User login
New studies expand on Aimovig for migraine
SAN FRANCISCO – Now that erenumab has won approval as the first-in-class calcitonin gene-related peptide inhibitor for prevention of episodic and chronic migraine, a flurry of new studies shedding light on how the drug might best be used in clinical practice emerged as a highlight of the annual meeting of the American Headache Society.
Erenumab (Aimovig) is a fully human monoclonal antibody targeting the calcitonin gene-related peptide (CGRP) receptor. It was approved by the Food and Drug Administration at 140 and 70 mg monthly by subcutaneous injection on the strength of the required 12-week, double-blind, placebo-controlled clinical trial data. Among the fresh data presented at the headache meeting was reassuring evidence of the monoclonal antibody’s continued high degree of safety and tolerability for episodic migraine after more than 3 years of therapy, as well as a suggestion that the biologic’s efficacy for chronic migraine was not only sustained but actually increased over the course of a 1-year study.
Also, erenumab appears to be a novel treatment option for patients who have previously failed adequate trials of two to four standard preventive therapies. In addition, it has now demonstrated efficacy in reducing overuse of acute migraine drugs such as triptans, ergots, and opioids.
Here are the highlights:
One-year safety and efficacy for chronic migraine
Stewart J. Tepper, MD, presented results of a 1-year, open-label extension study that began after participants completed a pivotal 12-week, placebo-controlled, double-blind, randomized trial whose findings have been published (Lancet Neurol. 2017 Jun;16[6]:425-34). At the end of the 12-week, double-blind period, the mean number of monthly migraine days (MMD) in the erenumab group had dropped by 6.6 days from 18.1 at baseline. Subsequently, after 52 weeks of open-label erenumab at 140 mg monthly, that figure had further improved to 10.5 fewer MMDs than at baseline.

“That’s encouraging for our patients in that we will be able to tell them that if you’re a responder, you’re likely to show some continued improvement over time,” observed Dr. Tepper, professor of neurology and director of the headache clinic at Dartmouth University in Hanover, N.H.
During 527 patient-years of open-label therapy, at least a 50% reduction in MMDs was achieved by 67% of patients in the 140-mg group and 53% in the 70-mg group.
“I think it’s also important to look at the rate of 75% or greater reduction in MMDs, which is the responder rate most closely linked to a drop in disability. That rate at 52 weeks was 42% at 140 mg and 27% at 70 mg,” the neurologist continued.
At week 52, 12.7% of patients on erenumab at 140 mg monthly had no migraines at all during the previous month.
The mean number of acute migraine-specific medication days per month decreased by about 4 days in the erenumab group during the 12-week, double-blind trial and continued to fall during the open-label extension study, with a mean reduction from baseline of 6.7 days at week 52.
“That’s clearly sustained efficacy, and it looks like some accumulating efficacy,” Dr. Tepper said.
Safety and tolerability continued to be excellent, as in the parent 12-week study.
“One of the very interesting findings we saw which differentiates erenumab from, say, onabotulinumtoxinA, is that if you looked at the patients who were on placebo during the 12 weeks of the double-blind study and then looked at the first month they went on erenumab, they caught up immediately to the patients who’d been receiving the monoclonal antibody from the beginning. That’s unlike onabotulinumtoxinA in the regulatory trials, where the patients who initially received placebo never quite caught up to those who received active therapy from the beginning,” he said.
Dr. Tepper emphasized that a clear-eyed view of an open-label study needs to recognize that only responders would have stayed in the erenumab study for a full 52 weeks, so the results paint an overly rosy picture of overall efficacy. Even so, he found the results impressive. Of 609 patients who enrolled in the study, 74% completed it.
“It’s a very encouraging 1-year study for erenumab,” he concluded.
Safety and tolerability for episodic migraine at 3-plus years
Among 383 episodic migraine patients who enrolled in an ongoing 5-year, open-label extension study of erenumab after completing a 12-week, placebo-controlled, double-blind clinical trial, there have been no new safety signals at a mean 3.2 years of follow-up. The incidence rates and types of adverse events remain indistinguishable from placebo as noted in the parent 12-week double-blind trial, with the exception of an increased rate of mild injection site reactions, reported Daniel D. Mikol, MD, PhD, executive medical director for global neuroscience development at Amgen in Thousand Oaks, Calif.

Erenumab proves effective in patients who have failed multiple preventive therapies
Jan Klatt, MD, presented the results of the 12-week, double-blind portion of the phase 3b LIBERTY study, the first clinical trial specifically designed to assess the effects of CGRP-directed therapy in patients who have previously failed to respond to and/or tolerate two to four currently available preventive medications at adequate doses for at least 2-3 months.
The rationale for the trial was straightforward: “There is a particularly high unmet need in patients who have failed currently available preventive therapies, and who are usually considered difficult to treat,” explained Dr. Klatt of Novartis in Basel, Switzerland.
LIBERTY included 246 such patients with episodic migraine who were randomized to 12 weeks of erenumab at 140 mg monthly or placebo. The primary endpoint – the proportion of patients with at least a 50% reduction in MMDs during weeks 9-12 from the baseline of 9.3 – occurred in 30.3% of the erenumab group, significantly better than the 13.7% rate with placebo, with an adjusted odds ratio of 2.73 for success with erenumab. An open-label extension study is ongoing.
A clear separation was already evident at week 4 of the randomized trial, a prespecified secondary endpoint. A 50% or greater reduction in MMDs was seen at that point in 23.5% of the erenumab group versus 4.8% in placebo-treated controls, for a response odds ratio of 6.16. A 75% or greater reduction in MMDs during weeks 9-12 occurred in 11.8% of the erenumab group and 4% of controls. A 100% response rate – that is, no migraines during weeks 9-12 – occurred in 5.9% of the active treatment arm and none of the controls. Outcomes on two novel patient-reported outcomes – the Migraine Physical Function Impact Diary–physical impairment and –everyday activities subscales – were also significantly better in the active treatment arm.
Treatment outcomes with erenumab were similar regardless of which drug classes patients had previously failed on, the three most common of which were beta-blockers, topiramate, and amitriptyline, which is approved for migraine prevention in Europe, where the LIBERTY trial was conducted.
One audience member observed that the 13.7% primary endpoint placebo response rate was far lower than typically seen in double-blind migraine trials.
“This is something we see quite consistently,” Dr. Klatt replied. “Once you proceed to this kind of more difficult-to-treat population, your placebo response rates drop to really low levels. It’s probably due to the fact that those patients have low expectations for any new therapy.”
Erenumab quells acute migraine medication overuse
Dr. Tepper presented a separate analysis of the 52-week, open-label extension study of erenumab, this time comparing outcomes in 252 chronic migraine patients who met criteria for acute medication overuse (AMO) at baseline and 357 who did not. Overuse was defined via International Headache Society criteria: The use of triptans, ergots, or combination analgesics on 10 or more days per month, or simple analgesics on at least 15 days monthly. Combination analgesics feature a simple analgesic plus opiates or butalbital.
“The bottom line here is that the drug worked about the same and in a very encouraging way in the AMO and nonoveruse patients,” he said. “This is obviously something that’s very, very important to us because we’d like to take our patients who are overusing acute migraine medications and use the monoclonal antibody as a way of getting them off overuse and into episodic migraine.”
After 52 weeks of open-label erenumab, 61% of the non-AMO group and 56% of those with baseline AMO had at least a 50% reduction from baseline in MMDs. The use of acute migraine-specific medications fell by 3.7 days in the non-AMO group and by 6.7 days in those with AMO at baseline.
Safety in patients with stable angina
Although no signal of any cardiovascular issues arose in four randomized, double-blind clinical trials or in the long-term extension studies, it was appropriate to take a more focused look at erenumab’s effects in a high-risk population with stable angina and proven ischemic coronary artery disease. That’s because CGRP is a neuropeptide that not only affects migraine, it also acts as a cytoprotective mediator released by cardiac nerve fibers during myocardial ischemia. Thus, erenumab posed a theoretic cardiovascular risk, explained Christophe Depre, MD, PhD, of Amgen.
He presented a 12-week, randomized, double-blind study in which 89 patients with stable angina were assigned to a single IV infusion of 140 mg of erenumab or placebo followed by an exercise treadmill test. Reassuringly, the CGRP inhibitor had no adverse effect on exercise time, time to onset of at least 1 mm of ST segment depression. Nor did it affect heart rate or blood pressure during or after exercise.
Dr. Tepper reported serving as a consultant to numerous pharmaceutical and medical device companies, including Amgen and Novartis, which sponsored the erenumab studies. Dr. Depre and Dr. Mikol are Amgen employees, while Dr. Klatt is employed by Novartis.
SAN FRANCISCO – Now that erenumab has won approval as the first-in-class calcitonin gene-related peptide inhibitor for prevention of episodic and chronic migraine, a flurry of new studies shedding light on how the drug might best be used in clinical practice emerged as a highlight of the annual meeting of the American Headache Society.
Erenumab (Aimovig) is a fully human monoclonal antibody targeting the calcitonin gene-related peptide (CGRP) receptor. It was approved by the Food and Drug Administration at 140 and 70 mg monthly by subcutaneous injection on the strength of the required 12-week, double-blind, placebo-controlled clinical trial data. Among the fresh data presented at the headache meeting was reassuring evidence of the monoclonal antibody’s continued high degree of safety and tolerability for episodic migraine after more than 3 years of therapy, as well as a suggestion that the biologic’s efficacy for chronic migraine was not only sustained but actually increased over the course of a 1-year study.
Also, erenumab appears to be a novel treatment option for patients who have previously failed adequate trials of two to four standard preventive therapies. In addition, it has now demonstrated efficacy in reducing overuse of acute migraine drugs such as triptans, ergots, and opioids.
Here are the highlights:
One-year safety and efficacy for chronic migraine
Stewart J. Tepper, MD, presented results of a 1-year, open-label extension study that began after participants completed a pivotal 12-week, placebo-controlled, double-blind, randomized trial whose findings have been published (Lancet Neurol. 2017 Jun;16[6]:425-34). At the end of the 12-week, double-blind period, the mean number of monthly migraine days (MMD) in the erenumab group had dropped by 6.6 days from 18.1 at baseline. Subsequently, after 52 weeks of open-label erenumab at 140 mg monthly, that figure had further improved to 10.5 fewer MMDs than at baseline.
“That’s encouraging for our patients in that we will be able to tell them that if you’re a responder, you’re likely to show some continued improvement over time,” observed Dr. Tepper, professor of neurology and director of the headache clinic at Dartmouth University in Hanover, N.H.
During 527 patient-years of open-label therapy, at least a 50% reduction in MMDs was achieved by 67% of patients in the 140-mg group and 53% in the 70-mg group.
“I think it’s also important to look at the rate of 75% or greater reduction in MMDs, which is the responder rate most closely linked to a drop in disability. That rate at 52 weeks was 42% at 140 mg and 27% at 70 mg,” the neurologist continued.
At week 52, 12.7% of patients on erenumab at 140 mg monthly had no migraines at all during the previous month.
The mean number of acute migraine-specific medication days per month decreased by about 4 days in the erenumab group during the 12-week, double-blind trial and continued to fall during the open-label extension study, with a mean reduction from baseline of 6.7 days at week 52.
“That’s clearly sustained efficacy, and it looks like some accumulating efficacy,” Dr. Tepper said.
Safety and tolerability continued to be excellent, as in the parent 12-week study.
“One of the very interesting findings we saw which differentiates erenumab from, say, onabotulinumtoxinA, is that if you looked at the patients who were on placebo during the 12 weeks of the double-blind study and then looked at the first month they went on erenumab, they caught up immediately to the patients who’d been receiving the monoclonal antibody from the beginning. That’s unlike onabotulinumtoxinA in the regulatory trials, where the patients who initially received placebo never quite caught up to those who received active therapy from the beginning,” he said.
Dr. Tepper emphasized that a clear-eyed view of an open-label study needs to recognize that only responders would have stayed in the erenumab study for a full 52 weeks, so the results paint an overly rosy picture of overall efficacy. Even so, he found the results impressive. Of 609 patients who enrolled in the study, 74% completed it.
“It’s a very encouraging 1-year study for erenumab,” he concluded.
Safety and tolerability for episodic migraine at 3-plus years
Among 383 episodic migraine patients who enrolled in an ongoing 5-year, open-label extension study of erenumab after completing a 12-week, placebo-controlled, double-blind clinical trial, there have been no new safety signals at a mean 3.2 years of follow-up. The incidence rates and types of adverse events remain indistinguishable from placebo as noted in the parent 12-week double-blind trial, with the exception of an increased rate of mild injection site reactions, reported Daniel D. Mikol, MD, PhD, executive medical director for global neuroscience development at Amgen in Thousand Oaks, Calif.
Erenumab proves effective in patients who have failed multiple preventive therapies
Jan Klatt, MD, presented the results of the 12-week, double-blind portion of the phase 3b LIBERTY study, the first clinical trial specifically designed to assess the effects of CGRP-directed therapy in patients who have previously failed to respond to and/or tolerate two to four currently available preventive medications at adequate doses for at least 2-3 months.
The rationale for the trial was straightforward: “There is a particularly high unmet need in patients who have failed currently available preventive therapies, and who are usually considered difficult to treat,” explained Dr. Klatt of Novartis in Basel, Switzerland.
LIBERTY included 246 such patients with episodic migraine who were randomized to 12 weeks of erenumab at 140 mg monthly or placebo. The primary endpoint – the proportion of patients with at least a 50% reduction in MMDs during weeks 9-12 from the baseline of 9.3 – occurred in 30.3% of the erenumab group, significantly better than the 13.7% rate with placebo, with an adjusted odds ratio of 2.73 for success with erenumab. An open-label extension study is ongoing.
A clear separation was already evident at week 4 of the randomized trial, a prespecified secondary endpoint. A 50% or greater reduction in MMDs was seen at that point in 23.5% of the erenumab group versus 4.8% in placebo-treated controls, for a response odds ratio of 6.16. A 75% or greater reduction in MMDs during weeks 9-12 occurred in 11.8% of the erenumab group and 4% of controls. A 100% response rate – that is, no migraines during weeks 9-12 – occurred in 5.9% of the active treatment arm and none of the controls. Outcomes on two novel patient-reported outcomes – the Migraine Physical Function Impact Diary–physical impairment and –everyday activities subscales – were also significantly better in the active treatment arm.
Treatment outcomes with erenumab were similar regardless of which drug classes patients had previously failed on, the three most common of which were beta-blockers, topiramate, and amitriptyline, which is approved for migraine prevention in Europe, where the LIBERTY trial was conducted.
One audience member observed that the 13.7% primary endpoint placebo response rate was far lower than typically seen in double-blind migraine trials.
“This is something we see quite consistently,” Dr. Klatt replied. “Once you proceed to this kind of more difficult-to-treat population, your placebo response rates drop to really low levels. It’s probably due to the fact that those patients have low expectations for any new therapy.”
Erenumab quells acute migraine medication overuse
Dr. Tepper presented a separate analysis of the 52-week, open-label extension study of erenumab, this time comparing outcomes in 252 chronic migraine patients who met criteria for acute medication overuse (AMO) at baseline and 357 who did not. Overuse was defined via International Headache Society criteria: The use of triptans, ergots, or combination analgesics on 10 or more days per month, or simple analgesics on at least 15 days monthly. Combination analgesics feature a simple analgesic plus opiates or butalbital.
“The bottom line here is that the drug worked about the same and in a very encouraging way in the AMO and nonoveruse patients,” he said. “This is obviously something that’s very, very important to us because we’d like to take our patients who are overusing acute migraine medications and use the monoclonal antibody as a way of getting them off overuse and into episodic migraine.”
After 52 weeks of open-label erenumab, 61% of the non-AMO group and 56% of those with baseline AMO had at least a 50% reduction from baseline in MMDs. The use of acute migraine-specific medications fell by 3.7 days in the non-AMO group and by 6.7 days in those with AMO at baseline.
Safety in patients with stable angina
Although no signal of any cardiovascular issues arose in four randomized, double-blind clinical trials or in the long-term extension studies, it was appropriate to take a more focused look at erenumab’s effects in a high-risk population with stable angina and proven ischemic coronary artery disease. That’s because CGRP is a neuropeptide that not only affects migraine, it also acts as a cytoprotective mediator released by cardiac nerve fibers during myocardial ischemia. Thus, erenumab posed a theoretic cardiovascular risk, explained Christophe Depre, MD, PhD, of Amgen.
He presented a 12-week, randomized, double-blind study in which 89 patients with stable angina were assigned to a single IV infusion of 140 mg of erenumab or placebo followed by an exercise treadmill test. Reassuringly, the CGRP inhibitor had no adverse effect on exercise time, time to onset of at least 1 mm of ST segment depression. Nor did it affect heart rate or blood pressure during or after exercise.
Dr. Tepper reported serving as a consultant to numerous pharmaceutical and medical device companies, including Amgen and Novartis, which sponsored the erenumab studies. Dr. Depre and Dr. Mikol are Amgen employees, while Dr. Klatt is employed by Novartis.
SAN FRANCISCO – Now that erenumab has won approval as the first-in-class calcitonin gene-related peptide inhibitor for prevention of episodic and chronic migraine, a flurry of new studies shedding light on how the drug might best be used in clinical practice emerged as a highlight of the annual meeting of the American Headache Society.
Erenumab (Aimovig) is a fully human monoclonal antibody targeting the calcitonin gene-related peptide (CGRP) receptor. It was approved by the Food and Drug Administration at 140 and 70 mg monthly by subcutaneous injection on the strength of the required 12-week, double-blind, placebo-controlled clinical trial data. Among the fresh data presented at the headache meeting was reassuring evidence of the monoclonal antibody’s continued high degree of safety and tolerability for episodic migraine after more than 3 years of therapy, as well as a suggestion that the biologic’s efficacy for chronic migraine was not only sustained but actually increased over the course of a 1-year study.
Also, erenumab appears to be a novel treatment option for patients who have previously failed adequate trials of two to four standard preventive therapies. In addition, it has now demonstrated efficacy in reducing overuse of acute migraine drugs such as triptans, ergots, and opioids.
Here are the highlights:
One-year safety and efficacy for chronic migraine
Stewart J. Tepper, MD, presented results of a 1-year, open-label extension study that began after participants completed a pivotal 12-week, placebo-controlled, double-blind, randomized trial whose findings have been published (Lancet Neurol. 2017 Jun;16[6]:425-34). At the end of the 12-week, double-blind period, the mean number of monthly migraine days (MMD) in the erenumab group had dropped by 6.6 days from 18.1 at baseline. Subsequently, after 52 weeks of open-label erenumab at 140 mg monthly, that figure had further improved to 10.5 fewer MMDs than at baseline.
“That’s encouraging for our patients in that we will be able to tell them that if you’re a responder, you’re likely to show some continued improvement over time,” observed Dr. Tepper, professor of neurology and director of the headache clinic at Dartmouth University in Hanover, N.H.
During 527 patient-years of open-label therapy, at least a 50% reduction in MMDs was achieved by 67% of patients in the 140-mg group and 53% in the 70-mg group.
“I think it’s also important to look at the rate of 75% or greater reduction in MMDs, which is the responder rate most closely linked to a drop in disability. That rate at 52 weeks was 42% at 140 mg and 27% at 70 mg,” the neurologist continued.
At week 52, 12.7% of patients on erenumab at 140 mg monthly had no migraines at all during the previous month.
The mean number of acute migraine-specific medication days per month decreased by about 4 days in the erenumab group during the 12-week, double-blind trial and continued to fall during the open-label extension study, with a mean reduction from baseline of 6.7 days at week 52.
“That’s clearly sustained efficacy, and it looks like some accumulating efficacy,” Dr. Tepper said.
Safety and tolerability continued to be excellent, as in the parent 12-week study.
“One of the very interesting findings we saw which differentiates erenumab from, say, onabotulinumtoxinA, is that if you looked at the patients who were on placebo during the 12 weeks of the double-blind study and then looked at the first month they went on erenumab, they caught up immediately to the patients who’d been receiving the monoclonal antibody from the beginning. That’s unlike onabotulinumtoxinA in the regulatory trials, where the patients who initially received placebo never quite caught up to those who received active therapy from the beginning,” he said.
Dr. Tepper emphasized that a clear-eyed view of an open-label study needs to recognize that only responders would have stayed in the erenumab study for a full 52 weeks, so the results paint an overly rosy picture of overall efficacy. Even so, he found the results impressive. Of 609 patients who enrolled in the study, 74% completed it.
“It’s a very encouraging 1-year study for erenumab,” he concluded.
Safety and tolerability for episodic migraine at 3-plus years
Among 383 episodic migraine patients who enrolled in an ongoing 5-year, open-label extension study of erenumab after completing a 12-week, placebo-controlled, double-blind clinical trial, there have been no new safety signals at a mean 3.2 years of follow-up. The incidence rates and types of adverse events remain indistinguishable from placebo as noted in the parent 12-week double-blind trial, with the exception of an increased rate of mild injection site reactions, reported Daniel D. Mikol, MD, PhD, executive medical director for global neuroscience development at Amgen in Thousand Oaks, Calif.
Erenumab proves effective in patients who have failed multiple preventive therapies
Jan Klatt, MD, presented the results of the 12-week, double-blind portion of the phase 3b LIBERTY study, the first clinical trial specifically designed to assess the effects of CGRP-directed therapy in patients who have previously failed to respond to and/or tolerate two to four currently available preventive medications at adequate doses for at least 2-3 months.
The rationale for the trial was straightforward: “There is a particularly high unmet need in patients who have failed currently available preventive therapies, and who are usually considered difficult to treat,” explained Dr. Klatt of Novartis in Basel, Switzerland.
LIBERTY included 246 such patients with episodic migraine who were randomized to 12 weeks of erenumab at 140 mg monthly or placebo. The primary endpoint – the proportion of patients with at least a 50% reduction in MMDs during weeks 9-12 from the baseline of 9.3 – occurred in 30.3% of the erenumab group, significantly better than the 13.7% rate with placebo, with an adjusted odds ratio of 2.73 for success with erenumab. An open-label extension study is ongoing.
A clear separation was already evident at week 4 of the randomized trial, a prespecified secondary endpoint. A 50% or greater reduction in MMDs was seen at that point in 23.5% of the erenumab group versus 4.8% in placebo-treated controls, for a response odds ratio of 6.16. A 75% or greater reduction in MMDs during weeks 9-12 occurred in 11.8% of the erenumab group and 4% of controls. A 100% response rate – that is, no migraines during weeks 9-12 – occurred in 5.9% of the active treatment arm and none of the controls. Outcomes on two novel patient-reported outcomes – the Migraine Physical Function Impact Diary–physical impairment and –everyday activities subscales – were also significantly better in the active treatment arm.
Treatment outcomes with erenumab were similar regardless of which drug classes patients had previously failed on, the three most common of which were beta-blockers, topiramate, and amitriptyline, which is approved for migraine prevention in Europe, where the LIBERTY trial was conducted.
One audience member observed that the 13.7% primary endpoint placebo response rate was far lower than typically seen in double-blind migraine trials.
“This is something we see quite consistently,” Dr. Klatt replied. “Once you proceed to this kind of more difficult-to-treat population, your placebo response rates drop to really low levels. It’s probably due to the fact that those patients have low expectations for any new therapy.”
Erenumab quells acute migraine medication overuse
Dr. Tepper presented a separate analysis of the 52-week, open-label extension study of erenumab, this time comparing outcomes in 252 chronic migraine patients who met criteria for acute medication overuse (AMO) at baseline and 357 who did not. Overuse was defined via International Headache Society criteria: The use of triptans, ergots, or combination analgesics on 10 or more days per month, or simple analgesics on at least 15 days monthly. Combination analgesics feature a simple analgesic plus opiates or butalbital.
“The bottom line here is that the drug worked about the same and in a very encouraging way in the AMO and nonoveruse patients,” he said. “This is obviously something that’s very, very important to us because we’d like to take our patients who are overusing acute migraine medications and use the monoclonal antibody as a way of getting them off overuse and into episodic migraine.”
After 52 weeks of open-label erenumab, 61% of the non-AMO group and 56% of those with baseline AMO had at least a 50% reduction from baseline in MMDs. The use of acute migraine-specific medications fell by 3.7 days in the non-AMO group and by 6.7 days in those with AMO at baseline.
Safety in patients with stable angina
Although no signal of any cardiovascular issues arose in four randomized, double-blind clinical trials or in the long-term extension studies, it was appropriate to take a more focused look at erenumab’s effects in a high-risk population with stable angina and proven ischemic coronary artery disease. That’s because CGRP is a neuropeptide that not only affects migraine, it also acts as a cytoprotective mediator released by cardiac nerve fibers during myocardial ischemia. Thus, erenumab posed a theoretic cardiovascular risk, explained Christophe Depre, MD, PhD, of Amgen.
He presented a 12-week, randomized, double-blind study in which 89 patients with stable angina were assigned to a single IV infusion of 140 mg of erenumab or placebo followed by an exercise treadmill test. Reassuringly, the CGRP inhibitor had no adverse effect on exercise time, time to onset of at least 1 mm of ST segment depression. Nor did it affect heart rate or blood pressure during or after exercise.
Dr. Tepper reported serving as a consultant to numerous pharmaceutical and medical device companies, including Amgen and Novartis, which sponsored the erenumab studies. Dr. Depre and Dr. Mikol are Amgen employees, while Dr. Klatt is employed by Novartis.
REPORTING FROM THE AHS ANNUAL MEETING
European experts envy U.S. pediatric flu vaccination approach
MALMO, SWEDEN – When American physicians think about health care in Europe, what typically comes to mind are government-funded, single-payer national health services with cradle-to-grave coverage of essential services, a strong public health bent, and perhaps some queuing.

“It’s complicated. There is no common strategic approach,” Hanna Nohynek, MD, PhD, observed at a session on childhood immunization against flu held during the annual meeting of the European Society for Paediatric Infectious Diseases.
“In real life, influenza coverage among [European] children is either not known or quite low. Impact assessments in children are done in only a few countries,” said Dr. Nohynek, chief physician in the infectious diseases control and vaccinations unit of the National Institute for Health and Welfare in Helsinki, Finland.
“The only country doing as well coverage-wise as the U.S. is the U.K., with rates of 50%-65%. In Finland it’s less than 40%,” according to Dr. Nohynek.
“We have 28 countries today in the E.U. [European Union], and we have 28 different recommendations in Europe. So where do we go from here? It’s really not easy,” observed session cochair Alberticus Osterhaus, DVM, PhD, emeritus professor of virology at Erasmus University in Rotterdam, the Netherlands.
For all the oft-cited shortcomings of health care in the United States, That’s why Jon S. Abramson, MD, a former chair of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices (ACIP), was invited to explain how the U.S. strategy was accomplished.
The U.S. approach
The current U.S. policy, implemented in 2010, is to recommend an annual flu shot for all persons older than 6 months of age.

Influenza vaccination has been part of the U.S. public health program since 1960. Children aged 6-23 months, as well as their household contacts and women who will be pregnant during flu season, were added in 2004. In 2006, the flu vaccine recommendation was expanded to include children aged 6-59 months as a result of persuasive data showing that the rate of flu-associated hospitalizations and deaths in children up to 4 years old was second only to the rate in the elderly population.
The rationale for expanding the recommendation to include all school-age children and adolescents stemmed from evidence that the highest average flu-related illness rate in the United States was in that age group, which confirmed that schools are a powerful vector for the spread of influenza. Vaccinating this age-group also was seen as having an indirect benefit for their household contacts.
The current policy of recommending vaccination of everyone over age 6 months was adopted because it checked off a lot of boxes: “It’s a single recommendation, easy to apply; it eliminates the need to look for indications and risk factors; it increases vaccination coverage rates; annual vaccination is safe and effective; and flu-related morbidity and mortality occur in all age groups,” Dr. Abramson continued.
The rate of influenza vaccine coverage in pregnant women has improved over time from less than 15% to about 50%. To place that in perspective, however, the rate in Argentina is 95%, the pediatrician noted.
“We’re doing better in children than we are in adults in terms of seasonal coverage rates,” he added. “In 2015, it was 59%, versus 42% in adults.”
Dr. Abramson said there remains some skepticism in the United States regarding the effectiveness of flu vaccines in preventing flu-related illness. That’s because of the difficulty in communicating that vaccine effectiveness varies from year to year, sometimes substantially, depending upon two factors: the transmission characteristics of the circulating strains and how well the vaccines match up against those strains.
“I think we have to learn to live with that. I don’t think we’ll see a universal flu vaccine that we can give once every 10 years,” he said.
“The bottom line is, even if a vaccine is only 50% efficacious overall, we’re still impacting huge numbers,” the pediatrician added.
Dr. Abramson cited a CDC estimate that, for the 2012-2013 season, where the vaccine was 49% efficacious, the result of vaccination was 6.6 million fewer cases of influenza-associated illnesses nationally, 3.2 million fewer flu-associated medical visits, and 79,000 hospitalizations avoided.
“I think we have a fairly good program in the United States. We’re doing well in children. We certainly could be doing better. Not having FluMist for the past 2 seasons probably hurt us some,” according to Dr. Abramson.
The FluMist experience
The FluMist episode is viewed by many European pediatric infectious disease experts as a debacle. Europeans eager to develop a pan-European strategy for seasonal immunization against influenza in children and adolescents viewed the U.S. FluMist episode with dismay. For the 2016-2017 and 2017-2018 flu seasons, the ACIP recommended against FluMist, a previously approved intranasally administered quadrivalent live attenuated virus vaccine, on the basis of a single study showing subpar effectiveness against influenza A H1NI. Then at its October 2017 meeting, ACIP reversed itself and reinstated FluMist for the 2018-2019 season after viewing data from Finland and several other countries demonstrating that, in countries where it hadn’t been taken off the market, the vaccine had performed as well as injectable inactivated influenza vaccines in the 2016-2017 flu season.
“I think from the European side, it’s been a bit of a sorry spectacle,” commented Dr. Osterhaus, referring to the ACIP’s waffling. After all, authorities in Canada and European countries where FluMist was available had looked at the same data that caused ACIP to derecommend the vaccine but hadn’t found it convincing.
“We’re very happy to see ACIP has reinstated the vaccine,” Dr. Nohynek said.
Dr. Abramson declined to defend the ACIP decision to drop FluMist.
“From my standpoint, knowing that influenza B kills more children than A does, if I had been on the ACIP committee – and I’m not anymore – that would not have been my vote,” he said. “Whatever you want to say about the live attenuated influenza vaccine, about how good it is against some A strains or not, it’s better than other vaccines against influenza B. And the death rate is higher from B than A in children, although that is not true in adults.”
Plus, FluMist was an important option for people avoiding immunization because they dislike shots.
“The vast majority of deaths due to flu in children in 2010-2016 have been in kids who didn’t get vaccinated,” he noted.
Dr. Nohynek said the Finnish real-world experience recorded in comprehensive national registries for the 2017-2018 flu season – a bad year for vaccine/virus mismatch in Europe – confirmed Dr. Abramson’s comments about the superiority of quadrivalent live attenuated influenza vaccine against influenza B. Among 54,611 Finnish children aged 24-35 months, the laboratory-confirmed vaccine effectiveness of trivalent inactivated virus vaccine, with 9% coverage, was 4.5% for influenza A and 12.2% for influenza B. In contrast, the vaccine effectiveness for the intranasal quadrivalent live attenuated influenza vaccine was 32% for A and a whopping 80% for B.
“It’s quite amazing, at least to me, to see figures like this in real world data,” she commented.
Session cochair Adam Finn, MD, PhD, said he has found it instructive to take a closer look at the U.K. data for the past several flu seasons.
“We’ve seen greater control of the epidemic in Scotland and Northern Ireland, where coverage in primary school kids was higher, in the 60%-70% area, and lower in England and Wales, where it was more like 50%. So we’re beginning to think that’s the kind of level of annual coverage in children we might need to suppress an epidemic. I think that’s a really important message that people should understand: We’re not looking for 95% coverage,” observed Dr. Finn, aprofessor of pediatrics at the University of Bristol (England).
Vaccine effectiveness will improve
Dr. Osterhaus predicted better times are coming in terms of vaccine effectiveness. Vaccine production times will become shorter as recombinant technologies replace the traditional lengthy chicken egg-based vaccine production; as a result, there will be less drift-associated mismatch. Improved surveillance, including the ability to follow strain mobility patterns and population-based antibody landscapes, are another important advance.
“We’ve always been looking at one side of the coin: the virus. Once or twice a year eminent gray people sitting together in Geneva at WHO decide which strains should be selected for the next vaccine. But if you know what antibodies are present in the population, this can be quite important information as well,” he said.
Dr. Nohynek reported receiving research funding from GlaxoSmithKline and Pfizer. The other speakers reported having no relevant financial conflicts of interest.
MALMO, SWEDEN – When American physicians think about health care in Europe, what typically comes to mind are government-funded, single-payer national health services with cradle-to-grave coverage of essential services, a strong public health bent, and perhaps some queuing.
“It’s complicated. There is no common strategic approach,” Hanna Nohynek, MD, PhD, observed at a session on childhood immunization against flu held during the annual meeting of the European Society for Paediatric Infectious Diseases.
“In real life, influenza coverage among [European] children is either not known or quite low. Impact assessments in children are done in only a few countries,” said Dr. Nohynek, chief physician in the infectious diseases control and vaccinations unit of the National Institute for Health and Welfare in Helsinki, Finland.
“The only country doing as well coverage-wise as the U.S. is the U.K., with rates of 50%-65%. In Finland it’s less than 40%,” according to Dr. Nohynek.
“We have 28 countries today in the E.U. [European Union], and we have 28 different recommendations in Europe. So where do we go from here? It’s really not easy,” observed session cochair Alberticus Osterhaus, DVM, PhD, emeritus professor of virology at Erasmus University in Rotterdam, the Netherlands.
For all the oft-cited shortcomings of health care in the United States, That’s why Jon S. Abramson, MD, a former chair of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices (ACIP), was invited to explain how the U.S. strategy was accomplished.
The U.S. approach
The current U.S. policy, implemented in 2010, is to recommend an annual flu shot for all persons older than 6 months of age.
Influenza vaccination has been part of the U.S. public health program since 1960. Children aged 6-23 months, as well as their household contacts and women who will be pregnant during flu season, were added in 2004. In 2006, the flu vaccine recommendation was expanded to include children aged 6-59 months as a result of persuasive data showing that the rate of flu-associated hospitalizations and deaths in children up to 4 years old was second only to the rate in the elderly population.
The rationale for expanding the recommendation to include all school-age children and adolescents stemmed from evidence that the highest average flu-related illness rate in the United States was in that age group, which confirmed that schools are a powerful vector for the spread of influenza. Vaccinating this age-group also was seen as having an indirect benefit for their household contacts.
The current policy of recommending vaccination of everyone over age 6 months was adopted because it checked off a lot of boxes: “It’s a single recommendation, easy to apply; it eliminates the need to look for indications and risk factors; it increases vaccination coverage rates; annual vaccination is safe and effective; and flu-related morbidity and mortality occur in all age groups,” Dr. Abramson continued.
The rate of influenza vaccine coverage in pregnant women has improved over time from less than 15% to about 50%. To place that in perspective, however, the rate in Argentina is 95%, the pediatrician noted.
“We’re doing better in children than we are in adults in terms of seasonal coverage rates,” he added. “In 2015, it was 59%, versus 42% in adults.”
Dr. Abramson said there remains some skepticism in the United States regarding the effectiveness of flu vaccines in preventing flu-related illness. That’s because of the difficulty in communicating that vaccine effectiveness varies from year to year, sometimes substantially, depending upon two factors: the transmission characteristics of the circulating strains and how well the vaccines match up against those strains.
“I think we have to learn to live with that. I don’t think we’ll see a universal flu vaccine that we can give once every 10 years,” he said.
“The bottom line is, even if a vaccine is only 50% efficacious overall, we’re still impacting huge numbers,” the pediatrician added.
Dr. Abramson cited a CDC estimate that, for the 2012-2013 season, where the vaccine was 49% efficacious, the result of vaccination was 6.6 million fewer cases of influenza-associated illnesses nationally, 3.2 million fewer flu-associated medical visits, and 79,000 hospitalizations avoided.
“I think we have a fairly good program in the United States. We’re doing well in children. We certainly could be doing better. Not having FluMist for the past 2 seasons probably hurt us some,” according to Dr. Abramson.
The FluMist experience
The FluMist episode is viewed by many European pediatric infectious disease experts as a debacle. Europeans eager to develop a pan-European strategy for seasonal immunization against influenza in children and adolescents viewed the U.S. FluMist episode with dismay. For the 2016-2017 and 2017-2018 flu seasons, the ACIP recommended against FluMist, a previously approved intranasally administered quadrivalent live attenuated virus vaccine, on the basis of a single study showing subpar effectiveness against influenza A H1NI. Then at its October 2017 meeting, ACIP reversed itself and reinstated FluMist for the 2018-2019 season after viewing data from Finland and several other countries demonstrating that, in countries where it hadn’t been taken off the market, the vaccine had performed as well as injectable inactivated influenza vaccines in the 2016-2017 flu season.
“I think from the European side, it’s been a bit of a sorry spectacle,” commented Dr. Osterhaus, referring to the ACIP’s waffling. After all, authorities in Canada and European countries where FluMist was available had looked at the same data that caused ACIP to derecommend the vaccine but hadn’t found it convincing.
“We’re very happy to see ACIP has reinstated the vaccine,” Dr. Nohynek said.
Dr. Abramson declined to defend the ACIP decision to drop FluMist.
“From my standpoint, knowing that influenza B kills more children than A does, if I had been on the ACIP committee – and I’m not anymore – that would not have been my vote,” he said. “Whatever you want to say about the live attenuated influenza vaccine, about how good it is against some A strains or not, it’s better than other vaccines against influenza B. And the death rate is higher from B than A in children, although that is not true in adults.”
Plus, FluMist was an important option for people avoiding immunization because they dislike shots.
“The vast majority of deaths due to flu in children in 2010-2016 have been in kids who didn’t get vaccinated,” he noted.
Dr. Nohynek said the Finnish real-world experience recorded in comprehensive national registries for the 2017-2018 flu season – a bad year for vaccine/virus mismatch in Europe – confirmed Dr. Abramson’s comments about the superiority of quadrivalent live attenuated influenza vaccine against influenza B. Among 54,611 Finnish children aged 24-35 months, the laboratory-confirmed vaccine effectiveness of trivalent inactivated virus vaccine, with 9% coverage, was 4.5% for influenza A and 12.2% for influenza B. In contrast, the vaccine effectiveness for the intranasal quadrivalent live attenuated influenza vaccine was 32% for A and a whopping 80% for B.
“It’s quite amazing, at least to me, to see figures like this in real world data,” she commented.
Session cochair Adam Finn, MD, PhD, said he has found it instructive to take a closer look at the U.K. data for the past several flu seasons.
“We’ve seen greater control of the epidemic in Scotland and Northern Ireland, where coverage in primary school kids was higher, in the 60%-70% area, and lower in England and Wales, where it was more like 50%. So we’re beginning to think that’s the kind of level of annual coverage in children we might need to suppress an epidemic. I think that’s a really important message that people should understand: We’re not looking for 95% coverage,” observed Dr. Finn, aprofessor of pediatrics at the University of Bristol (England).
Vaccine effectiveness will improve
Dr. Osterhaus predicted better times are coming in terms of vaccine effectiveness. Vaccine production times will become shorter as recombinant technologies replace the traditional lengthy chicken egg-based vaccine production; as a result, there will be less drift-associated mismatch. Improved surveillance, including the ability to follow strain mobility patterns and population-based antibody landscapes, are another important advance.
“We’ve always been looking at one side of the coin: the virus. Once or twice a year eminent gray people sitting together in Geneva at WHO decide which strains should be selected for the next vaccine. But if you know what antibodies are present in the population, this can be quite important information as well,” he said.
Dr. Nohynek reported receiving research funding from GlaxoSmithKline and Pfizer. The other speakers reported having no relevant financial conflicts of interest.
MALMO, SWEDEN – When American physicians think about health care in Europe, what typically comes to mind are government-funded, single-payer national health services with cradle-to-grave coverage of essential services, a strong public health bent, and perhaps some queuing.
“It’s complicated. There is no common strategic approach,” Hanna Nohynek, MD, PhD, observed at a session on childhood immunization against flu held during the annual meeting of the European Society for Paediatric Infectious Diseases.
“In real life, influenza coverage among [European] children is either not known or quite low. Impact assessments in children are done in only a few countries,” said Dr. Nohynek, chief physician in the infectious diseases control and vaccinations unit of the National Institute for Health and Welfare in Helsinki, Finland.
“The only country doing as well coverage-wise as the U.S. is the U.K., with rates of 50%-65%. In Finland it’s less than 40%,” according to Dr. Nohynek.
“We have 28 countries today in the E.U. [European Union], and we have 28 different recommendations in Europe. So where do we go from here? It’s really not easy,” observed session cochair Alberticus Osterhaus, DVM, PhD, emeritus professor of virology at Erasmus University in Rotterdam, the Netherlands.
For all the oft-cited shortcomings of health care in the United States, That’s why Jon S. Abramson, MD, a former chair of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices (ACIP), was invited to explain how the U.S. strategy was accomplished.
The U.S. approach
The current U.S. policy, implemented in 2010, is to recommend an annual flu shot for all persons older than 6 months of age.
Influenza vaccination has been part of the U.S. public health program since 1960. Children aged 6-23 months, as well as their household contacts and women who will be pregnant during flu season, were added in 2004. In 2006, the flu vaccine recommendation was expanded to include children aged 6-59 months as a result of persuasive data showing that the rate of flu-associated hospitalizations and deaths in children up to 4 years old was second only to the rate in the elderly population.
The rationale for expanding the recommendation to include all school-age children and adolescents stemmed from evidence that the highest average flu-related illness rate in the United States was in that age group, which confirmed that schools are a powerful vector for the spread of influenza. Vaccinating this age-group also was seen as having an indirect benefit for their household contacts.
The current policy of recommending vaccination of everyone over age 6 months was adopted because it checked off a lot of boxes: “It’s a single recommendation, easy to apply; it eliminates the need to look for indications and risk factors; it increases vaccination coverage rates; annual vaccination is safe and effective; and flu-related morbidity and mortality occur in all age groups,” Dr. Abramson continued.
The rate of influenza vaccine coverage in pregnant women has improved over time from less than 15% to about 50%. To place that in perspective, however, the rate in Argentina is 95%, the pediatrician noted.
“We’re doing better in children than we are in adults in terms of seasonal coverage rates,” he added. “In 2015, it was 59%, versus 42% in adults.”
Dr. Abramson said there remains some skepticism in the United States regarding the effectiveness of flu vaccines in preventing flu-related illness. That’s because of the difficulty in communicating that vaccine effectiveness varies from year to year, sometimes substantially, depending upon two factors: the transmission characteristics of the circulating strains and how well the vaccines match up against those strains.
“I think we have to learn to live with that. I don’t think we’ll see a universal flu vaccine that we can give once every 10 years,” he said.
“The bottom line is, even if a vaccine is only 50% efficacious overall, we’re still impacting huge numbers,” the pediatrician added.
Dr. Abramson cited a CDC estimate that, for the 2012-2013 season, where the vaccine was 49% efficacious, the result of vaccination was 6.6 million fewer cases of influenza-associated illnesses nationally, 3.2 million fewer flu-associated medical visits, and 79,000 hospitalizations avoided.
“I think we have a fairly good program in the United States. We’re doing well in children. We certainly could be doing better. Not having FluMist for the past 2 seasons probably hurt us some,” according to Dr. Abramson.
The FluMist experience
The FluMist episode is viewed by many European pediatric infectious disease experts as a debacle. Europeans eager to develop a pan-European strategy for seasonal immunization against influenza in children and adolescents viewed the U.S. FluMist episode with dismay. For the 2016-2017 and 2017-2018 flu seasons, the ACIP recommended against FluMist, a previously approved intranasally administered quadrivalent live attenuated virus vaccine, on the basis of a single study showing subpar effectiveness against influenza A H1NI. Then at its October 2017 meeting, ACIP reversed itself and reinstated FluMist for the 2018-2019 season after viewing data from Finland and several other countries demonstrating that, in countries where it hadn’t been taken off the market, the vaccine had performed as well as injectable inactivated influenza vaccines in the 2016-2017 flu season.
“I think from the European side, it’s been a bit of a sorry spectacle,” commented Dr. Osterhaus, referring to the ACIP’s waffling. After all, authorities in Canada and European countries where FluMist was available had looked at the same data that caused ACIP to derecommend the vaccine but hadn’t found it convincing.
“We’re very happy to see ACIP has reinstated the vaccine,” Dr. Nohynek said.
Dr. Abramson declined to defend the ACIP decision to drop FluMist.
“From my standpoint, knowing that influenza B kills more children than A does, if I had been on the ACIP committee – and I’m not anymore – that would not have been my vote,” he said. “Whatever you want to say about the live attenuated influenza vaccine, about how good it is against some A strains or not, it’s better than other vaccines against influenza B. And the death rate is higher from B than A in children, although that is not true in adults.”
Plus, FluMist was an important option for people avoiding immunization because they dislike shots.
“The vast majority of deaths due to flu in children in 2010-2016 have been in kids who didn’t get vaccinated,” he noted.
Dr. Nohynek said the Finnish real-world experience recorded in comprehensive national registries for the 2017-2018 flu season – a bad year for vaccine/virus mismatch in Europe – confirmed Dr. Abramson’s comments about the superiority of quadrivalent live attenuated influenza vaccine against influenza B. Among 54,611 Finnish children aged 24-35 months, the laboratory-confirmed vaccine effectiveness of trivalent inactivated virus vaccine, with 9% coverage, was 4.5% for influenza A and 12.2% for influenza B. In contrast, the vaccine effectiveness for the intranasal quadrivalent live attenuated influenza vaccine was 32% for A and a whopping 80% for B.
“It’s quite amazing, at least to me, to see figures like this in real world data,” she commented.
Session cochair Adam Finn, MD, PhD, said he has found it instructive to take a closer look at the U.K. data for the past several flu seasons.
“We’ve seen greater control of the epidemic in Scotland and Northern Ireland, where coverage in primary school kids was higher, in the 60%-70% area, and lower in England and Wales, where it was more like 50%. So we’re beginning to think that’s the kind of level of annual coverage in children we might need to suppress an epidemic. I think that’s a really important message that people should understand: We’re not looking for 95% coverage,” observed Dr. Finn, aprofessor of pediatrics at the University of Bristol (England).
Vaccine effectiveness will improve
Dr. Osterhaus predicted better times are coming in terms of vaccine effectiveness. Vaccine production times will become shorter as recombinant technologies replace the traditional lengthy chicken egg-based vaccine production; as a result, there will be less drift-associated mismatch. Improved surveillance, including the ability to follow strain mobility patterns and population-based antibody landscapes, are another important advance.
“We’ve always been looking at one side of the coin: the virus. Once or twice a year eminent gray people sitting together in Geneva at WHO decide which strains should be selected for the next vaccine. But if you know what antibodies are present in the population, this can be quite important information as well,” he said.
Dr. Nohynek reported receiving research funding from GlaxoSmithKline and Pfizer. The other speakers reported having no relevant financial conflicts of interest.
EXPERT ANALYSIS FROM ESPID 2018
Former Smokers Motivate Quitters
In 2012, the CDC launched the “Tips from Former Smokers” campaign. It was memorable and emotionally forceful—one woman who had oral and throat cancer delivered her ad through an artificial voicebox—but did it have an impact on actual quitting rates?
No study had been done to assess the campaign’s combined, multiyear impact until CDC researchers looked at sustained (6 month) cigarette abstinence during the first 4 years of the campaign (2012-2015).
They found that the Tips campaign led to about 9.15 million total quit attempts. Based on an assumed 5.7% abstinence rate for people attempting to quit, this amounts to approximately 522,000 sustained quits.
The researchers say their findings indicate that the comprehensive approach combining evidence-based messages with the promotion of cessation resources was highly successful. Their finding of more than half-million sustained quits underscores the critical role of national tobacco education campaigns as a “counterpoint” to the substantial pro-tobacco advertising and promotion.
In 2012, the CDC launched the “Tips from Former Smokers” campaign. It was memorable and emotionally forceful—one woman who had oral and throat cancer delivered her ad through an artificial voicebox—but did it have an impact on actual quitting rates?
No study had been done to assess the campaign’s combined, multiyear impact until CDC researchers looked at sustained (6 month) cigarette abstinence during the first 4 years of the campaign (2012-2015).
They found that the Tips campaign led to about 9.15 million total quit attempts. Based on an assumed 5.7% abstinence rate for people attempting to quit, this amounts to approximately 522,000 sustained quits.
The researchers say their findings indicate that the comprehensive approach combining evidence-based messages with the promotion of cessation resources was highly successful. Their finding of more than half-million sustained quits underscores the critical role of national tobacco education campaigns as a “counterpoint” to the substantial pro-tobacco advertising and promotion.
In 2012, the CDC launched the “Tips from Former Smokers” campaign. It was memorable and emotionally forceful—one woman who had oral and throat cancer delivered her ad through an artificial voicebox—but did it have an impact on actual quitting rates?
No study had been done to assess the campaign’s combined, multiyear impact until CDC researchers looked at sustained (6 month) cigarette abstinence during the first 4 years of the campaign (2012-2015).
They found that the Tips campaign led to about 9.15 million total quit attempts. Based on an assumed 5.7% abstinence rate for people attempting to quit, this amounts to approximately 522,000 sustained quits.
The researchers say their findings indicate that the comprehensive approach combining evidence-based messages with the promotion of cessation resources was highly successful. Their finding of more than half-million sustained quits underscores the critical role of national tobacco education campaigns as a “counterpoint” to the substantial pro-tobacco advertising and promotion.
Pros and Cons of Telemental Health Care
When telemental health care (TMH) works, it works well, the research agrees. For rural patients who often do not have easy access to health care TMH can be a lifesaver. The VA uses TMH to deliver care to veterans in rural VA medical centers, community-based outpatient clinics, and residential areas. However, TMH is still relatively new in many rural communities, say researchers from University of Mississippi in Oxford and Augusta University in Georgia, and few studies have examined the delivery tool from an administrative standpoint. The literature suggests TMH will save money—the exploratory study, however, suggests otherwise.
The researchers interviewed 6 providers selected from the 15 community mental health (MH) centers (CMHCs) in rural Mississippi as well as an independent MH counselor who develops policy for the Mississippi Counselors Association. They asked respondents about the feasibility of TMH in the Mississippi Delta; in particular, the benefits, the costs, and the role of the state in facilitating the service. The researchers also collected data from a grant-funded pilot project conducted in the Mississippi Delta region by the Delta Health Alliance, a nonprofit organization in partnership with the University of Mississippi Medical Center, which ran from 2008 to 2011. Telepsychiatry sessions are not currently being used in the region, but before the project ended, it was responsible for > 1,000 videoconferencing clinical sessions.
The initial counseling sessions were “awkward” for some patients, the interviewees said, and some clients felt the consultation was “less personal.” Getting used to the technology may take some time. Once clients acclimated the feedback was positive.
The health care providers were concerned by not being able to observe in-person nonverbal clues, such as poor hygiene, that would normally help them evaluate the client’s health. The nurse at the CMHC helped fill a gap, the researchers say, created by technology.
The researchers determined that the benefit side was weighty: For instance, patients had better access to well-trained MH professionals and to the state hospital, and family could visit inpatients via videoconferencing. Staff had better access to professional development and training.
However, cost issues were a definite concern. The project would not have been feasible without grant funding, the researchers say. Medicaid reimburses for TMH services but not for technology setup costs and maintenance. Moreover, the interviews with administrators, the researchers say, indicated that TMH did not save the organization money. Costs for the equipment, installation, rent, and other supplies were prohibitive.
Although start-up costs are high, overall systematic costs go down, with savings on travel-related costs, including fewer missed appointments. Broadband technology, videoconferencing software, webcams, and education all take money. “If policymakers are serious” about TMH, the researchers conclude, they should allocate appropriate funding and resources.
Source:
Holland J, Hatcher W, Meares WL. J Health Hum Serv Adm. 2018;41(1):52-86.
When telemental health care (TMH) works, it works well, the research agrees. For rural patients who often do not have easy access to health care TMH can be a lifesaver. The VA uses TMH to deliver care to veterans in rural VA medical centers, community-based outpatient clinics, and residential areas. However, TMH is still relatively new in many rural communities, say researchers from University of Mississippi in Oxford and Augusta University in Georgia, and few studies have examined the delivery tool from an administrative standpoint. The literature suggests TMH will save money—the exploratory study, however, suggests otherwise.
The researchers interviewed 6 providers selected from the 15 community mental health (MH) centers (CMHCs) in rural Mississippi as well as an independent MH counselor who develops policy for the Mississippi Counselors Association. They asked respondents about the feasibility of TMH in the Mississippi Delta; in particular, the benefits, the costs, and the role of the state in facilitating the service. The researchers also collected data from a grant-funded pilot project conducted in the Mississippi Delta region by the Delta Health Alliance, a nonprofit organization in partnership with the University of Mississippi Medical Center, which ran from 2008 to 2011. Telepsychiatry sessions are not currently being used in the region, but before the project ended, it was responsible for > 1,000 videoconferencing clinical sessions.
The initial counseling sessions were “awkward” for some patients, the interviewees said, and some clients felt the consultation was “less personal.” Getting used to the technology may take some time. Once clients acclimated the feedback was positive.
The health care providers were concerned by not being able to observe in-person nonverbal clues, such as poor hygiene, that would normally help them evaluate the client’s health. The nurse at the CMHC helped fill a gap, the researchers say, created by technology.
The researchers determined that the benefit side was weighty: For instance, patients had better access to well-trained MH professionals and to the state hospital, and family could visit inpatients via videoconferencing. Staff had better access to professional development and training.
However, cost issues were a definite concern. The project would not have been feasible without grant funding, the researchers say. Medicaid reimburses for TMH services but not for technology setup costs and maintenance. Moreover, the interviews with administrators, the researchers say, indicated that TMH did not save the organization money. Costs for the equipment, installation, rent, and other supplies were prohibitive.
Although start-up costs are high, overall systematic costs go down, with savings on travel-related costs, including fewer missed appointments. Broadband technology, videoconferencing software, webcams, and education all take money. “If policymakers are serious” about TMH, the researchers conclude, they should allocate appropriate funding and resources.
Source:
Holland J, Hatcher W, Meares WL. J Health Hum Serv Adm. 2018;41(1):52-86.
When telemental health care (TMH) works, it works well, the research agrees. For rural patients who often do not have easy access to health care TMH can be a lifesaver. The VA uses TMH to deliver care to veterans in rural VA medical centers, community-based outpatient clinics, and residential areas. However, TMH is still relatively new in many rural communities, say researchers from University of Mississippi in Oxford and Augusta University in Georgia, and few studies have examined the delivery tool from an administrative standpoint. The literature suggests TMH will save money—the exploratory study, however, suggests otherwise.
The researchers interviewed 6 providers selected from the 15 community mental health (MH) centers (CMHCs) in rural Mississippi as well as an independent MH counselor who develops policy for the Mississippi Counselors Association. They asked respondents about the feasibility of TMH in the Mississippi Delta; in particular, the benefits, the costs, and the role of the state in facilitating the service. The researchers also collected data from a grant-funded pilot project conducted in the Mississippi Delta region by the Delta Health Alliance, a nonprofit organization in partnership with the University of Mississippi Medical Center, which ran from 2008 to 2011. Telepsychiatry sessions are not currently being used in the region, but before the project ended, it was responsible for > 1,000 videoconferencing clinical sessions.
The initial counseling sessions were “awkward” for some patients, the interviewees said, and some clients felt the consultation was “less personal.” Getting used to the technology may take some time. Once clients acclimated the feedback was positive.
The health care providers were concerned by not being able to observe in-person nonverbal clues, such as poor hygiene, that would normally help them evaluate the client’s health. The nurse at the CMHC helped fill a gap, the researchers say, created by technology.
The researchers determined that the benefit side was weighty: For instance, patients had better access to well-trained MH professionals and to the state hospital, and family could visit inpatients via videoconferencing. Staff had better access to professional development and training.
However, cost issues were a definite concern. The project would not have been feasible without grant funding, the researchers say. Medicaid reimburses for TMH services but not for technology setup costs and maintenance. Moreover, the interviews with administrators, the researchers say, indicated that TMH did not save the organization money. Costs for the equipment, installation, rent, and other supplies were prohibitive.
Although start-up costs are high, overall systematic costs go down, with savings on travel-related costs, including fewer missed appointments. Broadband technology, videoconferencing software, webcams, and education all take money. “If policymakers are serious” about TMH, the researchers conclude, they should allocate appropriate funding and resources.
Source:
Holland J, Hatcher W, Meares WL. J Health Hum Serv Adm. 2018;41(1):52-86.
Drug approved for radical cure of P vivax malaria
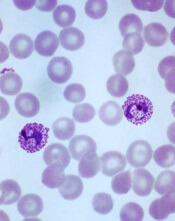
The US Food and Drug Administration (FDA) has approved tafenoquine (Krintafel) for the radical cure of Plasmodium vivax malaria.
Tafenoquine is a single-dose medicine that is now approved to prevent relapse of P vivax malaria in patients age 16 and older who are receiving appropriate antimalarial therapy for acute P vivax infection.
Tafenoquine is the first new treatment approved for P vivax malaria in more than 60 years.
Tafenoquine is an 8-aminoquinoline derivative with activity against all stages of the P vivax lifecycle, including hypnozoites. The product was first synthesized by scientists at the Walter Reed Army Institute of Research in 1978.
GSK began developing tafenoquine as a potential medicine for malaria more than 20 years ago. In 2008, GSK entered into a collaboration with Medicines for Malaria Venture to develop tafenoquine as an anti-relapse medicine for patients infected with P vivax.
The primary evidence for the clinical efficacy and safety of the 300 mg, single dose of tafenoquine was provided by a pair of phase 3 studies—DETECTIVE (NCT01376167, TAF112582) and GATHER (NCT02216123, TAF116564).
Results from these studies were presented at the 6th International Conference on Plasmodium vivax Research (ICPVR) in 2017 (abstract 63245 and abstract 63246).
DETECTIVE trial
In this double-blind, double-dummy study, researchers evaluated the efficacy, safety, and tolerability of tafenoquine. The trial included 522 patients with P vivax malaria who were randomized to receive one of the following:
- A single dose (1 day) of tafenoquine (300 mg)
- A 14-day course of primaquine (15 mg)
- Placebo.
All patients also received a 3-day course of chloroquine to treat the acute blood stage of the infection.
A significantly greater proportion of patients remained relapse-free over the 6-month follow-up period if they were treated with tafenoquine rather than placebo—60% and 26%, respectively—with an odds ratio for risk of relapse of 0.24 (P<0.001).
Likewise, a significantly greater proportion of patients were relapse-free when treated with primaquine rather than placebo—64% and 26%, respectively—with an odds ratio of 0.20 (P<0.001).
The frequency of adverse events (AEs) was 63% for the tafenoquine group, 59% for the primaquine group, and 65% for the placebo group. The frequency of serious AEs was 8%, 3%, and 5%, respectively.
GATHER trial
This study enrolled 251 patients, ages 16 and older, with microscopy-confirmed parasitemia.
Researchers compared how a single dose of tafenoquine (300 mg) and a 14-day course of primaquine (15 mg) affected hemoglobin levels in these patients. All patients also received a standard 3-day course of chloroquine.
The incidence of decline in hemoglobin (the primary endpoint) was similar between the 2 treatment groups—2.4% in the tafenoquine arm and 1.2% in the primaquine arm. The difference in proportion was 1.23% (95% CI, -4.16%, 4.98%).
None of the patients in this study required a blood transfusion.
The frequency of AEs was 72% for the tafenoquine group and 75% for the primaquine group. The frequency of serious AEs was 4% and 1%, respectively.
The US Food and Drug Administration (FDA) has approved tafenoquine (Krintafel) for the radical cure of Plasmodium vivax malaria.
Tafenoquine is a single-dose medicine that is now approved to prevent relapse of P vivax malaria in patients age 16 and older who are receiving appropriate antimalarial therapy for acute P vivax infection.
Tafenoquine is the first new treatment approved for P vivax malaria in more than 60 years.
Tafenoquine is an 8-aminoquinoline derivative with activity against all stages of the P vivax lifecycle, including hypnozoites. The product was first synthesized by scientists at the Walter Reed Army Institute of Research in 1978.
GSK began developing tafenoquine as a potential medicine for malaria more than 20 years ago. In 2008, GSK entered into a collaboration with Medicines for Malaria Venture to develop tafenoquine as an anti-relapse medicine for patients infected with P vivax.
The primary evidence for the clinical efficacy and safety of the 300 mg, single dose of tafenoquine was provided by a pair of phase 3 studies—DETECTIVE (NCT01376167, TAF112582) and GATHER (NCT02216123, TAF116564).
Results from these studies were presented at the 6th International Conference on Plasmodium vivax Research (ICPVR) in 2017 (abstract 63245 and abstract 63246).
DETECTIVE trial
In this double-blind, double-dummy study, researchers evaluated the efficacy, safety, and tolerability of tafenoquine. The trial included 522 patients with P vivax malaria who were randomized to receive one of the following:
- A single dose (1 day) of tafenoquine (300 mg)
- A 14-day course of primaquine (15 mg)
- Placebo.
All patients also received a 3-day course of chloroquine to treat the acute blood stage of the infection.
A significantly greater proportion of patients remained relapse-free over the 6-month follow-up period if they were treated with tafenoquine rather than placebo—60% and 26%, respectively—with an odds ratio for risk of relapse of 0.24 (P<0.001).
Likewise, a significantly greater proportion of patients were relapse-free when treated with primaquine rather than placebo—64% and 26%, respectively—with an odds ratio of 0.20 (P<0.001).
The frequency of adverse events (AEs) was 63% for the tafenoquine group, 59% for the primaquine group, and 65% for the placebo group. The frequency of serious AEs was 8%, 3%, and 5%, respectively.
GATHER trial
This study enrolled 251 patients, ages 16 and older, with microscopy-confirmed parasitemia.
Researchers compared how a single dose of tafenoquine (300 mg) and a 14-day course of primaquine (15 mg) affected hemoglobin levels in these patients. All patients also received a standard 3-day course of chloroquine.
The incidence of decline in hemoglobin (the primary endpoint) was similar between the 2 treatment groups—2.4% in the tafenoquine arm and 1.2% in the primaquine arm. The difference in proportion was 1.23% (95% CI, -4.16%, 4.98%).
None of the patients in this study required a blood transfusion.
The frequency of AEs was 72% for the tafenoquine group and 75% for the primaquine group. The frequency of serious AEs was 4% and 1%, respectively.
The US Food and Drug Administration (FDA) has approved tafenoquine (Krintafel) for the radical cure of Plasmodium vivax malaria.
Tafenoquine is a single-dose medicine that is now approved to prevent relapse of P vivax malaria in patients age 16 and older who are receiving appropriate antimalarial therapy for acute P vivax infection.
Tafenoquine is the first new treatment approved for P vivax malaria in more than 60 years.
Tafenoquine is an 8-aminoquinoline derivative with activity against all stages of the P vivax lifecycle, including hypnozoites. The product was first synthesized by scientists at the Walter Reed Army Institute of Research in 1978.
GSK began developing tafenoquine as a potential medicine for malaria more than 20 years ago. In 2008, GSK entered into a collaboration with Medicines for Malaria Venture to develop tafenoquine as an anti-relapse medicine for patients infected with P vivax.
The primary evidence for the clinical efficacy and safety of the 300 mg, single dose of tafenoquine was provided by a pair of phase 3 studies—DETECTIVE (NCT01376167, TAF112582) and GATHER (NCT02216123, TAF116564).
Results from these studies were presented at the 6th International Conference on Plasmodium vivax Research (ICPVR) in 2017 (abstract 63245 and abstract 63246).
DETECTIVE trial
In this double-blind, double-dummy study, researchers evaluated the efficacy, safety, and tolerability of tafenoquine. The trial included 522 patients with P vivax malaria who were randomized to receive one of the following:
- A single dose (1 day) of tafenoquine (300 mg)
- A 14-day course of primaquine (15 mg)
- Placebo.
All patients also received a 3-day course of chloroquine to treat the acute blood stage of the infection.
A significantly greater proportion of patients remained relapse-free over the 6-month follow-up period if they were treated with tafenoquine rather than placebo—60% and 26%, respectively—with an odds ratio for risk of relapse of 0.24 (P<0.001).
Likewise, a significantly greater proportion of patients were relapse-free when treated with primaquine rather than placebo—64% and 26%, respectively—with an odds ratio of 0.20 (P<0.001).
The frequency of adverse events (AEs) was 63% for the tafenoquine group, 59% for the primaquine group, and 65% for the placebo group. The frequency of serious AEs was 8%, 3%, and 5%, respectively.
GATHER trial
This study enrolled 251 patients, ages 16 and older, with microscopy-confirmed parasitemia.
Researchers compared how a single dose of tafenoquine (300 mg) and a 14-day course of primaquine (15 mg) affected hemoglobin levels in these patients. All patients also received a standard 3-day course of chloroquine.
The incidence of decline in hemoglobin (the primary endpoint) was similar between the 2 treatment groups—2.4% in the tafenoquine arm and 1.2% in the primaquine arm. The difference in proportion was 1.23% (95% CI, -4.16%, 4.98%).
None of the patients in this study required a blood transfusion.
The frequency of AEs was 72% for the tafenoquine group and 75% for the primaquine group. The frequency of serious AEs was 4% and 1%, respectively.
Team finds potential therapeutic targets for T-ALL

Researchers have found the NOTCH1 pathway “hijacks” heat shock transcription factor 1 (HSF1) signaling in T-cell acute lymphoblastic leukemia (T-ALL).
Therefore, blocking one or more genes in the HSF1 pathway could represent a new approach to treating T-ALL.
An experimental drug, PU-H71, is already in development against one of these targets, heat shock protein 90 (HSP90).
The researchers found that PU-H71 was active against T-ALL in vitro and in vivo.
The team recounted these findings in Nature Medicine.
“Our study shows how the NOTCH1 pathway hijacks the heat shock transcription factor 1 pathway to promote tumor growth,” said study author Iannis Aifantis, PhD, of NYU School of Medicine in New York, New York.
“The cancer cells are sending into overdrive a system that helps healthy cells respond to stress.”
Dr Aifantis and his colleagues found that HSF1 is involved in the pathogenesis of T-ALL. When they knocked down HSF1 in T-ALL cell lines, the researchers observed an increase in apoptosis, defective proteostasis, and a decrease in the growth of leukemic cells.
Similarly, HSF1 was deemed necessary for disease progression in mouse models of T-ALL. When the researchers deleted HSF1, mice experienced “striking” reductions in leukemic burden and “dramatic” improvements in survival. However, HSF1 deletion did not affect normal hematopoiesis.
Dr Aifantis and his colleagues also showed that NOTCH1 regulates the epichaperome in T-ALL. The team said previous studies have shown that, in the presence of oncogenic stress, heat shock proteins participate in a network nucleated by HSP90 and HSP70 chaperones—the epichaperome.
The researchers found that an intact epichaperome was critical for T-ALL by showing that pharmacologic inhibition of HSP90 and HSP70 significantly hindered the growth of human T-ALL in vitro. The team also found the HSP90 inhibitor PU-H71 reduced leukemic burden and extended survival in a NOTCH1-inducible T-ALL mouse model.
Then, the researchers found NOTCH1 levels could predict response to HSP90 inhibition in vitro. T-ALL patient samples expressing high levels of nuclear NOTCH1 and high levels of epichaperome were significantly more sensitive to treatment with PU-H71.
PU-H71 is already in early clinical trials of patients with breast cancer. If further testing proves successful, PU-H71 could be quickly adapted for trials in T-ALL patients, according to Dr Aifantis.
In the meantime, he and his colleagues plan to evaluate the effects of another 8 proteins produced by genes active in the HSF1 pathway to see if any show promising anticancer activity in T-ALL.
Researchers have found the NOTCH1 pathway “hijacks” heat shock transcription factor 1 (HSF1) signaling in T-cell acute lymphoblastic leukemia (T-ALL).
Therefore, blocking one or more genes in the HSF1 pathway could represent a new approach to treating T-ALL.
An experimental drug, PU-H71, is already in development against one of these targets, heat shock protein 90 (HSP90).
The researchers found that PU-H71 was active against T-ALL in vitro and in vivo.
The team recounted these findings in Nature Medicine.
“Our study shows how the NOTCH1 pathway hijacks the heat shock transcription factor 1 pathway to promote tumor growth,” said study author Iannis Aifantis, PhD, of NYU School of Medicine in New York, New York.
“The cancer cells are sending into overdrive a system that helps healthy cells respond to stress.”
Dr Aifantis and his colleagues found that HSF1 is involved in the pathogenesis of T-ALL. When they knocked down HSF1 in T-ALL cell lines, the researchers observed an increase in apoptosis, defective proteostasis, and a decrease in the growth of leukemic cells.
Similarly, HSF1 was deemed necessary for disease progression in mouse models of T-ALL. When the researchers deleted HSF1, mice experienced “striking” reductions in leukemic burden and “dramatic” improvements in survival. However, HSF1 deletion did not affect normal hematopoiesis.
Dr Aifantis and his colleagues also showed that NOTCH1 regulates the epichaperome in T-ALL. The team said previous studies have shown that, in the presence of oncogenic stress, heat shock proteins participate in a network nucleated by HSP90 and HSP70 chaperones—the epichaperome.
The researchers found that an intact epichaperome was critical for T-ALL by showing that pharmacologic inhibition of HSP90 and HSP70 significantly hindered the growth of human T-ALL in vitro. The team also found the HSP90 inhibitor PU-H71 reduced leukemic burden and extended survival in a NOTCH1-inducible T-ALL mouse model.
Then, the researchers found NOTCH1 levels could predict response to HSP90 inhibition in vitro. T-ALL patient samples expressing high levels of nuclear NOTCH1 and high levels of epichaperome were significantly more sensitive to treatment with PU-H71.
PU-H71 is already in early clinical trials of patients with breast cancer. If further testing proves successful, PU-H71 could be quickly adapted for trials in T-ALL patients, according to Dr Aifantis.
In the meantime, he and his colleagues plan to evaluate the effects of another 8 proteins produced by genes active in the HSF1 pathway to see if any show promising anticancer activity in T-ALL.
Researchers have found the NOTCH1 pathway “hijacks” heat shock transcription factor 1 (HSF1) signaling in T-cell acute lymphoblastic leukemia (T-ALL).
Therefore, blocking one or more genes in the HSF1 pathway could represent a new approach to treating T-ALL.
An experimental drug, PU-H71, is already in development against one of these targets, heat shock protein 90 (HSP90).
The researchers found that PU-H71 was active against T-ALL in vitro and in vivo.
The team recounted these findings in Nature Medicine.
“Our study shows how the NOTCH1 pathway hijacks the heat shock transcription factor 1 pathway to promote tumor growth,” said study author Iannis Aifantis, PhD, of NYU School of Medicine in New York, New York.
“The cancer cells are sending into overdrive a system that helps healthy cells respond to stress.”
Dr Aifantis and his colleagues found that HSF1 is involved in the pathogenesis of T-ALL. When they knocked down HSF1 in T-ALL cell lines, the researchers observed an increase in apoptosis, defective proteostasis, and a decrease in the growth of leukemic cells.
Similarly, HSF1 was deemed necessary for disease progression in mouse models of T-ALL. When the researchers deleted HSF1, mice experienced “striking” reductions in leukemic burden and “dramatic” improvements in survival. However, HSF1 deletion did not affect normal hematopoiesis.
Dr Aifantis and his colleagues also showed that NOTCH1 regulates the epichaperome in T-ALL. The team said previous studies have shown that, in the presence of oncogenic stress, heat shock proteins participate in a network nucleated by HSP90 and HSP70 chaperones—the epichaperome.
The researchers found that an intact epichaperome was critical for T-ALL by showing that pharmacologic inhibition of HSP90 and HSP70 significantly hindered the growth of human T-ALL in vitro. The team also found the HSP90 inhibitor PU-H71 reduced leukemic burden and extended survival in a NOTCH1-inducible T-ALL mouse model.
Then, the researchers found NOTCH1 levels could predict response to HSP90 inhibition in vitro. T-ALL patient samples expressing high levels of nuclear NOTCH1 and high levels of epichaperome were significantly more sensitive to treatment with PU-H71.
PU-H71 is already in early clinical trials of patients with breast cancer. If further testing proves successful, PU-H71 could be quickly adapted for trials in T-ALL patients, according to Dr Aifantis.
In the meantime, he and his colleagues plan to evaluate the effects of another 8 proteins produced by genes active in the HSF1 pathway to see if any show promising anticancer activity in T-ALL.
GPS receives fast track designation for MM
The US Food and Drug Administration (FDA) has granted fast track designation to the cancer vaccine galinpepimut-S (GPS) for the treatment of multiple myeloma (MM).
GPS consists of 4 modified peptide chains that induce an innate immune response (CD4+/CD8+ T cells) against the Wilms’ tumor 1 (WT1) antigen.
GPS is administered in combination with an adjuvant and an immune modulator to improve the immune response to the target.
GPS has been tested in a phase 2 trial of patients with MM. Results from this trial were presented at the 44th Annual Meeting of the EBMT in March.
Phase 2 trial
The study enrolled 19 MM patients. Fifteen of them had high-risk cytogenetics at diagnosis, and 18 were at least minimal residual disease-positive after autologous stem cell transplant (ASCT).
Patients began receiving GPS within 22 days of ASCT. Initially, they received 6 doses (administered subcutaneously with the oil emulsifier montanide) every 2 weeks. Injection sites were pre-stimulated with granulocyte-macrophage colony-stimulating factor (70 μg) on days -2 (± 1 day) and 0 of each GPS vaccination.
The patients underwent assessment 2 to 4 weeks after the 6th GPS dose. Then, they received 6 additional monthly doses of GPS in conjunction with lenalidomide maintenance (10 mg daily), starting on day 100 post-ASCT. Patients were assessed 2 to 4 weeks after the 12th GPS dose.
The researchers found that GPS stimulated time-dependent CD4+ or CD8+ T-cell immune responses specific for all 4 WT1 peptides within GPS, 2 of which are heteroclitic.
Immune responses were confirmed in up to 91% of patients, with multivalent immune responses in up to 64% of patients. Three-quarters of patients had multifunctional cross-epitope T-cell reactivity to antigenic epitopes against which the hosts were not specifically immunized, in a pattern akin to epitope spreading.
In patients who received all 12 doses of GPS (n=12), there was a “strong” association between clinical benefit—defined as complete response (CR) or very good partial response (VGPR)—and frequency of CD4/CD8 immune responses.
Of those patients who had achieved CR/VGPR upon completion of GPS treatment, 100% (n=11) had CD4 immune responses, and 81.8% (n=9) had CD8 immune responses.
The median progression-free survival was 23.6 months, and the median overall survival was not reached. At 18 months, the rate of progression-free survival was 62%, and the rate of overall survival was 88%.
About fast track designation
The FDA’s fast track development program is designed to expedite clinical development and submission of applications for products with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss the product’s development plan and written communications about issues such as trial design and use of biomarkers.
Products that receive fast track designation may be eligible for accelerated approval and priority review if relevant criteria are met. Such products may also be eligible for rolling review, which allows a developer to submit individual sections of a product’s application for review as they are ready, rather than waiting until all sections are complete.
The US Food and Drug Administration (FDA) has granted fast track designation to the cancer vaccine galinpepimut-S (GPS) for the treatment of multiple myeloma (MM).
GPS consists of 4 modified peptide chains that induce an innate immune response (CD4+/CD8+ T cells) against the Wilms’ tumor 1 (WT1) antigen.
GPS is administered in combination with an adjuvant and an immune modulator to improve the immune response to the target.
GPS has been tested in a phase 2 trial of patients with MM. Results from this trial were presented at the 44th Annual Meeting of the EBMT in March.
Phase 2 trial
The study enrolled 19 MM patients. Fifteen of them had high-risk cytogenetics at diagnosis, and 18 were at least minimal residual disease-positive after autologous stem cell transplant (ASCT).
Patients began receiving GPS within 22 days of ASCT. Initially, they received 6 doses (administered subcutaneously with the oil emulsifier montanide) every 2 weeks. Injection sites were pre-stimulated with granulocyte-macrophage colony-stimulating factor (70 μg) on days -2 (± 1 day) and 0 of each GPS vaccination.
The patients underwent assessment 2 to 4 weeks after the 6th GPS dose. Then, they received 6 additional monthly doses of GPS in conjunction with lenalidomide maintenance (10 mg daily), starting on day 100 post-ASCT. Patients were assessed 2 to 4 weeks after the 12th GPS dose.
The researchers found that GPS stimulated time-dependent CD4+ or CD8+ T-cell immune responses specific for all 4 WT1 peptides within GPS, 2 of which are heteroclitic.
Immune responses were confirmed in up to 91% of patients, with multivalent immune responses in up to 64% of patients. Three-quarters of patients had multifunctional cross-epitope T-cell reactivity to antigenic epitopes against which the hosts were not specifically immunized, in a pattern akin to epitope spreading.
In patients who received all 12 doses of GPS (n=12), there was a “strong” association between clinical benefit—defined as complete response (CR) or very good partial response (VGPR)—and frequency of CD4/CD8 immune responses.
Of those patients who had achieved CR/VGPR upon completion of GPS treatment, 100% (n=11) had CD4 immune responses, and 81.8% (n=9) had CD8 immune responses.
The median progression-free survival was 23.6 months, and the median overall survival was not reached. At 18 months, the rate of progression-free survival was 62%, and the rate of overall survival was 88%.
About fast track designation
The FDA’s fast track development program is designed to expedite clinical development and submission of applications for products with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss the product’s development plan and written communications about issues such as trial design and use of biomarkers.
Products that receive fast track designation may be eligible for accelerated approval and priority review if relevant criteria are met. Such products may also be eligible for rolling review, which allows a developer to submit individual sections of a product’s application for review as they are ready, rather than waiting until all sections are complete.
The US Food and Drug Administration (FDA) has granted fast track designation to the cancer vaccine galinpepimut-S (GPS) for the treatment of multiple myeloma (MM).
GPS consists of 4 modified peptide chains that induce an innate immune response (CD4+/CD8+ T cells) against the Wilms’ tumor 1 (WT1) antigen.
GPS is administered in combination with an adjuvant and an immune modulator to improve the immune response to the target.
GPS has been tested in a phase 2 trial of patients with MM. Results from this trial were presented at the 44th Annual Meeting of the EBMT in March.
Phase 2 trial
The study enrolled 19 MM patients. Fifteen of them had high-risk cytogenetics at diagnosis, and 18 were at least minimal residual disease-positive after autologous stem cell transplant (ASCT).
Patients began receiving GPS within 22 days of ASCT. Initially, they received 6 doses (administered subcutaneously with the oil emulsifier montanide) every 2 weeks. Injection sites were pre-stimulated with granulocyte-macrophage colony-stimulating factor (70 μg) on days -2 (± 1 day) and 0 of each GPS vaccination.
The patients underwent assessment 2 to 4 weeks after the 6th GPS dose. Then, they received 6 additional monthly doses of GPS in conjunction with lenalidomide maintenance (10 mg daily), starting on day 100 post-ASCT. Patients were assessed 2 to 4 weeks after the 12th GPS dose.
The researchers found that GPS stimulated time-dependent CD4+ or CD8+ T-cell immune responses specific for all 4 WT1 peptides within GPS, 2 of which are heteroclitic.
Immune responses were confirmed in up to 91% of patients, with multivalent immune responses in up to 64% of patients. Three-quarters of patients had multifunctional cross-epitope T-cell reactivity to antigenic epitopes against which the hosts were not specifically immunized, in a pattern akin to epitope spreading.
In patients who received all 12 doses of GPS (n=12), there was a “strong” association between clinical benefit—defined as complete response (CR) or very good partial response (VGPR)—and frequency of CD4/CD8 immune responses.
Of those patients who had achieved CR/VGPR upon completion of GPS treatment, 100% (n=11) had CD4 immune responses, and 81.8% (n=9) had CD8 immune responses.
The median progression-free survival was 23.6 months, and the median overall survival was not reached. At 18 months, the rate of progression-free survival was 62%, and the rate of overall survival was 88%.
About fast track designation
The FDA’s fast track development program is designed to expedite clinical development and submission of applications for products with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss the product’s development plan and written communications about issues such as trial design and use of biomarkers.
Products that receive fast track designation may be eligible for accelerated approval and priority review if relevant criteria are met. Such products may also be eligible for rolling review, which allows a developer to submit individual sections of a product’s application for review as they are ready, rather than waiting until all sections are complete.
Pregnancy and years of reproductive capability linked to dementia risk
CHICAGO – More pregnancies and a longer span of reproductive years appear to confer some level of protection against dementia, suggesting that lifetime estrogen exposure may be an important modulator of long-term cognitive health.
The study, presented at the Alzheimer’s Association International Conference, is the first to examine this question in a large cohort, comprising more than 14,500 women with up to 50 years of follow-up data. It is one of the first explorations in a renewed interest in the link between hormones and cognition, said Suzanne Craft, PhD, who moderated a press briefing on the topic.
“WHI [Women’s Heath Initiative] had a completely chilling effect on research into estrogen and cognition completely,” Dr. Craft of Wake Forest University, Winston-Salem, N.C., said in an interview. “Many well-regarded researchers simply couldn’t get funding and had to close their programs and move on to other areas. But now I think the pendulum is slowly moving back. It’s clear that something is going on, that there is a link. I’m glad we’re starting to explore this again.”
The investigation of women’s total reproductive experience affects dementia risk found several intriguing associations, said Paola Gilsanz, ScD, of Kaiser Permanente, Oakland, Calif. Earlier age of menarche, later menopause, and more completed pregnancies all independently reduced the risk of dementia in women.
Her study comprised 14,595 women, all members of the Kaiser Permanente health care database. All of them had completed a comprehensive health checkup sometime between 1964 and 1973, when they were 40-55 years old. They reported their number of miscarriages, number of children, ages at first and last menstrual period, and the total number of years in their reproductive period. Most of the group (68%) was white, but 16% were black, 6% Asian, and 5% Hispanic. Dr. Gilsanz looked at rates of dementia during 1996-2017, when the women were 62-86 years old.
A multivariate regression model controlled for age, race, education, midlife health issues (hypertension, smoking, and body mass index), hysterectomy, and late-life health issues (stroke, heart failure, and diabetes).
Half of the cohort (50%) had at least three children, and 75% had experienced at least one miscarriage. The average age at menarche was 13 years, and the average age at last natural menstrual period was 47 years. This equated to an average reproductive period of 34 years.
At the end of follow-up, 36% of the cohort had developed some type of dementia.
Women with at least three children were 12% less likely to develop dementia, compared with those with one child (hazard ratio, 0.88). The association remained significant even after researchers controlled for age, race, educational level, and hysterectomy.
Miscarriages also influenced the risk of dementia. Those who didn’t report one were 20% less likely to develop dementia than were those who had experienced at least one (HR, 0.81). The benefit of no miscarriage was even greater among women with at least three children, conferring a 28% reduced risk (HR, 0.72).
A shortened reproductive period increased the risk of dementia. Those who experienced menarche at 16 years or older had a 31% increased risk of dementia (HR, 1.31), and those who experienced their last period at age 45 or younger faced a 28% greater risk (HR, 1.28). Each additional year of reproductive capability was associated with a 2% decreased risk (HR, 0.98).
Women with a reproductive period of 21-30 years were 33% more likely to develop dementia than were those with a longer period (HR, 1.33)
“Reproductive events that signal different exposures to estrogen, like pregnancy and reproductive period may play a role in modulating dementia risk,” Dr. Gilsanz said. “Women who are less likely to have a miscarriage may have different hormonal milieu that may be neuroprotective. Underlying health conditions increasing the risk of miscarriages may also elevate risk of dementia.”
She had no financial disclosures.
SOURCE: Gilsanz P et al. AAIC 2018, Abstract P3-587
CHICAGO – More pregnancies and a longer span of reproductive years appear to confer some level of protection against dementia, suggesting that lifetime estrogen exposure may be an important modulator of long-term cognitive health.
The study, presented at the Alzheimer’s Association International Conference, is the first to examine this question in a large cohort, comprising more than 14,500 women with up to 50 years of follow-up data. It is one of the first explorations in a renewed interest in the link between hormones and cognition, said Suzanne Craft, PhD, who moderated a press briefing on the topic.
“WHI [Women’s Heath Initiative] had a completely chilling effect on research into estrogen and cognition completely,” Dr. Craft of Wake Forest University, Winston-Salem, N.C., said in an interview. “Many well-regarded researchers simply couldn’t get funding and had to close their programs and move on to other areas. But now I think the pendulum is slowly moving back. It’s clear that something is going on, that there is a link. I’m glad we’re starting to explore this again.”
The investigation of women’s total reproductive experience affects dementia risk found several intriguing associations, said Paola Gilsanz, ScD, of Kaiser Permanente, Oakland, Calif. Earlier age of menarche, later menopause, and more completed pregnancies all independently reduced the risk of dementia in women.
Her study comprised 14,595 women, all members of the Kaiser Permanente health care database. All of them had completed a comprehensive health checkup sometime between 1964 and 1973, when they were 40-55 years old. They reported their number of miscarriages, number of children, ages at first and last menstrual period, and the total number of years in their reproductive period. Most of the group (68%) was white, but 16% were black, 6% Asian, and 5% Hispanic. Dr. Gilsanz looked at rates of dementia during 1996-2017, when the women were 62-86 years old.
A multivariate regression model controlled for age, race, education, midlife health issues (hypertension, smoking, and body mass index), hysterectomy, and late-life health issues (stroke, heart failure, and diabetes).
Half of the cohort (50%) had at least three children, and 75% had experienced at least one miscarriage. The average age at menarche was 13 years, and the average age at last natural menstrual period was 47 years. This equated to an average reproductive period of 34 years.
At the end of follow-up, 36% of the cohort had developed some type of dementia.
Women with at least three children were 12% less likely to develop dementia, compared with those with one child (hazard ratio, 0.88). The association remained significant even after researchers controlled for age, race, educational level, and hysterectomy.
Miscarriages also influenced the risk of dementia. Those who didn’t report one were 20% less likely to develop dementia than were those who had experienced at least one (HR, 0.81). The benefit of no miscarriage was even greater among women with at least three children, conferring a 28% reduced risk (HR, 0.72).
A shortened reproductive period increased the risk of dementia. Those who experienced menarche at 16 years or older had a 31% increased risk of dementia (HR, 1.31), and those who experienced their last period at age 45 or younger faced a 28% greater risk (HR, 1.28). Each additional year of reproductive capability was associated with a 2% decreased risk (HR, 0.98).
Women with a reproductive period of 21-30 years were 33% more likely to develop dementia than were those with a longer period (HR, 1.33)
“Reproductive events that signal different exposures to estrogen, like pregnancy and reproductive period may play a role in modulating dementia risk,” Dr. Gilsanz said. “Women who are less likely to have a miscarriage may have different hormonal milieu that may be neuroprotective. Underlying health conditions increasing the risk of miscarriages may also elevate risk of dementia.”
She had no financial disclosures.
SOURCE: Gilsanz P et al. AAIC 2018, Abstract P3-587
CHICAGO – More pregnancies and a longer span of reproductive years appear to confer some level of protection against dementia, suggesting that lifetime estrogen exposure may be an important modulator of long-term cognitive health.
The study, presented at the Alzheimer’s Association International Conference, is the first to examine this question in a large cohort, comprising more than 14,500 women with up to 50 years of follow-up data. It is one of the first explorations in a renewed interest in the link between hormones and cognition, said Suzanne Craft, PhD, who moderated a press briefing on the topic.
“WHI [Women’s Heath Initiative] had a completely chilling effect on research into estrogen and cognition completely,” Dr. Craft of Wake Forest University, Winston-Salem, N.C., said in an interview. “Many well-regarded researchers simply couldn’t get funding and had to close their programs and move on to other areas. But now I think the pendulum is slowly moving back. It’s clear that something is going on, that there is a link. I’m glad we’re starting to explore this again.”
The investigation of women’s total reproductive experience affects dementia risk found several intriguing associations, said Paola Gilsanz, ScD, of Kaiser Permanente, Oakland, Calif. Earlier age of menarche, later menopause, and more completed pregnancies all independently reduced the risk of dementia in women.
Her study comprised 14,595 women, all members of the Kaiser Permanente health care database. All of them had completed a comprehensive health checkup sometime between 1964 and 1973, when they were 40-55 years old. They reported their number of miscarriages, number of children, ages at first and last menstrual period, and the total number of years in their reproductive period. Most of the group (68%) was white, but 16% were black, 6% Asian, and 5% Hispanic. Dr. Gilsanz looked at rates of dementia during 1996-2017, when the women were 62-86 years old.
A multivariate regression model controlled for age, race, education, midlife health issues (hypertension, smoking, and body mass index), hysterectomy, and late-life health issues (stroke, heart failure, and diabetes).
Half of the cohort (50%) had at least three children, and 75% had experienced at least one miscarriage. The average age at menarche was 13 years, and the average age at last natural menstrual period was 47 years. This equated to an average reproductive period of 34 years.
At the end of follow-up, 36% of the cohort had developed some type of dementia.
Women with at least three children were 12% less likely to develop dementia, compared with those with one child (hazard ratio, 0.88). The association remained significant even after researchers controlled for age, race, educational level, and hysterectomy.
Miscarriages also influenced the risk of dementia. Those who didn’t report one were 20% less likely to develop dementia than were those who had experienced at least one (HR, 0.81). The benefit of no miscarriage was even greater among women with at least three children, conferring a 28% reduced risk (HR, 0.72).
A shortened reproductive period increased the risk of dementia. Those who experienced menarche at 16 years or older had a 31% increased risk of dementia (HR, 1.31), and those who experienced their last period at age 45 or younger faced a 28% greater risk (HR, 1.28). Each additional year of reproductive capability was associated with a 2% decreased risk (HR, 0.98).
Women with a reproductive period of 21-30 years were 33% more likely to develop dementia than were those with a longer period (HR, 1.33)
“Reproductive events that signal different exposures to estrogen, like pregnancy and reproductive period may play a role in modulating dementia risk,” Dr. Gilsanz said. “Women who are less likely to have a miscarriage may have different hormonal milieu that may be neuroprotective. Underlying health conditions increasing the risk of miscarriages may also elevate risk of dementia.”
She had no financial disclosures.
SOURCE: Gilsanz P et al. AAIC 2018, Abstract P3-587
AT AAIC 2018
Key clinical point: More pregnancies and a longer reproductive period were associated with a significantly reduced risk of late-life dementia among women.
Major finding: Women with at least three children were 12% less likely to develop dementia than were those who had just one child.
Study details: The retrospective observational study comprised 14,595 women.
Disclosures: Dr. Gilsanz had no financial disclosures.
Source: Gilsanz P et al. AAIC 2018, Abstract P3-587.
Hypopigmentation on the Ear
The Diagnosis: Corticosteroid-Induced Hypopigmentation
This patient received several intralesional injections of triamcinolone acetonide once monthly for treatment of the keloid scar on the left ear at an outside institution. There was improvement in the size of the keloid over time. On physical examination during the most recent visit there was a prominent streak of hypopigmentation and atrophy near the corticosteroid injection site with extension to the postauricular region. There also was telangiectasia noted within the area of hypopigmentation. Intralesional triamcinolone injections were discontinued and the patient was advised to return for monitoring.
Intra-articular and intralesional corticosteroid injections frequently are used by clinicians. Cutaneous complications associated with these injections include atrophy, pigmentary changes, hypersensitivity reactions, flushing, cellulitis, and necrotizing fasciitis. Tendon rupture also has been reported.1
There are several case reports in the literature describing hypopigmentation and/or subcutaneous atrophy after intralesional or intra-articular corticosteroid injections. A variety of underlying conditions were treated including alopecia areata, keloids, rheumatoid arthritis, de Quervain tendonitis, and psoriasis.2-6 The lesions typically are described as linear rays of atrophy and hypopigmentation at or near the injection site, with some cases noting extension along lymph channels and proximal veins.4,6 There usually is no associated pruritus or pain.3 This phenomenon can be seen after single or multiple injections.4,6
Extension of hypopigmentation from the site of injection has been postulated to be due to venous or lymphatic uptake.2,4-6 The mechanism of hypopigmentation is not known. Biopsy of a previously described case showed intact melanocytes along the dermoepidermal junction.2 Biopsy from another case revealed a decrease in melanin staining, which suggests a decrease in number or activity of melanocytes.4 It was proposed that hypopigmentation was secondary to loss of melanocyte function instead of loss of melanocytes.2 Spontaneous improvement or resolution of the hypopigmentation were noted in some cases ranging from 1 month to 1 year after initial presentation, but the hypopigmentation also can be persistent.3-6
Hypopigmented sarcoidosis and hypopigmented mycosis fungoides, both often present on dark-skinned individuals, are included in the differential diagnosis. Hypopigmented sarcoidosis presents with hypopigmented macules or patches, some with central papules, and hypopigmented mycosis fungoides presents with hypopigmented patches or plaques with fine scale and onset often in childhood or adolescence.7,8 Morphea can present with an initial inflammatory stage that develops into a sclerotic firm plaque or nodule with hyperpigmentation or hypopigmentation.9 Vitiligo usually presents with depigmented macules or patches and depigmented hair within the lesion.10
- Brinks A, Koes BW, Volkers AC, et al. Adverse effects of extra-articular corticosteroid injections: a systematic review. BMC Musculoskelet Disord. 2010;11:206.
- Venkatesan P, Fangman WL. Linear hypopigmentation and cutaneous atrophy following intra-articular steroid injections for de Quervain's tendonitis. J Drugs Dermatol. 2009;8:492-493.
- Evans AV, McGibbon DH. Symmetrical hypopigmentation following triamcinolone injection for de Quervain's tenosynovitis. Clin Exp Dermatol. 2002;27:247-251.
- Friedman SJ, Butler DF, Pittelkow MR. Perilesional linear atrophy and hypopigmentation after intralesional corticosteroid therapy. report of two cases and review of the literature. J Am Acad Dermatol. 1988;19:537-541.
- van Vendeloo SN, Ettema HB. Skin depigmentation along lymph vessels of the lower leg following local corticosteroid injection for interdigital neuroma. Foot Ankle Surg. 2016;22:139-141.
- Kumar P, Adolph S. Hypopigmentation along subcutaneous veins following intrakeloid triamcinolone injection: a case report and review of literature. Burns. 1998;24:487-488.
- Elgart ML. Cutaneous sarcoidosis: definitions and types of lesions. Clin Dermatol. 1986;4:35-45.
- El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, et al. Hypopigmented mycosis fungoides: frequent expression of a CD8+ T-cell phenotype. Am J Surg Pathol. 2002;26:450-457.
- Marzano AV, Menni S, Parodi A, et al. Localized scleroderma in adults and children. clinical and laboratory investigations on 239 cases. Eur J Dermatol. 2003;13:171-176.
- Yaghoobi R, Omidian M, Bagherani N. Vitiligo: a review of the published work. J Dermatol. 2011;38:419-431.
The Diagnosis: Corticosteroid-Induced Hypopigmentation
This patient received several intralesional injections of triamcinolone acetonide once monthly for treatment of the keloid scar on the left ear at an outside institution. There was improvement in the size of the keloid over time. On physical examination during the most recent visit there was a prominent streak of hypopigmentation and atrophy near the corticosteroid injection site with extension to the postauricular region. There also was telangiectasia noted within the area of hypopigmentation. Intralesional triamcinolone injections were discontinued and the patient was advised to return for monitoring.
Intra-articular and intralesional corticosteroid injections frequently are used by clinicians. Cutaneous complications associated with these injections include atrophy, pigmentary changes, hypersensitivity reactions, flushing, cellulitis, and necrotizing fasciitis. Tendon rupture also has been reported.1
There are several case reports in the literature describing hypopigmentation and/or subcutaneous atrophy after intralesional or intra-articular corticosteroid injections. A variety of underlying conditions were treated including alopecia areata, keloids, rheumatoid arthritis, de Quervain tendonitis, and psoriasis.2-6 The lesions typically are described as linear rays of atrophy and hypopigmentation at or near the injection site, with some cases noting extension along lymph channels and proximal veins.4,6 There usually is no associated pruritus or pain.3 This phenomenon can be seen after single or multiple injections.4,6
Extension of hypopigmentation from the site of injection has been postulated to be due to venous or lymphatic uptake.2,4-6 The mechanism of hypopigmentation is not known. Biopsy of a previously described case showed intact melanocytes along the dermoepidermal junction.2 Biopsy from another case revealed a decrease in melanin staining, which suggests a decrease in number or activity of melanocytes.4 It was proposed that hypopigmentation was secondary to loss of melanocyte function instead of loss of melanocytes.2 Spontaneous improvement or resolution of the hypopigmentation were noted in some cases ranging from 1 month to 1 year after initial presentation, but the hypopigmentation also can be persistent.3-6
Hypopigmented sarcoidosis and hypopigmented mycosis fungoides, both often present on dark-skinned individuals, are included in the differential diagnosis. Hypopigmented sarcoidosis presents with hypopigmented macules or patches, some with central papules, and hypopigmented mycosis fungoides presents with hypopigmented patches or plaques with fine scale and onset often in childhood or adolescence.7,8 Morphea can present with an initial inflammatory stage that develops into a sclerotic firm plaque or nodule with hyperpigmentation or hypopigmentation.9 Vitiligo usually presents with depigmented macules or patches and depigmented hair within the lesion.10
The Diagnosis: Corticosteroid-Induced Hypopigmentation
This patient received several intralesional injections of triamcinolone acetonide once monthly for treatment of the keloid scar on the left ear at an outside institution. There was improvement in the size of the keloid over time. On physical examination during the most recent visit there was a prominent streak of hypopigmentation and atrophy near the corticosteroid injection site with extension to the postauricular region. There also was telangiectasia noted within the area of hypopigmentation. Intralesional triamcinolone injections were discontinued and the patient was advised to return for monitoring.
Intra-articular and intralesional corticosteroid injections frequently are used by clinicians. Cutaneous complications associated with these injections include atrophy, pigmentary changes, hypersensitivity reactions, flushing, cellulitis, and necrotizing fasciitis. Tendon rupture also has been reported.1
There are several case reports in the literature describing hypopigmentation and/or subcutaneous atrophy after intralesional or intra-articular corticosteroid injections. A variety of underlying conditions were treated including alopecia areata, keloids, rheumatoid arthritis, de Quervain tendonitis, and psoriasis.2-6 The lesions typically are described as linear rays of atrophy and hypopigmentation at or near the injection site, with some cases noting extension along lymph channels and proximal veins.4,6 There usually is no associated pruritus or pain.3 This phenomenon can be seen after single or multiple injections.4,6
Extension of hypopigmentation from the site of injection has been postulated to be due to venous or lymphatic uptake.2,4-6 The mechanism of hypopigmentation is not known. Biopsy of a previously described case showed intact melanocytes along the dermoepidermal junction.2 Biopsy from another case revealed a decrease in melanin staining, which suggests a decrease in number or activity of melanocytes.4 It was proposed that hypopigmentation was secondary to loss of melanocyte function instead of loss of melanocytes.2 Spontaneous improvement or resolution of the hypopigmentation were noted in some cases ranging from 1 month to 1 year after initial presentation, but the hypopigmentation also can be persistent.3-6
Hypopigmented sarcoidosis and hypopigmented mycosis fungoides, both often present on dark-skinned individuals, are included in the differential diagnosis. Hypopigmented sarcoidosis presents with hypopigmented macules or patches, some with central papules, and hypopigmented mycosis fungoides presents with hypopigmented patches or plaques with fine scale and onset often in childhood or adolescence.7,8 Morphea can present with an initial inflammatory stage that develops into a sclerotic firm plaque or nodule with hyperpigmentation or hypopigmentation.9 Vitiligo usually presents with depigmented macules or patches and depigmented hair within the lesion.10
- Brinks A, Koes BW, Volkers AC, et al. Adverse effects of extra-articular corticosteroid injections: a systematic review. BMC Musculoskelet Disord. 2010;11:206.
- Venkatesan P, Fangman WL. Linear hypopigmentation and cutaneous atrophy following intra-articular steroid injections for de Quervain's tendonitis. J Drugs Dermatol. 2009;8:492-493.
- Evans AV, McGibbon DH. Symmetrical hypopigmentation following triamcinolone injection for de Quervain's tenosynovitis. Clin Exp Dermatol. 2002;27:247-251.
- Friedman SJ, Butler DF, Pittelkow MR. Perilesional linear atrophy and hypopigmentation after intralesional corticosteroid therapy. report of two cases and review of the literature. J Am Acad Dermatol. 1988;19:537-541.
- van Vendeloo SN, Ettema HB. Skin depigmentation along lymph vessels of the lower leg following local corticosteroid injection for interdigital neuroma. Foot Ankle Surg. 2016;22:139-141.
- Kumar P, Adolph S. Hypopigmentation along subcutaneous veins following intrakeloid triamcinolone injection: a case report and review of literature. Burns. 1998;24:487-488.
- Elgart ML. Cutaneous sarcoidosis: definitions and types of lesions. Clin Dermatol. 1986;4:35-45.
- El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, et al. Hypopigmented mycosis fungoides: frequent expression of a CD8+ T-cell phenotype. Am J Surg Pathol. 2002;26:450-457.
- Marzano AV, Menni S, Parodi A, et al. Localized scleroderma in adults and children. clinical and laboratory investigations on 239 cases. Eur J Dermatol. 2003;13:171-176.
- Yaghoobi R, Omidian M, Bagherani N. Vitiligo: a review of the published work. J Dermatol. 2011;38:419-431.
- Brinks A, Koes BW, Volkers AC, et al. Adverse effects of extra-articular corticosteroid injections: a systematic review. BMC Musculoskelet Disord. 2010;11:206.
- Venkatesan P, Fangman WL. Linear hypopigmentation and cutaneous atrophy following intra-articular steroid injections for de Quervain's tendonitis. J Drugs Dermatol. 2009;8:492-493.
- Evans AV, McGibbon DH. Symmetrical hypopigmentation following triamcinolone injection for de Quervain's tenosynovitis. Clin Exp Dermatol. 2002;27:247-251.
- Friedman SJ, Butler DF, Pittelkow MR. Perilesional linear atrophy and hypopigmentation after intralesional corticosteroid therapy. report of two cases and review of the literature. J Am Acad Dermatol. 1988;19:537-541.
- van Vendeloo SN, Ettema HB. Skin depigmentation along lymph vessels of the lower leg following local corticosteroid injection for interdigital neuroma. Foot Ankle Surg. 2016;22:139-141.
- Kumar P, Adolph S. Hypopigmentation along subcutaneous veins following intrakeloid triamcinolone injection: a case report and review of literature. Burns. 1998;24:487-488.
- Elgart ML. Cutaneous sarcoidosis: definitions and types of lesions. Clin Dermatol. 1986;4:35-45.
- El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, et al. Hypopigmented mycosis fungoides: frequent expression of a CD8+ T-cell phenotype. Am J Surg Pathol. 2002;26:450-457.
- Marzano AV, Menni S, Parodi A, et al. Localized scleroderma in adults and children. clinical and laboratory investigations on 239 cases. Eur J Dermatol. 2003;13:171-176.
- Yaghoobi R, Omidian M, Bagherani N. Vitiligo: a review of the published work. J Dermatol. 2011;38:419-431.
A 20-year-old black woman underwent multiple intralesional corticosteroid injections for treatment of a keloid on the superior aspect of the left helix and subsequently presented with a streak of atrophy and hypopigmentation in the postauricular region of unknown duration due to the lesion location.
FDA: Krintafel approved as ‘radical cure’ for preventing malaria relapse
The Food and Drug Administration has approved, under priority review, single-dose tafenoquine (to be marketed as Krintafel) for the prevention of relapse in patients aged 16 years and older who are receiving appropriate antimalarial therapy for acute Plasmodium vivax infection, according to an announcement by research partners GSK and the nonprofit Medicines for Malaria Venture.
Clinical efficacy and safety of the 300-mg single-dose tablet was provided by three randomized, double-blind studies: DETECTIVE Part 1 and Part 2 (TAF112582) and GATHER (TAF116564). The results of the two phase III studies were announced in June 2017.
Tafenoquine is referred to as a “radical cure,” because it targets the dormant liver forms of P. vivax and is coadministered with currently available antimalarials such as chloroquine or artemisinin-based combination therapies.
“The world has waited decades for a new medicine to counter P. vivax malaria relapse. Today, we can say the wait is over. Moreover, as the first ever single-dose for this indication, Krintafel will help improve patient compliance,” David Reddy, MD, CEO of MMV stated.
The FDA approval letter and the Krintafel label information are available online.
The Food and Drug Administration has approved, under priority review, single-dose tafenoquine (to be marketed as Krintafel) for the prevention of relapse in patients aged 16 years and older who are receiving appropriate antimalarial therapy for acute Plasmodium vivax infection, according to an announcement by research partners GSK and the nonprofit Medicines for Malaria Venture.
Clinical efficacy and safety of the 300-mg single-dose tablet was provided by three randomized, double-blind studies: DETECTIVE Part 1 and Part 2 (TAF112582) and GATHER (TAF116564). The results of the two phase III studies were announced in June 2017.
Tafenoquine is referred to as a “radical cure,” because it targets the dormant liver forms of P. vivax and is coadministered with currently available antimalarials such as chloroquine or artemisinin-based combination therapies.
“The world has waited decades for a new medicine to counter P. vivax malaria relapse. Today, we can say the wait is over. Moreover, as the first ever single-dose for this indication, Krintafel will help improve patient compliance,” David Reddy, MD, CEO of MMV stated.
The FDA approval letter and the Krintafel label information are available online.
The Food and Drug Administration has approved, under priority review, single-dose tafenoquine (to be marketed as Krintafel) for the prevention of relapse in patients aged 16 years and older who are receiving appropriate antimalarial therapy for acute Plasmodium vivax infection, according to an announcement by research partners GSK and the nonprofit Medicines for Malaria Venture.
Clinical efficacy and safety of the 300-mg single-dose tablet was provided by three randomized, double-blind studies: DETECTIVE Part 1 and Part 2 (TAF112582) and GATHER (TAF116564). The results of the two phase III studies were announced in June 2017.
Tafenoquine is referred to as a “radical cure,” because it targets the dormant liver forms of P. vivax and is coadministered with currently available antimalarials such as chloroquine or artemisinin-based combination therapies.
“The world has waited decades for a new medicine to counter P. vivax malaria relapse. Today, we can say the wait is over. Moreover, as the first ever single-dose for this indication, Krintafel will help improve patient compliance,” David Reddy, MD, CEO of MMV stated.
The FDA approval letter and the Krintafel label information are available online.