User login
FDA approves first AI device to detect colon lesions
The GI Genius (Cosmo Artificial Intelligence) identifies areas of the colon where a colorectal polyp or tumor might be located. Clinicians then follow up with a closer examination and possible treatment.
“With the FDA’s authorization of this device today, clinicians now have a tool that could help improve their ability to detect gastrointestinal lesions they may have missed otherwise,” said Courtney H. Lias, PhD, acting director of the FDA’s gastrorenal, ob.gyn., general hospital, and urology devices office, in a media release.
The GI Genius consists of both hardware and software designed to work with an endoscope. It uses machine learning to recognize possible polyps during a colonoscopy. It marks these areas with green squares on the video generated by the endoscope’s camera and emits a short, low-volume sound. Clinicians decide if a lesion is truly present and whether to sample or remove such a lesion.
The device does not diagnose the lesions or recommend treatments and is not intended to take the place of laboratory sampling
The FDA based its approval on a trial in which 700 people aged 40-80 years underwent colonoscopies for colorectal cancer screening, surveillance, follow-up from positive results of a fecal occult blood test, or gastrointestinal symptoms of possible colon cancer.
Of these participants, 263 were being screened or surveilled every 3 years or more. The researchers randomly divided patients into a group of 136 who underwent white-light standard colonoscopy with the GI Genius, and 127 who underwent white-light standard colonoscopy without the GI Genius.
Using the GI Genius, clinicians identified adenomas or carcinomas that were later confirmed through lab results in 55.1% of patients. Without the GI Genius, the clinicians identified such lesions in 42.0% of patients.
The patients examined with the GI Genius received more biopsies, including slightly more that were not adenomas. But the biopsies did not lead to any adverse events such as perforations, infections, bleeding, or further biopsies.
More information on the GI Genius is available on the FDA website.
A version of this article first appeared on Medscape.com .
The GI Genius (Cosmo Artificial Intelligence) identifies areas of the colon where a colorectal polyp or tumor might be located. Clinicians then follow up with a closer examination and possible treatment.
“With the FDA’s authorization of this device today, clinicians now have a tool that could help improve their ability to detect gastrointestinal lesions they may have missed otherwise,” said Courtney H. Lias, PhD, acting director of the FDA’s gastrorenal, ob.gyn., general hospital, and urology devices office, in a media release.
The GI Genius consists of both hardware and software designed to work with an endoscope. It uses machine learning to recognize possible polyps during a colonoscopy. It marks these areas with green squares on the video generated by the endoscope’s camera and emits a short, low-volume sound. Clinicians decide if a lesion is truly present and whether to sample or remove such a lesion.
The device does not diagnose the lesions or recommend treatments and is not intended to take the place of laboratory sampling
The FDA based its approval on a trial in which 700 people aged 40-80 years underwent colonoscopies for colorectal cancer screening, surveillance, follow-up from positive results of a fecal occult blood test, or gastrointestinal symptoms of possible colon cancer.
Of these participants, 263 were being screened or surveilled every 3 years or more. The researchers randomly divided patients into a group of 136 who underwent white-light standard colonoscopy with the GI Genius, and 127 who underwent white-light standard colonoscopy without the GI Genius.
Using the GI Genius, clinicians identified adenomas or carcinomas that were later confirmed through lab results in 55.1% of patients. Without the GI Genius, the clinicians identified such lesions in 42.0% of patients.
The patients examined with the GI Genius received more biopsies, including slightly more that were not adenomas. But the biopsies did not lead to any adverse events such as perforations, infections, bleeding, or further biopsies.
More information on the GI Genius is available on the FDA website.
A version of this article first appeared on Medscape.com .
The GI Genius (Cosmo Artificial Intelligence) identifies areas of the colon where a colorectal polyp or tumor might be located. Clinicians then follow up with a closer examination and possible treatment.
“With the FDA’s authorization of this device today, clinicians now have a tool that could help improve their ability to detect gastrointestinal lesions they may have missed otherwise,” said Courtney H. Lias, PhD, acting director of the FDA’s gastrorenal, ob.gyn., general hospital, and urology devices office, in a media release.
The GI Genius consists of both hardware and software designed to work with an endoscope. It uses machine learning to recognize possible polyps during a colonoscopy. It marks these areas with green squares on the video generated by the endoscope’s camera and emits a short, low-volume sound. Clinicians decide if a lesion is truly present and whether to sample or remove such a lesion.
The device does not diagnose the lesions or recommend treatments and is not intended to take the place of laboratory sampling
The FDA based its approval on a trial in which 700 people aged 40-80 years underwent colonoscopies for colorectal cancer screening, surveillance, follow-up from positive results of a fecal occult blood test, or gastrointestinal symptoms of possible colon cancer.
Of these participants, 263 were being screened or surveilled every 3 years or more. The researchers randomly divided patients into a group of 136 who underwent white-light standard colonoscopy with the GI Genius, and 127 who underwent white-light standard colonoscopy without the GI Genius.
Using the GI Genius, clinicians identified adenomas or carcinomas that were later confirmed through lab results in 55.1% of patients. Without the GI Genius, the clinicians identified such lesions in 42.0% of patients.
The patients examined with the GI Genius received more biopsies, including slightly more that were not adenomas. But the biopsies did not lead to any adverse events such as perforations, infections, bleeding, or further biopsies.
More information on the GI Genius is available on the FDA website.
A version of this article first appeared on Medscape.com .
Multiple Sclerosis Medications in the VHA: Delivering Specialty, High-Cost, Pharmacy Care in a National System (FULL)
Prior to the first approved disease modifying therapy (DMT) in the 1990s, treatment approaches for multiple sclerosis (MS) were not well understood. The discovery that MS was an immune mediated inflammatory disease paved the way for the treatments we know today. In 1993, interferon β‐1b became the first DMT for MS approved by the US Food and Drug Administration (FDA). Approvals for interferon β‐1a as well as glatiramer acetate (GA) soon followed. Today, we consider these the mildest immunosuppressant DMTs; however, their success verified that suppressing the immune system had a positive effect on the MS disease process.
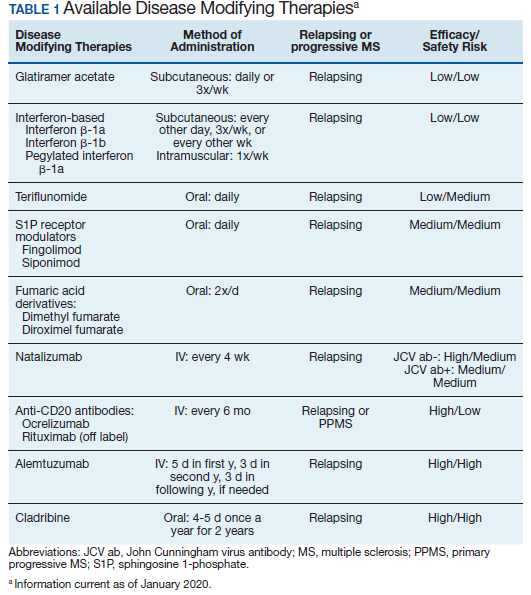
Following these approvals, the disease process in MS is now better understood. Recently approved therapies include monoclonal antibodies, which affect other immune pathways. Today, there are 14 approved DMTs (Table 1). Although the advent of these newer DMTs has revolutionized care for patients with MS, it has been accompanied by increasing costs for the agents. Direct medical costs associated with MS management, coupled with indirect costs from lost productivity, have been estimated to be $24.2 billion annually in the US.1 These increases have been seen across many levels of insurance coverage—private payer, Medicare, and the Veterans Health Administration (VHA).2,3
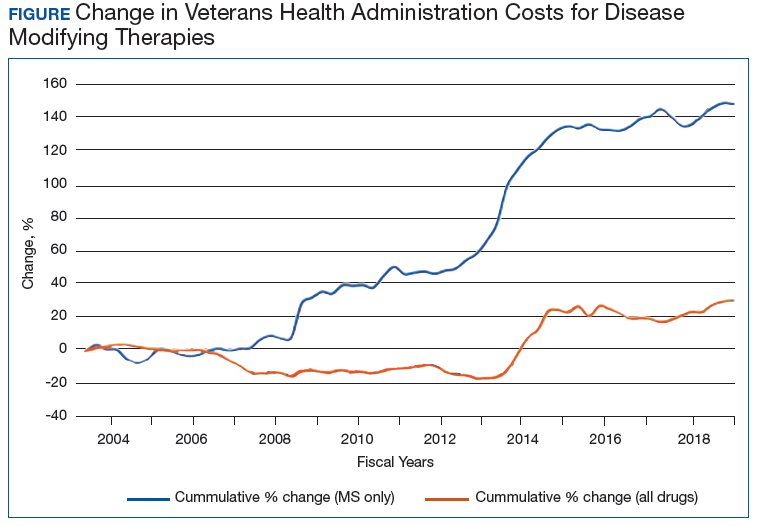
The Figure demonstrates the cost increase that have been seen across VHA between 2004 and 2019 for the DMTs identified in Table 1. Indeed, this compound annual growth rate may be an underestimate because infusion therapies (eg, natalizumab, ocrelizumab, and alemtuzumab) are difficult to track as they may be dispensed directly via a Risk Evaluation Medication Strategy (REMS) program. According to the VHA Pharmacy Benefit Management Service (PBM), in September 2019, dimethyl fumarate (DMF) had the 13th highest total outpatient drug cost for the US Department of Veterans Affairs (VA), interferon β‐1a ranked 62nd and 83rd (prefilled pen and syringe, respectively), and GA 40 mg ranked 89th.
The DMT landscape has demonstrated significant price fluctuations and given rise to a class of medications that requires extensive oversight in terms of efficacy, safety, and cost minimization. The purpose of this article is to show how delivery of this specialty group of medications can be optimized with safety, efficacy, and cost value within a large health care system.
Factors Impacting DMT Use
Recent changes to MS typing have impacted utilization of DMTs. Traditionally, there were 4 subtypes of MS: relapsing remitting (RRMS), secondary progressive (SPMS), progressive relapsing (PRMS), and primary progressive (PPMS). These subtypes are now viewed more broadly and grouped as either relapsing or progressive. The traditional subtypes fall under these broader definitions. Additionally, SPMS has been broken into active SPMS, characterized by continued worsening of disability unrelated to acute relapses, superimposed with activity that can be seen on magnetic resonance images (MRIs), and nonactive SPMS, which has the same disability progression as active SPMS but without MRI-visible activity.4-6 In 2019, these supplementary designations to SPMS made their first appearance in FDA-approved indications. All existing DMTs now include this terminology in their labelling and are indicated in active SPMS. There remain no DMTs that treat nonactive SPMS.
The current landscape of DMTs is highly varied in method of administration, risks, and benefits. As efficacy of these medications often is marked by how well they can prevent the immune system from attacking myelin, an inverse relationship between safety and efficacy results. The standard treatment outcomes in MS have evolved over time. The following are the commonly used primary outcomes in clinical trials: relapse reduction; increased time between relapses; decreased severity of relapses; prevention or extend time to disability milestones as measured by the Expanded Disability Status Scale (EDSS) and other disability measures; prevention or extension of time to onset of secondary progressive disease; prevention or reduction of the number and size of new and enhancing lesions on MRI; and limitation of overall MRI lesion burden in the central nervous system (CNS).
Newer treatment outcomes employed in more recent trials include: measures of axonal damage, CNS atrophy, evidence of microscopic disease via conventional MRI and advanced imaging modalities, biomarkers associated with inflammatory disease activity and neurodegeneration in MS, and the use of no evidence of disease activity (NEDA). These outcomes also must be evaluated by the safety concerns of each agent. Short- and long-term safety are critical factors in the selection of DMTs for MS. The injectable therapies for MS (interferon β‐1a, interferon β‐1b, and GA) have established long-term safety profiles from > 20 years of continuous use. The long-term safety profiles of oral immunomodulatory agents and monoclonal antibodies for these drugs in MS have yet to be determined. Safety concerns associated with some therapies and added requirements for safety monitoring may increase the complexity of a therapeutic selection.
Current cost minimization strategies for DMT include limiting DMT agents on formularies, tier systems that incentivize patients/prescribers to select the lowest priced agents on the formulary, negotiating arrangements with manufacturers to freeze prices or provide discounts in exchange for a priority position in the formulary, and requiring prior authorization to initiate or switch therapy. The use of generic medications and interchange to these agents from a brand name formulation can help reduce expense. Several of these strategies have been implemented in VHA.
Disease-Modifying Therapies
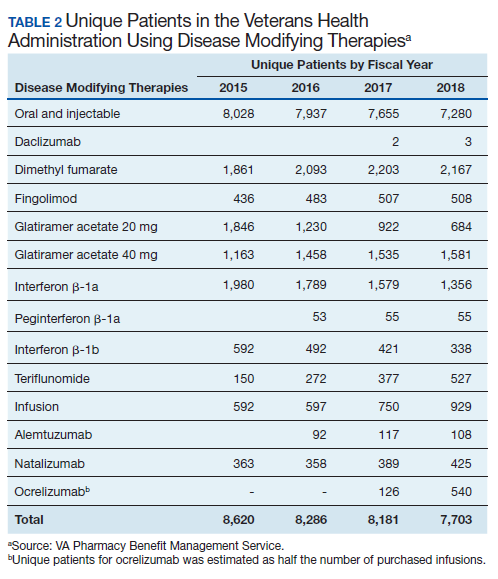
In 2019, 18,645 veterans with MS had either a MS-specific DMT or ≥ 1 annual encounters with a primary diagnosis of MS. Of this population, 4,720 were female and 13,357 were service connected according to VA data. About 50% of veterans with MS take a DMT. This percentage has remained stable over the past decade (Table 2). Although it appears the number of unique veterans prescribed an outpatient DMT is decreasing, this does not include the growing use of infused DMTs or DMTs obtained through the Veterans Choice Program (VCP)/Community Care (CC).
The overall outpatient pharmacy costs for veterans have remained constant despite the reduction in outpatient pharmacy prescription numbers. This may be due to increases in DMT cost to the VHA and the use of more expensive oral agents over the previously used platform injection DMTs.
Generic Conversion
GA is available in 20 mg daily and 40 mg3 times weekly subcutaneous injection dosing. The first evidence of clinical efficacy for a generic formulation for GA was evaluated by the GATE trial.7 This trial was a multicenter, randomized, double-blind, active- and placebo-controlled phase 3 trial. Eligible participants were randomized to receive daily SC injection for 9 months of 20 mg generic GA (n = 5,353), 20 mg brand GA (n = 5,357), or placebo (n = 584). The primary endpoint was the mean number of gadolinium (Gd1) lesions visible on MRIs during months 7, 8, and 9, which were significantly reduced in the combined GA-treated group and in each GA group individually when compared with the placebo group, confirming the study sensitivity (ie, GA was effective under the conditions of the study). Tolerability (including injection site reactions) and safety (incidence, spectrum, and severity of adverse events [AEs]) were similar in the generic and brand GA groups. These results demonstrated that generic and brand GA had equivalent efficacy, tolerability, and safety over a 9-month period.7
Results of a 15-month extension of the study were presented in 2015 and showed similar efficacy, safety, and tolerability in participants treated with generic GA for 2 years and patients switched from brand to generic GA.8 Multiple shifts for GA occurred, most notably the conversion from branded Copaxone (Teva Pharmaceutical Industries) to generic Glatopa (Sandoz). Subsequently, Sandoz released a generic 40 mg 3 times weekly formulation. Additionally, Mylan entered the generic GA market. With 3 competing manufacturers, internal data from the VHA indicated that it was able to negotiate a single source contract for this medication that provided a savings of $32,088,904.69 between September 2016 and May 2019.
The impact of generic conversions is just being realized. Soon, patents will begin to expire for oral DMTs, leading to an expected growth of generic alternatives. Already the FDA has approved 4 generic alternatives for teriflunomide, 3 for fingolimod (with 13 tentative approvals), and 15 generic alternatives for dimethyl fumarate (DMF). Implementation of therapeutic interchanges will be pursued by VHA as clinically supported by evidence.
Criteria for Use
PBM supports utilizing criteria to help guide providers on DMT options and promote safe, effective, and value-based selection of a DMT. The PBM creates monographs and criteria for use (CFU) for new medications. The monograph contains a literature evaluation of all studies available to date that concern both safety and efficacy of the new medication. Therapeutic alternatives also are presented and assessed for key elements that may determine the most safe and effective use. Additional safety areas for the new medications such as look-alike, sound-alike potential, special populations use (ie, those who are pregnant, the elderly, and those with liver or renal dysfunction), and drug-drug interactions are presented. Lastly, and possibly most importantly in an ever-growing growing world of DMTs, the monograph describes a reasonable place in therapy for the new DMT.
CFU are additional guidance for some DMTs. The development of CFU are based on several questions that arise during the monograph development for a DMT. These include, but are not limited to:
- Are there safety concerns that require the drug to receive a review to ensure safe prescribing (eg, agents with REMS programs, or safety concerns in specific populations)?
- Does the drug require a specialty provider type with knowledge and experience in those disease states to ensure appropriate and safe prescribing (eg restricted to infectious diseases)?
- Do VHA or non-VHA guidelines suggest alternative therapy be used prior to the agent?
- Is a review deemed necessary to ensure the preferred agent is used first (eg, second-line therapy)?
The CFU defines parameters of drug use consistent with high quality and evidence-based patient care. CFUs also serve as a basis for monitoring local, regional, and national patterns of pharmacologic care and help guide health care providers (HCPs) on appropriate use of medication.
CFUs are designed to ensure the HCP is safely starting a medication that has evidence for efficacy for their patient. For example, alemtuzumab is a high-risk, high-efficacy DMT. The alemtuzumab CFU acknowledges this by having exclusion criteria that prevent a veteran at high risk (ie, on another immunosuppressant) from being exposed to severe AEs (ie, severe leukopenia) that are associated with the medication. On the other hand, the inclusion criteria recognize the benefits of alemtuzumab and allows those with highly active MS who have failed other DMTs to receive the medication.
The drug monograph and CFU process is an important part of VHA efforts to optimize patient care. After a draft version is developed, HCPs can provide feedback on the exclusion/inclusion criteria and describe how they anticipate using the medication in their practice. This insight can be beneficial for MS treatment as diverse HCPs may have distinct viewpoints on how DMTs should be started. Pharmacists and physicians on a national level then discuss and decide together what to include in the final drafts of the drug monograph and CFU. Final documents are disseminated to all sites, which encourages consistent practices across the VHA.9 These documents are reviewed on a regular basis and updated as needed based on available literature evidence.
It is well accepted that early use of DMT correlates with lower accumulated long-term disability.10 However, discontinuation of DMT should be treated with equal importance. This benefits the patient by reducing their risk of AEs from DMTs and provides cost savings. Age and disease stability are factors to consider for DMT discontinuation. In a study with patients aged > 45 years and another with patients aged > 60 years, discontinuing DMT rarely had a negative impact and improved quality of life.11,12 A retrospective meta-analysis of age-dependent efficacy of current DMTs predicted that DMT loses efficacy at age 53 years. In addition, higher efficacy DMT only outperforms lower efficacy DMT in patients aged < 40.5 years.13 Stability of disease and lack of relapses for ≥ 2 years also may be a positive predictor to safely discontinue DMT.14,15 The growing literature to support safe discontinuation of DMT makes this a more convincing strategy to avoid unnecessary costs associated with current DMTs. With an average age of 59 years for veterans with MS, this may be one of the largest areas of cost avoidance to consider.
Off-Label Use
Other potential ways to reduce DMT costs is to consider off-label treatments. The OLYMPUS trial studied off-label use of rituximab, an anti-CD20 antibody like ocrelizumab. It did not meet statistical significance for its primary endpoint; however, in a subgroup analysis, off-label use was found to be more effective in a population aged < 51 years.16 Other case reports and smaller scale studies also describe rituximab’s efficacy in MS.17,18 In 2018, the FDA approved the first rituximab biosimilar.19 Further competition from biosimilars likely will make rituximab an even more cost-effective choice when compared with ocrelizumab.
Alternate Dosing Regimens
Extended interval dosing of natalizumab has been studied, extending the standard infusion interval from every 4 weeks to 5- to 8-week intervals. One recent article compared these interval extensions and found that all extended intervals of up to 56 days did not increase new or enhancing lesions on MRI when compared with standard interval dosing.20 Another larger randomized trial is underway to evaluate efficacy and safety of extended interval dosing of natalizumab (NCT03689972). Utilization of this dosing may reduce natalizumab annual costs by up to 50%.
Safety Monitoring
DMF is an oral DMT on the VHA formulary with CFU. Since leukopenia is a known AE, baseline and quarterly monitoring of the complete blood count (CBC) is recommended for patients taking DMF. Additionally, DMF should be held if white blood cell count (WBC) falls below 2,000/mm3.21 There have been recent reports of death secondary to progressive multifocal leukoencephalopathy (PML) among European patients taking DMF.22-24 This has raised concerns about adherence to recommended CBC monitoring in veterans taking DMF. The association of DMF and leukopenia has been evident since early clinical trials.25 Leukopenia in immunocompromised patients increases the risk of PML.
In the long-term extension study ENDORSE, 6% to 7% of patients continuing DMF had WBC counts of 3.0×109/L compared with 7% to 10% in the new to DMF group.26 In addition 6% to 8% of patients continuing DMF had lymphocyte counts of 0.5×109/L, compared with 5% to 9% in the new to DMF group. The cases of PML occurred in patients who had low lymphocyte counts over an extended period with no adjustment to DMF therapy, such as holding the drug until WBC counts returned to normal levels or stopping the drug. Discussion and review within VHA resulted in the recommendation for quarterly WBC monitoring criteria.
PBM and VA Center for Medication Safety (MedSafe) conducted a medication usage evaluation (MUE) on adherence to the WBC monitoring set forth in the CFU. Data collection began in fourth quarter of fiscal year (FY) 2015 with the most recent reporting period of fourth quarter of FY 2017. The Medication Utilization Evaluation Tool tracks patients with no reported WBC in 90 days and WBC < 2,000/mm3. Over the reporting period, 20% to 23% of patients have not received appropriate quarterly monitoring. Additionally, there have been 4 cases where the WBC decreased below the threshold limit. To ensure safe and effective use of DMF, it is important to adhere to the monitoring requirements set forth in the CFU.
Impact of REMS and Special Distribution
As DMTs increase in efficacy, there are often more risks associated with them. Some of these high-risk medications, including natalizumab and alemtuzumab, have REMS programs and/or have special distribution procedures. Although REMS are imperative for patient safety, the complexity of these programs can be difficult to navigate, which can create a barrier to access. The PBM helps to assist all sites with navigating and adhering to required actions to dispense and administer these medications through a national Special Handling Drugs Microsoft SharePoint site, which provides access to REMS forms and procurement information when drugs are dispensed from specialty pharmacies. Easing this process nationwide empowers more sites to be confident they can dispense specialty medications appropriately.
Clinical Pharmacists
The VHA is unique in its utilization of pharmacists in outpatient clinic settings. Utilization of an interdisciplinary team for medication management has been highly used in VHA for areas like primary care; however, pharmacist involvement in specialty areas is on the rise and MS is no exception. Pharmacists stationed in clinics, such as neurology or spinal cord injury, can impact care for veterans with MS. Interdisciplinary teams that include a pharmacist have been shown to increase patient adherence to DMTs.27 However, pharmacists often assist with medication education and monitoring, which adds an additional layer of safety to DMT treatment. At the VHA, pharmacists also can obtain a scope of practice that allows them to prescribe medications and increase access to care for veterans with MS.
Education
The VHA demonstrates how education on a disease state like MS can be distributed on a large, national scale through drug monographs, CFU, and Microsoft SharePoint sites. In addition, VHA has created the MS Centers of Excellence (MSCoE) that serve as a hub of specialized health care providers in all aspects of MS care.
A core function of the MSCoE is to provide education to both HCPs and patients. The MSCoE and its regional hubs support sites that may not have an HCP who specializes in MS by providing advice on DMT selection, how to obtain specialty medications, and monitoring that needs to be completed to ensure veterans’ safety. The MSCoE also has partnered with the National MS Society to hold a lecture series on topics in MS. This free series is available online to all HCPs who interact with patients who have MS and is a way that VA is extending its best practices and expertise beyond its own health care system. There also is a quarterly newsletter for veterans with MS that highlights new information on DMTs that can affect their care.
Conclusion
It is an exciting and challenging period in MS treatment. New DMTs are being approved and entering clinical trials at a rapid pace. These new DMT agents may offer increased efficacy, improvements in AE profiles, and the possibility of increased medication adherence—but often at a higher cost. The utilization of CFU and formulary management provides the ability to ensure the safe and appropriate use of medications by veterans, with a secondary outcome of controlling pharmacy expenditures.
The VHA had expenditures of $142,135,938 for DMT use in FY 2018. As the VHA sees the new contract prices for DMT in January 2020, we are reminded that costs will continue to rise with some pharmaceutical manufacturers implementing prices 8% to 11% higher than 2019 prices, when the consumer price index defines an increase of 1.0% for 2020 and 1.4% in 2021.28 It is imperative that the VHA formulary be managed judiciously and the necessary measures be in place for VHA practitioners to enable effective, safe and value-based care to the veteran population.
1. Gooch CL, Pracht E, Borenstein AR. The burden of neurological disease in the United States: a summary report and call to action. Ann Neurol. 2017;81(4):479-484.
2. Hartung DM, Bourdette DN, Ahmed SM, Whitham RH. The cost of multiple sclerosis drugs in the US and the pharmaceutical industry: too big to fail? [published correction appears in Neurology. 2015;85(19):1728]. Neurology. 2015;84(21):2185–2192.
3. San-Juan-Rodriguez A, Good CB, Heyman RA, Parekh N, Shrank WH, Hernandez I. Trends in prices, market share, and spending on self-administered disease-modifying therapies for multiple sclerosis in Medicare Part D. JAMA Neurol. 2019;76(11):1386-1390.
4. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278-286.
5. Eriksson M, Andersen O, Runmarker B. Long-term follow up of patients with clinically isolated syndromes, relapsing-remitting and secondary progressive multiple sclerosis [published correction appears in Mult Scler. 2003;9(6):641]. Mult Scler. 2003;9(3):260-274.
6. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173.
7. Cohen J, Belova A, Selmaj K, et al. Equivalence of generic glatiramer acetate in multiple sclerosis: a randomized clinical trial. JAMA Neurol. 2015;72(12):1433-1441.
8. Selmaj K, Barkhof F, Belova AN, et al; GATE study group. Switching from branded to generic glatiramer acetate: 15-month GATE trial extension results. Mult Scler. 2017;23(14):1909-1917.
9. Aspinall SL, Sales MM, Good CB, et al. Pharmacy benefits management in the Veterans Health Administration revisited: a decade of advancements, 2004-2014. J Manag Care Spec Pharm. 2016;22(9):1058-1063.
10. Brown JWL, Coles A, Horakova D, et al. Association of initial disease-modifying therapy with later conversion to secondary progressive multiple sclerosis. JAMA. 2019;321(2):175-187.
11. Hua LH, Harris H, Conway D, Thompson NR. Changes in patient-reported outcomes between continuers and discontinuers of disease modifying therapy in patients with multiple sclerosis over age 60 [published correction appears in Mult Scler Relat Disord. 2019;30:293]. Mult Scler Relat Disord. 2019;30:252-256.
12. Bsteh G, Feige J, Ehling R, et al. Discontinuation of disease-modifying therapies in multiple sclerosis - Clinical outcome and prognostic factors. Mult Scler. 2017;23(9):1241-1248.
13. Weideman AM, Tapia-Maltos MA, Johnson K, Greenwood M, Bielekova B. Meta-analysis of the age-dependent efficacy of multiple sclerosis treatments. Front Neurol. 2017;8:577.
14. Kister I, Spelman T, Alroughani R, et al; MSBase Study Group. Discontinuing disease-modifying therapy in MS after a prolonged relapse-free period: a propensity score-matched study [published correction appears in J Neurol Neurosurg Psychiatry. 2019;90(4):e2]. J Neurol Neurosurg Psychiatry. 2016;87(10):1133-1137.
15. Birnbaum G. Stopping disease-modifying therapy in nonrelapsing multiple sclerosis: experience from a clinical practice. Int J MS Care. 2017;19(1):11-14.
16. Hawker K, O’Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66(4):460-471.
17. Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–688.
18. Alping P, Frisell T, Novakova L, et al. Rituximab versus fingolimod after natalizumab in multiple sclerosis patients. Ann Neurol. 2016;79(6):950–958.
19. Rituximab-abbs [package insert]. North Wales, PA: Teva Pharmaceuticals; 2018.
20. Zhovtis Ryerson L, Frohman TC, Foley J, et al. Extended interval dosing of natalizumab in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2016;87(8):885-889.
21. Dimethyl fumarate [package insert]. Cambridge, MA: Biogen Inc; 2015.
22. van Kester MS, Bouwes Bavinck JN, Quint KD. PML in Patients treated with dimethyl fumarate. N Engl J Med. 2015;373(6):583-584.
23. Nieuwkamp DJ, Murk JL, van Oosten BW. PML in patients treated with dimethyl fumarate. N Engl J Med. 2015;373(6):584.
24. Rosenkranz T, Novas M, Terborg C. PML in a patient with lymphocytopenia treated with dimethyl fumarate. N Engl J Med. 2015;372(15):1476-1478.
25. Longbrake EE, Cross AH. Dimethyl fumarate associated lymphopenia in clinical practice. Mult Scler. 2015;21(6):796-797.
26. Gold R, Arnold DL, Bar-Or A, et al. Long-term effects of delayed-release dimethyl fumarate in multiple sclerosis: Interim analysis of ENDORSE, a randomized extension study. Mult Scler. 2017;23(2):253–265.
27. Hanson RL, Habibi M, Khamo N, Abdou S, Stubbings J. Integrated clinical and specialty pharmacy practice model for management of patients with multiple sclerosis. Am J Health Syst Pharm. 2014;71(6):463-469.
28. Federal Planning Bureau. Consumer Price Index - Inflation forecasts. https://www.plan.be/databases/17-en-consumer+price+index+inflation+forecasts. Updated March 3, 2020. Accessed March 9, 2020.
Prior to the first approved disease modifying therapy (DMT) in the 1990s, treatment approaches for multiple sclerosis (MS) were not well understood. The discovery that MS was an immune mediated inflammatory disease paved the way for the treatments we know today. In 1993, interferon β‐1b became the first DMT for MS approved by the US Food and Drug Administration (FDA). Approvals for interferon β‐1a as well as glatiramer acetate (GA) soon followed. Today, we consider these the mildest immunosuppressant DMTs; however, their success verified that suppressing the immune system had a positive effect on the MS disease process.
Following these approvals, the disease process in MS is now better understood. Recently approved therapies include monoclonal antibodies, which affect other immune pathways. Today, there are 14 approved DMTs (Table 1). Although the advent of these newer DMTs has revolutionized care for patients with MS, it has been accompanied by increasing costs for the agents. Direct medical costs associated with MS management, coupled with indirect costs from lost productivity, have been estimated to be $24.2 billion annually in the US.1 These increases have been seen across many levels of insurance coverage—private payer, Medicare, and the Veterans Health Administration (VHA).2,3
The Figure demonstrates the cost increase that have been seen across VHA between 2004 and 2019 for the DMTs identified in Table 1. Indeed, this compound annual growth rate may be an underestimate because infusion therapies (eg, natalizumab, ocrelizumab, and alemtuzumab) are difficult to track as they may be dispensed directly via a Risk Evaluation Medication Strategy (REMS) program. According to the VHA Pharmacy Benefit Management Service (PBM), in September 2019, dimethyl fumarate (DMF) had the 13th highest total outpatient drug cost for the US Department of Veterans Affairs (VA), interferon β‐1a ranked 62nd and 83rd (prefilled pen and syringe, respectively), and GA 40 mg ranked 89th.
The DMT landscape has demonstrated significant price fluctuations and given rise to a class of medications that requires extensive oversight in terms of efficacy, safety, and cost minimization. The purpose of this article is to show how delivery of this specialty group of medications can be optimized with safety, efficacy, and cost value within a large health care system.
Factors Impacting DMT Use
Recent changes to MS typing have impacted utilization of DMTs. Traditionally, there were 4 subtypes of MS: relapsing remitting (RRMS), secondary progressive (SPMS), progressive relapsing (PRMS), and primary progressive (PPMS). These subtypes are now viewed more broadly and grouped as either relapsing or progressive. The traditional subtypes fall under these broader definitions. Additionally, SPMS has been broken into active SPMS, characterized by continued worsening of disability unrelated to acute relapses, superimposed with activity that can be seen on magnetic resonance images (MRIs), and nonactive SPMS, which has the same disability progression as active SPMS but without MRI-visible activity.4-6 In 2019, these supplementary designations to SPMS made their first appearance in FDA-approved indications. All existing DMTs now include this terminology in their labelling and are indicated in active SPMS. There remain no DMTs that treat nonactive SPMS.
The current landscape of DMTs is highly varied in method of administration, risks, and benefits. As efficacy of these medications often is marked by how well they can prevent the immune system from attacking myelin, an inverse relationship between safety and efficacy results. The standard treatment outcomes in MS have evolved over time. The following are the commonly used primary outcomes in clinical trials: relapse reduction; increased time between relapses; decreased severity of relapses; prevention or extend time to disability milestones as measured by the Expanded Disability Status Scale (EDSS) and other disability measures; prevention or extension of time to onset of secondary progressive disease; prevention or reduction of the number and size of new and enhancing lesions on MRI; and limitation of overall MRI lesion burden in the central nervous system (CNS).
Newer treatment outcomes employed in more recent trials include: measures of axonal damage, CNS atrophy, evidence of microscopic disease via conventional MRI and advanced imaging modalities, biomarkers associated with inflammatory disease activity and neurodegeneration in MS, and the use of no evidence of disease activity (NEDA). These outcomes also must be evaluated by the safety concerns of each agent. Short- and long-term safety are critical factors in the selection of DMTs for MS. The injectable therapies for MS (interferon β‐1a, interferon β‐1b, and GA) have established long-term safety profiles from > 20 years of continuous use. The long-term safety profiles of oral immunomodulatory agents and monoclonal antibodies for these drugs in MS have yet to be determined. Safety concerns associated with some therapies and added requirements for safety monitoring may increase the complexity of a therapeutic selection.
Current cost minimization strategies for DMT include limiting DMT agents on formularies, tier systems that incentivize patients/prescribers to select the lowest priced agents on the formulary, negotiating arrangements with manufacturers to freeze prices or provide discounts in exchange for a priority position in the formulary, and requiring prior authorization to initiate or switch therapy. The use of generic medications and interchange to these agents from a brand name formulation can help reduce expense. Several of these strategies have been implemented in VHA.
Disease-Modifying Therapies
In 2019, 18,645 veterans with MS had either a MS-specific DMT or ≥ 1 annual encounters with a primary diagnosis of MS. Of this population, 4,720 were female and 13,357 were service connected according to VA data. About 50% of veterans with MS take a DMT. This percentage has remained stable over the past decade (Table 2). Although it appears the number of unique veterans prescribed an outpatient DMT is decreasing, this does not include the growing use of infused DMTs or DMTs obtained through the Veterans Choice Program (VCP)/Community Care (CC).
The overall outpatient pharmacy costs for veterans have remained constant despite the reduction in outpatient pharmacy prescription numbers. This may be due to increases in DMT cost to the VHA and the use of more expensive oral agents over the previously used platform injection DMTs.
Generic Conversion
GA is available in 20 mg daily and 40 mg3 times weekly subcutaneous injection dosing. The first evidence of clinical efficacy for a generic formulation for GA was evaluated by the GATE trial.7 This trial was a multicenter, randomized, double-blind, active- and placebo-controlled phase 3 trial. Eligible participants were randomized to receive daily SC injection for 9 months of 20 mg generic GA (n = 5,353), 20 mg brand GA (n = 5,357), or placebo (n = 584). The primary endpoint was the mean number of gadolinium (Gd1) lesions visible on MRIs during months 7, 8, and 9, which were significantly reduced in the combined GA-treated group and in each GA group individually when compared with the placebo group, confirming the study sensitivity (ie, GA was effective under the conditions of the study). Tolerability (including injection site reactions) and safety (incidence, spectrum, and severity of adverse events [AEs]) were similar in the generic and brand GA groups. These results demonstrated that generic and brand GA had equivalent efficacy, tolerability, and safety over a 9-month period.7
Results of a 15-month extension of the study were presented in 2015 and showed similar efficacy, safety, and tolerability in participants treated with generic GA for 2 years and patients switched from brand to generic GA.8 Multiple shifts for GA occurred, most notably the conversion from branded Copaxone (Teva Pharmaceutical Industries) to generic Glatopa (Sandoz). Subsequently, Sandoz released a generic 40 mg 3 times weekly formulation. Additionally, Mylan entered the generic GA market. With 3 competing manufacturers, internal data from the VHA indicated that it was able to negotiate a single source contract for this medication that provided a savings of $32,088,904.69 between September 2016 and May 2019.
The impact of generic conversions is just being realized. Soon, patents will begin to expire for oral DMTs, leading to an expected growth of generic alternatives. Already the FDA has approved 4 generic alternatives for teriflunomide, 3 for fingolimod (with 13 tentative approvals), and 15 generic alternatives for dimethyl fumarate (DMF). Implementation of therapeutic interchanges will be pursued by VHA as clinically supported by evidence.
Criteria for Use
PBM supports utilizing criteria to help guide providers on DMT options and promote safe, effective, and value-based selection of a DMT. The PBM creates monographs and criteria for use (CFU) for new medications. The monograph contains a literature evaluation of all studies available to date that concern both safety and efficacy of the new medication. Therapeutic alternatives also are presented and assessed for key elements that may determine the most safe and effective use. Additional safety areas for the new medications such as look-alike, sound-alike potential, special populations use (ie, those who are pregnant, the elderly, and those with liver or renal dysfunction), and drug-drug interactions are presented. Lastly, and possibly most importantly in an ever-growing growing world of DMTs, the monograph describes a reasonable place in therapy for the new DMT.
CFU are additional guidance for some DMTs. The development of CFU are based on several questions that arise during the monograph development for a DMT. These include, but are not limited to:
- Are there safety concerns that require the drug to receive a review to ensure safe prescribing (eg, agents with REMS programs, or safety concerns in specific populations)?
- Does the drug require a specialty provider type with knowledge and experience in those disease states to ensure appropriate and safe prescribing (eg restricted to infectious diseases)?
- Do VHA or non-VHA guidelines suggest alternative therapy be used prior to the agent?
- Is a review deemed necessary to ensure the preferred agent is used first (eg, second-line therapy)?
The CFU defines parameters of drug use consistent with high quality and evidence-based patient care. CFUs also serve as a basis for monitoring local, regional, and national patterns of pharmacologic care and help guide health care providers (HCPs) on appropriate use of medication.
CFUs are designed to ensure the HCP is safely starting a medication that has evidence for efficacy for their patient. For example, alemtuzumab is a high-risk, high-efficacy DMT. The alemtuzumab CFU acknowledges this by having exclusion criteria that prevent a veteran at high risk (ie, on another immunosuppressant) from being exposed to severe AEs (ie, severe leukopenia) that are associated with the medication. On the other hand, the inclusion criteria recognize the benefits of alemtuzumab and allows those with highly active MS who have failed other DMTs to receive the medication.
The drug monograph and CFU process is an important part of VHA efforts to optimize patient care. After a draft version is developed, HCPs can provide feedback on the exclusion/inclusion criteria and describe how they anticipate using the medication in their practice. This insight can be beneficial for MS treatment as diverse HCPs may have distinct viewpoints on how DMTs should be started. Pharmacists and physicians on a national level then discuss and decide together what to include in the final drafts of the drug monograph and CFU. Final documents are disseminated to all sites, which encourages consistent practices across the VHA.9 These documents are reviewed on a regular basis and updated as needed based on available literature evidence.
It is well accepted that early use of DMT correlates with lower accumulated long-term disability.10 However, discontinuation of DMT should be treated with equal importance. This benefits the patient by reducing their risk of AEs from DMTs and provides cost savings. Age and disease stability are factors to consider for DMT discontinuation. In a study with patients aged > 45 years and another with patients aged > 60 years, discontinuing DMT rarely had a negative impact and improved quality of life.11,12 A retrospective meta-analysis of age-dependent efficacy of current DMTs predicted that DMT loses efficacy at age 53 years. In addition, higher efficacy DMT only outperforms lower efficacy DMT in patients aged < 40.5 years.13 Stability of disease and lack of relapses for ≥ 2 years also may be a positive predictor to safely discontinue DMT.14,15 The growing literature to support safe discontinuation of DMT makes this a more convincing strategy to avoid unnecessary costs associated with current DMTs. With an average age of 59 years for veterans with MS, this may be one of the largest areas of cost avoidance to consider.
Off-Label Use
Other potential ways to reduce DMT costs is to consider off-label treatments. The OLYMPUS trial studied off-label use of rituximab, an anti-CD20 antibody like ocrelizumab. It did not meet statistical significance for its primary endpoint; however, in a subgroup analysis, off-label use was found to be more effective in a population aged < 51 years.16 Other case reports and smaller scale studies also describe rituximab’s efficacy in MS.17,18 In 2018, the FDA approved the first rituximab biosimilar.19 Further competition from biosimilars likely will make rituximab an even more cost-effective choice when compared with ocrelizumab.
Alternate Dosing Regimens
Extended interval dosing of natalizumab has been studied, extending the standard infusion interval from every 4 weeks to 5- to 8-week intervals. One recent article compared these interval extensions and found that all extended intervals of up to 56 days did not increase new or enhancing lesions on MRI when compared with standard interval dosing.20 Another larger randomized trial is underway to evaluate efficacy and safety of extended interval dosing of natalizumab (NCT03689972). Utilization of this dosing may reduce natalizumab annual costs by up to 50%.
Safety Monitoring
DMF is an oral DMT on the VHA formulary with CFU. Since leukopenia is a known AE, baseline and quarterly monitoring of the complete blood count (CBC) is recommended for patients taking DMF. Additionally, DMF should be held if white blood cell count (WBC) falls below 2,000/mm3.21 There have been recent reports of death secondary to progressive multifocal leukoencephalopathy (PML) among European patients taking DMF.22-24 This has raised concerns about adherence to recommended CBC monitoring in veterans taking DMF. The association of DMF and leukopenia has been evident since early clinical trials.25 Leukopenia in immunocompromised patients increases the risk of PML.
In the long-term extension study ENDORSE, 6% to 7% of patients continuing DMF had WBC counts of 3.0×109/L compared with 7% to 10% in the new to DMF group.26 In addition 6% to 8% of patients continuing DMF had lymphocyte counts of 0.5×109/L, compared with 5% to 9% in the new to DMF group. The cases of PML occurred in patients who had low lymphocyte counts over an extended period with no adjustment to DMF therapy, such as holding the drug until WBC counts returned to normal levels or stopping the drug. Discussion and review within VHA resulted in the recommendation for quarterly WBC monitoring criteria.
PBM and VA Center for Medication Safety (MedSafe) conducted a medication usage evaluation (MUE) on adherence to the WBC monitoring set forth in the CFU. Data collection began in fourth quarter of fiscal year (FY) 2015 with the most recent reporting period of fourth quarter of FY 2017. The Medication Utilization Evaluation Tool tracks patients with no reported WBC in 90 days and WBC < 2,000/mm3. Over the reporting period, 20% to 23% of patients have not received appropriate quarterly monitoring. Additionally, there have been 4 cases where the WBC decreased below the threshold limit. To ensure safe and effective use of DMF, it is important to adhere to the monitoring requirements set forth in the CFU.
Impact of REMS and Special Distribution
As DMTs increase in efficacy, there are often more risks associated with them. Some of these high-risk medications, including natalizumab and alemtuzumab, have REMS programs and/or have special distribution procedures. Although REMS are imperative for patient safety, the complexity of these programs can be difficult to navigate, which can create a barrier to access. The PBM helps to assist all sites with navigating and adhering to required actions to dispense and administer these medications through a national Special Handling Drugs Microsoft SharePoint site, which provides access to REMS forms and procurement information when drugs are dispensed from specialty pharmacies. Easing this process nationwide empowers more sites to be confident they can dispense specialty medications appropriately.
Clinical Pharmacists
The VHA is unique in its utilization of pharmacists in outpatient clinic settings. Utilization of an interdisciplinary team for medication management has been highly used in VHA for areas like primary care; however, pharmacist involvement in specialty areas is on the rise and MS is no exception. Pharmacists stationed in clinics, such as neurology or spinal cord injury, can impact care for veterans with MS. Interdisciplinary teams that include a pharmacist have been shown to increase patient adherence to DMTs.27 However, pharmacists often assist with medication education and monitoring, which adds an additional layer of safety to DMT treatment. At the VHA, pharmacists also can obtain a scope of practice that allows them to prescribe medications and increase access to care for veterans with MS.
Education
The VHA demonstrates how education on a disease state like MS can be distributed on a large, national scale through drug monographs, CFU, and Microsoft SharePoint sites. In addition, VHA has created the MS Centers of Excellence (MSCoE) that serve as a hub of specialized health care providers in all aspects of MS care.
A core function of the MSCoE is to provide education to both HCPs and patients. The MSCoE and its regional hubs support sites that may not have an HCP who specializes in MS by providing advice on DMT selection, how to obtain specialty medications, and monitoring that needs to be completed to ensure veterans’ safety. The MSCoE also has partnered with the National MS Society to hold a lecture series on topics in MS. This free series is available online to all HCPs who interact with patients who have MS and is a way that VA is extending its best practices and expertise beyond its own health care system. There also is a quarterly newsletter for veterans with MS that highlights new information on DMTs that can affect their care.
Conclusion
It is an exciting and challenging period in MS treatment. New DMTs are being approved and entering clinical trials at a rapid pace. These new DMT agents may offer increased efficacy, improvements in AE profiles, and the possibility of increased medication adherence—but often at a higher cost. The utilization of CFU and formulary management provides the ability to ensure the safe and appropriate use of medications by veterans, with a secondary outcome of controlling pharmacy expenditures.
The VHA had expenditures of $142,135,938 for DMT use in FY 2018. As the VHA sees the new contract prices for DMT in January 2020, we are reminded that costs will continue to rise with some pharmaceutical manufacturers implementing prices 8% to 11% higher than 2019 prices, when the consumer price index defines an increase of 1.0% for 2020 and 1.4% in 2021.28 It is imperative that the VHA formulary be managed judiciously and the necessary measures be in place for VHA practitioners to enable effective, safe and value-based care to the veteran population.
Prior to the first approved disease modifying therapy (DMT) in the 1990s, treatment approaches for multiple sclerosis (MS) were not well understood. The discovery that MS was an immune mediated inflammatory disease paved the way for the treatments we know today. In 1993, interferon β‐1b became the first DMT for MS approved by the US Food and Drug Administration (FDA). Approvals for interferon β‐1a as well as glatiramer acetate (GA) soon followed. Today, we consider these the mildest immunosuppressant DMTs; however, their success verified that suppressing the immune system had a positive effect on the MS disease process.
Following these approvals, the disease process in MS is now better understood. Recently approved therapies include monoclonal antibodies, which affect other immune pathways. Today, there are 14 approved DMTs (Table 1). Although the advent of these newer DMTs has revolutionized care for patients with MS, it has been accompanied by increasing costs for the agents. Direct medical costs associated with MS management, coupled with indirect costs from lost productivity, have been estimated to be $24.2 billion annually in the US.1 These increases have been seen across many levels of insurance coverage—private payer, Medicare, and the Veterans Health Administration (VHA).2,3
The Figure demonstrates the cost increase that have been seen across VHA between 2004 and 2019 for the DMTs identified in Table 1. Indeed, this compound annual growth rate may be an underestimate because infusion therapies (eg, natalizumab, ocrelizumab, and alemtuzumab) are difficult to track as they may be dispensed directly via a Risk Evaluation Medication Strategy (REMS) program. According to the VHA Pharmacy Benefit Management Service (PBM), in September 2019, dimethyl fumarate (DMF) had the 13th highest total outpatient drug cost for the US Department of Veterans Affairs (VA), interferon β‐1a ranked 62nd and 83rd (prefilled pen and syringe, respectively), and GA 40 mg ranked 89th.
The DMT landscape has demonstrated significant price fluctuations and given rise to a class of medications that requires extensive oversight in terms of efficacy, safety, and cost minimization. The purpose of this article is to show how delivery of this specialty group of medications can be optimized with safety, efficacy, and cost value within a large health care system.
Factors Impacting DMT Use
Recent changes to MS typing have impacted utilization of DMTs. Traditionally, there were 4 subtypes of MS: relapsing remitting (RRMS), secondary progressive (SPMS), progressive relapsing (PRMS), and primary progressive (PPMS). These subtypes are now viewed more broadly and grouped as either relapsing or progressive. The traditional subtypes fall under these broader definitions. Additionally, SPMS has been broken into active SPMS, characterized by continued worsening of disability unrelated to acute relapses, superimposed with activity that can be seen on magnetic resonance images (MRIs), and nonactive SPMS, which has the same disability progression as active SPMS but without MRI-visible activity.4-6 In 2019, these supplementary designations to SPMS made their first appearance in FDA-approved indications. All existing DMTs now include this terminology in their labelling and are indicated in active SPMS. There remain no DMTs that treat nonactive SPMS.
The current landscape of DMTs is highly varied in method of administration, risks, and benefits. As efficacy of these medications often is marked by how well they can prevent the immune system from attacking myelin, an inverse relationship between safety and efficacy results. The standard treatment outcomes in MS have evolved over time. The following are the commonly used primary outcomes in clinical trials: relapse reduction; increased time between relapses; decreased severity of relapses; prevention or extend time to disability milestones as measured by the Expanded Disability Status Scale (EDSS) and other disability measures; prevention or extension of time to onset of secondary progressive disease; prevention or reduction of the number and size of new and enhancing lesions on MRI; and limitation of overall MRI lesion burden in the central nervous system (CNS).
Newer treatment outcomes employed in more recent trials include: measures of axonal damage, CNS atrophy, evidence of microscopic disease via conventional MRI and advanced imaging modalities, biomarkers associated with inflammatory disease activity and neurodegeneration in MS, and the use of no evidence of disease activity (NEDA). These outcomes also must be evaluated by the safety concerns of each agent. Short- and long-term safety are critical factors in the selection of DMTs for MS. The injectable therapies for MS (interferon β‐1a, interferon β‐1b, and GA) have established long-term safety profiles from > 20 years of continuous use. The long-term safety profiles of oral immunomodulatory agents and monoclonal antibodies for these drugs in MS have yet to be determined. Safety concerns associated with some therapies and added requirements for safety monitoring may increase the complexity of a therapeutic selection.
Current cost minimization strategies for DMT include limiting DMT agents on formularies, tier systems that incentivize patients/prescribers to select the lowest priced agents on the formulary, negotiating arrangements with manufacturers to freeze prices or provide discounts in exchange for a priority position in the formulary, and requiring prior authorization to initiate or switch therapy. The use of generic medications and interchange to these agents from a brand name formulation can help reduce expense. Several of these strategies have been implemented in VHA.
Disease-Modifying Therapies
In 2019, 18,645 veterans with MS had either a MS-specific DMT or ≥ 1 annual encounters with a primary diagnosis of MS. Of this population, 4,720 were female and 13,357 were service connected according to VA data. About 50% of veterans with MS take a DMT. This percentage has remained stable over the past decade (Table 2). Although it appears the number of unique veterans prescribed an outpatient DMT is decreasing, this does not include the growing use of infused DMTs or DMTs obtained through the Veterans Choice Program (VCP)/Community Care (CC).
The overall outpatient pharmacy costs for veterans have remained constant despite the reduction in outpatient pharmacy prescription numbers. This may be due to increases in DMT cost to the VHA and the use of more expensive oral agents over the previously used platform injection DMTs.
Generic Conversion
GA is available in 20 mg daily and 40 mg3 times weekly subcutaneous injection dosing. The first evidence of clinical efficacy for a generic formulation for GA was evaluated by the GATE trial.7 This trial was a multicenter, randomized, double-blind, active- and placebo-controlled phase 3 trial. Eligible participants were randomized to receive daily SC injection for 9 months of 20 mg generic GA (n = 5,353), 20 mg brand GA (n = 5,357), or placebo (n = 584). The primary endpoint was the mean number of gadolinium (Gd1) lesions visible on MRIs during months 7, 8, and 9, which were significantly reduced in the combined GA-treated group and in each GA group individually when compared with the placebo group, confirming the study sensitivity (ie, GA was effective under the conditions of the study). Tolerability (including injection site reactions) and safety (incidence, spectrum, and severity of adverse events [AEs]) were similar in the generic and brand GA groups. These results demonstrated that generic and brand GA had equivalent efficacy, tolerability, and safety over a 9-month period.7
Results of a 15-month extension of the study were presented in 2015 and showed similar efficacy, safety, and tolerability in participants treated with generic GA for 2 years and patients switched from brand to generic GA.8 Multiple shifts for GA occurred, most notably the conversion from branded Copaxone (Teva Pharmaceutical Industries) to generic Glatopa (Sandoz). Subsequently, Sandoz released a generic 40 mg 3 times weekly formulation. Additionally, Mylan entered the generic GA market. With 3 competing manufacturers, internal data from the VHA indicated that it was able to negotiate a single source contract for this medication that provided a savings of $32,088,904.69 between September 2016 and May 2019.
The impact of generic conversions is just being realized. Soon, patents will begin to expire for oral DMTs, leading to an expected growth of generic alternatives. Already the FDA has approved 4 generic alternatives for teriflunomide, 3 for fingolimod (with 13 tentative approvals), and 15 generic alternatives for dimethyl fumarate (DMF). Implementation of therapeutic interchanges will be pursued by VHA as clinically supported by evidence.
Criteria for Use
PBM supports utilizing criteria to help guide providers on DMT options and promote safe, effective, and value-based selection of a DMT. The PBM creates monographs and criteria for use (CFU) for new medications. The monograph contains a literature evaluation of all studies available to date that concern both safety and efficacy of the new medication. Therapeutic alternatives also are presented and assessed for key elements that may determine the most safe and effective use. Additional safety areas for the new medications such as look-alike, sound-alike potential, special populations use (ie, those who are pregnant, the elderly, and those with liver or renal dysfunction), and drug-drug interactions are presented. Lastly, and possibly most importantly in an ever-growing growing world of DMTs, the monograph describes a reasonable place in therapy for the new DMT.
CFU are additional guidance for some DMTs. The development of CFU are based on several questions that arise during the monograph development for a DMT. These include, but are not limited to:
- Are there safety concerns that require the drug to receive a review to ensure safe prescribing (eg, agents with REMS programs, or safety concerns in specific populations)?
- Does the drug require a specialty provider type with knowledge and experience in those disease states to ensure appropriate and safe prescribing (eg restricted to infectious diseases)?
- Do VHA or non-VHA guidelines suggest alternative therapy be used prior to the agent?
- Is a review deemed necessary to ensure the preferred agent is used first (eg, second-line therapy)?
The CFU defines parameters of drug use consistent with high quality and evidence-based patient care. CFUs also serve as a basis for monitoring local, regional, and national patterns of pharmacologic care and help guide health care providers (HCPs) on appropriate use of medication.
CFUs are designed to ensure the HCP is safely starting a medication that has evidence for efficacy for their patient. For example, alemtuzumab is a high-risk, high-efficacy DMT. The alemtuzumab CFU acknowledges this by having exclusion criteria that prevent a veteran at high risk (ie, on another immunosuppressant) from being exposed to severe AEs (ie, severe leukopenia) that are associated with the medication. On the other hand, the inclusion criteria recognize the benefits of alemtuzumab and allows those with highly active MS who have failed other DMTs to receive the medication.
The drug monograph and CFU process is an important part of VHA efforts to optimize patient care. After a draft version is developed, HCPs can provide feedback on the exclusion/inclusion criteria and describe how they anticipate using the medication in their practice. This insight can be beneficial for MS treatment as diverse HCPs may have distinct viewpoints on how DMTs should be started. Pharmacists and physicians on a national level then discuss and decide together what to include in the final drafts of the drug monograph and CFU. Final documents are disseminated to all sites, which encourages consistent practices across the VHA.9 These documents are reviewed on a regular basis and updated as needed based on available literature evidence.
It is well accepted that early use of DMT correlates with lower accumulated long-term disability.10 However, discontinuation of DMT should be treated with equal importance. This benefits the patient by reducing their risk of AEs from DMTs and provides cost savings. Age and disease stability are factors to consider for DMT discontinuation. In a study with patients aged > 45 years and another with patients aged > 60 years, discontinuing DMT rarely had a negative impact and improved quality of life.11,12 A retrospective meta-analysis of age-dependent efficacy of current DMTs predicted that DMT loses efficacy at age 53 years. In addition, higher efficacy DMT only outperforms lower efficacy DMT in patients aged < 40.5 years.13 Stability of disease and lack of relapses for ≥ 2 years also may be a positive predictor to safely discontinue DMT.14,15 The growing literature to support safe discontinuation of DMT makes this a more convincing strategy to avoid unnecessary costs associated with current DMTs. With an average age of 59 years for veterans with MS, this may be one of the largest areas of cost avoidance to consider.
Off-Label Use
Other potential ways to reduce DMT costs is to consider off-label treatments. The OLYMPUS trial studied off-label use of rituximab, an anti-CD20 antibody like ocrelizumab. It did not meet statistical significance for its primary endpoint; however, in a subgroup analysis, off-label use was found to be more effective in a population aged < 51 years.16 Other case reports and smaller scale studies also describe rituximab’s efficacy in MS.17,18 In 2018, the FDA approved the first rituximab biosimilar.19 Further competition from biosimilars likely will make rituximab an even more cost-effective choice when compared with ocrelizumab.
Alternate Dosing Regimens
Extended interval dosing of natalizumab has been studied, extending the standard infusion interval from every 4 weeks to 5- to 8-week intervals. One recent article compared these interval extensions and found that all extended intervals of up to 56 days did not increase new or enhancing lesions on MRI when compared with standard interval dosing.20 Another larger randomized trial is underway to evaluate efficacy and safety of extended interval dosing of natalizumab (NCT03689972). Utilization of this dosing may reduce natalizumab annual costs by up to 50%.
Safety Monitoring
DMF is an oral DMT on the VHA formulary with CFU. Since leukopenia is a known AE, baseline and quarterly monitoring of the complete blood count (CBC) is recommended for patients taking DMF. Additionally, DMF should be held if white blood cell count (WBC) falls below 2,000/mm3.21 There have been recent reports of death secondary to progressive multifocal leukoencephalopathy (PML) among European patients taking DMF.22-24 This has raised concerns about adherence to recommended CBC monitoring in veterans taking DMF. The association of DMF and leukopenia has been evident since early clinical trials.25 Leukopenia in immunocompromised patients increases the risk of PML.
In the long-term extension study ENDORSE, 6% to 7% of patients continuing DMF had WBC counts of 3.0×109/L compared with 7% to 10% in the new to DMF group.26 In addition 6% to 8% of patients continuing DMF had lymphocyte counts of 0.5×109/L, compared with 5% to 9% in the new to DMF group. The cases of PML occurred in patients who had low lymphocyte counts over an extended period with no adjustment to DMF therapy, such as holding the drug until WBC counts returned to normal levels or stopping the drug. Discussion and review within VHA resulted in the recommendation for quarterly WBC monitoring criteria.
PBM and VA Center for Medication Safety (MedSafe) conducted a medication usage evaluation (MUE) on adherence to the WBC monitoring set forth in the CFU. Data collection began in fourth quarter of fiscal year (FY) 2015 with the most recent reporting period of fourth quarter of FY 2017. The Medication Utilization Evaluation Tool tracks patients with no reported WBC in 90 days and WBC < 2,000/mm3. Over the reporting period, 20% to 23% of patients have not received appropriate quarterly monitoring. Additionally, there have been 4 cases where the WBC decreased below the threshold limit. To ensure safe and effective use of DMF, it is important to adhere to the monitoring requirements set forth in the CFU.
Impact of REMS and Special Distribution
As DMTs increase in efficacy, there are often more risks associated with them. Some of these high-risk medications, including natalizumab and alemtuzumab, have REMS programs and/or have special distribution procedures. Although REMS are imperative for patient safety, the complexity of these programs can be difficult to navigate, which can create a barrier to access. The PBM helps to assist all sites with navigating and adhering to required actions to dispense and administer these medications through a national Special Handling Drugs Microsoft SharePoint site, which provides access to REMS forms and procurement information when drugs are dispensed from specialty pharmacies. Easing this process nationwide empowers more sites to be confident they can dispense specialty medications appropriately.
Clinical Pharmacists
The VHA is unique in its utilization of pharmacists in outpatient clinic settings. Utilization of an interdisciplinary team for medication management has been highly used in VHA for areas like primary care; however, pharmacist involvement in specialty areas is on the rise and MS is no exception. Pharmacists stationed in clinics, such as neurology or spinal cord injury, can impact care for veterans with MS. Interdisciplinary teams that include a pharmacist have been shown to increase patient adherence to DMTs.27 However, pharmacists often assist with medication education and monitoring, which adds an additional layer of safety to DMT treatment. At the VHA, pharmacists also can obtain a scope of practice that allows them to prescribe medications and increase access to care for veterans with MS.
Education
The VHA demonstrates how education on a disease state like MS can be distributed on a large, national scale through drug monographs, CFU, and Microsoft SharePoint sites. In addition, VHA has created the MS Centers of Excellence (MSCoE) that serve as a hub of specialized health care providers in all aspects of MS care.
A core function of the MSCoE is to provide education to both HCPs and patients. The MSCoE and its regional hubs support sites that may not have an HCP who specializes in MS by providing advice on DMT selection, how to obtain specialty medications, and monitoring that needs to be completed to ensure veterans’ safety. The MSCoE also has partnered with the National MS Society to hold a lecture series on topics in MS. This free series is available online to all HCPs who interact with patients who have MS and is a way that VA is extending its best practices and expertise beyond its own health care system. There also is a quarterly newsletter for veterans with MS that highlights new information on DMTs that can affect their care.
Conclusion
It is an exciting and challenging period in MS treatment. New DMTs are being approved and entering clinical trials at a rapid pace. These new DMT agents may offer increased efficacy, improvements in AE profiles, and the possibility of increased medication adherence—but often at a higher cost. The utilization of CFU and formulary management provides the ability to ensure the safe and appropriate use of medications by veterans, with a secondary outcome of controlling pharmacy expenditures.
The VHA had expenditures of $142,135,938 for DMT use in FY 2018. As the VHA sees the new contract prices for DMT in January 2020, we are reminded that costs will continue to rise with some pharmaceutical manufacturers implementing prices 8% to 11% higher than 2019 prices, when the consumer price index defines an increase of 1.0% for 2020 and 1.4% in 2021.28 It is imperative that the VHA formulary be managed judiciously and the necessary measures be in place for VHA practitioners to enable effective, safe and value-based care to the veteran population.
1. Gooch CL, Pracht E, Borenstein AR. The burden of neurological disease in the United States: a summary report and call to action. Ann Neurol. 2017;81(4):479-484.
2. Hartung DM, Bourdette DN, Ahmed SM, Whitham RH. The cost of multiple sclerosis drugs in the US and the pharmaceutical industry: too big to fail? [published correction appears in Neurology. 2015;85(19):1728]. Neurology. 2015;84(21):2185–2192.
3. San-Juan-Rodriguez A, Good CB, Heyman RA, Parekh N, Shrank WH, Hernandez I. Trends in prices, market share, and spending on self-administered disease-modifying therapies for multiple sclerosis in Medicare Part D. JAMA Neurol. 2019;76(11):1386-1390.
4. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278-286.
5. Eriksson M, Andersen O, Runmarker B. Long-term follow up of patients with clinically isolated syndromes, relapsing-remitting and secondary progressive multiple sclerosis [published correction appears in Mult Scler. 2003;9(6):641]. Mult Scler. 2003;9(3):260-274.
6. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173.
7. Cohen J, Belova A, Selmaj K, et al. Equivalence of generic glatiramer acetate in multiple sclerosis: a randomized clinical trial. JAMA Neurol. 2015;72(12):1433-1441.
8. Selmaj K, Barkhof F, Belova AN, et al; GATE study group. Switching from branded to generic glatiramer acetate: 15-month GATE trial extension results. Mult Scler. 2017;23(14):1909-1917.
9. Aspinall SL, Sales MM, Good CB, et al. Pharmacy benefits management in the Veterans Health Administration revisited: a decade of advancements, 2004-2014. J Manag Care Spec Pharm. 2016;22(9):1058-1063.
10. Brown JWL, Coles A, Horakova D, et al. Association of initial disease-modifying therapy with later conversion to secondary progressive multiple sclerosis. JAMA. 2019;321(2):175-187.
11. Hua LH, Harris H, Conway D, Thompson NR. Changes in patient-reported outcomes between continuers and discontinuers of disease modifying therapy in patients with multiple sclerosis over age 60 [published correction appears in Mult Scler Relat Disord. 2019;30:293]. Mult Scler Relat Disord. 2019;30:252-256.
12. Bsteh G, Feige J, Ehling R, et al. Discontinuation of disease-modifying therapies in multiple sclerosis - Clinical outcome and prognostic factors. Mult Scler. 2017;23(9):1241-1248.
13. Weideman AM, Tapia-Maltos MA, Johnson K, Greenwood M, Bielekova B. Meta-analysis of the age-dependent efficacy of multiple sclerosis treatments. Front Neurol. 2017;8:577.
14. Kister I, Spelman T, Alroughani R, et al; MSBase Study Group. Discontinuing disease-modifying therapy in MS after a prolonged relapse-free period: a propensity score-matched study [published correction appears in J Neurol Neurosurg Psychiatry. 2019;90(4):e2]. J Neurol Neurosurg Psychiatry. 2016;87(10):1133-1137.
15. Birnbaum G. Stopping disease-modifying therapy in nonrelapsing multiple sclerosis: experience from a clinical practice. Int J MS Care. 2017;19(1):11-14.
16. Hawker K, O’Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66(4):460-471.
17. Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–688.
18. Alping P, Frisell T, Novakova L, et al. Rituximab versus fingolimod after natalizumab in multiple sclerosis patients. Ann Neurol. 2016;79(6):950–958.
19. Rituximab-abbs [package insert]. North Wales, PA: Teva Pharmaceuticals; 2018.
20. Zhovtis Ryerson L, Frohman TC, Foley J, et al. Extended interval dosing of natalizumab in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2016;87(8):885-889.
21. Dimethyl fumarate [package insert]. Cambridge, MA: Biogen Inc; 2015.
22. van Kester MS, Bouwes Bavinck JN, Quint KD. PML in Patients treated with dimethyl fumarate. N Engl J Med. 2015;373(6):583-584.
23. Nieuwkamp DJ, Murk JL, van Oosten BW. PML in patients treated with dimethyl fumarate. N Engl J Med. 2015;373(6):584.
24. Rosenkranz T, Novas M, Terborg C. PML in a patient with lymphocytopenia treated with dimethyl fumarate. N Engl J Med. 2015;372(15):1476-1478.
25. Longbrake EE, Cross AH. Dimethyl fumarate associated lymphopenia in clinical practice. Mult Scler. 2015;21(6):796-797.
26. Gold R, Arnold DL, Bar-Or A, et al. Long-term effects of delayed-release dimethyl fumarate in multiple sclerosis: Interim analysis of ENDORSE, a randomized extension study. Mult Scler. 2017;23(2):253–265.
27. Hanson RL, Habibi M, Khamo N, Abdou S, Stubbings J. Integrated clinical and specialty pharmacy practice model for management of patients with multiple sclerosis. Am J Health Syst Pharm. 2014;71(6):463-469.
28. Federal Planning Bureau. Consumer Price Index - Inflation forecasts. https://www.plan.be/databases/17-en-consumer+price+index+inflation+forecasts. Updated March 3, 2020. Accessed March 9, 2020.
1. Gooch CL, Pracht E, Borenstein AR. The burden of neurological disease in the United States: a summary report and call to action. Ann Neurol. 2017;81(4):479-484.
2. Hartung DM, Bourdette DN, Ahmed SM, Whitham RH. The cost of multiple sclerosis drugs in the US and the pharmaceutical industry: too big to fail? [published correction appears in Neurology. 2015;85(19):1728]. Neurology. 2015;84(21):2185–2192.
3. San-Juan-Rodriguez A, Good CB, Heyman RA, Parekh N, Shrank WH, Hernandez I. Trends in prices, market share, and spending on self-administered disease-modifying therapies for multiple sclerosis in Medicare Part D. JAMA Neurol. 2019;76(11):1386-1390.
4. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278-286.
5. Eriksson M, Andersen O, Runmarker B. Long-term follow up of patients with clinically isolated syndromes, relapsing-remitting and secondary progressive multiple sclerosis [published correction appears in Mult Scler. 2003;9(6):641]. Mult Scler. 2003;9(3):260-274.
6. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173.
7. Cohen J, Belova A, Selmaj K, et al. Equivalence of generic glatiramer acetate in multiple sclerosis: a randomized clinical trial. JAMA Neurol. 2015;72(12):1433-1441.
8. Selmaj K, Barkhof F, Belova AN, et al; GATE study group. Switching from branded to generic glatiramer acetate: 15-month GATE trial extension results. Mult Scler. 2017;23(14):1909-1917.
9. Aspinall SL, Sales MM, Good CB, et al. Pharmacy benefits management in the Veterans Health Administration revisited: a decade of advancements, 2004-2014. J Manag Care Spec Pharm. 2016;22(9):1058-1063.
10. Brown JWL, Coles A, Horakova D, et al. Association of initial disease-modifying therapy with later conversion to secondary progressive multiple sclerosis. JAMA. 2019;321(2):175-187.
11. Hua LH, Harris H, Conway D, Thompson NR. Changes in patient-reported outcomes between continuers and discontinuers of disease modifying therapy in patients with multiple sclerosis over age 60 [published correction appears in Mult Scler Relat Disord. 2019;30:293]. Mult Scler Relat Disord. 2019;30:252-256.
12. Bsteh G, Feige J, Ehling R, et al. Discontinuation of disease-modifying therapies in multiple sclerosis - Clinical outcome and prognostic factors. Mult Scler. 2017;23(9):1241-1248.
13. Weideman AM, Tapia-Maltos MA, Johnson K, Greenwood M, Bielekova B. Meta-analysis of the age-dependent efficacy of multiple sclerosis treatments. Front Neurol. 2017;8:577.
14. Kister I, Spelman T, Alroughani R, et al; MSBase Study Group. Discontinuing disease-modifying therapy in MS after a prolonged relapse-free period: a propensity score-matched study [published correction appears in J Neurol Neurosurg Psychiatry. 2019;90(4):e2]. J Neurol Neurosurg Psychiatry. 2016;87(10):1133-1137.
15. Birnbaum G. Stopping disease-modifying therapy in nonrelapsing multiple sclerosis: experience from a clinical practice. Int J MS Care. 2017;19(1):11-14.
16. Hawker K, O’Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66(4):460-471.
17. Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–688.
18. Alping P, Frisell T, Novakova L, et al. Rituximab versus fingolimod after natalizumab in multiple sclerosis patients. Ann Neurol. 2016;79(6):950–958.
19. Rituximab-abbs [package insert]. North Wales, PA: Teva Pharmaceuticals; 2018.
20. Zhovtis Ryerson L, Frohman TC, Foley J, et al. Extended interval dosing of natalizumab in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2016;87(8):885-889.
21. Dimethyl fumarate [package insert]. Cambridge, MA: Biogen Inc; 2015.
22. van Kester MS, Bouwes Bavinck JN, Quint KD. PML in Patients treated with dimethyl fumarate. N Engl J Med. 2015;373(6):583-584.
23. Nieuwkamp DJ, Murk JL, van Oosten BW. PML in patients treated with dimethyl fumarate. N Engl J Med. 2015;373(6):584.
24. Rosenkranz T, Novas M, Terborg C. PML in a patient with lymphocytopenia treated with dimethyl fumarate. N Engl J Med. 2015;372(15):1476-1478.
25. Longbrake EE, Cross AH. Dimethyl fumarate associated lymphopenia in clinical practice. Mult Scler. 2015;21(6):796-797.
26. Gold R, Arnold DL, Bar-Or A, et al. Long-term effects of delayed-release dimethyl fumarate in multiple sclerosis: Interim analysis of ENDORSE, a randomized extension study. Mult Scler. 2017;23(2):253–265.
27. Hanson RL, Habibi M, Khamo N, Abdou S, Stubbings J. Integrated clinical and specialty pharmacy practice model for management of patients with multiple sclerosis. Am J Health Syst Pharm. 2014;71(6):463-469.
28. Federal Planning Bureau. Consumer Price Index - Inflation forecasts. https://www.plan.be/databases/17-en-consumer+price+index+inflation+forecasts. Updated March 3, 2020. Accessed March 9, 2020.
The Multiple Sclerosis Centers of Excellence: A Model of Excellence in the VA (FULL)
The Veterans Health Administration (VHA) has established a number of centers of excellence (CoEs), including centers focused on posttraumatic stress disorder, suicide prevention, epilepsy, and, most recently, the Senator Elizabeth Dole CoE for Veteran and Caregiver Research. Some VA CoE serve as centralized locations for specialty care. For example, the VA Epilepsy CoE is a network of 16 facilities that provide comprehensive epilepsy care for veterans with seizure disorders, including expert and presurgical evaluations and inpatient monitoring.
In contrast, other CoEs, including the multiple sclerosis (MS) CoE, achieve their missions by serving as a resource center to a network of regional and supporting various programs to optimize the care of veterans across the nation within their home US Department of Veterans Affairs (VA) medical center (VAMC). The MSCoE are charged, through VHA Directive 1011.06, with establishing at least 1 VA MS Regional Program in each of the 21 Veteran Integrated Service Networks (VISNs) across the country and integrating these and affiliated MS Support Programs into the MS National Network. Currently, there are 29 MS regional programs and 49 MS support programs across the US.1
Established in 2003, the MSCoE is dedicated to furthering the understanding of MS, its impact on veterans, and effective treatments to help manage the disease and its symptoms. In 2002, 2 coordinating centers were selected based on a competitive review process. The MSCoE-East is located at the Baltimore, Maryland and Washington, DC VAMC and serves VISNs 1 to 10. The MSCoE-West serves VISNs 11 to 23 and is jointly-based at VA Puget Sound Health Care System in Seattle, Washington and VA Portland Health Care System in Portland, Oregon. The MSCoEs were made permanent by The Veteran’ Benefits, Healthcare and Information Technology Act of 2006 (38 USC §7330). By partnering with veterans, caregivers, health care professionals, and other affiliates, the MSCoE endeavor to optimize health, activities, participation and quality of life for veterans with MS.
Core Functions
The MSCoE has a 3-part mission. First, the MSCoE seeks to expand care coordination between VAMCs by developing a national network of VA MSCoE Regional and Support Programs. Second, the MSCoE provides resources to VA health care providers (HCPs) through a collaborative approach to clinical care, education, research, and informatics. Third, the MSCoE improves the quality and consistency of health care services delivered to veterans diagnosed with MS nationwide. To meet its objectives, the MSCoE activities are organized around 4 functional cores: clinical care, research, education and training, and informatics and telemedicine.
Clinical Care
The MSCoE delivers high-quality clinical care by identifying veterans with MS who use VA services, understanding their needs, and facilitating appropriate interventions. Veterans with MS are a special cohort for many reasons including that about 70% are male. Men and women veterans not only have different genetics, but also may have different environmental exposures and other risk factors for MS. Since 1998, the VHA has evaluated > 50,000 veterans with MS. Over the past decade, between 18,000 and 20,000 veterans with MS have accessed care within the VHA annually.
The MSCoE advocates for appropriate and safe use of currently available MS disease modifying therapies through collaborations with the VA Pharmacy Benefits Management Service (PBM). The MSCoE partners with PBM to develop and disseminate Criteria For Use, safety, and economic monitoring of the impacts of the MS therapies. The MSCoE also provide national consultation services for complex MS cases, clinical education to VA HCPs, and mentors fellows, residents, and medical students.
The VA provides numerous resources that are not readily available in other health care systems and facilitate the care for patients with chronic diseases, including providing low or no co-pays to patients for MS disease modifying agents and other MS related medications, access to medically necessary adaptive equipment at no charge, the Home Improvement and Structural Alteration (HISA) grant for assistance with safe home ingress and egress, respite care, access to a homemaker/home health aide, and caregiver support programs. Eligible veterans also can access additional resources such as adaptive housing and an automobile grant. The VA also provides substantial hands-on assistance to veterans who are homeless. The clinical team and a veteran with MS can leverage VA resources through the National MS Society (NMSS) Navigator Program as well as other community resources.2
The VHA encourages physical activity and wellness through sports and leisure. Veterans with MS can participate in sports programs and special events, including the National Veterans Wheelchair Games, the National Disabled Veterans Winter Sports Clinic, the National Disabled Veterans TEE (Training, Exposure and Experience) golf tournament, the National Veterans Summer Sports Clinic, the National Veterans Golden Age Games, and the National Veterans Creative Sports Festival. HCPs or veterans who are not sure how to access any of these programs can contact the MSCoE or their local VA social workers.
Research
The primary goal of the MSCoE research core is to conduct clinical, health services, epidemiologic, and basic science research relevant to veterans with MS. The MSCoE serves to enhance collaboration among VAMCs, increase the participation of veterans in research, and provide research mentorship for the next generation of VA MS scientists. MSCoE research is carried out by investigators at the MSCoE and the MS Regional Programs, often in collaboration with investigators at academic institutions. This research is supported by competitive grant awards from a variety of funding agencies including the VA Research and Development Service (R&D) and the NMSS. Results from about 40 research grants in Fiscal Year 2019 were disseminated through 34 peer-reviewed publications, 30 posters, presentations, abstracts, and clinical practice guidelines.
There are many examples of recent high impact MS research performed by MSCoE investigators. For example, MSCoE researchers noted an increase in the estimated prevalence of MS to 1 million individuals in the US, about twice the previously estimated prevalence.3-5 In addition, a multicenter study highlighted the prevalence of MS misdiagnosis and common confounders for MS.6 Other research includes pilot clinical trials evaluating lipoic acid as a potential disease modifying therapy in people with secondary progressive MS and the impact of a multicomponent walking aid selection, fitting, and training program for preventing falls in people with MS.7,8 Clinical trial also are investigating telehealth counseling to improve physical activity in MS and a systematic review of rehabilitation interventions in MS.9,10
Education and Training
A unified program of education is essential to effective management of MS nationally. The primary goal of the education and training core is to provide a national program of MS education for HCPs, veterans, and caregivers to improve knowledge, enhance access to resources, and promote effective management strategies. The MSCoE collaborate with the Paralyzed Veterans of America (PVA), the Consortium of MS Centers (CMSC), the NMSS, and other national service organizations to increase educational opportunities, share knowledge, and expand participation.
The MSCoE education and training core produces a range of products both veterans, HCPs, and others affected by MS. The MSCoE sends a biannual patient newsletter to > 20,000 veterans and a monthly email to > 1,000 VA HCPs. Specific opportunities for HCP education include accredited multidisciplinary MS webinars, sponsored symposia and workshops at the CMSC and PVA Summit annual meetings, and presentations at other university and professional conferences. Enduring educational opportunities for veterans, caregivers, and HCPs can also be found by visiting www.va.gov/ms.
The MSCoE coordinate postdoctoral fellowship training programs to develop expertise in MS health care for the future. It offers VA physician fellowships for neurologists in Baltimore and Portland and for physiatrists in Seattle as well as NMSS fellowships for education and research. In 2019, MSCoE had 6 MD Fellows and 1 PhD Fellow.
Clinical Informatics and Telehealth
The primary goal of the informatics and telemedicine core is to employ state-of-the-art informatics, telemedicine technology, and the MSCoE website, to improve MS health care delivery. The VA has a integrated electronic health record and various data repositories are stored in the VHA Corporate Data Warehouse (CDW). MSCoE utilizes the CDW to maintain a national MS administrative data repository to understand the VHA care provided to veterans with MS. Data from the CDW have also served as an important resource to facilitate a wide range of veteran-focused MS research. This research has addressed clinical conditions like pain and obesity; health behaviors like smoking, alcohol use, and exercise as well as issues related to care delivery such as specialty care access, medication adherence, and appointment attendance.11-19
Monitoring the health of veterans with MS in the VA requires additional data not available in the CDW. To this end, we have developed the MS Surveillance Registry (MSSR), funded and maintained by the VA Office of Information Technology as part of their Veteran Integrated Registry Platform (VIRP). The purpose of the MSSR is to understand the unique characteristics and treatment patterns of veterans with MS in order to optimize their VHA care. HCPs input MS-specific clinical data on their patients into the MSSR, either through the MS Assessment Tool (MSAT) in the Computerized Patient Record System (CPRS) or through a secure online portal. Other data from existing databases from the CDW is also automatically fed into the MSSR. The MSSR continues to be developed and populated to serve as a resource for the future.

Neurologists, physiatrists, psychologists, and rehabilitation specialists can use telehealth to evaluate and treat veterans who have difficulty accessing outpatient clinics, either because of mobility limitations, or distance. Between 2012 and 2015, the VA MSCoE, together with the Epilepsy CoE and the Parkinson’s Disease Research and Clinical Centers in VISNs 5, 6 (mid-Atlantic) and 20 (Pacific Northwest) initiated an integrated teleneurology project. The goal of this project was to improve patient access to care at 4 tertiary and 12 regional VAMCs. A study team, with administrators and key clinical stakeholders, followed a traditional project management approach to design, plan, implement and evaluate an optimal model for communication and referrals with both live visits and telehealth (Table). Major outcomes of the project included: delivering subspecialty teleneurology to 47 patient sites, increasing interfacility consultation by 133% while reducing wait times by roughly 40%, and increasing telemedicine workload at these centers from 95 annual encounters in 2012 to 1,245 annual encounters in 2015 (Figure).
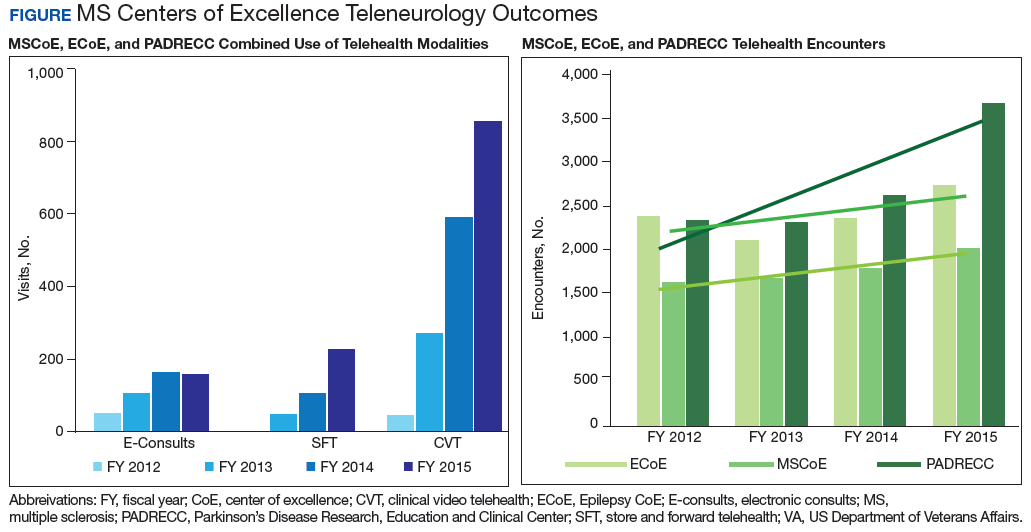
Today, telehealth for veterans with MS can be delivered to nearby VA facilities closer to their home, within their home, or anywhere else the veteran can use a cellphone or tablet. Telehealth visits can save travel time and expenses and optimize VA productivity and clinic use. The MSCoE and many of the MS regional programs are using telehealth for MS physician follow-up and therapies. The VA Office of Rural Health is also currently working with the MS network to use telehealth to increase access to physical therapy to those who have difficulty coming into clinic.
MSCoE Resources
The MSCoE is funded by VA Central Office through the Office of Specialty Care by Special Purpose funds. The directive specifies that funding for the regional and support programs is through Veterans Equitable Resource Allocation based on VISN and facility workload and complexity. Any research is funded separately through grants, some from VA R&D and others from other sources including the National Institutes of Health, the Patient Centered Outcome Research Institute, affiliated universities, the NMSS, the MS Society of Canada, the Consortium of MS Centers, foundations, and industry.
In 2019, MSCoE investigators received grants totaling > $18 million in funding. In-kind support also is provided by the PVA, the CMSC, the NMSS, and others. The first 3 foundations have been supporters since the inception of the MSCoE and have provided opportunities for the dissemination of education and research for HCPs, fellows, residents and medical students; travel; meeting rooms for MSCoE national meetings; exhibit space for HCP outreach; competitive research and educational grant support; programming and resources for veterans and significant others; organizational expertise; and opportunities for VA HCPs, veterans, and caregivers to learn how to navigate MS with others in the private sector.
Conclusion
The MSCoE had a tremendous impact on improving the consistency and quality of care for veterans with MS through clinical care, research, education and informatics and telehealth. Since opening in 2003, there has been an increase in the number of MS specialty clinics, served veterans with MS, and veterans receiving specialty neurologic and rehabilitation services in VA. Research programs in MS have been initiated to address key questions relevant to veterans with MS, including immunology, epidemiology, clinical care, and rehabilitation. Educational programs and products have evolved with technology and had a greater impact through partnerships with veteran and MS nonprofit organizations.
MSCoE strives to minimize impairment and maximize quality of life for veterans with MS by leveraging integrated electronic health records, data repositories, and telehealth services. These efforts have all improved veteran health, access and safety. We look forward to continuing into the next decade by bringing fresh ideas to the care of veterans with MS, their families and caregivers.
1. US Department of Veterans Affairs, Multiple Sclerosis Centers of Excellence. Multiple Sclerosis System of Care-VHA Directive 1101.06 and Multiple Sclerosis Centers of Excellence network facilities. https://www.va.gov/MS/veterans/find_a_clinic/index_clinics.asp. Updated February 26, 2020. Accessed March 6, 2020.
2. National MS Society. MS navigator program. https://www.nationalmssociety.org/For-Professionals/Clinical-Care/MS-Navigator-Program. Accessed March 6, 2020.
3. Wallin MT, Culpepper WJ, Campbell JD, et al. The prevalence of MS in the United States: a population-based estimate using health claims data. Neurology. 2019;92:e1029-e1040.
4. GBD 2016 Multiple Sclerosis Collaborators. Global, regional, and national burden of multiple sclerosis 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(3):269-285.
5. GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(5):459-480.
6. Solomon AJ, Bourdette DN, Cross AH, et al. The contemporary spectrum of multiple sclerosis misdiagnosis: a multicenter study. Neurology. 2016;87(13):1393-1399.
7. Spain R, Powers K, Murchison C, et al. Lipoic acid in secondary progressive MS: a randomized controlled pilot trial. Neurol Neuroimmunol Neuroinflamm. 2017;4(5):e374.
8. Martini DN, Zeeboer E, Hildebrand A, Fling BW, Hugos CL, Cameron MH. ADSTEP: preliminary investigation of a multicomponent walking aid program in people with multiple sclerosis. Arch Phys Med Rehabil. 2018;99(10):2050-2058.
9. Turner AP, Hartoonian N, Sloan AP, et al. Improving fatigue and depression in individuals with multiple sclerosis using telephone-administered physical activity counseling. J Consult Clin Psychol. 2016;84(4):297-309.
10. Haselkorn JK, Hughes C, Rae-Grant A, et al. Summary of comprehensive systematic review: rehabilitation in multiple sclerosis: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology. 2015;85(21):1896-1903.
11. Hirsh AT, Turner AP, Ehde DM, Haselkorn JK. Prevalence and impact of pain in multiple sclerosis: physical and psychologic contributors. Arch Phys Med Rehabil. 2009;90(4):646-651.
12. Khurana SR, Bamer AM, Turner AP, et al. The prevalence of overweight and obesity in veterans with multiple sclerosis. Am J Phys Med Rehabil. 2009;88(2):83-91.
13. Turner AP, Kivlahan DR, Kazis LE, Haselkorn JK. Smoking among veterans with multiple sclerosis: prevalence correlates, quit attempts, and unmet need for services. Arch Phys Med Rehabil. 2007;88(11):1394-1399.
14. Turner AP, Hawkins EJ, Haselkorn JK, Kivlahan DR. Alcohol misuse and multiple sclerosis. Arch Phys Med Rehabil. 2009;90(5):842-848.
15. Turner AP, Kivlahan DR, Haselkorn JK. Exercise and quality of life among people with multiple sclerosis: looking beyond physical functioning to mental health and participation in life. Arch Phys Med Rehabil. 2009;90(3):420-428.
16. Turner AP, Chapko MK, Yanez D, et al. Access to multiple sclerosis specialty care. PM R. 2013;5(12):1044-1050.
17. Gromisch ES, Turner AP, Leipertz SL, Beauvais J, Haselkorn JK. Risk factors for suboptimal medication adherence in persons with multiple sclerosis: development of an electronic health record-based explanatory model for disease-modifying therapy use [published online ahead of print, 2019 Dec 3]. Arch Phys Med Rehabil. 2019;S0003-9993(19)31430-3143.
18. Settle JR, Maloni H, Bedra M, Finkelstein J, Zhan M, Wallin M. Monitoring medication adherence in multiple sclerosis using a novel web-based tool. J Telemed Telecare. 2016;22:225-233.
19. Gromisch ES, Turner AP, Leipertz SL, Beauvais J, Haselkorn JK. Who is not coming to clinic? A predictive model of excessive missed appointments in persons with multiple sclerosis. Mult Scler Rel Dis. In Press.
The Veterans Health Administration (VHA) has established a number of centers of excellence (CoEs), including centers focused on posttraumatic stress disorder, suicide prevention, epilepsy, and, most recently, the Senator Elizabeth Dole CoE for Veteran and Caregiver Research. Some VA CoE serve as centralized locations for specialty care. For example, the VA Epilepsy CoE is a network of 16 facilities that provide comprehensive epilepsy care for veterans with seizure disorders, including expert and presurgical evaluations and inpatient monitoring.
In contrast, other CoEs, including the multiple sclerosis (MS) CoE, achieve their missions by serving as a resource center to a network of regional and supporting various programs to optimize the care of veterans across the nation within their home US Department of Veterans Affairs (VA) medical center (VAMC). The MSCoE are charged, through VHA Directive 1011.06, with establishing at least 1 VA MS Regional Program in each of the 21 Veteran Integrated Service Networks (VISNs) across the country and integrating these and affiliated MS Support Programs into the MS National Network. Currently, there are 29 MS regional programs and 49 MS support programs across the US.1
Established in 2003, the MSCoE is dedicated to furthering the understanding of MS, its impact on veterans, and effective treatments to help manage the disease and its symptoms. In 2002, 2 coordinating centers were selected based on a competitive review process. The MSCoE-East is located at the Baltimore, Maryland and Washington, DC VAMC and serves VISNs 1 to 10. The MSCoE-West serves VISNs 11 to 23 and is jointly-based at VA Puget Sound Health Care System in Seattle, Washington and VA Portland Health Care System in Portland, Oregon. The MSCoEs were made permanent by The Veteran’ Benefits, Healthcare and Information Technology Act of 2006 (38 USC §7330). By partnering with veterans, caregivers, health care professionals, and other affiliates, the MSCoE endeavor to optimize health, activities, participation and quality of life for veterans with MS.
Core Functions
The MSCoE has a 3-part mission. First, the MSCoE seeks to expand care coordination between VAMCs by developing a national network of VA MSCoE Regional and Support Programs. Second, the MSCoE provides resources to VA health care providers (HCPs) through a collaborative approach to clinical care, education, research, and informatics. Third, the MSCoE improves the quality and consistency of health care services delivered to veterans diagnosed with MS nationwide. To meet its objectives, the MSCoE activities are organized around 4 functional cores: clinical care, research, education and training, and informatics and telemedicine.
Clinical Care
The MSCoE delivers high-quality clinical care by identifying veterans with MS who use VA services, understanding their needs, and facilitating appropriate interventions. Veterans with MS are a special cohort for many reasons including that about 70% are male. Men and women veterans not only have different genetics, but also may have different environmental exposures and other risk factors for MS. Since 1998, the VHA has evaluated > 50,000 veterans with MS. Over the past decade, between 18,000 and 20,000 veterans with MS have accessed care within the VHA annually.
The MSCoE advocates for appropriate and safe use of currently available MS disease modifying therapies through collaborations with the VA Pharmacy Benefits Management Service (PBM). The MSCoE partners with PBM to develop and disseminate Criteria For Use, safety, and economic monitoring of the impacts of the MS therapies. The MSCoE also provide national consultation services for complex MS cases, clinical education to VA HCPs, and mentors fellows, residents, and medical students.
The VA provides numerous resources that are not readily available in other health care systems and facilitate the care for patients with chronic diseases, including providing low or no co-pays to patients for MS disease modifying agents and other MS related medications, access to medically necessary adaptive equipment at no charge, the Home Improvement and Structural Alteration (HISA) grant for assistance with safe home ingress and egress, respite care, access to a homemaker/home health aide, and caregiver support programs. Eligible veterans also can access additional resources such as adaptive housing and an automobile grant. The VA also provides substantial hands-on assistance to veterans who are homeless. The clinical team and a veteran with MS can leverage VA resources through the National MS Society (NMSS) Navigator Program as well as other community resources.2
The VHA encourages physical activity and wellness through sports and leisure. Veterans with MS can participate in sports programs and special events, including the National Veterans Wheelchair Games, the National Disabled Veterans Winter Sports Clinic, the National Disabled Veterans TEE (Training, Exposure and Experience) golf tournament, the National Veterans Summer Sports Clinic, the National Veterans Golden Age Games, and the National Veterans Creative Sports Festival. HCPs or veterans who are not sure how to access any of these programs can contact the MSCoE or their local VA social workers.
Research
The primary goal of the MSCoE research core is to conduct clinical, health services, epidemiologic, and basic science research relevant to veterans with MS. The MSCoE serves to enhance collaboration among VAMCs, increase the participation of veterans in research, and provide research mentorship for the next generation of VA MS scientists. MSCoE research is carried out by investigators at the MSCoE and the MS Regional Programs, often in collaboration with investigators at academic institutions. This research is supported by competitive grant awards from a variety of funding agencies including the VA Research and Development Service (R&D) and the NMSS. Results from about 40 research grants in Fiscal Year 2019 were disseminated through 34 peer-reviewed publications, 30 posters, presentations, abstracts, and clinical practice guidelines.
There are many examples of recent high impact MS research performed by MSCoE investigators. For example, MSCoE researchers noted an increase in the estimated prevalence of MS to 1 million individuals in the US, about twice the previously estimated prevalence.3-5 In addition, a multicenter study highlighted the prevalence of MS misdiagnosis and common confounders for MS.6 Other research includes pilot clinical trials evaluating lipoic acid as a potential disease modifying therapy in people with secondary progressive MS and the impact of a multicomponent walking aid selection, fitting, and training program for preventing falls in people with MS.7,8 Clinical trial also are investigating telehealth counseling to improve physical activity in MS and a systematic review of rehabilitation interventions in MS.9,10
Education and Training
A unified program of education is essential to effective management of MS nationally. The primary goal of the education and training core is to provide a national program of MS education for HCPs, veterans, and caregivers to improve knowledge, enhance access to resources, and promote effective management strategies. The MSCoE collaborate with the Paralyzed Veterans of America (PVA), the Consortium of MS Centers (CMSC), the NMSS, and other national service organizations to increase educational opportunities, share knowledge, and expand participation.
The MSCoE education and training core produces a range of products both veterans, HCPs, and others affected by MS. The MSCoE sends a biannual patient newsletter to > 20,000 veterans and a monthly email to > 1,000 VA HCPs. Specific opportunities for HCP education include accredited multidisciplinary MS webinars, sponsored symposia and workshops at the CMSC and PVA Summit annual meetings, and presentations at other university and professional conferences. Enduring educational opportunities for veterans, caregivers, and HCPs can also be found by visiting www.va.gov/ms.
The MSCoE coordinate postdoctoral fellowship training programs to develop expertise in MS health care for the future. It offers VA physician fellowships for neurologists in Baltimore and Portland and for physiatrists in Seattle as well as NMSS fellowships for education and research. In 2019, MSCoE had 6 MD Fellows and 1 PhD Fellow.
Clinical Informatics and Telehealth
The primary goal of the informatics and telemedicine core is to employ state-of-the-art informatics, telemedicine technology, and the MSCoE website, to improve MS health care delivery. The VA has a integrated electronic health record and various data repositories are stored in the VHA Corporate Data Warehouse (CDW). MSCoE utilizes the CDW to maintain a national MS administrative data repository to understand the VHA care provided to veterans with MS. Data from the CDW have also served as an important resource to facilitate a wide range of veteran-focused MS research. This research has addressed clinical conditions like pain and obesity; health behaviors like smoking, alcohol use, and exercise as well as issues related to care delivery such as specialty care access, medication adherence, and appointment attendance.11-19
Monitoring the health of veterans with MS in the VA requires additional data not available in the CDW. To this end, we have developed the MS Surveillance Registry (MSSR), funded and maintained by the VA Office of Information Technology as part of their Veteran Integrated Registry Platform (VIRP). The purpose of the MSSR is to understand the unique characteristics and treatment patterns of veterans with MS in order to optimize their VHA care. HCPs input MS-specific clinical data on their patients into the MSSR, either through the MS Assessment Tool (MSAT) in the Computerized Patient Record System (CPRS) or through a secure online portal. Other data from existing databases from the CDW is also automatically fed into the MSSR. The MSSR continues to be developed and populated to serve as a resource for the future.
Neurologists, physiatrists, psychologists, and rehabilitation specialists can use telehealth to evaluate and treat veterans who have difficulty accessing outpatient clinics, either because of mobility limitations, or distance. Between 2012 and 2015, the VA MSCoE, together with the Epilepsy CoE and the Parkinson’s Disease Research and Clinical Centers in VISNs 5, 6 (mid-Atlantic) and 20 (Pacific Northwest) initiated an integrated teleneurology project. The goal of this project was to improve patient access to care at 4 tertiary and 12 regional VAMCs. A study team, with administrators and key clinical stakeholders, followed a traditional project management approach to design, plan, implement and evaluate an optimal model for communication and referrals with both live visits and telehealth (Table). Major outcomes of the project included: delivering subspecialty teleneurology to 47 patient sites, increasing interfacility consultation by 133% while reducing wait times by roughly 40%, and increasing telemedicine workload at these centers from 95 annual encounters in 2012 to 1,245 annual encounters in 2015 (Figure).
Today, telehealth for veterans with MS can be delivered to nearby VA facilities closer to their home, within their home, or anywhere else the veteran can use a cellphone or tablet. Telehealth visits can save travel time and expenses and optimize VA productivity and clinic use. The MSCoE and many of the MS regional programs are using telehealth for MS physician follow-up and therapies. The VA Office of Rural Health is also currently working with the MS network to use telehealth to increase access to physical therapy to those who have difficulty coming into clinic.
MSCoE Resources
The MSCoE is funded by VA Central Office through the Office of Specialty Care by Special Purpose funds. The directive specifies that funding for the regional and support programs is through Veterans Equitable Resource Allocation based on VISN and facility workload and complexity. Any research is funded separately through grants, some from VA R&D and others from other sources including the National Institutes of Health, the Patient Centered Outcome Research Institute, affiliated universities, the NMSS, the MS Society of Canada, the Consortium of MS Centers, foundations, and industry.
In 2019, MSCoE investigators received grants totaling > $18 million in funding. In-kind support also is provided by the PVA, the CMSC, the NMSS, and others. The first 3 foundations have been supporters since the inception of the MSCoE and have provided opportunities for the dissemination of education and research for HCPs, fellows, residents and medical students; travel; meeting rooms for MSCoE national meetings; exhibit space for HCP outreach; competitive research and educational grant support; programming and resources for veterans and significant others; organizational expertise; and opportunities for VA HCPs, veterans, and caregivers to learn how to navigate MS with others in the private sector.
Conclusion
The MSCoE had a tremendous impact on improving the consistency and quality of care for veterans with MS through clinical care, research, education and informatics and telehealth. Since opening in 2003, there has been an increase in the number of MS specialty clinics, served veterans with MS, and veterans receiving specialty neurologic and rehabilitation services in VA. Research programs in MS have been initiated to address key questions relevant to veterans with MS, including immunology, epidemiology, clinical care, and rehabilitation. Educational programs and products have evolved with technology and had a greater impact through partnerships with veteran and MS nonprofit organizations.
MSCoE strives to minimize impairment and maximize quality of life for veterans with MS by leveraging integrated electronic health records, data repositories, and telehealth services. These efforts have all improved veteran health, access and safety. We look forward to continuing into the next decade by bringing fresh ideas to the care of veterans with MS, their families and caregivers.
The Veterans Health Administration (VHA) has established a number of centers of excellence (CoEs), including centers focused on posttraumatic stress disorder, suicide prevention, epilepsy, and, most recently, the Senator Elizabeth Dole CoE for Veteran and Caregiver Research. Some VA CoE serve as centralized locations for specialty care. For example, the VA Epilepsy CoE is a network of 16 facilities that provide comprehensive epilepsy care for veterans with seizure disorders, including expert and presurgical evaluations and inpatient monitoring.
In contrast, other CoEs, including the multiple sclerosis (MS) CoE, achieve their missions by serving as a resource center to a network of regional and supporting various programs to optimize the care of veterans across the nation within their home US Department of Veterans Affairs (VA) medical center (VAMC). The MSCoE are charged, through VHA Directive 1011.06, with establishing at least 1 VA MS Regional Program in each of the 21 Veteran Integrated Service Networks (VISNs) across the country and integrating these and affiliated MS Support Programs into the MS National Network. Currently, there are 29 MS regional programs and 49 MS support programs across the US.1
Established in 2003, the MSCoE is dedicated to furthering the understanding of MS, its impact on veterans, and effective treatments to help manage the disease and its symptoms. In 2002, 2 coordinating centers were selected based on a competitive review process. The MSCoE-East is located at the Baltimore, Maryland and Washington, DC VAMC and serves VISNs 1 to 10. The MSCoE-West serves VISNs 11 to 23 and is jointly-based at VA Puget Sound Health Care System in Seattle, Washington and VA Portland Health Care System in Portland, Oregon. The MSCoEs were made permanent by The Veteran’ Benefits, Healthcare and Information Technology Act of 2006 (38 USC §7330). By partnering with veterans, caregivers, health care professionals, and other affiliates, the MSCoE endeavor to optimize health, activities, participation and quality of life for veterans with MS.
Core Functions
The MSCoE has a 3-part mission. First, the MSCoE seeks to expand care coordination between VAMCs by developing a national network of VA MSCoE Regional and Support Programs. Second, the MSCoE provides resources to VA health care providers (HCPs) through a collaborative approach to clinical care, education, research, and informatics. Third, the MSCoE improves the quality and consistency of health care services delivered to veterans diagnosed with MS nationwide. To meet its objectives, the MSCoE activities are organized around 4 functional cores: clinical care, research, education and training, and informatics and telemedicine.
Clinical Care
The MSCoE delivers high-quality clinical care by identifying veterans with MS who use VA services, understanding their needs, and facilitating appropriate interventions. Veterans with MS are a special cohort for many reasons including that about 70% are male. Men and women veterans not only have different genetics, but also may have different environmental exposures and other risk factors for MS. Since 1998, the VHA has evaluated > 50,000 veterans with MS. Over the past decade, between 18,000 and 20,000 veterans with MS have accessed care within the VHA annually.
The MSCoE advocates for appropriate and safe use of currently available MS disease modifying therapies through collaborations with the VA Pharmacy Benefits Management Service (PBM). The MSCoE partners with PBM to develop and disseminate Criteria For Use, safety, and economic monitoring of the impacts of the MS therapies. The MSCoE also provide national consultation services for complex MS cases, clinical education to VA HCPs, and mentors fellows, residents, and medical students.
The VA provides numerous resources that are not readily available in other health care systems and facilitate the care for patients with chronic diseases, including providing low or no co-pays to patients for MS disease modifying agents and other MS related medications, access to medically necessary adaptive equipment at no charge, the Home Improvement and Structural Alteration (HISA) grant for assistance with safe home ingress and egress, respite care, access to a homemaker/home health aide, and caregiver support programs. Eligible veterans also can access additional resources such as adaptive housing and an automobile grant. The VA also provides substantial hands-on assistance to veterans who are homeless. The clinical team and a veteran with MS can leverage VA resources through the National MS Society (NMSS) Navigator Program as well as other community resources.2
The VHA encourages physical activity and wellness through sports and leisure. Veterans with MS can participate in sports programs and special events, including the National Veterans Wheelchair Games, the National Disabled Veterans Winter Sports Clinic, the National Disabled Veterans TEE (Training, Exposure and Experience) golf tournament, the National Veterans Summer Sports Clinic, the National Veterans Golden Age Games, and the National Veterans Creative Sports Festival. HCPs or veterans who are not sure how to access any of these programs can contact the MSCoE or their local VA social workers.
Research
The primary goal of the MSCoE research core is to conduct clinical, health services, epidemiologic, and basic science research relevant to veterans with MS. The MSCoE serves to enhance collaboration among VAMCs, increase the participation of veterans in research, and provide research mentorship for the next generation of VA MS scientists. MSCoE research is carried out by investigators at the MSCoE and the MS Regional Programs, often in collaboration with investigators at academic institutions. This research is supported by competitive grant awards from a variety of funding agencies including the VA Research and Development Service (R&D) and the NMSS. Results from about 40 research grants in Fiscal Year 2019 were disseminated through 34 peer-reviewed publications, 30 posters, presentations, abstracts, and clinical practice guidelines.
There are many examples of recent high impact MS research performed by MSCoE investigators. For example, MSCoE researchers noted an increase in the estimated prevalence of MS to 1 million individuals in the US, about twice the previously estimated prevalence.3-5 In addition, a multicenter study highlighted the prevalence of MS misdiagnosis and common confounders for MS.6 Other research includes pilot clinical trials evaluating lipoic acid as a potential disease modifying therapy in people with secondary progressive MS and the impact of a multicomponent walking aid selection, fitting, and training program for preventing falls in people with MS.7,8 Clinical trial also are investigating telehealth counseling to improve physical activity in MS and a systematic review of rehabilitation interventions in MS.9,10
Education and Training
A unified program of education is essential to effective management of MS nationally. The primary goal of the education and training core is to provide a national program of MS education for HCPs, veterans, and caregivers to improve knowledge, enhance access to resources, and promote effective management strategies. The MSCoE collaborate with the Paralyzed Veterans of America (PVA), the Consortium of MS Centers (CMSC), the NMSS, and other national service organizations to increase educational opportunities, share knowledge, and expand participation.
The MSCoE education and training core produces a range of products both veterans, HCPs, and others affected by MS. The MSCoE sends a biannual patient newsletter to > 20,000 veterans and a monthly email to > 1,000 VA HCPs. Specific opportunities for HCP education include accredited multidisciplinary MS webinars, sponsored symposia and workshops at the CMSC and PVA Summit annual meetings, and presentations at other university and professional conferences. Enduring educational opportunities for veterans, caregivers, and HCPs can also be found by visiting www.va.gov/ms.
The MSCoE coordinate postdoctoral fellowship training programs to develop expertise in MS health care for the future. It offers VA physician fellowships for neurologists in Baltimore and Portland and for physiatrists in Seattle as well as NMSS fellowships for education and research. In 2019, MSCoE had 6 MD Fellows and 1 PhD Fellow.
Clinical Informatics and Telehealth
The primary goal of the informatics and telemedicine core is to employ state-of-the-art informatics, telemedicine technology, and the MSCoE website, to improve MS health care delivery. The VA has a integrated electronic health record and various data repositories are stored in the VHA Corporate Data Warehouse (CDW). MSCoE utilizes the CDW to maintain a national MS administrative data repository to understand the VHA care provided to veterans with MS. Data from the CDW have also served as an important resource to facilitate a wide range of veteran-focused MS research. This research has addressed clinical conditions like pain and obesity; health behaviors like smoking, alcohol use, and exercise as well as issues related to care delivery such as specialty care access, medication adherence, and appointment attendance.11-19
Monitoring the health of veterans with MS in the VA requires additional data not available in the CDW. To this end, we have developed the MS Surveillance Registry (MSSR), funded and maintained by the VA Office of Information Technology as part of their Veteran Integrated Registry Platform (VIRP). The purpose of the MSSR is to understand the unique characteristics and treatment patterns of veterans with MS in order to optimize their VHA care. HCPs input MS-specific clinical data on their patients into the MSSR, either through the MS Assessment Tool (MSAT) in the Computerized Patient Record System (CPRS) or through a secure online portal. Other data from existing databases from the CDW is also automatically fed into the MSSR. The MSSR continues to be developed and populated to serve as a resource for the future.
Neurologists, physiatrists, psychologists, and rehabilitation specialists can use telehealth to evaluate and treat veterans who have difficulty accessing outpatient clinics, either because of mobility limitations, or distance. Between 2012 and 2015, the VA MSCoE, together with the Epilepsy CoE and the Parkinson’s Disease Research and Clinical Centers in VISNs 5, 6 (mid-Atlantic) and 20 (Pacific Northwest) initiated an integrated teleneurology project. The goal of this project was to improve patient access to care at 4 tertiary and 12 regional VAMCs. A study team, with administrators and key clinical stakeholders, followed a traditional project management approach to design, plan, implement and evaluate an optimal model for communication and referrals with both live visits and telehealth (Table). Major outcomes of the project included: delivering subspecialty teleneurology to 47 patient sites, increasing interfacility consultation by 133% while reducing wait times by roughly 40%, and increasing telemedicine workload at these centers from 95 annual encounters in 2012 to 1,245 annual encounters in 2015 (Figure).
Today, telehealth for veterans with MS can be delivered to nearby VA facilities closer to their home, within their home, or anywhere else the veteran can use a cellphone or tablet. Telehealth visits can save travel time and expenses and optimize VA productivity and clinic use. The MSCoE and many of the MS regional programs are using telehealth for MS physician follow-up and therapies. The VA Office of Rural Health is also currently working with the MS network to use telehealth to increase access to physical therapy to those who have difficulty coming into clinic.
MSCoE Resources
The MSCoE is funded by VA Central Office through the Office of Specialty Care by Special Purpose funds. The directive specifies that funding for the regional and support programs is through Veterans Equitable Resource Allocation based on VISN and facility workload and complexity. Any research is funded separately through grants, some from VA R&D and others from other sources including the National Institutes of Health, the Patient Centered Outcome Research Institute, affiliated universities, the NMSS, the MS Society of Canada, the Consortium of MS Centers, foundations, and industry.
In 2019, MSCoE investigators received grants totaling > $18 million in funding. In-kind support also is provided by the PVA, the CMSC, the NMSS, and others. The first 3 foundations have been supporters since the inception of the MSCoE and have provided opportunities for the dissemination of education and research for HCPs, fellows, residents and medical students; travel; meeting rooms for MSCoE national meetings; exhibit space for HCP outreach; competitive research and educational grant support; programming and resources for veterans and significant others; organizational expertise; and opportunities for VA HCPs, veterans, and caregivers to learn how to navigate MS with others in the private sector.
Conclusion
The MSCoE had a tremendous impact on improving the consistency and quality of care for veterans with MS through clinical care, research, education and informatics and telehealth. Since opening in 2003, there has been an increase in the number of MS specialty clinics, served veterans with MS, and veterans receiving specialty neurologic and rehabilitation services in VA. Research programs in MS have been initiated to address key questions relevant to veterans with MS, including immunology, epidemiology, clinical care, and rehabilitation. Educational programs and products have evolved with technology and had a greater impact through partnerships with veteran and MS nonprofit organizations.
MSCoE strives to minimize impairment and maximize quality of life for veterans with MS by leveraging integrated electronic health records, data repositories, and telehealth services. These efforts have all improved veteran health, access and safety. We look forward to continuing into the next decade by bringing fresh ideas to the care of veterans with MS, their families and caregivers.
1. US Department of Veterans Affairs, Multiple Sclerosis Centers of Excellence. Multiple Sclerosis System of Care-VHA Directive 1101.06 and Multiple Sclerosis Centers of Excellence network facilities. https://www.va.gov/MS/veterans/find_a_clinic/index_clinics.asp. Updated February 26, 2020. Accessed March 6, 2020.
2. National MS Society. MS navigator program. https://www.nationalmssociety.org/For-Professionals/Clinical-Care/MS-Navigator-Program. Accessed March 6, 2020.
3. Wallin MT, Culpepper WJ, Campbell JD, et al. The prevalence of MS in the United States: a population-based estimate using health claims data. Neurology. 2019;92:e1029-e1040.
4. GBD 2016 Multiple Sclerosis Collaborators. Global, regional, and national burden of multiple sclerosis 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(3):269-285.
5. GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(5):459-480.
6. Solomon AJ, Bourdette DN, Cross AH, et al. The contemporary spectrum of multiple sclerosis misdiagnosis: a multicenter study. Neurology. 2016;87(13):1393-1399.
7. Spain R, Powers K, Murchison C, et al. Lipoic acid in secondary progressive MS: a randomized controlled pilot trial. Neurol Neuroimmunol Neuroinflamm. 2017;4(5):e374.
8. Martini DN, Zeeboer E, Hildebrand A, Fling BW, Hugos CL, Cameron MH. ADSTEP: preliminary investigation of a multicomponent walking aid program in people with multiple sclerosis. Arch Phys Med Rehabil. 2018;99(10):2050-2058.
9. Turner AP, Hartoonian N, Sloan AP, et al. Improving fatigue and depression in individuals with multiple sclerosis using telephone-administered physical activity counseling. J Consult Clin Psychol. 2016;84(4):297-309.
10. Haselkorn JK, Hughes C, Rae-Grant A, et al. Summary of comprehensive systematic review: rehabilitation in multiple sclerosis: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology. 2015;85(21):1896-1903.
11. Hirsh AT, Turner AP, Ehde DM, Haselkorn JK. Prevalence and impact of pain in multiple sclerosis: physical and psychologic contributors. Arch Phys Med Rehabil. 2009;90(4):646-651.
12. Khurana SR, Bamer AM, Turner AP, et al. The prevalence of overweight and obesity in veterans with multiple sclerosis. Am J Phys Med Rehabil. 2009;88(2):83-91.
13. Turner AP, Kivlahan DR, Kazis LE, Haselkorn JK. Smoking among veterans with multiple sclerosis: prevalence correlates, quit attempts, and unmet need for services. Arch Phys Med Rehabil. 2007;88(11):1394-1399.
14. Turner AP, Hawkins EJ, Haselkorn JK, Kivlahan DR. Alcohol misuse and multiple sclerosis. Arch Phys Med Rehabil. 2009;90(5):842-848.
15. Turner AP, Kivlahan DR, Haselkorn JK. Exercise and quality of life among people with multiple sclerosis: looking beyond physical functioning to mental health and participation in life. Arch Phys Med Rehabil. 2009;90(3):420-428.
16. Turner AP, Chapko MK, Yanez D, et al. Access to multiple sclerosis specialty care. PM R. 2013;5(12):1044-1050.
17. Gromisch ES, Turner AP, Leipertz SL, Beauvais J, Haselkorn JK. Risk factors for suboptimal medication adherence in persons with multiple sclerosis: development of an electronic health record-based explanatory model for disease-modifying therapy use [published online ahead of print, 2019 Dec 3]. Arch Phys Med Rehabil. 2019;S0003-9993(19)31430-3143.
18. Settle JR, Maloni H, Bedra M, Finkelstein J, Zhan M, Wallin M. Monitoring medication adherence in multiple sclerosis using a novel web-based tool. J Telemed Telecare. 2016;22:225-233.
19. Gromisch ES, Turner AP, Leipertz SL, Beauvais J, Haselkorn JK. Who is not coming to clinic? A predictive model of excessive missed appointments in persons with multiple sclerosis. Mult Scler Rel Dis. In Press.
1. US Department of Veterans Affairs, Multiple Sclerosis Centers of Excellence. Multiple Sclerosis System of Care-VHA Directive 1101.06 and Multiple Sclerosis Centers of Excellence network facilities. https://www.va.gov/MS/veterans/find_a_clinic/index_clinics.asp. Updated February 26, 2020. Accessed March 6, 2020.
2. National MS Society. MS navigator program. https://www.nationalmssociety.org/For-Professionals/Clinical-Care/MS-Navigator-Program. Accessed March 6, 2020.
3. Wallin MT, Culpepper WJ, Campbell JD, et al. The prevalence of MS in the United States: a population-based estimate using health claims data. Neurology. 2019;92:e1029-e1040.
4. GBD 2016 Multiple Sclerosis Collaborators. Global, regional, and national burden of multiple sclerosis 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(3):269-285.
5. GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(5):459-480.
6. Solomon AJ, Bourdette DN, Cross AH, et al. The contemporary spectrum of multiple sclerosis misdiagnosis: a multicenter study. Neurology. 2016;87(13):1393-1399.
7. Spain R, Powers K, Murchison C, et al. Lipoic acid in secondary progressive MS: a randomized controlled pilot trial. Neurol Neuroimmunol Neuroinflamm. 2017;4(5):e374.
8. Martini DN, Zeeboer E, Hildebrand A, Fling BW, Hugos CL, Cameron MH. ADSTEP: preliminary investigation of a multicomponent walking aid program in people with multiple sclerosis. Arch Phys Med Rehabil. 2018;99(10):2050-2058.
9. Turner AP, Hartoonian N, Sloan AP, et al. Improving fatigue and depression in individuals with multiple sclerosis using telephone-administered physical activity counseling. J Consult Clin Psychol. 2016;84(4):297-309.
10. Haselkorn JK, Hughes C, Rae-Grant A, et al. Summary of comprehensive systematic review: rehabilitation in multiple sclerosis: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology. 2015;85(21):1896-1903.
11. Hirsh AT, Turner AP, Ehde DM, Haselkorn JK. Prevalence and impact of pain in multiple sclerosis: physical and psychologic contributors. Arch Phys Med Rehabil. 2009;90(4):646-651.
12. Khurana SR, Bamer AM, Turner AP, et al. The prevalence of overweight and obesity in veterans with multiple sclerosis. Am J Phys Med Rehabil. 2009;88(2):83-91.
13. Turner AP, Kivlahan DR, Kazis LE, Haselkorn JK. Smoking among veterans with multiple sclerosis: prevalence correlates, quit attempts, and unmet need for services. Arch Phys Med Rehabil. 2007;88(11):1394-1399.
14. Turner AP, Hawkins EJ, Haselkorn JK, Kivlahan DR. Alcohol misuse and multiple sclerosis. Arch Phys Med Rehabil. 2009;90(5):842-848.
15. Turner AP, Kivlahan DR, Haselkorn JK. Exercise and quality of life among people with multiple sclerosis: looking beyond physical functioning to mental health and participation in life. Arch Phys Med Rehabil. 2009;90(3):420-428.
16. Turner AP, Chapko MK, Yanez D, et al. Access to multiple sclerosis specialty care. PM R. 2013;5(12):1044-1050.
17. Gromisch ES, Turner AP, Leipertz SL, Beauvais J, Haselkorn JK. Risk factors for suboptimal medication adherence in persons with multiple sclerosis: development of an electronic health record-based explanatory model for disease-modifying therapy use [published online ahead of print, 2019 Dec 3]. Arch Phys Med Rehabil. 2019;S0003-9993(19)31430-3143.
18. Settle JR, Maloni H, Bedra M, Finkelstein J, Zhan M, Wallin M. Monitoring medication adherence in multiple sclerosis using a novel web-based tool. J Telemed Telecare. 2016;22:225-233.
19. Gromisch ES, Turner AP, Leipertz SL, Beauvais J, Haselkorn JK. Who is not coming to clinic? A predictive model of excessive missed appointments in persons with multiple sclerosis. Mult Scler Rel Dis. In Press.
Senate confirms Murthy as Surgeon General

Seven Republicans – Bill Cassidy (La.), Susan Collins (Maine), Roger Marshall (Kan.), Susan Murkowski (Alaska), Rob Portman (Ohio), Mitt Romney (Utah), and Dan Sullivan (Alaska) – joined all the Democrats and independents in the 57-43 vote approving Dr. Murthy’s nomination.
Dr. Murthy, 43, previously served as the 19th Surgeon General, from December 2014 to April 2017, when he was asked to step down by President Donald J. Trump.
Surgeons General serve 4-year terms.
During his first tenure, Dr. Murthy issued the first-ever Surgeon General’s report on the crisis of addiction and issued a call to action to doctors to help battle the opioid crisis.
When Dr. Murthy was nominated by President-elect Joseph R. Biden Jr. in December, he was acting as cochair of the incoming administration’s COVID-19 transition advisory board.
Early in 2020, before the COVID-19 pandemic hit, Dr. Murthy published a timely book: “Together: The Healing Power of Human Connection in a Sometimes Lonely World”.
He earned his bachelor’s degree from Harvard and his MD and MBA degrees from Yale. He completed his internal medicine residency at Brigham and Women’s Hospital in Boston, where he also served as a hospitalist, and later joined Harvard Medical School as a faculty member in internal medicine.
He is married to Alice Chen, MD. The couple have two children.
A version of this article first appeared on WebMD.com.
Seven Republicans – Bill Cassidy (La.), Susan Collins (Maine), Roger Marshall (Kan.), Susan Murkowski (Alaska), Rob Portman (Ohio), Mitt Romney (Utah), and Dan Sullivan (Alaska) – joined all the Democrats and independents in the 57-43 vote approving Dr. Murthy’s nomination.
Dr. Murthy, 43, previously served as the 19th Surgeon General, from December 2014 to April 2017, when he was asked to step down by President Donald J. Trump.
Surgeons General serve 4-year terms.
During his first tenure, Dr. Murthy issued the first-ever Surgeon General’s report on the crisis of addiction and issued a call to action to doctors to help battle the opioid crisis.
When Dr. Murthy was nominated by President-elect Joseph R. Biden Jr. in December, he was acting as cochair of the incoming administration’s COVID-19 transition advisory board.
Early in 2020, before the COVID-19 pandemic hit, Dr. Murthy published a timely book: “Together: The Healing Power of Human Connection in a Sometimes Lonely World”.
He earned his bachelor’s degree from Harvard and his MD and MBA degrees from Yale. He completed his internal medicine residency at Brigham and Women’s Hospital in Boston, where he also served as a hospitalist, and later joined Harvard Medical School as a faculty member in internal medicine.
He is married to Alice Chen, MD. The couple have two children.
A version of this article first appeared on WebMD.com.
Seven Republicans – Bill Cassidy (La.), Susan Collins (Maine), Roger Marshall (Kan.), Susan Murkowski (Alaska), Rob Portman (Ohio), Mitt Romney (Utah), and Dan Sullivan (Alaska) – joined all the Democrats and independents in the 57-43 vote approving Dr. Murthy’s nomination.
Dr. Murthy, 43, previously served as the 19th Surgeon General, from December 2014 to April 2017, when he was asked to step down by President Donald J. Trump.
Surgeons General serve 4-year terms.
During his first tenure, Dr. Murthy issued the first-ever Surgeon General’s report on the crisis of addiction and issued a call to action to doctors to help battle the opioid crisis.
When Dr. Murthy was nominated by President-elect Joseph R. Biden Jr. in December, he was acting as cochair of the incoming administration’s COVID-19 transition advisory board.
Early in 2020, before the COVID-19 pandemic hit, Dr. Murthy published a timely book: “Together: The Healing Power of Human Connection in a Sometimes Lonely World”.
He earned his bachelor’s degree from Harvard and his MD and MBA degrees from Yale. He completed his internal medicine residency at Brigham and Women’s Hospital in Boston, where he also served as a hospitalist, and later joined Harvard Medical School as a faculty member in internal medicine.
He is married to Alice Chen, MD. The couple have two children.
A version of this article first appeared on WebMD.com.

FDA warning letters target OTC cannabidiol product claims for pain relief
The Food and Drug Administration has warned two manufacturers about illegal marketing of drugs containing cannabidiol (CBD) for over-the-counter use without an approved new drug application, for using substandard manufacturing processes, and for failure to comply with current good manufacturing practices. These warnings add to 51 previous warning letters issued by the FDA since 2015 to other manufacturers of products containing CBD who were violating the Federal Food, Drug, and Cosmetic Act.

In a news release, the agency explained that its two most recent letters, sent to Honest Globe Inc. on March 15 and BioLyte Laboratories LLC on March 18, were issued because CBD has “known pharmacologic effects on humans, with demonstrated risks, it cannot be legally marketed as an inactive ingredient in OTC drug products that are not reviewed and approved by the FDA.” They also describe the companies’ failures to comply with current good manufacturing practices.
“The FDA continues to alert the public to potential safety and efficacy concerns with unapproved CBD products sold online and in stores across the country,” FDA Principal Deputy Commissioner Amy P. Abernethy, MD, PhD, said in the release. “It’s important that consumers understand that the FDA has only approved one drug containing CBD as an ingredient [Epidiolex]. These other, unapproved, CBD products may have dangerous health impacts and side effects. We remain focused on exploring potential pathways for CBD products to be lawfully marketed while also educating the public about these outstanding questions of CBD’s safety. Meanwhile, we will continue to monitor and take action, as needed, against companies that unlawfully market their products – prioritizing those that pose a risk to public health.”
The specific products from Santa Ana, Calif.–based Honest Globe that the FDA called unapproved new drugs and misbranded under the Federal Food, Drug, and Cosmetic Act included Elixicure Original Pain Relief and Elixicure Lavender Pain Relief, both of which were described as containing CBD. Products from Grand Rapids, Mich.–based BioLyte Laboratories LLC that the FDA similarly cited for violations included Silver Gel, Silver Gel with Aloe, Silver Liquid Supplement, Therapeutic Pain Gel, Pain Relief Cream, and Magnesium Oil Spray.
The agency has asked the two companies to respond to its letters within 15 working days, “stating how they will address these violations or providing their reasoning and supporting information as to why they believe these products are not in violation of the law. Failure to adequately address the violations promptly may result in legal action, including product seizure and/or injunction.”
The Food and Drug Administration has warned two manufacturers about illegal marketing of drugs containing cannabidiol (CBD) for over-the-counter use without an approved new drug application, for using substandard manufacturing processes, and for failure to comply with current good manufacturing practices. These warnings add to 51 previous warning letters issued by the FDA since 2015 to other manufacturers of products containing CBD who were violating the Federal Food, Drug, and Cosmetic Act.
In a news release, the agency explained that its two most recent letters, sent to Honest Globe Inc. on March 15 and BioLyte Laboratories LLC on March 18, were issued because CBD has “known pharmacologic effects on humans, with demonstrated risks, it cannot be legally marketed as an inactive ingredient in OTC drug products that are not reviewed and approved by the FDA.” They also describe the companies’ failures to comply with current good manufacturing practices.
“The FDA continues to alert the public to potential safety and efficacy concerns with unapproved CBD products sold online and in stores across the country,” FDA Principal Deputy Commissioner Amy P. Abernethy, MD, PhD, said in the release. “It’s important that consumers understand that the FDA has only approved one drug containing CBD as an ingredient [Epidiolex]. These other, unapproved, CBD products may have dangerous health impacts and side effects. We remain focused on exploring potential pathways for CBD products to be lawfully marketed while also educating the public about these outstanding questions of CBD’s safety. Meanwhile, we will continue to monitor and take action, as needed, against companies that unlawfully market their products – prioritizing those that pose a risk to public health.”
The specific products from Santa Ana, Calif.–based Honest Globe that the FDA called unapproved new drugs and misbranded under the Federal Food, Drug, and Cosmetic Act included Elixicure Original Pain Relief and Elixicure Lavender Pain Relief, both of which were described as containing CBD. Products from Grand Rapids, Mich.–based BioLyte Laboratories LLC that the FDA similarly cited for violations included Silver Gel, Silver Gel with Aloe, Silver Liquid Supplement, Therapeutic Pain Gel, Pain Relief Cream, and Magnesium Oil Spray.
The agency has asked the two companies to respond to its letters within 15 working days, “stating how they will address these violations or providing their reasoning and supporting information as to why they believe these products are not in violation of the law. Failure to adequately address the violations promptly may result in legal action, including product seizure and/or injunction.”
The Food and Drug Administration has warned two manufacturers about illegal marketing of drugs containing cannabidiol (CBD) for over-the-counter use without an approved new drug application, for using substandard manufacturing processes, and for failure to comply with current good manufacturing practices. These warnings add to 51 previous warning letters issued by the FDA since 2015 to other manufacturers of products containing CBD who were violating the Federal Food, Drug, and Cosmetic Act.
In a news release, the agency explained that its two most recent letters, sent to Honest Globe Inc. on March 15 and BioLyte Laboratories LLC on March 18, were issued because CBD has “known pharmacologic effects on humans, with demonstrated risks, it cannot be legally marketed as an inactive ingredient in OTC drug products that are not reviewed and approved by the FDA.” They also describe the companies’ failures to comply with current good manufacturing practices.
“The FDA continues to alert the public to potential safety and efficacy concerns with unapproved CBD products sold online and in stores across the country,” FDA Principal Deputy Commissioner Amy P. Abernethy, MD, PhD, said in the release. “It’s important that consumers understand that the FDA has only approved one drug containing CBD as an ingredient [Epidiolex]. These other, unapproved, CBD products may have dangerous health impacts and side effects. We remain focused on exploring potential pathways for CBD products to be lawfully marketed while also educating the public about these outstanding questions of CBD’s safety. Meanwhile, we will continue to monitor and take action, as needed, against companies that unlawfully market their products – prioritizing those that pose a risk to public health.”
The specific products from Santa Ana, Calif.–based Honest Globe that the FDA called unapproved new drugs and misbranded under the Federal Food, Drug, and Cosmetic Act included Elixicure Original Pain Relief and Elixicure Lavender Pain Relief, both of which were described as containing CBD. Products from Grand Rapids, Mich.–based BioLyte Laboratories LLC that the FDA similarly cited for violations included Silver Gel, Silver Gel with Aloe, Silver Liquid Supplement, Therapeutic Pain Gel, Pain Relief Cream, and Magnesium Oil Spray.
The agency has asked the two companies to respond to its letters within 15 working days, “stating how they will address these violations or providing their reasoning and supporting information as to why they believe these products are not in violation of the law. Failure to adequately address the violations promptly may result in legal action, including product seizure and/or injunction.”
Doctors Found Jet Fuel in Veteran’s Lungs. He Can’t Get Full Benefits.
The lungs Bill Thompson was born with told a gruesome, harrowing and unmistakable tale to Dr. Anthony Szema when he analyzed them and found the black spots, scarring, partially combusted jet fuel and metal inside.
The retired Army staff sergeant had suffered catastrophic lung damage from breathing incinerated waste burned in massive open-air pits and probably other irritants during his tour of duty in Iraq.
“There’s black spots that are burns, particles all over; there’s metal. It was all scarred,” said Szema, a pulmonologist and professor who studies toxic exposures and examined Thompson’s preserved lung tissue. “There was no gas exchange anywhere in that lung.”
Thompson is still alive, surviving on his second transplanted set of lungs. Yet the story burned into the veteran’s internal organs is not one that has been entirely convincing to the U.S. government.
The military has not linked the burn pits to illness. That means many who were exposed to burn pits and are sick do not qualify for benefits under any existing program.
Retirement and health benefits for members of the military depend on factors like length of service, active or reserve status, deployments to combat zones and whether the military considers specific injuries or illnesses to be service-related. Thompson has been able to get care through the Department of Veterans Affairs for his lung disease but has not been able to secure other benefits, like early retirement pay.
“I was denied my Army retirement because if it was not a combat action, then I don’t receive that retirement,” Thompson said at a Senate Veterans’ Affairs Committee hearing last week on service members’ exposures to toxic substances.
Thompson is one of at least 3.5 million veterans since 2001 who have served in war zones where the U.S. military decided to dispose of its trash by burning it, according to VA estimates.
It’s not clear how many people within that population have gotten sick from exposure. Only a small fraction — 234,000 — have enrolled in the VA’s online burn pit registry. Veterans’ advocacy groups have said the majority of claims to the agency stemming from toxic exposures are denied, even as most former service members report contacts with toxins in their deployments.
Soldiers returning from tours in the global war on terror have reported debilitating illnesses almost from its beginning, but got little traction with the military. This year, though, the likelihood of congressional action is high, with Democrats expressing interest and a president who suspects burn pits are to blame for his son’s death.
President Joe Biden’s son Beau died of brain cancer in 2015 at age 46. He had deployed to Iraq in two sites with burn pits — at Baghdad and Balad — around the same time Thompson was at Camp Striker, near the Baghdad airport.
“Because of exposure to burn pits — in my view, I can’t prove it yet — he came back with stage 4 glioblastoma,” Biden said in a 2019 speech.
In testimony at the March 10 hearing, Shane Liermann, who works for the group Disabled American Veterans, told the committee that 78% of burn pit claims are denied. “Part of the problem is VA is not recognizing that exposure as being toxic exposures,” Liermann said.
Aleks Morosky, with the Wounded Warrior Project, said that in his group’s survey of 28,000 veterans last year, 71% said they had “definitely” been exposed to toxic substances or hazardous chemicals, and 18% said they had “probably” been exposed. Half of those people rated their health as poor or fair. Only about 16% of the service members who believed they had suffered exposure said they got treatment from the VA, and 11% said they were denied treatment.
Thompson, who is 49, said care for his lung disease is often slow and sometimes denied. It took the VA three years to approve an air purifier for his home to filter out allergens, and the VA refused to help pay for the removal of dust-trapping carpets, he said.
Thompson’s presence at the hearing, though, was not just meant to put the spotlight on the VA. The military’s entire approach to toxic exposure is a morass that leaves ill soldiers and veterans like Thompson trying to navigate a bureaucracy more labyrinthine than the Pentagon’s corridors.
After Thompson was shipped back to Fort Stewart in Georgia, his medical ordeal was at first addressed within the military system, including a year at Walter Reed National Military Medical Center in Bethesda, Maryland, where doctors found his lungs filled with titanium, magnesium, iron and silica.
Yet he said he didn’t qualify for the Army’s traumatic-injury insurance program, which might have helped him pay to retrofit his home in West Virginia. And he can’t get his military retirement pay until he’s 60.
“I may not live to be age 60. I turn 50 this year,” Thompson said.
Illustrating the problem, several officials at the hearing with the Department of Defense, the Army and the National Guard were unable to explain why Thompson — with 23 years of service between the Guard and Army — might have such a hard time qualifying for retirement benefits when the evidence of his lungs and the findings of the Army’s own doctors are so vivid and extreme.
For advocates who have been working on the problem for decades, it reminds them all too vividly of Agent Orange, which the military is still coming to grips with.
“It’s already been, since the first Persian Gulf [War] — we’re talking 30 years — and since burn pits were again active, since 2001,” said Liermann. “We’re way behind the curve here.”
Although Congress has done relatively little to deal with burn pits, many members seem to at least be thinking along the same lines. The Senate Veterans’ Affairs hearing promised to be something of a kickoff to a year when lawmakers are poised to offer a slew of bills designed to confront the military’s inability to care for service members poisoned during their deployments.
“Make no mistake about it,” said the committee chairman, Sen. Jon Tester (D-Mont.). “We hold these hearings for two reasons: to gather information for the committee members and to help educate the VA that they might take action before Congress does.”
Republicans have also shown growing interest in the problem, offering targeted bills to ensure a handful of toxin-related diseases are covered by the VA.
At the hearing, conservative freshman Sen. Tommy Tuberville (R-Ala.) seemed especially moved.
“We got to do a better job of taking care of our young people,” Tuberville said. “If we’re going to go to war, we got to understand we got to pay the price for it on both ends.”
There is also likely to be high-profile support and attention when revised legislation starts rolling out this spring.
The broadest bill likely to be offered was first introduced by Sen. Kirsten Gillibrand (D-N.Y.) in the Senate and Rep. Raul Ruiz (D-Calif.) in the House in late 2019, with a boost from former “Daily Show” host Jon Stewart and a cadre of 9/11 responders who are turning their attention to toxic exposures.
Indeed, Ruiz and Gillibrand’s legislation is modeled in part on the 9/11 health act that passed in 2015. The burn pit bill would remove the burden of proving a service-related connection.
It would vastly simplify the lives of people like Thompson.
“I am a warrior of the United States of America. I gave my lungs for my country,” Thompson said.
He was cut off before he could finish, but his prepared remarks concluded, “Hopefully, after hearing my story, it will bring awareness for not only me but others who are battling the same or similar injuries related to burn pit exposures from Iraq or Afghanistan.”
KHN (Kaiser Health News) is a national newsroom that produces in-depth journalism about health issues. Together with Policy Analysis and Polling, KHN is one of the three major operating programs at KFF (Kaiser Family Foundation). KFF is an endowed nonprofit organization providing information on health issues to the nation.
USE OUR CONTENT
This story can be republished for free (details).
Subscribe to KHN's free Morning Briefing.
The lungs Bill Thompson was born with told a gruesome, harrowing and unmistakable tale to Dr. Anthony Szema when he analyzed them and found the black spots, scarring, partially combusted jet fuel and metal inside.
The retired Army staff sergeant had suffered catastrophic lung damage from breathing incinerated waste burned in massive open-air pits and probably other irritants during his tour of duty in Iraq.
“There’s black spots that are burns, particles all over; there’s metal. It was all scarred,” said Szema, a pulmonologist and professor who studies toxic exposures and examined Thompson’s preserved lung tissue. “There was no gas exchange anywhere in that lung.”
Thompson is still alive, surviving on his second transplanted set of lungs. Yet the story burned into the veteran’s internal organs is not one that has been entirely convincing to the U.S. government.
The military has not linked the burn pits to illness. That means many who were exposed to burn pits and are sick do not qualify for benefits under any existing program.
Retirement and health benefits for members of the military depend on factors like length of service, active or reserve status, deployments to combat zones and whether the military considers specific injuries or illnesses to be service-related. Thompson has been able to get care through the Department of Veterans Affairs for his lung disease but has not been able to secure other benefits, like early retirement pay.
“I was denied my Army retirement because if it was not a combat action, then I don’t receive that retirement,” Thompson said at a Senate Veterans’ Affairs Committee hearing last week on service members’ exposures to toxic substances.
Thompson is one of at least 3.5 million veterans since 2001 who have served in war zones where the U.S. military decided to dispose of its trash by burning it, according to VA estimates.
It’s not clear how many people within that population have gotten sick from exposure. Only a small fraction — 234,000 — have enrolled in the VA’s online burn pit registry. Veterans’ advocacy groups have said the majority of claims to the agency stemming from toxic exposures are denied, even as most former service members report contacts with toxins in their deployments.
Soldiers returning from tours in the global war on terror have reported debilitating illnesses almost from its beginning, but got little traction with the military. This year, though, the likelihood of congressional action is high, with Democrats expressing interest and a president who suspects burn pits are to blame for his son’s death.
President Joe Biden’s son Beau died of brain cancer in 2015 at age 46. He had deployed to Iraq in two sites with burn pits — at Baghdad and Balad — around the same time Thompson was at Camp Striker, near the Baghdad airport.
“Because of exposure to burn pits — in my view, I can’t prove it yet — he came back with stage 4 glioblastoma,” Biden said in a 2019 speech.
In testimony at the March 10 hearing, Shane Liermann, who works for the group Disabled American Veterans, told the committee that 78% of burn pit claims are denied. “Part of the problem is VA is not recognizing that exposure as being toxic exposures,” Liermann said.
Aleks Morosky, with the Wounded Warrior Project, said that in his group’s survey of 28,000 veterans last year, 71% said they had “definitely” been exposed to toxic substances or hazardous chemicals, and 18% said they had “probably” been exposed. Half of those people rated their health as poor or fair. Only about 16% of the service members who believed they had suffered exposure said they got treatment from the VA, and 11% said they were denied treatment.
Thompson, who is 49, said care for his lung disease is often slow and sometimes denied. It took the VA three years to approve an air purifier for his home to filter out allergens, and the VA refused to help pay for the removal of dust-trapping carpets, he said.
Thompson’s presence at the hearing, though, was not just meant to put the spotlight on the VA. The military’s entire approach to toxic exposure is a morass that leaves ill soldiers and veterans like Thompson trying to navigate a bureaucracy more labyrinthine than the Pentagon’s corridors.
After Thompson was shipped back to Fort Stewart in Georgia, his medical ordeal was at first addressed within the military system, including a year at Walter Reed National Military Medical Center in Bethesda, Maryland, where doctors found his lungs filled with titanium, magnesium, iron and silica.
Yet he said he didn’t qualify for the Army’s traumatic-injury insurance program, which might have helped him pay to retrofit his home in West Virginia. And he can’t get his military retirement pay until he’s 60.
“I may not live to be age 60. I turn 50 this year,” Thompson said.
Illustrating the problem, several officials at the hearing with the Department of Defense, the Army and the National Guard were unable to explain why Thompson — with 23 years of service between the Guard and Army — might have such a hard time qualifying for retirement benefits when the evidence of his lungs and the findings of the Army’s own doctors are so vivid and extreme.
For advocates who have been working on the problem for decades, it reminds them all too vividly of Agent Orange, which the military is still coming to grips with.
“It’s already been, since the first Persian Gulf [War] — we’re talking 30 years — and since burn pits were again active, since 2001,” said Liermann. “We’re way behind the curve here.”
Although Congress has done relatively little to deal with burn pits, many members seem to at least be thinking along the same lines. The Senate Veterans’ Affairs hearing promised to be something of a kickoff to a year when lawmakers are poised to offer a slew of bills designed to confront the military’s inability to care for service members poisoned during their deployments.
“Make no mistake about it,” said the committee chairman, Sen. Jon Tester (D-Mont.). “We hold these hearings for two reasons: to gather information for the committee members and to help educate the VA that they might take action before Congress does.”
Republicans have also shown growing interest in the problem, offering targeted bills to ensure a handful of toxin-related diseases are covered by the VA.
At the hearing, conservative freshman Sen. Tommy Tuberville (R-Ala.) seemed especially moved.
“We got to do a better job of taking care of our young people,” Tuberville said. “If we’re going to go to war, we got to understand we got to pay the price for it on both ends.”
There is also likely to be high-profile support and attention when revised legislation starts rolling out this spring.
The broadest bill likely to be offered was first introduced by Sen. Kirsten Gillibrand (D-N.Y.) in the Senate and Rep. Raul Ruiz (D-Calif.) in the House in late 2019, with a boost from former “Daily Show” host Jon Stewart and a cadre of 9/11 responders who are turning their attention to toxic exposures.
Indeed, Ruiz and Gillibrand’s legislation is modeled in part on the 9/11 health act that passed in 2015. The burn pit bill would remove the burden of proving a service-related connection.
It would vastly simplify the lives of people like Thompson.
“I am a warrior of the United States of America. I gave my lungs for my country,” Thompson said.
He was cut off before he could finish, but his prepared remarks concluded, “Hopefully, after hearing my story, it will bring awareness for not only me but others who are battling the same or similar injuries related to burn pit exposures from Iraq or Afghanistan.”
KHN (Kaiser Health News) is a national newsroom that produces in-depth journalism about health issues. Together with Policy Analysis and Polling, KHN is one of the three major operating programs at KFF (Kaiser Family Foundation). KFF is an endowed nonprofit organization providing information on health issues to the nation.
USE OUR CONTENT
This story can be republished for free (details).
Subscribe to KHN's free Morning Briefing.
The lungs Bill Thompson was born with told a gruesome, harrowing and unmistakable tale to Dr. Anthony Szema when he analyzed them and found the black spots, scarring, partially combusted jet fuel and metal inside.
The retired Army staff sergeant had suffered catastrophic lung damage from breathing incinerated waste burned in massive open-air pits and probably other irritants during his tour of duty in Iraq.
“There’s black spots that are burns, particles all over; there’s metal. It was all scarred,” said Szema, a pulmonologist and professor who studies toxic exposures and examined Thompson’s preserved lung tissue. “There was no gas exchange anywhere in that lung.”
Thompson is still alive, surviving on his second transplanted set of lungs. Yet the story burned into the veteran’s internal organs is not one that has been entirely convincing to the U.S. government.
The military has not linked the burn pits to illness. That means many who were exposed to burn pits and are sick do not qualify for benefits under any existing program.
Retirement and health benefits for members of the military depend on factors like length of service, active or reserve status, deployments to combat zones and whether the military considers specific injuries or illnesses to be service-related. Thompson has been able to get care through the Department of Veterans Affairs for his lung disease but has not been able to secure other benefits, like early retirement pay.
“I was denied my Army retirement because if it was not a combat action, then I don’t receive that retirement,” Thompson said at a Senate Veterans’ Affairs Committee hearing last week on service members’ exposures to toxic substances.
Thompson is one of at least 3.5 million veterans since 2001 who have served in war zones where the U.S. military decided to dispose of its trash by burning it, according to VA estimates.
It’s not clear how many people within that population have gotten sick from exposure. Only a small fraction — 234,000 — have enrolled in the VA’s online burn pit registry. Veterans’ advocacy groups have said the majority of claims to the agency stemming from toxic exposures are denied, even as most former service members report contacts with toxins in their deployments.
Soldiers returning from tours in the global war on terror have reported debilitating illnesses almost from its beginning, but got little traction with the military. This year, though, the likelihood of congressional action is high, with Democrats expressing interest and a president who suspects burn pits are to blame for his son’s death.
President Joe Biden’s son Beau died of brain cancer in 2015 at age 46. He had deployed to Iraq in two sites with burn pits — at Baghdad and Balad — around the same time Thompson was at Camp Striker, near the Baghdad airport.
“Because of exposure to burn pits — in my view, I can’t prove it yet — he came back with stage 4 glioblastoma,” Biden said in a 2019 speech.
In testimony at the March 10 hearing, Shane Liermann, who works for the group Disabled American Veterans, told the committee that 78% of burn pit claims are denied. “Part of the problem is VA is not recognizing that exposure as being toxic exposures,” Liermann said.
Aleks Morosky, with the Wounded Warrior Project, said that in his group’s survey of 28,000 veterans last year, 71% said they had “definitely” been exposed to toxic substances or hazardous chemicals, and 18% said they had “probably” been exposed. Half of those people rated their health as poor or fair. Only about 16% of the service members who believed they had suffered exposure said they got treatment from the VA, and 11% said they were denied treatment.
Thompson, who is 49, said care for his lung disease is often slow and sometimes denied. It took the VA three years to approve an air purifier for his home to filter out allergens, and the VA refused to help pay for the removal of dust-trapping carpets, he said.
Thompson’s presence at the hearing, though, was not just meant to put the spotlight on the VA. The military’s entire approach to toxic exposure is a morass that leaves ill soldiers and veterans like Thompson trying to navigate a bureaucracy more labyrinthine than the Pentagon’s corridors.
After Thompson was shipped back to Fort Stewart in Georgia, his medical ordeal was at first addressed within the military system, including a year at Walter Reed National Military Medical Center in Bethesda, Maryland, where doctors found his lungs filled with titanium, magnesium, iron and silica.
Yet he said he didn’t qualify for the Army’s traumatic-injury insurance program, which might have helped him pay to retrofit his home in West Virginia. And he can’t get his military retirement pay until he’s 60.
“I may not live to be age 60. I turn 50 this year,” Thompson said.
Illustrating the problem, several officials at the hearing with the Department of Defense, the Army and the National Guard were unable to explain why Thompson — with 23 years of service between the Guard and Army — might have such a hard time qualifying for retirement benefits when the evidence of his lungs and the findings of the Army’s own doctors are so vivid and extreme.
For advocates who have been working on the problem for decades, it reminds them all too vividly of Agent Orange, which the military is still coming to grips with.
“It’s already been, since the first Persian Gulf [War] — we’re talking 30 years — and since burn pits were again active, since 2001,” said Liermann. “We’re way behind the curve here.”
Although Congress has done relatively little to deal with burn pits, many members seem to at least be thinking along the same lines. The Senate Veterans’ Affairs hearing promised to be something of a kickoff to a year when lawmakers are poised to offer a slew of bills designed to confront the military’s inability to care for service members poisoned during their deployments.
“Make no mistake about it,” said the committee chairman, Sen. Jon Tester (D-Mont.). “We hold these hearings for two reasons: to gather information for the committee members and to help educate the VA that they might take action before Congress does.”
Republicans have also shown growing interest in the problem, offering targeted bills to ensure a handful of toxin-related diseases are covered by the VA.
At the hearing, conservative freshman Sen. Tommy Tuberville (R-Ala.) seemed especially moved.
“We got to do a better job of taking care of our young people,” Tuberville said. “If we’re going to go to war, we got to understand we got to pay the price for it on both ends.”
There is also likely to be high-profile support and attention when revised legislation starts rolling out this spring.
The broadest bill likely to be offered was first introduced by Sen. Kirsten Gillibrand (D-N.Y.) in the Senate and Rep. Raul Ruiz (D-Calif.) in the House in late 2019, with a boost from former “Daily Show” host Jon Stewart and a cadre of 9/11 responders who are turning their attention to toxic exposures.
Indeed, Ruiz and Gillibrand’s legislation is modeled in part on the 9/11 health act that passed in 2015. The burn pit bill would remove the burden of proving a service-related connection.
It would vastly simplify the lives of people like Thompson.
“I am a warrior of the United States of America. I gave my lungs for my country,” Thompson said.
He was cut off before he could finish, but his prepared remarks concluded, “Hopefully, after hearing my story, it will bring awareness for not only me but others who are battling the same or similar injuries related to burn pit exposures from Iraq or Afghanistan.”
KHN (Kaiser Health News) is a national newsroom that produces in-depth journalism about health issues. Together with Policy Analysis and Polling, KHN is one of the three major operating programs at KFF (Kaiser Family Foundation). KFF is an endowed nonprofit organization providing information on health issues to the nation.
USE OUR CONTENT
This story can be republished for free (details).
Subscribe to KHN's free Morning Briefing.
FDA scrutinizes cancer therapies granted accelerated approval
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
FDA authorizes first molecular at-home, OTC COVID-19 test
The U.S. Food and Drug Administration has granted emergency use authorization (EUA) for the Cue COVID-19 Test for Home and Over The Counter Use (Cue OTC Test, Cue Health).
The Cue OTC Test is the first molecular diagnostic test available to consumers without a prescription.
The test detects genetic material from SARS-CoV-2 present in the nostrils and delivers results in about 20 minutes to the user’s mobile smart device via the Cue Health app.
In testing, the Cue OTC Test correctly identified 96% of positive nasal swab samples from individuals known to have symptoms and correctly identified 100% of positive samples from individuals without symptoms.
The test is intended for use in people aged 2 years and older with and without symptoms.
“With this authorization, consumers can purchase and self-administer one of the easiest, fastest, and most accurate tests without a prescription,” Clint Sever, cofounder and chief product officer of Cue Health, said in a news release.
“This FDA authorization will help us improve patient outcomes with a solution that provides the accuracy of central lab tests, with the speed and accessibility required to address emergent global health issues,” he said.
Cue Health expects to produce more than 100,000 single-use test kits per day by this summer. Dena Cook, the company’s chief communications officer, told this news organization that the company hasn’t announced pricing information yet, but the price will be “comparable” to other price points and other products on the market.
“The FDA continues to prioritize the availability of more at-home testing options in response to the pandemic,” Jeff Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health, said in a statement.
“Cue COVID-19 Test for Home and Over-the-Counter Use provides access to accurate and reliable testing at home, without a prescription. The FDA will continue to work collaboratively with test developers to advance effective testing options for doctors, clinicians, and the public,” he said.
In June, the FDA granted an EUA to Cue Health’s COVID-19 test for use in clinical and point-of-care settings.
The test is currently being used in hospitals, physicians’ offices, and dental clinics, as well as schools, essential businesses, nursing homes, and other congregate-care facilities. The test is also being distributed through a program led by the U.S. Department of Defense and the U.S. Department of Health & Human Services across several states.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration has granted emergency use authorization (EUA) for the Cue COVID-19 Test for Home and Over The Counter Use (Cue OTC Test, Cue Health).
The Cue OTC Test is the first molecular diagnostic test available to consumers without a prescription.
The test detects genetic material from SARS-CoV-2 present in the nostrils and delivers results in about 20 minutes to the user’s mobile smart device via the Cue Health app.
In testing, the Cue OTC Test correctly identified 96% of positive nasal swab samples from individuals known to have symptoms and correctly identified 100% of positive samples from individuals without symptoms.
The test is intended for use in people aged 2 years and older with and without symptoms.
“With this authorization, consumers can purchase and self-administer one of the easiest, fastest, and most accurate tests without a prescription,” Clint Sever, cofounder and chief product officer of Cue Health, said in a news release.
“This FDA authorization will help us improve patient outcomes with a solution that provides the accuracy of central lab tests, with the speed and accessibility required to address emergent global health issues,” he said.
Cue Health expects to produce more than 100,000 single-use test kits per day by this summer. Dena Cook, the company’s chief communications officer, told this news organization that the company hasn’t announced pricing information yet, but the price will be “comparable” to other price points and other products on the market.
“The FDA continues to prioritize the availability of more at-home testing options in response to the pandemic,” Jeff Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health, said in a statement.
“Cue COVID-19 Test for Home and Over-the-Counter Use provides access to accurate and reliable testing at home, without a prescription. The FDA will continue to work collaboratively with test developers to advance effective testing options for doctors, clinicians, and the public,” he said.
In June, the FDA granted an EUA to Cue Health’s COVID-19 test for use in clinical and point-of-care settings.
The test is currently being used in hospitals, physicians’ offices, and dental clinics, as well as schools, essential businesses, nursing homes, and other congregate-care facilities. The test is also being distributed through a program led by the U.S. Department of Defense and the U.S. Department of Health & Human Services across several states.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration has granted emergency use authorization (EUA) for the Cue COVID-19 Test for Home and Over The Counter Use (Cue OTC Test, Cue Health).
The Cue OTC Test is the first molecular diagnostic test available to consumers without a prescription.
The test detects genetic material from SARS-CoV-2 present in the nostrils and delivers results in about 20 minutes to the user’s mobile smart device via the Cue Health app.
In testing, the Cue OTC Test correctly identified 96% of positive nasal swab samples from individuals known to have symptoms and correctly identified 100% of positive samples from individuals without symptoms.
The test is intended for use in people aged 2 years and older with and without symptoms.
“With this authorization, consumers can purchase and self-administer one of the easiest, fastest, and most accurate tests without a prescription,” Clint Sever, cofounder and chief product officer of Cue Health, said in a news release.
“This FDA authorization will help us improve patient outcomes with a solution that provides the accuracy of central lab tests, with the speed and accessibility required to address emergent global health issues,” he said.
Cue Health expects to produce more than 100,000 single-use test kits per day by this summer. Dena Cook, the company’s chief communications officer, told this news organization that the company hasn’t announced pricing information yet, but the price will be “comparable” to other price points and other products on the market.
“The FDA continues to prioritize the availability of more at-home testing options in response to the pandemic,” Jeff Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health, said in a statement.
“Cue COVID-19 Test for Home and Over-the-Counter Use provides access to accurate and reliable testing at home, without a prescription. The FDA will continue to work collaboratively with test developers to advance effective testing options for doctors, clinicians, and the public,” he said.
In June, the FDA granted an EUA to Cue Health’s COVID-19 test for use in clinical and point-of-care settings.
The test is currently being used in hospitals, physicians’ offices, and dental clinics, as well as schools, essential businesses, nursing homes, and other congregate-care facilities. The test is also being distributed through a program led by the U.S. Department of Defense and the U.S. Department of Health & Human Services across several states.
A version of this article first appeared on Medscape.com.
CDC chief lays out attack plan for COVID variants
earlier this week.
As part of JAMA’s Q&A series with JAMA editor in chief Howard Bauchner, MD, Dr. Walensky referenced the blueprint she coathored with Anthony Fauci, MD, the nation’s top infectious disease expert, and Henry T. Walke, MD, MPH, of the CDC, which was published on Feb. 17 in JAMA.
In the viewpoint article, they explain that the Department of Health & Human Services has established the SARS-CoV-2 Interagency Group to improve coordination among the CDC, the National Institutes of Health, the Food and Drug Administration, the Biomedical Advanced Research and Development Authority, the Department of Agriculture, and the Department of Defense.
Dr. Walensky said the first objective is to reinforce vigilance regarding public health mitigation strategies to decrease the amount of virus that’s circulating.
As part of that strategy, she said, the CDC strongly urges against nonessential travel.
In addition, public health leaders are working on a surveillance system to better understand the SARS-CoV-2 variants. That will take ramping up genome sequencing of the SARS-CoV-2 virus and ensuring that sampling is geographically representative.
She said the CDC is partnering with state health labs to obtain about 750 samples every week and is teaming up with commercial labs and academic centers to obtain an interim target of 6,000 samples per week.
She acknowledged the United States “is not where we need to be” with sequencing but has come a long way since January. At that time, they were sequencing 250 samples every week; they are currently sequencing thousands each week.
Data analysis is another concern: “We need to be able to understand at the basic science level what the information means,” Dr. Walensky said.
Researchers aren’t sure how the variants might affect use of convalescent plasma or monoclonal antibody treatments. It is expected that 5% of persons who are vaccinated against COVID-19 will nevertheless contract the disease. Sequencing will help answer whether such persons who have been vaccinated and who subsequently contract the virus are among those 5% or whether have been infected by a variant that evades the vaccine.
Accelerating vaccine administration globally and in the United States is essential, Dr. Walensky said.
As of Feb. 17, 56 million doses had been administered in the United States.
Top three threats
She updated the numbers on the three biggest variant threats.
Regarding B.1.1.7, which originated in the United Kingdom, she said: “So far, we’ve had over 1,200 cases in 41 states.” She noted that the variant is likely to be about 50% more transmissible and 30% to 50% more virulent.
“So far, it looks like that strain doesn’t have any real decrease in susceptibility to our vaccines,” she said.
The strain from South Africa (B.1.351) has been found in 19 cases in the United States.
The P.1. variant, which originated in Brazil, has been identified in two cases in two states.
Outlook for March and April
Dr. Bauchner asked Dr. Walensky what she envisions for March and April. He noted that public optimism is high in light of the continued reductions in COVID-19 case numbers, hospitalizations, and deaths, as well as the fact that warmer weather is coming and that more vaccinations are on the horizon.
“While I really am hopeful for what could happen in March and April,” Dr. Walensky said, “I really do know that this could go bad so fast. We saw it in November. We saw it in December.”
CDC models have projected that, by March, the more transmissible B.1.1.7 strain is likely to be the dominant strain, she reiterated.
“I worry that it will be spring, and we will all have had enough,” Dr. Walensky said. She noted that some states are already relaxing mask mandates.
“Around that time, life will look and feel a little better, and the motivation for those who might be vaccine hesitant may be diminished,” she said.
Dr. Bauchner also asked her to weigh in on whether a third vaccine, from Johnson & Johnson (J&J), may soon gain FDA emergency-use authorization – and whether its lower expected efficacy rate may result in a tiered system of vaccinations, with higher-risk populations receiving the more efficacious vaccines.
Dr. Walensky said more data are needed before that question can be answered.
“It may very well be that the data point us to the best populations in which to use this vaccine,” she said.
In phase 3 data, the J&J vaccine was shown to be 72% effective in the United States for moderate to severe disease.
Dr. Walensky said it’s important to remember that the projected efficacy for that vaccine is higher than that for the flu shot as well as many other vaccines currently in use for other diseases.
She said it also has several advantages. The vaccine has less-stringent storage requirements, requires just one dose, and protects against hospitalization and death, although it’s less efficacious in protecting against contracting the disease.
“I think many people would opt to get that one if they could get it sooner,” she said.
A version of this article first appeared on Medscape.com.
earlier this week.
As part of JAMA’s Q&A series with JAMA editor in chief Howard Bauchner, MD, Dr. Walensky referenced the blueprint she coathored with Anthony Fauci, MD, the nation’s top infectious disease expert, and Henry T. Walke, MD, MPH, of the CDC, which was published on Feb. 17 in JAMA.
In the viewpoint article, they explain that the Department of Health & Human Services has established the SARS-CoV-2 Interagency Group to improve coordination among the CDC, the National Institutes of Health, the Food and Drug Administration, the Biomedical Advanced Research and Development Authority, the Department of Agriculture, and the Department of Defense.
Dr. Walensky said the first objective is to reinforce vigilance regarding public health mitigation strategies to decrease the amount of virus that’s circulating.
As part of that strategy, she said, the CDC strongly urges against nonessential travel.
In addition, public health leaders are working on a surveillance system to better understand the SARS-CoV-2 variants. That will take ramping up genome sequencing of the SARS-CoV-2 virus and ensuring that sampling is geographically representative.
She said the CDC is partnering with state health labs to obtain about 750 samples every week and is teaming up with commercial labs and academic centers to obtain an interim target of 6,000 samples per week.
She acknowledged the United States “is not where we need to be” with sequencing but has come a long way since January. At that time, they were sequencing 250 samples every week; they are currently sequencing thousands each week.
Data analysis is another concern: “We need to be able to understand at the basic science level what the information means,” Dr. Walensky said.
Researchers aren’t sure how the variants might affect use of convalescent plasma or monoclonal antibody treatments. It is expected that 5% of persons who are vaccinated against COVID-19 will nevertheless contract the disease. Sequencing will help answer whether such persons who have been vaccinated and who subsequently contract the virus are among those 5% or whether have been infected by a variant that evades the vaccine.
Accelerating vaccine administration globally and in the United States is essential, Dr. Walensky said.
As of Feb. 17, 56 million doses had been administered in the United States.
Top three threats
She updated the numbers on the three biggest variant threats.
Regarding B.1.1.7, which originated in the United Kingdom, she said: “So far, we’ve had over 1,200 cases in 41 states.” She noted that the variant is likely to be about 50% more transmissible and 30% to 50% more virulent.
“So far, it looks like that strain doesn’t have any real decrease in susceptibility to our vaccines,” she said.
The strain from South Africa (B.1.351) has been found in 19 cases in the United States.
The P.1. variant, which originated in Brazil, has been identified in two cases in two states.
Outlook for March and April
Dr. Bauchner asked Dr. Walensky what she envisions for March and April. He noted that public optimism is high in light of the continued reductions in COVID-19 case numbers, hospitalizations, and deaths, as well as the fact that warmer weather is coming and that more vaccinations are on the horizon.
“While I really am hopeful for what could happen in March and April,” Dr. Walensky said, “I really do know that this could go bad so fast. We saw it in November. We saw it in December.”
CDC models have projected that, by March, the more transmissible B.1.1.7 strain is likely to be the dominant strain, she reiterated.
“I worry that it will be spring, and we will all have had enough,” Dr. Walensky said. She noted that some states are already relaxing mask mandates.
“Around that time, life will look and feel a little better, and the motivation for those who might be vaccine hesitant may be diminished,” she said.
Dr. Bauchner also asked her to weigh in on whether a third vaccine, from Johnson & Johnson (J&J), may soon gain FDA emergency-use authorization – and whether its lower expected efficacy rate may result in a tiered system of vaccinations, with higher-risk populations receiving the more efficacious vaccines.
Dr. Walensky said more data are needed before that question can be answered.
“It may very well be that the data point us to the best populations in which to use this vaccine,” she said.
In phase 3 data, the J&J vaccine was shown to be 72% effective in the United States for moderate to severe disease.
Dr. Walensky said it’s important to remember that the projected efficacy for that vaccine is higher than that for the flu shot as well as many other vaccines currently in use for other diseases.
She said it also has several advantages. The vaccine has less-stringent storage requirements, requires just one dose, and protects against hospitalization and death, although it’s less efficacious in protecting against contracting the disease.
“I think many people would opt to get that one if they could get it sooner,” she said.
A version of this article first appeared on Medscape.com.
earlier this week.
As part of JAMA’s Q&A series with JAMA editor in chief Howard Bauchner, MD, Dr. Walensky referenced the blueprint she coathored with Anthony Fauci, MD, the nation’s top infectious disease expert, and Henry T. Walke, MD, MPH, of the CDC, which was published on Feb. 17 in JAMA.
In the viewpoint article, they explain that the Department of Health & Human Services has established the SARS-CoV-2 Interagency Group to improve coordination among the CDC, the National Institutes of Health, the Food and Drug Administration, the Biomedical Advanced Research and Development Authority, the Department of Agriculture, and the Department of Defense.
Dr. Walensky said the first objective is to reinforce vigilance regarding public health mitigation strategies to decrease the amount of virus that’s circulating.
As part of that strategy, she said, the CDC strongly urges against nonessential travel.
In addition, public health leaders are working on a surveillance system to better understand the SARS-CoV-2 variants. That will take ramping up genome sequencing of the SARS-CoV-2 virus and ensuring that sampling is geographically representative.
She said the CDC is partnering with state health labs to obtain about 750 samples every week and is teaming up with commercial labs and academic centers to obtain an interim target of 6,000 samples per week.
She acknowledged the United States “is not where we need to be” with sequencing but has come a long way since January. At that time, they were sequencing 250 samples every week; they are currently sequencing thousands each week.
Data analysis is another concern: “We need to be able to understand at the basic science level what the information means,” Dr. Walensky said.
Researchers aren’t sure how the variants might affect use of convalescent plasma or monoclonal antibody treatments. It is expected that 5% of persons who are vaccinated against COVID-19 will nevertheless contract the disease. Sequencing will help answer whether such persons who have been vaccinated and who subsequently contract the virus are among those 5% or whether have been infected by a variant that evades the vaccine.
Accelerating vaccine administration globally and in the United States is essential, Dr. Walensky said.
As of Feb. 17, 56 million doses had been administered in the United States.
Top three threats
She updated the numbers on the three biggest variant threats.
Regarding B.1.1.7, which originated in the United Kingdom, she said: “So far, we’ve had over 1,200 cases in 41 states.” She noted that the variant is likely to be about 50% more transmissible and 30% to 50% more virulent.
“So far, it looks like that strain doesn’t have any real decrease in susceptibility to our vaccines,” she said.
The strain from South Africa (B.1.351) has been found in 19 cases in the United States.
The P.1. variant, which originated in Brazil, has been identified in two cases in two states.
Outlook for March and April
Dr. Bauchner asked Dr. Walensky what she envisions for March and April. He noted that public optimism is high in light of the continued reductions in COVID-19 case numbers, hospitalizations, and deaths, as well as the fact that warmer weather is coming and that more vaccinations are on the horizon.
“While I really am hopeful for what could happen in March and April,” Dr. Walensky said, “I really do know that this could go bad so fast. We saw it in November. We saw it in December.”
CDC models have projected that, by March, the more transmissible B.1.1.7 strain is likely to be the dominant strain, she reiterated.
“I worry that it will be spring, and we will all have had enough,” Dr. Walensky said. She noted that some states are already relaxing mask mandates.
“Around that time, life will look and feel a little better, and the motivation for those who might be vaccine hesitant may be diminished,” she said.
Dr. Bauchner also asked her to weigh in on whether a third vaccine, from Johnson & Johnson (J&J), may soon gain FDA emergency-use authorization – and whether its lower expected efficacy rate may result in a tiered system of vaccinations, with higher-risk populations receiving the more efficacious vaccines.
Dr. Walensky said more data are needed before that question can be answered.
“It may very well be that the data point us to the best populations in which to use this vaccine,” she said.
In phase 3 data, the J&J vaccine was shown to be 72% effective in the United States for moderate to severe disease.
Dr. Walensky said it’s important to remember that the projected efficacy for that vaccine is higher than that for the flu shot as well as many other vaccines currently in use for other diseases.
She said it also has several advantages. The vaccine has less-stringent storage requirements, requires just one dose, and protects against hospitalization and death, although it’s less efficacious in protecting against contracting the disease.
“I think many people would opt to get that one if they could get it sooner,” she said.
A version of this article first appeared on Medscape.com.
Mask mandates reduced COVID-19 hospitalizations
States that implemented mask mandates in 2020 saw a decline in the growth of COVID-19 hospitalizations between March and October 2020, according to a new study published Feb. 5 in the CDC’s Morbidity and Mortality Weekly Report.
Hospitalization growth rates declined by 5.5 percentage points for adults between ages 18-64 about 3 weeks after the mandates were implemented, compared with climbing growth rates in the 4 weeks before mandates.
CDC Director Rochelle Walensky said she was pleased to see the results, but that it’s “too early” to tell whether President Joe Biden’s recent mask orders have had an effect on cases and hospitalizations in 2021.
“We’re going to be watching the mask data very carefully,” she said during a news briefing with the White House COVID-19 Response Team on Feb. 5. “I think it’s probably still a bit too early to tell, but I’m encouraged with the decrease in case rates right now.”
In another study published Feb. 5 in the Morbidity and Mortality Weekly Report, trained observers tracked mask use at six universities with mask mandates between September and November 2020. Overall, observers reported that about 92% of people wore masks correctly indoors, which varied based on the type of mask.
About 97% of people used N95 masks correctly, compared with 92% who used cloth masks, and 79% who used bandanas, scarves, or neck gaiters. Cloth masks were most common, and bandanas and scarves were least common.
The Biden administration is considering whether to send masks directly to American households to encourage people to wear them, according to NBC News. The White House COVID-19 Response Team is debating the logistics of mailing out masks, including how many to send and what the mask material would be, the news outlet reported.
Wisconsin Gov. Tony Evers reissued a new statewide mask mandate on Feb. 4, just an hour after the Republican-controlled legislature voted to repeal his previous mandate, according to The Associated Press. Gov. Evers said his priority is to keep people safe and that wearing a mask is the easiest way to do so.
“If the legislature keeps playing politics and we don’t keep wearing masks, we’re going to see more preventable deaths,” he said. “It’s going to take even longer to get our state and our economy back on track.”
A version of this article first appeared on WebMD.com.
States that implemented mask mandates in 2020 saw a decline in the growth of COVID-19 hospitalizations between March and October 2020, according to a new study published Feb. 5 in the CDC’s Morbidity and Mortality Weekly Report.
Hospitalization growth rates declined by 5.5 percentage points for adults between ages 18-64 about 3 weeks after the mandates were implemented, compared with climbing growth rates in the 4 weeks before mandates.
CDC Director Rochelle Walensky said she was pleased to see the results, but that it’s “too early” to tell whether President Joe Biden’s recent mask orders have had an effect on cases and hospitalizations in 2021.
“We’re going to be watching the mask data very carefully,” she said during a news briefing with the White House COVID-19 Response Team on Feb. 5. “I think it’s probably still a bit too early to tell, but I’m encouraged with the decrease in case rates right now.”
In another study published Feb. 5 in the Morbidity and Mortality Weekly Report, trained observers tracked mask use at six universities with mask mandates between September and November 2020. Overall, observers reported that about 92% of people wore masks correctly indoors, which varied based on the type of mask.
About 97% of people used N95 masks correctly, compared with 92% who used cloth masks, and 79% who used bandanas, scarves, or neck gaiters. Cloth masks were most common, and bandanas and scarves were least common.
The Biden administration is considering whether to send masks directly to American households to encourage people to wear them, according to NBC News. The White House COVID-19 Response Team is debating the logistics of mailing out masks, including how many to send and what the mask material would be, the news outlet reported.
Wisconsin Gov. Tony Evers reissued a new statewide mask mandate on Feb. 4, just an hour after the Republican-controlled legislature voted to repeal his previous mandate, according to The Associated Press. Gov. Evers said his priority is to keep people safe and that wearing a mask is the easiest way to do so.
“If the legislature keeps playing politics and we don’t keep wearing masks, we’re going to see more preventable deaths,” he said. “It’s going to take even longer to get our state and our economy back on track.”
A version of this article first appeared on WebMD.com.
States that implemented mask mandates in 2020 saw a decline in the growth of COVID-19 hospitalizations between March and October 2020, according to a new study published Feb. 5 in the CDC’s Morbidity and Mortality Weekly Report.
Hospitalization growth rates declined by 5.5 percentage points for adults between ages 18-64 about 3 weeks after the mandates were implemented, compared with climbing growth rates in the 4 weeks before mandates.
CDC Director Rochelle Walensky said she was pleased to see the results, but that it’s “too early” to tell whether President Joe Biden’s recent mask orders have had an effect on cases and hospitalizations in 2021.
“We’re going to be watching the mask data very carefully,” she said during a news briefing with the White House COVID-19 Response Team on Feb. 5. “I think it’s probably still a bit too early to tell, but I’m encouraged with the decrease in case rates right now.”
In another study published Feb. 5 in the Morbidity and Mortality Weekly Report, trained observers tracked mask use at six universities with mask mandates between September and November 2020. Overall, observers reported that about 92% of people wore masks correctly indoors, which varied based on the type of mask.
About 97% of people used N95 masks correctly, compared with 92% who used cloth masks, and 79% who used bandanas, scarves, or neck gaiters. Cloth masks were most common, and bandanas and scarves were least common.
The Biden administration is considering whether to send masks directly to American households to encourage people to wear them, according to NBC News. The White House COVID-19 Response Team is debating the logistics of mailing out masks, including how many to send and what the mask material would be, the news outlet reported.
Wisconsin Gov. Tony Evers reissued a new statewide mask mandate on Feb. 4, just an hour after the Republican-controlled legislature voted to repeal his previous mandate, according to The Associated Press. Gov. Evers said his priority is to keep people safe and that wearing a mask is the easiest way to do so.
“If the legislature keeps playing politics and we don’t keep wearing masks, we’re going to see more preventable deaths,” he said. “It’s going to take even longer to get our state and our economy back on track.”
A version of this article first appeared on WebMD.com.