User login
Delayed diagnosis of breast cancer: $15M award
Delayed diagnosis of breast cancer: $15M award
A woman in her mid-50s had been seen by a breast surgeon for 16 years for regular mammograms and sonograms. In May 2009, the breast surgeon misinterpreted a mammogram as negative, as did a radiologist who re-read the mammogram weeks later. In December 2010, the patient returned to the breast surgeon with nipple discharge. No further testing was conducted. In October 2011, the patient was found to have Stage IIIA breast cancer involving 4 lymph nodes. She underwent left radical mastectomy, chemotherapy, radiation therapy, and breast reconstruction. At time of trial, the cancer had invaded her vertebrae, was Stage IV, and most likely incurable.
PATIENT'S CLAIM: Although the surgeon admittedly did not possess the qualifications required under the Mammography Quality Standards Act, he interpreted about 5,000 mammograms per year in his office. In this case, he failed to detect a small breast tumor in May 2009. He also failed to perform testing when the patient reported nipple discharge. A more timely diagnosis of breast cancer at Stage I would have provided a 90% chance of long-term survival.
DEFENDANTS' DEFENSE: The defense held the radiologist fully liable because the surgeon was not a qualified interpreter of mammography, therefore relying on the radiologist’s interpretation. The radiologist was legally responsible for the missed diagnosis.
VERDICT: A $15M New York verdict was reached, finding the breast surgeon 75% at fault and the radiologist 25%. The radiologist settled before the trial (the jury was not informed of this). The breast surgeon was responsible for $11.25M. The defense indicated intent to appeal.
Alleged failure to evacuate uterus after cesarean delivery
A 37-year-old woman underwent cesarean delivery (CD) performed by 2 ObGyns. After delivery, she began to hemorrhage and the uterus became atonic. Hysterectomy was performed but the bleeding did not stop. The ObGyns called in 3 other ObGyns. During exploratory laparotomy, the bleeding was halted.
PATIENT'S CLAIM: She and her husband had hoped to have more children but the hysterectomy precluded that. She sued all 5 ObGyns, alleging that the delivering ObGyns failed to properly perform the CD and that each physician failed to properly perform the laparotomy, causing a large scar. The claim was discontinued against the 3 surgical ObGyns; trial addressed the 2 delivering ObGyns.
The patient’s expert ObGyn remarked that the hemorrhage was caused by a small placental remnant that remained in the uterus as a result of inadequate evacuation following delivery. The presence of the remnant was indicated by the uterine atony and should have prompted immediate investigation. The physicians’ notes did not document exploration of the uterus prior to closure.
PHYSICIAN'S DEFENSE: The defense’s expert contended that atony would not be a result of a small remnant of placenta. The patient’s uterus was properly evacuated, the hemorrhage was an unforeseeable complication, and the ObGyns properly addressed the hemorrhage.
VERDICT: A New York defense verdict was returned.
Alleged bowel injury during hysterectomy
Two days after a woman underwent a hysterectomy performed by her ObGyn, she went to the emergency department with increasing pain. Her ObGyn admitted her to the hospital. A general surgeon performed an exploratory laparotomy the next day that revealed an abscess; a 1-cm perforation of the patient’s bowel was surgically repaired. The patient had a difficult recovery. She developed pneumonia and respiratory failure. She underwent multiple repair surgeries for recurrent abscesses and fistulas because the wound was slow to heal.
PATIENT'S CLAIM: The ObGyn’s surgical technique was negligent. He injured the bowel when inserting a trocar and did not identify the injury in a timely manner. The expert witness commented that such an injury can sometimes be a surgical complication, but not in this case: the ObGyn rushed the procedure because he had another patient waiting for CD at another hospital.
PHYSICIAN'S DEFENSE: The ObGyn denied negligence and contended that the trocar used in surgery was too blunt to have caused a perforation. It would have been obvious to the ObGyn during surgery if a perforation had occurred. The perforation developed days after surgery within an abscess.
VERDICT: A Mississippi defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to rbarbieri@mdedge.com. Please include your name and the city and state in which you practice.
Delayed diagnosis of breast cancer: $15M award
A woman in her mid-50s had been seen by a breast surgeon for 16 years for regular mammograms and sonograms. In May 2009, the breast surgeon misinterpreted a mammogram as negative, as did a radiologist who re-read the mammogram weeks later. In December 2010, the patient returned to the breast surgeon with nipple discharge. No further testing was conducted. In October 2011, the patient was found to have Stage IIIA breast cancer involving 4 lymph nodes. She underwent left radical mastectomy, chemotherapy, radiation therapy, and breast reconstruction. At time of trial, the cancer had invaded her vertebrae, was Stage IV, and most likely incurable.
PATIENT'S CLAIM: Although the surgeon admittedly did not possess the qualifications required under the Mammography Quality Standards Act, he interpreted about 5,000 mammograms per year in his office. In this case, he failed to detect a small breast tumor in May 2009. He also failed to perform testing when the patient reported nipple discharge. A more timely diagnosis of breast cancer at Stage I would have provided a 90% chance of long-term survival.
DEFENDANTS' DEFENSE: The defense held the radiologist fully liable because the surgeon was not a qualified interpreter of mammography, therefore relying on the radiologist’s interpretation. The radiologist was legally responsible for the missed diagnosis.
VERDICT: A $15M New York verdict was reached, finding the breast surgeon 75% at fault and the radiologist 25%. The radiologist settled before the trial (the jury was not informed of this). The breast surgeon was responsible for $11.25M. The defense indicated intent to appeal.
Alleged failure to evacuate uterus after cesarean delivery
A 37-year-old woman underwent cesarean delivery (CD) performed by 2 ObGyns. After delivery, she began to hemorrhage and the uterus became atonic. Hysterectomy was performed but the bleeding did not stop. The ObGyns called in 3 other ObGyns. During exploratory laparotomy, the bleeding was halted.
PATIENT'S CLAIM: She and her husband had hoped to have more children but the hysterectomy precluded that. She sued all 5 ObGyns, alleging that the delivering ObGyns failed to properly perform the CD and that each physician failed to properly perform the laparotomy, causing a large scar. The claim was discontinued against the 3 surgical ObGyns; trial addressed the 2 delivering ObGyns.
The patient’s expert ObGyn remarked that the hemorrhage was caused by a small placental remnant that remained in the uterus as a result of inadequate evacuation following delivery. The presence of the remnant was indicated by the uterine atony and should have prompted immediate investigation. The physicians’ notes did not document exploration of the uterus prior to closure.
PHYSICIAN'S DEFENSE: The defense’s expert contended that atony would not be a result of a small remnant of placenta. The patient’s uterus was properly evacuated, the hemorrhage was an unforeseeable complication, and the ObGyns properly addressed the hemorrhage.
VERDICT: A New York defense verdict was returned.
Alleged bowel injury during hysterectomy
Two days after a woman underwent a hysterectomy performed by her ObGyn, she went to the emergency department with increasing pain. Her ObGyn admitted her to the hospital. A general surgeon performed an exploratory laparotomy the next day that revealed an abscess; a 1-cm perforation of the patient’s bowel was surgically repaired. The patient had a difficult recovery. She developed pneumonia and respiratory failure. She underwent multiple repair surgeries for recurrent abscesses and fistulas because the wound was slow to heal.
PATIENT'S CLAIM: The ObGyn’s surgical technique was negligent. He injured the bowel when inserting a trocar and did not identify the injury in a timely manner. The expert witness commented that such an injury can sometimes be a surgical complication, but not in this case: the ObGyn rushed the procedure because he had another patient waiting for CD at another hospital.
PHYSICIAN'S DEFENSE: The ObGyn denied negligence and contended that the trocar used in surgery was too blunt to have caused a perforation. It would have been obvious to the ObGyn during surgery if a perforation had occurred. The perforation developed days after surgery within an abscess.
VERDICT: A Mississippi defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to rbarbieri@mdedge.com. Please include your name and the city and state in which you practice.
Delayed diagnosis of breast cancer: $15M award
A woman in her mid-50s had been seen by a breast surgeon for 16 years for regular mammograms and sonograms. In May 2009, the breast surgeon misinterpreted a mammogram as negative, as did a radiologist who re-read the mammogram weeks later. In December 2010, the patient returned to the breast surgeon with nipple discharge. No further testing was conducted. In October 2011, the patient was found to have Stage IIIA breast cancer involving 4 lymph nodes. She underwent left radical mastectomy, chemotherapy, radiation therapy, and breast reconstruction. At time of trial, the cancer had invaded her vertebrae, was Stage IV, and most likely incurable.
PATIENT'S CLAIM: Although the surgeon admittedly did not possess the qualifications required under the Mammography Quality Standards Act, he interpreted about 5,000 mammograms per year in his office. In this case, he failed to detect a small breast tumor in May 2009. He also failed to perform testing when the patient reported nipple discharge. A more timely diagnosis of breast cancer at Stage I would have provided a 90% chance of long-term survival.
DEFENDANTS' DEFENSE: The defense held the radiologist fully liable because the surgeon was not a qualified interpreter of mammography, therefore relying on the radiologist’s interpretation. The radiologist was legally responsible for the missed diagnosis.
VERDICT: A $15M New York verdict was reached, finding the breast surgeon 75% at fault and the radiologist 25%. The radiologist settled before the trial (the jury was not informed of this). The breast surgeon was responsible for $11.25M. The defense indicated intent to appeal.
Alleged failure to evacuate uterus after cesarean delivery
A 37-year-old woman underwent cesarean delivery (CD) performed by 2 ObGyns. After delivery, she began to hemorrhage and the uterus became atonic. Hysterectomy was performed but the bleeding did not stop. The ObGyns called in 3 other ObGyns. During exploratory laparotomy, the bleeding was halted.
PATIENT'S CLAIM: She and her husband had hoped to have more children but the hysterectomy precluded that. She sued all 5 ObGyns, alleging that the delivering ObGyns failed to properly perform the CD and that each physician failed to properly perform the laparotomy, causing a large scar. The claim was discontinued against the 3 surgical ObGyns; trial addressed the 2 delivering ObGyns.
The patient’s expert ObGyn remarked that the hemorrhage was caused by a small placental remnant that remained in the uterus as a result of inadequate evacuation following delivery. The presence of the remnant was indicated by the uterine atony and should have prompted immediate investigation. The physicians’ notes did not document exploration of the uterus prior to closure.
PHYSICIAN'S DEFENSE: The defense’s expert contended that atony would not be a result of a small remnant of placenta. The patient’s uterus was properly evacuated, the hemorrhage was an unforeseeable complication, and the ObGyns properly addressed the hemorrhage.
VERDICT: A New York defense verdict was returned.
Alleged bowel injury during hysterectomy
Two days after a woman underwent a hysterectomy performed by her ObGyn, she went to the emergency department with increasing pain. Her ObGyn admitted her to the hospital. A general surgeon performed an exploratory laparotomy the next day that revealed an abscess; a 1-cm perforation of the patient’s bowel was surgically repaired. The patient had a difficult recovery. She developed pneumonia and respiratory failure. She underwent multiple repair surgeries for recurrent abscesses and fistulas because the wound was slow to heal.
PATIENT'S CLAIM: The ObGyn’s surgical technique was negligent. He injured the bowel when inserting a trocar and did not identify the injury in a timely manner. The expert witness commented that such an injury can sometimes be a surgical complication, but not in this case: the ObGyn rushed the procedure because he had another patient waiting for CD at another hospital.
PHYSICIAN'S DEFENSE: The ObGyn denied negligence and contended that the trocar used in surgery was too blunt to have caused a perforation. It would have been obvious to the ObGyn during surgery if a perforation had occurred. The perforation developed days after surgery within an abscess.
VERDICT: A Mississippi defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to rbarbieri@mdedge.com. Please include your name and the city and state in which you practice.
Spinal Muscular Atrophy Added to Recommended Uniform Screening Panel
Screening will enable early detection, but the treatment’s exceptional cost could present a barrier to patients.
Spinal muscular atrophy (SMA) is now among the disorders officially included in the Recommended Uniform Screening Panel (RUSP), which state public health departments use to screen newborns for genetic disorders.
Secretary of the Department of Health and Human Services (HHS) Alex M. Azar II formally added SMA to the panel on July 2 on the recommendation of the Advisory Committee on Heritable Disorders in Newborns and Children.
“Adding SMA to the list will help ensure that babies born with SMA are identified, so that they have the opportunity to benefit from early treatment and intervention,” according to a statement from the Muscular Dystrophy Association about the decision. “This testing can also provide families with a genetic diagnosis—information that often is required to determine whether their child is eligible to participate in clinical trials.”
Adding SMA to the RUSP does not mean that states must screen newborns for the disorder. Each state’s public health apparatus decides independently whether to accept the recommendation and which disorders on the RUSP to screen for. Most states screen for most disorders on the RUSP. Evidence compiled by the advisory committee suggested a wide variation in resources, infrastructure, funding, and time to implementation among states.
Drug Approval Raised Ethical Questions
An estimated one in 11,000 newborns has SMA, a disorder caused by mutations in the survival motor neuron 1 (SMN1) gene. SMA affects motor neurons in the brainstem and spinal cord, thus leading to motor weakness and atrophy. The only treatment for SMA had been palliative care until the FDA approved nusinersen for the disorder in December 2016. The drug’s approval has raised ethical questions.1–3
After reviewing the evidence at its February 8 meeting, the advisory committee recommended adding SMA screening to the RUSP in a March 8 letter from committee chair Joseph A. Bocchini Jr, MD, Professor and Chair of Pediatrics at Louisiana State University Health in Shreveport.
Secretary Azar accepted the recommendation based on the evidence the committee provided; he also requested a follow-up report within two years “describing the status of implementing newborn screening for SMA and clinical outcomes of early treatment, including any potential harms, for infants diagnosed with SMA.”
The advisory committee makes its recommendations to HHS about which heritable disorders to include in the RUSP after it has assessed a systematic, evidence-based review conducted by an external, independent group. Alex R. Kemper, MD, MPH, Professor of Pediatrics at the Ohio State University and Division Chief of Ambulatory Pediatrics at Nationwide Children’s Hospital, both in Columbus, led the review group for SMA. Dr. Kemper is also deputy editor of Pediatrics and a member of the US Preventive Services Task Force.
According to Secretary Azar’s summary in his July 2 letter of acceptance, the evidence review suggested that “early screening and treatment can lead to decreased mortality for individuals with SMA and improved motor milestones.”
“SMA can be detected through newborn screening, and treatment is now available that can not only reduce the risk of death, but decrease the development of neurologic impairment,” he said in an interview. “As with adding any condition to newborn screening, public health laboratories will need to develop strategies to incorporate the screening test. The current FDA-approved treatment, nusinersen, is delivered by lumbar puncture into the spinal fluid. In addition, there are exciting advances in gene therapy leading to new treatment approaches.”
Symptom Onset Distinguishes the Types of SMA
Approximately 95% of SMA cases result from the deletion of exon 7 from both alleles of SMN1. Other, rarer cases are caused by mutations in different genes. Without the SMN protein produced by SMN1, a person gradually loses muscle function.
A similar gene, SMN2, also can produce the SMN protein, but in much lower amounts—typically less than 10% of what a person needs. People can, however, have multiple copies of SMN2, which can produce slightly more SMN protein and slow the disease process.
The five types of SMA are determined according to symptom onset, which directly correlates with disorder severity and prognosis. Approximately 54% of SMA cases are Type I, in which progressive weakness occurs over the first six months of life and results in early death. Only 18% of children with Type I live past age 4, and 68% die by age 2. Type 0 is rarer, but more severe, usually causing fetal loss or early infant death.
Type II represents 18% of SMA cases and causes progressive weakness by age 15 months. Most people with Type II survive to their 30s but later experience respiratory failure and rarely reach their 40s. Individuals with Types III and IV typically have a normal lifespan and only begin to see progressive muscle weakness after age 1 or in adulthood.
Dr. Kemper’s group focused on the three types diagnosed in infancy: Types I, II, and III. “It will be critical to make sure that infants diagnosed with SMA through newborn screening receive follow-up shortly afterward to determine whether they would benefit from nusinersen,” said Dr. Kemper. “More information is needed about the long-term outcomes of those infants who begin treatment following newborn screening, so we not only know about outcomes in later childhood and adolescence, but treatment approaches can be further refined and personalized.”
Long-Term Data on Nusinersen Are Lacking
Nusinersen alters the splicing of precursor messenger RNA in SMN2 so that the mRNA strands are longer, which increases the amount of SMN protein produced. Concerns about the medication, however, have included its cost—$750,000 in the first year and $375,000 every following year for life—and potential adverse events from repeated administration. Nusinersen is injected into the spinal canal four times in the first year and once annually thereafter, and the painful injections require patient immobilization. Potential adverse events include thrombocytopenia and nephrotoxicity, along with potential complications from repeated lumbar punctures over time.2
Other concerns about the drug include its limited evidence base, lack of long-term data, associated costs with administration (eg, travel costs), the potential for patients taking nusinersen to be excluded from future clinical trials on other treatments, and ensuring parents have enough information on the drug’s limitations and potential risks to provide adequate informed consent.2
Yet evidence to date is favorable in children with early onset SMA. Dr. Bocchini wrote in the letter to Secretary Azar that “limited data suggest that treatment effect is greater when the treatment is initiated before symptoms develop and when the individual has more copies of SMN2.”
Dr. Kemper’s group concluded that screening can detect SMA in newborns and that treatment can modify the disease course. “Grey literature suggests those with total disease duration less than or equal to 12 weeks before nusinersen treatment were more likely to have better outcomes than those with longer periods of disease duration.
“Presymptomatic treatment alters the natural history” of the disorder, the group found, although outcome data past age 1 are not yet available. Based on findings from a New York pilot program, they predicted that nationwide newborn screening would avert 33 deaths and 48 cases of children who were dependent on a ventilator among an annual cohort of four million births.
At the time of the evidence review, Massachusetts, Minnesota, Missouri, North Carolina, New York, Utah, and Wisconsin initiated pilot programs or whole-population mandated screening for SMA. Of the three states that reported costs, all reported costs of $1 or less per screen.
The research for the evidence review was funded by a Health Resources and Services Administration grant to Duke University in Durham, North Carolina. No disclosures were provided for evidence review group members.
—Tara Haelle
References
1. King NMP, Bishop CE. New treatments for serious conditions: ethical implications. Gene Ther. 2017;24(9):534-538.
2. Gerrity MS, Prasad V, Obley AJ. Concerns about the approval of nusinersen sodium by the US Food and Drug Administration. JAMA Intern Med. 2018;178(6):743-744.
3. Burgart AM, Magnus D, Tabor HK, et al. Ethical challenges confronted when providing nusinersen treatment for spinal muscular atrophy. JAMA Pediatr. 2018;172(2):188-192.
Screening will enable early detection, but the treatment’s exceptional cost could present a barrier to patients.
Screening will enable early detection, but the treatment’s exceptional cost could present a barrier to patients.
Spinal muscular atrophy (SMA) is now among the disorders officially included in the Recommended Uniform Screening Panel (RUSP), which state public health departments use to screen newborns for genetic disorders.
Secretary of the Department of Health and Human Services (HHS) Alex M. Azar II formally added SMA to the panel on July 2 on the recommendation of the Advisory Committee on Heritable Disorders in Newborns and Children.
“Adding SMA to the list will help ensure that babies born with SMA are identified, so that they have the opportunity to benefit from early treatment and intervention,” according to a statement from the Muscular Dystrophy Association about the decision. “This testing can also provide families with a genetic diagnosis—information that often is required to determine whether their child is eligible to participate in clinical trials.”
Adding SMA to the RUSP does not mean that states must screen newborns for the disorder. Each state’s public health apparatus decides independently whether to accept the recommendation and which disorders on the RUSP to screen for. Most states screen for most disorders on the RUSP. Evidence compiled by the advisory committee suggested a wide variation in resources, infrastructure, funding, and time to implementation among states.
Drug Approval Raised Ethical Questions
An estimated one in 11,000 newborns has SMA, a disorder caused by mutations in the survival motor neuron 1 (SMN1) gene. SMA affects motor neurons in the brainstem and spinal cord, thus leading to motor weakness and atrophy. The only treatment for SMA had been palliative care until the FDA approved nusinersen for the disorder in December 2016. The drug’s approval has raised ethical questions.1–3
After reviewing the evidence at its February 8 meeting, the advisory committee recommended adding SMA screening to the RUSP in a March 8 letter from committee chair Joseph A. Bocchini Jr, MD, Professor and Chair of Pediatrics at Louisiana State University Health in Shreveport.
Secretary Azar accepted the recommendation based on the evidence the committee provided; he also requested a follow-up report within two years “describing the status of implementing newborn screening for SMA and clinical outcomes of early treatment, including any potential harms, for infants diagnosed with SMA.”
The advisory committee makes its recommendations to HHS about which heritable disorders to include in the RUSP after it has assessed a systematic, evidence-based review conducted by an external, independent group. Alex R. Kemper, MD, MPH, Professor of Pediatrics at the Ohio State University and Division Chief of Ambulatory Pediatrics at Nationwide Children’s Hospital, both in Columbus, led the review group for SMA. Dr. Kemper is also deputy editor of Pediatrics and a member of the US Preventive Services Task Force.
According to Secretary Azar’s summary in his July 2 letter of acceptance, the evidence review suggested that “early screening and treatment can lead to decreased mortality for individuals with SMA and improved motor milestones.”
“SMA can be detected through newborn screening, and treatment is now available that can not only reduce the risk of death, but decrease the development of neurologic impairment,” he said in an interview. “As with adding any condition to newborn screening, public health laboratories will need to develop strategies to incorporate the screening test. The current FDA-approved treatment, nusinersen, is delivered by lumbar puncture into the spinal fluid. In addition, there are exciting advances in gene therapy leading to new treatment approaches.”
Symptom Onset Distinguishes the Types of SMA
Approximately 95% of SMA cases result from the deletion of exon 7 from both alleles of SMN1. Other, rarer cases are caused by mutations in different genes. Without the SMN protein produced by SMN1, a person gradually loses muscle function.
A similar gene, SMN2, also can produce the SMN protein, but in much lower amounts—typically less than 10% of what a person needs. People can, however, have multiple copies of SMN2, which can produce slightly more SMN protein and slow the disease process.
The five types of SMA are determined according to symptom onset, which directly correlates with disorder severity and prognosis. Approximately 54% of SMA cases are Type I, in which progressive weakness occurs over the first six months of life and results in early death. Only 18% of children with Type I live past age 4, and 68% die by age 2. Type 0 is rarer, but more severe, usually causing fetal loss or early infant death.
Type II represents 18% of SMA cases and causes progressive weakness by age 15 months. Most people with Type II survive to their 30s but later experience respiratory failure and rarely reach their 40s. Individuals with Types III and IV typically have a normal lifespan and only begin to see progressive muscle weakness after age 1 or in adulthood.
Dr. Kemper’s group focused on the three types diagnosed in infancy: Types I, II, and III. “It will be critical to make sure that infants diagnosed with SMA through newborn screening receive follow-up shortly afterward to determine whether they would benefit from nusinersen,” said Dr. Kemper. “More information is needed about the long-term outcomes of those infants who begin treatment following newborn screening, so we not only know about outcomes in later childhood and adolescence, but treatment approaches can be further refined and personalized.”
Long-Term Data on Nusinersen Are Lacking
Nusinersen alters the splicing of precursor messenger RNA in SMN2 so that the mRNA strands are longer, which increases the amount of SMN protein produced. Concerns about the medication, however, have included its cost—$750,000 in the first year and $375,000 every following year for life—and potential adverse events from repeated administration. Nusinersen is injected into the spinal canal four times in the first year and once annually thereafter, and the painful injections require patient immobilization. Potential adverse events include thrombocytopenia and nephrotoxicity, along with potential complications from repeated lumbar punctures over time.2
Other concerns about the drug include its limited evidence base, lack of long-term data, associated costs with administration (eg, travel costs), the potential for patients taking nusinersen to be excluded from future clinical trials on other treatments, and ensuring parents have enough information on the drug’s limitations and potential risks to provide adequate informed consent.2
Yet evidence to date is favorable in children with early onset SMA. Dr. Bocchini wrote in the letter to Secretary Azar that “limited data suggest that treatment effect is greater when the treatment is initiated before symptoms develop and when the individual has more copies of SMN2.”
Dr. Kemper’s group concluded that screening can detect SMA in newborns and that treatment can modify the disease course. “Grey literature suggests those with total disease duration less than or equal to 12 weeks before nusinersen treatment were more likely to have better outcomes than those with longer periods of disease duration.
“Presymptomatic treatment alters the natural history” of the disorder, the group found, although outcome data past age 1 are not yet available. Based on findings from a New York pilot program, they predicted that nationwide newborn screening would avert 33 deaths and 48 cases of children who were dependent on a ventilator among an annual cohort of four million births.
At the time of the evidence review, Massachusetts, Minnesota, Missouri, North Carolina, New York, Utah, and Wisconsin initiated pilot programs or whole-population mandated screening for SMA. Of the three states that reported costs, all reported costs of $1 or less per screen.
The research for the evidence review was funded by a Health Resources and Services Administration grant to Duke University in Durham, North Carolina. No disclosures were provided for evidence review group members.
—Tara Haelle
References
1. King NMP, Bishop CE. New treatments for serious conditions: ethical implications. Gene Ther. 2017;24(9):534-538.
2. Gerrity MS, Prasad V, Obley AJ. Concerns about the approval of nusinersen sodium by the US Food and Drug Administration. JAMA Intern Med. 2018;178(6):743-744.
3. Burgart AM, Magnus D, Tabor HK, et al. Ethical challenges confronted when providing nusinersen treatment for spinal muscular atrophy. JAMA Pediatr. 2018;172(2):188-192.
Spinal muscular atrophy (SMA) is now among the disorders officially included in the Recommended Uniform Screening Panel (RUSP), which state public health departments use to screen newborns for genetic disorders.
Secretary of the Department of Health and Human Services (HHS) Alex M. Azar II formally added SMA to the panel on July 2 on the recommendation of the Advisory Committee on Heritable Disorders in Newborns and Children.
“Adding SMA to the list will help ensure that babies born with SMA are identified, so that they have the opportunity to benefit from early treatment and intervention,” according to a statement from the Muscular Dystrophy Association about the decision. “This testing can also provide families with a genetic diagnosis—information that often is required to determine whether their child is eligible to participate in clinical trials.”
Adding SMA to the RUSP does not mean that states must screen newborns for the disorder. Each state’s public health apparatus decides independently whether to accept the recommendation and which disorders on the RUSP to screen for. Most states screen for most disorders on the RUSP. Evidence compiled by the advisory committee suggested a wide variation in resources, infrastructure, funding, and time to implementation among states.
Drug Approval Raised Ethical Questions
An estimated one in 11,000 newborns has SMA, a disorder caused by mutations in the survival motor neuron 1 (SMN1) gene. SMA affects motor neurons in the brainstem and spinal cord, thus leading to motor weakness and atrophy. The only treatment for SMA had been palliative care until the FDA approved nusinersen for the disorder in December 2016. The drug’s approval has raised ethical questions.1–3
After reviewing the evidence at its February 8 meeting, the advisory committee recommended adding SMA screening to the RUSP in a March 8 letter from committee chair Joseph A. Bocchini Jr, MD, Professor and Chair of Pediatrics at Louisiana State University Health in Shreveport.
Secretary Azar accepted the recommendation based on the evidence the committee provided; he also requested a follow-up report within two years “describing the status of implementing newborn screening for SMA and clinical outcomes of early treatment, including any potential harms, for infants diagnosed with SMA.”
The advisory committee makes its recommendations to HHS about which heritable disorders to include in the RUSP after it has assessed a systematic, evidence-based review conducted by an external, independent group. Alex R. Kemper, MD, MPH, Professor of Pediatrics at the Ohio State University and Division Chief of Ambulatory Pediatrics at Nationwide Children’s Hospital, both in Columbus, led the review group for SMA. Dr. Kemper is also deputy editor of Pediatrics and a member of the US Preventive Services Task Force.
According to Secretary Azar’s summary in his July 2 letter of acceptance, the evidence review suggested that “early screening and treatment can lead to decreased mortality for individuals with SMA and improved motor milestones.”
“SMA can be detected through newborn screening, and treatment is now available that can not only reduce the risk of death, but decrease the development of neurologic impairment,” he said in an interview. “As with adding any condition to newborn screening, public health laboratories will need to develop strategies to incorporate the screening test. The current FDA-approved treatment, nusinersen, is delivered by lumbar puncture into the spinal fluid. In addition, there are exciting advances in gene therapy leading to new treatment approaches.”
Symptom Onset Distinguishes the Types of SMA
Approximately 95% of SMA cases result from the deletion of exon 7 from both alleles of SMN1. Other, rarer cases are caused by mutations in different genes. Without the SMN protein produced by SMN1, a person gradually loses muscle function.
A similar gene, SMN2, also can produce the SMN protein, but in much lower amounts—typically less than 10% of what a person needs. People can, however, have multiple copies of SMN2, which can produce slightly more SMN protein and slow the disease process.
The five types of SMA are determined according to symptom onset, which directly correlates with disorder severity and prognosis. Approximately 54% of SMA cases are Type I, in which progressive weakness occurs over the first six months of life and results in early death. Only 18% of children with Type I live past age 4, and 68% die by age 2. Type 0 is rarer, but more severe, usually causing fetal loss or early infant death.
Type II represents 18% of SMA cases and causes progressive weakness by age 15 months. Most people with Type II survive to their 30s but later experience respiratory failure and rarely reach their 40s. Individuals with Types III and IV typically have a normal lifespan and only begin to see progressive muscle weakness after age 1 or in adulthood.
Dr. Kemper’s group focused on the three types diagnosed in infancy: Types I, II, and III. “It will be critical to make sure that infants diagnosed with SMA through newborn screening receive follow-up shortly afterward to determine whether they would benefit from nusinersen,” said Dr. Kemper. “More information is needed about the long-term outcomes of those infants who begin treatment following newborn screening, so we not only know about outcomes in later childhood and adolescence, but treatment approaches can be further refined and personalized.”
Long-Term Data on Nusinersen Are Lacking
Nusinersen alters the splicing of precursor messenger RNA in SMN2 so that the mRNA strands are longer, which increases the amount of SMN protein produced. Concerns about the medication, however, have included its cost—$750,000 in the first year and $375,000 every following year for life—and potential adverse events from repeated administration. Nusinersen is injected into the spinal canal four times in the first year and once annually thereafter, and the painful injections require patient immobilization. Potential adverse events include thrombocytopenia and nephrotoxicity, along with potential complications from repeated lumbar punctures over time.2
Other concerns about the drug include its limited evidence base, lack of long-term data, associated costs with administration (eg, travel costs), the potential for patients taking nusinersen to be excluded from future clinical trials on other treatments, and ensuring parents have enough information on the drug’s limitations and potential risks to provide adequate informed consent.2
Yet evidence to date is favorable in children with early onset SMA. Dr. Bocchini wrote in the letter to Secretary Azar that “limited data suggest that treatment effect is greater when the treatment is initiated before symptoms develop and when the individual has more copies of SMN2.”
Dr. Kemper’s group concluded that screening can detect SMA in newborns and that treatment can modify the disease course. “Grey literature suggests those with total disease duration less than or equal to 12 weeks before nusinersen treatment were more likely to have better outcomes than those with longer periods of disease duration.
“Presymptomatic treatment alters the natural history” of the disorder, the group found, although outcome data past age 1 are not yet available. Based on findings from a New York pilot program, they predicted that nationwide newborn screening would avert 33 deaths and 48 cases of children who were dependent on a ventilator among an annual cohort of four million births.
At the time of the evidence review, Massachusetts, Minnesota, Missouri, North Carolina, New York, Utah, and Wisconsin initiated pilot programs or whole-population mandated screening for SMA. Of the three states that reported costs, all reported costs of $1 or less per screen.
The research for the evidence review was funded by a Health Resources and Services Administration grant to Duke University in Durham, North Carolina. No disclosures were provided for evidence review group members.
—Tara Haelle
References
1. King NMP, Bishop CE. New treatments for serious conditions: ethical implications. Gene Ther. 2017;24(9):534-538.
2. Gerrity MS, Prasad V, Obley AJ. Concerns about the approval of nusinersen sodium by the US Food and Drug Administration. JAMA Intern Med. 2018;178(6):743-744.
3. Burgart AM, Magnus D, Tabor HK, et al. Ethical challenges confronted when providing nusinersen treatment for spinal muscular atrophy. JAMA Pediatr. 2018;172(2):188-192.
Does TBI Increase the Risk of Suicide?
Compared with the general population, people who seek medical attention for TBI may have almost twice the risk of suicide.
Residents of Denmark who seek medical attention for traumatic brain injury (TBI) have an increased risk of suicide, compared with the general Danish population without TBI, according to a study published in the August 14 issue of JAMA. “Additional analyses revealed that the risk of suicide was higher for individuals with severe TBI, numerous medical contacts, and longer hospital stays,” said lead author Trine Madsen, PhD. Individuals were at highest risk in the first six months after discharge, said Dr. Madsen, who is a postdoctoral fellow at the Danish Research Institute for Suicide Prevention in Hellerup.
A history of TBI previously has been associated with higher rates of self-harm, suicide, and death than are found in the general population. However, previous studies have been limited by methodological shortcomings, such as small sample sizes and low numbers of suicide cases with TBI. Dr. Madsen and colleagues conducted a retrospective cohort study using nationwide registers covering 7,418,391 individuals living in Denmark between 1980 and 2014 with 164,265,624 person-years’ follow-up. Of these people, 567,823 (7.6%) had a medical contact for TBI, which included mild TBI (ie, concussion), skull fracture without documented TBI, and severe TBI (ie, head injuries with evidence of structural brain injury).
Of 34,529 individuals who died by suicide, 3,536 (10.2%) had medical contact for TBI, including 2,701 for mild TBI, 174 for skull fracture without documented TBI, and 661 for severe TBI. The absolute suicide rate was 41 per 100,000 person-years among those with TBI versus 20 per 100,000 person-years among those with no diagnosis of TBI. After accounting for relevant covariates such as fractures not involving the skull, psychiatric diagnoses, and deliberate self-harm, the adjusted incidence ratio was 1.90.
This study “provides insights into the underappreciated relationship between TBI and suicide,” said Lee Goldstein, MD, PhD, and Ramon Diaz-Arrastia, MD, PhD, in an accompanying editorial. “The results … point to an important clinical triad—TBI history, recent injury (especially with long hospital stays), and more numerous postinjury medical contacts for TBI—that serves as a red flag for increased suicide risk,” said Dr. Goldstein, who is affiliated with Boston University School of Medicine, and Dr. Diaz-Arrastia, of the University of Pennsylvania’s Perelman School of Medicine in Philadelphia. The results “indicate that increased suicide risk is relevant across all TBI severity levels, including the far more common mild injuries. Clinicians, health care professionals, and mental health practitioners must take notice of this important information.”
—Glenn S. Williams
Suggested Reading
Goldstein L, Diaz-Arrastia R. Traumatic brain injury and risk of suicide. JAMA. 2018;320(6):554-556.
Madsen T, Erlangsen A, Orlovska S, et al. Association between traumatic brain injury and risk of suicide. JAMA. 2018;320(6):580-588.
Compared with the general population, people who seek medical attention for TBI may have almost twice the risk of suicide.
Compared with the general population, people who seek medical attention for TBI may have almost twice the risk of suicide.
Residents of Denmark who seek medical attention for traumatic brain injury (TBI) have an increased risk of suicide, compared with the general Danish population without TBI, according to a study published in the August 14 issue of JAMA. “Additional analyses revealed that the risk of suicide was higher for individuals with severe TBI, numerous medical contacts, and longer hospital stays,” said lead author Trine Madsen, PhD. Individuals were at highest risk in the first six months after discharge, said Dr. Madsen, who is a postdoctoral fellow at the Danish Research Institute for Suicide Prevention in Hellerup.
A history of TBI previously has been associated with higher rates of self-harm, suicide, and death than are found in the general population. However, previous studies have been limited by methodological shortcomings, such as small sample sizes and low numbers of suicide cases with TBI. Dr. Madsen and colleagues conducted a retrospective cohort study using nationwide registers covering 7,418,391 individuals living in Denmark between 1980 and 2014 with 164,265,624 person-years’ follow-up. Of these people, 567,823 (7.6%) had a medical contact for TBI, which included mild TBI (ie, concussion), skull fracture without documented TBI, and severe TBI (ie, head injuries with evidence of structural brain injury).
Of 34,529 individuals who died by suicide, 3,536 (10.2%) had medical contact for TBI, including 2,701 for mild TBI, 174 for skull fracture without documented TBI, and 661 for severe TBI. The absolute suicide rate was 41 per 100,000 person-years among those with TBI versus 20 per 100,000 person-years among those with no diagnosis of TBI. After accounting for relevant covariates such as fractures not involving the skull, psychiatric diagnoses, and deliberate self-harm, the adjusted incidence ratio was 1.90.
This study “provides insights into the underappreciated relationship between TBI and suicide,” said Lee Goldstein, MD, PhD, and Ramon Diaz-Arrastia, MD, PhD, in an accompanying editorial. “The results … point to an important clinical triad—TBI history, recent injury (especially with long hospital stays), and more numerous postinjury medical contacts for TBI—that serves as a red flag for increased suicide risk,” said Dr. Goldstein, who is affiliated with Boston University School of Medicine, and Dr. Diaz-Arrastia, of the University of Pennsylvania’s Perelman School of Medicine in Philadelphia. The results “indicate that increased suicide risk is relevant across all TBI severity levels, including the far more common mild injuries. Clinicians, health care professionals, and mental health practitioners must take notice of this important information.”
—Glenn S. Williams
Suggested Reading
Goldstein L, Diaz-Arrastia R. Traumatic brain injury and risk of suicide. JAMA. 2018;320(6):554-556.
Madsen T, Erlangsen A, Orlovska S, et al. Association between traumatic brain injury and risk of suicide. JAMA. 2018;320(6):580-588.
Residents of Denmark who seek medical attention for traumatic brain injury (TBI) have an increased risk of suicide, compared with the general Danish population without TBI, according to a study published in the August 14 issue of JAMA. “Additional analyses revealed that the risk of suicide was higher for individuals with severe TBI, numerous medical contacts, and longer hospital stays,” said lead author Trine Madsen, PhD. Individuals were at highest risk in the first six months after discharge, said Dr. Madsen, who is a postdoctoral fellow at the Danish Research Institute for Suicide Prevention in Hellerup.
A history of TBI previously has been associated with higher rates of self-harm, suicide, and death than are found in the general population. However, previous studies have been limited by methodological shortcomings, such as small sample sizes and low numbers of suicide cases with TBI. Dr. Madsen and colleagues conducted a retrospective cohort study using nationwide registers covering 7,418,391 individuals living in Denmark between 1980 and 2014 with 164,265,624 person-years’ follow-up. Of these people, 567,823 (7.6%) had a medical contact for TBI, which included mild TBI (ie, concussion), skull fracture without documented TBI, and severe TBI (ie, head injuries with evidence of structural brain injury).
Of 34,529 individuals who died by suicide, 3,536 (10.2%) had medical contact for TBI, including 2,701 for mild TBI, 174 for skull fracture without documented TBI, and 661 for severe TBI. The absolute suicide rate was 41 per 100,000 person-years among those with TBI versus 20 per 100,000 person-years among those with no diagnosis of TBI. After accounting for relevant covariates such as fractures not involving the skull, psychiatric diagnoses, and deliberate self-harm, the adjusted incidence ratio was 1.90.
This study “provides insights into the underappreciated relationship between TBI and suicide,” said Lee Goldstein, MD, PhD, and Ramon Diaz-Arrastia, MD, PhD, in an accompanying editorial. “The results … point to an important clinical triad—TBI history, recent injury (especially with long hospital stays), and more numerous postinjury medical contacts for TBI—that serves as a red flag for increased suicide risk,” said Dr. Goldstein, who is affiliated with Boston University School of Medicine, and Dr. Diaz-Arrastia, of the University of Pennsylvania’s Perelman School of Medicine in Philadelphia. The results “indicate that increased suicide risk is relevant across all TBI severity levels, including the far more common mild injuries. Clinicians, health care professionals, and mental health practitioners must take notice of this important information.”
—Glenn S. Williams
Suggested Reading
Goldstein L, Diaz-Arrastia R. Traumatic brain injury and risk of suicide. JAMA. 2018;320(6):554-556.
Madsen T, Erlangsen A, Orlovska S, et al. Association between traumatic brain injury and risk of suicide. JAMA. 2018;320(6):580-588.
Serum Nf-L shows promise as biomarker for BMT response in MS
LOS ANGELES – Serum neurofilament light chain shows promise as a biomarker for disease severity and response to treatment with autologous bone marrow transplant in patients with multiple sclerosis, according to findings from an analysis of paired samples.

Serum neurofilament light chain (Nf-L) levels were found to be significantly elevated in both the serum and cerebrospinal fluid (CSF) of 23 patients with aggressive multiple sclerosis (MS), compared with samples from 5 controls with noninflammatory neurologic conditions such as chronic fatigue syndrome or migraine. The levels in the patients with MS were significantly reduced following autologous bone marrow transplant (BMT), Simon Thebault, MD, reported in a poster at the annual meeting of the American Academy of Neurology.
Nf-L has been shown to be a biomarker for neuronal damage and to have potential for denoting disease severity and treatment response in MS; for this analysis, samples from patients were collected at baseline and annually for 3 years after transplant. Samples from controls were obtained for comparison.
“Our objective, really, was [to determine if we could] detect a treatment response in neurofilament light chain, and secondly, did the degree of neurofilament light chain being high at baseline predict anything about subsequent disease outcome?” Dr. Thebault, a third-year resident at the University of Ottawa, reported in the poster.
Indeed, a treatment response was detected; baseline levels were high, and levels in both serum and CSF were down significantly (P = .05) at both 1 and 3 years following bone marrow transplant, and they stayed down. “In fact, they were so low, they were not significantly different from [the levels] in noninflammatory controls,” he said, noting that serum and CSF NfL levels were highly correlated (r = .833; P less than .001).
Study subjects were patients with aggressive MS, characterized by early disease onset, rapid progression, frequent relapses, and poor treatment responses. Such patients who have received autologous BMT represent an interesting group for examining treatment responses, he said.
At baseline, these patients presumably have a high burden of tissue injury, which would be representative of high levels of Nf-L; good, prospective data show these patients have no evidence of ongoing inflammatory disease following an intensive regimen that includes chemoablation and reinfusion of autologous stem cells, which is representative of a significant reduction of neurofilament levels, he explained.
Importantly, serum and CSF levels were correlated in this study, he said, noting that most prior work has involved CSF, but “the real clinical utility in neurofilament light chain is probably in the serum, because patients don’t like having lumbar punctures too often.”
With respect to the second question pertaining to disease outcomes, the study did show that patients with baseline Nf-L levels above the median for the group had worse T1 lesion volumes at baseline and after transplant (P = .0021 at 36 months), and also had worsening brain atrophy even after transplant (P = .0389 at 60 months).
The curves appeared to be diverging, suggesting that, in the absence of inflammatory disease activity, patients with high Nf-L levels at baseline continue to have ongoing atrophy at a differential rate, compared with those with low baseline levels, Dr. Thebault said.
Other findings included better Expanded Disability Status Scale outcomes after transplant in patients with lower baseline Nf-L, and a trend toward worsening outcomes among those with higher baseline Nf-L levels, although the study may have been underpowered for this. Additionally, lower baseline Nf-L predicted significantly lower T1 and T2 lesion volume, number of gadolinium-enhancing lesions, and a reduction in brain atrophy, whereas higher baseline Nf-L predicted the opposite, he noted, adding that N-acetylaspartate-to-creatinine ratios also tended to vary inversely with Nf-L levels.
The most important findings of the study are “the sustained reduction in Nf-L levels after BMT, and that higher baseline levels predicted worse MRI outcomes,” Dr. Thebault noted. “Together these data add to a growing body of evidence that suggests that serum Nf-L has a role in monitoring treatment responses and even a predictive value in determining disease severity.”
This study was supported by Quanterix, which provided Nf-L assay kits. Dr. Thebault reported having no disclosures.
SOURCE: Thebault S et al. Neurology. 2018 Apr;90(15 Suppl.):P5.036.
LOS ANGELES – Serum neurofilament light chain shows promise as a biomarker for disease severity and response to treatment with autologous bone marrow transplant in patients with multiple sclerosis, according to findings from an analysis of paired samples.
Serum neurofilament light chain (Nf-L) levels were found to be significantly elevated in both the serum and cerebrospinal fluid (CSF) of 23 patients with aggressive multiple sclerosis (MS), compared with samples from 5 controls with noninflammatory neurologic conditions such as chronic fatigue syndrome or migraine. The levels in the patients with MS were significantly reduced following autologous bone marrow transplant (BMT), Simon Thebault, MD, reported in a poster at the annual meeting of the American Academy of Neurology.
Nf-L has been shown to be a biomarker for neuronal damage and to have potential for denoting disease severity and treatment response in MS; for this analysis, samples from patients were collected at baseline and annually for 3 years after transplant. Samples from controls were obtained for comparison.
“Our objective, really, was [to determine if we could] detect a treatment response in neurofilament light chain, and secondly, did the degree of neurofilament light chain being high at baseline predict anything about subsequent disease outcome?” Dr. Thebault, a third-year resident at the University of Ottawa, reported in the poster.
Indeed, a treatment response was detected; baseline levels were high, and levels in both serum and CSF were down significantly (P = .05) at both 1 and 3 years following bone marrow transplant, and they stayed down. “In fact, they were so low, they were not significantly different from [the levels] in noninflammatory controls,” he said, noting that serum and CSF NfL levels were highly correlated (r = .833; P less than .001).
Study subjects were patients with aggressive MS, characterized by early disease onset, rapid progression, frequent relapses, and poor treatment responses. Such patients who have received autologous BMT represent an interesting group for examining treatment responses, he said.
At baseline, these patients presumably have a high burden of tissue injury, which would be representative of high levels of Nf-L; good, prospective data show these patients have no evidence of ongoing inflammatory disease following an intensive regimen that includes chemoablation and reinfusion of autologous stem cells, which is representative of a significant reduction of neurofilament levels, he explained.
Importantly, serum and CSF levels were correlated in this study, he said, noting that most prior work has involved CSF, but “the real clinical utility in neurofilament light chain is probably in the serum, because patients don’t like having lumbar punctures too often.”
With respect to the second question pertaining to disease outcomes, the study did show that patients with baseline Nf-L levels above the median for the group had worse T1 lesion volumes at baseline and after transplant (P = .0021 at 36 months), and also had worsening brain atrophy even after transplant (P = .0389 at 60 months).
The curves appeared to be diverging, suggesting that, in the absence of inflammatory disease activity, patients with high Nf-L levels at baseline continue to have ongoing atrophy at a differential rate, compared with those with low baseline levels, Dr. Thebault said.
Other findings included better Expanded Disability Status Scale outcomes after transplant in patients with lower baseline Nf-L, and a trend toward worsening outcomes among those with higher baseline Nf-L levels, although the study may have been underpowered for this. Additionally, lower baseline Nf-L predicted significantly lower T1 and T2 lesion volume, number of gadolinium-enhancing lesions, and a reduction in brain atrophy, whereas higher baseline Nf-L predicted the opposite, he noted, adding that N-acetylaspartate-to-creatinine ratios also tended to vary inversely with Nf-L levels.
The most important findings of the study are “the sustained reduction in Nf-L levels after BMT, and that higher baseline levels predicted worse MRI outcomes,” Dr. Thebault noted. “Together these data add to a growing body of evidence that suggests that serum Nf-L has a role in monitoring treatment responses and even a predictive value in determining disease severity.”
This study was supported by Quanterix, which provided Nf-L assay kits. Dr. Thebault reported having no disclosures.
SOURCE: Thebault S et al. Neurology. 2018 Apr;90(15 Suppl.):P5.036.
LOS ANGELES – Serum neurofilament light chain shows promise as a biomarker for disease severity and response to treatment with autologous bone marrow transplant in patients with multiple sclerosis, according to findings from an analysis of paired samples.
Serum neurofilament light chain (Nf-L) levels were found to be significantly elevated in both the serum and cerebrospinal fluid (CSF) of 23 patients with aggressive multiple sclerosis (MS), compared with samples from 5 controls with noninflammatory neurologic conditions such as chronic fatigue syndrome or migraine. The levels in the patients with MS were significantly reduced following autologous bone marrow transplant (BMT), Simon Thebault, MD, reported in a poster at the annual meeting of the American Academy of Neurology.
Nf-L has been shown to be a biomarker for neuronal damage and to have potential for denoting disease severity and treatment response in MS; for this analysis, samples from patients were collected at baseline and annually for 3 years after transplant. Samples from controls were obtained for comparison.
“Our objective, really, was [to determine if we could] detect a treatment response in neurofilament light chain, and secondly, did the degree of neurofilament light chain being high at baseline predict anything about subsequent disease outcome?” Dr. Thebault, a third-year resident at the University of Ottawa, reported in the poster.
Indeed, a treatment response was detected; baseline levels were high, and levels in both serum and CSF were down significantly (P = .05) at both 1 and 3 years following bone marrow transplant, and they stayed down. “In fact, they were so low, they were not significantly different from [the levels] in noninflammatory controls,” he said, noting that serum and CSF NfL levels were highly correlated (r = .833; P less than .001).
Study subjects were patients with aggressive MS, characterized by early disease onset, rapid progression, frequent relapses, and poor treatment responses. Such patients who have received autologous BMT represent an interesting group for examining treatment responses, he said.
At baseline, these patients presumably have a high burden of tissue injury, which would be representative of high levels of Nf-L; good, prospective data show these patients have no evidence of ongoing inflammatory disease following an intensive regimen that includes chemoablation and reinfusion of autologous stem cells, which is representative of a significant reduction of neurofilament levels, he explained.
Importantly, serum and CSF levels were correlated in this study, he said, noting that most prior work has involved CSF, but “the real clinical utility in neurofilament light chain is probably in the serum, because patients don’t like having lumbar punctures too often.”
With respect to the second question pertaining to disease outcomes, the study did show that patients with baseline Nf-L levels above the median for the group had worse T1 lesion volumes at baseline and after transplant (P = .0021 at 36 months), and also had worsening brain atrophy even after transplant (P = .0389 at 60 months).
The curves appeared to be diverging, suggesting that, in the absence of inflammatory disease activity, patients with high Nf-L levels at baseline continue to have ongoing atrophy at a differential rate, compared with those with low baseline levels, Dr. Thebault said.
Other findings included better Expanded Disability Status Scale outcomes after transplant in patients with lower baseline Nf-L, and a trend toward worsening outcomes among those with higher baseline Nf-L levels, although the study may have been underpowered for this. Additionally, lower baseline Nf-L predicted significantly lower T1 and T2 lesion volume, number of gadolinium-enhancing lesions, and a reduction in brain atrophy, whereas higher baseline Nf-L predicted the opposite, he noted, adding that N-acetylaspartate-to-creatinine ratios also tended to vary inversely with Nf-L levels.
The most important findings of the study are “the sustained reduction in Nf-L levels after BMT, and that higher baseline levels predicted worse MRI outcomes,” Dr. Thebault noted. “Together these data add to a growing body of evidence that suggests that serum Nf-L has a role in monitoring treatment responses and even a predictive value in determining disease severity.”
This study was supported by Quanterix, which provided Nf-L assay kits. Dr. Thebault reported having no disclosures.
SOURCE: Thebault S et al. Neurology. 2018 Apr;90(15 Suppl.):P5.036.
REPORTING FROM AAN 2018
Key clinical point:
Major finding: Serum and cerebrospinal fluid Nf-L levels declines significantly after bone marrow transplant (P less than .05) and did not differ from the levels in controls.
Study details: An analysis of paired samples from 23 patients with multiple sclerosis and 5 controls.
Disclosures: This study was supported by Quanterix, which provided Nf-L assay kits. Dr. Thebault reported having no disclosures.
Source: Thebault S et al. Neurology. 2018 Apr;90(15 Suppl.):P5.036.
Inherited mutations drive 12% of Nigerian breast cancer
About one in eight Nigerian women with breast cancer has an inherited mutation of the BRCA1, BRCA2, PALB2, or TP53 gene.
A new analysis of the Nigerian Breast Cancer Study confirmed that these inherited mutations drive about 12% of the country’s breast cancer cases. The findings could pave the way for the first large-scale national breast cancer gene screening program, wrote Olufunmilayo I. Olopade, MD, and her colleagues. The report is in the Journal of Clinical Oncology.
“We suggest that genomic sequencing to identify women at extremely high risk of breast cancer could be a highly innovative approach to tailored risk management and life-saving interventions,” wrote Dr. Olopade, director of the Center for Clinical Cancer Genetics at the University of Chicago, and her colleagues. “Nigeria now has data to prioritize the integration of genetic testing into its cancer control plan. Women with an extremely high risk of breast cancer because of mutations in these genes can be identified inexpensively and unambiguously and offered interventions to reduce cancer risk.”
And, since about half of the sisters and daughters of affected women will carry the same mutation, such a screening program could reach far beyond every index patient identified, the investigators noted.
“If these women at very high risk can be identified either through their relatives with breast cancer or in the general population, resources can be focused particularly on their behalf. For as-yet unaffected women at high genetic risk, these resources would be intensive surveillance for early detection of breast cancer and, after childbearing is completed, the possibility of preventive salpingo-oophorectomy. Integrated population screening for cancer for all women is the goal, but focused outreach to women at extremely high risk represents an especially efficient use of resources and an attainable evidence-based global health approach.”
The Nigerian Breast Cancer Study enrolled 1,136 women with invasive breast cancer from 1998 to 2014. These were compared with 997 women without cancer, matched from the same communities. Genetic sequencing searched for mutations in both known and breast cancer genes.
Cases and controls were a mean of 47 years old; only 6% of cases reported a family history of breast cancer. Of 577 patients with information on tumor stage, 86% (497) were diagnosed at stage III (241) or IV (256).
Among the cases, 167 (14.7%) carried a mutation in a breast cancer risk gene, compared with 1.8% of controls. BRCA1 was the most common mutation, occurring in 7% of patients; these women were 23 times more likely to develop breast cancer than were those without the gene (odds ratio, 23.4). BRCA2 was the next most common, occurring in 4% of cases and conferring a nearly 11-fold increased risk (OR, 10.76). PALB2 occurred in 11 cases (1%) and no controls, and TP53 in four cases (0.4%).
Women with the BRCA1 mutation were diagnosed at a significantly younger age than were other patients (42.6 vs. 47.9 years), as were carriers of the TP53 mutation (32.8 vs. 47.6 years).
Ten other genes (ATM, BARD1, BRIP1, CHEK1, CHEK2, GEN1, NBN, RAD51C, RAD51D, and XRCC2) carried a mutation in at least one patient each. “When limited to mutations in the four high-risk genes, 11%-12% of cases in this study carried a loss-of-function variant.”
Dr. Olopade had no financial disclosures.
SOURCE: Olopade et al. J Clin Oncol. 2018 Aug 21. doi: 10.1200/JCO.2018.78.3977.
The findings of the Nigerian Breast Cancer Study make a case for large-scale breast cancer gene screening. But even in a wealthy country with good infrastructure, such a program would be dauntingly complex, Ophira Ginsburg, MD, and Paul Brennan, PhD, wrote in an accompanying editorial.
“Given the estimated 40,983 women in Nigeria younger than age 65 years who will be newly diagnosed with breast cancer in 2030, the estimated mutation carrier frequency for a high-risk gene of 11%-12% translates to approximately 5,000 women with breast cancer each year who might benefit directly from tailored risk-reducing strategies. Moreover, 50% of these women’s sisters and daughters would also stand to benefit,” they wrote.
However, 32 million women would need to be screened to find the 220,000 with one of the mutations – a task that is “clearly beyond the scope of most countries.
“Furthermore, women with pathogenic variants would require intensive follow-up and intervention strategies to reduce their risk of developing breast, ovarian/fallopian tube, and potentially other cancers depending on the gene involved. Importantly, this approach would not address the larger problem of the high breast cancer mortality among the vast majority of women without a pathogenic variant but who make up approximately 85% of the breast cancer burden.”
The World Health Organization recognizes this challenge; the agency doesn’t even recommend mammogram-based population screening unless there is a basic, reliable infrastructure including electricity, quality-assurance measures, referral and recall mechanisms, and monitoring and evaluation frameworks. But WHO does suggest some core elements to guide a country’s comprehensive cancer management strategy, including:
• Considering the whole continuum from prevention to palliation.
• Providing a sustainable strategic plan on the basis of the country’s cancer burden, risk factor prevalence, and the resources available to implement the plan.
• Developing an evidence-based approach generated by population-based cancer registries.
“As many countries improve their cancer systems, investing in human resources, infrastructure, monitoring, and evaluation, it is timely to consider how to evaluate readiness to undertake a population-level cancer genetics intervention and consider the core elements that should be in place to make a substantive effect on cancer mortality.”
Dr. Ginsburg is with the Perlmutter Cancer Center of New York University. Dr. Brennan is with the International Agency for Research on Cancer, Lyon, France.
The findings of the Nigerian Breast Cancer Study make a case for large-scale breast cancer gene screening. But even in a wealthy country with good infrastructure, such a program would be dauntingly complex, Ophira Ginsburg, MD, and Paul Brennan, PhD, wrote in an accompanying editorial.
“Given the estimated 40,983 women in Nigeria younger than age 65 years who will be newly diagnosed with breast cancer in 2030, the estimated mutation carrier frequency for a high-risk gene of 11%-12% translates to approximately 5,000 women with breast cancer each year who might benefit directly from tailored risk-reducing strategies. Moreover, 50% of these women’s sisters and daughters would also stand to benefit,” they wrote.
However, 32 million women would need to be screened to find the 220,000 with one of the mutations – a task that is “clearly beyond the scope of most countries.
“Furthermore, women with pathogenic variants would require intensive follow-up and intervention strategies to reduce their risk of developing breast, ovarian/fallopian tube, and potentially other cancers depending on the gene involved. Importantly, this approach would not address the larger problem of the high breast cancer mortality among the vast majority of women without a pathogenic variant but who make up approximately 85% of the breast cancer burden.”
The World Health Organization recognizes this challenge; the agency doesn’t even recommend mammogram-based population screening unless there is a basic, reliable infrastructure including electricity, quality-assurance measures, referral and recall mechanisms, and monitoring and evaluation frameworks. But WHO does suggest some core elements to guide a country’s comprehensive cancer management strategy, including:
• Considering the whole continuum from prevention to palliation.
• Providing a sustainable strategic plan on the basis of the country’s cancer burden, risk factor prevalence, and the resources available to implement the plan.
• Developing an evidence-based approach generated by population-based cancer registries.
“As many countries improve their cancer systems, investing in human resources, infrastructure, monitoring, and evaluation, it is timely to consider how to evaluate readiness to undertake a population-level cancer genetics intervention and consider the core elements that should be in place to make a substantive effect on cancer mortality.”
Dr. Ginsburg is with the Perlmutter Cancer Center of New York University. Dr. Brennan is with the International Agency for Research on Cancer, Lyon, France.
The findings of the Nigerian Breast Cancer Study make a case for large-scale breast cancer gene screening. But even in a wealthy country with good infrastructure, such a program would be dauntingly complex, Ophira Ginsburg, MD, and Paul Brennan, PhD, wrote in an accompanying editorial.
“Given the estimated 40,983 women in Nigeria younger than age 65 years who will be newly diagnosed with breast cancer in 2030, the estimated mutation carrier frequency for a high-risk gene of 11%-12% translates to approximately 5,000 women with breast cancer each year who might benefit directly from tailored risk-reducing strategies. Moreover, 50% of these women’s sisters and daughters would also stand to benefit,” they wrote.
However, 32 million women would need to be screened to find the 220,000 with one of the mutations – a task that is “clearly beyond the scope of most countries.
“Furthermore, women with pathogenic variants would require intensive follow-up and intervention strategies to reduce their risk of developing breast, ovarian/fallopian tube, and potentially other cancers depending on the gene involved. Importantly, this approach would not address the larger problem of the high breast cancer mortality among the vast majority of women without a pathogenic variant but who make up approximately 85% of the breast cancer burden.”
The World Health Organization recognizes this challenge; the agency doesn’t even recommend mammogram-based population screening unless there is a basic, reliable infrastructure including electricity, quality-assurance measures, referral and recall mechanisms, and monitoring and evaluation frameworks. But WHO does suggest some core elements to guide a country’s comprehensive cancer management strategy, including:
• Considering the whole continuum from prevention to palliation.
• Providing a sustainable strategic plan on the basis of the country’s cancer burden, risk factor prevalence, and the resources available to implement the plan.
• Developing an evidence-based approach generated by population-based cancer registries.
“As many countries improve their cancer systems, investing in human resources, infrastructure, monitoring, and evaluation, it is timely to consider how to evaluate readiness to undertake a population-level cancer genetics intervention and consider the core elements that should be in place to make a substantive effect on cancer mortality.”
Dr. Ginsburg is with the Perlmutter Cancer Center of New York University. Dr. Brennan is with the International Agency for Research on Cancer, Lyon, France.
About one in eight Nigerian women with breast cancer has an inherited mutation of the BRCA1, BRCA2, PALB2, or TP53 gene.
A new analysis of the Nigerian Breast Cancer Study confirmed that these inherited mutations drive about 12% of the country’s breast cancer cases. The findings could pave the way for the first large-scale national breast cancer gene screening program, wrote Olufunmilayo I. Olopade, MD, and her colleagues. The report is in the Journal of Clinical Oncology.
“We suggest that genomic sequencing to identify women at extremely high risk of breast cancer could be a highly innovative approach to tailored risk management and life-saving interventions,” wrote Dr. Olopade, director of the Center for Clinical Cancer Genetics at the University of Chicago, and her colleagues. “Nigeria now has data to prioritize the integration of genetic testing into its cancer control plan. Women with an extremely high risk of breast cancer because of mutations in these genes can be identified inexpensively and unambiguously and offered interventions to reduce cancer risk.”
And, since about half of the sisters and daughters of affected women will carry the same mutation, such a screening program could reach far beyond every index patient identified, the investigators noted.
“If these women at very high risk can be identified either through their relatives with breast cancer or in the general population, resources can be focused particularly on their behalf. For as-yet unaffected women at high genetic risk, these resources would be intensive surveillance for early detection of breast cancer and, after childbearing is completed, the possibility of preventive salpingo-oophorectomy. Integrated population screening for cancer for all women is the goal, but focused outreach to women at extremely high risk represents an especially efficient use of resources and an attainable evidence-based global health approach.”
The Nigerian Breast Cancer Study enrolled 1,136 women with invasive breast cancer from 1998 to 2014. These were compared with 997 women without cancer, matched from the same communities. Genetic sequencing searched for mutations in both known and breast cancer genes.
Cases and controls were a mean of 47 years old; only 6% of cases reported a family history of breast cancer. Of 577 patients with information on tumor stage, 86% (497) were diagnosed at stage III (241) or IV (256).
Among the cases, 167 (14.7%) carried a mutation in a breast cancer risk gene, compared with 1.8% of controls. BRCA1 was the most common mutation, occurring in 7% of patients; these women were 23 times more likely to develop breast cancer than were those without the gene (odds ratio, 23.4). BRCA2 was the next most common, occurring in 4% of cases and conferring a nearly 11-fold increased risk (OR, 10.76). PALB2 occurred in 11 cases (1%) and no controls, and TP53 in four cases (0.4%).
Women with the BRCA1 mutation were diagnosed at a significantly younger age than were other patients (42.6 vs. 47.9 years), as were carriers of the TP53 mutation (32.8 vs. 47.6 years).
Ten other genes (ATM, BARD1, BRIP1, CHEK1, CHEK2, GEN1, NBN, RAD51C, RAD51D, and XRCC2) carried a mutation in at least one patient each. “When limited to mutations in the four high-risk genes, 11%-12% of cases in this study carried a loss-of-function variant.”
Dr. Olopade had no financial disclosures.
SOURCE: Olopade et al. J Clin Oncol. 2018 Aug 21. doi: 10.1200/JCO.2018.78.3977.
About one in eight Nigerian women with breast cancer has an inherited mutation of the BRCA1, BRCA2, PALB2, or TP53 gene.
A new analysis of the Nigerian Breast Cancer Study confirmed that these inherited mutations drive about 12% of the country’s breast cancer cases. The findings could pave the way for the first large-scale national breast cancer gene screening program, wrote Olufunmilayo I. Olopade, MD, and her colleagues. The report is in the Journal of Clinical Oncology.
“We suggest that genomic sequencing to identify women at extremely high risk of breast cancer could be a highly innovative approach to tailored risk management and life-saving interventions,” wrote Dr. Olopade, director of the Center for Clinical Cancer Genetics at the University of Chicago, and her colleagues. “Nigeria now has data to prioritize the integration of genetic testing into its cancer control plan. Women with an extremely high risk of breast cancer because of mutations in these genes can be identified inexpensively and unambiguously and offered interventions to reduce cancer risk.”
And, since about half of the sisters and daughters of affected women will carry the same mutation, such a screening program could reach far beyond every index patient identified, the investigators noted.
“If these women at very high risk can be identified either through their relatives with breast cancer or in the general population, resources can be focused particularly on their behalf. For as-yet unaffected women at high genetic risk, these resources would be intensive surveillance for early detection of breast cancer and, after childbearing is completed, the possibility of preventive salpingo-oophorectomy. Integrated population screening for cancer for all women is the goal, but focused outreach to women at extremely high risk represents an especially efficient use of resources and an attainable evidence-based global health approach.”
The Nigerian Breast Cancer Study enrolled 1,136 women with invasive breast cancer from 1998 to 2014. These were compared with 997 women without cancer, matched from the same communities. Genetic sequencing searched for mutations in both known and breast cancer genes.
Cases and controls were a mean of 47 years old; only 6% of cases reported a family history of breast cancer. Of 577 patients with information on tumor stage, 86% (497) were diagnosed at stage III (241) or IV (256).
Among the cases, 167 (14.7%) carried a mutation in a breast cancer risk gene, compared with 1.8% of controls. BRCA1 was the most common mutation, occurring in 7% of patients; these women were 23 times more likely to develop breast cancer than were those without the gene (odds ratio, 23.4). BRCA2 was the next most common, occurring in 4% of cases and conferring a nearly 11-fold increased risk (OR, 10.76). PALB2 occurred in 11 cases (1%) and no controls, and TP53 in four cases (0.4%).
Women with the BRCA1 mutation were diagnosed at a significantly younger age than were other patients (42.6 vs. 47.9 years), as were carriers of the TP53 mutation (32.8 vs. 47.6 years).
Ten other genes (ATM, BARD1, BRIP1, CHEK1, CHEK2, GEN1, NBN, RAD51C, RAD51D, and XRCC2) carried a mutation in at least one patient each. “When limited to mutations in the four high-risk genes, 11%-12% of cases in this study carried a loss-of-function variant.”
Dr. Olopade had no financial disclosures.
SOURCE: Olopade et al. J Clin Oncol. 2018 Aug 21. doi: 10.1200/JCO.2018.78.3977.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Loss-of-function mutations in four breast cancer risk genes account for much of the disease among Nigerian women with the disease.
Major finding: Inherited mutations of the BRCA1, BRCA2, PALB2, or TP53 gene account for 12% of breast cancer in Nigerian women.
Study details: The Nigerian Breast Cancer Study comprised 1,136 women with invasive breast cancer and 997 controls.
Disclosures: Dr. Olopade had no financial disclosures. The study was largely funded by the National Institutes of Health and the Susan G Komen Foundation.
Source: Olopade et al. J Clin Oncol. 2018 Aug 21. doi: 10.1200/JCO.2018.78.3977.
Neratinib extends adjuvant treatment of patients with HER2-positive breast cancer
The small-molecule tyrosine kinase inhibitor neratinib is now approved for the extended adjuvant treatment of patients with early-stage HER2 [human epidermal growth factor receptor]-positive breast cancer following postoperative trastuzumab. Trastuzumab is a HER2-targeted monoclonal antibody that has become standard of care in combination with chemotherapy for the treatment of this patient population in which it significantly improves survival. However, disease recurrence will occur in about a quarter of trastuzumab-treated patients owing to the development of resistance.
Neratinib may help overcome trastuzumab resistance thanks to its potent inhibition of the downstream phosphorylation of HER2 and other members of the HER family. Its approval was based on the phase 3 ExteNET trial, in which extended adjuvant treatment with neratinib was compared with placebo among 2,840 patients who remained disease free after 1 year of adjuvant trastuzumab.1
The ExteNET trial was performed at 495 centers in Europe, Asia, Australia, New Zealand, and South America. Patients aged 18 years or older (≥20 years in Japan), with stage 1-3 HER2-positive breast cancer, who completed neoadjuvant and adjuvant trastuzumab therapy up to 1 year before randomization were eligible. Patients also had an Eastern Cooperative Oncology Group Performance Status of 0 or 1 (range, 0-5; 0, fully active, and 5, dead), normal organ function, and a left ventricular ejection fraction within normal institutional range. Patients with clinically significant cardiac, gastrointesintal or psychiatric comorbidities and those who were not able to swallow oral medication were excluded from the study.
Patients randomly received oral neratinib 240 mg per day or matching placebo, and randomization was stratified according to HR status (positive or negative), nodal status (0, 1-3, or ≥4) and trastuzumab-adjuvant regimen (sequentially or concurrently with chemotherapy).
The primary outcome was invasive disease-free survival (iDFS). The 2-year iDFS rate was 93.9% for neratinib, compared with 91.6% for placebo (hazard ratio [HR], 0.66; P < .008). Recently, a 5-year analysis of the ExteNET trial showed that after a median follow-up of 5.2 years, the iDFS rates were 90.2% vs 87.7% (HR, 0.73; P = .0083).2
Adverse events
The most common adverse event (AE) was diarrhea, in 95% of patients, 40% of whom had grade 3 diarrhea, leading to dose reduction in 26% of patients and discontinuation in 16.8% of patients. Serious AEs occurred in 7% of patients in the neratinib and 6% of those in the placebo arms. In the 5-year analysis, there was no evidence of increased risk of long-term toxicity or adverse consequences of neratinib-associated diarrhea. Furthermore, the ongoing, open-label phase 2 CONTROL trial suggests that the occurrence and severity of neratinib-associated diarrhea can be effectively controlled with antidiarrheal prophylaxis, with drugs such as loperamide.3
At the January 2017 cut-off, 137 patients treated with neratinib (240 mg/day) for 1 year had also received treatment with loperamide monotherapy, 64 patients had received loperamide and budesonide, and 10 patients had received loperamide and colestipol. The safety data from the loperamide monotherapy arm were compared with the safety data from the ExteNET trial, which was based in a similar population of patients who did not receive antidiarrheal prophylaxis. The incidence of all-grade diarrhea was 77% vs 95%, respectively, for those who received antidiarrheal prophylaxis in the CONTROL trial compared with those in the ExteNET trial who did not, and the repective rates of grade 3 diarrhea were 31% and 40%. The rate of dose reductions and holds owing to diarrhea were also lower among those who received antidiarrheal prophylaxis, but the rate of discontinuation due to diarrhea was higher in the loperamide-treated cohort.
Warnings and precautions
Neratinib is marketed as Nerlynx by Puma Biotechnology Inc. The prescribing information describes warnings and precautions relating to diarrhea, hepatotoxicity, and embryofetal toxicity. Patients should be monitored for diarrhea and treated with antidiarrheals as needed. Severe diarrhea with dehydration should be treated with fluids and electrolytes as needed, treatment should be interrupted and resumed at a reduced dose. For grade 3/4 diarrhea or diarrhea with complicating features (eg, dehydration, fever, neutropenia), stool cultures should be performed to rule out infectious causes.
Total bilirubin, aspartate and alanine aminotransferase, and alkaline phosphatase levels should be measured before starting treatment, every 3 months during treatment, or as clinically indicated. Neratinib can cause fetal harm, so pregnant women should be advised of the risk to the fetus and patients of reproductive potential should be counseled on the need for effective contraception during treatment and for at least 1 month after the last dose.4
1. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17: 367-377.
2. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab- based adjuvant therapy in HER2-positive breast cancer (ExteNET): a 5-year analysis of a randomised, double-blind, placebo- controlled, phase 3 trial. Lancet Oncol. 2017;18(12):1688-1700.
3. Ibrahim E, Tripathy D, Wilkinson M, et al. E£ects of adding budesonide or colestipol to loperamide prophylaxis on neratinib-associated diarrhea in patients (pts) with HER2+ early-stage breast cancer (EBC): The CONTROL trial. Cancer Res. 2017; 77(13 supplement): Abstract CT128.
4. Nerlynx (neratinib) tablets, for oral use. Prescribing information. Puma Biotechnology Inc. https://nerlynx.com/pdf/full-prescribinginformation. pdf. Revised July 2017. Accessed November 20th, 2017.
The small-molecule tyrosine kinase inhibitor neratinib is now approved for the extended adjuvant treatment of patients with early-stage HER2 [human epidermal growth factor receptor]-positive breast cancer following postoperative trastuzumab. Trastuzumab is a HER2-targeted monoclonal antibody that has become standard of care in combination with chemotherapy for the treatment of this patient population in which it significantly improves survival. However, disease recurrence will occur in about a quarter of trastuzumab-treated patients owing to the development of resistance.
Neratinib may help overcome trastuzumab resistance thanks to its potent inhibition of the downstream phosphorylation of HER2 and other members of the HER family. Its approval was based on the phase 3 ExteNET trial, in which extended adjuvant treatment with neratinib was compared with placebo among 2,840 patients who remained disease free after 1 year of adjuvant trastuzumab.1
The ExteNET trial was performed at 495 centers in Europe, Asia, Australia, New Zealand, and South America. Patients aged 18 years or older (≥20 years in Japan), with stage 1-3 HER2-positive breast cancer, who completed neoadjuvant and adjuvant trastuzumab therapy up to 1 year before randomization were eligible. Patients also had an Eastern Cooperative Oncology Group Performance Status of 0 or 1 (range, 0-5; 0, fully active, and 5, dead), normal organ function, and a left ventricular ejection fraction within normal institutional range. Patients with clinically significant cardiac, gastrointesintal or psychiatric comorbidities and those who were not able to swallow oral medication were excluded from the study.
Patients randomly received oral neratinib 240 mg per day or matching placebo, and randomization was stratified according to HR status (positive or negative), nodal status (0, 1-3, or ≥4) and trastuzumab-adjuvant regimen (sequentially or concurrently with chemotherapy).
The primary outcome was invasive disease-free survival (iDFS). The 2-year iDFS rate was 93.9% for neratinib, compared with 91.6% for placebo (hazard ratio [HR], 0.66; P < .008). Recently, a 5-year analysis of the ExteNET trial showed that after a median follow-up of 5.2 years, the iDFS rates were 90.2% vs 87.7% (HR, 0.73; P = .0083).2
Adverse events
The most common adverse event (AE) was diarrhea, in 95% of patients, 40% of whom had grade 3 diarrhea, leading to dose reduction in 26% of patients and discontinuation in 16.8% of patients. Serious AEs occurred in 7% of patients in the neratinib and 6% of those in the placebo arms. In the 5-year analysis, there was no evidence of increased risk of long-term toxicity or adverse consequences of neratinib-associated diarrhea. Furthermore, the ongoing, open-label phase 2 CONTROL trial suggests that the occurrence and severity of neratinib-associated diarrhea can be effectively controlled with antidiarrheal prophylaxis, with drugs such as loperamide.3
At the January 2017 cut-off, 137 patients treated with neratinib (240 mg/day) for 1 year had also received treatment with loperamide monotherapy, 64 patients had received loperamide and budesonide, and 10 patients had received loperamide and colestipol. The safety data from the loperamide monotherapy arm were compared with the safety data from the ExteNET trial, which was based in a similar population of patients who did not receive antidiarrheal prophylaxis. The incidence of all-grade diarrhea was 77% vs 95%, respectively, for those who received antidiarrheal prophylaxis in the CONTROL trial compared with those in the ExteNET trial who did not, and the repective rates of grade 3 diarrhea were 31% and 40%. The rate of dose reductions and holds owing to diarrhea were also lower among those who received antidiarrheal prophylaxis, but the rate of discontinuation due to diarrhea was higher in the loperamide-treated cohort.
Warnings and precautions
Neratinib is marketed as Nerlynx by Puma Biotechnology Inc. The prescribing information describes warnings and precautions relating to diarrhea, hepatotoxicity, and embryofetal toxicity. Patients should be monitored for diarrhea and treated with antidiarrheals as needed. Severe diarrhea with dehydration should be treated with fluids and electrolytes as needed, treatment should be interrupted and resumed at a reduced dose. For grade 3/4 diarrhea or diarrhea with complicating features (eg, dehydration, fever, neutropenia), stool cultures should be performed to rule out infectious causes.
Total bilirubin, aspartate and alanine aminotransferase, and alkaline phosphatase levels should be measured before starting treatment, every 3 months during treatment, or as clinically indicated. Neratinib can cause fetal harm, so pregnant women should be advised of the risk to the fetus and patients of reproductive potential should be counseled on the need for effective contraception during treatment and for at least 1 month after the last dose.4
The small-molecule tyrosine kinase inhibitor neratinib is now approved for the extended adjuvant treatment of patients with early-stage HER2 [human epidermal growth factor receptor]-positive breast cancer following postoperative trastuzumab. Trastuzumab is a HER2-targeted monoclonal antibody that has become standard of care in combination with chemotherapy for the treatment of this patient population in which it significantly improves survival. However, disease recurrence will occur in about a quarter of trastuzumab-treated patients owing to the development of resistance.
Neratinib may help overcome trastuzumab resistance thanks to its potent inhibition of the downstream phosphorylation of HER2 and other members of the HER family. Its approval was based on the phase 3 ExteNET trial, in which extended adjuvant treatment with neratinib was compared with placebo among 2,840 patients who remained disease free after 1 year of adjuvant trastuzumab.1
The ExteNET trial was performed at 495 centers in Europe, Asia, Australia, New Zealand, and South America. Patients aged 18 years or older (≥20 years in Japan), with stage 1-3 HER2-positive breast cancer, who completed neoadjuvant and adjuvant trastuzumab therapy up to 1 year before randomization were eligible. Patients also had an Eastern Cooperative Oncology Group Performance Status of 0 or 1 (range, 0-5; 0, fully active, and 5, dead), normal organ function, and a left ventricular ejection fraction within normal institutional range. Patients with clinically significant cardiac, gastrointesintal or psychiatric comorbidities and those who were not able to swallow oral medication were excluded from the study.
Patients randomly received oral neratinib 240 mg per day or matching placebo, and randomization was stratified according to HR status (positive or negative), nodal status (0, 1-3, or ≥4) and trastuzumab-adjuvant regimen (sequentially or concurrently with chemotherapy).
The primary outcome was invasive disease-free survival (iDFS). The 2-year iDFS rate was 93.9% for neratinib, compared with 91.6% for placebo (hazard ratio [HR], 0.66; P < .008). Recently, a 5-year analysis of the ExteNET trial showed that after a median follow-up of 5.2 years, the iDFS rates were 90.2% vs 87.7% (HR, 0.73; P = .0083).2
Adverse events
The most common adverse event (AE) was diarrhea, in 95% of patients, 40% of whom had grade 3 diarrhea, leading to dose reduction in 26% of patients and discontinuation in 16.8% of patients. Serious AEs occurred in 7% of patients in the neratinib and 6% of those in the placebo arms. In the 5-year analysis, there was no evidence of increased risk of long-term toxicity or adverse consequences of neratinib-associated diarrhea. Furthermore, the ongoing, open-label phase 2 CONTROL trial suggests that the occurrence and severity of neratinib-associated diarrhea can be effectively controlled with antidiarrheal prophylaxis, with drugs such as loperamide.3
At the January 2017 cut-off, 137 patients treated with neratinib (240 mg/day) for 1 year had also received treatment with loperamide monotherapy, 64 patients had received loperamide and budesonide, and 10 patients had received loperamide and colestipol. The safety data from the loperamide monotherapy arm were compared with the safety data from the ExteNET trial, which was based in a similar population of patients who did not receive antidiarrheal prophylaxis. The incidence of all-grade diarrhea was 77% vs 95%, respectively, for those who received antidiarrheal prophylaxis in the CONTROL trial compared with those in the ExteNET trial who did not, and the repective rates of grade 3 diarrhea were 31% and 40%. The rate of dose reductions and holds owing to diarrhea were also lower among those who received antidiarrheal prophylaxis, but the rate of discontinuation due to diarrhea was higher in the loperamide-treated cohort.
Warnings and precautions
Neratinib is marketed as Nerlynx by Puma Biotechnology Inc. The prescribing information describes warnings and precautions relating to diarrhea, hepatotoxicity, and embryofetal toxicity. Patients should be monitored for diarrhea and treated with antidiarrheals as needed. Severe diarrhea with dehydration should be treated with fluids and electrolytes as needed, treatment should be interrupted and resumed at a reduced dose. For grade 3/4 diarrhea or diarrhea with complicating features (eg, dehydration, fever, neutropenia), stool cultures should be performed to rule out infectious causes.
Total bilirubin, aspartate and alanine aminotransferase, and alkaline phosphatase levels should be measured before starting treatment, every 3 months during treatment, or as clinically indicated. Neratinib can cause fetal harm, so pregnant women should be advised of the risk to the fetus and patients of reproductive potential should be counseled on the need for effective contraception during treatment and for at least 1 month after the last dose.4
1. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17: 367-377.
2. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab- based adjuvant therapy in HER2-positive breast cancer (ExteNET): a 5-year analysis of a randomised, double-blind, placebo- controlled, phase 3 trial. Lancet Oncol. 2017;18(12):1688-1700.
3. Ibrahim E, Tripathy D, Wilkinson M, et al. E£ects of adding budesonide or colestipol to loperamide prophylaxis on neratinib-associated diarrhea in patients (pts) with HER2+ early-stage breast cancer (EBC): The CONTROL trial. Cancer Res. 2017; 77(13 supplement): Abstract CT128.
4. Nerlynx (neratinib) tablets, for oral use. Prescribing information. Puma Biotechnology Inc. https://nerlynx.com/pdf/full-prescribinginformation. pdf. Revised July 2017. Accessed November 20th, 2017.
1. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17: 367-377.
2. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab- based adjuvant therapy in HER2-positive breast cancer (ExteNET): a 5-year analysis of a randomised, double-blind, placebo- controlled, phase 3 trial. Lancet Oncol. 2017;18(12):1688-1700.
3. Ibrahim E, Tripathy D, Wilkinson M, et al. E£ects of adding budesonide or colestipol to loperamide prophylaxis on neratinib-associated diarrhea in patients (pts) with HER2+ early-stage breast cancer (EBC): The CONTROL trial. Cancer Res. 2017; 77(13 supplement): Abstract CT128.
4. Nerlynx (neratinib) tablets, for oral use. Prescribing information. Puma Biotechnology Inc. https://nerlynx.com/pdf/full-prescribinginformation. pdf. Revised July 2017. Accessed November 20th, 2017.
New MS subtype shows absence of cerebral white matter demyelination
A new subtype of multiple sclerosis called myelocortical multiple sclerosis is characterized by demyelination only in the spinal cord and cerebral cortex and not in the cerebral white matter.
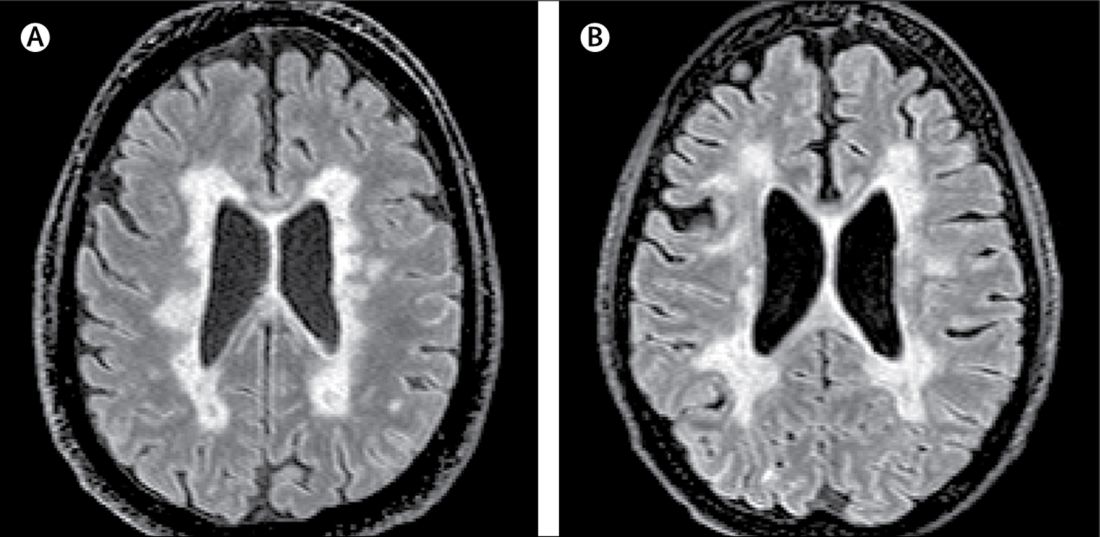
A paper published online Aug. 21 in Lancet Neurology presents the results of a study of the brains and spinal cords of 100 patients who died of multiple sclerosis.
Bruce D. Trapp, PhD, of the Lerner Research Institute at the Cleveland Clinic in Ohio, and his coauthors wrote that while the demyelination of cerebral white matter is a pathologic hallmark of multiple sclerosis, previous research has found only around half of cerebral T2-weighted hyperintense white matter lesions are demyelinated, and these lesions account for less than a third of variance in the rate of brain atrophy.
“In the absence of specific MRI metrics for demyelination, the relationship between cerebral white-matter demyelination and neurodegeneration remains speculative,” they wrote.
In this study, researchers scanned the brains with MRI before autopsy, then took centimeter-thick hemispheric slices to study the white-matter lesions. They identified 12 individuals as having what they describe as ‘myelocortical multiple sclerosis,’ characterized by the absence of areas of cerebral white-matter discoloration indicative of demyelinated lesions.
The authors then compared these individuals to 12 individuals with typical multiple sclerosis matched by age, sex, MRI protocol, multiple sclerosis disease subtype, disease duration, and Expanded Disability Status Scale.
They found that while individuals with myelocortical multiple sclerosis did not have demyelinated lesions in the cerebral white matter, they had similar areas of demyelinated lesions in the cerebral cortex to individuals with typical multiple sclerosis (median 4.45% vs. 9.74% respectively, P = .5512).
However, the individuals with myelocortical multiple sclerosis had a significantly smaller area of spinal cord demyelination (median 3.81% vs. 13.81%, P = .0083).
Individuals with myelocortical multiple sclerosis also had significantly lower mean cortical neuronal densities, compared with healthy control brains in layer III, layer V, and layer VI. But individuals with typical multiple sclerosis only had a lower cortical neuronal density in layer V when compared with controls.
Researchers also saw that in typical multiple sclerosis, neuronal density decreased as the area of brain white-matter demyelination increased. However, this negative linear correlation was not seen in myelocortical multiple sclerosis.
On MRI, researchers were still able to see abnormalities in the cerebral white matter in individuals with myelocortical multiple sclerosis, in T2-weighted, T1-weighted and magnetization transfer ratios (MTR) images.
They also found similar total T2-weighted and T1-weighted lesion volumes in individuals with myelocortical and with typical multiple sclerosis, although individuals with typical multiple sclerosis had significantly greater MTR lesion volumes.
“We propose that myelocortical multiple sclerosis is characterized by spinal cord demyelination, subpial cortical demyelination, and an absence of cerebral white-matter demyelination,” the authors wrote. “Our findings indicate that abnormal cerebral white-matter T2-T1-MTR regions of interest are not always demyelinated, and this pathological evidence suggests that cerebral white-matter demyelination and cortical neuronal degeneration can be independent events in myelocortical multiple sclerosis.”
The authors noted that their study may have been affected by selection bias, as all the patients in the study had died from complications of advanced multiple sclerosis. They suggested that it was therefore not appropriate to conclude that the prevalence of myelocortical multiple sclerosis seen in their sample would be similar across the multiple sclerosis population, nor were the findings likely to apply to people with earlier stage disease.
The study was funded by the U.S. National Institutes of Health and National Multiple Sclerosis Society. One author was an employee of Renovo Neural, and three authors were employees of Biogen. One author declared a pending patent related to automated lesion segmentation from MRI images, and four authors declared funding, fees, and non-financial support from pharmaceutical companies.
SOURCE: Trapp B et al. Lancet Neurol. 2018 Aug 21. doi: 10.1016/ S1474-4422(18)30245-X.
A new subtype of multiple sclerosis called myelocortical multiple sclerosis is characterized by demyelination only in the spinal cord and cerebral cortex and not in the cerebral white matter.
A paper published online Aug. 21 in Lancet Neurology presents the results of a study of the brains and spinal cords of 100 patients who died of multiple sclerosis.
Bruce D. Trapp, PhD, of the Lerner Research Institute at the Cleveland Clinic in Ohio, and his coauthors wrote that while the demyelination of cerebral white matter is a pathologic hallmark of multiple sclerosis, previous research has found only around half of cerebral T2-weighted hyperintense white matter lesions are demyelinated, and these lesions account for less than a third of variance in the rate of brain atrophy.
“In the absence of specific MRI metrics for demyelination, the relationship between cerebral white-matter demyelination and neurodegeneration remains speculative,” they wrote.
In this study, researchers scanned the brains with MRI before autopsy, then took centimeter-thick hemispheric slices to study the white-matter lesions. They identified 12 individuals as having what they describe as ‘myelocortical multiple sclerosis,’ characterized by the absence of areas of cerebral white-matter discoloration indicative of demyelinated lesions.
The authors then compared these individuals to 12 individuals with typical multiple sclerosis matched by age, sex, MRI protocol, multiple sclerosis disease subtype, disease duration, and Expanded Disability Status Scale.
They found that while individuals with myelocortical multiple sclerosis did not have demyelinated lesions in the cerebral white matter, they had similar areas of demyelinated lesions in the cerebral cortex to individuals with typical multiple sclerosis (median 4.45% vs. 9.74% respectively, P = .5512).
However, the individuals with myelocortical multiple sclerosis had a significantly smaller area of spinal cord demyelination (median 3.81% vs. 13.81%, P = .0083).
Individuals with myelocortical multiple sclerosis also had significantly lower mean cortical neuronal densities, compared with healthy control brains in layer III, layer V, and layer VI. But individuals with typical multiple sclerosis only had a lower cortical neuronal density in layer V when compared with controls.
Researchers also saw that in typical multiple sclerosis, neuronal density decreased as the area of brain white-matter demyelination increased. However, this negative linear correlation was not seen in myelocortical multiple sclerosis.
On MRI, researchers were still able to see abnormalities in the cerebral white matter in individuals with myelocortical multiple sclerosis, in T2-weighted, T1-weighted and magnetization transfer ratios (MTR) images.
They also found similar total T2-weighted and T1-weighted lesion volumes in individuals with myelocortical and with typical multiple sclerosis, although individuals with typical multiple sclerosis had significantly greater MTR lesion volumes.
“We propose that myelocortical multiple sclerosis is characterized by spinal cord demyelination, subpial cortical demyelination, and an absence of cerebral white-matter demyelination,” the authors wrote. “Our findings indicate that abnormal cerebral white-matter T2-T1-MTR regions of interest are not always demyelinated, and this pathological evidence suggests that cerebral white-matter demyelination and cortical neuronal degeneration can be independent events in myelocortical multiple sclerosis.”
The authors noted that their study may have been affected by selection bias, as all the patients in the study had died from complications of advanced multiple sclerosis. They suggested that it was therefore not appropriate to conclude that the prevalence of myelocortical multiple sclerosis seen in their sample would be similar across the multiple sclerosis population, nor were the findings likely to apply to people with earlier stage disease.
The study was funded by the U.S. National Institutes of Health and National Multiple Sclerosis Society. One author was an employee of Renovo Neural, and three authors were employees of Biogen. One author declared a pending patent related to automated lesion segmentation from MRI images, and four authors declared funding, fees, and non-financial support from pharmaceutical companies.
SOURCE: Trapp B et al. Lancet Neurol. 2018 Aug 21. doi: 10.1016/ S1474-4422(18)30245-X.
A new subtype of multiple sclerosis called myelocortical multiple sclerosis is characterized by demyelination only in the spinal cord and cerebral cortex and not in the cerebral white matter.
A paper published online Aug. 21 in Lancet Neurology presents the results of a study of the brains and spinal cords of 100 patients who died of multiple sclerosis.
Bruce D. Trapp, PhD, of the Lerner Research Institute at the Cleveland Clinic in Ohio, and his coauthors wrote that while the demyelination of cerebral white matter is a pathologic hallmark of multiple sclerosis, previous research has found only around half of cerebral T2-weighted hyperintense white matter lesions are demyelinated, and these lesions account for less than a third of variance in the rate of brain atrophy.
“In the absence of specific MRI metrics for demyelination, the relationship between cerebral white-matter demyelination and neurodegeneration remains speculative,” they wrote.
In this study, researchers scanned the brains with MRI before autopsy, then took centimeter-thick hemispheric slices to study the white-matter lesions. They identified 12 individuals as having what they describe as ‘myelocortical multiple sclerosis,’ characterized by the absence of areas of cerebral white-matter discoloration indicative of demyelinated lesions.
The authors then compared these individuals to 12 individuals with typical multiple sclerosis matched by age, sex, MRI protocol, multiple sclerosis disease subtype, disease duration, and Expanded Disability Status Scale.
They found that while individuals with myelocortical multiple sclerosis did not have demyelinated lesions in the cerebral white matter, they had similar areas of demyelinated lesions in the cerebral cortex to individuals with typical multiple sclerosis (median 4.45% vs. 9.74% respectively, P = .5512).
However, the individuals with myelocortical multiple sclerosis had a significantly smaller area of spinal cord demyelination (median 3.81% vs. 13.81%, P = .0083).
Individuals with myelocortical multiple sclerosis also had significantly lower mean cortical neuronal densities, compared with healthy control brains in layer III, layer V, and layer VI. But individuals with typical multiple sclerosis only had a lower cortical neuronal density in layer V when compared with controls.
Researchers also saw that in typical multiple sclerosis, neuronal density decreased as the area of brain white-matter demyelination increased. However, this negative linear correlation was not seen in myelocortical multiple sclerosis.
On MRI, researchers were still able to see abnormalities in the cerebral white matter in individuals with myelocortical multiple sclerosis, in T2-weighted, T1-weighted and magnetization transfer ratios (MTR) images.
They also found similar total T2-weighted and T1-weighted lesion volumes in individuals with myelocortical and with typical multiple sclerosis, although individuals with typical multiple sclerosis had significantly greater MTR lesion volumes.
“We propose that myelocortical multiple sclerosis is characterized by spinal cord demyelination, subpial cortical demyelination, and an absence of cerebral white-matter demyelination,” the authors wrote. “Our findings indicate that abnormal cerebral white-matter T2-T1-MTR regions of interest are not always demyelinated, and this pathological evidence suggests that cerebral white-matter demyelination and cortical neuronal degeneration can be independent events in myelocortical multiple sclerosis.”
The authors noted that their study may have been affected by selection bias, as all the patients in the study had died from complications of advanced multiple sclerosis. They suggested that it was therefore not appropriate to conclude that the prevalence of myelocortical multiple sclerosis seen in their sample would be similar across the multiple sclerosis population, nor were the findings likely to apply to people with earlier stage disease.
The study was funded by the U.S. National Institutes of Health and National Multiple Sclerosis Society. One author was an employee of Renovo Neural, and three authors were employees of Biogen. One author declared a pending patent related to automated lesion segmentation from MRI images, and four authors declared funding, fees, and non-financial support from pharmaceutical companies.
SOURCE: Trapp B et al. Lancet Neurol. 2018 Aug 21. doi: 10.1016/ S1474-4422(18)30245-X.
FROM LANCET NEUROLOGY
Key clinical point: Researchers have identified a new subtype of multiple sclerosis.
Major finding: Individuals with myelocortical multiple sclerosis show demyelination in the spinal cord and cortex only.
Study details: Post-mortem study of brains and spinal cords of 100 individuals with multiple sclerosis.
Disclosures: The study was funded by the U.S. National Institutes of Health and National Multiple Sclerosis Society. One author was an employee of Renovo Neural, three authors were employees of Biogen. One author declared a pending patent related to automated lesion segmentation from MRI images, and four authors declared funding, fees and non-financial support from pharmaceutical companies.
Source: Trapp B et al. Lancet Neurol. 2018 Aug 21. doi: 10.1016/ S1474-4422(18)30245-X.
Latex Allergy From Biologic Injectable Devices


Meeting the potential of immunotherapy: new targets provide rational combinations
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.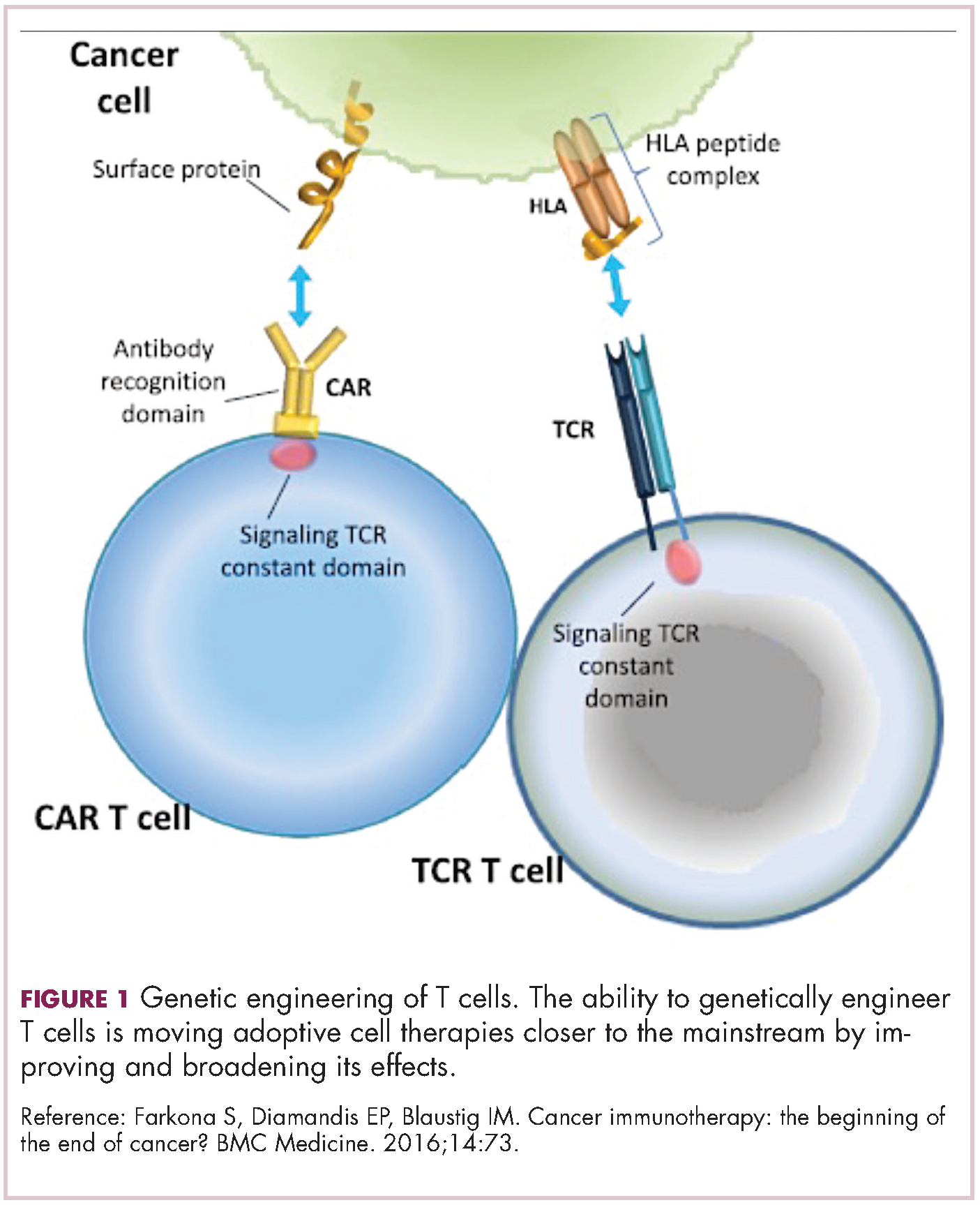
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17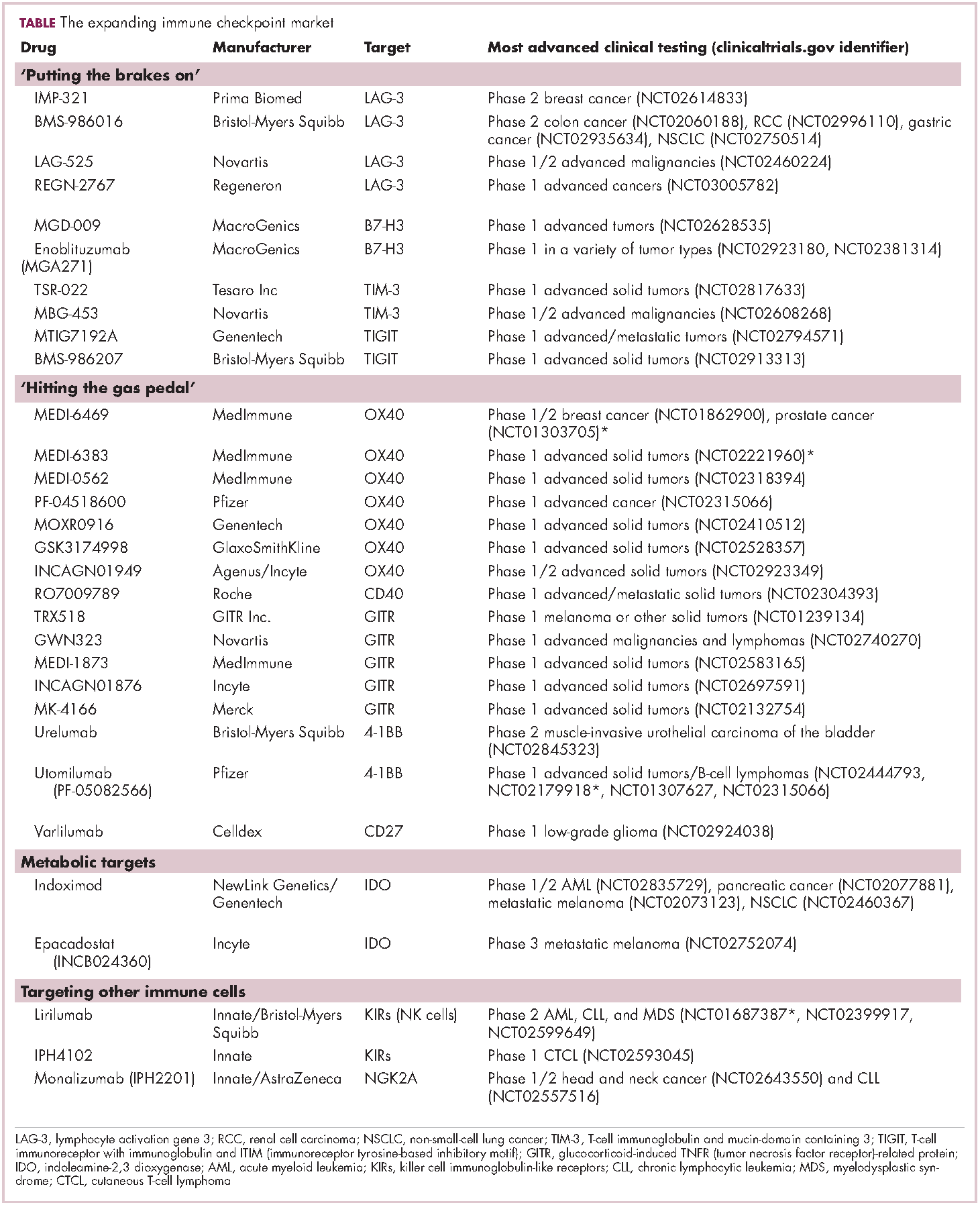
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
Innovations Lead to More Targeted Prostate Cancer Treatments (FULL)
The main treatment for prostate cancer—the third leading cause of cancer death in American men—often is “watchful waiting.” But what happens before, during, and after that waiting period has changed tremendously in recent years. Innovative and improved methods and drugs allow for a more precise diagnosis, better risk stratification, targeted treatment options, and longer survival.
Innovations in diagnosis include a revised histologic grading system, which was incorporated into the 2016 World Health Organization classification of tumors. The new grading system ranks prostate cancer on a 1-to-5 scale, making it more discriminating, as validated in a study of more than 25,000 men.
The use of new prognostic biomarkers has advanced risk stratification. According to a recent review, biopsy guided by ultrasound misses between 21% and 28% of prostate cancers and undergrades between 14% and 17%.1 But new serum-, tissue-, and image-based biomarkers may help identify potential false negatives. The prostate cancer antigen 3 test, for example, has an 88% negative predictive value for subsequent biopsy. Molecular biomarkers also can predict clinical progression, risk of adverse pathology, and metastatic risk.
Fortunately, biopsy guided by ultrasound is getting more precise. Advances in magnetic resonance imaging (MRI) now allow for “targeted biopsies.” The enhanced MRI has 89% sensitivity and 73% specificity for identifying prostate cancer. According to one study of 1,003 men, targeted prostate biopsy using MRI-ultrasound fusion identified 30% more cases of Gleason score ≥ 4 + 3 than did systematic prostate biopsy.1 Updates in positron emission tomography are garnering interest for improved staging because this technology allows for better detection of local recurrence, regional lymph node metastases, and distant metastases.
Once a prostate cancer diagnosis has been confirmed, the decision of what to do next may be watchful waiting (treating symptoms palliatively), but recent research suggests that active surveillance that includes regular prostate-specific antigen testing, physical examinations, and prostate biopsies may be a better choice, particularly for men with less aggressive cancer. One study of 1,298 men with mostly very low-risk disease followed for up to 60 months found metastasis in only 5; only 2 died. The Prostate Testing for Cancer and Treatment (ProtecT) trial found that the number of deaths in the active monitoring group did not differ significantly from those in the surgery or radiation groups.
What should be the contemporary standard of care? Androgen deprivation therapy (ADT) is still the go-to treatment for men with metastatic prostate cancer. Although ADT has been associated with toxicity, a meta-analysis found continuous ADT was better than intermittent in terms of disease progression and survival.1
Other research has focused on which types of prostate cancer respond best to specific therapies. Molecular subtyping (already available in bladder and breast cancer) is gaining popularity. Prostate cancer was thought to derive from glandular luminal cells, but recent evidence supports the idea that basal cells play a role as well. Researchers who analyzed nearly 4,000 samples suggest that luminal B tumors respond better to postoperative ADT than do nonluminal B cancers. These findings suggest that “personalized” ADT treatment may be possible.2
Several drugs have been shown to improve survival: Among them, docetaxel, abiraterone acetate, enzalutamide, and cabazitaxel. In the STAMPEDE trial, men with locally advanced or metastatic prostate cancer who received ADT plus abiraterone and prednisolone had significantly higher rates of overall and failure-free survival.3
Docetaxel, which can extend survival by 10 to 13 months compared with standard ADT, is taking on a bigger role for its ability to delay progression and recurrence while being well tolerated. Options for men whose cancer does not respond to ADT include abiraterone and enzalutamide. Both act on the androgen axis to slow progression and improve survival.
More than 30% of patients treated with radical prostatectomy will have recurrent cancer as will 50% of those treated with salvage radiation therapy. Bicalutamide has shown extremely promising action against recurrent cancer. In one study, the cumulative incidence of metastatic prostate cancer at 12 years was 14.5% in the bicalutamide group, compared with 23.0% in the placebo group.4
But while that study was going on, it was superseded by injectable gonadotropin-releasing hormone agonists as first-choice hormonal therapy with radiation. However, the researchers say that does not
Multimodal therapy and precision medicine are becoming bywords in prostate cancer treatment. Drugs on the horizon likely will be tailored to tumor molecular biology, with genetic information used to specifically guide diagnosis and treatment. Prostate cancer may still be a slow killer, but immunotherapies (like sipuleucel-T, the first FDA-approved cancer vaccine), hormonal therapies, and bone-targeting agents enable men with prostate cancer to not only live longer but also with a better quality of life.
Click here to read the digital edition.
1. Litwin MS, Tan HJ. The diagnosis and treatment of prostate cancer: a review. JAMA. 2017;317(24):2532-2542.
2. Zhao SG, Chang SL, Erho N, et al. Associations of luminal and basal subtyping of prostate cancer with prognosis and response to androgen deprivation therapy. JAMA Oncol. 2017. [Epub ahead of print.]
3. James ND, de Bono JS, Spears MR, et al; for the STAMPEDE Investigators. Abiraterone for prostate cancer not previously treated with hormone therapy. N Engl J Med. 2017. [Epub ahead of print.]
4. Shipley WU, Seiferheld W, Lukka HR, et al; NRG Oncology RTOG. Radiation with or without antiandrogen therapy in recurrent prostate cancer. N Engl J Med. 2017;376(5):417-428.
The main treatment for prostate cancer—the third leading cause of cancer death in American men—often is “watchful waiting.” But what happens before, during, and after that waiting period has changed tremendously in recent years. Innovative and improved methods and drugs allow for a more precise diagnosis, better risk stratification, targeted treatment options, and longer survival.
Innovations in diagnosis include a revised histologic grading system, which was incorporated into the 2016 World Health Organization classification of tumors. The new grading system ranks prostate cancer on a 1-to-5 scale, making it more discriminating, as validated in a study of more than 25,000 men.
The use of new prognostic biomarkers has advanced risk stratification. According to a recent review, biopsy guided by ultrasound misses between 21% and 28% of prostate cancers and undergrades between 14% and 17%.1 But new serum-, tissue-, and image-based biomarkers may help identify potential false negatives. The prostate cancer antigen 3 test, for example, has an 88% negative predictive value for subsequent biopsy. Molecular biomarkers also can predict clinical progression, risk of adverse pathology, and metastatic risk.
Fortunately, biopsy guided by ultrasound is getting more precise. Advances in magnetic resonance imaging (MRI) now allow for “targeted biopsies.” The enhanced MRI has 89% sensitivity and 73% specificity for identifying prostate cancer. According to one study of 1,003 men, targeted prostate biopsy using MRI-ultrasound fusion identified 30% more cases of Gleason score ≥ 4 + 3 than did systematic prostate biopsy.1 Updates in positron emission tomography are garnering interest for improved staging because this technology allows for better detection of local recurrence, regional lymph node metastases, and distant metastases.
Once a prostate cancer diagnosis has been confirmed, the decision of what to do next may be watchful waiting (treating symptoms palliatively), but recent research suggests that active surveillance that includes regular prostate-specific antigen testing, physical examinations, and prostate biopsies may be a better choice, particularly for men with less aggressive cancer. One study of 1,298 men with mostly very low-risk disease followed for up to 60 months found metastasis in only 5; only 2 died. The Prostate Testing for Cancer and Treatment (ProtecT) trial found that the number of deaths in the active monitoring group did not differ significantly from those in the surgery or radiation groups.
What should be the contemporary standard of care? Androgen deprivation therapy (ADT) is still the go-to treatment for men with metastatic prostate cancer. Although ADT has been associated with toxicity, a meta-analysis found continuous ADT was better than intermittent in terms of disease progression and survival.1
Other research has focused on which types of prostate cancer respond best to specific therapies. Molecular subtyping (already available in bladder and breast cancer) is gaining popularity. Prostate cancer was thought to derive from glandular luminal cells, but recent evidence supports the idea that basal cells play a role as well. Researchers who analyzed nearly 4,000 samples suggest that luminal B tumors respond better to postoperative ADT than do nonluminal B cancers. These findings suggest that “personalized” ADT treatment may be possible.2
Several drugs have been shown to improve survival: Among them, docetaxel, abiraterone acetate, enzalutamide, and cabazitaxel. In the STAMPEDE trial, men with locally advanced or metastatic prostate cancer who received ADT plus abiraterone and prednisolone had significantly higher rates of overall and failure-free survival.3
Docetaxel, which can extend survival by 10 to 13 months compared with standard ADT, is taking on a bigger role for its ability to delay progression and recurrence while being well tolerated. Options for men whose cancer does not respond to ADT include abiraterone and enzalutamide. Both act on the androgen axis to slow progression and improve survival.
More than 30% of patients treated with radical prostatectomy will have recurrent cancer as will 50% of those treated with salvage radiation therapy. Bicalutamide has shown extremely promising action against recurrent cancer. In one study, the cumulative incidence of metastatic prostate cancer at 12 years was 14.5% in the bicalutamide group, compared with 23.0% in the placebo group.4
But while that study was going on, it was superseded by injectable gonadotropin-releasing hormone agonists as first-choice hormonal therapy with radiation. However, the researchers say that does not
Multimodal therapy and precision medicine are becoming bywords in prostate cancer treatment. Drugs on the horizon likely will be tailored to tumor molecular biology, with genetic information used to specifically guide diagnosis and treatment. Prostate cancer may still be a slow killer, but immunotherapies (like sipuleucel-T, the first FDA-approved cancer vaccine), hormonal therapies, and bone-targeting agents enable men with prostate cancer to not only live longer but also with a better quality of life.
Click here to read the digital edition.
The main treatment for prostate cancer—the third leading cause of cancer death in American men—often is “watchful waiting.” But what happens before, during, and after that waiting period has changed tremendously in recent years. Innovative and improved methods and drugs allow for a more precise diagnosis, better risk stratification, targeted treatment options, and longer survival.
Innovations in diagnosis include a revised histologic grading system, which was incorporated into the 2016 World Health Organization classification of tumors. The new grading system ranks prostate cancer on a 1-to-5 scale, making it more discriminating, as validated in a study of more than 25,000 men.
The use of new prognostic biomarkers has advanced risk stratification. According to a recent review, biopsy guided by ultrasound misses between 21% and 28% of prostate cancers and undergrades between 14% and 17%.1 But new serum-, tissue-, and image-based biomarkers may help identify potential false negatives. The prostate cancer antigen 3 test, for example, has an 88% negative predictive value for subsequent biopsy. Molecular biomarkers also can predict clinical progression, risk of adverse pathology, and metastatic risk.
Fortunately, biopsy guided by ultrasound is getting more precise. Advances in magnetic resonance imaging (MRI) now allow for “targeted biopsies.” The enhanced MRI has 89% sensitivity and 73% specificity for identifying prostate cancer. According to one study of 1,003 men, targeted prostate biopsy using MRI-ultrasound fusion identified 30% more cases of Gleason score ≥ 4 + 3 than did systematic prostate biopsy.1 Updates in positron emission tomography are garnering interest for improved staging because this technology allows for better detection of local recurrence, regional lymph node metastases, and distant metastases.
Once a prostate cancer diagnosis has been confirmed, the decision of what to do next may be watchful waiting (treating symptoms palliatively), but recent research suggests that active surveillance that includes regular prostate-specific antigen testing, physical examinations, and prostate biopsies may be a better choice, particularly for men with less aggressive cancer. One study of 1,298 men with mostly very low-risk disease followed for up to 60 months found metastasis in only 5; only 2 died. The Prostate Testing for Cancer and Treatment (ProtecT) trial found that the number of deaths in the active monitoring group did not differ significantly from those in the surgery or radiation groups.
What should be the contemporary standard of care? Androgen deprivation therapy (ADT) is still the go-to treatment for men with metastatic prostate cancer. Although ADT has been associated with toxicity, a meta-analysis found continuous ADT was better than intermittent in terms of disease progression and survival.1
Other research has focused on which types of prostate cancer respond best to specific therapies. Molecular subtyping (already available in bladder and breast cancer) is gaining popularity. Prostate cancer was thought to derive from glandular luminal cells, but recent evidence supports the idea that basal cells play a role as well. Researchers who analyzed nearly 4,000 samples suggest that luminal B tumors respond better to postoperative ADT than do nonluminal B cancers. These findings suggest that “personalized” ADT treatment may be possible.2
Several drugs have been shown to improve survival: Among them, docetaxel, abiraterone acetate, enzalutamide, and cabazitaxel. In the STAMPEDE trial, men with locally advanced or metastatic prostate cancer who received ADT plus abiraterone and prednisolone had significantly higher rates of overall and failure-free survival.3
Docetaxel, which can extend survival by 10 to 13 months compared with standard ADT, is taking on a bigger role for its ability to delay progression and recurrence while being well tolerated. Options for men whose cancer does not respond to ADT include abiraterone and enzalutamide. Both act on the androgen axis to slow progression and improve survival.
More than 30% of patients treated with radical prostatectomy will have recurrent cancer as will 50% of those treated with salvage radiation therapy. Bicalutamide has shown extremely promising action against recurrent cancer. In one study, the cumulative incidence of metastatic prostate cancer at 12 years was 14.5% in the bicalutamide group, compared with 23.0% in the placebo group.4
But while that study was going on, it was superseded by injectable gonadotropin-releasing hormone agonists as first-choice hormonal therapy with radiation. However, the researchers say that does not
Multimodal therapy and precision medicine are becoming bywords in prostate cancer treatment. Drugs on the horizon likely will be tailored to tumor molecular biology, with genetic information used to specifically guide diagnosis and treatment. Prostate cancer may still be a slow killer, but immunotherapies (like sipuleucel-T, the first FDA-approved cancer vaccine), hormonal therapies, and bone-targeting agents enable men with prostate cancer to not only live longer but also with a better quality of life.
Click here to read the digital edition.
1. Litwin MS, Tan HJ. The diagnosis and treatment of prostate cancer: a review. JAMA. 2017;317(24):2532-2542.
2. Zhao SG, Chang SL, Erho N, et al. Associations of luminal and basal subtyping of prostate cancer with prognosis and response to androgen deprivation therapy. JAMA Oncol. 2017. [Epub ahead of print.]
3. James ND, de Bono JS, Spears MR, et al; for the STAMPEDE Investigators. Abiraterone for prostate cancer not previously treated with hormone therapy. N Engl J Med. 2017. [Epub ahead of print.]
4. Shipley WU, Seiferheld W, Lukka HR, et al; NRG Oncology RTOG. Radiation with or without antiandrogen therapy in recurrent prostate cancer. N Engl J Med. 2017;376(5):417-428.
1. Litwin MS, Tan HJ. The diagnosis and treatment of prostate cancer: a review. JAMA. 2017;317(24):2532-2542.
2. Zhao SG, Chang SL, Erho N, et al. Associations of luminal and basal subtyping of prostate cancer with prognosis and response to androgen deprivation therapy. JAMA Oncol. 2017. [Epub ahead of print.]
3. James ND, de Bono JS, Spears MR, et al; for the STAMPEDE Investigators. Abiraterone for prostate cancer not previously treated with hormone therapy. N Engl J Med. 2017. [Epub ahead of print.]
4. Shipley WU, Seiferheld W, Lukka HR, et al; NRG Oncology RTOG. Radiation with or without antiandrogen therapy in recurrent prostate cancer. N Engl J Med. 2017;376(5):417-428.