User login
Are There Differences in Efficacy and Safety Between 2nd-Generation Drug-Eluting Stents for Left Main Coronary Intervention?
Study Overview
Objective. To compare the effectiveness and safety profiles of various second-generation drug-eluting stents (DES) for left main coronary intervention.
Design. Retrospective study using 3 multicenter prospective registries (IRIS-DES, IRIS-MAIN, PRECOMBAT).
Setting and participants. Among the 4470 patients enrolled in the 3 registries treated between July 2007 and July 2015, the authors identified 2692 patients with significant left main coronary artery disease who received second-generation DES for inclusion in the study. The centers for IRIS-DES and PRECOMBAT are academic and community hospitals in South Korea, with IRIS-MAIN involving academic and community hospitals in South Korea, China, India, Indonesia, Japan, Malaysia, Taiwan, and Thailand. Of the patients in these registries, 1254 received cobalt-chromium everolimus-eluting stents (CoCr-EES), 232 biodegradable polymer biolimus-eluting stents (BP-BES), 616 platinum-chromium EES (PtCr-EES) and 590 Resolute zotarolimus-eluting stents (Re-ZES).
Main outcome measure. Target-vessel failure.
Main results. At 3 years, rates of target-vessel failure were not significantly different for the different types of stents (16.7% for the CoCr-EES, 13.2% for the BP-BES, 18.7% for the PtCr-EES, and 14.7% for the Re-ZES; P = 0.15). The adjusted hazard ratios (HRs) for target-vessel failure were similar in between-group comparisons of the different stents, except for the PtCr-EES versus the BP-BES (HR 1.60, 95% confidence interval 1.01 to 2.54; P = 0.046). There were no significant differences in risk of composite of all-cause death, any myocardial infarction, or any revascularization and its individual components according to the different types of stents.
Conclusion. There was no significant between-group differences in 3-year risk of target-vessel failure, except for a higher risk of primary outcome with PtCr-EES compared to BP-BES.
Commentary
Left main coronary artery disease is identified in 5% to 7% of the population and is one of the more perplexing lesions to treat given the poorer outcome compared to non–left main lesion and the importance of the vessels the left main supplies [1]. Historically, coronary artery bypass grafting (CABG) has been the standard of care on the basis of the survival benefit observed in early trials compared with medical therapy. Left main percutaneous coronary intervention (PCI) has evolved as an alternative to CABG over the past few decades. Early studies using balloon angioplasty or bare metal stents were limited primarily due to high restenosis rate [1]. In the DES era, results have been overall comparable to CABG. Unprotected left main PCI using first-generation DES was non-inferior compared to CABG in the pre-specified sub-study of SYNTAX trial and in PRECOMBAT trial using paclitaxel-eluting stents and sirolimus-eluting stents, respectively [2,3]. Largely based on these trials, the 2014 ACC/AHA guidelines give class IIa recommendation for patients with low-risk anatomy (Syntax score 0–22) and class IIb recommendation for patients with intermediate-risk anatomy (Syntax score 23–32) for left main PCI [4]. Moreover, European guidelines give class Ib recommendation for patients with low-risk anatomy, and class IIa recommendation for intermediate-risk anatomy for left main PCI [5]. However, the SYNTAX trial and PRECOMBAT trial were limited by not meeting non-inferiority (SYNTAX) and wide non-inferiority (PRECOMBAT) and selection bias due to large exclusion criteria. In addition, first-generation DES were used in these trials (tacrolimis-eluting stent for SYNTAX and sirolimus-eluting stent for PRECOMBAT). The standard of care has now shifted to wide use of second-generation DES [1].
Subsequently, 2 larger-scale clinical trials using second-generation DES were designed and results have been reported recently [6,7]. The EXCEL trial enrolled 1905 patients with significant left main coronary disease and compared CoCr-EES to CABG. At 3 years, the primary endpoint of a composite of death from any cause, stroke, or myocardial infarction occurred in 15.4% of the PCI patients and in 14.7% of the CABG patients (P = 0.02 for non-inferiority; P = 0.98 for superiority). Similarly, the NOBLE trial enrolled 1201 patients with significant left main coronary disease and compared PCI to CABG. In this trial, the biolimus-eluting second-generation stent became their preferred stent during the study period. At 5 years, the primary endpoint of a composite of all-cause mortality, non-procedural myocardial infarction, any repeat coronary intervention, and stroke was higher in PCI compared to CABG patients (28% vs 18%, HR 1.51, 95% CI 1.13–2.00), exceeding the limit of non-inferiority, and CABG was significantly better compared to PCI (P = 0.004). The difference in the results is likely due to trial design. The primary endpoint was different in the 2 studies—EXCEL did not include repeat coronary intervention in the composite endpoint. The NOBLE study had a longer enrollment period and earlier-generation stents (sirolimus-eluting) were used in the earlier stages of the trial. In addition, the NOBLE study did not assess for peri-procedural myocardial infarction as an endpoint, which is known to be associated with adverse outcome. In both trials, cardiovascular mortality and all-cause mortality were similar at the end of follow-up.
In this context, the Lee et al study compared 4 types of currently available second-generation stents by pooling data from 3 large registries in Asia [8]. The main finding from this study was that target-vessel failure, defined as the composite of cardiac death, target-vessel myocardial infarction, or target-vessel revascularization at 3 years follow-up was not different among the types of second-generation drug eluting stents (P = 0.15).
Another important finding from this study was that the stent thrombosis rate at follow-up was very low (< 1%). This is consistent with the EXCEL study, which reported a definite stent thrombosis rate of 0.7% and was lower than in the NOBLE study, which reported a rate of 3%. One of the possible explanations for this difference could be stent selection. In contrast to the EXCEL study, which exclusively used Co-Cr EES by study protocol, NOBLE
study included first-generation sirolimus-drug eluting stent (11%) and BP-BES (89%). However, there are multiple factors that contribute to stent thrombosis other than stent selection, such as lesion characteristics, adequate stent expansion, and use of dual antiplatelet therapy [9].
The observed finding of small increase in target-vessel failure in PtCr-EES versus the BP-BES needs to be interpreted with caution. First, this was an observational study, and the treatment strategy or choice of stent was determined by a local interventional cardiologist, which could lead to selection bias. Although the authors performed propensity analysis, residual cofounding is likely. Second, since there was no difference in the primary analysis, the subgroup analysis becomes less important. In addition, authors did not perform statistical correction for multiple comparisons.
Despite the above limitations, this large-scale observational study gives us important insights to the performance of each second-generation DES. All currently available second-generation DES appear to be an option for use for left main coronary intervention.
Applications for Clinical Practice
In patients presenting with significant left main disease, left main PCI using a contemporary second-generation stent is safe and effective and likely has equivalent outcomes to CABG. However, PCI may be associated with higher rate of repeat revascularization. The rate of target-vessel failure was similar between different types of second-generation DES.
—Taishi Hirai, MD, and John E.A. Blair, MD, University of Chicago Medical Center, Chicago, IL
1. Rab T, Sheiban I, Louvard Y, et al. Current interventions for the left main bifurcation. JACC Cardiovasc Interv 2017;10:849–65.
2. Morice MC, Serruys PW, Kappetein AP, et al. Outcomes in patients with de novo left main disease treated with either percutaneous coronary intervention using paclitaxel-eluting stents or coronary artery bypass graft treatment in the Synergy Between Percutaneous Coronary Intervention with TAXUS and Cardiac Surgery (SYNTAX) trial. Circulation 2010;121:2645–53.
3. Park SJ, Kim YH, Park DW, et al. Randomized trial of stents versus bypass surgery for left main coronary artery disease. N Engl J Med 2011;364:1718–27.
4. Fihn SD, Blankenship JC, Alexander KP, et al. 2014 ACC/AHA/AATS/PCNA/SCAI/STS focused update of the guideline for the diagnosis and management of patients with stable ischemic heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines, and the American Association for Thoracic Surgery, Preventive Cardiovascular Nurses Association, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol 2014;64:1929–49.
5. Windecker S, Kolh P, Alfonso F, et al. 2014 ESC/EACTS Guidelines on myocardial revascularization: The Task Force on Myocardial Revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS)Developed with the special contribution of the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur Heart J 2014;35:2541–619.
6. Stone GW, Sabik JF, Serruys PW, et al. Everolimus-eluting stents or bypass surgery for left main coronary artery disease. N Engl J Med 2016;375:2223–35.
7. Mäkikallio T, Holm NR, Lindsay M, et al. Percutaneous coronary angioplasty versus coronary artery bypass grafting in treatment of unprotected left main stenosis (NOBLE): a prospective, randomised, open-label, non-inferiority trial. Lancet 2016;388:2743–52.
8. Lee PH, Kwon O, Ahn JM, et al. Safety and effectiveness of second-generation drug-eluting stents in patients with left main coronary artery disease. J Am Coll Cardiol 2018;71:832–41.
9. Claessen BE, Henriques JP, Jaffer FA, et al. Stent thrombosis: a clinical perspective. JACC Cardiovasc Interv 2014;7:1081–92.
Study Overview
Objective. To compare the effectiveness and safety profiles of various second-generation drug-eluting stents (DES) for left main coronary intervention.
Design. Retrospective study using 3 multicenter prospective registries (IRIS-DES, IRIS-MAIN, PRECOMBAT).
Setting and participants. Among the 4470 patients enrolled in the 3 registries treated between July 2007 and July 2015, the authors identified 2692 patients with significant left main coronary artery disease who received second-generation DES for inclusion in the study. The centers for IRIS-DES and PRECOMBAT are academic and community hospitals in South Korea, with IRIS-MAIN involving academic and community hospitals in South Korea, China, India, Indonesia, Japan, Malaysia, Taiwan, and Thailand. Of the patients in these registries, 1254 received cobalt-chromium everolimus-eluting stents (CoCr-EES), 232 biodegradable polymer biolimus-eluting stents (BP-BES), 616 platinum-chromium EES (PtCr-EES) and 590 Resolute zotarolimus-eluting stents (Re-ZES).
Main outcome measure. Target-vessel failure.
Main results. At 3 years, rates of target-vessel failure were not significantly different for the different types of stents (16.7% for the CoCr-EES, 13.2% for the BP-BES, 18.7% for the PtCr-EES, and 14.7% for the Re-ZES; P = 0.15). The adjusted hazard ratios (HRs) for target-vessel failure were similar in between-group comparisons of the different stents, except for the PtCr-EES versus the BP-BES (HR 1.60, 95% confidence interval 1.01 to 2.54; P = 0.046). There were no significant differences in risk of composite of all-cause death, any myocardial infarction, or any revascularization and its individual components according to the different types of stents.
Conclusion. There was no significant between-group differences in 3-year risk of target-vessel failure, except for a higher risk of primary outcome with PtCr-EES compared to BP-BES.
Commentary
Left main coronary artery disease is identified in 5% to 7% of the population and is one of the more perplexing lesions to treat given the poorer outcome compared to non–left main lesion and the importance of the vessels the left main supplies [1]. Historically, coronary artery bypass grafting (CABG) has been the standard of care on the basis of the survival benefit observed in early trials compared with medical therapy. Left main percutaneous coronary intervention (PCI) has evolved as an alternative to CABG over the past few decades. Early studies using balloon angioplasty or bare metal stents were limited primarily due to high restenosis rate [1]. In the DES era, results have been overall comparable to CABG. Unprotected left main PCI using first-generation DES was non-inferior compared to CABG in the pre-specified sub-study of SYNTAX trial and in PRECOMBAT trial using paclitaxel-eluting stents and sirolimus-eluting stents, respectively [2,3]. Largely based on these trials, the 2014 ACC/AHA guidelines give class IIa recommendation for patients with low-risk anatomy (Syntax score 0–22) and class IIb recommendation for patients with intermediate-risk anatomy (Syntax score 23–32) for left main PCI [4]. Moreover, European guidelines give class Ib recommendation for patients with low-risk anatomy, and class IIa recommendation for intermediate-risk anatomy for left main PCI [5]. However, the SYNTAX trial and PRECOMBAT trial were limited by not meeting non-inferiority (SYNTAX) and wide non-inferiority (PRECOMBAT) and selection bias due to large exclusion criteria. In addition, first-generation DES were used in these trials (tacrolimis-eluting stent for SYNTAX and sirolimus-eluting stent for PRECOMBAT). The standard of care has now shifted to wide use of second-generation DES [1].
Subsequently, 2 larger-scale clinical trials using second-generation DES were designed and results have been reported recently [6,7]. The EXCEL trial enrolled 1905 patients with significant left main coronary disease and compared CoCr-EES to CABG. At 3 years, the primary endpoint of a composite of death from any cause, stroke, or myocardial infarction occurred in 15.4% of the PCI patients and in 14.7% of the CABG patients (P = 0.02 for non-inferiority; P = 0.98 for superiority). Similarly, the NOBLE trial enrolled 1201 patients with significant left main coronary disease and compared PCI to CABG. In this trial, the biolimus-eluting second-generation stent became their preferred stent during the study period. At 5 years, the primary endpoint of a composite of all-cause mortality, non-procedural myocardial infarction, any repeat coronary intervention, and stroke was higher in PCI compared to CABG patients (28% vs 18%, HR 1.51, 95% CI 1.13–2.00), exceeding the limit of non-inferiority, and CABG was significantly better compared to PCI (P = 0.004). The difference in the results is likely due to trial design. The primary endpoint was different in the 2 studies—EXCEL did not include repeat coronary intervention in the composite endpoint. The NOBLE study had a longer enrollment period and earlier-generation stents (sirolimus-eluting) were used in the earlier stages of the trial. In addition, the NOBLE study did not assess for peri-procedural myocardial infarction as an endpoint, which is known to be associated with adverse outcome. In both trials, cardiovascular mortality and all-cause mortality were similar at the end of follow-up.
In this context, the Lee et al study compared 4 types of currently available second-generation stents by pooling data from 3 large registries in Asia [8]. The main finding from this study was that target-vessel failure, defined as the composite of cardiac death, target-vessel myocardial infarction, or target-vessel revascularization at 3 years follow-up was not different among the types of second-generation drug eluting stents (P = 0.15).
Another important finding from this study was that the stent thrombosis rate at follow-up was very low (< 1%). This is consistent with the EXCEL study, which reported a definite stent thrombosis rate of 0.7% and was lower than in the NOBLE study, which reported a rate of 3%. One of the possible explanations for this difference could be stent selection. In contrast to the EXCEL study, which exclusively used Co-Cr EES by study protocol, NOBLE
study included first-generation sirolimus-drug eluting stent (11%) and BP-BES (89%). However, there are multiple factors that contribute to stent thrombosis other than stent selection, such as lesion characteristics, adequate stent expansion, and use of dual antiplatelet therapy [9].
The observed finding of small increase in target-vessel failure in PtCr-EES versus the BP-BES needs to be interpreted with caution. First, this was an observational study, and the treatment strategy or choice of stent was determined by a local interventional cardiologist, which could lead to selection bias. Although the authors performed propensity analysis, residual cofounding is likely. Second, since there was no difference in the primary analysis, the subgroup analysis becomes less important. In addition, authors did not perform statistical correction for multiple comparisons.
Despite the above limitations, this large-scale observational study gives us important insights to the performance of each second-generation DES. All currently available second-generation DES appear to be an option for use for left main coronary intervention.
Applications for Clinical Practice
In patients presenting with significant left main disease, left main PCI using a contemporary second-generation stent is safe and effective and likely has equivalent outcomes to CABG. However, PCI may be associated with higher rate of repeat revascularization. The rate of target-vessel failure was similar between different types of second-generation DES.
—Taishi Hirai, MD, and John E.A. Blair, MD, University of Chicago Medical Center, Chicago, IL
Study Overview
Objective. To compare the effectiveness and safety profiles of various second-generation drug-eluting stents (DES) for left main coronary intervention.
Design. Retrospective study using 3 multicenter prospective registries (IRIS-DES, IRIS-MAIN, PRECOMBAT).
Setting and participants. Among the 4470 patients enrolled in the 3 registries treated between July 2007 and July 2015, the authors identified 2692 patients with significant left main coronary artery disease who received second-generation DES for inclusion in the study. The centers for IRIS-DES and PRECOMBAT are academic and community hospitals in South Korea, with IRIS-MAIN involving academic and community hospitals in South Korea, China, India, Indonesia, Japan, Malaysia, Taiwan, and Thailand. Of the patients in these registries, 1254 received cobalt-chromium everolimus-eluting stents (CoCr-EES), 232 biodegradable polymer biolimus-eluting stents (BP-BES), 616 platinum-chromium EES (PtCr-EES) and 590 Resolute zotarolimus-eluting stents (Re-ZES).
Main outcome measure. Target-vessel failure.
Main results. At 3 years, rates of target-vessel failure were not significantly different for the different types of stents (16.7% for the CoCr-EES, 13.2% for the BP-BES, 18.7% for the PtCr-EES, and 14.7% for the Re-ZES; P = 0.15). The adjusted hazard ratios (HRs) for target-vessel failure were similar in between-group comparisons of the different stents, except for the PtCr-EES versus the BP-BES (HR 1.60, 95% confidence interval 1.01 to 2.54; P = 0.046). There were no significant differences in risk of composite of all-cause death, any myocardial infarction, or any revascularization and its individual components according to the different types of stents.
Conclusion. There was no significant between-group differences in 3-year risk of target-vessel failure, except for a higher risk of primary outcome with PtCr-EES compared to BP-BES.
Commentary
Left main coronary artery disease is identified in 5% to 7% of the population and is one of the more perplexing lesions to treat given the poorer outcome compared to non–left main lesion and the importance of the vessels the left main supplies [1]. Historically, coronary artery bypass grafting (CABG) has been the standard of care on the basis of the survival benefit observed in early trials compared with medical therapy. Left main percutaneous coronary intervention (PCI) has evolved as an alternative to CABG over the past few decades. Early studies using balloon angioplasty or bare metal stents were limited primarily due to high restenosis rate [1]. In the DES era, results have been overall comparable to CABG. Unprotected left main PCI using first-generation DES was non-inferior compared to CABG in the pre-specified sub-study of SYNTAX trial and in PRECOMBAT trial using paclitaxel-eluting stents and sirolimus-eluting stents, respectively [2,3]. Largely based on these trials, the 2014 ACC/AHA guidelines give class IIa recommendation for patients with low-risk anatomy (Syntax score 0–22) and class IIb recommendation for patients with intermediate-risk anatomy (Syntax score 23–32) for left main PCI [4]. Moreover, European guidelines give class Ib recommendation for patients with low-risk anatomy, and class IIa recommendation for intermediate-risk anatomy for left main PCI [5]. However, the SYNTAX trial and PRECOMBAT trial were limited by not meeting non-inferiority (SYNTAX) and wide non-inferiority (PRECOMBAT) and selection bias due to large exclusion criteria. In addition, first-generation DES were used in these trials (tacrolimis-eluting stent for SYNTAX and sirolimus-eluting stent for PRECOMBAT). The standard of care has now shifted to wide use of second-generation DES [1].
Subsequently, 2 larger-scale clinical trials using second-generation DES were designed and results have been reported recently [6,7]. The EXCEL trial enrolled 1905 patients with significant left main coronary disease and compared CoCr-EES to CABG. At 3 years, the primary endpoint of a composite of death from any cause, stroke, or myocardial infarction occurred in 15.4% of the PCI patients and in 14.7% of the CABG patients (P = 0.02 for non-inferiority; P = 0.98 for superiority). Similarly, the NOBLE trial enrolled 1201 patients with significant left main coronary disease and compared PCI to CABG. In this trial, the biolimus-eluting second-generation stent became their preferred stent during the study period. At 5 years, the primary endpoint of a composite of all-cause mortality, non-procedural myocardial infarction, any repeat coronary intervention, and stroke was higher in PCI compared to CABG patients (28% vs 18%, HR 1.51, 95% CI 1.13–2.00), exceeding the limit of non-inferiority, and CABG was significantly better compared to PCI (P = 0.004). The difference in the results is likely due to trial design. The primary endpoint was different in the 2 studies—EXCEL did not include repeat coronary intervention in the composite endpoint. The NOBLE study had a longer enrollment period and earlier-generation stents (sirolimus-eluting) were used in the earlier stages of the trial. In addition, the NOBLE study did not assess for peri-procedural myocardial infarction as an endpoint, which is known to be associated with adverse outcome. In both trials, cardiovascular mortality and all-cause mortality were similar at the end of follow-up.
In this context, the Lee et al study compared 4 types of currently available second-generation stents by pooling data from 3 large registries in Asia [8]. The main finding from this study was that target-vessel failure, defined as the composite of cardiac death, target-vessel myocardial infarction, or target-vessel revascularization at 3 years follow-up was not different among the types of second-generation drug eluting stents (P = 0.15).
Another important finding from this study was that the stent thrombosis rate at follow-up was very low (< 1%). This is consistent with the EXCEL study, which reported a definite stent thrombosis rate of 0.7% and was lower than in the NOBLE study, which reported a rate of 3%. One of the possible explanations for this difference could be stent selection. In contrast to the EXCEL study, which exclusively used Co-Cr EES by study protocol, NOBLE
study included first-generation sirolimus-drug eluting stent (11%) and BP-BES (89%). However, there are multiple factors that contribute to stent thrombosis other than stent selection, such as lesion characteristics, adequate stent expansion, and use of dual antiplatelet therapy [9].
The observed finding of small increase in target-vessel failure in PtCr-EES versus the BP-BES needs to be interpreted with caution. First, this was an observational study, and the treatment strategy or choice of stent was determined by a local interventional cardiologist, which could lead to selection bias. Although the authors performed propensity analysis, residual cofounding is likely. Second, since there was no difference in the primary analysis, the subgroup analysis becomes less important. In addition, authors did not perform statistical correction for multiple comparisons.
Despite the above limitations, this large-scale observational study gives us important insights to the performance of each second-generation DES. All currently available second-generation DES appear to be an option for use for left main coronary intervention.
Applications for Clinical Practice
In patients presenting with significant left main disease, left main PCI using a contemporary second-generation stent is safe and effective and likely has equivalent outcomes to CABG. However, PCI may be associated with higher rate of repeat revascularization. The rate of target-vessel failure was similar between different types of second-generation DES.
—Taishi Hirai, MD, and John E.A. Blair, MD, University of Chicago Medical Center, Chicago, IL
1. Rab T, Sheiban I, Louvard Y, et al. Current interventions for the left main bifurcation. JACC Cardiovasc Interv 2017;10:849–65.
2. Morice MC, Serruys PW, Kappetein AP, et al. Outcomes in patients with de novo left main disease treated with either percutaneous coronary intervention using paclitaxel-eluting stents or coronary artery bypass graft treatment in the Synergy Between Percutaneous Coronary Intervention with TAXUS and Cardiac Surgery (SYNTAX) trial. Circulation 2010;121:2645–53.
3. Park SJ, Kim YH, Park DW, et al. Randomized trial of stents versus bypass surgery for left main coronary artery disease. N Engl J Med 2011;364:1718–27.
4. Fihn SD, Blankenship JC, Alexander KP, et al. 2014 ACC/AHA/AATS/PCNA/SCAI/STS focused update of the guideline for the diagnosis and management of patients with stable ischemic heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines, and the American Association for Thoracic Surgery, Preventive Cardiovascular Nurses Association, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol 2014;64:1929–49.
5. Windecker S, Kolh P, Alfonso F, et al. 2014 ESC/EACTS Guidelines on myocardial revascularization: The Task Force on Myocardial Revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS)Developed with the special contribution of the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur Heart J 2014;35:2541–619.
6. Stone GW, Sabik JF, Serruys PW, et al. Everolimus-eluting stents or bypass surgery for left main coronary artery disease. N Engl J Med 2016;375:2223–35.
7. Mäkikallio T, Holm NR, Lindsay M, et al. Percutaneous coronary angioplasty versus coronary artery bypass grafting in treatment of unprotected left main stenosis (NOBLE): a prospective, randomised, open-label, non-inferiority trial. Lancet 2016;388:2743–52.
8. Lee PH, Kwon O, Ahn JM, et al. Safety and effectiveness of second-generation drug-eluting stents in patients with left main coronary artery disease. J Am Coll Cardiol 2018;71:832–41.
9. Claessen BE, Henriques JP, Jaffer FA, et al. Stent thrombosis: a clinical perspective. JACC Cardiovasc Interv 2014;7:1081–92.
1. Rab T, Sheiban I, Louvard Y, et al. Current interventions for the left main bifurcation. JACC Cardiovasc Interv 2017;10:849–65.
2. Morice MC, Serruys PW, Kappetein AP, et al. Outcomes in patients with de novo left main disease treated with either percutaneous coronary intervention using paclitaxel-eluting stents or coronary artery bypass graft treatment in the Synergy Between Percutaneous Coronary Intervention with TAXUS and Cardiac Surgery (SYNTAX) trial. Circulation 2010;121:2645–53.
3. Park SJ, Kim YH, Park DW, et al. Randomized trial of stents versus bypass surgery for left main coronary artery disease. N Engl J Med 2011;364:1718–27.
4. Fihn SD, Blankenship JC, Alexander KP, et al. 2014 ACC/AHA/AATS/PCNA/SCAI/STS focused update of the guideline for the diagnosis and management of patients with stable ischemic heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines, and the American Association for Thoracic Surgery, Preventive Cardiovascular Nurses Association, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol 2014;64:1929–49.
5. Windecker S, Kolh P, Alfonso F, et al. 2014 ESC/EACTS Guidelines on myocardial revascularization: The Task Force on Myocardial Revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS)Developed with the special contribution of the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur Heart J 2014;35:2541–619.
6. Stone GW, Sabik JF, Serruys PW, et al. Everolimus-eluting stents or bypass surgery for left main coronary artery disease. N Engl J Med 2016;375:2223–35.
7. Mäkikallio T, Holm NR, Lindsay M, et al. Percutaneous coronary angioplasty versus coronary artery bypass grafting in treatment of unprotected left main stenosis (NOBLE): a prospective, randomised, open-label, non-inferiority trial. Lancet 2016;388:2743–52.
8. Lee PH, Kwon O, Ahn JM, et al. Safety and effectiveness of second-generation drug-eluting stents in patients with left main coronary artery disease. J Am Coll Cardiol 2018;71:832–41.
9. Claessen BE, Henriques JP, Jaffer FA, et al. Stent thrombosis: a clinical perspective. JACC Cardiovasc Interv 2014;7:1081–92.
Usability and Patient Perceptions of the Sarilumab Pen for Treatment of RA
Study Overview
Objective. To assess usability and patient perceptions of the sarilumab auto-injector device (“sarilumab pen”) among patients with moderate-to-severe rheumatoid arthritis (RA).
Design. 12-week, randomized, parallel-group usability study.
Setting and participants. The study was conducted at 53 centers in 6 countries. Inclusion criteria were a diagnosis of RA (as defined by American College of Rheumatology/ European League Against Rheumatism 2010 Criteria) of ≥ 3-month disease duration, willing and able to self inject, continuous treatment with 1 or a combination of nonbiologic disease modifying antirheumatic drugs (except leflunomide in combination with methotrexate); and moderatly to severely active RA, defined as 4/66 swollen joint, 4/68 tender joints, and high-sensitivity C-reactive protein (hsCRP) measurement ≥ 4 mg/L. Exclusion criteria were age
Patients were randomized 1:1:1:1 to sarilumamb 150 or 200 mg every 2 weeks administered by single-use, disposable, prefilled pen or pre-filled syringe. Randomization method was not reported.
Main outcomes measures. The primary endpoint was number of “product technical failures” (PTFs). Patients randomized to the pen were given a diary that had questions related to their ability to remove the cap, start the injection, and complete the injection. Participants were asked to answer the questions each time they used the pen. If the response was “no” to any of the 3 questions, this was considered a “product technical complaint” (PTC). PTCs that had a validated technical cause based on pen evaluation and analysis were considered PTFs.
In addition, patient perceptions and satisfaction with the pen were assessed via questionnaire. At baseline, patients were asked about injections and prior experience with self-injection, and at 12 weeks they were asked about their experiences in using the pen. Other outcomes assessed included adverse events and pharmokinetic parameters.
Results. 217 participants were enrolled: 108 patients were in the pen group (56 randomized to 150 mg and 52 randomized to 200 mg) and 109 were in the syringe group (53 randomized to 150 mg and 56 randomized to 200 mg). Completion rates were similar among groups. Sixteen patients discontinued due to treatment-emergent adverse events. There were no PTFs. There was one PTC, in which the user accidently bumped the pen, which expelled the drug onto the floor.
At baseline, before the first injection, the majority of patients reported that they were not afraid of needles (58%), had past experience with self-injections (55%), and were either “very confident” or “extremely confident” regarding self-injections (55%). After the 12-week assessment phase, when asked about their overall level of satisfaction, 98% of patients reported they were “satisfied” or “very satisfied” with the sarilumab pen.
Treatment emergent adverse events occurred in 66% of patients, with no clinically meaningful differences leading to discontinuation in the pen and syringe groups. The most common adverse events were infections and neutropenia.
Conclusion. Patients successfully completed self-injections with the sarilumab pen and found it easy to use.
Commentary
Rheumatoid arthritis (RA) is a common immune-mediated disease characterized by chronically progressive inflammation and destruction of joints and associated structures, resulting in significant morbidity, mortality, and disability. Improved understanding of RA disease pathogenesis in recent years has led to the development of new biologic treatments designed to target specific elements of the RA inflammatory response.
Sarilumab is an interleukin-6 blocker that was approved in the US in 2017 for the treatment of adult patients with moderately to severely active RA who have had an inadequate response or intolerance to one or more disease-modifying antirheumatic drugs. While a syringe form of this drug is currently available, at the time of this writing the pen has not yet been released.
In this real-world usability study sponsored by Sanofi, there were no technical difficulties with using the pen. Most patients thought the pen was easy or very easy to use, and safety and effeicacy appeared to be generally comparable between the pen and syringe. The pen also offers safety protection features that prevent needlestick injury.
The authors of the current study noted that results from previous studies have shown that patients with RA favor treatment devices that are easy to use, convenient, less painful, and take less time to use, and patients have demonstrated a preference for autoinjector devices over more conventional methods of treatment administration [1–3], such as syringes. Pens have been well accepted for the treatment of other chronic health conditions, including diabetes mellitus, migraine headaches, and growth hormone deficiency, and subcutaneous administration of a tumor necrosis factor (TNF) inhibitor via pen has also been accepted for the treatment of RA [1]. As RA requires lifelong treatment, the use of a pen that is ergonomically designed to take into account the manual dexterity issues relevant to this patient population could potentially enhance compliance.
Applications for Clinical Practice
A prefilled pen was well accepted and associated with favorable patient perceptions,
1. Kivitz A, Cohen S, Dowd JE, et al. Clinical assessment of pain, tolerability, and preference of an autoinjection pen versus a prefilled syringe for patient self-administration of the fully human, monoclonal antibody adalimumab: the TOUCH trial. Clin Ther 2006;28:1619–29.
2. Demary W, Schwenke H, Rockwitz K, et al. Subcutaneously administered methotrexate for rheumatoid arthritis, by prefilled syringes versus prefilled pens: patient preference and comparison of the self-injection experience. Patient Prefer Adherence 2014;8:1061–71.
3. Thakur K, Biberger A, Handrich A, Rezk MF. Patient perceptions and preferences of two etanercept autoinjectors for rheumatoid arthritis: findings from a patient survey in Europe. Rheumatol Ther 2016;3:245–56.
Study Overview
Objective. To assess usability and patient perceptions of the sarilumab auto-injector device (“sarilumab pen”) among patients with moderate-to-severe rheumatoid arthritis (RA).
Design. 12-week, randomized, parallel-group usability study.
Setting and participants. The study was conducted at 53 centers in 6 countries. Inclusion criteria were a diagnosis of RA (as defined by American College of Rheumatology/ European League Against Rheumatism 2010 Criteria) of ≥ 3-month disease duration, willing and able to self inject, continuous treatment with 1 or a combination of nonbiologic disease modifying antirheumatic drugs (except leflunomide in combination with methotrexate); and moderatly to severely active RA, defined as 4/66 swollen joint, 4/68 tender joints, and high-sensitivity C-reactive protein (hsCRP) measurement ≥ 4 mg/L. Exclusion criteria were age
Patients were randomized 1:1:1:1 to sarilumamb 150 or 200 mg every 2 weeks administered by single-use, disposable, prefilled pen or pre-filled syringe. Randomization method was not reported.
Main outcomes measures. The primary endpoint was number of “product technical failures” (PTFs). Patients randomized to the pen were given a diary that had questions related to their ability to remove the cap, start the injection, and complete the injection. Participants were asked to answer the questions each time they used the pen. If the response was “no” to any of the 3 questions, this was considered a “product technical complaint” (PTC). PTCs that had a validated technical cause based on pen evaluation and analysis were considered PTFs.
In addition, patient perceptions and satisfaction with the pen were assessed via questionnaire. At baseline, patients were asked about injections and prior experience with self-injection, and at 12 weeks they were asked about their experiences in using the pen. Other outcomes assessed included adverse events and pharmokinetic parameters.
Results. 217 participants were enrolled: 108 patients were in the pen group (56 randomized to 150 mg and 52 randomized to 200 mg) and 109 were in the syringe group (53 randomized to 150 mg and 56 randomized to 200 mg). Completion rates were similar among groups. Sixteen patients discontinued due to treatment-emergent adverse events. There were no PTFs. There was one PTC, in which the user accidently bumped the pen, which expelled the drug onto the floor.
At baseline, before the first injection, the majority of patients reported that they were not afraid of needles (58%), had past experience with self-injections (55%), and were either “very confident” or “extremely confident” regarding self-injections (55%). After the 12-week assessment phase, when asked about their overall level of satisfaction, 98% of patients reported they were “satisfied” or “very satisfied” with the sarilumab pen.
Treatment emergent adverse events occurred in 66% of patients, with no clinically meaningful differences leading to discontinuation in the pen and syringe groups. The most common adverse events were infections and neutropenia.
Conclusion. Patients successfully completed self-injections with the sarilumab pen and found it easy to use.
Commentary
Rheumatoid arthritis (RA) is a common immune-mediated disease characterized by chronically progressive inflammation and destruction of joints and associated structures, resulting in significant morbidity, mortality, and disability. Improved understanding of RA disease pathogenesis in recent years has led to the development of new biologic treatments designed to target specific elements of the RA inflammatory response.
Sarilumab is an interleukin-6 blocker that was approved in the US in 2017 for the treatment of adult patients with moderately to severely active RA who have had an inadequate response or intolerance to one or more disease-modifying antirheumatic drugs. While a syringe form of this drug is currently available, at the time of this writing the pen has not yet been released.
In this real-world usability study sponsored by Sanofi, there were no technical difficulties with using the pen. Most patients thought the pen was easy or very easy to use, and safety and effeicacy appeared to be generally comparable between the pen and syringe. The pen also offers safety protection features that prevent needlestick injury.
The authors of the current study noted that results from previous studies have shown that patients with RA favor treatment devices that are easy to use, convenient, less painful, and take less time to use, and patients have demonstrated a preference for autoinjector devices over more conventional methods of treatment administration [1–3], such as syringes. Pens have been well accepted for the treatment of other chronic health conditions, including diabetes mellitus, migraine headaches, and growth hormone deficiency, and subcutaneous administration of a tumor necrosis factor (TNF) inhibitor via pen has also been accepted for the treatment of RA [1]. As RA requires lifelong treatment, the use of a pen that is ergonomically designed to take into account the manual dexterity issues relevant to this patient population could potentially enhance compliance.
Applications for Clinical Practice
A prefilled pen was well accepted and associated with favorable patient perceptions,
Study Overview
Objective. To assess usability and patient perceptions of the sarilumab auto-injector device (“sarilumab pen”) among patients with moderate-to-severe rheumatoid arthritis (RA).
Design. 12-week, randomized, parallel-group usability study.
Setting and participants. The study was conducted at 53 centers in 6 countries. Inclusion criteria were a diagnosis of RA (as defined by American College of Rheumatology/ European League Against Rheumatism 2010 Criteria) of ≥ 3-month disease duration, willing and able to self inject, continuous treatment with 1 or a combination of nonbiologic disease modifying antirheumatic drugs (except leflunomide in combination with methotrexate); and moderatly to severely active RA, defined as 4/66 swollen joint, 4/68 tender joints, and high-sensitivity C-reactive protein (hsCRP) measurement ≥ 4 mg/L. Exclusion criteria were age
Patients were randomized 1:1:1:1 to sarilumamb 150 or 200 mg every 2 weeks administered by single-use, disposable, prefilled pen or pre-filled syringe. Randomization method was not reported.
Main outcomes measures. The primary endpoint was number of “product technical failures” (PTFs). Patients randomized to the pen were given a diary that had questions related to their ability to remove the cap, start the injection, and complete the injection. Participants were asked to answer the questions each time they used the pen. If the response was “no” to any of the 3 questions, this was considered a “product technical complaint” (PTC). PTCs that had a validated technical cause based on pen evaluation and analysis were considered PTFs.
In addition, patient perceptions and satisfaction with the pen were assessed via questionnaire. At baseline, patients were asked about injections and prior experience with self-injection, and at 12 weeks they were asked about their experiences in using the pen. Other outcomes assessed included adverse events and pharmokinetic parameters.
Results. 217 participants were enrolled: 108 patients were in the pen group (56 randomized to 150 mg and 52 randomized to 200 mg) and 109 were in the syringe group (53 randomized to 150 mg and 56 randomized to 200 mg). Completion rates were similar among groups. Sixteen patients discontinued due to treatment-emergent adverse events. There were no PTFs. There was one PTC, in which the user accidently bumped the pen, which expelled the drug onto the floor.
At baseline, before the first injection, the majority of patients reported that they were not afraid of needles (58%), had past experience with self-injections (55%), and were either “very confident” or “extremely confident” regarding self-injections (55%). After the 12-week assessment phase, when asked about their overall level of satisfaction, 98% of patients reported they were “satisfied” or “very satisfied” with the sarilumab pen.
Treatment emergent adverse events occurred in 66% of patients, with no clinically meaningful differences leading to discontinuation in the pen and syringe groups. The most common adverse events were infections and neutropenia.
Conclusion. Patients successfully completed self-injections with the sarilumab pen and found it easy to use.
Commentary
Rheumatoid arthritis (RA) is a common immune-mediated disease characterized by chronically progressive inflammation and destruction of joints and associated structures, resulting in significant morbidity, mortality, and disability. Improved understanding of RA disease pathogenesis in recent years has led to the development of new biologic treatments designed to target specific elements of the RA inflammatory response.
Sarilumab is an interleukin-6 blocker that was approved in the US in 2017 for the treatment of adult patients with moderately to severely active RA who have had an inadequate response or intolerance to one or more disease-modifying antirheumatic drugs. While a syringe form of this drug is currently available, at the time of this writing the pen has not yet been released.
In this real-world usability study sponsored by Sanofi, there were no technical difficulties with using the pen. Most patients thought the pen was easy or very easy to use, and safety and effeicacy appeared to be generally comparable between the pen and syringe. The pen also offers safety protection features that prevent needlestick injury.
The authors of the current study noted that results from previous studies have shown that patients with RA favor treatment devices that are easy to use, convenient, less painful, and take less time to use, and patients have demonstrated a preference for autoinjector devices over more conventional methods of treatment administration [1–3], such as syringes. Pens have been well accepted for the treatment of other chronic health conditions, including diabetes mellitus, migraine headaches, and growth hormone deficiency, and subcutaneous administration of a tumor necrosis factor (TNF) inhibitor via pen has also been accepted for the treatment of RA [1]. As RA requires lifelong treatment, the use of a pen that is ergonomically designed to take into account the manual dexterity issues relevant to this patient population could potentially enhance compliance.
Applications for Clinical Practice
A prefilled pen was well accepted and associated with favorable patient perceptions,
1. Kivitz A, Cohen S, Dowd JE, et al. Clinical assessment of pain, tolerability, and preference of an autoinjection pen versus a prefilled syringe for patient self-administration of the fully human, monoclonal antibody adalimumab: the TOUCH trial. Clin Ther 2006;28:1619–29.
2. Demary W, Schwenke H, Rockwitz K, et al. Subcutaneously administered methotrexate for rheumatoid arthritis, by prefilled syringes versus prefilled pens: patient preference and comparison of the self-injection experience. Patient Prefer Adherence 2014;8:1061–71.
3. Thakur K, Biberger A, Handrich A, Rezk MF. Patient perceptions and preferences of two etanercept autoinjectors for rheumatoid arthritis: findings from a patient survey in Europe. Rheumatol Ther 2016;3:245–56.
1. Kivitz A, Cohen S, Dowd JE, et al. Clinical assessment of pain, tolerability, and preference of an autoinjection pen versus a prefilled syringe for patient self-administration of the fully human, monoclonal antibody adalimumab: the TOUCH trial. Clin Ther 2006;28:1619–29.
2. Demary W, Schwenke H, Rockwitz K, et al. Subcutaneously administered methotrexate for rheumatoid arthritis, by prefilled syringes versus prefilled pens: patient preference and comparison of the self-injection experience. Patient Prefer Adherence 2014;8:1061–71.
3. Thakur K, Biberger A, Handrich A, Rezk MF. Patient perceptions and preferences of two etanercept autoinjectors for rheumatoid arthritis: findings from a patient survey in Europe. Rheumatol Ther 2016;3:245–56.
Are PTSD Responses Inherited or Acquired?
Neuroimaging studies have consistently reported reduced activation of the medial prefrontal cortex (mPFC) in patients with posttraumatic stress disorder (PTSD) while they recall and imagine stressful personal events. During script-driven imagery (SDI) sessions, patients with PTSD exhibit increased psychophysiologic (eg, heart rate, skin conductance, and facial electromyographic) responses to trauma-related memories. However, the origin of the responses remained unclear. Are they familial, acquired, or resulting from trauma exposure?
Researchers from Harvard University, University of California Los Angeles, and University of New England conducted a study of 26 male identical twin pairs to help find the answer. The participants were divided into 4 groups: combat-exposed with PTSD (ExP+), their combat-unexposed twins without PTSD, combat-exposed participants without PTSD, and their combat-unexposed twins without PTSD. They engaged in SDI during functional magnetic resonance (fMRI) imaging and concurrent skin conductance measurement.
The results of the fMRI tests showed diminished activation in the medial prefrontal cortex of the patients with PTSD compared with the other groups. The SC response scores did not correlate significantly with PTSD symptom severity.
Contrary to the researchers’ predictions, mPFC activation was not inversely correlated with PTSD symptom severity. However, they say their finding of reduced mPFC activation in the ExP+ group provides evidence that the abnormality is an acquired characteristic. If those findings are replicated, such objectively measured biologic characteristics could potentially aid in diagnosing PTSD or assessing treatment response.
Source:
Dahlgren MK, Laifer LM, VanElzakker MB, et al. Psychol Med. 2018;48(7):1128-1138.
doi: 10.1017/S003329171700263X.
Neuroimaging studies have consistently reported reduced activation of the medial prefrontal cortex (mPFC) in patients with posttraumatic stress disorder (PTSD) while they recall and imagine stressful personal events. During script-driven imagery (SDI) sessions, patients with PTSD exhibit increased psychophysiologic (eg, heart rate, skin conductance, and facial electromyographic) responses to trauma-related memories. However, the origin of the responses remained unclear. Are they familial, acquired, or resulting from trauma exposure?
Researchers from Harvard University, University of California Los Angeles, and University of New England conducted a study of 26 male identical twin pairs to help find the answer. The participants were divided into 4 groups: combat-exposed with PTSD (ExP+), their combat-unexposed twins without PTSD, combat-exposed participants without PTSD, and their combat-unexposed twins without PTSD. They engaged in SDI during functional magnetic resonance (fMRI) imaging and concurrent skin conductance measurement.
The results of the fMRI tests showed diminished activation in the medial prefrontal cortex of the patients with PTSD compared with the other groups. The SC response scores did not correlate significantly with PTSD symptom severity.
Contrary to the researchers’ predictions, mPFC activation was not inversely correlated with PTSD symptom severity. However, they say their finding of reduced mPFC activation in the ExP+ group provides evidence that the abnormality is an acquired characteristic. If those findings are replicated, such objectively measured biologic characteristics could potentially aid in diagnosing PTSD or assessing treatment response.
Source:
Dahlgren MK, Laifer LM, VanElzakker MB, et al. Psychol Med. 2018;48(7):1128-1138.
doi: 10.1017/S003329171700263X.
Neuroimaging studies have consistently reported reduced activation of the medial prefrontal cortex (mPFC) in patients with posttraumatic stress disorder (PTSD) while they recall and imagine stressful personal events. During script-driven imagery (SDI) sessions, patients with PTSD exhibit increased psychophysiologic (eg, heart rate, skin conductance, and facial electromyographic) responses to trauma-related memories. However, the origin of the responses remained unclear. Are they familial, acquired, or resulting from trauma exposure?
Researchers from Harvard University, University of California Los Angeles, and University of New England conducted a study of 26 male identical twin pairs to help find the answer. The participants were divided into 4 groups: combat-exposed with PTSD (ExP+), their combat-unexposed twins without PTSD, combat-exposed participants without PTSD, and their combat-unexposed twins without PTSD. They engaged in SDI during functional magnetic resonance (fMRI) imaging and concurrent skin conductance measurement.
The results of the fMRI tests showed diminished activation in the medial prefrontal cortex of the patients with PTSD compared with the other groups. The SC response scores did not correlate significantly with PTSD symptom severity.
Contrary to the researchers’ predictions, mPFC activation was not inversely correlated with PTSD symptom severity. However, they say their finding of reduced mPFC activation in the ExP+ group provides evidence that the abnormality is an acquired characteristic. If those findings are replicated, such objectively measured biologic characteristics could potentially aid in diagnosing PTSD or assessing treatment response.
Source:
Dahlgren MK, Laifer LM, VanElzakker MB, et al. Psychol Med. 2018;48(7):1128-1138.
doi: 10.1017/S003329171700263X.
Federal Health Data Trends:Vietnam Era Veterans (FULL)
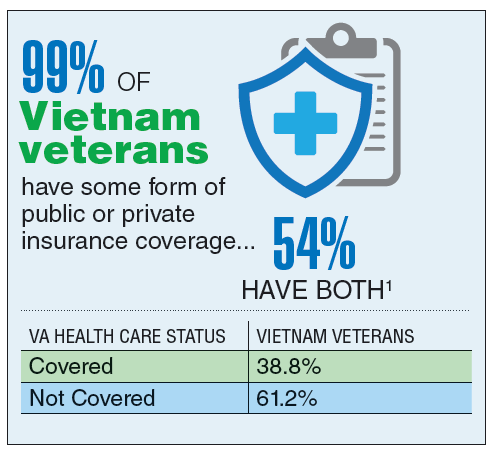
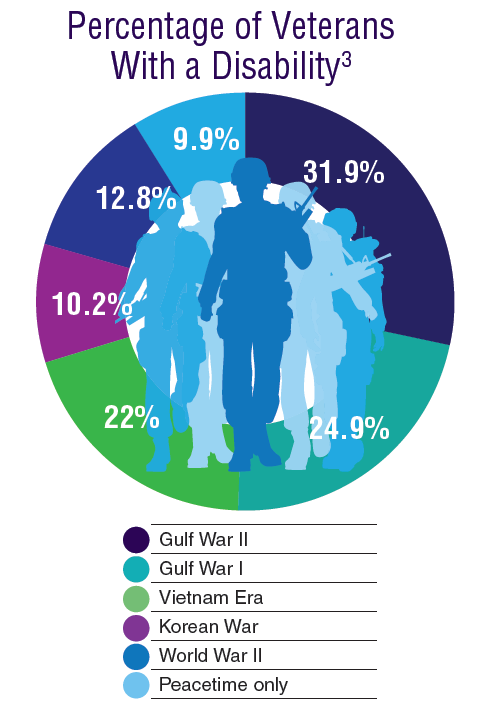
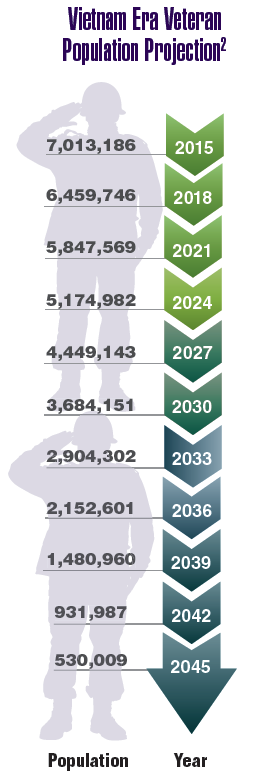
According to the VA, 8,744,000 veterans served in the Armed Forces in the time between the Gulf of Tonkin incident and the signing of the Paris Peace Accords.1 Of those, 3,403,000 were deployed to Southeast Asia. Those who served during this period often were exposed to unique environmental hazards, such as commonly used pesticides and herbicides, as well as diseases attributed to the tropical environment, such as fungal infections, and there were more than 40,000 reported cases of malaria. Upon returning home, these veterans faced a tough readjustment that often magnified the stress associated with combat.
Today, the VA recognizes 8 conditions related to service in Vietnam for the purposes of establishing service-connection: soft tissue sarcoma, non-Hodgkin lymphoma, Hodgkin disease, chloracne, porphyria cutanea tarda, respiratory cancers, multiple myeloma, prostate cancer, acute peripheral neuropathy, and spina bifida in offspring. As the veterans of this conflict age into their retirement years, the VA now also faces the challenge of providing care to this growing senior population.

According to the VA, 8,744,000 veterans served in the Armed Forces in the time between the Gulf of Tonkin incident and the signing of the Paris Peace Accords.1 Of those, 3,403,000 were deployed to Southeast Asia. Those who served during this period often were exposed to unique environmental hazards, such as commonly used pesticides and herbicides, as well as diseases attributed to the tropical environment, such as fungal infections, and there were more than 40,000 reported cases of malaria. Upon returning home, these veterans faced a tough readjustment that often magnified the stress associated with combat.
Today, the VA recognizes 8 conditions related to service in Vietnam for the purposes of establishing service-connection: soft tissue sarcoma, non-Hodgkin lymphoma, Hodgkin disease, chloracne, porphyria cutanea tarda, respiratory cancers, multiple myeloma, prostate cancer, acute peripheral neuropathy, and spina bifida in offspring. As the veterans of this conflict age into their retirement years, the VA now also faces the challenge of providing care to this growing senior population.
According to the VA, 8,744,000 veterans served in the Armed Forces in the time between the Gulf of Tonkin incident and the signing of the Paris Peace Accords.1 Of those, 3,403,000 were deployed to Southeast Asia. Those who served during this period often were exposed to unique environmental hazards, such as commonly used pesticides and herbicides, as well as diseases attributed to the tropical environment, such as fungal infections, and there were more than 40,000 reported cases of malaria. Upon returning home, these veterans faced a tough readjustment that often magnified the stress associated with combat.
Today, the VA recognizes 8 conditions related to service in Vietnam for the purposes of establishing service-connection: soft tissue sarcoma, non-Hodgkin lymphoma, Hodgkin disease, chloracne, porphyria cutanea tarda, respiratory cancers, multiple myeloma, prostate cancer, acute peripheral neuropathy, and spina bifida in offspring. As the veterans of this conflict age into their retirement years, the VA now also faces the challenge of providing care to this growing senior population.
Health Canada expands approval of obinutuzumab

Health Canada has expanded the approved use of obinutuzumab (Gazyva®).
The anti-CD20 monoclonal antibody is now approved for use in combination with chemotherapy to treat patients with previously untreated follicular lymphoma (FL) that is advanced (stage II bulky, stage III, or stage IV).
In patients who respond to this treatment, obinutuzumab monotherapy can be given as maintenance.
Health Canada previously approved obinutuzumab for the following indications:
- In combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia
- First in combination with bendamustine, then as monotherapy, in FL patients who relapsed after or are refractory to a rituximab-containing regimen.
Phase 3 results
Health Canada’s latest approval of obinutuzumab is based on results from the phase 3 GALLIUM study, which were published in NEJM in October 2017. The following are updated data from the product monograph.
GALLIUM included 1385 patients with previously untreated non-Hodgkin lymphoma, and 1202 of these patients had previously untreated, advanced FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), and bendamustine.
At a median observation time of 41.1 months, the overall response rate was 91% in the obinutuzumab arm and 88% in the rituximab arm. The complete response rates were 28% and 27%, respectively.
The median progression-free survival was not reached in either arm. The hazard ratio, for obinutuzumab compared to rituximab, was 0.72 (95% CI, 0.56-0.93, P=0.0118).
The estimated 3-year progression-free survival was 78.9% in the rituximab arm and 83.4% in the obinutuzumab arm.
Safety was evaluated based on all 1385 patients in the study, 86% of whom had FL and 14% of whom had marginal zone lymphoma.
Serious adverse events (AEs) occurred in 50% of patients in the obinutuzumab arm and 43% in the rituximab arm. Fatal AEs occurred in 5% and 4%, respectively. Infections and second malignancies were the leading causes of these deaths.
During the monotherapy period, the most common AEs (≥ 5%) in patients treated with obinutuzumab were cough (21%), neutropenia (19%), upper respiratory tract infection (15%), viral upper respiratory tract infection (15%), diarrhea (13%), arthralgia (10%), fatigue (9%), sinusitis (9%), infusion reactions (8%), pneumonia (8%), herpes zoster (8%), lower respiratory tract infection (7%), pyrexia (7%), back pain (6%), headache (6%), urinary tract infection (6%), nausea (6%), bronchitis (5%), and vomiting (5%).
Grade 3-4 AEs (≥1%) in patients treated with obinutuzumab included neutropenia (17%), pneumonia (3%), and febrile neutropenia (2%). There were 2 deaths due to pneumonia in the obinutuzumab arm.
Health Canada has expanded the approved use of obinutuzumab (Gazyva®).
The anti-CD20 monoclonal antibody is now approved for use in combination with chemotherapy to treat patients with previously untreated follicular lymphoma (FL) that is advanced (stage II bulky, stage III, or stage IV).
In patients who respond to this treatment, obinutuzumab monotherapy can be given as maintenance.
Health Canada previously approved obinutuzumab for the following indications:
- In combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia
- First in combination with bendamustine, then as monotherapy, in FL patients who relapsed after or are refractory to a rituximab-containing regimen.
Phase 3 results
Health Canada’s latest approval of obinutuzumab is based on results from the phase 3 GALLIUM study, which were published in NEJM in October 2017. The following are updated data from the product monograph.
GALLIUM included 1385 patients with previously untreated non-Hodgkin lymphoma, and 1202 of these patients had previously untreated, advanced FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), and bendamustine.
At a median observation time of 41.1 months, the overall response rate was 91% in the obinutuzumab arm and 88% in the rituximab arm. The complete response rates were 28% and 27%, respectively.
The median progression-free survival was not reached in either arm. The hazard ratio, for obinutuzumab compared to rituximab, was 0.72 (95% CI, 0.56-0.93, P=0.0118).
The estimated 3-year progression-free survival was 78.9% in the rituximab arm and 83.4% in the obinutuzumab arm.
Safety was evaluated based on all 1385 patients in the study, 86% of whom had FL and 14% of whom had marginal zone lymphoma.
Serious adverse events (AEs) occurred in 50% of patients in the obinutuzumab arm and 43% in the rituximab arm. Fatal AEs occurred in 5% and 4%, respectively. Infections and second malignancies were the leading causes of these deaths.
During the monotherapy period, the most common AEs (≥ 5%) in patients treated with obinutuzumab were cough (21%), neutropenia (19%), upper respiratory tract infection (15%), viral upper respiratory tract infection (15%), diarrhea (13%), arthralgia (10%), fatigue (9%), sinusitis (9%), infusion reactions (8%), pneumonia (8%), herpes zoster (8%), lower respiratory tract infection (7%), pyrexia (7%), back pain (6%), headache (6%), urinary tract infection (6%), nausea (6%), bronchitis (5%), and vomiting (5%).
Grade 3-4 AEs (≥1%) in patients treated with obinutuzumab included neutropenia (17%), pneumonia (3%), and febrile neutropenia (2%). There were 2 deaths due to pneumonia in the obinutuzumab arm.
Health Canada has expanded the approved use of obinutuzumab (Gazyva®).
The anti-CD20 monoclonal antibody is now approved for use in combination with chemotherapy to treat patients with previously untreated follicular lymphoma (FL) that is advanced (stage II bulky, stage III, or stage IV).
In patients who respond to this treatment, obinutuzumab monotherapy can be given as maintenance.
Health Canada previously approved obinutuzumab for the following indications:
- In combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia
- First in combination with bendamustine, then as monotherapy, in FL patients who relapsed after or are refractory to a rituximab-containing regimen.
Phase 3 results
Health Canada’s latest approval of obinutuzumab is based on results from the phase 3 GALLIUM study, which were published in NEJM in October 2017. The following are updated data from the product monograph.
GALLIUM included 1385 patients with previously untreated non-Hodgkin lymphoma, and 1202 of these patients had previously untreated, advanced FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), and bendamustine.
At a median observation time of 41.1 months, the overall response rate was 91% in the obinutuzumab arm and 88% in the rituximab arm. The complete response rates were 28% and 27%, respectively.
The median progression-free survival was not reached in either arm. The hazard ratio, for obinutuzumab compared to rituximab, was 0.72 (95% CI, 0.56-0.93, P=0.0118).
The estimated 3-year progression-free survival was 78.9% in the rituximab arm and 83.4% in the obinutuzumab arm.
Safety was evaluated based on all 1385 patients in the study, 86% of whom had FL and 14% of whom had marginal zone lymphoma.
Serious adverse events (AEs) occurred in 50% of patients in the obinutuzumab arm and 43% in the rituximab arm. Fatal AEs occurred in 5% and 4%, respectively. Infections and second malignancies were the leading causes of these deaths.
During the monotherapy period, the most common AEs (≥ 5%) in patients treated with obinutuzumab were cough (21%), neutropenia (19%), upper respiratory tract infection (15%), viral upper respiratory tract infection (15%), diarrhea (13%), arthralgia (10%), fatigue (9%), sinusitis (9%), infusion reactions (8%), pneumonia (8%), herpes zoster (8%), lower respiratory tract infection (7%), pyrexia (7%), back pain (6%), headache (6%), urinary tract infection (6%), nausea (6%), bronchitis (5%), and vomiting (5%).
Grade 3-4 AEs (≥1%) in patients treated with obinutuzumab included neutropenia (17%), pneumonia (3%), and febrile neutropenia (2%). There were 2 deaths due to pneumonia in the obinutuzumab arm.
A new use for ibrutinib?

Preclinical research suggests ibrutinib could treat G-CSFR-mutant myeloid disorders.
“Mutations in G-CSFR have a harmful effect on the production of neutrophils and are reported in patients with several blood disorders, including severe congenital neutropenia, chronic neutrophilic leukemia, and acute myeloid leukemia,” said Ken Greis, PhD, of the University of Cincinnati in Ohio.
“Unfortunately, despite years of research, the malignant signaling of the mutated G-CSFRs is not well understood.”
With this in mind, Dr Greis and his colleagues created a comprehensive signaling network of normal and mutated G-CSFR. Their goal was to understand how abnormal cellular signaling from the mutant receptors results in disease development.
The researchers described this work in Leukemia.
“We are able to look at . . . phosphorylation that results in phosphate groups being attached to the amino acid tyrosine (Tyr) in proteins,” Dr Greis explained. “These phosphorylation events (pTyr) can act as switches to activate or inactivate proteins and/or specific cellular processes.”
“By evaluating pTyr activity in the normal versus mutant receptor cells, we can produce a network similar to a wiring diagram of cellular regulation. Observed disruptions at any of the nodes in the network for the mutated receptors can then be investigated further to understand and perhaps target the abnormal signaling corresponding to the disease.”
This analysis of pTyr activity revealed that G-CSFR mutants had aberrant activation of BTK, as well as abnormal kinetics of canonical STAT3, STAT5, and MAPK phosphorylation.
“When we first got these results, one of the most exciting things was that BTK was already the target of an FDA-approved drug, ibrutinib . . .,” said study author H. Leighton Grimes, PhD, of the University of Cincinnati.
The researchers tested ibrutinib in cells with mutant and wild-type G-CSFR and found the drug killed the mutant cells but not the wild-type cells. This was the case in myeloid progenitor 32D cell lines and primary human CD34+ umbilical cord blood cells.
“Progenitor cells expressing mutated G-CSFR in animal models and in human blood cells also showed enhanced sensitivity to ibrutinib compared to the normal G-CSFR, thus confirming that the mutated cells could likely be eliminated by treatment with ibrutinib and may represent an effective therapy for these patients,” Dr Grimes said.
Ibrutinib also demonstrated synergy with the JAK1/2 inhibitor ruxolitinib. G-CSFR-mutant CD34+ cells were sensitive to each drug alone, but combining them “dramatically enhanced” the sensitivity, according to the researchers.
“These data demonstrate the strength of global proteomics approaches, like the pTyr profiling used here, in dissecting cancer-forming pathways and points to the possibility that ibrutinib could be an effective therapy for myeloid leukemias with G-CSFR mutations,” Dr Greis said.
“Further studies are needed to determine if these findings will be applicable in patient samples, but the hope is that clinical trials are just around the corner, since we’re investigating a drug that has already been found to be safe by the FDA.”
Preclinical research suggests ibrutinib could treat G-CSFR-mutant myeloid disorders.
“Mutations in G-CSFR have a harmful effect on the production of neutrophils and are reported in patients with several blood disorders, including severe congenital neutropenia, chronic neutrophilic leukemia, and acute myeloid leukemia,” said Ken Greis, PhD, of the University of Cincinnati in Ohio.
“Unfortunately, despite years of research, the malignant signaling of the mutated G-CSFRs is not well understood.”
With this in mind, Dr Greis and his colleagues created a comprehensive signaling network of normal and mutated G-CSFR. Their goal was to understand how abnormal cellular signaling from the mutant receptors results in disease development.
The researchers described this work in Leukemia.
“We are able to look at . . . phosphorylation that results in phosphate groups being attached to the amino acid tyrosine (Tyr) in proteins,” Dr Greis explained. “These phosphorylation events (pTyr) can act as switches to activate or inactivate proteins and/or specific cellular processes.”
“By evaluating pTyr activity in the normal versus mutant receptor cells, we can produce a network similar to a wiring diagram of cellular regulation. Observed disruptions at any of the nodes in the network for the mutated receptors can then be investigated further to understand and perhaps target the abnormal signaling corresponding to the disease.”
This analysis of pTyr activity revealed that G-CSFR mutants had aberrant activation of BTK, as well as abnormal kinetics of canonical STAT3, STAT5, and MAPK phosphorylation.
“When we first got these results, one of the most exciting things was that BTK was already the target of an FDA-approved drug, ibrutinib . . .,” said study author H. Leighton Grimes, PhD, of the University of Cincinnati.
The researchers tested ibrutinib in cells with mutant and wild-type G-CSFR and found the drug killed the mutant cells but not the wild-type cells. This was the case in myeloid progenitor 32D cell lines and primary human CD34+ umbilical cord blood cells.
“Progenitor cells expressing mutated G-CSFR in animal models and in human blood cells also showed enhanced sensitivity to ibrutinib compared to the normal G-CSFR, thus confirming that the mutated cells could likely be eliminated by treatment with ibrutinib and may represent an effective therapy for these patients,” Dr Grimes said.
Ibrutinib also demonstrated synergy with the JAK1/2 inhibitor ruxolitinib. G-CSFR-mutant CD34+ cells were sensitive to each drug alone, but combining them “dramatically enhanced” the sensitivity, according to the researchers.
“These data demonstrate the strength of global proteomics approaches, like the pTyr profiling used here, in dissecting cancer-forming pathways and points to the possibility that ibrutinib could be an effective therapy for myeloid leukemias with G-CSFR mutations,” Dr Greis said.
“Further studies are needed to determine if these findings will be applicable in patient samples, but the hope is that clinical trials are just around the corner, since we’re investigating a drug that has already been found to be safe by the FDA.”
Preclinical research suggests ibrutinib could treat G-CSFR-mutant myeloid disorders.
“Mutations in G-CSFR have a harmful effect on the production of neutrophils and are reported in patients with several blood disorders, including severe congenital neutropenia, chronic neutrophilic leukemia, and acute myeloid leukemia,” said Ken Greis, PhD, of the University of Cincinnati in Ohio.
“Unfortunately, despite years of research, the malignant signaling of the mutated G-CSFRs is not well understood.”
With this in mind, Dr Greis and his colleagues created a comprehensive signaling network of normal and mutated G-CSFR. Their goal was to understand how abnormal cellular signaling from the mutant receptors results in disease development.
The researchers described this work in Leukemia.
“We are able to look at . . . phosphorylation that results in phosphate groups being attached to the amino acid tyrosine (Tyr) in proteins,” Dr Greis explained. “These phosphorylation events (pTyr) can act as switches to activate or inactivate proteins and/or specific cellular processes.”
“By evaluating pTyr activity in the normal versus mutant receptor cells, we can produce a network similar to a wiring diagram of cellular regulation. Observed disruptions at any of the nodes in the network for the mutated receptors can then be investigated further to understand and perhaps target the abnormal signaling corresponding to the disease.”
This analysis of pTyr activity revealed that G-CSFR mutants had aberrant activation of BTK, as well as abnormal kinetics of canonical STAT3, STAT5, and MAPK phosphorylation.
“When we first got these results, one of the most exciting things was that BTK was already the target of an FDA-approved drug, ibrutinib . . .,” said study author H. Leighton Grimes, PhD, of the University of Cincinnati.
The researchers tested ibrutinib in cells with mutant and wild-type G-CSFR and found the drug killed the mutant cells but not the wild-type cells. This was the case in myeloid progenitor 32D cell lines and primary human CD34+ umbilical cord blood cells.
“Progenitor cells expressing mutated G-CSFR in animal models and in human blood cells also showed enhanced sensitivity to ibrutinib compared to the normal G-CSFR, thus confirming that the mutated cells could likely be eliminated by treatment with ibrutinib and may represent an effective therapy for these patients,” Dr Grimes said.
Ibrutinib also demonstrated synergy with the JAK1/2 inhibitor ruxolitinib. G-CSFR-mutant CD34+ cells were sensitive to each drug alone, but combining them “dramatically enhanced” the sensitivity, according to the researchers.
“These data demonstrate the strength of global proteomics approaches, like the pTyr profiling used here, in dissecting cancer-forming pathways and points to the possibility that ibrutinib could be an effective therapy for myeloid leukemias with G-CSFR mutations,” Dr Greis said.
“Further studies are needed to determine if these findings will be applicable in patient samples, but the hope is that clinical trials are just around the corner, since we’re investigating a drug that has already been found to be safe by the FDA.”
Conflicts of interest among FDA advisers

An investigative report has unearthed potential conflicts of interest among physicians who serve on advisory panels for the US Food and Drug Administration (FDA).
The investigation revealed that some FDA advisers are receiving significant post-hoc payments from the makers of drugs they reviewed.
The investigation also uncovered relationships between advisers and drug companies that predate drug reviews.
Journalist Charles Piller and his colleagues conducted this investigation and detailed the results in Science.
The report includes data—from the federal Open Payments website—on 107 physicians who voted on FDA advisory committees between 2013 and 2016.
Forty of these advisers received more than $10,000 in post-hoc earnings or research support from the makers of drugs they reviewed or from competing drug companies.
Twenty-six advisers received more than $100,000, and 7 advisers received more than $1 million.
The 17 top earners received more than $300,000 each. For these advisers, 94% of their earnings came from the makers of drugs they previously reviewed or from those companies’ competitors.
The data also show that some advisers received funds from drug companies concurrent with or in the year before their advisory service.
Of the 17 top-earning advisers, 11 received financial support from competing companies on one or more of the drugs they reviewed. Five advisers also received support from the makers of one or more of the drugs reviewed.
The FDA did not disclose this information to the public or issue waivers for these potential conflicts. The FDA can issue a waiver to allow the participation of an adviser with an active conflict or one that ended in the year before a vote, as long as the adviser in question can provide expertise that cannot be provided by someone else.
It is possible that the FDA dismissed the aforementioned financial ties that predated drug reviews, deciding these relationships were not conflicts and did not require a waiver. However, it is also possible that the FDA did not know about these potential conflicts.
Piller and his colleagues were unable to determine what the FDA knew, as the agency refused to release disclosure documents, discuss individual advisers, or explain what steps, if any, the FDA takes to validate advisers’ disclosures.
An investigative report has unearthed potential conflicts of interest among physicians who serve on advisory panels for the US Food and Drug Administration (FDA).
The investigation revealed that some FDA advisers are receiving significant post-hoc payments from the makers of drugs they reviewed.
The investigation also uncovered relationships between advisers and drug companies that predate drug reviews.
Journalist Charles Piller and his colleagues conducted this investigation and detailed the results in Science.
The report includes data—from the federal Open Payments website—on 107 physicians who voted on FDA advisory committees between 2013 and 2016.
Forty of these advisers received more than $10,000 in post-hoc earnings or research support from the makers of drugs they reviewed or from competing drug companies.
Twenty-six advisers received more than $100,000, and 7 advisers received more than $1 million.
The 17 top earners received more than $300,000 each. For these advisers, 94% of their earnings came from the makers of drugs they previously reviewed or from those companies’ competitors.
The data also show that some advisers received funds from drug companies concurrent with or in the year before their advisory service.
Of the 17 top-earning advisers, 11 received financial support from competing companies on one or more of the drugs they reviewed. Five advisers also received support from the makers of one or more of the drugs reviewed.
The FDA did not disclose this information to the public or issue waivers for these potential conflicts. The FDA can issue a waiver to allow the participation of an adviser with an active conflict or one that ended in the year before a vote, as long as the adviser in question can provide expertise that cannot be provided by someone else.
It is possible that the FDA dismissed the aforementioned financial ties that predated drug reviews, deciding these relationships were not conflicts and did not require a waiver. However, it is also possible that the FDA did not know about these potential conflicts.
Piller and his colleagues were unable to determine what the FDA knew, as the agency refused to release disclosure documents, discuss individual advisers, or explain what steps, if any, the FDA takes to validate advisers’ disclosures.
An investigative report has unearthed potential conflicts of interest among physicians who serve on advisory panels for the US Food and Drug Administration (FDA).
The investigation revealed that some FDA advisers are receiving significant post-hoc payments from the makers of drugs they reviewed.
The investigation also uncovered relationships between advisers and drug companies that predate drug reviews.
Journalist Charles Piller and his colleagues conducted this investigation and detailed the results in Science.
The report includes data—from the federal Open Payments website—on 107 physicians who voted on FDA advisory committees between 2013 and 2016.
Forty of these advisers received more than $10,000 in post-hoc earnings or research support from the makers of drugs they reviewed or from competing drug companies.
Twenty-six advisers received more than $100,000, and 7 advisers received more than $1 million.
The 17 top earners received more than $300,000 each. For these advisers, 94% of their earnings came from the makers of drugs they previously reviewed or from those companies’ competitors.
The data also show that some advisers received funds from drug companies concurrent with or in the year before their advisory service.
Of the 17 top-earning advisers, 11 received financial support from competing companies on one or more of the drugs they reviewed. Five advisers also received support from the makers of one or more of the drugs reviewed.
The FDA did not disclose this information to the public or issue waivers for these potential conflicts. The FDA can issue a waiver to allow the participation of an adviser with an active conflict or one that ended in the year before a vote, as long as the adviser in question can provide expertise that cannot be provided by someone else.
It is possible that the FDA dismissed the aforementioned financial ties that predated drug reviews, deciding these relationships were not conflicts and did not require a waiver. However, it is also possible that the FDA did not know about these potential conflicts.
Piller and his colleagues were unable to determine what the FDA knew, as the agency refused to release disclosure documents, discuss individual advisers, or explain what steps, if any, the FDA takes to validate advisers’ disclosures.
Long-term follow-up of monoclonal gammopathy of undetermined significance
Clinical question: What is the expected clinical progression of patients with monoclonal gammopathy of undetermined significance (MGUS)?

Study design: Prospective, observational cohort study.
Setting: Single institution in Minnesota.
Synopsis: Investigators identified 1,395 patients with MGUS during 1960-1994, with a median follow-up of 34 years. Progression to multiple myeloma, plasma cell disorders, or lymphoid disorders was noted in 147 patients (11%), which represents a 6.5-times higher risk for these disorders, compared with the age/sex–adjusted control population.
Two risk factors were associated with progression of disease: elevated serum M protein (greater than 1.5 g/dL) and an abnormal serum free light chain ratio. Risk of progression at 20 years in patients with both of these risk factors was 55% in patients with IgM subtypes and 30% in patients with non-IgM subtypes. With a single risk factor, risk of progression at 20 years was 41% and 20%, respectively. With no risk factors the risk of progression at 20 years was 19% and 7%. Overall expected survival was shorter in patients with MGUS versus that in the age/sex–matched control population.
Bottom line: Patients with MGUS have a shorter life expectancy than the general population, and the IgM subtype is associated with a greater risk of progression at 20 years, compared with the non-IgM subtype.
Citation: Kyle RA et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Eng J Med. 2018 Jan 18;378(3):241-9.
Dr. Thota is a hospitalist at UC San Diego Health and an assistant clinical professor at the University of California, San Diego.
Clinical question: What is the expected clinical progression of patients with monoclonal gammopathy of undetermined significance (MGUS)?
Study design: Prospective, observational cohort study.
Setting: Single institution in Minnesota.
Synopsis: Investigators identified 1,395 patients with MGUS during 1960-1994, with a median follow-up of 34 years. Progression to multiple myeloma, plasma cell disorders, or lymphoid disorders was noted in 147 patients (11%), which represents a 6.5-times higher risk for these disorders, compared with the age/sex–adjusted control population.
Two risk factors were associated with progression of disease: elevated serum M protein (greater than 1.5 g/dL) and an abnormal serum free light chain ratio. Risk of progression at 20 years in patients with both of these risk factors was 55% in patients with IgM subtypes and 30% in patients with non-IgM subtypes. With a single risk factor, risk of progression at 20 years was 41% and 20%, respectively. With no risk factors the risk of progression at 20 years was 19% and 7%. Overall expected survival was shorter in patients with MGUS versus that in the age/sex–matched control population.
Bottom line: Patients with MGUS have a shorter life expectancy than the general population, and the IgM subtype is associated with a greater risk of progression at 20 years, compared with the non-IgM subtype.
Citation: Kyle RA et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Eng J Med. 2018 Jan 18;378(3):241-9.
Dr. Thota is a hospitalist at UC San Diego Health and an assistant clinical professor at the University of California, San Diego.
Clinical question: What is the expected clinical progression of patients with monoclonal gammopathy of undetermined significance (MGUS)?
Study design: Prospective, observational cohort study.
Setting: Single institution in Minnesota.
Synopsis: Investigators identified 1,395 patients with MGUS during 1960-1994, with a median follow-up of 34 years. Progression to multiple myeloma, plasma cell disorders, or lymphoid disorders was noted in 147 patients (11%), which represents a 6.5-times higher risk for these disorders, compared with the age/sex–adjusted control population.
Two risk factors were associated with progression of disease: elevated serum M protein (greater than 1.5 g/dL) and an abnormal serum free light chain ratio. Risk of progression at 20 years in patients with both of these risk factors was 55% in patients with IgM subtypes and 30% in patients with non-IgM subtypes. With a single risk factor, risk of progression at 20 years was 41% and 20%, respectively. With no risk factors the risk of progression at 20 years was 19% and 7%. Overall expected survival was shorter in patients with MGUS versus that in the age/sex–matched control population.
Bottom line: Patients with MGUS have a shorter life expectancy than the general population, and the IgM subtype is associated with a greater risk of progression at 20 years, compared with the non-IgM subtype.
Citation: Kyle RA et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Eng J Med. 2018 Jan 18;378(3):241-9.
Dr. Thota is a hospitalist at UC San Diego Health and an assistant clinical professor at the University of California, San Diego.
More than 16% of ED sepsis patients not admitted to hospital
SAN DIEGO – More than 16% of emergency department sepsis patients are not admitted to the hospital, preliminary results from a large retrospective cohort study found.

“Nothing is really known about this topic,” lead study author Ithan D. Peltan, MD, said in an interview at an international conference of the American Thoracic Society. “In previous research, we’ve been focused on patients with sepsis who are admitted to the hospital. We have never thoroughly recognized that a fair number of patients who meet clinical criteria for sepsis in the emergency department are actually triaged to outpatient management. We don’t really know anything about these patients. What are their clinical characteristics and what are their outcomes like? And what are the factors that are leading them to be discharged from the ED rather than be admitted to the hospital?”
To find out, he and his associates retrospectively reviewed the medical records of 12,002 adult ED patients who met criteria for sepsis at two tertiary hospitals and two community hospitals in Utah between July 2013 and December 2016. They excluded trauma patients, those who left the ED against medical advice, those who were discharged to hospice or who died in the ED, and eligible patients’ repeat ED encounters. Patients transferred to another acute care facility were considered admitted, while transfers to non-acute care such as skilled nursing or psychiatric facilities were classified as discharges. Next, Dr. Peltan and his associates employed inverse probability weights using a propensity score for ED discharge based on age, sex, Charlson score, ED acuity score, initial ED vital signs, white blood cell count, lactate, sequential organ failure assessment (SOFA)score, busyness of the ED, and study hospital to compare 30-day mortality between patients admitted to the hospital versus those discharged from the ED.
Of the 12,002 patients included in the analysis, 10,032 (83.6%) were admitted, while 1,970 (16.4%) were discharged. Compared with admitted patients, discharged patients were younger (a mean of 53 vs. 60 years, respectively; P less than .001); more likely to be female (65% vs. 55%; P less than .001); more likely to be nonwhite or Hispanic (21% vs 17%; P less than .001), and had fewer comorbidities and physiologic derangements. In addition, crude mortality at 30 days was lower in discharged versus admitted patients (1.0% vs. 6.2%, respectively; P less than .001). After the propensity-adjusted analysis, there was no significant difference in 30-day mortality for discharged vs. admitted sepsis patients (adjusted odds ratio 1.0).
“We were worried that discharged ED sepsis patients were being mismanaged and weren’t going to do well as similar patients who were admitted to the hospital,” Dr. Peltan said. “This analysis is still a work in progress, but with that caveat, our findings so far suggest that physicians are making pretty good decisions overall.”
The researchers also found that, among 89 ED physicians who cared for 20 or more eligible patients, some did not discharge any of their sepsis patients, while others discharged 39% of their sepsis patients. “That was surprising,” Dr. Peltan said. “This could mean that some hospital sepsis admissions depend on physician practice style more than the patient’s condition or treatment needs.”
Researchers emphasized that they do not recommend routine outpatient management for individual sepsis patients. “Almost certainly, some of the discharged patients should have been admitted to the hospital.” Dr. Peltan said. “I think there’s still a lot of opportunity to understand who these patients are, understand why there is so much physician variation, and to develop tools to further optimize triage decisions.”
The study was funded in part by the Intermountain Research and Medical Foundation in Salt Lake City. Dr. Peltan reported having no financial disclosures.
SOURCE: Peltan ID et al. ATS 2018, Abstract A5994/702.
SAN DIEGO – More than 16% of emergency department sepsis patients are not admitted to the hospital, preliminary results from a large retrospective cohort study found.
“Nothing is really known about this topic,” lead study author Ithan D. Peltan, MD, said in an interview at an international conference of the American Thoracic Society. “In previous research, we’ve been focused on patients with sepsis who are admitted to the hospital. We have never thoroughly recognized that a fair number of patients who meet clinical criteria for sepsis in the emergency department are actually triaged to outpatient management. We don’t really know anything about these patients. What are their clinical characteristics and what are their outcomes like? And what are the factors that are leading them to be discharged from the ED rather than be admitted to the hospital?”
To find out, he and his associates retrospectively reviewed the medical records of 12,002 adult ED patients who met criteria for sepsis at two tertiary hospitals and two community hospitals in Utah between July 2013 and December 2016. They excluded trauma patients, those who left the ED against medical advice, those who were discharged to hospice or who died in the ED, and eligible patients’ repeat ED encounters. Patients transferred to another acute care facility were considered admitted, while transfers to non-acute care such as skilled nursing or psychiatric facilities were classified as discharges. Next, Dr. Peltan and his associates employed inverse probability weights using a propensity score for ED discharge based on age, sex, Charlson score, ED acuity score, initial ED vital signs, white blood cell count, lactate, sequential organ failure assessment (SOFA)score, busyness of the ED, and study hospital to compare 30-day mortality between patients admitted to the hospital versus those discharged from the ED.
Of the 12,002 patients included in the analysis, 10,032 (83.6%) were admitted, while 1,970 (16.4%) were discharged. Compared with admitted patients, discharged patients were younger (a mean of 53 vs. 60 years, respectively; P less than .001); more likely to be female (65% vs. 55%; P less than .001); more likely to be nonwhite or Hispanic (21% vs 17%; P less than .001), and had fewer comorbidities and physiologic derangements. In addition, crude mortality at 30 days was lower in discharged versus admitted patients (1.0% vs. 6.2%, respectively; P less than .001). After the propensity-adjusted analysis, there was no significant difference in 30-day mortality for discharged vs. admitted sepsis patients (adjusted odds ratio 1.0).
“We were worried that discharged ED sepsis patients were being mismanaged and weren’t going to do well as similar patients who were admitted to the hospital,” Dr. Peltan said. “This analysis is still a work in progress, but with that caveat, our findings so far suggest that physicians are making pretty good decisions overall.”
The researchers also found that, among 89 ED physicians who cared for 20 or more eligible patients, some did not discharge any of their sepsis patients, while others discharged 39% of their sepsis patients. “That was surprising,” Dr. Peltan said. “This could mean that some hospital sepsis admissions depend on physician practice style more than the patient’s condition or treatment needs.”
Researchers emphasized that they do not recommend routine outpatient management for individual sepsis patients. “Almost certainly, some of the discharged patients should have been admitted to the hospital.” Dr. Peltan said. “I think there’s still a lot of opportunity to understand who these patients are, understand why there is so much physician variation, and to develop tools to further optimize triage decisions.”
The study was funded in part by the Intermountain Research and Medical Foundation in Salt Lake City. Dr. Peltan reported having no financial disclosures.
SOURCE: Peltan ID et al. ATS 2018, Abstract A5994/702.
SAN DIEGO – More than 16% of emergency department sepsis patients are not admitted to the hospital, preliminary results from a large retrospective cohort study found.
“Nothing is really known about this topic,” lead study author Ithan D. Peltan, MD, said in an interview at an international conference of the American Thoracic Society. “In previous research, we’ve been focused on patients with sepsis who are admitted to the hospital. We have never thoroughly recognized that a fair number of patients who meet clinical criteria for sepsis in the emergency department are actually triaged to outpatient management. We don’t really know anything about these patients. What are their clinical characteristics and what are their outcomes like? And what are the factors that are leading them to be discharged from the ED rather than be admitted to the hospital?”
To find out, he and his associates retrospectively reviewed the medical records of 12,002 adult ED patients who met criteria for sepsis at two tertiary hospitals and two community hospitals in Utah between July 2013 and December 2016. They excluded trauma patients, those who left the ED against medical advice, those who were discharged to hospice or who died in the ED, and eligible patients’ repeat ED encounters. Patients transferred to another acute care facility were considered admitted, while transfers to non-acute care such as skilled nursing or psychiatric facilities were classified as discharges. Next, Dr. Peltan and his associates employed inverse probability weights using a propensity score for ED discharge based on age, sex, Charlson score, ED acuity score, initial ED vital signs, white blood cell count, lactate, sequential organ failure assessment (SOFA)score, busyness of the ED, and study hospital to compare 30-day mortality between patients admitted to the hospital versus those discharged from the ED.
Of the 12,002 patients included in the analysis, 10,032 (83.6%) were admitted, while 1,970 (16.4%) were discharged. Compared with admitted patients, discharged patients were younger (a mean of 53 vs. 60 years, respectively; P less than .001); more likely to be female (65% vs. 55%; P less than .001); more likely to be nonwhite or Hispanic (21% vs 17%; P less than .001), and had fewer comorbidities and physiologic derangements. In addition, crude mortality at 30 days was lower in discharged versus admitted patients (1.0% vs. 6.2%, respectively; P less than .001). After the propensity-adjusted analysis, there was no significant difference in 30-day mortality for discharged vs. admitted sepsis patients (adjusted odds ratio 1.0).
“We were worried that discharged ED sepsis patients were being mismanaged and weren’t going to do well as similar patients who were admitted to the hospital,” Dr. Peltan said. “This analysis is still a work in progress, but with that caveat, our findings so far suggest that physicians are making pretty good decisions overall.”
The researchers also found that, among 89 ED physicians who cared for 20 or more eligible patients, some did not discharge any of their sepsis patients, while others discharged 39% of their sepsis patients. “That was surprising,” Dr. Peltan said. “This could mean that some hospital sepsis admissions depend on physician practice style more than the patient’s condition or treatment needs.”
Researchers emphasized that they do not recommend routine outpatient management for individual sepsis patients. “Almost certainly, some of the discharged patients should have been admitted to the hospital.” Dr. Peltan said. “I think there’s still a lot of opportunity to understand who these patients are, understand why there is so much physician variation, and to develop tools to further optimize triage decisions.”
The study was funded in part by the Intermountain Research and Medical Foundation in Salt Lake City. Dr. Peltan reported having no financial disclosures.
SOURCE: Peltan ID et al. ATS 2018, Abstract A5994/702.
AT ATS 2018
Key clinical point: More research is needed to optimize triage decisions for ED sepsis patients and to understand possible disparities in ED disposition.
Major finding: Among adult patients who met clinical criteria for sepsis in the emergency department, 16.4% were not admitted to the hospital.
Study details: A retrospective study of 12,002 adult ED patients who met criteria for sepsis at two tertiary hospitals and two community hospitals in Utah.
Disclosures: The study was funded in part by the Intermountain Research and Medical Foundation in Salt Lake City. Dr. Peltan reported having no financial disclosures.
Source: Peltan ID et al. Abstract 5994/702, ATS 2018.
Biosimilar switch accepted by most rheumatic disease patients
LIVERPOOL, ENGLAND – , although the biosimilar they are being switched to may be important, according to data from three separate poster presentations at the British Society for Rheumatology annual conference.

Of 35 patients who expressed concerns about the switch, most (n = 27) were concerned about the efficacy of the biosimilar, with others were mainly concerned about safety (n = 5), side effects (n = 3), or other factors (n = 5).
“This is the population of patients we were worried about, because we had got them on a drug that had finally worked for them,” poster presenter Joanne Kitchen, MBChB, said in an interview.
“It’s hard enough to get on the biologic, and we were concerned about whether they would lose response. ... There wasn’t a lot of evidence about if they didn’t respond and we switched back, would it still work for them,” explained Dr. Kitchen, a consultant rheumatologist who works at the Royal Berkshire Hospital in Reading, England.
Biosimilar etanercept became available in the United Kingdom in April 2016, and many rheumatology centers had to make the switch to its use at the behest of their health trusts in a cost-saving effort. The switch at the Royal Berkshire occurred in August 2016, and Dr. Kitchen explained that prior to the switch, letters were sent out to inform patients, who were then seen in the clinic. There also was an understanding between the medical team and the patients that, if things did not work out, patients could switch back to the originator etanercept.
Between August 2016 and February 2017, 113 patients had switched to biosimilar etanercept for their rheumatoid arthritis (RA), spondyloarthritis, or psoriatic arthritis.
Although worsening joint pain or stiffness (n = 12) or increased fatigue (n = 4) were reported by some patients, the fact that 88% of those who responded to the survey in October 2017 were still taking the drug 6-12 months after initiation suggests that these side effects were minor or manageable. Adherence to medication was not checked, however, which might have been a factor in any flare ups.
Medication changes occurred for four patients who switched back to originator etanercept, three to an alternative biologic, and four who discontinued biologics.
Other adverse effects reported by patients were more painful injections (n = 5), infections (n = 2), and others incidents such as individual cases of rash and headache in the remainder.
“We know our biologic costs are incrementally increasing, but it’s still very hard for some patients to get onto these drugs,” Dr. Kitchen said. She hopes that with the cost-savings being made from the switch, it could help with negotiations to lower the threshold at which patients become eligible for biologic/biosimilar use, thus enabling more patients in need to be treated.
“I think these data have given confidence that patients can switch onto a biosimilar, and that the real-world experience matches what we’re seeing in trials,” Dr. Kitchen said. “We haven’t had a negative experience, and that’s what patients and we were worried about.”
In a separate poster presentation, Kavina Shah, MBBS, and her associates from Northwick Park Hospital, London, reported their experience of switching 115 patients with RA from etanercept to the biosimilar Benepali between January and June 2017.
They conducted a prospective study in which patients were offered an education session and then attended a clinic appointment set up to manage the switch. Patients were assessed by various objective and subjective means before and 4 months after the switch.
Dr. Shah and her associates found that 43% of patients were pleased with the switch. Part of the reason patients might have been happy with the switch was the easier mode of administration, they observed: “Patients commented on the easier technique and less manual dexterity required.”
However, almost a quarter (23%) of patients were not happy with the switch, with others being indifferent (7%) or unsure (8%).
Patients were also asked how they felt their RA was after the switch, and 75% responded that it was no different, 11% said it had improved, and 17% said it was worse.
The mean Disease Activity Score in 28 joints (DAS28) values were significantly lower in patients after the switch than before (2.66 vs. 2.97; P = .0019). “This could be explained by the lower levels of immunogenicity with Benepali,” Dr. Shah and her coauthors wrote on their poster. Alternatively, it could be an artifact introduced by lower rates of anxiety at follow-up, they said.
There were also statistically nonsignificant improvements in health assessment questionnaire (HAQ) and European Quality of Life-5 Dimensions (EQ-5D) scores.
Taken together, these findings are “reassuring,” Dr. Shah and her associates noted, and “should positively encourage clinicians and patients to switch to biosimilars in order to optimize the cost saving to the NHS.”
Not all biosimilar switches may go as smoothly as switching from TNF inhibitors, as Muhammad K. Nisar, MBBS, reported in another poster presentation at the conference. Dr. Nisar, a consultant rheumatologist for Luton (England) and Dunstable Hospital University Trust, reported his center’s experience of switching patients on rituximab (Rituxan) to biosimilar rituximab (Truxima).
Of 44 patients who were established on rituximab, 39 were eligible to make the switch. Four patients had stopped taking rituximab before the switch took place and one patient remained on the originator. As of October 2017, 24 (61.5%) of patients had actually made the switch.
“All were happy to switch after receiving a letter and having the opportunity to contact if necessary,” Dr. Nisar reported. “At group level there were no major differences in disease outcomes and 80% reported no issues.”
However, five (20%) patients developed a severe serum sickness reaction early on with loss of efficacy. This happened in the first week after the second dose of the biosimilar was given, Dr. Nisar explained. No obvious reason could be found, but two patients required emergency hospital treatment within 24 hours.
“Our experience of switching rituximab patients is certainly not as smooth as it was for infliximab or and etanercept,” Dr. Nisar said. While he said “they support routine switching from originator to biosimilar,” he noted that “close monitoring is required, certainly in the first week of dose administration.”
All authors had nothing to disclose.
SOURCES: Hoque T et al. Rheumatology. 2018 Apr 25;57(Suppl. 3):key075.296. Shah K et al. Rheumatology. 2018 Apr 25;57(Suppl. 3):key075.456. Nisar MK. Rheumatology. 2018 Apr 1;57(Suppl. 3):key075.516.
LIVERPOOL, ENGLAND – , although the biosimilar they are being switched to may be important, according to data from three separate poster presentations at the British Society for Rheumatology annual conference.
Of 35 patients who expressed concerns about the switch, most (n = 27) were concerned about the efficacy of the biosimilar, with others were mainly concerned about safety (n = 5), side effects (n = 3), or other factors (n = 5).
“This is the population of patients we were worried about, because we had got them on a drug that had finally worked for them,” poster presenter Joanne Kitchen, MBChB, said in an interview.
“It’s hard enough to get on the biologic, and we were concerned about whether they would lose response. ... There wasn’t a lot of evidence about if they didn’t respond and we switched back, would it still work for them,” explained Dr. Kitchen, a consultant rheumatologist who works at the Royal Berkshire Hospital in Reading, England.
Biosimilar etanercept became available in the United Kingdom in April 2016, and many rheumatology centers had to make the switch to its use at the behest of their health trusts in a cost-saving effort. The switch at the Royal Berkshire occurred in August 2016, and Dr. Kitchen explained that prior to the switch, letters were sent out to inform patients, who were then seen in the clinic. There also was an understanding between the medical team and the patients that, if things did not work out, patients could switch back to the originator etanercept.
Between August 2016 and February 2017, 113 patients had switched to biosimilar etanercept for their rheumatoid arthritis (RA), spondyloarthritis, or psoriatic arthritis.
Although worsening joint pain or stiffness (n = 12) or increased fatigue (n = 4) were reported by some patients, the fact that 88% of those who responded to the survey in October 2017 were still taking the drug 6-12 months after initiation suggests that these side effects were minor or manageable. Adherence to medication was not checked, however, which might have been a factor in any flare ups.
Medication changes occurred for four patients who switched back to originator etanercept, three to an alternative biologic, and four who discontinued biologics.
Other adverse effects reported by patients were more painful injections (n = 5), infections (n = 2), and others incidents such as individual cases of rash and headache in the remainder.
“We know our biologic costs are incrementally increasing, but it’s still very hard for some patients to get onto these drugs,” Dr. Kitchen said. She hopes that with the cost-savings being made from the switch, it could help with negotiations to lower the threshold at which patients become eligible for biologic/biosimilar use, thus enabling more patients in need to be treated.
“I think these data have given confidence that patients can switch onto a biosimilar, and that the real-world experience matches what we’re seeing in trials,” Dr. Kitchen said. “We haven’t had a negative experience, and that’s what patients and we were worried about.”
In a separate poster presentation, Kavina Shah, MBBS, and her associates from Northwick Park Hospital, London, reported their experience of switching 115 patients with RA from etanercept to the biosimilar Benepali between January and June 2017.
They conducted a prospective study in which patients were offered an education session and then attended a clinic appointment set up to manage the switch. Patients were assessed by various objective and subjective means before and 4 months after the switch.
Dr. Shah and her associates found that 43% of patients were pleased with the switch. Part of the reason patients might have been happy with the switch was the easier mode of administration, they observed: “Patients commented on the easier technique and less manual dexterity required.”
However, almost a quarter (23%) of patients were not happy with the switch, with others being indifferent (7%) or unsure (8%).
Patients were also asked how they felt their RA was after the switch, and 75% responded that it was no different, 11% said it had improved, and 17% said it was worse.
The mean Disease Activity Score in 28 joints (DAS28) values were significantly lower in patients after the switch than before (2.66 vs. 2.97; P = .0019). “This could be explained by the lower levels of immunogenicity with Benepali,” Dr. Shah and her coauthors wrote on their poster. Alternatively, it could be an artifact introduced by lower rates of anxiety at follow-up, they said.
There were also statistically nonsignificant improvements in health assessment questionnaire (HAQ) and European Quality of Life-5 Dimensions (EQ-5D) scores.
Taken together, these findings are “reassuring,” Dr. Shah and her associates noted, and “should positively encourage clinicians and patients to switch to biosimilars in order to optimize the cost saving to the NHS.”
Not all biosimilar switches may go as smoothly as switching from TNF inhibitors, as Muhammad K. Nisar, MBBS, reported in another poster presentation at the conference. Dr. Nisar, a consultant rheumatologist for Luton (England) and Dunstable Hospital University Trust, reported his center’s experience of switching patients on rituximab (Rituxan) to biosimilar rituximab (Truxima).
Of 44 patients who were established on rituximab, 39 were eligible to make the switch. Four patients had stopped taking rituximab before the switch took place and one patient remained on the originator. As of October 2017, 24 (61.5%) of patients had actually made the switch.
“All were happy to switch after receiving a letter and having the opportunity to contact if necessary,” Dr. Nisar reported. “At group level there were no major differences in disease outcomes and 80% reported no issues.”
However, five (20%) patients developed a severe serum sickness reaction early on with loss of efficacy. This happened in the first week after the second dose of the biosimilar was given, Dr. Nisar explained. No obvious reason could be found, but two patients required emergency hospital treatment within 24 hours.
“Our experience of switching rituximab patients is certainly not as smooth as it was for infliximab or and etanercept,” Dr. Nisar said. While he said “they support routine switching from originator to biosimilar,” he noted that “close monitoring is required, certainly in the first week of dose administration.”
All authors had nothing to disclose.
SOURCES: Hoque T et al. Rheumatology. 2018 Apr 25;57(Suppl. 3):key075.296. Shah K et al. Rheumatology. 2018 Apr 25;57(Suppl. 3):key075.456. Nisar MK. Rheumatology. 2018 Apr 1;57(Suppl. 3):key075.516.
LIVERPOOL, ENGLAND – , although the biosimilar they are being switched to may be important, according to data from three separate poster presentations at the British Society for Rheumatology annual conference.
Of 35 patients who expressed concerns about the switch, most (n = 27) were concerned about the efficacy of the biosimilar, with others were mainly concerned about safety (n = 5), side effects (n = 3), or other factors (n = 5).
“This is the population of patients we were worried about, because we had got them on a drug that had finally worked for them,” poster presenter Joanne Kitchen, MBChB, said in an interview.
“It’s hard enough to get on the biologic, and we were concerned about whether they would lose response. ... There wasn’t a lot of evidence about if they didn’t respond and we switched back, would it still work for them,” explained Dr. Kitchen, a consultant rheumatologist who works at the Royal Berkshire Hospital in Reading, England.
Biosimilar etanercept became available in the United Kingdom in April 2016, and many rheumatology centers had to make the switch to its use at the behest of their health trusts in a cost-saving effort. The switch at the Royal Berkshire occurred in August 2016, and Dr. Kitchen explained that prior to the switch, letters were sent out to inform patients, who were then seen in the clinic. There also was an understanding between the medical team and the patients that, if things did not work out, patients could switch back to the originator etanercept.
Between August 2016 and February 2017, 113 patients had switched to biosimilar etanercept for their rheumatoid arthritis (RA), spondyloarthritis, or psoriatic arthritis.
Although worsening joint pain or stiffness (n = 12) or increased fatigue (n = 4) were reported by some patients, the fact that 88% of those who responded to the survey in October 2017 were still taking the drug 6-12 months after initiation suggests that these side effects were minor or manageable. Adherence to medication was not checked, however, which might have been a factor in any flare ups.
Medication changes occurred for four patients who switched back to originator etanercept, three to an alternative biologic, and four who discontinued biologics.
Other adverse effects reported by patients were more painful injections (n = 5), infections (n = 2), and others incidents such as individual cases of rash and headache in the remainder.
“We know our biologic costs are incrementally increasing, but it’s still very hard for some patients to get onto these drugs,” Dr. Kitchen said. She hopes that with the cost-savings being made from the switch, it could help with negotiations to lower the threshold at which patients become eligible for biologic/biosimilar use, thus enabling more patients in need to be treated.
“I think these data have given confidence that patients can switch onto a biosimilar, and that the real-world experience matches what we’re seeing in trials,” Dr. Kitchen said. “We haven’t had a negative experience, and that’s what patients and we were worried about.”
In a separate poster presentation, Kavina Shah, MBBS, and her associates from Northwick Park Hospital, London, reported their experience of switching 115 patients with RA from etanercept to the biosimilar Benepali between January and June 2017.
They conducted a prospective study in which patients were offered an education session and then attended a clinic appointment set up to manage the switch. Patients were assessed by various objective and subjective means before and 4 months after the switch.
Dr. Shah and her associates found that 43% of patients were pleased with the switch. Part of the reason patients might have been happy with the switch was the easier mode of administration, they observed: “Patients commented on the easier technique and less manual dexterity required.”
However, almost a quarter (23%) of patients were not happy with the switch, with others being indifferent (7%) or unsure (8%).
Patients were also asked how they felt their RA was after the switch, and 75% responded that it was no different, 11% said it had improved, and 17% said it was worse.
The mean Disease Activity Score in 28 joints (DAS28) values were significantly lower in patients after the switch than before (2.66 vs. 2.97; P = .0019). “This could be explained by the lower levels of immunogenicity with Benepali,” Dr. Shah and her coauthors wrote on their poster. Alternatively, it could be an artifact introduced by lower rates of anxiety at follow-up, they said.
There were also statistically nonsignificant improvements in health assessment questionnaire (HAQ) and European Quality of Life-5 Dimensions (EQ-5D) scores.
Taken together, these findings are “reassuring,” Dr. Shah and her associates noted, and “should positively encourage clinicians and patients to switch to biosimilars in order to optimize the cost saving to the NHS.”
Not all biosimilar switches may go as smoothly as switching from TNF inhibitors, as Muhammad K. Nisar, MBBS, reported in another poster presentation at the conference. Dr. Nisar, a consultant rheumatologist for Luton (England) and Dunstable Hospital University Trust, reported his center’s experience of switching patients on rituximab (Rituxan) to biosimilar rituximab (Truxima).
Of 44 patients who were established on rituximab, 39 were eligible to make the switch. Four patients had stopped taking rituximab before the switch took place and one patient remained on the originator. As of October 2017, 24 (61.5%) of patients had actually made the switch.
“All were happy to switch after receiving a letter and having the opportunity to contact if necessary,” Dr. Nisar reported. “At group level there were no major differences in disease outcomes and 80% reported no issues.”
However, five (20%) patients developed a severe serum sickness reaction early on with loss of efficacy. This happened in the first week after the second dose of the biosimilar was given, Dr. Nisar explained. No obvious reason could be found, but two patients required emergency hospital treatment within 24 hours.
“Our experience of switching rituximab patients is certainly not as smooth as it was for infliximab or and etanercept,” Dr. Nisar said. While he said “they support routine switching from originator to biosimilar,” he noted that “close monitoring is required, certainly in the first week of dose administration.”
All authors had nothing to disclose.
SOURCES: Hoque T et al. Rheumatology. 2018 Apr 25;57(Suppl. 3):key075.296. Shah K et al. Rheumatology. 2018 Apr 25;57(Suppl. 3):key075.456. Nisar MK. Rheumatology. 2018 Apr 1;57(Suppl. 3):key075.516.
REPORTING FROM BSR 2018