User login
Bevacizumab fails to improve survival in newly diagnosed glioblastoma
The addition of bevacizumab to standard treatment did not improve survival in patients with newly diagnosed glioblastoma, and in some cases, worsened quality of life and led to cognitive decline.
Bevacizumab (Avastin) added to frontline radiation and temozolomide therapy extended progression-free survival, but did not improve overall survival in the Radiation Therapy Oncology Group (RTOG) 0825 study, a double-blind, placebo-controlled phase III trial, Dr. Mark R. Gilbert of the University of Texas M.D. Anderson Cancer Center, Houston, and his associates reported Feb. 19 in the New England Journal of Medicine.
Among 637 patients with centrally confirmed glioblastoma who were randomized, median overall survival reached 16.1 months in those assigned to radiation, temozolomide, and placebo, compared with 15.7 months in patients assigned to radiation, temozolomide, and bevacizumab. (N. Engl. J. Med. 2014; 370:699-708.)
Median overall survival data were virtually identical in a second similarly designed study, also reported Feb. 19 in the New England Journal of Medicine.
Median survival in Avaglio, which ran parallel to RTOG 0825, was 16.8 months in 458 patients in its radiation, temozolomide, and bevacizumab arm, vs. 16.7 months in 463 patients in its radiation, temozolomide, and placebo arm, Dr. Olivier L. Chinot of Aix-Marseille University, Marseille, France, and his associates reported (N. Engl. J. Med. 2014; 370:709-22).
Median progression-free survival in Avaglio reached 10.6 months in the bevacizumab arm vs. 6.2 months in the placebo arm, and the difference was significant (hazard ratio, 0.64; P less than .0001).
The Avaglio trial showed a benefit or maintenance of quality of life measures, but did not look at neurocognitive outcomes. More patients in the bevacizumab group than in the placebo group had grade 3 or higher adverse events (66.8% vs. 51.3%) and grade 3 or higher adverse events often associated with bevacizumab (32.5% vs. 15.8%), reported Dr. Chinot and his associates.
Both studies were presented last year at the annual meeting of the American Society of Clinical Oncology.
The RTOG 0285 study was supported by the National Cancer Institute, with additional support from Genentech. Avaglio was supported by Roche. Dr. Gilbert disclosed consulting for, and receiving honoraria and research support from, Genentech. Dr. Chinot disclosed receiving financial and nonfinancial support from Roche.
lnikolaides@frontlinemedcom.com
On Twitter @NikolaidesLaura
The addition of bevacizumab to standard treatment did not improve survival in patients with newly diagnosed glioblastoma, and in some cases, worsened quality of life and led to cognitive decline.
Bevacizumab (Avastin) added to frontline radiation and temozolomide therapy extended progression-free survival, but did not improve overall survival in the Radiation Therapy Oncology Group (RTOG) 0825 study, a double-blind, placebo-controlled phase III trial, Dr. Mark R. Gilbert of the University of Texas M.D. Anderson Cancer Center, Houston, and his associates reported Feb. 19 in the New England Journal of Medicine.
Among 637 patients with centrally confirmed glioblastoma who were randomized, median overall survival reached 16.1 months in those assigned to radiation, temozolomide, and placebo, compared with 15.7 months in patients assigned to radiation, temozolomide, and bevacizumab. (N. Engl. J. Med. 2014; 370:699-708.)
Median overall survival data were virtually identical in a second similarly designed study, also reported Feb. 19 in the New England Journal of Medicine.
Median survival in Avaglio, which ran parallel to RTOG 0825, was 16.8 months in 458 patients in its radiation, temozolomide, and bevacizumab arm, vs. 16.7 months in 463 patients in its radiation, temozolomide, and placebo arm, Dr. Olivier L. Chinot of Aix-Marseille University, Marseille, France, and his associates reported (N. Engl. J. Med. 2014; 370:709-22).
Median progression-free survival in Avaglio reached 10.6 months in the bevacizumab arm vs. 6.2 months in the placebo arm, and the difference was significant (hazard ratio, 0.64; P less than .0001).
The Avaglio trial showed a benefit or maintenance of quality of life measures, but did not look at neurocognitive outcomes. More patients in the bevacizumab group than in the placebo group had grade 3 or higher adverse events (66.8% vs. 51.3%) and grade 3 or higher adverse events often associated with bevacizumab (32.5% vs. 15.8%), reported Dr. Chinot and his associates.
Both studies were presented last year at the annual meeting of the American Society of Clinical Oncology.
The RTOG 0285 study was supported by the National Cancer Institute, with additional support from Genentech. Avaglio was supported by Roche. Dr. Gilbert disclosed consulting for, and receiving honoraria and research support from, Genentech. Dr. Chinot disclosed receiving financial and nonfinancial support from Roche.
lnikolaides@frontlinemedcom.com
On Twitter @NikolaidesLaura
The addition of bevacizumab to standard treatment did not improve survival in patients with newly diagnosed glioblastoma, and in some cases, worsened quality of life and led to cognitive decline.
Bevacizumab (Avastin) added to frontline radiation and temozolomide therapy extended progression-free survival, but did not improve overall survival in the Radiation Therapy Oncology Group (RTOG) 0825 study, a double-blind, placebo-controlled phase III trial, Dr. Mark R. Gilbert of the University of Texas M.D. Anderson Cancer Center, Houston, and his associates reported Feb. 19 in the New England Journal of Medicine.
Among 637 patients with centrally confirmed glioblastoma who were randomized, median overall survival reached 16.1 months in those assigned to radiation, temozolomide, and placebo, compared with 15.7 months in patients assigned to radiation, temozolomide, and bevacizumab. (N. Engl. J. Med. 2014; 370:699-708.)
Median overall survival data were virtually identical in a second similarly designed study, also reported Feb. 19 in the New England Journal of Medicine.
Median survival in Avaglio, which ran parallel to RTOG 0825, was 16.8 months in 458 patients in its radiation, temozolomide, and bevacizumab arm, vs. 16.7 months in 463 patients in its radiation, temozolomide, and placebo arm, Dr. Olivier L. Chinot of Aix-Marseille University, Marseille, France, and his associates reported (N. Engl. J. Med. 2014; 370:709-22).
Median progression-free survival in Avaglio reached 10.6 months in the bevacizumab arm vs. 6.2 months in the placebo arm, and the difference was significant (hazard ratio, 0.64; P less than .0001).
The Avaglio trial showed a benefit or maintenance of quality of life measures, but did not look at neurocognitive outcomes. More patients in the bevacizumab group than in the placebo group had grade 3 or higher adverse events (66.8% vs. 51.3%) and grade 3 or higher adverse events often associated with bevacizumab (32.5% vs. 15.8%), reported Dr. Chinot and his associates.
Both studies were presented last year at the annual meeting of the American Society of Clinical Oncology.
The RTOG 0285 study was supported by the National Cancer Institute, with additional support from Genentech. Avaglio was supported by Roche. Dr. Gilbert disclosed consulting for, and receiving honoraria and research support from, Genentech. Dr. Chinot disclosed receiving financial and nonfinancial support from Roche.
lnikolaides@frontlinemedcom.com
On Twitter @NikolaidesLaura
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Major finding: Two similar studies found no difference in median survival with the addition of bevacizumab to standard therapy for patients with newly diagnosed glioblastoma. Median overall survival was 15.7 months in the bevacizumab arm vs. 16.1 months in the placebo arm in one study, and16.8 months in the bevacizumab arm vs. 16.7 months in the placebo arm in a second, similar study.
Data source: Two randomized, double-blind placebo-controlled phase III trials; the Radiation Therapy Oncology Group (RCOG) 0825 trial included 637 patients and the Avastin in Glioblastoma (Avaglia) trial involved 921 patients.
Disclosures: The RTOG 0285 study was supported by the National Cancer Institute, with additional support from Genentech. Avaglio was supported by Roche. Dr. Gilbert disclosed consulting for, and receiving honoraria and research support from, Genentech. Dr. Chinot disclosed receiving financial and nonfinancial support from Roche.
Treating female pattern hair loss
Female pattern hair loss, which affects over 21 million women in the United States, is a nonscarring hair loss primarily involving the frontal and vertex scalp. FPHS causes women significant emotional and psychological distress. We see, and will continue to see, a lot of these cases in primary care. If left untreated or unaddressed, FPHL results in a slow, progressive decline in the density of scalp hair.
FPHL is characterized by the production of shorter and finer hairs and shortening of the growth phase of hair follicles. One may find it important to rule out secondary causes of hair loss, such as hyperandrogenism. So after excluding secondary causes, what are the best treatment options for treatment?
Researchers conducted a systematic review (Cochrane Database Syst. Rev. 2012 May, CD007628 [doi:10.1002/14651858.CD007628.pub3]) assessing the effectiveness of interventions for female pattern hair loss. Studies were included if they compared any type of monotherapy or combination therapy to treat FPHL. Studies evaluating treatments in women with increased circulating androgens (such as polycystic ovarian syndrome) were included. Primary outcomes included self-reported hair regrowth, quality of life, and adverse effects.
The Cochrane review included 22 studies that enrolled a total of 2,349 subjects. Ten studies evaluated minoxidil, four evaluated finasteride, two cyproterone acetate, and two flutamide. A variety of other exotic interventions was evaluated, including topical melatonin-alcohol solution, adenosine lotion, and pulsed electrostatic field.
The best data continue to exist for minoxidil. "Pooled data from 4 studies indicated that a greater proportion of participants (121/488) treated with minoxidil reported a moderate increase in their hair regrowth when compared with placebo (64/476) (risk ratio, 1.86; 95% confidence interval [CI], 1.42 to 2.43). In 7 studies, there was an important increase of 13.28 in total hair count per cm² in the minoxidil group compared to the placebo group (95% CI, 10.89 to 15.68). There was no difference in the number of adverse events in the twice daily minoxidil and placebo intervention groups, with the exception of a reported increase of adverse events (additional hair growth on areas other than the scalp) with minoxidil (5%) twice daily," according to the Cochrane report.
Other promising agents might be octyl nicotinate (0.5%), myristyl nicotinate (5%), and flutamide. Fulvestrant, adenosine, pulsed electrostatic field, and estradiol valerate are ineffective.
Minoxidil it is. It may be important to remind patients that 2% twice daily may be as effective and safe as 5% once a day.
Dr. Ebbert is professor of medicine and a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author. He reports no conflicts of interest.
Female pattern hair loss, which affects over 21 million women in the United States, is a nonscarring hair loss primarily involving the frontal and vertex scalp. FPHS causes women significant emotional and psychological distress. We see, and will continue to see, a lot of these cases in primary care. If left untreated or unaddressed, FPHL results in a slow, progressive decline in the density of scalp hair.
FPHL is characterized by the production of shorter and finer hairs and shortening of the growth phase of hair follicles. One may find it important to rule out secondary causes of hair loss, such as hyperandrogenism. So after excluding secondary causes, what are the best treatment options for treatment?
Researchers conducted a systematic review (Cochrane Database Syst. Rev. 2012 May, CD007628 [doi:10.1002/14651858.CD007628.pub3]) assessing the effectiveness of interventions for female pattern hair loss. Studies were included if they compared any type of monotherapy or combination therapy to treat FPHL. Studies evaluating treatments in women with increased circulating androgens (such as polycystic ovarian syndrome) were included. Primary outcomes included self-reported hair regrowth, quality of life, and adverse effects.
The Cochrane review included 22 studies that enrolled a total of 2,349 subjects. Ten studies evaluated minoxidil, four evaluated finasteride, two cyproterone acetate, and two flutamide. A variety of other exotic interventions was evaluated, including topical melatonin-alcohol solution, adenosine lotion, and pulsed electrostatic field.
The best data continue to exist for minoxidil. "Pooled data from 4 studies indicated that a greater proportion of participants (121/488) treated with minoxidil reported a moderate increase in their hair regrowth when compared with placebo (64/476) (risk ratio, 1.86; 95% confidence interval [CI], 1.42 to 2.43). In 7 studies, there was an important increase of 13.28 in total hair count per cm² in the minoxidil group compared to the placebo group (95% CI, 10.89 to 15.68). There was no difference in the number of adverse events in the twice daily minoxidil and placebo intervention groups, with the exception of a reported increase of adverse events (additional hair growth on areas other than the scalp) with minoxidil (5%) twice daily," according to the Cochrane report.
Other promising agents might be octyl nicotinate (0.5%), myristyl nicotinate (5%), and flutamide. Fulvestrant, adenosine, pulsed electrostatic field, and estradiol valerate are ineffective.
Minoxidil it is. It may be important to remind patients that 2% twice daily may be as effective and safe as 5% once a day.
Dr. Ebbert is professor of medicine and a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author. He reports no conflicts of interest.
Female pattern hair loss, which affects over 21 million women in the United States, is a nonscarring hair loss primarily involving the frontal and vertex scalp. FPHS causes women significant emotional and psychological distress. We see, and will continue to see, a lot of these cases in primary care. If left untreated or unaddressed, FPHL results in a slow, progressive decline in the density of scalp hair.
FPHL is characterized by the production of shorter and finer hairs and shortening of the growth phase of hair follicles. One may find it important to rule out secondary causes of hair loss, such as hyperandrogenism. So after excluding secondary causes, what are the best treatment options for treatment?
Researchers conducted a systematic review (Cochrane Database Syst. Rev. 2012 May, CD007628 [doi:10.1002/14651858.CD007628.pub3]) assessing the effectiveness of interventions for female pattern hair loss. Studies were included if they compared any type of monotherapy or combination therapy to treat FPHL. Studies evaluating treatments in women with increased circulating androgens (such as polycystic ovarian syndrome) were included. Primary outcomes included self-reported hair regrowth, quality of life, and adverse effects.
The Cochrane review included 22 studies that enrolled a total of 2,349 subjects. Ten studies evaluated minoxidil, four evaluated finasteride, two cyproterone acetate, and two flutamide. A variety of other exotic interventions was evaluated, including topical melatonin-alcohol solution, adenosine lotion, and pulsed electrostatic field.
The best data continue to exist for minoxidil. "Pooled data from 4 studies indicated that a greater proportion of participants (121/488) treated with minoxidil reported a moderate increase in their hair regrowth when compared with placebo (64/476) (risk ratio, 1.86; 95% confidence interval [CI], 1.42 to 2.43). In 7 studies, there was an important increase of 13.28 in total hair count per cm² in the minoxidil group compared to the placebo group (95% CI, 10.89 to 15.68). There was no difference in the number of adverse events in the twice daily minoxidil and placebo intervention groups, with the exception of a reported increase of adverse events (additional hair growth on areas other than the scalp) with minoxidil (5%) twice daily," according to the Cochrane report.
Other promising agents might be octyl nicotinate (0.5%), myristyl nicotinate (5%), and flutamide. Fulvestrant, adenosine, pulsed electrostatic field, and estradiol valerate are ineffective.
Minoxidil it is. It may be important to remind patients that 2% twice daily may be as effective and safe as 5% once a day.
Dr. Ebbert is professor of medicine and a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author. He reports no conflicts of interest.
Digital Dermatology: VisualDx
"You should always consider three differential diagnoses for each patient." This was sound advice from my dermatology residency director, but advice I often neglect to take. What about you?
If you are like most of us, then your brain in clinic is on autopilot. It instantly selects the diagnosis and moves on. But when pushed by a confusing rash or a disease unresponsive to our standard treatment, we quickly encounter the limits of the human brain.
When stumped, we all do the same thing: Recruit more eyeballs (and brains). We find a colleague nearby and pull him into the room. "So, what do you think this is? What would you do?" Although often helpful, this method is inefficient and fails to capitalize on the most important of all medical tools: the computer.
Unlike our brains, computers in the form of clinical decision support (CDS) tools, are not prone to cognitive errors. Good CDS tools aren’t subject to top-of-mind biases. Their ability to generate differential diagnoses exceeds even the masters among us. Fortunately, there is such a CDS for skin disease: VisualDx.
VisualDx is a CDS tool focused on dermatologic conditions. It covers common and rare skin conditions and has more than 25,000 professional images.
What’s unique here is that VisualDx is more than a database of images. "It is truly a diagnostic decision support tool that allows you to search by multiple factors at once – symptoms, diagnoses, medications, medical history, travel, skin color, etc.," said Dr. Noah Craft, practicing dermatologist and chief medical officer of VisualDx.
In contrast to textbooks and other medical knowledge databases, this system is designed for easy, point-of-care use – it makes a dermatologist’s or even a primary care physician’s work easier. By quickly reviewing photos and diagnostic pearls, our brains are supercharged with deep differentials and management ideas.
For example, I recently had a patient who presented with papulosquamous eruptions that involved his body and hands. Among other diagnoses was secondary syphilis. Yes, I had thought of that, but a quick scan through VisualDx prompted me to ask about other symptoms, including vision changes (which he had). The patient also had HIV. Quick, which test is the best for me to order? Too slow, it’s already there in front of me on my screen.
In addition to improving quality, tools such as these also can improve access. Studies from the company show that the average user saves between 15 and 26 minutes per day using their product. For the working dermatologist, that means being able to see two additional patients a day.
VisualDx also educates and empowers patients. Don’t believe those bumps you have are molluscum? You can see here that these photos look exactly like the bumps you have. Rather than explain conditions through difficult doctor-speak, physicians can show complex knowledge to patients visually. As Dr. Craft notes and many of us have experienced: "For many patients, seeing is believing."
Whether it’s corroborating a diagnosis or exploring treatment options, having the doctor and patient share the same screen is an effective way to increase comprehension and build trust. No matter how good our drawings on the back of a prescription pad may be, they are not as accurate or helpful as curated digital photos. Our screen-savvy patients will soon expect this type of technology with every visit.
Good digital medicine tools also will help remedy one of medicine’s oldest and most glaring defects: We don’t account for the fact that the vast majority of health care happens in between doctor visits. Now patient education doesn’t stop at the culmination of the visit. Physicians can either print or e-mail images and information to patients so that they can have an accurate record at home to share with family members and caregivers.
VisualDx is a leading technology in what will be the future of medicine: Digital tools that serve doctors with everything they need to diagnose and treat patients with a click or flick of the screen. Having ten thousand treatment options instantly in your pocket – try that with any lab coat reference book.
Oftentimes, technology is more sparkle than substance. Not so with VisualDx. Have you used it in your practice? Let us know what you think about it.
For more information and to learn how to subscribe, visit www.visualdx.com. VisualDx is a paid subscription service.
Dr. Benabio is a practicing dermatologist and physician director of health care transformation at Kaiser Permanente in San Diego. Dr. Benabio said he has no financial interest in VisualDx, but he has had complimentary access. Connect with him on Twitter @Dermdoc or drop him a line at benabio@gmail.com.
"You should always consider three differential diagnoses for each patient." This was sound advice from my dermatology residency director, but advice I often neglect to take. What about you?
If you are like most of us, then your brain in clinic is on autopilot. It instantly selects the diagnosis and moves on. But when pushed by a confusing rash or a disease unresponsive to our standard treatment, we quickly encounter the limits of the human brain.
When stumped, we all do the same thing: Recruit more eyeballs (and brains). We find a colleague nearby and pull him into the room. "So, what do you think this is? What would you do?" Although often helpful, this method is inefficient and fails to capitalize on the most important of all medical tools: the computer.
Unlike our brains, computers in the form of clinical decision support (CDS) tools, are not prone to cognitive errors. Good CDS tools aren’t subject to top-of-mind biases. Their ability to generate differential diagnoses exceeds even the masters among us. Fortunately, there is such a CDS for skin disease: VisualDx.
VisualDx is a CDS tool focused on dermatologic conditions. It covers common and rare skin conditions and has more than 25,000 professional images.
What’s unique here is that VisualDx is more than a database of images. "It is truly a diagnostic decision support tool that allows you to search by multiple factors at once – symptoms, diagnoses, medications, medical history, travel, skin color, etc.," said Dr. Noah Craft, practicing dermatologist and chief medical officer of VisualDx.
In contrast to textbooks and other medical knowledge databases, this system is designed for easy, point-of-care use – it makes a dermatologist’s or even a primary care physician’s work easier. By quickly reviewing photos and diagnostic pearls, our brains are supercharged with deep differentials and management ideas.
For example, I recently had a patient who presented with papulosquamous eruptions that involved his body and hands. Among other diagnoses was secondary syphilis. Yes, I had thought of that, but a quick scan through VisualDx prompted me to ask about other symptoms, including vision changes (which he had). The patient also had HIV. Quick, which test is the best for me to order? Too slow, it’s already there in front of me on my screen.
In addition to improving quality, tools such as these also can improve access. Studies from the company show that the average user saves between 15 and 26 minutes per day using their product. For the working dermatologist, that means being able to see two additional patients a day.
VisualDx also educates and empowers patients. Don’t believe those bumps you have are molluscum? You can see here that these photos look exactly like the bumps you have. Rather than explain conditions through difficult doctor-speak, physicians can show complex knowledge to patients visually. As Dr. Craft notes and many of us have experienced: "For many patients, seeing is believing."
Whether it’s corroborating a diagnosis or exploring treatment options, having the doctor and patient share the same screen is an effective way to increase comprehension and build trust. No matter how good our drawings on the back of a prescription pad may be, they are not as accurate or helpful as curated digital photos. Our screen-savvy patients will soon expect this type of technology with every visit.
Good digital medicine tools also will help remedy one of medicine’s oldest and most glaring defects: We don’t account for the fact that the vast majority of health care happens in between doctor visits. Now patient education doesn’t stop at the culmination of the visit. Physicians can either print or e-mail images and information to patients so that they can have an accurate record at home to share with family members and caregivers.
VisualDx is a leading technology in what will be the future of medicine: Digital tools that serve doctors with everything they need to diagnose and treat patients with a click or flick of the screen. Having ten thousand treatment options instantly in your pocket – try that with any lab coat reference book.
Oftentimes, technology is more sparkle than substance. Not so with VisualDx. Have you used it in your practice? Let us know what you think about it.
For more information and to learn how to subscribe, visit www.visualdx.com. VisualDx is a paid subscription service.
Dr. Benabio is a practicing dermatologist and physician director of health care transformation at Kaiser Permanente in San Diego. Dr. Benabio said he has no financial interest in VisualDx, but he has had complimentary access. Connect with him on Twitter @Dermdoc or drop him a line at benabio@gmail.com.
"You should always consider three differential diagnoses for each patient." This was sound advice from my dermatology residency director, but advice I often neglect to take. What about you?
If you are like most of us, then your brain in clinic is on autopilot. It instantly selects the diagnosis and moves on. But when pushed by a confusing rash or a disease unresponsive to our standard treatment, we quickly encounter the limits of the human brain.
When stumped, we all do the same thing: Recruit more eyeballs (and brains). We find a colleague nearby and pull him into the room. "So, what do you think this is? What would you do?" Although often helpful, this method is inefficient and fails to capitalize on the most important of all medical tools: the computer.
Unlike our brains, computers in the form of clinical decision support (CDS) tools, are not prone to cognitive errors. Good CDS tools aren’t subject to top-of-mind biases. Their ability to generate differential diagnoses exceeds even the masters among us. Fortunately, there is such a CDS for skin disease: VisualDx.
VisualDx is a CDS tool focused on dermatologic conditions. It covers common and rare skin conditions and has more than 25,000 professional images.
What’s unique here is that VisualDx is more than a database of images. "It is truly a diagnostic decision support tool that allows you to search by multiple factors at once – symptoms, diagnoses, medications, medical history, travel, skin color, etc.," said Dr. Noah Craft, practicing dermatologist and chief medical officer of VisualDx.
In contrast to textbooks and other medical knowledge databases, this system is designed for easy, point-of-care use – it makes a dermatologist’s or even a primary care physician’s work easier. By quickly reviewing photos and diagnostic pearls, our brains are supercharged with deep differentials and management ideas.
For example, I recently had a patient who presented with papulosquamous eruptions that involved his body and hands. Among other diagnoses was secondary syphilis. Yes, I had thought of that, but a quick scan through VisualDx prompted me to ask about other symptoms, including vision changes (which he had). The patient also had HIV. Quick, which test is the best for me to order? Too slow, it’s already there in front of me on my screen.
In addition to improving quality, tools such as these also can improve access. Studies from the company show that the average user saves between 15 and 26 minutes per day using their product. For the working dermatologist, that means being able to see two additional patients a day.
VisualDx also educates and empowers patients. Don’t believe those bumps you have are molluscum? You can see here that these photos look exactly like the bumps you have. Rather than explain conditions through difficult doctor-speak, physicians can show complex knowledge to patients visually. As Dr. Craft notes and many of us have experienced: "For many patients, seeing is believing."
Whether it’s corroborating a diagnosis or exploring treatment options, having the doctor and patient share the same screen is an effective way to increase comprehension and build trust. No matter how good our drawings on the back of a prescription pad may be, they are not as accurate or helpful as curated digital photos. Our screen-savvy patients will soon expect this type of technology with every visit.
Good digital medicine tools also will help remedy one of medicine’s oldest and most glaring defects: We don’t account for the fact that the vast majority of health care happens in between doctor visits. Now patient education doesn’t stop at the culmination of the visit. Physicians can either print or e-mail images and information to patients so that they can have an accurate record at home to share with family members and caregivers.
VisualDx is a leading technology in what will be the future of medicine: Digital tools that serve doctors with everything they need to diagnose and treat patients with a click or flick of the screen. Having ten thousand treatment options instantly in your pocket – try that with any lab coat reference book.
Oftentimes, technology is more sparkle than substance. Not so with VisualDx. Have you used it in your practice? Let us know what you think about it.
For more information and to learn how to subscribe, visit www.visualdx.com. VisualDx is a paid subscription service.
Dr. Benabio is a practicing dermatologist and physician director of health care transformation at Kaiser Permanente in San Diego. Dr. Benabio said he has no financial interest in VisualDx, but he has had complimentary access. Connect with him on Twitter @Dermdoc or drop him a line at benabio@gmail.com.

MRI method appears comparable to PET/CT
A modified MRI technique can effectively detect tumors in young cancer patients without exposing them to radiation, according to a small study published in The Lancet Oncology.
The method, called whole-body diffusion-weighted MRI, employs a contrast agent consisting of iron oxide nanoparticles.
This technique proved roughly as effective as 18F-FDG-PET/CT scans for detecting lymphoma and sarcoma in pediatric and young adult patients.
Researchers also noted that, as the MRI technique does not employ ionizing radiation, it might help prevent some of the adverse effects typically observed in patients who have undergone radiographic staging, particularly, secondary malignancies.
“I’m excited about having an imaging test for cancer patients that requires zero radiation exposure,” said senior study author Heike Daldrup-Link, MD, of the Stanford University School of Medicine in California.
She and her colleagues pointed out that, in the past, certain obstacles prevented physicians from using whole-body MRIs. For one, the scans take up to 2 hours, whereas a whole-body PET/CT takes only a few minutes.
In addition, in many organs, MRI does not distinguish healthy tissue from cancerous tissue. And existing contrast agents leave the tissues too quickly to be used in a lengthy, whole-body MRI.
In an attempt to overcome these obstacles, Dr Daldrup-Link and her colleagues used a contrast agent consisting of ferumoxytol nanoparticles. Injections of these iron oxide nanoparticles are approved by the US Food and Drug Administration (FDA) to treat anemia, and the researchers obtained FDA permission for use in their study.
The nanoparticles are retained in the body for days. On MRIs, they cause blood vessels to appear brighter, providing anatomic landmarks. The nanoparticles also cause healthy bone marrow, lymph nodes, livers, and spleens to appear darker, which makes tumors stand out.
The researchers compared the whole-body diffusion-weighted MRI method to PET/CTs in 22 patients, ages 8 to 33, who had lymphoma or sarcoma. Fourteen of the patients had Hodgkin lymphoma, 5 had non-Hodgkin lymphoma, 1 had Burkitt leukemia, 1 had Ewing’s sarcoma, and 1 had osteosarcoma.
The team found the MRI scans and PET/CT scans provided comparable information, although tumor detection was slightly better with PET/CT. The PET/CTs detected 163 of the 174 total tumors, and the MRIs detected 158.
The two methods had similar levels of sensitivity, specificity, and diagnostic accuracy. Sensitivity was 93.7% with PET/CT and 90.8% with MRI. Specificity was 97.7% with PET/CT and 99.5% with MRI. And diagnostic accuracy was 97.2% with PET/CT and 98.3% with MRI.
The researchers also noted that none of the patients experienced adverse reactions to the ferumoxytol nanoparticles, although the FDA previously observed a small risk of allergic reaction to the nanoparticles’ coating.
Dr Daldrup-Link said future research will aim to validate the MRI method in larger, more diverse groups of cancer patients, as well as examine its possible use for monitoring tumors over the course of cancer treatment. The technique also holds promise for scanning patients after their treatment is complete. ![]()
A modified MRI technique can effectively detect tumors in young cancer patients without exposing them to radiation, according to a small study published in The Lancet Oncology.
The method, called whole-body diffusion-weighted MRI, employs a contrast agent consisting of iron oxide nanoparticles.
This technique proved roughly as effective as 18F-FDG-PET/CT scans for detecting lymphoma and sarcoma in pediatric and young adult patients.
Researchers also noted that, as the MRI technique does not employ ionizing radiation, it might help prevent some of the adverse effects typically observed in patients who have undergone radiographic staging, particularly, secondary malignancies.
“I’m excited about having an imaging test for cancer patients that requires zero radiation exposure,” said senior study author Heike Daldrup-Link, MD, of the Stanford University School of Medicine in California.
She and her colleagues pointed out that, in the past, certain obstacles prevented physicians from using whole-body MRIs. For one, the scans take up to 2 hours, whereas a whole-body PET/CT takes only a few minutes.
In addition, in many organs, MRI does not distinguish healthy tissue from cancerous tissue. And existing contrast agents leave the tissues too quickly to be used in a lengthy, whole-body MRI.
In an attempt to overcome these obstacles, Dr Daldrup-Link and her colleagues used a contrast agent consisting of ferumoxytol nanoparticles. Injections of these iron oxide nanoparticles are approved by the US Food and Drug Administration (FDA) to treat anemia, and the researchers obtained FDA permission for use in their study.
The nanoparticles are retained in the body for days. On MRIs, they cause blood vessels to appear brighter, providing anatomic landmarks. The nanoparticles also cause healthy bone marrow, lymph nodes, livers, and spleens to appear darker, which makes tumors stand out.
The researchers compared the whole-body diffusion-weighted MRI method to PET/CTs in 22 patients, ages 8 to 33, who had lymphoma or sarcoma. Fourteen of the patients had Hodgkin lymphoma, 5 had non-Hodgkin lymphoma, 1 had Burkitt leukemia, 1 had Ewing’s sarcoma, and 1 had osteosarcoma.
The team found the MRI scans and PET/CT scans provided comparable information, although tumor detection was slightly better with PET/CT. The PET/CTs detected 163 of the 174 total tumors, and the MRIs detected 158.
The two methods had similar levels of sensitivity, specificity, and diagnostic accuracy. Sensitivity was 93.7% with PET/CT and 90.8% with MRI. Specificity was 97.7% with PET/CT and 99.5% with MRI. And diagnostic accuracy was 97.2% with PET/CT and 98.3% with MRI.
The researchers also noted that none of the patients experienced adverse reactions to the ferumoxytol nanoparticles, although the FDA previously observed a small risk of allergic reaction to the nanoparticles’ coating.
Dr Daldrup-Link said future research will aim to validate the MRI method in larger, more diverse groups of cancer patients, as well as examine its possible use for monitoring tumors over the course of cancer treatment. The technique also holds promise for scanning patients after their treatment is complete. ![]()
A modified MRI technique can effectively detect tumors in young cancer patients without exposing them to radiation, according to a small study published in The Lancet Oncology.
The method, called whole-body diffusion-weighted MRI, employs a contrast agent consisting of iron oxide nanoparticles.
This technique proved roughly as effective as 18F-FDG-PET/CT scans for detecting lymphoma and sarcoma in pediatric and young adult patients.
Researchers also noted that, as the MRI technique does not employ ionizing radiation, it might help prevent some of the adverse effects typically observed in patients who have undergone radiographic staging, particularly, secondary malignancies.
“I’m excited about having an imaging test for cancer patients that requires zero radiation exposure,” said senior study author Heike Daldrup-Link, MD, of the Stanford University School of Medicine in California.
She and her colleagues pointed out that, in the past, certain obstacles prevented physicians from using whole-body MRIs. For one, the scans take up to 2 hours, whereas a whole-body PET/CT takes only a few minutes.
In addition, in many organs, MRI does not distinguish healthy tissue from cancerous tissue. And existing contrast agents leave the tissues too quickly to be used in a lengthy, whole-body MRI.
In an attempt to overcome these obstacles, Dr Daldrup-Link and her colleagues used a contrast agent consisting of ferumoxytol nanoparticles. Injections of these iron oxide nanoparticles are approved by the US Food and Drug Administration (FDA) to treat anemia, and the researchers obtained FDA permission for use in their study.
The nanoparticles are retained in the body for days. On MRIs, they cause blood vessels to appear brighter, providing anatomic landmarks. The nanoparticles also cause healthy bone marrow, lymph nodes, livers, and spleens to appear darker, which makes tumors stand out.
The researchers compared the whole-body diffusion-weighted MRI method to PET/CTs in 22 patients, ages 8 to 33, who had lymphoma or sarcoma. Fourteen of the patients had Hodgkin lymphoma, 5 had non-Hodgkin lymphoma, 1 had Burkitt leukemia, 1 had Ewing’s sarcoma, and 1 had osteosarcoma.
The team found the MRI scans and PET/CT scans provided comparable information, although tumor detection was slightly better with PET/CT. The PET/CTs detected 163 of the 174 total tumors, and the MRIs detected 158.
The two methods had similar levels of sensitivity, specificity, and diagnostic accuracy. Sensitivity was 93.7% with PET/CT and 90.8% with MRI. Specificity was 97.7% with PET/CT and 99.5% with MRI. And diagnostic accuracy was 97.2% with PET/CT and 98.3% with MRI.
The researchers also noted that none of the patients experienced adverse reactions to the ferumoxytol nanoparticles, although the FDA previously observed a small risk of allergic reaction to the nanoparticles’ coating.
Dr Daldrup-Link said future research will aim to validate the MRI method in larger, more diverse groups of cancer patients, as well as examine its possible use for monitoring tumors over the course of cancer treatment. The technique also holds promise for scanning patients after their treatment is complete. ![]()
Compression device can prevent VTE after surgery
Credit: Piotr Bodzek
A mobile compression device can prevent venous thromboembolism (VTE) after joint replacement surgery, according to research published in the Journal of Bone and Joint Surgery.
The device, called ActiveCare+S.F.T., delivers compressions to the leg that coordinate with a patient’s respiration rate, and this improves blood flow.
Of more than 3000 patients who used the device, with or without aspirin, less than 1% developed VTE.
When the researchers compared this rate to VTE rates observed in previous studies of warfarin, enoxaparin, rivaroxaban, and dabigatran, they found the device to be noninferior to anticoagulant therapy.
“Blood thinners have long been considered the standard of care to prevent blood clots after orthopedic surgery, but they can have side effects that are concerning for many patients,” said study author Clifford Colwell, MD, of the Scripps Clinic in La Jolla, California.
“Through this research, we have found and established an equally effective means of accomplishing the same goal, with an added layer of safety for patients.”
Dr Colwell and his colleagues established a registry of 3060 patients to determine the rate of symptomatic VTE after primary knee arthroplasty (n=1551) or hip arthroplasty (n=1509) performed at 10 different sites.
All of the patients enrolled were 18 years of age or older. They had no known history of VTE, coagulation disorders, or solid tumor malignancies.
Patients wore the ActiveCare+S.F.T device both during and after surgery, for a minimum of 10 days. The researchers evaluated patients at 3 months after their surgery to document evidence of deep vein thrombosis (DVT) or pulmonary embolism (PE).
In all, 28 patients (0.92%) developed VTE. Twenty patients had distal DVT, 3 had proximal DVT, and 5 had PE. One patient died of coronary failure, but there was no autopsy, so it is not clear if the patient developed a PE.
Overall, the rate of VTE with the compression device—0.92%—was considered noninferior to rates previously observed with anticoagulants—2.2% for warfarin, 1.1% for enoxaparin, 0.64% for rivaroxaban, and 1.2% for dabigatran.
However, among patients who underwent knee arthroplasty, the device fell short of the noninferiority margin (1.0%) for rivaroxaban by 0.06%.
The device’s manufacturer, Medical Compression Systems Inc., funded the registry used in this study but did not have a role in the registry design or protocol. And the researchers did not receive compensation from the manufacturer. ![]()
Credit: Piotr Bodzek
A mobile compression device can prevent venous thromboembolism (VTE) after joint replacement surgery, according to research published in the Journal of Bone and Joint Surgery.
The device, called ActiveCare+S.F.T., delivers compressions to the leg that coordinate with a patient’s respiration rate, and this improves blood flow.
Of more than 3000 patients who used the device, with or without aspirin, less than 1% developed VTE.
When the researchers compared this rate to VTE rates observed in previous studies of warfarin, enoxaparin, rivaroxaban, and dabigatran, they found the device to be noninferior to anticoagulant therapy.
“Blood thinners have long been considered the standard of care to prevent blood clots after orthopedic surgery, but they can have side effects that are concerning for many patients,” said study author Clifford Colwell, MD, of the Scripps Clinic in La Jolla, California.
“Through this research, we have found and established an equally effective means of accomplishing the same goal, with an added layer of safety for patients.”
Dr Colwell and his colleagues established a registry of 3060 patients to determine the rate of symptomatic VTE after primary knee arthroplasty (n=1551) or hip arthroplasty (n=1509) performed at 10 different sites.
All of the patients enrolled were 18 years of age or older. They had no known history of VTE, coagulation disorders, or solid tumor malignancies.
Patients wore the ActiveCare+S.F.T device both during and after surgery, for a minimum of 10 days. The researchers evaluated patients at 3 months after their surgery to document evidence of deep vein thrombosis (DVT) or pulmonary embolism (PE).
In all, 28 patients (0.92%) developed VTE. Twenty patients had distal DVT, 3 had proximal DVT, and 5 had PE. One patient died of coronary failure, but there was no autopsy, so it is not clear if the patient developed a PE.
Overall, the rate of VTE with the compression device—0.92%—was considered noninferior to rates previously observed with anticoagulants—2.2% for warfarin, 1.1% for enoxaparin, 0.64% for rivaroxaban, and 1.2% for dabigatran.
However, among patients who underwent knee arthroplasty, the device fell short of the noninferiority margin (1.0%) for rivaroxaban by 0.06%.
The device’s manufacturer, Medical Compression Systems Inc., funded the registry used in this study but did not have a role in the registry design or protocol. And the researchers did not receive compensation from the manufacturer. ![]()
Credit: Piotr Bodzek
A mobile compression device can prevent venous thromboembolism (VTE) after joint replacement surgery, according to research published in the Journal of Bone and Joint Surgery.
The device, called ActiveCare+S.F.T., delivers compressions to the leg that coordinate with a patient’s respiration rate, and this improves blood flow.
Of more than 3000 patients who used the device, with or without aspirin, less than 1% developed VTE.
When the researchers compared this rate to VTE rates observed in previous studies of warfarin, enoxaparin, rivaroxaban, and dabigatran, they found the device to be noninferior to anticoagulant therapy.
“Blood thinners have long been considered the standard of care to prevent blood clots after orthopedic surgery, but they can have side effects that are concerning for many patients,” said study author Clifford Colwell, MD, of the Scripps Clinic in La Jolla, California.
“Through this research, we have found and established an equally effective means of accomplishing the same goal, with an added layer of safety for patients.”
Dr Colwell and his colleagues established a registry of 3060 patients to determine the rate of symptomatic VTE after primary knee arthroplasty (n=1551) or hip arthroplasty (n=1509) performed at 10 different sites.
All of the patients enrolled were 18 years of age or older. They had no known history of VTE, coagulation disorders, or solid tumor malignancies.
Patients wore the ActiveCare+S.F.T device both during and after surgery, for a minimum of 10 days. The researchers evaluated patients at 3 months after their surgery to document evidence of deep vein thrombosis (DVT) or pulmonary embolism (PE).
In all, 28 patients (0.92%) developed VTE. Twenty patients had distal DVT, 3 had proximal DVT, and 5 had PE. One patient died of coronary failure, but there was no autopsy, so it is not clear if the patient developed a PE.
Overall, the rate of VTE with the compression device—0.92%—was considered noninferior to rates previously observed with anticoagulants—2.2% for warfarin, 1.1% for enoxaparin, 0.64% for rivaroxaban, and 1.2% for dabigatran.
However, among patients who underwent knee arthroplasty, the device fell short of the noninferiority margin (1.0%) for rivaroxaban by 0.06%.
The device’s manufacturer, Medical Compression Systems Inc., funded the registry used in this study but did not have a role in the registry design or protocol. And the researchers did not receive compensation from the manufacturer. ![]()
Environment may play role in malaria transmission
the wall of a mosquito midgut
Credit: Krijn Paaijmans
Researchers have found the environment can significantly influence whether or not Wolbachia bacteria will prevent mosquitoes from transmitting malaria.
“Bacteria in the genus Wolbachia represent a promising new tool for controlling malaria due to their demonstrated ability to block the development of the pathogen within Anopheles mosquitoes,” said study investigator Courtney Murdock, PhD, of Pennsylvania State University.
“However, much of the work on the Wolbachia-malaria interaction has been conducted under highly simplified laboratory conditions. In this study, we investigated the ability of Wolbachia to block transmission of malaria—Plasmodium—parasites across variable environmental conditions, which are more reflective of conditions in the field.”
Dr Murdock and her colleagues described this research in Nature Scientific Reports.
The researchers used the malaria parasite Plasmodium yoelii, which affects rodents, and the mosquito Anopheles stephensi as a model system to investigate whether Wolbachia would block the ability of the malaria parasite to infect the mosquitoes.
The team divided the mosquitoes into an uninfected control group and a group infected with Wolbachia. Next, they raised all groups of mosquitoes in incubators set to different experimental temperatures—68, 72, 75, 79, and 82 degrees Fahrenheit.
At 82 degrees, Wolbachia reduced the number of mosquitoes infected by malaria parasites, the number of malaria parasites within each mosquito, and the intensity of oocysts.
At 75 degrees, Wolbachia had no effect on the prevalence of malaria parasites but increased oocyst intensity. At 68 degrees, Wolbachia had no effect on the prevalence of parasites or the intensity of oocysts.
The researchers also identified a previously undiscovered effect of Wolbachia. Infection with the bacterium reduced the development of sporozoites across all temperatures. This suggests that Wolbachia and malaria parasites may compete for similar hosts.
“Typically, the more oocysts a mosquito has on its midgut, the more sporozoites it produces,” Dr Murdock said. “So, depending on the environmental temperature, Wolbachia either reduced, enhanced, or had no effect on the number of oocysts.”
“At 75 degrees Fahrenheit, Wolbachia-infected mosquitos had 3 times the numbers of oocysts relative to uninfected mosquitoes. Thus, we would predict these mosquitoes to produce more sporozoites. But instead, we see that this is not the case, and that is because Wolbachia infection significantly reduces the number of sporozoites produced per oocyst, regardless of the environmental temperature.”
“This effect counteracts the enhancement we see at 75 degrees Fahrenheit. How the influence of Wolbachia on parasite establishment and the production of sporozoites under different temperatures plays out to ultimately affect transmission remains to be determined.”
Dr Murdock and her colleagues plan to duplicate their experiment using a species of malaria parasite that affects humans to determine whether or not the temperature effects they observed occur in humans as well.
The team also intends to explore the effects of additional environmental variation—such as daily temperature fluctuation and differential access to food resources in the mosquito larval and adult environments—on the transmission-blocking ability of Wolbachia. ![]()
the wall of a mosquito midgut
Credit: Krijn Paaijmans
Researchers have found the environment can significantly influence whether or not Wolbachia bacteria will prevent mosquitoes from transmitting malaria.
“Bacteria in the genus Wolbachia represent a promising new tool for controlling malaria due to their demonstrated ability to block the development of the pathogen within Anopheles mosquitoes,” said study investigator Courtney Murdock, PhD, of Pennsylvania State University.
“However, much of the work on the Wolbachia-malaria interaction has been conducted under highly simplified laboratory conditions. In this study, we investigated the ability of Wolbachia to block transmission of malaria—Plasmodium—parasites across variable environmental conditions, which are more reflective of conditions in the field.”
Dr Murdock and her colleagues described this research in Nature Scientific Reports.
The researchers used the malaria parasite Plasmodium yoelii, which affects rodents, and the mosquito Anopheles stephensi as a model system to investigate whether Wolbachia would block the ability of the malaria parasite to infect the mosquitoes.
The team divided the mosquitoes into an uninfected control group and a group infected with Wolbachia. Next, they raised all groups of mosquitoes in incubators set to different experimental temperatures—68, 72, 75, 79, and 82 degrees Fahrenheit.
At 82 degrees, Wolbachia reduced the number of mosquitoes infected by malaria parasites, the number of malaria parasites within each mosquito, and the intensity of oocysts.
At 75 degrees, Wolbachia had no effect on the prevalence of malaria parasites but increased oocyst intensity. At 68 degrees, Wolbachia had no effect on the prevalence of parasites or the intensity of oocysts.
The researchers also identified a previously undiscovered effect of Wolbachia. Infection with the bacterium reduced the development of sporozoites across all temperatures. This suggests that Wolbachia and malaria parasites may compete for similar hosts.
“Typically, the more oocysts a mosquito has on its midgut, the more sporozoites it produces,” Dr Murdock said. “So, depending on the environmental temperature, Wolbachia either reduced, enhanced, or had no effect on the number of oocysts.”
“At 75 degrees Fahrenheit, Wolbachia-infected mosquitos had 3 times the numbers of oocysts relative to uninfected mosquitoes. Thus, we would predict these mosquitoes to produce more sporozoites. But instead, we see that this is not the case, and that is because Wolbachia infection significantly reduces the number of sporozoites produced per oocyst, regardless of the environmental temperature.”
“This effect counteracts the enhancement we see at 75 degrees Fahrenheit. How the influence of Wolbachia on parasite establishment and the production of sporozoites under different temperatures plays out to ultimately affect transmission remains to be determined.”
Dr Murdock and her colleagues plan to duplicate their experiment using a species of malaria parasite that affects humans to determine whether or not the temperature effects they observed occur in humans as well.
The team also intends to explore the effects of additional environmental variation—such as daily temperature fluctuation and differential access to food resources in the mosquito larval and adult environments—on the transmission-blocking ability of Wolbachia. ![]()
the wall of a mosquito midgut
Credit: Krijn Paaijmans
Researchers have found the environment can significantly influence whether or not Wolbachia bacteria will prevent mosquitoes from transmitting malaria.
“Bacteria in the genus Wolbachia represent a promising new tool for controlling malaria due to their demonstrated ability to block the development of the pathogen within Anopheles mosquitoes,” said study investigator Courtney Murdock, PhD, of Pennsylvania State University.
“However, much of the work on the Wolbachia-malaria interaction has been conducted under highly simplified laboratory conditions. In this study, we investigated the ability of Wolbachia to block transmission of malaria—Plasmodium—parasites across variable environmental conditions, which are more reflective of conditions in the field.”
Dr Murdock and her colleagues described this research in Nature Scientific Reports.
The researchers used the malaria parasite Plasmodium yoelii, which affects rodents, and the mosquito Anopheles stephensi as a model system to investigate whether Wolbachia would block the ability of the malaria parasite to infect the mosquitoes.
The team divided the mosquitoes into an uninfected control group and a group infected with Wolbachia. Next, they raised all groups of mosquitoes in incubators set to different experimental temperatures—68, 72, 75, 79, and 82 degrees Fahrenheit.
At 82 degrees, Wolbachia reduced the number of mosquitoes infected by malaria parasites, the number of malaria parasites within each mosquito, and the intensity of oocysts.
At 75 degrees, Wolbachia had no effect on the prevalence of malaria parasites but increased oocyst intensity. At 68 degrees, Wolbachia had no effect on the prevalence of parasites or the intensity of oocysts.
The researchers also identified a previously undiscovered effect of Wolbachia. Infection with the bacterium reduced the development of sporozoites across all temperatures. This suggests that Wolbachia and malaria parasites may compete for similar hosts.
“Typically, the more oocysts a mosquito has on its midgut, the more sporozoites it produces,” Dr Murdock said. “So, depending on the environmental temperature, Wolbachia either reduced, enhanced, or had no effect on the number of oocysts.”
“At 75 degrees Fahrenheit, Wolbachia-infected mosquitos had 3 times the numbers of oocysts relative to uninfected mosquitoes. Thus, we would predict these mosquitoes to produce more sporozoites. But instead, we see that this is not the case, and that is because Wolbachia infection significantly reduces the number of sporozoites produced per oocyst, regardless of the environmental temperature.”
“This effect counteracts the enhancement we see at 75 degrees Fahrenheit. How the influence of Wolbachia on parasite establishment and the production of sporozoites under different temperatures plays out to ultimately affect transmission remains to be determined.”
Dr Murdock and her colleagues plan to duplicate their experiment using a species of malaria parasite that affects humans to determine whether or not the temperature effects they observed occur in humans as well.
The team also intends to explore the effects of additional environmental variation—such as daily temperature fluctuation and differential access to food resources in the mosquito larval and adult environments—on the transmission-blocking ability of Wolbachia. ![]()
Group finds progenitors of ILCs

with ILCs (green), epithelial
cells (red), and nuclei (blue)
University of Pennsylvania
Scientists say they’ve discovered the progenitors of innate lymphoid cells (ILCs) in the liver of fetal mice and the bone marrow of adult mice.
ILCs are among the first components of the immune system to confront certain pathogens, yet the cells went undetected by researchers for a century.
“Scientists tend to look for immune cells in the blood, lymph nodes, or spleen,” said Albert Bendelac, PhD, of the University of Chicago in Illinois.
“That is precisely where you would not find these cells. Once they mature, they directly go to tissues, such as the gut or the skin. You seldom see them in blood.”
To understand how ILCs fit into the ecosystem of cells that fight off infections and cancers, Dr Bendelac’s team focused on finding ILCs’ source.
And they reported their findings in a letter to Nature.
The team noted that ILCs, which were first recognized 5 years ago, are rare. A mouse might have 200 million lymphocytes and only a few thousand ILCs.
But previous work on natural killer (NK) cells showed that ILCs express the transcription factor PLZF during their development.
So Dr Bendelac and his colleagues created mice with the gene for green fluorescent protein inserted into mouse DNA, just downstream from the PLZF gene. As a result, cells from mice that expressed PLZF appeared bright green under the microscope.
Nevertheless, finding the precursors to ILCs was not easy. The precursors are not in the blood, and, by the time they migrate to the lungs or gut, they have already matured into ILCs.
The researchers eventually found the precursor cells—known as ILCPs—in the liver of fetal mice and in the bone marrow of adult mice.
When the team purified the ILCPs, which still contained the GFP gene, and transferred them into mice that lacked ILCs, the precursors were able to reconstitute the 3 known types of ILCs—ILCs 1, 2, and 3.
“There were no B cells or T cells or myeloid cells—no other immune cells, just these,” Dr Bendelac said. “So we think the ILCP really is a committed precursor to innate lymphoid cells.”
To confirm their finding, the researchers designed mice in which PLZF gene expression was tied to the gene for diphtheria toxin. When the cells expressed PLZF, they also produced the toxin, which was lethal for those cells. The result was a mouse that had a normal immune system except that it completely lacked ILCs.
“ILCs are found in the most exposed tissues,” Dr Bendelac noted. “They are one of your first lines of defense. We now suspect they may also influence the ensuing adaptive immune response, priming the pump, influencing how T-helper cells respond.”
Each of the 3 types of ILCs has different properties and serves different functions. ILC1 cells help prevent viral infections and can detect and remove some cancerous cells. They are similar to NK cells, except that NK cells circulate in the blood, and ILC1s live in the gut and the liver.
ILC2s are found in the lungs, where they can detect and respond to parasites. But they can also initiate an allergic reaction and mucus hyper-secretion.
ILC3 cells cluster in the gut, where they help mediate interactions between the bowel and bacteria. When that balance is disturbed, they can accelerate inflammation and may play a role in inflammatory bowel disease.
Dr Bendelac said his group’s research provides “one more tool for understanding this complex system,” and it could help generate a “powerful new way to assess the function of innate lymphocytes.” ![]()
with ILCs (green), epithelial
cells (red), and nuclei (blue)
University of Pennsylvania
Scientists say they’ve discovered the progenitors of innate lymphoid cells (ILCs) in the liver of fetal mice and the bone marrow of adult mice.
ILCs are among the first components of the immune system to confront certain pathogens, yet the cells went undetected by researchers for a century.
“Scientists tend to look for immune cells in the blood, lymph nodes, or spleen,” said Albert Bendelac, PhD, of the University of Chicago in Illinois.
“That is precisely where you would not find these cells. Once they mature, they directly go to tissues, such as the gut or the skin. You seldom see them in blood.”
To understand how ILCs fit into the ecosystem of cells that fight off infections and cancers, Dr Bendelac’s team focused on finding ILCs’ source.
And they reported their findings in a letter to Nature.
The team noted that ILCs, which were first recognized 5 years ago, are rare. A mouse might have 200 million lymphocytes and only a few thousand ILCs.
But previous work on natural killer (NK) cells showed that ILCs express the transcription factor PLZF during their development.
So Dr Bendelac and his colleagues created mice with the gene for green fluorescent protein inserted into mouse DNA, just downstream from the PLZF gene. As a result, cells from mice that expressed PLZF appeared bright green under the microscope.
Nevertheless, finding the precursors to ILCs was not easy. The precursors are not in the blood, and, by the time they migrate to the lungs or gut, they have already matured into ILCs.
The researchers eventually found the precursor cells—known as ILCPs—in the liver of fetal mice and in the bone marrow of adult mice.
When the team purified the ILCPs, which still contained the GFP gene, and transferred them into mice that lacked ILCs, the precursors were able to reconstitute the 3 known types of ILCs—ILCs 1, 2, and 3.
“There were no B cells or T cells or myeloid cells—no other immune cells, just these,” Dr Bendelac said. “So we think the ILCP really is a committed precursor to innate lymphoid cells.”
To confirm their finding, the researchers designed mice in which PLZF gene expression was tied to the gene for diphtheria toxin. When the cells expressed PLZF, they also produced the toxin, which was lethal for those cells. The result was a mouse that had a normal immune system except that it completely lacked ILCs.
“ILCs are found in the most exposed tissues,” Dr Bendelac noted. “They are one of your first lines of defense. We now suspect they may also influence the ensuing adaptive immune response, priming the pump, influencing how T-helper cells respond.”
Each of the 3 types of ILCs has different properties and serves different functions. ILC1 cells help prevent viral infections and can detect and remove some cancerous cells. They are similar to NK cells, except that NK cells circulate in the blood, and ILC1s live in the gut and the liver.
ILC2s are found in the lungs, where they can detect and respond to parasites. But they can also initiate an allergic reaction and mucus hyper-secretion.
ILC3 cells cluster in the gut, where they help mediate interactions between the bowel and bacteria. When that balance is disturbed, they can accelerate inflammation and may play a role in inflammatory bowel disease.
Dr Bendelac said his group’s research provides “one more tool for understanding this complex system,” and it could help generate a “powerful new way to assess the function of innate lymphocytes.” ![]()
with ILCs (green), epithelial
cells (red), and nuclei (blue)
University of Pennsylvania
Scientists say they’ve discovered the progenitors of innate lymphoid cells (ILCs) in the liver of fetal mice and the bone marrow of adult mice.
ILCs are among the first components of the immune system to confront certain pathogens, yet the cells went undetected by researchers for a century.
“Scientists tend to look for immune cells in the blood, lymph nodes, or spleen,” said Albert Bendelac, PhD, of the University of Chicago in Illinois.
“That is precisely where you would not find these cells. Once they mature, they directly go to tissues, such as the gut or the skin. You seldom see them in blood.”
To understand how ILCs fit into the ecosystem of cells that fight off infections and cancers, Dr Bendelac’s team focused on finding ILCs’ source.
And they reported their findings in a letter to Nature.
The team noted that ILCs, which were first recognized 5 years ago, are rare. A mouse might have 200 million lymphocytes and only a few thousand ILCs.
But previous work on natural killer (NK) cells showed that ILCs express the transcription factor PLZF during their development.
So Dr Bendelac and his colleagues created mice with the gene for green fluorescent protein inserted into mouse DNA, just downstream from the PLZF gene. As a result, cells from mice that expressed PLZF appeared bright green under the microscope.
Nevertheless, finding the precursors to ILCs was not easy. The precursors are not in the blood, and, by the time they migrate to the lungs or gut, they have already matured into ILCs.
The researchers eventually found the precursor cells—known as ILCPs—in the liver of fetal mice and in the bone marrow of adult mice.
When the team purified the ILCPs, which still contained the GFP gene, and transferred them into mice that lacked ILCs, the precursors were able to reconstitute the 3 known types of ILCs—ILCs 1, 2, and 3.
“There were no B cells or T cells or myeloid cells—no other immune cells, just these,” Dr Bendelac said. “So we think the ILCP really is a committed precursor to innate lymphoid cells.”
To confirm their finding, the researchers designed mice in which PLZF gene expression was tied to the gene for diphtheria toxin. When the cells expressed PLZF, they also produced the toxin, which was lethal for those cells. The result was a mouse that had a normal immune system except that it completely lacked ILCs.
“ILCs are found in the most exposed tissues,” Dr Bendelac noted. “They are one of your first lines of defense. We now suspect they may also influence the ensuing adaptive immune response, priming the pump, influencing how T-helper cells respond.”
Each of the 3 types of ILCs has different properties and serves different functions. ILC1 cells help prevent viral infections and can detect and remove some cancerous cells. They are similar to NK cells, except that NK cells circulate in the blood, and ILC1s live in the gut and the liver.
ILC2s are found in the lungs, where they can detect and respond to parasites. But they can also initiate an allergic reaction and mucus hyper-secretion.
ILC3 cells cluster in the gut, where they help mediate interactions between the bowel and bacteria. When that balance is disturbed, they can accelerate inflammation and may play a role in inflammatory bowel disease.
Dr Bendelac said his group’s research provides “one more tool for understanding this complex system,” and it could help generate a “powerful new way to assess the function of innate lymphocytes.” ![]()
In‐hospital CPR Practices
An estimated 200,000 adult patients suffer cardiac arrest in US hospitals each year, of which <20% survive to hospital discharge.[1, 2] Patient survival from in‐hospital cardiac arrest (IHCA), however, varies widely across hospitals, and may be partly attributed to differences in hospital practices.[3, 4, 5] Although there are data to support specific patient‐level practices in the hospital, such as delivery of electrical shock for ventricular fibrillation within 2 minutes of onset of the lethal rhythm,[6] little is known about in‐hospital systems‐level factors. Similar to patient‐level practices, some organizational and systems level practices are supported by international consensus and guideline recommendations.[7, 8] However, the adoption of these practices is poorly understood. As such, we sought to gain a better understanding of current US hospital practices with regard to IHCA and resuscitation with the hopes of identifying potential targets for improvement in quality and outcomes.
METHODS
We conducted a nationally representative mail survey between May 2011 and November 2011, targeting a stratified random sample of 1000 hospitals. We utilized the US Acute‐Care Hospitals (FY2008) database from the American Hospital Association to determine the total population of 3809 community hospitals (ie, nonfederal government, nonpsychiatric, and nonlong‐term care hospitals).[9] This included general medical and surgical, surgical, cancer, heart, orthopedic, and children's hospitals. These hospitals were stratified into tertiles by annual in‐patient days and teaching status (major, minor, nonteaching), from which our sample was randomly selected (Table 1). We identified each hospital's cardiopulmonary resuscitation (CPR) committee (sometimes known as code committee, code blue committee, or cardiac arrest committee) chair or chief medical/quality officer, to whom the paper‐based survey was addressed, with instructions to forward to the most appropriate person if someone other than the recipient. This study was evaluated by the University of Chicago institutional review board and deemed exempt from further review.
Survey
The survey content was developed by the study investigators and iteratively adapted by consensus and beta testing to require approximately 10 minutes to complete. Questions were edited and formatted by the University of Chicago Survey Lab (Chicago, IL) to be more precise and generalizable. Surveys were mailed in May 2011 and resent twice to nonresponders. A $10 incentive was included in the second mailing. When more than 1 response from a hospital was received, the more complete survey was used, or if equally complete, the responses were combined. All printing, mailing, receipt control, and data entry were performed by the University of Chicago Survey Lab, and data entry was double‐keyed to ensure accuracy.
Response rate was calculated based on the American Association for Public Opinion Research standard response rate formula.[10] It was assumed that the portion of nonresponding cases were ineligible at the same rate of cases for which eligibility was determined. A survey was considered complete if at least 75% of individual questions contained a valid response, partially complete if at least 40% but less than 75% of questions contained a valid response, and a nonresponse if less than 40% was completed. Nonresponses were excluded from the analysis.
Statistical Analysis
Analyses were performed using a statistical software application (Stata version 11.0; StataCorp, College Station, TX). Descriptive statistics were calculated and presented as number (%) or median (interquartile range). A [2] statistic was used to assess bias in response rate. We determined a priori 2 indicators of resource allocation (availability of a CPR committee and dedicated personnel for resuscitation quality improvement) and tested their association with quality improvement initiatives, using logistic regression to adjust for hospital teaching status and number of admissions as potential confounders. All tests of significance used a 2‐sided P<0.05.
RESULTS
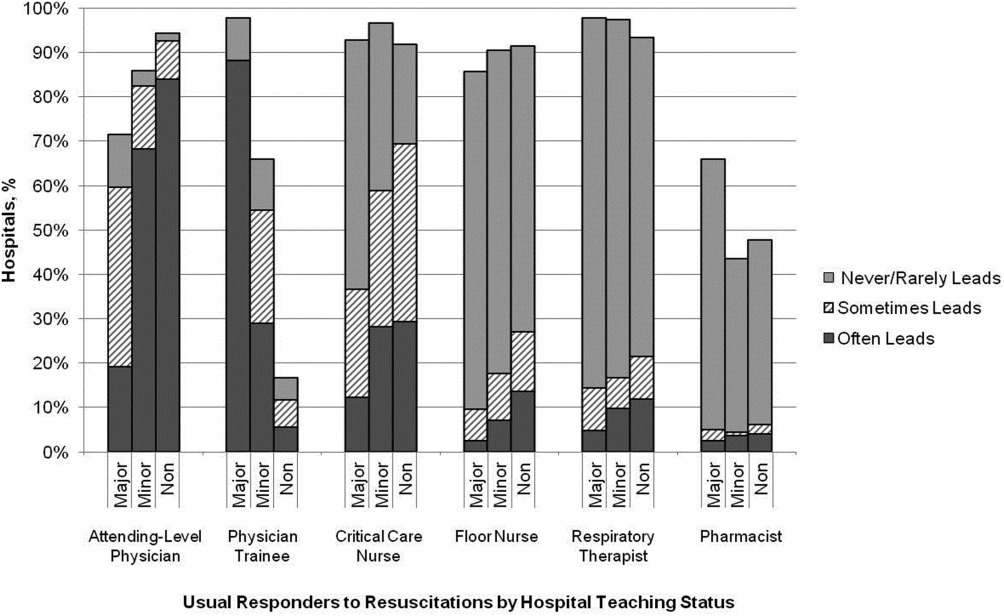
Responses were received from 439 hospitals (425 complete and 14 partially complete), yielding a response rate of 44%. One subject ID was removed from the survey and could not be identified, so it was excluded from any analyses. Hospital demographics were similar between responders and nonresponders (P=0.50) (Table 1). Respondents who filled out the surveys included chief medical/quality officers (n=143 [33%]), chairs of CPR committees (n=64 [15%]), members of CPR committees (n=29 [7%]), chiefs of staff (n=33 [8%]), resuscitation officers/nurses (n=27 [6%]), chief nursing officers (n=13 [3%]), and others (n=131 [30%]).
| Teaching Status | Annual Inpatient Days | Total | ||
|---|---|---|---|---|
| <17,695 | 17,695‐52,500 | >52,500 | ||
| ||||
| Major | 1/2 (50) | 1/8 (13) | 40/82 (49) | 42/92 (46) |
| Minor | 13/39 (33) | 40/89 (45) | 62/133 (47) | 115/261 (44) |
| Nonteaching | 141/293 (48) | 100/236 (42) | 40/118 (34) | 281/647 (43) |
| Total | 156/335 (47) | 143/335 (43) | 145/336 (43) | 438/1,000 (44) |
Table 2 summarizes structure, equipment, quality improvement, and pre‐ and postarrest practices across the hospitals. Of note, 77% of hospitals (n=334) reported having a predesignated, dedicated code team, and 66% (n=281) reported standardized defibrillator make and model throughout their hospital. However, less than one‐third of hospitals utilized any CPR assist technology (eg, CPR quality sensor or mechanical CPR device). The majority of hospitals reported having a rapid response team (RRT) (n=391 [91%]). Although a therapeutic hypothermia protocol for postarrest care was in place in over half of hospitals (n=252 [58%]), utilization of hypothermia for patients with return of spontaneous circulation was infrequent.
| Value | 2010 AHA Guidelines | |
|---|---|---|
| ||
| Structure | ||
| Existing CPR committee | 270 (66) | |
| CPR chair | ||
| Physician only | 129 (48) | |
| Nurse only | 90 (34) | |
| Nurse/physician co‐chair | 31 (12) | |
| Other | 17 (6) | |
| Clinical specialty of chaira | ||
| Pulmonary/critical care | 79 (35) | |
| Emergency medicine | 71 (31) | |
| Anesthesia/critical care | 43 (19) | |
| Cardiology | 38 (17) | |
| Other | 32 (14) | |
| Hospital medicine | 23 (10) | |
| Predetermined cardiac arrest team structure | 334 (77) | |
| Notifications of respondersa | ||
| Hospital‐wide PA system | 406 (93) | |
| Pager/calls to individuals | 230 (53) | |
| Local alarm | 49 (11) | |
| Equipment | ||
| AEDs used as primary defibrillator by location | ||
| High‐acuity inpatient areas | 69 (16) | |
| Low‐acuity inpatient areas | 109 (26) | |
| Outpatient areas | 206 (51) | Class IIb, LOE Cb |
| Public areas | 263 (78) | Class IIb, LOE Cb |
| Defibrillator throughout hospital | ||
| Same brand and model | 281 (66) | |
| Same brand, different models | 93 (22) | |
| Different brands | 54 (13) | |
| CPR assist technology useda | ||
| None | 291 (70) | |
| Capnography | 106 (25) | Class IIb, LOE Cb |
| Mechanical CPR | 25 (6) | Class IIb, LOE B/Cbc |
| Feedback device | 17 (4) | Class IIa, LOE B |
| Quality improvement | ||
| IHCA tracked | 336 (82) | Supportedbd |
| Data reviewed | Supportedbd | |
| Data not tracked/never reviewed | 85 (20) | |
| Intermittently | 53 (12) | |
| Routinely | 287 (68) | |
| Routine cardiac arrest case reviews/debriefing | 149 (34) | Class IIa, LOE C |
| Dedicated staff to resuscitation QI | 196 (49) | |
| Full‐time equivalent staffing, median (IQR) | 0.5 (0.251.2) | |
| Routine simulated resuscitation training | 268 (62) | |
| Pre‐ and postarrest measures | ||
| Hospitals with RRT | 391 (91) | Class I, LOE Cb |
| Formal RRT‐specific training | ||
| Never | 50 (14) | |
| Once | 110 (30) | |
| Recurrent | 163 (45) | |
| TH protocol/order set in place | 252 (58) | |
| Percent of patients with ROSC receiving TH | Class IIb, LOE Bb | |
| <5% | 309 (74) | |
| 5%25% | 68 (16) | |
| 26%50% | 11 (3) | |
| 51%75% | 10 (2) | |
| >75% | 18 (4) | |
Hospitals reported that routine responders to IHCA events included respiratory therapists (n=414 [95%]), critical care nurses (n=406 [93%]), floor nurses (n=396 [90%]), attending physicians (n=392 [89%]), physician trainees (n=162 [37%]), and pharmacists (n=210 [48%]). Figure 1 shows the distribution of responders and team leaders by hospital type. Of the nonteaching hospitals, attending‐level physicians were likely to respond at 94% (265/281) and routinely lead the resuscitations at 84% (236/281), whereas, of major teaching hospitals, attending physicians were only likely to respond at 71% (30/42) and routinely lead at 19% (8/42).
Two‐thirds of the hospitals had a CPR committee (n=270 [66%]), and 196 (49%) had some staff time dedicated to resuscitation quality improvement. Hospitals with a specific committee dedicated to resuscitation and/or dedicated staff for resuscitation quality improvement were more likely to routinely track cardiac arrest data (odds ratio [OR]: 3.64, 95% confidence interval [CI]: 2.056.47 and OR: 2.02, 95% CI: 1.16‐3.54, respectively) and review the data (OR: 2.67, 95% CI: 1.45‐4.92 and OR: 2.18, 95% CI: 1.22‐3.89, respectively), after adjusting for teaching status and hospital size. These hospitals were also more likely to engage in simulation training and debriefing (Table 3).
| CPR Committee, n=406 | Dedicated QI Staff, n=398 | |
|---|---|---|
| ||
| IHCA tracking | 3.64 (2.056.47) | 2.02 (1.16‐3.54) |
| Routinely review | 2.67 (1.45‐4.92) | 2.18 (1.22‐3.89) |
| Simulation training | 2.63 (1.66‐4.18) | 1.89 (1.24‐2.89) |
| Debriefing | 3.19 (1.89‐5.36) | 2.14 (1.39‐3.32) |
Ninety percent (n=391) of respondents agreed that there is room for improvement in resuscitation practice at my hospital, and 70% (n=302) agreed that improved resuscitation would translate into improved patient outcomes. Overall, 78% (n=338) cited at least 1 barrier to improved resuscitation quality, of which the lack of adequate training (n=233 [54%]) and the lack of an appropriate champion (n=230 [53%]) were the most common. In subgroup analysis, nonteaching hospitals were significantly more likely to report the lack of a champion than their teaching counterparts (P=0.001) (Figure 2). In addition, we analyzed the data by hospitals that reported lack of a champion was not a barrier and compared them to those for whom it was, and found significantly higher adherence across all the measures in Table 2 supported by the 2010 guidelines, with the exception of real‐time feedback (data not shown).
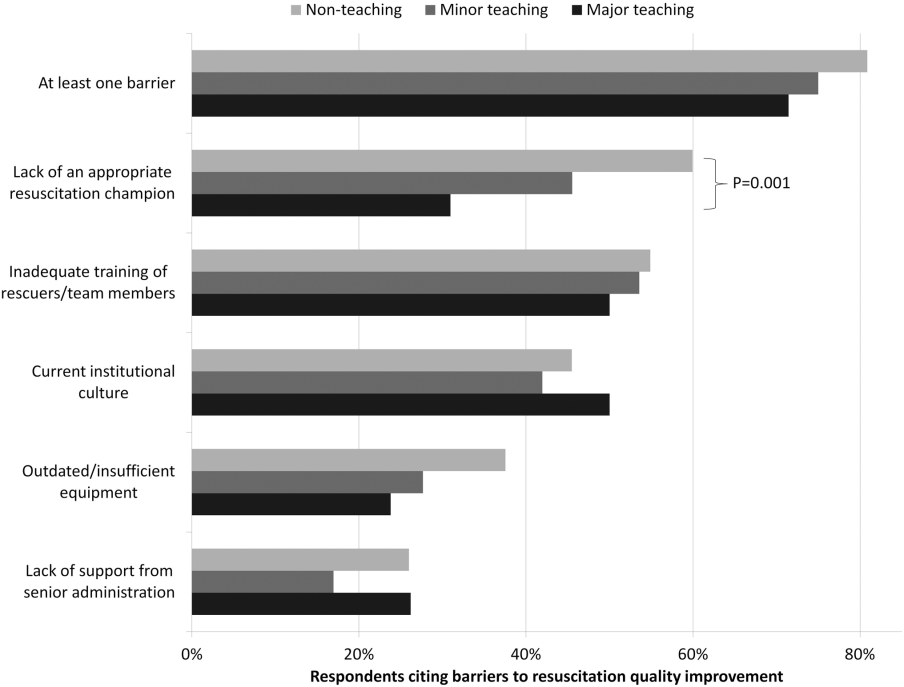
DISCUSSION
In this nationally representative sample of hospitals, we found considerable variability in cardiac arrest and resuscitation structures and processes, suggesting potential areas to target for improvement. Some practices, including use of RRTs and defibrillator standardization, were fairly routine, whereas others, such as therapeutic hypothermia and CPR assist technology, were rarely utilized. Quality initiatives, such as data tracking and review, simulation training, and debriefing were variable.
Several factors likely contribute to the variable implementation of evidence‐based practices. Guidelines alone have been shown to have little impact on practice by physicians in general.[11] This is supported by the lack of correlation we found between the presence, absence or strength of specific American Heart Association (AHA) emergency cardiovascular care treatment recommendations and the percent of hospitals reporting performing that measure. It is possible that other factors, such as a lack of familiarity or agreement with those guidelines, or the presence of external barriers, may be contributing.[12, 13] Specifically, the importance of a clinical champion was supported by our finding that hospitals reporting lack of a champion as a barrier were less likely to be adherent with guidelines. However, because the study did not directly test the impact of a champion, we wanted to be careful to avoid overstating or editorializing our results.
Some of the variability may also be related to the resource intensiveness of the practice. Routine simulation training and debriefing interventions, for example, are time intensive and require trained personnel to institute. That may explain the correlation we noted between these practices and the presence of CPR committee and dedicated personnel. The use of dedicated personnel was rare in this study, with less than half of respondents reporting any dedicated staff and a median of 0.5 full‐time equivalents for those reporting positively. This is in stark contrast to the routine use of resuscitation officers (primarily nurses dedicated to overseeing resuscitation practices and education at the hospital) in the United Kingdom.[14] Such a resuscitation officer model adopted by US hospitals could improve the quality and intensity of resuscitation care approaches.
Particularly surprising was the high rate of respondents (70%) reporting that they do not utilize any CPR assist technology. In the patient who does not have an arterial line, use of quantitative capnography is the best measure of cardiac output during cardiac arrest, yet only one‐quarter of hospitals reported using it, with no discrepancy between hospital type or size. A recent summit of national resuscitation experts expounded on the AHA guidelines suggesting that end‐tidal carbon dioxide should be used in all arrests to guide the quality of CPR with a goal value of >20.[8] Similarly, CPR feedback devices have an even higher level of evidence recommendation in the 2010 AHA guidelines than capnography, yet only 4% of hospitals reported utilizing them. Although it is true that introducing these CPR assist technologies into a hospital would require some effort on the part of hospital leadership, it is important to recognize the potential role such devices might play in the larger context of a resuscitation quality program to optimize clinical outcomes from IHCA.
Several differences were noted between hospitals based on teaching status. Although all hospitals were more likely to rely on physicians to lead resuscitations, nonteaching hospitals were more likely to report routine leadership by nurses and pharmacists. Nonteaching hospitals were also less likely to have a CPR committee, even after adjusting for hospital size. In addition, these hospitals were also more likely to report the lack of a clinical champion as a barrier to quality improvement.
There were several limitations to this study. First, this was a descriptive survey that was not tied to outcomes. As such, we are unable to draw conclusions about which practices correlate with decreased incidence of cardiac arrest and improved survival. Second, this was an optional survey with a somewhat limited response rate. Even though the characteristics of the nonresponding hospitals were similar to the responding hospitals, we cannot rule out the possibility that a selection bias was introduced, which would likely overestimate adherence to the guidelines. Self‐reported responses may have introduced additional errors. Finally, the short interval between the release of the 2010 guidelines and the administration of the first survey may have contributed to the variability in implementation of some practices, but many of the recommendations had been previously included in the 2005 guidelines.
We conclude that there is wide variability between hospitals and within practices for resuscitation care. Future work should seek to understand which practices are associated with improved patient outcomes and how best to implement these practices in a more uniform fashion.
Acknowledgements
The authors thank Nancy Hinckley, who championed the study; David Chearo, Christelle Marpaud, and Martha Van Haitsma of the University of Chicago Survey Lab for their assistance in formulating and distributing the survey; and JoAnne Resnic, Nicole Twu, and Frank Zadravecz for administrative support.
Disclosures: This study was supported by the Society of Hospital Medicine with a grant from Philips Healthcare (Andover, MA). Dr. Edelson is supported by a career development award from the National Heart, Lung, and Blood Institute (K23 HL097157). In addition, she has received research support and honoraria from Philips Healthcare (Andover, MA), research support from the American Heart Association (Dallas, TX) and Laerdal Medical (Stavanger, Norway), and an honorarium from Early Sense (Tel Aviv, Israel). Dr. Hunt has received research support from the Laerdal Foundation for Acute Medicine (Stavanger, Norway), the Hartwell Foundation (Memphis, TN), and the Arthur Vining Davis Foundation (Jacksonville, FL), and honoraria from the Kansas University Endowment (Kansas City, KS), JCCC (Overland Park, KS), and the UVA School of Medicine (Charlottesville, VA) and the European School of Management (Berlin, Germany). Dr. Mancini is supported in part by an Agency for Healthcare Research and Quality grant (R18HS020416). In addition, she has received research support from the American Heart Association (Dallas, TX) and Laerdal Medical (Stavanger, Norway), and honoraria from Sotera Wireless, Inc. (San Diego, CA). Dr. Abella has received research support from the National Institutes of Health (NIH), Medtronic Foundation (Minneapolis, MN), and Philips Healthcare (Andover, MA); has volunteered with the American Heart Association; and received honoraria from Heartsine (Belfast, Ireland), Velomedix (Menlo Park, CA), and Stryker (Kalamazoo, MI). Mr. Miller is employed by the Society of Hospital Medicine.
- , , , , , . Trends in survival after in‐hospital cardiac arrest. N Engl J Med. 2012;367(20):1912–1920.
- , , , et al. Incidence of treated cardiac arrest in hospitalized patients in the United States. Crit Care Med. 2011;39(11):2401–2406.
- , , , et al. Racial differences in survival after in‐hospital cardiac arrest. JAMA. 2009;302(11):1195–1201.
- , , , , . Hospital variation in time to defibrillation after in‐hospital cardiac arrest. Arch Intern Med. 2009;169(14):1265–1273.
- , , , et al. Duration of resuscitation efforts and survival after in‐hospital cardiac arrest: an observational study. Lancet. 2012;380(9852):1473–1481.
- , , , . Delayed time to defibrillation after in‐hospital cardiac arrest. N Engl J Med. 2008;358(1):9–17.
- 2010 American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care science. Circulation. 2010;122(18 suppl 3):S640–S946.
- , , , et al. Cardiopulmonary resuscitation quality: improving cardiac resuscitation outcomes both inside and outside the hospital: a consensus statement from the American Heart Association. Circulation. 2013;128(4):417–435.
- American Hospital Association. 2008 AHA annual survey. AHA data viewer: survey instruments. 2012; Available at: http://www.ahadataviewer.com/about/hospital‐database. Accessed October 11, 2013.
- The American Association for Public Opinion Research. Standard Definitions: Final Dispositions of Case Codes and Outcome Rates for Surveys. 7th ed. Deerfield, IL: AAPOR; 2011.
- , , , , , . Do practice guidelines guide practice? The effect of a consensus statement on the practice of physicians. N Engl J Med. 1989;321(19):1306–1311.
- , , , et al. Why don't physicians follow clinical practice guidelines? A framework for improvement. JAMA. 1999;282(15):1458–1465.
- , . From best evidence to best practice: effective implementation of change in patients' care. Lancet. 2003;362(9391):1225–1230.
- , , , et al. Cardiopulmonary resuscitation standards for clinical practice and training in the UK. Accid Emerg Nurs. 2005;13(3):171–179.
An estimated 200,000 adult patients suffer cardiac arrest in US hospitals each year, of which <20% survive to hospital discharge.[1, 2] Patient survival from in‐hospital cardiac arrest (IHCA), however, varies widely across hospitals, and may be partly attributed to differences in hospital practices.[3, 4, 5] Although there are data to support specific patient‐level practices in the hospital, such as delivery of electrical shock for ventricular fibrillation within 2 minutes of onset of the lethal rhythm,[6] little is known about in‐hospital systems‐level factors. Similar to patient‐level practices, some organizational and systems level practices are supported by international consensus and guideline recommendations.[7, 8] However, the adoption of these practices is poorly understood. As such, we sought to gain a better understanding of current US hospital practices with regard to IHCA and resuscitation with the hopes of identifying potential targets for improvement in quality and outcomes.
METHODS
We conducted a nationally representative mail survey between May 2011 and November 2011, targeting a stratified random sample of 1000 hospitals. We utilized the US Acute‐Care Hospitals (FY2008) database from the American Hospital Association to determine the total population of 3809 community hospitals (ie, nonfederal government, nonpsychiatric, and nonlong‐term care hospitals).[9] This included general medical and surgical, surgical, cancer, heart, orthopedic, and children's hospitals. These hospitals were stratified into tertiles by annual in‐patient days and teaching status (major, minor, nonteaching), from which our sample was randomly selected (Table 1). We identified each hospital's cardiopulmonary resuscitation (CPR) committee (sometimes known as code committee, code blue committee, or cardiac arrest committee) chair or chief medical/quality officer, to whom the paper‐based survey was addressed, with instructions to forward to the most appropriate person if someone other than the recipient. This study was evaluated by the University of Chicago institutional review board and deemed exempt from further review.
Survey
The survey content was developed by the study investigators and iteratively adapted by consensus and beta testing to require approximately 10 minutes to complete. Questions were edited and formatted by the University of Chicago Survey Lab (Chicago, IL) to be more precise and generalizable. Surveys were mailed in May 2011 and resent twice to nonresponders. A $10 incentive was included in the second mailing. When more than 1 response from a hospital was received, the more complete survey was used, or if equally complete, the responses were combined. All printing, mailing, receipt control, and data entry were performed by the University of Chicago Survey Lab, and data entry was double‐keyed to ensure accuracy.
Response rate was calculated based on the American Association for Public Opinion Research standard response rate formula.[10] It was assumed that the portion of nonresponding cases were ineligible at the same rate of cases for which eligibility was determined. A survey was considered complete if at least 75% of individual questions contained a valid response, partially complete if at least 40% but less than 75% of questions contained a valid response, and a nonresponse if less than 40% was completed. Nonresponses were excluded from the analysis.
Statistical Analysis
Analyses were performed using a statistical software application (Stata version 11.0; StataCorp, College Station, TX). Descriptive statistics were calculated and presented as number (%) or median (interquartile range). A [2] statistic was used to assess bias in response rate. We determined a priori 2 indicators of resource allocation (availability of a CPR committee and dedicated personnel for resuscitation quality improvement) and tested their association with quality improvement initiatives, using logistic regression to adjust for hospital teaching status and number of admissions as potential confounders. All tests of significance used a 2‐sided P<0.05.
RESULTS
Responses were received from 439 hospitals (425 complete and 14 partially complete), yielding a response rate of 44%. One subject ID was removed from the survey and could not be identified, so it was excluded from any analyses. Hospital demographics were similar between responders and nonresponders (P=0.50) (Table 1). Respondents who filled out the surveys included chief medical/quality officers (n=143 [33%]), chairs of CPR committees (n=64 [15%]), members of CPR committees (n=29 [7%]), chiefs of staff (n=33 [8%]), resuscitation officers/nurses (n=27 [6%]), chief nursing officers (n=13 [3%]), and others (n=131 [30%]).
| Teaching Status | Annual Inpatient Days | Total | ||
|---|---|---|---|---|
| <17,695 | 17,695‐52,500 | >52,500 | ||
| ||||
| Major | 1/2 (50) | 1/8 (13) | 40/82 (49) | 42/92 (46) |
| Minor | 13/39 (33) | 40/89 (45) | 62/133 (47) | 115/261 (44) |
| Nonteaching | 141/293 (48) | 100/236 (42) | 40/118 (34) | 281/647 (43) |
| Total | 156/335 (47) | 143/335 (43) | 145/336 (43) | 438/1,000 (44) |
Table 2 summarizes structure, equipment, quality improvement, and pre‐ and postarrest practices across the hospitals. Of note, 77% of hospitals (n=334) reported having a predesignated, dedicated code team, and 66% (n=281) reported standardized defibrillator make and model throughout their hospital. However, less than one‐third of hospitals utilized any CPR assist technology (eg, CPR quality sensor or mechanical CPR device). The majority of hospitals reported having a rapid response team (RRT) (n=391 [91%]). Although a therapeutic hypothermia protocol for postarrest care was in place in over half of hospitals (n=252 [58%]), utilization of hypothermia for patients with return of spontaneous circulation was infrequent.
| Value | 2010 AHA Guidelines | |
|---|---|---|
| ||
| Structure | ||
| Existing CPR committee | 270 (66) | |
| CPR chair | ||
| Physician only | 129 (48) | |
| Nurse only | 90 (34) | |
| Nurse/physician co‐chair | 31 (12) | |
| Other | 17 (6) | |
| Clinical specialty of chaira | ||
| Pulmonary/critical care | 79 (35) | |
| Emergency medicine | 71 (31) | |
| Anesthesia/critical care | 43 (19) | |
| Cardiology | 38 (17) | |
| Other | 32 (14) | |
| Hospital medicine | 23 (10) | |
| Predetermined cardiac arrest team structure | 334 (77) | |
| Notifications of respondersa | ||
| Hospital‐wide PA system | 406 (93) | |
| Pager/calls to individuals | 230 (53) | |
| Local alarm | 49 (11) | |
| Equipment | ||
| AEDs used as primary defibrillator by location | ||
| High‐acuity inpatient areas | 69 (16) | |
| Low‐acuity inpatient areas | 109 (26) | |
| Outpatient areas | 206 (51) | Class IIb, LOE Cb |
| Public areas | 263 (78) | Class IIb, LOE Cb |
| Defibrillator throughout hospital | ||
| Same brand and model | 281 (66) | |
| Same brand, different models | 93 (22) | |
| Different brands | 54 (13) | |
| CPR assist technology useda | ||
| None | 291 (70) | |
| Capnography | 106 (25) | Class IIb, LOE Cb |
| Mechanical CPR | 25 (6) | Class IIb, LOE B/Cbc |
| Feedback device | 17 (4) | Class IIa, LOE B |
| Quality improvement | ||
| IHCA tracked | 336 (82) | Supportedbd |
| Data reviewed | Supportedbd | |
| Data not tracked/never reviewed | 85 (20) | |
| Intermittently | 53 (12) | |
| Routinely | 287 (68) | |
| Routine cardiac arrest case reviews/debriefing | 149 (34) | Class IIa, LOE C |
| Dedicated staff to resuscitation QI | 196 (49) | |
| Full‐time equivalent staffing, median (IQR) | 0.5 (0.251.2) | |
| Routine simulated resuscitation training | 268 (62) | |
| Pre‐ and postarrest measures | ||
| Hospitals with RRT | 391 (91) | Class I, LOE Cb |
| Formal RRT‐specific training | ||
| Never | 50 (14) | |
| Once | 110 (30) | |
| Recurrent | 163 (45) | |
| TH protocol/order set in place | 252 (58) | |
| Percent of patients with ROSC receiving TH | Class IIb, LOE Bb | |
| <5% | 309 (74) | |
| 5%25% | 68 (16) | |
| 26%50% | 11 (3) | |
| 51%75% | 10 (2) | |
| >75% | 18 (4) | |
Hospitals reported that routine responders to IHCA events included respiratory therapists (n=414 [95%]), critical care nurses (n=406 [93%]), floor nurses (n=396 [90%]), attending physicians (n=392 [89%]), physician trainees (n=162 [37%]), and pharmacists (n=210 [48%]). Figure 1 shows the distribution of responders and team leaders by hospital type. Of the nonteaching hospitals, attending‐level physicians were likely to respond at 94% (265/281) and routinely lead the resuscitations at 84% (236/281), whereas, of major teaching hospitals, attending physicians were only likely to respond at 71% (30/42) and routinely lead at 19% (8/42).
Two‐thirds of the hospitals had a CPR committee (n=270 [66%]), and 196 (49%) had some staff time dedicated to resuscitation quality improvement. Hospitals with a specific committee dedicated to resuscitation and/or dedicated staff for resuscitation quality improvement were more likely to routinely track cardiac arrest data (odds ratio [OR]: 3.64, 95% confidence interval [CI]: 2.056.47 and OR: 2.02, 95% CI: 1.16‐3.54, respectively) and review the data (OR: 2.67, 95% CI: 1.45‐4.92 and OR: 2.18, 95% CI: 1.22‐3.89, respectively), after adjusting for teaching status and hospital size. These hospitals were also more likely to engage in simulation training and debriefing (Table 3).
| CPR Committee, n=406 | Dedicated QI Staff, n=398 | |
|---|---|---|
| ||
| IHCA tracking | 3.64 (2.056.47) | 2.02 (1.16‐3.54) |
| Routinely review | 2.67 (1.45‐4.92) | 2.18 (1.22‐3.89) |
| Simulation training | 2.63 (1.66‐4.18) | 1.89 (1.24‐2.89) |
| Debriefing | 3.19 (1.89‐5.36) | 2.14 (1.39‐3.32) |
Ninety percent (n=391) of respondents agreed that there is room for improvement in resuscitation practice at my hospital, and 70% (n=302) agreed that improved resuscitation would translate into improved patient outcomes. Overall, 78% (n=338) cited at least 1 barrier to improved resuscitation quality, of which the lack of adequate training (n=233 [54%]) and the lack of an appropriate champion (n=230 [53%]) were the most common. In subgroup analysis, nonteaching hospitals were significantly more likely to report the lack of a champion than their teaching counterparts (P=0.001) (Figure 2). In addition, we analyzed the data by hospitals that reported lack of a champion was not a barrier and compared them to those for whom it was, and found significantly higher adherence across all the measures in Table 2 supported by the 2010 guidelines, with the exception of real‐time feedback (data not shown).
DISCUSSION
In this nationally representative sample of hospitals, we found considerable variability in cardiac arrest and resuscitation structures and processes, suggesting potential areas to target for improvement. Some practices, including use of RRTs and defibrillator standardization, were fairly routine, whereas others, such as therapeutic hypothermia and CPR assist technology, were rarely utilized. Quality initiatives, such as data tracking and review, simulation training, and debriefing were variable.
Several factors likely contribute to the variable implementation of evidence‐based practices. Guidelines alone have been shown to have little impact on practice by physicians in general.[11] This is supported by the lack of correlation we found between the presence, absence or strength of specific American Heart Association (AHA) emergency cardiovascular care treatment recommendations and the percent of hospitals reporting performing that measure. It is possible that other factors, such as a lack of familiarity or agreement with those guidelines, or the presence of external barriers, may be contributing.[12, 13] Specifically, the importance of a clinical champion was supported by our finding that hospitals reporting lack of a champion as a barrier were less likely to be adherent with guidelines. However, because the study did not directly test the impact of a champion, we wanted to be careful to avoid overstating or editorializing our results.
Some of the variability may also be related to the resource intensiveness of the practice. Routine simulation training and debriefing interventions, for example, are time intensive and require trained personnel to institute. That may explain the correlation we noted between these practices and the presence of CPR committee and dedicated personnel. The use of dedicated personnel was rare in this study, with less than half of respondents reporting any dedicated staff and a median of 0.5 full‐time equivalents for those reporting positively. This is in stark contrast to the routine use of resuscitation officers (primarily nurses dedicated to overseeing resuscitation practices and education at the hospital) in the United Kingdom.[14] Such a resuscitation officer model adopted by US hospitals could improve the quality and intensity of resuscitation care approaches.
Particularly surprising was the high rate of respondents (70%) reporting that they do not utilize any CPR assist technology. In the patient who does not have an arterial line, use of quantitative capnography is the best measure of cardiac output during cardiac arrest, yet only one‐quarter of hospitals reported using it, with no discrepancy between hospital type or size. A recent summit of national resuscitation experts expounded on the AHA guidelines suggesting that end‐tidal carbon dioxide should be used in all arrests to guide the quality of CPR with a goal value of >20.[8] Similarly, CPR feedback devices have an even higher level of evidence recommendation in the 2010 AHA guidelines than capnography, yet only 4% of hospitals reported utilizing them. Although it is true that introducing these CPR assist technologies into a hospital would require some effort on the part of hospital leadership, it is important to recognize the potential role such devices might play in the larger context of a resuscitation quality program to optimize clinical outcomes from IHCA.
Several differences were noted between hospitals based on teaching status. Although all hospitals were more likely to rely on physicians to lead resuscitations, nonteaching hospitals were more likely to report routine leadership by nurses and pharmacists. Nonteaching hospitals were also less likely to have a CPR committee, even after adjusting for hospital size. In addition, these hospitals were also more likely to report the lack of a clinical champion as a barrier to quality improvement.
There were several limitations to this study. First, this was a descriptive survey that was not tied to outcomes. As such, we are unable to draw conclusions about which practices correlate with decreased incidence of cardiac arrest and improved survival. Second, this was an optional survey with a somewhat limited response rate. Even though the characteristics of the nonresponding hospitals were similar to the responding hospitals, we cannot rule out the possibility that a selection bias was introduced, which would likely overestimate adherence to the guidelines. Self‐reported responses may have introduced additional errors. Finally, the short interval between the release of the 2010 guidelines and the administration of the first survey may have contributed to the variability in implementation of some practices, but many of the recommendations had been previously included in the 2005 guidelines.
We conclude that there is wide variability between hospitals and within practices for resuscitation care. Future work should seek to understand which practices are associated with improved patient outcomes and how best to implement these practices in a more uniform fashion.
Acknowledgements
The authors thank Nancy Hinckley, who championed the study; David Chearo, Christelle Marpaud, and Martha Van Haitsma of the University of Chicago Survey Lab for their assistance in formulating and distributing the survey; and JoAnne Resnic, Nicole Twu, and Frank Zadravecz for administrative support.
Disclosures: This study was supported by the Society of Hospital Medicine with a grant from Philips Healthcare (Andover, MA). Dr. Edelson is supported by a career development award from the National Heart, Lung, and Blood Institute (K23 HL097157). In addition, she has received research support and honoraria from Philips Healthcare (Andover, MA), research support from the American Heart Association (Dallas, TX) and Laerdal Medical (Stavanger, Norway), and an honorarium from Early Sense (Tel Aviv, Israel). Dr. Hunt has received research support from the Laerdal Foundation for Acute Medicine (Stavanger, Norway), the Hartwell Foundation (Memphis, TN), and the Arthur Vining Davis Foundation (Jacksonville, FL), and honoraria from the Kansas University Endowment (Kansas City, KS), JCCC (Overland Park, KS), and the UVA School of Medicine (Charlottesville, VA) and the European School of Management (Berlin, Germany). Dr. Mancini is supported in part by an Agency for Healthcare Research and Quality grant (R18HS020416). In addition, she has received research support from the American Heart Association (Dallas, TX) and Laerdal Medical (Stavanger, Norway), and honoraria from Sotera Wireless, Inc. (San Diego, CA). Dr. Abella has received research support from the National Institutes of Health (NIH), Medtronic Foundation (Minneapolis, MN), and Philips Healthcare (Andover, MA); has volunteered with the American Heart Association; and received honoraria from Heartsine (Belfast, Ireland), Velomedix (Menlo Park, CA), and Stryker (Kalamazoo, MI). Mr. Miller is employed by the Society of Hospital Medicine.
An estimated 200,000 adult patients suffer cardiac arrest in US hospitals each year, of which <20% survive to hospital discharge.[1, 2] Patient survival from in‐hospital cardiac arrest (IHCA), however, varies widely across hospitals, and may be partly attributed to differences in hospital practices.[3, 4, 5] Although there are data to support specific patient‐level practices in the hospital, such as delivery of electrical shock for ventricular fibrillation within 2 minutes of onset of the lethal rhythm,[6] little is known about in‐hospital systems‐level factors. Similar to patient‐level practices, some organizational and systems level practices are supported by international consensus and guideline recommendations.[7, 8] However, the adoption of these practices is poorly understood. As such, we sought to gain a better understanding of current US hospital practices with regard to IHCA and resuscitation with the hopes of identifying potential targets for improvement in quality and outcomes.
METHODS
We conducted a nationally representative mail survey between May 2011 and November 2011, targeting a stratified random sample of 1000 hospitals. We utilized the US Acute‐Care Hospitals (FY2008) database from the American Hospital Association to determine the total population of 3809 community hospitals (ie, nonfederal government, nonpsychiatric, and nonlong‐term care hospitals).[9] This included general medical and surgical, surgical, cancer, heart, orthopedic, and children's hospitals. These hospitals were stratified into tertiles by annual in‐patient days and teaching status (major, minor, nonteaching), from which our sample was randomly selected (Table 1). We identified each hospital's cardiopulmonary resuscitation (CPR) committee (sometimes known as code committee, code blue committee, or cardiac arrest committee) chair or chief medical/quality officer, to whom the paper‐based survey was addressed, with instructions to forward to the most appropriate person if someone other than the recipient. This study was evaluated by the University of Chicago institutional review board and deemed exempt from further review.
Survey
The survey content was developed by the study investigators and iteratively adapted by consensus and beta testing to require approximately 10 minutes to complete. Questions were edited and formatted by the University of Chicago Survey Lab (Chicago, IL) to be more precise and generalizable. Surveys were mailed in May 2011 and resent twice to nonresponders. A $10 incentive was included in the second mailing. When more than 1 response from a hospital was received, the more complete survey was used, or if equally complete, the responses were combined. All printing, mailing, receipt control, and data entry were performed by the University of Chicago Survey Lab, and data entry was double‐keyed to ensure accuracy.
Response rate was calculated based on the American Association for Public Opinion Research standard response rate formula.[10] It was assumed that the portion of nonresponding cases were ineligible at the same rate of cases for which eligibility was determined. A survey was considered complete if at least 75% of individual questions contained a valid response, partially complete if at least 40% but less than 75% of questions contained a valid response, and a nonresponse if less than 40% was completed. Nonresponses were excluded from the analysis.
Statistical Analysis
Analyses were performed using a statistical software application (Stata version 11.0; StataCorp, College Station, TX). Descriptive statistics were calculated and presented as number (%) or median (interquartile range). A [2] statistic was used to assess bias in response rate. We determined a priori 2 indicators of resource allocation (availability of a CPR committee and dedicated personnel for resuscitation quality improvement) and tested their association with quality improvement initiatives, using logistic regression to adjust for hospital teaching status and number of admissions as potential confounders. All tests of significance used a 2‐sided P<0.05.
RESULTS
Responses were received from 439 hospitals (425 complete and 14 partially complete), yielding a response rate of 44%. One subject ID was removed from the survey and could not be identified, so it was excluded from any analyses. Hospital demographics were similar between responders and nonresponders (P=0.50) (Table 1). Respondents who filled out the surveys included chief medical/quality officers (n=143 [33%]), chairs of CPR committees (n=64 [15%]), members of CPR committees (n=29 [7%]), chiefs of staff (n=33 [8%]), resuscitation officers/nurses (n=27 [6%]), chief nursing officers (n=13 [3%]), and others (n=131 [30%]).
| Teaching Status | Annual Inpatient Days | Total | ||
|---|---|---|---|---|
| <17,695 | 17,695‐52,500 | >52,500 | ||
| ||||
| Major | 1/2 (50) | 1/8 (13) | 40/82 (49) | 42/92 (46) |
| Minor | 13/39 (33) | 40/89 (45) | 62/133 (47) | 115/261 (44) |
| Nonteaching | 141/293 (48) | 100/236 (42) | 40/118 (34) | 281/647 (43) |
| Total | 156/335 (47) | 143/335 (43) | 145/336 (43) | 438/1,000 (44) |
Table 2 summarizes structure, equipment, quality improvement, and pre‐ and postarrest practices across the hospitals. Of note, 77% of hospitals (n=334) reported having a predesignated, dedicated code team, and 66% (n=281) reported standardized defibrillator make and model throughout their hospital. However, less than one‐third of hospitals utilized any CPR assist technology (eg, CPR quality sensor or mechanical CPR device). The majority of hospitals reported having a rapid response team (RRT) (n=391 [91%]). Although a therapeutic hypothermia protocol for postarrest care was in place in over half of hospitals (n=252 [58%]), utilization of hypothermia for patients with return of spontaneous circulation was infrequent.
| Value | 2010 AHA Guidelines | |
|---|---|---|
| ||
| Structure | ||
| Existing CPR committee | 270 (66) | |
| CPR chair | ||
| Physician only | 129 (48) | |
| Nurse only | 90 (34) | |
| Nurse/physician co‐chair | 31 (12) | |
| Other | 17 (6) | |
| Clinical specialty of chaira | ||
| Pulmonary/critical care | 79 (35) | |
| Emergency medicine | 71 (31) | |
| Anesthesia/critical care | 43 (19) | |
| Cardiology | 38 (17) | |
| Other | 32 (14) | |
| Hospital medicine | 23 (10) | |
| Predetermined cardiac arrest team structure | 334 (77) | |
| Notifications of respondersa | ||
| Hospital‐wide PA system | 406 (93) | |
| Pager/calls to individuals | 230 (53) | |
| Local alarm | 49 (11) | |
| Equipment | ||
| AEDs used as primary defibrillator by location | ||
| High‐acuity inpatient areas | 69 (16) | |
| Low‐acuity inpatient areas | 109 (26) | |
| Outpatient areas | 206 (51) | Class IIb, LOE Cb |
| Public areas | 263 (78) | Class IIb, LOE Cb |
| Defibrillator throughout hospital | ||
| Same brand and model | 281 (66) | |
| Same brand, different models | 93 (22) | |
| Different brands | 54 (13) | |
| CPR assist technology useda | ||
| None | 291 (70) | |
| Capnography | 106 (25) | Class IIb, LOE Cb |
| Mechanical CPR | 25 (6) | Class IIb, LOE B/Cbc |
| Feedback device | 17 (4) | Class IIa, LOE B |
| Quality improvement | ||
| IHCA tracked | 336 (82) | Supportedbd |
| Data reviewed | Supportedbd | |
| Data not tracked/never reviewed | 85 (20) | |
| Intermittently | 53 (12) | |
| Routinely | 287 (68) | |
| Routine cardiac arrest case reviews/debriefing | 149 (34) | Class IIa, LOE C |
| Dedicated staff to resuscitation QI | 196 (49) | |
| Full‐time equivalent staffing, median (IQR) | 0.5 (0.251.2) | |
| Routine simulated resuscitation training | 268 (62) | |
| Pre‐ and postarrest measures | ||
| Hospitals with RRT | 391 (91) | Class I, LOE Cb |
| Formal RRT‐specific training | ||
| Never | 50 (14) | |
| Once | 110 (30) | |
| Recurrent | 163 (45) | |
| TH protocol/order set in place | 252 (58) | |
| Percent of patients with ROSC receiving TH | Class IIb, LOE Bb | |
| <5% | 309 (74) | |
| 5%25% | 68 (16) | |
| 26%50% | 11 (3) | |
| 51%75% | 10 (2) | |
| >75% | 18 (4) | |
Hospitals reported that routine responders to IHCA events included respiratory therapists (n=414 [95%]), critical care nurses (n=406 [93%]), floor nurses (n=396 [90%]), attending physicians (n=392 [89%]), physician trainees (n=162 [37%]), and pharmacists (n=210 [48%]). Figure 1 shows the distribution of responders and team leaders by hospital type. Of the nonteaching hospitals, attending‐level physicians were likely to respond at 94% (265/281) and routinely lead the resuscitations at 84% (236/281), whereas, of major teaching hospitals, attending physicians were only likely to respond at 71% (30/42) and routinely lead at 19% (8/42).
Two‐thirds of the hospitals had a CPR committee (n=270 [66%]), and 196 (49%) had some staff time dedicated to resuscitation quality improvement. Hospitals with a specific committee dedicated to resuscitation and/or dedicated staff for resuscitation quality improvement were more likely to routinely track cardiac arrest data (odds ratio [OR]: 3.64, 95% confidence interval [CI]: 2.056.47 and OR: 2.02, 95% CI: 1.16‐3.54, respectively) and review the data (OR: 2.67, 95% CI: 1.45‐4.92 and OR: 2.18, 95% CI: 1.22‐3.89, respectively), after adjusting for teaching status and hospital size. These hospitals were also more likely to engage in simulation training and debriefing (Table 3).
| CPR Committee, n=406 | Dedicated QI Staff, n=398 | |
|---|---|---|
| ||
| IHCA tracking | 3.64 (2.056.47) | 2.02 (1.16‐3.54) |
| Routinely review | 2.67 (1.45‐4.92) | 2.18 (1.22‐3.89) |
| Simulation training | 2.63 (1.66‐4.18) | 1.89 (1.24‐2.89) |
| Debriefing | 3.19 (1.89‐5.36) | 2.14 (1.39‐3.32) |
Ninety percent (n=391) of respondents agreed that there is room for improvement in resuscitation practice at my hospital, and 70% (n=302) agreed that improved resuscitation would translate into improved patient outcomes. Overall, 78% (n=338) cited at least 1 barrier to improved resuscitation quality, of which the lack of adequate training (n=233 [54%]) and the lack of an appropriate champion (n=230 [53%]) were the most common. In subgroup analysis, nonteaching hospitals were significantly more likely to report the lack of a champion than their teaching counterparts (P=0.001) (Figure 2). In addition, we analyzed the data by hospitals that reported lack of a champion was not a barrier and compared them to those for whom it was, and found significantly higher adherence across all the measures in Table 2 supported by the 2010 guidelines, with the exception of real‐time feedback (data not shown).
DISCUSSION
In this nationally representative sample of hospitals, we found considerable variability in cardiac arrest and resuscitation structures and processes, suggesting potential areas to target for improvement. Some practices, including use of RRTs and defibrillator standardization, were fairly routine, whereas others, such as therapeutic hypothermia and CPR assist technology, were rarely utilized. Quality initiatives, such as data tracking and review, simulation training, and debriefing were variable.
Several factors likely contribute to the variable implementation of evidence‐based practices. Guidelines alone have been shown to have little impact on practice by physicians in general.[11] This is supported by the lack of correlation we found between the presence, absence or strength of specific American Heart Association (AHA) emergency cardiovascular care treatment recommendations and the percent of hospitals reporting performing that measure. It is possible that other factors, such as a lack of familiarity or agreement with those guidelines, or the presence of external barriers, may be contributing.[12, 13] Specifically, the importance of a clinical champion was supported by our finding that hospitals reporting lack of a champion as a barrier were less likely to be adherent with guidelines. However, because the study did not directly test the impact of a champion, we wanted to be careful to avoid overstating or editorializing our results.
Some of the variability may also be related to the resource intensiveness of the practice. Routine simulation training and debriefing interventions, for example, are time intensive and require trained personnel to institute. That may explain the correlation we noted between these practices and the presence of CPR committee and dedicated personnel. The use of dedicated personnel was rare in this study, with less than half of respondents reporting any dedicated staff and a median of 0.5 full‐time equivalents for those reporting positively. This is in stark contrast to the routine use of resuscitation officers (primarily nurses dedicated to overseeing resuscitation practices and education at the hospital) in the United Kingdom.[14] Such a resuscitation officer model adopted by US hospitals could improve the quality and intensity of resuscitation care approaches.
Particularly surprising was the high rate of respondents (70%) reporting that they do not utilize any CPR assist technology. In the patient who does not have an arterial line, use of quantitative capnography is the best measure of cardiac output during cardiac arrest, yet only one‐quarter of hospitals reported using it, with no discrepancy between hospital type or size. A recent summit of national resuscitation experts expounded on the AHA guidelines suggesting that end‐tidal carbon dioxide should be used in all arrests to guide the quality of CPR with a goal value of >20.[8] Similarly, CPR feedback devices have an even higher level of evidence recommendation in the 2010 AHA guidelines than capnography, yet only 4% of hospitals reported utilizing them. Although it is true that introducing these CPR assist technologies into a hospital would require some effort on the part of hospital leadership, it is important to recognize the potential role such devices might play in the larger context of a resuscitation quality program to optimize clinical outcomes from IHCA.
Several differences were noted between hospitals based on teaching status. Although all hospitals were more likely to rely on physicians to lead resuscitations, nonteaching hospitals were more likely to report routine leadership by nurses and pharmacists. Nonteaching hospitals were also less likely to have a CPR committee, even after adjusting for hospital size. In addition, these hospitals were also more likely to report the lack of a clinical champion as a barrier to quality improvement.
There were several limitations to this study. First, this was a descriptive survey that was not tied to outcomes. As such, we are unable to draw conclusions about which practices correlate with decreased incidence of cardiac arrest and improved survival. Second, this was an optional survey with a somewhat limited response rate. Even though the characteristics of the nonresponding hospitals were similar to the responding hospitals, we cannot rule out the possibility that a selection bias was introduced, which would likely overestimate adherence to the guidelines. Self‐reported responses may have introduced additional errors. Finally, the short interval between the release of the 2010 guidelines and the administration of the first survey may have contributed to the variability in implementation of some practices, but many of the recommendations had been previously included in the 2005 guidelines.
We conclude that there is wide variability between hospitals and within practices for resuscitation care. Future work should seek to understand which practices are associated with improved patient outcomes and how best to implement these practices in a more uniform fashion.
Acknowledgements
The authors thank Nancy Hinckley, who championed the study; David Chearo, Christelle Marpaud, and Martha Van Haitsma of the University of Chicago Survey Lab for their assistance in formulating and distributing the survey; and JoAnne Resnic, Nicole Twu, and Frank Zadravecz for administrative support.
Disclosures: This study was supported by the Society of Hospital Medicine with a grant from Philips Healthcare (Andover, MA). Dr. Edelson is supported by a career development award from the National Heart, Lung, and Blood Institute (K23 HL097157). In addition, she has received research support and honoraria from Philips Healthcare (Andover, MA), research support from the American Heart Association (Dallas, TX) and Laerdal Medical (Stavanger, Norway), and an honorarium from Early Sense (Tel Aviv, Israel). Dr. Hunt has received research support from the Laerdal Foundation for Acute Medicine (Stavanger, Norway), the Hartwell Foundation (Memphis, TN), and the Arthur Vining Davis Foundation (Jacksonville, FL), and honoraria from the Kansas University Endowment (Kansas City, KS), JCCC (Overland Park, KS), and the UVA School of Medicine (Charlottesville, VA) and the European School of Management (Berlin, Germany). Dr. Mancini is supported in part by an Agency for Healthcare Research and Quality grant (R18HS020416). In addition, she has received research support from the American Heart Association (Dallas, TX) and Laerdal Medical (Stavanger, Norway), and honoraria from Sotera Wireless, Inc. (San Diego, CA). Dr. Abella has received research support from the National Institutes of Health (NIH), Medtronic Foundation (Minneapolis, MN), and Philips Healthcare (Andover, MA); has volunteered with the American Heart Association; and received honoraria from Heartsine (Belfast, Ireland), Velomedix (Menlo Park, CA), and Stryker (Kalamazoo, MI). Mr. Miller is employed by the Society of Hospital Medicine.
- , , , , , . Trends in survival after in‐hospital cardiac arrest. N Engl J Med. 2012;367(20):1912–1920.
- , , , et al. Incidence of treated cardiac arrest in hospitalized patients in the United States. Crit Care Med. 2011;39(11):2401–2406.
- , , , et al. Racial differences in survival after in‐hospital cardiac arrest. JAMA. 2009;302(11):1195–1201.
- , , , , . Hospital variation in time to defibrillation after in‐hospital cardiac arrest. Arch Intern Med. 2009;169(14):1265–1273.
- , , , et al. Duration of resuscitation efforts and survival after in‐hospital cardiac arrest: an observational study. Lancet. 2012;380(9852):1473–1481.
- , , , . Delayed time to defibrillation after in‐hospital cardiac arrest. N Engl J Med. 2008;358(1):9–17.
- 2010 American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care science. Circulation. 2010;122(18 suppl 3):S640–S946.
- , , , et al. Cardiopulmonary resuscitation quality: improving cardiac resuscitation outcomes both inside and outside the hospital: a consensus statement from the American Heart Association. Circulation. 2013;128(4):417–435.
- American Hospital Association. 2008 AHA annual survey. AHA data viewer: survey instruments. 2012; Available at: http://www.ahadataviewer.com/about/hospital‐database. Accessed October 11, 2013.
- The American Association for Public Opinion Research. Standard Definitions: Final Dispositions of Case Codes and Outcome Rates for Surveys. 7th ed. Deerfield, IL: AAPOR; 2011.
- , , , , , . Do practice guidelines guide practice? The effect of a consensus statement on the practice of physicians. N Engl J Med. 1989;321(19):1306–1311.
- , , , et al. Why don't physicians follow clinical practice guidelines? A framework for improvement. JAMA. 1999;282(15):1458–1465.
- , . From best evidence to best practice: effective implementation of change in patients' care. Lancet. 2003;362(9391):1225–1230.
- , , , et al. Cardiopulmonary resuscitation standards for clinical practice and training in the UK. Accid Emerg Nurs. 2005;13(3):171–179.
- , , , , , . Trends in survival after in‐hospital cardiac arrest. N Engl J Med. 2012;367(20):1912–1920.
- , , , et al. Incidence of treated cardiac arrest in hospitalized patients in the United States. Crit Care Med. 2011;39(11):2401–2406.
- , , , et al. Racial differences in survival after in‐hospital cardiac arrest. JAMA. 2009;302(11):1195–1201.
- , , , , . Hospital variation in time to defibrillation after in‐hospital cardiac arrest. Arch Intern Med. 2009;169(14):1265–1273.
- , , , et al. Duration of resuscitation efforts and survival after in‐hospital cardiac arrest: an observational study. Lancet. 2012;380(9852):1473–1481.
- , , , . Delayed time to defibrillation after in‐hospital cardiac arrest. N Engl J Med. 2008;358(1):9–17.
- 2010 American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care science. Circulation. 2010;122(18 suppl 3):S640–S946.
- , , , et al. Cardiopulmonary resuscitation quality: improving cardiac resuscitation outcomes both inside and outside the hospital: a consensus statement from the American Heart Association. Circulation. 2013;128(4):417–435.
- American Hospital Association. 2008 AHA annual survey. AHA data viewer: survey instruments. 2012; Available at: http://www.ahadataviewer.com/about/hospital‐database. Accessed October 11, 2013.
- The American Association for Public Opinion Research. Standard Definitions: Final Dispositions of Case Codes and Outcome Rates for Surveys. 7th ed. Deerfield, IL: AAPOR; 2011.
- , , , , , . Do practice guidelines guide practice? The effect of a consensus statement on the practice of physicians. N Engl J Med. 1989;321(19):1306–1311.
- , , , et al. Why don't physicians follow clinical practice guidelines? A framework for improvement. JAMA. 1999;282(15):1458–1465.
- , . From best evidence to best practice: effective implementation of change in patients' care. Lancet. 2003;362(9391):1225–1230.
- , , , et al. Cardiopulmonary resuscitation standards for clinical practice and training in the UK. Accid Emerg Nurs. 2005;13(3):171–179.
© 2014 Society of Hospital Medicine
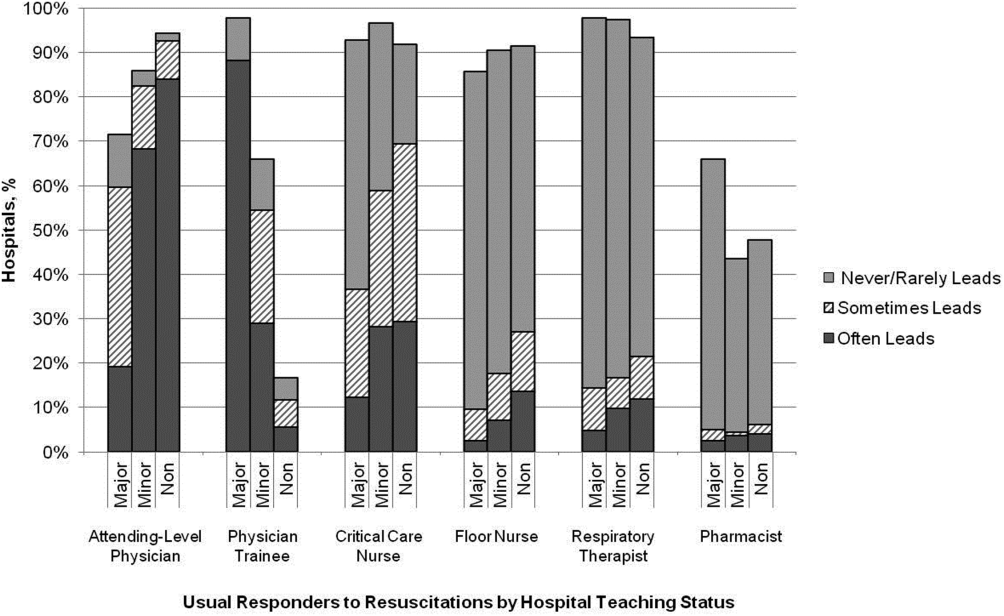
The Nonmotor Symptoms of Parkinson’s Disease: Update on Diagnosis and Treatment
From the Department of Neurology, Movement Disorders Division, University of Pittsburgh Medical Center, Pittsburgh, PA.
Abstract
- Objective: To review the prevalence, diagnosis, and treatment of the nonmotor symptoms (NMS) associated with Parkinson’s disease (PD).
- Methods: Narrative review of the literature.
- Results: The NMS of PD are becoming increasingly recognized as having a critical role in the impact of this neurodegenerative movement disorder. This has led to significant investigative efforts to identify new or better NMS therapies. The preponderance of PD patients will be diagnosed with 1 or multiple NMS during the course of their disease, with many of these symptoms occurring months or even years prior to receiving the PD diagnosis. Despite the high prevalence and impact on disease burden, NMS often go undetected due to a lack of reporting by patients or insufficient interrogation by physicians. Further complicating NMS management is that only a few therapies have the level of evidence needed to support their use in the treatment of NMS.
- Conclusion: The practitioner needs to be aware of NMS and conduct thorough patient questioning in order to recognize, diagnose, and address NMS in PD patients.
Parkinson’s disease (PD) is a neurodegenerative movement disorder with an estimated prevalence of 1% to 2% among  the population over the age of 65 years [1]. Recognition and clinical diagnosis of PD is primarily made based on the cardinal motor features, including rigidity, tremor, bradykinesia, and postural instability. The motor symptoms are neuropathologically associated with accumulation of alpha-synuclein with Lewy body formation and neurodegeneration of the nigrostriatal dopamine system. Postmortem evaluation of the brains of PD patients has revealed more widespread degeneration in nondopaminergic systems, including several brainstem nuclei (raphe nucleus, locus ceruleus, dorsal vagal nucleus), limbic and neocortical structures, as well as the peripheral autonomic system [2,3].
the population over the age of 65 years [1]. Recognition and clinical diagnosis of PD is primarily made based on the cardinal motor features, including rigidity, tremor, bradykinesia, and postural instability. The motor symptoms are neuropathologically associated with accumulation of alpha-synuclein with Lewy body formation and neurodegeneration of the nigrostriatal dopamine system. Postmortem evaluation of the brains of PD patients has revealed more widespread degeneration in nondopaminergic systems, including several brainstem nuclei (raphe nucleus, locus ceruleus, dorsal vagal nucleus), limbic and neocortical structures, as well as the peripheral autonomic system [2,3].
 The nonmotor symptoms (NMS) of PD are the clinical manifestations of this extensive degeneration, which suggests that NMS are intrinsic and fundamental features of PD. NMS are exceedingly common, and up to 90% of PD patients will experience nonmotor features, including depression, anxiety, sleep disturbances, cognitive impairment, and dysautonomia [4,5] (Table).
The nonmotor symptoms (NMS) of PD are the clinical manifestations of this extensive degeneration, which suggests that NMS are intrinsic and fundamental features of PD. NMS are exceedingly common, and up to 90% of PD patients will experience nonmotor features, including depression, anxiety, sleep disturbances, cognitive impairment, and dysautonomia [4,5] (Table).
NMS have a greater impact on quality of life as compared to the motor symptoms [6,7], but are frequently underrecognized [8]. Evidence suggests that unless there is systematic and specific interrogation by practioners, NMS will elude recognition [9–11]. Recognizing NMS as part of PD is complicated by the fact that these symptoms are common in the general population and not specific for PD [12,13]. NMS can occur at any stage of the disease and may predate diagnosis [12], although as PD progresses the NMS become more prevalent, with a greater impact on health care costs and institutionalization rates than motor features [14,15].
Neuropsychiatric Symptoms
Depression
Epidemiology and Diagnosis
Depression is one of the most common neuropsychiatric manifestations observed in PD patients, with prevalence reports between 4% and 72%, though likely to be closer to 30% to 45% [16–20]. The severity of depression in the PD population has been shown to be greater than in patients with matched chronic disabilities [21,22] and also greater than in the general population over the age of 65 years [23]. The onset of depression can occur at any stage of the disease, even predating the diagnosis. Additionally, depression has more than twice the impact on health status than motor symptoms [24].
Though the mechanisms are not fully understood, it is suspected that psychosocial as well as neuropathological changes contribute to the pathogenesis of depression in PD. In a study comparing 104 PD patients and 61 patients with equivalent disability scores, functional disability was found to be responsible for only 9% of the variation of depression scores [22]. The increased prevalence of depression in PD patients can in part be explained by the neuropathological changes seen in post-mortem studies. Two neurotransmitters that are fundamental in the pathogenesis of depression are serotonin, from the raphe nuclei, and norepinepherine, from the locus ceruleus [20]. Both of these brainstem structures demonstrate alpha-synucleinopathy-associated degeneration and these changes can precede the development of motor dysfunction [3].
Diagnosing depression in PD is complicated by the fact that there is overlap between other PD symptoms and clinical features of depression (ie, amotivation, bradykinesia, fatigue, and sleep disturbances). However, many depressed PD patients are less likely to report feelings of guilt or failure and tend to have higher rates of anxiety [9,20,25]. Typically, PD patients are more likely to be diagnosed with minor depression or dysthymia rather than a major depressive disorder [19,20]. Formal testing through systematic questionnaires are diagnostically useful in the clinic, and serial testing can reveal changes over time to guide more effective treatment. Validated tools to evaluate depression in PD include the Beck Depression Inventory, Hamilton Depression Rating Scale, Montgomery-Asberg Depression Rating Scale, Geriatric DRS, and Hospital Anxiety and Depression scale [20].
Treatment Options
Treatment of depression in PD demonstrates generally poorer responses to typical antidepressants and side effects that may worsen other PD symptoms. Selective serotonin reuptake inhibitors (SSRIs) have been widely used as there are generally few drug-drug interactions and minimal effect on motor symptoms; however, several studies have demonstrated little benefit on depression in PD [26]. In a randomized, double-blind, placebo-controlled trial of the antidepressants paroxetine and venlafaxine, both were found to be effective and well tolerated [27]. Tricyclic anti-depressants (TCAs) have also demonstrated efficacy. In randomized controlled trials comparing TCAs to SSRIs, a greater benefit on depression symptoms has been found with TCAs [28–30]. The use of TCAs, however, is limited by anticholinergic side effects that occasionally worsen orthostatic hypotension or cognitive impairment [15,31]. Dopamine agonists have also been studied in depressed PD patients. In a randomized, double-blind, placebo-controlled trial [32] and a prospective observational study [33], pramipexole demonstrated significant improvements in depression symptoms. Ropinirole also demonstrated significant symptomatic improvement [34]. These studies suggest that while SSRIs are commonly used, evidence is accumulating to support the role of TCAs, SNRIs, and dopamine agonists in the treatment of depression in PD.
Other therapies have also been tried in pharmacologic-resistant patients. Electroconvulsive therapy has been reported to improve both depression and motor symptoms [35,36]; however, this is a treatment reserved for patients with severe and drug-refractory depression. A randomized controlled trial investigating cognitive behavioral therapy has also demonstrated improvement of depression scores [37]. The role of physical activity as treatment for depression in PD patients is unclear. As described in a recent review by Loprinzi et al [38], the literature is contradictory, with one group experiencing reduced depression but with no signficant effect in several other studies.
Anxiety
Epidemiology and Diagnosis
The prevalence of anxiety in PD patients is about 40% [39], which is 2 times greater than in the general population [9]. Anxiety may worsen PD symptoms, especially tremor and cognition. Risk factors for anxiety include the female gender, greater motor fluctuations, prior history of anxiety, and younger age of PD onset [40]. As with depression, some patients also report worsening of anxious symptoms during “off” states [41]. Screening tools that have been validated to help practitioners identify anxiety in PD include the Hospital Anxiety and Depression Scale, Beck Anxiety Inventory, Zung Self-rating Anxiety Scale, Spielberger State Trait Anxiety Inventory, and Hamilton Anxiety Rating Scale [15].
Treatment Options
The treatment of diagnosed anxiety in PD is primarily with benzodiazepines, which are particularly beneficial in patients whose tremors are exacerbated by anxiety or stress. The use of benzodiazepines has not been evaluated by a randomized controlled trial and use should be limited given the potential risks of sedation, cognitive effects, and psychomotor agitation. Other case studies have found benefit with serotonergic medications like fluoxetine or citalopram (especially with concomitant depression) or with optimization of levodopa therapy [42,43].
Hallucinations, Delusions, and Psychosis
Epidemiology
The prevalence of visual hallucinations in PD patients is about 20% to 40% [44,45]. Risk factors for psychotic symptoms include cognitive impairment, advanced age, prolonged duration of disease, depression, severe dysautonomia, and sleep disorders [46–48]. Early recognition of hallucinations is critical because of a strong correlation between the manifestation of psychosis and the need for nursing home placement or hospitalization. With early and effective treatment there is a decreased need for placement and a reduction on caregiver burden [44,49].
Treatment Options
Hallucinations can occur in delirium and it is important to first rule out an underlying infection or an offending medication, especially if there is a sudden onset or worsening of symptoms. Psychotic symptoms have been reported in drug-naive patients, though they are often iatrogenically induced with dopaminergic agents. All antiparkinsonian medications are capable of inducing or exacerbating hallucinations [9,50]. Additionally, psychotic symptoms tend to improve when dopaminergic agonists are reduced or eliminated. However, there is no clear relationship between the dose of dopaminergic agents and manifestation of hallucinations [48,51,52]. If hallucinations persist or there are motor complications that arise from reduction of dopaminergic agents, initiation of clozapine has been demonstrated to be efficacious in a rater-blinded prospective study and in a retrospective analysis [53–55]; however, regular monitoring for neutropenia is required. Quetiapine has demonstrated similar benefit without significant effects on motor symptoms in a randomized, rater-blinded study and in an evidence-based review [56,57]. It is also important to review or eliminate other medications that may contribute to hallucinations.
Cognitive Impairment
Epidemiology
The prevalence of dementia in the PD population is 20% to 40% [58], though almost 80% of PD patients ultimately develop cognitive decline [59]. Overall, a PD patient is 6 times more likely to develop dementia than someone in the general population [60]. There may be parallel progression of cognitive impairment and motor symptoms, but there is no correlation with overall duration of disease [60,61]. Risk factors linked with the presence of dementia include older age at onset of PD, presence of hallucinations, and male gender [62,63].
Cognitive dysfunction can be detected early in PD through neuropsychological testing; however, impairment of cognition is often insidious and may not be appreciated until symptoms become severe. Several screening tools have been used to evaluate for cognitive impairment in PD including the Mini-Mental State Exam (MMSE), Montreal Cognitive Assessment (MoCA), Mini-Mental Parkinson, Scales for Outcomes of Parkinson’s disease–Cognition, and others. Accumulating evidence, however, is suggestive of the superiority of the MoCA in the detection of cognitive deficits associated
with PD [64].
Dementia is a substantial burden for the caregiver and is a significant contributor to mortality in PD patients [65]. Cognitive impairment often presents with other behavioral symptoms, which further hastens placement outside the home and increases cost of caring for PD patients [49,66].
Cognitive impairment in Parkinson’s disease is typically associated with degeneration of primarily subcortical structures. PD patients with mild cognitive impairment were found to have deficits most significantly in memory, executive function, memory, and language abilities [67]. A recent study by Mak et al evaluated grey matter volumes by structural MRI in PD patients with evidence of mild cognitive impairment by MMSE and MoCA as compared with findings in cognitively intact patients. This demonstrated decreased brain volumes in areas that correlate with affected cognitive domains including the left insula, left superior frontal and left middle temporal areas [68].
Treatment Options
Prior to initiation of therapy, it is important to evaluate the patient for depression and to rule out pseudodementia. Bradyphrenia, or slowness of thought, should also be considered, as this symptom may also lead to an incorrect dementia diagnosis. Lastly, a thorough review of medications should be performed and offending agents including anticholinergics, TCAs, dopamine agonists, and amantadine should be discontinued as these can worsen cognition.
Rivastigmine has demonstrated modest improvement in cognitive performance in PD patients with dementia in a large multicenter, placebo-controlled study [69]. Other cholinesterase inhibitors (ie, donepezil or galantamine) are not recommended at this time due to limited studies or contradictory results in the literature [31,54]. Caution is advised with use of cholinesterase inhibitors as they may worsen tremor or autonomic dysfunction; also, use is limited by nausea or other gastrointestinal symptoms. Memantine, an NMDA receptor antagonist, has also been investigated in randomized, double-blind, placebo-controlled trials and demonstrated modest improvement of cognition and is generally well tolerated [70,71].
Nonpharmacologic therapy includes physical exercise, which has demonstrated improvement in memory tasks and processing speed [72]. Cognitive training has been less rigorously studied; however, a recent single-blinded controlled study demonstrated significant improvement of learning and memory in PD patients who completed computer-based cognitive training [73].
Compulsive Disorders
Impulse Control Disorders
Impulse control disorders (ICDs) are inappropriate behaviors resulting from a failure to resist an impulse, which leads to pleasure-seeking activities at the expense of relationships and ability to function socially. In PD, ICDs are expressed as pathologic gambling, hypersexuality, binge eating, compulsive shopping, and excessive spending [9,66]. The prevalence of all ICDs in PD is 15% to 20% and a patient may be diagnosed with multiple ICDs [74]. Dopamine agonist use has been implicated in the development of ICDs and this risk is further increased with the addition of levodopa [75,76]. Clinical features associated with ICDs include young age of onset, male gender, family history of addiction, depression or anxiety, and disinhibition or impulsive traits [77,78].
Traditionally, treatment consists of reduction or elimination of dopamine agonists, though adjustment of levodopa therapy may also be necessary. Amantadine as an adjunct therapy has been shown in a randomized, double-blind crossover study to reduce impulsivity in a few patients with pathologic gambling [79].
Dopamine Dysregulation Syndrome
Dopamine dysregulation syndrome (DDS) is characterized by compulsive use of dopaminergic medications beyond what is needed to treat parkinsonian symptoms, and is associated with social impairment. Patients describe addictive symptoms like craving or intense desire to obtain more dopaminergic medication [9,74]. Like ICDs, treatment of DDS consists of modification to dopaminergic medications, though patients with DDS may also require psychiatric evaluation and treatment.
Punding
Punding is another compulsive disorder that is defined as an intense fascination with objects and is associated with repetitive handling, manipulation, sorting, or arrangement of the items [80]. Occurrence of punding has been associated with higher total daily levels of levodopa, although one study has also implicated dopamine agonists [15,81]. As with the other compulsive disorders, punding also tends to respond well to reduction or discontinuation of levodopa. Studies have demonstrated modest benefit with SSRIs or atypical antipsychotics in long-term follow-up [82,83], though one study reported worsening of punding with quetiapine [84].
Apathy
Epidemiology and Treatment
Apathy is often characterized by a loss of motivation or inability to initiate goal-directed behavior, which results in dependence on others for activities of daily living and increases caregiver burden [85]. Patients demonstrate indifference, lack of interest, or inability to express or describe emotion. The apathetic patient may lack spontaneous and voluntary activity, and their affect display is often flattened [86].
With a prevalence of 30% to 50% [87], apathy is as common as depression in PD patients [66,88]. Risk factors associated with apathy include advanced age, severity of depression, severity of motor dysfunction, and dementia [89]. Apathy is frequently mistaken for depression given the significant overlap in symptoms; however, the patient with pure apathy will deny sadness or depressed feelings. It is also important to distinguish apathy from motor impairment or cognitive dysfunction that could explain the behavioral changes. No medications have reliably been shown to improve apathy, though it may be improved with initiation of dopaminergic therapy, especially early in the course [86,90].
Sleep Disorders
The original report of PD by James Parkinson describes sleep disturbances and daytime somnolence [91], which suggests that sleep disorders may be an intrinsic feature of the neurodegenerative process of PD itself.
REM Behavioral Disorder
Epidemiology and Diagnosis
Rapid eye movement behavioral disorder (RBD) is a parasomnia characterized by vocalizations and motor activity during dreaming due to loss of normal atonia associated with rapid eye movement (REM) sleep. Patients enact their dreams, which may lead to violent behaviors that can injure the patient or their bed partner. RBD is seen in 25% to 50% of PD patients [92,93], with variability depending on diagnostic technique and patient selection. Polysomnography is the most important diagnostic tool and demonstrates increased chin tone and limb movements during REM sleep in RBD [94,95]. Diagnosis can also be made clinically with patient and bed partner reports, though sensitivity is only approximately 30% [15].
Interestingly, many studies are now investigating the relationship between presence of RBD and later onset of neurodegenerative disorders. Multiple studies have shown that 40% to 65% of patients diagnosed with idiopathic RBD later develop an alpha-synucleinopathy, which includes PD, dementia with Lewy bodies, or multiple system atrophy within 10 years [92,95]. Prior studies report that as many as 90% of patients with idiopathic RBD develop neurodegenerative synucleinopathy when followed over 14 years [96]. Idiopathic RBD is currently being investigated as a potential clinical marker of pre-symptomatic PD in a multicenter observational study. If RBD is an early marker for neurodegenerative disease, it may be used to identify patients for neuroprotective trials as treatments are developed.
Treatment Options
Low-dose clonazepam (0.25–1 mg) is the mainstay of therapy, especially for patients that injure themselves or bed partners [97]; however, the use of benzodiazepines is historical and there remain no randomized controlled double-blind studies to evaluate the efficacy of clonazepam. Use of clonazepam may be limited by daytime sedation, confusion, or psychomotor agitation [31,97,98]. Melatonin (doses between 3–12 mg at bedtime) has also demonstrated benefit in RBD in a double-blind, placebo-controlled trial and in a small case series, with fewer side effects and no addiction potential as compared to clonazepam [99,100]. Case reports also support the use of several other effective medications, including cholinesterase inhibitors (rivastigmine and donepezil) and dopaminergic agents (pramipexole and levodopa) [15,20].
Restless Leg Syndrome and Periodic Limb Movements in Sleep
Epidemiology
Restless leg syndrome (RLS) and periodic limb movements in sleep (PLMS) cause disruptions of sleep and have an important impact on quality of sleep in PD patients. RLS is described as a strong urge to move the legs, accompanied by an uncomfortable sensation that is exacerbated at rest and relieved by movement. RLS is more frequently diagnosed in patients with PD, though prevalence reports vary widely [15]. Secondary causes for RLS should be investigated including iron deficiency, uremia and polyneuropathy. Several case reports demonstrate onset or worsening of RLS with use of antidepressants [101, 102] or antipsychotics like risperidone, aripiprazole, and quetiapine [103,104].
PLMS occurs in approximately 80% to 90% of patients with RLS, though may be present independently, and when seen on polysomnography is supportive of RLS [105]. PLMS is characterized by repetitive dorsiflexion of the foot, extension of the great toe, and may be accompanied by flexion of the knee and hip. The prevalence of PLMS in PD is approximately 60% and correlates with severity of PD motor features [106].
Treatment Options
Treatment of RLS should be initiated with nonpharmacologic therapies including good sleep hygiene, exercise, leg massage, and heat or ice packs [105,107]. Dopamine (DA) agonists are the primary treatment for RLS; however, even modest adjustments in levodopa can be helpful. One drawback to levodopa therapy is augmentation (a worsening or reappearance of symptoms) when serum levels fall due to the short half-life of levodopa [107,108]. DA agonists are less likely to cause augmentation. Both pramipexole and ropinirole have been extensively investigated in controlled, randomized, double-blind studies with benefits in 70% to 90% of patients with RLS and PLMS; however, there is a risk of developing compulsive behaviors [109–112]. Another option for PD patients is rotigotine, which has demonstrated improvement of RLS symptoms in a randomized, double-blind, placebo-controlled trial and has the added benefit that it may also help with motor symptoms [113,114].
More recently, gabapentin enacarbil has demonstrated improvement of moderate to severe RLS and was well tolerated in multiple randomized, double-blind, placebo-controlled trials [107,115,116]. Lastly, opioids (tramadol, oxycodone, codeine) have been shown to be effective, especially in the treatment of RLS that is refractory to other treatments [105,107].
Insomnia
Epidemiology
The most common sleep disorder in PD is insomnia, with a prevalence between 37% to 88% [14,117]. Insomnia is associated with difficulty in initiation or maintenance of sleep. Disruption of sleep typically leads to daytime somnolence and patient reports of a strong impact on motor disability and overall quality of life. There are several contributors to insomnia in PD patients including nocturia, depression, RLS, dystonia, and akinesia/rigidity/difficulty turning in bed [118].
Treatment Options
The use of carbidopa/levodopa controlled-release formulations at bedtime is associated with improved sleep duration and nocturnal akinesia, although it does not demonstrate a significant improvement in overall sleep ratings [54]. Hypnotics like eszopiclone and zolpidem have also demonstrated improved quality of sleep in limited controlled trials and a meta-analysis, but use is limited by sedation, dizziness, and falls [54,119]. Benzodiazepines improve sleep latency, but there is a risk of cognitive impairment, tolerance, and falls [117,120]. Melatonin at 3 to 5 mg and 50 mg doses have been investigated in 2 randomized, double-blind, placebo-controlled trials; however, there was a modest benefit and it was concluded that there is insufficient evidence to support the use of melatonin [54]. Nevertheless, melatonin is well tolerated and may be tried with minimal risk [54]. More recently, a randomized controlled trial using doxepin has demonstrated improvement of insomnia scores and was generally well tolerated [121].
Excessive Daytime Sleepiness and Abrupt Sleep Onset
EDS and Fatigue: Epidemiology and Treatment
A common complaint by PD patients is excessive daytime sleepiness (EDS), which can be verified with multiple sleep latency testing. EDS frequency varies in the literature, but is seen in approximately 15% to 50% of PD patients [4,122]. The etiology is usually multifactorial, with insomnia, dysautonomia, and depression as contributing factors [117]. A longer duration of symptoms, greater total load of levodopa, cognitive decline, and male gender are all risk factors for EDS [122,123]. It has been proposed that EDS is an intrinsic feature of PD; however, there is also an association with the use of antiparkinsonian medications. A randomized controlled trial demonstrated that use of the dopamine agonist pramipexole was associated with greater somnolence as compared to levodopa therapy (35% vs. 13%); however, this difference was only seen during the initial escalation phase [124]. Additionally, the combined use of dopamine agonists and levodopa has shown an even greater risk of EDS [125]. The evidence for the use of stimulants for EDS is lacking. The few studies conducted with modafinil have not demonstrated a robust improvement of EDS [126–128]. Other stimulants like methylphenidate have been studied with improvement of Epworth Sleepiness Score, though no randomized control trials have been undertaken [129].
It is important to distinguish EDS, a propensity for daytime sleep, from fatigue or excessive tiredness associated with mental or physical exertion [117]. Fatigue is often multifactorial and may be related to insomnia, sleep apnea, sedating effects of medications, frequent awakenings from nocturia, and degeneration of brain areas regulating sleep/wake cycles related to the underlying disease process [20, 117]. It is also important to consider depression and dementia in the differential, as these disorders may be erroneously be diagnosed as fatigue. Treatment of fatigue should include regular mild exercise, maintenance of a stimulating environment, removal of sedating medications, and management of intrinsic sleep disorders if present [117]. The use of stimulants for fatigue is controversial. A small randomized controlled trial (n = 48) using modafinil demonstrated improvement on the global clinical impression scale for fatigue but no significant change on the Fatigue Severity Scale; this study was limited by the power and points to the need for a larger study [130].
Sleep Attacks: Epidemiology and Treatment
Abrupt sleep onset, or “sleep attacks,” occurs when transition from wake to sleep is unavoidable and may occur without warning. Sleep attacks are threefold more likely to occur in patients using DA agonists, with an associated dose-related increase in risk [131]. Adjustment or elimination of DA agonists often improves sleep attacks, though it is important to address concurrent EDS if present. Nonpharmacologic treatments to consider include mild exercise, early morning bright light exposure, and a stimulating environment [117].
Sleep-Disordered Breathing/Obstructive Sleep Apnea
Epidemiology and Treatment
Sleep-disordered breathing (SDB) consists of either a deficit in the drive to breathe as in central sleep apnea, or may be due to an blockage of the airway as seen in obstructive sleep apnea (OSA). Apnea leads to oxygen desaturations that consequently trigger awakenings throughout the night, which in turn is experienced by the patient as daytime somnolence [117]. The prevalence of SDB and OSA is variable in the literature, ranging from no increased risk in PD patients [132,133] to 50% prevalence in PD patients [134,135]. Discussions with bed partners, history of snoring, and clinical reports of EDS or daytime fatigue are important indicators of SDB. Polysomnography confirms the diagnosis and can direct treatment, which frequently includes application of CPAP devices during sleep.
Autinomic Dysfunction
Orthostatic Hypotension
Epidemiology and Diagnosis
Orthostatic hypotension (OH) is defined as a 20-mm Hg fall in systolic blood pressure or 10-mm Hg drop in diastolic blood pressure within 3 minutes of a change in position. The prevalence of OH in PD patients is 30% to 60% [136,137]. Symptoms of OH can occur early in the disease and may precede diagnosis of PD [137]. Patients experience OH as dizziness, drowsiness, palpitations, nausea, or loss of consciousness. Additionally, falls and supine hypertension that accompany OH are associated with increased risk of morbidity and mortality in PD patients [138]. Several medications used in the treatment of PD can exacerbate OH, including levodopa, DA agonists, MAO-B inhibitors, and TCAs [139].
Treatment Options
First-line therapies for OH include nonpharmacologic methods such as compression stockings, sleeping with head elevated to 30 degrees, increased water and salt intake, more frequent small meals, and slowly changing position [140]. Additionally, it is important to discuss the removal or reduction of all antihypertensives with the patient’s PCP. Fludrocortisone (a mineralacorticoid) and domperidone (a peripheral dopamine antagonist not currently approved for use in the United States) modestly improved OH in a 2-phase, randomized, controlled, double-blind, crossover trial [141]. Pyridostigmine has also demonstrated improvement of standing blood pressure and OH symptoms in a double-blind, randomized cross-over study and has the additional benefit of not worsening supine hypertension [142]. Other effective treatments include midodrine, per a randomized, double-blind multicenter study [143], as well as droxidopa in a double-blind, crossover, placebo-controlled study [144]. Currently there is insufficient evidence to support the preferential use of any specific agent in the treatment of OH in PD.
Gastrointestinal Dysmotility
Constipation: Epidemiology and Treatment
Constipation is reported by nearly 60% of PD patients [145]. Constipation can precede the development of motor symptoms of PD, and the prevalence of GI disturbances increases with age and longer duration of disease. Nearly one third of patients will have been diagnosed with a GI disturbance within the year prior to PD diagnosis [146], which is associated with an increased risk for the development PD [147]. People with constipation (defined as < 1 bowel movement per day) but without a PD diagnosis had more nigral Lewy body degeneration postmortem [148] compared with people without constipation.
Treatments for constipation include dietary modification, increased fluid intake, and mild exercise. Macrogol significantly improved constipation in PD patients and was very well tolerated in a randomized placebo-controlled study [149]. Lubiprostone, a GI active prostaglandin, is also effective in the short-term treatment of constipation in a placebo-controlled trial [150].
Gastroparesis: Epidemiology and Treatment
Gastroparesis, like constipation, is related to enteric dopaminergic cell loss and degeneration of the dorsal motor nucleus of the vagus [151]. Patients experience gastroparesis as early satiety, full sensation, and nausea. Decreased gastric motility leads to retention of food as well as medications, which can slow absorption and delay onset of action for many medications including levodopa. Domperidone has both prokinetic and antiemetic properties, which have been beneficial in the treatment of gastroparesis [152], but its use is not currently approved in the United States.
Dysphagia: Epidemiology and Treatment
Dysphagia is associated with more advanced stages of PD as well as a significant increase in morbidity. Swallow exercises have demonstrated improvement of dysphagia [153]. The impact of levodopa therapy on dysphagia in the literature is controversial. Videofluoroscopic examination is the most common method for evaluation of swallowing disorders and provides important information for speech-language pathologists regarding recommendations for dietary modifications [154]. Adjustment of medication regimens to avoid an oral route is also helpful. This includes Parcopa, orally disintegrating carbidopa/levodopa tablets, and transdermal approaches like the rotigotine patch. For some patients, enteral nutrition is needed and placement of nasogastric tubes or percutaneous endoscopic gastrostomy tubes are an option.
Sialorrhea (Drooling)
Epidemiology
Difficulty handling oral secretions due to impaired or infrequent swallowing results in sialorrhea in up to 75% of PD patients [155], which is a significant embarrassment for most patients [156]. PD patients with drooling have difficulty speaking, eating, and engaging in social interactions, which significantly impacts perceived quality of life [157].
Treatment Options
Botulinum toxin (A and B) injections into the submandibular or parotid glands have demonstrated efficacy in multiple double-blind, randomized, placebo-controlled studies for the treatment of sialorrhea in PD patients; however, injections are associated with greater invasiveness and cost [158–160]. Glycopyrrolate, an anticholinergic drug, was also efficacious in the treatment of sialorrhea in the short term in a double-blind, randomized, placebo-controlled study [161]. Alternatively, gum chewing increases swallow frequency, improves drooling, and also shows a benefit with dysphagia [162].
Genitourinary Disturbances
Bladdery dysfunction: Epidemiology and Treatment
Bladder dysfunction in PD is often secondary to hyperactivity of the detrusor muscle leading to urinary urgency, increased urinary frequency, and nocturia. Less commonly, hypoactive detrusor muscle causes difficulty with initiation of urination, delayed bladder emptying, and recurrent infections. Urinary disturbances may occur before the onset of motor symptoms or early on in the disease course [12]. Disease severity is associated with greater urinary disturbances, and more than 50% of advanced PD patients report severe bladder symptoms [163].
Anticholinergic medications such as oxybutynin, solifenacin, and tolterodine are commonly used in the treatment of detrusor hyperactivity and demonstrate significant improvement in detrusor pressure in a recent systemic review and meta-analysis [164]. PD patients on these agents should be closely monitored for side effects including cognitive impairment, somnolence, hallucinations, confusion, and blurred vision. Other treatments include botulinum toxin injections into the detrusor muscle, which has demonstrated safety and efficacy in a recent systematic review [165].
Erectile dysfunction: Epidemiology and Treatment
Erectile dysfunction (ED) is reported by more than 60% of male PD patients [145] and is thought to be related to hypothalamic dysfunction and modification of the dopamine-oxytocin pathway [166]. Effects of PD medications, cognitive impairment, fatigue, apathy, and low testosterone contribute to loss of libido and ED [20,167]. Phosphodiesterase inhibitors such as sildenafil, vardenafil, and tadalafil are possibly useful in the treatment of ED in PD patients, though randomized trials have been limited [166,168]. Apomorphine sublingually is another medication that has demonstrated improvement of ED in a double-blind, crossover study and can be considered for patients with contraindications to phosphodiesterase inhibitors [169].
Sensory Symptoms
Pain
Epidemiology
Sensory disturbances in PD include diminished ability to identify odors, visual abnormalities (blurred vision, abnormal color perception, double vision), and pain. Pain is the most disabling sensory disturbance, though frequently underreported. Nearly two thirds of PD patients report pain, [170], though only half of patients receive any treatment [171]. Pain may also be a presenting symptom that precedes the clinical diagnosis of PD [172,173].
Treatment Options
There are several types of pain described by PD patients, the most common of which is musculoskeletal, typically involving the shoulder. Other types include dystonic, radicular, and central pain [174]. First-line treatment of musculoskeletal complaints includes nonsteroidal anti-inflammatory drugs (NSAIDs) and physiotherapy. Modification of levodopa regimen (including altering timing and frequency or adding controlled release formulations) can often provide relief for dystonic pain, and also for central pain for some patients [173, 174]. Deep brain stimulation, with subthalamic nucleus or globus pallidus targets, has demonstrated improvement with dystonic, central, and musculoskeletal pain in a small clinical study [175].
Conclusion
NMS are an intrinsic part of PD, may predate diagnosis, and substantially affect the majority of patients with PD. For many of these patients, NMS have a greater impact on quality of life and health care costs than the cardinal motor symptoms that define the disease. Many of these symptoms are not recognized by practioners and often are not volunteered by PD patients, making it important for practitioners to routinely and directly inquire about NMS. Treatment of NMS in PD is challenging, and only a few therapies have the level of evidence needed to support their use in the treatment of these problems. Nevertheless, proper recognition and addressing of these symptoms afford the clinician an opportunity to make a positive and potentially significant impact on the PD patient’s quality of life.
Corresponding author: Samay Jain, MD, MS, Dept of Neurology, 811 Kaufmann Bldg, Pittsburgh, PA 15213, jains@upmc.edu.
Financial disclosures: None.
1. Alves G, Forsaa EB, Pedersen KF, et al. Epidemiology of Parkinson's disease. J Neurol 2008;255 Suppl 5:18–32.
2. Stern MB, Lang A, Poewe W. Toward a redefinition of Parkinson's disease. Mov Disord 2012;27:54–60.
3. Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003;24:197–211.
4. Tandberg E, Larsen JP, Karlsen K. A community-based study of sleep disorders in patients with Parkinson's disease. Mov Disord 1998;13:895–9.
5. Shulman LM, Taback RL, Bean J, Weiner WJ. Comorbidity of the nonmotor symptoms of Parkinson's disease. Mov Disord 2001;16:507–10.
6. Park A, Stacy M. Non-motor symptoms in Parkinson's disease. J Neurol 2009;256 Suppl 3:293–8.
7. Chaudhuri KR, Schapira AH. Non-motor symptoms of Parkinson's disease: dopaminergic pathophysiology and treatment. Lancet Neurol 2009;8:464–74.
8. Shulman LM, Taback RL, Rabinstein AA, Weiner WJ. Non-recognition of depression and other non-motor symptoms in Parkinson's disease. Parkinsonism Relat Disord 2002;8:193–7.
9. Bonnet AM, Jutras MF, Czernecki V, et al. Nonmotor symptoms in Parkinson's disease in 2012: relevant clinical aspects. Parkinsons Dis 2012:2012:198316.
10. Chaudhuri KR, Healy DG, Schapira AH. Non-motor symptoms of Parkinson's disease: diagnosis and management. Lancet Neurol 2006;5:235–45.
11. Hussl AK, Seppi K, Poewe W. Nonmotor symptoms in Parkinson's disease. Expert Rev Neurother 2013;13:581–3.
12. O'Sullivan SS, Williams DR, Gallagher DA, et al. Nonmotor symptoms as presenting complaints in Parkinson's disease: a clinicopathological study. Mov Disord 2008;23:101–6.
13. Lang AE. A critical appraisal of the premotor symptoms of Parkinson's disease: potential usefulness in early diagnosis and design of neuroprotective trials. Mov Disord 2011;26:775–83.
14. Barone P, Antonini A, Colosimo C, et al. The PRIAMO study: A multicenter assessment of nonmotor symptoms and their impact on quality of life in Parkinson's disease. Mov Disord 2009;24:1641–9.
15. Bernal-Pacheco O, Limotai N, Go CL, Fernandez HH. Nonmotor manifestations in Parkinson disease. Neurologist 2012;18:1–16.
16. Lemke MR, Fuchs G, Gemende I, et al. Depression and Parkinson's disease. J Neurol 2004;251 Suppl 6: VI/24–7.
17. Ravina, B, Camicioli R, Como PG, et al, The impact of depressive symptoms in early Parkinson disease. Neurology 2007;69:342–7.
18. Jasinska-Myga B, Putzke JD Wider C, et al. Depression in Parkinson's disease. Can J Neurol Sci 2010;37:61–6.
19. Slaughter JR, Slaughter KA, Nichols D, et al. Prevalence, clinical manifestations, etiology, and treatment of depression in Parkinson's disease. J Neuropsychiatry Clin Neurosci 2001;13:187–96.
20. Simuni T, Sethi K. Nonmotor manifestations of Parkinson's disease. Ann Neurol 2008;64 Suppl 2:S65–80.
21. Ehmann TS, Beninger RJ, Gawel MJ, Riopelle RJ. Depressive symptoms in Parkinson's disease: a comparison with disabled control subjects. J Geriatr Psychiatry Neurol 1990;3:3–9.
22. Menza MA, Mark MH. Parkinson's disease and depression: the relationship to disability and personality. J Neuropsychiatry Clin Neurosci 1994;6:165–9.
23. CDC. Current depression among adults–United States, 2006 and 2008. Morb Mort Weekly Rep 2010;59:1229–35.
24. Hinnell C, Hurt CS, Landau S, et al. Nonmotor versus motor symptoms: how much do they matter to health status in Parkinson's disease? Mov Disord 2012;27:236–41.
25. Cummings JL. Depression and Parkinson's disease: a review. Am J Psychiatry 1992;149:443–54.
26. Weintraub D, Morales KH, Moberg PJ, et al. Antidepressant studies in Parkinson's disease: a review and meta-analysis. Mov Disord 2005;20:1161–9.
27. Richard IH, McDermott MP, Kurlan R, et al. A randomized, double-blind, placebo-controlled trial of antidepressants in Parkinson disease. Neurology 2012;78:1229–36.
28. Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009;72:886–92.
29. Okun MS, Fernandez HH. Will tricyclic antidepressants make a comeback for depressed Parkinson disease patients? Neurology 2009;72:868–9.
30. Antonini A, Tesei S, Zecchinelli A, et al. Randomized study of sertraline and low-dose amitriptyline in patients with Parkinson's disease and depression: effect on quality of life. Mov Disord 2006;21:1119–22.
31. Pedrosa DJ, Timmermann L. Review: management of Parkinson's disease. Neuropsychiatr Dis Treat 2013;9: 321–40.
32. Barone P, Poewe W, Albrecht S, et al. Pramipexole for the treatment of depressive symptoms in patients with Parkinson's disease: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2010;9:573–80.
33. Lemke MR, Brecht HM, Koester J, et al. Anhedonia, depression, and motor functioning in Parkinson's disease during treatment with pramipexole. J Neuropsychiatry Clin Neurosci 2005;17:214–20.
34. Rektorova I, Balaz M, Svatova J, et al. Effects of ropinirole on nonmotor symptoms of Parkinson disease: a prospective multicenter study. Clin Neuropharmacol 2008;31:261–6.
35. Van der Wurff FB, Stek ML, Hoogendijk WL, Beekman AT. Electroconvulsive therapy for the depressed elderly. Cochrane Database Syst Rev 2003(2):CD003593.
36. Okun MS, Watts RL. Depression associated with Parkinson's disease: clinical features and treatment. Neurology 2002;58(4 Suppl 1):S63–70.
37. Dobkin RD, Menza M, Allen LA, et al. Cognitive-behavioral therapy for depression in Parkinson's disease: a randomized, controlled trial. Am J Psychiatry 2013;168:1066–74.
38. Loprinzi PD, Herod SM, Cardinal BJ, Noakes TD. Physical activity and the brain: A review of this dynamic, bi-directional relationship. Brain Res 2013;1539:95–104.
39. Richard IH. Anxiety disorders in Parkinson's disease. Adv Neurol 2005;96:42–55.
40. Leentjens AF, Dujardin K, Marsh L, et al. Symptomatology and markers of anxiety disorders in Parkinson's disease: a cross-sectional study. Mov Disord 2011;26:484–92.
41. Witjas T, Kaphan E, Azulay JP, et al. Nonmotor fluctuations in Parkinson's disease: frequent and disabling. Neurology 2002;59:408–13.
42. Olanow CW, Stern MB, Sethi K. The scientific and clinical basis for the treatment of Parkinson disease Neurology 2009;72(21 Suppl 4):S1–136.
43. Richard IH, Schiffer RB, Kurlan R. Anxiety and Parkinson's disease. J Neuropsychiatry Clin Neurosci 1996;8:383–92.
44. Fenelon G, Alves G. Epidemiology of psychosis in Parkinson's disease. J Neurol Sci 2010;289:12–7.
45. Papapetropoulos S, Katzen H, Schrag A, et al. A questionnaire-based (UM-PDHQ) study of hallucinations in Parkinson's disease. BMC Neurol 2008;8:21.
46. Fenelon G, Mahieux F, Huon R, Ziegler M. Hallucinations in Parkinson's disease: prevalence, phenomenology and risk factors. Brain 2000;123 (Pt 4):733–45.
47. Papapetropoulos S, Mash DC. Psychotic symptoms in Parkinson's disease. From description to etiology. J Neurol 2005;252:753–64.
48. Aarsland D, Larsen JP, Cummins JL, Laake K. Prevalence and clinical correlates of psychotic symptoms in Parkinson disease: a community-based study. Arch Neurol 1999;56:595–601.
49. Aarsland D, Larsen JP, Tandberg E, Laake K. Predictors of nursing home placement in Parkinson's disease: a population-based, prospective study. J Am Geriatr Soc 2000;48:938–42.
50. Lohle M, Storch A, Reichmann H, Beyond tremor and rigidity: non-motor features of Parkinson's disease. J Neural Transm 2009;116:1483–92.
51. Fenelon G. Psychosis in Parkinson's disease: phenomenology, frequency, risk factors, and current understanding of pathophysiologic mechanisms. CNS Spectr 2008;13(3 Suppl 4):18–25.
52. Merims D, Shabtai H, Korczyn AD, et al. Antiparkinsonian medication is not a risk factor for the development of hallucinations in Parkinson's disease. J Neural Transm 2004;111:1447–53.
53. Merims D, Balas M, Peretz C, et al. Rater-blinded, prospective comparison: quetiapine versus clozapine for Parkinson's disease psychosis. Clin Neuropharmacol 2006;29:331–7.
54. Seppi K, Weintraub D, Coelho M, et al. The Movement Disorder Society Evidence-Based Medicine Review Update: Treatments for the non-motor symptoms of Parkinson's disease. Mov Disord 2011;26 Suppl 3:S42–80.
55. Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004;19:831–3.
56. Morgante L, Epifanio A, Spina E, et al. Quetiapine and clozapine in parkinsonian patients with dopaminergic psychosis. Clin Neuropharmacol 2004;27:153–6.
57. Miyasaki JM, Shannon K, Voon V, et al. Practice parameter: evaluation and treatment of depression, psychosis, and dementia in Parkinson disease (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2006;66:996–1002.
58. Riedel O, Klotsche J, Spottke A, et al. Cognitive impairment in 873 patients with idiopathic Parkinson's disease. Results from the German Study on Epidemiology of Parkinson's Disease with Dementia (GEPAD). J Neurol 2008;255:255–64.
59. Aarsland D, Andersen K, Larsen JP, et al. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol 2003;60:387–92.
60. Aarsland D, Andersen K, Larsen JP, et al. Risk of dementia in Parkinson's disease: a community-based, prospective study. Neurology 2001;56:730–6.
61. Riggeal BD, Crucian GP, Seignourel P, et al. Cognitive decline tracks motor progression and not disease duration in Parkinson patients. Neuropsychiatr Dis Treat 2007;3:955–8.
62. Hughes TA, Ross HF, Musa S, et al. A 10-year study of the incidence of and factors predicting dementia in Parkinson's disease. Neurology 2000;54:1596–602.
63. Hobson P, Meara J. Risk and incidence of dementia in a cohort of older subjects with Parkinson's disease in the United Kingdom. Mov Disord 2004;19:1043–9.
64. Hoops S, Nazem S, Siderowf AD, et al. Validity of the MoCA and MMSE in the detection of MCI and dementia in Parkinson disease. Neurology 2009;73:1738–45.
65. Louis ED, Marder K, Cote L, et al. Mortality from Parkinson disease. Arch Neurol 1997;54:260–4.
66. Fernandez HH. Nonmotor complications of Parkinson disease. Cleve Clin J Med 2012;79 Suppl 2:S14–8.
67. Sollinger AB, Goldstein FC, Lah JJ, et al. Mild cognitive impairment in Parkinson's disease: subtypes and motor characteristics. Parkinsonism Relat Disord 2010;16:177–80.
68. Mak E, Zhou J, Tan LC, et al. Cognitive deficits in mild Parkinson's disease are associated with distinct areas of grey matter atrophy. J Neurol Neurosurg Psychiatry 2013 Oct 16.
69. Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson's disease. N Engl J Med 2004;351:2509–18.
70. Aarsland D, Ballard C, Walker Z, et al. Memantine in patients with Parkinson's disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial. Lancet Neurol 2009;8:613–8.
71. Leroi I, Overshott R, Byrne EJ, et al. Randomized controlled trial of memantine in dementia associated with Parkinson's disease. Mov Disord 2009;24:1217–21.
72. Speelman AD, van de Warrenburg BP, van Nimwegen M, et al. How might physical activity benefit patients with Parkinson disease? Nat Rev Neurol 2013;7:528–34.
73. Naismith SL, Mowszowski L, Diamond K, Lewis SJ. Improving memory in Parkinson's disease: a healthy brain ageing cognitive training program. Mov Disord 2013;28:1097–103.
74. Weintraub D, Koester J, Potenza MN, et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol 2010;67:589–95.
75. Voon V, Reynolds B, Brezing C, et al. Impulsive choice and response in dopamine agonist-related impulse control behaviors. Psychopharmacology (Berl) 2010;207:645–59.
76. Weintraub D, Siderowf AD, Potenza MN, et al. Association of dopamine agonist use with impulse control disorders in Parkinson disease. Arch Neurol 2006; 63:969–73.
77. Pontone G, Williams JR, Bassett SS, Marsh L. Clinical features associated with impulse control disorders in Parkinson disease. Neurology 2006;67:1258–61.
78. Voon V, Mehta AR, Hallett M. Impulse control disorders in Parkinson's disease: recent advances. Curr Opin Neurol 2011;24:324–30.
79. Thomas A, Bonanni L, Gambi F, et al. Pathological gambling in Parkinson disease is reduced by amantadine. Ann Neurol 2010;68:400–4.
80. Miyasaki JM, Al Hassan K, Lang AE, Voon V. Punding prevalence in Parkinson's disease. Mov Disord 2007;22:1179–81.
81. Nguyen FN, Chang YL, Okun MS, et al. Prevalence and characteristics of punding and repetitive behaviors among Parkinson patients in North-Central Florida. Int J Geriatr Psychiatry 2010;25:540–1.
82. Sohtaoglu M, Demiray DY, Kenangil G, et al. Long term follow-up of Parkinson's disease patients with impulse control disorders. Parkinsonism Relat Disord 2010;16:334–7.
83. Antonini A, Cilia R. Behavioural adverse effects of dopaminergic treatments in Parkinson's disease: incidence, neurobiological basis, management and prevention. Drug Saf 2009;
32:475–88.
84. Miwa H, Morita S, Nakanishi I, Kondo T, Stereotyped behaviors or punding after quetiapine administration in Parkinson's disease. Parkinsonism Relat Disord 2004;10:177–80.
85. Skorvanek M, Rosenberger J, Gdovinova Z, et al. Apathy in elderly nondemented patients with parkinson's disease: clinical determinants and relationship to quality of life. J Geriatr Psychiatry Neurol 2013;26:237–43.
86. Marin RS, Fogel BS, Hawkins J, et al. Apathy: a treatable syndrome. J Neuropsychiatry Clin Neurosci 1995;7:23–30.
87. Pluck GC, Brown RG. Apathy in Parkinson's disease. J Neurol Neurosurg Psychiatry 2002;73:636–42.
88. Kirsch-Darrow L, Fernandez HH, Marsiske M, et al. Dissociating apathy and depression in Parkinson disease. Neurology 2006;67:33–8.
89. Pedersen KF, Alves G, Aarsland D, Larsen JP. Occurrence and risk factors for apathy in Parkinson disease: a 4-year prospective longitudinal study. J Neurol Neurosurg Psychiatry 2009;80:1279–82.
90. Czernecki V, Pillon B, Houeto JL, et al. Motivation, reward, and Parkinson's disease: influence of dopatherapy. Neuropsychologia 2002;40:2257–67.
91. Parkinson J. An essay on the shaking palsy. Sherwood, Neely, and Jones; 1817.
92. Schenck CH, Mahowald WM. REM sleep behavior disorder: clinical, developmental, and neuroscience perspectives 16 years after its formal identification in SLEEP. Sleep 2002;25:120–38.
93. Sixel-Doring F, Trautmann E, Mollenhauer B, Trenkwalder C. Associated factors for REM sleep behavior disorder in Parkinson disease. Neurology 2011;77:1048–54.
94. Eisensehr I, v Lindeiner H, Jager M, Noachtar S. REM sleep behavior disorder in sleep-disordered patients with versus without Parkinson's disease: is there a need for polysomnography? J Neurol Sci 2001;186:7–11.
95. Postuma RB, Gagnon JF, Montplaisir JY. REM sleep behavior disorder and prodromal neurodegeneration - where are we headed? Tremor Other Hyperkinet Mov (N Y) 2013;3.
96. Iranzo A, Tolosa E, Gelpi E, et al. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: an observational cohort study. Lancet Neurol 2013;12: 443–53.
97. Schenck CH, Mahowald MW. Rapid eye movement sleep parasomnias. Neurol Clin 2005;23:1107–26.
98. Anderson KN, Shneerson JM. Drug treatment of REM sleep behavior disorder: the use of drug therapies other than clonazepam. J Clin Sleep Med 2009;5:235–9.
99. Boeve BF, Silber MH, Ferman TJ. Melatonin for treatment of REM sleep behavior disorder in neurologic disorders: results in 14 patients. Sleep Med 2003;4:281–4.
100. Kunz D, Mahlberg R. A two-part, double-blind, placebo-controlled trial of exogenous melatonin in REM sleep behaviour disorder. J Sleep Res 2010;19:591–6.
101. Perroud N, Lazignac C, Baleydier B, et al. Restless legs syndrome induced by citalopram: a psychiatric emergency? Gen Hosp Psychiatry 2007;29:72–4.
102. Buskova J, Vorlova T, Pisko J, Sonka K, Severe sleep-related movement disorder induced by sertraline. Sleep Med 2012;13:769–70.
103. Rittmannsberger H, Werl R. Restless legs syndrome induced by quetiapine: report of seven cases and review of the literature. Int J Neuropsychopharmacol 2013;16:1427–31.
104. Perez-Lloret S, Rey MV, Bondon-Guitton E, et al. Drugs associated with restless legs syndrome: a case/noncase study in the French pharmacovigilance database. J Clin Psychopharmacol 2012;32:824–7.
105. Aurora RN, Kristo DA, Bista SR, et al. The treatment of restless legs syndrome and periodic limb movement disorder in adults-an update for 2012: practice parameters with an evidence-based systematic review and meta-analyses: an American Academy of Sleep Medicine Clinical Practice Guideline. Sleep 2012;35:1039–62.
106. Covassin N, Neikrug AB, Liu L, et al. Clinical correlates of periodic limb movements in sleep in Parkinson's disease. J Neurol Sci 2012;316:131–6.
107. Rios Romenets S, Postuma RB. Treatment of restless legs syndrome. Curr Treat Options Neurol 2013;15:396-409.
108. Hogl B, Paulus W, Clarenbach P, Trenkwalder C, Restless legs syndrome: diagnostic assessment and the advantages and risks of dopaminergic treatment. J Neurol 2006;253 Suppl 4:IV22-8.
109. Garcia-Borreguero D, Kohnen R, Silber MH, et al. The long-term treatment of restless legs syndrome/Willis-Ekbom disease: evidence-based guidelines and clinical consensus best practice guidance: a report from the International Restless Legs Syndrome Study Group. Sleep Med 2013;14:675–84.
110. Montagna P, Hornyak M, Ulfberg J, et al. Randomized trial of pramipexole for patients with restless legs syndrome (RLS) and RLS-related impairment of mood. Sleep Med 2011;12:34–40.
111. Partinen M, Hirvonen K, Jama L, et al. Efficacy and safety of pramipexole in idiopathic restless legs syndrome: a polysomnographic dose-finding study--the PRELUDE study. Sleep Med 2006;7:407–17.
112. Trenkwalder C Garcia-Borreguero D, Montagna P, et al. Ropinirole in the treatment of restless legs syndrome: results from the TREAT RLS 1 study, a 12 week, randomised, placebo controlled study in 10 European countries. J Neurol Neurosurg Psychiatry 2004;75:92–7.
113. Hening WA, Allen RP, Ondo WG, et al. Rotigotine improves restless legs syndrome: a 6-month randomized, double-blind, placebo-controlled trial in the United States. Mov Disord 2010;25:1675–83.
114. Oertel WH, Benes H, Garcia-Borreguero D, et al. Rotigotine transdermal patch in moderate to severe idiopathic restless legs syndrome: a randomized, placebo-controlled polysomnographic study. Sleep Med 2010;11:848–56.
115. Lee DO, Ziman RB, Perkins AT, et al. A randomized, double-blind, placebo-controlled study to assess the efficacy and tolerability of gabapentin enacarbil in subjects with restless legs syndrome. J Clin Sleep Med 2011;7:282–92.
116. Kushida CA, Walters AS, Becker P, et al. A randomized, double-blind, placebo-controlled, crossover study of XP13512/GSK1838262 in the treatment of patients with primary restless legs syndrome. Sleep 2009;32:159–68.
117. Menza M, Dobkin RD, Marin H, Bienfait K. Sleep disturbances in Parkinson's disease. Mov Disord 2010;25 Suppl 1:S117–22.
118. Louter M, van Sloun RJ, Pevernagie DA, et al. Subjectively impaired bed mobility in Parkinson disease affects sleep efficiency. Sleep Med 2013;14:668–74.
119. Menza M, Dobkin RD, Marin H, et al. Treatment of insomnia in Parkinson's disease: a controlled trial of eszopiclone and placebo. Mov Disord 2010;25:1708–14.
120. Nowell PD, Mazumdar S, Buysse DJ, et al. Benzodiazepines and zolpidem for chronic insomnia: a meta-analysis of treatment efficacy. JAMA 1997;278:2170–7.
121. Rios Romenets S, Creti L, Fichten C, et al. Doxepin and cognitive behavioural therapy for insomnia in patients with Parkinson's disease – a randomized study. Parkinsonism Relat Disord 2013;19:670–5.
122. Ondo WG, Dat Vuong K, Khan H, et al. Daytime sleepiness and other sleep disorders in Parkinson's disease. Neurology 2001;57:1392–6.
123. Razmy A, Lang AE, Shapiro CM, Predictors of impaired daytime sleep and wakefulness in patients with Parkinson disease treated with older (ergot) vs newer (nonergot) dopamine agonists. Arch Neurol 2004;61:97–102.
124. Holloway RG, Shoulson I, Fahn S, et al. Pramipexole vs levodopa as initial treatment for Parkinson disease: a 4-year randomized controlled trial. Arch Neurol 2004;61:1044–53.
125. Paus S, Brecht HM, Koster J, et al. Sleep attacks, daytime sleepiness, and dopamine agonists in Parkinson's disease. Mov Disord 2003;18:659–67.
126. Hogl B, Saletu M, Brandauer E, et al. Modafinil for the treatment of daytime sleepiness in Parkinson's disease: a double-blind, randomized, crossover, placebo-controlled polygraphic trial. Sleep 2002;25:905–9.
127. Ondo WG, Fayle R, Atassi F, Jankovic J. Modafinil for daytime somnolence in Parkinson's disease: double blind, placebo controlled parallel trial. J Neurol Neurosurg Psychiatry 2005;76:1636-–9.
128. Adler CH, Caviness JN, Hentz JG, et al. Randomized trial of modafinil for treating subjective daytime sleepiness in patients with Parkinson's disease. Mov Disord 2003;18:287–93.
129. Devos D, Krystkowiak P, Clement F, et al. Improvement of gait by chronic, high doses of methylphenidate in patients with advanced Parkinson's disease. J Neurol Neurosurg Psychiatry 2007;78:470–5.
130. Tyne HL, Taylor J, Baker GA, Steiger MJ. Modafinil for Parkinson's disease fatigue. J Neurol 2010;257:452–6.
131. Avorn J, Schneeweiss S, Sudarsky LR, et al. Sudden uncontrollable somnolence and medication use in Parkinson disease. Arch Neurol 2005;62:1242–8.
132. Trotti LM, Bliwise DL. No increased risk of obstructive sleep apnea in Parkinson's disease. Mov Disord 2010;25:2246–9.
133. da Silva-Junior FP, do Prado GF, Barbosa ER, et al. Sleep disordered breathing in Parkinson's disease: A critical appraisal. Sleep Med Rev 2013 Jul 22.
134. Noradina AT, Karim NA, Hamidon BB, et al. Sleep-disordered breathing in patients with Parkinson's disease. Singapore Med J 2010;51:60–4.
135. Oerlemans WG, de Weerd AW. The prevalence of sleep disorders in patients with Parkinson's disease. A self-reported, community-based survey. Sleep Med 2002;3:147–9.
136. Low PA. Prevalence of orthostatic hypotension. Clin Auton Res 2008;18 Suppl 1:8–13.
137. Goldstein DS. Orthostatic hypotension as an early finding in Parkinson's disease. Clin Auton Res 2006;16:46–54.
138. Sharabi Y, Goldstein DS. Mechanisms of orthostatic hypotension and supine hypertension in Parkinson disease. J Neurol Sci 2011;310:123–8.
139. Sanchez-Ferro A, Benito-Leon J, Gomez-Esteban JC. The management of orthostatic hypotension in Parkinson's disease. Front Neurol 2013;4:64.
140. Lahrmann H, Cortelli P, Hilz M, et al. EFNS guidelines on the diagnosis and management of orthostatic hypotension. Eur J Neurol 2006;13:930–6.
141. Schoffer KL, Henderson RD, O'Maley K, O'Sullivan JD. Nonpharmacological treatment, fludrocortisone, and domperidone for orthostatic hypotension in Parkinson's disease. Mov Disord 2007;22:1543–9.
142. Singer W, Sandroni P, Opfer-Gehrking TL, et al. Pyridostigmine treatment trial in neurogenic orthostatic hypotension. Arch Neurol 2006;63:513–8.
143. Low PA, Gilden JL, Freeman R, et al. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA 1997;277:1046–51.
144. Kaufmann H. L-dihydroxyphenylserine (Droxidopa): a new therapy for neurogenic orthostatic hypotension: the US experience. Clin Auton Res 2008;18 Suppl 1:19–24.
145. Magerkurth C, Schnitzer R, Braune S. Symptoms of autonomic failure in Parkinson's disease: prevalence and impact on daily life. Clin Auton Res 2005;15:76–82.
146. Makaroff L, Gunn A, Gervasoni C, Richy F. Gastrointestinal Disorders in Parkinson's Disease: Prevalence and Health Outcomes in a US Claims Database. J Parkinsons Dis 2011;1:65–74.
147. Abbott RD, Petrovitch H, White LR, et al. Frequency of bowel movements and the future risk of Parkinson's disease. Neurology 2001;57:456–62.
148. Abbott RD, Ross GW, Petrovitch H, et al. Bowel movement frequency in late-life and incidental Lewy bodies. Mov Disord 2007;22:1581–6.
149. Zangaglia R, Martignoni E, Glorioso M, et al. Macrogol for the treatment of constipation in Parkinson's disease. A randomized placebo-controlled study. Mov Disord 2007;22:1239–44.
150. Ondo WG, Kenney C, Sullivan K, et al. Placebo-controlled trial of lubiprostone for constipation associated with Parkinson disease. Neurology 2012;78:1650–4.
151. Cersosimo MG, Benarroch EE. Neural control of the gastrointestinal tract: implications for Parkinson disease. Mov Disord 2008;23:1065–75.
152. Reddymasu SC, Soykan I, McCallum RW. Domperidone: review of pharmacology and clinical applications in gastroenterology. Am J Gastroenterol 2007;102:2036–45.
153. Argolo N, Sampaio M, Pinho P, et al. Do swallowing exercises improve swallowing dynamic and quality of life in Parkinson's disease? NeuroRehabilitation 2013;32:949–55.
154. Troche MS, Sapienza CM, Rosenbek JC. Effects of bolus consistency on timing and safety of swallow in patients with Parkinson's disease. Dysphagia 2008;23:26–32.
155. Edwards LL, Quigley EM, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson's disease: frequency and pathophysiology. Neurology 1992;42:726–32.
156. Kalf JG, Smit AM, Bloem BR, et al. Impact of drooling in Parkinson's disease. J Neurol 2007;254:1227–32.
157. Leibner J, Ramjit A, Sedig L, et al. The impact of and the factors associated with drooling in Parkinson's disease. Parkinsonism Relat Disord 2010;16:475–7.
158. Mancini F, Zangaglia R, Cristina S, et al. Double-blind, placebo-controlled study to evaluate the efficacy and safety of botulinum toxin type A in the treatment of drooling in parkinsonism. Mov Disord 2003;18:685–8.
159. Lagalla G, Millevolte M, Capecci M, et al. Long-lasting benefits of botulinum toxin type B in Parkinson's disease-related drooling. J Neurol 2009;256:563–7.
160. Lagalla G, Millevolte N, Capecci M, et al. Botulinum toxin type A for drooling in Parkinson's disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2006;21:704–7.
161. Arbouw ME, Movig KL, Koopmann M, et al. Glycopyrrolate for sialorrhea in Parkinson disease: a randomized, double-blind, crossover trial. Neurology 2010; 74:1203–7.
162. South AR, Somers, Jog MS. Gum chewing improves swallow frequency and latency in Parkinson patients: a preliminary study. Neurology 2010;74:1198–202.
163. Winge K, Nielsen KK. Bladder dysfunction in advanced Parkinson's disease. Neurourol Urodyn 2012;31:1279–83.
164. Madhuvrata P, Singh PM, Hasafa Z, Abdel-Fattah M. Anticholinergic drugs for adult neurogenic detrusor overactivity: a systematic review and meta-analysis. Eur Urol 2012;62:816–30.
165. Soljanik I. Efficacy and safety of botulinum toxin a intradetrusor injections in adults with neurogenic detrusor overactivity/neurogenic overactive bladder: a systematic review. Drugs 2013;73:1055–66.
166. Sakakibara R, Uchiyama T, Yamanishi T, Kishi M. Genitourinary dysfunction in Parkinson's disease. Mov Disord 2010;25:2–12.
167. Kummer A, Cardoso F, Teixeira AL. Loss of libido in Parkinson's disease. J Sex Med 2009;6:1024–31.
168. Lombardi G, Nelli F, Celso M, et al. Treating erectile dysfunction and central neurological diseases with oral phosphodiesterase type 5 inhibitors. Review of the literature. J Sex Med 2012 9(4): 970–85.
169. Dula E, Bukofzer S, Perdok R, George M. Double-blind, crossover comparison of 3 mg apomorphine SL with placebo and with 4 mg apomorphine SL in male erectile dysfunction. Eur Urol 2001;39(5): 558–3
170. Negre-Pages L, Regragui W, Bouhassira D, et al. Chronic pain in Parkinson's disease: the cross-sectional French DoPaMiP survey. Mov Disord 2008;23:1361–9.
171. Beiske AG, Loge JH, Ronningen A, Svensson E. Pain in Parkinson's disease: Prevalence and characteristics. Pain 2009. 141:173–7.
172. Lin CH, Wu RM, H.Y. Chang HY, et al. Preceding pain symptoms and Parkinson's disease: a nationwide population-based cohort study. Eur J Neurol 2013;20:1398–404.
173. Ha AD, Jankovic J. Pain in Parkinson's disease. Mov Disord 2012; 27:485–91.
174. Ford B. Pain in Parkinson's disease. Mov Disord 2010;25 Suppl 1:S98–103.
175. Loher TJ, Burgunder JM, Weber S, et al. Effect of chronic pallidal deep brain stimulation on off period dystonia and sensory symptoms in advanced Parkinson's disease. J Neurol Neurosurg Psychiatry 2002;73:395–9.
176. Ramirez-Ruiz B, Marti MJ, Tolosa E, et al. Longitudinal evaluation of cerebral morphological changes in Parkinson's disease with and without dementia. J Neurol 2005;252:1345–52.
177. Jellinger KA. Formation and development of Lewy pathology: a critical update. J Neurol 2009;256 Suppl 3:270–9.
178. Levy R, Dubois B. Apathy and the functional anatomy of the prefrontal cortex-basal ganglia circuits. Cereb Cortex 2006;16:916–28.
179. Connor JR, Boyer PJ, Menzies SL, et al. Neuropathological examination suggests impaired brain iron acquisition in restless legs syndrome. Neurology 2003;61: 304–9.
180. Earley CJ, Barker P, Horská A, Allen RP. MRI-determined regional brain iron concentrations in early- and late-onset restless legs syndrome. Sleep Med 2006;7: 458–61.
181. Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci 2001;24:726–31.
182. Boeve BF, Silber MH, Saper CB, et al. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain 2007;130(Pt 11): 2770–88.
183. Benarroch EE, Schmeichel AM, Paris J. Involvement of the ventrolateral medulla in parkinsonism with autonomic failure. Neurology 2000;54:963–8.
184. Chudler EH, Dong WK. The role of the basal ganglia in nociception and pain. Pain 1995;60:3–38.
185. Wolters E. Non-motor extranigral signs and symptoms in Parkinson's disease. Parkinsonism Relat Disord 2009;15 Suppl 3:S6–12.
186. Davidsdottir S, Cronin-Golomb A, Lee A. Visual and spatial symptoms in Parkinson's disease. Vision Res 2005;45:1285–96.
From the Department of Neurology, Movement Disorders Division, University of Pittsburgh Medical Center, Pittsburgh, PA.
Abstract
- Objective: To review the prevalence, diagnosis, and treatment of the nonmotor symptoms (NMS) associated with Parkinson’s disease (PD).
- Methods: Narrative review of the literature.
- Results: The NMS of PD are becoming increasingly recognized as having a critical role in the impact of this neurodegenerative movement disorder. This has led to significant investigative efforts to identify new or better NMS therapies. The preponderance of PD patients will be diagnosed with 1 or multiple NMS during the course of their disease, with many of these symptoms occurring months or even years prior to receiving the PD diagnosis. Despite the high prevalence and impact on disease burden, NMS often go undetected due to a lack of reporting by patients or insufficient interrogation by physicians. Further complicating NMS management is that only a few therapies have the level of evidence needed to support their use in the treatment of NMS.
- Conclusion: The practitioner needs to be aware of NMS and conduct thorough patient questioning in order to recognize, diagnose, and address NMS in PD patients.
Parkinson’s disease (PD) is a neurodegenerative movement disorder with an estimated prevalence of 1% to 2% among the population over the age of 65 years [1]. Recognition and clinical diagnosis of PD is primarily made based on the cardinal motor features, including rigidity, tremor, bradykinesia, and postural instability. The motor symptoms are neuropathologically associated with accumulation of alpha-synuclein with Lewy body formation and neurodegeneration of the nigrostriatal dopamine system. Postmortem evaluation of the brains of PD patients has revealed more widespread degeneration in nondopaminergic systems, including several brainstem nuclei (raphe nucleus, locus ceruleus, dorsal vagal nucleus), limbic and neocortical structures, as well as the peripheral autonomic system [2,3].
The nonmotor symptoms (NMS) of PD are the clinical manifestations of this extensive degeneration, which suggests that NMS are intrinsic and fundamental features of PD. NMS are exceedingly common, and up to 90% of PD patients will experience nonmotor features, including depression, anxiety, sleep disturbances, cognitive impairment, and dysautonomia [4,5] (Table).
NMS have a greater impact on quality of life as compared to the motor symptoms [6,7], but are frequently underrecognized [8]. Evidence suggests that unless there is systematic and specific interrogation by practioners, NMS will elude recognition [9–11]. Recognizing NMS as part of PD is complicated by the fact that these symptoms are common in the general population and not specific for PD [12,13]. NMS can occur at any stage of the disease and may predate diagnosis [12], although as PD progresses the NMS become more prevalent, with a greater impact on health care costs and institutionalization rates than motor features [14,15].
Neuropsychiatric Symptoms
Depression
Epidemiology and Diagnosis
Depression is one of the most common neuropsychiatric manifestations observed in PD patients, with prevalence reports between 4% and 72%, though likely to be closer to 30% to 45% [16–20]. The severity of depression in the PD population has been shown to be greater than in patients with matched chronic disabilities [21,22] and also greater than in the general population over the age of 65 years [23]. The onset of depression can occur at any stage of the disease, even predating the diagnosis. Additionally, depression has more than twice the impact on health status than motor symptoms [24].
Though the mechanisms are not fully understood, it is suspected that psychosocial as well as neuropathological changes contribute to the pathogenesis of depression in PD. In a study comparing 104 PD patients and 61 patients with equivalent disability scores, functional disability was found to be responsible for only 9% of the variation of depression scores [22]. The increased prevalence of depression in PD patients can in part be explained by the neuropathological changes seen in post-mortem studies. Two neurotransmitters that are fundamental in the pathogenesis of depression are serotonin, from the raphe nuclei, and norepinepherine, from the locus ceruleus [20]. Both of these brainstem structures demonstrate alpha-synucleinopathy-associated degeneration and these changes can precede the development of motor dysfunction [3].
Diagnosing depression in PD is complicated by the fact that there is overlap between other PD symptoms and clinical features of depression (ie, amotivation, bradykinesia, fatigue, and sleep disturbances). However, many depressed PD patients are less likely to report feelings of guilt or failure and tend to have higher rates of anxiety [9,20,25]. Typically, PD patients are more likely to be diagnosed with minor depression or dysthymia rather than a major depressive disorder [19,20]. Formal testing through systematic questionnaires are diagnostically useful in the clinic, and serial testing can reveal changes over time to guide more effective treatment. Validated tools to evaluate depression in PD include the Beck Depression Inventory, Hamilton Depression Rating Scale, Montgomery-Asberg Depression Rating Scale, Geriatric DRS, and Hospital Anxiety and Depression scale [20].
Treatment Options
Treatment of depression in PD demonstrates generally poorer responses to typical antidepressants and side effects that may worsen other PD symptoms. Selective serotonin reuptake inhibitors (SSRIs) have been widely used as there are generally few drug-drug interactions and minimal effect on motor symptoms; however, several studies have demonstrated little benefit on depression in PD [26]. In a randomized, double-blind, placebo-controlled trial of the antidepressants paroxetine and venlafaxine, both were found to be effective and well tolerated [27]. Tricyclic anti-depressants (TCAs) have also demonstrated efficacy. In randomized controlled trials comparing TCAs to SSRIs, a greater benefit on depression symptoms has been found with TCAs [28–30]. The use of TCAs, however, is limited by anticholinergic side effects that occasionally worsen orthostatic hypotension or cognitive impairment [15,31]. Dopamine agonists have also been studied in depressed PD patients. In a randomized, double-blind, placebo-controlled trial [32] and a prospective observational study [33], pramipexole demonstrated significant improvements in depression symptoms. Ropinirole also demonstrated significant symptomatic improvement [34]. These studies suggest that while SSRIs are commonly used, evidence is accumulating to support the role of TCAs, SNRIs, and dopamine agonists in the treatment of depression in PD.
Other therapies have also been tried in pharmacologic-resistant patients. Electroconvulsive therapy has been reported to improve both depression and motor symptoms [35,36]; however, this is a treatment reserved for patients with severe and drug-refractory depression. A randomized controlled trial investigating cognitive behavioral therapy has also demonstrated improvement of depression scores [37]. The role of physical activity as treatment for depression in PD patients is unclear. As described in a recent review by Loprinzi et al [38], the literature is contradictory, with one group experiencing reduced depression but with no signficant effect in several other studies.
Anxiety
Epidemiology and Diagnosis
The prevalence of anxiety in PD patients is about 40% [39], which is 2 times greater than in the general population [9]. Anxiety may worsen PD symptoms, especially tremor and cognition. Risk factors for anxiety include the female gender, greater motor fluctuations, prior history of anxiety, and younger age of PD onset [40]. As with depression, some patients also report worsening of anxious symptoms during “off” states [41]. Screening tools that have been validated to help practitioners identify anxiety in PD include the Hospital Anxiety and Depression Scale, Beck Anxiety Inventory, Zung Self-rating Anxiety Scale, Spielberger State Trait Anxiety Inventory, and Hamilton Anxiety Rating Scale [15].
Treatment Options
The treatment of diagnosed anxiety in PD is primarily with benzodiazepines, which are particularly beneficial in patients whose tremors are exacerbated by anxiety or stress. The use of benzodiazepines has not been evaluated by a randomized controlled trial and use should be limited given the potential risks of sedation, cognitive effects, and psychomotor agitation. Other case studies have found benefit with serotonergic medications like fluoxetine or citalopram (especially with concomitant depression) or with optimization of levodopa therapy [42,43].
Hallucinations, Delusions, and Psychosis
Epidemiology
The prevalence of visual hallucinations in PD patients is about 20% to 40% [44,45]. Risk factors for psychotic symptoms include cognitive impairment, advanced age, prolonged duration of disease, depression, severe dysautonomia, and sleep disorders [46–48]. Early recognition of hallucinations is critical because of a strong correlation between the manifestation of psychosis and the need for nursing home placement or hospitalization. With early and effective treatment there is a decreased need for placement and a reduction on caregiver burden [44,49].
Treatment Options
Hallucinations can occur in delirium and it is important to first rule out an underlying infection or an offending medication, especially if there is a sudden onset or worsening of symptoms. Psychotic symptoms have been reported in drug-naive patients, though they are often iatrogenically induced with dopaminergic agents. All antiparkinsonian medications are capable of inducing or exacerbating hallucinations [9,50]. Additionally, psychotic symptoms tend to improve when dopaminergic agonists are reduced or eliminated. However, there is no clear relationship between the dose of dopaminergic agents and manifestation of hallucinations [48,51,52]. If hallucinations persist or there are motor complications that arise from reduction of dopaminergic agents, initiation of clozapine has been demonstrated to be efficacious in a rater-blinded prospective study and in a retrospective analysis [53–55]; however, regular monitoring for neutropenia is required. Quetiapine has demonstrated similar benefit without significant effects on motor symptoms in a randomized, rater-blinded study and in an evidence-based review [56,57]. It is also important to review or eliminate other medications that may contribute to hallucinations.
Cognitive Impairment
Epidemiology
The prevalence of dementia in the PD population is 20% to 40% [58], though almost 80% of PD patients ultimately develop cognitive decline [59]. Overall, a PD patient is 6 times more likely to develop dementia than someone in the general population [60]. There may be parallel progression of cognitive impairment and motor symptoms, but there is no correlation with overall duration of disease [60,61]. Risk factors linked with the presence of dementia include older age at onset of PD, presence of hallucinations, and male gender [62,63].
Cognitive dysfunction can be detected early in PD through neuropsychological testing; however, impairment of cognition is often insidious and may not be appreciated until symptoms become severe. Several screening tools have been used to evaluate for cognitive impairment in PD including the Mini-Mental State Exam (MMSE), Montreal Cognitive Assessment (MoCA), Mini-Mental Parkinson, Scales for Outcomes of Parkinson’s disease–Cognition, and others. Accumulating evidence, however, is suggestive of the superiority of the MoCA in the detection of cognitive deficits associated
with PD [64].
Dementia is a substantial burden for the caregiver and is a significant contributor to mortality in PD patients [65]. Cognitive impairment often presents with other behavioral symptoms, which further hastens placement outside the home and increases cost of caring for PD patients [49,66].
Cognitive impairment in Parkinson’s disease is typically associated with degeneration of primarily subcortical structures. PD patients with mild cognitive impairment were found to have deficits most significantly in memory, executive function, memory, and language abilities [67]. A recent study by Mak et al evaluated grey matter volumes by structural MRI in PD patients with evidence of mild cognitive impairment by MMSE and MoCA as compared with findings in cognitively intact patients. This demonstrated decreased brain volumes in areas that correlate with affected cognitive domains including the left insula, left superior frontal and left middle temporal areas [68].
Treatment Options
Prior to initiation of therapy, it is important to evaluate the patient for depression and to rule out pseudodementia. Bradyphrenia, or slowness of thought, should also be considered, as this symptom may also lead to an incorrect dementia diagnosis. Lastly, a thorough review of medications should be performed and offending agents including anticholinergics, TCAs, dopamine agonists, and amantadine should be discontinued as these can worsen cognition.
Rivastigmine has demonstrated modest improvement in cognitive performance in PD patients with dementia in a large multicenter, placebo-controlled study [69]. Other cholinesterase inhibitors (ie, donepezil or galantamine) are not recommended at this time due to limited studies or contradictory results in the literature [31,54]. Caution is advised with use of cholinesterase inhibitors as they may worsen tremor or autonomic dysfunction; also, use is limited by nausea or other gastrointestinal symptoms. Memantine, an NMDA receptor antagonist, has also been investigated in randomized, double-blind, placebo-controlled trials and demonstrated modest improvement of cognition and is generally well tolerated [70,71].
Nonpharmacologic therapy includes physical exercise, which has demonstrated improvement in memory tasks and processing speed [72]. Cognitive training has been less rigorously studied; however, a recent single-blinded controlled study demonstrated significant improvement of learning and memory in PD patients who completed computer-based cognitive training [73].
Compulsive Disorders
Impulse Control Disorders
Impulse control disorders (ICDs) are inappropriate behaviors resulting from a failure to resist an impulse, which leads to pleasure-seeking activities at the expense of relationships and ability to function socially. In PD, ICDs are expressed as pathologic gambling, hypersexuality, binge eating, compulsive shopping, and excessive spending [9,66]. The prevalence of all ICDs in PD is 15% to 20% and a patient may be diagnosed with multiple ICDs [74]. Dopamine agonist use has been implicated in the development of ICDs and this risk is further increased with the addition of levodopa [75,76]. Clinical features associated with ICDs include young age of onset, male gender, family history of addiction, depression or anxiety, and disinhibition or impulsive traits [77,78].
Traditionally, treatment consists of reduction or elimination of dopamine agonists, though adjustment of levodopa therapy may also be necessary. Amantadine as an adjunct therapy has been shown in a randomized, double-blind crossover study to reduce impulsivity in a few patients with pathologic gambling [79].
Dopamine Dysregulation Syndrome
Dopamine dysregulation syndrome (DDS) is characterized by compulsive use of dopaminergic medications beyond what is needed to treat parkinsonian symptoms, and is associated with social impairment. Patients describe addictive symptoms like craving or intense desire to obtain more dopaminergic medication [9,74]. Like ICDs, treatment of DDS consists of modification to dopaminergic medications, though patients with DDS may also require psychiatric evaluation and treatment.
Punding
Punding is another compulsive disorder that is defined as an intense fascination with objects and is associated with repetitive handling, manipulation, sorting, or arrangement of the items [80]. Occurrence of punding has been associated with higher total daily levels of levodopa, although one study has also implicated dopamine agonists [15,81]. As with the other compulsive disorders, punding also tends to respond well to reduction or discontinuation of levodopa. Studies have demonstrated modest benefit with SSRIs or atypical antipsychotics in long-term follow-up [82,83], though one study reported worsening of punding with quetiapine [84].
Apathy
Epidemiology and Treatment
Apathy is often characterized by a loss of motivation or inability to initiate goal-directed behavior, which results in dependence on others for activities of daily living and increases caregiver burden [85]. Patients demonstrate indifference, lack of interest, or inability to express or describe emotion. The apathetic patient may lack spontaneous and voluntary activity, and their affect display is often flattened [86].
With a prevalence of 30% to 50% [87], apathy is as common as depression in PD patients [66,88]. Risk factors associated with apathy include advanced age, severity of depression, severity of motor dysfunction, and dementia [89]. Apathy is frequently mistaken for depression given the significant overlap in symptoms; however, the patient with pure apathy will deny sadness or depressed feelings. It is also important to distinguish apathy from motor impairment or cognitive dysfunction that could explain the behavioral changes. No medications have reliably been shown to improve apathy, though it may be improved with initiation of dopaminergic therapy, especially early in the course [86,90].
Sleep Disorders
The original report of PD by James Parkinson describes sleep disturbances and daytime somnolence [91], which suggests that sleep disorders may be an intrinsic feature of the neurodegenerative process of PD itself.
REM Behavioral Disorder
Epidemiology and Diagnosis
Rapid eye movement behavioral disorder (RBD) is a parasomnia characterized by vocalizations and motor activity during dreaming due to loss of normal atonia associated with rapid eye movement (REM) sleep. Patients enact their dreams, which may lead to violent behaviors that can injure the patient or their bed partner. RBD is seen in 25% to 50% of PD patients [92,93], with variability depending on diagnostic technique and patient selection. Polysomnography is the most important diagnostic tool and demonstrates increased chin tone and limb movements during REM sleep in RBD [94,95]. Diagnosis can also be made clinically with patient and bed partner reports, though sensitivity is only approximately 30% [15].
Interestingly, many studies are now investigating the relationship between presence of RBD and later onset of neurodegenerative disorders. Multiple studies have shown that 40% to 65% of patients diagnosed with idiopathic RBD later develop an alpha-synucleinopathy, which includes PD, dementia with Lewy bodies, or multiple system atrophy within 10 years [92,95]. Prior studies report that as many as 90% of patients with idiopathic RBD develop neurodegenerative synucleinopathy when followed over 14 years [96]. Idiopathic RBD is currently being investigated as a potential clinical marker of pre-symptomatic PD in a multicenter observational study. If RBD is an early marker for neurodegenerative disease, it may be used to identify patients for neuroprotective trials as treatments are developed.
Treatment Options
Low-dose clonazepam (0.25–1 mg) is the mainstay of therapy, especially for patients that injure themselves or bed partners [97]; however, the use of benzodiazepines is historical and there remain no randomized controlled double-blind studies to evaluate the efficacy of clonazepam. Use of clonazepam may be limited by daytime sedation, confusion, or psychomotor agitation [31,97,98]. Melatonin (doses between 3–12 mg at bedtime) has also demonstrated benefit in RBD in a double-blind, placebo-controlled trial and in a small case series, with fewer side effects and no addiction potential as compared to clonazepam [99,100]. Case reports also support the use of several other effective medications, including cholinesterase inhibitors (rivastigmine and donepezil) and dopaminergic agents (pramipexole and levodopa) [15,20].
Restless Leg Syndrome and Periodic Limb Movements in Sleep
Epidemiology
Restless leg syndrome (RLS) and periodic limb movements in sleep (PLMS) cause disruptions of sleep and have an important impact on quality of sleep in PD patients. RLS is described as a strong urge to move the legs, accompanied by an uncomfortable sensation that is exacerbated at rest and relieved by movement. RLS is more frequently diagnosed in patients with PD, though prevalence reports vary widely [15]. Secondary causes for RLS should be investigated including iron deficiency, uremia and polyneuropathy. Several case reports demonstrate onset or worsening of RLS with use of antidepressants [101, 102] or antipsychotics like risperidone, aripiprazole, and quetiapine [103,104].
PLMS occurs in approximately 80% to 90% of patients with RLS, though may be present independently, and when seen on polysomnography is supportive of RLS [105]. PLMS is characterized by repetitive dorsiflexion of the foot, extension of the great toe, and may be accompanied by flexion of the knee and hip. The prevalence of PLMS in PD is approximately 60% and correlates with severity of PD motor features [106].
Treatment Options
Treatment of RLS should be initiated with nonpharmacologic therapies including good sleep hygiene, exercise, leg massage, and heat or ice packs [105,107]. Dopamine (DA) agonists are the primary treatment for RLS; however, even modest adjustments in levodopa can be helpful. One drawback to levodopa therapy is augmentation (a worsening or reappearance of symptoms) when serum levels fall due to the short half-life of levodopa [107,108]. DA agonists are less likely to cause augmentation. Both pramipexole and ropinirole have been extensively investigated in controlled, randomized, double-blind studies with benefits in 70% to 90% of patients with RLS and PLMS; however, there is a risk of developing compulsive behaviors [109–112]. Another option for PD patients is rotigotine, which has demonstrated improvement of RLS symptoms in a randomized, double-blind, placebo-controlled trial and has the added benefit that it may also help with motor symptoms [113,114].
More recently, gabapentin enacarbil has demonstrated improvement of moderate to severe RLS and was well tolerated in multiple randomized, double-blind, placebo-controlled trials [107,115,116]. Lastly, opioids (tramadol, oxycodone, codeine) have been shown to be effective, especially in the treatment of RLS that is refractory to other treatments [105,107].
Insomnia
Epidemiology
The most common sleep disorder in PD is insomnia, with a prevalence between 37% to 88% [14,117]. Insomnia is associated with difficulty in initiation or maintenance of sleep. Disruption of sleep typically leads to daytime somnolence and patient reports of a strong impact on motor disability and overall quality of life. There are several contributors to insomnia in PD patients including nocturia, depression, RLS, dystonia, and akinesia/rigidity/difficulty turning in bed [118].
Treatment Options
The use of carbidopa/levodopa controlled-release formulations at bedtime is associated with improved sleep duration and nocturnal akinesia, although it does not demonstrate a significant improvement in overall sleep ratings [54]. Hypnotics like eszopiclone and zolpidem have also demonstrated improved quality of sleep in limited controlled trials and a meta-analysis, but use is limited by sedation, dizziness, and falls [54,119]. Benzodiazepines improve sleep latency, but there is a risk of cognitive impairment, tolerance, and falls [117,120]. Melatonin at 3 to 5 mg and 50 mg doses have been investigated in 2 randomized, double-blind, placebo-controlled trials; however, there was a modest benefit and it was concluded that there is insufficient evidence to support the use of melatonin [54]. Nevertheless, melatonin is well tolerated and may be tried with minimal risk [54]. More recently, a randomized controlled trial using doxepin has demonstrated improvement of insomnia scores and was generally well tolerated [121].
Excessive Daytime Sleepiness and Abrupt Sleep Onset
EDS and Fatigue: Epidemiology and Treatment
A common complaint by PD patients is excessive daytime sleepiness (EDS), which can be verified with multiple sleep latency testing. EDS frequency varies in the literature, but is seen in approximately 15% to 50% of PD patients [4,122]. The etiology is usually multifactorial, with insomnia, dysautonomia, and depression as contributing factors [117]. A longer duration of symptoms, greater total load of levodopa, cognitive decline, and male gender are all risk factors for EDS [122,123]. It has been proposed that EDS is an intrinsic feature of PD; however, there is also an association with the use of antiparkinsonian medications. A randomized controlled trial demonstrated that use of the dopamine agonist pramipexole was associated with greater somnolence as compared to levodopa therapy (35% vs. 13%); however, this difference was only seen during the initial escalation phase [124]. Additionally, the combined use of dopamine agonists and levodopa has shown an even greater risk of EDS [125]. The evidence for the use of stimulants for EDS is lacking. The few studies conducted with modafinil have not demonstrated a robust improvement of EDS [126–128]. Other stimulants like methylphenidate have been studied with improvement of Epworth Sleepiness Score, though no randomized control trials have been undertaken [129].
It is important to distinguish EDS, a propensity for daytime sleep, from fatigue or excessive tiredness associated with mental or physical exertion [117]. Fatigue is often multifactorial and may be related to insomnia, sleep apnea, sedating effects of medications, frequent awakenings from nocturia, and degeneration of brain areas regulating sleep/wake cycles related to the underlying disease process [20, 117]. It is also important to consider depression and dementia in the differential, as these disorders may be erroneously be diagnosed as fatigue. Treatment of fatigue should include regular mild exercise, maintenance of a stimulating environment, removal of sedating medications, and management of intrinsic sleep disorders if present [117]. The use of stimulants for fatigue is controversial. A small randomized controlled trial (n = 48) using modafinil demonstrated improvement on the global clinical impression scale for fatigue but no significant change on the Fatigue Severity Scale; this study was limited by the power and points to the need for a larger study [130].
Sleep Attacks: Epidemiology and Treatment
Abrupt sleep onset, or “sleep attacks,” occurs when transition from wake to sleep is unavoidable and may occur without warning. Sleep attacks are threefold more likely to occur in patients using DA agonists, with an associated dose-related increase in risk [131]. Adjustment or elimination of DA agonists often improves sleep attacks, though it is important to address concurrent EDS if present. Nonpharmacologic treatments to consider include mild exercise, early morning bright light exposure, and a stimulating environment [117].
Sleep-Disordered Breathing/Obstructive Sleep Apnea
Epidemiology and Treatment
Sleep-disordered breathing (SDB) consists of either a deficit in the drive to breathe as in central sleep apnea, or may be due to an blockage of the airway as seen in obstructive sleep apnea (OSA). Apnea leads to oxygen desaturations that consequently trigger awakenings throughout the night, which in turn is experienced by the patient as daytime somnolence [117]. The prevalence of SDB and OSA is variable in the literature, ranging from no increased risk in PD patients [132,133] to 50% prevalence in PD patients [134,135]. Discussions with bed partners, history of snoring, and clinical reports of EDS or daytime fatigue are important indicators of SDB. Polysomnography confirms the diagnosis and can direct treatment, which frequently includes application of CPAP devices during sleep.
Autinomic Dysfunction
Orthostatic Hypotension
Epidemiology and Diagnosis
Orthostatic hypotension (OH) is defined as a 20-mm Hg fall in systolic blood pressure or 10-mm Hg drop in diastolic blood pressure within 3 minutes of a change in position. The prevalence of OH in PD patients is 30% to 60% [136,137]. Symptoms of OH can occur early in the disease and may precede diagnosis of PD [137]. Patients experience OH as dizziness, drowsiness, palpitations, nausea, or loss of consciousness. Additionally, falls and supine hypertension that accompany OH are associated with increased risk of morbidity and mortality in PD patients [138]. Several medications used in the treatment of PD can exacerbate OH, including levodopa, DA agonists, MAO-B inhibitors, and TCAs [139].
Treatment Options
First-line therapies for OH include nonpharmacologic methods such as compression stockings, sleeping with head elevated to 30 degrees, increased water and salt intake, more frequent small meals, and slowly changing position [140]. Additionally, it is important to discuss the removal or reduction of all antihypertensives with the patient’s PCP. Fludrocortisone (a mineralacorticoid) and domperidone (a peripheral dopamine antagonist not currently approved for use in the United States) modestly improved OH in a 2-phase, randomized, controlled, double-blind, crossover trial [141]. Pyridostigmine has also demonstrated improvement of standing blood pressure and OH symptoms in a double-blind, randomized cross-over study and has the additional benefit of not worsening supine hypertension [142]. Other effective treatments include midodrine, per a randomized, double-blind multicenter study [143], as well as droxidopa in a double-blind, crossover, placebo-controlled study [144]. Currently there is insufficient evidence to support the preferential use of any specific agent in the treatment of OH in PD.
Gastrointestinal Dysmotility
Constipation: Epidemiology and Treatment
Constipation is reported by nearly 60% of PD patients [145]. Constipation can precede the development of motor symptoms of PD, and the prevalence of GI disturbances increases with age and longer duration of disease. Nearly one third of patients will have been diagnosed with a GI disturbance within the year prior to PD diagnosis [146], which is associated with an increased risk for the development PD [147]. People with constipation (defined as < 1 bowel movement per day) but without a PD diagnosis had more nigral Lewy body degeneration postmortem [148] compared with people without constipation.
Treatments for constipation include dietary modification, increased fluid intake, and mild exercise. Macrogol significantly improved constipation in PD patients and was very well tolerated in a randomized placebo-controlled study [149]. Lubiprostone, a GI active prostaglandin, is also effective in the short-term treatment of constipation in a placebo-controlled trial [150].
Gastroparesis: Epidemiology and Treatment
Gastroparesis, like constipation, is related to enteric dopaminergic cell loss and degeneration of the dorsal motor nucleus of the vagus [151]. Patients experience gastroparesis as early satiety, full sensation, and nausea. Decreased gastric motility leads to retention of food as well as medications, which can slow absorption and delay onset of action for many medications including levodopa. Domperidone has both prokinetic and antiemetic properties, which have been beneficial in the treatment of gastroparesis [152], but its use is not currently approved in the United States.
Dysphagia: Epidemiology and Treatment
Dysphagia is associated with more advanced stages of PD as well as a significant increase in morbidity. Swallow exercises have demonstrated improvement of dysphagia [153]. The impact of levodopa therapy on dysphagia in the literature is controversial. Videofluoroscopic examination is the most common method for evaluation of swallowing disorders and provides important information for speech-language pathologists regarding recommendations for dietary modifications [154]. Adjustment of medication regimens to avoid an oral route is also helpful. This includes Parcopa, orally disintegrating carbidopa/levodopa tablets, and transdermal approaches like the rotigotine patch. For some patients, enteral nutrition is needed and placement of nasogastric tubes or percutaneous endoscopic gastrostomy tubes are an option.
Sialorrhea (Drooling)
Epidemiology
Difficulty handling oral secretions due to impaired or infrequent swallowing results in sialorrhea in up to 75% of PD patients [155], which is a significant embarrassment for most patients [156]. PD patients with drooling have difficulty speaking, eating, and engaging in social interactions, which significantly impacts perceived quality of life [157].
Treatment Options
Botulinum toxin (A and B) injections into the submandibular or parotid glands have demonstrated efficacy in multiple double-blind, randomized, placebo-controlled studies for the treatment of sialorrhea in PD patients; however, injections are associated with greater invasiveness and cost [158–160]. Glycopyrrolate, an anticholinergic drug, was also efficacious in the treatment of sialorrhea in the short term in a double-blind, randomized, placebo-controlled study [161]. Alternatively, gum chewing increases swallow frequency, improves drooling, and also shows a benefit with dysphagia [162].
Genitourinary Disturbances
Bladdery dysfunction: Epidemiology and Treatment
Bladder dysfunction in PD is often secondary to hyperactivity of the detrusor muscle leading to urinary urgency, increased urinary frequency, and nocturia. Less commonly, hypoactive detrusor muscle causes difficulty with initiation of urination, delayed bladder emptying, and recurrent infections. Urinary disturbances may occur before the onset of motor symptoms or early on in the disease course [12]. Disease severity is associated with greater urinary disturbances, and more than 50% of advanced PD patients report severe bladder symptoms [163].
Anticholinergic medications such as oxybutynin, solifenacin, and tolterodine are commonly used in the treatment of detrusor hyperactivity and demonstrate significant improvement in detrusor pressure in a recent systemic review and meta-analysis [164]. PD patients on these agents should be closely monitored for side effects including cognitive impairment, somnolence, hallucinations, confusion, and blurred vision. Other treatments include botulinum toxin injections into the detrusor muscle, which has demonstrated safety and efficacy in a recent systematic review [165].
Erectile dysfunction: Epidemiology and Treatment
Erectile dysfunction (ED) is reported by more than 60% of male PD patients [145] and is thought to be related to hypothalamic dysfunction and modification of the dopamine-oxytocin pathway [166]. Effects of PD medications, cognitive impairment, fatigue, apathy, and low testosterone contribute to loss of libido and ED [20,167]. Phosphodiesterase inhibitors such as sildenafil, vardenafil, and tadalafil are possibly useful in the treatment of ED in PD patients, though randomized trials have been limited [166,168]. Apomorphine sublingually is another medication that has demonstrated improvement of ED in a double-blind, crossover study and can be considered for patients with contraindications to phosphodiesterase inhibitors [169].
Sensory Symptoms
Pain
Epidemiology
Sensory disturbances in PD include diminished ability to identify odors, visual abnormalities (blurred vision, abnormal color perception, double vision), and pain. Pain is the most disabling sensory disturbance, though frequently underreported. Nearly two thirds of PD patients report pain, [170], though only half of patients receive any treatment [171]. Pain may also be a presenting symptom that precedes the clinical diagnosis of PD [172,173].
Treatment Options
There are several types of pain described by PD patients, the most common of which is musculoskeletal, typically involving the shoulder. Other types include dystonic, radicular, and central pain [174]. First-line treatment of musculoskeletal complaints includes nonsteroidal anti-inflammatory drugs (NSAIDs) and physiotherapy. Modification of levodopa regimen (including altering timing and frequency or adding controlled release formulations) can often provide relief for dystonic pain, and also for central pain for some patients [173, 174]. Deep brain stimulation, with subthalamic nucleus or globus pallidus targets, has demonstrated improvement with dystonic, central, and musculoskeletal pain in a small clinical study [175].
Conclusion
NMS are an intrinsic part of PD, may predate diagnosis, and substantially affect the majority of patients with PD. For many of these patients, NMS have a greater impact on quality of life and health care costs than the cardinal motor symptoms that define the disease. Many of these symptoms are not recognized by practioners and often are not volunteered by PD patients, making it important for practitioners to routinely and directly inquire about NMS. Treatment of NMS in PD is challenging, and only a few therapies have the level of evidence needed to support their use in the treatment of these problems. Nevertheless, proper recognition and addressing of these symptoms afford the clinician an opportunity to make a positive and potentially significant impact on the PD patient’s quality of life.
Corresponding author: Samay Jain, MD, MS, Dept of Neurology, 811 Kaufmann Bldg, Pittsburgh, PA 15213, jains@upmc.edu.
Financial disclosures: None.
From the Department of Neurology, Movement Disorders Division, University of Pittsburgh Medical Center, Pittsburgh, PA.
Abstract
- Objective: To review the prevalence, diagnosis, and treatment of the nonmotor symptoms (NMS) associated with Parkinson’s disease (PD).
- Methods: Narrative review of the literature.
- Results: The NMS of PD are becoming increasingly recognized as having a critical role in the impact of this neurodegenerative movement disorder. This has led to significant investigative efforts to identify new or better NMS therapies. The preponderance of PD patients will be diagnosed with 1 or multiple NMS during the course of their disease, with many of these symptoms occurring months or even years prior to receiving the PD diagnosis. Despite the high prevalence and impact on disease burden, NMS often go undetected due to a lack of reporting by patients or insufficient interrogation by physicians. Further complicating NMS management is that only a few therapies have the level of evidence needed to support their use in the treatment of NMS.
- Conclusion: The practitioner needs to be aware of NMS and conduct thorough patient questioning in order to recognize, diagnose, and address NMS in PD patients.
Parkinson’s disease (PD) is a neurodegenerative movement disorder with an estimated prevalence of 1% to 2% among the population over the age of 65 years [1]. Recognition and clinical diagnosis of PD is primarily made based on the cardinal motor features, including rigidity, tremor, bradykinesia, and postural instability. The motor symptoms are neuropathologically associated with accumulation of alpha-synuclein with Lewy body formation and neurodegeneration of the nigrostriatal dopamine system. Postmortem evaluation of the brains of PD patients has revealed more widespread degeneration in nondopaminergic systems, including several brainstem nuclei (raphe nucleus, locus ceruleus, dorsal vagal nucleus), limbic and neocortical structures, as well as the peripheral autonomic system [2,3].
The nonmotor symptoms (NMS) of PD are the clinical manifestations of this extensive degeneration, which suggests that NMS are intrinsic and fundamental features of PD. NMS are exceedingly common, and up to 90% of PD patients will experience nonmotor features, including depression, anxiety, sleep disturbances, cognitive impairment, and dysautonomia [4,5] (Table).
NMS have a greater impact on quality of life as compared to the motor symptoms [6,7], but are frequently underrecognized [8]. Evidence suggests that unless there is systematic and specific interrogation by practioners, NMS will elude recognition [9–11]. Recognizing NMS as part of PD is complicated by the fact that these symptoms are common in the general population and not specific for PD [12,13]. NMS can occur at any stage of the disease and may predate diagnosis [12], although as PD progresses the NMS become more prevalent, with a greater impact on health care costs and institutionalization rates than motor features [14,15].
Neuropsychiatric Symptoms
Depression
Epidemiology and Diagnosis
Depression is one of the most common neuropsychiatric manifestations observed in PD patients, with prevalence reports between 4% and 72%, though likely to be closer to 30% to 45% [16–20]. The severity of depression in the PD population has been shown to be greater than in patients with matched chronic disabilities [21,22] and also greater than in the general population over the age of 65 years [23]. The onset of depression can occur at any stage of the disease, even predating the diagnosis. Additionally, depression has more than twice the impact on health status than motor symptoms [24].
Though the mechanisms are not fully understood, it is suspected that psychosocial as well as neuropathological changes contribute to the pathogenesis of depression in PD. In a study comparing 104 PD patients and 61 patients with equivalent disability scores, functional disability was found to be responsible for only 9% of the variation of depression scores [22]. The increased prevalence of depression in PD patients can in part be explained by the neuropathological changes seen in post-mortem studies. Two neurotransmitters that are fundamental in the pathogenesis of depression are serotonin, from the raphe nuclei, and norepinepherine, from the locus ceruleus [20]. Both of these brainstem structures demonstrate alpha-synucleinopathy-associated degeneration and these changes can precede the development of motor dysfunction [3].
Diagnosing depression in PD is complicated by the fact that there is overlap between other PD symptoms and clinical features of depression (ie, amotivation, bradykinesia, fatigue, and sleep disturbances). However, many depressed PD patients are less likely to report feelings of guilt or failure and tend to have higher rates of anxiety [9,20,25]. Typically, PD patients are more likely to be diagnosed with minor depression or dysthymia rather than a major depressive disorder [19,20]. Formal testing through systematic questionnaires are diagnostically useful in the clinic, and serial testing can reveal changes over time to guide more effective treatment. Validated tools to evaluate depression in PD include the Beck Depression Inventory, Hamilton Depression Rating Scale, Montgomery-Asberg Depression Rating Scale, Geriatric DRS, and Hospital Anxiety and Depression scale [20].
Treatment Options
Treatment of depression in PD demonstrates generally poorer responses to typical antidepressants and side effects that may worsen other PD symptoms. Selective serotonin reuptake inhibitors (SSRIs) have been widely used as there are generally few drug-drug interactions and minimal effect on motor symptoms; however, several studies have demonstrated little benefit on depression in PD [26]. In a randomized, double-blind, placebo-controlled trial of the antidepressants paroxetine and venlafaxine, both were found to be effective and well tolerated [27]. Tricyclic anti-depressants (TCAs) have also demonstrated efficacy. In randomized controlled trials comparing TCAs to SSRIs, a greater benefit on depression symptoms has been found with TCAs [28–30]. The use of TCAs, however, is limited by anticholinergic side effects that occasionally worsen orthostatic hypotension or cognitive impairment [15,31]. Dopamine agonists have also been studied in depressed PD patients. In a randomized, double-blind, placebo-controlled trial [32] and a prospective observational study [33], pramipexole demonstrated significant improvements in depression symptoms. Ropinirole also demonstrated significant symptomatic improvement [34]. These studies suggest that while SSRIs are commonly used, evidence is accumulating to support the role of TCAs, SNRIs, and dopamine agonists in the treatment of depression in PD.
Other therapies have also been tried in pharmacologic-resistant patients. Electroconvulsive therapy has been reported to improve both depression and motor symptoms [35,36]; however, this is a treatment reserved for patients with severe and drug-refractory depression. A randomized controlled trial investigating cognitive behavioral therapy has also demonstrated improvement of depression scores [37]. The role of physical activity as treatment for depression in PD patients is unclear. As described in a recent review by Loprinzi et al [38], the literature is contradictory, with one group experiencing reduced depression but with no signficant effect in several other studies.
Anxiety
Epidemiology and Diagnosis
The prevalence of anxiety in PD patients is about 40% [39], which is 2 times greater than in the general population [9]. Anxiety may worsen PD symptoms, especially tremor and cognition. Risk factors for anxiety include the female gender, greater motor fluctuations, prior history of anxiety, and younger age of PD onset [40]. As with depression, some patients also report worsening of anxious symptoms during “off” states [41]. Screening tools that have been validated to help practitioners identify anxiety in PD include the Hospital Anxiety and Depression Scale, Beck Anxiety Inventory, Zung Self-rating Anxiety Scale, Spielberger State Trait Anxiety Inventory, and Hamilton Anxiety Rating Scale [15].
Treatment Options
The treatment of diagnosed anxiety in PD is primarily with benzodiazepines, which are particularly beneficial in patients whose tremors are exacerbated by anxiety or stress. The use of benzodiazepines has not been evaluated by a randomized controlled trial and use should be limited given the potential risks of sedation, cognitive effects, and psychomotor agitation. Other case studies have found benefit with serotonergic medications like fluoxetine or citalopram (especially with concomitant depression) or with optimization of levodopa therapy [42,43].
Hallucinations, Delusions, and Psychosis
Epidemiology
The prevalence of visual hallucinations in PD patients is about 20% to 40% [44,45]. Risk factors for psychotic symptoms include cognitive impairment, advanced age, prolonged duration of disease, depression, severe dysautonomia, and sleep disorders [46–48]. Early recognition of hallucinations is critical because of a strong correlation between the manifestation of psychosis and the need for nursing home placement or hospitalization. With early and effective treatment there is a decreased need for placement and a reduction on caregiver burden [44,49].
Treatment Options
Hallucinations can occur in delirium and it is important to first rule out an underlying infection or an offending medication, especially if there is a sudden onset or worsening of symptoms. Psychotic symptoms have been reported in drug-naive patients, though they are often iatrogenically induced with dopaminergic agents. All antiparkinsonian medications are capable of inducing or exacerbating hallucinations [9,50]. Additionally, psychotic symptoms tend to improve when dopaminergic agonists are reduced or eliminated. However, there is no clear relationship between the dose of dopaminergic agents and manifestation of hallucinations [48,51,52]. If hallucinations persist or there are motor complications that arise from reduction of dopaminergic agents, initiation of clozapine has been demonstrated to be efficacious in a rater-blinded prospective study and in a retrospective analysis [53–55]; however, regular monitoring for neutropenia is required. Quetiapine has demonstrated similar benefit without significant effects on motor symptoms in a randomized, rater-blinded study and in an evidence-based review [56,57]. It is also important to review or eliminate other medications that may contribute to hallucinations.
Cognitive Impairment
Epidemiology
The prevalence of dementia in the PD population is 20% to 40% [58], though almost 80% of PD patients ultimately develop cognitive decline [59]. Overall, a PD patient is 6 times more likely to develop dementia than someone in the general population [60]. There may be parallel progression of cognitive impairment and motor symptoms, but there is no correlation with overall duration of disease [60,61]. Risk factors linked with the presence of dementia include older age at onset of PD, presence of hallucinations, and male gender [62,63].
Cognitive dysfunction can be detected early in PD through neuropsychological testing; however, impairment of cognition is often insidious and may not be appreciated until symptoms become severe. Several screening tools have been used to evaluate for cognitive impairment in PD including the Mini-Mental State Exam (MMSE), Montreal Cognitive Assessment (MoCA), Mini-Mental Parkinson, Scales for Outcomes of Parkinson’s disease–Cognition, and others. Accumulating evidence, however, is suggestive of the superiority of the MoCA in the detection of cognitive deficits associated
with PD [64].
Dementia is a substantial burden for the caregiver and is a significant contributor to mortality in PD patients [65]. Cognitive impairment often presents with other behavioral symptoms, which further hastens placement outside the home and increases cost of caring for PD patients [49,66].
Cognitive impairment in Parkinson’s disease is typically associated with degeneration of primarily subcortical structures. PD patients with mild cognitive impairment were found to have deficits most significantly in memory, executive function, memory, and language abilities [67]. A recent study by Mak et al evaluated grey matter volumes by structural MRI in PD patients with evidence of mild cognitive impairment by MMSE and MoCA as compared with findings in cognitively intact patients. This demonstrated decreased brain volumes in areas that correlate with affected cognitive domains including the left insula, left superior frontal and left middle temporal areas [68].
Treatment Options
Prior to initiation of therapy, it is important to evaluate the patient for depression and to rule out pseudodementia. Bradyphrenia, or slowness of thought, should also be considered, as this symptom may also lead to an incorrect dementia diagnosis. Lastly, a thorough review of medications should be performed and offending agents including anticholinergics, TCAs, dopamine agonists, and amantadine should be discontinued as these can worsen cognition.
Rivastigmine has demonstrated modest improvement in cognitive performance in PD patients with dementia in a large multicenter, placebo-controlled study [69]. Other cholinesterase inhibitors (ie, donepezil or galantamine) are not recommended at this time due to limited studies or contradictory results in the literature [31,54]. Caution is advised with use of cholinesterase inhibitors as they may worsen tremor or autonomic dysfunction; also, use is limited by nausea or other gastrointestinal symptoms. Memantine, an NMDA receptor antagonist, has also been investigated in randomized, double-blind, placebo-controlled trials and demonstrated modest improvement of cognition and is generally well tolerated [70,71].
Nonpharmacologic therapy includes physical exercise, which has demonstrated improvement in memory tasks and processing speed [72]. Cognitive training has been less rigorously studied; however, a recent single-blinded controlled study demonstrated significant improvement of learning and memory in PD patients who completed computer-based cognitive training [73].
Compulsive Disorders
Impulse Control Disorders
Impulse control disorders (ICDs) are inappropriate behaviors resulting from a failure to resist an impulse, which leads to pleasure-seeking activities at the expense of relationships and ability to function socially. In PD, ICDs are expressed as pathologic gambling, hypersexuality, binge eating, compulsive shopping, and excessive spending [9,66]. The prevalence of all ICDs in PD is 15% to 20% and a patient may be diagnosed with multiple ICDs [74]. Dopamine agonist use has been implicated in the development of ICDs and this risk is further increased with the addition of levodopa [75,76]. Clinical features associated with ICDs include young age of onset, male gender, family history of addiction, depression or anxiety, and disinhibition or impulsive traits [77,78].
Traditionally, treatment consists of reduction or elimination of dopamine agonists, though adjustment of levodopa therapy may also be necessary. Amantadine as an adjunct therapy has been shown in a randomized, double-blind crossover study to reduce impulsivity in a few patients with pathologic gambling [79].
Dopamine Dysregulation Syndrome
Dopamine dysregulation syndrome (DDS) is characterized by compulsive use of dopaminergic medications beyond what is needed to treat parkinsonian symptoms, and is associated with social impairment. Patients describe addictive symptoms like craving or intense desire to obtain more dopaminergic medication [9,74]. Like ICDs, treatment of DDS consists of modification to dopaminergic medications, though patients with DDS may also require psychiatric evaluation and treatment.
Punding
Punding is another compulsive disorder that is defined as an intense fascination with objects and is associated with repetitive handling, manipulation, sorting, or arrangement of the items [80]. Occurrence of punding has been associated with higher total daily levels of levodopa, although one study has also implicated dopamine agonists [15,81]. As with the other compulsive disorders, punding also tends to respond well to reduction or discontinuation of levodopa. Studies have demonstrated modest benefit with SSRIs or atypical antipsychotics in long-term follow-up [82,83], though one study reported worsening of punding with quetiapine [84].
Apathy
Epidemiology and Treatment
Apathy is often characterized by a loss of motivation or inability to initiate goal-directed behavior, which results in dependence on others for activities of daily living and increases caregiver burden [85]. Patients demonstrate indifference, lack of interest, or inability to express or describe emotion. The apathetic patient may lack spontaneous and voluntary activity, and their affect display is often flattened [86].
With a prevalence of 30% to 50% [87], apathy is as common as depression in PD patients [66,88]. Risk factors associated with apathy include advanced age, severity of depression, severity of motor dysfunction, and dementia [89]. Apathy is frequently mistaken for depression given the significant overlap in symptoms; however, the patient with pure apathy will deny sadness or depressed feelings. It is also important to distinguish apathy from motor impairment or cognitive dysfunction that could explain the behavioral changes. No medications have reliably been shown to improve apathy, though it may be improved with initiation of dopaminergic therapy, especially early in the course [86,90].
Sleep Disorders
The original report of PD by James Parkinson describes sleep disturbances and daytime somnolence [91], which suggests that sleep disorders may be an intrinsic feature of the neurodegenerative process of PD itself.
REM Behavioral Disorder
Epidemiology and Diagnosis
Rapid eye movement behavioral disorder (RBD) is a parasomnia characterized by vocalizations and motor activity during dreaming due to loss of normal atonia associated with rapid eye movement (REM) sleep. Patients enact their dreams, which may lead to violent behaviors that can injure the patient or their bed partner. RBD is seen in 25% to 50% of PD patients [92,93], with variability depending on diagnostic technique and patient selection. Polysomnography is the most important diagnostic tool and demonstrates increased chin tone and limb movements during REM sleep in RBD [94,95]. Diagnosis can also be made clinically with patient and bed partner reports, though sensitivity is only approximately 30% [15].
Interestingly, many studies are now investigating the relationship between presence of RBD and later onset of neurodegenerative disorders. Multiple studies have shown that 40% to 65% of patients diagnosed with idiopathic RBD later develop an alpha-synucleinopathy, which includes PD, dementia with Lewy bodies, or multiple system atrophy within 10 years [92,95]. Prior studies report that as many as 90% of patients with idiopathic RBD develop neurodegenerative synucleinopathy when followed over 14 years [96]. Idiopathic RBD is currently being investigated as a potential clinical marker of pre-symptomatic PD in a multicenter observational study. If RBD is an early marker for neurodegenerative disease, it may be used to identify patients for neuroprotective trials as treatments are developed.
Treatment Options
Low-dose clonazepam (0.25–1 mg) is the mainstay of therapy, especially for patients that injure themselves or bed partners [97]; however, the use of benzodiazepines is historical and there remain no randomized controlled double-blind studies to evaluate the efficacy of clonazepam. Use of clonazepam may be limited by daytime sedation, confusion, or psychomotor agitation [31,97,98]. Melatonin (doses between 3–12 mg at bedtime) has also demonstrated benefit in RBD in a double-blind, placebo-controlled trial and in a small case series, with fewer side effects and no addiction potential as compared to clonazepam [99,100]. Case reports also support the use of several other effective medications, including cholinesterase inhibitors (rivastigmine and donepezil) and dopaminergic agents (pramipexole and levodopa) [15,20].
Restless Leg Syndrome and Periodic Limb Movements in Sleep
Epidemiology
Restless leg syndrome (RLS) and periodic limb movements in sleep (PLMS) cause disruptions of sleep and have an important impact on quality of sleep in PD patients. RLS is described as a strong urge to move the legs, accompanied by an uncomfortable sensation that is exacerbated at rest and relieved by movement. RLS is more frequently diagnosed in patients with PD, though prevalence reports vary widely [15]. Secondary causes for RLS should be investigated including iron deficiency, uremia and polyneuropathy. Several case reports demonstrate onset or worsening of RLS with use of antidepressants [101, 102] or antipsychotics like risperidone, aripiprazole, and quetiapine [103,104].
PLMS occurs in approximately 80% to 90% of patients with RLS, though may be present independently, and when seen on polysomnography is supportive of RLS [105]. PLMS is characterized by repetitive dorsiflexion of the foot, extension of the great toe, and may be accompanied by flexion of the knee and hip. The prevalence of PLMS in PD is approximately 60% and correlates with severity of PD motor features [106].
Treatment Options
Treatment of RLS should be initiated with nonpharmacologic therapies including good sleep hygiene, exercise, leg massage, and heat or ice packs [105,107]. Dopamine (DA) agonists are the primary treatment for RLS; however, even modest adjustments in levodopa can be helpful. One drawback to levodopa therapy is augmentation (a worsening or reappearance of symptoms) when serum levels fall due to the short half-life of levodopa [107,108]. DA agonists are less likely to cause augmentation. Both pramipexole and ropinirole have been extensively investigated in controlled, randomized, double-blind studies with benefits in 70% to 90% of patients with RLS and PLMS; however, there is a risk of developing compulsive behaviors [109–112]. Another option for PD patients is rotigotine, which has demonstrated improvement of RLS symptoms in a randomized, double-blind, placebo-controlled trial and has the added benefit that it may also help with motor symptoms [113,114].
More recently, gabapentin enacarbil has demonstrated improvement of moderate to severe RLS and was well tolerated in multiple randomized, double-blind, placebo-controlled trials [107,115,116]. Lastly, opioids (tramadol, oxycodone, codeine) have been shown to be effective, especially in the treatment of RLS that is refractory to other treatments [105,107].
Insomnia
Epidemiology
The most common sleep disorder in PD is insomnia, with a prevalence between 37% to 88% [14,117]. Insomnia is associated with difficulty in initiation or maintenance of sleep. Disruption of sleep typically leads to daytime somnolence and patient reports of a strong impact on motor disability and overall quality of life. There are several contributors to insomnia in PD patients including nocturia, depression, RLS, dystonia, and akinesia/rigidity/difficulty turning in bed [118].
Treatment Options
The use of carbidopa/levodopa controlled-release formulations at bedtime is associated with improved sleep duration and nocturnal akinesia, although it does not demonstrate a significant improvement in overall sleep ratings [54]. Hypnotics like eszopiclone and zolpidem have also demonstrated improved quality of sleep in limited controlled trials and a meta-analysis, but use is limited by sedation, dizziness, and falls [54,119]. Benzodiazepines improve sleep latency, but there is a risk of cognitive impairment, tolerance, and falls [117,120]. Melatonin at 3 to 5 mg and 50 mg doses have been investigated in 2 randomized, double-blind, placebo-controlled trials; however, there was a modest benefit and it was concluded that there is insufficient evidence to support the use of melatonin [54]. Nevertheless, melatonin is well tolerated and may be tried with minimal risk [54]. More recently, a randomized controlled trial using doxepin has demonstrated improvement of insomnia scores and was generally well tolerated [121].
Excessive Daytime Sleepiness and Abrupt Sleep Onset
EDS and Fatigue: Epidemiology and Treatment
A common complaint by PD patients is excessive daytime sleepiness (EDS), which can be verified with multiple sleep latency testing. EDS frequency varies in the literature, but is seen in approximately 15% to 50% of PD patients [4,122]. The etiology is usually multifactorial, with insomnia, dysautonomia, and depression as contributing factors [117]. A longer duration of symptoms, greater total load of levodopa, cognitive decline, and male gender are all risk factors for EDS [122,123]. It has been proposed that EDS is an intrinsic feature of PD; however, there is also an association with the use of antiparkinsonian medications. A randomized controlled trial demonstrated that use of the dopamine agonist pramipexole was associated with greater somnolence as compared to levodopa therapy (35% vs. 13%); however, this difference was only seen during the initial escalation phase [124]. Additionally, the combined use of dopamine agonists and levodopa has shown an even greater risk of EDS [125]. The evidence for the use of stimulants for EDS is lacking. The few studies conducted with modafinil have not demonstrated a robust improvement of EDS [126–128]. Other stimulants like methylphenidate have been studied with improvement of Epworth Sleepiness Score, though no randomized control trials have been undertaken [129].
It is important to distinguish EDS, a propensity for daytime sleep, from fatigue or excessive tiredness associated with mental or physical exertion [117]. Fatigue is often multifactorial and may be related to insomnia, sleep apnea, sedating effects of medications, frequent awakenings from nocturia, and degeneration of brain areas regulating sleep/wake cycles related to the underlying disease process [20, 117]. It is also important to consider depression and dementia in the differential, as these disorders may be erroneously be diagnosed as fatigue. Treatment of fatigue should include regular mild exercise, maintenance of a stimulating environment, removal of sedating medications, and management of intrinsic sleep disorders if present [117]. The use of stimulants for fatigue is controversial. A small randomized controlled trial (n = 48) using modafinil demonstrated improvement on the global clinical impression scale for fatigue but no significant change on the Fatigue Severity Scale; this study was limited by the power and points to the need for a larger study [130].
Sleep Attacks: Epidemiology and Treatment
Abrupt sleep onset, or “sleep attacks,” occurs when transition from wake to sleep is unavoidable and may occur without warning. Sleep attacks are threefold more likely to occur in patients using DA agonists, with an associated dose-related increase in risk [131]. Adjustment or elimination of DA agonists often improves sleep attacks, though it is important to address concurrent EDS if present. Nonpharmacologic treatments to consider include mild exercise, early morning bright light exposure, and a stimulating environment [117].
Sleep-Disordered Breathing/Obstructive Sleep Apnea
Epidemiology and Treatment
Sleep-disordered breathing (SDB) consists of either a deficit in the drive to breathe as in central sleep apnea, or may be due to an blockage of the airway as seen in obstructive sleep apnea (OSA). Apnea leads to oxygen desaturations that consequently trigger awakenings throughout the night, which in turn is experienced by the patient as daytime somnolence [117]. The prevalence of SDB and OSA is variable in the literature, ranging from no increased risk in PD patients [132,133] to 50% prevalence in PD patients [134,135]. Discussions with bed partners, history of snoring, and clinical reports of EDS or daytime fatigue are important indicators of SDB. Polysomnography confirms the diagnosis and can direct treatment, which frequently includes application of CPAP devices during sleep.
Autinomic Dysfunction
Orthostatic Hypotension
Epidemiology and Diagnosis
Orthostatic hypotension (OH) is defined as a 20-mm Hg fall in systolic blood pressure or 10-mm Hg drop in diastolic blood pressure within 3 minutes of a change in position. The prevalence of OH in PD patients is 30% to 60% [136,137]. Symptoms of OH can occur early in the disease and may precede diagnosis of PD [137]. Patients experience OH as dizziness, drowsiness, palpitations, nausea, or loss of consciousness. Additionally, falls and supine hypertension that accompany OH are associated with increased risk of morbidity and mortality in PD patients [138]. Several medications used in the treatment of PD can exacerbate OH, including levodopa, DA agonists, MAO-B inhibitors, and TCAs [139].
Treatment Options
First-line therapies for OH include nonpharmacologic methods such as compression stockings, sleeping with head elevated to 30 degrees, increased water and salt intake, more frequent small meals, and slowly changing position [140]. Additionally, it is important to discuss the removal or reduction of all antihypertensives with the patient’s PCP. Fludrocortisone (a mineralacorticoid) and domperidone (a peripheral dopamine antagonist not currently approved for use in the United States) modestly improved OH in a 2-phase, randomized, controlled, double-blind, crossover trial [141]. Pyridostigmine has also demonstrated improvement of standing blood pressure and OH symptoms in a double-blind, randomized cross-over study and has the additional benefit of not worsening supine hypertension [142]. Other effective treatments include midodrine, per a randomized, double-blind multicenter study [143], as well as droxidopa in a double-blind, crossover, placebo-controlled study [144]. Currently there is insufficient evidence to support the preferential use of any specific agent in the treatment of OH in PD.
Gastrointestinal Dysmotility
Constipation: Epidemiology and Treatment
Constipation is reported by nearly 60% of PD patients [145]. Constipation can precede the development of motor symptoms of PD, and the prevalence of GI disturbances increases with age and longer duration of disease. Nearly one third of patients will have been diagnosed with a GI disturbance within the year prior to PD diagnosis [146], which is associated with an increased risk for the development PD [147]. People with constipation (defined as < 1 bowel movement per day) but without a PD diagnosis had more nigral Lewy body degeneration postmortem [148] compared with people without constipation.
Treatments for constipation include dietary modification, increased fluid intake, and mild exercise. Macrogol significantly improved constipation in PD patients and was very well tolerated in a randomized placebo-controlled study [149]. Lubiprostone, a GI active prostaglandin, is also effective in the short-term treatment of constipation in a placebo-controlled trial [150].
Gastroparesis: Epidemiology and Treatment
Gastroparesis, like constipation, is related to enteric dopaminergic cell loss and degeneration of the dorsal motor nucleus of the vagus [151]. Patients experience gastroparesis as early satiety, full sensation, and nausea. Decreased gastric motility leads to retention of food as well as medications, which can slow absorption and delay onset of action for many medications including levodopa. Domperidone has both prokinetic and antiemetic properties, which have been beneficial in the treatment of gastroparesis [152], but its use is not currently approved in the United States.
Dysphagia: Epidemiology and Treatment
Dysphagia is associated with more advanced stages of PD as well as a significant increase in morbidity. Swallow exercises have demonstrated improvement of dysphagia [153]. The impact of levodopa therapy on dysphagia in the literature is controversial. Videofluoroscopic examination is the most common method for evaluation of swallowing disorders and provides important information for speech-language pathologists regarding recommendations for dietary modifications [154]. Adjustment of medication regimens to avoid an oral route is also helpful. This includes Parcopa, orally disintegrating carbidopa/levodopa tablets, and transdermal approaches like the rotigotine patch. For some patients, enteral nutrition is needed and placement of nasogastric tubes or percutaneous endoscopic gastrostomy tubes are an option.
Sialorrhea (Drooling)
Epidemiology
Difficulty handling oral secretions due to impaired or infrequent swallowing results in sialorrhea in up to 75% of PD patients [155], which is a significant embarrassment for most patients [156]. PD patients with drooling have difficulty speaking, eating, and engaging in social interactions, which significantly impacts perceived quality of life [157].
Treatment Options
Botulinum toxin (A and B) injections into the submandibular or parotid glands have demonstrated efficacy in multiple double-blind, randomized, placebo-controlled studies for the treatment of sialorrhea in PD patients; however, injections are associated with greater invasiveness and cost [158–160]. Glycopyrrolate, an anticholinergic drug, was also efficacious in the treatment of sialorrhea in the short term in a double-blind, randomized, placebo-controlled study [161]. Alternatively, gum chewing increases swallow frequency, improves drooling, and also shows a benefit with dysphagia [162].
Genitourinary Disturbances
Bladdery dysfunction: Epidemiology and Treatment
Bladder dysfunction in PD is often secondary to hyperactivity of the detrusor muscle leading to urinary urgency, increased urinary frequency, and nocturia. Less commonly, hypoactive detrusor muscle causes difficulty with initiation of urination, delayed bladder emptying, and recurrent infections. Urinary disturbances may occur before the onset of motor symptoms or early on in the disease course [12]. Disease severity is associated with greater urinary disturbances, and more than 50% of advanced PD patients report severe bladder symptoms [163].
Anticholinergic medications such as oxybutynin, solifenacin, and tolterodine are commonly used in the treatment of detrusor hyperactivity and demonstrate significant improvement in detrusor pressure in a recent systemic review and meta-analysis [164]. PD patients on these agents should be closely monitored for side effects including cognitive impairment, somnolence, hallucinations, confusion, and blurred vision. Other treatments include botulinum toxin injections into the detrusor muscle, which has demonstrated safety and efficacy in a recent systematic review [165].
Erectile dysfunction: Epidemiology and Treatment
Erectile dysfunction (ED) is reported by more than 60% of male PD patients [145] and is thought to be related to hypothalamic dysfunction and modification of the dopamine-oxytocin pathway [166]. Effects of PD medications, cognitive impairment, fatigue, apathy, and low testosterone contribute to loss of libido and ED [20,167]. Phosphodiesterase inhibitors such as sildenafil, vardenafil, and tadalafil are possibly useful in the treatment of ED in PD patients, though randomized trials have been limited [166,168]. Apomorphine sublingually is another medication that has demonstrated improvement of ED in a double-blind, crossover study and can be considered for patients with contraindications to phosphodiesterase inhibitors [169].
Sensory Symptoms
Pain
Epidemiology
Sensory disturbances in PD include diminished ability to identify odors, visual abnormalities (blurred vision, abnormal color perception, double vision), and pain. Pain is the most disabling sensory disturbance, though frequently underreported. Nearly two thirds of PD patients report pain, [170], though only half of patients receive any treatment [171]. Pain may also be a presenting symptom that precedes the clinical diagnosis of PD [172,173].
Treatment Options
There are several types of pain described by PD patients, the most common of which is musculoskeletal, typically involving the shoulder. Other types include dystonic, radicular, and central pain [174]. First-line treatment of musculoskeletal complaints includes nonsteroidal anti-inflammatory drugs (NSAIDs) and physiotherapy. Modification of levodopa regimen (including altering timing and frequency or adding controlled release formulations) can often provide relief for dystonic pain, and also for central pain for some patients [173, 174]. Deep brain stimulation, with subthalamic nucleus or globus pallidus targets, has demonstrated improvement with dystonic, central, and musculoskeletal pain in a small clinical study [175].
Conclusion
NMS are an intrinsic part of PD, may predate diagnosis, and substantially affect the majority of patients with PD. For many of these patients, NMS have a greater impact on quality of life and health care costs than the cardinal motor symptoms that define the disease. Many of these symptoms are not recognized by practioners and often are not volunteered by PD patients, making it important for practitioners to routinely and directly inquire about NMS. Treatment of NMS in PD is challenging, and only a few therapies have the level of evidence needed to support their use in the treatment of these problems. Nevertheless, proper recognition and addressing of these symptoms afford the clinician an opportunity to make a positive and potentially significant impact on the PD patient’s quality of life.
Corresponding author: Samay Jain, MD, MS, Dept of Neurology, 811 Kaufmann Bldg, Pittsburgh, PA 15213, jains@upmc.edu.
Financial disclosures: None.
1. Alves G, Forsaa EB, Pedersen KF, et al. Epidemiology of Parkinson's disease. J Neurol 2008;255 Suppl 5:18–32.
2. Stern MB, Lang A, Poewe W. Toward a redefinition of Parkinson's disease. Mov Disord 2012;27:54–60.
3. Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003;24:197–211.
4. Tandberg E, Larsen JP, Karlsen K. A community-based study of sleep disorders in patients with Parkinson's disease. Mov Disord 1998;13:895–9.
5. Shulman LM, Taback RL, Bean J, Weiner WJ. Comorbidity of the nonmotor symptoms of Parkinson's disease. Mov Disord 2001;16:507–10.
6. Park A, Stacy M. Non-motor symptoms in Parkinson's disease. J Neurol 2009;256 Suppl 3:293–8.
7. Chaudhuri KR, Schapira AH. Non-motor symptoms of Parkinson's disease: dopaminergic pathophysiology and treatment. Lancet Neurol 2009;8:464–74.
8. Shulman LM, Taback RL, Rabinstein AA, Weiner WJ. Non-recognition of depression and other non-motor symptoms in Parkinson's disease. Parkinsonism Relat Disord 2002;8:193–7.
9. Bonnet AM, Jutras MF, Czernecki V, et al. Nonmotor symptoms in Parkinson's disease in 2012: relevant clinical aspects. Parkinsons Dis 2012:2012:198316.
10. Chaudhuri KR, Healy DG, Schapira AH. Non-motor symptoms of Parkinson's disease: diagnosis and management. Lancet Neurol 2006;5:235–45.
11. Hussl AK, Seppi K, Poewe W. Nonmotor symptoms in Parkinson's disease. Expert Rev Neurother 2013;13:581–3.
12. O'Sullivan SS, Williams DR, Gallagher DA, et al. Nonmotor symptoms as presenting complaints in Parkinson's disease: a clinicopathological study. Mov Disord 2008;23:101–6.
13. Lang AE. A critical appraisal of the premotor symptoms of Parkinson's disease: potential usefulness in early diagnosis and design of neuroprotective trials. Mov Disord 2011;26:775–83.
14. Barone P, Antonini A, Colosimo C, et al. The PRIAMO study: A multicenter assessment of nonmotor symptoms and their impact on quality of life in Parkinson's disease. Mov Disord 2009;24:1641–9.
15. Bernal-Pacheco O, Limotai N, Go CL, Fernandez HH. Nonmotor manifestations in Parkinson disease. Neurologist 2012;18:1–16.
16. Lemke MR, Fuchs G, Gemende I, et al. Depression and Parkinson's disease. J Neurol 2004;251 Suppl 6: VI/24–7.
17. Ravina, B, Camicioli R, Como PG, et al, The impact of depressive symptoms in early Parkinson disease. Neurology 2007;69:342–7.
18. Jasinska-Myga B, Putzke JD Wider C, et al. Depression in Parkinson's disease. Can J Neurol Sci 2010;37:61–6.
19. Slaughter JR, Slaughter KA, Nichols D, et al. Prevalence, clinical manifestations, etiology, and treatment of depression in Parkinson's disease. J Neuropsychiatry Clin Neurosci 2001;13:187–96.
20. Simuni T, Sethi K. Nonmotor manifestations of Parkinson's disease. Ann Neurol 2008;64 Suppl 2:S65–80.
21. Ehmann TS, Beninger RJ, Gawel MJ, Riopelle RJ. Depressive symptoms in Parkinson's disease: a comparison with disabled control subjects. J Geriatr Psychiatry Neurol 1990;3:3–9.
22. Menza MA, Mark MH. Parkinson's disease and depression: the relationship to disability and personality. J Neuropsychiatry Clin Neurosci 1994;6:165–9.
23. CDC. Current depression among adults–United States, 2006 and 2008. Morb Mort Weekly Rep 2010;59:1229–35.
24. Hinnell C, Hurt CS, Landau S, et al. Nonmotor versus motor symptoms: how much do they matter to health status in Parkinson's disease? Mov Disord 2012;27:236–41.
25. Cummings JL. Depression and Parkinson's disease: a review. Am J Psychiatry 1992;149:443–54.
26. Weintraub D, Morales KH, Moberg PJ, et al. Antidepressant studies in Parkinson's disease: a review and meta-analysis. Mov Disord 2005;20:1161–9.
27. Richard IH, McDermott MP, Kurlan R, et al. A randomized, double-blind, placebo-controlled trial of antidepressants in Parkinson disease. Neurology 2012;78:1229–36.
28. Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009;72:886–92.
29. Okun MS, Fernandez HH. Will tricyclic antidepressants make a comeback for depressed Parkinson disease patients? Neurology 2009;72:868–9.
30. Antonini A, Tesei S, Zecchinelli A, et al. Randomized study of sertraline and low-dose amitriptyline in patients with Parkinson's disease and depression: effect on quality of life. Mov Disord 2006;21:1119–22.
31. Pedrosa DJ, Timmermann L. Review: management of Parkinson's disease. Neuropsychiatr Dis Treat 2013;9: 321–40.
32. Barone P, Poewe W, Albrecht S, et al. Pramipexole for the treatment of depressive symptoms in patients with Parkinson's disease: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2010;9:573–80.
33. Lemke MR, Brecht HM, Koester J, et al. Anhedonia, depression, and motor functioning in Parkinson's disease during treatment with pramipexole. J Neuropsychiatry Clin Neurosci 2005;17:214–20.
34. Rektorova I, Balaz M, Svatova J, et al. Effects of ropinirole on nonmotor symptoms of Parkinson disease: a prospective multicenter study. Clin Neuropharmacol 2008;31:261–6.
35. Van der Wurff FB, Stek ML, Hoogendijk WL, Beekman AT. Electroconvulsive therapy for the depressed elderly. Cochrane Database Syst Rev 2003(2):CD003593.
36. Okun MS, Watts RL. Depression associated with Parkinson's disease: clinical features and treatment. Neurology 2002;58(4 Suppl 1):S63–70.
37. Dobkin RD, Menza M, Allen LA, et al. Cognitive-behavioral therapy for depression in Parkinson's disease: a randomized, controlled trial. Am J Psychiatry 2013;168:1066–74.
38. Loprinzi PD, Herod SM, Cardinal BJ, Noakes TD. Physical activity and the brain: A review of this dynamic, bi-directional relationship. Brain Res 2013;1539:95–104.
39. Richard IH. Anxiety disorders in Parkinson's disease. Adv Neurol 2005;96:42–55.
40. Leentjens AF, Dujardin K, Marsh L, et al. Symptomatology and markers of anxiety disorders in Parkinson's disease: a cross-sectional study. Mov Disord 2011;26:484–92.
41. Witjas T, Kaphan E, Azulay JP, et al. Nonmotor fluctuations in Parkinson's disease: frequent and disabling. Neurology 2002;59:408–13.
42. Olanow CW, Stern MB, Sethi K. The scientific and clinical basis for the treatment of Parkinson disease Neurology 2009;72(21 Suppl 4):S1–136.
43. Richard IH, Schiffer RB, Kurlan R. Anxiety and Parkinson's disease. J Neuropsychiatry Clin Neurosci 1996;8:383–92.
44. Fenelon G, Alves G. Epidemiology of psychosis in Parkinson's disease. J Neurol Sci 2010;289:12–7.
45. Papapetropoulos S, Katzen H, Schrag A, et al. A questionnaire-based (UM-PDHQ) study of hallucinations in Parkinson's disease. BMC Neurol 2008;8:21.
46. Fenelon G, Mahieux F, Huon R, Ziegler M. Hallucinations in Parkinson's disease: prevalence, phenomenology and risk factors. Brain 2000;123 (Pt 4):733–45.
47. Papapetropoulos S, Mash DC. Psychotic symptoms in Parkinson's disease. From description to etiology. J Neurol 2005;252:753–64.
48. Aarsland D, Larsen JP, Cummins JL, Laake K. Prevalence and clinical correlates of psychotic symptoms in Parkinson disease: a community-based study. Arch Neurol 1999;56:595–601.
49. Aarsland D, Larsen JP, Tandberg E, Laake K. Predictors of nursing home placement in Parkinson's disease: a population-based, prospective study. J Am Geriatr Soc 2000;48:938–42.
50. Lohle M, Storch A, Reichmann H, Beyond tremor and rigidity: non-motor features of Parkinson's disease. J Neural Transm 2009;116:1483–92.
51. Fenelon G. Psychosis in Parkinson's disease: phenomenology, frequency, risk factors, and current understanding of pathophysiologic mechanisms. CNS Spectr 2008;13(3 Suppl 4):18–25.
52. Merims D, Shabtai H, Korczyn AD, et al. Antiparkinsonian medication is not a risk factor for the development of hallucinations in Parkinson's disease. J Neural Transm 2004;111:1447–53.
53. Merims D, Balas M, Peretz C, et al. Rater-blinded, prospective comparison: quetiapine versus clozapine for Parkinson's disease psychosis. Clin Neuropharmacol 2006;29:331–7.
54. Seppi K, Weintraub D, Coelho M, et al. The Movement Disorder Society Evidence-Based Medicine Review Update: Treatments for the non-motor symptoms of Parkinson's disease. Mov Disord 2011;26 Suppl 3:S42–80.
55. Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004;19:831–3.
56. Morgante L, Epifanio A, Spina E, et al. Quetiapine and clozapine in parkinsonian patients with dopaminergic psychosis. Clin Neuropharmacol 2004;27:153–6.
57. Miyasaki JM, Shannon K, Voon V, et al. Practice parameter: evaluation and treatment of depression, psychosis, and dementia in Parkinson disease (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2006;66:996–1002.
58. Riedel O, Klotsche J, Spottke A, et al. Cognitive impairment in 873 patients with idiopathic Parkinson's disease. Results from the German Study on Epidemiology of Parkinson's Disease with Dementia (GEPAD). J Neurol 2008;255:255–64.
59. Aarsland D, Andersen K, Larsen JP, et al. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol 2003;60:387–92.
60. Aarsland D, Andersen K, Larsen JP, et al. Risk of dementia in Parkinson's disease: a community-based, prospective study. Neurology 2001;56:730–6.
61. Riggeal BD, Crucian GP, Seignourel P, et al. Cognitive decline tracks motor progression and not disease duration in Parkinson patients. Neuropsychiatr Dis Treat 2007;3:955–8.
62. Hughes TA, Ross HF, Musa S, et al. A 10-year study of the incidence of and factors predicting dementia in Parkinson's disease. Neurology 2000;54:1596–602.
63. Hobson P, Meara J. Risk and incidence of dementia in a cohort of older subjects with Parkinson's disease in the United Kingdom. Mov Disord 2004;19:1043–9.
64. Hoops S, Nazem S, Siderowf AD, et al. Validity of the MoCA and MMSE in the detection of MCI and dementia in Parkinson disease. Neurology 2009;73:1738–45.
65. Louis ED, Marder K, Cote L, et al. Mortality from Parkinson disease. Arch Neurol 1997;54:260–4.
66. Fernandez HH. Nonmotor complications of Parkinson disease. Cleve Clin J Med 2012;79 Suppl 2:S14–8.
67. Sollinger AB, Goldstein FC, Lah JJ, et al. Mild cognitive impairment in Parkinson's disease: subtypes and motor characteristics. Parkinsonism Relat Disord 2010;16:177–80.
68. Mak E, Zhou J, Tan LC, et al. Cognitive deficits in mild Parkinson's disease are associated with distinct areas of grey matter atrophy. J Neurol Neurosurg Psychiatry 2013 Oct 16.
69. Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson's disease. N Engl J Med 2004;351:2509–18.
70. Aarsland D, Ballard C, Walker Z, et al. Memantine in patients with Parkinson's disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial. Lancet Neurol 2009;8:613–8.
71. Leroi I, Overshott R, Byrne EJ, et al. Randomized controlled trial of memantine in dementia associated with Parkinson's disease. Mov Disord 2009;24:1217–21.
72. Speelman AD, van de Warrenburg BP, van Nimwegen M, et al. How might physical activity benefit patients with Parkinson disease? Nat Rev Neurol 2013;7:528–34.
73. Naismith SL, Mowszowski L, Diamond K, Lewis SJ. Improving memory in Parkinson's disease: a healthy brain ageing cognitive training program. Mov Disord 2013;28:1097–103.
74. Weintraub D, Koester J, Potenza MN, et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol 2010;67:589–95.
75. Voon V, Reynolds B, Brezing C, et al. Impulsive choice and response in dopamine agonist-related impulse control behaviors. Psychopharmacology (Berl) 2010;207:645–59.
76. Weintraub D, Siderowf AD, Potenza MN, et al. Association of dopamine agonist use with impulse control disorders in Parkinson disease. Arch Neurol 2006; 63:969–73.
77. Pontone G, Williams JR, Bassett SS, Marsh L. Clinical features associated with impulse control disorders in Parkinson disease. Neurology 2006;67:1258–61.
78. Voon V, Mehta AR, Hallett M. Impulse control disorders in Parkinson's disease: recent advances. Curr Opin Neurol 2011;24:324–30.
79. Thomas A, Bonanni L, Gambi F, et al. Pathological gambling in Parkinson disease is reduced by amantadine. Ann Neurol 2010;68:400–4.
80. Miyasaki JM, Al Hassan K, Lang AE, Voon V. Punding prevalence in Parkinson's disease. Mov Disord 2007;22:1179–81.
81. Nguyen FN, Chang YL, Okun MS, et al. Prevalence and characteristics of punding and repetitive behaviors among Parkinson patients in North-Central Florida. Int J Geriatr Psychiatry 2010;25:540–1.
82. Sohtaoglu M, Demiray DY, Kenangil G, et al. Long term follow-up of Parkinson's disease patients with impulse control disorders. Parkinsonism Relat Disord 2010;16:334–7.
83. Antonini A, Cilia R. Behavioural adverse effects of dopaminergic treatments in Parkinson's disease: incidence, neurobiological basis, management and prevention. Drug Saf 2009;
32:475–88.
84. Miwa H, Morita S, Nakanishi I, Kondo T, Stereotyped behaviors or punding after quetiapine administration in Parkinson's disease. Parkinsonism Relat Disord 2004;10:177–80.
85. Skorvanek M, Rosenberger J, Gdovinova Z, et al. Apathy in elderly nondemented patients with parkinson's disease: clinical determinants and relationship to quality of life. J Geriatr Psychiatry Neurol 2013;26:237–43.
86. Marin RS, Fogel BS, Hawkins J, et al. Apathy: a treatable syndrome. J Neuropsychiatry Clin Neurosci 1995;7:23–30.
87. Pluck GC, Brown RG. Apathy in Parkinson's disease. J Neurol Neurosurg Psychiatry 2002;73:636–42.
88. Kirsch-Darrow L, Fernandez HH, Marsiske M, et al. Dissociating apathy and depression in Parkinson disease. Neurology 2006;67:33–8.
89. Pedersen KF, Alves G, Aarsland D, Larsen JP. Occurrence and risk factors for apathy in Parkinson disease: a 4-year prospective longitudinal study. J Neurol Neurosurg Psychiatry 2009;80:1279–82.
90. Czernecki V, Pillon B, Houeto JL, et al. Motivation, reward, and Parkinson's disease: influence of dopatherapy. Neuropsychologia 2002;40:2257–67.
91. Parkinson J. An essay on the shaking palsy. Sherwood, Neely, and Jones; 1817.
92. Schenck CH, Mahowald WM. REM sleep behavior disorder: clinical, developmental, and neuroscience perspectives 16 years after its formal identification in SLEEP. Sleep 2002;25:120–38.
93. Sixel-Doring F, Trautmann E, Mollenhauer B, Trenkwalder C. Associated factors for REM sleep behavior disorder in Parkinson disease. Neurology 2011;77:1048–54.
94. Eisensehr I, v Lindeiner H, Jager M, Noachtar S. REM sleep behavior disorder in sleep-disordered patients with versus without Parkinson's disease: is there a need for polysomnography? J Neurol Sci 2001;186:7–11.
95. Postuma RB, Gagnon JF, Montplaisir JY. REM sleep behavior disorder and prodromal neurodegeneration - where are we headed? Tremor Other Hyperkinet Mov (N Y) 2013;3.
96. Iranzo A, Tolosa E, Gelpi E, et al. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: an observational cohort study. Lancet Neurol 2013;12: 443–53.
97. Schenck CH, Mahowald MW. Rapid eye movement sleep parasomnias. Neurol Clin 2005;23:1107–26.
98. Anderson KN, Shneerson JM. Drug treatment of REM sleep behavior disorder: the use of drug therapies other than clonazepam. J Clin Sleep Med 2009;5:235–9.
99. Boeve BF, Silber MH, Ferman TJ. Melatonin for treatment of REM sleep behavior disorder in neurologic disorders: results in 14 patients. Sleep Med 2003;4:281–4.
100. Kunz D, Mahlberg R. A two-part, double-blind, placebo-controlled trial of exogenous melatonin in REM sleep behaviour disorder. J Sleep Res 2010;19:591–6.
101. Perroud N, Lazignac C, Baleydier B, et al. Restless legs syndrome induced by citalopram: a psychiatric emergency? Gen Hosp Psychiatry 2007;29:72–4.
102. Buskova J, Vorlova T, Pisko J, Sonka K, Severe sleep-related movement disorder induced by sertraline. Sleep Med 2012;13:769–70.
103. Rittmannsberger H, Werl R. Restless legs syndrome induced by quetiapine: report of seven cases and review of the literature. Int J Neuropsychopharmacol 2013;16:1427–31.
104. Perez-Lloret S, Rey MV, Bondon-Guitton E, et al. Drugs associated with restless legs syndrome: a case/noncase study in the French pharmacovigilance database. J Clin Psychopharmacol 2012;32:824–7.
105. Aurora RN, Kristo DA, Bista SR, et al. The treatment of restless legs syndrome and periodic limb movement disorder in adults-an update for 2012: practice parameters with an evidence-based systematic review and meta-analyses: an American Academy of Sleep Medicine Clinical Practice Guideline. Sleep 2012;35:1039–62.
106. Covassin N, Neikrug AB, Liu L, et al. Clinical correlates of periodic limb movements in sleep in Parkinson's disease. J Neurol Sci 2012;316:131–6.
107. Rios Romenets S, Postuma RB. Treatment of restless legs syndrome. Curr Treat Options Neurol 2013;15:396-409.
108. Hogl B, Paulus W, Clarenbach P, Trenkwalder C, Restless legs syndrome: diagnostic assessment and the advantages and risks of dopaminergic treatment. J Neurol 2006;253 Suppl 4:IV22-8.
109. Garcia-Borreguero D, Kohnen R, Silber MH, et al. The long-term treatment of restless legs syndrome/Willis-Ekbom disease: evidence-based guidelines and clinical consensus best practice guidance: a report from the International Restless Legs Syndrome Study Group. Sleep Med 2013;14:675–84.
110. Montagna P, Hornyak M, Ulfberg J, et al. Randomized trial of pramipexole for patients with restless legs syndrome (RLS) and RLS-related impairment of mood. Sleep Med 2011;12:34–40.
111. Partinen M, Hirvonen K, Jama L, et al. Efficacy and safety of pramipexole in idiopathic restless legs syndrome: a polysomnographic dose-finding study--the PRELUDE study. Sleep Med 2006;7:407–17.
112. Trenkwalder C Garcia-Borreguero D, Montagna P, et al. Ropinirole in the treatment of restless legs syndrome: results from the TREAT RLS 1 study, a 12 week, randomised, placebo controlled study in 10 European countries. J Neurol Neurosurg Psychiatry 2004;75:92–7.
113. Hening WA, Allen RP, Ondo WG, et al. Rotigotine improves restless legs syndrome: a 6-month randomized, double-blind, placebo-controlled trial in the United States. Mov Disord 2010;25:1675–83.
114. Oertel WH, Benes H, Garcia-Borreguero D, et al. Rotigotine transdermal patch in moderate to severe idiopathic restless legs syndrome: a randomized, placebo-controlled polysomnographic study. Sleep Med 2010;11:848–56.
115. Lee DO, Ziman RB, Perkins AT, et al. A randomized, double-blind, placebo-controlled study to assess the efficacy and tolerability of gabapentin enacarbil in subjects with restless legs syndrome. J Clin Sleep Med 2011;7:282–92.
116. Kushida CA, Walters AS, Becker P, et al. A randomized, double-blind, placebo-controlled, crossover study of XP13512/GSK1838262 in the treatment of patients with primary restless legs syndrome. Sleep 2009;32:159–68.
117. Menza M, Dobkin RD, Marin H, Bienfait K. Sleep disturbances in Parkinson's disease. Mov Disord 2010;25 Suppl 1:S117–22.
118. Louter M, van Sloun RJ, Pevernagie DA, et al. Subjectively impaired bed mobility in Parkinson disease affects sleep efficiency. Sleep Med 2013;14:668–74.
119. Menza M, Dobkin RD, Marin H, et al. Treatment of insomnia in Parkinson's disease: a controlled trial of eszopiclone and placebo. Mov Disord 2010;25:1708–14.
120. Nowell PD, Mazumdar S, Buysse DJ, et al. Benzodiazepines and zolpidem for chronic insomnia: a meta-analysis of treatment efficacy. JAMA 1997;278:2170–7.
121. Rios Romenets S, Creti L, Fichten C, et al. Doxepin and cognitive behavioural therapy for insomnia in patients with Parkinson's disease – a randomized study. Parkinsonism Relat Disord 2013;19:670–5.
122. Ondo WG, Dat Vuong K, Khan H, et al. Daytime sleepiness and other sleep disorders in Parkinson's disease. Neurology 2001;57:1392–6.
123. Razmy A, Lang AE, Shapiro CM, Predictors of impaired daytime sleep and wakefulness in patients with Parkinson disease treated with older (ergot) vs newer (nonergot) dopamine agonists. Arch Neurol 2004;61:97–102.
124. Holloway RG, Shoulson I, Fahn S, et al. Pramipexole vs levodopa as initial treatment for Parkinson disease: a 4-year randomized controlled trial. Arch Neurol 2004;61:1044–53.
125. Paus S, Brecht HM, Koster J, et al. Sleep attacks, daytime sleepiness, and dopamine agonists in Parkinson's disease. Mov Disord 2003;18:659–67.
126. Hogl B, Saletu M, Brandauer E, et al. Modafinil for the treatment of daytime sleepiness in Parkinson's disease: a double-blind, randomized, crossover, placebo-controlled polygraphic trial. Sleep 2002;25:905–9.
127. Ondo WG, Fayle R, Atassi F, Jankovic J. Modafinil for daytime somnolence in Parkinson's disease: double blind, placebo controlled parallel trial. J Neurol Neurosurg Psychiatry 2005;76:1636-–9.
128. Adler CH, Caviness JN, Hentz JG, et al. Randomized trial of modafinil for treating subjective daytime sleepiness in patients with Parkinson's disease. Mov Disord 2003;18:287–93.
129. Devos D, Krystkowiak P, Clement F, et al. Improvement of gait by chronic, high doses of methylphenidate in patients with advanced Parkinson's disease. J Neurol Neurosurg Psychiatry 2007;78:470–5.
130. Tyne HL, Taylor J, Baker GA, Steiger MJ. Modafinil for Parkinson's disease fatigue. J Neurol 2010;257:452–6.
131. Avorn J, Schneeweiss S, Sudarsky LR, et al. Sudden uncontrollable somnolence and medication use in Parkinson disease. Arch Neurol 2005;62:1242–8.
132. Trotti LM, Bliwise DL. No increased risk of obstructive sleep apnea in Parkinson's disease. Mov Disord 2010;25:2246–9.
133. da Silva-Junior FP, do Prado GF, Barbosa ER, et al. Sleep disordered breathing in Parkinson's disease: A critical appraisal. Sleep Med Rev 2013 Jul 22.
134. Noradina AT, Karim NA, Hamidon BB, et al. Sleep-disordered breathing in patients with Parkinson's disease. Singapore Med J 2010;51:60–4.
135. Oerlemans WG, de Weerd AW. The prevalence of sleep disorders in patients with Parkinson's disease. A self-reported, community-based survey. Sleep Med 2002;3:147–9.
136. Low PA. Prevalence of orthostatic hypotension. Clin Auton Res 2008;18 Suppl 1:8–13.
137. Goldstein DS. Orthostatic hypotension as an early finding in Parkinson's disease. Clin Auton Res 2006;16:46–54.
138. Sharabi Y, Goldstein DS. Mechanisms of orthostatic hypotension and supine hypertension in Parkinson disease. J Neurol Sci 2011;310:123–8.
139. Sanchez-Ferro A, Benito-Leon J, Gomez-Esteban JC. The management of orthostatic hypotension in Parkinson's disease. Front Neurol 2013;4:64.
140. Lahrmann H, Cortelli P, Hilz M, et al. EFNS guidelines on the diagnosis and management of orthostatic hypotension. Eur J Neurol 2006;13:930–6.
141. Schoffer KL, Henderson RD, O'Maley K, O'Sullivan JD. Nonpharmacological treatment, fludrocortisone, and domperidone for orthostatic hypotension in Parkinson's disease. Mov Disord 2007;22:1543–9.
142. Singer W, Sandroni P, Opfer-Gehrking TL, et al. Pyridostigmine treatment trial in neurogenic orthostatic hypotension. Arch Neurol 2006;63:513–8.
143. Low PA, Gilden JL, Freeman R, et al. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA 1997;277:1046–51.
144. Kaufmann H. L-dihydroxyphenylserine (Droxidopa): a new therapy for neurogenic orthostatic hypotension: the US experience. Clin Auton Res 2008;18 Suppl 1:19–24.
145. Magerkurth C, Schnitzer R, Braune S. Symptoms of autonomic failure in Parkinson's disease: prevalence and impact on daily life. Clin Auton Res 2005;15:76–82.
146. Makaroff L, Gunn A, Gervasoni C, Richy F. Gastrointestinal Disorders in Parkinson's Disease: Prevalence and Health Outcomes in a US Claims Database. J Parkinsons Dis 2011;1:65–74.
147. Abbott RD, Petrovitch H, White LR, et al. Frequency of bowel movements and the future risk of Parkinson's disease. Neurology 2001;57:456–62.
148. Abbott RD, Ross GW, Petrovitch H, et al. Bowel movement frequency in late-life and incidental Lewy bodies. Mov Disord 2007;22:1581–6.
149. Zangaglia R, Martignoni E, Glorioso M, et al. Macrogol for the treatment of constipation in Parkinson's disease. A randomized placebo-controlled study. Mov Disord 2007;22:1239–44.
150. Ondo WG, Kenney C, Sullivan K, et al. Placebo-controlled trial of lubiprostone for constipation associated with Parkinson disease. Neurology 2012;78:1650–4.
151. Cersosimo MG, Benarroch EE. Neural control of the gastrointestinal tract: implications for Parkinson disease. Mov Disord 2008;23:1065–75.
152. Reddymasu SC, Soykan I, McCallum RW. Domperidone: review of pharmacology and clinical applications in gastroenterology. Am J Gastroenterol 2007;102:2036–45.
153. Argolo N, Sampaio M, Pinho P, et al. Do swallowing exercises improve swallowing dynamic and quality of life in Parkinson's disease? NeuroRehabilitation 2013;32:949–55.
154. Troche MS, Sapienza CM, Rosenbek JC. Effects of bolus consistency on timing and safety of swallow in patients with Parkinson's disease. Dysphagia 2008;23:26–32.
155. Edwards LL, Quigley EM, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson's disease: frequency and pathophysiology. Neurology 1992;42:726–32.
156. Kalf JG, Smit AM, Bloem BR, et al. Impact of drooling in Parkinson's disease. J Neurol 2007;254:1227–32.
157. Leibner J, Ramjit A, Sedig L, et al. The impact of and the factors associated with drooling in Parkinson's disease. Parkinsonism Relat Disord 2010;16:475–7.
158. Mancini F, Zangaglia R, Cristina S, et al. Double-blind, placebo-controlled study to evaluate the efficacy and safety of botulinum toxin type A in the treatment of drooling in parkinsonism. Mov Disord 2003;18:685–8.
159. Lagalla G, Millevolte M, Capecci M, et al. Long-lasting benefits of botulinum toxin type B in Parkinson's disease-related drooling. J Neurol 2009;256:563–7.
160. Lagalla G, Millevolte N, Capecci M, et al. Botulinum toxin type A for drooling in Parkinson's disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2006;21:704–7.
161. Arbouw ME, Movig KL, Koopmann M, et al. Glycopyrrolate for sialorrhea in Parkinson disease: a randomized, double-blind, crossover trial. Neurology 2010; 74:1203–7.
162. South AR, Somers, Jog MS. Gum chewing improves swallow frequency and latency in Parkinson patients: a preliminary study. Neurology 2010;74:1198–202.
163. Winge K, Nielsen KK. Bladder dysfunction in advanced Parkinson's disease. Neurourol Urodyn 2012;31:1279–83.
164. Madhuvrata P, Singh PM, Hasafa Z, Abdel-Fattah M. Anticholinergic drugs for adult neurogenic detrusor overactivity: a systematic review and meta-analysis. Eur Urol 2012;62:816–30.
165. Soljanik I. Efficacy and safety of botulinum toxin a intradetrusor injections in adults with neurogenic detrusor overactivity/neurogenic overactive bladder: a systematic review. Drugs 2013;73:1055–66.
166. Sakakibara R, Uchiyama T, Yamanishi T, Kishi M. Genitourinary dysfunction in Parkinson's disease. Mov Disord 2010;25:2–12.
167. Kummer A, Cardoso F, Teixeira AL. Loss of libido in Parkinson's disease. J Sex Med 2009;6:1024–31.
168. Lombardi G, Nelli F, Celso M, et al. Treating erectile dysfunction and central neurological diseases with oral phosphodiesterase type 5 inhibitors. Review of the literature. J Sex Med 2012 9(4): 970–85.
169. Dula E, Bukofzer S, Perdok R, George M. Double-blind, crossover comparison of 3 mg apomorphine SL with placebo and with 4 mg apomorphine SL in male erectile dysfunction. Eur Urol 2001;39(5): 558–3
170. Negre-Pages L, Regragui W, Bouhassira D, et al. Chronic pain in Parkinson's disease: the cross-sectional French DoPaMiP survey. Mov Disord 2008;23:1361–9.
171. Beiske AG, Loge JH, Ronningen A, Svensson E. Pain in Parkinson's disease: Prevalence and characteristics. Pain 2009. 141:173–7.
172. Lin CH, Wu RM, H.Y. Chang HY, et al. Preceding pain symptoms and Parkinson's disease: a nationwide population-based cohort study. Eur J Neurol 2013;20:1398–404.
173. Ha AD, Jankovic J. Pain in Parkinson's disease. Mov Disord 2012; 27:485–91.
174. Ford B. Pain in Parkinson's disease. Mov Disord 2010;25 Suppl 1:S98–103.
175. Loher TJ, Burgunder JM, Weber S, et al. Effect of chronic pallidal deep brain stimulation on off period dystonia and sensory symptoms in advanced Parkinson's disease. J Neurol Neurosurg Psychiatry 2002;73:395–9.
176. Ramirez-Ruiz B, Marti MJ, Tolosa E, et al. Longitudinal evaluation of cerebral morphological changes in Parkinson's disease with and without dementia. J Neurol 2005;252:1345–52.
177. Jellinger KA. Formation and development of Lewy pathology: a critical update. J Neurol 2009;256 Suppl 3:270–9.
178. Levy R, Dubois B. Apathy and the functional anatomy of the prefrontal cortex-basal ganglia circuits. Cereb Cortex 2006;16:916–28.
179. Connor JR, Boyer PJ, Menzies SL, et al. Neuropathological examination suggests impaired brain iron acquisition in restless legs syndrome. Neurology 2003;61: 304–9.
180. Earley CJ, Barker P, Horská A, Allen RP. MRI-determined regional brain iron concentrations in early- and late-onset restless legs syndrome. Sleep Med 2006;7: 458–61.
181. Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci 2001;24:726–31.
182. Boeve BF, Silber MH, Saper CB, et al. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain 2007;130(Pt 11): 2770–88.
183. Benarroch EE, Schmeichel AM, Paris J. Involvement of the ventrolateral medulla in parkinsonism with autonomic failure. Neurology 2000;54:963–8.
184. Chudler EH, Dong WK. The role of the basal ganglia in nociception and pain. Pain 1995;60:3–38.
185. Wolters E. Non-motor extranigral signs and symptoms in Parkinson's disease. Parkinsonism Relat Disord 2009;15 Suppl 3:S6–12.
186. Davidsdottir S, Cronin-Golomb A, Lee A. Visual and spatial symptoms in Parkinson's disease. Vision Res 2005;45:1285–96.
1. Alves G, Forsaa EB, Pedersen KF, et al. Epidemiology of Parkinson's disease. J Neurol 2008;255 Suppl 5:18–32.
2. Stern MB, Lang A, Poewe W. Toward a redefinition of Parkinson's disease. Mov Disord 2012;27:54–60.
3. Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003;24:197–211.
4. Tandberg E, Larsen JP, Karlsen K. A community-based study of sleep disorders in patients with Parkinson's disease. Mov Disord 1998;13:895–9.
5. Shulman LM, Taback RL, Bean J, Weiner WJ. Comorbidity of the nonmotor symptoms of Parkinson's disease. Mov Disord 2001;16:507–10.
6. Park A, Stacy M. Non-motor symptoms in Parkinson's disease. J Neurol 2009;256 Suppl 3:293–8.
7. Chaudhuri KR, Schapira AH. Non-motor symptoms of Parkinson's disease: dopaminergic pathophysiology and treatment. Lancet Neurol 2009;8:464–74.
8. Shulman LM, Taback RL, Rabinstein AA, Weiner WJ. Non-recognition of depression and other non-motor symptoms in Parkinson's disease. Parkinsonism Relat Disord 2002;8:193–7.
9. Bonnet AM, Jutras MF, Czernecki V, et al. Nonmotor symptoms in Parkinson's disease in 2012: relevant clinical aspects. Parkinsons Dis 2012:2012:198316.
10. Chaudhuri KR, Healy DG, Schapira AH. Non-motor symptoms of Parkinson's disease: diagnosis and management. Lancet Neurol 2006;5:235–45.
11. Hussl AK, Seppi K, Poewe W. Nonmotor symptoms in Parkinson's disease. Expert Rev Neurother 2013;13:581–3.
12. O'Sullivan SS, Williams DR, Gallagher DA, et al. Nonmotor symptoms as presenting complaints in Parkinson's disease: a clinicopathological study. Mov Disord 2008;23:101–6.
13. Lang AE. A critical appraisal of the premotor symptoms of Parkinson's disease: potential usefulness in early diagnosis and design of neuroprotective trials. Mov Disord 2011;26:775–83.
14. Barone P, Antonini A, Colosimo C, et al. The PRIAMO study: A multicenter assessment of nonmotor symptoms and their impact on quality of life in Parkinson's disease. Mov Disord 2009;24:1641–9.
15. Bernal-Pacheco O, Limotai N, Go CL, Fernandez HH. Nonmotor manifestations in Parkinson disease. Neurologist 2012;18:1–16.
16. Lemke MR, Fuchs G, Gemende I, et al. Depression and Parkinson's disease. J Neurol 2004;251 Suppl 6: VI/24–7.
17. Ravina, B, Camicioli R, Como PG, et al, The impact of depressive symptoms in early Parkinson disease. Neurology 2007;69:342–7.
18. Jasinska-Myga B, Putzke JD Wider C, et al. Depression in Parkinson's disease. Can J Neurol Sci 2010;37:61–6.
19. Slaughter JR, Slaughter KA, Nichols D, et al. Prevalence, clinical manifestations, etiology, and treatment of depression in Parkinson's disease. J Neuropsychiatry Clin Neurosci 2001;13:187–96.
20. Simuni T, Sethi K. Nonmotor manifestations of Parkinson's disease. Ann Neurol 2008;64 Suppl 2:S65–80.
21. Ehmann TS, Beninger RJ, Gawel MJ, Riopelle RJ. Depressive symptoms in Parkinson's disease: a comparison with disabled control subjects. J Geriatr Psychiatry Neurol 1990;3:3–9.
22. Menza MA, Mark MH. Parkinson's disease and depression: the relationship to disability and personality. J Neuropsychiatry Clin Neurosci 1994;6:165–9.
23. CDC. Current depression among adults–United States, 2006 and 2008. Morb Mort Weekly Rep 2010;59:1229–35.
24. Hinnell C, Hurt CS, Landau S, et al. Nonmotor versus motor symptoms: how much do they matter to health status in Parkinson's disease? Mov Disord 2012;27:236–41.
25. Cummings JL. Depression and Parkinson's disease: a review. Am J Psychiatry 1992;149:443–54.
26. Weintraub D, Morales KH, Moberg PJ, et al. Antidepressant studies in Parkinson's disease: a review and meta-analysis. Mov Disord 2005;20:1161–9.
27. Richard IH, McDermott MP, Kurlan R, et al. A randomized, double-blind, placebo-controlled trial of antidepressants in Parkinson disease. Neurology 2012;78:1229–36.
28. Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009;72:886–92.
29. Okun MS, Fernandez HH. Will tricyclic antidepressants make a comeback for depressed Parkinson disease patients? Neurology 2009;72:868–9.
30. Antonini A, Tesei S, Zecchinelli A, et al. Randomized study of sertraline and low-dose amitriptyline in patients with Parkinson's disease and depression: effect on quality of life. Mov Disord 2006;21:1119–22.
31. Pedrosa DJ, Timmermann L. Review: management of Parkinson's disease. Neuropsychiatr Dis Treat 2013;9: 321–40.
32. Barone P, Poewe W, Albrecht S, et al. Pramipexole for the treatment of depressive symptoms in patients with Parkinson's disease: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2010;9:573–80.
33. Lemke MR, Brecht HM, Koester J, et al. Anhedonia, depression, and motor functioning in Parkinson's disease during treatment with pramipexole. J Neuropsychiatry Clin Neurosci 2005;17:214–20.
34. Rektorova I, Balaz M, Svatova J, et al. Effects of ropinirole on nonmotor symptoms of Parkinson disease: a prospective multicenter study. Clin Neuropharmacol 2008;31:261–6.
35. Van der Wurff FB, Stek ML, Hoogendijk WL, Beekman AT. Electroconvulsive therapy for the depressed elderly. Cochrane Database Syst Rev 2003(2):CD003593.
36. Okun MS, Watts RL. Depression associated with Parkinson's disease: clinical features and treatment. Neurology 2002;58(4 Suppl 1):S63–70.
37. Dobkin RD, Menza M, Allen LA, et al. Cognitive-behavioral therapy for depression in Parkinson's disease: a randomized, controlled trial. Am J Psychiatry 2013;168:1066–74.
38. Loprinzi PD, Herod SM, Cardinal BJ, Noakes TD. Physical activity and the brain: A review of this dynamic, bi-directional relationship. Brain Res 2013;1539:95–104.
39. Richard IH. Anxiety disorders in Parkinson's disease. Adv Neurol 2005;96:42–55.
40. Leentjens AF, Dujardin K, Marsh L, et al. Symptomatology and markers of anxiety disorders in Parkinson's disease: a cross-sectional study. Mov Disord 2011;26:484–92.
41. Witjas T, Kaphan E, Azulay JP, et al. Nonmotor fluctuations in Parkinson's disease: frequent and disabling. Neurology 2002;59:408–13.
42. Olanow CW, Stern MB, Sethi K. The scientific and clinical basis for the treatment of Parkinson disease Neurology 2009;72(21 Suppl 4):S1–136.
43. Richard IH, Schiffer RB, Kurlan R. Anxiety and Parkinson's disease. J Neuropsychiatry Clin Neurosci 1996;8:383–92.
44. Fenelon G, Alves G. Epidemiology of psychosis in Parkinson's disease. J Neurol Sci 2010;289:12–7.
45. Papapetropoulos S, Katzen H, Schrag A, et al. A questionnaire-based (UM-PDHQ) study of hallucinations in Parkinson's disease. BMC Neurol 2008;8:21.
46. Fenelon G, Mahieux F, Huon R, Ziegler M. Hallucinations in Parkinson's disease: prevalence, phenomenology and risk factors. Brain 2000;123 (Pt 4):733–45.
47. Papapetropoulos S, Mash DC. Psychotic symptoms in Parkinson's disease. From description to etiology. J Neurol 2005;252:753–64.
48. Aarsland D, Larsen JP, Cummins JL, Laake K. Prevalence and clinical correlates of psychotic symptoms in Parkinson disease: a community-based study. Arch Neurol 1999;56:595–601.
49. Aarsland D, Larsen JP, Tandberg E, Laake K. Predictors of nursing home placement in Parkinson's disease: a population-based, prospective study. J Am Geriatr Soc 2000;48:938–42.
50. Lohle M, Storch A, Reichmann H, Beyond tremor and rigidity: non-motor features of Parkinson's disease. J Neural Transm 2009;116:1483–92.
51. Fenelon G. Psychosis in Parkinson's disease: phenomenology, frequency, risk factors, and current understanding of pathophysiologic mechanisms. CNS Spectr 2008;13(3 Suppl 4):18–25.
52. Merims D, Shabtai H, Korczyn AD, et al. Antiparkinsonian medication is not a risk factor for the development of hallucinations in Parkinson's disease. J Neural Transm 2004;111:1447–53.
53. Merims D, Balas M, Peretz C, et al. Rater-blinded, prospective comparison: quetiapine versus clozapine for Parkinson's disease psychosis. Clin Neuropharmacol 2006;29:331–7.
54. Seppi K, Weintraub D, Coelho M, et al. The Movement Disorder Society Evidence-Based Medicine Review Update: Treatments for the non-motor symptoms of Parkinson's disease. Mov Disord 2011;26 Suppl 3:S42–80.
55. Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004;19:831–3.
56. Morgante L, Epifanio A, Spina E, et al. Quetiapine and clozapine in parkinsonian patients with dopaminergic psychosis. Clin Neuropharmacol 2004;27:153–6.
57. Miyasaki JM, Shannon K, Voon V, et al. Practice parameter: evaluation and treatment of depression, psychosis, and dementia in Parkinson disease (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2006;66:996–1002.
58. Riedel O, Klotsche J, Spottke A, et al. Cognitive impairment in 873 patients with idiopathic Parkinson's disease. Results from the German Study on Epidemiology of Parkinson's Disease with Dementia (GEPAD). J Neurol 2008;255:255–64.
59. Aarsland D, Andersen K, Larsen JP, et al. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol 2003;60:387–92.
60. Aarsland D, Andersen K, Larsen JP, et al. Risk of dementia in Parkinson's disease: a community-based, prospective study. Neurology 2001;56:730–6.
61. Riggeal BD, Crucian GP, Seignourel P, et al. Cognitive decline tracks motor progression and not disease duration in Parkinson patients. Neuropsychiatr Dis Treat 2007;3:955–8.
62. Hughes TA, Ross HF, Musa S, et al. A 10-year study of the incidence of and factors predicting dementia in Parkinson's disease. Neurology 2000;54:1596–602.
63. Hobson P, Meara J. Risk and incidence of dementia in a cohort of older subjects with Parkinson's disease in the United Kingdom. Mov Disord 2004;19:1043–9.
64. Hoops S, Nazem S, Siderowf AD, et al. Validity of the MoCA and MMSE in the detection of MCI and dementia in Parkinson disease. Neurology 2009;73:1738–45.
65. Louis ED, Marder K, Cote L, et al. Mortality from Parkinson disease. Arch Neurol 1997;54:260–4.
66. Fernandez HH. Nonmotor complications of Parkinson disease. Cleve Clin J Med 2012;79 Suppl 2:S14–8.
67. Sollinger AB, Goldstein FC, Lah JJ, et al. Mild cognitive impairment in Parkinson's disease: subtypes and motor characteristics. Parkinsonism Relat Disord 2010;16:177–80.
68. Mak E, Zhou J, Tan LC, et al. Cognitive deficits in mild Parkinson's disease are associated with distinct areas of grey matter atrophy. J Neurol Neurosurg Psychiatry 2013 Oct 16.
69. Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson's disease. N Engl J Med 2004;351:2509–18.
70. Aarsland D, Ballard C, Walker Z, et al. Memantine in patients with Parkinson's disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial. Lancet Neurol 2009;8:613–8.
71. Leroi I, Overshott R, Byrne EJ, et al. Randomized controlled trial of memantine in dementia associated with Parkinson's disease. Mov Disord 2009;24:1217–21.
72. Speelman AD, van de Warrenburg BP, van Nimwegen M, et al. How might physical activity benefit patients with Parkinson disease? Nat Rev Neurol 2013;7:528–34.
73. Naismith SL, Mowszowski L, Diamond K, Lewis SJ. Improving memory in Parkinson's disease: a healthy brain ageing cognitive training program. Mov Disord 2013;28:1097–103.
74. Weintraub D, Koester J, Potenza MN, et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol 2010;67:589–95.
75. Voon V, Reynolds B, Brezing C, et al. Impulsive choice and response in dopamine agonist-related impulse control behaviors. Psychopharmacology (Berl) 2010;207:645–59.
76. Weintraub D, Siderowf AD, Potenza MN, et al. Association of dopamine agonist use with impulse control disorders in Parkinson disease. Arch Neurol 2006; 63:969–73.
77. Pontone G, Williams JR, Bassett SS, Marsh L. Clinical features associated with impulse control disorders in Parkinson disease. Neurology 2006;67:1258–61.
78. Voon V, Mehta AR, Hallett M. Impulse control disorders in Parkinson's disease: recent advances. Curr Opin Neurol 2011;24:324–30.
79. Thomas A, Bonanni L, Gambi F, et al. Pathological gambling in Parkinson disease is reduced by amantadine. Ann Neurol 2010;68:400–4.
80. Miyasaki JM, Al Hassan K, Lang AE, Voon V. Punding prevalence in Parkinson's disease. Mov Disord 2007;22:1179–81.
81. Nguyen FN, Chang YL, Okun MS, et al. Prevalence and characteristics of punding and repetitive behaviors among Parkinson patients in North-Central Florida. Int J Geriatr Psychiatry 2010;25:540–1.
82. Sohtaoglu M, Demiray DY, Kenangil G, et al. Long term follow-up of Parkinson's disease patients with impulse control disorders. Parkinsonism Relat Disord 2010;16:334–7.
83. Antonini A, Cilia R. Behavioural adverse effects of dopaminergic treatments in Parkinson's disease: incidence, neurobiological basis, management and prevention. Drug Saf 2009;
32:475–88.
84. Miwa H, Morita S, Nakanishi I, Kondo T, Stereotyped behaviors or punding after quetiapine administration in Parkinson's disease. Parkinsonism Relat Disord 2004;10:177–80.
85. Skorvanek M, Rosenberger J, Gdovinova Z, et al. Apathy in elderly nondemented patients with parkinson's disease: clinical determinants and relationship to quality of life. J Geriatr Psychiatry Neurol 2013;26:237–43.
86. Marin RS, Fogel BS, Hawkins J, et al. Apathy: a treatable syndrome. J Neuropsychiatry Clin Neurosci 1995;7:23–30.
87. Pluck GC, Brown RG. Apathy in Parkinson's disease. J Neurol Neurosurg Psychiatry 2002;73:636–42.
88. Kirsch-Darrow L, Fernandez HH, Marsiske M, et al. Dissociating apathy and depression in Parkinson disease. Neurology 2006;67:33–8.
89. Pedersen KF, Alves G, Aarsland D, Larsen JP. Occurrence and risk factors for apathy in Parkinson disease: a 4-year prospective longitudinal study. J Neurol Neurosurg Psychiatry 2009;80:1279–82.
90. Czernecki V, Pillon B, Houeto JL, et al. Motivation, reward, and Parkinson's disease: influence of dopatherapy. Neuropsychologia 2002;40:2257–67.
91. Parkinson J. An essay on the shaking palsy. Sherwood, Neely, and Jones; 1817.
92. Schenck CH, Mahowald WM. REM sleep behavior disorder: clinical, developmental, and neuroscience perspectives 16 years after its formal identification in SLEEP. Sleep 2002;25:120–38.
93. Sixel-Doring F, Trautmann E, Mollenhauer B, Trenkwalder C. Associated factors for REM sleep behavior disorder in Parkinson disease. Neurology 2011;77:1048–54.
94. Eisensehr I, v Lindeiner H, Jager M, Noachtar S. REM sleep behavior disorder in sleep-disordered patients with versus without Parkinson's disease: is there a need for polysomnography? J Neurol Sci 2001;186:7–11.
95. Postuma RB, Gagnon JF, Montplaisir JY. REM sleep behavior disorder and prodromal neurodegeneration - where are we headed? Tremor Other Hyperkinet Mov (N Y) 2013;3.
96. Iranzo A, Tolosa E, Gelpi E, et al. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: an observational cohort study. Lancet Neurol 2013;12: 443–53.
97. Schenck CH, Mahowald MW. Rapid eye movement sleep parasomnias. Neurol Clin 2005;23:1107–26.
98. Anderson KN, Shneerson JM. Drug treatment of REM sleep behavior disorder: the use of drug therapies other than clonazepam. J Clin Sleep Med 2009;5:235–9.
99. Boeve BF, Silber MH, Ferman TJ. Melatonin for treatment of REM sleep behavior disorder in neurologic disorders: results in 14 patients. Sleep Med 2003;4:281–4.
100. Kunz D, Mahlberg R. A two-part, double-blind, placebo-controlled trial of exogenous melatonin in REM sleep behaviour disorder. J Sleep Res 2010;19:591–6.
101. Perroud N, Lazignac C, Baleydier B, et al. Restless legs syndrome induced by citalopram: a psychiatric emergency? Gen Hosp Psychiatry 2007;29:72–4.
102. Buskova J, Vorlova T, Pisko J, Sonka K, Severe sleep-related movement disorder induced by sertraline. Sleep Med 2012;13:769–70.
103. Rittmannsberger H, Werl R. Restless legs syndrome induced by quetiapine: report of seven cases and review of the literature. Int J Neuropsychopharmacol 2013;16:1427–31.
104. Perez-Lloret S, Rey MV, Bondon-Guitton E, et al. Drugs associated with restless legs syndrome: a case/noncase study in the French pharmacovigilance database. J Clin Psychopharmacol 2012;32:824–7.
105. Aurora RN, Kristo DA, Bista SR, et al. The treatment of restless legs syndrome and periodic limb movement disorder in adults-an update for 2012: practice parameters with an evidence-based systematic review and meta-analyses: an American Academy of Sleep Medicine Clinical Practice Guideline. Sleep 2012;35:1039–62.
106. Covassin N, Neikrug AB, Liu L, et al. Clinical correlates of periodic limb movements in sleep in Parkinson's disease. J Neurol Sci 2012;316:131–6.
107. Rios Romenets S, Postuma RB. Treatment of restless legs syndrome. Curr Treat Options Neurol 2013;15:396-409.
108. Hogl B, Paulus W, Clarenbach P, Trenkwalder C, Restless legs syndrome: diagnostic assessment and the advantages and risks of dopaminergic treatment. J Neurol 2006;253 Suppl 4:IV22-8.
109. Garcia-Borreguero D, Kohnen R, Silber MH, et al. The long-term treatment of restless legs syndrome/Willis-Ekbom disease: evidence-based guidelines and clinical consensus best practice guidance: a report from the International Restless Legs Syndrome Study Group. Sleep Med 2013;14:675–84.
110. Montagna P, Hornyak M, Ulfberg J, et al. Randomized trial of pramipexole for patients with restless legs syndrome (RLS) and RLS-related impairment of mood. Sleep Med 2011;12:34–40.
111. Partinen M, Hirvonen K, Jama L, et al. Efficacy and safety of pramipexole in idiopathic restless legs syndrome: a polysomnographic dose-finding study--the PRELUDE study. Sleep Med 2006;7:407–17.
112. Trenkwalder C Garcia-Borreguero D, Montagna P, et al. Ropinirole in the treatment of restless legs syndrome: results from the TREAT RLS 1 study, a 12 week, randomised, placebo controlled study in 10 European countries. J Neurol Neurosurg Psychiatry 2004;75:92–7.
113. Hening WA, Allen RP, Ondo WG, et al. Rotigotine improves restless legs syndrome: a 6-month randomized, double-blind, placebo-controlled trial in the United States. Mov Disord 2010;25:1675–83.
114. Oertel WH, Benes H, Garcia-Borreguero D, et al. Rotigotine transdermal patch in moderate to severe idiopathic restless legs syndrome: a randomized, placebo-controlled polysomnographic study. Sleep Med 2010;11:848–56.
115. Lee DO, Ziman RB, Perkins AT, et al. A randomized, double-blind, placebo-controlled study to assess the efficacy and tolerability of gabapentin enacarbil in subjects with restless legs syndrome. J Clin Sleep Med 2011;7:282–92.
116. Kushida CA, Walters AS, Becker P, et al. A randomized, double-blind, placebo-controlled, crossover study of XP13512/GSK1838262 in the treatment of patients with primary restless legs syndrome. Sleep 2009;32:159–68.
117. Menza M, Dobkin RD, Marin H, Bienfait K. Sleep disturbances in Parkinson's disease. Mov Disord 2010;25 Suppl 1:S117–22.
118. Louter M, van Sloun RJ, Pevernagie DA, et al. Subjectively impaired bed mobility in Parkinson disease affects sleep efficiency. Sleep Med 2013;14:668–74.
119. Menza M, Dobkin RD, Marin H, et al. Treatment of insomnia in Parkinson's disease: a controlled trial of eszopiclone and placebo. Mov Disord 2010;25:1708–14.
120. Nowell PD, Mazumdar S, Buysse DJ, et al. Benzodiazepines and zolpidem for chronic insomnia: a meta-analysis of treatment efficacy. JAMA 1997;278:2170–7.
121. Rios Romenets S, Creti L, Fichten C, et al. Doxepin and cognitive behavioural therapy for insomnia in patients with Parkinson's disease – a randomized study. Parkinsonism Relat Disord 2013;19:670–5.
122. Ondo WG, Dat Vuong K, Khan H, et al. Daytime sleepiness and other sleep disorders in Parkinson's disease. Neurology 2001;57:1392–6.
123. Razmy A, Lang AE, Shapiro CM, Predictors of impaired daytime sleep and wakefulness in patients with Parkinson disease treated with older (ergot) vs newer (nonergot) dopamine agonists. Arch Neurol 2004;61:97–102.
124. Holloway RG, Shoulson I, Fahn S, et al. Pramipexole vs levodopa as initial treatment for Parkinson disease: a 4-year randomized controlled trial. Arch Neurol 2004;61:1044–53.
125. Paus S, Brecht HM, Koster J, et al. Sleep attacks, daytime sleepiness, and dopamine agonists in Parkinson's disease. Mov Disord 2003;18:659–67.
126. Hogl B, Saletu M, Brandauer E, et al. Modafinil for the treatment of daytime sleepiness in Parkinson's disease: a double-blind, randomized, crossover, placebo-controlled polygraphic trial. Sleep 2002;25:905–9.
127. Ondo WG, Fayle R, Atassi F, Jankovic J. Modafinil for daytime somnolence in Parkinson's disease: double blind, placebo controlled parallel trial. J Neurol Neurosurg Psychiatry 2005;76:1636-–9.
128. Adler CH, Caviness JN, Hentz JG, et al. Randomized trial of modafinil for treating subjective daytime sleepiness in patients with Parkinson's disease. Mov Disord 2003;18:287–93.
129. Devos D, Krystkowiak P, Clement F, et al. Improvement of gait by chronic, high doses of methylphenidate in patients with advanced Parkinson's disease. J Neurol Neurosurg Psychiatry 2007;78:470–5.
130. Tyne HL, Taylor J, Baker GA, Steiger MJ. Modafinil for Parkinson's disease fatigue. J Neurol 2010;257:452–6.
131. Avorn J, Schneeweiss S, Sudarsky LR, et al. Sudden uncontrollable somnolence and medication use in Parkinson disease. Arch Neurol 2005;62:1242–8.
132. Trotti LM, Bliwise DL. No increased risk of obstructive sleep apnea in Parkinson's disease. Mov Disord 2010;25:2246–9.
133. da Silva-Junior FP, do Prado GF, Barbosa ER, et al. Sleep disordered breathing in Parkinson's disease: A critical appraisal. Sleep Med Rev 2013 Jul 22.
134. Noradina AT, Karim NA, Hamidon BB, et al. Sleep-disordered breathing in patients with Parkinson's disease. Singapore Med J 2010;51:60–4.
135. Oerlemans WG, de Weerd AW. The prevalence of sleep disorders in patients with Parkinson's disease. A self-reported, community-based survey. Sleep Med 2002;3:147–9.
136. Low PA. Prevalence of orthostatic hypotension. Clin Auton Res 2008;18 Suppl 1:8–13.
137. Goldstein DS. Orthostatic hypotension as an early finding in Parkinson's disease. Clin Auton Res 2006;16:46–54.
138. Sharabi Y, Goldstein DS. Mechanisms of orthostatic hypotension and supine hypertension in Parkinson disease. J Neurol Sci 2011;310:123–8.
139. Sanchez-Ferro A, Benito-Leon J, Gomez-Esteban JC. The management of orthostatic hypotension in Parkinson's disease. Front Neurol 2013;4:64.
140. Lahrmann H, Cortelli P, Hilz M, et al. EFNS guidelines on the diagnosis and management of orthostatic hypotension. Eur J Neurol 2006;13:930–6.
141. Schoffer KL, Henderson RD, O'Maley K, O'Sullivan JD. Nonpharmacological treatment, fludrocortisone, and domperidone for orthostatic hypotension in Parkinson's disease. Mov Disord 2007;22:1543–9.
142. Singer W, Sandroni P, Opfer-Gehrking TL, et al. Pyridostigmine treatment trial in neurogenic orthostatic hypotension. Arch Neurol 2006;63:513–8.
143. Low PA, Gilden JL, Freeman R, et al. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA 1997;277:1046–51.
144. Kaufmann H. L-dihydroxyphenylserine (Droxidopa): a new therapy for neurogenic orthostatic hypotension: the US experience. Clin Auton Res 2008;18 Suppl 1:19–24.
145. Magerkurth C, Schnitzer R, Braune S. Symptoms of autonomic failure in Parkinson's disease: prevalence and impact on daily life. Clin Auton Res 2005;15:76–82.
146. Makaroff L, Gunn A, Gervasoni C, Richy F. Gastrointestinal Disorders in Parkinson's Disease: Prevalence and Health Outcomes in a US Claims Database. J Parkinsons Dis 2011;1:65–74.
147. Abbott RD, Petrovitch H, White LR, et al. Frequency of bowel movements and the future risk of Parkinson's disease. Neurology 2001;57:456–62.
148. Abbott RD, Ross GW, Petrovitch H, et al. Bowel movement frequency in late-life and incidental Lewy bodies. Mov Disord 2007;22:1581–6.
149. Zangaglia R, Martignoni E, Glorioso M, et al. Macrogol for the treatment of constipation in Parkinson's disease. A randomized placebo-controlled study. Mov Disord 2007;22:1239–44.
150. Ondo WG, Kenney C, Sullivan K, et al. Placebo-controlled trial of lubiprostone for constipation associated with Parkinson disease. Neurology 2012;78:1650–4.
151. Cersosimo MG, Benarroch EE. Neural control of the gastrointestinal tract: implications for Parkinson disease. Mov Disord 2008;23:1065–75.
152. Reddymasu SC, Soykan I, McCallum RW. Domperidone: review of pharmacology and clinical applications in gastroenterology. Am J Gastroenterol 2007;102:2036–45.
153. Argolo N, Sampaio M, Pinho P, et al. Do swallowing exercises improve swallowing dynamic and quality of life in Parkinson's disease? NeuroRehabilitation 2013;32:949–55.
154. Troche MS, Sapienza CM, Rosenbek JC. Effects of bolus consistency on timing and safety of swallow in patients with Parkinson's disease. Dysphagia 2008;23:26–32.
155. Edwards LL, Quigley EM, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson's disease: frequency and pathophysiology. Neurology 1992;42:726–32.
156. Kalf JG, Smit AM, Bloem BR, et al. Impact of drooling in Parkinson's disease. J Neurol 2007;254:1227–32.
157. Leibner J, Ramjit A, Sedig L, et al. The impact of and the factors associated with drooling in Parkinson's disease. Parkinsonism Relat Disord 2010;16:475–7.
158. Mancini F, Zangaglia R, Cristina S, et al. Double-blind, placebo-controlled study to evaluate the efficacy and safety of botulinum toxin type A in the treatment of drooling in parkinsonism. Mov Disord 2003;18:685–8.
159. Lagalla G, Millevolte M, Capecci M, et al. Long-lasting benefits of botulinum toxin type B in Parkinson's disease-related drooling. J Neurol 2009;256:563–7.
160. Lagalla G, Millevolte N, Capecci M, et al. Botulinum toxin type A for drooling in Parkinson's disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2006;21:704–7.
161. Arbouw ME, Movig KL, Koopmann M, et al. Glycopyrrolate for sialorrhea in Parkinson disease: a randomized, double-blind, crossover trial. Neurology 2010; 74:1203–7.
162. South AR, Somers, Jog MS. Gum chewing improves swallow frequency and latency in Parkinson patients: a preliminary study. Neurology 2010;74:1198–202.
163. Winge K, Nielsen KK. Bladder dysfunction in advanced Parkinson's disease. Neurourol Urodyn 2012;31:1279–83.
164. Madhuvrata P, Singh PM, Hasafa Z, Abdel-Fattah M. Anticholinergic drugs for adult neurogenic detrusor overactivity: a systematic review and meta-analysis. Eur Urol 2012;62:816–30.
165. Soljanik I. Efficacy and safety of botulinum toxin a intradetrusor injections in adults with neurogenic detrusor overactivity/neurogenic overactive bladder: a systematic review. Drugs 2013;73:1055–66.
166. Sakakibara R, Uchiyama T, Yamanishi T, Kishi M. Genitourinary dysfunction in Parkinson's disease. Mov Disord 2010;25:2–12.
167. Kummer A, Cardoso F, Teixeira AL. Loss of libido in Parkinson's disease. J Sex Med 2009;6:1024–31.
168. Lombardi G, Nelli F, Celso M, et al. Treating erectile dysfunction and central neurological diseases with oral phosphodiesterase type 5 inhibitors. Review of the literature. J Sex Med 2012 9(4): 970–85.
169. Dula E, Bukofzer S, Perdok R, George M. Double-blind, crossover comparison of 3 mg apomorphine SL with placebo and with 4 mg apomorphine SL in male erectile dysfunction. Eur Urol 2001;39(5): 558–3
170. Negre-Pages L, Regragui W, Bouhassira D, et al. Chronic pain in Parkinson's disease: the cross-sectional French DoPaMiP survey. Mov Disord 2008;23:1361–9.
171. Beiske AG, Loge JH, Ronningen A, Svensson E. Pain in Parkinson's disease: Prevalence and characteristics. Pain 2009. 141:173–7.
172. Lin CH, Wu RM, H.Y. Chang HY, et al. Preceding pain symptoms and Parkinson's disease: a nationwide population-based cohort study. Eur J Neurol 2013;20:1398–404.
173. Ha AD, Jankovic J. Pain in Parkinson's disease. Mov Disord 2012; 27:485–91.
174. Ford B. Pain in Parkinson's disease. Mov Disord 2010;25 Suppl 1:S98–103.
175. Loher TJ, Burgunder JM, Weber S, et al. Effect of chronic pallidal deep brain stimulation on off period dystonia and sensory symptoms in advanced Parkinson's disease. J Neurol Neurosurg Psychiatry 2002;73:395–9.
176. Ramirez-Ruiz B, Marti MJ, Tolosa E, et al. Longitudinal evaluation of cerebral morphological changes in Parkinson's disease with and without dementia. J Neurol 2005;252:1345–52.
177. Jellinger KA. Formation and development of Lewy pathology: a critical update. J Neurol 2009;256 Suppl 3:270–9.
178. Levy R, Dubois B. Apathy and the functional anatomy of the prefrontal cortex-basal ganglia circuits. Cereb Cortex 2006;16:916–28.
179. Connor JR, Boyer PJ, Menzies SL, et al. Neuropathological examination suggests impaired brain iron acquisition in restless legs syndrome. Neurology 2003;61: 304–9.
180. Earley CJ, Barker P, Horská A, Allen RP. MRI-determined regional brain iron concentrations in early- and late-onset restless legs syndrome. Sleep Med 2006;7: 458–61.
181. Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci 2001;24:726–31.
182. Boeve BF, Silber MH, Saper CB, et al. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain 2007;130(Pt 11): 2770–88.
183. Benarroch EE, Schmeichel AM, Paris J. Involvement of the ventrolateral medulla in parkinsonism with autonomic failure. Neurology 2000;54:963–8.
184. Chudler EH, Dong WK. The role of the basal ganglia in nociception and pain. Pain 1995;60:3–38.
185. Wolters E. Non-motor extranigral signs and symptoms in Parkinson's disease. Parkinsonism Relat Disord 2009;15 Suppl 3:S6–12.
186. Davidsdottir S, Cronin-Golomb A, Lee A. Visual and spatial symptoms in Parkinson's disease. Vision Res 2005;45:1285–96.
What Is the Global Burden of Unsafe Medical Care?
Study Overview
Objective. To examine the global burden of unsafe medical care and its comparative frequency in low/middle-income vs. high-income countries.
Design. Analytical modeling of aggregated data from observational studies.
Data. Two primary sources of data were used. First, the team conducted a search of over 16,000 articles written in English after 1976 that aimed for a comprehensive exam-ination of both peer-reviewed and non–peer-reviewed studies that focused on 7 inpatient adverse events (see below), and the clinical features of the patients who were injured from them. Two separate literature reviews were conducted in 2007 through early 2008 and then repeated in 2011. Discussions with international experts in each topic area informed the selection process. The second source of data was epidemiological studies commissioned by the World Health Organization (WHO). These aimed to identify inpatient adverse events using a 2-stage medical record review in 26 hospitals across 8 low- and middle-income countries (LMICs) in the Eastern Mediterranean and North Africanregions, and 35 hospitals across 5 countries in Latin America.
Main outcome measures. 7 types of adverse events were evaluated in the analysis: (1) adverse drug events, (2) catheter-related urinary tract infection, (3) catheter-related blood stream infections, (4) nosocomial pneumonia, (5) venous thromboembolism, (6) falls, and (7) pressure ulcers (decubiti). The global burden of disease (GBD) is a standard metric that uses disability-adjusted life years (DALYs) as a proxy measure of morbidity and mortality related to a specific condition. The GBD DALYs model requires several key inputs: the number of people affected, the age at which they are affected, and the clinical consequence of the adverse events. In this study, a single average age per event was used instead of the standard GBD calculations by age and sex. Each input of GBD and DALYs was calculated separately for high-income countries (HICs) versus LMICs. The World Bank sets the income categorization for countries and adjusts the information on an annual basis. Countries in each category share common characteristics of socioeconomic development and epidemiological profiles.
Main results. The rate of hospitalization in HICs was higher than in LMICs: 10.8 vs. 3.7 per 100 citizens per year. There were large variations in the reported incidence of adverse events in both HICs and LMICs. Of the 7 adverse events assessed, adverse drug events were the most common type in HICs, with an incidence rate of 5.0%. In LMICs, venous thromboembolism was most common, with an incidence rate of 3.0%. Catheter-related blood stream infection, venous thromboembolism, and pressure ulcers had comparable rates between HICs and LMIC . The authors estimated that for every 100 hospitalizations, approximately 14.2 adverse events in HICs and 12.7 in LMICs. This is roughly 16.8 million injuries annually among hospitalized patients in HICs. LMICs and experienced approximately 50% more adverse events than HICs. Of note, LMICs had 5 times the population of HICs but the authors did not calculate proportional incidence rates.
The authors estimated 22.6 million DALYs lost due to these adverse events in 2009 globally. Unsurprisingly, the number of DALYs lost were more than twice as high as in LMICs as they were in HICs. This is likely due to the combination of weaker health systems and human resources for health shortages in those countries. In LMICs, venous thromboembolism was the main source of lost DALYs. Although incidences of hospital-acquired infections--such as nosocomial pneumonia, catheter-related blood stream and urinary tract infections--were smaller, they caused a comparable number of DALYs lost. Premature death from adverse events was the primary source of DALYs lost for all countries.
Conclusion. Adverse events from unsafe care is a significant problem across all countries.
Commentary
Globally, the efforts to improve health care delivery for diseases that cause substantial morbidity and mortality have been largely successful. For example, antimalarial drugs and antiretroviral therapies have become more accessible to patients in need [1,2]. However, in order to create more sustainable model, the health care systems of developing countries need sustainable investments to care for their growing populations and increasing medical needs [3,4]. Allengranzi et al [5] concluded from a systemic review that health care–associated infections are ubiquitous and occur at much higher rates in LMICs than in HICs. Findings from this study support those from Allengranzi’s review.
This study helped further our understanding of and explored the impact of unsafe medical care on GBD and DALYs. Several other adverse events related to unsafe care, such as unsafe surgery, harms due to counterfeit drugs, unsafe childbirth and unsafe blood use, were not included in this study due to data limitations. The estimated lost DALYs would be much higher if these events were counted.
This study has several strengths. First, the authors sought out the best available data from a large number of sources. Evidence selected for the analysis came from studies with good quality ratings. The 7 outcome measures used in this study are now standard minimum reporting data internationally. Nonetheless, several limitations are present. As the authors noted, the lack of availability high-quality data is common in international analyses. There can be reporting delays, data collection errors due to a lack of technical capacity, and corruption problems that may influence data quality. Poor reporting practices may exclude or underreport adverse events. Also, the paucity of data for some variables limited the calculation of estimates Second, few studies used standardized approaches in their data collection and analysis, contributing to data inconsistencies that may affect the reliability of the results. Third, the same life expectancy value (the WHO standard) was used for all individuals regardless of their countries’ life expectancy. The authors acknowledged that this approach was controversial and may have resulted in a different number of DALYs lost. Finally, only English-language publications were used, which may have influenced the findings. Latin America, the former Soviet Union states, and many Asian countries have growing bodies of research published in their native languages.
Despite the limitations, the study is one of the first systematic analyses of GBD, the outcomes of unsafe medical care, and associated lost DALYs. The analysis identified that a majority of the harms from adverse events occur in LMICs. Policies addressing, supporting, and enforcing patient safety measures during the health care experience will help ensure reductions in mortality and morbidity in LMICs. Improving the safety of the healthcare system should be a major policy and research emphasis across the globe.
Applications for Clinical Practice
Even though patient safety initiatives have been at the forefront of many organizational policies and health care provider education since the 1999 Institute of Medicine report “Crossing the Quality Chasm,” this study reminds practitioners that safe clinical practice is essential for reducing domestic disease burden. The cost of adverse events from unsafe practice in the United States was estimated to be around $16.6 billion in 2004 alone [6]. With the World Health Organization calling for strengthened research infrastructure across the globe and LMICs now seeing the value of data for health systems policymaking and management, future research will help to further refine the methods developed in this study.
—Jin Jun, MSN, APRN-BC, CCRN, and Allison Squires, PhD, RN
1. Kaplan J, Hanson D, Dworkin M, et al. Epidemiology of human immunodeficiency virus-associated opportunistic infections in the United States in the era of highly active antiretroviral therapy, Clin Infect Dis 2000;30 Suppl 1:S5–14.
2. Eaton J, et al. Health benefits, costs, and cost-effectiveness of earlier eligibility for adult antiretroviral therapy and expanded treatment coverage: a cmbined analysis of 12 mathematical models, Lancet Global Health 2014;2:e23–34.
3. Mills A, Brugha R, Hanson K, et al. What can be done about the private health sector in low-income countries? Bull World Health Org 2002;80:325–30.
4. Schlein K, De La Cruz A, Gopalakrishnan T, Montagu D. Private sector delivery of health services in developing countries: a mixed-methods study on quality assurance in social franchises, BMC Health Serv Res 2013;13:4.
5. Allegranzi B, Bagheri N, Combescure C, et al. Burden of endemic health-care-associated infection in developing countries: systematic review and meta-analysis, Lancet 2011;377;
228–41.
6. Jha A, Chan D, Ridgway A, et al. Improving safety and eliminating redundant tests: cutting costs in US hospitals. Health Affairs 2009;28:1475–84.
Study Overview
Objective. To examine the global burden of unsafe medical care and its comparative frequency in low/middle-income vs. high-income countries.
Design. Analytical modeling of aggregated data from observational studies.
Data. Two primary sources of data were used. First, the team conducted a search of over 16,000 articles written in English after 1976 that aimed for a comprehensive exam-ination of both peer-reviewed and non–peer-reviewed studies that focused on 7 inpatient adverse events (see below), and the clinical features of the patients who were injured from them. Two separate literature reviews were conducted in 2007 through early 2008 and then repeated in 2011. Discussions with international experts in each topic area informed the selection process. The second source of data was epidemiological studies commissioned by the World Health Organization (WHO). These aimed to identify inpatient adverse events using a 2-stage medical record review in 26 hospitals across 8 low- and middle-income countries (LMICs) in the Eastern Mediterranean and North Africanregions, and 35 hospitals across 5 countries in Latin America.
Main outcome measures. 7 types of adverse events were evaluated in the analysis: (1) adverse drug events, (2) catheter-related urinary tract infection, (3) catheter-related blood stream infections, (4) nosocomial pneumonia, (5) venous thromboembolism, (6) falls, and (7) pressure ulcers (decubiti). The global burden of disease (GBD) is a standard metric that uses disability-adjusted life years (DALYs) as a proxy measure of morbidity and mortality related to a specific condition. The GBD DALYs model requires several key inputs: the number of people affected, the age at which they are affected, and the clinical consequence of the adverse events. In this study, a single average age per event was used instead of the standard GBD calculations by age and sex. Each input of GBD and DALYs was calculated separately for high-income countries (HICs) versus LMICs. The World Bank sets the income categorization for countries and adjusts the information on an annual basis. Countries in each category share common characteristics of socioeconomic development and epidemiological profiles.
Main results. The rate of hospitalization in HICs was higher than in LMICs: 10.8 vs. 3.7 per 100 citizens per year. There were large variations in the reported incidence of adverse events in both HICs and LMICs. Of the 7 adverse events assessed, adverse drug events were the most common type in HICs, with an incidence rate of 5.0%. In LMICs, venous thromboembolism was most common, with an incidence rate of 3.0%. Catheter-related blood stream infection, venous thromboembolism, and pressure ulcers had comparable rates between HICs and LMIC . The authors estimated that for every 100 hospitalizations, approximately 14.2 adverse events in HICs and 12.7 in LMICs. This is roughly 16.8 million injuries annually among hospitalized patients in HICs. LMICs and experienced approximately 50% more adverse events than HICs. Of note, LMICs had 5 times the population of HICs but the authors did not calculate proportional incidence rates.
The authors estimated 22.6 million DALYs lost due to these adverse events in 2009 globally. Unsurprisingly, the number of DALYs lost were more than twice as high as in LMICs as they were in HICs. This is likely due to the combination of weaker health systems and human resources for health shortages in those countries. In LMICs, venous thromboembolism was the main source of lost DALYs. Although incidences of hospital-acquired infections--such as nosocomial pneumonia, catheter-related blood stream and urinary tract infections--were smaller, they caused a comparable number of DALYs lost. Premature death from adverse events was the primary source of DALYs lost for all countries.
Conclusion. Adverse events from unsafe care is a significant problem across all countries.
Commentary
Globally, the efforts to improve health care delivery for diseases that cause substantial morbidity and mortality have been largely successful. For example, antimalarial drugs and antiretroviral therapies have become more accessible to patients in need [1,2]. However, in order to create more sustainable model, the health care systems of developing countries need sustainable investments to care for their growing populations and increasing medical needs [3,4]. Allengranzi et al [5] concluded from a systemic review that health care–associated infections are ubiquitous and occur at much higher rates in LMICs than in HICs. Findings from this study support those from Allengranzi’s review.
This study helped further our understanding of and explored the impact of unsafe medical care on GBD and DALYs. Several other adverse events related to unsafe care, such as unsafe surgery, harms due to counterfeit drugs, unsafe childbirth and unsafe blood use, were not included in this study due to data limitations. The estimated lost DALYs would be much higher if these events were counted.
This study has several strengths. First, the authors sought out the best available data from a large number of sources. Evidence selected for the analysis came from studies with good quality ratings. The 7 outcome measures used in this study are now standard minimum reporting data internationally. Nonetheless, several limitations are present. As the authors noted, the lack of availability high-quality data is common in international analyses. There can be reporting delays, data collection errors due to a lack of technical capacity, and corruption problems that may influence data quality. Poor reporting practices may exclude or underreport adverse events. Also, the paucity of data for some variables limited the calculation of estimates Second, few studies used standardized approaches in their data collection and analysis, contributing to data inconsistencies that may affect the reliability of the results. Third, the same life expectancy value (the WHO standard) was used for all individuals regardless of their countries’ life expectancy. The authors acknowledged that this approach was controversial and may have resulted in a different number of DALYs lost. Finally, only English-language publications were used, which may have influenced the findings. Latin America, the former Soviet Union states, and many Asian countries have growing bodies of research published in their native languages.
Despite the limitations, the study is one of the first systematic analyses of GBD, the outcomes of unsafe medical care, and associated lost DALYs. The analysis identified that a majority of the harms from adverse events occur in LMICs. Policies addressing, supporting, and enforcing patient safety measures during the health care experience will help ensure reductions in mortality and morbidity in LMICs. Improving the safety of the healthcare system should be a major policy and research emphasis across the globe.
Applications for Clinical Practice
Even though patient safety initiatives have been at the forefront of many organizational policies and health care provider education since the 1999 Institute of Medicine report “Crossing the Quality Chasm,” this study reminds practitioners that safe clinical practice is essential for reducing domestic disease burden. The cost of adverse events from unsafe practice in the United States was estimated to be around $16.6 billion in 2004 alone [6]. With the World Health Organization calling for strengthened research infrastructure across the globe and LMICs now seeing the value of data for health systems policymaking and management, future research will help to further refine the methods developed in this study.
—Jin Jun, MSN, APRN-BC, CCRN, and Allison Squires, PhD, RN
Study Overview
Objective. To examine the global burden of unsafe medical care and its comparative frequency in low/middle-income vs. high-income countries.
Design. Analytical modeling of aggregated data from observational studies.
Data. Two primary sources of data were used. First, the team conducted a search of over 16,000 articles written in English after 1976 that aimed for a comprehensive exam-ination of both peer-reviewed and non–peer-reviewed studies that focused on 7 inpatient adverse events (see below), and the clinical features of the patients who were injured from them. Two separate literature reviews were conducted in 2007 through early 2008 and then repeated in 2011. Discussions with international experts in each topic area informed the selection process. The second source of data was epidemiological studies commissioned by the World Health Organization (WHO). These aimed to identify inpatient adverse events using a 2-stage medical record review in 26 hospitals across 8 low- and middle-income countries (LMICs) in the Eastern Mediterranean and North Africanregions, and 35 hospitals across 5 countries in Latin America.
Main outcome measures. 7 types of adverse events were evaluated in the analysis: (1) adverse drug events, (2) catheter-related urinary tract infection, (3) catheter-related blood stream infections, (4) nosocomial pneumonia, (5) venous thromboembolism, (6) falls, and (7) pressure ulcers (decubiti). The global burden of disease (GBD) is a standard metric that uses disability-adjusted life years (DALYs) as a proxy measure of morbidity and mortality related to a specific condition. The GBD DALYs model requires several key inputs: the number of people affected, the age at which they are affected, and the clinical consequence of the adverse events. In this study, a single average age per event was used instead of the standard GBD calculations by age and sex. Each input of GBD and DALYs was calculated separately for high-income countries (HICs) versus LMICs. The World Bank sets the income categorization for countries and adjusts the information on an annual basis. Countries in each category share common characteristics of socioeconomic development and epidemiological profiles.
Main results. The rate of hospitalization in HICs was higher than in LMICs: 10.8 vs. 3.7 per 100 citizens per year. There were large variations in the reported incidence of adverse events in both HICs and LMICs. Of the 7 adverse events assessed, adverse drug events were the most common type in HICs, with an incidence rate of 5.0%. In LMICs, venous thromboembolism was most common, with an incidence rate of 3.0%. Catheter-related blood stream infection, venous thromboembolism, and pressure ulcers had comparable rates between HICs and LMIC . The authors estimated that for every 100 hospitalizations, approximately 14.2 adverse events in HICs and 12.7 in LMICs. This is roughly 16.8 million injuries annually among hospitalized patients in HICs. LMICs and experienced approximately 50% more adverse events than HICs. Of note, LMICs had 5 times the population of HICs but the authors did not calculate proportional incidence rates.
The authors estimated 22.6 million DALYs lost due to these adverse events in 2009 globally. Unsurprisingly, the number of DALYs lost were more than twice as high as in LMICs as they were in HICs. This is likely due to the combination of weaker health systems and human resources for health shortages in those countries. In LMICs, venous thromboembolism was the main source of lost DALYs. Although incidences of hospital-acquired infections--such as nosocomial pneumonia, catheter-related blood stream and urinary tract infections--were smaller, they caused a comparable number of DALYs lost. Premature death from adverse events was the primary source of DALYs lost for all countries.
Conclusion. Adverse events from unsafe care is a significant problem across all countries.
Commentary
Globally, the efforts to improve health care delivery for diseases that cause substantial morbidity and mortality have been largely successful. For example, antimalarial drugs and antiretroviral therapies have become more accessible to patients in need [1,2]. However, in order to create more sustainable model, the health care systems of developing countries need sustainable investments to care for their growing populations and increasing medical needs [3,4]. Allengranzi et al [5] concluded from a systemic review that health care–associated infections are ubiquitous and occur at much higher rates in LMICs than in HICs. Findings from this study support those from Allengranzi’s review.
This study helped further our understanding of and explored the impact of unsafe medical care on GBD and DALYs. Several other adverse events related to unsafe care, such as unsafe surgery, harms due to counterfeit drugs, unsafe childbirth and unsafe blood use, were not included in this study due to data limitations. The estimated lost DALYs would be much higher if these events were counted.
This study has several strengths. First, the authors sought out the best available data from a large number of sources. Evidence selected for the analysis came from studies with good quality ratings. The 7 outcome measures used in this study are now standard minimum reporting data internationally. Nonetheless, several limitations are present. As the authors noted, the lack of availability high-quality data is common in international analyses. There can be reporting delays, data collection errors due to a lack of technical capacity, and corruption problems that may influence data quality. Poor reporting practices may exclude or underreport adverse events. Also, the paucity of data for some variables limited the calculation of estimates Second, few studies used standardized approaches in their data collection and analysis, contributing to data inconsistencies that may affect the reliability of the results. Third, the same life expectancy value (the WHO standard) was used for all individuals regardless of their countries’ life expectancy. The authors acknowledged that this approach was controversial and may have resulted in a different number of DALYs lost. Finally, only English-language publications were used, which may have influenced the findings. Latin America, the former Soviet Union states, and many Asian countries have growing bodies of research published in their native languages.
Despite the limitations, the study is one of the first systematic analyses of GBD, the outcomes of unsafe medical care, and associated lost DALYs. The analysis identified that a majority of the harms from adverse events occur in LMICs. Policies addressing, supporting, and enforcing patient safety measures during the health care experience will help ensure reductions in mortality and morbidity in LMICs. Improving the safety of the healthcare system should be a major policy and research emphasis across the globe.
Applications for Clinical Practice
Even though patient safety initiatives have been at the forefront of many organizational policies and health care provider education since the 1999 Institute of Medicine report “Crossing the Quality Chasm,” this study reminds practitioners that safe clinical practice is essential for reducing domestic disease burden. The cost of adverse events from unsafe practice in the United States was estimated to be around $16.6 billion in 2004 alone [6]. With the World Health Organization calling for strengthened research infrastructure across the globe and LMICs now seeing the value of data for health systems policymaking and management, future research will help to further refine the methods developed in this study.
—Jin Jun, MSN, APRN-BC, CCRN, and Allison Squires, PhD, RN
1. Kaplan J, Hanson D, Dworkin M, et al. Epidemiology of human immunodeficiency virus-associated opportunistic infections in the United States in the era of highly active antiretroviral therapy, Clin Infect Dis 2000;30 Suppl 1:S5–14.
2. Eaton J, et al. Health benefits, costs, and cost-effectiveness of earlier eligibility for adult antiretroviral therapy and expanded treatment coverage: a cmbined analysis of 12 mathematical models, Lancet Global Health 2014;2:e23–34.
3. Mills A, Brugha R, Hanson K, et al. What can be done about the private health sector in low-income countries? Bull World Health Org 2002;80:325–30.
4. Schlein K, De La Cruz A, Gopalakrishnan T, Montagu D. Private sector delivery of health services in developing countries: a mixed-methods study on quality assurance in social franchises, BMC Health Serv Res 2013;13:4.
5. Allegranzi B, Bagheri N, Combescure C, et al. Burden of endemic health-care-associated infection in developing countries: systematic review and meta-analysis, Lancet 2011;377;
228–41.
6. Jha A, Chan D, Ridgway A, et al. Improving safety and eliminating redundant tests: cutting costs in US hospitals. Health Affairs 2009;28:1475–84.
1. Kaplan J, Hanson D, Dworkin M, et al. Epidemiology of human immunodeficiency virus-associated opportunistic infections in the United States in the era of highly active antiretroviral therapy, Clin Infect Dis 2000;30 Suppl 1:S5–14.
2. Eaton J, et al. Health benefits, costs, and cost-effectiveness of earlier eligibility for adult antiretroviral therapy and expanded treatment coverage: a cmbined analysis of 12 mathematical models, Lancet Global Health 2014;2:e23–34.
3. Mills A, Brugha R, Hanson K, et al. What can be done about the private health sector in low-income countries? Bull World Health Org 2002;80:325–30.
4. Schlein K, De La Cruz A, Gopalakrishnan T, Montagu D. Private sector delivery of health services in developing countries: a mixed-methods study on quality assurance in social franchises, BMC Health Serv Res 2013;13:4.
5. Allegranzi B, Bagheri N, Combescure C, et al. Burden of endemic health-care-associated infection in developing countries: systematic review and meta-analysis, Lancet 2011;377;
228–41.
6. Jha A, Chan D, Ridgway A, et al. Improving safety and eliminating redundant tests: cutting costs in US hospitals. Health Affairs 2009;28:1475–84.