User login
Accidental bowel injury occurs in 2% of hernia repairs
occurring in only about 2% of these cases, a database review has determined. But patients who experience this kind of injury have a significantly longer length of stay and are at increased risk for fistulas, sepsis, reoperations and readmissions, and even death, David M. Krpata, MD, of the Cleveland Clinic and his colleagues wrote in Surgery.
“When these events occur, the surgeon must decide whether to repair primarily or resect the bowel, proceed with definitive hernia repair with mesh, or abort the procedure and repair primarily the hernia defect,” they wrote. The lack of published studies on this injury prompted the research team to look into prevalence and outcomes in order to offer some data to guide surgical decision making.
The research team examined surgical outcomes among 5,916 patients who underwent a ventral hernia repair during 2013-2017 and were included in the Americas Hernia Society Quality Collaborative, a national hernia surgery database. The database included information from the records of 180 surgeons.
The multivariate analysis controlled for sex, race, elective case, wound status, hernia width, immunosuppressants, subcutaneous flaps, myofascial release, drains, smoking, body mass index, age, diabetes, laparoscopic surgery, mesh type, and concomitant procedure.
Among the cohort, there were 110 full-thickness bowel injuries (1.9%). Three patients also had a bladder injury. Most of the enterotomies were small-bowel injuries (85%); the rest were colon injuries. The majority of patients (64%) underwent a primary repair; 36% required bowel resection. Injuries were most common among patients with larger hernia defects, recurrent repairs, mesh or active infection, a history of abdominal wound infection, and older age.
Patients with the accidental enterotomies were less likely to get a mesh repair (85% vs. 94%). When they did, their surgeons were less likely to use a permanent synthetic barrier–coated mesh and more likely to use biologic mesh, absorbable mesh, and/or uncoated synthetic mesh. But the investigators wrote: “Further data are necessary to address specifically what is the most appropriate mesh to utilize (if any) after an inadvertent enterotomy has occurred and which compartment within the abdominal wall is safest.”
In the fully adjusted analysis, injured patients were no more likely to experience surgical site infections, but they were significantly more likely to develop an enterocutaneous fistula (4% vs. 1%), sepsis (2% vs. 1%), and to die (3% vs 1%) after a bowel injury. They also had a significantly longer length of stay (7 vs. 4 days), and more reoperations (6% vs. 3%). Major wound complication was the most common reason for reoperation (43%) and readmission (58%).
The limitations of this study mostly reflect variables not captured by the database. For example, the tenacity of adhesions and the duration of adhesiolysis are not accounted for. The investigators noted that “patients with an enterotomy likely had more tenacious adhesions, given that an operative time greater than 2 hours had a greater association with an enterotomy (91% vs. 71%).” These patients were more likely to be older and have COPD. In addition, unrecognized enterotomies were not accounted for in the data, but inclusion of those injuries would likely have meant worse outcomes.
“Although definitive hernia repair with mesh can be safely performed, surgeons should consider multiple factors, including type of mesh and location of mesh in the abdominal wall, before proceeding with definitive repair in any case of an enterotomy,” Dr. Krpata and his coauthors concluded.
The investigators reported no financial disclosures.
SOURCE: Krpata et al. Surg. 2018. doi: 10.1016/j.surg.2018.04.003
occurring in only about 2% of these cases, a database review has determined. But patients who experience this kind of injury have a significantly longer length of stay and are at increased risk for fistulas, sepsis, reoperations and readmissions, and even death, David M. Krpata, MD, of the Cleveland Clinic and his colleagues wrote in Surgery.
“When these events occur, the surgeon must decide whether to repair primarily or resect the bowel, proceed with definitive hernia repair with mesh, or abort the procedure and repair primarily the hernia defect,” they wrote. The lack of published studies on this injury prompted the research team to look into prevalence and outcomes in order to offer some data to guide surgical decision making.
The research team examined surgical outcomes among 5,916 patients who underwent a ventral hernia repair during 2013-2017 and were included in the Americas Hernia Society Quality Collaborative, a national hernia surgery database. The database included information from the records of 180 surgeons.
The multivariate analysis controlled for sex, race, elective case, wound status, hernia width, immunosuppressants, subcutaneous flaps, myofascial release, drains, smoking, body mass index, age, diabetes, laparoscopic surgery, mesh type, and concomitant procedure.
Among the cohort, there were 110 full-thickness bowel injuries (1.9%). Three patients also had a bladder injury. Most of the enterotomies were small-bowel injuries (85%); the rest were colon injuries. The majority of patients (64%) underwent a primary repair; 36% required bowel resection. Injuries were most common among patients with larger hernia defects, recurrent repairs, mesh or active infection, a history of abdominal wound infection, and older age.
Patients with the accidental enterotomies were less likely to get a mesh repair (85% vs. 94%). When they did, their surgeons were less likely to use a permanent synthetic barrier–coated mesh and more likely to use biologic mesh, absorbable mesh, and/or uncoated synthetic mesh. But the investigators wrote: “Further data are necessary to address specifically what is the most appropriate mesh to utilize (if any) after an inadvertent enterotomy has occurred and which compartment within the abdominal wall is safest.”
In the fully adjusted analysis, injured patients were no more likely to experience surgical site infections, but they were significantly more likely to develop an enterocutaneous fistula (4% vs. 1%), sepsis (2% vs. 1%), and to die (3% vs 1%) after a bowel injury. They also had a significantly longer length of stay (7 vs. 4 days), and more reoperations (6% vs. 3%). Major wound complication was the most common reason for reoperation (43%) and readmission (58%).
The limitations of this study mostly reflect variables not captured by the database. For example, the tenacity of adhesions and the duration of adhesiolysis are not accounted for. The investigators noted that “patients with an enterotomy likely had more tenacious adhesions, given that an operative time greater than 2 hours had a greater association with an enterotomy (91% vs. 71%).” These patients were more likely to be older and have COPD. In addition, unrecognized enterotomies were not accounted for in the data, but inclusion of those injuries would likely have meant worse outcomes.
“Although definitive hernia repair with mesh can be safely performed, surgeons should consider multiple factors, including type of mesh and location of mesh in the abdominal wall, before proceeding with definitive repair in any case of an enterotomy,” Dr. Krpata and his coauthors concluded.
The investigators reported no financial disclosures.
SOURCE: Krpata et al. Surg. 2018. doi: 10.1016/j.surg.2018.04.003
occurring in only about 2% of these cases, a database review has determined. But patients who experience this kind of injury have a significantly longer length of stay and are at increased risk for fistulas, sepsis, reoperations and readmissions, and even death, David M. Krpata, MD, of the Cleveland Clinic and his colleagues wrote in Surgery.
“When these events occur, the surgeon must decide whether to repair primarily or resect the bowel, proceed with definitive hernia repair with mesh, or abort the procedure and repair primarily the hernia defect,” they wrote. The lack of published studies on this injury prompted the research team to look into prevalence and outcomes in order to offer some data to guide surgical decision making.
The research team examined surgical outcomes among 5,916 patients who underwent a ventral hernia repair during 2013-2017 and were included in the Americas Hernia Society Quality Collaborative, a national hernia surgery database. The database included information from the records of 180 surgeons.
The multivariate analysis controlled for sex, race, elective case, wound status, hernia width, immunosuppressants, subcutaneous flaps, myofascial release, drains, smoking, body mass index, age, diabetes, laparoscopic surgery, mesh type, and concomitant procedure.
Among the cohort, there were 110 full-thickness bowel injuries (1.9%). Three patients also had a bladder injury. Most of the enterotomies were small-bowel injuries (85%); the rest were colon injuries. The majority of patients (64%) underwent a primary repair; 36% required bowel resection. Injuries were most common among patients with larger hernia defects, recurrent repairs, mesh or active infection, a history of abdominal wound infection, and older age.
Patients with the accidental enterotomies were less likely to get a mesh repair (85% vs. 94%). When they did, their surgeons were less likely to use a permanent synthetic barrier–coated mesh and more likely to use biologic mesh, absorbable mesh, and/or uncoated synthetic mesh. But the investigators wrote: “Further data are necessary to address specifically what is the most appropriate mesh to utilize (if any) after an inadvertent enterotomy has occurred and which compartment within the abdominal wall is safest.”
In the fully adjusted analysis, injured patients were no more likely to experience surgical site infections, but they were significantly more likely to develop an enterocutaneous fistula (4% vs. 1%), sepsis (2% vs. 1%), and to die (3% vs 1%) after a bowel injury. They also had a significantly longer length of stay (7 vs. 4 days), and more reoperations (6% vs. 3%). Major wound complication was the most common reason for reoperation (43%) and readmission (58%).
The limitations of this study mostly reflect variables not captured by the database. For example, the tenacity of adhesions and the duration of adhesiolysis are not accounted for. The investigators noted that “patients with an enterotomy likely had more tenacious adhesions, given that an operative time greater than 2 hours had a greater association with an enterotomy (91% vs. 71%).” These patients were more likely to be older and have COPD. In addition, unrecognized enterotomies were not accounted for in the data, but inclusion of those injuries would likely have meant worse outcomes.
“Although definitive hernia repair with mesh can be safely performed, surgeons should consider multiple factors, including type of mesh and location of mesh in the abdominal wall, before proceeding with definitive repair in any case of an enterotomy,” Dr. Krpata and his coauthors concluded.
The investigators reported no financial disclosures.
SOURCE: Krpata et al. Surg. 2018. doi: 10.1016/j.surg.2018.04.003
FROM SURGERY
Key clinical point: Accidental bowel injuries during ventral hernia increase risk for longer hospital stays, fistula, sepsis, and readmissions.
Major finding: The overall rate of accidental enterotomy during ventral hernia repair was 2%.
Study details: The database review included 5,916 hernia repair patients.
Disclosures: None of the authors reported any financial disclosures.
Source: Krpata D et al. Surg 2018; doi.org/10.1016/j.surg.2018.04.003
Chilblain Lupus Erythematosus Presenting With Bilateral Hemorrhagic Bullae of Distal Halluces
To the Editor:
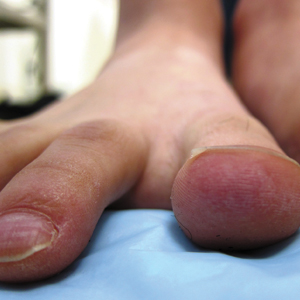
A 20-year-old man with no notable medical history presented to our dermatology clinic for evaluation of mildly painful, hemorrhagic bullae on the bilateral halluces of 1 month’s duration. On initial presentation the patient reported the lesions developed after wearing a new pair of tight-fitting shoes, suggesting a diagnosis of trauma-induced bullae. The patient was instructed to wear loose-fitting shoes and to follow up in 6 weeks to assess for improvement. At follow-up the bullae had resolved with residual violaceous patches on the bilateral distal halluces. He additionally developed a faint retiform erythematous patch on the left distal toe (Figure 1). The patient also had reticulate erythematous patches on the dorsal aspects of the hands extending to the forearms and legs resembling livedo reticularis. The patient was unsure if the skin lesions were triggered or worsened by cold exposure and reported that he smoked half a pack of cigarettes daily. At this time, the differential diagnosis still included trauma; however, there was concern for either embolic, thrombotic, or connective-tissue disease. A 4-mm punch biopsy of the left distal hallux demonstrated basal vacuolar interface dermatitis with superficial and deep perivascular inflammation and deep periadnexal mucin deposition (Figure 2) consistent with lupus dermatitis.

(H&E, original magnification ×400) with deep periadnexal mucin deposition (B)(colloidal iron, original magnification ×40).")
Serologic workup revealed increased antinuclear antibody titers of 1:320 (reference range, <1:40) and anti-Ro/Sjögren syndrome antigen antibodies of 86 (reference range, <20). There was no elevation in anti–double-stranded DNA, anti-Smith, antiribonucleoprotein, or anticardiolipin antibodies. Complement levels also were within reference range. Furthermore, the patient denied a history of Raynaud phenomenon, photosensitivity, oral ulcers, joint pain, shortness of breath, pleuritic chest pain, arthritis, blood clots, or any other systemic symptoms. Additional evaluation by the rheumatology department did not support criteria for systemic lupus erythematosus (SLE). In the context of the clinical presentation, histologic findings, and serologic markers, a diagnosis of chilblain lupus erythematosus (CHLE) was made. He was counseled on sun protection and smoking cessation and declined systemic therapy citing concern for side effects. Follow-up with the dermatology and rheumatology departments was advised.
Cutaneous lupus erythematosus (CLE) comprises various forms of lupus, including acute cutaneous lupus, subacute cutaneous lupus, and chronic cutaneous lupus. Chilblain lupus erythematosus is a rare subset of chronic CLE that first was described in 18881 and is characterized by tender violaceous papules and plaques that typically present in an acral distribution (ie, fingers, toes, nose, cheeks, ears). The skin lesions often are triggered or exacerbated by cold temperatures and dampness. As the lesions evolve, they can ulcerate, fissure, become hyperkeratotic, or result in atrophic plaques with scarring.2,3 A subset of patients also may have concurrent Raynaud phenomenon.1 Up to 20% of patients will eventually develop SLE, especially those patients with concurrent discoid lupus erythematosus, warranting close long-term follow-up.3 Serologic studies can reveal antinuclear antibodies, anti-Ro/Sjögren syndrome antigen antibodies, rheumatic factor, and anti–double-stranded DNA antibodies.1,4 Hypergammaglobulinemia also is a common finding in patients with CHLE, affecting more than two-thirds of patients.1 Typical features of CHLE seen on histopathology include interface dermatitis, perivascular lymphocytic infiltrate, apoptotic keratinocytes, lichenoid tissue reaction, and increased dermal mucin.1,4
Chilblain lupus erythematosus most commonly presents sporadically; however, there is a familial form that has been previously described.5 Sporadic CHLE usually occurs in middle-aged females, in contrast to familial CHLE, which presents in early childhood.1 The pathogenesis of the sporadic form is poorly understood, but it is thought to be stimulated by vasoconstriction or microvascular injury provoked by cold exposure. Furthermore, hypergammaglobulinemia and the presence of autoantibodies may contribute to the pathogenesis by increasing blood viscosity.1 The
Several drugs including thiazides, terbinafine, calcium channel blockers, angiotensin-converting enzyme inhibitors, and chemotherapeutic agents have been reported to trigger CHLE.4 Tumor necrosis factor α inhibitors have been shown to precipitate CHLE.6 Of note, drug-induced CHLE usually is limited to the skin and has not been shown to progress to SLE.6 Lebeau et al4 described a patient with breast cancer and preexisting CHLE that flared while the patient received docetaxel therapy, suggesting that certain drugs may not only induce but also may aggravate CHLE.
Many of the therapies that are effective in SLE such as antimalarial agents (ie, chloroquine, hydroxychloroquine) often are less efficacious in treating the lesions of CHLE.1 However, these patients often can be managed successfully by physical protection from the cold environment.1 Calcium channel blockers such as nifedipine also have been implicated, as they counteract vasoconstriction, which is thought to contribute to the pathogenesis of CHLE.1 Topical and systemic steroids also have been used to treat CHLE. Dapsone and pentoxifylline are other treatment modalities that have been effective in select cases of CHLE.5 Boehm and Bieber7 reported near resolution of CHLE with mycophenolate mofetil in an elderly woman with skin lesions that had been refractory to systemic steroids, antimalarial agents, azathioprine, dapsone, and pentoxifylline, suggesting that mycophenolate mofetil may be a therapeutic option for recalcitrant cases of CHLE. Local immunosuppressive agents such as tacrolimus also can be considered in treatment-refractory disease.
Chilblain lupus erythematosus is a rare chronic form of CLE that typically occurs sporadically but also has a familial form that has been described in several families. It most commonly is observed in middle-aged women, but we describe a case in a young man. Although CHLE typically does not respond well to traditional lupus therapies used in the management of SLE, good effects have been observed with cold avoidance, calcium channel blockers, and topical or oral steroids. For treatment-refractory cases, mycophenolate mofetil and other immunosuppressive agents have been shown to be effective.
- Hedrich CM, Fiebig B, Hauck FH, et al. Chilblain lupus erythematosus—a review of literature. Clin Rheumatol. 2008;27:949-954.
- Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. Berlin, Germany: Springer; 2005.
- Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010;19:1050-1070.
- Lebeau S, També S, Sallam MA, et al. Docetaxel-induced relapse of subacute cutaneous lupus erythematosus and chilblain lupus. J Dtsch Dermatol Ges. 2013;11:871-874.
- Günther C, Hillebrand M, Brunk J, et al. Systemic involvement in TREX1-associated familial chilblain lupus. J Am Acad Dermatol. 2013;69:179-181.
- Sifuentes Giraldo WA, Ahijón Lana M, García Villanueva MJ, et al. Chilblain lupus induced by TNF-α antagonists: a case report and literature review. Clin Rheumatol. 2012;31:563-568.
- Boehm I, Bieber T. Chilblain lupus erythematosus Hutchinson: successful treatment with mycophenolate mofetil. Arch Dermatol. 2001;137:235-236.
To the Editor:
A 20-year-old man with no notable medical history presented to our dermatology clinic for evaluation of mildly painful, hemorrhagic bullae on the bilateral halluces of 1 month’s duration. On initial presentation the patient reported the lesions developed after wearing a new pair of tight-fitting shoes, suggesting a diagnosis of trauma-induced bullae. The patient was instructed to wear loose-fitting shoes and to follow up in 6 weeks to assess for improvement. At follow-up the bullae had resolved with residual violaceous patches on the bilateral distal halluces. He additionally developed a faint retiform erythematous patch on the left distal toe (Figure 1). The patient also had reticulate erythematous patches on the dorsal aspects of the hands extending to the forearms and legs resembling livedo reticularis. The patient was unsure if the skin lesions were triggered or worsened by cold exposure and reported that he smoked half a pack of cigarettes daily. At this time, the differential diagnosis still included trauma; however, there was concern for either embolic, thrombotic, or connective-tissue disease. A 4-mm punch biopsy of the left distal hallux demonstrated basal vacuolar interface dermatitis with superficial and deep perivascular inflammation and deep periadnexal mucin deposition (Figure 2) consistent with lupus dermatitis.
Serologic workup revealed increased antinuclear antibody titers of 1:320 (reference range, <1:40) and anti-Ro/Sjögren syndrome antigen antibodies of 86 (reference range, <20). There was no elevation in anti–double-stranded DNA, anti-Smith, antiribonucleoprotein, or anticardiolipin antibodies. Complement levels also were within reference range. Furthermore, the patient denied a history of Raynaud phenomenon, photosensitivity, oral ulcers, joint pain, shortness of breath, pleuritic chest pain, arthritis, blood clots, or any other systemic symptoms. Additional evaluation by the rheumatology department did not support criteria for systemic lupus erythematosus (SLE). In the context of the clinical presentation, histologic findings, and serologic markers, a diagnosis of chilblain lupus erythematosus (CHLE) was made. He was counseled on sun protection and smoking cessation and declined systemic therapy citing concern for side effects. Follow-up with the dermatology and rheumatology departments was advised.
Cutaneous lupus erythematosus (CLE) comprises various forms of lupus, including acute cutaneous lupus, subacute cutaneous lupus, and chronic cutaneous lupus. Chilblain lupus erythematosus is a rare subset of chronic CLE that first was described in 18881 and is characterized by tender violaceous papules and plaques that typically present in an acral distribution (ie, fingers, toes, nose, cheeks, ears). The skin lesions often are triggered or exacerbated by cold temperatures and dampness. As the lesions evolve, they can ulcerate, fissure, become hyperkeratotic, or result in atrophic plaques with scarring.2,3 A subset of patients also may have concurrent Raynaud phenomenon.1 Up to 20% of patients will eventually develop SLE, especially those patients with concurrent discoid lupus erythematosus, warranting close long-term follow-up.3 Serologic studies can reveal antinuclear antibodies, anti-Ro/Sjögren syndrome antigen antibodies, rheumatic factor, and anti–double-stranded DNA antibodies.1,4 Hypergammaglobulinemia also is a common finding in patients with CHLE, affecting more than two-thirds of patients.1 Typical features of CHLE seen on histopathology include interface dermatitis, perivascular lymphocytic infiltrate, apoptotic keratinocytes, lichenoid tissue reaction, and increased dermal mucin.1,4
Chilblain lupus erythematosus most commonly presents sporadically; however, there is a familial form that has been previously described.5 Sporadic CHLE usually occurs in middle-aged females, in contrast to familial CHLE, which presents in early childhood.1 The pathogenesis of the sporadic form is poorly understood, but it is thought to be stimulated by vasoconstriction or microvascular injury provoked by cold exposure. Furthermore, hypergammaglobulinemia and the presence of autoantibodies may contribute to the pathogenesis by increasing blood viscosity.1 The
Several drugs including thiazides, terbinafine, calcium channel blockers, angiotensin-converting enzyme inhibitors, and chemotherapeutic agents have been reported to trigger CHLE.4 Tumor necrosis factor α inhibitors have been shown to precipitate CHLE.6 Of note, drug-induced CHLE usually is limited to the skin and has not been shown to progress to SLE.6 Lebeau et al4 described a patient with breast cancer and preexisting CHLE that flared while the patient received docetaxel therapy, suggesting that certain drugs may not only induce but also may aggravate CHLE.
Many of the therapies that are effective in SLE such as antimalarial agents (ie, chloroquine, hydroxychloroquine) often are less efficacious in treating the lesions of CHLE.1 However, these patients often can be managed successfully by physical protection from the cold environment.1 Calcium channel blockers such as nifedipine also have been implicated, as they counteract vasoconstriction, which is thought to contribute to the pathogenesis of CHLE.1 Topical and systemic steroids also have been used to treat CHLE. Dapsone and pentoxifylline are other treatment modalities that have been effective in select cases of CHLE.5 Boehm and Bieber7 reported near resolution of CHLE with mycophenolate mofetil in an elderly woman with skin lesions that had been refractory to systemic steroids, antimalarial agents, azathioprine, dapsone, and pentoxifylline, suggesting that mycophenolate mofetil may be a therapeutic option for recalcitrant cases of CHLE. Local immunosuppressive agents such as tacrolimus also can be considered in treatment-refractory disease.
Chilblain lupus erythematosus is a rare chronic form of CLE that typically occurs sporadically but also has a familial form that has been described in several families. It most commonly is observed in middle-aged women, but we describe a case in a young man. Although CHLE typically does not respond well to traditional lupus therapies used in the management of SLE, good effects have been observed with cold avoidance, calcium channel blockers, and topical or oral steroids. For treatment-refractory cases, mycophenolate mofetil and other immunosuppressive agents have been shown to be effective.
To the Editor:
A 20-year-old man with no notable medical history presented to our dermatology clinic for evaluation of mildly painful, hemorrhagic bullae on the bilateral halluces of 1 month’s duration. On initial presentation the patient reported the lesions developed after wearing a new pair of tight-fitting shoes, suggesting a diagnosis of trauma-induced bullae. The patient was instructed to wear loose-fitting shoes and to follow up in 6 weeks to assess for improvement. At follow-up the bullae had resolved with residual violaceous patches on the bilateral distal halluces. He additionally developed a faint retiform erythematous patch on the left distal toe (Figure 1). The patient also had reticulate erythematous patches on the dorsal aspects of the hands extending to the forearms and legs resembling livedo reticularis. The patient was unsure if the skin lesions were triggered or worsened by cold exposure and reported that he smoked half a pack of cigarettes daily. At this time, the differential diagnosis still included trauma; however, there was concern for either embolic, thrombotic, or connective-tissue disease. A 4-mm punch biopsy of the left distal hallux demonstrated basal vacuolar interface dermatitis with superficial and deep perivascular inflammation and deep periadnexal mucin deposition (Figure 2) consistent with lupus dermatitis.
Serologic workup revealed increased antinuclear antibody titers of 1:320 (reference range, <1:40) and anti-Ro/Sjögren syndrome antigen antibodies of 86 (reference range, <20). There was no elevation in anti–double-stranded DNA, anti-Smith, antiribonucleoprotein, or anticardiolipin antibodies. Complement levels also were within reference range. Furthermore, the patient denied a history of Raynaud phenomenon, photosensitivity, oral ulcers, joint pain, shortness of breath, pleuritic chest pain, arthritis, blood clots, or any other systemic symptoms. Additional evaluation by the rheumatology department did not support criteria for systemic lupus erythematosus (SLE). In the context of the clinical presentation, histologic findings, and serologic markers, a diagnosis of chilblain lupus erythematosus (CHLE) was made. He was counseled on sun protection and smoking cessation and declined systemic therapy citing concern for side effects. Follow-up with the dermatology and rheumatology departments was advised.
Cutaneous lupus erythematosus (CLE) comprises various forms of lupus, including acute cutaneous lupus, subacute cutaneous lupus, and chronic cutaneous lupus. Chilblain lupus erythematosus is a rare subset of chronic CLE that first was described in 18881 and is characterized by tender violaceous papules and plaques that typically present in an acral distribution (ie, fingers, toes, nose, cheeks, ears). The skin lesions often are triggered or exacerbated by cold temperatures and dampness. As the lesions evolve, they can ulcerate, fissure, become hyperkeratotic, or result in atrophic plaques with scarring.2,3 A subset of patients also may have concurrent Raynaud phenomenon.1 Up to 20% of patients will eventually develop SLE, especially those patients with concurrent discoid lupus erythematosus, warranting close long-term follow-up.3 Serologic studies can reveal antinuclear antibodies, anti-Ro/Sjögren syndrome antigen antibodies, rheumatic factor, and anti–double-stranded DNA antibodies.1,4 Hypergammaglobulinemia also is a common finding in patients with CHLE, affecting more than two-thirds of patients.1 Typical features of CHLE seen on histopathology include interface dermatitis, perivascular lymphocytic infiltrate, apoptotic keratinocytes, lichenoid tissue reaction, and increased dermal mucin.1,4
Chilblain lupus erythematosus most commonly presents sporadically; however, there is a familial form that has been previously described.5 Sporadic CHLE usually occurs in middle-aged females, in contrast to familial CHLE, which presents in early childhood.1 The pathogenesis of the sporadic form is poorly understood, but it is thought to be stimulated by vasoconstriction or microvascular injury provoked by cold exposure. Furthermore, hypergammaglobulinemia and the presence of autoantibodies may contribute to the pathogenesis by increasing blood viscosity.1 The
Several drugs including thiazides, terbinafine, calcium channel blockers, angiotensin-converting enzyme inhibitors, and chemotherapeutic agents have been reported to trigger CHLE.4 Tumor necrosis factor α inhibitors have been shown to precipitate CHLE.6 Of note, drug-induced CHLE usually is limited to the skin and has not been shown to progress to SLE.6 Lebeau et al4 described a patient with breast cancer and preexisting CHLE that flared while the patient received docetaxel therapy, suggesting that certain drugs may not only induce but also may aggravate CHLE.
Many of the therapies that are effective in SLE such as antimalarial agents (ie, chloroquine, hydroxychloroquine) often are less efficacious in treating the lesions of CHLE.1 However, these patients often can be managed successfully by physical protection from the cold environment.1 Calcium channel blockers such as nifedipine also have been implicated, as they counteract vasoconstriction, which is thought to contribute to the pathogenesis of CHLE.1 Topical and systemic steroids also have been used to treat CHLE. Dapsone and pentoxifylline are other treatment modalities that have been effective in select cases of CHLE.5 Boehm and Bieber7 reported near resolution of CHLE with mycophenolate mofetil in an elderly woman with skin lesions that had been refractory to systemic steroids, antimalarial agents, azathioprine, dapsone, and pentoxifylline, suggesting that mycophenolate mofetil may be a therapeutic option for recalcitrant cases of CHLE. Local immunosuppressive agents such as tacrolimus also can be considered in treatment-refractory disease.
Chilblain lupus erythematosus is a rare chronic form of CLE that typically occurs sporadically but also has a familial form that has been described in several families. It most commonly is observed in middle-aged women, but we describe a case in a young man. Although CHLE typically does not respond well to traditional lupus therapies used in the management of SLE, good effects have been observed with cold avoidance, calcium channel blockers, and topical or oral steroids. For treatment-refractory cases, mycophenolate mofetil and other immunosuppressive agents have been shown to be effective.
- Hedrich CM, Fiebig B, Hauck FH, et al. Chilblain lupus erythematosus—a review of literature. Clin Rheumatol. 2008;27:949-954.
- Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. Berlin, Germany: Springer; 2005.
- Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010;19:1050-1070.
- Lebeau S, També S, Sallam MA, et al. Docetaxel-induced relapse of subacute cutaneous lupus erythematosus and chilblain lupus. J Dtsch Dermatol Ges. 2013;11:871-874.
- Günther C, Hillebrand M, Brunk J, et al. Systemic involvement in TREX1-associated familial chilblain lupus. J Am Acad Dermatol. 2013;69:179-181.
- Sifuentes Giraldo WA, Ahijón Lana M, García Villanueva MJ, et al. Chilblain lupus induced by TNF-α antagonists: a case report and literature review. Clin Rheumatol. 2012;31:563-568.
- Boehm I, Bieber T. Chilblain lupus erythematosus Hutchinson: successful treatment with mycophenolate mofetil. Arch Dermatol. 2001;137:235-236.
- Hedrich CM, Fiebig B, Hauck FH, et al. Chilblain lupus erythematosus—a review of literature. Clin Rheumatol. 2008;27:949-954.
- Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. Berlin, Germany: Springer; 2005.
- Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010;19:1050-1070.
- Lebeau S, També S, Sallam MA, et al. Docetaxel-induced relapse of subacute cutaneous lupus erythematosus and chilblain lupus. J Dtsch Dermatol Ges. 2013;11:871-874.
- Günther C, Hillebrand M, Brunk J, et al. Systemic involvement in TREX1-associated familial chilblain lupus. J Am Acad Dermatol. 2013;69:179-181.
- Sifuentes Giraldo WA, Ahijón Lana M, García Villanueva MJ, et al. Chilblain lupus induced by TNF-α antagonists: a case report and literature review. Clin Rheumatol. 2012;31:563-568.
- Boehm I, Bieber T. Chilblain lupus erythematosus Hutchinson: successful treatment with mycophenolate mofetil. Arch Dermatol. 2001;137:235-236.
Practice Points
- Up to 20% of patients with chilblain lupus erythematosus (CHLE) will develop systemic lupus erythematosus (SLE), necessitating close long-term follow-up.
- Medications such as antihypertensives, antifungals, chemotherapeutic agents, and tumor necrosis factor 11α inhibitors have been reported to trigger CHLE.
- Chilblain lupus erythematosus is less responsive to traditional antimalarial agents commonly used to treat SLE.
- Management of CHLE includes physical protection from cold environments, calcium channel blockers, topical and systemic steroids, and pentoxifylline, among other treatment modalities.
Epidermolysis Bullosa Acquisita in Association With Mantle Cell Lymphoma
To the Editor:
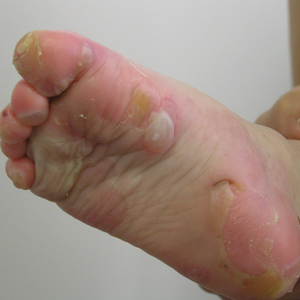
A 46-year-old man presented with multiple tense bullae and denuded patches on the palms (Figure 1A) and soles (Figure 1B). The blisters first appeared 2 months prior to presentation, shortly after he was diagnosed with stage IVB mantle cell lymphoma, and waxed and waned in intensity since then. He denied antecedent trauma or friction and reported that all sites were painful. He had no family or personal history of blistering disorders.
 and multiple bullae on the sole (B).")
The mantle cell lymphoma initially was treated with 4 cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy more than 2.5 years prior to the current presentation, which resulted in partial remission, followed by R-ICE (rituximab, ifosfamide, carboplatin, etoposide) therapy as well as autologous stem cell transplantation; complete remission was achieved. His recovery was complicated by a necrotic small bowel leading to resection. Eighteen months following the second course of chemotherapy, a mass was noted on the neck; biopsy performed by an outside dermatologist revealed mantle cell lymphoma.
Punch biopsy revealed a subepidermal bulla. Six weeks later, biopsy of a newly developed hand lesion performed at our office revealed a subepidermal cleft with minimal dermal infiltrate (Figure 2). Direct immunofluorescence was negative for immunoglobulin and complement deposition. Porphyrin elevation was not detected with a 24-hour urine assay. New lesions were drained and injected with triamcinolone, which appeared to hasten healing.
.")
Mantle cell lymphoma is a distinct lymphoproliferative disorder of B cells that represents less than 7% of non-Hodgkin lymphoma cases.1 The tumor cells originate in the mantle zone of the lymph nodes. Most patients present with advanced disease involving lymph nodes and other organs. The disease is characterized by male predominance and an aggressive course with a median overall survival of less than 5 years.1
Epidermolysis bullosa acquisita is a rare blistering disease that usually develops in adulthood. It is a subepidermal disorder characterized by the appearance of fragile tense bullae. Epidermolysis bullosa acquisita can be divided into 2 subtypes: inflammatory and mechanobullous (classic EBA).2 Inflammatory EBA presents similarly to bullous pemphigoid and other subepithelial autoimmune blistering diseases. Vesiculobullous lesions predominate on the trunk and extremities and often are accompanied by intense pruritus. The less common mechanobullous noninflammatory subtype, illustrated in our case, presents in trauma-prone areas with skin fragility and tense noninflamed vesicles and bullae that rupture leaving erosions. Associated findings may include milia and scarring. Lesions appear in areas exposed to friction and trauma such as the hands, feet, elbows, knees, and lower back. The differential diagnosis includes dystrophic epidermolysis bullosa, porphyria cutanea tarda, and pseudoporphyria. Dystrophic epidermolysis bullosa is ruled out by family history and disease onset at birth. The lesions of porphyria cutanea tarda and pseudoporphyria occur on sun-exposed areas; porphyrin levels are elevated in the former. Direct immunofluorescence of a perilesional EBA site usually reveals IgG deposition.3 Negative direct immunofluorescence in our case could have resulted from technical error, sample location, or response to systemic immunosuppressive treatment.4
Epidermolysis bullosa acquisita is caused by autoantibodies against type VII collagen.2,3 After the autoantibodies bind, a complement cascade reaction is activated, leading to deposition of C3a and C5a, which recruit leukocytes and mast cells. The anchoring fibrils in the basement membrane zones of the skin and mucosa are disrupted.5,6 Injection of anti–type VII collagen antibodies into mice induces a blistering disease resembling EBA.7 In a study of 14 patients with EBA, disease severity was correlated to levels of anticollagen autoantibodies measured by enzyme-linked immunosorbent assay.8
Epidermolysis bullosa acquisita has been linked to Crohn disease and approximately 30% of EBA cases occur in patients with this disease.9,10 Two case reports document an association with multiple myeloma.11,12 Treatment often proves challenging and unsatisfactory; valid controlled clinical trials are impossible given the paucity of cases. Successful therapeutic outcomes have been reported with oral prednisone,13 colchicine,14 cyclosporine,15 dapsone,16 and rituximab.17 Our patient received 2 separate courses of rituximab as part of chemotherapy for mantle cell lymphoma without measurable improvement. He was lost to follow-up after recurrence of the lymphoma and we learned from his wife that he had died.
- Hitz F, Bargetzi M, Cogliatti S, et al. Diagnosis and treatment of mantle cell lymphoma. Swiss Med Wkly. 2013;143:w13868.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822.
- Woodley DT, Briggaman RA, O’Keefe EJ. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310:1007-1013.
- Hashimoto T, Ishii N, Ohata C, et al. Pathogenesis of epidermolysis bullosa acquisita, an autoimmune subepidermal bullous disease. J Pathol. 2012;228:1-7.
- Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. 2006;177:3461-3468.
- Marzano AV, Cozzani E, Fanoni D, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol. 2013;168:80-84.
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have autoantibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-229.
- Radfar L, Fatahzadeh M, Shahamat Y, et al. Paraneoplastic epidermolysis bullosa acquisita associated with multiple myeloma. Spec Care Dentist. 2006;26:159-163.
- Engineer L, Dow EC, Braverman IM, et al. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol. 2002;47:943-946.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230.
- Megahed M, Scharffetter-Kochanek K. Epidermolysis bullosa acquisita—successful treatment with colchicine. Arch Dermatol Res. 1994;286:35-46.
- Khatri ML, Benghazeil M, Shafi M. Epidermolysis bullosa acquisita responsive to cyclosporin therapy. J Eur Acad Dermatol Venereol. 2001;15:182-184.
- Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg. 2001;5:397-399.
- Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. 2012;39:477-479.
To the Editor:
A 46-year-old man presented with multiple tense bullae and denuded patches on the palms (Figure 1A) and soles (Figure 1B). The blisters first appeared 2 months prior to presentation, shortly after he was diagnosed with stage IVB mantle cell lymphoma, and waxed and waned in intensity since then. He denied antecedent trauma or friction and reported that all sites were painful. He had no family or personal history of blistering disorders.
The mantle cell lymphoma initially was treated with 4 cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy more than 2.5 years prior to the current presentation, which resulted in partial remission, followed by R-ICE (rituximab, ifosfamide, carboplatin, etoposide) therapy as well as autologous stem cell transplantation; complete remission was achieved. His recovery was complicated by a necrotic small bowel leading to resection. Eighteen months following the second course of chemotherapy, a mass was noted on the neck; biopsy performed by an outside dermatologist revealed mantle cell lymphoma.
Punch biopsy revealed a subepidermal bulla. Six weeks later, biopsy of a newly developed hand lesion performed at our office revealed a subepidermal cleft with minimal dermal infiltrate (Figure 2). Direct immunofluorescence was negative for immunoglobulin and complement deposition. Porphyrin elevation was not detected with a 24-hour urine assay. New lesions were drained and injected with triamcinolone, which appeared to hasten healing.
Mantle cell lymphoma is a distinct lymphoproliferative disorder of B cells that represents less than 7% of non-Hodgkin lymphoma cases.1 The tumor cells originate in the mantle zone of the lymph nodes. Most patients present with advanced disease involving lymph nodes and other organs. The disease is characterized by male predominance and an aggressive course with a median overall survival of less than 5 years.1
Epidermolysis bullosa acquisita is a rare blistering disease that usually develops in adulthood. It is a subepidermal disorder characterized by the appearance of fragile tense bullae. Epidermolysis bullosa acquisita can be divided into 2 subtypes: inflammatory and mechanobullous (classic EBA).2 Inflammatory EBA presents similarly to bullous pemphigoid and other subepithelial autoimmune blistering diseases. Vesiculobullous lesions predominate on the trunk and extremities and often are accompanied by intense pruritus. The less common mechanobullous noninflammatory subtype, illustrated in our case, presents in trauma-prone areas with skin fragility and tense noninflamed vesicles and bullae that rupture leaving erosions. Associated findings may include milia and scarring. Lesions appear in areas exposed to friction and trauma such as the hands, feet, elbows, knees, and lower back. The differential diagnosis includes dystrophic epidermolysis bullosa, porphyria cutanea tarda, and pseudoporphyria. Dystrophic epidermolysis bullosa is ruled out by family history and disease onset at birth. The lesions of porphyria cutanea tarda and pseudoporphyria occur on sun-exposed areas; porphyrin levels are elevated in the former. Direct immunofluorescence of a perilesional EBA site usually reveals IgG deposition.3 Negative direct immunofluorescence in our case could have resulted from technical error, sample location, or response to systemic immunosuppressive treatment.4
Epidermolysis bullosa acquisita is caused by autoantibodies against type VII collagen.2,3 After the autoantibodies bind, a complement cascade reaction is activated, leading to deposition of C3a and C5a, which recruit leukocytes and mast cells. The anchoring fibrils in the basement membrane zones of the skin and mucosa are disrupted.5,6 Injection of anti–type VII collagen antibodies into mice induces a blistering disease resembling EBA.7 In a study of 14 patients with EBA, disease severity was correlated to levels of anticollagen autoantibodies measured by enzyme-linked immunosorbent assay.8
Epidermolysis bullosa acquisita has been linked to Crohn disease and approximately 30% of EBA cases occur in patients with this disease.9,10 Two case reports document an association with multiple myeloma.11,12 Treatment often proves challenging and unsatisfactory; valid controlled clinical trials are impossible given the paucity of cases. Successful therapeutic outcomes have been reported with oral prednisone,13 colchicine,14 cyclosporine,15 dapsone,16 and rituximab.17 Our patient received 2 separate courses of rituximab as part of chemotherapy for mantle cell lymphoma without measurable improvement. He was lost to follow-up after recurrence of the lymphoma and we learned from his wife that he had died.
To the Editor:
A 46-year-old man presented with multiple tense bullae and denuded patches on the palms (Figure 1A) and soles (Figure 1B). The blisters first appeared 2 months prior to presentation, shortly after he was diagnosed with stage IVB mantle cell lymphoma, and waxed and waned in intensity since then. He denied antecedent trauma or friction and reported that all sites were painful. He had no family or personal history of blistering disorders.
The mantle cell lymphoma initially was treated with 4 cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy more than 2.5 years prior to the current presentation, which resulted in partial remission, followed by R-ICE (rituximab, ifosfamide, carboplatin, etoposide) therapy as well as autologous stem cell transplantation; complete remission was achieved. His recovery was complicated by a necrotic small bowel leading to resection. Eighteen months following the second course of chemotherapy, a mass was noted on the neck; biopsy performed by an outside dermatologist revealed mantle cell lymphoma.
Punch biopsy revealed a subepidermal bulla. Six weeks later, biopsy of a newly developed hand lesion performed at our office revealed a subepidermal cleft with minimal dermal infiltrate (Figure 2). Direct immunofluorescence was negative for immunoglobulin and complement deposition. Porphyrin elevation was not detected with a 24-hour urine assay. New lesions were drained and injected with triamcinolone, which appeared to hasten healing.
Mantle cell lymphoma is a distinct lymphoproliferative disorder of B cells that represents less than 7% of non-Hodgkin lymphoma cases.1 The tumor cells originate in the mantle zone of the lymph nodes. Most patients present with advanced disease involving lymph nodes and other organs. The disease is characterized by male predominance and an aggressive course with a median overall survival of less than 5 years.1
Epidermolysis bullosa acquisita is a rare blistering disease that usually develops in adulthood. It is a subepidermal disorder characterized by the appearance of fragile tense bullae. Epidermolysis bullosa acquisita can be divided into 2 subtypes: inflammatory and mechanobullous (classic EBA).2 Inflammatory EBA presents similarly to bullous pemphigoid and other subepithelial autoimmune blistering diseases. Vesiculobullous lesions predominate on the trunk and extremities and often are accompanied by intense pruritus. The less common mechanobullous noninflammatory subtype, illustrated in our case, presents in trauma-prone areas with skin fragility and tense noninflamed vesicles and bullae that rupture leaving erosions. Associated findings may include milia and scarring. Lesions appear in areas exposed to friction and trauma such as the hands, feet, elbows, knees, and lower back. The differential diagnosis includes dystrophic epidermolysis bullosa, porphyria cutanea tarda, and pseudoporphyria. Dystrophic epidermolysis bullosa is ruled out by family history and disease onset at birth. The lesions of porphyria cutanea tarda and pseudoporphyria occur on sun-exposed areas; porphyrin levels are elevated in the former. Direct immunofluorescence of a perilesional EBA site usually reveals IgG deposition.3 Negative direct immunofluorescence in our case could have resulted from technical error, sample location, or response to systemic immunosuppressive treatment.4
Epidermolysis bullosa acquisita is caused by autoantibodies against type VII collagen.2,3 After the autoantibodies bind, a complement cascade reaction is activated, leading to deposition of C3a and C5a, which recruit leukocytes and mast cells. The anchoring fibrils in the basement membrane zones of the skin and mucosa are disrupted.5,6 Injection of anti–type VII collagen antibodies into mice induces a blistering disease resembling EBA.7 In a study of 14 patients with EBA, disease severity was correlated to levels of anticollagen autoantibodies measured by enzyme-linked immunosorbent assay.8
Epidermolysis bullosa acquisita has been linked to Crohn disease and approximately 30% of EBA cases occur in patients with this disease.9,10 Two case reports document an association with multiple myeloma.11,12 Treatment often proves challenging and unsatisfactory; valid controlled clinical trials are impossible given the paucity of cases. Successful therapeutic outcomes have been reported with oral prednisone,13 colchicine,14 cyclosporine,15 dapsone,16 and rituximab.17 Our patient received 2 separate courses of rituximab as part of chemotherapy for mantle cell lymphoma without measurable improvement. He was lost to follow-up after recurrence of the lymphoma and we learned from his wife that he had died.
- Hitz F, Bargetzi M, Cogliatti S, et al. Diagnosis and treatment of mantle cell lymphoma. Swiss Med Wkly. 2013;143:w13868.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822.
- Woodley DT, Briggaman RA, O’Keefe EJ. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310:1007-1013.
- Hashimoto T, Ishii N, Ohata C, et al. Pathogenesis of epidermolysis bullosa acquisita, an autoimmune subepidermal bullous disease. J Pathol. 2012;228:1-7.
- Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. 2006;177:3461-3468.
- Marzano AV, Cozzani E, Fanoni D, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol. 2013;168:80-84.
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have autoantibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-229.
- Radfar L, Fatahzadeh M, Shahamat Y, et al. Paraneoplastic epidermolysis bullosa acquisita associated with multiple myeloma. Spec Care Dentist. 2006;26:159-163.
- Engineer L, Dow EC, Braverman IM, et al. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol. 2002;47:943-946.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230.
- Megahed M, Scharffetter-Kochanek K. Epidermolysis bullosa acquisita—successful treatment with colchicine. Arch Dermatol Res. 1994;286:35-46.
- Khatri ML, Benghazeil M, Shafi M. Epidermolysis bullosa acquisita responsive to cyclosporin therapy. J Eur Acad Dermatol Venereol. 2001;15:182-184.
- Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg. 2001;5:397-399.
- Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. 2012;39:477-479.
- Hitz F, Bargetzi M, Cogliatti S, et al. Diagnosis and treatment of mantle cell lymphoma. Swiss Med Wkly. 2013;143:w13868.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822.
- Woodley DT, Briggaman RA, O’Keefe EJ. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310:1007-1013.
- Hashimoto T, Ishii N, Ohata C, et al. Pathogenesis of epidermolysis bullosa acquisita, an autoimmune subepidermal bullous disease. J Pathol. 2012;228:1-7.
- Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. 2006;177:3461-3468.
- Marzano AV, Cozzani E, Fanoni D, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol. 2013;168:80-84.
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have autoantibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-229.
- Radfar L, Fatahzadeh M, Shahamat Y, et al. Paraneoplastic epidermolysis bullosa acquisita associated with multiple myeloma. Spec Care Dentist. 2006;26:159-163.
- Engineer L, Dow EC, Braverman IM, et al. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol. 2002;47:943-946.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230.
- Megahed M, Scharffetter-Kochanek K. Epidermolysis bullosa acquisita—successful treatment with colchicine. Arch Dermatol Res. 1994;286:35-46.
- Khatri ML, Benghazeil M, Shafi M. Epidermolysis bullosa acquisita responsive to cyclosporin therapy. J Eur Acad Dermatol Venereol. 2001;15:182-184.
- Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg. 2001;5:397-399.
- Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. 2012;39:477-479.
Practice Points
- Epidermolysis bullosa acquisita (EBA) is an uncommon blistering disorder and few cases have been associated with malignancy.
- Diagnosis of EBA is challenging and requires exclusion of other blistering diseases.
Slow-growing, Asymptomatic, Annular Plaques on the Bilateral Palms
The Diagnosis: Circumscribed Palmar Hypokeratosis
Circumscribed palmar hypokeratosis is a rare, benign, acquired dermatosis that was first described by Pérez et al1 in 2002 and is characterized by annular plaques with an atrophic center and hyperkeratotic edges. Classically, the lesions present on the thenar and hypothenar eminences of the palms.2 The condition predominantly affects women (4:1 ratio), with a mean age of onset of 65 years.3
Although the pathogenesis of circumscribed palmar hypokeratosis is unknown, local trauma generally is considered to be the causative factor. Other hypotheses include human papillomaviruses 4 and 6 infection and primary abnormal keratinization in the epidermis.3 Immunohistochemical studies have demonstrated increased expression of keratin 16 and Ki-67 in cutaneous lesions, which is postulated to be responsible for keratinocyte fragility associated with epidermal hyperproliferation. Other reported cases have shown diminished keratin 9, keratin 2e, and connexin 26 expression, which normally are abundant in the acral epidermis. Abnormal expression of antigens associated with epidermal proliferation and differentiation also have been reported,3 suggesting that there is an altered regulation of the cutaneous desquamation process.
Histologically, circumscribed palmar hypokeratosis is characterized by an abrupt reduction in the stratum corneum (Figure), forming a step between the lesion and the perilesional normal skin.2,3 The clinical appearance of erythema is due to visualization of dermal blood circulation in the area of corneal thinning and is not a result of vasodilation. The dermis is uninvolved, and inflammation is absent. The differential diagnosis includes psoriasis, Bowen disease, porokeratosis, and dermatophytosis.3
(H&E, original magnification ×4). No notable inflammation was evident in the dermis (B)(H&E, original magnification ×10).")
Circumscribed palmar hypokeratosis is a chronic condition, and there are no known reports of development of malignancy. Treatment is not required but may include cryotherapy; topical therapy with corticosteroids, retinoids, urea, and calcipotriene; and photodynamic therapy. Circumscribed hypokeratosis should be included in the differential diagnosis of palmar lesions.
- Pérez A, Rütten A, Gold R, et al. Circumscribed palmar or plantar hypokeratosis: a distinctive epidermal malformation of the palms or soles. J Am Acad Dermatol. 2002;47:21-27.
- Mitkov M, Balagula Y, Lockshin B. Case report: circumscribed plantar hypokeratosis. Int J Dermatol. 2015;54:E203-E205.
- Rocha L, Nico M. Circumscribed palmoplantar hypokeratosis: report of two Brazilian cases. An Bras Dermatol. 2013;88:623-626.
The Diagnosis: Circumscribed Palmar Hypokeratosis
Circumscribed palmar hypokeratosis is a rare, benign, acquired dermatosis that was first described by Pérez et al1 in 2002 and is characterized by annular plaques with an atrophic center and hyperkeratotic edges. Classically, the lesions present on the thenar and hypothenar eminences of the palms.2 The condition predominantly affects women (4:1 ratio), with a mean age of onset of 65 years.3
Although the pathogenesis of circumscribed palmar hypokeratosis is unknown, local trauma generally is considered to be the causative factor. Other hypotheses include human papillomaviruses 4 and 6 infection and primary abnormal keratinization in the epidermis.3 Immunohistochemical studies have demonstrated increased expression of keratin 16 and Ki-67 in cutaneous lesions, which is postulated to be responsible for keratinocyte fragility associated with epidermal hyperproliferation. Other reported cases have shown diminished keratin 9, keratin 2e, and connexin 26 expression, which normally are abundant in the acral epidermis. Abnormal expression of antigens associated with epidermal proliferation and differentiation also have been reported,3 suggesting that there is an altered regulation of the cutaneous desquamation process.
Histologically, circumscribed palmar hypokeratosis is characterized by an abrupt reduction in the stratum corneum (Figure), forming a step between the lesion and the perilesional normal skin.2,3 The clinical appearance of erythema is due to visualization of dermal blood circulation in the area of corneal thinning and is not a result of vasodilation. The dermis is uninvolved, and inflammation is absent. The differential diagnosis includes psoriasis, Bowen disease, porokeratosis, and dermatophytosis.3
Circumscribed palmar hypokeratosis is a chronic condition, and there are no known reports of development of malignancy. Treatment is not required but may include cryotherapy; topical therapy with corticosteroids, retinoids, urea, and calcipotriene; and photodynamic therapy. Circumscribed hypokeratosis should be included in the differential diagnosis of palmar lesions.
The Diagnosis: Circumscribed Palmar Hypokeratosis
Circumscribed palmar hypokeratosis is a rare, benign, acquired dermatosis that was first described by Pérez et al1 in 2002 and is characterized by annular plaques with an atrophic center and hyperkeratotic edges. Classically, the lesions present on the thenar and hypothenar eminences of the palms.2 The condition predominantly affects women (4:1 ratio), with a mean age of onset of 65 years.3
Although the pathogenesis of circumscribed palmar hypokeratosis is unknown, local trauma generally is considered to be the causative factor. Other hypotheses include human papillomaviruses 4 and 6 infection and primary abnormal keratinization in the epidermis.3 Immunohistochemical studies have demonstrated increased expression of keratin 16 and Ki-67 in cutaneous lesions, which is postulated to be responsible for keratinocyte fragility associated with epidermal hyperproliferation. Other reported cases have shown diminished keratin 9, keratin 2e, and connexin 26 expression, which normally are abundant in the acral epidermis. Abnormal expression of antigens associated with epidermal proliferation and differentiation also have been reported,3 suggesting that there is an altered regulation of the cutaneous desquamation process.
Histologically, circumscribed palmar hypokeratosis is characterized by an abrupt reduction in the stratum corneum (Figure), forming a step between the lesion and the perilesional normal skin.2,3 The clinical appearance of erythema is due to visualization of dermal blood circulation in the area of corneal thinning and is not a result of vasodilation. The dermis is uninvolved, and inflammation is absent. The differential diagnosis includes psoriasis, Bowen disease, porokeratosis, and dermatophytosis.3
Circumscribed palmar hypokeratosis is a chronic condition, and there are no known reports of development of malignancy. Treatment is not required but may include cryotherapy; topical therapy with corticosteroids, retinoids, urea, and calcipotriene; and photodynamic therapy. Circumscribed hypokeratosis should be included in the differential diagnosis of palmar lesions.
- Pérez A, Rütten A, Gold R, et al. Circumscribed palmar or plantar hypokeratosis: a distinctive epidermal malformation of the palms or soles. J Am Acad Dermatol. 2002;47:21-27.
- Mitkov M, Balagula Y, Lockshin B. Case report: circumscribed plantar hypokeratosis. Int J Dermatol. 2015;54:E203-E205.
- Rocha L, Nico M. Circumscribed palmoplantar hypokeratosis: report of two Brazilian cases. An Bras Dermatol. 2013;88:623-626.
- Pérez A, Rütten A, Gold R, et al. Circumscribed palmar or plantar hypokeratosis: a distinctive epidermal malformation of the palms or soles. J Am Acad Dermatol. 2002;47:21-27.
- Mitkov M, Balagula Y, Lockshin B. Case report: circumscribed plantar hypokeratosis. Int J Dermatol. 2015;54:E203-E205.
- Rocha L, Nico M. Circumscribed palmoplantar hypokeratosis: report of two Brazilian cases. An Bras Dermatol. 2013;88:623-626.
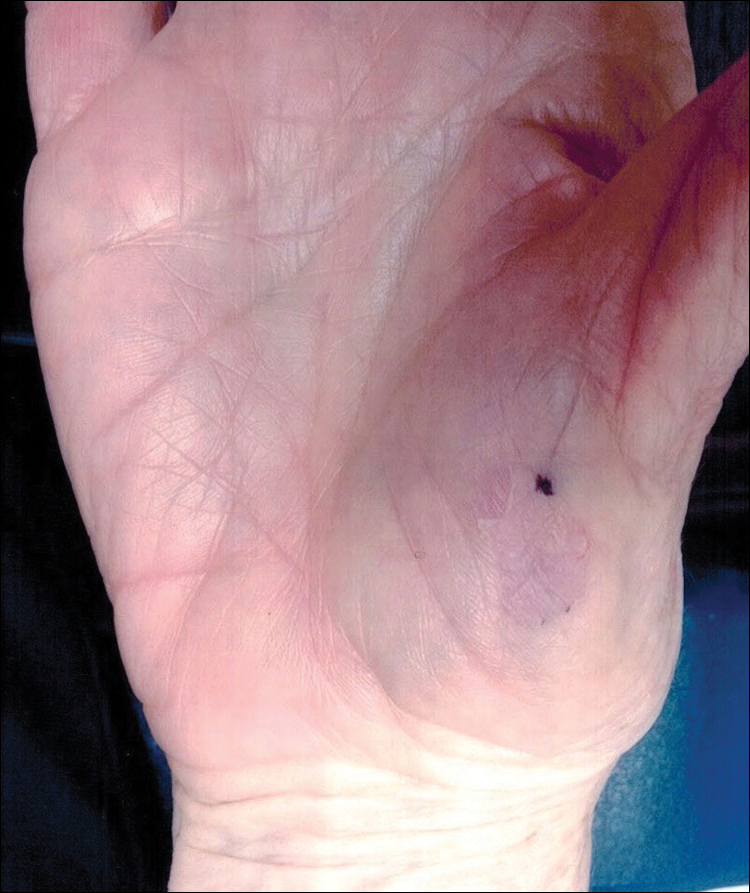
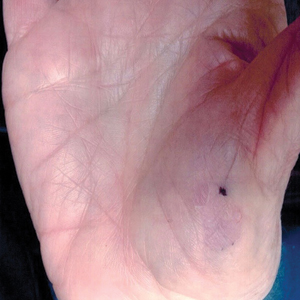
A 77-year-old woman presented with slow-growing, asymptomatic, annular plaques on the bilateral palms of many years' duration. There was no history of trauma or local infection. Prior treatment with over-the-counter creams was unsuccessful. A 3-mm punch biopsy of the lesion on the right palm was performed.
Intensive treatment for T2D pays off in the long run
ORLANDO – Intensified multifactorial treatment proved cost effective over time in type 2 diabetes patients in the practice-changing Steno-2 study, according to 21-year follow-up data from the randomized Danish study.
Cumulative direct health care costs from the start of the trial in 1993 through 2014 were about $13 million in 24 patients in the intensive treatment group who were available for follow-up, and about $12.3 million in 42 patients in the conventional treatment group. The difference in costs between the groups was not statistically significant, Joachim Gaede reported at the annual scientific sessions of the American Diabetes Association.
Costs per patient-year during 1996-2014, however, were significantly lower in the intensive treatment group ($9,648 vs. $10, 681, respectively), said Mr. Gaede, a graduate student in the medicine program at the University of Copenhagen.
Furthermore, patients in the intensified treatment group lived a median of 7.9 years longer than did those who were in the conventional treatment group, suggesting that while costs might be higher early on, investing in early intensified treatment of all known modifiable risk factors in high-risk patients will prolong life and still save money over time thanks to reduced complication-related costs, he noted.
Steno-2 was an open, parallel group study initiated in 1993 to compare conventional multifactorial treatment of type 2 diabetes mellitus (T2DM) with an intensified approach over an 8-year period. Enrollment included 160 patients with high-risk type 2 diabetes. After the primary composite cardiovascular endpoint was assessed, the trial continued as an observational study, with all patients given the intensified, multifactorial treatment that consisted of lifestyle measures and medications targeting hyperglycemia, hypertension, hypercholesterolemia, and hypercoagulation.
Reports from the study over the years led to changes in treatment guidelines to promote more intensive multifactorial treatment, Mr. Gaede said. For example, the initial results reported in 1999 showed a 50% relative risk reduction in kidney, eye, and nerve complications after 4 years with intensive versus conventional treatment; a 2003 report showed a 53% relative risk reduction in MI, stroke, and amputation after 8 years; and a 2008 report demonstrated a 46% relative risk reduction in death after 13 years. Finally, in 2016 a 7.9-year gain in lifespan after 21 years with intensive versus conventional treatment was reported.
In this video interview, Mr. Gaede, junior lead study author, discusses the Steno-2 study findings and the current cost analysis data.
“The bottom line is that ... you can actually, as a patient, be treated at a specialized diabetes clinic ... and, in the long run, it doesn’t cost you anything” more, he said, explaining that the up-front costs of intensive treatment are offset by the money saved because of the reduced complications over time.
Mr. Gaede reported having no disclosures.
sworcester@frontlinemedcom.com
SOURCE: Gaede J et al. ADA 2018, Abstract 162-OR.
ORLANDO – Intensified multifactorial treatment proved cost effective over time in type 2 diabetes patients in the practice-changing Steno-2 study, according to 21-year follow-up data from the randomized Danish study.
Cumulative direct health care costs from the start of the trial in 1993 through 2014 were about $13 million in 24 patients in the intensive treatment group who were available for follow-up, and about $12.3 million in 42 patients in the conventional treatment group. The difference in costs between the groups was not statistically significant, Joachim Gaede reported at the annual scientific sessions of the American Diabetes Association.
Costs per patient-year during 1996-2014, however, were significantly lower in the intensive treatment group ($9,648 vs. $10, 681, respectively), said Mr. Gaede, a graduate student in the medicine program at the University of Copenhagen.
Furthermore, patients in the intensified treatment group lived a median of 7.9 years longer than did those who were in the conventional treatment group, suggesting that while costs might be higher early on, investing in early intensified treatment of all known modifiable risk factors in high-risk patients will prolong life and still save money over time thanks to reduced complication-related costs, he noted.
Steno-2 was an open, parallel group study initiated in 1993 to compare conventional multifactorial treatment of type 2 diabetes mellitus (T2DM) with an intensified approach over an 8-year period. Enrollment included 160 patients with high-risk type 2 diabetes. After the primary composite cardiovascular endpoint was assessed, the trial continued as an observational study, with all patients given the intensified, multifactorial treatment that consisted of lifestyle measures and medications targeting hyperglycemia, hypertension, hypercholesterolemia, and hypercoagulation.
Reports from the study over the years led to changes in treatment guidelines to promote more intensive multifactorial treatment, Mr. Gaede said. For example, the initial results reported in 1999 showed a 50% relative risk reduction in kidney, eye, and nerve complications after 4 years with intensive versus conventional treatment; a 2003 report showed a 53% relative risk reduction in MI, stroke, and amputation after 8 years; and a 2008 report demonstrated a 46% relative risk reduction in death after 13 years. Finally, in 2016 a 7.9-year gain in lifespan after 21 years with intensive versus conventional treatment was reported.
In this video interview, Mr. Gaede, junior lead study author, discusses the Steno-2 study findings and the current cost analysis data.
“The bottom line is that ... you can actually, as a patient, be treated at a specialized diabetes clinic ... and, in the long run, it doesn’t cost you anything” more, he said, explaining that the up-front costs of intensive treatment are offset by the money saved because of the reduced complications over time.
Mr. Gaede reported having no disclosures.
sworcester@frontlinemedcom.com
SOURCE: Gaede J et al. ADA 2018, Abstract 162-OR.
ORLANDO – Intensified multifactorial treatment proved cost effective over time in type 2 diabetes patients in the practice-changing Steno-2 study, according to 21-year follow-up data from the randomized Danish study.
Cumulative direct health care costs from the start of the trial in 1993 through 2014 were about $13 million in 24 patients in the intensive treatment group who were available for follow-up, and about $12.3 million in 42 patients in the conventional treatment group. The difference in costs between the groups was not statistically significant, Joachim Gaede reported at the annual scientific sessions of the American Diabetes Association.
Costs per patient-year during 1996-2014, however, were significantly lower in the intensive treatment group ($9,648 vs. $10, 681, respectively), said Mr. Gaede, a graduate student in the medicine program at the University of Copenhagen.
Furthermore, patients in the intensified treatment group lived a median of 7.9 years longer than did those who were in the conventional treatment group, suggesting that while costs might be higher early on, investing in early intensified treatment of all known modifiable risk factors in high-risk patients will prolong life and still save money over time thanks to reduced complication-related costs, he noted.
Steno-2 was an open, parallel group study initiated in 1993 to compare conventional multifactorial treatment of type 2 diabetes mellitus (T2DM) with an intensified approach over an 8-year period. Enrollment included 160 patients with high-risk type 2 diabetes. After the primary composite cardiovascular endpoint was assessed, the trial continued as an observational study, with all patients given the intensified, multifactorial treatment that consisted of lifestyle measures and medications targeting hyperglycemia, hypertension, hypercholesterolemia, and hypercoagulation.
Reports from the study over the years led to changes in treatment guidelines to promote more intensive multifactorial treatment, Mr. Gaede said. For example, the initial results reported in 1999 showed a 50% relative risk reduction in kidney, eye, and nerve complications after 4 years with intensive versus conventional treatment; a 2003 report showed a 53% relative risk reduction in MI, stroke, and amputation after 8 years; and a 2008 report demonstrated a 46% relative risk reduction in death after 13 years. Finally, in 2016 a 7.9-year gain in lifespan after 21 years with intensive versus conventional treatment was reported.
In this video interview, Mr. Gaede, junior lead study author, discusses the Steno-2 study findings and the current cost analysis data.
“The bottom line is that ... you can actually, as a patient, be treated at a specialized diabetes clinic ... and, in the long run, it doesn’t cost you anything” more, he said, explaining that the up-front costs of intensive treatment are offset by the money saved because of the reduced complications over time.
Mr. Gaede reported having no disclosures.
sworcester@frontlinemedcom.com
SOURCE: Gaede J et al. ADA 2018, Abstract 162-OR.
REPORTING FROM ADA 2018

Better stent technology needed for diabetes patients
PARIS – Interventional cardiologists are hopeful that a new generation of investigational coronary stents designed specifically for use in diabetes patients will improve upon the relatively poor current outcomes of percutaneous coronary intervention in that population.
The operative word here is “abluminal.” Both of the novel drug-eluting stents featured at the annual meeting of the European Association of Percutaneous Cardiovascular Interventions position their antirestenosis drugs abluminally: that is, aimed toward the vessel wall surface, not the lumen.
“The hypothesis is that ,” explained Luca Testa, MD, PhD, head of the coronary revascularization unit at San Donato Hospital in Milan.
There is a major unmet need for improved stent technology that addresses the special needs of diabetes patients, who tend to have more diffuse and rapidly progressive coronary artery disease (CAD) with longer lesions. Target lesion revascularization rates at 5 years of follow-up in diabetes patients with current generation drug-eluting stents (DES) remain high, at 20% or more. And diabetes patients are roughly 3.5-fold more likely to have nonfocal, diffuse coronary lesions than are nondiabetic patients with CAD, the cardiologist noted.

The sense of urgency surrounding this unmet need stems from the ongoing worldwide epidemic of diabetes. The global prevalence of diabetes was estimated at 382 million in 2013 and is projected to climb to nearly 600 million by 2035. Diabetes patients are two to four times more likely to develop CAD than are those without the disease. Because of the current suboptimal results with percutaneous coronary intervention (PCI), many of them are being referred for coronary artery bypass surgery.
Dr. Testa presented the 1-year results of the ongoing en-ABL e-Registry, a 5-year, multicenter, prospective, all-comers registry of 859 diabetic and 1,641 nondiabetic CAD patients who received the Abluminus DES at 31 centers in India. The novel stent, developed by Envision Scientific of India, is coated with sirolimus on the abluminal side. The device is actually both a DES and a drug-coated balloon. The balloon, including its proximal and distal ends, are also sirolimus coated to maximize exposure of diseased artery to the drug. The balloon needs to be inflated in position for at least 30 seconds to deliver its portion of sirolimus. The stent is composed of a biodegradable polymer matrix that is metabolized within 6-8 months.
The primary endpoint at 1 year of follow-up was the composite of cardiac death, target vessel MI, and target lesion or vessel revascularization. The rate was 3.12% in the diabetic population, which wasn’t significantly different from the 2.1% rate in nondiabetic patients. Of note, the rate was 5.17% in the 138 insulin-dependent diabetes patients, compared with 2.76% in 721 non–insulin-dependent patients.
Among diabetes patients, the composite endpoint occurred in 2.82% of those who underwent primary PCI with the Abluminus DES for an acute MI, 3.96% of those treated for lesions in small vessels 2.75 mm or less in diameter, 3.75% in diabetes patients treated for long lesions, and 4.18% in the subgroup with long lesions in small vessels.
On the basis of these encouraging results, Dr. Testa has been named the principal investigator for the new prospective, multicenter, observational DEDICATE registry, restricted to diabetic patients treated with the Abluminus DES.
Also getting underway is a randomized, investigator-initiated, multicenter, single-blind pilot study involving 165 diabetes patients assigned 2:1 to the Abluminus DES or the Xience everolimus-eluting stent, widely considered the current gold standard DES. The study, known as the ABILITY trial, has as its primary endpoint the in-stent neointimal volume as measured by optical coherence tomography 6 months post PCI. The medical director of the study is Antonio Colombo, MD, director of the cardiac catheterization laboratory and the interventional cardiology unit at San Raffaele Hospital in Milan.
Elsewhere at EuroPCR 2018, officials at Alvimedica Medical Technologies announced that the company’s abluminal stent, known as the Cre8 EVO, will be pitted against the everolimus-eluting stent in a 55-center trial of 3,040 diabetes patients. The hypothesis of the Diab8 trial, based on preliminary data from pilot studies, is that the abluminal stent will show clinical superiority – not merely equivalence – at 1 year.
The Cre8 EVO stent utilizes a proprietary, polymer-free, drug-release technology involving reservoirs located on the stent’s outer surface that direct the controlled release of a mixture of sirolimus and fatty acids that the company calls the amphilimus formulation. The drug mixture is designed to enhance tissue permeation and sirolimus bioavailability. The body of the stent is cobalt, which was used based upon a conviction that polymers are more proinflammatory.
Dr. Colombo is also the principal investigator of the Diab8 trial, sponsored by Alvimedica.
Dr. Testa reported having no financial conflicts regarding his work on the en-ABL e-Registry, funded by a nonprofit Italian cardiovascular research foundation.
PARIS – Interventional cardiologists are hopeful that a new generation of investigational coronary stents designed specifically for use in diabetes patients will improve upon the relatively poor current outcomes of percutaneous coronary intervention in that population.
The operative word here is “abluminal.” Both of the novel drug-eluting stents featured at the annual meeting of the European Association of Percutaneous Cardiovascular Interventions position their antirestenosis drugs abluminally: that is, aimed toward the vessel wall surface, not the lumen.
“The hypothesis is that ,” explained Luca Testa, MD, PhD, head of the coronary revascularization unit at San Donato Hospital in Milan.
There is a major unmet need for improved stent technology that addresses the special needs of diabetes patients, who tend to have more diffuse and rapidly progressive coronary artery disease (CAD) with longer lesions. Target lesion revascularization rates at 5 years of follow-up in diabetes patients with current generation drug-eluting stents (DES) remain high, at 20% or more. And diabetes patients are roughly 3.5-fold more likely to have nonfocal, diffuse coronary lesions than are nondiabetic patients with CAD, the cardiologist noted.
The sense of urgency surrounding this unmet need stems from the ongoing worldwide epidemic of diabetes. The global prevalence of diabetes was estimated at 382 million in 2013 and is projected to climb to nearly 600 million by 2035. Diabetes patients are two to four times more likely to develop CAD than are those without the disease. Because of the current suboptimal results with percutaneous coronary intervention (PCI), many of them are being referred for coronary artery bypass surgery.
Dr. Testa presented the 1-year results of the ongoing en-ABL e-Registry, a 5-year, multicenter, prospective, all-comers registry of 859 diabetic and 1,641 nondiabetic CAD patients who received the Abluminus DES at 31 centers in India. The novel stent, developed by Envision Scientific of India, is coated with sirolimus on the abluminal side. The device is actually both a DES and a drug-coated balloon. The balloon, including its proximal and distal ends, are also sirolimus coated to maximize exposure of diseased artery to the drug. The balloon needs to be inflated in position for at least 30 seconds to deliver its portion of sirolimus. The stent is composed of a biodegradable polymer matrix that is metabolized within 6-8 months.
The primary endpoint at 1 year of follow-up was the composite of cardiac death, target vessel MI, and target lesion or vessel revascularization. The rate was 3.12% in the diabetic population, which wasn’t significantly different from the 2.1% rate in nondiabetic patients. Of note, the rate was 5.17% in the 138 insulin-dependent diabetes patients, compared with 2.76% in 721 non–insulin-dependent patients.
Among diabetes patients, the composite endpoint occurred in 2.82% of those who underwent primary PCI with the Abluminus DES for an acute MI, 3.96% of those treated for lesions in small vessels 2.75 mm or less in diameter, 3.75% in diabetes patients treated for long lesions, and 4.18% in the subgroup with long lesions in small vessels.
On the basis of these encouraging results, Dr. Testa has been named the principal investigator for the new prospective, multicenter, observational DEDICATE registry, restricted to diabetic patients treated with the Abluminus DES.
Also getting underway is a randomized, investigator-initiated, multicenter, single-blind pilot study involving 165 diabetes patients assigned 2:1 to the Abluminus DES or the Xience everolimus-eluting stent, widely considered the current gold standard DES. The study, known as the ABILITY trial, has as its primary endpoint the in-stent neointimal volume as measured by optical coherence tomography 6 months post PCI. The medical director of the study is Antonio Colombo, MD, director of the cardiac catheterization laboratory and the interventional cardiology unit at San Raffaele Hospital in Milan.
Elsewhere at EuroPCR 2018, officials at Alvimedica Medical Technologies announced that the company’s abluminal stent, known as the Cre8 EVO, will be pitted against the everolimus-eluting stent in a 55-center trial of 3,040 diabetes patients. The hypothesis of the Diab8 trial, based on preliminary data from pilot studies, is that the abluminal stent will show clinical superiority – not merely equivalence – at 1 year.
The Cre8 EVO stent utilizes a proprietary, polymer-free, drug-release technology involving reservoirs located on the stent’s outer surface that direct the controlled release of a mixture of sirolimus and fatty acids that the company calls the amphilimus formulation. The drug mixture is designed to enhance tissue permeation and sirolimus bioavailability. The body of the stent is cobalt, which was used based upon a conviction that polymers are more proinflammatory.
Dr. Colombo is also the principal investigator of the Diab8 trial, sponsored by Alvimedica.
Dr. Testa reported having no financial conflicts regarding his work on the en-ABL e-Registry, funded by a nonprofit Italian cardiovascular research foundation.
PARIS – Interventional cardiologists are hopeful that a new generation of investigational coronary stents designed specifically for use in diabetes patients will improve upon the relatively poor current outcomes of percutaneous coronary intervention in that population.
The operative word here is “abluminal.” Both of the novel drug-eluting stents featured at the annual meeting of the European Association of Percutaneous Cardiovascular Interventions position their antirestenosis drugs abluminally: that is, aimed toward the vessel wall surface, not the lumen.
“The hypothesis is that ,” explained Luca Testa, MD, PhD, head of the coronary revascularization unit at San Donato Hospital in Milan.
There is a major unmet need for improved stent technology that addresses the special needs of diabetes patients, who tend to have more diffuse and rapidly progressive coronary artery disease (CAD) with longer lesions. Target lesion revascularization rates at 5 years of follow-up in diabetes patients with current generation drug-eluting stents (DES) remain high, at 20% or more. And diabetes patients are roughly 3.5-fold more likely to have nonfocal, diffuse coronary lesions than are nondiabetic patients with CAD, the cardiologist noted.
The sense of urgency surrounding this unmet need stems from the ongoing worldwide epidemic of diabetes. The global prevalence of diabetes was estimated at 382 million in 2013 and is projected to climb to nearly 600 million by 2035. Diabetes patients are two to four times more likely to develop CAD than are those without the disease. Because of the current suboptimal results with percutaneous coronary intervention (PCI), many of them are being referred for coronary artery bypass surgery.
Dr. Testa presented the 1-year results of the ongoing en-ABL e-Registry, a 5-year, multicenter, prospective, all-comers registry of 859 diabetic and 1,641 nondiabetic CAD patients who received the Abluminus DES at 31 centers in India. The novel stent, developed by Envision Scientific of India, is coated with sirolimus on the abluminal side. The device is actually both a DES and a drug-coated balloon. The balloon, including its proximal and distal ends, are also sirolimus coated to maximize exposure of diseased artery to the drug. The balloon needs to be inflated in position for at least 30 seconds to deliver its portion of sirolimus. The stent is composed of a biodegradable polymer matrix that is metabolized within 6-8 months.
The primary endpoint at 1 year of follow-up was the composite of cardiac death, target vessel MI, and target lesion or vessel revascularization. The rate was 3.12% in the diabetic population, which wasn’t significantly different from the 2.1% rate in nondiabetic patients. Of note, the rate was 5.17% in the 138 insulin-dependent diabetes patients, compared with 2.76% in 721 non–insulin-dependent patients.
Among diabetes patients, the composite endpoint occurred in 2.82% of those who underwent primary PCI with the Abluminus DES for an acute MI, 3.96% of those treated for lesions in small vessels 2.75 mm or less in diameter, 3.75% in diabetes patients treated for long lesions, and 4.18% in the subgroup with long lesions in small vessels.
On the basis of these encouraging results, Dr. Testa has been named the principal investigator for the new prospective, multicenter, observational DEDICATE registry, restricted to diabetic patients treated with the Abluminus DES.
Also getting underway is a randomized, investigator-initiated, multicenter, single-blind pilot study involving 165 diabetes patients assigned 2:1 to the Abluminus DES or the Xience everolimus-eluting stent, widely considered the current gold standard DES. The study, known as the ABILITY trial, has as its primary endpoint the in-stent neointimal volume as measured by optical coherence tomography 6 months post PCI. The medical director of the study is Antonio Colombo, MD, director of the cardiac catheterization laboratory and the interventional cardiology unit at San Raffaele Hospital in Milan.
Elsewhere at EuroPCR 2018, officials at Alvimedica Medical Technologies announced that the company’s abluminal stent, known as the Cre8 EVO, will be pitted against the everolimus-eluting stent in a 55-center trial of 3,040 diabetes patients. The hypothesis of the Diab8 trial, based on preliminary data from pilot studies, is that the abluminal stent will show clinical superiority – not merely equivalence – at 1 year.
The Cre8 EVO stent utilizes a proprietary, polymer-free, drug-release technology involving reservoirs located on the stent’s outer surface that direct the controlled release of a mixture of sirolimus and fatty acids that the company calls the amphilimus formulation. The drug mixture is designed to enhance tissue permeation and sirolimus bioavailability. The body of the stent is cobalt, which was used based upon a conviction that polymers are more proinflammatory.
Dr. Colombo is also the principal investigator of the Diab8 trial, sponsored by Alvimedica.
Dr. Testa reported having no financial conflicts regarding his work on the en-ABL e-Registry, funded by a nonprofit Italian cardiovascular research foundation.
REPORTING FROM EUROPCR 2018
Health care, technology, and the future
Major forces combining to reshape care delivery
What will be the role of humans in the future health system?
At first blush, this is a peculiar question. Health care is all about humans. How could one doubt their presence or role? It is working with and for people that attracted many to this profession.
On the cusp of a significant health system reformulation, it is the very question that hospitalists now must ponder. Just as ATMs replaced bank cashiers, online shopping replaced retail stores, and autonomous cars will soon replace drivers, the human landscape of health care is about to change. What pressures will force the changes?
On one hand, there is increasing demand. The Affordable Care Act opened the insurance door for people previously uncovered. Aging is delivering the baby boomer bubble into their sicker years. Hospitalists witness this phenomenon every day in the ballooning parade of patients they serve. At times, those pressures can overwhelm.
On the other hand, the political will to provide government subsidized health coverage is waning. Washington is tripping over itself to dismantle Obamacare with glancing concern for how it will inflate the ranks of the uninsured. Employers are eager to free themselves from the burden of providing increasingly expensive health coverage benefits. By removing the mandate to buy health care insurance, the current political health system architects are liberating the healthy paying population from their contributions to the overall insurance pool. Simply put, there is and will be less money and less of all that it buys.
Combine building demand with decreasing budget into a system that does not follow general market forces: You get that earthquake. A consumer can forgo that new phone in hard times but not that cardiac procedure. People will be caught in the fissures of the system. Waits, quality, burnout, morale problems, and financial losses will all trend in the wrong directions. The process will evolve in slow motion. Some might argue that we have already arrived.
Enter entrepreneurs, technologic advances, and a growing savvy and willingness to engage tech solutions to everyday problems. If Alexa can turn on your toaster, could it take your blood pressure? If a robot can vacuum your rug, could a different robot provide personal care services? And, if an algorithm can drive your car, could it similarly diagnose what ails you?
On Jan. 30, 2018, one of the greatest disrupters of all time, Amazon, announced that it is joining forces with Berkshire Hathaway and JPMorgan Chase to leap into health care. While they are initially experimenting with health care changes for their corporate employees, the ultimate marketwide goal is to apply technology to both reduce costs and improve patient care. Warren Buffet, Berkshire Hathaway’s founder, said in a statement, “The ballooning costs of health care act as a hungry tapeworm on the American economy.” (And yes, I imagine that many hospitalists would take umbrage with that characterization.) In addition to the Amazon alliance, CVS Health and Aetna also recently agreed to join forces.
The rising health care interest by Amazon begs the imagination. Technology already is far along in automating routine procedures, elevating patient safety protocols, and recalculating patient flows and information. This added corporate interest and investment will further expand new ideas and innovative technologies. And, for sure, it will challenge long held beliefs and practices that shape the health system we have today.
Hospitalist insight needed
What is the role of hospitalist leaders in this shifting equation? Hospitalists already can claim significant credit for introducing major changes in the landscape of hospital care in this country, with all the concomitant improvements in the efficiencies and quality of more integrated service delivery. Can you also guide the system in strategically selecting where and how technology can best be applied to automate and reconfigure service delivery?
The most important questions are: What is it that humans in health care uniquely do that cannot otherwise be accomplished? Are we able to hold onto the humane sides of health care, even as we seek to introduce cost-saving efficiencies?
Top of mind come the most personal sides of health service delivery: touch, empathy, understanding, and care itself. Next come human analysis, understanding, and translation. And beyond that, leadership, direction, and the vision to craft a health care system that meets our societal expectations – not just for the wealthy who cannot afford it – but for everyone.
It would be easy to dismiss this conversation. Society never decided whether those bank tellers, travel agents, or journalists were critical to our functioning. Along these same lines, you and your patients are more than mere algorithms.
As I often share in my leadership seminars, one key function of leaders is to identify and ask the right questions and to be at the decision-making table. What are those questions?
As a hospitalist leader, which part of your work and your activities could be eased by automation? Where might technology ease pressures and enhance your interactions with patients? How do we improve the efficiencies and effectiveness of health service delivery while we preserve the very human qualities that are fundamental to its values? No patient wants to speak to a physician who stares at a computer screen without eye contact, reassurance, or genuine interest. We can do better than that.
Business stakeholders in the system – and clearly, they are positioning and are powerful – will hold great sway on the contours of our future health care system. They could see humans – with all their costs, imperfections, and distractions – as replaceable.
Know that as you lead and pose your questions, there are people interested in listening. Certainly, the tech industry is looking for opportunities to generate broad market appeal. Similarly, health system decision makers looking to enhance how the system functions likewise seek guidance on what could – and could not – work. And who knows: Those decision makers could very well be you.
This is a conversation the country deserves. There is nothing more intimate, more personally important, and more professionally satisfying than the genuine person-to-person quality of what we do in health care. What we arrive at in the end should be achieved by intent, not by accident.
Dr. Marcus is coauthor of “Renegotiating Health Care: Resolving Conflict to Build Collaboration,” 2nd ed. (San Francisco: Jossey-Bass Publishers, 2011) and is director of the program for health care negotiation and conflict resolution, Harvard T.H. Chan School of Public Health, Boston. Dr. Marcus teaches regularly in the SHM Leadership Academy. He can be reached at ljmarcus@hsph.harvard.edu.
Major forces combining to reshape care delivery
Major forces combining to reshape care delivery
What will be the role of humans in the future health system?
At first blush, this is a peculiar question. Health care is all about humans. How could one doubt their presence or role? It is working with and for people that attracted many to this profession.
On the cusp of a significant health system reformulation, it is the very question that hospitalists now must ponder. Just as ATMs replaced bank cashiers, online shopping replaced retail stores, and autonomous cars will soon replace drivers, the human landscape of health care is about to change. What pressures will force the changes?
On one hand, there is increasing demand. The Affordable Care Act opened the insurance door for people previously uncovered. Aging is delivering the baby boomer bubble into their sicker years. Hospitalists witness this phenomenon every day in the ballooning parade of patients they serve. At times, those pressures can overwhelm.
On the other hand, the political will to provide government subsidized health coverage is waning. Washington is tripping over itself to dismantle Obamacare with glancing concern for how it will inflate the ranks of the uninsured. Employers are eager to free themselves from the burden of providing increasingly expensive health coverage benefits. By removing the mandate to buy health care insurance, the current political health system architects are liberating the healthy paying population from their contributions to the overall insurance pool. Simply put, there is and will be less money and less of all that it buys.
Combine building demand with decreasing budget into a system that does not follow general market forces: You get that earthquake. A consumer can forgo that new phone in hard times but not that cardiac procedure. People will be caught in the fissures of the system. Waits, quality, burnout, morale problems, and financial losses will all trend in the wrong directions. The process will evolve in slow motion. Some might argue that we have already arrived.
Enter entrepreneurs, technologic advances, and a growing savvy and willingness to engage tech solutions to everyday problems. If Alexa can turn on your toaster, could it take your blood pressure? If a robot can vacuum your rug, could a different robot provide personal care services? And, if an algorithm can drive your car, could it similarly diagnose what ails you?
On Jan. 30, 2018, one of the greatest disrupters of all time, Amazon, announced that it is joining forces with Berkshire Hathaway and JPMorgan Chase to leap into health care. While they are initially experimenting with health care changes for their corporate employees, the ultimate marketwide goal is to apply technology to both reduce costs and improve patient care. Warren Buffet, Berkshire Hathaway’s founder, said in a statement, “The ballooning costs of health care act as a hungry tapeworm on the American economy.” (And yes, I imagine that many hospitalists would take umbrage with that characterization.) In addition to the Amazon alliance, CVS Health and Aetna also recently agreed to join forces.
The rising health care interest by Amazon begs the imagination. Technology already is far along in automating routine procedures, elevating patient safety protocols, and recalculating patient flows and information. This added corporate interest and investment will further expand new ideas and innovative technologies. And, for sure, it will challenge long held beliefs and practices that shape the health system we have today.
Hospitalist insight needed
What is the role of hospitalist leaders in this shifting equation? Hospitalists already can claim significant credit for introducing major changes in the landscape of hospital care in this country, with all the concomitant improvements in the efficiencies and quality of more integrated service delivery. Can you also guide the system in strategically selecting where and how technology can best be applied to automate and reconfigure service delivery?
The most important questions are: What is it that humans in health care uniquely do that cannot otherwise be accomplished? Are we able to hold onto the humane sides of health care, even as we seek to introduce cost-saving efficiencies?
Top of mind come the most personal sides of health service delivery: touch, empathy, understanding, and care itself. Next come human analysis, understanding, and translation. And beyond that, leadership, direction, and the vision to craft a health care system that meets our societal expectations – not just for the wealthy who cannot afford it – but for everyone.
It would be easy to dismiss this conversation. Society never decided whether those bank tellers, travel agents, or journalists were critical to our functioning. Along these same lines, you and your patients are more than mere algorithms.
As I often share in my leadership seminars, one key function of leaders is to identify and ask the right questions and to be at the decision-making table. What are those questions?
As a hospitalist leader, which part of your work and your activities could be eased by automation? Where might technology ease pressures and enhance your interactions with patients? How do we improve the efficiencies and effectiveness of health service delivery while we preserve the very human qualities that are fundamental to its values? No patient wants to speak to a physician who stares at a computer screen without eye contact, reassurance, or genuine interest. We can do better than that.
Business stakeholders in the system – and clearly, they are positioning and are powerful – will hold great sway on the contours of our future health care system. They could see humans – with all their costs, imperfections, and distractions – as replaceable.
Know that as you lead and pose your questions, there are people interested in listening. Certainly, the tech industry is looking for opportunities to generate broad market appeal. Similarly, health system decision makers looking to enhance how the system functions likewise seek guidance on what could – and could not – work. And who knows: Those decision makers could very well be you.
This is a conversation the country deserves. There is nothing more intimate, more personally important, and more professionally satisfying than the genuine person-to-person quality of what we do in health care. What we arrive at in the end should be achieved by intent, not by accident.
Dr. Marcus is coauthor of “Renegotiating Health Care: Resolving Conflict to Build Collaboration,” 2nd ed. (San Francisco: Jossey-Bass Publishers, 2011) and is director of the program for health care negotiation and conflict resolution, Harvard T.H. Chan School of Public Health, Boston. Dr. Marcus teaches regularly in the SHM Leadership Academy. He can be reached at ljmarcus@hsph.harvard.edu.
What will be the role of humans in the future health system?
At first blush, this is a peculiar question. Health care is all about humans. How could one doubt their presence or role? It is working with and for people that attracted many to this profession.
On the cusp of a significant health system reformulation, it is the very question that hospitalists now must ponder. Just as ATMs replaced bank cashiers, online shopping replaced retail stores, and autonomous cars will soon replace drivers, the human landscape of health care is about to change. What pressures will force the changes?
On one hand, there is increasing demand. The Affordable Care Act opened the insurance door for people previously uncovered. Aging is delivering the baby boomer bubble into their sicker years. Hospitalists witness this phenomenon every day in the ballooning parade of patients they serve. At times, those pressures can overwhelm.
On the other hand, the political will to provide government subsidized health coverage is waning. Washington is tripping over itself to dismantle Obamacare with glancing concern for how it will inflate the ranks of the uninsured. Employers are eager to free themselves from the burden of providing increasingly expensive health coverage benefits. By removing the mandate to buy health care insurance, the current political health system architects are liberating the healthy paying population from their contributions to the overall insurance pool. Simply put, there is and will be less money and less of all that it buys.
Combine building demand with decreasing budget into a system that does not follow general market forces: You get that earthquake. A consumer can forgo that new phone in hard times but not that cardiac procedure. People will be caught in the fissures of the system. Waits, quality, burnout, morale problems, and financial losses will all trend in the wrong directions. The process will evolve in slow motion. Some might argue that we have already arrived.
Enter entrepreneurs, technologic advances, and a growing savvy and willingness to engage tech solutions to everyday problems. If Alexa can turn on your toaster, could it take your blood pressure? If a robot can vacuum your rug, could a different robot provide personal care services? And, if an algorithm can drive your car, could it similarly diagnose what ails you?
On Jan. 30, 2018, one of the greatest disrupters of all time, Amazon, announced that it is joining forces with Berkshire Hathaway and JPMorgan Chase to leap into health care. While they are initially experimenting with health care changes for their corporate employees, the ultimate marketwide goal is to apply technology to both reduce costs and improve patient care. Warren Buffet, Berkshire Hathaway’s founder, said in a statement, “The ballooning costs of health care act as a hungry tapeworm on the American economy.” (And yes, I imagine that many hospitalists would take umbrage with that characterization.) In addition to the Amazon alliance, CVS Health and Aetna also recently agreed to join forces.
The rising health care interest by Amazon begs the imagination. Technology already is far along in automating routine procedures, elevating patient safety protocols, and recalculating patient flows and information. This added corporate interest and investment will further expand new ideas and innovative technologies. And, for sure, it will challenge long held beliefs and practices that shape the health system we have today.
Hospitalist insight needed
What is the role of hospitalist leaders in this shifting equation? Hospitalists already can claim significant credit for introducing major changes in the landscape of hospital care in this country, with all the concomitant improvements in the efficiencies and quality of more integrated service delivery. Can you also guide the system in strategically selecting where and how technology can best be applied to automate and reconfigure service delivery?
The most important questions are: What is it that humans in health care uniquely do that cannot otherwise be accomplished? Are we able to hold onto the humane sides of health care, even as we seek to introduce cost-saving efficiencies?
Top of mind come the most personal sides of health service delivery: touch, empathy, understanding, and care itself. Next come human analysis, understanding, and translation. And beyond that, leadership, direction, and the vision to craft a health care system that meets our societal expectations – not just for the wealthy who cannot afford it – but for everyone.
It would be easy to dismiss this conversation. Society never decided whether those bank tellers, travel agents, or journalists were critical to our functioning. Along these same lines, you and your patients are more than mere algorithms.
As I often share in my leadership seminars, one key function of leaders is to identify and ask the right questions and to be at the decision-making table. What are those questions?
As a hospitalist leader, which part of your work and your activities could be eased by automation? Where might technology ease pressures and enhance your interactions with patients? How do we improve the efficiencies and effectiveness of health service delivery while we preserve the very human qualities that are fundamental to its values? No patient wants to speak to a physician who stares at a computer screen without eye contact, reassurance, or genuine interest. We can do better than that.
Business stakeholders in the system – and clearly, they are positioning and are powerful – will hold great sway on the contours of our future health care system. They could see humans – with all their costs, imperfections, and distractions – as replaceable.
Know that as you lead and pose your questions, there are people interested in listening. Certainly, the tech industry is looking for opportunities to generate broad market appeal. Similarly, health system decision makers looking to enhance how the system functions likewise seek guidance on what could – and could not – work. And who knows: Those decision makers could very well be you.
This is a conversation the country deserves. There is nothing more intimate, more personally important, and more professionally satisfying than the genuine person-to-person quality of what we do in health care. What we arrive at in the end should be achieved by intent, not by accident.
Dr. Marcus is coauthor of “Renegotiating Health Care: Resolving Conflict to Build Collaboration,” 2nd ed. (San Francisco: Jossey-Bass Publishers, 2011) and is director of the program for health care negotiation and conflict resolution, Harvard T.H. Chan School of Public Health, Boston. Dr. Marcus teaches regularly in the SHM Leadership Academy. He can be reached at ljmarcus@hsph.harvard.edu.
ERRATUM TO: Cardiac Troponins in Low-Risk Pulmonary Embolism Patients: A Systematic Review and Meta-Analysis
The authors would like to make the following corrections to their manuscript, Cardiac Troponins in Low-Risk Pulmonary Embolism Patients: A Systematic Review and Meta-Analysis (doi: 10.12788/jhm.2961), published online first April 25, 2018 (all corrections in bold):
- The last sentence of the results section in the abstract should read: The pooled likelihood ratios (LRs) for all-cause mortality were positive LR 2.04 [95% CI, 1.53 to 2.72] and negative LR 0.72 [95% CI, 0.37 to 1.40].
- In the "All studies pooled" of the last row of Table 2, Tn+ is corrected to 463. See revised table below.
- On page E5, the first paragraph in the "Outcomes of Studies with Corresponding Troponin+ and Troponin-" section beginning with the fifth sentence should read as follows):
"In the pooled data, 463 (67%) patients tested negative for troponin and 228 (33%) tested positive. The overall mortality (from sensitivity analysis) including in-hospital, 30-day, and 90-day mortalities was 1.2%. The NPVs for all individual studies and the overall NPV are 1 or approximately 1. The overall PPVs and by study were low, ranging from 0 to 0.60. The PLRs and NLRs were not estimated for an outcome within an individual study if none of the patients experienced the outcome. When outcomes were only observed among troponin-negative patients, such as in the study of Moore (2009) who used 30-day all-cause mortality, the PLR had a value of zero. When outcomes were only observed among troponin-positive patients, as for 30-day all-cause mortality in the Hakemi9(2015), Lauque10 (2014), and Lankeit16 (2011) studies, the NLR had a value of zero. For zero cells, a continuity correction of 0.5 was applied. The pooled likelihood ratios (LRs) for all-cause mortality were positive LR 2.04 [95% CI, 1.53 to 2.72] and negative LR 0.72 [95% CI, 0.37 to 1.40]. The OR for all-cause mortality was 4.79 [95% CI 1.11 to 20.68, P = .0357].

The authors would like to make the following corrections to their manuscript, Cardiac Troponins in Low-Risk Pulmonary Embolism Patients: A Systematic Review and Meta-Analysis (doi: 10.12788/jhm.2961), published online first April 25, 2018 (all corrections in bold):
- The last sentence of the results section in the abstract should read: The pooled likelihood ratios (LRs) for all-cause mortality were positive LR 2.04 [95% CI, 1.53 to 2.72] and negative LR 0.72 [95% CI, 0.37 to 1.40].
- In the "All studies pooled" of the last row of Table 2, Tn+ is corrected to 463. See revised table below.
- On page E5, the first paragraph in the "Outcomes of Studies with Corresponding Troponin+ and Troponin-" section beginning with the fifth sentence should read as follows):
"In the pooled data, 463 (67%) patients tested negative for troponin and 228 (33%) tested positive. The overall mortality (from sensitivity analysis) including in-hospital, 30-day, and 90-day mortalities was 1.2%. The NPVs for all individual studies and the overall NPV are 1 or approximately 1. The overall PPVs and by study were low, ranging from 0 to 0.60. The PLRs and NLRs were not estimated for an outcome within an individual study if none of the patients experienced the outcome. When outcomes were only observed among troponin-negative patients, such as in the study of Moore (2009) who used 30-day all-cause mortality, the PLR had a value of zero. When outcomes were only observed among troponin-positive patients, as for 30-day all-cause mortality in the Hakemi9(2015), Lauque10 (2014), and Lankeit16 (2011) studies, the NLR had a value of zero. For zero cells, a continuity correction of 0.5 was applied. The pooled likelihood ratios (LRs) for all-cause mortality were positive LR 2.04 [95% CI, 1.53 to 2.72] and negative LR 0.72 [95% CI, 0.37 to 1.40]. The OR for all-cause mortality was 4.79 [95% CI 1.11 to 20.68, P = .0357].
The authors would like to make the following corrections to their manuscript, Cardiac Troponins in Low-Risk Pulmonary Embolism Patients: A Systematic Review and Meta-Analysis (doi: 10.12788/jhm.2961), published online first April 25, 2018 (all corrections in bold):
- The last sentence of the results section in the abstract should read: The pooled likelihood ratios (LRs) for all-cause mortality were positive LR 2.04 [95% CI, 1.53 to 2.72] and negative LR 0.72 [95% CI, 0.37 to 1.40].
- In the "All studies pooled" of the last row of Table 2, Tn+ is corrected to 463. See revised table below.
- On page E5, the first paragraph in the "Outcomes of Studies with Corresponding Troponin+ and Troponin-" section beginning with the fifth sentence should read as follows):
"In the pooled data, 463 (67%) patients tested negative for troponin and 228 (33%) tested positive. The overall mortality (from sensitivity analysis) including in-hospital, 30-day, and 90-day mortalities was 1.2%. The NPVs for all individual studies and the overall NPV are 1 or approximately 1. The overall PPVs and by study were low, ranging from 0 to 0.60. The PLRs and NLRs were not estimated for an outcome within an individual study if none of the patients experienced the outcome. When outcomes were only observed among troponin-negative patients, such as in the study of Moore (2009) who used 30-day all-cause mortality, the PLR had a value of zero. When outcomes were only observed among troponin-positive patients, as for 30-day all-cause mortality in the Hakemi9(2015), Lauque10 (2014), and Lankeit16 (2011) studies, the NLR had a value of zero. For zero cells, a continuity correction of 0.5 was applied. The pooled likelihood ratios (LRs) for all-cause mortality were positive LR 2.04 [95% CI, 1.53 to 2.72] and negative LR 0.72 [95% CI, 0.37 to 1.40]. The OR for all-cause mortality was 4.79 [95% CI 1.11 to 20.68, P = .0357].
© 2018 Society of Hospital Medicine
Salivary gland ultrasound is accurate diagnostic tool for Sjögren’s
AMSTERDAM – Ultrasound of the salivary glands is a readily available and inexpensive tool for the diagnosis of Sjögren’s syndrome, according to a study that evaluated this test in relation to the recent American College of Rheumatology and European League Against Rheumatism (ACR/EULAR) classification criteria.
In a video interview, Esther-Jellina Mossel reported that the sensitivity and specificity of a Sjögren’s syndrome diagnosis is essentially unchanged when ultrasound replaces a positive ocular staining score, the Schirmer test, or an unstimulated whole saliva flow test, without reducing diagnostic accuracy.
The sensitivity of the diagnosis is reduced only if ultrasound is used to replace either of the two remaining ACR/EULAR criteria, which are a labial gland biopsy or an anti-SSA antibody test. In relation to the three criteria that it can replace without loss of diagnostic accuracy, ultrasound might have advantages.
“People who don’t have access to an ophthalmologist performing an ocular staining score, for instance, could use an ultrasound of the salivary glands instead of the ocular staining score and still make a diagnosis,” said Ms. Mossel, a PhD student in the department of rheumatology at the University of Groningen (the Netherlands).
Ultrasound, which is commonly used to evaluate joints of patients with inflammatory diseases, is available in the offices of most rheumatologists, according to Ms. Mossel. She estimated that the evaluation of the salivary glands, which reveals characteristic hypoechogenic areas when Sjögren’s syndrome is present, takes about 10 minutes.
At Ms. Mossel’s center, ultrasound has already become a standard tool for the diagnosis of Sjögren’s syndrome. She said that other centers have also found this imaging tool to be accurate and useful for Sjögren’s syndrome diagnosis.
Based on the experience at the University of Groningen, Ms. Mossel believes that ultrasound will eventually be widely adopted for Sjögren’s syndrome diagnosis. Indeed, she expects that this strategy is likely to be added to the ACR/EULAR diagnostic criteria when its accuracy becomes more generally recognized.
AMSTERDAM – Ultrasound of the salivary glands is a readily available and inexpensive tool for the diagnosis of Sjögren’s syndrome, according to a study that evaluated this test in relation to the recent American College of Rheumatology and European League Against Rheumatism (ACR/EULAR) classification criteria.
In a video interview, Esther-Jellina Mossel reported that the sensitivity and specificity of a Sjögren’s syndrome diagnosis is essentially unchanged when ultrasound replaces a positive ocular staining score, the Schirmer test, or an unstimulated whole saliva flow test, without reducing diagnostic accuracy.
The sensitivity of the diagnosis is reduced only if ultrasound is used to replace either of the two remaining ACR/EULAR criteria, which are a labial gland biopsy or an anti-SSA antibody test. In relation to the three criteria that it can replace without loss of diagnostic accuracy, ultrasound might have advantages.
“People who don’t have access to an ophthalmologist performing an ocular staining score, for instance, could use an ultrasound of the salivary glands instead of the ocular staining score and still make a diagnosis,” said Ms. Mossel, a PhD student in the department of rheumatology at the University of Groningen (the Netherlands).
Ultrasound, which is commonly used to evaluate joints of patients with inflammatory diseases, is available in the offices of most rheumatologists, according to Ms. Mossel. She estimated that the evaluation of the salivary glands, which reveals characteristic hypoechogenic areas when Sjögren’s syndrome is present, takes about 10 minutes.
At Ms. Mossel’s center, ultrasound has already become a standard tool for the diagnosis of Sjögren’s syndrome. She said that other centers have also found this imaging tool to be accurate and useful for Sjögren’s syndrome diagnosis.
Based on the experience at the University of Groningen, Ms. Mossel believes that ultrasound will eventually be widely adopted for Sjögren’s syndrome diagnosis. Indeed, she expects that this strategy is likely to be added to the ACR/EULAR diagnostic criteria when its accuracy becomes more generally recognized.
AMSTERDAM – Ultrasound of the salivary glands is a readily available and inexpensive tool for the diagnosis of Sjögren’s syndrome, according to a study that evaluated this test in relation to the recent American College of Rheumatology and European League Against Rheumatism (ACR/EULAR) classification criteria.
In a video interview, Esther-Jellina Mossel reported that the sensitivity and specificity of a Sjögren’s syndrome diagnosis is essentially unchanged when ultrasound replaces a positive ocular staining score, the Schirmer test, or an unstimulated whole saliva flow test, without reducing diagnostic accuracy.
The sensitivity of the diagnosis is reduced only if ultrasound is used to replace either of the two remaining ACR/EULAR criteria, which are a labial gland biopsy or an anti-SSA antibody test. In relation to the three criteria that it can replace without loss of diagnostic accuracy, ultrasound might have advantages.
“People who don’t have access to an ophthalmologist performing an ocular staining score, for instance, could use an ultrasound of the salivary glands instead of the ocular staining score and still make a diagnosis,” said Ms. Mossel, a PhD student in the department of rheumatology at the University of Groningen (the Netherlands).
Ultrasound, which is commonly used to evaluate joints of patients with inflammatory diseases, is available in the offices of most rheumatologists, according to Ms. Mossel. She estimated that the evaluation of the salivary glands, which reveals characteristic hypoechogenic areas when Sjögren’s syndrome is present, takes about 10 minutes.
At Ms. Mossel’s center, ultrasound has already become a standard tool for the diagnosis of Sjögren’s syndrome. She said that other centers have also found this imaging tool to be accurate and useful for Sjögren’s syndrome diagnosis.
Based on the experience at the University of Groningen, Ms. Mossel believes that ultrasound will eventually be widely adopted for Sjögren’s syndrome diagnosis. Indeed, she expects that this strategy is likely to be added to the ACR/EULAR diagnostic criteria when its accuracy becomes more generally recognized.
REPORTING FROM THE EULAR 2018 CONGRESS

FDA approves Epidiolex for Lennox-Gastaut syndrome and Dravet syndrome
The Food and Drug Administration has approved cannabidiol oral solution (Epidiolex, GW Pharmaceuticals) for the treatment of two rare pediatric seizure disorders.

“This product approval demonstrates that advancing sound scientific research to investigate ingredients derived from marijuana can lead to important therapies. This new treatment provides new options for patients,” said FDA Commissioner Scott Gottlieb, MD, in a statement.
However, he cautioned, “This is an important medical advance. But it’s also important to note that this is not an approval of marijuana or all of its components. This is the approval of one specific CBD medication for a specific use. And it was based on well-controlled clinical trials evaluating the use of this compound in the treatment of a specific condition.”
The FDA Peripheral and Central Nervous System Drugs Advisory Committee’s earlier positive recommendation was based on three randomized, double-blind, placebo-controlled clinical trials. These trials showed a 50% reduction of drop seizure frequency in 40%-44% of patients with Lennox-Gastaut syndrome, and a 39% decrease in convulsive seizure frequency for trial participants with Dravet Syndrome. A total of 516 patients with one of the two seizure disorders participated in the clinical trials.
“In addition to another important treatment option for Lennox-Gastaut patients, this first-ever approval of a drug specifically for Dravet patients will provide a significant and needed improvement in the therapeutic approach to caring for people with this condition,” said Billy Dunn, MD, director of the Division of Neurology Products in the FDA Center for Drug Evaluation and Research, in a statement.
After reviewing information provided by the drug’s sponsor and the FDA, the advisory committee judged that CBD-OS, derived from a non-psychoactive chemical found in marijuana, was very unlikely to have potential for abuse.
Sedation, sleepiness, and lethargy were among the most frequently reported adverse events for the patients taking CBD-OS. In data pooled from the clinical trials, 16.3% of patients taking CBD-OS at the higher dose of 20 mg/kg/day had liver transaminase elevations above three times the upper limit of normal; this level of transaminase elevation was seen in 0.9% of patients taking placebo.
A patient medication guide detailing risks and how the drug should be used will accompany CBD-OS when it is dispensed, according to the FDA approval.
In his statement, Dr. Gottlieb put the approval in the context of the FDA’s broader efforts to encourage a strong clinical development program for marijuana-derived drugs that does not compromise standards for ensuring safety and efficacy of drugs approved by the agency. He also noted that ongoing efforts to support high quality research into marijuana-based therapies involve other federal agencies, including the National Institute on Drug Abuse and the Drug Enforcement Administration.
The FDA’s actions against companies distributing unapproved products that contain cannabidiol and making unproven marketing claims will continue, said Dr. Gottlieb. Still, “Today’s approval demonstrates our commitment to the scientific process and working with product developers to bring marijuana-based products to market,” he said.
The Food and Drug Administration has approved cannabidiol oral solution (Epidiolex, GW Pharmaceuticals) for the treatment of two rare pediatric seizure disorders.
“This product approval demonstrates that advancing sound scientific research to investigate ingredients derived from marijuana can lead to important therapies. This new treatment provides new options for patients,” said FDA Commissioner Scott Gottlieb, MD, in a statement.
However, he cautioned, “This is an important medical advance. But it’s also important to note that this is not an approval of marijuana or all of its components. This is the approval of one specific CBD medication for a specific use. And it was based on well-controlled clinical trials evaluating the use of this compound in the treatment of a specific condition.”
The FDA Peripheral and Central Nervous System Drugs Advisory Committee’s earlier positive recommendation was based on three randomized, double-blind, placebo-controlled clinical trials. These trials showed a 50% reduction of drop seizure frequency in 40%-44% of patients with Lennox-Gastaut syndrome, and a 39% decrease in convulsive seizure frequency for trial participants with Dravet Syndrome. A total of 516 patients with one of the two seizure disorders participated in the clinical trials.
“In addition to another important treatment option for Lennox-Gastaut patients, this first-ever approval of a drug specifically for Dravet patients will provide a significant and needed improvement in the therapeutic approach to caring for people with this condition,” said Billy Dunn, MD, director of the Division of Neurology Products in the FDA Center for Drug Evaluation and Research, in a statement.
After reviewing information provided by the drug’s sponsor and the FDA, the advisory committee judged that CBD-OS, derived from a non-psychoactive chemical found in marijuana, was very unlikely to have potential for abuse.
Sedation, sleepiness, and lethargy were among the most frequently reported adverse events for the patients taking CBD-OS. In data pooled from the clinical trials, 16.3% of patients taking CBD-OS at the higher dose of 20 mg/kg/day had liver transaminase elevations above three times the upper limit of normal; this level of transaminase elevation was seen in 0.9% of patients taking placebo.
A patient medication guide detailing risks and how the drug should be used will accompany CBD-OS when it is dispensed, according to the FDA approval.
In his statement, Dr. Gottlieb put the approval in the context of the FDA’s broader efforts to encourage a strong clinical development program for marijuana-derived drugs that does not compromise standards for ensuring safety and efficacy of drugs approved by the agency. He also noted that ongoing efforts to support high quality research into marijuana-based therapies involve other federal agencies, including the National Institute on Drug Abuse and the Drug Enforcement Administration.
The FDA’s actions against companies distributing unapproved products that contain cannabidiol and making unproven marketing claims will continue, said Dr. Gottlieb. Still, “Today’s approval demonstrates our commitment to the scientific process and working with product developers to bring marijuana-based products to market,” he said.
The Food and Drug Administration has approved cannabidiol oral solution (Epidiolex, GW Pharmaceuticals) for the treatment of two rare pediatric seizure disorders.
“This product approval demonstrates that advancing sound scientific research to investigate ingredients derived from marijuana can lead to important therapies. This new treatment provides new options for patients,” said FDA Commissioner Scott Gottlieb, MD, in a statement.
However, he cautioned, “This is an important medical advance. But it’s also important to note that this is not an approval of marijuana or all of its components. This is the approval of one specific CBD medication for a specific use. And it was based on well-controlled clinical trials evaluating the use of this compound in the treatment of a specific condition.”
The FDA Peripheral and Central Nervous System Drugs Advisory Committee’s earlier positive recommendation was based on three randomized, double-blind, placebo-controlled clinical trials. These trials showed a 50% reduction of drop seizure frequency in 40%-44% of patients with Lennox-Gastaut syndrome, and a 39% decrease in convulsive seizure frequency for trial participants with Dravet Syndrome. A total of 516 patients with one of the two seizure disorders participated in the clinical trials.
“In addition to another important treatment option for Lennox-Gastaut patients, this first-ever approval of a drug specifically for Dravet patients will provide a significant and needed improvement in the therapeutic approach to caring for people with this condition,” said Billy Dunn, MD, director of the Division of Neurology Products in the FDA Center for Drug Evaluation and Research, in a statement.
After reviewing information provided by the drug’s sponsor and the FDA, the advisory committee judged that CBD-OS, derived from a non-psychoactive chemical found in marijuana, was very unlikely to have potential for abuse.
Sedation, sleepiness, and lethargy were among the most frequently reported adverse events for the patients taking CBD-OS. In data pooled from the clinical trials, 16.3% of patients taking CBD-OS at the higher dose of 20 mg/kg/day had liver transaminase elevations above three times the upper limit of normal; this level of transaminase elevation was seen in 0.9% of patients taking placebo.
A patient medication guide detailing risks and how the drug should be used will accompany CBD-OS when it is dispensed, according to the FDA approval.
In his statement, Dr. Gottlieb put the approval in the context of the FDA’s broader efforts to encourage a strong clinical development program for marijuana-derived drugs that does not compromise standards for ensuring safety and efficacy of drugs approved by the agency. He also noted that ongoing efforts to support high quality research into marijuana-based therapies involve other federal agencies, including the National Institute on Drug Abuse and the Drug Enforcement Administration.
The FDA’s actions against companies distributing unapproved products that contain cannabidiol and making unproven marketing claims will continue, said Dr. Gottlieb. Still, “Today’s approval demonstrates our commitment to the scientific process and working with product developers to bring marijuana-based products to market,” he said.