User login
Trio of biosimilars have good showing
CHICAGO – investigators reported at the annual meeting of the American Society of Clinical Oncology. The findings further advance the promise of new agents that have no clinically meaningful differences in efficacy and safety when compared with their reference drugs but have substantially lower cost.

Bevacizumab biosimilar
The REFLECTIONS trial (NCT02364999) was a multinational, first-line, randomized, controlled trial among 719 patients with advanced nonsquamous NSCLC. Patients were randomized to paclitaxel and carboplatin chemotherapy plus either bevacizumab (sourced from the European Union) or the candidate bevacizumab biosimilar PF-06439535 on a double-blind basis, followed by monotherapy with the same assigned agent.
The confidence interval for the risk difference fell within the equivalence margins set by European Union regulators (–13% and +13% for the 95% confidence interval). And the confidence interval for the risk ratio fell within the equivalence margins set by the Food and Drug Administration (0.73 and 1.37 for the 90% CI) and Japanese regulators (0.729 and 1.371 for the 95% CI).
Median progression-free survival was 9.0 months with the biosimilar and 7.7 months with bevacizumab (hazard ratio, 0.974; P = .814), and corresponding 1-year rates were 30.8% and 29.3%, Dr. Socinski reported. Median overall survival was 18.4 months and 17.8 months (HR, 1.001; P = .991), and corresponding 1-year rates were 66.4% and 68.8%.
Rates of grade 3 or higher hypertension, cardiac disorders, and bleeding did not differ significantly with the two agents. Patients also had similar rates of grade 3 or higher serious adverse events and of fatal (grade 5) serious adverse events (5.3% with the biosimilar and 5.9% with bevacizumab).
“Similarity between PF-06439535 and bevacizumab-EU was demonstrated for the primary efficacy endpoint of overall response rate. ... There were no clinically meaningful differences in safety profile shown in this trial, and similar pharmacokinetic and immunogenicity results were seen across treatment groups,” Dr. Socinski summarized.
“These results confirm similarity demonstrated in earlier analytical, nonclinical, and clinical studies of PF-06439535 with bevacizumab-EU,” he concluded.
Trastuzumab biosimilar
The phase 3 HERITAGE trial was a first-line, randomized, controlled trial that compared biosimilar trastuzumab-dkst (Ogivri) with trastuzumab in combination with taxane chemotherapy and then as maintenance monotherapy in 458 patients with HER2+ advanced breast cancer.
The 24-week results, previously reported (JAMA. 2017 Jan 3;317[1]:37-47), showed a similar overall response rate with each agent when combined with chemotherapy. Rates of various adverse events were essentially the same.
Presence of overall response at 24 weeks correlated with duration of progression-free survival at 48 weeks (biserial r = .752). “Additional patients achieved a response during the monotherapy portion of the treatment, which is intriguing and clearly emphasizes the importance of monotherapy, as well as the importance of having alternate agents at lower cost available,” Dr. Rugo commented.
Common adverse events through week 48 were much the same as those seen at week 24, with few additional ones occurring during monotherapy. “No new safety issues were observed, and in fact, toxicity during monotherapy was quite minor,” she noted. “One thing that’s interesting here is that there was more arthralgia during the first 24 weeks with trastuzumab-dkst than with trastuzumab, but in monotherapy, this fell down to a very low number and was identical between the two arms. Paclitaxel, which people stayed on for longer [with the biosimilar], may have been the cause of this.”
The 48-week rates of adverse events of special interest – respiratory events, cardiac disorders, and infusion-related adverse events – and of serious adverse events were similar for the two agents.
“We didn’t see any additional serious cardiac events during monotherapy,” Dr. Rugo noted. Mean and median left ventricular ejection fraction over 48 weeks were similar, as was the rate of LVEF, which dropped below 50% (4.0% with trastuzumab-dkst and 3.3% with trastuzumab). The incidences of antidrug antibody and neutralizing antibody were also comparably low in both groups.
“HERITAGE data, now at week 48, supports trastuzumab-dkst as a biosimilar to trastuzumab in all approved indications,” Dr. Rugo said. “Final overall survival will be assessed after 36 months or after 240 deaths, whichever occurs first. Based on current data, this is predicted to conclude by the end of 2018, with final overall survival data available next year.
“Trastuzumab-dkst provides an additional high-quality treatment option for patients with HER2+ breast cancers in any setting,” she added. “This study indeed shows that biosimilars offer the potential for worldwide cost savings and improved access to life-saving therapies. It’s sobering to think that the patients enrolled in this study would not otherwise have had access to continued trastuzumab therapy, and so many of them are still alive with longer follow-up.”
Filgrastim biosimilar
Investigators led by Nadia Harbeck, MD, PhD, head of the Breast Center and chair for Conservative Oncology in the department of ob&gyn at the University of Munich (Germany), compared efficacy of filgrastim-sndz (Zarxio), a biosimilar of filgrastim (recombinant granulocyte colony–stimulating factor, or G-CSF), in a trial population with that of a real-world population of women receiving chemotherapy for breast cancer.
Dr. Harbeck and her colleagues compared 217 women who had nonmetastatic breast cancer from the trial with 466 women who had any-stage breast cancer (42% metastatic) from the real-world cohort.
Results showed that the 6.2% rate of chemotherapy-induced febrile neutropenia in any cycle seen in the real-world population was much the same as the 5.1% rate seen previously in the trial population. Findings were similar for temperature exceeding 38.5˚ C in any cycle: 3.4% and 5.6%. The real-world population had a lower rate of severe neutropenia than did the trial population (19.5% vs. 74.3%) and higher rates of infection (15.5% vs. 7.9%) and hospitalization caused by febrile neutropenia (3.9% vs. 1.8%). Findings were essentially the same in cycle-level analyses.
The real-world cohort had many fewer any-severity safety events of special interest than did the trial cohort, such as musculoskeletal/connective tissue disorders (20 vs. 261 events, respectively) and skin/subcutaneous tissue disorders (5 vs. 258 events). “Seeing these data, you have to keep in mind first of all that the patients received totally different chemotherapy. TAC chemotherapy has a lot of chemotherapy-associated side effects,” Dr. Harbeck noted. “The other thing is that MONITOR was a real-world database, and one could assume that there is some underreporting of events that are not directly correlated to the events that are of particular interest.”
Additional results available only from the trial showed that no patients developed binding or neutralizing antibodies against G-CSF.
“From a clinician’s point of view, it is very reassuring that we did not see any other safety signals in the real-world data than we saw in the randomized controlled trial and the efficacy was very, very similar,” Dr. Harbeck commented. “Having seen the discrepancies in the data … I think it’s important to have randomized controlled trials to assess and monitor adverse events for registration purposes and real-world evidence to reflect the daily clinical routine,” she concluded.
Dr. Socinski disclosed that his institution receives research funding from Pfizer, among other disclosures; the REFLECTIONS trial was sponsored by Pfizer. Dr. Rugo disclosed that she receives travel, accommodations, and/or expenses from Mylan, among other disclosures; the HERITAGE trial was sponsored by Mylan. Dr. Harbeck disclosed that she has a consulting or advisory role with Sandoz, among other disclosures; the PIONEER and MONITOR-GCSF trials were both sponsored by Sandoz.
SOURCE: Socinski MA et al. ASCO 2018, Abstract 109. Manikhas A et al. ASCO 2018, Abstract 110. Harbeck N et al. ASCO 2018, Abstract 111.
A variety of issues are influencing whether and how clinicians incorporate biosimilars into cancer care, according to Michael A. Thompson, MD, PhD, medical director of the Early Phase Cancer Research Program and the Oncology Precision Medicine Program at Aurora Health Care in Milwaukee, Wis., who spoke at the annual meeting of the American Society of Clinical Oncology.
“The issue of competition is highly relevant to biosimilars,” he said. Among important questions here: Is the oncology drug market a free market? Who owns the biosimilar companies? Does competition lower drug prices? And if biosimilars don’t decrease drug cost, why bother pursuing them? “We are seeing examples where the biosimilars have been developed, they appear to work, they appear safe, and really the proof will be how much is this pushing the market to decrease cost,” he noted.
Real-world data provide some insight into how biosimilars are being incorporated into oncology care. For example, in patients with non-Hodgkin lymphoma, hematologists tend to use rituximab (Rituxan) biosimilars in later lines of therapy, in patients with a better performance status and fewer comorbidities, and in cases of indolent or incurable disease (J Clin Oncol. 2018;36[suppl; abstr 112]). “So it appears that prescribers are acting tentatively to cautiously test the waters,” Dr. Thompson said.
Use will be influenced by clinical decision support and pathways, whether those are developed by institutions or insurers. These tools generally look at efficacy first, safety second, and cost third.
The relevance of patient choice (especially when physicians decreasingly have a choice) and perception of biosimilars may, or may not, be important, according to Dr. Thompson. In some areas of medicine, there is evidence of a nocebo effect: Patients perceive worsening of symptoms when they believe they are getting a nonbranded medication. But “I am not sure if this is valid in oncology, where we are already using many older chemotherapy drugs, the generics,” he said.
The American Society of Clinical Oncology recently published a statement on the use of biosimilars and related issues, such as safety and efficacy; naming and labeling; interchangeability, switching, and substitution; and the value proposition of these agents (J Clin Oncol. 2018 Apr 20;36[12]:1260-5). “The ASCO statement and guidelines are a great resource for really digging deeply into this area,” Dr. Thompson commented.
One concern surrounding uptake of biosimilars is the possibility of an actual increase in patient cost related to single sources and potentially differing reimbursement rates, which could diminish the financial benefit of these drugs. Technically, if biosimilars have similar efficacy and safety, and lower cost, they provide greater value than the reference drugs.
But there may still be reasons for not using a higher-value drug, according to Dr. Thompson. Clinicians may have lingering questions about efficacy and safety despite trial data, a situation that is being addressed in Europe by postmarketing pharmacovigilance. Other issues include delays in pathway implementation, the contracting of pharmacies with companies, and creation of new chemotherapy builds in electronic medical records. “These are all minor but potential barriers to as fast an implementation as possible,” he said.
A variety of issues are influencing whether and how clinicians incorporate biosimilars into cancer care, according to Michael A. Thompson, MD, PhD, medical director of the Early Phase Cancer Research Program and the Oncology Precision Medicine Program at Aurora Health Care in Milwaukee, Wis., who spoke at the annual meeting of the American Society of Clinical Oncology.
“The issue of competition is highly relevant to biosimilars,” he said. Among important questions here: Is the oncology drug market a free market? Who owns the biosimilar companies? Does competition lower drug prices? And if biosimilars don’t decrease drug cost, why bother pursuing them? “We are seeing examples where the biosimilars have been developed, they appear to work, they appear safe, and really the proof will be how much is this pushing the market to decrease cost,” he noted.
Real-world data provide some insight into how biosimilars are being incorporated into oncology care. For example, in patients with non-Hodgkin lymphoma, hematologists tend to use rituximab (Rituxan) biosimilars in later lines of therapy, in patients with a better performance status and fewer comorbidities, and in cases of indolent or incurable disease (J Clin Oncol. 2018;36[suppl; abstr 112]). “So it appears that prescribers are acting tentatively to cautiously test the waters,” Dr. Thompson said.
Use will be influenced by clinical decision support and pathways, whether those are developed by institutions or insurers. These tools generally look at efficacy first, safety second, and cost third.
The relevance of patient choice (especially when physicians decreasingly have a choice) and perception of biosimilars may, or may not, be important, according to Dr. Thompson. In some areas of medicine, there is evidence of a nocebo effect: Patients perceive worsening of symptoms when they believe they are getting a nonbranded medication. But “I am not sure if this is valid in oncology, where we are already using many older chemotherapy drugs, the generics,” he said.
The American Society of Clinical Oncology recently published a statement on the use of biosimilars and related issues, such as safety and efficacy; naming and labeling; interchangeability, switching, and substitution; and the value proposition of these agents (J Clin Oncol. 2018 Apr 20;36[12]:1260-5). “The ASCO statement and guidelines are a great resource for really digging deeply into this area,” Dr. Thompson commented.
One concern surrounding uptake of biosimilars is the possibility of an actual increase in patient cost related to single sources and potentially differing reimbursement rates, which could diminish the financial benefit of these drugs. Technically, if biosimilars have similar efficacy and safety, and lower cost, they provide greater value than the reference drugs.
But there may still be reasons for not using a higher-value drug, according to Dr. Thompson. Clinicians may have lingering questions about efficacy and safety despite trial data, a situation that is being addressed in Europe by postmarketing pharmacovigilance. Other issues include delays in pathway implementation, the contracting of pharmacies with companies, and creation of new chemotherapy builds in electronic medical records. “These are all minor but potential barriers to as fast an implementation as possible,” he said.
A variety of issues are influencing whether and how clinicians incorporate biosimilars into cancer care, according to Michael A. Thompson, MD, PhD, medical director of the Early Phase Cancer Research Program and the Oncology Precision Medicine Program at Aurora Health Care in Milwaukee, Wis., who spoke at the annual meeting of the American Society of Clinical Oncology.
“The issue of competition is highly relevant to biosimilars,” he said. Among important questions here: Is the oncology drug market a free market? Who owns the biosimilar companies? Does competition lower drug prices? And if biosimilars don’t decrease drug cost, why bother pursuing them? “We are seeing examples where the biosimilars have been developed, they appear to work, they appear safe, and really the proof will be how much is this pushing the market to decrease cost,” he noted.
Real-world data provide some insight into how biosimilars are being incorporated into oncology care. For example, in patients with non-Hodgkin lymphoma, hematologists tend to use rituximab (Rituxan) biosimilars in later lines of therapy, in patients with a better performance status and fewer comorbidities, and in cases of indolent or incurable disease (J Clin Oncol. 2018;36[suppl; abstr 112]). “So it appears that prescribers are acting tentatively to cautiously test the waters,” Dr. Thompson said.
Use will be influenced by clinical decision support and pathways, whether those are developed by institutions or insurers. These tools generally look at efficacy first, safety second, and cost third.
The relevance of patient choice (especially when physicians decreasingly have a choice) and perception of biosimilars may, or may not, be important, according to Dr. Thompson. In some areas of medicine, there is evidence of a nocebo effect: Patients perceive worsening of symptoms when they believe they are getting a nonbranded medication. But “I am not sure if this is valid in oncology, where we are already using many older chemotherapy drugs, the generics,” he said.
The American Society of Clinical Oncology recently published a statement on the use of biosimilars and related issues, such as safety and efficacy; naming and labeling; interchangeability, switching, and substitution; and the value proposition of these agents (J Clin Oncol. 2018 Apr 20;36[12]:1260-5). “The ASCO statement and guidelines are a great resource for really digging deeply into this area,” Dr. Thompson commented.
One concern surrounding uptake of biosimilars is the possibility of an actual increase in patient cost related to single sources and potentially differing reimbursement rates, which could diminish the financial benefit of these drugs. Technically, if biosimilars have similar efficacy and safety, and lower cost, they provide greater value than the reference drugs.
But there may still be reasons for not using a higher-value drug, according to Dr. Thompson. Clinicians may have lingering questions about efficacy and safety despite trial data, a situation that is being addressed in Europe by postmarketing pharmacovigilance. Other issues include delays in pathway implementation, the contracting of pharmacies with companies, and creation of new chemotherapy builds in electronic medical records. “These are all minor but potential barriers to as fast an implementation as possible,” he said.
CHICAGO – investigators reported at the annual meeting of the American Society of Clinical Oncology. The findings further advance the promise of new agents that have no clinically meaningful differences in efficacy and safety when compared with their reference drugs but have substantially lower cost.
Bevacizumab biosimilar
The REFLECTIONS trial (NCT02364999) was a multinational, first-line, randomized, controlled trial among 719 patients with advanced nonsquamous NSCLC. Patients were randomized to paclitaxel and carboplatin chemotherapy plus either bevacizumab (sourced from the European Union) or the candidate bevacizumab biosimilar PF-06439535 on a double-blind basis, followed by monotherapy with the same assigned agent.
The confidence interval for the risk difference fell within the equivalence margins set by European Union regulators (–13% and +13% for the 95% confidence interval). And the confidence interval for the risk ratio fell within the equivalence margins set by the Food and Drug Administration (0.73 and 1.37 for the 90% CI) and Japanese regulators (0.729 and 1.371 for the 95% CI).
Median progression-free survival was 9.0 months with the biosimilar and 7.7 months with bevacizumab (hazard ratio, 0.974; P = .814), and corresponding 1-year rates were 30.8% and 29.3%, Dr. Socinski reported. Median overall survival was 18.4 months and 17.8 months (HR, 1.001; P = .991), and corresponding 1-year rates were 66.4% and 68.8%.
Rates of grade 3 or higher hypertension, cardiac disorders, and bleeding did not differ significantly with the two agents. Patients also had similar rates of grade 3 or higher serious adverse events and of fatal (grade 5) serious adverse events (5.3% with the biosimilar and 5.9% with bevacizumab).
“Similarity between PF-06439535 and bevacizumab-EU was demonstrated for the primary efficacy endpoint of overall response rate. ... There were no clinically meaningful differences in safety profile shown in this trial, and similar pharmacokinetic and immunogenicity results were seen across treatment groups,” Dr. Socinski summarized.
“These results confirm similarity demonstrated in earlier analytical, nonclinical, and clinical studies of PF-06439535 with bevacizumab-EU,” he concluded.
Trastuzumab biosimilar
The phase 3 HERITAGE trial was a first-line, randomized, controlled trial that compared biosimilar trastuzumab-dkst (Ogivri) with trastuzumab in combination with taxane chemotherapy and then as maintenance monotherapy in 458 patients with HER2+ advanced breast cancer.
The 24-week results, previously reported (JAMA. 2017 Jan 3;317[1]:37-47), showed a similar overall response rate with each agent when combined with chemotherapy. Rates of various adverse events were essentially the same.
Presence of overall response at 24 weeks correlated with duration of progression-free survival at 48 weeks (biserial r = .752). “Additional patients achieved a response during the monotherapy portion of the treatment, which is intriguing and clearly emphasizes the importance of monotherapy, as well as the importance of having alternate agents at lower cost available,” Dr. Rugo commented.
Common adverse events through week 48 were much the same as those seen at week 24, with few additional ones occurring during monotherapy. “No new safety issues were observed, and in fact, toxicity during monotherapy was quite minor,” she noted. “One thing that’s interesting here is that there was more arthralgia during the first 24 weeks with trastuzumab-dkst than with trastuzumab, but in monotherapy, this fell down to a very low number and was identical between the two arms. Paclitaxel, which people stayed on for longer [with the biosimilar], may have been the cause of this.”
The 48-week rates of adverse events of special interest – respiratory events, cardiac disorders, and infusion-related adverse events – and of serious adverse events were similar for the two agents.
“We didn’t see any additional serious cardiac events during monotherapy,” Dr. Rugo noted. Mean and median left ventricular ejection fraction over 48 weeks were similar, as was the rate of LVEF, which dropped below 50% (4.0% with trastuzumab-dkst and 3.3% with trastuzumab). The incidences of antidrug antibody and neutralizing antibody were also comparably low in both groups.
“HERITAGE data, now at week 48, supports trastuzumab-dkst as a biosimilar to trastuzumab in all approved indications,” Dr. Rugo said. “Final overall survival will be assessed after 36 months or after 240 deaths, whichever occurs first. Based on current data, this is predicted to conclude by the end of 2018, with final overall survival data available next year.
“Trastuzumab-dkst provides an additional high-quality treatment option for patients with HER2+ breast cancers in any setting,” she added. “This study indeed shows that biosimilars offer the potential for worldwide cost savings and improved access to life-saving therapies. It’s sobering to think that the patients enrolled in this study would not otherwise have had access to continued trastuzumab therapy, and so many of them are still alive with longer follow-up.”
Filgrastim biosimilar
Investigators led by Nadia Harbeck, MD, PhD, head of the Breast Center and chair for Conservative Oncology in the department of ob&gyn at the University of Munich (Germany), compared efficacy of filgrastim-sndz (Zarxio), a biosimilar of filgrastim (recombinant granulocyte colony–stimulating factor, or G-CSF), in a trial population with that of a real-world population of women receiving chemotherapy for breast cancer.
Dr. Harbeck and her colleagues compared 217 women who had nonmetastatic breast cancer from the trial with 466 women who had any-stage breast cancer (42% metastatic) from the real-world cohort.
Results showed that the 6.2% rate of chemotherapy-induced febrile neutropenia in any cycle seen in the real-world population was much the same as the 5.1% rate seen previously in the trial population. Findings were similar for temperature exceeding 38.5˚ C in any cycle: 3.4% and 5.6%. The real-world population had a lower rate of severe neutropenia than did the trial population (19.5% vs. 74.3%) and higher rates of infection (15.5% vs. 7.9%) and hospitalization caused by febrile neutropenia (3.9% vs. 1.8%). Findings were essentially the same in cycle-level analyses.
The real-world cohort had many fewer any-severity safety events of special interest than did the trial cohort, such as musculoskeletal/connective tissue disorders (20 vs. 261 events, respectively) and skin/subcutaneous tissue disorders (5 vs. 258 events). “Seeing these data, you have to keep in mind first of all that the patients received totally different chemotherapy. TAC chemotherapy has a lot of chemotherapy-associated side effects,” Dr. Harbeck noted. “The other thing is that MONITOR was a real-world database, and one could assume that there is some underreporting of events that are not directly correlated to the events that are of particular interest.”
Additional results available only from the trial showed that no patients developed binding or neutralizing antibodies against G-CSF.
“From a clinician’s point of view, it is very reassuring that we did not see any other safety signals in the real-world data than we saw in the randomized controlled trial and the efficacy was very, very similar,” Dr. Harbeck commented. “Having seen the discrepancies in the data … I think it’s important to have randomized controlled trials to assess and monitor adverse events for registration purposes and real-world evidence to reflect the daily clinical routine,” she concluded.
Dr. Socinski disclosed that his institution receives research funding from Pfizer, among other disclosures; the REFLECTIONS trial was sponsored by Pfizer. Dr. Rugo disclosed that she receives travel, accommodations, and/or expenses from Mylan, among other disclosures; the HERITAGE trial was sponsored by Mylan. Dr. Harbeck disclosed that she has a consulting or advisory role with Sandoz, among other disclosures; the PIONEER and MONITOR-GCSF trials were both sponsored by Sandoz.
SOURCE: Socinski MA et al. ASCO 2018, Abstract 109. Manikhas A et al. ASCO 2018, Abstract 110. Harbeck N et al. ASCO 2018, Abstract 111.
CHICAGO – investigators reported at the annual meeting of the American Society of Clinical Oncology. The findings further advance the promise of new agents that have no clinically meaningful differences in efficacy and safety when compared with their reference drugs but have substantially lower cost.
Bevacizumab biosimilar
The REFLECTIONS trial (NCT02364999) was a multinational, first-line, randomized, controlled trial among 719 patients with advanced nonsquamous NSCLC. Patients were randomized to paclitaxel and carboplatin chemotherapy plus either bevacizumab (sourced from the European Union) or the candidate bevacizumab biosimilar PF-06439535 on a double-blind basis, followed by monotherapy with the same assigned agent.
The confidence interval for the risk difference fell within the equivalence margins set by European Union regulators (–13% and +13% for the 95% confidence interval). And the confidence interval for the risk ratio fell within the equivalence margins set by the Food and Drug Administration (0.73 and 1.37 for the 90% CI) and Japanese regulators (0.729 and 1.371 for the 95% CI).
Median progression-free survival was 9.0 months with the biosimilar and 7.7 months with bevacizumab (hazard ratio, 0.974; P = .814), and corresponding 1-year rates were 30.8% and 29.3%, Dr. Socinski reported. Median overall survival was 18.4 months and 17.8 months (HR, 1.001; P = .991), and corresponding 1-year rates were 66.4% and 68.8%.
Rates of grade 3 or higher hypertension, cardiac disorders, and bleeding did not differ significantly with the two agents. Patients also had similar rates of grade 3 or higher serious adverse events and of fatal (grade 5) serious adverse events (5.3% with the biosimilar and 5.9% with bevacizumab).
“Similarity between PF-06439535 and bevacizumab-EU was demonstrated for the primary efficacy endpoint of overall response rate. ... There were no clinically meaningful differences in safety profile shown in this trial, and similar pharmacokinetic and immunogenicity results were seen across treatment groups,” Dr. Socinski summarized.
“These results confirm similarity demonstrated in earlier analytical, nonclinical, and clinical studies of PF-06439535 with bevacizumab-EU,” he concluded.
Trastuzumab biosimilar
The phase 3 HERITAGE trial was a first-line, randomized, controlled trial that compared biosimilar trastuzumab-dkst (Ogivri) with trastuzumab in combination with taxane chemotherapy and then as maintenance monotherapy in 458 patients with HER2+ advanced breast cancer.
The 24-week results, previously reported (JAMA. 2017 Jan 3;317[1]:37-47), showed a similar overall response rate with each agent when combined with chemotherapy. Rates of various adverse events were essentially the same.
Presence of overall response at 24 weeks correlated with duration of progression-free survival at 48 weeks (biserial r = .752). “Additional patients achieved a response during the monotherapy portion of the treatment, which is intriguing and clearly emphasizes the importance of monotherapy, as well as the importance of having alternate agents at lower cost available,” Dr. Rugo commented.
Common adverse events through week 48 were much the same as those seen at week 24, with few additional ones occurring during monotherapy. “No new safety issues were observed, and in fact, toxicity during monotherapy was quite minor,” she noted. “One thing that’s interesting here is that there was more arthralgia during the first 24 weeks with trastuzumab-dkst than with trastuzumab, but in monotherapy, this fell down to a very low number and was identical between the two arms. Paclitaxel, which people stayed on for longer [with the biosimilar], may have been the cause of this.”
The 48-week rates of adverse events of special interest – respiratory events, cardiac disorders, and infusion-related adverse events – and of serious adverse events were similar for the two agents.
“We didn’t see any additional serious cardiac events during monotherapy,” Dr. Rugo noted. Mean and median left ventricular ejection fraction over 48 weeks were similar, as was the rate of LVEF, which dropped below 50% (4.0% with trastuzumab-dkst and 3.3% with trastuzumab). The incidences of antidrug antibody and neutralizing antibody were also comparably low in both groups.
“HERITAGE data, now at week 48, supports trastuzumab-dkst as a biosimilar to trastuzumab in all approved indications,” Dr. Rugo said. “Final overall survival will be assessed after 36 months or after 240 deaths, whichever occurs first. Based on current data, this is predicted to conclude by the end of 2018, with final overall survival data available next year.
“Trastuzumab-dkst provides an additional high-quality treatment option for patients with HER2+ breast cancers in any setting,” she added. “This study indeed shows that biosimilars offer the potential for worldwide cost savings and improved access to life-saving therapies. It’s sobering to think that the patients enrolled in this study would not otherwise have had access to continued trastuzumab therapy, and so many of them are still alive with longer follow-up.”
Filgrastim biosimilar
Investigators led by Nadia Harbeck, MD, PhD, head of the Breast Center and chair for Conservative Oncology in the department of ob&gyn at the University of Munich (Germany), compared efficacy of filgrastim-sndz (Zarxio), a biosimilar of filgrastim (recombinant granulocyte colony–stimulating factor, or G-CSF), in a trial population with that of a real-world population of women receiving chemotherapy for breast cancer.
Dr. Harbeck and her colleagues compared 217 women who had nonmetastatic breast cancer from the trial with 466 women who had any-stage breast cancer (42% metastatic) from the real-world cohort.
Results showed that the 6.2% rate of chemotherapy-induced febrile neutropenia in any cycle seen in the real-world population was much the same as the 5.1% rate seen previously in the trial population. Findings were similar for temperature exceeding 38.5˚ C in any cycle: 3.4% and 5.6%. The real-world population had a lower rate of severe neutropenia than did the trial population (19.5% vs. 74.3%) and higher rates of infection (15.5% vs. 7.9%) and hospitalization caused by febrile neutropenia (3.9% vs. 1.8%). Findings were essentially the same in cycle-level analyses.
The real-world cohort had many fewer any-severity safety events of special interest than did the trial cohort, such as musculoskeletal/connective tissue disorders (20 vs. 261 events, respectively) and skin/subcutaneous tissue disorders (5 vs. 258 events). “Seeing these data, you have to keep in mind first of all that the patients received totally different chemotherapy. TAC chemotherapy has a lot of chemotherapy-associated side effects,” Dr. Harbeck noted. “The other thing is that MONITOR was a real-world database, and one could assume that there is some underreporting of events that are not directly correlated to the events that are of particular interest.”
Additional results available only from the trial showed that no patients developed binding or neutralizing antibodies against G-CSF.
“From a clinician’s point of view, it is very reassuring that we did not see any other safety signals in the real-world data than we saw in the randomized controlled trial and the efficacy was very, very similar,” Dr. Harbeck commented. “Having seen the discrepancies in the data … I think it’s important to have randomized controlled trials to assess and monitor adverse events for registration purposes and real-world evidence to reflect the daily clinical routine,” she concluded.
Dr. Socinski disclosed that his institution receives research funding from Pfizer, among other disclosures; the REFLECTIONS trial was sponsored by Pfizer. Dr. Rugo disclosed that she receives travel, accommodations, and/or expenses from Mylan, among other disclosures; the HERITAGE trial was sponsored by Mylan. Dr. Harbeck disclosed that she has a consulting or advisory role with Sandoz, among other disclosures; the PIONEER and MONITOR-GCSF trials were both sponsored by Sandoz.
SOURCE: Socinski MA et al. ASCO 2018, Abstract 109. Manikhas A et al. ASCO 2018, Abstract 110. Harbeck N et al. ASCO 2018, Abstract 111.
REPORTING FROM ASCO 2018
Key clinical point: Biosimilars for bevacizumab, trastuzumab, and filgrastim showed similar efficacy and safety.
Major finding: In patients with advanced nonsquamous NSCLC, the overall response rate was 45.3% with a candidate bevacizumab biosimilar and 44.6% with bevacizumab. In patients with HER2+ advanced breast cancer, 48-week median progression-free survival was 11.1 months for both trastuzumab-dkst and trastuzumab. The rate of chemotherapy-induced febrile neutropenia among breast cancer patients given a biosimilar for filgrastim was 5.1% in a trial population and 6.2% in a real-world population.
Study details: Randomized, controlled trials of first-line therapy among 719 patients with advanced nonsquamous NSCLC (REFLECTIONS trial) and among 458 patients with HER2+ advanced breast cancer (HERITAGE trial). Comparison of outcomes in a randomized, controlled trial among 217 patients with nonmetastatic breast cancer (PIONEER trial) and a real-world cohort study of 466 patients with any-stage breast cancer (MONITOR-GCSF).
Disclosures: Dr. Socinski disclosed that his institution receives research funding from Pfizer, among other disclosures; the REFLECTIONS trial was sponsored by Pfizer. Dr. Rugo disclosed that she receives travel, accommodations, and/or expenses from Mylan, among other disclosures; the HERITAGE trial was sponsored by Mylan. Dr. Harbeck disclosed that she has a consulting or advisory role with Sandoz, among other disclosures; the PIONEER and MONITOR-GCSF trials were sponsored by Sandoz.
Source: Socinski MA et al. ASCO 2018, Abstract 109. Manikhas A et al. ASCO 2018, Abstract 110. Harbeck N et al. ASCO 2018, Abstract 111.
Could High BMI Reduce Premenopausal Breast Cancer Risk?
Young women may not want to hear it, but fat could be their friend. Researchers from the Premenopausal Breast Cancer Collaborative Group have found that women aged 18 – 24 years with high body fat have a lower risk of developing breast cancer before menopause.
The researchers pooled data from 19 different studies, involving about 800,000 women from around the world. Overall, 1.7% of the women developed breast cancer. The researchers found that the relative risk of premenopausal breast cancer dropped 12% to 23% for each 5-unit increase in body mass index, depending on age. They saw the strongest effect at ages 18 – 24 years: Very obese women in this age group were 4.2 times less likely to develop premenopausal breast cancer than women with low body mass index (BMI) at the same age.
The researchers do not know why high BMI might protect against breast cancer in some women. Breast cancer is relatively rare before menopause, although previous studies have suggested that the risk factors might be different for younger vs older women, says Dale Sandler, PhD, co-author of the group and head of the Epidemiology Branch at the National Institute of Environmental Health Sciences. For instance, it is well known that women who gain weight, particularly after menopause, have a higher risk. The fact that this study found that the risk not only is not increased, but actually decreased, in younger women points to the possibility that different biologic mechanisms are at work, Sandler says.
Nonetheless, the researchers caution that young women should not intentionally gain weight to offset the risk.
Source:
National Institutes of Health. https://www.nih.gov/news-events/news-releases/nih-study-associates-obesity-lower-breast-cancer-risk-young-women. Published June 27, 2018. Accessed July 18, 2018.
Young women may not want to hear it, but fat could be their friend. Researchers from the Premenopausal Breast Cancer Collaborative Group have found that women aged 18 – 24 years with high body fat have a lower risk of developing breast cancer before menopause.
The researchers pooled data from 19 different studies, involving about 800,000 women from around the world. Overall, 1.7% of the women developed breast cancer. The researchers found that the relative risk of premenopausal breast cancer dropped 12% to 23% for each 5-unit increase in body mass index, depending on age. They saw the strongest effect at ages 18 – 24 years: Very obese women in this age group were 4.2 times less likely to develop premenopausal breast cancer than women with low body mass index (BMI) at the same age.
The researchers do not know why high BMI might protect against breast cancer in some women. Breast cancer is relatively rare before menopause, although previous studies have suggested that the risk factors might be different for younger vs older women, says Dale Sandler, PhD, co-author of the group and head of the Epidemiology Branch at the National Institute of Environmental Health Sciences. For instance, it is well known that women who gain weight, particularly after menopause, have a higher risk. The fact that this study found that the risk not only is not increased, but actually decreased, in younger women points to the possibility that different biologic mechanisms are at work, Sandler says.
Nonetheless, the researchers caution that young women should not intentionally gain weight to offset the risk.
Source:
National Institutes of Health. https://www.nih.gov/news-events/news-releases/nih-study-associates-obesity-lower-breast-cancer-risk-young-women. Published June 27, 2018. Accessed July 18, 2018.
Young women may not want to hear it, but fat could be their friend. Researchers from the Premenopausal Breast Cancer Collaborative Group have found that women aged 18 – 24 years with high body fat have a lower risk of developing breast cancer before menopause.
The researchers pooled data from 19 different studies, involving about 800,000 women from around the world. Overall, 1.7% of the women developed breast cancer. The researchers found that the relative risk of premenopausal breast cancer dropped 12% to 23% for each 5-unit increase in body mass index, depending on age. They saw the strongest effect at ages 18 – 24 years: Very obese women in this age group were 4.2 times less likely to develop premenopausal breast cancer than women with low body mass index (BMI) at the same age.
The researchers do not know why high BMI might protect against breast cancer in some women. Breast cancer is relatively rare before menopause, although previous studies have suggested that the risk factors might be different for younger vs older women, says Dale Sandler, PhD, co-author of the group and head of the Epidemiology Branch at the National Institute of Environmental Health Sciences. For instance, it is well known that women who gain weight, particularly after menopause, have a higher risk. The fact that this study found that the risk not only is not increased, but actually decreased, in younger women points to the possibility that different biologic mechanisms are at work, Sandler says.
Nonetheless, the researchers caution that young women should not intentionally gain weight to offset the risk.
Source:
National Institutes of Health. https://www.nih.gov/news-events/news-releases/nih-study-associates-obesity-lower-breast-cancer-risk-young-women. Published June 27, 2018. Accessed July 18, 2018.
FDA expands indication for ribociclib for advanced breast cancer
The Food and Drug Administration has approved ribociclib (Kisqali) in combination with an aromatase inhibitor (AI) for the treatment of pre/perimenopausal or postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)–negative advanced or metastatic breast cancer, as initial endocrine-based therapy.

Ribociclib was first approved in March 2017 for use with an AI to treat HR-positive, HER2-negative advanced breast cancer in postmenopausal women.
Approval for ribociclib in combination with an AI for pre/perimenopausal women was based on progression-free survival (PFS) in MONALEESA-7, a trial of premenopausal women with HR-positive, HER2-negative, advanced breast cancer. The women received either ribociclib and an AI, or placebo and an AI, and all also received ovarian suppression with goserelin (Zoladex). Of 495 women who received nonsteroidal AIs, median PFS was 27.5 months for women also receiving ribociclib, versus 13.8 months for women who received placebo plus the AI.
Approval for ribociclib in combination with fulvestrant in treating advanced or metastatic breast cancer was based on PFS results from MONALEESA-3, which enrolled 726 women with HR-positive, HER2-negative, advanced breast cancer who received no or up to one line of prior endocrine therapy. Median PFS was 20.5 months for women randomized to receive ribociclib and fulvestrant, compared with 12.8 months for women randomized to receive placebo plus fulvestrant.
The common side effects of ribociclib are infections, neutropenia, leukopenia, headache, cough, nausea, fatigue, diarrhea, vomiting, constipation, hair loss, and rash. Warnings include the risk of QT prolongation, serious liver problems, low white blood cell counts, and fetal harm, the FDA said.
This is the first FDA approval as part of two new pilot programs announced earlier this year: Real-Time Oncology Review allows for the FDA to review much of the data earlier, before the information is formally submitted to the FDA, and the Assessment Aid is a structured template that offers a more streamlined approach.
“With today’s approval, the FDA used these new approaches to allow the review team to start analyzing data before the actual submission of the application and help guide the sponsor’s analysis of the top-line data to tease out the most relevant information,” FDA Commissioner Scott Gottlieb, MD, said in the press statement. “This enabled our approval less than 1 month after the June 28 submission date and several months ahead of the goal date.”
The two pilot programs are currently being used for supplemental applications for already approved cancer drugs and could later be expanded to original drugs and biologics, the FDA said.
Ribociclib is marketed as Kisqali by Novartis Pharmaceuticals Corporation.
The Food and Drug Administration has approved ribociclib (Kisqali) in combination with an aromatase inhibitor (AI) for the treatment of pre/perimenopausal or postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)–negative advanced or metastatic breast cancer, as initial endocrine-based therapy.
Ribociclib was first approved in March 2017 for use with an AI to treat HR-positive, HER2-negative advanced breast cancer in postmenopausal women.
Approval for ribociclib in combination with an AI for pre/perimenopausal women was based on progression-free survival (PFS) in MONALEESA-7, a trial of premenopausal women with HR-positive, HER2-negative, advanced breast cancer. The women received either ribociclib and an AI, or placebo and an AI, and all also received ovarian suppression with goserelin (Zoladex). Of 495 women who received nonsteroidal AIs, median PFS was 27.5 months for women also receiving ribociclib, versus 13.8 months for women who received placebo plus the AI.
Approval for ribociclib in combination with fulvestrant in treating advanced or metastatic breast cancer was based on PFS results from MONALEESA-3, which enrolled 726 women with HR-positive, HER2-negative, advanced breast cancer who received no or up to one line of prior endocrine therapy. Median PFS was 20.5 months for women randomized to receive ribociclib and fulvestrant, compared with 12.8 months for women randomized to receive placebo plus fulvestrant.
The common side effects of ribociclib are infections, neutropenia, leukopenia, headache, cough, nausea, fatigue, diarrhea, vomiting, constipation, hair loss, and rash. Warnings include the risk of QT prolongation, serious liver problems, low white blood cell counts, and fetal harm, the FDA said.
This is the first FDA approval as part of two new pilot programs announced earlier this year: Real-Time Oncology Review allows for the FDA to review much of the data earlier, before the information is formally submitted to the FDA, and the Assessment Aid is a structured template that offers a more streamlined approach.
“With today’s approval, the FDA used these new approaches to allow the review team to start analyzing data before the actual submission of the application and help guide the sponsor’s analysis of the top-line data to tease out the most relevant information,” FDA Commissioner Scott Gottlieb, MD, said in the press statement. “This enabled our approval less than 1 month after the June 28 submission date and several months ahead of the goal date.”
The two pilot programs are currently being used for supplemental applications for already approved cancer drugs and could later be expanded to original drugs and biologics, the FDA said.
Ribociclib is marketed as Kisqali by Novartis Pharmaceuticals Corporation.
The Food and Drug Administration has approved ribociclib (Kisqali) in combination with an aromatase inhibitor (AI) for the treatment of pre/perimenopausal or postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)–negative advanced or metastatic breast cancer, as initial endocrine-based therapy.
Ribociclib was first approved in March 2017 for use with an AI to treat HR-positive, HER2-negative advanced breast cancer in postmenopausal women.
Approval for ribociclib in combination with an AI for pre/perimenopausal women was based on progression-free survival (PFS) in MONALEESA-7, a trial of premenopausal women with HR-positive, HER2-negative, advanced breast cancer. The women received either ribociclib and an AI, or placebo and an AI, and all also received ovarian suppression with goserelin (Zoladex). Of 495 women who received nonsteroidal AIs, median PFS was 27.5 months for women also receiving ribociclib, versus 13.8 months for women who received placebo plus the AI.
Approval for ribociclib in combination with fulvestrant in treating advanced or metastatic breast cancer was based on PFS results from MONALEESA-3, which enrolled 726 women with HR-positive, HER2-negative, advanced breast cancer who received no or up to one line of prior endocrine therapy. Median PFS was 20.5 months for women randomized to receive ribociclib and fulvestrant, compared with 12.8 months for women randomized to receive placebo plus fulvestrant.
The common side effects of ribociclib are infections, neutropenia, leukopenia, headache, cough, nausea, fatigue, diarrhea, vomiting, constipation, hair loss, and rash. Warnings include the risk of QT prolongation, serious liver problems, low white blood cell counts, and fetal harm, the FDA said.
This is the first FDA approval as part of two new pilot programs announced earlier this year: Real-Time Oncology Review allows for the FDA to review much of the data earlier, before the information is formally submitted to the FDA, and the Assessment Aid is a structured template that offers a more streamlined approach.
“With today’s approval, the FDA used these new approaches to allow the review team to start analyzing data before the actual submission of the application and help guide the sponsor’s analysis of the top-line data to tease out the most relevant information,” FDA Commissioner Scott Gottlieb, MD, said in the press statement. “This enabled our approval less than 1 month after the June 28 submission date and several months ahead of the goal date.”
The two pilot programs are currently being used for supplemental applications for already approved cancer drugs and could later be expanded to original drugs and biologics, the FDA said.
Ribociclib is marketed as Kisqali by Novartis Pharmaceuticals Corporation.
ABP 980 similar to trastuzumab in HER2+ breast cancer in all but name
In women with HER2-positive early breast cancer, the anti-HER2 biosimilar agent ABP-980 was clinically similar in efficacy and safety to the original drug trastuzumab (Herceptin).
Although ABP 980 was associated with a higher pathologic complete response (pCR) rate in breast tissues and axillary lymph nodes compared with trastuzumab, the trial technically failed to meet its coprimary endpoints of risk ratio and risk difference because of a statistical nicety involving local lab review of tissue samples vs. centralized review, reported Gunter von Minckwitz, MD, PhD, of the German Breast Group in Neu-Isenburg, Germany, and his colleagues.
“In our sensitivity analyses based on central laboratory evaluation of tumor samples, estimates for the two drugs were contained within the predefined equivalence margins, indicating similar efficacy. ABP 980 and trastuzumab had similar safety outcomes in both the neoadjuvant and adjuvant phases of the study,” the researchers wrote. The report was published in The Lancet Oncology.
ABP 980 is one of several contenders for trastuzumab biosimilar making their way through clinical trials. In phase 1 studies, it was shown to be similar in its structure, pharmacodynamics, and pharmacokinetics to the reference agent trastuzumab. In the LILAC trial Dr. von Minckwitz and his associates put the biosimilar through its paces to see whether it would also be equivalent in efficacy and safety, including in patients switched from the original drug to the copy-cat agent.
Investigators for the randomized phase 3 trial, conducted in 97 centers in 20 countries in Europe, South America, and Canada, enrolled 827 women age and 18 and older with HER2-positive breast cancer, 725 of whom were randomly assigned to neoadjuvant therapy with either ABP 980 or trastuzumab plus paclitaxel after a four-cycle run-in of anthracycline-based chemotherapy,
Neoadjuvant therapy was followed 3-7 weeks later by surgery and adjuvant therapy with either of the HER2 inhibitors. At baseline, patients were randomly assigned to either continue adjuvant therapy with their original HER2 inhibitor, or to switch from trastuzumab in the neoadjuvant setting to ABP 980 in the adjuvant setting.
In all, 696 patients were evaluable for the primary endpoint, 358 of whom received the biosimilar, and 338 of whom received trastuzumab. In all, 48% of patients randomly assigned to ABP 980 had a pCR in breast and axillary lymph node tissues assessed at a local laboratory, compared with 41% assigned to trastuzumab.
The risk difference was 7.3%, (90% confidence interval [CI] 1.2-13.4), The risk ratio was 1.188 (90% CI, 1.033-1.366). Although the lower bounds of the confidence intervals showed that ABP 980 was noninferior to trastuzumab, the upper bounds exceeded the predefined equivalence margins of a 13% risk difference and 1.318 risk ratio, respectively, meaning that technically the trial did not meet its coprimary endpoints.
However, in central laboratory review pCR was seen in 48% of patients assigned to ABP 980 at baseline and 42% of those assigned to trastuzumab at baseline. The risk difference was 5.8% (90% CI, –0.5-12.0), and risk ratio was 1.142 (90% CI, 0.993-1.312), and both the lower and upper bounds of the confidence intervals fell within prespecified limits.
The safety analysis showed a similar incidence of grade 3 or greater adverse events during neoadjuvant therapy (15% of patients on ABP 980 vs. 14% on trastuzumab). Grade 3 or greater neutropenia occurred in 6% of patients in each group.
During adjuvant therapy, grade 3 or greater adverse events occurred in 9% of patients continuing ABP 980, 6% continuing trastuzumab, and 8% of these switched from trastuzumab to ABP 980. The most frequent grade 3 or greater events of interest were infections and neutropenia, all occurring in 1% of patients in each arm, and infusion reaction, which occurred in 1% of patients who stayed on the assigned HER2 inhibitor and in 2% of patients who were switched to ABP 980.
There were two patient deaths from adverse events, each deemed to be unrelated to treatment. One patient died from pneumonia during neoadjuvant ABP 980 therapy, and one died from septic shock during adjuvant therapy with ABP 980 after being switch from trastuzumab.
“To our knowledge, this is the first study of a trastuzumab biosimilar encompassing a single-switch design from the reference product to a biosimilar, which allowed us to assess the clinical safety and immunogenicity of this approach to treatment. Safety and immunogenicity were similar in patients who were switched and in those who continued to receive trastuzumab as adjuvant therapy,” the investigators wrote.
SOURCE: von Minckwitz G et al. Lancet Oncol 2018 Jun 4. doi: 10.1016/S1470-2045(18)30241-9.
The LILAC trial has some strengths and weaknesses and raises a curious regulatory issue. To begin with the weaknesses, only 696 of 725 randomized patients were evaluable for pathological complete response after surgery. No data about the outcomes, characteristics, or allocated treatment of the patients who did not reach surgery were provided. These lost patients should have been included in the intention-to-treat analysis and their responses classified when possible (e.g., those who did not reach surgery due to progressive disease should have been classified as nonpathological complete response). The effect of these few patients on the overall results is unknown, although it is possibly small.
Among the strengths of LILAC were that the trial was done in a sensitive population (i.e., a population in which differences in safety, immunogenicity, and efficacy could be attributed to the biosimilar or reference drug rather than patient-related or disease-related factors). Two chemotherapy choices were included that are broadly used worldwide, and thus mimicked routine clinical practice, and the study had a sensitive primary endpoint (pathological complete response). The aim of clinical trials in the regulatory pathway of biosimilars is to show an acceptable degree of similarity in clinical efficacy and safety to the reference product. For original products, endpoints in clinical trials must show benefits to patients, such as progression-free survival, disease-free survival, or overall survival, whereas for biosimilars, surrogate endpoints, such as the proportion of patients with pathological response in breast cancer neoadjuvant trials, are appropriate. The study design of LILAC, therefore, meets the main clinical requirements demanded by medicine agencies for the registration of biosimilars.
Miguel Martin, MD, PhD is with Instituto de Investigación Sanitaria Gregorio Marañón, Universidad Complutense, Madrid. Dr. Martin’s remarks are adapted and condensed from an editorial in The Lancet Oncology accompanying the study by von Minckwitz G et al. He disclosed grants from Novartis and Roche and personal fees from AstraZeneca, Lilly, Pfizer, and Roche.
The LILAC trial has some strengths and weaknesses and raises a curious regulatory issue. To begin with the weaknesses, only 696 of 725 randomized patients were evaluable for pathological complete response after surgery. No data about the outcomes, characteristics, or allocated treatment of the patients who did not reach surgery were provided. These lost patients should have been included in the intention-to-treat analysis and their responses classified when possible (e.g., those who did not reach surgery due to progressive disease should have been classified as nonpathological complete response). The effect of these few patients on the overall results is unknown, although it is possibly small.
Among the strengths of LILAC were that the trial was done in a sensitive population (i.e., a population in which differences in safety, immunogenicity, and efficacy could be attributed to the biosimilar or reference drug rather than patient-related or disease-related factors). Two chemotherapy choices were included that are broadly used worldwide, and thus mimicked routine clinical practice, and the study had a sensitive primary endpoint (pathological complete response). The aim of clinical trials in the regulatory pathway of biosimilars is to show an acceptable degree of similarity in clinical efficacy and safety to the reference product. For original products, endpoints in clinical trials must show benefits to patients, such as progression-free survival, disease-free survival, or overall survival, whereas for biosimilars, surrogate endpoints, such as the proportion of patients with pathological response in breast cancer neoadjuvant trials, are appropriate. The study design of LILAC, therefore, meets the main clinical requirements demanded by medicine agencies for the registration of biosimilars.
Miguel Martin, MD, PhD is with Instituto de Investigación Sanitaria Gregorio Marañón, Universidad Complutense, Madrid. Dr. Martin’s remarks are adapted and condensed from an editorial in The Lancet Oncology accompanying the study by von Minckwitz G et al. He disclosed grants from Novartis and Roche and personal fees from AstraZeneca, Lilly, Pfizer, and Roche.
The LILAC trial has some strengths and weaknesses and raises a curious regulatory issue. To begin with the weaknesses, only 696 of 725 randomized patients were evaluable for pathological complete response after surgery. No data about the outcomes, characteristics, or allocated treatment of the patients who did not reach surgery were provided. These lost patients should have been included in the intention-to-treat analysis and their responses classified when possible (e.g., those who did not reach surgery due to progressive disease should have been classified as nonpathological complete response). The effect of these few patients on the overall results is unknown, although it is possibly small.
Among the strengths of LILAC were that the trial was done in a sensitive population (i.e., a population in which differences in safety, immunogenicity, and efficacy could be attributed to the biosimilar or reference drug rather than patient-related or disease-related factors). Two chemotherapy choices were included that are broadly used worldwide, and thus mimicked routine clinical practice, and the study had a sensitive primary endpoint (pathological complete response). The aim of clinical trials in the regulatory pathway of biosimilars is to show an acceptable degree of similarity in clinical efficacy and safety to the reference product. For original products, endpoints in clinical trials must show benefits to patients, such as progression-free survival, disease-free survival, or overall survival, whereas for biosimilars, surrogate endpoints, such as the proportion of patients with pathological response in breast cancer neoadjuvant trials, are appropriate. The study design of LILAC, therefore, meets the main clinical requirements demanded by medicine agencies for the registration of biosimilars.
Miguel Martin, MD, PhD is with Instituto de Investigación Sanitaria Gregorio Marañón, Universidad Complutense, Madrid. Dr. Martin’s remarks are adapted and condensed from an editorial in The Lancet Oncology accompanying the study by von Minckwitz G et al. He disclosed grants from Novartis and Roche and personal fees from AstraZeneca, Lilly, Pfizer, and Roche.
In women with HER2-positive early breast cancer, the anti-HER2 biosimilar agent ABP-980 was clinically similar in efficacy and safety to the original drug trastuzumab (Herceptin).
Although ABP 980 was associated with a higher pathologic complete response (pCR) rate in breast tissues and axillary lymph nodes compared with trastuzumab, the trial technically failed to meet its coprimary endpoints of risk ratio and risk difference because of a statistical nicety involving local lab review of tissue samples vs. centralized review, reported Gunter von Minckwitz, MD, PhD, of the German Breast Group in Neu-Isenburg, Germany, and his colleagues.
“In our sensitivity analyses based on central laboratory evaluation of tumor samples, estimates for the two drugs were contained within the predefined equivalence margins, indicating similar efficacy. ABP 980 and trastuzumab had similar safety outcomes in both the neoadjuvant and adjuvant phases of the study,” the researchers wrote. The report was published in The Lancet Oncology.
ABP 980 is one of several contenders for trastuzumab biosimilar making their way through clinical trials. In phase 1 studies, it was shown to be similar in its structure, pharmacodynamics, and pharmacokinetics to the reference agent trastuzumab. In the LILAC trial Dr. von Minckwitz and his associates put the biosimilar through its paces to see whether it would also be equivalent in efficacy and safety, including in patients switched from the original drug to the copy-cat agent.
Investigators for the randomized phase 3 trial, conducted in 97 centers in 20 countries in Europe, South America, and Canada, enrolled 827 women age and 18 and older with HER2-positive breast cancer, 725 of whom were randomly assigned to neoadjuvant therapy with either ABP 980 or trastuzumab plus paclitaxel after a four-cycle run-in of anthracycline-based chemotherapy,
Neoadjuvant therapy was followed 3-7 weeks later by surgery and adjuvant therapy with either of the HER2 inhibitors. At baseline, patients were randomly assigned to either continue adjuvant therapy with their original HER2 inhibitor, or to switch from trastuzumab in the neoadjuvant setting to ABP 980 in the adjuvant setting.
In all, 696 patients were evaluable for the primary endpoint, 358 of whom received the biosimilar, and 338 of whom received trastuzumab. In all, 48% of patients randomly assigned to ABP 980 had a pCR in breast and axillary lymph node tissues assessed at a local laboratory, compared with 41% assigned to trastuzumab.
The risk difference was 7.3%, (90% confidence interval [CI] 1.2-13.4), The risk ratio was 1.188 (90% CI, 1.033-1.366). Although the lower bounds of the confidence intervals showed that ABP 980 was noninferior to trastuzumab, the upper bounds exceeded the predefined equivalence margins of a 13% risk difference and 1.318 risk ratio, respectively, meaning that technically the trial did not meet its coprimary endpoints.
However, in central laboratory review pCR was seen in 48% of patients assigned to ABP 980 at baseline and 42% of those assigned to trastuzumab at baseline. The risk difference was 5.8% (90% CI, –0.5-12.0), and risk ratio was 1.142 (90% CI, 0.993-1.312), and both the lower and upper bounds of the confidence intervals fell within prespecified limits.
The safety analysis showed a similar incidence of grade 3 or greater adverse events during neoadjuvant therapy (15% of patients on ABP 980 vs. 14% on trastuzumab). Grade 3 or greater neutropenia occurred in 6% of patients in each group.
During adjuvant therapy, grade 3 or greater adverse events occurred in 9% of patients continuing ABP 980, 6% continuing trastuzumab, and 8% of these switched from trastuzumab to ABP 980. The most frequent grade 3 or greater events of interest were infections and neutropenia, all occurring in 1% of patients in each arm, and infusion reaction, which occurred in 1% of patients who stayed on the assigned HER2 inhibitor and in 2% of patients who were switched to ABP 980.
There were two patient deaths from adverse events, each deemed to be unrelated to treatment. One patient died from pneumonia during neoadjuvant ABP 980 therapy, and one died from septic shock during adjuvant therapy with ABP 980 after being switch from trastuzumab.
“To our knowledge, this is the first study of a trastuzumab biosimilar encompassing a single-switch design from the reference product to a biosimilar, which allowed us to assess the clinical safety and immunogenicity of this approach to treatment. Safety and immunogenicity were similar in patients who were switched and in those who continued to receive trastuzumab as adjuvant therapy,” the investigators wrote.
SOURCE: von Minckwitz G et al. Lancet Oncol 2018 Jun 4. doi: 10.1016/S1470-2045(18)30241-9.
In women with HER2-positive early breast cancer, the anti-HER2 biosimilar agent ABP-980 was clinically similar in efficacy and safety to the original drug trastuzumab (Herceptin).
Although ABP 980 was associated with a higher pathologic complete response (pCR) rate in breast tissues and axillary lymph nodes compared with trastuzumab, the trial technically failed to meet its coprimary endpoints of risk ratio and risk difference because of a statistical nicety involving local lab review of tissue samples vs. centralized review, reported Gunter von Minckwitz, MD, PhD, of the German Breast Group in Neu-Isenburg, Germany, and his colleagues.
“In our sensitivity analyses based on central laboratory evaluation of tumor samples, estimates for the two drugs were contained within the predefined equivalence margins, indicating similar efficacy. ABP 980 and trastuzumab had similar safety outcomes in both the neoadjuvant and adjuvant phases of the study,” the researchers wrote. The report was published in The Lancet Oncology.
ABP 980 is one of several contenders for trastuzumab biosimilar making their way through clinical trials. In phase 1 studies, it was shown to be similar in its structure, pharmacodynamics, and pharmacokinetics to the reference agent trastuzumab. In the LILAC trial Dr. von Minckwitz and his associates put the biosimilar through its paces to see whether it would also be equivalent in efficacy and safety, including in patients switched from the original drug to the copy-cat agent.
Investigators for the randomized phase 3 trial, conducted in 97 centers in 20 countries in Europe, South America, and Canada, enrolled 827 women age and 18 and older with HER2-positive breast cancer, 725 of whom were randomly assigned to neoadjuvant therapy with either ABP 980 or trastuzumab plus paclitaxel after a four-cycle run-in of anthracycline-based chemotherapy,
Neoadjuvant therapy was followed 3-7 weeks later by surgery and adjuvant therapy with either of the HER2 inhibitors. At baseline, patients were randomly assigned to either continue adjuvant therapy with their original HER2 inhibitor, or to switch from trastuzumab in the neoadjuvant setting to ABP 980 in the adjuvant setting.
In all, 696 patients were evaluable for the primary endpoint, 358 of whom received the biosimilar, and 338 of whom received trastuzumab. In all, 48% of patients randomly assigned to ABP 980 had a pCR in breast and axillary lymph node tissues assessed at a local laboratory, compared with 41% assigned to trastuzumab.
The risk difference was 7.3%, (90% confidence interval [CI] 1.2-13.4), The risk ratio was 1.188 (90% CI, 1.033-1.366). Although the lower bounds of the confidence intervals showed that ABP 980 was noninferior to trastuzumab, the upper bounds exceeded the predefined equivalence margins of a 13% risk difference and 1.318 risk ratio, respectively, meaning that technically the trial did not meet its coprimary endpoints.
However, in central laboratory review pCR was seen in 48% of patients assigned to ABP 980 at baseline and 42% of those assigned to trastuzumab at baseline. The risk difference was 5.8% (90% CI, –0.5-12.0), and risk ratio was 1.142 (90% CI, 0.993-1.312), and both the lower and upper bounds of the confidence intervals fell within prespecified limits.
The safety analysis showed a similar incidence of grade 3 or greater adverse events during neoadjuvant therapy (15% of patients on ABP 980 vs. 14% on trastuzumab). Grade 3 or greater neutropenia occurred in 6% of patients in each group.
During adjuvant therapy, grade 3 or greater adverse events occurred in 9% of patients continuing ABP 980, 6% continuing trastuzumab, and 8% of these switched from trastuzumab to ABP 980. The most frequent grade 3 or greater events of interest were infections and neutropenia, all occurring in 1% of patients in each arm, and infusion reaction, which occurred in 1% of patients who stayed on the assigned HER2 inhibitor and in 2% of patients who were switched to ABP 980.
There were two patient deaths from adverse events, each deemed to be unrelated to treatment. One patient died from pneumonia during neoadjuvant ABP 980 therapy, and one died from septic shock during adjuvant therapy with ABP 980 after being switch from trastuzumab.
“To our knowledge, this is the first study of a trastuzumab biosimilar encompassing a single-switch design from the reference product to a biosimilar, which allowed us to assess the clinical safety and immunogenicity of this approach to treatment. Safety and immunogenicity were similar in patients who were switched and in those who continued to receive trastuzumab as adjuvant therapy,” the investigators wrote.
SOURCE: von Minckwitz G et al. Lancet Oncol 2018 Jun 4. doi: 10.1016/S1470-2045(18)30241-9.
FROM THE LANCET ONCOLOGY
Key clinical point: The biosimilar ABP 980 appears to be comparable in efficacy and safety to trastuzumab in women with early HER2-positive breast cancer.
Major finding: According to local lab assessments, 48% of patients assigned to ABP 980 had a pathologic complete response, compared with 41% assigned to trastuzumab.
Study details: Randomized, double-blind, phase 3 trial of 696 adult women with HER2-positive breast cancer.
Disclosures: Dr. von Minckwitz is a consultant for Amgen, which funded the study. Two coauthors are employees of the company and stockholders. Other coauthors disclosed relationships with various companies.
Source: von Minckwitz G et al. Lancet Oncol 2018 Jun 4. doi: 10.1016/S1470-2045(18)30241-9.
Scalp Psoriasis With Increased Hair Density
Case Report
A 19-year-old man first presented to our outpatient dermatology clinic for evaluation of a rash on the elbows and knees of 2 to 3 months’ duration. The lesions were asymptomatic. A review of symptoms including joint pain was largely negative. His medical history was remarkable for terminal ileitis, Crohn disease, anal fissure, rhabdomyolysis, and viral gastroenteritis. Physical examination revealed a well-nourished man with red, scaly, indurated papules and plaques involving approximately 0.5% of the body surface area. A diagnosis of plaque psoriasis was made, and he was treated with topical corticosteroids for 2 weeks and as needed thereafter.
The patient remained stable for 5 years before presenting again to the dermatology clinic for psoriasis that had now spread to the scalp. Clinical examination revealed a very thin, faintly erythematous, scaly patch associated with increased hair density of the right frontal and parietal scalp (Figure). The patient denied any trauma or injury to the area or application of hair dye. We prescribed clobetasol solution 0.05% twice daily to the affected area of the scalp for 2 weeks, which resulted in minimal resolution of the psoriatic scalp lesion.

Comment
The scalp is a site of predilection in psoriasis, as approximately 80% of psoriasis patients report involvement of the scalp.1 Scalp involvement can dramatically affect a patient’s quality of life and often poses considerable therapeutic challenges for dermatologists.1 Alopecia in the setting of scalp psoriasis is common but is not well understood.2 First described by Shuster3 in 1972, psoriatic alopecia is associated with diminished hair density, follicular miniaturization, sebaceous gland atrophy, and an increased number of dystrophic bulbs in psoriatic plaques.4 It clinically presents as pink scaly plaques consistent with psoriasis with overlying alopecia. There are few instances of psoriatic alopecia reported as cicatricial hair loss and generalized telogen effluvium.2 It is known that a higher proportion of telogen and catagen hairs exist in patients with psoriatic alopecia.5 Additionally, psoriasis patients have more dystrophic hairs in affected and unaffected skin despite no differences in skin when compared to unaffected patients. Many patients achieve hair regrowth following treatment of psoriasis.2
We described a patient with scalp psoriasis who had increased and preserved hair density. Our case suggests that while most patients with scalp psoriasis experience psoriatic alopecia of the lesional skin, some may unconventionally experience increased hair density, which is contradictory to propositions that the friction associated with the application of topical treatments results in breakage of telogen hairs.2 Additionally, the presence of increased hair density in scalp psoriasis can further complicate antipsoriatic treatment by making the scalp inaccessible and topical therapies even more difficult to apply.
- Krueger G, Koo J, Lebwohl M, et al. The impact of psoriasis on quality of life: results of a 1998 National Psoriasis Foundation patient-membership survey. Arch Dermatol. 2001;137:280-284.
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721.
- Shuster S. Psoriatic alopecia. Br J Dermatol. 1972;87:73-77.
- Wyatt E, Bottoms E, Comaish S. Abnormal hair shafts in psoriasis on scanning electron microscopy. Br J Dermatol. 1972;87:368-373.
- Schoorl WJ, van Baar HJ, van de Kerkhof PC. The hair root pattern in psoriasis of the scalp. Acta Derm Venereol. 1992;72:141-142.
Case Report
A 19-year-old man first presented to our outpatient dermatology clinic for evaluation of a rash on the elbows and knees of 2 to 3 months’ duration. The lesions were asymptomatic. A review of symptoms including joint pain was largely negative. His medical history was remarkable for terminal ileitis, Crohn disease, anal fissure, rhabdomyolysis, and viral gastroenteritis. Physical examination revealed a well-nourished man with red, scaly, indurated papules and plaques involving approximately 0.5% of the body surface area. A diagnosis of plaque psoriasis was made, and he was treated with topical corticosteroids for 2 weeks and as needed thereafter.
The patient remained stable for 5 years before presenting again to the dermatology clinic for psoriasis that had now spread to the scalp. Clinical examination revealed a very thin, faintly erythematous, scaly patch associated with increased hair density of the right frontal and parietal scalp (Figure). The patient denied any trauma or injury to the area or application of hair dye. We prescribed clobetasol solution 0.05% twice daily to the affected area of the scalp for 2 weeks, which resulted in minimal resolution of the psoriatic scalp lesion.
Comment
The scalp is a site of predilection in psoriasis, as approximately 80% of psoriasis patients report involvement of the scalp.1 Scalp involvement can dramatically affect a patient’s quality of life and often poses considerable therapeutic challenges for dermatologists.1 Alopecia in the setting of scalp psoriasis is common but is not well understood.2 First described by Shuster3 in 1972, psoriatic alopecia is associated with diminished hair density, follicular miniaturization, sebaceous gland atrophy, and an increased number of dystrophic bulbs in psoriatic plaques.4 It clinically presents as pink scaly plaques consistent with psoriasis with overlying alopecia. There are few instances of psoriatic alopecia reported as cicatricial hair loss and generalized telogen effluvium.2 It is known that a higher proportion of telogen and catagen hairs exist in patients with psoriatic alopecia.5 Additionally, psoriasis patients have more dystrophic hairs in affected and unaffected skin despite no differences in skin when compared to unaffected patients. Many patients achieve hair regrowth following treatment of psoriasis.2
We described a patient with scalp psoriasis who had increased and preserved hair density. Our case suggests that while most patients with scalp psoriasis experience psoriatic alopecia of the lesional skin, some may unconventionally experience increased hair density, which is contradictory to propositions that the friction associated with the application of topical treatments results in breakage of telogen hairs.2 Additionally, the presence of increased hair density in scalp psoriasis can further complicate antipsoriatic treatment by making the scalp inaccessible and topical therapies even more difficult to apply.
Case Report
A 19-year-old man first presented to our outpatient dermatology clinic for evaluation of a rash on the elbows and knees of 2 to 3 months’ duration. The lesions were asymptomatic. A review of symptoms including joint pain was largely negative. His medical history was remarkable for terminal ileitis, Crohn disease, anal fissure, rhabdomyolysis, and viral gastroenteritis. Physical examination revealed a well-nourished man with red, scaly, indurated papules and plaques involving approximately 0.5% of the body surface area. A diagnosis of plaque psoriasis was made, and he was treated with topical corticosteroids for 2 weeks and as needed thereafter.
The patient remained stable for 5 years before presenting again to the dermatology clinic for psoriasis that had now spread to the scalp. Clinical examination revealed a very thin, faintly erythematous, scaly patch associated with increased hair density of the right frontal and parietal scalp (Figure). The patient denied any trauma or injury to the area or application of hair dye. We prescribed clobetasol solution 0.05% twice daily to the affected area of the scalp for 2 weeks, which resulted in minimal resolution of the psoriatic scalp lesion.
Comment
The scalp is a site of predilection in psoriasis, as approximately 80% of psoriasis patients report involvement of the scalp.1 Scalp involvement can dramatically affect a patient’s quality of life and often poses considerable therapeutic challenges for dermatologists.1 Alopecia in the setting of scalp psoriasis is common but is not well understood.2 First described by Shuster3 in 1972, psoriatic alopecia is associated with diminished hair density, follicular miniaturization, sebaceous gland atrophy, and an increased number of dystrophic bulbs in psoriatic plaques.4 It clinically presents as pink scaly plaques consistent with psoriasis with overlying alopecia. There are few instances of psoriatic alopecia reported as cicatricial hair loss and generalized telogen effluvium.2 It is known that a higher proportion of telogen and catagen hairs exist in patients with psoriatic alopecia.5 Additionally, psoriasis patients have more dystrophic hairs in affected and unaffected skin despite no differences in skin when compared to unaffected patients. Many patients achieve hair regrowth following treatment of psoriasis.2
We described a patient with scalp psoriasis who had increased and preserved hair density. Our case suggests that while most patients with scalp psoriasis experience psoriatic alopecia of the lesional skin, some may unconventionally experience increased hair density, which is contradictory to propositions that the friction associated with the application of topical treatments results in breakage of telogen hairs.2 Additionally, the presence of increased hair density in scalp psoriasis can further complicate antipsoriatic treatment by making the scalp inaccessible and topical therapies even more difficult to apply.
- Krueger G, Koo J, Lebwohl M, et al. The impact of psoriasis on quality of life: results of a 1998 National Psoriasis Foundation patient-membership survey. Arch Dermatol. 2001;137:280-284.
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721.
- Shuster S. Psoriatic alopecia. Br J Dermatol. 1972;87:73-77.
- Wyatt E, Bottoms E, Comaish S. Abnormal hair shafts in psoriasis on scanning electron microscopy. Br J Dermatol. 1972;87:368-373.
- Schoorl WJ, van Baar HJ, van de Kerkhof PC. The hair root pattern in psoriasis of the scalp. Acta Derm Venereol. 1992;72:141-142.
- Krueger G, Koo J, Lebwohl M, et al. The impact of psoriasis on quality of life: results of a 1998 National Psoriasis Foundation patient-membership survey. Arch Dermatol. 2001;137:280-284.
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721.
- Shuster S. Psoriatic alopecia. Br J Dermatol. 1972;87:73-77.
- Wyatt E, Bottoms E, Comaish S. Abnormal hair shafts in psoriasis on scanning electron microscopy. Br J Dermatol. 1972;87:368-373.
- Schoorl WJ, van Baar HJ, van de Kerkhof PC. The hair root pattern in psoriasis of the scalp. Acta Derm Venereol. 1992;72:141-142.
Practice Points
- Scalp psoriasis may present with hair loss or increased hair density.
- Psoriasis with increased hair density may make topical medications more difficult to apply.
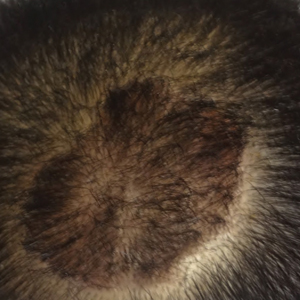
Trastuzumab biosimilar is equivalent on central review
CHICAGO – according to a new analysis of the phase 3 LILAC trial.
The 725 women in the multinational trial received run-in, anthracycline-based chemotherapy and were then evenly randomized to receive ABP 980 or trastuzumab, each with paclitaxel, followed by surgery.
The difference in pathologic complete response (pCR) rate assessed by local pathologists has been previously reported (Lancet Oncol. 2018 Jun 4. doi: 10.1016/S1470-2045(18)30241-9); those findings established non-inferiority of the biosimilar but left the matter of non-superiority inconclusive. However, in the new analysis, reported in a poster session at the ASCO Annual Meeting, the difference in pCR rate when instead assessed by a central pathologist fell within all bounds for equivalence.
“This is part of the totality of evidence in the course of approval of ABP 980,” lead author Hans-Christian Kolberg, MD, head of the department of gynecology and obstetrics of the Breast Cancer Center of the Gynecologic Cancer Center at Marien Hospital Bottrop (Germany), commented in an interview.
The new data prompted European regulators to authorize marketing of the biosimilar (branded as Kanjinti) for HER2+ early breast cancer and metastatic breast cancer, as well as HER2+ metastatic gastric cancer. (In the United States, the Food and Drug Administration recently rejected the application for ABP 980 market approval.)
“Breast cancer therapy is getting more and more expensive, and we somehow have to raise the money to pay for it. If we have a chance to make an antibody that is 20%-30% cheaper, which is what we hope it will be in Europe, we have that money for other things,” Dr. Kolberg said, reflecting on the bigger picture.
“I am also a visiting professor at a university in China, where patients who are HER2+ don’t get Herceptin because they can’t afford it. We always have to remember that in Europe and the U.S., we are kind of living on an island. If you look at Africa, Asia, and South America, making things affordable is important,” he added. “I hope and believe that this is just the beginning of the price fight. I hope that the biosimilar companies really will fight to see who will have the lowest price because that will be good for the patients. The lower the price, the better for the patients.”
Study details
Research leading up to the LILAC trial established that ABP 980 had analytic characteristics, nonclinical attributes, and pharmacokinetics similar to those of trastuzumab. The trial, conducted in 97 centers in 20 countries in western Europe, eastern Europe, and other world regions, assessed clinical similarity.
“I think central review was done in the study because we had so many centers all over the world that it was questionable as to how we could monitor the quality in dozens and dozens of pathology labs,” Dr. Kolberg explained. “So the idea was that we make it a little bit more difficult, a little bit more expensive, but more reliable if we use one pathologist.”
The central review was not without logistical issues, he acknowledged. In particular, it was challenging to ensure that all centers – including some doing so for the first time – followed a standardized procedure for sending tissue to the central lab.
The previously reported locally assessed pCR rates in breast tissue and axillary lymph nodes were 48.0% with ABP 980 and 40.5% with trastuzumab. The risk difference was 7.3% (90% confidence interval, 1.2%-13.4%) and the risk ratio was 1.188 (90% CI, 1.033-1.366), with the upper bounds of the confidence intervals exceeding the predefined equivalence margins of 13% and 1.318, respectively.
The centrally assessed pCR rates were 47.8% with ABP 980 and 41.8% with trastuzumab. The risk difference was 5.8% (90% CI, –0.5% to 12.0%), and the risk ratio was 1.14 (90% CI, 0.993 to 1.312), with the upper bounds of the confidence intervals now falling within the equivalence margins.
“This is the first study ever that used central pathology review for pCR in a neoadjuvant breast cancer study. We were really skeptical at the beginning as to whether that would work because we had a lot of centers all over the world, from Russia, Brazil, the U.S., Germany,” Dr. Kolberg commented.
“It worked, and we were very lucky that it worked because in the local review, we did not reach our biosimilar margins, our equivalence margins. In the central review, we were well within the margins,” he said. “So if we had not in the beginning planned a coprimary endpoint with local and central pathology review, the medication would never have been approved.”
Dr. Kolberg disclosed that he is a consultant for Amgen, Carl Zeiss Meditec, Genomic Health, GlaxoSmithKline, Janssen, LIV Pharma, Novartis, Pfizer, Roche, SurgVision, Teva Pharmaceutical Industries, and Theraclion. The trial was sponsored by Amgen.
SOURCE: Kolberg HC et al. ASCO Annual Meeting, Abstract 583.
CHICAGO – according to a new analysis of the phase 3 LILAC trial.
The 725 women in the multinational trial received run-in, anthracycline-based chemotherapy and were then evenly randomized to receive ABP 980 or trastuzumab, each with paclitaxel, followed by surgery.
The difference in pathologic complete response (pCR) rate assessed by local pathologists has been previously reported (Lancet Oncol. 2018 Jun 4. doi: 10.1016/S1470-2045(18)30241-9); those findings established non-inferiority of the biosimilar but left the matter of non-superiority inconclusive. However, in the new analysis, reported in a poster session at the ASCO Annual Meeting, the difference in pCR rate when instead assessed by a central pathologist fell within all bounds for equivalence.
“This is part of the totality of evidence in the course of approval of ABP 980,” lead author Hans-Christian Kolberg, MD, head of the department of gynecology and obstetrics of the Breast Cancer Center of the Gynecologic Cancer Center at Marien Hospital Bottrop (Germany), commented in an interview.
The new data prompted European regulators to authorize marketing of the biosimilar (branded as Kanjinti) for HER2+ early breast cancer and metastatic breast cancer, as well as HER2+ metastatic gastric cancer. (In the United States, the Food and Drug Administration recently rejected the application for ABP 980 market approval.)
“Breast cancer therapy is getting more and more expensive, and we somehow have to raise the money to pay for it. If we have a chance to make an antibody that is 20%-30% cheaper, which is what we hope it will be in Europe, we have that money for other things,” Dr. Kolberg said, reflecting on the bigger picture.
“I am also a visiting professor at a university in China, where patients who are HER2+ don’t get Herceptin because they can’t afford it. We always have to remember that in Europe and the U.S., we are kind of living on an island. If you look at Africa, Asia, and South America, making things affordable is important,” he added. “I hope and believe that this is just the beginning of the price fight. I hope that the biosimilar companies really will fight to see who will have the lowest price because that will be good for the patients. The lower the price, the better for the patients.”
Study details
Research leading up to the LILAC trial established that ABP 980 had analytic characteristics, nonclinical attributes, and pharmacokinetics similar to those of trastuzumab. The trial, conducted in 97 centers in 20 countries in western Europe, eastern Europe, and other world regions, assessed clinical similarity.
“I think central review was done in the study because we had so many centers all over the world that it was questionable as to how we could monitor the quality in dozens and dozens of pathology labs,” Dr. Kolberg explained. “So the idea was that we make it a little bit more difficult, a little bit more expensive, but more reliable if we use one pathologist.”
The central review was not without logistical issues, he acknowledged. In particular, it was challenging to ensure that all centers – including some doing so for the first time – followed a standardized procedure for sending tissue to the central lab.
The previously reported locally assessed pCR rates in breast tissue and axillary lymph nodes were 48.0% with ABP 980 and 40.5% with trastuzumab. The risk difference was 7.3% (90% confidence interval, 1.2%-13.4%) and the risk ratio was 1.188 (90% CI, 1.033-1.366), with the upper bounds of the confidence intervals exceeding the predefined equivalence margins of 13% and 1.318, respectively.
The centrally assessed pCR rates were 47.8% with ABP 980 and 41.8% with trastuzumab. The risk difference was 5.8% (90% CI, –0.5% to 12.0%), and the risk ratio was 1.14 (90% CI, 0.993 to 1.312), with the upper bounds of the confidence intervals now falling within the equivalence margins.
“This is the first study ever that used central pathology review for pCR in a neoadjuvant breast cancer study. We were really skeptical at the beginning as to whether that would work because we had a lot of centers all over the world, from Russia, Brazil, the U.S., Germany,” Dr. Kolberg commented.
“It worked, and we were very lucky that it worked because in the local review, we did not reach our biosimilar margins, our equivalence margins. In the central review, we were well within the margins,” he said. “So if we had not in the beginning planned a coprimary endpoint with local and central pathology review, the medication would never have been approved.”
Dr. Kolberg disclosed that he is a consultant for Amgen, Carl Zeiss Meditec, Genomic Health, GlaxoSmithKline, Janssen, LIV Pharma, Novartis, Pfizer, Roche, SurgVision, Teva Pharmaceutical Industries, and Theraclion. The trial was sponsored by Amgen.
SOURCE: Kolberg HC et al. ASCO Annual Meeting, Abstract 583.
CHICAGO – according to a new analysis of the phase 3 LILAC trial.
The 725 women in the multinational trial received run-in, anthracycline-based chemotherapy and were then evenly randomized to receive ABP 980 or trastuzumab, each with paclitaxel, followed by surgery.
The difference in pathologic complete response (pCR) rate assessed by local pathologists has been previously reported (Lancet Oncol. 2018 Jun 4. doi: 10.1016/S1470-2045(18)30241-9); those findings established non-inferiority of the biosimilar but left the matter of non-superiority inconclusive. However, in the new analysis, reported in a poster session at the ASCO Annual Meeting, the difference in pCR rate when instead assessed by a central pathologist fell within all bounds for equivalence.
“This is part of the totality of evidence in the course of approval of ABP 980,” lead author Hans-Christian Kolberg, MD, head of the department of gynecology and obstetrics of the Breast Cancer Center of the Gynecologic Cancer Center at Marien Hospital Bottrop (Germany), commented in an interview.
The new data prompted European regulators to authorize marketing of the biosimilar (branded as Kanjinti) for HER2+ early breast cancer and metastatic breast cancer, as well as HER2+ metastatic gastric cancer. (In the United States, the Food and Drug Administration recently rejected the application for ABP 980 market approval.)
“Breast cancer therapy is getting more and more expensive, and we somehow have to raise the money to pay for it. If we have a chance to make an antibody that is 20%-30% cheaper, which is what we hope it will be in Europe, we have that money for other things,” Dr. Kolberg said, reflecting on the bigger picture.
“I am also a visiting professor at a university in China, where patients who are HER2+ don’t get Herceptin because they can’t afford it. We always have to remember that in Europe and the U.S., we are kind of living on an island. If you look at Africa, Asia, and South America, making things affordable is important,” he added. “I hope and believe that this is just the beginning of the price fight. I hope that the biosimilar companies really will fight to see who will have the lowest price because that will be good for the patients. The lower the price, the better for the patients.”
Study details
Research leading up to the LILAC trial established that ABP 980 had analytic characteristics, nonclinical attributes, and pharmacokinetics similar to those of trastuzumab. The trial, conducted in 97 centers in 20 countries in western Europe, eastern Europe, and other world regions, assessed clinical similarity.
“I think central review was done in the study because we had so many centers all over the world that it was questionable as to how we could monitor the quality in dozens and dozens of pathology labs,” Dr. Kolberg explained. “So the idea was that we make it a little bit more difficult, a little bit more expensive, but more reliable if we use one pathologist.”
The central review was not without logistical issues, he acknowledged. In particular, it was challenging to ensure that all centers – including some doing so for the first time – followed a standardized procedure for sending tissue to the central lab.
The previously reported locally assessed pCR rates in breast tissue and axillary lymph nodes were 48.0% with ABP 980 and 40.5% with trastuzumab. The risk difference was 7.3% (90% confidence interval, 1.2%-13.4%) and the risk ratio was 1.188 (90% CI, 1.033-1.366), with the upper bounds of the confidence intervals exceeding the predefined equivalence margins of 13% and 1.318, respectively.
The centrally assessed pCR rates were 47.8% with ABP 980 and 41.8% with trastuzumab. The risk difference was 5.8% (90% CI, –0.5% to 12.0%), and the risk ratio was 1.14 (90% CI, 0.993 to 1.312), with the upper bounds of the confidence intervals now falling within the equivalence margins.
“This is the first study ever that used central pathology review for pCR in a neoadjuvant breast cancer study. We were really skeptical at the beginning as to whether that would work because we had a lot of centers all over the world, from Russia, Brazil, the U.S., Germany,” Dr. Kolberg commented.
“It worked, and we were very lucky that it worked because in the local review, we did not reach our biosimilar margins, our equivalence margins. In the central review, we were well within the margins,” he said. “So if we had not in the beginning planned a coprimary endpoint with local and central pathology review, the medication would never have been approved.”
Dr. Kolberg disclosed that he is a consultant for Amgen, Carl Zeiss Meditec, Genomic Health, GlaxoSmithKline, Janssen, LIV Pharma, Novartis, Pfizer, Roche, SurgVision, Teva Pharmaceutical Industries, and Theraclion. The trial was sponsored by Amgen.
SOURCE: Kolberg HC et al. ASCO Annual Meeting, Abstract 583.
REPORTING FROM THE ASCO ANNUAL MEETING
Key clinical point: Central review determined that ABP 980 was neither inferior nor superior to trastuzumab in breast cancer patients.
Major finding: The centrally determined pCR rates were 47.8% with ABP 980 and 41.8% with trastuzumab, with bounds of the confidence intervals for risk difference and for risk ratio falling within the predefined equivalence margins.
Study details: An analysis of a phase 3 randomized controlled trial of neoadjuvant (and adjuvant) therapy among 725 patients with HER2+ early breast cancer (LILAC trial).
Disclosures: Dr. Kolberg disclosed that he is a consultant for Amgen, Carl Zeiss Meditec, Genomic Health, GlaxoSmithKline, Janssen, LIV Pharma, Novartis, Pfizer, Roche, SurgVision, Teva Pharmaceutical Industries, and Theraclion. The trial was sponsored by Amgen.
Source: Kolberg HC et al. ASCO Annual Meeting, Abstract 583.
Research supports cannabis in MS, but legal, clinical pictures are murky
The medical marijuana landscape is changing so fast that Colorado Neurological Institute neurologist Allen C. Bowling, MD, PhD, already needs to update a presentation he gave about cannabis in multiple sclerosis in late May.

Since then, both those facts became history over a span of 2 days.
First, on June 25, the FDA announced its approval of Epidiolex (cannabidiol) for the treatment of seizures in two rare forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome. It’s the first time the FDA has approved a drug with a purified ingredient – cannabidiol, a nonpsychoactive substance – that’s derived from marijuana.
Then, on June 26, voters in Oklahoma approved a ballot measure that allows the possession of marijuana for medical use; users must register with the state. Thirty states and the District of Columbia have made medical marijuana legal, according to the procon.org website, although the two newest ones (Oklahoma and West Virginia) are still developing procedures.
The laws vary widely. Some states don’t allow patients to smoke medical marijuana, and some don’t allow visitors to use out-of-state registry ID cards. And certain states limit the use of medical marijuana to specific conditions. Medical marijuana use by patients with MS is specifically allowed in many states, including Alaska, Arizona, Florida, Minnesota, and several others.
There’s another complexity: According to procon.org, 17 states have laws about the use of cannabidiol. In Georgia, for instance, the use of some cannabis oil is allowed for the treatment of MS and other conditions.
In the wake of the FDA ruling, Dr. Bowling spoke in an interview about cannabis, MS, and the questions that neurologists should be asking themselves.
Q: What are studies telling us about cannabis and MS?
A: There are lots of clinical studies – 19 randomized controlled trials. A consistent finding is that there’s benefit in terms of pain and people’s subjective sense of spasticity (Neurology. 2014 Apr 29;82(17):1556-63).
Q: During your CMSC presentation, you talked about how “fidelity” has been a problem in cannabis research. Could you elaborate on what you mean?
A: The products used in these studies are generally standardized, research-grade products that you can’t buy in any U.S. dispensary.
Cannabis is complex and contains more than 100 different potentially pharmacologically active molecules. You can’t conclude that if you see a product in clinical trials, you’ll then be able to walk into a dispensary for recreational or medical cannabis and get a product that produces the same effect.
Q: What have you seen in your own patient population in terms of cannabis use?
A: I find what’s been found with the studies: It helps with pain and people’s sense of muscle stiffness.
It’s especially helpful in people with pain and spasticity that breaks through in the late afternoon or at night when they’re trying to go to sleep. Just a little bit of cannabis can get them through those difficult times and improve their quality of life.
Q: What choices do patients make regarding whether to get high from the cannabis they use?
A: Some have absolutely zero interest in getting high, and they try to avoid the THC-containing products. Other like getting high in addition to getting help with pain and spasticity.
Q: Who should not use medical marijuana in the MS community?
A: Patients who don’t have symptoms that could respond.
I’m also very concerned about patients who are 25 years and younger because of the effects that cannabis can have on brain development out to age 25 and the higher risk of addiction in people who are younger.
Q: What do you think the future will hold on the cannabis front?
A: Now that it’s less of a taboo topic, there’s an ever-growing number of trials each year, including very high-quality studies.
Pharmaceutically produced, cannabis-based medicines will be a growing area. Epidiolex is a perfect example of that.
It’s important for physicians to know that the way cannabis-based medicine is produced by a pharmaceutical company is different in so many levels than the cannabis in states with recreational and medical marijuana.
Q: What are some ways that the pharmaceutical products are different?
A: The rigor of the production process, the standardization, the purity, the correct labeling and expiration dates. Plus, the lack of the use of pesticides and other contaminants. And they’re distributed by pharmacists.
Q: What should neurologists be thinking if they’re considering whether to recommend cannabis to their patients?
A: This is a very complex topic, and it’s not something that most of us have training in. You can’t sit down for 1 or 2 hours, get up to speed, and have your own well-informed opinion on it. You really need to put more time and effort.
Q: What are some issues that neurologists should consider?
A: You really need to find out what your state is doing about it and see how you feel about that.
How is your state administering medical and/or recreational marijuana? The administration of these programs is extremely different from state to state. Do these details satisfy you, and are you content having your patients interface with these programs?
Dr. Bowling reports no relevant disclosures.
The medical marijuana landscape is changing so fast that Colorado Neurological Institute neurologist Allen C. Bowling, MD, PhD, already needs to update a presentation he gave about cannabis in multiple sclerosis in late May.
Since then, both those facts became history over a span of 2 days.
First, on June 25, the FDA announced its approval of Epidiolex (cannabidiol) for the treatment of seizures in two rare forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome. It’s the first time the FDA has approved a drug with a purified ingredient – cannabidiol, a nonpsychoactive substance – that’s derived from marijuana.
Then, on June 26, voters in Oklahoma approved a ballot measure that allows the possession of marijuana for medical use; users must register with the state. Thirty states and the District of Columbia have made medical marijuana legal, according to the procon.org website, although the two newest ones (Oklahoma and West Virginia) are still developing procedures.
The laws vary widely. Some states don’t allow patients to smoke medical marijuana, and some don’t allow visitors to use out-of-state registry ID cards. And certain states limit the use of medical marijuana to specific conditions. Medical marijuana use by patients with MS is specifically allowed in many states, including Alaska, Arizona, Florida, Minnesota, and several others.
There’s another complexity: According to procon.org, 17 states have laws about the use of cannabidiol. In Georgia, for instance, the use of some cannabis oil is allowed for the treatment of MS and other conditions.
In the wake of the FDA ruling, Dr. Bowling spoke in an interview about cannabis, MS, and the questions that neurologists should be asking themselves.
Q: What are studies telling us about cannabis and MS?
A: There are lots of clinical studies – 19 randomized controlled trials. A consistent finding is that there’s benefit in terms of pain and people’s subjective sense of spasticity (Neurology. 2014 Apr 29;82(17):1556-63).
Q: During your CMSC presentation, you talked about how “fidelity” has been a problem in cannabis research. Could you elaborate on what you mean?
A: The products used in these studies are generally standardized, research-grade products that you can’t buy in any U.S. dispensary.
Cannabis is complex and contains more than 100 different potentially pharmacologically active molecules. You can’t conclude that if you see a product in clinical trials, you’ll then be able to walk into a dispensary for recreational or medical cannabis and get a product that produces the same effect.
Q: What have you seen in your own patient population in terms of cannabis use?
A: I find what’s been found with the studies: It helps with pain and people’s sense of muscle stiffness.
It’s especially helpful in people with pain and spasticity that breaks through in the late afternoon or at night when they’re trying to go to sleep. Just a little bit of cannabis can get them through those difficult times and improve their quality of life.
Q: What choices do patients make regarding whether to get high from the cannabis they use?
A: Some have absolutely zero interest in getting high, and they try to avoid the THC-containing products. Other like getting high in addition to getting help with pain and spasticity.
Q: Who should not use medical marijuana in the MS community?
A: Patients who don’t have symptoms that could respond.
I’m also very concerned about patients who are 25 years and younger because of the effects that cannabis can have on brain development out to age 25 and the higher risk of addiction in people who are younger.
Q: What do you think the future will hold on the cannabis front?
A: Now that it’s less of a taboo topic, there’s an ever-growing number of trials each year, including very high-quality studies.
Pharmaceutically produced, cannabis-based medicines will be a growing area. Epidiolex is a perfect example of that.
It’s important for physicians to know that the way cannabis-based medicine is produced by a pharmaceutical company is different in so many levels than the cannabis in states with recreational and medical marijuana.
Q: What are some ways that the pharmaceutical products are different?
A: The rigor of the production process, the standardization, the purity, the correct labeling and expiration dates. Plus, the lack of the use of pesticides and other contaminants. And they’re distributed by pharmacists.
Q: What should neurologists be thinking if they’re considering whether to recommend cannabis to their patients?
A: This is a very complex topic, and it’s not something that most of us have training in. You can’t sit down for 1 or 2 hours, get up to speed, and have your own well-informed opinion on it. You really need to put more time and effort.
Q: What are some issues that neurologists should consider?
A: You really need to find out what your state is doing about it and see how you feel about that.
How is your state administering medical and/or recreational marijuana? The administration of these programs is extremely different from state to state. Do these details satisfy you, and are you content having your patients interface with these programs?
Dr. Bowling reports no relevant disclosures.
The medical marijuana landscape is changing so fast that Colorado Neurological Institute neurologist Allen C. Bowling, MD, PhD, already needs to update a presentation he gave about cannabis in multiple sclerosis in late May.
Since then, both those facts became history over a span of 2 days.
First, on June 25, the FDA announced its approval of Epidiolex (cannabidiol) for the treatment of seizures in two rare forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome. It’s the first time the FDA has approved a drug with a purified ingredient – cannabidiol, a nonpsychoactive substance – that’s derived from marijuana.
Then, on June 26, voters in Oklahoma approved a ballot measure that allows the possession of marijuana for medical use; users must register with the state. Thirty states and the District of Columbia have made medical marijuana legal, according to the procon.org website, although the two newest ones (Oklahoma and West Virginia) are still developing procedures.
The laws vary widely. Some states don’t allow patients to smoke medical marijuana, and some don’t allow visitors to use out-of-state registry ID cards. And certain states limit the use of medical marijuana to specific conditions. Medical marijuana use by patients with MS is specifically allowed in many states, including Alaska, Arizona, Florida, Minnesota, and several others.
There’s another complexity: According to procon.org, 17 states have laws about the use of cannabidiol. In Georgia, for instance, the use of some cannabis oil is allowed for the treatment of MS and other conditions.
In the wake of the FDA ruling, Dr. Bowling spoke in an interview about cannabis, MS, and the questions that neurologists should be asking themselves.
Q: What are studies telling us about cannabis and MS?
A: There are lots of clinical studies – 19 randomized controlled trials. A consistent finding is that there’s benefit in terms of pain and people’s subjective sense of spasticity (Neurology. 2014 Apr 29;82(17):1556-63).
Q: During your CMSC presentation, you talked about how “fidelity” has been a problem in cannabis research. Could you elaborate on what you mean?
A: The products used in these studies are generally standardized, research-grade products that you can’t buy in any U.S. dispensary.
Cannabis is complex and contains more than 100 different potentially pharmacologically active molecules. You can’t conclude that if you see a product in clinical trials, you’ll then be able to walk into a dispensary for recreational or medical cannabis and get a product that produces the same effect.
Q: What have you seen in your own patient population in terms of cannabis use?
A: I find what’s been found with the studies: It helps with pain and people’s sense of muscle stiffness.
It’s especially helpful in people with pain and spasticity that breaks through in the late afternoon or at night when they’re trying to go to sleep. Just a little bit of cannabis can get them through those difficult times and improve their quality of life.
Q: What choices do patients make regarding whether to get high from the cannabis they use?
A: Some have absolutely zero interest in getting high, and they try to avoid the THC-containing products. Other like getting high in addition to getting help with pain and spasticity.
Q: Who should not use medical marijuana in the MS community?
A: Patients who don’t have symptoms that could respond.
I’m also very concerned about patients who are 25 years and younger because of the effects that cannabis can have on brain development out to age 25 and the higher risk of addiction in people who are younger.
Q: What do you think the future will hold on the cannabis front?
A: Now that it’s less of a taboo topic, there’s an ever-growing number of trials each year, including very high-quality studies.
Pharmaceutically produced, cannabis-based medicines will be a growing area. Epidiolex is a perfect example of that.
It’s important for physicians to know that the way cannabis-based medicine is produced by a pharmaceutical company is different in so many levels than the cannabis in states with recreational and medical marijuana.
Q: What are some ways that the pharmaceutical products are different?
A: The rigor of the production process, the standardization, the purity, the correct labeling and expiration dates. Plus, the lack of the use of pesticides and other contaminants. And they’re distributed by pharmacists.
Q: What should neurologists be thinking if they’re considering whether to recommend cannabis to their patients?
A: This is a very complex topic, and it’s not something that most of us have training in. You can’t sit down for 1 or 2 hours, get up to speed, and have your own well-informed opinion on it. You really need to put more time and effort.
Q: What are some issues that neurologists should consider?
A: You really need to find out what your state is doing about it and see how you feel about that.
How is your state administering medical and/or recreational marijuana? The administration of these programs is extremely different from state to state. Do these details satisfy you, and are you content having your patients interface with these programs?
Dr. Bowling reports no relevant disclosures.
Diet and Dermatology: Google Search Results for Acne, Psoriasis, and Eczema
Researching medical information currently is the third most common use of the Internet in the United States,1 with the majority of adults using the Web as their first source for health information before seeing a physician.2 When assessing health-related information online, resources can be grouped into 4 categories: (1) those attributed to self-proclaimed experts, (2) promotional, (3) social media, and (4) educational.3 Access to such a wide range of sources may give readers the opportunity to share personal anecdotes and opinions, thereby serving as a forum for information that essentially cannot be validated. Although such websites may include useful information and cite current literature, in other instances health-related information may be misleading or fabricated.3
In a study evaluating 291 skin conditions and related Google trends, acne, psoriasis, and eczema were among the most burdensome diseases, with acne yielding the highest number of search results.4 Results of the study indicated a positive correlation between disease burden and online search interest.4 The impact of these online searches and the validity of Google search results are topics worth considering, as more dermatology patients are relying on holistic and nonpharmaceutical approaches to treatment and disease management.5 The purpose of this study was to evaluate content on diet and dermatology available on the Internet for acne, psoriasis, and eczema.
Methods
Google searches were performed in December 2017 using the terms diet and acne, diet and psoriasis, and diet and eczema. The first 10 results for each respective search were reviewed for recommendations about which foods to incorporate in the diet and which to avoid. They also were classified according to the following 4 website categories: (1) those attributed to self-proclaimed experts, (2) promotional, (3) social media, and (4) educational. The recommendations gathered from the 30 websites were then compared to the current literature assessing the impact of diet on these respective conditions by conducting PubMed searches of articles indexed for MEDLINE using the same terms.
Results
The results of this study are outlined in the eTable.

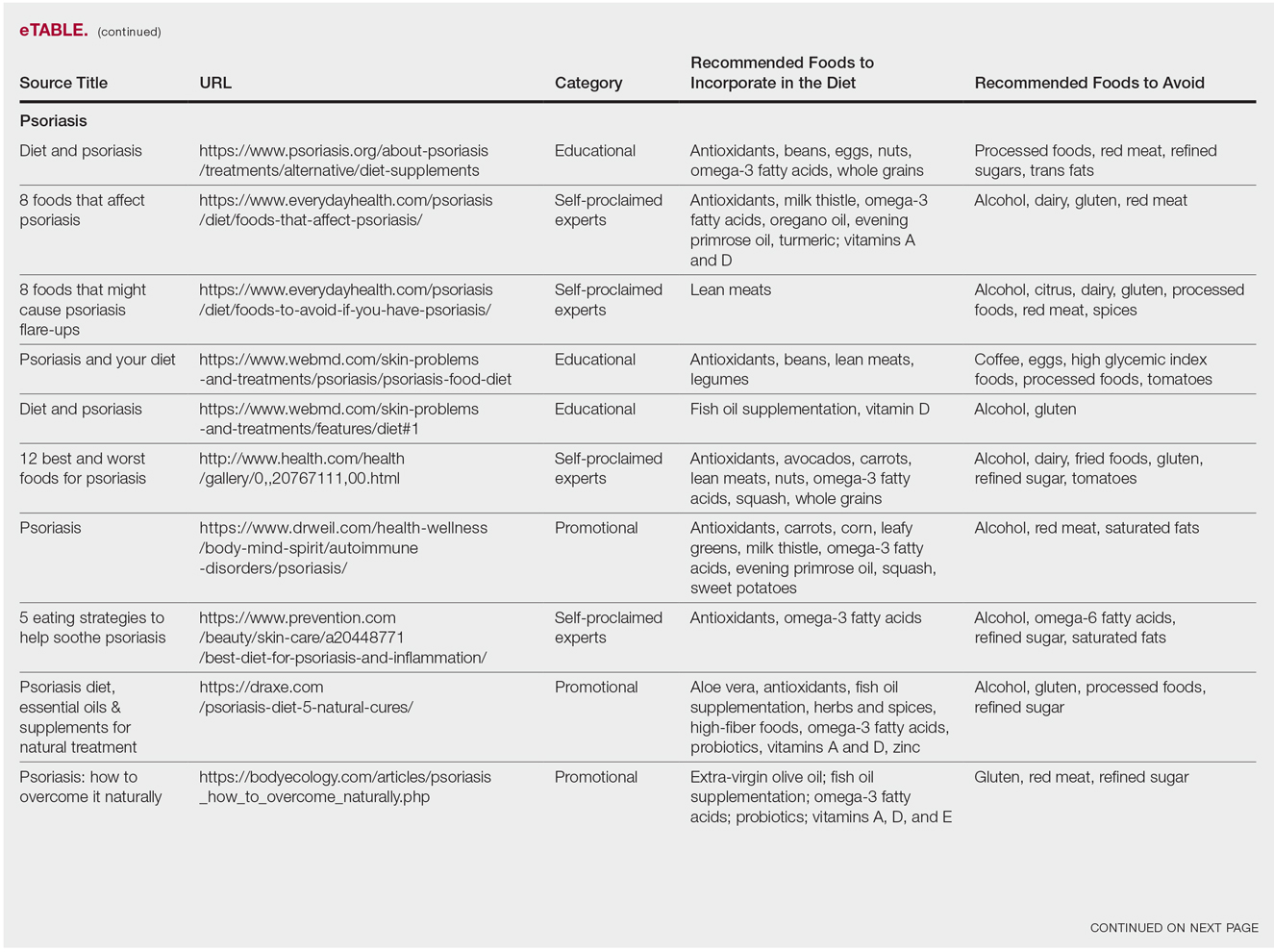
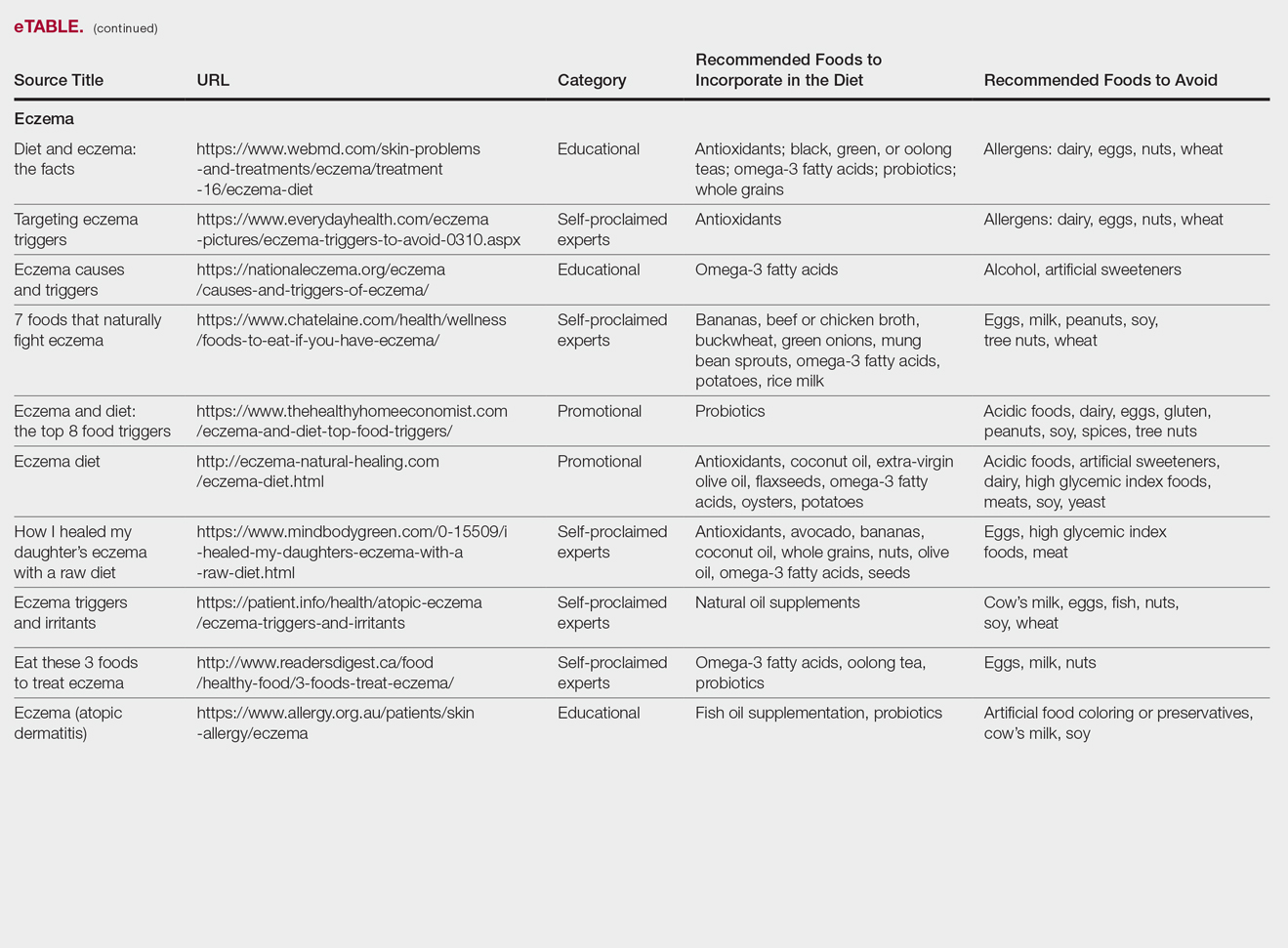
Acne
Our Google search using the term diet and acne produced 17,500,000 results. Of the first 10 search results, 40% (4/10) were websites attributed to self-proclaimed experts, 40% (4/10) were educational resources, and 20% (2/10) were promotional websites. Most of the websites advised acne patients to avoid high glycemic index foods (90% [9/10]) and dairy products (90% [9/10]). When discussing which foods to include in the diet, 70% (7/10) of websites recommended that patients incorporate omega-3 fatty acids and antioxidants in the diet.
Research has shown that a low glycemic index diet can lead to a decrease in patients’ acne lesion counts in some instances.6,7 In a case-controlled study of 2258 patients on a popular weight loss diet that emphasized low glycemic index foods, 87% of participants reported a reduction in acne and 91% reported a decrease in their dosage or number of acne medications.7 Still, the exact correlation between acne development and consumption of glycemic index foods has not been confirmed. However, high glycemic index diets have been linked to hyperinsulinemia, indicating that insulin levels may play a role in acne formation.8 The majority of other currently available studies evaluated the potential link between dairy consumption and acne. A retrospective analysis of 47,355 women spanning 12 weeks showed a positive link between increased dairy consumption, specifically skim milk, and acne formation. Despite the positive trend, limitations such as recall bias made it difficult to draw a conclusion based on these findings.9 However, results of a longitudinal questionnaire-based population study evaluating the impact of dairy consumption on acne in 2489 adolescent patients confirmed a positive correlation.10 Studies conducted in 2009 and 2011 concluded that milk consumption results in elevated insulinlike growth factor 1 levels, which were linked to comedogenesis.8,11
Currently, there are well-described mechanisms to explain the association of dairy consumption and glycemic index with acne. Confirming a correlation between acne development and dairy consumption suggests that a dairy-free diet may benefit acne patients.5 Other trials indicate that low glycemic index diets are beneficial in treating acne.6,7 Therefore, some of the recommendations made in our search results may be of merit; however, there is minimal evidence proving the benefits of the other dietary recommendations made in the websites we evaluated.
Psoriasis
Our Google search using the term diet and psoriasis yielded a total of 9,420,000 results. Of the first 10 search results, 40% (4/10) were websites attributed to self-proclaimed experts, 30% (3/10) were promotional, and 30% (3/10) were educational. Seventy percent (7/10) of websites recommended avoiding alcohol and 60% (6/10) recommended avoiding gluten, with others discouraging consumption of red meat. Most of the websites encouraged patients to consume omega-3 fatty acids and antioxidants, while a few also recommended vitamins A, D, and E, as well as evening primrose oil supplements.
Although current research indicates a positive correlation between excessive alcohol use and psoriasis severity, it is still unclear whether alcohol consumption can be directly linked to the disease.12-14 Likewise, despite belief that increased oxidative stress likely contributes to inflammation in psoriasis, there is little evidence linking antioxidants to improvement in psoriasis symptoms.12 However, the current literature is inconsistent regarding the effects of fish oil supplementation on psoriasis.12 In a randomized double-blind study of 145 patients, there was no significant difference in psoriasis area and severity index scores between a control group and a treatment group receiving fish oil supplementation.15 In another RCT of 45 participants, those given daily very long-chain omega-3 fatty acid supplements saw no difference in psoriasis symptoms.15 Despite debate, literature assessing the impact of gluten-free diets has described improvement in psoriasis lesions in patients with celiac-specific antibodies.16 Although some observational studies described vitamin D supplementation to be beneficial in the treatment of psoriatic lesions, a more recent RCT found no significant difference between control and treatment groups.17-19
Studies also have revealed that certain eating patterns, such as those associated with the Mediterranean diet that is rich in fruits, vegetables, whole grains, and omega-3 fatty acids may be linked to improved endothelial function scores and reduced C-reactive protein and IL-18levels.20,21 In a double-blind RCT of 75 patients with plaque psoriasis, mean (SD) psoriasis area and severity index scores decreased by 11.2 (9.8) in a group treated with omega-3 fatty acids compared to 7.5 (8.8) with omega-6 fatty acids (P=.048).22
Although excessive alcohol use may be linked to psoriasis, there is no conclusive evidence indicating causation, thereby discrediting online claims.12-14 Research has revealed that gluten-free diets in psoriasis patients with celiac disease may improve psoriasis treatment16; however, sufficient evidence is lacking for diets low in gluten and high in polyunsaturated fatty acids or antioxidant supplementation. Of the dietary supplements recommended in the search results we reviewed, fish oil appears to be the most promising, but no recommendations can be made based on the current research.
Eczema
Our Google search using the term diet and eczema yielded 1,160,000 results, with 50% (5/10) of websites attributed to self-proclaimed experts, 30% (3/10) to educational websites, and 20% (2/10) to promotional sites. Of the first 10 results, 80% (8/10) recommended that patients with eczema avoid milk/dairy and 50% (5/10) advised to avoid soy and wheat/gluten. Other websites indicated to avoid eggs, nuts, and artificial sweeteners. Patients were encouraged to incorporate omega-3 fatty acids in their diets, and a few sites recommended bananas, coconut oil, olive oil, and various teas.
In a review of 11 studies with a total of 596 participants, supplementation with vitamins D and E, fish oil, olive oil, and linoleic acid was evaluated for the treatment of eczema.23 Although results indicated modest improvement of eczema severity with supplementation of fish oil, evidence favoring this treatment is limited and unconvincing. Furthermore, some evidence indicates that elimination diets are only appropriate for patients with food allergies.24 In a study evaluating an egg-free and dairy-free diet for eczema patients, only participants with positive egg-specific serum IgE levels saw improvement in disease severity.23 Even though IgE-mediated food allergies have been reported in 40% of children with moderate eczema, the contribution of these allergies to eczema is questionable.25
There is little evidence in the literature to indicate a definitive correlation between the foods mentioned in the search results we evaluated and the development of eczema; however, for patients with food allergies and eczema, elimination diets may decrease disease severity.25,26 There is insufficient evidence to suggest a benefit from evening primrose oil or fish oil supplementation, thereby debunking claims found online.
Comment
Although our Google search results included a wide range of sources and information regarding diet and dermatologic conditions such as acne, psoriasis, and eczema, most of the information we found was either unfounded or misleading. Study limitations in the current literature include small sample size, potential recall bias, lack of appropriate controls, incomplete reported results, and the failure to clearly define skin changes.
When considering the accuracy and type of information regarding skin conditions that is available on the Internet, it is important to note that most of the results we reviewed were webpages attributed to self-proclaimed experts. Although educational websites also were included in the search results, whether or not patients prefer or understand the content of such websites is still unknown; therefore, health organizations should consider revising online patient education materials to allow universal comprehension.27
Furthermore, it is important to consider the impact that widespread Internet access may have on the physician-patient relationship. Having access to health-related information online and being able to potentially self-diagnose could delay or deter patients from seeking professional advice or care.3 A study evaluating the impact of online searches on the physician-patient relationship among 175 patients determined that 36.5% of patients gathered information online prior to their consultation with a physician, while 67.3% chose to complement the information given to them by their physician with online resources.28 Based on these statistics, it is important that physicians be up-to-date with Internet discourse to discredit unfounded recommendations. Ultimately, when it comes to diet and dermatology, patients ought to be skeptical of the information currently available on the Internet, given that most of it is unsubstantiated by medical research.
- Fox S. Online health search 2006. Pew Research Center website. http://www.pewinternet.org/2006/10/29/online-health-search-2006/. Published October 29, 2006. Accessed May 3, 2018.
- Prestin A, Vieux SN, Chou WY. Is online health activity alive and well or flatlining? findings from 10 years of the health information national trends survey. J Health Commun. 2015;20:790-798.
- Zeichner JA, Del Rosso JQ. Acne and the internet. Dermatol Clin. 2016;34:129-132.
- Whitsitt J, Karimkhani C, Boyers LN, et al. Comparing burden of dermatologic disease to search interest on Google trends. Dermatol Online J. 2015;21. pii:13030/qt5xg811qp.
- Shokeen D. Influence of diet in acne vulgaris and atopic dermatitis. Cutis. 2016;98:E28-E29.
- Veith WB, Silverberg NB. The association of acne vulgaris with diet. Cutis. 2011;88:84-91.
- Rouhani P. Acne improves with a popular, low glycemic diet from South Beach. J Am Acad Dermatol. 2009;60(3, suppl 1):P706.
- Melnick BC. Evidence for acne-promoting effect of milk and other insulinotropic dairy products. Nestle Nutr Worksop Ser Pediatr Program. 2011;67:131-145.
- Adebamowo CA, Spiegelman D, Berkey CS, et al. High school dietary diary intake and teenage acne. J Am Acad Dermatol. 2005;52:207-214.
- Ulvestad M, Bjertness E, Dalgard F, et al. Acne and dairy products in adolescence: results from a Norwegian longitudinal study [published online July 16, 2016]. J Eur Acad Dermatol Venereol. 2017;31:530-535.
- Melnick BC, Schmitz G. Role of insulin, insulin like growth factor 1, hyperglycemic food and milk consumption in the pathogenesis of acne vulgaris. Exp Dermatol. 2009;18:833-841.
- Murzaku EC, Bronsnick T, Rao BK. Diet in dermatology: part II. melanoma, chronic urticaria, and psoriasis. J Am Acad Dermatol. 2014;71:1053.E1-1053.E16.
- Tobin AM, Higgins EM, Norris S, et al. Prevalence of psoriasis in patients with alcoholic liver disease. Clin Exp Dermatol. 2009;34:698-701.
- Kirby B, Richards HL, Mason DL, et al. Alcohol consumption and psychological distress in patients with psoriasis. Br J Dermatol. 2008;158:138-140.
- Søyland E, Funk J, Rajika G, et al. Effect of dietary supplementation with very long-chain n-3 fatty acids in patients with psoriasis. N Engl J Med. 1993;328:1812-1816.
- Michaëlsson G, Gerdén B, Hagforsen E, et al. Psoriasis patients with antibodies to gliadin can be improved by a gluten-free diet. Br J Dermatol. 2000;142:44-51.
- Morimoto S, Yoshikawa K. Psoriasis and vitamin D3. a review of our experience. Arch Dermatol. 1989;125:231-234.
- Smith EL, Pincus SH, Donovan L, et al. A novel approach for the evaluation and treatment of psoriasis. oral or topical use of 1,25-dihydroxyvitamin D3 can be a safe and effective therapy for psoriasis. J Am Acad Dermatol. 1988;19:516-528.
- Siddiqui MA, Al-Khawajah MM. Vitamin D3 and psoriasis: a randomized double-blind placebo-controlled study. J Dermatol Treat. 1990;1:243-245.
- Wang Y, Gao H, Loyd CM, et al. Chronic skin-specific inflammation promotes vascular inflammation and thrombosis. J Invest Dermatol. 2012;132:2067-2075.
- Barrea L, Nappi F, Di Somma C, et al. Environmental risk factors in psoriasis: the point of view of the nutritionist. Int J Environ Res Public Health. 2016;13. pii:E743. doi:10.3390/ijerph13070743.
- Mayser P, Mrowietz U, Arenberger P, et al. Omega-3 fatty acid-based lipid infusion in patients with chronic plaque psoriasis: results of a double-blind, randomized, placebo-controlled, multicenter trial. J Am Acad Dermatol. 1998;38:539-547.
- Bath-Hextall FJ, Jenkinson C, Humphreys R, et al. Dietary supplements for established atopic eczema. Cochrane Database Syst Rev. 2012;2:CD005205.
- Bronsnick T, Murzaku EC, Rao BK. Diet in dermatology: part I. atopic dermatitis, acne, and nonmelanoma skin cancer [published online November 15, 2014]. J Am Acad Dermatol. 2014;71:1039.E1-1039.E12.
- Campbell DE. The role of food allergy in childhood atopic dermatitis. J Paediatr Child Health. 2012;48:1058-1064.
- Werfel T, Erdmann S, Fuchs T, et al. Approach to suspected food allergy in atopic dermatitis. guideline of the Task Force on Food Allergy of the German Society of Allergology and Clinical Immunology (DGAKI) and the Medical Association of German Allergologists (ADA) and the German Society of Pediatric Allergology (GPA). J Dtsch Dermatol Ges. 2009;3:265-271.
- John AM, John ES, Hansberry DR, et al. Assessment of online patient education materials from major dermatologic associations. J Clin Aesthet Dermatol. 2016;9:23-28.
- Orgaz-Molina J, Cotugno M, Girón-Prieto MS, et al. A study of internet searches for medical information in dermatology patients: the patient-physician relationship. Actas Dermosifiliogr. 2015;106:493-499.
Researching medical information currently is the third most common use of the Internet in the United States,1 with the majority of adults using the Web as their first source for health information before seeing a physician.2 When assessing health-related information online, resources can be grouped into 4 categories: (1) those attributed to self-proclaimed experts, (2) promotional, (3) social media, and (4) educational.3 Access to such a wide range of sources may give readers the opportunity to share personal anecdotes and opinions, thereby serving as a forum for information that essentially cannot be validated. Although such websites may include useful information and cite current literature, in other instances health-related information may be misleading or fabricated.3
In a study evaluating 291 skin conditions and related Google trends, acne, psoriasis, and eczema were among the most burdensome diseases, with acne yielding the highest number of search results.4 Results of the study indicated a positive correlation between disease burden and online search interest.4 The impact of these online searches and the validity of Google search results are topics worth considering, as more dermatology patients are relying on holistic and nonpharmaceutical approaches to treatment and disease management.5 The purpose of this study was to evaluate content on diet and dermatology available on the Internet for acne, psoriasis, and eczema.
Methods
Google searches were performed in December 2017 using the terms diet and acne, diet and psoriasis, and diet and eczema. The first 10 results for each respective search were reviewed for recommendations about which foods to incorporate in the diet and which to avoid. They also were classified according to the following 4 website categories: (1) those attributed to self-proclaimed experts, (2) promotional, (3) social media, and (4) educational. The recommendations gathered from the 30 websites were then compared to the current literature assessing the impact of diet on these respective conditions by conducting PubMed searches of articles indexed for MEDLINE using the same terms.
Results
The results of this study are outlined in the eTable.
Acne
Our Google search using the term diet and acne produced 17,500,000 results. Of the first 10 search results, 40% (4/10) were websites attributed to self-proclaimed experts, 40% (4/10) were educational resources, and 20% (2/10) were promotional websites. Most of the websites advised acne patients to avoid high glycemic index foods (90% [9/10]) and dairy products (90% [9/10]). When discussing which foods to include in the diet, 70% (7/10) of websites recommended that patients incorporate omega-3 fatty acids and antioxidants in the diet.
Research has shown that a low glycemic index diet can lead to a decrease in patients’ acne lesion counts in some instances.6,7 In a case-controlled study of 2258 patients on a popular weight loss diet that emphasized low glycemic index foods, 87% of participants reported a reduction in acne and 91% reported a decrease in their dosage or number of acne medications.7 Still, the exact correlation between acne development and consumption of glycemic index foods has not been confirmed. However, high glycemic index diets have been linked to hyperinsulinemia, indicating that insulin levels may play a role in acne formation.8 The majority of other currently available studies evaluated the potential link between dairy consumption and acne. A retrospective analysis of 47,355 women spanning 12 weeks showed a positive link between increased dairy consumption, specifically skim milk, and acne formation. Despite the positive trend, limitations such as recall bias made it difficult to draw a conclusion based on these findings.9 However, results of a longitudinal questionnaire-based population study evaluating the impact of dairy consumption on acne in 2489 adolescent patients confirmed a positive correlation.10 Studies conducted in 2009 and 2011 concluded that milk consumption results in elevated insulinlike growth factor 1 levels, which were linked to comedogenesis.8,11
Currently, there are well-described mechanisms to explain the association of dairy consumption and glycemic index with acne. Confirming a correlation between acne development and dairy consumption suggests that a dairy-free diet may benefit acne patients.5 Other trials indicate that low glycemic index diets are beneficial in treating acne.6,7 Therefore, some of the recommendations made in our search results may be of merit; however, there is minimal evidence proving the benefits of the other dietary recommendations made in the websites we evaluated.
Psoriasis
Our Google search using the term diet and psoriasis yielded a total of 9,420,000 results. Of the first 10 search results, 40% (4/10) were websites attributed to self-proclaimed experts, 30% (3/10) were promotional, and 30% (3/10) were educational. Seventy percent (7/10) of websites recommended avoiding alcohol and 60% (6/10) recommended avoiding gluten, with others discouraging consumption of red meat. Most of the websites encouraged patients to consume omega-3 fatty acids and antioxidants, while a few also recommended vitamins A, D, and E, as well as evening primrose oil supplements.
Although current research indicates a positive correlation between excessive alcohol use and psoriasis severity, it is still unclear whether alcohol consumption can be directly linked to the disease.12-14 Likewise, despite belief that increased oxidative stress likely contributes to inflammation in psoriasis, there is little evidence linking antioxidants to improvement in psoriasis symptoms.12 However, the current literature is inconsistent regarding the effects of fish oil supplementation on psoriasis.12 In a randomized double-blind study of 145 patients, there was no significant difference in psoriasis area and severity index scores between a control group and a treatment group receiving fish oil supplementation.15 In another RCT of 45 participants, those given daily very long-chain omega-3 fatty acid supplements saw no difference in psoriasis symptoms.15 Despite debate, literature assessing the impact of gluten-free diets has described improvement in psoriasis lesions in patients with celiac-specific antibodies.16 Although some observational studies described vitamin D supplementation to be beneficial in the treatment of psoriatic lesions, a more recent RCT found no significant difference between control and treatment groups.17-19
Studies also have revealed that certain eating patterns, such as those associated with the Mediterranean diet that is rich in fruits, vegetables, whole grains, and omega-3 fatty acids may be linked to improved endothelial function scores and reduced C-reactive protein and IL-18levels.20,21 In a double-blind RCT of 75 patients with plaque psoriasis, mean (SD) psoriasis area and severity index scores decreased by 11.2 (9.8) in a group treated with omega-3 fatty acids compared to 7.5 (8.8) with omega-6 fatty acids (P=.048).22
Although excessive alcohol use may be linked to psoriasis, there is no conclusive evidence indicating causation, thereby discrediting online claims.12-14 Research has revealed that gluten-free diets in psoriasis patients with celiac disease may improve psoriasis treatment16; however, sufficient evidence is lacking for diets low in gluten and high in polyunsaturated fatty acids or antioxidant supplementation. Of the dietary supplements recommended in the search results we reviewed, fish oil appears to be the most promising, but no recommendations can be made based on the current research.
Eczema
Our Google search using the term diet and eczema yielded 1,160,000 results, with 50% (5/10) of websites attributed to self-proclaimed experts, 30% (3/10) to educational websites, and 20% (2/10) to promotional sites. Of the first 10 results, 80% (8/10) recommended that patients with eczema avoid milk/dairy and 50% (5/10) advised to avoid soy and wheat/gluten. Other websites indicated to avoid eggs, nuts, and artificial sweeteners. Patients were encouraged to incorporate omega-3 fatty acids in their diets, and a few sites recommended bananas, coconut oil, olive oil, and various teas.
In a review of 11 studies with a total of 596 participants, supplementation with vitamins D and E, fish oil, olive oil, and linoleic acid was evaluated for the treatment of eczema.23 Although results indicated modest improvement of eczema severity with supplementation of fish oil, evidence favoring this treatment is limited and unconvincing. Furthermore, some evidence indicates that elimination diets are only appropriate for patients with food allergies.24 In a study evaluating an egg-free and dairy-free diet for eczema patients, only participants with positive egg-specific serum IgE levels saw improvement in disease severity.23 Even though IgE-mediated food allergies have been reported in 40% of children with moderate eczema, the contribution of these allergies to eczema is questionable.25
There is little evidence in the literature to indicate a definitive correlation between the foods mentioned in the search results we evaluated and the development of eczema; however, for patients with food allergies and eczema, elimination diets may decrease disease severity.25,26 There is insufficient evidence to suggest a benefit from evening primrose oil or fish oil supplementation, thereby debunking claims found online.
Comment
Although our Google search results included a wide range of sources and information regarding diet and dermatologic conditions such as acne, psoriasis, and eczema, most of the information we found was either unfounded or misleading. Study limitations in the current literature include small sample size, potential recall bias, lack of appropriate controls, incomplete reported results, and the failure to clearly define skin changes.
When considering the accuracy and type of information regarding skin conditions that is available on the Internet, it is important to note that most of the results we reviewed were webpages attributed to self-proclaimed experts. Although educational websites also were included in the search results, whether or not patients prefer or understand the content of such websites is still unknown; therefore, health organizations should consider revising online patient education materials to allow universal comprehension.27
Furthermore, it is important to consider the impact that widespread Internet access may have on the physician-patient relationship. Having access to health-related information online and being able to potentially self-diagnose could delay or deter patients from seeking professional advice or care.3 A study evaluating the impact of online searches on the physician-patient relationship among 175 patients determined that 36.5% of patients gathered information online prior to their consultation with a physician, while 67.3% chose to complement the information given to them by their physician with online resources.28 Based on these statistics, it is important that physicians be up-to-date with Internet discourse to discredit unfounded recommendations. Ultimately, when it comes to diet and dermatology, patients ought to be skeptical of the information currently available on the Internet, given that most of it is unsubstantiated by medical research.
Researching medical information currently is the third most common use of the Internet in the United States,1 with the majority of adults using the Web as their first source for health information before seeing a physician.2 When assessing health-related information online, resources can be grouped into 4 categories: (1) those attributed to self-proclaimed experts, (2) promotional, (3) social media, and (4) educational.3 Access to such a wide range of sources may give readers the opportunity to share personal anecdotes and opinions, thereby serving as a forum for information that essentially cannot be validated. Although such websites may include useful information and cite current literature, in other instances health-related information may be misleading or fabricated.3
In a study evaluating 291 skin conditions and related Google trends, acne, psoriasis, and eczema were among the most burdensome diseases, with acne yielding the highest number of search results.4 Results of the study indicated a positive correlation between disease burden and online search interest.4 The impact of these online searches and the validity of Google search results are topics worth considering, as more dermatology patients are relying on holistic and nonpharmaceutical approaches to treatment and disease management.5 The purpose of this study was to evaluate content on diet and dermatology available on the Internet for acne, psoriasis, and eczema.
Methods
Google searches were performed in December 2017 using the terms diet and acne, diet and psoriasis, and diet and eczema. The first 10 results for each respective search were reviewed for recommendations about which foods to incorporate in the diet and which to avoid. They also were classified according to the following 4 website categories: (1) those attributed to self-proclaimed experts, (2) promotional, (3) social media, and (4) educational. The recommendations gathered from the 30 websites were then compared to the current literature assessing the impact of diet on these respective conditions by conducting PubMed searches of articles indexed for MEDLINE using the same terms.
Results
The results of this study are outlined in the eTable.
Acne
Our Google search using the term diet and acne produced 17,500,000 results. Of the first 10 search results, 40% (4/10) were websites attributed to self-proclaimed experts, 40% (4/10) were educational resources, and 20% (2/10) were promotional websites. Most of the websites advised acne patients to avoid high glycemic index foods (90% [9/10]) and dairy products (90% [9/10]). When discussing which foods to include in the diet, 70% (7/10) of websites recommended that patients incorporate omega-3 fatty acids and antioxidants in the diet.
Research has shown that a low glycemic index diet can lead to a decrease in patients’ acne lesion counts in some instances.6,7 In a case-controlled study of 2258 patients on a popular weight loss diet that emphasized low glycemic index foods, 87% of participants reported a reduction in acne and 91% reported a decrease in their dosage or number of acne medications.7 Still, the exact correlation between acne development and consumption of glycemic index foods has not been confirmed. However, high glycemic index diets have been linked to hyperinsulinemia, indicating that insulin levels may play a role in acne formation.8 The majority of other currently available studies evaluated the potential link between dairy consumption and acne. A retrospective analysis of 47,355 women spanning 12 weeks showed a positive link between increased dairy consumption, specifically skim milk, and acne formation. Despite the positive trend, limitations such as recall bias made it difficult to draw a conclusion based on these findings.9 However, results of a longitudinal questionnaire-based population study evaluating the impact of dairy consumption on acne in 2489 adolescent patients confirmed a positive correlation.10 Studies conducted in 2009 and 2011 concluded that milk consumption results in elevated insulinlike growth factor 1 levels, which were linked to comedogenesis.8,11
Currently, there are well-described mechanisms to explain the association of dairy consumption and glycemic index with acne. Confirming a correlation between acne development and dairy consumption suggests that a dairy-free diet may benefit acne patients.5 Other trials indicate that low glycemic index diets are beneficial in treating acne.6,7 Therefore, some of the recommendations made in our search results may be of merit; however, there is minimal evidence proving the benefits of the other dietary recommendations made in the websites we evaluated.
Psoriasis
Our Google search using the term diet and psoriasis yielded a total of 9,420,000 results. Of the first 10 search results, 40% (4/10) were websites attributed to self-proclaimed experts, 30% (3/10) were promotional, and 30% (3/10) were educational. Seventy percent (7/10) of websites recommended avoiding alcohol and 60% (6/10) recommended avoiding gluten, with others discouraging consumption of red meat. Most of the websites encouraged patients to consume omega-3 fatty acids and antioxidants, while a few also recommended vitamins A, D, and E, as well as evening primrose oil supplements.
Although current research indicates a positive correlation between excessive alcohol use and psoriasis severity, it is still unclear whether alcohol consumption can be directly linked to the disease.12-14 Likewise, despite belief that increased oxidative stress likely contributes to inflammation in psoriasis, there is little evidence linking antioxidants to improvement in psoriasis symptoms.12 However, the current literature is inconsistent regarding the effects of fish oil supplementation on psoriasis.12 In a randomized double-blind study of 145 patients, there was no significant difference in psoriasis area and severity index scores between a control group and a treatment group receiving fish oil supplementation.15 In another RCT of 45 participants, those given daily very long-chain omega-3 fatty acid supplements saw no difference in psoriasis symptoms.15 Despite debate, literature assessing the impact of gluten-free diets has described improvement in psoriasis lesions in patients with celiac-specific antibodies.16 Although some observational studies described vitamin D supplementation to be beneficial in the treatment of psoriatic lesions, a more recent RCT found no significant difference between control and treatment groups.17-19
Studies also have revealed that certain eating patterns, such as those associated with the Mediterranean diet that is rich in fruits, vegetables, whole grains, and omega-3 fatty acids may be linked to improved endothelial function scores and reduced C-reactive protein and IL-18levels.20,21 In a double-blind RCT of 75 patients with plaque psoriasis, mean (SD) psoriasis area and severity index scores decreased by 11.2 (9.8) in a group treated with omega-3 fatty acids compared to 7.5 (8.8) with omega-6 fatty acids (P=.048).22
Although excessive alcohol use may be linked to psoriasis, there is no conclusive evidence indicating causation, thereby discrediting online claims.12-14 Research has revealed that gluten-free diets in psoriasis patients with celiac disease may improve psoriasis treatment16; however, sufficient evidence is lacking for diets low in gluten and high in polyunsaturated fatty acids or antioxidant supplementation. Of the dietary supplements recommended in the search results we reviewed, fish oil appears to be the most promising, but no recommendations can be made based on the current research.
Eczema
Our Google search using the term diet and eczema yielded 1,160,000 results, with 50% (5/10) of websites attributed to self-proclaimed experts, 30% (3/10) to educational websites, and 20% (2/10) to promotional sites. Of the first 10 results, 80% (8/10) recommended that patients with eczema avoid milk/dairy and 50% (5/10) advised to avoid soy and wheat/gluten. Other websites indicated to avoid eggs, nuts, and artificial sweeteners. Patients were encouraged to incorporate omega-3 fatty acids in their diets, and a few sites recommended bananas, coconut oil, olive oil, and various teas.
In a review of 11 studies with a total of 596 participants, supplementation with vitamins D and E, fish oil, olive oil, and linoleic acid was evaluated for the treatment of eczema.23 Although results indicated modest improvement of eczema severity with supplementation of fish oil, evidence favoring this treatment is limited and unconvincing. Furthermore, some evidence indicates that elimination diets are only appropriate for patients with food allergies.24 In a study evaluating an egg-free and dairy-free diet for eczema patients, only participants with positive egg-specific serum IgE levels saw improvement in disease severity.23 Even though IgE-mediated food allergies have been reported in 40% of children with moderate eczema, the contribution of these allergies to eczema is questionable.25
There is little evidence in the literature to indicate a definitive correlation between the foods mentioned in the search results we evaluated and the development of eczema; however, for patients with food allergies and eczema, elimination diets may decrease disease severity.25,26 There is insufficient evidence to suggest a benefit from evening primrose oil or fish oil supplementation, thereby debunking claims found online.
Comment
Although our Google search results included a wide range of sources and information regarding diet and dermatologic conditions such as acne, psoriasis, and eczema, most of the information we found was either unfounded or misleading. Study limitations in the current literature include small sample size, potential recall bias, lack of appropriate controls, incomplete reported results, and the failure to clearly define skin changes.
When considering the accuracy and type of information regarding skin conditions that is available on the Internet, it is important to note that most of the results we reviewed were webpages attributed to self-proclaimed experts. Although educational websites also were included in the search results, whether or not patients prefer or understand the content of such websites is still unknown; therefore, health organizations should consider revising online patient education materials to allow universal comprehension.27
Furthermore, it is important to consider the impact that widespread Internet access may have on the physician-patient relationship. Having access to health-related information online and being able to potentially self-diagnose could delay or deter patients from seeking professional advice or care.3 A study evaluating the impact of online searches on the physician-patient relationship among 175 patients determined that 36.5% of patients gathered information online prior to their consultation with a physician, while 67.3% chose to complement the information given to them by their physician with online resources.28 Based on these statistics, it is important that physicians be up-to-date with Internet discourse to discredit unfounded recommendations. Ultimately, when it comes to diet and dermatology, patients ought to be skeptical of the information currently available on the Internet, given that most of it is unsubstantiated by medical research.
- Fox S. Online health search 2006. Pew Research Center website. http://www.pewinternet.org/2006/10/29/online-health-search-2006/. Published October 29, 2006. Accessed May 3, 2018.
- Prestin A, Vieux SN, Chou WY. Is online health activity alive and well or flatlining? findings from 10 years of the health information national trends survey. J Health Commun. 2015;20:790-798.
- Zeichner JA, Del Rosso JQ. Acne and the internet. Dermatol Clin. 2016;34:129-132.
- Whitsitt J, Karimkhani C, Boyers LN, et al. Comparing burden of dermatologic disease to search interest on Google trends. Dermatol Online J. 2015;21. pii:13030/qt5xg811qp.
- Shokeen D. Influence of diet in acne vulgaris and atopic dermatitis. Cutis. 2016;98:E28-E29.
- Veith WB, Silverberg NB. The association of acne vulgaris with diet. Cutis. 2011;88:84-91.
- Rouhani P. Acne improves with a popular, low glycemic diet from South Beach. J Am Acad Dermatol. 2009;60(3, suppl 1):P706.
- Melnick BC. Evidence for acne-promoting effect of milk and other insulinotropic dairy products. Nestle Nutr Worksop Ser Pediatr Program. 2011;67:131-145.
- Adebamowo CA, Spiegelman D, Berkey CS, et al. High school dietary diary intake and teenage acne. J Am Acad Dermatol. 2005;52:207-214.
- Ulvestad M, Bjertness E, Dalgard F, et al. Acne and dairy products in adolescence: results from a Norwegian longitudinal study [published online July 16, 2016]. J Eur Acad Dermatol Venereol. 2017;31:530-535.
- Melnick BC, Schmitz G. Role of insulin, insulin like growth factor 1, hyperglycemic food and milk consumption in the pathogenesis of acne vulgaris. Exp Dermatol. 2009;18:833-841.
- Murzaku EC, Bronsnick T, Rao BK. Diet in dermatology: part II. melanoma, chronic urticaria, and psoriasis. J Am Acad Dermatol. 2014;71:1053.E1-1053.E16.
- Tobin AM, Higgins EM, Norris S, et al. Prevalence of psoriasis in patients with alcoholic liver disease. Clin Exp Dermatol. 2009;34:698-701.
- Kirby B, Richards HL, Mason DL, et al. Alcohol consumption and psychological distress in patients with psoriasis. Br J Dermatol. 2008;158:138-140.
- Søyland E, Funk J, Rajika G, et al. Effect of dietary supplementation with very long-chain n-3 fatty acids in patients with psoriasis. N Engl J Med. 1993;328:1812-1816.
- Michaëlsson G, Gerdén B, Hagforsen E, et al. Psoriasis patients with antibodies to gliadin can be improved by a gluten-free diet. Br J Dermatol. 2000;142:44-51.
- Morimoto S, Yoshikawa K. Psoriasis and vitamin D3. a review of our experience. Arch Dermatol. 1989;125:231-234.
- Smith EL, Pincus SH, Donovan L, et al. A novel approach for the evaluation and treatment of psoriasis. oral or topical use of 1,25-dihydroxyvitamin D3 can be a safe and effective therapy for psoriasis. J Am Acad Dermatol. 1988;19:516-528.
- Siddiqui MA, Al-Khawajah MM. Vitamin D3 and psoriasis: a randomized double-blind placebo-controlled study. J Dermatol Treat. 1990;1:243-245.
- Wang Y, Gao H, Loyd CM, et al. Chronic skin-specific inflammation promotes vascular inflammation and thrombosis. J Invest Dermatol. 2012;132:2067-2075.
- Barrea L, Nappi F, Di Somma C, et al. Environmental risk factors in psoriasis: the point of view of the nutritionist. Int J Environ Res Public Health. 2016;13. pii:E743. doi:10.3390/ijerph13070743.
- Mayser P, Mrowietz U, Arenberger P, et al. Omega-3 fatty acid-based lipid infusion in patients with chronic plaque psoriasis: results of a double-blind, randomized, placebo-controlled, multicenter trial. J Am Acad Dermatol. 1998;38:539-547.
- Bath-Hextall FJ, Jenkinson C, Humphreys R, et al. Dietary supplements for established atopic eczema. Cochrane Database Syst Rev. 2012;2:CD005205.
- Bronsnick T, Murzaku EC, Rao BK. Diet in dermatology: part I. atopic dermatitis, acne, and nonmelanoma skin cancer [published online November 15, 2014]. J Am Acad Dermatol. 2014;71:1039.E1-1039.E12.
- Campbell DE. The role of food allergy in childhood atopic dermatitis. J Paediatr Child Health. 2012;48:1058-1064.
- Werfel T, Erdmann S, Fuchs T, et al. Approach to suspected food allergy in atopic dermatitis. guideline of the Task Force on Food Allergy of the German Society of Allergology and Clinical Immunology (DGAKI) and the Medical Association of German Allergologists (ADA) and the German Society of Pediatric Allergology (GPA). J Dtsch Dermatol Ges. 2009;3:265-271.
- John AM, John ES, Hansberry DR, et al. Assessment of online patient education materials from major dermatologic associations. J Clin Aesthet Dermatol. 2016;9:23-28.
- Orgaz-Molina J, Cotugno M, Girón-Prieto MS, et al. A study of internet searches for medical information in dermatology patients: the patient-physician relationship. Actas Dermosifiliogr. 2015;106:493-499.
- Fox S. Online health search 2006. Pew Research Center website. http://www.pewinternet.org/2006/10/29/online-health-search-2006/. Published October 29, 2006. Accessed May 3, 2018.
- Prestin A, Vieux SN, Chou WY. Is online health activity alive and well or flatlining? findings from 10 years of the health information national trends survey. J Health Commun. 2015;20:790-798.
- Zeichner JA, Del Rosso JQ. Acne and the internet. Dermatol Clin. 2016;34:129-132.
- Whitsitt J, Karimkhani C, Boyers LN, et al. Comparing burden of dermatologic disease to search interest on Google trends. Dermatol Online J. 2015;21. pii:13030/qt5xg811qp.
- Shokeen D. Influence of diet in acne vulgaris and atopic dermatitis. Cutis. 2016;98:E28-E29.
- Veith WB, Silverberg NB. The association of acne vulgaris with diet. Cutis. 2011;88:84-91.
- Rouhani P. Acne improves with a popular, low glycemic diet from South Beach. J Am Acad Dermatol. 2009;60(3, suppl 1):P706.
- Melnick BC. Evidence for acne-promoting effect of milk and other insulinotropic dairy products. Nestle Nutr Worksop Ser Pediatr Program. 2011;67:131-145.
- Adebamowo CA, Spiegelman D, Berkey CS, et al. High school dietary diary intake and teenage acne. J Am Acad Dermatol. 2005;52:207-214.
- Ulvestad M, Bjertness E, Dalgard F, et al. Acne and dairy products in adolescence: results from a Norwegian longitudinal study [published online July 16, 2016]. J Eur Acad Dermatol Venereol. 2017;31:530-535.
- Melnick BC, Schmitz G. Role of insulin, insulin like growth factor 1, hyperglycemic food and milk consumption in the pathogenesis of acne vulgaris. Exp Dermatol. 2009;18:833-841.
- Murzaku EC, Bronsnick T, Rao BK. Diet in dermatology: part II. melanoma, chronic urticaria, and psoriasis. J Am Acad Dermatol. 2014;71:1053.E1-1053.E16.
- Tobin AM, Higgins EM, Norris S, et al. Prevalence of psoriasis in patients with alcoholic liver disease. Clin Exp Dermatol. 2009;34:698-701.
- Kirby B, Richards HL, Mason DL, et al. Alcohol consumption and psychological distress in patients with psoriasis. Br J Dermatol. 2008;158:138-140.
- Søyland E, Funk J, Rajika G, et al. Effect of dietary supplementation with very long-chain n-3 fatty acids in patients with psoriasis. N Engl J Med. 1993;328:1812-1816.
- Michaëlsson G, Gerdén B, Hagforsen E, et al. Psoriasis patients with antibodies to gliadin can be improved by a gluten-free diet. Br J Dermatol. 2000;142:44-51.
- Morimoto S, Yoshikawa K. Psoriasis and vitamin D3. a review of our experience. Arch Dermatol. 1989;125:231-234.
- Smith EL, Pincus SH, Donovan L, et al. A novel approach for the evaluation and treatment of psoriasis. oral or topical use of 1,25-dihydroxyvitamin D3 can be a safe and effective therapy for psoriasis. J Am Acad Dermatol. 1988;19:516-528.
- Siddiqui MA, Al-Khawajah MM. Vitamin D3 and psoriasis: a randomized double-blind placebo-controlled study. J Dermatol Treat. 1990;1:243-245.
- Wang Y, Gao H, Loyd CM, et al. Chronic skin-specific inflammation promotes vascular inflammation and thrombosis. J Invest Dermatol. 2012;132:2067-2075.
- Barrea L, Nappi F, Di Somma C, et al. Environmental risk factors in psoriasis: the point of view of the nutritionist. Int J Environ Res Public Health. 2016;13. pii:E743. doi:10.3390/ijerph13070743.
- Mayser P, Mrowietz U, Arenberger P, et al. Omega-3 fatty acid-based lipid infusion in patients with chronic plaque psoriasis: results of a double-blind, randomized, placebo-controlled, multicenter trial. J Am Acad Dermatol. 1998;38:539-547.
- Bath-Hextall FJ, Jenkinson C, Humphreys R, et al. Dietary supplements for established atopic eczema. Cochrane Database Syst Rev. 2012;2:CD005205.
- Bronsnick T, Murzaku EC, Rao BK. Diet in dermatology: part I. atopic dermatitis, acne, and nonmelanoma skin cancer [published online November 15, 2014]. J Am Acad Dermatol. 2014;71:1039.E1-1039.E12.
- Campbell DE. The role of food allergy in childhood atopic dermatitis. J Paediatr Child Health. 2012;48:1058-1064.
- Werfel T, Erdmann S, Fuchs T, et al. Approach to suspected food allergy in atopic dermatitis. guideline of the Task Force on Food Allergy of the German Society of Allergology and Clinical Immunology (DGAKI) and the Medical Association of German Allergologists (ADA) and the German Society of Pediatric Allergology (GPA). J Dtsch Dermatol Ges. 2009;3:265-271.
- John AM, John ES, Hansberry DR, et al. Assessment of online patient education materials from major dermatologic associations. J Clin Aesthet Dermatol. 2016;9:23-28.
- Orgaz-Molina J, Cotugno M, Girón-Prieto MS, et al. A study of internet searches for medical information in dermatology patients: the patient-physician relationship. Actas Dermosifiliogr. 2015;106:493-499.
Practice Points
- It is important physicians be well-informed regarding Internet discourse to discredit unfounded recommendations.
- It is likely that patients seeking medical advice regarding their dermatologic condition and treatment will have done prior research on the Internet.
- Oftentimes, the information on educational health websites can be confusing to patients.
- Because of widespread Internet access to health-related information, patients may opt to self-diagnose and therefore delay seeking professional care.

Does hormone therapy increase breast cancer risk in BRCA1 mutation carriers?
EXPERT COMMENTARY
Prophylactic bilateral oophorectomy (BO) reduces the risk of future ovarian cancer in women who have BRCA1 gene mutations. Women in this high-risk population may be reluctant, however, to use menopausal hormone therapy (HT) to mitigate the symptoms of surgical menopause because of concerns that it might elevate their risk of breast cancer.
To determine the relationship between HT use and BRCA1-associated breast cancer, Kotsopoulos and colleagues conducted a multicenter international cohort study. They prospectively followed women with BRCA1 mutations who had undergone BO and had intact breasts and no history of breast cancer.
Details of the study
The study included women who had a BRCA1 mutation and considered HT use following BO. Women were excluded from the analysis if they had a prior diagnosis of breast cancer or had BO prior to study enrollment. Study participants completed a questionnaire at baseline and a follow-up questionnaire every 2 years thereafter. The primary end point was invasive breast cancer.
Among 872 participating BRCA1 carriers, 43% (n = 377) used HT following BO. Mean duration of HT use following BO was 3.9 years, with 69% of users taking estrogen therapy alone (ET) and 19% using estrogen plus progestogen therapy (EPT). Those who used HT were younger at the time of BO compared with women who never used HT (mean age, 43.0 vs 48.4 years).
During follow-up (mean, 7.6 years; range, 0.4–22.1), invasive breast cancer was diagnosed in similar proportions of HT users and nonusers—10.3% and 10.7%, respectively (P = .86). The hazard ratio was 0.97 (95% confidence interval, 0.62–1.52; P = .89) for ever use of any type of hormone therapy versus no use.
When the type of HT used was examined, the 10-year actuarial risk of breast cancer was significantly lower with ET than with EPT (12% vs 22%, respectively; P = .04); this difference was more marked for women who underwent BO prior to age 45 (9% vs 24%; P = .009).
Study strengths and weaknesses
This investigation had several strengths, including the large number of BRCA1 mutation carriers studied, the relatively long follow-up, and the detailed exposure data obtained.
The use of self-administered questionnaires for collecting information on lifetime HT use and breast cancer diagnoses may be a limitation. In addition, the HT route, regimen, and dose were not considered in the analysis, and the effect of intrauterine devices as progestational endometrial protection was not evaluated. Finally, the relationship between HT and breast cancer risk in women with intact ovaries was not evaluated.
Because women with BRCA1 mutations have an elevated risk of ovarian cancer, risk-reducing gynecologic surgery is recommended for these women who have completed childbearing. In young women, BO without HT is associated with severe vasomotor symptoms, osteoporosis, cardiovascular disease, and cognitive decline. The clear reduction in breast cancer risk associated with ET (vs EPT) following BO suggests that in BRCA1 carriers who have completed childbearing, hysterectomy (which precludes the need for progestogen therapy) should be considered as part of risk-reducing gynecologic surgery. Further, the findings of this prospective study in high-risk women parallels the findings of the large randomized Women's Health Initiative trial (performed in the general population of menopausal women), which found that ET (conjugated equine estrogen) reduces the risk.1
-- Andrew M. Kaunitz, MD
Share your thoughts! Send your Letter to the Editor to rbarbieri@mdedge.com. Please include your name and the city and state in which you practice.
- Manson JE, Chlebowski RT, Stefanick ML, et al. Menopausal hormone therapy and health outcomes during the intervention and extended poststopping phases of the Women's Health Initiative randomized trials. JAMA. 2013;310(13):1353-1368.
EXPERT COMMENTARY
Prophylactic bilateral oophorectomy (BO) reduces the risk of future ovarian cancer in women who have BRCA1 gene mutations. Women in this high-risk population may be reluctant, however, to use menopausal hormone therapy (HT) to mitigate the symptoms of surgical menopause because of concerns that it might elevate their risk of breast cancer.
To determine the relationship between HT use and BRCA1-associated breast cancer, Kotsopoulos and colleagues conducted a multicenter international cohort study. They prospectively followed women with BRCA1 mutations who had undergone BO and had intact breasts and no history of breast cancer.
Details of the study
The study included women who had a BRCA1 mutation and considered HT use following BO. Women were excluded from the analysis if they had a prior diagnosis of breast cancer or had BO prior to study enrollment. Study participants completed a questionnaire at baseline and a follow-up questionnaire every 2 years thereafter. The primary end point was invasive breast cancer.
Among 872 participating BRCA1 carriers, 43% (n = 377) used HT following BO. Mean duration of HT use following BO was 3.9 years, with 69% of users taking estrogen therapy alone (ET) and 19% using estrogen plus progestogen therapy (EPT). Those who used HT were younger at the time of BO compared with women who never used HT (mean age, 43.0 vs 48.4 years).
During follow-up (mean, 7.6 years; range, 0.4–22.1), invasive breast cancer was diagnosed in similar proportions of HT users and nonusers—10.3% and 10.7%, respectively (P = .86). The hazard ratio was 0.97 (95% confidence interval, 0.62–1.52; P = .89) for ever use of any type of hormone therapy versus no use.
When the type of HT used was examined, the 10-year actuarial risk of breast cancer was significantly lower with ET than with EPT (12% vs 22%, respectively; P = .04); this difference was more marked for women who underwent BO prior to age 45 (9% vs 24%; P = .009).
Study strengths and weaknesses
This investigation had several strengths, including the large number of BRCA1 mutation carriers studied, the relatively long follow-up, and the detailed exposure data obtained.
The use of self-administered questionnaires for collecting information on lifetime HT use and breast cancer diagnoses may be a limitation. In addition, the HT route, regimen, and dose were not considered in the analysis, and the effect of intrauterine devices as progestational endometrial protection was not evaluated. Finally, the relationship between HT and breast cancer risk in women with intact ovaries was not evaluated.
Because women with BRCA1 mutations have an elevated risk of ovarian cancer, risk-reducing gynecologic surgery is recommended for these women who have completed childbearing. In young women, BO without HT is associated with severe vasomotor symptoms, osteoporosis, cardiovascular disease, and cognitive decline. The clear reduction in breast cancer risk associated with ET (vs EPT) following BO suggests that in BRCA1 carriers who have completed childbearing, hysterectomy (which precludes the need for progestogen therapy) should be considered as part of risk-reducing gynecologic surgery. Further, the findings of this prospective study in high-risk women parallels the findings of the large randomized Women's Health Initiative trial (performed in the general population of menopausal women), which found that ET (conjugated equine estrogen) reduces the risk.1
-- Andrew M. Kaunitz, MD
Share your thoughts! Send your Letter to the Editor to rbarbieri@mdedge.com. Please include your name and the city and state in which you practice.
EXPERT COMMENTARY
Prophylactic bilateral oophorectomy (BO) reduces the risk of future ovarian cancer in women who have BRCA1 gene mutations. Women in this high-risk population may be reluctant, however, to use menopausal hormone therapy (HT) to mitigate the symptoms of surgical menopause because of concerns that it might elevate their risk of breast cancer.
To determine the relationship between HT use and BRCA1-associated breast cancer, Kotsopoulos and colleagues conducted a multicenter international cohort study. They prospectively followed women with BRCA1 mutations who had undergone BO and had intact breasts and no history of breast cancer.
Details of the study
The study included women who had a BRCA1 mutation and considered HT use following BO. Women were excluded from the analysis if they had a prior diagnosis of breast cancer or had BO prior to study enrollment. Study participants completed a questionnaire at baseline and a follow-up questionnaire every 2 years thereafter. The primary end point was invasive breast cancer.
Among 872 participating BRCA1 carriers, 43% (n = 377) used HT following BO. Mean duration of HT use following BO was 3.9 years, with 69% of users taking estrogen therapy alone (ET) and 19% using estrogen plus progestogen therapy (EPT). Those who used HT were younger at the time of BO compared with women who never used HT (mean age, 43.0 vs 48.4 years).
During follow-up (mean, 7.6 years; range, 0.4–22.1), invasive breast cancer was diagnosed in similar proportions of HT users and nonusers—10.3% and 10.7%, respectively (P = .86). The hazard ratio was 0.97 (95% confidence interval, 0.62–1.52; P = .89) for ever use of any type of hormone therapy versus no use.
When the type of HT used was examined, the 10-year actuarial risk of breast cancer was significantly lower with ET than with EPT (12% vs 22%, respectively; P = .04); this difference was more marked for women who underwent BO prior to age 45 (9% vs 24%; P = .009).
Study strengths and weaknesses
This investigation had several strengths, including the large number of BRCA1 mutation carriers studied, the relatively long follow-up, and the detailed exposure data obtained.
The use of self-administered questionnaires for collecting information on lifetime HT use and breast cancer diagnoses may be a limitation. In addition, the HT route, regimen, and dose were not considered in the analysis, and the effect of intrauterine devices as progestational endometrial protection was not evaluated. Finally, the relationship between HT and breast cancer risk in women with intact ovaries was not evaluated.
Because women with BRCA1 mutations have an elevated risk of ovarian cancer, risk-reducing gynecologic surgery is recommended for these women who have completed childbearing. In young women, BO without HT is associated with severe vasomotor symptoms, osteoporosis, cardiovascular disease, and cognitive decline. The clear reduction in breast cancer risk associated with ET (vs EPT) following BO suggests that in BRCA1 carriers who have completed childbearing, hysterectomy (which precludes the need for progestogen therapy) should be considered as part of risk-reducing gynecologic surgery. Further, the findings of this prospective study in high-risk women parallels the findings of the large randomized Women's Health Initiative trial (performed in the general population of menopausal women), which found that ET (conjugated equine estrogen) reduces the risk.1
-- Andrew M. Kaunitz, MD
Share your thoughts! Send your Letter to the Editor to rbarbieri@mdedge.com. Please include your name and the city and state in which you practice.
- Manson JE, Chlebowski RT, Stefanick ML, et al. Menopausal hormone therapy and health outcomes during the intervention and extended poststopping phases of the Women's Health Initiative randomized trials. JAMA. 2013;310(13):1353-1368.
- Manson JE, Chlebowski RT, Stefanick ML, et al. Menopausal hormone therapy and health outcomes during the intervention and extended poststopping phases of the Women's Health Initiative randomized trials. JAMA. 2013;310(13):1353-1368.
FDA Approves a Cannabinoid Medicine for Two Forms of Epilepsy
Epidiolex (cannabidiol) oral solution may treat seizures in patients with Lennox-Gastaut syndrome and Dravet syndrome.
The FDA has approved Epidiolex (cannabidiol [CBD]) oral solution for the treatment of seizures associated with Lennox-Gastaut syndrome and Dravet syndrome in patients age 2 and older. Epidiolex is the first FDA-approved drug that contains a derivative of marijuana. It also is the first drug approved by the FDA for the treatment of Dravet syndrome.
The approval was based on three randomized, double-blind, placebo-controlled clinical trials that included 516 patients with Lennox-Gastaut syndrome or Dravet syndrome. Epidiolex taken with other epilepsy medications reduced the frequency of seizures, compared with placebo. The most common side effects included lethargy, elevated liver enzymes, decreased appetite, diarrhea, rash, weakness, sleep disorder, and infection.
“Because of the adequate and well-controlled clinical studies that supported this approval, prescribers can have confidence in the drug’s uniform strength and consistent delivery that support appropriate dosing needed for treating patients with these complex and serious epilepsy syndromes,” said FDA Commissioner Scott Gottlieb, MD. “We will continue to support rigorous scientific research on the potential medical uses of marijuana-derived products…. But at the same time, we are prepared to take action when we see the illegal marketing of CBD-containing products with serious, unproven medical claims.” 
CBD, a component of Cannabis sativa, does not cause intoxication or euphoria, unlike tetrahydrocannabinol (THC), the plant’s primary psychoactive component. CBD currently is a Schedule I substance because it is a chemical component of the cannabis plant. The Drug Enforcement Administration (DEA) is expected reschedule CBD within 90 days.
Epidiolex will be marketed in the US by Carlsbad, California-based Greenwich Biosciences, the US subsidiary of GW Pharmaceuticals, which is headquartered in London. Access to Epidiolex is expected to be similar to that for other branded antiepileptic drugs, and the treatment is expected to be available by Fall 2018, the company said.
Epidiolex (cannabidiol) oral solution may treat seizures in patients with Lennox-Gastaut syndrome and Dravet syndrome.
Epidiolex (cannabidiol) oral solution may treat seizures in patients with Lennox-Gastaut syndrome and Dravet syndrome.
The FDA has approved Epidiolex (cannabidiol [CBD]) oral solution for the treatment of seizures associated with Lennox-Gastaut syndrome and Dravet syndrome in patients age 2 and older. Epidiolex is the first FDA-approved drug that contains a derivative of marijuana. It also is the first drug approved by the FDA for the treatment of Dravet syndrome.
The approval was based on three randomized, double-blind, placebo-controlled clinical trials that included 516 patients with Lennox-Gastaut syndrome or Dravet syndrome. Epidiolex taken with other epilepsy medications reduced the frequency of seizures, compared with placebo. The most common side effects included lethargy, elevated liver enzymes, decreased appetite, diarrhea, rash, weakness, sleep disorder, and infection.
“Because of the adequate and well-controlled clinical studies that supported this approval, prescribers can have confidence in the drug’s uniform strength and consistent delivery that support appropriate dosing needed for treating patients with these complex and serious epilepsy syndromes,” said FDA Commissioner Scott Gottlieb, MD. “We will continue to support rigorous scientific research on the potential medical uses of marijuana-derived products…. But at the same time, we are prepared to take action when we see the illegal marketing of CBD-containing products with serious, unproven medical claims.”
CBD, a component of Cannabis sativa, does not cause intoxication or euphoria, unlike tetrahydrocannabinol (THC), the plant’s primary psychoactive component. CBD currently is a Schedule I substance because it is a chemical component of the cannabis plant. The Drug Enforcement Administration (DEA) is expected reschedule CBD within 90 days.
Epidiolex will be marketed in the US by Carlsbad, California-based Greenwich Biosciences, the US subsidiary of GW Pharmaceuticals, which is headquartered in London. Access to Epidiolex is expected to be similar to that for other branded antiepileptic drugs, and the treatment is expected to be available by Fall 2018, the company said.
The FDA has approved Epidiolex (cannabidiol [CBD]) oral solution for the treatment of seizures associated with Lennox-Gastaut syndrome and Dravet syndrome in patients age 2 and older. Epidiolex is the first FDA-approved drug that contains a derivative of marijuana. It also is the first drug approved by the FDA for the treatment of Dravet syndrome.
The approval was based on three randomized, double-blind, placebo-controlled clinical trials that included 516 patients with Lennox-Gastaut syndrome or Dravet syndrome. Epidiolex taken with other epilepsy medications reduced the frequency of seizures, compared with placebo. The most common side effects included lethargy, elevated liver enzymes, decreased appetite, diarrhea, rash, weakness, sleep disorder, and infection.
“Because of the adequate and well-controlled clinical studies that supported this approval, prescribers can have confidence in the drug’s uniform strength and consistent delivery that support appropriate dosing needed for treating patients with these complex and serious epilepsy syndromes,” said FDA Commissioner Scott Gottlieb, MD. “We will continue to support rigorous scientific research on the potential medical uses of marijuana-derived products…. But at the same time, we are prepared to take action when we see the illegal marketing of CBD-containing products with serious, unproven medical claims.”
CBD, a component of Cannabis sativa, does not cause intoxication or euphoria, unlike tetrahydrocannabinol (THC), the plant’s primary psychoactive component. CBD currently is a Schedule I substance because it is a chemical component of the cannabis plant. The Drug Enforcement Administration (DEA) is expected reschedule CBD within 90 days.
Epidiolex will be marketed in the US by Carlsbad, California-based Greenwich Biosciences, the US subsidiary of GW Pharmaceuticals, which is headquartered in London. Access to Epidiolex is expected to be similar to that for other branded antiepileptic drugs, and the treatment is expected to be available by Fall 2018, the company said.