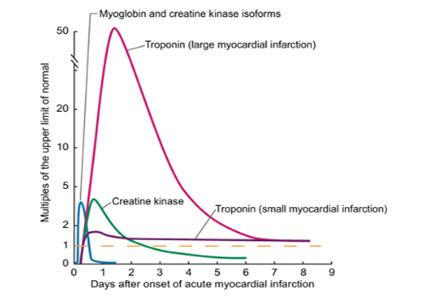
User login
In reply: Sleep apnea ABCs
In Reply: We thank Dr. Abouda for underscoring the role of arousals in the pathophysiology of obstructive sleep apnea (OSA). Although the focus of the referenced article was to provide a general overview of the epidemiology, diagnostic testing, and cardiovascular ramifications of untreated OSA and not a detailed summary of the underlying pathophysiology, we welcome the comments from Dr. Abouda to highlight the importance of cortical or microarousals in OSA.
Whether cortical arousal during sleep is bad or good is controversial. During the development of the American Academy of Sleep Medicine respiratory event guidelines, the assignment of detriment or benefit to the arousal when considering defining and scoring of a hypopnea event was a topic of much discussion.1,2 Supporters of including arousal in the hypopnea definition cite data that sleep fragmentation without attendant hypoxia is associated with symptoms such as excessive daytime somnolence, which is recognized to be effectively addressed with OSA treatment.3,4 Moreover, experimental data indicate that arousals lead to activation of the sympathetic nervous system.5 On the other hand, those who question the inclusion of cortical arousal in the hypopnea definition cite large-scale epidemiologic studies that have failed to find a significantly increased cardiovascular risk in relation to increasing arousal index, as well as the enhanced potential to introduce measurement variability.1
The effects of cortical arousals as a purported source of sympathetic activation may operate in concert with hypoxic influences, the latter resulting in sustained increases in blood pressure in both animal models and human studies.6,7 Gottlieb et al8 examined the effect of supplemental oxygen vs continuous positive airway pressure (CPAP) on 24-hour mean arterial pressure in a multicenter randomized controlled trial. Although CPAP reduced blood pressure, as expected, the somewhat unanticipated finding that supplemental oxygen did not suggests that other factors such as hypercapnia and cortical arousals with attendant sympathetic activation may represent potential culprits. Along these lines, in patients with OSA and increased loop gain, benefit in response to sedative hypnotics has been shown to reduce ventilatory instability through an increase in arousal threshold.9 A genetic predisposition may influence the intensity of cortical arousals and accompanying cardiovascular influences that appear to be consistent within individuals but that are heterogeneous within populations.10
Few studies have identified increased cortical arousals as a cardiovascular risk factor. In the Cleveland Family Study, an elevated arousal index was associated with hypertension, but respiratory event-specific arousals was not specifically examined.11 Not only have large-scale epidemiologic studies failed to identify an association between arousal index and cardiovascular outcomes, existing data appear to support the contrary. For example, the extent of incident white matter disease identified on brain magnetic resonance imaging was inversely related to the arousal index in a subset of participants of the Sleep Heart Health Study, a large population-based study focused on sleep and cardiovascular outcomes.12 Furthermore, elevated arousal indices in women were associated with reduced incidence of stroke in the Sleep Heart Health Study.13 These data suggest that arousals may represent beneficial, protective biomarkers reflecting truncation of respiratory events translating into reduced duration of hypoxic exposure and decreased work of breathing.
Needed is further investigation dedicated to understanding the impact of cortical arousals on health outcomes in population-based studies and elucidating the mechanistic role of cortical arousals in the autonomic nervous system physiology in various subtypes of sleep-disordered breathing (eg, obstructive vs central sleep apnea) as well as periodic limb movements.
As the upper Airway is central to the pathophysiology of OSA leading to compromise in Breathing and Circulatory or Cardiovascular ramifications, we think it logical that the “A” in ABCs should stand for “airway.” Hopefully, future research will allow us to better understand the associated benefit vs detriment of cortical arousals as they pertain to subgroup susceptibilities and enhance our ability to tailor a personalized medicine approach to the treatment of sleep disorders.
- Berry RB, Budhiraja R, Gottlieb DJ, et al. Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med 2012; 8:597–619.
- Ruehland WR, Rochford PD, O’Donoghue FJ, Pierce RJ, Singh P, Thornton AT. The new AASM criteria for scoring hypopneas: impact on the apnea hypopnea index. Sleep 2009; 32:150-157.
- Guilleminault C, Stoohs R, Clerk A, Cetel M, Maistros P. A cause of excessive daytime sleepiness. The upper airway resistance syndrome. Chest 1993; 104:781–787.
- Bonnet MH, Doghramji K, Roehrs T, et al. The scoring of arousal in sleep: reliability, validity, and alternatives. J Clin Sleep Med 2007; 3:133–145.
- Loredo JS, Ziegler MG, Ancoli-Israel S, Clausen JL, Dimsdale JE. Relationship of arousals from sleep to sympathetic nervous system activity and BP in obstructive sleep apnea. Chest J 1999; 116:655–659.
- Fletcher EC, Lesske J, Culman J, Miller CC, Unger T. Sympathetic denervation blocks blood pressure elevation in episodic hypoxia. Hypertension 1992; 20:612–619.
- Tamisier R, Pépin JL, Rémy J, et al. 14 nights of intermittent hypoxia elevate daytime blood pressure and sympathetic activity in healthy humans. Eur Respir J 2011; 37:119–128.
- Gottlieb DJ, Punjabi NM, Mehra R, et al. CPAP versus oxygen in obstructive sleep apnea. N Engl J Med 2014; 370:2276–2285.
- Eckert DJ, Owens RL, Kehlmann GB, et al. Eszopiclone increases the respiratory arousal threshold and lowers the apnoea/hypopnoea index in obstructive sleep apnoea patients with a low arousal threshold. Clin Sci Lond Engl 1979. 2011; 120:505–514.
- Azarbarzin A, Ostrowski M, Hanly P, Younes M. Relationship between arousal intensity and heart rate response to arousal. Sleep 2014; 37:645–653.
- Sulit L, Storfer-Isser A, Kirchner HL, Redline S. Differences in polysomnography predictors for hypertension and impaired glucose tolerance. Sleep 2006; 29:777–783.
- Ding J, Nieto FJ, Beauchamp NJ, et al. Sleep-disordered breathing and white matter disease in the brainstem in older adults. Sleep 2004; 27:474–479.
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the Sleep Heart Health Study. Am J Respir Crit Care Med 2010; 182:269–277.
In Reply: We thank Dr. Abouda for underscoring the role of arousals in the pathophysiology of obstructive sleep apnea (OSA). Although the focus of the referenced article was to provide a general overview of the epidemiology, diagnostic testing, and cardiovascular ramifications of untreated OSA and not a detailed summary of the underlying pathophysiology, we welcome the comments from Dr. Abouda to highlight the importance of cortical or microarousals in OSA.
Whether cortical arousal during sleep is bad or good is controversial. During the development of the American Academy of Sleep Medicine respiratory event guidelines, the assignment of detriment or benefit to the arousal when considering defining and scoring of a hypopnea event was a topic of much discussion.1,2 Supporters of including arousal in the hypopnea definition cite data that sleep fragmentation without attendant hypoxia is associated with symptoms such as excessive daytime somnolence, which is recognized to be effectively addressed with OSA treatment.3,4 Moreover, experimental data indicate that arousals lead to activation of the sympathetic nervous system.5 On the other hand, those who question the inclusion of cortical arousal in the hypopnea definition cite large-scale epidemiologic studies that have failed to find a significantly increased cardiovascular risk in relation to increasing arousal index, as well as the enhanced potential to introduce measurement variability.1
The effects of cortical arousals as a purported source of sympathetic activation may operate in concert with hypoxic influences, the latter resulting in sustained increases in blood pressure in both animal models and human studies.6,7 Gottlieb et al8 examined the effect of supplemental oxygen vs continuous positive airway pressure (CPAP) on 24-hour mean arterial pressure in a multicenter randomized controlled trial. Although CPAP reduced blood pressure, as expected, the somewhat unanticipated finding that supplemental oxygen did not suggests that other factors such as hypercapnia and cortical arousals with attendant sympathetic activation may represent potential culprits. Along these lines, in patients with OSA and increased loop gain, benefit in response to sedative hypnotics has been shown to reduce ventilatory instability through an increase in arousal threshold.9 A genetic predisposition may influence the intensity of cortical arousals and accompanying cardiovascular influences that appear to be consistent within individuals but that are heterogeneous within populations.10
Few studies have identified increased cortical arousals as a cardiovascular risk factor. In the Cleveland Family Study, an elevated arousal index was associated with hypertension, but respiratory event-specific arousals was not specifically examined.11 Not only have large-scale epidemiologic studies failed to identify an association between arousal index and cardiovascular outcomes, existing data appear to support the contrary. For example, the extent of incident white matter disease identified on brain magnetic resonance imaging was inversely related to the arousal index in a subset of participants of the Sleep Heart Health Study, a large population-based study focused on sleep and cardiovascular outcomes.12 Furthermore, elevated arousal indices in women were associated with reduced incidence of stroke in the Sleep Heart Health Study.13 These data suggest that arousals may represent beneficial, protective biomarkers reflecting truncation of respiratory events translating into reduced duration of hypoxic exposure and decreased work of breathing.
Needed is further investigation dedicated to understanding the impact of cortical arousals on health outcomes in population-based studies and elucidating the mechanistic role of cortical arousals in the autonomic nervous system physiology in various subtypes of sleep-disordered breathing (eg, obstructive vs central sleep apnea) as well as periodic limb movements.
As the upper Airway is central to the pathophysiology of OSA leading to compromise in Breathing and Circulatory or Cardiovascular ramifications, we think it logical that the “A” in ABCs should stand for “airway.” Hopefully, future research will allow us to better understand the associated benefit vs detriment of cortical arousals as they pertain to subgroup susceptibilities and enhance our ability to tailor a personalized medicine approach to the treatment of sleep disorders.
In Reply: We thank Dr. Abouda for underscoring the role of arousals in the pathophysiology of obstructive sleep apnea (OSA). Although the focus of the referenced article was to provide a general overview of the epidemiology, diagnostic testing, and cardiovascular ramifications of untreated OSA and not a detailed summary of the underlying pathophysiology, we welcome the comments from Dr. Abouda to highlight the importance of cortical or microarousals in OSA.
Whether cortical arousal during sleep is bad or good is controversial. During the development of the American Academy of Sleep Medicine respiratory event guidelines, the assignment of detriment or benefit to the arousal when considering defining and scoring of a hypopnea event was a topic of much discussion.1,2 Supporters of including arousal in the hypopnea definition cite data that sleep fragmentation without attendant hypoxia is associated with symptoms such as excessive daytime somnolence, which is recognized to be effectively addressed with OSA treatment.3,4 Moreover, experimental data indicate that arousals lead to activation of the sympathetic nervous system.5 On the other hand, those who question the inclusion of cortical arousal in the hypopnea definition cite large-scale epidemiologic studies that have failed to find a significantly increased cardiovascular risk in relation to increasing arousal index, as well as the enhanced potential to introduce measurement variability.1
The effects of cortical arousals as a purported source of sympathetic activation may operate in concert with hypoxic influences, the latter resulting in sustained increases in blood pressure in both animal models and human studies.6,7 Gottlieb et al8 examined the effect of supplemental oxygen vs continuous positive airway pressure (CPAP) on 24-hour mean arterial pressure in a multicenter randomized controlled trial. Although CPAP reduced blood pressure, as expected, the somewhat unanticipated finding that supplemental oxygen did not suggests that other factors such as hypercapnia and cortical arousals with attendant sympathetic activation may represent potential culprits. Along these lines, in patients with OSA and increased loop gain, benefit in response to sedative hypnotics has been shown to reduce ventilatory instability through an increase in arousal threshold.9 A genetic predisposition may influence the intensity of cortical arousals and accompanying cardiovascular influences that appear to be consistent within individuals but that are heterogeneous within populations.10
Few studies have identified increased cortical arousals as a cardiovascular risk factor. In the Cleveland Family Study, an elevated arousal index was associated with hypertension, but respiratory event-specific arousals was not specifically examined.11 Not only have large-scale epidemiologic studies failed to identify an association between arousal index and cardiovascular outcomes, existing data appear to support the contrary. For example, the extent of incident white matter disease identified on brain magnetic resonance imaging was inversely related to the arousal index in a subset of participants of the Sleep Heart Health Study, a large population-based study focused on sleep and cardiovascular outcomes.12 Furthermore, elevated arousal indices in women were associated with reduced incidence of stroke in the Sleep Heart Health Study.13 These data suggest that arousals may represent beneficial, protective biomarkers reflecting truncation of respiratory events translating into reduced duration of hypoxic exposure and decreased work of breathing.
Needed is further investigation dedicated to understanding the impact of cortical arousals on health outcomes in population-based studies and elucidating the mechanistic role of cortical arousals in the autonomic nervous system physiology in various subtypes of sleep-disordered breathing (eg, obstructive vs central sleep apnea) as well as periodic limb movements.
As the upper Airway is central to the pathophysiology of OSA leading to compromise in Breathing and Circulatory or Cardiovascular ramifications, we think it logical that the “A” in ABCs should stand for “airway.” Hopefully, future research will allow us to better understand the associated benefit vs detriment of cortical arousals as they pertain to subgroup susceptibilities and enhance our ability to tailor a personalized medicine approach to the treatment of sleep disorders.
- Berry RB, Budhiraja R, Gottlieb DJ, et al. Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med 2012; 8:597–619.
- Ruehland WR, Rochford PD, O’Donoghue FJ, Pierce RJ, Singh P, Thornton AT. The new AASM criteria for scoring hypopneas: impact on the apnea hypopnea index. Sleep 2009; 32:150-157.
- Guilleminault C, Stoohs R, Clerk A, Cetel M, Maistros P. A cause of excessive daytime sleepiness. The upper airway resistance syndrome. Chest 1993; 104:781–787.
- Bonnet MH, Doghramji K, Roehrs T, et al. The scoring of arousal in sleep: reliability, validity, and alternatives. J Clin Sleep Med 2007; 3:133–145.
- Loredo JS, Ziegler MG, Ancoli-Israel S, Clausen JL, Dimsdale JE. Relationship of arousals from sleep to sympathetic nervous system activity and BP in obstructive sleep apnea. Chest J 1999; 116:655–659.
- Fletcher EC, Lesske J, Culman J, Miller CC, Unger T. Sympathetic denervation blocks blood pressure elevation in episodic hypoxia. Hypertension 1992; 20:612–619.
- Tamisier R, Pépin JL, Rémy J, et al. 14 nights of intermittent hypoxia elevate daytime blood pressure and sympathetic activity in healthy humans. Eur Respir J 2011; 37:119–128.
- Gottlieb DJ, Punjabi NM, Mehra R, et al. CPAP versus oxygen in obstructive sleep apnea. N Engl J Med 2014; 370:2276–2285.
- Eckert DJ, Owens RL, Kehlmann GB, et al. Eszopiclone increases the respiratory arousal threshold and lowers the apnoea/hypopnoea index in obstructive sleep apnoea patients with a low arousal threshold. Clin Sci Lond Engl 1979. 2011; 120:505–514.
- Azarbarzin A, Ostrowski M, Hanly P, Younes M. Relationship between arousal intensity and heart rate response to arousal. Sleep 2014; 37:645–653.
- Sulit L, Storfer-Isser A, Kirchner HL, Redline S. Differences in polysomnography predictors for hypertension and impaired glucose tolerance. Sleep 2006; 29:777–783.
- Ding J, Nieto FJ, Beauchamp NJ, et al. Sleep-disordered breathing and white matter disease in the brainstem in older adults. Sleep 2004; 27:474–479.
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the Sleep Heart Health Study. Am J Respir Crit Care Med 2010; 182:269–277.
- Berry RB, Budhiraja R, Gottlieb DJ, et al. Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med 2012; 8:597–619.
- Ruehland WR, Rochford PD, O’Donoghue FJ, Pierce RJ, Singh P, Thornton AT. The new AASM criteria for scoring hypopneas: impact on the apnea hypopnea index. Sleep 2009; 32:150-157.
- Guilleminault C, Stoohs R, Clerk A, Cetel M, Maistros P. A cause of excessive daytime sleepiness. The upper airway resistance syndrome. Chest 1993; 104:781–787.
- Bonnet MH, Doghramji K, Roehrs T, et al. The scoring of arousal in sleep: reliability, validity, and alternatives. J Clin Sleep Med 2007; 3:133–145.
- Loredo JS, Ziegler MG, Ancoli-Israel S, Clausen JL, Dimsdale JE. Relationship of arousals from sleep to sympathetic nervous system activity and BP in obstructive sleep apnea. Chest J 1999; 116:655–659.
- Fletcher EC, Lesske J, Culman J, Miller CC, Unger T. Sympathetic denervation blocks blood pressure elevation in episodic hypoxia. Hypertension 1992; 20:612–619.
- Tamisier R, Pépin JL, Rémy J, et al. 14 nights of intermittent hypoxia elevate daytime blood pressure and sympathetic activity in healthy humans. Eur Respir J 2011; 37:119–128.
- Gottlieb DJ, Punjabi NM, Mehra R, et al. CPAP versus oxygen in obstructive sleep apnea. N Engl J Med 2014; 370:2276–2285.
- Eckert DJ, Owens RL, Kehlmann GB, et al. Eszopiclone increases the respiratory arousal threshold and lowers the apnoea/hypopnoea index in obstructive sleep apnoea patients with a low arousal threshold. Clin Sci Lond Engl 1979. 2011; 120:505–514.
- Azarbarzin A, Ostrowski M, Hanly P, Younes M. Relationship between arousal intensity and heart rate response to arousal. Sleep 2014; 37:645–653.
- Sulit L, Storfer-Isser A, Kirchner HL, Redline S. Differences in polysomnography predictors for hypertension and impaired glucose tolerance. Sleep 2006; 29:777–783.
- Ding J, Nieto FJ, Beauchamp NJ, et al. Sleep-disordered breathing and white matter disease in the brainstem in older adults. Sleep 2004; 27:474–479.
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the Sleep Heart Health Study. Am J Respir Crit Care Med 2010; 182:269–277.
Lactic acidosis: Clinical implications and management strategies
Physicians are paying more attention to serum lactate levels in hospitalized patients than in the past, especially with the advent of point-of-care testing. Elevated lactate levels are associated with tissue hypoxia and hypoperfusion but can also be found in a number of other conditions. Therefore, confusion can arise as to how to interpret elevated levels and subsequently manage these patients in a variety of settings.
In this review, we discuss the mechanisms underlying lactic acidosis, its prognostic implications, and its use as a therapeutic target in treating patients in septic shock and other serious disorders.
LACTATE IS A PRODUCT OF ANAEROBIC RESPIRATION
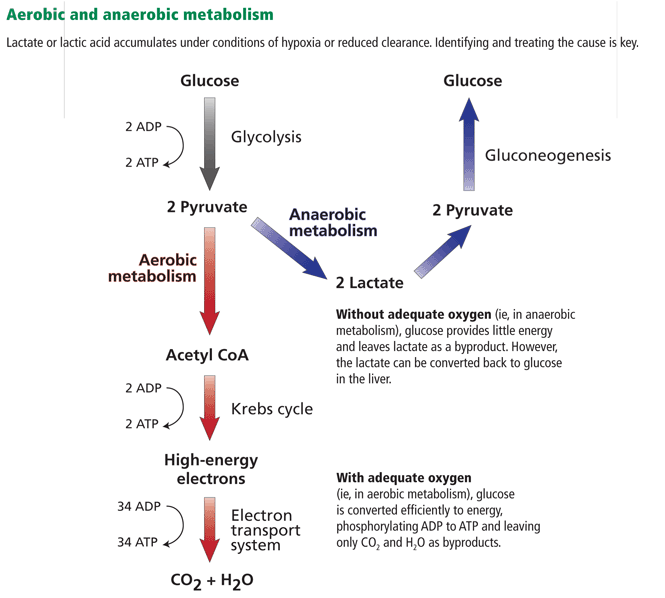
Lactate, or lactic acid, is produced from pyruvate as an end product of glycolysis under anaerobic conditions (Figure 1). It is produced in most tissues in the body, but primarily in skeletal muscle, brain, intestine, and red blood cells. During times of stress, lactate is also produced in the lungs, white blood cells, and splanchnic organs.
Most lactate in the blood is cleared by the liver, where it is the substrate for gluconeogenesis, and a small amount is cleared by the kidneys.1,2 The entire pathway by which lactate is produced and converted back to glucose is called the Cori cycle.
NORMAL LEVELS ARE LESS THAN ABOUT 2.0 MMOL/L
In this review, we will present lactate levels in the SI units of mmol/L (1 mmol/L = 9 mg/dL).
Basal lactate production is approximately 0.8 mmol/kg body weight/hour. The average normal arterial blood lactate level is approximately 0.620 mmol/L and the venous level is slightly higher at 0.997 mmol/L,3 but overall, arterial and venous lactate levels correlate well.
Normal lactate levels are less than 2 mmol/L,4 intermediate levels range from 2 to less than 4 mmol/L, and high levels are 4 mmol/L or higher.5
To minimize variations in measurement, blood samples should be drawn without a tourniquet into tubes containing fluoride, placed on ice, and processed quickly (ideally within 15 minutes).
INCREASED PRODUCTION, DECREASED CLEARANCE, OR BOTH
An elevated lactate level can be the result of increased production, decreased clearance, or both (as in liver dysfunction).
Type A lactic acidosis—due to hypoperfusion and hypoxia—occurs when there is a mismatch between oxygen delivery and consumption, with resultant anaerobic glycolysis.

The guidelines from the Surviving Sepsis Campaign6 emphasize using lactate levels to diagnose patients with sepsis-induced hypoperfusion. However, hyperlactatemia can indicate inadequate oxygen delivery due to any type of shock (Table 1).
Type B lactic acidosis—not due to hypoperfusion—occurs in a variety of conditions (Table 1), including liver disease, malignancy, use of certain medications (eg, metformin, epinephrine), total parenteral nutrition, human immunodeficiency virus infection, thiamine deficiency, mitochondrial myopathies, and congenital lactic acidosis.1–3,7 Yet other causes include trauma, excessive exercise, diabetic ketoacidosis, ethanol intoxication, dysfunction of the enzyme pyruvate dehydrogenase, and increased muscle degradation leading to increased production of pyruvate. In these latter scenarios, glucose metabolism exceeds the oxidation capacity of the mitochondria, and the rise in pyruvate concentration drives lactate production.8,9 Mitochondrial dysfunction and subsequent deficits in cellular oxygen use can also result in persistently high lactate levels.10
In some situations, patients with mildly elevated lactic acid levels in type B lactic acidosis can be monitored to ensure stability, rather than be treated aggressively.
HIGHER LEVELS AND LOWER CLEARANCE PREDICT DEATH
The higher the lactate level and the slower the rate of normalization (lactate clearance), the higher the risk of death.
Lactate levels and mortality rate
Shapiro et al11 showed that increases in lactate level are associated with proportional increases in the mortality rate. Mikkelsen et al12 showed that intermediate levels (2.0–3.9 mmol/L) and high levels (≥ 4 mmol/L) of serum lactate are associated with increased risk of death independent of organ failure and shock. Patients with mildly elevated and intermediate lactate levels and sepsis have higher rates of in-hospital and 30-day mortality, which correlate with the baseline lactate level.13
In a post hoc analysis of a randomized controlled trial, patients with septic shock who presented to the emergency department with hypotension and a lactate level higher than 2 mmol/L had a significantly higher in-hospital mortality rate than those who presented with hypotension and a lactate level of 2 mmol/L or less (26% vs 9%, P < .0001).14 These data suggest that elevated lactate levels may have a significant prognostic role, independent of blood pressure.
Slower clearance
The prognostic implications of lactate clearance (reductions in lactate levels over time, as opposed to a single value in time), have also been evaluated.
Lactate clearance of at least 10% at 6 hours after presentation has been associated with a lower mortality rate than nonclearance (19% vs 60%) in patients with sepsis or septic shock with elevated levels.15–17 Similar findings have been reported in a general intensive care unit population,18 as well as a surgical intensive care population.sup>19
Puskarich et al20 have also shown that lactate normalization to less than 2 mmol/L during early sepsis resuscitation is the strongest predictor of survival (odds ratio [OR] 5.2), followed by lactate clearance of 50% (OR 4.0) within the first 6 hours of presentation. Not only is lactate clearance associated with improved outcomes, but a faster rate of clearance after initial presentation is also beneficial.15,16,18
Lactate clearance over a longer period (> 6 hours) has not been studied in patients with septic shock. However, in the general intensive care unit population, therapy guided by lactate clearance for the first 8 hours after presentation has shown a reduction in mortality rate.18 There are no data available on outcomes of lactate-directed therapy beyond 8 hours, but lactate concentration and lactate clearance at 24 hours correlate with the 28-day mortality rate.21
Cryptic shock
Cryptic shock describes a state in a subgroup of patients who have elevated lactate levels and global tissue hypoxia despite being normotensive or even hypertensive. These patients have a higher mortality rate independent of blood pressure. Jansen et al18 found that patients with a lactate level higher than 4 mmol/L and preserved blood pressure had a mortality rate of 15%, while those without shock or hyperlactatemia had a mortality rate of 2.5%. In addition, patients with an elevated lactate level in the absence of hypotension have mortality rates similar to those in patients with high lactate levels and hypotension refractory to fluid boluses, suggesting the presence of tissue hypoxia even in these normotensive patients.6
HOW TO APPROACH AN ELEVATED LACTATE LEVEL
An elevated lactate level should prompt an evaluation for causes of decreased oxygen delivery, due either to a systemic low-flow state (as a result of decreased cardiac output) or severe anemia, or to regionally decreased perfusion, (eg, limb or mesenteric ischemia). If tissue hypoxia is ruled out after an exhaustive workup, consideration should be given to causes of hyperlactatemia without concomitant tissue hypoxia (type B acidosis).
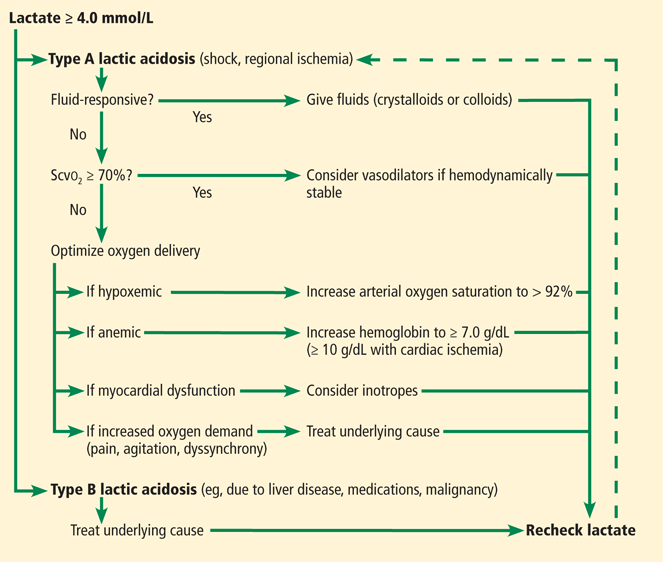
Treatment differs depending on the underlying mechanism of the lactate elevation; nevertheless, treatment is mostly related to optimizing oxygen delivery by giving fluids, packed red blood cells, and vasopressors or inotropic agents, or both (Figure 2). The specific treatment differs based on the shock state, but there are similarities that can guide the clinician.
FLUID SUPPORT
Giving fluids, with a goal of improving cardiac output, remains a cornerstone of initial therapy for most shock states.22,23
How much fluid?
Fluids should be given until the patient is no longer preload-dependent, although there is much debate about which assessment strategy should be used to determine if cardiac output will improve with more fluid (ie, fluid-responsiveness).24 In many cases, fluid resuscitation alone may be enough to restore hemodynamic stability, improve tissue perfusion, and reduce elevated lactate concentrations.25
The decision to give more fluids should not be made lightly, though, as a more positive fluid balance early in the course of septic shock and over 4 days has been associated with a higher mortality rate.26 Additionally, pushing fluids in patients with cardiogenic shock due to impaired left ventricular systolic function may lead to or worsen pulmonary edema. Therefore, the indiscriminate use of fluids should be avoided.
Which fluids?
Despite years of research, controversy persists about whether crystalloids or colloids are better for resuscitation. Randomized trials in heterogeneous intensive care unit patients have not detected differences in 28-day mortality rates between those allocated to crystalloids or 4% albumin27 and those allocated to crystalloids or hydroxyethyl starch.28
Hydroxyethyl starch may not be best. In a study of patients with severe sepsis, those randomized to receive hydroxyethyl starch had a higher 90-day mortality rate than patients randomized to crystalloids (51% vs 43%, P = .03).29 A sequential prospective before-and-after study did not detect a difference in the time to normalization (< 2.2 mmol/L) of lactate (P = .68) or cessation of vasopressors (P = .11) in patients with severe sepsis who received fluid resuscitation with crystalloids, gelatin, or hydroxyethyl starch. More patients who received hydroxyethyl starch in these studies developed acute kidney injury than those receiving crystalloids.28–30
Taken together, these data strongly suggest hydroxyethyl starch should not be used for fluid resuscitation in the intensive care unit.
Normal saline or albumin? Although some data suggest that albumin may be preferable to 0.9% sodium chloride in patients with severe sepsis,31,32 these analyses should be viewed as hypothesis-generating. There do not seem to be differences between fluid types in terms of subsequent serum lactate concentrations or achievement of lactate clearance goals.28–30 Until further studies are completed, both albumin and crystalloids are reasonable for resuscitation.
Caironi et al33 performed an open-label study comparing albumin replacement (with a goal serum albumin concentration of 3 g/dL) plus a crystalloid solution vs a crystalloid solution alone in patients with severe sepsis or septic shock. They detected no difference between the albumin and crystalloid groups in mortality rates at 28 days (31.8% vs 32.0%, P = .94) or 90 days (41.1% vs 43.6%, P = .29). However, patients in the albumin group had a shorter time to cessation of vasoactive agents (median 3 vs 4 days, P = .007) and lower cardiovascular Sequential Organ Failure Assessment subscores (median 1.20 vs 1.42, P = .03), and more frequently achieved a mean arterial pressure of at least 65 mm Hg within 6 hours of randomization (86.0% vs 82.5%, P = .04).
Although serum lactate levels were lower in the albumin group at baseline (1.7 mmol/L vs 1.8 mmol/L, P = .05), inspection of the data appears to show a similar daily lactate clearance rate between groups over the first 7 study days (although these data were not analyzed by the authors). Achievement of a lactate level lower than 2 mmol/L on the first day of therapy was not significantly different between groups (73.4% vs 72.5%, P = .11).33
In a post hoc subgroup analysis, patients with septic shock at baseline randomized to albumin had a lower 90-day mortality rate than patients randomized to crystalloid solutions (RR 0.87, 95% CI 0.77–0.99). There was no difference in the 90-day mortality rate in patients without septic shock (RR 1.13, 95% CI 0.92–1.39, P = .03 for heterogeneity).33
These data suggest that albumin replacement may not improve outcomes in patients with severe sepsis, but may have advantages in terms of hemodynamic variables (and potentially mortality) in patients with septic shock. The role of albumin replacement in patients with septic shock warrants further study.
VASOPRESSORS
Vasopressors, inotropes, or both should be given to patients who have signs of hypoperfusion (including elevated lactate levels) despite preload optimization or ongoing fluid administration. The most appropriate drug depends on the goal: vasopressors are used to increase systemic vascular resistance, while inotropes are used to improve cardiac output and oxygen delivery.
Blood pressure target
The Surviving Sepsis Campaign guidelines recommend a mean arterial blood pressure target of at least 65 mm Hg during initial resuscitation and when vasopressors are applied for patients with septic shock.22 This recommendation is based on small studies that did not show differences in serum lactate levels or regional blood flow when the mean arterial pressure was elevated above 65 mm Hg with norepinephrine.34,35 However, the campaign guidelines note that the mean arterial pressure goal must be individualized in order to achieve optimal perfusion.
A large, open-label trial36 detected no difference in 28-day mortality rates in patients with septic shock between those allocated to a mean arterial pressure goal of 80 to 85 mm Hg or 65 to 70 mm Hg (36.6% vs 34.0%, P = .57). Although lactate levels did not differ between groups, the incidence of new-onset atrial fibrillation was higher in the higher-target group (6.7% vs 2.8%, P = .02). Fewer patients with chronic hypertension needed renal replacement therapy in the higher pressure group, further emphasizing the need to individualize the mean arterial pressure goal for patients in shock.36
Which vasopressor agent?
Dopamine and norepinephrine have traditionally been the preferred initial vasopressors for patients with shock. Until recently there were few data to guide selection between the two, but this is changing.
In a 2010 study of 1,679 patients with shock requiring vasopressors, there was no difference in the 28-day mortality rate between patients randomized to dopamine or norepinephrine (53% vs 49%, P = .10).37 Patients allocated to dopamine, though, had a higher incidence of arrhythmias (24% vs 12%, P < .001) and more frequently required open-label norepinephrine (26% vs 20%, P < .001). Although lactate levels and the time to achievement of a mean arterial pressure of 65 mm Hg were similar between groups, patients allocated to norepinephrine had more vasopressor-free days through day 28.
An a priori-planned subgroup analysis evaluated the influence of the type of shock on patient outcome. Patients with cardiogenic shock randomized to dopamine had a higher mortality rate than those randomized to norepinephrine (P = .03). However, the overall effect of treatment did not differ among the shock subgroups (interaction P = .87), suggesting that the reported differences in mortality according to subgroup may be spurious.
In a 2012 meta-analysis of patients with septic shock, dopamine use was associated with a higher mortality rate than norepinephrine use.38
In light of these data, norepinephrine should be preferred over dopamine as the initial vasopressor in most types of shock.
Epinephrine does not offer an outcome advantage over norepinephrine and may be associated with a higher incidence of adverse events.39–42 Indeed, in a study of patients with septic shock, lactate concentrations on the first day after randomization were significantly higher in patients allocated to epinephrine than in patients allocated to norepinephrine plus dobutamine.39 Similar effects on lactate concentrations with epinephrine were seen in patients with various types of shock40 and in those with cardiogenic shock.42
These differences in lactate concentrations may be directly attributable to epinephrine. Epinephrine can increase lactate concentrations through glycolysis and pyruvate dehydrogenase activation by stimulation of sodium-potassium ATPase activity via beta-2 adrenergic receptors in skeletal muscles,43 as well as decrease splanchnic perfusion.42,44,45 These effects may preclude using lactate clearance as a resuscitation goal in patients receiving epinephrine. Epinephrine is likely best reserved for patients with refractory shock,22 particularly those in whom cardiac output is known to be low.
Phenylephrine, essentially a pure vasoconstrictor, should be avoided in low cardiac output states and is best reserved for patients who develop a tachyarrhythmia on norepinephrine.22
Vasopressin, also a pure vasoconstrictor that should be avoided in low cardiac output states, has been best studied in patients with vasodilatory shock. Although controversy exists on the mortality benefits of vasopressin in vasodilatory shock, it is a relatively safe drug with consistent norepinephrine-sparing effects when added to existing norepinephrine therapy.46,47 In patients with less severe septic shock, including those with low lactate concentrations, adding vasopressin to norepinephrine instead of continuing norepinephrine alone may confer a mortality advantage.48
OTHER MEASURES TO OPTIMIZE OXYGEN DELIVERY
In circulatory shock from any cause, tissue oxygen demand exceeds oxygen delivery. Once arterial oxygenation and hemoglobin levels (by packed red blood cell transfusion) have been optimized, cardiac output is the critical determinant of oxygen delivery. Cardiac output may be augmented by ensuring adequate preload (by fluid resuscitation) or by giving inotropes or vasodilators.
The optimal cardiac output is difficult to define, and the exact marker for determining when cardiac output should be augmented is unclear. A strategy of increasing cardiac output to predefined “supranormal” levels was not associated with a lower mortality rate.49 Therefore, the decision to augment cardiac output must be individualized and will likely vary in the same patient over time.23
A reasonable approach to determining when augmentation of cardiac output is necessary was proposed in a study by Rivers et al.50 In that study, in patients randomized to early goal-directed therapy, inotropes were recommended when the central venous oxygenation saturation (Scvo2) was below 70% despite adequate fluid resuscitation (central venous pressure ≥ 8 mm Hg) and hematocrits were higher than 30%.
When an inotrope is indicated to improve cardiac output, dobutamine is usually the preferred agent. Dobutamine has a shorter half-life (allowing for easier titration) and causes less hypotension (assuming preload has been optimized) than phosphodiesterase type III inhibitors such as milrinone.
Mechanical support devices, such as intra-aortic balloon counterpulsation, and vasodilators can also be used to improve tissue perfusion in selected patients with low cardiac output syndromes.
USING LACTATE LEVELS TO GUIDE THERAPY
Lactate levels above 4.0 mmol/L
Lactate may be a useful marker for determining whether organ dysfunction is present and, hence, what course of therapy should be given, especially in sepsis. A serum lactate level higher than 4.0 mmol/L has been used as the trigger to start aggressive resuscitation in patients with sepsis.50,51
Traditionally, as delineated by Rivers et al50 in their landmark study of early goal-directed therapy, this entailed placing an arterial line and a central line for hemodynamic monitoring, with specific interventions directed at increasing the central venous pressure, mean arterial pressure, and central venous oxygen saturation.50 However, a recent study in a similar population of patients with sepsis with elevated lactate found no significant advantage of protocol-based resuscitation over care provided according to physician judgment, and no significant benefit in central venous catheterization and hemodynamic monitoring in all patients.51
Lactate clearance: 10% or above at 8 hours?
Regardless of the approach chosen, decreasing lactate levels can be interpreted as an adequate response to the interventions provided. As a matter of fact, several groups of investigators have also demonstrated the merits of lactate clearance alone as a prognostic indicator in patients requiring hemodynamic support.
McNelis et al52 retrospectively evaluated 95 postsurgical patients who required hemodynamic monitoring.52,53 The authors found that the slower the lactate clearance, the higher the mortality rate.
Given the prognostic implications of lactate clearance, investigators have evaluated whether lactate clearance could be used as a surrogate resuscitation goal for optimizing oxygen delivery. Using lactate clearance may have significant practical advantages over using central venous oxygen saturation, since it does not require a central venous catheter or continuous oximetric monitoring.
In a study comparing these two resuscitation end points, patients were randomized to a goal of either central venous oxygen saturation of 70% or more or lactate clearance of 10% or more within the first 6 hours after presentation as a marker of oxygen delivery.53 Mortality rates were similar with either strategy. Of note, only 10% of the patients actually required therapies to improve their oxygen delivery. Furthermore, there were no differences in the treatments given (including fluids, vasopressors, inotropes, packed red blood cells) throughout the treatment period.
These findings provide several insights. First, few patients admitted to the emergency department with severe sepsis and treated with an initial quantitative resuscitation protocol require additional therapy for augmenting oxygen delivery. Second, lactate clearance, in a setting where initial resuscitation with fluids and vasopressors restores adequate oxygen delivery for the majority of patients, is likely as good a target for resuscitation as central venous oxygen saturation.
This study, however, does not address the question of whether lactate clearance is useful as an additional marker of oxygen delivery (in conjunction with central venous oxygen saturation). Indeed, caution should be taken to target central venous oxygen saturation goals alone, as patients with septic shock presenting with venous hyperoxia (central venous oxygen saturation > 89%) have been shown to have a higher mortality rate than patients with normoxia (central venous oxygen saturation 71%–89%).54
This was further demonstrated by Arnold et al in a study of patients presenting to the emergency department with severe sepsis.15 In this study, significant discordance between central venous oxygen saturation and lactate clearance was seen, where 79% of patients with less than 10% lactate clearance had concomitant central venous oxygen saturation of 70% or greater.
Jansen et al18 evaluated the role of targeting lactate clearance in conjunction with central venous oxygen saturation monitoring. In this study, critically ill patients with elevated lactate and inadequate lactate clearance were randomized to usual care or to resuscitation to adequate lactate clearance (20% or more). The therapies to optimize oxygen delivery were given according to the central venous oxygen saturation. Overall, after adjustment for predefined risk factors, the in-hospital mortality rate was lower in the lactate clearance group. This may signify that patients with sepsis and central venous oxygen saturation of 70% or more may continue to have poor lactate clearance, warranting further treatment.
Taken together, serum lactate may be helpful for prognostication, determination of course of therapy, and quantification for tissue hypoperfusion for targeted therapies. Figure 2 presents our approach to an elevated lactate level. As performed in the study by Jansen et al,18 it seems reasonable to measure lactate levels every 2 hours for the first 8 hours of resuscitation in patients with type A lactic acidosis. These levels should be interpreted in the context of lactate clearance (at least 10%, but preferably 20%) and normalization, and should be treated with an approach similar to the one outlined in Figure 2.
TREATING TYPE B LACTIC ACIDOSIS (NORMAL PERFUSION AND OXYGENATION)
Treating type B lactic acidosis is quite different because the goal is not to correct mismatches in oxygen consumption and delivery. Since most cases are due to underlying conditions such as malignancy or medications, treatment should be centered around eliminating the cause (eg, treat the malignancy, discontinue the offending medication). The main reason for treatment is to alleviate the harmful effects of acidosis. For example, acidosis can result in a negative inotropic effect.
Sodium bicarbonate, dichloroacetate, carbicarb, and tromethamine have all been studied in the management of type B lactic acidosis, with little success.55,56
Renal replacement therapy has had some success in drug-induced lactic acidosis.57,58
l-carnitine has had promising results in treating patients with human immunodeficiency virus infection, since these patients are carnitine-deficient and carnitine plays an important role in mitochondrial function.59
Thiamine and biotin deficiencies can occur in patients receiving total parenteral nutrition without vitamins and in patients who drink alcohol heavily and can cause lactic acidosis. These nutrients should be supplemented accordingly.
Treatment of mitochondrial disorders includes antioxidants (coenzyme Q10, vitamin C, vitamin E) and amino acids (l-arginine).60
- Andersen LW, Mackenhauer J, Roberts JC, Berg KM, Cocchi MN, Donnino MW. Etiology and therapeutic approach to elevated lactate levels. Mayo Clin Proc 2013; 88:1127–1140.
- Fuller BM, Dellinger RP. Lactate as a hemodynamic marker in the critically ill. Curr Opin Crit Care 2012; 18:267–272.
- Fall PJ, Szerlip HM. Lactic acidosis: from sour milk to septic shock. J Intensive Care Med 2005; 20:255–271.
- Kruse O, Grunnet N, Barfod C. Blood lactate as a predictor for in-hospital mortality in patients admitted acutely to hospital: a systematic review. Scand J Trauma Resusc Emerg Med 2011;19:74.
- Howell MD, Donnino M, Clardy P, Talmor D, Shapiro NI. Occult hypoperfusion and mortality in patients with suspected infection. Intensive Care Med 2007; 33:1892–1899.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Outcomes of patients undergoing early sepsis resuscitation for cryptic shock compared with overt shock. Resuscitation 2011; 82:1289–1293.
- Bakker J, Nijsten MW, Jansen TC. Clinical use of lactate monitoring in critically ill patients. Ann Intensive Care 2013; 3:12.
- Levy B, Gibot S, Franck P, Cravoisy A, Bollaert PE. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet 2005; 365:871–875.
- Vary TC. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 1996; 6:89–94.
- Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360:219–223.
- Shapiro NI, Howell MD, Talmor D, et al. Serum lactate as a predictor of mortality in emergency department patients with infection. Ann Emerg Med 2005; 45:524–528.
- Mikkelsen ME, Miltiades AN, Gaieski DF, et al. Serum lactate is associated with mortality in severe sepsis independent of organ failure and shock. Crit Care Med 2009; 37:1670–1677.
- Liu V, Morehouse JW, Soule J, Whippy A, Escobar GJ. Fluid volume, lactate values, and mortality in sepsis patients with intermediate lactate values. Ann Am Thorac Soc 2013; 10:466–473.
- Sterling SA, Puskarich MA, Shapiro NI, et al; Emergency Medicine Shock Research Network (EMShockNET). Characteristics and outcomes of patients with vasoplegic versus tissue dysoxic septic shock. Shock 2013; 40:11–14.
- Arnold RC, Shapiro NI, Jones AE, et al; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of early lactate clearance as a determinant of survival in patients with presumed sepsis. Shock 2009; 32:35–39.
- Jones AE. Lactate clearance for assessing response to resuscitation in severe sepsis. Acad Emerg Med 2013; 20:844–847.
- Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med 2004; 32:1637–1642.
- Jansen TC, van Bommel J, Schoonderbeek FJ, et al; LACTATE study group. Early lactate-guided therapy in intensive care unit patients: a multicenter, open-label, randomized controlled trial. Am J Respir Crit Care Med 2010; 182:752–761.
- Husain FA, Martin MJ, Mullenix PS, Steele SR, Elliott DC. Serum lactate and base deficit as predictors of mortality and morbidity. Am J Surg 2003; 185:485–491.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Whole blood lactate kinetics in patients undergoing quantitative resuscitation for severe sepsis and septic shock. Chest 2013; 143:1548–1553.
- Marty P, Roquilly A, Vallee F, et al. Lactate clearance for death prediction in severe sepsis or septic shock patients during the first 24 hours in intensive care unit: an observational study. Ann Intensive Care 2013; 3:3.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: International guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Vincent JL, De Backer D. Circulatory shock. N Engl J Med 2013; 369:1726–1734.
- Durairaj L, Schmidt GA. Fluid therapy in resuscitated sepsis: less is more. Chest 2008; 133:252–263.
- Vincent JL, Dufaye P, Berré J, Leeman M, Degaute JP, Kahn RJ. Serial lactate determinations during circulatory shock. Crit Care Med 1983; 11:449–451.
- Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med 2011; 39:259–265.
- Finfer S, Bellomo R, Boyce N, French J, Myburgh J, Norton R; SAFE Study Investigators. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med 2004; 350:2247–2256.
- Myburgh JA, Finfer S, Bellomo R, et al; CHEST Investigators; Australian and New Zealand Intensive Care Society Clinical Trials Group. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med 2012; 367:1901–1911.
- Perner A, Haase N, Guttormsen AB, et al; 6S Trial Group; Scandinavian Critical Care Trials Group. Hydroxyethyl starch 130/0.42 versus Ringer’s acetate in severe sepsis. N Engl J Med 2012; 367:124–134.
- Bayer O, Reinhart K, Kohl M, et al. Effects of fluid resuscitation with synthetic colloids or crystalloids alone on shock reversal, fluid balance, and patient outcomes in patients with severe sepsis: a prospective sequential analysis. Crit Care Med 2012; 40:2543–2551.
- Delaney AP, Dan A, McCaffrey J, Finfer S. The role of albumin as a resuscitation fluid for patients with sepsis: a systematic review and meta-analysis. Crit Care Med 2011; 39:386–391.
- SAFE Study Investigators; Finfer S, McEvoy S, Bellomo R, McArthur C, Myburgh J, Norton R. Impact of albumin compared to saline on organ function and mortality of patients with severe sepsis. Intensive Care Med 2011; 37:86–96.
- Caironi P, Tognoni G, Masson S, et al; ALBIOS Study Investigators. Albumin replacement in patients with severe sepsis or septic shock. N Engl J Med 2014; 370:1412–1421.
- Bourgoin A, Leone M, Delmas A, Garnier F, Albanèse J, Martin C. Increasing mean arterial pressure in patients with septic shock: effects on oxygen variables and renal function. Crit Care Med 2005; 33:780–786.
- LeDoux D, Astiz ME, Carpati CM, Rackow EC. Effects of perfusion pressure on tissue perfusion in septic shock. Crit Care Med 2000; 28:2729–2732.
- Asfar P, Meziani F, Hamel JF, et al; SEPSISPAM Investigators. High versus low blood-pressure target in patients with septic shock. N Engl J Med 2014; 370:1583–1593.
- De Backer D, Biston P, Devriendt J, et al; SOAP II Investigators. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789.
- De Backer D, Aldecoa C, Njimi H, Vincent JL. Dopamine versus norepinephrine in the treatment of septic shock: a meta-analysis. Crit Care Med 2012; 40:725–730.
- Annane D, Vignon P, Renault A, et al: CATS Study Group. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet 2007; 370:676–684.
- Myburgh JA, Higgins A, Jovanovska A, Lipman J, Ramakrishnan N, Santamaria J; CAT Study investigators. A comparison of epinephrine and norepinephrine in critically ill patients. Intensive Care Med 2008; 34:2226–2234.
- Schmittinger CA, Torgersen C, Luckner G, Schröder DC, Lorenz I, Dünser MW. Adverse cardiac events during catecholamine vasopressor therapy: a prospective observational study. Intensive Care Med 2012; 38:950–958.
- Levy B, Perez P, Perny J, Thivilier C, Gerard A. Comparison of norepinephrine-dobutamine to epinephrine for hemodynamics, lactate metabolism, and organ function variables in cardiogenic shock. A prospective, randomized pilot study. Crit Care Med 2011; 39:450–455.
- Watt MJ, Howlett KF, Febbraio MA, Spriet LL, Hargreaves M. Adrenaline increases skeletal muscle glycogenolysis, pyruvate dehydrogenase activation and carbohydrate oxidation during moderate exercise in humans. J Physiol 2001; 534:269–278.
- De Backer D, Creteur J, Silva E, Vincent JL. Effects of dopamine, norepinephrine, and epinephrine on the splanchnic circulation in septic shock: which is best? Crit Care Med 2003; 31:1659–1667.
- Levy B, Bollaert PE, Charpentier C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med 1997; 23:282–287.
- Polito A, Parisini E, Ricci Z, Picardo S, Annane D. Vasopressin for treatment of vasodilatory shock: an ESICM systematic review and meta-analysis. Intensive Care Med 2012; 38:9–19.
- Serpa Neto A, Nassar APJ, Cardoso SO, et al. Vasopressin and terlipressin in adult vasodilatory shock: a systematic review and meta-analysis of nine randomized controlled trials. Crit Care 2012; 16:R154.
- Russell JA, Walley KR, Singer J, et al; VASST Investigators. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008; 358:877–887.
- Gattinoni L, Brazzi L, Pelosi P, et al; for the SvO2 Collaborative Group. A trial of goal-oriented hemodynamic therapy in critically ill patients. N Engl J Med 1995; 333:1025–1032.
- Rivers E, Nguyen B, Havstad S, et al; Early Goal-Directed Therapy Collaborative Group. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- ProCESS Investigators; Yealy DM, Kellum JA, Huang DT, et al. A randomized trial of protocol-based care for early septic shock. N Engl J Med 2014; 370:1683–1693.
- McNelis J, Marini CP, Jurkiewicz A, et al. Prolonged lactate clearance is associated with increased mortality in the surgical intensive care unit. Am J Surg 2001; 182:481–485.
- Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA 2010; 303:739–746.
- Pope JV, Jones AE, Gaieski DF, Arnold RC, Trzeciak S, Shapiro NI; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of central venous oxygen saturation (ScvO2) as a predictor of mortality in patients with sepsis. Ann Emerg Med 2010; 55:40–46.e1
- Kraut JA, Kurtz I. Use of base in the treatment of severe acidemic states. Am J Kidney Dis 2001; 38:703–727.
- Levraut J, Grimaud D. Treatment of metabolic acidosis. Curr Opin Crit Care 2003; 9:260–265.
- Orija AA, Jenks CL. Nucleoside analog reverse transcriptase inhibitor induced lactic acidosis treated with continuous renal replacement in the medical intensive care unit. Crit Care & Shock 2012; 15:9–11.
- Friesecke S, Abel P, Kraft M, Gerner A, Runge S. Combined renal replacement therapy for severe metformin-induced lactic acidosis. Nephrol Dial Transplant 2006; 21:2038–2039.
- Claessens YE, Cariou A, Monchi M, et al. Detecting life-threatening lactic acidosis related to nucleoside-analog treatment of human immunodeficiency virus-infected patients, and treatment with l-carnitine. Crit Care Med 2003; 31:1042–1047.
- Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R; Medicine Society TM. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol 2009; 11:414–430.
Physicians are paying more attention to serum lactate levels in hospitalized patients than in the past, especially with the advent of point-of-care testing. Elevated lactate levels are associated with tissue hypoxia and hypoperfusion but can also be found in a number of other conditions. Therefore, confusion can arise as to how to interpret elevated levels and subsequently manage these patients in a variety of settings.
In this review, we discuss the mechanisms underlying lactic acidosis, its prognostic implications, and its use as a therapeutic target in treating patients in septic shock and other serious disorders.
LACTATE IS A PRODUCT OF ANAEROBIC RESPIRATION
Lactate, or lactic acid, is produced from pyruvate as an end product of glycolysis under anaerobic conditions (Figure 1). It is produced in most tissues in the body, but primarily in skeletal muscle, brain, intestine, and red blood cells. During times of stress, lactate is also produced in the lungs, white blood cells, and splanchnic organs.
Most lactate in the blood is cleared by the liver, where it is the substrate for gluconeogenesis, and a small amount is cleared by the kidneys.1,2 The entire pathway by which lactate is produced and converted back to glucose is called the Cori cycle.
NORMAL LEVELS ARE LESS THAN ABOUT 2.0 MMOL/L
In this review, we will present lactate levels in the SI units of mmol/L (1 mmol/L = 9 mg/dL).
Basal lactate production is approximately 0.8 mmol/kg body weight/hour. The average normal arterial blood lactate level is approximately 0.620 mmol/L and the venous level is slightly higher at 0.997 mmol/L,3 but overall, arterial and venous lactate levels correlate well.
Normal lactate levels are less than 2 mmol/L,4 intermediate levels range from 2 to less than 4 mmol/L, and high levels are 4 mmol/L or higher.5
To minimize variations in measurement, blood samples should be drawn without a tourniquet into tubes containing fluoride, placed on ice, and processed quickly (ideally within 15 minutes).
INCREASED PRODUCTION, DECREASED CLEARANCE, OR BOTH
An elevated lactate level can be the result of increased production, decreased clearance, or both (as in liver dysfunction).
Type A lactic acidosis—due to hypoperfusion and hypoxia—occurs when there is a mismatch between oxygen delivery and consumption, with resultant anaerobic glycolysis.
The guidelines from the Surviving Sepsis Campaign6 emphasize using lactate levels to diagnose patients with sepsis-induced hypoperfusion. However, hyperlactatemia can indicate inadequate oxygen delivery due to any type of shock (Table 1).
Type B lactic acidosis—not due to hypoperfusion—occurs in a variety of conditions (Table 1), including liver disease, malignancy, use of certain medications (eg, metformin, epinephrine), total parenteral nutrition, human immunodeficiency virus infection, thiamine deficiency, mitochondrial myopathies, and congenital lactic acidosis.1–3,7 Yet other causes include trauma, excessive exercise, diabetic ketoacidosis, ethanol intoxication, dysfunction of the enzyme pyruvate dehydrogenase, and increased muscle degradation leading to increased production of pyruvate. In these latter scenarios, glucose metabolism exceeds the oxidation capacity of the mitochondria, and the rise in pyruvate concentration drives lactate production.8,9 Mitochondrial dysfunction and subsequent deficits in cellular oxygen use can also result in persistently high lactate levels.10
In some situations, patients with mildly elevated lactic acid levels in type B lactic acidosis can be monitored to ensure stability, rather than be treated aggressively.
HIGHER LEVELS AND LOWER CLEARANCE PREDICT DEATH
The higher the lactate level and the slower the rate of normalization (lactate clearance), the higher the risk of death.
Lactate levels and mortality rate
Shapiro et al11 showed that increases in lactate level are associated with proportional increases in the mortality rate. Mikkelsen et al12 showed that intermediate levels (2.0–3.9 mmol/L) and high levels (≥ 4 mmol/L) of serum lactate are associated with increased risk of death independent of organ failure and shock. Patients with mildly elevated and intermediate lactate levels and sepsis have higher rates of in-hospital and 30-day mortality, which correlate with the baseline lactate level.13
In a post hoc analysis of a randomized controlled trial, patients with septic shock who presented to the emergency department with hypotension and a lactate level higher than 2 mmol/L had a significantly higher in-hospital mortality rate than those who presented with hypotension and a lactate level of 2 mmol/L or less (26% vs 9%, P < .0001).14 These data suggest that elevated lactate levels may have a significant prognostic role, independent of blood pressure.
Slower clearance
The prognostic implications of lactate clearance (reductions in lactate levels over time, as opposed to a single value in time), have also been evaluated.
Lactate clearance of at least 10% at 6 hours after presentation has been associated with a lower mortality rate than nonclearance (19% vs 60%) in patients with sepsis or septic shock with elevated levels.15–17 Similar findings have been reported in a general intensive care unit population,18 as well as a surgical intensive care population.sup>19
Puskarich et al20 have also shown that lactate normalization to less than 2 mmol/L during early sepsis resuscitation is the strongest predictor of survival (odds ratio [OR] 5.2), followed by lactate clearance of 50% (OR 4.0) within the first 6 hours of presentation. Not only is lactate clearance associated with improved outcomes, but a faster rate of clearance after initial presentation is also beneficial.15,16,18
Lactate clearance over a longer period (> 6 hours) has not been studied in patients with septic shock. However, in the general intensive care unit population, therapy guided by lactate clearance for the first 8 hours after presentation has shown a reduction in mortality rate.18 There are no data available on outcomes of lactate-directed therapy beyond 8 hours, but lactate concentration and lactate clearance at 24 hours correlate with the 28-day mortality rate.21
Cryptic shock
Cryptic shock describes a state in a subgroup of patients who have elevated lactate levels and global tissue hypoxia despite being normotensive or even hypertensive. These patients have a higher mortality rate independent of blood pressure. Jansen et al18 found that patients with a lactate level higher than 4 mmol/L and preserved blood pressure had a mortality rate of 15%, while those without shock or hyperlactatemia had a mortality rate of 2.5%. In addition, patients with an elevated lactate level in the absence of hypotension have mortality rates similar to those in patients with high lactate levels and hypotension refractory to fluid boluses, suggesting the presence of tissue hypoxia even in these normotensive patients.6
HOW TO APPROACH AN ELEVATED LACTATE LEVEL
An elevated lactate level should prompt an evaluation for causes of decreased oxygen delivery, due either to a systemic low-flow state (as a result of decreased cardiac output) or severe anemia, or to regionally decreased perfusion, (eg, limb or mesenteric ischemia). If tissue hypoxia is ruled out after an exhaustive workup, consideration should be given to causes of hyperlactatemia without concomitant tissue hypoxia (type B acidosis).
Treatment differs depending on the underlying mechanism of the lactate elevation; nevertheless, treatment is mostly related to optimizing oxygen delivery by giving fluids, packed red blood cells, and vasopressors or inotropic agents, or both (Figure 2). The specific treatment differs based on the shock state, but there are similarities that can guide the clinician.
FLUID SUPPORT
Giving fluids, with a goal of improving cardiac output, remains a cornerstone of initial therapy for most shock states.22,23
How much fluid?
Fluids should be given until the patient is no longer preload-dependent, although there is much debate about which assessment strategy should be used to determine if cardiac output will improve with more fluid (ie, fluid-responsiveness).24 In many cases, fluid resuscitation alone may be enough to restore hemodynamic stability, improve tissue perfusion, and reduce elevated lactate concentrations.25
The decision to give more fluids should not be made lightly, though, as a more positive fluid balance early in the course of septic shock and over 4 days has been associated with a higher mortality rate.26 Additionally, pushing fluids in patients with cardiogenic shock due to impaired left ventricular systolic function may lead to or worsen pulmonary edema. Therefore, the indiscriminate use of fluids should be avoided.
Which fluids?
Despite years of research, controversy persists about whether crystalloids or colloids are better for resuscitation. Randomized trials in heterogeneous intensive care unit patients have not detected differences in 28-day mortality rates between those allocated to crystalloids or 4% albumin27 and those allocated to crystalloids or hydroxyethyl starch.28
Hydroxyethyl starch may not be best. In a study of patients with severe sepsis, those randomized to receive hydroxyethyl starch had a higher 90-day mortality rate than patients randomized to crystalloids (51% vs 43%, P = .03).29 A sequential prospective before-and-after study did not detect a difference in the time to normalization (< 2.2 mmol/L) of lactate (P = .68) or cessation of vasopressors (P = .11) in patients with severe sepsis who received fluid resuscitation with crystalloids, gelatin, or hydroxyethyl starch. More patients who received hydroxyethyl starch in these studies developed acute kidney injury than those receiving crystalloids.28–30
Taken together, these data strongly suggest hydroxyethyl starch should not be used for fluid resuscitation in the intensive care unit.
Normal saline or albumin? Although some data suggest that albumin may be preferable to 0.9% sodium chloride in patients with severe sepsis,31,32 these analyses should be viewed as hypothesis-generating. There do not seem to be differences between fluid types in terms of subsequent serum lactate concentrations or achievement of lactate clearance goals.28–30 Until further studies are completed, both albumin and crystalloids are reasonable for resuscitation.
Caironi et al33 performed an open-label study comparing albumin replacement (with a goal serum albumin concentration of 3 g/dL) plus a crystalloid solution vs a crystalloid solution alone in patients with severe sepsis or septic shock. They detected no difference between the albumin and crystalloid groups in mortality rates at 28 days (31.8% vs 32.0%, P = .94) or 90 days (41.1% vs 43.6%, P = .29). However, patients in the albumin group had a shorter time to cessation of vasoactive agents (median 3 vs 4 days, P = .007) and lower cardiovascular Sequential Organ Failure Assessment subscores (median 1.20 vs 1.42, P = .03), and more frequently achieved a mean arterial pressure of at least 65 mm Hg within 6 hours of randomization (86.0% vs 82.5%, P = .04).
Although serum lactate levels were lower in the albumin group at baseline (1.7 mmol/L vs 1.8 mmol/L, P = .05), inspection of the data appears to show a similar daily lactate clearance rate between groups over the first 7 study days (although these data were not analyzed by the authors). Achievement of a lactate level lower than 2 mmol/L on the first day of therapy was not significantly different between groups (73.4% vs 72.5%, P = .11).33
In a post hoc subgroup analysis, patients with septic shock at baseline randomized to albumin had a lower 90-day mortality rate than patients randomized to crystalloid solutions (RR 0.87, 95% CI 0.77–0.99). There was no difference in the 90-day mortality rate in patients without septic shock (RR 1.13, 95% CI 0.92–1.39, P = .03 for heterogeneity).33
These data suggest that albumin replacement may not improve outcomes in patients with severe sepsis, but may have advantages in terms of hemodynamic variables (and potentially mortality) in patients with septic shock. The role of albumin replacement in patients with septic shock warrants further study.
VASOPRESSORS
Vasopressors, inotropes, or both should be given to patients who have signs of hypoperfusion (including elevated lactate levels) despite preload optimization or ongoing fluid administration. The most appropriate drug depends on the goal: vasopressors are used to increase systemic vascular resistance, while inotropes are used to improve cardiac output and oxygen delivery.
Blood pressure target
The Surviving Sepsis Campaign guidelines recommend a mean arterial blood pressure target of at least 65 mm Hg during initial resuscitation and when vasopressors are applied for patients with septic shock.22 This recommendation is based on small studies that did not show differences in serum lactate levels or regional blood flow when the mean arterial pressure was elevated above 65 mm Hg with norepinephrine.34,35 However, the campaign guidelines note that the mean arterial pressure goal must be individualized in order to achieve optimal perfusion.
A large, open-label trial36 detected no difference in 28-day mortality rates in patients with septic shock between those allocated to a mean arterial pressure goal of 80 to 85 mm Hg or 65 to 70 mm Hg (36.6% vs 34.0%, P = .57). Although lactate levels did not differ between groups, the incidence of new-onset atrial fibrillation was higher in the higher-target group (6.7% vs 2.8%, P = .02). Fewer patients with chronic hypertension needed renal replacement therapy in the higher pressure group, further emphasizing the need to individualize the mean arterial pressure goal for patients in shock.36
Which vasopressor agent?
Dopamine and norepinephrine have traditionally been the preferred initial vasopressors for patients with shock. Until recently there were few data to guide selection between the two, but this is changing.
In a 2010 study of 1,679 patients with shock requiring vasopressors, there was no difference in the 28-day mortality rate between patients randomized to dopamine or norepinephrine (53% vs 49%, P = .10).37 Patients allocated to dopamine, though, had a higher incidence of arrhythmias (24% vs 12%, P < .001) and more frequently required open-label norepinephrine (26% vs 20%, P < .001). Although lactate levels and the time to achievement of a mean arterial pressure of 65 mm Hg were similar between groups, patients allocated to norepinephrine had more vasopressor-free days through day 28.
An a priori-planned subgroup analysis evaluated the influence of the type of shock on patient outcome. Patients with cardiogenic shock randomized to dopamine had a higher mortality rate than those randomized to norepinephrine (P = .03). However, the overall effect of treatment did not differ among the shock subgroups (interaction P = .87), suggesting that the reported differences in mortality according to subgroup may be spurious.
In a 2012 meta-analysis of patients with septic shock, dopamine use was associated with a higher mortality rate than norepinephrine use.38
In light of these data, norepinephrine should be preferred over dopamine as the initial vasopressor in most types of shock.
Epinephrine does not offer an outcome advantage over norepinephrine and may be associated with a higher incidence of adverse events.39–42 Indeed, in a study of patients with septic shock, lactate concentrations on the first day after randomization were significantly higher in patients allocated to epinephrine than in patients allocated to norepinephrine plus dobutamine.39 Similar effects on lactate concentrations with epinephrine were seen in patients with various types of shock40 and in those with cardiogenic shock.42
These differences in lactate concentrations may be directly attributable to epinephrine. Epinephrine can increase lactate concentrations through glycolysis and pyruvate dehydrogenase activation by stimulation of sodium-potassium ATPase activity via beta-2 adrenergic receptors in skeletal muscles,43 as well as decrease splanchnic perfusion.42,44,45 These effects may preclude using lactate clearance as a resuscitation goal in patients receiving epinephrine. Epinephrine is likely best reserved for patients with refractory shock,22 particularly those in whom cardiac output is known to be low.
Phenylephrine, essentially a pure vasoconstrictor, should be avoided in low cardiac output states and is best reserved for patients who develop a tachyarrhythmia on norepinephrine.22
Vasopressin, also a pure vasoconstrictor that should be avoided in low cardiac output states, has been best studied in patients with vasodilatory shock. Although controversy exists on the mortality benefits of vasopressin in vasodilatory shock, it is a relatively safe drug with consistent norepinephrine-sparing effects when added to existing norepinephrine therapy.46,47 In patients with less severe septic shock, including those with low lactate concentrations, adding vasopressin to norepinephrine instead of continuing norepinephrine alone may confer a mortality advantage.48
OTHER MEASURES TO OPTIMIZE OXYGEN DELIVERY
In circulatory shock from any cause, tissue oxygen demand exceeds oxygen delivery. Once arterial oxygenation and hemoglobin levels (by packed red blood cell transfusion) have been optimized, cardiac output is the critical determinant of oxygen delivery. Cardiac output may be augmented by ensuring adequate preload (by fluid resuscitation) or by giving inotropes or vasodilators.
The optimal cardiac output is difficult to define, and the exact marker for determining when cardiac output should be augmented is unclear. A strategy of increasing cardiac output to predefined “supranormal” levels was not associated with a lower mortality rate.49 Therefore, the decision to augment cardiac output must be individualized and will likely vary in the same patient over time.23
A reasonable approach to determining when augmentation of cardiac output is necessary was proposed in a study by Rivers et al.50 In that study, in patients randomized to early goal-directed therapy, inotropes were recommended when the central venous oxygenation saturation (Scvo2) was below 70% despite adequate fluid resuscitation (central venous pressure ≥ 8 mm Hg) and hematocrits were higher than 30%.
When an inotrope is indicated to improve cardiac output, dobutamine is usually the preferred agent. Dobutamine has a shorter half-life (allowing for easier titration) and causes less hypotension (assuming preload has been optimized) than phosphodiesterase type III inhibitors such as milrinone.
Mechanical support devices, such as intra-aortic balloon counterpulsation, and vasodilators can also be used to improve tissue perfusion in selected patients with low cardiac output syndromes.
USING LACTATE LEVELS TO GUIDE THERAPY
Lactate levels above 4.0 mmol/L
Lactate may be a useful marker for determining whether organ dysfunction is present and, hence, what course of therapy should be given, especially in sepsis. A serum lactate level higher than 4.0 mmol/L has been used as the trigger to start aggressive resuscitation in patients with sepsis.50,51
Traditionally, as delineated by Rivers et al50 in their landmark study of early goal-directed therapy, this entailed placing an arterial line and a central line for hemodynamic monitoring, with specific interventions directed at increasing the central venous pressure, mean arterial pressure, and central venous oxygen saturation.50 However, a recent study in a similar population of patients with sepsis with elevated lactate found no significant advantage of protocol-based resuscitation over care provided according to physician judgment, and no significant benefit in central venous catheterization and hemodynamic monitoring in all patients.51
Lactate clearance: 10% or above at 8 hours?
Regardless of the approach chosen, decreasing lactate levels can be interpreted as an adequate response to the interventions provided. As a matter of fact, several groups of investigators have also demonstrated the merits of lactate clearance alone as a prognostic indicator in patients requiring hemodynamic support.
McNelis et al52 retrospectively evaluated 95 postsurgical patients who required hemodynamic monitoring.52,53 The authors found that the slower the lactate clearance, the higher the mortality rate.
Given the prognostic implications of lactate clearance, investigators have evaluated whether lactate clearance could be used as a surrogate resuscitation goal for optimizing oxygen delivery. Using lactate clearance may have significant practical advantages over using central venous oxygen saturation, since it does not require a central venous catheter or continuous oximetric monitoring.
In a study comparing these two resuscitation end points, patients were randomized to a goal of either central venous oxygen saturation of 70% or more or lactate clearance of 10% or more within the first 6 hours after presentation as a marker of oxygen delivery.53 Mortality rates were similar with either strategy. Of note, only 10% of the patients actually required therapies to improve their oxygen delivery. Furthermore, there were no differences in the treatments given (including fluids, vasopressors, inotropes, packed red blood cells) throughout the treatment period.
These findings provide several insights. First, few patients admitted to the emergency department with severe sepsis and treated with an initial quantitative resuscitation protocol require additional therapy for augmenting oxygen delivery. Second, lactate clearance, in a setting where initial resuscitation with fluids and vasopressors restores adequate oxygen delivery for the majority of patients, is likely as good a target for resuscitation as central venous oxygen saturation.
This study, however, does not address the question of whether lactate clearance is useful as an additional marker of oxygen delivery (in conjunction with central venous oxygen saturation). Indeed, caution should be taken to target central venous oxygen saturation goals alone, as patients with septic shock presenting with venous hyperoxia (central venous oxygen saturation > 89%) have been shown to have a higher mortality rate than patients with normoxia (central venous oxygen saturation 71%–89%).54
This was further demonstrated by Arnold et al in a study of patients presenting to the emergency department with severe sepsis.15 In this study, significant discordance between central venous oxygen saturation and lactate clearance was seen, where 79% of patients with less than 10% lactate clearance had concomitant central venous oxygen saturation of 70% or greater.
Jansen et al18 evaluated the role of targeting lactate clearance in conjunction with central venous oxygen saturation monitoring. In this study, critically ill patients with elevated lactate and inadequate lactate clearance were randomized to usual care or to resuscitation to adequate lactate clearance (20% or more). The therapies to optimize oxygen delivery were given according to the central venous oxygen saturation. Overall, after adjustment for predefined risk factors, the in-hospital mortality rate was lower in the lactate clearance group. This may signify that patients with sepsis and central venous oxygen saturation of 70% or more may continue to have poor lactate clearance, warranting further treatment.
Taken together, serum lactate may be helpful for prognostication, determination of course of therapy, and quantification for tissue hypoperfusion for targeted therapies. Figure 2 presents our approach to an elevated lactate level. As performed in the study by Jansen et al,18 it seems reasonable to measure lactate levels every 2 hours for the first 8 hours of resuscitation in patients with type A lactic acidosis. These levels should be interpreted in the context of lactate clearance (at least 10%, but preferably 20%) and normalization, and should be treated with an approach similar to the one outlined in Figure 2.
TREATING TYPE B LACTIC ACIDOSIS (NORMAL PERFUSION AND OXYGENATION)
Treating type B lactic acidosis is quite different because the goal is not to correct mismatches in oxygen consumption and delivery. Since most cases are due to underlying conditions such as malignancy or medications, treatment should be centered around eliminating the cause (eg, treat the malignancy, discontinue the offending medication). The main reason for treatment is to alleviate the harmful effects of acidosis. For example, acidosis can result in a negative inotropic effect.
Sodium bicarbonate, dichloroacetate, carbicarb, and tromethamine have all been studied in the management of type B lactic acidosis, with little success.55,56
Renal replacement therapy has had some success in drug-induced lactic acidosis.57,58
l-carnitine has had promising results in treating patients with human immunodeficiency virus infection, since these patients are carnitine-deficient and carnitine plays an important role in mitochondrial function.59
Thiamine and biotin deficiencies can occur in patients receiving total parenteral nutrition without vitamins and in patients who drink alcohol heavily and can cause lactic acidosis. These nutrients should be supplemented accordingly.
Treatment of mitochondrial disorders includes antioxidants (coenzyme Q10, vitamin C, vitamin E) and amino acids (l-arginine).60
Physicians are paying more attention to serum lactate levels in hospitalized patients than in the past, especially with the advent of point-of-care testing. Elevated lactate levels are associated with tissue hypoxia and hypoperfusion but can also be found in a number of other conditions. Therefore, confusion can arise as to how to interpret elevated levels and subsequently manage these patients in a variety of settings.
In this review, we discuss the mechanisms underlying lactic acidosis, its prognostic implications, and its use as a therapeutic target in treating patients in septic shock and other serious disorders.
LACTATE IS A PRODUCT OF ANAEROBIC RESPIRATION
Lactate, or lactic acid, is produced from pyruvate as an end product of glycolysis under anaerobic conditions (Figure 1). It is produced in most tissues in the body, but primarily in skeletal muscle, brain, intestine, and red blood cells. During times of stress, lactate is also produced in the lungs, white blood cells, and splanchnic organs.
Most lactate in the blood is cleared by the liver, where it is the substrate for gluconeogenesis, and a small amount is cleared by the kidneys.1,2 The entire pathway by which lactate is produced and converted back to glucose is called the Cori cycle.
NORMAL LEVELS ARE LESS THAN ABOUT 2.0 MMOL/L
In this review, we will present lactate levels in the SI units of mmol/L (1 mmol/L = 9 mg/dL).
Basal lactate production is approximately 0.8 mmol/kg body weight/hour. The average normal arterial blood lactate level is approximately 0.620 mmol/L and the venous level is slightly higher at 0.997 mmol/L,3 but overall, arterial and venous lactate levels correlate well.
Normal lactate levels are less than 2 mmol/L,4 intermediate levels range from 2 to less than 4 mmol/L, and high levels are 4 mmol/L or higher.5
To minimize variations in measurement, blood samples should be drawn without a tourniquet into tubes containing fluoride, placed on ice, and processed quickly (ideally within 15 minutes).
INCREASED PRODUCTION, DECREASED CLEARANCE, OR BOTH
An elevated lactate level can be the result of increased production, decreased clearance, or both (as in liver dysfunction).
Type A lactic acidosis—due to hypoperfusion and hypoxia—occurs when there is a mismatch between oxygen delivery and consumption, with resultant anaerobic glycolysis.
The guidelines from the Surviving Sepsis Campaign6 emphasize using lactate levels to diagnose patients with sepsis-induced hypoperfusion. However, hyperlactatemia can indicate inadequate oxygen delivery due to any type of shock (Table 1).
Type B lactic acidosis—not due to hypoperfusion—occurs in a variety of conditions (Table 1), including liver disease, malignancy, use of certain medications (eg, metformin, epinephrine), total parenteral nutrition, human immunodeficiency virus infection, thiamine deficiency, mitochondrial myopathies, and congenital lactic acidosis.1–3,7 Yet other causes include trauma, excessive exercise, diabetic ketoacidosis, ethanol intoxication, dysfunction of the enzyme pyruvate dehydrogenase, and increased muscle degradation leading to increased production of pyruvate. In these latter scenarios, glucose metabolism exceeds the oxidation capacity of the mitochondria, and the rise in pyruvate concentration drives lactate production.8,9 Mitochondrial dysfunction and subsequent deficits in cellular oxygen use can also result in persistently high lactate levels.10
In some situations, patients with mildly elevated lactic acid levels in type B lactic acidosis can be monitored to ensure stability, rather than be treated aggressively.
HIGHER LEVELS AND LOWER CLEARANCE PREDICT DEATH
The higher the lactate level and the slower the rate of normalization (lactate clearance), the higher the risk of death.
Lactate levels and mortality rate
Shapiro et al11 showed that increases in lactate level are associated with proportional increases in the mortality rate. Mikkelsen et al12 showed that intermediate levels (2.0–3.9 mmol/L) and high levels (≥ 4 mmol/L) of serum lactate are associated with increased risk of death independent of organ failure and shock. Patients with mildly elevated and intermediate lactate levels and sepsis have higher rates of in-hospital and 30-day mortality, which correlate with the baseline lactate level.13
In a post hoc analysis of a randomized controlled trial, patients with septic shock who presented to the emergency department with hypotension and a lactate level higher than 2 mmol/L had a significantly higher in-hospital mortality rate than those who presented with hypotension and a lactate level of 2 mmol/L or less (26% vs 9%, P < .0001).14 These data suggest that elevated lactate levels may have a significant prognostic role, independent of blood pressure.
Slower clearance
The prognostic implications of lactate clearance (reductions in lactate levels over time, as opposed to a single value in time), have also been evaluated.
Lactate clearance of at least 10% at 6 hours after presentation has been associated with a lower mortality rate than nonclearance (19% vs 60%) in patients with sepsis or septic shock with elevated levels.15–17 Similar findings have been reported in a general intensive care unit population,18 as well as a surgical intensive care population.sup>19
Puskarich et al20 have also shown that lactate normalization to less than 2 mmol/L during early sepsis resuscitation is the strongest predictor of survival (odds ratio [OR] 5.2), followed by lactate clearance of 50% (OR 4.0) within the first 6 hours of presentation. Not only is lactate clearance associated with improved outcomes, but a faster rate of clearance after initial presentation is also beneficial.15,16,18
Lactate clearance over a longer period (> 6 hours) has not been studied in patients with septic shock. However, in the general intensive care unit population, therapy guided by lactate clearance for the first 8 hours after presentation has shown a reduction in mortality rate.18 There are no data available on outcomes of lactate-directed therapy beyond 8 hours, but lactate concentration and lactate clearance at 24 hours correlate with the 28-day mortality rate.21
Cryptic shock
Cryptic shock describes a state in a subgroup of patients who have elevated lactate levels and global tissue hypoxia despite being normotensive or even hypertensive. These patients have a higher mortality rate independent of blood pressure. Jansen et al18 found that patients with a lactate level higher than 4 mmol/L and preserved blood pressure had a mortality rate of 15%, while those without shock or hyperlactatemia had a mortality rate of 2.5%. In addition, patients with an elevated lactate level in the absence of hypotension have mortality rates similar to those in patients with high lactate levels and hypotension refractory to fluid boluses, suggesting the presence of tissue hypoxia even in these normotensive patients.6
HOW TO APPROACH AN ELEVATED LACTATE LEVEL
An elevated lactate level should prompt an evaluation for causes of decreased oxygen delivery, due either to a systemic low-flow state (as a result of decreased cardiac output) or severe anemia, or to regionally decreased perfusion, (eg, limb or mesenteric ischemia). If tissue hypoxia is ruled out after an exhaustive workup, consideration should be given to causes of hyperlactatemia without concomitant tissue hypoxia (type B acidosis).
Treatment differs depending on the underlying mechanism of the lactate elevation; nevertheless, treatment is mostly related to optimizing oxygen delivery by giving fluids, packed red blood cells, and vasopressors or inotropic agents, or both (Figure 2). The specific treatment differs based on the shock state, but there are similarities that can guide the clinician.
FLUID SUPPORT
Giving fluids, with a goal of improving cardiac output, remains a cornerstone of initial therapy for most shock states.22,23
How much fluid?
Fluids should be given until the patient is no longer preload-dependent, although there is much debate about which assessment strategy should be used to determine if cardiac output will improve with more fluid (ie, fluid-responsiveness).24 In many cases, fluid resuscitation alone may be enough to restore hemodynamic stability, improve tissue perfusion, and reduce elevated lactate concentrations.25
The decision to give more fluids should not be made lightly, though, as a more positive fluid balance early in the course of septic shock and over 4 days has been associated with a higher mortality rate.26 Additionally, pushing fluids in patients with cardiogenic shock due to impaired left ventricular systolic function may lead to or worsen pulmonary edema. Therefore, the indiscriminate use of fluids should be avoided.
Which fluids?
Despite years of research, controversy persists about whether crystalloids or colloids are better for resuscitation. Randomized trials in heterogeneous intensive care unit patients have not detected differences in 28-day mortality rates between those allocated to crystalloids or 4% albumin27 and those allocated to crystalloids or hydroxyethyl starch.28
Hydroxyethyl starch may not be best. In a study of patients with severe sepsis, those randomized to receive hydroxyethyl starch had a higher 90-day mortality rate than patients randomized to crystalloids (51% vs 43%, P = .03).29 A sequential prospective before-and-after study did not detect a difference in the time to normalization (< 2.2 mmol/L) of lactate (P = .68) or cessation of vasopressors (P = .11) in patients with severe sepsis who received fluid resuscitation with crystalloids, gelatin, or hydroxyethyl starch. More patients who received hydroxyethyl starch in these studies developed acute kidney injury than those receiving crystalloids.28–30
Taken together, these data strongly suggest hydroxyethyl starch should not be used for fluid resuscitation in the intensive care unit.
Normal saline or albumin? Although some data suggest that albumin may be preferable to 0.9% sodium chloride in patients with severe sepsis,31,32 these analyses should be viewed as hypothesis-generating. There do not seem to be differences between fluid types in terms of subsequent serum lactate concentrations or achievement of lactate clearance goals.28–30 Until further studies are completed, both albumin and crystalloids are reasonable for resuscitation.
Caironi et al33 performed an open-label study comparing albumin replacement (with a goal serum albumin concentration of 3 g/dL) plus a crystalloid solution vs a crystalloid solution alone in patients with severe sepsis or septic shock. They detected no difference between the albumin and crystalloid groups in mortality rates at 28 days (31.8% vs 32.0%, P = .94) or 90 days (41.1% vs 43.6%, P = .29). However, patients in the albumin group had a shorter time to cessation of vasoactive agents (median 3 vs 4 days, P = .007) and lower cardiovascular Sequential Organ Failure Assessment subscores (median 1.20 vs 1.42, P = .03), and more frequently achieved a mean arterial pressure of at least 65 mm Hg within 6 hours of randomization (86.0% vs 82.5%, P = .04).
Although serum lactate levels were lower in the albumin group at baseline (1.7 mmol/L vs 1.8 mmol/L, P = .05), inspection of the data appears to show a similar daily lactate clearance rate between groups over the first 7 study days (although these data were not analyzed by the authors). Achievement of a lactate level lower than 2 mmol/L on the first day of therapy was not significantly different between groups (73.4% vs 72.5%, P = .11).33
In a post hoc subgroup analysis, patients with septic shock at baseline randomized to albumin had a lower 90-day mortality rate than patients randomized to crystalloid solutions (RR 0.87, 95% CI 0.77–0.99). There was no difference in the 90-day mortality rate in patients without septic shock (RR 1.13, 95% CI 0.92–1.39, P = .03 for heterogeneity).33
These data suggest that albumin replacement may not improve outcomes in patients with severe sepsis, but may have advantages in terms of hemodynamic variables (and potentially mortality) in patients with septic shock. The role of albumin replacement in patients with septic shock warrants further study.
VASOPRESSORS
Vasopressors, inotropes, or both should be given to patients who have signs of hypoperfusion (including elevated lactate levels) despite preload optimization or ongoing fluid administration. The most appropriate drug depends on the goal: vasopressors are used to increase systemic vascular resistance, while inotropes are used to improve cardiac output and oxygen delivery.
Blood pressure target
The Surviving Sepsis Campaign guidelines recommend a mean arterial blood pressure target of at least 65 mm Hg during initial resuscitation and when vasopressors are applied for patients with septic shock.22 This recommendation is based on small studies that did not show differences in serum lactate levels or regional blood flow when the mean arterial pressure was elevated above 65 mm Hg with norepinephrine.34,35 However, the campaign guidelines note that the mean arterial pressure goal must be individualized in order to achieve optimal perfusion.
A large, open-label trial36 detected no difference in 28-day mortality rates in patients with septic shock between those allocated to a mean arterial pressure goal of 80 to 85 mm Hg or 65 to 70 mm Hg (36.6% vs 34.0%, P = .57). Although lactate levels did not differ between groups, the incidence of new-onset atrial fibrillation was higher in the higher-target group (6.7% vs 2.8%, P = .02). Fewer patients with chronic hypertension needed renal replacement therapy in the higher pressure group, further emphasizing the need to individualize the mean arterial pressure goal for patients in shock.36
Which vasopressor agent?
Dopamine and norepinephrine have traditionally been the preferred initial vasopressors for patients with shock. Until recently there were few data to guide selection between the two, but this is changing.
In a 2010 study of 1,679 patients with shock requiring vasopressors, there was no difference in the 28-day mortality rate between patients randomized to dopamine or norepinephrine (53% vs 49%, P = .10).37 Patients allocated to dopamine, though, had a higher incidence of arrhythmias (24% vs 12%, P < .001) and more frequently required open-label norepinephrine (26% vs 20%, P < .001). Although lactate levels and the time to achievement of a mean arterial pressure of 65 mm Hg were similar between groups, patients allocated to norepinephrine had more vasopressor-free days through day 28.
An a priori-planned subgroup analysis evaluated the influence of the type of shock on patient outcome. Patients with cardiogenic shock randomized to dopamine had a higher mortality rate than those randomized to norepinephrine (P = .03). However, the overall effect of treatment did not differ among the shock subgroups (interaction P = .87), suggesting that the reported differences in mortality according to subgroup may be spurious.
In a 2012 meta-analysis of patients with septic shock, dopamine use was associated with a higher mortality rate than norepinephrine use.38
In light of these data, norepinephrine should be preferred over dopamine as the initial vasopressor in most types of shock.
Epinephrine does not offer an outcome advantage over norepinephrine and may be associated with a higher incidence of adverse events.39–42 Indeed, in a study of patients with septic shock, lactate concentrations on the first day after randomization were significantly higher in patients allocated to epinephrine than in patients allocated to norepinephrine plus dobutamine.39 Similar effects on lactate concentrations with epinephrine were seen in patients with various types of shock40 and in those with cardiogenic shock.42
These differences in lactate concentrations may be directly attributable to epinephrine. Epinephrine can increase lactate concentrations through glycolysis and pyruvate dehydrogenase activation by stimulation of sodium-potassium ATPase activity via beta-2 adrenergic receptors in skeletal muscles,43 as well as decrease splanchnic perfusion.42,44,45 These effects may preclude using lactate clearance as a resuscitation goal in patients receiving epinephrine. Epinephrine is likely best reserved for patients with refractory shock,22 particularly those in whom cardiac output is known to be low.
Phenylephrine, essentially a pure vasoconstrictor, should be avoided in low cardiac output states and is best reserved for patients who develop a tachyarrhythmia on norepinephrine.22
Vasopressin, also a pure vasoconstrictor that should be avoided in low cardiac output states, has been best studied in patients with vasodilatory shock. Although controversy exists on the mortality benefits of vasopressin in vasodilatory shock, it is a relatively safe drug with consistent norepinephrine-sparing effects when added to existing norepinephrine therapy.46,47 In patients with less severe septic shock, including those with low lactate concentrations, adding vasopressin to norepinephrine instead of continuing norepinephrine alone may confer a mortality advantage.48
OTHER MEASURES TO OPTIMIZE OXYGEN DELIVERY
In circulatory shock from any cause, tissue oxygen demand exceeds oxygen delivery. Once arterial oxygenation and hemoglobin levels (by packed red blood cell transfusion) have been optimized, cardiac output is the critical determinant of oxygen delivery. Cardiac output may be augmented by ensuring adequate preload (by fluid resuscitation) or by giving inotropes or vasodilators.
The optimal cardiac output is difficult to define, and the exact marker for determining when cardiac output should be augmented is unclear. A strategy of increasing cardiac output to predefined “supranormal” levels was not associated with a lower mortality rate.49 Therefore, the decision to augment cardiac output must be individualized and will likely vary in the same patient over time.23
A reasonable approach to determining when augmentation of cardiac output is necessary was proposed in a study by Rivers et al.50 In that study, in patients randomized to early goal-directed therapy, inotropes were recommended when the central venous oxygenation saturation (Scvo2) was below 70% despite adequate fluid resuscitation (central venous pressure ≥ 8 mm Hg) and hematocrits were higher than 30%.
When an inotrope is indicated to improve cardiac output, dobutamine is usually the preferred agent. Dobutamine has a shorter half-life (allowing for easier titration) and causes less hypotension (assuming preload has been optimized) than phosphodiesterase type III inhibitors such as milrinone.
Mechanical support devices, such as intra-aortic balloon counterpulsation, and vasodilators can also be used to improve tissue perfusion in selected patients with low cardiac output syndromes.
USING LACTATE LEVELS TO GUIDE THERAPY
Lactate levels above 4.0 mmol/L
Lactate may be a useful marker for determining whether organ dysfunction is present and, hence, what course of therapy should be given, especially in sepsis. A serum lactate level higher than 4.0 mmol/L has been used as the trigger to start aggressive resuscitation in patients with sepsis.50,51
Traditionally, as delineated by Rivers et al50 in their landmark study of early goal-directed therapy, this entailed placing an arterial line and a central line for hemodynamic monitoring, with specific interventions directed at increasing the central venous pressure, mean arterial pressure, and central venous oxygen saturation.50 However, a recent study in a similar population of patients with sepsis with elevated lactate found no significant advantage of protocol-based resuscitation over care provided according to physician judgment, and no significant benefit in central venous catheterization and hemodynamic monitoring in all patients.51
Lactate clearance: 10% or above at 8 hours?
Regardless of the approach chosen, decreasing lactate levels can be interpreted as an adequate response to the interventions provided. As a matter of fact, several groups of investigators have also demonstrated the merits of lactate clearance alone as a prognostic indicator in patients requiring hemodynamic support.
McNelis et al52 retrospectively evaluated 95 postsurgical patients who required hemodynamic monitoring.52,53 The authors found that the slower the lactate clearance, the higher the mortality rate.
Given the prognostic implications of lactate clearance, investigators have evaluated whether lactate clearance could be used as a surrogate resuscitation goal for optimizing oxygen delivery. Using lactate clearance may have significant practical advantages over using central venous oxygen saturation, since it does not require a central venous catheter or continuous oximetric monitoring.
In a study comparing these two resuscitation end points, patients were randomized to a goal of either central venous oxygen saturation of 70% or more or lactate clearance of 10% or more within the first 6 hours after presentation as a marker of oxygen delivery.53 Mortality rates were similar with either strategy. Of note, only 10% of the patients actually required therapies to improve their oxygen delivery. Furthermore, there were no differences in the treatments given (including fluids, vasopressors, inotropes, packed red blood cells) throughout the treatment period.
These findings provide several insights. First, few patients admitted to the emergency department with severe sepsis and treated with an initial quantitative resuscitation protocol require additional therapy for augmenting oxygen delivery. Second, lactate clearance, in a setting where initial resuscitation with fluids and vasopressors restores adequate oxygen delivery for the majority of patients, is likely as good a target for resuscitation as central venous oxygen saturation.
This study, however, does not address the question of whether lactate clearance is useful as an additional marker of oxygen delivery (in conjunction with central venous oxygen saturation). Indeed, caution should be taken to target central venous oxygen saturation goals alone, as patients with septic shock presenting with venous hyperoxia (central venous oxygen saturation > 89%) have been shown to have a higher mortality rate than patients with normoxia (central venous oxygen saturation 71%–89%).54
This was further demonstrated by Arnold et al in a study of patients presenting to the emergency department with severe sepsis.15 In this study, significant discordance between central venous oxygen saturation and lactate clearance was seen, where 79% of patients with less than 10% lactate clearance had concomitant central venous oxygen saturation of 70% or greater.
Jansen et al18 evaluated the role of targeting lactate clearance in conjunction with central venous oxygen saturation monitoring. In this study, critically ill patients with elevated lactate and inadequate lactate clearance were randomized to usual care or to resuscitation to adequate lactate clearance (20% or more). The therapies to optimize oxygen delivery were given according to the central venous oxygen saturation. Overall, after adjustment for predefined risk factors, the in-hospital mortality rate was lower in the lactate clearance group. This may signify that patients with sepsis and central venous oxygen saturation of 70% or more may continue to have poor lactate clearance, warranting further treatment.
Taken together, serum lactate may be helpful for prognostication, determination of course of therapy, and quantification for tissue hypoperfusion for targeted therapies. Figure 2 presents our approach to an elevated lactate level. As performed in the study by Jansen et al,18 it seems reasonable to measure lactate levels every 2 hours for the first 8 hours of resuscitation in patients with type A lactic acidosis. These levels should be interpreted in the context of lactate clearance (at least 10%, but preferably 20%) and normalization, and should be treated with an approach similar to the one outlined in Figure 2.
TREATING TYPE B LACTIC ACIDOSIS (NORMAL PERFUSION AND OXYGENATION)
Treating type B lactic acidosis is quite different because the goal is not to correct mismatches in oxygen consumption and delivery. Since most cases are due to underlying conditions such as malignancy or medications, treatment should be centered around eliminating the cause (eg, treat the malignancy, discontinue the offending medication). The main reason for treatment is to alleviate the harmful effects of acidosis. For example, acidosis can result in a negative inotropic effect.
Sodium bicarbonate, dichloroacetate, carbicarb, and tromethamine have all been studied in the management of type B lactic acidosis, with little success.55,56
Renal replacement therapy has had some success in drug-induced lactic acidosis.57,58
l-carnitine has had promising results in treating patients with human immunodeficiency virus infection, since these patients are carnitine-deficient and carnitine plays an important role in mitochondrial function.59
Thiamine and biotin deficiencies can occur in patients receiving total parenteral nutrition without vitamins and in patients who drink alcohol heavily and can cause lactic acidosis. These nutrients should be supplemented accordingly.
Treatment of mitochondrial disorders includes antioxidants (coenzyme Q10, vitamin C, vitamin E) and amino acids (l-arginine).60
- Andersen LW, Mackenhauer J, Roberts JC, Berg KM, Cocchi MN, Donnino MW. Etiology and therapeutic approach to elevated lactate levels. Mayo Clin Proc 2013; 88:1127–1140.
- Fuller BM, Dellinger RP. Lactate as a hemodynamic marker in the critically ill. Curr Opin Crit Care 2012; 18:267–272.
- Fall PJ, Szerlip HM. Lactic acidosis: from sour milk to septic shock. J Intensive Care Med 2005; 20:255–271.
- Kruse O, Grunnet N, Barfod C. Blood lactate as a predictor for in-hospital mortality in patients admitted acutely to hospital: a systematic review. Scand J Trauma Resusc Emerg Med 2011;19:74.
- Howell MD, Donnino M, Clardy P, Talmor D, Shapiro NI. Occult hypoperfusion and mortality in patients with suspected infection. Intensive Care Med 2007; 33:1892–1899.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Outcomes of patients undergoing early sepsis resuscitation for cryptic shock compared with overt shock. Resuscitation 2011; 82:1289–1293.
- Bakker J, Nijsten MW, Jansen TC. Clinical use of lactate monitoring in critically ill patients. Ann Intensive Care 2013; 3:12.
- Levy B, Gibot S, Franck P, Cravoisy A, Bollaert PE. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet 2005; 365:871–875.
- Vary TC. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 1996; 6:89–94.
- Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360:219–223.
- Shapiro NI, Howell MD, Talmor D, et al. Serum lactate as a predictor of mortality in emergency department patients with infection. Ann Emerg Med 2005; 45:524–528.
- Mikkelsen ME, Miltiades AN, Gaieski DF, et al. Serum lactate is associated with mortality in severe sepsis independent of organ failure and shock. Crit Care Med 2009; 37:1670–1677.
- Liu V, Morehouse JW, Soule J, Whippy A, Escobar GJ. Fluid volume, lactate values, and mortality in sepsis patients with intermediate lactate values. Ann Am Thorac Soc 2013; 10:466–473.
- Sterling SA, Puskarich MA, Shapiro NI, et al; Emergency Medicine Shock Research Network (EMShockNET). Characteristics and outcomes of patients with vasoplegic versus tissue dysoxic septic shock. Shock 2013; 40:11–14.
- Arnold RC, Shapiro NI, Jones AE, et al; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of early lactate clearance as a determinant of survival in patients with presumed sepsis. Shock 2009; 32:35–39.
- Jones AE. Lactate clearance for assessing response to resuscitation in severe sepsis. Acad Emerg Med 2013; 20:844–847.
- Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med 2004; 32:1637–1642.
- Jansen TC, van Bommel J, Schoonderbeek FJ, et al; LACTATE study group. Early lactate-guided therapy in intensive care unit patients: a multicenter, open-label, randomized controlled trial. Am J Respir Crit Care Med 2010; 182:752–761.
- Husain FA, Martin MJ, Mullenix PS, Steele SR, Elliott DC. Serum lactate and base deficit as predictors of mortality and morbidity. Am J Surg 2003; 185:485–491.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Whole blood lactate kinetics in patients undergoing quantitative resuscitation for severe sepsis and septic shock. Chest 2013; 143:1548–1553.
- Marty P, Roquilly A, Vallee F, et al. Lactate clearance for death prediction in severe sepsis or septic shock patients during the first 24 hours in intensive care unit: an observational study. Ann Intensive Care 2013; 3:3.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: International guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Vincent JL, De Backer D. Circulatory shock. N Engl J Med 2013; 369:1726–1734.
- Durairaj L, Schmidt GA. Fluid therapy in resuscitated sepsis: less is more. Chest 2008; 133:252–263.
- Vincent JL, Dufaye P, Berré J, Leeman M, Degaute JP, Kahn RJ. Serial lactate determinations during circulatory shock. Crit Care Med 1983; 11:449–451.
- Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med 2011; 39:259–265.
- Finfer S, Bellomo R, Boyce N, French J, Myburgh J, Norton R; SAFE Study Investigators. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med 2004; 350:2247–2256.
- Myburgh JA, Finfer S, Bellomo R, et al; CHEST Investigators; Australian and New Zealand Intensive Care Society Clinical Trials Group. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med 2012; 367:1901–1911.
- Perner A, Haase N, Guttormsen AB, et al; 6S Trial Group; Scandinavian Critical Care Trials Group. Hydroxyethyl starch 130/0.42 versus Ringer’s acetate in severe sepsis. N Engl J Med 2012; 367:124–134.
- Bayer O, Reinhart K, Kohl M, et al. Effects of fluid resuscitation with synthetic colloids or crystalloids alone on shock reversal, fluid balance, and patient outcomes in patients with severe sepsis: a prospective sequential analysis. Crit Care Med 2012; 40:2543–2551.
- Delaney AP, Dan A, McCaffrey J, Finfer S. The role of albumin as a resuscitation fluid for patients with sepsis: a systematic review and meta-analysis. Crit Care Med 2011; 39:386–391.
- SAFE Study Investigators; Finfer S, McEvoy S, Bellomo R, McArthur C, Myburgh J, Norton R. Impact of albumin compared to saline on organ function and mortality of patients with severe sepsis. Intensive Care Med 2011; 37:86–96.
- Caironi P, Tognoni G, Masson S, et al; ALBIOS Study Investigators. Albumin replacement in patients with severe sepsis or septic shock. N Engl J Med 2014; 370:1412–1421.
- Bourgoin A, Leone M, Delmas A, Garnier F, Albanèse J, Martin C. Increasing mean arterial pressure in patients with septic shock: effects on oxygen variables and renal function. Crit Care Med 2005; 33:780–786.
- LeDoux D, Astiz ME, Carpati CM, Rackow EC. Effects of perfusion pressure on tissue perfusion in septic shock. Crit Care Med 2000; 28:2729–2732.
- Asfar P, Meziani F, Hamel JF, et al; SEPSISPAM Investigators. High versus low blood-pressure target in patients with septic shock. N Engl J Med 2014; 370:1583–1593.
- De Backer D, Biston P, Devriendt J, et al; SOAP II Investigators. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789.
- De Backer D, Aldecoa C, Njimi H, Vincent JL. Dopamine versus norepinephrine in the treatment of septic shock: a meta-analysis. Crit Care Med 2012; 40:725–730.
- Annane D, Vignon P, Renault A, et al: CATS Study Group. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet 2007; 370:676–684.
- Myburgh JA, Higgins A, Jovanovska A, Lipman J, Ramakrishnan N, Santamaria J; CAT Study investigators. A comparison of epinephrine and norepinephrine in critically ill patients. Intensive Care Med 2008; 34:2226–2234.
- Schmittinger CA, Torgersen C, Luckner G, Schröder DC, Lorenz I, Dünser MW. Adverse cardiac events during catecholamine vasopressor therapy: a prospective observational study. Intensive Care Med 2012; 38:950–958.
- Levy B, Perez P, Perny J, Thivilier C, Gerard A. Comparison of norepinephrine-dobutamine to epinephrine for hemodynamics, lactate metabolism, and organ function variables in cardiogenic shock. A prospective, randomized pilot study. Crit Care Med 2011; 39:450–455.
- Watt MJ, Howlett KF, Febbraio MA, Spriet LL, Hargreaves M. Adrenaline increases skeletal muscle glycogenolysis, pyruvate dehydrogenase activation and carbohydrate oxidation during moderate exercise in humans. J Physiol 2001; 534:269–278.
- De Backer D, Creteur J, Silva E, Vincent JL. Effects of dopamine, norepinephrine, and epinephrine on the splanchnic circulation in septic shock: which is best? Crit Care Med 2003; 31:1659–1667.
- Levy B, Bollaert PE, Charpentier C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med 1997; 23:282–287.
- Polito A, Parisini E, Ricci Z, Picardo S, Annane D. Vasopressin for treatment of vasodilatory shock: an ESICM systematic review and meta-analysis. Intensive Care Med 2012; 38:9–19.
- Serpa Neto A, Nassar APJ, Cardoso SO, et al. Vasopressin and terlipressin in adult vasodilatory shock: a systematic review and meta-analysis of nine randomized controlled trials. Crit Care 2012; 16:R154.
- Russell JA, Walley KR, Singer J, et al; VASST Investigators. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008; 358:877–887.
- Gattinoni L, Brazzi L, Pelosi P, et al; for the SvO2 Collaborative Group. A trial of goal-oriented hemodynamic therapy in critically ill patients. N Engl J Med 1995; 333:1025–1032.
- Rivers E, Nguyen B, Havstad S, et al; Early Goal-Directed Therapy Collaborative Group. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- ProCESS Investigators; Yealy DM, Kellum JA, Huang DT, et al. A randomized trial of protocol-based care for early septic shock. N Engl J Med 2014; 370:1683–1693.
- McNelis J, Marini CP, Jurkiewicz A, et al. Prolonged lactate clearance is associated with increased mortality in the surgical intensive care unit. Am J Surg 2001; 182:481–485.
- Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA 2010; 303:739–746.
- Pope JV, Jones AE, Gaieski DF, Arnold RC, Trzeciak S, Shapiro NI; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of central venous oxygen saturation (ScvO2) as a predictor of mortality in patients with sepsis. Ann Emerg Med 2010; 55:40–46.e1
- Kraut JA, Kurtz I. Use of base in the treatment of severe acidemic states. Am J Kidney Dis 2001; 38:703–727.
- Levraut J, Grimaud D. Treatment of metabolic acidosis. Curr Opin Crit Care 2003; 9:260–265.
- Orija AA, Jenks CL. Nucleoside analog reverse transcriptase inhibitor induced lactic acidosis treated with continuous renal replacement in the medical intensive care unit. Crit Care & Shock 2012; 15:9–11.
- Friesecke S, Abel P, Kraft M, Gerner A, Runge S. Combined renal replacement therapy for severe metformin-induced lactic acidosis. Nephrol Dial Transplant 2006; 21:2038–2039.
- Claessens YE, Cariou A, Monchi M, et al. Detecting life-threatening lactic acidosis related to nucleoside-analog treatment of human immunodeficiency virus-infected patients, and treatment with l-carnitine. Crit Care Med 2003; 31:1042–1047.
- Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R; Medicine Society TM. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol 2009; 11:414–430.
- Andersen LW, Mackenhauer J, Roberts JC, Berg KM, Cocchi MN, Donnino MW. Etiology and therapeutic approach to elevated lactate levels. Mayo Clin Proc 2013; 88:1127–1140.
- Fuller BM, Dellinger RP. Lactate as a hemodynamic marker in the critically ill. Curr Opin Crit Care 2012; 18:267–272.
- Fall PJ, Szerlip HM. Lactic acidosis: from sour milk to septic shock. J Intensive Care Med 2005; 20:255–271.
- Kruse O, Grunnet N, Barfod C. Blood lactate as a predictor for in-hospital mortality in patients admitted acutely to hospital: a systematic review. Scand J Trauma Resusc Emerg Med 2011;19:74.
- Howell MD, Donnino M, Clardy P, Talmor D, Shapiro NI. Occult hypoperfusion and mortality in patients with suspected infection. Intensive Care Med 2007; 33:1892–1899.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Outcomes of patients undergoing early sepsis resuscitation for cryptic shock compared with overt shock. Resuscitation 2011; 82:1289–1293.
- Bakker J, Nijsten MW, Jansen TC. Clinical use of lactate monitoring in critically ill patients. Ann Intensive Care 2013; 3:12.
- Levy B, Gibot S, Franck P, Cravoisy A, Bollaert PE. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet 2005; 365:871–875.
- Vary TC. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 1996; 6:89–94.
- Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360:219–223.
- Shapiro NI, Howell MD, Talmor D, et al. Serum lactate as a predictor of mortality in emergency department patients with infection. Ann Emerg Med 2005; 45:524–528.
- Mikkelsen ME, Miltiades AN, Gaieski DF, et al. Serum lactate is associated with mortality in severe sepsis independent of organ failure and shock. Crit Care Med 2009; 37:1670–1677.
- Liu V, Morehouse JW, Soule J, Whippy A, Escobar GJ. Fluid volume, lactate values, and mortality in sepsis patients with intermediate lactate values. Ann Am Thorac Soc 2013; 10:466–473.
- Sterling SA, Puskarich MA, Shapiro NI, et al; Emergency Medicine Shock Research Network (EMShockNET). Characteristics and outcomes of patients with vasoplegic versus tissue dysoxic septic shock. Shock 2013; 40:11–14.
- Arnold RC, Shapiro NI, Jones AE, et al; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of early lactate clearance as a determinant of survival in patients with presumed sepsis. Shock 2009; 32:35–39.
- Jones AE. Lactate clearance for assessing response to resuscitation in severe sepsis. Acad Emerg Med 2013; 20:844–847.
- Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med 2004; 32:1637–1642.
- Jansen TC, van Bommel J, Schoonderbeek FJ, et al; LACTATE study group. Early lactate-guided therapy in intensive care unit patients: a multicenter, open-label, randomized controlled trial. Am J Respir Crit Care Med 2010; 182:752–761.
- Husain FA, Martin MJ, Mullenix PS, Steele SR, Elliott DC. Serum lactate and base deficit as predictors of mortality and morbidity. Am J Surg 2003; 185:485–491.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Whole blood lactate kinetics in patients undergoing quantitative resuscitation for severe sepsis and septic shock. Chest 2013; 143:1548–1553.
- Marty P, Roquilly A, Vallee F, et al. Lactate clearance for death prediction in severe sepsis or septic shock patients during the first 24 hours in intensive care unit: an observational study. Ann Intensive Care 2013; 3:3.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: International guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Vincent JL, De Backer D. Circulatory shock. N Engl J Med 2013; 369:1726–1734.
- Durairaj L, Schmidt GA. Fluid therapy in resuscitated sepsis: less is more. Chest 2008; 133:252–263.
- Vincent JL, Dufaye P, Berré J, Leeman M, Degaute JP, Kahn RJ. Serial lactate determinations during circulatory shock. Crit Care Med 1983; 11:449–451.
- Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med 2011; 39:259–265.
- Finfer S, Bellomo R, Boyce N, French J, Myburgh J, Norton R; SAFE Study Investigators. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med 2004; 350:2247–2256.
- Myburgh JA, Finfer S, Bellomo R, et al; CHEST Investigators; Australian and New Zealand Intensive Care Society Clinical Trials Group. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med 2012; 367:1901–1911.
- Perner A, Haase N, Guttormsen AB, et al; 6S Trial Group; Scandinavian Critical Care Trials Group. Hydroxyethyl starch 130/0.42 versus Ringer’s acetate in severe sepsis. N Engl J Med 2012; 367:124–134.
- Bayer O, Reinhart K, Kohl M, et al. Effects of fluid resuscitation with synthetic colloids or crystalloids alone on shock reversal, fluid balance, and patient outcomes in patients with severe sepsis: a prospective sequential analysis. Crit Care Med 2012; 40:2543–2551.
- Delaney AP, Dan A, McCaffrey J, Finfer S. The role of albumin as a resuscitation fluid for patients with sepsis: a systematic review and meta-analysis. Crit Care Med 2011; 39:386–391.
- SAFE Study Investigators; Finfer S, McEvoy S, Bellomo R, McArthur C, Myburgh J, Norton R. Impact of albumin compared to saline on organ function and mortality of patients with severe sepsis. Intensive Care Med 2011; 37:86–96.
- Caironi P, Tognoni G, Masson S, et al; ALBIOS Study Investigators. Albumin replacement in patients with severe sepsis or septic shock. N Engl J Med 2014; 370:1412–1421.
- Bourgoin A, Leone M, Delmas A, Garnier F, Albanèse J, Martin C. Increasing mean arterial pressure in patients with septic shock: effects on oxygen variables and renal function. Crit Care Med 2005; 33:780–786.
- LeDoux D, Astiz ME, Carpati CM, Rackow EC. Effects of perfusion pressure on tissue perfusion in septic shock. Crit Care Med 2000; 28:2729–2732.
- Asfar P, Meziani F, Hamel JF, et al; SEPSISPAM Investigators. High versus low blood-pressure target in patients with septic shock. N Engl J Med 2014; 370:1583–1593.
- De Backer D, Biston P, Devriendt J, et al; SOAP II Investigators. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789.
- De Backer D, Aldecoa C, Njimi H, Vincent JL. Dopamine versus norepinephrine in the treatment of septic shock: a meta-analysis. Crit Care Med 2012; 40:725–730.
- Annane D, Vignon P, Renault A, et al: CATS Study Group. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet 2007; 370:676–684.
- Myburgh JA, Higgins A, Jovanovska A, Lipman J, Ramakrishnan N, Santamaria J; CAT Study investigators. A comparison of epinephrine and norepinephrine in critically ill patients. Intensive Care Med 2008; 34:2226–2234.
- Schmittinger CA, Torgersen C, Luckner G, Schröder DC, Lorenz I, Dünser MW. Adverse cardiac events during catecholamine vasopressor therapy: a prospective observational study. Intensive Care Med 2012; 38:950–958.
- Levy B, Perez P, Perny J, Thivilier C, Gerard A. Comparison of norepinephrine-dobutamine to epinephrine for hemodynamics, lactate metabolism, and organ function variables in cardiogenic shock. A prospective, randomized pilot study. Crit Care Med 2011; 39:450–455.
- Watt MJ, Howlett KF, Febbraio MA, Spriet LL, Hargreaves M. Adrenaline increases skeletal muscle glycogenolysis, pyruvate dehydrogenase activation and carbohydrate oxidation during moderate exercise in humans. J Physiol 2001; 534:269–278.
- De Backer D, Creteur J, Silva E, Vincent JL. Effects of dopamine, norepinephrine, and epinephrine on the splanchnic circulation in septic shock: which is best? Crit Care Med 2003; 31:1659–1667.
- Levy B, Bollaert PE, Charpentier C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med 1997; 23:282–287.
- Polito A, Parisini E, Ricci Z, Picardo S, Annane D. Vasopressin for treatment of vasodilatory shock: an ESICM systematic review and meta-analysis. Intensive Care Med 2012; 38:9–19.
- Serpa Neto A, Nassar APJ, Cardoso SO, et al. Vasopressin and terlipressin in adult vasodilatory shock: a systematic review and meta-analysis of nine randomized controlled trials. Crit Care 2012; 16:R154.
- Russell JA, Walley KR, Singer J, et al; VASST Investigators. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008; 358:877–887.
- Gattinoni L, Brazzi L, Pelosi P, et al; for the SvO2 Collaborative Group. A trial of goal-oriented hemodynamic therapy in critically ill patients. N Engl J Med 1995; 333:1025–1032.
- Rivers E, Nguyen B, Havstad S, et al; Early Goal-Directed Therapy Collaborative Group. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- ProCESS Investigators; Yealy DM, Kellum JA, Huang DT, et al. A randomized trial of protocol-based care for early septic shock. N Engl J Med 2014; 370:1683–1693.
- McNelis J, Marini CP, Jurkiewicz A, et al. Prolonged lactate clearance is associated with increased mortality in the surgical intensive care unit. Am J Surg 2001; 182:481–485.
- Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA 2010; 303:739–746.
- Pope JV, Jones AE, Gaieski DF, Arnold RC, Trzeciak S, Shapiro NI; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of central venous oxygen saturation (ScvO2) as a predictor of mortality in patients with sepsis. Ann Emerg Med 2010; 55:40–46.e1
- Kraut JA, Kurtz I. Use of base in the treatment of severe acidemic states. Am J Kidney Dis 2001; 38:703–727.
- Levraut J, Grimaud D. Treatment of metabolic acidosis. Curr Opin Crit Care 2003; 9:260–265.
- Orija AA, Jenks CL. Nucleoside analog reverse transcriptase inhibitor induced lactic acidosis treated with continuous renal replacement in the medical intensive care unit. Crit Care & Shock 2012; 15:9–11.
- Friesecke S, Abel P, Kraft M, Gerner A, Runge S. Combined renal replacement therapy for severe metformin-induced lactic acidosis. Nephrol Dial Transplant 2006; 21:2038–2039.
- Claessens YE, Cariou A, Monchi M, et al. Detecting life-threatening lactic acidosis related to nucleoside-analog treatment of human immunodeficiency virus-infected patients, and treatment with l-carnitine. Crit Care Med 2003; 31:1042–1047.
- Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R; Medicine Society TM. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol 2009; 11:414–430.
KEY POINTS
- Serum lactate levels can become elevated by a variety of underlying processes, categorized as increased production in conditions of hypoperfusion and hypoxia (type A lactic acidosis), or as increased production or decreased clearance not due to hypoperfusion and hypoxia (type B).
- The higher the lactate level and the slower the rate of normalization (lactate clearance), the higher the risk of death.
- Treatments differ depending on the underlying mechanism of the lactate elevation. Thus, identifying the reason for hyperlactatemia and differentiating between type A and B lactic acidosis are of the utmost importance.
- Treatment of type A lactic acidosis aims to improve perfusion and match oxygen consumption with oxygen delivery by giving fluids, packed red blood cells, and vasopressors or inotropic agents, or both.
- Treatment of type B involves more specific management, such as discontinuing offending medications or supplementing key cofactors for anaerobic metabolism.
Middle East respiratory syndrome: SARS redux?
Middle East respiratory syndrome (MERS) is a potentially lethal illness caused by the Middle East respiratory syndrome coronavirus (MERS-CoV). The virus was first reported in 2012, when it was isolated from the sputum of a previously healthy man in Saudi Arabia who presented with acute pneumonia and subsequent renal failure with a fatal outcome.1 Retrospective studies subsequently identified an earlier outbreak that year involving 13 patients in Jordan, and since then cases have been reported in 25 countries across the Arabian Peninsula and in Asia, Europe, Africa, and the United States, with over 1,000 confirmed cases and 450 related deaths.2,3
At the time of this writing, two cases of MERS have been reported in the United States, both in May 2014. Both reported cases involved patients who had traveled from Saudi Arabia, and which did not result in secondary cases.4 Beginning in May 2015, the Republic of Korea had experienced the largest known outbreak of MERS outside the Arabian Peninsula, with over 100 cases.5
THE VIRUS
MERS-CoV is classified as a coronavirus, which is a family of single-stranded RNA viruses. In 2003, a previously unknown coronavirus (SARS-CoV) caused a global outbreak of pneumonia that resulted in approximately 800 deaths.6 The MERS-CoV virus attaches to dipeptidyl peptidase 4 to enter cells, and this receptor is believed to be critical for pathogenesis, as infection does not occur in its absence.7
The source and mode of transmission to humans is not completely defined. Early reports suggested that MERS-CoV originated in bats, as RNA sequences related to MERS-CoV have been found in several bat species, but the virus itself has not been isolated from bats.8 Camels have been found to have a high rate of anti-MERS-CoV antibodies and to have the virus in nose swabs, and evidence for camel-to-human transmission has been presented.9–11 However, the precise role of camels and other animals as reservoirs or vectors of infection is still under investigation.
The incubation period from exposure to the development of clinical disease is estimated at 5 to 14 days.
For MERS-CoV, the basic reproduction ratio (R0), which measures the average number of secondary cases from each infected person, is estimated12 to be less than 0.7. In diseases in which the R0 is less than 1.0, infections occur in isolated clusters as limited chains of transmission, and thus the sustained transmission of MERS-CoV resulting in a large epidemic is thought to be unlikely. As a comparison, the median R0 value for seasonal influenza is estimated13 at 1.28. “Superspreading” may result in limited outbreaks of secondary cases; however, the continued epidemic spread of infection is thought to be unlikely.14 Nevertheless, viral adaptation with increased transmissibility remains a concern and a potential threat.
CLINICAL PRESENTATION
MERS most commonly presents as a respiratory illness, although asymptomatic infection occurs. The percentage of patients who experience asymptomatic infection is unknown. A recent survey of 255 patients with laboratory-confirmed MERS-CoV found that 64 (25.1%) were reported as asymptomatic at time of specimen collection. However, when 33 (52%) of those patients were interviewed, 26 (79%) reported at least one symptom that was consistent with a viral respiratory illness.15
For symptomatic patients, the initial complaints are nonspecific, beginning with fever, cough, sore throat, chills, and myalgia. Patients experiencing severe infection progress to dyspnea and pneumonia, with requirements for ventilatory support, vasopressors, and renal replacement therapy.16 Gastrointestinal symptoms such as vomiting and diarrhea have been reported in about one-third of patients.17
In a study of 47 patients with MERS-CoV, most of whom had underlying medical illnesses, 42 (89%) required intensive care and 34 (72%) required mechanical ventilation.17 The case-fatality rate in this study was 60%, but other studies have reported rates closer to 30%.15
Laboratory findings in patients with MERS-CoV infection usually include leukopenia and thrombocytopenia. Severely ill patients may have evidence of acute kidney injury.
Radiographic findings of MERS are those of viral pneumonitis and acute respiratory distress syndrome. Computed tomographic findings include ground-glass opacities, with peripheral lower-lobe preference.18
DIAGNOSIS
As MERS is a respiratory illness, sampling of respiratory secretions provides the highest yield for diagnosis. A study of 112 patients with MERS-CoV reported that polymerase chain reaction (PCR) testing of tracheal aspirates and bronchoalveolar lavage samples yielded significantly higher MERS-CoV loads than nasopharyngeal swab samples and sputum samples.19 However, upper respiratory tract testing is less invasive, and a positive nasopharyngeal swab result may obviate the need for further testing.
The US Centers for Disease Control and Prevention (CDC) recommends collecting multiple specimens from different sites at different times after the onset of symptoms in order to increase the diagnostic yield. Specifically, it recommends testing a lower respiratory specimen (eg, sputum, bronchoalveolar lavage fluid, tracheal aspirate), a nasopharyngeal and oropharyngeal swab, and serum, using the CDC MERS-CoV rRT-PCR assay. In addition, for patients whose symptoms began more than 14 days earlier, the CDC also recommends testing a serum specimen with the CDC MERS-CoV serologic assay. As these guidelines are updated frequently, clinicians are advised to check the CDC website for the most up-to-date information (www.cdc.gov/coronavirus/mers/guidelines-clinical-specimens.html).20 The identification of MERS-CoV by virus isolation in cell culture is not recommended and, if pursued, must be performed in a biosafety level 3 facility. (Level 3 is the second-highest level of biosafety. The highest, level 4, is reserved for extremely dangerous agents such as Ebola virus).20
Given the nonspecific clinical presentation of MERS-CoV, clinicians may consider testing for other respiratory pathogens. A recent review of 54 travelers to California from MERS-CoV-affected areas found that while none tested positive for MERS-CoV, 32 (62%) of 52 travelers had other respiratory viruses.21 When testing for alternative pathogens, clinicians should order molecular or antigen-based detection methods.
TREATMENT
Unfortunately, treatment for MERS is primarily supportive.
Ribavirin and interferon alfa-2b demonstrated activity in an animal model, but the regimen was ineffective when given a median of 19 (range 10–22) days after admission in 5 critically ill patients who subsequently died.22 A retrospective analysis comparing 20 patients with severe MERS-CoV who received ribavirin and interferon alfa-2a with 24 patients who did not reported that while survival was improved at 14 days, the mortality rates were similar at 28 days.23
A systematic review of treatments used for severe acute respiratory syndrome (SARS) reported that most studies investigating steroid use were inconclusive and some showed possible harm, suggesting that systemic steroids should be avoided in coronavirus infections.24
PREVENTION
Healthcare-associated outbreaks of MERS are well described, and thus recognition of potential cases and prompt institution of appropriate infection control measures are critical.15,25

Healthcare providers should ask patients about recent travel history and ascertain if they meet the CDC criteria for a “patient under investigation” (PUI), ie, if they have both clinical features and an epidemiologic risk of MERS (Table 1). However, these recommendations for identification will assuredly change as the outbreak matures, and healthcare providers should refer to the CDC website for the most up-to-date information.
Once a PUI is identified, standard, contact, and airborne precautions are advised. These measures include performing hand hygiene and donning personal protective equipment, including gloves, gowns, eye protection, and respiratory protection (ie, a respirator) that is at least as protective as a fit-tested National Institute for Occupational Safety and Health-certified N95 filtering face-piece respirator. In addition, a patient with possible MERS should be placed in an airborne infection isolation room.
Traveler’s advice
The CDC does not currently recommend that Americans change their travel plans because of MERS. Clinicians performing pretravel evaluations should advise patients of current information on MERS. Patients at risk for MERS who develop a respiratory illness within 14 days of return should seek medical attention and inform healthcare providers of their travel history.
SUMMARY
Recent experience with SARS, Ebola virus disease, and now MERS-CoV highlights the impact of global air travel as a vector for the rapid worldwide dissemination of communicable diseases. Healthcare providers should elicit a travel history in all patients presenting with a febrile illness, as an infection acquired in one continent may not become manifest until the patient presents in another.
The scope of the current MERS-CoV outbreak is still evolving, with concerns that viral evolution could result in a SARS-like outbreak, as experienced almost a decade ago.
Healthcare providers are advised to screen patients at risk for MERS-CoV for respiratory symptoms, and to institute appropriate infection control measures. Through recognition and isolation, healthcare providers are at the front line in limiting the spread of this potentially lethal virus.
- Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 2012; 367:1814–1820.
- Al-Abdallat MM, Payne DC, Alqasrawi S, et al. Hospital-associated outbreak of Middle East respiratory syndrome coronavirus: a serologic, epidemiologic, and clinical description. Clin Infect Dis 2014; 59:1225–1233.
- World Health Organization. Frequently asked questions on Middle East respiratory syndrome coronavirus (MERS-CoV). www.who.int/csr/disease/coronavirus_infections/faq/en/. Accessed July 29, 2015.
- Bialek SR, Allen D, Alvarado-Ramy F, et al; Centers for Disease Control and Prevention (CDC). First confirmed cases of Middle East respiratory syndrome coronavirus (MERS-CoV) infection in the United States, updated information on the epidemiology of MERS-CoV infection, and guidance for the public, clinicians, and public health authorities—May 2014. MMWR Morb Mortal Wkly Rep 2014; 63:431–436.
- World Health Organization. Middle East respiratory syndrome coronavirus (MERS-CoV) – Republic of Korea. www.who.int/csr/don/12-june-2015-mers-korea/en/. Accessed July 29, 2015.
- Peiris JSM, Guan Y, Yuen KY. Severe acute respiratory syndrome. Nat Med 2004; 10:S88–S97.
- van Doremalen N, Miazqowicz KL, Milne-Price S, et al. Host species restriction of Middle East respiratory syndrome coronavirus through its receptor, dipeptidyl peptidase 4. J Virol 2014; 88:9220–9232.
- Zumla A, Hui DS, Perlman S. Middle East respiratory syndrome. Lancet 2015; S0140-6736(15)60454-604548 (Epub ahead of print).
- Meyer B, Muller MA, Corman WM, et al. Antibodies against MERS coronavirus in dromedary camels, United Arab Emirates, 2003 and 2013. Emerg Infect Dis 2014; 20:552–559.
- Haagmans BL, Al Dhahiry SH, Reusken CB, et al. Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect Dis 2014; 14:140–145.
- Azhar EI, El-Kafrawy SA, Farraj SA, et al. Evidence for camel-to-human transmission of MERS coronavirus. N Engl J Med 2014; 370:2499–2505.
- Chowell G, Blumberg S, Simonsen L, Miller MA, Viboud C. Synthesizing data and models for the spread of MERS-CoV, 2013: key role of index cases and hospital transmission. Epidemics 2014; 9:40–51.
- Biggerstaff M, Chauchemez S, Reed C, Gambhir M, Finelli L. Estimates of the reproduction number for seasonal, pandemic, and zoonotic influenza: a systematic review of the literature. BMC Infect Dis 2014: 14:480.
- Kucharski AJ, Althaus CL. The role of superspreading in Middle East respiratory syndrome coronavirus (MERS-CoV) transmission. Euro Surveill 2015; 20.
- Oboho I, Tomczyk S, Al-Asmari A, et al. 2014 MERS-CoV outbreak in Jeddah—a link to health care facilities. N Engl J Med 2015; 372:846–854.
- Arabi YM, Arifi AA, Balkhy HH, et al. Clinical course and outcomes of critically ill patients with Middle East respiratory syndrome coronavirus infection. Ann Intern Med 2014; 160:389–397.
- Assiri A, Al-Tawfig JA, Al-Rabeeah AA, et al. Epidemiological, demographic, and clinical characteristics of 47 cases of Middle East respiratory syndrome coronavirus disease from Saudi Arabia: a descriptive study. Lancet Infect Dis 2013; 13:752–761.
- Das KM, Lee EY, Enani MA, et al. CT correlation with outcomes in 15 patients with acute Middle East respiratory syndrome coronavirus. AJR Am J Roentgenol 2015; 204:736–742.
- Memish ZA, Al-Tawfiq JA, Makhdoom HQ, et al. Respiratory tract samples, viral load, and genome fraction yield in patients with Middle East respiratory syndrome. J Infect Dis 2014; 210:1590–1594.
- Centers for Disease Control and Prevention. Middle East respiratory syndrome (MERS). Interim guidelines for collecting, handling, and testing clinical specimens from patients under investigation (PUIs) for Middle East respiratory syndrome coronavirus (MERS-CoV)—version 2.1. www.cdc.gov/coronavirus/mers/guidelines-clinical-specimens.html. Accessed July 29, 2015.
- Shakhkarami M, Yen C, Glaser CA, Xia D, Watt J, Wadford DA. Laboratory testing for Middle East respiratory syndrome coronavirus, California, USA, 2013–2014. Emerg Infect Dis 2015; 21: E-pub ahead of print. wwwnc.cdc.gov/eid/article/21/9/15-0476_article. Accessed July 29, 2015.
- Al-Tawfiq JA, Momattin H, Dib J, Memish ZA. Ribavirin and interferon therapy in patients infected with the Middle East respiratory syndrome coronavirus: an observational study. Int J Infect Dis 2014; 20:42–46.
- Omrani AS, Saad MM, Baig K, et al. Ribavirin and interferon alfa-2a for severe Middle East respiratory syndrome coronavirus infection: a retrospective cohort study. Lancet Infect Dis 2014; 14:1090–1095.
- Stockman LJ, Bellamy R, Garner, P. SARS: systematic review of treatment effects. PLoS Med 2006; 3:e343.
- Assiri A, McGeer A, Perl TM, et al; KSA MERS-CoV Investigation Team. Hospital outbreak of Middle East respiratory syndrome coronavirus. N Engl J Med 2013; 369:407–416.
Middle East respiratory syndrome (MERS) is a potentially lethal illness caused by the Middle East respiratory syndrome coronavirus (MERS-CoV). The virus was first reported in 2012, when it was isolated from the sputum of a previously healthy man in Saudi Arabia who presented with acute pneumonia and subsequent renal failure with a fatal outcome.1 Retrospective studies subsequently identified an earlier outbreak that year involving 13 patients in Jordan, and since then cases have been reported in 25 countries across the Arabian Peninsula and in Asia, Europe, Africa, and the United States, with over 1,000 confirmed cases and 450 related deaths.2,3
At the time of this writing, two cases of MERS have been reported in the United States, both in May 2014. Both reported cases involved patients who had traveled from Saudi Arabia, and which did not result in secondary cases.4 Beginning in May 2015, the Republic of Korea had experienced the largest known outbreak of MERS outside the Arabian Peninsula, with over 100 cases.5
THE VIRUS
MERS-CoV is classified as a coronavirus, which is a family of single-stranded RNA viruses. In 2003, a previously unknown coronavirus (SARS-CoV) caused a global outbreak of pneumonia that resulted in approximately 800 deaths.6 The MERS-CoV virus attaches to dipeptidyl peptidase 4 to enter cells, and this receptor is believed to be critical for pathogenesis, as infection does not occur in its absence.7
The source and mode of transmission to humans is not completely defined. Early reports suggested that MERS-CoV originated in bats, as RNA sequences related to MERS-CoV have been found in several bat species, but the virus itself has not been isolated from bats.8 Camels have been found to have a high rate of anti-MERS-CoV antibodies and to have the virus in nose swabs, and evidence for camel-to-human transmission has been presented.9–11 However, the precise role of camels and other animals as reservoirs or vectors of infection is still under investigation.
The incubation period from exposure to the development of clinical disease is estimated at 5 to 14 days.
For MERS-CoV, the basic reproduction ratio (R0), which measures the average number of secondary cases from each infected person, is estimated12 to be less than 0.7. In diseases in which the R0 is less than 1.0, infections occur in isolated clusters as limited chains of transmission, and thus the sustained transmission of MERS-CoV resulting in a large epidemic is thought to be unlikely. As a comparison, the median R0 value for seasonal influenza is estimated13 at 1.28. “Superspreading” may result in limited outbreaks of secondary cases; however, the continued epidemic spread of infection is thought to be unlikely.14 Nevertheless, viral adaptation with increased transmissibility remains a concern and a potential threat.
CLINICAL PRESENTATION
MERS most commonly presents as a respiratory illness, although asymptomatic infection occurs. The percentage of patients who experience asymptomatic infection is unknown. A recent survey of 255 patients with laboratory-confirmed MERS-CoV found that 64 (25.1%) were reported as asymptomatic at time of specimen collection. However, when 33 (52%) of those patients were interviewed, 26 (79%) reported at least one symptom that was consistent with a viral respiratory illness.15
For symptomatic patients, the initial complaints are nonspecific, beginning with fever, cough, sore throat, chills, and myalgia. Patients experiencing severe infection progress to dyspnea and pneumonia, with requirements for ventilatory support, vasopressors, and renal replacement therapy.16 Gastrointestinal symptoms such as vomiting and diarrhea have been reported in about one-third of patients.17
In a study of 47 patients with MERS-CoV, most of whom had underlying medical illnesses, 42 (89%) required intensive care and 34 (72%) required mechanical ventilation.17 The case-fatality rate in this study was 60%, but other studies have reported rates closer to 30%.15
Laboratory findings in patients with MERS-CoV infection usually include leukopenia and thrombocytopenia. Severely ill patients may have evidence of acute kidney injury.
Radiographic findings of MERS are those of viral pneumonitis and acute respiratory distress syndrome. Computed tomographic findings include ground-glass opacities, with peripheral lower-lobe preference.18
DIAGNOSIS
As MERS is a respiratory illness, sampling of respiratory secretions provides the highest yield for diagnosis. A study of 112 patients with MERS-CoV reported that polymerase chain reaction (PCR) testing of tracheal aspirates and bronchoalveolar lavage samples yielded significantly higher MERS-CoV loads than nasopharyngeal swab samples and sputum samples.19 However, upper respiratory tract testing is less invasive, and a positive nasopharyngeal swab result may obviate the need for further testing.
The US Centers for Disease Control and Prevention (CDC) recommends collecting multiple specimens from different sites at different times after the onset of symptoms in order to increase the diagnostic yield. Specifically, it recommends testing a lower respiratory specimen (eg, sputum, bronchoalveolar lavage fluid, tracheal aspirate), a nasopharyngeal and oropharyngeal swab, and serum, using the CDC MERS-CoV rRT-PCR assay. In addition, for patients whose symptoms began more than 14 days earlier, the CDC also recommends testing a serum specimen with the CDC MERS-CoV serologic assay. As these guidelines are updated frequently, clinicians are advised to check the CDC website for the most up-to-date information (www.cdc.gov/coronavirus/mers/guidelines-clinical-specimens.html).20 The identification of MERS-CoV by virus isolation in cell culture is not recommended and, if pursued, must be performed in a biosafety level 3 facility. (Level 3 is the second-highest level of biosafety. The highest, level 4, is reserved for extremely dangerous agents such as Ebola virus).20
Given the nonspecific clinical presentation of MERS-CoV, clinicians may consider testing for other respiratory pathogens. A recent review of 54 travelers to California from MERS-CoV-affected areas found that while none tested positive for MERS-CoV, 32 (62%) of 52 travelers had other respiratory viruses.21 When testing for alternative pathogens, clinicians should order molecular or antigen-based detection methods.
TREATMENT
Unfortunately, treatment for MERS is primarily supportive.
Ribavirin and interferon alfa-2b demonstrated activity in an animal model, but the regimen was ineffective when given a median of 19 (range 10–22) days after admission in 5 critically ill patients who subsequently died.22 A retrospective analysis comparing 20 patients with severe MERS-CoV who received ribavirin and interferon alfa-2a with 24 patients who did not reported that while survival was improved at 14 days, the mortality rates were similar at 28 days.23
A systematic review of treatments used for severe acute respiratory syndrome (SARS) reported that most studies investigating steroid use were inconclusive and some showed possible harm, suggesting that systemic steroids should be avoided in coronavirus infections.24
PREVENTION
Healthcare-associated outbreaks of MERS are well described, and thus recognition of potential cases and prompt institution of appropriate infection control measures are critical.15,25
Healthcare providers should ask patients about recent travel history and ascertain if they meet the CDC criteria for a “patient under investigation” (PUI), ie, if they have both clinical features and an epidemiologic risk of MERS (Table 1). However, these recommendations for identification will assuredly change as the outbreak matures, and healthcare providers should refer to the CDC website for the most up-to-date information.
Once a PUI is identified, standard, contact, and airborne precautions are advised. These measures include performing hand hygiene and donning personal protective equipment, including gloves, gowns, eye protection, and respiratory protection (ie, a respirator) that is at least as protective as a fit-tested National Institute for Occupational Safety and Health-certified N95 filtering face-piece respirator. In addition, a patient with possible MERS should be placed in an airborne infection isolation room.
Traveler’s advice
The CDC does not currently recommend that Americans change their travel plans because of MERS. Clinicians performing pretravel evaluations should advise patients of current information on MERS. Patients at risk for MERS who develop a respiratory illness within 14 days of return should seek medical attention and inform healthcare providers of their travel history.
SUMMARY
Recent experience with SARS, Ebola virus disease, and now MERS-CoV highlights the impact of global air travel as a vector for the rapid worldwide dissemination of communicable diseases. Healthcare providers should elicit a travel history in all patients presenting with a febrile illness, as an infection acquired in one continent may not become manifest until the patient presents in another.
The scope of the current MERS-CoV outbreak is still evolving, with concerns that viral evolution could result in a SARS-like outbreak, as experienced almost a decade ago.
Healthcare providers are advised to screen patients at risk for MERS-CoV for respiratory symptoms, and to institute appropriate infection control measures. Through recognition and isolation, healthcare providers are at the front line in limiting the spread of this potentially lethal virus.
Middle East respiratory syndrome (MERS) is a potentially lethal illness caused by the Middle East respiratory syndrome coronavirus (MERS-CoV). The virus was first reported in 2012, when it was isolated from the sputum of a previously healthy man in Saudi Arabia who presented with acute pneumonia and subsequent renal failure with a fatal outcome.1 Retrospective studies subsequently identified an earlier outbreak that year involving 13 patients in Jordan, and since then cases have been reported in 25 countries across the Arabian Peninsula and in Asia, Europe, Africa, and the United States, with over 1,000 confirmed cases and 450 related deaths.2,3
At the time of this writing, two cases of MERS have been reported in the United States, both in May 2014. Both reported cases involved patients who had traveled from Saudi Arabia, and which did not result in secondary cases.4 Beginning in May 2015, the Republic of Korea had experienced the largest known outbreak of MERS outside the Arabian Peninsula, with over 100 cases.5
THE VIRUS
MERS-CoV is classified as a coronavirus, which is a family of single-stranded RNA viruses. In 2003, a previously unknown coronavirus (SARS-CoV) caused a global outbreak of pneumonia that resulted in approximately 800 deaths.6 The MERS-CoV virus attaches to dipeptidyl peptidase 4 to enter cells, and this receptor is believed to be critical for pathogenesis, as infection does not occur in its absence.7
The source and mode of transmission to humans is not completely defined. Early reports suggested that MERS-CoV originated in bats, as RNA sequences related to MERS-CoV have been found in several bat species, but the virus itself has not been isolated from bats.8 Camels have been found to have a high rate of anti-MERS-CoV antibodies and to have the virus in nose swabs, and evidence for camel-to-human transmission has been presented.9–11 However, the precise role of camels and other animals as reservoirs or vectors of infection is still under investigation.
The incubation period from exposure to the development of clinical disease is estimated at 5 to 14 days.
For MERS-CoV, the basic reproduction ratio (R0), which measures the average number of secondary cases from each infected person, is estimated12 to be less than 0.7. In diseases in which the R0 is less than 1.0, infections occur in isolated clusters as limited chains of transmission, and thus the sustained transmission of MERS-CoV resulting in a large epidemic is thought to be unlikely. As a comparison, the median R0 value for seasonal influenza is estimated13 at 1.28. “Superspreading” may result in limited outbreaks of secondary cases; however, the continued epidemic spread of infection is thought to be unlikely.14 Nevertheless, viral adaptation with increased transmissibility remains a concern and a potential threat.
CLINICAL PRESENTATION
MERS most commonly presents as a respiratory illness, although asymptomatic infection occurs. The percentage of patients who experience asymptomatic infection is unknown. A recent survey of 255 patients with laboratory-confirmed MERS-CoV found that 64 (25.1%) were reported as asymptomatic at time of specimen collection. However, when 33 (52%) of those patients were interviewed, 26 (79%) reported at least one symptom that was consistent with a viral respiratory illness.15
For symptomatic patients, the initial complaints are nonspecific, beginning with fever, cough, sore throat, chills, and myalgia. Patients experiencing severe infection progress to dyspnea and pneumonia, with requirements for ventilatory support, vasopressors, and renal replacement therapy.16 Gastrointestinal symptoms such as vomiting and diarrhea have been reported in about one-third of patients.17
In a study of 47 patients with MERS-CoV, most of whom had underlying medical illnesses, 42 (89%) required intensive care and 34 (72%) required mechanical ventilation.17 The case-fatality rate in this study was 60%, but other studies have reported rates closer to 30%.15
Laboratory findings in patients with MERS-CoV infection usually include leukopenia and thrombocytopenia. Severely ill patients may have evidence of acute kidney injury.
Radiographic findings of MERS are those of viral pneumonitis and acute respiratory distress syndrome. Computed tomographic findings include ground-glass opacities, with peripheral lower-lobe preference.18
DIAGNOSIS
As MERS is a respiratory illness, sampling of respiratory secretions provides the highest yield for diagnosis. A study of 112 patients with MERS-CoV reported that polymerase chain reaction (PCR) testing of tracheal aspirates and bronchoalveolar lavage samples yielded significantly higher MERS-CoV loads than nasopharyngeal swab samples and sputum samples.19 However, upper respiratory tract testing is less invasive, and a positive nasopharyngeal swab result may obviate the need for further testing.
The US Centers for Disease Control and Prevention (CDC) recommends collecting multiple specimens from different sites at different times after the onset of symptoms in order to increase the diagnostic yield. Specifically, it recommends testing a lower respiratory specimen (eg, sputum, bronchoalveolar lavage fluid, tracheal aspirate), a nasopharyngeal and oropharyngeal swab, and serum, using the CDC MERS-CoV rRT-PCR assay. In addition, for patients whose symptoms began more than 14 days earlier, the CDC also recommends testing a serum specimen with the CDC MERS-CoV serologic assay. As these guidelines are updated frequently, clinicians are advised to check the CDC website for the most up-to-date information (www.cdc.gov/coronavirus/mers/guidelines-clinical-specimens.html).20 The identification of MERS-CoV by virus isolation in cell culture is not recommended and, if pursued, must be performed in a biosafety level 3 facility. (Level 3 is the second-highest level of biosafety. The highest, level 4, is reserved for extremely dangerous agents such as Ebola virus).20
Given the nonspecific clinical presentation of MERS-CoV, clinicians may consider testing for other respiratory pathogens. A recent review of 54 travelers to California from MERS-CoV-affected areas found that while none tested positive for MERS-CoV, 32 (62%) of 52 travelers had other respiratory viruses.21 When testing for alternative pathogens, clinicians should order molecular or antigen-based detection methods.
TREATMENT
Unfortunately, treatment for MERS is primarily supportive.
Ribavirin and interferon alfa-2b demonstrated activity in an animal model, but the regimen was ineffective when given a median of 19 (range 10–22) days after admission in 5 critically ill patients who subsequently died.22 A retrospective analysis comparing 20 patients with severe MERS-CoV who received ribavirin and interferon alfa-2a with 24 patients who did not reported that while survival was improved at 14 days, the mortality rates were similar at 28 days.23
A systematic review of treatments used for severe acute respiratory syndrome (SARS) reported that most studies investigating steroid use were inconclusive and some showed possible harm, suggesting that systemic steroids should be avoided in coronavirus infections.24
PREVENTION
Healthcare-associated outbreaks of MERS are well described, and thus recognition of potential cases and prompt institution of appropriate infection control measures are critical.15,25
Healthcare providers should ask patients about recent travel history and ascertain if they meet the CDC criteria for a “patient under investigation” (PUI), ie, if they have both clinical features and an epidemiologic risk of MERS (Table 1). However, these recommendations for identification will assuredly change as the outbreak matures, and healthcare providers should refer to the CDC website for the most up-to-date information.
Once a PUI is identified, standard, contact, and airborne precautions are advised. These measures include performing hand hygiene and donning personal protective equipment, including gloves, gowns, eye protection, and respiratory protection (ie, a respirator) that is at least as protective as a fit-tested National Institute for Occupational Safety and Health-certified N95 filtering face-piece respirator. In addition, a patient with possible MERS should be placed in an airborne infection isolation room.
Traveler’s advice
The CDC does not currently recommend that Americans change their travel plans because of MERS. Clinicians performing pretravel evaluations should advise patients of current information on MERS. Patients at risk for MERS who develop a respiratory illness within 14 days of return should seek medical attention and inform healthcare providers of their travel history.
SUMMARY
Recent experience with SARS, Ebola virus disease, and now MERS-CoV highlights the impact of global air travel as a vector for the rapid worldwide dissemination of communicable diseases. Healthcare providers should elicit a travel history in all patients presenting with a febrile illness, as an infection acquired in one continent may not become manifest until the patient presents in another.
The scope of the current MERS-CoV outbreak is still evolving, with concerns that viral evolution could result in a SARS-like outbreak, as experienced almost a decade ago.
Healthcare providers are advised to screen patients at risk for MERS-CoV for respiratory symptoms, and to institute appropriate infection control measures. Through recognition and isolation, healthcare providers are at the front line in limiting the spread of this potentially lethal virus.
- Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 2012; 367:1814–1820.
- Al-Abdallat MM, Payne DC, Alqasrawi S, et al. Hospital-associated outbreak of Middle East respiratory syndrome coronavirus: a serologic, epidemiologic, and clinical description. Clin Infect Dis 2014; 59:1225–1233.
- World Health Organization. Frequently asked questions on Middle East respiratory syndrome coronavirus (MERS-CoV). www.who.int/csr/disease/coronavirus_infections/faq/en/. Accessed July 29, 2015.
- Bialek SR, Allen D, Alvarado-Ramy F, et al; Centers for Disease Control and Prevention (CDC). First confirmed cases of Middle East respiratory syndrome coronavirus (MERS-CoV) infection in the United States, updated information on the epidemiology of MERS-CoV infection, and guidance for the public, clinicians, and public health authorities—May 2014. MMWR Morb Mortal Wkly Rep 2014; 63:431–436.
- World Health Organization. Middle East respiratory syndrome coronavirus (MERS-CoV) – Republic of Korea. www.who.int/csr/don/12-june-2015-mers-korea/en/. Accessed July 29, 2015.
- Peiris JSM, Guan Y, Yuen KY. Severe acute respiratory syndrome. Nat Med 2004; 10:S88–S97.
- van Doremalen N, Miazqowicz KL, Milne-Price S, et al. Host species restriction of Middle East respiratory syndrome coronavirus through its receptor, dipeptidyl peptidase 4. J Virol 2014; 88:9220–9232.
- Zumla A, Hui DS, Perlman S. Middle East respiratory syndrome. Lancet 2015; S0140-6736(15)60454-604548 (Epub ahead of print).
- Meyer B, Muller MA, Corman WM, et al. Antibodies against MERS coronavirus in dromedary camels, United Arab Emirates, 2003 and 2013. Emerg Infect Dis 2014; 20:552–559.
- Haagmans BL, Al Dhahiry SH, Reusken CB, et al. Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect Dis 2014; 14:140–145.
- Azhar EI, El-Kafrawy SA, Farraj SA, et al. Evidence for camel-to-human transmission of MERS coronavirus. N Engl J Med 2014; 370:2499–2505.
- Chowell G, Blumberg S, Simonsen L, Miller MA, Viboud C. Synthesizing data and models for the spread of MERS-CoV, 2013: key role of index cases and hospital transmission. Epidemics 2014; 9:40–51.
- Biggerstaff M, Chauchemez S, Reed C, Gambhir M, Finelli L. Estimates of the reproduction number for seasonal, pandemic, and zoonotic influenza: a systematic review of the literature. BMC Infect Dis 2014: 14:480.
- Kucharski AJ, Althaus CL. The role of superspreading in Middle East respiratory syndrome coronavirus (MERS-CoV) transmission. Euro Surveill 2015; 20.
- Oboho I, Tomczyk S, Al-Asmari A, et al. 2014 MERS-CoV outbreak in Jeddah—a link to health care facilities. N Engl J Med 2015; 372:846–854.
- Arabi YM, Arifi AA, Balkhy HH, et al. Clinical course and outcomes of critically ill patients with Middle East respiratory syndrome coronavirus infection. Ann Intern Med 2014; 160:389–397.
- Assiri A, Al-Tawfig JA, Al-Rabeeah AA, et al. Epidemiological, demographic, and clinical characteristics of 47 cases of Middle East respiratory syndrome coronavirus disease from Saudi Arabia: a descriptive study. Lancet Infect Dis 2013; 13:752–761.
- Das KM, Lee EY, Enani MA, et al. CT correlation with outcomes in 15 patients with acute Middle East respiratory syndrome coronavirus. AJR Am J Roentgenol 2015; 204:736–742.
- Memish ZA, Al-Tawfiq JA, Makhdoom HQ, et al. Respiratory tract samples, viral load, and genome fraction yield in patients with Middle East respiratory syndrome. J Infect Dis 2014; 210:1590–1594.
- Centers for Disease Control and Prevention. Middle East respiratory syndrome (MERS). Interim guidelines for collecting, handling, and testing clinical specimens from patients under investigation (PUIs) for Middle East respiratory syndrome coronavirus (MERS-CoV)—version 2.1. www.cdc.gov/coronavirus/mers/guidelines-clinical-specimens.html. Accessed July 29, 2015.
- Shakhkarami M, Yen C, Glaser CA, Xia D, Watt J, Wadford DA. Laboratory testing for Middle East respiratory syndrome coronavirus, California, USA, 2013–2014. Emerg Infect Dis 2015; 21: E-pub ahead of print. wwwnc.cdc.gov/eid/article/21/9/15-0476_article. Accessed July 29, 2015.
- Al-Tawfiq JA, Momattin H, Dib J, Memish ZA. Ribavirin and interferon therapy in patients infected with the Middle East respiratory syndrome coronavirus: an observational study. Int J Infect Dis 2014; 20:42–46.
- Omrani AS, Saad MM, Baig K, et al. Ribavirin and interferon alfa-2a for severe Middle East respiratory syndrome coronavirus infection: a retrospective cohort study. Lancet Infect Dis 2014; 14:1090–1095.
- Stockman LJ, Bellamy R, Garner, P. SARS: systematic review of treatment effects. PLoS Med 2006; 3:e343.
- Assiri A, McGeer A, Perl TM, et al; KSA MERS-CoV Investigation Team. Hospital outbreak of Middle East respiratory syndrome coronavirus. N Engl J Med 2013; 369:407–416.
- Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 2012; 367:1814–1820.
- Al-Abdallat MM, Payne DC, Alqasrawi S, et al. Hospital-associated outbreak of Middle East respiratory syndrome coronavirus: a serologic, epidemiologic, and clinical description. Clin Infect Dis 2014; 59:1225–1233.
- World Health Organization. Frequently asked questions on Middle East respiratory syndrome coronavirus (MERS-CoV). www.who.int/csr/disease/coronavirus_infections/faq/en/. Accessed July 29, 2015.
- Bialek SR, Allen D, Alvarado-Ramy F, et al; Centers for Disease Control and Prevention (CDC). First confirmed cases of Middle East respiratory syndrome coronavirus (MERS-CoV) infection in the United States, updated information on the epidemiology of MERS-CoV infection, and guidance for the public, clinicians, and public health authorities—May 2014. MMWR Morb Mortal Wkly Rep 2014; 63:431–436.
- World Health Organization. Middle East respiratory syndrome coronavirus (MERS-CoV) – Republic of Korea. www.who.int/csr/don/12-june-2015-mers-korea/en/. Accessed July 29, 2015.
- Peiris JSM, Guan Y, Yuen KY. Severe acute respiratory syndrome. Nat Med 2004; 10:S88–S97.
- van Doremalen N, Miazqowicz KL, Milne-Price S, et al. Host species restriction of Middle East respiratory syndrome coronavirus through its receptor, dipeptidyl peptidase 4. J Virol 2014; 88:9220–9232.
- Zumla A, Hui DS, Perlman S. Middle East respiratory syndrome. Lancet 2015; S0140-6736(15)60454-604548 (Epub ahead of print).
- Meyer B, Muller MA, Corman WM, et al. Antibodies against MERS coronavirus in dromedary camels, United Arab Emirates, 2003 and 2013. Emerg Infect Dis 2014; 20:552–559.
- Haagmans BL, Al Dhahiry SH, Reusken CB, et al. Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect Dis 2014; 14:140–145.
- Azhar EI, El-Kafrawy SA, Farraj SA, et al. Evidence for camel-to-human transmission of MERS coronavirus. N Engl J Med 2014; 370:2499–2505.
- Chowell G, Blumberg S, Simonsen L, Miller MA, Viboud C. Synthesizing data and models for the spread of MERS-CoV, 2013: key role of index cases and hospital transmission. Epidemics 2014; 9:40–51.
- Biggerstaff M, Chauchemez S, Reed C, Gambhir M, Finelli L. Estimates of the reproduction number for seasonal, pandemic, and zoonotic influenza: a systematic review of the literature. BMC Infect Dis 2014: 14:480.
- Kucharski AJ, Althaus CL. The role of superspreading in Middle East respiratory syndrome coronavirus (MERS-CoV) transmission. Euro Surveill 2015; 20.
- Oboho I, Tomczyk S, Al-Asmari A, et al. 2014 MERS-CoV outbreak in Jeddah—a link to health care facilities. N Engl J Med 2015; 372:846–854.
- Arabi YM, Arifi AA, Balkhy HH, et al. Clinical course and outcomes of critically ill patients with Middle East respiratory syndrome coronavirus infection. Ann Intern Med 2014; 160:389–397.
- Assiri A, Al-Tawfig JA, Al-Rabeeah AA, et al. Epidemiological, demographic, and clinical characteristics of 47 cases of Middle East respiratory syndrome coronavirus disease from Saudi Arabia: a descriptive study. Lancet Infect Dis 2013; 13:752–761.
- Das KM, Lee EY, Enani MA, et al. CT correlation with outcomes in 15 patients with acute Middle East respiratory syndrome coronavirus. AJR Am J Roentgenol 2015; 204:736–742.
- Memish ZA, Al-Tawfiq JA, Makhdoom HQ, et al. Respiratory tract samples, viral load, and genome fraction yield in patients with Middle East respiratory syndrome. J Infect Dis 2014; 210:1590–1594.
- Centers for Disease Control and Prevention. Middle East respiratory syndrome (MERS). Interim guidelines for collecting, handling, and testing clinical specimens from patients under investigation (PUIs) for Middle East respiratory syndrome coronavirus (MERS-CoV)—version 2.1. www.cdc.gov/coronavirus/mers/guidelines-clinical-specimens.html. Accessed July 29, 2015.
- Shakhkarami M, Yen C, Glaser CA, Xia D, Watt J, Wadford DA. Laboratory testing for Middle East respiratory syndrome coronavirus, California, USA, 2013–2014. Emerg Infect Dis 2015; 21: E-pub ahead of print. wwwnc.cdc.gov/eid/article/21/9/15-0476_article. Accessed July 29, 2015.
- Al-Tawfiq JA, Momattin H, Dib J, Memish ZA. Ribavirin and interferon therapy in patients infected with the Middle East respiratory syndrome coronavirus: an observational study. Int J Infect Dis 2014; 20:42–46.
- Omrani AS, Saad MM, Baig K, et al. Ribavirin and interferon alfa-2a for severe Middle East respiratory syndrome coronavirus infection: a retrospective cohort study. Lancet Infect Dis 2014; 14:1090–1095.
- Stockman LJ, Bellamy R, Garner, P. SARS: systematic review of treatment effects. PLoS Med 2006; 3:e343.
- Assiri A, McGeer A, Perl TM, et al; KSA MERS-CoV Investigation Team. Hospital outbreak of Middle East respiratory syndrome coronavirus. N Engl J Med 2013; 369:407–416.
KEY POINTS
- In MERS, initial complaints are of fever, cough, chills and myalgia. In a subset of patients, usually those with underlying illnesses, the disease can progress to fulminant sepsis with respiratory and renal failure and death.
- Healthcare providers should regularly visit the US Centers for Disease Control and Prevention website for current information on countries experiencing a MERS outbreak, and for advice on how to identify a potentially infected patient.
- MERS-CoV has caused several healthcare-related outbreaks, so prompt identification and isolation of infected patients is critical to limiting the spread of infection. A “patient under identification” (ie, a person who has both clinical features and an epidemiologic risk) should be cared for under standard, contact, and airborne precautions.
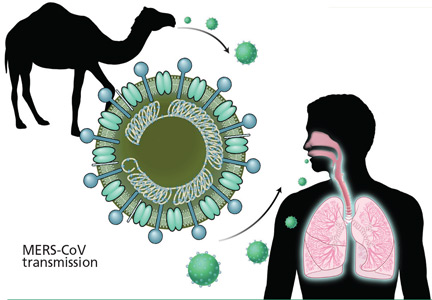
Herpes zoster triplex
A 77-year-old man presented with a 4-day history of painful eruptions on the left chest, right lower groin, and left thigh. He had been taking oral prednisolone 16 mg daily for interstitial pneumonia for 5 years. Ten days earlier, he had started to feel a stinging pain in these areas, but without eruptions.
![]()
Physical examination showed several grouped erythematous vesicles in the T2 dermatome of the left chest, L1 dermatome of the right groin, and L2 dermatome of the left upper anterior thigh (Figure 1).
Based on the presentation and a Tzanck smear of the lesions, a diagnosis of preherpetic neuralgia with herpes zoster triplex was made. The patient received intravenous acyclovir 750 mg/day for 7 days and continued to take the prednisolone. The lesions improved within 1 month, leaving scarring but no postherpetic neuralgia.
PREHERPETIC NEURALGIA
Herpes zoster usually occurs unilaterally in a single dermatome, with dermatomal pain appearing before the rash.1 Preherpetic neuralgia may be misdiagnosed as myocardial infarction or renal colic, especially in a case of zoster sine herpete.
Making the diagnosis of preherpetic neuralgia in our patient was difficult because it occurred simultaneously in three dermatomes. At first, his symptoms were suspected of being a recurrence of his past illnesses, including aortic dissection, gallstones, and diverticulitis. Anti-varicella-zoster virus immunoglobulin (Ig) M antibody was not detected, and the IgG antibody titer did not increase.
It has been suggested that cellular immunity is more important than humoral immunity for the surveillance and control of reactivations of herpes viruses. Risk factors for reactivation are increasing age, cancer, acquired immunodeficiency syndrome, and immunosuppressive medications.2,3 In addition, varicella-zoster virus can cause atypical lesions, including recurrent chickenpox, single-dermatomal herpes zoster with scattered rash, and herpes zoster in multiple dermatomes.4
Clinical suspicion for herpes zoster is important in the differential diagnosis of acute pain of uncertain origin, even if it occurs in multiple dermatomes in an immunocompromised patient.
- James WD, Berger TG, Elston DM. Andrew’s Diseases of the Skin. Clinical Dermatology, 10th ed. Philadelphia, PA: WB Saunders; 2006.
- Vu AQ, Radonich MA, Heald PW. Herpes zoster in seven disparate dermatomes (zoster multiplex): report of a case and review of literature. J Am Acad Dermatol 1999; 40:868–869.
- Failla V, Jacques J, Castronovo C, Nikkels AF. Herpes zoster in patients treated with biologicals. Dermatology 2012; 224:251–256.
- Kennedy PG, Steiner I. A molecular and cellular model to explain the differences in reactivation from latency by herpes simplex and varicella-zoster viruses. Neuropathol Appl Neurobiol 1994; 20:368–374.
A 77-year-old man presented with a 4-day history of painful eruptions on the left chest, right lower groin, and left thigh. He had been taking oral prednisolone 16 mg daily for interstitial pneumonia for 5 years. Ten days earlier, he had started to feel a stinging pain in these areas, but without eruptions.
![]()
Physical examination showed several grouped erythematous vesicles in the T2 dermatome of the left chest, L1 dermatome of the right groin, and L2 dermatome of the left upper anterior thigh (Figure 1).
Based on the presentation and a Tzanck smear of the lesions, a diagnosis of preherpetic neuralgia with herpes zoster triplex was made. The patient received intravenous acyclovir 750 mg/day for 7 days and continued to take the prednisolone. The lesions improved within 1 month, leaving scarring but no postherpetic neuralgia.
PREHERPETIC NEURALGIA
Herpes zoster usually occurs unilaterally in a single dermatome, with dermatomal pain appearing before the rash.1 Preherpetic neuralgia may be misdiagnosed as myocardial infarction or renal colic, especially in a case of zoster sine herpete.
Making the diagnosis of preherpetic neuralgia in our patient was difficult because it occurred simultaneously in three dermatomes. At first, his symptoms were suspected of being a recurrence of his past illnesses, including aortic dissection, gallstones, and diverticulitis. Anti-varicella-zoster virus immunoglobulin (Ig) M antibody was not detected, and the IgG antibody titer did not increase.
It has been suggested that cellular immunity is more important than humoral immunity for the surveillance and control of reactivations of herpes viruses. Risk factors for reactivation are increasing age, cancer, acquired immunodeficiency syndrome, and immunosuppressive medications.2,3 In addition, varicella-zoster virus can cause atypical lesions, including recurrent chickenpox, single-dermatomal herpes zoster with scattered rash, and herpes zoster in multiple dermatomes.4
Clinical suspicion for herpes zoster is important in the differential diagnosis of acute pain of uncertain origin, even if it occurs in multiple dermatomes in an immunocompromised patient.
A 77-year-old man presented with a 4-day history of painful eruptions on the left chest, right lower groin, and left thigh. He had been taking oral prednisolone 16 mg daily for interstitial pneumonia for 5 years. Ten days earlier, he had started to feel a stinging pain in these areas, but without eruptions.
![]()
Physical examination showed several grouped erythematous vesicles in the T2 dermatome of the left chest, L1 dermatome of the right groin, and L2 dermatome of the left upper anterior thigh (Figure 1).
Based on the presentation and a Tzanck smear of the lesions, a diagnosis of preherpetic neuralgia with herpes zoster triplex was made. The patient received intravenous acyclovir 750 mg/day for 7 days and continued to take the prednisolone. The lesions improved within 1 month, leaving scarring but no postherpetic neuralgia.
PREHERPETIC NEURALGIA
Herpes zoster usually occurs unilaterally in a single dermatome, with dermatomal pain appearing before the rash.1 Preherpetic neuralgia may be misdiagnosed as myocardial infarction or renal colic, especially in a case of zoster sine herpete.
Making the diagnosis of preherpetic neuralgia in our patient was difficult because it occurred simultaneously in three dermatomes. At first, his symptoms were suspected of being a recurrence of his past illnesses, including aortic dissection, gallstones, and diverticulitis. Anti-varicella-zoster virus immunoglobulin (Ig) M antibody was not detected, and the IgG antibody titer did not increase.
It has been suggested that cellular immunity is more important than humoral immunity for the surveillance and control of reactivations of herpes viruses. Risk factors for reactivation are increasing age, cancer, acquired immunodeficiency syndrome, and immunosuppressive medications.2,3 In addition, varicella-zoster virus can cause atypical lesions, including recurrent chickenpox, single-dermatomal herpes zoster with scattered rash, and herpes zoster in multiple dermatomes.4
Clinical suspicion for herpes zoster is important in the differential diagnosis of acute pain of uncertain origin, even if it occurs in multiple dermatomes in an immunocompromised patient.
- James WD, Berger TG, Elston DM. Andrew’s Diseases of the Skin. Clinical Dermatology, 10th ed. Philadelphia, PA: WB Saunders; 2006.
- Vu AQ, Radonich MA, Heald PW. Herpes zoster in seven disparate dermatomes (zoster multiplex): report of a case and review of literature. J Am Acad Dermatol 1999; 40:868–869.
- Failla V, Jacques J, Castronovo C, Nikkels AF. Herpes zoster in patients treated with biologicals. Dermatology 2012; 224:251–256.
- Kennedy PG, Steiner I. A molecular and cellular model to explain the differences in reactivation from latency by herpes simplex and varicella-zoster viruses. Neuropathol Appl Neurobiol 1994; 20:368–374.
- James WD, Berger TG, Elston DM. Andrew’s Diseases of the Skin. Clinical Dermatology, 10th ed. Philadelphia, PA: WB Saunders; 2006.
- Vu AQ, Radonich MA, Heald PW. Herpes zoster in seven disparate dermatomes (zoster multiplex): report of a case and review of literature. J Am Acad Dermatol 1999; 40:868–869.
- Failla V, Jacques J, Castronovo C, Nikkels AF. Herpes zoster in patients treated with biologicals. Dermatology 2012; 224:251–256.
- Kennedy PG, Steiner I. A molecular and cellular model to explain the differences in reactivation from latency by herpes simplex and varicella-zoster viruses. Neuropathol Appl Neurobiol 1994; 20:368–374.
What is the difference between palliative care and hospice care?
Hospice care generally falls under the category of palliative care, despite being an older subspecialty. However, the two have different indications and goals and are often provided in different settings.
ORIGINS OF PALLIATIVE CARE
Prompted by what he perceived as neglect of dying patients in the acute care setting, Dr. Balfour Mount opened the first acute inpatient palliative care unit in Royal Victoria Hospital in Montréal, Québec, in 1976.1 His purpose was to provide a crisis-intervention service for patients who were actively dying, and this continues to be the main reason for consulting palliative care services in the hospital.
Palliative care has evolved since the 1970s and is now used in a variety of situations:
- A life-limiting illness in a patient who is not terminally ill
- A life-threatening illness in a patient who has symptoms but with the potential to recover
- A chronic illness such as heart failure or chronic obstructive pulmonary disease in a patient who is on disease-modifying therapy but has symptoms and will eventually succumb to the illness, but is expected to live longer than someone with advanced cancer.2
PALLIATIVE CARE IN CANCER PATIENTS
In patients with advanced cancer, palliative care is utilized earlier in the course of serious and life-limiting illness and is even involved in patient care when cure is the goal. Importantly, it now includes outpatient clinics to provide patients seamless care in conjunction with their oncologist’s care.3
Because palliative care focuses on the patient’s experience of the illness (sickness) rather than on disease itself (pathology), symptom management, psychosocial support, and assistance in decision-making are foremost. Initiating palliative care early in advanced cancer improves multiple outcomes and limits overly aggressive, ineffective therapies at the end of life (eg, late chemotherapy, late referral to hospice care, death in the intensive care unit), without hastening death. In fact, it may prolong life.3,4
Palliative care is indicated in a number of situations in oncology:
- Symptomatic presentations of cancer, even when curative treatments are available
- At the time of a sentinel event such as recurrence or unanticipated hospitalization
- When palliative radiation is needed
- When changes in chemotherapy are needed because of disease progression.
Also, cancer patients may develop symptoms that require a palliative procedure such as thoracentesis for pleural effusion, paracentesis for ascites, or surgery for a fracture or spinal cord compression. A palliative care consultation is also appropriate when patients change their goals of care (ie, palliation rather than cure), and when an oncologic crisis occurs and there is a need to offer support to the family and to clarify the goals of care.
PALLIATIVE CARE IN OTHER DISEASES
For patients with illnesses other than cancer, palliative care may be helpful when disease-modifying therapy becomes burdensome or ineffective, or when patients are symptomatic despite maximum therapy. Palliative care should also be considered when goals of care need to be explored, when a second opinion is needed on goals of care, or if the primary care provider and family are at odds.
WHEN A CONSULT IS INAPPROPRIATE
Palliative care consultation is inappropriate when used in lieu of an oncology consult in advanced cancer. Palliative care specialists are not experts in cancer care, whereas oncologists are familiar with rapid advancements in cancer care, including targeted agents that may offer benefit to patients with advanced cancer.
Palliative care consultation is also inappropriate if the patient does not want to see a palliative care specialist, or if the consult is used as a way to convince a patient to change advance directives or to choose not to be resuscitated. Also, cancer patients who are asymptomatic are unlikely to benefit from palliative care initially. The decision to consult palliative care should not depend on prognosis, and palliative care is more cost-effective when utilized early rather than as a crisis intervention near the end of life.3
THE PALLIATIVE CARE EVALUATION
The initial palliative care consultation usually involves an evaluation of the patient’s symptoms and concerns. Symptoms are targeted based on the patient’s priorities and on an assessment using validated questionnaires. A validated questionnaire is a better way to comprehensively gauge symptom burden than depending on patients to volunteer symptoms.5
As the relationship develops between patient, family, and palliative care specialist and as the disease takes its course, advance directives, prognosis, and end-of-life care goals can be addressed in follow-up consultations.3 Patients want to know about their prognosis, and they usually complete advance directives based on clinical circumstances rather than viewing them as an extension of patient autonomy, as originally intended.6
REIMBURSEMENT FOR PALLIATIVE CARE
Reimbursement for palliative care is similar to that for acute care and falls within the All Patient Refined Diagnosis-Related Group, or APR-DRG, system, and palliative care has its own V code for identification. Codes are used to designate disease, stage or location of metastases, disease complications, and symptoms, as well as for the discussion of goals of care.
WHAT PALLIATIVE CARE IS NOT
Palliative care has too often been tied to end-of-life care.7 The two often appear together in titles of reports in the literature. As a result, patients and physicians may be confused and, thus, reluctant to utilize palliative care services. To avoid the confusion, certain programs have included the term “supportive” oncology care in their title. This appears to facilitate palliative care referral, but may be misleading.8
WHAT IS HOSPICE CARE?
Hospice care is a service funded and capitated under Medicare part A and is largely provided as outpatient home care for those deemed terminally ill.9 An illness must be certified as terminal by two physicians. Medicare defines terminal illness as a life expectancy of 6 months or less if the illness runs its normal course.
The philosophy of hospice care is to provide comfort through intensive nurse management and home-based follow-up. In some cases, disease-modifying therapies are continued to control symptoms—eg, continuing angiotensin-converting enzyme inhibitors in heart failure patients. Hospice care is typically delivered at home, but it is also delivered in nursing homes, in hospital inpatient units, and at private or nonprofit hospice facilities.
Inpatient palliative care units are often mistaken for hospices. The purpose of hospice care is to provide quality of life and comfort and to avoid overly aggressive, expensive, and futile care at the end of life. The focus is on intensive, hands-on, personalized symptom care and family support at home. The goal is to provide a comfortable and dignified death among friends and family. The use of palliative radiation, transfusions, and antibiotics in hospice varies among hospice programs and is considered on a case-by-case basis.10
The Medicare per diem payment limits what hospices can afford, so they must be fiscally responsible. Hospice agencies are capitated and are responsible for providing medications and durable equipment necessary to treat symptoms related to the terminal illness. They also provide bereavement services for family members at no charge. Enrollment in hospice care can be revoked depending on circumstances and then reinstituted later as the goals of care change.
Care for nonterminal comorbid illnesses can be continued by a general practitioner or internist. This care is not covered under the Medicare hospice benefit, but it is covered under Medicare part B.
The patient and family can choose the hospice physician, who may be a family practitioner, internist, oncologist, or palliative care specialist, or may designate the hospice medical director as the hospice physician.
Criteria for hospice admission have been established for noncancer terminal illnesses and should be considered when practitioners decide to consult hospice.11–13
HOME-BASED PALLIATIVE CARE
Programs such as advanced illness management or home-based palliative care aim to improve the quality of care at home and prevent rehospitalization, particularly for patients with repeated hospitalizations.14 Home-based palliative care services are provided either by a clinician who makes home visits or by a certified home health care agency. Services are particularly useful for patients with serious illnesses who do not qualify for hospice services but are homebound. Consultations are obtained for ongoing supportive care at home, assessment for medication compliance, and disease monitoring at home. Consultations are scheduled at the time of hospital discharge.
Unlike hospice care, home-based palliative care does not include 24-hour on-call service. Comprehensive services (eg, home health aide, durable equipment, medications) are not provided as they are under hospice care: patients must qualify under Medicare stipulations for such services outside of hospice care. For example, home oxygen can only be supplied if the patient's oxygen saturation is less than 90%, while under the hospice benefit it is provided without regard to oxygen saturation and is based on symptom need. For home-based palliative care, patients must be largely homebound or unable to be seen regularly in the outpatient clinic. This type of care can be a bridge to hospice care for patients who feel they are not ready for hospice care at the time of discharge from acute care. Those who receive palliative care at home are less likely to be hospitalized at the end of life, are more likely to be transitioned to hospice at an appropriate time, and are more likely to have relief of symptoms.15
- Mount BM. The problem of caring for the dying in a general hospital; the palliative care unit as a possible solution. Can Med Assoc J 1976; 115:119–121.
- Higginson I. Palliative care: a review of past changes and future trends. J Public Health Med 1993; 15:3–8.
- Temel JS, Greer JA, Muzikansky A, et al. Early palliative care for patients with metastatic non-small-cell lung cancer. N Engl J Med 2010; 363:733–742.
- Zimmermann C, Riechelmann R, Krzyzanowska M, Rodin G, Tannock I. Effectiveness of specialized palliative care: a systematic review. JAMA 2008; 299:1698–1709.
- Homsi J, Walsh D, Rivera N, et al. Symptom evaluation in palliative medicine: patient report vs systematic assessment. Support Care Cancer 2006; 14:444–453.
- Tang ST, Liu TW, Lai MS, Liu LN, Chen CH, Koong SL. Congruence of knowledge, experiences, and p for disclosure of diagnosis and prognosis between terminally-ill cancer patients and their family caregivers in Taiwan. Cancer Invest 2006; 24:360–366.
- Bakitas M, Lyons KD, Hegel MT, Ahles T. Oncologists’ perspectives on concurrent palliative care in a National Cancer Institute-designated comprehensive cancer center. Palliat Support Care 2013; 11:415–423.
- Fadul N, Elsayem A, Palmer JL, et al. Supportive versus palliative care: what’s in a name: a survey of medical oncologists and midlevel providers at a comprehensive cancer center. Cancer 2009; 115:2013–2021.
- Rinaldo MJ. Medicare to cover hospice services. J Med Soc NJ 1982; 79:1015–1016.
- Enck RE. Palliative radiation therapy in hospice care. Am J Hosp Palliat Care 2002; 19:151–152.
- Luchins DJ, Hanrahan P, Murphy K. Criteria for enrolling dementia patients in hospice. J Am Geriatr Soc 1997; 45:1054–1059.
- Fox E, Landrum-McNiff K, Zhong Z, Dawson NV, Wu AW, Lynn J. Evaluation of prognostic criteria for determining hospice eligibility in patients with advanced lung, heart, or liver disease. JAMA 1999; 282:1638–1645.
- Stuart B. The NHO medical guidelines for non-cancer disease and local medical review policy: hospice access for patients with diseases other than cancer. Hosp J 1999; 14:139–154.
- McKinney M. Beyond hospice. New models of care focus on advanced illnesses. Mod Healthc 2013; 43:14–15.
- Gomes B, Calanzani N, Curiale V, McCrone P, Higginson IJ. Effectiveness and cost-effectiveness of home palliative care services for adults with advanced illness and their caregivers. Cochrane Database Syst Rev 2013; 6:CD007760.
Hospice care generally falls under the category of palliative care, despite being an older subspecialty. However, the two have different indications and goals and are often provided in different settings.
ORIGINS OF PALLIATIVE CARE
Prompted by what he perceived as neglect of dying patients in the acute care setting, Dr. Balfour Mount opened the first acute inpatient palliative care unit in Royal Victoria Hospital in Montréal, Québec, in 1976.1 His purpose was to provide a crisis-intervention service for patients who were actively dying, and this continues to be the main reason for consulting palliative care services in the hospital.
Palliative care has evolved since the 1970s and is now used in a variety of situations:
- A life-limiting illness in a patient who is not terminally ill
- A life-threatening illness in a patient who has symptoms but with the potential to recover
- A chronic illness such as heart failure or chronic obstructive pulmonary disease in a patient who is on disease-modifying therapy but has symptoms and will eventually succumb to the illness, but is expected to live longer than someone with advanced cancer.2
PALLIATIVE CARE IN CANCER PATIENTS
In patients with advanced cancer, palliative care is utilized earlier in the course of serious and life-limiting illness and is even involved in patient care when cure is the goal. Importantly, it now includes outpatient clinics to provide patients seamless care in conjunction with their oncologist’s care.3
Because palliative care focuses on the patient’s experience of the illness (sickness) rather than on disease itself (pathology), symptom management, psychosocial support, and assistance in decision-making are foremost. Initiating palliative care early in advanced cancer improves multiple outcomes and limits overly aggressive, ineffective therapies at the end of life (eg, late chemotherapy, late referral to hospice care, death in the intensive care unit), without hastening death. In fact, it may prolong life.3,4
Palliative care is indicated in a number of situations in oncology:
- Symptomatic presentations of cancer, even when curative treatments are available
- At the time of a sentinel event such as recurrence or unanticipated hospitalization
- When palliative radiation is needed
- When changes in chemotherapy are needed because of disease progression.
Also, cancer patients may develop symptoms that require a palliative procedure such as thoracentesis for pleural effusion, paracentesis for ascites, or surgery for a fracture or spinal cord compression. A palliative care consultation is also appropriate when patients change their goals of care (ie, palliation rather than cure), and when an oncologic crisis occurs and there is a need to offer support to the family and to clarify the goals of care.
PALLIATIVE CARE IN OTHER DISEASES
For patients with illnesses other than cancer, palliative care may be helpful when disease-modifying therapy becomes burdensome or ineffective, or when patients are symptomatic despite maximum therapy. Palliative care should also be considered when goals of care need to be explored, when a second opinion is needed on goals of care, or if the primary care provider and family are at odds.
WHEN A CONSULT IS INAPPROPRIATE
Palliative care consultation is inappropriate when used in lieu of an oncology consult in advanced cancer. Palliative care specialists are not experts in cancer care, whereas oncologists are familiar with rapid advancements in cancer care, including targeted agents that may offer benefit to patients with advanced cancer.
Palliative care consultation is also inappropriate if the patient does not want to see a palliative care specialist, or if the consult is used as a way to convince a patient to change advance directives or to choose not to be resuscitated. Also, cancer patients who are asymptomatic are unlikely to benefit from palliative care initially. The decision to consult palliative care should not depend on prognosis, and palliative care is more cost-effective when utilized early rather than as a crisis intervention near the end of life.3
THE PALLIATIVE CARE EVALUATION
The initial palliative care consultation usually involves an evaluation of the patient’s symptoms and concerns. Symptoms are targeted based on the patient’s priorities and on an assessment using validated questionnaires. A validated questionnaire is a better way to comprehensively gauge symptom burden than depending on patients to volunteer symptoms.5
As the relationship develops between patient, family, and palliative care specialist and as the disease takes its course, advance directives, prognosis, and end-of-life care goals can be addressed in follow-up consultations.3 Patients want to know about their prognosis, and they usually complete advance directives based on clinical circumstances rather than viewing them as an extension of patient autonomy, as originally intended.6
REIMBURSEMENT FOR PALLIATIVE CARE
Reimbursement for palliative care is similar to that for acute care and falls within the All Patient Refined Diagnosis-Related Group, or APR-DRG, system, and palliative care has its own V code for identification. Codes are used to designate disease, stage or location of metastases, disease complications, and symptoms, as well as for the discussion of goals of care.
WHAT PALLIATIVE CARE IS NOT
Palliative care has too often been tied to end-of-life care.7 The two often appear together in titles of reports in the literature. As a result, patients and physicians may be confused and, thus, reluctant to utilize palliative care services. To avoid the confusion, certain programs have included the term “supportive” oncology care in their title. This appears to facilitate palliative care referral, but may be misleading.8
WHAT IS HOSPICE CARE?
Hospice care is a service funded and capitated under Medicare part A and is largely provided as outpatient home care for those deemed terminally ill.9 An illness must be certified as terminal by two physicians. Medicare defines terminal illness as a life expectancy of 6 months or less if the illness runs its normal course.
The philosophy of hospice care is to provide comfort through intensive nurse management and home-based follow-up. In some cases, disease-modifying therapies are continued to control symptoms—eg, continuing angiotensin-converting enzyme inhibitors in heart failure patients. Hospice care is typically delivered at home, but it is also delivered in nursing homes, in hospital inpatient units, and at private or nonprofit hospice facilities.
Inpatient palliative care units are often mistaken for hospices. The purpose of hospice care is to provide quality of life and comfort and to avoid overly aggressive, expensive, and futile care at the end of life. The focus is on intensive, hands-on, personalized symptom care and family support at home. The goal is to provide a comfortable and dignified death among friends and family. The use of palliative radiation, transfusions, and antibiotics in hospice varies among hospice programs and is considered on a case-by-case basis.10
The Medicare per diem payment limits what hospices can afford, so they must be fiscally responsible. Hospice agencies are capitated and are responsible for providing medications and durable equipment necessary to treat symptoms related to the terminal illness. They also provide bereavement services for family members at no charge. Enrollment in hospice care can be revoked depending on circumstances and then reinstituted later as the goals of care change.
Care for nonterminal comorbid illnesses can be continued by a general practitioner or internist. This care is not covered under the Medicare hospice benefit, but it is covered under Medicare part B.
The patient and family can choose the hospice physician, who may be a family practitioner, internist, oncologist, or palliative care specialist, or may designate the hospice medical director as the hospice physician.
Criteria for hospice admission have been established for noncancer terminal illnesses and should be considered when practitioners decide to consult hospice.11–13
HOME-BASED PALLIATIVE CARE
Programs such as advanced illness management or home-based palliative care aim to improve the quality of care at home and prevent rehospitalization, particularly for patients with repeated hospitalizations.14 Home-based palliative care services are provided either by a clinician who makes home visits or by a certified home health care agency. Services are particularly useful for patients with serious illnesses who do not qualify for hospice services but are homebound. Consultations are obtained for ongoing supportive care at home, assessment for medication compliance, and disease monitoring at home. Consultations are scheduled at the time of hospital discharge.
Unlike hospice care, home-based palliative care does not include 24-hour on-call service. Comprehensive services (eg, home health aide, durable equipment, medications) are not provided as they are under hospice care: patients must qualify under Medicare stipulations for such services outside of hospice care. For example, home oxygen can only be supplied if the patient's oxygen saturation is less than 90%, while under the hospice benefit it is provided without regard to oxygen saturation and is based on symptom need. For home-based palliative care, patients must be largely homebound or unable to be seen regularly in the outpatient clinic. This type of care can be a bridge to hospice care for patients who feel they are not ready for hospice care at the time of discharge from acute care. Those who receive palliative care at home are less likely to be hospitalized at the end of life, are more likely to be transitioned to hospice at an appropriate time, and are more likely to have relief of symptoms.15
Hospice care generally falls under the category of palliative care, despite being an older subspecialty. However, the two have different indications and goals and are often provided in different settings.
ORIGINS OF PALLIATIVE CARE
Prompted by what he perceived as neglect of dying patients in the acute care setting, Dr. Balfour Mount opened the first acute inpatient palliative care unit in Royal Victoria Hospital in Montréal, Québec, in 1976.1 His purpose was to provide a crisis-intervention service for patients who were actively dying, and this continues to be the main reason for consulting palliative care services in the hospital.
Palliative care has evolved since the 1970s and is now used in a variety of situations:
- A life-limiting illness in a patient who is not terminally ill
- A life-threatening illness in a patient who has symptoms but with the potential to recover
- A chronic illness such as heart failure or chronic obstructive pulmonary disease in a patient who is on disease-modifying therapy but has symptoms and will eventually succumb to the illness, but is expected to live longer than someone with advanced cancer.2
PALLIATIVE CARE IN CANCER PATIENTS
In patients with advanced cancer, palliative care is utilized earlier in the course of serious and life-limiting illness and is even involved in patient care when cure is the goal. Importantly, it now includes outpatient clinics to provide patients seamless care in conjunction with their oncologist’s care.3
Because palliative care focuses on the patient’s experience of the illness (sickness) rather than on disease itself (pathology), symptom management, psychosocial support, and assistance in decision-making are foremost. Initiating palliative care early in advanced cancer improves multiple outcomes and limits overly aggressive, ineffective therapies at the end of life (eg, late chemotherapy, late referral to hospice care, death in the intensive care unit), without hastening death. In fact, it may prolong life.3,4
Palliative care is indicated in a number of situations in oncology:
- Symptomatic presentations of cancer, even when curative treatments are available
- At the time of a sentinel event such as recurrence or unanticipated hospitalization
- When palliative radiation is needed
- When changes in chemotherapy are needed because of disease progression.
Also, cancer patients may develop symptoms that require a palliative procedure such as thoracentesis for pleural effusion, paracentesis for ascites, or surgery for a fracture or spinal cord compression. A palliative care consultation is also appropriate when patients change their goals of care (ie, palliation rather than cure), and when an oncologic crisis occurs and there is a need to offer support to the family and to clarify the goals of care.
PALLIATIVE CARE IN OTHER DISEASES
For patients with illnesses other than cancer, palliative care may be helpful when disease-modifying therapy becomes burdensome or ineffective, or when patients are symptomatic despite maximum therapy. Palliative care should also be considered when goals of care need to be explored, when a second opinion is needed on goals of care, or if the primary care provider and family are at odds.
WHEN A CONSULT IS INAPPROPRIATE
Palliative care consultation is inappropriate when used in lieu of an oncology consult in advanced cancer. Palliative care specialists are not experts in cancer care, whereas oncologists are familiar with rapid advancements in cancer care, including targeted agents that may offer benefit to patients with advanced cancer.
Palliative care consultation is also inappropriate if the patient does not want to see a palliative care specialist, or if the consult is used as a way to convince a patient to change advance directives or to choose not to be resuscitated. Also, cancer patients who are asymptomatic are unlikely to benefit from palliative care initially. The decision to consult palliative care should not depend on prognosis, and palliative care is more cost-effective when utilized early rather than as a crisis intervention near the end of life.3
THE PALLIATIVE CARE EVALUATION
The initial palliative care consultation usually involves an evaluation of the patient’s symptoms and concerns. Symptoms are targeted based on the patient’s priorities and on an assessment using validated questionnaires. A validated questionnaire is a better way to comprehensively gauge symptom burden than depending on patients to volunteer symptoms.5
As the relationship develops between patient, family, and palliative care specialist and as the disease takes its course, advance directives, prognosis, and end-of-life care goals can be addressed in follow-up consultations.3 Patients want to know about their prognosis, and they usually complete advance directives based on clinical circumstances rather than viewing them as an extension of patient autonomy, as originally intended.6
REIMBURSEMENT FOR PALLIATIVE CARE
Reimbursement for palliative care is similar to that for acute care and falls within the All Patient Refined Diagnosis-Related Group, or APR-DRG, system, and palliative care has its own V code for identification. Codes are used to designate disease, stage or location of metastases, disease complications, and symptoms, as well as for the discussion of goals of care.
WHAT PALLIATIVE CARE IS NOT
Palliative care has too often been tied to end-of-life care.7 The two often appear together in titles of reports in the literature. As a result, patients and physicians may be confused and, thus, reluctant to utilize palliative care services. To avoid the confusion, certain programs have included the term “supportive” oncology care in their title. This appears to facilitate palliative care referral, but may be misleading.8
WHAT IS HOSPICE CARE?
Hospice care is a service funded and capitated under Medicare part A and is largely provided as outpatient home care for those deemed terminally ill.9 An illness must be certified as terminal by two physicians. Medicare defines terminal illness as a life expectancy of 6 months or less if the illness runs its normal course.
The philosophy of hospice care is to provide comfort through intensive nurse management and home-based follow-up. In some cases, disease-modifying therapies are continued to control symptoms—eg, continuing angiotensin-converting enzyme inhibitors in heart failure patients. Hospice care is typically delivered at home, but it is also delivered in nursing homes, in hospital inpatient units, and at private or nonprofit hospice facilities.
Inpatient palliative care units are often mistaken for hospices. The purpose of hospice care is to provide quality of life and comfort and to avoid overly aggressive, expensive, and futile care at the end of life. The focus is on intensive, hands-on, personalized symptom care and family support at home. The goal is to provide a comfortable and dignified death among friends and family. The use of palliative radiation, transfusions, and antibiotics in hospice varies among hospice programs and is considered on a case-by-case basis.10
The Medicare per diem payment limits what hospices can afford, so they must be fiscally responsible. Hospice agencies are capitated and are responsible for providing medications and durable equipment necessary to treat symptoms related to the terminal illness. They also provide bereavement services for family members at no charge. Enrollment in hospice care can be revoked depending on circumstances and then reinstituted later as the goals of care change.
Care for nonterminal comorbid illnesses can be continued by a general practitioner or internist. This care is not covered under the Medicare hospice benefit, but it is covered under Medicare part B.
The patient and family can choose the hospice physician, who may be a family practitioner, internist, oncologist, or palliative care specialist, or may designate the hospice medical director as the hospice physician.
Criteria for hospice admission have been established for noncancer terminal illnesses and should be considered when practitioners decide to consult hospice.11–13
HOME-BASED PALLIATIVE CARE
Programs such as advanced illness management or home-based palliative care aim to improve the quality of care at home and prevent rehospitalization, particularly for patients with repeated hospitalizations.14 Home-based palliative care services are provided either by a clinician who makes home visits or by a certified home health care agency. Services are particularly useful for patients with serious illnesses who do not qualify for hospice services but are homebound. Consultations are obtained for ongoing supportive care at home, assessment for medication compliance, and disease monitoring at home. Consultations are scheduled at the time of hospital discharge.
Unlike hospice care, home-based palliative care does not include 24-hour on-call service. Comprehensive services (eg, home health aide, durable equipment, medications) are not provided as they are under hospice care: patients must qualify under Medicare stipulations for such services outside of hospice care. For example, home oxygen can only be supplied if the patient's oxygen saturation is less than 90%, while under the hospice benefit it is provided without regard to oxygen saturation and is based on symptom need. For home-based palliative care, patients must be largely homebound or unable to be seen regularly in the outpatient clinic. This type of care can be a bridge to hospice care for patients who feel they are not ready for hospice care at the time of discharge from acute care. Those who receive palliative care at home are less likely to be hospitalized at the end of life, are more likely to be transitioned to hospice at an appropriate time, and are more likely to have relief of symptoms.15
- Mount BM. The problem of caring for the dying in a general hospital; the palliative care unit as a possible solution. Can Med Assoc J 1976; 115:119–121.
- Higginson I. Palliative care: a review of past changes and future trends. J Public Health Med 1993; 15:3–8.
- Temel JS, Greer JA, Muzikansky A, et al. Early palliative care for patients with metastatic non-small-cell lung cancer. N Engl J Med 2010; 363:733–742.
- Zimmermann C, Riechelmann R, Krzyzanowska M, Rodin G, Tannock I. Effectiveness of specialized palliative care: a systematic review. JAMA 2008; 299:1698–1709.
- Homsi J, Walsh D, Rivera N, et al. Symptom evaluation in palliative medicine: patient report vs systematic assessment. Support Care Cancer 2006; 14:444–453.
- Tang ST, Liu TW, Lai MS, Liu LN, Chen CH, Koong SL. Congruence of knowledge, experiences, and p for disclosure of diagnosis and prognosis between terminally-ill cancer patients and their family caregivers in Taiwan. Cancer Invest 2006; 24:360–366.
- Bakitas M, Lyons KD, Hegel MT, Ahles T. Oncologists’ perspectives on concurrent palliative care in a National Cancer Institute-designated comprehensive cancer center. Palliat Support Care 2013; 11:415–423.
- Fadul N, Elsayem A, Palmer JL, et al. Supportive versus palliative care: what’s in a name: a survey of medical oncologists and midlevel providers at a comprehensive cancer center. Cancer 2009; 115:2013–2021.
- Rinaldo MJ. Medicare to cover hospice services. J Med Soc NJ 1982; 79:1015–1016.
- Enck RE. Palliative radiation therapy in hospice care. Am J Hosp Palliat Care 2002; 19:151–152.
- Luchins DJ, Hanrahan P, Murphy K. Criteria for enrolling dementia patients in hospice. J Am Geriatr Soc 1997; 45:1054–1059.
- Fox E, Landrum-McNiff K, Zhong Z, Dawson NV, Wu AW, Lynn J. Evaluation of prognostic criteria for determining hospice eligibility in patients with advanced lung, heart, or liver disease. JAMA 1999; 282:1638–1645.
- Stuart B. The NHO medical guidelines for non-cancer disease and local medical review policy: hospice access for patients with diseases other than cancer. Hosp J 1999; 14:139–154.
- McKinney M. Beyond hospice. New models of care focus on advanced illnesses. Mod Healthc 2013; 43:14–15.
- Gomes B, Calanzani N, Curiale V, McCrone P, Higginson IJ. Effectiveness and cost-effectiveness of home palliative care services for adults with advanced illness and their caregivers. Cochrane Database Syst Rev 2013; 6:CD007760.
- Mount BM. The problem of caring for the dying in a general hospital; the palliative care unit as a possible solution. Can Med Assoc J 1976; 115:119–121.
- Higginson I. Palliative care: a review of past changes and future trends. J Public Health Med 1993; 15:3–8.
- Temel JS, Greer JA, Muzikansky A, et al. Early palliative care for patients with metastatic non-small-cell lung cancer. N Engl J Med 2010; 363:733–742.
- Zimmermann C, Riechelmann R, Krzyzanowska M, Rodin G, Tannock I. Effectiveness of specialized palliative care: a systematic review. JAMA 2008; 299:1698–1709.
- Homsi J, Walsh D, Rivera N, et al. Symptom evaluation in palliative medicine: patient report vs systematic assessment. Support Care Cancer 2006; 14:444–453.
- Tang ST, Liu TW, Lai MS, Liu LN, Chen CH, Koong SL. Congruence of knowledge, experiences, and p for disclosure of diagnosis and prognosis between terminally-ill cancer patients and their family caregivers in Taiwan. Cancer Invest 2006; 24:360–366.
- Bakitas M, Lyons KD, Hegel MT, Ahles T. Oncologists’ perspectives on concurrent palliative care in a National Cancer Institute-designated comprehensive cancer center. Palliat Support Care 2013; 11:415–423.
- Fadul N, Elsayem A, Palmer JL, et al. Supportive versus palliative care: what’s in a name: a survey of medical oncologists and midlevel providers at a comprehensive cancer center. Cancer 2009; 115:2013–2021.
- Rinaldo MJ. Medicare to cover hospice services. J Med Soc NJ 1982; 79:1015–1016.
- Enck RE. Palliative radiation therapy in hospice care. Am J Hosp Palliat Care 2002; 19:151–152.
- Luchins DJ, Hanrahan P, Murphy K. Criteria for enrolling dementia patients in hospice. J Am Geriatr Soc 1997; 45:1054–1059.
- Fox E, Landrum-McNiff K, Zhong Z, Dawson NV, Wu AW, Lynn J. Evaluation of prognostic criteria for determining hospice eligibility in patients with advanced lung, heart, or liver disease. JAMA 1999; 282:1638–1645.
- Stuart B. The NHO medical guidelines for non-cancer disease and local medical review policy: hospice access for patients with diseases other than cancer. Hosp J 1999; 14:139–154.
- McKinney M. Beyond hospice. New models of care focus on advanced illnesses. Mod Healthc 2013; 43:14–15.
- Gomes B, Calanzani N, Curiale V, McCrone P, Higginson IJ. Effectiveness and cost-effectiveness of home palliative care services for adults with advanced illness and their caregivers. Cochrane Database Syst Rev 2013; 6:CD007760.
Occult satellite metastasis of an auricular melanoma
A 90-year-old man presented to our clinic with a dark, exophytic, hemorrhagic mass on the helix of his right auricle (Figure 1A). He had first noticed the lesion 6 months before.
Evaluation of the lesion with the standard ABCDE criteria (Asymmetry, Border irregularity, Color variation, Diameter > 6 mm, Evolution/elevation) raised our suspicion of melanoma.1 We performed a wide, full-thickness, auricular wedge resection, which revealed a second dark lesion in the subcutaneous tissue of the upper border of the resected specimen. The rest of the second lesion was evident on the corresponding location of the edge of the remaining auricle (Figure 1B). Thus, we excised an additional strip of auricular tissue. The aesthetic result of the auricular reconstruction was quite good (Figure 1C).
Histopathologic study confirmed cutaneous melanoma and showed the second lesion to be a satellite melanoma metastasis (Figure 2). The patient refused to undergo staging investigations for lymph node and distant metastases. He died 1 year later of ischemic stroke.
IN-TRANSIT AND SATELLITE METASTASES
Melanoma is highly metastatic. In addition to regional lymph node and distant metastases, patients may develop in-transit metastases and satellite metastases.
In-transit metastases grow more than 2 cm away from the primary tumor but not beyond the regional lymph node basin. Satellite lesions are found within 2 cm of the primary melanoma.
As seen in our patient, satellite metastases are not always cutaneous and evident. This is also true of in-transit melanoma lesions. They can also be located in subcutaneous tissue, making them difficult to detect. The presence of satellite lesions is a sign of aggressive disease and requires a thorough evaluation for metastases.2
- Thomas L, Tranchand P, Berard F, Secchi T, Colin C, Moulin G. Semiological value of ABCDE criteria in the diagnosis of cutaneous pigmented tumors. Dermatology 1998; 197:11–17.
- Homsi J, Kashani-Sabet M, Messina JL, Daud A. Cutaneous melanoma: prognostic factors. Cancer Control 2005; 12:223–229.
A 90-year-old man presented to our clinic with a dark, exophytic, hemorrhagic mass on the helix of his right auricle (Figure 1A). He had first noticed the lesion 6 months before.
Evaluation of the lesion with the standard ABCDE criteria (Asymmetry, Border irregularity, Color variation, Diameter > 6 mm, Evolution/elevation) raised our suspicion of melanoma.1 We performed a wide, full-thickness, auricular wedge resection, which revealed a second dark lesion in the subcutaneous tissue of the upper border of the resected specimen. The rest of the second lesion was evident on the corresponding location of the edge of the remaining auricle (Figure 1B). Thus, we excised an additional strip of auricular tissue. The aesthetic result of the auricular reconstruction was quite good (Figure 1C).
Histopathologic study confirmed cutaneous melanoma and showed the second lesion to be a satellite melanoma metastasis (Figure 2). The patient refused to undergo staging investigations for lymph node and distant metastases. He died 1 year later of ischemic stroke.
IN-TRANSIT AND SATELLITE METASTASES
Melanoma is highly metastatic. In addition to regional lymph node and distant metastases, patients may develop in-transit metastases and satellite metastases.
In-transit metastases grow more than 2 cm away from the primary tumor but not beyond the regional lymph node basin. Satellite lesions are found within 2 cm of the primary melanoma.
As seen in our patient, satellite metastases are not always cutaneous and evident. This is also true of in-transit melanoma lesions. They can also be located in subcutaneous tissue, making them difficult to detect. The presence of satellite lesions is a sign of aggressive disease and requires a thorough evaluation for metastases.2
A 90-year-old man presented to our clinic with a dark, exophytic, hemorrhagic mass on the helix of his right auricle (Figure 1A). He had first noticed the lesion 6 months before.
Evaluation of the lesion with the standard ABCDE criteria (Asymmetry, Border irregularity, Color variation, Diameter > 6 mm, Evolution/elevation) raised our suspicion of melanoma.1 We performed a wide, full-thickness, auricular wedge resection, which revealed a second dark lesion in the subcutaneous tissue of the upper border of the resected specimen. The rest of the second lesion was evident on the corresponding location of the edge of the remaining auricle (Figure 1B). Thus, we excised an additional strip of auricular tissue. The aesthetic result of the auricular reconstruction was quite good (Figure 1C).
Histopathologic study confirmed cutaneous melanoma and showed the second lesion to be a satellite melanoma metastasis (Figure 2). The patient refused to undergo staging investigations for lymph node and distant metastases. He died 1 year later of ischemic stroke.
IN-TRANSIT AND SATELLITE METASTASES
Melanoma is highly metastatic. In addition to regional lymph node and distant metastases, patients may develop in-transit metastases and satellite metastases.
In-transit metastases grow more than 2 cm away from the primary tumor but not beyond the regional lymph node basin. Satellite lesions are found within 2 cm of the primary melanoma.
As seen in our patient, satellite metastases are not always cutaneous and evident. This is also true of in-transit melanoma lesions. They can also be located in subcutaneous tissue, making them difficult to detect. The presence of satellite lesions is a sign of aggressive disease and requires a thorough evaluation for metastases.2
- Thomas L, Tranchand P, Berard F, Secchi T, Colin C, Moulin G. Semiological value of ABCDE criteria in the diagnosis of cutaneous pigmented tumors. Dermatology 1998; 197:11–17.
- Homsi J, Kashani-Sabet M, Messina JL, Daud A. Cutaneous melanoma: prognostic factors. Cancer Control 2005; 12:223–229.
- Thomas L, Tranchand P, Berard F, Secchi T, Colin C, Moulin G. Semiological value of ABCDE criteria in the diagnosis of cutaneous pigmented tumors. Dermatology 1998; 197:11–17.
- Homsi J, Kashani-Sabet M, Messina JL, Daud A. Cutaneous melanoma: prognostic factors. Cancer Control 2005; 12:223–229.
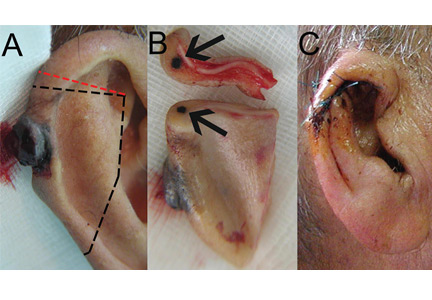
2015 Update on Parkinson disease
This has been a boom year for Parkinson disease, with the US Food and Drug Administration (FDA) approving two new therapies, and with others in the pipeline.
This article details clinical signs of Parkinson disease, discusses functional imaging, provides an update on current thinking on disease pathogenesis, and gives an overview of managing parkinsonian symptoms and dyskinesias.
DIAGNOSIS REMAINS CLINICAL
Although a better understanding of Parkinson disease has been gained in recent years, with the recognition of several premotor features and potential biomarkers, its diagnosis is still primarily based on clinical motor findings. The four cardinal motor features have the mnemonic TRAP:
- Tremor at rest can be subtle, involving just the thumb, best observed when the patient is sitting with the hand resting on the lap; or it can be obvious, involving the entire hand, arm, feet, lips, and chin.
- Rigidity can be felt rather than seen, by slowly passively rotating the patient’s wrist or elbow and feeling resistance. The right and left sides often differ.
- Akinesia or bradykinesia (slowness or lack of movement) can be observed by having the patient walk down a hallway. One may observe reduced arm swing and hesitation in initiating movement.
- Postural instability usually develops later rather than sooner in the disease progression. The patient may need to hold onto someone to maintain balance when getting up or walking.
At least two features must be present to make the diagnosis of parkinsonism. One feature must be tremor or rigidity.
Although the criteria for parkinsonism appear simple, the diagnosis of Parkinson disease is not always clear-cut. For example, shaking can be secondary to a dopamine receptor-blocking medication, to anxiety, or to essential tremor; rigidity and slowness may be due to arthritis; and postural instability can result from a neuropathy. Moreover, other neurodegenerative parkinsonian disorders may respond to levodopa (at least initially) and may present with levodopa-induced dyskinesias. Robust response to levodopa and the occurrence of dyskinesias are two additional features that strongly suggest the diagnosis of Parkinson disease.
Supporting parkinsonian features include stooped posture, masked facies, micrographia (small handwriting), drooling, speech changes (eg, hypophonia or soft speech, stuttering, slurring, monotonic speech), and a shuffling, festinating gait (quick short steps as if falling forward).
PARKINSON MIMICS
Parkinsonism is a broader term than Parkinson disease or idiopathic Parkinson disease. It is characterized by akinetic rigidity and impaired motor activity that leads to reduced function and falls; behavioral changes also may occur.
In the United States, Parkinson disease is the most common cause of parkinsonism. Other nonneurodegenerative causes are drug-induced parkinsonism (due to dopamine receptor antagonists such as antipsychotic or antiemetic drugs), stroke (in the basal ganglia or frontal lobe), and normal-pressure hydrocephalus (causing lower-body parkinsonism). Mimics of parkinsonism include essential tremor and psychogenic parkinsonism.
Parkinsonism can also be caused by Parkinson-plus disorders, ie, neurodegenerative conditions characterized by parkinsonism along with additional signs and symptoms, as listed below. Parkinson-plus disorders include progressive supranuclear palsy, multiple system atrophy, corticobasal degeneration, and Lewy body disease.
Clinical features that suggest a diagnosis other than Parkinson disease include1:
- Poor response to adequate dosages of levodopa
- Early onset of postural instability and falls
- Axial rigidity (eg, stiff neck) more than appendicular rigidity
- Early dementia
- Supranuclear gaze palsy
- Unusual movements besides tremor, eg, limb dystonia, myoclonus, limb levitation or alien limb syndrome
- Profound autonomic dysfunction
- Psychotic symptoms before taking levodopa or dopaminergic medication.
The precise diagnosis of Parkinson-plus disorders is not critical, as the treatment is generally the same for all of them: ie, levodopa (if it shows some efficacy and is well tolerated), with additional symptomatic treatment for features such as depression, cognitive impairment, and autonomic dysfunction, and supportive therapy including physical, occupational, speech, and swallowing therapy.
IMAGING MAY ASSIST IN THE DIAGNOSIS
Dopamine transporter single-photon emission computed tomography (SPECT) is a functional imaging technique that supposedly reflects dopamine uptake by surviving presynaptic dopaminergic neurons in the striate bodies of the basal ganglia. Normal uptake shows distinct cashew-shaped enhancement bilaterally. In Parkinson disease, the enhanced areas are smaller and asymmetric, first with diminution of the tail (representing the putamen), then later involving the head (representing the caudate) along with the other striate bodies (Figure 1).

Dopamine transporter SPECT does not distinguish one neurodegenerative parkinsonian disorder from another. Therefore, it should not be used to distinguish Parkinson disease from other Parkinson-plus syndromes. But it does distinguish neurodegenerative parkinsonian disorders from nonneurodegenerative conditions and mimics, which have a normal result on dopamine transporter SPECT (Table 1).
SLOWING DISEASE PROGRESSION
Current treatments for Parkinson disease can significantly improve symptoms but, unfortunately, do not cure the disease or slow its progression. Testing whether agents modify the disease course is particularly difficult with Parkinson disease, because it affects individuals differently, has a wide spectrum of symptoms, has a long time course, and lacks definitive markers to monitor progression. Some agents have shown promise:
Caffeine. People who drink coffee are less likely to develop Parkinson disease, with the risk declining with the number of cups per day.2 For those who have the disease, drinking coffee is associated with reduced symptoms.
Exercise improves Parkinson disease and may prevent it, and some studies suggest that it can delay its progression.3 Exercise has been shown in an animal model to reduce the vulnerability of dopamine neurons to the toxic agent 6-hydroxydopamine.4 Functional magnetic resonance imaging studies have shown blood flow patterns before and after exercise that are similar to those seen in patients with and without Parkinson medication.3
Rasagiline, a monoamine oxidase B (MAO-B) inhibitor used for symptomatic treatment of Parkinson disease, had conflicting results in a neuroprotective clinical trial. Patients who received rasagiline 1 mg daily—but not those who received 2 mg daily—at the beginning of the trial had better Parkinson motor scores compared with patients who received rasagiline 9 months later.5
Inosine is a urate precursor that elevates urate levels in serum and the central nervous system. For unknown reasons, patients with Parkinson disease tend to have a low uric acid level, and higher levels are associated with milder disease. It is hoped that raising the uric acid level to a “pre-gout level” may slow the progression of Parkinson disease.
Isradipine, a calcium channel blocker, was found in an epidemiologic study of elderly patients to be associated with reduced likelihood of developing Parkinson disease.6 The drug is now undergoing clinical trials.
Smoking. Although cigarette smokers have long been recognized as having a very low risk of developing Parkinson disease, smoking is not recommended.
Agents found ineffective. Agents that have been tested and found ineffective in modifying the course of Parkinson disease include vitamin E, coenzyme Q10, riluzole, GPI-1485, pramipexole, cogane, CEP-1347, TCH-346, and creatine.
NOT JUST DOPAMINE—OR TREMORS
Dopamine deficiency is central to the current understanding of the pathogenesis of Parkinson disease and the focus of treatment efforts, but if dopamine deficiency were the only problem, replacing it should completely ameliorate all parkinsonian features. Other neurotransmitters also play roles: norepinephrine is implicated in orthostatic symptoms and apathy, acetylcholine in cognitive behaviors, glutamate in dyskinesias, and serotonin in depression, anxiety, and sleep abnormalities.
The most recognized area of involvement in the brain has traditionally been the substantia nigra in the midbrain. However, current thinking is that the disease starts lower in the caudal area of the brainstem (along with the olfactory tubercle), moves through the pons to the midbrain, then spreads across the cerebrum with extensive neocortical involvement.
Early premotor indicators are now recognized to occur 15 to 20 years before a tremor appears. The first signs are often hyposmia (diminished sense of smell, reflecting involvement of the olfactory tubercle) and constipation (reflecting involvement of the medulla and the vagus nucleus). With pons involvement, the patient can develop rapid eye movement sleep behavior disorder, depression, or anxiety. Only then does the disease spread to the midbrain and cause resting tremor, rigidity, and bradykinesia.7
Identifying the preclinical stages and starting disease-modifying treatments before the onset of motor symptoms may one day prove important, but at this point, the premotor symptoms (anosmia, constipation, depression) are too nonspecific to be useful, and such treatments have not yet been identified.
TREATMENT: LEVODOPA STILL PRIMARY
When to start drug treatment depends primarily on how much the symptoms bother the patient. Regardless of the clinician’s (or patient’s) belief in the benefits of delaying symptomatic treatment, it is universally considered necessary to start medication when gait problems develop because of the danger of a fall and resulting disability.
Carbidopa-levodopa combination therapy remains the most effective treatment; if it is not effective, another diagnosis may need to be considered. Carbidopa-levodopa improves tremor, rigidity, and bradykinesia, particularly in the early stages of Parkinson disease. It is well tolerated, has rapid onset, reduces the risk of death, and is the least expensive of the medications for Parkinson disease.
Immediate-release and continued-release formulations are available, as well as one that dissolves rapidly on the tongue and can be taken without water. An oral extended-release carbidopa-levodopa formulation (Rytary) was approved by the FDA in January 2015. Tablets are filled with drug-containing microbeads that dissolve at different rates to achieve therapeutic levodopa levels as quickly as the immediate-release formulation and maintain them for an extended time.8
The development of dyskinesias is the major psychological drawback of levodopa, occurring in 80% of patients after 5 to 10 years of treatment. Although many patients fear this side effect, most patients who develop it find it preferable to the rigidity and bradykinesia of Parkinson disease. In most cases, bothersome dyskinesias can be controlled by adjusting medications.9,10
Dopamine agonists include pramipexole, ropinirole, and rotigotine. They are available in generic form as three-times-daily dosing; once-daily dosing is also available, but not as a generic formulation. Dopamine agonists have the advantage of potentially improving depression and delaying the onset of dyskinesias.
However, dopamine agonists have a number of disadvantages compared with levodopa: they have a longer titration period, are less effective, and are less well tolerated, especially in the elderly. Side effects occur more frequently than with levodopa and include general and peripheral edema, hallucinations, nausea, lightheadedness, and sleepiness.11,12 These drugs are also associated with “sleep attacks” (sudden falling asleep while active, such as while driving or eating) and with compulsive and impulsive behaviors such as hypersexuality, buying, binge eating, and gambling. Although these behaviors occur in fewer than 10% of patients, they can be devastating, leading to marital, financial, and legal problems. A bothersome clinical state termed dopamine agonist withdrawal syndrome is characterized by anxiety, depression, jitteriness, and palpitations when dopamine agonists are tapered or discontinued because of a side effect.13
MAO-B inhibitors delay the breakdown of dopamine, allowing it to “stay” in the brain for a longer period of time. Rasagiline for early monotherapy has the advantages of once-daily dosing, no titration, and excellent tolerability, even in the elderly. Potential drug interactions should be considered when using this drug. Early warnings about interactions with tyramine-rich foods were lifted after trials showed that this was not a problem.14
Amantadine is an N-methyl-d-aspartate (NMDA) receptor antagonist often used in early Parkinson disease and for treatment of dyskinesias and fatigue. It is the only drug that is intrinsically antidyskinetic and also improves Parkinson symptoms.15 Side effects include leg swelling, livedo reticularis, and neuropsychiatric and anticholinergic effects.
Anticholinergic agents (eg, trihexyphenidyl) improve tremor but are not as useful for bradykinesia or rigidity, and often have anticholinergic effects such as mental dullness, dry mouth, dry eye, and urinary hesitancy, especially in the elderly, so they have a limited role in Parkinson treatment.
MOTOR COMPLICATIONS: FLUCTUATIONS AND DYSKINESIAS
Motor fluctuations are changes between the akinetic and mobile phases of Parkinson disease, or the off-periods and on-periods of drug treatment. A patient who is “off” is generally rigid and feels that the medication is not working. A patient who is “on” feels loose and mobile and that the medication is working. Variants of motor fluctuations include:
- End-of-dose deterioration
- Delayed onset of response (more than half an hour after taking medication)
- Drug-resistant offs—medication has become ineffective
- Random oscillation—on-off phenomenon
- Freezing—unpredictable inability to start or finish a movement.
Dyskinesias are abnormal involuntary movements such as writhing and twisting. They are associated with dopaminergic therapy at peak dose, when the drug starts to turn on or wear off (termed diphasic dyskinesias).16
The storage hypothesis provides a plausible explanation for the development of motor complications as the disease progresses. Although the half-life of levodopa is only 60 to 90 minutes, it is effective in early disease when given three times a day. It is believed that at this stage of the disease, enough dopaminergic neurons survive to “store” dopamine and release it as needed. As the disease progresses and dopaminergic neurons die, storage capacity diminishes, and the clinical effect slowly starts to approximate the pharmacokinetic profile of the drug. Upon taking the medication, the patient gets a surge of drug, causing dyskinesias, followed later by rigidity as the effect wears off since there are fewer surviving dopaminergic cells to store dopamine.
MANAGING DYSKINESIAS
Patients with dyskinesias should first be asked if they are bothered by them; not all patients are troubled by dyskinesias. If the movements only bother others (eg, family members), then education is often the only treatment needed. If the patient is uncomfortable, the following measures can be tried:
- Taking lower, more frequent doses of levodopa (however, risk of wearing off becomes a problem)
- Adding a dopamine agonist or MAO-B inhibitor while lowering the levodopa dose (however, MAO-B inhibitors pose a risk of side effects in elderly patients)
- Adding clozapine (periodic laboratory testing is required to monitor blood levels and liver and kidney function)
- Adding amantadine (however, this poses a risk of cognitive side effects).
Deep-brain-stimulation surgery is appropriate for select patients who are generally physically healthy, cognitively intact, and emotionally stable, with a strong family support system, but who are bothered by symptoms of parkinsonism (such as tremors), motor fluctuations, or dyskinesias.17
Infusion pump. In January 2015, the FDA approved a new system that continuously delivers levodopa-carbidopa in a 4:1 ratio in gel suspension for 16 hours directly into the small intestine, minimizing motor fluctuations. The patient changes the cartridge daily and turns it off at bedtime.
*Dr. Fernandez has received research support from AbbVie, Acadia, Auspex, Biotie Therapies, Civitas, Kyowa/ProStrakan, Michael J. Fox Foundation, Movement Disorders Society, NIH/NINDS, Parkinson Study Group, Rhythm, Synosia, and Teva. He also has received honoraria from Carling Communications, International Parkinson and Movement Disorders Society, The Ohio State University, and PRIME Education, Inc as a speaker in CME events. He has received honoraria from Biogen, GE Health Care, Lundbeck, Merz Pharmaceuticals, and Pfizer as a consultant. He has received royalty payments from Demos Publishing for serving as a book author/editor. Cleveland Clinic has contracts with AbbVie and Merz Pharmaceuticals for Dr. Fernandez’s role as a member of the Global Steering Committee for LCIG studies and as a consultant or speaker, and as Head Principal Investigator for the Xeomin Registry Study. Dr. Fernandez has received a stipend from International Parkinson and Movement Disorders Society for serving as medical editor of the Movement Disorders Society website.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Hernán MA, Takkouche B, Caamaño-Isoma F, et al. A meta-analysis of coffee drinking, cigarette smoking, and risk of Parkinson’s disease. Ann Neurol 2002; 52:276–84.
- Ridgel A, Thota A, Vitek JL, Alberts JL. Forced, not voluntary, exercise improves motor function in Parkinson’s disease patients. Neurorehabil Neural Repair 2009; 23:600–608.
- Smith AD, Zigmond MJ. Can the brain be protected through exercise? Lessons from an animal model of parkinsonism. Exp Neurol 2003; 184:31–39.
- Olanow CW, Rascol O, Hauser R, et al, for the ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278.
- Pasternak B, Svanström H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson’s disease. Am J Epidemiol 2012; 175:627-635.
- Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24:197–211.
- Hauser RA, Ellenbogen AL, Metman LV, et al. Crossover comparison of IPX066 and a standard levodopa formulation in advanced Parkinson’s disease. Mov Disord 2011; 26:2246–2252.
- Hely MA, Morris JG, Reid WG, Trafficante R. Sydney Multicenter Study of Parkinson’s disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord 2005; 20:190–199.
- Hung SW, Adeli GM, Arenovich T, Fox SH, Lang AE. Patient perception of dyskinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2010; 81:1112–1115.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N Engl J Med 2000; 342:1484–1491.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Nirenberg MJ. Dopamine agonist withdrawal syndrome: implications for patient care. Drugs Aging 2013; 30:587–592.
- Teva Neuroscience, Inc. Azilect prescribing information. https://www.azilect.com/Content/pdf/azi-40850-azilect-electronic-pi.pdf. Accessed June 29, 2015.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Adler CH, Ahlskog JE, eds. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for the Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Machado A, Fernandez HH, Deogaonkar M. Deep brain stimulation: what can patients expect from it? Cleve Clin J Med 2012; 79:113–120.
This has been a boom year for Parkinson disease, with the US Food and Drug Administration (FDA) approving two new therapies, and with others in the pipeline.
This article details clinical signs of Parkinson disease, discusses functional imaging, provides an update on current thinking on disease pathogenesis, and gives an overview of managing parkinsonian symptoms and dyskinesias.
DIAGNOSIS REMAINS CLINICAL
Although a better understanding of Parkinson disease has been gained in recent years, with the recognition of several premotor features and potential biomarkers, its diagnosis is still primarily based on clinical motor findings. The four cardinal motor features have the mnemonic TRAP:
- Tremor at rest can be subtle, involving just the thumb, best observed when the patient is sitting with the hand resting on the lap; or it can be obvious, involving the entire hand, arm, feet, lips, and chin.
- Rigidity can be felt rather than seen, by slowly passively rotating the patient’s wrist or elbow and feeling resistance. The right and left sides often differ.
- Akinesia or bradykinesia (slowness or lack of movement) can be observed by having the patient walk down a hallway. One may observe reduced arm swing and hesitation in initiating movement.
- Postural instability usually develops later rather than sooner in the disease progression. The patient may need to hold onto someone to maintain balance when getting up or walking.
At least two features must be present to make the diagnosis of parkinsonism. One feature must be tremor or rigidity.
Although the criteria for parkinsonism appear simple, the diagnosis of Parkinson disease is not always clear-cut. For example, shaking can be secondary to a dopamine receptor-blocking medication, to anxiety, or to essential tremor; rigidity and slowness may be due to arthritis; and postural instability can result from a neuropathy. Moreover, other neurodegenerative parkinsonian disorders may respond to levodopa (at least initially) and may present with levodopa-induced dyskinesias. Robust response to levodopa and the occurrence of dyskinesias are two additional features that strongly suggest the diagnosis of Parkinson disease.
Supporting parkinsonian features include stooped posture, masked facies, micrographia (small handwriting), drooling, speech changes (eg, hypophonia or soft speech, stuttering, slurring, monotonic speech), and a shuffling, festinating gait (quick short steps as if falling forward).
PARKINSON MIMICS
Parkinsonism is a broader term than Parkinson disease or idiopathic Parkinson disease. It is characterized by akinetic rigidity and impaired motor activity that leads to reduced function and falls; behavioral changes also may occur.
In the United States, Parkinson disease is the most common cause of parkinsonism. Other nonneurodegenerative causes are drug-induced parkinsonism (due to dopamine receptor antagonists such as antipsychotic or antiemetic drugs), stroke (in the basal ganglia or frontal lobe), and normal-pressure hydrocephalus (causing lower-body parkinsonism). Mimics of parkinsonism include essential tremor and psychogenic parkinsonism.
Parkinsonism can also be caused by Parkinson-plus disorders, ie, neurodegenerative conditions characterized by parkinsonism along with additional signs and symptoms, as listed below. Parkinson-plus disorders include progressive supranuclear palsy, multiple system atrophy, corticobasal degeneration, and Lewy body disease.
Clinical features that suggest a diagnosis other than Parkinson disease include1:
- Poor response to adequate dosages of levodopa
- Early onset of postural instability and falls
- Axial rigidity (eg, stiff neck) more than appendicular rigidity
- Early dementia
- Supranuclear gaze palsy
- Unusual movements besides tremor, eg, limb dystonia, myoclonus, limb levitation or alien limb syndrome
- Profound autonomic dysfunction
- Psychotic symptoms before taking levodopa or dopaminergic medication.
The precise diagnosis of Parkinson-plus disorders is not critical, as the treatment is generally the same for all of them: ie, levodopa (if it shows some efficacy and is well tolerated), with additional symptomatic treatment for features such as depression, cognitive impairment, and autonomic dysfunction, and supportive therapy including physical, occupational, speech, and swallowing therapy.
IMAGING MAY ASSIST IN THE DIAGNOSIS
Dopamine transporter single-photon emission computed tomography (SPECT) is a functional imaging technique that supposedly reflects dopamine uptake by surviving presynaptic dopaminergic neurons in the striate bodies of the basal ganglia. Normal uptake shows distinct cashew-shaped enhancement bilaterally. In Parkinson disease, the enhanced areas are smaller and asymmetric, first with diminution of the tail (representing the putamen), then later involving the head (representing the caudate) along with the other striate bodies (Figure 1).
Dopamine transporter SPECT does not distinguish one neurodegenerative parkinsonian disorder from another. Therefore, it should not be used to distinguish Parkinson disease from other Parkinson-plus syndromes. But it does distinguish neurodegenerative parkinsonian disorders from nonneurodegenerative conditions and mimics, which have a normal result on dopamine transporter SPECT (Table 1).
SLOWING DISEASE PROGRESSION
Current treatments for Parkinson disease can significantly improve symptoms but, unfortunately, do not cure the disease or slow its progression. Testing whether agents modify the disease course is particularly difficult with Parkinson disease, because it affects individuals differently, has a wide spectrum of symptoms, has a long time course, and lacks definitive markers to monitor progression. Some agents have shown promise:
Caffeine. People who drink coffee are less likely to develop Parkinson disease, with the risk declining with the number of cups per day.2 For those who have the disease, drinking coffee is associated with reduced symptoms.
Exercise improves Parkinson disease and may prevent it, and some studies suggest that it can delay its progression.3 Exercise has been shown in an animal model to reduce the vulnerability of dopamine neurons to the toxic agent 6-hydroxydopamine.4 Functional magnetic resonance imaging studies have shown blood flow patterns before and after exercise that are similar to those seen in patients with and without Parkinson medication.3
Rasagiline, a monoamine oxidase B (MAO-B) inhibitor used for symptomatic treatment of Parkinson disease, had conflicting results in a neuroprotective clinical trial. Patients who received rasagiline 1 mg daily—but not those who received 2 mg daily—at the beginning of the trial had better Parkinson motor scores compared with patients who received rasagiline 9 months later.5
Inosine is a urate precursor that elevates urate levels in serum and the central nervous system. For unknown reasons, patients with Parkinson disease tend to have a low uric acid level, and higher levels are associated with milder disease. It is hoped that raising the uric acid level to a “pre-gout level” may slow the progression of Parkinson disease.
Isradipine, a calcium channel blocker, was found in an epidemiologic study of elderly patients to be associated with reduced likelihood of developing Parkinson disease.6 The drug is now undergoing clinical trials.
Smoking. Although cigarette smokers have long been recognized as having a very low risk of developing Parkinson disease, smoking is not recommended.
Agents found ineffective. Agents that have been tested and found ineffective in modifying the course of Parkinson disease include vitamin E, coenzyme Q10, riluzole, GPI-1485, pramipexole, cogane, CEP-1347, TCH-346, and creatine.
NOT JUST DOPAMINE—OR TREMORS
Dopamine deficiency is central to the current understanding of the pathogenesis of Parkinson disease and the focus of treatment efforts, but if dopamine deficiency were the only problem, replacing it should completely ameliorate all parkinsonian features. Other neurotransmitters also play roles: norepinephrine is implicated in orthostatic symptoms and apathy, acetylcholine in cognitive behaviors, glutamate in dyskinesias, and serotonin in depression, anxiety, and sleep abnormalities.
The most recognized area of involvement in the brain has traditionally been the substantia nigra in the midbrain. However, current thinking is that the disease starts lower in the caudal area of the brainstem (along with the olfactory tubercle), moves through the pons to the midbrain, then spreads across the cerebrum with extensive neocortical involvement.
Early premotor indicators are now recognized to occur 15 to 20 years before a tremor appears. The first signs are often hyposmia (diminished sense of smell, reflecting involvement of the olfactory tubercle) and constipation (reflecting involvement of the medulla and the vagus nucleus). With pons involvement, the patient can develop rapid eye movement sleep behavior disorder, depression, or anxiety. Only then does the disease spread to the midbrain and cause resting tremor, rigidity, and bradykinesia.7
Identifying the preclinical stages and starting disease-modifying treatments before the onset of motor symptoms may one day prove important, but at this point, the premotor symptoms (anosmia, constipation, depression) are too nonspecific to be useful, and such treatments have not yet been identified.
TREATMENT: LEVODOPA STILL PRIMARY
When to start drug treatment depends primarily on how much the symptoms bother the patient. Regardless of the clinician’s (or patient’s) belief in the benefits of delaying symptomatic treatment, it is universally considered necessary to start medication when gait problems develop because of the danger of a fall and resulting disability.
Carbidopa-levodopa combination therapy remains the most effective treatment; if it is not effective, another diagnosis may need to be considered. Carbidopa-levodopa improves tremor, rigidity, and bradykinesia, particularly in the early stages of Parkinson disease. It is well tolerated, has rapid onset, reduces the risk of death, and is the least expensive of the medications for Parkinson disease.
Immediate-release and continued-release formulations are available, as well as one that dissolves rapidly on the tongue and can be taken without water. An oral extended-release carbidopa-levodopa formulation (Rytary) was approved by the FDA in January 2015. Tablets are filled with drug-containing microbeads that dissolve at different rates to achieve therapeutic levodopa levels as quickly as the immediate-release formulation and maintain them for an extended time.8
The development of dyskinesias is the major psychological drawback of levodopa, occurring in 80% of patients after 5 to 10 years of treatment. Although many patients fear this side effect, most patients who develop it find it preferable to the rigidity and bradykinesia of Parkinson disease. In most cases, bothersome dyskinesias can be controlled by adjusting medications.9,10
Dopamine agonists include pramipexole, ropinirole, and rotigotine. They are available in generic form as three-times-daily dosing; once-daily dosing is also available, but not as a generic formulation. Dopamine agonists have the advantage of potentially improving depression and delaying the onset of dyskinesias.
However, dopamine agonists have a number of disadvantages compared with levodopa: they have a longer titration period, are less effective, and are less well tolerated, especially in the elderly. Side effects occur more frequently than with levodopa and include general and peripheral edema, hallucinations, nausea, lightheadedness, and sleepiness.11,12 These drugs are also associated with “sleep attacks” (sudden falling asleep while active, such as while driving or eating) and with compulsive and impulsive behaviors such as hypersexuality, buying, binge eating, and gambling. Although these behaviors occur in fewer than 10% of patients, they can be devastating, leading to marital, financial, and legal problems. A bothersome clinical state termed dopamine agonist withdrawal syndrome is characterized by anxiety, depression, jitteriness, and palpitations when dopamine agonists are tapered or discontinued because of a side effect.13
MAO-B inhibitors delay the breakdown of dopamine, allowing it to “stay” in the brain for a longer period of time. Rasagiline for early monotherapy has the advantages of once-daily dosing, no titration, and excellent tolerability, even in the elderly. Potential drug interactions should be considered when using this drug. Early warnings about interactions with tyramine-rich foods were lifted after trials showed that this was not a problem.14
Amantadine is an N-methyl-d-aspartate (NMDA) receptor antagonist often used in early Parkinson disease and for treatment of dyskinesias and fatigue. It is the only drug that is intrinsically antidyskinetic and also improves Parkinson symptoms.15 Side effects include leg swelling, livedo reticularis, and neuropsychiatric and anticholinergic effects.
Anticholinergic agents (eg, trihexyphenidyl) improve tremor but are not as useful for bradykinesia or rigidity, and often have anticholinergic effects such as mental dullness, dry mouth, dry eye, and urinary hesitancy, especially in the elderly, so they have a limited role in Parkinson treatment.
MOTOR COMPLICATIONS: FLUCTUATIONS AND DYSKINESIAS
Motor fluctuations are changes between the akinetic and mobile phases of Parkinson disease, or the off-periods and on-periods of drug treatment. A patient who is “off” is generally rigid and feels that the medication is not working. A patient who is “on” feels loose and mobile and that the medication is working. Variants of motor fluctuations include:
- End-of-dose deterioration
- Delayed onset of response (more than half an hour after taking medication)
- Drug-resistant offs—medication has become ineffective
- Random oscillation—on-off phenomenon
- Freezing—unpredictable inability to start or finish a movement.
Dyskinesias are abnormal involuntary movements such as writhing and twisting. They are associated with dopaminergic therapy at peak dose, when the drug starts to turn on or wear off (termed diphasic dyskinesias).16
The storage hypothesis provides a plausible explanation for the development of motor complications as the disease progresses. Although the half-life of levodopa is only 60 to 90 minutes, it is effective in early disease when given three times a day. It is believed that at this stage of the disease, enough dopaminergic neurons survive to “store” dopamine and release it as needed. As the disease progresses and dopaminergic neurons die, storage capacity diminishes, and the clinical effect slowly starts to approximate the pharmacokinetic profile of the drug. Upon taking the medication, the patient gets a surge of drug, causing dyskinesias, followed later by rigidity as the effect wears off since there are fewer surviving dopaminergic cells to store dopamine.
MANAGING DYSKINESIAS
Patients with dyskinesias should first be asked if they are bothered by them; not all patients are troubled by dyskinesias. If the movements only bother others (eg, family members), then education is often the only treatment needed. If the patient is uncomfortable, the following measures can be tried:
- Taking lower, more frequent doses of levodopa (however, risk of wearing off becomes a problem)
- Adding a dopamine agonist or MAO-B inhibitor while lowering the levodopa dose (however, MAO-B inhibitors pose a risk of side effects in elderly patients)
- Adding clozapine (periodic laboratory testing is required to monitor blood levels and liver and kidney function)
- Adding amantadine (however, this poses a risk of cognitive side effects).
Deep-brain-stimulation surgery is appropriate for select patients who are generally physically healthy, cognitively intact, and emotionally stable, with a strong family support system, but who are bothered by symptoms of parkinsonism (such as tremors), motor fluctuations, or dyskinesias.17
Infusion pump. In January 2015, the FDA approved a new system that continuously delivers levodopa-carbidopa in a 4:1 ratio in gel suspension for 16 hours directly into the small intestine, minimizing motor fluctuations. The patient changes the cartridge daily and turns it off at bedtime.
*Dr. Fernandez has received research support from AbbVie, Acadia, Auspex, Biotie Therapies, Civitas, Kyowa/ProStrakan, Michael J. Fox Foundation, Movement Disorders Society, NIH/NINDS, Parkinson Study Group, Rhythm, Synosia, and Teva. He also has received honoraria from Carling Communications, International Parkinson and Movement Disorders Society, The Ohio State University, and PRIME Education, Inc as a speaker in CME events. He has received honoraria from Biogen, GE Health Care, Lundbeck, Merz Pharmaceuticals, and Pfizer as a consultant. He has received royalty payments from Demos Publishing for serving as a book author/editor. Cleveland Clinic has contracts with AbbVie and Merz Pharmaceuticals for Dr. Fernandez’s role as a member of the Global Steering Committee for LCIG studies and as a consultant or speaker, and as Head Principal Investigator for the Xeomin Registry Study. Dr. Fernandez has received a stipend from International Parkinson and Movement Disorders Society for serving as medical editor of the Movement Disorders Society website.
This has been a boom year for Parkinson disease, with the US Food and Drug Administration (FDA) approving two new therapies, and with others in the pipeline.
This article details clinical signs of Parkinson disease, discusses functional imaging, provides an update on current thinking on disease pathogenesis, and gives an overview of managing parkinsonian symptoms and dyskinesias.
DIAGNOSIS REMAINS CLINICAL
Although a better understanding of Parkinson disease has been gained in recent years, with the recognition of several premotor features and potential biomarkers, its diagnosis is still primarily based on clinical motor findings. The four cardinal motor features have the mnemonic TRAP:
- Tremor at rest can be subtle, involving just the thumb, best observed when the patient is sitting with the hand resting on the lap; or it can be obvious, involving the entire hand, arm, feet, lips, and chin.
- Rigidity can be felt rather than seen, by slowly passively rotating the patient’s wrist or elbow and feeling resistance. The right and left sides often differ.
- Akinesia or bradykinesia (slowness or lack of movement) can be observed by having the patient walk down a hallway. One may observe reduced arm swing and hesitation in initiating movement.
- Postural instability usually develops later rather than sooner in the disease progression. The patient may need to hold onto someone to maintain balance when getting up or walking.
At least two features must be present to make the diagnosis of parkinsonism. One feature must be tremor or rigidity.
Although the criteria for parkinsonism appear simple, the diagnosis of Parkinson disease is not always clear-cut. For example, shaking can be secondary to a dopamine receptor-blocking medication, to anxiety, or to essential tremor; rigidity and slowness may be due to arthritis; and postural instability can result from a neuropathy. Moreover, other neurodegenerative parkinsonian disorders may respond to levodopa (at least initially) and may present with levodopa-induced dyskinesias. Robust response to levodopa and the occurrence of dyskinesias are two additional features that strongly suggest the diagnosis of Parkinson disease.
Supporting parkinsonian features include stooped posture, masked facies, micrographia (small handwriting), drooling, speech changes (eg, hypophonia or soft speech, stuttering, slurring, monotonic speech), and a shuffling, festinating gait (quick short steps as if falling forward).
PARKINSON MIMICS
Parkinsonism is a broader term than Parkinson disease or idiopathic Parkinson disease. It is characterized by akinetic rigidity and impaired motor activity that leads to reduced function and falls; behavioral changes also may occur.
In the United States, Parkinson disease is the most common cause of parkinsonism. Other nonneurodegenerative causes are drug-induced parkinsonism (due to dopamine receptor antagonists such as antipsychotic or antiemetic drugs), stroke (in the basal ganglia or frontal lobe), and normal-pressure hydrocephalus (causing lower-body parkinsonism). Mimics of parkinsonism include essential tremor and psychogenic parkinsonism.
Parkinsonism can also be caused by Parkinson-plus disorders, ie, neurodegenerative conditions characterized by parkinsonism along with additional signs and symptoms, as listed below. Parkinson-plus disorders include progressive supranuclear palsy, multiple system atrophy, corticobasal degeneration, and Lewy body disease.
Clinical features that suggest a diagnosis other than Parkinson disease include1:
- Poor response to adequate dosages of levodopa
- Early onset of postural instability and falls
- Axial rigidity (eg, stiff neck) more than appendicular rigidity
- Early dementia
- Supranuclear gaze palsy
- Unusual movements besides tremor, eg, limb dystonia, myoclonus, limb levitation or alien limb syndrome
- Profound autonomic dysfunction
- Psychotic symptoms before taking levodopa or dopaminergic medication.
The precise diagnosis of Parkinson-plus disorders is not critical, as the treatment is generally the same for all of them: ie, levodopa (if it shows some efficacy and is well tolerated), with additional symptomatic treatment for features such as depression, cognitive impairment, and autonomic dysfunction, and supportive therapy including physical, occupational, speech, and swallowing therapy.
IMAGING MAY ASSIST IN THE DIAGNOSIS
Dopamine transporter single-photon emission computed tomography (SPECT) is a functional imaging technique that supposedly reflects dopamine uptake by surviving presynaptic dopaminergic neurons in the striate bodies of the basal ganglia. Normal uptake shows distinct cashew-shaped enhancement bilaterally. In Parkinson disease, the enhanced areas are smaller and asymmetric, first with diminution of the tail (representing the putamen), then later involving the head (representing the caudate) along with the other striate bodies (Figure 1).
Dopamine transporter SPECT does not distinguish one neurodegenerative parkinsonian disorder from another. Therefore, it should not be used to distinguish Parkinson disease from other Parkinson-plus syndromes. But it does distinguish neurodegenerative parkinsonian disorders from nonneurodegenerative conditions and mimics, which have a normal result on dopamine transporter SPECT (Table 1).
SLOWING DISEASE PROGRESSION
Current treatments for Parkinson disease can significantly improve symptoms but, unfortunately, do not cure the disease or slow its progression. Testing whether agents modify the disease course is particularly difficult with Parkinson disease, because it affects individuals differently, has a wide spectrum of symptoms, has a long time course, and lacks definitive markers to monitor progression. Some agents have shown promise:
Caffeine. People who drink coffee are less likely to develop Parkinson disease, with the risk declining with the number of cups per day.2 For those who have the disease, drinking coffee is associated with reduced symptoms.
Exercise improves Parkinson disease and may prevent it, and some studies suggest that it can delay its progression.3 Exercise has been shown in an animal model to reduce the vulnerability of dopamine neurons to the toxic agent 6-hydroxydopamine.4 Functional magnetic resonance imaging studies have shown blood flow patterns before and after exercise that are similar to those seen in patients with and without Parkinson medication.3
Rasagiline, a monoamine oxidase B (MAO-B) inhibitor used for symptomatic treatment of Parkinson disease, had conflicting results in a neuroprotective clinical trial. Patients who received rasagiline 1 mg daily—but not those who received 2 mg daily—at the beginning of the trial had better Parkinson motor scores compared with patients who received rasagiline 9 months later.5
Inosine is a urate precursor that elevates urate levels in serum and the central nervous system. For unknown reasons, patients with Parkinson disease tend to have a low uric acid level, and higher levels are associated with milder disease. It is hoped that raising the uric acid level to a “pre-gout level” may slow the progression of Parkinson disease.
Isradipine, a calcium channel blocker, was found in an epidemiologic study of elderly patients to be associated with reduced likelihood of developing Parkinson disease.6 The drug is now undergoing clinical trials.
Smoking. Although cigarette smokers have long been recognized as having a very low risk of developing Parkinson disease, smoking is not recommended.
Agents found ineffective. Agents that have been tested and found ineffective in modifying the course of Parkinson disease include vitamin E, coenzyme Q10, riluzole, GPI-1485, pramipexole, cogane, CEP-1347, TCH-346, and creatine.
NOT JUST DOPAMINE—OR TREMORS
Dopamine deficiency is central to the current understanding of the pathogenesis of Parkinson disease and the focus of treatment efforts, but if dopamine deficiency were the only problem, replacing it should completely ameliorate all parkinsonian features. Other neurotransmitters also play roles: norepinephrine is implicated in orthostatic symptoms and apathy, acetylcholine in cognitive behaviors, glutamate in dyskinesias, and serotonin in depression, anxiety, and sleep abnormalities.
The most recognized area of involvement in the brain has traditionally been the substantia nigra in the midbrain. However, current thinking is that the disease starts lower in the caudal area of the brainstem (along with the olfactory tubercle), moves through the pons to the midbrain, then spreads across the cerebrum with extensive neocortical involvement.
Early premotor indicators are now recognized to occur 15 to 20 years before a tremor appears. The first signs are often hyposmia (diminished sense of smell, reflecting involvement of the olfactory tubercle) and constipation (reflecting involvement of the medulla and the vagus nucleus). With pons involvement, the patient can develop rapid eye movement sleep behavior disorder, depression, or anxiety. Only then does the disease spread to the midbrain and cause resting tremor, rigidity, and bradykinesia.7
Identifying the preclinical stages and starting disease-modifying treatments before the onset of motor symptoms may one day prove important, but at this point, the premotor symptoms (anosmia, constipation, depression) are too nonspecific to be useful, and such treatments have not yet been identified.
TREATMENT: LEVODOPA STILL PRIMARY
When to start drug treatment depends primarily on how much the symptoms bother the patient. Regardless of the clinician’s (or patient’s) belief in the benefits of delaying symptomatic treatment, it is universally considered necessary to start medication when gait problems develop because of the danger of a fall and resulting disability.
Carbidopa-levodopa combination therapy remains the most effective treatment; if it is not effective, another diagnosis may need to be considered. Carbidopa-levodopa improves tremor, rigidity, and bradykinesia, particularly in the early stages of Parkinson disease. It is well tolerated, has rapid onset, reduces the risk of death, and is the least expensive of the medications for Parkinson disease.
Immediate-release and continued-release formulations are available, as well as one that dissolves rapidly on the tongue and can be taken without water. An oral extended-release carbidopa-levodopa formulation (Rytary) was approved by the FDA in January 2015. Tablets are filled with drug-containing microbeads that dissolve at different rates to achieve therapeutic levodopa levels as quickly as the immediate-release formulation and maintain them for an extended time.8
The development of dyskinesias is the major psychological drawback of levodopa, occurring in 80% of patients after 5 to 10 years of treatment. Although many patients fear this side effect, most patients who develop it find it preferable to the rigidity and bradykinesia of Parkinson disease. In most cases, bothersome dyskinesias can be controlled by adjusting medications.9,10
Dopamine agonists include pramipexole, ropinirole, and rotigotine. They are available in generic form as three-times-daily dosing; once-daily dosing is also available, but not as a generic formulation. Dopamine agonists have the advantage of potentially improving depression and delaying the onset of dyskinesias.
However, dopamine agonists have a number of disadvantages compared with levodopa: they have a longer titration period, are less effective, and are less well tolerated, especially in the elderly. Side effects occur more frequently than with levodopa and include general and peripheral edema, hallucinations, nausea, lightheadedness, and sleepiness.11,12 These drugs are also associated with “sleep attacks” (sudden falling asleep while active, such as while driving or eating) and with compulsive and impulsive behaviors such as hypersexuality, buying, binge eating, and gambling. Although these behaviors occur in fewer than 10% of patients, they can be devastating, leading to marital, financial, and legal problems. A bothersome clinical state termed dopamine agonist withdrawal syndrome is characterized by anxiety, depression, jitteriness, and palpitations when dopamine agonists are tapered or discontinued because of a side effect.13
MAO-B inhibitors delay the breakdown of dopamine, allowing it to “stay” in the brain for a longer period of time. Rasagiline for early monotherapy has the advantages of once-daily dosing, no titration, and excellent tolerability, even in the elderly. Potential drug interactions should be considered when using this drug. Early warnings about interactions with tyramine-rich foods were lifted after trials showed that this was not a problem.14
Amantadine is an N-methyl-d-aspartate (NMDA) receptor antagonist often used in early Parkinson disease and for treatment of dyskinesias and fatigue. It is the only drug that is intrinsically antidyskinetic and also improves Parkinson symptoms.15 Side effects include leg swelling, livedo reticularis, and neuropsychiatric and anticholinergic effects.
Anticholinergic agents (eg, trihexyphenidyl) improve tremor but are not as useful for bradykinesia or rigidity, and often have anticholinergic effects such as mental dullness, dry mouth, dry eye, and urinary hesitancy, especially in the elderly, so they have a limited role in Parkinson treatment.
MOTOR COMPLICATIONS: FLUCTUATIONS AND DYSKINESIAS
Motor fluctuations are changes between the akinetic and mobile phases of Parkinson disease, or the off-periods and on-periods of drug treatment. A patient who is “off” is generally rigid and feels that the medication is not working. A patient who is “on” feels loose and mobile and that the medication is working. Variants of motor fluctuations include:
- End-of-dose deterioration
- Delayed onset of response (more than half an hour after taking medication)
- Drug-resistant offs—medication has become ineffective
- Random oscillation—on-off phenomenon
- Freezing—unpredictable inability to start or finish a movement.
Dyskinesias are abnormal involuntary movements such as writhing and twisting. They are associated with dopaminergic therapy at peak dose, when the drug starts to turn on or wear off (termed diphasic dyskinesias).16
The storage hypothesis provides a plausible explanation for the development of motor complications as the disease progresses. Although the half-life of levodopa is only 60 to 90 minutes, it is effective in early disease when given three times a day. It is believed that at this stage of the disease, enough dopaminergic neurons survive to “store” dopamine and release it as needed. As the disease progresses and dopaminergic neurons die, storage capacity diminishes, and the clinical effect slowly starts to approximate the pharmacokinetic profile of the drug. Upon taking the medication, the patient gets a surge of drug, causing dyskinesias, followed later by rigidity as the effect wears off since there are fewer surviving dopaminergic cells to store dopamine.
MANAGING DYSKINESIAS
Patients with dyskinesias should first be asked if they are bothered by them; not all patients are troubled by dyskinesias. If the movements only bother others (eg, family members), then education is often the only treatment needed. If the patient is uncomfortable, the following measures can be tried:
- Taking lower, more frequent doses of levodopa (however, risk of wearing off becomes a problem)
- Adding a dopamine agonist or MAO-B inhibitor while lowering the levodopa dose (however, MAO-B inhibitors pose a risk of side effects in elderly patients)
- Adding clozapine (periodic laboratory testing is required to monitor blood levels and liver and kidney function)
- Adding amantadine (however, this poses a risk of cognitive side effects).
Deep-brain-stimulation surgery is appropriate for select patients who are generally physically healthy, cognitively intact, and emotionally stable, with a strong family support system, but who are bothered by symptoms of parkinsonism (such as tremors), motor fluctuations, or dyskinesias.17
Infusion pump. In January 2015, the FDA approved a new system that continuously delivers levodopa-carbidopa in a 4:1 ratio in gel suspension for 16 hours directly into the small intestine, minimizing motor fluctuations. The patient changes the cartridge daily and turns it off at bedtime.
*Dr. Fernandez has received research support from AbbVie, Acadia, Auspex, Biotie Therapies, Civitas, Kyowa/ProStrakan, Michael J. Fox Foundation, Movement Disorders Society, NIH/NINDS, Parkinson Study Group, Rhythm, Synosia, and Teva. He also has received honoraria from Carling Communications, International Parkinson and Movement Disorders Society, The Ohio State University, and PRIME Education, Inc as a speaker in CME events. He has received honoraria from Biogen, GE Health Care, Lundbeck, Merz Pharmaceuticals, and Pfizer as a consultant. He has received royalty payments from Demos Publishing for serving as a book author/editor. Cleveland Clinic has contracts with AbbVie and Merz Pharmaceuticals for Dr. Fernandez’s role as a member of the Global Steering Committee for LCIG studies and as a consultant or speaker, and as Head Principal Investigator for the Xeomin Registry Study. Dr. Fernandez has received a stipend from International Parkinson and Movement Disorders Society for serving as medical editor of the Movement Disorders Society website.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Hernán MA, Takkouche B, Caamaño-Isoma F, et al. A meta-analysis of coffee drinking, cigarette smoking, and risk of Parkinson’s disease. Ann Neurol 2002; 52:276–84.
- Ridgel A, Thota A, Vitek JL, Alberts JL. Forced, not voluntary, exercise improves motor function in Parkinson’s disease patients. Neurorehabil Neural Repair 2009; 23:600–608.
- Smith AD, Zigmond MJ. Can the brain be protected through exercise? Lessons from an animal model of parkinsonism. Exp Neurol 2003; 184:31–39.
- Olanow CW, Rascol O, Hauser R, et al, for the ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278.
- Pasternak B, Svanström H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson’s disease. Am J Epidemiol 2012; 175:627-635.
- Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24:197–211.
- Hauser RA, Ellenbogen AL, Metman LV, et al. Crossover comparison of IPX066 and a standard levodopa formulation in advanced Parkinson’s disease. Mov Disord 2011; 26:2246–2252.
- Hely MA, Morris JG, Reid WG, Trafficante R. Sydney Multicenter Study of Parkinson’s disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord 2005; 20:190–199.
- Hung SW, Adeli GM, Arenovich T, Fox SH, Lang AE. Patient perception of dyskinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2010; 81:1112–1115.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N Engl J Med 2000; 342:1484–1491.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Nirenberg MJ. Dopamine agonist withdrawal syndrome: implications for patient care. Drugs Aging 2013; 30:587–592.
- Teva Neuroscience, Inc. Azilect prescribing information. https://www.azilect.com/Content/pdf/azi-40850-azilect-electronic-pi.pdf. Accessed June 29, 2015.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Adler CH, Ahlskog JE, eds. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for the Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Machado A, Fernandez HH, Deogaonkar M. Deep brain stimulation: what can patients expect from it? Cleve Clin J Med 2012; 79:113–120.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Hernán MA, Takkouche B, Caamaño-Isoma F, et al. A meta-analysis of coffee drinking, cigarette smoking, and risk of Parkinson’s disease. Ann Neurol 2002; 52:276–84.
- Ridgel A, Thota A, Vitek JL, Alberts JL. Forced, not voluntary, exercise improves motor function in Parkinson’s disease patients. Neurorehabil Neural Repair 2009; 23:600–608.
- Smith AD, Zigmond MJ. Can the brain be protected through exercise? Lessons from an animal model of parkinsonism. Exp Neurol 2003; 184:31–39.
- Olanow CW, Rascol O, Hauser R, et al, for the ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278.
- Pasternak B, Svanström H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson’s disease. Am J Epidemiol 2012; 175:627-635.
- Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24:197–211.
- Hauser RA, Ellenbogen AL, Metman LV, et al. Crossover comparison of IPX066 and a standard levodopa formulation in advanced Parkinson’s disease. Mov Disord 2011; 26:2246–2252.
- Hely MA, Morris JG, Reid WG, Trafficante R. Sydney Multicenter Study of Parkinson’s disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord 2005; 20:190–199.
- Hung SW, Adeli GM, Arenovich T, Fox SH, Lang AE. Patient perception of dyskinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2010; 81:1112–1115.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N Engl J Med 2000; 342:1484–1491.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Nirenberg MJ. Dopamine agonist withdrawal syndrome: implications for patient care. Drugs Aging 2013; 30:587–592.
- Teva Neuroscience, Inc. Azilect prescribing information. https://www.azilect.com/Content/pdf/azi-40850-azilect-electronic-pi.pdf. Accessed June 29, 2015.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Adler CH, Ahlskog JE, eds. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for the Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Machado A, Fernandez HH, Deogaonkar M. Deep brain stimulation: what can patients expect from it? Cleve Clin J Med 2012; 79:113–120.
KEY POINTS
- Parkinson disease is diagnosed by clinical signs with the mnemonic TRAP: Tremor at rest, Rigidity, Akinesia or bradykinesia, and Postural/gait instability.
- A dopamine transporter functional scan can distinguish neurodegenerative parkinsonian disorders from nonneurodegenerative etiologies such as drug-induced parkinsonism and vascular parkinsonism, and from mimics such as psychogenic parkinsonism and essential tremor.
- Coffee consumption and exercise may benefit patients with Parkinson disease.
- Carbidopa-levodopa combination therapy is still the most effective treatment, but most patients develop dyskinesia after 5 to 10 years of treatment.
- Dyskinesias can be managed by adjusting or changing medications, switching to the new levodopa infusion pump system, or with deep-brain-stimulation surgery.
Perioperative MI: Data, practice, and questions

Except in emergency or specific high-risk surgery, or for extremely fragile high-risk patients, we anticipate a successful outcome from noncardiac surgery. The skills and tools of our anesthesiology colleagues have advanced to the point that severe intraoperative and immediate postoperative complications are rare.
Preoperative risk assessment and perioperative medical management in large medical centers are now largely done by hospital-based physicians with interest and expertise in this subspecialty, and are integrated into the care of the surgical patient. This has likely contributed to improved patient outcomes. Yet postoperative cardiovascular events still cause significant morbidity (although they generally occur in less than 10% of patients).
The entity of perioperative myocardial infarction (MI) has an interesting history. We have recognized for several decades that its presentation is often different than the usually diagnosed MI: perioperative MI is often painless and may manifest as unexplained sinus tachycardia, subtle changes in mental status, or mild dyspnea. These symptoms, if they occurred while the patient was at home, would often be mild enough that the patient would not seek immediate medical attention. Autopsy studies suggested that many of these MIs result from a different pathophysiology than the garden variety MI; plaque rupture with or without secondary thrombosis may be less common than myocardial injury resulting from an imbalance between cardiac demand and blood flow. Studies initially suggested that postoperative MI occurred many days after the surgery. But as tests to diagnose myocyte injury became more sensitive (electrocardiography, creatine kinase, creatine kinase MB, and now troponin), it was recognized that cardiac injury actually occurred very soon after or even during surgery.
With the advent of highly sensitive and fairly specific troponin assays, it seems that perioperative cardiac injury is extremely common, perhaps occurring in up to 20% of patients (if we include patients at high risk based on traditional criteria). This has led to the newly described entity of “myocardial injury after noncardiac surgery” (MINS). MINS patients, diagnosed by troponin elevations, usually are asymptomatic, and many do not meet criteria for any type of MI. But strikingly, as discussed in this issue of the Journal by Horr et al, simply having a postoperative troponin elevation predicts an increased risk of clinical cardiovascular events and a decreased 30-day survival rate.
Adding postoperative troponin measurement to the usual preoperative screening protocol significantly increases our ability to predict delayed cardiovascular events and mortality. As pointed out by Cohn in his accompanying editorial, the benefit, if any, of screening low-risk patients remains to be defined. But an even more important issue, as commented upon in both papers, is what to do when an elevated troponin is detected in a postoperative patient who is otherwise doing perfectly well. Given our current knowledge of the pathophysiology of postoperative MI and the still overall low mortality, it seems unreasonable to immediately take all of these patients to the catheterization suite. Yet with current knowledge of the prognostic significance of troponin elevation, this can’t be ignored. Should all patients receive immediate high-intensity statin therapy, antiplatelet therapy if safe in the specific perioperative setting, and postdischarge physiologic stress studies, or should we “just” take it as a potential high-impact teaching moment and advise patients of their increased cardiovascular risk and offer our usual heart-healthy admonitions?
The confirmed observation that postoperative troponin elevation predicts morbidity and mortality over the subsequent 30 days, and perhaps even longer, has triggered the start of several interventional trials. The results of these will, hopefully, help us to further improve perioperative outcomes.
Except in emergency or specific high-risk surgery, or for extremely fragile high-risk patients, we anticipate a successful outcome from noncardiac surgery. The skills and tools of our anesthesiology colleagues have advanced to the point that severe intraoperative and immediate postoperative complications are rare.
Preoperative risk assessment and perioperative medical management in large medical centers are now largely done by hospital-based physicians with interest and expertise in this subspecialty, and are integrated into the care of the surgical patient. This has likely contributed to improved patient outcomes. Yet postoperative cardiovascular events still cause significant morbidity (although they generally occur in less than 10% of patients).
The entity of perioperative myocardial infarction (MI) has an interesting history. We have recognized for several decades that its presentation is often different than the usually diagnosed MI: perioperative MI is often painless and may manifest as unexplained sinus tachycardia, subtle changes in mental status, or mild dyspnea. These symptoms, if they occurred while the patient was at home, would often be mild enough that the patient would not seek immediate medical attention. Autopsy studies suggested that many of these MIs result from a different pathophysiology than the garden variety MI; plaque rupture with or without secondary thrombosis may be less common than myocardial injury resulting from an imbalance between cardiac demand and blood flow. Studies initially suggested that postoperative MI occurred many days after the surgery. But as tests to diagnose myocyte injury became more sensitive (electrocardiography, creatine kinase, creatine kinase MB, and now troponin), it was recognized that cardiac injury actually occurred very soon after or even during surgery.
With the advent of highly sensitive and fairly specific troponin assays, it seems that perioperative cardiac injury is extremely common, perhaps occurring in up to 20% of patients (if we include patients at high risk based on traditional criteria). This has led to the newly described entity of “myocardial injury after noncardiac surgery” (MINS). MINS patients, diagnosed by troponin elevations, usually are asymptomatic, and many do not meet criteria for any type of MI. But strikingly, as discussed in this issue of the Journal by Horr et al, simply having a postoperative troponin elevation predicts an increased risk of clinical cardiovascular events and a decreased 30-day survival rate.
Adding postoperative troponin measurement to the usual preoperative screening protocol significantly increases our ability to predict delayed cardiovascular events and mortality. As pointed out by Cohn in his accompanying editorial, the benefit, if any, of screening low-risk patients remains to be defined. But an even more important issue, as commented upon in both papers, is what to do when an elevated troponin is detected in a postoperative patient who is otherwise doing perfectly well. Given our current knowledge of the pathophysiology of postoperative MI and the still overall low mortality, it seems unreasonable to immediately take all of these patients to the catheterization suite. Yet with current knowledge of the prognostic significance of troponin elevation, this can’t be ignored. Should all patients receive immediate high-intensity statin therapy, antiplatelet therapy if safe in the specific perioperative setting, and postdischarge physiologic stress studies, or should we “just” take it as a potential high-impact teaching moment and advise patients of their increased cardiovascular risk and offer our usual heart-healthy admonitions?
The confirmed observation that postoperative troponin elevation predicts morbidity and mortality over the subsequent 30 days, and perhaps even longer, has triggered the start of several interventional trials. The results of these will, hopefully, help us to further improve perioperative outcomes.
Except in emergency or specific high-risk surgery, or for extremely fragile high-risk patients, we anticipate a successful outcome from noncardiac surgery. The skills and tools of our anesthesiology colleagues have advanced to the point that severe intraoperative and immediate postoperative complications are rare.
Preoperative risk assessment and perioperative medical management in large medical centers are now largely done by hospital-based physicians with interest and expertise in this subspecialty, and are integrated into the care of the surgical patient. This has likely contributed to improved patient outcomes. Yet postoperative cardiovascular events still cause significant morbidity (although they generally occur in less than 10% of patients).
The entity of perioperative myocardial infarction (MI) has an interesting history. We have recognized for several decades that its presentation is often different than the usually diagnosed MI: perioperative MI is often painless and may manifest as unexplained sinus tachycardia, subtle changes in mental status, or mild dyspnea. These symptoms, if they occurred while the patient was at home, would often be mild enough that the patient would not seek immediate medical attention. Autopsy studies suggested that many of these MIs result from a different pathophysiology than the garden variety MI; plaque rupture with or without secondary thrombosis may be less common than myocardial injury resulting from an imbalance between cardiac demand and blood flow. Studies initially suggested that postoperative MI occurred many days after the surgery. But as tests to diagnose myocyte injury became more sensitive (electrocardiography, creatine kinase, creatine kinase MB, and now troponin), it was recognized that cardiac injury actually occurred very soon after or even during surgery.
With the advent of highly sensitive and fairly specific troponin assays, it seems that perioperative cardiac injury is extremely common, perhaps occurring in up to 20% of patients (if we include patients at high risk based on traditional criteria). This has led to the newly described entity of “myocardial injury after noncardiac surgery” (MINS). MINS patients, diagnosed by troponin elevations, usually are asymptomatic, and many do not meet criteria for any type of MI. But strikingly, as discussed in this issue of the Journal by Horr et al, simply having a postoperative troponin elevation predicts an increased risk of clinical cardiovascular events and a decreased 30-day survival rate.
Adding postoperative troponin measurement to the usual preoperative screening protocol significantly increases our ability to predict delayed cardiovascular events and mortality. As pointed out by Cohn in his accompanying editorial, the benefit, if any, of screening low-risk patients remains to be defined. But an even more important issue, as commented upon in both papers, is what to do when an elevated troponin is detected in a postoperative patient who is otherwise doing perfectly well. Given our current knowledge of the pathophysiology of postoperative MI and the still overall low mortality, it seems unreasonable to immediately take all of these patients to the catheterization suite. Yet with current knowledge of the prognostic significance of troponin elevation, this can’t be ignored. Should all patients receive immediate high-intensity statin therapy, antiplatelet therapy if safe in the specific perioperative setting, and postdischarge physiologic stress studies, or should we “just” take it as a potential high-impact teaching moment and advise patients of their increased cardiovascular risk and offer our usual heart-healthy admonitions?
The confirmed observation that postoperative troponin elevation predicts morbidity and mortality over the subsequent 30 days, and perhaps even longer, has triggered the start of several interventional trials. The results of these will, hopefully, help us to further improve perioperative outcomes.
Postoperative troponin surveillance: A diagnostic dilemma
A major goal of perioperative medicine is to prevent, detect, and treat postoperative complications—in particular, cardiovascular complications. In the Perioperative Ischemic Evaluation (POISE) study,1 the 30-day mortality rate was four times higher in patients who had a perioperative myocardial infarction (MI) than in those who did not.1 Yet fewer than half of patients who have a postoperative MI have ischemic symptoms, suggesting that routine monitoring of cardiac biomarkers could detect these events and allow early intervention.
From 10% to 20% of patients have troponin elevations after noncardiac surgery.2 But until recently, many of these elevations were thought to be of minor importance and were ignored unless the patient met diagnostic criteria for MI. A new entity called MINS (myocardial injury after noncardiac surgery)3 was defined as a troponin level exceeding the upper limit of normal with or without ischemic symptoms or electrocardiographic changes and excluding noncardiac causes such as stroke, sepsis, and pulmonary embolism. Because elevations of troponin at any level have been associated with increased 30-day mortality rates, the question of the value of routine screening of asymptomatic postoperative patients for troponin elevation has been raised.
In this issue of Cleveland Clinic Journal of Medicine, Horr et al4 review the controversy of postoperative screening using troponin measurement and propose an algorithm for management.
QUESTIONS TO CONSIDER
Before recommending screening asymptomatic patients for troponin elevation, we need to consider a number of questions:
- Which patients should be screened?
- How should troponin elevations be treated?
- Would casting a wider net improve outcomes?
- What are the possible harms of troponin screening?
The bottom line is, will postoperative troponin screening change management and result in improved outcomes?
WHICH PATIENTS SHOULD BE SCREENED?
Why routine screening may be indicated
Elevated or even just detectable troponin levels are associated with adverse outcomes. A systematic review and meta-analysis of 3,318 patients2 demonstrated that high troponin levels after noncardiac surgery were independently associated with a risk of death three times higher than in patients with normal troponin levels.
In the Vascular Events in Noncardiac Surgery Patients Cohort Evaluation (VISION) study,5 troponin T was measured in 15,133 patients after surgery. The overall mortality rate was 1.9%, and the higher the peak troponin T level the higher the risk of death.
In a single-center Canadian retrospective cohort analysis of 51,701 consecutive patients by Beattie et al,6 the peak postoperative level of troponin I improved the ability of a multivariable model to predict the risk of death. As in the VISION study, the mortality rate rose with the troponin level.6
In a study by van Waes et al7 in 2,232 consecutive noncardiac surgery patients over age 60 at intermediate to high risk, the all-cause mortality rate was 3%, and troponin I was elevated in 19% of patients. As in VISION and the Canadian retrospective study, the mortality rate increased with the troponin level.
Why routine screening may not help
In VISION,5 the probability of detecting myocardial injury was three times higher if patients were screened for 3 days after surgery than if they were tested only if clinical signs or symptoms indicated it.
However, in deciding whether to screen troponin levels in postoperative patients, we must take into account the patient’s clinical risk as well as the risk of the surgical procedure. Troponin elevation in low-risk patients is associated with a low mortality rate, and troponin elevations often are secondary to causes other than myocardial ischemia. In the study by van Waes et al,7 the association was stronger with all-cause mortality than with myocardial infarction, and in VISION5 there were more nonvascular deaths than vascular deaths, suggesting that troponin elevation is a nonspecific marker of adverse events.
Beattie et al6 found that the probability that a patient’s postoperative troponin level would be elevated increased as the patient’s clinical risk increased, but the yield was very low and the mortality rate was less than 1% in patients in risk classes 1 through 3 (of a possible 5 classes). In risk class 4, troponin I was elevated in 21.8%, and the mortality rate was 2.5%; in risk class 5 troponin I was elevated in 18.6%, and the mortality rate was 11.9%. Analyzing the data according to the type of surgery, mortality rates were highest in patients undergoing vascular surgery, neurosurgery, general surgery, and thoracic procedures, with all-cause mortality rates ranging from 2.6% to 5.2%.6
Screening should depend on risk
If postoperative troponin screening is to be recommended, it should not be routine for all patients but should be restricted to those with high clinical risk and those undergoing high-risk surgical procedures.
Rodseth and Devereaux8 recommended routine postoperative troponin measurement not only after vascular surgery, but also after high-risk surgery (general, neurosurgery, emergency surgery), as well as in patients over age 65 and patients with established atherosclerotic disease or risk factors for it. However, I believe this latter group may not be at high enough risk to justify routine screening.
Beattie et al6 advocated limiting postoperative troponin screening to patients with at least a moderate risk of MI and also suggested an international consensus conference to define criteria for postoperative MI, populations who should have routine postoperative screening, and consensus on treatment of patients with troponin elevations but not meeting the criteria for MI. Without this consensus on treatment, it is unclear if protocols for universal postoperative screening would improve outcomes.
For these reasons, the 2014 joint guidelines of the American College of Cardiology and American Heart Association9 (ACC/AHA) stated that the benefit of postoperative screening of troponin levels in patients with a high perioperative risk of MI but no signs or symptoms of myocardial ischemia or MI is “uncertain in the absence of established risks and benefits of a defined management strategy.” This recommendation was given a class IIb rating (may be considered) and level of evidence B (usefulness or efficacy less well established). On the other hand, the guidelines recommend measuring troponin levels if signs or symptoms suggest myocardial ischemia or MI (class I recommendation, level of evidence A) but state there is no benefit in routine screening of unselected patients without signs or symptoms of ischemia (class III recommendation, level of evidence B).
HOW SHOULD ELEVATIONS BE TREATED?
Because a troponin elevation in a patient without signs or symptoms of ischemia does not predict a specific type of death, physicians need to treat patients individually. Perioperative ischemia and inflammation could lead to injury of other organs, and death could result from multiorgan injury rather than from myocardial injury. Treating these troponin elevations in the same way we treat MI—ie, with antiplatelet therapy and anticoagulation—may result in increased bleeding or unnecessary cardiac catheterization, and starting beta-blockers in the perioperative period may be harmful. Because it is unclear how to manage these patients, cardiac medications have not routinely been given in previous studies.
POISE provided some evidence that patients with postoperative MI who were given aspirin and a statin did better.1 And the results of a smaller study10 suggested that intensification of drug therapy (aspirin, statin, beta-blocker, angiotensin-converting enzyme inhibitor) in patients with postoperative troponin I elevations was associated with improved outcomes at 1 year. If the bleeding risk is low, I believe that there is potential benefit in prescribing aspirin and statins for these patients.
CASTING A WIDER NET
Further complicating matters in the near future is the issue of using fifth-generation high-sensitivity troponin T assays. The European Society of Cardiology guidelines11 are somewhat more liberal than the ACC/AHA guidelines, stating that measuring high-sensitivity troponin after surgery “may be considered in high-risk patients to improve risk stratification.” This is a class IIB recommendation, level of evidence B.
With fifth-generation high-sensitivity troponin assays, troponin may be elevated in as many as 20% of patients preoperatively and 40% postoperatively, significantly increasing the number of patients said to have a complication. Besides potentially subjecting these patients to unproven treatments, such results would give the false impression that hospitals and surgeons using the screening tools actually had higher complication rates than those that did not screen.
POSSIBLE HARMS OF SCREENING
Elevated postoperative troponin may identify patients at higher risk of any adverse event but not specifically of cardiac-specific events. In an editorial, Beckman12 stated that routine measurement of troponin “is more likely to cause harm than to provide benefit and should not be used as a screening modality” because of the lack of a proven beneficial treatment strategy, because of the possible harm from applying the standard treatment for type 1 MI, and because it could divert attention from a true cause of an adverse event to a false one—ie, from a nonvascular condition to MI.11
There is clearly a need for clinical trials to determine which treatment, if any, can improve outcomes in these patients, and several trials have been started. But until we have evidence, we can only speculate as to whether screening postoperative patients for troponin elevation is beneficial or detrimental.
- Devereaux PJ, Xavier D, Pogue J, et al. Characteristics and short-term prognosis of perioperative myocardial infarction in patients undergoing noncardiac surgery: a cohort study. Ann Intern Med 2011; 154:523–528.
- Levy M, Heels-Ansdell D, Hiralal R, et al. Prognostic value of troponin and creatine kinase muscle and brain isoenzyme measurement after noncardiac surgery: a systematic review and meta-analysis. Anesthesiology 2011; 114:796–806.
- Botto F, Alonso-Coello P, Chan MT, et al. Myocardial injury after noncardiac surgery: a large, international, prospective cohort study establishing diagnostic criteria, characteristics, predictors, and 30-day outcomes. Anesthesiology 2014; 120:564–578.
- Horr S, Reed G, Menon V. Troponin elevation after noncardiac surgery: significance and management. Cleve Clin J Med 2015; 82:595–602.
- Vascular Events In Noncardiac Surgery Patients Cohort Evaluation Study I, Devereaux PJ, Chan MT, Alonso-Coello P, et al. Association between postoperative troponin levels and 30-day mortality among patients undergoing noncardiac surgery. JAMA 2012; 307:2295–2304.
- Beattie WS, Karkouti K, Tait G, et al. Use of clinically based troponin underestimates the cardiac injury in non-cardiac surgery: a single-centre cohort study in 51,701 consecutive patients. Can J Anaesth 2012; 59:1013–1022.
- van Waes JA, Nathoe HM, de Graaff JC, et al. Myocardial injury after noncardiac surgery and its association with short-term mortality. Circulation 2013; 127:2264–2271.
- Rodseth R, Devereaux PJ. Should we measure troponin routinely in patients after vascular surgery? American College of Cardiology. www.acc.org/latest-in-cardiology/articles/2014/07/18/14/46/should-we-measure-troponin-routinely-in-patients-after-vascular-surgery?w_nav=LC. Accessed August 5, 2015.
- Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; 64:e77–e137.
- Foucrier A, Rodseth R, Aissaoui M, et al. The long-term impact of early cardiovascular therapy intensification for postoperative troponin elevation after major vascular surgery. Anesth Analg 2014; 119:1053–1063.
- Kristensen SD, Knuuti J, Saraste A, et al. 2014 ESC/ESA Guidelines on non-cardiac surgery: cardiovascular assessment and management: The Joint Task Force on non-cardiac surgery: cardiovascular assessment and management of the European Society of Cardiology (ESC) and the European Society of Anaesthesiology (ESA). Eur Heart J 2014; 35:2383–2431.
- Beckman JA. Postoperative troponin screening: a cardiac Cassandra? Circulation 2013; 127:2253–2266.
A major goal of perioperative medicine is to prevent, detect, and treat postoperative complications—in particular, cardiovascular complications. In the Perioperative Ischemic Evaluation (POISE) study,1 the 30-day mortality rate was four times higher in patients who had a perioperative myocardial infarction (MI) than in those who did not.1 Yet fewer than half of patients who have a postoperative MI have ischemic symptoms, suggesting that routine monitoring of cardiac biomarkers could detect these events and allow early intervention.
From 10% to 20% of patients have troponin elevations after noncardiac surgery.2 But until recently, many of these elevations were thought to be of minor importance and were ignored unless the patient met diagnostic criteria for MI. A new entity called MINS (myocardial injury after noncardiac surgery)3 was defined as a troponin level exceeding the upper limit of normal with or without ischemic symptoms or electrocardiographic changes and excluding noncardiac causes such as stroke, sepsis, and pulmonary embolism. Because elevations of troponin at any level have been associated with increased 30-day mortality rates, the question of the value of routine screening of asymptomatic postoperative patients for troponin elevation has been raised.
In this issue of Cleveland Clinic Journal of Medicine, Horr et al4 review the controversy of postoperative screening using troponin measurement and propose an algorithm for management.
QUESTIONS TO CONSIDER
Before recommending screening asymptomatic patients for troponin elevation, we need to consider a number of questions:
- Which patients should be screened?
- How should troponin elevations be treated?
- Would casting a wider net improve outcomes?
- What are the possible harms of troponin screening?
The bottom line is, will postoperative troponin screening change management and result in improved outcomes?
WHICH PATIENTS SHOULD BE SCREENED?
Why routine screening may be indicated
Elevated or even just detectable troponin levels are associated with adverse outcomes. A systematic review and meta-analysis of 3,318 patients2 demonstrated that high troponin levels after noncardiac surgery were independently associated with a risk of death three times higher than in patients with normal troponin levels.
In the Vascular Events in Noncardiac Surgery Patients Cohort Evaluation (VISION) study,5 troponin T was measured in 15,133 patients after surgery. The overall mortality rate was 1.9%, and the higher the peak troponin T level the higher the risk of death.
In a single-center Canadian retrospective cohort analysis of 51,701 consecutive patients by Beattie et al,6 the peak postoperative level of troponin I improved the ability of a multivariable model to predict the risk of death. As in the VISION study, the mortality rate rose with the troponin level.6
In a study by van Waes et al7 in 2,232 consecutive noncardiac surgery patients over age 60 at intermediate to high risk, the all-cause mortality rate was 3%, and troponin I was elevated in 19% of patients. As in VISION and the Canadian retrospective study, the mortality rate increased with the troponin level.
Why routine screening may not help
In VISION,5 the probability of detecting myocardial injury was three times higher if patients were screened for 3 days after surgery than if they were tested only if clinical signs or symptoms indicated it.
However, in deciding whether to screen troponin levels in postoperative patients, we must take into account the patient’s clinical risk as well as the risk of the surgical procedure. Troponin elevation in low-risk patients is associated with a low mortality rate, and troponin elevations often are secondary to causes other than myocardial ischemia. In the study by van Waes et al,7 the association was stronger with all-cause mortality than with myocardial infarction, and in VISION5 there were more nonvascular deaths than vascular deaths, suggesting that troponin elevation is a nonspecific marker of adverse events.
Beattie et al6 found that the probability that a patient’s postoperative troponin level would be elevated increased as the patient’s clinical risk increased, but the yield was very low and the mortality rate was less than 1% in patients in risk classes 1 through 3 (of a possible 5 classes). In risk class 4, troponin I was elevated in 21.8%, and the mortality rate was 2.5%; in risk class 5 troponin I was elevated in 18.6%, and the mortality rate was 11.9%. Analyzing the data according to the type of surgery, mortality rates were highest in patients undergoing vascular surgery, neurosurgery, general surgery, and thoracic procedures, with all-cause mortality rates ranging from 2.6% to 5.2%.6
Screening should depend on risk
If postoperative troponin screening is to be recommended, it should not be routine for all patients but should be restricted to those with high clinical risk and those undergoing high-risk surgical procedures.
Rodseth and Devereaux8 recommended routine postoperative troponin measurement not only after vascular surgery, but also after high-risk surgery (general, neurosurgery, emergency surgery), as well as in patients over age 65 and patients with established atherosclerotic disease or risk factors for it. However, I believe this latter group may not be at high enough risk to justify routine screening.
Beattie et al6 advocated limiting postoperative troponin screening to patients with at least a moderate risk of MI and also suggested an international consensus conference to define criteria for postoperative MI, populations who should have routine postoperative screening, and consensus on treatment of patients with troponin elevations but not meeting the criteria for MI. Without this consensus on treatment, it is unclear if protocols for universal postoperative screening would improve outcomes.
For these reasons, the 2014 joint guidelines of the American College of Cardiology and American Heart Association9 (ACC/AHA) stated that the benefit of postoperative screening of troponin levels in patients with a high perioperative risk of MI but no signs or symptoms of myocardial ischemia or MI is “uncertain in the absence of established risks and benefits of a defined management strategy.” This recommendation was given a class IIb rating (may be considered) and level of evidence B (usefulness or efficacy less well established). On the other hand, the guidelines recommend measuring troponin levels if signs or symptoms suggest myocardial ischemia or MI (class I recommendation, level of evidence A) but state there is no benefit in routine screening of unselected patients without signs or symptoms of ischemia (class III recommendation, level of evidence B).
HOW SHOULD ELEVATIONS BE TREATED?
Because a troponin elevation in a patient without signs or symptoms of ischemia does not predict a specific type of death, physicians need to treat patients individually. Perioperative ischemia and inflammation could lead to injury of other organs, and death could result from multiorgan injury rather than from myocardial injury. Treating these troponin elevations in the same way we treat MI—ie, with antiplatelet therapy and anticoagulation—may result in increased bleeding or unnecessary cardiac catheterization, and starting beta-blockers in the perioperative period may be harmful. Because it is unclear how to manage these patients, cardiac medications have not routinely been given in previous studies.
POISE provided some evidence that patients with postoperative MI who were given aspirin and a statin did better.1 And the results of a smaller study10 suggested that intensification of drug therapy (aspirin, statin, beta-blocker, angiotensin-converting enzyme inhibitor) in patients with postoperative troponin I elevations was associated with improved outcomes at 1 year. If the bleeding risk is low, I believe that there is potential benefit in prescribing aspirin and statins for these patients.
CASTING A WIDER NET
Further complicating matters in the near future is the issue of using fifth-generation high-sensitivity troponin T assays. The European Society of Cardiology guidelines11 are somewhat more liberal than the ACC/AHA guidelines, stating that measuring high-sensitivity troponin after surgery “may be considered in high-risk patients to improve risk stratification.” This is a class IIB recommendation, level of evidence B.
With fifth-generation high-sensitivity troponin assays, troponin may be elevated in as many as 20% of patients preoperatively and 40% postoperatively, significantly increasing the number of patients said to have a complication. Besides potentially subjecting these patients to unproven treatments, such results would give the false impression that hospitals and surgeons using the screening tools actually had higher complication rates than those that did not screen.
POSSIBLE HARMS OF SCREENING
Elevated postoperative troponin may identify patients at higher risk of any adverse event but not specifically of cardiac-specific events. In an editorial, Beckman12 stated that routine measurement of troponin “is more likely to cause harm than to provide benefit and should not be used as a screening modality” because of the lack of a proven beneficial treatment strategy, because of the possible harm from applying the standard treatment for type 1 MI, and because it could divert attention from a true cause of an adverse event to a false one—ie, from a nonvascular condition to MI.11
There is clearly a need for clinical trials to determine which treatment, if any, can improve outcomes in these patients, and several trials have been started. But until we have evidence, we can only speculate as to whether screening postoperative patients for troponin elevation is beneficial or detrimental.
A major goal of perioperative medicine is to prevent, detect, and treat postoperative complications—in particular, cardiovascular complications. In the Perioperative Ischemic Evaluation (POISE) study,1 the 30-day mortality rate was four times higher in patients who had a perioperative myocardial infarction (MI) than in those who did not.1 Yet fewer than half of patients who have a postoperative MI have ischemic symptoms, suggesting that routine monitoring of cardiac biomarkers could detect these events and allow early intervention.
From 10% to 20% of patients have troponin elevations after noncardiac surgery.2 But until recently, many of these elevations were thought to be of minor importance and were ignored unless the patient met diagnostic criteria for MI. A new entity called MINS (myocardial injury after noncardiac surgery)3 was defined as a troponin level exceeding the upper limit of normal with or without ischemic symptoms or electrocardiographic changes and excluding noncardiac causes such as stroke, sepsis, and pulmonary embolism. Because elevations of troponin at any level have been associated with increased 30-day mortality rates, the question of the value of routine screening of asymptomatic postoperative patients for troponin elevation has been raised.
In this issue of Cleveland Clinic Journal of Medicine, Horr et al4 review the controversy of postoperative screening using troponin measurement and propose an algorithm for management.
QUESTIONS TO CONSIDER
Before recommending screening asymptomatic patients for troponin elevation, we need to consider a number of questions:
- Which patients should be screened?
- How should troponin elevations be treated?
- Would casting a wider net improve outcomes?
- What are the possible harms of troponin screening?
The bottom line is, will postoperative troponin screening change management and result in improved outcomes?
WHICH PATIENTS SHOULD BE SCREENED?
Why routine screening may be indicated
Elevated or even just detectable troponin levels are associated with adverse outcomes. A systematic review and meta-analysis of 3,318 patients2 demonstrated that high troponin levels after noncardiac surgery were independently associated with a risk of death three times higher than in patients with normal troponin levels.
In the Vascular Events in Noncardiac Surgery Patients Cohort Evaluation (VISION) study,5 troponin T was measured in 15,133 patients after surgery. The overall mortality rate was 1.9%, and the higher the peak troponin T level the higher the risk of death.
In a single-center Canadian retrospective cohort analysis of 51,701 consecutive patients by Beattie et al,6 the peak postoperative level of troponin I improved the ability of a multivariable model to predict the risk of death. As in the VISION study, the mortality rate rose with the troponin level.6
In a study by van Waes et al7 in 2,232 consecutive noncardiac surgery patients over age 60 at intermediate to high risk, the all-cause mortality rate was 3%, and troponin I was elevated in 19% of patients. As in VISION and the Canadian retrospective study, the mortality rate increased with the troponin level.
Why routine screening may not help
In VISION,5 the probability of detecting myocardial injury was three times higher if patients were screened for 3 days after surgery than if they were tested only if clinical signs or symptoms indicated it.
However, in deciding whether to screen troponin levels in postoperative patients, we must take into account the patient’s clinical risk as well as the risk of the surgical procedure. Troponin elevation in low-risk patients is associated with a low mortality rate, and troponin elevations often are secondary to causes other than myocardial ischemia. In the study by van Waes et al,7 the association was stronger with all-cause mortality than with myocardial infarction, and in VISION5 there were more nonvascular deaths than vascular deaths, suggesting that troponin elevation is a nonspecific marker of adverse events.
Beattie et al6 found that the probability that a patient’s postoperative troponin level would be elevated increased as the patient’s clinical risk increased, but the yield was very low and the mortality rate was less than 1% in patients in risk classes 1 through 3 (of a possible 5 classes). In risk class 4, troponin I was elevated in 21.8%, and the mortality rate was 2.5%; in risk class 5 troponin I was elevated in 18.6%, and the mortality rate was 11.9%. Analyzing the data according to the type of surgery, mortality rates were highest in patients undergoing vascular surgery, neurosurgery, general surgery, and thoracic procedures, with all-cause mortality rates ranging from 2.6% to 5.2%.6
Screening should depend on risk
If postoperative troponin screening is to be recommended, it should not be routine for all patients but should be restricted to those with high clinical risk and those undergoing high-risk surgical procedures.
Rodseth and Devereaux8 recommended routine postoperative troponin measurement not only after vascular surgery, but also after high-risk surgery (general, neurosurgery, emergency surgery), as well as in patients over age 65 and patients with established atherosclerotic disease or risk factors for it. However, I believe this latter group may not be at high enough risk to justify routine screening.
Beattie et al6 advocated limiting postoperative troponin screening to patients with at least a moderate risk of MI and also suggested an international consensus conference to define criteria for postoperative MI, populations who should have routine postoperative screening, and consensus on treatment of patients with troponin elevations but not meeting the criteria for MI. Without this consensus on treatment, it is unclear if protocols for universal postoperative screening would improve outcomes.
For these reasons, the 2014 joint guidelines of the American College of Cardiology and American Heart Association9 (ACC/AHA) stated that the benefit of postoperative screening of troponin levels in patients with a high perioperative risk of MI but no signs or symptoms of myocardial ischemia or MI is “uncertain in the absence of established risks and benefits of a defined management strategy.” This recommendation was given a class IIb rating (may be considered) and level of evidence B (usefulness or efficacy less well established). On the other hand, the guidelines recommend measuring troponin levels if signs or symptoms suggest myocardial ischemia or MI (class I recommendation, level of evidence A) but state there is no benefit in routine screening of unselected patients without signs or symptoms of ischemia (class III recommendation, level of evidence B).
HOW SHOULD ELEVATIONS BE TREATED?
Because a troponin elevation in a patient without signs or symptoms of ischemia does not predict a specific type of death, physicians need to treat patients individually. Perioperative ischemia and inflammation could lead to injury of other organs, and death could result from multiorgan injury rather than from myocardial injury. Treating these troponin elevations in the same way we treat MI—ie, with antiplatelet therapy and anticoagulation—may result in increased bleeding or unnecessary cardiac catheterization, and starting beta-blockers in the perioperative period may be harmful. Because it is unclear how to manage these patients, cardiac medications have not routinely been given in previous studies.
POISE provided some evidence that patients with postoperative MI who were given aspirin and a statin did better.1 And the results of a smaller study10 suggested that intensification of drug therapy (aspirin, statin, beta-blocker, angiotensin-converting enzyme inhibitor) in patients with postoperative troponin I elevations was associated with improved outcomes at 1 year. If the bleeding risk is low, I believe that there is potential benefit in prescribing aspirin and statins for these patients.
CASTING A WIDER NET
Further complicating matters in the near future is the issue of using fifth-generation high-sensitivity troponin T assays. The European Society of Cardiology guidelines11 are somewhat more liberal than the ACC/AHA guidelines, stating that measuring high-sensitivity troponin after surgery “may be considered in high-risk patients to improve risk stratification.” This is a class IIB recommendation, level of evidence B.
With fifth-generation high-sensitivity troponin assays, troponin may be elevated in as many as 20% of patients preoperatively and 40% postoperatively, significantly increasing the number of patients said to have a complication. Besides potentially subjecting these patients to unproven treatments, such results would give the false impression that hospitals and surgeons using the screening tools actually had higher complication rates than those that did not screen.
POSSIBLE HARMS OF SCREENING
Elevated postoperative troponin may identify patients at higher risk of any adverse event but not specifically of cardiac-specific events. In an editorial, Beckman12 stated that routine measurement of troponin “is more likely to cause harm than to provide benefit and should not be used as a screening modality” because of the lack of a proven beneficial treatment strategy, because of the possible harm from applying the standard treatment for type 1 MI, and because it could divert attention from a true cause of an adverse event to a false one—ie, from a nonvascular condition to MI.11
There is clearly a need for clinical trials to determine which treatment, if any, can improve outcomes in these patients, and several trials have been started. But until we have evidence, we can only speculate as to whether screening postoperative patients for troponin elevation is beneficial or detrimental.
- Devereaux PJ, Xavier D, Pogue J, et al. Characteristics and short-term prognosis of perioperative myocardial infarction in patients undergoing noncardiac surgery: a cohort study. Ann Intern Med 2011; 154:523–528.
- Levy M, Heels-Ansdell D, Hiralal R, et al. Prognostic value of troponin and creatine kinase muscle and brain isoenzyme measurement after noncardiac surgery: a systematic review and meta-analysis. Anesthesiology 2011; 114:796–806.
- Botto F, Alonso-Coello P, Chan MT, et al. Myocardial injury after noncardiac surgery: a large, international, prospective cohort study establishing diagnostic criteria, characteristics, predictors, and 30-day outcomes. Anesthesiology 2014; 120:564–578.
- Horr S, Reed G, Menon V. Troponin elevation after noncardiac surgery: significance and management. Cleve Clin J Med 2015; 82:595–602.
- Vascular Events In Noncardiac Surgery Patients Cohort Evaluation Study I, Devereaux PJ, Chan MT, Alonso-Coello P, et al. Association between postoperative troponin levels and 30-day mortality among patients undergoing noncardiac surgery. JAMA 2012; 307:2295–2304.
- Beattie WS, Karkouti K, Tait G, et al. Use of clinically based troponin underestimates the cardiac injury in non-cardiac surgery: a single-centre cohort study in 51,701 consecutive patients. Can J Anaesth 2012; 59:1013–1022.
- van Waes JA, Nathoe HM, de Graaff JC, et al. Myocardial injury after noncardiac surgery and its association with short-term mortality. Circulation 2013; 127:2264–2271.
- Rodseth R, Devereaux PJ. Should we measure troponin routinely in patients after vascular surgery? American College of Cardiology. www.acc.org/latest-in-cardiology/articles/2014/07/18/14/46/should-we-measure-troponin-routinely-in-patients-after-vascular-surgery?w_nav=LC. Accessed August 5, 2015.
- Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; 64:e77–e137.
- Foucrier A, Rodseth R, Aissaoui M, et al. The long-term impact of early cardiovascular therapy intensification for postoperative troponin elevation after major vascular surgery. Anesth Analg 2014; 119:1053–1063.
- Kristensen SD, Knuuti J, Saraste A, et al. 2014 ESC/ESA Guidelines on non-cardiac surgery: cardiovascular assessment and management: The Joint Task Force on non-cardiac surgery: cardiovascular assessment and management of the European Society of Cardiology (ESC) and the European Society of Anaesthesiology (ESA). Eur Heart J 2014; 35:2383–2431.
- Beckman JA. Postoperative troponin screening: a cardiac Cassandra? Circulation 2013; 127:2253–2266.
- Devereaux PJ, Xavier D, Pogue J, et al. Characteristics and short-term prognosis of perioperative myocardial infarction in patients undergoing noncardiac surgery: a cohort study. Ann Intern Med 2011; 154:523–528.
- Levy M, Heels-Ansdell D, Hiralal R, et al. Prognostic value of troponin and creatine kinase muscle and brain isoenzyme measurement after noncardiac surgery: a systematic review and meta-analysis. Anesthesiology 2011; 114:796–806.
- Botto F, Alonso-Coello P, Chan MT, et al. Myocardial injury after noncardiac surgery: a large, international, prospective cohort study establishing diagnostic criteria, characteristics, predictors, and 30-day outcomes. Anesthesiology 2014; 120:564–578.
- Horr S, Reed G, Menon V. Troponin elevation after noncardiac surgery: significance and management. Cleve Clin J Med 2015; 82:595–602.
- Vascular Events In Noncardiac Surgery Patients Cohort Evaluation Study I, Devereaux PJ, Chan MT, Alonso-Coello P, et al. Association between postoperative troponin levels and 30-day mortality among patients undergoing noncardiac surgery. JAMA 2012; 307:2295–2304.
- Beattie WS, Karkouti K, Tait G, et al. Use of clinically based troponin underestimates the cardiac injury in non-cardiac surgery: a single-centre cohort study in 51,701 consecutive patients. Can J Anaesth 2012; 59:1013–1022.
- van Waes JA, Nathoe HM, de Graaff JC, et al. Myocardial injury after noncardiac surgery and its association with short-term mortality. Circulation 2013; 127:2264–2271.
- Rodseth R, Devereaux PJ. Should we measure troponin routinely in patients after vascular surgery? American College of Cardiology. www.acc.org/latest-in-cardiology/articles/2014/07/18/14/46/should-we-measure-troponin-routinely-in-patients-after-vascular-surgery?w_nav=LC. Accessed August 5, 2015.
- Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; 64:e77–e137.
- Foucrier A, Rodseth R, Aissaoui M, et al. The long-term impact of early cardiovascular therapy intensification for postoperative troponin elevation after major vascular surgery. Anesth Analg 2014; 119:1053–1063.
- Kristensen SD, Knuuti J, Saraste A, et al. 2014 ESC/ESA Guidelines on non-cardiac surgery: cardiovascular assessment and management: The Joint Task Force on non-cardiac surgery: cardiovascular assessment and management of the European Society of Cardiology (ESC) and the European Society of Anaesthesiology (ESA). Eur Heart J 2014; 35:2383–2431.
- Beckman JA. Postoperative troponin screening: a cardiac Cassandra? Circulation 2013; 127:2253–2266.

Troponin elevation after noncardiac surgery: Significance and management
More than 200 million patients undergo noncardiac surgery each year, and the volume is increasing.1 Cardiovascular complications are a major cause of morbidity and mortality in the perioperative period.
Before the advent of modern cardiac biomarkers, an estimated 2% to 3% of all patients undergoing noncardiac surgery had a major adverse cardiac event.2 However, more recent studies suggest that 5% to 25% of patients have troponin elevations after noncardiac surgery, depending on the patient population,3–6 and many are asymptomatic, suggesting that many patients are sustaining undetected myocardial injury. Those who suffer a myocardial infarction or myocardial injury have elevated morbidity and mortality rates, not only perioperatively, but also at 30 days and even at up to 1 year.3–5,7–11
Yet there are almost no data on how best to manage these patients; the available guidelines, therefore, do not provide sufficient recommendations for clinical practice.
To address the lack of guidelines, we examine the incidence and proposed mechanisms of myocardial injury after noncardiac surgery, suggest an approach to identifying patients at risk, recommend treatment strategies, and consider future directions.
CARDIAC BIOMARKERS
When cardiac cellular injury from ischemia, direct trauma, or other cause disrupts the cell membrane, intracellular contents enter the extracellular space, including the blood stream. If the myocyte damage is extensive enough, biochemical assays can detect these substances.

Troponin, creatine kinase, myoglobin, and lactate dehydrogenase are common biomarkers of necrosis that, when detected in the plasma, may indicate cardiac injury. Each can be detected at varying times after cardiac injury (Figure 1).12
Cardiac troponins I and T
Of the biomarkers, cardiac troponin I and cardiac troponin T are now the most widely used and are the most specific for myocyte injury.
Troponins are proteins that regulate the calcium-induced interaction between myosin and actin that results in muscle contraction. Troponin is a complex consisting of three subunits: troponin C, troponin I, and troponin T. The cardiac troponin I and T isoforms are distinct from those found in skeletal muscle, making them specific for myocyte injury, and they are currently the recommended markers for diagnosing acute myocardial infarction.13
The troponin immunoassays currently available are not standardized among laboratories and point-of-care methods, and thus, levels cannot be compared across testing centers.14 Each assay has unique performance characteristics, but guidelines recommend using the 99th percentile value from a normal reference population for a given assay to define whether myocardial injury is present.13
Troponin elevation has prognostic value in patients presenting with acute coronary syndromes,15–18 and the degree of elevation correlates with infarct size.19–21
Controversy exists as to whether troponin and other biomarkers are released only after myocardial necrosis or after reversible injury as well. Using newer, highly sensitive assays, troponin elevations have been detected after short periods of ischemia during stress testing22,23 and in patients with stable angina,24 suggesting that reversible cardiac stress and injury can lead to troponin release. This mechanism may play an important role during the myocardial injury that can occur in patients undergoing noncardiac surgery.
MYOCARDIAL INFARCTION vs MYOCARDIAL INJURY
In 2000, the Joint Task Force of the European Society of Cardiology, American College of Cardiology Foundation, American Heart Association, and World Heart Federation revised the criteria for the diagnosis of myocardial infarction created by the World Health Organization in 1979. The definition was revised again in 2007 and once more in 2012 to create the third universal definition of myocardial infarction.
Acute myocardial infarction
Acute myocardial infarction is defined as evidence of myocardial necrosis in a setting of myocardial ischemia, not related to causes such as trauma or pulmonary embolism, with a rise or a fall (or a rise and a fall) of cardiac biomarkers (at least one value being above the 99th percentile in the reference population) and any of the following:
- Symptoms of ischemia
- New ST-segment changes or new left bundle branch block
- Pathologic Q waves
- Imaging evidence of new loss of viable myocardium or new regional wall-motion abnormality
- Intracoronary thrombus by angiography or autopsy.13
Myocardial injury after noncardiac surgery
Studies10,11 have shown that many patients undergoing noncardiac surgery have evidence of cardiac biomarker release but do not meet the universal definition of myocardial infarction.
The Perioperative Ischemic Evaluation (POISE) trial10 reported that 415 (5%) of its patients met the definition of myocardial infarction, of whom only about 35% had symptoms of ischemia. Another 697 patients (8.3%) had isolated elevations in biomarkers without meeting the definition of myocardial infarction.
The VISION study11 (Vascular Events in Noncardiac Surgery Patients Cohort Evaluation) prospectively screened more than 15,000 patients in several countries for troponin elevation during the first 3 postoperative days and for ischemic symptoms and features. Of the patients screened, approximately 1,200 (8%) had troponin elevations, with fewer than half fulfilling the criteria for myocardial infarction.
In another study, van Waes et al6 prospectively screened 2,232 patients ages 60 and older undergoing intermediate- to high-risk noncardiac surgery. Troponin levels were elevated in 19% of the patients, but only 10 of these patients met the universal definition of myocardial infarction.
In all of these studies, patients with isolated elevation in myocardial biomarkers had worse short-term and long-term outcomes than those without. These observations led to a proposed definition of “myocardial injury after noncardiac surgery” that is broader than that of myocardial infarction and requires only elevation of cardiac biomarkers judged to be due to myocardial ischemia (ie, not from another obvious cause such as pulmonary embolism or myocarditis).3
FIVE TYPES OF MYOCARDIAL INFARCTION
The Joint Task Force13 categorizes myocardial infarction into five distinct types:
- Type 1—due to plaque rupture
- Type 2—due to imbalance between oxygen supply and demand
- Type 3—sudden cardiac death
- Type 4a—associated with percutaneous coronary intervention
- Type 4b—associated with stent thrombosis
- Type 5—associated with coronary artery bypass surgery.
Types 1 and 2 have both been implicated in perioperative myocardial infarction and injury. Patient characteristics and the physiologic response to surgical and anesthetic stressors likely contribute to the development of myocardial infarction and injury after noncardiac surgery.
Plaque rupture as a cause of postoperative myocardial infarction
The mechanism of type 1 myocardial infarction—plaque rupture or erosion leading to thrombosis and infarction—plays a significant role in most cases of acute coronary syndromes. Its role in perioperative and postoperative myocardial infarction or injury, however, is less clear.
In an autopsy study of 26 patients who died of myocardial infarction after noncardiac surgery, plaque rupture was evident in 12 (46%).25 A prospective angiographic study of 120 patients with acute coronary syndromes after noncardiac surgery found that nearly 50% had evidence of plaque rupture.26
Higher levels of catecholamines, cortisol,27,28 platelet reactivity,29 procoagulant factors,30 and coronary artery shear stress31 are all present in the postoperative period and may contribute to an increased propensity for plaque rupture or erosion. Whether plaque rupture is present in patients who have isolated troponin elevation but do not meet the criteria for myocardial infarction has not been investigated.
Oxygen supply-demand imbalance during and after surgery
Oxygen supply-demand imbalance (the mechanism in type 2 myocardial infarction) leading to myocyte stress, ischemia, and subsequent infarction is likely common in the perioperative and postoperative periods. As previously discussed, this imbalance may be present with or without symptoms.
Oxygen demand may increase in this period as a result of tachycardia32 caused by bleeding, pain, and catecholamines or increased wall stress from hypertension due to vasoconstriction or pain.33 Oxygen supply can be decreased secondary to tachycardia, anemia,34 hypotension, hypoxemia, hypercarbia, intravascular fluid shifts (bleeding or volume overload), or coronary vasoconstriction.33,35
These mechanisms of myocardial injury, infarction, or both can occur with or without underlying significant obstructive coronary artery disease. However, severe coronary artery disease is more common in those who have had a perioperative myocardial infarction.36
POSTOPERATIVE TROPONIN ELEVATION CARRIES A WORSE PROGNOSIS
Patients who suffer a myocardial infarction after noncardiac surgery have worse short- and long-term outcomes than their counterparts.4,5,7, 8,10,33 In the POISE trial,10 the 30-day mortality rate was 11.6% in those who had had a perioperative myocardial infarction, compared with 2.2% in those who did not (P < .001). The patients who had had a myocardial infarction were also more likely to have nonfatal cardiac arrest, coronary revascularization, and congestive heart failure.
Myocardial injury not fulfilling the criteria for myocardial infarction after noncardiac surgery is also associated with worse short-term and long-term outcomes.3,6,10,11,37,38 POISE patients with isolated elevations in cardiac biomarkers had a higher 30-day risk of coronary revascularization and nonfatal arrest.10 In the VISION trial, an elevation in troponin was the strongest predictor of death within 30 days after noncardiac surgery. This analysis also showed that the higher the peak troponin value, the greater the risk of death and the shorter the median time until death.11
A meta-analysis of 14 studies in 3,139 patients found that elevated troponin after noncardiac surgery was an independent predictor of death within 1 year (odds ratio [OR] 6.7, 95% confidence interval [CI] 4.1–10.9) and beyond 1 year (OR 1.8, 95% CI 1.4–2.3).37
SHOULD SCREENING BE ROUTINE AFTER NONCARDIAC SURGERY?
Since patients suffering myocardial infarction or injury after noncardiac surgery have a worse prognosis, many experts advocate routinely screening all high-risk patients and those undergoing moderate- to high-risk surgery. Many tools exist to determine which patients undergoing noncardiac surgery are at high risk of cardiac complications.
The revised Goldman Cardiac Risk Index is commonly used and well validated. Variables in this index that predict major cardiac complications are:
- High-risk surgery (vascular surgery, orthopedic surgery, and intraperitoneal or intrathoracic surgery)
- History of ischemic heart disease
- History of congestive heart failure
- History of cerebrovascular disease
- Diabetes requiring insulin therapy
- Chronic kidney disease with a creatinine > 2.0 mg/dL.
The more of these variables that are present, the higher the risk of perioperative cardiac events2,4:
- No risk factors: 0.4% risk (95% CI 0.1–0.8)
- One risk factor: 1.0% risk (95% CI 0.5–1.4)
- Two risk factors: 2.4% risk (95% CI 1.3–3.5)
- Three or more risk factors: 5.4% risk (95% CI 2.7–7.9).
Current guidelines from the American College of Cardiology and the American Heart Association give a class I recommendation (the highest) for measuring troponin levels after noncardiac surgery in patients who have symptoms or signs suggesting myocardial ischemia. They give a class IIb recommendation (usefulness is less well established) for screening those at high risk but without symptoms or signs of ischemia, despite the previously cited evidence that patients with troponin elevation are at increased risk. The IIb recommendation is due to a lack of validated treatment strategies to modify and attenuate the recognized risk with troponin elevation in this setting.39
LITTLE EVIDENCE TO GUIDE TREATMENT
In current practice, internists and cardiologists are often asked to consult on patients with troponin elevations noted after noncardiac surgery. Although published and ongoing studies examine strategies to prevent cardiovascular events during noncardiac surgery, we lack data on managing the cases of myocardial infarction and injury that actually occur after noncardiac surgery.
When managing a patient who has a troponin elevation after surgery, many clinical factors must be weighed, including hemodynamic and clinical stability and risk of bleeding. Confronted with ST-segment elevation myocardial infarction or high-risk non–ST-segment elevation myocardial infarction, most clinicians would favor an early invasive reperfusion strategy in accordance with guidelines on managing acute coronary syndrome. Fibrinolytic drugs for ST-segment elevation myocardial infarction are likely to be contraindicated in the postoperative period because they pose an unacceptable risk of bleeding.
Guideline-directed medical therapies for those suffering perioperative myocardial infarction may lower the risk of future cardiovascular events, as suggested by a retrospective study of 66 patients diagnosed with perioperative myocardial infarction after vascular surgery.40 Those in whom medical therapy for coronary artery disease was not intensified—defined as adding or increasing the dose of antiplatelet agent, statin, beta-blocker, or angiotensin-converting enzyme inhibitor—had higher rates of cardiovascular events at 12 months (hazard ratio [HR] 2.80, 95% CI 1.05–24.2).40
In those with asymptomatic myocardial infarction or isolated elevation in cardiac biomarkers, no treatment strategies have been assessed prospectively or in randomized trials. However, statins and aspirin have been suggested as providing some benefit. In a substudy of the POISE trial, the use of aspirin was associated with a 46% reduction in the 30-day mortality rate in those suffering a perioperative myocardial infarction, and statins were associated with a 76% reduction.10 In a single-center retrospective analysis of 337 patients undergoing moderate- to high-risk vascular surgery, statin therapy was associated with a lower 1-year mortality rate (OR 0.63, 95% CI 0.40–0.98).38
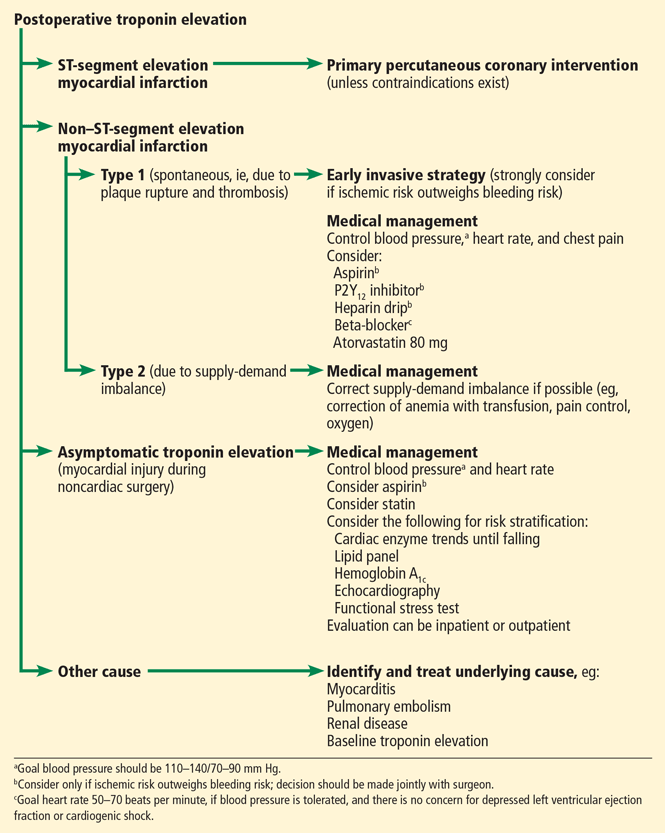
We propose a treatment algorithm for patients identified as having cardiovascular events after noncardiac surgery (Figure 2), based on current evidence and guidelines. Ultimately, treatment decisions should be tailored to the individual patient. Discussion of the risks and benefits of therapeutic options should include the patient and surgeon.
Ongoing and future trials
Ongoing and future trials are aimed at addressing definitive treatment strategies in this patient population.
The MANAGE trial (Management of Myocardial Injury After Non-cardiac Surgery Trial) is randomizing patients suffering myocardial injury after noncardiac surgery to receive either dabigatran and omeprazole or placebo to assess the efficacy of these agents in preventing major adverse cardiac events and the safety of anticoagulation (ClinicalTrials.gov Identifier: NCT01661101).
The INTREPID trial (Study of Ticagrelor Versus Aspirin Treatment in Patients With Myocardial Injury Post Major Non-Cardiac Surgery) will assess the efficacy and safety of ticagrelor treatment compared with aspirin in a similar population (ClinicalTrial.gov Identifier: NCT02291419). The trial will enroll approximately 1,000 patients identified as having a postoperative troponin elevation more than two times the upper limit of normal of the assay during the index hospitalization (Figure 3). Enrollment was to have begun in mid-2015.
- Weiser TG, Regenbogen SE, Thompson KD, et al. An estimation of the global volume of surgery: a modelling strategy based on available data. Lancet 2008; 372:139–144.
- Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation 1999; 100:1043–1049.
- Botto F, Alonso-Coello P, Chan MT, et al. Myocardial injury after noncardiac surgery: a large, international, prospective cohort study establishing diagnostic criteria, characteristics, predictors, and 30-day outcomes. Anesthesiology 2014; 120:564–578.
- Devereaux PJ, Goldman L, Cook DJ, Gilbert K, Leslie K, Guyatt GH. Perioperative cardiac events in patients undergoing noncardiac surgery: a review of the magnitude of the problem, the pathophysiology of the events and methods to estimate and communicate risk. CMAJ 2005; 173:627–634.
- McFalls EO, Ward HB, Moritz TE, et al. Predictors and outcomes of a perioperative myocardial infarction following elective vascular surgery in patients with documented coronary artery disease: results of the CARP trial. Eur Heart J 2008; 29:394–401.
- van Waes JA, Nathoe HM, de Graaff JC, et al. Myocardial injury after noncardiac surgery and its association with short-term mortality. Circulation 2013; 127:2264–2271.
- Badner NH, Knill RL, Brown JE, Novick TV, Gelb AW. Myocardial infarction after noncardiac surgery. Anesthesiology 1998; 88:572–578.
- Kim LJ, Martinez EA, Faraday N, et al. Cardiac troponin I predicts short-term mortality in vascular surgery patients. Circulation 2002; 106:2366–2371.
- Landesberg G, Shatz V, Akopnik I, et al. Association of cardiac troponin, CK-MB, and postoperative myocardial ischemia with long-term survival after major vascular surgery. J Am Coll Cardiol 2003; 42:1547–1554.
- Devereaux PJ, Xavier D, Pogue J, et al. Characteristics and short-term prognosis of perioperative myocardial infarction in patients undergoing noncardiac surgery: a cohort study. Ann Intern Med 2011; 154:523–528.
- Devereaux PJ, Chan MT, Alonso-Coello P, et al. Association between postoperative troponin levels and 30-day mortality among patients undergoing noncardiac surgery. JAMA 2012; 307:2295–2304.
- Kumar A, Cannon CP. Acute coronary syndromes: diagnosis and management, part I. Mayo Clin Proc 2009; 84:917–938.
- Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Circulation 2012; 126:2020–2035.
- Apple FS, Quist HE, Doyle PJ, Otto AP, Murakami MM. Plasma 99th percentile reference limits for cardiac troponin and creatine kinase MB mass for use with European Society of Cardiology/American College of Cardiology consensus recommendations. Clin Chem 2003; 49:1331–1336.
- Ottani F, Galvani M, Nicolini FA, et al. Elevated cardiac troponin levels predict the risk of adverse outcome in patients with acute coronary syndromes. Am Heart J 2000; 140:917–927.
- Ohman EM, Armstrong PW, White HD, et al. Risk stratification with a point-of-care cardiac troponin T test in acute myocardial infarction. GUSTO III investigators. Global Use of Strategies to Open Occluded Coronary Arteries. Am J Cardiol 1999; 84:1281–1286.
- deFilippi CR, Tocchi M, Parmar RJ, et al. Cardiac troponin T in chest pain unit patients without ischemic electrocardiographic changes: angiographic correlates and long-term clinical outcomes. J Am Coll Cardiol 2000; 35:1827–1834.
- Heidenreich PA, Alloggiamento T, Melsop K, McDonald KM, Go AS, Hlatky MA. The prognostic value of troponin in patients with non-ST elevation acute coronary syndromes: a meta-analysis. J Am Coll Cardiol 2001; 38:478–485.
- Steen H, Giannitsis E, Futterer S, Merten C, Juenger C, Katus HA. Cardiac troponin T at 96 hours after acute myocardial infarction correlates with infarct size and cardiac function. J Am Coll Cardiol 2006; 48:2192–2194.
- Licka M, Zimmermann R, Zehelein J, Dengler TJ, Katus HA, Kubler W. Troponin T concentrations 72 hours after myocardial infarction as a serological estimate of infarct size. Heart 2002; 87:520–524.
- Vasile VC, Babuin L, Giannitsis E, Katus HA, Jaffe AS. Relationship of MRI-determined infarct size and cTnI measurements in patients with ST-elevation myocardial infarction. Clin Chem 2008; 54:617–619.
- Sabatine MS, Morrow DA, de Lemos JA, Jarolim P, Braunwald E. Detection of acute changes in circulating troponin in the setting of transient stress test-induced myocardial ischaemia using an ultrasensitive assay: results from TIMI 35. Eur Heart J 2009; 30:162–169.
- Siriwardena M, Campbell V, Richards AM, Pemberton CJ. Cardiac biomarker responses to dobutamine stress echocardiography in healthy volunteers and patients with coronary artery disease. Clin Chem 2012; 58:1492–1494.
- Turer AT, Addo TA, Martin JL, et al. Myocardial ischemia induced by rapid atrial pacing causes troponin T release detectable by a highly sensitive assay: insights from a coronary sinus sampling study. J Am Coll Cardiol 2011; 57:2398–2405.
- Cohen MC, Aretz TH. Histological analysis of coronary artery lesions in fatal postoperative myocardial infarction. Cardiovasc Pathol 1999; 8:133–139.
- Gualandro DM, Campos CA, Calderaro D, et al. Coronary plaque rupture in patients with myocardial infarction after noncardiac surgery: frequent and dangerous. Atherosclerosis 2012; 222:191–195.
- Sametz W, Metzler H, Gries M, et al. Perioperative catecholamine changes in cardiac risk patients. Eur J Clin Invest 1999; 29:582–587.
- Frank SM, Higgins MS, Breslow MJ, et al. The catecholamine, cortisol, and hemodynamic responses to mild perioperative hypothermia. A randomized clinical trial. Anesthesiology 1995; 82:83–93.
- Rosenfeld BA, Faraday N, Campbell D, et al. Perioperative platelet reactivity and the effects of clonidine. Anesthesiology 1993; 79:255–261.
- Lison S, Weiss G, Spannagl M, Heindl B. Postoperative changes in procoagulant factors after major surgery. Blood Coagul Fibrinolysis 2011; 22:190–196.
- Fukumoto Y, Hiro T, Fujii T, et al. Localized elevation of shear stress is related to coronary plaque rupture: a 3-dimensional intravascular ultrasound study with in-vivo color mapping of shear stress distribution. J Am Coll Cardiol 2008; 51:645–650.
- Feringa HH, Bax JJ, Boersma E, et al. High-dose beta-blockers and tight heart rate control reduce myocardial ischemia and troponin T release in vascular surgery patients. Circulation 2006; 114:I-344–I-349.
- Landesberg G. The pathophysiology of perioperative myocardial infarction: facts and perspectives. J Cardiothorac Vasc Anesth 2003; 17:90–100.
- Nelson AH, Fleisher LA, Rosenbaum SH. Relationship between postoperative anemia and cardiac morbidity in high-risk vascular patients in the intensive care unit. Crit Care Med 1993; 21:860–866.
- Landesberg G, Beattie WS, Mosseri M, Jaffe AS, Alpert JS. Perioperative myocardial infarction. Circulation 2009; 119:2936–2944.
- Ellis SG, Hertzer NR, Young JR, Brener S. Angiographic correlates of cardiac death and myocardial infarction complicating major nonthoracic vascular surgery. Am J Cardiol 1996; 77:1126–1128.
- Levy M, Heels-Ansdell D, Hiralal R, et al. Prognostic value of troponin and creatine kinase muscle and brain isoenzyme measurement after noncardiac surgery: a systematic review and meta-analysis. Anesthesiology 2011; 114:796–806.
- Garcia S, Marston N, Sandoval Y, et al. Prognostic value of 12-lead electrocardiogram and peak troponin I level after vascular surgery. J Vasc Surg 2013; 57:166–172.
- Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association task force on practice guidelines. J Am Coll Cardiol 2014; 64:e77–e137.
- Foucrier A, Rodseth R, Aissaoui M, et al. The long-term impact of early cardiovascular therapy intensification for postoperative troponin elevation after major vascular surgery. Anesth Analg 2014; 119:1053–1063.
More than 200 million patients undergo noncardiac surgery each year, and the volume is increasing.1 Cardiovascular complications are a major cause of morbidity and mortality in the perioperative period.
Before the advent of modern cardiac biomarkers, an estimated 2% to 3% of all patients undergoing noncardiac surgery had a major adverse cardiac event.2 However, more recent studies suggest that 5% to 25% of patients have troponin elevations after noncardiac surgery, depending on the patient population,3–6 and many are asymptomatic, suggesting that many patients are sustaining undetected myocardial injury. Those who suffer a myocardial infarction or myocardial injury have elevated morbidity and mortality rates, not only perioperatively, but also at 30 days and even at up to 1 year.3–5,7–11
Yet there are almost no data on how best to manage these patients; the available guidelines, therefore, do not provide sufficient recommendations for clinical practice.
To address the lack of guidelines, we examine the incidence and proposed mechanisms of myocardial injury after noncardiac surgery, suggest an approach to identifying patients at risk, recommend treatment strategies, and consider future directions.
CARDIAC BIOMARKERS
When cardiac cellular injury from ischemia, direct trauma, or other cause disrupts the cell membrane, intracellular contents enter the extracellular space, including the blood stream. If the myocyte damage is extensive enough, biochemical assays can detect these substances.
Troponin, creatine kinase, myoglobin, and lactate dehydrogenase are common biomarkers of necrosis that, when detected in the plasma, may indicate cardiac injury. Each can be detected at varying times after cardiac injury (Figure 1).12
Cardiac troponins I and T
Of the biomarkers, cardiac troponin I and cardiac troponin T are now the most widely used and are the most specific for myocyte injury.
Troponins are proteins that regulate the calcium-induced interaction between myosin and actin that results in muscle contraction. Troponin is a complex consisting of three subunits: troponin C, troponin I, and troponin T. The cardiac troponin I and T isoforms are distinct from those found in skeletal muscle, making them specific for myocyte injury, and they are currently the recommended markers for diagnosing acute myocardial infarction.13
The troponin immunoassays currently available are not standardized among laboratories and point-of-care methods, and thus, levels cannot be compared across testing centers.14 Each assay has unique performance characteristics, but guidelines recommend using the 99th percentile value from a normal reference population for a given assay to define whether myocardial injury is present.13
Troponin elevation has prognostic value in patients presenting with acute coronary syndromes,15–18 and the degree of elevation correlates with infarct size.19–21
Controversy exists as to whether troponin and other biomarkers are released only after myocardial necrosis or after reversible injury as well. Using newer, highly sensitive assays, troponin elevations have been detected after short periods of ischemia during stress testing22,23 and in patients with stable angina,24 suggesting that reversible cardiac stress and injury can lead to troponin release. This mechanism may play an important role during the myocardial injury that can occur in patients undergoing noncardiac surgery.
MYOCARDIAL INFARCTION vs MYOCARDIAL INJURY
In 2000, the Joint Task Force of the European Society of Cardiology, American College of Cardiology Foundation, American Heart Association, and World Heart Federation revised the criteria for the diagnosis of myocardial infarction created by the World Health Organization in 1979. The definition was revised again in 2007 and once more in 2012 to create the third universal definition of myocardial infarction.
Acute myocardial infarction
Acute myocardial infarction is defined as evidence of myocardial necrosis in a setting of myocardial ischemia, not related to causes such as trauma or pulmonary embolism, with a rise or a fall (or a rise and a fall) of cardiac biomarkers (at least one value being above the 99th percentile in the reference population) and any of the following:
- Symptoms of ischemia
- New ST-segment changes or new left bundle branch block
- Pathologic Q waves
- Imaging evidence of new loss of viable myocardium or new regional wall-motion abnormality
- Intracoronary thrombus by angiography or autopsy.13
Myocardial injury after noncardiac surgery
Studies10,11 have shown that many patients undergoing noncardiac surgery have evidence of cardiac biomarker release but do not meet the universal definition of myocardial infarction.
The Perioperative Ischemic Evaluation (POISE) trial10 reported that 415 (5%) of its patients met the definition of myocardial infarction, of whom only about 35% had symptoms of ischemia. Another 697 patients (8.3%) had isolated elevations in biomarkers without meeting the definition of myocardial infarction.
The VISION study11 (Vascular Events in Noncardiac Surgery Patients Cohort Evaluation) prospectively screened more than 15,000 patients in several countries for troponin elevation during the first 3 postoperative days and for ischemic symptoms and features. Of the patients screened, approximately 1,200 (8%) had troponin elevations, with fewer than half fulfilling the criteria for myocardial infarction.
In another study, van Waes et al6 prospectively screened 2,232 patients ages 60 and older undergoing intermediate- to high-risk noncardiac surgery. Troponin levels were elevated in 19% of the patients, but only 10 of these patients met the universal definition of myocardial infarction.
In all of these studies, patients with isolated elevation in myocardial biomarkers had worse short-term and long-term outcomes than those without. These observations led to a proposed definition of “myocardial injury after noncardiac surgery” that is broader than that of myocardial infarction and requires only elevation of cardiac biomarkers judged to be due to myocardial ischemia (ie, not from another obvious cause such as pulmonary embolism or myocarditis).3
FIVE TYPES OF MYOCARDIAL INFARCTION
The Joint Task Force13 categorizes myocardial infarction into five distinct types:
- Type 1—due to plaque rupture
- Type 2—due to imbalance between oxygen supply and demand
- Type 3—sudden cardiac death
- Type 4a—associated with percutaneous coronary intervention
- Type 4b—associated with stent thrombosis
- Type 5—associated with coronary artery bypass surgery.
Types 1 and 2 have both been implicated in perioperative myocardial infarction and injury. Patient characteristics and the physiologic response to surgical and anesthetic stressors likely contribute to the development of myocardial infarction and injury after noncardiac surgery.
Plaque rupture as a cause of postoperative myocardial infarction
The mechanism of type 1 myocardial infarction—plaque rupture or erosion leading to thrombosis and infarction—plays a significant role in most cases of acute coronary syndromes. Its role in perioperative and postoperative myocardial infarction or injury, however, is less clear.
In an autopsy study of 26 patients who died of myocardial infarction after noncardiac surgery, plaque rupture was evident in 12 (46%).25 A prospective angiographic study of 120 patients with acute coronary syndromes after noncardiac surgery found that nearly 50% had evidence of plaque rupture.26
Higher levels of catecholamines, cortisol,27,28 platelet reactivity,29 procoagulant factors,30 and coronary artery shear stress31 are all present in the postoperative period and may contribute to an increased propensity for plaque rupture or erosion. Whether plaque rupture is present in patients who have isolated troponin elevation but do not meet the criteria for myocardial infarction has not been investigated.
Oxygen supply-demand imbalance during and after surgery
Oxygen supply-demand imbalance (the mechanism in type 2 myocardial infarction) leading to myocyte stress, ischemia, and subsequent infarction is likely common in the perioperative and postoperative periods. As previously discussed, this imbalance may be present with or without symptoms.
Oxygen demand may increase in this period as a result of tachycardia32 caused by bleeding, pain, and catecholamines or increased wall stress from hypertension due to vasoconstriction or pain.33 Oxygen supply can be decreased secondary to tachycardia, anemia,34 hypotension, hypoxemia, hypercarbia, intravascular fluid shifts (bleeding or volume overload), or coronary vasoconstriction.33,35
These mechanisms of myocardial injury, infarction, or both can occur with or without underlying significant obstructive coronary artery disease. However, severe coronary artery disease is more common in those who have had a perioperative myocardial infarction.36
POSTOPERATIVE TROPONIN ELEVATION CARRIES A WORSE PROGNOSIS
Patients who suffer a myocardial infarction after noncardiac surgery have worse short- and long-term outcomes than their counterparts.4,5,7, 8,10,33 In the POISE trial,10 the 30-day mortality rate was 11.6% in those who had had a perioperative myocardial infarction, compared with 2.2% in those who did not (P < .001). The patients who had had a myocardial infarction were also more likely to have nonfatal cardiac arrest, coronary revascularization, and congestive heart failure.
Myocardial injury not fulfilling the criteria for myocardial infarction after noncardiac surgery is also associated with worse short-term and long-term outcomes.3,6,10,11,37,38 POISE patients with isolated elevations in cardiac biomarkers had a higher 30-day risk of coronary revascularization and nonfatal arrest.10 In the VISION trial, an elevation in troponin was the strongest predictor of death within 30 days after noncardiac surgery. This analysis also showed that the higher the peak troponin value, the greater the risk of death and the shorter the median time until death.11
A meta-analysis of 14 studies in 3,139 patients found that elevated troponin after noncardiac surgery was an independent predictor of death within 1 year (odds ratio [OR] 6.7, 95% confidence interval [CI] 4.1–10.9) and beyond 1 year (OR 1.8, 95% CI 1.4–2.3).37
SHOULD SCREENING BE ROUTINE AFTER NONCARDIAC SURGERY?
Since patients suffering myocardial infarction or injury after noncardiac surgery have a worse prognosis, many experts advocate routinely screening all high-risk patients and those undergoing moderate- to high-risk surgery. Many tools exist to determine which patients undergoing noncardiac surgery are at high risk of cardiac complications.
The revised Goldman Cardiac Risk Index is commonly used and well validated. Variables in this index that predict major cardiac complications are:
- High-risk surgery (vascular surgery, orthopedic surgery, and intraperitoneal or intrathoracic surgery)
- History of ischemic heart disease
- History of congestive heart failure
- History of cerebrovascular disease
- Diabetes requiring insulin therapy
- Chronic kidney disease with a creatinine > 2.0 mg/dL.
The more of these variables that are present, the higher the risk of perioperative cardiac events2,4:
- No risk factors: 0.4% risk (95% CI 0.1–0.8)
- One risk factor: 1.0% risk (95% CI 0.5–1.4)
- Two risk factors: 2.4% risk (95% CI 1.3–3.5)
- Three or more risk factors: 5.4% risk (95% CI 2.7–7.9).
Current guidelines from the American College of Cardiology and the American Heart Association give a class I recommendation (the highest) for measuring troponin levels after noncardiac surgery in patients who have symptoms or signs suggesting myocardial ischemia. They give a class IIb recommendation (usefulness is less well established) for screening those at high risk but without symptoms or signs of ischemia, despite the previously cited evidence that patients with troponin elevation are at increased risk. The IIb recommendation is due to a lack of validated treatment strategies to modify and attenuate the recognized risk with troponin elevation in this setting.39
LITTLE EVIDENCE TO GUIDE TREATMENT
In current practice, internists and cardiologists are often asked to consult on patients with troponin elevations noted after noncardiac surgery. Although published and ongoing studies examine strategies to prevent cardiovascular events during noncardiac surgery, we lack data on managing the cases of myocardial infarction and injury that actually occur after noncardiac surgery.
When managing a patient who has a troponin elevation after surgery, many clinical factors must be weighed, including hemodynamic and clinical stability and risk of bleeding. Confronted with ST-segment elevation myocardial infarction or high-risk non–ST-segment elevation myocardial infarction, most clinicians would favor an early invasive reperfusion strategy in accordance with guidelines on managing acute coronary syndrome. Fibrinolytic drugs for ST-segment elevation myocardial infarction are likely to be contraindicated in the postoperative period because they pose an unacceptable risk of bleeding.
Guideline-directed medical therapies for those suffering perioperative myocardial infarction may lower the risk of future cardiovascular events, as suggested by a retrospective study of 66 patients diagnosed with perioperative myocardial infarction after vascular surgery.40 Those in whom medical therapy for coronary artery disease was not intensified—defined as adding or increasing the dose of antiplatelet agent, statin, beta-blocker, or angiotensin-converting enzyme inhibitor—had higher rates of cardiovascular events at 12 months (hazard ratio [HR] 2.80, 95% CI 1.05–24.2).40
In those with asymptomatic myocardial infarction or isolated elevation in cardiac biomarkers, no treatment strategies have been assessed prospectively or in randomized trials. However, statins and aspirin have been suggested as providing some benefit. In a substudy of the POISE trial, the use of aspirin was associated with a 46% reduction in the 30-day mortality rate in those suffering a perioperative myocardial infarction, and statins were associated with a 76% reduction.10 In a single-center retrospective analysis of 337 patients undergoing moderate- to high-risk vascular surgery, statin therapy was associated with a lower 1-year mortality rate (OR 0.63, 95% CI 0.40–0.98).38
We propose a treatment algorithm for patients identified as having cardiovascular events after noncardiac surgery (Figure 2), based on current evidence and guidelines. Ultimately, treatment decisions should be tailored to the individual patient. Discussion of the risks and benefits of therapeutic options should include the patient and surgeon.
Ongoing and future trials
Ongoing and future trials are aimed at addressing definitive treatment strategies in this patient population.
The MANAGE trial (Management of Myocardial Injury After Non-cardiac Surgery Trial) is randomizing patients suffering myocardial injury after noncardiac surgery to receive either dabigatran and omeprazole or placebo to assess the efficacy of these agents in preventing major adverse cardiac events and the safety of anticoagulation (ClinicalTrials.gov Identifier: NCT01661101).
The INTREPID trial (Study of Ticagrelor Versus Aspirin Treatment in Patients With Myocardial Injury Post Major Non-Cardiac Surgery) will assess the efficacy and safety of ticagrelor treatment compared with aspirin in a similar population (ClinicalTrial.gov Identifier: NCT02291419). The trial will enroll approximately 1,000 patients identified as having a postoperative troponin elevation more than two times the upper limit of normal of the assay during the index hospitalization (Figure 3). Enrollment was to have begun in mid-2015.
More than 200 million patients undergo noncardiac surgery each year, and the volume is increasing.1 Cardiovascular complications are a major cause of morbidity and mortality in the perioperative period.
Before the advent of modern cardiac biomarkers, an estimated 2% to 3% of all patients undergoing noncardiac surgery had a major adverse cardiac event.2 However, more recent studies suggest that 5% to 25% of patients have troponin elevations after noncardiac surgery, depending on the patient population,3–6 and many are asymptomatic, suggesting that many patients are sustaining undetected myocardial injury. Those who suffer a myocardial infarction or myocardial injury have elevated morbidity and mortality rates, not only perioperatively, but also at 30 days and even at up to 1 year.3–5,7–11
Yet there are almost no data on how best to manage these patients; the available guidelines, therefore, do not provide sufficient recommendations for clinical practice.
To address the lack of guidelines, we examine the incidence and proposed mechanisms of myocardial injury after noncardiac surgery, suggest an approach to identifying patients at risk, recommend treatment strategies, and consider future directions.
CARDIAC BIOMARKERS
When cardiac cellular injury from ischemia, direct trauma, or other cause disrupts the cell membrane, intracellular contents enter the extracellular space, including the blood stream. If the myocyte damage is extensive enough, biochemical assays can detect these substances.
Troponin, creatine kinase, myoglobin, and lactate dehydrogenase are common biomarkers of necrosis that, when detected in the plasma, may indicate cardiac injury. Each can be detected at varying times after cardiac injury (Figure 1).12
Cardiac troponins I and T
Of the biomarkers, cardiac troponin I and cardiac troponin T are now the most widely used and are the most specific for myocyte injury.
Troponins are proteins that regulate the calcium-induced interaction between myosin and actin that results in muscle contraction. Troponin is a complex consisting of three subunits: troponin C, troponin I, and troponin T. The cardiac troponin I and T isoforms are distinct from those found in skeletal muscle, making them specific for myocyte injury, and they are currently the recommended markers for diagnosing acute myocardial infarction.13
The troponin immunoassays currently available are not standardized among laboratories and point-of-care methods, and thus, levels cannot be compared across testing centers.14 Each assay has unique performance characteristics, but guidelines recommend using the 99th percentile value from a normal reference population for a given assay to define whether myocardial injury is present.13
Troponin elevation has prognostic value in patients presenting with acute coronary syndromes,15–18 and the degree of elevation correlates with infarct size.19–21
Controversy exists as to whether troponin and other biomarkers are released only after myocardial necrosis or after reversible injury as well. Using newer, highly sensitive assays, troponin elevations have been detected after short periods of ischemia during stress testing22,23 and in patients with stable angina,24 suggesting that reversible cardiac stress and injury can lead to troponin release. This mechanism may play an important role during the myocardial injury that can occur in patients undergoing noncardiac surgery.
MYOCARDIAL INFARCTION vs MYOCARDIAL INJURY
In 2000, the Joint Task Force of the European Society of Cardiology, American College of Cardiology Foundation, American Heart Association, and World Heart Federation revised the criteria for the diagnosis of myocardial infarction created by the World Health Organization in 1979. The definition was revised again in 2007 and once more in 2012 to create the third universal definition of myocardial infarction.
Acute myocardial infarction
Acute myocardial infarction is defined as evidence of myocardial necrosis in a setting of myocardial ischemia, not related to causes such as trauma or pulmonary embolism, with a rise or a fall (or a rise and a fall) of cardiac biomarkers (at least one value being above the 99th percentile in the reference population) and any of the following:
- Symptoms of ischemia
- New ST-segment changes or new left bundle branch block
- Pathologic Q waves
- Imaging evidence of new loss of viable myocardium or new regional wall-motion abnormality
- Intracoronary thrombus by angiography or autopsy.13
Myocardial injury after noncardiac surgery
Studies10,11 have shown that many patients undergoing noncardiac surgery have evidence of cardiac biomarker release but do not meet the universal definition of myocardial infarction.
The Perioperative Ischemic Evaluation (POISE) trial10 reported that 415 (5%) of its patients met the definition of myocardial infarction, of whom only about 35% had symptoms of ischemia. Another 697 patients (8.3%) had isolated elevations in biomarkers without meeting the definition of myocardial infarction.
The VISION study11 (Vascular Events in Noncardiac Surgery Patients Cohort Evaluation) prospectively screened more than 15,000 patients in several countries for troponin elevation during the first 3 postoperative days and for ischemic symptoms and features. Of the patients screened, approximately 1,200 (8%) had troponin elevations, with fewer than half fulfilling the criteria for myocardial infarction.
In another study, van Waes et al6 prospectively screened 2,232 patients ages 60 and older undergoing intermediate- to high-risk noncardiac surgery. Troponin levels were elevated in 19% of the patients, but only 10 of these patients met the universal definition of myocardial infarction.
In all of these studies, patients with isolated elevation in myocardial biomarkers had worse short-term and long-term outcomes than those without. These observations led to a proposed definition of “myocardial injury after noncardiac surgery” that is broader than that of myocardial infarction and requires only elevation of cardiac biomarkers judged to be due to myocardial ischemia (ie, not from another obvious cause such as pulmonary embolism or myocarditis).3
FIVE TYPES OF MYOCARDIAL INFARCTION
The Joint Task Force13 categorizes myocardial infarction into five distinct types:
- Type 1—due to plaque rupture
- Type 2—due to imbalance between oxygen supply and demand
- Type 3—sudden cardiac death
- Type 4a—associated with percutaneous coronary intervention
- Type 4b—associated with stent thrombosis
- Type 5—associated with coronary artery bypass surgery.
Types 1 and 2 have both been implicated in perioperative myocardial infarction and injury. Patient characteristics and the physiologic response to surgical and anesthetic stressors likely contribute to the development of myocardial infarction and injury after noncardiac surgery.
Plaque rupture as a cause of postoperative myocardial infarction
The mechanism of type 1 myocardial infarction—plaque rupture or erosion leading to thrombosis and infarction—plays a significant role in most cases of acute coronary syndromes. Its role in perioperative and postoperative myocardial infarction or injury, however, is less clear.
In an autopsy study of 26 patients who died of myocardial infarction after noncardiac surgery, plaque rupture was evident in 12 (46%).25 A prospective angiographic study of 120 patients with acute coronary syndromes after noncardiac surgery found that nearly 50% had evidence of plaque rupture.26
Higher levels of catecholamines, cortisol,27,28 platelet reactivity,29 procoagulant factors,30 and coronary artery shear stress31 are all present in the postoperative period and may contribute to an increased propensity for plaque rupture or erosion. Whether plaque rupture is present in patients who have isolated troponin elevation but do not meet the criteria for myocardial infarction has not been investigated.
Oxygen supply-demand imbalance during and after surgery
Oxygen supply-demand imbalance (the mechanism in type 2 myocardial infarction) leading to myocyte stress, ischemia, and subsequent infarction is likely common in the perioperative and postoperative periods. As previously discussed, this imbalance may be present with or without symptoms.
Oxygen demand may increase in this period as a result of tachycardia32 caused by bleeding, pain, and catecholamines or increased wall stress from hypertension due to vasoconstriction or pain.33 Oxygen supply can be decreased secondary to tachycardia, anemia,34 hypotension, hypoxemia, hypercarbia, intravascular fluid shifts (bleeding or volume overload), or coronary vasoconstriction.33,35
These mechanisms of myocardial injury, infarction, or both can occur with or without underlying significant obstructive coronary artery disease. However, severe coronary artery disease is more common in those who have had a perioperative myocardial infarction.36
POSTOPERATIVE TROPONIN ELEVATION CARRIES A WORSE PROGNOSIS
Patients who suffer a myocardial infarction after noncardiac surgery have worse short- and long-term outcomes than their counterparts.4,5,7, 8,10,33 In the POISE trial,10 the 30-day mortality rate was 11.6% in those who had had a perioperative myocardial infarction, compared with 2.2% in those who did not (P < .001). The patients who had had a myocardial infarction were also more likely to have nonfatal cardiac arrest, coronary revascularization, and congestive heart failure.
Myocardial injury not fulfilling the criteria for myocardial infarction after noncardiac surgery is also associated with worse short-term and long-term outcomes.3,6,10,11,37,38 POISE patients with isolated elevations in cardiac biomarkers had a higher 30-day risk of coronary revascularization and nonfatal arrest.10 In the VISION trial, an elevation in troponin was the strongest predictor of death within 30 days after noncardiac surgery. This analysis also showed that the higher the peak troponin value, the greater the risk of death and the shorter the median time until death.11
A meta-analysis of 14 studies in 3,139 patients found that elevated troponin after noncardiac surgery was an independent predictor of death within 1 year (odds ratio [OR] 6.7, 95% confidence interval [CI] 4.1–10.9) and beyond 1 year (OR 1.8, 95% CI 1.4–2.3).37
SHOULD SCREENING BE ROUTINE AFTER NONCARDIAC SURGERY?
Since patients suffering myocardial infarction or injury after noncardiac surgery have a worse prognosis, many experts advocate routinely screening all high-risk patients and those undergoing moderate- to high-risk surgery. Many tools exist to determine which patients undergoing noncardiac surgery are at high risk of cardiac complications.
The revised Goldman Cardiac Risk Index is commonly used and well validated. Variables in this index that predict major cardiac complications are:
- High-risk surgery (vascular surgery, orthopedic surgery, and intraperitoneal or intrathoracic surgery)
- History of ischemic heart disease
- History of congestive heart failure
- History of cerebrovascular disease
- Diabetes requiring insulin therapy
- Chronic kidney disease with a creatinine > 2.0 mg/dL.
The more of these variables that are present, the higher the risk of perioperative cardiac events2,4:
- No risk factors: 0.4% risk (95% CI 0.1–0.8)
- One risk factor: 1.0% risk (95% CI 0.5–1.4)
- Two risk factors: 2.4% risk (95% CI 1.3–3.5)
- Three or more risk factors: 5.4% risk (95% CI 2.7–7.9).
Current guidelines from the American College of Cardiology and the American Heart Association give a class I recommendation (the highest) for measuring troponin levels after noncardiac surgery in patients who have symptoms or signs suggesting myocardial ischemia. They give a class IIb recommendation (usefulness is less well established) for screening those at high risk but without symptoms or signs of ischemia, despite the previously cited evidence that patients with troponin elevation are at increased risk. The IIb recommendation is due to a lack of validated treatment strategies to modify and attenuate the recognized risk with troponin elevation in this setting.39
LITTLE EVIDENCE TO GUIDE TREATMENT
In current practice, internists and cardiologists are often asked to consult on patients with troponin elevations noted after noncardiac surgery. Although published and ongoing studies examine strategies to prevent cardiovascular events during noncardiac surgery, we lack data on managing the cases of myocardial infarction and injury that actually occur after noncardiac surgery.
When managing a patient who has a troponin elevation after surgery, many clinical factors must be weighed, including hemodynamic and clinical stability and risk of bleeding. Confronted with ST-segment elevation myocardial infarction or high-risk non–ST-segment elevation myocardial infarction, most clinicians would favor an early invasive reperfusion strategy in accordance with guidelines on managing acute coronary syndrome. Fibrinolytic drugs for ST-segment elevation myocardial infarction are likely to be contraindicated in the postoperative period because they pose an unacceptable risk of bleeding.
Guideline-directed medical therapies for those suffering perioperative myocardial infarction may lower the risk of future cardiovascular events, as suggested by a retrospective study of 66 patients diagnosed with perioperative myocardial infarction after vascular surgery.40 Those in whom medical therapy for coronary artery disease was not intensified—defined as adding or increasing the dose of antiplatelet agent, statin, beta-blocker, or angiotensin-converting enzyme inhibitor—had higher rates of cardiovascular events at 12 months (hazard ratio [HR] 2.80, 95% CI 1.05–24.2).40
In those with asymptomatic myocardial infarction or isolated elevation in cardiac biomarkers, no treatment strategies have been assessed prospectively or in randomized trials. However, statins and aspirin have been suggested as providing some benefit. In a substudy of the POISE trial, the use of aspirin was associated with a 46% reduction in the 30-day mortality rate in those suffering a perioperative myocardial infarction, and statins were associated with a 76% reduction.10 In a single-center retrospective analysis of 337 patients undergoing moderate- to high-risk vascular surgery, statin therapy was associated with a lower 1-year mortality rate (OR 0.63, 95% CI 0.40–0.98).38
We propose a treatment algorithm for patients identified as having cardiovascular events after noncardiac surgery (Figure 2), based on current evidence and guidelines. Ultimately, treatment decisions should be tailored to the individual patient. Discussion of the risks and benefits of therapeutic options should include the patient and surgeon.
Ongoing and future trials
Ongoing and future trials are aimed at addressing definitive treatment strategies in this patient population.
The MANAGE trial (Management of Myocardial Injury After Non-cardiac Surgery Trial) is randomizing patients suffering myocardial injury after noncardiac surgery to receive either dabigatran and omeprazole or placebo to assess the efficacy of these agents in preventing major adverse cardiac events and the safety of anticoagulation (ClinicalTrials.gov Identifier: NCT01661101).
The INTREPID trial (Study of Ticagrelor Versus Aspirin Treatment in Patients With Myocardial Injury Post Major Non-Cardiac Surgery) will assess the efficacy and safety of ticagrelor treatment compared with aspirin in a similar population (ClinicalTrial.gov Identifier: NCT02291419). The trial will enroll approximately 1,000 patients identified as having a postoperative troponin elevation more than two times the upper limit of normal of the assay during the index hospitalization (Figure 3). Enrollment was to have begun in mid-2015.
- Weiser TG, Regenbogen SE, Thompson KD, et al. An estimation of the global volume of surgery: a modelling strategy based on available data. Lancet 2008; 372:139–144.
- Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation 1999; 100:1043–1049.
- Botto F, Alonso-Coello P, Chan MT, et al. Myocardial injury after noncardiac surgery: a large, international, prospective cohort study establishing diagnostic criteria, characteristics, predictors, and 30-day outcomes. Anesthesiology 2014; 120:564–578.
- Devereaux PJ, Goldman L, Cook DJ, Gilbert K, Leslie K, Guyatt GH. Perioperative cardiac events in patients undergoing noncardiac surgery: a review of the magnitude of the problem, the pathophysiology of the events and methods to estimate and communicate risk. CMAJ 2005; 173:627–634.
- McFalls EO, Ward HB, Moritz TE, et al. Predictors and outcomes of a perioperative myocardial infarction following elective vascular surgery in patients with documented coronary artery disease: results of the CARP trial. Eur Heart J 2008; 29:394–401.
- van Waes JA, Nathoe HM, de Graaff JC, et al. Myocardial injury after noncardiac surgery and its association with short-term mortality. Circulation 2013; 127:2264–2271.
- Badner NH, Knill RL, Brown JE, Novick TV, Gelb AW. Myocardial infarction after noncardiac surgery. Anesthesiology 1998; 88:572–578.
- Kim LJ, Martinez EA, Faraday N, et al. Cardiac troponin I predicts short-term mortality in vascular surgery patients. Circulation 2002; 106:2366–2371.
- Landesberg G, Shatz V, Akopnik I, et al. Association of cardiac troponin, CK-MB, and postoperative myocardial ischemia with long-term survival after major vascular surgery. J Am Coll Cardiol 2003; 42:1547–1554.
- Devereaux PJ, Xavier D, Pogue J, et al. Characteristics and short-term prognosis of perioperative myocardial infarction in patients undergoing noncardiac surgery: a cohort study. Ann Intern Med 2011; 154:523–528.
- Devereaux PJ, Chan MT, Alonso-Coello P, et al. Association between postoperative troponin levels and 30-day mortality among patients undergoing noncardiac surgery. JAMA 2012; 307:2295–2304.
- Kumar A, Cannon CP. Acute coronary syndromes: diagnosis and management, part I. Mayo Clin Proc 2009; 84:917–938.
- Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Circulation 2012; 126:2020–2035.
- Apple FS, Quist HE, Doyle PJ, Otto AP, Murakami MM. Plasma 99th percentile reference limits for cardiac troponin and creatine kinase MB mass for use with European Society of Cardiology/American College of Cardiology consensus recommendations. Clin Chem 2003; 49:1331–1336.
- Ottani F, Galvani M, Nicolini FA, et al. Elevated cardiac troponin levels predict the risk of adverse outcome in patients with acute coronary syndromes. Am Heart J 2000; 140:917–927.
- Ohman EM, Armstrong PW, White HD, et al. Risk stratification with a point-of-care cardiac troponin T test in acute myocardial infarction. GUSTO III investigators. Global Use of Strategies to Open Occluded Coronary Arteries. Am J Cardiol 1999; 84:1281–1286.
- deFilippi CR, Tocchi M, Parmar RJ, et al. Cardiac troponin T in chest pain unit patients without ischemic electrocardiographic changes: angiographic correlates and long-term clinical outcomes. J Am Coll Cardiol 2000; 35:1827–1834.
- Heidenreich PA, Alloggiamento T, Melsop K, McDonald KM, Go AS, Hlatky MA. The prognostic value of troponin in patients with non-ST elevation acute coronary syndromes: a meta-analysis. J Am Coll Cardiol 2001; 38:478–485.
- Steen H, Giannitsis E, Futterer S, Merten C, Juenger C, Katus HA. Cardiac troponin T at 96 hours after acute myocardial infarction correlates with infarct size and cardiac function. J Am Coll Cardiol 2006; 48:2192–2194.
- Licka M, Zimmermann R, Zehelein J, Dengler TJ, Katus HA, Kubler W. Troponin T concentrations 72 hours after myocardial infarction as a serological estimate of infarct size. Heart 2002; 87:520–524.
- Vasile VC, Babuin L, Giannitsis E, Katus HA, Jaffe AS. Relationship of MRI-determined infarct size and cTnI measurements in patients with ST-elevation myocardial infarction. Clin Chem 2008; 54:617–619.
- Sabatine MS, Morrow DA, de Lemos JA, Jarolim P, Braunwald E. Detection of acute changes in circulating troponin in the setting of transient stress test-induced myocardial ischaemia using an ultrasensitive assay: results from TIMI 35. Eur Heart J 2009; 30:162–169.
- Siriwardena M, Campbell V, Richards AM, Pemberton CJ. Cardiac biomarker responses to dobutamine stress echocardiography in healthy volunteers and patients with coronary artery disease. Clin Chem 2012; 58:1492–1494.
- Turer AT, Addo TA, Martin JL, et al. Myocardial ischemia induced by rapid atrial pacing causes troponin T release detectable by a highly sensitive assay: insights from a coronary sinus sampling study. J Am Coll Cardiol 2011; 57:2398–2405.
- Cohen MC, Aretz TH. Histological analysis of coronary artery lesions in fatal postoperative myocardial infarction. Cardiovasc Pathol 1999; 8:133–139.
- Gualandro DM, Campos CA, Calderaro D, et al. Coronary plaque rupture in patients with myocardial infarction after noncardiac surgery: frequent and dangerous. Atherosclerosis 2012; 222:191–195.
- Sametz W, Metzler H, Gries M, et al. Perioperative catecholamine changes in cardiac risk patients. Eur J Clin Invest 1999; 29:582–587.
- Frank SM, Higgins MS, Breslow MJ, et al. The catecholamine, cortisol, and hemodynamic responses to mild perioperative hypothermia. A randomized clinical trial. Anesthesiology 1995; 82:83–93.
- Rosenfeld BA, Faraday N, Campbell D, et al. Perioperative platelet reactivity and the effects of clonidine. Anesthesiology 1993; 79:255–261.
- Lison S, Weiss G, Spannagl M, Heindl B. Postoperative changes in procoagulant factors after major surgery. Blood Coagul Fibrinolysis 2011; 22:190–196.
- Fukumoto Y, Hiro T, Fujii T, et al. Localized elevation of shear stress is related to coronary plaque rupture: a 3-dimensional intravascular ultrasound study with in-vivo color mapping of shear stress distribution. J Am Coll Cardiol 2008; 51:645–650.
- Feringa HH, Bax JJ, Boersma E, et al. High-dose beta-blockers and tight heart rate control reduce myocardial ischemia and troponin T release in vascular surgery patients. Circulation 2006; 114:I-344–I-349.
- Landesberg G. The pathophysiology of perioperative myocardial infarction: facts and perspectives. J Cardiothorac Vasc Anesth 2003; 17:90–100.
- Nelson AH, Fleisher LA, Rosenbaum SH. Relationship between postoperative anemia and cardiac morbidity in high-risk vascular patients in the intensive care unit. Crit Care Med 1993; 21:860–866.
- Landesberg G, Beattie WS, Mosseri M, Jaffe AS, Alpert JS. Perioperative myocardial infarction. Circulation 2009; 119:2936–2944.
- Ellis SG, Hertzer NR, Young JR, Brener S. Angiographic correlates of cardiac death and myocardial infarction complicating major nonthoracic vascular surgery. Am J Cardiol 1996; 77:1126–1128.
- Levy M, Heels-Ansdell D, Hiralal R, et al. Prognostic value of troponin and creatine kinase muscle and brain isoenzyme measurement after noncardiac surgery: a systematic review and meta-analysis. Anesthesiology 2011; 114:796–806.
- Garcia S, Marston N, Sandoval Y, et al. Prognostic value of 12-lead electrocardiogram and peak troponin I level after vascular surgery. J Vasc Surg 2013; 57:166–172.
- Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association task force on practice guidelines. J Am Coll Cardiol 2014; 64:e77–e137.
- Foucrier A, Rodseth R, Aissaoui M, et al. The long-term impact of early cardiovascular therapy intensification for postoperative troponin elevation after major vascular surgery. Anesth Analg 2014; 119:1053–1063.
- Weiser TG, Regenbogen SE, Thompson KD, et al. An estimation of the global volume of surgery: a modelling strategy based on available data. Lancet 2008; 372:139–144.
- Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation 1999; 100:1043–1049.
- Botto F, Alonso-Coello P, Chan MT, et al. Myocardial injury after noncardiac surgery: a large, international, prospective cohort study establishing diagnostic criteria, characteristics, predictors, and 30-day outcomes. Anesthesiology 2014; 120:564–578.
- Devereaux PJ, Goldman L, Cook DJ, Gilbert K, Leslie K, Guyatt GH. Perioperative cardiac events in patients undergoing noncardiac surgery: a review of the magnitude of the problem, the pathophysiology of the events and methods to estimate and communicate risk. CMAJ 2005; 173:627–634.
- McFalls EO, Ward HB, Moritz TE, et al. Predictors and outcomes of a perioperative myocardial infarction following elective vascular surgery in patients with documented coronary artery disease: results of the CARP trial. Eur Heart J 2008; 29:394–401.
- van Waes JA, Nathoe HM, de Graaff JC, et al. Myocardial injury after noncardiac surgery and its association with short-term mortality. Circulation 2013; 127:2264–2271.
- Badner NH, Knill RL, Brown JE, Novick TV, Gelb AW. Myocardial infarction after noncardiac surgery. Anesthesiology 1998; 88:572–578.
- Kim LJ, Martinez EA, Faraday N, et al. Cardiac troponin I predicts short-term mortality in vascular surgery patients. Circulation 2002; 106:2366–2371.
- Landesberg G, Shatz V, Akopnik I, et al. Association of cardiac troponin, CK-MB, and postoperative myocardial ischemia with long-term survival after major vascular surgery. J Am Coll Cardiol 2003; 42:1547–1554.
- Devereaux PJ, Xavier D, Pogue J, et al. Characteristics and short-term prognosis of perioperative myocardial infarction in patients undergoing noncardiac surgery: a cohort study. Ann Intern Med 2011; 154:523–528.
- Devereaux PJ, Chan MT, Alonso-Coello P, et al. Association between postoperative troponin levels and 30-day mortality among patients undergoing noncardiac surgery. JAMA 2012; 307:2295–2304.
- Kumar A, Cannon CP. Acute coronary syndromes: diagnosis and management, part I. Mayo Clin Proc 2009; 84:917–938.
- Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Circulation 2012; 126:2020–2035.
- Apple FS, Quist HE, Doyle PJ, Otto AP, Murakami MM. Plasma 99th percentile reference limits for cardiac troponin and creatine kinase MB mass for use with European Society of Cardiology/American College of Cardiology consensus recommendations. Clin Chem 2003; 49:1331–1336.
- Ottani F, Galvani M, Nicolini FA, et al. Elevated cardiac troponin levels predict the risk of adverse outcome in patients with acute coronary syndromes. Am Heart J 2000; 140:917–927.
- Ohman EM, Armstrong PW, White HD, et al. Risk stratification with a point-of-care cardiac troponin T test in acute myocardial infarction. GUSTO III investigators. Global Use of Strategies to Open Occluded Coronary Arteries. Am J Cardiol 1999; 84:1281–1286.
- deFilippi CR, Tocchi M, Parmar RJ, et al. Cardiac troponin T in chest pain unit patients without ischemic electrocardiographic changes: angiographic correlates and long-term clinical outcomes. J Am Coll Cardiol 2000; 35:1827–1834.
- Heidenreich PA, Alloggiamento T, Melsop K, McDonald KM, Go AS, Hlatky MA. The prognostic value of troponin in patients with non-ST elevation acute coronary syndromes: a meta-analysis. J Am Coll Cardiol 2001; 38:478–485.
- Steen H, Giannitsis E, Futterer S, Merten C, Juenger C, Katus HA. Cardiac troponin T at 96 hours after acute myocardial infarction correlates with infarct size and cardiac function. J Am Coll Cardiol 2006; 48:2192–2194.
- Licka M, Zimmermann R, Zehelein J, Dengler TJ, Katus HA, Kubler W. Troponin T concentrations 72 hours after myocardial infarction as a serological estimate of infarct size. Heart 2002; 87:520–524.
- Vasile VC, Babuin L, Giannitsis E, Katus HA, Jaffe AS. Relationship of MRI-determined infarct size and cTnI measurements in patients with ST-elevation myocardial infarction. Clin Chem 2008; 54:617–619.
- Sabatine MS, Morrow DA, de Lemos JA, Jarolim P, Braunwald E. Detection of acute changes in circulating troponin in the setting of transient stress test-induced myocardial ischaemia using an ultrasensitive assay: results from TIMI 35. Eur Heart J 2009; 30:162–169.
- Siriwardena M, Campbell V, Richards AM, Pemberton CJ. Cardiac biomarker responses to dobutamine stress echocardiography in healthy volunteers and patients with coronary artery disease. Clin Chem 2012; 58:1492–1494.
- Turer AT, Addo TA, Martin JL, et al. Myocardial ischemia induced by rapid atrial pacing causes troponin T release detectable by a highly sensitive assay: insights from a coronary sinus sampling study. J Am Coll Cardiol 2011; 57:2398–2405.
- Cohen MC, Aretz TH. Histological analysis of coronary artery lesions in fatal postoperative myocardial infarction. Cardiovasc Pathol 1999; 8:133–139.
- Gualandro DM, Campos CA, Calderaro D, et al. Coronary plaque rupture in patients with myocardial infarction after noncardiac surgery: frequent and dangerous. Atherosclerosis 2012; 222:191–195.
- Sametz W, Metzler H, Gries M, et al. Perioperative catecholamine changes in cardiac risk patients. Eur J Clin Invest 1999; 29:582–587.
- Frank SM, Higgins MS, Breslow MJ, et al. The catecholamine, cortisol, and hemodynamic responses to mild perioperative hypothermia. A randomized clinical trial. Anesthesiology 1995; 82:83–93.
- Rosenfeld BA, Faraday N, Campbell D, et al. Perioperative platelet reactivity and the effects of clonidine. Anesthesiology 1993; 79:255–261.
- Lison S, Weiss G, Spannagl M, Heindl B. Postoperative changes in procoagulant factors after major surgery. Blood Coagul Fibrinolysis 2011; 22:190–196.
- Fukumoto Y, Hiro T, Fujii T, et al. Localized elevation of shear stress is related to coronary plaque rupture: a 3-dimensional intravascular ultrasound study with in-vivo color mapping of shear stress distribution. J Am Coll Cardiol 2008; 51:645–650.
- Feringa HH, Bax JJ, Boersma E, et al. High-dose beta-blockers and tight heart rate control reduce myocardial ischemia and troponin T release in vascular surgery patients. Circulation 2006; 114:I-344–I-349.
- Landesberg G. The pathophysiology of perioperative myocardial infarction: facts and perspectives. J Cardiothorac Vasc Anesth 2003; 17:90–100.
- Nelson AH, Fleisher LA, Rosenbaum SH. Relationship between postoperative anemia and cardiac morbidity in high-risk vascular patients in the intensive care unit. Crit Care Med 1993; 21:860–866.
- Landesberg G, Beattie WS, Mosseri M, Jaffe AS, Alpert JS. Perioperative myocardial infarction. Circulation 2009; 119:2936–2944.
- Ellis SG, Hertzer NR, Young JR, Brener S. Angiographic correlates of cardiac death and myocardial infarction complicating major nonthoracic vascular surgery. Am J Cardiol 1996; 77:1126–1128.
- Levy M, Heels-Ansdell D, Hiralal R, et al. Prognostic value of troponin and creatine kinase muscle and brain isoenzyme measurement after noncardiac surgery: a systematic review and meta-analysis. Anesthesiology 2011; 114:796–806.
- Garcia S, Marston N, Sandoval Y, et al. Prognostic value of 12-lead electrocardiogram and peak troponin I level after vascular surgery. J Vasc Surg 2013; 57:166–172.
- Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association task force on practice guidelines. J Am Coll Cardiol 2014; 64:e77–e137.
- Foucrier A, Rodseth R, Aissaoui M, et al. The long-term impact of early cardiovascular therapy intensification for postoperative troponin elevation after major vascular surgery. Anesth Analg 2014; 119:1053–1063.
KEY POINTS
- Cardiovascular events are a major cause of morbidity and mortality in patients undergoing noncardiac surgery and occur frequently, especially in high-risk patients.
- Myocardial injury or infarction after noncardiac surgery heightens the short- and long-term risk of mortality and major adverse cardiac events.
- The dominant mechanism of myocardial injury after noncardiac surgery remains uncertain.
- In the absence of therapies proven to affect the outcome, the benefit of screening to identify these patients remains uncertain.
- Clinical trials are under way to help clinicians provide optimal care to this at-risk population.