User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Exuberant Lymphomatoid Papulosis of the Head and Upper Trunk
To the Editor:
Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder. This disease affects patients of all ages but most commonly presents in the fifth decade with a slight male predominance.1 The estimated worldwide incidence is 1.2 to 1.9 cases per 1,000,000 individuals, and the 10-year survival rate is close to 100%.1 Clinically, LyP presents as a few to more than 100 red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution. They most commonly are distributed on the trunk and extremities; however, the face, scalp, and oral mucosa rarely may be involved. Each lesion may last on average 3 to 8 weeks, with residual hyperpigmentation or hypopigmentation of the skin or superficial varioliform scars. The clinical characteristic of spontaneous regression is crucial for distinguishing LyP from other forms of cutaneous lymphoma.2 The disease course is variable, lasting anywhere from a few months to decades. Histopathologically, LyP consists of a frequently CD30+ lymphocytic proliferation in multiple described patterns.1 We report a case of LyP in a patient who initially presented with pink edematous papules and vesicles that progressed to crusted ulcerations, nodules, and deep necrotic eschars on the scalp, neck, and upper trunk. Multiple biopsies and T-cell gene rearrangement studies were necessary to make the diagnosis.
A 73-year-old man presented with edematous crusted papules and nodules as well as scarring with serous drainage on the scalp and upper trunk of several months’ duration. He also reported pain and pruritus. He had a medical history of B-cell CD20− chronic lymphocytic leukemia (CLL) that was treated with fludarabine, cyclophosphamide, rituximab, and intravenous immunoglobulin approximately one year prior and currently was in remission; prostate cancer treated with prostatectomy; hypertension; and type 2 diabetes mellitus. His medications included metoprolol, valsartan, and glipizide.
Histopathology revealed a hypersensitivity reaction, and the clinicopathologic correlation was believed to represent an exuberant arthropod bite reaction in the setting of CLL. The eruption responded well to oral prednisone and topical corticosteroids but recurred when the medications were withdrawn. A repeat biopsy resulted in a diagnosis of atypical eosinophil-predominant Sweet syndrome. The condition resolved.
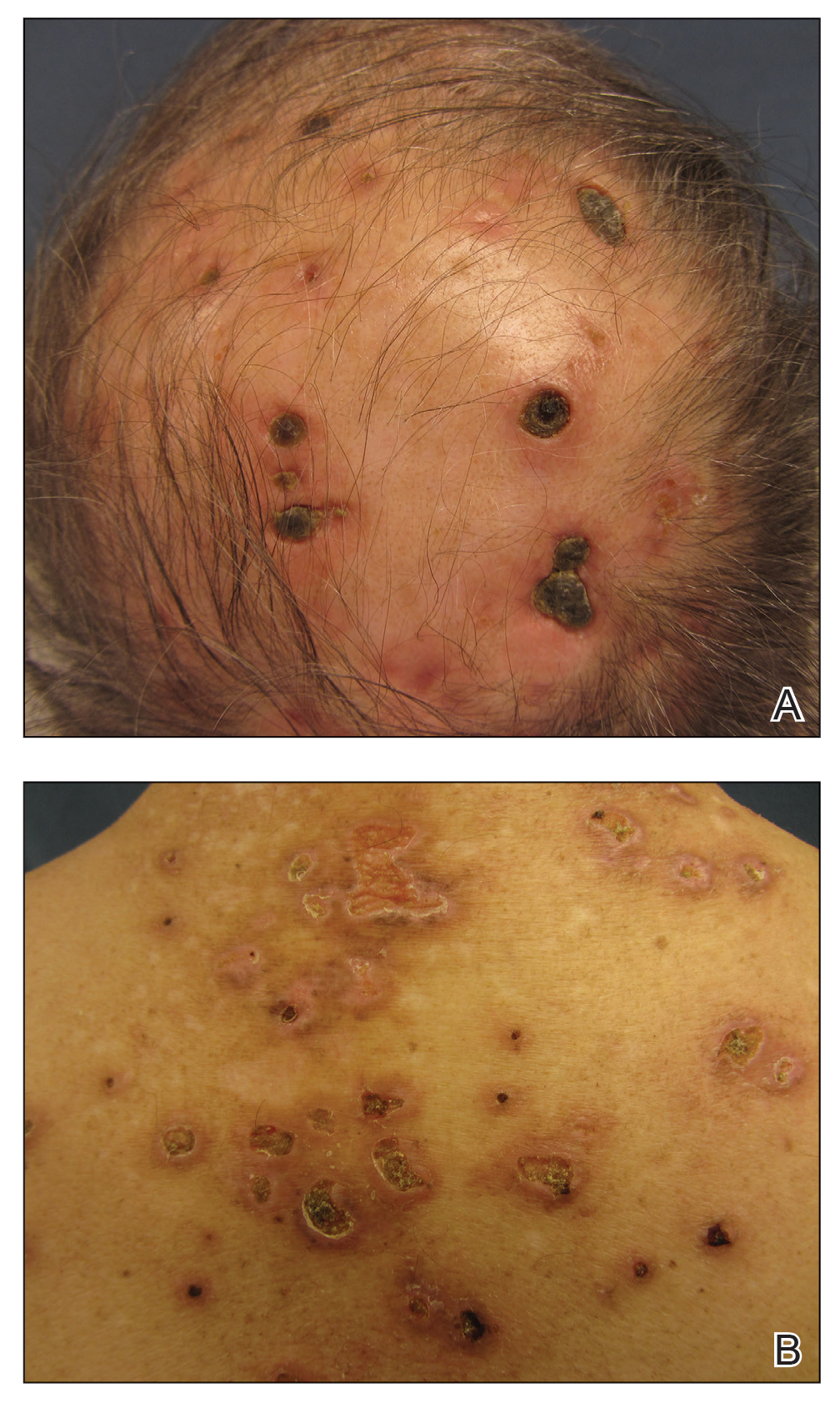
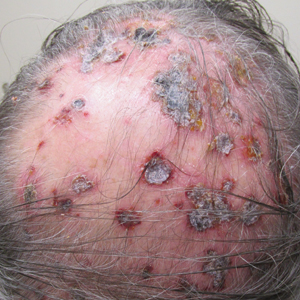
Three years later he developed multiple honey-crusted, superficial ulcers as well as serous, fluid-filled vesiculobullae on the head. A tissue culture revealed Proteus mirabilis, Staphylococcus aureus, and Enterococcus faecalis, and was negative for acid-fast bacteria and fungus. Biopsy of these lesions revealed dermal ulceration with a mixed inflammatory infiltrate and numerous eosinophils as well as a few clustered CD30+ cells; direct immunofluorescence was negative. An extensive laboratory workup including bullous pemphigoid antigens, C-reactive protein, antinuclear antibodies comprehensive profile, antineutrophil cytoplasmic antibodies, rheumatoid factor, anticyclic citrullinated peptide antibodies, serum protein electrophoresis, lactate dehydrogenase, complete blood cell count with differential, complete metabolic profile, thyroid-stimulating hormone, uric acid, C3, C4, immunoglobulin profile, angiotensin-converting enzyme level, and urinalysis was unremarkable. He improved with courses of minocycline, prednisone, and topical clobetasol, but he had periodic and progressive flares over several months with punched-out crusted ulcerations developing on the scalp (Figure 1A) and neck (Figure 1B). The oral and ocular mucosae were uninvolved, but the nasal mucosa had some involvement.
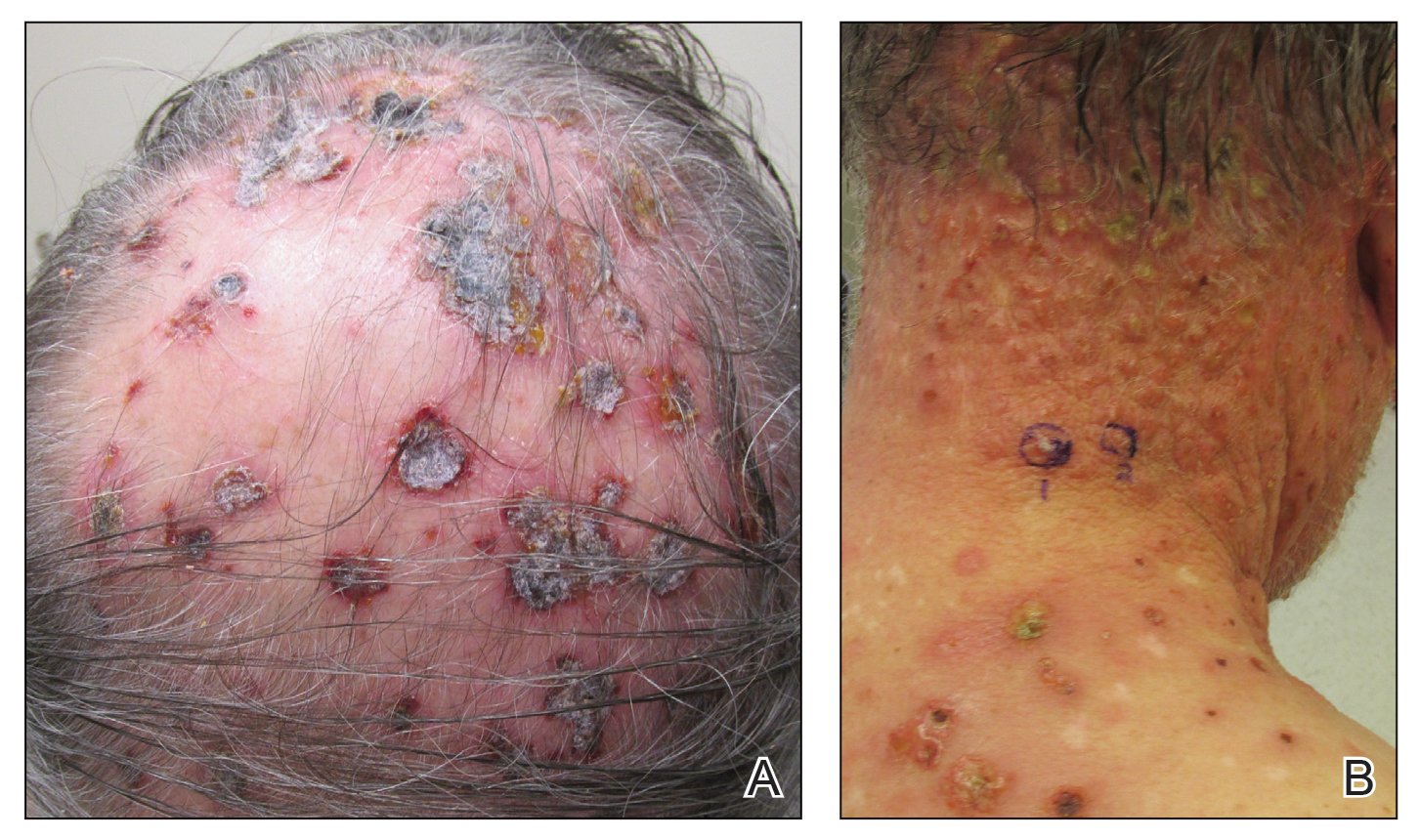
A repeat biopsy demonstrated an atypical CD30+ lymphoid infiltrate favoring LyP. T-cell clonality performed on this specimen and the prior biopsy demonstrated identical T-cell receptor β and γ clones. CD3, CD5, CD7, and CD4 immunostains highlighted the perivascular, perifollicular, and folliculotropic lymphocytic infiltrate. CD8 highlighted occasional background small T cells with only a few folliculotropic forms. A CD30 study revealed several scattered enlarged lymphocytes, and CD20 displayed a few dispersed B cells. A repeat perilesional direct immunofluorescence study was again negative. With treatment, he later formed multiple dry punched-out ulcers with dark eschars on the scalp, posterior neck, and upper back. There were multiple scars on the head, chest, and back, and no vesicles or bullae were present (Figure 2). The patient was presented at a meeting of the Philadelphia Dermatological Society and a consensus diagnosis of LyP was reached. The patient has continued to improve with oral minocycline 100 mg twice daily, topical clobetasol, and topical mupirocin.
Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy, with some disagreement in the literature concerning the exact percentage.3 In some studies, lymphoma has been estimated to occur in less than 20% of cases.4,5 Wieser et al1 reported a retrospective analysis of 180 patients with LyP that revealed a secondary malignancy in 52% of patients. They also reported that the number of lesions and the symptom severity were not associated with lymphoma development.1 Similarly, Cordel et al6 reported a diagnosis of lymphoma in 41% of 106 patients. These analyses reveal that the association with lymphoma may be higher than previously thought, but referral bias may be a confounding factor in these numbers.1,5,6 Associated malignancies may occur prior to, concomitantly, or years after the diagnosis of LyP. The most frequently reported malignancies include mycosis fungoides, Hodgkin lymphoma, and primary cutaneous anaplastic large cell lymphoma.1,4
Nicolaou et al3 indicated that head involvement was more likely associated with lymphoma. Our patient had a history of CLL prior to the development of LyP, and it continues to be in remission. The incidence of CLL in patients with LyP is reported to be 0.8%.4 Our patient had an exuberant case of LyP predominantly involving the head, neck, and upper torso, which is an unusual distribution. Vesiculobullous lesions also are uncharacteristic of LyP and may have represented concomitant bullous impetigo, but bullous variants of LyP also have been reported.7 Due to the unique distribution and characteristic scarring, Brunsting-Perry cicatricial pemphigoid also was considered in the clinical differential diagnosis.
The pathogenesis of LyP associated with malignancy is not definitively known. Theories propose that progression to a malignant clonal T-cell population may come from cytogenetic events, inadequate host response, or persistent antigenic or viral stimulation.4 Studies have demonstrated overlapping T-cell receptor gene rearrangement clones in lesions in patients with both LyP and mycosis fungoides, suggesting a common origin between the diseases.8 Other theories suggest that LyP may arise from an early, reactive, polyclonal lymphoid expansion that evolves into a clonal neoplastic process.4 Interestingly, LyP is a clonal T-cell disorder, while Hodgkin lymphoma and CLL are B-cell disorders. Thus, reports of CLL occurring with LyP, as in our patient, may support the theory that LyP arises from an early stem-cell or precursor-cell defect.4
There is no cure for LyP and data regarding the potential of aggressive therapy on the prevention of secondary lymphomas is lacking. Wieser et al1 reported that treatment did not prevent the progression to lymphoma in their retrospective analysis of 180 patients. The number of lesions, frequency of outbreaks, and extent of the scarring can dictate the treatment approach for LyP. Conservative topical therapies include corticosteroids, bexarotene, and imiquimod. Mupirocin may help to prevent infection of ulcerated lesions.1,2 Low-dose methotrexate has been shown to be the most efficacious treatment in reducing the number of lesions, particularly for scarring or cosmetically sensitive areas. Oral methotrexate at a dosage of 10 mg to 25 mg weekly tapered to the lowest effective dose may suppress outbreaks of LyP lesions.1,2 Other therapies include psoralen plus UVA, UVB, interferon alfa-2a, oral bexarotene, oral acyclovir or valacyclovir, etretinate, mycophenolic acid, photodynamic therapy, oral antibiotics, excision, and radiotherapy.1,2 Systemic chemotherapy and total-skin electron beam therapy have shown efficacy in clearing the lesions; however, the disease recurs after discontinuation of therapy.2 Systemic chemotherapy is not recommended for the treatment of LyP, as risks outweigh the benefits and it does not reduce the risk for developing lymphoma.1 The prognosis generally is good, though long-term follow-up is imperative to monitor for the development of other lymphomas.
Our patient presented with LyP a few months after completing chemotherapy for his CLL. It is unknown if he developed LyP just before the time of presentation, or if he may have developed it at the same time as his CLL by a common inciting event. In the latter case, it is speculative that the LyP may have been controlled by chemotherapy for his CLL, only to become clinically apparent after discontinuation, then naturally remit for a longer period. Case reports such as ours with unusual clinical presentations, B-cell lymphoma associations, and unique timing of lymphoma onset may help to provide insight into the pathogenesis of this disease.
We highlighted an unusual case of LyP that presented clinically with crusted ulcerations as well as vesiculobullous and edematous papules that progressed into deep punched-out ulcers with eschars, nodules, and scarring on the head and upper trunk. Lymphomatoid papulosis can be difficult to diagnose histopathologically at the early stages, and multiple repeat biopsies may be necessary to confirm the diagnosis. T-cell gene rearrangement and immunohistochemistry studies are helpful along with clinical correlation to establish a diagnosis in these cases. We recommend that physicians keep LyP on the differential diagnosis for patients with similar clinical presentations and remain vigilant in monitoring for the development of secondary lymphoma.
- Wieser I, Oh C, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Duvic M. CD30+ neoplasms of the skin. Curr Hematol Malig Rep. 2011;6:245-250.
- Nicolaou V, Papadavid E, Ekonomise A, et al. Association of clinicopathological characteristics with secondary neoplastic lymphoproliferative disorders in patients with lymphomatoid papulosis. Leuk Lymphoma. 2015;56:1303-1307.
- Ahn C, Orscheln C, Huang W. Lymphomatoid papulosis as a harbinger of chronic lymphocytic leukemia. Ann Hematol. 2014;93:1923-1925.
- Kunishige J, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-5781.
- Cordelet al. Frequency and risk factors for associated lymphomas in patients with lymphomatoid papulosis. Oncologist. 2016;21:76-83.
- Sureda N, Thomas L, Bathelier E, et al. Bullous lymphomatoid papulosis. Clin Exp Dermatol. 2011;36:800-801.
- de la Garza Bravo M, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin. Hum Pathol. 2015;46:558-569.
To the Editor:
Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder. This disease affects patients of all ages but most commonly presents in the fifth decade with a slight male predominance.1 The estimated worldwide incidence is 1.2 to 1.9 cases per 1,000,000 individuals, and the 10-year survival rate is close to 100%.1 Clinically, LyP presents as a few to more than 100 red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution. They most commonly are distributed on the trunk and extremities; however, the face, scalp, and oral mucosa rarely may be involved. Each lesion may last on average 3 to 8 weeks, with residual hyperpigmentation or hypopigmentation of the skin or superficial varioliform scars. The clinical characteristic of spontaneous regression is crucial for distinguishing LyP from other forms of cutaneous lymphoma.2 The disease course is variable, lasting anywhere from a few months to decades. Histopathologically, LyP consists of a frequently CD30+ lymphocytic proliferation in multiple described patterns.1 We report a case of LyP in a patient who initially presented with pink edematous papules and vesicles that progressed to crusted ulcerations, nodules, and deep necrotic eschars on the scalp, neck, and upper trunk. Multiple biopsies and T-cell gene rearrangement studies were necessary to make the diagnosis.
A 73-year-old man presented with edematous crusted papules and nodules as well as scarring with serous drainage on the scalp and upper trunk of several months’ duration. He also reported pain and pruritus. He had a medical history of B-cell CD20− chronic lymphocytic leukemia (CLL) that was treated with fludarabine, cyclophosphamide, rituximab, and intravenous immunoglobulin approximately one year prior and currently was in remission; prostate cancer treated with prostatectomy; hypertension; and type 2 diabetes mellitus. His medications included metoprolol, valsartan, and glipizide.
Histopathology revealed a hypersensitivity reaction, and the clinicopathologic correlation was believed to represent an exuberant arthropod bite reaction in the setting of CLL. The eruption responded well to oral prednisone and topical corticosteroids but recurred when the medications were withdrawn. A repeat biopsy resulted in a diagnosis of atypical eosinophil-predominant Sweet syndrome. The condition resolved.
Three years later he developed multiple honey-crusted, superficial ulcers as well as serous, fluid-filled vesiculobullae on the head. A tissue culture revealed Proteus mirabilis, Staphylococcus aureus, and Enterococcus faecalis, and was negative for acid-fast bacteria and fungus. Biopsy of these lesions revealed dermal ulceration with a mixed inflammatory infiltrate and numerous eosinophils as well as a few clustered CD30+ cells; direct immunofluorescence was negative. An extensive laboratory workup including bullous pemphigoid antigens, C-reactive protein, antinuclear antibodies comprehensive profile, antineutrophil cytoplasmic antibodies, rheumatoid factor, anticyclic citrullinated peptide antibodies, serum protein electrophoresis, lactate dehydrogenase, complete blood cell count with differential, complete metabolic profile, thyroid-stimulating hormone, uric acid, C3, C4, immunoglobulin profile, angiotensin-converting enzyme level, and urinalysis was unremarkable. He improved with courses of minocycline, prednisone, and topical clobetasol, but he had periodic and progressive flares over several months with punched-out crusted ulcerations developing on the scalp (Figure 1A) and neck (Figure 1B). The oral and ocular mucosae were uninvolved, but the nasal mucosa had some involvement.
A repeat biopsy demonstrated an atypical CD30+ lymphoid infiltrate favoring LyP. T-cell clonality performed on this specimen and the prior biopsy demonstrated identical T-cell receptor β and γ clones. CD3, CD5, CD7, and CD4 immunostains highlighted the perivascular, perifollicular, and folliculotropic lymphocytic infiltrate. CD8 highlighted occasional background small T cells with only a few folliculotropic forms. A CD30 study revealed several scattered enlarged lymphocytes, and CD20 displayed a few dispersed B cells. A repeat perilesional direct immunofluorescence study was again negative. With treatment, he later formed multiple dry punched-out ulcers with dark eschars on the scalp, posterior neck, and upper back. There were multiple scars on the head, chest, and back, and no vesicles or bullae were present (Figure 2). The patient was presented at a meeting of the Philadelphia Dermatological Society and a consensus diagnosis of LyP was reached. The patient has continued to improve with oral minocycline 100 mg twice daily, topical clobetasol, and topical mupirocin.
Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy, with some disagreement in the literature concerning the exact percentage.3 In some studies, lymphoma has been estimated to occur in less than 20% of cases.4,5 Wieser et al1 reported a retrospective analysis of 180 patients with LyP that revealed a secondary malignancy in 52% of patients. They also reported that the number of lesions and the symptom severity were not associated with lymphoma development.1 Similarly, Cordel et al6 reported a diagnosis of lymphoma in 41% of 106 patients. These analyses reveal that the association with lymphoma may be higher than previously thought, but referral bias may be a confounding factor in these numbers.1,5,6 Associated malignancies may occur prior to, concomitantly, or years after the diagnosis of LyP. The most frequently reported malignancies include mycosis fungoides, Hodgkin lymphoma, and primary cutaneous anaplastic large cell lymphoma.1,4
Nicolaou et al3 indicated that head involvement was more likely associated with lymphoma. Our patient had a history of CLL prior to the development of LyP, and it continues to be in remission. The incidence of CLL in patients with LyP is reported to be 0.8%.4 Our patient had an exuberant case of LyP predominantly involving the head, neck, and upper torso, which is an unusual distribution. Vesiculobullous lesions also are uncharacteristic of LyP and may have represented concomitant bullous impetigo, but bullous variants of LyP also have been reported.7 Due to the unique distribution and characteristic scarring, Brunsting-Perry cicatricial pemphigoid also was considered in the clinical differential diagnosis.
The pathogenesis of LyP associated with malignancy is not definitively known. Theories propose that progression to a malignant clonal T-cell population may come from cytogenetic events, inadequate host response, or persistent antigenic or viral stimulation.4 Studies have demonstrated overlapping T-cell receptor gene rearrangement clones in lesions in patients with both LyP and mycosis fungoides, suggesting a common origin between the diseases.8 Other theories suggest that LyP may arise from an early, reactive, polyclonal lymphoid expansion that evolves into a clonal neoplastic process.4 Interestingly, LyP is a clonal T-cell disorder, while Hodgkin lymphoma and CLL are B-cell disorders. Thus, reports of CLL occurring with LyP, as in our patient, may support the theory that LyP arises from an early stem-cell or precursor-cell defect.4
There is no cure for LyP and data regarding the potential of aggressive therapy on the prevention of secondary lymphomas is lacking. Wieser et al1 reported that treatment did not prevent the progression to lymphoma in their retrospective analysis of 180 patients. The number of lesions, frequency of outbreaks, and extent of the scarring can dictate the treatment approach for LyP. Conservative topical therapies include corticosteroids, bexarotene, and imiquimod. Mupirocin may help to prevent infection of ulcerated lesions.1,2 Low-dose methotrexate has been shown to be the most efficacious treatment in reducing the number of lesions, particularly for scarring or cosmetically sensitive areas. Oral methotrexate at a dosage of 10 mg to 25 mg weekly tapered to the lowest effective dose may suppress outbreaks of LyP lesions.1,2 Other therapies include psoralen plus UVA, UVB, interferon alfa-2a, oral bexarotene, oral acyclovir or valacyclovir, etretinate, mycophenolic acid, photodynamic therapy, oral antibiotics, excision, and radiotherapy.1,2 Systemic chemotherapy and total-skin electron beam therapy have shown efficacy in clearing the lesions; however, the disease recurs after discontinuation of therapy.2 Systemic chemotherapy is not recommended for the treatment of LyP, as risks outweigh the benefits and it does not reduce the risk for developing lymphoma.1 The prognosis generally is good, though long-term follow-up is imperative to monitor for the development of other lymphomas.
Our patient presented with LyP a few months after completing chemotherapy for his CLL. It is unknown if he developed LyP just before the time of presentation, or if he may have developed it at the same time as his CLL by a common inciting event. In the latter case, it is speculative that the LyP may have been controlled by chemotherapy for his CLL, only to become clinically apparent after discontinuation, then naturally remit for a longer period. Case reports such as ours with unusual clinical presentations, B-cell lymphoma associations, and unique timing of lymphoma onset may help to provide insight into the pathogenesis of this disease.
We highlighted an unusual case of LyP that presented clinically with crusted ulcerations as well as vesiculobullous and edematous papules that progressed into deep punched-out ulcers with eschars, nodules, and scarring on the head and upper trunk. Lymphomatoid papulosis can be difficult to diagnose histopathologically at the early stages, and multiple repeat biopsies may be necessary to confirm the diagnosis. T-cell gene rearrangement and immunohistochemistry studies are helpful along with clinical correlation to establish a diagnosis in these cases. We recommend that physicians keep LyP on the differential diagnosis for patients with similar clinical presentations and remain vigilant in monitoring for the development of secondary lymphoma.
To the Editor:
Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder. This disease affects patients of all ages but most commonly presents in the fifth decade with a slight male predominance.1 The estimated worldwide incidence is 1.2 to 1.9 cases per 1,000,000 individuals, and the 10-year survival rate is close to 100%.1 Clinically, LyP presents as a few to more than 100 red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution. They most commonly are distributed on the trunk and extremities; however, the face, scalp, and oral mucosa rarely may be involved. Each lesion may last on average 3 to 8 weeks, with residual hyperpigmentation or hypopigmentation of the skin or superficial varioliform scars. The clinical characteristic of spontaneous regression is crucial for distinguishing LyP from other forms of cutaneous lymphoma.2 The disease course is variable, lasting anywhere from a few months to decades. Histopathologically, LyP consists of a frequently CD30+ lymphocytic proliferation in multiple described patterns.1 We report a case of LyP in a patient who initially presented with pink edematous papules and vesicles that progressed to crusted ulcerations, nodules, and deep necrotic eschars on the scalp, neck, and upper trunk. Multiple biopsies and T-cell gene rearrangement studies were necessary to make the diagnosis.
A 73-year-old man presented with edematous crusted papules and nodules as well as scarring with serous drainage on the scalp and upper trunk of several months’ duration. He also reported pain and pruritus. He had a medical history of B-cell CD20− chronic lymphocytic leukemia (CLL) that was treated with fludarabine, cyclophosphamide, rituximab, and intravenous immunoglobulin approximately one year prior and currently was in remission; prostate cancer treated with prostatectomy; hypertension; and type 2 diabetes mellitus. His medications included metoprolol, valsartan, and glipizide.
Histopathology revealed a hypersensitivity reaction, and the clinicopathologic correlation was believed to represent an exuberant arthropod bite reaction in the setting of CLL. The eruption responded well to oral prednisone and topical corticosteroids but recurred when the medications were withdrawn. A repeat biopsy resulted in a diagnosis of atypical eosinophil-predominant Sweet syndrome. The condition resolved.
Three years later he developed multiple honey-crusted, superficial ulcers as well as serous, fluid-filled vesiculobullae on the head. A tissue culture revealed Proteus mirabilis, Staphylococcus aureus, and Enterococcus faecalis, and was negative for acid-fast bacteria and fungus. Biopsy of these lesions revealed dermal ulceration with a mixed inflammatory infiltrate and numerous eosinophils as well as a few clustered CD30+ cells; direct immunofluorescence was negative. An extensive laboratory workup including bullous pemphigoid antigens, C-reactive protein, antinuclear antibodies comprehensive profile, antineutrophil cytoplasmic antibodies, rheumatoid factor, anticyclic citrullinated peptide antibodies, serum protein electrophoresis, lactate dehydrogenase, complete blood cell count with differential, complete metabolic profile, thyroid-stimulating hormone, uric acid, C3, C4, immunoglobulin profile, angiotensin-converting enzyme level, and urinalysis was unremarkable. He improved with courses of minocycline, prednisone, and topical clobetasol, but he had periodic and progressive flares over several months with punched-out crusted ulcerations developing on the scalp (Figure 1A) and neck (Figure 1B). The oral and ocular mucosae were uninvolved, but the nasal mucosa had some involvement.
A repeat biopsy demonstrated an atypical CD30+ lymphoid infiltrate favoring LyP. T-cell clonality performed on this specimen and the prior biopsy demonstrated identical T-cell receptor β and γ clones. CD3, CD5, CD7, and CD4 immunostains highlighted the perivascular, perifollicular, and folliculotropic lymphocytic infiltrate. CD8 highlighted occasional background small T cells with only a few folliculotropic forms. A CD30 study revealed several scattered enlarged lymphocytes, and CD20 displayed a few dispersed B cells. A repeat perilesional direct immunofluorescence study was again negative. With treatment, he later formed multiple dry punched-out ulcers with dark eschars on the scalp, posterior neck, and upper back. There were multiple scars on the head, chest, and back, and no vesicles or bullae were present (Figure 2). The patient was presented at a meeting of the Philadelphia Dermatological Society and a consensus diagnosis of LyP was reached. The patient has continued to improve with oral minocycline 100 mg twice daily, topical clobetasol, and topical mupirocin.
Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy, with some disagreement in the literature concerning the exact percentage.3 In some studies, lymphoma has been estimated to occur in less than 20% of cases.4,5 Wieser et al1 reported a retrospective analysis of 180 patients with LyP that revealed a secondary malignancy in 52% of patients. They also reported that the number of lesions and the symptom severity were not associated with lymphoma development.1 Similarly, Cordel et al6 reported a diagnosis of lymphoma in 41% of 106 patients. These analyses reveal that the association with lymphoma may be higher than previously thought, but referral bias may be a confounding factor in these numbers.1,5,6 Associated malignancies may occur prior to, concomitantly, or years after the diagnosis of LyP. The most frequently reported malignancies include mycosis fungoides, Hodgkin lymphoma, and primary cutaneous anaplastic large cell lymphoma.1,4
Nicolaou et al3 indicated that head involvement was more likely associated with lymphoma. Our patient had a history of CLL prior to the development of LyP, and it continues to be in remission. The incidence of CLL in patients with LyP is reported to be 0.8%.4 Our patient had an exuberant case of LyP predominantly involving the head, neck, and upper torso, which is an unusual distribution. Vesiculobullous lesions also are uncharacteristic of LyP and may have represented concomitant bullous impetigo, but bullous variants of LyP also have been reported.7 Due to the unique distribution and characteristic scarring, Brunsting-Perry cicatricial pemphigoid also was considered in the clinical differential diagnosis.
The pathogenesis of LyP associated with malignancy is not definitively known. Theories propose that progression to a malignant clonal T-cell population may come from cytogenetic events, inadequate host response, or persistent antigenic or viral stimulation.4 Studies have demonstrated overlapping T-cell receptor gene rearrangement clones in lesions in patients with both LyP and mycosis fungoides, suggesting a common origin between the diseases.8 Other theories suggest that LyP may arise from an early, reactive, polyclonal lymphoid expansion that evolves into a clonal neoplastic process.4 Interestingly, LyP is a clonal T-cell disorder, while Hodgkin lymphoma and CLL are B-cell disorders. Thus, reports of CLL occurring with LyP, as in our patient, may support the theory that LyP arises from an early stem-cell or precursor-cell defect.4
There is no cure for LyP and data regarding the potential of aggressive therapy on the prevention of secondary lymphomas is lacking. Wieser et al1 reported that treatment did not prevent the progression to lymphoma in their retrospective analysis of 180 patients. The number of lesions, frequency of outbreaks, and extent of the scarring can dictate the treatment approach for LyP. Conservative topical therapies include corticosteroids, bexarotene, and imiquimod. Mupirocin may help to prevent infection of ulcerated lesions.1,2 Low-dose methotrexate has been shown to be the most efficacious treatment in reducing the number of lesions, particularly for scarring or cosmetically sensitive areas. Oral methotrexate at a dosage of 10 mg to 25 mg weekly tapered to the lowest effective dose may suppress outbreaks of LyP lesions.1,2 Other therapies include psoralen plus UVA, UVB, interferon alfa-2a, oral bexarotene, oral acyclovir or valacyclovir, etretinate, mycophenolic acid, photodynamic therapy, oral antibiotics, excision, and radiotherapy.1,2 Systemic chemotherapy and total-skin electron beam therapy have shown efficacy in clearing the lesions; however, the disease recurs after discontinuation of therapy.2 Systemic chemotherapy is not recommended for the treatment of LyP, as risks outweigh the benefits and it does not reduce the risk for developing lymphoma.1 The prognosis generally is good, though long-term follow-up is imperative to monitor for the development of other lymphomas.
Our patient presented with LyP a few months after completing chemotherapy for his CLL. It is unknown if he developed LyP just before the time of presentation, or if he may have developed it at the same time as his CLL by a common inciting event. In the latter case, it is speculative that the LyP may have been controlled by chemotherapy for his CLL, only to become clinically apparent after discontinuation, then naturally remit for a longer period. Case reports such as ours with unusual clinical presentations, B-cell lymphoma associations, and unique timing of lymphoma onset may help to provide insight into the pathogenesis of this disease.
We highlighted an unusual case of LyP that presented clinically with crusted ulcerations as well as vesiculobullous and edematous papules that progressed into deep punched-out ulcers with eschars, nodules, and scarring on the head and upper trunk. Lymphomatoid papulosis can be difficult to diagnose histopathologically at the early stages, and multiple repeat biopsies may be necessary to confirm the diagnosis. T-cell gene rearrangement and immunohistochemistry studies are helpful along with clinical correlation to establish a diagnosis in these cases. We recommend that physicians keep LyP on the differential diagnosis for patients with similar clinical presentations and remain vigilant in monitoring for the development of secondary lymphoma.
- Wieser I, Oh C, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Duvic M. CD30+ neoplasms of the skin. Curr Hematol Malig Rep. 2011;6:245-250.
- Nicolaou V, Papadavid E, Ekonomise A, et al. Association of clinicopathological characteristics with secondary neoplastic lymphoproliferative disorders in patients with lymphomatoid papulosis. Leuk Lymphoma. 2015;56:1303-1307.
- Ahn C, Orscheln C, Huang W. Lymphomatoid papulosis as a harbinger of chronic lymphocytic leukemia. Ann Hematol. 2014;93:1923-1925.
- Kunishige J, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-5781.
- Cordelet al. Frequency and risk factors for associated lymphomas in patients with lymphomatoid papulosis. Oncologist. 2016;21:76-83.
- Sureda N, Thomas L, Bathelier E, et al. Bullous lymphomatoid papulosis. Clin Exp Dermatol. 2011;36:800-801.
- de la Garza Bravo M, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin. Hum Pathol. 2015;46:558-569.
- Wieser I, Oh C, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Duvic M. CD30+ neoplasms of the skin. Curr Hematol Malig Rep. 2011;6:245-250.
- Nicolaou V, Papadavid E, Ekonomise A, et al. Association of clinicopathological characteristics with secondary neoplastic lymphoproliferative disorders in patients with lymphomatoid papulosis. Leuk Lymphoma. 2015;56:1303-1307.
- Ahn C, Orscheln C, Huang W. Lymphomatoid papulosis as a harbinger of chronic lymphocytic leukemia. Ann Hematol. 2014;93:1923-1925.
- Kunishige J, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-5781.
- Cordelet al. Frequency and risk factors for associated lymphomas in patients with lymphomatoid papulosis. Oncologist. 2016;21:76-83.
- Sureda N, Thomas L, Bathelier E, et al. Bullous lymphomatoid papulosis. Clin Exp Dermatol. 2011;36:800-801.
- de la Garza Bravo M, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin. Hum Pathol. 2015;46:558-569.
Practice Points
- Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder characterized by red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution.
- Histopathologically, LyP consists of a frequently CD30Mathematical Pi LT Std+ lymphocytic proliferation in multiple described patterns.
- Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy.
Ulcerative Heliotrope Rash in Antimelanoma Differentiation–Associated Gene 5 Dermatomyositis
Dermatomyositis (DM) is an autoimmune condition characterized by skin and muscle inflammation with an estimated incidence of 9 cases per 1 million people. The incidence of amyopathic DM, which includes antimelanoma differentiation–associated gene 5 (anti-MDA5) DM, is approximately 2 cases per 1 million people.1 Classic cutaneous manifestations of DM include a heliotrope rash, Gottron papules, and the shawl sign.
Case Reports
Patient 1
A woman in her 30s presented with diffuse arthralgias, bilateral eyelid edema, fatigue, and a progressive diffuse exanthem of 3 months’ duration. A review of systems was notable for the absence of myalgias. Physical examination revealed periorbital poikilodermatous patches with erythematous-to-violaceous plaques along the eyelid margins, violaceous papules on the dorsal knuckles, and edematous eroded plaques on the palmar fingertips. The patient was found to have a positive antinuclear antibody titer of 1:320 (reference range, <1:80) with a speckled pattern. A computed tomography (CT) scan of the chest showed patchy bilateral ground-glass opacities that were concerning for ILD. The cutaneous erosions, absence of myalgias, considerable proximal weakness, radiographic evidence of ILD, and positive antinuclear antibody test were clinically suggestive of anti-MDA5 DM. Further workup confirmed this diagnosis with positive reactivity to MDA5 by line immunoassay. The patient was treated with intravenous corticosteroids and was discharged after a 17-day hospitalization; however, she presented 2 months later to outpatient dermatology for progression of the cutaneous ulcerations, at which time an ulcerative heliotrope rash (Figure 1) was identified. Despite compliance with oral corticosteroids (1 mg/kg/d), she was hospitalized 1 month later for progressive respiratory insufficiency. A chest CT showed ground-glass linear opacities centrally located in all lobes of both lungs, consistent with rapidly progressive ILD. Over the course of her 5-day hospitalization, she was treated with corticosteroids, intravenous immunoglobulin (IVIG), and mycophenolate mofetil. The patient responded well to these therapies, leading to resolution of the respiratory symptoms, and she was discharged with plans to continue this regimen as an outpatient.
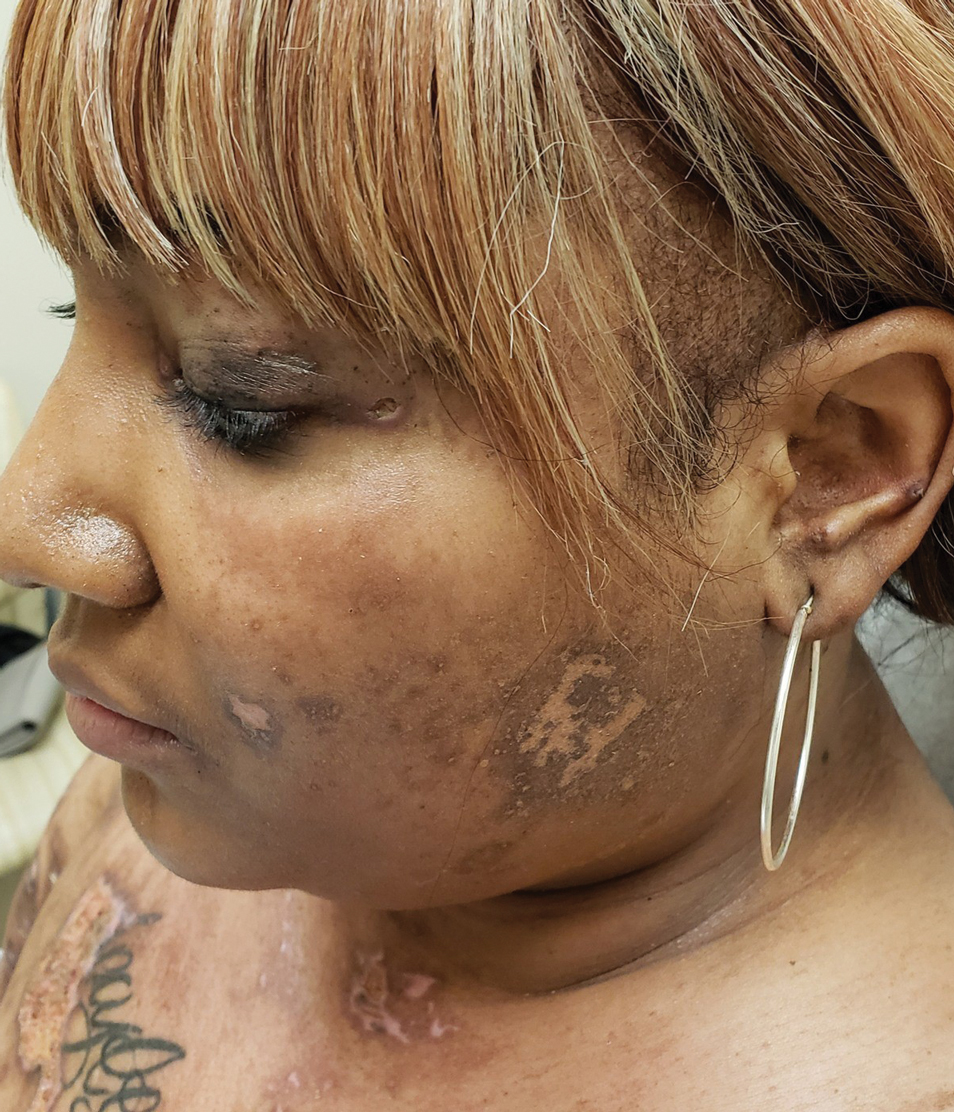
Patient 2
A woman in her late 30s with a history of known anti-MDA5 DM confirmed by line immunoassay 1 year prior presented to the emergency department with shortness of breath due to progressive ILD and a worsening exanthem. Dermatology was consulted to provide treatment recommendations. The treatment team was concerned for infection or anti-MDA5 DM disease progression. Physical examination revealed an ulcerative heliotrope rash (Figure 2) in addition to cutaneous findings classic for anti-MDA5 DM. Despite interventions, including high-dose corticosteroids, rituximab, IVIG, and plasma exchange, the ILD continued to progress, and the patient and her family elected to de-escalate aggressive medical care and pursue comfort care. The patient later died in in patient hospice.
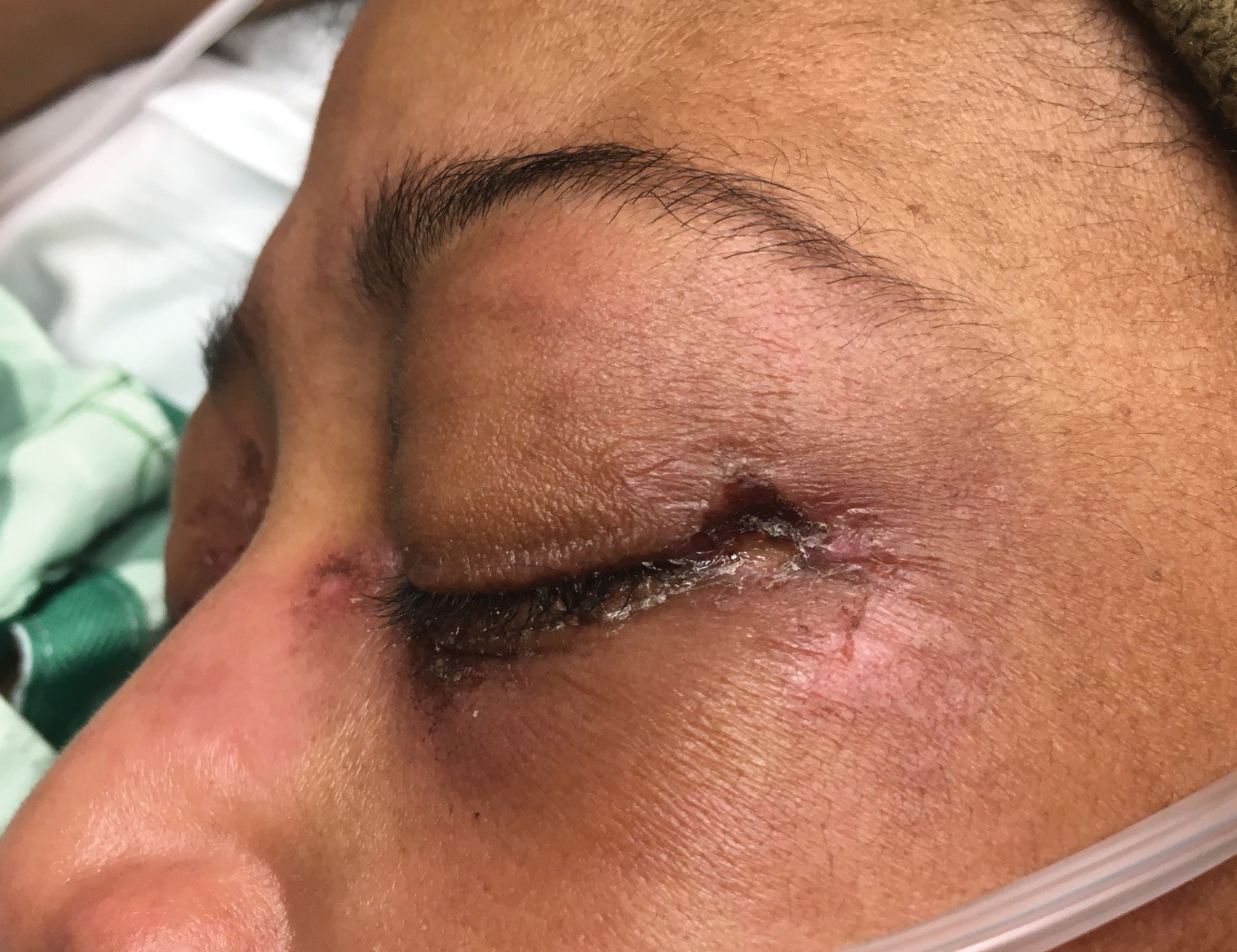
Comment
Clinical Presentation of Anti-MDA5 DM
Dermatomyositis classically presents with cutaneous manifestations including a heliotropic erythematous rash and Gottron papules as well as accompanying muscle weakness.2 However, a subtype known as amyopathic DM, which includes anti-MDA5 DM, usually presents without muscle involvement.3 Clinical muscle weakness has been reported in cases of anti-MDA5 DM, though it is less likely in these patients.4 The characteristic cutaneous phenotype of
While a heliotrope rash is classic for DM, and ulcerations are a hallmark of the anti-MDA5 DM subtype, overlap of these cutaneous manifestations is not commonly reported. In both cases presented here, ulcerations of the lateral canthi were associated with progression of ILD.
Diagnosis of Anti-MDA5 DM
Anti-MDA5 DM is defined by the presence of the anti-MDA5 antibody in the serum, named for its reactivity against the RNA helicase encoded by MDA5, within the clinical context of cutaneous signs of DM as described above.12
As described by Rider et al,13 a thorough laboratory analysis, including complete blood cell count, serum electrolytes, calcium, magnesium, phosphorus, and thyroid-stimulating hormone, is necessary to rule out conditions with similar presentations. Additionally, serum analysis for elevated muscle enzymes (creatinine phosphokinase, aldolase, lactate dehydrogenase, alanine aminotransferase, and aspartate aminotransferase) is necessary to assess for subclinical muscle involvement. Serologic evidence of myositis usually denotes an alternative diagnosis.13 Antinuclear antibodies and myositis-specific antibody positivity are much less frequent in the anti-MDA5 DM subtype than in other forms of DM.6
Anti-MDA5 antibody titer, ferritin, and IL-18 can be trended and may be useful in the evaluation of the response to treatment and ILD status in patients with anti-MDA5 DM.14,15 Elevated alveolar-arterial gradient, serum ferritin, serum chitotriosidase, and serum chitinase-3-like protein 1 (YKL-40) have each been associated with poorer prognosis of anti-MDA5 DM. The aforementioned serologies therefore may be helpful in determination of risk stratification and treatment aggressiveness.16-19
Because of its strong association with RP-ILD, screening for pulmonary disease is necessary in all patients with confirmed or strongly suspected anti-MDA5 DM. Screening can be performed with pulmonary function testing; however, high-resolution chest CT is the gold standard for diagnosis of ILD.20
Finally, all patients with a new diagnosis of DM should be evaluated for underlying malignancy through cancer screenings, given the propensity for DM to present as a paraneoplastic process.21 However, reports have indicated that the anti-MDA5 DM subtype may have a reduced risk for or an inverse relationship with underlying malignancy.5
Treatment Options for Anti-MDA5 DM
Early and aggressive therapy should be considered in the treatment of anti-MDA5 DM because of its association with RP-ILD. No treatment protocol is well established; thus, an individualized therapeutic approach may be guided by symptom severity and the clinical, radiographic, or functional evidence of ILD.6 High-dose systemic corticosteroids are first line, either in combination with or as a bridge to corticosteroid-sparing agents for immunosuppression. Many steroid-sparing medications have been employed with varying success. Mycophenolate mofetil is a reasonable first-line corticosteroid-sparing immunosuppressant agent, given its added benefit of attenuating ILD progression.6 A combination of high-dose corticosteroids, cyclosporine, and cyclophosphamide is utilized by some initially in the treatment of anti-MDA5 with ILD.22,23 While others have used combinations of these immunomodulatory agents with mycophenolate mofetil, IVIG, rituximab, azathioprine, tofacitinib, and polymyxin B, direct hemoperfusion has been added, leading to successful remission.23-28
Conclusion
We present 2 patients with anti-MDA5 DM who demonstrated a rare cutaneous manifestation of an ulcerative heliotrope rash. In both cases, this cutaneous finding was associated with the development of RP-ILD. Because of the strong association with and rapid progression of ILD seen in anti-MDA5 DM, early identification and aggressive treatment of this subtype are imperative. The clinician should recognize nonacral locations of cutaneous ulcerations, including an ulcerated heliotrope rash, to optimize diagnosis and management.
- Bendewald MJ, Wetter DA, Li X, et al. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146:26-30. doi:10.1001/archdermatol.2009.328
- Bogdanov I, Kazandjieva J, Darlenski R, et al. Dermatomyositis: current concepts. Clin Dermatol. 2018;36:450-458. doi:10.1016/j.clindermatol.2018.04.003
- Caproni M, Cardinali C, Parodi A, et al. Amyopathic dermatomyositis: a review by the Italian Group of Immunodermatology. Arch Dermatol. 2002;138:23-27. doi:10.1001/archderm.138.1.23
- Li J, Liu Y, Li Y, et al. Associations between anti-melanoma differentiation-associated gene 5 antibody and demographics, clinical characteristics and laboratory results of patients with dermatomyositis: a systematic meta-analysis. J Dermatol. 2018;45:46-52. doi:10.1111/1346-8138.14092
- Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34. doi:10.1016/j.jaad.2010.09.016
- Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation–associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785. doi:10.1016/j.jaad.2017.12.010
- Narang NS, Casciola-Rosen L, Li S, et al. Cutaneous ulceration in dermatomyositis: association with anti-melanoma differentiation-associated gene 5 antibodies and interstitial lung disease: analysis of skin ulcers in dermatomyositis. Arthritis Care Res. 2015;67:667-672. doi:10.1002/acr.22498
- Charrow A, Vleugels RA. Cutaneous ulcerations in anti-MDA5 dermatomyositis. N Engl J Med. 2019;381:465. doi:10.1056/NEJMicm1816147
- Cao H, Xia Q, Pan M, et al. Gottron papules and Gottron sign with ulceration: a distinctive cutaneous feature in a subset of patients with classic dermatomyositis and clinically amyopathic dermatomyositis. J Rheumatol. 2016;43:1735-1742. doi:10.3899/jrheum.160024
- Moghadam-Kia S, Oddis CV, Sato S, et al. Antimelanoma differentiation-associated gene 5 antibody: expanding the clinical spectrum in North American patients with dermatomyositis. J Rheumatol. 2017;44:319-325. doi:10.3899/jrheum.160682
- Li L, Wang Q, Wen X, et al. Assessment of anti-MDA5 antibody as a diagnostic biomarker in patients with dermatomyositis-associated interstitial lung disease or rapidly progressive interstitial lung disease. Oncotarget. 2017;876129-76140. doi:10.18632/oncotarget.19050
- Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60:2193-2200. doi:10.1002/art.24621
- Rider LG, Miller FW. Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathies. JAMA. 2011;305:183-190. doi:10.1001/jama.2010.1977
- Nishioka A, Tsunoda S, Abe T, et al. Serum neopterin as well as ferritin, soluble interleukin-2 receptor, KL-6 and anti-MDA5 antibody titer provide markers of the response to therapy in patients with interstitial lung disease complicating anti-MDA5 antibody-positive dermatomyositis. Mod Rheumatol. 2019;29:814-820. doi:10.1080/14397595.2018.1548918
- Gono T, Sato S, Kawaguchi Y, et al. Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatology. 2012;51:1563-1570. doi:10.1093/rheumatology/kes102
- Jiang L, Wang Y, Peng Q, et al. Serum YKL-40 level is associated with severity of interstitial lung disease and poor prognosis in dermatomyositis with anti-MDA5 antibody. Clin Rheumatol. 2019;38:1655-1663. doi:10.1007/s10067-019-04457-w
- Fujisawa T, Hozumi H, Yasui H, et al. Clinical significance of serum chitotriosidase level in anti-MDA5 antibody–positive dermatomyositis-associated interstitial lung disease. J Rheumatol. 2019;46:935-942. doi:10.3899/jrheum.180825
- Enomoto N, Oyama Y, Enomoto Y, et al. Prognostic evaluation of serum ferritin in acute exacerbation of idiopathic pulmonary fibrosis. Clin Resp J. 2018;12:2378-2389. doi:10.1111/crj.12918
- Fujiki Y, Kotani T, Isoda K, et al. Evaluation of clinical prognostic factors for interstitial pneumonia in anti-MDA5 antibody-positive dermatomyositis patients. Mod Rheumatol. 2018;28:133-140. doi:10.1080/14397595.2017.1318468
- Raghu G, Remy-Jardin M, Myers JL, et al; American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of idiopathic pulmonary fibrosis. an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:E44-E68. doi:10.1164/rccm.201807-1255ST
- Yang Z, Lin F, Qin B, et al. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42:282-291. doi:10.3899/jrheum.140566
- Hisanaga J, Kotani T, Fujiki Y, et al. Successful multi-target therapy including rituximab and mycophenolate mofetil in anti-melanoma differentiation-associated gene 5 antibody-positive rapidly progressive interstitial lung disease with clinically amyopathic dermatomyositis. Int J Rheumatic Dis. 2017;20:2182-2185. doi:10.1111/1756-185X.13136
- Kameda H, Nagasawa H, Ogawa H, et al. Combination therapy with corticosteroids, cyclosporin A, and intravenous pulse cyclophosphamide for acute/subacute interstitial pneumonia in patients with dermatomyositis. J Rheumatol. 2005;32:1719-1726.
- Endo Y, Koga T, Suzuki T, et al. Successful treatment of plasma exchange for rapidly progressive interstitial lung disease with anti–MDA5 antibody–positive dermatomyositis: a case report. Medicine. 2018;97:e0436. doi:10.1097/MD.0000000000010436
- So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989. doi:10.1007/s10067-018-4122-2
- Kurasawa K, Arai S, Namiki Y, et al. Tofacitinib for refractory interstitial lung diseases in anti-melanoma differentiation-associated 5 gene antibody-positive dermatomyositis. Rheumatology. 2018;57:2114-2119. doi:10.1093/rheumatology/key188
- Nawata T, Kubo M, Okuda S, et al. Successful treatment with intravenous cyclophosphamide for anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis associated with myelodysplastic syndrome. Scand J Rheumatol. 2017;46:496-498. doi:10.1080/03009742.2016.1253770
- Griger Z, Nagy-Vincze M, Dankó K. Pharmacological management of dermatomyositis. Exp Rev Clin Pharmacol. 2017;10:1109-1118. doi:10.1080/17512433.2017.1353910
Dermatomyositis (DM) is an autoimmune condition characterized by skin and muscle inflammation with an estimated incidence of 9 cases per 1 million people. The incidence of amyopathic DM, which includes antimelanoma differentiation–associated gene 5 (anti-MDA5) DM, is approximately 2 cases per 1 million people.1 Classic cutaneous manifestations of DM include a heliotrope rash, Gottron papules, and the shawl sign.
Case Reports
Patient 1
A woman in her 30s presented with diffuse arthralgias, bilateral eyelid edema, fatigue, and a progressive diffuse exanthem of 3 months’ duration. A review of systems was notable for the absence of myalgias. Physical examination revealed periorbital poikilodermatous patches with erythematous-to-violaceous plaques along the eyelid margins, violaceous papules on the dorsal knuckles, and edematous eroded plaques on the palmar fingertips. The patient was found to have a positive antinuclear antibody titer of 1:320 (reference range, <1:80) with a speckled pattern. A computed tomography (CT) scan of the chest showed patchy bilateral ground-glass opacities that were concerning for ILD. The cutaneous erosions, absence of myalgias, considerable proximal weakness, radiographic evidence of ILD, and positive antinuclear antibody test were clinically suggestive of anti-MDA5 DM. Further workup confirmed this diagnosis with positive reactivity to MDA5 by line immunoassay. The patient was treated with intravenous corticosteroids and was discharged after a 17-day hospitalization; however, she presented 2 months later to outpatient dermatology for progression of the cutaneous ulcerations, at which time an ulcerative heliotrope rash (Figure 1) was identified. Despite compliance with oral corticosteroids (1 mg/kg/d), she was hospitalized 1 month later for progressive respiratory insufficiency. A chest CT showed ground-glass linear opacities centrally located in all lobes of both lungs, consistent with rapidly progressive ILD. Over the course of her 5-day hospitalization, she was treated with corticosteroids, intravenous immunoglobulin (IVIG), and mycophenolate mofetil. The patient responded well to these therapies, leading to resolution of the respiratory symptoms, and she was discharged with plans to continue this regimen as an outpatient.
Patient 2
A woman in her late 30s with a history of known anti-MDA5 DM confirmed by line immunoassay 1 year prior presented to the emergency department with shortness of breath due to progressive ILD and a worsening exanthem. Dermatology was consulted to provide treatment recommendations. The treatment team was concerned for infection or anti-MDA5 DM disease progression. Physical examination revealed an ulcerative heliotrope rash (Figure 2) in addition to cutaneous findings classic for anti-MDA5 DM. Despite interventions, including high-dose corticosteroids, rituximab, IVIG, and plasma exchange, the ILD continued to progress, and the patient and her family elected to de-escalate aggressive medical care and pursue comfort care. The patient later died in in patient hospice.
Comment
Clinical Presentation of Anti-MDA5 DM
Dermatomyositis classically presents with cutaneous manifestations including a heliotropic erythematous rash and Gottron papules as well as accompanying muscle weakness.2 However, a subtype known as amyopathic DM, which includes anti-MDA5 DM, usually presents without muscle involvement.3 Clinical muscle weakness has been reported in cases of anti-MDA5 DM, though it is less likely in these patients.4 The characteristic cutaneous phenotype of
While a heliotrope rash is classic for DM, and ulcerations are a hallmark of the anti-MDA5 DM subtype, overlap of these cutaneous manifestations is not commonly reported. In both cases presented here, ulcerations of the lateral canthi were associated with progression of ILD.
Diagnosis of Anti-MDA5 DM
Anti-MDA5 DM is defined by the presence of the anti-MDA5 antibody in the serum, named for its reactivity against the RNA helicase encoded by MDA5, within the clinical context of cutaneous signs of DM as described above.12
As described by Rider et al,13 a thorough laboratory analysis, including complete blood cell count, serum electrolytes, calcium, magnesium, phosphorus, and thyroid-stimulating hormone, is necessary to rule out conditions with similar presentations. Additionally, serum analysis for elevated muscle enzymes (creatinine phosphokinase, aldolase, lactate dehydrogenase, alanine aminotransferase, and aspartate aminotransferase) is necessary to assess for subclinical muscle involvement. Serologic evidence of myositis usually denotes an alternative diagnosis.13 Antinuclear antibodies and myositis-specific antibody positivity are much less frequent in the anti-MDA5 DM subtype than in other forms of DM.6
Anti-MDA5 antibody titer, ferritin, and IL-18 can be trended and may be useful in the evaluation of the response to treatment and ILD status in patients with anti-MDA5 DM.14,15 Elevated alveolar-arterial gradient, serum ferritin, serum chitotriosidase, and serum chitinase-3-like protein 1 (YKL-40) have each been associated with poorer prognosis of anti-MDA5 DM. The aforementioned serologies therefore may be helpful in determination of risk stratification and treatment aggressiveness.16-19
Because of its strong association with RP-ILD, screening for pulmonary disease is necessary in all patients with confirmed or strongly suspected anti-MDA5 DM. Screening can be performed with pulmonary function testing; however, high-resolution chest CT is the gold standard for diagnosis of ILD.20
Finally, all patients with a new diagnosis of DM should be evaluated for underlying malignancy through cancer screenings, given the propensity for DM to present as a paraneoplastic process.21 However, reports have indicated that the anti-MDA5 DM subtype may have a reduced risk for or an inverse relationship with underlying malignancy.5
Treatment Options for Anti-MDA5 DM
Early and aggressive therapy should be considered in the treatment of anti-MDA5 DM because of its association with RP-ILD. No treatment protocol is well established; thus, an individualized therapeutic approach may be guided by symptom severity and the clinical, radiographic, or functional evidence of ILD.6 High-dose systemic corticosteroids are first line, either in combination with or as a bridge to corticosteroid-sparing agents for immunosuppression. Many steroid-sparing medications have been employed with varying success. Mycophenolate mofetil is a reasonable first-line corticosteroid-sparing immunosuppressant agent, given its added benefit of attenuating ILD progression.6 A combination of high-dose corticosteroids, cyclosporine, and cyclophosphamide is utilized by some initially in the treatment of anti-MDA5 with ILD.22,23 While others have used combinations of these immunomodulatory agents with mycophenolate mofetil, IVIG, rituximab, azathioprine, tofacitinib, and polymyxin B, direct hemoperfusion has been added, leading to successful remission.23-28
Conclusion
We present 2 patients with anti-MDA5 DM who demonstrated a rare cutaneous manifestation of an ulcerative heliotrope rash. In both cases, this cutaneous finding was associated with the development of RP-ILD. Because of the strong association with and rapid progression of ILD seen in anti-MDA5 DM, early identification and aggressive treatment of this subtype are imperative. The clinician should recognize nonacral locations of cutaneous ulcerations, including an ulcerated heliotrope rash, to optimize diagnosis and management.
Dermatomyositis (DM) is an autoimmune condition characterized by skin and muscle inflammation with an estimated incidence of 9 cases per 1 million people. The incidence of amyopathic DM, which includes antimelanoma differentiation–associated gene 5 (anti-MDA5) DM, is approximately 2 cases per 1 million people.1 Classic cutaneous manifestations of DM include a heliotrope rash, Gottron papules, and the shawl sign.
Case Reports
Patient 1
A woman in her 30s presented with diffuse arthralgias, bilateral eyelid edema, fatigue, and a progressive diffuse exanthem of 3 months’ duration. A review of systems was notable for the absence of myalgias. Physical examination revealed periorbital poikilodermatous patches with erythematous-to-violaceous plaques along the eyelid margins, violaceous papules on the dorsal knuckles, and edematous eroded plaques on the palmar fingertips. The patient was found to have a positive antinuclear antibody titer of 1:320 (reference range, <1:80) with a speckled pattern. A computed tomography (CT) scan of the chest showed patchy bilateral ground-glass opacities that were concerning for ILD. The cutaneous erosions, absence of myalgias, considerable proximal weakness, radiographic evidence of ILD, and positive antinuclear antibody test were clinically suggestive of anti-MDA5 DM. Further workup confirmed this diagnosis with positive reactivity to MDA5 by line immunoassay. The patient was treated with intravenous corticosteroids and was discharged after a 17-day hospitalization; however, she presented 2 months later to outpatient dermatology for progression of the cutaneous ulcerations, at which time an ulcerative heliotrope rash (Figure 1) was identified. Despite compliance with oral corticosteroids (1 mg/kg/d), she was hospitalized 1 month later for progressive respiratory insufficiency. A chest CT showed ground-glass linear opacities centrally located in all lobes of both lungs, consistent with rapidly progressive ILD. Over the course of her 5-day hospitalization, she was treated with corticosteroids, intravenous immunoglobulin (IVIG), and mycophenolate mofetil. The patient responded well to these therapies, leading to resolution of the respiratory symptoms, and she was discharged with plans to continue this regimen as an outpatient.
Patient 2
A woman in her late 30s with a history of known anti-MDA5 DM confirmed by line immunoassay 1 year prior presented to the emergency department with shortness of breath due to progressive ILD and a worsening exanthem. Dermatology was consulted to provide treatment recommendations. The treatment team was concerned for infection or anti-MDA5 DM disease progression. Physical examination revealed an ulcerative heliotrope rash (Figure 2) in addition to cutaneous findings classic for anti-MDA5 DM. Despite interventions, including high-dose corticosteroids, rituximab, IVIG, and plasma exchange, the ILD continued to progress, and the patient and her family elected to de-escalate aggressive medical care and pursue comfort care. The patient later died in in patient hospice.
Comment
Clinical Presentation of Anti-MDA5 DM
Dermatomyositis classically presents with cutaneous manifestations including a heliotropic erythematous rash and Gottron papules as well as accompanying muscle weakness.2 However, a subtype known as amyopathic DM, which includes anti-MDA5 DM, usually presents without muscle involvement.3 Clinical muscle weakness has been reported in cases of anti-MDA5 DM, though it is less likely in these patients.4 The characteristic cutaneous phenotype of
While a heliotrope rash is classic for DM, and ulcerations are a hallmark of the anti-MDA5 DM subtype, overlap of these cutaneous manifestations is not commonly reported. In both cases presented here, ulcerations of the lateral canthi were associated with progression of ILD.
Diagnosis of Anti-MDA5 DM
Anti-MDA5 DM is defined by the presence of the anti-MDA5 antibody in the serum, named for its reactivity against the RNA helicase encoded by MDA5, within the clinical context of cutaneous signs of DM as described above.12
As described by Rider et al,13 a thorough laboratory analysis, including complete blood cell count, serum electrolytes, calcium, magnesium, phosphorus, and thyroid-stimulating hormone, is necessary to rule out conditions with similar presentations. Additionally, serum analysis for elevated muscle enzymes (creatinine phosphokinase, aldolase, lactate dehydrogenase, alanine aminotransferase, and aspartate aminotransferase) is necessary to assess for subclinical muscle involvement. Serologic evidence of myositis usually denotes an alternative diagnosis.13 Antinuclear antibodies and myositis-specific antibody positivity are much less frequent in the anti-MDA5 DM subtype than in other forms of DM.6
Anti-MDA5 antibody titer, ferritin, and IL-18 can be trended and may be useful in the evaluation of the response to treatment and ILD status in patients with anti-MDA5 DM.14,15 Elevated alveolar-arterial gradient, serum ferritin, serum chitotriosidase, and serum chitinase-3-like protein 1 (YKL-40) have each been associated with poorer prognosis of anti-MDA5 DM. The aforementioned serologies therefore may be helpful in determination of risk stratification and treatment aggressiveness.16-19
Because of its strong association with RP-ILD, screening for pulmonary disease is necessary in all patients with confirmed or strongly suspected anti-MDA5 DM. Screening can be performed with pulmonary function testing; however, high-resolution chest CT is the gold standard for diagnosis of ILD.20
Finally, all patients with a new diagnosis of DM should be evaluated for underlying malignancy through cancer screenings, given the propensity for DM to present as a paraneoplastic process.21 However, reports have indicated that the anti-MDA5 DM subtype may have a reduced risk for or an inverse relationship with underlying malignancy.5
Treatment Options for Anti-MDA5 DM
Early and aggressive therapy should be considered in the treatment of anti-MDA5 DM because of its association with RP-ILD. No treatment protocol is well established; thus, an individualized therapeutic approach may be guided by symptom severity and the clinical, radiographic, or functional evidence of ILD.6 High-dose systemic corticosteroids are first line, either in combination with or as a bridge to corticosteroid-sparing agents for immunosuppression. Many steroid-sparing medications have been employed with varying success. Mycophenolate mofetil is a reasonable first-line corticosteroid-sparing immunosuppressant agent, given its added benefit of attenuating ILD progression.6 A combination of high-dose corticosteroids, cyclosporine, and cyclophosphamide is utilized by some initially in the treatment of anti-MDA5 with ILD.22,23 While others have used combinations of these immunomodulatory agents with mycophenolate mofetil, IVIG, rituximab, azathioprine, tofacitinib, and polymyxin B, direct hemoperfusion has been added, leading to successful remission.23-28
Conclusion
We present 2 patients with anti-MDA5 DM who demonstrated a rare cutaneous manifestation of an ulcerative heliotrope rash. In both cases, this cutaneous finding was associated with the development of RP-ILD. Because of the strong association with and rapid progression of ILD seen in anti-MDA5 DM, early identification and aggressive treatment of this subtype are imperative. The clinician should recognize nonacral locations of cutaneous ulcerations, including an ulcerated heliotrope rash, to optimize diagnosis and management.
- Bendewald MJ, Wetter DA, Li X, et al. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146:26-30. doi:10.1001/archdermatol.2009.328
- Bogdanov I, Kazandjieva J, Darlenski R, et al. Dermatomyositis: current concepts. Clin Dermatol. 2018;36:450-458. doi:10.1016/j.clindermatol.2018.04.003
- Caproni M, Cardinali C, Parodi A, et al. Amyopathic dermatomyositis: a review by the Italian Group of Immunodermatology. Arch Dermatol. 2002;138:23-27. doi:10.1001/archderm.138.1.23
- Li J, Liu Y, Li Y, et al. Associations between anti-melanoma differentiation-associated gene 5 antibody and demographics, clinical characteristics and laboratory results of patients with dermatomyositis: a systematic meta-analysis. J Dermatol. 2018;45:46-52. doi:10.1111/1346-8138.14092
- Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34. doi:10.1016/j.jaad.2010.09.016
- Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation–associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785. doi:10.1016/j.jaad.2017.12.010
- Narang NS, Casciola-Rosen L, Li S, et al. Cutaneous ulceration in dermatomyositis: association with anti-melanoma differentiation-associated gene 5 antibodies and interstitial lung disease: analysis of skin ulcers in dermatomyositis. Arthritis Care Res. 2015;67:667-672. doi:10.1002/acr.22498
- Charrow A, Vleugels RA. Cutaneous ulcerations in anti-MDA5 dermatomyositis. N Engl J Med. 2019;381:465. doi:10.1056/NEJMicm1816147
- Cao H, Xia Q, Pan M, et al. Gottron papules and Gottron sign with ulceration: a distinctive cutaneous feature in a subset of patients with classic dermatomyositis and clinically amyopathic dermatomyositis. J Rheumatol. 2016;43:1735-1742. doi:10.3899/jrheum.160024
- Moghadam-Kia S, Oddis CV, Sato S, et al. Antimelanoma differentiation-associated gene 5 antibody: expanding the clinical spectrum in North American patients with dermatomyositis. J Rheumatol. 2017;44:319-325. doi:10.3899/jrheum.160682
- Li L, Wang Q, Wen X, et al. Assessment of anti-MDA5 antibody as a diagnostic biomarker in patients with dermatomyositis-associated interstitial lung disease or rapidly progressive interstitial lung disease. Oncotarget. 2017;876129-76140. doi:10.18632/oncotarget.19050
- Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60:2193-2200. doi:10.1002/art.24621
- Rider LG, Miller FW. Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathies. JAMA. 2011;305:183-190. doi:10.1001/jama.2010.1977
- Nishioka A, Tsunoda S, Abe T, et al. Serum neopterin as well as ferritin, soluble interleukin-2 receptor, KL-6 and anti-MDA5 antibody titer provide markers of the response to therapy in patients with interstitial lung disease complicating anti-MDA5 antibody-positive dermatomyositis. Mod Rheumatol. 2019;29:814-820. doi:10.1080/14397595.2018.1548918
- Gono T, Sato S, Kawaguchi Y, et al. Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatology. 2012;51:1563-1570. doi:10.1093/rheumatology/kes102
- Jiang L, Wang Y, Peng Q, et al. Serum YKL-40 level is associated with severity of interstitial lung disease and poor prognosis in dermatomyositis with anti-MDA5 antibody. Clin Rheumatol. 2019;38:1655-1663. doi:10.1007/s10067-019-04457-w
- Fujisawa T, Hozumi H, Yasui H, et al. Clinical significance of serum chitotriosidase level in anti-MDA5 antibody–positive dermatomyositis-associated interstitial lung disease. J Rheumatol. 2019;46:935-942. doi:10.3899/jrheum.180825
- Enomoto N, Oyama Y, Enomoto Y, et al. Prognostic evaluation of serum ferritin in acute exacerbation of idiopathic pulmonary fibrosis. Clin Resp J. 2018;12:2378-2389. doi:10.1111/crj.12918
- Fujiki Y, Kotani T, Isoda K, et al. Evaluation of clinical prognostic factors for interstitial pneumonia in anti-MDA5 antibody-positive dermatomyositis patients. Mod Rheumatol. 2018;28:133-140. doi:10.1080/14397595.2017.1318468
- Raghu G, Remy-Jardin M, Myers JL, et al; American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of idiopathic pulmonary fibrosis. an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:E44-E68. doi:10.1164/rccm.201807-1255ST
- Yang Z, Lin F, Qin B, et al. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42:282-291. doi:10.3899/jrheum.140566
- Hisanaga J, Kotani T, Fujiki Y, et al. Successful multi-target therapy including rituximab and mycophenolate mofetil in anti-melanoma differentiation-associated gene 5 antibody-positive rapidly progressive interstitial lung disease with clinically amyopathic dermatomyositis. Int J Rheumatic Dis. 2017;20:2182-2185. doi:10.1111/1756-185X.13136
- Kameda H, Nagasawa H, Ogawa H, et al. Combination therapy with corticosteroids, cyclosporin A, and intravenous pulse cyclophosphamide for acute/subacute interstitial pneumonia in patients with dermatomyositis. J Rheumatol. 2005;32:1719-1726.
- Endo Y, Koga T, Suzuki T, et al. Successful treatment of plasma exchange for rapidly progressive interstitial lung disease with anti–MDA5 antibody–positive dermatomyositis: a case report. Medicine. 2018;97:e0436. doi:10.1097/MD.0000000000010436
- So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989. doi:10.1007/s10067-018-4122-2
- Kurasawa K, Arai S, Namiki Y, et al. Tofacitinib for refractory interstitial lung diseases in anti-melanoma differentiation-associated 5 gene antibody-positive dermatomyositis. Rheumatology. 2018;57:2114-2119. doi:10.1093/rheumatology/key188
- Nawata T, Kubo M, Okuda S, et al. Successful treatment with intravenous cyclophosphamide for anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis associated with myelodysplastic syndrome. Scand J Rheumatol. 2017;46:496-498. doi:10.1080/03009742.2016.1253770
- Griger Z, Nagy-Vincze M, Dankó K. Pharmacological management of dermatomyositis. Exp Rev Clin Pharmacol. 2017;10:1109-1118. doi:10.1080/17512433.2017.1353910
- Bendewald MJ, Wetter DA, Li X, et al. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146:26-30. doi:10.1001/archdermatol.2009.328
- Bogdanov I, Kazandjieva J, Darlenski R, et al. Dermatomyositis: current concepts. Clin Dermatol. 2018;36:450-458. doi:10.1016/j.clindermatol.2018.04.003
- Caproni M, Cardinali C, Parodi A, et al. Amyopathic dermatomyositis: a review by the Italian Group of Immunodermatology. Arch Dermatol. 2002;138:23-27. doi:10.1001/archderm.138.1.23
- Li J, Liu Y, Li Y, et al. Associations between anti-melanoma differentiation-associated gene 5 antibody and demographics, clinical characteristics and laboratory results of patients with dermatomyositis: a systematic meta-analysis. J Dermatol. 2018;45:46-52. doi:10.1111/1346-8138.14092
- Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34. doi:10.1016/j.jaad.2010.09.016
- Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation–associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785. doi:10.1016/j.jaad.2017.12.010
- Narang NS, Casciola-Rosen L, Li S, et al. Cutaneous ulceration in dermatomyositis: association with anti-melanoma differentiation-associated gene 5 antibodies and interstitial lung disease: analysis of skin ulcers in dermatomyositis. Arthritis Care Res. 2015;67:667-672. doi:10.1002/acr.22498
- Charrow A, Vleugels RA. Cutaneous ulcerations in anti-MDA5 dermatomyositis. N Engl J Med. 2019;381:465. doi:10.1056/NEJMicm1816147
- Cao H, Xia Q, Pan M, et al. Gottron papules and Gottron sign with ulceration: a distinctive cutaneous feature in a subset of patients with classic dermatomyositis and clinically amyopathic dermatomyositis. J Rheumatol. 2016;43:1735-1742. doi:10.3899/jrheum.160024
- Moghadam-Kia S, Oddis CV, Sato S, et al. Antimelanoma differentiation-associated gene 5 antibody: expanding the clinical spectrum in North American patients with dermatomyositis. J Rheumatol. 2017;44:319-325. doi:10.3899/jrheum.160682
- Li L, Wang Q, Wen X, et al. Assessment of anti-MDA5 antibody as a diagnostic biomarker in patients with dermatomyositis-associated interstitial lung disease or rapidly progressive interstitial lung disease. Oncotarget. 2017;876129-76140. doi:10.18632/oncotarget.19050
- Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60:2193-2200. doi:10.1002/art.24621
- Rider LG, Miller FW. Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathies. JAMA. 2011;305:183-190. doi:10.1001/jama.2010.1977
- Nishioka A, Tsunoda S, Abe T, et al. Serum neopterin as well as ferritin, soluble interleukin-2 receptor, KL-6 and anti-MDA5 antibody titer provide markers of the response to therapy in patients with interstitial lung disease complicating anti-MDA5 antibody-positive dermatomyositis. Mod Rheumatol. 2019;29:814-820. doi:10.1080/14397595.2018.1548918
- Gono T, Sato S, Kawaguchi Y, et al. Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatology. 2012;51:1563-1570. doi:10.1093/rheumatology/kes102
- Jiang L, Wang Y, Peng Q, et al. Serum YKL-40 level is associated with severity of interstitial lung disease and poor prognosis in dermatomyositis with anti-MDA5 antibody. Clin Rheumatol. 2019;38:1655-1663. doi:10.1007/s10067-019-04457-w
- Fujisawa T, Hozumi H, Yasui H, et al. Clinical significance of serum chitotriosidase level in anti-MDA5 antibody–positive dermatomyositis-associated interstitial lung disease. J Rheumatol. 2019;46:935-942. doi:10.3899/jrheum.180825
- Enomoto N, Oyama Y, Enomoto Y, et al. Prognostic evaluation of serum ferritin in acute exacerbation of idiopathic pulmonary fibrosis. Clin Resp J. 2018;12:2378-2389. doi:10.1111/crj.12918
- Fujiki Y, Kotani T, Isoda K, et al. Evaluation of clinical prognostic factors for interstitial pneumonia in anti-MDA5 antibody-positive dermatomyositis patients. Mod Rheumatol. 2018;28:133-140. doi:10.1080/14397595.2017.1318468
- Raghu G, Remy-Jardin M, Myers JL, et al; American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of idiopathic pulmonary fibrosis. an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:E44-E68. doi:10.1164/rccm.201807-1255ST
- Yang Z, Lin F, Qin B, et al. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42:282-291. doi:10.3899/jrheum.140566
- Hisanaga J, Kotani T, Fujiki Y, et al. Successful multi-target therapy including rituximab and mycophenolate mofetil in anti-melanoma differentiation-associated gene 5 antibody-positive rapidly progressive interstitial lung disease with clinically amyopathic dermatomyositis. Int J Rheumatic Dis. 2017;20:2182-2185. doi:10.1111/1756-185X.13136
- Kameda H, Nagasawa H, Ogawa H, et al. Combination therapy with corticosteroids, cyclosporin A, and intravenous pulse cyclophosphamide for acute/subacute interstitial pneumonia in patients with dermatomyositis. J Rheumatol. 2005;32:1719-1726.
- Endo Y, Koga T, Suzuki T, et al. Successful treatment of plasma exchange for rapidly progressive interstitial lung disease with anti–MDA5 antibody–positive dermatomyositis: a case report. Medicine. 2018;97:e0436. doi:10.1097/MD.0000000000010436
- So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989. doi:10.1007/s10067-018-4122-2
- Kurasawa K, Arai S, Namiki Y, et al. Tofacitinib for refractory interstitial lung diseases in anti-melanoma differentiation-associated 5 gene antibody-positive dermatomyositis. Rheumatology. 2018;57:2114-2119. doi:10.1093/rheumatology/key188
- Nawata T, Kubo M, Okuda S, et al. Successful treatment with intravenous cyclophosphamide for anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis associated with myelodysplastic syndrome. Scand J Rheumatol. 2017;46:496-498. doi:10.1080/03009742.2016.1253770
- Griger Z, Nagy-Vincze M, Dankó K. Pharmacological management of dermatomyositis. Exp Rev Clin Pharmacol. 2017;10:1109-1118. doi:10.1080/17512433.2017.1353910
Practice Points
- Antimelanoma differentiation–associated gene 5 dermatomyositis (anti-MDA5 DM) can present with an ulcerative heliotrope rash.
- Ulceration of the heliotrope rash in anti-MDA5 DM may indicate disease progression.
- Rapidly progressive interstitial lung disease is highly associated with anti-MDA5 DM.
Microaggressions in Medicine
As manifestations of overt racism and macroaggressions have gained increased visibility, there is a need for discussion of another expression of racism: microaggressions. Although racism classically is viewed as blatant structural, attitudinal, and behavioral prejudice, experts pose that the face of racism has evolved into a more covert insidious form. This form of racism was originally coined racial microaggressions by psychiatrist Chester M. Pierce, MD, 50 years ago.1,2 Since that time, microaggressions have further expanded to describe “brief and commonplace daily verbal, behavioral, and environmental indignities, whether intentional or unintentional, that communicate hostile, derogatory, or negative racial, gender, sexual-orientation, and religious slights and insults to the target person or group.” 3 This article aims to define and depict examples of microaggressions in medicine, discuss the resulting harmful effects, and offer strategies to minimize and counter these negative ramifications.
What are microaggressions?
Microaggressions are behaviors that stem from implicit bias and occur at an interpersonal level. Implicit bias refers to unconscious stereotypes, assumptions, and beliefs held about an individual’s identity. One of the earliest microaggressions—invisibility—was characterized by Ralph Ellison in his novel Invisible Man. Ellison states, “I am invisible, understand, simply because people refuse to see me . . . When they approach me they see only my surroundings, themselves, or figments of their imagination—indeed, everything and anything except me.”4 This concept of invisibility is a primary microaggression faced by people of color.
In medicine, microaggressions and implicit bias may be encountered throughout medical training and clinical practice in interactions with colleagues, superiors, patients, and patients’ families.5,6 Examples of microaggressions in medicine include demeaning comments, nonverbal disrespect, generalizations of social identity, assumption of nonphysician status, role- or credential-questioning behavior, explicit epithets, rejection of care, questioning or inquiries of ethnic/racial origin, and sexual harassment.7
An example of microaggressions in medicine was fully displayed when physician Tamika Cross described her experience of being turned away from helping an unresponsive passenger during a flight emergency.
[T]he flight attendant yells “call overhead for a physician on board.” I raised my hand to grab her attention. She said to me “oh no sweetie put [your] hand down, we are looking for actual physicians or nurses or some type of medical personnel, we don’t have time to talk to you” . . . Another “seasoned” white male approaches the row and says he is a physician as well. She says to me “thanks for your help but he can help us, and he has his credentials.”8
What are the effects of microaggressions?
Although microaggressions may be unconscious and unintentional by the offender, the negative ramifications are notable. Recent studies report that women and underrepresented minority (URM) medical students, residents, and physicians experience microaggressions and implicit bias at a higher prevalence and frequency compared with their male and non-URM counterparts.7,9 Repetitive microaggressions are harmful to the health and safety of women and URM medical students, residents, physicians, other providers, and patients. The Table provides example scenarios of microaggressions in medicine categorized according to Berk.10
Microaggressions negatively impact physical, mental, and emotional well-being. Current data support that medical students and residents who experience microaggressions are more likely to report associated symptoms of burnout, depression, and suicidal thoughts.11,12 Subjection to persistent bias can lead to minority status stress and racial battle fatigue, creating feelings of invisibility, isolation, exclusion, and loneliness for those impacted.13,14
In the book Black Man in a White Coat: A Doctor’s Reflections on Race and Medicine, Damon Tweedy, MD, reflects on race in medicine. Tweedy notes his experience as a medical student when a professor mistakenly assumed he was a maintenance worker in the classroom. Tweedy describes how he internalized the exchange and, despite his success throughout the course of his medical training, combatted feelings of anxiety, self-doubt, and implied inferiority.15
Although microaggressions are harmful to one’s health, they also undermine the learning and teaching experience for students, residents, and faculty, and they detract from the larger goal of providing care for patients.11 Frequent devaluing and questioning of an individual’s contributions, qualifications, and credentials based on identity can lower productivity and problem-solving abilities. These behaviors cultivate an unwelcome and hostile work/learning environment that is stressful and polarizing for the recipient.
Despite the heavy burden of microaggressions, most students, residents, and faculty physicians do not report incidents to their institutions and feel that training, resources, and policies to respond to bias adequately are lacking.7 As a result of implicit bias and microaggressions, women and URM medical students and providers are unable to focus solely on the practice of medicine. They are tasked with the additional burden of shouldering the emotional and cognitive complexities that microaggressions produce.16
What are strategies to reduce microaggressions in medicine?
To minimize the harmful effects of microaggressions, intervention strategies must be implemented that reduce the likelihood of the occurrence of microaggressions and challenge the stereotypes that undergird implicit bias. These strategies include cultivating allies, followed by demanding structural accountability. Allies are members of the majority group who collectively collaborate with members of the nonmajority group to effect change through the promotion of diversity, equity, and inclusion efforts.17 Cultivating allies involves building a network of collaboration among these groups and emphasizes education. Education is critical for allies to address microaggressions at the interpersonal level. This process of education involves personal reflection and self-awareness in exploring one’s biases, fears, and assumptions. Integral to this step is broadening one’s acceptance of different cultures, racial/ethnic groups, and identities. There must be a willingness to engage in difficult or uncomfortable conversations and a readiness to actively listen to concerns rather than perpetuating further harm through avoidance and dismissive or defensive behavior.18
Demanding structural accountability facilitates deconstruction of bias and microaggression at the larger systemic level. This strategy involves implicit bias and antiracism training, development of retention plans, and identification of mentors for women and URM providers and students. Implicit bias and microaggression training and policies should be incorporated into medical education and resident curriculums. Similarly, educational resources and training must be made available to practicing physicians, faculty, and other providers through their institutions and places of employment. Equipping students and providers with the tools needed when microaggressions are witnessed or experienced demonstrates systemic-level accountability and communicates the importance of the issue. Furthermore, the development of retention plans and identification of mentors provide a support system and foster a culture of inclusion where recipients of microaggressions feel protected and valued. Increased feelings of inclusivity and belonging help bridge the gap created through microaggressions and implicit bias.
Final Thoughts
Despite an often covert nature, the detrimental effects of microaggressions are tangible and far reaching. As providers, we must strive to understand all categories of racism and expose the many ways prejudice manifests within medical training and clinical practice. It is our obligation to undertake the challenge of “making the ‘invisible’ visible” as we confront microaggressions and implicit bias to promote a safer and more inclusive medical community and workforce.19
- Torres MB, Salles A, Cochran A. Recognizing and reacting to microaggressions in medicine and surgery. JAMA Surg. 2019;154:868-872. doi:10.1001/jamasurg.2019.1648
- Williams MT. Microaggressions: clarification, evidence, and impact. Perspect Psychol Sci. 2020;15:3-26. doi:10.1177/1745691619827499
- Sue DW. Microaggressions in Everyday Life: Race, Gender, and Sexual Orientation. Wiley; 2010.
- Ellison R. Invisible Man. Random House; 1952.
- Molina MF, Landry AI, Chary AN, et al. Addressing the elephant in the room: microaggressions in medicine. Ann Emerg Med. 2020;76:387-391. doi:10.1016/j.annemergmed.2020.04.009
- Overland MK, Zumsteg JM, Lindo EG, et al. Microaggressions in clinical training and practice. PM R. 2019;11:1004-1012. doi:10.1002/pmrj.12229
- de Bourmont SS, Burra A, Nouri SS, et al. Resident physician experiences with and responses to biased patients. JAMA Netw Open. 2020;3:e2021769. doi:10.1001/jamanetworkopen.2020.21769
- TK Cross Facebook page. October 9, 2016. Accessed April 19, 2021. https://www.facebook.com/tamika.cross.52/posts/658443077654049
- Periyakoil VS, Chaudron L, Hill EV, et al. Common types of gender-based microaggressions in medicine. Acad Med. 2020;95:450-457. doi:10.1097/ACM.0000000000003057
- Berk RA. Microaggressions trilogy: part 1. why do microaggressions matter? J Fac Dev. 2017;31:63-73.
- Chisholm LP, Jackson KR, Davidson HA, et al. Evaluation of racial microaggressions experienced during medical school training and the effect on medical student education and burnout: a validation study. J Natl Med Assoc. 2020:S0027-9684(20)30428-4. doi:10.1016/j.jnma.2020.11.009
- Hu YY, Ellis RJ, Hewitt DB, et al. Discrimination, abuse, harassment, and burnout in surgical residency training. N Engl J Med. 2019;381:1741-1752. doi:10.1056/NEJMsa1903759
- Acholonu RG, Oyeku SO. Addressing microaggressions in the health care workforce-a path toward achieving equity and inclusion. JAMA Netw Open. 2020;3:E2021770. doi:10.1001/jamanetworkopen.2020.21770
- O’Keefe VM, Wingate LR, Cole AB, et al. Seemingly harmless racial communications are not so harmless: racial microaggressions lead to suicidal ideation by way of depression symptoms. Suicide Life Threat Behav. 2015;45:567-576. doi:10.1111/sltb.12150
- Tweedy D. Black Man in a White Coat: A Doctor’s Reflections on Race and Medicine. Picador; 2016.
- Osseo-Asare A, Balasuriya L, Huot SJ, et al. Minority resident physicians’ views on the role of race/ethnicity in their training experiences in the workplace. JAMA Netw Open. 2018;1:E182723. doi: 10.1001/jamanetworkopen.2018.2723
- Melaku TM, Beeman A, Smith DG, et al. Be a better ally. Harvard Business Review. Published November-December 2020. Accessed April 23, 2021. https://hbr.org/2020/11/be-a-better-ally
- Sue DW, Capodilupo CM, Torino GC, et al. Racial microaggressions in everyday life: implications for clinical practice. Am Psychol. 2007;62:271-286. doi:10.1037/0003-066X.62.4.271
- Sue DW. Whiteness and ethnocentric monoculturalism: making the “invisible” visible. Am Psychol. 2004;59:761-769. doi:10.1037/0003-066X.59.8.761
As manifestations of overt racism and macroaggressions have gained increased visibility, there is a need for discussion of another expression of racism: microaggressions. Although racism classically is viewed as blatant structural, attitudinal, and behavioral prejudice, experts pose that the face of racism has evolved into a more covert insidious form. This form of racism was originally coined racial microaggressions by psychiatrist Chester M. Pierce, MD, 50 years ago.1,2 Since that time, microaggressions have further expanded to describe “brief and commonplace daily verbal, behavioral, and environmental indignities, whether intentional or unintentional, that communicate hostile, derogatory, or negative racial, gender, sexual-orientation, and religious slights and insults to the target person or group.” 3 This article aims to define and depict examples of microaggressions in medicine, discuss the resulting harmful effects, and offer strategies to minimize and counter these negative ramifications.
What are microaggressions?
Microaggressions are behaviors that stem from implicit bias and occur at an interpersonal level. Implicit bias refers to unconscious stereotypes, assumptions, and beliefs held about an individual’s identity. One of the earliest microaggressions—invisibility—was characterized by Ralph Ellison in his novel Invisible Man. Ellison states, “I am invisible, understand, simply because people refuse to see me . . . When they approach me they see only my surroundings, themselves, or figments of their imagination—indeed, everything and anything except me.”4 This concept of invisibility is a primary microaggression faced by people of color.
In medicine, microaggressions and implicit bias may be encountered throughout medical training and clinical practice in interactions with colleagues, superiors, patients, and patients’ families.5,6 Examples of microaggressions in medicine include demeaning comments, nonverbal disrespect, generalizations of social identity, assumption of nonphysician status, role- or credential-questioning behavior, explicit epithets, rejection of care, questioning or inquiries of ethnic/racial origin, and sexual harassment.7
An example of microaggressions in medicine was fully displayed when physician Tamika Cross described her experience of being turned away from helping an unresponsive passenger during a flight emergency.
[T]he flight attendant yells “call overhead for a physician on board.” I raised my hand to grab her attention. She said to me “oh no sweetie put [your] hand down, we are looking for actual physicians or nurses or some type of medical personnel, we don’t have time to talk to you” . . . Another “seasoned” white male approaches the row and says he is a physician as well. She says to me “thanks for your help but he can help us, and he has his credentials.”8
What are the effects of microaggressions?
Although microaggressions may be unconscious and unintentional by the offender, the negative ramifications are notable. Recent studies report that women and underrepresented minority (URM) medical students, residents, and physicians experience microaggressions and implicit bias at a higher prevalence and frequency compared with their male and non-URM counterparts.7,9 Repetitive microaggressions are harmful to the health and safety of women and URM medical students, residents, physicians, other providers, and patients. The Table provides example scenarios of microaggressions in medicine categorized according to Berk.10
Microaggressions negatively impact physical, mental, and emotional well-being. Current data support that medical students and residents who experience microaggressions are more likely to report associated symptoms of burnout, depression, and suicidal thoughts.11,12 Subjection to persistent bias can lead to minority status stress and racial battle fatigue, creating feelings of invisibility, isolation, exclusion, and loneliness for those impacted.13,14
In the book Black Man in a White Coat: A Doctor’s Reflections on Race and Medicine, Damon Tweedy, MD, reflects on race in medicine. Tweedy notes his experience as a medical student when a professor mistakenly assumed he was a maintenance worker in the classroom. Tweedy describes how he internalized the exchange and, despite his success throughout the course of his medical training, combatted feelings of anxiety, self-doubt, and implied inferiority.15
Although microaggressions are harmful to one’s health, they also undermine the learning and teaching experience for students, residents, and faculty, and they detract from the larger goal of providing care for patients.11 Frequent devaluing and questioning of an individual’s contributions, qualifications, and credentials based on identity can lower productivity and problem-solving abilities. These behaviors cultivate an unwelcome and hostile work/learning environment that is stressful and polarizing for the recipient.
Despite the heavy burden of microaggressions, most students, residents, and faculty physicians do not report incidents to their institutions and feel that training, resources, and policies to respond to bias adequately are lacking.7 As a result of implicit bias and microaggressions, women and URM medical students and providers are unable to focus solely on the practice of medicine. They are tasked with the additional burden of shouldering the emotional and cognitive complexities that microaggressions produce.16
What are strategies to reduce microaggressions in medicine?
To minimize the harmful effects of microaggressions, intervention strategies must be implemented that reduce the likelihood of the occurrence of microaggressions and challenge the stereotypes that undergird implicit bias. These strategies include cultivating allies, followed by demanding structural accountability. Allies are members of the majority group who collectively collaborate with members of the nonmajority group to effect change through the promotion of diversity, equity, and inclusion efforts.17 Cultivating allies involves building a network of collaboration among these groups and emphasizes education. Education is critical for allies to address microaggressions at the interpersonal level. This process of education involves personal reflection and self-awareness in exploring one’s biases, fears, and assumptions. Integral to this step is broadening one’s acceptance of different cultures, racial/ethnic groups, and identities. There must be a willingness to engage in difficult or uncomfortable conversations and a readiness to actively listen to concerns rather than perpetuating further harm through avoidance and dismissive or defensive behavior.18
Demanding structural accountability facilitates deconstruction of bias and microaggression at the larger systemic level. This strategy involves implicit bias and antiracism training, development of retention plans, and identification of mentors for women and URM providers and students. Implicit bias and microaggression training and policies should be incorporated into medical education and resident curriculums. Similarly, educational resources and training must be made available to practicing physicians, faculty, and other providers through their institutions and places of employment. Equipping students and providers with the tools needed when microaggressions are witnessed or experienced demonstrates systemic-level accountability and communicates the importance of the issue. Furthermore, the development of retention plans and identification of mentors provide a support system and foster a culture of inclusion where recipients of microaggressions feel protected and valued. Increased feelings of inclusivity and belonging help bridge the gap created through microaggressions and implicit bias.
Final Thoughts
Despite an often covert nature, the detrimental effects of microaggressions are tangible and far reaching. As providers, we must strive to understand all categories of racism and expose the many ways prejudice manifests within medical training and clinical practice. It is our obligation to undertake the challenge of “making the ‘invisible’ visible” as we confront microaggressions and implicit bias to promote a safer and more inclusive medical community and workforce.19
As manifestations of overt racism and macroaggressions have gained increased visibility, there is a need for discussion of another expression of racism: microaggressions. Although racism classically is viewed as blatant structural, attitudinal, and behavioral prejudice, experts pose that the face of racism has evolved into a more covert insidious form. This form of racism was originally coined racial microaggressions by psychiatrist Chester M. Pierce, MD, 50 years ago.1,2 Since that time, microaggressions have further expanded to describe “brief and commonplace daily verbal, behavioral, and environmental indignities, whether intentional or unintentional, that communicate hostile, derogatory, or negative racial, gender, sexual-orientation, and religious slights and insults to the target person or group.” 3 This article aims to define and depict examples of microaggressions in medicine, discuss the resulting harmful effects, and offer strategies to minimize and counter these negative ramifications.
What are microaggressions?
Microaggressions are behaviors that stem from implicit bias and occur at an interpersonal level. Implicit bias refers to unconscious stereotypes, assumptions, and beliefs held about an individual’s identity. One of the earliest microaggressions—invisibility—was characterized by Ralph Ellison in his novel Invisible Man. Ellison states, “I am invisible, understand, simply because people refuse to see me . . . When they approach me they see only my surroundings, themselves, or figments of their imagination—indeed, everything and anything except me.”4 This concept of invisibility is a primary microaggression faced by people of color.
In medicine, microaggressions and implicit bias may be encountered throughout medical training and clinical practice in interactions with colleagues, superiors, patients, and patients’ families.5,6 Examples of microaggressions in medicine include demeaning comments, nonverbal disrespect, generalizations of social identity, assumption of nonphysician status, role- or credential-questioning behavior, explicit epithets, rejection of care, questioning or inquiries of ethnic/racial origin, and sexual harassment.7
An example of microaggressions in medicine was fully displayed when physician Tamika Cross described her experience of being turned away from helping an unresponsive passenger during a flight emergency.
[T]he flight attendant yells “call overhead for a physician on board.” I raised my hand to grab her attention. She said to me “oh no sweetie put [your] hand down, we are looking for actual physicians or nurses or some type of medical personnel, we don’t have time to talk to you” . . . Another “seasoned” white male approaches the row and says he is a physician as well. She says to me “thanks for your help but he can help us, and he has his credentials.”8
What are the effects of microaggressions?
Although microaggressions may be unconscious and unintentional by the offender, the negative ramifications are notable. Recent studies report that women and underrepresented minority (URM) medical students, residents, and physicians experience microaggressions and implicit bias at a higher prevalence and frequency compared with their male and non-URM counterparts.7,9 Repetitive microaggressions are harmful to the health and safety of women and URM medical students, residents, physicians, other providers, and patients. The Table provides example scenarios of microaggressions in medicine categorized according to Berk.10
Microaggressions negatively impact physical, mental, and emotional well-being. Current data support that medical students and residents who experience microaggressions are more likely to report associated symptoms of burnout, depression, and suicidal thoughts.11,12 Subjection to persistent bias can lead to minority status stress and racial battle fatigue, creating feelings of invisibility, isolation, exclusion, and loneliness for those impacted.13,14
In the book Black Man in a White Coat: A Doctor’s Reflections on Race and Medicine, Damon Tweedy, MD, reflects on race in medicine. Tweedy notes his experience as a medical student when a professor mistakenly assumed he was a maintenance worker in the classroom. Tweedy describes how he internalized the exchange and, despite his success throughout the course of his medical training, combatted feelings of anxiety, self-doubt, and implied inferiority.15
Although microaggressions are harmful to one’s health, they also undermine the learning and teaching experience for students, residents, and faculty, and they detract from the larger goal of providing care for patients.11 Frequent devaluing and questioning of an individual’s contributions, qualifications, and credentials based on identity can lower productivity and problem-solving abilities. These behaviors cultivate an unwelcome and hostile work/learning environment that is stressful and polarizing for the recipient.
Despite the heavy burden of microaggressions, most students, residents, and faculty physicians do not report incidents to their institutions and feel that training, resources, and policies to respond to bias adequately are lacking.7 As a result of implicit bias and microaggressions, women and URM medical students and providers are unable to focus solely on the practice of medicine. They are tasked with the additional burden of shouldering the emotional and cognitive complexities that microaggressions produce.16
What are strategies to reduce microaggressions in medicine?
To minimize the harmful effects of microaggressions, intervention strategies must be implemented that reduce the likelihood of the occurrence of microaggressions and challenge the stereotypes that undergird implicit bias. These strategies include cultivating allies, followed by demanding structural accountability. Allies are members of the majority group who collectively collaborate with members of the nonmajority group to effect change through the promotion of diversity, equity, and inclusion efforts.17 Cultivating allies involves building a network of collaboration among these groups and emphasizes education. Education is critical for allies to address microaggressions at the interpersonal level. This process of education involves personal reflection and self-awareness in exploring one’s biases, fears, and assumptions. Integral to this step is broadening one’s acceptance of different cultures, racial/ethnic groups, and identities. There must be a willingness to engage in difficult or uncomfortable conversations and a readiness to actively listen to concerns rather than perpetuating further harm through avoidance and dismissive or defensive behavior.18
Demanding structural accountability facilitates deconstruction of bias and microaggression at the larger systemic level. This strategy involves implicit bias and antiracism training, development of retention plans, and identification of mentors for women and URM providers and students. Implicit bias and microaggression training and policies should be incorporated into medical education and resident curriculums. Similarly, educational resources and training must be made available to practicing physicians, faculty, and other providers through their institutions and places of employment. Equipping students and providers with the tools needed when microaggressions are witnessed or experienced demonstrates systemic-level accountability and communicates the importance of the issue. Furthermore, the development of retention plans and identification of mentors provide a support system and foster a culture of inclusion where recipients of microaggressions feel protected and valued. Increased feelings of inclusivity and belonging help bridge the gap created through microaggressions and implicit bias.
Final Thoughts
Despite an often covert nature, the detrimental effects of microaggressions are tangible and far reaching. As providers, we must strive to understand all categories of racism and expose the many ways prejudice manifests within medical training and clinical practice. It is our obligation to undertake the challenge of “making the ‘invisible’ visible” as we confront microaggressions and implicit bias to promote a safer and more inclusive medical community and workforce.19
- Torres MB, Salles A, Cochran A. Recognizing and reacting to microaggressions in medicine and surgery. JAMA Surg. 2019;154:868-872. doi:10.1001/jamasurg.2019.1648
- Williams MT. Microaggressions: clarification, evidence, and impact. Perspect Psychol Sci. 2020;15:3-26. doi:10.1177/1745691619827499
- Sue DW. Microaggressions in Everyday Life: Race, Gender, and Sexual Orientation. Wiley; 2010.
- Ellison R. Invisible Man. Random House; 1952.
- Molina MF, Landry AI, Chary AN, et al. Addressing the elephant in the room: microaggressions in medicine. Ann Emerg Med. 2020;76:387-391. doi:10.1016/j.annemergmed.2020.04.009
- Overland MK, Zumsteg JM, Lindo EG, et al. Microaggressions in clinical training and practice. PM R. 2019;11:1004-1012. doi:10.1002/pmrj.12229
- de Bourmont SS, Burra A, Nouri SS, et al. Resident physician experiences with and responses to biased patients. JAMA Netw Open. 2020;3:e2021769. doi:10.1001/jamanetworkopen.2020.21769
- TK Cross Facebook page. October 9, 2016. Accessed April 19, 2021. https://www.facebook.com/tamika.cross.52/posts/658443077654049
- Periyakoil VS, Chaudron L, Hill EV, et al. Common types of gender-based microaggressions in medicine. Acad Med. 2020;95:450-457. doi:10.1097/ACM.0000000000003057
- Berk RA. Microaggressions trilogy: part 1. why do microaggressions matter? J Fac Dev. 2017;31:63-73.
- Chisholm LP, Jackson KR, Davidson HA, et al. Evaluation of racial microaggressions experienced during medical school training and the effect on medical student education and burnout: a validation study. J Natl Med Assoc. 2020:S0027-9684(20)30428-4. doi:10.1016/j.jnma.2020.11.009
- Hu YY, Ellis RJ, Hewitt DB, et al. Discrimination, abuse, harassment, and burnout in surgical residency training. N Engl J Med. 2019;381:1741-1752. doi:10.1056/NEJMsa1903759
- Acholonu RG, Oyeku SO. Addressing microaggressions in the health care workforce-a path toward achieving equity and inclusion. JAMA Netw Open. 2020;3:E2021770. doi:10.1001/jamanetworkopen.2020.21770
- O’Keefe VM, Wingate LR, Cole AB, et al. Seemingly harmless racial communications are not so harmless: racial microaggressions lead to suicidal ideation by way of depression symptoms. Suicide Life Threat Behav. 2015;45:567-576. doi:10.1111/sltb.12150
- Tweedy D. Black Man in a White Coat: A Doctor’s Reflections on Race and Medicine. Picador; 2016.
- Osseo-Asare A, Balasuriya L, Huot SJ, et al. Minority resident physicians’ views on the role of race/ethnicity in their training experiences in the workplace. JAMA Netw Open. 2018;1:E182723. doi: 10.1001/jamanetworkopen.2018.2723
- Melaku TM, Beeman A, Smith DG, et al. Be a better ally. Harvard Business Review. Published November-December 2020. Accessed April 23, 2021. https://hbr.org/2020/11/be-a-better-ally
- Sue DW, Capodilupo CM, Torino GC, et al. Racial microaggressions in everyday life: implications for clinical practice. Am Psychol. 2007;62:271-286. doi:10.1037/0003-066X.62.4.271
- Sue DW. Whiteness and ethnocentric monoculturalism: making the “invisible” visible. Am Psychol. 2004;59:761-769. doi:10.1037/0003-066X.59.8.761
- Torres MB, Salles A, Cochran A. Recognizing and reacting to microaggressions in medicine and surgery. JAMA Surg. 2019;154:868-872. doi:10.1001/jamasurg.2019.1648
- Williams MT. Microaggressions: clarification, evidence, and impact. Perspect Psychol Sci. 2020;15:3-26. doi:10.1177/1745691619827499
- Sue DW. Microaggressions in Everyday Life: Race, Gender, and Sexual Orientation. Wiley; 2010.
- Ellison R. Invisible Man. Random House; 1952.
- Molina MF, Landry AI, Chary AN, et al. Addressing the elephant in the room: microaggressions in medicine. Ann Emerg Med. 2020;76:387-391. doi:10.1016/j.annemergmed.2020.04.009
- Overland MK, Zumsteg JM, Lindo EG, et al. Microaggressions in clinical training and practice. PM R. 2019;11:1004-1012. doi:10.1002/pmrj.12229
- de Bourmont SS, Burra A, Nouri SS, et al. Resident physician experiences with and responses to biased patients. JAMA Netw Open. 2020;3:e2021769. doi:10.1001/jamanetworkopen.2020.21769
- TK Cross Facebook page. October 9, 2016. Accessed April 19, 2021. https://www.facebook.com/tamika.cross.52/posts/658443077654049
- Periyakoil VS, Chaudron L, Hill EV, et al. Common types of gender-based microaggressions in medicine. Acad Med. 2020;95:450-457. doi:10.1097/ACM.0000000000003057
- Berk RA. Microaggressions trilogy: part 1. why do microaggressions matter? J Fac Dev. 2017;31:63-73.
- Chisholm LP, Jackson KR, Davidson HA, et al. Evaluation of racial microaggressions experienced during medical school training and the effect on medical student education and burnout: a validation study. J Natl Med Assoc. 2020:S0027-9684(20)30428-4. doi:10.1016/j.jnma.2020.11.009
- Hu YY, Ellis RJ, Hewitt DB, et al. Discrimination, abuse, harassment, and burnout in surgical residency training. N Engl J Med. 2019;381:1741-1752. doi:10.1056/NEJMsa1903759
- Acholonu RG, Oyeku SO. Addressing microaggressions in the health care workforce-a path toward achieving equity and inclusion. JAMA Netw Open. 2020;3:E2021770. doi:10.1001/jamanetworkopen.2020.21770
- O’Keefe VM, Wingate LR, Cole AB, et al. Seemingly harmless racial communications are not so harmless: racial microaggressions lead to suicidal ideation by way of depression symptoms. Suicide Life Threat Behav. 2015;45:567-576. doi:10.1111/sltb.12150
- Tweedy D. Black Man in a White Coat: A Doctor’s Reflections on Race and Medicine. Picador; 2016.
- Osseo-Asare A, Balasuriya L, Huot SJ, et al. Minority resident physicians’ views on the role of race/ethnicity in their training experiences in the workplace. JAMA Netw Open. 2018;1:E182723. doi: 10.1001/jamanetworkopen.2018.2723
- Melaku TM, Beeman A, Smith DG, et al. Be a better ally. Harvard Business Review. Published November-December 2020. Accessed April 23, 2021. https://hbr.org/2020/11/be-a-better-ally
- Sue DW, Capodilupo CM, Torino GC, et al. Racial microaggressions in everyday life: implications for clinical practice. Am Psychol. 2007;62:271-286. doi:10.1037/0003-066X.62.4.271
- Sue DW. Whiteness and ethnocentric monoculturalism: making the “invisible” visible. Am Psychol. 2004;59:761-769. doi:10.1037/0003-066X.59.8.761
Practice Points
- As providers, we must strive to understand all categories of racism and expose the many ways prejudice manifests within medical training and clinical practice.
- Intervention strategies must be implemented to reduce the likelihood of the occurrence of microaggressions in medicine and challenge the stereotypes that undergird implicit bias.
- It is important to promote collaboration in diversity, equity, and inclusion efforts to demonstrate support for women and underrepresented minority medical students, residents, physicians, providers, and patients.
Widespread Hyperkeratotic Papules in a Transplant Recipient
The Diagnosis: Trichodysplasia Spinulosa
Trichodysplasia spinulosa has been described in case reports over the last several decades, with its causative virus trichodysplasia spinulosa-associated polyomavirus (TSPyV) identified in 2010 by van der Meijden et al.1 Trichodysplasia spinulosa-associated polyomavirus is a small, nonenveloped, double-stranded DNA virus in the Polyomaviridae family, among several other known cutaneous polyomaviruses including Merkel cell polyomavirus, human polyomavirus (HPyV) 6, HPyV7, HPyV10, and possibly HPyV13.2 The primary target of TSPyV is follicular keratinocytes, and it is believed to cause trichodysplasia spinulosa by primary infection rather than by reactivation. Trichodysplasia spinulosa presents in immunosuppressed patients as a folliculocentric eruption of papules with keratinous spines on the face, often with concurrent alopecia, eventually spreading to the trunk and extremities.3 The diagnosis often is clinical, but a biopsy may be performed for histopathologic confirmation. Alternatively, lesional spicules can be painlessly collected manually and submitted for viral polymerase chain reaction (PCR).4 The diagnosis of trichodysplasia spinulosa can be difficult due to similarities with other more common conditions such as keratosis pilaris, milia, filiform warts, or lichen spinulosus.
Similar to trichodysplasia spinulosa, keratosis pilaris also presents with folliculocentric and often erythematous papules.5 Keratosis pilaris most frequently affects the posterior upper arms and thighs but also may affect the cheeks, as seen in trichodysplasia spinulosa. Differentiation between the 2 diagnoses can be made on a clinical basis, as keratosis pilaris lacks the characteristic keratinous spines and often spares the central face and nose, locations that commonly are affected in trichodysplasia spinulosa.3
Milia typically appear as white to yellow papules, often on the cheeks, eyelids, nose, and chin.6 Given their predilection for the face, milia can appear similarly to trichodysplasia spinulosa. Differentiation can be made clinically, as milia typically are not as numerous as the spiculed papules seen in trichodysplasia spinulosa. Morphologically, milia will present as smooth, dome-shaped papules as opposed to the keratinous spicules seen in trichodysplasia spinulosa. The diagnosis of milia can be confirmed by incision and removal of the white chalky keratin core, a feature absent in trichodysplasia spinulosa.
Filiform warts are benign epidermal proliferations caused by human papillomavirus infection that manifest as flesh-colored, verrucous, hyperkeratotic papules.7 They can appear on virtually any skin surface, including the face, and thus may be mistaken for trichodysplasia spinulosa. Close inspection usually will reveal tiny black dots that represent thrombosed capillaries, a feature lacking in trichodysplasia spinulosa. In long-standing lesions or immunocompromised patients, confluent verrucous plaques may develop.8 Diagnosis of filiform warts can be confirmed with biopsy, which will demonstrate a compact stratum corneum, coarse hypergranulosis, and papillomatosis curving inward, while biopsy of a trichodysplasia spinulosa lesion would show polyomavirus infection of the hair follicle and characteristic eosinophilic inclusion bodies.9
Lichen spinulosus may appear as multiple folliculocentric scaly papules with hairlike horny spines.10 Lichen spinulosus differs from trichodysplasia spinulosa in that it commonly appears on the neck, abdomen, trochanteric region, arms, elbows, or knees. Lichen spinulosus also classically appears as a concrete cluster of papules, often localized to a certain region, in contrast to trichodysplasia spinulosa, which will be widespread, often spreading over time. Finally, clinical history may help differentiate the 2 entities. Lichen spinulosus most often appears in children and adolescents and often has an indolent course, typically resolving during puberty, while trichodysplasia spinulosa is seen in immunocompromised patients.
In our patient, the dermatology team made a diagnosis of trichodysplasia spinulosa based on the characteristic clinical presentation, which was confirmed after approximately 10 lesional spicules were removed by tissue forceps and submitted for PCR analysis showing TSPyV (Figure). Two other cases utilized spicule PCR analysis for confirmation of TSPyV.11,12 This technique may represent a viable option for diagnostic confirmation in pediatric cases.
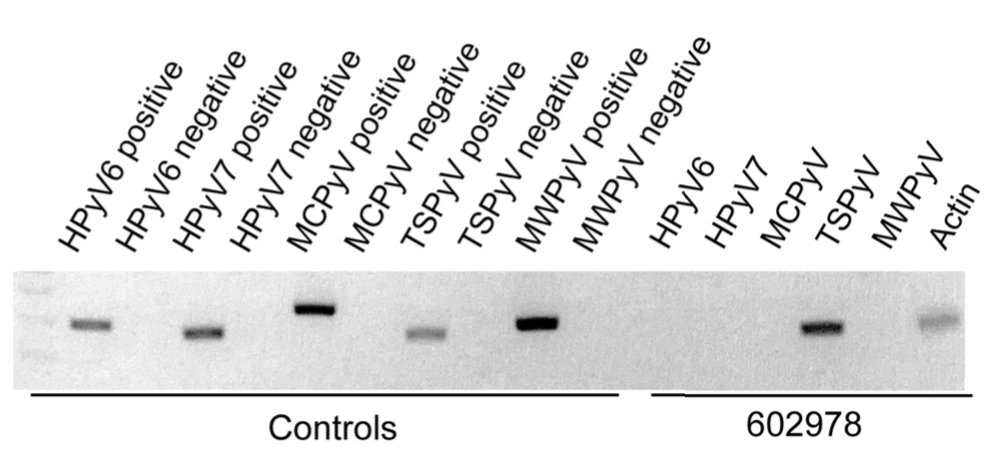
Although some articles have examined the molecular and biologic features of trichodysplasia spinulosa, literature on clinical presentation and management is limited to isolated case reports with no comprehensive studies to establish a standardized treatment. Of these reports, oral valganciclovir 900 mg daily, topical retinoids, cidofovir cream 1% to 3%, and decreasing or altering the immunosuppressive regimen all have been noted to provide clinical improvement.13,14 Other therapies including leflunomide and routine manual extraction of spicules also have shown effectiveness in the treatment of trichodysplasia spinulosa.15
In our patient, treatment included decreasing immunosuppression, as she was getting recurrent sinus and upper respiratory infections. Mycophenolate mofetil was discontinued, and the patient was continued solely on tacrolimus therapy. She demonstrated notable improvement after 3 months, with approximately 50% clearance of the eruption. A mutual decision was made at that visit to initiate therapy with compounded cidofovir cream 1% daily to the lesions until the next follow-up visit. Unfortunately, the patient did not return for her scheduled dermatology visits and was lost to long-term follow-up.
Acknowledgment
We thank Richard C. Wang, MD, PhD (Dallas, Texas), for his dermatologic expertise and assistance in analysis of lesional samples for TSPyV.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromised patient. PLoS Pathog. 2010;6:E1001024.
- Sheu JC, Tran J, Rady PL, et al. Polyomaviruses of the skin: integrating molecular and clinical advances in an emerging class of viruses. Br J Dermatol. 2019;180:1302-1311.
- Sperling LC, Tomaszewski MM, Thomas DA. Viral-associated trichodysplasia in patients who are immunocompromised. J Am Acad Dermatol. 2004;50:318-322.
- Wu JH, Nguyen HP, Rady PL, et al. Molecular insight into the viral biology and clinical features of trichodysplasia spinulosa. Br J Dermatol. 2016;174:490-498.
- Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Micali G, Dall'Oglio F, Nasca MR, et al. Management of cutaneous warts: an evidence-based approach. Am J Clin Dermatol. 2004;5:311-317.
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier; 2018.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Tilly JJ, Drolet BA, Esterly NB. Lichenoid eruptions in children. J Am Acad Dermatol. 2004;51:606-624.
- Chamseddin BH, Tran BAPD, Lee EE, et al. Trichodysplasia spinulosa in a child: identification of trichodysplasia spinulosa-associated polyomavirus in skin, serum, and urine. Pediatr Dermatol. 2019;36:723-724.
- Sonstegard A, Grossman M, Garg A. Trichodysplasia spinulosa in a kidney transplant recipient. JAMA Dermatol. 2021;157:105.
- Leitenberger JJ, Abdelmalek M, Wang RC, et al. Two cases of trichodysplasia spinulosa responsive to compounded topical cidofovir 3% cream. JAAD Case Rep. 2015;1:S33-S35.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Nguyen KD, Chamseddin BH, Cockerell CJ, et al. The biology and clinical features of cutaneous polyomaviruses. J Invest Dermatol. 2019;139:285-292.
The Diagnosis: Trichodysplasia Spinulosa
Trichodysplasia spinulosa has been described in case reports over the last several decades, with its causative virus trichodysplasia spinulosa-associated polyomavirus (TSPyV) identified in 2010 by van der Meijden et al.1 Trichodysplasia spinulosa-associated polyomavirus is a small, nonenveloped, double-stranded DNA virus in the Polyomaviridae family, among several other known cutaneous polyomaviruses including Merkel cell polyomavirus, human polyomavirus (HPyV) 6, HPyV7, HPyV10, and possibly HPyV13.2 The primary target of TSPyV is follicular keratinocytes, and it is believed to cause trichodysplasia spinulosa by primary infection rather than by reactivation. Trichodysplasia spinulosa presents in immunosuppressed patients as a folliculocentric eruption of papules with keratinous spines on the face, often with concurrent alopecia, eventually spreading to the trunk and extremities.3 The diagnosis often is clinical, but a biopsy may be performed for histopathologic confirmation. Alternatively, lesional spicules can be painlessly collected manually and submitted for viral polymerase chain reaction (PCR).4 The diagnosis of trichodysplasia spinulosa can be difficult due to similarities with other more common conditions such as keratosis pilaris, milia, filiform warts, or lichen spinulosus.
Similar to trichodysplasia spinulosa, keratosis pilaris also presents with folliculocentric and often erythematous papules.5 Keratosis pilaris most frequently affects the posterior upper arms and thighs but also may affect the cheeks, as seen in trichodysplasia spinulosa. Differentiation between the 2 diagnoses can be made on a clinical basis, as keratosis pilaris lacks the characteristic keratinous spines and often spares the central face and nose, locations that commonly are affected in trichodysplasia spinulosa.3
Milia typically appear as white to yellow papules, often on the cheeks, eyelids, nose, and chin.6 Given their predilection for the face, milia can appear similarly to trichodysplasia spinulosa. Differentiation can be made clinically, as milia typically are not as numerous as the spiculed papules seen in trichodysplasia spinulosa. Morphologically, milia will present as smooth, dome-shaped papules as opposed to the keratinous spicules seen in trichodysplasia spinulosa. The diagnosis of milia can be confirmed by incision and removal of the white chalky keratin core, a feature absent in trichodysplasia spinulosa.
Filiform warts are benign epidermal proliferations caused by human papillomavirus infection that manifest as flesh-colored, verrucous, hyperkeratotic papules.7 They can appear on virtually any skin surface, including the face, and thus may be mistaken for trichodysplasia spinulosa. Close inspection usually will reveal tiny black dots that represent thrombosed capillaries, a feature lacking in trichodysplasia spinulosa. In long-standing lesions or immunocompromised patients, confluent verrucous plaques may develop.8 Diagnosis of filiform warts can be confirmed with biopsy, which will demonstrate a compact stratum corneum, coarse hypergranulosis, and papillomatosis curving inward, while biopsy of a trichodysplasia spinulosa lesion would show polyomavirus infection of the hair follicle and characteristic eosinophilic inclusion bodies.9
Lichen spinulosus may appear as multiple folliculocentric scaly papules with hairlike horny spines.10 Lichen spinulosus differs from trichodysplasia spinulosa in that it commonly appears on the neck, abdomen, trochanteric region, arms, elbows, or knees. Lichen spinulosus also classically appears as a concrete cluster of papules, often localized to a certain region, in contrast to trichodysplasia spinulosa, which will be widespread, often spreading over time. Finally, clinical history may help differentiate the 2 entities. Lichen spinulosus most often appears in children and adolescents and often has an indolent course, typically resolving during puberty, while trichodysplasia spinulosa is seen in immunocompromised patients.
In our patient, the dermatology team made a diagnosis of trichodysplasia spinulosa based on the characteristic clinical presentation, which was confirmed after approximately 10 lesional spicules were removed by tissue forceps and submitted for PCR analysis showing TSPyV (Figure). Two other cases utilized spicule PCR analysis for confirmation of TSPyV.11,12 This technique may represent a viable option for diagnostic confirmation in pediatric cases.
Although some articles have examined the molecular and biologic features of trichodysplasia spinulosa, literature on clinical presentation and management is limited to isolated case reports with no comprehensive studies to establish a standardized treatment. Of these reports, oral valganciclovir 900 mg daily, topical retinoids, cidofovir cream 1% to 3%, and decreasing or altering the immunosuppressive regimen all have been noted to provide clinical improvement.13,14 Other therapies including leflunomide and routine manual extraction of spicules also have shown effectiveness in the treatment of trichodysplasia spinulosa.15
In our patient, treatment included decreasing immunosuppression, as she was getting recurrent sinus and upper respiratory infections. Mycophenolate mofetil was discontinued, and the patient was continued solely on tacrolimus therapy. She demonstrated notable improvement after 3 months, with approximately 50% clearance of the eruption. A mutual decision was made at that visit to initiate therapy with compounded cidofovir cream 1% daily to the lesions until the next follow-up visit. Unfortunately, the patient did not return for her scheduled dermatology visits and was lost to long-term follow-up.
Acknowledgment
We thank Richard C. Wang, MD, PhD (Dallas, Texas), for his dermatologic expertise and assistance in analysis of lesional samples for TSPyV.
The Diagnosis: Trichodysplasia Spinulosa
Trichodysplasia spinulosa has been described in case reports over the last several decades, with its causative virus trichodysplasia spinulosa-associated polyomavirus (TSPyV) identified in 2010 by van der Meijden et al.1 Trichodysplasia spinulosa-associated polyomavirus is a small, nonenveloped, double-stranded DNA virus in the Polyomaviridae family, among several other known cutaneous polyomaviruses including Merkel cell polyomavirus, human polyomavirus (HPyV) 6, HPyV7, HPyV10, and possibly HPyV13.2 The primary target of TSPyV is follicular keratinocytes, and it is believed to cause trichodysplasia spinulosa by primary infection rather than by reactivation. Trichodysplasia spinulosa presents in immunosuppressed patients as a folliculocentric eruption of papules with keratinous spines on the face, often with concurrent alopecia, eventually spreading to the trunk and extremities.3 The diagnosis often is clinical, but a biopsy may be performed for histopathologic confirmation. Alternatively, lesional spicules can be painlessly collected manually and submitted for viral polymerase chain reaction (PCR).4 The diagnosis of trichodysplasia spinulosa can be difficult due to similarities with other more common conditions such as keratosis pilaris, milia, filiform warts, or lichen spinulosus.
Similar to trichodysplasia spinulosa, keratosis pilaris also presents with folliculocentric and often erythematous papules.5 Keratosis pilaris most frequently affects the posterior upper arms and thighs but also may affect the cheeks, as seen in trichodysplasia spinulosa. Differentiation between the 2 diagnoses can be made on a clinical basis, as keratosis pilaris lacks the characteristic keratinous spines and often spares the central face and nose, locations that commonly are affected in trichodysplasia spinulosa.3
Milia typically appear as white to yellow papules, often on the cheeks, eyelids, nose, and chin.6 Given their predilection for the face, milia can appear similarly to trichodysplasia spinulosa. Differentiation can be made clinically, as milia typically are not as numerous as the spiculed papules seen in trichodysplasia spinulosa. Morphologically, milia will present as smooth, dome-shaped papules as opposed to the keratinous spicules seen in trichodysplasia spinulosa. The diagnosis of milia can be confirmed by incision and removal of the white chalky keratin core, a feature absent in trichodysplasia spinulosa.
Filiform warts are benign epidermal proliferations caused by human papillomavirus infection that manifest as flesh-colored, verrucous, hyperkeratotic papules.7 They can appear on virtually any skin surface, including the face, and thus may be mistaken for trichodysplasia spinulosa. Close inspection usually will reveal tiny black dots that represent thrombosed capillaries, a feature lacking in trichodysplasia spinulosa. In long-standing lesions or immunocompromised patients, confluent verrucous plaques may develop.8 Diagnosis of filiform warts can be confirmed with biopsy, which will demonstrate a compact stratum corneum, coarse hypergranulosis, and papillomatosis curving inward, while biopsy of a trichodysplasia spinulosa lesion would show polyomavirus infection of the hair follicle and characteristic eosinophilic inclusion bodies.9
Lichen spinulosus may appear as multiple folliculocentric scaly papules with hairlike horny spines.10 Lichen spinulosus differs from trichodysplasia spinulosa in that it commonly appears on the neck, abdomen, trochanteric region, arms, elbows, or knees. Lichen spinulosus also classically appears as a concrete cluster of papules, often localized to a certain region, in contrast to trichodysplasia spinulosa, which will be widespread, often spreading over time. Finally, clinical history may help differentiate the 2 entities. Lichen spinulosus most often appears in children and adolescents and often has an indolent course, typically resolving during puberty, while trichodysplasia spinulosa is seen in immunocompromised patients.
In our patient, the dermatology team made a diagnosis of trichodysplasia spinulosa based on the characteristic clinical presentation, which was confirmed after approximately 10 lesional spicules were removed by tissue forceps and submitted for PCR analysis showing TSPyV (Figure). Two other cases utilized spicule PCR analysis for confirmation of TSPyV.11,12 This technique may represent a viable option for diagnostic confirmation in pediatric cases.
Although some articles have examined the molecular and biologic features of trichodysplasia spinulosa, literature on clinical presentation and management is limited to isolated case reports with no comprehensive studies to establish a standardized treatment. Of these reports, oral valganciclovir 900 mg daily, topical retinoids, cidofovir cream 1% to 3%, and decreasing or altering the immunosuppressive regimen all have been noted to provide clinical improvement.13,14 Other therapies including leflunomide and routine manual extraction of spicules also have shown effectiveness in the treatment of trichodysplasia spinulosa.15
In our patient, treatment included decreasing immunosuppression, as she was getting recurrent sinus and upper respiratory infections. Mycophenolate mofetil was discontinued, and the patient was continued solely on tacrolimus therapy. She demonstrated notable improvement after 3 months, with approximately 50% clearance of the eruption. A mutual decision was made at that visit to initiate therapy with compounded cidofovir cream 1% daily to the lesions until the next follow-up visit. Unfortunately, the patient did not return for her scheduled dermatology visits and was lost to long-term follow-up.
Acknowledgment
We thank Richard C. Wang, MD, PhD (Dallas, Texas), for his dermatologic expertise and assistance in analysis of lesional samples for TSPyV.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromised patient. PLoS Pathog. 2010;6:E1001024.
- Sheu JC, Tran J, Rady PL, et al. Polyomaviruses of the skin: integrating molecular and clinical advances in an emerging class of viruses. Br J Dermatol. 2019;180:1302-1311.
- Sperling LC, Tomaszewski MM, Thomas DA. Viral-associated trichodysplasia in patients who are immunocompromised. J Am Acad Dermatol. 2004;50:318-322.
- Wu JH, Nguyen HP, Rady PL, et al. Molecular insight into the viral biology and clinical features of trichodysplasia spinulosa. Br J Dermatol. 2016;174:490-498.
- Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Micali G, Dall'Oglio F, Nasca MR, et al. Management of cutaneous warts: an evidence-based approach. Am J Clin Dermatol. 2004;5:311-317.
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier; 2018.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Tilly JJ, Drolet BA, Esterly NB. Lichenoid eruptions in children. J Am Acad Dermatol. 2004;51:606-624.
- Chamseddin BH, Tran BAPD, Lee EE, et al. Trichodysplasia spinulosa in a child: identification of trichodysplasia spinulosa-associated polyomavirus in skin, serum, and urine. Pediatr Dermatol. 2019;36:723-724.
- Sonstegard A, Grossman M, Garg A. Trichodysplasia spinulosa in a kidney transplant recipient. JAMA Dermatol. 2021;157:105.
- Leitenberger JJ, Abdelmalek M, Wang RC, et al. Two cases of trichodysplasia spinulosa responsive to compounded topical cidofovir 3% cream. JAAD Case Rep. 2015;1:S33-S35.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Nguyen KD, Chamseddin BH, Cockerell CJ, et al. The biology and clinical features of cutaneous polyomaviruses. J Invest Dermatol. 2019;139:285-292.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromised patient. PLoS Pathog. 2010;6:E1001024.
- Sheu JC, Tran J, Rady PL, et al. Polyomaviruses of the skin: integrating molecular and clinical advances in an emerging class of viruses. Br J Dermatol. 2019;180:1302-1311.
- Sperling LC, Tomaszewski MM, Thomas DA. Viral-associated trichodysplasia in patients who are immunocompromised. J Am Acad Dermatol. 2004;50:318-322.
- Wu JH, Nguyen HP, Rady PL, et al. Molecular insight into the viral biology and clinical features of trichodysplasia spinulosa. Br J Dermatol. 2016;174:490-498.
- Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Micali G, Dall'Oglio F, Nasca MR, et al. Management of cutaneous warts: an evidence-based approach. Am J Clin Dermatol. 2004;5:311-317.
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 4th ed. Elsevier; 2018.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Tilly JJ, Drolet BA, Esterly NB. Lichenoid eruptions in children. J Am Acad Dermatol. 2004;51:606-624.
- Chamseddin BH, Tran BAPD, Lee EE, et al. Trichodysplasia spinulosa in a child: identification of trichodysplasia spinulosa-associated polyomavirus in skin, serum, and urine. Pediatr Dermatol. 2019;36:723-724.
- Sonstegard A, Grossman M, Garg A. Trichodysplasia spinulosa in a kidney transplant recipient. JAMA Dermatol. 2021;157:105.
- Leitenberger JJ, Abdelmalek M, Wang RC, et al. Two cases of trichodysplasia spinulosa responsive to compounded topical cidofovir 3% cream. JAAD Case Rep. 2015;1:S33-S35.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Nguyen KD, Chamseddin BH, Cockerell CJ, et al. The biology and clinical features of cutaneous polyomaviruses. J Invest Dermatol. 2019;139:285-292.
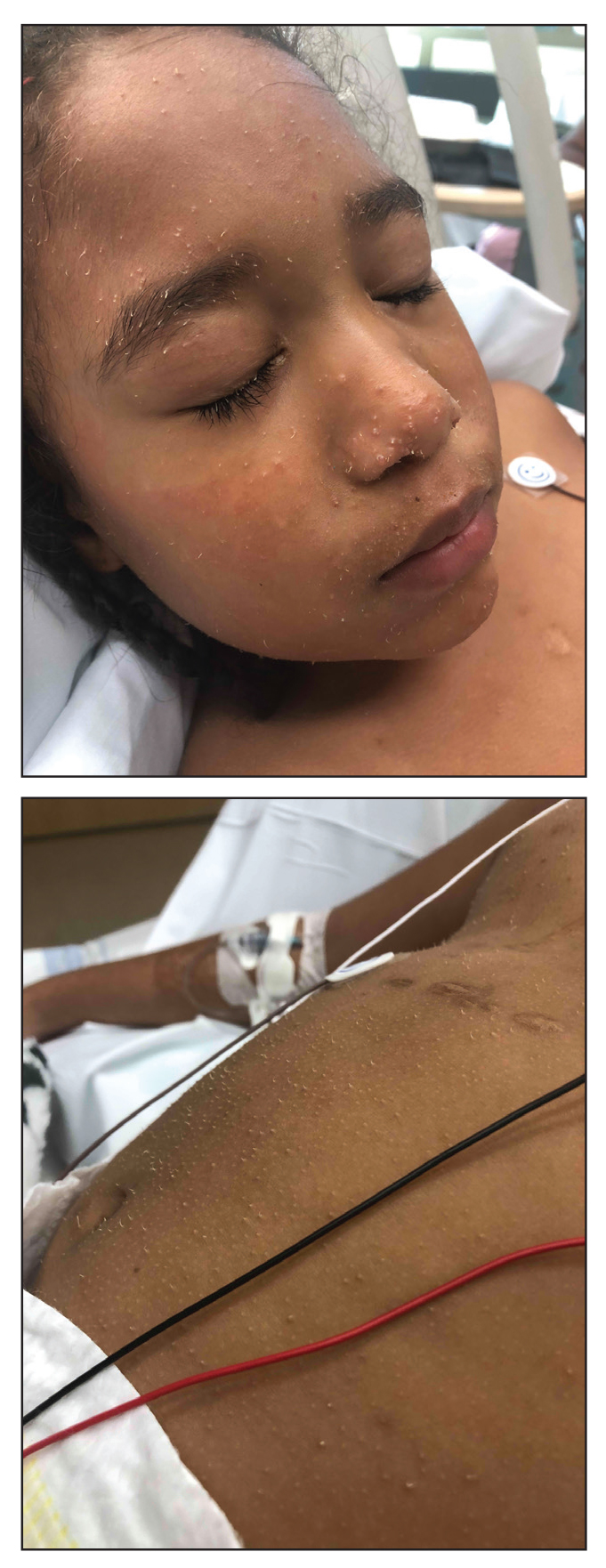
A 4-year-old girl with a history of cardiac transplantation 1 year prior for dilated cardiomyopathy presented to the dermatology consultation service with widespread hyperkeratotic papules of 2 months’ duration. The eruption initially had appeared on the face with subsequent involvement of the trunk and extremities. Her immunosuppressive medications included oral tacrolimus and mycophenolate mofetil. No over-the-counter or prescription treatments had been used for the eruption; the patient’s mother had been manually extracting the spicules from the nose, cheeks, and forehead with tweezers. The lesions were asymptomatic with only mild follicular erythema. Physical examination revealed multiple folliculocentric keratinous spicules on the nose, cheeks, forehead (top), trunk (bottom), arms, and legs.
Psoriatic Alopecia in a Patient With Crohn Disease: An Uncommon Manifestation of Tumor Necrosis Factor α Inhibitors
Tumor necrosis factor α (TNF-α) inhibitor–induced psoriasis is a known paradoxical adverse effect of this family of medications, which includes infliximab, adalimumab, etanercept, golimumab, and certolizumab. In the pediatric population, these therapies recently gained approval for nondermatologic conditions—meaning that this phenomenon is encountered more frequently.1 In a systematic review of TNF-α inhibitor–induced psoriasis, severe scalp involvement was associated with alopecia in 7.5% of cases.2 Onset of scalp psoriasis with alopecia in patients being treated with a TNF-α inhibitor should lead to consideration of this condition.
Psoriatic alopecia is an uncommon presentation of psoriasis. Although well described, alopecia as a clinical manifestation of scalp psoriasis is not a well-known concept among clinicians and has never been widely accepted. Adding to the diagnostic challenge is that psoriatic alopecia secondary to TNF-α inhibitor–induced psoriasis rarely has been reported in adults or children.3-5 Including our case, our review of the literature yielded 7 pediatric cases (≤18 years) of TNF-α inhibitor–induced psoriatic alopecia.6,7 A primary literature search of PubMed articles indexed for MEDLINE was conducted using the terms psoriatic alopecia, psoriasiform alopecia, TNF-α inhibitors, infliximab, adalimumab, etanercept, golimumab, and certolizumab.
We present the case of a pediatric patient with psoriatic alopecia secondary to treatment with adalimumab for Crohn disease (CD). We also provide a review of reported cases of psoriatic alopecia induced by a TNF-α inhibitor in the literature.
Case Report
A 12-year-old girl presented to our dermatology clinic with erythematous scaly plaques on the trunk, scalp, arms, and legs of 2 months’ duration. The lesions involved approximately 15% of the body surface area. The patient’s medical history was remarkable for CD diagnosed 4 years prior to presentation of the skin lesions. She had been treated for the past 2 years with adalimumab 40 mg once every 2 weeks and azathioprine 100 mg once daily. Because her CD was poorly controlled, the dosage of adalimumab was increased to 40 mg once weekly 6 months prior to the current presentation.
Our diagnosis was TNF-α inhibitor-induced psoriasis secondary to treatment with adalimumab.
The patient was treated with mometasone lotion 0.1% for the scalp lesions and triamcinolone cream 0.1% for the body lesions. Because of the extent of the psoriasis, we recommended changing adalimumab to ustekinumab, which is approved for CD in adults but is off label in children.
At 1-month follow-up, after receiving the induction dose of ustekinumab, the patient presented with partial improvement of the skin lesions but had developed a large, alopecic, erythematous plaque with thick yellowish scales on the scalp (Figure 1). She also had a positive hair pull test. The presumptive initial diagnosis of the alopecic scalp lesion was tinea capitis, for which multiple potassium hydroxide preparations of scales were performed, all yielding negative results. In addition, histopathologic examination with hematoxylin and eosin staining was performed (Figures 2A and 2B). Sterile tissue cultures for bacteria, fungi, and acid-fast bacilli were obtained and showed no growth. Periodic acid–Schiff staining was negative for fungal structures.
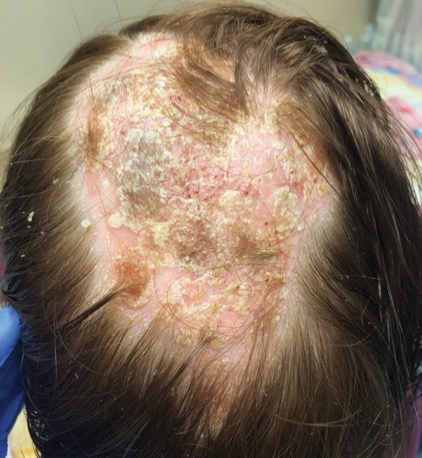
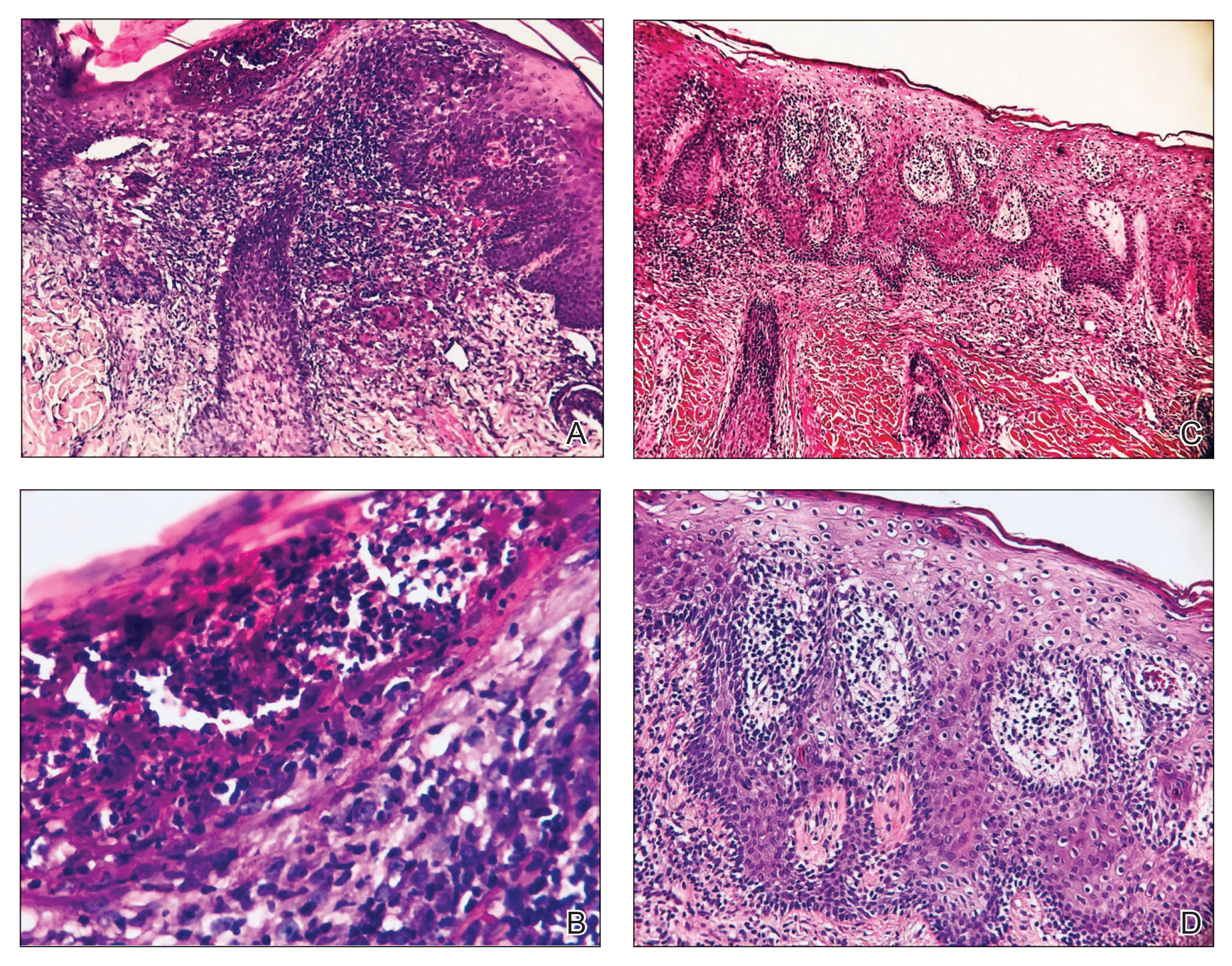
A second biopsy showed a psoriasiform pattern, parakeratosis, and hypogranulosis, highly suggestive of psoriasis (Figure 2C and 2D). Based on those findings, a diagnosis of psoriatic alopecia was made. The mometasone was switched to clobetasol lotion 0.05%. The patient continued treatment with ustekinumab. At 6-month follow-up, her CD was well controlled and she showed hair regrowth in previously alopecic areas (Figure 3).

Comment
Psoriatic alopecia induced by a TNF-α inhibitor was first reported in 2007 in a 30-year-old woman with ankylosing spondylitis who was being treated with adalimumab.8 She had erythematous, scaly, alopecic plaques on the scalp and palmoplantar pustulosis. Findings on skin biopsy were compatible with psoriasis. The patient’s severe scalp psoriasis failed to respond to topical steroid treatment and adalimumab cessation. The extensive hair loss responded to cyclosporine 3 mg/kg daily.8
After conducting an extensive literature review, we found 26 cases of TNF-α–induced psoriatic alopecia, including the current case (Table).6-16 The mean age at diagnosis was 27.8 years (SD, 13.6 years; range, 7–60 years). The female-to-male ratio was 3.3:1. The most common underlying condition for which TNF-α inhibitors were prescribed was CD (77% [20/26]). Psoriatic alopecia most commonly was reported secondary to treatment with infliximab (54% [14/26]), followed by adalimumab (42% [11/26]). Golimumab was the causative drug in 1 (4%) case. We did not find reports of etanercept or certolizumab having induced this manifestation. The onset of the scalp lesions occurred 2 to 46 months after starting treatment with the causative medication.
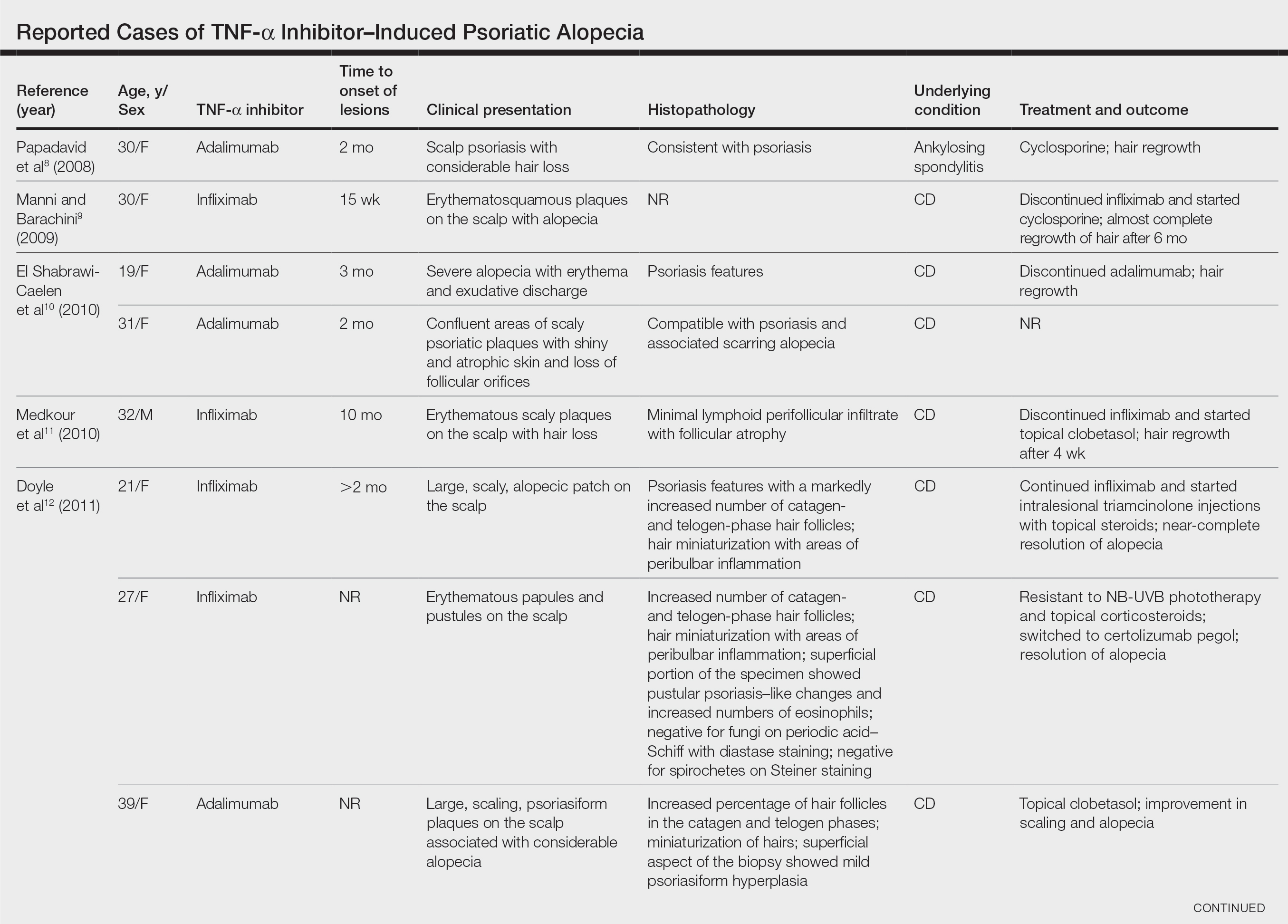
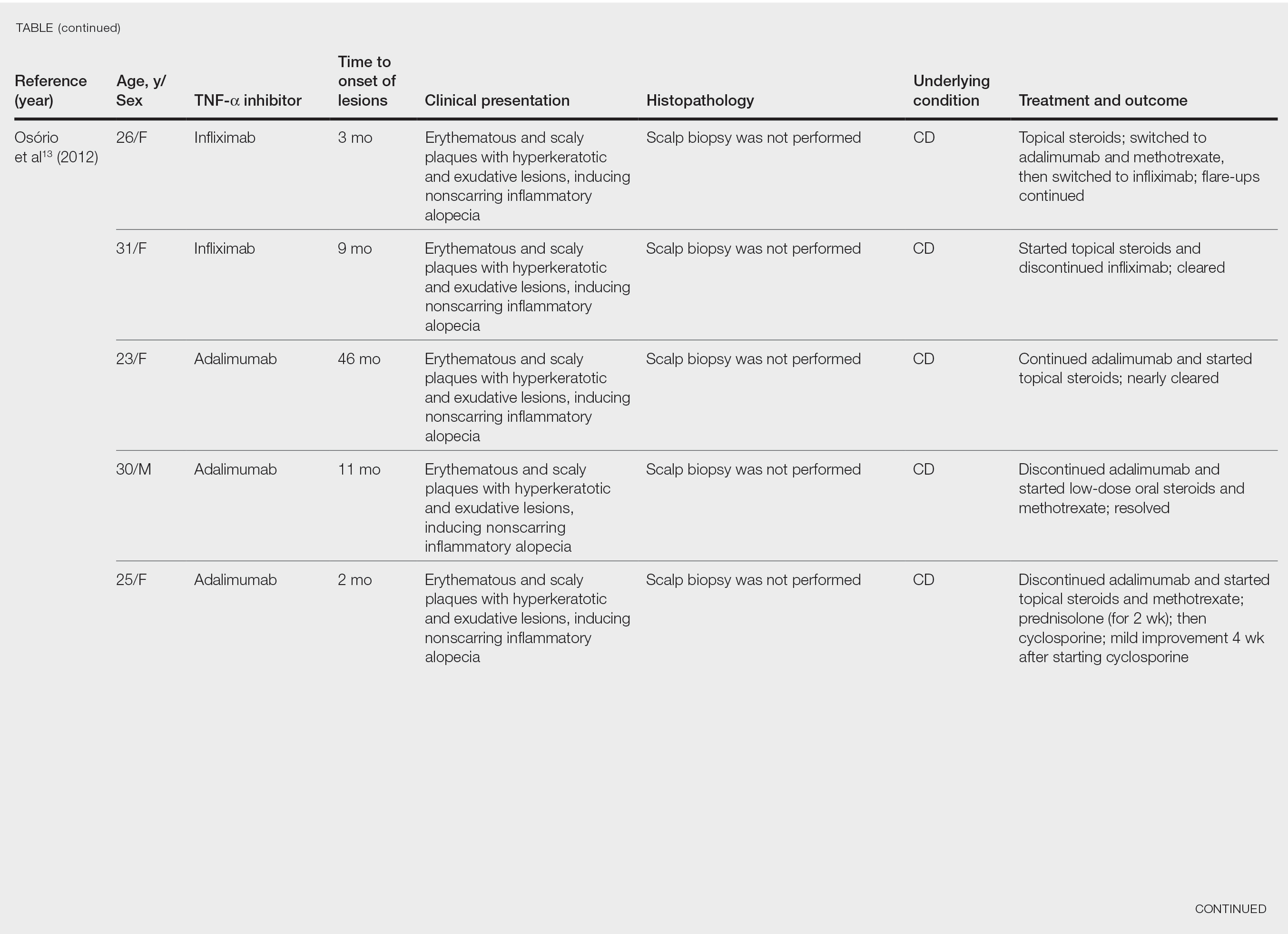
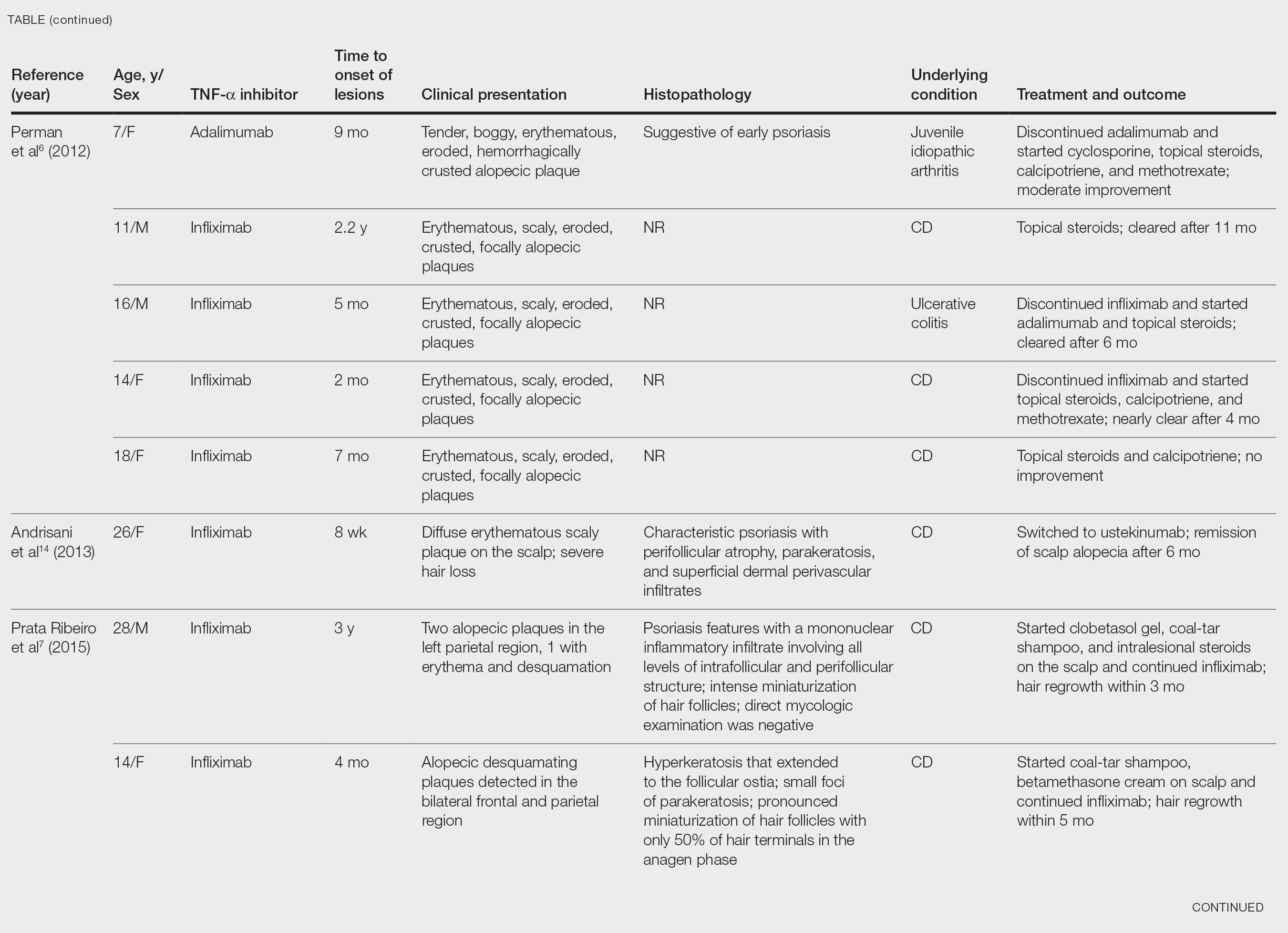
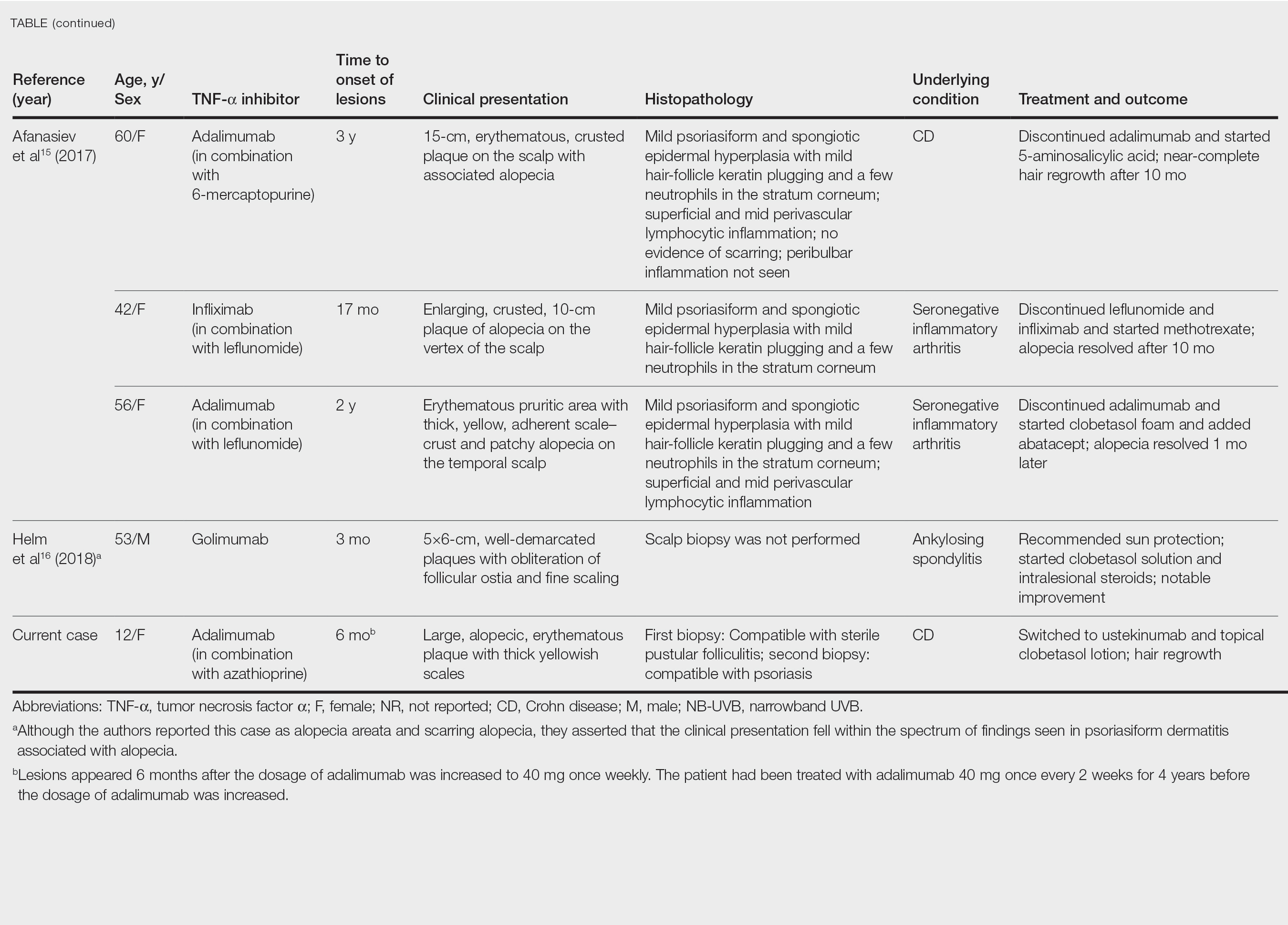
Laga et al17 reported that TNF-α inhibitor–induced psoriasis can have a variety of histopathologic findings, including typical findings of various stages of psoriasis, a lichenoid pattern mimicking remnants of lichen planus, and sterile pustular folliculitis. Our patient’s 2 scalp biopsies demonstrated results consistent with findings reported by Laga et al.17 In the first biopsy, findings were consistent with a dense neutrophilic infiltrate with negative sterile cultures and negative periodic acid–Schiff stain (sterile folliculitis), with crust and areas of parakeratosis. The second biopsy demonstrated psoriasiform hyperplasia, parakeratosis, and an absent granular layer, all typical features of psoriasis (Figure 2).
Including the current case, our review of the literature yielded 7 pediatric (ie, 0–18 years of age) cases of TNF-α inhibitor–induced psoriatic alopecia. Of the 6 previously reported pediatric cases, 5 occurred after administration of infliximab.6,7
Similar to our case, TNF-α inhibitor–induced psoriatic alopecia was reported in a 7-year-old girl who was treated with adalimumab for juvenile idiopathic arthritis.6 Nine months after starting treatment, that patient presented with a tender, erythematous, eroded, and crusted alopecic plaque along with scaly plaques on the scalp. Adalimumab was discontinued, and cyclosporine and topical steroids were started. Cyclosporine was then discontinued due to partial resolution of the psoriasis; the patient was started on abatacept, with persistence of the psoriasis and alopecia. The patient was then started on oral methotrexate 12.5 mg once weekly with moderate improvement and mild to moderate exacerbations.
Tumor necrosis factor α inhibitor–induced psoriasis may occur as a result of a cytokine imbalance. A TNF-α blockade leads to upregulation of interferon α (IFN-α) and TNF-α production by plasmacytoid dendritic cells (pDCs), usually in genetically susceptible people.6,7,9-15 The IFN-α induces maturation of myeloid dendritic cells (mDCs) responsible for increasing proinflammatory cytokines that contribute to psoriasis.11 Generation of TNF-α by pDCs leads to mature or activated dendritic cells derived from pDCs through autocrine TNF-α production and paracrine IFN-α production from immature mDCs.9 Once pDCs mature, they are incapable of producing IFN-α; TNF-α then inhibits IFN-α production by inducing pDC maturation.11 Overproduction of IFN-α during TNF-α inhibition induces expression of the chemokine receptor CXCR3 on T cells, which recruits T cells to the dermis. The T cells then produce TNF-α, causing psoriatic skin lesions.10,11,13,14
Although TNF-α inhibitor–induced psoriatic alopecia is uncommon, the condition should be considered in female patients with underlying proinflammatory disease—CD in particular. Perman et al6 reported 5 cases of psoriatic alopecia in which 3 patients initially were treated with griseofulvin because of suspected tinea capitis.
Conditions with similar clinical findings should be ruled out before making a diagnosis of TNF-α inhibitor–induced psoriatic alopecia. Although clinicopathologic correlation is essential for making the diagnosis, it is possible that the histologic findings will not be specific for psoriasis.17 It is important to be aware of this condition in patients being treated with a TNF-α inhibitor as early as 2 months to 4 years or longer after starting treatment.
Previously reported cases have demonstrated various treatment options that yielded improvement or resolution of TNF-α inhibitor–induced psoriatic alopecia. These include either continuation or discontinuation of the TNF-α inhibitor combined with topical or intralesional steroids, methotrexate, or cyclosporine. Another option is to switch the TNF-α inhibitor to another biologic. Outcomes vary from patient to patient, making the physician’s clinical judgment crucial in deciding which treatment route to take. Our patient showed notable improvement when she was switched from adalimumab to ustekinumab as well as the combination of ustekinumab and clobetasol lotion 0.05%.
Conclusion
We recommend an individualized approach that provides patients with the safest and least invasive treatment option for TNF-α inhibitor–induced psoriatic alopecia. In most reported cases, the problem resolved with treatment, thereby classifying this form of alopecia as noncicatricial alopecia.
- Horneff G, Seyger MMB, Arikan D, et al. Safety of adalimumab in pediatric patients with polyarticular juvenile idiopathic arthritis, enthesitis-related arthritis, psoriasis, and Crohn’s disease. J Pediatr. 2018;201:166-175.e3. doi:10.1016/j.jpeds.2018.05.042
- Brown G, Wang E, Leon A, et al. Tumor necrosis factor-α inhibitor-induced psoriasis: systematic review of clinical features, histopathological findings, and management experience. J Am Acad Dermatol. 2017;76:334-341. doi:10.1016/j.jaad.2016.08.012
- George SMC, Taylor MR, Farrant PBJ. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721. doi:10.1111/ced.12715
- Shuster S. Psoriatic alopecia. Br J Dermatol. 1972;87:73-77. doi:10.1111/j.1365-2133.1972.tb05103.x
- Silva CY, Brown KL, Kurban AK, et al. Psoriatic alopecia—fact or fiction? a clinicohistopathologic reappraisal. Indian J Dermatol Venereol Leprol. 2012;78:611-619. doi:10.4103/0378-6323.100574
- Perman MJ, Lovell DJ, Denson LA, et al. Five cases of anti-tumor necrosis factor alpha-induced psoriasis presenting with severe scalp involvement in children. Pediatr Dermatol. 2012;29:454-459. doi:10.1111/j.1525-1470.2011.01521.x
- Prata Ribeiro LB, Gonçalves Rego JC, Duque Estrada B, et al. Alopecia secondary to anti-tumor necrosis factor-alpha therapy. An Bras Dermatol. 2015;90:232–235. doi:10.1590/abd1806-4841.20153084
- Papadavid E, Gazi S, Dalamaga M, et al. Palmoplantar and scalp psoriasis occurring during anti-tumour necrosis factor-alpha therapy: a case series of four patients and guidelines for management. J Eur Acad Dermatol Venereol. 2008;22:380-382. doi:10.1111/j.1468-3083.2007.02335.x
- Manni E, Barachini P. Psoriasis induced by infliximab in a patient suffering from Crohn’s disease. Int J Immunopathol Pharmacol. 2009;22:841-844. doi:10.1177/039463200902200331
- El Shabrawi-Caelen L, La Placa M, Vincenzi C, et al. Adalimumab-induced psoriasis of the scalp with diffuse alopecia: a severe potentially irreversible cutaneous side effect of TNF-alpha blockers. Inflamm Bowel Dis. 2010;16:182-183. doi:10.1002/ibd.20954
- Medkour F, Babai S, Chanteloup E, et al. Development of diffuse psoriasis with alopecia during treatment of Crohn’s disease with infliximab. Gastroenterol Clin Biol. 2010;34:140-141. doi:10.1016/j.gcb.2009.10.021
- Doyle LA, Sperling LC, Baksh S, et al. Psoriatic alopecia/alopecia areata-like reactions secondary to anti-tumor necrosis factor-α therapy: a novel cause of noncicatricial alopecia. Am J Dermatopathol. 2011;33:161-166. doi:10.1097/DAD.0b013e3181ef7403
- Osório F, Magro F, Lisboa C, et al. Anti-TNF-alpha induced psoriasiform eruptions with severe scalp involvement and alopecia: report of five cases and review of the literature. Dermatology. 2012;225:163-167. doi:10.1159/000342503
- Andrisani G, Marzo M, Celleno L, et al. Development of psoriasis scalp with alopecia during treatment of Crohn’s disease with infliximab and rapid response to both diseases to ustekinumab. Eur Rev Med Pharmacol Sci. 2013;17:2831-2836.
- Afanasiev OK, Zhang CZ, Ruhoy SM. TNF-inhibitor associated psoriatic alopecia: diagnostic utility of sebaceous lobule atrophy. J Cutan Pathol. 2017;44:563-569. doi:10.1111/cup.12932
- Helm MM, Haddad S. Alopecia areata and scarring alopecia presenting during golimumab therapy for ankylosing spondylitis. N Am J Med Sci. 2018;11:22-24. doi:10.7156/najms.2018.110122
- Laga AC, Vleugels RA, Qureshi AA, et al. Histopathologic spectrum of psoriasiform skin reactions associated with tumor necrosis factor-a inhibitor therapy. a study of 16 biopsies. Am J Dermatopathol. 2010;32:568-573. doi:10.1097/DAD.0b013e3181cb3ff7
Tumor necrosis factor α (TNF-α) inhibitor–induced psoriasis is a known paradoxical adverse effect of this family of medications, which includes infliximab, adalimumab, etanercept, golimumab, and certolizumab. In the pediatric population, these therapies recently gained approval for nondermatologic conditions—meaning that this phenomenon is encountered more frequently.1 In a systematic review of TNF-α inhibitor–induced psoriasis, severe scalp involvement was associated with alopecia in 7.5% of cases.2 Onset of scalp psoriasis with alopecia in patients being treated with a TNF-α inhibitor should lead to consideration of this condition.
Psoriatic alopecia is an uncommon presentation of psoriasis. Although well described, alopecia as a clinical manifestation of scalp psoriasis is not a well-known concept among clinicians and has never been widely accepted. Adding to the diagnostic challenge is that psoriatic alopecia secondary to TNF-α inhibitor–induced psoriasis rarely has been reported in adults or children.3-5 Including our case, our review of the literature yielded 7 pediatric cases (≤18 years) of TNF-α inhibitor–induced psoriatic alopecia.6,7 A primary literature search of PubMed articles indexed for MEDLINE was conducted using the terms psoriatic alopecia, psoriasiform alopecia, TNF-α inhibitors, infliximab, adalimumab, etanercept, golimumab, and certolizumab.
We present the case of a pediatric patient with psoriatic alopecia secondary to treatment with adalimumab for Crohn disease (CD). We also provide a review of reported cases of psoriatic alopecia induced by a TNF-α inhibitor in the literature.
Case Report
A 12-year-old girl presented to our dermatology clinic with erythematous scaly plaques on the trunk, scalp, arms, and legs of 2 months’ duration. The lesions involved approximately 15% of the body surface area. The patient’s medical history was remarkable for CD diagnosed 4 years prior to presentation of the skin lesions. She had been treated for the past 2 years with adalimumab 40 mg once every 2 weeks and azathioprine 100 mg once daily. Because her CD was poorly controlled, the dosage of adalimumab was increased to 40 mg once weekly 6 months prior to the current presentation.
Our diagnosis was TNF-α inhibitor-induced psoriasis secondary to treatment with adalimumab.
The patient was treated with mometasone lotion 0.1% for the scalp lesions and triamcinolone cream 0.1% for the body lesions. Because of the extent of the psoriasis, we recommended changing adalimumab to ustekinumab, which is approved for CD in adults but is off label in children.
At 1-month follow-up, after receiving the induction dose of ustekinumab, the patient presented with partial improvement of the skin lesions but had developed a large, alopecic, erythematous plaque with thick yellowish scales on the scalp (Figure 1). She also had a positive hair pull test. The presumptive initial diagnosis of the alopecic scalp lesion was tinea capitis, for which multiple potassium hydroxide preparations of scales were performed, all yielding negative results. In addition, histopathologic examination with hematoxylin and eosin staining was performed (Figures 2A and 2B). Sterile tissue cultures for bacteria, fungi, and acid-fast bacilli were obtained and showed no growth. Periodic acid–Schiff staining was negative for fungal structures.
A second biopsy showed a psoriasiform pattern, parakeratosis, and hypogranulosis, highly suggestive of psoriasis (Figure 2C and 2D). Based on those findings, a diagnosis of psoriatic alopecia was made. The mometasone was switched to clobetasol lotion 0.05%. The patient continued treatment with ustekinumab. At 6-month follow-up, her CD was well controlled and she showed hair regrowth in previously alopecic areas (Figure 3).
Comment
Psoriatic alopecia induced by a TNF-α inhibitor was first reported in 2007 in a 30-year-old woman with ankylosing spondylitis who was being treated with adalimumab.8 She had erythematous, scaly, alopecic plaques on the scalp and palmoplantar pustulosis. Findings on skin biopsy were compatible with psoriasis. The patient’s severe scalp psoriasis failed to respond to topical steroid treatment and adalimumab cessation. The extensive hair loss responded to cyclosporine 3 mg/kg daily.8
After conducting an extensive literature review, we found 26 cases of TNF-α–induced psoriatic alopecia, including the current case (Table).6-16 The mean age at diagnosis was 27.8 years (SD, 13.6 years; range, 7–60 years). The female-to-male ratio was 3.3:1. The most common underlying condition for which TNF-α inhibitors were prescribed was CD (77% [20/26]). Psoriatic alopecia most commonly was reported secondary to treatment with infliximab (54% [14/26]), followed by adalimumab (42% [11/26]). Golimumab was the causative drug in 1 (4%) case. We did not find reports of etanercept or certolizumab having induced this manifestation. The onset of the scalp lesions occurred 2 to 46 months after starting treatment with the causative medication.
Laga et al17 reported that TNF-α inhibitor–induced psoriasis can have a variety of histopathologic findings, including typical findings of various stages of psoriasis, a lichenoid pattern mimicking remnants of lichen planus, and sterile pustular folliculitis. Our patient’s 2 scalp biopsies demonstrated results consistent with findings reported by Laga et al.17 In the first biopsy, findings were consistent with a dense neutrophilic infiltrate with negative sterile cultures and negative periodic acid–Schiff stain (sterile folliculitis), with crust and areas of parakeratosis. The second biopsy demonstrated psoriasiform hyperplasia, parakeratosis, and an absent granular layer, all typical features of psoriasis (Figure 2).
Including the current case, our review of the literature yielded 7 pediatric (ie, 0–18 years of age) cases of TNF-α inhibitor–induced psoriatic alopecia. Of the 6 previously reported pediatric cases, 5 occurred after administration of infliximab.6,7
Similar to our case, TNF-α inhibitor–induced psoriatic alopecia was reported in a 7-year-old girl who was treated with adalimumab for juvenile idiopathic arthritis.6 Nine months after starting treatment, that patient presented with a tender, erythematous, eroded, and crusted alopecic plaque along with scaly plaques on the scalp. Adalimumab was discontinued, and cyclosporine and topical steroids were started. Cyclosporine was then discontinued due to partial resolution of the psoriasis; the patient was started on abatacept, with persistence of the psoriasis and alopecia. The patient was then started on oral methotrexate 12.5 mg once weekly with moderate improvement and mild to moderate exacerbations.
Tumor necrosis factor α inhibitor–induced psoriasis may occur as a result of a cytokine imbalance. A TNF-α blockade leads to upregulation of interferon α (IFN-α) and TNF-α production by plasmacytoid dendritic cells (pDCs), usually in genetically susceptible people.6,7,9-15 The IFN-α induces maturation of myeloid dendritic cells (mDCs) responsible for increasing proinflammatory cytokines that contribute to psoriasis.11 Generation of TNF-α by pDCs leads to mature or activated dendritic cells derived from pDCs through autocrine TNF-α production and paracrine IFN-α production from immature mDCs.9 Once pDCs mature, they are incapable of producing IFN-α; TNF-α then inhibits IFN-α production by inducing pDC maturation.11 Overproduction of IFN-α during TNF-α inhibition induces expression of the chemokine receptor CXCR3 on T cells, which recruits T cells to the dermis. The T cells then produce TNF-α, causing psoriatic skin lesions.10,11,13,14
Although TNF-α inhibitor–induced psoriatic alopecia is uncommon, the condition should be considered in female patients with underlying proinflammatory disease—CD in particular. Perman et al6 reported 5 cases of psoriatic alopecia in which 3 patients initially were treated with griseofulvin because of suspected tinea capitis.
Conditions with similar clinical findings should be ruled out before making a diagnosis of TNF-α inhibitor–induced psoriatic alopecia. Although clinicopathologic correlation is essential for making the diagnosis, it is possible that the histologic findings will not be specific for psoriasis.17 It is important to be aware of this condition in patients being treated with a TNF-α inhibitor as early as 2 months to 4 years or longer after starting treatment.
Previously reported cases have demonstrated various treatment options that yielded improvement or resolution of TNF-α inhibitor–induced psoriatic alopecia. These include either continuation or discontinuation of the TNF-α inhibitor combined with topical or intralesional steroids, methotrexate, or cyclosporine. Another option is to switch the TNF-α inhibitor to another biologic. Outcomes vary from patient to patient, making the physician’s clinical judgment crucial in deciding which treatment route to take. Our patient showed notable improvement when she was switched from adalimumab to ustekinumab as well as the combination of ustekinumab and clobetasol lotion 0.05%.
Conclusion
We recommend an individualized approach that provides patients with the safest and least invasive treatment option for TNF-α inhibitor–induced psoriatic alopecia. In most reported cases, the problem resolved with treatment, thereby classifying this form of alopecia as noncicatricial alopecia.
Tumor necrosis factor α (TNF-α) inhibitor–induced psoriasis is a known paradoxical adverse effect of this family of medications, which includes infliximab, adalimumab, etanercept, golimumab, and certolizumab. In the pediatric population, these therapies recently gained approval for nondermatologic conditions—meaning that this phenomenon is encountered more frequently.1 In a systematic review of TNF-α inhibitor–induced psoriasis, severe scalp involvement was associated with alopecia in 7.5% of cases.2 Onset of scalp psoriasis with alopecia in patients being treated with a TNF-α inhibitor should lead to consideration of this condition.
Psoriatic alopecia is an uncommon presentation of psoriasis. Although well described, alopecia as a clinical manifestation of scalp psoriasis is not a well-known concept among clinicians and has never been widely accepted. Adding to the diagnostic challenge is that psoriatic alopecia secondary to TNF-α inhibitor–induced psoriasis rarely has been reported in adults or children.3-5 Including our case, our review of the literature yielded 7 pediatric cases (≤18 years) of TNF-α inhibitor–induced psoriatic alopecia.6,7 A primary literature search of PubMed articles indexed for MEDLINE was conducted using the terms psoriatic alopecia, psoriasiform alopecia, TNF-α inhibitors, infliximab, adalimumab, etanercept, golimumab, and certolizumab.
We present the case of a pediatric patient with psoriatic alopecia secondary to treatment with adalimumab for Crohn disease (CD). We also provide a review of reported cases of psoriatic alopecia induced by a TNF-α inhibitor in the literature.
Case Report
A 12-year-old girl presented to our dermatology clinic with erythematous scaly plaques on the trunk, scalp, arms, and legs of 2 months’ duration. The lesions involved approximately 15% of the body surface area. The patient’s medical history was remarkable for CD diagnosed 4 years prior to presentation of the skin lesions. She had been treated for the past 2 years with adalimumab 40 mg once every 2 weeks and azathioprine 100 mg once daily. Because her CD was poorly controlled, the dosage of adalimumab was increased to 40 mg once weekly 6 months prior to the current presentation.
Our diagnosis was TNF-α inhibitor-induced psoriasis secondary to treatment with adalimumab.
The patient was treated with mometasone lotion 0.1% for the scalp lesions and triamcinolone cream 0.1% for the body lesions. Because of the extent of the psoriasis, we recommended changing adalimumab to ustekinumab, which is approved for CD in adults but is off label in children.
At 1-month follow-up, after receiving the induction dose of ustekinumab, the patient presented with partial improvement of the skin lesions but had developed a large, alopecic, erythematous plaque with thick yellowish scales on the scalp (Figure 1). She also had a positive hair pull test. The presumptive initial diagnosis of the alopecic scalp lesion was tinea capitis, for which multiple potassium hydroxide preparations of scales were performed, all yielding negative results. In addition, histopathologic examination with hematoxylin and eosin staining was performed (Figures 2A and 2B). Sterile tissue cultures for bacteria, fungi, and acid-fast bacilli were obtained and showed no growth. Periodic acid–Schiff staining was negative for fungal structures.
A second biopsy showed a psoriasiform pattern, parakeratosis, and hypogranulosis, highly suggestive of psoriasis (Figure 2C and 2D). Based on those findings, a diagnosis of psoriatic alopecia was made. The mometasone was switched to clobetasol lotion 0.05%. The patient continued treatment with ustekinumab. At 6-month follow-up, her CD was well controlled and she showed hair regrowth in previously alopecic areas (Figure 3).
Comment
Psoriatic alopecia induced by a TNF-α inhibitor was first reported in 2007 in a 30-year-old woman with ankylosing spondylitis who was being treated with adalimumab.8 She had erythematous, scaly, alopecic plaques on the scalp and palmoplantar pustulosis. Findings on skin biopsy were compatible with psoriasis. The patient’s severe scalp psoriasis failed to respond to topical steroid treatment and adalimumab cessation. The extensive hair loss responded to cyclosporine 3 mg/kg daily.8
After conducting an extensive literature review, we found 26 cases of TNF-α–induced psoriatic alopecia, including the current case (Table).6-16 The mean age at diagnosis was 27.8 years (SD, 13.6 years; range, 7–60 years). The female-to-male ratio was 3.3:1. The most common underlying condition for which TNF-α inhibitors were prescribed was CD (77% [20/26]). Psoriatic alopecia most commonly was reported secondary to treatment with infliximab (54% [14/26]), followed by adalimumab (42% [11/26]). Golimumab was the causative drug in 1 (4%) case. We did not find reports of etanercept or certolizumab having induced this manifestation. The onset of the scalp lesions occurred 2 to 46 months after starting treatment with the causative medication.
Laga et al17 reported that TNF-α inhibitor–induced psoriasis can have a variety of histopathologic findings, including typical findings of various stages of psoriasis, a lichenoid pattern mimicking remnants of lichen planus, and sterile pustular folliculitis. Our patient’s 2 scalp biopsies demonstrated results consistent with findings reported by Laga et al.17 In the first biopsy, findings were consistent with a dense neutrophilic infiltrate with negative sterile cultures and negative periodic acid–Schiff stain (sterile folliculitis), with crust and areas of parakeratosis. The second biopsy demonstrated psoriasiform hyperplasia, parakeratosis, and an absent granular layer, all typical features of psoriasis (Figure 2).
Including the current case, our review of the literature yielded 7 pediatric (ie, 0–18 years of age) cases of TNF-α inhibitor–induced psoriatic alopecia. Of the 6 previously reported pediatric cases, 5 occurred after administration of infliximab.6,7
Similar to our case, TNF-α inhibitor–induced psoriatic alopecia was reported in a 7-year-old girl who was treated with adalimumab for juvenile idiopathic arthritis.6 Nine months after starting treatment, that patient presented with a tender, erythematous, eroded, and crusted alopecic plaque along with scaly plaques on the scalp. Adalimumab was discontinued, and cyclosporine and topical steroids were started. Cyclosporine was then discontinued due to partial resolution of the psoriasis; the patient was started on abatacept, with persistence of the psoriasis and alopecia. The patient was then started on oral methotrexate 12.5 mg once weekly with moderate improvement and mild to moderate exacerbations.
Tumor necrosis factor α inhibitor–induced psoriasis may occur as a result of a cytokine imbalance. A TNF-α blockade leads to upregulation of interferon α (IFN-α) and TNF-α production by plasmacytoid dendritic cells (pDCs), usually in genetically susceptible people.6,7,9-15 The IFN-α induces maturation of myeloid dendritic cells (mDCs) responsible for increasing proinflammatory cytokines that contribute to psoriasis.11 Generation of TNF-α by pDCs leads to mature or activated dendritic cells derived from pDCs through autocrine TNF-α production and paracrine IFN-α production from immature mDCs.9 Once pDCs mature, they are incapable of producing IFN-α; TNF-α then inhibits IFN-α production by inducing pDC maturation.11 Overproduction of IFN-α during TNF-α inhibition induces expression of the chemokine receptor CXCR3 on T cells, which recruits T cells to the dermis. The T cells then produce TNF-α, causing psoriatic skin lesions.10,11,13,14
Although TNF-α inhibitor–induced psoriatic alopecia is uncommon, the condition should be considered in female patients with underlying proinflammatory disease—CD in particular. Perman et al6 reported 5 cases of psoriatic alopecia in which 3 patients initially were treated with griseofulvin because of suspected tinea capitis.
Conditions with similar clinical findings should be ruled out before making a diagnosis of TNF-α inhibitor–induced psoriatic alopecia. Although clinicopathologic correlation is essential for making the diagnosis, it is possible that the histologic findings will not be specific for psoriasis.17 It is important to be aware of this condition in patients being treated with a TNF-α inhibitor as early as 2 months to 4 years or longer after starting treatment.
Previously reported cases have demonstrated various treatment options that yielded improvement or resolution of TNF-α inhibitor–induced psoriatic alopecia. These include either continuation or discontinuation of the TNF-α inhibitor combined with topical or intralesional steroids, methotrexate, or cyclosporine. Another option is to switch the TNF-α inhibitor to another biologic. Outcomes vary from patient to patient, making the physician’s clinical judgment crucial in deciding which treatment route to take. Our patient showed notable improvement when she was switched from adalimumab to ustekinumab as well as the combination of ustekinumab and clobetasol lotion 0.05%.
Conclusion
We recommend an individualized approach that provides patients with the safest and least invasive treatment option for TNF-α inhibitor–induced psoriatic alopecia. In most reported cases, the problem resolved with treatment, thereby classifying this form of alopecia as noncicatricial alopecia.
- Horneff G, Seyger MMB, Arikan D, et al. Safety of adalimumab in pediatric patients with polyarticular juvenile idiopathic arthritis, enthesitis-related arthritis, psoriasis, and Crohn’s disease. J Pediatr. 2018;201:166-175.e3. doi:10.1016/j.jpeds.2018.05.042
- Brown G, Wang E, Leon A, et al. Tumor necrosis factor-α inhibitor-induced psoriasis: systematic review of clinical features, histopathological findings, and management experience. J Am Acad Dermatol. 2017;76:334-341. doi:10.1016/j.jaad.2016.08.012
- George SMC, Taylor MR, Farrant PBJ. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721. doi:10.1111/ced.12715
- Shuster S. Psoriatic alopecia. Br J Dermatol. 1972;87:73-77. doi:10.1111/j.1365-2133.1972.tb05103.x
- Silva CY, Brown KL, Kurban AK, et al. Psoriatic alopecia—fact or fiction? a clinicohistopathologic reappraisal. Indian J Dermatol Venereol Leprol. 2012;78:611-619. doi:10.4103/0378-6323.100574
- Perman MJ, Lovell DJ, Denson LA, et al. Five cases of anti-tumor necrosis factor alpha-induced psoriasis presenting with severe scalp involvement in children. Pediatr Dermatol. 2012;29:454-459. doi:10.1111/j.1525-1470.2011.01521.x
- Prata Ribeiro LB, Gonçalves Rego JC, Duque Estrada B, et al. Alopecia secondary to anti-tumor necrosis factor-alpha therapy. An Bras Dermatol. 2015;90:232–235. doi:10.1590/abd1806-4841.20153084
- Papadavid E, Gazi S, Dalamaga M, et al. Palmoplantar and scalp psoriasis occurring during anti-tumour necrosis factor-alpha therapy: a case series of four patients and guidelines for management. J Eur Acad Dermatol Venereol. 2008;22:380-382. doi:10.1111/j.1468-3083.2007.02335.x
- Manni E, Barachini P. Psoriasis induced by infliximab in a patient suffering from Crohn’s disease. Int J Immunopathol Pharmacol. 2009;22:841-844. doi:10.1177/039463200902200331
- El Shabrawi-Caelen L, La Placa M, Vincenzi C, et al. Adalimumab-induced psoriasis of the scalp with diffuse alopecia: a severe potentially irreversible cutaneous side effect of TNF-alpha blockers. Inflamm Bowel Dis. 2010;16:182-183. doi:10.1002/ibd.20954
- Medkour F, Babai S, Chanteloup E, et al. Development of diffuse psoriasis with alopecia during treatment of Crohn’s disease with infliximab. Gastroenterol Clin Biol. 2010;34:140-141. doi:10.1016/j.gcb.2009.10.021
- Doyle LA, Sperling LC, Baksh S, et al. Psoriatic alopecia/alopecia areata-like reactions secondary to anti-tumor necrosis factor-α therapy: a novel cause of noncicatricial alopecia. Am J Dermatopathol. 2011;33:161-166. doi:10.1097/DAD.0b013e3181ef7403
- Osório F, Magro F, Lisboa C, et al. Anti-TNF-alpha induced psoriasiform eruptions with severe scalp involvement and alopecia: report of five cases and review of the literature. Dermatology. 2012;225:163-167. doi:10.1159/000342503
- Andrisani G, Marzo M, Celleno L, et al. Development of psoriasis scalp with alopecia during treatment of Crohn’s disease with infliximab and rapid response to both diseases to ustekinumab. Eur Rev Med Pharmacol Sci. 2013;17:2831-2836.
- Afanasiev OK, Zhang CZ, Ruhoy SM. TNF-inhibitor associated psoriatic alopecia: diagnostic utility of sebaceous lobule atrophy. J Cutan Pathol. 2017;44:563-569. doi:10.1111/cup.12932
- Helm MM, Haddad S. Alopecia areata and scarring alopecia presenting during golimumab therapy for ankylosing spondylitis. N Am J Med Sci. 2018;11:22-24. doi:10.7156/najms.2018.110122
- Laga AC, Vleugels RA, Qureshi AA, et al. Histopathologic spectrum of psoriasiform skin reactions associated with tumor necrosis factor-a inhibitor therapy. a study of 16 biopsies. Am J Dermatopathol. 2010;32:568-573. doi:10.1097/DAD.0b013e3181cb3ff7
- Horneff G, Seyger MMB, Arikan D, et al. Safety of adalimumab in pediatric patients with polyarticular juvenile idiopathic arthritis, enthesitis-related arthritis, psoriasis, and Crohn’s disease. J Pediatr. 2018;201:166-175.e3. doi:10.1016/j.jpeds.2018.05.042
- Brown G, Wang E, Leon A, et al. Tumor necrosis factor-α inhibitor-induced psoriasis: systematic review of clinical features, histopathological findings, and management experience. J Am Acad Dermatol. 2017;76:334-341. doi:10.1016/j.jaad.2016.08.012
- George SMC, Taylor MR, Farrant PBJ. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721. doi:10.1111/ced.12715
- Shuster S. Psoriatic alopecia. Br J Dermatol. 1972;87:73-77. doi:10.1111/j.1365-2133.1972.tb05103.x
- Silva CY, Brown KL, Kurban AK, et al. Psoriatic alopecia—fact or fiction? a clinicohistopathologic reappraisal. Indian J Dermatol Venereol Leprol. 2012;78:611-619. doi:10.4103/0378-6323.100574
- Perman MJ, Lovell DJ, Denson LA, et al. Five cases of anti-tumor necrosis factor alpha-induced psoriasis presenting with severe scalp involvement in children. Pediatr Dermatol. 2012;29:454-459. doi:10.1111/j.1525-1470.2011.01521.x
- Prata Ribeiro LB, Gonçalves Rego JC, Duque Estrada B, et al. Alopecia secondary to anti-tumor necrosis factor-alpha therapy. An Bras Dermatol. 2015;90:232–235. doi:10.1590/abd1806-4841.20153084
- Papadavid E, Gazi S, Dalamaga M, et al. Palmoplantar and scalp psoriasis occurring during anti-tumour necrosis factor-alpha therapy: a case series of four patients and guidelines for management. J Eur Acad Dermatol Venereol. 2008;22:380-382. doi:10.1111/j.1468-3083.2007.02335.x
- Manni E, Barachini P. Psoriasis induced by infliximab in a patient suffering from Crohn’s disease. Int J Immunopathol Pharmacol. 2009;22:841-844. doi:10.1177/039463200902200331
- El Shabrawi-Caelen L, La Placa M, Vincenzi C, et al. Adalimumab-induced psoriasis of the scalp with diffuse alopecia: a severe potentially irreversible cutaneous side effect of TNF-alpha blockers. Inflamm Bowel Dis. 2010;16:182-183. doi:10.1002/ibd.20954
- Medkour F, Babai S, Chanteloup E, et al. Development of diffuse psoriasis with alopecia during treatment of Crohn’s disease with infliximab. Gastroenterol Clin Biol. 2010;34:140-141. doi:10.1016/j.gcb.2009.10.021
- Doyle LA, Sperling LC, Baksh S, et al. Psoriatic alopecia/alopecia areata-like reactions secondary to anti-tumor necrosis factor-α therapy: a novel cause of noncicatricial alopecia. Am J Dermatopathol. 2011;33:161-166. doi:10.1097/DAD.0b013e3181ef7403
- Osório F, Magro F, Lisboa C, et al. Anti-TNF-alpha induced psoriasiform eruptions with severe scalp involvement and alopecia: report of five cases and review of the literature. Dermatology. 2012;225:163-167. doi:10.1159/000342503
- Andrisani G, Marzo M, Celleno L, et al. Development of psoriasis scalp with alopecia during treatment of Crohn’s disease with infliximab and rapid response to both diseases to ustekinumab. Eur Rev Med Pharmacol Sci. 2013;17:2831-2836.
- Afanasiev OK, Zhang CZ, Ruhoy SM. TNF-inhibitor associated psoriatic alopecia: diagnostic utility of sebaceous lobule atrophy. J Cutan Pathol. 2017;44:563-569. doi:10.1111/cup.12932
- Helm MM, Haddad S. Alopecia areata and scarring alopecia presenting during golimumab therapy for ankylosing spondylitis. N Am J Med Sci. 2018;11:22-24. doi:10.7156/najms.2018.110122
- Laga AC, Vleugels RA, Qureshi AA, et al. Histopathologic spectrum of psoriasiform skin reactions associated with tumor necrosis factor-a inhibitor therapy. a study of 16 biopsies. Am J Dermatopathol. 2010;32:568-573. doi:10.1097/DAD.0b013e3181cb3ff7
Practice Points
- Psoriatic alopecia is a rare nonscarring alopecia that can present as a complication of treatment with tumor necrosis factor α inhibitors.
- This finding commonly is seen in females undergoing treatment with infliximab or adalimumab, usually for Crohn disease.
- Histopathologic findings can show a psoriasiform-pattern, neutrophil-rich, inflammatory infiltrate involving hair follicles or a lichenoid pattern.
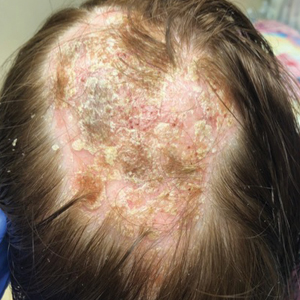
Subcutaneous, Mucocutaneous, and Mucous Membrane Tumors
The Diagnosis: Granular Cell Tumor
Histopathologic analysis from the axillary excision demonstrated cords and sheets of large polygonal cells in the dermis with uniform, oval, hyperchromatic nuclei and ample pink granular-staining cytoplasm (quiz images). An infiltrative growth pattern was noted; however, there was no evidence of conspicuous mitoses, nuclear pleomorphism, or necrosis. These results in conjunction with the immunohistochemistry findings were consistent with a benign granular cell tumor (GCT), a rare neoplasm considered to have neural/Schwann cell origin.1-3
Our case demonstrates the difficulty in clinically diagnosing cutaneous GCTs. The tumor often presents as a solitary, 0.5- to 3-cm, asymptomatic, firm nodule4,5; however, GCTs also can appear verrucous, eroded, or with other variable morphologies, which can create diagnostic challenges.5,6 Accordingly, a 1980 study of 110 patients with GCTs found that the preoperative clinical diagnosis was incorrect in all but 3 cases,7 emphasizing the need for histologic evaluation. Benign GCTs tend to exhibit sheets of polygonal tumor cells with eosinophilic granular cytoplasm and small central nuclei.3,5 The cytoplasmic granules are periodic acid-Schiff positive and diastase resistant.6 Many cases feature pseudoepitheliomatous hyperplasia, which can misleadingly resemble squamous cell carcinoma.3,5,6 Of note, invasive growth patterns on histology can occur with benign GCTs, as in our patient's case, and do not impact prognosis.3,4 On immunohistochemistry, benign, atypical, and malignant GCTs often stain positive for S-100 protein, vimentin, neuron-specific enolase, SOX10, and CD68.1,3
Although our patient's GCTs were benign, an estimated 1% to 2% are malignant.1,4 In 1998, Fanburg-Smith et al1 defined 6 histologic criteria that characterize malignant GCTs: necrosis, tumor cell spindling, vesicular nuclei with large nucleoli, high nuclear to cytoplasmic ratio, increased mitosis, and pleomorphism. Neoplasms with 3 or more of these features are classified as malignant, those with 1 or 2 are considered atypical, and those with only pleomorphism or no other criteria met are diagnosed as benign.1
Multiple GCTs have been reported in 10% to 25% of cases and, as highlighted in our case, can occur in both a metachronous and synchronous manner.2-4,6 Our patient developed a solitary GCT on the inferior lip 3 years prior to the appearance of 2 additional GCTs within 6 months of each other. The presence of multiple GCTs has been associated with genetic syndromes, such as neurofibromatosis type 1 and Noonan syndrome with multiple lentigines3,8; however, as our case demonstrates, multiple GCTs can occur in nonsyndromic patients as well. When multiple GCTs develop at distant sites, they can resemble metastasis.3 To differentiate these clinical scenarios, Machado et al3 proposed utilizing histology and anatomic location. Multiple tumors with benign characteristics on histology likely represent multiple GCTs, whereas tumors arising at sites common to GCT metastasis, such as lymph node, bone, or viscera, are more concerning for metastatic disease. It has been suggested that patients with multiple GCTs should be monitored with physical examination and repeat magnetic resonance imaging or computed tomography every 6 to 12 months.2 Given our patient's presentation with new tumors arising within 6 months of one another, we recommended a 6-month follow-up interval rather than 1 year. Due to the rarity of GCTs, clinical trials to define treatment guidelines and recommendations have not been performed.3 However, the most commonly utilized treatment modality is wide local excision, as performed in our patient.2,4
Melanoma, atypical fibroxanthoma (AFX), xanthoma, and leiomyosarcoma may be difficult to distinguish from GCT.1,3,4 Melanoma incidence has increased dramatically over the last several decades, with rates in the United States rising from 6.8 cases per 100,000 individuals in the 1970s to 20.1 in the early 2000s. Risk factors for its development include UV radiation exposure and particularly severe sunburns during childhood, along with a number of host risk factors such as total number of melanocytic nevi, family history, and fair complexion.9 Histologically, it often demonstrates irregularly distributed, poorly defined melanocytes with pagetoid spread and dyscohesive nests (Figure 1).10 Melanoma metastasis occasionally can present as a soft-tissue mass and often stains positive for S-100 and vimentin, thus resembling GCT1,4; however, unlike melanoma, GCTs lack melanosomes and stain negative for more specific melanocyte markers, such as melanoma antigen recognized by T cells 1 (MART-1).1,3,4
Atypical fibroxanthoma is a cutaneous neoplasm with fibrohistiocytic mesenchymal origin.11 These tumors typically arise on the head and neck in elderly individuals, particularly men with sun-damaged skin. They often present as superficial, rapidly growing nodules with the potential to ulcerate and bleed.11,12 Histologic features include pleomorphic spindle and epithelioid cells, whose nuclei appear hyperchromatic with atypical mitoses (Figure 2).12 Granular cell changes occur infrequently with AFXs, but in such cases immunohistochemistry can readily distinguish AFX from GCT. Although both tend to stain positive for CD68 and vimentin, AFXs lack S-100 protein and SOX10 expression that frequently is observed in GCTs.3,12
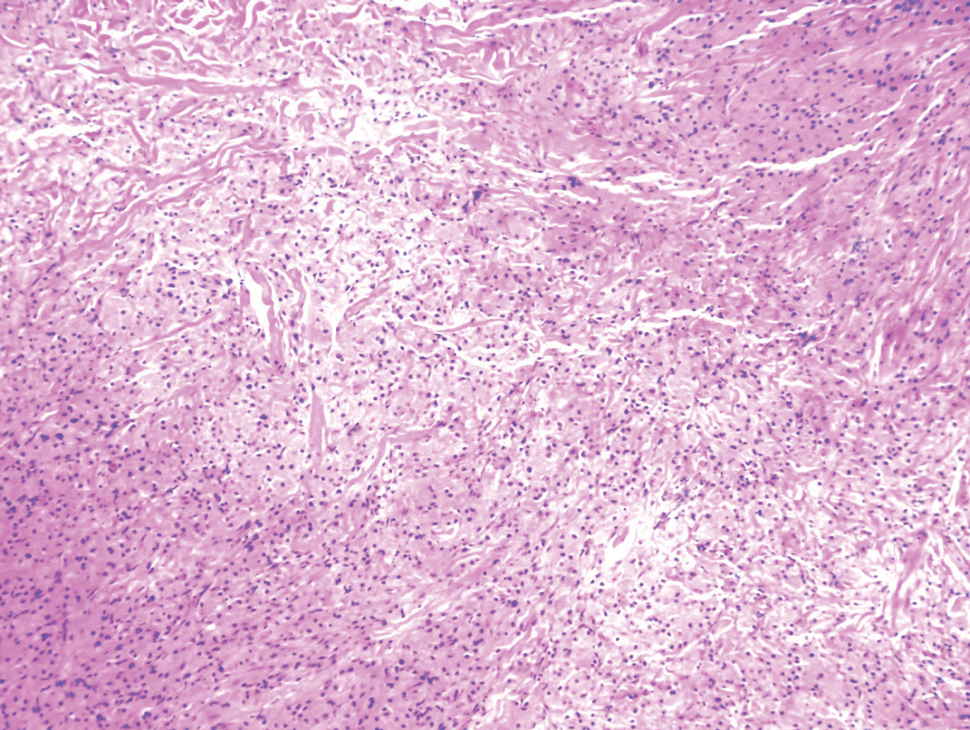

Xanthomas are localized lipid deposits in the connective tissue of the skin that often arise in association with dyslipidemia.13 They typically present as soft to semisolid yellow papules, plaques, or nodules. Their clinical appearance can resemble GCTs; however, histologic analysis enables differentiation with ease, as xanthomas demonstrate characteristic foam cells, consisting of lipid-laden macrophages (Figure 3).13
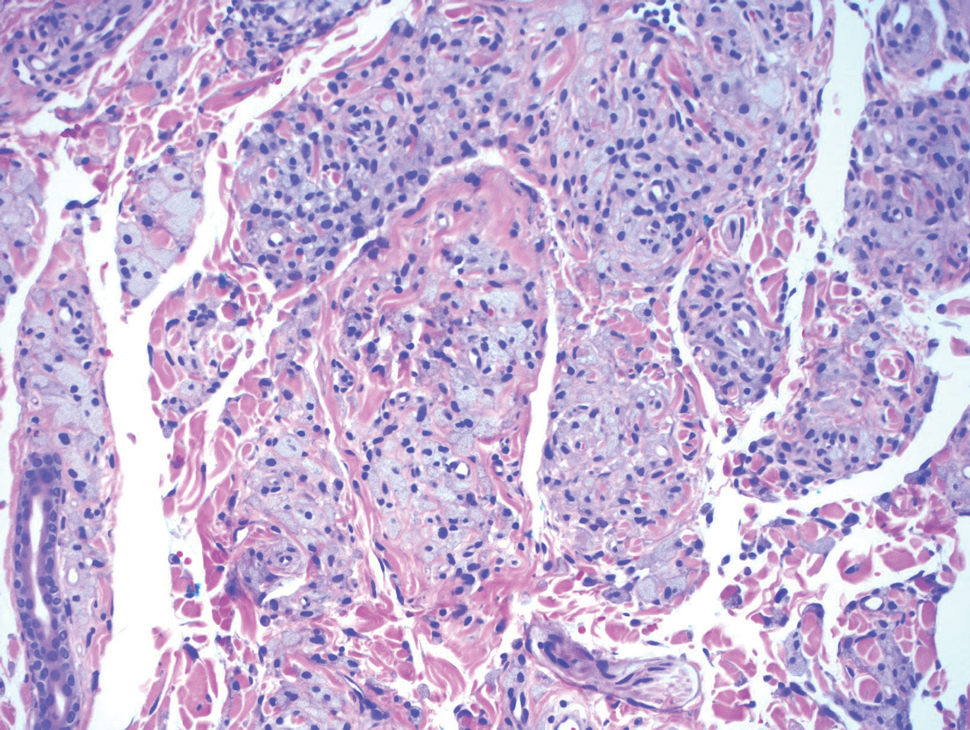
Cutaneous leiomyosarcoma is a rare dermal neoplasm, accounting for 2% to 3% of all sarcomas.14 They typically occur in White males during the fifth to seventh decades of life and often present as asymptomatic lesions on the lower extremities. They frequently arise from pilar smooth muscle. Unlike uterine and soft-tissue leiomyosarcoma, cutaneous leiomyosarcoma tends to follow an indolent course and rarely metastasizes.14 Histologically, these tumors display intersecting, well-defined, spindle-cell fascicles with abundant eosinophilic cytoplasm and cigar-shaped, blunt-ended nuclei (Figure 4).15 Occasionally, leiomyosarcomas can demonstrate cytoplasmic granularity due to lysosome accumulation4; nevertheless, the diagnosis usually can be elucidated by examining more typical histologic areas and utilizing immunohistochemistry, which often stains positive for α-smooth muscle actin, desmin, and h-caldesmon.4,15
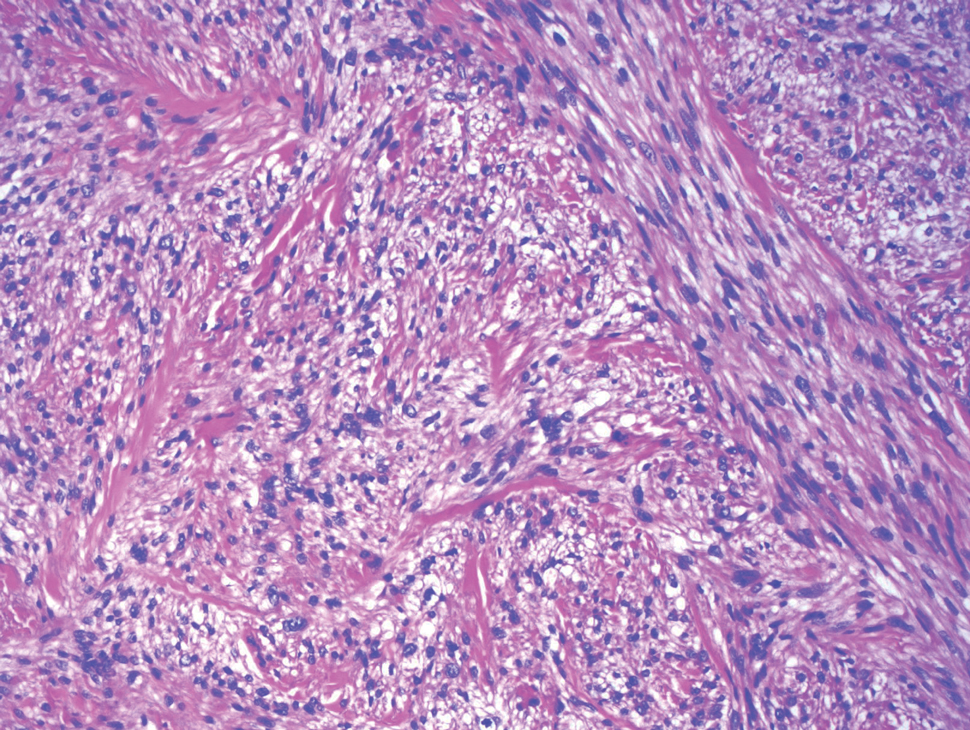
- Fanburg-Smith JC, Meis-Kindblom JM, Fante R, et al. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol. 1998;22:779-794.
- Moten AS, Movva S, von Mehren M, et al. Granular cell tumor experience at a comprehensive cancer center. J Surg Res. 2018;226:1-7.
- Machado I, Cruz J, Lavernia J, et al. Solitary, multiple, benign, atypical, or malignant: the "granular cell tumor" puzzle. Virchows Arch. 2016;468:527-538.
- Ordóñez NG. Granular cell tumor: a review and update. Adv Anat Pathol. 1999;6:186-203.
- Vaughan V, Ferringer T. Granular cell tumor. Cutis. 2014;94:275, 279-280.
- Van L, Parker SR. Multiple morphologically distinct cutaneous granular cell tumors occurring in a single patient. Cutis. 2016;97:E26-E29.
- Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
- Bamps S, Oyen T, Legius E, et al. Multiple granular cell tumors in a child with Noonan syndrome. Eur J Pediatr Surg. 2013;23:257-259.
- Rastrelli M, Tropea S, Rossi CR, et al. Melanoma: epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo. 2014;28:1005-1011.
- Smoller BR. Histologic criteria for diagnosing primary cutaneousmalignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Cardis MA, Ni J, Bhawan J. Granular cell differentiation: a review of the published work. J Dermatol. 2017;44:251-258.
- Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations [published online April 29, 2014]. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188.
- Sandhu N, Sauvageau AP, Groman A, et al. Cutaneous leiomyosarcoma: a SEER database analysis. Dermatol Surg. 2020;46:159-164.
- George S, Serrano C, Hensley ML, et al. Soft tissue and uterine leiomyosarcoma. J Clin Oncol. 2018;36:144-150.
The Diagnosis: Granular Cell Tumor
Histopathologic analysis from the axillary excision demonstrated cords and sheets of large polygonal cells in the dermis with uniform, oval, hyperchromatic nuclei and ample pink granular-staining cytoplasm (quiz images). An infiltrative growth pattern was noted; however, there was no evidence of conspicuous mitoses, nuclear pleomorphism, or necrosis. These results in conjunction with the immunohistochemistry findings were consistent with a benign granular cell tumor (GCT), a rare neoplasm considered to have neural/Schwann cell origin.1-3
Our case demonstrates the difficulty in clinically diagnosing cutaneous GCTs. The tumor often presents as a solitary, 0.5- to 3-cm, asymptomatic, firm nodule4,5; however, GCTs also can appear verrucous, eroded, or with other variable morphologies, which can create diagnostic challenges.5,6 Accordingly, a 1980 study of 110 patients with GCTs found that the preoperative clinical diagnosis was incorrect in all but 3 cases,7 emphasizing the need for histologic evaluation. Benign GCTs tend to exhibit sheets of polygonal tumor cells with eosinophilic granular cytoplasm and small central nuclei.3,5 The cytoplasmic granules are periodic acid-Schiff positive and diastase resistant.6 Many cases feature pseudoepitheliomatous hyperplasia, which can misleadingly resemble squamous cell carcinoma.3,5,6 Of note, invasive growth patterns on histology can occur with benign GCTs, as in our patient's case, and do not impact prognosis.3,4 On immunohistochemistry, benign, atypical, and malignant GCTs often stain positive for S-100 protein, vimentin, neuron-specific enolase, SOX10, and CD68.1,3
Although our patient's GCTs were benign, an estimated 1% to 2% are malignant.1,4 In 1998, Fanburg-Smith et al1 defined 6 histologic criteria that characterize malignant GCTs: necrosis, tumor cell spindling, vesicular nuclei with large nucleoli, high nuclear to cytoplasmic ratio, increased mitosis, and pleomorphism. Neoplasms with 3 or more of these features are classified as malignant, those with 1 or 2 are considered atypical, and those with only pleomorphism or no other criteria met are diagnosed as benign.1
Multiple GCTs have been reported in 10% to 25% of cases and, as highlighted in our case, can occur in both a metachronous and synchronous manner.2-4,6 Our patient developed a solitary GCT on the inferior lip 3 years prior to the appearance of 2 additional GCTs within 6 months of each other. The presence of multiple GCTs has been associated with genetic syndromes, such as neurofibromatosis type 1 and Noonan syndrome with multiple lentigines3,8; however, as our case demonstrates, multiple GCTs can occur in nonsyndromic patients as well. When multiple GCTs develop at distant sites, they can resemble metastasis.3 To differentiate these clinical scenarios, Machado et al3 proposed utilizing histology and anatomic location. Multiple tumors with benign characteristics on histology likely represent multiple GCTs, whereas tumors arising at sites common to GCT metastasis, such as lymph node, bone, or viscera, are more concerning for metastatic disease. It has been suggested that patients with multiple GCTs should be monitored with physical examination and repeat magnetic resonance imaging or computed tomography every 6 to 12 months.2 Given our patient's presentation with new tumors arising within 6 months of one another, we recommended a 6-month follow-up interval rather than 1 year. Due to the rarity of GCTs, clinical trials to define treatment guidelines and recommendations have not been performed.3 However, the most commonly utilized treatment modality is wide local excision, as performed in our patient.2,4
Melanoma, atypical fibroxanthoma (AFX), xanthoma, and leiomyosarcoma may be difficult to distinguish from GCT.1,3,4 Melanoma incidence has increased dramatically over the last several decades, with rates in the United States rising from 6.8 cases per 100,000 individuals in the 1970s to 20.1 in the early 2000s. Risk factors for its development include UV radiation exposure and particularly severe sunburns during childhood, along with a number of host risk factors such as total number of melanocytic nevi, family history, and fair complexion.9 Histologically, it often demonstrates irregularly distributed, poorly defined melanocytes with pagetoid spread and dyscohesive nests (Figure 1).10 Melanoma metastasis occasionally can present as a soft-tissue mass and often stains positive for S-100 and vimentin, thus resembling GCT1,4; however, unlike melanoma, GCTs lack melanosomes and stain negative for more specific melanocyte markers, such as melanoma antigen recognized by T cells 1 (MART-1).1,3,4
Atypical fibroxanthoma is a cutaneous neoplasm with fibrohistiocytic mesenchymal origin.11 These tumors typically arise on the head and neck in elderly individuals, particularly men with sun-damaged skin. They often present as superficial, rapidly growing nodules with the potential to ulcerate and bleed.11,12 Histologic features include pleomorphic spindle and epithelioid cells, whose nuclei appear hyperchromatic with atypical mitoses (Figure 2).12 Granular cell changes occur infrequently with AFXs, but in such cases immunohistochemistry can readily distinguish AFX from GCT. Although both tend to stain positive for CD68 and vimentin, AFXs lack S-100 protein and SOX10 expression that frequently is observed in GCTs.3,12
Xanthomas are localized lipid deposits in the connective tissue of the skin that often arise in association with dyslipidemia.13 They typically present as soft to semisolid yellow papules, plaques, or nodules. Their clinical appearance can resemble GCTs; however, histologic analysis enables differentiation with ease, as xanthomas demonstrate characteristic foam cells, consisting of lipid-laden macrophages (Figure 3).13
Cutaneous leiomyosarcoma is a rare dermal neoplasm, accounting for 2% to 3% of all sarcomas.14 They typically occur in White males during the fifth to seventh decades of life and often present as asymptomatic lesions on the lower extremities. They frequently arise from pilar smooth muscle. Unlike uterine and soft-tissue leiomyosarcoma, cutaneous leiomyosarcoma tends to follow an indolent course and rarely metastasizes.14 Histologically, these tumors display intersecting, well-defined, spindle-cell fascicles with abundant eosinophilic cytoplasm and cigar-shaped, blunt-ended nuclei (Figure 4).15 Occasionally, leiomyosarcomas can demonstrate cytoplasmic granularity due to lysosome accumulation4; nevertheless, the diagnosis usually can be elucidated by examining more typical histologic areas and utilizing immunohistochemistry, which often stains positive for α-smooth muscle actin, desmin, and h-caldesmon.4,15
The Diagnosis: Granular Cell Tumor
Histopathologic analysis from the axillary excision demonstrated cords and sheets of large polygonal cells in the dermis with uniform, oval, hyperchromatic nuclei and ample pink granular-staining cytoplasm (quiz images). An infiltrative growth pattern was noted; however, there was no evidence of conspicuous mitoses, nuclear pleomorphism, or necrosis. These results in conjunction with the immunohistochemistry findings were consistent with a benign granular cell tumor (GCT), a rare neoplasm considered to have neural/Schwann cell origin.1-3
Our case demonstrates the difficulty in clinically diagnosing cutaneous GCTs. The tumor often presents as a solitary, 0.5- to 3-cm, asymptomatic, firm nodule4,5; however, GCTs also can appear verrucous, eroded, or with other variable morphologies, which can create diagnostic challenges.5,6 Accordingly, a 1980 study of 110 patients with GCTs found that the preoperative clinical diagnosis was incorrect in all but 3 cases,7 emphasizing the need for histologic evaluation. Benign GCTs tend to exhibit sheets of polygonal tumor cells with eosinophilic granular cytoplasm and small central nuclei.3,5 The cytoplasmic granules are periodic acid-Schiff positive and diastase resistant.6 Many cases feature pseudoepitheliomatous hyperplasia, which can misleadingly resemble squamous cell carcinoma.3,5,6 Of note, invasive growth patterns on histology can occur with benign GCTs, as in our patient's case, and do not impact prognosis.3,4 On immunohistochemistry, benign, atypical, and malignant GCTs often stain positive for S-100 protein, vimentin, neuron-specific enolase, SOX10, and CD68.1,3
Although our patient's GCTs were benign, an estimated 1% to 2% are malignant.1,4 In 1998, Fanburg-Smith et al1 defined 6 histologic criteria that characterize malignant GCTs: necrosis, tumor cell spindling, vesicular nuclei with large nucleoli, high nuclear to cytoplasmic ratio, increased mitosis, and pleomorphism. Neoplasms with 3 or more of these features are classified as malignant, those with 1 or 2 are considered atypical, and those with only pleomorphism or no other criteria met are diagnosed as benign.1
Multiple GCTs have been reported in 10% to 25% of cases and, as highlighted in our case, can occur in both a metachronous and synchronous manner.2-4,6 Our patient developed a solitary GCT on the inferior lip 3 years prior to the appearance of 2 additional GCTs within 6 months of each other. The presence of multiple GCTs has been associated with genetic syndromes, such as neurofibromatosis type 1 and Noonan syndrome with multiple lentigines3,8; however, as our case demonstrates, multiple GCTs can occur in nonsyndromic patients as well. When multiple GCTs develop at distant sites, they can resemble metastasis.3 To differentiate these clinical scenarios, Machado et al3 proposed utilizing histology and anatomic location. Multiple tumors with benign characteristics on histology likely represent multiple GCTs, whereas tumors arising at sites common to GCT metastasis, such as lymph node, bone, or viscera, are more concerning for metastatic disease. It has been suggested that patients with multiple GCTs should be monitored with physical examination and repeat magnetic resonance imaging or computed tomography every 6 to 12 months.2 Given our patient's presentation with new tumors arising within 6 months of one another, we recommended a 6-month follow-up interval rather than 1 year. Due to the rarity of GCTs, clinical trials to define treatment guidelines and recommendations have not been performed.3 However, the most commonly utilized treatment modality is wide local excision, as performed in our patient.2,4
Melanoma, atypical fibroxanthoma (AFX), xanthoma, and leiomyosarcoma may be difficult to distinguish from GCT.1,3,4 Melanoma incidence has increased dramatically over the last several decades, with rates in the United States rising from 6.8 cases per 100,000 individuals in the 1970s to 20.1 in the early 2000s. Risk factors for its development include UV radiation exposure and particularly severe sunburns during childhood, along with a number of host risk factors such as total number of melanocytic nevi, family history, and fair complexion.9 Histologically, it often demonstrates irregularly distributed, poorly defined melanocytes with pagetoid spread and dyscohesive nests (Figure 1).10 Melanoma metastasis occasionally can present as a soft-tissue mass and often stains positive for S-100 and vimentin, thus resembling GCT1,4; however, unlike melanoma, GCTs lack melanosomes and stain negative for more specific melanocyte markers, such as melanoma antigen recognized by T cells 1 (MART-1).1,3,4
Atypical fibroxanthoma is a cutaneous neoplasm with fibrohistiocytic mesenchymal origin.11 These tumors typically arise on the head and neck in elderly individuals, particularly men with sun-damaged skin. They often present as superficial, rapidly growing nodules with the potential to ulcerate and bleed.11,12 Histologic features include pleomorphic spindle and epithelioid cells, whose nuclei appear hyperchromatic with atypical mitoses (Figure 2).12 Granular cell changes occur infrequently with AFXs, but in such cases immunohistochemistry can readily distinguish AFX from GCT. Although both tend to stain positive for CD68 and vimentin, AFXs lack S-100 protein and SOX10 expression that frequently is observed in GCTs.3,12
Xanthomas are localized lipid deposits in the connective tissue of the skin that often arise in association with dyslipidemia.13 They typically present as soft to semisolid yellow papules, plaques, or nodules. Their clinical appearance can resemble GCTs; however, histologic analysis enables differentiation with ease, as xanthomas demonstrate characteristic foam cells, consisting of lipid-laden macrophages (Figure 3).13
Cutaneous leiomyosarcoma is a rare dermal neoplasm, accounting for 2% to 3% of all sarcomas.14 They typically occur in White males during the fifth to seventh decades of life and often present as asymptomatic lesions on the lower extremities. They frequently arise from pilar smooth muscle. Unlike uterine and soft-tissue leiomyosarcoma, cutaneous leiomyosarcoma tends to follow an indolent course and rarely metastasizes.14 Histologically, these tumors display intersecting, well-defined, spindle-cell fascicles with abundant eosinophilic cytoplasm and cigar-shaped, blunt-ended nuclei (Figure 4).15 Occasionally, leiomyosarcomas can demonstrate cytoplasmic granularity due to lysosome accumulation4; nevertheless, the diagnosis usually can be elucidated by examining more typical histologic areas and utilizing immunohistochemistry, which often stains positive for α-smooth muscle actin, desmin, and h-caldesmon.4,15
- Fanburg-Smith JC, Meis-Kindblom JM, Fante R, et al. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol. 1998;22:779-794.
- Moten AS, Movva S, von Mehren M, et al. Granular cell tumor experience at a comprehensive cancer center. J Surg Res. 2018;226:1-7.
- Machado I, Cruz J, Lavernia J, et al. Solitary, multiple, benign, atypical, or malignant: the "granular cell tumor" puzzle. Virchows Arch. 2016;468:527-538.
- Ordóñez NG. Granular cell tumor: a review and update. Adv Anat Pathol. 1999;6:186-203.
- Vaughan V, Ferringer T. Granular cell tumor. Cutis. 2014;94:275, 279-280.
- Van L, Parker SR. Multiple morphologically distinct cutaneous granular cell tumors occurring in a single patient. Cutis. 2016;97:E26-E29.
- Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
- Bamps S, Oyen T, Legius E, et al. Multiple granular cell tumors in a child with Noonan syndrome. Eur J Pediatr Surg. 2013;23:257-259.
- Rastrelli M, Tropea S, Rossi CR, et al. Melanoma: epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo. 2014;28:1005-1011.
- Smoller BR. Histologic criteria for diagnosing primary cutaneousmalignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Cardis MA, Ni J, Bhawan J. Granular cell differentiation: a review of the published work. J Dermatol. 2017;44:251-258.
- Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations [published online April 29, 2014]. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188.
- Sandhu N, Sauvageau AP, Groman A, et al. Cutaneous leiomyosarcoma: a SEER database analysis. Dermatol Surg. 2020;46:159-164.
- George S, Serrano C, Hensley ML, et al. Soft tissue and uterine leiomyosarcoma. J Clin Oncol. 2018;36:144-150.
- Fanburg-Smith JC, Meis-Kindblom JM, Fante R, et al. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol. 1998;22:779-794.
- Moten AS, Movva S, von Mehren M, et al. Granular cell tumor experience at a comprehensive cancer center. J Surg Res. 2018;226:1-7.
- Machado I, Cruz J, Lavernia J, et al. Solitary, multiple, benign, atypical, or malignant: the "granular cell tumor" puzzle. Virchows Arch. 2016;468:527-538.
- Ordóñez NG. Granular cell tumor: a review and update. Adv Anat Pathol. 1999;6:186-203.
- Vaughan V, Ferringer T. Granular cell tumor. Cutis. 2014;94:275, 279-280.
- Van L, Parker SR. Multiple morphologically distinct cutaneous granular cell tumors occurring in a single patient. Cutis. 2016;97:E26-E29.
- Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
- Bamps S, Oyen T, Legius E, et al. Multiple granular cell tumors in a child with Noonan syndrome. Eur J Pediatr Surg. 2013;23:257-259.
- Rastrelli M, Tropea S, Rossi CR, et al. Melanoma: epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo. 2014;28:1005-1011.
- Smoller BR. Histologic criteria for diagnosing primary cutaneousmalignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Cardis MA, Ni J, Bhawan J. Granular cell differentiation: a review of the published work. J Dermatol. 2017;44:251-258.
- Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations [published online April 29, 2014]. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188.
- Sandhu N, Sauvageau AP, Groman A, et al. Cutaneous leiomyosarcoma: a SEER database analysis. Dermatol Surg. 2020;46:159-164.
- George S, Serrano C, Hensley ML, et al. Soft tissue and uterine leiomyosarcoma. J Clin Oncol. 2018;36:144-150.

A 26-year-old woman with a history of dysplastic nevi with severe atypia presented with a growth on the lower lip of 3 years’ duration. She denied any inciting event, such as prior trauma to the area, and reported that the lesion had been asymptomatic without a notable change in size. Physical examination revealed a translucent, soft, compressible cystic papule on the left inferior vermilion lip. Wide local excision following incisional biopsy was performed. Six months later, the patient returned to our clinic with a lesion on the right lateral tongue of 6 weeks’ duration as well as a 1-cm subcutaneous cyst in the left axilla of 6 months’ duration. Excisional biopsies of both lesions were performed for histopathologic analysis. Pathology results were similar among the lip, tongue, and axillary lesions. Immunohistochemistry revealed strong positive staining with antibodies to S-100 protein, SOX10, and CD68.
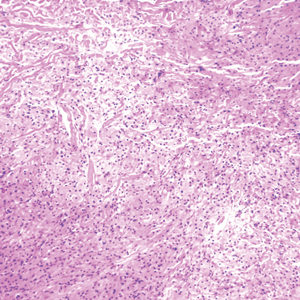
Recommendations for Pregnant Members of Dermatology Health Care Teams During the COVID-19 Pandemic
Information is scarce regarding the impact of COVID-19 on pregnant women and newborns; health care workers (HCWs), particularly pregnant women,1 who are caring for patients during the pandemic might experience concern and uncertainty. The American College of Obstetricians and Gynecologists (ACOG) released recommendations, based on expert consensus, regarding pregnant HCWs on December 14, 2020.2 We propose an appropriation of the ACOG recommendations for dermatologists and their teams caring for patients during the COVID-19 pandemic.
Risks to Pregnant HCWs
Worldwide, viral pneumonia is a leading cause of death during pregnancy,3 with higher mortality documented among pregnant patients during the 1918 influenza pandemic and the 2003 severe acute respiratory syndrome–associated coronavirus pandemic,3 and an increased rate of hospital admission documented among these patients compared to the general population during the 2009 H1N1 influenza pandemic.4
Data from the Centers for Disease Control and Prevention (CDC) suggest that pregnant women with symptomatic COVID-19 (n=30,415) are at increased risk for the following (compared to nonpregnant women with symptomatic COVID-19 [n=431,410])5:
• Admission to the intensive care unit (10.5 of every 1000 cases vs 3.9 of every 1000 cases; adjusted risk ratio [aRR]=3.0; 95% CI, 2.6-3.4)
• Receipt of invasive ventilation (2.9 of every 1000 cases vs 1.1 of every 1000 cases; aRR=2.9; 95% CI, 2.2-3.8)
• Receipt of extracorporeal membrane oxygenation (0.7 of every 1000 cases vs 0.3 of every 1000 cases; aRR=2.4; 95% CI, 1.5-4.0)
• Death (1.5 of every 1000 cases vs 1.2 of every 1000 cases; aRR=1.7; 95% CI, 1.2-2.4).
Although the absolute risk of severe COVID-19–related outcomes is low, the CDC includes pregnant women in its increased risk category for COVID-19. Furthermore, in a systematic review of 61 studies comprising 790 COVID-19–positive pregnant women and 548 newborns, the rates of cesarean delivery, premature birth, low birth weight, and adverse pregnancy events (the latter comprising preterm birth, death or stillbirth, and early termination of pregnancy) were estimated to be 72%, 23%, 7%, and 27%, respectively.6 In a systematic review of 39 studies (case series and cohort studies), comprising 936 SARS-CoV-2–tested newborns of mothers with COVID-19, mother-to-fetus transmission of SARS-CoV-2 occurred during the third trimester in approximately 3.2% of infected mothers.7
In pregnant women with COVID-19 who develop cytokine storm syndrome, a fetal inflammatory response syndrome can ensue, which has been shown to cause ventricular expansion and bleeding in animal models.8 In addition, underlying conditions, such as cardiovascular disease, diabetes mellitus, pre-existing lung disease, and obesity, which are well-established risks factors for severe COVID-19 in nonpregnant patients, can increase the severity of COVID-19 in pregnant women.5,9-11
Recommendations From ACOG for Pregnant HCWs
The American College of Obstetricians and Gynecologists recommends that health care facilities consider limiting the exposure of pregnant HCWs to patients with confirmed or suspected COVID-19. They also recommend that pregnant women continue to work in patient-facing roles if they want to, if recommended personal protective equipment (PPE) is available for them to wear.2 The US Food and Drug Administration issued an Emergency Use Authorization for 2 messenger RNA COVID-19 vaccines. Although these vaccines have not been tested in pregnant women, ACOG recommends that COVID-19 vaccines not be withheld from pregnant women who fulfill the criteria for vaccination; pregnant women who decline vaccination should be supported in their decision.12 In dermatology, telemedicine is an effective alternative to face-to-face visits, reducing the risk of transmitting SARS-CoV-2 to physicians and patients.
Ideally, pregnant dermatology attending physicians and residents can continue to provide care through teledermatology. They also can continue to provide in-person care, if they choose to; however, higher-risk procedures should be avoided.12 In dermatology, that might include ablative laser procedures to the face, prolonged surgery, such as hair transplantation, and intraoral or intranasal procedures. Alternatively, pregnant dermatology residents can be allocated to clinical rotations in which face-to-face contact with patients is not required such as dermatopathology and a research rotation. Likewise, telework options can be encouraged for other pregnant members of dermatology teams, including front-desk staff, nurses, medical assistants, and remaining ancillary staff.
Guidance on Face Masks for Pregnant HCWs
Universal masking of HCWs has been shown to reduce the rate of health care–related acquisition of SARS-CoV-2.13 However, extended use or reuse of N95 respirators might contribute to SARS-CoV-2 transmission.14 The American College of Obstetricians and Gynecologists recommends that all HCWs wear a face mask at all times while working in a health care facility, even if patients are wearing a face covering or face mask.2 Based on CDC guidelines,15 HCWs in regions where community transmission is moderate or substantial should wear eye protection in addition to a face mask, and they should wear an N95, N95-equivalent, or higher-level respirator instead of a face mask when performing aerosol-generating procedures and surgical procedures. If working in a patient-facing role caring for patients with suspected or confirmed COVID-19, HCWs should wear an N95, N95-equivalent, or higher-level respirator; gown; gloves; and eye protection (goggles or a disposable face shield).15
Final Thoughts
COVID-19 has brought about acute and likely permanent changes to the US health care system. Dermatologists are integral members of that system and are essential to the treatment of patients with skin, hair, and nail disorders. Pregnant dermatologists and residents should refrain from patient-facing roles when feasible; however, when all recommended PPE are available, they may continue to work in patient-facing roles until they give birth if they desire to do so. Alternatively, teledermatology and non–face-to-face rotations should be encouraged. Higher-risk and aerosol-generating procedures are of particular concern regarding the risk for transmitting SARS-CoV-2 and should be avoided. Correct and universal use of PPE is paramount; when all recommended PPE is not available, pregnant HCWs should avoid exposure to patients with suspected or confirmed COVID-19. These recommendations will help safeguard pregnant members of dermatology teams during the COVID-19 pandemic while maximizing patient care.
- Rashidi Fakari F, Simbar M. Coronavirus pandemic and worries during pregnancy; a letter to editor. Arch Acad Emerg Med. 2020;8:E21.
- The American College of Obstetricians and Gynecologists. COVID-19 FAQs for obstetrician-gynecologists, obstetrics. 2020. Accessed April 21, 2021. https://www.acog.org/clinical-information/physician-faqs/covid-19-faqs-for-ob-gyns-obstetrics
- Schwartz DA, Graham AL. Potential maternal and infant outcomes from (Wuhan) coronavirus 2019-nCoV infecting pregnant women: lessons from SARS, MERS, and other human coronavirus infections. Viruses. 2020;12:194. doi:10.3390/v12020194
- Yan J, Guo J, Fan C, et al. Coronavirus disease 2019 in pregnant women: a report based on 116 cases. Am J Obstet Gynecol. 2020;223:111.e1-111.e14. doi:10.1016/j.ajog.2020.04.014
- Zambrano LD, Ellington S, Strid P, et al; . Update: characteristics of symptomatic women of reproductive age with laboratory-confirmed SARS-CoV-2 infection by pregnancy status—United States, January 22–October 3, 2020. MMWR Morb Mortal Wkly Rep. 2020;69:1641-1647. doi:10.15585/mmwr.mm6944e3
- Dubey P, Reddy SY, Manuel S, et al. Maternal and neonatal characteristics and outcomes among COVID-19 infected women: an updated systematic review and meta-analysis. Eur J Obstet Gynecol Reprod Biol. 2020;252:490-501. doi:10.1016/j.ejogrb.2020.07.034
- Kotlyar AM, Grechukhina O, Chen A, et al. Vertical transmission of coronavirus disease 2019: a systematic review and meta-analysis. Am J Obstet Gynecol. 2020;224:35-53.e3. doi:10.1016/j.ajog.2020.07.049
- Mitchell T, MacDonald JW, Srinouanpranchanh S, et al. Evidence of cardiac involvement in the fetal inflammatory response syndrome: disruption of gene networks programming cardiac development in nonhuman primates. Am J Obstet Gynecol. 2018;218:438.e1-438.e16. doi:10.1016/j.ajog.2018.01.009
- Ellington S, Strid P, Tong VT, et al. Characteristics of women of reproductive age with laboratory-confirmed SARS-CoV-2 infection by pregnancy status—United States, January 22–June 7, 2020. MMWR Morb Mortal Wkly Rep. 2020;69:769-775. doi:10.15585/mmwr.mm6925a1
- Panagiotakopoulos L, Myers TR, Gee J, et al. SARS-CoV-2 infection among hospitalized pregnant women: reasons for admission and pregnancy characteristics—eight U.S. health care centers, March 1–May 30, 2020. 2020. doi:10.15585/mmwr.mm6938e2
- Knight M, Bunch K, Vousden N, et al; . Characteristics and outcomes of pregnant women admitted to hospital with confirmed SARS-CoV-2 infection in UK: national population based cohort study. BMJ. 2020;369:m2107. doi:10.1136/bmj.m2107
- The American College of Obstetricians and Gynecologists. Vaccinating pregnant and lactating patients against COVID-19. December 2020. Updated March 24, 2021. Accessed April 28, 2021. https://www.acog.org/clinical/clinical-guidance/practice-advisory/articles/2020/12/vaccinating-pregnant-and-lactating-patients-against-covid-19
- Seidelman JL, Lewis SS, Advani SD, et al. Universal masking is an effective strategy to flatten the severe acute respiratory coronavirus virus 2 (SARS-CoV-2) healthcare worker epidemiologic curve. Infect Control Hosp Epidemiol. 2020;41:1466-1467. doi:10.1017/ice.2020.31314.
- Degesys NF, Wang RC, Kwan E, et al. Correlation between N95 extended use and reuse and fit failure in an emergency department. JAMA. 2020;324:94-96. doi:10.1001/jama.2020.9843
- Centers for Disease Control and Prevention. Interim infection prevention and control recommendations for healthcare personnel during the coronavirus disease 2019 (COVID-19) pandemic 2020. Updated February 23, 2021. Accessed April 21, 2021. https://www.cdc.gov/coronavirus/2019-ncov/hcp/infection-control-recommendations.html?CDC_AA_refVal=https%3A%2F%2Fwww.cdc.gov%2Fcoronavirus%2F2019-ncov%2Finfection-control%2Fcontrol-recommendations.html
Information is scarce regarding the impact of COVID-19 on pregnant women and newborns; health care workers (HCWs), particularly pregnant women,1 who are caring for patients during the pandemic might experience concern and uncertainty. The American College of Obstetricians and Gynecologists (ACOG) released recommendations, based on expert consensus, regarding pregnant HCWs on December 14, 2020.2 We propose an appropriation of the ACOG recommendations for dermatologists and their teams caring for patients during the COVID-19 pandemic.
Risks to Pregnant HCWs
Worldwide, viral pneumonia is a leading cause of death during pregnancy,3 with higher mortality documented among pregnant patients during the 1918 influenza pandemic and the 2003 severe acute respiratory syndrome–associated coronavirus pandemic,3 and an increased rate of hospital admission documented among these patients compared to the general population during the 2009 H1N1 influenza pandemic.4
Data from the Centers for Disease Control and Prevention (CDC) suggest that pregnant women with symptomatic COVID-19 (n=30,415) are at increased risk for the following (compared to nonpregnant women with symptomatic COVID-19 [n=431,410])5:
• Admission to the intensive care unit (10.5 of every 1000 cases vs 3.9 of every 1000 cases; adjusted risk ratio [aRR]=3.0; 95% CI, 2.6-3.4)
• Receipt of invasive ventilation (2.9 of every 1000 cases vs 1.1 of every 1000 cases; aRR=2.9; 95% CI, 2.2-3.8)
• Receipt of extracorporeal membrane oxygenation (0.7 of every 1000 cases vs 0.3 of every 1000 cases; aRR=2.4; 95% CI, 1.5-4.0)
• Death (1.5 of every 1000 cases vs 1.2 of every 1000 cases; aRR=1.7; 95% CI, 1.2-2.4).
Although the absolute risk of severe COVID-19–related outcomes is low, the CDC includes pregnant women in its increased risk category for COVID-19. Furthermore, in a systematic review of 61 studies comprising 790 COVID-19–positive pregnant women and 548 newborns, the rates of cesarean delivery, premature birth, low birth weight, and adverse pregnancy events (the latter comprising preterm birth, death or stillbirth, and early termination of pregnancy) were estimated to be 72%, 23%, 7%, and 27%, respectively.6 In a systematic review of 39 studies (case series and cohort studies), comprising 936 SARS-CoV-2–tested newborns of mothers with COVID-19, mother-to-fetus transmission of SARS-CoV-2 occurred during the third trimester in approximately 3.2% of infected mothers.7
In pregnant women with COVID-19 who develop cytokine storm syndrome, a fetal inflammatory response syndrome can ensue, which has been shown to cause ventricular expansion and bleeding in animal models.8 In addition, underlying conditions, such as cardiovascular disease, diabetes mellitus, pre-existing lung disease, and obesity, which are well-established risks factors for severe COVID-19 in nonpregnant patients, can increase the severity of COVID-19 in pregnant women.5,9-11
Recommendations From ACOG for Pregnant HCWs
The American College of Obstetricians and Gynecologists recommends that health care facilities consider limiting the exposure of pregnant HCWs to patients with confirmed or suspected COVID-19. They also recommend that pregnant women continue to work in patient-facing roles if they want to, if recommended personal protective equipment (PPE) is available for them to wear.2 The US Food and Drug Administration issued an Emergency Use Authorization for 2 messenger RNA COVID-19 vaccines. Although these vaccines have not been tested in pregnant women, ACOG recommends that COVID-19 vaccines not be withheld from pregnant women who fulfill the criteria for vaccination; pregnant women who decline vaccination should be supported in their decision.12 In dermatology, telemedicine is an effective alternative to face-to-face visits, reducing the risk of transmitting SARS-CoV-2 to physicians and patients.
Ideally, pregnant dermatology attending physicians and residents can continue to provide care through teledermatology. They also can continue to provide in-person care, if they choose to; however, higher-risk procedures should be avoided.12 In dermatology, that might include ablative laser procedures to the face, prolonged surgery, such as hair transplantation, and intraoral or intranasal procedures. Alternatively, pregnant dermatology residents can be allocated to clinical rotations in which face-to-face contact with patients is not required such as dermatopathology and a research rotation. Likewise, telework options can be encouraged for other pregnant members of dermatology teams, including front-desk staff, nurses, medical assistants, and remaining ancillary staff.
Guidance on Face Masks for Pregnant HCWs
Universal masking of HCWs has been shown to reduce the rate of health care–related acquisition of SARS-CoV-2.13 However, extended use or reuse of N95 respirators might contribute to SARS-CoV-2 transmission.14 The American College of Obstetricians and Gynecologists recommends that all HCWs wear a face mask at all times while working in a health care facility, even if patients are wearing a face covering or face mask.2 Based on CDC guidelines,15 HCWs in regions where community transmission is moderate or substantial should wear eye protection in addition to a face mask, and they should wear an N95, N95-equivalent, or higher-level respirator instead of a face mask when performing aerosol-generating procedures and surgical procedures. If working in a patient-facing role caring for patients with suspected or confirmed COVID-19, HCWs should wear an N95, N95-equivalent, or higher-level respirator; gown; gloves; and eye protection (goggles or a disposable face shield).15
Final Thoughts
COVID-19 has brought about acute and likely permanent changes to the US health care system. Dermatologists are integral members of that system and are essential to the treatment of patients with skin, hair, and nail disorders. Pregnant dermatologists and residents should refrain from patient-facing roles when feasible; however, when all recommended PPE are available, they may continue to work in patient-facing roles until they give birth if they desire to do so. Alternatively, teledermatology and non–face-to-face rotations should be encouraged. Higher-risk and aerosol-generating procedures are of particular concern regarding the risk for transmitting SARS-CoV-2 and should be avoided. Correct and universal use of PPE is paramount; when all recommended PPE is not available, pregnant HCWs should avoid exposure to patients with suspected or confirmed COVID-19. These recommendations will help safeguard pregnant members of dermatology teams during the COVID-19 pandemic while maximizing patient care.
Information is scarce regarding the impact of COVID-19 on pregnant women and newborns; health care workers (HCWs), particularly pregnant women,1 who are caring for patients during the pandemic might experience concern and uncertainty. The American College of Obstetricians and Gynecologists (ACOG) released recommendations, based on expert consensus, regarding pregnant HCWs on December 14, 2020.2 We propose an appropriation of the ACOG recommendations for dermatologists and their teams caring for patients during the COVID-19 pandemic.
Risks to Pregnant HCWs
Worldwide, viral pneumonia is a leading cause of death during pregnancy,3 with higher mortality documented among pregnant patients during the 1918 influenza pandemic and the 2003 severe acute respiratory syndrome–associated coronavirus pandemic,3 and an increased rate of hospital admission documented among these patients compared to the general population during the 2009 H1N1 influenza pandemic.4
Data from the Centers for Disease Control and Prevention (CDC) suggest that pregnant women with symptomatic COVID-19 (n=30,415) are at increased risk for the following (compared to nonpregnant women with symptomatic COVID-19 [n=431,410])5:
• Admission to the intensive care unit (10.5 of every 1000 cases vs 3.9 of every 1000 cases; adjusted risk ratio [aRR]=3.0; 95% CI, 2.6-3.4)
• Receipt of invasive ventilation (2.9 of every 1000 cases vs 1.1 of every 1000 cases; aRR=2.9; 95% CI, 2.2-3.8)
• Receipt of extracorporeal membrane oxygenation (0.7 of every 1000 cases vs 0.3 of every 1000 cases; aRR=2.4; 95% CI, 1.5-4.0)
• Death (1.5 of every 1000 cases vs 1.2 of every 1000 cases; aRR=1.7; 95% CI, 1.2-2.4).
Although the absolute risk of severe COVID-19–related outcomes is low, the CDC includes pregnant women in its increased risk category for COVID-19. Furthermore, in a systematic review of 61 studies comprising 790 COVID-19–positive pregnant women and 548 newborns, the rates of cesarean delivery, premature birth, low birth weight, and adverse pregnancy events (the latter comprising preterm birth, death or stillbirth, and early termination of pregnancy) were estimated to be 72%, 23%, 7%, and 27%, respectively.6 In a systematic review of 39 studies (case series and cohort studies), comprising 936 SARS-CoV-2–tested newborns of mothers with COVID-19, mother-to-fetus transmission of SARS-CoV-2 occurred during the third trimester in approximately 3.2% of infected mothers.7
In pregnant women with COVID-19 who develop cytokine storm syndrome, a fetal inflammatory response syndrome can ensue, which has been shown to cause ventricular expansion and bleeding in animal models.8 In addition, underlying conditions, such as cardiovascular disease, diabetes mellitus, pre-existing lung disease, and obesity, which are well-established risks factors for severe COVID-19 in nonpregnant patients, can increase the severity of COVID-19 in pregnant women.5,9-11
Recommendations From ACOG for Pregnant HCWs
The American College of Obstetricians and Gynecologists recommends that health care facilities consider limiting the exposure of pregnant HCWs to patients with confirmed or suspected COVID-19. They also recommend that pregnant women continue to work in patient-facing roles if they want to, if recommended personal protective equipment (PPE) is available for them to wear.2 The US Food and Drug Administration issued an Emergency Use Authorization for 2 messenger RNA COVID-19 vaccines. Although these vaccines have not been tested in pregnant women, ACOG recommends that COVID-19 vaccines not be withheld from pregnant women who fulfill the criteria for vaccination; pregnant women who decline vaccination should be supported in their decision.12 In dermatology, telemedicine is an effective alternative to face-to-face visits, reducing the risk of transmitting SARS-CoV-2 to physicians and patients.
Ideally, pregnant dermatology attending physicians and residents can continue to provide care through teledermatology. They also can continue to provide in-person care, if they choose to; however, higher-risk procedures should be avoided.12 In dermatology, that might include ablative laser procedures to the face, prolonged surgery, such as hair transplantation, and intraoral or intranasal procedures. Alternatively, pregnant dermatology residents can be allocated to clinical rotations in which face-to-face contact with patients is not required such as dermatopathology and a research rotation. Likewise, telework options can be encouraged for other pregnant members of dermatology teams, including front-desk staff, nurses, medical assistants, and remaining ancillary staff.
Guidance on Face Masks for Pregnant HCWs
Universal masking of HCWs has been shown to reduce the rate of health care–related acquisition of SARS-CoV-2.13 However, extended use or reuse of N95 respirators might contribute to SARS-CoV-2 transmission.14 The American College of Obstetricians and Gynecologists recommends that all HCWs wear a face mask at all times while working in a health care facility, even if patients are wearing a face covering or face mask.2 Based on CDC guidelines,15 HCWs in regions where community transmission is moderate or substantial should wear eye protection in addition to a face mask, and they should wear an N95, N95-equivalent, or higher-level respirator instead of a face mask when performing aerosol-generating procedures and surgical procedures. If working in a patient-facing role caring for patients with suspected or confirmed COVID-19, HCWs should wear an N95, N95-equivalent, or higher-level respirator; gown; gloves; and eye protection (goggles or a disposable face shield).15
Final Thoughts
COVID-19 has brought about acute and likely permanent changes to the US health care system. Dermatologists are integral members of that system and are essential to the treatment of patients with skin, hair, and nail disorders. Pregnant dermatologists and residents should refrain from patient-facing roles when feasible; however, when all recommended PPE are available, they may continue to work in patient-facing roles until they give birth if they desire to do so. Alternatively, teledermatology and non–face-to-face rotations should be encouraged. Higher-risk and aerosol-generating procedures are of particular concern regarding the risk for transmitting SARS-CoV-2 and should be avoided. Correct and universal use of PPE is paramount; when all recommended PPE is not available, pregnant HCWs should avoid exposure to patients with suspected or confirmed COVID-19. These recommendations will help safeguard pregnant members of dermatology teams during the COVID-19 pandemic while maximizing patient care.
- Rashidi Fakari F, Simbar M. Coronavirus pandemic and worries during pregnancy; a letter to editor. Arch Acad Emerg Med. 2020;8:E21.
- The American College of Obstetricians and Gynecologists. COVID-19 FAQs for obstetrician-gynecologists, obstetrics. 2020. Accessed April 21, 2021. https://www.acog.org/clinical-information/physician-faqs/covid-19-faqs-for-ob-gyns-obstetrics
- Schwartz DA, Graham AL. Potential maternal and infant outcomes from (Wuhan) coronavirus 2019-nCoV infecting pregnant women: lessons from SARS, MERS, and other human coronavirus infections. Viruses. 2020;12:194. doi:10.3390/v12020194
- Yan J, Guo J, Fan C, et al. Coronavirus disease 2019 in pregnant women: a report based on 116 cases. Am J Obstet Gynecol. 2020;223:111.e1-111.e14. doi:10.1016/j.ajog.2020.04.014
- Zambrano LD, Ellington S, Strid P, et al; . Update: characteristics of symptomatic women of reproductive age with laboratory-confirmed SARS-CoV-2 infection by pregnancy status—United States, January 22–October 3, 2020. MMWR Morb Mortal Wkly Rep. 2020;69:1641-1647. doi:10.15585/mmwr.mm6944e3
- Dubey P, Reddy SY, Manuel S, et al. Maternal and neonatal characteristics and outcomes among COVID-19 infected women: an updated systematic review and meta-analysis. Eur J Obstet Gynecol Reprod Biol. 2020;252:490-501. doi:10.1016/j.ejogrb.2020.07.034
- Kotlyar AM, Grechukhina O, Chen A, et al. Vertical transmission of coronavirus disease 2019: a systematic review and meta-analysis. Am J Obstet Gynecol. 2020;224:35-53.e3. doi:10.1016/j.ajog.2020.07.049
- Mitchell T, MacDonald JW, Srinouanpranchanh S, et al. Evidence of cardiac involvement in the fetal inflammatory response syndrome: disruption of gene networks programming cardiac development in nonhuman primates. Am J Obstet Gynecol. 2018;218:438.e1-438.e16. doi:10.1016/j.ajog.2018.01.009
- Ellington S, Strid P, Tong VT, et al. Characteristics of women of reproductive age with laboratory-confirmed SARS-CoV-2 infection by pregnancy status—United States, January 22–June 7, 2020. MMWR Morb Mortal Wkly Rep. 2020;69:769-775. doi:10.15585/mmwr.mm6925a1
- Panagiotakopoulos L, Myers TR, Gee J, et al. SARS-CoV-2 infection among hospitalized pregnant women: reasons for admission and pregnancy characteristics—eight U.S. health care centers, March 1–May 30, 2020. 2020. doi:10.15585/mmwr.mm6938e2
- Knight M, Bunch K, Vousden N, et al; . Characteristics and outcomes of pregnant women admitted to hospital with confirmed SARS-CoV-2 infection in UK: national population based cohort study. BMJ. 2020;369:m2107. doi:10.1136/bmj.m2107
- The American College of Obstetricians and Gynecologists. Vaccinating pregnant and lactating patients against COVID-19. December 2020. Updated March 24, 2021. Accessed April 28, 2021. https://www.acog.org/clinical/clinical-guidance/practice-advisory/articles/2020/12/vaccinating-pregnant-and-lactating-patients-against-covid-19
- Seidelman JL, Lewis SS, Advani SD, et al. Universal masking is an effective strategy to flatten the severe acute respiratory coronavirus virus 2 (SARS-CoV-2) healthcare worker epidemiologic curve. Infect Control Hosp Epidemiol. 2020;41:1466-1467. doi:10.1017/ice.2020.31314.
- Degesys NF, Wang RC, Kwan E, et al. Correlation between N95 extended use and reuse and fit failure in an emergency department. JAMA. 2020;324:94-96. doi:10.1001/jama.2020.9843
- Centers for Disease Control and Prevention. Interim infection prevention and control recommendations for healthcare personnel during the coronavirus disease 2019 (COVID-19) pandemic 2020. Updated February 23, 2021. Accessed April 21, 2021. https://www.cdc.gov/coronavirus/2019-ncov/hcp/infection-control-recommendations.html?CDC_AA_refVal=https%3A%2F%2Fwww.cdc.gov%2Fcoronavirus%2F2019-ncov%2Finfection-control%2Fcontrol-recommendations.html
- Rashidi Fakari F, Simbar M. Coronavirus pandemic and worries during pregnancy; a letter to editor. Arch Acad Emerg Med. 2020;8:E21.
- The American College of Obstetricians and Gynecologists. COVID-19 FAQs for obstetrician-gynecologists, obstetrics. 2020. Accessed April 21, 2021. https://www.acog.org/clinical-information/physician-faqs/covid-19-faqs-for-ob-gyns-obstetrics
- Schwartz DA, Graham AL. Potential maternal and infant outcomes from (Wuhan) coronavirus 2019-nCoV infecting pregnant women: lessons from SARS, MERS, and other human coronavirus infections. Viruses. 2020;12:194. doi:10.3390/v12020194
- Yan J, Guo J, Fan C, et al. Coronavirus disease 2019 in pregnant women: a report based on 116 cases. Am J Obstet Gynecol. 2020;223:111.e1-111.e14. doi:10.1016/j.ajog.2020.04.014
- Zambrano LD, Ellington S, Strid P, et al; . Update: characteristics of symptomatic women of reproductive age with laboratory-confirmed SARS-CoV-2 infection by pregnancy status—United States, January 22–October 3, 2020. MMWR Morb Mortal Wkly Rep. 2020;69:1641-1647. doi:10.15585/mmwr.mm6944e3
- Dubey P, Reddy SY, Manuel S, et al. Maternal and neonatal characteristics and outcomes among COVID-19 infected women: an updated systematic review and meta-analysis. Eur J Obstet Gynecol Reprod Biol. 2020;252:490-501. doi:10.1016/j.ejogrb.2020.07.034
- Kotlyar AM, Grechukhina O, Chen A, et al. Vertical transmission of coronavirus disease 2019: a systematic review and meta-analysis. Am J Obstet Gynecol. 2020;224:35-53.e3. doi:10.1016/j.ajog.2020.07.049
- Mitchell T, MacDonald JW, Srinouanpranchanh S, et al. Evidence of cardiac involvement in the fetal inflammatory response syndrome: disruption of gene networks programming cardiac development in nonhuman primates. Am J Obstet Gynecol. 2018;218:438.e1-438.e16. doi:10.1016/j.ajog.2018.01.009
- Ellington S, Strid P, Tong VT, et al. Characteristics of women of reproductive age with laboratory-confirmed SARS-CoV-2 infection by pregnancy status—United States, January 22–June 7, 2020. MMWR Morb Mortal Wkly Rep. 2020;69:769-775. doi:10.15585/mmwr.mm6925a1
- Panagiotakopoulos L, Myers TR, Gee J, et al. SARS-CoV-2 infection among hospitalized pregnant women: reasons for admission and pregnancy characteristics—eight U.S. health care centers, March 1–May 30, 2020. 2020. doi:10.15585/mmwr.mm6938e2
- Knight M, Bunch K, Vousden N, et al; . Characteristics and outcomes of pregnant women admitted to hospital with confirmed SARS-CoV-2 infection in UK: national population based cohort study. BMJ. 2020;369:m2107. doi:10.1136/bmj.m2107
- The American College of Obstetricians and Gynecologists. Vaccinating pregnant and lactating patients against COVID-19. December 2020. Updated March 24, 2021. Accessed April 28, 2021. https://www.acog.org/clinical/clinical-guidance/practice-advisory/articles/2020/12/vaccinating-pregnant-and-lactating-patients-against-covid-19
- Seidelman JL, Lewis SS, Advani SD, et al. Universal masking is an effective strategy to flatten the severe acute respiratory coronavirus virus 2 (SARS-CoV-2) healthcare worker epidemiologic curve. Infect Control Hosp Epidemiol. 2020;41:1466-1467. doi:10.1017/ice.2020.31314.
- Degesys NF, Wang RC, Kwan E, et al. Correlation between N95 extended use and reuse and fit failure in an emergency department. JAMA. 2020;324:94-96. doi:10.1001/jama.2020.9843
- Centers for Disease Control and Prevention. Interim infection prevention and control recommendations for healthcare personnel during the coronavirus disease 2019 (COVID-19) pandemic 2020. Updated February 23, 2021. Accessed April 21, 2021. https://www.cdc.gov/coronavirus/2019-ncov/hcp/infection-control-recommendations.html?CDC_AA_refVal=https%3A%2F%2Fwww.cdc.gov%2Fcoronavirus%2F2019-ncov%2Finfection-control%2Fcontrol-recommendations.html
Practice Points
- Pregnant women are at an increased risk for severe illness due to COVID-19 compared with nonpregnant women; therefore, it is important to protect pregnant health care workers who are caring for patients during the current pandemic.
- Although currently available COVID-19 vaccines have not been tested in pregnant women, they should not be withheld from pregnant individuals.
- Pregnant attending physicians and residents in dermatology can continue to provide care through telemedicine; if they choose to, and if all recommended personal protective equipment (PPE) are available, they can continue to provide in-person care.
- Correct and comprehensive use of PPE by pregnant health care workers is paramount to minimizing exposure to SARS-CoV-2.
Acetophenone Azine: The 2021 American Contact Dermatitis Society Allergen of the Year
It’s time for the American Contact Dermatitis Society (ACDS) Allergen of the Year! For 2021, the esteemed award goes to acetophenone azine (AA). If you have never heard of this chemical, you are not alone. Acetophenone azine has been identified in foam materials made of the copolymer ethyl-vinyl acetate (EVA). Contact allergy to AA initially was reported in 2016.1 There are only a few European and Canadian case reports and one case series of AA contact allergy in the literature, all of which are associated with foam shin pads or shin guards, shoe insoles, and/or flip-flops.2-6 Acetophenone azine is an important emerging allergen, and in this column, we will introduce you to AA and the sneaky places it can lurk and cause allergic contact dermatitis (ACD). We also highlight diagnosis, management, and patch testing for AA contact allergy.
AA Contact Allergy in the Literature
The first case of AA contact allergy was reported in Europe in 2016 when a 13-year-old male soccer player developed severe lower leg dermatitis and later generalized dermatitis associated with wearing foam shin guards.1 Patch testing to standard and supplemental trays was negative or not relevant; however, the patient exhibited strong reactions when patch tested directly to a piece of the shin guard soaked in acetone, water, and ethanol. Additional testing with AA diluted in acetone, water, and petrolatum resulted in positive patch test reactions to acetone dilutions of 1%, 0.1%, 0.01%, and 0.001% and aqueous solutions of 1% and 0.1%. Chromatographic analyses with high-performance liquid chromatography (HPLC) of shin guard extracts confirmed the culprit allergen to be AA.1
In the following months, the same clinic saw 2 more cases of AA contact allergy.2 An 11-year-old male soccer player developed lower leg dermatitis and later generalized dermatitis from wearing shin guards. Months later, he also developed dermatitis on the soles of the feet, which was attributed to wearing flip-flops. Patch tests to pieces of the shin guards and flip-flops were positive; AA in acetone 0.1% and 0.01% also was positive. As you might expect, HPLC again confirmed the presence of AA in the shin guards and flip-flops. The third patient was a 12-year-old boy with dermatitis on the soles of both feet; later he also developed a generalized dermatitis. Patch testing to pieces of the insoles of his sneakers and AA in acetone 0.1% and 0.01% was positive. Again, HPLC was positive for the presence of AA in the insoles of his sneakers.2
Several more cases of AA contact allergy have been reported in the literature. A 29-year-old European male hockey player demonstrated contact allergy to the gray foam of his shin pads as well as localized leg dermatitis followed by generalized dermatitis (are you noticing a trend yet?), and later dermatitis on the soles of the feet with positive patch-test reactions to pieces of his shin pads and shoe insoles as well as AA 0.1% and 0.01% in acetone.3 A 6-year-old Canadian male soccer player presented with leg dermatitis and later generalized dermatitis and dermatitis on the soles of the feet with positive reactions to pieces of his shin pads and shoe insoles as well as to AA 1% and 0.1% in petrolatum.4 A 17-year-old British male (another trend, all males so far!) hockey player developed dermatitis localized to the legs and positive patch tests to the worn foam inner lining of his shin pads as well as to AA 0.1%, 0.01%, and 0.001% in acetone.5Finally, Darrigade et al6 published a case series of 6 European children with AA contact allergy associated with shin pads and shoes; all had localized leg dermatitis, and some had generalized dermatitis. Patch testing to pieces of shin pads and shoe parts as well as to AA 0.1% in petrolatum and/or acetone showed with positive reactions to the foam pieces and AA in all 6 patients.
What’s the Deal With AA?
Acetophenone azine (also known as methylphenylketazine or bis[1-phenylethylidene]hydrazine) is composed of 2 acetophenone structures and a hydrazine moiety. It has been identified in EVA foam, which can be found in sports equipment such as shin pads or shin guards, shoes, and flip-flops. Raison-Peyron et al1 confirmed the presence of AA in EVA foam but reported that they did not know the exact reason for its presence. The authors theorized that AA might be a catalyst during EVA polymerization and also noted that it has antimicrobial and antihelminthic activity.1 Several authors noted that AA could be a by-product of EVA synthesis and that sports equipment manufacturers might not be aware of its presence in EVA.2,4-6 Some noted that AA concentration was higher in shin guards than in shoe insoles; they thought this explained why patients reacted first to their shin guards and were perhaps even initially sensitized to the shin guards, as well as why shoe insole contact allergy commonly was reported later or only after allergy to shin guards had already developed.4,6
Differential Diagnosis of Shin Pad or Shin Guard Dermatitis
We would be remiss if we did not mention the appropriate differential diagnosis when shin pad or shin guard dermatitis is identified. In fact, in most cases, shin guard dermatitis results from irritant contact dermatitis from friction, heat, and/or perspiration. Acetophenone azine contact allergy is not the most likely diagnosis when your sports-savvy, shin guard–wearing patient presents with anterior lower leg dermatitis. However, when conservative therapy (eg, barrier between the shin guard and the skin, control or management of perspiration, topical corticosteroid therapy) fails, patch testing to evaluate for ACD is indicated.
Management of AA Contact Allergy
As astute readers of this column are already aware, treatment of ACD requires strict allergen avoidance. You will find that we have the same recommendations for AA contact allergy. Given that there are only a handful of cases in the literature, there are limited recommendations on practical allergen avoidance other than “don’t wear the problem shin guards, shoe insoles, or flip-flops.” However, Darrigade et al6 recommended wearing polyurethane shin guards and leather insoles as alternatives when AA contact allergy is suspected or confirmed. They also made it clear that thick socks worn between shin guards and the skin often are not good enough to avoid ACD because the relevant allergens may achieve skin contact despite the barrier.6
Patch Testing for AA Contact Allergy
Historically, ACD to shin guards or shin pads, insoles of shoes, and even flip-flops has been associated with rubber-related chemicals such as mercapto mix, thiuram mix, N-isopropyl-N’-phenyl-p-phenylenediamine, thioureas, and carbamates, as well as dyes, benzoyl peroxide, and urea formaldehyde or phenol formaldehyde resins.1 Most of these chemicals can be tested with standard screening series or supplemental series. Patients with contact allergy to AA may have negative patch testing to screening series and/or supplemental series and may have strong positive reactions to pieces of suspected foam shin pads or shin guards, shoes, and/or flip-flops. Although Koumaki et al5 recommended patch testing for AA contact allergy with AA 0.1% in acetone, Besner Morin et al4 mentioned that petrolatum may be a more desirable vehicle because it could maintain stability for a longer period of time. In fact, a 2021 article highlighting the American Contact Dermatitis Society Allergen of the Year recommends testing with either AA 0.1% in acetone or AA 0.1% in petrolatum.7 Unfortunately, AA is not commercially available for purchase at the time of publication. We are hopeful that this will change in the near future.
Final Interpretation
Acetophenone azine is an emerging allergen commonly identified in EVA foam and attributed to contact allergy to shin guards or pads, soles of shoes, and flip-flops. Most cases have been reported in Europe and Canada and have been identified in young male athletes. In addition to standard patch testing, athletes with lower leg dermatitis and/or dermatitis of the soles of the feet should undergo patch testing with AA 0.1% in acetone or petrolatum and pieces of the equipment and/or footwear.
- Raison-Peyron N, Bergendorff O, Bourrain JL, et al. Acetophenone azine: a new allergen responsible for severe contact dermatitis from shin pads. Contact Dermatitis. 2016;75:106-110.
- Raison-Peyron N, Bergendorff O, Du-Thanh A, et al. Two new cases of severe allergic contact dermatitis caused by acetophenone azine. Contact Dermatitis. 2017;76:380-381.
- De Fré C, Bergendorff O, Raison-Peyron N, et al. Acetophenone azine: a new shoe allergen causing severe foot dermatitis. Contact Dermatitis. 2017;77:416-417.
- Besner Morin C, Stanciu M, Miedzybrodzki B, et al. Allergic contact dermatitis from acetophenone azine in a Canadian child. Contact Dermatitis. 2020;83:41-42.
- Koumaki D, Bergendorff O, Bruze M, et al. Allergic contact dermatitis to shin pads in a hockey player: acetophenone is an emerging allergen. Dermatitis. 2019;30:162-163.
- Darrigade AS, Raison-Peyron N, Courouge-Dorcier D, et al. The chemical acetophenone azine: an important cause of shin and foot dermatitis in children. J Eur Acad Dermatol Venereol. 2020;34:E61-E62.
- Raison-Peyron N, Sasseville D. Acetophenone azine. Dermatitis. 2021;32:5-9.
It’s time for the American Contact Dermatitis Society (ACDS) Allergen of the Year! For 2021, the esteemed award goes to acetophenone azine (AA). If you have never heard of this chemical, you are not alone. Acetophenone azine has been identified in foam materials made of the copolymer ethyl-vinyl acetate (EVA). Contact allergy to AA initially was reported in 2016.1 There are only a few European and Canadian case reports and one case series of AA contact allergy in the literature, all of which are associated with foam shin pads or shin guards, shoe insoles, and/or flip-flops.2-6 Acetophenone azine is an important emerging allergen, and in this column, we will introduce you to AA and the sneaky places it can lurk and cause allergic contact dermatitis (ACD). We also highlight diagnosis, management, and patch testing for AA contact allergy.
AA Contact Allergy in the Literature
The first case of AA contact allergy was reported in Europe in 2016 when a 13-year-old male soccer player developed severe lower leg dermatitis and later generalized dermatitis associated with wearing foam shin guards.1 Patch testing to standard and supplemental trays was negative or not relevant; however, the patient exhibited strong reactions when patch tested directly to a piece of the shin guard soaked in acetone, water, and ethanol. Additional testing with AA diluted in acetone, water, and petrolatum resulted in positive patch test reactions to acetone dilutions of 1%, 0.1%, 0.01%, and 0.001% and aqueous solutions of 1% and 0.1%. Chromatographic analyses with high-performance liquid chromatography (HPLC) of shin guard extracts confirmed the culprit allergen to be AA.1
In the following months, the same clinic saw 2 more cases of AA contact allergy.2 An 11-year-old male soccer player developed lower leg dermatitis and later generalized dermatitis from wearing shin guards. Months later, he also developed dermatitis on the soles of the feet, which was attributed to wearing flip-flops. Patch tests to pieces of the shin guards and flip-flops were positive; AA in acetone 0.1% and 0.01% also was positive. As you might expect, HPLC again confirmed the presence of AA in the shin guards and flip-flops. The third patient was a 12-year-old boy with dermatitis on the soles of both feet; later he also developed a generalized dermatitis. Patch testing to pieces of the insoles of his sneakers and AA in acetone 0.1% and 0.01% was positive. Again, HPLC was positive for the presence of AA in the insoles of his sneakers.2
Several more cases of AA contact allergy have been reported in the literature. A 29-year-old European male hockey player demonstrated contact allergy to the gray foam of his shin pads as well as localized leg dermatitis followed by generalized dermatitis (are you noticing a trend yet?), and later dermatitis on the soles of the feet with positive patch-test reactions to pieces of his shin pads and shoe insoles as well as AA 0.1% and 0.01% in acetone.3 A 6-year-old Canadian male soccer player presented with leg dermatitis and later generalized dermatitis and dermatitis on the soles of the feet with positive reactions to pieces of his shin pads and shoe insoles as well as to AA 1% and 0.1% in petrolatum.4 A 17-year-old British male (another trend, all males so far!) hockey player developed dermatitis localized to the legs and positive patch tests to the worn foam inner lining of his shin pads as well as to AA 0.1%, 0.01%, and 0.001% in acetone.5Finally, Darrigade et al6 published a case series of 6 European children with AA contact allergy associated with shin pads and shoes; all had localized leg dermatitis, and some had generalized dermatitis. Patch testing to pieces of shin pads and shoe parts as well as to AA 0.1% in petrolatum and/or acetone showed with positive reactions to the foam pieces and AA in all 6 patients.
What’s the Deal With AA?
Acetophenone azine (also known as methylphenylketazine or bis[1-phenylethylidene]hydrazine) is composed of 2 acetophenone structures and a hydrazine moiety. It has been identified in EVA foam, which can be found in sports equipment such as shin pads or shin guards, shoes, and flip-flops. Raison-Peyron et al1 confirmed the presence of AA in EVA foam but reported that they did not know the exact reason for its presence. The authors theorized that AA might be a catalyst during EVA polymerization and also noted that it has antimicrobial and antihelminthic activity.1 Several authors noted that AA could be a by-product of EVA synthesis and that sports equipment manufacturers might not be aware of its presence in EVA.2,4-6 Some noted that AA concentration was higher in shin guards than in shoe insoles; they thought this explained why patients reacted first to their shin guards and were perhaps even initially sensitized to the shin guards, as well as why shoe insole contact allergy commonly was reported later or only after allergy to shin guards had already developed.4,6
Differential Diagnosis of Shin Pad or Shin Guard Dermatitis
We would be remiss if we did not mention the appropriate differential diagnosis when shin pad or shin guard dermatitis is identified. In fact, in most cases, shin guard dermatitis results from irritant contact dermatitis from friction, heat, and/or perspiration. Acetophenone azine contact allergy is not the most likely diagnosis when your sports-savvy, shin guard–wearing patient presents with anterior lower leg dermatitis. However, when conservative therapy (eg, barrier between the shin guard and the skin, control or management of perspiration, topical corticosteroid therapy) fails, patch testing to evaluate for ACD is indicated.
Management of AA Contact Allergy
As astute readers of this column are already aware, treatment of ACD requires strict allergen avoidance. You will find that we have the same recommendations for AA contact allergy. Given that there are only a handful of cases in the literature, there are limited recommendations on practical allergen avoidance other than “don’t wear the problem shin guards, shoe insoles, or flip-flops.” However, Darrigade et al6 recommended wearing polyurethane shin guards and leather insoles as alternatives when AA contact allergy is suspected or confirmed. They also made it clear that thick socks worn between shin guards and the skin often are not good enough to avoid ACD because the relevant allergens may achieve skin contact despite the barrier.6
Patch Testing for AA Contact Allergy
Historically, ACD to shin guards or shin pads, insoles of shoes, and even flip-flops has been associated with rubber-related chemicals such as mercapto mix, thiuram mix, N-isopropyl-N’-phenyl-p-phenylenediamine, thioureas, and carbamates, as well as dyes, benzoyl peroxide, and urea formaldehyde or phenol formaldehyde resins.1 Most of these chemicals can be tested with standard screening series or supplemental series. Patients with contact allergy to AA may have negative patch testing to screening series and/or supplemental series and may have strong positive reactions to pieces of suspected foam shin pads or shin guards, shoes, and/or flip-flops. Although Koumaki et al5 recommended patch testing for AA contact allergy with AA 0.1% in acetone, Besner Morin et al4 mentioned that petrolatum may be a more desirable vehicle because it could maintain stability for a longer period of time. In fact, a 2021 article highlighting the American Contact Dermatitis Society Allergen of the Year recommends testing with either AA 0.1% in acetone or AA 0.1% in petrolatum.7 Unfortunately, AA is not commercially available for purchase at the time of publication. We are hopeful that this will change in the near future.
Final Interpretation
Acetophenone azine is an emerging allergen commonly identified in EVA foam and attributed to contact allergy to shin guards or pads, soles of shoes, and flip-flops. Most cases have been reported in Europe and Canada and have been identified in young male athletes. In addition to standard patch testing, athletes with lower leg dermatitis and/or dermatitis of the soles of the feet should undergo patch testing with AA 0.1% in acetone or petrolatum and pieces of the equipment and/or footwear.
It’s time for the American Contact Dermatitis Society (ACDS) Allergen of the Year! For 2021, the esteemed award goes to acetophenone azine (AA). If you have never heard of this chemical, you are not alone. Acetophenone azine has been identified in foam materials made of the copolymer ethyl-vinyl acetate (EVA). Contact allergy to AA initially was reported in 2016.1 There are only a few European and Canadian case reports and one case series of AA contact allergy in the literature, all of which are associated with foam shin pads or shin guards, shoe insoles, and/or flip-flops.2-6 Acetophenone azine is an important emerging allergen, and in this column, we will introduce you to AA and the sneaky places it can lurk and cause allergic contact dermatitis (ACD). We also highlight diagnosis, management, and patch testing for AA contact allergy.
AA Contact Allergy in the Literature
The first case of AA contact allergy was reported in Europe in 2016 when a 13-year-old male soccer player developed severe lower leg dermatitis and later generalized dermatitis associated with wearing foam shin guards.1 Patch testing to standard and supplemental trays was negative or not relevant; however, the patient exhibited strong reactions when patch tested directly to a piece of the shin guard soaked in acetone, water, and ethanol. Additional testing with AA diluted in acetone, water, and petrolatum resulted in positive patch test reactions to acetone dilutions of 1%, 0.1%, 0.01%, and 0.001% and aqueous solutions of 1% and 0.1%. Chromatographic analyses with high-performance liquid chromatography (HPLC) of shin guard extracts confirmed the culprit allergen to be AA.1
In the following months, the same clinic saw 2 more cases of AA contact allergy.2 An 11-year-old male soccer player developed lower leg dermatitis and later generalized dermatitis from wearing shin guards. Months later, he also developed dermatitis on the soles of the feet, which was attributed to wearing flip-flops. Patch tests to pieces of the shin guards and flip-flops were positive; AA in acetone 0.1% and 0.01% also was positive. As you might expect, HPLC again confirmed the presence of AA in the shin guards and flip-flops. The third patient was a 12-year-old boy with dermatitis on the soles of both feet; later he also developed a generalized dermatitis. Patch testing to pieces of the insoles of his sneakers and AA in acetone 0.1% and 0.01% was positive. Again, HPLC was positive for the presence of AA in the insoles of his sneakers.2
Several more cases of AA contact allergy have been reported in the literature. A 29-year-old European male hockey player demonstrated contact allergy to the gray foam of his shin pads as well as localized leg dermatitis followed by generalized dermatitis (are you noticing a trend yet?), and later dermatitis on the soles of the feet with positive patch-test reactions to pieces of his shin pads and shoe insoles as well as AA 0.1% and 0.01% in acetone.3 A 6-year-old Canadian male soccer player presented with leg dermatitis and later generalized dermatitis and dermatitis on the soles of the feet with positive reactions to pieces of his shin pads and shoe insoles as well as to AA 1% and 0.1% in petrolatum.4 A 17-year-old British male (another trend, all males so far!) hockey player developed dermatitis localized to the legs and positive patch tests to the worn foam inner lining of his shin pads as well as to AA 0.1%, 0.01%, and 0.001% in acetone.5Finally, Darrigade et al6 published a case series of 6 European children with AA contact allergy associated with shin pads and shoes; all had localized leg dermatitis, and some had generalized dermatitis. Patch testing to pieces of shin pads and shoe parts as well as to AA 0.1% in petrolatum and/or acetone showed with positive reactions to the foam pieces and AA in all 6 patients.
What’s the Deal With AA?
Acetophenone azine (also known as methylphenylketazine or bis[1-phenylethylidene]hydrazine) is composed of 2 acetophenone structures and a hydrazine moiety. It has been identified in EVA foam, which can be found in sports equipment such as shin pads or shin guards, shoes, and flip-flops. Raison-Peyron et al1 confirmed the presence of AA in EVA foam but reported that they did not know the exact reason for its presence. The authors theorized that AA might be a catalyst during EVA polymerization and also noted that it has antimicrobial and antihelminthic activity.1 Several authors noted that AA could be a by-product of EVA synthesis and that sports equipment manufacturers might not be aware of its presence in EVA.2,4-6 Some noted that AA concentration was higher in shin guards than in shoe insoles; they thought this explained why patients reacted first to their shin guards and were perhaps even initially sensitized to the shin guards, as well as why shoe insole contact allergy commonly was reported later or only after allergy to shin guards had already developed.4,6
Differential Diagnosis of Shin Pad or Shin Guard Dermatitis
We would be remiss if we did not mention the appropriate differential diagnosis when shin pad or shin guard dermatitis is identified. In fact, in most cases, shin guard dermatitis results from irritant contact dermatitis from friction, heat, and/or perspiration. Acetophenone azine contact allergy is not the most likely diagnosis when your sports-savvy, shin guard–wearing patient presents with anterior lower leg dermatitis. However, when conservative therapy (eg, barrier between the shin guard and the skin, control or management of perspiration, topical corticosteroid therapy) fails, patch testing to evaluate for ACD is indicated.
Management of AA Contact Allergy
As astute readers of this column are already aware, treatment of ACD requires strict allergen avoidance. You will find that we have the same recommendations for AA contact allergy. Given that there are only a handful of cases in the literature, there are limited recommendations on practical allergen avoidance other than “don’t wear the problem shin guards, shoe insoles, or flip-flops.” However, Darrigade et al6 recommended wearing polyurethane shin guards and leather insoles as alternatives when AA contact allergy is suspected or confirmed. They also made it clear that thick socks worn between shin guards and the skin often are not good enough to avoid ACD because the relevant allergens may achieve skin contact despite the barrier.6
Patch Testing for AA Contact Allergy
Historically, ACD to shin guards or shin pads, insoles of shoes, and even flip-flops has been associated with rubber-related chemicals such as mercapto mix, thiuram mix, N-isopropyl-N’-phenyl-p-phenylenediamine, thioureas, and carbamates, as well as dyes, benzoyl peroxide, and urea formaldehyde or phenol formaldehyde resins.1 Most of these chemicals can be tested with standard screening series or supplemental series. Patients with contact allergy to AA may have negative patch testing to screening series and/or supplemental series and may have strong positive reactions to pieces of suspected foam shin pads or shin guards, shoes, and/or flip-flops. Although Koumaki et al5 recommended patch testing for AA contact allergy with AA 0.1% in acetone, Besner Morin et al4 mentioned that petrolatum may be a more desirable vehicle because it could maintain stability for a longer period of time. In fact, a 2021 article highlighting the American Contact Dermatitis Society Allergen of the Year recommends testing with either AA 0.1% in acetone or AA 0.1% in petrolatum.7 Unfortunately, AA is not commercially available for purchase at the time of publication. We are hopeful that this will change in the near future.
Final Interpretation
Acetophenone azine is an emerging allergen commonly identified in EVA foam and attributed to contact allergy to shin guards or pads, soles of shoes, and flip-flops. Most cases have been reported in Europe and Canada and have been identified in young male athletes. In addition to standard patch testing, athletes with lower leg dermatitis and/or dermatitis of the soles of the feet should undergo patch testing with AA 0.1% in acetone or petrolatum and pieces of the equipment and/or footwear.
- Raison-Peyron N, Bergendorff O, Bourrain JL, et al. Acetophenone azine: a new allergen responsible for severe contact dermatitis from shin pads. Contact Dermatitis. 2016;75:106-110.
- Raison-Peyron N, Bergendorff O, Du-Thanh A, et al. Two new cases of severe allergic contact dermatitis caused by acetophenone azine. Contact Dermatitis. 2017;76:380-381.
- De Fré C, Bergendorff O, Raison-Peyron N, et al. Acetophenone azine: a new shoe allergen causing severe foot dermatitis. Contact Dermatitis. 2017;77:416-417.
- Besner Morin C, Stanciu M, Miedzybrodzki B, et al. Allergic contact dermatitis from acetophenone azine in a Canadian child. Contact Dermatitis. 2020;83:41-42.
- Koumaki D, Bergendorff O, Bruze M, et al. Allergic contact dermatitis to shin pads in a hockey player: acetophenone is an emerging allergen. Dermatitis. 2019;30:162-163.
- Darrigade AS, Raison-Peyron N, Courouge-Dorcier D, et al. The chemical acetophenone azine: an important cause of shin and foot dermatitis in children. J Eur Acad Dermatol Venereol. 2020;34:E61-E62.
- Raison-Peyron N, Sasseville D. Acetophenone azine. Dermatitis. 2021;32:5-9.
- Raison-Peyron N, Bergendorff O, Bourrain JL, et al. Acetophenone azine: a new allergen responsible for severe contact dermatitis from shin pads. Contact Dermatitis. 2016;75:106-110.
- Raison-Peyron N, Bergendorff O, Du-Thanh A, et al. Two new cases of severe allergic contact dermatitis caused by acetophenone azine. Contact Dermatitis. 2017;76:380-381.
- De Fré C, Bergendorff O, Raison-Peyron N, et al. Acetophenone azine: a new shoe allergen causing severe foot dermatitis. Contact Dermatitis. 2017;77:416-417.
- Besner Morin C, Stanciu M, Miedzybrodzki B, et al. Allergic contact dermatitis from acetophenone azine in a Canadian child. Contact Dermatitis. 2020;83:41-42.
- Koumaki D, Bergendorff O, Bruze M, et al. Allergic contact dermatitis to shin pads in a hockey player: acetophenone is an emerging allergen. Dermatitis. 2019;30:162-163.
- Darrigade AS, Raison-Peyron N, Courouge-Dorcier D, et al. The chemical acetophenone azine: an important cause of shin and foot dermatitis in children. J Eur Acad Dermatol Venereol. 2020;34:E61-E62.
- Raison-Peyron N, Sasseville D. Acetophenone azine. Dermatitis. 2021;32:5-9.
Practice Points
- Acetophenone azine is an emerging allergen identified in ethyl-vinyl acetate foam used in shin guards, shoe soles, and flip-flops.
- Cases have been reported in young male athletes in Europe and Canada.
- Patch testing can be completed with acetophenone azine 0.1% in acetone or petrolatum.
Desmoplastic Melanoma Masquerading as Neurofibroma
Desmoplastic melanoma (DMM) is a rare variant of melanoma that presents major challenges to both clinicians and pathologists.1 Clinically, the lesions may appear as subtle bland papules, nodules, or plaques. They can be easily mistaken for benign growths, leading to a delayed diagnosis. Consequently, most DMMs at the time of diagnosis tend to be thick, with a mean Breslow depth ranging from 2.0 to 6.5 mm.2 Histopathologic evaluation has its difficulties. At scanning magnification, these tumors may show low cellularity, mimicking a benign proliferation. It is well recognized that S-100 and other tumor markers lack specificity for DMM, which can be positive in a range of neural tumors and other cell types.2 In some amelanotic tumors, DMM becomes virtually indistinguishable from benign peripheral sheath tumors such as neurofribroma.3
Desmoplastic melanoma is exceedingly uncommon in the United States, with an estimated annual incidence rate of 2.0 cases per million.2 Typical locations of presentation include sun-exposed skin, with the head and neck regions representing more than half of reported cases.2 Desmoplastic melanoma largely is a disease of fair-skinned patients, with 95.5% of cases in the United States occurring in white non-Hispanic individuals. Advancing age, male gender, and head and neck location are associated with an increased risk for DMM-specific death.2 It is important that new or changing lesions in the correct cohort and location are biopsied promptly. We present this case to highlight the ongoing challenges of diagnosing DMM both clinically and histologically and to review the salient features of this often benign-appearing tumor.
Case Report
A 51-year-old White man with a history of prostate cancer, a personal and family history of melanoma, and benign neurofibromas presented with a 6-mm, pink, well-demarcated, soft papule on the left lateral neck (Figure 1). The lesion had been stable for many years but began growing more rapidly 1 to 2 years prior to presentation. The lesion was asymptomatic, and he denied changes in color or texture. There also was no bleeding or ulceration. A review of systems was unremarkable. A shave biopsy of the lesion revealed a nodular spindle cell tumor in the dermis resembling a neurofibroma on low power (Figure 2). However, overlying the tumor was a confluent proliferation positive for MART-1 and S-100, which was consistent with a diagnosis of melanoma in situ (Figure 3). Higher-power evaluation of the dermal proliferation showed both bland and hyperchromatic spindled and epithelioid cells (Figure 4), with rare mitotic figures highlighted by PHH3, an uncommon finding in neurofibromas (Figure 5). The dermal spindle cells were positive for S-100 and p75 and negative for Melan-A. Epithelial membrane antigen highlighted a faint sheath surrounding the dermal component. Ki-67 revealed a mildly increased proliferative index in the dermal component. The diagnosis of DMM was made after outside dermatopathology consultation was in agreement. However, the possibility of a melanoma in situ growing in association with an underlying neurofibroma remained a diagnostic consideration histologically. The lesion was widely excised.
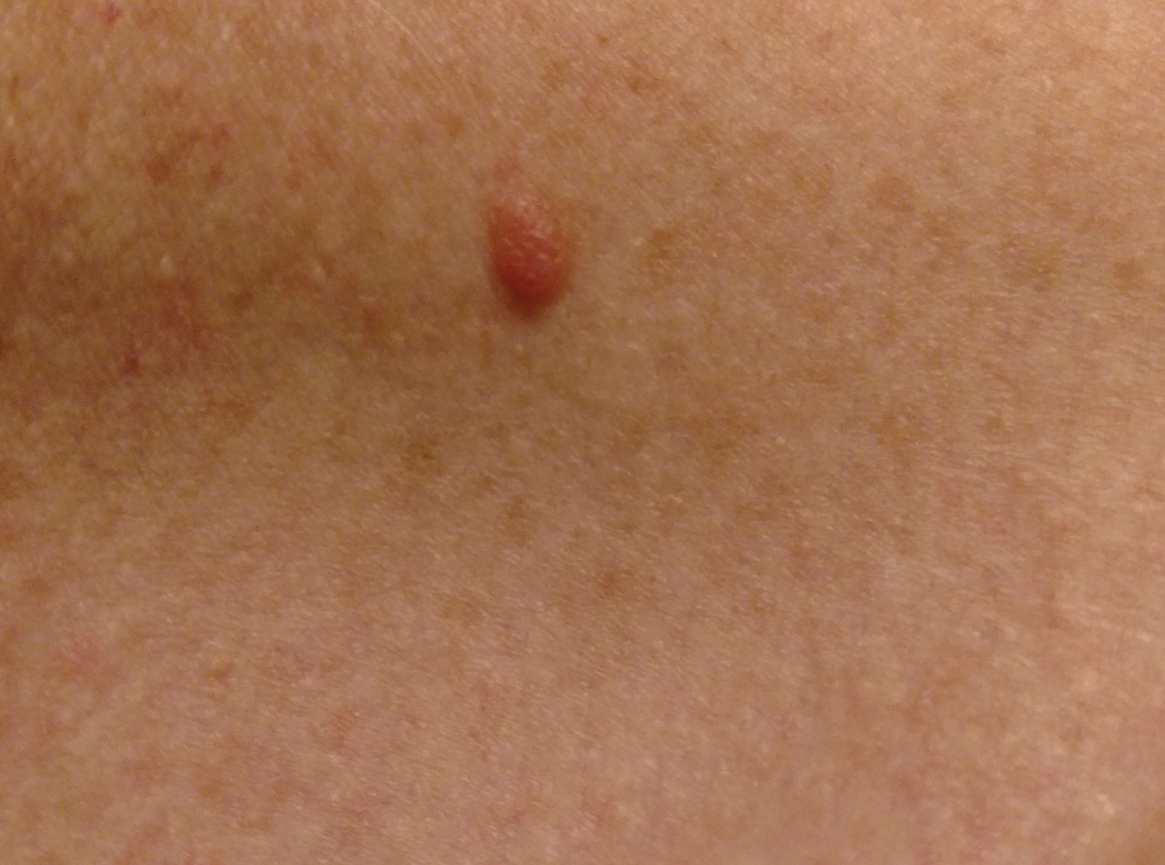
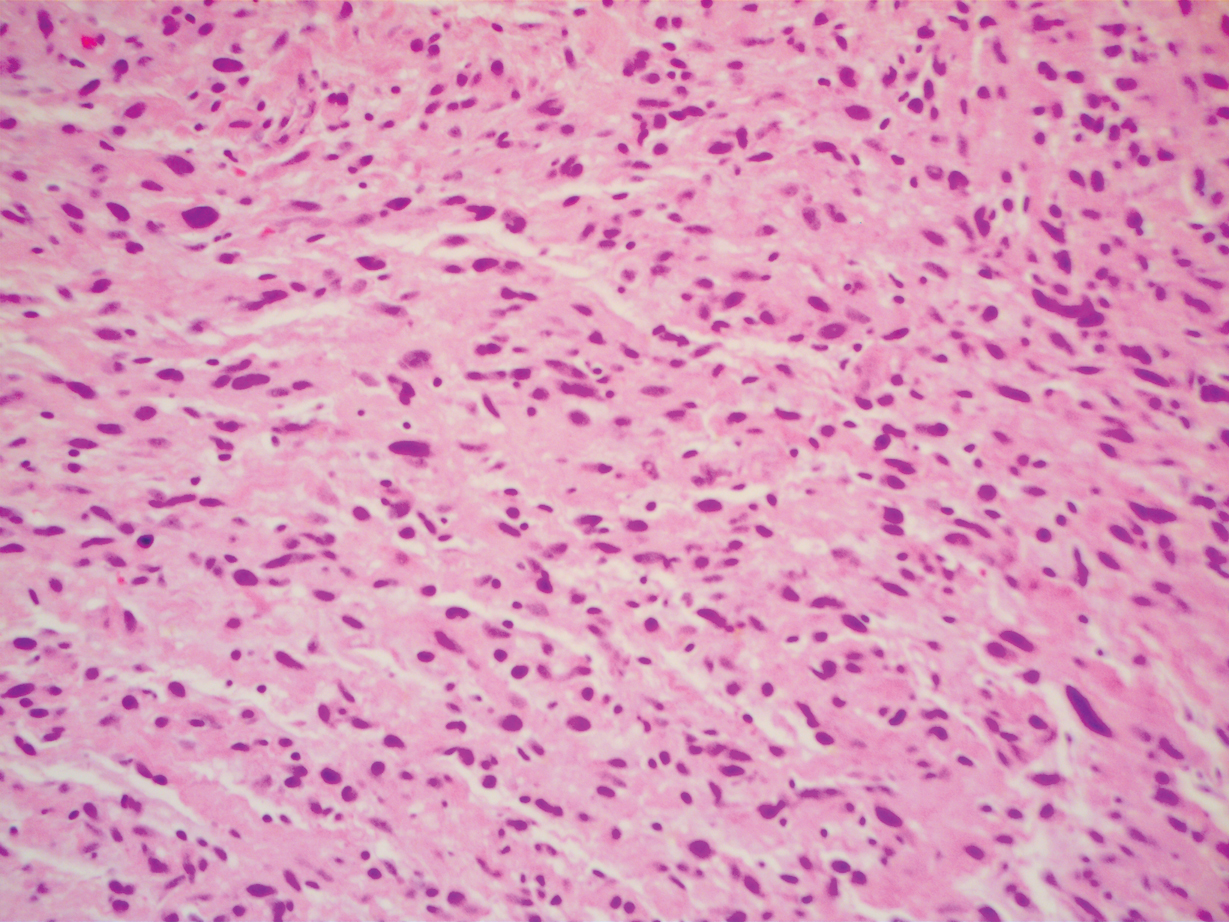
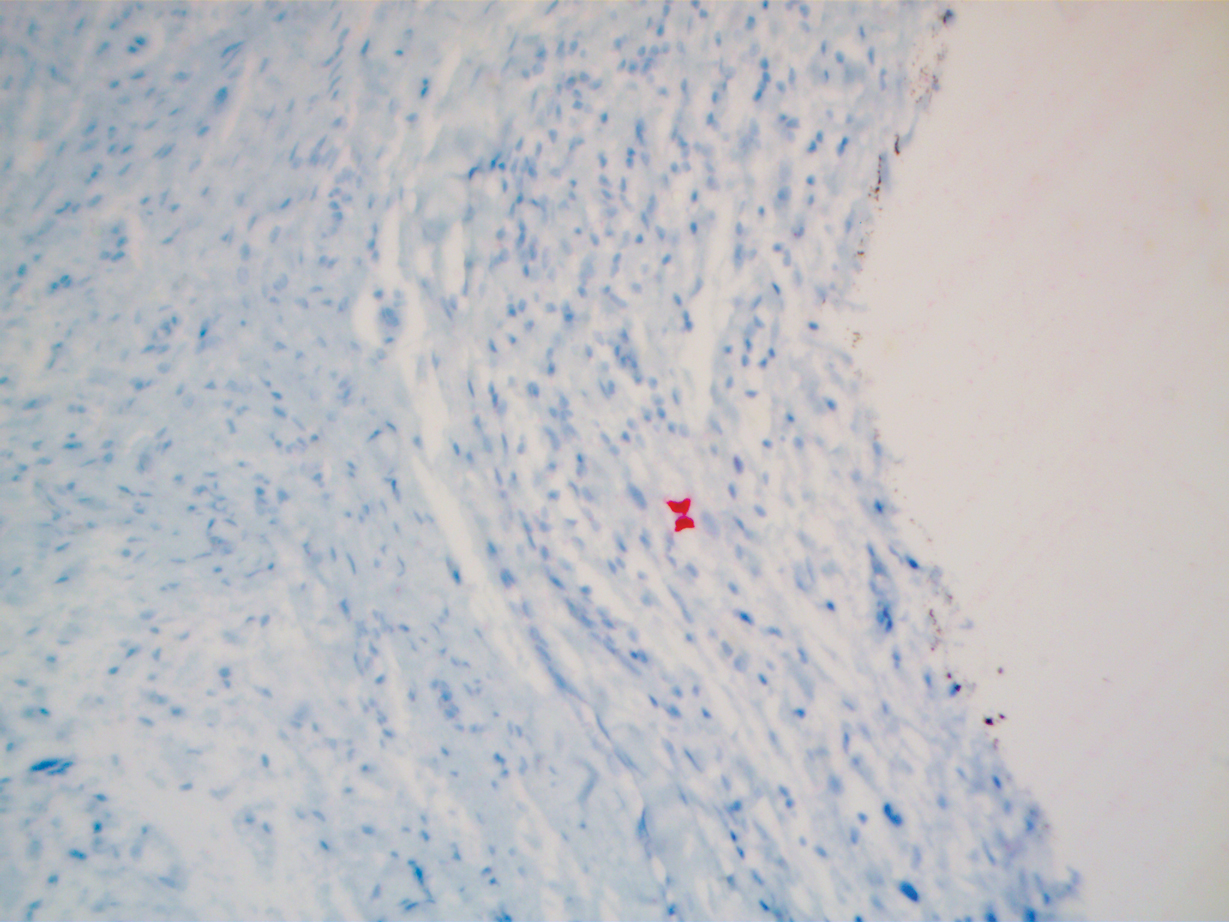
Comment
Differential for DMM
Early DMMs may not show sufficient cytologic atypia to permit obvious distinction from neurofibromas, which becomes problematic when encountering a spindle cell proliferation within severely sun-damaged skin, or even more so when an intraepidermal population of melanocytes is situated above a dermal population of slender, spindled, S-100–positive cells, as seen in our patient.4 For these challenging scenarios, Yeh and McCalmont4 have proposed evaluating for a CD34 “fingerprint” pattern. This pattern typically is widespread in neurofibroma but absent or limited in DMM, and it is a useful adjunct in the differential diagnosis when conventional immunohistochemistry has little contribution.
There are several case reports in the literature of DMM mimicking other benign or malignant proliferations. In 2012, Jou et al5 described a case of a 62-year-old White man who presented with an oral nodule consistent with fibrous inflammatory hyperplasia clinically. Incisional biopsy later confirmed the diagnosis of amelanotic DMM.5 Similar case reports have been described in which the diagnosis of DMM was later found to resemble a sarcoma and malignant peripheral nerve sheath tumor.6,7
Diagnosis of DMM
The prototypical DMM is an asymmetrical and deeply infiltrative spindle cell lesion in severely sun-damaged skin. By definition, the individual melanocytes are separated by connective tissue components, giving the tumor a paucicellular appearance.1 Although the low cellularity can give a deceptively bland scanning aspect, on high-power examination there usually are identifiable atypical spindled cells with enlarged, elongated, and hyperchromatic nuclei. S-100 typically is diffusely positive in DMM, though occasional cases show more limited staining.8 Other commonly used and more specific markers of melanocytic differentiation, including HMB45 and Melan-A, typically are negative in the paucicellular spindle cell components.9 Desmoplastic melanoma can be further categorized by the degree of fibrosis within a particular tumor. If fibrosis is prominent throughout the entire tumor, it is named pure DMM. On the other hand, fibrosis may only represent a portion of an otherwise nondesmoplastic melanoma, which is known as combined DMM.10
Conclusion
We present this case to highlight the ongoing challenges of diagnosing DMM both clinically and histologically. Although a bland-appearing lesion, key clinical features prompting a biopsy in our patient included recent growth of the lesion, a personal history of melanoma, the patient’s fair skin type, a history of heavy sun exposure, and the location of the lesion. According to Busam,11 an associated melanoma in situ component is identified in 80% to 85% of DMM cases. Detection of a melanoma in situ component associated with a malignant spindle cell tumor can help establish the diagnosis of DMM. In the absence of melanoma in situ, a strong diffuse immunoreactivity for S-100 and lack of epithelial markers support the diagnosis.11 After review of the literature, our case likely represents DMM as opposed to a melanoma in situ developing within a neurofibroma.
- Wood BA. Desmoplastic melanoma: recent advances and persisting challenges. Pathology. 2013;45:453-463.
- Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
- Machado I, Llombart B, Cruz J, et al. Desmoplastic melanoma may mimic a cutaneous peripheral nerve sheath tumor: report of 3 challenging cases. J Cutan Pathol. 2017;4:632-638.
- Yeh I, McCalmont, TH. Distinguishing neurofibroma from desmoplastic melanoma: the value of the CD34 fingerprint. J Cutan Pathol. 2011;38:625-630.
- Jou A, Miranda FV, Oliveira MG, et al. Oral desmoplastic melanoma mimicking inflammatory hyperplasia. Gerodontology. 2012;29:E1163-E1167.
- Ishikura H, Kojo T, Ichimura H, et al. Desmoplastic malignant melanoma of the uterine cervix: a rare primary malignancy in the uterus mimicking a sarcoma. Histopathology. 1998;33:93-94.
- Barnett SL, Wells MJ, Mickey B, et al. Perineural extension of cutaneous desmoplastic melanoma mimicking an intracranial malignant peripheral nerve sheath tumor. case report. J Neurosurg. 2011;115:273-277.
- Jain S, Allen PW. Desmoplastic malignant melanoma and its variants. a study of 45 cases. Am J Surg Pathol. 1989;13:358-373.
- Skelton HG, Maceira J, Smith KJ, et al. HMB45 negative spindle cell malignant melanoma. Am J Dermatopathol. 1997;19:580-584.
- George E, McClain SE, Slingluff CL, et al. Subclassification of desmoplastic melanoma: pure and mixed variants have significantly different capacities for lymph node metastasis. J Cutan Pathol. 2009;36:425-432.
- Busam KJ. Desmoplastic melanoma. Clin Lab Med. 2011;31:321-330.
Desmoplastic melanoma (DMM) is a rare variant of melanoma that presents major challenges to both clinicians and pathologists.1 Clinically, the lesions may appear as subtle bland papules, nodules, or plaques. They can be easily mistaken for benign growths, leading to a delayed diagnosis. Consequently, most DMMs at the time of diagnosis tend to be thick, with a mean Breslow depth ranging from 2.0 to 6.5 mm.2 Histopathologic evaluation has its difficulties. At scanning magnification, these tumors may show low cellularity, mimicking a benign proliferation. It is well recognized that S-100 and other tumor markers lack specificity for DMM, which can be positive in a range of neural tumors and other cell types.2 In some amelanotic tumors, DMM becomes virtually indistinguishable from benign peripheral sheath tumors such as neurofribroma.3
Desmoplastic melanoma is exceedingly uncommon in the United States, with an estimated annual incidence rate of 2.0 cases per million.2 Typical locations of presentation include sun-exposed skin, with the head and neck regions representing more than half of reported cases.2 Desmoplastic melanoma largely is a disease of fair-skinned patients, with 95.5% of cases in the United States occurring in white non-Hispanic individuals. Advancing age, male gender, and head and neck location are associated with an increased risk for DMM-specific death.2 It is important that new or changing lesions in the correct cohort and location are biopsied promptly. We present this case to highlight the ongoing challenges of diagnosing DMM both clinically and histologically and to review the salient features of this often benign-appearing tumor.
Case Report
A 51-year-old White man with a history of prostate cancer, a personal and family history of melanoma, and benign neurofibromas presented with a 6-mm, pink, well-demarcated, soft papule on the left lateral neck (Figure 1). The lesion had been stable for many years but began growing more rapidly 1 to 2 years prior to presentation. The lesion was asymptomatic, and he denied changes in color or texture. There also was no bleeding or ulceration. A review of systems was unremarkable. A shave biopsy of the lesion revealed a nodular spindle cell tumor in the dermis resembling a neurofibroma on low power (Figure 2). However, overlying the tumor was a confluent proliferation positive for MART-1 and S-100, which was consistent with a diagnosis of melanoma in situ (Figure 3). Higher-power evaluation of the dermal proliferation showed both bland and hyperchromatic spindled and epithelioid cells (Figure 4), with rare mitotic figures highlighted by PHH3, an uncommon finding in neurofibromas (Figure 5). The dermal spindle cells were positive for S-100 and p75 and negative for Melan-A. Epithelial membrane antigen highlighted a faint sheath surrounding the dermal component. Ki-67 revealed a mildly increased proliferative index in the dermal component. The diagnosis of DMM was made after outside dermatopathology consultation was in agreement. However, the possibility of a melanoma in situ growing in association with an underlying neurofibroma remained a diagnostic consideration histologically. The lesion was widely excised.
Comment
Differential for DMM
Early DMMs may not show sufficient cytologic atypia to permit obvious distinction from neurofibromas, which becomes problematic when encountering a spindle cell proliferation within severely sun-damaged skin, or even more so when an intraepidermal population of melanocytes is situated above a dermal population of slender, spindled, S-100–positive cells, as seen in our patient.4 For these challenging scenarios, Yeh and McCalmont4 have proposed evaluating for a CD34 “fingerprint” pattern. This pattern typically is widespread in neurofibroma but absent or limited in DMM, and it is a useful adjunct in the differential diagnosis when conventional immunohistochemistry has little contribution.
There are several case reports in the literature of DMM mimicking other benign or malignant proliferations. In 2012, Jou et al5 described a case of a 62-year-old White man who presented with an oral nodule consistent with fibrous inflammatory hyperplasia clinically. Incisional biopsy later confirmed the diagnosis of amelanotic DMM.5 Similar case reports have been described in which the diagnosis of DMM was later found to resemble a sarcoma and malignant peripheral nerve sheath tumor.6,7
Diagnosis of DMM
The prototypical DMM is an asymmetrical and deeply infiltrative spindle cell lesion in severely sun-damaged skin. By definition, the individual melanocytes are separated by connective tissue components, giving the tumor a paucicellular appearance.1 Although the low cellularity can give a deceptively bland scanning aspect, on high-power examination there usually are identifiable atypical spindled cells with enlarged, elongated, and hyperchromatic nuclei. S-100 typically is diffusely positive in DMM, though occasional cases show more limited staining.8 Other commonly used and more specific markers of melanocytic differentiation, including HMB45 and Melan-A, typically are negative in the paucicellular spindle cell components.9 Desmoplastic melanoma can be further categorized by the degree of fibrosis within a particular tumor. If fibrosis is prominent throughout the entire tumor, it is named pure DMM. On the other hand, fibrosis may only represent a portion of an otherwise nondesmoplastic melanoma, which is known as combined DMM.10
Conclusion
We present this case to highlight the ongoing challenges of diagnosing DMM both clinically and histologically. Although a bland-appearing lesion, key clinical features prompting a biopsy in our patient included recent growth of the lesion, a personal history of melanoma, the patient’s fair skin type, a history of heavy sun exposure, and the location of the lesion. According to Busam,11 an associated melanoma in situ component is identified in 80% to 85% of DMM cases. Detection of a melanoma in situ component associated with a malignant spindle cell tumor can help establish the diagnosis of DMM. In the absence of melanoma in situ, a strong diffuse immunoreactivity for S-100 and lack of epithelial markers support the diagnosis.11 After review of the literature, our case likely represents DMM as opposed to a melanoma in situ developing within a neurofibroma.
Desmoplastic melanoma (DMM) is a rare variant of melanoma that presents major challenges to both clinicians and pathologists.1 Clinically, the lesions may appear as subtle bland papules, nodules, or plaques. They can be easily mistaken for benign growths, leading to a delayed diagnosis. Consequently, most DMMs at the time of diagnosis tend to be thick, with a mean Breslow depth ranging from 2.0 to 6.5 mm.2 Histopathologic evaluation has its difficulties. At scanning magnification, these tumors may show low cellularity, mimicking a benign proliferation. It is well recognized that S-100 and other tumor markers lack specificity for DMM, which can be positive in a range of neural tumors and other cell types.2 In some amelanotic tumors, DMM becomes virtually indistinguishable from benign peripheral sheath tumors such as neurofribroma.3
Desmoplastic melanoma is exceedingly uncommon in the United States, with an estimated annual incidence rate of 2.0 cases per million.2 Typical locations of presentation include sun-exposed skin, with the head and neck regions representing more than half of reported cases.2 Desmoplastic melanoma largely is a disease of fair-skinned patients, with 95.5% of cases in the United States occurring in white non-Hispanic individuals. Advancing age, male gender, and head and neck location are associated with an increased risk for DMM-specific death.2 It is important that new or changing lesions in the correct cohort and location are biopsied promptly. We present this case to highlight the ongoing challenges of diagnosing DMM both clinically and histologically and to review the salient features of this often benign-appearing tumor.
Case Report
A 51-year-old White man with a history of prostate cancer, a personal and family history of melanoma, and benign neurofibromas presented with a 6-mm, pink, well-demarcated, soft papule on the left lateral neck (Figure 1). The lesion had been stable for many years but began growing more rapidly 1 to 2 years prior to presentation. The lesion was asymptomatic, and he denied changes in color or texture. There also was no bleeding or ulceration. A review of systems was unremarkable. A shave biopsy of the lesion revealed a nodular spindle cell tumor in the dermis resembling a neurofibroma on low power (Figure 2). However, overlying the tumor was a confluent proliferation positive for MART-1 and S-100, which was consistent with a diagnosis of melanoma in situ (Figure 3). Higher-power evaluation of the dermal proliferation showed both bland and hyperchromatic spindled and epithelioid cells (Figure 4), with rare mitotic figures highlighted by PHH3, an uncommon finding in neurofibromas (Figure 5). The dermal spindle cells were positive for S-100 and p75 and negative for Melan-A. Epithelial membrane antigen highlighted a faint sheath surrounding the dermal component. Ki-67 revealed a mildly increased proliferative index in the dermal component. The diagnosis of DMM was made after outside dermatopathology consultation was in agreement. However, the possibility of a melanoma in situ growing in association with an underlying neurofibroma remained a diagnostic consideration histologically. The lesion was widely excised.
Comment
Differential for DMM
Early DMMs may not show sufficient cytologic atypia to permit obvious distinction from neurofibromas, which becomes problematic when encountering a spindle cell proliferation within severely sun-damaged skin, or even more so when an intraepidermal population of melanocytes is situated above a dermal population of slender, spindled, S-100–positive cells, as seen in our patient.4 For these challenging scenarios, Yeh and McCalmont4 have proposed evaluating for a CD34 “fingerprint” pattern. This pattern typically is widespread in neurofibroma but absent or limited in DMM, and it is a useful adjunct in the differential diagnosis when conventional immunohistochemistry has little contribution.
There are several case reports in the literature of DMM mimicking other benign or malignant proliferations. In 2012, Jou et al5 described a case of a 62-year-old White man who presented with an oral nodule consistent with fibrous inflammatory hyperplasia clinically. Incisional biopsy later confirmed the diagnosis of amelanotic DMM.5 Similar case reports have been described in which the diagnosis of DMM was later found to resemble a sarcoma and malignant peripheral nerve sheath tumor.6,7
Diagnosis of DMM
The prototypical DMM is an asymmetrical and deeply infiltrative spindle cell lesion in severely sun-damaged skin. By definition, the individual melanocytes are separated by connective tissue components, giving the tumor a paucicellular appearance.1 Although the low cellularity can give a deceptively bland scanning aspect, on high-power examination there usually are identifiable atypical spindled cells with enlarged, elongated, and hyperchromatic nuclei. S-100 typically is diffusely positive in DMM, though occasional cases show more limited staining.8 Other commonly used and more specific markers of melanocytic differentiation, including HMB45 and Melan-A, typically are negative in the paucicellular spindle cell components.9 Desmoplastic melanoma can be further categorized by the degree of fibrosis within a particular tumor. If fibrosis is prominent throughout the entire tumor, it is named pure DMM. On the other hand, fibrosis may only represent a portion of an otherwise nondesmoplastic melanoma, which is known as combined DMM.10
Conclusion
We present this case to highlight the ongoing challenges of diagnosing DMM both clinically and histologically. Although a bland-appearing lesion, key clinical features prompting a biopsy in our patient included recent growth of the lesion, a personal history of melanoma, the patient’s fair skin type, a history of heavy sun exposure, and the location of the lesion. According to Busam,11 an associated melanoma in situ component is identified in 80% to 85% of DMM cases. Detection of a melanoma in situ component associated with a malignant spindle cell tumor can help establish the diagnosis of DMM. In the absence of melanoma in situ, a strong diffuse immunoreactivity for S-100 and lack of epithelial markers support the diagnosis.11 After review of the literature, our case likely represents DMM as opposed to a melanoma in situ developing within a neurofibroma.
- Wood BA. Desmoplastic melanoma: recent advances and persisting challenges. Pathology. 2013;45:453-463.
- Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
- Machado I, Llombart B, Cruz J, et al. Desmoplastic melanoma may mimic a cutaneous peripheral nerve sheath tumor: report of 3 challenging cases. J Cutan Pathol. 2017;4:632-638.
- Yeh I, McCalmont, TH. Distinguishing neurofibroma from desmoplastic melanoma: the value of the CD34 fingerprint. J Cutan Pathol. 2011;38:625-630.
- Jou A, Miranda FV, Oliveira MG, et al. Oral desmoplastic melanoma mimicking inflammatory hyperplasia. Gerodontology. 2012;29:E1163-E1167.
- Ishikura H, Kojo T, Ichimura H, et al. Desmoplastic malignant melanoma of the uterine cervix: a rare primary malignancy in the uterus mimicking a sarcoma. Histopathology. 1998;33:93-94.
- Barnett SL, Wells MJ, Mickey B, et al. Perineural extension of cutaneous desmoplastic melanoma mimicking an intracranial malignant peripheral nerve sheath tumor. case report. J Neurosurg. 2011;115:273-277.
- Jain S, Allen PW. Desmoplastic malignant melanoma and its variants. a study of 45 cases. Am J Surg Pathol. 1989;13:358-373.
- Skelton HG, Maceira J, Smith KJ, et al. HMB45 negative spindle cell malignant melanoma. Am J Dermatopathol. 1997;19:580-584.
- George E, McClain SE, Slingluff CL, et al. Subclassification of desmoplastic melanoma: pure and mixed variants have significantly different capacities for lymph node metastasis. J Cutan Pathol. 2009;36:425-432.
- Busam KJ. Desmoplastic melanoma. Clin Lab Med. 2011;31:321-330.
- Wood BA. Desmoplastic melanoma: recent advances and persisting challenges. Pathology. 2013;45:453-463.
- Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
- Machado I, Llombart B, Cruz J, et al. Desmoplastic melanoma may mimic a cutaneous peripheral nerve sheath tumor: report of 3 challenging cases. J Cutan Pathol. 2017;4:632-638.
- Yeh I, McCalmont, TH. Distinguishing neurofibroma from desmoplastic melanoma: the value of the CD34 fingerprint. J Cutan Pathol. 2011;38:625-630.
- Jou A, Miranda FV, Oliveira MG, et al. Oral desmoplastic melanoma mimicking inflammatory hyperplasia. Gerodontology. 2012;29:E1163-E1167.
- Ishikura H, Kojo T, Ichimura H, et al. Desmoplastic malignant melanoma of the uterine cervix: a rare primary malignancy in the uterus mimicking a sarcoma. Histopathology. 1998;33:93-94.
- Barnett SL, Wells MJ, Mickey B, et al. Perineural extension of cutaneous desmoplastic melanoma mimicking an intracranial malignant peripheral nerve sheath tumor. case report. J Neurosurg. 2011;115:273-277.
- Jain S, Allen PW. Desmoplastic malignant melanoma and its variants. a study of 45 cases. Am J Surg Pathol. 1989;13:358-373.
- Skelton HG, Maceira J, Smith KJ, et al. HMB45 negative spindle cell malignant melanoma. Am J Dermatopathol. 1997;19:580-584.
- George E, McClain SE, Slingluff CL, et al. Subclassification of desmoplastic melanoma: pure and mixed variants have significantly different capacities for lymph node metastasis. J Cutan Pathol. 2009;36:425-432.
- Busam KJ. Desmoplastic melanoma. Clin Lab Med. 2011;31:321-330.
Practice Points
- Desmoplastic melanoma remains a diagnostic challenge both clinically and histologically.
- New or changing lesions on sun-exposed sites of elderly patients with fair skin types should have a low threshold for biopsy.
- Consensus between more than one dermatopathologist is sometimes required to make the diagnosis histologically.
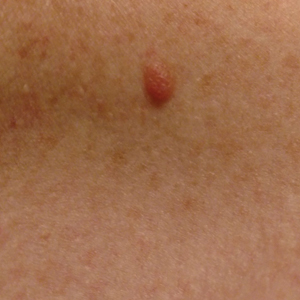
What’s Eating You? Culex Mosquitoes and West Nile Virus
What is West Nile virus? How is it contracted, and who can become infected?
West Nile virus (WNV) is a single-stranded RNA virus of the Flaviviridae family and Flavivirus genus, a lineage that also includes the yellow fever, dengue, Zika, Japanese encephalitis, and Saint Louis encephalitis viruses.1 Birds serve as the reservoir hosts of WNV, and mosquitoes acquire the virus during feeding.2 West Nile virus then is transmitted to humans primarily by bites from Culex mosquitoes, which are especially prevalent in wooded areas during peak mosquito season (summer through early fall in North America).1 Mosquitoes also can infect horses; however, humans and horses are dead-end hosts, meaning they do not pass the virus on to other biting mosquitoes.3 There also have been rare reports of transmission of WNV through blood and donation as well as mother-to-baby transmission.2
What is the epidemiology of WNV in the United States?
Since the introduction of WNV to the United States in 1999, it has become an important public health concern, with 48,183 cases and 2163 deaths reported since 1999.2,3 In 2018, Nebraska had the highest number of cases of WNV (n=251), followed by California (n=217), North Dakota (n=204), Illinois (n=176), and South Dakota (n=169).3 West Nile virus is endemic to all 48 contiguous states and Canada, though the Great Plains region is especially affected by WNV due to several factors, such as a greater percentage of rural land, forests, and irrigated areas.4 The Great Plains region also has been thought to be an ecological niche for a more virulent species (Culex tarsalis) compared to other regions in the United States.5
The annual incidence of WNV in the United States peaked in 2003 at 9862 cases (up from 62 cases in 1999), then declined gradually until 2008 to 2011, during which the incidence was stable at 700 to 1100 new cases per year. However, there was a resurgence of cases (n=5674) in 2012 that steadied at around 2200 cases annually in subsequent years.6 Although there likely are several factors affecting WNV incidence trends in the United States, interannual changes in temperature and precipitation have been described. An increased mean annual temperature (from September through October, the end of peak mosquito season) and an increased temperature in winter months (from January through March, prior to peak mosquito season) have both been associated with an increased incidence of WNV.7 An increased temperature is thought to increase population numbers of mosquitoes both by increasing reproductive rates and creating ideal breeding environments via pooled water areas.8 Depending on the region, both above average and below average precipitation levels in the United States can increase WNV incidence the following year.7,9
What are the signs and symptoms of WNV infection?
Up to 80% of those infected with WNV are asymptomatic.3 After an incubation period of roughly 2 to 14 days, the remaining 20% may develop symptoms of West Nile fever (WNF), typically a self-limited illness that consists of 3 to 10 days of nonspecific symptoms such as fever, headache, fatigue, muscle pain and/or weakness, eye pain, gastrointestinal tract upset, and a macular rash that usually presents on the trunk or extremities.1,3 Less than 1% of patients affected by WNV develop neuroinvasive disease, including meningitis, encephalitis, and/or acute flaccid paralysis.10 West Nile virus neuroinvasive disease can cause permanent neurologic sequelae such as muscle weakness, confusion, memory loss, and fatigue; it carries a mortality rate of 10% to 30%, which is mainly dependent on older age and immunosuppression status.1,10
What is the reported spectrum of cutaneous findings in WNV?
Of the roughly 20% of patients infected with WNV that develop WNF, approximately 25% to 50% will develop an associated rash.1 It most commonly is described as a morbilliform or maculopapular rash located on the chest, back, and arms, usually sparing the palms and soles, though 1 case report noted involvement with these areas (Figure).11,12 It typically appears 5 days after symptom onset, can be associated with defervescence, and lasts less than a week.1,13 Pruritus and dysesthesia are sometimes present.13 Other rare presentations that have been reported include an ill-defined pseudovesicular rash with erythematous papules on the palms and pink, scaly, psoriasiform papules on the feet and thighs, as well as neuroinvasive WNV leading to purpura fulminans.14,15 A diffuse, erythematous, petechial rash on the face, neck, trunk, and extremities was reported in a pediatric patient, but there have been no reports of a petechial rash associated with WNV in adult patients.16 These findings suggest some potential variability in the presentation of the WNV rash.
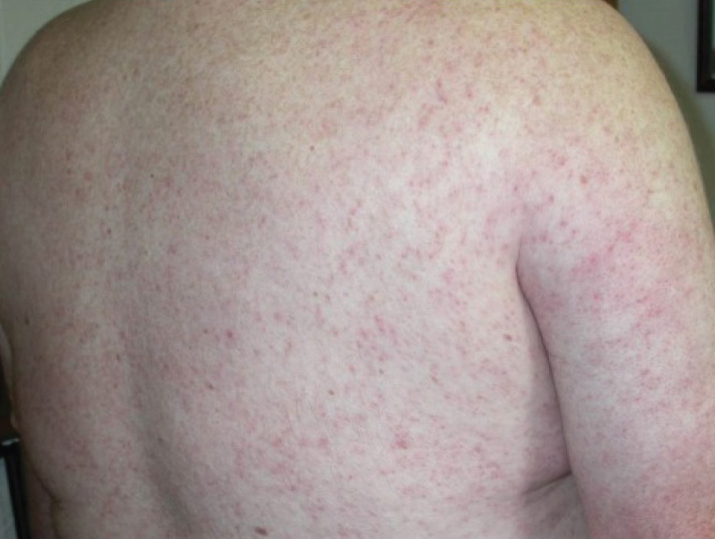
What role does the presence of rash play diagnostically and prognostically?
The rash of WNV has been implicated as a potential prognostic factor in predicting more favorable outcomes.17 Using 2002 data from the Illinois Department of Public Health and 2003 data from the Colorado Department of Public Health, Huhn and Dworkin17 found the age-adjusted risk of encephalitis and death to be decreased in WNV patients with a rash (relative risk, 0.44; 95% CI, 0.21-0.92). The reasons for this are not definitively known, but we hypothesize that the rash may prompt patients to seek earlier medical attention or indicate a more robust immune response. Additionally, a rash in WNV more commonly is seen in younger patients, whereas WNV neuroinvasive disease is more common in older patients, who also tend to have worse outcomes.10 One study found rash to be the only symptom that demonstrated a significant association with seropositivity (overall risk=6.35; P<.05; 95% CI, 3.75-10.80) by multivariate analysis.18
How is WNV diagnosed? What are the downsides to WNV testing?
Given that the presenting symptoms of WNV and WNF are nonspecific, it becomes challenging to arrive at the diagnosis based solely on physical examination. As such, the patient’s clinical and epidemiologic history, such as timing, pattern, and appearance of the rash or recent history of mosquito bites, is key to arriving at the correct diagnosis. With clinical suspicion, possible diagnostic tests include an IgM enzyme-linked immunosorbent assay (ELISA) for WNV, a plaque reduction neutralization test (PNRT), and blood polymerase chain reaction (PCR).
An ELISA is a confirmatory test to detect IgM antibodies to WNV in the serum. Because IgM seroconversion typically occurs between days 4 and 10 of symptom onset, there is a high probability of initial false-negative testing within the first 8 days after symptom onset.19,20 Clinical understanding of this fact is imperative, as an initial negative ELISA does not rule out WNV, and a retest is warranted if clinical suspicion is high. In addition to a high initial false-negative rate with ELISA, there are several other limitations to note. IgM antibodies remain elevated for 1 to 3 months or possibly up to a year in immunocompromised patients.1 Due to this, false positives may be present if there was a recent prior infection. Enzyme-linked immunosorbent assay may not distinguish from different flaviviruses, including the yellow fever, dengue, Zika, Japanese encephalitis, and Saint Louis encephalitis viruses. Seropositivity has been estimated in some states, including 1999 data from New York (2.6%), 2003 data from Nebraska (9.5%), and 2012-2014 data from Connecticut (8.5%).21-23 Regional variance may be expected, as there also were significant differences in WNV seropositivity between different regions in Nebraska (P<.001).23
Because ELISA testing for WNV has readily apparent flaws, other tests have been utilized in its diagnosis. The PNRT is the most specific test, and it works by measuring neutralizing antibody titers for different flaviviruses. It has the ability to determine cross-reactivity with other flaviviruses; however, it does not discriminate between a current infection and a prior infection or prior flavivirus vaccine (ie, yellow fever vaccine). Despite this, a positive PNRT can lend credibility to a positive ELISA test and determine specificity for WNV for those with no prior flavivirus exposure.24 According to the Centers for Disease Control and Prevention (CDC), this test can be performed by the CDC or in reference laboratories designated by the CDC.3 Additionally, some state health laboratories may perform PRNTs.
Viral detection with PCR currently is used to screen blood donations and may be beneficial for immunocompromised patients that lack the ability to form a robust antibody response or if a patient presents early, as PCR works best within the first week of symptom onset.1 Tilley et al25 showed that a combination of PCR and ELISA were able to accurately predict 94.2% of patients (180/191) with documented WNV on a first blood sample compared to 45% and 58.1% for only viral detection or ELISA, respectively. Based on costs from a Midwest academic center, antibody detection tests are around $100 while PCR may range from $500 to $1000 and is only performed in reference laboratories. Although these tests remain in the repertoire for WNV diagnosis, financial stewardship is important.
If there are symptoms of photophobia, phonophobia, nuchal rigidity, loss of consciousness, or marked personality changes, a lumbar puncture for WNV IgM in the cerebrospinal fluid can be performed. As with most viral infections, cerebrospinal fluid findings normally include an elevated protein and lymphocyte count, but neutrophils may be predominantly elevated if the infection is early in its course.26
What are the management options?
To date, there is no curative treatment for WNV, and management is largely supportive. For WNF, over-the-counter pain medications may be helpful to reduce fever and pain. If more severe disease develops, hospitalization for further supportive care may be needed.27 If meningitis or encephalitis is suspected, broad-spectrum antibiotics may need to be started until other common etiologies are ruled out.28
How can you prevent WNV infection?
Disease prevention largely consists of educating the public to avoid heavily wooded areas, especially in areas of high prevalence and during peak months, and to use protective clothing and insect repellant that has been approved by the Environmental Protection Agency.3 Insect repellants approved by the Environmental Protection Agency contain ingredients such as DEET (N, N-diethyl-meta-toluamide), picaridin, IR3535 (ethyl butylacetylaminopropionate), and oil of lemon eucalyptus, which have been proven safe and effective.29 Patients also can protect their homes by using window screens and promptly repairing screens with holes.3
What is the differential diagnosis for WNV?
The differential diagnosis for fever with generalized maculopapular rash broadly ranges from viral etiologies (eg, WNV, Zika, measles), to tick bites (eg, Rocky Mountain spotted fever, ehrlichiosis), to drug-induced rashes. A detailed patient history inquiring on recent sick contacts, travel (WNV in the Midwest, ehrlichiosis in the Southeast), environmental exposures (ticks, mosquitoes), and new medications (typically 7–10 days after starting) is imperative to narrow the differential.30 In addition, the distribution, timing, and clinical characteristics of the rash may aid in diagnosis, along with an appropriately correlated clinical picture. West Nile virus likely will present in the summer in mid central geographic locations and often develops on the trunk and extremities as a blanching, generalized, maculopapular rash around 5 days after symptom onset or with defervescence.1
- Petersen LR. Clinical manifestations and diagnosis of West Nile virus infection. UpToDate website. Updated August 7, 2020. Accessed April 16, 2021. https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-west-nile-virus-infection?search=clinical-manifestations-and-diagnosis-of-west-nile-virusinfection.&source=search_result&selectedTitle=1~78&usage_type=default&display_rank=1
- Sampathkumar P. West Nile virus: epidemiology, clinical presentation, diagnosis, and prevention. Mayo Clin Proc. 2003;78:1137-1144.
- Centers for Disease Control and Prevention. West Nile virus. Updated June 3, 2020. Accessed April 16, 2021. https://www.cdc.gov/westnile/index.html
- Chuang TW, Hockett CW, Kightlinger L, et al. Landscape-level spatial patterns of West Nile virus risk in the northern Great Plains. Am J Trop Med Hyg. 2012;86:724-731.
- Wimberly MC, Hildreth MB, Boyte SP, et al. Ecological niche of the 2003 West Nile virus epidemic in the northern great plains of the United States. PLoS One. 2008;3:E3744. doi:10.1371/journal.pone.0003744
- Centers for Disease Control and Prevention. West Nile virus disease cases reported to CDC by state of residence, 1999-2019. Accessed April 26, 2021. https://www.cdc.gov/westnile/resources/pdfs/data/West-Nile-virus-disease-cases-by-state_1999-2019-P.pdf
- Hahn MB, Monaghan AJ, Hayden MH, et al. Meteorological conditions associated with increased incidence of West Nile virus disease in the United States, 2004–2012. Am J Trop Med Hyg. 2015;92:1013-1022.
- Brown CM, DeMaria A Jr. The resurgence of West Nile virus. Ann Intern Med. 2012;157:823-824.
- Landesman WJ, Allan BF, Langerhans RB, et al. Inter-annual associations between precipitation and human incidence of West Nile virus in the United States. Vector Borne Zoonotic Dis. 2007;7:337-343.
- Hart J Jr, Tillman G, Kraut MA, et al. West Nile virus neuroinvasive disease: neurological manifestations and prospective longitudinal outcomes. BMC Infect Dis. 2014;14:248.
- Wu JJ, Huang DB, Tyring SK. West Nile virus rash on the palms and soles of the feet. J Eur Acad Dermatol Venereol. 2006;20:1393-1394.
- Sejvar J. Clinical manifestations and outcomes of West Nile virus infection. Viruses. 2014;6:606-623.
- Ferguson DD, Gershman K, LeBailly A, et al. Characteristics of the rash associated with West Nile virus fever. Clin Infect Dis. 2005;41:1204-1207.
- Marszalek R, Chen A, Gjede J. Psoriasiform eruption in the setting of West Nile virus. J Am Acad Dermatol. 2014;70:AB4. doi:10.1016/j.jaad.2014.01.017
- Shah S, Fite LP, Lane N, et al. Purpura fulminans associated with acute West Nile virus encephalitis. J Clin Virol. 2016;75:1-4.
- Civen R, Villacorte F, Robles DT, et al. West Nile virus infection in the pediatric population. Pediatr Infect Dis J. 2006;25:75-78.
- Huhn GD, Dworkin MS. Rash as a prognostic factor in West Nile virus disease. Clin Infect Dis. 2006;43:388-389.
- Murphy TD, Grandpre J, Novick SL, et al. West Nile virus infection among health-fair participants, Wyoming 2003: assessment of symptoms and risk factors. Vector Borne Zoonotic Dis. 2005;5:246-251.
- Prince HE, Tobler LH, Lapé-Nixon M, et al. Development and persistence of West Nile virus–specific immunoglobulin M (IgM), IgA, and IgG in viremic blood donors. J Clin Microbiol. 2005;43:4316-4320.
- Busch MP, Kleinman SH, Tobler LH, et al. Virus and antibody dynamics in acute West Nile Virus infection. J Infect Dis. 2008;198:984-993.
- Mostashari F, Bunning ML, Kitsutani PT, et al. Epidemic West Nile encephalitis, New York, 1999: results of a household-based seroepidemiological survey. Lancet. 2001;358:261-264.
- Cahill ME, Yao Y, Nock D, et al. West Nile virus seroprevalence, Connecticut, USA, 2000–2014. Emerg Infect Dis. 2017;23:708-710.
- Schweitzer BK, Kramer WL, Sambol AR, et al. Geographic factors contributing to a high seroprevalence of West Nile virus-specific antibodies in humans following an epidemic. Clin Vaccine Immunol. 2006;13:314-318.
- Maeda A, Maeda J. Review of diagnostic plaque reduction neutralization tests for flavivirus infection. Vet J. 2013;195:33-40.
- Tilley PA, Fox JD, Jayaraman GC, et al. Nucleic acid testing for west nile virus RNA in plasma enhances rapid diagnosis of acute infection in symptomatic patients. J Infect Dis. 2006;193:1361-1364.
- Petersen LR, Brault AC, Nasci RS. West Nile virus: review of the literature. JAMA. 2013;310:308-315.
- Yu A, Ferenczi E, Moussa K, et al. Clinical spectrum of West Nile virus neuroinvasive disease. Neurohospitalist. 2020;10:43-47.
- Michaelis M, Kleinschmidt MC, Doerr HW, et al. Minocycline inhibits West Nile virus replication and apoptosis in human neuronal cells. J Antimicrob Chemother. 2007;60:981-986.
- United State Environmental Protection Agency. Skin-applied repellent ingredients. https://www.epa.gov/insect-repellents/skin-applied-repellent-ingredients. Accessed April 16, 2021.
- Muzumdar S, Rothe MJ, Grant-Kels JM. The rash with maculopapules and fever in adults. Clin Dermatol. 2019;37:109-118.
What is West Nile virus? How is it contracted, and who can become infected?
West Nile virus (WNV) is a single-stranded RNA virus of the Flaviviridae family and Flavivirus genus, a lineage that also includes the yellow fever, dengue, Zika, Japanese encephalitis, and Saint Louis encephalitis viruses.1 Birds serve as the reservoir hosts of WNV, and mosquitoes acquire the virus during feeding.2 West Nile virus then is transmitted to humans primarily by bites from Culex mosquitoes, which are especially prevalent in wooded areas during peak mosquito season (summer through early fall in North America).1 Mosquitoes also can infect horses; however, humans and horses are dead-end hosts, meaning they do not pass the virus on to other biting mosquitoes.3 There also have been rare reports of transmission of WNV through blood and donation as well as mother-to-baby transmission.2
What is the epidemiology of WNV in the United States?
Since the introduction of WNV to the United States in 1999, it has become an important public health concern, with 48,183 cases and 2163 deaths reported since 1999.2,3 In 2018, Nebraska had the highest number of cases of WNV (n=251), followed by California (n=217), North Dakota (n=204), Illinois (n=176), and South Dakota (n=169).3 West Nile virus is endemic to all 48 contiguous states and Canada, though the Great Plains region is especially affected by WNV due to several factors, such as a greater percentage of rural land, forests, and irrigated areas.4 The Great Plains region also has been thought to be an ecological niche for a more virulent species (Culex tarsalis) compared to other regions in the United States.5
The annual incidence of WNV in the United States peaked in 2003 at 9862 cases (up from 62 cases in 1999), then declined gradually until 2008 to 2011, during which the incidence was stable at 700 to 1100 new cases per year. However, there was a resurgence of cases (n=5674) in 2012 that steadied at around 2200 cases annually in subsequent years.6 Although there likely are several factors affecting WNV incidence trends in the United States, interannual changes in temperature and precipitation have been described. An increased mean annual temperature (from September through October, the end of peak mosquito season) and an increased temperature in winter months (from January through March, prior to peak mosquito season) have both been associated with an increased incidence of WNV.7 An increased temperature is thought to increase population numbers of mosquitoes both by increasing reproductive rates and creating ideal breeding environments via pooled water areas.8 Depending on the region, both above average and below average precipitation levels in the United States can increase WNV incidence the following year.7,9
What are the signs and symptoms of WNV infection?
Up to 80% of those infected with WNV are asymptomatic.3 After an incubation period of roughly 2 to 14 days, the remaining 20% may develop symptoms of West Nile fever (WNF), typically a self-limited illness that consists of 3 to 10 days of nonspecific symptoms such as fever, headache, fatigue, muscle pain and/or weakness, eye pain, gastrointestinal tract upset, and a macular rash that usually presents on the trunk or extremities.1,3 Less than 1% of patients affected by WNV develop neuroinvasive disease, including meningitis, encephalitis, and/or acute flaccid paralysis.10 West Nile virus neuroinvasive disease can cause permanent neurologic sequelae such as muscle weakness, confusion, memory loss, and fatigue; it carries a mortality rate of 10% to 30%, which is mainly dependent on older age and immunosuppression status.1,10
What is the reported spectrum of cutaneous findings in WNV?
Of the roughly 20% of patients infected with WNV that develop WNF, approximately 25% to 50% will develop an associated rash.1 It most commonly is described as a morbilliform or maculopapular rash located on the chest, back, and arms, usually sparing the palms and soles, though 1 case report noted involvement with these areas (Figure).11,12 It typically appears 5 days after symptom onset, can be associated with defervescence, and lasts less than a week.1,13 Pruritus and dysesthesia are sometimes present.13 Other rare presentations that have been reported include an ill-defined pseudovesicular rash with erythematous papules on the palms and pink, scaly, psoriasiform papules on the feet and thighs, as well as neuroinvasive WNV leading to purpura fulminans.14,15 A diffuse, erythematous, petechial rash on the face, neck, trunk, and extremities was reported in a pediatric patient, but there have been no reports of a petechial rash associated with WNV in adult patients.16 These findings suggest some potential variability in the presentation of the WNV rash.
What role does the presence of rash play diagnostically and prognostically?
The rash of WNV has been implicated as a potential prognostic factor in predicting more favorable outcomes.17 Using 2002 data from the Illinois Department of Public Health and 2003 data from the Colorado Department of Public Health, Huhn and Dworkin17 found the age-adjusted risk of encephalitis and death to be decreased in WNV patients with a rash (relative risk, 0.44; 95% CI, 0.21-0.92). The reasons for this are not definitively known, but we hypothesize that the rash may prompt patients to seek earlier medical attention or indicate a more robust immune response. Additionally, a rash in WNV more commonly is seen in younger patients, whereas WNV neuroinvasive disease is more common in older patients, who also tend to have worse outcomes.10 One study found rash to be the only symptom that demonstrated a significant association with seropositivity (overall risk=6.35; P<.05; 95% CI, 3.75-10.80) by multivariate analysis.18
How is WNV diagnosed? What are the downsides to WNV testing?
Given that the presenting symptoms of WNV and WNF are nonspecific, it becomes challenging to arrive at the diagnosis based solely on physical examination. As such, the patient’s clinical and epidemiologic history, such as timing, pattern, and appearance of the rash or recent history of mosquito bites, is key to arriving at the correct diagnosis. With clinical suspicion, possible diagnostic tests include an IgM enzyme-linked immunosorbent assay (ELISA) for WNV, a plaque reduction neutralization test (PNRT), and blood polymerase chain reaction (PCR).
An ELISA is a confirmatory test to detect IgM antibodies to WNV in the serum. Because IgM seroconversion typically occurs between days 4 and 10 of symptom onset, there is a high probability of initial false-negative testing within the first 8 days after symptom onset.19,20 Clinical understanding of this fact is imperative, as an initial negative ELISA does not rule out WNV, and a retest is warranted if clinical suspicion is high. In addition to a high initial false-negative rate with ELISA, there are several other limitations to note. IgM antibodies remain elevated for 1 to 3 months or possibly up to a year in immunocompromised patients.1 Due to this, false positives may be present if there was a recent prior infection. Enzyme-linked immunosorbent assay may not distinguish from different flaviviruses, including the yellow fever, dengue, Zika, Japanese encephalitis, and Saint Louis encephalitis viruses. Seropositivity has been estimated in some states, including 1999 data from New York (2.6%), 2003 data from Nebraska (9.5%), and 2012-2014 data from Connecticut (8.5%).21-23 Regional variance may be expected, as there also were significant differences in WNV seropositivity between different regions in Nebraska (P<.001).23
Because ELISA testing for WNV has readily apparent flaws, other tests have been utilized in its diagnosis. The PNRT is the most specific test, and it works by measuring neutralizing antibody titers for different flaviviruses. It has the ability to determine cross-reactivity with other flaviviruses; however, it does not discriminate between a current infection and a prior infection or prior flavivirus vaccine (ie, yellow fever vaccine). Despite this, a positive PNRT can lend credibility to a positive ELISA test and determine specificity for WNV for those with no prior flavivirus exposure.24 According to the Centers for Disease Control and Prevention (CDC), this test can be performed by the CDC or in reference laboratories designated by the CDC.3 Additionally, some state health laboratories may perform PRNTs.
Viral detection with PCR currently is used to screen blood donations and may be beneficial for immunocompromised patients that lack the ability to form a robust antibody response or if a patient presents early, as PCR works best within the first week of symptom onset.1 Tilley et al25 showed that a combination of PCR and ELISA were able to accurately predict 94.2% of patients (180/191) with documented WNV on a first blood sample compared to 45% and 58.1% for only viral detection or ELISA, respectively. Based on costs from a Midwest academic center, antibody detection tests are around $100 while PCR may range from $500 to $1000 and is only performed in reference laboratories. Although these tests remain in the repertoire for WNV diagnosis, financial stewardship is important.
If there are symptoms of photophobia, phonophobia, nuchal rigidity, loss of consciousness, or marked personality changes, a lumbar puncture for WNV IgM in the cerebrospinal fluid can be performed. As with most viral infections, cerebrospinal fluid findings normally include an elevated protein and lymphocyte count, but neutrophils may be predominantly elevated if the infection is early in its course.26
What are the management options?
To date, there is no curative treatment for WNV, and management is largely supportive. For WNF, over-the-counter pain medications may be helpful to reduce fever and pain. If more severe disease develops, hospitalization for further supportive care may be needed.27 If meningitis or encephalitis is suspected, broad-spectrum antibiotics may need to be started until other common etiologies are ruled out.28
How can you prevent WNV infection?
Disease prevention largely consists of educating the public to avoid heavily wooded areas, especially in areas of high prevalence and during peak months, and to use protective clothing and insect repellant that has been approved by the Environmental Protection Agency.3 Insect repellants approved by the Environmental Protection Agency contain ingredients such as DEET (N, N-diethyl-meta-toluamide), picaridin, IR3535 (ethyl butylacetylaminopropionate), and oil of lemon eucalyptus, which have been proven safe and effective.29 Patients also can protect their homes by using window screens and promptly repairing screens with holes.3
What is the differential diagnosis for WNV?
The differential diagnosis for fever with generalized maculopapular rash broadly ranges from viral etiologies (eg, WNV, Zika, measles), to tick bites (eg, Rocky Mountain spotted fever, ehrlichiosis), to drug-induced rashes. A detailed patient history inquiring on recent sick contacts, travel (WNV in the Midwest, ehrlichiosis in the Southeast), environmental exposures (ticks, mosquitoes), and new medications (typically 7–10 days after starting) is imperative to narrow the differential.30 In addition, the distribution, timing, and clinical characteristics of the rash may aid in diagnosis, along with an appropriately correlated clinical picture. West Nile virus likely will present in the summer in mid central geographic locations and often develops on the trunk and extremities as a blanching, generalized, maculopapular rash around 5 days after symptom onset or with defervescence.1
What is West Nile virus? How is it contracted, and who can become infected?
West Nile virus (WNV) is a single-stranded RNA virus of the Flaviviridae family and Flavivirus genus, a lineage that also includes the yellow fever, dengue, Zika, Japanese encephalitis, and Saint Louis encephalitis viruses.1 Birds serve as the reservoir hosts of WNV, and mosquitoes acquire the virus during feeding.2 West Nile virus then is transmitted to humans primarily by bites from Culex mosquitoes, which are especially prevalent in wooded areas during peak mosquito season (summer through early fall in North America).1 Mosquitoes also can infect horses; however, humans and horses are dead-end hosts, meaning they do not pass the virus on to other biting mosquitoes.3 There also have been rare reports of transmission of WNV through blood and donation as well as mother-to-baby transmission.2
What is the epidemiology of WNV in the United States?
Since the introduction of WNV to the United States in 1999, it has become an important public health concern, with 48,183 cases and 2163 deaths reported since 1999.2,3 In 2018, Nebraska had the highest number of cases of WNV (n=251), followed by California (n=217), North Dakota (n=204), Illinois (n=176), and South Dakota (n=169).3 West Nile virus is endemic to all 48 contiguous states and Canada, though the Great Plains region is especially affected by WNV due to several factors, such as a greater percentage of rural land, forests, and irrigated areas.4 The Great Plains region also has been thought to be an ecological niche for a more virulent species (Culex tarsalis) compared to other regions in the United States.5
The annual incidence of WNV in the United States peaked in 2003 at 9862 cases (up from 62 cases in 1999), then declined gradually until 2008 to 2011, during which the incidence was stable at 700 to 1100 new cases per year. However, there was a resurgence of cases (n=5674) in 2012 that steadied at around 2200 cases annually in subsequent years.6 Although there likely are several factors affecting WNV incidence trends in the United States, interannual changes in temperature and precipitation have been described. An increased mean annual temperature (from September through October, the end of peak mosquito season) and an increased temperature in winter months (from January through March, prior to peak mosquito season) have both been associated with an increased incidence of WNV.7 An increased temperature is thought to increase population numbers of mosquitoes both by increasing reproductive rates and creating ideal breeding environments via pooled water areas.8 Depending on the region, both above average and below average precipitation levels in the United States can increase WNV incidence the following year.7,9
What are the signs and symptoms of WNV infection?
Up to 80% of those infected with WNV are asymptomatic.3 After an incubation period of roughly 2 to 14 days, the remaining 20% may develop symptoms of West Nile fever (WNF), typically a self-limited illness that consists of 3 to 10 days of nonspecific symptoms such as fever, headache, fatigue, muscle pain and/or weakness, eye pain, gastrointestinal tract upset, and a macular rash that usually presents on the trunk or extremities.1,3 Less than 1% of patients affected by WNV develop neuroinvasive disease, including meningitis, encephalitis, and/or acute flaccid paralysis.10 West Nile virus neuroinvasive disease can cause permanent neurologic sequelae such as muscle weakness, confusion, memory loss, and fatigue; it carries a mortality rate of 10% to 30%, which is mainly dependent on older age and immunosuppression status.1,10
What is the reported spectrum of cutaneous findings in WNV?
Of the roughly 20% of patients infected with WNV that develop WNF, approximately 25% to 50% will develop an associated rash.1 It most commonly is described as a morbilliform or maculopapular rash located on the chest, back, and arms, usually sparing the palms and soles, though 1 case report noted involvement with these areas (Figure).11,12 It typically appears 5 days after symptom onset, can be associated with defervescence, and lasts less than a week.1,13 Pruritus and dysesthesia are sometimes present.13 Other rare presentations that have been reported include an ill-defined pseudovesicular rash with erythematous papules on the palms and pink, scaly, psoriasiform papules on the feet and thighs, as well as neuroinvasive WNV leading to purpura fulminans.14,15 A diffuse, erythematous, petechial rash on the face, neck, trunk, and extremities was reported in a pediatric patient, but there have been no reports of a petechial rash associated with WNV in adult patients.16 These findings suggest some potential variability in the presentation of the WNV rash.
What role does the presence of rash play diagnostically and prognostically?
The rash of WNV has been implicated as a potential prognostic factor in predicting more favorable outcomes.17 Using 2002 data from the Illinois Department of Public Health and 2003 data from the Colorado Department of Public Health, Huhn and Dworkin17 found the age-adjusted risk of encephalitis and death to be decreased in WNV patients with a rash (relative risk, 0.44; 95% CI, 0.21-0.92). The reasons for this are not definitively known, but we hypothesize that the rash may prompt patients to seek earlier medical attention or indicate a more robust immune response. Additionally, a rash in WNV more commonly is seen in younger patients, whereas WNV neuroinvasive disease is more common in older patients, who also tend to have worse outcomes.10 One study found rash to be the only symptom that demonstrated a significant association with seropositivity (overall risk=6.35; P<.05; 95% CI, 3.75-10.80) by multivariate analysis.18
How is WNV diagnosed? What are the downsides to WNV testing?
Given that the presenting symptoms of WNV and WNF are nonspecific, it becomes challenging to arrive at the diagnosis based solely on physical examination. As such, the patient’s clinical and epidemiologic history, such as timing, pattern, and appearance of the rash or recent history of mosquito bites, is key to arriving at the correct diagnosis. With clinical suspicion, possible diagnostic tests include an IgM enzyme-linked immunosorbent assay (ELISA) for WNV, a plaque reduction neutralization test (PNRT), and blood polymerase chain reaction (PCR).
An ELISA is a confirmatory test to detect IgM antibodies to WNV in the serum. Because IgM seroconversion typically occurs between days 4 and 10 of symptom onset, there is a high probability of initial false-negative testing within the first 8 days after symptom onset.19,20 Clinical understanding of this fact is imperative, as an initial negative ELISA does not rule out WNV, and a retest is warranted if clinical suspicion is high. In addition to a high initial false-negative rate with ELISA, there are several other limitations to note. IgM antibodies remain elevated for 1 to 3 months or possibly up to a year in immunocompromised patients.1 Due to this, false positives may be present if there was a recent prior infection. Enzyme-linked immunosorbent assay may not distinguish from different flaviviruses, including the yellow fever, dengue, Zika, Japanese encephalitis, and Saint Louis encephalitis viruses. Seropositivity has been estimated in some states, including 1999 data from New York (2.6%), 2003 data from Nebraska (9.5%), and 2012-2014 data from Connecticut (8.5%).21-23 Regional variance may be expected, as there also were significant differences in WNV seropositivity between different regions in Nebraska (P<.001).23
Because ELISA testing for WNV has readily apparent flaws, other tests have been utilized in its diagnosis. The PNRT is the most specific test, and it works by measuring neutralizing antibody titers for different flaviviruses. It has the ability to determine cross-reactivity with other flaviviruses; however, it does not discriminate between a current infection and a prior infection or prior flavivirus vaccine (ie, yellow fever vaccine). Despite this, a positive PNRT can lend credibility to a positive ELISA test and determine specificity for WNV for those with no prior flavivirus exposure.24 According to the Centers for Disease Control and Prevention (CDC), this test can be performed by the CDC or in reference laboratories designated by the CDC.3 Additionally, some state health laboratories may perform PRNTs.
Viral detection with PCR currently is used to screen blood donations and may be beneficial for immunocompromised patients that lack the ability to form a robust antibody response or if a patient presents early, as PCR works best within the first week of symptom onset.1 Tilley et al25 showed that a combination of PCR and ELISA were able to accurately predict 94.2% of patients (180/191) with documented WNV on a first blood sample compared to 45% and 58.1% for only viral detection or ELISA, respectively. Based on costs from a Midwest academic center, antibody detection tests are around $100 while PCR may range from $500 to $1000 and is only performed in reference laboratories. Although these tests remain in the repertoire for WNV diagnosis, financial stewardship is important.
If there are symptoms of photophobia, phonophobia, nuchal rigidity, loss of consciousness, or marked personality changes, a lumbar puncture for WNV IgM in the cerebrospinal fluid can be performed. As with most viral infections, cerebrospinal fluid findings normally include an elevated protein and lymphocyte count, but neutrophils may be predominantly elevated if the infection is early in its course.26
What are the management options?
To date, there is no curative treatment for WNV, and management is largely supportive. For WNF, over-the-counter pain medications may be helpful to reduce fever and pain. If more severe disease develops, hospitalization for further supportive care may be needed.27 If meningitis or encephalitis is suspected, broad-spectrum antibiotics may need to be started until other common etiologies are ruled out.28
How can you prevent WNV infection?
Disease prevention largely consists of educating the public to avoid heavily wooded areas, especially in areas of high prevalence and during peak months, and to use protective clothing and insect repellant that has been approved by the Environmental Protection Agency.3 Insect repellants approved by the Environmental Protection Agency contain ingredients such as DEET (N, N-diethyl-meta-toluamide), picaridin, IR3535 (ethyl butylacetylaminopropionate), and oil of lemon eucalyptus, which have been proven safe and effective.29 Patients also can protect their homes by using window screens and promptly repairing screens with holes.3
What is the differential diagnosis for WNV?
The differential diagnosis for fever with generalized maculopapular rash broadly ranges from viral etiologies (eg, WNV, Zika, measles), to tick bites (eg, Rocky Mountain spotted fever, ehrlichiosis), to drug-induced rashes. A detailed patient history inquiring on recent sick contacts, travel (WNV in the Midwest, ehrlichiosis in the Southeast), environmental exposures (ticks, mosquitoes), and new medications (typically 7–10 days after starting) is imperative to narrow the differential.30 In addition, the distribution, timing, and clinical characteristics of the rash may aid in diagnosis, along with an appropriately correlated clinical picture. West Nile virus likely will present in the summer in mid central geographic locations and often develops on the trunk and extremities as a blanching, generalized, maculopapular rash around 5 days after symptom onset or with defervescence.1
- Petersen LR. Clinical manifestations and diagnosis of West Nile virus infection. UpToDate website. Updated August 7, 2020. Accessed April 16, 2021. https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-west-nile-virus-infection?search=clinical-manifestations-and-diagnosis-of-west-nile-virusinfection.&source=search_result&selectedTitle=1~78&usage_type=default&display_rank=1
- Sampathkumar P. West Nile virus: epidemiology, clinical presentation, diagnosis, and prevention. Mayo Clin Proc. 2003;78:1137-1144.
- Centers for Disease Control and Prevention. West Nile virus. Updated June 3, 2020. Accessed April 16, 2021. https://www.cdc.gov/westnile/index.html
- Chuang TW, Hockett CW, Kightlinger L, et al. Landscape-level spatial patterns of West Nile virus risk in the northern Great Plains. Am J Trop Med Hyg. 2012;86:724-731.
- Wimberly MC, Hildreth MB, Boyte SP, et al. Ecological niche of the 2003 West Nile virus epidemic in the northern great plains of the United States. PLoS One. 2008;3:E3744. doi:10.1371/journal.pone.0003744
- Centers for Disease Control and Prevention. West Nile virus disease cases reported to CDC by state of residence, 1999-2019. Accessed April 26, 2021. https://www.cdc.gov/westnile/resources/pdfs/data/West-Nile-virus-disease-cases-by-state_1999-2019-P.pdf
- Hahn MB, Monaghan AJ, Hayden MH, et al. Meteorological conditions associated with increased incidence of West Nile virus disease in the United States, 2004–2012. Am J Trop Med Hyg. 2015;92:1013-1022.
- Brown CM, DeMaria A Jr. The resurgence of West Nile virus. Ann Intern Med. 2012;157:823-824.
- Landesman WJ, Allan BF, Langerhans RB, et al. Inter-annual associations between precipitation and human incidence of West Nile virus in the United States. Vector Borne Zoonotic Dis. 2007;7:337-343.
- Hart J Jr, Tillman G, Kraut MA, et al. West Nile virus neuroinvasive disease: neurological manifestations and prospective longitudinal outcomes. BMC Infect Dis. 2014;14:248.
- Wu JJ, Huang DB, Tyring SK. West Nile virus rash on the palms and soles of the feet. J Eur Acad Dermatol Venereol. 2006;20:1393-1394.
- Sejvar J. Clinical manifestations and outcomes of West Nile virus infection. Viruses. 2014;6:606-623.
- Ferguson DD, Gershman K, LeBailly A, et al. Characteristics of the rash associated with West Nile virus fever. Clin Infect Dis. 2005;41:1204-1207.
- Marszalek R, Chen A, Gjede J. Psoriasiform eruption in the setting of West Nile virus. J Am Acad Dermatol. 2014;70:AB4. doi:10.1016/j.jaad.2014.01.017
- Shah S, Fite LP, Lane N, et al. Purpura fulminans associated with acute West Nile virus encephalitis. J Clin Virol. 2016;75:1-4.
- Civen R, Villacorte F, Robles DT, et al. West Nile virus infection in the pediatric population. Pediatr Infect Dis J. 2006;25:75-78.
- Huhn GD, Dworkin MS. Rash as a prognostic factor in West Nile virus disease. Clin Infect Dis. 2006;43:388-389.
- Murphy TD, Grandpre J, Novick SL, et al. West Nile virus infection among health-fair participants, Wyoming 2003: assessment of symptoms and risk factors. Vector Borne Zoonotic Dis. 2005;5:246-251.
- Prince HE, Tobler LH, Lapé-Nixon M, et al. Development and persistence of West Nile virus–specific immunoglobulin M (IgM), IgA, and IgG in viremic blood donors. J Clin Microbiol. 2005;43:4316-4320.
- Busch MP, Kleinman SH, Tobler LH, et al. Virus and antibody dynamics in acute West Nile Virus infection. J Infect Dis. 2008;198:984-993.
- Mostashari F, Bunning ML, Kitsutani PT, et al. Epidemic West Nile encephalitis, New York, 1999: results of a household-based seroepidemiological survey. Lancet. 2001;358:261-264.
- Cahill ME, Yao Y, Nock D, et al. West Nile virus seroprevalence, Connecticut, USA, 2000–2014. Emerg Infect Dis. 2017;23:708-710.
- Schweitzer BK, Kramer WL, Sambol AR, et al. Geographic factors contributing to a high seroprevalence of West Nile virus-specific antibodies in humans following an epidemic. Clin Vaccine Immunol. 2006;13:314-318.
- Maeda A, Maeda J. Review of diagnostic plaque reduction neutralization tests for flavivirus infection. Vet J. 2013;195:33-40.
- Tilley PA, Fox JD, Jayaraman GC, et al. Nucleic acid testing for west nile virus RNA in plasma enhances rapid diagnosis of acute infection in symptomatic patients. J Infect Dis. 2006;193:1361-1364.
- Petersen LR, Brault AC, Nasci RS. West Nile virus: review of the literature. JAMA. 2013;310:308-315.
- Yu A, Ferenczi E, Moussa K, et al. Clinical spectrum of West Nile virus neuroinvasive disease. Neurohospitalist. 2020;10:43-47.
- Michaelis M, Kleinschmidt MC, Doerr HW, et al. Minocycline inhibits West Nile virus replication and apoptosis in human neuronal cells. J Antimicrob Chemother. 2007;60:981-986.
- United State Environmental Protection Agency. Skin-applied repellent ingredients. https://www.epa.gov/insect-repellents/skin-applied-repellent-ingredients. Accessed April 16, 2021.
- Muzumdar S, Rothe MJ, Grant-Kels JM. The rash with maculopapules and fever in adults. Clin Dermatol. 2019;37:109-118.
- Petersen LR. Clinical manifestations and diagnosis of West Nile virus infection. UpToDate website. Updated August 7, 2020. Accessed April 16, 2021. https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-west-nile-virus-infection?search=clinical-manifestations-and-diagnosis-of-west-nile-virusinfection.&source=search_result&selectedTitle=1~78&usage_type=default&display_rank=1
- Sampathkumar P. West Nile virus: epidemiology, clinical presentation, diagnosis, and prevention. Mayo Clin Proc. 2003;78:1137-1144.
- Centers for Disease Control and Prevention. West Nile virus. Updated June 3, 2020. Accessed April 16, 2021. https://www.cdc.gov/westnile/index.html
- Chuang TW, Hockett CW, Kightlinger L, et al. Landscape-level spatial patterns of West Nile virus risk in the northern Great Plains. Am J Trop Med Hyg. 2012;86:724-731.
- Wimberly MC, Hildreth MB, Boyte SP, et al. Ecological niche of the 2003 West Nile virus epidemic in the northern great plains of the United States. PLoS One. 2008;3:E3744. doi:10.1371/journal.pone.0003744
- Centers for Disease Control and Prevention. West Nile virus disease cases reported to CDC by state of residence, 1999-2019. Accessed April 26, 2021. https://www.cdc.gov/westnile/resources/pdfs/data/West-Nile-virus-disease-cases-by-state_1999-2019-P.pdf
- Hahn MB, Monaghan AJ, Hayden MH, et al. Meteorological conditions associated with increased incidence of West Nile virus disease in the United States, 2004–2012. Am J Trop Med Hyg. 2015;92:1013-1022.
- Brown CM, DeMaria A Jr. The resurgence of West Nile virus. Ann Intern Med. 2012;157:823-824.
- Landesman WJ, Allan BF, Langerhans RB, et al. Inter-annual associations between precipitation and human incidence of West Nile virus in the United States. Vector Borne Zoonotic Dis. 2007;7:337-343.
- Hart J Jr, Tillman G, Kraut MA, et al. West Nile virus neuroinvasive disease: neurological manifestations and prospective longitudinal outcomes. BMC Infect Dis. 2014;14:248.
- Wu JJ, Huang DB, Tyring SK. West Nile virus rash on the palms and soles of the feet. J Eur Acad Dermatol Venereol. 2006;20:1393-1394.
- Sejvar J. Clinical manifestations and outcomes of West Nile virus infection. Viruses. 2014;6:606-623.
- Ferguson DD, Gershman K, LeBailly A, et al. Characteristics of the rash associated with West Nile virus fever. Clin Infect Dis. 2005;41:1204-1207.
- Marszalek R, Chen A, Gjede J. Psoriasiform eruption in the setting of West Nile virus. J Am Acad Dermatol. 2014;70:AB4. doi:10.1016/j.jaad.2014.01.017
- Shah S, Fite LP, Lane N, et al. Purpura fulminans associated with acute West Nile virus encephalitis. J Clin Virol. 2016;75:1-4.
- Civen R, Villacorte F, Robles DT, et al. West Nile virus infection in the pediatric population. Pediatr Infect Dis J. 2006;25:75-78.
- Huhn GD, Dworkin MS. Rash as a prognostic factor in West Nile virus disease. Clin Infect Dis. 2006;43:388-389.
- Murphy TD, Grandpre J, Novick SL, et al. West Nile virus infection among health-fair participants, Wyoming 2003: assessment of symptoms and risk factors. Vector Borne Zoonotic Dis. 2005;5:246-251.
- Prince HE, Tobler LH, Lapé-Nixon M, et al. Development and persistence of West Nile virus–specific immunoglobulin M (IgM), IgA, and IgG in viremic blood donors. J Clin Microbiol. 2005;43:4316-4320.
- Busch MP, Kleinman SH, Tobler LH, et al. Virus and antibody dynamics in acute West Nile Virus infection. J Infect Dis. 2008;198:984-993.
- Mostashari F, Bunning ML, Kitsutani PT, et al. Epidemic West Nile encephalitis, New York, 1999: results of a household-based seroepidemiological survey. Lancet. 2001;358:261-264.
- Cahill ME, Yao Y, Nock D, et al. West Nile virus seroprevalence, Connecticut, USA, 2000–2014. Emerg Infect Dis. 2017;23:708-710.
- Schweitzer BK, Kramer WL, Sambol AR, et al. Geographic factors contributing to a high seroprevalence of West Nile virus-specific antibodies in humans following an epidemic. Clin Vaccine Immunol. 2006;13:314-318.
- Maeda A, Maeda J. Review of diagnostic plaque reduction neutralization tests for flavivirus infection. Vet J. 2013;195:33-40.
- Tilley PA, Fox JD, Jayaraman GC, et al. Nucleic acid testing for west nile virus RNA in plasma enhances rapid diagnosis of acute infection in symptomatic patients. J Infect Dis. 2006;193:1361-1364.
- Petersen LR, Brault AC, Nasci RS. West Nile virus: review of the literature. JAMA. 2013;310:308-315.
- Yu A, Ferenczi E, Moussa K, et al. Clinical spectrum of West Nile virus neuroinvasive disease. Neurohospitalist. 2020;10:43-47.
- Michaelis M, Kleinschmidt MC, Doerr HW, et al. Minocycline inhibits West Nile virus replication and apoptosis in human neuronal cells. J Antimicrob Chemother. 2007;60:981-986.
- United State Environmental Protection Agency. Skin-applied repellent ingredients. https://www.epa.gov/insect-repellents/skin-applied-repellent-ingredients. Accessed April 16, 2021.
- Muzumdar S, Rothe MJ, Grant-Kels JM. The rash with maculopapules and fever in adults. Clin Dermatol. 2019;37:109-118.
Practice Points
- Dermatologists should be aware of the most common rash associated with West Nile virus (WNV), which is a nonspecific maculopapular rash appearing on the trunk and extremities around 5 days after the onset of fever, fatigue, and other nonspecific symptoms.
- Rash may serve as a prognostic indicator for improved outcomes in WNV due to its association with decreased risk of encephalitis and death.
- An IgM enzyme-linked immunosorbent assay for WNV initially may yield false-negative results, as the development of detectable antibodies against the virus may take up to 8 days after symptom onset.