re going to have better, safer, and more available options for patients,” she said in an interview. “It’s exciting.”
Dr. Allegretti is an author on three presentations of results from PRISM3, a phase 2 trial of CP101. They will be presented this week at the annual meeting of the American College of Gastroenterology. These results extend out to 24 weeks, whereas the 8-week results of this trial were presented a year ago at the same meeting.
Study details
The study enrolled 198 people who received antibiotics for recurrent C. difficile infections. Some patients had two or more recurrences, while others had only one recurrence but were 65 years of age or older.
“That was a unique aspect of this study, to see the effect of bringing a therapy like CP101 earlier in the treatment paradigm,” said Dr. Allegretti. “You can imagine for an older, frail, or more fragile patient that you would want to get rid of this [infection] earlier.”
After waiting 2-6 days for the antibiotics to wash out, the researchers randomly assigned 102 of these patients to take the CP101 pills orally and 96 to take placebo pills, both without bowel preparation.
The two groups were not significantly different in age, gender, comorbidities, the number of C. difficile recurrences, or the type of test used to diagnose the infection (PCR-based vs. toxin EIA-based).
After 8 weeks, 74.5% of those given the CP101 pills had not had a recurrence, compared with 61.5% of those given the placebo. The difference was just barely statistically significant (P = .0488).
Sixteen weeks later, the effect endured, with 73.5% of the CP101 group and 59.4% of the placebo group still free of recurrence. The statistical significance of the difference improved slightly (P = .0347).
Drug-related emergent adverse events were similar between the two groups: 16.3% for the CP101 group vs. 19.2% for the placebo group. These were mostly gastrointestinal symptoms, and none were serious.
Some of the patients received vancomycin as a first-line treatment for C. difficile infections, and the researchers wondered if the washout period was not sufficient to purge that antibiotic, leaving enough to interfere with the effectiveness of CP101.
Therefore, they separately analyzed 40 patients treated with fidaxomicin, which they expected to wash out more quickly. Among these patients, 81% who received CP101 were free of recurrences, at 8 weeks and 24 weeks. This compared with 42.1% of those who received the placebo, at both time points. This difference was more statistically significant (P = .0211).
Understanding how it works
To understand better how CP101 achieves its effects, the researchers collected stool samples from the patients and counted the number of different kinds organisms in each sample.
At baseline, the patients had about the same number, but after a week the diversity was greater in the patients treated with CP101, and that difference had increased at week 8. The researchers also found much less diversity of organisms in the stools of those patients who had recurrences of C. difficile infection.
The diversity of microbes in the successfully treated patients appeared to have been introduced by CP101. Dr. Allegretti and colleagues measured the number of organisms in the stool samples that came from CP101. They found that 96% of patients colonized by the CP101 organisms had avoided recurrence of the C. difficile infections, compared with 54.2% of those patients not colonized by these microbes.
“We now have some microbiome-based markers that show us as early as week 1 that the patient is going to be cured or not,” Dr. Allegretti said.
Based on these results, Finch plans to launch a phase 3 trial soon, she said.
The data on colonization is interesting because it has not been found with fecal microbiota transplants, said Purna Kashyap, MBBS, codirector of the Microbiome Program at the Mayo Clinic College of Medicine in Rochester, Minn., who was not involved in the study.
But to better interpret the data, it would be helpful to know more about how the placebo and CP101 groups compared at baseline with regard to medications, immunosuppression, and antibiotics used to treat the C. difficile infections, Dr. Kashyap said. He was struck by the lower cure rate in the portion of the placebo group treated with fidaxomicin.
“Overall, I think these are exciting observations based on the data but require careful review of the entire data to make sense of [them], which will happen when it goes through peer review,” he told this news organization in an email.
Several other standardized microbiota restoration products are under development, including at least two other capsules. In contrast to CP101, which is made up of whole stool, VE303 (Vedanta Biosciences) is a “rationally defined bacterial consortium,” and SER-109 (Seres Therapeutics) is a “consortium of highly purified Firmicutes spores.” VE303 has completed a phase 2 trial, and SER-109 has completed a phase 3 trial.
Dr. Allegretti is a consultant for Finch Therapeutics, which funded the trial. Dr. Kashyap has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AT ACG 2021
FDA approves first nasal spray for dry eye
The first nasal spray to treat dry eye disease has won approval from the Food and Drug Administration.
Sprayed twice daily into the nostrils, 0.03-mg varenicline solution (Tyrvaya) improves signs and symptoms of dry eye disease. It provides an alternative to the immunomodulators currently available as prescription treatments, according to Marian Macsai, MD, chief medical officer for the drug’s maker, Oyster Point Pharma.
“We’re super excited to bring a new treatment for dry eye disease to patients and eye care practitioners,” she told this news organization.
The company plans to make the drug available to wholesalers in November in cartons containing two multidose nasal spray bottles. Each bottle supplies treatment for 15 days. Samples will be made available to eye care practitioners.
The company is working with payers on reimbursement codes and will supply the drug for $10 or less to patients who are not insured, said Dr. Macsai.
Varenicline can be prescribed for anyone with dry eye disease who has not gotten relief from artificial tears or who needs to use artificial tears “more than three or four times a day,” she said.
“In our pivotal trials, we enrolled patients with mild, moderate, and severe disease,” said Dr. Macsai. “And in each subgroup, we reached statistical significance. So with this new route of administration and a new mechanism of action, I’m hopeful that this will provide relief to many of the dry eye patients out there that are currently suffering.”
The causes of dry eye disease are multifactorial, and it can prove difficult to treat. Varenicline appears to work by stimulating the trigeminal nerve, causing natural tears to form.
Marketed as the oral drug Chantix by Pfizer, varenicline is prescribed to reduce cigarette cravings. Administered as a nasal spray for dry eye, much less of it enters the bloodstream, according to Michael Raizman, MD, an associate professor of ophthalmology at Tufts University, Boston, who was an investigator in the phase 3 ONSET-2 trial of the drug.
The spray acts in as little as 14 days, rather than the 3-6 months required for prescription immunomodulators, and it doesn’t irritate the eyes, he said.
In the ONSET-2 trial, basal tear production and symptoms were assessed. Schirmer test scores increased by10 mm or more for 47% of the patients treated with varenicline vs. 28% of patients treated with placebo.
The mean change from baseline in Eye Dryness Score at week 4 was –10.3 mm for varenicline-treated patients, compared with –7.4 mm for vehicle-treated patients. The difference was not statistically significant. However, that test was disrupted by COVID-19 precautions, Dr. Macsai said. The phase 2b ONSET-1 trial showed a statistically significant advantage in Eye Dryness Score for patients treated with varenicline in comparison with those treated with placebo.
Almost everyone who took varenicline sneezed, but only about 12% experienced any ocular adverse events, which was similar to the placebo group. No one reported burning or stinging in the eyes.
A few patients coughed or felt throat or nose irritation. In the group that received 1.2 mg/mL, eight people discontinued the drug because of adverse reactions, compared with five in the group that received 0.6 mg/mL and four in the placebo group.
“This approval is exciting for the ophthalmic community, as it gives us a new therapeutic agent that can be used alone or in combination with existing therapies to treat individuals who fall under the umbrella term ‘dry eye,’ “ said Anat Galor, MD, MSPH, clinical spokesperson for the American Academy of Ophthalmology and associate professor at University of Miami.
Some idea of what to expect from Tyrvaya comes from TrueTear, a device made by Allergan that caused tearing by electrically stimulating the anterior ethmoidal nerve through the nasal passage. It provided benefit to some patients who had not gotten relief through medication, but was expensive and was eventually discontinued, Dr. Galor said.
A new device, the iTear100, from Olympic Ophthalmics, stimulates the anterior ethmoidal nerve through the side of the nose. It received FDA clearance last year.
ONSET-2 was funded by Oyster Point Pharma. Dr. Macsai is an employee of Oyster Point. Dr. Raizman is a consultant to Oyster Point Pharma. Dr. Galor reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The first nasal spray to treat dry eye disease has won approval from the Food and Drug Administration.
Sprayed twice daily into the nostrils, 0.03-mg varenicline solution (Tyrvaya) improves signs and symptoms of dry eye disease. It provides an alternative to the immunomodulators currently available as prescription treatments, according to Marian Macsai, MD, chief medical officer for the drug’s maker, Oyster Point Pharma.
“We’re super excited to bring a new treatment for dry eye disease to patients and eye care practitioners,” she told this news organization.
The company plans to make the drug available to wholesalers in November in cartons containing two multidose nasal spray bottles. Each bottle supplies treatment for 15 days. Samples will be made available to eye care practitioners.
The company is working with payers on reimbursement codes and will supply the drug for $10 or less to patients who are not insured, said Dr. Macsai.
Varenicline can be prescribed for anyone with dry eye disease who has not gotten relief from artificial tears or who needs to use artificial tears “more than three or four times a day,” she said.
“In our pivotal trials, we enrolled patients with mild, moderate, and severe disease,” said Dr. Macsai. “And in each subgroup, we reached statistical significance. So with this new route of administration and a new mechanism of action, I’m hopeful that this will provide relief to many of the dry eye patients out there that are currently suffering.”
The causes of dry eye disease are multifactorial, and it can prove difficult to treat. Varenicline appears to work by stimulating the trigeminal nerve, causing natural tears to form.
Marketed as the oral drug Chantix by Pfizer, varenicline is prescribed to reduce cigarette cravings. Administered as a nasal spray for dry eye, much less of it enters the bloodstream, according to Michael Raizman, MD, an associate professor of ophthalmology at Tufts University, Boston, who was an investigator in the phase 3 ONSET-2 trial of the drug.
The spray acts in as little as 14 days, rather than the 3-6 months required for prescription immunomodulators, and it doesn’t irritate the eyes, he said.
In the ONSET-2 trial, basal tear production and symptoms were assessed. Schirmer test scores increased by10 mm or more for 47% of the patients treated with varenicline vs. 28% of patients treated with placebo.
The mean change from baseline in Eye Dryness Score at week 4 was –10.3 mm for varenicline-treated patients, compared with –7.4 mm for vehicle-treated patients. The difference was not statistically significant. However, that test was disrupted by COVID-19 precautions, Dr. Macsai said. The phase 2b ONSET-1 trial showed a statistically significant advantage in Eye Dryness Score for patients treated with varenicline in comparison with those treated with placebo.
Almost everyone who took varenicline sneezed, but only about 12% experienced any ocular adverse events, which was similar to the placebo group. No one reported burning or stinging in the eyes.
A few patients coughed or felt throat or nose irritation. In the group that received 1.2 mg/mL, eight people discontinued the drug because of adverse reactions, compared with five in the group that received 0.6 mg/mL and four in the placebo group.
“This approval is exciting for the ophthalmic community, as it gives us a new therapeutic agent that can be used alone or in combination with existing therapies to treat individuals who fall under the umbrella term ‘dry eye,’ “ said Anat Galor, MD, MSPH, clinical spokesperson for the American Academy of Ophthalmology and associate professor at University of Miami.
Some idea of what to expect from Tyrvaya comes from TrueTear, a device made by Allergan that caused tearing by electrically stimulating the anterior ethmoidal nerve through the nasal passage. It provided benefit to some patients who had not gotten relief through medication, but was expensive and was eventually discontinued, Dr. Galor said.
A new device, the iTear100, from Olympic Ophthalmics, stimulates the anterior ethmoidal nerve through the side of the nose. It received FDA clearance last year.
ONSET-2 was funded by Oyster Point Pharma. Dr. Macsai is an employee of Oyster Point. Dr. Raizman is a consultant to Oyster Point Pharma. Dr. Galor reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The first nasal spray to treat dry eye disease has won approval from the Food and Drug Administration.
Sprayed twice daily into the nostrils, 0.03-mg varenicline solution (Tyrvaya) improves signs and symptoms of dry eye disease. It provides an alternative to the immunomodulators currently available as prescription treatments, according to Marian Macsai, MD, chief medical officer for the drug’s maker, Oyster Point Pharma.
“We’re super excited to bring a new treatment for dry eye disease to patients and eye care practitioners,” she told this news organization.
The company plans to make the drug available to wholesalers in November in cartons containing two multidose nasal spray bottles. Each bottle supplies treatment for 15 days. Samples will be made available to eye care practitioners.
The company is working with payers on reimbursement codes and will supply the drug for $10 or less to patients who are not insured, said Dr. Macsai.
Varenicline can be prescribed for anyone with dry eye disease who has not gotten relief from artificial tears or who needs to use artificial tears “more than three or four times a day,” she said.
“In our pivotal trials, we enrolled patients with mild, moderate, and severe disease,” said Dr. Macsai. “And in each subgroup, we reached statistical significance. So with this new route of administration and a new mechanism of action, I’m hopeful that this will provide relief to many of the dry eye patients out there that are currently suffering.”
The causes of dry eye disease are multifactorial, and it can prove difficult to treat. Varenicline appears to work by stimulating the trigeminal nerve, causing natural tears to form.
Marketed as the oral drug Chantix by Pfizer, varenicline is prescribed to reduce cigarette cravings. Administered as a nasal spray for dry eye, much less of it enters the bloodstream, according to Michael Raizman, MD, an associate professor of ophthalmology at Tufts University, Boston, who was an investigator in the phase 3 ONSET-2 trial of the drug.
The spray acts in as little as 14 days, rather than the 3-6 months required for prescription immunomodulators, and it doesn’t irritate the eyes, he said.
In the ONSET-2 trial, basal tear production and symptoms were assessed. Schirmer test scores increased by10 mm or more for 47% of the patients treated with varenicline vs. 28% of patients treated with placebo.
The mean change from baseline in Eye Dryness Score at week 4 was –10.3 mm for varenicline-treated patients, compared with –7.4 mm for vehicle-treated patients. The difference was not statistically significant. However, that test was disrupted by COVID-19 precautions, Dr. Macsai said. The phase 2b ONSET-1 trial showed a statistically significant advantage in Eye Dryness Score for patients treated with varenicline in comparison with those treated with placebo.
Almost everyone who took varenicline sneezed, but only about 12% experienced any ocular adverse events, which was similar to the placebo group. No one reported burning or stinging in the eyes.
A few patients coughed or felt throat or nose irritation. In the group that received 1.2 mg/mL, eight people discontinued the drug because of adverse reactions, compared with five in the group that received 0.6 mg/mL and four in the placebo group.
“This approval is exciting for the ophthalmic community, as it gives us a new therapeutic agent that can be used alone or in combination with existing therapies to treat individuals who fall under the umbrella term ‘dry eye,’ “ said Anat Galor, MD, MSPH, clinical spokesperson for the American Academy of Ophthalmology and associate professor at University of Miami.
Some idea of what to expect from Tyrvaya comes from TrueTear, a device made by Allergan that caused tearing by electrically stimulating the anterior ethmoidal nerve through the nasal passage. It provided benefit to some patients who had not gotten relief through medication, but was expensive and was eventually discontinued, Dr. Galor said.
A new device, the iTear100, from Olympic Ophthalmics, stimulates the anterior ethmoidal nerve through the side of the nose. It received FDA clearance last year.
ONSET-2 was funded by Oyster Point Pharma. Dr. Macsai is an employee of Oyster Point. Dr. Raizman is a consultant to Oyster Point Pharma. Dr. Galor reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.