User login
Arboviral and other vector-borne diseases
May has arrived, and for the majority of your patients it signals the end of the school year and the beginning of summer vacation. Zika virus is on the minds of most people since its arrival to the Western Hemisphere in March 2015. With the fluidity of this outbreak and almost daily news updates and recommendations, many parents have voiced or will be voicing concerns regarding summer travel destinations.
Many concerns about Zika virus have been previously addressed in this column (“Zika virus: More questions than answers?” by Dr. Kristina Bryant). However, if the decision is to avoid international travel because of the ongoing Zika outbreak, it doesn’t mean your patients get a free pass and will not have to be concerned about acquiring any infectious diseases. They still need to be vigilant about avoiding those pesky vectors that transmit arboviruses and other vector-borne diseases that occur in the United States.

Arboviruses are transmitted by mosquitoes, ticks, or fleas. Most infections are subclinical. If symptoms develop, they are manifested by a generalized febrile illness including fever, headache, myalgia, arthralgia, and rash. Hemorrhagic fever (dengue) or neuroinvasive disease can include aseptic meningitis, encephalitis, or acute flaccid paralysis. Neuroinvasive disease rarely occurs with dengue, Colorado tick fever, and chikungunya infections.
While more than 100 arboviruses can cause infection, some of the more common arboviruses associated with human disease include West Nile, first detected in the United States in 1999 and chikungunya, first reported in the Americas in 2013 with local transmission documented in Florida, Puerto Rico, and the U.S. Virgin Islands in 2014. It is estimated that dengue causes over 100 million cases worldwide annually. Almost 40% of the world’s inhabitants live in endemic areas. The majority of cases on the U.S. mainland are imported. However, it is endemic in all U.S. territories including Guam, American Samoa, the U.S. Virgin Islands, and Puerto Rico. Between September 2015 and March 2016, Hawaii experienced a dengue outbreak involving 264 individuals including 46 children. As of April 16, 2016, there were no infectious individuals on the island.

Other domestic arboviruses causing disease include St. Louis, Eastern, and Western Equine encephalitis, La Crosse encephalitis, Colorado tick fever, and Powassan virus. All are transmitted by mosquitoes with the exception of Powassan and Colorado tick fever, which are transmitted by ticks. The numbers of cases nationally are much lower for these diseases, compared with West Nile, dengue, and chikungunya. National and state-specific information is available for domestic arboviruses at diseasemaps.usgs.gov/mapviewer. Data is compiled by ArboNET, a national arboviral surveillance system that is managed by the Centers for Disease Control and Prevention (CDC) in conjunction with state health departments. Not only is human disease monitored, but it also maintains data on viremic blood donors, dead birds, mosquitoes, veterinary disease cases, and sentinel animals.
Spring and summer are the most active seasons for ticks. Bacterial and spirochetal diseases transmitted by them include rickettsial diseases such as Rocky Mountain Spotted Fever, ehrlichiosis, and anaplasmosis. Tularemia in addition to Lyme and tick-borne relapsing fever are also transmitted by ticks. Babesiosis, which is due to a parasite, and southern tick-associated rash illness (STARI), whose causative agent is yet to be determined, are two additional tick-related diagnoses.
Of note, dengue, chikungunya, and Zika are all transmitted by infected Aedes mosquitoes. There is no enzootic cycle. Just human-mosquito-human transmission. In contrast, West Nile virus is transmitted by Culex mosquitoes in an enzootic cycle between an avian reservoir and humans.
Treatment
There is no specific treatment for arboviral infections. The primary goal is relief of symptoms with fluids, bed rest, and analgesics. For bacterial vector-borne diseases, antibiotic therapy is indicated and is based on the specific pathogen. Doxycycline is the drug of choice for treatment of suspected and confirmed Rocky Mountain Spotted Fever, ehrlichiosis, and anaplasmosis even in children less than 8 years of age. Delay in initiation of antimicrobial therapy pending definitive diagnosis may lead to an adverse outcome. It is also the drug of choice for tick-borne relapsing fever.
Lyme disease is also responsive to antibiotic treatment. Therapy is based on the disease category. (Lyme disease in “Red Book: 2015 Report of the Committee on Infectious Diseases,” [Elk Grove Village, Ill.: American Academy of Pediatrics, 2015, pp. 516-25]).
STARI clinically presents with a lesion that resembles erythema migrans in southern and southeastern states. However, it has not been associated with any of the complications reported with disseminated Lyme disease. Treatment is not recommended.
Tularemia and babesiosis are both responsive to antimicrobial therapy and would best be managed in consultation with an infectious disease physician.
A handy, concise, up to date reference guide about all of the tick-borne diseases including photographs is available at the App Store. The Tickborne Diseases App was developed by the CDC and it is free!
Prevention
The cornerstone of disease prevention is avoidance of mosquito and tick bites, in addition to eliminating mosquito breeding sites. Ticks are generally found near the ground, in brushy or wooded areas. They usually wait for a potential host to brush against them. When this happens, they climb onto the host and find a site to attach.
Is there a role for antimicrobial prophylaxis once a tick has been discovered? There is no data to support antimicrobial prophylaxis to prevent Rocky Mountain spotted fever, ehrlichiosis, and anaplasmosis. Prophylaxis with doxycycline or ciprofloxacin is recommended for children and adults after exposure to an intentional release of tularemia and for laboratory workers after inadvertent exposure. For prevention of Lyme disease, a single dose of doxycycline (4 mg/kg, max dose 200 mg) may be offered under limited conditions: The patient is at least 8 years of age, resides in an area where Lyme is highly endemic, the tick removed was engorged, therapy can be initiated within 72 hours after tick removal, and the estimated time of attachment was at least 36 hours. There is inadequate data on the use of amoxicillin.
Remember, not all mosquitoes are alike. Those that transmit chikungunya, dengue, and Zika (Aedes mosquitoes) are primarily daytime mosquitoes, but also can bite at night. West Nile is transmitted by Culex mosquitoes, which feed from dusk to dawn.
Here are some tips to share with your patients that should decrease their chances of acquiring a mosquito or tick-borne disease:
• Apply mosquito repellent only to intact exposed skin when outdoors. Most repellents can be safely used on children at least 2 months of age and older. Avoid applying repellent directly on the child’s hand. Use at least a 20% DEET (N,N-diethyl-meta-toluamide) containing product. Other Environmental Protection Agency–registered repellents are an alternative (Additional information is available at http://www2.epa.gov/insect-repellents). Products containing oil of lemon eucalyptus (OLE) or p-Menthane-3,8-diol (PMD) should not be used on children under 3 years of age.
• Apply permethrin to clothing, hats, boots, and so on. It is designed to repel mosquitoes and ticks. It can last for several washings. It is ideal to spray over nets covering carriers in children younger than 2 months of age.
• Wear long-sleeved shirts and long pants tucked inside of socks when hiking.
• Check for ticks daily, especially under the arms, behind the ears, around the waist, behind the knees, and inside belly buttons after outdoor activities.
• Have your patients learn how to effectively remove a tick. With a fine tipped tweezer, grasp the tick as close to the skin as possible and pull straight up with even pressure. Do not twist or jerk the tick. Do not squash the tick. Place it in a bag and dispose of it. Clean the site after removal with alcohol, iodine, or soap and water.
• Encourage families to mosquito proof their home by using screens on windows and doors, and using air conditioning when available.
• Empty and scrub all items that contain water such as birdbaths, planters, or wading pools around the outside of the home at least weekly because mosquitoes lay eggs in or near free standing water.
• Dogs and cats should be treated for ticks as recommended by the veterinarian.
The impact of the ongoing Zika virus outbreak is uncertain. While it may have an impact on those planning international travel now and in the near future, several arboviral and vector-borne diseases currently exist in the United States. Encouraging our patients to practice interventions to prevent mosquito and tick bites now will also serve to protect them if Zika virus becomes established in the Aedes mosquitoes here in the future and/or if they have plans for international travel. For up to date information on Zika virus for yourself and your patients, visit www.cdc.gov/zika.
Bonnie M. Word, M.D., is a pediatric infectious disease specialist and director of the Houston Travel Medicine Clinic. She said she had no relevant financial disclosures. Email Dr. Word at pdnews@frontlinemedcom.com.
May has arrived, and for the majority of your patients it signals the end of the school year and the beginning of summer vacation. Zika virus is on the minds of most people since its arrival to the Western Hemisphere in March 2015. With the fluidity of this outbreak and almost daily news updates and recommendations, many parents have voiced or will be voicing concerns regarding summer travel destinations.
Many concerns about Zika virus have been previously addressed in this column (“Zika virus: More questions than answers?” by Dr. Kristina Bryant). However, if the decision is to avoid international travel because of the ongoing Zika outbreak, it doesn’t mean your patients get a free pass and will not have to be concerned about acquiring any infectious diseases. They still need to be vigilant about avoiding those pesky vectors that transmit arboviruses and other vector-borne diseases that occur in the United States.
Arboviruses are transmitted by mosquitoes, ticks, or fleas. Most infections are subclinical. If symptoms develop, they are manifested by a generalized febrile illness including fever, headache, myalgia, arthralgia, and rash. Hemorrhagic fever (dengue) or neuroinvasive disease can include aseptic meningitis, encephalitis, or acute flaccid paralysis. Neuroinvasive disease rarely occurs with dengue, Colorado tick fever, and chikungunya infections.
While more than 100 arboviruses can cause infection, some of the more common arboviruses associated with human disease include West Nile, first detected in the United States in 1999 and chikungunya, first reported in the Americas in 2013 with local transmission documented in Florida, Puerto Rico, and the U.S. Virgin Islands in 2014. It is estimated that dengue causes over 100 million cases worldwide annually. Almost 40% of the world’s inhabitants live in endemic areas. The majority of cases on the U.S. mainland are imported. However, it is endemic in all U.S. territories including Guam, American Samoa, the U.S. Virgin Islands, and Puerto Rico. Between September 2015 and March 2016, Hawaii experienced a dengue outbreak involving 264 individuals including 46 children. As of April 16, 2016, there were no infectious individuals on the island.
Other domestic arboviruses causing disease include St. Louis, Eastern, and Western Equine encephalitis, La Crosse encephalitis, Colorado tick fever, and Powassan virus. All are transmitted by mosquitoes with the exception of Powassan and Colorado tick fever, which are transmitted by ticks. The numbers of cases nationally are much lower for these diseases, compared with West Nile, dengue, and chikungunya. National and state-specific information is available for domestic arboviruses at diseasemaps.usgs.gov/mapviewer. Data is compiled by ArboNET, a national arboviral surveillance system that is managed by the Centers for Disease Control and Prevention (CDC) in conjunction with state health departments. Not only is human disease monitored, but it also maintains data on viremic blood donors, dead birds, mosquitoes, veterinary disease cases, and sentinel animals.
Spring and summer are the most active seasons for ticks. Bacterial and spirochetal diseases transmitted by them include rickettsial diseases such as Rocky Mountain Spotted Fever, ehrlichiosis, and anaplasmosis. Tularemia in addition to Lyme and tick-borne relapsing fever are also transmitted by ticks. Babesiosis, which is due to a parasite, and southern tick-associated rash illness (STARI), whose causative agent is yet to be determined, are two additional tick-related diagnoses.
Of note, dengue, chikungunya, and Zika are all transmitted by infected Aedes mosquitoes. There is no enzootic cycle. Just human-mosquito-human transmission. In contrast, West Nile virus is transmitted by Culex mosquitoes in an enzootic cycle between an avian reservoir and humans.
Treatment
There is no specific treatment for arboviral infections. The primary goal is relief of symptoms with fluids, bed rest, and analgesics. For bacterial vector-borne diseases, antibiotic therapy is indicated and is based on the specific pathogen. Doxycycline is the drug of choice for treatment of suspected and confirmed Rocky Mountain Spotted Fever, ehrlichiosis, and anaplasmosis even in children less than 8 years of age. Delay in initiation of antimicrobial therapy pending definitive diagnosis may lead to an adverse outcome. It is also the drug of choice for tick-borne relapsing fever.
Lyme disease is also responsive to antibiotic treatment. Therapy is based on the disease category. (Lyme disease in “Red Book: 2015 Report of the Committee on Infectious Diseases,” [Elk Grove Village, Ill.: American Academy of Pediatrics, 2015, pp. 516-25]).
STARI clinically presents with a lesion that resembles erythema migrans in southern and southeastern states. However, it has not been associated with any of the complications reported with disseminated Lyme disease. Treatment is not recommended.
Tularemia and babesiosis are both responsive to antimicrobial therapy and would best be managed in consultation with an infectious disease physician.
A handy, concise, up to date reference guide about all of the tick-borne diseases including photographs is available at the App Store. The Tickborne Diseases App was developed by the CDC and it is free!
Prevention
The cornerstone of disease prevention is avoidance of mosquito and tick bites, in addition to eliminating mosquito breeding sites. Ticks are generally found near the ground, in brushy or wooded areas. They usually wait for a potential host to brush against them. When this happens, they climb onto the host and find a site to attach.
Is there a role for antimicrobial prophylaxis once a tick has been discovered? There is no data to support antimicrobial prophylaxis to prevent Rocky Mountain spotted fever, ehrlichiosis, and anaplasmosis. Prophylaxis with doxycycline or ciprofloxacin is recommended for children and adults after exposure to an intentional release of tularemia and for laboratory workers after inadvertent exposure. For prevention of Lyme disease, a single dose of doxycycline (4 mg/kg, max dose 200 mg) may be offered under limited conditions: The patient is at least 8 years of age, resides in an area where Lyme is highly endemic, the tick removed was engorged, therapy can be initiated within 72 hours after tick removal, and the estimated time of attachment was at least 36 hours. There is inadequate data on the use of amoxicillin.
Remember, not all mosquitoes are alike. Those that transmit chikungunya, dengue, and Zika (Aedes mosquitoes) are primarily daytime mosquitoes, but also can bite at night. West Nile is transmitted by Culex mosquitoes, which feed from dusk to dawn.
Here are some tips to share with your patients that should decrease their chances of acquiring a mosquito or tick-borne disease:
• Apply mosquito repellent only to intact exposed skin when outdoors. Most repellents can be safely used on children at least 2 months of age and older. Avoid applying repellent directly on the child’s hand. Use at least a 20% DEET (N,N-diethyl-meta-toluamide) containing product. Other Environmental Protection Agency–registered repellents are an alternative (Additional information is available at http://www2.epa.gov/insect-repellents). Products containing oil of lemon eucalyptus (OLE) or p-Menthane-3,8-diol (PMD) should not be used on children under 3 years of age.
• Apply permethrin to clothing, hats, boots, and so on. It is designed to repel mosquitoes and ticks. It can last for several washings. It is ideal to spray over nets covering carriers in children younger than 2 months of age.
• Wear long-sleeved shirts and long pants tucked inside of socks when hiking.
• Check for ticks daily, especially under the arms, behind the ears, around the waist, behind the knees, and inside belly buttons after outdoor activities.
• Have your patients learn how to effectively remove a tick. With a fine tipped tweezer, grasp the tick as close to the skin as possible and pull straight up with even pressure. Do not twist or jerk the tick. Do not squash the tick. Place it in a bag and dispose of it. Clean the site after removal with alcohol, iodine, or soap and water.
• Encourage families to mosquito proof their home by using screens on windows and doors, and using air conditioning when available.
• Empty and scrub all items that contain water such as birdbaths, planters, or wading pools around the outside of the home at least weekly because mosquitoes lay eggs in or near free standing water.
• Dogs and cats should be treated for ticks as recommended by the veterinarian.
The impact of the ongoing Zika virus outbreak is uncertain. While it may have an impact on those planning international travel now and in the near future, several arboviral and vector-borne diseases currently exist in the United States. Encouraging our patients to practice interventions to prevent mosquito and tick bites now will also serve to protect them if Zika virus becomes established in the Aedes mosquitoes here in the future and/or if they have plans for international travel. For up to date information on Zika virus for yourself and your patients, visit www.cdc.gov/zika.
Bonnie M. Word, M.D., is a pediatric infectious disease specialist and director of the Houston Travel Medicine Clinic. She said she had no relevant financial disclosures. Email Dr. Word at pdnews@frontlinemedcom.com.
May has arrived, and for the majority of your patients it signals the end of the school year and the beginning of summer vacation. Zika virus is on the minds of most people since its arrival to the Western Hemisphere in March 2015. With the fluidity of this outbreak and almost daily news updates and recommendations, many parents have voiced or will be voicing concerns regarding summer travel destinations.
Many concerns about Zika virus have been previously addressed in this column (“Zika virus: More questions than answers?” by Dr. Kristina Bryant). However, if the decision is to avoid international travel because of the ongoing Zika outbreak, it doesn’t mean your patients get a free pass and will not have to be concerned about acquiring any infectious diseases. They still need to be vigilant about avoiding those pesky vectors that transmit arboviruses and other vector-borne diseases that occur in the United States.
Arboviruses are transmitted by mosquitoes, ticks, or fleas. Most infections are subclinical. If symptoms develop, they are manifested by a generalized febrile illness including fever, headache, myalgia, arthralgia, and rash. Hemorrhagic fever (dengue) or neuroinvasive disease can include aseptic meningitis, encephalitis, or acute flaccid paralysis. Neuroinvasive disease rarely occurs with dengue, Colorado tick fever, and chikungunya infections.
While more than 100 arboviruses can cause infection, some of the more common arboviruses associated with human disease include West Nile, first detected in the United States in 1999 and chikungunya, first reported in the Americas in 2013 with local transmission documented in Florida, Puerto Rico, and the U.S. Virgin Islands in 2014. It is estimated that dengue causes over 100 million cases worldwide annually. Almost 40% of the world’s inhabitants live in endemic areas. The majority of cases on the U.S. mainland are imported. However, it is endemic in all U.S. territories including Guam, American Samoa, the U.S. Virgin Islands, and Puerto Rico. Between September 2015 and March 2016, Hawaii experienced a dengue outbreak involving 264 individuals including 46 children. As of April 16, 2016, there were no infectious individuals on the island.
Other domestic arboviruses causing disease include St. Louis, Eastern, and Western Equine encephalitis, La Crosse encephalitis, Colorado tick fever, and Powassan virus. All are transmitted by mosquitoes with the exception of Powassan and Colorado tick fever, which are transmitted by ticks. The numbers of cases nationally are much lower for these diseases, compared with West Nile, dengue, and chikungunya. National and state-specific information is available for domestic arboviruses at diseasemaps.usgs.gov/mapviewer. Data is compiled by ArboNET, a national arboviral surveillance system that is managed by the Centers for Disease Control and Prevention (CDC) in conjunction with state health departments. Not only is human disease monitored, but it also maintains data on viremic blood donors, dead birds, mosquitoes, veterinary disease cases, and sentinel animals.
Spring and summer are the most active seasons for ticks. Bacterial and spirochetal diseases transmitted by them include rickettsial diseases such as Rocky Mountain Spotted Fever, ehrlichiosis, and anaplasmosis. Tularemia in addition to Lyme and tick-borne relapsing fever are also transmitted by ticks. Babesiosis, which is due to a parasite, and southern tick-associated rash illness (STARI), whose causative agent is yet to be determined, are two additional tick-related diagnoses.
Of note, dengue, chikungunya, and Zika are all transmitted by infected Aedes mosquitoes. There is no enzootic cycle. Just human-mosquito-human transmission. In contrast, West Nile virus is transmitted by Culex mosquitoes in an enzootic cycle between an avian reservoir and humans.
Treatment
There is no specific treatment for arboviral infections. The primary goal is relief of symptoms with fluids, bed rest, and analgesics. For bacterial vector-borne diseases, antibiotic therapy is indicated and is based on the specific pathogen. Doxycycline is the drug of choice for treatment of suspected and confirmed Rocky Mountain Spotted Fever, ehrlichiosis, and anaplasmosis even in children less than 8 years of age. Delay in initiation of antimicrobial therapy pending definitive diagnosis may lead to an adverse outcome. It is also the drug of choice for tick-borne relapsing fever.
Lyme disease is also responsive to antibiotic treatment. Therapy is based on the disease category. (Lyme disease in “Red Book: 2015 Report of the Committee on Infectious Diseases,” [Elk Grove Village, Ill.: American Academy of Pediatrics, 2015, pp. 516-25]).
STARI clinically presents with a lesion that resembles erythema migrans in southern and southeastern states. However, it has not been associated with any of the complications reported with disseminated Lyme disease. Treatment is not recommended.
Tularemia and babesiosis are both responsive to antimicrobial therapy and would best be managed in consultation with an infectious disease physician.
A handy, concise, up to date reference guide about all of the tick-borne diseases including photographs is available at the App Store. The Tickborne Diseases App was developed by the CDC and it is free!
Prevention
The cornerstone of disease prevention is avoidance of mosquito and tick bites, in addition to eliminating mosquito breeding sites. Ticks are generally found near the ground, in brushy or wooded areas. They usually wait for a potential host to brush against them. When this happens, they climb onto the host and find a site to attach.
Is there a role for antimicrobial prophylaxis once a tick has been discovered? There is no data to support antimicrobial prophylaxis to prevent Rocky Mountain spotted fever, ehrlichiosis, and anaplasmosis. Prophylaxis with doxycycline or ciprofloxacin is recommended for children and adults after exposure to an intentional release of tularemia and for laboratory workers after inadvertent exposure. For prevention of Lyme disease, a single dose of doxycycline (4 mg/kg, max dose 200 mg) may be offered under limited conditions: The patient is at least 8 years of age, resides in an area where Lyme is highly endemic, the tick removed was engorged, therapy can be initiated within 72 hours after tick removal, and the estimated time of attachment was at least 36 hours. There is inadequate data on the use of amoxicillin.
Remember, not all mosquitoes are alike. Those that transmit chikungunya, dengue, and Zika (Aedes mosquitoes) are primarily daytime mosquitoes, but also can bite at night. West Nile is transmitted by Culex mosquitoes, which feed from dusk to dawn.
Here are some tips to share with your patients that should decrease their chances of acquiring a mosquito or tick-borne disease:
• Apply mosquito repellent only to intact exposed skin when outdoors. Most repellents can be safely used on children at least 2 months of age and older. Avoid applying repellent directly on the child’s hand. Use at least a 20% DEET (N,N-diethyl-meta-toluamide) containing product. Other Environmental Protection Agency–registered repellents are an alternative (Additional information is available at http://www2.epa.gov/insect-repellents). Products containing oil of lemon eucalyptus (OLE) or p-Menthane-3,8-diol (PMD) should not be used on children under 3 years of age.
• Apply permethrin to clothing, hats, boots, and so on. It is designed to repel mosquitoes and ticks. It can last for several washings. It is ideal to spray over nets covering carriers in children younger than 2 months of age.
• Wear long-sleeved shirts and long pants tucked inside of socks when hiking.
• Check for ticks daily, especially under the arms, behind the ears, around the waist, behind the knees, and inside belly buttons after outdoor activities.
• Have your patients learn how to effectively remove a tick. With a fine tipped tweezer, grasp the tick as close to the skin as possible and pull straight up with even pressure. Do not twist or jerk the tick. Do not squash the tick. Place it in a bag and dispose of it. Clean the site after removal with alcohol, iodine, or soap and water.
• Encourage families to mosquito proof their home by using screens on windows and doors, and using air conditioning when available.
• Empty and scrub all items that contain water such as birdbaths, planters, or wading pools around the outside of the home at least weekly because mosquitoes lay eggs in or near free standing water.
• Dogs and cats should be treated for ticks as recommended by the veterinarian.
The impact of the ongoing Zika virus outbreak is uncertain. While it may have an impact on those planning international travel now and in the near future, several arboviral and vector-borne diseases currently exist in the United States. Encouraging our patients to practice interventions to prevent mosquito and tick bites now will also serve to protect them if Zika virus becomes established in the Aedes mosquitoes here in the future and/or if they have plans for international travel. For up to date information on Zika virus for yourself and your patients, visit www.cdc.gov/zika.
Bonnie M. Word, M.D., is a pediatric infectious disease specialist and director of the Houston Travel Medicine Clinic. She said she had no relevant financial disclosures. Email Dr. Word at pdnews@frontlinemedcom.com.

Allergen-specific IgE serologic assays define sensitization, not disease
To the Editor: I read with great interest the commentary by Lau and Naugler1 regarding how much allergen-specific immunoglobulin E (IgE) testing is too much. The authors made a number of important conclusions that directly contradict the international consensus statement on IgE antibody test performance published by the Clinical Laboratory Standards Institute (CLSI) in 2009 (2nd edition)2 and updated in 2016 (3rd edition) in the I/LA-20 guidance document.3
The most important conclusion of the CLSI I/LA-20 panel was to reaffirm the golden rule of diagnostic allergy testing, which states that allergen-specific IgE antibody detected by either skin testing or serology methods is simply a marker for sensitization and thus only one of many risk factors for allergic disease. IgE positivity is not synonymous with the presence of allergic disease without a positive clinical history.4 Clinicians, since the time that IgE was discovered as the reagin in 1967, have tried to use the presence of IgE antibody as detected either by skin testing or serology as the definitive indicator of allergic disease. This is simply inappropriate. Both skin testing and serology are diagnostic tests that indicate sensitization (the presence of IgE antibody) and not disease. The clinician using a positive clinical history of allergic symptoms, objectively collected, must make the link between sensitization (IgE antibody positivity) and allergic disease.
Lau and Naugler make this same mistake and conclude from their Figure 1 data that “serum antigen-specific IgE testing is not a reliable diagnostic tool.” They use the Wians criterion5 of the summed diagnostic sensitivity and specificity of 170 to indicate if a test is clinically useful. They determined the sums of the diagnostic sensitivity and specificity for 89 allergen specificities, most of which they report as below 170. Among the specificities they cover are select aeroallergens, food allergens, venoms, and drugs. Importantly, they use a positive threshold of 0.35 kU/L for only some of their specificities, and they consider a sum of the diagnostic sensitivity and specificity equal to or greater than 170 as clinically relevant.
While Wians’ analysis may have been appropriate for laboratory tests like glucose and even prostate-specific antigen that associate closely with defining a disease state, this criterion is inappropriate for IgE antibody tests that do not directly identify allergic disease. There is peer-reviewed literature on nonreactors based on their clinical history with a validated positive IgE skin test, IgE antibody serology, or food challenge tests.6,7 Thus, the data in their Figure 1 have no value in defining the performance of IgE antibody tests of sensitization.
Moreover, their report is vague on the actual IgE antibody assay method that was used. This information is important because we know that different IgE assay methods measure different populations of IgE antibody.2,3 Also, the report does not define whether the participants who provided sera for testing actually had physician-defined allergic disease based on an objective clinical history.
The act of determining optimal cutoff values to maximize the “diagnostic” sensitivity and specificity is appropriate for many laboratory tests, but for allergen-specific IgE antibody analyses, it should be considered inappropriate. These are tests of sensitization, not disease. The IgE antibody result should be reported down to the regulatory-cleared and manufacturer-defined analytical sensitivity, which for the principal IgE antibody autoanalyzers used worldwide is 0.1 kU/L.8 These concerns essentially invalidate the conclusions of their report. Unfortunately, they leave the reader with misleading negative impressions about the utility of IgE antibody analyses that are extensively validated methods.
Finally, contrary to the assertions of the authors, current commentaries on the topic of relative diagnostic performance of skin testing and autoanalyzer-based IgE serology tests support the conclusion that, especially for aeroallergens, both the in vivo skin test and the current autoanalyzer-based in vitro serology tests provide overlapping, indistinguishable, and thus comparable diagnostic sensitivity and specificity results.9,10 Unfortunately, the authors refer to the 2008 Bernstein practice parameter that is out of date in relation to autoanalyzer technology, which has advanced by 2016.
Thus, contrary to the assertions of Lau and Naugler, IgE antibody serology has a clear, well-defined, and positive role in defining sensitization as a key part of the diagnostic workup of a patient who is suspected of having allergic disease. As with any laboratory test, IgE antibody measurements need to be judiciously ordered and used by the clinician only when there is a strong pretest likelihood, based on the patient’s clinical history, of allergic disease.
- Lau CK, Naugler C. Serum allergen-specific IgE testing: how much is too much? Cleve Clin J Med 2016; 83;21–24.
- Matsson P, Hamilton RG, Esch RE, et al. Analytical Performance Characteristics and Clinical Utility of Immunological Assays for Human Immunoglobulin E (IgE) Antibodies of Defined Allergen Specificities; Approved Guideline—Second Edition. CLSI document I/LA20-A2. Clinical and Laboratory Standards Institute, Wayne, Pennsylvania USA, 2009
- Hamilton RG, Matsson P, Chan S, et al. Analytical Performance Characteristics, Quality Assurance and Clinical Utility of Immunological Assays for Human Immunoglobulin E (IgE) Antibodies of Defined Allergen Specificities; Approved Guideline—Third Edition. CLSI document I/LA20-A3. Clinical and Laboratory Standards Institute, Wayne, Pennsylvania, USA, 2016.
- Hamilton RG. Allergic sensitization is a key risk factor for but not synonymous with allergic disease. J Allergy Clin Immunol 2014; 134:360–361.
- Wians FH Jr. Clinical laboratory tests: which, why and what do the results mean? Lab Medicine 2009; 40:105–113.
- Chokshi NY, Sicherer SH. Interpreting IgE sensitization tests in food allergy. Expert Rev Clin Immunol 2015; 15:1–15.
- Sicherer SH, Wood RA, Vickery BP, et al. Impact of allergic reactions on food-specific IgE concentrations and skin test results. J Allergy Clin Immunol Pract. 2015 Dec 21. pii: S2213-2198(15)00658-3. doi: 10.1016/j.jaip.2015.11.015. [Epub ahead of print]
- Hamilton RG. Clinical laboratories worldwide need to report IgE antibody results on clinical specimens as analytical results and not use differential positive thresholds (letter). J Allergy Clin Immunol 2015; 136:811–812.
- Adkinson NF Jr, Hamilton RG. Clinical history-driven diagnosis of allergic diseases: utilizing in vitro IgE testing. Allergy Clin Immunol Pract 2015; 3:871–876.
- Kleine-Tebbe J, Matricardi PM, Hamilton RG. Allergy work-up including component-resolved diagnosis: how to make allergen-specific immunotherapy more specific. Immunol Allergy Clin North Am 2016; 36:191–203.
To the Editor: I read with great interest the commentary by Lau and Naugler1 regarding how much allergen-specific immunoglobulin E (IgE) testing is too much. The authors made a number of important conclusions that directly contradict the international consensus statement on IgE antibody test performance published by the Clinical Laboratory Standards Institute (CLSI) in 2009 (2nd edition)2 and updated in 2016 (3rd edition) in the I/LA-20 guidance document.3
The most important conclusion of the CLSI I/LA-20 panel was to reaffirm the golden rule of diagnostic allergy testing, which states that allergen-specific IgE antibody detected by either skin testing or serology methods is simply a marker for sensitization and thus only one of many risk factors for allergic disease. IgE positivity is not synonymous with the presence of allergic disease without a positive clinical history.4 Clinicians, since the time that IgE was discovered as the reagin in 1967, have tried to use the presence of IgE antibody as detected either by skin testing or serology as the definitive indicator of allergic disease. This is simply inappropriate. Both skin testing and serology are diagnostic tests that indicate sensitization (the presence of IgE antibody) and not disease. The clinician using a positive clinical history of allergic symptoms, objectively collected, must make the link between sensitization (IgE antibody positivity) and allergic disease.
Lau and Naugler make this same mistake and conclude from their Figure 1 data that “serum antigen-specific IgE testing is not a reliable diagnostic tool.” They use the Wians criterion5 of the summed diagnostic sensitivity and specificity of 170 to indicate if a test is clinically useful. They determined the sums of the diagnostic sensitivity and specificity for 89 allergen specificities, most of which they report as below 170. Among the specificities they cover are select aeroallergens, food allergens, venoms, and drugs. Importantly, they use a positive threshold of 0.35 kU/L for only some of their specificities, and they consider a sum of the diagnostic sensitivity and specificity equal to or greater than 170 as clinically relevant.
While Wians’ analysis may have been appropriate for laboratory tests like glucose and even prostate-specific antigen that associate closely with defining a disease state, this criterion is inappropriate for IgE antibody tests that do not directly identify allergic disease. There is peer-reviewed literature on nonreactors based on their clinical history with a validated positive IgE skin test, IgE antibody serology, or food challenge tests.6,7 Thus, the data in their Figure 1 have no value in defining the performance of IgE antibody tests of sensitization.
Moreover, their report is vague on the actual IgE antibody assay method that was used. This information is important because we know that different IgE assay methods measure different populations of IgE antibody.2,3 Also, the report does not define whether the participants who provided sera for testing actually had physician-defined allergic disease based on an objective clinical history.
The act of determining optimal cutoff values to maximize the “diagnostic” sensitivity and specificity is appropriate for many laboratory tests, but for allergen-specific IgE antibody analyses, it should be considered inappropriate. These are tests of sensitization, not disease. The IgE antibody result should be reported down to the regulatory-cleared and manufacturer-defined analytical sensitivity, which for the principal IgE antibody autoanalyzers used worldwide is 0.1 kU/L.8 These concerns essentially invalidate the conclusions of their report. Unfortunately, they leave the reader with misleading negative impressions about the utility of IgE antibody analyses that are extensively validated methods.
Finally, contrary to the assertions of the authors, current commentaries on the topic of relative diagnostic performance of skin testing and autoanalyzer-based IgE serology tests support the conclusion that, especially for aeroallergens, both the in vivo skin test and the current autoanalyzer-based in vitro serology tests provide overlapping, indistinguishable, and thus comparable diagnostic sensitivity and specificity results.9,10 Unfortunately, the authors refer to the 2008 Bernstein practice parameter that is out of date in relation to autoanalyzer technology, which has advanced by 2016.
Thus, contrary to the assertions of Lau and Naugler, IgE antibody serology has a clear, well-defined, and positive role in defining sensitization as a key part of the diagnostic workup of a patient who is suspected of having allergic disease. As with any laboratory test, IgE antibody measurements need to be judiciously ordered and used by the clinician only when there is a strong pretest likelihood, based on the patient’s clinical history, of allergic disease.
To the Editor: I read with great interest the commentary by Lau and Naugler1 regarding how much allergen-specific immunoglobulin E (IgE) testing is too much. The authors made a number of important conclusions that directly contradict the international consensus statement on IgE antibody test performance published by the Clinical Laboratory Standards Institute (CLSI) in 2009 (2nd edition)2 and updated in 2016 (3rd edition) in the I/LA-20 guidance document.3
The most important conclusion of the CLSI I/LA-20 panel was to reaffirm the golden rule of diagnostic allergy testing, which states that allergen-specific IgE antibody detected by either skin testing or serology methods is simply a marker for sensitization and thus only one of many risk factors for allergic disease. IgE positivity is not synonymous with the presence of allergic disease without a positive clinical history.4 Clinicians, since the time that IgE was discovered as the reagin in 1967, have tried to use the presence of IgE antibody as detected either by skin testing or serology as the definitive indicator of allergic disease. This is simply inappropriate. Both skin testing and serology are diagnostic tests that indicate sensitization (the presence of IgE antibody) and not disease. The clinician using a positive clinical history of allergic symptoms, objectively collected, must make the link between sensitization (IgE antibody positivity) and allergic disease.
Lau and Naugler make this same mistake and conclude from their Figure 1 data that “serum antigen-specific IgE testing is not a reliable diagnostic tool.” They use the Wians criterion5 of the summed diagnostic sensitivity and specificity of 170 to indicate if a test is clinically useful. They determined the sums of the diagnostic sensitivity and specificity for 89 allergen specificities, most of which they report as below 170. Among the specificities they cover are select aeroallergens, food allergens, venoms, and drugs. Importantly, they use a positive threshold of 0.35 kU/L for only some of their specificities, and they consider a sum of the diagnostic sensitivity and specificity equal to or greater than 170 as clinically relevant.
While Wians’ analysis may have been appropriate for laboratory tests like glucose and even prostate-specific antigen that associate closely with defining a disease state, this criterion is inappropriate for IgE antibody tests that do not directly identify allergic disease. There is peer-reviewed literature on nonreactors based on their clinical history with a validated positive IgE skin test, IgE antibody serology, or food challenge tests.6,7 Thus, the data in their Figure 1 have no value in defining the performance of IgE antibody tests of sensitization.
Moreover, their report is vague on the actual IgE antibody assay method that was used. This information is important because we know that different IgE assay methods measure different populations of IgE antibody.2,3 Also, the report does not define whether the participants who provided sera for testing actually had physician-defined allergic disease based on an objective clinical history.
The act of determining optimal cutoff values to maximize the “diagnostic” sensitivity and specificity is appropriate for many laboratory tests, but for allergen-specific IgE antibody analyses, it should be considered inappropriate. These are tests of sensitization, not disease. The IgE antibody result should be reported down to the regulatory-cleared and manufacturer-defined analytical sensitivity, which for the principal IgE antibody autoanalyzers used worldwide is 0.1 kU/L.8 These concerns essentially invalidate the conclusions of their report. Unfortunately, they leave the reader with misleading negative impressions about the utility of IgE antibody analyses that are extensively validated methods.
Finally, contrary to the assertions of the authors, current commentaries on the topic of relative diagnostic performance of skin testing and autoanalyzer-based IgE serology tests support the conclusion that, especially for aeroallergens, both the in vivo skin test and the current autoanalyzer-based in vitro serology tests provide overlapping, indistinguishable, and thus comparable diagnostic sensitivity and specificity results.9,10 Unfortunately, the authors refer to the 2008 Bernstein practice parameter that is out of date in relation to autoanalyzer technology, which has advanced by 2016.
Thus, contrary to the assertions of Lau and Naugler, IgE antibody serology has a clear, well-defined, and positive role in defining sensitization as a key part of the diagnostic workup of a patient who is suspected of having allergic disease. As with any laboratory test, IgE antibody measurements need to be judiciously ordered and used by the clinician only when there is a strong pretest likelihood, based on the patient’s clinical history, of allergic disease.
- Lau CK, Naugler C. Serum allergen-specific IgE testing: how much is too much? Cleve Clin J Med 2016; 83;21–24.
- Matsson P, Hamilton RG, Esch RE, et al. Analytical Performance Characteristics and Clinical Utility of Immunological Assays for Human Immunoglobulin E (IgE) Antibodies of Defined Allergen Specificities; Approved Guideline—Second Edition. CLSI document I/LA20-A2. Clinical and Laboratory Standards Institute, Wayne, Pennsylvania USA, 2009
- Hamilton RG, Matsson P, Chan S, et al. Analytical Performance Characteristics, Quality Assurance and Clinical Utility of Immunological Assays for Human Immunoglobulin E (IgE) Antibodies of Defined Allergen Specificities; Approved Guideline—Third Edition. CLSI document I/LA20-A3. Clinical and Laboratory Standards Institute, Wayne, Pennsylvania, USA, 2016.
- Hamilton RG. Allergic sensitization is a key risk factor for but not synonymous with allergic disease. J Allergy Clin Immunol 2014; 134:360–361.
- Wians FH Jr. Clinical laboratory tests: which, why and what do the results mean? Lab Medicine 2009; 40:105–113.
- Chokshi NY, Sicherer SH. Interpreting IgE sensitization tests in food allergy. Expert Rev Clin Immunol 2015; 15:1–15.
- Sicherer SH, Wood RA, Vickery BP, et al. Impact of allergic reactions on food-specific IgE concentrations and skin test results. J Allergy Clin Immunol Pract. 2015 Dec 21. pii: S2213-2198(15)00658-3. doi: 10.1016/j.jaip.2015.11.015. [Epub ahead of print]
- Hamilton RG. Clinical laboratories worldwide need to report IgE antibody results on clinical specimens as analytical results and not use differential positive thresholds (letter). J Allergy Clin Immunol 2015; 136:811–812.
- Adkinson NF Jr, Hamilton RG. Clinical history-driven diagnosis of allergic diseases: utilizing in vitro IgE testing. Allergy Clin Immunol Pract 2015; 3:871–876.
- Kleine-Tebbe J, Matricardi PM, Hamilton RG. Allergy work-up including component-resolved diagnosis: how to make allergen-specific immunotherapy more specific. Immunol Allergy Clin North Am 2016; 36:191–203.
- Lau CK, Naugler C. Serum allergen-specific IgE testing: how much is too much? Cleve Clin J Med 2016; 83;21–24.
- Matsson P, Hamilton RG, Esch RE, et al. Analytical Performance Characteristics and Clinical Utility of Immunological Assays for Human Immunoglobulin E (IgE) Antibodies of Defined Allergen Specificities; Approved Guideline—Second Edition. CLSI document I/LA20-A2. Clinical and Laboratory Standards Institute, Wayne, Pennsylvania USA, 2009
- Hamilton RG, Matsson P, Chan S, et al. Analytical Performance Characteristics, Quality Assurance and Clinical Utility of Immunological Assays for Human Immunoglobulin E (IgE) Antibodies of Defined Allergen Specificities; Approved Guideline—Third Edition. CLSI document I/LA20-A3. Clinical and Laboratory Standards Institute, Wayne, Pennsylvania, USA, 2016.
- Hamilton RG. Allergic sensitization is a key risk factor for but not synonymous with allergic disease. J Allergy Clin Immunol 2014; 134:360–361.
- Wians FH Jr. Clinical laboratory tests: which, why and what do the results mean? Lab Medicine 2009; 40:105–113.
- Chokshi NY, Sicherer SH. Interpreting IgE sensitization tests in food allergy. Expert Rev Clin Immunol 2015; 15:1–15.
- Sicherer SH, Wood RA, Vickery BP, et al. Impact of allergic reactions on food-specific IgE concentrations and skin test results. J Allergy Clin Immunol Pract. 2015 Dec 21. pii: S2213-2198(15)00658-3. doi: 10.1016/j.jaip.2015.11.015. [Epub ahead of print]
- Hamilton RG. Clinical laboratories worldwide need to report IgE antibody results on clinical specimens as analytical results and not use differential positive thresholds (letter). J Allergy Clin Immunol 2015; 136:811–812.
- Adkinson NF Jr, Hamilton RG. Clinical history-driven diagnosis of allergic diseases: utilizing in vitro IgE testing. Allergy Clin Immunol Pract 2015; 3:871–876.
- Kleine-Tebbe J, Matricardi PM, Hamilton RG. Allergy work-up including component-resolved diagnosis: how to make allergen-specific immunotherapy more specific. Immunol Allergy Clin North Am 2016; 36:191–203.
In reply: Allergen-specific IgE serologic assays define sensitization, not disease
In Reply: We thank Dr. Hamilton for his interest in our article and for providing more recent literature than was available at the time we submitted our manuscript.
There are multiple points of view toward allergy testing. But the bottom line, as emphasized by Dr. Hamilton and in our article, is that serum IgE testing should not be used as the sole diagnostic tool because it is an indicator of sensitization, not disease, and that clinical history should always be used in conjunction to ensure proper diagnosis.
It is our experience that some clinicians indiscriminately order large panels of serum IgE tests. As Dr. Hamilton indicates, patients can have positive serum IgE results but not display allergy symptoms, which can lead to unnecessary food avoidance. In addition, false-negative results from injudiciously ordered tests (ie, not based on pretest probability) can lead to missed diagnoses. All of these points should be kept in mind in delivering good clinical care, and as such, Choosing Wisely has highlighted the importance of using this test appropriately.
In response to the origin of the sensitivities and specificities used to calculate the sum, the values were curated from available literature and thus limited the number of allergens that could be profiled. A cutoff of 0.35 kU/L was used because this was the cutoff used by the references.
In Reply: We thank Dr. Hamilton for his interest in our article and for providing more recent literature than was available at the time we submitted our manuscript.
There are multiple points of view toward allergy testing. But the bottom line, as emphasized by Dr. Hamilton and in our article, is that serum IgE testing should not be used as the sole diagnostic tool because it is an indicator of sensitization, not disease, and that clinical history should always be used in conjunction to ensure proper diagnosis.
It is our experience that some clinicians indiscriminately order large panels of serum IgE tests. As Dr. Hamilton indicates, patients can have positive serum IgE results but not display allergy symptoms, which can lead to unnecessary food avoidance. In addition, false-negative results from injudiciously ordered tests (ie, not based on pretest probability) can lead to missed diagnoses. All of these points should be kept in mind in delivering good clinical care, and as such, Choosing Wisely has highlighted the importance of using this test appropriately.
In response to the origin of the sensitivities and specificities used to calculate the sum, the values were curated from available literature and thus limited the number of allergens that could be profiled. A cutoff of 0.35 kU/L was used because this was the cutoff used by the references.
In Reply: We thank Dr. Hamilton for his interest in our article and for providing more recent literature than was available at the time we submitted our manuscript.
There are multiple points of view toward allergy testing. But the bottom line, as emphasized by Dr. Hamilton and in our article, is that serum IgE testing should not be used as the sole diagnostic tool because it is an indicator of sensitization, not disease, and that clinical history should always be used in conjunction to ensure proper diagnosis.
It is our experience that some clinicians indiscriminately order large panels of serum IgE tests. As Dr. Hamilton indicates, patients can have positive serum IgE results but not display allergy symptoms, which can lead to unnecessary food avoidance. In addition, false-negative results from injudiciously ordered tests (ie, not based on pretest probability) can lead to missed diagnoses. All of these points should be kept in mind in delivering good clinical care, and as such, Choosing Wisely has highlighted the importance of using this test appropriately.
In response to the origin of the sensitivities and specificities used to calculate the sum, the values were curated from available literature and thus limited the number of allergens that could be profiled. A cutoff of 0.35 kU/L was used because this was the cutoff used by the references.
Evaluation of nail lines: Color and shape hold clues
Inspection of the fingernails and toenails should be part of a complete physical examination. A basic understanding of nail anatomy and recognition of several basic types of nail lines and bands allow the clinician to properly diagnose and treat the nail disease, to recognize possible underlying systemic diseases, and to know when to refer the patient to a dermatologist for specialized evaluation and biopsy.
In this review, we delineate the three basic types of nail lines—white lines (leukonychia striata), brown-black lines (longitudinal melanonychia), and red lines (longitudinal erythronychia)—and the differential diagnosis for each type. We also discuss grooves in the nail plate, or Beau lines.
BASIC NAIL ANATOMY
A fundamental understanding of the anatomy of the nail unit is necessary to understand the origin of nail diseases and underlying pathologic conditions.
The nail unit includes the nail matrix, the lunula, the nail fold, the nail plate, and the nail bed. The nail matrix extends from under the proximal nail fold to the half-moon-shaped area (ie, the lunula) and is responsible for nail plate production. The nail bed lies under the nail plate and on top of the distal phalanx and extends from the lunula to just proximal to the free edge of the nail; its rich blood supply gives it its reddish color.
Nails grow slowly, and this should be kept in mind during the examination. Regrowth of a fingernail takes at least 6 months, and regrowth of a toenail may take 12 to 18 months. Therefore, a defect in the nail plate may reveal an injury that occurred—or a condition that began—several months before.1
NAIL EXAMINATION ESSENTIALS
A complete examination includes all 20 nail units and the periungual skin. Patients should be instructed to remove nail polish from all nails, as it may camouflage dystrophy or disease of the nail. Photography and careful measurement help document changes over time.
LEUKONYCHIA STRIATA: WHITE NAIL LINES
White nail lines or leukonychia is classified as true or apparent, depending on whether the origin is in the nail matrix or the nail bed.
In true leukonychia, there is abnormal keratinization of the underlying nail matrix, resulting in parakeratosis within the nail plate and an opaque appearance on examination.2 The white discoloration is unaffected by pressure, and the opacity moves distally as the nail grows out, which can be documented by serial photography on subsequent visits.
Apparent leukonychia involves abnormal nail bed vasculature, which changes the translucency of the nail plate. The whiteness disappears with pressure, is unaffected by nail growth, and will likely show no change on later visits with serial photography.3
True leukonychia
Leukonychia striata, a subtype of true leukonychia, is characterized by transverse or longitudinal bands. It is most often associated with microtrauma, such as from a manicure.4 Lines due to trauma are typically more apparent in the central part of the nail plate; they spare the lateral portion and lie parallel to the edge of the proximal nail fold.5
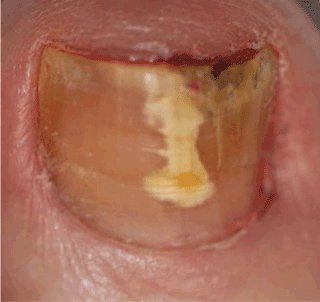
Onychomycosis. White longitudinal bands may also be seen in onychomycosis, a fungal infection of the nail accounting for up to 50% of all cases of nail disease. The infection may present as irregular dense longitudinal white or yellowish bands or “spikes” on the nail plate with associated hyperkeratosis, known as a dermatophytoma (Figure 1).
If a fungal infection is suspected, a potassium hydroxide stain can be performed on the subungual debris, which is then examined with direct microscopy.6 Alternatively, the physician can send a nail plate clipping in a 10% buffered formalin container with a request for a fungal stain such as periodic acid-Schiff.7 Microscopic examination of a dermatophytoma shows a dense mass of dermatophyte hyphae, otherwise known as a fungal abscess.8
The physician can play an important role in diagnosis because clinical findings suggestive of a dermatophytoma are associated with a poor response to antifungal therapy.9
Inherited diseases. White longitudinal bands are also an important clue to the rare autosomal dominant genodermatoses Hailey-Hailey disease (from mutations of the ATP2A2 gene) and Darier disease (from mutations of the ATP2C1 gene). Patients with Hailey-Hailey disease may have nails with multiple parallel longitudinal white stripes of variable width originating in the lunula and most prominent on the thumbs.10–12 These patients also have recurrent vesicular eruptions in flexural skin areas, such as the groin, axilla, neck, and periumbilical area causing significant morbidity.
Patients with Darier disease may have nails with alternating red and white longitudinal streaks, described as “candy-cane,”13 as well as wedge-shaped distal subungual keratosis accompanied by flat keratotic papules on the proximal nail fold.14 These nail changes are reported in 92% to 95% of patients with Darier disease.15,16 Patients typically have skin findings characterized by keratotic papules and plaques predominantly in seborrheic areas and palmoplantar pits, as well as secondary infections and malodor causing significant morbidity.15 Therefore, knowing the characteristic nail findings in these diseases may lead to more rapid diagnosis and treatment.
Mees lines. Leukonychia striata can present as transverse white lines, commonly known as Mees lines. They are 1- to 2-mm wide horizontal parallel white bands that span the width of the nail plate, usually affecting all fingernails.17 They are not a common finding and are most often associated with arsenic poisoning. They can also be used to identify the time of poisoning, since they tend to appear 2 months after the initial insult.
Mees lines are also associated with acute systemic stresses, such as acute renal failure, heart failure, ulcerative colitis, breast cancer, infections such as measles and tuberculosis, and systemic lupus erythematosus, and with exposure to toxic metals such as thallium.3
Apparent leukonychia
Apparent leukonychia can alert the physician to systemic diseases, infections, drug side effects, and nutrient deficiencies. Specific nail findings include Muehrcke lines, “half-and-half” nails, and Terry nails.
Muehrcke lines are paired white transverse bands that span the width of the nail bed and run parallel to the distal lunula. They were first described in the fingernails of patients with severe hypoalbuminemia, some of whom also had nephrotic syndrome, which resolved with normalization of the serum albumin level. Muehrcke lines have since been reported in patients with liver disease, malnutrition, chemotherapy, organ transplant, human immunodeficiency virus (HIV) infection, and acquired immunodeficiency syndrome.3,18 They are associated with periods of metabolic stress, ie, when the body’s capacity to synthesize proteins is diminished.19
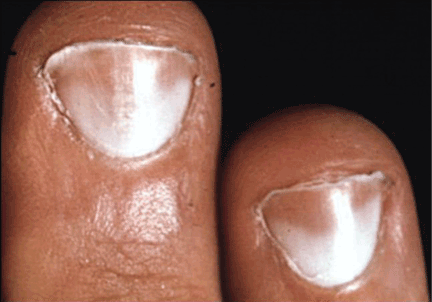
Half-and-half nails, or Lindsay nails, are characterized by a white band proximally, a pink or red-brown band distally, and a sharp demarcation between the two (Figure 2). They were originally described in association with chronic renal disease,20 and surprisingly, they resolve with kidney transplant but not with hemodialysis treatment or improvement in hemoglobin or albumin levels.21–23 Half-and-half nails have been reported with Kawasaki disease, hepatic cirrhosis, Crohn disease, zinc deficiency, chemotherapy, Behçet disease, and pellagra.3,24,25 They should be distinguished from Terry nails, which are characterized by leukonychia involving more than 80% of the total nail length.26
Terry nails were originally reported in association with hepatic cirrhosis, usually secondary to alcoholism27 but have since been found with heart failure, type 2 diabetes mellitus, pulmonary tuberculosis, reactive arthritis, older age, Hansen disease, and peripheral vascular disease.3,26,28,29
LONGITUDINAL MELANONYCHIA: VERTICAL BROWN-BLACK NAIL LINES
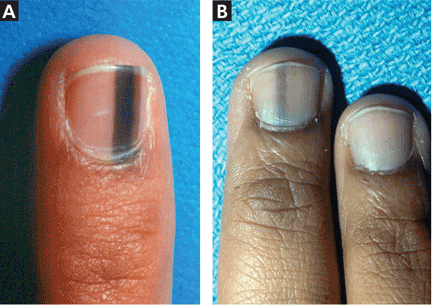
Longitudinal melanonychia is the presence of black-brown vertical lines in the nail plate. They have a variety of causes, including blood from trauma; bacterial, fungal, or HIV infection; drug therapy (eg, from minocycline); endocrine disorders (Addison disease); exogenous pigmentation; or excess melanin production within the nail matrix.30–32 They may also be a sign of a benign condition such as benign melanocytic activation, lentigines, or nevi, or a malignant condition such as melanoma (Figure 3).33,34
When to suspect melanoma and refer
Although melanoma is less commonly associated with brown-black vertical nail lines, awareness of melanoma-associated longitudinal melanonychia reduces the likelihood of delayed diagnosis and improves patient outcomes.35 Also, it is important to remember that although nail melanoma is more common in the 5th and 6th decades of life, it can occur at any age, even in children.36

Findings that raise suspicion of nail melanoma (Table 1)33,37 and that should prompt referral to a dermatologist who specializes in nails include the following:
- A personal or family history of melanoma
- Involvement of a “high-risk” digit (thumb, index finger, great toe),30,31,38 although nail melanoma can occur in any digit
- Any new vertical brown-black nail pigmentation in a fair-skinned patient
- Only one nail affected: involvement of more than one nail is common in people with darker skin, and nearly all patients with darker skin exhibit longitudinal melanonychia by age 5031
- Changes in the band such as darkening, widening, and bleeding
- A bandwidth greater than 6 mm33
- A band that is wider proximally than distally34
- Nonuniform color of the line
- Indistinct lateral borders
- Associated with pigmentation of the nail fold (the Hutchinson sign, representing subungual melanoma),31,39 nail plate dystrophy, bleeding, or ulceration.33
While these features may help distinguish benign from malignant causes of longitudinal melanonychia, the clinical examination alone may not provide a definitive diagnosis. Delayed diagnosis of nail melanoma carries a high mortality rate; the internist can promote early diagnosis by recognizing the risk factors and clinical signs and referring the patient to a dermatologist for further evaluation with nail biopsy.
LONGITUDINAL ERYTHRONYCHIA: VERTICAL RED NAIL LINES

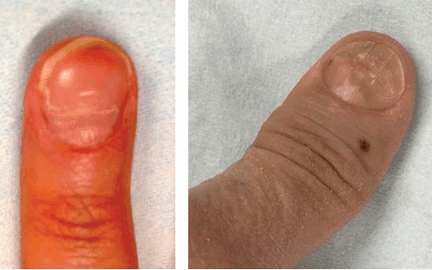
Longitudinal erythronychia—the presence of one or more linear red bands in the nail unit—can be localized (involving only one nail) or polydactylous (involving more than one nail). The localized form is usually due to a neoplastic process, whereas involvement of more than one nail may indicate an underlying regional or systemic disease.13Table 2 lists indications for referral to a nail specialist.
General features on examination

Clinical examination reveals one or more linear, pink-red streaks extending from the proximal nail fold to the distal free edge of the nail plate (Figure 4). The width of the band typically ranges from less than 1 mm to 3 mm.40 Other features may include splinter hemorrhages within a red band, a semitransparent distal matrix, distal V-shaped chipping, splitting, onycholysis of the nail plate, and reactive distal nail bed and hyponychial hyperkeratosis. These features can be visible to the naked eye but may be better viewed with a magnifying glass, a 7× loupe, or a dermatoscope.13
Localized longitudinal erythronychia is usually seen in middle-aged individuals and is most commonly found on the thumbnail, followed by the index finger.41,42 The condition may be asymptomatic, but the patient may present with pain or with concern that the split end of the nail catches on fabrics or small objects.42
Glomus tumor
Intense, pulsatile pain with sensitivity to cold and tenderness to palpation is highly suggestive of glomus tumor,43 a benign neoplasm that originates from a neuromyoarterial glomus body. Glomus bodies are located throughout the body but are more highly concentrated in the fingertips, especially beneath the nails, and they regulate skin circulation. Therefore, the nail unit is the most common site for glomus tumor.44,45 A characteristic feature of subungual glomus tumor is demonstration of tenderness after pin-point palpation of the suspected tumor (positive Love sign).45 While it is typical for glomus tumor to affect only one nail, multiple tumors are associated with neurofibromatosis type 1.46 Confirmation of this diagnosis requires referral to a dermatologist.
Other causes of localized red nail lines
Onychopapilloma, a benign idiopathic tumor, is the most common cause of localized longitudinal erythronychia. Unlike glomus tumor, it is usually asymptomatic.42,47 Less common benign conditions are warts, warty dyskeratoma, benign vascular proliferation, a solitary lesion of lichen planus, hemiplegia, and postsurgical scarring of the nail matrix. In some cases, the lines are idiopathic.42,43
Malignant diseases that can present as localized longitudinal erythronychia include invasive squamous cell carcinoma, squamous cell carcinoma in situ (Bowen disease), and, less frequently, amelanotic melanoma in situ, malignant melanoma, and basal cell carcinoma.42 Squamous cell carcinoma in situ most commonly presents in the 5th decade of life and is the malignancy most commonly associated with localized longitudinal erythronychia. Clinically, there is also often nail dystrophy, such as distal subungual keratosis or onycholysis.43
Patients with asymptomatic, stable localized longitudinal erythronychia may be followed closely with photography and measurements. However, any new lesion or a change in an existing lesion should prompt referral to a dermatologist for biopsy.13
Red streaks on more than one nail
Polydactylous longitudinal erythronychia usually presents in adults as red streaks on multiple nails and, depending on the presence or absence of symptoms (eg, pain, splitting), may be the patient’s chief complaint or an incidental finding noted by the astute clinician. Often, it is associated with systemic disease, most commonly lichen planus or Darier disease.
Lichen planus is a papulosquamous skin disease with nail involvement in 10% of patients and permanent nail dystrophy in 4%. Common nail findings include thinning, longitudinal ridging, and fissuring, as well as scarring of the nail matrix resulting in pterygium. Linear red streaks may accompany these more typical nail findings.13 Patients with Darier disease present with alternating red and white linear bands on multiple nails as in leukonychia striata.
Less frequently, polydactylous longitudinal erythronychia is associated with primary and systemic amyloidosis, hemiplegia, graft-vs-host disease, acantholytic epidermolysis bullosa, acantholytic dyskeratotic epidermal nevus, acrokeratosis verruciformis of Hopf, or pseudobulbar syndrome, or is idiopathic.13,42,48 Therefore, the physician evaluating a patient with these nail findings should focus on a workup for regional or systemic disease or refer the patient to a dermatologist who specializes in nails.
BEAU LINES
Beau lines are a common finding in clinical practice. They are not true lines, but transverse grooves in the nail plate that arise from the temporary suppression of nail growth within the nail matrix that can occur during periods of acute or chronic stress or systemic illness (Figure 5).49
The precipitating event may be local trauma or paronychia, chemotherapeutic agents cytotoxic to the nail matrix, or the abrupt onset of systemic disease.18,50 The grooves have also been associated with rheumatic fever, malaria, pemphigus, Raynaud disease, and myocardial infarction, as well as following deep-sea dives.51–53 The distance of a Beau line from the proximal nail fold can provide an estimate of the time of the acute stress, based on an average growth rate of 3 mm per month for fingernails and 1 mm per month for toenails.49
- Scher RK, Rich P, Pariser D, Elewski B. The epidemiology, etiology, and pathophysiology of onychomycosis. Semin Cutan Med Surg 2013; 32(suppl 1):S2–S4.
- Lawry MA, Haneke E, Strobeck K, Martin S, Zimmer B, Romano PS. Methods for diagnosing onychomycosis: a comparative study and review of the literature. Arch Dermatol 2000; 136:1112–1116.
- Zaiac MN, Walker A. Nail abnormalities associated with systemic pathologies. Clin Dermatol 2013; 31:627–649.
- Zaiac MN, Daniel CR. Nails in systemic disease. Dermatol Ther 2002; 15:99–106.
- Tosti A, Iorizzo M, Piraccini BM, Starace M. The nail in systemic diseases. Dermatol Clin 2006; 24:341–347.
- Scher RK, Daniel CR, eds. Nails Diagnosis, Therapy, Surgery. 3rd ed. Oxford: Elsevier Saunders; 2005.
- Smith MB, McGinnis MR. Diagnostic histopathology. In: Hospenthal DR, Rinaldi MG, eds. Diagnosis and Treatment of Human Mycoses. Totowa, NJ: Humana Press; 2008:37–51.
- Roberts DT, Evans EG. Subungual dermatophytoma complicating dermatophyte onychomycosis. Br J Dermatol 1998; 138:189–190.
- Sigurgeirsson B. Prognostic factors for cure following treatment of onychomycosis. J Eur Acad Dermatol Venereol 2010; 24:679–684.
- Kumar R, Zawar V. Longitudinal leukonychia in Hailey-Hailey disease: a sign not to be missed. Dermatol Online J 2008; 14:17.
- Burge SM. Hailey-Hailey disease: the clinical features, response to treatment and prognosis. Br J Dermatol 1992; 126:275–282.
- Kirtschig G, Effendy I, Happle R. Leukonychia longitudinalis as the primary symptom of Hailey-Hailey disease. Hautarzt 1992; 43:451–452. German.
- Jellinek NJ. Longitudinal erythronychia: suggestions for evaluation and management. J Am Acad Dermatol 2011; 64:167.e1–167.e11
- Zaias N, Ackerman AB. The nail in Darier-White disease. Arch Dermatol 1973; 107:193–199.
- Burge SM, Wilkinson JD. Darier-White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol 1992; 27:40–50.
- Munro CS. The phenotype of Darier's disease: penetrance and expressivity in adults and children. Br J Dermatol 1992; 127:126–130.
- Schwartz RA. Arsenic and the skin. Int J Dermatol 1997; 36:241–250.
- Fawcett RS, Linford S, Stulberg DL. Nail abnormalities: clues to systemic disease. Am Fam Physician 2004; 69:1417–1424.
- Morrison-Bryant M, Gradon JD. Images in clinical medicine. Muehrcke's lines. N Engl J Med 2007; 357:917.
- Daniel CR 3rd, Bower JD, Daniel CR Jr. The “half and half fingernail”: the most significant onychopathological indicator of chronic renal failure. J Miss State Med Assoc 1975; 16:367–370.
- Saray Y, Seckin D, Gulec AT, Akgun S, Haberal M. Nail disorders in hemodialysis patients and renal transplant recipients: a case-control study. J Am Acad Dermatol 2004; 50:197–202.
- Dyachenko P, Monselise A, Shustak A, Ziv M, Rozenman D. Nail disorders in patients with chronic renal failure and undergoing haemodialysis treatment: a case-control study. J Eur Acad Dermatol Venereol 2007; 21:340–344.
- Salem A, Al Mokadem S, Attwa E, Abd El Raoof S, Ebrahim HM, Faheem KT. Nail changes in chronic renal failure patients under haemodialysis. J Eur Acad Dermatol Venereol 2008; 22:1326–1331.
- Zagoni T, Sipos F, Tarjan Z, Peter Z. The half-and-half nail: a new sign of Crohn's disease? Report of four cases. Dis Colon Rectum 2006; 49:1071–1073.
- Nixon DW, Pirozzi D, York RM, Black M, Lawson DH. Dermatologic changes after systemic cancer therapy. Cutis 1981; 27:181–194.
- Holzberg M, Walker HK. Terry's nails: revised definition and new correlations. Lancet 1984; 1:896–899.
- Terry R. White nails in hepatic cirrhosis. Lancet 1954; 266:757–759.
- Coskun BK, Saral Y, Ozturk P, Coskun N. Reiter syndrome accompanied by Terry nail. J Eur Acad Dermatol Venereol 2005; 19:87–89.
- Blyumin M, Khachemoune A, Bourelly P. What is your diagnosis? Terry nails. Cutis 2005; 76:201–202.
- Haneke E, Baran R. Longitudinal melanonychia. Dermatol Surg 2001; 27:580–584.
- Andre J, Lateur N. Pigmented nail disorders. Dermatol Clin 2006; 24:329–339.
- Braun RP, Baran R, Le Gal FA, et al. Diagnosis and management of nail pigmentations. J Am Acad Dermatol 2007; 56:835–847.
- Mannava KA, Mannava S, Koman LA, Robinson-Bostom L, Jellinek N. Longitudinal melanonychia: detection and management of nail melanoma. Hand Surg 2013; 18:133–139.
- Ruben BS. Pigmented lesions of the nail unit: clinical and histopathologic features. Semin Cutan Med Surg 2010; 29:148–158.
- Cohen T, Busam KJ, Patel A, Brady MS. Subungual melanoma: management considerations. Am J Surg 2008; 195:244–248.
- Iorizzo M, Tosti A, Di Chiacchio N, et al. Nail melanoma in children: differential diagnosis and management. Dermatol Surg 2008; 34:974–978.
- Jellinek N. Nail matrix biopsy of longitudinal melanonychia: diagnostic algorithm including the matrix shave biopsy. J Am Acad Dermatol 2007; 56:803–810.
- Husain S, Scher RK, Silvers DN, Ackerman AB. Melanotic macule of nail unit and its clinicopathologic spectrum. J Am Acad Dermatol 2006; 54:664–667.
- Baran R, Kechijian P. Hutchinson's sign: a reappraisal. J Am Acad Dermatol 1996; 34:87–90.
- Baran R. Red nails. Dermatol Online 2005; 11:29.
- Baran R, Perrin C. Longitudinal erythronychia with distal subungual keratosis: onychopapilloma of the nail bed and Bowen’s disease. Br J Dermatol 2000; 143:132–135.
- de Berker DA, Perrin C, Baran R. Localized longitudinal erythronychia: diagnostic significance and physical explanation. Arch Dermatol 2004; 140:1253–1257.
- Cohen PR. Longitudinal erythronychia: individual or multiple linear red bands of the nail plate: a review of clinical features and associated conditions. Am J Clin Dermatol 2011; 12:217–231.
- Van Geertruyden J, Lorea P, Goldschmidt D, et al. Glomus tumours of the hand. A retrospective study of 51 cases. J Hand Surg Br 1996; 21:257–260.
- Moon SE, Won JH, Kwon OS, Kim JA. Subungual glomus tumor: clinical manifestations and outcome of surgical treatment. J Dermatol 2004; 31:993–997.
- Okada O, Demitsu T, Manabe M, Yoneda K. A case of multiple subungual glomus tumors associated with neurofibromatosis type 1. J Dermatol 1999; 26:535–537.
- Gee BC, Millard PR, Dawber RP. Onychopapilloma is not a distinct clinicopathological entity. Br J Dermatol 2002; 146:156–157.
- Siragusa M, Del Gracco S, Ferri R, Schepis C. Longitudinal red streaks on the big toenails in a patient with pseudobulbar syndrome. J Eur Acad Dermatol Venereol 2001; 15:85–86.
- Lipner S, Scher RK. Nails. In: Callen J, Jorizzo JL, eds. Dermatological Signs of Systemic Disease. 5th ed: Elsevier; in press.
- Mortimer NJ, Mills J. Images in clinical medicine. Beau’s lines. N Engl J Med 2004; 351:1778.
- Schwartz H. Clinical observation: Beau’s lines on fingernails after deep saturation dives. Undersea Hyperb Med 2006; 33:5–10.
- Gugelmann HM, Gaieski DF. Beau’s lines after cardiac arrest. Ther Hypothermia Temp Manag 2013; 3:199–202.
- Lauber J, Turk K. Beau’s lines and pemphigus vulgaris. Int J Dermatol 1990; 29:309.
Inspection of the fingernails and toenails should be part of a complete physical examination. A basic understanding of nail anatomy and recognition of several basic types of nail lines and bands allow the clinician to properly diagnose and treat the nail disease, to recognize possible underlying systemic diseases, and to know when to refer the patient to a dermatologist for specialized evaluation and biopsy.
In this review, we delineate the three basic types of nail lines—white lines (leukonychia striata), brown-black lines (longitudinal melanonychia), and red lines (longitudinal erythronychia)—and the differential diagnosis for each type. We also discuss grooves in the nail plate, or Beau lines.
BASIC NAIL ANATOMY
A fundamental understanding of the anatomy of the nail unit is necessary to understand the origin of nail diseases and underlying pathologic conditions.
The nail unit includes the nail matrix, the lunula, the nail fold, the nail plate, and the nail bed. The nail matrix extends from under the proximal nail fold to the half-moon-shaped area (ie, the lunula) and is responsible for nail plate production. The nail bed lies under the nail plate and on top of the distal phalanx and extends from the lunula to just proximal to the free edge of the nail; its rich blood supply gives it its reddish color.
Nails grow slowly, and this should be kept in mind during the examination. Regrowth of a fingernail takes at least 6 months, and regrowth of a toenail may take 12 to 18 months. Therefore, a defect in the nail plate may reveal an injury that occurred—or a condition that began—several months before.1
NAIL EXAMINATION ESSENTIALS
A complete examination includes all 20 nail units and the periungual skin. Patients should be instructed to remove nail polish from all nails, as it may camouflage dystrophy or disease of the nail. Photography and careful measurement help document changes over time.
LEUKONYCHIA STRIATA: WHITE NAIL LINES
White nail lines or leukonychia is classified as true or apparent, depending on whether the origin is in the nail matrix or the nail bed.
In true leukonychia, there is abnormal keratinization of the underlying nail matrix, resulting in parakeratosis within the nail plate and an opaque appearance on examination.2 The white discoloration is unaffected by pressure, and the opacity moves distally as the nail grows out, which can be documented by serial photography on subsequent visits.
Apparent leukonychia involves abnormal nail bed vasculature, which changes the translucency of the nail plate. The whiteness disappears with pressure, is unaffected by nail growth, and will likely show no change on later visits with serial photography.3
True leukonychia
Leukonychia striata, a subtype of true leukonychia, is characterized by transverse or longitudinal bands. It is most often associated with microtrauma, such as from a manicure.4 Lines due to trauma are typically more apparent in the central part of the nail plate; they spare the lateral portion and lie parallel to the edge of the proximal nail fold.5
Onychomycosis. White longitudinal bands may also be seen in onychomycosis, a fungal infection of the nail accounting for up to 50% of all cases of nail disease. The infection may present as irregular dense longitudinal white or yellowish bands or “spikes” on the nail plate with associated hyperkeratosis, known as a dermatophytoma (Figure 1).
If a fungal infection is suspected, a potassium hydroxide stain can be performed on the subungual debris, which is then examined with direct microscopy.6 Alternatively, the physician can send a nail plate clipping in a 10% buffered formalin container with a request for a fungal stain such as periodic acid-Schiff.7 Microscopic examination of a dermatophytoma shows a dense mass of dermatophyte hyphae, otherwise known as a fungal abscess.8
The physician can play an important role in diagnosis because clinical findings suggestive of a dermatophytoma are associated with a poor response to antifungal therapy.9
Inherited diseases. White longitudinal bands are also an important clue to the rare autosomal dominant genodermatoses Hailey-Hailey disease (from mutations of the ATP2A2 gene) and Darier disease (from mutations of the ATP2C1 gene). Patients with Hailey-Hailey disease may have nails with multiple parallel longitudinal white stripes of variable width originating in the lunula and most prominent on the thumbs.10–12 These patients also have recurrent vesicular eruptions in flexural skin areas, such as the groin, axilla, neck, and periumbilical area causing significant morbidity.
Patients with Darier disease may have nails with alternating red and white longitudinal streaks, described as “candy-cane,”13 as well as wedge-shaped distal subungual keratosis accompanied by flat keratotic papules on the proximal nail fold.14 These nail changes are reported in 92% to 95% of patients with Darier disease.15,16 Patients typically have skin findings characterized by keratotic papules and plaques predominantly in seborrheic areas and palmoplantar pits, as well as secondary infections and malodor causing significant morbidity.15 Therefore, knowing the characteristic nail findings in these diseases may lead to more rapid diagnosis and treatment.
Mees lines. Leukonychia striata can present as transverse white lines, commonly known as Mees lines. They are 1- to 2-mm wide horizontal parallel white bands that span the width of the nail plate, usually affecting all fingernails.17 They are not a common finding and are most often associated with arsenic poisoning. They can also be used to identify the time of poisoning, since they tend to appear 2 months after the initial insult.
Mees lines are also associated with acute systemic stresses, such as acute renal failure, heart failure, ulcerative colitis, breast cancer, infections such as measles and tuberculosis, and systemic lupus erythematosus, and with exposure to toxic metals such as thallium.3
Apparent leukonychia
Apparent leukonychia can alert the physician to systemic diseases, infections, drug side effects, and nutrient deficiencies. Specific nail findings include Muehrcke lines, “half-and-half” nails, and Terry nails.
Muehrcke lines are paired white transverse bands that span the width of the nail bed and run parallel to the distal lunula. They were first described in the fingernails of patients with severe hypoalbuminemia, some of whom also had nephrotic syndrome, which resolved with normalization of the serum albumin level. Muehrcke lines have since been reported in patients with liver disease, malnutrition, chemotherapy, organ transplant, human immunodeficiency virus (HIV) infection, and acquired immunodeficiency syndrome.3,18 They are associated with periods of metabolic stress, ie, when the body’s capacity to synthesize proteins is diminished.19
Half-and-half nails, or Lindsay nails, are characterized by a white band proximally, a pink or red-brown band distally, and a sharp demarcation between the two (Figure 2). They were originally described in association with chronic renal disease,20 and surprisingly, they resolve with kidney transplant but not with hemodialysis treatment or improvement in hemoglobin or albumin levels.21–23 Half-and-half nails have been reported with Kawasaki disease, hepatic cirrhosis, Crohn disease, zinc deficiency, chemotherapy, Behçet disease, and pellagra.3,24,25 They should be distinguished from Terry nails, which are characterized by leukonychia involving more than 80% of the total nail length.26
Terry nails were originally reported in association with hepatic cirrhosis, usually secondary to alcoholism27 but have since been found with heart failure, type 2 diabetes mellitus, pulmonary tuberculosis, reactive arthritis, older age, Hansen disease, and peripheral vascular disease.3,26,28,29
LONGITUDINAL MELANONYCHIA: VERTICAL BROWN-BLACK NAIL LINES
Longitudinal melanonychia is the presence of black-brown vertical lines in the nail plate. They have a variety of causes, including blood from trauma; bacterial, fungal, or HIV infection; drug therapy (eg, from minocycline); endocrine disorders (Addison disease); exogenous pigmentation; or excess melanin production within the nail matrix.30–32 They may also be a sign of a benign condition such as benign melanocytic activation, lentigines, or nevi, or a malignant condition such as melanoma (Figure 3).33,34
When to suspect melanoma and refer
Although melanoma is less commonly associated with brown-black vertical nail lines, awareness of melanoma-associated longitudinal melanonychia reduces the likelihood of delayed diagnosis and improves patient outcomes.35 Also, it is important to remember that although nail melanoma is more common in the 5th and 6th decades of life, it can occur at any age, even in children.36
Findings that raise suspicion of nail melanoma (Table 1)33,37 and that should prompt referral to a dermatologist who specializes in nails include the following:
- A personal or family history of melanoma
- Involvement of a “high-risk” digit (thumb, index finger, great toe),30,31,38 although nail melanoma can occur in any digit
- Any new vertical brown-black nail pigmentation in a fair-skinned patient
- Only one nail affected: involvement of more than one nail is common in people with darker skin, and nearly all patients with darker skin exhibit longitudinal melanonychia by age 5031
- Changes in the band such as darkening, widening, and bleeding
- A bandwidth greater than 6 mm33
- A band that is wider proximally than distally34
- Nonuniform color of the line
- Indistinct lateral borders
- Associated with pigmentation of the nail fold (the Hutchinson sign, representing subungual melanoma),31,39 nail plate dystrophy, bleeding, or ulceration.33
While these features may help distinguish benign from malignant causes of longitudinal melanonychia, the clinical examination alone may not provide a definitive diagnosis. Delayed diagnosis of nail melanoma carries a high mortality rate; the internist can promote early diagnosis by recognizing the risk factors and clinical signs and referring the patient to a dermatologist for further evaluation with nail biopsy.
LONGITUDINAL ERYTHRONYCHIA: VERTICAL RED NAIL LINES
Longitudinal erythronychia—the presence of one or more linear red bands in the nail unit—can be localized (involving only one nail) or polydactylous (involving more than one nail). The localized form is usually due to a neoplastic process, whereas involvement of more than one nail may indicate an underlying regional or systemic disease.13Table 2 lists indications for referral to a nail specialist.
General features on examination
Clinical examination reveals one or more linear, pink-red streaks extending from the proximal nail fold to the distal free edge of the nail plate (Figure 4). The width of the band typically ranges from less than 1 mm to 3 mm.40 Other features may include splinter hemorrhages within a red band, a semitransparent distal matrix, distal V-shaped chipping, splitting, onycholysis of the nail plate, and reactive distal nail bed and hyponychial hyperkeratosis. These features can be visible to the naked eye but may be better viewed with a magnifying glass, a 7× loupe, or a dermatoscope.13
Localized longitudinal erythronychia is usually seen in middle-aged individuals and is most commonly found on the thumbnail, followed by the index finger.41,42 The condition may be asymptomatic, but the patient may present with pain or with concern that the split end of the nail catches on fabrics or small objects.42
Glomus tumor
Intense, pulsatile pain with sensitivity to cold and tenderness to palpation is highly suggestive of glomus tumor,43 a benign neoplasm that originates from a neuromyoarterial glomus body. Glomus bodies are located throughout the body but are more highly concentrated in the fingertips, especially beneath the nails, and they regulate skin circulation. Therefore, the nail unit is the most common site for glomus tumor.44,45 A characteristic feature of subungual glomus tumor is demonstration of tenderness after pin-point palpation of the suspected tumor (positive Love sign).45 While it is typical for glomus tumor to affect only one nail, multiple tumors are associated with neurofibromatosis type 1.46 Confirmation of this diagnosis requires referral to a dermatologist.
Other causes of localized red nail lines
Onychopapilloma, a benign idiopathic tumor, is the most common cause of localized longitudinal erythronychia. Unlike glomus tumor, it is usually asymptomatic.42,47 Less common benign conditions are warts, warty dyskeratoma, benign vascular proliferation, a solitary lesion of lichen planus, hemiplegia, and postsurgical scarring of the nail matrix. In some cases, the lines are idiopathic.42,43
Malignant diseases that can present as localized longitudinal erythronychia include invasive squamous cell carcinoma, squamous cell carcinoma in situ (Bowen disease), and, less frequently, amelanotic melanoma in situ, malignant melanoma, and basal cell carcinoma.42 Squamous cell carcinoma in situ most commonly presents in the 5th decade of life and is the malignancy most commonly associated with localized longitudinal erythronychia. Clinically, there is also often nail dystrophy, such as distal subungual keratosis or onycholysis.43
Patients with asymptomatic, stable localized longitudinal erythronychia may be followed closely with photography and measurements. However, any new lesion or a change in an existing lesion should prompt referral to a dermatologist for biopsy.13
Red streaks on more than one nail
Polydactylous longitudinal erythronychia usually presents in adults as red streaks on multiple nails and, depending on the presence or absence of symptoms (eg, pain, splitting), may be the patient’s chief complaint or an incidental finding noted by the astute clinician. Often, it is associated with systemic disease, most commonly lichen planus or Darier disease.
Lichen planus is a papulosquamous skin disease with nail involvement in 10% of patients and permanent nail dystrophy in 4%. Common nail findings include thinning, longitudinal ridging, and fissuring, as well as scarring of the nail matrix resulting in pterygium. Linear red streaks may accompany these more typical nail findings.13 Patients with Darier disease present with alternating red and white linear bands on multiple nails as in leukonychia striata.
Less frequently, polydactylous longitudinal erythronychia is associated with primary and systemic amyloidosis, hemiplegia, graft-vs-host disease, acantholytic epidermolysis bullosa, acantholytic dyskeratotic epidermal nevus, acrokeratosis verruciformis of Hopf, or pseudobulbar syndrome, or is idiopathic.13,42,48 Therefore, the physician evaluating a patient with these nail findings should focus on a workup for regional or systemic disease or refer the patient to a dermatologist who specializes in nails.
BEAU LINES
Beau lines are a common finding in clinical practice. They are not true lines, but transverse grooves in the nail plate that arise from the temporary suppression of nail growth within the nail matrix that can occur during periods of acute or chronic stress or systemic illness (Figure 5).49
The precipitating event may be local trauma or paronychia, chemotherapeutic agents cytotoxic to the nail matrix, or the abrupt onset of systemic disease.18,50 The grooves have also been associated with rheumatic fever, malaria, pemphigus, Raynaud disease, and myocardial infarction, as well as following deep-sea dives.51–53 The distance of a Beau line from the proximal nail fold can provide an estimate of the time of the acute stress, based on an average growth rate of 3 mm per month for fingernails and 1 mm per month for toenails.49
Inspection of the fingernails and toenails should be part of a complete physical examination. A basic understanding of nail anatomy and recognition of several basic types of nail lines and bands allow the clinician to properly diagnose and treat the nail disease, to recognize possible underlying systemic diseases, and to know when to refer the patient to a dermatologist for specialized evaluation and biopsy.
In this review, we delineate the three basic types of nail lines—white lines (leukonychia striata), brown-black lines (longitudinal melanonychia), and red lines (longitudinal erythronychia)—and the differential diagnosis for each type. We also discuss grooves in the nail plate, or Beau lines.
BASIC NAIL ANATOMY
A fundamental understanding of the anatomy of the nail unit is necessary to understand the origin of nail diseases and underlying pathologic conditions.
The nail unit includes the nail matrix, the lunula, the nail fold, the nail plate, and the nail bed. The nail matrix extends from under the proximal nail fold to the half-moon-shaped area (ie, the lunula) and is responsible for nail plate production. The nail bed lies under the nail plate and on top of the distal phalanx and extends from the lunula to just proximal to the free edge of the nail; its rich blood supply gives it its reddish color.
Nails grow slowly, and this should be kept in mind during the examination. Regrowth of a fingernail takes at least 6 months, and regrowth of a toenail may take 12 to 18 months. Therefore, a defect in the nail plate may reveal an injury that occurred—or a condition that began—several months before.1
NAIL EXAMINATION ESSENTIALS
A complete examination includes all 20 nail units and the periungual skin. Patients should be instructed to remove nail polish from all nails, as it may camouflage dystrophy or disease of the nail. Photography and careful measurement help document changes over time.
LEUKONYCHIA STRIATA: WHITE NAIL LINES
White nail lines or leukonychia is classified as true or apparent, depending on whether the origin is in the nail matrix or the nail bed.
In true leukonychia, there is abnormal keratinization of the underlying nail matrix, resulting in parakeratosis within the nail plate and an opaque appearance on examination.2 The white discoloration is unaffected by pressure, and the opacity moves distally as the nail grows out, which can be documented by serial photography on subsequent visits.
Apparent leukonychia involves abnormal nail bed vasculature, which changes the translucency of the nail plate. The whiteness disappears with pressure, is unaffected by nail growth, and will likely show no change on later visits with serial photography.3
True leukonychia
Leukonychia striata, a subtype of true leukonychia, is characterized by transverse or longitudinal bands. It is most often associated with microtrauma, such as from a manicure.4 Lines due to trauma are typically more apparent in the central part of the nail plate; they spare the lateral portion and lie parallel to the edge of the proximal nail fold.5
Onychomycosis. White longitudinal bands may also be seen in onychomycosis, a fungal infection of the nail accounting for up to 50% of all cases of nail disease. The infection may present as irregular dense longitudinal white or yellowish bands or “spikes” on the nail plate with associated hyperkeratosis, known as a dermatophytoma (Figure 1).
If a fungal infection is suspected, a potassium hydroxide stain can be performed on the subungual debris, which is then examined with direct microscopy.6 Alternatively, the physician can send a nail plate clipping in a 10% buffered formalin container with a request for a fungal stain such as periodic acid-Schiff.7 Microscopic examination of a dermatophytoma shows a dense mass of dermatophyte hyphae, otherwise known as a fungal abscess.8
The physician can play an important role in diagnosis because clinical findings suggestive of a dermatophytoma are associated with a poor response to antifungal therapy.9
Inherited diseases. White longitudinal bands are also an important clue to the rare autosomal dominant genodermatoses Hailey-Hailey disease (from mutations of the ATP2A2 gene) and Darier disease (from mutations of the ATP2C1 gene). Patients with Hailey-Hailey disease may have nails with multiple parallel longitudinal white stripes of variable width originating in the lunula and most prominent on the thumbs.10–12 These patients also have recurrent vesicular eruptions in flexural skin areas, such as the groin, axilla, neck, and periumbilical area causing significant morbidity.
Patients with Darier disease may have nails with alternating red and white longitudinal streaks, described as “candy-cane,”13 as well as wedge-shaped distal subungual keratosis accompanied by flat keratotic papules on the proximal nail fold.14 These nail changes are reported in 92% to 95% of patients with Darier disease.15,16 Patients typically have skin findings characterized by keratotic papules and plaques predominantly in seborrheic areas and palmoplantar pits, as well as secondary infections and malodor causing significant morbidity.15 Therefore, knowing the characteristic nail findings in these diseases may lead to more rapid diagnosis and treatment.
Mees lines. Leukonychia striata can present as transverse white lines, commonly known as Mees lines. They are 1- to 2-mm wide horizontal parallel white bands that span the width of the nail plate, usually affecting all fingernails.17 They are not a common finding and are most often associated with arsenic poisoning. They can also be used to identify the time of poisoning, since they tend to appear 2 months after the initial insult.
Mees lines are also associated with acute systemic stresses, such as acute renal failure, heart failure, ulcerative colitis, breast cancer, infections such as measles and tuberculosis, and systemic lupus erythematosus, and with exposure to toxic metals such as thallium.3
Apparent leukonychia
Apparent leukonychia can alert the physician to systemic diseases, infections, drug side effects, and nutrient deficiencies. Specific nail findings include Muehrcke lines, “half-and-half” nails, and Terry nails.
Muehrcke lines are paired white transverse bands that span the width of the nail bed and run parallel to the distal lunula. They were first described in the fingernails of patients with severe hypoalbuminemia, some of whom also had nephrotic syndrome, which resolved with normalization of the serum albumin level. Muehrcke lines have since been reported in patients with liver disease, malnutrition, chemotherapy, organ transplant, human immunodeficiency virus (HIV) infection, and acquired immunodeficiency syndrome.3,18 They are associated with periods of metabolic stress, ie, when the body’s capacity to synthesize proteins is diminished.19
Half-and-half nails, or Lindsay nails, are characterized by a white band proximally, a pink or red-brown band distally, and a sharp demarcation between the two (Figure 2). They were originally described in association with chronic renal disease,20 and surprisingly, they resolve with kidney transplant but not with hemodialysis treatment or improvement in hemoglobin or albumin levels.21–23 Half-and-half nails have been reported with Kawasaki disease, hepatic cirrhosis, Crohn disease, zinc deficiency, chemotherapy, Behçet disease, and pellagra.3,24,25 They should be distinguished from Terry nails, which are characterized by leukonychia involving more than 80% of the total nail length.26
Terry nails were originally reported in association with hepatic cirrhosis, usually secondary to alcoholism27 but have since been found with heart failure, type 2 diabetes mellitus, pulmonary tuberculosis, reactive arthritis, older age, Hansen disease, and peripheral vascular disease.3,26,28,29
LONGITUDINAL MELANONYCHIA: VERTICAL BROWN-BLACK NAIL LINES
Longitudinal melanonychia is the presence of black-brown vertical lines in the nail plate. They have a variety of causes, including blood from trauma; bacterial, fungal, or HIV infection; drug therapy (eg, from minocycline); endocrine disorders (Addison disease); exogenous pigmentation; or excess melanin production within the nail matrix.30–32 They may also be a sign of a benign condition such as benign melanocytic activation, lentigines, or nevi, or a malignant condition such as melanoma (Figure 3).33,34
When to suspect melanoma and refer
Although melanoma is less commonly associated with brown-black vertical nail lines, awareness of melanoma-associated longitudinal melanonychia reduces the likelihood of delayed diagnosis and improves patient outcomes.35 Also, it is important to remember that although nail melanoma is more common in the 5th and 6th decades of life, it can occur at any age, even in children.36
Findings that raise suspicion of nail melanoma (Table 1)33,37 and that should prompt referral to a dermatologist who specializes in nails include the following:
- A personal or family history of melanoma
- Involvement of a “high-risk” digit (thumb, index finger, great toe),30,31,38 although nail melanoma can occur in any digit
- Any new vertical brown-black nail pigmentation in a fair-skinned patient
- Only one nail affected: involvement of more than one nail is common in people with darker skin, and nearly all patients with darker skin exhibit longitudinal melanonychia by age 5031
- Changes in the band such as darkening, widening, and bleeding
- A bandwidth greater than 6 mm33
- A band that is wider proximally than distally34
- Nonuniform color of the line
- Indistinct lateral borders
- Associated with pigmentation of the nail fold (the Hutchinson sign, representing subungual melanoma),31,39 nail plate dystrophy, bleeding, or ulceration.33
While these features may help distinguish benign from malignant causes of longitudinal melanonychia, the clinical examination alone may not provide a definitive diagnosis. Delayed diagnosis of nail melanoma carries a high mortality rate; the internist can promote early diagnosis by recognizing the risk factors and clinical signs and referring the patient to a dermatologist for further evaluation with nail biopsy.
LONGITUDINAL ERYTHRONYCHIA: VERTICAL RED NAIL LINES
Longitudinal erythronychia—the presence of one or more linear red bands in the nail unit—can be localized (involving only one nail) or polydactylous (involving more than one nail). The localized form is usually due to a neoplastic process, whereas involvement of more than one nail may indicate an underlying regional or systemic disease.13Table 2 lists indications for referral to a nail specialist.
General features on examination
Clinical examination reveals one or more linear, pink-red streaks extending from the proximal nail fold to the distal free edge of the nail plate (Figure 4). The width of the band typically ranges from less than 1 mm to 3 mm.40 Other features may include splinter hemorrhages within a red band, a semitransparent distal matrix, distal V-shaped chipping, splitting, onycholysis of the nail plate, and reactive distal nail bed and hyponychial hyperkeratosis. These features can be visible to the naked eye but may be better viewed with a magnifying glass, a 7× loupe, or a dermatoscope.13
Localized longitudinal erythronychia is usually seen in middle-aged individuals and is most commonly found on the thumbnail, followed by the index finger.41,42 The condition may be asymptomatic, but the patient may present with pain or with concern that the split end of the nail catches on fabrics or small objects.42
Glomus tumor
Intense, pulsatile pain with sensitivity to cold and tenderness to palpation is highly suggestive of glomus tumor,43 a benign neoplasm that originates from a neuromyoarterial glomus body. Glomus bodies are located throughout the body but are more highly concentrated in the fingertips, especially beneath the nails, and they regulate skin circulation. Therefore, the nail unit is the most common site for glomus tumor.44,45 A characteristic feature of subungual glomus tumor is demonstration of tenderness after pin-point palpation of the suspected tumor (positive Love sign).45 While it is typical for glomus tumor to affect only one nail, multiple tumors are associated with neurofibromatosis type 1.46 Confirmation of this diagnosis requires referral to a dermatologist.
Other causes of localized red nail lines
Onychopapilloma, a benign idiopathic tumor, is the most common cause of localized longitudinal erythronychia. Unlike glomus tumor, it is usually asymptomatic.42,47 Less common benign conditions are warts, warty dyskeratoma, benign vascular proliferation, a solitary lesion of lichen planus, hemiplegia, and postsurgical scarring of the nail matrix. In some cases, the lines are idiopathic.42,43
Malignant diseases that can present as localized longitudinal erythronychia include invasive squamous cell carcinoma, squamous cell carcinoma in situ (Bowen disease), and, less frequently, amelanotic melanoma in situ, malignant melanoma, and basal cell carcinoma.42 Squamous cell carcinoma in situ most commonly presents in the 5th decade of life and is the malignancy most commonly associated with localized longitudinal erythronychia. Clinically, there is also often nail dystrophy, such as distal subungual keratosis or onycholysis.43
Patients with asymptomatic, stable localized longitudinal erythronychia may be followed closely with photography and measurements. However, any new lesion or a change in an existing lesion should prompt referral to a dermatologist for biopsy.13
Red streaks on more than one nail
Polydactylous longitudinal erythronychia usually presents in adults as red streaks on multiple nails and, depending on the presence or absence of symptoms (eg, pain, splitting), may be the patient’s chief complaint or an incidental finding noted by the astute clinician. Often, it is associated with systemic disease, most commonly lichen planus or Darier disease.
Lichen planus is a papulosquamous skin disease with nail involvement in 10% of patients and permanent nail dystrophy in 4%. Common nail findings include thinning, longitudinal ridging, and fissuring, as well as scarring of the nail matrix resulting in pterygium. Linear red streaks may accompany these more typical nail findings.13 Patients with Darier disease present with alternating red and white linear bands on multiple nails as in leukonychia striata.
Less frequently, polydactylous longitudinal erythronychia is associated with primary and systemic amyloidosis, hemiplegia, graft-vs-host disease, acantholytic epidermolysis bullosa, acantholytic dyskeratotic epidermal nevus, acrokeratosis verruciformis of Hopf, or pseudobulbar syndrome, or is idiopathic.13,42,48 Therefore, the physician evaluating a patient with these nail findings should focus on a workup for regional or systemic disease or refer the patient to a dermatologist who specializes in nails.
BEAU LINES
Beau lines are a common finding in clinical practice. They are not true lines, but transverse grooves in the nail plate that arise from the temporary suppression of nail growth within the nail matrix that can occur during periods of acute or chronic stress or systemic illness (Figure 5).49
The precipitating event may be local trauma or paronychia, chemotherapeutic agents cytotoxic to the nail matrix, or the abrupt onset of systemic disease.18,50 The grooves have also been associated with rheumatic fever, malaria, pemphigus, Raynaud disease, and myocardial infarction, as well as following deep-sea dives.51–53 The distance of a Beau line from the proximal nail fold can provide an estimate of the time of the acute stress, based on an average growth rate of 3 mm per month for fingernails and 1 mm per month for toenails.49
- Scher RK, Rich P, Pariser D, Elewski B. The epidemiology, etiology, and pathophysiology of onychomycosis. Semin Cutan Med Surg 2013; 32(suppl 1):S2–S4.
- Lawry MA, Haneke E, Strobeck K, Martin S, Zimmer B, Romano PS. Methods for diagnosing onychomycosis: a comparative study and review of the literature. Arch Dermatol 2000; 136:1112–1116.
- Zaiac MN, Walker A. Nail abnormalities associated with systemic pathologies. Clin Dermatol 2013; 31:627–649.
- Zaiac MN, Daniel CR. Nails in systemic disease. Dermatol Ther 2002; 15:99–106.
- Tosti A, Iorizzo M, Piraccini BM, Starace M. The nail in systemic diseases. Dermatol Clin 2006; 24:341–347.
- Scher RK, Daniel CR, eds. Nails Diagnosis, Therapy, Surgery. 3rd ed. Oxford: Elsevier Saunders; 2005.
- Smith MB, McGinnis MR. Diagnostic histopathology. In: Hospenthal DR, Rinaldi MG, eds. Diagnosis and Treatment of Human Mycoses. Totowa, NJ: Humana Press; 2008:37–51.
- Roberts DT, Evans EG. Subungual dermatophytoma complicating dermatophyte onychomycosis. Br J Dermatol 1998; 138:189–190.
- Sigurgeirsson B. Prognostic factors for cure following treatment of onychomycosis. J Eur Acad Dermatol Venereol 2010; 24:679–684.
- Kumar R, Zawar V. Longitudinal leukonychia in Hailey-Hailey disease: a sign not to be missed. Dermatol Online J 2008; 14:17.
- Burge SM. Hailey-Hailey disease: the clinical features, response to treatment and prognosis. Br J Dermatol 1992; 126:275–282.
- Kirtschig G, Effendy I, Happle R. Leukonychia longitudinalis as the primary symptom of Hailey-Hailey disease. Hautarzt 1992; 43:451–452. German.
- Jellinek NJ. Longitudinal erythronychia: suggestions for evaluation and management. J Am Acad Dermatol 2011; 64:167.e1–167.e11
- Zaias N, Ackerman AB. The nail in Darier-White disease. Arch Dermatol 1973; 107:193–199.
- Burge SM, Wilkinson JD. Darier-White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol 1992; 27:40–50.
- Munro CS. The phenotype of Darier's disease: penetrance and expressivity in adults and children. Br J Dermatol 1992; 127:126–130.
- Schwartz RA. Arsenic and the skin. Int J Dermatol 1997; 36:241–250.
- Fawcett RS, Linford S, Stulberg DL. Nail abnormalities: clues to systemic disease. Am Fam Physician 2004; 69:1417–1424.
- Morrison-Bryant M, Gradon JD. Images in clinical medicine. Muehrcke's lines. N Engl J Med 2007; 357:917.
- Daniel CR 3rd, Bower JD, Daniel CR Jr. The “half and half fingernail”: the most significant onychopathological indicator of chronic renal failure. J Miss State Med Assoc 1975; 16:367–370.
- Saray Y, Seckin D, Gulec AT, Akgun S, Haberal M. Nail disorders in hemodialysis patients and renal transplant recipients: a case-control study. J Am Acad Dermatol 2004; 50:197–202.
- Dyachenko P, Monselise A, Shustak A, Ziv M, Rozenman D. Nail disorders in patients with chronic renal failure and undergoing haemodialysis treatment: a case-control study. J Eur Acad Dermatol Venereol 2007; 21:340–344.
- Salem A, Al Mokadem S, Attwa E, Abd El Raoof S, Ebrahim HM, Faheem KT. Nail changes in chronic renal failure patients under haemodialysis. J Eur Acad Dermatol Venereol 2008; 22:1326–1331.
- Zagoni T, Sipos F, Tarjan Z, Peter Z. The half-and-half nail: a new sign of Crohn's disease? Report of four cases. Dis Colon Rectum 2006; 49:1071–1073.
- Nixon DW, Pirozzi D, York RM, Black M, Lawson DH. Dermatologic changes after systemic cancer therapy. Cutis 1981; 27:181–194.
- Holzberg M, Walker HK. Terry's nails: revised definition and new correlations. Lancet 1984; 1:896–899.
- Terry R. White nails in hepatic cirrhosis. Lancet 1954; 266:757–759.
- Coskun BK, Saral Y, Ozturk P, Coskun N. Reiter syndrome accompanied by Terry nail. J Eur Acad Dermatol Venereol 2005; 19:87–89.
- Blyumin M, Khachemoune A, Bourelly P. What is your diagnosis? Terry nails. Cutis 2005; 76:201–202.
- Haneke E, Baran R. Longitudinal melanonychia. Dermatol Surg 2001; 27:580–584.
- Andre J, Lateur N. Pigmented nail disorders. Dermatol Clin 2006; 24:329–339.
- Braun RP, Baran R, Le Gal FA, et al. Diagnosis and management of nail pigmentations. J Am Acad Dermatol 2007; 56:835–847.
- Mannava KA, Mannava S, Koman LA, Robinson-Bostom L, Jellinek N. Longitudinal melanonychia: detection and management of nail melanoma. Hand Surg 2013; 18:133–139.
- Ruben BS. Pigmented lesions of the nail unit: clinical and histopathologic features. Semin Cutan Med Surg 2010; 29:148–158.
- Cohen T, Busam KJ, Patel A, Brady MS. Subungual melanoma: management considerations. Am J Surg 2008; 195:244–248.
- Iorizzo M, Tosti A, Di Chiacchio N, et al. Nail melanoma in children: differential diagnosis and management. Dermatol Surg 2008; 34:974–978.
- Jellinek N. Nail matrix biopsy of longitudinal melanonychia: diagnostic algorithm including the matrix shave biopsy. J Am Acad Dermatol 2007; 56:803–810.
- Husain S, Scher RK, Silvers DN, Ackerman AB. Melanotic macule of nail unit and its clinicopathologic spectrum. J Am Acad Dermatol 2006; 54:664–667.
- Baran R, Kechijian P. Hutchinson's sign: a reappraisal. J Am Acad Dermatol 1996; 34:87–90.
- Baran R. Red nails. Dermatol Online 2005; 11:29.
- Baran R, Perrin C. Longitudinal erythronychia with distal subungual keratosis: onychopapilloma of the nail bed and Bowen’s disease. Br J Dermatol 2000; 143:132–135.
- de Berker DA, Perrin C, Baran R. Localized longitudinal erythronychia: diagnostic significance and physical explanation. Arch Dermatol 2004; 140:1253–1257.
- Cohen PR. Longitudinal erythronychia: individual or multiple linear red bands of the nail plate: a review of clinical features and associated conditions. Am J Clin Dermatol 2011; 12:217–231.
- Van Geertruyden J, Lorea P, Goldschmidt D, et al. Glomus tumours of the hand. A retrospective study of 51 cases. J Hand Surg Br 1996; 21:257–260.
- Moon SE, Won JH, Kwon OS, Kim JA. Subungual glomus tumor: clinical manifestations and outcome of surgical treatment. J Dermatol 2004; 31:993–997.
- Okada O, Demitsu T, Manabe M, Yoneda K. A case of multiple subungual glomus tumors associated with neurofibromatosis type 1. J Dermatol 1999; 26:535–537.
- Gee BC, Millard PR, Dawber RP. Onychopapilloma is not a distinct clinicopathological entity. Br J Dermatol 2002; 146:156–157.
- Siragusa M, Del Gracco S, Ferri R, Schepis C. Longitudinal red streaks on the big toenails in a patient with pseudobulbar syndrome. J Eur Acad Dermatol Venereol 2001; 15:85–86.
- Lipner S, Scher RK. Nails. In: Callen J, Jorizzo JL, eds. Dermatological Signs of Systemic Disease. 5th ed: Elsevier; in press.
- Mortimer NJ, Mills J. Images in clinical medicine. Beau’s lines. N Engl J Med 2004; 351:1778.
- Schwartz H. Clinical observation: Beau’s lines on fingernails after deep saturation dives. Undersea Hyperb Med 2006; 33:5–10.
- Gugelmann HM, Gaieski DF. Beau’s lines after cardiac arrest. Ther Hypothermia Temp Manag 2013; 3:199–202.
- Lauber J, Turk K. Beau’s lines and pemphigus vulgaris. Int J Dermatol 1990; 29:309.
- Scher RK, Rich P, Pariser D, Elewski B. The epidemiology, etiology, and pathophysiology of onychomycosis. Semin Cutan Med Surg 2013; 32(suppl 1):S2–S4.
- Lawry MA, Haneke E, Strobeck K, Martin S, Zimmer B, Romano PS. Methods for diagnosing onychomycosis: a comparative study and review of the literature. Arch Dermatol 2000; 136:1112–1116.
- Zaiac MN, Walker A. Nail abnormalities associated with systemic pathologies. Clin Dermatol 2013; 31:627–649.
- Zaiac MN, Daniel CR. Nails in systemic disease. Dermatol Ther 2002; 15:99–106.
- Tosti A, Iorizzo M, Piraccini BM, Starace M. The nail in systemic diseases. Dermatol Clin 2006; 24:341–347.
- Scher RK, Daniel CR, eds. Nails Diagnosis, Therapy, Surgery. 3rd ed. Oxford: Elsevier Saunders; 2005.
- Smith MB, McGinnis MR. Diagnostic histopathology. In: Hospenthal DR, Rinaldi MG, eds. Diagnosis and Treatment of Human Mycoses. Totowa, NJ: Humana Press; 2008:37–51.
- Roberts DT, Evans EG. Subungual dermatophytoma complicating dermatophyte onychomycosis. Br J Dermatol 1998; 138:189–190.
- Sigurgeirsson B. Prognostic factors for cure following treatment of onychomycosis. J Eur Acad Dermatol Venereol 2010; 24:679–684.
- Kumar R, Zawar V. Longitudinal leukonychia in Hailey-Hailey disease: a sign not to be missed. Dermatol Online J 2008; 14:17.
- Burge SM. Hailey-Hailey disease: the clinical features, response to treatment and prognosis. Br J Dermatol 1992; 126:275–282.
- Kirtschig G, Effendy I, Happle R. Leukonychia longitudinalis as the primary symptom of Hailey-Hailey disease. Hautarzt 1992; 43:451–452. German.
- Jellinek NJ. Longitudinal erythronychia: suggestions for evaluation and management. J Am Acad Dermatol 2011; 64:167.e1–167.e11
- Zaias N, Ackerman AB. The nail in Darier-White disease. Arch Dermatol 1973; 107:193–199.
- Burge SM, Wilkinson JD. Darier-White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol 1992; 27:40–50.
- Munro CS. The phenotype of Darier's disease: penetrance and expressivity in adults and children. Br J Dermatol 1992; 127:126–130.
- Schwartz RA. Arsenic and the skin. Int J Dermatol 1997; 36:241–250.
- Fawcett RS, Linford S, Stulberg DL. Nail abnormalities: clues to systemic disease. Am Fam Physician 2004; 69:1417–1424.
- Morrison-Bryant M, Gradon JD. Images in clinical medicine. Muehrcke's lines. N Engl J Med 2007; 357:917.
- Daniel CR 3rd, Bower JD, Daniel CR Jr. The “half and half fingernail”: the most significant onychopathological indicator of chronic renal failure. J Miss State Med Assoc 1975; 16:367–370.
- Saray Y, Seckin D, Gulec AT, Akgun S, Haberal M. Nail disorders in hemodialysis patients and renal transplant recipients: a case-control study. J Am Acad Dermatol 2004; 50:197–202.
- Dyachenko P, Monselise A, Shustak A, Ziv M, Rozenman D. Nail disorders in patients with chronic renal failure and undergoing haemodialysis treatment: a case-control study. J Eur Acad Dermatol Venereol 2007; 21:340–344.
- Salem A, Al Mokadem S, Attwa E, Abd El Raoof S, Ebrahim HM, Faheem KT. Nail changes in chronic renal failure patients under haemodialysis. J Eur Acad Dermatol Venereol 2008; 22:1326–1331.
- Zagoni T, Sipos F, Tarjan Z, Peter Z. The half-and-half nail: a new sign of Crohn's disease? Report of four cases. Dis Colon Rectum 2006; 49:1071–1073.
- Nixon DW, Pirozzi D, York RM, Black M, Lawson DH. Dermatologic changes after systemic cancer therapy. Cutis 1981; 27:181–194.
- Holzberg M, Walker HK. Terry's nails: revised definition and new correlations. Lancet 1984; 1:896–899.
- Terry R. White nails in hepatic cirrhosis. Lancet 1954; 266:757–759.
- Coskun BK, Saral Y, Ozturk P, Coskun N. Reiter syndrome accompanied by Terry nail. J Eur Acad Dermatol Venereol 2005; 19:87–89.
- Blyumin M, Khachemoune A, Bourelly P. What is your diagnosis? Terry nails. Cutis 2005; 76:201–202.
- Haneke E, Baran R. Longitudinal melanonychia. Dermatol Surg 2001; 27:580–584.
- Andre J, Lateur N. Pigmented nail disorders. Dermatol Clin 2006; 24:329–339.
- Braun RP, Baran R, Le Gal FA, et al. Diagnosis and management of nail pigmentations. J Am Acad Dermatol 2007; 56:835–847.
- Mannava KA, Mannava S, Koman LA, Robinson-Bostom L, Jellinek N. Longitudinal melanonychia: detection and management of nail melanoma. Hand Surg 2013; 18:133–139.
- Ruben BS. Pigmented lesions of the nail unit: clinical and histopathologic features. Semin Cutan Med Surg 2010; 29:148–158.
- Cohen T, Busam KJ, Patel A, Brady MS. Subungual melanoma: management considerations. Am J Surg 2008; 195:244–248.
- Iorizzo M, Tosti A, Di Chiacchio N, et al. Nail melanoma in children: differential diagnosis and management. Dermatol Surg 2008; 34:974–978.
- Jellinek N. Nail matrix biopsy of longitudinal melanonychia: diagnostic algorithm including the matrix shave biopsy. J Am Acad Dermatol 2007; 56:803–810.
- Husain S, Scher RK, Silvers DN, Ackerman AB. Melanotic macule of nail unit and its clinicopathologic spectrum. J Am Acad Dermatol 2006; 54:664–667.
- Baran R, Kechijian P. Hutchinson's sign: a reappraisal. J Am Acad Dermatol 1996; 34:87–90.
- Baran R. Red nails. Dermatol Online 2005; 11:29.
- Baran R, Perrin C. Longitudinal erythronychia with distal subungual keratosis: onychopapilloma of the nail bed and Bowen’s disease. Br J Dermatol 2000; 143:132–135.
- de Berker DA, Perrin C, Baran R. Localized longitudinal erythronychia: diagnostic significance and physical explanation. Arch Dermatol 2004; 140:1253–1257.
- Cohen PR. Longitudinal erythronychia: individual or multiple linear red bands of the nail plate: a review of clinical features and associated conditions. Am J Clin Dermatol 2011; 12:217–231.
- Van Geertruyden J, Lorea P, Goldschmidt D, et al. Glomus tumours of the hand. A retrospective study of 51 cases. J Hand Surg Br 1996; 21:257–260.
- Moon SE, Won JH, Kwon OS, Kim JA. Subungual glomus tumor: clinical manifestations and outcome of surgical treatment. J Dermatol 2004; 31:993–997.
- Okada O, Demitsu T, Manabe M, Yoneda K. A case of multiple subungual glomus tumors associated with neurofibromatosis type 1. J Dermatol 1999; 26:535–537.
- Gee BC, Millard PR, Dawber RP. Onychopapilloma is not a distinct clinicopathological entity. Br J Dermatol 2002; 146:156–157.
- Siragusa M, Del Gracco S, Ferri R, Schepis C. Longitudinal red streaks on the big toenails in a patient with pseudobulbar syndrome. J Eur Acad Dermatol Venereol 2001; 15:85–86.
- Lipner S, Scher RK. Nails. In: Callen J, Jorizzo JL, eds. Dermatological Signs of Systemic Disease. 5th ed: Elsevier; in press.
- Mortimer NJ, Mills J. Images in clinical medicine. Beau’s lines. N Engl J Med 2004; 351:1778.
- Schwartz H. Clinical observation: Beau’s lines on fingernails after deep saturation dives. Undersea Hyperb Med 2006; 33:5–10.
- Gugelmann HM, Gaieski DF. Beau’s lines after cardiac arrest. Ther Hypothermia Temp Manag 2013; 3:199–202.
- Lauber J, Turk K. Beau’s lines and pemphigus vulgaris. Int J Dermatol 1990; 29:309.
KEY POINTS
- Transverse white nail lines, or Mees lines, have been associated with acute systemic stress, such as from acute renal failure, heart failure, ulcerative colitis, breast cancer, infection (measles, tuberculosis), and systemic lupus erythematosus, and with exposure to toxic metals such as thallium.
- In true leukonychia, there is abnormal keratinization of the underlying nail matrix, resulting in a white discoloration that is unaffected by pressure. In apparent leukonychia, the white discoloration is due to abnormal nail bed vasculature, and the whiteness disappears with pressure.
- Brown-black nail lines may represent blood from trauma; bacterial, fungal, or viral infection; drug reaction; endocrine disorders; exogenous pigmentation; excess melanin production within the nail matrix; nevi; or melanoma.
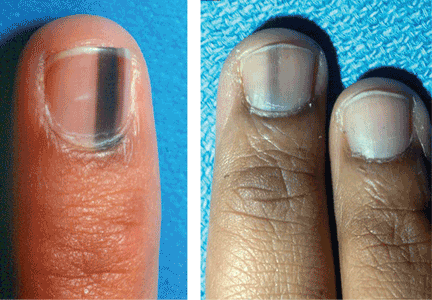
Pseudomembranous colitis: Not always Clostridium difficile
Pseudomembranous colitis is most often due to Clostridium difficile infection, but it has a variety of other causes, including other infections, ischemia, medications, and inflammatory mucosal diseases (Table 1). When pseudomembranes are found, one should consider these other causes if tests for C difficile are negative or if anti-C difficile therapy does not produce a response.

These less common causes are important to consider to avoid needlessly escalating anti-C difficile antibiotic therapy and to provide appropriate treatment. Pseudomembranous colitis is a nonspecific finding that suggests a larger disease process. Associated signs and symptoms, including fever, abdominal pain, leukocytosis, diarrhea, toxic megacolon, and electrolyte imbalances, may portend a life-threatening condition.1 Awareness of causes of pseudomembranous colitis other than C difficile infection, the focus of this review, is key to prompt diagnosis and potentially life-saving patient care.
PSEUDOMEMBRANES ARE NONSPECIFIC
A pseudomembrane is a layer of fibropurulent exudate composed of acute inflammatory cells and mucus originating from inflamed and erupting crypts.2 Although most often seen in C difficile infection, pseudomembranous colitis is a nonspecific pattern of injury resulting from decreased oxygenation, endothelial damage, and impaired blood flow to the mucosa that can be triggered by a number of disease states.2
On endoscopy, pseudomembranes appear as raised whitish or yellowish plaques that may be scattered or confluent in distribution (Figure 1).2 They are usually found in the rectosigmoid colon but may be isolated to more proximal segments.3 Lower endoscopy is often performed in the diagnostic evaluation of patients with unexplained diarrhea, hematochezia, and abnormal abdominal computed tomographic findings (eg, colonic thickening).
CAUSES OF NON-C DIFFICLE PSEUDOMEMBRANOUS COLITIS
When pseudomembranous colitis is confirmed endoscopically, C difficile infection naturally comes to mind, but the two terms are not interchangeable. A wide differential diagnosis should be maintained, especially when there are clues that C difficile infection may not be the correct diagnosis.
Chemicals and medications
Several chemicals and medications can injure the bowel and predispose to pseudomembrane formation.
Glutaraldehyde has long been used to sanitize endoscopes because of its broad antimicrobial activity. Nevertheless, if the disinfecting solution is not adequately rinsed off the endoscope, direct contact with colonic mucosa can produce an allergic and a chemical reaction, resulting in an acute self-limited colitis with pseudomembrane formation.4
Chemotherapeutic and antiproliferative agents can be toxic to the bowel, generally through production of free radicals and up-regulation of inflammatory cytokines. The colonic epithelium is then more susceptible to ulceration and mucosal necrosis with pseudomembrane development. Cisplatin, cyclosporine A, docetaxel, and 5-fluorouracil are prominent examples.5–8
Nonsteroidal anti-inflammatory drugs can damage the mucosa at all levels of the gastrointestinal tract. Although gastric ulcerations are more typical (from nonselective cyclooxygenase inhibition), colonic ulcerations and colitis can occur.9 These drugs, particularly diclofenac and indomethacin, have been associated with non-C difficile pseudomembranous colitis when used by themselves or in conjunction with other agents such as cyclosporine A.8,10,11
Infections
C difficile is the organism most commonly linked to pseudomembranous colitis, but other bacterial, viral, and parasitic pathogens have also been implicated.
Staphylococcus aureus was believed to be responsible for enterocolitis in a series of 155 surgical patients between 1958 and 1962 receiving antibiotic therapy. All had a positive stool culture for S aureus, and nine were found to have pseudomembranes at autopsy.12
Although this finding has been disputed as a misdiagnosis, since C difficile infection was not widely recognized until the 1970s, there is evidence that S aureus may indeed be a cause of non-C difficile pseudomembranous colitis. In a review of 36 cases of methicillin-resistant S aureus bacteremia in Japan, four patients were documented to have intestinal pseudomembranes either by endoscopy or autopsy. In two of these patients, biopsies of the pseudomembranes were positive for methicillin-resistant S aureus.13
Escherichia coli O157:H7. Pseudomembranes have been seen endoscopically in several adults and children with enterohemorrhagic E coli O157:H7 infection.14,15 This invasive gram-negative rod normally resides in the gastrointestinal tract of cattle, sheep, and other animals and can be pathogenic to people who eat undercooked beef. The organism attaches to and effaces intestinal epithelial cells, and bacterial proteins and the Shiga toxin then damage the vasculature, precipitating bloody diarrhea. Colonic damage can range from mild hemorrhagic colitis to severe colitis with ischemic changes. In patients with enterohemorrhagic E coli O157:H7 infection, pseudomembrane formation results from colon ischemia due to microvascular thrombosis or from destructive effects of bacterial enterotoxin.1,15
Cytomegalovirus is a ubiquitous human herpes virus that can affect nearly all organ systems. Infection is often reported in immunocompromised patients, eg, those with acquired immunodeficiency syndrome, chronic corticosteroid use, inflammatory bowel disease, malignancy, or solid-organ transplants. Gastrointestinal manifestations can be nonspecific and range from abdominal discomfort to diarrhea to tenesmus. Pseudomembranous colitis can be a presenting feature of cytomegalovirus involvement.16,17 Ulcerative lesions in the colonic mucosa are a frequent accompanying finding on endoscopy.18,19
Other rarer infectious causes of pseudomembranous colitis identified in case reports include Clostridium ramosum, Entamoeba histolytica, Klebsiella oxytoca, Plesiomonas shigelloides, Schistosomiasis mansoni, and Salmonella and Shigella species.20–26 Additionally, there have been two reports of Strongyloides stercoralis hyperinfection manifesting as pseudomembranous colitis.27,28
Ischemia
Colon ischemia usually affects elderly or debilitated patients who have multiple comorbidities. Known risk factors include aortoiliac surgery, cardiovascular disease, diabetes mellitus, hemodialysis, and pulmonary disease. The ischemia can be related to an occlusive arterial or venous thromboembolism, but hypoperfusion without occlusion of the mesenteric or internal iliac arteries is the primary mechanism. Low blood flow states such as atherosclerosis and septic shock can affect the watershed areas, typically the splenic flexure and rectosigmoid junction.
We reported a case of a patient with vascular disease who was incorrectly diagnosed and treated as having refractory C difficile infection when pseudomembranes were seen on flexible sigmoidoscopy. Further investigation revealed ischemic colitis secondary to a high-grade inferior mesenteric artery stenosis as the true cause.29
Microvascular thrombosis is the likely mechanism in a number of non-C difficile causes of pseudomembranous colitis. For example, in most patients with enterohemorrhagic E coli O157:H7 infection, histologic review of colonic mucosal biopsies has revealed fibrin and platelet thrombi in the capillaries, suggesting microvascular thrombosis.15,30
Cocaine has been associated with pseudomembranes in the setting of ischemia in the cecum and ascending colon. Cocaine can cause vasoconstriction after stimulation of alpha-adrenergic receptors and hence intestinal ischemia, thrombosis of vessels in the large and small intestines, and direct toxic effects.31
Inflammatory conditions
Collagenous colitis is an inflammatory disease that often affects middle-aged women and presents with copious watery diarrhea. It is a type of microscopic colitis—the endoscopic appearance is often normal, while the histologic appearance is abnormal and characterized by collagen deposition in the lamina propria. Medications that have been implicated in microscopic colitis include acid-suppressive agents (eg, histamine receptor antagonists, proton pump inhibitors) and nonsteroidal anti-inflammatory drugs.
An increasing number of cases of pseudomembranous changes are being reported in patients diagnosed with collagenous colitis.32–36 Although the pathophysiologic mechanism is unknown, some authors have suggested that pseudomembrane formation is actually part of the presenting spectrum of collagenous colitis.36
Inflammatory bowel disease. Crohn disease and ulcerative colitis have been associated with pseudomembranous colitis. Pseudomembranes can be found on endoscopy in patients with inflammatory bowel disease during a disease exacerbation with or without C difficile.37,38 In patients with inflammatory bowel disease and C difficile infection, pseudomembranes can be found endoscopically in up to 13% of cases.39 Pseudomembranous colitis has been reported in a patient with ulcerative colitis exacerbation in association with cytomegalovirus colitis.40
Behçet disease is a rare, immune-mediated small-vessel systemic vasculitis. It usually presents with mucous membrane ulcerations and ocular disease but can affect any organ.41 Pseudomembranous colitis can occur in Behçet disease in the absence of C difficile infection or any infectious colitis. Treatment includes corticosteroids and immunosuppressants such as azathioprine and anti-tumor necrosis factor agents.41
INITIAL EVALUATION
The initial evaluation of a patient with suspected or confirmed pseudomembranous colitis should include a comprehensive medical history with information on recent hospitalizations or procedures, antibiotic use, infections, exposure to sick contacts, recent travel, and medications taken.
Testing for C difficile
As most patients with pseudomembranous colitis have C difficile infection, it should be excluded first. Empiric anti-C difficile treatment is recommended in seriously ill-appearing patients, ideally starting after a stool sample is obtained.
Diagnosis of C difficile infection requires laboratory demonstration of the toxin or detection of toxigenic organisms. The gold standard test is the cell culture cytotoxicity assay, but it is labor- and time-intensive.42 More widely available tests are polymerase chain reaction for the toxin gene or genes, enzyme immunoassay, and stool evaluation for glutamate dehydrogenase, which can yield results readily within hours.
Polymerase chain reaction has a sensitivity of 97% and a specificity of 93%. Results can be falsely positive if empiric treatment is started before specimen collection, in which case C difficile DNA may still be present and detectable, but not the organism itself.43
Enzyme immunoassay for toxins A and B carries a sensitivity of 75% and a specificity of 99%, but 100 to 1,000 pg of toxin must be present for a positive result.44,45
If the initial enzyme immunoassay or polymerase chain reaction result is negative, current guidelines do not recommend repeat testing, which has limited value.44,46 Repeat testing after a negative result is positive in less than 5% of samples and greatly increases the chances of false-positive results.44,46 Nevertheless, if a laboratory’s enzyme immunoassay test has a low sensitivity, repeating negative tests may improve its sensitivity.
Glutamate dehydrogenase is an enzyme produced by both toxigenic and nontoxigenic strains of C difficile. As a result, stool testing for glutamate dehydrogenase is sensitive but not specific for C difficile infection, although multistep testing sequences (glutamate dehydrogenase followed by polymerase chain reaction) have proven to be useful screening tools.44
Treatment for C difficile infection
If testing for C difficile is positive, treatment is generally based on the severity and the complications of the illness46:
- Mild or moderate C difficile infection should be treated with oral metronidazole 500 mg three times per day for 10 to 14 days.
- Severe infection, which is defined as a white blood cell count of 15.0 × 109/L or higher or a serum creatinine level greater than or equal to 1.5 times the premorbid level, should be treated with oral vancomycin 125 mg four times per day for 10 to 14 days.
- Severe C difficile infection complicated by hypotension, shock, ileus, or megacolon should be treated with a combination of high-dose oral vancomycin (and possibly rectal vancomycin as well) at 500 mg four times per day plus intravenous metronidazole.
Additional treatment recommendations for individualized situations, recurrent C difficile infection, and comorbid conditions are discussed elsewhere.46
ENDOSCOPIC EVALUATION
Colonoscopy or flexible sigmoidoscopy is the primary means by which pseudomembranous colitis is diagnosed. Lower endoscopy should be pursued as an adjunctive tool when C difficile infection remains strongly suspected despite negative testing, when presumed C difficile infection does not respond to medical therapy, and when non-C difficile diagnoses are considered. If pseudomembranes are demonstrated on lower endoscopy, obtaining biopsy samples of normal- and abnormal-appearing mucosa is recommended. Pseudomembranes are suggestive but not diagnostic of C difficile infection, and microscopic evaluation of the mucosa is warranted to explore causes of pseudomembranous colitis not related to C difficile.
The pattern and distribution of pseudomembranes may provide clues to the etiology and the degree of mucosal injury. In intestinal ischemia, for example, a localized segment of the bowel is typically involved, and mucosal changes are often well delineated from normal mucosa. On endoscopic examination, mild ischemia is characterized by granular mucosa with decreased vascularity, whereas friable, edematous, ulcerated, and at times necrotic mucosa is evident in severe cases. Punctate pseudomembrane formation is seen in early ischemia, but as injury progresses, the pseudomembranes may grow and merge. In fact, diffuse involvement of the mucosal surface of the biopsy specimen by pseudomembranes has been shown to be more closely associated with ischemic colitis than C difficile infection.47
MICROSCOPIC EVALUATION
Histologic study can differentiate the various causes of pseudomembranous colitis.
In C difficile infection and drug reaction, there is acute crypt injury and dilation. The upper lamina propria is usually involved, and affected crypts are filled with an exudate similar to that found in pseudomembranes.1 However, in drug reaction, there is also prominent apoptosis and increased intraepithelial lymphocytosis.9
In colon ischemia, hyalinization of the lamina propria is a sensitive and specific marker.47 This has been shown in a study comparing histologic characteristics of colonic biopsies in patients with pseudomembranous colitis due to either known colon ischemia or C difficile infection.47 Crypt atrophy, lamina propria hemorrhage, full-thickness mucosal necrosis, and layering of pseudomembranes would further favor the diagnosis.
In collagenous colitis, a thickened subepithelial collagen band and intraepithelial lymphocytosis are often seen.
In inflammatory bowel disease, even with secondary pseudomembranes, ulcerative colitis and Crohn disease retain the characteristics of inflammatory bowel disease with crypt architectural distortion and focal or diffuse basal lymphoplasmacytosis on microscopy.48
- Gebhard RL, Gerding DN, Olson MM, et al. Clinical and endoscopic findings in patients early in the course of clostridium difficile-associated pseudomembranous colitis. Am J Med 1985; 78:45–48.
- Carpenter HA, Talley NJ. The importance of clinicopathological correlation in the diagnosis of inflammatory conditions of the colon: histological patterns with clinical implications. Am J Gastroenterol 2000; 95:878–896.
- Seppälä K. Colonoscopy in the diagnosis of pseudomembranous colitis. Br Med J 1978; 2:435.
- Stein BL, Lamoureux E, Miller M, Vasilevsky CA, Julien L, Gordon PH. Glutaraldehyde-induced colitis. Can J Surg 2001; 44:113–116.
- Trevisani F, Simoncini M, Alampi G, Bernardi M. Colitis associated to chemotherapy with 5-fluorouracil. Hepatogastroenterology 1997; 44:710–712.
- Takao T, Nishida M, Maeda Y, Takao K, Oka M. The study of continuous infusion chemotherapy with low-dose cisplatin and 5-fluorouracil for patients with primary liver cancer. Gan To Kagaku Ryoho 1997; 24:1724–1727. Japanese.
- Carrion AF, Hosein PJ, Cooper EM, Lopes G, Pelaez L, Rocha-Lima CM. Severe colitis associated with docetaxel use: a report of four cases. World J Gastrointest Oncol 2010; 2:390-394.
- Constantopoulos A. Colitis induced by interaction of cyclosporine A and non-steroidal anti-inflammatory drugs. Pediatr Int 1999; 41:184–186.
- Price AB. Pathology of drug-associated gastrointestinal disease. Br J Clin Pharmacol 2003; 56:477–482.
- Gentric A, Pennec YL. Diclofenac-induced pseudomembranous colitis. Lancet 1992; 340:126–127.
- Romero-Gómez M, Suárez García E, Castro Fernández M. Pseudomembranous colitis induced by diclofenac. J Clin Gastroenterol 1998; 26:228.
- Altemeier WA, Hummel RP, Hill EO. Staphylococcal enterocolitis following antibiotic therapy. Ann Surg 1963; 157:847–858.
- Ogawa Y, Saraya T, Koide T, et al. Methicillin-resistant Staphylococcus aureus enterocolitis sequentially complicated with septic arthritis: a case report and review of the literature. BMC Res Notes 2014; 7:21.
- Griffin PM, Olmstead LC, Petras RE. Escherichia coli O157:H7-associated colitis. A clinical and histological study of 11 cases. Gastroenterology 1990; 99:142–149.
- Uc A, Mitros FA, Kao SC, Sanders KD. Pseudomembranous colitis with Escherichia coli O157:H7. J Pediatr Gastroenterol Nutr 1997; 24:590–593.
- Battaglino MP, Rockey DC. Cytomegalovirus colitis presenting with the endoscopic appearance of pseudomembranous colitis. Gastrointest Endosc 1999; 50:697–700.
- Olofinlade O, Chiang C. Cytomegalovirus infection as a cause of pseudomembrane colitis: a report of four cases. J Clin Gastroenterol 2001; 32:82–84.
- Wilcox CM, Chalasani N, Lazenby A, Schwartz DA. Cytomegalovirus colitis in acquired immunodeficiency syndrome: a clinical and endoscopic study. Gastrointest Endosc 1998; 48:39–43.
- Seo TH, Kim JH, Ko SY, et al. Cytomegalovirus colitis in immunocompetent patients: a clinical and endoscopic study. Hepatogastroenterology 2012; 59:2137–2141.
- Högenauer C, Langner C, Beubler E, et al. Klebsiella oxytoca as a causative organism of antibiotic-associated hemorrhagic colitis. N Engl J Med 2006; 355:2418–2426.
- Hovius SE, Rietra PJ. Salmonella colitis clinically presenting as a pseudomembranous colitis. Neth J Surg 1982; 34:81–82.
- Kelber M, Ament ME. Shigella dysenteriae I: a forgotten cause of pseudomembranous colitis. J Pediatr 1976; 89:595–596.
- Neves J, Raso P, Pinto Dde M, da Silva SP, Alvarenga RJ. Ischaemic colitis (necrotizing colitis, pseudomembranous colitis) in acute schistosomiasis mansoni: report of two cases. Trans R Soc Trop Med Hyg 1993; 87:449–452.
- Alcalde-Vargas A, Trigo-Salado C, Leo Carnerero E, De-la-Cruz-Ramírez D, Herrera-Justiniano JM. Pseudomembranous colitis and bacteremia in an immunocompetent patient associated with a rare specie of Clostridium (C. ramosum). Rev Esp Enferm Dig 2012; 104:498–499.
- Koo JS, Choi WS, Park DW. Fulminant amebic colitis mimicking pseudomembranous colitis. Gastrointest Endosc 2010; 71:400–401.
- van Loon FP, Rahim Z, Chowdhury KA, Kay BA, Rahman SA. Case report of Plesiomonas shigelloides-associated persistent dysentery and pseudomembranous colitis. J Clin Microbiol 1989; 27:1913–1915.
- Janvier J, Kuhn S, Church D. Not all pseudomembranous colitis is caused by Clostridium difficile. Can J Infect Dis Med Microbiol 2008; 19:256–257.
- Jain AK, Agarwal SK, el-Sadr W. Streptococcus bovis bacteremia and meningitis associated with Strongyloides stercoralis colitis in a patient infected with human immunodeficiency virus. Clin Infect Dis 1994; 18:253–254.
- Tang DM, Urrunaga NH, De Groot H, von Rosenvinge EC, Xie G, Ghazi LJ. Pseudomembranous colitis: not always caused by Clostridium difficile. Case Rep Med 2014; 2014:812704.
- Kendrick JB, Risbano M, Groshong SD, Frankel SK. A rare presentation of ischemic pseudomembranous colitis due to Escherichia coli O157:H7. Clin Infect Dis 2007; 45:217–219.
- Fishel R, Hamamoto G, Barbul A, Jiji V, Efron G. Cocaine colitis. Is this a new syndrome? Dis Colon Rectum 1985; 28:264–266.
- Khan-Kheil AM, Disney B, Ruban E, Wood G. Pseudomembranous collagenous colitis: an unusual cause of chronic diarrhoea. BMJ Case Rep 2014; 2014.
- Villanacci V, Cristina S, Muscarà M, et al. Pseudomembranous collagenous colitis with superimposed drug damage. Pathol Res Pract 2013; 209:735–739.
- Denız K, Coban G, Ozbakir O, Denız E. Pseudomembranous collagenous colitis. Turk J Gastroenterol 2012; 23:93–95.
- Vesoulis Z, Lozanski G, Loiudice T. Synchronous occurrence of collagenous colitis and pseudomembranous colitis. Can J Gastroenterol 2000; 14:353–358.
- Yuan S, Reyes V, Bronner MP. Pseudomembranous collagenous colitis. Am J Surg Pathol 2003; 27:1375–1379.
- Berdichevski T, Barshack I, Bar-Meir S, Ben-Horin S. Pseudomembranes in a patient with flare-up of inflammatory bowel disease (IBD): is it only Clostridium difficile or is it still an IBD exacerbation? Endoscopy 2010; 42(suppl 2):E131.
- Kilinçalp S, Altinbas A, Basar O, Deveci M, Yüksel O. A case of ulcerative colitis co-existing with pseudo-membranous enterocolitis. J Crohns Colitis 2011; 5:506–507.
- Ben-Horin S, Margalit M, Bossuyt P, et al; European Crohn’s and Colitis Organization (ECCO). Prevalence and clinical impact of endoscopic pseudomembranes in patients with inflammatory bowel disease and Clostridium difficile infection. J Crohns Colitis 2010; 4:194–198.
- Chiba M, Abe T, Tsuda S, Ono I. Cytomegalovirus infection associated with onset of ulcerative colitis. BMC Res Notes 2013; 6:40.
- Shukla A, Tolan RW. Behcet disease presenting with pseudomembranous colitis and progression to neurological involvement: case report and review of the literature. Clin Pediatr (Phila) 2012; 51:1197–1201.
- Shanholtzer CJ, Willard KE, Holter JJ, Olson MM, Gerding DN, Peterson LR. Comparison of the VIDAS Clostridium difficile toxin A immunoassay with C. difficile culture and cytotoxin and latex tests. J Clin Microbiol 1992; 30:1837–1840.
- Huang H, Weintraub A, Fang H, Nord CE. Comparison of a commercial multiplex real-time PCR to the cell cytotoxicity neutralization assay for diagnosis of Clostridium difficile infections. J Clin Microbiol 2009; 47:3729–3731.
- Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society For Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol 2010; 31:431–455.
- Bartlett JG. Clinical practice. Antibiotic-associated diarrhea. N Engl J Med 2002; 346:334–339.
- Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol 2013; 108:478–498.
- Dignan CR, Greenson JK. Can ischemic colitis be differentiated from C difficile colitis in biopsy specimens? Am J Surg Pathol 1997; 21:706–710.
- Jenkins D, Balsitis M, Gallivan S, et al. Guidelines for the initial biopsy diagnosis of suspected chronic idiopathic inflammatory bowel disease. The British Society of Gastroenterology Initiative. J Clin Pathol 1997; 50:93–105.
Pseudomembranous colitis is most often due to Clostridium difficile infection, but it has a variety of other causes, including other infections, ischemia, medications, and inflammatory mucosal diseases (Table 1). When pseudomembranes are found, one should consider these other causes if tests for C difficile are negative or if anti-C difficile therapy does not produce a response.
These less common causes are important to consider to avoid needlessly escalating anti-C difficile antibiotic therapy and to provide appropriate treatment. Pseudomembranous colitis is a nonspecific finding that suggests a larger disease process. Associated signs and symptoms, including fever, abdominal pain, leukocytosis, diarrhea, toxic megacolon, and electrolyte imbalances, may portend a life-threatening condition.1 Awareness of causes of pseudomembranous colitis other than C difficile infection, the focus of this review, is key to prompt diagnosis and potentially life-saving patient care.
PSEUDOMEMBRANES ARE NONSPECIFIC
A pseudomembrane is a layer of fibropurulent exudate composed of acute inflammatory cells and mucus originating from inflamed and erupting crypts.2 Although most often seen in C difficile infection, pseudomembranous colitis is a nonspecific pattern of injury resulting from decreased oxygenation, endothelial damage, and impaired blood flow to the mucosa that can be triggered by a number of disease states.2
On endoscopy, pseudomembranes appear as raised whitish or yellowish plaques that may be scattered or confluent in distribution (Figure 1).2 They are usually found in the rectosigmoid colon but may be isolated to more proximal segments.3 Lower endoscopy is often performed in the diagnostic evaluation of patients with unexplained diarrhea, hematochezia, and abnormal abdominal computed tomographic findings (eg, colonic thickening).
CAUSES OF NON-C DIFFICLE PSEUDOMEMBRANOUS COLITIS
When pseudomembranous colitis is confirmed endoscopically, C difficile infection naturally comes to mind, but the two terms are not interchangeable. A wide differential diagnosis should be maintained, especially when there are clues that C difficile infection may not be the correct diagnosis.
Chemicals and medications
Several chemicals and medications can injure the bowel and predispose to pseudomembrane formation.
Glutaraldehyde has long been used to sanitize endoscopes because of its broad antimicrobial activity. Nevertheless, if the disinfecting solution is not adequately rinsed off the endoscope, direct contact with colonic mucosa can produce an allergic and a chemical reaction, resulting in an acute self-limited colitis with pseudomembrane formation.4
Chemotherapeutic and antiproliferative agents can be toxic to the bowel, generally through production of free radicals and up-regulation of inflammatory cytokines. The colonic epithelium is then more susceptible to ulceration and mucosal necrosis with pseudomembrane development. Cisplatin, cyclosporine A, docetaxel, and 5-fluorouracil are prominent examples.5–8
Nonsteroidal anti-inflammatory drugs can damage the mucosa at all levels of the gastrointestinal tract. Although gastric ulcerations are more typical (from nonselective cyclooxygenase inhibition), colonic ulcerations and colitis can occur.9 These drugs, particularly diclofenac and indomethacin, have been associated with non-C difficile pseudomembranous colitis when used by themselves or in conjunction with other agents such as cyclosporine A.8,10,11
Infections
C difficile is the organism most commonly linked to pseudomembranous colitis, but other bacterial, viral, and parasitic pathogens have also been implicated.
Staphylococcus aureus was believed to be responsible for enterocolitis in a series of 155 surgical patients between 1958 and 1962 receiving antibiotic therapy. All had a positive stool culture for S aureus, and nine were found to have pseudomembranes at autopsy.12
Although this finding has been disputed as a misdiagnosis, since C difficile infection was not widely recognized until the 1970s, there is evidence that S aureus may indeed be a cause of non-C difficile pseudomembranous colitis. In a review of 36 cases of methicillin-resistant S aureus bacteremia in Japan, four patients were documented to have intestinal pseudomembranes either by endoscopy or autopsy. In two of these patients, biopsies of the pseudomembranes were positive for methicillin-resistant S aureus.13
Escherichia coli O157:H7. Pseudomembranes have been seen endoscopically in several adults and children with enterohemorrhagic E coli O157:H7 infection.14,15 This invasive gram-negative rod normally resides in the gastrointestinal tract of cattle, sheep, and other animals and can be pathogenic to people who eat undercooked beef. The organism attaches to and effaces intestinal epithelial cells, and bacterial proteins and the Shiga toxin then damage the vasculature, precipitating bloody diarrhea. Colonic damage can range from mild hemorrhagic colitis to severe colitis with ischemic changes. In patients with enterohemorrhagic E coli O157:H7 infection, pseudomembrane formation results from colon ischemia due to microvascular thrombosis or from destructive effects of bacterial enterotoxin.1,15
Cytomegalovirus is a ubiquitous human herpes virus that can affect nearly all organ systems. Infection is often reported in immunocompromised patients, eg, those with acquired immunodeficiency syndrome, chronic corticosteroid use, inflammatory bowel disease, malignancy, or solid-organ transplants. Gastrointestinal manifestations can be nonspecific and range from abdominal discomfort to diarrhea to tenesmus. Pseudomembranous colitis can be a presenting feature of cytomegalovirus involvement.16,17 Ulcerative lesions in the colonic mucosa are a frequent accompanying finding on endoscopy.18,19
Other rarer infectious causes of pseudomembranous colitis identified in case reports include Clostridium ramosum, Entamoeba histolytica, Klebsiella oxytoca, Plesiomonas shigelloides, Schistosomiasis mansoni, and Salmonella and Shigella species.20–26 Additionally, there have been two reports of Strongyloides stercoralis hyperinfection manifesting as pseudomembranous colitis.27,28
Ischemia
Colon ischemia usually affects elderly or debilitated patients who have multiple comorbidities. Known risk factors include aortoiliac surgery, cardiovascular disease, diabetes mellitus, hemodialysis, and pulmonary disease. The ischemia can be related to an occlusive arterial or venous thromboembolism, but hypoperfusion without occlusion of the mesenteric or internal iliac arteries is the primary mechanism. Low blood flow states such as atherosclerosis and septic shock can affect the watershed areas, typically the splenic flexure and rectosigmoid junction.
We reported a case of a patient with vascular disease who was incorrectly diagnosed and treated as having refractory C difficile infection when pseudomembranes were seen on flexible sigmoidoscopy. Further investigation revealed ischemic colitis secondary to a high-grade inferior mesenteric artery stenosis as the true cause.29
Microvascular thrombosis is the likely mechanism in a number of non-C difficile causes of pseudomembranous colitis. For example, in most patients with enterohemorrhagic E coli O157:H7 infection, histologic review of colonic mucosal biopsies has revealed fibrin and platelet thrombi in the capillaries, suggesting microvascular thrombosis.15,30
Cocaine has been associated with pseudomembranes in the setting of ischemia in the cecum and ascending colon. Cocaine can cause vasoconstriction after stimulation of alpha-adrenergic receptors and hence intestinal ischemia, thrombosis of vessels in the large and small intestines, and direct toxic effects.31
Inflammatory conditions
Collagenous colitis is an inflammatory disease that often affects middle-aged women and presents with copious watery diarrhea. It is a type of microscopic colitis—the endoscopic appearance is often normal, while the histologic appearance is abnormal and characterized by collagen deposition in the lamina propria. Medications that have been implicated in microscopic colitis include acid-suppressive agents (eg, histamine receptor antagonists, proton pump inhibitors) and nonsteroidal anti-inflammatory drugs.
An increasing number of cases of pseudomembranous changes are being reported in patients diagnosed with collagenous colitis.32–36 Although the pathophysiologic mechanism is unknown, some authors have suggested that pseudomembrane formation is actually part of the presenting spectrum of collagenous colitis.36
Inflammatory bowel disease. Crohn disease and ulcerative colitis have been associated with pseudomembranous colitis. Pseudomembranes can be found on endoscopy in patients with inflammatory bowel disease during a disease exacerbation with or without C difficile.37,38 In patients with inflammatory bowel disease and C difficile infection, pseudomembranes can be found endoscopically in up to 13% of cases.39 Pseudomembranous colitis has been reported in a patient with ulcerative colitis exacerbation in association with cytomegalovirus colitis.40
Behçet disease is a rare, immune-mediated small-vessel systemic vasculitis. It usually presents with mucous membrane ulcerations and ocular disease but can affect any organ.41 Pseudomembranous colitis can occur in Behçet disease in the absence of C difficile infection or any infectious colitis. Treatment includes corticosteroids and immunosuppressants such as azathioprine and anti-tumor necrosis factor agents.41
INITIAL EVALUATION
The initial evaluation of a patient with suspected or confirmed pseudomembranous colitis should include a comprehensive medical history with information on recent hospitalizations or procedures, antibiotic use, infections, exposure to sick contacts, recent travel, and medications taken.
Testing for C difficile
As most patients with pseudomembranous colitis have C difficile infection, it should be excluded first. Empiric anti-C difficile treatment is recommended in seriously ill-appearing patients, ideally starting after a stool sample is obtained.
Diagnosis of C difficile infection requires laboratory demonstration of the toxin or detection of toxigenic organisms. The gold standard test is the cell culture cytotoxicity assay, but it is labor- and time-intensive.42 More widely available tests are polymerase chain reaction for the toxin gene or genes, enzyme immunoassay, and stool evaluation for glutamate dehydrogenase, which can yield results readily within hours.
Polymerase chain reaction has a sensitivity of 97% and a specificity of 93%. Results can be falsely positive if empiric treatment is started before specimen collection, in which case C difficile DNA may still be present and detectable, but not the organism itself.43
Enzyme immunoassay for toxins A and B carries a sensitivity of 75% and a specificity of 99%, but 100 to 1,000 pg of toxin must be present for a positive result.44,45
If the initial enzyme immunoassay or polymerase chain reaction result is negative, current guidelines do not recommend repeat testing, which has limited value.44,46 Repeat testing after a negative result is positive in less than 5% of samples and greatly increases the chances of false-positive results.44,46 Nevertheless, if a laboratory’s enzyme immunoassay test has a low sensitivity, repeating negative tests may improve its sensitivity.
Glutamate dehydrogenase is an enzyme produced by both toxigenic and nontoxigenic strains of C difficile. As a result, stool testing for glutamate dehydrogenase is sensitive but not specific for C difficile infection, although multistep testing sequences (glutamate dehydrogenase followed by polymerase chain reaction) have proven to be useful screening tools.44
Treatment for C difficile infection
If testing for C difficile is positive, treatment is generally based on the severity and the complications of the illness46:
- Mild or moderate C difficile infection should be treated with oral metronidazole 500 mg three times per day for 10 to 14 days.
- Severe infection, which is defined as a white blood cell count of 15.0 × 109/L or higher or a serum creatinine level greater than or equal to 1.5 times the premorbid level, should be treated with oral vancomycin 125 mg four times per day for 10 to 14 days.
- Severe C difficile infection complicated by hypotension, shock, ileus, or megacolon should be treated with a combination of high-dose oral vancomycin (and possibly rectal vancomycin as well) at 500 mg four times per day plus intravenous metronidazole.
Additional treatment recommendations for individualized situations, recurrent C difficile infection, and comorbid conditions are discussed elsewhere.46
ENDOSCOPIC EVALUATION
Colonoscopy or flexible sigmoidoscopy is the primary means by which pseudomembranous colitis is diagnosed. Lower endoscopy should be pursued as an adjunctive tool when C difficile infection remains strongly suspected despite negative testing, when presumed C difficile infection does not respond to medical therapy, and when non-C difficile diagnoses are considered. If pseudomembranes are demonstrated on lower endoscopy, obtaining biopsy samples of normal- and abnormal-appearing mucosa is recommended. Pseudomembranes are suggestive but not diagnostic of C difficile infection, and microscopic evaluation of the mucosa is warranted to explore causes of pseudomembranous colitis not related to C difficile.
The pattern and distribution of pseudomembranes may provide clues to the etiology and the degree of mucosal injury. In intestinal ischemia, for example, a localized segment of the bowel is typically involved, and mucosal changes are often well delineated from normal mucosa. On endoscopic examination, mild ischemia is characterized by granular mucosa with decreased vascularity, whereas friable, edematous, ulcerated, and at times necrotic mucosa is evident in severe cases. Punctate pseudomembrane formation is seen in early ischemia, but as injury progresses, the pseudomembranes may grow and merge. In fact, diffuse involvement of the mucosal surface of the biopsy specimen by pseudomembranes has been shown to be more closely associated with ischemic colitis than C difficile infection.47
MICROSCOPIC EVALUATION
Histologic study can differentiate the various causes of pseudomembranous colitis.
In C difficile infection and drug reaction, there is acute crypt injury and dilation. The upper lamina propria is usually involved, and affected crypts are filled with an exudate similar to that found in pseudomembranes.1 However, in drug reaction, there is also prominent apoptosis and increased intraepithelial lymphocytosis.9
In colon ischemia, hyalinization of the lamina propria is a sensitive and specific marker.47 This has been shown in a study comparing histologic characteristics of colonic biopsies in patients with pseudomembranous colitis due to either known colon ischemia or C difficile infection.47 Crypt atrophy, lamina propria hemorrhage, full-thickness mucosal necrosis, and layering of pseudomembranes would further favor the diagnosis.
In collagenous colitis, a thickened subepithelial collagen band and intraepithelial lymphocytosis are often seen.
In inflammatory bowel disease, even with secondary pseudomembranes, ulcerative colitis and Crohn disease retain the characteristics of inflammatory bowel disease with crypt architectural distortion and focal or diffuse basal lymphoplasmacytosis on microscopy.48
Pseudomembranous colitis is most often due to Clostridium difficile infection, but it has a variety of other causes, including other infections, ischemia, medications, and inflammatory mucosal diseases (Table 1). When pseudomembranes are found, one should consider these other causes if tests for C difficile are negative or if anti-C difficile therapy does not produce a response.
These less common causes are important to consider to avoid needlessly escalating anti-C difficile antibiotic therapy and to provide appropriate treatment. Pseudomembranous colitis is a nonspecific finding that suggests a larger disease process. Associated signs and symptoms, including fever, abdominal pain, leukocytosis, diarrhea, toxic megacolon, and electrolyte imbalances, may portend a life-threatening condition.1 Awareness of causes of pseudomembranous colitis other than C difficile infection, the focus of this review, is key to prompt diagnosis and potentially life-saving patient care.
PSEUDOMEMBRANES ARE NONSPECIFIC
A pseudomembrane is a layer of fibropurulent exudate composed of acute inflammatory cells and mucus originating from inflamed and erupting crypts.2 Although most often seen in C difficile infection, pseudomembranous colitis is a nonspecific pattern of injury resulting from decreased oxygenation, endothelial damage, and impaired blood flow to the mucosa that can be triggered by a number of disease states.2
On endoscopy, pseudomembranes appear as raised whitish or yellowish plaques that may be scattered or confluent in distribution (Figure 1).2 They are usually found in the rectosigmoid colon but may be isolated to more proximal segments.3 Lower endoscopy is often performed in the diagnostic evaluation of patients with unexplained diarrhea, hematochezia, and abnormal abdominal computed tomographic findings (eg, colonic thickening).
CAUSES OF NON-C DIFFICLE PSEUDOMEMBRANOUS COLITIS
When pseudomembranous colitis is confirmed endoscopically, C difficile infection naturally comes to mind, but the two terms are not interchangeable. A wide differential diagnosis should be maintained, especially when there are clues that C difficile infection may not be the correct diagnosis.
Chemicals and medications
Several chemicals and medications can injure the bowel and predispose to pseudomembrane formation.
Glutaraldehyde has long been used to sanitize endoscopes because of its broad antimicrobial activity. Nevertheless, if the disinfecting solution is not adequately rinsed off the endoscope, direct contact with colonic mucosa can produce an allergic and a chemical reaction, resulting in an acute self-limited colitis with pseudomembrane formation.4
Chemotherapeutic and antiproliferative agents can be toxic to the bowel, generally through production of free radicals and up-regulation of inflammatory cytokines. The colonic epithelium is then more susceptible to ulceration and mucosal necrosis with pseudomembrane development. Cisplatin, cyclosporine A, docetaxel, and 5-fluorouracil are prominent examples.5–8
Nonsteroidal anti-inflammatory drugs can damage the mucosa at all levels of the gastrointestinal tract. Although gastric ulcerations are more typical (from nonselective cyclooxygenase inhibition), colonic ulcerations and colitis can occur.9 These drugs, particularly diclofenac and indomethacin, have been associated with non-C difficile pseudomembranous colitis when used by themselves or in conjunction with other agents such as cyclosporine A.8,10,11
Infections
C difficile is the organism most commonly linked to pseudomembranous colitis, but other bacterial, viral, and parasitic pathogens have also been implicated.
Staphylococcus aureus was believed to be responsible for enterocolitis in a series of 155 surgical patients between 1958 and 1962 receiving antibiotic therapy. All had a positive stool culture for S aureus, and nine were found to have pseudomembranes at autopsy.12
Although this finding has been disputed as a misdiagnosis, since C difficile infection was not widely recognized until the 1970s, there is evidence that S aureus may indeed be a cause of non-C difficile pseudomembranous colitis. In a review of 36 cases of methicillin-resistant S aureus bacteremia in Japan, four patients were documented to have intestinal pseudomembranes either by endoscopy or autopsy. In two of these patients, biopsies of the pseudomembranes were positive for methicillin-resistant S aureus.13
Escherichia coli O157:H7. Pseudomembranes have been seen endoscopically in several adults and children with enterohemorrhagic E coli O157:H7 infection.14,15 This invasive gram-negative rod normally resides in the gastrointestinal tract of cattle, sheep, and other animals and can be pathogenic to people who eat undercooked beef. The organism attaches to and effaces intestinal epithelial cells, and bacterial proteins and the Shiga toxin then damage the vasculature, precipitating bloody diarrhea. Colonic damage can range from mild hemorrhagic colitis to severe colitis with ischemic changes. In patients with enterohemorrhagic E coli O157:H7 infection, pseudomembrane formation results from colon ischemia due to microvascular thrombosis or from destructive effects of bacterial enterotoxin.1,15
Cytomegalovirus is a ubiquitous human herpes virus that can affect nearly all organ systems. Infection is often reported in immunocompromised patients, eg, those with acquired immunodeficiency syndrome, chronic corticosteroid use, inflammatory bowel disease, malignancy, or solid-organ transplants. Gastrointestinal manifestations can be nonspecific and range from abdominal discomfort to diarrhea to tenesmus. Pseudomembranous colitis can be a presenting feature of cytomegalovirus involvement.16,17 Ulcerative lesions in the colonic mucosa are a frequent accompanying finding on endoscopy.18,19
Other rarer infectious causes of pseudomembranous colitis identified in case reports include Clostridium ramosum, Entamoeba histolytica, Klebsiella oxytoca, Plesiomonas shigelloides, Schistosomiasis mansoni, and Salmonella and Shigella species.20–26 Additionally, there have been two reports of Strongyloides stercoralis hyperinfection manifesting as pseudomembranous colitis.27,28
Ischemia
Colon ischemia usually affects elderly or debilitated patients who have multiple comorbidities. Known risk factors include aortoiliac surgery, cardiovascular disease, diabetes mellitus, hemodialysis, and pulmonary disease. The ischemia can be related to an occlusive arterial or venous thromboembolism, but hypoperfusion without occlusion of the mesenteric or internal iliac arteries is the primary mechanism. Low blood flow states such as atherosclerosis and septic shock can affect the watershed areas, typically the splenic flexure and rectosigmoid junction.
We reported a case of a patient with vascular disease who was incorrectly diagnosed and treated as having refractory C difficile infection when pseudomembranes were seen on flexible sigmoidoscopy. Further investigation revealed ischemic colitis secondary to a high-grade inferior mesenteric artery stenosis as the true cause.29
Microvascular thrombosis is the likely mechanism in a number of non-C difficile causes of pseudomembranous colitis. For example, in most patients with enterohemorrhagic E coli O157:H7 infection, histologic review of colonic mucosal biopsies has revealed fibrin and platelet thrombi in the capillaries, suggesting microvascular thrombosis.15,30
Cocaine has been associated with pseudomembranes in the setting of ischemia in the cecum and ascending colon. Cocaine can cause vasoconstriction after stimulation of alpha-adrenergic receptors and hence intestinal ischemia, thrombosis of vessels in the large and small intestines, and direct toxic effects.31
Inflammatory conditions
Collagenous colitis is an inflammatory disease that often affects middle-aged women and presents with copious watery diarrhea. It is a type of microscopic colitis—the endoscopic appearance is often normal, while the histologic appearance is abnormal and characterized by collagen deposition in the lamina propria. Medications that have been implicated in microscopic colitis include acid-suppressive agents (eg, histamine receptor antagonists, proton pump inhibitors) and nonsteroidal anti-inflammatory drugs.
An increasing number of cases of pseudomembranous changes are being reported in patients diagnosed with collagenous colitis.32–36 Although the pathophysiologic mechanism is unknown, some authors have suggested that pseudomembrane formation is actually part of the presenting spectrum of collagenous colitis.36
Inflammatory bowel disease. Crohn disease and ulcerative colitis have been associated with pseudomembranous colitis. Pseudomembranes can be found on endoscopy in patients with inflammatory bowel disease during a disease exacerbation with or without C difficile.37,38 In patients with inflammatory bowel disease and C difficile infection, pseudomembranes can be found endoscopically in up to 13% of cases.39 Pseudomembranous colitis has been reported in a patient with ulcerative colitis exacerbation in association with cytomegalovirus colitis.40
Behçet disease is a rare, immune-mediated small-vessel systemic vasculitis. It usually presents with mucous membrane ulcerations and ocular disease but can affect any organ.41 Pseudomembranous colitis can occur in Behçet disease in the absence of C difficile infection or any infectious colitis. Treatment includes corticosteroids and immunosuppressants such as azathioprine and anti-tumor necrosis factor agents.41
INITIAL EVALUATION
The initial evaluation of a patient with suspected or confirmed pseudomembranous colitis should include a comprehensive medical history with information on recent hospitalizations or procedures, antibiotic use, infections, exposure to sick contacts, recent travel, and medications taken.
Testing for C difficile
As most patients with pseudomembranous colitis have C difficile infection, it should be excluded first. Empiric anti-C difficile treatment is recommended in seriously ill-appearing patients, ideally starting after a stool sample is obtained.
Diagnosis of C difficile infection requires laboratory demonstration of the toxin or detection of toxigenic organisms. The gold standard test is the cell culture cytotoxicity assay, but it is labor- and time-intensive.42 More widely available tests are polymerase chain reaction for the toxin gene or genes, enzyme immunoassay, and stool evaluation for glutamate dehydrogenase, which can yield results readily within hours.
Polymerase chain reaction has a sensitivity of 97% and a specificity of 93%. Results can be falsely positive if empiric treatment is started before specimen collection, in which case C difficile DNA may still be present and detectable, but not the organism itself.43
Enzyme immunoassay for toxins A and B carries a sensitivity of 75% and a specificity of 99%, but 100 to 1,000 pg of toxin must be present for a positive result.44,45
If the initial enzyme immunoassay or polymerase chain reaction result is negative, current guidelines do not recommend repeat testing, which has limited value.44,46 Repeat testing after a negative result is positive in less than 5% of samples and greatly increases the chances of false-positive results.44,46 Nevertheless, if a laboratory’s enzyme immunoassay test has a low sensitivity, repeating negative tests may improve its sensitivity.
Glutamate dehydrogenase is an enzyme produced by both toxigenic and nontoxigenic strains of C difficile. As a result, stool testing for glutamate dehydrogenase is sensitive but not specific for C difficile infection, although multistep testing sequences (glutamate dehydrogenase followed by polymerase chain reaction) have proven to be useful screening tools.44
Treatment for C difficile infection
If testing for C difficile is positive, treatment is generally based on the severity and the complications of the illness46:
- Mild or moderate C difficile infection should be treated with oral metronidazole 500 mg three times per day for 10 to 14 days.
- Severe infection, which is defined as a white blood cell count of 15.0 × 109/L or higher or a serum creatinine level greater than or equal to 1.5 times the premorbid level, should be treated with oral vancomycin 125 mg four times per day for 10 to 14 days.
- Severe C difficile infection complicated by hypotension, shock, ileus, or megacolon should be treated with a combination of high-dose oral vancomycin (and possibly rectal vancomycin as well) at 500 mg four times per day plus intravenous metronidazole.
Additional treatment recommendations for individualized situations, recurrent C difficile infection, and comorbid conditions are discussed elsewhere.46
ENDOSCOPIC EVALUATION
Colonoscopy or flexible sigmoidoscopy is the primary means by which pseudomembranous colitis is diagnosed. Lower endoscopy should be pursued as an adjunctive tool when C difficile infection remains strongly suspected despite negative testing, when presumed C difficile infection does not respond to medical therapy, and when non-C difficile diagnoses are considered. If pseudomembranes are demonstrated on lower endoscopy, obtaining biopsy samples of normal- and abnormal-appearing mucosa is recommended. Pseudomembranes are suggestive but not diagnostic of C difficile infection, and microscopic evaluation of the mucosa is warranted to explore causes of pseudomembranous colitis not related to C difficile.
The pattern and distribution of pseudomembranes may provide clues to the etiology and the degree of mucosal injury. In intestinal ischemia, for example, a localized segment of the bowel is typically involved, and mucosal changes are often well delineated from normal mucosa. On endoscopic examination, mild ischemia is characterized by granular mucosa with decreased vascularity, whereas friable, edematous, ulcerated, and at times necrotic mucosa is evident in severe cases. Punctate pseudomembrane formation is seen in early ischemia, but as injury progresses, the pseudomembranes may grow and merge. In fact, diffuse involvement of the mucosal surface of the biopsy specimen by pseudomembranes has been shown to be more closely associated with ischemic colitis than C difficile infection.47
MICROSCOPIC EVALUATION
Histologic study can differentiate the various causes of pseudomembranous colitis.
In C difficile infection and drug reaction, there is acute crypt injury and dilation. The upper lamina propria is usually involved, and affected crypts are filled with an exudate similar to that found in pseudomembranes.1 However, in drug reaction, there is also prominent apoptosis and increased intraepithelial lymphocytosis.9
In colon ischemia, hyalinization of the lamina propria is a sensitive and specific marker.47 This has been shown in a study comparing histologic characteristics of colonic biopsies in patients with pseudomembranous colitis due to either known colon ischemia or C difficile infection.47 Crypt atrophy, lamina propria hemorrhage, full-thickness mucosal necrosis, and layering of pseudomembranes would further favor the diagnosis.
In collagenous colitis, a thickened subepithelial collagen band and intraepithelial lymphocytosis are often seen.
In inflammatory bowel disease, even with secondary pseudomembranes, ulcerative colitis and Crohn disease retain the characteristics of inflammatory bowel disease with crypt architectural distortion and focal or diffuse basal lymphoplasmacytosis on microscopy.48
- Gebhard RL, Gerding DN, Olson MM, et al. Clinical and endoscopic findings in patients early in the course of clostridium difficile-associated pseudomembranous colitis. Am J Med 1985; 78:45–48.
- Carpenter HA, Talley NJ. The importance of clinicopathological correlation in the diagnosis of inflammatory conditions of the colon: histological patterns with clinical implications. Am J Gastroenterol 2000; 95:878–896.
- Seppälä K. Colonoscopy in the diagnosis of pseudomembranous colitis. Br Med J 1978; 2:435.
- Stein BL, Lamoureux E, Miller M, Vasilevsky CA, Julien L, Gordon PH. Glutaraldehyde-induced colitis. Can J Surg 2001; 44:113–116.
- Trevisani F, Simoncini M, Alampi G, Bernardi M. Colitis associated to chemotherapy with 5-fluorouracil. Hepatogastroenterology 1997; 44:710–712.
- Takao T, Nishida M, Maeda Y, Takao K, Oka M. The study of continuous infusion chemotherapy with low-dose cisplatin and 5-fluorouracil for patients with primary liver cancer. Gan To Kagaku Ryoho 1997; 24:1724–1727. Japanese.
- Carrion AF, Hosein PJ, Cooper EM, Lopes G, Pelaez L, Rocha-Lima CM. Severe colitis associated with docetaxel use: a report of four cases. World J Gastrointest Oncol 2010; 2:390-394.
- Constantopoulos A. Colitis induced by interaction of cyclosporine A and non-steroidal anti-inflammatory drugs. Pediatr Int 1999; 41:184–186.
- Price AB. Pathology of drug-associated gastrointestinal disease. Br J Clin Pharmacol 2003; 56:477–482.
- Gentric A, Pennec YL. Diclofenac-induced pseudomembranous colitis. Lancet 1992; 340:126–127.
- Romero-Gómez M, Suárez García E, Castro Fernández M. Pseudomembranous colitis induced by diclofenac. J Clin Gastroenterol 1998; 26:228.
- Altemeier WA, Hummel RP, Hill EO. Staphylococcal enterocolitis following antibiotic therapy. Ann Surg 1963; 157:847–858.
- Ogawa Y, Saraya T, Koide T, et al. Methicillin-resistant Staphylococcus aureus enterocolitis sequentially complicated with septic arthritis: a case report and review of the literature. BMC Res Notes 2014; 7:21.
- Griffin PM, Olmstead LC, Petras RE. Escherichia coli O157:H7-associated colitis. A clinical and histological study of 11 cases. Gastroenterology 1990; 99:142–149.
- Uc A, Mitros FA, Kao SC, Sanders KD. Pseudomembranous colitis with Escherichia coli O157:H7. J Pediatr Gastroenterol Nutr 1997; 24:590–593.
- Battaglino MP, Rockey DC. Cytomegalovirus colitis presenting with the endoscopic appearance of pseudomembranous colitis. Gastrointest Endosc 1999; 50:697–700.
- Olofinlade O, Chiang C. Cytomegalovirus infection as a cause of pseudomembrane colitis: a report of four cases. J Clin Gastroenterol 2001; 32:82–84.
- Wilcox CM, Chalasani N, Lazenby A, Schwartz DA. Cytomegalovirus colitis in acquired immunodeficiency syndrome: a clinical and endoscopic study. Gastrointest Endosc 1998; 48:39–43.
- Seo TH, Kim JH, Ko SY, et al. Cytomegalovirus colitis in immunocompetent patients: a clinical and endoscopic study. Hepatogastroenterology 2012; 59:2137–2141.
- Högenauer C, Langner C, Beubler E, et al. Klebsiella oxytoca as a causative organism of antibiotic-associated hemorrhagic colitis. N Engl J Med 2006; 355:2418–2426.
- Hovius SE, Rietra PJ. Salmonella colitis clinically presenting as a pseudomembranous colitis. Neth J Surg 1982; 34:81–82.
- Kelber M, Ament ME. Shigella dysenteriae I: a forgotten cause of pseudomembranous colitis. J Pediatr 1976; 89:595–596.
- Neves J, Raso P, Pinto Dde M, da Silva SP, Alvarenga RJ. Ischaemic colitis (necrotizing colitis, pseudomembranous colitis) in acute schistosomiasis mansoni: report of two cases. Trans R Soc Trop Med Hyg 1993; 87:449–452.
- Alcalde-Vargas A, Trigo-Salado C, Leo Carnerero E, De-la-Cruz-Ramírez D, Herrera-Justiniano JM. Pseudomembranous colitis and bacteremia in an immunocompetent patient associated with a rare specie of Clostridium (C. ramosum). Rev Esp Enferm Dig 2012; 104:498–499.
- Koo JS, Choi WS, Park DW. Fulminant amebic colitis mimicking pseudomembranous colitis. Gastrointest Endosc 2010; 71:400–401.
- van Loon FP, Rahim Z, Chowdhury KA, Kay BA, Rahman SA. Case report of Plesiomonas shigelloides-associated persistent dysentery and pseudomembranous colitis. J Clin Microbiol 1989; 27:1913–1915.
- Janvier J, Kuhn S, Church D. Not all pseudomembranous colitis is caused by Clostridium difficile. Can J Infect Dis Med Microbiol 2008; 19:256–257.
- Jain AK, Agarwal SK, el-Sadr W. Streptococcus bovis bacteremia and meningitis associated with Strongyloides stercoralis colitis in a patient infected with human immunodeficiency virus. Clin Infect Dis 1994; 18:253–254.
- Tang DM, Urrunaga NH, De Groot H, von Rosenvinge EC, Xie G, Ghazi LJ. Pseudomembranous colitis: not always caused by Clostridium difficile. Case Rep Med 2014; 2014:812704.
- Kendrick JB, Risbano M, Groshong SD, Frankel SK. A rare presentation of ischemic pseudomembranous colitis due to Escherichia coli O157:H7. Clin Infect Dis 2007; 45:217–219.
- Fishel R, Hamamoto G, Barbul A, Jiji V, Efron G. Cocaine colitis. Is this a new syndrome? Dis Colon Rectum 1985; 28:264–266.
- Khan-Kheil AM, Disney B, Ruban E, Wood G. Pseudomembranous collagenous colitis: an unusual cause of chronic diarrhoea. BMJ Case Rep 2014; 2014.
- Villanacci V, Cristina S, Muscarà M, et al. Pseudomembranous collagenous colitis with superimposed drug damage. Pathol Res Pract 2013; 209:735–739.
- Denız K, Coban G, Ozbakir O, Denız E. Pseudomembranous collagenous colitis. Turk J Gastroenterol 2012; 23:93–95.
- Vesoulis Z, Lozanski G, Loiudice T. Synchronous occurrence of collagenous colitis and pseudomembranous colitis. Can J Gastroenterol 2000; 14:353–358.
- Yuan S, Reyes V, Bronner MP. Pseudomembranous collagenous colitis. Am J Surg Pathol 2003; 27:1375–1379.
- Berdichevski T, Barshack I, Bar-Meir S, Ben-Horin S. Pseudomembranes in a patient with flare-up of inflammatory bowel disease (IBD): is it only Clostridium difficile or is it still an IBD exacerbation? Endoscopy 2010; 42(suppl 2):E131.
- Kilinçalp S, Altinbas A, Basar O, Deveci M, Yüksel O. A case of ulcerative colitis co-existing with pseudo-membranous enterocolitis. J Crohns Colitis 2011; 5:506–507.
- Ben-Horin S, Margalit M, Bossuyt P, et al; European Crohn’s and Colitis Organization (ECCO). Prevalence and clinical impact of endoscopic pseudomembranes in patients with inflammatory bowel disease and Clostridium difficile infection. J Crohns Colitis 2010; 4:194–198.
- Chiba M, Abe T, Tsuda S, Ono I. Cytomegalovirus infection associated with onset of ulcerative colitis. BMC Res Notes 2013; 6:40.
- Shukla A, Tolan RW. Behcet disease presenting with pseudomembranous colitis and progression to neurological involvement: case report and review of the literature. Clin Pediatr (Phila) 2012; 51:1197–1201.
- Shanholtzer CJ, Willard KE, Holter JJ, Olson MM, Gerding DN, Peterson LR. Comparison of the VIDAS Clostridium difficile toxin A immunoassay with C. difficile culture and cytotoxin and latex tests. J Clin Microbiol 1992; 30:1837–1840.
- Huang H, Weintraub A, Fang H, Nord CE. Comparison of a commercial multiplex real-time PCR to the cell cytotoxicity neutralization assay for diagnosis of Clostridium difficile infections. J Clin Microbiol 2009; 47:3729–3731.
- Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society For Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol 2010; 31:431–455.
- Bartlett JG. Clinical practice. Antibiotic-associated diarrhea. N Engl J Med 2002; 346:334–339.
- Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol 2013; 108:478–498.
- Dignan CR, Greenson JK. Can ischemic colitis be differentiated from C difficile colitis in biopsy specimens? Am J Surg Pathol 1997; 21:706–710.
- Jenkins D, Balsitis M, Gallivan S, et al. Guidelines for the initial biopsy diagnosis of suspected chronic idiopathic inflammatory bowel disease. The British Society of Gastroenterology Initiative. J Clin Pathol 1997; 50:93–105.
- Gebhard RL, Gerding DN, Olson MM, et al. Clinical and endoscopic findings in patients early in the course of clostridium difficile-associated pseudomembranous colitis. Am J Med 1985; 78:45–48.
- Carpenter HA, Talley NJ. The importance of clinicopathological correlation in the diagnosis of inflammatory conditions of the colon: histological patterns with clinical implications. Am J Gastroenterol 2000; 95:878–896.
- Seppälä K. Colonoscopy in the diagnosis of pseudomembranous colitis. Br Med J 1978; 2:435.
- Stein BL, Lamoureux E, Miller M, Vasilevsky CA, Julien L, Gordon PH. Glutaraldehyde-induced colitis. Can J Surg 2001; 44:113–116.
- Trevisani F, Simoncini M, Alampi G, Bernardi M. Colitis associated to chemotherapy with 5-fluorouracil. Hepatogastroenterology 1997; 44:710–712.
- Takao T, Nishida M, Maeda Y, Takao K, Oka M. The study of continuous infusion chemotherapy with low-dose cisplatin and 5-fluorouracil for patients with primary liver cancer. Gan To Kagaku Ryoho 1997; 24:1724–1727. Japanese.
- Carrion AF, Hosein PJ, Cooper EM, Lopes G, Pelaez L, Rocha-Lima CM. Severe colitis associated with docetaxel use: a report of four cases. World J Gastrointest Oncol 2010; 2:390-394.
- Constantopoulos A. Colitis induced by interaction of cyclosporine A and non-steroidal anti-inflammatory drugs. Pediatr Int 1999; 41:184–186.
- Price AB. Pathology of drug-associated gastrointestinal disease. Br J Clin Pharmacol 2003; 56:477–482.
- Gentric A, Pennec YL. Diclofenac-induced pseudomembranous colitis. Lancet 1992; 340:126–127.
- Romero-Gómez M, Suárez García E, Castro Fernández M. Pseudomembranous colitis induced by diclofenac. J Clin Gastroenterol 1998; 26:228.
- Altemeier WA, Hummel RP, Hill EO. Staphylococcal enterocolitis following antibiotic therapy. Ann Surg 1963; 157:847–858.
- Ogawa Y, Saraya T, Koide T, et al. Methicillin-resistant Staphylococcus aureus enterocolitis sequentially complicated with septic arthritis: a case report and review of the literature. BMC Res Notes 2014; 7:21.
- Griffin PM, Olmstead LC, Petras RE. Escherichia coli O157:H7-associated colitis. A clinical and histological study of 11 cases. Gastroenterology 1990; 99:142–149.
- Uc A, Mitros FA, Kao SC, Sanders KD. Pseudomembranous colitis with Escherichia coli O157:H7. J Pediatr Gastroenterol Nutr 1997; 24:590–593.
- Battaglino MP, Rockey DC. Cytomegalovirus colitis presenting with the endoscopic appearance of pseudomembranous colitis. Gastrointest Endosc 1999; 50:697–700.
- Olofinlade O, Chiang C. Cytomegalovirus infection as a cause of pseudomembrane colitis: a report of four cases. J Clin Gastroenterol 2001; 32:82–84.
- Wilcox CM, Chalasani N, Lazenby A, Schwartz DA. Cytomegalovirus colitis in acquired immunodeficiency syndrome: a clinical and endoscopic study. Gastrointest Endosc 1998; 48:39–43.
- Seo TH, Kim JH, Ko SY, et al. Cytomegalovirus colitis in immunocompetent patients: a clinical and endoscopic study. Hepatogastroenterology 2012; 59:2137–2141.
- Högenauer C, Langner C, Beubler E, et al. Klebsiella oxytoca as a causative organism of antibiotic-associated hemorrhagic colitis. N Engl J Med 2006; 355:2418–2426.
- Hovius SE, Rietra PJ. Salmonella colitis clinically presenting as a pseudomembranous colitis. Neth J Surg 1982; 34:81–82.
- Kelber M, Ament ME. Shigella dysenteriae I: a forgotten cause of pseudomembranous colitis. J Pediatr 1976; 89:595–596.
- Neves J, Raso P, Pinto Dde M, da Silva SP, Alvarenga RJ. Ischaemic colitis (necrotizing colitis, pseudomembranous colitis) in acute schistosomiasis mansoni: report of two cases. Trans R Soc Trop Med Hyg 1993; 87:449–452.
- Alcalde-Vargas A, Trigo-Salado C, Leo Carnerero E, De-la-Cruz-Ramírez D, Herrera-Justiniano JM. Pseudomembranous colitis and bacteremia in an immunocompetent patient associated with a rare specie of Clostridium (C. ramosum). Rev Esp Enferm Dig 2012; 104:498–499.
- Koo JS, Choi WS, Park DW. Fulminant amebic colitis mimicking pseudomembranous colitis. Gastrointest Endosc 2010; 71:400–401.
- van Loon FP, Rahim Z, Chowdhury KA, Kay BA, Rahman SA. Case report of Plesiomonas shigelloides-associated persistent dysentery and pseudomembranous colitis. J Clin Microbiol 1989; 27:1913–1915.
- Janvier J, Kuhn S, Church D. Not all pseudomembranous colitis is caused by Clostridium difficile. Can J Infect Dis Med Microbiol 2008; 19:256–257.
- Jain AK, Agarwal SK, el-Sadr W. Streptococcus bovis bacteremia and meningitis associated with Strongyloides stercoralis colitis in a patient infected with human immunodeficiency virus. Clin Infect Dis 1994; 18:253–254.
- Tang DM, Urrunaga NH, De Groot H, von Rosenvinge EC, Xie G, Ghazi LJ. Pseudomembranous colitis: not always caused by Clostridium difficile. Case Rep Med 2014; 2014:812704.
- Kendrick JB, Risbano M, Groshong SD, Frankel SK. A rare presentation of ischemic pseudomembranous colitis due to Escherichia coli O157:H7. Clin Infect Dis 2007; 45:217–219.
- Fishel R, Hamamoto G, Barbul A, Jiji V, Efron G. Cocaine colitis. Is this a new syndrome? Dis Colon Rectum 1985; 28:264–266.
- Khan-Kheil AM, Disney B, Ruban E, Wood G. Pseudomembranous collagenous colitis: an unusual cause of chronic diarrhoea. BMJ Case Rep 2014; 2014.
- Villanacci V, Cristina S, Muscarà M, et al. Pseudomembranous collagenous colitis with superimposed drug damage. Pathol Res Pract 2013; 209:735–739.
- Denız K, Coban G, Ozbakir O, Denız E. Pseudomembranous collagenous colitis. Turk J Gastroenterol 2012; 23:93–95.
- Vesoulis Z, Lozanski G, Loiudice T. Synchronous occurrence of collagenous colitis and pseudomembranous colitis. Can J Gastroenterol 2000; 14:353–358.
- Yuan S, Reyes V, Bronner MP. Pseudomembranous collagenous colitis. Am J Surg Pathol 2003; 27:1375–1379.
- Berdichevski T, Barshack I, Bar-Meir S, Ben-Horin S. Pseudomembranes in a patient with flare-up of inflammatory bowel disease (IBD): is it only Clostridium difficile or is it still an IBD exacerbation? Endoscopy 2010; 42(suppl 2):E131.
- Kilinçalp S, Altinbas A, Basar O, Deveci M, Yüksel O. A case of ulcerative colitis co-existing with pseudo-membranous enterocolitis. J Crohns Colitis 2011; 5:506–507.
- Ben-Horin S, Margalit M, Bossuyt P, et al; European Crohn’s and Colitis Organization (ECCO). Prevalence and clinical impact of endoscopic pseudomembranes in patients with inflammatory bowel disease and Clostridium difficile infection. J Crohns Colitis 2010; 4:194–198.
- Chiba M, Abe T, Tsuda S, Ono I. Cytomegalovirus infection associated with onset of ulcerative colitis. BMC Res Notes 2013; 6:40.
- Shukla A, Tolan RW. Behcet disease presenting with pseudomembranous colitis and progression to neurological involvement: case report and review of the literature. Clin Pediatr (Phila) 2012; 51:1197–1201.
- Shanholtzer CJ, Willard KE, Holter JJ, Olson MM, Gerding DN, Peterson LR. Comparison of the VIDAS Clostridium difficile toxin A immunoassay with C. difficile culture and cytotoxin and latex tests. J Clin Microbiol 1992; 30:1837–1840.
- Huang H, Weintraub A, Fang H, Nord CE. Comparison of a commercial multiplex real-time PCR to the cell cytotoxicity neutralization assay for diagnosis of Clostridium difficile infections. J Clin Microbiol 2009; 47:3729–3731.
- Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society For Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol 2010; 31:431–455.
- Bartlett JG. Clinical practice. Antibiotic-associated diarrhea. N Engl J Med 2002; 346:334–339.
- Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol 2013; 108:478–498.
- Dignan CR, Greenson JK. Can ischemic colitis be differentiated from C difficile colitis in biopsy specimens? Am J Surg Pathol 1997; 21:706–710.
- Jenkins D, Balsitis M, Gallivan S, et al. Guidelines for the initial biopsy diagnosis of suspected chronic idiopathic inflammatory bowel disease. The British Society of Gastroenterology Initiative. J Clin Pathol 1997; 50:93–105.
KEY POINTS
- Pseudomembranous colitis is a nonspecific pattern of injury resulting from decreased oxygenation, endothelial damage, and impaired blood flow to the mucosa that can be triggered by a number of disease states.
- Chemicals, medications, ischemia, microscopic colitis, other infectious organisms, and inflammatory conditions can all predispose to pseudomembrane formation and should be included in the differential diagnosis.
- As most patients with pseudomembranous colitis have C difficile infection, it should be excluded first. Empiric treatment for C difficile should be started if the patient is seriously ill.
- Testing for C difficile is with polymerase chain reaction, enzyme immunoassay for toxins A and B, and glutamate dehydrogenase measurement.
Extracorporeal membrane oxygenation in adults: A practical guide for internists
Extracorporeal membrance oxygenation (ECMO) provides temporary cardiorespiratory support for patients with severe respiratory or cardiac failure refractory to conventional therapy.1 It can be configured to provide oxygen, remove carbon dioxide, support perfusion, or all of the above. It may provide a bridge to recovery in patients with acute cardiopulmonary failure or to heart or lung transplant.
Developed in the 1970s, ECMO has proven effective and is widely used in children with respiratory and cardiopulmonary failure.2 However, it remained little used in adults, as early randomized trials showed higher rates of complications in adults who received it and no survival advantage.3,4 Proponents of using it in adult patients believe that these poor outcomes were at least partially due to limited training, intensive anticoagulation, and excessive volume and pressure during mechanical ventilation. Although ECMO technology has improved substantially in the last decade and survival rates have improved (www.elso.org), evidence to support its routine use in adults remains limited.
Nevertheless, about 14,000 adult patients received ECMO between 1990 and 2014, with a rate of survival to discharge of 57% for those in respiratory failure and 41% for those in cardiac failure.5 Its use increased 433% in the United States from 2006 to 2011.6
A national survey of critical care physicians and trainees in the United States found they had limited knowledge about ECMO technology and wanted to include specific educational objectives about it in their training.7
This article summarizes the principles of ECMO, including practical aspects such as patient selection, monitoring, and complications.
LIMITED EVIDENCE OF BENEFIT FROM CONTROLLED TRIALS
There is limited evidence from randomized controlled trials that ECMO is beneficial in adults.
In acute respiratory failure, the first randomized trial of ECMO in adults was conducted in 1979 in multiple medical centers.3 The survival rate was no higher with ECMO than with mechanical ventilation alone, and complication rates were very high.
Similarly, Morris et al4 performed a single-center trial comparing pressure-controlled inverse-ratio ventilation and extracorporeal carbon dioxide removal in patients with acute respiratory distress syndrome, which showed no survival benefit.

After these two early trials, ECMO was largely abandoned, and not until 2009 did a multicenter randomized trial in acute respiratory distress syndrome8 rejuvenate interest in its use. Although the trial did not conclusively prove that ECMO was more effective than conventional mechanical ventilation, the findings supported early referral to tertiary care centers with ECMO expertise, and the survival rate was substantially higher than in previous studies. A concise summary of randomized trials and retrospective studies utilizing ECMO in respiratory failure is shown in Table 1.8–14
During the global pandemic of influenza H1N1 in 2009–2010, several centers reported survival benefits from ECMO in patients with severe acute respiratory distress syndrome secondary to influenza.9–12,15–19 Two retrospective case-control studies reported lower mortality rates when H1N1 patients were transferred to ECMO centers10 and among younger patients with H1N1 who received ECMO.12
Ongoing trials (ClinicalTrials.gov identifier NCT01470703) may provide definitive evidence for the effectiveness of ECMO as a rescue therapy in acute respiratory distress syndrome.
In cardiogenic shock, single-center retrospective and observational studies have reported better outcomes for patients who received ECMO for cardiogenic shock secondary to myocardial infarction, pulmonary embolism, sepsis-related cardiomyopathy, and even extracorporeal cardiopulmonary resuscitation.20
WHAT IS ECMO?

In ECMO, venous blood is shunted through a machine to add oxygen, remove carbon dioxide, and regulate temperature (Figure 1). The components of an ECMO circuit are as follows:
- Blood pump
- Membrane oxygenator
- Gas mixer
- Cannulas
- Heater/cooler
- Console.
TWO BASIC CONFIGURATIONS

Two basic ECMO configurations are used in adults: venoarterial and venovenous,21 although combinations of the two—hybrid configurations—are sometimes used (Figure 2).
Venoarterial ECMO
Venoarterial ECMO provides complete or partial support to the heart and lungs and is the configuration of choice in patients with isolated cardiac failure that is refractory to other treatments. It takes deoxygenated blood from the venous system and returns oxygenated blood to the arterial circulation.
In the central venoarterial configuration, the intake cannula is most often surgically placed in the right atrium and the return cannula is placed in the proximal ascending aorta.
In the peripheral femoral configuration, the drainage cannula is placed in the femoral vein and advanced to the right atrium, and the return cannula is placed in either the ipsilateral or contralateral femoral artery. However, this configuration provides the patient with retrograde flow (against the native cardiac output), and oxygen delivery to the upper body may be impeded.
Axillary cannulation, in which the return cannula is placed directly into the axillary artery to provide antegrade flow, has been used recently in patients with pulmonary hypertension or right ventricular failure.22
Venovenous ECMO
Venovenous ECMO provides complete or partial support to the lungs and is the configuration of choice in isolated respiratory failure when cardiac function is preserved. It takes deoxygenated blood from the central venous system—either the femoral vein or internal jugular vein—and returns oxygenated blood to the venous circulation directed into the right atrium. It can be delivered by different cannula configurations based on the patient’s size and clinical requirements.
In the past, the most commonly used configuration was the femoral-atrial, in which the drainage cannula was placed in the femoral vein with the tip advanced to the level of the diaphragm in the inferior vena cava, and the return cannula was placed in the right internal jugular vein with its tip at the junction of the superior vena cava and right atrium. In this configuration, some of the oxygenated blood delivered by the superior vena cava cannula reaches the inferior vena cava cannula, creating a “shunt,” also known as “recirculation.”
Currently, a double-lumen cannula is preferred. This type of cannula is placed in the right internal jugular vein with the tip advanced to the inferior vena cava so that blood is drained through one lumen from both the inferior and superior vena cavas and returned via the other lumen with the jet directed over the tricuspid valve. Advantages of this system are that as it delivers more oxygen to the pulmonary arteries it reduces recirculation, it requires only a single cannula to be inserted, and it facilitates ambulation and rehabilitation in patients requiring long-term ECMO.
A newer double-lumen cannula designed to drain venous blood from the right atrium and reinfuse it directly into the pulmonary artery may provide an alternative for patients with right ventricular failure.
Extracorporeal removal of carbon dioxide
ECMO can remove carbon dioxide in patients with hypercapneic respiratory failure. Early technology used a variation of venovenous ECMO with very low blood flow rates through the pump, which allowed use of smaller cannulas while efficiently removing carbon dioxide.23
Since then, a pumpless extracorporeal lung-assist device has been developed that uses an arteriovenous configuration with two smaller cannulas inserted into the femoral artery and vein (Novalung, Germany).24 Lacking a pump, it avoids the complications associated with pumps such as hemolysis and clotting. It effectively removes carbon dioxide and helps reduce the frequency and intensity of mechanical ventilation. Since the flow is driven by the patient’s arteriovenous pressure gradient, good cardiac output is a prerequisite for its use.
A portable low-blood-flow machine that uses a very small (ie, 15-F) catheter in the venovenous configuration is under investigation (Hemolung RAS, Alung Technologies).
WHO CAN BENEFIT FROM ECMO?

Although evidence to support the routine use of ECMO is limited, tools and guidelines have been developed to help clinicians decide if a patient might benefit from it. Indications for and contraindications to ECMO are shown in Table 2.
The Extracorporeal Life Support Organization recommends considering ECMO if the predicted risk of death is greater than 50% without it, and says ECMO is indicated if the predicted risk exceeds 80%. A scoring system has been developed to help predict the risk of death in patients on ECMO.14 This system has been validated using a historical cohort of patients, and current studies are ongoing for prospective validation.
Many centers are now using ECMO as a salvage therapy in patients with severe respiratory failure when conventional mechanical ventilation and adjunctive therapies such as neuromuscular blockade, inhaled nitric oxide, steroids, prone positioning, and high-frequency oscillation therapy fail to improve gas exchange.25,26
ECMO is also indicated in hypercapneic respiratory failure secondary to status asthmaticus and exacerbation of chronic obstructive pulmonary disease, permissive hypercapnea with a Paco2 greater than 80 mm Hg, or inability to achieve safe inflation pressures with plateau pressures of 30 cm H2O or higher, refractory to conventional therapy.27
Sometimes, delay in referral leads to irreversible ventilator-induced lung injury due to intense mechanical ventilation, thus limiting the utility of ECMO.8 Early referral should be considered if the patient does not improve after a few days on optimal ventilator settings. In centers where this technology is not available, referral to the nearest ECMO center should be considered. A list of certified ECMO centers is available at www.elso.org/Members/CenterDirectory.aspx.
Contraindications to ECMO
Advanced age, comorbid conditions such as malignancy, nonpulmonary organ dysfunction (including complications of critical illness), and immunodeficiency or pharmacologic immune suppression have been associated with poor outcomes in ECMO patients.28 Severe aortic incompetence and aortic dissection are contraindications, since ventricular end-diastolic pressure can be increased with resultant ventricular distention, compromised myocardial oxygenation, and worsening of left heart failure.
ECMO is increasingly being used in situations in which it was previously considered contraindicated. Pregnant and postpartum patients with cardiorespiratory failure were previously not considered for ECMO because of a possible increased risk of coagulopathy and complications. However, a recent review showed that the outcomes of ECMO in pregnancy and postpartum were similar to those in nonpregnant patients, and the risk of catastrophic bleeding was minor.29
Similarly, ECMO is also being used increasingly in posttrauma patients and patients with other bleeding risks.30

Morbid obesity was once considered a contraindication because of difficulty in cannulation, but with newer types of cannulas, even patients with a body mass index greater than 60 kg/m2 are receiving ECMO.31
HOW DO YOU DO IT?

Figures 3 and 4 depict clinical decision-making in starting and weaning from ECMO in respiratory failure and cardiogenic shock, respectively.
Management of patients on ECMO
Appropriate patient selection and initiation of ECMO are only the beginning of a tough journey. Successful management requires minimizing lung injury from mechanical ventilation, careful monitoring of anticoagulation, and instituting adequate physical therapy, including ambulation when possible (Table 3).
Initial ECMO settings and monitoring

The cannulas for venovenous ECMO are frequently inserted under fluoroscopic or transesophageal echocardiographic guidance, whereas venoarterial ECMO cannulation does not require imaging and can be performed at the bedside in the intensive care unit or operating room.
The initial ECMO settings are titrated according to the patient’s hemodynamic and respiratory needs. There are three main variables: blood flow, fraction of oxygen in the sweep gas, and sweep gas flow rate. These are adjusted to achieve desirable levels of oxygen and carbon dioxide in the blood.
Blood flow is determined by the revolutions per minute of the pump, preload, and afterload of the circuit. Common patient conditions that may reduce flow are systemic hypertension, hypovolemia, cardiac tamponade, and tension pneumothorax, depending on the modality. In addition, mechanical factors such as clots in the oxygenator or kinks in the circuit can increase resistance and reduce flow. Resistance to flow is directly proportional to cannula lengths and inversely proportional to cannula radius to the fourth power. The greater the flow, the greater the oxygen delivery.
Fraction of oxygen in the sweep gas. The oxygenator has a gas blender that mixes air and oxygen and allows for a range of oxygen concentrations. Increases in fraction of oxygen increase the partial pressure of oxygen in the blood.
Sweep gas flow rate. Venous blood in the extracorporeal circuit is exposed to fresh gas (or sweep gas) that oxygenates the blood and removes carbon dioxide by diffusion. Increasing the sweep gas flow rate results in greater carbon dioxide elimination from the blood.
Laboratory monitoring. During ECMO, the following values are monitored frequently:
- Arterial blood gases
- Blood gases in the ECMO circuit before and after going through the oxygenator— to monitor the efficacy of the oxygenator membrane
- Lactic acid—to monitor for tissue hypoxia
- Plasma free hemoglobin (a marker of hemolysis)—to monitor for hemolysis.
Mechanical ventilation on ECMO
Low tidal volume ventilation greatly reduces the risk of death in patients on ECMO by reducing ventilator-induced lung injury. Proponents of ECMO believe that ECMO provides “lung rest,” and thus it is imperative that lung-protective ventilation strategies be followed in patients on ECMO.8 In most cases, after ECMO is started, low tidal volume ventilation (6 mL/kg) is possible and should be used—or even very low tidal volume ventilation (3–6 mL/kg).32,33 Many cases have also been described in which patients have been safely extubated while on ECMO to prevent ventilator-induced lung injury.34,35
If hypoxemia persists
Despite full support with venovenous ECMO, some patients remain hypoxemic due to inadequate blood flow to match metabolic demands, eg, patients with morbid obesity or severe sepsis and fever. The physician should ensure there is no recirculation, maximize blood flow, optimize the hematocrit to increase oxygen delivery, and consider ways to decrease oxygen consumption, including sedation, paralysis, and hypothermia.
Recirculation can be calculated by measuring the oxygen saturation of the blood in the ECMO machine before and after it goes through the oxygenator, and also in the central venous blood. Recirculation has been reduced by using double-lumen cannulas but can also be reduced by manipulation of the reinfusion cannula or increasing the distance between drainage and reinfusion ports in other configurations of venovenous ECMO.
Expert opinion suggests that oxygen saturation of 86% or more and Pao2 of 55 mm Hg or more in patients on venovenous ECMO are sufficient to prevent hypoxia-related end-organ injury.36 Venoarterial ECMO should be considered in patients on venovenous ECMO with refractory hypoxemia with the above measures.
Harlequin syndrome is characterized by upper body hypoxia resulting in cerebral hypoxemia due to poorly oxygenated blood in the coronary and cerebral circulations, especially in patients on peripheral venoarterial ECMO. It can be detected by sampling the blood in the arm (where the oxygen isn’t going) instead of the leg (where the oxygen is going), and it can be corrected by adjusting the Fio2, using positive end-expiratory pressure, or both to increase oxygenation. If ventilator settings do not improve this syndrome, the arterial cannulation site can be switched from the femoral artery to the axillary or carotid artery.
Alternatively, a mixed-configuration venoarterial-venous ECMO can also be created, in which a portion of arterialized blood from the arterial outflow cannula is diverted via the right internal jugular artery to the right heart. This enriches the blood traveling through the pulmonary circulation and to the left ventricle to provide better oxygen delivery to the coronary and cerebral circulations.
Anticoagulation monitoring and transfusions
Anticoagulation is necessary to maintain a clot-free and functional circuit. Most clots develop in the oxygenator membrane, where they can prevent optimal gas exchange and, rarely, lead to embolization to the systemic circulation. However, reports have suggested that anticoagulation can be held for short periods on ECMO if necessary.
Unfractionated heparin is usually used for anticoagulation. Commonly used tests to monitor anticoagulation are the augmented partial thromboplastin time, activated clotting time, and anti-factor Xa levels. Lately, thromboelastography analysis is being used to comprehensively monitor various components of the coagulation cascade.37 Anticoagulation is usually tailored to whether there are clots in the circuit, coagulopathy, and bleeding while on ECMO.38
Traditionally, blood products were used liberally during ECMO to maintain a normal hematocrit and improve oxygen delivery, although recent data suggest that outcomes may be similar with conservative use of blood products.39,40
Fluid management on ECMO
ECMO patients are fluid-overloaded due to a profound inflammatory response, cardiac failure, or both. Studies have shown that conservative fluid management improve lung function and shortens the duration of mechanical ventilation and intensive care in patients with lung injury.41 Hence, the patient’s net fluid balance should be kept negative, provided renal and hemodynamic parameters remain stable.
There is a high incidence of acute kidney injury in ECMO patients, and fluid overload is one of the main indications for renal replacement therapy.42 Continuous renal replacement therapy can be provided either by an in-line hemofilter or by incorporating a standard continuous renal replacement therapy machine into the ECMO circuit. There are no studies comparing the efficacy of these techniques, but they allow for rapid improvement in fluid balance and electrolyte disturbances and are commonly used in ECMO patients.42,43
Physical rehabilitation and ambulation on ECMO
Physical rehabilitation in mechanically ventilated patients has been shown to reduce ventilator days and stay in the intensive care unit.44 With the use of internal jugular double-lumen cannulas for venovenous ECMO and improvement in durability of the ECMO circuit, several centers are implementing physical rehabilitation and ambulation for patients while on ECMO. Current data suggest that physical therapy is safe for patients receiving ECMO and may accelerate the weaning process and shorten length of stay in the hospital after ECMO.45,46 Aggressive rehabilitation is especially beneficial in patients awaiting lung transplant and may improve posttransplant recovery and outcomes.47
Weaning from ECMO
There are no standard guidelines for weaning from venovenous or venoarterial ECMO. Once the underlying condition for which ECMO was initiated has improved, weaning can begin by reducing the blood flow rate, the flow rate of the sweep gas, or both.
Weaning from venovenous ECMO should be started when there is improvement in lung compliance, tidal volumes, and oxygenation. Once the circuit flow rate is reduced to less than 3 L/minute, ventilator settings are adjusted to standard lung-protective settings. ECMO support is gradually decreased by reducing the flow rate of sweep gas to less than 2 L/minute. If tidal volumes, respiratory rate, and gas exchange remain adequate after approximately 2 to 4 hours on a low rate of sweep gas, the patient can be weaned off the venovenous ECMO circuit.
Weaning from venoarterial ECMO should be considered when there is myocardial recovery with improved pulse pressure and contractility on echocardiography. This is done by reducing flow rates in increments of 0.5 to 2 L/minute over 24 to 36 hours and monitoring mean arterial pressures, central venous pressure, and myocardial contractility. When acceptable, patients are mostly weaned in a surgical setting. Prolonged periods on a low rate of blood flow are avoided to prevent thrombus formation in the circuit.
COMPLICATIONS OF ECMO
ECMO use can be associated with a myriad of patient and mechanical complications.
Hemorrhage is the most common complication encountered in ECMO, occurring in approximately 43% of patients.29 It occurs most frequently from cannulation and surgical sites. Although rare, potentially life-threatening pulmonary hemorrhage (including bleeding at the tracheostomy site), intracranial hemorrhage, and gastrointestinal hemorrhage have also been reported.30
Infections, including new infection and worsening sepsis in patients with acute respiratory distress syndrome secondary to infection, are common in patients on ECMO.48
Renal failure secondary to acute tubular necrosis requiring hemodialysis has been reported to occur in 13% of patients on ECMO.30
Other complications of concern, especially in patients on venoarterial ECMO, are lower limb ischemia and thromboembolism associated with site of cannulation and direction of blood flow.49 Mechanical complications include inappropriate placement of the cannula leading to insufficient oxygenation, injury to vessel walls, and rarely myocardial wall rupture; thrombus formation within the circuit causing failure of the oxygenator and sometimes, pulmonary or systemic embolism; and air embolism from the circuit.36
NOT SUITED FOR ALL
Despite limited data to support its use, there has been a recent increase in utilization of ECMO to support critically ill adult patients with cardiopulmonary failure. ECMO support is not suited for all patients. Careful selection of patients should be done to optimize resource utilization and provide the best opportunity for recovery or transplant.
- MacLaren G, Combes A, Bartlett RH. Contemporary extracorporeal membrane oxygenation for adult respiratory failure: life support in the new era. Intensive Care Med 2012; 38:210–220.
- Maslach-Hubbard A, Bratton SL. Extracorporeal membrane oxygenation for pediatric respiratory failure: history, development and current status. World J Crit Care Med 2013; 2:29–39.
- Zapol WM, Snider MT, Hill JD, et al. Extracorporeal membrane oxygenation in severe acute respiratory failure. A randomized prospective study. JAMA 1979; 242:2193–2196.
- Morris AH, Wallace CJ, Menlove RL, et al. Randomized clinical trial of pressure-controlled inverse ratio ventilation and extracorporeal CO2 removal for adult respiratory distress syndrome. Am J Respir Crit Care Med 1994; 149:295–305.
- Extracorporeal Life Support Organization. ECLS registry report. International Summary. January 2016. https://www.elso.org/Registry.aspx. Accessed March 17, 2016.
- Sauer CM, Yuh DD, Bonde P. Extracorporeal membrane oxygenation use has increased by 433% in adults in the United States from 2006 to 2011. ASAIO J 2015; 61:31–36.
- Sharma N, Wille K, Bellot S, Brodie D, Diaz-Guzman E. Role of extracorporeal membrane oxygenation in management of refractory ARDS in the intensive care unit: a national survey on perspectives of the adult critical care physicians and trainees. Chest 2014. http://journal.publications.chestnet.org/article.aspx?articleid=1913336. Accessed March 17, 2016.
- Peek GJ, Mugford M, Tiruvoipati R, et al; CESAR trial collaboration. Efficacy and economic assessment of conventional ventilatory support versus extracorporeal membrane oxygenation for severe adult respiratory failure (CESAR): a multicentre randomised controlled trial. Lancet 2009; 374:1351–1363.
- Australia and New Zealand Extracorporeal Membrane Oxygenation (ANZ ECMO) Influenza Investigators; Davies A, Jones D, Bailey M, et al. Extracorporeal membrane oxygenation for 2009 influenza A(H1N1) acute respiratory distress syndrome. JAMA 2009; 302:1888–1895.
- Noah MA, Peek GJ, Finney SJ, et al. Referral to an extracorporeal membrane oxygenation center and mortality among patients with severe 2009 influenza A(H1N1). JAMA 2011; 306:1659–1668.
- Patroniti N, Zangrillo A, Pappalardo F, et al. The Italian ECMO network experience during the 2009 influenza A(H1N1) pandemic: preparation for severe respiratory emergency outbreaks. Intensive Care Med 2011; 37:1447–1457.
- Pham T, Combes A, Roze H, et al; REVA Research Network. Extracorporeal membrane oxygenation for pandemic influenza A(H1N1)-induced acute respiratory distress syndrome: a cohort study and propensity-matched analysis. Am J Respir Crit Care Med 2013; 187:276–285.
- Schmidt M, Zogheib E, Roze H, et al. The PRESERVE mortality risk score and analysis of long-term outcomes after extracorporeal membrane oxygenation for severe acute respiratory distress syndrome. Intensive Care Med 2013; 39; 532:1704–1713.
- Schmidt M, Bailey M, Sheldrake J, et al. Predicting survival after extracorporeal membrane oxygenation for severe acute respiratory failure. The Respiratory Extracorporeal Membrane Oxygenation Survival Prediction (RESP) score. Am J Respir Crit Care Med 2014; 189:1374–1382.
- Chan KK, Lee KL, Lam PK, Law KI, Joynt GM, Yan WW. Hong Kong's experience on the use of extracorporeal membrane oxygenation for the treatment of influenza A (H1N1). Hong Kong Med J 2010; 16:447–454.
- Freed DH, Henzler D, White CW, et al; Canadian Critical Care Trials Group. Extracorporeal lung support for patients who had severe respiratory failure secondary to influenza A (H1N1) 2009 infection in Canada. Can J Anaesth 2010; 57:240–247.
- Nair P, Davies AR, Beca J, et al. Extracorporeal membrane oxygenation for severe ARDS in pregnant and postpartum women during the 2009 H1N1 pandemic. Intensive Care Med 2011; 37:648–654.
- Turner DA, Rehder KJ, Peterson-Carmichael SL, et al. Extracorporeal membrane oxygenation for severe refractory respiratory failure secondary to 2009 H1N1 influenza A. Respir Care 2011; 56:941–946.
- Kumar A, Zarychanski R, Pinto R, et al; Canadian Critical Care Trials Group H1N1 Collaborative. Critically ill patients with 2009 influenza A(H1N1) infection in Canada. JAMA 2009; 302:1872–1879.
- Abrams D, Combes A, Brodie D. Extracorporeal membrane oxygenation in cardiopulmonary disease in adults. J Am Coll Cardiol 2014; 63:2769–2778.
- Marasco SF, Lukas G, McDonald M, McMillan J, Ihle B. Review of ECMO (extra corporeal membrane oxygenation) support in critically ill adult patients. Heart Lung Circ 2008; 17(suppl 4):S41–S47.
- Hysi I, Fabre O, Renaut C, Guesnier L. Extracorporeal membrane oxygenation with direct axillary artery perfusion. J Card Surg 2014; 29:268–269.
- Gattinoni L, Kolobow T, Agostoni A, et al. Clinical application of low frequency positive pressure ventilation with extracorporeal CO2 removal (LFPPV-ECCO2R) in treatment of adult respiratory distress syndrome (ARDS). Int J Artif Organs 1979; 2:282–283.
- Liebold A, Philipp A, Kaiser M, Merk J, Schmid FX, Birnbaum DE. Pumpless extracorporeal lung assist using an arterio-venous shunt. Applications and limitations. Minerva Anestesiol 2002; 68:387–391.
- Paden ML, Conrad SA, Rycus PT, Thiagarajan RR; ELSO Registry. Extracorporeal Life Support Organization Registry Report 2012. ASAIO J 2013; 59:202–210.
- Shekar K, Davies AR, Mullany DV, Tiruvoipati R, Fraser JF. To ventilate, oscillate, or cannulate? J Crit Care 2013; 28:655–662.
- Mikkelsen ME, Woo YJ, Sager JS, Fuchs BD, Christie JD. Outcomes using extracorporeal life support for adult respiratory failure due to status asthmaticus. ASAIO J 2009; 55:47–52.
- Turner DA, Cheifetz IM. Extracorporeal membrane oxygenation for adult respiratory failure. Respir Care 2013; 58:1038–1052.
- Sharma NS, Wille KM, Bellot SC, Diaz-Guzman E. Modern use of extracorporeal life support in pregnancy and postpartum. ASAIO J 2015; 61:110–114.
- Ried M, Bein T, Philipp A, et al. Extracorporeal lung support in trauma patients with severe chest injury and acute lung failure: a 10-year institutional experience. Crit Care 2013; 17:R110.
- Al-Soufi S, Buscher H, Nguyen ND, Rycus P, Nair P. Lack of association between body weight and mortality in patients on veno-venous extracorporeal membrane oxygenation. Intensive Care Med 2013; 39:1995–2002.
- Marhong JD, Telesnicki T, Munshi L, Del Sorbo L, Detsky M, Fan E. Mechanical ventilation during extracorporeal membrane oxygenation. An international survey. Ann Am Thorac Soc 2014; 11:956–961.
- Schmidt M, Pellegrino V, Combes A, Scheinkestel C, Cooper DJ, Hodgson C. Mechanical ventilation during extracorporeal membrane oxygenation. Crit Care 2014; 18:203.
- Bein T, Wittmann S, Philipp A, Nerlich M, Kuehnel T, Schlitt HJ. Successful extubation of an "unweanable" patient with severe ankylosing spondylitis (Bechterew's disease) using a pumpless extracorporeal lung assist. Intensive Care Med 2008; 34:2313–2314.
- Anton-Martin P, Thompson MT, Sheeran PD, Fischer AC, Taylor D, Thomas JA. Extubation during pediatric extracorporeal membrane oxygenation: a single-center experience. Pediatr Crit Care Med 2014; 15:861–869.
- Sidebotham D, McGeorge A, McGuinness S, Edwards M, Willcox T, Beca J. Extracorporeal membrane oxygenation for treating severe cardiac and respiratory failure in adults: part 2-technical considerations. J Cardiothorac Vasc Anesth 2010; 24:164–172.
- Stammers AH, Willett L, Fristoe L, et al. Coagulation monitoring during extracorporeal membrane oxygenation: the role of thrombelastography. J Extra Corpor Technol 1995; 27:137–145.
- Bembea MM, Schwartz JM, Shah N, et al. Anticoagulation monitoring during pediatric extracorporeal membrane oxygenation. ASAIO J 2013; 59:63–68.
- Agerstrand CL, Burkart KM, Abrams DC, Bacchetta MD, Brodie D. Blood conservation in extracorporeal membrane oxygenation for acute respiratory distress syndrome. Ann Thorac Surg 2015; 99:590–595.
- Voelker MT, Busch T, Bercker S, Fichtner F, Kaisers UX, Laudi S. Restrictive transfusion practice during extracorporeal membrane oxygenation therapy for severe acute respiratory distress syndrome. Artif Organs 2015; 39:374–378.
- National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network; Wiedemann HP, Wheeler AP, Bernard GR, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med 2006; 354:2564–2575.
- Fleming GM, Askenazi DJ, Bridges BC, et al. A multicenter international survey of renal supportive therapy during ECMO: the Kidney Intervention During Extracorporeal Membrane Oxygenation (KIDMO) group. ASAIO J 2012; 58:407–414.
- Askenazi DJ, Selewski DT, Paden ML, et al. Renal replacement therapy in critically ill patients receiving extracorporeal membrane oxygenation. Clin J Am Soc Nephrol 2012; 7:1328–1336.
- Schweickert WD, Pohlman MC, Pohlman AS, et al. Early physical and occupational therapy in mechanically ventilated, critically ill patients: a randomised controlled trial. Lancet 2009; 373:1874–1882.
- Abrams D, Javidfar J, Farrand E, et al. Early mobilization of patients receiving extracorporeal membrane oxygenation: a retrospective cohort study. Crit Care 2014; 18:R38.
- Thiagarajan RR, Teele SA, Teele KP, Beke DM. Physical therapy and rehabilitation issues for patients supported with extracorporeal membrane oxygenation. J Pediatr Rehabil Med 2012; 5:47–52.
- Hoopes CW, Kukreja J, Golden J, Davenport DL, Diaz-Guzman E, Zwischenberger JB. Extracorporeal membrane oxygenation as a bridge to pulmonary transplantation. J Thorac Cardiovasc Surg 2013; 145:862–868.
- Burket JS, Bartlett RH, Vander Hyde K, Chenoweth CE. Nosocomial infections in adult patients undergoing extracorporeal membrane oxygenation. Clin Infect Dis 1999; 28:828–833.
- Lafç G, Budak AB, Yener AÜ, Cicek OF. Use of extracorporeal membrane oxygenation in adults. Heart Lung Circ 2014; 23:10–23.
Extracorporeal membrance oxygenation (ECMO) provides temporary cardiorespiratory support for patients with severe respiratory or cardiac failure refractory to conventional therapy.1 It can be configured to provide oxygen, remove carbon dioxide, support perfusion, or all of the above. It may provide a bridge to recovery in patients with acute cardiopulmonary failure or to heart or lung transplant.
Developed in the 1970s, ECMO has proven effective and is widely used in children with respiratory and cardiopulmonary failure.2 However, it remained little used in adults, as early randomized trials showed higher rates of complications in adults who received it and no survival advantage.3,4 Proponents of using it in adult patients believe that these poor outcomes were at least partially due to limited training, intensive anticoagulation, and excessive volume and pressure during mechanical ventilation. Although ECMO technology has improved substantially in the last decade and survival rates have improved (www.elso.org), evidence to support its routine use in adults remains limited.
Nevertheless, about 14,000 adult patients received ECMO between 1990 and 2014, with a rate of survival to discharge of 57% for those in respiratory failure and 41% for those in cardiac failure.5 Its use increased 433% in the United States from 2006 to 2011.6
A national survey of critical care physicians and trainees in the United States found they had limited knowledge about ECMO technology and wanted to include specific educational objectives about it in their training.7
This article summarizes the principles of ECMO, including practical aspects such as patient selection, monitoring, and complications.
LIMITED EVIDENCE OF BENEFIT FROM CONTROLLED TRIALS
There is limited evidence from randomized controlled trials that ECMO is beneficial in adults.
In acute respiratory failure, the first randomized trial of ECMO in adults was conducted in 1979 in multiple medical centers.3 The survival rate was no higher with ECMO than with mechanical ventilation alone, and complication rates were very high.
Similarly, Morris et al4 performed a single-center trial comparing pressure-controlled inverse-ratio ventilation and extracorporeal carbon dioxide removal in patients with acute respiratory distress syndrome, which showed no survival benefit.
After these two early trials, ECMO was largely abandoned, and not until 2009 did a multicenter randomized trial in acute respiratory distress syndrome8 rejuvenate interest in its use. Although the trial did not conclusively prove that ECMO was more effective than conventional mechanical ventilation, the findings supported early referral to tertiary care centers with ECMO expertise, and the survival rate was substantially higher than in previous studies. A concise summary of randomized trials and retrospective studies utilizing ECMO in respiratory failure is shown in Table 1.8–14
During the global pandemic of influenza H1N1 in 2009–2010, several centers reported survival benefits from ECMO in patients with severe acute respiratory distress syndrome secondary to influenza.9–12,15–19 Two retrospective case-control studies reported lower mortality rates when H1N1 patients were transferred to ECMO centers10 and among younger patients with H1N1 who received ECMO.12
Ongoing trials (ClinicalTrials.gov identifier NCT01470703) may provide definitive evidence for the effectiveness of ECMO as a rescue therapy in acute respiratory distress syndrome.
In cardiogenic shock, single-center retrospective and observational studies have reported better outcomes for patients who received ECMO for cardiogenic shock secondary to myocardial infarction, pulmonary embolism, sepsis-related cardiomyopathy, and even extracorporeal cardiopulmonary resuscitation.20
WHAT IS ECMO?
In ECMO, venous blood is shunted through a machine to add oxygen, remove carbon dioxide, and regulate temperature (Figure 1). The components of an ECMO circuit are as follows:
- Blood pump
- Membrane oxygenator
- Gas mixer
- Cannulas
- Heater/cooler
- Console.
TWO BASIC CONFIGURATIONS
Two basic ECMO configurations are used in adults: venoarterial and venovenous,21 although combinations of the two—hybrid configurations—are sometimes used (Figure 2).
Venoarterial ECMO
Venoarterial ECMO provides complete or partial support to the heart and lungs and is the configuration of choice in patients with isolated cardiac failure that is refractory to other treatments. It takes deoxygenated blood from the venous system and returns oxygenated blood to the arterial circulation.
In the central venoarterial configuration, the intake cannula is most often surgically placed in the right atrium and the return cannula is placed in the proximal ascending aorta.
In the peripheral femoral configuration, the drainage cannula is placed in the femoral vein and advanced to the right atrium, and the return cannula is placed in either the ipsilateral or contralateral femoral artery. However, this configuration provides the patient with retrograde flow (against the native cardiac output), and oxygen delivery to the upper body may be impeded.
Axillary cannulation, in which the return cannula is placed directly into the axillary artery to provide antegrade flow, has been used recently in patients with pulmonary hypertension or right ventricular failure.22
Venovenous ECMO
Venovenous ECMO provides complete or partial support to the lungs and is the configuration of choice in isolated respiratory failure when cardiac function is preserved. It takes deoxygenated blood from the central venous system—either the femoral vein or internal jugular vein—and returns oxygenated blood to the venous circulation directed into the right atrium. It can be delivered by different cannula configurations based on the patient’s size and clinical requirements.
In the past, the most commonly used configuration was the femoral-atrial, in which the drainage cannula was placed in the femoral vein with the tip advanced to the level of the diaphragm in the inferior vena cava, and the return cannula was placed in the right internal jugular vein with its tip at the junction of the superior vena cava and right atrium. In this configuration, some of the oxygenated blood delivered by the superior vena cava cannula reaches the inferior vena cava cannula, creating a “shunt,” also known as “recirculation.”
Currently, a double-lumen cannula is preferred. This type of cannula is placed in the right internal jugular vein with the tip advanced to the inferior vena cava so that blood is drained through one lumen from both the inferior and superior vena cavas and returned via the other lumen with the jet directed over the tricuspid valve. Advantages of this system are that as it delivers more oxygen to the pulmonary arteries it reduces recirculation, it requires only a single cannula to be inserted, and it facilitates ambulation and rehabilitation in patients requiring long-term ECMO.
A newer double-lumen cannula designed to drain venous blood from the right atrium and reinfuse it directly into the pulmonary artery may provide an alternative for patients with right ventricular failure.
Extracorporeal removal of carbon dioxide
ECMO can remove carbon dioxide in patients with hypercapneic respiratory failure. Early technology used a variation of venovenous ECMO with very low blood flow rates through the pump, which allowed use of smaller cannulas while efficiently removing carbon dioxide.23
Since then, a pumpless extracorporeal lung-assist device has been developed that uses an arteriovenous configuration with two smaller cannulas inserted into the femoral artery and vein (Novalung, Germany).24 Lacking a pump, it avoids the complications associated with pumps such as hemolysis and clotting. It effectively removes carbon dioxide and helps reduce the frequency and intensity of mechanical ventilation. Since the flow is driven by the patient’s arteriovenous pressure gradient, good cardiac output is a prerequisite for its use.
A portable low-blood-flow machine that uses a very small (ie, 15-F) catheter in the venovenous configuration is under investigation (Hemolung RAS, Alung Technologies).
WHO CAN BENEFIT FROM ECMO?
Although evidence to support the routine use of ECMO is limited, tools and guidelines have been developed to help clinicians decide if a patient might benefit from it. Indications for and contraindications to ECMO are shown in Table 2.
The Extracorporeal Life Support Organization recommends considering ECMO if the predicted risk of death is greater than 50% without it, and says ECMO is indicated if the predicted risk exceeds 80%. A scoring system has been developed to help predict the risk of death in patients on ECMO.14 This system has been validated using a historical cohort of patients, and current studies are ongoing for prospective validation.
Many centers are now using ECMO as a salvage therapy in patients with severe respiratory failure when conventional mechanical ventilation and adjunctive therapies such as neuromuscular blockade, inhaled nitric oxide, steroids, prone positioning, and high-frequency oscillation therapy fail to improve gas exchange.25,26
ECMO is also indicated in hypercapneic respiratory failure secondary to status asthmaticus and exacerbation of chronic obstructive pulmonary disease, permissive hypercapnea with a Paco2 greater than 80 mm Hg, or inability to achieve safe inflation pressures with plateau pressures of 30 cm H2O or higher, refractory to conventional therapy.27
Sometimes, delay in referral leads to irreversible ventilator-induced lung injury due to intense mechanical ventilation, thus limiting the utility of ECMO.8 Early referral should be considered if the patient does not improve after a few days on optimal ventilator settings. In centers where this technology is not available, referral to the nearest ECMO center should be considered. A list of certified ECMO centers is available at www.elso.org/Members/CenterDirectory.aspx.
Contraindications to ECMO
Advanced age, comorbid conditions such as malignancy, nonpulmonary organ dysfunction (including complications of critical illness), and immunodeficiency or pharmacologic immune suppression have been associated with poor outcomes in ECMO patients.28 Severe aortic incompetence and aortic dissection are contraindications, since ventricular end-diastolic pressure can be increased with resultant ventricular distention, compromised myocardial oxygenation, and worsening of left heart failure.
ECMO is increasingly being used in situations in which it was previously considered contraindicated. Pregnant and postpartum patients with cardiorespiratory failure were previously not considered for ECMO because of a possible increased risk of coagulopathy and complications. However, a recent review showed that the outcomes of ECMO in pregnancy and postpartum were similar to those in nonpregnant patients, and the risk of catastrophic bleeding was minor.29
Similarly, ECMO is also being used increasingly in posttrauma patients and patients with other bleeding risks.30
Morbid obesity was once considered a contraindication because of difficulty in cannulation, but with newer types of cannulas, even patients with a body mass index greater than 60 kg/m2 are receiving ECMO.31
HOW DO YOU DO IT?
Figures 3 and 4 depict clinical decision-making in starting and weaning from ECMO in respiratory failure and cardiogenic shock, respectively.
Management of patients on ECMO
Appropriate patient selection and initiation of ECMO are only the beginning of a tough journey. Successful management requires minimizing lung injury from mechanical ventilation, careful monitoring of anticoagulation, and instituting adequate physical therapy, including ambulation when possible (Table 3).
Initial ECMO settings and monitoring
The cannulas for venovenous ECMO are frequently inserted under fluoroscopic or transesophageal echocardiographic guidance, whereas venoarterial ECMO cannulation does not require imaging and can be performed at the bedside in the intensive care unit or operating room.
The initial ECMO settings are titrated according to the patient’s hemodynamic and respiratory needs. There are three main variables: blood flow, fraction of oxygen in the sweep gas, and sweep gas flow rate. These are adjusted to achieve desirable levels of oxygen and carbon dioxide in the blood.
Blood flow is determined by the revolutions per minute of the pump, preload, and afterload of the circuit. Common patient conditions that may reduce flow are systemic hypertension, hypovolemia, cardiac tamponade, and tension pneumothorax, depending on the modality. In addition, mechanical factors such as clots in the oxygenator or kinks in the circuit can increase resistance and reduce flow. Resistance to flow is directly proportional to cannula lengths and inversely proportional to cannula radius to the fourth power. The greater the flow, the greater the oxygen delivery.
Fraction of oxygen in the sweep gas. The oxygenator has a gas blender that mixes air and oxygen and allows for a range of oxygen concentrations. Increases in fraction of oxygen increase the partial pressure of oxygen in the blood.
Sweep gas flow rate. Venous blood in the extracorporeal circuit is exposed to fresh gas (or sweep gas) that oxygenates the blood and removes carbon dioxide by diffusion. Increasing the sweep gas flow rate results in greater carbon dioxide elimination from the blood.
Laboratory monitoring. During ECMO, the following values are monitored frequently:
- Arterial blood gases
- Blood gases in the ECMO circuit before and after going through the oxygenator— to monitor the efficacy of the oxygenator membrane
- Lactic acid—to monitor for tissue hypoxia
- Plasma free hemoglobin (a marker of hemolysis)—to monitor for hemolysis.
Mechanical ventilation on ECMO
Low tidal volume ventilation greatly reduces the risk of death in patients on ECMO by reducing ventilator-induced lung injury. Proponents of ECMO believe that ECMO provides “lung rest,” and thus it is imperative that lung-protective ventilation strategies be followed in patients on ECMO.8 In most cases, after ECMO is started, low tidal volume ventilation (6 mL/kg) is possible and should be used—or even very low tidal volume ventilation (3–6 mL/kg).32,33 Many cases have also been described in which patients have been safely extubated while on ECMO to prevent ventilator-induced lung injury.34,35
If hypoxemia persists
Despite full support with venovenous ECMO, some patients remain hypoxemic due to inadequate blood flow to match metabolic demands, eg, patients with morbid obesity or severe sepsis and fever. The physician should ensure there is no recirculation, maximize blood flow, optimize the hematocrit to increase oxygen delivery, and consider ways to decrease oxygen consumption, including sedation, paralysis, and hypothermia.
Recirculation can be calculated by measuring the oxygen saturation of the blood in the ECMO machine before and after it goes through the oxygenator, and also in the central venous blood. Recirculation has been reduced by using double-lumen cannulas but can also be reduced by manipulation of the reinfusion cannula or increasing the distance between drainage and reinfusion ports in other configurations of venovenous ECMO.
Expert opinion suggests that oxygen saturation of 86% or more and Pao2 of 55 mm Hg or more in patients on venovenous ECMO are sufficient to prevent hypoxia-related end-organ injury.36 Venoarterial ECMO should be considered in patients on venovenous ECMO with refractory hypoxemia with the above measures.
Harlequin syndrome is characterized by upper body hypoxia resulting in cerebral hypoxemia due to poorly oxygenated blood in the coronary and cerebral circulations, especially in patients on peripheral venoarterial ECMO. It can be detected by sampling the blood in the arm (where the oxygen isn’t going) instead of the leg (where the oxygen is going), and it can be corrected by adjusting the Fio2, using positive end-expiratory pressure, or both to increase oxygenation. If ventilator settings do not improve this syndrome, the arterial cannulation site can be switched from the femoral artery to the axillary or carotid artery.
Alternatively, a mixed-configuration venoarterial-venous ECMO can also be created, in which a portion of arterialized blood from the arterial outflow cannula is diverted via the right internal jugular artery to the right heart. This enriches the blood traveling through the pulmonary circulation and to the left ventricle to provide better oxygen delivery to the coronary and cerebral circulations.
Anticoagulation monitoring and transfusions
Anticoagulation is necessary to maintain a clot-free and functional circuit. Most clots develop in the oxygenator membrane, where they can prevent optimal gas exchange and, rarely, lead to embolization to the systemic circulation. However, reports have suggested that anticoagulation can be held for short periods on ECMO if necessary.
Unfractionated heparin is usually used for anticoagulation. Commonly used tests to monitor anticoagulation are the augmented partial thromboplastin time, activated clotting time, and anti-factor Xa levels. Lately, thromboelastography analysis is being used to comprehensively monitor various components of the coagulation cascade.37 Anticoagulation is usually tailored to whether there are clots in the circuit, coagulopathy, and bleeding while on ECMO.38
Traditionally, blood products were used liberally during ECMO to maintain a normal hematocrit and improve oxygen delivery, although recent data suggest that outcomes may be similar with conservative use of blood products.39,40
Fluid management on ECMO
ECMO patients are fluid-overloaded due to a profound inflammatory response, cardiac failure, or both. Studies have shown that conservative fluid management improve lung function and shortens the duration of mechanical ventilation and intensive care in patients with lung injury.41 Hence, the patient’s net fluid balance should be kept negative, provided renal and hemodynamic parameters remain stable.
There is a high incidence of acute kidney injury in ECMO patients, and fluid overload is one of the main indications for renal replacement therapy.42 Continuous renal replacement therapy can be provided either by an in-line hemofilter or by incorporating a standard continuous renal replacement therapy machine into the ECMO circuit. There are no studies comparing the efficacy of these techniques, but they allow for rapid improvement in fluid balance and electrolyte disturbances and are commonly used in ECMO patients.42,43
Physical rehabilitation and ambulation on ECMO
Physical rehabilitation in mechanically ventilated patients has been shown to reduce ventilator days and stay in the intensive care unit.44 With the use of internal jugular double-lumen cannulas for venovenous ECMO and improvement in durability of the ECMO circuit, several centers are implementing physical rehabilitation and ambulation for patients while on ECMO. Current data suggest that physical therapy is safe for patients receiving ECMO and may accelerate the weaning process and shorten length of stay in the hospital after ECMO.45,46 Aggressive rehabilitation is especially beneficial in patients awaiting lung transplant and may improve posttransplant recovery and outcomes.47
Weaning from ECMO
There are no standard guidelines for weaning from venovenous or venoarterial ECMO. Once the underlying condition for which ECMO was initiated has improved, weaning can begin by reducing the blood flow rate, the flow rate of the sweep gas, or both.
Weaning from venovenous ECMO should be started when there is improvement in lung compliance, tidal volumes, and oxygenation. Once the circuit flow rate is reduced to less than 3 L/minute, ventilator settings are adjusted to standard lung-protective settings. ECMO support is gradually decreased by reducing the flow rate of sweep gas to less than 2 L/minute. If tidal volumes, respiratory rate, and gas exchange remain adequate after approximately 2 to 4 hours on a low rate of sweep gas, the patient can be weaned off the venovenous ECMO circuit.
Weaning from venoarterial ECMO should be considered when there is myocardial recovery with improved pulse pressure and contractility on echocardiography. This is done by reducing flow rates in increments of 0.5 to 2 L/minute over 24 to 36 hours and monitoring mean arterial pressures, central venous pressure, and myocardial contractility. When acceptable, patients are mostly weaned in a surgical setting. Prolonged periods on a low rate of blood flow are avoided to prevent thrombus formation in the circuit.
COMPLICATIONS OF ECMO
ECMO use can be associated with a myriad of patient and mechanical complications.
Hemorrhage is the most common complication encountered in ECMO, occurring in approximately 43% of patients.29 It occurs most frequently from cannulation and surgical sites. Although rare, potentially life-threatening pulmonary hemorrhage (including bleeding at the tracheostomy site), intracranial hemorrhage, and gastrointestinal hemorrhage have also been reported.30
Infections, including new infection and worsening sepsis in patients with acute respiratory distress syndrome secondary to infection, are common in patients on ECMO.48
Renal failure secondary to acute tubular necrosis requiring hemodialysis has been reported to occur in 13% of patients on ECMO.30
Other complications of concern, especially in patients on venoarterial ECMO, are lower limb ischemia and thromboembolism associated with site of cannulation and direction of blood flow.49 Mechanical complications include inappropriate placement of the cannula leading to insufficient oxygenation, injury to vessel walls, and rarely myocardial wall rupture; thrombus formation within the circuit causing failure of the oxygenator and sometimes, pulmonary or systemic embolism; and air embolism from the circuit.36
NOT SUITED FOR ALL
Despite limited data to support its use, there has been a recent increase in utilization of ECMO to support critically ill adult patients with cardiopulmonary failure. ECMO support is not suited for all patients. Careful selection of patients should be done to optimize resource utilization and provide the best opportunity for recovery or transplant.
Extracorporeal membrance oxygenation (ECMO) provides temporary cardiorespiratory support for patients with severe respiratory or cardiac failure refractory to conventional therapy.1 It can be configured to provide oxygen, remove carbon dioxide, support perfusion, or all of the above. It may provide a bridge to recovery in patients with acute cardiopulmonary failure or to heart or lung transplant.
Developed in the 1970s, ECMO has proven effective and is widely used in children with respiratory and cardiopulmonary failure.2 However, it remained little used in adults, as early randomized trials showed higher rates of complications in adults who received it and no survival advantage.3,4 Proponents of using it in adult patients believe that these poor outcomes were at least partially due to limited training, intensive anticoagulation, and excessive volume and pressure during mechanical ventilation. Although ECMO technology has improved substantially in the last decade and survival rates have improved (www.elso.org), evidence to support its routine use in adults remains limited.
Nevertheless, about 14,000 adult patients received ECMO between 1990 and 2014, with a rate of survival to discharge of 57% for those in respiratory failure and 41% for those in cardiac failure.5 Its use increased 433% in the United States from 2006 to 2011.6
A national survey of critical care physicians and trainees in the United States found they had limited knowledge about ECMO technology and wanted to include specific educational objectives about it in their training.7
This article summarizes the principles of ECMO, including practical aspects such as patient selection, monitoring, and complications.
LIMITED EVIDENCE OF BENEFIT FROM CONTROLLED TRIALS
There is limited evidence from randomized controlled trials that ECMO is beneficial in adults.
In acute respiratory failure, the first randomized trial of ECMO in adults was conducted in 1979 in multiple medical centers.3 The survival rate was no higher with ECMO than with mechanical ventilation alone, and complication rates were very high.
Similarly, Morris et al4 performed a single-center trial comparing pressure-controlled inverse-ratio ventilation and extracorporeal carbon dioxide removal in patients with acute respiratory distress syndrome, which showed no survival benefit.
After these two early trials, ECMO was largely abandoned, and not until 2009 did a multicenter randomized trial in acute respiratory distress syndrome8 rejuvenate interest in its use. Although the trial did not conclusively prove that ECMO was more effective than conventional mechanical ventilation, the findings supported early referral to tertiary care centers with ECMO expertise, and the survival rate was substantially higher than in previous studies. A concise summary of randomized trials and retrospective studies utilizing ECMO in respiratory failure is shown in Table 1.8–14
During the global pandemic of influenza H1N1 in 2009–2010, several centers reported survival benefits from ECMO in patients with severe acute respiratory distress syndrome secondary to influenza.9–12,15–19 Two retrospective case-control studies reported lower mortality rates when H1N1 patients were transferred to ECMO centers10 and among younger patients with H1N1 who received ECMO.12
Ongoing trials (ClinicalTrials.gov identifier NCT01470703) may provide definitive evidence for the effectiveness of ECMO as a rescue therapy in acute respiratory distress syndrome.
In cardiogenic shock, single-center retrospective and observational studies have reported better outcomes for patients who received ECMO for cardiogenic shock secondary to myocardial infarction, pulmonary embolism, sepsis-related cardiomyopathy, and even extracorporeal cardiopulmonary resuscitation.20
WHAT IS ECMO?
In ECMO, venous blood is shunted through a machine to add oxygen, remove carbon dioxide, and regulate temperature (Figure 1). The components of an ECMO circuit are as follows:
- Blood pump
- Membrane oxygenator
- Gas mixer
- Cannulas
- Heater/cooler
- Console.
TWO BASIC CONFIGURATIONS
Two basic ECMO configurations are used in adults: venoarterial and venovenous,21 although combinations of the two—hybrid configurations—are sometimes used (Figure 2).
Venoarterial ECMO
Venoarterial ECMO provides complete or partial support to the heart and lungs and is the configuration of choice in patients with isolated cardiac failure that is refractory to other treatments. It takes deoxygenated blood from the venous system and returns oxygenated blood to the arterial circulation.
In the central venoarterial configuration, the intake cannula is most often surgically placed in the right atrium and the return cannula is placed in the proximal ascending aorta.
In the peripheral femoral configuration, the drainage cannula is placed in the femoral vein and advanced to the right atrium, and the return cannula is placed in either the ipsilateral or contralateral femoral artery. However, this configuration provides the patient with retrograde flow (against the native cardiac output), and oxygen delivery to the upper body may be impeded.
Axillary cannulation, in which the return cannula is placed directly into the axillary artery to provide antegrade flow, has been used recently in patients with pulmonary hypertension or right ventricular failure.22
Venovenous ECMO
Venovenous ECMO provides complete or partial support to the lungs and is the configuration of choice in isolated respiratory failure when cardiac function is preserved. It takes deoxygenated blood from the central venous system—either the femoral vein or internal jugular vein—and returns oxygenated blood to the venous circulation directed into the right atrium. It can be delivered by different cannula configurations based on the patient’s size and clinical requirements.
In the past, the most commonly used configuration was the femoral-atrial, in which the drainage cannula was placed in the femoral vein with the tip advanced to the level of the diaphragm in the inferior vena cava, and the return cannula was placed in the right internal jugular vein with its tip at the junction of the superior vena cava and right atrium. In this configuration, some of the oxygenated blood delivered by the superior vena cava cannula reaches the inferior vena cava cannula, creating a “shunt,” also known as “recirculation.”
Currently, a double-lumen cannula is preferred. This type of cannula is placed in the right internal jugular vein with the tip advanced to the inferior vena cava so that blood is drained through one lumen from both the inferior and superior vena cavas and returned via the other lumen with the jet directed over the tricuspid valve. Advantages of this system are that as it delivers more oxygen to the pulmonary arteries it reduces recirculation, it requires only a single cannula to be inserted, and it facilitates ambulation and rehabilitation in patients requiring long-term ECMO.
A newer double-lumen cannula designed to drain venous blood from the right atrium and reinfuse it directly into the pulmonary artery may provide an alternative for patients with right ventricular failure.
Extracorporeal removal of carbon dioxide
ECMO can remove carbon dioxide in patients with hypercapneic respiratory failure. Early technology used a variation of venovenous ECMO with very low blood flow rates through the pump, which allowed use of smaller cannulas while efficiently removing carbon dioxide.23
Since then, a pumpless extracorporeal lung-assist device has been developed that uses an arteriovenous configuration with two smaller cannulas inserted into the femoral artery and vein (Novalung, Germany).24 Lacking a pump, it avoids the complications associated with pumps such as hemolysis and clotting. It effectively removes carbon dioxide and helps reduce the frequency and intensity of mechanical ventilation. Since the flow is driven by the patient’s arteriovenous pressure gradient, good cardiac output is a prerequisite for its use.
A portable low-blood-flow machine that uses a very small (ie, 15-F) catheter in the venovenous configuration is under investigation (Hemolung RAS, Alung Technologies).
WHO CAN BENEFIT FROM ECMO?
Although evidence to support the routine use of ECMO is limited, tools and guidelines have been developed to help clinicians decide if a patient might benefit from it. Indications for and contraindications to ECMO are shown in Table 2.
The Extracorporeal Life Support Organization recommends considering ECMO if the predicted risk of death is greater than 50% without it, and says ECMO is indicated if the predicted risk exceeds 80%. A scoring system has been developed to help predict the risk of death in patients on ECMO.14 This system has been validated using a historical cohort of patients, and current studies are ongoing for prospective validation.
Many centers are now using ECMO as a salvage therapy in patients with severe respiratory failure when conventional mechanical ventilation and adjunctive therapies such as neuromuscular blockade, inhaled nitric oxide, steroids, prone positioning, and high-frequency oscillation therapy fail to improve gas exchange.25,26
ECMO is also indicated in hypercapneic respiratory failure secondary to status asthmaticus and exacerbation of chronic obstructive pulmonary disease, permissive hypercapnea with a Paco2 greater than 80 mm Hg, or inability to achieve safe inflation pressures with plateau pressures of 30 cm H2O or higher, refractory to conventional therapy.27
Sometimes, delay in referral leads to irreversible ventilator-induced lung injury due to intense mechanical ventilation, thus limiting the utility of ECMO.8 Early referral should be considered if the patient does not improve after a few days on optimal ventilator settings. In centers where this technology is not available, referral to the nearest ECMO center should be considered. A list of certified ECMO centers is available at www.elso.org/Members/CenterDirectory.aspx.
Contraindications to ECMO
Advanced age, comorbid conditions such as malignancy, nonpulmonary organ dysfunction (including complications of critical illness), and immunodeficiency or pharmacologic immune suppression have been associated with poor outcomes in ECMO patients.28 Severe aortic incompetence and aortic dissection are contraindications, since ventricular end-diastolic pressure can be increased with resultant ventricular distention, compromised myocardial oxygenation, and worsening of left heart failure.
ECMO is increasingly being used in situations in which it was previously considered contraindicated. Pregnant and postpartum patients with cardiorespiratory failure were previously not considered for ECMO because of a possible increased risk of coagulopathy and complications. However, a recent review showed that the outcomes of ECMO in pregnancy and postpartum were similar to those in nonpregnant patients, and the risk of catastrophic bleeding was minor.29
Similarly, ECMO is also being used increasingly in posttrauma patients and patients with other bleeding risks.30
Morbid obesity was once considered a contraindication because of difficulty in cannulation, but with newer types of cannulas, even patients with a body mass index greater than 60 kg/m2 are receiving ECMO.31
HOW DO YOU DO IT?
Figures 3 and 4 depict clinical decision-making in starting and weaning from ECMO in respiratory failure and cardiogenic shock, respectively.
Management of patients on ECMO
Appropriate patient selection and initiation of ECMO are only the beginning of a tough journey. Successful management requires minimizing lung injury from mechanical ventilation, careful monitoring of anticoagulation, and instituting adequate physical therapy, including ambulation when possible (Table 3).
Initial ECMO settings and monitoring
The cannulas for venovenous ECMO are frequently inserted under fluoroscopic or transesophageal echocardiographic guidance, whereas venoarterial ECMO cannulation does not require imaging and can be performed at the bedside in the intensive care unit or operating room.
The initial ECMO settings are titrated according to the patient’s hemodynamic and respiratory needs. There are three main variables: blood flow, fraction of oxygen in the sweep gas, and sweep gas flow rate. These are adjusted to achieve desirable levels of oxygen and carbon dioxide in the blood.
Blood flow is determined by the revolutions per minute of the pump, preload, and afterload of the circuit. Common patient conditions that may reduce flow are systemic hypertension, hypovolemia, cardiac tamponade, and tension pneumothorax, depending on the modality. In addition, mechanical factors such as clots in the oxygenator or kinks in the circuit can increase resistance and reduce flow. Resistance to flow is directly proportional to cannula lengths and inversely proportional to cannula radius to the fourth power. The greater the flow, the greater the oxygen delivery.
Fraction of oxygen in the sweep gas. The oxygenator has a gas blender that mixes air and oxygen and allows for a range of oxygen concentrations. Increases in fraction of oxygen increase the partial pressure of oxygen in the blood.
Sweep gas flow rate. Venous blood in the extracorporeal circuit is exposed to fresh gas (or sweep gas) that oxygenates the blood and removes carbon dioxide by diffusion. Increasing the sweep gas flow rate results in greater carbon dioxide elimination from the blood.
Laboratory monitoring. During ECMO, the following values are monitored frequently:
- Arterial blood gases
- Blood gases in the ECMO circuit before and after going through the oxygenator— to monitor the efficacy of the oxygenator membrane
- Lactic acid—to monitor for tissue hypoxia
- Plasma free hemoglobin (a marker of hemolysis)—to monitor for hemolysis.
Mechanical ventilation on ECMO
Low tidal volume ventilation greatly reduces the risk of death in patients on ECMO by reducing ventilator-induced lung injury. Proponents of ECMO believe that ECMO provides “lung rest,” and thus it is imperative that lung-protective ventilation strategies be followed in patients on ECMO.8 In most cases, after ECMO is started, low tidal volume ventilation (6 mL/kg) is possible and should be used—or even very low tidal volume ventilation (3–6 mL/kg).32,33 Many cases have also been described in which patients have been safely extubated while on ECMO to prevent ventilator-induced lung injury.34,35
If hypoxemia persists
Despite full support with venovenous ECMO, some patients remain hypoxemic due to inadequate blood flow to match metabolic demands, eg, patients with morbid obesity or severe sepsis and fever. The physician should ensure there is no recirculation, maximize blood flow, optimize the hematocrit to increase oxygen delivery, and consider ways to decrease oxygen consumption, including sedation, paralysis, and hypothermia.
Recirculation can be calculated by measuring the oxygen saturation of the blood in the ECMO machine before and after it goes through the oxygenator, and also in the central venous blood. Recirculation has been reduced by using double-lumen cannulas but can also be reduced by manipulation of the reinfusion cannula or increasing the distance between drainage and reinfusion ports in other configurations of venovenous ECMO.
Expert opinion suggests that oxygen saturation of 86% or more and Pao2 of 55 mm Hg or more in patients on venovenous ECMO are sufficient to prevent hypoxia-related end-organ injury.36 Venoarterial ECMO should be considered in patients on venovenous ECMO with refractory hypoxemia with the above measures.
Harlequin syndrome is characterized by upper body hypoxia resulting in cerebral hypoxemia due to poorly oxygenated blood in the coronary and cerebral circulations, especially in patients on peripheral venoarterial ECMO. It can be detected by sampling the blood in the arm (where the oxygen isn’t going) instead of the leg (where the oxygen is going), and it can be corrected by adjusting the Fio2, using positive end-expiratory pressure, or both to increase oxygenation. If ventilator settings do not improve this syndrome, the arterial cannulation site can be switched from the femoral artery to the axillary or carotid artery.
Alternatively, a mixed-configuration venoarterial-venous ECMO can also be created, in which a portion of arterialized blood from the arterial outflow cannula is diverted via the right internal jugular artery to the right heart. This enriches the blood traveling through the pulmonary circulation and to the left ventricle to provide better oxygen delivery to the coronary and cerebral circulations.
Anticoagulation monitoring and transfusions
Anticoagulation is necessary to maintain a clot-free and functional circuit. Most clots develop in the oxygenator membrane, where they can prevent optimal gas exchange and, rarely, lead to embolization to the systemic circulation. However, reports have suggested that anticoagulation can be held for short periods on ECMO if necessary.
Unfractionated heparin is usually used for anticoagulation. Commonly used tests to monitor anticoagulation are the augmented partial thromboplastin time, activated clotting time, and anti-factor Xa levels. Lately, thromboelastography analysis is being used to comprehensively monitor various components of the coagulation cascade.37 Anticoagulation is usually tailored to whether there are clots in the circuit, coagulopathy, and bleeding while on ECMO.38
Traditionally, blood products were used liberally during ECMO to maintain a normal hematocrit and improve oxygen delivery, although recent data suggest that outcomes may be similar with conservative use of blood products.39,40
Fluid management on ECMO
ECMO patients are fluid-overloaded due to a profound inflammatory response, cardiac failure, or both. Studies have shown that conservative fluid management improve lung function and shortens the duration of mechanical ventilation and intensive care in patients with lung injury.41 Hence, the patient’s net fluid balance should be kept negative, provided renal and hemodynamic parameters remain stable.
There is a high incidence of acute kidney injury in ECMO patients, and fluid overload is one of the main indications for renal replacement therapy.42 Continuous renal replacement therapy can be provided either by an in-line hemofilter or by incorporating a standard continuous renal replacement therapy machine into the ECMO circuit. There are no studies comparing the efficacy of these techniques, but they allow for rapid improvement in fluid balance and electrolyte disturbances and are commonly used in ECMO patients.42,43
Physical rehabilitation and ambulation on ECMO
Physical rehabilitation in mechanically ventilated patients has been shown to reduce ventilator days and stay in the intensive care unit.44 With the use of internal jugular double-lumen cannulas for venovenous ECMO and improvement in durability of the ECMO circuit, several centers are implementing physical rehabilitation and ambulation for patients while on ECMO. Current data suggest that physical therapy is safe for patients receiving ECMO and may accelerate the weaning process and shorten length of stay in the hospital after ECMO.45,46 Aggressive rehabilitation is especially beneficial in patients awaiting lung transplant and may improve posttransplant recovery and outcomes.47
Weaning from ECMO
There are no standard guidelines for weaning from venovenous or venoarterial ECMO. Once the underlying condition for which ECMO was initiated has improved, weaning can begin by reducing the blood flow rate, the flow rate of the sweep gas, or both.
Weaning from venovenous ECMO should be started when there is improvement in lung compliance, tidal volumes, and oxygenation. Once the circuit flow rate is reduced to less than 3 L/minute, ventilator settings are adjusted to standard lung-protective settings. ECMO support is gradually decreased by reducing the flow rate of sweep gas to less than 2 L/minute. If tidal volumes, respiratory rate, and gas exchange remain adequate after approximately 2 to 4 hours on a low rate of sweep gas, the patient can be weaned off the venovenous ECMO circuit.
Weaning from venoarterial ECMO should be considered when there is myocardial recovery with improved pulse pressure and contractility on echocardiography. This is done by reducing flow rates in increments of 0.5 to 2 L/minute over 24 to 36 hours and monitoring mean arterial pressures, central venous pressure, and myocardial contractility. When acceptable, patients are mostly weaned in a surgical setting. Prolonged periods on a low rate of blood flow are avoided to prevent thrombus formation in the circuit.
COMPLICATIONS OF ECMO
ECMO use can be associated with a myriad of patient and mechanical complications.
Hemorrhage is the most common complication encountered in ECMO, occurring in approximately 43% of patients.29 It occurs most frequently from cannulation and surgical sites. Although rare, potentially life-threatening pulmonary hemorrhage (including bleeding at the tracheostomy site), intracranial hemorrhage, and gastrointestinal hemorrhage have also been reported.30
Infections, including new infection and worsening sepsis in patients with acute respiratory distress syndrome secondary to infection, are common in patients on ECMO.48
Renal failure secondary to acute tubular necrosis requiring hemodialysis has been reported to occur in 13% of patients on ECMO.30
Other complications of concern, especially in patients on venoarterial ECMO, are lower limb ischemia and thromboembolism associated with site of cannulation and direction of blood flow.49 Mechanical complications include inappropriate placement of the cannula leading to insufficient oxygenation, injury to vessel walls, and rarely myocardial wall rupture; thrombus formation within the circuit causing failure of the oxygenator and sometimes, pulmonary or systemic embolism; and air embolism from the circuit.36
NOT SUITED FOR ALL
Despite limited data to support its use, there has been a recent increase in utilization of ECMO to support critically ill adult patients with cardiopulmonary failure. ECMO support is not suited for all patients. Careful selection of patients should be done to optimize resource utilization and provide the best opportunity for recovery or transplant.
- MacLaren G, Combes A, Bartlett RH. Contemporary extracorporeal membrane oxygenation for adult respiratory failure: life support in the new era. Intensive Care Med 2012; 38:210–220.
- Maslach-Hubbard A, Bratton SL. Extracorporeal membrane oxygenation for pediatric respiratory failure: history, development and current status. World J Crit Care Med 2013; 2:29–39.
- Zapol WM, Snider MT, Hill JD, et al. Extracorporeal membrane oxygenation in severe acute respiratory failure. A randomized prospective study. JAMA 1979; 242:2193–2196.
- Morris AH, Wallace CJ, Menlove RL, et al. Randomized clinical trial of pressure-controlled inverse ratio ventilation and extracorporeal CO2 removal for adult respiratory distress syndrome. Am J Respir Crit Care Med 1994; 149:295–305.
- Extracorporeal Life Support Organization. ECLS registry report. International Summary. January 2016. https://www.elso.org/Registry.aspx. Accessed March 17, 2016.
- Sauer CM, Yuh DD, Bonde P. Extracorporeal membrane oxygenation use has increased by 433% in adults in the United States from 2006 to 2011. ASAIO J 2015; 61:31–36.
- Sharma N, Wille K, Bellot S, Brodie D, Diaz-Guzman E. Role of extracorporeal membrane oxygenation in management of refractory ARDS in the intensive care unit: a national survey on perspectives of the adult critical care physicians and trainees. Chest 2014. http://journal.publications.chestnet.org/article.aspx?articleid=1913336. Accessed March 17, 2016.
- Peek GJ, Mugford M, Tiruvoipati R, et al; CESAR trial collaboration. Efficacy and economic assessment of conventional ventilatory support versus extracorporeal membrane oxygenation for severe adult respiratory failure (CESAR): a multicentre randomised controlled trial. Lancet 2009; 374:1351–1363.
- Australia and New Zealand Extracorporeal Membrane Oxygenation (ANZ ECMO) Influenza Investigators; Davies A, Jones D, Bailey M, et al. Extracorporeal membrane oxygenation for 2009 influenza A(H1N1) acute respiratory distress syndrome. JAMA 2009; 302:1888–1895.
- Noah MA, Peek GJ, Finney SJ, et al. Referral to an extracorporeal membrane oxygenation center and mortality among patients with severe 2009 influenza A(H1N1). JAMA 2011; 306:1659–1668.
- Patroniti N, Zangrillo A, Pappalardo F, et al. The Italian ECMO network experience during the 2009 influenza A(H1N1) pandemic: preparation for severe respiratory emergency outbreaks. Intensive Care Med 2011; 37:1447–1457.
- Pham T, Combes A, Roze H, et al; REVA Research Network. Extracorporeal membrane oxygenation for pandemic influenza A(H1N1)-induced acute respiratory distress syndrome: a cohort study and propensity-matched analysis. Am J Respir Crit Care Med 2013; 187:276–285.
- Schmidt M, Zogheib E, Roze H, et al. The PRESERVE mortality risk score and analysis of long-term outcomes after extracorporeal membrane oxygenation for severe acute respiratory distress syndrome. Intensive Care Med 2013; 39; 532:1704–1713.
- Schmidt M, Bailey M, Sheldrake J, et al. Predicting survival after extracorporeal membrane oxygenation for severe acute respiratory failure. The Respiratory Extracorporeal Membrane Oxygenation Survival Prediction (RESP) score. Am J Respir Crit Care Med 2014; 189:1374–1382.
- Chan KK, Lee KL, Lam PK, Law KI, Joynt GM, Yan WW. Hong Kong's experience on the use of extracorporeal membrane oxygenation for the treatment of influenza A (H1N1). Hong Kong Med J 2010; 16:447–454.
- Freed DH, Henzler D, White CW, et al; Canadian Critical Care Trials Group. Extracorporeal lung support for patients who had severe respiratory failure secondary to influenza A (H1N1) 2009 infection in Canada. Can J Anaesth 2010; 57:240–247.
- Nair P, Davies AR, Beca J, et al. Extracorporeal membrane oxygenation for severe ARDS in pregnant and postpartum women during the 2009 H1N1 pandemic. Intensive Care Med 2011; 37:648–654.
- Turner DA, Rehder KJ, Peterson-Carmichael SL, et al. Extracorporeal membrane oxygenation for severe refractory respiratory failure secondary to 2009 H1N1 influenza A. Respir Care 2011; 56:941–946.
- Kumar A, Zarychanski R, Pinto R, et al; Canadian Critical Care Trials Group H1N1 Collaborative. Critically ill patients with 2009 influenza A(H1N1) infection in Canada. JAMA 2009; 302:1872–1879.
- Abrams D, Combes A, Brodie D. Extracorporeal membrane oxygenation in cardiopulmonary disease in adults. J Am Coll Cardiol 2014; 63:2769–2778.
- Marasco SF, Lukas G, McDonald M, McMillan J, Ihle B. Review of ECMO (extra corporeal membrane oxygenation) support in critically ill adult patients. Heart Lung Circ 2008; 17(suppl 4):S41–S47.
- Hysi I, Fabre O, Renaut C, Guesnier L. Extracorporeal membrane oxygenation with direct axillary artery perfusion. J Card Surg 2014; 29:268–269.
- Gattinoni L, Kolobow T, Agostoni A, et al. Clinical application of low frequency positive pressure ventilation with extracorporeal CO2 removal (LFPPV-ECCO2R) in treatment of adult respiratory distress syndrome (ARDS). Int J Artif Organs 1979; 2:282–283.
- Liebold A, Philipp A, Kaiser M, Merk J, Schmid FX, Birnbaum DE. Pumpless extracorporeal lung assist using an arterio-venous shunt. Applications and limitations. Minerva Anestesiol 2002; 68:387–391.
- Paden ML, Conrad SA, Rycus PT, Thiagarajan RR; ELSO Registry. Extracorporeal Life Support Organization Registry Report 2012. ASAIO J 2013; 59:202–210.
- Shekar K, Davies AR, Mullany DV, Tiruvoipati R, Fraser JF. To ventilate, oscillate, or cannulate? J Crit Care 2013; 28:655–662.
- Mikkelsen ME, Woo YJ, Sager JS, Fuchs BD, Christie JD. Outcomes using extracorporeal life support for adult respiratory failure due to status asthmaticus. ASAIO J 2009; 55:47–52.
- Turner DA, Cheifetz IM. Extracorporeal membrane oxygenation for adult respiratory failure. Respir Care 2013; 58:1038–1052.
- Sharma NS, Wille KM, Bellot SC, Diaz-Guzman E. Modern use of extracorporeal life support in pregnancy and postpartum. ASAIO J 2015; 61:110–114.
- Ried M, Bein T, Philipp A, et al. Extracorporeal lung support in trauma patients with severe chest injury and acute lung failure: a 10-year institutional experience. Crit Care 2013; 17:R110.
- Al-Soufi S, Buscher H, Nguyen ND, Rycus P, Nair P. Lack of association between body weight and mortality in patients on veno-venous extracorporeal membrane oxygenation. Intensive Care Med 2013; 39:1995–2002.
- Marhong JD, Telesnicki T, Munshi L, Del Sorbo L, Detsky M, Fan E. Mechanical ventilation during extracorporeal membrane oxygenation. An international survey. Ann Am Thorac Soc 2014; 11:956–961.
- Schmidt M, Pellegrino V, Combes A, Scheinkestel C, Cooper DJ, Hodgson C. Mechanical ventilation during extracorporeal membrane oxygenation. Crit Care 2014; 18:203.
- Bein T, Wittmann S, Philipp A, Nerlich M, Kuehnel T, Schlitt HJ. Successful extubation of an "unweanable" patient with severe ankylosing spondylitis (Bechterew's disease) using a pumpless extracorporeal lung assist. Intensive Care Med 2008; 34:2313–2314.
- Anton-Martin P, Thompson MT, Sheeran PD, Fischer AC, Taylor D, Thomas JA. Extubation during pediatric extracorporeal membrane oxygenation: a single-center experience. Pediatr Crit Care Med 2014; 15:861–869.
- Sidebotham D, McGeorge A, McGuinness S, Edwards M, Willcox T, Beca J. Extracorporeal membrane oxygenation for treating severe cardiac and respiratory failure in adults: part 2-technical considerations. J Cardiothorac Vasc Anesth 2010; 24:164–172.
- Stammers AH, Willett L, Fristoe L, et al. Coagulation monitoring during extracorporeal membrane oxygenation: the role of thrombelastography. J Extra Corpor Technol 1995; 27:137–145.
- Bembea MM, Schwartz JM, Shah N, et al. Anticoagulation monitoring during pediatric extracorporeal membrane oxygenation. ASAIO J 2013; 59:63–68.
- Agerstrand CL, Burkart KM, Abrams DC, Bacchetta MD, Brodie D. Blood conservation in extracorporeal membrane oxygenation for acute respiratory distress syndrome. Ann Thorac Surg 2015; 99:590–595.
- Voelker MT, Busch T, Bercker S, Fichtner F, Kaisers UX, Laudi S. Restrictive transfusion practice during extracorporeal membrane oxygenation therapy for severe acute respiratory distress syndrome. Artif Organs 2015; 39:374–378.
- National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network; Wiedemann HP, Wheeler AP, Bernard GR, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med 2006; 354:2564–2575.
- Fleming GM, Askenazi DJ, Bridges BC, et al. A multicenter international survey of renal supportive therapy during ECMO: the Kidney Intervention During Extracorporeal Membrane Oxygenation (KIDMO) group. ASAIO J 2012; 58:407–414.
- Askenazi DJ, Selewski DT, Paden ML, et al. Renal replacement therapy in critically ill patients receiving extracorporeal membrane oxygenation. Clin J Am Soc Nephrol 2012; 7:1328–1336.
- Schweickert WD, Pohlman MC, Pohlman AS, et al. Early physical and occupational therapy in mechanically ventilated, critically ill patients: a randomised controlled trial. Lancet 2009; 373:1874–1882.
- Abrams D, Javidfar J, Farrand E, et al. Early mobilization of patients receiving extracorporeal membrane oxygenation: a retrospective cohort study. Crit Care 2014; 18:R38.
- Thiagarajan RR, Teele SA, Teele KP, Beke DM. Physical therapy and rehabilitation issues for patients supported with extracorporeal membrane oxygenation. J Pediatr Rehabil Med 2012; 5:47–52.
- Hoopes CW, Kukreja J, Golden J, Davenport DL, Diaz-Guzman E, Zwischenberger JB. Extracorporeal membrane oxygenation as a bridge to pulmonary transplantation. J Thorac Cardiovasc Surg 2013; 145:862–868.
- Burket JS, Bartlett RH, Vander Hyde K, Chenoweth CE. Nosocomial infections in adult patients undergoing extracorporeal membrane oxygenation. Clin Infect Dis 1999; 28:828–833.
- Lafç G, Budak AB, Yener AÜ, Cicek OF. Use of extracorporeal membrane oxygenation in adults. Heart Lung Circ 2014; 23:10–23.
- MacLaren G, Combes A, Bartlett RH. Contemporary extracorporeal membrane oxygenation for adult respiratory failure: life support in the new era. Intensive Care Med 2012; 38:210–220.
- Maslach-Hubbard A, Bratton SL. Extracorporeal membrane oxygenation for pediatric respiratory failure: history, development and current status. World J Crit Care Med 2013; 2:29–39.
- Zapol WM, Snider MT, Hill JD, et al. Extracorporeal membrane oxygenation in severe acute respiratory failure. A randomized prospective study. JAMA 1979; 242:2193–2196.
- Morris AH, Wallace CJ, Menlove RL, et al. Randomized clinical trial of pressure-controlled inverse ratio ventilation and extracorporeal CO2 removal for adult respiratory distress syndrome. Am J Respir Crit Care Med 1994; 149:295–305.
- Extracorporeal Life Support Organization. ECLS registry report. International Summary. January 2016. https://www.elso.org/Registry.aspx. Accessed March 17, 2016.
- Sauer CM, Yuh DD, Bonde P. Extracorporeal membrane oxygenation use has increased by 433% in adults in the United States from 2006 to 2011. ASAIO J 2015; 61:31–36.
- Sharma N, Wille K, Bellot S, Brodie D, Diaz-Guzman E. Role of extracorporeal membrane oxygenation in management of refractory ARDS in the intensive care unit: a national survey on perspectives of the adult critical care physicians and trainees. Chest 2014. http://journal.publications.chestnet.org/article.aspx?articleid=1913336. Accessed March 17, 2016.
- Peek GJ, Mugford M, Tiruvoipati R, et al; CESAR trial collaboration. Efficacy and economic assessment of conventional ventilatory support versus extracorporeal membrane oxygenation for severe adult respiratory failure (CESAR): a multicentre randomised controlled trial. Lancet 2009; 374:1351–1363.
- Australia and New Zealand Extracorporeal Membrane Oxygenation (ANZ ECMO) Influenza Investigators; Davies A, Jones D, Bailey M, et al. Extracorporeal membrane oxygenation for 2009 influenza A(H1N1) acute respiratory distress syndrome. JAMA 2009; 302:1888–1895.
- Noah MA, Peek GJ, Finney SJ, et al. Referral to an extracorporeal membrane oxygenation center and mortality among patients with severe 2009 influenza A(H1N1). JAMA 2011; 306:1659–1668.
- Patroniti N, Zangrillo A, Pappalardo F, et al. The Italian ECMO network experience during the 2009 influenza A(H1N1) pandemic: preparation for severe respiratory emergency outbreaks. Intensive Care Med 2011; 37:1447–1457.
- Pham T, Combes A, Roze H, et al; REVA Research Network. Extracorporeal membrane oxygenation for pandemic influenza A(H1N1)-induced acute respiratory distress syndrome: a cohort study and propensity-matched analysis. Am J Respir Crit Care Med 2013; 187:276–285.
- Schmidt M, Zogheib E, Roze H, et al. The PRESERVE mortality risk score and analysis of long-term outcomes after extracorporeal membrane oxygenation for severe acute respiratory distress syndrome. Intensive Care Med 2013; 39; 532:1704–1713.
- Schmidt M, Bailey M, Sheldrake J, et al. Predicting survival after extracorporeal membrane oxygenation for severe acute respiratory failure. The Respiratory Extracorporeal Membrane Oxygenation Survival Prediction (RESP) score. Am J Respir Crit Care Med 2014; 189:1374–1382.
- Chan KK, Lee KL, Lam PK, Law KI, Joynt GM, Yan WW. Hong Kong's experience on the use of extracorporeal membrane oxygenation for the treatment of influenza A (H1N1). Hong Kong Med J 2010; 16:447–454.
- Freed DH, Henzler D, White CW, et al; Canadian Critical Care Trials Group. Extracorporeal lung support for patients who had severe respiratory failure secondary to influenza A (H1N1) 2009 infection in Canada. Can J Anaesth 2010; 57:240–247.
- Nair P, Davies AR, Beca J, et al. Extracorporeal membrane oxygenation for severe ARDS in pregnant and postpartum women during the 2009 H1N1 pandemic. Intensive Care Med 2011; 37:648–654.
- Turner DA, Rehder KJ, Peterson-Carmichael SL, et al. Extracorporeal membrane oxygenation for severe refractory respiratory failure secondary to 2009 H1N1 influenza A. Respir Care 2011; 56:941–946.
- Kumar A, Zarychanski R, Pinto R, et al; Canadian Critical Care Trials Group H1N1 Collaborative. Critically ill patients with 2009 influenza A(H1N1) infection in Canada. JAMA 2009; 302:1872–1879.
- Abrams D, Combes A, Brodie D. Extracorporeal membrane oxygenation in cardiopulmonary disease in adults. J Am Coll Cardiol 2014; 63:2769–2778.
- Marasco SF, Lukas G, McDonald M, McMillan J, Ihle B. Review of ECMO (extra corporeal membrane oxygenation) support in critically ill adult patients. Heart Lung Circ 2008; 17(suppl 4):S41–S47.
- Hysi I, Fabre O, Renaut C, Guesnier L. Extracorporeal membrane oxygenation with direct axillary artery perfusion. J Card Surg 2014; 29:268–269.
- Gattinoni L, Kolobow T, Agostoni A, et al. Clinical application of low frequency positive pressure ventilation with extracorporeal CO2 removal (LFPPV-ECCO2R) in treatment of adult respiratory distress syndrome (ARDS). Int J Artif Organs 1979; 2:282–283.
- Liebold A, Philipp A, Kaiser M, Merk J, Schmid FX, Birnbaum DE. Pumpless extracorporeal lung assist using an arterio-venous shunt. Applications and limitations. Minerva Anestesiol 2002; 68:387–391.
- Paden ML, Conrad SA, Rycus PT, Thiagarajan RR; ELSO Registry. Extracorporeal Life Support Organization Registry Report 2012. ASAIO J 2013; 59:202–210.
- Shekar K, Davies AR, Mullany DV, Tiruvoipati R, Fraser JF. To ventilate, oscillate, or cannulate? J Crit Care 2013; 28:655–662.
- Mikkelsen ME, Woo YJ, Sager JS, Fuchs BD, Christie JD. Outcomes using extracorporeal life support for adult respiratory failure due to status asthmaticus. ASAIO J 2009; 55:47–52.
- Turner DA, Cheifetz IM. Extracorporeal membrane oxygenation for adult respiratory failure. Respir Care 2013; 58:1038–1052.
- Sharma NS, Wille KM, Bellot SC, Diaz-Guzman E. Modern use of extracorporeal life support in pregnancy and postpartum. ASAIO J 2015; 61:110–114.
- Ried M, Bein T, Philipp A, et al. Extracorporeal lung support in trauma patients with severe chest injury and acute lung failure: a 10-year institutional experience. Crit Care 2013; 17:R110.
- Al-Soufi S, Buscher H, Nguyen ND, Rycus P, Nair P. Lack of association between body weight and mortality in patients on veno-venous extracorporeal membrane oxygenation. Intensive Care Med 2013; 39:1995–2002.
- Marhong JD, Telesnicki T, Munshi L, Del Sorbo L, Detsky M, Fan E. Mechanical ventilation during extracorporeal membrane oxygenation. An international survey. Ann Am Thorac Soc 2014; 11:956–961.
- Schmidt M, Pellegrino V, Combes A, Scheinkestel C, Cooper DJ, Hodgson C. Mechanical ventilation during extracorporeal membrane oxygenation. Crit Care 2014; 18:203.
- Bein T, Wittmann S, Philipp A, Nerlich M, Kuehnel T, Schlitt HJ. Successful extubation of an "unweanable" patient with severe ankylosing spondylitis (Bechterew's disease) using a pumpless extracorporeal lung assist. Intensive Care Med 2008; 34:2313–2314.
- Anton-Martin P, Thompson MT, Sheeran PD, Fischer AC, Taylor D, Thomas JA. Extubation during pediatric extracorporeal membrane oxygenation: a single-center experience. Pediatr Crit Care Med 2014; 15:861–869.
- Sidebotham D, McGeorge A, McGuinness S, Edwards M, Willcox T, Beca J. Extracorporeal membrane oxygenation for treating severe cardiac and respiratory failure in adults: part 2-technical considerations. J Cardiothorac Vasc Anesth 2010; 24:164–172.
- Stammers AH, Willett L, Fristoe L, et al. Coagulation monitoring during extracorporeal membrane oxygenation: the role of thrombelastography. J Extra Corpor Technol 1995; 27:137–145.
- Bembea MM, Schwartz JM, Shah N, et al. Anticoagulation monitoring during pediatric extracorporeal membrane oxygenation. ASAIO J 2013; 59:63–68.
- Agerstrand CL, Burkart KM, Abrams DC, Bacchetta MD, Brodie D. Blood conservation in extracorporeal membrane oxygenation for acute respiratory distress syndrome. Ann Thorac Surg 2015; 99:590–595.
- Voelker MT, Busch T, Bercker S, Fichtner F, Kaisers UX, Laudi S. Restrictive transfusion practice during extracorporeal membrane oxygenation therapy for severe acute respiratory distress syndrome. Artif Organs 2015; 39:374–378.
- National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network; Wiedemann HP, Wheeler AP, Bernard GR, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med 2006; 354:2564–2575.
- Fleming GM, Askenazi DJ, Bridges BC, et al. A multicenter international survey of renal supportive therapy during ECMO: the Kidney Intervention During Extracorporeal Membrane Oxygenation (KIDMO) group. ASAIO J 2012; 58:407–414.
- Askenazi DJ, Selewski DT, Paden ML, et al. Renal replacement therapy in critically ill patients receiving extracorporeal membrane oxygenation. Clin J Am Soc Nephrol 2012; 7:1328–1336.
- Schweickert WD, Pohlman MC, Pohlman AS, et al. Early physical and occupational therapy in mechanically ventilated, critically ill patients: a randomised controlled trial. Lancet 2009; 373:1874–1882.
- Abrams D, Javidfar J, Farrand E, et al. Early mobilization of patients receiving extracorporeal membrane oxygenation: a retrospective cohort study. Crit Care 2014; 18:R38.
- Thiagarajan RR, Teele SA, Teele KP, Beke DM. Physical therapy and rehabilitation issues for patients supported with extracorporeal membrane oxygenation. J Pediatr Rehabil Med 2012; 5:47–52.
- Hoopes CW, Kukreja J, Golden J, Davenport DL, Diaz-Guzman E, Zwischenberger JB. Extracorporeal membrane oxygenation as a bridge to pulmonary transplantation. J Thorac Cardiovasc Surg 2013; 145:862–868.
- Burket JS, Bartlett RH, Vander Hyde K, Chenoweth CE. Nosocomial infections in adult patients undergoing extracorporeal membrane oxygenation. Clin Infect Dis 1999; 28:828–833.
- Lafç G, Budak AB, Yener AÜ, Cicek OF. Use of extracorporeal membrane oxygenation in adults. Heart Lung Circ 2014; 23:10–23.
KEY POINTS
- Two basic configurations of ECMO are used in adults: venoarterial, which can provide cardiac or cardiopulmonary support; and venovenous, which provides respiratory support only.
- ECMO is used in adults who are at very high risk of death without it.
- Because ECMO patients must receive anticoagulation, bleeding is a common complication. Others are infection, renal failure, and thrombosis.
- ECMO may provide “lung rest,” allowing lower tidal volumes and pressures and lower fractions of inspired oxygen to be used in mechanical ventilation, strategies associated with lower mortality rates.
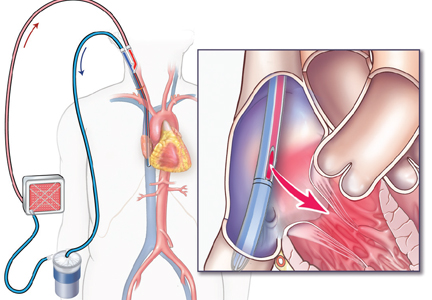
Reproductive health and the environment: Counseling patients about risks
A 28-year-old woman presents for routine follow-up of asthma. She has a 1-year-old son and is considering a second pregnancy. She says she read on the Internet that the US Food and Drug Administration recently banned baby bottles and “sippy” cups that contain bisphenol A (BPA), as recommended by the American Medical Association. She wonders if there are other sources of BPA and if they pose a health risk to her, her son, and possible future children.
Environmental toxins have been linked to pregnancy complications and poor birth outcomes. The past several decades have seen a significant rise in reproductive disorders such as early onset of puberty, low sperm count, and birth defects such as cryptorchidism and hypospadias.1–4 These changes may be partly explained by other trends over time, such as older maternal age, rising incidence of obesity and maternal diabetes, and demographic changes that exacerbate health disparities. However, these well-recognized factors explain only some of the wide range of reproductive health problems that have been identified.
This review focuses on emerging evidence of the adverse reproductive effects of human-produced endocrine-disrupting chemicals (EDCs).
ENDOCRINE-DISRUPTING CHEMICALS ARE UBIQUITOUS
EDCs are exogenous substances that alter normal functioning of the endocrine system and consequently have the potential to cause adverse health effects in an intact organism and its progeny.2,3,5 Substances classified as EDCs are divided into two groups:
- Human-produced compounds, such as pesticides, industrial solvents and lubricants, plastics (eg, BPA), and plasticizers (eg, phthalates)
- Phytoestrogens, which are naturally occurring plant compounds that bind to estrogen receptors. The major dietary source of phytoestrogens is soy.6
Many human-produced EDCs are lipid-soluble. Some bioconcentrate in fat. The dietary route of exposure is the most common, primarily from fat-containing foods such as meats and seafood. Common predatory fish such as tuna and swordfish, as well as other large fish, are more likely to have high levels of EDCs accumulated from contaminated water.7 Water, air, soil, and dust in communities, schools, and workplaces may also carry EDCs.8
Some human-produced EDCs have a very long half-life and can remain in the environment for years or even decades. Because of their long half-life, some EDCs can “travel” and become part of the food chain, even in “pristine” areas where manufactured substances are not normally found.2
Women of childbearing age and pregnant women come in contact with EDCs on a daily basis. According to the Fourth National Report on Human Exposure to Environmental Chemicals, conducted by the US Centers for Disease Control and Prevention (CDC) in 2009, nearly all pregnant women in the United States have detectable serum levels of EDCs, including BPA, perchlorates, phthalates, polybrominated diphenyl ethers, and pesticides.9 Furthermore, BPA was found in more than 90% of the urine samples tested from random subsamples of 2,500 participants in the 2009 National Health and Nutrition Examination Survey report.10
EDCs AFFECT MULTIPLE PATHWAYS
Laboratory and animal research suggests that both manufactured and naturally occurring EDCs primarily affect sex steroid hormone pathways. However, some EDCs can affect adrenal, thyroid, and other endocrine pathways.
EDCs can potentially alter normal endocrine functioning by several mechanisms. They can bind to nuclear hormone receptors such as estrogen receptors, androgen receptors, progesterone receptors, thyroid hormone receptors, and peroxisome proliferator-activated receptor gamma. They can also bind to nonnuclear steroid hormone receptors (membrane estrogen receptors), nonsteroid receptors such as those for dopamine, serotonin, and norepinephrine, and orphan receptors such as aryl hydrocarbon receptor. In addition, some affect enzymatic pathways involved in steroid biosynthesis and metabolism and disrupt centralized endocrine pathways through positive and negative feedback.2 There is also evidence that EDCs have epigenetic and transgenerational effects, as they seem to be able to influence DNA methylation and histone acetylation.11
EVIDENCE OF HARM, LIMITATIONS OF EVIDENCE
In 2009, the Endocrine Society found convincing evidence that EDCs affect the male and female reproductive systems, breast health, oncogenesis, neuroendocrine function, thyroid function, metabolism, and cardiovascular health.2 The first evidence of harm from EDCs came from long-term follow-up of randomized trials of diethylstilbestrol, a synthetic estrogen used as a pharmacologic agent between 1938 and 1971 to prevent miscarriage.12
In 2013, the American College of Obstetricians and Gynecologists and the American Society for Reproductive Medicine issued a joint opinion report on exposure to toxic environmental agents. The report acknowledged that environmental toxins are ubiquitous and that they can negatively affect health from preconception through adult life. “[We] join leading scientists and other clinical practitioners in calling for timely action to identify and reduce exposure to toxic environmental agents while addressing the consequences of such exposure.”13
Because the effects of endocrine disruption can occur during fetal and embryonic development, EDC exposure before and during pregnancy is of special concern.14 Long-term effects of exposure to hazardous chemicals such as cadmium, lead, and mercury in breast milk can lead to negative consequences in adulthood.15,16
Animal studies have pointed to the negative health effects of human-produced EDCs. For example, animals exposed to BPA have aberrant breast and genitourinary development and exhibit abnormal endometrial stimulation and early puberty.2
Despite these suggestive findings, the health effects of environmental toxins on humans are difficult to determine definitively. Evidence comes mainly from epidemiologic studies showing distribution of particular medical conditions based on different levels of exposure. More research is needed on human exposure to environmental toxicants and their effects.2,17,18 The National Children’s Study, a cohort collaborative study being conducted by the Environmental Protection Agency, CDC, and National Institutes of Health, is examining the effects of environmental exposures on more than 100,000 children across the United States, following them from before birth until age 21.19
THE PRECAUTIONARY PRINCIPLE
The precautionary principle is a fairly new concept in environmental science, although clinicians have been adhering to it for a long time. As proposed in a meeting of scientists, lawyers, policy makers, and environmentalists in 1998, “When an activity raises threats of harm to human health or the environment, precautionary measures should be taken even if some cause and effect relationships are not fully established scientifically.”20
For example, studies show that breast milk in the United States commonly contains many chemicals that do not meet US Food and Drug Administration standards for baby food.15,16 Although medical ethics prohibits randomized clinical trials in humans to examine the potential harms of these chemicals, proponents of the precautionary principle advocate that it is reasonable to be concerned that these chemicals may be harmful.21–23
Applying the precautionary principle: Recommendations for patients
Without categorizing all chemicals as toxicants, nor all toxicants as worrisome for reproductive health, physicians can provide access to a growing body of information about reasonable strategies to reduce risk.

Educating patients about potential risks of exposure needs to be tempered with an appreciation of the economic and social barriers to avoiding or reducing risks. Often, people do not make conscious decisions about exposure. Rather, they are unknowingly exposed or have no access to safe, equivalent alternatives. Substituting suspected or known harmful products with potentially safer ones can be financially prohibitive. Where a patient lives and works may result in unavoidable exposure to environmental toxins.
Nevertheless, there are simple, economical alternatives that reduce or eliminate exposure to EDCs (Table 1).24–26 These include avoiding plastic and metal containers that are not marked “BPA-free,” eating locally grown fruits and vegetables, controlling pests through frequent cleaning and trapping, and avoiding pesticides.
Additionally, the flame retardant tris (1,3-dichloro-2-propyl) phosphate may be an endocrine disrupter. It is found in numerous products for infants and young children, as well as in dust, automobiles, and furniture. Flame retardants are no longer used in infant clothing but may still be found in foam products such as upholstered furniture, automobiles, and children’s nap pads.27,28

National organizations that provide resources for counseling women about human-produced EDCs as well as patient education materials are listed in Table 2.
CLINICAL CASE RESOLUTION
You counsel the patient that, although more research in humans is needed, because of safety concerns, avoiding exposing herself and her baby to BPA when possible is a reasonable approach. You review recommendations including avoiding BPA-containing plastics for food preparation (recycle code #7) and BPA-containing toys that might be mouthed by the baby, as well as minimizing the consumption of canned foods when possible. You further advise her to avoid products with flame retardants.
- Sutton P, Giudice LC, Woodruff TJ. Reproductive environmental health. Curr Opin Obstet Gynecol 2010; 22:517–524.
- Diamanti-Kandarakis E, Bouguignon JP, Giudice LC, et al. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev 2009; 30:293–342.
- Friedrich MJ. Endocrine-disrupting chemicals. JAMA 2013; 309:1578.
- Schettler T. Environmental exposures, infertility, and related reproductive disorders: an update. October 2011. The Collaborative on Health and the Environment. www.healthandenvironment.org/infertility/peer_reviewed. Accessed March 9, 2016.
- Woodruff TJ, Carlson A, Schwartz JM, Giudice LC. Proceedings of the Summit on the Environmental Challenges to Reproductive Health and Fertility: executive summary. Fertil Steril 2008; 89:281–300.
- Cao Y, Calafat AM, Doerge DR, et al. Isoflavones in urine, saliva, and blood of infants: data from a pilot study on the estrogenic activity of soy formula. J Expo Sci Environ Epidemiol 2009; 19:223–234.
- Endocrine-disrupting chemicals. The Collaborative on Health and the Environment. Available at www.healthandenvironment.org/initiatives/learning/r/prevention. Accessed March 9, 2016.
- Environmental impacts on reproductive health: clinical proceedings. Association of Reproductive Health Professionals. www.arhp.org/uploadDocs/CPRHE.pdf. Accessed March 9, 2016.
- Fourth national report on human exposure to environmental chemicals. Centers for Disease Control and Prevention (CDC). www.cdc.gov/exposurereport/pdf/FourthReport.pdf. Accessed March 9, 2016.
- Woodruff TJ, Zota AR, Schwartz JM. Environmental chemicals in pregnant women in the United States: NHANES 2003-2004. Environ Health Perspect 2011; 119:878–885.
- Anway MD, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors. Endocrinology 2006; 147(suppl 6):S43–S49.
- About DES. Centers for Disease Control and Prevention (CDC). www.cdc.gov/des/consumers/about/index.html. Accessed March 9, 2016.
- Exposure to toxic environmental agents. American Congress of Obstetricians and Gynecologists (ACOG). www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Health-Care-for-Underserved-Women/Exposure-to-Toxic-Environmental-Agents. Accessed March 9, 2016.
- Bergman A, Heindel JJ, Jobling S, Kidd KA, Zoeller RT. United Nations Environment Programme (UNEP). State of the science of endocrine disrupting chemicals: 2012. www.who.int/ceh/publications/endocrine/en/. Accessed March 9, 2016.
- Pohl HR, Hibbs BF. Breast-feeding exposure of infants to environmental contaminants—a public health risk assessment viewpoint: chlorinated dibenzodioxins and chlorinated dibenzofurans. Toxicol Ind Health 1996; 12:593–611.
- Abadin HG, Hibbs BF, Pohl HR. Breast-feeding exposure of infants to cadmium, lead and mercury: a public health viewpoint. Toxicol Ind Health 1997; 13:495–517.
- Bretveld RW, Thomas CM, Scheepers PT, Zielhuis GA, Roeleveld N. Pesticide exposure: the hormonal function of the female reproductive system disrupted? Reprod Biol Endocrinol 2006; 4:30.
- Mendola P, Messer LC, Rappazzo K. Science linking environmental contaminant exposures with fertility and reproductive health impacts in the adult female. Fertil Steril 2008; 89(suppl 2):e81–e94.
- National Institute of Health (NIH). National Children’s Study (NCS). www.nationalchildrensstudy.gov/Pages/default.aspx. Accessed March 9, 2016.
- Raffensperger C, Tickner J, editors. Protecting public health and the environment: implementing the precautionary principle. Washington, DC: Island Press; 1999:8.
- Woodruff TJ, Sutton P. The navigation guide systematic review methodology: a rigorous and transparent method for translating environmental health science into better health outcomes. Environ Health Perspect 2014; 122:1007–1014.
- Sutton P, Woodruff TJ. Risk communication and decision tools for children’s health protection. Birth Defects Res C Embryo Today 2013; 99:45–49.
- Woodruff TJ. Bridging epidemiology and model organisms to increase understanding of endocrine disrupting chemicals and human health effects. J Steroid Biochem Mol Biol 2011; 127:108–117.
- National Institute of Environmental Health Sciences. Bisphenol A (BPA). www.niehs.nih.gov/health/topics/agents/sya-bpa/index.cfm. Accessed March 9, 2016.
- Environmental Working Group. Shoppers guide to presticides in produce. www.ewg.org/foodnews/. Accessed March 9, 2016.
- Schettler T. Human exposure to phthalates via consumer products. Int J Androl 2006; 29:134–139.
- Betts KS. Exposure to TDCPP appears widespread. Environ Health Perspect 2013; 121:a150.
- Environmental Working Group. Healthy home tips. www.ewg.org/research/healthy-home-tips/tip-4-avoid-fire-retardants. Accessed March 9, 2016.
A 28-year-old woman presents for routine follow-up of asthma. She has a 1-year-old son and is considering a second pregnancy. She says she read on the Internet that the US Food and Drug Administration recently banned baby bottles and “sippy” cups that contain bisphenol A (BPA), as recommended by the American Medical Association. She wonders if there are other sources of BPA and if they pose a health risk to her, her son, and possible future children.
Environmental toxins have been linked to pregnancy complications and poor birth outcomes. The past several decades have seen a significant rise in reproductive disorders such as early onset of puberty, low sperm count, and birth defects such as cryptorchidism and hypospadias.1–4 These changes may be partly explained by other trends over time, such as older maternal age, rising incidence of obesity and maternal diabetes, and demographic changes that exacerbate health disparities. However, these well-recognized factors explain only some of the wide range of reproductive health problems that have been identified.
This review focuses on emerging evidence of the adverse reproductive effects of human-produced endocrine-disrupting chemicals (EDCs).
ENDOCRINE-DISRUPTING CHEMICALS ARE UBIQUITOUS
EDCs are exogenous substances that alter normal functioning of the endocrine system and consequently have the potential to cause adverse health effects in an intact organism and its progeny.2,3,5 Substances classified as EDCs are divided into two groups:
- Human-produced compounds, such as pesticides, industrial solvents and lubricants, plastics (eg, BPA), and plasticizers (eg, phthalates)
- Phytoestrogens, which are naturally occurring plant compounds that bind to estrogen receptors. The major dietary source of phytoestrogens is soy.6
Many human-produced EDCs are lipid-soluble. Some bioconcentrate in fat. The dietary route of exposure is the most common, primarily from fat-containing foods such as meats and seafood. Common predatory fish such as tuna and swordfish, as well as other large fish, are more likely to have high levels of EDCs accumulated from contaminated water.7 Water, air, soil, and dust in communities, schools, and workplaces may also carry EDCs.8
Some human-produced EDCs have a very long half-life and can remain in the environment for years or even decades. Because of their long half-life, some EDCs can “travel” and become part of the food chain, even in “pristine” areas where manufactured substances are not normally found.2
Women of childbearing age and pregnant women come in contact with EDCs on a daily basis. According to the Fourth National Report on Human Exposure to Environmental Chemicals, conducted by the US Centers for Disease Control and Prevention (CDC) in 2009, nearly all pregnant women in the United States have detectable serum levels of EDCs, including BPA, perchlorates, phthalates, polybrominated diphenyl ethers, and pesticides.9 Furthermore, BPA was found in more than 90% of the urine samples tested from random subsamples of 2,500 participants in the 2009 National Health and Nutrition Examination Survey report.10
EDCs AFFECT MULTIPLE PATHWAYS
Laboratory and animal research suggests that both manufactured and naturally occurring EDCs primarily affect sex steroid hormone pathways. However, some EDCs can affect adrenal, thyroid, and other endocrine pathways.
EDCs can potentially alter normal endocrine functioning by several mechanisms. They can bind to nuclear hormone receptors such as estrogen receptors, androgen receptors, progesterone receptors, thyroid hormone receptors, and peroxisome proliferator-activated receptor gamma. They can also bind to nonnuclear steroid hormone receptors (membrane estrogen receptors), nonsteroid receptors such as those for dopamine, serotonin, and norepinephrine, and orphan receptors such as aryl hydrocarbon receptor. In addition, some affect enzymatic pathways involved in steroid biosynthesis and metabolism and disrupt centralized endocrine pathways through positive and negative feedback.2 There is also evidence that EDCs have epigenetic and transgenerational effects, as they seem to be able to influence DNA methylation and histone acetylation.11
EVIDENCE OF HARM, LIMITATIONS OF EVIDENCE
In 2009, the Endocrine Society found convincing evidence that EDCs affect the male and female reproductive systems, breast health, oncogenesis, neuroendocrine function, thyroid function, metabolism, and cardiovascular health.2 The first evidence of harm from EDCs came from long-term follow-up of randomized trials of diethylstilbestrol, a synthetic estrogen used as a pharmacologic agent between 1938 and 1971 to prevent miscarriage.12
In 2013, the American College of Obstetricians and Gynecologists and the American Society for Reproductive Medicine issued a joint opinion report on exposure to toxic environmental agents. The report acknowledged that environmental toxins are ubiquitous and that they can negatively affect health from preconception through adult life. “[We] join leading scientists and other clinical practitioners in calling for timely action to identify and reduce exposure to toxic environmental agents while addressing the consequences of such exposure.”13
Because the effects of endocrine disruption can occur during fetal and embryonic development, EDC exposure before and during pregnancy is of special concern.14 Long-term effects of exposure to hazardous chemicals such as cadmium, lead, and mercury in breast milk can lead to negative consequences in adulthood.15,16
Animal studies have pointed to the negative health effects of human-produced EDCs. For example, animals exposed to BPA have aberrant breast and genitourinary development and exhibit abnormal endometrial stimulation and early puberty.2
Despite these suggestive findings, the health effects of environmental toxins on humans are difficult to determine definitively. Evidence comes mainly from epidemiologic studies showing distribution of particular medical conditions based on different levels of exposure. More research is needed on human exposure to environmental toxicants and their effects.2,17,18 The National Children’s Study, a cohort collaborative study being conducted by the Environmental Protection Agency, CDC, and National Institutes of Health, is examining the effects of environmental exposures on more than 100,000 children across the United States, following them from before birth until age 21.19
THE PRECAUTIONARY PRINCIPLE
The precautionary principle is a fairly new concept in environmental science, although clinicians have been adhering to it for a long time. As proposed in a meeting of scientists, lawyers, policy makers, and environmentalists in 1998, “When an activity raises threats of harm to human health or the environment, precautionary measures should be taken even if some cause and effect relationships are not fully established scientifically.”20
For example, studies show that breast milk in the United States commonly contains many chemicals that do not meet US Food and Drug Administration standards for baby food.15,16 Although medical ethics prohibits randomized clinical trials in humans to examine the potential harms of these chemicals, proponents of the precautionary principle advocate that it is reasonable to be concerned that these chemicals may be harmful.21–23
Applying the precautionary principle: Recommendations for patients
Without categorizing all chemicals as toxicants, nor all toxicants as worrisome for reproductive health, physicians can provide access to a growing body of information about reasonable strategies to reduce risk.
Educating patients about potential risks of exposure needs to be tempered with an appreciation of the economic and social barriers to avoiding or reducing risks. Often, people do not make conscious decisions about exposure. Rather, they are unknowingly exposed or have no access to safe, equivalent alternatives. Substituting suspected or known harmful products with potentially safer ones can be financially prohibitive. Where a patient lives and works may result in unavoidable exposure to environmental toxins.
Nevertheless, there are simple, economical alternatives that reduce or eliminate exposure to EDCs (Table 1).24–26 These include avoiding plastic and metal containers that are not marked “BPA-free,” eating locally grown fruits and vegetables, controlling pests through frequent cleaning and trapping, and avoiding pesticides.
Additionally, the flame retardant tris (1,3-dichloro-2-propyl) phosphate may be an endocrine disrupter. It is found in numerous products for infants and young children, as well as in dust, automobiles, and furniture. Flame retardants are no longer used in infant clothing but may still be found in foam products such as upholstered furniture, automobiles, and children’s nap pads.27,28
National organizations that provide resources for counseling women about human-produced EDCs as well as patient education materials are listed in Table 2.
CLINICAL CASE RESOLUTION
You counsel the patient that, although more research in humans is needed, because of safety concerns, avoiding exposing herself and her baby to BPA when possible is a reasonable approach. You review recommendations including avoiding BPA-containing plastics for food preparation (recycle code #7) and BPA-containing toys that might be mouthed by the baby, as well as minimizing the consumption of canned foods when possible. You further advise her to avoid products with flame retardants.
A 28-year-old woman presents for routine follow-up of asthma. She has a 1-year-old son and is considering a second pregnancy. She says she read on the Internet that the US Food and Drug Administration recently banned baby bottles and “sippy” cups that contain bisphenol A (BPA), as recommended by the American Medical Association. She wonders if there are other sources of BPA and if they pose a health risk to her, her son, and possible future children.
Environmental toxins have been linked to pregnancy complications and poor birth outcomes. The past several decades have seen a significant rise in reproductive disorders such as early onset of puberty, low sperm count, and birth defects such as cryptorchidism and hypospadias.1–4 These changes may be partly explained by other trends over time, such as older maternal age, rising incidence of obesity and maternal diabetes, and demographic changes that exacerbate health disparities. However, these well-recognized factors explain only some of the wide range of reproductive health problems that have been identified.
This review focuses on emerging evidence of the adverse reproductive effects of human-produced endocrine-disrupting chemicals (EDCs).
ENDOCRINE-DISRUPTING CHEMICALS ARE UBIQUITOUS
EDCs are exogenous substances that alter normal functioning of the endocrine system and consequently have the potential to cause adverse health effects in an intact organism and its progeny.2,3,5 Substances classified as EDCs are divided into two groups:
- Human-produced compounds, such as pesticides, industrial solvents and lubricants, plastics (eg, BPA), and plasticizers (eg, phthalates)
- Phytoestrogens, which are naturally occurring plant compounds that bind to estrogen receptors. The major dietary source of phytoestrogens is soy.6
Many human-produced EDCs are lipid-soluble. Some bioconcentrate in fat. The dietary route of exposure is the most common, primarily from fat-containing foods such as meats and seafood. Common predatory fish such as tuna and swordfish, as well as other large fish, are more likely to have high levels of EDCs accumulated from contaminated water.7 Water, air, soil, and dust in communities, schools, and workplaces may also carry EDCs.8
Some human-produced EDCs have a very long half-life and can remain in the environment for years or even decades. Because of their long half-life, some EDCs can “travel” and become part of the food chain, even in “pristine” areas where manufactured substances are not normally found.2
Women of childbearing age and pregnant women come in contact with EDCs on a daily basis. According to the Fourth National Report on Human Exposure to Environmental Chemicals, conducted by the US Centers for Disease Control and Prevention (CDC) in 2009, nearly all pregnant women in the United States have detectable serum levels of EDCs, including BPA, perchlorates, phthalates, polybrominated diphenyl ethers, and pesticides.9 Furthermore, BPA was found in more than 90% of the urine samples tested from random subsamples of 2,500 participants in the 2009 National Health and Nutrition Examination Survey report.10
EDCs AFFECT MULTIPLE PATHWAYS
Laboratory and animal research suggests that both manufactured and naturally occurring EDCs primarily affect sex steroid hormone pathways. However, some EDCs can affect adrenal, thyroid, and other endocrine pathways.
EDCs can potentially alter normal endocrine functioning by several mechanisms. They can bind to nuclear hormone receptors such as estrogen receptors, androgen receptors, progesterone receptors, thyroid hormone receptors, and peroxisome proliferator-activated receptor gamma. They can also bind to nonnuclear steroid hormone receptors (membrane estrogen receptors), nonsteroid receptors such as those for dopamine, serotonin, and norepinephrine, and orphan receptors such as aryl hydrocarbon receptor. In addition, some affect enzymatic pathways involved in steroid biosynthesis and metabolism and disrupt centralized endocrine pathways through positive and negative feedback.2 There is also evidence that EDCs have epigenetic and transgenerational effects, as they seem to be able to influence DNA methylation and histone acetylation.11
EVIDENCE OF HARM, LIMITATIONS OF EVIDENCE
In 2009, the Endocrine Society found convincing evidence that EDCs affect the male and female reproductive systems, breast health, oncogenesis, neuroendocrine function, thyroid function, metabolism, and cardiovascular health.2 The first evidence of harm from EDCs came from long-term follow-up of randomized trials of diethylstilbestrol, a synthetic estrogen used as a pharmacologic agent between 1938 and 1971 to prevent miscarriage.12
In 2013, the American College of Obstetricians and Gynecologists and the American Society for Reproductive Medicine issued a joint opinion report on exposure to toxic environmental agents. The report acknowledged that environmental toxins are ubiquitous and that they can negatively affect health from preconception through adult life. “[We] join leading scientists and other clinical practitioners in calling for timely action to identify and reduce exposure to toxic environmental agents while addressing the consequences of such exposure.”13
Because the effects of endocrine disruption can occur during fetal and embryonic development, EDC exposure before and during pregnancy is of special concern.14 Long-term effects of exposure to hazardous chemicals such as cadmium, lead, and mercury in breast milk can lead to negative consequences in adulthood.15,16
Animal studies have pointed to the negative health effects of human-produced EDCs. For example, animals exposed to BPA have aberrant breast and genitourinary development and exhibit abnormal endometrial stimulation and early puberty.2
Despite these suggestive findings, the health effects of environmental toxins on humans are difficult to determine definitively. Evidence comes mainly from epidemiologic studies showing distribution of particular medical conditions based on different levels of exposure. More research is needed on human exposure to environmental toxicants and their effects.2,17,18 The National Children’s Study, a cohort collaborative study being conducted by the Environmental Protection Agency, CDC, and National Institutes of Health, is examining the effects of environmental exposures on more than 100,000 children across the United States, following them from before birth until age 21.19
THE PRECAUTIONARY PRINCIPLE
The precautionary principle is a fairly new concept in environmental science, although clinicians have been adhering to it for a long time. As proposed in a meeting of scientists, lawyers, policy makers, and environmentalists in 1998, “When an activity raises threats of harm to human health or the environment, precautionary measures should be taken even if some cause and effect relationships are not fully established scientifically.”20
For example, studies show that breast milk in the United States commonly contains many chemicals that do not meet US Food and Drug Administration standards for baby food.15,16 Although medical ethics prohibits randomized clinical trials in humans to examine the potential harms of these chemicals, proponents of the precautionary principle advocate that it is reasonable to be concerned that these chemicals may be harmful.21–23
Applying the precautionary principle: Recommendations for patients
Without categorizing all chemicals as toxicants, nor all toxicants as worrisome for reproductive health, physicians can provide access to a growing body of information about reasonable strategies to reduce risk.
Educating patients about potential risks of exposure needs to be tempered with an appreciation of the economic and social barriers to avoiding or reducing risks. Often, people do not make conscious decisions about exposure. Rather, they are unknowingly exposed or have no access to safe, equivalent alternatives. Substituting suspected or known harmful products with potentially safer ones can be financially prohibitive. Where a patient lives and works may result in unavoidable exposure to environmental toxins.
Nevertheless, there are simple, economical alternatives that reduce or eliminate exposure to EDCs (Table 1).24–26 These include avoiding plastic and metal containers that are not marked “BPA-free,” eating locally grown fruits and vegetables, controlling pests through frequent cleaning and trapping, and avoiding pesticides.
Additionally, the flame retardant tris (1,3-dichloro-2-propyl) phosphate may be an endocrine disrupter. It is found in numerous products for infants and young children, as well as in dust, automobiles, and furniture. Flame retardants are no longer used in infant clothing but may still be found in foam products such as upholstered furniture, automobiles, and children’s nap pads.27,28
National organizations that provide resources for counseling women about human-produced EDCs as well as patient education materials are listed in Table 2.
CLINICAL CASE RESOLUTION
You counsel the patient that, although more research in humans is needed, because of safety concerns, avoiding exposing herself and her baby to BPA when possible is a reasonable approach. You review recommendations including avoiding BPA-containing plastics for food preparation (recycle code #7) and BPA-containing toys that might be mouthed by the baby, as well as minimizing the consumption of canned foods when possible. You further advise her to avoid products with flame retardants.
- Sutton P, Giudice LC, Woodruff TJ. Reproductive environmental health. Curr Opin Obstet Gynecol 2010; 22:517–524.
- Diamanti-Kandarakis E, Bouguignon JP, Giudice LC, et al. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev 2009; 30:293–342.
- Friedrich MJ. Endocrine-disrupting chemicals. JAMA 2013; 309:1578.
- Schettler T. Environmental exposures, infertility, and related reproductive disorders: an update. October 2011. The Collaborative on Health and the Environment. www.healthandenvironment.org/infertility/peer_reviewed. Accessed March 9, 2016.
- Woodruff TJ, Carlson A, Schwartz JM, Giudice LC. Proceedings of the Summit on the Environmental Challenges to Reproductive Health and Fertility: executive summary. Fertil Steril 2008; 89:281–300.
- Cao Y, Calafat AM, Doerge DR, et al. Isoflavones in urine, saliva, and blood of infants: data from a pilot study on the estrogenic activity of soy formula. J Expo Sci Environ Epidemiol 2009; 19:223–234.
- Endocrine-disrupting chemicals. The Collaborative on Health and the Environment. Available at www.healthandenvironment.org/initiatives/learning/r/prevention. Accessed March 9, 2016.
- Environmental impacts on reproductive health: clinical proceedings. Association of Reproductive Health Professionals. www.arhp.org/uploadDocs/CPRHE.pdf. Accessed March 9, 2016.
- Fourth national report on human exposure to environmental chemicals. Centers for Disease Control and Prevention (CDC). www.cdc.gov/exposurereport/pdf/FourthReport.pdf. Accessed March 9, 2016.
- Woodruff TJ, Zota AR, Schwartz JM. Environmental chemicals in pregnant women in the United States: NHANES 2003-2004. Environ Health Perspect 2011; 119:878–885.
- Anway MD, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors. Endocrinology 2006; 147(suppl 6):S43–S49.
- About DES. Centers for Disease Control and Prevention (CDC). www.cdc.gov/des/consumers/about/index.html. Accessed March 9, 2016.
- Exposure to toxic environmental agents. American Congress of Obstetricians and Gynecologists (ACOG). www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Health-Care-for-Underserved-Women/Exposure-to-Toxic-Environmental-Agents. Accessed March 9, 2016.
- Bergman A, Heindel JJ, Jobling S, Kidd KA, Zoeller RT. United Nations Environment Programme (UNEP). State of the science of endocrine disrupting chemicals: 2012. www.who.int/ceh/publications/endocrine/en/. Accessed March 9, 2016.
- Pohl HR, Hibbs BF. Breast-feeding exposure of infants to environmental contaminants—a public health risk assessment viewpoint: chlorinated dibenzodioxins and chlorinated dibenzofurans. Toxicol Ind Health 1996; 12:593–611.
- Abadin HG, Hibbs BF, Pohl HR. Breast-feeding exposure of infants to cadmium, lead and mercury: a public health viewpoint. Toxicol Ind Health 1997; 13:495–517.
- Bretveld RW, Thomas CM, Scheepers PT, Zielhuis GA, Roeleveld N. Pesticide exposure: the hormonal function of the female reproductive system disrupted? Reprod Biol Endocrinol 2006; 4:30.
- Mendola P, Messer LC, Rappazzo K. Science linking environmental contaminant exposures with fertility and reproductive health impacts in the adult female. Fertil Steril 2008; 89(suppl 2):e81–e94.
- National Institute of Health (NIH). National Children’s Study (NCS). www.nationalchildrensstudy.gov/Pages/default.aspx. Accessed March 9, 2016.
- Raffensperger C, Tickner J, editors. Protecting public health and the environment: implementing the precautionary principle. Washington, DC: Island Press; 1999:8.
- Woodruff TJ, Sutton P. The navigation guide systematic review methodology: a rigorous and transparent method for translating environmental health science into better health outcomes. Environ Health Perspect 2014; 122:1007–1014.
- Sutton P, Woodruff TJ. Risk communication and decision tools for children’s health protection. Birth Defects Res C Embryo Today 2013; 99:45–49.
- Woodruff TJ. Bridging epidemiology and model organisms to increase understanding of endocrine disrupting chemicals and human health effects. J Steroid Biochem Mol Biol 2011; 127:108–117.
- National Institute of Environmental Health Sciences. Bisphenol A (BPA). www.niehs.nih.gov/health/topics/agents/sya-bpa/index.cfm. Accessed March 9, 2016.
- Environmental Working Group. Shoppers guide to presticides in produce. www.ewg.org/foodnews/. Accessed March 9, 2016.
- Schettler T. Human exposure to phthalates via consumer products. Int J Androl 2006; 29:134–139.
- Betts KS. Exposure to TDCPP appears widespread. Environ Health Perspect 2013; 121:a150.
- Environmental Working Group. Healthy home tips. www.ewg.org/research/healthy-home-tips/tip-4-avoid-fire-retardants. Accessed March 9, 2016.
- Sutton P, Giudice LC, Woodruff TJ. Reproductive environmental health. Curr Opin Obstet Gynecol 2010; 22:517–524.
- Diamanti-Kandarakis E, Bouguignon JP, Giudice LC, et al. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev 2009; 30:293–342.
- Friedrich MJ. Endocrine-disrupting chemicals. JAMA 2013; 309:1578.
- Schettler T. Environmental exposures, infertility, and related reproductive disorders: an update. October 2011. The Collaborative on Health and the Environment. www.healthandenvironment.org/infertility/peer_reviewed. Accessed March 9, 2016.
- Woodruff TJ, Carlson A, Schwartz JM, Giudice LC. Proceedings of the Summit on the Environmental Challenges to Reproductive Health and Fertility: executive summary. Fertil Steril 2008; 89:281–300.
- Cao Y, Calafat AM, Doerge DR, et al. Isoflavones in urine, saliva, and blood of infants: data from a pilot study on the estrogenic activity of soy formula. J Expo Sci Environ Epidemiol 2009; 19:223–234.
- Endocrine-disrupting chemicals. The Collaborative on Health and the Environment. Available at www.healthandenvironment.org/initiatives/learning/r/prevention. Accessed March 9, 2016.
- Environmental impacts on reproductive health: clinical proceedings. Association of Reproductive Health Professionals. www.arhp.org/uploadDocs/CPRHE.pdf. Accessed March 9, 2016.
- Fourth national report on human exposure to environmental chemicals. Centers for Disease Control and Prevention (CDC). www.cdc.gov/exposurereport/pdf/FourthReport.pdf. Accessed March 9, 2016.
- Woodruff TJ, Zota AR, Schwartz JM. Environmental chemicals in pregnant women in the United States: NHANES 2003-2004. Environ Health Perspect 2011; 119:878–885.
- Anway MD, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors. Endocrinology 2006; 147(suppl 6):S43–S49.
- About DES. Centers for Disease Control and Prevention (CDC). www.cdc.gov/des/consumers/about/index.html. Accessed March 9, 2016.
- Exposure to toxic environmental agents. American Congress of Obstetricians and Gynecologists (ACOG). www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Health-Care-for-Underserved-Women/Exposure-to-Toxic-Environmental-Agents. Accessed March 9, 2016.
- Bergman A, Heindel JJ, Jobling S, Kidd KA, Zoeller RT. United Nations Environment Programme (UNEP). State of the science of endocrine disrupting chemicals: 2012. www.who.int/ceh/publications/endocrine/en/. Accessed March 9, 2016.
- Pohl HR, Hibbs BF. Breast-feeding exposure of infants to environmental contaminants—a public health risk assessment viewpoint: chlorinated dibenzodioxins and chlorinated dibenzofurans. Toxicol Ind Health 1996; 12:593–611.
- Abadin HG, Hibbs BF, Pohl HR. Breast-feeding exposure of infants to cadmium, lead and mercury: a public health viewpoint. Toxicol Ind Health 1997; 13:495–517.
- Bretveld RW, Thomas CM, Scheepers PT, Zielhuis GA, Roeleveld N. Pesticide exposure: the hormonal function of the female reproductive system disrupted? Reprod Biol Endocrinol 2006; 4:30.
- Mendola P, Messer LC, Rappazzo K. Science linking environmental contaminant exposures with fertility and reproductive health impacts in the adult female. Fertil Steril 2008; 89(suppl 2):e81–e94.
- National Institute of Health (NIH). National Children’s Study (NCS). www.nationalchildrensstudy.gov/Pages/default.aspx. Accessed March 9, 2016.
- Raffensperger C, Tickner J, editors. Protecting public health and the environment: implementing the precautionary principle. Washington, DC: Island Press; 1999:8.
- Woodruff TJ, Sutton P. The navigation guide systematic review methodology: a rigorous and transparent method for translating environmental health science into better health outcomes. Environ Health Perspect 2014; 122:1007–1014.
- Sutton P, Woodruff TJ. Risk communication and decision tools for children’s health protection. Birth Defects Res C Embryo Today 2013; 99:45–49.
- Woodruff TJ. Bridging epidemiology and model organisms to increase understanding of endocrine disrupting chemicals and human health effects. J Steroid Biochem Mol Biol 2011; 127:108–117.
- National Institute of Environmental Health Sciences. Bisphenol A (BPA). www.niehs.nih.gov/health/topics/agents/sya-bpa/index.cfm. Accessed March 9, 2016.
- Environmental Working Group. Shoppers guide to presticides in produce. www.ewg.org/foodnews/. Accessed March 9, 2016.
- Schettler T. Human exposure to phthalates via consumer products. Int J Androl 2006; 29:134–139.
- Betts KS. Exposure to TDCPP appears widespread. Environ Health Perspect 2013; 121:a150.
- Environmental Working Group. Healthy home tips. www.ewg.org/research/healthy-home-tips/tip-4-avoid-fire-retardants. Accessed March 9, 2016.
KEY POINTS
- Although EDCs primarily affect sex steroid hormone pathways, some can affect adrenal, thyroid, and other endocrine pathways.
- Human-produced EDCs vary widely in their properties. Many, but not all, concentrate in fat, and some have a very long half-life.
- Because it would be impossible to perform randomized, controlled trials of the health effects of the thousands of manufactured EDCs encountered in daily life, physicians should follow the precautionary principal when counseling patients: ie, tell them to avoid chemicals when possible, especially those that have proven or plausible health risks.
- On the other hand, physicians need to keep in mind the economic hardships patients may face in switching to potentially safer products or foods and unavoidable exposures at work and at home.

Epiglottic cysts in clinical practice
A 50-year-old man presented to the otolaryngology clinic, complaining of throat discomfort for the past 3 months that worsened in the supine position. The clinical examination revealed one cystic lesion on either side of the epiglottis (Figure 1). The larger cyst measured 23 × 13 × 12 mm. The man’s symptoms resolved completely after endoscopic removal of the cysts under general anesthesia (Figure 2).
CLINICAL IMPLICATIONS
Epiglottic cysts are benign lesions on the lingual or laryngeal aspect of the epiglottis and are often a result of mucus retention. Otolaryngologists, anesthesiologists, and endoscopists are usually the first to discover them. Because the cysts are usually asymptomatic, it is difficult to calculate their prevalence in the general population.
If the cysts are symptomatic, patients usually describe nonspecific complaints such as pharyngeal discomfort and foreign-body sensation. Voluminous cysts discovered incidentally in patients requiring general anesthesia pose a challenge to intubation and increase the risk of airway obstruction.
In addition, epiglottic cysts have been reported in a proportion of adult patients with acute epiglottitis (25% in one series)1; They may be discovered either during the acute infection or when inflammation subsides.2 These patients have been reported to have a higher risk of acute airway obstruction, with a sixfold increase in the need for acute airway management in one series.1 Moreover, epiglottitis was reported to recur in 12.5% and 17% of patients with cysts in two different series,1,2 corresponding to a recurrence rate 15 times higher than in patients with no cysts.1 Hence, it would be useful to rule out their presence after any episode of acute epiglottitis.
Once an epiglottic cyst is discovered in a symptomatic patient, it can be managed either by elective endoscopic resection or by marsupialization. Either procedure is safe, with good long-term results. Simple evacuation of the cyst should be considered in cases of acute airway management.
- Yoon TM, Choi JO, Lim SC, Lee JK. The incidence of epiglottic cysts in a cohort of adults with acute epiglottitis. Clin Otolaryngol 2010; 35:18–24.
- Berger G, Averbuch E, Zilka K, Berger R, Ophir D. Adult vallecular cyst: thirteen-year experience. Otolaryngol Head Neck Surg 2008; 138:321–327.
A 50-year-old man presented to the otolaryngology clinic, complaining of throat discomfort for the past 3 months that worsened in the supine position. The clinical examination revealed one cystic lesion on either side of the epiglottis (Figure 1). The larger cyst measured 23 × 13 × 12 mm. The man’s symptoms resolved completely after endoscopic removal of the cysts under general anesthesia (Figure 2).
CLINICAL IMPLICATIONS
Epiglottic cysts are benign lesions on the lingual or laryngeal aspect of the epiglottis and are often a result of mucus retention. Otolaryngologists, anesthesiologists, and endoscopists are usually the first to discover them. Because the cysts are usually asymptomatic, it is difficult to calculate their prevalence in the general population.
If the cysts are symptomatic, patients usually describe nonspecific complaints such as pharyngeal discomfort and foreign-body sensation. Voluminous cysts discovered incidentally in patients requiring general anesthesia pose a challenge to intubation and increase the risk of airway obstruction.
In addition, epiglottic cysts have been reported in a proportion of adult patients with acute epiglottitis (25% in one series)1; They may be discovered either during the acute infection or when inflammation subsides.2 These patients have been reported to have a higher risk of acute airway obstruction, with a sixfold increase in the need for acute airway management in one series.1 Moreover, epiglottitis was reported to recur in 12.5% and 17% of patients with cysts in two different series,1,2 corresponding to a recurrence rate 15 times higher than in patients with no cysts.1 Hence, it would be useful to rule out their presence after any episode of acute epiglottitis.
Once an epiglottic cyst is discovered in a symptomatic patient, it can be managed either by elective endoscopic resection or by marsupialization. Either procedure is safe, with good long-term results. Simple evacuation of the cyst should be considered in cases of acute airway management.
A 50-year-old man presented to the otolaryngology clinic, complaining of throat discomfort for the past 3 months that worsened in the supine position. The clinical examination revealed one cystic lesion on either side of the epiglottis (Figure 1). The larger cyst measured 23 × 13 × 12 mm. The man’s symptoms resolved completely after endoscopic removal of the cysts under general anesthesia (Figure 2).
CLINICAL IMPLICATIONS
Epiglottic cysts are benign lesions on the lingual or laryngeal aspect of the epiglottis and are often a result of mucus retention. Otolaryngologists, anesthesiologists, and endoscopists are usually the first to discover them. Because the cysts are usually asymptomatic, it is difficult to calculate their prevalence in the general population.
If the cysts are symptomatic, patients usually describe nonspecific complaints such as pharyngeal discomfort and foreign-body sensation. Voluminous cysts discovered incidentally in patients requiring general anesthesia pose a challenge to intubation and increase the risk of airway obstruction.
In addition, epiglottic cysts have been reported in a proportion of adult patients with acute epiglottitis (25% in one series)1; They may be discovered either during the acute infection or when inflammation subsides.2 These patients have been reported to have a higher risk of acute airway obstruction, with a sixfold increase in the need for acute airway management in one series.1 Moreover, epiglottitis was reported to recur in 12.5% and 17% of patients with cysts in two different series,1,2 corresponding to a recurrence rate 15 times higher than in patients with no cysts.1 Hence, it would be useful to rule out their presence after any episode of acute epiglottitis.
Once an epiglottic cyst is discovered in a symptomatic patient, it can be managed either by elective endoscopic resection or by marsupialization. Either procedure is safe, with good long-term results. Simple evacuation of the cyst should be considered in cases of acute airway management.
- Yoon TM, Choi JO, Lim SC, Lee JK. The incidence of epiglottic cysts in a cohort of adults with acute epiglottitis. Clin Otolaryngol 2010; 35:18–24.
- Berger G, Averbuch E, Zilka K, Berger R, Ophir D. Adult vallecular cyst: thirteen-year experience. Otolaryngol Head Neck Surg 2008; 138:321–327.
- Yoon TM, Choi JO, Lim SC, Lee JK. The incidence of epiglottic cysts in a cohort of adults with acute epiglottitis. Clin Otolaryngol 2010; 35:18–24.
- Berger G, Averbuch E, Zilka K, Berger R, Ophir D. Adult vallecular cyst: thirteen-year experience. Otolaryngol Head Neck Surg 2008; 138:321–327.

Measles: Back again
Measles continues to rear its head in the United States. Because it is so contagious, even the few cases introduced by travelers quickly spread to susceptible contacts. Life-threatening and severely disabling complications can occur, although this is rare. Widespread immunization and prompt recognition and isolation of contacts are key to controlling outbreaks.
This article reviews the epidemiology of measles, describes its distinctive clinical picture, and provides recommendations for infection control and prevention, including in immunosuppressed populations.
MEASLES IS SERIOUS AND HIGHLY CONTAGIOUS
Up to 90% of susceptible people develop measles after exposure, making it one of the most contagious of infections. The virus is transmitted by airborne spread when an infected person coughs or sneezes, or by direct contact with infectious droplets. The virus can remain infectious in the air or on a surface for up to 2 hours.1
Worldwide, an estimated 20 million people are infected with measles each year, and 146,000 die of complications. In 1980, before widespread vaccination, 2.6 million deaths were attributable to measles annually. In the United States before the introduction of measles vaccine in 1963, measles was a significant cause of disease and death: an estimated 3 to 4 million people were infected annually, although only about 549,000 were reported. There were 48,000 hospitalizations, 1,000 cases of permanent brain damage from measles encephalitis, and 495 deaths annually.2
Outbreaks still occur regularly
In 2000, measles was declared eliminated from the United States,3 but annual outbreaks have occurred since then as a result of cases imported from other countries and their subsequent transmission to unvaccinated people. From 2001 to 2012, a median of four outbreaks and 60 cases were reported annually to the US Centers for Disease Control and Prevention.4
In January 2015, a multistate measles outbreak originating in Disneyland in California was recognized. As of April 17, when the outbreak was declared over, 111 measles cases from seven states had been linked to this outbreak.5 Of the evaluable cases, 44% were in unvaccinated people and 38% were in those whose vaccination status was unknown or undocumented. The median age of patients was 21, and 20% required hospitalization.
This outbreak, as well as four other smaller US outbreaks the same year, underscores the transmissibility of the virus in populations containing only a small percentage of unvaccinated people.6
DISTINCTIVE CLINICAL PICTURE
The incubation period for measles infection is 7 to 21 days, with most cases becoming apparent 10 to 12 days after exposure. Measles should be suspected in a patient with the following clinical features whose history indicates susceptibility and exposure (ie, an unimmunized person with a history of exposure or travel):
Severe acute respiratory illness. Measles usually presents as an acute respiratory viral illness, which typically lasts 2 to 4 days. The illness involves high fevers, malaise, anorexia, and the “three Cs”: cough, coryza (rhinitis), and conjunctivitis. Patients usually appear sicker than those with more common viral illnesses.

Koplik spots, which are pathognomonic for measles, are seen in the first few days of illness. They are bluish-white, slightly raised lesions on an erythematous base on the buccal mucosa, usually opposite the first molar (Figure 1). Spots can also be seen on the soft palate, conjunctiva, and vaginal mucosa. Koplik spots usually disappear after a few days and often are not appreciable at the time of evaluation.

Discrete erythematous patches develop on the face and neck a day after the appearance of Koplik spots. This rash becomes more confluent as it spreads to involve the entire body (Figure 2). It typically lasts for 3 to 7 days, then fades in a similar pattern. The confluent nature of this rash and its spread from the face and neck to the entire body are characteristic of measles. Patients are highly contagious from 4 days before the onset of the rash to 4 days after.
COMPLICATIONS CAN BE SEVERE
Those at highest risk for measles complications are infants, children under age 5, adults over age 20, pregnant women, and immunosuppressed individuals.7
Pneumonia—either a primary measles pneumonia or a secondary viral or bacterial pneumonia—is the most common cause of death.8,9 Viruses complicating measles are typically adenovirus and herpes simplex virus. Bacteria causing secondary infection are usually Staphylococcus aureus and Streptococcus pneumoniae and, less commonly, gram-negative bacteria.
Laryngotracheobronchitis (croup) is the second most common cause of death, with bacteria and viruses similar to those causing measles-related pneumonia.
Otitis media is the most common complication of measles. Other respiratory complications include mastoiditis, pneumothorax, and mediastinal emphysema.
Acute measles encephalitis occurs in 1 measles case per 1,000 and often results in permanent brain damage. During the convalescent phase of the illness, fever again emerges, with the development of headaches, seizures, and altered consciousness.10
Subacute sclerosing panencephalitis is a rare fatal degenerative disease of the central nervous system caused by a persistent infection with a defective measles virus. The precise pathophysiology is unclear, but it is thought that mutations of the viral genome lead to altered cellular immunity.11 The condition typically occurs 7 to 10 years after the initial measles infection, particularly in those who developed measles before age 2. Clinical manifestations include behavioral disturbances, intellectual deterioration, and myoclonic seizures, slowly progressing to a vegetative state and death.12
Other complications of measles include diarrhea and stomatitis, which are associated with malnutrition in developing countries, and subclinical hepatitis, thrombocytopenia, appendicitis, ileocolitis, hypokalemia, and myocarditis.
During pregnancy, measles infection can be complicated by primary measles pneumonia and is associated with an increased risk of miscarriage and premature birth.13
Patients with a cell-mediated immunodeficiency who develop measles are particularly susceptible to fatal measles pneumonia and acute progressive encephalitis.14
ATYPICAL MEASLES IN THOSE WHO RECEIVED KILLED VACCINE
From 1963 to 1967, a killed measles vaccine was available in the United States. Those who received this vaccine are susceptible to an atypical form of measles when exposed to the virus,15 characterized by a 1- to 2-day prodrome, followed by the appearance of a maculopapular or petechial rash on the distal extremities that spreads centripetally. Patients develop high fever and edema of the hands and feet, and have a more prolonged course than with classic measles. It is believed not to be contagious.16
LABORATORY CONFIRMATION
Laboratory confirmation of measles is recommended for suspected cases. Because viral isolation is technically difficult and is not readily available in most laboratories, measles-specific immunoglobulin M antibody serologic testing is most commonly used. It is almost 100% sensitive when done 2 to 3 days after the onset of the rash.17
Measles RNA testing by real-time polymerase chain reaction to detect measles virus in the blood, throat, or urine is more specific and if available may be preferred over serologic testing.18
SUPPORTIVE MANAGEMENT AND VITAMIN A SUPPLEMENTATION
No specific antiviral therapy for measles is available. Management involves supportive measures and monitoring for secondary bacterial complications.
The World Health Organization and the American Academy of Pediatrics recommend vitamin A supplementation for all children with acute measles.19 In developing countries, it has been shown to reduce rates of morbidity and death in measles-infected children.20 In the United States, children with measles have been found to have low serum levels of vitamin A, with lower levels associated with more severe disease.
VACCINATION RECOMMENDATIONS
The only measles vaccine available in the United States is a live further-attenuated strain prepared in chick embryo cell culture and combined with mumps and rubella vaccine (MMR) or with measles, mumps, rubella, and varicella vaccine (MMRV).
Healthy children. Two doses of measles vaccine are recommended, as a single dose is associated with a 5% failure rate. The recommended schedule is:
- First dose at age 12 to 15 months
- Second dose at the time of school entry (ages 4 to 6), or at any time at least 28 days after the first dose.19
More than 99% of children who receive two doses of vaccine according to this schedule develop serologic evidence of measles immunity. Vaccination provides long-term immunity, and many epidemiologic studies have documented that waning immunity after vaccination occurs only very rarely.21
All school-age children, including elementary, middle, and high school students, who received only one dose of measles vaccine should receive the second dose.
Adults born in 1957 or later should receive at least one dose of measles vaccine unless they have other acceptable evidence of immunity, such as:4
- Documentation of age-appropriate live measles vaccine, ie, one dose of vaccine for adults not at high risk, or two doses for those at high risk (see below)
- Laboratory evidence of immunity (ie, measles immunoglobulin G in serum)
- Laboratory confirmation of disease.
Adults born before 1957 can be considered to be immune to measles, although MMR vaccine can be administered in those without contraindications.
Adults at increased risk of exposure or transmission of measles and who do not have evidence of immunity should receive two doses of MMR vaccine, given at least 28 days apart. This high-risk group includes:
- Students attending college or other post-high school educational institution
- Healthcare personnel
- International travelers.
During measles outbreaks, every effort should be made to ensure that those at high risk are vaccinated with two doses of MMR or have other acceptable evidence of immunity.
LIVE VACCINE IS SAFE FOR MOST PEOPLE
Mild side effects. A transient fever, which may be accompanied by a discrete or confluent rash, occurs in 5% to 15% of recipients 5 to 12 days after vaccination.
Transmission does not occur. People who have been newly vaccinated do not transmit the virus to susceptible contacts, even if they develop a vaccine-associated rash. The vaccine can safely be given to close contacts of immunocompromised and other susceptible people.
Egg allergy not a concern. Measles vaccine is produced in chick embryo cell culture but has been shown to be safe for people with egg allergy and is recommended without the need for egg allergy testing.19
Autism link debunked. No scientific evidence shows that the risk of autism is higher in children who receive MMR vaccine than in those who do not. In 2001, an Institute of Medicine report rejected a causal relationship between MMR vaccine and autism spectrum disorders.22
CONTRAINDICATIONS
Measles vaccine is contraindicated for:
- Patients who have cell-mediated immune deficiencies, except human immunodeficiency virus (HIV) infection
- Pregnant women
- Those who have had a severe allergic reaction to a vaccine component in the past
- Those with moderate or severe acute illness
- Those who have recently received immunoglobulin products.
People with HIV infection who are severely immunosuppressed should not receive live measles vaccine. However, because of the risk of severe measles in HIV-infected patients and because the vaccine has been shown to be safe for patients with HIV without severe immunosuppression, the vaccine is recommended for those with asymptomatic or mildly symptomatic HIV infection who do not have evidence of severe immunosuppression (ie, CD4 lymphocytes < 15% or < 200 cells/µL).3,4
INFECTION CONTROL AND PREVENTION
Healthcare workers should maintain a high index of suspicion for measles and implement isolation procedures promptly in patients with a febrile illness, rash, and a history of travel abroad or contact with travelers from abroad.23 Suspected cases should be reported promptly to local health agencies to help limit spread.
Patients with measles should be placed in airborne isolation (eg, use of an N95 or higher level respirator and an airborne infection isolation room) for 4 days after the onset of the rash in a normal host and for the duration of the illness in an immunocompromised patient. Healthcare staff, regardless of their immunity status, should adhere to these precautions when entering the room of infected patients.
Immunization programs should be established to ensure that everyone who works or volunteers in healthcare facilities is protected against measles.4
Postexposure prophylaxis. Measles vaccination given to susceptible contacts within 72 hours of exposure may provide protection against infection and induces protection against subsequent measles exposures.24,25
Vaccination is the best intervention for susceptible contacts older than 12 months who do not have a contraindication to measles vaccination, and for those who have received only one dose of measles vaccine.
Passive immunization. Active immunization is the best strategy for controlling measles outbreaks. Passive immunization with intramuscularly or intravenously administered immunoglobulin given within 6 days of exposure can be used to prevent transmission or modify the clinical course of infection for susceptible contacts at high risk of developing severe or fatal measles. This includes people who are being treated with immunosuppressive agents, HIV-infected, pregnant, or younger than 1 year of age.
- Stokes J Jr, Reilly CM, Buynak EB, Hilleman MR. Immunologic studies of measles. Am J Hyg 1961; 74:293–303.
- Bloch AB, Orenstein WA, Stetler HC, et al. Health impact of measles vaccination in the United States. Pediatrics 1985; 76:524–532.
- Katz SL, Hinman AR. Summary and conclusions: measles elimination meeting, 16-17 March 2000. J Infect Dis 2004; 189(suppl 1):S43–S47.
- McLean HQ, Fiebelkorn AP, Temte JL, Wallace GS; Centers for Disease Control and Prevention (CDC). Prevention of measles, rubella, congenital rubella syndrome, and mumps, 2013: summary recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2013; 62:1–34.
- Clemmons NS, Gastanaduy PA, Fiebelkorn AP, Redd SB, Wallace GS; Centers for Disease Control and Prevention (CDC). Measles—United States, January 4 - April 2, 2015. MMWR Morb Mortal Wkly Rep 2015; 64:373–376.
- Gay NJ. The theory of measles elimination: implications for the design of elimination strategies. J Infect Dis 2004; 189(suppl 1):S27–S35.
- Perry RT, Halsey NA. The clinical significance of measles: a review. J Infect Dis 2004; 189(suppl 1):S4–S16).
- Gremillion DH, Crawford GE. Measles pneumonia in young adults. An analysis of 106 cases. Am J Med 1981; 71:539–542.
- Quiambao BP, Gatchalian SR, Halonen P, et al. Coinfection is common in measles-associated pneumonia. Pediatr Infect Dis J 1998; 17:89–93.
- Johnson RT, Griffin D, Hirsch R, et al. Measles encephalomyelitis—clinical and immunologic studies. N Engl J Med 1984; 310:137–141.
- Garg RK. Subacute sclerosing panencephalitis. Postgrad Med J 2002; 78:63–70.
- Sever JL. Persistent measles infection of the central nervous system: subacute sclerosing panencephalitis. Rev Infect Dis 1983; 5:467–473.
- Atmar RL, Englund JA, Hammill H. Complications of measles during pregnancy. Clin Infect Dis 1992; 14:217–226.
- Kaplan LJ, Daum RS, Smaron M, McCarthy CA. Severe measles in immunocompromised patients. JAMA 1992; 267:1237–1241.
- Frey HM, Krugman S. Atypical measles syndrome: unusual hepatic, pulmonary, and immunologic aspects. Am J Med Sci 1981; 281:51–55.
- Fulginiti VA, Eller JJ, Downie AW, Kempe CH. Altered reactivity to measles virus. Atypical measles in children previously immunized with inactivated measles virus vaccines. JAMA 1967; 202:1075–1080.
- Bellini WJ, Helfand RF. The challenges and strategies for laboratory diagnosis of measles in an international setting. J Infect Dis 2003; 187(suppl 1):S283–S290.
- Riddell MA, Chibo D, Kelly HA, Catton MG, Birch CJ. Investigation of optimal specimen type and sampling time for detection of measles virus RNA during a measles epidemic. J Clin Microbiol 2001; 39:375–376.
- Measles. In: Pickering LK, Baker CJ, Kimberlin DW, Long SS, editors. Red Book: 2012 Report of the Committee on Infectious Diseases. 29th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2012:489-499.
- Huiming Y, Chaomin W, Meng M. Vitamin A for treating measles in children. Cochrane Database Syst Rev 2005; 4:CD001479.
- Markowitz LE, Preblud SR, Fine PE, Orenstein WA. Duration of live measles vaccine-induced immunity. Pediatr Infect Dis J 1990; 9:101–110.
- Stratton K, Gable A, Shetty P, McCormick M. Immunization safety review: measles-mumps-rubella vaccine and autism. Washington, DC: National Academy Press; 2001.
- Centers for Disease Control and Prevention (CDC). 2007 guideline for isolation precautions: preventing transmission of infectious agents in healthcare settings. www.cdc.gov/hicpac/2007ip/2007isolationprecautions.html. Accessed April 8, 2016.
- Berkovich S, Starr S. Use of live-measles-virus vaccine to abort an expected outbreak of measles within a closed population. N Engl J Med 1963; 269:75–77.
- Ruuskanen O, Salmi TT, Halonen P. Measles vaccination after exposure to natural measles. J Pediatr 1978; 93:43–46.
Measles continues to rear its head in the United States. Because it is so contagious, even the few cases introduced by travelers quickly spread to susceptible contacts. Life-threatening and severely disabling complications can occur, although this is rare. Widespread immunization and prompt recognition and isolation of contacts are key to controlling outbreaks.
This article reviews the epidemiology of measles, describes its distinctive clinical picture, and provides recommendations for infection control and prevention, including in immunosuppressed populations.
MEASLES IS SERIOUS AND HIGHLY CONTAGIOUS
Up to 90% of susceptible people develop measles after exposure, making it one of the most contagious of infections. The virus is transmitted by airborne spread when an infected person coughs or sneezes, or by direct contact with infectious droplets. The virus can remain infectious in the air or on a surface for up to 2 hours.1
Worldwide, an estimated 20 million people are infected with measles each year, and 146,000 die of complications. In 1980, before widespread vaccination, 2.6 million deaths were attributable to measles annually. In the United States before the introduction of measles vaccine in 1963, measles was a significant cause of disease and death: an estimated 3 to 4 million people were infected annually, although only about 549,000 were reported. There were 48,000 hospitalizations, 1,000 cases of permanent brain damage from measles encephalitis, and 495 deaths annually.2
Outbreaks still occur regularly
In 2000, measles was declared eliminated from the United States,3 but annual outbreaks have occurred since then as a result of cases imported from other countries and their subsequent transmission to unvaccinated people. From 2001 to 2012, a median of four outbreaks and 60 cases were reported annually to the US Centers for Disease Control and Prevention.4
In January 2015, a multistate measles outbreak originating in Disneyland in California was recognized. As of April 17, when the outbreak was declared over, 111 measles cases from seven states had been linked to this outbreak.5 Of the evaluable cases, 44% were in unvaccinated people and 38% were in those whose vaccination status was unknown or undocumented. The median age of patients was 21, and 20% required hospitalization.
This outbreak, as well as four other smaller US outbreaks the same year, underscores the transmissibility of the virus in populations containing only a small percentage of unvaccinated people.6
DISTINCTIVE CLINICAL PICTURE
The incubation period for measles infection is 7 to 21 days, with most cases becoming apparent 10 to 12 days after exposure. Measles should be suspected in a patient with the following clinical features whose history indicates susceptibility and exposure (ie, an unimmunized person with a history of exposure or travel):
Severe acute respiratory illness. Measles usually presents as an acute respiratory viral illness, which typically lasts 2 to 4 days. The illness involves high fevers, malaise, anorexia, and the “three Cs”: cough, coryza (rhinitis), and conjunctivitis. Patients usually appear sicker than those with more common viral illnesses.
Koplik spots, which are pathognomonic for measles, are seen in the first few days of illness. They are bluish-white, slightly raised lesions on an erythematous base on the buccal mucosa, usually opposite the first molar (Figure 1). Spots can also be seen on the soft palate, conjunctiva, and vaginal mucosa. Koplik spots usually disappear after a few days and often are not appreciable at the time of evaluation.
Discrete erythematous patches develop on the face and neck a day after the appearance of Koplik spots. This rash becomes more confluent as it spreads to involve the entire body (Figure 2). It typically lasts for 3 to 7 days, then fades in a similar pattern. The confluent nature of this rash and its spread from the face and neck to the entire body are characteristic of measles. Patients are highly contagious from 4 days before the onset of the rash to 4 days after.
COMPLICATIONS CAN BE SEVERE
Those at highest risk for measles complications are infants, children under age 5, adults over age 20, pregnant women, and immunosuppressed individuals.7
Pneumonia—either a primary measles pneumonia or a secondary viral or bacterial pneumonia—is the most common cause of death.8,9 Viruses complicating measles are typically adenovirus and herpes simplex virus. Bacteria causing secondary infection are usually Staphylococcus aureus and Streptococcus pneumoniae and, less commonly, gram-negative bacteria.
Laryngotracheobronchitis (croup) is the second most common cause of death, with bacteria and viruses similar to those causing measles-related pneumonia.
Otitis media is the most common complication of measles. Other respiratory complications include mastoiditis, pneumothorax, and mediastinal emphysema.
Acute measles encephalitis occurs in 1 measles case per 1,000 and often results in permanent brain damage. During the convalescent phase of the illness, fever again emerges, with the development of headaches, seizures, and altered consciousness.10
Subacute sclerosing panencephalitis is a rare fatal degenerative disease of the central nervous system caused by a persistent infection with a defective measles virus. The precise pathophysiology is unclear, but it is thought that mutations of the viral genome lead to altered cellular immunity.11 The condition typically occurs 7 to 10 years after the initial measles infection, particularly in those who developed measles before age 2. Clinical manifestations include behavioral disturbances, intellectual deterioration, and myoclonic seizures, slowly progressing to a vegetative state and death.12
Other complications of measles include diarrhea and stomatitis, which are associated with malnutrition in developing countries, and subclinical hepatitis, thrombocytopenia, appendicitis, ileocolitis, hypokalemia, and myocarditis.
During pregnancy, measles infection can be complicated by primary measles pneumonia and is associated with an increased risk of miscarriage and premature birth.13
Patients with a cell-mediated immunodeficiency who develop measles are particularly susceptible to fatal measles pneumonia and acute progressive encephalitis.14
ATYPICAL MEASLES IN THOSE WHO RECEIVED KILLED VACCINE
From 1963 to 1967, a killed measles vaccine was available in the United States. Those who received this vaccine are susceptible to an atypical form of measles when exposed to the virus,15 characterized by a 1- to 2-day prodrome, followed by the appearance of a maculopapular or petechial rash on the distal extremities that spreads centripetally. Patients develop high fever and edema of the hands and feet, and have a more prolonged course than with classic measles. It is believed not to be contagious.16
LABORATORY CONFIRMATION
Laboratory confirmation of measles is recommended for suspected cases. Because viral isolation is technically difficult and is not readily available in most laboratories, measles-specific immunoglobulin M antibody serologic testing is most commonly used. It is almost 100% sensitive when done 2 to 3 days after the onset of the rash.17
Measles RNA testing by real-time polymerase chain reaction to detect measles virus in the blood, throat, or urine is more specific and if available may be preferred over serologic testing.18
SUPPORTIVE MANAGEMENT AND VITAMIN A SUPPLEMENTATION
No specific antiviral therapy for measles is available. Management involves supportive measures and monitoring for secondary bacterial complications.
The World Health Organization and the American Academy of Pediatrics recommend vitamin A supplementation for all children with acute measles.19 In developing countries, it has been shown to reduce rates of morbidity and death in measles-infected children.20 In the United States, children with measles have been found to have low serum levels of vitamin A, with lower levels associated with more severe disease.
VACCINATION RECOMMENDATIONS
The only measles vaccine available in the United States is a live further-attenuated strain prepared in chick embryo cell culture and combined with mumps and rubella vaccine (MMR) or with measles, mumps, rubella, and varicella vaccine (MMRV).
Healthy children. Two doses of measles vaccine are recommended, as a single dose is associated with a 5% failure rate. The recommended schedule is:
- First dose at age 12 to 15 months
- Second dose at the time of school entry (ages 4 to 6), or at any time at least 28 days after the first dose.19
More than 99% of children who receive two doses of vaccine according to this schedule develop serologic evidence of measles immunity. Vaccination provides long-term immunity, and many epidemiologic studies have documented that waning immunity after vaccination occurs only very rarely.21
All school-age children, including elementary, middle, and high school students, who received only one dose of measles vaccine should receive the second dose.
Adults born in 1957 or later should receive at least one dose of measles vaccine unless they have other acceptable evidence of immunity, such as:4
- Documentation of age-appropriate live measles vaccine, ie, one dose of vaccine for adults not at high risk, or two doses for those at high risk (see below)
- Laboratory evidence of immunity (ie, measles immunoglobulin G in serum)
- Laboratory confirmation of disease.
Adults born before 1957 can be considered to be immune to measles, although MMR vaccine can be administered in those without contraindications.
Adults at increased risk of exposure or transmission of measles and who do not have evidence of immunity should receive two doses of MMR vaccine, given at least 28 days apart. This high-risk group includes:
- Students attending college or other post-high school educational institution
- Healthcare personnel
- International travelers.
During measles outbreaks, every effort should be made to ensure that those at high risk are vaccinated with two doses of MMR or have other acceptable evidence of immunity.
LIVE VACCINE IS SAFE FOR MOST PEOPLE
Mild side effects. A transient fever, which may be accompanied by a discrete or confluent rash, occurs in 5% to 15% of recipients 5 to 12 days after vaccination.
Transmission does not occur. People who have been newly vaccinated do not transmit the virus to susceptible contacts, even if they develop a vaccine-associated rash. The vaccine can safely be given to close contacts of immunocompromised and other susceptible people.
Egg allergy not a concern. Measles vaccine is produced in chick embryo cell culture but has been shown to be safe for people with egg allergy and is recommended without the need for egg allergy testing.19
Autism link debunked. No scientific evidence shows that the risk of autism is higher in children who receive MMR vaccine than in those who do not. In 2001, an Institute of Medicine report rejected a causal relationship between MMR vaccine and autism spectrum disorders.22
CONTRAINDICATIONS
Measles vaccine is contraindicated for:
- Patients who have cell-mediated immune deficiencies, except human immunodeficiency virus (HIV) infection
- Pregnant women
- Those who have had a severe allergic reaction to a vaccine component in the past
- Those with moderate or severe acute illness
- Those who have recently received immunoglobulin products.
People with HIV infection who are severely immunosuppressed should not receive live measles vaccine. However, because of the risk of severe measles in HIV-infected patients and because the vaccine has been shown to be safe for patients with HIV without severe immunosuppression, the vaccine is recommended for those with asymptomatic or mildly symptomatic HIV infection who do not have evidence of severe immunosuppression (ie, CD4 lymphocytes < 15% or < 200 cells/µL).3,4
INFECTION CONTROL AND PREVENTION
Healthcare workers should maintain a high index of suspicion for measles and implement isolation procedures promptly in patients with a febrile illness, rash, and a history of travel abroad or contact with travelers from abroad.23 Suspected cases should be reported promptly to local health agencies to help limit spread.
Patients with measles should be placed in airborne isolation (eg, use of an N95 or higher level respirator and an airborne infection isolation room) for 4 days after the onset of the rash in a normal host and for the duration of the illness in an immunocompromised patient. Healthcare staff, regardless of their immunity status, should adhere to these precautions when entering the room of infected patients.
Immunization programs should be established to ensure that everyone who works or volunteers in healthcare facilities is protected against measles.4
Postexposure prophylaxis. Measles vaccination given to susceptible contacts within 72 hours of exposure may provide protection against infection and induces protection against subsequent measles exposures.24,25
Vaccination is the best intervention for susceptible contacts older than 12 months who do not have a contraindication to measles vaccination, and for those who have received only one dose of measles vaccine.
Passive immunization. Active immunization is the best strategy for controlling measles outbreaks. Passive immunization with intramuscularly or intravenously administered immunoglobulin given within 6 days of exposure can be used to prevent transmission or modify the clinical course of infection for susceptible contacts at high risk of developing severe or fatal measles. This includes people who are being treated with immunosuppressive agents, HIV-infected, pregnant, or younger than 1 year of age.
Measles continues to rear its head in the United States. Because it is so contagious, even the few cases introduced by travelers quickly spread to susceptible contacts. Life-threatening and severely disabling complications can occur, although this is rare. Widespread immunization and prompt recognition and isolation of contacts are key to controlling outbreaks.
This article reviews the epidemiology of measles, describes its distinctive clinical picture, and provides recommendations for infection control and prevention, including in immunosuppressed populations.
MEASLES IS SERIOUS AND HIGHLY CONTAGIOUS
Up to 90% of susceptible people develop measles after exposure, making it one of the most contagious of infections. The virus is transmitted by airborne spread when an infected person coughs or sneezes, or by direct contact with infectious droplets. The virus can remain infectious in the air or on a surface for up to 2 hours.1
Worldwide, an estimated 20 million people are infected with measles each year, and 146,000 die of complications. In 1980, before widespread vaccination, 2.6 million deaths were attributable to measles annually. In the United States before the introduction of measles vaccine in 1963, measles was a significant cause of disease and death: an estimated 3 to 4 million people were infected annually, although only about 549,000 were reported. There were 48,000 hospitalizations, 1,000 cases of permanent brain damage from measles encephalitis, and 495 deaths annually.2
Outbreaks still occur regularly
In 2000, measles was declared eliminated from the United States,3 but annual outbreaks have occurred since then as a result of cases imported from other countries and their subsequent transmission to unvaccinated people. From 2001 to 2012, a median of four outbreaks and 60 cases were reported annually to the US Centers for Disease Control and Prevention.4
In January 2015, a multistate measles outbreak originating in Disneyland in California was recognized. As of April 17, when the outbreak was declared over, 111 measles cases from seven states had been linked to this outbreak.5 Of the evaluable cases, 44% were in unvaccinated people and 38% were in those whose vaccination status was unknown or undocumented. The median age of patients was 21, and 20% required hospitalization.
This outbreak, as well as four other smaller US outbreaks the same year, underscores the transmissibility of the virus in populations containing only a small percentage of unvaccinated people.6
DISTINCTIVE CLINICAL PICTURE
The incubation period for measles infection is 7 to 21 days, with most cases becoming apparent 10 to 12 days after exposure. Measles should be suspected in a patient with the following clinical features whose history indicates susceptibility and exposure (ie, an unimmunized person with a history of exposure or travel):
Severe acute respiratory illness. Measles usually presents as an acute respiratory viral illness, which typically lasts 2 to 4 days. The illness involves high fevers, malaise, anorexia, and the “three Cs”: cough, coryza (rhinitis), and conjunctivitis. Patients usually appear sicker than those with more common viral illnesses.
Koplik spots, which are pathognomonic for measles, are seen in the first few days of illness. They are bluish-white, slightly raised lesions on an erythematous base on the buccal mucosa, usually opposite the first molar (Figure 1). Spots can also be seen on the soft palate, conjunctiva, and vaginal mucosa. Koplik spots usually disappear after a few days and often are not appreciable at the time of evaluation.
Discrete erythematous patches develop on the face and neck a day after the appearance of Koplik spots. This rash becomes more confluent as it spreads to involve the entire body (Figure 2). It typically lasts for 3 to 7 days, then fades in a similar pattern. The confluent nature of this rash and its spread from the face and neck to the entire body are characteristic of measles. Patients are highly contagious from 4 days before the onset of the rash to 4 days after.
COMPLICATIONS CAN BE SEVERE
Those at highest risk for measles complications are infants, children under age 5, adults over age 20, pregnant women, and immunosuppressed individuals.7
Pneumonia—either a primary measles pneumonia or a secondary viral or bacterial pneumonia—is the most common cause of death.8,9 Viruses complicating measles are typically adenovirus and herpes simplex virus. Bacteria causing secondary infection are usually Staphylococcus aureus and Streptococcus pneumoniae and, less commonly, gram-negative bacteria.
Laryngotracheobronchitis (croup) is the second most common cause of death, with bacteria and viruses similar to those causing measles-related pneumonia.
Otitis media is the most common complication of measles. Other respiratory complications include mastoiditis, pneumothorax, and mediastinal emphysema.
Acute measles encephalitis occurs in 1 measles case per 1,000 and often results in permanent brain damage. During the convalescent phase of the illness, fever again emerges, with the development of headaches, seizures, and altered consciousness.10
Subacute sclerosing panencephalitis is a rare fatal degenerative disease of the central nervous system caused by a persistent infection with a defective measles virus. The precise pathophysiology is unclear, but it is thought that mutations of the viral genome lead to altered cellular immunity.11 The condition typically occurs 7 to 10 years after the initial measles infection, particularly in those who developed measles before age 2. Clinical manifestations include behavioral disturbances, intellectual deterioration, and myoclonic seizures, slowly progressing to a vegetative state and death.12
Other complications of measles include diarrhea and stomatitis, which are associated with malnutrition in developing countries, and subclinical hepatitis, thrombocytopenia, appendicitis, ileocolitis, hypokalemia, and myocarditis.
During pregnancy, measles infection can be complicated by primary measles pneumonia and is associated with an increased risk of miscarriage and premature birth.13
Patients with a cell-mediated immunodeficiency who develop measles are particularly susceptible to fatal measles pneumonia and acute progressive encephalitis.14
ATYPICAL MEASLES IN THOSE WHO RECEIVED KILLED VACCINE
From 1963 to 1967, a killed measles vaccine was available in the United States. Those who received this vaccine are susceptible to an atypical form of measles when exposed to the virus,15 characterized by a 1- to 2-day prodrome, followed by the appearance of a maculopapular or petechial rash on the distal extremities that spreads centripetally. Patients develop high fever and edema of the hands and feet, and have a more prolonged course than with classic measles. It is believed not to be contagious.16
LABORATORY CONFIRMATION
Laboratory confirmation of measles is recommended for suspected cases. Because viral isolation is technically difficult and is not readily available in most laboratories, measles-specific immunoglobulin M antibody serologic testing is most commonly used. It is almost 100% sensitive when done 2 to 3 days after the onset of the rash.17
Measles RNA testing by real-time polymerase chain reaction to detect measles virus in the blood, throat, or urine is more specific and if available may be preferred over serologic testing.18
SUPPORTIVE MANAGEMENT AND VITAMIN A SUPPLEMENTATION
No specific antiviral therapy for measles is available. Management involves supportive measures and monitoring for secondary bacterial complications.
The World Health Organization and the American Academy of Pediatrics recommend vitamin A supplementation for all children with acute measles.19 In developing countries, it has been shown to reduce rates of morbidity and death in measles-infected children.20 In the United States, children with measles have been found to have low serum levels of vitamin A, with lower levels associated with more severe disease.
VACCINATION RECOMMENDATIONS
The only measles vaccine available in the United States is a live further-attenuated strain prepared in chick embryo cell culture and combined with mumps and rubella vaccine (MMR) or with measles, mumps, rubella, and varicella vaccine (MMRV).
Healthy children. Two doses of measles vaccine are recommended, as a single dose is associated with a 5% failure rate. The recommended schedule is:
- First dose at age 12 to 15 months
- Second dose at the time of school entry (ages 4 to 6), or at any time at least 28 days after the first dose.19
More than 99% of children who receive two doses of vaccine according to this schedule develop serologic evidence of measles immunity. Vaccination provides long-term immunity, and many epidemiologic studies have documented that waning immunity after vaccination occurs only very rarely.21
All school-age children, including elementary, middle, and high school students, who received only one dose of measles vaccine should receive the second dose.
Adults born in 1957 or later should receive at least one dose of measles vaccine unless they have other acceptable evidence of immunity, such as:4
- Documentation of age-appropriate live measles vaccine, ie, one dose of vaccine for adults not at high risk, or two doses for those at high risk (see below)
- Laboratory evidence of immunity (ie, measles immunoglobulin G in serum)
- Laboratory confirmation of disease.
Adults born before 1957 can be considered to be immune to measles, although MMR vaccine can be administered in those without contraindications.
Adults at increased risk of exposure or transmission of measles and who do not have evidence of immunity should receive two doses of MMR vaccine, given at least 28 days apart. This high-risk group includes:
- Students attending college or other post-high school educational institution
- Healthcare personnel
- International travelers.
During measles outbreaks, every effort should be made to ensure that those at high risk are vaccinated with two doses of MMR or have other acceptable evidence of immunity.
LIVE VACCINE IS SAFE FOR MOST PEOPLE
Mild side effects. A transient fever, which may be accompanied by a discrete or confluent rash, occurs in 5% to 15% of recipients 5 to 12 days after vaccination.
Transmission does not occur. People who have been newly vaccinated do not transmit the virus to susceptible contacts, even if they develop a vaccine-associated rash. The vaccine can safely be given to close contacts of immunocompromised and other susceptible people.
Egg allergy not a concern. Measles vaccine is produced in chick embryo cell culture but has been shown to be safe for people with egg allergy and is recommended without the need for egg allergy testing.19
Autism link debunked. No scientific evidence shows that the risk of autism is higher in children who receive MMR vaccine than in those who do not. In 2001, an Institute of Medicine report rejected a causal relationship between MMR vaccine and autism spectrum disorders.22
CONTRAINDICATIONS
Measles vaccine is contraindicated for:
- Patients who have cell-mediated immune deficiencies, except human immunodeficiency virus (HIV) infection
- Pregnant women
- Those who have had a severe allergic reaction to a vaccine component in the past
- Those with moderate or severe acute illness
- Those who have recently received immunoglobulin products.
People with HIV infection who are severely immunosuppressed should not receive live measles vaccine. However, because of the risk of severe measles in HIV-infected patients and because the vaccine has been shown to be safe for patients with HIV without severe immunosuppression, the vaccine is recommended for those with asymptomatic or mildly symptomatic HIV infection who do not have evidence of severe immunosuppression (ie, CD4 lymphocytes < 15% or < 200 cells/µL).3,4
INFECTION CONTROL AND PREVENTION
Healthcare workers should maintain a high index of suspicion for measles and implement isolation procedures promptly in patients with a febrile illness, rash, and a history of travel abroad or contact with travelers from abroad.23 Suspected cases should be reported promptly to local health agencies to help limit spread.
Patients with measles should be placed in airborne isolation (eg, use of an N95 or higher level respirator and an airborne infection isolation room) for 4 days after the onset of the rash in a normal host and for the duration of the illness in an immunocompromised patient. Healthcare staff, regardless of their immunity status, should adhere to these precautions when entering the room of infected patients.
Immunization programs should be established to ensure that everyone who works or volunteers in healthcare facilities is protected against measles.4
Postexposure prophylaxis. Measles vaccination given to susceptible contacts within 72 hours of exposure may provide protection against infection and induces protection against subsequent measles exposures.24,25
Vaccination is the best intervention for susceptible contacts older than 12 months who do not have a contraindication to measles vaccination, and for those who have received only one dose of measles vaccine.
Passive immunization. Active immunization is the best strategy for controlling measles outbreaks. Passive immunization with intramuscularly or intravenously administered immunoglobulin given within 6 days of exposure can be used to prevent transmission or modify the clinical course of infection for susceptible contacts at high risk of developing severe or fatal measles. This includes people who are being treated with immunosuppressive agents, HIV-infected, pregnant, or younger than 1 year of age.
- Stokes J Jr, Reilly CM, Buynak EB, Hilleman MR. Immunologic studies of measles. Am J Hyg 1961; 74:293–303.
- Bloch AB, Orenstein WA, Stetler HC, et al. Health impact of measles vaccination in the United States. Pediatrics 1985; 76:524–532.
- Katz SL, Hinman AR. Summary and conclusions: measles elimination meeting, 16-17 March 2000. J Infect Dis 2004; 189(suppl 1):S43–S47.
- McLean HQ, Fiebelkorn AP, Temte JL, Wallace GS; Centers for Disease Control and Prevention (CDC). Prevention of measles, rubella, congenital rubella syndrome, and mumps, 2013: summary recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2013; 62:1–34.
- Clemmons NS, Gastanaduy PA, Fiebelkorn AP, Redd SB, Wallace GS; Centers for Disease Control and Prevention (CDC). Measles—United States, January 4 - April 2, 2015. MMWR Morb Mortal Wkly Rep 2015; 64:373–376.
- Gay NJ. The theory of measles elimination: implications for the design of elimination strategies. J Infect Dis 2004; 189(suppl 1):S27–S35.
- Perry RT, Halsey NA. The clinical significance of measles: a review. J Infect Dis 2004; 189(suppl 1):S4–S16).
- Gremillion DH, Crawford GE. Measles pneumonia in young adults. An analysis of 106 cases. Am J Med 1981; 71:539–542.
- Quiambao BP, Gatchalian SR, Halonen P, et al. Coinfection is common in measles-associated pneumonia. Pediatr Infect Dis J 1998; 17:89–93.
- Johnson RT, Griffin D, Hirsch R, et al. Measles encephalomyelitis—clinical and immunologic studies. N Engl J Med 1984; 310:137–141.
- Garg RK. Subacute sclerosing panencephalitis. Postgrad Med J 2002; 78:63–70.
- Sever JL. Persistent measles infection of the central nervous system: subacute sclerosing panencephalitis. Rev Infect Dis 1983; 5:467–473.
- Atmar RL, Englund JA, Hammill H. Complications of measles during pregnancy. Clin Infect Dis 1992; 14:217–226.
- Kaplan LJ, Daum RS, Smaron M, McCarthy CA. Severe measles in immunocompromised patients. JAMA 1992; 267:1237–1241.
- Frey HM, Krugman S. Atypical measles syndrome: unusual hepatic, pulmonary, and immunologic aspects. Am J Med Sci 1981; 281:51–55.
- Fulginiti VA, Eller JJ, Downie AW, Kempe CH. Altered reactivity to measles virus. Atypical measles in children previously immunized with inactivated measles virus vaccines. JAMA 1967; 202:1075–1080.
- Bellini WJ, Helfand RF. The challenges and strategies for laboratory diagnosis of measles in an international setting. J Infect Dis 2003; 187(suppl 1):S283–S290.
- Riddell MA, Chibo D, Kelly HA, Catton MG, Birch CJ. Investigation of optimal specimen type and sampling time for detection of measles virus RNA during a measles epidemic. J Clin Microbiol 2001; 39:375–376.
- Measles. In: Pickering LK, Baker CJ, Kimberlin DW, Long SS, editors. Red Book: 2012 Report of the Committee on Infectious Diseases. 29th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2012:489-499.
- Huiming Y, Chaomin W, Meng M. Vitamin A for treating measles in children. Cochrane Database Syst Rev 2005; 4:CD001479.
- Markowitz LE, Preblud SR, Fine PE, Orenstein WA. Duration of live measles vaccine-induced immunity. Pediatr Infect Dis J 1990; 9:101–110.
- Stratton K, Gable A, Shetty P, McCormick M. Immunization safety review: measles-mumps-rubella vaccine and autism. Washington, DC: National Academy Press; 2001.
- Centers for Disease Control and Prevention (CDC). 2007 guideline for isolation precautions: preventing transmission of infectious agents in healthcare settings. www.cdc.gov/hicpac/2007ip/2007isolationprecautions.html. Accessed April 8, 2016.
- Berkovich S, Starr S. Use of live-measles-virus vaccine to abort an expected outbreak of measles within a closed population. N Engl J Med 1963; 269:75–77.
- Ruuskanen O, Salmi TT, Halonen P. Measles vaccination after exposure to natural measles. J Pediatr 1978; 93:43–46.
- Stokes J Jr, Reilly CM, Buynak EB, Hilleman MR. Immunologic studies of measles. Am J Hyg 1961; 74:293–303.
- Bloch AB, Orenstein WA, Stetler HC, et al. Health impact of measles vaccination in the United States. Pediatrics 1985; 76:524–532.
- Katz SL, Hinman AR. Summary and conclusions: measles elimination meeting, 16-17 March 2000. J Infect Dis 2004; 189(suppl 1):S43–S47.
- McLean HQ, Fiebelkorn AP, Temte JL, Wallace GS; Centers for Disease Control and Prevention (CDC). Prevention of measles, rubella, congenital rubella syndrome, and mumps, 2013: summary recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2013; 62:1–34.
- Clemmons NS, Gastanaduy PA, Fiebelkorn AP, Redd SB, Wallace GS; Centers for Disease Control and Prevention (CDC). Measles—United States, January 4 - April 2, 2015. MMWR Morb Mortal Wkly Rep 2015; 64:373–376.
- Gay NJ. The theory of measles elimination: implications for the design of elimination strategies. J Infect Dis 2004; 189(suppl 1):S27–S35.
- Perry RT, Halsey NA. The clinical significance of measles: a review. J Infect Dis 2004; 189(suppl 1):S4–S16).
- Gremillion DH, Crawford GE. Measles pneumonia in young adults. An analysis of 106 cases. Am J Med 1981; 71:539–542.
- Quiambao BP, Gatchalian SR, Halonen P, et al. Coinfection is common in measles-associated pneumonia. Pediatr Infect Dis J 1998; 17:89–93.
- Johnson RT, Griffin D, Hirsch R, et al. Measles encephalomyelitis—clinical and immunologic studies. N Engl J Med 1984; 310:137–141.
- Garg RK. Subacute sclerosing panencephalitis. Postgrad Med J 2002; 78:63–70.
- Sever JL. Persistent measles infection of the central nervous system: subacute sclerosing panencephalitis. Rev Infect Dis 1983; 5:467–473.
- Atmar RL, Englund JA, Hammill H. Complications of measles during pregnancy. Clin Infect Dis 1992; 14:217–226.
- Kaplan LJ, Daum RS, Smaron M, McCarthy CA. Severe measles in immunocompromised patients. JAMA 1992; 267:1237–1241.
- Frey HM, Krugman S. Atypical measles syndrome: unusual hepatic, pulmonary, and immunologic aspects. Am J Med Sci 1981; 281:51–55.
- Fulginiti VA, Eller JJ, Downie AW, Kempe CH. Altered reactivity to measles virus. Atypical measles in children previously immunized with inactivated measles virus vaccines. JAMA 1967; 202:1075–1080.
- Bellini WJ, Helfand RF. The challenges and strategies for laboratory diagnosis of measles in an international setting. J Infect Dis 2003; 187(suppl 1):S283–S290.
- Riddell MA, Chibo D, Kelly HA, Catton MG, Birch CJ. Investigation of optimal specimen type and sampling time for detection of measles virus RNA during a measles epidemic. J Clin Microbiol 2001; 39:375–376.
- Measles. In: Pickering LK, Baker CJ, Kimberlin DW, Long SS, editors. Red Book: 2012 Report of the Committee on Infectious Diseases. 29th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2012:489-499.
- Huiming Y, Chaomin W, Meng M. Vitamin A for treating measles in children. Cochrane Database Syst Rev 2005; 4:CD001479.
- Markowitz LE, Preblud SR, Fine PE, Orenstein WA. Duration of live measles vaccine-induced immunity. Pediatr Infect Dis J 1990; 9:101–110.
- Stratton K, Gable A, Shetty P, McCormick M. Immunization safety review: measles-mumps-rubella vaccine and autism. Washington, DC: National Academy Press; 2001.
- Centers for Disease Control and Prevention (CDC). 2007 guideline for isolation precautions: preventing transmission of infectious agents in healthcare settings. www.cdc.gov/hicpac/2007ip/2007isolationprecautions.html. Accessed April 8, 2016.
- Berkovich S, Starr S. Use of live-measles-virus vaccine to abort an expected outbreak of measles within a closed population. N Engl J Med 1963; 269:75–77.
- Ruuskanen O, Salmi TT, Halonen P. Measles vaccination after exposure to natural measles. J Pediatr 1978; 93:43–46.
KEY POINTS
- Patients with measles are usually very sick with high fever, cough, rhinitis, and conjunctivitis.
- Koplik spots—small bluish-white lesions on the buccal mucosa—are usually evident only in the first few days of illness. Soon after, a patchy red rash develops, starting with the face and neck, then spreading to the entire body.
- Measles can lead to pneumonia, encephalitis, brain damage, and death.
- Suspected cases should be isolated and susceptible contacts vaccinated or given immunoglobulin if at high risk of developing severe disease.
- The diagnosis should be confirmed by serologic testing with measles-specific immunoglobulin M antibody.
- Vaccination confers lifelong immunity and is recommended for all healthy children in two doses: the first at 12 to 15 months of age and the second at the time of school entry.

This is not an acute coronary syndrome
A 72-year-old woman presented to the emergency room with persistent substernal chest discomfort. The initial electrocardiogram (ECG) showed 2.0-mm ST-segment elevation in leads II, III, and aVF, and 1.0-mm ST-segment elevation in lead V6 (Figure 1). The index troponin T level was 1.5 ng/mL (reference range < 0.01). ST-elevation myocardial infarction protocols were activated, and she was taken for urgent catheterization.
Coronary angiography showed normal coronary arteries. However, intraprocedural left ventriculography identified circumferential midventricular and apical akinesis with compensatory basal hyperkinesis (Figure 2).
Further inquiry into the patient’s medical history revealed that she had been experiencing psychological distress brought on by the failure of businesses she owned.
Transthoracic echocardiography subsequently verified a depressed ejection fraction (30%) with prominent apical and midventricular wall akinesis, thus confirming the diagnosis of stress cardiomyopathy. She was discharged home on a low-dose beta-blocker and an angiotensin-converting enzyme inhibitor, and within 6 weeks her systolic function had completely returned to normal.
STRESS CARDIOMYOPATHY: CAUSES, DIAGNOSIS, PROGNOSIS
Stress cardiomyopathy—also called broken heart syndrome, stress-induced cardiomyopathy, takotsubo cardiomyopathy, and apical ballooning syndrome—is an increasingly recognized acquired cardiomyopathy that typically affects older postmenopausal women exposed to a triggering stressor such as severe medical illness, major surgery, or a psychologically stressful life event.1,2
Our patient’s acute presentation is a classic example of how stress cardiomyopathy can be indistinguishable from acute coronary syndrome. It has been estimated that 1% to 2% of patients presenting with an initial diagnosis of acute coronary syndrome actually have stress cardiomyopathy.2
Diagnostic criteria
The diagnosis of stress cardiomyopathy can be established when coronary angiography reveals nonobstructive coronary arteries in patients with abnormal ventricular wall motion identified on echocardiography or ventriculography, or both. These findings are part of the proposed diagnostic criteria2:
- Transient hypokinesis, akinesis, or dyskinesis of the left ventricle midsegments, with or without apical involvement; regional wall motion abnormalities extending beyond a single epicardial vascular distribution; usually, a psychological or physiologic stressor is present
- No obstructive coronary disease or no angiographic evidence of acute plaque rupture
- New abnormalities on ECG, or modest elevation in cardiac enzymes
- No evidence of pheochromocytoma or myocarditis.2
Other characteristics that help to differentiate stress cardiomyopathy from acute coronary syndrome include a prolonged QTc interval, attenuation of the QRS amplitude, and a decreased troponin-ejection fraction product.3–5
Prognosis
The prognosis is generally excellent, with most patients achieving full recovery of myocardial function within several weeks.2 However, in the acute setting, there are relatively high rates of acute heart failure (44% to 46%), left ventricular outflow tract obstruction (19%), and unstable ventricular arrhythmias (3.4%), including torsades de pointes.1,2,6,7
Stress cardiomyopathy recurs in approximately 11% of patients within 4 years.8 Death is considered a rare complication but has occurred in as many as 8% of reported cases.1
- Bybee KA, Kara T, Prasad A, et al. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann Intern Med 2004; 141:858–865.
- Prasad A, Lerman A, Rihal CS. Apical ballooning syndrome (takotsubo or stress cardiomyopathy): a mimic of acute myocardial infarction. Am Heart J 2008; 155:408–417.
- Bennett J, Ferdinande B, Kayaert P, et al. Time course of electrocardiographic changes in transient left ventricular ballooning syndrome. Int J Cardiol 2013; 169:276–280.
- Madias JE. Transient attenuation of the amplitude of the QRS complexes in the diagnosis of takotsubo syndrome. Eur Heart J Acute Cardiovasc Care 2014; 3:28–36.
- Nascimento FO, Yang S, Larrauri-Reyes M, et al. Usefulness of the troponin-ejection fraction product to differentiate stress cardiomyopathy from ST-segment elevation myocardial infarction. Am J Cardiol 2014; 113:429–433.
- De Backer O, Debonnaire P, Gevaert S, Missault L, Gheeraert P, Muyldermans L. Prevalence, associated factors and management implications of left ventricular outflow tract obstruction in takotsubo cardiomyopathy: a two-year, two-center experience. BMC Cardiovasc Disord 2014; 14:147.
- Syed FF, Asirvatham SJ, Francis J. Arrhythmia occurrence with takotsubo cardiomyopathy: a literature review. Europace 2011; 13:780–788.
- Elesber AA, Prasad A, Lennon RJ, Wright RS, Lerman A, Rihal CS. Four-year recurrence rate and prognosis of the apical ballooning syndrome. J Am Coll Cardiol 2007; 50:448–452.
A 72-year-old woman presented to the emergency room with persistent substernal chest discomfort. The initial electrocardiogram (ECG) showed 2.0-mm ST-segment elevation in leads II, III, and aVF, and 1.0-mm ST-segment elevation in lead V6 (Figure 1). The index troponin T level was 1.5 ng/mL (reference range < 0.01). ST-elevation myocardial infarction protocols were activated, and she was taken for urgent catheterization.
Coronary angiography showed normal coronary arteries. However, intraprocedural left ventriculography identified circumferential midventricular and apical akinesis with compensatory basal hyperkinesis (Figure 2).
Further inquiry into the patient’s medical history revealed that she had been experiencing psychological distress brought on by the failure of businesses she owned.
Transthoracic echocardiography subsequently verified a depressed ejection fraction (30%) with prominent apical and midventricular wall akinesis, thus confirming the diagnosis of stress cardiomyopathy. She was discharged home on a low-dose beta-blocker and an angiotensin-converting enzyme inhibitor, and within 6 weeks her systolic function had completely returned to normal.
STRESS CARDIOMYOPATHY: CAUSES, DIAGNOSIS, PROGNOSIS
Stress cardiomyopathy—also called broken heart syndrome, stress-induced cardiomyopathy, takotsubo cardiomyopathy, and apical ballooning syndrome—is an increasingly recognized acquired cardiomyopathy that typically affects older postmenopausal women exposed to a triggering stressor such as severe medical illness, major surgery, or a psychologically stressful life event.1,2
Our patient’s acute presentation is a classic example of how stress cardiomyopathy can be indistinguishable from acute coronary syndrome. It has been estimated that 1% to 2% of patients presenting with an initial diagnosis of acute coronary syndrome actually have stress cardiomyopathy.2
Diagnostic criteria
The diagnosis of stress cardiomyopathy can be established when coronary angiography reveals nonobstructive coronary arteries in patients with abnormal ventricular wall motion identified on echocardiography or ventriculography, or both. These findings are part of the proposed diagnostic criteria2:
- Transient hypokinesis, akinesis, or dyskinesis of the left ventricle midsegments, with or without apical involvement; regional wall motion abnormalities extending beyond a single epicardial vascular distribution; usually, a psychological or physiologic stressor is present
- No obstructive coronary disease or no angiographic evidence of acute plaque rupture
- New abnormalities on ECG, or modest elevation in cardiac enzymes
- No evidence of pheochromocytoma or myocarditis.2
Other characteristics that help to differentiate stress cardiomyopathy from acute coronary syndrome include a prolonged QTc interval, attenuation of the QRS amplitude, and a decreased troponin-ejection fraction product.3–5
Prognosis
The prognosis is generally excellent, with most patients achieving full recovery of myocardial function within several weeks.2 However, in the acute setting, there are relatively high rates of acute heart failure (44% to 46%), left ventricular outflow tract obstruction (19%), and unstable ventricular arrhythmias (3.4%), including torsades de pointes.1,2,6,7
Stress cardiomyopathy recurs in approximately 11% of patients within 4 years.8 Death is considered a rare complication but has occurred in as many as 8% of reported cases.1
A 72-year-old woman presented to the emergency room with persistent substernal chest discomfort. The initial electrocardiogram (ECG) showed 2.0-mm ST-segment elevation in leads II, III, and aVF, and 1.0-mm ST-segment elevation in lead V6 (Figure 1). The index troponin T level was 1.5 ng/mL (reference range < 0.01). ST-elevation myocardial infarction protocols were activated, and she was taken for urgent catheterization.
Coronary angiography showed normal coronary arteries. However, intraprocedural left ventriculography identified circumferential midventricular and apical akinesis with compensatory basal hyperkinesis (Figure 2).
Further inquiry into the patient’s medical history revealed that she had been experiencing psychological distress brought on by the failure of businesses she owned.
Transthoracic echocardiography subsequently verified a depressed ejection fraction (30%) with prominent apical and midventricular wall akinesis, thus confirming the diagnosis of stress cardiomyopathy. She was discharged home on a low-dose beta-blocker and an angiotensin-converting enzyme inhibitor, and within 6 weeks her systolic function had completely returned to normal.
STRESS CARDIOMYOPATHY: CAUSES, DIAGNOSIS, PROGNOSIS
Stress cardiomyopathy—also called broken heart syndrome, stress-induced cardiomyopathy, takotsubo cardiomyopathy, and apical ballooning syndrome—is an increasingly recognized acquired cardiomyopathy that typically affects older postmenopausal women exposed to a triggering stressor such as severe medical illness, major surgery, or a psychologically stressful life event.1,2
Our patient’s acute presentation is a classic example of how stress cardiomyopathy can be indistinguishable from acute coronary syndrome. It has been estimated that 1% to 2% of patients presenting with an initial diagnosis of acute coronary syndrome actually have stress cardiomyopathy.2
Diagnostic criteria
The diagnosis of stress cardiomyopathy can be established when coronary angiography reveals nonobstructive coronary arteries in patients with abnormal ventricular wall motion identified on echocardiography or ventriculography, or both. These findings are part of the proposed diagnostic criteria2:
- Transient hypokinesis, akinesis, or dyskinesis of the left ventricle midsegments, with or without apical involvement; regional wall motion abnormalities extending beyond a single epicardial vascular distribution; usually, a psychological or physiologic stressor is present
- No obstructive coronary disease or no angiographic evidence of acute plaque rupture
- New abnormalities on ECG, or modest elevation in cardiac enzymes
- No evidence of pheochromocytoma or myocarditis.2
Other characteristics that help to differentiate stress cardiomyopathy from acute coronary syndrome include a prolonged QTc interval, attenuation of the QRS amplitude, and a decreased troponin-ejection fraction product.3–5
Prognosis
The prognosis is generally excellent, with most patients achieving full recovery of myocardial function within several weeks.2 However, in the acute setting, there are relatively high rates of acute heart failure (44% to 46%), left ventricular outflow tract obstruction (19%), and unstable ventricular arrhythmias (3.4%), including torsades de pointes.1,2,6,7
Stress cardiomyopathy recurs in approximately 11% of patients within 4 years.8 Death is considered a rare complication but has occurred in as many as 8% of reported cases.1
- Bybee KA, Kara T, Prasad A, et al. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann Intern Med 2004; 141:858–865.
- Prasad A, Lerman A, Rihal CS. Apical ballooning syndrome (takotsubo or stress cardiomyopathy): a mimic of acute myocardial infarction. Am Heart J 2008; 155:408–417.
- Bennett J, Ferdinande B, Kayaert P, et al. Time course of electrocardiographic changes in transient left ventricular ballooning syndrome. Int J Cardiol 2013; 169:276–280.
- Madias JE. Transient attenuation of the amplitude of the QRS complexes in the diagnosis of takotsubo syndrome. Eur Heart J Acute Cardiovasc Care 2014; 3:28–36.
- Nascimento FO, Yang S, Larrauri-Reyes M, et al. Usefulness of the troponin-ejection fraction product to differentiate stress cardiomyopathy from ST-segment elevation myocardial infarction. Am J Cardiol 2014; 113:429–433.
- De Backer O, Debonnaire P, Gevaert S, Missault L, Gheeraert P, Muyldermans L. Prevalence, associated factors and management implications of left ventricular outflow tract obstruction in takotsubo cardiomyopathy: a two-year, two-center experience. BMC Cardiovasc Disord 2014; 14:147.
- Syed FF, Asirvatham SJ, Francis J. Arrhythmia occurrence with takotsubo cardiomyopathy: a literature review. Europace 2011; 13:780–788.
- Elesber AA, Prasad A, Lennon RJ, Wright RS, Lerman A, Rihal CS. Four-year recurrence rate and prognosis of the apical ballooning syndrome. J Am Coll Cardiol 2007; 50:448–452.
- Bybee KA, Kara T, Prasad A, et al. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann Intern Med 2004; 141:858–865.
- Prasad A, Lerman A, Rihal CS. Apical ballooning syndrome (takotsubo or stress cardiomyopathy): a mimic of acute myocardial infarction. Am Heart J 2008; 155:408–417.
- Bennett J, Ferdinande B, Kayaert P, et al. Time course of electrocardiographic changes in transient left ventricular ballooning syndrome. Int J Cardiol 2013; 169:276–280.
- Madias JE. Transient attenuation of the amplitude of the QRS complexes in the diagnosis of takotsubo syndrome. Eur Heart J Acute Cardiovasc Care 2014; 3:28–36.
- Nascimento FO, Yang S, Larrauri-Reyes M, et al. Usefulness of the troponin-ejection fraction product to differentiate stress cardiomyopathy from ST-segment elevation myocardial infarction. Am J Cardiol 2014; 113:429–433.
- De Backer O, Debonnaire P, Gevaert S, Missault L, Gheeraert P, Muyldermans L. Prevalence, associated factors and management implications of left ventricular outflow tract obstruction in takotsubo cardiomyopathy: a two-year, two-center experience. BMC Cardiovasc Disord 2014; 14:147.
- Syed FF, Asirvatham SJ, Francis J. Arrhythmia occurrence with takotsubo cardiomyopathy: a literature review. Europace 2011; 13:780–788.
- Elesber AA, Prasad A, Lennon RJ, Wright RS, Lerman A, Rihal CS. Four-year recurrence rate and prognosis of the apical ballooning syndrome. J Am Coll Cardiol 2007; 50:448–452.