User login
RA Prevention: A Decade of Trials Provides Insights on What’s to Come
With the discovery of autoantibodies and other risk factors for rheumatoid arthritis (RA), researchers developed clinical trials to see whether the disease can be prevented entirely. In the past 10 years, a number of these trials have concluded, with variable results.
While some trials demonstrated no effect at all, others showed that medical intervention can delay the onset of disease in certain populations and even reduce the rates of progression to RA. These completed trials also offer researchers the chance to identify opportunities to improve RA prevention trials moving forward.
“We’re looking at all that data and trying to figure out what the next step is going to be,” said Kevin Deane, MD, PhD, a professor of medicine and a rheumatologist at the University of Colorado School of Medicine, Aurora.
Key lessons include the need for improved risk stratification tools and better understanding of RA pathogenesis, he said.
The Research So Far
All RA prevention trials except for one have been completed and/or published within the past decade, bringing valuable insights to the field. (See chart below.)
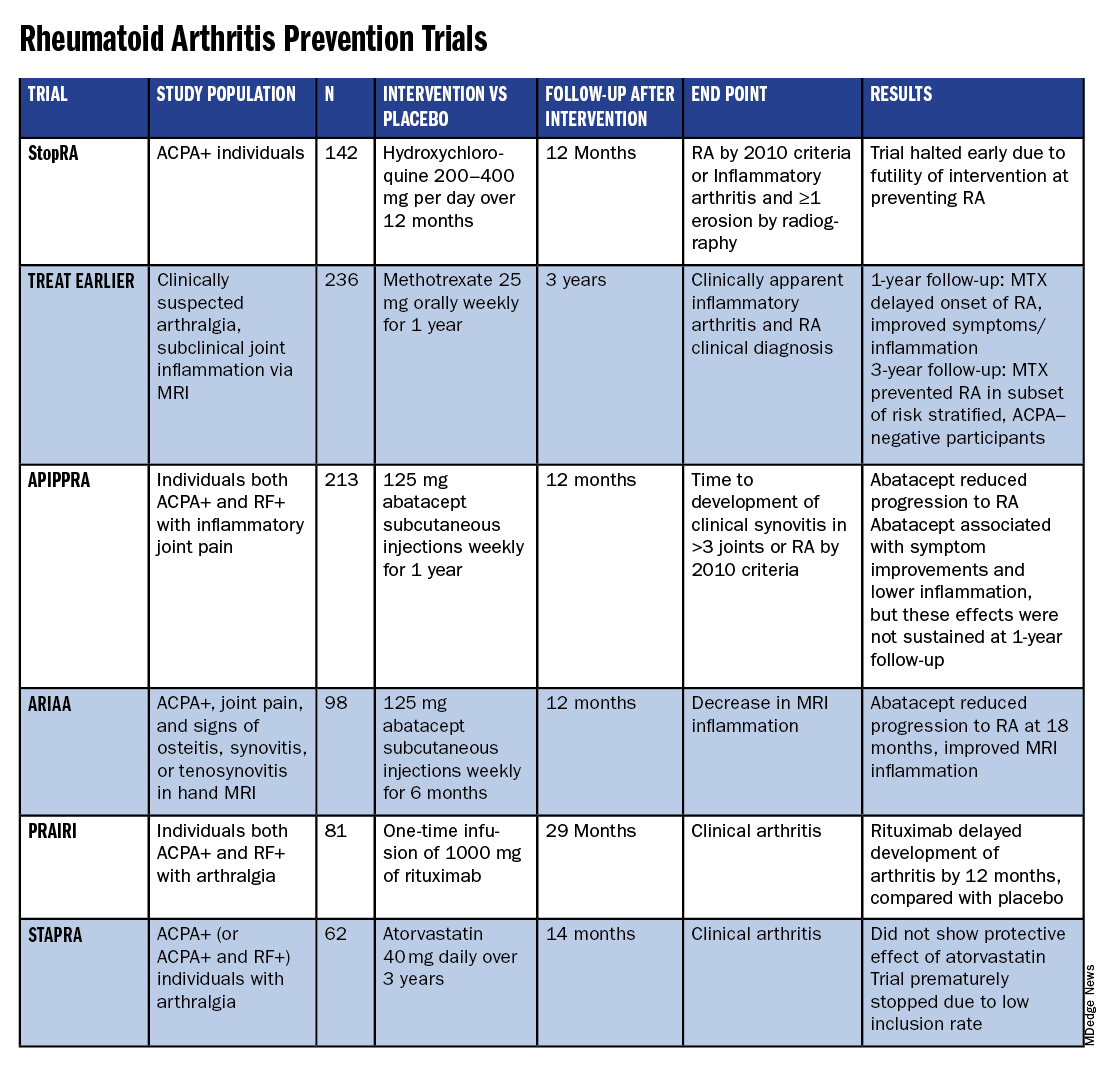
Atorvastatin (STAPRA) and hydroxychloroquine (StopRA) proved ineffective in preventing the onset of RA, and both trials were stopped early. Rituximab and methotrexate (MTX) both delayed the onset of RA, but the effect disappeared by the end of the follow-up periods.
However, the 2-year results from the TREAT EARLIER trial showed that compared with patients given placebo, those given MTX showed improved MRI-detected joint inflammation, physical functioning, and reported symptoms.
The 4-year analysis of the trial further risk stratified participants and found that MTX showed a preventive effect in anti–citrullinated protein antibody (ACPA)–negative participants at an increased risk for RA.
Abatacept also showed promise in preventing RA in two separate trials. In the ARIAA trial, compared with placebo, 6 months of treatment with abatacept reduced MRI inflammation and symptoms and lowered the rates of progression to RA. This treatment effect lessened during the 1-year follow-up period, but the difference between the two groups was still significant at 18 months.
In the APIPPRA trial, 12 months of treatment with abatacept improved subclinical inflammation and quality-of-life measures in participants and reduced the rates of progression to RA through another 12 months of observation. However, during this post-treatment follow-up period, the treatment effect began to diminish.
While there have been some promising findings — not only in disease prevention but also in disease modification — these studies all looked at different patient groups, noted Kulveer Mankia, MA, DM, an associate professor and consulting rheumatologist at the Leeds Institute of Rheumatic and Musculoskeletal Medicine, University of Leeds in England.
“You have disparate, different inclusion criteria in different studies, all of which take years to complete,” he said. For example, while the TREAT EARLIER trial recruited patients with joint pain and subclinical joint inflammation via MRI, regardless of autoantibody status, the APIPPRA trial enrolled patients that were both ACPA+ and rheumatoid factor (RF)+ with joint pain.
“You’re left extrapolating as to whether [these interventions] will work in different at-risk populations,” he said.
Even with specific inclusion criteria in each study, there can still be heterogeneity in risk within a study group, Deane said. In the TREAT EARLIER study, 18%-20% of participants ultimately developed RA over the study period, which is lower than expected.
“While it seemed like a pretty high-risk group, it wasn’t as high risk as we thought,” he said, “and that’s why we’ve gone back to the drawing board.”
Risk Stratification Efforts
There are now two ongoing joint efforts by the American College of Rheumatology (ACR) and the European Alliance of Associations for Rheumatology (EULAR) to define these populations and “bring some consensus to the field,” Mankia said.

The first aims to create a unanimous risk stratification tool for future RA prevention studies. The proposed system, devised for individuals with new joint symptoms who are at a risk for RA, was presented at the EULAR 2024 annual meeting and will be further discussed at the upcoming ACR 2024 annual meeting in Washington, DC.
The system uses a point system based on six criteria — three lab tests and three criteria commonly assessed in clinical practice:
- Morning stiffness
- Patient-reported joint swelling
- Difficulty making a fist
- Increased C-reactive protein
- RF positivity
- ACPA positivity
These criteria were picked so that the risk stratification tool can be used without imaging; however, the inclusion of MRI can further refine the score.
The ACR-EULAR task force that created the tool has emphasized that this criterion is specifically designed for research purposes and should not be used in clinical practice. Using this stratification tool should allow future clinical studies to group patients by similar risk, Deane said.
“Not that all studies have to look at exactly the same people, but each study should have similar risk stratification,” he said.
The second ACR-EULAR joint effort is taking a population-based approach to risk stratification, Deane said, to better predict RA risk in individuals without common symptoms like joint pain.
The aim is to create something analogous to the Framingham Risk Score in predicting cardiovascular disease, in which simple variables like total cholesterol, high-density lipoprotein cholesterol, systolic blood pressure, and smoking status can be used to calculate an individual’s 10-year risk for CVD, Deane explained.
The second approach could also identify patients earlier in the progression to RA, which may be easier to treat than later stages of disease.
Understanding RA Origins
However, treating an earlier stage of disease might require a different approach. Up to this point, medical interventions for RA prevention used drugs approved to treat RA, but inventions during the pre-RA stage — before any joint symptoms appear — might require targeting different immunologic pathways.
“The general concept is if there is a pre-RA stage when joints are not involved, that means all the immunologic abnormalities are probably happening somewhere else in the body,” he said. “The big question is: Where is that, and how exactly is that happening?”
One theory is that RA begins to develop in mucosal sites, such as the intestines or lungs, before it involves synovial joints.
“In the absence of resolution, these localized immune processes transition into a systemic process that targets the joints, either by direct effects of microbiota, molecular mimicry, and/or immune amplification,” wrote Deane and coauthors in a recent review article in Annals of the Rheumatic Diseases. “This, in turn, leads to inappropriate engagement of a range of effector mechanisms in both synovium and periarticular sites.”
Following this logic, the progression of the at-risk stage of RA could be considered a continuum along which there are multiple possible points for intervention. It’s also probable that the disease can develop through multiple pathways, Deane said.
“If you look at all the people who get rheumatoid arthritis, there’s probably no way those could have the same exact pathways,” he said. “There’s probably going to be different endotypes and understanding that is going to help us prevent disease in a better way.”
Looking Forward
Beyond improving risk stratification and understanding RA pathogenesis, researchers are also considering novel therapeutic approaches for future trials. Glucagon-like peptide 1 (GLP-1) receptor agonists could be worth exploring in RA prevention and treatment, said Jeffrey A. Sparks, MD, MMSc, a rheumatologist at Brigham and Women’s Hospital, Boston, Massachusetts.
These drugs — initially developed for diabetes — have already shown anti-inflammatory effects, and one study suggested that GLP-1s lowered the risk for major adverse cardiovascular events and all-cause mortality in individuals with immune-mediated inflammatory diseases. Obesity is a known risk factor for RA, so weight loss aided by GLP-1 drugs could also help reduce risk in certain patients. Clinical trials are needed to explore GLP-1s for both RA prevention and treatment, he said.
While prevention trials up to this point have used one-time, time-limited interventions, longer durations of medication or multiple rounds of therapy may be more efficacious. Even for trials that demonstrated the intervention arms had less progression to RA, this effect diminished once participants stopped the medication. In the ARIAA and APIPPRA trials using abatacept, “it wasn’t like we hit a reset button and [patients] just permanently now did not get rheumatoid arthritis,” Deane said, suggesting that alternative approaches should be explored.
“Future studies need to look at potentially longer doses of drug or lower doses of drug, or some combination that might be effective,” he said.
Deane received honoraria from Bristol-Myers Squibb, Thermo Fisher, and Werfen and grant funding from Janssen Research and Development and Gilead Sciences. Mankia received grant support from Gilead, Lilly, AstraZeneca, and Serac Life Sciences and honoraria or consultant fees from AbbVie, UCB, Lilly, Galapagos, DeepCure, Serac Life Sciences, AstraZeneca, and Zura Bio. Sparks received research support from Boehringer Ingelheim, Bristol-Myers Squibb, Janssen, and Sonoma Biotherapeutics. He consulted for AbbVie, Amgen, Boehringer Ingelheim, Bristol Myers Squibb, Gilead, Inova Diagnostics, Janssen, Merck, Mustang, Optum, Pfizer, ReCor Medical, Sana, Sobi, and UCB.
A version of this article first appeared on Medscape.com.
With the discovery of autoantibodies and other risk factors for rheumatoid arthritis (RA), researchers developed clinical trials to see whether the disease can be prevented entirely. In the past 10 years, a number of these trials have concluded, with variable results.
While some trials demonstrated no effect at all, others showed that medical intervention can delay the onset of disease in certain populations and even reduce the rates of progression to RA. These completed trials also offer researchers the chance to identify opportunities to improve RA prevention trials moving forward.
“We’re looking at all that data and trying to figure out what the next step is going to be,” said Kevin Deane, MD, PhD, a professor of medicine and a rheumatologist at the University of Colorado School of Medicine, Aurora.
Key lessons include the need for improved risk stratification tools and better understanding of RA pathogenesis, he said.
The Research So Far
All RA prevention trials except for one have been completed and/or published within the past decade, bringing valuable insights to the field. (See chart below.)
Atorvastatin (STAPRA) and hydroxychloroquine (StopRA) proved ineffective in preventing the onset of RA, and both trials were stopped early. Rituximab and methotrexate (MTX) both delayed the onset of RA, but the effect disappeared by the end of the follow-up periods.
However, the 2-year results from the TREAT EARLIER trial showed that compared with patients given placebo, those given MTX showed improved MRI-detected joint inflammation, physical functioning, and reported symptoms.
The 4-year analysis of the trial further risk stratified participants and found that MTX showed a preventive effect in anti–citrullinated protein antibody (ACPA)–negative participants at an increased risk for RA.
Abatacept also showed promise in preventing RA in two separate trials. In the ARIAA trial, compared with placebo, 6 months of treatment with abatacept reduced MRI inflammation and symptoms and lowered the rates of progression to RA. This treatment effect lessened during the 1-year follow-up period, but the difference between the two groups was still significant at 18 months.
In the APIPPRA trial, 12 months of treatment with abatacept improved subclinical inflammation and quality-of-life measures in participants and reduced the rates of progression to RA through another 12 months of observation. However, during this post-treatment follow-up period, the treatment effect began to diminish.
While there have been some promising findings — not only in disease prevention but also in disease modification — these studies all looked at different patient groups, noted Kulveer Mankia, MA, DM, an associate professor and consulting rheumatologist at the Leeds Institute of Rheumatic and Musculoskeletal Medicine, University of Leeds in England.
“You have disparate, different inclusion criteria in different studies, all of which take years to complete,” he said. For example, while the TREAT EARLIER trial recruited patients with joint pain and subclinical joint inflammation via MRI, regardless of autoantibody status, the APIPPRA trial enrolled patients that were both ACPA+ and rheumatoid factor (RF)+ with joint pain.
“You’re left extrapolating as to whether [these interventions] will work in different at-risk populations,” he said.
Even with specific inclusion criteria in each study, there can still be heterogeneity in risk within a study group, Deane said. In the TREAT EARLIER study, 18%-20% of participants ultimately developed RA over the study period, which is lower than expected.
“While it seemed like a pretty high-risk group, it wasn’t as high risk as we thought,” he said, “and that’s why we’ve gone back to the drawing board.”
Risk Stratification Efforts
There are now two ongoing joint efforts by the American College of Rheumatology (ACR) and the European Alliance of Associations for Rheumatology (EULAR) to define these populations and “bring some consensus to the field,” Mankia said.
The first aims to create a unanimous risk stratification tool for future RA prevention studies. The proposed system, devised for individuals with new joint symptoms who are at a risk for RA, was presented at the EULAR 2024 annual meeting and will be further discussed at the upcoming ACR 2024 annual meeting in Washington, DC.
The system uses a point system based on six criteria — three lab tests and three criteria commonly assessed in clinical practice:
- Morning stiffness
- Patient-reported joint swelling
- Difficulty making a fist
- Increased C-reactive protein
- RF positivity
- ACPA positivity
These criteria were picked so that the risk stratification tool can be used without imaging; however, the inclusion of MRI can further refine the score.
The ACR-EULAR task force that created the tool has emphasized that this criterion is specifically designed for research purposes and should not be used in clinical practice. Using this stratification tool should allow future clinical studies to group patients by similar risk, Deane said.
“Not that all studies have to look at exactly the same people, but each study should have similar risk stratification,” he said.
The second ACR-EULAR joint effort is taking a population-based approach to risk stratification, Deane said, to better predict RA risk in individuals without common symptoms like joint pain.
The aim is to create something analogous to the Framingham Risk Score in predicting cardiovascular disease, in which simple variables like total cholesterol, high-density lipoprotein cholesterol, systolic blood pressure, and smoking status can be used to calculate an individual’s 10-year risk for CVD, Deane explained.
The second approach could also identify patients earlier in the progression to RA, which may be easier to treat than later stages of disease.
Understanding RA Origins
However, treating an earlier stage of disease might require a different approach. Up to this point, medical interventions for RA prevention used drugs approved to treat RA, but inventions during the pre-RA stage — before any joint symptoms appear — might require targeting different immunologic pathways.
“The general concept is if there is a pre-RA stage when joints are not involved, that means all the immunologic abnormalities are probably happening somewhere else in the body,” he said. “The big question is: Where is that, and how exactly is that happening?”
One theory is that RA begins to develop in mucosal sites, such as the intestines or lungs, before it involves synovial joints.
“In the absence of resolution, these localized immune processes transition into a systemic process that targets the joints, either by direct effects of microbiota, molecular mimicry, and/or immune amplification,” wrote Deane and coauthors in a recent review article in Annals of the Rheumatic Diseases. “This, in turn, leads to inappropriate engagement of a range of effector mechanisms in both synovium and periarticular sites.”
Following this logic, the progression of the at-risk stage of RA could be considered a continuum along which there are multiple possible points for intervention. It’s also probable that the disease can develop through multiple pathways, Deane said.
“If you look at all the people who get rheumatoid arthritis, there’s probably no way those could have the same exact pathways,” he said. “There’s probably going to be different endotypes and understanding that is going to help us prevent disease in a better way.”
Looking Forward
Beyond improving risk stratification and understanding RA pathogenesis, researchers are also considering novel therapeutic approaches for future trials. Glucagon-like peptide 1 (GLP-1) receptor agonists could be worth exploring in RA prevention and treatment, said Jeffrey A. Sparks, MD, MMSc, a rheumatologist at Brigham and Women’s Hospital, Boston, Massachusetts.
These drugs — initially developed for diabetes — have already shown anti-inflammatory effects, and one study suggested that GLP-1s lowered the risk for major adverse cardiovascular events and all-cause mortality in individuals with immune-mediated inflammatory diseases. Obesity is a known risk factor for RA, so weight loss aided by GLP-1 drugs could also help reduce risk in certain patients. Clinical trials are needed to explore GLP-1s for both RA prevention and treatment, he said.
While prevention trials up to this point have used one-time, time-limited interventions, longer durations of medication or multiple rounds of therapy may be more efficacious. Even for trials that demonstrated the intervention arms had less progression to RA, this effect diminished once participants stopped the medication. In the ARIAA and APIPPRA trials using abatacept, “it wasn’t like we hit a reset button and [patients] just permanently now did not get rheumatoid arthritis,” Deane said, suggesting that alternative approaches should be explored.
“Future studies need to look at potentially longer doses of drug or lower doses of drug, or some combination that might be effective,” he said.
Deane received honoraria from Bristol-Myers Squibb, Thermo Fisher, and Werfen and grant funding from Janssen Research and Development and Gilead Sciences. Mankia received grant support from Gilead, Lilly, AstraZeneca, and Serac Life Sciences and honoraria or consultant fees from AbbVie, UCB, Lilly, Galapagos, DeepCure, Serac Life Sciences, AstraZeneca, and Zura Bio. Sparks received research support from Boehringer Ingelheim, Bristol-Myers Squibb, Janssen, and Sonoma Biotherapeutics. He consulted for AbbVie, Amgen, Boehringer Ingelheim, Bristol Myers Squibb, Gilead, Inova Diagnostics, Janssen, Merck, Mustang, Optum, Pfizer, ReCor Medical, Sana, Sobi, and UCB.
A version of this article first appeared on Medscape.com.
With the discovery of autoantibodies and other risk factors for rheumatoid arthritis (RA), researchers developed clinical trials to see whether the disease can be prevented entirely. In the past 10 years, a number of these trials have concluded, with variable results.
While some trials demonstrated no effect at all, others showed that medical intervention can delay the onset of disease in certain populations and even reduce the rates of progression to RA. These completed trials also offer researchers the chance to identify opportunities to improve RA prevention trials moving forward.
“We’re looking at all that data and trying to figure out what the next step is going to be,” said Kevin Deane, MD, PhD, a professor of medicine and a rheumatologist at the University of Colorado School of Medicine, Aurora.
Key lessons include the need for improved risk stratification tools and better understanding of RA pathogenesis, he said.
The Research So Far
All RA prevention trials except for one have been completed and/or published within the past decade, bringing valuable insights to the field. (See chart below.)
Atorvastatin (STAPRA) and hydroxychloroquine (StopRA) proved ineffective in preventing the onset of RA, and both trials were stopped early. Rituximab and methotrexate (MTX) both delayed the onset of RA, but the effect disappeared by the end of the follow-up periods.
However, the 2-year results from the TREAT EARLIER trial showed that compared with patients given placebo, those given MTX showed improved MRI-detected joint inflammation, physical functioning, and reported symptoms.
The 4-year analysis of the trial further risk stratified participants and found that MTX showed a preventive effect in anti–citrullinated protein antibody (ACPA)–negative participants at an increased risk for RA.
Abatacept also showed promise in preventing RA in two separate trials. In the ARIAA trial, compared with placebo, 6 months of treatment with abatacept reduced MRI inflammation and symptoms and lowered the rates of progression to RA. This treatment effect lessened during the 1-year follow-up period, but the difference between the two groups was still significant at 18 months.
In the APIPPRA trial, 12 months of treatment with abatacept improved subclinical inflammation and quality-of-life measures in participants and reduced the rates of progression to RA through another 12 months of observation. However, during this post-treatment follow-up period, the treatment effect began to diminish.
While there have been some promising findings — not only in disease prevention but also in disease modification — these studies all looked at different patient groups, noted Kulveer Mankia, MA, DM, an associate professor and consulting rheumatologist at the Leeds Institute of Rheumatic and Musculoskeletal Medicine, University of Leeds in England.
“You have disparate, different inclusion criteria in different studies, all of which take years to complete,” he said. For example, while the TREAT EARLIER trial recruited patients with joint pain and subclinical joint inflammation via MRI, regardless of autoantibody status, the APIPPRA trial enrolled patients that were both ACPA+ and rheumatoid factor (RF)+ with joint pain.
“You’re left extrapolating as to whether [these interventions] will work in different at-risk populations,” he said.
Even with specific inclusion criteria in each study, there can still be heterogeneity in risk within a study group, Deane said. In the TREAT EARLIER study, 18%-20% of participants ultimately developed RA over the study period, which is lower than expected.
“While it seemed like a pretty high-risk group, it wasn’t as high risk as we thought,” he said, “and that’s why we’ve gone back to the drawing board.”
Risk Stratification Efforts
There are now two ongoing joint efforts by the American College of Rheumatology (ACR) and the European Alliance of Associations for Rheumatology (EULAR) to define these populations and “bring some consensus to the field,” Mankia said.
The first aims to create a unanimous risk stratification tool for future RA prevention studies. The proposed system, devised for individuals with new joint symptoms who are at a risk for RA, was presented at the EULAR 2024 annual meeting and will be further discussed at the upcoming ACR 2024 annual meeting in Washington, DC.
The system uses a point system based on six criteria — three lab tests and three criteria commonly assessed in clinical practice:
- Morning stiffness
- Patient-reported joint swelling
- Difficulty making a fist
- Increased C-reactive protein
- RF positivity
- ACPA positivity
These criteria were picked so that the risk stratification tool can be used without imaging; however, the inclusion of MRI can further refine the score.
The ACR-EULAR task force that created the tool has emphasized that this criterion is specifically designed for research purposes and should not be used in clinical practice. Using this stratification tool should allow future clinical studies to group patients by similar risk, Deane said.
“Not that all studies have to look at exactly the same people, but each study should have similar risk stratification,” he said.
The second ACR-EULAR joint effort is taking a population-based approach to risk stratification, Deane said, to better predict RA risk in individuals without common symptoms like joint pain.
The aim is to create something analogous to the Framingham Risk Score in predicting cardiovascular disease, in which simple variables like total cholesterol, high-density lipoprotein cholesterol, systolic blood pressure, and smoking status can be used to calculate an individual’s 10-year risk for CVD, Deane explained.
The second approach could also identify patients earlier in the progression to RA, which may be easier to treat than later stages of disease.
Understanding RA Origins
However, treating an earlier stage of disease might require a different approach. Up to this point, medical interventions for RA prevention used drugs approved to treat RA, but inventions during the pre-RA stage — before any joint symptoms appear — might require targeting different immunologic pathways.
“The general concept is if there is a pre-RA stage when joints are not involved, that means all the immunologic abnormalities are probably happening somewhere else in the body,” he said. “The big question is: Where is that, and how exactly is that happening?”
One theory is that RA begins to develop in mucosal sites, such as the intestines or lungs, before it involves synovial joints.
“In the absence of resolution, these localized immune processes transition into a systemic process that targets the joints, either by direct effects of microbiota, molecular mimicry, and/or immune amplification,” wrote Deane and coauthors in a recent review article in Annals of the Rheumatic Diseases. “This, in turn, leads to inappropriate engagement of a range of effector mechanisms in both synovium and periarticular sites.”
Following this logic, the progression of the at-risk stage of RA could be considered a continuum along which there are multiple possible points for intervention. It’s also probable that the disease can develop through multiple pathways, Deane said.
“If you look at all the people who get rheumatoid arthritis, there’s probably no way those could have the same exact pathways,” he said. “There’s probably going to be different endotypes and understanding that is going to help us prevent disease in a better way.”
Looking Forward
Beyond improving risk stratification and understanding RA pathogenesis, researchers are also considering novel therapeutic approaches for future trials. Glucagon-like peptide 1 (GLP-1) receptor agonists could be worth exploring in RA prevention and treatment, said Jeffrey A. Sparks, MD, MMSc, a rheumatologist at Brigham and Women’s Hospital, Boston, Massachusetts.
These drugs — initially developed for diabetes — have already shown anti-inflammatory effects, and one study suggested that GLP-1s lowered the risk for major adverse cardiovascular events and all-cause mortality in individuals with immune-mediated inflammatory diseases. Obesity is a known risk factor for RA, so weight loss aided by GLP-1 drugs could also help reduce risk in certain patients. Clinical trials are needed to explore GLP-1s for both RA prevention and treatment, he said.
While prevention trials up to this point have used one-time, time-limited interventions, longer durations of medication or multiple rounds of therapy may be more efficacious. Even for trials that demonstrated the intervention arms had less progression to RA, this effect diminished once participants stopped the medication. In the ARIAA and APIPPRA trials using abatacept, “it wasn’t like we hit a reset button and [patients] just permanently now did not get rheumatoid arthritis,” Deane said, suggesting that alternative approaches should be explored.
“Future studies need to look at potentially longer doses of drug or lower doses of drug, or some combination that might be effective,” he said.
Deane received honoraria from Bristol-Myers Squibb, Thermo Fisher, and Werfen and grant funding from Janssen Research and Development and Gilead Sciences. Mankia received grant support from Gilead, Lilly, AstraZeneca, and Serac Life Sciences and honoraria or consultant fees from AbbVie, UCB, Lilly, Galapagos, DeepCure, Serac Life Sciences, AstraZeneca, and Zura Bio. Sparks received research support from Boehringer Ingelheim, Bristol-Myers Squibb, Janssen, and Sonoma Biotherapeutics. He consulted for AbbVie, Amgen, Boehringer Ingelheim, Bristol Myers Squibb, Gilead, Inova Diagnostics, Janssen, Merck, Mustang, Optum, Pfizer, ReCor Medical, Sana, Sobi, and UCB.
A version of this article first appeared on Medscape.com.
New Scanner Creates Highly Detailed, 3D Images of Blood Vessels in Seconds
A new scanner can provide three-dimensional (3D) photoacoustic images of millimeter-scale veins and arteries in seconds.
The scanner, developed by researchers at University College London (UCL) in England, could help clinicians better visualize and track microvascular changes for a wide range of diseases, including cancer, rheumatoid arthritis (RA), and peripheral vascular disease (PVD).
The case studies “illustrate potential areas of application that warrant future, more comprehensive clinical studies,” the authors wrote. “Moreover, they demonstrate the feasibility of using the scanner on a real-world patient cohort where imaging is more challenging due to frailty, comorbidity, or pain that may limit their ability to tolerate prolonged scan times.”
The work was published online in Nature Biomedical Engineering.
Improving Photoacoustic Imaging
PAT works using the photoacoustic effect, a phenomenon where sound waves are generated when light is absorbed by a material. When pulsed light from a laser is directed at tissue, some of that light is absorbed and causes an increase in heat in the targeted area. This localized heat also increases pressure, which generates ultrasound waves that can be detected by specialized sensors.
While previous PAT scanners translated these sound waves to electric signals directly to generate imaging, UCL engineers developed a sensor in the early 2000s that can detect these ultrasound waves using light. The result was much clearer, 3D images.
“That was great, but the problem was it was very slow, and it would take 5 minutes to get an image,” explained Paul Beard, PhD, professor of biomedical photoacoustics at UCL and senior author of the study. “That’s fine if you’re imaging a dead mouse or an anesthetized mouse, but not so useful for human imaging,” he continued, where motion would blur the image.
In this new paper, Beard and colleagues outlined how they cut scanning times to an order of seconds (or fraction of a second) rather than minutes. While previous iterations could detect only acoustic waves from one point at a time, this new scanner can detect waves from multiple points simultaneously. The scanner can visualize veins and arteries up to 15 mm deep in human tissue and can also provide dynamic, 3D images of “time-varying tissue perfusion and other hemodynamic events,” the authors wrote.
With these types of scanners, there is always a trade-off between imaging quality and imaging speed, explained Srivalleesha Mallidi, PhD, an assistant professor of biomedical engineering at Tufts University in Medford, Massachusetts. She was not involved with the work.
“With the resolution that [the authors] are providing and the depth at which they are seeing the signals, it is one of the fastest systems,” she said.
Clinical Utility
Beard and colleagues also tested the scanner to visualize blood vessels in participants with RA, suspected PVD, and skin inflammation. The scanning images “illustrated how vascular abnormalities such as increased vessel tortuosity, which has previously been linked to PVD, and the neovascularization associated with inflammation can be visualized and quantified,” the authors wrote.
The next step, Beard noted, is testing whether these characteristics can be used as a marker for the progression of disease.
Nehal Mehta, MD, a cardiologist and professor of medicine at the George Washington University, Washington, DC, agreed that more longitudinal research is needed to understand how the abnormalities captured in these images can inform detection and diagnosis of various diseases.
“You don’t know whether these images look bad because of reverse causation — the disease is doing this — or true causation — that this is actually detecting the root cause of the disease,” he explained. “Until we have a bank of normal and abnormal scans, we don’t know what any of these things mean.”
Though still some time away from entering the clinic, Mehta likened the technology to the introduction of optical coherence tomography in the 1980s. Before being adapted for clinical use, researchers first needed to visualize differences between normal coronary vasculature and myocardial infarction.
“I think this is an amazingly strong first proof of concept,” Mehta said. “This technology is showing a true promise in the field imaging.”
The work was funded by grants from Cancer Research UK, the Engineering & Physical Sciences Research Council, Wellcome Trust, the European Research Council, and the National Institute for Health and Care Research University College London Hospitals Biomedical Research Centre. Beard and two coauthors are shareholders of DeepColor Imaging to which the intellectual property associated with the new scanner has been licensed, but the company was not involved in any of this research. Mallidi and Mehta had no relevant disclosures.
A version of this article first appeared on Medscape.com.
A new scanner can provide three-dimensional (3D) photoacoustic images of millimeter-scale veins and arteries in seconds.
The scanner, developed by researchers at University College London (UCL) in England, could help clinicians better visualize and track microvascular changes for a wide range of diseases, including cancer, rheumatoid arthritis (RA), and peripheral vascular disease (PVD).
The case studies “illustrate potential areas of application that warrant future, more comprehensive clinical studies,” the authors wrote. “Moreover, they demonstrate the feasibility of using the scanner on a real-world patient cohort where imaging is more challenging due to frailty, comorbidity, or pain that may limit their ability to tolerate prolonged scan times.”
The work was published online in Nature Biomedical Engineering.
Improving Photoacoustic Imaging
PAT works using the photoacoustic effect, a phenomenon where sound waves are generated when light is absorbed by a material. When pulsed light from a laser is directed at tissue, some of that light is absorbed and causes an increase in heat in the targeted area. This localized heat also increases pressure, which generates ultrasound waves that can be detected by specialized sensors.
While previous PAT scanners translated these sound waves to electric signals directly to generate imaging, UCL engineers developed a sensor in the early 2000s that can detect these ultrasound waves using light. The result was much clearer, 3D images.
“That was great, but the problem was it was very slow, and it would take 5 minutes to get an image,” explained Paul Beard, PhD, professor of biomedical photoacoustics at UCL and senior author of the study. “That’s fine if you’re imaging a dead mouse or an anesthetized mouse, but not so useful for human imaging,” he continued, where motion would blur the image.
In this new paper, Beard and colleagues outlined how they cut scanning times to an order of seconds (or fraction of a second) rather than minutes. While previous iterations could detect only acoustic waves from one point at a time, this new scanner can detect waves from multiple points simultaneously. The scanner can visualize veins and arteries up to 15 mm deep in human tissue and can also provide dynamic, 3D images of “time-varying tissue perfusion and other hemodynamic events,” the authors wrote.
With these types of scanners, there is always a trade-off between imaging quality and imaging speed, explained Srivalleesha Mallidi, PhD, an assistant professor of biomedical engineering at Tufts University in Medford, Massachusetts. She was not involved with the work.
“With the resolution that [the authors] are providing and the depth at which they are seeing the signals, it is one of the fastest systems,” she said.
Clinical Utility
Beard and colleagues also tested the scanner to visualize blood vessels in participants with RA, suspected PVD, and skin inflammation. The scanning images “illustrated how vascular abnormalities such as increased vessel tortuosity, which has previously been linked to PVD, and the neovascularization associated with inflammation can be visualized and quantified,” the authors wrote.
The next step, Beard noted, is testing whether these characteristics can be used as a marker for the progression of disease.
Nehal Mehta, MD, a cardiologist and professor of medicine at the George Washington University, Washington, DC, agreed that more longitudinal research is needed to understand how the abnormalities captured in these images can inform detection and diagnosis of various diseases.
“You don’t know whether these images look bad because of reverse causation — the disease is doing this — or true causation — that this is actually detecting the root cause of the disease,” he explained. “Until we have a bank of normal and abnormal scans, we don’t know what any of these things mean.”
Though still some time away from entering the clinic, Mehta likened the technology to the introduction of optical coherence tomography in the 1980s. Before being adapted for clinical use, researchers first needed to visualize differences between normal coronary vasculature and myocardial infarction.
“I think this is an amazingly strong first proof of concept,” Mehta said. “This technology is showing a true promise in the field imaging.”
The work was funded by grants from Cancer Research UK, the Engineering & Physical Sciences Research Council, Wellcome Trust, the European Research Council, and the National Institute for Health and Care Research University College London Hospitals Biomedical Research Centre. Beard and two coauthors are shareholders of DeepColor Imaging to which the intellectual property associated with the new scanner has been licensed, but the company was not involved in any of this research. Mallidi and Mehta had no relevant disclosures.
A version of this article first appeared on Medscape.com.
A new scanner can provide three-dimensional (3D) photoacoustic images of millimeter-scale veins and arteries in seconds.
The scanner, developed by researchers at University College London (UCL) in England, could help clinicians better visualize and track microvascular changes for a wide range of diseases, including cancer, rheumatoid arthritis (RA), and peripheral vascular disease (PVD).
The case studies “illustrate potential areas of application that warrant future, more comprehensive clinical studies,” the authors wrote. “Moreover, they demonstrate the feasibility of using the scanner on a real-world patient cohort where imaging is more challenging due to frailty, comorbidity, or pain that may limit their ability to tolerate prolonged scan times.”
The work was published online in Nature Biomedical Engineering.
Improving Photoacoustic Imaging
PAT works using the photoacoustic effect, a phenomenon where sound waves are generated when light is absorbed by a material. When pulsed light from a laser is directed at tissue, some of that light is absorbed and causes an increase in heat in the targeted area. This localized heat also increases pressure, which generates ultrasound waves that can be detected by specialized sensors.
While previous PAT scanners translated these sound waves to electric signals directly to generate imaging, UCL engineers developed a sensor in the early 2000s that can detect these ultrasound waves using light. The result was much clearer, 3D images.
“That was great, but the problem was it was very slow, and it would take 5 minutes to get an image,” explained Paul Beard, PhD, professor of biomedical photoacoustics at UCL and senior author of the study. “That’s fine if you’re imaging a dead mouse or an anesthetized mouse, but not so useful for human imaging,” he continued, where motion would blur the image.
In this new paper, Beard and colleagues outlined how they cut scanning times to an order of seconds (or fraction of a second) rather than minutes. While previous iterations could detect only acoustic waves from one point at a time, this new scanner can detect waves from multiple points simultaneously. The scanner can visualize veins and arteries up to 15 mm deep in human tissue and can also provide dynamic, 3D images of “time-varying tissue perfusion and other hemodynamic events,” the authors wrote.
With these types of scanners, there is always a trade-off between imaging quality and imaging speed, explained Srivalleesha Mallidi, PhD, an assistant professor of biomedical engineering at Tufts University in Medford, Massachusetts. She was not involved with the work.
“With the resolution that [the authors] are providing and the depth at which they are seeing the signals, it is one of the fastest systems,” she said.
Clinical Utility
Beard and colleagues also tested the scanner to visualize blood vessels in participants with RA, suspected PVD, and skin inflammation. The scanning images “illustrated how vascular abnormalities such as increased vessel tortuosity, which has previously been linked to PVD, and the neovascularization associated with inflammation can be visualized and quantified,” the authors wrote.
The next step, Beard noted, is testing whether these characteristics can be used as a marker for the progression of disease.
Nehal Mehta, MD, a cardiologist and professor of medicine at the George Washington University, Washington, DC, agreed that more longitudinal research is needed to understand how the abnormalities captured in these images can inform detection and diagnosis of various diseases.
“You don’t know whether these images look bad because of reverse causation — the disease is doing this — or true causation — that this is actually detecting the root cause of the disease,” he explained. “Until we have a bank of normal and abnormal scans, we don’t know what any of these things mean.”
Though still some time away from entering the clinic, Mehta likened the technology to the introduction of optical coherence tomography in the 1980s. Before being adapted for clinical use, researchers first needed to visualize differences between normal coronary vasculature and myocardial infarction.
“I think this is an amazingly strong first proof of concept,” Mehta said. “This technology is showing a true promise in the field imaging.”
The work was funded by grants from Cancer Research UK, the Engineering & Physical Sciences Research Council, Wellcome Trust, the European Research Council, and the National Institute for Health and Care Research University College London Hospitals Biomedical Research Centre. Beard and two coauthors are shareholders of DeepColor Imaging to which the intellectual property associated with the new scanner has been licensed, but the company was not involved in any of this research. Mallidi and Mehta had no relevant disclosures.
A version of this article first appeared on Medscape.com.
FROM NATURE BIOMEDICAL ENGINEERING
Methotrexate in Preventing RA: Benefits in ACPA-Negative Patients, and Is It Cost Effective?
A 1-year course of methotrexate (MTX) in clinically suspected arthralgia may prevent the development of rheumatoid arthritis (RA) in at-risk individuals who test negative for anti-citrullinated protein antibody (ACPA), according to 4-year results from the TREAT EARLIER study.
While 2-year data did not show a preventive effect, researchers risk-stratified patients in this most recent data. The previous study also grouped all individuals together, while this new analysis separated patients by seropositivity.
“Heterogeneity in risk of rheumatoid arthritis development in ACPA–negative participants with clinically suspect arthralgia might have concealed a treatment effect due to dilution,” wrote senior author Annette H. van der Helm-van Mil, MD, PhD, professor of rheumatology at Leiden University Medical Center, Leiden, the Netherlands, and colleagues. “Therefore, risk-stratified analyses are required to adequately assess the possibility of prevention of rheumatoid arthritis in people with clinically suspect arthralgia who are ACPA–negative.”

To qualify for the study, participants needed to have recent-onset joint pain that a treating rheumatologist suspected of progressing to RA. Second, participants had to have subclinical joint inflammation, detected via MRI.
These are “promising results” for a group where predicting risk for RA has been more difficult than for their ACPA–positive counterparts, said Kevin Deane, MD, PhD, a professor of medicine and rheumatologist at the University of Colorado School of Medicine, Aurora, who was not involved with the study. However, additional research is necessary to investigate these findings.
The clinical utility of this finding is also unclear, he noted, as it would be an “extensive process” for all ACPA–negative individuals with joint pain to undergo MRI screening, he continued.
“It’s hard to find people who would meet these criteria, and healthcare systems need to understand how ultimately this could be implemented in clinical care,” he said.
Adding Risk Stratification
The TREAT EARLIER trial included 236 participants; nearly two thirds were women, and 77% were ACPA–negative (specifically for anti-cyclic citrullinated peptide 2). Patients randomly assigned to active treatment received a single intramuscular glucocorticoid injection (methylprednisolone 120 mg) upon inclusion and then completed a 1-year course of MTX. The comparator group received a single placebo injection at the beginning of the trial and a 1-year course of placebo tablets. All trial screenings and visits were conducted at the Leiden University Medical Center.
At the 2-year mark, there was no difference in the development of RA between the treatment and placebo groups, although there was improvement in joint pain, physical functioning, and MRI-detected joint inflammation in all at-risk groups given MTX — ACPA–positive patients and those at high risk for clinical arthritis development.
MTX delayed the onset of RA, with a statistically significant difference between treatment and placebo at 6 and 12 months, but not at 24 months.
For this 4-year analysis, published in The Lancet Rheumatology, authors stratified patients at their time of enrollment according to their risk of developing RA based on a published model for predicting inflammatory arthritis. Predictors included ACPA positivity (2 points), rheumatoid factor positivity (1 point), more than two locations of subclinical inflammation on MRI (2 points), and presence of metacarpophalangeal extensor tenosynovitis on MRI (1 point).
Patients with at least 4 points were classified as “high-risk,” with a 70% or higher predicted risk of developing RA. Participants with 2-3 points were at “increased risk” — translating to a 25%-70% higher likelihood of developing the condition. Low-risk patients, with 0-1 points, had < 25% chance of developing RA.
Of the 182 ACPA–negative participants in the study, none were considered high risk, 66 (36%) were at increased risk, and 116 (64%) were at low risk.
Decreased Rates of RA Development
Of these ACPA–negative patients stratified as increased risk, 3 of 35 (9%) in the treatment group developed RA, compared with 9 of 31 (29%) in the placebo arm (hazard ratio [HR], 0.27; P = .034).
All 54 ACPA–positive patients enrolled in the study were classified as either increased risk or high risk, but the treatment showed no difference in the rate of RA development in this group, and more than half (56%) developed RA during 4 years of follow-up. However, Dr. van der Helm-van Mil noted that the 2-year data showed treatment improved the severity of subclinical inflammation and symptoms over time in these patients.
The 5-year data from the trial, including physical function and other measures of disease burden, will be analyzed in 2025, she said, and will reveal whether ACPA–negative patients treated with MTX had sustained improvements in these measures.
Additional studies are needed to validate these findings, Dr. van der Helm-van Mil said, but the results indicate that ACPA–positive and ACPA–negative patients “are different populations, and we should evaluate them separately.”
Future RA prevention studies should also risk stratify patients before enrollment so that patients at low risk of developing the disease are not included in the interventions. “You can’t expect a treatment effect if there is no risk for disease,” she added.
Is It Cost-Effective?
In a separate analysis, published in Annals of the Rheumatic Diseases, Dr. van der Helm-van Mil and colleagues sought to investigate the cost-effectiveness of the TREAT EARLIER intervention at 2 years of follow-up.
“There is an ongoing debate whether people with arthralgia at risk for RA should be treated with DMARDs [disease-modifying antirheumatic drugs]; however, the economic effects of an intervention in the arthralgia at risk phase are unknown.”
The analysis calculated healthcare productivity and work productivity costs from enrollment to 2 years of follow-up. To demonstrate effect, they also calculated change in quality-adjusted life years (QALYs).
Over the course of 2 years, estimated costs for the treatment arm were €4809 lower (−$5304) than the placebo arm per patient. Lower productivity costs accounted for 97% of this difference.
The treatment arm also resulted in a small improvement (+0.041) in QALYs, compared with placebo.
“These data provide the first evidence that first-line treatment aiming at secondary prevention in arthralgia at-risk for RA is cost-effective,” the authors wrote.
Dr. van der Helm-van Mil emphasized that this cost analysis used only 2-year follow-up data (rather than the newly published 4-year data) and did not differentiate ACPA–negative patients. Despite including a greater heterogeneity of patients, the intervention was still cost-effective. A future analysis that includes 5-year follow-up data and stratifies patients by ACPA status and excludes low-risk individuals could demonstrate more cost benefits to a temporary MTX regimen, she said.
Considering the costs of these preventive interventions is important, added Dr. Deane, who agreed that future analyses should examine cost-effectiveness in groups at high risk of developing RA. (Dr. Deane noted that he had reviewed this article for publication.) However, this analysis did not include the costs of screening patients before enrollment in the study.

“Additional factors that need to be considered are costs to find individuals who would meet the criteria for treatment,” he said, which would include getting an MRI to detect subclinical joint inflammation.
However, Dr. van der Helm-van Mil noted that both placebo and treatment groups received MRI scans, which would therefore not affect cost differences between the groups.
Future studies should also focus on longer-term outcomes, both Dr. Deane and Dr. van der Helm-van Mil agreed.
“Since RA is a chronic disease for which part of the patients require expensive biologicals, future cost-effectiveness analyses should also consider a lifetime horizon,” Dr. van der Helm-van Mil and colleagues wrote.
The TREAT EARLIER trial was funded by the Dutch Research Council and the Dutch Arthritis Society. The cost analysis study was funded by ZonMw and the Dutch Arthritis Society. Dr. Deane is a member of an American College of Rheumatology/European Alliance of Associations for Rheumatology task force for risk stratification in RA. He has received payments as a speaker for Werfen and Thermo Fisher Scientific and low-cost biomarker assays for research from Werfen. He has also received grant funding from Thermo Fisher Scientific, Gilead, and Boehringer Ingelheim. Dr. van der Helm-van Mil reported no disclosures.
A version of this article first appeared on Medscape.com.
A 1-year course of methotrexate (MTX) in clinically suspected arthralgia may prevent the development of rheumatoid arthritis (RA) in at-risk individuals who test negative for anti-citrullinated protein antibody (ACPA), according to 4-year results from the TREAT EARLIER study.
While 2-year data did not show a preventive effect, researchers risk-stratified patients in this most recent data. The previous study also grouped all individuals together, while this new analysis separated patients by seropositivity.
“Heterogeneity in risk of rheumatoid arthritis development in ACPA–negative participants with clinically suspect arthralgia might have concealed a treatment effect due to dilution,” wrote senior author Annette H. van der Helm-van Mil, MD, PhD, professor of rheumatology at Leiden University Medical Center, Leiden, the Netherlands, and colleagues. “Therefore, risk-stratified analyses are required to adequately assess the possibility of prevention of rheumatoid arthritis in people with clinically suspect arthralgia who are ACPA–negative.”
To qualify for the study, participants needed to have recent-onset joint pain that a treating rheumatologist suspected of progressing to RA. Second, participants had to have subclinical joint inflammation, detected via MRI.
These are “promising results” for a group where predicting risk for RA has been more difficult than for their ACPA–positive counterparts, said Kevin Deane, MD, PhD, a professor of medicine and rheumatologist at the University of Colorado School of Medicine, Aurora, who was not involved with the study. However, additional research is necessary to investigate these findings.
The clinical utility of this finding is also unclear, he noted, as it would be an “extensive process” for all ACPA–negative individuals with joint pain to undergo MRI screening, he continued.
“It’s hard to find people who would meet these criteria, and healthcare systems need to understand how ultimately this could be implemented in clinical care,” he said.
Adding Risk Stratification
The TREAT EARLIER trial included 236 participants; nearly two thirds were women, and 77% were ACPA–negative (specifically for anti-cyclic citrullinated peptide 2). Patients randomly assigned to active treatment received a single intramuscular glucocorticoid injection (methylprednisolone 120 mg) upon inclusion and then completed a 1-year course of MTX. The comparator group received a single placebo injection at the beginning of the trial and a 1-year course of placebo tablets. All trial screenings and visits were conducted at the Leiden University Medical Center.
At the 2-year mark, there was no difference in the development of RA between the treatment and placebo groups, although there was improvement in joint pain, physical functioning, and MRI-detected joint inflammation in all at-risk groups given MTX — ACPA–positive patients and those at high risk for clinical arthritis development.
MTX delayed the onset of RA, with a statistically significant difference between treatment and placebo at 6 and 12 months, but not at 24 months.
For this 4-year analysis, published in The Lancet Rheumatology, authors stratified patients at their time of enrollment according to their risk of developing RA based on a published model for predicting inflammatory arthritis. Predictors included ACPA positivity (2 points), rheumatoid factor positivity (1 point), more than two locations of subclinical inflammation on MRI (2 points), and presence of metacarpophalangeal extensor tenosynovitis on MRI (1 point).
Patients with at least 4 points were classified as “high-risk,” with a 70% or higher predicted risk of developing RA. Participants with 2-3 points were at “increased risk” — translating to a 25%-70% higher likelihood of developing the condition. Low-risk patients, with 0-1 points, had < 25% chance of developing RA.
Of the 182 ACPA–negative participants in the study, none were considered high risk, 66 (36%) were at increased risk, and 116 (64%) were at low risk.
Decreased Rates of RA Development
Of these ACPA–negative patients stratified as increased risk, 3 of 35 (9%) in the treatment group developed RA, compared with 9 of 31 (29%) in the placebo arm (hazard ratio [HR], 0.27; P = .034).
All 54 ACPA–positive patients enrolled in the study were classified as either increased risk or high risk, but the treatment showed no difference in the rate of RA development in this group, and more than half (56%) developed RA during 4 years of follow-up. However, Dr. van der Helm-van Mil noted that the 2-year data showed treatment improved the severity of subclinical inflammation and symptoms over time in these patients.
The 5-year data from the trial, including physical function and other measures of disease burden, will be analyzed in 2025, she said, and will reveal whether ACPA–negative patients treated with MTX had sustained improvements in these measures.
Additional studies are needed to validate these findings, Dr. van der Helm-van Mil said, but the results indicate that ACPA–positive and ACPA–negative patients “are different populations, and we should evaluate them separately.”
Future RA prevention studies should also risk stratify patients before enrollment so that patients at low risk of developing the disease are not included in the interventions. “You can’t expect a treatment effect if there is no risk for disease,” she added.
Is It Cost-Effective?
In a separate analysis, published in Annals of the Rheumatic Diseases, Dr. van der Helm-van Mil and colleagues sought to investigate the cost-effectiveness of the TREAT EARLIER intervention at 2 years of follow-up.
“There is an ongoing debate whether people with arthralgia at risk for RA should be treated with DMARDs [disease-modifying antirheumatic drugs]; however, the economic effects of an intervention in the arthralgia at risk phase are unknown.”
The analysis calculated healthcare productivity and work productivity costs from enrollment to 2 years of follow-up. To demonstrate effect, they also calculated change in quality-adjusted life years (QALYs).
Over the course of 2 years, estimated costs for the treatment arm were €4809 lower (−$5304) than the placebo arm per patient. Lower productivity costs accounted for 97% of this difference.
The treatment arm also resulted in a small improvement (+0.041) in QALYs, compared with placebo.
“These data provide the first evidence that first-line treatment aiming at secondary prevention in arthralgia at-risk for RA is cost-effective,” the authors wrote.
Dr. van der Helm-van Mil emphasized that this cost analysis used only 2-year follow-up data (rather than the newly published 4-year data) and did not differentiate ACPA–negative patients. Despite including a greater heterogeneity of patients, the intervention was still cost-effective. A future analysis that includes 5-year follow-up data and stratifies patients by ACPA status and excludes low-risk individuals could demonstrate more cost benefits to a temporary MTX regimen, she said.
Considering the costs of these preventive interventions is important, added Dr. Deane, who agreed that future analyses should examine cost-effectiveness in groups at high risk of developing RA. (Dr. Deane noted that he had reviewed this article for publication.) However, this analysis did not include the costs of screening patients before enrollment in the study.
“Additional factors that need to be considered are costs to find individuals who would meet the criteria for treatment,” he said, which would include getting an MRI to detect subclinical joint inflammation.
However, Dr. van der Helm-van Mil noted that both placebo and treatment groups received MRI scans, which would therefore not affect cost differences between the groups.
Future studies should also focus on longer-term outcomes, both Dr. Deane and Dr. van der Helm-van Mil agreed.
“Since RA is a chronic disease for which part of the patients require expensive biologicals, future cost-effectiveness analyses should also consider a lifetime horizon,” Dr. van der Helm-van Mil and colleagues wrote.
The TREAT EARLIER trial was funded by the Dutch Research Council and the Dutch Arthritis Society. The cost analysis study was funded by ZonMw and the Dutch Arthritis Society. Dr. Deane is a member of an American College of Rheumatology/European Alliance of Associations for Rheumatology task force for risk stratification in RA. He has received payments as a speaker for Werfen and Thermo Fisher Scientific and low-cost biomarker assays for research from Werfen. He has also received grant funding from Thermo Fisher Scientific, Gilead, and Boehringer Ingelheim. Dr. van der Helm-van Mil reported no disclosures.
A version of this article first appeared on Medscape.com.
A 1-year course of methotrexate (MTX) in clinically suspected arthralgia may prevent the development of rheumatoid arthritis (RA) in at-risk individuals who test negative for anti-citrullinated protein antibody (ACPA), according to 4-year results from the TREAT EARLIER study.
While 2-year data did not show a preventive effect, researchers risk-stratified patients in this most recent data. The previous study also grouped all individuals together, while this new analysis separated patients by seropositivity.
“Heterogeneity in risk of rheumatoid arthritis development in ACPA–negative participants with clinically suspect arthralgia might have concealed a treatment effect due to dilution,” wrote senior author Annette H. van der Helm-van Mil, MD, PhD, professor of rheumatology at Leiden University Medical Center, Leiden, the Netherlands, and colleagues. “Therefore, risk-stratified analyses are required to adequately assess the possibility of prevention of rheumatoid arthritis in people with clinically suspect arthralgia who are ACPA–negative.”
To qualify for the study, participants needed to have recent-onset joint pain that a treating rheumatologist suspected of progressing to RA. Second, participants had to have subclinical joint inflammation, detected via MRI.
These are “promising results” for a group where predicting risk for RA has been more difficult than for their ACPA–positive counterparts, said Kevin Deane, MD, PhD, a professor of medicine and rheumatologist at the University of Colorado School of Medicine, Aurora, who was not involved with the study. However, additional research is necessary to investigate these findings.
The clinical utility of this finding is also unclear, he noted, as it would be an “extensive process” for all ACPA–negative individuals with joint pain to undergo MRI screening, he continued.
“It’s hard to find people who would meet these criteria, and healthcare systems need to understand how ultimately this could be implemented in clinical care,” he said.
Adding Risk Stratification
The TREAT EARLIER trial included 236 participants; nearly two thirds were women, and 77% were ACPA–negative (specifically for anti-cyclic citrullinated peptide 2). Patients randomly assigned to active treatment received a single intramuscular glucocorticoid injection (methylprednisolone 120 mg) upon inclusion and then completed a 1-year course of MTX. The comparator group received a single placebo injection at the beginning of the trial and a 1-year course of placebo tablets. All trial screenings and visits were conducted at the Leiden University Medical Center.
At the 2-year mark, there was no difference in the development of RA between the treatment and placebo groups, although there was improvement in joint pain, physical functioning, and MRI-detected joint inflammation in all at-risk groups given MTX — ACPA–positive patients and those at high risk for clinical arthritis development.
MTX delayed the onset of RA, with a statistically significant difference between treatment and placebo at 6 and 12 months, but not at 24 months.
For this 4-year analysis, published in The Lancet Rheumatology, authors stratified patients at their time of enrollment according to their risk of developing RA based on a published model for predicting inflammatory arthritis. Predictors included ACPA positivity (2 points), rheumatoid factor positivity (1 point), more than two locations of subclinical inflammation on MRI (2 points), and presence of metacarpophalangeal extensor tenosynovitis on MRI (1 point).
Patients with at least 4 points were classified as “high-risk,” with a 70% or higher predicted risk of developing RA. Participants with 2-3 points were at “increased risk” — translating to a 25%-70% higher likelihood of developing the condition. Low-risk patients, with 0-1 points, had < 25% chance of developing RA.
Of the 182 ACPA–negative participants in the study, none were considered high risk, 66 (36%) were at increased risk, and 116 (64%) were at low risk.
Decreased Rates of RA Development
Of these ACPA–negative patients stratified as increased risk, 3 of 35 (9%) in the treatment group developed RA, compared with 9 of 31 (29%) in the placebo arm (hazard ratio [HR], 0.27; P = .034).
All 54 ACPA–positive patients enrolled in the study were classified as either increased risk or high risk, but the treatment showed no difference in the rate of RA development in this group, and more than half (56%) developed RA during 4 years of follow-up. However, Dr. van der Helm-van Mil noted that the 2-year data showed treatment improved the severity of subclinical inflammation and symptoms over time in these patients.
The 5-year data from the trial, including physical function and other measures of disease burden, will be analyzed in 2025, she said, and will reveal whether ACPA–negative patients treated with MTX had sustained improvements in these measures.
Additional studies are needed to validate these findings, Dr. van der Helm-van Mil said, but the results indicate that ACPA–positive and ACPA–negative patients “are different populations, and we should evaluate them separately.”
Future RA prevention studies should also risk stratify patients before enrollment so that patients at low risk of developing the disease are not included in the interventions. “You can’t expect a treatment effect if there is no risk for disease,” she added.
Is It Cost-Effective?
In a separate analysis, published in Annals of the Rheumatic Diseases, Dr. van der Helm-van Mil and colleagues sought to investigate the cost-effectiveness of the TREAT EARLIER intervention at 2 years of follow-up.
“There is an ongoing debate whether people with arthralgia at risk for RA should be treated with DMARDs [disease-modifying antirheumatic drugs]; however, the economic effects of an intervention in the arthralgia at risk phase are unknown.”
The analysis calculated healthcare productivity and work productivity costs from enrollment to 2 years of follow-up. To demonstrate effect, they also calculated change in quality-adjusted life years (QALYs).
Over the course of 2 years, estimated costs for the treatment arm were €4809 lower (−$5304) than the placebo arm per patient. Lower productivity costs accounted for 97% of this difference.
The treatment arm also resulted in a small improvement (+0.041) in QALYs, compared with placebo.
“These data provide the first evidence that first-line treatment aiming at secondary prevention in arthralgia at-risk for RA is cost-effective,” the authors wrote.
Dr. van der Helm-van Mil emphasized that this cost analysis used only 2-year follow-up data (rather than the newly published 4-year data) and did not differentiate ACPA–negative patients. Despite including a greater heterogeneity of patients, the intervention was still cost-effective. A future analysis that includes 5-year follow-up data and stratifies patients by ACPA status and excludes low-risk individuals could demonstrate more cost benefits to a temporary MTX regimen, she said.
Considering the costs of these preventive interventions is important, added Dr. Deane, who agreed that future analyses should examine cost-effectiveness in groups at high risk of developing RA. (Dr. Deane noted that he had reviewed this article for publication.) However, this analysis did not include the costs of screening patients before enrollment in the study.
“Additional factors that need to be considered are costs to find individuals who would meet the criteria for treatment,” he said, which would include getting an MRI to detect subclinical joint inflammation.
However, Dr. van der Helm-van Mil noted that both placebo and treatment groups received MRI scans, which would therefore not affect cost differences between the groups.
Future studies should also focus on longer-term outcomes, both Dr. Deane and Dr. van der Helm-van Mil agreed.
“Since RA is a chronic disease for which part of the patients require expensive biologicals, future cost-effectiveness analyses should also consider a lifetime horizon,” Dr. van der Helm-van Mil and colleagues wrote.
The TREAT EARLIER trial was funded by the Dutch Research Council and the Dutch Arthritis Society. The cost analysis study was funded by ZonMw and the Dutch Arthritis Society. Dr. Deane is a member of an American College of Rheumatology/European Alliance of Associations for Rheumatology task force for risk stratification in RA. He has received payments as a speaker for Werfen and Thermo Fisher Scientific and low-cost biomarker assays for research from Werfen. He has also received grant funding from Thermo Fisher Scientific, Gilead, and Boehringer Ingelheim. Dr. van der Helm-van Mil reported no disclosures.
A version of this article first appeared on Medscape.com.

FDA Approves Ustekinumab Biosimilar Otulfi
This is the fourth ustekinumab biosimilar approved in the United States. Like the reference product, ustekinumab-aauz is indicated for:
- Patients 6 years or older with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy
- Patients 6 years or older with active psoriatic arthritis
- Adult patients with moderately to severely active Crohn’s disease
- Adult patients with moderately to severely active ulcerative colitis
Ustekinumab-aauz, produced by a partnership between Fresenius Kabi and Formycon, has two formulations: subcutaneous injection (45 mg/0.5 mL or 90 mg/mL solution in a single-dose prefilled syringe) or intravenous infusion (130 mg/26 mL solution in a single-dose vial).
The biosimilar will launch in the United States “no later than February 22, 2025,” according to the press release, “in accordance with the patent settlement between Fresenius Kabi, Formycon, and Johnson & Johnson.”
Ustekinumab-aauz is Fresenius Kabi’s fourth biosimilar granted US approval, behind adalimumab-aacf (Idacio), tocilizumab-aazg (Tyenne), and pegfilgrastim-fpgk (Stimufend).
A version of this article first appeared on Medscape.com.
This is the fourth ustekinumab biosimilar approved in the United States. Like the reference product, ustekinumab-aauz is indicated for:
- Patients 6 years or older with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy
- Patients 6 years or older with active psoriatic arthritis
- Adult patients with moderately to severely active Crohn’s disease
- Adult patients with moderately to severely active ulcerative colitis
Ustekinumab-aauz, produced by a partnership between Fresenius Kabi and Formycon, has two formulations: subcutaneous injection (45 mg/0.5 mL or 90 mg/mL solution in a single-dose prefilled syringe) or intravenous infusion (130 mg/26 mL solution in a single-dose vial).
The biosimilar will launch in the United States “no later than February 22, 2025,” according to the press release, “in accordance with the patent settlement between Fresenius Kabi, Formycon, and Johnson & Johnson.”
Ustekinumab-aauz is Fresenius Kabi’s fourth biosimilar granted US approval, behind adalimumab-aacf (Idacio), tocilizumab-aazg (Tyenne), and pegfilgrastim-fpgk (Stimufend).
A version of this article first appeared on Medscape.com.
This is the fourth ustekinumab biosimilar approved in the United States. Like the reference product, ustekinumab-aauz is indicated for:
- Patients 6 years or older with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy
- Patients 6 years or older with active psoriatic arthritis
- Adult patients with moderately to severely active Crohn’s disease
- Adult patients with moderately to severely active ulcerative colitis
Ustekinumab-aauz, produced by a partnership between Fresenius Kabi and Formycon, has two formulations: subcutaneous injection (45 mg/0.5 mL or 90 mg/mL solution in a single-dose prefilled syringe) or intravenous infusion (130 mg/26 mL solution in a single-dose vial).
The biosimilar will launch in the United States “no later than February 22, 2025,” according to the press release, “in accordance with the patent settlement between Fresenius Kabi, Formycon, and Johnson & Johnson.”
Ustekinumab-aauz is Fresenius Kabi’s fourth biosimilar granted US approval, behind adalimumab-aacf (Idacio), tocilizumab-aazg (Tyenne), and pegfilgrastim-fpgk (Stimufend).
A version of this article first appeared on Medscape.com.
Bimekizumab Gains FDA Approval for Psoriatic Arthritis, Axial Spondyloarthritis
The Food and Drug Administration has approved bimekizumab-bkzx (Bimzelx; UCB) for adult patients with active psoriatic arthritis (PsA), active nonradiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation, and active ankylosing spondylitis (AS).
The drug, an interleukin (IL)–17A and IL-17F inhibitor, was first approved in October 2023 for treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy.
“In psoriatic arthritis and across the spectrum of axSpA, clinical study results and real-world experience outside the US have highlighted that Bimzelx can help patients achieve high thresholds of clinical response that are rapid in onset and sustained up to 2 years,” said Emmanuel Caeymaex, executive vice president, head of patient impact, and chief commercial officer of UCB in a press release.
The recommended dosage of bimekizumab for adult patients with active PsA, nr-axSpA, or AS is 160 mg by subcutaneous injection every 4 weeks. For patients with PsA and coexistent moderate to severe plaque psoriasis, the dosage is the same as for patients with plaque psoriasis. The dosing for plaque psoriasis is to administer 320 mg (two 160-mg injections) by subcutaneous injection at weeks 0, 4, 8, 12, and 16, then every 8 weeks thereafter. For patients weighing ≥ 120 kg, consider a dose of 320 mg every 4 weeks after week 16.
PsA Clinical Trials
The approval for PsA was based on data from two phase 3 clinical trials, including 852 participants naive to biologics (BE OPTIMAL) and 400 participants with inadequate response to treatment with one or two tumor necrosis factor (TNF) inhibitors (BE COMPLETE). Both studies met their primary endpoint, 50% improvement in American College of Rheumatology response criteria (ACR50) at 16 weeks, as well as ranked secondary endpoints. Secondary endpoints included minimal disease activity (MDA) and Psoriasis Area and Severity Index 100 (complete skin clearance) at week 16.
At 16 weeks:
- About 44% of both the biologic-naive (189 of 431) and TNF inhibitor–resistant (116 of 267) groups receiving bimekizumab achieved ACR50 response, compared with 10% (28 of 281) and 7% (9 of 133) receiving placebo, respectively.
- About 45% of all patients treated with bimekizumab achieved MDA.
- Nearly 60% of TNF inhibitor–resistant patients had complete skin clearance.
These responses generally were sustained for 1 year. The most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, and urinary tract infection.
NR-axSpA and AS Clinical Trials
The approval for active nr-axSpA and active AS was based on data from two clinical studies, BE MOBILE 1 (nr-axSpA) and BE MOBILE 2 (AS). Both studies met their primary endpoint, 40% improvement in Assessment of Spondyloarthritis International Society response criteria (ASAS40) at 16 weeks.
Key findings included:
- In nr-axSpA patients, 47.7% (61 of 128) receiving bimekizumab achieved ASAS40 at week 16, compared with 21.4% (27 of 126) receiving placebo.
- In AS patients, 44.8% (99 of 221) in the bimekizumab group achieved ASAS40 response at week 16 vs 22.5% (25 of 111) receiving placebo.
- At 1 year in both groups, 60% treated with bimekizumab achieved an Ankylosing Spondylitis Disease Activity Score < 2.1.
In nr-axSpA, the most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, cough, fatigue, musculoskeletal pain, myalgia, tonsillitis, increase in transaminase, and urinary tract infection. In AS, the most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, injection-site pain, rash, and vulvovaginal mycotic infection.
Bimekizumab was approved by the European Commission for the same rheumatologic indications in June 2023.
Bimekizumab is currently available to eligible patients in the United States, according to the press release.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved bimekizumab-bkzx (Bimzelx; UCB) for adult patients with active psoriatic arthritis (PsA), active nonradiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation, and active ankylosing spondylitis (AS).
The drug, an interleukin (IL)–17A and IL-17F inhibitor, was first approved in October 2023 for treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy.
“In psoriatic arthritis and across the spectrum of axSpA, clinical study results and real-world experience outside the US have highlighted that Bimzelx can help patients achieve high thresholds of clinical response that are rapid in onset and sustained up to 2 years,” said Emmanuel Caeymaex, executive vice president, head of patient impact, and chief commercial officer of UCB in a press release.
The recommended dosage of bimekizumab for adult patients with active PsA, nr-axSpA, or AS is 160 mg by subcutaneous injection every 4 weeks. For patients with PsA and coexistent moderate to severe plaque psoriasis, the dosage is the same as for patients with plaque psoriasis. The dosing for plaque psoriasis is to administer 320 mg (two 160-mg injections) by subcutaneous injection at weeks 0, 4, 8, 12, and 16, then every 8 weeks thereafter. For patients weighing ≥ 120 kg, consider a dose of 320 mg every 4 weeks after week 16.
PsA Clinical Trials
The approval for PsA was based on data from two phase 3 clinical trials, including 852 participants naive to biologics (BE OPTIMAL) and 400 participants with inadequate response to treatment with one or two tumor necrosis factor (TNF) inhibitors (BE COMPLETE). Both studies met their primary endpoint, 50% improvement in American College of Rheumatology response criteria (ACR50) at 16 weeks, as well as ranked secondary endpoints. Secondary endpoints included minimal disease activity (MDA) and Psoriasis Area and Severity Index 100 (complete skin clearance) at week 16.
At 16 weeks:
- About 44% of both the biologic-naive (189 of 431) and TNF inhibitor–resistant (116 of 267) groups receiving bimekizumab achieved ACR50 response, compared with 10% (28 of 281) and 7% (9 of 133) receiving placebo, respectively.
- About 45% of all patients treated with bimekizumab achieved MDA.
- Nearly 60% of TNF inhibitor–resistant patients had complete skin clearance.
These responses generally were sustained for 1 year. The most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, and urinary tract infection.
NR-axSpA and AS Clinical Trials
The approval for active nr-axSpA and active AS was based on data from two clinical studies, BE MOBILE 1 (nr-axSpA) and BE MOBILE 2 (AS). Both studies met their primary endpoint, 40% improvement in Assessment of Spondyloarthritis International Society response criteria (ASAS40) at 16 weeks.
Key findings included:
- In nr-axSpA patients, 47.7% (61 of 128) receiving bimekizumab achieved ASAS40 at week 16, compared with 21.4% (27 of 126) receiving placebo.
- In AS patients, 44.8% (99 of 221) in the bimekizumab group achieved ASAS40 response at week 16 vs 22.5% (25 of 111) receiving placebo.
- At 1 year in both groups, 60% treated with bimekizumab achieved an Ankylosing Spondylitis Disease Activity Score < 2.1.
In nr-axSpA, the most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, cough, fatigue, musculoskeletal pain, myalgia, tonsillitis, increase in transaminase, and urinary tract infection. In AS, the most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, injection-site pain, rash, and vulvovaginal mycotic infection.
Bimekizumab was approved by the European Commission for the same rheumatologic indications in June 2023.
Bimekizumab is currently available to eligible patients in the United States, according to the press release.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved bimekizumab-bkzx (Bimzelx; UCB) for adult patients with active psoriatic arthritis (PsA), active nonradiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation, and active ankylosing spondylitis (AS).
The drug, an interleukin (IL)–17A and IL-17F inhibitor, was first approved in October 2023 for treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy.
“In psoriatic arthritis and across the spectrum of axSpA, clinical study results and real-world experience outside the US have highlighted that Bimzelx can help patients achieve high thresholds of clinical response that are rapid in onset and sustained up to 2 years,” said Emmanuel Caeymaex, executive vice president, head of patient impact, and chief commercial officer of UCB in a press release.
The recommended dosage of bimekizumab for adult patients with active PsA, nr-axSpA, or AS is 160 mg by subcutaneous injection every 4 weeks. For patients with PsA and coexistent moderate to severe plaque psoriasis, the dosage is the same as for patients with plaque psoriasis. The dosing for plaque psoriasis is to administer 320 mg (two 160-mg injections) by subcutaneous injection at weeks 0, 4, 8, 12, and 16, then every 8 weeks thereafter. For patients weighing ≥ 120 kg, consider a dose of 320 mg every 4 weeks after week 16.
PsA Clinical Trials
The approval for PsA was based on data from two phase 3 clinical trials, including 852 participants naive to biologics (BE OPTIMAL) and 400 participants with inadequate response to treatment with one or two tumor necrosis factor (TNF) inhibitors (BE COMPLETE). Both studies met their primary endpoint, 50% improvement in American College of Rheumatology response criteria (ACR50) at 16 weeks, as well as ranked secondary endpoints. Secondary endpoints included minimal disease activity (MDA) and Psoriasis Area and Severity Index 100 (complete skin clearance) at week 16.
At 16 weeks:
- About 44% of both the biologic-naive (189 of 431) and TNF inhibitor–resistant (116 of 267) groups receiving bimekizumab achieved ACR50 response, compared with 10% (28 of 281) and 7% (9 of 133) receiving placebo, respectively.
- About 45% of all patients treated with bimekizumab achieved MDA.
- Nearly 60% of TNF inhibitor–resistant patients had complete skin clearance.
These responses generally were sustained for 1 year. The most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, and urinary tract infection.
NR-axSpA and AS Clinical Trials
The approval for active nr-axSpA and active AS was based on data from two clinical studies, BE MOBILE 1 (nr-axSpA) and BE MOBILE 2 (AS). Both studies met their primary endpoint, 40% improvement in Assessment of Spondyloarthritis International Society response criteria (ASAS40) at 16 weeks.
Key findings included:
- In nr-axSpA patients, 47.7% (61 of 128) receiving bimekizumab achieved ASAS40 at week 16, compared with 21.4% (27 of 126) receiving placebo.
- In AS patients, 44.8% (99 of 221) in the bimekizumab group achieved ASAS40 response at week 16 vs 22.5% (25 of 111) receiving placebo.
- At 1 year in both groups, 60% treated with bimekizumab achieved an Ankylosing Spondylitis Disease Activity Score < 2.1.
In nr-axSpA, the most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, cough, fatigue, musculoskeletal pain, myalgia, tonsillitis, increase in transaminase, and urinary tract infection. In AS, the most common adverse reactions are upper respiratory tract infections, oral candidiasis, headache, diarrhea, injection-site pain, rash, and vulvovaginal mycotic infection.
Bimekizumab was approved by the European Commission for the same rheumatologic indications in June 2023.
Bimekizumab is currently available to eligible patients in the United States, according to the press release.
A version of this article first appeared on Medscape.com.
Benralizumab Now FDA Approved to Treat EGPA Vasculitis
The Food and Drug Administration (FDA) has approved benralizumab (Fasenra) for the treatment of adults with eosinophilic granulomatosis with polyangiitis (EGPA), formerly known as Churg-Strauss syndrome.
The drug is the second approved biologic for the treatment of EGPA. The first, mepolizumab (Nucala), was approved in 2017.
“This disease has a devastating impact on patients and the quality of their life, and they need more treatment options. The approval of another treatment in EGPA is welcome news to the approximately 15,000 patients living in the US with this difficult-to-treat rare disease,” said Joyce Kullman, executive director of the Vasculitis Foundation, in a press release on September 18. 
Benralizumab, developed by AstraZeneca, is a monoclonal antibody against the interleukin-5 alpha receptor expressed on eosinophils. The drug was first approved in 2017 as an add-on treatment for patients 12 years and older with severe eosinophilic asthma, and is now approved for use in children aged 6 years and older.
The new indication was based on positive results from a noninferiority trial comparing benralizumab and mepolizumab. For the trial, published in the New England Journal of Medicine earlier in 2024, 140 adults with relapsing or refractory EGPA were randomized to a 30-mg subcutaneous injection of benralizumab or three separate 100-mg mepolizumab injections every 4 weeks for 1 year. At weeks 36 and 48, 59% of patients in the benralizumab group and 56% of patients in the mepolizumab group achieved remission (95% CI, –13 to 18; P = .73 for superiority). From week 42 to 52, 41% of patients who received benralizumab completely stopped taking oral glucocorticoids, compared with 26% of those who received mepolizumab.
“Patients often rely on long-term oral corticosteroids, which can cause serious and lasting side effects. Benralizumab is a much-needed treatment option, with data showing that not only is remission an achievable goal for EGPA patients, but benralizumab can also help patients taper off steroid therapy,” Michael Wechsler, MD, director of The Asthma Institute at National Jewish Health in Denver, Colorado, and the international coordinating investigator for the clinical trial, said in the press release.
Benralizumab is administered via subcutaneous injection. In adults with EGPA, the recommended dosage is 30 mg every 4 weeks for the first three doses, then once every 8 weeks.
The most common adverse reactions include headache and pharyngitis, according to the prescribing information.
Benralizumab is also in development for the treatment of chronic obstructive pulmonary disease, chronic rhinosinusitis with nasal polyps, and hypereosinophilic syndrome.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration (FDA) has approved benralizumab (Fasenra) for the treatment of adults with eosinophilic granulomatosis with polyangiitis (EGPA), formerly known as Churg-Strauss syndrome.
The drug is the second approved biologic for the treatment of EGPA. The first, mepolizumab (Nucala), was approved in 2017.
“This disease has a devastating impact on patients and the quality of their life, and they need more treatment options. The approval of another treatment in EGPA is welcome news to the approximately 15,000 patients living in the US with this difficult-to-treat rare disease,” said Joyce Kullman, executive director of the Vasculitis Foundation, in a press release on September 18.
Benralizumab, developed by AstraZeneca, is a monoclonal antibody against the interleukin-5 alpha receptor expressed on eosinophils. The drug was first approved in 2017 as an add-on treatment for patients 12 years and older with severe eosinophilic asthma, and is now approved for use in children aged 6 years and older.
The new indication was based on positive results from a noninferiority trial comparing benralizumab and mepolizumab. For the trial, published in the New England Journal of Medicine earlier in 2024, 140 adults with relapsing or refractory EGPA were randomized to a 30-mg subcutaneous injection of benralizumab or three separate 100-mg mepolizumab injections every 4 weeks for 1 year. At weeks 36 and 48, 59% of patients in the benralizumab group and 56% of patients in the mepolizumab group achieved remission (95% CI, –13 to 18; P = .73 for superiority). From week 42 to 52, 41% of patients who received benralizumab completely stopped taking oral glucocorticoids, compared with 26% of those who received mepolizumab.
“Patients often rely on long-term oral corticosteroids, which can cause serious and lasting side effects. Benralizumab is a much-needed treatment option, with data showing that not only is remission an achievable goal for EGPA patients, but benralizumab can also help patients taper off steroid therapy,” Michael Wechsler, MD, director of The Asthma Institute at National Jewish Health in Denver, Colorado, and the international coordinating investigator for the clinical trial, said in the press release.
Benralizumab is administered via subcutaneous injection. In adults with EGPA, the recommended dosage is 30 mg every 4 weeks for the first three doses, then once every 8 weeks.
The most common adverse reactions include headache and pharyngitis, according to the prescribing information.
Benralizumab is also in development for the treatment of chronic obstructive pulmonary disease, chronic rhinosinusitis with nasal polyps, and hypereosinophilic syndrome.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration (FDA) has approved benralizumab (Fasenra) for the treatment of adults with eosinophilic granulomatosis with polyangiitis (EGPA), formerly known as Churg-Strauss syndrome.
The drug is the second approved biologic for the treatment of EGPA. The first, mepolizumab (Nucala), was approved in 2017.
“This disease has a devastating impact on patients and the quality of their life, and they need more treatment options. The approval of another treatment in EGPA is welcome news to the approximately 15,000 patients living in the US with this difficult-to-treat rare disease,” said Joyce Kullman, executive director of the Vasculitis Foundation, in a press release on September 18.
Benralizumab, developed by AstraZeneca, is a monoclonal antibody against the interleukin-5 alpha receptor expressed on eosinophils. The drug was first approved in 2017 as an add-on treatment for patients 12 years and older with severe eosinophilic asthma, and is now approved for use in children aged 6 years and older.
The new indication was based on positive results from a noninferiority trial comparing benralizumab and mepolizumab. For the trial, published in the New England Journal of Medicine earlier in 2024, 140 adults with relapsing or refractory EGPA were randomized to a 30-mg subcutaneous injection of benralizumab or three separate 100-mg mepolizumab injections every 4 weeks for 1 year. At weeks 36 and 48, 59% of patients in the benralizumab group and 56% of patients in the mepolizumab group achieved remission (95% CI, –13 to 18; P = .73 for superiority). From week 42 to 52, 41% of patients who received benralizumab completely stopped taking oral glucocorticoids, compared with 26% of those who received mepolizumab.
“Patients often rely on long-term oral corticosteroids, which can cause serious and lasting side effects. Benralizumab is a much-needed treatment option, with data showing that not only is remission an achievable goal for EGPA patients, but benralizumab can also help patients taper off steroid therapy,” Michael Wechsler, MD, director of The Asthma Institute at National Jewish Health in Denver, Colorado, and the international coordinating investigator for the clinical trial, said in the press release.
Benralizumab is administered via subcutaneous injection. In adults with EGPA, the recommended dosage is 30 mg every 4 weeks for the first three doses, then once every 8 weeks.
The most common adverse reactions include headache and pharyngitis, according to the prescribing information.
Benralizumab is also in development for the treatment of chronic obstructive pulmonary disease, chronic rhinosinusitis with nasal polyps, and hypereosinophilic syndrome.
A version of this article first appeared on Medscape.com.
Rheumatology Clinic Interventions for Smoking, Blood Pressure ‘Make a Big Difference’
Two relatively simple interventions — addressing high blood pressure (BP) and smoking cessation — could make a huge difference for patients with rheumatic disease. Patients with autoimmune disease are up to three times more likely to develop cardiovascular disease (CVD) than the general population. In addition to compounding CVD, smoking is tied to the development of certain autoimmune conditions, as well as worse outcomes. Christie Bartels, MD, chief of the Division of Rheumatology at the University of Wisconsin School of Medicine and Public Health, Madison, has focused her research on improving cardiac health in inflammatory diseases. This news organization spoke with Bartels about two short interventions she developed that tackle hypertension and smoking cessation during regular visits, each taking less than 3 minutes.
How Do These Programs Address Cardiac Disease Prevention?
The BP and Quit Connect programs help clinics systematically address the two most modifiable risk factors for CVD: high BP and smoking. There’s also evidence that addressing these two risk factors improves outcomes in rheumatic diseases. Hypertension predicts an increase in lupus damage. Particularly in lupus nephritis, hypertension will increase the risk for CVD and kidney failure. People who use tobacco have worse outcomes in diseases like rheumatoid arthritis, psoriatic arthritis, and lupus, as well as more CVD, and antirheumatic drugs may not work as well.

In 90 seconds to 3 minutes, staff can do protocol-based care, which we’ve done across 20,000-plus visits. We showed we can improve population level rates of high BP and BP control, as well as increase smoking quitting rates across different patient settings.
What Is the Quit Connect Program?
The Quit Connect program is a 10- to 90-second point of care intervention. During rooming, staff (medical assistants and nurses) ask patients: “A) Do you smoke? and B) Have you thought about cutting back or quitting in the next 30 days?”
It turns out, when you ask the question that way, between a third and a half of people say that they’ve thought about cutting back or quitting. Then, we can get patients connected directly to Quitline, a free public service across all 50 states that smokers can use to get cessation support.
If patients are ready, we ask if we can arrange for them to receive a call from a Quitline coach about setting a quit date or receiving free nicotine replacement therapy. The beautiful thing is when that all happens, A) it’s free to the patient, and B) the results from the Quitline can be recorded right back to the electronic health record.
In our most recent publication in Arthritis Care & Research, we documented bringing Quit Connect to Grady Hospital in downtown Atlanta. It’s a safety net hospital, where 80% patients are Black and 70%-80% patients are on public insurance or uninsured. Using this protocol, we improved Quitline referrals 20-fold.
What Is the BP Connect Program?
At least half of the encounters in United States happen in specialty clinics. Unfortunately, when patients get their BP measured in a specialty clinic that’s not a cardiology or a vascular clinic, often, even if the pressure is high, the clinic doesn’t give patients feedback on that. The problem is because we haven’t said anything, that gives people the false reassurance that their BP is okay.
We’ve developed a 3-minute protocol to ask, advise, and connect. The idea is that if we measure a high BP, then we remeasure and confirm that it’s high. Then, we advise why it matters in rheumatic disease: Patients with rheumatic diseases are already at an increased risk for heart disease, and controlling BP can make a big difference. Then, we connect patients with high BP back to primary care.
Specifically, a SmartSet — an electronic medical record feature — prompts different actions based on confirmed high BP readings:
- If systolic BP ≥ 140-159, the SmartSet directs scheduling a visit to a nurse or primary care provider.
- If systolic BP ≥ 160-179, the next primary care visit anticipates the need to see a prescriber.
- If systolic BP ≥ 180, then the medical assistant or nurse at the visit is instructed to notify the provider who can arrange a provider-to-provider handoff for safety to exclude a hypertensive emergency.
That order goes to the scheduler to call primary care to coordinate follow-up. BP Connect doubled the likelihood of a guideline-recommended follow-up in primary care within 30 days. All patients benefited, and disparities decreased. BP Connect has had 1100 downloads, and both BP and Quit Connect programs are endorsed by the Centers for Disease Control and Prevention and Million Hearts.
How Do These Programs Affect Clinical Practice?
We developed these interventions with a health system engineer, and we time stamped everything. Part of the sustainability of this model is that it fits within a regular workflow. As a practicing rheumatologist, I understand that time is a precious commodity.
The interventions are in partnership with frontline staff. We’ve received feedback that they feel pride participating in these initiatives. They can say, because of me, 30 patients followed up last month for high BP, or 10 patients took a referral to the Quitline last year. We celebrate these accomplishments with the staff.
What Are the Next Steps for These Programs?
Public-facing toolkits for both BP and Quit Connect programs are available online. We have implemented [these programs] in a rural setting, in an urban setting, in Milwaukee and in Atlanta, and we are looking in the future to do a larger, multistate implementation study. If folks are interested, we’d love to partner with them to look at disseminating this further.
A version of this article appeared on Medscape.com.
Two relatively simple interventions — addressing high blood pressure (BP) and smoking cessation — could make a huge difference for patients with rheumatic disease. Patients with autoimmune disease are up to three times more likely to develop cardiovascular disease (CVD) than the general population. In addition to compounding CVD, smoking is tied to the development of certain autoimmune conditions, as well as worse outcomes. Christie Bartels, MD, chief of the Division of Rheumatology at the University of Wisconsin School of Medicine and Public Health, Madison, has focused her research on improving cardiac health in inflammatory diseases. This news organization spoke with Bartels about two short interventions she developed that tackle hypertension and smoking cessation during regular visits, each taking less than 3 minutes.
How Do These Programs Address Cardiac Disease Prevention?
The BP and Quit Connect programs help clinics systematically address the two most modifiable risk factors for CVD: high BP and smoking. There’s also evidence that addressing these two risk factors improves outcomes in rheumatic diseases. Hypertension predicts an increase in lupus damage. Particularly in lupus nephritis, hypertension will increase the risk for CVD and kidney failure. People who use tobacco have worse outcomes in diseases like rheumatoid arthritis, psoriatic arthritis, and lupus, as well as more CVD, and antirheumatic drugs may not work as well.
In 90 seconds to 3 minutes, staff can do protocol-based care, which we’ve done across 20,000-plus visits. We showed we can improve population level rates of high BP and BP control, as well as increase smoking quitting rates across different patient settings.
What Is the Quit Connect Program?
The Quit Connect program is a 10- to 90-second point of care intervention. During rooming, staff (medical assistants and nurses) ask patients: “A) Do you smoke? and B) Have you thought about cutting back or quitting in the next 30 days?”
It turns out, when you ask the question that way, between a third and a half of people say that they’ve thought about cutting back or quitting. Then, we can get patients connected directly to Quitline, a free public service across all 50 states that smokers can use to get cessation support.
If patients are ready, we ask if we can arrange for them to receive a call from a Quitline coach about setting a quit date or receiving free nicotine replacement therapy. The beautiful thing is when that all happens, A) it’s free to the patient, and B) the results from the Quitline can be recorded right back to the electronic health record.
In our most recent publication in Arthritis Care & Research, we documented bringing Quit Connect to Grady Hospital in downtown Atlanta. It’s a safety net hospital, where 80% patients are Black and 70%-80% patients are on public insurance or uninsured. Using this protocol, we improved Quitline referrals 20-fold.
What Is the BP Connect Program?
At least half of the encounters in United States happen in specialty clinics. Unfortunately, when patients get their BP measured in a specialty clinic that’s not a cardiology or a vascular clinic, often, even if the pressure is high, the clinic doesn’t give patients feedback on that. The problem is because we haven’t said anything, that gives people the false reassurance that their BP is okay.
We’ve developed a 3-minute protocol to ask, advise, and connect. The idea is that if we measure a high BP, then we remeasure and confirm that it’s high. Then, we advise why it matters in rheumatic disease: Patients with rheumatic diseases are already at an increased risk for heart disease, and controlling BP can make a big difference. Then, we connect patients with high BP back to primary care.
Specifically, a SmartSet — an electronic medical record feature — prompts different actions based on confirmed high BP readings:
- If systolic BP ≥ 140-159, the SmartSet directs scheduling a visit to a nurse or primary care provider.
- If systolic BP ≥ 160-179, the next primary care visit anticipates the need to see a prescriber.
- If systolic BP ≥ 180, then the medical assistant or nurse at the visit is instructed to notify the provider who can arrange a provider-to-provider handoff for safety to exclude a hypertensive emergency.
That order goes to the scheduler to call primary care to coordinate follow-up. BP Connect doubled the likelihood of a guideline-recommended follow-up in primary care within 30 days. All patients benefited, and disparities decreased. BP Connect has had 1100 downloads, and both BP and Quit Connect programs are endorsed by the Centers for Disease Control and Prevention and Million Hearts.
How Do These Programs Affect Clinical Practice?
We developed these interventions with a health system engineer, and we time stamped everything. Part of the sustainability of this model is that it fits within a regular workflow. As a practicing rheumatologist, I understand that time is a precious commodity.
The interventions are in partnership with frontline staff. We’ve received feedback that they feel pride participating in these initiatives. They can say, because of me, 30 patients followed up last month for high BP, or 10 patients took a referral to the Quitline last year. We celebrate these accomplishments with the staff.
What Are the Next Steps for These Programs?
Public-facing toolkits for both BP and Quit Connect programs are available online. We have implemented [these programs] in a rural setting, in an urban setting, in Milwaukee and in Atlanta, and we are looking in the future to do a larger, multistate implementation study. If folks are interested, we’d love to partner with them to look at disseminating this further.
A version of this article appeared on Medscape.com.
Two relatively simple interventions — addressing high blood pressure (BP) and smoking cessation — could make a huge difference for patients with rheumatic disease. Patients with autoimmune disease are up to three times more likely to develop cardiovascular disease (CVD) than the general population. In addition to compounding CVD, smoking is tied to the development of certain autoimmune conditions, as well as worse outcomes. Christie Bartels, MD, chief of the Division of Rheumatology at the University of Wisconsin School of Medicine and Public Health, Madison, has focused her research on improving cardiac health in inflammatory diseases. This news organization spoke with Bartels about two short interventions she developed that tackle hypertension and smoking cessation during regular visits, each taking less than 3 minutes.
How Do These Programs Address Cardiac Disease Prevention?
The BP and Quit Connect programs help clinics systematically address the two most modifiable risk factors for CVD: high BP and smoking. There’s also evidence that addressing these two risk factors improves outcomes in rheumatic diseases. Hypertension predicts an increase in lupus damage. Particularly in lupus nephritis, hypertension will increase the risk for CVD and kidney failure. People who use tobacco have worse outcomes in diseases like rheumatoid arthritis, psoriatic arthritis, and lupus, as well as more CVD, and antirheumatic drugs may not work as well.
In 90 seconds to 3 minutes, staff can do protocol-based care, which we’ve done across 20,000-plus visits. We showed we can improve population level rates of high BP and BP control, as well as increase smoking quitting rates across different patient settings.
What Is the Quit Connect Program?
The Quit Connect program is a 10- to 90-second point of care intervention. During rooming, staff (medical assistants and nurses) ask patients: “A) Do you smoke? and B) Have you thought about cutting back or quitting in the next 30 days?”
It turns out, when you ask the question that way, between a third and a half of people say that they’ve thought about cutting back or quitting. Then, we can get patients connected directly to Quitline, a free public service across all 50 states that smokers can use to get cessation support.
If patients are ready, we ask if we can arrange for them to receive a call from a Quitline coach about setting a quit date or receiving free nicotine replacement therapy. The beautiful thing is when that all happens, A) it’s free to the patient, and B) the results from the Quitline can be recorded right back to the electronic health record.
In our most recent publication in Arthritis Care & Research, we documented bringing Quit Connect to Grady Hospital in downtown Atlanta. It’s a safety net hospital, where 80% patients are Black and 70%-80% patients are on public insurance or uninsured. Using this protocol, we improved Quitline referrals 20-fold.
What Is the BP Connect Program?
At least half of the encounters in United States happen in specialty clinics. Unfortunately, when patients get their BP measured in a specialty clinic that’s not a cardiology or a vascular clinic, often, even if the pressure is high, the clinic doesn’t give patients feedback on that. The problem is because we haven’t said anything, that gives people the false reassurance that their BP is okay.
We’ve developed a 3-minute protocol to ask, advise, and connect. The idea is that if we measure a high BP, then we remeasure and confirm that it’s high. Then, we advise why it matters in rheumatic disease: Patients with rheumatic diseases are already at an increased risk for heart disease, and controlling BP can make a big difference. Then, we connect patients with high BP back to primary care.
Specifically, a SmartSet — an electronic medical record feature — prompts different actions based on confirmed high BP readings:
- If systolic BP ≥ 140-159, the SmartSet directs scheduling a visit to a nurse or primary care provider.
- If systolic BP ≥ 160-179, the next primary care visit anticipates the need to see a prescriber.
- If systolic BP ≥ 180, then the medical assistant or nurse at the visit is instructed to notify the provider who can arrange a provider-to-provider handoff for safety to exclude a hypertensive emergency.
That order goes to the scheduler to call primary care to coordinate follow-up. BP Connect doubled the likelihood of a guideline-recommended follow-up in primary care within 30 days. All patients benefited, and disparities decreased. BP Connect has had 1100 downloads, and both BP and Quit Connect programs are endorsed by the Centers for Disease Control and Prevention and Million Hearts.
How Do These Programs Affect Clinical Practice?
We developed these interventions with a health system engineer, and we time stamped everything. Part of the sustainability of this model is that it fits within a regular workflow. As a practicing rheumatologist, I understand that time is a precious commodity.
The interventions are in partnership with frontline staff. We’ve received feedback that they feel pride participating in these initiatives. They can say, because of me, 30 patients followed up last month for high BP, or 10 patients took a referral to the Quitline last year. We celebrate these accomplishments with the staff.
What Are the Next Steps for These Programs?
Public-facing toolkits for both BP and Quit Connect programs are available online. We have implemented [these programs] in a rural setting, in an urban setting, in Milwaukee and in Atlanta, and we are looking in the future to do a larger, multistate implementation study. If folks are interested, we’d love to partner with them to look at disseminating this further.
A version of this article appeared on Medscape.com.
What’s Causing Raynaud Phenomenon Severity to Rise With High Temperatures?
TOPLINE:
In systemic sclerosis, Raynaud phenomenon is more severe at both high and low temperature extremes, according to new research.
BACKGROUND:
- Raynaud phenomenon, a condition that causes decreased blood flow to extremities, occurs in about 95% of individuals with systemic sclerosis.
- Episodes of Raynaud phenomenon can be triggered by cold exposure and ambient temperature changes.
- In severe cases, it can cause permanent damage to tissues of the fingers and toes.
METHODOLOGY:
- Researchers analyzed data from 2243 participants with Raynaud phenomenon secondary to systemic sclerosis from the Scleroderma Patient-centered Intervention Network (SPIN) Cohort.
- Participants completed past-week Raynaud phenomenon severity assessments using a 0-10 numerical rating scale at enrollment and every 3 months.
- The study included data from 20,233 Raynaud phenomenon severity assessments between April 15, 2014, and August 1, 2023.
- Researchers used average daily temperature from a weather site close to the participant’s recruiting center and mapped these ambient temperature changes to Raynaud’s phenomenon outcomes.
TAKEAWAY:
- Raynaud’s phenomenon severity was highest at –25 °C (–13 °F), with assessment scores at 6.8 points out of 10.0, and lowest at 25 °C (77 °F), with scores at 2.6.
- Severity scores increased again at temperatures above 35 °C (95 °F), reaching a high of 5.6 out of 10 at 40 °C (104 °F).
- This spike at higher temperatures is presumably due to air conditioning, the authors said.
- In an accompanying commentary, Cutolo et al. posited that increased sweating and hypotension could also lead to a relative hypovolemic state in patients, causing Raynaud-like symptoms.
IN PRACTICE:
“Temperature-related variations in Raynaud’s phenomenon severity scores should be considered in clinical trials to account for normal within-season temperature fluctuations, enhancing the accuracy of treatment outcomes,” wrote Cutolo and colleagues in their commentary.
SOURCE:
The study was led by Gabrielle Virgili-Gervais, MSc, McGill University Health Centre in Montreal, Quebec, Canada. It was published online on August 28 in The Lancet Rheumatology. The accompanying commentary, also published on August 28, was authored by Maurizio Cutolo, MD, and Elvis Hysa, MD, both of University of Genova, Italy, as well as Vanessa Smith, MD, PhD, of Ghent University in Ghent, Belgium.
LIMITATIONS:
The lower number of assessments at extreme temperatures (–25 °C and 40 °C) may affect the robustness of the findings at these ranges. The study did not account for vasodilator use, which could influence participants’ response to temperature. The study also did not account for other potential confounding factors such as sex, smoking status, psychosocial factors, and comorbid conditions like cardiovascular disease.
DISCLOSURES:
A variety of scleroderma-related patient advocacy groups helped to fund research on the SPIN cohort, in addition to the Canadian Institutes of Health Research, the Arthritis Society, the Lady Davis Institute for Medical Research of the Jewish General Hospital, the Jewish General Hospital Foundation, and McGill University. Two authors reported having financial ties with pharmaceutical companies. Dr. Cutolo, Dr. Smith, and Dr. Hysa had no disclosures.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
In systemic sclerosis, Raynaud phenomenon is more severe at both high and low temperature extremes, according to new research.
BACKGROUND:
- Raynaud phenomenon, a condition that causes decreased blood flow to extremities, occurs in about 95% of individuals with systemic sclerosis.
- Episodes of Raynaud phenomenon can be triggered by cold exposure and ambient temperature changes.
- In severe cases, it can cause permanent damage to tissues of the fingers and toes.
METHODOLOGY:
- Researchers analyzed data from 2243 participants with Raynaud phenomenon secondary to systemic sclerosis from the Scleroderma Patient-centered Intervention Network (SPIN) Cohort.
- Participants completed past-week Raynaud phenomenon severity assessments using a 0-10 numerical rating scale at enrollment and every 3 months.
- The study included data from 20,233 Raynaud phenomenon severity assessments between April 15, 2014, and August 1, 2023.
- Researchers used average daily temperature from a weather site close to the participant’s recruiting center and mapped these ambient temperature changes to Raynaud’s phenomenon outcomes.
TAKEAWAY:
- Raynaud’s phenomenon severity was highest at –25 °C (–13 °F), with assessment scores at 6.8 points out of 10.0, and lowest at 25 °C (77 °F), with scores at 2.6.
- Severity scores increased again at temperatures above 35 °C (95 °F), reaching a high of 5.6 out of 10 at 40 °C (104 °F).
- This spike at higher temperatures is presumably due to air conditioning, the authors said.
- In an accompanying commentary, Cutolo et al. posited that increased sweating and hypotension could also lead to a relative hypovolemic state in patients, causing Raynaud-like symptoms.
IN PRACTICE:
“Temperature-related variations in Raynaud’s phenomenon severity scores should be considered in clinical trials to account for normal within-season temperature fluctuations, enhancing the accuracy of treatment outcomes,” wrote Cutolo and colleagues in their commentary.
SOURCE:
The study was led by Gabrielle Virgili-Gervais, MSc, McGill University Health Centre in Montreal, Quebec, Canada. It was published online on August 28 in The Lancet Rheumatology. The accompanying commentary, also published on August 28, was authored by Maurizio Cutolo, MD, and Elvis Hysa, MD, both of University of Genova, Italy, as well as Vanessa Smith, MD, PhD, of Ghent University in Ghent, Belgium.
LIMITATIONS:
The lower number of assessments at extreme temperatures (–25 °C and 40 °C) may affect the robustness of the findings at these ranges. The study did not account for vasodilator use, which could influence participants’ response to temperature. The study also did not account for other potential confounding factors such as sex, smoking status, psychosocial factors, and comorbid conditions like cardiovascular disease.
DISCLOSURES:
A variety of scleroderma-related patient advocacy groups helped to fund research on the SPIN cohort, in addition to the Canadian Institutes of Health Research, the Arthritis Society, the Lady Davis Institute for Medical Research of the Jewish General Hospital, the Jewish General Hospital Foundation, and McGill University. Two authors reported having financial ties with pharmaceutical companies. Dr. Cutolo, Dr. Smith, and Dr. Hysa had no disclosures.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
In systemic sclerosis, Raynaud phenomenon is more severe at both high and low temperature extremes, according to new research.
BACKGROUND:
- Raynaud phenomenon, a condition that causes decreased blood flow to extremities, occurs in about 95% of individuals with systemic sclerosis.
- Episodes of Raynaud phenomenon can be triggered by cold exposure and ambient temperature changes.
- In severe cases, it can cause permanent damage to tissues of the fingers and toes.
METHODOLOGY:
- Researchers analyzed data from 2243 participants with Raynaud phenomenon secondary to systemic sclerosis from the Scleroderma Patient-centered Intervention Network (SPIN) Cohort.
- Participants completed past-week Raynaud phenomenon severity assessments using a 0-10 numerical rating scale at enrollment and every 3 months.
- The study included data from 20,233 Raynaud phenomenon severity assessments between April 15, 2014, and August 1, 2023.
- Researchers used average daily temperature from a weather site close to the participant’s recruiting center and mapped these ambient temperature changes to Raynaud’s phenomenon outcomes.
TAKEAWAY:
- Raynaud’s phenomenon severity was highest at –25 °C (–13 °F), with assessment scores at 6.8 points out of 10.0, and lowest at 25 °C (77 °F), with scores at 2.6.
- Severity scores increased again at temperatures above 35 °C (95 °F), reaching a high of 5.6 out of 10 at 40 °C (104 °F).
- This spike at higher temperatures is presumably due to air conditioning, the authors said.
- In an accompanying commentary, Cutolo et al. posited that increased sweating and hypotension could also lead to a relative hypovolemic state in patients, causing Raynaud-like symptoms.
IN PRACTICE:
“Temperature-related variations in Raynaud’s phenomenon severity scores should be considered in clinical trials to account for normal within-season temperature fluctuations, enhancing the accuracy of treatment outcomes,” wrote Cutolo and colleagues in their commentary.
SOURCE:
The study was led by Gabrielle Virgili-Gervais, MSc, McGill University Health Centre in Montreal, Quebec, Canada. It was published online on August 28 in The Lancet Rheumatology. The accompanying commentary, also published on August 28, was authored by Maurizio Cutolo, MD, and Elvis Hysa, MD, both of University of Genova, Italy, as well as Vanessa Smith, MD, PhD, of Ghent University in Ghent, Belgium.
LIMITATIONS:
The lower number of assessments at extreme temperatures (–25 °C and 40 °C) may affect the robustness of the findings at these ranges. The study did not account for vasodilator use, which could influence participants’ response to temperature. The study also did not account for other potential confounding factors such as sex, smoking status, psychosocial factors, and comorbid conditions like cardiovascular disease.
DISCLOSURES:
A variety of scleroderma-related patient advocacy groups helped to fund research on the SPIN cohort, in addition to the Canadian Institutes of Health Research, the Arthritis Society, the Lady Davis Institute for Medical Research of the Jewish General Hospital, the Jewish General Hospital Foundation, and McGill University. Two authors reported having financial ties with pharmaceutical companies. Dr. Cutolo, Dr. Smith, and Dr. Hysa had no disclosures.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Do You Have Patients With JAKne — JAK Inhibitor–Associated Acne? Here’s What to Know
Since the first Food and Drug Administration approval of a Janus kinase (JAK) inhibitor in 2011, the number of these medications available — and their treatment indications — have continued to grow. Prescribing physicians are familiar with the benefits and risks for these drugs, including higher risk for cardiac events and malignancy; however, one adverse effect may be overlooked, especially by specialties outside of dermatology: acne. Though less serious than some other side effects, JAK inhibitor–associated acne — JAKne, for short — can be a concern for patients.
“Your physical appearance and how you present yourself to the world is an important part of your self-confidence and living life on your own terms,” said Arash Mostaghimi, MD, the director of inpatient dermatology at Brigham and Women’s Hospital in Boston, Massachusetts. “I think letting people know about [JAKne] and then addressing it when it occurs should be a normal part of managing these medications.”
What Is JAKne?
JAKne generally looks like other kinds of acne, explained Janelle Nassim, MD, director of laser and cosmetic dermatology at the Indiana University School of Medicine, Indianapolis. “It can affect the same areas that typical acne affects, including the face, chest, back, neck, and upper shoulders.”
Though it appears like typical forms of acne, it is not clear what drives these skin eruptions in patients taking JAK inhibitors.

“We don’t understand the underlying pathophysiology,” Dr. Mostaghimi said. “It looks like acne, but we don’t know if the exact underlying inflammatory process is the same or if it’s different.”
In a 2023 systematic review of clinical studies, Dr. Mostaghimi and colleagues found that patients on any JAK inhibitor were nearly four times more likely to experience acne than patients who received placebo, but risk varied between medications. Patients taking JAK inhibitors for skin conditions had higher risk for acne than those given the medications for other indications. However, Dr. Mostaghimi thinks this finding is the result of selection bias.
Participants may not mention side effects like acne in trials for rheumatologic or gastrointestinal conditions, he said, unlike in trials for skin conditions. “Clinically, I’ve seen it in patients across every indication.”
Patients with a history of acne seem to be more likely to develop this side effect, though formal studies looking into risk factors are lacking. In Dr. Mostaghimi’s own clinical experience, JAKne is also more common in younger patients, but it can happen to anyone. “I’ve seen 70-year-olds develop acne — patients who’ve never had an issue their whole life — when they’re taking a JAK inhibitor.”
This issue also appears to be more common earlier in treatment, he added, and may improve over time as a patient continues with the medication.
How Do You Treat It?
“I think in other specialties, you will often feel awkward addressing skin conditions or pointing out acne,” Dr. Mostaghimi said. The most important steps are being aware of this potential side effect, and if you see it practice, to bring it up.
“Say: I’m noticing there’s some changes in your skin. Some patients on JAK inhibitors develop more acne. Have you noticed this? And if so, is this bothering you?”
Generally, JAKne is mild to moderate, explained Dr. Nassim, and if non-dermatologists are comfortable, they can prescribe a first-line topical regimen for patients. Dr. Mostaghimi recommends prescribing a clindamycin 1% lotion or gel. In addition, patients can use a benzoyl peroxide wash (4% or 10%) combined with a gentle retinoid, such as adapalene. (Both of these treatments are now available over the counter.)

In patients with scalp or hairline involvement, he often prescribes a ketoconazole 2% shampoo, which patients can use to wash their scalp, face, chest, and back in the shower.
If they aren’t responding to these initial treatments, then refer to a dermatologist for further assessment.
“Ultimately, referring to a dermatologist is the best course of action,” Dr. Nassim said. “I have had patients on JAK inhibitors who improved with topical acne treatments, and some that required more aggressive treatment with oral medications.”
Dr. Mostaghimi reported consulting fees from AbbVie, Concert Pharmaceuticals, Pfizer, and 3Derm Systems; research funding from Incyte, Aclaris Therapeutics, Eli Lilly, and Concert Pharmaceuticals; personal fees from Equillium, ASLAN Pharmaceuticals, ACOM, and Boehringer Ingelheim; and advisory board fees from Fig.1 Beauty, Eli Lilly, Pfizer, and Hims & Hers Health. Dr. Nassim had no relevant disclosures.
A version of this article first appeared on Medscape.com.
Since the first Food and Drug Administration approval of a Janus kinase (JAK) inhibitor in 2011, the number of these medications available — and their treatment indications — have continued to grow. Prescribing physicians are familiar with the benefits and risks for these drugs, including higher risk for cardiac events and malignancy; however, one adverse effect may be overlooked, especially by specialties outside of dermatology: acne. Though less serious than some other side effects, JAK inhibitor–associated acne — JAKne, for short — can be a concern for patients.
“Your physical appearance and how you present yourself to the world is an important part of your self-confidence and living life on your own terms,” said Arash Mostaghimi, MD, the director of inpatient dermatology at Brigham and Women’s Hospital in Boston, Massachusetts. “I think letting people know about [JAKne] and then addressing it when it occurs should be a normal part of managing these medications.”
What Is JAKne?
JAKne generally looks like other kinds of acne, explained Janelle Nassim, MD, director of laser and cosmetic dermatology at the Indiana University School of Medicine, Indianapolis. “It can affect the same areas that typical acne affects, including the face, chest, back, neck, and upper shoulders.”
Though it appears like typical forms of acne, it is not clear what drives these skin eruptions in patients taking JAK inhibitors.
“We don’t understand the underlying pathophysiology,” Dr. Mostaghimi said. “It looks like acne, but we don’t know if the exact underlying inflammatory process is the same or if it’s different.”
In a 2023 systematic review of clinical studies, Dr. Mostaghimi and colleagues found that patients on any JAK inhibitor were nearly four times more likely to experience acne than patients who received placebo, but risk varied between medications. Patients taking JAK inhibitors for skin conditions had higher risk for acne than those given the medications for other indications. However, Dr. Mostaghimi thinks this finding is the result of selection bias.
Participants may not mention side effects like acne in trials for rheumatologic or gastrointestinal conditions, he said, unlike in trials for skin conditions. “Clinically, I’ve seen it in patients across every indication.”
Patients with a history of acne seem to be more likely to develop this side effect, though formal studies looking into risk factors are lacking. In Dr. Mostaghimi’s own clinical experience, JAKne is also more common in younger patients, but it can happen to anyone. “I’ve seen 70-year-olds develop acne — patients who’ve never had an issue their whole life — when they’re taking a JAK inhibitor.”
This issue also appears to be more common earlier in treatment, he added, and may improve over time as a patient continues with the medication.
How Do You Treat It?
“I think in other specialties, you will often feel awkward addressing skin conditions or pointing out acne,” Dr. Mostaghimi said. The most important steps are being aware of this potential side effect, and if you see it practice, to bring it up.
“Say: I’m noticing there’s some changes in your skin. Some patients on JAK inhibitors develop more acne. Have you noticed this? And if so, is this bothering you?”
Generally, JAKne is mild to moderate, explained Dr. Nassim, and if non-dermatologists are comfortable, they can prescribe a first-line topical regimen for patients. Dr. Mostaghimi recommends prescribing a clindamycin 1% lotion or gel. In addition, patients can use a benzoyl peroxide wash (4% or 10%) combined with a gentle retinoid, such as adapalene. (Both of these treatments are now available over the counter.)
In patients with scalp or hairline involvement, he often prescribes a ketoconazole 2% shampoo, which patients can use to wash their scalp, face, chest, and back in the shower.
If they aren’t responding to these initial treatments, then refer to a dermatologist for further assessment.
“Ultimately, referring to a dermatologist is the best course of action,” Dr. Nassim said. “I have had patients on JAK inhibitors who improved with topical acne treatments, and some that required more aggressive treatment with oral medications.”
Dr. Mostaghimi reported consulting fees from AbbVie, Concert Pharmaceuticals, Pfizer, and 3Derm Systems; research funding from Incyte, Aclaris Therapeutics, Eli Lilly, and Concert Pharmaceuticals; personal fees from Equillium, ASLAN Pharmaceuticals, ACOM, and Boehringer Ingelheim; and advisory board fees from Fig.1 Beauty, Eli Lilly, Pfizer, and Hims & Hers Health. Dr. Nassim had no relevant disclosures.
A version of this article first appeared on Medscape.com.
Since the first Food and Drug Administration approval of a Janus kinase (JAK) inhibitor in 2011, the number of these medications available — and their treatment indications — have continued to grow. Prescribing physicians are familiar with the benefits and risks for these drugs, including higher risk for cardiac events and malignancy; however, one adverse effect may be overlooked, especially by specialties outside of dermatology: acne. Though less serious than some other side effects, JAK inhibitor–associated acne — JAKne, for short — can be a concern for patients.
“Your physical appearance and how you present yourself to the world is an important part of your self-confidence and living life on your own terms,” said Arash Mostaghimi, MD, the director of inpatient dermatology at Brigham and Women’s Hospital in Boston, Massachusetts. “I think letting people know about [JAKne] and then addressing it when it occurs should be a normal part of managing these medications.”
What Is JAKne?
JAKne generally looks like other kinds of acne, explained Janelle Nassim, MD, director of laser and cosmetic dermatology at the Indiana University School of Medicine, Indianapolis. “It can affect the same areas that typical acne affects, including the face, chest, back, neck, and upper shoulders.”
Though it appears like typical forms of acne, it is not clear what drives these skin eruptions in patients taking JAK inhibitors.
“We don’t understand the underlying pathophysiology,” Dr. Mostaghimi said. “It looks like acne, but we don’t know if the exact underlying inflammatory process is the same or if it’s different.”
In a 2023 systematic review of clinical studies, Dr. Mostaghimi and colleagues found that patients on any JAK inhibitor were nearly four times more likely to experience acne than patients who received placebo, but risk varied between medications. Patients taking JAK inhibitors for skin conditions had higher risk for acne than those given the medications for other indications. However, Dr. Mostaghimi thinks this finding is the result of selection bias.
Participants may not mention side effects like acne in trials for rheumatologic or gastrointestinal conditions, he said, unlike in trials for skin conditions. “Clinically, I’ve seen it in patients across every indication.”
Patients with a history of acne seem to be more likely to develop this side effect, though formal studies looking into risk factors are lacking. In Dr. Mostaghimi’s own clinical experience, JAKne is also more common in younger patients, but it can happen to anyone. “I’ve seen 70-year-olds develop acne — patients who’ve never had an issue their whole life — when they’re taking a JAK inhibitor.”
This issue also appears to be more common earlier in treatment, he added, and may improve over time as a patient continues with the medication.
How Do You Treat It?
“I think in other specialties, you will often feel awkward addressing skin conditions or pointing out acne,” Dr. Mostaghimi said. The most important steps are being aware of this potential side effect, and if you see it practice, to bring it up.
“Say: I’m noticing there’s some changes in your skin. Some patients on JAK inhibitors develop more acne. Have you noticed this? And if so, is this bothering you?”
Generally, JAKne is mild to moderate, explained Dr. Nassim, and if non-dermatologists are comfortable, they can prescribe a first-line topical regimen for patients. Dr. Mostaghimi recommends prescribing a clindamycin 1% lotion or gel. In addition, patients can use a benzoyl peroxide wash (4% or 10%) combined with a gentle retinoid, such as adapalene. (Both of these treatments are now available over the counter.)
In patients with scalp or hairline involvement, he often prescribes a ketoconazole 2% shampoo, which patients can use to wash their scalp, face, chest, and back in the shower.
If they aren’t responding to these initial treatments, then refer to a dermatologist for further assessment.
“Ultimately, referring to a dermatologist is the best course of action,” Dr. Nassim said. “I have had patients on JAK inhibitors who improved with topical acne treatments, and some that required more aggressive treatment with oral medications.”
Dr. Mostaghimi reported consulting fees from AbbVie, Concert Pharmaceuticals, Pfizer, and 3Derm Systems; research funding from Incyte, Aclaris Therapeutics, Eli Lilly, and Concert Pharmaceuticals; personal fees from Equillium, ASLAN Pharmaceuticals, ACOM, and Boehringer Ingelheim; and advisory board fees from Fig.1 Beauty, Eli Lilly, Pfizer, and Hims & Hers Health. Dr. Nassim had no relevant disclosures.
A version of this article first appeared on Medscape.com.
Trends in Rheumatic Disease Pain Management Show Decline in Opioid Use
TOPLINE:
Since 2014, opioid use for autoimmune rheumatic diseases decreased by 15% annually while other management modalities increased or stabilized.
METHODOLOGY:
- Researchers analyzed de-identified US claims data from the MarketScan Database from 2007-2021.
- The study included nearly 142,000 patients with autoimmune rheumatic diseases: 10,927 with ankylosing spondylitis (AS); 21,438 with psoriatic arthritis (PsA); 71,393 with rheumatoid arthritis (RA); 16,718 with Sjögren disease; 18,018 with systemic lupus erythematosus; and 3468 with systemic sclerosis.
- Primary outcome was opioid use annual trends, with secondary outcomes including trends in the use of anticonvulsants, antidepressants, skeletal muscle relaxants, nonsteroidal anti-inflammatory drugs (NSAIDs), topical pain medications, and physical or occupational therapy.
TAKEAWAY:
- The incidence of opioid use increased annually by 4% until 2014 and decreased annually by 15% after 2014.
- NSAID use increased 2% annually until 2014, then declined by 5% afterward.
- The proportion of patients utilizing physical therapy or anticonvulsants doubled from 2008 to 2020.
- NSAID prescriptions were highest in AS, PsA, and RA, while they were lowest in Sjögren disease and systemic sclerosis.
IN PRACTICE:
“Our work, along with the published literature, highlights the need for future studies to evaluate the effectiveness of pain management modality changes over time and to understand the possible effects that changes have had on outcomes such as quality of life, disability, health status, and function,” wrote the authors of the study.
SOURCE:
The study was led by Titilola Falasinnu, PhD, Stanford University School of Medicine, Stanford, California. It was published online in The Lancet Rheumatology.
LIMITATIONS:
The study relied on administrative claims data, which did not contain information on use of over-the-counter medications like NSAIDs and topical analgesics. The study did not include the duration of pain treatment modalities, making it difficult to differentiate between acute and chronic use. The analysis did not include race or ethnicity, which is important for understanding pain outcomes across different sociodemographic groups.
DISCLOSURES:
The study was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Since 2014, opioid use for autoimmune rheumatic diseases decreased by 15% annually while other management modalities increased or stabilized.
METHODOLOGY:
- Researchers analyzed de-identified US claims data from the MarketScan Database from 2007-2021.
- The study included nearly 142,000 patients with autoimmune rheumatic diseases: 10,927 with ankylosing spondylitis (AS); 21,438 with psoriatic arthritis (PsA); 71,393 with rheumatoid arthritis (RA); 16,718 with Sjögren disease; 18,018 with systemic lupus erythematosus; and 3468 with systemic sclerosis.
- Primary outcome was opioid use annual trends, with secondary outcomes including trends in the use of anticonvulsants, antidepressants, skeletal muscle relaxants, nonsteroidal anti-inflammatory drugs (NSAIDs), topical pain medications, and physical or occupational therapy.
TAKEAWAY:
- The incidence of opioid use increased annually by 4% until 2014 and decreased annually by 15% after 2014.
- NSAID use increased 2% annually until 2014, then declined by 5% afterward.
- The proportion of patients utilizing physical therapy or anticonvulsants doubled from 2008 to 2020.
- NSAID prescriptions were highest in AS, PsA, and RA, while they were lowest in Sjögren disease and systemic sclerosis.
IN PRACTICE:
“Our work, along with the published literature, highlights the need for future studies to evaluate the effectiveness of pain management modality changes over time and to understand the possible effects that changes have had on outcomes such as quality of life, disability, health status, and function,” wrote the authors of the study.
SOURCE:
The study was led by Titilola Falasinnu, PhD, Stanford University School of Medicine, Stanford, California. It was published online in The Lancet Rheumatology.
LIMITATIONS:
The study relied on administrative claims data, which did not contain information on use of over-the-counter medications like NSAIDs and topical analgesics. The study did not include the duration of pain treatment modalities, making it difficult to differentiate between acute and chronic use. The analysis did not include race or ethnicity, which is important for understanding pain outcomes across different sociodemographic groups.
DISCLOSURES:
The study was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Since 2014, opioid use for autoimmune rheumatic diseases decreased by 15% annually while other management modalities increased or stabilized.
METHODOLOGY:
- Researchers analyzed de-identified US claims data from the MarketScan Database from 2007-2021.
- The study included nearly 142,000 patients with autoimmune rheumatic diseases: 10,927 with ankylosing spondylitis (AS); 21,438 with psoriatic arthritis (PsA); 71,393 with rheumatoid arthritis (RA); 16,718 with Sjögren disease; 18,018 with systemic lupus erythematosus; and 3468 with systemic sclerosis.
- Primary outcome was opioid use annual trends, with secondary outcomes including trends in the use of anticonvulsants, antidepressants, skeletal muscle relaxants, nonsteroidal anti-inflammatory drugs (NSAIDs), topical pain medications, and physical or occupational therapy.
TAKEAWAY:
- The incidence of opioid use increased annually by 4% until 2014 and decreased annually by 15% after 2014.
- NSAID use increased 2% annually until 2014, then declined by 5% afterward.
- The proportion of patients utilizing physical therapy or anticonvulsants doubled from 2008 to 2020.
- NSAID prescriptions were highest in AS, PsA, and RA, while they were lowest in Sjögren disease and systemic sclerosis.
IN PRACTICE:
“Our work, along with the published literature, highlights the need for future studies to evaluate the effectiveness of pain management modality changes over time and to understand the possible effects that changes have had on outcomes such as quality of life, disability, health status, and function,” wrote the authors of the study.
SOURCE:
The study was led by Titilola Falasinnu, PhD, Stanford University School of Medicine, Stanford, California. It was published online in The Lancet Rheumatology.
LIMITATIONS:
The study relied on administrative claims data, which did not contain information on use of over-the-counter medications like NSAIDs and topical analgesics. The study did not include the duration of pain treatment modalities, making it difficult to differentiate between acute and chronic use. The analysis did not include race or ethnicity, which is important for understanding pain outcomes across different sociodemographic groups.
DISCLOSURES:
The study was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.