User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Dermatofibrosarcoma Protuberans More Common In Black Patients, Analysis Finds
TOPLINE:
that also found that larger tumor size and older age were associated with survival outcomes.
METHODOLOGY:
- Researchers used the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) registry from 2000 through 2018 to provide a comprehensive report on the incidence of DFSP, a rare, low-grade cutaneous soft tissue sarcoma, and factors associated with metastatic progression, overall survival (OS), and cancer-specific survival.
- A total of 7748 patients (mean age, 43.5 years; 53.3% women; 52% non-Hispanic White) were diagnosed with histologically confirmed DFSP of the skin and connective tissue and were included in the study.
- DFSP incidence was reported as cases per million person-years and age-adjusted to the 2000 US Standard Population, and factors influencing metastasis were assessed.
TAKEAWAY:
- The overall DFSP incidence rate was 6.25 cases per million person-years, with a higher incidence in Black individuals than in White individuals (8.74 vs 4.53).
- The 5-year OS rate was 95.8%. Older age (≥ 60 years; hazard ratio [HR], 6.66), male gender assigned at birth (HR, 1.79), and larger tumor size (≥ 3 cm; HR, 2.02) were associated with poorer OS (P < .001 for all).
- The 1-year and 5-year DFSP-specific survival rates were 99.9% and 99.2%, respectively. Older age (HR, 3.47; P < .001) and larger tumor size (≥ 3 cm; HR, 5.34; P = .002) were associated with significantly worse cancer-specific survival.
- Large tumor size (odds ratio [OR], 2.24) and DFSP located on the head and neck (OR, 4.88), or genitalia (OR, 3.16) were significantly associated with increased metastasis risk. Higher socioeconomic status was linked to a lower risk for metastasis.
IN PRACTICE:
“Our findings highlight the increased incidence rates of DFSP among Black patients. We demonstrate the interplay between patient demographics and clinical factors in influencing DFSP metastasis, OS, and cancer-specific survival,” the authors wrote. The results, they added, “may be useful for further evaluation of proposed causes, which will ultimately lead to further understanding and prevention of this disease.”
SOURCE:
The study was led by Jalal Maghfour, MD, Department of Dermatology, Henry Ford Health, Detroit, and was published online on June 20 in the Journal of the American Academy of Dermatology.
LIMITATIONS:
Details on specific cases in the SEER registry are limited. For 1752 patients, tumor size was not included, increasing the risk for misclassification bias. Because specific pathology reports were not available, the analysis did not address histologic grade.
DISCLOSURES:
The study did not receive any funding support. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
that also found that larger tumor size and older age were associated with survival outcomes.
METHODOLOGY:
- Researchers used the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) registry from 2000 through 2018 to provide a comprehensive report on the incidence of DFSP, a rare, low-grade cutaneous soft tissue sarcoma, and factors associated with metastatic progression, overall survival (OS), and cancer-specific survival.
- A total of 7748 patients (mean age, 43.5 years; 53.3% women; 52% non-Hispanic White) were diagnosed with histologically confirmed DFSP of the skin and connective tissue and were included in the study.
- DFSP incidence was reported as cases per million person-years and age-adjusted to the 2000 US Standard Population, and factors influencing metastasis were assessed.
TAKEAWAY:
- The overall DFSP incidence rate was 6.25 cases per million person-years, with a higher incidence in Black individuals than in White individuals (8.74 vs 4.53).
- The 5-year OS rate was 95.8%. Older age (≥ 60 years; hazard ratio [HR], 6.66), male gender assigned at birth (HR, 1.79), and larger tumor size (≥ 3 cm; HR, 2.02) were associated with poorer OS (P < .001 for all).
- The 1-year and 5-year DFSP-specific survival rates were 99.9% and 99.2%, respectively. Older age (HR, 3.47; P < .001) and larger tumor size (≥ 3 cm; HR, 5.34; P = .002) were associated with significantly worse cancer-specific survival.
- Large tumor size (odds ratio [OR], 2.24) and DFSP located on the head and neck (OR, 4.88), or genitalia (OR, 3.16) were significantly associated with increased metastasis risk. Higher socioeconomic status was linked to a lower risk for metastasis.
IN PRACTICE:
“Our findings highlight the increased incidence rates of DFSP among Black patients. We demonstrate the interplay between patient demographics and clinical factors in influencing DFSP metastasis, OS, and cancer-specific survival,” the authors wrote. The results, they added, “may be useful for further evaluation of proposed causes, which will ultimately lead to further understanding and prevention of this disease.”
SOURCE:
The study was led by Jalal Maghfour, MD, Department of Dermatology, Henry Ford Health, Detroit, and was published online on June 20 in the Journal of the American Academy of Dermatology.
LIMITATIONS:
Details on specific cases in the SEER registry are limited. For 1752 patients, tumor size was not included, increasing the risk for misclassification bias. Because specific pathology reports were not available, the analysis did not address histologic grade.
DISCLOSURES:
The study did not receive any funding support. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
that also found that larger tumor size and older age were associated with survival outcomes.
METHODOLOGY:
- Researchers used the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) registry from 2000 through 2018 to provide a comprehensive report on the incidence of DFSP, a rare, low-grade cutaneous soft tissue sarcoma, and factors associated with metastatic progression, overall survival (OS), and cancer-specific survival.
- A total of 7748 patients (mean age, 43.5 years; 53.3% women; 52% non-Hispanic White) were diagnosed with histologically confirmed DFSP of the skin and connective tissue and were included in the study.
- DFSP incidence was reported as cases per million person-years and age-adjusted to the 2000 US Standard Population, and factors influencing metastasis were assessed.
TAKEAWAY:
- The overall DFSP incidence rate was 6.25 cases per million person-years, with a higher incidence in Black individuals than in White individuals (8.74 vs 4.53).
- The 5-year OS rate was 95.8%. Older age (≥ 60 years; hazard ratio [HR], 6.66), male gender assigned at birth (HR, 1.79), and larger tumor size (≥ 3 cm; HR, 2.02) were associated with poorer OS (P < .001 for all).
- The 1-year and 5-year DFSP-specific survival rates were 99.9% and 99.2%, respectively. Older age (HR, 3.47; P < .001) and larger tumor size (≥ 3 cm; HR, 5.34; P = .002) were associated with significantly worse cancer-specific survival.
- Large tumor size (odds ratio [OR], 2.24) and DFSP located on the head and neck (OR, 4.88), or genitalia (OR, 3.16) were significantly associated with increased metastasis risk. Higher socioeconomic status was linked to a lower risk for metastasis.
IN PRACTICE:
“Our findings highlight the increased incidence rates of DFSP among Black patients. We demonstrate the interplay between patient demographics and clinical factors in influencing DFSP metastasis, OS, and cancer-specific survival,” the authors wrote. The results, they added, “may be useful for further evaluation of proposed causes, which will ultimately lead to further understanding and prevention of this disease.”
SOURCE:
The study was led by Jalal Maghfour, MD, Department of Dermatology, Henry Ford Health, Detroit, and was published online on June 20 in the Journal of the American Academy of Dermatology.
LIMITATIONS:
Details on specific cases in the SEER registry are limited. For 1752 patients, tumor size was not included, increasing the risk for misclassification bias. Because specific pathology reports were not available, the analysis did not address histologic grade.
DISCLOSURES:
The study did not receive any funding support. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Debate Over Axial Involvement in Psoriatic Arthritis Still Unresolved Despite New Studies
VIENNA — While there is no doubt that some people with psoriatic arthritis (PsA) have axial symptoms, data presented at the annual European Congress of Rheumatology do not appear to add much to what is already known about axial PsA or to further the cause of differentiating it from axial spondyloarthritis (axSpA).
In both the AXIS study and Reuma.pt, around one in three patients with PsA were found to have axial involvement. Notably, the percentage of people with axial PsA was found to vary according to how imaging information was interpreted in the AXIS study. Both studies were discussed during the Axial Involvement in PsA and SpA session at EULAR 2024.
The One-Million-Dollar Question
“So, the one-million-dollar question: What is it, really?” Philippe Carron, MD, PhD, Ghent University Hospital, Ghent, Belgium, said in the presentation that started the session. Despite PsA being described more than 60 years ago, “we still have no internationally accepted definition or a consensus on how we should define these patients and how we should screen them,” he said.
“There are some believers that it is just a form of axial SpA with concomitant psoriasis, but also some people that think that the axial PsA is a typical disease, with typical characteristics which are different from axial disease,” Dr. Carron said.
The lack of consensus makes it difficult to estimate just how many people have axial PsA. Reported prevalences range from 5% to 70%, “all caused by which criteria that you’re using to define axial involvement,” Dr. Carron added.
There are, however, two things that can be agreed upon, according to Dr. Carron. First, the prevalence of axial involvement in people with early PsA is “much, much lower” than that of more established disease. Second, exclusive axial involvement is seen in “just a minority of PsA patients.” Most people with axial disease also have peripheral disease, he added.
Imaging findings in axial PsA “are quite similar to those seen in axial SpA,” although Dr. Carron also said that there were some distinct differences. Radiographic sacroiliitis occurs in around 25%-50% of people with axial PsA, and atypical syndesmophytes are more often found in people with axial PsA than in those with axSpA.
Shared Characteristics
But are axial PsA and axSpA separate diseases or part of the same disease continuum? That’s a question that is still very much open for debate, said Sofia Ramiro, MD, PhD, a senior researcher at Leiden University Medical Center, Leiden, the Netherlands, and rheumatology consultant at Zuyderland Medical Center in Heerlen, the Netherlands.
While many studies have looked to answer this question, there is a big methodological problem — the studies largely cannot be compared as they have used different definitions of axSpA.
Take a patient with inflammatory back pain, psoriasis, and oligoarthritis, Dr. Ramiro said. If the patient goes to one rheumatologist, they may get a diagnosis of axSpA, but if they go to a different rheumatologist, they may get a diagnosis of axial PsA.
“This is influenced by training, expertise, by beliefs, and by belonging to ASAS [Assessment of Spondyloarthritis International Society] or to GRAPPA [Group for Research and Assessment of Psoriasis and Psoriatic Arthritis],” Dr. Ramiro suggested. It’s “a diagnostic bias” that is very difficult to overcome and makes direct comparisons between patient populations recruited into clinical studies “extremely challenging.”
To confuse matters more, axial PsA and axSpA share common characteristics: Inflammatory back pain, HLA-B27 positivity, elevated levels of C-reactive protein (CRP) or a higher erythrocyte sedimentation rate, and structural lesions in the sacroiliac joints and spine.
AXIS Study ‘Gives Answers’
More research into factors associated with axial PsA need to be performed to try to help define the condition and enable classification and ultimately treatment guidelines. This is where the AXIS study comes in.
The AXIS study is a joint project of ASAS and GRAPPA that was started in January 2019 with the aim of defining a homogeneous subgroup of patients who could be studied.
“The objectives of the AXIS study are to determine the frequency of axial involvement in patients with PsA; to identify the frequency of active inflammatory and structural changes on imaging; and to identify factors associated with the presence of axial involvement in PsA,” Murat Torgutalp, MD, of Charité – Universitätsmedizin Berlin, Berlin, Germany, said at EULAR 2024.
The study population consisted of 409 consecutively recruited patients diagnosed with PsA according to CASPAR (Classification for Psoriatic Arthritis) criteria; all have had PsA for up to 10 years and were untreated with biologic or targeted synthetic disease modifying drugs at the time of inclusion.
Dr. Torgutalp, who is the study’s primary research coordinator, reported that a diagnosis of PsA was made in 37% of the population when local investigators considered available clinical, laboratory, and imaging data. However, patients’ imaging data were also centrally assessed, and when the local investigators were party to the expert imaging interpretations, the percentage of people diagnosed with PsA dropped to 27%.
“When we looked at the clinical characteristics, the presence of the back pain, particularly inflammatory back pain, HLA-B27 positivity, elevated CRP, and presence of active, inflammatory and structural changes in the sacroiliac joints and spine were associated with the final conclusion on the presence of axial involvement,” Dr. Torgutalp said.
Despite the title of his presentation being “The Axis Study Gives Answers,” Dr. Torgutalp presented lots of data without giving much insight into how they might be used. He concluded that “overall, there was a trend toward overestimation of the presence of imaging changes indicative of axial involvement across all imaging modalities” by the local investigators.
Dennis McGonagle, MB, MCH, BAO, PhD, of the University of Leeds, Leeds, England,said in an interview that the AXIS study “is a noble, international effort across multiple countries to try and better understand axial PsA.”
Dr. McGonagle, who was not involved in the study, added: “A lot of data are being generated, and a lot of analysis needs to be done to drill down to get a clear message that could influence practice.”
Axial PsA in the Portuguese Population
Separately, Catarina Abreu, a rheumatology intern at Hospital Garcia de Orta, Almada, Portugal, presented some real-world data on axial PsA from Reuma.pt.
Of 2304 patients, 854 (37.1%) reportedly had axial PsA, which had been defined as physician-reported spondylitis or the presence of imaging findings suggestive of axial involvement. This included radiographic- or MRI-detected sacroiliitis or syndesmophytes seen on axial x-rays.
The majority (78.2%) of those with an axial PsA diagnosis had concomitant peripheral involvement, with 8.1% having exclusive axial disease.
About 70% of the axial PsA diagnoses had been made using clinical or laboratory findings alone, and 30% of diagnoses was based on imaging results. Of the latter, Ms. Abreu noted that patients who had imaging data available were more likely to be HLA-B27 positive and less likely to have dactylitis, with respective odds ratios (ORs) of 3.10 and 2.42.
Individuals with axial PsA were more likely to have enthesitis (OR, 1.92), although no data were available on whether this was axial or peripheral enthesitis. Tobacco exposure was also linked to an increased chance of having axial PsA (OR, 1.66).
Ms. Abreu noted that the “scarce number of available imaging exams” and other missing data in Reuma.pt may have led to an underdiagnosis of axial PsA.
“The difference that we found between axial and peripheral [PsA] are similar to the differences found in other studies that compared axial psoriatic arthritis with axial spondyloarthritis,” Ms. Abreu said.
“So, we leave with the question that was already left before here: If these are different diseases or just different phenotypes of the same disease, and what implications will this have in the future?” Ms. Abreu concluded.
Dr. Carron received educational grants, speaker fees, or honoraria for other consultancy work from AbbVie, UCB, Pfizer, Eli Lilly, Novartis, Janssen, and Galapagos/Alfasigma. Dr. Ramiro is an ASAS executive committee member and received research grants or consulting/speaker fees from AbbVie, Eli Lilly, Galapagos, Janssen, Merck Sharp and Dohme, Novartis, Pfizer, Sanofi, and UCB. AXIS is supported by unrestricted research grants from AbbVie, Galapagos, Janssen, Eli Lilly, Novartis, Pfizer, and UCB. Dr. Torgutalp is the primary research coordinator for the study; he reported no financial conflicts of interest. The Reuma.pt registry was developed with the financial support of the pharmaceutical industry and is currently supported by AbbVie, Amgen, AstraZeneca, Boehringer Ingelheim, Eli Lilly, Merck Sharp and Dohme, Novartis, Pfizer, and Sobi. Ms. Abreu reported no financial conflicts of interest.
A version of this article appeared on Medscape.com.
VIENNA — While there is no doubt that some people with psoriatic arthritis (PsA) have axial symptoms, data presented at the annual European Congress of Rheumatology do not appear to add much to what is already known about axial PsA or to further the cause of differentiating it from axial spondyloarthritis (axSpA).
In both the AXIS study and Reuma.pt, around one in three patients with PsA were found to have axial involvement. Notably, the percentage of people with axial PsA was found to vary according to how imaging information was interpreted in the AXIS study. Both studies were discussed during the Axial Involvement in PsA and SpA session at EULAR 2024.
The One-Million-Dollar Question
“So, the one-million-dollar question: What is it, really?” Philippe Carron, MD, PhD, Ghent University Hospital, Ghent, Belgium, said in the presentation that started the session. Despite PsA being described more than 60 years ago, “we still have no internationally accepted definition or a consensus on how we should define these patients and how we should screen them,” he said.
“There are some believers that it is just a form of axial SpA with concomitant psoriasis, but also some people that think that the axial PsA is a typical disease, with typical characteristics which are different from axial disease,” Dr. Carron said.
The lack of consensus makes it difficult to estimate just how many people have axial PsA. Reported prevalences range from 5% to 70%, “all caused by which criteria that you’re using to define axial involvement,” Dr. Carron added.
There are, however, two things that can be agreed upon, according to Dr. Carron. First, the prevalence of axial involvement in people with early PsA is “much, much lower” than that of more established disease. Second, exclusive axial involvement is seen in “just a minority of PsA patients.” Most people with axial disease also have peripheral disease, he added.
Imaging findings in axial PsA “are quite similar to those seen in axial SpA,” although Dr. Carron also said that there were some distinct differences. Radiographic sacroiliitis occurs in around 25%-50% of people with axial PsA, and atypical syndesmophytes are more often found in people with axial PsA than in those with axSpA.
Shared Characteristics
But are axial PsA and axSpA separate diseases or part of the same disease continuum? That’s a question that is still very much open for debate, said Sofia Ramiro, MD, PhD, a senior researcher at Leiden University Medical Center, Leiden, the Netherlands, and rheumatology consultant at Zuyderland Medical Center in Heerlen, the Netherlands.
While many studies have looked to answer this question, there is a big methodological problem — the studies largely cannot be compared as they have used different definitions of axSpA.
Take a patient with inflammatory back pain, psoriasis, and oligoarthritis, Dr. Ramiro said. If the patient goes to one rheumatologist, they may get a diagnosis of axSpA, but if they go to a different rheumatologist, they may get a diagnosis of axial PsA.
“This is influenced by training, expertise, by beliefs, and by belonging to ASAS [Assessment of Spondyloarthritis International Society] or to GRAPPA [Group for Research and Assessment of Psoriasis and Psoriatic Arthritis],” Dr. Ramiro suggested. It’s “a diagnostic bias” that is very difficult to overcome and makes direct comparisons between patient populations recruited into clinical studies “extremely challenging.”
To confuse matters more, axial PsA and axSpA share common characteristics: Inflammatory back pain, HLA-B27 positivity, elevated levels of C-reactive protein (CRP) or a higher erythrocyte sedimentation rate, and structural lesions in the sacroiliac joints and spine.
AXIS Study ‘Gives Answers’
More research into factors associated with axial PsA need to be performed to try to help define the condition and enable classification and ultimately treatment guidelines. This is where the AXIS study comes in.
The AXIS study is a joint project of ASAS and GRAPPA that was started in January 2019 with the aim of defining a homogeneous subgroup of patients who could be studied.
“The objectives of the AXIS study are to determine the frequency of axial involvement in patients with PsA; to identify the frequency of active inflammatory and structural changes on imaging; and to identify factors associated with the presence of axial involvement in PsA,” Murat Torgutalp, MD, of Charité – Universitätsmedizin Berlin, Berlin, Germany, said at EULAR 2024.
The study population consisted of 409 consecutively recruited patients diagnosed with PsA according to CASPAR (Classification for Psoriatic Arthritis) criteria; all have had PsA for up to 10 years and were untreated with biologic or targeted synthetic disease modifying drugs at the time of inclusion.
Dr. Torgutalp, who is the study’s primary research coordinator, reported that a diagnosis of PsA was made in 37% of the population when local investigators considered available clinical, laboratory, and imaging data. However, patients’ imaging data were also centrally assessed, and when the local investigators were party to the expert imaging interpretations, the percentage of people diagnosed with PsA dropped to 27%.
“When we looked at the clinical characteristics, the presence of the back pain, particularly inflammatory back pain, HLA-B27 positivity, elevated CRP, and presence of active, inflammatory and structural changes in the sacroiliac joints and spine were associated with the final conclusion on the presence of axial involvement,” Dr. Torgutalp said.
Despite the title of his presentation being “The Axis Study Gives Answers,” Dr. Torgutalp presented lots of data without giving much insight into how they might be used. He concluded that “overall, there was a trend toward overestimation of the presence of imaging changes indicative of axial involvement across all imaging modalities” by the local investigators.
Dennis McGonagle, MB, MCH, BAO, PhD, of the University of Leeds, Leeds, England,said in an interview that the AXIS study “is a noble, international effort across multiple countries to try and better understand axial PsA.”
Dr. McGonagle, who was not involved in the study, added: “A lot of data are being generated, and a lot of analysis needs to be done to drill down to get a clear message that could influence practice.”
Axial PsA in the Portuguese Population
Separately, Catarina Abreu, a rheumatology intern at Hospital Garcia de Orta, Almada, Portugal, presented some real-world data on axial PsA from Reuma.pt.
Of 2304 patients, 854 (37.1%) reportedly had axial PsA, which had been defined as physician-reported spondylitis or the presence of imaging findings suggestive of axial involvement. This included radiographic- or MRI-detected sacroiliitis or syndesmophytes seen on axial x-rays.
The majority (78.2%) of those with an axial PsA diagnosis had concomitant peripheral involvement, with 8.1% having exclusive axial disease.
About 70% of the axial PsA diagnoses had been made using clinical or laboratory findings alone, and 30% of diagnoses was based on imaging results. Of the latter, Ms. Abreu noted that patients who had imaging data available were more likely to be HLA-B27 positive and less likely to have dactylitis, with respective odds ratios (ORs) of 3.10 and 2.42.
Individuals with axial PsA were more likely to have enthesitis (OR, 1.92), although no data were available on whether this was axial or peripheral enthesitis. Tobacco exposure was also linked to an increased chance of having axial PsA (OR, 1.66).
Ms. Abreu noted that the “scarce number of available imaging exams” and other missing data in Reuma.pt may have led to an underdiagnosis of axial PsA.
“The difference that we found between axial and peripheral [PsA] are similar to the differences found in other studies that compared axial psoriatic arthritis with axial spondyloarthritis,” Ms. Abreu said.
“So, we leave with the question that was already left before here: If these are different diseases or just different phenotypes of the same disease, and what implications will this have in the future?” Ms. Abreu concluded.
Dr. Carron received educational grants, speaker fees, or honoraria for other consultancy work from AbbVie, UCB, Pfizer, Eli Lilly, Novartis, Janssen, and Galapagos/Alfasigma. Dr. Ramiro is an ASAS executive committee member and received research grants or consulting/speaker fees from AbbVie, Eli Lilly, Galapagos, Janssen, Merck Sharp and Dohme, Novartis, Pfizer, Sanofi, and UCB. AXIS is supported by unrestricted research grants from AbbVie, Galapagos, Janssen, Eli Lilly, Novartis, Pfizer, and UCB. Dr. Torgutalp is the primary research coordinator for the study; he reported no financial conflicts of interest. The Reuma.pt registry was developed with the financial support of the pharmaceutical industry and is currently supported by AbbVie, Amgen, AstraZeneca, Boehringer Ingelheim, Eli Lilly, Merck Sharp and Dohme, Novartis, Pfizer, and Sobi. Ms. Abreu reported no financial conflicts of interest.
A version of this article appeared on Medscape.com.
VIENNA — While there is no doubt that some people with psoriatic arthritis (PsA) have axial symptoms, data presented at the annual European Congress of Rheumatology do not appear to add much to what is already known about axial PsA or to further the cause of differentiating it from axial spondyloarthritis (axSpA).
In both the AXIS study and Reuma.pt, around one in three patients with PsA were found to have axial involvement. Notably, the percentage of people with axial PsA was found to vary according to how imaging information was interpreted in the AXIS study. Both studies were discussed during the Axial Involvement in PsA and SpA session at EULAR 2024.
The One-Million-Dollar Question
“So, the one-million-dollar question: What is it, really?” Philippe Carron, MD, PhD, Ghent University Hospital, Ghent, Belgium, said in the presentation that started the session. Despite PsA being described more than 60 years ago, “we still have no internationally accepted definition or a consensus on how we should define these patients and how we should screen them,” he said.
“There are some believers that it is just a form of axial SpA with concomitant psoriasis, but also some people that think that the axial PsA is a typical disease, with typical characteristics which are different from axial disease,” Dr. Carron said.
The lack of consensus makes it difficult to estimate just how many people have axial PsA. Reported prevalences range from 5% to 70%, “all caused by which criteria that you’re using to define axial involvement,” Dr. Carron added.
There are, however, two things that can be agreed upon, according to Dr. Carron. First, the prevalence of axial involvement in people with early PsA is “much, much lower” than that of more established disease. Second, exclusive axial involvement is seen in “just a minority of PsA patients.” Most people with axial disease also have peripheral disease, he added.
Imaging findings in axial PsA “are quite similar to those seen in axial SpA,” although Dr. Carron also said that there were some distinct differences. Radiographic sacroiliitis occurs in around 25%-50% of people with axial PsA, and atypical syndesmophytes are more often found in people with axial PsA than in those with axSpA.
Shared Characteristics
But are axial PsA and axSpA separate diseases or part of the same disease continuum? That’s a question that is still very much open for debate, said Sofia Ramiro, MD, PhD, a senior researcher at Leiden University Medical Center, Leiden, the Netherlands, and rheumatology consultant at Zuyderland Medical Center in Heerlen, the Netherlands.
While many studies have looked to answer this question, there is a big methodological problem — the studies largely cannot be compared as they have used different definitions of axSpA.
Take a patient with inflammatory back pain, psoriasis, and oligoarthritis, Dr. Ramiro said. If the patient goes to one rheumatologist, they may get a diagnosis of axSpA, but if they go to a different rheumatologist, they may get a diagnosis of axial PsA.
“This is influenced by training, expertise, by beliefs, and by belonging to ASAS [Assessment of Spondyloarthritis International Society] or to GRAPPA [Group for Research and Assessment of Psoriasis and Psoriatic Arthritis],” Dr. Ramiro suggested. It’s “a diagnostic bias” that is very difficult to overcome and makes direct comparisons between patient populations recruited into clinical studies “extremely challenging.”
To confuse matters more, axial PsA and axSpA share common characteristics: Inflammatory back pain, HLA-B27 positivity, elevated levels of C-reactive protein (CRP) or a higher erythrocyte sedimentation rate, and structural lesions in the sacroiliac joints and spine.
AXIS Study ‘Gives Answers’
More research into factors associated with axial PsA need to be performed to try to help define the condition and enable classification and ultimately treatment guidelines. This is where the AXIS study comes in.
The AXIS study is a joint project of ASAS and GRAPPA that was started in January 2019 with the aim of defining a homogeneous subgroup of patients who could be studied.
“The objectives of the AXIS study are to determine the frequency of axial involvement in patients with PsA; to identify the frequency of active inflammatory and structural changes on imaging; and to identify factors associated with the presence of axial involvement in PsA,” Murat Torgutalp, MD, of Charité – Universitätsmedizin Berlin, Berlin, Germany, said at EULAR 2024.
The study population consisted of 409 consecutively recruited patients diagnosed with PsA according to CASPAR (Classification for Psoriatic Arthritis) criteria; all have had PsA for up to 10 years and were untreated with biologic or targeted synthetic disease modifying drugs at the time of inclusion.
Dr. Torgutalp, who is the study’s primary research coordinator, reported that a diagnosis of PsA was made in 37% of the population when local investigators considered available clinical, laboratory, and imaging data. However, patients’ imaging data were also centrally assessed, and when the local investigators were party to the expert imaging interpretations, the percentage of people diagnosed with PsA dropped to 27%.
“When we looked at the clinical characteristics, the presence of the back pain, particularly inflammatory back pain, HLA-B27 positivity, elevated CRP, and presence of active, inflammatory and structural changes in the sacroiliac joints and spine were associated with the final conclusion on the presence of axial involvement,” Dr. Torgutalp said.
Despite the title of his presentation being “The Axis Study Gives Answers,” Dr. Torgutalp presented lots of data without giving much insight into how they might be used. He concluded that “overall, there was a trend toward overestimation of the presence of imaging changes indicative of axial involvement across all imaging modalities” by the local investigators.
Dennis McGonagle, MB, MCH, BAO, PhD, of the University of Leeds, Leeds, England,said in an interview that the AXIS study “is a noble, international effort across multiple countries to try and better understand axial PsA.”
Dr. McGonagle, who was not involved in the study, added: “A lot of data are being generated, and a lot of analysis needs to be done to drill down to get a clear message that could influence practice.”
Axial PsA in the Portuguese Population
Separately, Catarina Abreu, a rheumatology intern at Hospital Garcia de Orta, Almada, Portugal, presented some real-world data on axial PsA from Reuma.pt.
Of 2304 patients, 854 (37.1%) reportedly had axial PsA, which had been defined as physician-reported spondylitis or the presence of imaging findings suggestive of axial involvement. This included radiographic- or MRI-detected sacroiliitis or syndesmophytes seen on axial x-rays.
The majority (78.2%) of those with an axial PsA diagnosis had concomitant peripheral involvement, with 8.1% having exclusive axial disease.
About 70% of the axial PsA diagnoses had been made using clinical or laboratory findings alone, and 30% of diagnoses was based on imaging results. Of the latter, Ms. Abreu noted that patients who had imaging data available were more likely to be HLA-B27 positive and less likely to have dactylitis, with respective odds ratios (ORs) of 3.10 and 2.42.
Individuals with axial PsA were more likely to have enthesitis (OR, 1.92), although no data were available on whether this was axial or peripheral enthesitis. Tobacco exposure was also linked to an increased chance of having axial PsA (OR, 1.66).
Ms. Abreu noted that the “scarce number of available imaging exams” and other missing data in Reuma.pt may have led to an underdiagnosis of axial PsA.
“The difference that we found between axial and peripheral [PsA] are similar to the differences found in other studies that compared axial psoriatic arthritis with axial spondyloarthritis,” Ms. Abreu said.
“So, we leave with the question that was already left before here: If these are different diseases or just different phenotypes of the same disease, and what implications will this have in the future?” Ms. Abreu concluded.
Dr. Carron received educational grants, speaker fees, or honoraria for other consultancy work from AbbVie, UCB, Pfizer, Eli Lilly, Novartis, Janssen, and Galapagos/Alfasigma. Dr. Ramiro is an ASAS executive committee member and received research grants or consulting/speaker fees from AbbVie, Eli Lilly, Galapagos, Janssen, Merck Sharp and Dohme, Novartis, Pfizer, Sanofi, and UCB. AXIS is supported by unrestricted research grants from AbbVie, Galapagos, Janssen, Eli Lilly, Novartis, Pfizer, and UCB. Dr. Torgutalp is the primary research coordinator for the study; he reported no financial conflicts of interest. The Reuma.pt registry was developed with the financial support of the pharmaceutical industry and is currently supported by AbbVie, Amgen, AstraZeneca, Boehringer Ingelheim, Eli Lilly, Merck Sharp and Dohme, Novartis, Pfizer, and Sobi. Ms. Abreu reported no financial conflicts of interest.
A version of this article appeared on Medscape.com.
FROM EULAR 2024
Two Techniques to Avoid Cyst Spray During Excision
Practice Gap
Epidermoid cysts are asymptomatic, well-circumscribed, mobile, subcutaneous masses that elevate the skin. Also known as epidermal, keratin, or infundibular cysts, epidermoid cysts are caused by proliferation of surface epidermoid cells within the dermis and can arise anywhere on the body, most commonly on the face, neck, and trunk.1 Cutaneous cysts often contain fluid or semifluid contents and can be aesthetically displeasing or cause mild pain, prompting patients to seek removal. Definitive treatment of epidermoid cysts is complete surgical removal,2 which can be performed in office in a sterile or clean manner by either dermatologists or primary care providers.
Prior to incision, a local anesthetic—commonly lidocaine with epinephrine—is injected in the region surrounding the cyst sac so as not to rupture the cyst wall. Maintaining the cyst wall throughout the procedure ensures total cyst removal and minimizes the risk for recurrence. However, it often is difficult to approximate the cyst border because it cannot be visualized prior to incision.
Throughout the duration of the procedure, cyst contents may suddenly spray out of the area and pose a risk to providers and their staff (Figure, A). Even with careful application around the periphery, either puncture or pericystic anesthesia between the cyst wall and the dermis can lead to splatter. Larger and wider peripheral anesthesia may not be possible given a shortage of lidocaine and a desire to minimize injection. Even with meticulous use of personal protective equipment in cutaneous surgery, infectious organisms found in ruptured cysts and abscesses may spray the surgical field.3 Therefore, it is in our best interest to minimize the trajectory of cyst spray contents.
The Tools
We have employed 2 simple techniques using equipment normally found on a standard surgical tray for easy safe injection of cysts. Supplies needed include 4×4-inch gauze pads, alcohol and chlorhexidine, a marker, all instruments necessary for cyst excision, and a small clear biohazard bag.
The Technique
Prior to covering the cyst, care is taken to locate the cyst opening. At times, a comedo or punctum can be seen overlying the cyst bulge. We mark the lumen and cyst opening with a surgical marker. If the pore is not easily identified, we draw an 8-mm circle around the mound of the cyst.
One option is to apply a gauze pad over the cyst to allow for stabilization of the surgical field and blanket the area from splatter (Figure, B). Then we cover the cyst using antiseptic-soaked gauze as a protective barrier to avoid potentially contaminated spray. This tool can be constructed from a 4×4-inch gauze pad with the addition of alcohol and chlorhexidine. When the cyst is covered, the surgeon can inject the lesion and surrounding tissue without biohazard splatter.
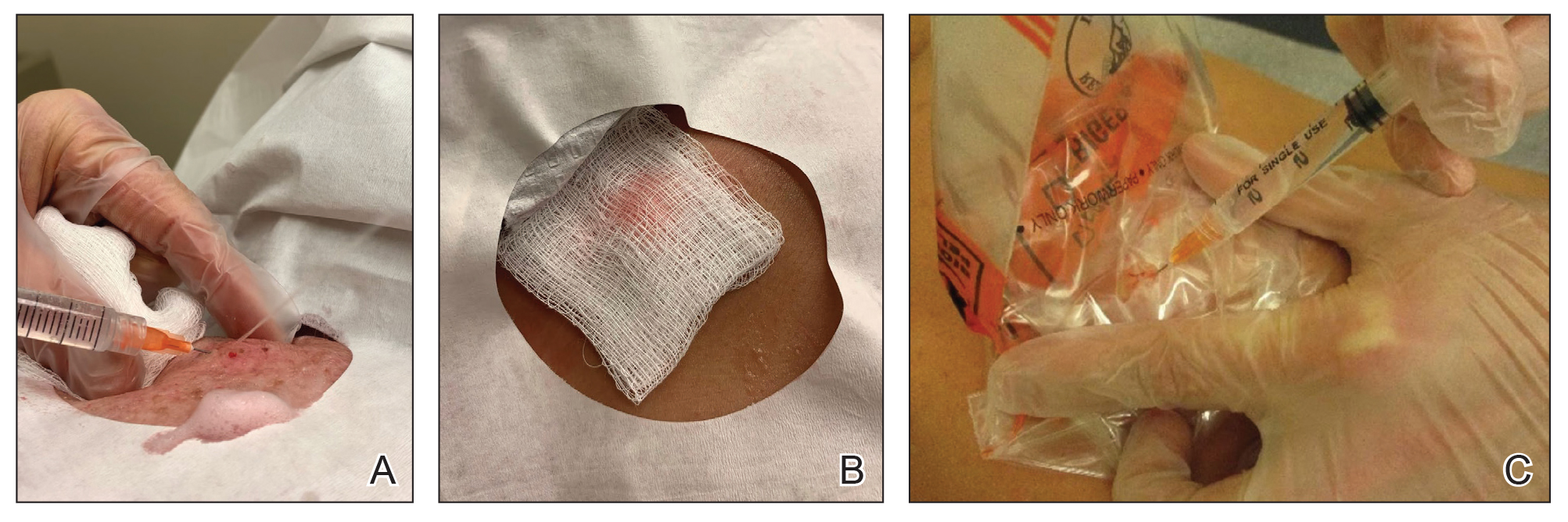
Another method is to cover the cyst with a small clear biohazard bag (Figure, C). When injecting anesthetic through the bag, the spray is captured by the bag and does not reach the surgeon or staff. This method is potentially more effective given that the cyst can still be visualized fully for more accurate injection.
Practice Implications
Outpatient surgical excision is a common effective procedure for epidermoid cysts. However, it is not uncommon for cyst contents to spray during the injection of anesthetic, posing a nuisance to the surgeon, health care staff, and patient. The technique of covering the lesion with antiseptic-soaked gauze or a small clear biohazard bag prevents cyst contents from spraying and reduces risk for contamination. In addition to these protective benefits, the use of readily available items replaces the need to order a splatter control shield.
Limitations—Although we seldom see spray using our technique, covering the cyst with gauze may disguise the region of interest and interfere with accurate incision. Marking the lesion prior to anesthesia administration or using a clear biohazard bag minimizes difficulty visualizing the cyst opening.
- Zito PM, Scharf R. Epidermoid cyst. StatPearls [Internet]. Updated August 8, 2023. Accessed June 13, 2024. https://www.ncbi.nlm.nih.gov/books/NBK499974
- Weir CB, St. Hilaire NJ. Epidermal inclusion cyst. StatPearls [Internet]. Updated August 8, 2023. Accessed June3, 2024. https://www.ncbi.nlm.nih.gov/books/NBK532310/
- Kuniyuki S, Yoshida Y, Maekawa N, et al. Bacteriological study of epidermal cysts. Acta Derm Venereol. 2018;88:23-25. doi:10.2340/00015555-0348
Practice Gap
Epidermoid cysts are asymptomatic, well-circumscribed, mobile, subcutaneous masses that elevate the skin. Also known as epidermal, keratin, or infundibular cysts, epidermoid cysts are caused by proliferation of surface epidermoid cells within the dermis and can arise anywhere on the body, most commonly on the face, neck, and trunk.1 Cutaneous cysts often contain fluid or semifluid contents and can be aesthetically displeasing or cause mild pain, prompting patients to seek removal. Definitive treatment of epidermoid cysts is complete surgical removal,2 which can be performed in office in a sterile or clean manner by either dermatologists or primary care providers.
Prior to incision, a local anesthetic—commonly lidocaine with epinephrine—is injected in the region surrounding the cyst sac so as not to rupture the cyst wall. Maintaining the cyst wall throughout the procedure ensures total cyst removal and minimizes the risk for recurrence. However, it often is difficult to approximate the cyst border because it cannot be visualized prior to incision.
Throughout the duration of the procedure, cyst contents may suddenly spray out of the area and pose a risk to providers and their staff (Figure, A). Even with careful application around the periphery, either puncture or pericystic anesthesia between the cyst wall and the dermis can lead to splatter. Larger and wider peripheral anesthesia may not be possible given a shortage of lidocaine and a desire to minimize injection. Even with meticulous use of personal protective equipment in cutaneous surgery, infectious organisms found in ruptured cysts and abscesses may spray the surgical field.3 Therefore, it is in our best interest to minimize the trajectory of cyst spray contents.
The Tools
We have employed 2 simple techniques using equipment normally found on a standard surgical tray for easy safe injection of cysts. Supplies needed include 4×4-inch gauze pads, alcohol and chlorhexidine, a marker, all instruments necessary for cyst excision, and a small clear biohazard bag.
The Technique
Prior to covering the cyst, care is taken to locate the cyst opening. At times, a comedo or punctum can be seen overlying the cyst bulge. We mark the lumen and cyst opening with a surgical marker. If the pore is not easily identified, we draw an 8-mm circle around the mound of the cyst.
One option is to apply a gauze pad over the cyst to allow for stabilization of the surgical field and blanket the area from splatter (Figure, B). Then we cover the cyst using antiseptic-soaked gauze as a protective barrier to avoid potentially contaminated spray. This tool can be constructed from a 4×4-inch gauze pad with the addition of alcohol and chlorhexidine. When the cyst is covered, the surgeon can inject the lesion and surrounding tissue without biohazard splatter.
Another method is to cover the cyst with a small clear biohazard bag (Figure, C). When injecting anesthetic through the bag, the spray is captured by the bag and does not reach the surgeon or staff. This method is potentially more effective given that the cyst can still be visualized fully for more accurate injection.
Practice Implications
Outpatient surgical excision is a common effective procedure for epidermoid cysts. However, it is not uncommon for cyst contents to spray during the injection of anesthetic, posing a nuisance to the surgeon, health care staff, and patient. The technique of covering the lesion with antiseptic-soaked gauze or a small clear biohazard bag prevents cyst contents from spraying and reduces risk for contamination. In addition to these protective benefits, the use of readily available items replaces the need to order a splatter control shield.
Limitations—Although we seldom see spray using our technique, covering the cyst with gauze may disguise the region of interest and interfere with accurate incision. Marking the lesion prior to anesthesia administration or using a clear biohazard bag minimizes difficulty visualizing the cyst opening.
Practice Gap
Epidermoid cysts are asymptomatic, well-circumscribed, mobile, subcutaneous masses that elevate the skin. Also known as epidermal, keratin, or infundibular cysts, epidermoid cysts are caused by proliferation of surface epidermoid cells within the dermis and can arise anywhere on the body, most commonly on the face, neck, and trunk.1 Cutaneous cysts often contain fluid or semifluid contents and can be aesthetically displeasing or cause mild pain, prompting patients to seek removal. Definitive treatment of epidermoid cysts is complete surgical removal,2 which can be performed in office in a sterile or clean manner by either dermatologists or primary care providers.
Prior to incision, a local anesthetic—commonly lidocaine with epinephrine—is injected in the region surrounding the cyst sac so as not to rupture the cyst wall. Maintaining the cyst wall throughout the procedure ensures total cyst removal and minimizes the risk for recurrence. However, it often is difficult to approximate the cyst border because it cannot be visualized prior to incision.
Throughout the duration of the procedure, cyst contents may suddenly spray out of the area and pose a risk to providers and their staff (Figure, A). Even with careful application around the periphery, either puncture or pericystic anesthesia between the cyst wall and the dermis can lead to splatter. Larger and wider peripheral anesthesia may not be possible given a shortage of lidocaine and a desire to minimize injection. Even with meticulous use of personal protective equipment in cutaneous surgery, infectious organisms found in ruptured cysts and abscesses may spray the surgical field.3 Therefore, it is in our best interest to minimize the trajectory of cyst spray contents.
The Tools
We have employed 2 simple techniques using equipment normally found on a standard surgical tray for easy safe injection of cysts. Supplies needed include 4×4-inch gauze pads, alcohol and chlorhexidine, a marker, all instruments necessary for cyst excision, and a small clear biohazard bag.
The Technique
Prior to covering the cyst, care is taken to locate the cyst opening. At times, a comedo or punctum can be seen overlying the cyst bulge. We mark the lumen and cyst opening with a surgical marker. If the pore is not easily identified, we draw an 8-mm circle around the mound of the cyst.
One option is to apply a gauze pad over the cyst to allow for stabilization of the surgical field and blanket the area from splatter (Figure, B). Then we cover the cyst using antiseptic-soaked gauze as a protective barrier to avoid potentially contaminated spray. This tool can be constructed from a 4×4-inch gauze pad with the addition of alcohol and chlorhexidine. When the cyst is covered, the surgeon can inject the lesion and surrounding tissue without biohazard splatter.
Another method is to cover the cyst with a small clear biohazard bag (Figure, C). When injecting anesthetic through the bag, the spray is captured by the bag and does not reach the surgeon or staff. This method is potentially more effective given that the cyst can still be visualized fully for more accurate injection.
Practice Implications
Outpatient surgical excision is a common effective procedure for epidermoid cysts. However, it is not uncommon for cyst contents to spray during the injection of anesthetic, posing a nuisance to the surgeon, health care staff, and patient. The technique of covering the lesion with antiseptic-soaked gauze or a small clear biohazard bag prevents cyst contents from spraying and reduces risk for contamination. In addition to these protective benefits, the use of readily available items replaces the need to order a splatter control shield.
Limitations—Although we seldom see spray using our technique, covering the cyst with gauze may disguise the region of interest and interfere with accurate incision. Marking the lesion prior to anesthesia administration or using a clear biohazard bag minimizes difficulty visualizing the cyst opening.
- Zito PM, Scharf R. Epidermoid cyst. StatPearls [Internet]. Updated August 8, 2023. Accessed June 13, 2024. https://www.ncbi.nlm.nih.gov/books/NBK499974
- Weir CB, St. Hilaire NJ. Epidermal inclusion cyst. StatPearls [Internet]. Updated August 8, 2023. Accessed June3, 2024. https://www.ncbi.nlm.nih.gov/books/NBK532310/
- Kuniyuki S, Yoshida Y, Maekawa N, et al. Bacteriological study of epidermal cysts. Acta Derm Venereol. 2018;88:23-25. doi:10.2340/00015555-0348
- Zito PM, Scharf R. Epidermoid cyst. StatPearls [Internet]. Updated August 8, 2023. Accessed June 13, 2024. https://www.ncbi.nlm.nih.gov/books/NBK499974
- Weir CB, St. Hilaire NJ. Epidermal inclusion cyst. StatPearls [Internet]. Updated August 8, 2023. Accessed June3, 2024. https://www.ncbi.nlm.nih.gov/books/NBK532310/
- Kuniyuki S, Yoshida Y, Maekawa N, et al. Bacteriological study of epidermal cysts. Acta Derm Venereol. 2018;88:23-25. doi:10.2340/00015555-0348
Pyzchiva Receives FDA Approval as Third Ustekinumab Biosimilar
The Food and Drug Administration has approved ustekinumab-ttwe (Pyzchiva) as a biosimilar to ustekinumab (Stelara) for the treatment of multiple inflammatory conditions.
In addition, the agency “provisionally determined” that the medication would be interchangeable with the reference product but that designation would not take hold until the interchangeability exclusivity period for the first approved biosimilar ustekinumab-auub (Wezlana) expires, according to a press release. This designation would, depending on state law, allow a pharmacist to substitute the biosimilar for the reference product without involving the prescribing clinician. It’s unclear when ustekinumab-auub’s interchangeability exclusivity ends.
Ustekinumab-ttwe, a human interleukin (IL)-12 and IL-23 antagonist, is indicated for the treatment of:
- Moderate to severe plaque psoriasis in adults and pediatric patients aged 6 years or older who are candidates for phototherapy or systemic therapy
- Active psoriatic arthritis in adults and pediatric patients aged 6 years or older with moderately to severely active Crohn’s disease or ulcerative colitis
It is administered via subcutaneous injection in 45 mg/0.5 mL and 90 mg/mL prefilled syringes or via intravenous infusion in 130 mg/26 mL (5 mg/mL) single-dose vial.
Developed by Samsung Bioepis, ustekinumab-ttwe will be commercialized by Sandoz in the United States. Besides ustekinumab-auub, the other ustekinumab biosimilar is ustekinumab-aekn (Selarsdi).
Ustekinumab-ttwe is expected to launch in February 2025 “in accordance with the settlement and license agreement with Janssen Biotech,” which manufacturers the reference product, Sandoz said. The other approved ustekinumab biosimilars will launch within a similar time frame.
A version of this article appeared on Medscape.com.
The Food and Drug Administration has approved ustekinumab-ttwe (Pyzchiva) as a biosimilar to ustekinumab (Stelara) for the treatment of multiple inflammatory conditions.
In addition, the agency “provisionally determined” that the medication would be interchangeable with the reference product but that designation would not take hold until the interchangeability exclusivity period for the first approved biosimilar ustekinumab-auub (Wezlana) expires, according to a press release. This designation would, depending on state law, allow a pharmacist to substitute the biosimilar for the reference product without involving the prescribing clinician. It’s unclear when ustekinumab-auub’s interchangeability exclusivity ends.
Ustekinumab-ttwe, a human interleukin (IL)-12 and IL-23 antagonist, is indicated for the treatment of:
- Moderate to severe plaque psoriasis in adults and pediatric patients aged 6 years or older who are candidates for phototherapy or systemic therapy
- Active psoriatic arthritis in adults and pediatric patients aged 6 years or older with moderately to severely active Crohn’s disease or ulcerative colitis
It is administered via subcutaneous injection in 45 mg/0.5 mL and 90 mg/mL prefilled syringes or via intravenous infusion in 130 mg/26 mL (5 mg/mL) single-dose vial.
Developed by Samsung Bioepis, ustekinumab-ttwe will be commercialized by Sandoz in the United States. Besides ustekinumab-auub, the other ustekinumab biosimilar is ustekinumab-aekn (Selarsdi).
Ustekinumab-ttwe is expected to launch in February 2025 “in accordance with the settlement and license agreement with Janssen Biotech,” which manufacturers the reference product, Sandoz said. The other approved ustekinumab biosimilars will launch within a similar time frame.
A version of this article appeared on Medscape.com.
The Food and Drug Administration has approved ustekinumab-ttwe (Pyzchiva) as a biosimilar to ustekinumab (Stelara) for the treatment of multiple inflammatory conditions.
In addition, the agency “provisionally determined” that the medication would be interchangeable with the reference product but that designation would not take hold until the interchangeability exclusivity period for the first approved biosimilar ustekinumab-auub (Wezlana) expires, according to a press release. This designation would, depending on state law, allow a pharmacist to substitute the biosimilar for the reference product without involving the prescribing clinician. It’s unclear when ustekinumab-auub’s interchangeability exclusivity ends.
Ustekinumab-ttwe, a human interleukin (IL)-12 and IL-23 antagonist, is indicated for the treatment of:
- Moderate to severe plaque psoriasis in adults and pediatric patients aged 6 years or older who are candidates for phototherapy or systemic therapy
- Active psoriatic arthritis in adults and pediatric patients aged 6 years or older with moderately to severely active Crohn’s disease or ulcerative colitis
It is administered via subcutaneous injection in 45 mg/0.5 mL and 90 mg/mL prefilled syringes or via intravenous infusion in 130 mg/26 mL (5 mg/mL) single-dose vial.
Developed by Samsung Bioepis, ustekinumab-ttwe will be commercialized by Sandoz in the United States. Besides ustekinumab-auub, the other ustekinumab biosimilar is ustekinumab-aekn (Selarsdi).
Ustekinumab-ttwe is expected to launch in February 2025 “in accordance with the settlement and license agreement with Janssen Biotech,” which manufacturers the reference product, Sandoz said. The other approved ustekinumab biosimilars will launch within a similar time frame.
A version of this article appeared on Medscape.com.
Vascular Mass on the Posterior Neck in a Newborn
The Diagnosis: Congenital Hemangioma
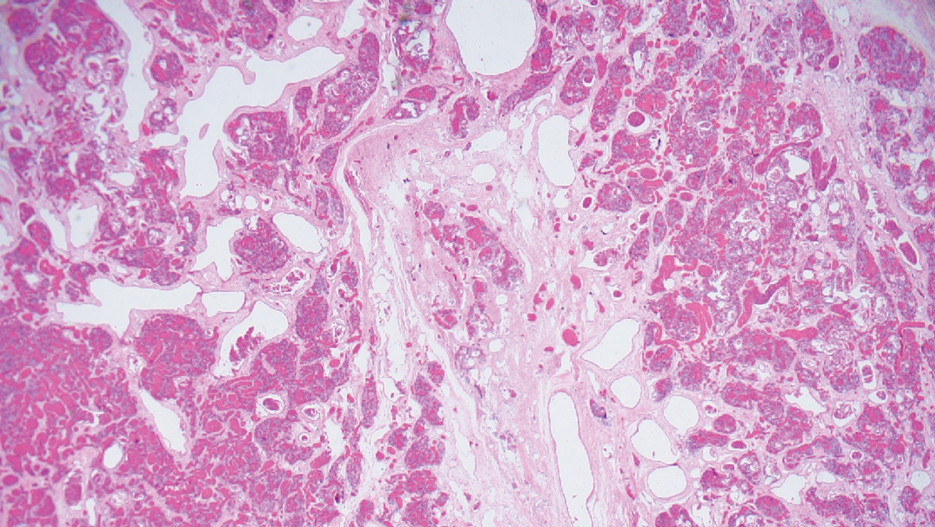
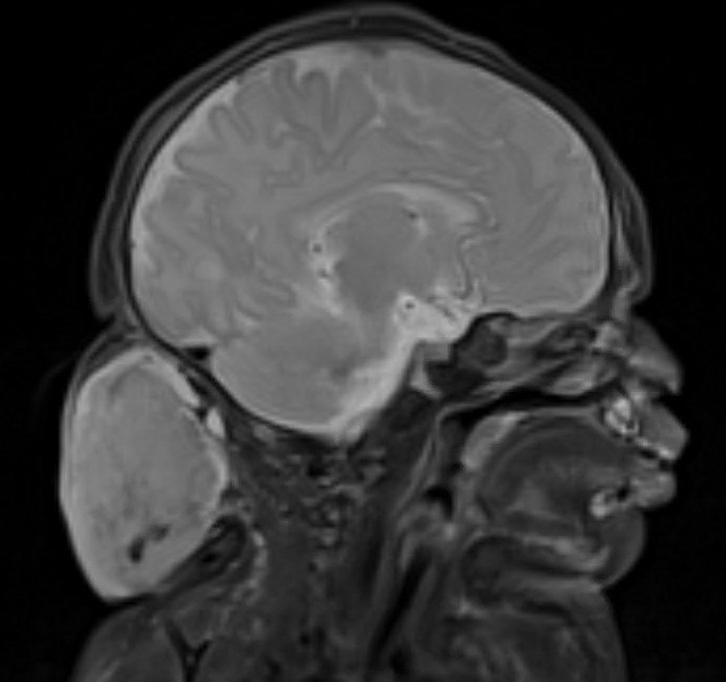
Surgical resection of the mass was performed at 4 months of age without complication (Figure 1). Histopathology revealed a lobular endothelial cell proliferation within a densely fibrotic stroma, multiple thin-walled vessels, and negative immunoreactivity to glucose transporter type 1 (GLUT-1)(Figures 2 and 3). Combined with the patient’s clinical history and findings on imaging (Figure 4), the most accurate diagnosis was a congenital hemangioma (CH). The mass was determined to be a noninvoluting congenital hemangioma (NICH).
A variety of vascular anomalies manifest in newborns and can be differentiated by the patient’s clinical history—particularly whether the lesion is present at birth or develops after birth. Imaging and histopathology of the lesion(s) may be utilized when clinical examination alone is not sufficient to make a diagnosis. Histopathology and immunohistochemistry further aid in differentiating the type of vascular lesion.

Overall, vascular anomalies are classified broadly into 2 categories based on their pathogenesis: tumors and malformations. Vascular tumors are composed of proliferating endothelial cells that have the potential to resolve spontaneously over time. Examples include CH, infantile hemangioma (IH), kaposiform hemangioendothelioma (KHE), and tufted angioma (TA). In contrast, vascular malformations (ie, arteriovenous malformations) are composed of dysplastic vessels with normal endothelial cell turnover and do not resolve without intervention.1
Congenital hemangiomas are rare vascular tumors that are fully developed at birth. These tumors proliferate in utero, enabling prenatal detection via ultrasonography as early as 12 weeks’ gestation for large heterogeneous vascular masses.2-4 Congenital hemangiomas are described as solitary, well-circumscribed, raised, violaceous lesions most commonly located in the head and neck region.4-6 Histopathologically, they are characterized by lobules of proliferating capillaries surrounded by fibrous stroma and dysplastic vascular channels.6,7
Congenital hemangiomas are categorized based on their postnatal involution patterns.2 Fetally involuting CH both develops and begins regression in utero and often is completely regressed at birth.8 Rapidly involuting CH begins regression in the first few weeks of life and usually is completely involuted by 14 months of age.6,9-11 Conversely, NICH does not regress, often requiring surgical excision due to functional and cosmetic issues.12,13 Partially involuting CH is intermediary, beginning as rapidly involuting but not involuting completely and persisting as lesions that resemble NICH.14-16 Although generally benign and asymptomatic, these tumors can cause transient thrombocytopenia and coagulopathy at birth, as seen in our patient.17,18
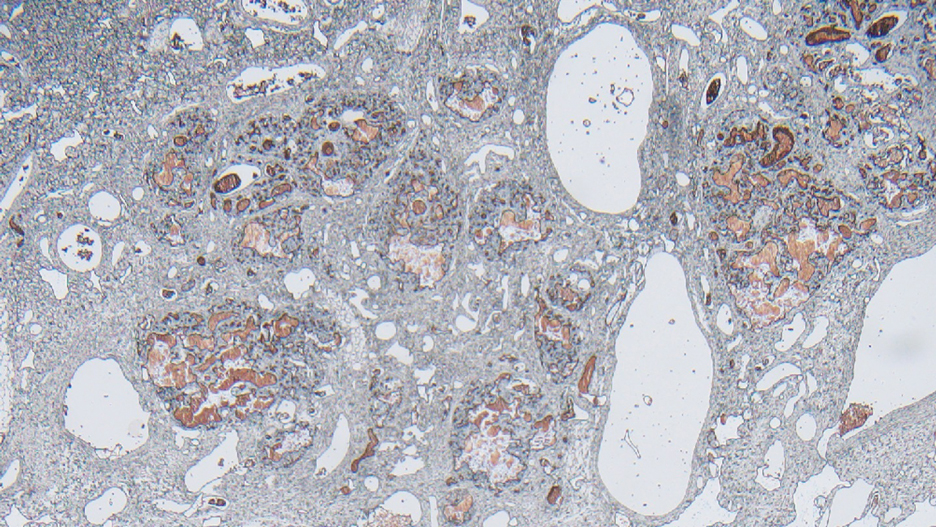
Infantile hemangioma is the most common vascular tumor of infancy.19-21 Although a precursor lesion may be present at birth, generally this tumor becomes apparent after the first few weeks of life as a solitary vascular plaque or nodule with a predilection for the head and neck.22-25 Once it arises, IH quickly enters a period of rapid growth, followed by a period of slower continued growth, with most reaching maximum size by 3 months.22 Thereafter, IH enters a slow period of involution (range, 3–9 years)26; more recent data suggest near resolution by 5 years of age.27 Infantile hemangioma is categorized based on its depth in the skin and subcutaneous tissues and can be classified as superficial, mixed, or deep.22,24,28,29 Superficial IH appears as a red plaque and may exhibit lobulation, while deep IH can be identified as flesh-colored or blue subcutaneous masses. Mixed IH may manifest with both superficial and deep features depending on the extent of its involvement in the dermal and subcutaneous layers. The pattern of involvement may be focal, segmental, or indeterminate.24 In contrast, CH typically is a solitary vascular mass with prominent telangiectases, nodules, and radiating veins.6 Histologically, IH is composed of proliferative plump endothelial cells that form capillaries, and the lesion stains positively for GLUT-1, whereas CH does not.30
Kaposiform hemangioendothelioma is classified as a locally aggressive vascular tumor that manifests either prenatally or in early infancy.31 It is described as a solitary, ill-defined, firm, purple plaque most commonly located on the extremities and retroperitoneum.32-34 Histopathologically, these lesions are characterized by dilated lymphatic channels and irregular sheets or lobules of spindle-shaped endothelial cells infiltrating the dermis and subcutaneous fat.33,35 In contrast to CH, KHE lesions show immunoreactivity to the markers podoplanin, lymphatic vessel endothelial receptor 1, and prospero homeobox 1 protein.36,37 Notably, 70% of these tumors are complicated by the presence of Kasabach-Merritt phenomenon, a potentially life-threatening emergency that occurs when platelets are trapped within a vascular tumor, leading to the consumption of clotting factors, intralesional bleeding, and rapid enlargement of the tumor.32 The Kasabach-Merritt phenomenon manifests clinically as microangiopathic hemolytic anemia, severe thrombocytopenia, and disseminated intravascular coagulation. 38 Although CH lesions also can be associated with thrombocytopenia and coagulopathy, they generally are mild and self-limited.18
Tufted angioma is a vascular tumor that arises within the first 5 years of life as firm violaceous papules or plaques, often with associated hyperhidrosis or hypertrichosis.39,40 Although TA grows slowly for a period of time, it eventually stabilizes and persists, rarely regressing completely.41 These tumors share many similarities with KHE, and it has been suggested that they may be part of the same spectrum. 42 As with KHE, TA lesions show immunoreactivity to the markers podoplanin, lymphatic vessel endothelial receptor 1, and prospero homeobox 1 protein, which are negative in CH.36,37 Although TA also can be complicated by Kasabach-Merritt phenomenon, the incidence is much lower (up to 38%).43,44 As such, TAs tend to be recognized as more superficial benign lesions. However, they still can cause notable cosmetic and functional impairment and should be monitored closely, especially in the presence of associated symptoms or complications.
Arteriovenous malformation is a vascular lesion that results from errors during the embryonic development of vascular channels.45 Although present at birth, it may not become clinically apparent until later in life. Arteriovenous malformations enlarge postnatally, and their growth is proportional to the developmental growth of the affected individual rather than the result of endothelial proliferation.46 In infants, AVM may manifest as a faint vascular stain that can evolve over time into a pink patch associated with a palpable thrill during adolescence. 4 On Doppler flow imaging, AVMs are identified as fast-flow anomalies arising from an abnormal communication between high-pressure arterial systems and low-pressure venous systems without the presence of a capillary bed.47 One of the differentiating factors between AVM and CH is that AVMs do not regress spontaneously and tend to have high recurrence rates, even with intervention. 48 In contrast, CH can be categorized based on its postnatal involution pattern. Another distinguishing factor is that AVMs tend to be larger and more invasive than CHs.46 Therefore, early diagnosis and intervention are crucial to prevent complications such as bleeding, seizures, or neurologic deficits associated with AVMs.1
- Enjolras O, Wassef M, Chapot R. Introduction: ISSVA Classification. In: Enjolras O, Wassef M, Chapot R, eds. Color Atlas of Vascular Tumors and Vascular Malformations. Cambridge University Press; 2007:3-11.
- Fadell MF, Jones BV, Adams DM. Prenatal diagnosis and postnatal follow-up of rapidly involuting congenital hemangioma (RICH). Pediatr Radiol. 2011;41:1057-1060.
- Feygin T, Khalek N, Moldenhauer JS. Fetal brain, head, and neck tumors: prenatal imaging and management. Prenat Diagn. 2020;40:1203-1219.
- Foley LS, Kulungowski AM. Vascular anomalies in pediatrics. Adv Pediatr. 2015;62:227-255.
- Bruder E, Alaggio R, Kozakewich HPW, et al. Vascular and perivascular lesions of skin and soft tissues in children and adolescents. Pediatr Dev Pathol. 2012;15:26-61.
- Berenguer B, Mulliken JB, Enjolras O, et al. Rapidly involuting congenital hemangioma: clinical and histopathologic features. Pediatr Dev Pathol. 2003;6:495-510.
- North PE, Waner M, James CA, et al. Congenital nonprogressive hemangioma: a distinct clinicopathologic entity unlike infantile hemangioma. Arch Dermatol. 2001;137:1607-1620.
- Maguiness S, Uihlein LC, Liang MG, et al. Rapidly involuting congenital hemangioma with fetal involution. Pediatr Dermatol. 2015;32:321-326.
- Keating LJ, Soares GM, Muratore CS. Rapidly involuting congenital hemangioma. Med Health R I. 2012;95:149-152.
- Schafer F, Tapia M, Pinto C. Rapidly involuting congenital haemangioma. Arch Dis Child Fetal Neonatal Ed. 2014;99:F422.
- Boon LM, Enjolras O, Mulliken JB. Congenital hemangioma: evidence of accelerated involution. J Pediatr. 1996;128:329-335.
- Liang MG, Frieden IJ. Infantile and congenital hemangiomas. Semin Pediatr Surg. 2014;23:162-167.
- Enjolras O, Mulliken JB, Boon LM, et al. Noninvoluting congenital hemangioma: a rare cutaneous vascular anomaly. Plast Reconstr Surg. 2001;107:1647-1654.
- Nasseri E, Piram M, McCuaig CC, et al. Partially involuting congenital hemangiomas: a report of 8 cases and review of the literature. J Am Acad Dermatol. 2014;70:75-79.
- Wassef M, Blei F, Adams D, et al. Vascular anomalies classification: recommendations from the International Society for the Study of Vascular Anomalies. Pediatrics. 2015;136:E203-E214.
- Boull C, Maguiness SM. Congenital hemangiomas. Semin Cutan Med Surg. 2016;35:124-127.
- Drolet BA, Frommelt PC, Chamlin SL, et al. Initiation and use of propranolol for infantile hemangioma: report of a consensus conference. Pediatrics. 2013;131:128-140.
- Baselga E, Cordisco MR, Garzon M, et al. Rapidly involuting congenital haemangioma associated with transient thrombocytopenia and coagulopathy: a case series. Br J Dermatol. 2008;158:1363-1370.
- Kanada KN, Merin MR, Munden A, et al. A prospective study of cutaneous findings in newborns in the United States: correlation with race, ethnicity, and gestational status using updated classification and nomenclature. J Pediatr. 2012;161:240-245.
- Munden A, Butschek R, Tom WL, et al. Prospective study of infantile haemangiomas: incidence, clinical characteristics and association with placental anomalies. Br J Dermatol. 2014;170:907-913.
- Léauté-Labrèze C, Harper JI, Hoeger PH. Infantile haemangioma. Lancet. 2017;390:85-94.
- Chang LC, Haggstrom AN, Drolet BA, et al. Growth characteristics of infantile hemangiomas: implications for management. Pediatrics. 2008;122:360-367.
- Hidano A, Nakajima S. Earliest features of the strawberry mark in the newborn. Br J Dermatol. 1972;87:138-144.
- Martinez-Perez D, Fein NA, Boon LM, et al. Not all hemangiomas look like strawberries: uncommon presentations of the most common tumor of infancy. Pediatr Dermatol. 1995;12:1-6.
- Payne MM, Moyer F, Marcks KM, et al. The precursor to the hemangioma. Plast Reconstr Surg. 1966;38:64-67.
- Bowers RE, Graham EA, Tomlinson KM. The natural history of the strawberry nevus. Arch Dermatol. 1960;82:667-680.
- Couto RA, Maclellan RA, Zurakowski D, et al. Infantile hemangioma: clinical assessment of the involuting phase and implications for management. Plast Reconstr Surg. 2012;130:619-624.
- Drolet BA, Esterly NB, Frieden IJ. Hemangiomas in children. N Engl J Med. 1999;341:173-181.
- Chiller KG, Passaro D, Frieden IJ. Hemangiomas of infancy: clinical characteristics, morphologic subtypes, and their relationship to race, ethnicity, and sex. Arch Dermatol. 2002;138:1567-1576.
- North PE, Waner M, Mizeracki A, et al. GLUT1: a newly discovered immunohistochemical marker for juvenile hemangiomas. Hum Pathol. 2000;31:11-22.
- Gruman A, Liang MG, Mulliken JB, et al. Kaposiform hemangioendothelioma without Kasabach-Merritt phenomenon. J Am Acad Dermatol. 2005;52:616-622.
- Croteau SE, Liang MG, Kozakewich HP, et al. Kaposiform hemangioendothelioma: atypical features and risks of Kasabach- Merritt phenomenon in 107 referrals. J Pediatr. 2013;162:142-147.
- Zukerberg LR, Nickoloff BJ, Weiss SW. Kaposiform hemangioendothelioma of infancy and childhood. an aggressive neoplasm associated with Kasabach-Merritt syndrome and lymphangiomatosis. Am J Surg Pathol. 1993;17:321-328.
- Mac-Moune Lai F, To KF, Choi PC, et al. Kaposiform hemangioendothelioma: five patients with cutaneous lesion and long follow-up. Mod Pathol. 2001;14:1087-1092.
- O’Rafferty C, O’Regan GM, Irvine AD, et al. Recent advances in the pathobiology and management of Kasabach-Merritt phenomenon. Br J Haematol. 2015;171:38-51.
- Le Huu AR, Jokinen CH, Rubin BP, et al. Expression of prox1, lymphatic endothelial nuclear transcription factor, in kaposiform hemangioendothelioma and tufted angioma. Am J Surg Pathol. 2010;34:1563-1573.
- Debelenko LV, Perez-Atayde AR, Mulliken JB, et al. D2-40 immuno-histochemical analysis of pediatric vascular tumors reveals positivity in kaposiform hemangioendothelioma. Mod Pathol. 2005;18:1454-1460.
- Haisley-Royster C, Enjolras O, Frieden IJ, et al. Kasabach-Merritt phenomenon: a retrospective study of treatment with vincristine. J Pediatr Hematol Oncol. 2002;24:459-462.
- Wilmer A, Kaatz M, Bocker T, et al. Tufted angioma. Eur J Dermatol. 1999;9:51-53.
- Herron MD, Coffin CM, Vanderhooft SL. Tufted angiomas: variability of the clinical morphology. Pediatr Dermatol. 2002;19:394-401.
- North PE. Pediatric vascular tumors and malformations. Surg Pathol Clin. 2010,3:455-494.
- Chu CY, Hsiao CH, Chiu HC. Transformation between kaposiform hemangioendothelioma and tufted angioma. Dermatology. 2003;206:334-337.
- Osio A, Fraitag S, Hadj-Rabia S, et al. Clinical spectrum of tufted angiomas in childhood: a report of 13 cases and a review of the literature. Arch Dermatol. 2010;146:758-763.
- Johnson EF, Davis DM, Tollefson MM, et al. Vascular tumors in infants: case report and review of clinical, histopathologic, and immunohistochemical characteristics of infantile hemangioma, pyogenic granuloma, noninvoluting congenital hemangioma, tufted angioma, and kaposiform hemangioendothelioma. Am J Dermatopathol. 2018;40:231-239.
- Christison-Lagay ER, Fishman SJ. Vascular anomalies. Surg Clin North Am. 2006;86:393-425.
- Liu AS, Mulliken JB, Zurakowski D, et al. Extracranial arteriovenous malformations: natural progression and recurrence after treatment. Plast Reconstr Surg. 2010;125:1185-1194.
- Young AE, Mulliken JB. Arteriovenous malformations. In: Mulliken JB, Young AE, eds. Vascular Birthmarks: Haemangiomas and Malformations. WB Saunders; 1988:228-245.
- Duggan EM, Fishman SJ. Vascular anomalies. In: Holcomb GW III, Murphy JP, St Peter SD, eds. Holcomb and Ashcraft’s Pediatric Surgery. 7th edition. Elsevier; 2019:1147-1170.
The Diagnosis: Congenital Hemangioma
Surgical resection of the mass was performed at 4 months of age without complication (Figure 1). Histopathology revealed a lobular endothelial cell proliferation within a densely fibrotic stroma, multiple thin-walled vessels, and negative immunoreactivity to glucose transporter type 1 (GLUT-1)(Figures 2 and 3). Combined with the patient’s clinical history and findings on imaging (Figure 4), the most accurate diagnosis was a congenital hemangioma (CH). The mass was determined to be a noninvoluting congenital hemangioma (NICH).
A variety of vascular anomalies manifest in newborns and can be differentiated by the patient’s clinical history—particularly whether the lesion is present at birth or develops after birth. Imaging and histopathology of the lesion(s) may be utilized when clinical examination alone is not sufficient to make a diagnosis. Histopathology and immunohistochemistry further aid in differentiating the type of vascular lesion.
Overall, vascular anomalies are classified broadly into 2 categories based on their pathogenesis: tumors and malformations. Vascular tumors are composed of proliferating endothelial cells that have the potential to resolve spontaneously over time. Examples include CH, infantile hemangioma (IH), kaposiform hemangioendothelioma (KHE), and tufted angioma (TA). In contrast, vascular malformations (ie, arteriovenous malformations) are composed of dysplastic vessels with normal endothelial cell turnover and do not resolve without intervention.1
Congenital hemangiomas are rare vascular tumors that are fully developed at birth. These tumors proliferate in utero, enabling prenatal detection via ultrasonography as early as 12 weeks’ gestation for large heterogeneous vascular masses.2-4 Congenital hemangiomas are described as solitary, well-circumscribed, raised, violaceous lesions most commonly located in the head and neck region.4-6 Histopathologically, they are characterized by lobules of proliferating capillaries surrounded by fibrous stroma and dysplastic vascular channels.6,7
Congenital hemangiomas are categorized based on their postnatal involution patterns.2 Fetally involuting CH both develops and begins regression in utero and often is completely regressed at birth.8 Rapidly involuting CH begins regression in the first few weeks of life and usually is completely involuted by 14 months of age.6,9-11 Conversely, NICH does not regress, often requiring surgical excision due to functional and cosmetic issues.12,13 Partially involuting CH is intermediary, beginning as rapidly involuting but not involuting completely and persisting as lesions that resemble NICH.14-16 Although generally benign and asymptomatic, these tumors can cause transient thrombocytopenia and coagulopathy at birth, as seen in our patient.17,18
Infantile hemangioma is the most common vascular tumor of infancy.19-21 Although a precursor lesion may be present at birth, generally this tumor becomes apparent after the first few weeks of life as a solitary vascular plaque or nodule with a predilection for the head and neck.22-25 Once it arises, IH quickly enters a period of rapid growth, followed by a period of slower continued growth, with most reaching maximum size by 3 months.22 Thereafter, IH enters a slow period of involution (range, 3–9 years)26; more recent data suggest near resolution by 5 years of age.27 Infantile hemangioma is categorized based on its depth in the skin and subcutaneous tissues and can be classified as superficial, mixed, or deep.22,24,28,29 Superficial IH appears as a red plaque and may exhibit lobulation, while deep IH can be identified as flesh-colored or blue subcutaneous masses. Mixed IH may manifest with both superficial and deep features depending on the extent of its involvement in the dermal and subcutaneous layers. The pattern of involvement may be focal, segmental, or indeterminate.24 In contrast, CH typically is a solitary vascular mass with prominent telangiectases, nodules, and radiating veins.6 Histologically, IH is composed of proliferative plump endothelial cells that form capillaries, and the lesion stains positively for GLUT-1, whereas CH does not.30
Kaposiform hemangioendothelioma is classified as a locally aggressive vascular tumor that manifests either prenatally or in early infancy.31 It is described as a solitary, ill-defined, firm, purple plaque most commonly located on the extremities and retroperitoneum.32-34 Histopathologically, these lesions are characterized by dilated lymphatic channels and irregular sheets or lobules of spindle-shaped endothelial cells infiltrating the dermis and subcutaneous fat.33,35 In contrast to CH, KHE lesions show immunoreactivity to the markers podoplanin, lymphatic vessel endothelial receptor 1, and prospero homeobox 1 protein.36,37 Notably, 70% of these tumors are complicated by the presence of Kasabach-Merritt phenomenon, a potentially life-threatening emergency that occurs when platelets are trapped within a vascular tumor, leading to the consumption of clotting factors, intralesional bleeding, and rapid enlargement of the tumor.32 The Kasabach-Merritt phenomenon manifests clinically as microangiopathic hemolytic anemia, severe thrombocytopenia, and disseminated intravascular coagulation. 38 Although CH lesions also can be associated with thrombocytopenia and coagulopathy, they generally are mild and self-limited.18
Tufted angioma is a vascular tumor that arises within the first 5 years of life as firm violaceous papules or plaques, often with associated hyperhidrosis or hypertrichosis.39,40 Although TA grows slowly for a period of time, it eventually stabilizes and persists, rarely regressing completely.41 These tumors share many similarities with KHE, and it has been suggested that they may be part of the same spectrum. 42 As with KHE, TA lesions show immunoreactivity to the markers podoplanin, lymphatic vessel endothelial receptor 1, and prospero homeobox 1 protein, which are negative in CH.36,37 Although TA also can be complicated by Kasabach-Merritt phenomenon, the incidence is much lower (up to 38%).43,44 As such, TAs tend to be recognized as more superficial benign lesions. However, they still can cause notable cosmetic and functional impairment and should be monitored closely, especially in the presence of associated symptoms or complications.
Arteriovenous malformation is a vascular lesion that results from errors during the embryonic development of vascular channels.45 Although present at birth, it may not become clinically apparent until later in life. Arteriovenous malformations enlarge postnatally, and their growth is proportional to the developmental growth of the affected individual rather than the result of endothelial proliferation.46 In infants, AVM may manifest as a faint vascular stain that can evolve over time into a pink patch associated with a palpable thrill during adolescence. 4 On Doppler flow imaging, AVMs are identified as fast-flow anomalies arising from an abnormal communication between high-pressure arterial systems and low-pressure venous systems without the presence of a capillary bed.47 One of the differentiating factors between AVM and CH is that AVMs do not regress spontaneously and tend to have high recurrence rates, even with intervention. 48 In contrast, CH can be categorized based on its postnatal involution pattern. Another distinguishing factor is that AVMs tend to be larger and more invasive than CHs.46 Therefore, early diagnosis and intervention are crucial to prevent complications such as bleeding, seizures, or neurologic deficits associated with AVMs.1
The Diagnosis: Congenital Hemangioma
Surgical resection of the mass was performed at 4 months of age without complication (Figure 1). Histopathology revealed a lobular endothelial cell proliferation within a densely fibrotic stroma, multiple thin-walled vessels, and negative immunoreactivity to glucose transporter type 1 (GLUT-1)(Figures 2 and 3). Combined with the patient’s clinical history and findings on imaging (Figure 4), the most accurate diagnosis was a congenital hemangioma (CH). The mass was determined to be a noninvoluting congenital hemangioma (NICH).
A variety of vascular anomalies manifest in newborns and can be differentiated by the patient’s clinical history—particularly whether the lesion is present at birth or develops after birth. Imaging and histopathology of the lesion(s) may be utilized when clinical examination alone is not sufficient to make a diagnosis. Histopathology and immunohistochemistry further aid in differentiating the type of vascular lesion.
Overall, vascular anomalies are classified broadly into 2 categories based on their pathogenesis: tumors and malformations. Vascular tumors are composed of proliferating endothelial cells that have the potential to resolve spontaneously over time. Examples include CH, infantile hemangioma (IH), kaposiform hemangioendothelioma (KHE), and tufted angioma (TA). In contrast, vascular malformations (ie, arteriovenous malformations) are composed of dysplastic vessels with normal endothelial cell turnover and do not resolve without intervention.1
Congenital hemangiomas are rare vascular tumors that are fully developed at birth. These tumors proliferate in utero, enabling prenatal detection via ultrasonography as early as 12 weeks’ gestation for large heterogeneous vascular masses.2-4 Congenital hemangiomas are described as solitary, well-circumscribed, raised, violaceous lesions most commonly located in the head and neck region.4-6 Histopathologically, they are characterized by lobules of proliferating capillaries surrounded by fibrous stroma and dysplastic vascular channels.6,7
Congenital hemangiomas are categorized based on their postnatal involution patterns.2 Fetally involuting CH both develops and begins regression in utero and often is completely regressed at birth.8 Rapidly involuting CH begins regression in the first few weeks of life and usually is completely involuted by 14 months of age.6,9-11 Conversely, NICH does not regress, often requiring surgical excision due to functional and cosmetic issues.12,13 Partially involuting CH is intermediary, beginning as rapidly involuting but not involuting completely and persisting as lesions that resemble NICH.14-16 Although generally benign and asymptomatic, these tumors can cause transient thrombocytopenia and coagulopathy at birth, as seen in our patient.17,18
Infantile hemangioma is the most common vascular tumor of infancy.19-21 Although a precursor lesion may be present at birth, generally this tumor becomes apparent after the first few weeks of life as a solitary vascular plaque or nodule with a predilection for the head and neck.22-25 Once it arises, IH quickly enters a period of rapid growth, followed by a period of slower continued growth, with most reaching maximum size by 3 months.22 Thereafter, IH enters a slow period of involution (range, 3–9 years)26; more recent data suggest near resolution by 5 years of age.27 Infantile hemangioma is categorized based on its depth in the skin and subcutaneous tissues and can be classified as superficial, mixed, or deep.22,24,28,29 Superficial IH appears as a red plaque and may exhibit lobulation, while deep IH can be identified as flesh-colored or blue subcutaneous masses. Mixed IH may manifest with both superficial and deep features depending on the extent of its involvement in the dermal and subcutaneous layers. The pattern of involvement may be focal, segmental, or indeterminate.24 In contrast, CH typically is a solitary vascular mass with prominent telangiectases, nodules, and radiating veins.6 Histologically, IH is composed of proliferative plump endothelial cells that form capillaries, and the lesion stains positively for GLUT-1, whereas CH does not.30
Kaposiform hemangioendothelioma is classified as a locally aggressive vascular tumor that manifests either prenatally or in early infancy.31 It is described as a solitary, ill-defined, firm, purple plaque most commonly located on the extremities and retroperitoneum.32-34 Histopathologically, these lesions are characterized by dilated lymphatic channels and irregular sheets or lobules of spindle-shaped endothelial cells infiltrating the dermis and subcutaneous fat.33,35 In contrast to CH, KHE lesions show immunoreactivity to the markers podoplanin, lymphatic vessel endothelial receptor 1, and prospero homeobox 1 protein.36,37 Notably, 70% of these tumors are complicated by the presence of Kasabach-Merritt phenomenon, a potentially life-threatening emergency that occurs when platelets are trapped within a vascular tumor, leading to the consumption of clotting factors, intralesional bleeding, and rapid enlargement of the tumor.32 The Kasabach-Merritt phenomenon manifests clinically as microangiopathic hemolytic anemia, severe thrombocytopenia, and disseminated intravascular coagulation. 38 Although CH lesions also can be associated with thrombocytopenia and coagulopathy, they generally are mild and self-limited.18
Tufted angioma is a vascular tumor that arises within the first 5 years of life as firm violaceous papules or plaques, often with associated hyperhidrosis or hypertrichosis.39,40 Although TA grows slowly for a period of time, it eventually stabilizes and persists, rarely regressing completely.41 These tumors share many similarities with KHE, and it has been suggested that they may be part of the same spectrum. 42 As with KHE, TA lesions show immunoreactivity to the markers podoplanin, lymphatic vessel endothelial receptor 1, and prospero homeobox 1 protein, which are negative in CH.36,37 Although TA also can be complicated by Kasabach-Merritt phenomenon, the incidence is much lower (up to 38%).43,44 As such, TAs tend to be recognized as more superficial benign lesions. However, they still can cause notable cosmetic and functional impairment and should be monitored closely, especially in the presence of associated symptoms or complications.
Arteriovenous malformation is a vascular lesion that results from errors during the embryonic development of vascular channels.45 Although present at birth, it may not become clinically apparent until later in life. Arteriovenous malformations enlarge postnatally, and their growth is proportional to the developmental growth of the affected individual rather than the result of endothelial proliferation.46 In infants, AVM may manifest as a faint vascular stain that can evolve over time into a pink patch associated with a palpable thrill during adolescence. 4 On Doppler flow imaging, AVMs are identified as fast-flow anomalies arising from an abnormal communication between high-pressure arterial systems and low-pressure venous systems without the presence of a capillary bed.47 One of the differentiating factors between AVM and CH is that AVMs do not regress spontaneously and tend to have high recurrence rates, even with intervention. 48 In contrast, CH can be categorized based on its postnatal involution pattern. Another distinguishing factor is that AVMs tend to be larger and more invasive than CHs.46 Therefore, early diagnosis and intervention are crucial to prevent complications such as bleeding, seizures, or neurologic deficits associated with AVMs.1
- Enjolras O, Wassef M, Chapot R. Introduction: ISSVA Classification. In: Enjolras O, Wassef M, Chapot R, eds. Color Atlas of Vascular Tumors and Vascular Malformations. Cambridge University Press; 2007:3-11.
- Fadell MF, Jones BV, Adams DM. Prenatal diagnosis and postnatal follow-up of rapidly involuting congenital hemangioma (RICH). Pediatr Radiol. 2011;41:1057-1060.
- Feygin T, Khalek N, Moldenhauer JS. Fetal brain, head, and neck tumors: prenatal imaging and management. Prenat Diagn. 2020;40:1203-1219.
- Foley LS, Kulungowski AM. Vascular anomalies in pediatrics. Adv Pediatr. 2015;62:227-255.
- Bruder E, Alaggio R, Kozakewich HPW, et al. Vascular and perivascular lesions of skin and soft tissues in children and adolescents. Pediatr Dev Pathol. 2012;15:26-61.
- Berenguer B, Mulliken JB, Enjolras O, et al. Rapidly involuting congenital hemangioma: clinical and histopathologic features. Pediatr Dev Pathol. 2003;6:495-510.
- North PE, Waner M, James CA, et al. Congenital nonprogressive hemangioma: a distinct clinicopathologic entity unlike infantile hemangioma. Arch Dermatol. 2001;137:1607-1620.
- Maguiness S, Uihlein LC, Liang MG, et al. Rapidly involuting congenital hemangioma with fetal involution. Pediatr Dermatol. 2015;32:321-326.
- Keating LJ, Soares GM, Muratore CS. Rapidly involuting congenital hemangioma. Med Health R I. 2012;95:149-152.
- Schafer F, Tapia M, Pinto C. Rapidly involuting congenital haemangioma. Arch Dis Child Fetal Neonatal Ed. 2014;99:F422.
- Boon LM, Enjolras O, Mulliken JB. Congenital hemangioma: evidence of accelerated involution. J Pediatr. 1996;128:329-335.
- Liang MG, Frieden IJ. Infantile and congenital hemangiomas. Semin Pediatr Surg. 2014;23:162-167.
- Enjolras O, Mulliken JB, Boon LM, et al. Noninvoluting congenital hemangioma: a rare cutaneous vascular anomaly. Plast Reconstr Surg. 2001;107:1647-1654.
- Nasseri E, Piram M, McCuaig CC, et al. Partially involuting congenital hemangiomas: a report of 8 cases and review of the literature. J Am Acad Dermatol. 2014;70:75-79.
- Wassef M, Blei F, Adams D, et al. Vascular anomalies classification: recommendations from the International Society for the Study of Vascular Anomalies. Pediatrics. 2015;136:E203-E214.
- Boull C, Maguiness SM. Congenital hemangiomas. Semin Cutan Med Surg. 2016;35:124-127.
- Drolet BA, Frommelt PC, Chamlin SL, et al. Initiation and use of propranolol for infantile hemangioma: report of a consensus conference. Pediatrics. 2013;131:128-140.
- Baselga E, Cordisco MR, Garzon M, et al. Rapidly involuting congenital haemangioma associated with transient thrombocytopenia and coagulopathy: a case series. Br J Dermatol. 2008;158:1363-1370.
- Kanada KN, Merin MR, Munden A, et al. A prospective study of cutaneous findings in newborns in the United States: correlation with race, ethnicity, and gestational status using updated classification and nomenclature. J Pediatr. 2012;161:240-245.
- Munden A, Butschek R, Tom WL, et al. Prospective study of infantile haemangiomas: incidence, clinical characteristics and association with placental anomalies. Br J Dermatol. 2014;170:907-913.
- Léauté-Labrèze C, Harper JI, Hoeger PH. Infantile haemangioma. Lancet. 2017;390:85-94.
- Chang LC, Haggstrom AN, Drolet BA, et al. Growth characteristics of infantile hemangiomas: implications for management. Pediatrics. 2008;122:360-367.
- Hidano A, Nakajima S. Earliest features of the strawberry mark in the newborn. Br J Dermatol. 1972;87:138-144.
- Martinez-Perez D, Fein NA, Boon LM, et al. Not all hemangiomas look like strawberries: uncommon presentations of the most common tumor of infancy. Pediatr Dermatol. 1995;12:1-6.
- Payne MM, Moyer F, Marcks KM, et al. The precursor to the hemangioma. Plast Reconstr Surg. 1966;38:64-67.
- Bowers RE, Graham EA, Tomlinson KM. The natural history of the strawberry nevus. Arch Dermatol. 1960;82:667-680.
- Couto RA, Maclellan RA, Zurakowski D, et al. Infantile hemangioma: clinical assessment of the involuting phase and implications for management. Plast Reconstr Surg. 2012;130:619-624.
- Drolet BA, Esterly NB, Frieden IJ. Hemangiomas in children. N Engl J Med. 1999;341:173-181.
- Chiller KG, Passaro D, Frieden IJ. Hemangiomas of infancy: clinical characteristics, morphologic subtypes, and their relationship to race, ethnicity, and sex. Arch Dermatol. 2002;138:1567-1576.
- North PE, Waner M, Mizeracki A, et al. GLUT1: a newly discovered immunohistochemical marker for juvenile hemangiomas. Hum Pathol. 2000;31:11-22.
- Gruman A, Liang MG, Mulliken JB, et al. Kaposiform hemangioendothelioma without Kasabach-Merritt phenomenon. J Am Acad Dermatol. 2005;52:616-622.
- Croteau SE, Liang MG, Kozakewich HP, et al. Kaposiform hemangioendothelioma: atypical features and risks of Kasabach- Merritt phenomenon in 107 referrals. J Pediatr. 2013;162:142-147.
- Zukerberg LR, Nickoloff BJ, Weiss SW. Kaposiform hemangioendothelioma of infancy and childhood. an aggressive neoplasm associated with Kasabach-Merritt syndrome and lymphangiomatosis. Am J Surg Pathol. 1993;17:321-328.
- Mac-Moune Lai F, To KF, Choi PC, et al. Kaposiform hemangioendothelioma: five patients with cutaneous lesion and long follow-up. Mod Pathol. 2001;14:1087-1092.
- O’Rafferty C, O’Regan GM, Irvine AD, et al. Recent advances in the pathobiology and management of Kasabach-Merritt phenomenon. Br J Haematol. 2015;171:38-51.
- Le Huu AR, Jokinen CH, Rubin BP, et al. Expression of prox1, lymphatic endothelial nuclear transcription factor, in kaposiform hemangioendothelioma and tufted angioma. Am J Surg Pathol. 2010;34:1563-1573.
- Debelenko LV, Perez-Atayde AR, Mulliken JB, et al. D2-40 immuno-histochemical analysis of pediatric vascular tumors reveals positivity in kaposiform hemangioendothelioma. Mod Pathol. 2005;18:1454-1460.
- Haisley-Royster C, Enjolras O, Frieden IJ, et al. Kasabach-Merritt phenomenon: a retrospective study of treatment with vincristine. J Pediatr Hematol Oncol. 2002;24:459-462.
- Wilmer A, Kaatz M, Bocker T, et al. Tufted angioma. Eur J Dermatol. 1999;9:51-53.
- Herron MD, Coffin CM, Vanderhooft SL. Tufted angiomas: variability of the clinical morphology. Pediatr Dermatol. 2002;19:394-401.
- North PE. Pediatric vascular tumors and malformations. Surg Pathol Clin. 2010,3:455-494.
- Chu CY, Hsiao CH, Chiu HC. Transformation between kaposiform hemangioendothelioma and tufted angioma. Dermatology. 2003;206:334-337.
- Osio A, Fraitag S, Hadj-Rabia S, et al. Clinical spectrum of tufted angiomas in childhood: a report of 13 cases and a review of the literature. Arch Dermatol. 2010;146:758-763.
- Johnson EF, Davis DM, Tollefson MM, et al. Vascular tumors in infants: case report and review of clinical, histopathologic, and immunohistochemical characteristics of infantile hemangioma, pyogenic granuloma, noninvoluting congenital hemangioma, tufted angioma, and kaposiform hemangioendothelioma. Am J Dermatopathol. 2018;40:231-239.
- Christison-Lagay ER, Fishman SJ. Vascular anomalies. Surg Clin North Am. 2006;86:393-425.
- Liu AS, Mulliken JB, Zurakowski D, et al. Extracranial arteriovenous malformations: natural progression and recurrence after treatment. Plast Reconstr Surg. 2010;125:1185-1194.
- Young AE, Mulliken JB. Arteriovenous malformations. In: Mulliken JB, Young AE, eds. Vascular Birthmarks: Haemangiomas and Malformations. WB Saunders; 1988:228-245.
- Duggan EM, Fishman SJ. Vascular anomalies. In: Holcomb GW III, Murphy JP, St Peter SD, eds. Holcomb and Ashcraft’s Pediatric Surgery. 7th edition. Elsevier; 2019:1147-1170.
- Enjolras O, Wassef M, Chapot R. Introduction: ISSVA Classification. In: Enjolras O, Wassef M, Chapot R, eds. Color Atlas of Vascular Tumors and Vascular Malformations. Cambridge University Press; 2007:3-11.
- Fadell MF, Jones BV, Adams DM. Prenatal diagnosis and postnatal follow-up of rapidly involuting congenital hemangioma (RICH). Pediatr Radiol. 2011;41:1057-1060.
- Feygin T, Khalek N, Moldenhauer JS. Fetal brain, head, and neck tumors: prenatal imaging and management. Prenat Diagn. 2020;40:1203-1219.
- Foley LS, Kulungowski AM. Vascular anomalies in pediatrics. Adv Pediatr. 2015;62:227-255.
- Bruder E, Alaggio R, Kozakewich HPW, et al. Vascular and perivascular lesions of skin and soft tissues in children and adolescents. Pediatr Dev Pathol. 2012;15:26-61.
- Berenguer B, Mulliken JB, Enjolras O, et al. Rapidly involuting congenital hemangioma: clinical and histopathologic features. Pediatr Dev Pathol. 2003;6:495-510.
- North PE, Waner M, James CA, et al. Congenital nonprogressive hemangioma: a distinct clinicopathologic entity unlike infantile hemangioma. Arch Dermatol. 2001;137:1607-1620.
- Maguiness S, Uihlein LC, Liang MG, et al. Rapidly involuting congenital hemangioma with fetal involution. Pediatr Dermatol. 2015;32:321-326.
- Keating LJ, Soares GM, Muratore CS. Rapidly involuting congenital hemangioma. Med Health R I. 2012;95:149-152.
- Schafer F, Tapia M, Pinto C. Rapidly involuting congenital haemangioma. Arch Dis Child Fetal Neonatal Ed. 2014;99:F422.
- Boon LM, Enjolras O, Mulliken JB. Congenital hemangioma: evidence of accelerated involution. J Pediatr. 1996;128:329-335.
- Liang MG, Frieden IJ. Infantile and congenital hemangiomas. Semin Pediatr Surg. 2014;23:162-167.
- Enjolras O, Mulliken JB, Boon LM, et al. Noninvoluting congenital hemangioma: a rare cutaneous vascular anomaly. Plast Reconstr Surg. 2001;107:1647-1654.
- Nasseri E, Piram M, McCuaig CC, et al. Partially involuting congenital hemangiomas: a report of 8 cases and review of the literature. J Am Acad Dermatol. 2014;70:75-79.
- Wassef M, Blei F, Adams D, et al. Vascular anomalies classification: recommendations from the International Society for the Study of Vascular Anomalies. Pediatrics. 2015;136:E203-E214.
- Boull C, Maguiness SM. Congenital hemangiomas. Semin Cutan Med Surg. 2016;35:124-127.
- Drolet BA, Frommelt PC, Chamlin SL, et al. Initiation and use of propranolol for infantile hemangioma: report of a consensus conference. Pediatrics. 2013;131:128-140.
- Baselga E, Cordisco MR, Garzon M, et al. Rapidly involuting congenital haemangioma associated with transient thrombocytopenia and coagulopathy: a case series. Br J Dermatol. 2008;158:1363-1370.
- Kanada KN, Merin MR, Munden A, et al. A prospective study of cutaneous findings in newborns in the United States: correlation with race, ethnicity, and gestational status using updated classification and nomenclature. J Pediatr. 2012;161:240-245.
- Munden A, Butschek R, Tom WL, et al. Prospective study of infantile haemangiomas: incidence, clinical characteristics and association with placental anomalies. Br J Dermatol. 2014;170:907-913.
- Léauté-Labrèze C, Harper JI, Hoeger PH. Infantile haemangioma. Lancet. 2017;390:85-94.
- Chang LC, Haggstrom AN, Drolet BA, et al. Growth characteristics of infantile hemangiomas: implications for management. Pediatrics. 2008;122:360-367.
- Hidano A, Nakajima S. Earliest features of the strawberry mark in the newborn. Br J Dermatol. 1972;87:138-144.
- Martinez-Perez D, Fein NA, Boon LM, et al. Not all hemangiomas look like strawberries: uncommon presentations of the most common tumor of infancy. Pediatr Dermatol. 1995;12:1-6.
- Payne MM, Moyer F, Marcks KM, et al. The precursor to the hemangioma. Plast Reconstr Surg. 1966;38:64-67.
- Bowers RE, Graham EA, Tomlinson KM. The natural history of the strawberry nevus. Arch Dermatol. 1960;82:667-680.
- Couto RA, Maclellan RA, Zurakowski D, et al. Infantile hemangioma: clinical assessment of the involuting phase and implications for management. Plast Reconstr Surg. 2012;130:619-624.
- Drolet BA, Esterly NB, Frieden IJ. Hemangiomas in children. N Engl J Med. 1999;341:173-181.
- Chiller KG, Passaro D, Frieden IJ. Hemangiomas of infancy: clinical characteristics, morphologic subtypes, and their relationship to race, ethnicity, and sex. Arch Dermatol. 2002;138:1567-1576.
- North PE, Waner M, Mizeracki A, et al. GLUT1: a newly discovered immunohistochemical marker for juvenile hemangiomas. Hum Pathol. 2000;31:11-22.
- Gruman A, Liang MG, Mulliken JB, et al. Kaposiform hemangioendothelioma without Kasabach-Merritt phenomenon. J Am Acad Dermatol. 2005;52:616-622.
- Croteau SE, Liang MG, Kozakewich HP, et al. Kaposiform hemangioendothelioma: atypical features and risks of Kasabach- Merritt phenomenon in 107 referrals. J Pediatr. 2013;162:142-147.
- Zukerberg LR, Nickoloff BJ, Weiss SW. Kaposiform hemangioendothelioma of infancy and childhood. an aggressive neoplasm associated with Kasabach-Merritt syndrome and lymphangiomatosis. Am J Surg Pathol. 1993;17:321-328.
- Mac-Moune Lai F, To KF, Choi PC, et al. Kaposiform hemangioendothelioma: five patients with cutaneous lesion and long follow-up. Mod Pathol. 2001;14:1087-1092.
- O’Rafferty C, O’Regan GM, Irvine AD, et al. Recent advances in the pathobiology and management of Kasabach-Merritt phenomenon. Br J Haematol. 2015;171:38-51.
- Le Huu AR, Jokinen CH, Rubin BP, et al. Expression of prox1, lymphatic endothelial nuclear transcription factor, in kaposiform hemangioendothelioma and tufted angioma. Am J Surg Pathol. 2010;34:1563-1573.
- Debelenko LV, Perez-Atayde AR, Mulliken JB, et al. D2-40 immuno-histochemical analysis of pediatric vascular tumors reveals positivity in kaposiform hemangioendothelioma. Mod Pathol. 2005;18:1454-1460.
- Haisley-Royster C, Enjolras O, Frieden IJ, et al. Kasabach-Merritt phenomenon: a retrospective study of treatment with vincristine. J Pediatr Hematol Oncol. 2002;24:459-462.
- Wilmer A, Kaatz M, Bocker T, et al. Tufted angioma. Eur J Dermatol. 1999;9:51-53.
- Herron MD, Coffin CM, Vanderhooft SL. Tufted angiomas: variability of the clinical morphology. Pediatr Dermatol. 2002;19:394-401.
- North PE. Pediatric vascular tumors and malformations. Surg Pathol Clin. 2010,3:455-494.
- Chu CY, Hsiao CH, Chiu HC. Transformation between kaposiform hemangioendothelioma and tufted angioma. Dermatology. 2003;206:334-337.
- Osio A, Fraitag S, Hadj-Rabia S, et al. Clinical spectrum of tufted angiomas in childhood: a report of 13 cases and a review of the literature. Arch Dermatol. 2010;146:758-763.
- Johnson EF, Davis DM, Tollefson MM, et al. Vascular tumors in infants: case report and review of clinical, histopathologic, and immunohistochemical characteristics of infantile hemangioma, pyogenic granuloma, noninvoluting congenital hemangioma, tufted angioma, and kaposiform hemangioendothelioma. Am J Dermatopathol. 2018;40:231-239.
- Christison-Lagay ER, Fishman SJ. Vascular anomalies. Surg Clin North Am. 2006;86:393-425.
- Liu AS, Mulliken JB, Zurakowski D, et al. Extracranial arteriovenous malformations: natural progression and recurrence after treatment. Plast Reconstr Surg. 2010;125:1185-1194.
- Young AE, Mulliken JB. Arteriovenous malformations. In: Mulliken JB, Young AE, eds. Vascular Birthmarks: Haemangiomas and Malformations. WB Saunders; 1988:228-245.
- Duggan EM, Fishman SJ. Vascular anomalies. In: Holcomb GW III, Murphy JP, St Peter SD, eds. Holcomb and Ashcraft’s Pediatric Surgery. 7th edition. Elsevier; 2019:1147-1170.
A newborn male was delivered via cesarean section at 38 weeks 5 days’ gestation with a large vascular mass on the posterior neck. The mass previously had been identified on a 23-week prenatal ultrasound. Physical examination by dermatology at birth revealed a well-defined violaceous mass measuring 6×5 cm with prominent radiating veins, coarse telangiectases, and a pale rim. Magnetic resonance imaging demonstrated a well-circumscribed mass with avid arterial phase enhancement. The patient experienced transient thrombocytopenia that resolved following administration of methylprednisolone. No evidence of rapid involution was noted after 3 months of observation.
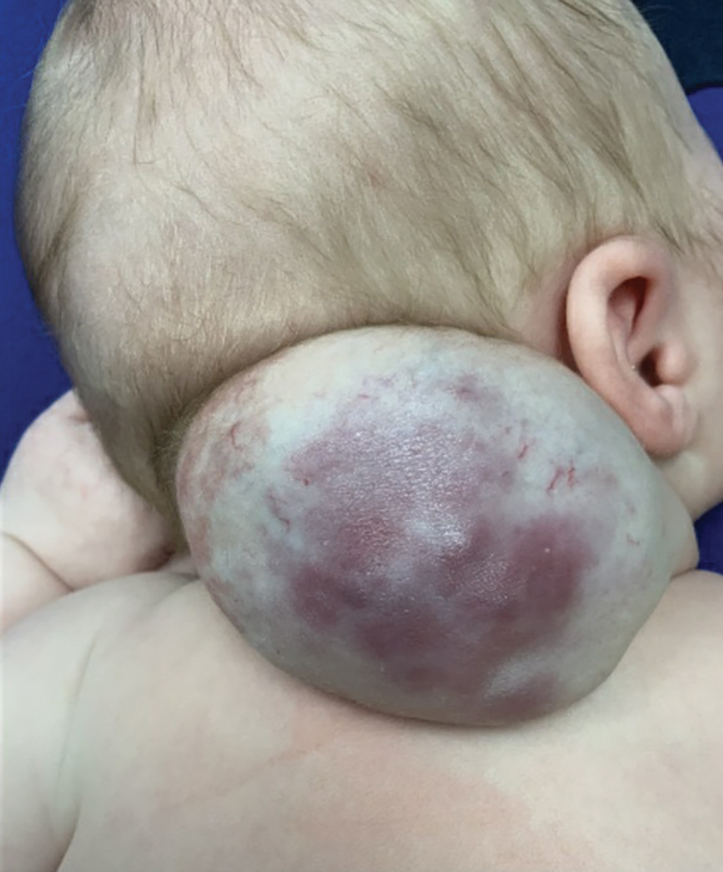
Psoriatic Arthritis Symptoms Relieved with TYK2 Inhibitor in Phase 2 Trial
TOPLINE:
The tyrosine kinase 2 (TYK2) inhibitor TAK-279 demonstrated superiority to placebo in patients with active psoriatic arthritis (PsA), according to phase 2 trial results.
METHODOLOGY:
- Eligible patients were over 18 years old, had PsA for over 6 months, met the classification criteria for PsA, and had at least three swollen and tender joints despite prior nonsteroidal anti-inflammatory drug, disease-modifying antirheumatic drug, or biologic treatment.
- A total of 290 patients were randomized 1:1:1:1 to receive placebo, oral TAK-279 5 mg, 15 mg, or 30 mg once daily.
- The primary endpoint was a 20% improvement in the American College of Rheumatology response criteria (ACR20) at 12 weeks.
TAKEAWAY:
- More than half of patients assigned to TAK-279 15 mg (53.3%) and TAK-279 30 mg (54.2%) achieved ACR20 at 12 weeks, compared with 29.2% of those assigned to placebo.
- Psoriasis Area and Severity Index 75 response rates were also higher in patients assigned to TAK-279 30 mg (45.7%) or 15 mg (28.3%) than those in placebo (15.4%).
- Treatment-emergent adverse events (TEAEs) of any kind were numerically higher in the 30-mg group, though serious TEAEs were similar across all treatment arms.
- The most frequent adverse events were nasal pharyngitis, upper respiratory tract infections, headache, and rash, with rash being most common in the TAK-279 30-mg group.
IN PRACTICE:
“There are few targeted oral therapies for active PSA available currently,” said lead author Alan Kivitz, MD, Altoona Center for Clinical Research, Duncansville, Pennsylvania, “and [TAK-279], which was well tolerated and demonstrated superior efficacy versus placebo, may be a promising targeted oral therapy for patients with PsA.”
SOURCE:
Dr. Kivitz presented the study findings at the European Alliance of Associations for Rheumatology (EULAR) 2024 Annual Meeting, held in Vienna.
LIMITATIONS:
The study was a phase 2 trial, and larger studies in active PsA are needed (and currently being planned).
DISCLOSURES:
The phase 2 trial was funded by Nimbus and Takeda. Dr. Kivitz has received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from AbbVie, Amgen, Eli Lilly, GlaxoSmithKline, Pfizer, and UCB. He has stock or stock options in Pfizer, Amgen, GlaxoSmithKline, Gilead, Novartis, and Pfizer and has received consultant fees from Fresenius Kabi, Genzyme, Gilead, Grunenthal, GlaxoSmithKline, Horizon, Janssen, Pfizer, Selecta, SynAct Pharma, and Takeda. He has been part of a board or advisory board for ChemoCentryx, Horizon, Janssen, Novartis, Princeton Biopartners, and UCB. Other authors also disclosed many relationships with pharmaceutical companies.
A version of this article first appeared on Medscape.com.
TOPLINE:
The tyrosine kinase 2 (TYK2) inhibitor TAK-279 demonstrated superiority to placebo in patients with active psoriatic arthritis (PsA), according to phase 2 trial results.
METHODOLOGY:
- Eligible patients were over 18 years old, had PsA for over 6 months, met the classification criteria for PsA, and had at least three swollen and tender joints despite prior nonsteroidal anti-inflammatory drug, disease-modifying antirheumatic drug, or biologic treatment.
- A total of 290 patients were randomized 1:1:1:1 to receive placebo, oral TAK-279 5 mg, 15 mg, or 30 mg once daily.
- The primary endpoint was a 20% improvement in the American College of Rheumatology response criteria (ACR20) at 12 weeks.
TAKEAWAY:
- More than half of patients assigned to TAK-279 15 mg (53.3%) and TAK-279 30 mg (54.2%) achieved ACR20 at 12 weeks, compared with 29.2% of those assigned to placebo.
- Psoriasis Area and Severity Index 75 response rates were also higher in patients assigned to TAK-279 30 mg (45.7%) or 15 mg (28.3%) than those in placebo (15.4%).
- Treatment-emergent adverse events (TEAEs) of any kind were numerically higher in the 30-mg group, though serious TEAEs were similar across all treatment arms.
- The most frequent adverse events were nasal pharyngitis, upper respiratory tract infections, headache, and rash, with rash being most common in the TAK-279 30-mg group.
IN PRACTICE:
“There are few targeted oral therapies for active PSA available currently,” said lead author Alan Kivitz, MD, Altoona Center for Clinical Research, Duncansville, Pennsylvania, “and [TAK-279], which was well tolerated and demonstrated superior efficacy versus placebo, may be a promising targeted oral therapy for patients with PsA.”
SOURCE:
Dr. Kivitz presented the study findings at the European Alliance of Associations for Rheumatology (EULAR) 2024 Annual Meeting, held in Vienna.
LIMITATIONS:
The study was a phase 2 trial, and larger studies in active PsA are needed (and currently being planned).
DISCLOSURES:
The phase 2 trial was funded by Nimbus and Takeda. Dr. Kivitz has received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from AbbVie, Amgen, Eli Lilly, GlaxoSmithKline, Pfizer, and UCB. He has stock or stock options in Pfizer, Amgen, GlaxoSmithKline, Gilead, Novartis, and Pfizer and has received consultant fees from Fresenius Kabi, Genzyme, Gilead, Grunenthal, GlaxoSmithKline, Horizon, Janssen, Pfizer, Selecta, SynAct Pharma, and Takeda. He has been part of a board or advisory board for ChemoCentryx, Horizon, Janssen, Novartis, Princeton Biopartners, and UCB. Other authors also disclosed many relationships with pharmaceutical companies.
A version of this article first appeared on Medscape.com.
TOPLINE:
The tyrosine kinase 2 (TYK2) inhibitor TAK-279 demonstrated superiority to placebo in patients with active psoriatic arthritis (PsA), according to phase 2 trial results.
METHODOLOGY:
- Eligible patients were over 18 years old, had PsA for over 6 months, met the classification criteria for PsA, and had at least three swollen and tender joints despite prior nonsteroidal anti-inflammatory drug, disease-modifying antirheumatic drug, or biologic treatment.
- A total of 290 patients were randomized 1:1:1:1 to receive placebo, oral TAK-279 5 mg, 15 mg, or 30 mg once daily.
- The primary endpoint was a 20% improvement in the American College of Rheumatology response criteria (ACR20) at 12 weeks.
TAKEAWAY:
- More than half of patients assigned to TAK-279 15 mg (53.3%) and TAK-279 30 mg (54.2%) achieved ACR20 at 12 weeks, compared with 29.2% of those assigned to placebo.
- Psoriasis Area and Severity Index 75 response rates were also higher in patients assigned to TAK-279 30 mg (45.7%) or 15 mg (28.3%) than those in placebo (15.4%).
- Treatment-emergent adverse events (TEAEs) of any kind were numerically higher in the 30-mg group, though serious TEAEs were similar across all treatment arms.
- The most frequent adverse events were nasal pharyngitis, upper respiratory tract infections, headache, and rash, with rash being most common in the TAK-279 30-mg group.
IN PRACTICE:
“There are few targeted oral therapies for active PSA available currently,” said lead author Alan Kivitz, MD, Altoona Center for Clinical Research, Duncansville, Pennsylvania, “and [TAK-279], which was well tolerated and demonstrated superior efficacy versus placebo, may be a promising targeted oral therapy for patients with PsA.”
SOURCE:
Dr. Kivitz presented the study findings at the European Alliance of Associations for Rheumatology (EULAR) 2024 Annual Meeting, held in Vienna.
LIMITATIONS:
The study was a phase 2 trial, and larger studies in active PsA are needed (and currently being planned).
DISCLOSURES:
The phase 2 trial was funded by Nimbus and Takeda. Dr. Kivitz has received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from AbbVie, Amgen, Eli Lilly, GlaxoSmithKline, Pfizer, and UCB. He has stock or stock options in Pfizer, Amgen, GlaxoSmithKline, Gilead, Novartis, and Pfizer and has received consultant fees from Fresenius Kabi, Genzyme, Gilead, Grunenthal, GlaxoSmithKline, Horizon, Janssen, Pfizer, Selecta, SynAct Pharma, and Takeda. He has been part of a board or advisory board for ChemoCentryx, Horizon, Janssen, Novartis, Princeton Biopartners, and UCB. Other authors also disclosed many relationships with pharmaceutical companies.
A version of this article first appeared on Medscape.com.
Psoriatic Arthritis Drug Candidate Sonelokimab Yields Significant Improvements in Phase 2 Trial
TOPLINE:
Treatment of patients with active psoriatic arthritis with sonelokimab — an interleukin (IL)-17A- and IL-17F-inhibiting nanobody — led to a higher percentage of patients with 50% or greater improvement in American College of Rheumatology response criteria (ACR50) compared with the placebo in a phase 2 trial.
METHODOLOGY:
- Sonelokimab is a 40-kDa nanobody that binds to IL-17A, IL-17F, and albumin.
- Eligible patients were at least 18 years old with active PsA (at least three swollen and three tender joints) and had a psoriasis diagnosis.
- A total of 207 patients were randomized 1:1:1:1 to every 4 weeks receive placebo, sonelokimab 60 mg with no induction (NI) period, sonelokimab 60 mg with induction, and sonelokimab 120 mg with induction.
- Induction was once every 2 weeks up to week 8 of the trial.
- The primary endpoint was meeting ACR20 response criteria at 12 weeks.
TAKEAWAY:
- About 46% of patients in the sonelokimab 120-mg and 60-mg groups achieved ACR50, compared with 36.6% in the sonelokimab 60-mg NI group and 20% of those assigned to placebo.
- ACR20 and 90% or greater reduction in Psoriasis Area and Severity Index score response rates were higher in all three sonelokimab groups than in the placebo group.
- There were no unexpected safety findings during the trial, and no cases of inflammatory bowel disease or major cardiovascular events.
- There were two cases of oral candidiasis, which did not lead to study discontinuation.
IN PRACTICE:
These data “support further exploration in phase 3 trials of sonelokimab to evaluate its potential for the treatment of PsA,” the authors noted in the presentation.
SOURCE:
Iain B. McInnes, MD, PhD, of the University of Glasgow, Glasgow, Scotland, presented these phase 2 trial results at the European Alliance of Associations for Rheumatology (EULAR) 2024 Annual Congress, held in Vienna.
LIMITATIONS:
The results are from a phase 2 trial, and more research is needed.
DISCLOSURES:
MoonLake Immunotherapeutics funded the research. Dr. McInnes disclosed relationships with AbbVie, Amgen, AstraZeneca, Bristol Myers Squibb, Causeway Therapeutics, Cabaletta Bio, Compugen, Evelo, Gilead, GlaxoSmithKline, Janssen, Eli Lilly, Novartis, MoonLake Immunotherapeutics, Pfizer, Sanofi Regeneron, and UCB. Other authors also disclosed many relationships with pharmaceutical companies.
A version of this article first appeared on Medscape.com.
TOPLINE:
Treatment of patients with active psoriatic arthritis with sonelokimab — an interleukin (IL)-17A- and IL-17F-inhibiting nanobody — led to a higher percentage of patients with 50% or greater improvement in American College of Rheumatology response criteria (ACR50) compared with the placebo in a phase 2 trial.
METHODOLOGY:
- Sonelokimab is a 40-kDa nanobody that binds to IL-17A, IL-17F, and albumin.
- Eligible patients were at least 18 years old with active PsA (at least three swollen and three tender joints) and had a psoriasis diagnosis.
- A total of 207 patients were randomized 1:1:1:1 to every 4 weeks receive placebo, sonelokimab 60 mg with no induction (NI) period, sonelokimab 60 mg with induction, and sonelokimab 120 mg with induction.
- Induction was once every 2 weeks up to week 8 of the trial.
- The primary endpoint was meeting ACR20 response criteria at 12 weeks.
TAKEAWAY:
- About 46% of patients in the sonelokimab 120-mg and 60-mg groups achieved ACR50, compared with 36.6% in the sonelokimab 60-mg NI group and 20% of those assigned to placebo.
- ACR20 and 90% or greater reduction in Psoriasis Area and Severity Index score response rates were higher in all three sonelokimab groups than in the placebo group.
- There were no unexpected safety findings during the trial, and no cases of inflammatory bowel disease or major cardiovascular events.
- There were two cases of oral candidiasis, which did not lead to study discontinuation.
IN PRACTICE:
These data “support further exploration in phase 3 trials of sonelokimab to evaluate its potential for the treatment of PsA,” the authors noted in the presentation.
SOURCE:
Iain B. McInnes, MD, PhD, of the University of Glasgow, Glasgow, Scotland, presented these phase 2 trial results at the European Alliance of Associations for Rheumatology (EULAR) 2024 Annual Congress, held in Vienna.
LIMITATIONS:
The results are from a phase 2 trial, and more research is needed.
DISCLOSURES:
MoonLake Immunotherapeutics funded the research. Dr. McInnes disclosed relationships with AbbVie, Amgen, AstraZeneca, Bristol Myers Squibb, Causeway Therapeutics, Cabaletta Bio, Compugen, Evelo, Gilead, GlaxoSmithKline, Janssen, Eli Lilly, Novartis, MoonLake Immunotherapeutics, Pfizer, Sanofi Regeneron, and UCB. Other authors also disclosed many relationships with pharmaceutical companies.
A version of this article first appeared on Medscape.com.
TOPLINE:
Treatment of patients with active psoriatic arthritis with sonelokimab — an interleukin (IL)-17A- and IL-17F-inhibiting nanobody — led to a higher percentage of patients with 50% or greater improvement in American College of Rheumatology response criteria (ACR50) compared with the placebo in a phase 2 trial.
METHODOLOGY:
- Sonelokimab is a 40-kDa nanobody that binds to IL-17A, IL-17F, and albumin.
- Eligible patients were at least 18 years old with active PsA (at least three swollen and three tender joints) and had a psoriasis diagnosis.
- A total of 207 patients were randomized 1:1:1:1 to every 4 weeks receive placebo, sonelokimab 60 mg with no induction (NI) period, sonelokimab 60 mg with induction, and sonelokimab 120 mg with induction.
- Induction was once every 2 weeks up to week 8 of the trial.
- The primary endpoint was meeting ACR20 response criteria at 12 weeks.
TAKEAWAY:
- About 46% of patients in the sonelokimab 120-mg and 60-mg groups achieved ACR50, compared with 36.6% in the sonelokimab 60-mg NI group and 20% of those assigned to placebo.
- ACR20 and 90% or greater reduction in Psoriasis Area and Severity Index score response rates were higher in all three sonelokimab groups than in the placebo group.
- There were no unexpected safety findings during the trial, and no cases of inflammatory bowel disease or major cardiovascular events.
- There were two cases of oral candidiasis, which did not lead to study discontinuation.
IN PRACTICE:
These data “support further exploration in phase 3 trials of sonelokimab to evaluate its potential for the treatment of PsA,” the authors noted in the presentation.
SOURCE:
Iain B. McInnes, MD, PhD, of the University of Glasgow, Glasgow, Scotland, presented these phase 2 trial results at the European Alliance of Associations for Rheumatology (EULAR) 2024 Annual Congress, held in Vienna.
LIMITATIONS:
The results are from a phase 2 trial, and more research is needed.
DISCLOSURES:
MoonLake Immunotherapeutics funded the research. Dr. McInnes disclosed relationships with AbbVie, Amgen, AstraZeneca, Bristol Myers Squibb, Causeway Therapeutics, Cabaletta Bio, Compugen, Evelo, Gilead, GlaxoSmithKline, Janssen, Eli Lilly, Novartis, MoonLake Immunotherapeutics, Pfizer, Sanofi Regeneron, and UCB. Other authors also disclosed many relationships with pharmaceutical companies.
A version of this article first appeared on Medscape.com.
FDA Proposes that Interchangeability Status for Biosimilars Doesn’t Need Switching Studies
The Food and Drug Administration (FDA) has issued new draft guidance that does not require additional switching studies for biosimilars seeking interchangeability. These studies were previously recommended to demonstrate that switching between the biosimilar and its reference product showed no greater risk than using the reference product alone.
“The recommendations in today’s draft guidance, when finalized, will provide clarity and transparency about the FDA’s thinking and align the review and approval process with existing and emerging science,” said Sarah Yim, MD, director of the FDA’s Office of Therapeutic Biologics and Biosimilars in a statement on June 20. “We have gained valuable experience reviewing both biosimilar and interchangeable biosimilar medications over the past 10 years. Both biosimilars and interchangeable biosimilars meet the same high standard of biosimilarity for FDA approval and both are as safe and effective as the reference product.”
An interchangeable status allows a biosimilar product to be swapped with the reference product without involvement from the prescribing provider, depending on state law.
While switching studies were not required under previous FDA guidance, the 2019 document did state that the agency “expects that applications generally will include data from a switching study or studies in one or more appropriate conditions of use.”
However, of the 13 biosimilars that received interchangeability status, 9 did not include switching study data.
“Experience has shown that, for the products approved as biosimilars to date, the risk in terms of safety or diminished efficacy is insignificant following single or multiple switches between a reference product and a biosimilar product,” the FDA stated. The agency’s investigators also conducted a systematic review of switching studies, which found no differences in risk for death, serious adverse events, and treatment discontinuations in participants switched between biosimilars and reference products and those that remained on reference products.
“Additionally, today’s analytical tools can accurately evaluate the structure and effects [of] biologic products, both in the lab (in vitro) and in living organisms (in vivo) with more precision and sensitivity than switching studies,” the agency noted.
The FDA is now calling for commentary on these draft recommendations to be submitted by Aug. 20, 2024.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration (FDA) has issued new draft guidance that does not require additional switching studies for biosimilars seeking interchangeability. These studies were previously recommended to demonstrate that switching between the biosimilar and its reference product showed no greater risk than using the reference product alone.
“The recommendations in today’s draft guidance, when finalized, will provide clarity and transparency about the FDA’s thinking and align the review and approval process with existing and emerging science,” said Sarah Yim, MD, director of the FDA’s Office of Therapeutic Biologics and Biosimilars in a statement on June 20. “We have gained valuable experience reviewing both biosimilar and interchangeable biosimilar medications over the past 10 years. Both biosimilars and interchangeable biosimilars meet the same high standard of biosimilarity for FDA approval and both are as safe and effective as the reference product.”
An interchangeable status allows a biosimilar product to be swapped with the reference product without involvement from the prescribing provider, depending on state law.
While switching studies were not required under previous FDA guidance, the 2019 document did state that the agency “expects that applications generally will include data from a switching study or studies in one or more appropriate conditions of use.”
However, of the 13 biosimilars that received interchangeability status, 9 did not include switching study data.
“Experience has shown that, for the products approved as biosimilars to date, the risk in terms of safety or diminished efficacy is insignificant following single or multiple switches between a reference product and a biosimilar product,” the FDA stated. The agency’s investigators also conducted a systematic review of switching studies, which found no differences in risk for death, serious adverse events, and treatment discontinuations in participants switched between biosimilars and reference products and those that remained on reference products.
“Additionally, today’s analytical tools can accurately evaluate the structure and effects [of] biologic products, both in the lab (in vitro) and in living organisms (in vivo) with more precision and sensitivity than switching studies,” the agency noted.
The FDA is now calling for commentary on these draft recommendations to be submitted by Aug. 20, 2024.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration (FDA) has issued new draft guidance that does not require additional switching studies for biosimilars seeking interchangeability. These studies were previously recommended to demonstrate that switching between the biosimilar and its reference product showed no greater risk than using the reference product alone.
“The recommendations in today’s draft guidance, when finalized, will provide clarity and transparency about the FDA’s thinking and align the review and approval process with existing and emerging science,” said Sarah Yim, MD, director of the FDA’s Office of Therapeutic Biologics and Biosimilars in a statement on June 20. “We have gained valuable experience reviewing both biosimilar and interchangeable biosimilar medications over the past 10 years. Both biosimilars and interchangeable biosimilars meet the same high standard of biosimilarity for FDA approval and both are as safe and effective as the reference product.”
An interchangeable status allows a biosimilar product to be swapped with the reference product without involvement from the prescribing provider, depending on state law.
While switching studies were not required under previous FDA guidance, the 2019 document did state that the agency “expects that applications generally will include data from a switching study or studies in one or more appropriate conditions of use.”
However, of the 13 biosimilars that received interchangeability status, 9 did not include switching study data.
“Experience has shown that, for the products approved as biosimilars to date, the risk in terms of safety or diminished efficacy is insignificant following single or multiple switches between a reference product and a biosimilar product,” the FDA stated. The agency’s investigators also conducted a systematic review of switching studies, which found no differences in risk for death, serious adverse events, and treatment discontinuations in participants switched between biosimilars and reference products and those that remained on reference products.
“Additionally, today’s analytical tools can accurately evaluate the structure and effects [of] biologic products, both in the lab (in vitro) and in living organisms (in vivo) with more precision and sensitivity than switching studies,” the agency noted.
The FDA is now calling for commentary on these draft recommendations to be submitted by Aug. 20, 2024.
A version of this article first appeared on Medscape.com.
Commentary: Topical Treatments for AD and Possible Lifestyle Adjustments, July 2024

In this real-life study, Patruno and colleagues found that dupilumab worked well but more slowly in patients with a higher body mass index (BMI). On the basis of these findings, if patients are not in a hurry, the standard dose of dupilumab should eventually work, regardless of BMI. If patients are in a hurry to see improvement, perhaps dose escalation could be considered for patients with a high BMI, or perhaps topical triamcinolone could be used to speed time-to–initial resolution in the high-BMI population.
In the very well-done study by Silverberg and colleagues, tapinarof was effective, well tolerated, and generally safe for atopic dermatitis in adults and children. Great! Topical tapinarof should soon be another good option for our patients with atopic dermatitis. How valuable will it be? We already have topical corticosteroids that are very effective for atopic dermatitis, and we have multiple other nonsteroidal topical agents, including topical calcineurin inhibitors and topical ruxolitinib.
Perhaps the biggest limitation of all these treatments is poor adherence to topical treatment. I'm not sure how effective even highly effective nonsteroidal topicals will be for patients who did not respond to topical steroids when the primary reason for topical steroid failure is poor treatment adherence. I'd love to see the development of a once-a-week or once-a-month topical therapy that would address the poor-adherence hurdle.
Abrocitinib is an effective treatment for improving atopic dermatitis. Although atopic dermatitis is a chronic condition requiring long-term management, we'd like to minimize exposure to the drug to avoid side effects. Thyssen and colleagues described the effectiveness of two maintenance treatment regimens: continuing 200 mg/d or reducing the dose to 100 mg/d. Both regimens prevented flares more than did placebo. This study also provided information on safety of the maintenance regimens. Rates of herpetic infections were low across all the groups, but unlike the two treatment groups, there were no cases of herpes simplex infection in the patients in the placebo arm.
In this real-life study, Patruno and colleagues found that dupilumab worked well but more slowly in patients with a higher body mass index (BMI). On the basis of these findings, if patients are not in a hurry, the standard dose of dupilumab should eventually work, regardless of BMI. If patients are in a hurry to see improvement, perhaps dose escalation could be considered for patients with a high BMI, or perhaps topical triamcinolone could be used to speed time-to–initial resolution in the high-BMI population.
In the very well-done study by Silverberg and colleagues, tapinarof was effective, well tolerated, and generally safe for atopic dermatitis in adults and children. Great! Topical tapinarof should soon be another good option for our patients with atopic dermatitis. How valuable will it be? We already have topical corticosteroids that are very effective for atopic dermatitis, and we have multiple other nonsteroidal topical agents, including topical calcineurin inhibitors and topical ruxolitinib.
Perhaps the biggest limitation of all these treatments is poor adherence to topical treatment. I'm not sure how effective even highly effective nonsteroidal topicals will be for patients who did not respond to topical steroids when the primary reason for topical steroid failure is poor treatment adherence. I'd love to see the development of a once-a-week or once-a-month topical therapy that would address the poor-adherence hurdle.
Abrocitinib is an effective treatment for improving atopic dermatitis. Although atopic dermatitis is a chronic condition requiring long-term management, we'd like to minimize exposure to the drug to avoid side effects. Thyssen and colleagues described the effectiveness of two maintenance treatment regimens: continuing 200 mg/d or reducing the dose to 100 mg/d. Both regimens prevented flares more than did placebo. This study also provided information on safety of the maintenance regimens. Rates of herpetic infections were low across all the groups, but unlike the two treatment groups, there were no cases of herpes simplex infection in the patients in the placebo arm.
In this real-life study, Patruno and colleagues found that dupilumab worked well but more slowly in patients with a higher body mass index (BMI). On the basis of these findings, if patients are not in a hurry, the standard dose of dupilumab should eventually work, regardless of BMI. If patients are in a hurry to see improvement, perhaps dose escalation could be considered for patients with a high BMI, or perhaps topical triamcinolone could be used to speed time-to–initial resolution in the high-BMI population.
In the very well-done study by Silverberg and colleagues, tapinarof was effective, well tolerated, and generally safe for atopic dermatitis in adults and children. Great! Topical tapinarof should soon be another good option for our patients with atopic dermatitis. How valuable will it be? We already have topical corticosteroids that are very effective for atopic dermatitis, and we have multiple other nonsteroidal topical agents, including topical calcineurin inhibitors and topical ruxolitinib.
Perhaps the biggest limitation of all these treatments is poor adherence to topical treatment. I'm not sure how effective even highly effective nonsteroidal topicals will be for patients who did not respond to topical steroids when the primary reason for topical steroid failure is poor treatment adherence. I'd love to see the development of a once-a-week or once-a-month topical therapy that would address the poor-adherence hurdle.
Abrocitinib is an effective treatment for improving atopic dermatitis. Although atopic dermatitis is a chronic condition requiring long-term management, we'd like to minimize exposure to the drug to avoid side effects. Thyssen and colleagues described the effectiveness of two maintenance treatment regimens: continuing 200 mg/d or reducing the dose to 100 mg/d. Both regimens prevented flares more than did placebo. This study also provided information on safety of the maintenance regimens. Rates of herpetic infections were low across all the groups, but unlike the two treatment groups, there were no cases of herpes simplex infection in the patients in the placebo arm.
Dengue Surge in US Cases This Year
Federal health officials with the US Centers for Disease Control and Prevention (CDC) have issued an alert, warning health professionals and the public about an increased risk for dengue virus infections in the United States.
The global incidence of dengue in 2024 is the highest on record, reported the agency.
In the United States, Puerto Rico has declared a public health emergency, with 1498 dengue cases reported so far and a “higher-than-expected” number of dengue cases having been identified among US travelers in the first half of this year at 745 cases, according to the alert.
The CDC reports 197 dengue cases in Florida, 134 in New York, 50 in Massachusetts, 40 in California, 14 in Colorado, nine in Arizona, and eight in the District of Columbia, among others.
Transmitted by infected Aedes genus mosquitoes, dengue is the most common arboviral disease globally and is a nationally notifiable disease in the United States.
The six US territories and freely associated states with frequent or continuous dengue transmission are Puerto Rico, American Samoa, the US Virgin Islands, the Federated States of Micronesia, the Republic of the Marshall Islands, and the Republic of Palau.
Monitoring for Dengue
With rising global and domestic cases of dengue, the CDC urges healthcare providers to monitor for dengue:
- Maintain a high index of suspicion in patients with fever who have been in areas with frequent or continuous dengue transmission within 14 days before illness onset.
- Order diagnostic tests for acute dengue infection such as reverse transcription polymerase chain reaction and immunoglobulin M (IgM) antibody tests or nonstructural protein 1 antigen tests and IgM antibody tests.
- Ensure timely reporting of dengue cases to public health authorities.
- Promote mosquito bite prevention measures among people living in or visiting areas with frequent or continuous dengue transmission.
Roughly one in four dengue virus infections are symptomatic and can be mild or severe. Symptoms begin after an incubation period of about 5-7 days.
Symptoms include fever accompanied by nonspecific signs and symptoms such as nausea, vomiting, rash, muscle aches, joint pain, bone pain, pain behind the eyes, headache, or low white blood cell counts.
Disease Progression
Warning signs that may predict progression to severe disease include abdominal pain or tenderness, persistent vomiting, clinical fluid accumulation, mucosal bleeding, lethargy or restlessness, and progressive increase in hematocrit or liver enlargement.
One in 20 people with symptomatic dengue will develop severe disease, with bleeding, shock, or respiratory distress caused by plasma leakage or end-organ impairment.
Infants aged a year or younger, pregnant people, adults aged 65 years or older, people with certain medical conditions, and those with previous dengue infections are at increased risk for severe dengue.
“Healthcare providers should be prepared to recognize, diagnose, manage, and report dengue cases to health authorities; public health partners should investigate cases and disseminate clear prevention messages to the public,” the alert stated.
The CDC is actively implementing several strategies to address the increase in cases of dengue in the United States. In early April, the agency launched a program-led emergency response and is providing monthly situational updates on dengue to partners, stakeholders, and jurisdictions.
The CDC is also expanding laboratory capacity to improve laboratory testing approaches; collaborating with state, tribal, local, and territorial health departments to strengthen dengue surveillance and recommend prevention strategies; and working to educate the public on dengue prevention.
A version of this article first appeared on Medscape.com.
Federal health officials with the US Centers for Disease Control and Prevention (CDC) have issued an alert, warning health professionals and the public about an increased risk for dengue virus infections in the United States.
The global incidence of dengue in 2024 is the highest on record, reported the agency.
In the United States, Puerto Rico has declared a public health emergency, with 1498 dengue cases reported so far and a “higher-than-expected” number of dengue cases having been identified among US travelers in the first half of this year at 745 cases, according to the alert.
The CDC reports 197 dengue cases in Florida, 134 in New York, 50 in Massachusetts, 40 in California, 14 in Colorado, nine in Arizona, and eight in the District of Columbia, among others.
Transmitted by infected Aedes genus mosquitoes, dengue is the most common arboviral disease globally and is a nationally notifiable disease in the United States.
The six US territories and freely associated states with frequent or continuous dengue transmission are Puerto Rico, American Samoa, the US Virgin Islands, the Federated States of Micronesia, the Republic of the Marshall Islands, and the Republic of Palau.
Monitoring for Dengue
With rising global and domestic cases of dengue, the CDC urges healthcare providers to monitor for dengue:
- Maintain a high index of suspicion in patients with fever who have been in areas with frequent or continuous dengue transmission within 14 days before illness onset.
- Order diagnostic tests for acute dengue infection such as reverse transcription polymerase chain reaction and immunoglobulin M (IgM) antibody tests or nonstructural protein 1 antigen tests and IgM antibody tests.
- Ensure timely reporting of dengue cases to public health authorities.
- Promote mosquito bite prevention measures among people living in or visiting areas with frequent or continuous dengue transmission.
Roughly one in four dengue virus infections are symptomatic and can be mild or severe. Symptoms begin after an incubation period of about 5-7 days.
Symptoms include fever accompanied by nonspecific signs and symptoms such as nausea, vomiting, rash, muscle aches, joint pain, bone pain, pain behind the eyes, headache, or low white blood cell counts.
Disease Progression
Warning signs that may predict progression to severe disease include abdominal pain or tenderness, persistent vomiting, clinical fluid accumulation, mucosal bleeding, lethargy or restlessness, and progressive increase in hematocrit or liver enlargement.
One in 20 people with symptomatic dengue will develop severe disease, with bleeding, shock, or respiratory distress caused by plasma leakage or end-organ impairment.
Infants aged a year or younger, pregnant people, adults aged 65 years or older, people with certain medical conditions, and those with previous dengue infections are at increased risk for severe dengue.
“Healthcare providers should be prepared to recognize, diagnose, manage, and report dengue cases to health authorities; public health partners should investigate cases and disseminate clear prevention messages to the public,” the alert stated.
The CDC is actively implementing several strategies to address the increase in cases of dengue in the United States. In early April, the agency launched a program-led emergency response and is providing monthly situational updates on dengue to partners, stakeholders, and jurisdictions.
The CDC is also expanding laboratory capacity to improve laboratory testing approaches; collaborating with state, tribal, local, and territorial health departments to strengthen dengue surveillance and recommend prevention strategies; and working to educate the public on dengue prevention.
A version of this article first appeared on Medscape.com.
Federal health officials with the US Centers for Disease Control and Prevention (CDC) have issued an alert, warning health professionals and the public about an increased risk for dengue virus infections in the United States.
The global incidence of dengue in 2024 is the highest on record, reported the agency.
In the United States, Puerto Rico has declared a public health emergency, with 1498 dengue cases reported so far and a “higher-than-expected” number of dengue cases having been identified among US travelers in the first half of this year at 745 cases, according to the alert.
The CDC reports 197 dengue cases in Florida, 134 in New York, 50 in Massachusetts, 40 in California, 14 in Colorado, nine in Arizona, and eight in the District of Columbia, among others.
Transmitted by infected Aedes genus mosquitoes, dengue is the most common arboviral disease globally and is a nationally notifiable disease in the United States.
The six US territories and freely associated states with frequent or continuous dengue transmission are Puerto Rico, American Samoa, the US Virgin Islands, the Federated States of Micronesia, the Republic of the Marshall Islands, and the Republic of Palau.
Monitoring for Dengue
With rising global and domestic cases of dengue, the CDC urges healthcare providers to monitor for dengue:
- Maintain a high index of suspicion in patients with fever who have been in areas with frequent or continuous dengue transmission within 14 days before illness onset.
- Order diagnostic tests for acute dengue infection such as reverse transcription polymerase chain reaction and immunoglobulin M (IgM) antibody tests or nonstructural protein 1 antigen tests and IgM antibody tests.
- Ensure timely reporting of dengue cases to public health authorities.
- Promote mosquito bite prevention measures among people living in or visiting areas with frequent or continuous dengue transmission.
Roughly one in four dengue virus infections are symptomatic and can be mild or severe. Symptoms begin after an incubation period of about 5-7 days.
Symptoms include fever accompanied by nonspecific signs and symptoms such as nausea, vomiting, rash, muscle aches, joint pain, bone pain, pain behind the eyes, headache, or low white blood cell counts.
Disease Progression
Warning signs that may predict progression to severe disease include abdominal pain or tenderness, persistent vomiting, clinical fluid accumulation, mucosal bleeding, lethargy or restlessness, and progressive increase in hematocrit or liver enlargement.
One in 20 people with symptomatic dengue will develop severe disease, with bleeding, shock, or respiratory distress caused by plasma leakage or end-organ impairment.
Infants aged a year or younger, pregnant people, adults aged 65 years or older, people with certain medical conditions, and those with previous dengue infections are at increased risk for severe dengue.
“Healthcare providers should be prepared to recognize, diagnose, manage, and report dengue cases to health authorities; public health partners should investigate cases and disseminate clear prevention messages to the public,” the alert stated.
The CDC is actively implementing several strategies to address the increase in cases of dengue in the United States. In early April, the agency launched a program-led emergency response and is providing monthly situational updates on dengue to partners, stakeholders, and jurisdictions.
The CDC is also expanding laboratory capacity to improve laboratory testing approaches; collaborating with state, tribal, local, and territorial health departments to strengthen dengue surveillance and recommend prevention strategies; and working to educate the public on dengue prevention.
A version of this article first appeared on Medscape.com.