User login
Unusual Presentation of Erythema Elevatum Diutinum With Underlying Hepatitis B Infection
Erythema elevatum diutinum (EED) manifests on a clinicopathologic spectrum of chronic cutaneous small vessel vasculitis. The lesions typically present as persistent, symmetric, firm, red to purple papules or nodules on the extensor arms and dorsal hands.1,2 Underlying infectious, malignant, or autoimmune processes are commonly associated with the disease, notably Streptococcus infection and IgA monoclonal gammopathy.2,3 Hepatitis virus also is often implicated in association with EED. Cases of EED have been seen with concomitant human immunodeficiency virus (HIV) infection.4-6 We report a case of EED presenting in various stages of evolution associated with underlying hepatitis B infection alone.
Case Report
A 57-year-old man originally presented to an outpatient dermatology practice with a nodular, painful, episodic rash on the trunk and upper and lower extremities. A biopsy revealed leukocytoclastic vasculitis (LCV) with prominent eosinophils. At the time, the skin findings were believed to be a manifestation of drug hypersensitivity, likely to opioid use. The patient was lost to follow-up.
Seven years later, the patient was admitted to the hospital with new-onset burning and stinging red nodules on the dorsum of the hands and persistence of the original episodic rash over the lower legs and bilateral flanks. In the interim, he was briefly treated with an oral prednisone taper and topical corticosteroids including triamcinolone cream 0.1% and clobetasol cream 0.05% without improvement.
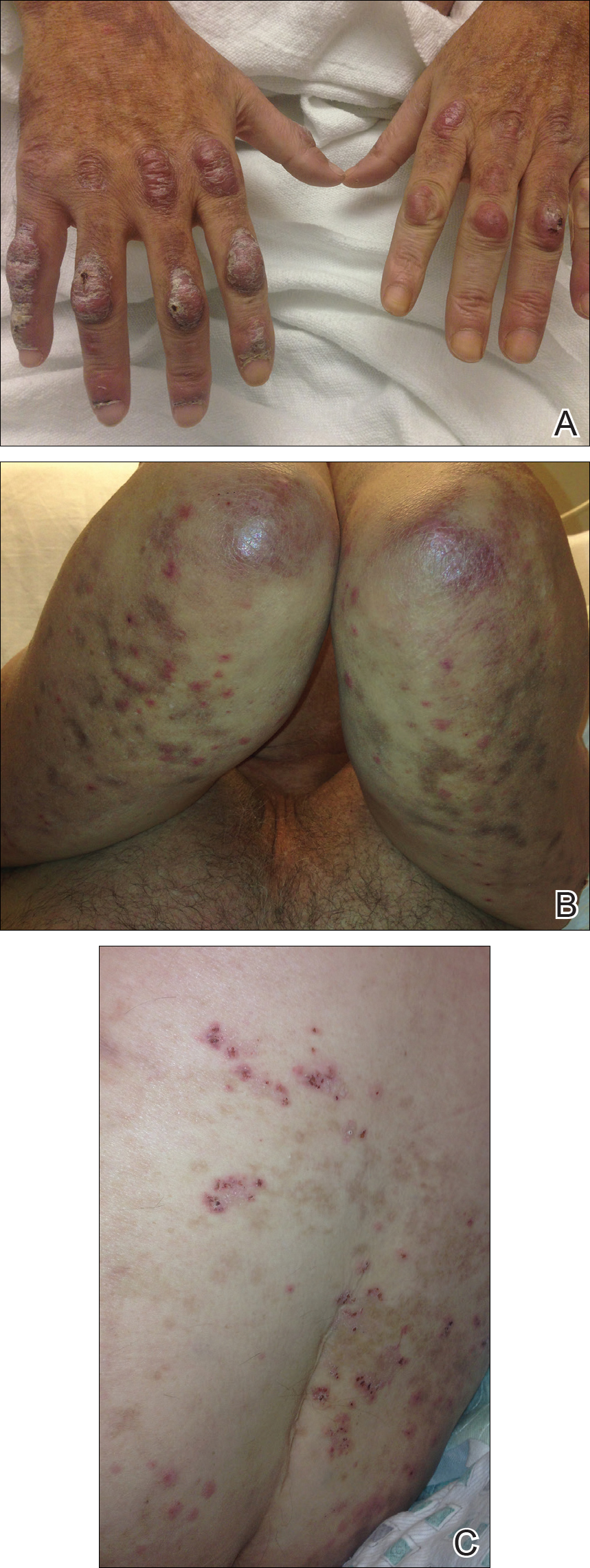
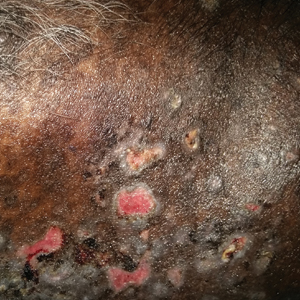
On examination deep red to violaceous discrete nodules and plaques with overlying hyperkeratosis involving all distal and proximal interphalangeal joints of the hands and extensor elbows were seen (Figure 1A). On the bilateral posterior arms (Figure 1B), anterior legs, and periumbilical area were deeply erythematous papules and plaques with background hyperpigmentation. Across his lower back and bilateral flanks were erythematous papules with central hemorrhagic crusting (Figure 1C).
Pertinent laboratory findings included a positive hepatitis B surface antigen with hepatitis B DNA value 4,313,876 IU/mL and a hepatitis B virus quantitative polymerase chain reaction value of 6.64 U. The etiology was suspected to be intravenous drug abuse; however, the patient denied recreational drug use.
An additional infectious workup was negative for hepatitis C, streptococcus, syphilis, tuberculosis, and HIV. A complete blood cell count, complete metabolic panel, urinalysis, complement, cryoglobulins, and serum protein electrophoresis were within reference range. Autoimmune serologies were negative including antinuclear antibody, rheumatoid factor, anti-Sjögren syndrome–related antigen A and B, anticyclic citrullinated peptide, anti-Smith, and antineutrophilic cytoplasmic antibodies. Peripheral blood immunophenotyping, lactate dehydrogenase, quantitative immunoglobulins, and age-appropriate cancer screens did not demonstrate evidence for malignancy underlying the disease. Bilateral hand radiographs showed mild periostitis of the proximal phalanges without obvious erosions.
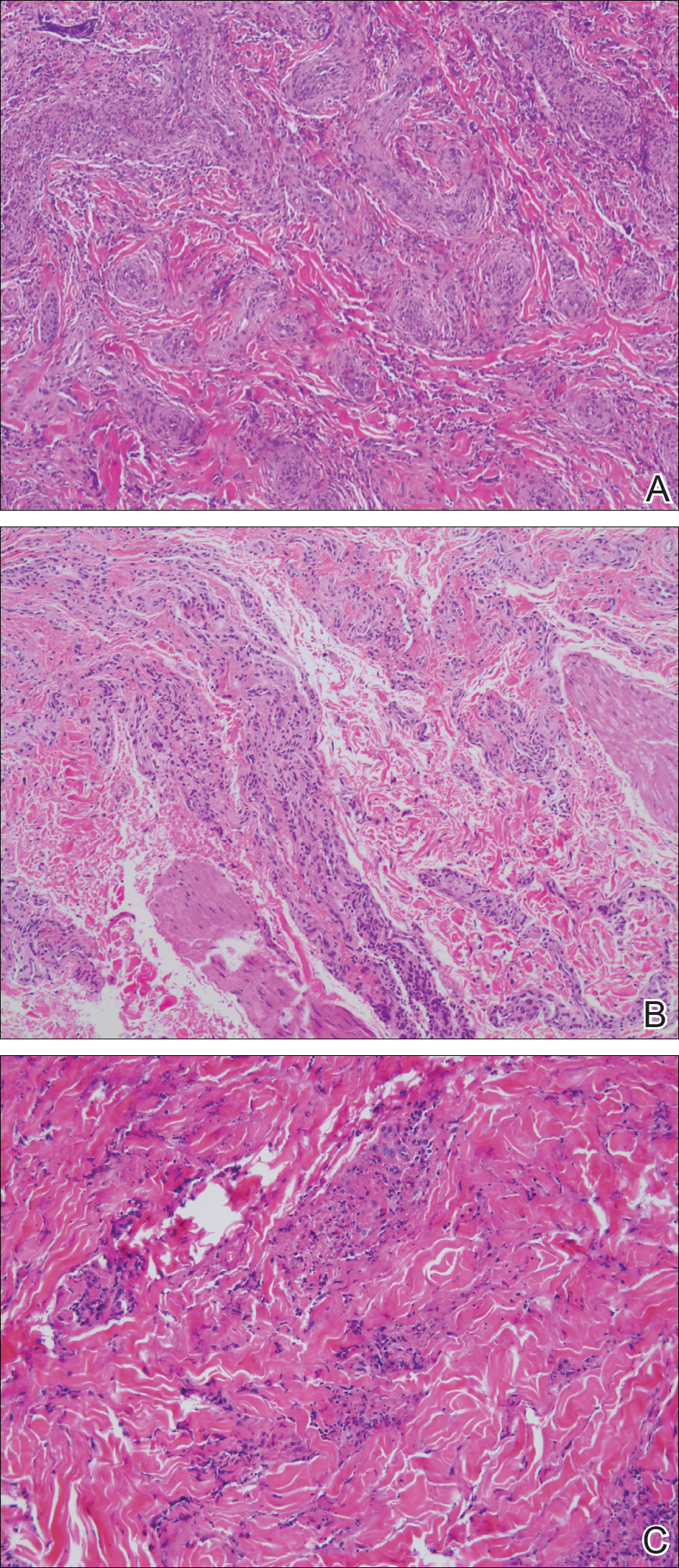
Three 4-mm punch biopsies were performed from the left fifth digit, left posterior arm, and left flank. Tissue of the left fifth digit showed an intradermal vascular proliferation with a concentric pattern resembling onion skin in a background of increased fibrosis. The blood vessels showed focal fibrinoid necrosis (Figure 2A). The biopsy of the left posterior arm showed an intradermal vascular proliferation with an associated mild acute and chronic perivascular inflammation (Figure 2B). The left flank biopsy showed LCV with focal epidermal necrosis (Figure 2C).
The constellation of clinical findings together with the histopathologic changes represented EED in various stages of evolution. The patient was started on dapsone 100 mg daily and referred to the infectious disease service for treatment of chronic hepatitis B; however, he was subsequently lost to follow-up.
Comment
Overview of EED
Erythema elevatum diutinum represents a rare form of chronic cutaneous small vessel vasculitis. Originally described by Hutchinson7 and Bury8 as symmetric purpuric nodules of the skin, it was later named by Crocker and Williams9 in 1894. The disease classically presents as firm, fixed, red-brown to violaceous papules, plaques, and nodules affecting the extensor upper or lower extremities.1 Lesions are most commonly found symmetrically overlying joints of the hands, feet, elbows, and knees, as well as the Achilles tendon and buttocks.3 Less common locations include the palms and soles, face,10,11 trunk,12 and periauricular region.1 Although they are typically asymptomatic, sensations such as burning, stinging, and pruritus have been noted.1 Our patient was unique because in addition to typical lesions of EED, he presented with crusted papules on the flanks and violaceous papules of the lower legs and periumbilicus.
Etiology
Originally associated with Streptococcus as isolated from EED lesions,3,13 additional infectious etiologies include viral hepatitis,4-6 human herpesvirus 6,14 and rarely HIV.1,15 Hepatitis B and C are well known to be associated with EED, with only rare reports in patients with concomitant HIV infection. Erythema elevatum diutinum also has been described in relationship to myeloproliferative disorders and hematologic malignancies such as IgA myeloma,16 non-Hodgkin lymphoma,17 chronic lymphocytic leukemia,18 and hypergammaglobulinemia.19 In a study of 13 patients with EED, 4 had associated underlying IgA monoclonal gammopathy.2 Autoimmune conditions such as rheumatoid arthritis,20 ulcerative colitis,21 relapsing polychondritis,22 and systemic lupus erythematosus23 also have been implicated.
Pathogenesis
Although the precise pathogenesis of EED remains unknown, it has been suggested that a complement cascade initiated by immune-complex deposition in postcapillary venules induces an LCV.24,25 Chronic antigenic exposure or high antibody levels26 in the face of infections, autoimmune disease, or malignancy may incite this immune-complex reaction. Skin lesions seen in association with hepatitis reflect circulating immune-complex deposition in vessel walls causing destruction. It has been postulated that the duration of immune complexemia may be sufficient to account for the differences in the type of vascular injury seen in acute versus chronic infection.27
Histopathology
Erythema elevatum diutinum may present on a histopathologic spectrum of LCV, as manifested in our patient. Early lesions show predominantly polymorphonuclear cells with nuclear dust pattern in a wedge-shaped infiltrate with fibrin deposition in the superficial and mid dermis.2,3 Later lesions show vasculitis in addition to dermal aggregates of lymphocytes, neutrophils, fibrosis, and areas of granulation tissue. The fibrosis may be dense and comprised of fibroblasts and myofibroblasts.28 Newly formed vessels within the granulation tissue have been postulated to be more susceptible to immune-complex deposition, thus potentiating the process.1,29
Management
Spontaneous resolution of EED may occur, albeit after a prolonged and recurrent course of up to 5 to 10 years.30 Treatment of the underlying cause, when identified, remains paramount. First-line therapy includes dapsone, shown to be effective in reducing lesion size to complete resolution in 80% of the 47 cases reviewed by Momen et al.31 Dapsone monotherapy tends to be less effective in treating nodular lesions associated with HIV-positivity, likely due to the extensive fibrosis.4,31 Combination therapy with dapsone and a sulfonamide,32 niacinamide and tetracycline,33 colchicine,34 or surgical excision35 may be necessary in more resistant cases.
Conclusion
Our case exemplifies the clinical histologic spectrum that EED can present. The constellation of clinical findings was histologically confirmed to be manifestations of the disease in various stages of evolution. When typical lesions of EED present along with cutaneous findings in less common locations, performing multiple biopsies can be helpful. The clinician should retain a high index of suspicion for an underlying etiology and perform a complete workup for infection, malignancy, or autoimmune disease.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Yiannias JA, el-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol. 1992;26:38-44.
- Wilkinson SM, English JS, Smith NP, et al. Erythema elevatum diutinum: a clinicopathological study. Clin Exp Dermatol. 1992;17:87-93.
- Fakheri A, Gupta SM, White SM, et al. Erythema elevatum diutinum in a patient with human immunodeficiency virus. Cutis. 2001;68:41-42, 55.
- Kim H. Erythema elevatum diutinum in an HIV-positive patient. J Drugs Dermatol. 2003;2:411-412.
- Revenga F, Vera A, Muñoz A, et al. Erythema elevatum diutinum and AIDS: are they related? Clin Exp Dermatol. 1997;22:250-251.
- Hutchinson J. On two remarkable cases of symmetrieal purple congestion of the skin in patches, with induration. Br J Dermatol. 1888;1:10-15.
- Bury JS. A case of erythema with remarkable nodular thickening and induration of the skin associated with intermittent albuminuria. Illustrated Medical News. 1889;3:145-149.
- Crocker HR, Williams C. Erythema elevatum diutinum. Br J Dermatol. 1894;6:33-38.
- Barzegar M, Davatchi CC, Akhyani M, et al. An atypical presentation of erythema elevatum diutinum involving palms and soles. Int J Dermatol. 2009;48:73-75.
- Futei Y, Konohana I. A case of erythema elevatum diutinum associated with B-cell lymphoma: a rare distribution involving palms, soles and nails. Br J Dermatol. 2000;142:116-119.
- Ben-Zvi GT, Bardsley V, Burrows NP. An atypical distribution of erythema elevatum diutinum. Clin Exp Dermatol. 2014;39:269-270.
- Weidman FD, Besancon JH. Erythema elevatum diutinum. role of streptococci, and relationship to other rheumatic dermatoses. Arch Dermatol Syphilol. 1929;20:593-620.
- Drago F, Semino M, Rampini P, et al. Erythema elevatum diutinum in a patient with human herpesvirus 6 infection. Acta Derm Venereol. 1999;79:91-92.
- Muratori S, Carrera C, Gorani A, et al. Erythema elevatum diutinum and HIV infection: a report of five cases. Br J Dermatol. 1999;141:335-338.
- Archimandritis AJ, Fertakis A, Alegakis G, et al. Erythema elevatum diutinum and IgA myeloma: an interesting association. Br Med J. 1977;2:613-614.
- Hatzitolios A, Tzellos TG, Savopoulos C, et al. Erythema elevatum diutinum with rare distribution as a first clinical sign of non-Hodgkin’s lymphoma: a novel association? J Dermatol. 2008;35:297-300.
- Delaporte E, Alfandari S, Fenaux P, et al. Erythema elevatum diutinum and chronic lymphocytic leukaemia. Clin Exp Dermatol. 1994;19:188-189.
- Miyagawa S, Kitamura W, Morita K, et al. Association of hyperimmunoglobulinaemia D syndrome with erythema elevatum diutinum. Br J Dermatol. 1993;128:572-574.
- Collier PM, Neill SM, Branfoot AC, et al. Erythema elevatum diutinum—a solitary lesion in a patient with rheumatoid arthritis. Clin Exp Dermatol. 1990;15:394-395.
- Buahene K, Hudson M, Mowat A, et al. Erythema elevatum diutinum—an unusual association with ulcerative colitis. Clin Exp Dermatol. 1991;16:204-206.
- Bernard P, Bedane C, Delrous JL, et al. Erythema elevatum diutinum in a patient with relapsing polychondritis. J Am Acad Dermatol. 1992;26:312-315.
- Hancox JG, Wallace CA, Sangueza OP, et al. Erythema elevatum diutinum associated with lupus panniculitis in a patient with discoid lesions of chronic cutaneous lupus erythematosus. J Am Acad Dermatol. 2004;50:652-653.
- Haber H. Erythema elevatum diutinum. Br J Dermatol. 1955;67:121-145.
- Katz SI, Gallin JL, Hertz KC, et al. Erythema elevatum diutinum: skin and systemic manifestations, immunologic studies, and successful treatment with dapsone. Medicine (Baltimore). 1977;56:443-455.
- Walker KD, Badame AJ. Erythema elevatum diutinum in a patient with Crohn’s disease. J Am Acad Dermatol. 1990;22:948-952.
- Popp JW, Harrist T, Dienstag JL, et al. Cutaneous vasculitis associated with acute and chronic hepatitis. Arch Intern Med. 1981;141:623-629.
- Lee AY, Nakagawa H, Nogita T, et al. Erythema elevatum diutinum: an ultrastructural case study. J Cutan Pathol. 1989;16:211-217.
- LeBoit PE, Yen TS, Wintroub B. The evolution of lesions in erythema elevatum diutinum. Am J Dermatopathol. 1986;8:392-402.
- Soubeiran E, Wacker J, Hausser I, et al. Erythema elevatum diutinum with unusual clinical appearance. J Dtsch Dermatol Ges. 2008;6:303-305.
- Momen SE, Jorizzo J, Al-Niaimi F. Erythema elevatum diutinum: a review of presentation and treatment. J Eur Acad Dermatol Venereol. 2014;28:1594-1602.
- Vollum DI. Erythema elevatum diutinum—vesicular lesions and sulfone response. Br J Dermatol. 1968;80:178-183.
- Kohler IK, Lorincz AL. Erythema elevatum diutinum treated with niacinamide and tetracycline. Arch Dermatol. 1980;116:693-695.
- Henriksson R, Hofor PA, Hörngvist R. Erythema elevatum diutinum—a case successfully treated with colchicine. Clin Exp Dermatol. 1989;14:451-453.
- Zacaron LH, Gonçalves JC, Curty VM, et al. Clinical and surgical therapeutic approach in erythema elevatum diutinum—case report. An Bras Dermatol. 2013;88(6, suppl 1):15-18.
Erythema elevatum diutinum (EED) manifests on a clinicopathologic spectrum of chronic cutaneous small vessel vasculitis. The lesions typically present as persistent, symmetric, firm, red to purple papules or nodules on the extensor arms and dorsal hands.1,2 Underlying infectious, malignant, or autoimmune processes are commonly associated with the disease, notably Streptococcus infection and IgA monoclonal gammopathy.2,3 Hepatitis virus also is often implicated in association with EED. Cases of EED have been seen with concomitant human immunodeficiency virus (HIV) infection.4-6 We report a case of EED presenting in various stages of evolution associated with underlying hepatitis B infection alone.
Case Report
A 57-year-old man originally presented to an outpatient dermatology practice with a nodular, painful, episodic rash on the trunk and upper and lower extremities. A biopsy revealed leukocytoclastic vasculitis (LCV) with prominent eosinophils. At the time, the skin findings were believed to be a manifestation of drug hypersensitivity, likely to opioid use. The patient was lost to follow-up.
Seven years later, the patient was admitted to the hospital with new-onset burning and stinging red nodules on the dorsum of the hands and persistence of the original episodic rash over the lower legs and bilateral flanks. In the interim, he was briefly treated with an oral prednisone taper and topical corticosteroids including triamcinolone cream 0.1% and clobetasol cream 0.05% without improvement.
On examination deep red to violaceous discrete nodules and plaques with overlying hyperkeratosis involving all distal and proximal interphalangeal joints of the hands and extensor elbows were seen (Figure 1A). On the bilateral posterior arms (Figure 1B), anterior legs, and periumbilical area were deeply erythematous papules and plaques with background hyperpigmentation. Across his lower back and bilateral flanks were erythematous papules with central hemorrhagic crusting (Figure 1C).
Pertinent laboratory findings included a positive hepatitis B surface antigen with hepatitis B DNA value 4,313,876 IU/mL and a hepatitis B virus quantitative polymerase chain reaction value of 6.64 U. The etiology was suspected to be intravenous drug abuse; however, the patient denied recreational drug use.
An additional infectious workup was negative for hepatitis C, streptococcus, syphilis, tuberculosis, and HIV. A complete blood cell count, complete metabolic panel, urinalysis, complement, cryoglobulins, and serum protein electrophoresis were within reference range. Autoimmune serologies were negative including antinuclear antibody, rheumatoid factor, anti-Sjögren syndrome–related antigen A and B, anticyclic citrullinated peptide, anti-Smith, and antineutrophilic cytoplasmic antibodies. Peripheral blood immunophenotyping, lactate dehydrogenase, quantitative immunoglobulins, and age-appropriate cancer screens did not demonstrate evidence for malignancy underlying the disease. Bilateral hand radiographs showed mild periostitis of the proximal phalanges without obvious erosions.
Three 4-mm punch biopsies were performed from the left fifth digit, left posterior arm, and left flank. Tissue of the left fifth digit showed an intradermal vascular proliferation with a concentric pattern resembling onion skin in a background of increased fibrosis. The blood vessels showed focal fibrinoid necrosis (Figure 2A). The biopsy of the left posterior arm showed an intradermal vascular proliferation with an associated mild acute and chronic perivascular inflammation (Figure 2B). The left flank biopsy showed LCV with focal epidermal necrosis (Figure 2C).
The constellation of clinical findings together with the histopathologic changes represented EED in various stages of evolution. The patient was started on dapsone 100 mg daily and referred to the infectious disease service for treatment of chronic hepatitis B; however, he was subsequently lost to follow-up.
Comment
Overview of EED
Erythema elevatum diutinum represents a rare form of chronic cutaneous small vessel vasculitis. Originally described by Hutchinson7 and Bury8 as symmetric purpuric nodules of the skin, it was later named by Crocker and Williams9 in 1894. The disease classically presents as firm, fixed, red-brown to violaceous papules, plaques, and nodules affecting the extensor upper or lower extremities.1 Lesions are most commonly found symmetrically overlying joints of the hands, feet, elbows, and knees, as well as the Achilles tendon and buttocks.3 Less common locations include the palms and soles, face,10,11 trunk,12 and periauricular region.1 Although they are typically asymptomatic, sensations such as burning, stinging, and pruritus have been noted.1 Our patient was unique because in addition to typical lesions of EED, he presented with crusted papules on the flanks and violaceous papules of the lower legs and periumbilicus.
Etiology
Originally associated with Streptococcus as isolated from EED lesions,3,13 additional infectious etiologies include viral hepatitis,4-6 human herpesvirus 6,14 and rarely HIV.1,15 Hepatitis B and C are well known to be associated with EED, with only rare reports in patients with concomitant HIV infection. Erythema elevatum diutinum also has been described in relationship to myeloproliferative disorders and hematologic malignancies such as IgA myeloma,16 non-Hodgkin lymphoma,17 chronic lymphocytic leukemia,18 and hypergammaglobulinemia.19 In a study of 13 patients with EED, 4 had associated underlying IgA monoclonal gammopathy.2 Autoimmune conditions such as rheumatoid arthritis,20 ulcerative colitis,21 relapsing polychondritis,22 and systemic lupus erythematosus23 also have been implicated.
Pathogenesis
Although the precise pathogenesis of EED remains unknown, it has been suggested that a complement cascade initiated by immune-complex deposition in postcapillary venules induces an LCV.24,25 Chronic antigenic exposure or high antibody levels26 in the face of infections, autoimmune disease, or malignancy may incite this immune-complex reaction. Skin lesions seen in association with hepatitis reflect circulating immune-complex deposition in vessel walls causing destruction. It has been postulated that the duration of immune complexemia may be sufficient to account for the differences in the type of vascular injury seen in acute versus chronic infection.27
Histopathology
Erythema elevatum diutinum may present on a histopathologic spectrum of LCV, as manifested in our patient. Early lesions show predominantly polymorphonuclear cells with nuclear dust pattern in a wedge-shaped infiltrate with fibrin deposition in the superficial and mid dermis.2,3 Later lesions show vasculitis in addition to dermal aggregates of lymphocytes, neutrophils, fibrosis, and areas of granulation tissue. The fibrosis may be dense and comprised of fibroblasts and myofibroblasts.28 Newly formed vessels within the granulation tissue have been postulated to be more susceptible to immune-complex deposition, thus potentiating the process.1,29
Management
Spontaneous resolution of EED may occur, albeit after a prolonged and recurrent course of up to 5 to 10 years.30 Treatment of the underlying cause, when identified, remains paramount. First-line therapy includes dapsone, shown to be effective in reducing lesion size to complete resolution in 80% of the 47 cases reviewed by Momen et al.31 Dapsone monotherapy tends to be less effective in treating nodular lesions associated with HIV-positivity, likely due to the extensive fibrosis.4,31 Combination therapy with dapsone and a sulfonamide,32 niacinamide and tetracycline,33 colchicine,34 or surgical excision35 may be necessary in more resistant cases.
Conclusion
Our case exemplifies the clinical histologic spectrum that EED can present. The constellation of clinical findings was histologically confirmed to be manifestations of the disease in various stages of evolution. When typical lesions of EED present along with cutaneous findings in less common locations, performing multiple biopsies can be helpful. The clinician should retain a high index of suspicion for an underlying etiology and perform a complete workup for infection, malignancy, or autoimmune disease.
Erythema elevatum diutinum (EED) manifests on a clinicopathologic spectrum of chronic cutaneous small vessel vasculitis. The lesions typically present as persistent, symmetric, firm, red to purple papules or nodules on the extensor arms and dorsal hands.1,2 Underlying infectious, malignant, or autoimmune processes are commonly associated with the disease, notably Streptococcus infection and IgA monoclonal gammopathy.2,3 Hepatitis virus also is often implicated in association with EED. Cases of EED have been seen with concomitant human immunodeficiency virus (HIV) infection.4-6 We report a case of EED presenting in various stages of evolution associated with underlying hepatitis B infection alone.
Case Report
A 57-year-old man originally presented to an outpatient dermatology practice with a nodular, painful, episodic rash on the trunk and upper and lower extremities. A biopsy revealed leukocytoclastic vasculitis (LCV) with prominent eosinophils. At the time, the skin findings were believed to be a manifestation of drug hypersensitivity, likely to opioid use. The patient was lost to follow-up.
Seven years later, the patient was admitted to the hospital with new-onset burning and stinging red nodules on the dorsum of the hands and persistence of the original episodic rash over the lower legs and bilateral flanks. In the interim, he was briefly treated with an oral prednisone taper and topical corticosteroids including triamcinolone cream 0.1% and clobetasol cream 0.05% without improvement.
On examination deep red to violaceous discrete nodules and plaques with overlying hyperkeratosis involving all distal and proximal interphalangeal joints of the hands and extensor elbows were seen (Figure 1A). On the bilateral posterior arms (Figure 1B), anterior legs, and periumbilical area were deeply erythematous papules and plaques with background hyperpigmentation. Across his lower back and bilateral flanks were erythematous papules with central hemorrhagic crusting (Figure 1C).
Pertinent laboratory findings included a positive hepatitis B surface antigen with hepatitis B DNA value 4,313,876 IU/mL and a hepatitis B virus quantitative polymerase chain reaction value of 6.64 U. The etiology was suspected to be intravenous drug abuse; however, the patient denied recreational drug use.
An additional infectious workup was negative for hepatitis C, streptococcus, syphilis, tuberculosis, and HIV. A complete blood cell count, complete metabolic panel, urinalysis, complement, cryoglobulins, and serum protein electrophoresis were within reference range. Autoimmune serologies were negative including antinuclear antibody, rheumatoid factor, anti-Sjögren syndrome–related antigen A and B, anticyclic citrullinated peptide, anti-Smith, and antineutrophilic cytoplasmic antibodies. Peripheral blood immunophenotyping, lactate dehydrogenase, quantitative immunoglobulins, and age-appropriate cancer screens did not demonstrate evidence for malignancy underlying the disease. Bilateral hand radiographs showed mild periostitis of the proximal phalanges without obvious erosions.
Three 4-mm punch biopsies were performed from the left fifth digit, left posterior arm, and left flank. Tissue of the left fifth digit showed an intradermal vascular proliferation with a concentric pattern resembling onion skin in a background of increased fibrosis. The blood vessels showed focal fibrinoid necrosis (Figure 2A). The biopsy of the left posterior arm showed an intradermal vascular proliferation with an associated mild acute and chronic perivascular inflammation (Figure 2B). The left flank biopsy showed LCV with focal epidermal necrosis (Figure 2C).
The constellation of clinical findings together with the histopathologic changes represented EED in various stages of evolution. The patient was started on dapsone 100 mg daily and referred to the infectious disease service for treatment of chronic hepatitis B; however, he was subsequently lost to follow-up.
Comment
Overview of EED
Erythema elevatum diutinum represents a rare form of chronic cutaneous small vessel vasculitis. Originally described by Hutchinson7 and Bury8 as symmetric purpuric nodules of the skin, it was later named by Crocker and Williams9 in 1894. The disease classically presents as firm, fixed, red-brown to violaceous papules, plaques, and nodules affecting the extensor upper or lower extremities.1 Lesions are most commonly found symmetrically overlying joints of the hands, feet, elbows, and knees, as well as the Achilles tendon and buttocks.3 Less common locations include the palms and soles, face,10,11 trunk,12 and periauricular region.1 Although they are typically asymptomatic, sensations such as burning, stinging, and pruritus have been noted.1 Our patient was unique because in addition to typical lesions of EED, he presented with crusted papules on the flanks and violaceous papules of the lower legs and periumbilicus.
Etiology
Originally associated with Streptococcus as isolated from EED lesions,3,13 additional infectious etiologies include viral hepatitis,4-6 human herpesvirus 6,14 and rarely HIV.1,15 Hepatitis B and C are well known to be associated with EED, with only rare reports in patients with concomitant HIV infection. Erythema elevatum diutinum also has been described in relationship to myeloproliferative disorders and hematologic malignancies such as IgA myeloma,16 non-Hodgkin lymphoma,17 chronic lymphocytic leukemia,18 and hypergammaglobulinemia.19 In a study of 13 patients with EED, 4 had associated underlying IgA monoclonal gammopathy.2 Autoimmune conditions such as rheumatoid arthritis,20 ulcerative colitis,21 relapsing polychondritis,22 and systemic lupus erythematosus23 also have been implicated.
Pathogenesis
Although the precise pathogenesis of EED remains unknown, it has been suggested that a complement cascade initiated by immune-complex deposition in postcapillary venules induces an LCV.24,25 Chronic antigenic exposure or high antibody levels26 in the face of infections, autoimmune disease, or malignancy may incite this immune-complex reaction. Skin lesions seen in association with hepatitis reflect circulating immune-complex deposition in vessel walls causing destruction. It has been postulated that the duration of immune complexemia may be sufficient to account for the differences in the type of vascular injury seen in acute versus chronic infection.27
Histopathology
Erythema elevatum diutinum may present on a histopathologic spectrum of LCV, as manifested in our patient. Early lesions show predominantly polymorphonuclear cells with nuclear dust pattern in a wedge-shaped infiltrate with fibrin deposition in the superficial and mid dermis.2,3 Later lesions show vasculitis in addition to dermal aggregates of lymphocytes, neutrophils, fibrosis, and areas of granulation tissue. The fibrosis may be dense and comprised of fibroblasts and myofibroblasts.28 Newly formed vessels within the granulation tissue have been postulated to be more susceptible to immune-complex deposition, thus potentiating the process.1,29
Management
Spontaneous resolution of EED may occur, albeit after a prolonged and recurrent course of up to 5 to 10 years.30 Treatment of the underlying cause, when identified, remains paramount. First-line therapy includes dapsone, shown to be effective in reducing lesion size to complete resolution in 80% of the 47 cases reviewed by Momen et al.31 Dapsone monotherapy tends to be less effective in treating nodular lesions associated with HIV-positivity, likely due to the extensive fibrosis.4,31 Combination therapy with dapsone and a sulfonamide,32 niacinamide and tetracycline,33 colchicine,34 or surgical excision35 may be necessary in more resistant cases.
Conclusion
Our case exemplifies the clinical histologic spectrum that EED can present. The constellation of clinical findings was histologically confirmed to be manifestations of the disease in various stages of evolution. When typical lesions of EED present along with cutaneous findings in less common locations, performing multiple biopsies can be helpful. The clinician should retain a high index of suspicion for an underlying etiology and perform a complete workup for infection, malignancy, or autoimmune disease.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Yiannias JA, el-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol. 1992;26:38-44.
- Wilkinson SM, English JS, Smith NP, et al. Erythema elevatum diutinum: a clinicopathological study. Clin Exp Dermatol. 1992;17:87-93.
- Fakheri A, Gupta SM, White SM, et al. Erythema elevatum diutinum in a patient with human immunodeficiency virus. Cutis. 2001;68:41-42, 55.
- Kim H. Erythema elevatum diutinum in an HIV-positive patient. J Drugs Dermatol. 2003;2:411-412.
- Revenga F, Vera A, Muñoz A, et al. Erythema elevatum diutinum and AIDS: are they related? Clin Exp Dermatol. 1997;22:250-251.
- Hutchinson J. On two remarkable cases of symmetrieal purple congestion of the skin in patches, with induration. Br J Dermatol. 1888;1:10-15.
- Bury JS. A case of erythema with remarkable nodular thickening and induration of the skin associated with intermittent albuminuria. Illustrated Medical News. 1889;3:145-149.
- Crocker HR, Williams C. Erythema elevatum diutinum. Br J Dermatol. 1894;6:33-38.
- Barzegar M, Davatchi CC, Akhyani M, et al. An atypical presentation of erythema elevatum diutinum involving palms and soles. Int J Dermatol. 2009;48:73-75.
- Futei Y, Konohana I. A case of erythema elevatum diutinum associated with B-cell lymphoma: a rare distribution involving palms, soles and nails. Br J Dermatol. 2000;142:116-119.
- Ben-Zvi GT, Bardsley V, Burrows NP. An atypical distribution of erythema elevatum diutinum. Clin Exp Dermatol. 2014;39:269-270.
- Weidman FD, Besancon JH. Erythema elevatum diutinum. role of streptococci, and relationship to other rheumatic dermatoses. Arch Dermatol Syphilol. 1929;20:593-620.
- Drago F, Semino M, Rampini P, et al. Erythema elevatum diutinum in a patient with human herpesvirus 6 infection. Acta Derm Venereol. 1999;79:91-92.
- Muratori S, Carrera C, Gorani A, et al. Erythema elevatum diutinum and HIV infection: a report of five cases. Br J Dermatol. 1999;141:335-338.
- Archimandritis AJ, Fertakis A, Alegakis G, et al. Erythema elevatum diutinum and IgA myeloma: an interesting association. Br Med J. 1977;2:613-614.
- Hatzitolios A, Tzellos TG, Savopoulos C, et al. Erythema elevatum diutinum with rare distribution as a first clinical sign of non-Hodgkin’s lymphoma: a novel association? J Dermatol. 2008;35:297-300.
- Delaporte E, Alfandari S, Fenaux P, et al. Erythema elevatum diutinum and chronic lymphocytic leukaemia. Clin Exp Dermatol. 1994;19:188-189.
- Miyagawa S, Kitamura W, Morita K, et al. Association of hyperimmunoglobulinaemia D syndrome with erythema elevatum diutinum. Br J Dermatol. 1993;128:572-574.
- Collier PM, Neill SM, Branfoot AC, et al. Erythema elevatum diutinum—a solitary lesion in a patient with rheumatoid arthritis. Clin Exp Dermatol. 1990;15:394-395.
- Buahene K, Hudson M, Mowat A, et al. Erythema elevatum diutinum—an unusual association with ulcerative colitis. Clin Exp Dermatol. 1991;16:204-206.
- Bernard P, Bedane C, Delrous JL, et al. Erythema elevatum diutinum in a patient with relapsing polychondritis. J Am Acad Dermatol. 1992;26:312-315.
- Hancox JG, Wallace CA, Sangueza OP, et al. Erythema elevatum diutinum associated with lupus panniculitis in a patient with discoid lesions of chronic cutaneous lupus erythematosus. J Am Acad Dermatol. 2004;50:652-653.
- Haber H. Erythema elevatum diutinum. Br J Dermatol. 1955;67:121-145.
- Katz SI, Gallin JL, Hertz KC, et al. Erythema elevatum diutinum: skin and systemic manifestations, immunologic studies, and successful treatment with dapsone. Medicine (Baltimore). 1977;56:443-455.
- Walker KD, Badame AJ. Erythema elevatum diutinum in a patient with Crohn’s disease. J Am Acad Dermatol. 1990;22:948-952.
- Popp JW, Harrist T, Dienstag JL, et al. Cutaneous vasculitis associated with acute and chronic hepatitis. Arch Intern Med. 1981;141:623-629.
- Lee AY, Nakagawa H, Nogita T, et al. Erythema elevatum diutinum: an ultrastructural case study. J Cutan Pathol. 1989;16:211-217.
- LeBoit PE, Yen TS, Wintroub B. The evolution of lesions in erythema elevatum diutinum. Am J Dermatopathol. 1986;8:392-402.
- Soubeiran E, Wacker J, Hausser I, et al. Erythema elevatum diutinum with unusual clinical appearance. J Dtsch Dermatol Ges. 2008;6:303-305.
- Momen SE, Jorizzo J, Al-Niaimi F. Erythema elevatum diutinum: a review of presentation and treatment. J Eur Acad Dermatol Venereol. 2014;28:1594-1602.
- Vollum DI. Erythema elevatum diutinum—vesicular lesions and sulfone response. Br J Dermatol. 1968;80:178-183.
- Kohler IK, Lorincz AL. Erythema elevatum diutinum treated with niacinamide and tetracycline. Arch Dermatol. 1980;116:693-695.
- Henriksson R, Hofor PA, Hörngvist R. Erythema elevatum diutinum—a case successfully treated with colchicine. Clin Exp Dermatol. 1989;14:451-453.
- Zacaron LH, Gonçalves JC, Curty VM, et al. Clinical and surgical therapeutic approach in erythema elevatum diutinum—case report. An Bras Dermatol. 2013;88(6, suppl 1):15-18.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Yiannias JA, el-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol. 1992;26:38-44.
- Wilkinson SM, English JS, Smith NP, et al. Erythema elevatum diutinum: a clinicopathological study. Clin Exp Dermatol. 1992;17:87-93.
- Fakheri A, Gupta SM, White SM, et al. Erythema elevatum diutinum in a patient with human immunodeficiency virus. Cutis. 2001;68:41-42, 55.
- Kim H. Erythema elevatum diutinum in an HIV-positive patient. J Drugs Dermatol. 2003;2:411-412.
- Revenga F, Vera A, Muñoz A, et al. Erythema elevatum diutinum and AIDS: are they related? Clin Exp Dermatol. 1997;22:250-251.
- Hutchinson J. On two remarkable cases of symmetrieal purple congestion of the skin in patches, with induration. Br J Dermatol. 1888;1:10-15.
- Bury JS. A case of erythema with remarkable nodular thickening and induration of the skin associated with intermittent albuminuria. Illustrated Medical News. 1889;3:145-149.
- Crocker HR, Williams C. Erythema elevatum diutinum. Br J Dermatol. 1894;6:33-38.
- Barzegar M, Davatchi CC, Akhyani M, et al. An atypical presentation of erythema elevatum diutinum involving palms and soles. Int J Dermatol. 2009;48:73-75.
- Futei Y, Konohana I. A case of erythema elevatum diutinum associated with B-cell lymphoma: a rare distribution involving palms, soles and nails. Br J Dermatol. 2000;142:116-119.
- Ben-Zvi GT, Bardsley V, Burrows NP. An atypical distribution of erythema elevatum diutinum. Clin Exp Dermatol. 2014;39:269-270.
- Weidman FD, Besancon JH. Erythema elevatum diutinum. role of streptococci, and relationship to other rheumatic dermatoses. Arch Dermatol Syphilol. 1929;20:593-620.
- Drago F, Semino M, Rampini P, et al. Erythema elevatum diutinum in a patient with human herpesvirus 6 infection. Acta Derm Venereol. 1999;79:91-92.
- Muratori S, Carrera C, Gorani A, et al. Erythema elevatum diutinum and HIV infection: a report of five cases. Br J Dermatol. 1999;141:335-338.
- Archimandritis AJ, Fertakis A, Alegakis G, et al. Erythema elevatum diutinum and IgA myeloma: an interesting association. Br Med J. 1977;2:613-614.
- Hatzitolios A, Tzellos TG, Savopoulos C, et al. Erythema elevatum diutinum with rare distribution as a first clinical sign of non-Hodgkin’s lymphoma: a novel association? J Dermatol. 2008;35:297-300.
- Delaporte E, Alfandari S, Fenaux P, et al. Erythema elevatum diutinum and chronic lymphocytic leukaemia. Clin Exp Dermatol. 1994;19:188-189.
- Miyagawa S, Kitamura W, Morita K, et al. Association of hyperimmunoglobulinaemia D syndrome with erythema elevatum diutinum. Br J Dermatol. 1993;128:572-574.
- Collier PM, Neill SM, Branfoot AC, et al. Erythema elevatum diutinum—a solitary lesion in a patient with rheumatoid arthritis. Clin Exp Dermatol. 1990;15:394-395.
- Buahene K, Hudson M, Mowat A, et al. Erythema elevatum diutinum—an unusual association with ulcerative colitis. Clin Exp Dermatol. 1991;16:204-206.
- Bernard P, Bedane C, Delrous JL, et al. Erythema elevatum diutinum in a patient with relapsing polychondritis. J Am Acad Dermatol. 1992;26:312-315.
- Hancox JG, Wallace CA, Sangueza OP, et al. Erythema elevatum diutinum associated with lupus panniculitis in a patient with discoid lesions of chronic cutaneous lupus erythematosus. J Am Acad Dermatol. 2004;50:652-653.
- Haber H. Erythema elevatum diutinum. Br J Dermatol. 1955;67:121-145.
- Katz SI, Gallin JL, Hertz KC, et al. Erythema elevatum diutinum: skin and systemic manifestations, immunologic studies, and successful treatment with dapsone. Medicine (Baltimore). 1977;56:443-455.
- Walker KD, Badame AJ. Erythema elevatum diutinum in a patient with Crohn’s disease. J Am Acad Dermatol. 1990;22:948-952.
- Popp JW, Harrist T, Dienstag JL, et al. Cutaneous vasculitis associated with acute and chronic hepatitis. Arch Intern Med. 1981;141:623-629.
- Lee AY, Nakagawa H, Nogita T, et al. Erythema elevatum diutinum: an ultrastructural case study. J Cutan Pathol. 1989;16:211-217.
- LeBoit PE, Yen TS, Wintroub B. The evolution of lesions in erythema elevatum diutinum. Am J Dermatopathol. 1986;8:392-402.
- Soubeiran E, Wacker J, Hausser I, et al. Erythema elevatum diutinum with unusual clinical appearance. J Dtsch Dermatol Ges. 2008;6:303-305.
- Momen SE, Jorizzo J, Al-Niaimi F. Erythema elevatum diutinum: a review of presentation and treatment. J Eur Acad Dermatol Venereol. 2014;28:1594-1602.
- Vollum DI. Erythema elevatum diutinum—vesicular lesions and sulfone response. Br J Dermatol. 1968;80:178-183.
- Kohler IK, Lorincz AL. Erythema elevatum diutinum treated with niacinamide and tetracycline. Arch Dermatol. 1980;116:693-695.
- Henriksson R, Hofor PA, Hörngvist R. Erythema elevatum diutinum—a case successfully treated with colchicine. Clin Exp Dermatol. 1989;14:451-453.
- Zacaron LH, Gonçalves JC, Curty VM, et al. Clinical and surgical therapeutic approach in erythema elevatum diutinum—case report. An Bras Dermatol. 2013;88(6, suppl 1):15-18.
Practice Points
- Erythema elevatum diutinum (EED) often is associated with an underlying infectious process, including hepatitis B and hepatitis C, or a hematologic or autoimmune condition.
- If EED is suspected clinically, it may be beneficial to perform multiple biopsies from lesions at different stages of evolution to establish the diagnosis.
- First-line therapy includes treatment of any underlying condition and dapsone.

MRSA in Dermatology Inpatients With a Vesiculobullous Disorder
Methicillin, cloxacillin, flucloxacillin, and cefoxitin are stable, penicillinase-producing β-lactam antibiotics; Staphylococcus aureus strains resistant to these agents are designated as methicillin-resistant S aureus (MRSA). Based on genotypic and phenotypic differences there are 2 strains of MRSA: hospital acquired and community acquired.
The potential for nosocomial transmission and the limited number of antibiotics available to treat MRSA are problematic. Moreover, MRSA has emerged worldwide as a major nosocomial pathogen that contributes to morbidity and mortality. Methicillin-resistant S aureus infection in vesiculobullous disorders such as pemphigus vulgaris (PV) and toxic epidermal necrolysis (TEN) is known to contribute to mortality.1
The reported prevalence of MRSA in India ranges from 12% to 38.44%.2-4 We frequently encounter MRSA in dermatology inpatients, especially those with a vesiculobullous disorder. The primary objective of this study was to determine the prevalence of MRSA in dermatology inpatients with a vesiculobullous disorder; the secondary objective was to determine if MRSA contributes to mortality.
Materials and Methods
A 1-year prospective, cross-sectional, descriptive study was conducted in a tertiary-care center. The study population included all dermatology inpatients with a vesiculobullous disorder. Patients with a vesiculobullous disorder secondary to a primary viral or bacterial disorder were excluded. Permission to conduct the study was granted by the institution’s Human Ethics Committee.
All patients underwent a detailed history and clinical examination. Routine hematology testing, urinalysis, measurement of the blood glucose level, and other investigations relevant to the vesiculobullous disorder were performed. Special investigations were Gram staining, culture, and susceptibility testing of material from a nasal swab and a swab of a representative skin lesion.
Detection of MRSA
Skin lesions were thoroughly cleaned with sterile normal saline. Specimens of pus were drawn with a sterile swab for Gram staining, culture, and susceptibility testing and were analyzed in the institution’s microbiology department. A direct colony suspension (equivalent to McFarland Standard No. 0.5) was inoculated on a Mueller-Hinton agar plate, incorporating cefoxitin, linezolid, vancomycin, amikacin, and rifampicin supplemented with sodium chloride 2% and incubated at 37°C for 24 hours. Staphylococcus aureus colonies were identified by their smooth, convex, shiny, and opaque appearance with a golden yellow pigment, as well as by coagulase positivity, mannitol fermentation, and production of phosphatase.
Methicillin-resistant S aureus was defined as an isolate having a minimum inhibitory concentration of more than 2 μg/mL of cefoxitin; a methicillin-sensitive S aureus isolate was defined as having a minimum inhibitory concentration of less than or equal to 2 μg/mL of cefoxitin. Specimens showing moderate to heavy growth of MRSA were included in the study. For specimens showing mild growth, testing was repeated; if no growth was seen on repeat testing, results were interpreted as negative.
Data were collected and analyzed for frequency and percentage; P<.05 was considered significant.
Results
The number of patients analyzed in the study period was 43. Table 1 shows their salient demographic characteristics, clinical features, and findings of the investigation. The youngest patient was aged 13 years; the oldest was aged 80 years. The male to female ratio was 0.65 to 1. The most common primary lesion was a combined vesicle and bulla (34 patients [79.1%]); the most common secondary lesion was a combination of erosion with crusting (22 patients [51.2%]).
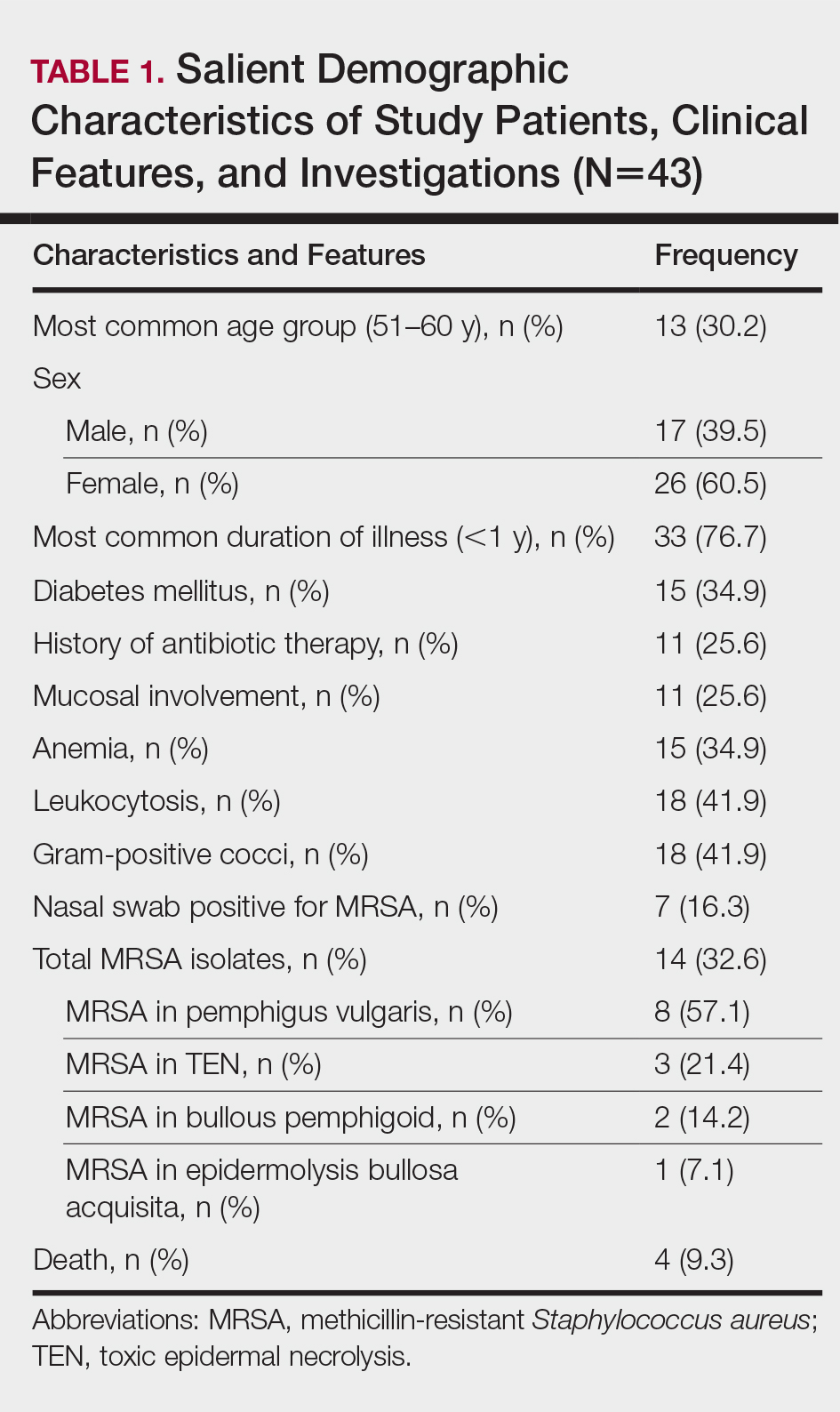
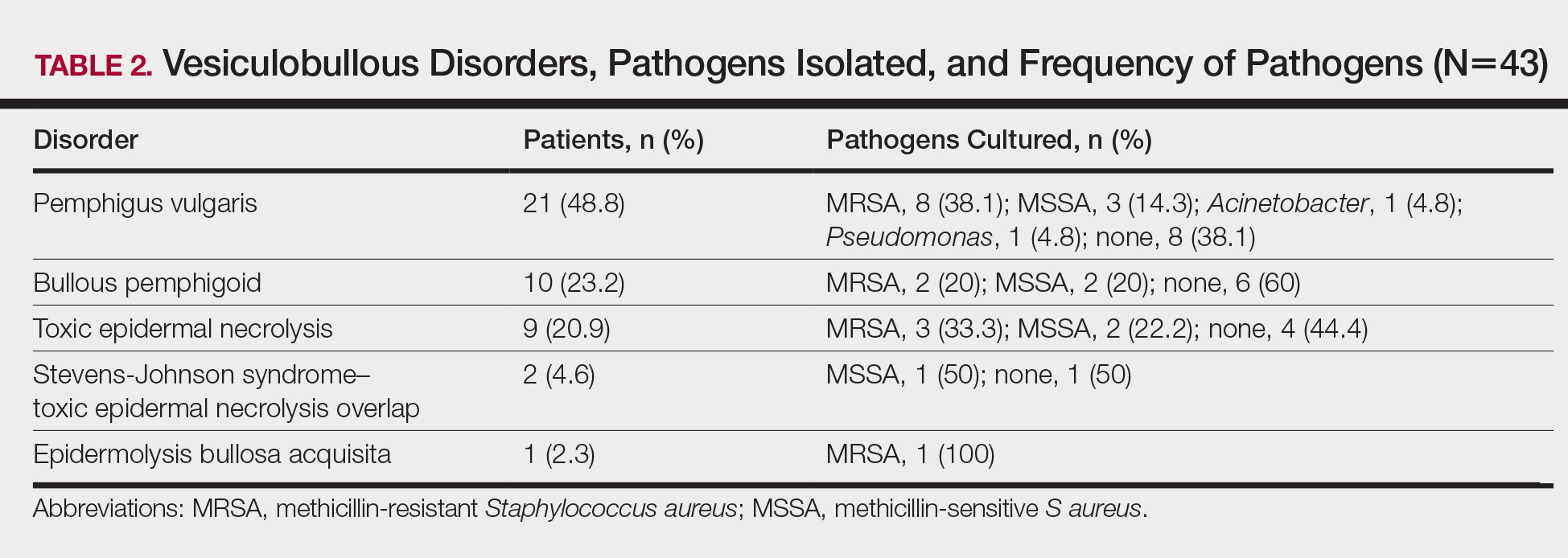
Table 2 lists the types of vesiculobullous disorders seen in this study. Pemphigus vulgaris was the most common (21 patients [48.8%])(Figure 1). Drug-induced vesiculobullous disorders (eg, TEN) were noted in 11 patients (25.6%)(Figure 2).


Table 2 also lists pathogens cultured in the study group. There were 24 bacterial isolates, of which S aureus accounted for 22 (91.7%). Methicillin-resistant S aureus was cultured in 14 patients (32.6%); culture was sterile in 19 patients (44.2%).
Among the 22 cultured staphylococcal species, MRSA accounted for 14 (63.6%) and constituted 58.3% (14/24) of all bacterial isolates. The nasal swab for MRSA was positive in 4 PV patients (9.3%), 2 TEN patients (4.6%), and 1 bullous pemphigoid patient (2.3%). Methicillin-resistant S aureus was most commonly cultured in PV patients (8/14 [57.1%]).
All MRSA strains (100%) were sensitive to vancomycin and linezolid; 34 (79.1%) were sensitive to amikacin. Additionally, 100% of MRSA strains were resistant to oxacillin, cloxacillin, and cefoxitin.
Three patients with PV (7.0%) and 1 patient with TEN (2.3%) died during the course of the study; only 1 death (2.3%) occurred in a patient who had a positive MRSA culture.
Comment
In this 1-year study, we tested and followed 43 patients with autoimmune and drug-induced vesiculobullous disorders. Vesiculobullous disorders in dermatology inpatients are a cause of great concern. When lesions rupture, they leave behind a large area of erosion that forms a nidus of bacterial colonization; often, these bacteria cause severe infection, including septicemia, and result in death.5 Moreover, autoimmune bullous disorders usually require a prolonged hospital stay and powerful immunosuppressive drugs, which contributes to bacterial infection, especially MRSA.6
The age of patients in this study ranged from 13 to 80 years; most patients were in the 6th decade, a pattern seen in studies worldwide.5 In a study by Kanwar and De,7 however, most cases were aged 20 to 40 years.7 In our study, there was a female preponderance (male to female ratio of 0.65 to 1).
Studies have shown that the duration of illness in vesiculobullous disorder is directly associated with MRSA infection. However, in our study with MRSA detected in 14 patients, most patients had a duration of illness less than 1 year (statistically insignificant [P>.05]), a finding similar to Shafi et al.8
The symptomatic nature of these diseases, their unsightly appearance, and mucous-membrane involvement of vesiculobullous disorders prompts these patients to present to the hospital early. However, a prolonged hospital stay by patients with an autoimmune vesiculobullous disorders sets the stage for MRSA colonization.
In this study, diabetes mellitus (DM) was seen in 15 patients (34.9%); 5 of them had MRSA infection (statistically insignificant [P>.05]). Diabetes mellitus contributing to sepsis and MRSA infection, which in turn contributes to morbidity and mortality, has been well-documented.2,4,9
Methicillin-resistant S aureus in this study was isolated most often from blisters and erosions. Vesiculobullous disorders and drug reactions (eg, Stevens-Johnson syndrome, TEN) are characterized by blisters that rupture to form erosions and crusting, which form fissures in the epidermal barrier function that are nidi for colonization by microbes, especially S aureus and MRSA in particular; later, these bacteria can enter dermal vessels and then the bloodstream, leading to septicemia.10
The prevalence of MRSA in this study was 32.6% (14/43), which is high compared to other studies.2-4 Pemphigus vulgaris was the most common disorder infected by MRSA in this study (57.1% [8/14] of MRSA isolates)(Table 1), a finding that reveals that the incidence of MRSA is high among staphylococcal isolates in vesiculobullous disorders. However, the high incidence of MRSA in this study could be a reflection of the number of patients with a severe and chronic vesiculobullous disorder, such as PV, and serious drug reactions such as TEN referred to our tertiary-care center, where we get a large number of patients affected by autoimmune and drug-induced vesiculobullous disorders. Similar findings have been reported by Stryjewski et al.11
A high prevalence of MRSA in a dermatology unit has grave consequences, contributing to morbidity and mortality in particular among patients with a vesiculobullous disorder. Immunosuppressive therapy and comorbidities such as DM contribute to MRSA colonization in vesiculobullous disorders.12 Overcrowding and poor sterilization techniques in public hospitals in India may contribute to the high prevalence of MRSA seen in hospital units.
Patients with a vesiculobullous disorder who are chronic nasal carriers of MRSA are at risk for cutaneous MRSA infection, which in turn can lead to MRSA septicemia and an elevated risk of death. In this study, however, a nasal swab was positive for MRSA in only 7 patients. One patient with MRSA colonization died, which was statistically insignificant (P=1).
In this study, all MRSA strains (100%) were resistant to first-line antibiotics, such as oxacillin, cloxacillin, and cefoxitin; all strains were susceptible to vancomycin and linezolid.
Conclusion
Our study shows that MRSA is becoming the prominent pathogen in nosocomial infections, especially in bedridden patients, which has grave implications. The use of a prophylactic S aureus conjugate vaccine in patients with a chronic vesiculobullous disorder might be justified in the future.15 We found a high prevalence (32.6%) of MRSA in vesiculobullous disorders, no relationship between DM and MRSA colonization, PV was the most common disorder complicated by MRSA, no relationship between nasal colonization and MRSA infection, no relationship between death during the study period and MRSA infection, 100% of MRSA strains were susceptible to vancomycin and linezolid, and 79.1% of MRSA strains were susceptible to amikacin.
- Nair SP. A retrospective study of mortality of pemphigus patients in a tertiary care hospital. Indian J Dermatol Venereol Leprol. 2013;79:706-709.
- Sachdev D, Amladi S, Natarj G, et al. An outbreak of methicillin-resistant Staphylococcus aureus (MRSA) infection in dermatology inpatients. Indian J Dermatol Venereol Leprol. 2003;69:377-380.
- Vijayamohan N, Nair SP. A study of the prevalence of methicillin-resistant Staphylococcus aureus in dermatology inpatients. Indian Dermatol Online J. 2014;5:441-445.
- Malhotra SK, Malhotra S, Dhaliwal GS, et al. Bacterial study of pyodermas in a tertiary care dermatological center. Indian J Dermatol. 2012;57:358-361.
- Valencia IC, Kirsner RS, Kerdel FA. Microbiological evaluation of skin wounds: alarming trends towards antibiotic resistance in an inpatient dermatology service during a 10-year period. J Am Acad Dermatol. 2004;50:845-849.
- Lehman JS, Murell DF, Camilleri MJ, et al. Infection and infection prevention in patients treated with immunosuppressive medications for autoimmune bullous disorders. Dermatol Clin. 2011;29:591-598.
- Kanwar AJ, De D. Pemphigus in India. Indian J Dermatol Venereol Leprol. 2011;77:439-449.
- Shafi M, Khatri ML, Mashima M, et al. Pemphigus: a clinical study of 109 cases from Tripoli, Libya. Indian J Dermatol Venereol Leprol. 1994;60:140-143.
- Torres K, Sampathkumar P. Predictors of methicillin-resistant Staphylococcus aureus colonization at hospital admission. Am J Infect Control. 2013;41:1043-1047.
- Miller LG, Quan C, Shay A, et al. A prospective investigation of outcomes after hospital discharge for endemic, community-acquired methicillin-resistant Staphylococcus aureus skin infection. Clin Infect Dis. 2007;44:483-492.
- Stryjewski M, Chambers HF. Skin and soft-tissue infections caused by community-acquired methicillin-resistant Staphylococcus aureus. Clin Infect Dis. 2008;46(suppl 5):S368-S377.
- Mutasim DF. Management of autoimmune bullous diseases: pharmacology and therapeutics. J Am Acad Dermatol. 2004;51:859-877.
- Cohen PR. Community-acquired methicillin-resistant Staphylococcus aureus skin infections: a review of epidemiology, clinical features, management, and prevention. Int J Dermatol. 2007;46:1-11.
- Elston DM. Methicillin-sensitive and methicillin-resistant Staphylococcus aureus: management principles and selection of antibiotic therapy. Dermatol Clin. 2007;25:157-164.
- Shinefield H, Black S, Fattom A, et al. Use of a Staphylococcus aureus conjugate vaccine in patients receiving hemodialysis. N Engl J Med. 2001;346:491-496.
Methicillin, cloxacillin, flucloxacillin, and cefoxitin are stable, penicillinase-producing β-lactam antibiotics; Staphylococcus aureus strains resistant to these agents are designated as methicillin-resistant S aureus (MRSA). Based on genotypic and phenotypic differences there are 2 strains of MRSA: hospital acquired and community acquired.
The potential for nosocomial transmission and the limited number of antibiotics available to treat MRSA are problematic. Moreover, MRSA has emerged worldwide as a major nosocomial pathogen that contributes to morbidity and mortality. Methicillin-resistant S aureus infection in vesiculobullous disorders such as pemphigus vulgaris (PV) and toxic epidermal necrolysis (TEN) is known to contribute to mortality.1
The reported prevalence of MRSA in India ranges from 12% to 38.44%.2-4 We frequently encounter MRSA in dermatology inpatients, especially those with a vesiculobullous disorder. The primary objective of this study was to determine the prevalence of MRSA in dermatology inpatients with a vesiculobullous disorder; the secondary objective was to determine if MRSA contributes to mortality.
Materials and Methods
A 1-year prospective, cross-sectional, descriptive study was conducted in a tertiary-care center. The study population included all dermatology inpatients with a vesiculobullous disorder. Patients with a vesiculobullous disorder secondary to a primary viral or bacterial disorder were excluded. Permission to conduct the study was granted by the institution’s Human Ethics Committee.
All patients underwent a detailed history and clinical examination. Routine hematology testing, urinalysis, measurement of the blood glucose level, and other investigations relevant to the vesiculobullous disorder were performed. Special investigations were Gram staining, culture, and susceptibility testing of material from a nasal swab and a swab of a representative skin lesion.
Detection of MRSA
Skin lesions were thoroughly cleaned with sterile normal saline. Specimens of pus were drawn with a sterile swab for Gram staining, culture, and susceptibility testing and were analyzed in the institution’s microbiology department. A direct colony suspension (equivalent to McFarland Standard No. 0.5) was inoculated on a Mueller-Hinton agar plate, incorporating cefoxitin, linezolid, vancomycin, amikacin, and rifampicin supplemented with sodium chloride 2% and incubated at 37°C for 24 hours. Staphylococcus aureus colonies were identified by their smooth, convex, shiny, and opaque appearance with a golden yellow pigment, as well as by coagulase positivity, mannitol fermentation, and production of phosphatase.
Methicillin-resistant S aureus was defined as an isolate having a minimum inhibitory concentration of more than 2 μg/mL of cefoxitin; a methicillin-sensitive S aureus isolate was defined as having a minimum inhibitory concentration of less than or equal to 2 μg/mL of cefoxitin. Specimens showing moderate to heavy growth of MRSA were included in the study. For specimens showing mild growth, testing was repeated; if no growth was seen on repeat testing, results were interpreted as negative.
Data were collected and analyzed for frequency and percentage; P<.05 was considered significant.
Results
The number of patients analyzed in the study period was 43. Table 1 shows their salient demographic characteristics, clinical features, and findings of the investigation. The youngest patient was aged 13 years; the oldest was aged 80 years. The male to female ratio was 0.65 to 1. The most common primary lesion was a combined vesicle and bulla (34 patients [79.1%]); the most common secondary lesion was a combination of erosion with crusting (22 patients [51.2%]).
Table 2 lists the types of vesiculobullous disorders seen in this study. Pemphigus vulgaris was the most common (21 patients [48.8%])(Figure 1). Drug-induced vesiculobullous disorders (eg, TEN) were noted in 11 patients (25.6%)(Figure 2).
Table 2 also lists pathogens cultured in the study group. There were 24 bacterial isolates, of which S aureus accounted for 22 (91.7%). Methicillin-resistant S aureus was cultured in 14 patients (32.6%); culture was sterile in 19 patients (44.2%).
Among the 22 cultured staphylococcal species, MRSA accounted for 14 (63.6%) and constituted 58.3% (14/24) of all bacterial isolates. The nasal swab for MRSA was positive in 4 PV patients (9.3%), 2 TEN patients (4.6%), and 1 bullous pemphigoid patient (2.3%). Methicillin-resistant S aureus was most commonly cultured in PV patients (8/14 [57.1%]).
All MRSA strains (100%) were sensitive to vancomycin and linezolid; 34 (79.1%) were sensitive to amikacin. Additionally, 100% of MRSA strains were resistant to oxacillin, cloxacillin, and cefoxitin.
Three patients with PV (7.0%) and 1 patient with TEN (2.3%) died during the course of the study; only 1 death (2.3%) occurred in a patient who had a positive MRSA culture.
Comment
In this 1-year study, we tested and followed 43 patients with autoimmune and drug-induced vesiculobullous disorders. Vesiculobullous disorders in dermatology inpatients are a cause of great concern. When lesions rupture, they leave behind a large area of erosion that forms a nidus of bacterial colonization; often, these bacteria cause severe infection, including septicemia, and result in death.5 Moreover, autoimmune bullous disorders usually require a prolonged hospital stay and powerful immunosuppressive drugs, which contributes to bacterial infection, especially MRSA.6
The age of patients in this study ranged from 13 to 80 years; most patients were in the 6th decade, a pattern seen in studies worldwide.5 In a study by Kanwar and De,7 however, most cases were aged 20 to 40 years.7 In our study, there was a female preponderance (male to female ratio of 0.65 to 1).
Studies have shown that the duration of illness in vesiculobullous disorder is directly associated with MRSA infection. However, in our study with MRSA detected in 14 patients, most patients had a duration of illness less than 1 year (statistically insignificant [P>.05]), a finding similar to Shafi et al.8
The symptomatic nature of these diseases, their unsightly appearance, and mucous-membrane involvement of vesiculobullous disorders prompts these patients to present to the hospital early. However, a prolonged hospital stay by patients with an autoimmune vesiculobullous disorders sets the stage for MRSA colonization.
In this study, diabetes mellitus (DM) was seen in 15 patients (34.9%); 5 of them had MRSA infection (statistically insignificant [P>.05]). Diabetes mellitus contributing to sepsis and MRSA infection, which in turn contributes to morbidity and mortality, has been well-documented.2,4,9
Methicillin-resistant S aureus in this study was isolated most often from blisters and erosions. Vesiculobullous disorders and drug reactions (eg, Stevens-Johnson syndrome, TEN) are characterized by blisters that rupture to form erosions and crusting, which form fissures in the epidermal barrier function that are nidi for colonization by microbes, especially S aureus and MRSA in particular; later, these bacteria can enter dermal vessels and then the bloodstream, leading to septicemia.10
The prevalence of MRSA in this study was 32.6% (14/43), which is high compared to other studies.2-4 Pemphigus vulgaris was the most common disorder infected by MRSA in this study (57.1% [8/14] of MRSA isolates)(Table 1), a finding that reveals that the incidence of MRSA is high among staphylococcal isolates in vesiculobullous disorders. However, the high incidence of MRSA in this study could be a reflection of the number of patients with a severe and chronic vesiculobullous disorder, such as PV, and serious drug reactions such as TEN referred to our tertiary-care center, where we get a large number of patients affected by autoimmune and drug-induced vesiculobullous disorders. Similar findings have been reported by Stryjewski et al.11
A high prevalence of MRSA in a dermatology unit has grave consequences, contributing to morbidity and mortality in particular among patients with a vesiculobullous disorder. Immunosuppressive therapy and comorbidities such as DM contribute to MRSA colonization in vesiculobullous disorders.12 Overcrowding and poor sterilization techniques in public hospitals in India may contribute to the high prevalence of MRSA seen in hospital units.
Patients with a vesiculobullous disorder who are chronic nasal carriers of MRSA are at risk for cutaneous MRSA infection, which in turn can lead to MRSA septicemia and an elevated risk of death. In this study, however, a nasal swab was positive for MRSA in only 7 patients. One patient with MRSA colonization died, which was statistically insignificant (P=1).
In this study, all MRSA strains (100%) were resistant to first-line antibiotics, such as oxacillin, cloxacillin, and cefoxitin; all strains were susceptible to vancomycin and linezolid.
Conclusion
Our study shows that MRSA is becoming the prominent pathogen in nosocomial infections, especially in bedridden patients, which has grave implications. The use of a prophylactic S aureus conjugate vaccine in patients with a chronic vesiculobullous disorder might be justified in the future.15 We found a high prevalence (32.6%) of MRSA in vesiculobullous disorders, no relationship between DM and MRSA colonization, PV was the most common disorder complicated by MRSA, no relationship between nasal colonization and MRSA infection, no relationship between death during the study period and MRSA infection, 100% of MRSA strains were susceptible to vancomycin and linezolid, and 79.1% of MRSA strains were susceptible to amikacin.
Methicillin, cloxacillin, flucloxacillin, and cefoxitin are stable, penicillinase-producing β-lactam antibiotics; Staphylococcus aureus strains resistant to these agents are designated as methicillin-resistant S aureus (MRSA). Based on genotypic and phenotypic differences there are 2 strains of MRSA: hospital acquired and community acquired.
The potential for nosocomial transmission and the limited number of antibiotics available to treat MRSA are problematic. Moreover, MRSA has emerged worldwide as a major nosocomial pathogen that contributes to morbidity and mortality. Methicillin-resistant S aureus infection in vesiculobullous disorders such as pemphigus vulgaris (PV) and toxic epidermal necrolysis (TEN) is known to contribute to mortality.1
The reported prevalence of MRSA in India ranges from 12% to 38.44%.2-4 We frequently encounter MRSA in dermatology inpatients, especially those with a vesiculobullous disorder. The primary objective of this study was to determine the prevalence of MRSA in dermatology inpatients with a vesiculobullous disorder; the secondary objective was to determine if MRSA contributes to mortality.
Materials and Methods
A 1-year prospective, cross-sectional, descriptive study was conducted in a tertiary-care center. The study population included all dermatology inpatients with a vesiculobullous disorder. Patients with a vesiculobullous disorder secondary to a primary viral or bacterial disorder were excluded. Permission to conduct the study was granted by the institution’s Human Ethics Committee.
All patients underwent a detailed history and clinical examination. Routine hematology testing, urinalysis, measurement of the blood glucose level, and other investigations relevant to the vesiculobullous disorder were performed. Special investigations were Gram staining, culture, and susceptibility testing of material from a nasal swab and a swab of a representative skin lesion.
Detection of MRSA
Skin lesions were thoroughly cleaned with sterile normal saline. Specimens of pus were drawn with a sterile swab for Gram staining, culture, and susceptibility testing and were analyzed in the institution’s microbiology department. A direct colony suspension (equivalent to McFarland Standard No. 0.5) was inoculated on a Mueller-Hinton agar plate, incorporating cefoxitin, linezolid, vancomycin, amikacin, and rifampicin supplemented with sodium chloride 2% and incubated at 37°C for 24 hours. Staphylococcus aureus colonies were identified by their smooth, convex, shiny, and opaque appearance with a golden yellow pigment, as well as by coagulase positivity, mannitol fermentation, and production of phosphatase.
Methicillin-resistant S aureus was defined as an isolate having a minimum inhibitory concentration of more than 2 μg/mL of cefoxitin; a methicillin-sensitive S aureus isolate was defined as having a minimum inhibitory concentration of less than or equal to 2 μg/mL of cefoxitin. Specimens showing moderate to heavy growth of MRSA were included in the study. For specimens showing mild growth, testing was repeated; if no growth was seen on repeat testing, results were interpreted as negative.
Data were collected and analyzed for frequency and percentage; P<.05 was considered significant.
Results
The number of patients analyzed in the study period was 43. Table 1 shows their salient demographic characteristics, clinical features, and findings of the investigation. The youngest patient was aged 13 years; the oldest was aged 80 years. The male to female ratio was 0.65 to 1. The most common primary lesion was a combined vesicle and bulla (34 patients [79.1%]); the most common secondary lesion was a combination of erosion with crusting (22 patients [51.2%]).
Table 2 lists the types of vesiculobullous disorders seen in this study. Pemphigus vulgaris was the most common (21 patients [48.8%])(Figure 1). Drug-induced vesiculobullous disorders (eg, TEN) were noted in 11 patients (25.6%)(Figure 2).
Table 2 also lists pathogens cultured in the study group. There were 24 bacterial isolates, of which S aureus accounted for 22 (91.7%). Methicillin-resistant S aureus was cultured in 14 patients (32.6%); culture was sterile in 19 patients (44.2%).
Among the 22 cultured staphylococcal species, MRSA accounted for 14 (63.6%) and constituted 58.3% (14/24) of all bacterial isolates. The nasal swab for MRSA was positive in 4 PV patients (9.3%), 2 TEN patients (4.6%), and 1 bullous pemphigoid patient (2.3%). Methicillin-resistant S aureus was most commonly cultured in PV patients (8/14 [57.1%]).
All MRSA strains (100%) were sensitive to vancomycin and linezolid; 34 (79.1%) were sensitive to amikacin. Additionally, 100% of MRSA strains were resistant to oxacillin, cloxacillin, and cefoxitin.
Three patients with PV (7.0%) and 1 patient with TEN (2.3%) died during the course of the study; only 1 death (2.3%) occurred in a patient who had a positive MRSA culture.
Comment
In this 1-year study, we tested and followed 43 patients with autoimmune and drug-induced vesiculobullous disorders. Vesiculobullous disorders in dermatology inpatients are a cause of great concern. When lesions rupture, they leave behind a large area of erosion that forms a nidus of bacterial colonization; often, these bacteria cause severe infection, including septicemia, and result in death.5 Moreover, autoimmune bullous disorders usually require a prolonged hospital stay and powerful immunosuppressive drugs, which contributes to bacterial infection, especially MRSA.6
The age of patients in this study ranged from 13 to 80 years; most patients were in the 6th decade, a pattern seen in studies worldwide.5 In a study by Kanwar and De,7 however, most cases were aged 20 to 40 years.7 In our study, there was a female preponderance (male to female ratio of 0.65 to 1).
Studies have shown that the duration of illness in vesiculobullous disorder is directly associated with MRSA infection. However, in our study with MRSA detected in 14 patients, most patients had a duration of illness less than 1 year (statistically insignificant [P>.05]), a finding similar to Shafi et al.8
The symptomatic nature of these diseases, their unsightly appearance, and mucous-membrane involvement of vesiculobullous disorders prompts these patients to present to the hospital early. However, a prolonged hospital stay by patients with an autoimmune vesiculobullous disorders sets the stage for MRSA colonization.
In this study, diabetes mellitus (DM) was seen in 15 patients (34.9%); 5 of them had MRSA infection (statistically insignificant [P>.05]). Diabetes mellitus contributing to sepsis and MRSA infection, which in turn contributes to morbidity and mortality, has been well-documented.2,4,9
Methicillin-resistant S aureus in this study was isolated most often from blisters and erosions. Vesiculobullous disorders and drug reactions (eg, Stevens-Johnson syndrome, TEN) are characterized by blisters that rupture to form erosions and crusting, which form fissures in the epidermal barrier function that are nidi for colonization by microbes, especially S aureus and MRSA in particular; later, these bacteria can enter dermal vessels and then the bloodstream, leading to septicemia.10
The prevalence of MRSA in this study was 32.6% (14/43), which is high compared to other studies.2-4 Pemphigus vulgaris was the most common disorder infected by MRSA in this study (57.1% [8/14] of MRSA isolates)(Table 1), a finding that reveals that the incidence of MRSA is high among staphylococcal isolates in vesiculobullous disorders. However, the high incidence of MRSA in this study could be a reflection of the number of patients with a severe and chronic vesiculobullous disorder, such as PV, and serious drug reactions such as TEN referred to our tertiary-care center, where we get a large number of patients affected by autoimmune and drug-induced vesiculobullous disorders. Similar findings have been reported by Stryjewski et al.11
A high prevalence of MRSA in a dermatology unit has grave consequences, contributing to morbidity and mortality in particular among patients with a vesiculobullous disorder. Immunosuppressive therapy and comorbidities such as DM contribute to MRSA colonization in vesiculobullous disorders.12 Overcrowding and poor sterilization techniques in public hospitals in India may contribute to the high prevalence of MRSA seen in hospital units.
Patients with a vesiculobullous disorder who are chronic nasal carriers of MRSA are at risk for cutaneous MRSA infection, which in turn can lead to MRSA septicemia and an elevated risk of death. In this study, however, a nasal swab was positive for MRSA in only 7 patients. One patient with MRSA colonization died, which was statistically insignificant (P=1).
In this study, all MRSA strains (100%) were resistant to first-line antibiotics, such as oxacillin, cloxacillin, and cefoxitin; all strains were susceptible to vancomycin and linezolid.
Conclusion
Our study shows that MRSA is becoming the prominent pathogen in nosocomial infections, especially in bedridden patients, which has grave implications. The use of a prophylactic S aureus conjugate vaccine in patients with a chronic vesiculobullous disorder might be justified in the future.15 We found a high prevalence (32.6%) of MRSA in vesiculobullous disorders, no relationship between DM and MRSA colonization, PV was the most common disorder complicated by MRSA, no relationship between nasal colonization and MRSA infection, no relationship between death during the study period and MRSA infection, 100% of MRSA strains were susceptible to vancomycin and linezolid, and 79.1% of MRSA strains were susceptible to amikacin.
- Nair SP. A retrospective study of mortality of pemphigus patients in a tertiary care hospital. Indian J Dermatol Venereol Leprol. 2013;79:706-709.
- Sachdev D, Amladi S, Natarj G, et al. An outbreak of methicillin-resistant Staphylococcus aureus (MRSA) infection in dermatology inpatients. Indian J Dermatol Venereol Leprol. 2003;69:377-380.
- Vijayamohan N, Nair SP. A study of the prevalence of methicillin-resistant Staphylococcus aureus in dermatology inpatients. Indian Dermatol Online J. 2014;5:441-445.
- Malhotra SK, Malhotra S, Dhaliwal GS, et al. Bacterial study of pyodermas in a tertiary care dermatological center. Indian J Dermatol. 2012;57:358-361.
- Valencia IC, Kirsner RS, Kerdel FA. Microbiological evaluation of skin wounds: alarming trends towards antibiotic resistance in an inpatient dermatology service during a 10-year period. J Am Acad Dermatol. 2004;50:845-849.
- Lehman JS, Murell DF, Camilleri MJ, et al. Infection and infection prevention in patients treated with immunosuppressive medications for autoimmune bullous disorders. Dermatol Clin. 2011;29:591-598.
- Kanwar AJ, De D. Pemphigus in India. Indian J Dermatol Venereol Leprol. 2011;77:439-449.
- Shafi M, Khatri ML, Mashima M, et al. Pemphigus: a clinical study of 109 cases from Tripoli, Libya. Indian J Dermatol Venereol Leprol. 1994;60:140-143.
- Torres K, Sampathkumar P. Predictors of methicillin-resistant Staphylococcus aureus colonization at hospital admission. Am J Infect Control. 2013;41:1043-1047.
- Miller LG, Quan C, Shay A, et al. A prospective investigation of outcomes after hospital discharge for endemic, community-acquired methicillin-resistant Staphylococcus aureus skin infection. Clin Infect Dis. 2007;44:483-492.
- Stryjewski M, Chambers HF. Skin and soft-tissue infections caused by community-acquired methicillin-resistant Staphylococcus aureus. Clin Infect Dis. 2008;46(suppl 5):S368-S377.
- Mutasim DF. Management of autoimmune bullous diseases: pharmacology and therapeutics. J Am Acad Dermatol. 2004;51:859-877.
- Cohen PR. Community-acquired methicillin-resistant Staphylococcus aureus skin infections: a review of epidemiology, clinical features, management, and prevention. Int J Dermatol. 2007;46:1-11.
- Elston DM. Methicillin-sensitive and methicillin-resistant Staphylococcus aureus: management principles and selection of antibiotic therapy. Dermatol Clin. 2007;25:157-164.
- Shinefield H, Black S, Fattom A, et al. Use of a Staphylococcus aureus conjugate vaccine in patients receiving hemodialysis. N Engl J Med. 2001;346:491-496.
- Nair SP. A retrospective study of mortality of pemphigus patients in a tertiary care hospital. Indian J Dermatol Venereol Leprol. 2013;79:706-709.
- Sachdev D, Amladi S, Natarj G, et al. An outbreak of methicillin-resistant Staphylococcus aureus (MRSA) infection in dermatology inpatients. Indian J Dermatol Venereol Leprol. 2003;69:377-380.
- Vijayamohan N, Nair SP. A study of the prevalence of methicillin-resistant Staphylococcus aureus in dermatology inpatients. Indian Dermatol Online J. 2014;5:441-445.
- Malhotra SK, Malhotra S, Dhaliwal GS, et al. Bacterial study of pyodermas in a tertiary care dermatological center. Indian J Dermatol. 2012;57:358-361.
- Valencia IC, Kirsner RS, Kerdel FA. Microbiological evaluation of skin wounds: alarming trends towards antibiotic resistance in an inpatient dermatology service during a 10-year period. J Am Acad Dermatol. 2004;50:845-849.
- Lehman JS, Murell DF, Camilleri MJ, et al. Infection and infection prevention in patients treated with immunosuppressive medications for autoimmune bullous disorders. Dermatol Clin. 2011;29:591-598.
- Kanwar AJ, De D. Pemphigus in India. Indian J Dermatol Venereol Leprol. 2011;77:439-449.
- Shafi M, Khatri ML, Mashima M, et al. Pemphigus: a clinical study of 109 cases from Tripoli, Libya. Indian J Dermatol Venereol Leprol. 1994;60:140-143.
- Torres K, Sampathkumar P. Predictors of methicillin-resistant Staphylococcus aureus colonization at hospital admission. Am J Infect Control. 2013;41:1043-1047.
- Miller LG, Quan C, Shay A, et al. A prospective investigation of outcomes after hospital discharge for endemic, community-acquired methicillin-resistant Staphylococcus aureus skin infection. Clin Infect Dis. 2007;44:483-492.
- Stryjewski M, Chambers HF. Skin and soft-tissue infections caused by community-acquired methicillin-resistant Staphylococcus aureus. Clin Infect Dis. 2008;46(suppl 5):S368-S377.
- Mutasim DF. Management of autoimmune bullous diseases: pharmacology and therapeutics. J Am Acad Dermatol. 2004;51:859-877.
- Cohen PR. Community-acquired methicillin-resistant Staphylococcus aureus skin infections: a review of epidemiology, clinical features, management, and prevention. Int J Dermatol. 2007;46:1-11.
- Elston DM. Methicillin-sensitive and methicillin-resistant Staphylococcus aureus: management principles and selection of antibiotic therapy. Dermatol Clin. 2007;25:157-164.
- Shinefield H, Black S, Fattom A, et al. Use of a Staphylococcus aureus conjugate vaccine in patients receiving hemodialysis. N Engl J Med. 2001;346:491-496.
Practice Points
- Methicillin-resistant Staphylococcus aureus (MRSA) infection in vesiculobullous disorders such as pemphigus vulgaris and toxic epidermal necrolysis is known to contribute to an increase in disease-related mortality.
- Methicillin-resistant S aureus is becoming the prominent pathogen in nosocomial infections, especially in bedridden patients.
- The prevalence of MRSA in vesiculobullous disorders is high; pemphigus vulgaris is the most common vesiculobullous disorder complicated by MRSA.
- Early diagnosis of MRSA helps reduce morbidity and mortality and improves the patient’s prognosis.
Sweet Syndrome With Aseptic Splenic Abscesses and Multiple Myeloma
To the Editor:
An 84-year-old man was admitted to the hospital with 5 erythematous cutaneous nodules of several days’ duration on the legs ranging in size from 1.0 to 1.5 cm. Upon admission, the patient also had a chest radiograph suspicious for pneumonia. The patient had received sulfamethoxazole/trimethoprim for a urinary tract infection as an outpatient 5 days prior to presentation, but he stopped the medication due to the appearance of the cutaneous nodules. Of note, the patient also reported unintentional weight loss of 15 pounds over the last few months.
New nodules had developed at a rate of 1 to 2 lesions daily in the 3 days prior to presentation and continued to develop after admission to the hospital. The nodules appeared as tender, erythematous lesions that evolved to form pustules and developed overlying crusts in later stages (Figure 1). They were limited to the arms and legs, primarily involving the lower legs. There was no evidence of oral or ocular involvement. A hemoglobin count of 10.9 g/dL (reference range, 14.0–17.5 g/dL), white blood cell count of 8.8×109/L (reference range, 4.5–11.0×109/L), and erythrocyte sedimentation rate of 69 mm/h (reference range, 0–20 mm/h) were noted on admission.
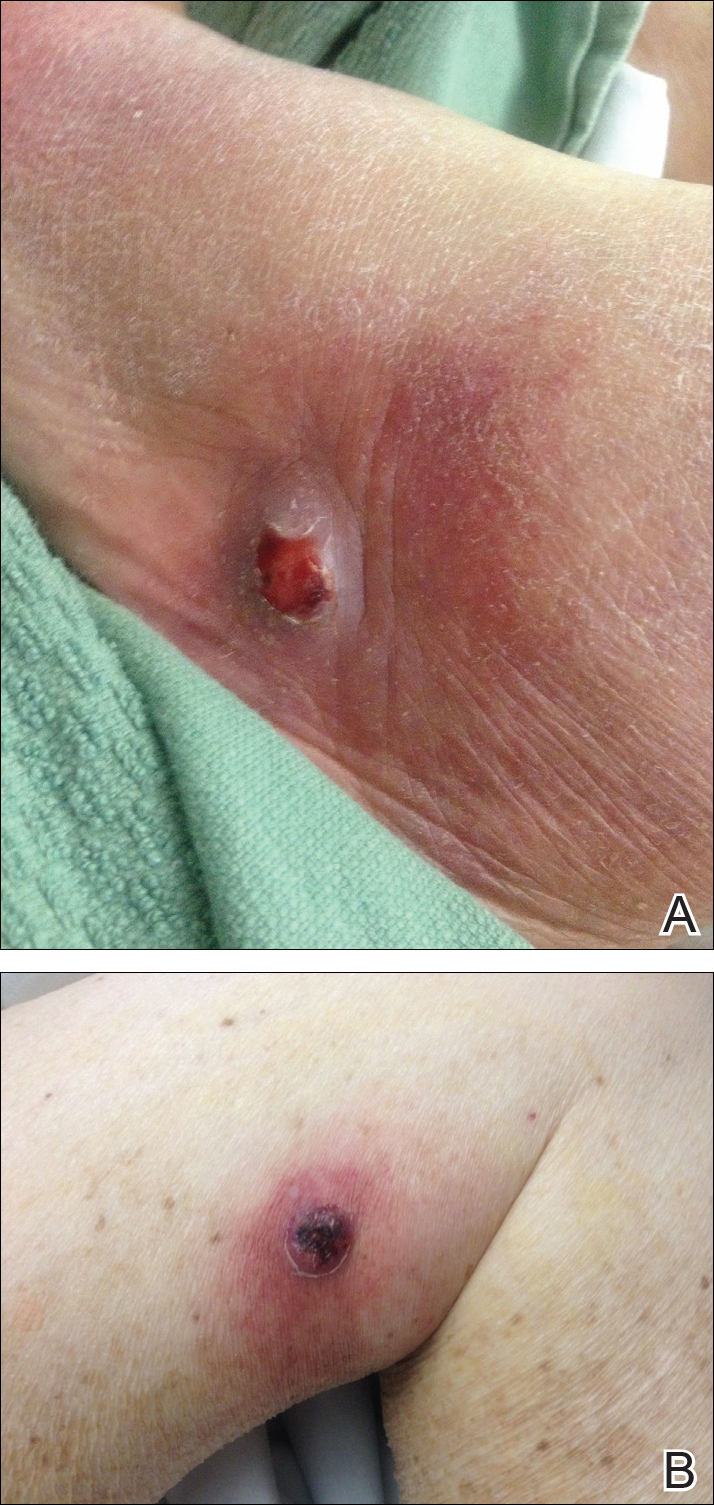
The patient was started on ceftriaxone and azithromycin for suspected pneumonia. The differential diagnosis for the cutaneous nodules included lymphoma, acid-fast bacilli (AFB) infection, deep fungal infection, pyoderma gangrenosum, Sweet syndrome (SS), panniculitis, erythema elevatum diutinum, and polyarteritis nodosa. A punch biopsy of a nodule on the left foot was performed. Histopathology demonstrated a neutrophilic panniculitis (Figure 2) with an epidermal abscess. No vasculitis was identified, and periodic acid–Schiff and AFB staining of the skin biopsy were negative. These findings were consistent with SS. Computed tomography scans of the chest, abdomen, and pelvis, which were completed early in the course of hospitalization due to concern for underlying malignancy, revealed pericardial and pleural effusions as well as cystic lesions in the lungs, spleen, kidneys, and prostate, with the largest lesion on the spleen measuring 5.6×4.8 cm (Figure 3). Computed tomography scanning was negative for areas of consolidation in the lungs. A splenic biopsy was performed by an interventional radiologist during the patient's hospitalization that identified an aseptic, neutrophilic process. Fungal, bacterial, and AFB cultures of the splenic tissue and cystic contents were negative. Bilateral pleural effusions also were identified, and a thoracentesis was performed. The pleural fluid indicated rare mesothelial cells in the background of acute inflammation with no growth of the bacterial, fungal, or AFB cultures.
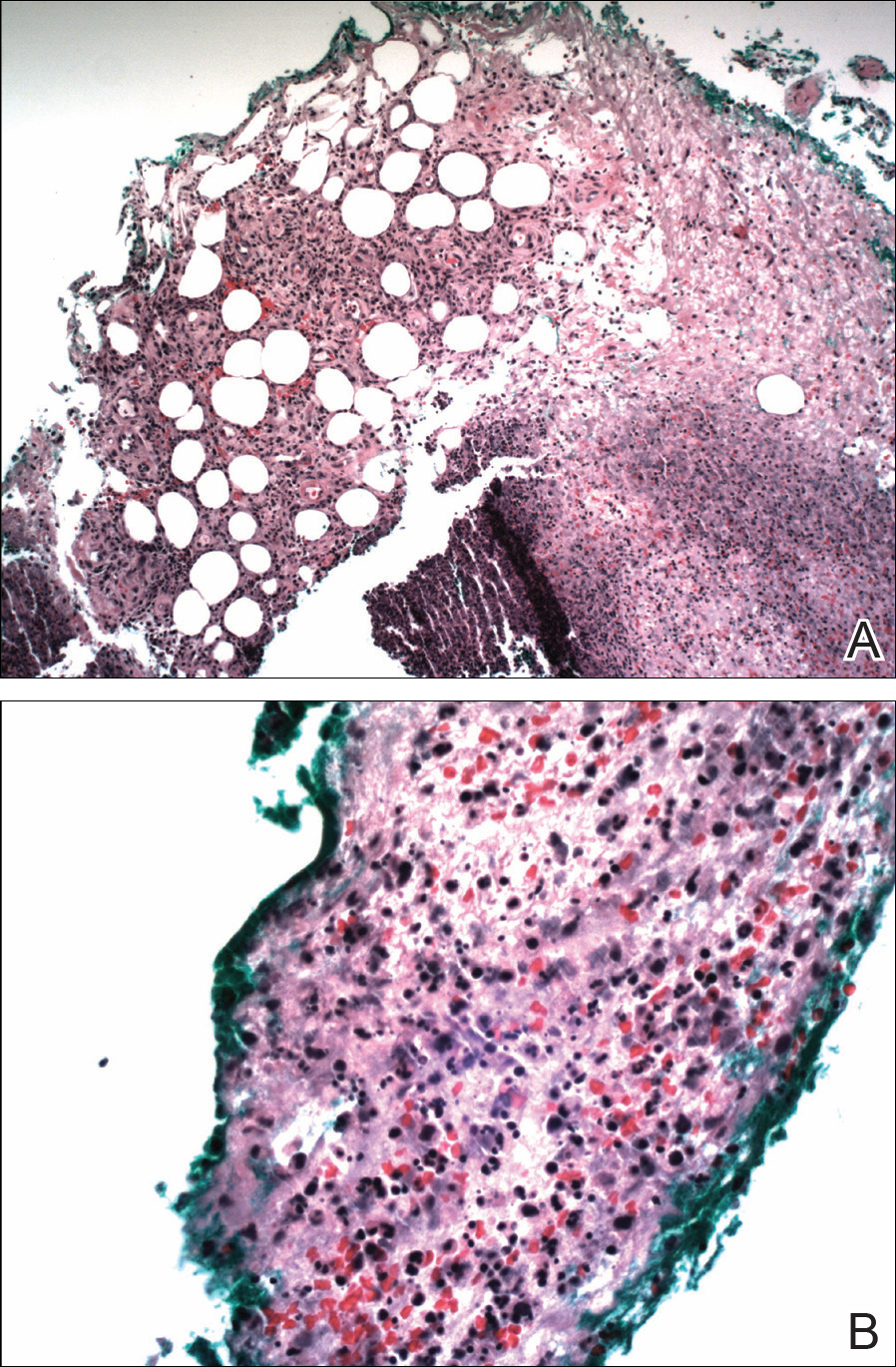
Due to the association of hematologic malignances with SS, a bone marrow biopsy was performed, which revealed multiple myeloma. Serum protein electrophoresis demonstrated monoclonal gammopathy of κ light chains. During the course of his hospitalization, new skin lesions continued to develop on the hands, face, and trunk. The patient was discharged from the hospital shortly after diagnosis to receive outpatient treatment for multiple myeloma with lenalidomide and dexamethasone. Upon follow-up with the patient’s family via telephone 3 weeks into treatment, his son confirmed that the nodules were resolving.
Our case could be consistent with either drug-induced or malignancy-associated SS. Sweet syndrome initially was described in 1964 in 8 female patients with leukocytosis and cutaneous plaques infiltrated by neutrophils.1 The skin lesions typically are red and painful, ranging in size from 0.5 cm to 12.0 cm, and can last weeks to years if not treated.2 Variations of skin lesions include bullous and pustular morphologies.3
Diagnostic criteria for SS have been established.4 Both of the major criteria must be met as well as 2 of 4 minor criteria. Major criteria include abrupt onset of tender erythematous plaques and nodules; secondly, a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis must be seen on histopathology. Minor criteria include pyrexia, association with underlying condition (malignancy, pregnancy, drug exposure, inflammatory disorder), responsiveness to systemic steroids, and abnormal laboratory values (erythrocyte sedimentation rate, white blood cell count, C-reactive protein, neutrophilia).4
Sweet syndrome can be divided into 3 classifications: classical or idiopathic, drug-induced, or malignancy-associated.4 Classical SS most commonly is seen in middle-aged women after an upper respiratory or gastrointestinal infection. Drug-induced SS most often is associated with granulocyte-stimulating factor colony therapy4; however, it has been associated with use of trimethoprim/sulfamethoxazole.5 Malignancy-associated SS most commonly is seen in individuals with hematologic malignancy, specifically acute myeloid leukemia. Although its association with multiple myeloma is not as frequent, cases of malignancy-associated SS identifying this association have been reported.6,7 Mucosal involvement in the form of aphthouslike lesions more frequently is seen in malignancy-associated SS.8 Differing from classical SS, which has a female predilection of around 4:1, the malignancy-associated disorder has a 1:1 female-to-male ratio.4
In the majority of cases of SS, the neutrophilic infiltrate is in the papillary and upper reticular dermis; however, if the neutrophilic infiltrate is predominately in the subcutaneous tissue (known as subcutaneous SS), there is a strong association with malignancy.9 The histopathology in our case demonstrated a neutrophilic infiltrate in the subcutaneous tissue.
Fever is the most common systemic manifestation of SS and is present in 54% to 65% of patients.8,10 Besides the skin, the most common site affected is the eye, with 13% to 75% of patients reporting ocular involvement, usually conjunctivitis.4,10 Although infrequent, extracutaneous SS has been identified in the bones, central nervous system, kidneys, heart, liver, spleen, lungs, ears, eyes, and intestines.4 A case of SS with splenic involvement in the form of sterile abscesses also was reported.11 This case was related to parvovirus B19.
Sweet syndrome is a condition characterized by tender, erythematous cutaneous lesions with histopathology demonstrating neutrophilic infiltrate in the absence of vasculitis. We report a case of suspected extracutaneous SS in the form of splenic cysts in a patient whose SS was associated with malignancy and/or drug ingestion.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome and malignancy. Am J Med. 1987;82:1220-1226.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2002;41:182-184.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Belhadjali H, Chaabane S, Njim L, et al. Sweet’s syndrome associated with multiple myeloma. Acta Dermatovenerol Alp Pannonica Adriat. 2008;17:31-33.
- Bayer-Garner IB, Cottler-Fox M, Smoller BR. Sweet syndrome in multiple myeloma: a series of six cases. J Cutan Pathol. 2003;30:261-264.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556; quiz 557-560.
- Neoh CY, Tan AW, Ng SK. Sweet’s syndrome: a spectrum of unusual clinical presentation and associations. Br J Dermatol. 2007;156:480-485.
- Fortna RR, Toporcer M, Elder DE, et al. A case of sweet syndrome with spleen and lymph node involvement preceded by parvovirus B19 infection, and review of the literature on extracutaneous Sweet syndrome. Am J Dermatopathol. 2010;32:621-627.
To the Editor:
An 84-year-old man was admitted to the hospital with 5 erythematous cutaneous nodules of several days’ duration on the legs ranging in size from 1.0 to 1.5 cm. Upon admission, the patient also had a chest radiograph suspicious for pneumonia. The patient had received sulfamethoxazole/trimethoprim for a urinary tract infection as an outpatient 5 days prior to presentation, but he stopped the medication due to the appearance of the cutaneous nodules. Of note, the patient also reported unintentional weight loss of 15 pounds over the last few months.
New nodules had developed at a rate of 1 to 2 lesions daily in the 3 days prior to presentation and continued to develop after admission to the hospital. The nodules appeared as tender, erythematous lesions that evolved to form pustules and developed overlying crusts in later stages (Figure 1). They were limited to the arms and legs, primarily involving the lower legs. There was no evidence of oral or ocular involvement. A hemoglobin count of 10.9 g/dL (reference range, 14.0–17.5 g/dL), white blood cell count of 8.8×109/L (reference range, 4.5–11.0×109/L), and erythrocyte sedimentation rate of 69 mm/h (reference range, 0–20 mm/h) were noted on admission.
The patient was started on ceftriaxone and azithromycin for suspected pneumonia. The differential diagnosis for the cutaneous nodules included lymphoma, acid-fast bacilli (AFB) infection, deep fungal infection, pyoderma gangrenosum, Sweet syndrome (SS), panniculitis, erythema elevatum diutinum, and polyarteritis nodosa. A punch biopsy of a nodule on the left foot was performed. Histopathology demonstrated a neutrophilic panniculitis (Figure 2) with an epidermal abscess. No vasculitis was identified, and periodic acid–Schiff and AFB staining of the skin biopsy were negative. These findings were consistent with SS. Computed tomography scans of the chest, abdomen, and pelvis, which were completed early in the course of hospitalization due to concern for underlying malignancy, revealed pericardial and pleural effusions as well as cystic lesions in the lungs, spleen, kidneys, and prostate, with the largest lesion on the spleen measuring 5.6×4.8 cm (Figure 3). Computed tomography scanning was negative for areas of consolidation in the lungs. A splenic biopsy was performed by an interventional radiologist during the patient's hospitalization that identified an aseptic, neutrophilic process. Fungal, bacterial, and AFB cultures of the splenic tissue and cystic contents were negative. Bilateral pleural effusions also were identified, and a thoracentesis was performed. The pleural fluid indicated rare mesothelial cells in the background of acute inflammation with no growth of the bacterial, fungal, or AFB cultures.
Due to the association of hematologic malignances with SS, a bone marrow biopsy was performed, which revealed multiple myeloma. Serum protein electrophoresis demonstrated monoclonal gammopathy of κ light chains. During the course of his hospitalization, new skin lesions continued to develop on the hands, face, and trunk. The patient was discharged from the hospital shortly after diagnosis to receive outpatient treatment for multiple myeloma with lenalidomide and dexamethasone. Upon follow-up with the patient’s family via telephone 3 weeks into treatment, his son confirmed that the nodules were resolving.
Our case could be consistent with either drug-induced or malignancy-associated SS. Sweet syndrome initially was described in 1964 in 8 female patients with leukocytosis and cutaneous plaques infiltrated by neutrophils.1 The skin lesions typically are red and painful, ranging in size from 0.5 cm to 12.0 cm, and can last weeks to years if not treated.2 Variations of skin lesions include bullous and pustular morphologies.3
Diagnostic criteria for SS have been established.4 Both of the major criteria must be met as well as 2 of 4 minor criteria. Major criteria include abrupt onset of tender erythematous plaques and nodules; secondly, a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis must be seen on histopathology. Minor criteria include pyrexia, association with underlying condition (malignancy, pregnancy, drug exposure, inflammatory disorder), responsiveness to systemic steroids, and abnormal laboratory values (erythrocyte sedimentation rate, white blood cell count, C-reactive protein, neutrophilia).4
Sweet syndrome can be divided into 3 classifications: classical or idiopathic, drug-induced, or malignancy-associated.4 Classical SS most commonly is seen in middle-aged women after an upper respiratory or gastrointestinal infection. Drug-induced SS most often is associated with granulocyte-stimulating factor colony therapy4; however, it has been associated with use of trimethoprim/sulfamethoxazole.5 Malignancy-associated SS most commonly is seen in individuals with hematologic malignancy, specifically acute myeloid leukemia. Although its association with multiple myeloma is not as frequent, cases of malignancy-associated SS identifying this association have been reported.6,7 Mucosal involvement in the form of aphthouslike lesions more frequently is seen in malignancy-associated SS.8 Differing from classical SS, which has a female predilection of around 4:1, the malignancy-associated disorder has a 1:1 female-to-male ratio.4
In the majority of cases of SS, the neutrophilic infiltrate is in the papillary and upper reticular dermis; however, if the neutrophilic infiltrate is predominately in the subcutaneous tissue (known as subcutaneous SS), there is a strong association with malignancy.9 The histopathology in our case demonstrated a neutrophilic infiltrate in the subcutaneous tissue.
Fever is the most common systemic manifestation of SS and is present in 54% to 65% of patients.8,10 Besides the skin, the most common site affected is the eye, with 13% to 75% of patients reporting ocular involvement, usually conjunctivitis.4,10 Although infrequent, extracutaneous SS has been identified in the bones, central nervous system, kidneys, heart, liver, spleen, lungs, ears, eyes, and intestines.4 A case of SS with splenic involvement in the form of sterile abscesses also was reported.11 This case was related to parvovirus B19.
Sweet syndrome is a condition characterized by tender, erythematous cutaneous lesions with histopathology demonstrating neutrophilic infiltrate in the absence of vasculitis. We report a case of suspected extracutaneous SS in the form of splenic cysts in a patient whose SS was associated with malignancy and/or drug ingestion.
To the Editor:
An 84-year-old man was admitted to the hospital with 5 erythematous cutaneous nodules of several days’ duration on the legs ranging in size from 1.0 to 1.5 cm. Upon admission, the patient also had a chest radiograph suspicious for pneumonia. The patient had received sulfamethoxazole/trimethoprim for a urinary tract infection as an outpatient 5 days prior to presentation, but he stopped the medication due to the appearance of the cutaneous nodules. Of note, the patient also reported unintentional weight loss of 15 pounds over the last few months.
New nodules had developed at a rate of 1 to 2 lesions daily in the 3 days prior to presentation and continued to develop after admission to the hospital. The nodules appeared as tender, erythematous lesions that evolved to form pustules and developed overlying crusts in later stages (Figure 1). They were limited to the arms and legs, primarily involving the lower legs. There was no evidence of oral or ocular involvement. A hemoglobin count of 10.9 g/dL (reference range, 14.0–17.5 g/dL), white blood cell count of 8.8×109/L (reference range, 4.5–11.0×109/L), and erythrocyte sedimentation rate of 69 mm/h (reference range, 0–20 mm/h) were noted on admission.
The patient was started on ceftriaxone and azithromycin for suspected pneumonia. The differential diagnosis for the cutaneous nodules included lymphoma, acid-fast bacilli (AFB) infection, deep fungal infection, pyoderma gangrenosum, Sweet syndrome (SS), panniculitis, erythema elevatum diutinum, and polyarteritis nodosa. A punch biopsy of a nodule on the left foot was performed. Histopathology demonstrated a neutrophilic panniculitis (Figure 2) with an epidermal abscess. No vasculitis was identified, and periodic acid–Schiff and AFB staining of the skin biopsy were negative. These findings were consistent with SS. Computed tomography scans of the chest, abdomen, and pelvis, which were completed early in the course of hospitalization due to concern for underlying malignancy, revealed pericardial and pleural effusions as well as cystic lesions in the lungs, spleen, kidneys, and prostate, with the largest lesion on the spleen measuring 5.6×4.8 cm (Figure 3). Computed tomography scanning was negative for areas of consolidation in the lungs. A splenic biopsy was performed by an interventional radiologist during the patient's hospitalization that identified an aseptic, neutrophilic process. Fungal, bacterial, and AFB cultures of the splenic tissue and cystic contents were negative. Bilateral pleural effusions also were identified, and a thoracentesis was performed. The pleural fluid indicated rare mesothelial cells in the background of acute inflammation with no growth of the bacterial, fungal, or AFB cultures.
Due to the association of hematologic malignances with SS, a bone marrow biopsy was performed, which revealed multiple myeloma. Serum protein electrophoresis demonstrated monoclonal gammopathy of κ light chains. During the course of his hospitalization, new skin lesions continued to develop on the hands, face, and trunk. The patient was discharged from the hospital shortly after diagnosis to receive outpatient treatment for multiple myeloma with lenalidomide and dexamethasone. Upon follow-up with the patient’s family via telephone 3 weeks into treatment, his son confirmed that the nodules were resolving.
Our case could be consistent with either drug-induced or malignancy-associated SS. Sweet syndrome initially was described in 1964 in 8 female patients with leukocytosis and cutaneous plaques infiltrated by neutrophils.1 The skin lesions typically are red and painful, ranging in size from 0.5 cm to 12.0 cm, and can last weeks to years if not treated.2 Variations of skin lesions include bullous and pustular morphologies.3
Diagnostic criteria for SS have been established.4 Both of the major criteria must be met as well as 2 of 4 minor criteria. Major criteria include abrupt onset of tender erythematous plaques and nodules; secondly, a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis must be seen on histopathology. Minor criteria include pyrexia, association with underlying condition (malignancy, pregnancy, drug exposure, inflammatory disorder), responsiveness to systemic steroids, and abnormal laboratory values (erythrocyte sedimentation rate, white blood cell count, C-reactive protein, neutrophilia).4
Sweet syndrome can be divided into 3 classifications: classical or idiopathic, drug-induced, or malignancy-associated.4 Classical SS most commonly is seen in middle-aged women after an upper respiratory or gastrointestinal infection. Drug-induced SS most often is associated with granulocyte-stimulating factor colony therapy4; however, it has been associated with use of trimethoprim/sulfamethoxazole.5 Malignancy-associated SS most commonly is seen in individuals with hematologic malignancy, specifically acute myeloid leukemia. Although its association with multiple myeloma is not as frequent, cases of malignancy-associated SS identifying this association have been reported.6,7 Mucosal involvement in the form of aphthouslike lesions more frequently is seen in malignancy-associated SS.8 Differing from classical SS, which has a female predilection of around 4:1, the malignancy-associated disorder has a 1:1 female-to-male ratio.4
In the majority of cases of SS, the neutrophilic infiltrate is in the papillary and upper reticular dermis; however, if the neutrophilic infiltrate is predominately in the subcutaneous tissue (known as subcutaneous SS), there is a strong association with malignancy.9 The histopathology in our case demonstrated a neutrophilic infiltrate in the subcutaneous tissue.
Fever is the most common systemic manifestation of SS and is present in 54% to 65% of patients.8,10 Besides the skin, the most common site affected is the eye, with 13% to 75% of patients reporting ocular involvement, usually conjunctivitis.4,10 Although infrequent, extracutaneous SS has been identified in the bones, central nervous system, kidneys, heart, liver, spleen, lungs, ears, eyes, and intestines.4 A case of SS with splenic involvement in the form of sterile abscesses also was reported.11 This case was related to parvovirus B19.
Sweet syndrome is a condition characterized by tender, erythematous cutaneous lesions with histopathology demonstrating neutrophilic infiltrate in the absence of vasculitis. We report a case of suspected extracutaneous SS in the form of splenic cysts in a patient whose SS was associated with malignancy and/or drug ingestion.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome and malignancy. Am J Med. 1987;82:1220-1226.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2002;41:182-184.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Belhadjali H, Chaabane S, Njim L, et al. Sweet’s syndrome associated with multiple myeloma. Acta Dermatovenerol Alp Pannonica Adriat. 2008;17:31-33.
- Bayer-Garner IB, Cottler-Fox M, Smoller BR. Sweet syndrome in multiple myeloma: a series of six cases. J Cutan Pathol. 2003;30:261-264.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556; quiz 557-560.
- Neoh CY, Tan AW, Ng SK. Sweet’s syndrome: a spectrum of unusual clinical presentation and associations. Br J Dermatol. 2007;156:480-485.
- Fortna RR, Toporcer M, Elder DE, et al. A case of sweet syndrome with spleen and lymph node involvement preceded by parvovirus B19 infection, and review of the literature on extracutaneous Sweet syndrome. Am J Dermatopathol. 2010;32:621-627.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome and malignancy. Am J Med. 1987;82:1220-1226.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2002;41:182-184.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Belhadjali H, Chaabane S, Njim L, et al. Sweet’s syndrome associated with multiple myeloma. Acta Dermatovenerol Alp Pannonica Adriat. 2008;17:31-33.
- Bayer-Garner IB, Cottler-Fox M, Smoller BR. Sweet syndrome in multiple myeloma: a series of six cases. J Cutan Pathol. 2003;30:261-264.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556; quiz 557-560.
- Neoh CY, Tan AW, Ng SK. Sweet’s syndrome: a spectrum of unusual clinical presentation and associations. Br J Dermatol. 2007;156:480-485.
- Fortna RR, Toporcer M, Elder DE, et al. A case of sweet syndrome with spleen and lymph node involvement preceded by parvovirus B19 infection, and review of the literature on extracutaneous Sweet syndrome. Am J Dermatopathol. 2010;32:621-627.
Practice Points
- Sweet syndrome (SS), also known as acute febrile neutrophilic dermatosis, is an inflammatory process characterized by a diffuse dermal neutrophilic infiltrate in the absence of vasculitis.
- A diagnosis of SS warrants further investigation due to its association with malignancy, especially hematologic malignancy.
- Other organs in SS also may have aseptic involvement.
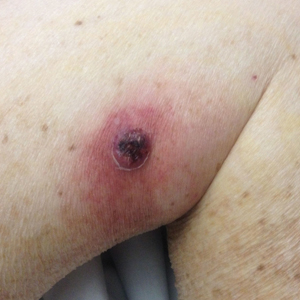
Eosinophilic Pustular Folliculitis With Underlying Mantle Cell Lymphoma
Eosinophilic pustular folliculitis (EPF) was originally described in 1965 and has since evolved into 3 distinct subtypes: classic, immunosuppressed (IS), and infantile types. Immunosuppressed EPF can be further subdivided into human immunodeficiency virus (HIV) associated (IS-HIV) and non-HIV associated. Human immunodeficiency virus–seronegative cases have been associated with underlying malignancies (IS-heme) or chronic immunosuppression, such as that seen in transplant patients.
Case Report
A 52-year-old man with a medical history limited to prostate adenocarcinoma treated with a robotic prostatectomy presented with a pruritic red rash on the face, neck, shoulders, and chest of 1 month’s duration. The patient previously completed a course of azithromycin 250 mg, intramuscular triamcinolone, and oral prednisone with only minor improvement. Physical examination demonstrated multiple pink folliculocentric papules and pustules scattered on the head (Figure 1A), neck, and chest (Figure 1B), as well as edematous pink papules and plaques on the forehead (Figures 1C and 1D). The palms, soles, and oral mucosa were clear.
, neck, and chest (B), as well as edematous pink papules and plaques on the forehead (C and D).")
Initial biopsy of the right side of the chest was nonspecific and most consistent with a reaction to an arthropod bite. The patient was started on oral doxycycline 100 mg twice daily for 2 weeks. With no improvement seen, additional biopsies were obtained from the left side of the chest and forehead. The biopsy of the chest showed ruptured folliculitis with evidence of acute and chronic inflammation. The biopsy of the forehead demonstrated eosinophilic follicular spongiosis with intrafollicular Langerhans cell microgranulomas along with abundant eosinophils adjacent to follicles, consistent with EPF (Figure 2). Serum HIV testing was negative. Serum white blood cell count was normal at 6400/µL (reference range, 4500–11,000/µL) with mild elevation of eosinophils (8%). The remaining complete blood cell count and comprehensive metabolic panel were within reference range. The patient was subsequently started on oral indomethacin 25 mg twice daily and triamcinolone cream 0.1%. Within a few days he experienced initial improvement in his symptoms of pruritus and diminution in the number of inflammatory follicular papules.
(H&E, original magnifications ×10 and ×20).")
Approximately 1 month after presentation, he began to experience symptoms of dysphagia and fatigue. In addition, tonsillar hypertrophy and palpable neck and axillary lymphadenopathy were present. Computed tomography of the neck, chest, and abdomen showed diffuse lymphadenopathy. Full-body positron emission tomography–computed tomography demonstrated extensive metabolically active lymphoma in multiple nodal groups above and below the diaphragm. There also was lymphomatous involvement of the spleen. An axillary lymph node biopsy was diagnostic for mantle cell lymphoma (CD4:CD8, 1:1; CD45 negative; CD20 positive; CD5 positive). He
Comment
Subtypes of EPF
Eosinophilic pustular folliculitis was first described in a Japanese female presenting with folliculocentric pustules distributed on the face, torso, and arms.1 This noninfectious eosinophilic infiltration of hair follicles predominantly seen in the Japanese population is now regarded as the classic form. Three distinct subtypes of EPF now exist, including the originally described classic variant (Ofuji disease), an IS variant, and a rare infantile form.1
All 3 subtypes of EPF are more commonly seen in men than women. The classic form has a peak incidence between the third and fourth decades of life. It presents as chronic annular papules and sterile pustules exhibiting peripheral extension, with individual lesions lasting for approximately 7 to 10 days with frequent relapses. The face is the most common area of involvement, followed by the trunk, extremities, and more rarely the palmoplantar surfaces. Concomitant leukocytosis with eosinophilia is seen in up to 35% of patients.1 The infantile type represents the rarest EPF form. The average age of onset is 5 months, with most cases resolving by 14 months of age.1
Clinically, EPF is characterized by recurrent papules and pustules predominantly on the scalp without annular or polycyclic ring formation, as seen in the classic type. The palms and soles may be involved, which can clinically mimic infantile acropustulosis and scabies infection. Most patients exhibit a concomitant peripheral eosinophilia.1,2
In the late 1980s, the IS variant of EPF was recognized in HIV-positive (IS-HIV) and HIV-negative malignancy-associated (IS-heme) populations.1,3 This newly characterized form differs morphologically and biologically from the classic and infantile subtypes. The IS subtype has a unique presentation including intensely pruritic, discrete, erythematous, follicular papules with palmoplantar sparing and infrequent annular or circinate plaque forms.1 Frequently, with the IS-HIV form, CD4+ T-cell counts are below 300 cells/mL, and 25% to 50% of patients have lymphopenia with eosinophilia.3 Highly active antiretroviral therapy has been associated with EPF resolution in HIV-positive individuals; however, it also has been shown to induce transient EPF during the first 3 to 6 months of initiation.1,3,4
Unlike the IS-HIV form, the IS-heme form has occurred solely in males and is predominantly associated with hematologic malignancies (eg, non-Hodgkin lymphoma, acute lymphoblastic leukemia, acute myeloid leukemia, myelodysplastic syndrome) 30 to 90 days following bone marrow transplant, peripheral blood stem cell transplant, or chemotherapy treatment.5,6 Unlike the chronic and persistent IS-HIV form, prior cases of IS-heme EPF have been predominantly self-limited. Interestingly, only 2 reported cases of EPF have occurred prior to the diagnosis of malignancy including B-cell leukemia and myelodysplastic syndrome.5
Histopathology
All 3 identified forms of EPF histopathologically show acute and chronic lymphoeosinophilic infiltrate concentrated at the follicular isthmus, which can lead to follicular destruction. Scattered mononuclear cells, eosinophils, and neutrophils are found within the pilar outer root sheath, sebaceous glands, and ducts. Approximately 40% of cases demonstrate follicular mucinosis.1 Histopathology of lesional palmar skin in classic-type EPF demonstrates intraepidermal pustule formation with abundant eosinophils and neutrophils adjacent to the acrosyringium.7,8
Pathogenesis
Although the pathophysiology of EPF is largely unknown, it is thought to represent a helper T cell (TH2) response involving IL-4, IL-5, and IL-13 cytokines.9 Chemoattractant receptor homologous molecule 2, which is expressed on eosinophils and lymphocytes, is believed to play a role in the pruritus, edema, and inflammatory response seen adjacent to pilosebaceous units in EPF.10 Moreover, immunohistochemical and flow cytometry analysis has revealed a prevalence of prostaglandin D2 within the perisebocyte infiltrate in EPF.9 Prostaglandin D2 induces eotaxin-3 production within sebocytes via peroxisome proliferator-activated receptor γ, which enhances chemoattraction of eosinophils. This pathogenesis represents a prostaglandin-based mechanism and potentially explains the efficacy of indomethacin treatment of EPF through its cyclooxygenase inhibition and reduction of chemoattractant receptor homologous molecule 2 expression.9-11
Treatment
Multiple therapeutic modalities have been reported for the treatment of EPF. For all 3 subtypes, moderate- to high-potency topical corticosteroids are considered first-line therapy. UVB phototherapy 2 to 3 times weekly remains the gold standard, given its consistent efficacy.1,12 Indomethacin (50–75 mg daily) remains first-line treatment of classic EPF.4,12 Previously reported cases of classic EPF and IS-EPF have responded well to oral prednisone (1 mg/kg daily).12,13 In a retrospective review of EPF treatment data, the following treatments also have been reported to be successful: psoralen plus UVA, oral cetirizine (20–40 mg daily, particularly for IS-EPF cases), metronidazole (250 mg 3 times daily), minocycline (150 mg daily), itraconazole (200–400 mg daily, dapsone (50–200 mg daily), systemic retinoids, tacrolimus ointment 0.1%, and permethrin cream.4,12
Malignancy
Although the entity of IS-heme EPF is rare, the morphology and treatment are unique and can potentially unmask an underlying hematologic malignancy. In patients with EPF and associated malignancy, such as our patient, a differential diagnosis to consider is eosinophilic dermatosis of hematologic malignancy (EDHM). Eosinophilic dermatosis of hematologic malignancy is most commonly associated with chronic lymphocytic leukemia and can be differentiated from EPF clinically, histopathologically, and by treatment response. Eosinophilic dermatosis of hematologic malignancy clinically presents with nonspecific papules, pustules, and/or vesicles on the head, trunk, and extremities. On histopathology, EDHM shows a superficial and deep perivascular and interstitial lymphoeosinophilic infiltration. Furthermore, EDHM patients typically exhibit a poor treatment response to oral indomethacin.14
Conclusion
Eosinophilic pustular folliculitis is a noninfectious folliculocentric process comprised of 3 distinct types. The histopathology shows follicular spongiosis with increased eosinophils. The pathogenesis is most likely related to a multifactorial immune system dysregulation involving TH2 T cells, prostaglandin D2, and eotaxin-3. The treatment of EPF may involve topical corticosteroids, UVB phototherapy, or most notably oral indomethacin. In patients with EPF and malignancy, EDHM is a differential diagnosis to consider. Our case serves as a reminder that rare eosinophilic dermatoses may represent manifestations of underlying hematopoietic malignancy and, when investigated early, can lead to appropriate life-saving treatment.
- Nervi J, Stephen. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- Hernández-Martín Á, Nuño-González A, Colmenero I, et al. Eosinophilic pustular folliculitis of infancy: a series of 15 cases and review of the literature [published online July 21, 2012]. J Am Acad Dermatol. 2013;68:150-155.
- Soepr
ono F, Schinella R. Eosinophilic pustular folliculitis in patients with acquired immunodeficiency syndrome. report of three cases. J Am Acad Dermatol. 1986;14:1020-1022. - Katoh
M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. - Keida
T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplant. J Dermatol. 2004;31:21-26. - Goiriz R, Gul-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36.
- Satoh T, Ikeda H, Yokozeki H. Acrosyringeal involvement of palmoplantar lesions of eosinophilic pustular folliculitis. Acta Derm Venereol. 2013;93:99.
- Tsuboi H, Wakita K, Fujimura T, et al. Acral variant of eosinophilic pustular folliculitis (Ofuji’s disease). Clin Exp Dermatol. 2003;28:321-324.
- Nakahig
ashi K, Doi H, Otsuka A, et al. PGD2 induces eotaxin-3 via PPARgamma from sebocytes: a possible pathogenesis of eosinophilic pustular folliculitis. J Allergy Clin Immunol. 2012;129:536-543. - Satoh
T, Shimura C, Miyagishi C, et al. Indomethacin-induced reduction in CRTH2 in eosinophilic pustular folliculitis (Ofuji’s disease): a proposed mechanism of action. Acta Derm Venereol. 2010;90:18-22. - Hagiwara A, Fujimura T, Furudate S, et al. Induction of CD163(+)M2 macrophages in the lesional skin of eosinophilic pustular folliculitis. Acta Derm Venereol. 2014;94:104-106.
- Ellis
E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197. - Bull R
H, Harland CA, Fallowfield ME, et al. Eosinophilic folliculitis: a self-limiting illness in patients being treated for haematological malignancy. Br J Dermatol. 1993;129:178-182. - Farber M, Forgia S, Sahu J, et al. Eosinophilic dermatosis of hematologic malignancy. J Cutan Pathol. 2012;39:690-695.
Eosinophilic pustular folliculitis (EPF) was originally described in 1965 and has since evolved into 3 distinct subtypes: classic, immunosuppressed (IS), and infantile types. Immunosuppressed EPF can be further subdivided into human immunodeficiency virus (HIV) associated (IS-HIV) and non-HIV associated. Human immunodeficiency virus–seronegative cases have been associated with underlying malignancies (IS-heme) or chronic immunosuppression, such as that seen in transplant patients.
Case Report
A 52-year-old man with a medical history limited to prostate adenocarcinoma treated with a robotic prostatectomy presented with a pruritic red rash on the face, neck, shoulders, and chest of 1 month’s duration. The patient previously completed a course of azithromycin 250 mg, intramuscular triamcinolone, and oral prednisone with only minor improvement. Physical examination demonstrated multiple pink folliculocentric papules and pustules scattered on the head (Figure 1A), neck, and chest (Figure 1B), as well as edematous pink papules and plaques on the forehead (Figures 1C and 1D). The palms, soles, and oral mucosa were clear.
Initial biopsy of the right side of the chest was nonspecific and most consistent with a reaction to an arthropod bite. The patient was started on oral doxycycline 100 mg twice daily for 2 weeks. With no improvement seen, additional biopsies were obtained from the left side of the chest and forehead. The biopsy of the chest showed ruptured folliculitis with evidence of acute and chronic inflammation. The biopsy of the forehead demonstrated eosinophilic follicular spongiosis with intrafollicular Langerhans cell microgranulomas along with abundant eosinophils adjacent to follicles, consistent with EPF (Figure 2). Serum HIV testing was negative. Serum white blood cell count was normal at 6400/µL (reference range, 4500–11,000/µL) with mild elevation of eosinophils (8%). The remaining complete blood cell count and comprehensive metabolic panel were within reference range. The patient was subsequently started on oral indomethacin 25 mg twice daily and triamcinolone cream 0.1%. Within a few days he experienced initial improvement in his symptoms of pruritus and diminution in the number of inflammatory follicular papules.
Approximately 1 month after presentation, he began to experience symptoms of dysphagia and fatigue. In addition, tonsillar hypertrophy and palpable neck and axillary lymphadenopathy were present. Computed tomography of the neck, chest, and abdomen showed diffuse lymphadenopathy. Full-body positron emission tomography–computed tomography demonstrated extensive metabolically active lymphoma in multiple nodal groups above and below the diaphragm. There also was lymphomatous involvement of the spleen. An axillary lymph node biopsy was diagnostic for mantle cell lymphoma (CD4:CD8, 1:1; CD45 negative; CD20 positive; CD5 positive). He
Comment
Subtypes of EPF
Eosinophilic pustular folliculitis was first described in a Japanese female presenting with folliculocentric pustules distributed on the face, torso, and arms.1 This noninfectious eosinophilic infiltration of hair follicles predominantly seen in the Japanese population is now regarded as the classic form. Three distinct subtypes of EPF now exist, including the originally described classic variant (Ofuji disease), an IS variant, and a rare infantile form.1
All 3 subtypes of EPF are more commonly seen in men than women. The classic form has a peak incidence between the third and fourth decades of life. It presents as chronic annular papules and sterile pustules exhibiting peripheral extension, with individual lesions lasting for approximately 7 to 10 days with frequent relapses. The face is the most common area of involvement, followed by the trunk, extremities, and more rarely the palmoplantar surfaces. Concomitant leukocytosis with eosinophilia is seen in up to 35% of patients.1 The infantile type represents the rarest EPF form. The average age of onset is 5 months, with most cases resolving by 14 months of age.1
Clinically, EPF is characterized by recurrent papules and pustules predominantly on the scalp without annular or polycyclic ring formation, as seen in the classic type. The palms and soles may be involved, which can clinically mimic infantile acropustulosis and scabies infection. Most patients exhibit a concomitant peripheral eosinophilia.1,2
In the late 1980s, the IS variant of EPF was recognized in HIV-positive (IS-HIV) and HIV-negative malignancy-associated (IS-heme) populations.1,3 This newly characterized form differs morphologically and biologically from the classic and infantile subtypes. The IS subtype has a unique presentation including intensely pruritic, discrete, erythematous, follicular papules with palmoplantar sparing and infrequent annular or circinate plaque forms.1 Frequently, with the IS-HIV form, CD4+ T-cell counts are below 300 cells/mL, and 25% to 50% of patients have lymphopenia with eosinophilia.3 Highly active antiretroviral therapy has been associated with EPF resolution in HIV-positive individuals; however, it also has been shown to induce transient EPF during the first 3 to 6 months of initiation.1,3,4
Unlike the IS-HIV form, the IS-heme form has occurred solely in males and is predominantly associated with hematologic malignancies (eg, non-Hodgkin lymphoma, acute lymphoblastic leukemia, acute myeloid leukemia, myelodysplastic syndrome) 30 to 90 days following bone marrow transplant, peripheral blood stem cell transplant, or chemotherapy treatment.5,6 Unlike the chronic and persistent IS-HIV form, prior cases of IS-heme EPF have been predominantly self-limited. Interestingly, only 2 reported cases of EPF have occurred prior to the diagnosis of malignancy including B-cell leukemia and myelodysplastic syndrome.5
Histopathology
All 3 identified forms of EPF histopathologically show acute and chronic lymphoeosinophilic infiltrate concentrated at the follicular isthmus, which can lead to follicular destruction. Scattered mononuclear cells, eosinophils, and neutrophils are found within the pilar outer root sheath, sebaceous glands, and ducts. Approximately 40% of cases demonstrate follicular mucinosis.1 Histopathology of lesional palmar skin in classic-type EPF demonstrates intraepidermal pustule formation with abundant eosinophils and neutrophils adjacent to the acrosyringium.7,8
Pathogenesis
Although the pathophysiology of EPF is largely unknown, it is thought to represent a helper T cell (TH2) response involving IL-4, IL-5, and IL-13 cytokines.9 Chemoattractant receptor homologous molecule 2, which is expressed on eosinophils and lymphocytes, is believed to play a role in the pruritus, edema, and inflammatory response seen adjacent to pilosebaceous units in EPF.10 Moreover, immunohistochemical and flow cytometry analysis has revealed a prevalence of prostaglandin D2 within the perisebocyte infiltrate in EPF.9 Prostaglandin D2 induces eotaxin-3 production within sebocytes via peroxisome proliferator-activated receptor γ, which enhances chemoattraction of eosinophils. This pathogenesis represents a prostaglandin-based mechanism and potentially explains the efficacy of indomethacin treatment of EPF through its cyclooxygenase inhibition and reduction of chemoattractant receptor homologous molecule 2 expression.9-11
Treatment
Multiple therapeutic modalities have been reported for the treatment of EPF. For all 3 subtypes, moderate- to high-potency topical corticosteroids are considered first-line therapy. UVB phototherapy 2 to 3 times weekly remains the gold standard, given its consistent efficacy.1,12 Indomethacin (50–75 mg daily) remains first-line treatment of classic EPF.4,12 Previously reported cases of classic EPF and IS-EPF have responded well to oral prednisone (1 mg/kg daily).12,13 In a retrospective review of EPF treatment data, the following treatments also have been reported to be successful: psoralen plus UVA, oral cetirizine (20–40 mg daily, particularly for IS-EPF cases), metronidazole (250 mg 3 times daily), minocycline (150 mg daily), itraconazole (200–400 mg daily, dapsone (50–200 mg daily), systemic retinoids, tacrolimus ointment 0.1%, and permethrin cream.4,12
Malignancy
Although the entity of IS-heme EPF is rare, the morphology and treatment are unique and can potentially unmask an underlying hematologic malignancy. In patients with EPF and associated malignancy, such as our patient, a differential diagnosis to consider is eosinophilic dermatosis of hematologic malignancy (EDHM). Eosinophilic dermatosis of hematologic malignancy is most commonly associated with chronic lymphocytic leukemia and can be differentiated from EPF clinically, histopathologically, and by treatment response. Eosinophilic dermatosis of hematologic malignancy clinically presents with nonspecific papules, pustules, and/or vesicles on the head, trunk, and extremities. On histopathology, EDHM shows a superficial and deep perivascular and interstitial lymphoeosinophilic infiltration. Furthermore, EDHM patients typically exhibit a poor treatment response to oral indomethacin.14
Conclusion
Eosinophilic pustular folliculitis is a noninfectious folliculocentric process comprised of 3 distinct types. The histopathology shows follicular spongiosis with increased eosinophils. The pathogenesis is most likely related to a multifactorial immune system dysregulation involving TH2 T cells, prostaglandin D2, and eotaxin-3. The treatment of EPF may involve topical corticosteroids, UVB phototherapy, or most notably oral indomethacin. In patients with EPF and malignancy, EDHM is a differential diagnosis to consider. Our case serves as a reminder that rare eosinophilic dermatoses may represent manifestations of underlying hematopoietic malignancy and, when investigated early, can lead to appropriate life-saving treatment.
Eosinophilic pustular folliculitis (EPF) was originally described in 1965 and has since evolved into 3 distinct subtypes: classic, immunosuppressed (IS), and infantile types. Immunosuppressed EPF can be further subdivided into human immunodeficiency virus (HIV) associated (IS-HIV) and non-HIV associated. Human immunodeficiency virus–seronegative cases have been associated with underlying malignancies (IS-heme) or chronic immunosuppression, such as that seen in transplant patients.
Case Report
A 52-year-old man with a medical history limited to prostate adenocarcinoma treated with a robotic prostatectomy presented with a pruritic red rash on the face, neck, shoulders, and chest of 1 month’s duration. The patient previously completed a course of azithromycin 250 mg, intramuscular triamcinolone, and oral prednisone with only minor improvement. Physical examination demonstrated multiple pink folliculocentric papules and pustules scattered on the head (Figure 1A), neck, and chest (Figure 1B), as well as edematous pink papules and plaques on the forehead (Figures 1C and 1D). The palms, soles, and oral mucosa were clear.
Initial biopsy of the right side of the chest was nonspecific and most consistent with a reaction to an arthropod bite. The patient was started on oral doxycycline 100 mg twice daily for 2 weeks. With no improvement seen, additional biopsies were obtained from the left side of the chest and forehead. The biopsy of the chest showed ruptured folliculitis with evidence of acute and chronic inflammation. The biopsy of the forehead demonstrated eosinophilic follicular spongiosis with intrafollicular Langerhans cell microgranulomas along with abundant eosinophils adjacent to follicles, consistent with EPF (Figure 2). Serum HIV testing was negative. Serum white blood cell count was normal at 6400/µL (reference range, 4500–11,000/µL) with mild elevation of eosinophils (8%). The remaining complete blood cell count and comprehensive metabolic panel were within reference range. The patient was subsequently started on oral indomethacin 25 mg twice daily and triamcinolone cream 0.1%. Within a few days he experienced initial improvement in his symptoms of pruritus and diminution in the number of inflammatory follicular papules.
Approximately 1 month after presentation, he began to experience symptoms of dysphagia and fatigue. In addition, tonsillar hypertrophy and palpable neck and axillary lymphadenopathy were present. Computed tomography of the neck, chest, and abdomen showed diffuse lymphadenopathy. Full-body positron emission tomography–computed tomography demonstrated extensive metabolically active lymphoma in multiple nodal groups above and below the diaphragm. There also was lymphomatous involvement of the spleen. An axillary lymph node biopsy was diagnostic for mantle cell lymphoma (CD4:CD8, 1:1; CD45 negative; CD20 positive; CD5 positive). He
Comment
Subtypes of EPF
Eosinophilic pustular folliculitis was first described in a Japanese female presenting with folliculocentric pustules distributed on the face, torso, and arms.1 This noninfectious eosinophilic infiltration of hair follicles predominantly seen in the Japanese population is now regarded as the classic form. Three distinct subtypes of EPF now exist, including the originally described classic variant (Ofuji disease), an IS variant, and a rare infantile form.1
All 3 subtypes of EPF are more commonly seen in men than women. The classic form has a peak incidence between the third and fourth decades of life. It presents as chronic annular papules and sterile pustules exhibiting peripheral extension, with individual lesions lasting for approximately 7 to 10 days with frequent relapses. The face is the most common area of involvement, followed by the trunk, extremities, and more rarely the palmoplantar surfaces. Concomitant leukocytosis with eosinophilia is seen in up to 35% of patients.1 The infantile type represents the rarest EPF form. The average age of onset is 5 months, with most cases resolving by 14 months of age.1
Clinically, EPF is characterized by recurrent papules and pustules predominantly on the scalp without annular or polycyclic ring formation, as seen in the classic type. The palms and soles may be involved, which can clinically mimic infantile acropustulosis and scabies infection. Most patients exhibit a concomitant peripheral eosinophilia.1,2
In the late 1980s, the IS variant of EPF was recognized in HIV-positive (IS-HIV) and HIV-negative malignancy-associated (IS-heme) populations.1,3 This newly characterized form differs morphologically and biologically from the classic and infantile subtypes. The IS subtype has a unique presentation including intensely pruritic, discrete, erythematous, follicular papules with palmoplantar sparing and infrequent annular or circinate plaque forms.1 Frequently, with the IS-HIV form, CD4+ T-cell counts are below 300 cells/mL, and 25% to 50% of patients have lymphopenia with eosinophilia.3 Highly active antiretroviral therapy has been associated with EPF resolution in HIV-positive individuals; however, it also has been shown to induce transient EPF during the first 3 to 6 months of initiation.1,3,4
Unlike the IS-HIV form, the IS-heme form has occurred solely in males and is predominantly associated with hematologic malignancies (eg, non-Hodgkin lymphoma, acute lymphoblastic leukemia, acute myeloid leukemia, myelodysplastic syndrome) 30 to 90 days following bone marrow transplant, peripheral blood stem cell transplant, or chemotherapy treatment.5,6 Unlike the chronic and persistent IS-HIV form, prior cases of IS-heme EPF have been predominantly self-limited. Interestingly, only 2 reported cases of EPF have occurred prior to the diagnosis of malignancy including B-cell leukemia and myelodysplastic syndrome.5
Histopathology
All 3 identified forms of EPF histopathologically show acute and chronic lymphoeosinophilic infiltrate concentrated at the follicular isthmus, which can lead to follicular destruction. Scattered mononuclear cells, eosinophils, and neutrophils are found within the pilar outer root sheath, sebaceous glands, and ducts. Approximately 40% of cases demonstrate follicular mucinosis.1 Histopathology of lesional palmar skin in classic-type EPF demonstrates intraepidermal pustule formation with abundant eosinophils and neutrophils adjacent to the acrosyringium.7,8
Pathogenesis
Although the pathophysiology of EPF is largely unknown, it is thought to represent a helper T cell (TH2) response involving IL-4, IL-5, and IL-13 cytokines.9 Chemoattractant receptor homologous molecule 2, which is expressed on eosinophils and lymphocytes, is believed to play a role in the pruritus, edema, and inflammatory response seen adjacent to pilosebaceous units in EPF.10 Moreover, immunohistochemical and flow cytometry analysis has revealed a prevalence of prostaglandin D2 within the perisebocyte infiltrate in EPF.9 Prostaglandin D2 induces eotaxin-3 production within sebocytes via peroxisome proliferator-activated receptor γ, which enhances chemoattraction of eosinophils. This pathogenesis represents a prostaglandin-based mechanism and potentially explains the efficacy of indomethacin treatment of EPF through its cyclooxygenase inhibition and reduction of chemoattractant receptor homologous molecule 2 expression.9-11
Treatment
Multiple therapeutic modalities have been reported for the treatment of EPF. For all 3 subtypes, moderate- to high-potency topical corticosteroids are considered first-line therapy. UVB phototherapy 2 to 3 times weekly remains the gold standard, given its consistent efficacy.1,12 Indomethacin (50–75 mg daily) remains first-line treatment of classic EPF.4,12 Previously reported cases of classic EPF and IS-EPF have responded well to oral prednisone (1 mg/kg daily).12,13 In a retrospective review of EPF treatment data, the following treatments also have been reported to be successful: psoralen plus UVA, oral cetirizine (20–40 mg daily, particularly for IS-EPF cases), metronidazole (250 mg 3 times daily), minocycline (150 mg daily), itraconazole (200–400 mg daily, dapsone (50–200 mg daily), systemic retinoids, tacrolimus ointment 0.1%, and permethrin cream.4,12
Malignancy
Although the entity of IS-heme EPF is rare, the morphology and treatment are unique and can potentially unmask an underlying hematologic malignancy. In patients with EPF and associated malignancy, such as our patient, a differential diagnosis to consider is eosinophilic dermatosis of hematologic malignancy (EDHM). Eosinophilic dermatosis of hematologic malignancy is most commonly associated with chronic lymphocytic leukemia and can be differentiated from EPF clinically, histopathologically, and by treatment response. Eosinophilic dermatosis of hematologic malignancy clinically presents with nonspecific papules, pustules, and/or vesicles on the head, trunk, and extremities. On histopathology, EDHM shows a superficial and deep perivascular and interstitial lymphoeosinophilic infiltration. Furthermore, EDHM patients typically exhibit a poor treatment response to oral indomethacin.14
Conclusion
Eosinophilic pustular folliculitis is a noninfectious folliculocentric process comprised of 3 distinct types. The histopathology shows follicular spongiosis with increased eosinophils. The pathogenesis is most likely related to a multifactorial immune system dysregulation involving TH2 T cells, prostaglandin D2, and eotaxin-3. The treatment of EPF may involve topical corticosteroids, UVB phototherapy, or most notably oral indomethacin. In patients with EPF and malignancy, EDHM is a differential diagnosis to consider. Our case serves as a reminder that rare eosinophilic dermatoses may represent manifestations of underlying hematopoietic malignancy and, when investigated early, can lead to appropriate life-saving treatment.
- Nervi J, Stephen. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- Hernández-Martín Á, Nuño-González A, Colmenero I, et al. Eosinophilic pustular folliculitis of infancy: a series of 15 cases and review of the literature [published online July 21, 2012]. J Am Acad Dermatol. 2013;68:150-155.
- Soepr
ono F, Schinella R. Eosinophilic pustular folliculitis in patients with acquired immunodeficiency syndrome. report of three cases. J Am Acad Dermatol. 1986;14:1020-1022. - Katoh
M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. - Keida
T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplant. J Dermatol. 2004;31:21-26. - Goiriz R, Gul-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36.
- Satoh T, Ikeda H, Yokozeki H. Acrosyringeal involvement of palmoplantar lesions of eosinophilic pustular folliculitis. Acta Derm Venereol. 2013;93:99.
- Tsuboi H, Wakita K, Fujimura T, et al. Acral variant of eosinophilic pustular folliculitis (Ofuji’s disease). Clin Exp Dermatol. 2003;28:321-324.
- Nakahig
ashi K, Doi H, Otsuka A, et al. PGD2 induces eotaxin-3 via PPARgamma from sebocytes: a possible pathogenesis of eosinophilic pustular folliculitis. J Allergy Clin Immunol. 2012;129:536-543. - Satoh
T, Shimura C, Miyagishi C, et al. Indomethacin-induced reduction in CRTH2 in eosinophilic pustular folliculitis (Ofuji’s disease): a proposed mechanism of action. Acta Derm Venereol. 2010;90:18-22. - Hagiwara A, Fujimura T, Furudate S, et al. Induction of CD163(+)M2 macrophages in the lesional skin of eosinophilic pustular folliculitis. Acta Derm Venereol. 2014;94:104-106.
- Ellis
E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197. - Bull R
H, Harland CA, Fallowfield ME, et al. Eosinophilic folliculitis: a self-limiting illness in patients being treated for haematological malignancy. Br J Dermatol. 1993;129:178-182. - Farber M, Forgia S, Sahu J, et al. Eosinophilic dermatosis of hematologic malignancy. J Cutan Pathol. 2012;39:690-695.
- Nervi J, Stephen. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- Hernández-Martín Á, Nuño-González A, Colmenero I, et al. Eosinophilic pustular folliculitis of infancy: a series of 15 cases and review of the literature [published online July 21, 2012]. J Am Acad Dermatol. 2013;68:150-155.
- Soepr
ono F, Schinella R. Eosinophilic pustular folliculitis in patients with acquired immunodeficiency syndrome. report of three cases. J Am Acad Dermatol. 1986;14:1020-1022. - Katoh
M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. - Keida
T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplant. J Dermatol. 2004;31:21-26. - Goiriz R, Gul-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36.
- Satoh T, Ikeda H, Yokozeki H. Acrosyringeal involvement of palmoplantar lesions of eosinophilic pustular folliculitis. Acta Derm Venereol. 2013;93:99.
- Tsuboi H, Wakita K, Fujimura T, et al. Acral variant of eosinophilic pustular folliculitis (Ofuji’s disease). Clin Exp Dermatol. 2003;28:321-324.
- Nakahig
ashi K, Doi H, Otsuka A, et al. PGD2 induces eotaxin-3 via PPARgamma from sebocytes: a possible pathogenesis of eosinophilic pustular folliculitis. J Allergy Clin Immunol. 2012;129:536-543. - Satoh
T, Shimura C, Miyagishi C, et al. Indomethacin-induced reduction in CRTH2 in eosinophilic pustular folliculitis (Ofuji’s disease): a proposed mechanism of action. Acta Derm Venereol. 2010;90:18-22. - Hagiwara A, Fujimura T, Furudate S, et al. Induction of CD163(+)M2 macrophages in the lesional skin of eosinophilic pustular folliculitis. Acta Derm Venereol. 2014;94:104-106.
- Ellis
E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197. - Bull R
H, Harland CA, Fallowfield ME, et al. Eosinophilic folliculitis: a self-limiting illness in patients being treated for haematological malignancy. Br J Dermatol. 1993;129:178-182. - Farber M, Forgia S, Sahu J, et al. Eosinophilic dermatosis of hematologic malignancy. J Cutan Pathol. 2012;39:690-695.
Practice Points
- Recalcitrant folliculocentric papules and pustules involving the head, trunk, arms, and legs should raise suspicion of possible eosinophilic pustular folliculitis (EPF).
- Underlying hematopoietic malignancy may be associated with cases of EPF.
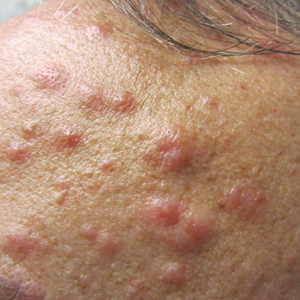
Acrodermatitis Enteropathica From Zinc-Deficient Total Parenteral Nutrition
Case Report
A 54-year-old woman presented with a pruritic and slightly painful skin eruption that began perinasally and progressed over 1 week to involve the labial commissures, finger webs, dorsal surfaces of the feet, heels, and bilateral gluteal folds. In addition, the eruption involved the left thigh at the donor site of a prior skin graft. She received no relief after an intramuscular steroid injection and hydrocortisone cream 1% prescribed by a primary care physician who diagnosed the rash as poison ivy contact dermatitis despite no exposure to plants. Review of systems was negative and she denied any new medication use. Her medical history was notable for extensive mesenteric injury secondary to a motor vehicle accident. She subsequently had multiple enterocutaneous fistulas that resulted in a complete small bowel enterectomy 10 months prior to presentation, which caused her to become dependent on total parenteral nutrition (TPN).
Physical examination revealed sharply demarcated, erythematous, scaly plaques perinasally, periorally, and on the bilateral gluteal folds (Figure 1). There were sharply demarcated, erythematous, scaly plaques on the right and left finger webs, dorsal surface of the right foot, and left upper thigh. Hemorrhagic bullae were appreciated on the left finger webs. Large flaccid bullae were present on the bilateral heels and dorsum of the right foot (Figure 2).

 and dorsum of the right foot (B).")
Suspecting a diagnosis of acrodermatitis enteropathica (AE), laboratory testing included a serum zinc level, which was 42 µg/dL (reference range, 70–130 µg/dL). The copper and selenium levels also were low with values of 71 µg/dL (reference range, 80–155 µg/dL) and 31 µg/dL (reference range, 79–326 µg/dL), respectively. No additional vitamin or mineral deficiencies were discovered. A complete blood cell count and comprehensive metabolic panel were performed and showed no abnormalities other than a mildly elevated sodium level of 147 mEq/L (reference range, 136–142 mEq/L).
A punch biopsy was performed. Histopathology revealed subcorneal neutrophils and neutrophilic crust, mild spongiosis, and a dense upper dermal mixed neutrophilic and lymphohistiocytic infiltrate. The specimen also exhibited mild intercellular edema and prominent capillaries (Figure 3).

After further investigation, the company providing the patient’s TPN confirmed that zinc had been removed several weeks prior to the onset of symptoms due to a critical national shortage of trace element additives. Zinc was supplemented at 15 mg daily to the TPN solution. Three days later a skin examination revealed dramatic changes with notable improvement of the finger web plaques and complete resolution of the facial lesions. The plaques and bullae on the lower extremities also had resolved (Figure 4).

Comment
Background
Acrodermatitis enteropathica is a rare autosomal-recessive disorder of zinc metabolism characterized by skin lesions predominantly distributed in acral and periorificial sites as well as alopecia and diarrhea. Acrodermatitis enteropathica was first described by Brandt1 in 1936 and later characterized by Danbolt and Closs2 in 1942 as a unique and often fatal disease of unknown etiology. More than 30 years later, the link between zinc deficiency and AE was illustrated by Moynahan3 who demonstrated clinical improvement with zinc supplementation. It was not until 2002 that the molecular pathogenesis of hypozincemia in patients with inherited AE was described. Küry et al4 identified a mutation in the SLC39A4 gene responsible for encoding the Zip4 protein, a zinc transporter found on enterocytes, particularly in the proximal small intestine.5,6 Classically, patients with inherited AE are children who present within days of birth or days to weeks after being weaned from breast milk to cow’s milk. The zinc in bovine milk is less bioavailable than breast milk, though both have similar total zinc concentrations, which results in the decreased plasma zinc levels seen in children with inherited AE.5-8 Occasionally, children present before weaning due to decreased maternal mammary zinc secretion (lactogenic AE).9,10
Clinical Presentation
Similar clinical findings are seen in patients with noninherited forms of zinc deficiency known as acquired AE. Acquired zinc deficiency may be broadly categorized as being from inadequate intake, deficient absorption, excess demand, or overexcretion.8 Such disturbances of zinc balance are most frequently seen in patients with restrictive diets, anorexia nervosa, intestinal bypass procedures, Crohn disease, pancreatic insufficiency, alcoholism, human immunodeficiency virus, and extensive cutaneous burns. Premature infants, mothers who are breastfeeding, and those dependent on TPN are at risk for developing acquired zinc deficiency.7-9,11
Differentiating Characteristics
Both acquired and inherited AE present as erythematous or pink eczematous scaly plaques with the variable presence of vesicular or bullous lesions involving periorificial, acral, and anogenital regions. Early manifestations of AE may include angular cheilitis and paronychia. Alopecia and diarrhea are characteristics of later disease. In fact, the complete triad of dermatitis, alopecia, and diarrhea is seen in only 20% of cases.7Without treatment, patients may develop blepharitis, conjunctivitis, photophobia, irritability, anorexia, apathy, growth retardation, hypogonadism, hypogeusia, and mental slowing. Skin lesions frequently become secondarily infected with Candida albicans and/or bacteria.5,7,11
Histopathology
Histopathologic examination of skin biopsy specimens from AE lesions demonstrates nonspecific findings similar to other deficiency dermatoses, such as pellagra and glucagonoma-associated necrolytic migratory erythema. Histology typically reveals cytoplasmic pallor with vacuolization and ballooning degeneration of keratinocytes, followed by confluent keratinocyte necrosis within the stratum granulosum and stratum spinosum of the epidermis.5 Confluent parakeratosis with hypogranulosis variably associated with neutrophil crust also is seen. Scattered dyskeratotic keratinocytes may be found within all levels of the epidermis. In resolving or chronic AE lesions, psoriasiform hyperplasia is prevalent, though necrolysis may be minimal or absent.5,11
Diagnosis
Evaluation includes measurement of plasma zinc levels. Zinc levels less than 50 µg/dL are suggestive but not diagnostic of AE.5 Although plasma zinc measurement is the most useful indicator of zinc status, its utility in assessing the true total body store of zinc is limited. Plasma zinc is tightly regulated and only represents 0.1% of body stores.5,6 Additionally, zinc levels may decrease in proinflammatory states.12 Beyond zinc measurement, evaluation of alkaline phosphatase, a zinc-dependent enzyme, can provide useful diagnostic information.5,6
Zinc and TPN
Patients on TPN are at a unique risk for developing zinc and other nutritional deficiencies. Because the daily recommended dietary allowance for zinc is low (8 mg daily for adult women and 11 mg daily for adult men)5 and the element is found in a wide variety of foods, maintaining adequate zinc levels is easily achieved in healthy individuals with normal diets. Kay et al13 described 4 patients on parenteral nutrition who developed hypozincemia and an AE-like syndrome within weeks of TPN induction. The authors described rapid and drastic clinical improvement after initiating zinc supplementation, accentuating the importance of including zinc as a component of TPN.13,14 Brazin et al15 also reported a case of an AE-like syndrome from zinc-deficient hyperalimentation in a patient receiving TPN for short bowel syndrome. Chun et al16 described another case of acquired AE in a patient on TPN for acute pancreatitis. Both cases demonstrated prompt improvement of skin lesions after treatment with zinc supplementation. Other nutrient deficiencies may reveal themselves through similar dermatologic manifestations. For example, cases of scaly dermatitis secondary to the development of essential fatty acid deficiency from TPN formulations lacking adequate quantities of linoleic acid have been reported.Similar to our case, the resolution of skin lesions was seen after TPN was supplemented with the deficient nutrient.17 These cases exemplify the importance in considering deficiency dermatoses in the TPN-dependent patient population.
Conclusion
In our case, the development of skin lesions directly coincided with a recent removal of zinc from the patient’s TPN, which provided us with a unique opportunity to observe the causal relationship between decreased zinc intake and the development of clinical signs of acquired AE. This association was further elucidated by laboratory confirmation of low serum zinc levels and rapid improvement in all skin lesions after zinc supplementation was initiated.
- Brandt T. Dermatitis in children with disturbances of general condition and absorption of food. Acta Derm Venereol. 1936;17:513-537.
- Danbolt N, Closs K. Acrodermatitis enteropathica. Acta Derm Venereol. 1942;23:127-169.
- Moynahan E. Acrodermatitis enteropathica: a lethal inherited human zinc deficiency disorder. Lancet. 1974;2:299-400.
- Küry S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:238-240.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease: part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33; quiz 244-246.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kumar P, Ranjan NR, Mondal AK. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Neldner K, Hambidge K, Walravens P. Acrodermatitis enteropathica.Int J Dermatol. 1978;17:380-387.
- Gehrig K, Dinulos J. Acrodermatitis due to nutritional deficiency. Curr Opin Pediatr. 2010;22:107-112.
- Liuzzi JP, Lichten LA, Rivera S, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to hypozincemia of the acute-phase response. Proct Natl Acad Sci U S A. 2005;102:6843-6848.
- Kay RG, Tasman-Jones C, Pybus J, et al. A syndrome of acute zinc deficiency during total parenteral nutrition in man. Ann Surg. 1976;183:331-340.
- Jeejeebhoy K. Zinc: an essential trace element for parenteral nutrition. Gastroenterology. 2009;137(5 suppl):S7-S12.
- Brazin SA, Johnson WT, Abramson LJ. The acrodermatitis enteropathica-like syndrome. Arch Dermatol. 1979;115:597-599.
- Chun JH, Baek JH, Chung NG. Development of bullous acrodermatitis enteropathica during the course of chemotherapy for acute lymphocytic leukemia. Ann Dermatol. 2011;23(suppl 3):S326-S328.
- Roongpisuthipong W, Phanachet P, Roongpisuthipong C, et al. Essential fatty acid deficiency while a patient receiving fat regimen total parenteral nutrition [published June 14, 2012]. BMJ Case Rep. doi:10.1136/bcr.07.2011.4475.
Case Report
A 54-year-old woman presented with a pruritic and slightly painful skin eruption that began perinasally and progressed over 1 week to involve the labial commissures, finger webs, dorsal surfaces of the feet, heels, and bilateral gluteal folds. In addition, the eruption involved the left thigh at the donor site of a prior skin graft. She received no relief after an intramuscular steroid injection and hydrocortisone cream 1% prescribed by a primary care physician who diagnosed the rash as poison ivy contact dermatitis despite no exposure to plants. Review of systems was negative and she denied any new medication use. Her medical history was notable for extensive mesenteric injury secondary to a motor vehicle accident. She subsequently had multiple enterocutaneous fistulas that resulted in a complete small bowel enterectomy 10 months prior to presentation, which caused her to become dependent on total parenteral nutrition (TPN).
Physical examination revealed sharply demarcated, erythematous, scaly plaques perinasally, periorally, and on the bilateral gluteal folds (Figure 1). There were sharply demarcated, erythematous, scaly plaques on the right and left finger webs, dorsal surface of the right foot, and left upper thigh. Hemorrhagic bullae were appreciated on the left finger webs. Large flaccid bullae were present on the bilateral heels and dorsum of the right foot (Figure 2).
Suspecting a diagnosis of acrodermatitis enteropathica (AE), laboratory testing included a serum zinc level, which was 42 µg/dL (reference range, 70–130 µg/dL). The copper and selenium levels also were low with values of 71 µg/dL (reference range, 80–155 µg/dL) and 31 µg/dL (reference range, 79–326 µg/dL), respectively. No additional vitamin or mineral deficiencies were discovered. A complete blood cell count and comprehensive metabolic panel were performed and showed no abnormalities other than a mildly elevated sodium level of 147 mEq/L (reference range, 136–142 mEq/L).
A punch biopsy was performed. Histopathology revealed subcorneal neutrophils and neutrophilic crust, mild spongiosis, and a dense upper dermal mixed neutrophilic and lymphohistiocytic infiltrate. The specimen also exhibited mild intercellular edema and prominent capillaries (Figure 3).
After further investigation, the company providing the patient’s TPN confirmed that zinc had been removed several weeks prior to the onset of symptoms due to a critical national shortage of trace element additives. Zinc was supplemented at 15 mg daily to the TPN solution. Three days later a skin examination revealed dramatic changes with notable improvement of the finger web plaques and complete resolution of the facial lesions. The plaques and bullae on the lower extremities also had resolved (Figure 4).
Comment
Background
Acrodermatitis enteropathica is a rare autosomal-recessive disorder of zinc metabolism characterized by skin lesions predominantly distributed in acral and periorificial sites as well as alopecia and diarrhea. Acrodermatitis enteropathica was first described by Brandt1 in 1936 and later characterized by Danbolt and Closs2 in 1942 as a unique and often fatal disease of unknown etiology. More than 30 years later, the link between zinc deficiency and AE was illustrated by Moynahan3 who demonstrated clinical improvement with zinc supplementation. It was not until 2002 that the molecular pathogenesis of hypozincemia in patients with inherited AE was described. Küry et al4 identified a mutation in the SLC39A4 gene responsible for encoding the Zip4 protein, a zinc transporter found on enterocytes, particularly in the proximal small intestine.5,6 Classically, patients with inherited AE are children who present within days of birth or days to weeks after being weaned from breast milk to cow’s milk. The zinc in bovine milk is less bioavailable than breast milk, though both have similar total zinc concentrations, which results in the decreased plasma zinc levels seen in children with inherited AE.5-8 Occasionally, children present before weaning due to decreased maternal mammary zinc secretion (lactogenic AE).9,10
Clinical Presentation
Similar clinical findings are seen in patients with noninherited forms of zinc deficiency known as acquired AE. Acquired zinc deficiency may be broadly categorized as being from inadequate intake, deficient absorption, excess demand, or overexcretion.8 Such disturbances of zinc balance are most frequently seen in patients with restrictive diets, anorexia nervosa, intestinal bypass procedures, Crohn disease, pancreatic insufficiency, alcoholism, human immunodeficiency virus, and extensive cutaneous burns. Premature infants, mothers who are breastfeeding, and those dependent on TPN are at risk for developing acquired zinc deficiency.7-9,11
Differentiating Characteristics
Both acquired and inherited AE present as erythematous or pink eczematous scaly plaques with the variable presence of vesicular or bullous lesions involving periorificial, acral, and anogenital regions. Early manifestations of AE may include angular cheilitis and paronychia. Alopecia and diarrhea are characteristics of later disease. In fact, the complete triad of dermatitis, alopecia, and diarrhea is seen in only 20% of cases.7Without treatment, patients may develop blepharitis, conjunctivitis, photophobia, irritability, anorexia, apathy, growth retardation, hypogonadism, hypogeusia, and mental slowing. Skin lesions frequently become secondarily infected with Candida albicans and/or bacteria.5,7,11
Histopathology
Histopathologic examination of skin biopsy specimens from AE lesions demonstrates nonspecific findings similar to other deficiency dermatoses, such as pellagra and glucagonoma-associated necrolytic migratory erythema. Histology typically reveals cytoplasmic pallor with vacuolization and ballooning degeneration of keratinocytes, followed by confluent keratinocyte necrosis within the stratum granulosum and stratum spinosum of the epidermis.5 Confluent parakeratosis with hypogranulosis variably associated with neutrophil crust also is seen. Scattered dyskeratotic keratinocytes may be found within all levels of the epidermis. In resolving or chronic AE lesions, psoriasiform hyperplasia is prevalent, though necrolysis may be minimal or absent.5,11
Diagnosis
Evaluation includes measurement of plasma zinc levels. Zinc levels less than 50 µg/dL are suggestive but not diagnostic of AE.5 Although plasma zinc measurement is the most useful indicator of zinc status, its utility in assessing the true total body store of zinc is limited. Plasma zinc is tightly regulated and only represents 0.1% of body stores.5,6 Additionally, zinc levels may decrease in proinflammatory states.12 Beyond zinc measurement, evaluation of alkaline phosphatase, a zinc-dependent enzyme, can provide useful diagnostic information.5,6
Zinc and TPN
Patients on TPN are at a unique risk for developing zinc and other nutritional deficiencies. Because the daily recommended dietary allowance for zinc is low (8 mg daily for adult women and 11 mg daily for adult men)5 and the element is found in a wide variety of foods, maintaining adequate zinc levels is easily achieved in healthy individuals with normal diets. Kay et al13 described 4 patients on parenteral nutrition who developed hypozincemia and an AE-like syndrome within weeks of TPN induction. The authors described rapid and drastic clinical improvement after initiating zinc supplementation, accentuating the importance of including zinc as a component of TPN.13,14 Brazin et al15 also reported a case of an AE-like syndrome from zinc-deficient hyperalimentation in a patient receiving TPN for short bowel syndrome. Chun et al16 described another case of acquired AE in a patient on TPN for acute pancreatitis. Both cases demonstrated prompt improvement of skin lesions after treatment with zinc supplementation. Other nutrient deficiencies may reveal themselves through similar dermatologic manifestations. For example, cases of scaly dermatitis secondary to the development of essential fatty acid deficiency from TPN formulations lacking adequate quantities of linoleic acid have been reported.Similar to our case, the resolution of skin lesions was seen after TPN was supplemented with the deficient nutrient.17 These cases exemplify the importance in considering deficiency dermatoses in the TPN-dependent patient population.
Conclusion
In our case, the development of skin lesions directly coincided with a recent removal of zinc from the patient’s TPN, which provided us with a unique opportunity to observe the causal relationship between decreased zinc intake and the development of clinical signs of acquired AE. This association was further elucidated by laboratory confirmation of low serum zinc levels and rapid improvement in all skin lesions after zinc supplementation was initiated.
Case Report
A 54-year-old woman presented with a pruritic and slightly painful skin eruption that began perinasally and progressed over 1 week to involve the labial commissures, finger webs, dorsal surfaces of the feet, heels, and bilateral gluteal folds. In addition, the eruption involved the left thigh at the donor site of a prior skin graft. She received no relief after an intramuscular steroid injection and hydrocortisone cream 1% prescribed by a primary care physician who diagnosed the rash as poison ivy contact dermatitis despite no exposure to plants. Review of systems was negative and she denied any new medication use. Her medical history was notable for extensive mesenteric injury secondary to a motor vehicle accident. She subsequently had multiple enterocutaneous fistulas that resulted in a complete small bowel enterectomy 10 months prior to presentation, which caused her to become dependent on total parenteral nutrition (TPN).
Physical examination revealed sharply demarcated, erythematous, scaly plaques perinasally, periorally, and on the bilateral gluteal folds (Figure 1). There were sharply demarcated, erythematous, scaly plaques on the right and left finger webs, dorsal surface of the right foot, and left upper thigh. Hemorrhagic bullae were appreciated on the left finger webs. Large flaccid bullae were present on the bilateral heels and dorsum of the right foot (Figure 2).
Suspecting a diagnosis of acrodermatitis enteropathica (AE), laboratory testing included a serum zinc level, which was 42 µg/dL (reference range, 70–130 µg/dL). The copper and selenium levels also were low with values of 71 µg/dL (reference range, 80–155 µg/dL) and 31 µg/dL (reference range, 79–326 µg/dL), respectively. No additional vitamin or mineral deficiencies were discovered. A complete blood cell count and comprehensive metabolic panel were performed and showed no abnormalities other than a mildly elevated sodium level of 147 mEq/L (reference range, 136–142 mEq/L).
A punch biopsy was performed. Histopathology revealed subcorneal neutrophils and neutrophilic crust, mild spongiosis, and a dense upper dermal mixed neutrophilic and lymphohistiocytic infiltrate. The specimen also exhibited mild intercellular edema and prominent capillaries (Figure 3).
After further investigation, the company providing the patient’s TPN confirmed that zinc had been removed several weeks prior to the onset of symptoms due to a critical national shortage of trace element additives. Zinc was supplemented at 15 mg daily to the TPN solution. Three days later a skin examination revealed dramatic changes with notable improvement of the finger web plaques and complete resolution of the facial lesions. The plaques and bullae on the lower extremities also had resolved (Figure 4).
Comment
Background
Acrodermatitis enteropathica is a rare autosomal-recessive disorder of zinc metabolism characterized by skin lesions predominantly distributed in acral and periorificial sites as well as alopecia and diarrhea. Acrodermatitis enteropathica was first described by Brandt1 in 1936 and later characterized by Danbolt and Closs2 in 1942 as a unique and often fatal disease of unknown etiology. More than 30 years later, the link between zinc deficiency and AE was illustrated by Moynahan3 who demonstrated clinical improvement with zinc supplementation. It was not until 2002 that the molecular pathogenesis of hypozincemia in patients with inherited AE was described. Küry et al4 identified a mutation in the SLC39A4 gene responsible for encoding the Zip4 protein, a zinc transporter found on enterocytes, particularly in the proximal small intestine.5,6 Classically, patients with inherited AE are children who present within days of birth or days to weeks after being weaned from breast milk to cow’s milk. The zinc in bovine milk is less bioavailable than breast milk, though both have similar total zinc concentrations, which results in the decreased plasma zinc levels seen in children with inherited AE.5-8 Occasionally, children present before weaning due to decreased maternal mammary zinc secretion (lactogenic AE).9,10
Clinical Presentation
Similar clinical findings are seen in patients with noninherited forms of zinc deficiency known as acquired AE. Acquired zinc deficiency may be broadly categorized as being from inadequate intake, deficient absorption, excess demand, or overexcretion.8 Such disturbances of zinc balance are most frequently seen in patients with restrictive diets, anorexia nervosa, intestinal bypass procedures, Crohn disease, pancreatic insufficiency, alcoholism, human immunodeficiency virus, and extensive cutaneous burns. Premature infants, mothers who are breastfeeding, and those dependent on TPN are at risk for developing acquired zinc deficiency.7-9,11
Differentiating Characteristics
Both acquired and inherited AE present as erythematous or pink eczematous scaly plaques with the variable presence of vesicular or bullous lesions involving periorificial, acral, and anogenital regions. Early manifestations of AE may include angular cheilitis and paronychia. Alopecia and diarrhea are characteristics of later disease. In fact, the complete triad of dermatitis, alopecia, and diarrhea is seen in only 20% of cases.7Without treatment, patients may develop blepharitis, conjunctivitis, photophobia, irritability, anorexia, apathy, growth retardation, hypogonadism, hypogeusia, and mental slowing. Skin lesions frequently become secondarily infected with Candida albicans and/or bacteria.5,7,11
Histopathology
Histopathologic examination of skin biopsy specimens from AE lesions demonstrates nonspecific findings similar to other deficiency dermatoses, such as pellagra and glucagonoma-associated necrolytic migratory erythema. Histology typically reveals cytoplasmic pallor with vacuolization and ballooning degeneration of keratinocytes, followed by confluent keratinocyte necrosis within the stratum granulosum and stratum spinosum of the epidermis.5 Confluent parakeratosis with hypogranulosis variably associated with neutrophil crust also is seen. Scattered dyskeratotic keratinocytes may be found within all levels of the epidermis. In resolving or chronic AE lesions, psoriasiform hyperplasia is prevalent, though necrolysis may be minimal or absent.5,11
Diagnosis
Evaluation includes measurement of plasma zinc levels. Zinc levels less than 50 µg/dL are suggestive but not diagnostic of AE.5 Although plasma zinc measurement is the most useful indicator of zinc status, its utility in assessing the true total body store of zinc is limited. Plasma zinc is tightly regulated and only represents 0.1% of body stores.5,6 Additionally, zinc levels may decrease in proinflammatory states.12 Beyond zinc measurement, evaluation of alkaline phosphatase, a zinc-dependent enzyme, can provide useful diagnostic information.5,6
Zinc and TPN
Patients on TPN are at a unique risk for developing zinc and other nutritional deficiencies. Because the daily recommended dietary allowance for zinc is low (8 mg daily for adult women and 11 mg daily for adult men)5 and the element is found in a wide variety of foods, maintaining adequate zinc levels is easily achieved in healthy individuals with normal diets. Kay et al13 described 4 patients on parenteral nutrition who developed hypozincemia and an AE-like syndrome within weeks of TPN induction. The authors described rapid and drastic clinical improvement after initiating zinc supplementation, accentuating the importance of including zinc as a component of TPN.13,14 Brazin et al15 also reported a case of an AE-like syndrome from zinc-deficient hyperalimentation in a patient receiving TPN for short bowel syndrome. Chun et al16 described another case of acquired AE in a patient on TPN for acute pancreatitis. Both cases demonstrated prompt improvement of skin lesions after treatment with zinc supplementation. Other nutrient deficiencies may reveal themselves through similar dermatologic manifestations. For example, cases of scaly dermatitis secondary to the development of essential fatty acid deficiency from TPN formulations lacking adequate quantities of linoleic acid have been reported.Similar to our case, the resolution of skin lesions was seen after TPN was supplemented with the deficient nutrient.17 These cases exemplify the importance in considering deficiency dermatoses in the TPN-dependent patient population.
Conclusion
In our case, the development of skin lesions directly coincided with a recent removal of zinc from the patient’s TPN, which provided us with a unique opportunity to observe the causal relationship between decreased zinc intake and the development of clinical signs of acquired AE. This association was further elucidated by laboratory confirmation of low serum zinc levels and rapid improvement in all skin lesions after zinc supplementation was initiated.
- Brandt T. Dermatitis in children with disturbances of general condition and absorption of food. Acta Derm Venereol. 1936;17:513-537.
- Danbolt N, Closs K. Acrodermatitis enteropathica. Acta Derm Venereol. 1942;23:127-169.
- Moynahan E. Acrodermatitis enteropathica: a lethal inherited human zinc deficiency disorder. Lancet. 1974;2:299-400.
- Küry S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:238-240.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease: part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33; quiz 244-246.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kumar P, Ranjan NR, Mondal AK. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Neldner K, Hambidge K, Walravens P. Acrodermatitis enteropathica.Int J Dermatol. 1978;17:380-387.
- Gehrig K, Dinulos J. Acrodermatitis due to nutritional deficiency. Curr Opin Pediatr. 2010;22:107-112.
- Liuzzi JP, Lichten LA, Rivera S, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to hypozincemia of the acute-phase response. Proct Natl Acad Sci U S A. 2005;102:6843-6848.
- Kay RG, Tasman-Jones C, Pybus J, et al. A syndrome of acute zinc deficiency during total parenteral nutrition in man. Ann Surg. 1976;183:331-340.
- Jeejeebhoy K. Zinc: an essential trace element for parenteral nutrition. Gastroenterology. 2009;137(5 suppl):S7-S12.
- Brazin SA, Johnson WT, Abramson LJ. The acrodermatitis enteropathica-like syndrome. Arch Dermatol. 1979;115:597-599.
- Chun JH, Baek JH, Chung NG. Development of bullous acrodermatitis enteropathica during the course of chemotherapy for acute lymphocytic leukemia. Ann Dermatol. 2011;23(suppl 3):S326-S328.
- Roongpisuthipong W, Phanachet P, Roongpisuthipong C, et al. Essential fatty acid deficiency while a patient receiving fat regimen total parenteral nutrition [published June 14, 2012]. BMJ Case Rep. doi:10.1136/bcr.07.2011.4475.
- Brandt T. Dermatitis in children with disturbances of general condition and absorption of food. Acta Derm Venereol. 1936;17:513-537.
- Danbolt N, Closs K. Acrodermatitis enteropathica. Acta Derm Venereol. 1942;23:127-169.
- Moynahan E. Acrodermatitis enteropathica: a lethal inherited human zinc deficiency disorder. Lancet. 1974;2:299-400.
- Küry S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:238-240.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease: part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33; quiz 244-246.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kumar P, Ranjan NR, Mondal AK. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Neldner K, Hambidge K, Walravens P. Acrodermatitis enteropathica.Int J Dermatol. 1978;17:380-387.
- Gehrig K, Dinulos J. Acrodermatitis due to nutritional deficiency. Curr Opin Pediatr. 2010;22:107-112.
- Liuzzi JP, Lichten LA, Rivera S, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to hypozincemia of the acute-phase response. Proct Natl Acad Sci U S A. 2005;102:6843-6848.
- Kay RG, Tasman-Jones C, Pybus J, et al. A syndrome of acute zinc deficiency during total parenteral nutrition in man. Ann Surg. 1976;183:331-340.
- Jeejeebhoy K. Zinc: an essential trace element for parenteral nutrition. Gastroenterology. 2009;137(5 suppl):S7-S12.
- Brazin SA, Johnson WT, Abramson LJ. The acrodermatitis enteropathica-like syndrome. Arch Dermatol. 1979;115:597-599.
- Chun JH, Baek JH, Chung NG. Development of bullous acrodermatitis enteropathica during the course of chemotherapy for acute lymphocytic leukemia. Ann Dermatol. 2011;23(suppl 3):S326-S328.
- Roongpisuthipong W, Phanachet P, Roongpisuthipong C, et al. Essential fatty acid deficiency while a patient receiving fat regimen total parenteral nutrition [published June 14, 2012]. BMJ Case Rep. doi:10.1136/bcr.07.2011.4475.
Practice Points
- Acrodermatitis enteropathica (AE) may be acquired or due to a rare autosomal-recessive disorder of zinc absorption.
- Hereditary AE typically becomes symptomatic during infancy, while acquired AE may develop during hypozincemia in patients of any age.
- Both acquired and hereditary AE improve with zinc supplementation.
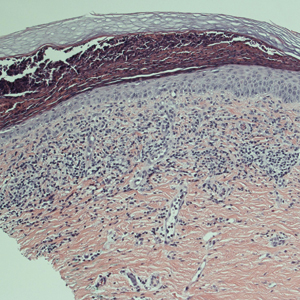
Statin effect in prostate cancer may be caused by reduced inflammation
Also today, Verma unveils Medicaid scorecard but refuses to judge efforts, the AMA reports that opioid prescriptions are down since 2013, and a prior knee injury creates a ‘distinct group’ in osteoarthritis.
Listen to the MDedge Daily News podcast for all the details on today’s top news.
Also today, Verma unveils Medicaid scorecard but refuses to judge efforts, the AMA reports that opioid prescriptions are down since 2013, and a prior knee injury creates a ‘distinct group’ in osteoarthritis.
Listen to the MDedge Daily News podcast for all the details on today’s top news.
Also today, Verma unveils Medicaid scorecard but refuses to judge efforts, the AMA reports that opioid prescriptions are down since 2013, and a prior knee injury creates a ‘distinct group’ in osteoarthritis.
Listen to the MDedge Daily News podcast for all the details on today’s top news.

Uncommon Presentation of Chromoblastomycosis
Case Report
A 25-year-old man who was a dairy farmer in Ahmednagar, Maharashtra, India, presented with a history of slowly growing, occasionally itchy lesions on both cheeks of 20 years’ duration. Most of the right cheek was covered by a well-defined, lobulated, gray-brown verrucous mass with a cerebriform surface (Figure 1). The left cheek was covered with a gray-brown infiltrated plaque surrounded by brown-tinged monomorphic papules.

Routine investigations were normal at presentation. Tests for purified protein derivative (tuberculin) and antibodies to human immunodeficiency virus were negative. Magnetic resonance imaging of the head showed soft tissue thickening with ulcerations involving the skin, subcutaneous tissue, and underlying facial muscles of the right cheek.
On histopathology, a hematoxylin and eosin–stained section showed hyperkeratosis, parakeratosis, pseudoepitheliomatous hyperplasia, and follicular plugs in the epidermis, as well as a mixed cellular infiltrate with Langhans giant cells and sclerotic bodies in the dermis (Figure 2). Periodic acid–Schiff and methenamine silver special stains revealed sclerotic bodies.
 in the dermis (H&E, original magnification ×100).")
Fungal culture on Sabouraud dextrose agar at 25°C and 37°C grew olive green, rugose, velvety, leathery colonies within 48 hours, with pigmentation front and reverse (Figure 3). A panfungal polymerase chain reaction assay was positive. Direct microscopic examination of a 10% potassium hydroxide mount of the colonies showed mycelia with dematiaceous septate hyphae (Figure 4), apical branching, branching conidiophores, elliptical conidia in long chains, and pathognomonic round yeastlike bodies resembling copper pennies known as sclerotic cells (also called muriform cells and medlar bodies).1,2 The causative organism was identified as Cladosporium carrionii. A final diagnosis of chromoblastomycosis was made.
 grew olive green, rugose, velvety, leathery colonies within 48 hours.")
.")
After 2 months of treatment with oral itraconazole 400 mg daily, there was no notable clinical improvement and fungal elements were still seen on culture. Four treatment cycles of intravenous liposomal amphotericin B 50 mg daily (1 mg/kg daily) for 15 days followed by itraconazole 200 mg daily for another 15 days caused substantial reduction and flattening of the lesion on the right side and resolution of the lesions on the left side. Healing was accompanied by central erythema and depigmentation (Figure 5). With a suspicion of continuing C carrionii activity on the right cheek, intralesional liposomal amphotericin B 0.2 mL (in a dilution of 5 mg in 1 mL) was given weekly in the peripheral hyperpigmented raised margin, which resulted in further flattening and reduction in tissue resistance. Fungal elements were absent on repeat biopsy and culture after 4 weeks.

Six months after negative culture, further cosmetic correction of the scar on the right cheek was performed with a patterned full-thickness graft for the upper half and excision with approximation of the edges for the lower half (Figure 6). Cultures have been negative for the last 20 months; as of this writing, there has been no recurrence of lesions.

Comment
Distribution
Chromoblastomycosis, also known as chromomycosis and verrucous dermatitis,3 is a chronic subcutaneous mycosis found in tropical and subtropical regions.3,4 It is caused by traumatic inoculation of any of several members of a specific group of dematiaceous fungi through the skin.2,3 Common causative organisms include Fonsecaea pedrosoi, C carrionii, Fonsecaea compacta, and Phialophora verrucosa, all of which are saprophytes in soil and plants. Fonsecaea pedrosoi is the most common causative agent worldwide (70%–90% of cases).2Cladosporium carrionii tends to be the predominant pathogen isolated in patients who present in drier climates, with F pedrosoi in humid forests.1-4
In India, chromoblastomycosis has been reported from the sub-Himalayan belt and western and eastern coasts.1,5 Our patient resided in Ahmednagar, Maharashtra, India, which has a predominantly hot and dry climate. The history might include vegetational trauma, such as a thorn prick. Time between inoculation and development of disease is believed to be years.
Clinical Presentation
Chromoblastomycosis is characterized by a slowly enlarging lesion at the site of inoculation. Five morphological variants are known: nodular, tumoral, verrucous, plaque, and cicatricial; verrucous and nodular types are most common.3,4
The disease is limited to the skin and subcutaneous tissue, growing in extent rather than in depth and not directly invading muscle or bone.4 Lymphatic and hematogenous dissemination can occur.3,4 Secondary bacterial infection is common. The most common affected site is the lower limb, especially the foot.1,3 The upper limb and rarely the ear, trunk, face, and breast can be affected.
Diagnosis
Routine laboratory investigations are usually within reference range. Diagnosis is made by histopathological and mycological studies. Preferably, scrapings or biopsy material are taken from lesions that are covered with what is described as “black dots” (an area of transepidermal elimination of the fungus) where there is a better diagnostic yield.2-4 Routine histopathology shows hyperkeratosis, pseudoepitheliomatous hyperplasia of the epidermis, a mixed granulomatous neutrophil response with multinucleated giant cells and neutrophil abscesses, refractile fungal spores, typical sclerotic cells around abscesses or granulomas, and a dense fibrous response in the dermis and subcutaneous tissue.
Extensive fibrosis, coupled with a chronic inflammatory infiltrate and increased susceptibility to secondary infection, leads to obstruction of lymphatic flow and lymphedema below the affected site.2-4 Periodic acid–Schiff and Gomori methenamine silver stains confirm the presence of fungus. Direct microscopic examination of a 10% potassium hydroxide mount of scrapings reveals spherical, thick-walled, darkly pigmented, multiseptate sclerotic cells known as medlar bodies, copper pennies, and muriform cells that are pathognomonic for chromoblastomycosis.1-4Cladosporium carrionii culture on Sabouraud dextrose agar at 37°C shows olive green, dark, rugose, smooth, hairy, leathery or velvety colonies with pigmentation front and reverse. Direct microscopic examination of the colonies shows dematiaceous septate hyphae and sparsely branching conidiophores bearing ellipsoidal, smooth-walled conidia in long acropetal chains.1,4
Treatment
Treatment options for chromoblastomycosis can be divided into antifungal agents and physical methods.Antifungal agents include itraconazole (200–400 mg daily),3 terbinafine (250–500 mg daily),3 5-fluorocytosine (100–150 mg/kg daily),3 amphotericin B (intravenous/intralesional), and others (eg, fluconazole, ketoconazole, posaconazole [800 mg daily],6,7 potassium iodide, voriconazole). Physical methods include CO2 laser, cryosurgery, local heat therapy, Mohs micrographic surgery, and standard surgery.3 There is no evidence-based treatment protocol. Itraconazole and terbinafine are considered drugs of first choice1,8; however, combination therapy is the best option.9
- Ajanta S, Naba KH, Deepak G. Chromoblastomycosis in sub-tropical regions of India. Mycopathologia. 2010;169:381-386.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Flavio QT, Phillippe E, Maigualida PB, et al. Chromoblastomycosis: an overview of clinical manifestations, diagnosis and treatment. Med Mycol. 2009;47:3-15.
- López Martínez R, Méndez Tovar LJ. Chromoblastomycosis. Clin Dermatol. 2007;25:188-194.
- Pradhan SV, Talwar OP, Ghosh A, et al. Chromoblastomycosis in Nepal: a study of 13 cases. Indian J Dermatol Venereol Leprol. 2007;73:176-178.
- Krzys´ciak PM, Pindycka-Piaszczys´ska M, Piaszczys´ski M. Chromoblastomycosis [published online October 22, 2014]. Postepy Dermatol Alergol. 2014;31:310-321.
- Negroni R, Tobón A, Bustamante B, et al. Posaconazole treatment of refractory eumycetoma and chromoblastomycosis. Rev Inst Med Trop Sao Paulo. 2005;47:339-346.
- Mohanty L, Mohanty P, Padhi T, et al. Verrucous growth on leg. Indian J Dermatol Venereol Leprol. 2006;72:399-400.
- Najafzadeh MJ, Rezusta A, Cameo MI, et al. Successful treatment of chromoblastomycosis of 36 years duration caused by Fonsecaea monophora. Med Mycol. 2010;48:390-393.
Case Report
A 25-year-old man who was a dairy farmer in Ahmednagar, Maharashtra, India, presented with a history of slowly growing, occasionally itchy lesions on both cheeks of 20 years’ duration. Most of the right cheek was covered by a well-defined, lobulated, gray-brown verrucous mass with a cerebriform surface (Figure 1). The left cheek was covered with a gray-brown infiltrated plaque surrounded by brown-tinged monomorphic papules.
Routine investigations were normal at presentation. Tests for purified protein derivative (tuberculin) and antibodies to human immunodeficiency virus were negative. Magnetic resonance imaging of the head showed soft tissue thickening with ulcerations involving the skin, subcutaneous tissue, and underlying facial muscles of the right cheek.
On histopathology, a hematoxylin and eosin–stained section showed hyperkeratosis, parakeratosis, pseudoepitheliomatous hyperplasia, and follicular plugs in the epidermis, as well as a mixed cellular infiltrate with Langhans giant cells and sclerotic bodies in the dermis (Figure 2). Periodic acid–Schiff and methenamine silver special stains revealed sclerotic bodies.
Fungal culture on Sabouraud dextrose agar at 25°C and 37°C grew olive green, rugose, velvety, leathery colonies within 48 hours, with pigmentation front and reverse (Figure 3). A panfungal polymerase chain reaction assay was positive. Direct microscopic examination of a 10% potassium hydroxide mount of the colonies showed mycelia with dematiaceous septate hyphae (Figure 4), apical branching, branching conidiophores, elliptical conidia in long chains, and pathognomonic round yeastlike bodies resembling copper pennies known as sclerotic cells (also called muriform cells and medlar bodies).1,2 The causative organism was identified as Cladosporium carrionii. A final diagnosis of chromoblastomycosis was made.
After 2 months of treatment with oral itraconazole 400 mg daily, there was no notable clinical improvement and fungal elements were still seen on culture. Four treatment cycles of intravenous liposomal amphotericin B 50 mg daily (1 mg/kg daily) for 15 days followed by itraconazole 200 mg daily for another 15 days caused substantial reduction and flattening of the lesion on the right side and resolution of the lesions on the left side. Healing was accompanied by central erythema and depigmentation (Figure 5). With a suspicion of continuing C carrionii activity on the right cheek, intralesional liposomal amphotericin B 0.2 mL (in a dilution of 5 mg in 1 mL) was given weekly in the peripheral hyperpigmented raised margin, which resulted in further flattening and reduction in tissue resistance. Fungal elements were absent on repeat biopsy and culture after 4 weeks.
Six months after negative culture, further cosmetic correction of the scar on the right cheek was performed with a patterned full-thickness graft for the upper half and excision with approximation of the edges for the lower half (Figure 6). Cultures have been negative for the last 20 months; as of this writing, there has been no recurrence of lesions.
Comment
Distribution
Chromoblastomycosis, also known as chromomycosis and verrucous dermatitis,3 is a chronic subcutaneous mycosis found in tropical and subtropical regions.3,4 It is caused by traumatic inoculation of any of several members of a specific group of dematiaceous fungi through the skin.2,3 Common causative organisms include Fonsecaea pedrosoi, C carrionii, Fonsecaea compacta, and Phialophora verrucosa, all of which are saprophytes in soil and plants. Fonsecaea pedrosoi is the most common causative agent worldwide (70%–90% of cases).2Cladosporium carrionii tends to be the predominant pathogen isolated in patients who present in drier climates, with F pedrosoi in humid forests.1-4
In India, chromoblastomycosis has been reported from the sub-Himalayan belt and western and eastern coasts.1,5 Our patient resided in Ahmednagar, Maharashtra, India, which has a predominantly hot and dry climate. The history might include vegetational trauma, such as a thorn prick. Time between inoculation and development of disease is believed to be years.
Clinical Presentation
Chromoblastomycosis is characterized by a slowly enlarging lesion at the site of inoculation. Five morphological variants are known: nodular, tumoral, verrucous, plaque, and cicatricial; verrucous and nodular types are most common.3,4
The disease is limited to the skin and subcutaneous tissue, growing in extent rather than in depth and not directly invading muscle or bone.4 Lymphatic and hematogenous dissemination can occur.3,4 Secondary bacterial infection is common. The most common affected site is the lower limb, especially the foot.1,3 The upper limb and rarely the ear, trunk, face, and breast can be affected.
Diagnosis
Routine laboratory investigations are usually within reference range. Diagnosis is made by histopathological and mycological studies. Preferably, scrapings or biopsy material are taken from lesions that are covered with what is described as “black dots” (an area of transepidermal elimination of the fungus) where there is a better diagnostic yield.2-4 Routine histopathology shows hyperkeratosis, pseudoepitheliomatous hyperplasia of the epidermis, a mixed granulomatous neutrophil response with multinucleated giant cells and neutrophil abscesses, refractile fungal spores, typical sclerotic cells around abscesses or granulomas, and a dense fibrous response in the dermis and subcutaneous tissue.
Extensive fibrosis, coupled with a chronic inflammatory infiltrate and increased susceptibility to secondary infection, leads to obstruction of lymphatic flow and lymphedema below the affected site.2-4 Periodic acid–Schiff and Gomori methenamine silver stains confirm the presence of fungus. Direct microscopic examination of a 10% potassium hydroxide mount of scrapings reveals spherical, thick-walled, darkly pigmented, multiseptate sclerotic cells known as medlar bodies, copper pennies, and muriform cells that are pathognomonic for chromoblastomycosis.1-4Cladosporium carrionii culture on Sabouraud dextrose agar at 37°C shows olive green, dark, rugose, smooth, hairy, leathery or velvety colonies with pigmentation front and reverse. Direct microscopic examination of the colonies shows dematiaceous septate hyphae and sparsely branching conidiophores bearing ellipsoidal, smooth-walled conidia in long acropetal chains.1,4
Treatment
Treatment options for chromoblastomycosis can be divided into antifungal agents and physical methods.Antifungal agents include itraconazole (200–400 mg daily),3 terbinafine (250–500 mg daily),3 5-fluorocytosine (100–150 mg/kg daily),3 amphotericin B (intravenous/intralesional), and others (eg, fluconazole, ketoconazole, posaconazole [800 mg daily],6,7 potassium iodide, voriconazole). Physical methods include CO2 laser, cryosurgery, local heat therapy, Mohs micrographic surgery, and standard surgery.3 There is no evidence-based treatment protocol. Itraconazole and terbinafine are considered drugs of first choice1,8; however, combination therapy is the best option.9
Case Report
A 25-year-old man who was a dairy farmer in Ahmednagar, Maharashtra, India, presented with a history of slowly growing, occasionally itchy lesions on both cheeks of 20 years’ duration. Most of the right cheek was covered by a well-defined, lobulated, gray-brown verrucous mass with a cerebriform surface (Figure 1). The left cheek was covered with a gray-brown infiltrated plaque surrounded by brown-tinged monomorphic papules.
Routine investigations were normal at presentation. Tests for purified protein derivative (tuberculin) and antibodies to human immunodeficiency virus were negative. Magnetic resonance imaging of the head showed soft tissue thickening with ulcerations involving the skin, subcutaneous tissue, and underlying facial muscles of the right cheek.
On histopathology, a hematoxylin and eosin–stained section showed hyperkeratosis, parakeratosis, pseudoepitheliomatous hyperplasia, and follicular plugs in the epidermis, as well as a mixed cellular infiltrate with Langhans giant cells and sclerotic bodies in the dermis (Figure 2). Periodic acid–Schiff and methenamine silver special stains revealed sclerotic bodies.
Fungal culture on Sabouraud dextrose agar at 25°C and 37°C grew olive green, rugose, velvety, leathery colonies within 48 hours, with pigmentation front and reverse (Figure 3). A panfungal polymerase chain reaction assay was positive. Direct microscopic examination of a 10% potassium hydroxide mount of the colonies showed mycelia with dematiaceous septate hyphae (Figure 4), apical branching, branching conidiophores, elliptical conidia in long chains, and pathognomonic round yeastlike bodies resembling copper pennies known as sclerotic cells (also called muriform cells and medlar bodies).1,2 The causative organism was identified as Cladosporium carrionii. A final diagnosis of chromoblastomycosis was made.
After 2 months of treatment with oral itraconazole 400 mg daily, there was no notable clinical improvement and fungal elements were still seen on culture. Four treatment cycles of intravenous liposomal amphotericin B 50 mg daily (1 mg/kg daily) for 15 days followed by itraconazole 200 mg daily for another 15 days caused substantial reduction and flattening of the lesion on the right side and resolution of the lesions on the left side. Healing was accompanied by central erythema and depigmentation (Figure 5). With a suspicion of continuing C carrionii activity on the right cheek, intralesional liposomal amphotericin B 0.2 mL (in a dilution of 5 mg in 1 mL) was given weekly in the peripheral hyperpigmented raised margin, which resulted in further flattening and reduction in tissue resistance. Fungal elements were absent on repeat biopsy and culture after 4 weeks.
Six months after negative culture, further cosmetic correction of the scar on the right cheek was performed with a patterned full-thickness graft for the upper half and excision with approximation of the edges for the lower half (Figure 6). Cultures have been negative for the last 20 months; as of this writing, there has been no recurrence of lesions.
Comment
Distribution
Chromoblastomycosis, also known as chromomycosis and verrucous dermatitis,3 is a chronic subcutaneous mycosis found in tropical and subtropical regions.3,4 It is caused by traumatic inoculation of any of several members of a specific group of dematiaceous fungi through the skin.2,3 Common causative organisms include Fonsecaea pedrosoi, C carrionii, Fonsecaea compacta, and Phialophora verrucosa, all of which are saprophytes in soil and plants. Fonsecaea pedrosoi is the most common causative agent worldwide (70%–90% of cases).2Cladosporium carrionii tends to be the predominant pathogen isolated in patients who present in drier climates, with F pedrosoi in humid forests.1-4
In India, chromoblastomycosis has been reported from the sub-Himalayan belt and western and eastern coasts.1,5 Our patient resided in Ahmednagar, Maharashtra, India, which has a predominantly hot and dry climate. The history might include vegetational trauma, such as a thorn prick. Time between inoculation and development of disease is believed to be years.
Clinical Presentation
Chromoblastomycosis is characterized by a slowly enlarging lesion at the site of inoculation. Five morphological variants are known: nodular, tumoral, verrucous, plaque, and cicatricial; verrucous and nodular types are most common.3,4
The disease is limited to the skin and subcutaneous tissue, growing in extent rather than in depth and not directly invading muscle or bone.4 Lymphatic and hematogenous dissemination can occur.3,4 Secondary bacterial infection is common. The most common affected site is the lower limb, especially the foot.1,3 The upper limb and rarely the ear, trunk, face, and breast can be affected.
Diagnosis
Routine laboratory investigations are usually within reference range. Diagnosis is made by histopathological and mycological studies. Preferably, scrapings or biopsy material are taken from lesions that are covered with what is described as “black dots” (an area of transepidermal elimination of the fungus) where there is a better diagnostic yield.2-4 Routine histopathology shows hyperkeratosis, pseudoepitheliomatous hyperplasia of the epidermis, a mixed granulomatous neutrophil response with multinucleated giant cells and neutrophil abscesses, refractile fungal spores, typical sclerotic cells around abscesses or granulomas, and a dense fibrous response in the dermis and subcutaneous tissue.
Extensive fibrosis, coupled with a chronic inflammatory infiltrate and increased susceptibility to secondary infection, leads to obstruction of lymphatic flow and lymphedema below the affected site.2-4 Periodic acid–Schiff and Gomori methenamine silver stains confirm the presence of fungus. Direct microscopic examination of a 10% potassium hydroxide mount of scrapings reveals spherical, thick-walled, darkly pigmented, multiseptate sclerotic cells known as medlar bodies, copper pennies, and muriform cells that are pathognomonic for chromoblastomycosis.1-4Cladosporium carrionii culture on Sabouraud dextrose agar at 37°C shows olive green, dark, rugose, smooth, hairy, leathery or velvety colonies with pigmentation front and reverse. Direct microscopic examination of the colonies shows dematiaceous septate hyphae and sparsely branching conidiophores bearing ellipsoidal, smooth-walled conidia in long acropetal chains.1,4
Treatment
Treatment options for chromoblastomycosis can be divided into antifungal agents and physical methods.Antifungal agents include itraconazole (200–400 mg daily),3 terbinafine (250–500 mg daily),3 5-fluorocytosine (100–150 mg/kg daily),3 amphotericin B (intravenous/intralesional), and others (eg, fluconazole, ketoconazole, posaconazole [800 mg daily],6,7 potassium iodide, voriconazole). Physical methods include CO2 laser, cryosurgery, local heat therapy, Mohs micrographic surgery, and standard surgery.3 There is no evidence-based treatment protocol. Itraconazole and terbinafine are considered drugs of first choice1,8; however, combination therapy is the best option.9
- Ajanta S, Naba KH, Deepak G. Chromoblastomycosis in sub-tropical regions of India. Mycopathologia. 2010;169:381-386.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Flavio QT, Phillippe E, Maigualida PB, et al. Chromoblastomycosis: an overview of clinical manifestations, diagnosis and treatment. Med Mycol. 2009;47:3-15.
- López Martínez R, Méndez Tovar LJ. Chromoblastomycosis. Clin Dermatol. 2007;25:188-194.
- Pradhan SV, Talwar OP, Ghosh A, et al. Chromoblastomycosis in Nepal: a study of 13 cases. Indian J Dermatol Venereol Leprol. 2007;73:176-178.
- Krzys´ciak PM, Pindycka-Piaszczys´ska M, Piaszczys´ski M. Chromoblastomycosis [published online October 22, 2014]. Postepy Dermatol Alergol. 2014;31:310-321.
- Negroni R, Tobón A, Bustamante B, et al. Posaconazole treatment of refractory eumycetoma and chromoblastomycosis. Rev Inst Med Trop Sao Paulo. 2005;47:339-346.
- Mohanty L, Mohanty P, Padhi T, et al. Verrucous growth on leg. Indian J Dermatol Venereol Leprol. 2006;72:399-400.
- Najafzadeh MJ, Rezusta A, Cameo MI, et al. Successful treatment of chromoblastomycosis of 36 years duration caused by Fonsecaea monophora. Med Mycol. 2010;48:390-393.
- Ajanta S, Naba KH, Deepak G. Chromoblastomycosis in sub-tropical regions of India. Mycopathologia. 2010;169:381-386.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Flavio QT, Phillippe E, Maigualida PB, et al. Chromoblastomycosis: an overview of clinical manifestations, diagnosis and treatment. Med Mycol. 2009;47:3-15.
- López Martínez R, Méndez Tovar LJ. Chromoblastomycosis. Clin Dermatol. 2007;25:188-194.
- Pradhan SV, Talwar OP, Ghosh A, et al. Chromoblastomycosis in Nepal: a study of 13 cases. Indian J Dermatol Venereol Leprol. 2007;73:176-178.
- Krzys´ciak PM, Pindycka-Piaszczys´ska M, Piaszczys´ski M. Chromoblastomycosis [published online October 22, 2014]. Postepy Dermatol Alergol. 2014;31:310-321.
- Negroni R, Tobón A, Bustamante B, et al. Posaconazole treatment of refractory eumycetoma and chromoblastomycosis. Rev Inst Med Trop Sao Paulo. 2005;47:339-346.
- Mohanty L, Mohanty P, Padhi T, et al. Verrucous growth on leg. Indian J Dermatol Venereol Leprol. 2006;72:399-400.
- Najafzadeh MJ, Rezusta A, Cameo MI, et al. Successful treatment of chromoblastomycosis of 36 years duration caused by Fonsecaea monophora. Med Mycol. 2010;48:390-393.
Practice Points
- Chromoblastomycosis is limited to skin and subcutaneous tissue, most commonly of the lower limb, especially the foot; it does not directly invade muscle or bone. Secondary bacterial infection is common.
- Chromoblastomycosis is a therapeutic challenge due to its recalcitrant nature. Itraconazole and terbinafine are considered drugs of first choice, but consensus and evidence are lacking on a standard of care.
Marijuana’s perceived approval ratings on the rise
Parents’ disapproval of marijuana use has dropped since 1979 – at least that’s what their teenage children say, according to results of the 2017 Monitoring the Future survey.
The approximately 13,500 12th graders involved in the 2017 survey believe that their parents are much less likely to disapprove of marijuana use, compared with the students who responded to the survey during 1976-1979. At that time, 15% of the 12th graders said that their parents would not disapprove of using marijuana once or twice, but by 2017 the number had made a statistically significant rise to 23%, Richard A. Miech, PhD, and his associates said in their report on the 2017 survey.
Perceived approval of occasional marijuana use, which had garnered only an 8% share of respondents in 1976-1979, was up to a significantly higher 17% in 2017, and regular use went from 4% to 13%, said Dr. Miech and his associates, of the University of Michigan Institute for Social Research, Ann Arbor.
Parents’ increased acceptance of marijuana, as perceived by the 12th-grade students, was not matched for other substances. Disapproval for smoking one or more packs of cigarettes a day, for example, climbed from 89% in 1976-1979 to 92% in 2017, while disapproval of weekend binge drinking rose just a bit, going from 85% to 86%, they said.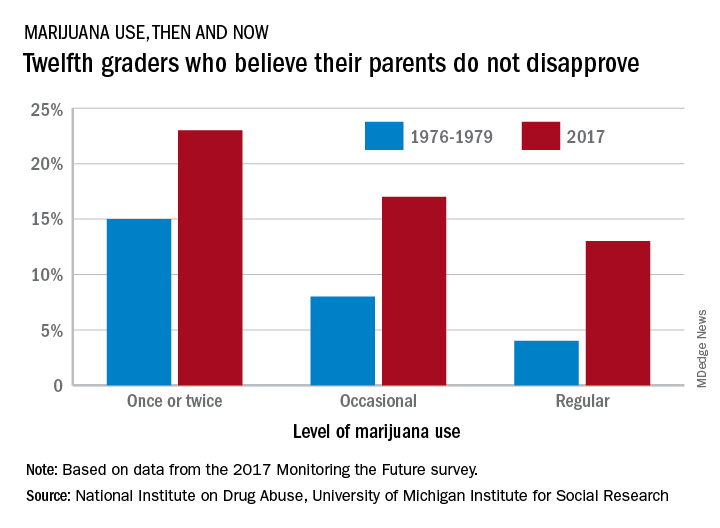
Measures of parental disapproval were reintroduced into the survey in 2017 after being removed in 1979 – the survey began in 1975 – “because students’ responses varied little over time and across drugs,” Dr. Miech and his associates noted. “Today’s parents of 12th graders have more experience with drug use than did parents in the late 1970s [and] population attitudes toward marijuana use across all ages are becoming more lenient,” they wrote.
The 2017 edition of the annual survey, which is funded by the National Institute on Drug Abuse, is based on reports from almost 44,000 students in 8th, 10th, and 12th grade in 360 public and private secondary schools across the country.
Parents’ disapproval of marijuana use has dropped since 1979 – at least that’s what their teenage children say, according to results of the 2017 Monitoring the Future survey.
The approximately 13,500 12th graders involved in the 2017 survey believe that their parents are much less likely to disapprove of marijuana use, compared with the students who responded to the survey during 1976-1979. At that time, 15% of the 12th graders said that their parents would not disapprove of using marijuana once or twice, but by 2017 the number had made a statistically significant rise to 23%, Richard A. Miech, PhD, and his associates said in their report on the 2017 survey.
Perceived approval of occasional marijuana use, which had garnered only an 8% share of respondents in 1976-1979, was up to a significantly higher 17% in 2017, and regular use went from 4% to 13%, said Dr. Miech and his associates, of the University of Michigan Institute for Social Research, Ann Arbor.
Parents’ increased acceptance of marijuana, as perceived by the 12th-grade students, was not matched for other substances. Disapproval for smoking one or more packs of cigarettes a day, for example, climbed from 89% in 1976-1979 to 92% in 2017, while disapproval of weekend binge drinking rose just a bit, going from 85% to 86%, they said.
Measures of parental disapproval were reintroduced into the survey in 2017 after being removed in 1979 – the survey began in 1975 – “because students’ responses varied little over time and across drugs,” Dr. Miech and his associates noted. “Today’s parents of 12th graders have more experience with drug use than did parents in the late 1970s [and] population attitudes toward marijuana use across all ages are becoming more lenient,” they wrote.
The 2017 edition of the annual survey, which is funded by the National Institute on Drug Abuse, is based on reports from almost 44,000 students in 8th, 10th, and 12th grade in 360 public and private secondary schools across the country.
Parents’ disapproval of marijuana use has dropped since 1979 – at least that’s what their teenage children say, according to results of the 2017 Monitoring the Future survey.
The approximately 13,500 12th graders involved in the 2017 survey believe that their parents are much less likely to disapprove of marijuana use, compared with the students who responded to the survey during 1976-1979. At that time, 15% of the 12th graders said that their parents would not disapprove of using marijuana once or twice, but by 2017 the number had made a statistically significant rise to 23%, Richard A. Miech, PhD, and his associates said in their report on the 2017 survey.
Perceived approval of occasional marijuana use, which had garnered only an 8% share of respondents in 1976-1979, was up to a significantly higher 17% in 2017, and regular use went from 4% to 13%, said Dr. Miech and his associates, of the University of Michigan Institute for Social Research, Ann Arbor.
Parents’ increased acceptance of marijuana, as perceived by the 12th-grade students, was not matched for other substances. Disapproval for smoking one or more packs of cigarettes a day, for example, climbed from 89% in 1976-1979 to 92% in 2017, while disapproval of weekend binge drinking rose just a bit, going from 85% to 86%, they said.
Measures of parental disapproval were reintroduced into the survey in 2017 after being removed in 1979 – the survey began in 1975 – “because students’ responses varied little over time and across drugs,” Dr. Miech and his associates noted. “Today’s parents of 12th graders have more experience with drug use than did parents in the late 1970s [and] population attitudes toward marijuana use across all ages are becoming more lenient,” they wrote.
The 2017 edition of the annual survey, which is funded by the National Institute on Drug Abuse, is based on reports from almost 44,000 students in 8th, 10th, and 12th grade in 360 public and private secondary schools across the country.
Two agents could take AML therapy in new directions
CHICAGO—Two agents targeting novel pathways in myeloid malignancies—mivebresib and bencentinib—are showing promise in early studies, according to a speaker at the 2018 ASCO Annual Meeting.
“Both BET and AXL inhibition appear to be new and exciting targets in myeloid malignancies,” said Alice S. Mims, MD, and both have produced responses as single agents.
Dr Mims, of Ohio State University Wexner Medical Center in Columbus, made these observations in a poster discussion presentation that included commentary on the two agents.
Mivebresib (ABBV-075), an inhibitor of bromodomain and extra terminal (BET) proteins, yielded some responses in relapsed/refractory acute myeloid leukemia (AML) patients in a first-in-human study presented at the meeting (abstract 7019*).
Bemcentinib (BGB324), a first-in class selective inhibitor of the AXL tyrosine kinase, also showed activity in preliminary results of a study including patients with relapsed/refractory disease (abstract 7020*).
“It will be important to know individual patient characteristics to determine the potential response predictors,” Dr Mims said.
Mivebresib (NCT02391480)
Mivebresib is the subject of an ongoing phase 1 dose-escalation study in which 23 patients have been treated. That includes 12 who received the BET inhibitor as monotherapy, and 11 who got it in combination with the BCL-2 inhibitor venetoclax, which is indicated in CLL and has breakthrough therapy designation for AML.
Investigators observed responses in 3 of 17 evaluable patients (17.6%), including 1 complete remission with incomplete blood count recovery in a patient on mivebresib monotherapy, plus 1 partial response and 1 patient achieving a morphologic leukemia-free state with the combination.
The most common grade 3/4 treatment-emergent adverse events included anemia in 52%, thrombocytopenia in 44%, and febrile neutropenia in 26% of patients, with no dose-limiting toxicities noted as of this report.
Bemcentinib (NCT02488408)
Bemcentinib is being evaluated in a phase 1/2 trial including patients with relapsed/refractory AML and myelodysplastic syndromes (MDS).
For 32 patients treated so far, 3 patients achieved a complete remission, including 1 AML and 2 MDS patients.
In addition, 3 patients achieved partial response, including 1 MDS and 2 AML patients.
Treatment with bemcentinib was generally well-tolerated, and most adverse events were mild or moderate, investigators reported in their poster.
Pre-treatment levels of soluble AXL were lower in responders compared with non-responders, investigators also noted.
“Soluble AXL levels may be a predictive biomarker for AXL inhibition, but further assessment is necessary,” Dr Mims said.
*Data presented at the meeting differ from the abstracts.
CHICAGO—Two agents targeting novel pathways in myeloid malignancies—mivebresib and bencentinib—are showing promise in early studies, according to a speaker at the 2018 ASCO Annual Meeting.
“Both BET and AXL inhibition appear to be new and exciting targets in myeloid malignancies,” said Alice S. Mims, MD, and both have produced responses as single agents.
Dr Mims, of Ohio State University Wexner Medical Center in Columbus, made these observations in a poster discussion presentation that included commentary on the two agents.
Mivebresib (ABBV-075), an inhibitor of bromodomain and extra terminal (BET) proteins, yielded some responses in relapsed/refractory acute myeloid leukemia (AML) patients in a first-in-human study presented at the meeting (abstract 7019*).
Bemcentinib (BGB324), a first-in class selective inhibitor of the AXL tyrosine kinase, also showed activity in preliminary results of a study including patients with relapsed/refractory disease (abstract 7020*).
“It will be important to know individual patient characteristics to determine the potential response predictors,” Dr Mims said.
Mivebresib (NCT02391480)
Mivebresib is the subject of an ongoing phase 1 dose-escalation study in which 23 patients have been treated. That includes 12 who received the BET inhibitor as monotherapy, and 11 who got it in combination with the BCL-2 inhibitor venetoclax, which is indicated in CLL and has breakthrough therapy designation for AML.
Investigators observed responses in 3 of 17 evaluable patients (17.6%), including 1 complete remission with incomplete blood count recovery in a patient on mivebresib monotherapy, plus 1 partial response and 1 patient achieving a morphologic leukemia-free state with the combination.
The most common grade 3/4 treatment-emergent adverse events included anemia in 52%, thrombocytopenia in 44%, and febrile neutropenia in 26% of patients, with no dose-limiting toxicities noted as of this report.
Bemcentinib (NCT02488408)
Bemcentinib is being evaluated in a phase 1/2 trial including patients with relapsed/refractory AML and myelodysplastic syndromes (MDS).
For 32 patients treated so far, 3 patients achieved a complete remission, including 1 AML and 2 MDS patients.
In addition, 3 patients achieved partial response, including 1 MDS and 2 AML patients.
Treatment with bemcentinib was generally well-tolerated, and most adverse events were mild or moderate, investigators reported in their poster.
Pre-treatment levels of soluble AXL were lower in responders compared with non-responders, investigators also noted.
“Soluble AXL levels may be a predictive biomarker for AXL inhibition, but further assessment is necessary,” Dr Mims said.
*Data presented at the meeting differ from the abstracts.
CHICAGO—Two agents targeting novel pathways in myeloid malignancies—mivebresib and bencentinib—are showing promise in early studies, according to a speaker at the 2018 ASCO Annual Meeting.
“Both BET and AXL inhibition appear to be new and exciting targets in myeloid malignancies,” said Alice S. Mims, MD, and both have produced responses as single agents.
Dr Mims, of Ohio State University Wexner Medical Center in Columbus, made these observations in a poster discussion presentation that included commentary on the two agents.
Mivebresib (ABBV-075), an inhibitor of bromodomain and extra terminal (BET) proteins, yielded some responses in relapsed/refractory acute myeloid leukemia (AML) patients in a first-in-human study presented at the meeting (abstract 7019*).
Bemcentinib (BGB324), a first-in class selective inhibitor of the AXL tyrosine kinase, also showed activity in preliminary results of a study including patients with relapsed/refractory disease (abstract 7020*).
“It will be important to know individual patient characteristics to determine the potential response predictors,” Dr Mims said.
Mivebresib (NCT02391480)
Mivebresib is the subject of an ongoing phase 1 dose-escalation study in which 23 patients have been treated. That includes 12 who received the BET inhibitor as monotherapy, and 11 who got it in combination with the BCL-2 inhibitor venetoclax, which is indicated in CLL and has breakthrough therapy designation for AML.
Investigators observed responses in 3 of 17 evaluable patients (17.6%), including 1 complete remission with incomplete blood count recovery in a patient on mivebresib monotherapy, plus 1 partial response and 1 patient achieving a morphologic leukemia-free state with the combination.
The most common grade 3/4 treatment-emergent adverse events included anemia in 52%, thrombocytopenia in 44%, and febrile neutropenia in 26% of patients, with no dose-limiting toxicities noted as of this report.
Bemcentinib (NCT02488408)
Bemcentinib is being evaluated in a phase 1/2 trial including patients with relapsed/refractory AML and myelodysplastic syndromes (MDS).
For 32 patients treated so far, 3 patients achieved a complete remission, including 1 AML and 2 MDS patients.
In addition, 3 patients achieved partial response, including 1 MDS and 2 AML patients.
Treatment with bemcentinib was generally well-tolerated, and most adverse events were mild or moderate, investigators reported in their poster.
Pre-treatment levels of soluble AXL were lower in responders compared with non-responders, investigators also noted.
“Soluble AXL levels may be a predictive biomarker for AXL inhibition, but further assessment is necessary,” Dr Mims said.
*Data presented at the meeting differ from the abstracts.
Emicizumab granted priority review for hemophilia A without inhibitors
The US Food and Drug Administration (FDA) has granted priority review for emicizumab (Hemlibra®) for adults and children with hemophilia A without factor VIII inhibitors.
Earlier this year, the agency awarded emicizumab breakthrough therapy designation for the same population.
Emicizumab is a bispecific factor IXa- and factor X-directed antibody approved by the FDA for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children who have hemophilia A with factor VIII inhibitors.
The FDA based its decision to grant emicizumab priority review on the phase 3 HAVEN 3 study, results of which were presented recently at the World Federation of Hemophilia congress.
In HAVEN 3, emicizumab demonstrated a 68% reduction (P<0.0001) in treated bleeds based on an intra-patient comparison in patients who were previously enrolled in a prospective non-interventional study.
According to Genentech, co-developer of the drug, this makes emicizumab the first medicine to show superior efficacy to prior treatment with factor VIII prophylaxis, the current standard of care for people with hemophilia A without factor VIII inhibitors.
About HAVEN 3
The randomized, multicenter, open-label trial evaluated prophylaxis versus no prophylaxis in patients without factor VIII inhibitors.
The study included 152 patients 12 years or older who were previously treated with factor VIII therapy on-demand or as prophylaxis.
Patients previously treated with on-demand factor VIII were randomized in a 2:2:1 fashion to 1 of 3 treatment groups:
- Arm A received emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 1.5 mg/kg/wk until the end of study.
- Arm B received emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 3 mg/kg/2wks for at least 24 weeks.
- Arm C received no prophylaxis
Patients previously treated prophylactically with factor VIII were enrolled in Arm D and received emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 1.5 mg/kg/wk until the end of study.
The protocol permitted episodic treatment of breakthrough bleeds with factor VIII therapy.
Patients in the prophylaxis groups achieved a 96% (P<0.0001) and 97% (P<0.0001) reduction in treated bleeds, respectively, compared to those who received no prophylaxis.
Additionally, 55.6% of patients treated weekly and 60% treated every 2 weeks had no treated bleeds. In contrast, 0% in the prophylaxis group achieved zero treated bleeds.
Investigators observed no unexpected or serious adverse events (AEs), no thrombotic events, and no cases of thrombotic microangiopathy.
The most common AEs occurring in 5% or more of patients were injection site reactions, arthralgia, nasopharyngitis, headache, upper respiratory tract infection, and influenza.
The FDA is expected to make a decision regarding approval by October 4.
The US Food and Drug Administration (FDA) has granted priority review for emicizumab (Hemlibra®) for adults and children with hemophilia A without factor VIII inhibitors.
Earlier this year, the agency awarded emicizumab breakthrough therapy designation for the same population.
Emicizumab is a bispecific factor IXa- and factor X-directed antibody approved by the FDA for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children who have hemophilia A with factor VIII inhibitors.
The FDA based its decision to grant emicizumab priority review on the phase 3 HAVEN 3 study, results of which were presented recently at the World Federation of Hemophilia congress.
In HAVEN 3, emicizumab demonstrated a 68% reduction (P<0.0001) in treated bleeds based on an intra-patient comparison in patients who were previously enrolled in a prospective non-interventional study.
According to Genentech, co-developer of the drug, this makes emicizumab the first medicine to show superior efficacy to prior treatment with factor VIII prophylaxis, the current standard of care for people with hemophilia A without factor VIII inhibitors.
About HAVEN 3
The randomized, multicenter, open-label trial evaluated prophylaxis versus no prophylaxis in patients without factor VIII inhibitors.
The study included 152 patients 12 years or older who were previously treated with factor VIII therapy on-demand or as prophylaxis.
Patients previously treated with on-demand factor VIII were randomized in a 2:2:1 fashion to 1 of 3 treatment groups:
- Arm A received emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 1.5 mg/kg/wk until the end of study.
- Arm B received emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 3 mg/kg/2wks for at least 24 weeks.
- Arm C received no prophylaxis
Patients previously treated prophylactically with factor VIII were enrolled in Arm D and received emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 1.5 mg/kg/wk until the end of study.
The protocol permitted episodic treatment of breakthrough bleeds with factor VIII therapy.
Patients in the prophylaxis groups achieved a 96% (P<0.0001) and 97% (P<0.0001) reduction in treated bleeds, respectively, compared to those who received no prophylaxis.
Additionally, 55.6% of patients treated weekly and 60% treated every 2 weeks had no treated bleeds. In contrast, 0% in the prophylaxis group achieved zero treated bleeds.
Investigators observed no unexpected or serious adverse events (AEs), no thrombotic events, and no cases of thrombotic microangiopathy.
The most common AEs occurring in 5% or more of patients were injection site reactions, arthralgia, nasopharyngitis, headache, upper respiratory tract infection, and influenza.
The FDA is expected to make a decision regarding approval by October 4.
The US Food and Drug Administration (FDA) has granted priority review for emicizumab (Hemlibra®) for adults and children with hemophilia A without factor VIII inhibitors.
Earlier this year, the agency awarded emicizumab breakthrough therapy designation for the same population.
Emicizumab is a bispecific factor IXa- and factor X-directed antibody approved by the FDA for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children who have hemophilia A with factor VIII inhibitors.
The FDA based its decision to grant emicizumab priority review on the phase 3 HAVEN 3 study, results of which were presented recently at the World Federation of Hemophilia congress.
In HAVEN 3, emicizumab demonstrated a 68% reduction (P<0.0001) in treated bleeds based on an intra-patient comparison in patients who were previously enrolled in a prospective non-interventional study.
According to Genentech, co-developer of the drug, this makes emicizumab the first medicine to show superior efficacy to prior treatment with factor VIII prophylaxis, the current standard of care for people with hemophilia A without factor VIII inhibitors.
About HAVEN 3
The randomized, multicenter, open-label trial evaluated prophylaxis versus no prophylaxis in patients without factor VIII inhibitors.
The study included 152 patients 12 years or older who were previously treated with factor VIII therapy on-demand or as prophylaxis.
Patients previously treated with on-demand factor VIII were randomized in a 2:2:1 fashion to 1 of 3 treatment groups:
- Arm A received emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 1.5 mg/kg/wk until the end of study.
- Arm B received emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 3 mg/kg/2wks for at least 24 weeks.
- Arm C received no prophylaxis
Patients previously treated prophylactically with factor VIII were enrolled in Arm D and received emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 1.5 mg/kg/wk until the end of study.
The protocol permitted episodic treatment of breakthrough bleeds with factor VIII therapy.
Patients in the prophylaxis groups achieved a 96% (P<0.0001) and 97% (P<0.0001) reduction in treated bleeds, respectively, compared to those who received no prophylaxis.
Additionally, 55.6% of patients treated weekly and 60% treated every 2 weeks had no treated bleeds. In contrast, 0% in the prophylaxis group achieved zero treated bleeds.
Investigators observed no unexpected or serious adverse events (AEs), no thrombotic events, and no cases of thrombotic microangiopathy.
The most common AEs occurring in 5% or more of patients were injection site reactions, arthralgia, nasopharyngitis, headache, upper respiratory tract infection, and influenza.
The FDA is expected to make a decision regarding approval by October 4.