User login
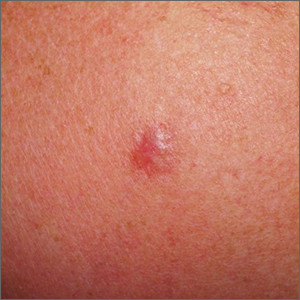
Shoulder lesion

A punch biopsy of the lesion was performed and the results were consistent with a dermatofibroma, which is a benign growth.
Dermatofibromas may manifest as a pink papule on fair-skinned individuals or a darker brown papule in patients of color. Clinically, the texture can be helpful to discern an etiology—dermatofibromas may dimple when pinched laterally, while melanocytic nevi or melanomas tend to be somewhat softer on palpation. Cutaneous sarcoma, while exceedingly rare, may be firmer and chaotic, and varied with multiple colors and topographical changes.
The dermoscopic pattern of a dermatofibroma includes central scar-like areas, a peripheral pigment network, occasional shiny white lines, and confluent circular brown macules. Other less frequent dermoscopic structures may also be seen. A prospective study of the dermoscopic morphology of 412 dermatofibromas found 10 distinct dermoscopic patterns, but also noted that 25% of the dermatofibromas exhibited an atypical pattern.1 Atypical pigment, multiple scar-like areas, and dotted vessels can occur in a dermatofibroma, as well as in a Spitz nevus, and melanoma. Thus, such findings should prompt a biopsy.
Dermatofibromas are safe to observe, but they can be surgically excised if they cause pain or cosmetic concerns.
This patient was reassured to know that the lesion would not require surgical intervention and was unlikely to enlarge or change significantly over time.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
1. Zaballos P, Puig S, Llambrich A, Malvehy J. Dermoscopy of dermatofibromas: a prospective morphological study of 412 cases. Arch Dermatol. 2008;144:75-83. doi: 10.1001/archdermatol.2007.8
A punch biopsy of the lesion was performed and the results were consistent with a dermatofibroma, which is a benign growth.
Dermatofibromas may manifest as a pink papule on fair-skinned individuals or a darker brown papule in patients of color. Clinically, the texture can be helpful to discern an etiology—dermatofibromas may dimple when pinched laterally, while melanocytic nevi or melanomas tend to be somewhat softer on palpation. Cutaneous sarcoma, while exceedingly rare, may be firmer and chaotic, and varied with multiple colors and topographical changes.
The dermoscopic pattern of a dermatofibroma includes central scar-like areas, a peripheral pigment network, occasional shiny white lines, and confluent circular brown macules. Other less frequent dermoscopic structures may also be seen. A prospective study of the dermoscopic morphology of 412 dermatofibromas found 10 distinct dermoscopic patterns, but also noted that 25% of the dermatofibromas exhibited an atypical pattern.1 Atypical pigment, multiple scar-like areas, and dotted vessels can occur in a dermatofibroma, as well as in a Spitz nevus, and melanoma. Thus, such findings should prompt a biopsy.
Dermatofibromas are safe to observe, but they can be surgically excised if they cause pain or cosmetic concerns.
This patient was reassured to know that the lesion would not require surgical intervention and was unlikely to enlarge or change significantly over time.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
A punch biopsy of the lesion was performed and the results were consistent with a dermatofibroma, which is a benign growth.
Dermatofibromas may manifest as a pink papule on fair-skinned individuals or a darker brown papule in patients of color. Clinically, the texture can be helpful to discern an etiology—dermatofibromas may dimple when pinched laterally, while melanocytic nevi or melanomas tend to be somewhat softer on palpation. Cutaneous sarcoma, while exceedingly rare, may be firmer and chaotic, and varied with multiple colors and topographical changes.
The dermoscopic pattern of a dermatofibroma includes central scar-like areas, a peripheral pigment network, occasional shiny white lines, and confluent circular brown macules. Other less frequent dermoscopic structures may also be seen. A prospective study of the dermoscopic morphology of 412 dermatofibromas found 10 distinct dermoscopic patterns, but also noted that 25% of the dermatofibromas exhibited an atypical pattern.1 Atypical pigment, multiple scar-like areas, and dotted vessels can occur in a dermatofibroma, as well as in a Spitz nevus, and melanoma. Thus, such findings should prompt a biopsy.
Dermatofibromas are safe to observe, but they can be surgically excised if they cause pain or cosmetic concerns.
This patient was reassured to know that the lesion would not require surgical intervention and was unlikely to enlarge or change significantly over time.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
1. Zaballos P, Puig S, Llambrich A, Malvehy J. Dermoscopy of dermatofibromas: a prospective morphological study of 412 cases. Arch Dermatol. 2008;144:75-83. doi: 10.1001/archdermatol.2007.8
1. Zaballos P, Puig S, Llambrich A, Malvehy J. Dermoscopy of dermatofibromas: a prospective morphological study of 412 cases. Arch Dermatol. 2008;144:75-83. doi: 10.1001/archdermatol.2007.8
AI and reality – diagnosing otitis media is a real challenge
Let’s pretend for a moment that you receive a call from one of your college roommates who thanks to his family connections has become a venture capitalist in California. His group is considering investing in a start-up that is developing a handheld instrument that it claims will use artificial intelligence to diagnose ear infections far more accurately than the human eye. He wonders if you would like to help him evaluate the company’s proposal and offers you a small percentage of the profits for your efforts should they choose to invest.
Your former roommate has done enough research on his own to understand that otitis media makes up a large chunk of a pediatrician’s workload and that making an accurate diagnosis can often be difficult in a struggling child. He describes his own experience watching a frustrated pediatrician attempting to remove wax from his child’s ear and eventually prescribing antibiotics “to be safe.”

You agree and review the prospectus, which includes a paper from a peer-reviewed journal. What you discover is that the investigators used more than 600 high-resolution images of tympanic membranes taken “during operative myringotomy and tympanostomy tube placement” and the findings at tympanocentesis to train a neural network.
Once trained, the model they developed could differentiate with 95% accuracy between an image of a tympanic membrane that covered a normal middle ear from one that merely contained fluid and from one that contained infected fluid. When these same images were shown to 39 clinicians, more than half of which were pediatricians and included both faculty-level staff and trainees, the average diagnostic accuracy was 65%.
The prospectus includes prediction that this technology could easily be developed into a handheld instrument similar to a traditional otoscope, which could then be linked to the operator’s smartphone, giving the clinician an instant treat or no-treat answer.
Now, remember you have nothing to lose except maybe a friendship. How would you advise your old college roommate?
My advice to your college buddy would be one of caution! Yes, there is a potential big upside because there is a real need for a device that could provide a diagnostic accuracy that this AI model promises. While I suspect that AI will always be more accurate in diagnosis using static images, I bet that most people, clinicians and nonclinicians, could improve their accuracy by linking photos with diagnoses with an hour of practice.
However, evaluating a high-resolution photograph taken through an operative scope inserted into the cerumenless ear canal of a sedated, afrebrile child is several orders of magnitude less difficult than the real-world environment in which the diagnosis of otitis media is usually made.
If the venture capitalists were still interested in getting into the otitis media marketplace, you might suggest they look into companies that have already developed image capture otoscopes. At this point I could only find one on the Internet that was portable and it certainly isn’t small-child friendly. Once we have a tool that can capture images in real-world situations, the next step is to train AI systems to interpret them using the approach these researchers have developed. I bet it can be done. It will be only a matter of time ... and money.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at pdnews@mdedge.com.
Let’s pretend for a moment that you receive a call from one of your college roommates who thanks to his family connections has become a venture capitalist in California. His group is considering investing in a start-up that is developing a handheld instrument that it claims will use artificial intelligence to diagnose ear infections far more accurately than the human eye. He wonders if you would like to help him evaluate the company’s proposal and offers you a small percentage of the profits for your efforts should they choose to invest.
Your former roommate has done enough research on his own to understand that otitis media makes up a large chunk of a pediatrician’s workload and that making an accurate diagnosis can often be difficult in a struggling child. He describes his own experience watching a frustrated pediatrician attempting to remove wax from his child’s ear and eventually prescribing antibiotics “to be safe.”
You agree and review the prospectus, which includes a paper from a peer-reviewed journal. What you discover is that the investigators used more than 600 high-resolution images of tympanic membranes taken “during operative myringotomy and tympanostomy tube placement” and the findings at tympanocentesis to train a neural network.
Once trained, the model they developed could differentiate with 95% accuracy between an image of a tympanic membrane that covered a normal middle ear from one that merely contained fluid and from one that contained infected fluid. When these same images were shown to 39 clinicians, more than half of which were pediatricians and included both faculty-level staff and trainees, the average diagnostic accuracy was 65%.
The prospectus includes prediction that this technology could easily be developed into a handheld instrument similar to a traditional otoscope, which could then be linked to the operator’s smartphone, giving the clinician an instant treat or no-treat answer.
Now, remember you have nothing to lose except maybe a friendship. How would you advise your old college roommate?
My advice to your college buddy would be one of caution! Yes, there is a potential big upside because there is a real need for a device that could provide a diagnostic accuracy that this AI model promises. While I suspect that AI will always be more accurate in diagnosis using static images, I bet that most people, clinicians and nonclinicians, could improve their accuracy by linking photos with diagnoses with an hour of practice.
However, evaluating a high-resolution photograph taken through an operative scope inserted into the cerumenless ear canal of a sedated, afrebrile child is several orders of magnitude less difficult than the real-world environment in which the diagnosis of otitis media is usually made.
If the venture capitalists were still interested in getting into the otitis media marketplace, you might suggest they look into companies that have already developed image capture otoscopes. At this point I could only find one on the Internet that was portable and it certainly isn’t small-child friendly. Once we have a tool that can capture images in real-world situations, the next step is to train AI systems to interpret them using the approach these researchers have developed. I bet it can be done. It will be only a matter of time ... and money.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at pdnews@mdedge.com.
Let’s pretend for a moment that you receive a call from one of your college roommates who thanks to his family connections has become a venture capitalist in California. His group is considering investing in a start-up that is developing a handheld instrument that it claims will use artificial intelligence to diagnose ear infections far more accurately than the human eye. He wonders if you would like to help him evaluate the company’s proposal and offers you a small percentage of the profits for your efforts should they choose to invest.
Your former roommate has done enough research on his own to understand that otitis media makes up a large chunk of a pediatrician’s workload and that making an accurate diagnosis can often be difficult in a struggling child. He describes his own experience watching a frustrated pediatrician attempting to remove wax from his child’s ear and eventually prescribing antibiotics “to be safe.”
You agree and review the prospectus, which includes a paper from a peer-reviewed journal. What you discover is that the investigators used more than 600 high-resolution images of tympanic membranes taken “during operative myringotomy and tympanostomy tube placement” and the findings at tympanocentesis to train a neural network.
Once trained, the model they developed could differentiate with 95% accuracy between an image of a tympanic membrane that covered a normal middle ear from one that merely contained fluid and from one that contained infected fluid. When these same images were shown to 39 clinicians, more than half of which were pediatricians and included both faculty-level staff and trainees, the average diagnostic accuracy was 65%.
The prospectus includes prediction that this technology could easily be developed into a handheld instrument similar to a traditional otoscope, which could then be linked to the operator’s smartphone, giving the clinician an instant treat or no-treat answer.
Now, remember you have nothing to lose except maybe a friendship. How would you advise your old college roommate?
My advice to your college buddy would be one of caution! Yes, there is a potential big upside because there is a real need for a device that could provide a diagnostic accuracy that this AI model promises. While I suspect that AI will always be more accurate in diagnosis using static images, I bet that most people, clinicians and nonclinicians, could improve their accuracy by linking photos with diagnoses with an hour of practice.
However, evaluating a high-resolution photograph taken through an operative scope inserted into the cerumenless ear canal of a sedated, afrebrile child is several orders of magnitude less difficult than the real-world environment in which the diagnosis of otitis media is usually made.
If the venture capitalists were still interested in getting into the otitis media marketplace, you might suggest they look into companies that have already developed image capture otoscopes. At this point I could only find one on the Internet that was portable and it certainly isn’t small-child friendly. Once we have a tool that can capture images in real-world situations, the next step is to train AI systems to interpret them using the approach these researchers have developed. I bet it can be done. It will be only a matter of time ... and money.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at pdnews@mdedge.com.
FDA approves Botox challenger Daxxify for frown lines
The U.S. .
According to a company news release, Daxxify, an acetylcholine release inhibitor and neuromuscular blocking agent, is the first peptide-formulated, long-acting neuromodulator approved for this indication.
The approval of Daxxify, manufactured by Revance Therapeutics, was based on the data from the SAKURA phase 3 clinical trial program, which included more than 2,700 adults who received roughly 4,200 treatments, according to the company.
About three-quarters of participants achieved at least a two-grade improvement in glabellar lines at week 4 as judged by both investigator and patient, and 98% achieved “none or mild wrinkle severity” at week 4 per investigator assessment, the company said.
The median duration of treatment effect was 6 months, with some patients maintaining treatment results at 9 months, compared with a 3- to 4-month duration of treatment effect with conventional neuromodulators.
“Compelling data from the largest phase 3 clinical program ever conducted for glabellar lines demonstrated that Daxxify was well tolerated and achieved clinically significant improvement with long-lasting results and high patient satisfaction,” SAKURA investigator Jeffrey Dover, MD, co-director of SkinCare Physicians, Chestnut Hill, Mass., said in the news release.
“Notably,” said Dr. Dover, “Daxxify was able to demonstrate a long duration of effect while only utilizing 0.18 ng of core active ingredient in the 40-unit labeled indication for glabellar lines.”
Daxxify has a safety profile in line with other currently available neuromodulators in the aesthetics market, the company said, with no serious treatment-related adverse events reported in clinical trial participants.
The most common treatment-related adverse events in the pivotal studies were headache (6%), eyelid ptosis (2%) and facial paresis, including facial asymmetry (1%).
Daxxify is contraindicated in adults with hypersensitivity to any botulinum toxin preparation or any of the components in the formulation and infection at the injection sites.
Full prescribing information is available online.
A version of this article first appeared on Medscape.com.
The U.S. .
According to a company news release, Daxxify, an acetylcholine release inhibitor and neuromuscular blocking agent, is the first peptide-formulated, long-acting neuromodulator approved for this indication.
The approval of Daxxify, manufactured by Revance Therapeutics, was based on the data from the SAKURA phase 3 clinical trial program, which included more than 2,700 adults who received roughly 4,200 treatments, according to the company.
About three-quarters of participants achieved at least a two-grade improvement in glabellar lines at week 4 as judged by both investigator and patient, and 98% achieved “none or mild wrinkle severity” at week 4 per investigator assessment, the company said.
The median duration of treatment effect was 6 months, with some patients maintaining treatment results at 9 months, compared with a 3- to 4-month duration of treatment effect with conventional neuromodulators.
“Compelling data from the largest phase 3 clinical program ever conducted for glabellar lines demonstrated that Daxxify was well tolerated and achieved clinically significant improvement with long-lasting results and high patient satisfaction,” SAKURA investigator Jeffrey Dover, MD, co-director of SkinCare Physicians, Chestnut Hill, Mass., said in the news release.
“Notably,” said Dr. Dover, “Daxxify was able to demonstrate a long duration of effect while only utilizing 0.18 ng of core active ingredient in the 40-unit labeled indication for glabellar lines.”
Daxxify has a safety profile in line with other currently available neuromodulators in the aesthetics market, the company said, with no serious treatment-related adverse events reported in clinical trial participants.
The most common treatment-related adverse events in the pivotal studies were headache (6%), eyelid ptosis (2%) and facial paresis, including facial asymmetry (1%).
Daxxify is contraindicated in adults with hypersensitivity to any botulinum toxin preparation or any of the components in the formulation and infection at the injection sites.
Full prescribing information is available online.
A version of this article first appeared on Medscape.com.
The U.S. .
According to a company news release, Daxxify, an acetylcholine release inhibitor and neuromuscular blocking agent, is the first peptide-formulated, long-acting neuromodulator approved for this indication.
The approval of Daxxify, manufactured by Revance Therapeutics, was based on the data from the SAKURA phase 3 clinical trial program, which included more than 2,700 adults who received roughly 4,200 treatments, according to the company.
About three-quarters of participants achieved at least a two-grade improvement in glabellar lines at week 4 as judged by both investigator and patient, and 98% achieved “none or mild wrinkle severity” at week 4 per investigator assessment, the company said.
The median duration of treatment effect was 6 months, with some patients maintaining treatment results at 9 months, compared with a 3- to 4-month duration of treatment effect with conventional neuromodulators.
“Compelling data from the largest phase 3 clinical program ever conducted for glabellar lines demonstrated that Daxxify was well tolerated and achieved clinically significant improvement with long-lasting results and high patient satisfaction,” SAKURA investigator Jeffrey Dover, MD, co-director of SkinCare Physicians, Chestnut Hill, Mass., said in the news release.
“Notably,” said Dr. Dover, “Daxxify was able to demonstrate a long duration of effect while only utilizing 0.18 ng of core active ingredient in the 40-unit labeled indication for glabellar lines.”
Daxxify has a safety profile in line with other currently available neuromodulators in the aesthetics market, the company said, with no serious treatment-related adverse events reported in clinical trial participants.
The most common treatment-related adverse events in the pivotal studies were headache (6%), eyelid ptosis (2%) and facial paresis, including facial asymmetry (1%).
Daxxify is contraindicated in adults with hypersensitivity to any botulinum toxin preparation or any of the components in the formulation and infection at the injection sites.
Full prescribing information is available online.
A version of this article first appeared on Medscape.com.
Subtle visual dysfunctions often precede early-stage psychosis
A multinational group of investigators found that VisDys were reported considerably more often by patients with recent-onset psychosis and CHR than by those with recent-onset depression or a group acting as healthy control participants.
In addition, vision problems of higher severity were associated with less functional remission both for patients at CHR and those with recent-onset psychosis. Among patients with CHR, VisDys was also linked to lower quality of life (QOL), higher depressiveness, and more severe impairment of visuospatial constructability.
The researchers used fMRI imaging to compare resting-state functional brain connectivity in participants with recent-onset psychosis, CHR, and recent-onset depression. They found that the occipital (ON) and frontoparietal (FPN) subnetworks were particularly implicated in VisDys.
“Subtle VisDys should be regarded as a frequent phenomenon across the psychosis spectrum, impinging negatively on patients’ current ability to function in several settings of their daily and social life, their QOL, and visuospatial abilities,” write investigators led by Johanna Schwarzer, Institute for Translational Psychiatry, University of Muenster (Germany).
“These large-sample study findings suggest that VisDys are clinically highly relevant not only in [recent-onset psychosis] but especially in CHR,” they stated.
The findings were published online in Neuropsychopharmacology.
Subtle, underrecognized
Unlike patients with nonpsychotic disorders, approximately 50%-60% of patients diagnosed with schizophrenia report VisDys involving brightness, motion, form, color perception, or distorted perception of their own face, the researchers reported.
These “subtle” VisDys are “often underrecognized during clinical examination, despite their clinical relevance related to suicidal ideation, cognitive impairment, or poorer treatment response,” they wrote.
Most research into these vision problems in patients with schizophrenia has focused on patients in which the illness is in a stable, chronic state – although VisDys often appear years before the diagnosis of a psychotic disorder.
Moreover, there has been little research into the neurobiological underpinnings of VisDys, specifically in early states of psychosis and/or in comparison to other disorders, such as depression.
The Personalised Prognostic Indicators for Early Psychosis Management (PRONIA) Consortium studied the psychophysiological phenomenon of VisDys in a large sample of adolescents and young adults. The sample consisted of three diagnostic groups: those with recent-onset psychosis, those with CHR, and those with recent-onset depression.
VisDys in daily life were measured using the Schizophrenia Proneness Instrument–Adult Scale (SPI-A), which assesses basic symptoms that indicate increased risk for psychosis.
Visual information processing
Resting-state imaging data on intrinsic brain networks were also assessed in the PRONIA sample and were analyzed across 12,720 functional connectivities between 160 regions of interest across the whole brain.
In particular, the researchers were interested in the primary networks involved in visual information processing, especially the dorsal visual stream, with further focus on the ON and FPN intrinsic subnetworks.
The ON was chosen because it comprises “primary visual processing pathways,” while the FPN is “widely suggested to modulate attention related to visual information processing at higher cognitive levels.”
The investigators used a machine-learning multivariate pattern analysis approach that “enables the consideration of multiple interactions within brain systems.”
The current study involved 721 participants from the PRONIA database, including 147 participants with recent-onset psychosis (mean age, 28.45 years; 60.5% men), 143 with CHR (mean age, 26.97 years; about 50% men), 151 with recent-onset depression (mean age, 29.13 years; 47% men), and 280 in the healthy-controls group (mean age, 28.54 years; 39.4% men).
The researchers selected 14 items to assess from the SPI-A that represented different aspects of VisDys. Severity was defined by the maximum frequency within the past 3 months – from 1 (never) to 6 (daily).
The 14 items were as follows: oversensitivity to light and/or certain visual perception objects, photopsia, micropsia/macropsia, near and tele-vision, metamorphopsia, changes in color vision, altered perception of a patient’s own face, pseudomovements of optic stimuli, diplopia or oblique vision, disturbances of the estimation of distances or sizes, disturbances of the perception of straight lines/contours, maintenance of optic stimuli “visual echoes,” partial seeing (including tubular vision), and captivation of attention by details of the visual field.
Participants also completed the Beck Depression Inventory–II scale (BDI-II), the Positive and Negative Syndrome Scale (PANSS), the Functional Remission in General Schizophrenia, and several other scales that measure global and social functioning.
Other assessments included QOL and the Rey-Osterrieth Complex Figure Test, which is a neuropsychological measurement of visuospatial constructability.
Specific to early-stage psychosis?
Results showed that VisDys were reported more frequently in both recent-onset psychosis and CHR groups compared with the recent-onset depression and healthy control groups (50.34% and 55.94% vs. 16.56% and 4.28%, respectively).
The investigators noted that VisDys sum scores “showed high internal consistency” (Cronbachs alpha, 0.78 over all participants).
Among those with recent-onset psychosis, a higher VisDys sum score was correlated with lower scores for functional remission (P = .036) and social functioning (P = .014).
In CHR, higher VisDys sum scores were associated with lower scores for health-related functional remission (P = .024), lower physical and psychological QOL (P = .004 and P = .015, respectively), more severe depression on the BDI-II (P = .021), and more impaired visuospatial constructability (P = .027).
Among those with recent-onset depression and their healthy peers, “no relevant correlations were found between VisDys sum scores and any parameters representing functional remission, QOL, depressiveness, or visuospatial constructability,” the researchers wrote.
A total of 135 participants with recent-onset psychosis, 128 with CHR, and 134 with recent-onset depression also underwent resting-state fMRI.
ON functional connectivity predicted presence of VisDys in patients with recent-onset psychosis and those with CHR, with a balanced accuracy of 60.17% (P = .0001) and 67.38% (P = .029), respectively. In the combined recent-onset psychosis plus CHR sample, VisDys were predicted by FPN functional connectivity (balanced accuracy, 61.1%; P = .006).
“Findings from multivariate pattern analysis support a model of functional integrity within ON and FPN driving the VisDys phenomenon and being implicated in core disease mechanisms of early psychosis states,” the investigators noted.
“The main findings from this large sample study support the idea of VisDys being specific to the psychosis spectrum already at early stages,” while being less frequently reported in recent-onset depression, they wrote. VisDys also “appeared negligible” among those without psychiatric disorders.
Regular assessment needed
Steven Silverstein, PhD, professor of biopsychosocial medicine and professor of psychiatry, neuroscience, and ophthalmology, Center for Visual Science, University of Rochester (N.Y.) Medical Center, called the findings “important” because “they will increase appreciation in the field of mental health for the frequency and disabling nature of visual symptoms and the need for regular assessment in routine clinical practice with people at risk for or with psychotic disorders.”

In addition, “the brain imaging findings are providing needed information that could lead to treatments that target the brain networks generating the visual symptoms,” such as neurofeedback or brain stimulation, said Dr. Silverstein, who was not involved with the research.
The study was funded by a grant for the PRONIA Consortium. Individual researchers received funding from NARSAD Young Investigator Award of the Brain and Behavior Research Foundation, the Koeln Fortune Program/Faculty of Medicine, the University of Cologne, and the European Union’s Horizon 2020 research and innovation program. Open Access funding was enabled and organized by Projekt DEAL. Ms. Schwarzer and Dr. Silverstein reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A multinational group of investigators found that VisDys were reported considerably more often by patients with recent-onset psychosis and CHR than by those with recent-onset depression or a group acting as healthy control participants.
In addition, vision problems of higher severity were associated with less functional remission both for patients at CHR and those with recent-onset psychosis. Among patients with CHR, VisDys was also linked to lower quality of life (QOL), higher depressiveness, and more severe impairment of visuospatial constructability.
The researchers used fMRI imaging to compare resting-state functional brain connectivity in participants with recent-onset psychosis, CHR, and recent-onset depression. They found that the occipital (ON) and frontoparietal (FPN) subnetworks were particularly implicated in VisDys.
“Subtle VisDys should be regarded as a frequent phenomenon across the psychosis spectrum, impinging negatively on patients’ current ability to function in several settings of their daily and social life, their QOL, and visuospatial abilities,” write investigators led by Johanna Schwarzer, Institute for Translational Psychiatry, University of Muenster (Germany).
“These large-sample study findings suggest that VisDys are clinically highly relevant not only in [recent-onset psychosis] but especially in CHR,” they stated.
The findings were published online in Neuropsychopharmacology.
Subtle, underrecognized
Unlike patients with nonpsychotic disorders, approximately 50%-60% of patients diagnosed with schizophrenia report VisDys involving brightness, motion, form, color perception, or distorted perception of their own face, the researchers reported.
These “subtle” VisDys are “often underrecognized during clinical examination, despite their clinical relevance related to suicidal ideation, cognitive impairment, or poorer treatment response,” they wrote.
Most research into these vision problems in patients with schizophrenia has focused on patients in which the illness is in a stable, chronic state – although VisDys often appear years before the diagnosis of a psychotic disorder.
Moreover, there has been little research into the neurobiological underpinnings of VisDys, specifically in early states of psychosis and/or in comparison to other disorders, such as depression.
The Personalised Prognostic Indicators for Early Psychosis Management (PRONIA) Consortium studied the psychophysiological phenomenon of VisDys in a large sample of adolescents and young adults. The sample consisted of three diagnostic groups: those with recent-onset psychosis, those with CHR, and those with recent-onset depression.
VisDys in daily life were measured using the Schizophrenia Proneness Instrument–Adult Scale (SPI-A), which assesses basic symptoms that indicate increased risk for psychosis.
Visual information processing
Resting-state imaging data on intrinsic brain networks were also assessed in the PRONIA sample and were analyzed across 12,720 functional connectivities between 160 regions of interest across the whole brain.
In particular, the researchers were interested in the primary networks involved in visual information processing, especially the dorsal visual stream, with further focus on the ON and FPN intrinsic subnetworks.
The ON was chosen because it comprises “primary visual processing pathways,” while the FPN is “widely suggested to modulate attention related to visual information processing at higher cognitive levels.”
The investigators used a machine-learning multivariate pattern analysis approach that “enables the consideration of multiple interactions within brain systems.”
The current study involved 721 participants from the PRONIA database, including 147 participants with recent-onset psychosis (mean age, 28.45 years; 60.5% men), 143 with CHR (mean age, 26.97 years; about 50% men), 151 with recent-onset depression (mean age, 29.13 years; 47% men), and 280 in the healthy-controls group (mean age, 28.54 years; 39.4% men).
The researchers selected 14 items to assess from the SPI-A that represented different aspects of VisDys. Severity was defined by the maximum frequency within the past 3 months – from 1 (never) to 6 (daily).
The 14 items were as follows: oversensitivity to light and/or certain visual perception objects, photopsia, micropsia/macropsia, near and tele-vision, metamorphopsia, changes in color vision, altered perception of a patient’s own face, pseudomovements of optic stimuli, diplopia or oblique vision, disturbances of the estimation of distances or sizes, disturbances of the perception of straight lines/contours, maintenance of optic stimuli “visual echoes,” partial seeing (including tubular vision), and captivation of attention by details of the visual field.
Participants also completed the Beck Depression Inventory–II scale (BDI-II), the Positive and Negative Syndrome Scale (PANSS), the Functional Remission in General Schizophrenia, and several other scales that measure global and social functioning.
Other assessments included QOL and the Rey-Osterrieth Complex Figure Test, which is a neuropsychological measurement of visuospatial constructability.
Specific to early-stage psychosis?
Results showed that VisDys were reported more frequently in both recent-onset psychosis and CHR groups compared with the recent-onset depression and healthy control groups (50.34% and 55.94% vs. 16.56% and 4.28%, respectively).
The investigators noted that VisDys sum scores “showed high internal consistency” (Cronbachs alpha, 0.78 over all participants).
Among those with recent-onset psychosis, a higher VisDys sum score was correlated with lower scores for functional remission (P = .036) and social functioning (P = .014).
In CHR, higher VisDys sum scores were associated with lower scores for health-related functional remission (P = .024), lower physical and psychological QOL (P = .004 and P = .015, respectively), more severe depression on the BDI-II (P = .021), and more impaired visuospatial constructability (P = .027).
Among those with recent-onset depression and their healthy peers, “no relevant correlations were found between VisDys sum scores and any parameters representing functional remission, QOL, depressiveness, or visuospatial constructability,” the researchers wrote.
A total of 135 participants with recent-onset psychosis, 128 with CHR, and 134 with recent-onset depression also underwent resting-state fMRI.
ON functional connectivity predicted presence of VisDys in patients with recent-onset psychosis and those with CHR, with a balanced accuracy of 60.17% (P = .0001) and 67.38% (P = .029), respectively. In the combined recent-onset psychosis plus CHR sample, VisDys were predicted by FPN functional connectivity (balanced accuracy, 61.1%; P = .006).
“Findings from multivariate pattern analysis support a model of functional integrity within ON and FPN driving the VisDys phenomenon and being implicated in core disease mechanisms of early psychosis states,” the investigators noted.
“The main findings from this large sample study support the idea of VisDys being specific to the psychosis spectrum already at early stages,” while being less frequently reported in recent-onset depression, they wrote. VisDys also “appeared negligible” among those without psychiatric disorders.
Regular assessment needed
Steven Silverstein, PhD, professor of biopsychosocial medicine and professor of psychiatry, neuroscience, and ophthalmology, Center for Visual Science, University of Rochester (N.Y.) Medical Center, called the findings “important” because “they will increase appreciation in the field of mental health for the frequency and disabling nature of visual symptoms and the need for regular assessment in routine clinical practice with people at risk for or with psychotic disorders.”
In addition, “the brain imaging findings are providing needed information that could lead to treatments that target the brain networks generating the visual symptoms,” such as neurofeedback or brain stimulation, said Dr. Silverstein, who was not involved with the research.
The study was funded by a grant for the PRONIA Consortium. Individual researchers received funding from NARSAD Young Investigator Award of the Brain and Behavior Research Foundation, the Koeln Fortune Program/Faculty of Medicine, the University of Cologne, and the European Union’s Horizon 2020 research and innovation program. Open Access funding was enabled and organized by Projekt DEAL. Ms. Schwarzer and Dr. Silverstein reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A multinational group of investigators found that VisDys were reported considerably more often by patients with recent-onset psychosis and CHR than by those with recent-onset depression or a group acting as healthy control participants.
In addition, vision problems of higher severity were associated with less functional remission both for patients at CHR and those with recent-onset psychosis. Among patients with CHR, VisDys was also linked to lower quality of life (QOL), higher depressiveness, and more severe impairment of visuospatial constructability.
The researchers used fMRI imaging to compare resting-state functional brain connectivity in participants with recent-onset psychosis, CHR, and recent-onset depression. They found that the occipital (ON) and frontoparietal (FPN) subnetworks were particularly implicated in VisDys.
“Subtle VisDys should be regarded as a frequent phenomenon across the psychosis spectrum, impinging negatively on patients’ current ability to function in several settings of their daily and social life, their QOL, and visuospatial abilities,” write investigators led by Johanna Schwarzer, Institute for Translational Psychiatry, University of Muenster (Germany).
“These large-sample study findings suggest that VisDys are clinically highly relevant not only in [recent-onset psychosis] but especially in CHR,” they stated.
The findings were published online in Neuropsychopharmacology.
Subtle, underrecognized
Unlike patients with nonpsychotic disorders, approximately 50%-60% of patients diagnosed with schizophrenia report VisDys involving brightness, motion, form, color perception, or distorted perception of their own face, the researchers reported.
These “subtle” VisDys are “often underrecognized during clinical examination, despite their clinical relevance related to suicidal ideation, cognitive impairment, or poorer treatment response,” they wrote.
Most research into these vision problems in patients with schizophrenia has focused on patients in which the illness is in a stable, chronic state – although VisDys often appear years before the diagnosis of a psychotic disorder.
Moreover, there has been little research into the neurobiological underpinnings of VisDys, specifically in early states of psychosis and/or in comparison to other disorders, such as depression.
The Personalised Prognostic Indicators for Early Psychosis Management (PRONIA) Consortium studied the psychophysiological phenomenon of VisDys in a large sample of adolescents and young adults. The sample consisted of three diagnostic groups: those with recent-onset psychosis, those with CHR, and those with recent-onset depression.
VisDys in daily life were measured using the Schizophrenia Proneness Instrument–Adult Scale (SPI-A), which assesses basic symptoms that indicate increased risk for psychosis.
Visual information processing
Resting-state imaging data on intrinsic brain networks were also assessed in the PRONIA sample and were analyzed across 12,720 functional connectivities between 160 regions of interest across the whole brain.
In particular, the researchers were interested in the primary networks involved in visual information processing, especially the dorsal visual stream, with further focus on the ON and FPN intrinsic subnetworks.
The ON was chosen because it comprises “primary visual processing pathways,” while the FPN is “widely suggested to modulate attention related to visual information processing at higher cognitive levels.”
The investigators used a machine-learning multivariate pattern analysis approach that “enables the consideration of multiple interactions within brain systems.”
The current study involved 721 participants from the PRONIA database, including 147 participants with recent-onset psychosis (mean age, 28.45 years; 60.5% men), 143 with CHR (mean age, 26.97 years; about 50% men), 151 with recent-onset depression (mean age, 29.13 years; 47% men), and 280 in the healthy-controls group (mean age, 28.54 years; 39.4% men).
The researchers selected 14 items to assess from the SPI-A that represented different aspects of VisDys. Severity was defined by the maximum frequency within the past 3 months – from 1 (never) to 6 (daily).
The 14 items were as follows: oversensitivity to light and/or certain visual perception objects, photopsia, micropsia/macropsia, near and tele-vision, metamorphopsia, changes in color vision, altered perception of a patient’s own face, pseudomovements of optic stimuli, diplopia or oblique vision, disturbances of the estimation of distances or sizes, disturbances of the perception of straight lines/contours, maintenance of optic stimuli “visual echoes,” partial seeing (including tubular vision), and captivation of attention by details of the visual field.
Participants also completed the Beck Depression Inventory–II scale (BDI-II), the Positive and Negative Syndrome Scale (PANSS), the Functional Remission in General Schizophrenia, and several other scales that measure global and social functioning.
Other assessments included QOL and the Rey-Osterrieth Complex Figure Test, which is a neuropsychological measurement of visuospatial constructability.
Specific to early-stage psychosis?
Results showed that VisDys were reported more frequently in both recent-onset psychosis and CHR groups compared with the recent-onset depression and healthy control groups (50.34% and 55.94% vs. 16.56% and 4.28%, respectively).
The investigators noted that VisDys sum scores “showed high internal consistency” (Cronbachs alpha, 0.78 over all participants).
Among those with recent-onset psychosis, a higher VisDys sum score was correlated with lower scores for functional remission (P = .036) and social functioning (P = .014).
In CHR, higher VisDys sum scores were associated with lower scores for health-related functional remission (P = .024), lower physical and psychological QOL (P = .004 and P = .015, respectively), more severe depression on the BDI-II (P = .021), and more impaired visuospatial constructability (P = .027).
Among those with recent-onset depression and their healthy peers, “no relevant correlations were found between VisDys sum scores and any parameters representing functional remission, QOL, depressiveness, or visuospatial constructability,” the researchers wrote.
A total of 135 participants with recent-onset psychosis, 128 with CHR, and 134 with recent-onset depression also underwent resting-state fMRI.
ON functional connectivity predicted presence of VisDys in patients with recent-onset psychosis and those with CHR, with a balanced accuracy of 60.17% (P = .0001) and 67.38% (P = .029), respectively. In the combined recent-onset psychosis plus CHR sample, VisDys were predicted by FPN functional connectivity (balanced accuracy, 61.1%; P = .006).
“Findings from multivariate pattern analysis support a model of functional integrity within ON and FPN driving the VisDys phenomenon and being implicated in core disease mechanisms of early psychosis states,” the investigators noted.
“The main findings from this large sample study support the idea of VisDys being specific to the psychosis spectrum already at early stages,” while being less frequently reported in recent-onset depression, they wrote. VisDys also “appeared negligible” among those without psychiatric disorders.
Regular assessment needed
Steven Silverstein, PhD, professor of biopsychosocial medicine and professor of psychiatry, neuroscience, and ophthalmology, Center for Visual Science, University of Rochester (N.Y.) Medical Center, called the findings “important” because “they will increase appreciation in the field of mental health for the frequency and disabling nature of visual symptoms and the need for regular assessment in routine clinical practice with people at risk for or with psychotic disorders.”
In addition, “the brain imaging findings are providing needed information that could lead to treatments that target the brain networks generating the visual symptoms,” such as neurofeedback or brain stimulation, said Dr. Silverstein, who was not involved with the research.
The study was funded by a grant for the PRONIA Consortium. Individual researchers received funding from NARSAD Young Investigator Award of the Brain and Behavior Research Foundation, the Koeln Fortune Program/Faculty of Medicine, the University of Cologne, and the European Union’s Horizon 2020 research and innovation program. Open Access funding was enabled and organized by Projekt DEAL. Ms. Schwarzer and Dr. Silverstein reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM NEUROPSYCHOPHARMACOLOGY
AGA Clinical Practice Update: Expert review on endoscopic management for recurrent acute and chronic pancreatitis
Endoscopy plays an integral role in the evaluation and management of patients with recurrent acute pancreatitis and chronic pancreatitis, according to a new American Gastroenterological Association clinical practice update published in Gastroenterology.
Acute pancreatitis remains the leading cause of inpatient care among gastrointestinal conditions, with about 10%-30% of patients developing recurrent acute pancreatitis, wrote co–first authors Daniel Strand, MD, from the University of Virginia Health System, Charlottesville, and Ryan J. Law, MD, from the Mayo Clinic, Rochester, Minn., and colleagues. About 35% of patients with recurrent acute pancreatitis will progress to chronic pancreatitis. Both conditions are associated with significant morbidity and mortality.
“Interventions aimed to better evaluate, mitigate the progression of, and treat symptoms related to [acute pancreatitis] and [chronic pancreatitis] are critical to improve patients’ quality of life and other long-term outcomes,” the authors of the expert review wrote.
The authors reviewed randomized controlled trials, observational studies, systematic reviews and meta-analyses, and expert consensus in the field to develop eight clinical practice advice statements.
First, when the initial evaluation reveals no clear explanation for acute or recurrent pancreatitis, endoscopic ultrasound is the preferred diagnostic test. The authors noted that, although there isn’t a concretely defined optimal timing for EUS defined, most experts advise a short delay of 2-6 weeks after resolution of acute pancreatitis. MRI with contrast and cholangiopancreatography can be a reasonable complementary or alternative test, based on local expertise and availability.
Second, the role of ERCP remains controversial for reducing the frequency of acute pancreatitis episodes in patients with pancreas divisum, the most common congenital pancreatic anomaly, the authors wrote. However, minor papilla endotherapy may be useful, particularly for those with objective signs of outflow obstruction, such as a dilated dorsal pancreatic duct or santorinicele. However, there is no role for ERCP in treating pain alone in patients with pancreas divisum.
Third, ERCP remains even more controversial for reducing the frequency of pancreatitis episodes in patients with unexplained recurrent acute pancreatitis and standard pancreatic ductal anatomy, according to the authors. It should only be considered after a comprehensive discussion of the uncertain benefits and potentially severe procedure-related adverse events. When used, ERCP with biliary sphincterotomy alone may be preferable to dual sphincterotomy.
Fourth, for long-term treatment of patients with painful obstructive chronic pancreatitis, surgical intervention should be considered over endoscopic therapy, the study authors wrote. Pain is the most common symptom and important driver of impaired quality of life in patients with chronic pancreatitis, among whom a subset will be affected by intraductal hypertension from an obstructed pancreatic duct. The authors noted that endoscopic intervention remains a reasonable alternative to surgery for suboptimal operative candidates or patients who want a less-invasive approach, as long as they are clearly informed that the best practice advice primarily favors surgery.
Fifth, when using ERCP for pancreatic duct stones, small main pancreatic duct stones of 5 mm or less can be treated with pancreatography and conventional stone extraction maneuvers. For larger stones, however, extracorporeal shockwave lithotripsy or pancreatoscopy with intraductal lithotripsy can be considered, although the former is not widely available in the United States and the success rates for the latter vary.
Sixth, when using ERCP for pancreatic duct strictures, prolonged stent therapy for 6-12 months is effective for treating symptoms and remodeling main pancreatic duct strictures. The preferred approach is to place and sequentially add multiple plastic stents in parallel, or up-sizing. Emerging evidence suggests that fully covered self-expanding metal stents may be useful in this case, but additional research is needed. For example, one study suggested that patients treated with these self-expanding stents required fewer ERCPs, but their adverse event rate was significantly higher (39% vs. 14%).
Seventh, ERCP with stent insertion is the preferred treatment for benign biliary stricture caused by chronic pancreatitis. Fully covered self-expanding metal stents are favored over placing multiple plastic stents when feasible, given the similar efficacy but significantly lower need for stent exchange procedures during the treatment course.
Eighth, celiac plexus block shouldn’t be routinely performed for the management of pain caused by chronic pancreatitis. Celiac plexus block could be considered in certain patients on a case-by-case basis if they have debilitating pain that hasn’t responded to other therapeutic measures. However, this should only be considered after a discussion about the unclear outcomes and its procedural risks.
“Given the current lack of evidence, additional well-designed prospective comparative studies are needed to support a more unified diagnostic and therapeutic pathway for the treatment of these complex cases,” the authors concluded.
The authors reported no grant support or funding sources for this report. Several authors disclosed financial relationships with companies such as Olympus America, Medtronic, and Microtech.
Endoscopy plays an integral role in the evaluation and management of patients with recurrent acute pancreatitis and chronic pancreatitis, according to a new American Gastroenterological Association clinical practice update published in Gastroenterology.
Acute pancreatitis remains the leading cause of inpatient care among gastrointestinal conditions, with about 10%-30% of patients developing recurrent acute pancreatitis, wrote co–first authors Daniel Strand, MD, from the University of Virginia Health System, Charlottesville, and Ryan J. Law, MD, from the Mayo Clinic, Rochester, Minn., and colleagues. About 35% of patients with recurrent acute pancreatitis will progress to chronic pancreatitis. Both conditions are associated with significant morbidity and mortality.
“Interventions aimed to better evaluate, mitigate the progression of, and treat symptoms related to [acute pancreatitis] and [chronic pancreatitis] are critical to improve patients’ quality of life and other long-term outcomes,” the authors of the expert review wrote.
The authors reviewed randomized controlled trials, observational studies, systematic reviews and meta-analyses, and expert consensus in the field to develop eight clinical practice advice statements.
First, when the initial evaluation reveals no clear explanation for acute or recurrent pancreatitis, endoscopic ultrasound is the preferred diagnostic test. The authors noted that, although there isn’t a concretely defined optimal timing for EUS defined, most experts advise a short delay of 2-6 weeks after resolution of acute pancreatitis. MRI with contrast and cholangiopancreatography can be a reasonable complementary or alternative test, based on local expertise and availability.
Second, the role of ERCP remains controversial for reducing the frequency of acute pancreatitis episodes in patients with pancreas divisum, the most common congenital pancreatic anomaly, the authors wrote. However, minor papilla endotherapy may be useful, particularly for those with objective signs of outflow obstruction, such as a dilated dorsal pancreatic duct or santorinicele. However, there is no role for ERCP in treating pain alone in patients with pancreas divisum.
Third, ERCP remains even more controversial for reducing the frequency of pancreatitis episodes in patients with unexplained recurrent acute pancreatitis and standard pancreatic ductal anatomy, according to the authors. It should only be considered after a comprehensive discussion of the uncertain benefits and potentially severe procedure-related adverse events. When used, ERCP with biliary sphincterotomy alone may be preferable to dual sphincterotomy.
Fourth, for long-term treatment of patients with painful obstructive chronic pancreatitis, surgical intervention should be considered over endoscopic therapy, the study authors wrote. Pain is the most common symptom and important driver of impaired quality of life in patients with chronic pancreatitis, among whom a subset will be affected by intraductal hypertension from an obstructed pancreatic duct. The authors noted that endoscopic intervention remains a reasonable alternative to surgery for suboptimal operative candidates or patients who want a less-invasive approach, as long as they are clearly informed that the best practice advice primarily favors surgery.
Fifth, when using ERCP for pancreatic duct stones, small main pancreatic duct stones of 5 mm or less can be treated with pancreatography and conventional stone extraction maneuvers. For larger stones, however, extracorporeal shockwave lithotripsy or pancreatoscopy with intraductal lithotripsy can be considered, although the former is not widely available in the United States and the success rates for the latter vary.
Sixth, when using ERCP for pancreatic duct strictures, prolonged stent therapy for 6-12 months is effective for treating symptoms and remodeling main pancreatic duct strictures. The preferred approach is to place and sequentially add multiple plastic stents in parallel, or up-sizing. Emerging evidence suggests that fully covered self-expanding metal stents may be useful in this case, but additional research is needed. For example, one study suggested that patients treated with these self-expanding stents required fewer ERCPs, but their adverse event rate was significantly higher (39% vs. 14%).
Seventh, ERCP with stent insertion is the preferred treatment for benign biliary stricture caused by chronic pancreatitis. Fully covered self-expanding metal stents are favored over placing multiple plastic stents when feasible, given the similar efficacy but significantly lower need for stent exchange procedures during the treatment course.
Eighth, celiac plexus block shouldn’t be routinely performed for the management of pain caused by chronic pancreatitis. Celiac plexus block could be considered in certain patients on a case-by-case basis if they have debilitating pain that hasn’t responded to other therapeutic measures. However, this should only be considered after a discussion about the unclear outcomes and its procedural risks.
“Given the current lack of evidence, additional well-designed prospective comparative studies are needed to support a more unified diagnostic and therapeutic pathway for the treatment of these complex cases,” the authors concluded.
The authors reported no grant support or funding sources for this report. Several authors disclosed financial relationships with companies such as Olympus America, Medtronic, and Microtech.
Endoscopy plays an integral role in the evaluation and management of patients with recurrent acute pancreatitis and chronic pancreatitis, according to a new American Gastroenterological Association clinical practice update published in Gastroenterology.
Acute pancreatitis remains the leading cause of inpatient care among gastrointestinal conditions, with about 10%-30% of patients developing recurrent acute pancreatitis, wrote co–first authors Daniel Strand, MD, from the University of Virginia Health System, Charlottesville, and Ryan J. Law, MD, from the Mayo Clinic, Rochester, Minn., and colleagues. About 35% of patients with recurrent acute pancreatitis will progress to chronic pancreatitis. Both conditions are associated with significant morbidity and mortality.
“Interventions aimed to better evaluate, mitigate the progression of, and treat symptoms related to [acute pancreatitis] and [chronic pancreatitis] are critical to improve patients’ quality of life and other long-term outcomes,” the authors of the expert review wrote.
The authors reviewed randomized controlled trials, observational studies, systematic reviews and meta-analyses, and expert consensus in the field to develop eight clinical practice advice statements.
First, when the initial evaluation reveals no clear explanation for acute or recurrent pancreatitis, endoscopic ultrasound is the preferred diagnostic test. The authors noted that, although there isn’t a concretely defined optimal timing for EUS defined, most experts advise a short delay of 2-6 weeks after resolution of acute pancreatitis. MRI with contrast and cholangiopancreatography can be a reasonable complementary or alternative test, based on local expertise and availability.
Second, the role of ERCP remains controversial for reducing the frequency of acute pancreatitis episodes in patients with pancreas divisum, the most common congenital pancreatic anomaly, the authors wrote. However, minor papilla endotherapy may be useful, particularly for those with objective signs of outflow obstruction, such as a dilated dorsal pancreatic duct or santorinicele. However, there is no role for ERCP in treating pain alone in patients with pancreas divisum.
Third, ERCP remains even more controversial for reducing the frequency of pancreatitis episodes in patients with unexplained recurrent acute pancreatitis and standard pancreatic ductal anatomy, according to the authors. It should only be considered after a comprehensive discussion of the uncertain benefits and potentially severe procedure-related adverse events. When used, ERCP with biliary sphincterotomy alone may be preferable to dual sphincterotomy.
Fourth, for long-term treatment of patients with painful obstructive chronic pancreatitis, surgical intervention should be considered over endoscopic therapy, the study authors wrote. Pain is the most common symptom and important driver of impaired quality of life in patients with chronic pancreatitis, among whom a subset will be affected by intraductal hypertension from an obstructed pancreatic duct. The authors noted that endoscopic intervention remains a reasonable alternative to surgery for suboptimal operative candidates or patients who want a less-invasive approach, as long as they are clearly informed that the best practice advice primarily favors surgery.
Fifth, when using ERCP for pancreatic duct stones, small main pancreatic duct stones of 5 mm or less can be treated with pancreatography and conventional stone extraction maneuvers. For larger stones, however, extracorporeal shockwave lithotripsy or pancreatoscopy with intraductal lithotripsy can be considered, although the former is not widely available in the United States and the success rates for the latter vary.
Sixth, when using ERCP for pancreatic duct strictures, prolonged stent therapy for 6-12 months is effective for treating symptoms and remodeling main pancreatic duct strictures. The preferred approach is to place and sequentially add multiple plastic stents in parallel, or up-sizing. Emerging evidence suggests that fully covered self-expanding metal stents may be useful in this case, but additional research is needed. For example, one study suggested that patients treated with these self-expanding stents required fewer ERCPs, but their adverse event rate was significantly higher (39% vs. 14%).
Seventh, ERCP with stent insertion is the preferred treatment for benign biliary stricture caused by chronic pancreatitis. Fully covered self-expanding metal stents are favored over placing multiple plastic stents when feasible, given the similar efficacy but significantly lower need for stent exchange procedures during the treatment course.
Eighth, celiac plexus block shouldn’t be routinely performed for the management of pain caused by chronic pancreatitis. Celiac plexus block could be considered in certain patients on a case-by-case basis if they have debilitating pain that hasn’t responded to other therapeutic measures. However, this should only be considered after a discussion about the unclear outcomes and its procedural risks.
“Given the current lack of evidence, additional well-designed prospective comparative studies are needed to support a more unified diagnostic and therapeutic pathway for the treatment of these complex cases,” the authors concluded.
The authors reported no grant support or funding sources for this report. Several authors disclosed financial relationships with companies such as Olympus America, Medtronic, and Microtech.
FROM GASTROENTEROLOGY
Psychiatrists’ views on psychoactive drugs clash with U.S. policy
“The consensus among experts, including psychiatrists, about specific drugs is not consistent or congruent with the schedule of these drugs” in the United States, lead author Adam Levin, MD, third-year psychiatry resident, Ohio State University, Columbus, and affiliate scholar at the Center for Psychedelic Drug Research and Education, Ohio State College of Social Work, told this news organization.

Dr. Levin stressed the importance of appropriate drug scheduling to improve access to treatments such as psilocybin (psychedelic mushrooms) and 4-methylenedioxy methamphetamine (MDMA), which are now being tested for psychiatric disorders.
“We are in the middle of a mental health crisis so having any new tools would be really important,” he said.
The survey findings were published online in the International Journal of Drug Policy.
Five drug schedules
The Controlled Substances Act of 1970 created five “schedules” that organized drugs from most to least dangerous (schedule I-V). However, Dr. Levin said that the schedules do not accurately reflect the harms or therapeutic benefits of the various drugs.
Some drugs in lower, less restrictive schedules have greater potential for harm than do those in higher schedules, he noted. For example, methamphetamine, which has been recalled in multiple formulations because of concerns about abuse and limited medical use, remains a schedule II drug.
In addition, several schedule I drugs, including psilocybin and MDMA that are deemed dangerous and of no medical value, have shown therapeutic potential and low rates of misuse, addiction, or physical harm, the investigators noted.
In fact, the Food and Drug Administration has granted breakthrough therapy status to psilocybin for treatment-resistant depression and major depressive disorder (MDD) and to MDMA for posttraumatic stress disorder. This has positioned these drugs for possible FDA approval within the next few years.
Access to schedule I drugs for research purposes is tightly controlled. “Once psilocybin was placed in schedule I, there was this massive drop-off in the research funding and amount of research; and we’re just now starting to understand the potential therapeutic value of this drug,” said Dr. Levin.
Even with a recent research resurgence, most studies are funded by charitable donations or for-profit companies because of continued hesitancy on the part of grant-making organizations, he added.
Apparent contradictions
Given the pending approval of several schedule I drugs and escalating abuse of drugs in lower schedules, there is a growing need to understand physician attitudes surrounding the apparent contradictions in the drug schedule, the investigators noted.
Their survey included a geographically diverse group of 181 mostly middle-aged psychiatrists (65.2% were men) with an average of 16.2 years of practice after residency.
Participants were randomly assigned to respond to a vignette depicting a clinical scenario where a patient wants one of four drugs to help treat severe depression: psilocybin, a schedule I drug; methamphetamine (Desoxyn), a schedule II drug; ketamine, a Schedule III drug; or alprazolam (Xanax), a schedule IV drug.
Each of these therapies has established antidepressant properties, but none are FDA approved for treatment of MDD. However, an intranasal formulation of the ketamine enantiomer Spravato (esketamine) was recently approved for treatment-resistant depression.
There were significant differences among the groups presented with different vignettes. Participants were more likely to warn against repeated use of and development of a new psychiatric problem with methamphetamine and alprazolam compared with psilocybin or ketamine.
Respondents were most concerned about increased suicide risk after the nonprescribed use of alprazolam compared with psilocybin and ketamine.
Compared with all other drugs, ketamine was more likely to be integrated into treatment plans.
Therapeutic value, abuse potential
Participants were asked to rate the safety, therapeutic value, and abuse potential of the four drugs as well as alcohol, a nonscheduled legal drug, if used properly or as directed.
Respondents viewed psilocybin and ketamine as similarly safe – and safer than methamphetamine and alprazolam. They considered ketamine as having the highest therapeutic potential, followed by psilocybin, and then alprazolam and methamphetamine. “Last was alcohol, which we expected because alcohol is not used therapeutically,” said Dr. Levin.
Survey completers viewed methamphetamine, alprazolam, and alcohol as having similarly high abuse potential, and ketamine as having mid-level abuse potential. Psilocybin was rated as having the lowest abuse potential, “which is exactly the opposite of what is implied by its schedule I status,” noted Dr. Levin.
The results provide evidence these drugs “are incorrectly scheduled,” he said.
“This suggests the schedule does not reflect current evidence, which I think is really important to understand because there are consequences to the drug schedule,” including criminal justice and research consequences, he added.
Dr. Levin pointed out that possession of drugs in more harmful schedules is linked to sometimes lengthy prison sentences.
The psychiatrists’ perceptions of the drugs “overlaps pretty significantly” with recent surveys of other mental health professionals, including psychologists and addiction experts, he noted.
The study was funded by the Drug Enforcement and Policy Center, Moritz College of Law, and The Ohio State University. Dr. Levin reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
“The consensus among experts, including psychiatrists, about specific drugs is not consistent or congruent with the schedule of these drugs” in the United States, lead author Adam Levin, MD, third-year psychiatry resident, Ohio State University, Columbus, and affiliate scholar at the Center for Psychedelic Drug Research and Education, Ohio State College of Social Work, told this news organization.
Dr. Levin stressed the importance of appropriate drug scheduling to improve access to treatments such as psilocybin (psychedelic mushrooms) and 4-methylenedioxy methamphetamine (MDMA), which are now being tested for psychiatric disorders.
“We are in the middle of a mental health crisis so having any new tools would be really important,” he said.
The survey findings were published online in the International Journal of Drug Policy.
Five drug schedules
The Controlled Substances Act of 1970 created five “schedules” that organized drugs from most to least dangerous (schedule I-V). However, Dr. Levin said that the schedules do not accurately reflect the harms or therapeutic benefits of the various drugs.
Some drugs in lower, less restrictive schedules have greater potential for harm than do those in higher schedules, he noted. For example, methamphetamine, which has been recalled in multiple formulations because of concerns about abuse and limited medical use, remains a schedule II drug.
In addition, several schedule I drugs, including psilocybin and MDMA that are deemed dangerous and of no medical value, have shown therapeutic potential and low rates of misuse, addiction, or physical harm, the investigators noted.
In fact, the Food and Drug Administration has granted breakthrough therapy status to psilocybin for treatment-resistant depression and major depressive disorder (MDD) and to MDMA for posttraumatic stress disorder. This has positioned these drugs for possible FDA approval within the next few years.
Access to schedule I drugs for research purposes is tightly controlled. “Once psilocybin was placed in schedule I, there was this massive drop-off in the research funding and amount of research; and we’re just now starting to understand the potential therapeutic value of this drug,” said Dr. Levin.
Even with a recent research resurgence, most studies are funded by charitable donations or for-profit companies because of continued hesitancy on the part of grant-making organizations, he added.
Apparent contradictions
Given the pending approval of several schedule I drugs and escalating abuse of drugs in lower schedules, there is a growing need to understand physician attitudes surrounding the apparent contradictions in the drug schedule, the investigators noted.
Their survey included a geographically diverse group of 181 mostly middle-aged psychiatrists (65.2% were men) with an average of 16.2 years of practice after residency.
Participants were randomly assigned to respond to a vignette depicting a clinical scenario where a patient wants one of four drugs to help treat severe depression: psilocybin, a schedule I drug; methamphetamine (Desoxyn), a schedule II drug; ketamine, a Schedule III drug; or alprazolam (Xanax), a schedule IV drug.
Each of these therapies has established antidepressant properties, but none are FDA approved for treatment of MDD. However, an intranasal formulation of the ketamine enantiomer Spravato (esketamine) was recently approved for treatment-resistant depression.
There were significant differences among the groups presented with different vignettes. Participants were more likely to warn against repeated use of and development of a new psychiatric problem with methamphetamine and alprazolam compared with psilocybin or ketamine.
Respondents were most concerned about increased suicide risk after the nonprescribed use of alprazolam compared with psilocybin and ketamine.
Compared with all other drugs, ketamine was more likely to be integrated into treatment plans.
Therapeutic value, abuse potential
Participants were asked to rate the safety, therapeutic value, and abuse potential of the four drugs as well as alcohol, a nonscheduled legal drug, if used properly or as directed.
Respondents viewed psilocybin and ketamine as similarly safe – and safer than methamphetamine and alprazolam. They considered ketamine as having the highest therapeutic potential, followed by psilocybin, and then alprazolam and methamphetamine. “Last was alcohol, which we expected because alcohol is not used therapeutically,” said Dr. Levin.
Survey completers viewed methamphetamine, alprazolam, and alcohol as having similarly high abuse potential, and ketamine as having mid-level abuse potential. Psilocybin was rated as having the lowest abuse potential, “which is exactly the opposite of what is implied by its schedule I status,” noted Dr. Levin.
The results provide evidence these drugs “are incorrectly scheduled,” he said.
“This suggests the schedule does not reflect current evidence, which I think is really important to understand because there are consequences to the drug schedule,” including criminal justice and research consequences, he added.
Dr. Levin pointed out that possession of drugs in more harmful schedules is linked to sometimes lengthy prison sentences.
The psychiatrists’ perceptions of the drugs “overlaps pretty significantly” with recent surveys of other mental health professionals, including psychologists and addiction experts, he noted.
The study was funded by the Drug Enforcement and Policy Center, Moritz College of Law, and The Ohio State University. Dr. Levin reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
“The consensus among experts, including psychiatrists, about specific drugs is not consistent or congruent with the schedule of these drugs” in the United States, lead author Adam Levin, MD, third-year psychiatry resident, Ohio State University, Columbus, and affiliate scholar at the Center for Psychedelic Drug Research and Education, Ohio State College of Social Work, told this news organization.
Dr. Levin stressed the importance of appropriate drug scheduling to improve access to treatments such as psilocybin (psychedelic mushrooms) and 4-methylenedioxy methamphetamine (MDMA), which are now being tested for psychiatric disorders.
“We are in the middle of a mental health crisis so having any new tools would be really important,” he said.
The survey findings were published online in the International Journal of Drug Policy.
Five drug schedules
The Controlled Substances Act of 1970 created five “schedules” that organized drugs from most to least dangerous (schedule I-V). However, Dr. Levin said that the schedules do not accurately reflect the harms or therapeutic benefits of the various drugs.
Some drugs in lower, less restrictive schedules have greater potential for harm than do those in higher schedules, he noted. For example, methamphetamine, which has been recalled in multiple formulations because of concerns about abuse and limited medical use, remains a schedule II drug.
In addition, several schedule I drugs, including psilocybin and MDMA that are deemed dangerous and of no medical value, have shown therapeutic potential and low rates of misuse, addiction, or physical harm, the investigators noted.
In fact, the Food and Drug Administration has granted breakthrough therapy status to psilocybin for treatment-resistant depression and major depressive disorder (MDD) and to MDMA for posttraumatic stress disorder. This has positioned these drugs for possible FDA approval within the next few years.
Access to schedule I drugs for research purposes is tightly controlled. “Once psilocybin was placed in schedule I, there was this massive drop-off in the research funding and amount of research; and we’re just now starting to understand the potential therapeutic value of this drug,” said Dr. Levin.
Even with a recent research resurgence, most studies are funded by charitable donations or for-profit companies because of continued hesitancy on the part of grant-making organizations, he added.
Apparent contradictions
Given the pending approval of several schedule I drugs and escalating abuse of drugs in lower schedules, there is a growing need to understand physician attitudes surrounding the apparent contradictions in the drug schedule, the investigators noted.
Their survey included a geographically diverse group of 181 mostly middle-aged psychiatrists (65.2% were men) with an average of 16.2 years of practice after residency.
Participants were randomly assigned to respond to a vignette depicting a clinical scenario where a patient wants one of four drugs to help treat severe depression: psilocybin, a schedule I drug; methamphetamine (Desoxyn), a schedule II drug; ketamine, a Schedule III drug; or alprazolam (Xanax), a schedule IV drug.
Each of these therapies has established antidepressant properties, but none are FDA approved for treatment of MDD. However, an intranasal formulation of the ketamine enantiomer Spravato (esketamine) was recently approved for treatment-resistant depression.
There were significant differences among the groups presented with different vignettes. Participants were more likely to warn against repeated use of and development of a new psychiatric problem with methamphetamine and alprazolam compared with psilocybin or ketamine.
Respondents were most concerned about increased suicide risk after the nonprescribed use of alprazolam compared with psilocybin and ketamine.
Compared with all other drugs, ketamine was more likely to be integrated into treatment plans.
Therapeutic value, abuse potential
Participants were asked to rate the safety, therapeutic value, and abuse potential of the four drugs as well as alcohol, a nonscheduled legal drug, if used properly or as directed.
Respondents viewed psilocybin and ketamine as similarly safe – and safer than methamphetamine and alprazolam. They considered ketamine as having the highest therapeutic potential, followed by psilocybin, and then alprazolam and methamphetamine. “Last was alcohol, which we expected because alcohol is not used therapeutically,” said Dr. Levin.
Survey completers viewed methamphetamine, alprazolam, and alcohol as having similarly high abuse potential, and ketamine as having mid-level abuse potential. Psilocybin was rated as having the lowest abuse potential, “which is exactly the opposite of what is implied by its schedule I status,” noted Dr. Levin.
The results provide evidence these drugs “are incorrectly scheduled,” he said.
“This suggests the schedule does not reflect current evidence, which I think is really important to understand because there are consequences to the drug schedule,” including criminal justice and research consequences, he added.
Dr. Levin pointed out that possession of drugs in more harmful schedules is linked to sometimes lengthy prison sentences.
The psychiatrists’ perceptions of the drugs “overlaps pretty significantly” with recent surveys of other mental health professionals, including psychologists and addiction experts, he noted.
The study was funded by the Drug Enforcement and Policy Center, Moritz College of Law, and The Ohio State University. Dr. Levin reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE INTERNATIONAL JOURNAL OF DRUG POLICY
AI algorithm detects erosions, ankylosis with high accuracy in patients with sacroiliitis
Erosions and ankylosis in patients with sacroiliitis are detectable to a high degree of accuracy on CT images using an artificial intelligence (AI)–based algorithm, according to research presented at the 13th International Congress on Spondyloarthritides.
Lennart Jans, MD, head of clinics in musculoskeletal imaging in the department of radiology at Ghent (Belgium) University Hospital, shared data on the development and validation of the algorithm for automatic detection of erosion and ankylosis on CT images of the sacroiliac (SI) joints.

“Essentially, in terms of statistics, this AI algorithm has 95% sensitivity for picking up erosions in patients with clinical symptoms of sacroiliitis, and if this is further developed as a tool, it could aid detection in people with erosions that would otherwise go undetected and undiagnosed,” Dr. Jans said in an interview, stressing that the results were still preliminary.
“We want to move from reporting one patient at a time to a system that detects and helps to diagnose larger numbers of patients and makes a larger impact on patient outcomes.”
He stressed that, with thousands of images per patient, it is an impossible workload for any radiology department to read every image necessary to inform diagnoses, and this is only exacerbated by the shortage of rheumatologists, especially in the United States.
Denis Poddubnyy, MD, head of rheumatology at Charité University Hospital, Berlin, acknowledged that AI has potential to improve the recognition of changes indicative of spondyloarthritis (SpA) on imaging. “A standardized, valid, and reliable detection of those changes is relevant for both diagnosis, including differential diagnosis, and classification of SpA.”
Dr. Poddubnyy added that the AI-based algorithm developed by Dr. Jans and associates is designed to detect very specific SpA structural changes in the SI joints on CT. “CT is usually applied in the clinical practice after MRI ... normally in cases where MRI does not provide conclusive results,” he said. Since MRI scans have also been recently used to develop an AI-based algorithm for the detection of active inflammation – not captured by CT – and structural changes in SI joints, he noted that the “generated data on CT should be, therefore, seen in a broader context toward standardization of imaging findings detection.”
Proof-of-concept findings are due for scale-up
Dr. Jans acknowledged that the current data only establish proof of concept. Among the study’s 145 patients, 60% were used for training the AI algorithm and 40% for testing it. All patients who had clinical symptoms of sacroiliitis and had undergone a SI joint CT scan were included from two hospitals: Ghent University Hospital and the University of Alberta Hospital, Edmonton. The majority of patients were female (81 of 145). They had a mean age of 40 years, 84 had diagnosed axial SpA, 15 had mechanical back pain, and 46 did not have a final diagnosis.
CT images were examined by three independent and blinded radiologists who annotated erosions more than 1 mm and ankylosis more than 2 mm, while a type of AI algorithm known as a neural network pipeline was developed to segment the SI joints and detect structural lesions.
In the first instance, Dr. Jans explained, examination of CT images using the AI algorithm from patients who enter the hospital for other reasons, such as trauma, rheumatic diseases, kidney stones, or appendicitis, might lead to the detection of otherwise unknown erosions. “Often patients have complained of backache for years, seeing various physiotherapists and similar, but had no idea what might be causing it,” he said. “We just don’t have the time for examining all the thousands of images separately. We need some kind of aid here. We need an extra pair of eyes. This is what AI software does.”
Dr. Jans said rheumatologists who ultimately want to detect and diagnose patients with SI erosions want to reduce the false-negative findings. “They want the system to pick up all the patients who have erosions. Here, the most important parameter is sensitivity, and we find that our algorithm shows a very high sensitivity. Optimization of the AI algorithm to reduce false negatives resulted in a sensitivity of 95% for detection of erosions on CT of the sacroiliac joints on a patient level.”
While overall accuracy was over 90%, Dr. Jans acknowledged that the algorithm was run in a relatively select population of dedicated CT scans of the joints. He is also aware that a good AI algorithm needs to work well across locations and populations. “If you make something within your institution alone, it will not work in a hospital on the other side of the street.”
However, he added, the researchers used images from four different CT scanners and images from two different institutions – one in Canada and their own in Belgium, providing a case mix that makes their algorithm more refined.
Next step: Test in an unselected population
When asked to comment on the study, Mikael Boesen, MD, PhD, of Bispebjerg and Frederiksberg Hospital, Copenhagen, congratulated Dr. Jans on the work and remarked that he found the research potentially clinically useful.
“The next steps would be to test the performance of the model in an unselected population of patients who have CT scans of the abdomen for other reasons to test the model’s ability to flag potential SI joint disease to the reader, which is often overlooked, as well as [to see] how the model performs in larger datasets from other hospitals, vendors, and CT-reconstruction algorithms.”
Finally, Dr. Boesen pointed out that it would be interesting to see if the AI algorithm can detect different reasons for erosions. “Especially [for] separation between mechanical and inflammatory courses. This could potentially be done by automatically mapping the location of the erosions in the SI joints.”
Dr. Jans has now opened up the project to other radiologists to collaborate and provide images to train and test the algorithm further. “We now have 2.4 million images that have been enriched, and we will use these in the near future as we move beyond the proof-of-concept stage.
He is looking for as for as many partners as possible to help collect enriched images and develop this into a real tool for use in hospitals worldwide on clinical patients. “We have joined forces with several hospitals but continue looking for further collaborations.
“We need, just like self-driving cars, not just thousands, but tens of thousands or millions of images to develop this.”
Dr. Jans declared receiving speaker fees from UCB, AbbVie, Lilly, and Novartis, and that he is cofounder of a future spin-off of Ghent University RheumaFinder. Dr. Poddubnyy and Dr. Boesen declared no relevant disclosures.
Erosions and ankylosis in patients with sacroiliitis are detectable to a high degree of accuracy on CT images using an artificial intelligence (AI)–based algorithm, according to research presented at the 13th International Congress on Spondyloarthritides.
Lennart Jans, MD, head of clinics in musculoskeletal imaging in the department of radiology at Ghent (Belgium) University Hospital, shared data on the development and validation of the algorithm for automatic detection of erosion and ankylosis on CT images of the sacroiliac (SI) joints.
“Essentially, in terms of statistics, this AI algorithm has 95% sensitivity for picking up erosions in patients with clinical symptoms of sacroiliitis, and if this is further developed as a tool, it could aid detection in people with erosions that would otherwise go undetected and undiagnosed,” Dr. Jans said in an interview, stressing that the results were still preliminary.
“We want to move from reporting one patient at a time to a system that detects and helps to diagnose larger numbers of patients and makes a larger impact on patient outcomes.”
He stressed that, with thousands of images per patient, it is an impossible workload for any radiology department to read every image necessary to inform diagnoses, and this is only exacerbated by the shortage of rheumatologists, especially in the United States.
Denis Poddubnyy, MD, head of rheumatology at Charité University Hospital, Berlin, acknowledged that AI has potential to improve the recognition of changes indicative of spondyloarthritis (SpA) on imaging. “A standardized, valid, and reliable detection of those changes is relevant for both diagnosis, including differential diagnosis, and classification of SpA.”
Dr. Poddubnyy added that the AI-based algorithm developed by Dr. Jans and associates is designed to detect very specific SpA structural changes in the SI joints on CT. “CT is usually applied in the clinical practice after MRI ... normally in cases where MRI does not provide conclusive results,” he said. Since MRI scans have also been recently used to develop an AI-based algorithm for the detection of active inflammation – not captured by CT – and structural changes in SI joints, he noted that the “generated data on CT should be, therefore, seen in a broader context toward standardization of imaging findings detection.”
Proof-of-concept findings are due for scale-up
Dr. Jans acknowledged that the current data only establish proof of concept. Among the study’s 145 patients, 60% were used for training the AI algorithm and 40% for testing it. All patients who had clinical symptoms of sacroiliitis and had undergone a SI joint CT scan were included from two hospitals: Ghent University Hospital and the University of Alberta Hospital, Edmonton. The majority of patients were female (81 of 145). They had a mean age of 40 years, 84 had diagnosed axial SpA, 15 had mechanical back pain, and 46 did not have a final diagnosis.
CT images were examined by three independent and blinded radiologists who annotated erosions more than 1 mm and ankylosis more than 2 mm, while a type of AI algorithm known as a neural network pipeline was developed to segment the SI joints and detect structural lesions.
In the first instance, Dr. Jans explained, examination of CT images using the AI algorithm from patients who enter the hospital for other reasons, such as trauma, rheumatic diseases, kidney stones, or appendicitis, might lead to the detection of otherwise unknown erosions. “Often patients have complained of backache for years, seeing various physiotherapists and similar, but had no idea what might be causing it,” he said. “We just don’t have the time for examining all the thousands of images separately. We need some kind of aid here. We need an extra pair of eyes. This is what AI software does.”
Dr. Jans said rheumatologists who ultimately want to detect and diagnose patients with SI erosions want to reduce the false-negative findings. “They want the system to pick up all the patients who have erosions. Here, the most important parameter is sensitivity, and we find that our algorithm shows a very high sensitivity. Optimization of the AI algorithm to reduce false negatives resulted in a sensitivity of 95% for detection of erosions on CT of the sacroiliac joints on a patient level.”
While overall accuracy was over 90%, Dr. Jans acknowledged that the algorithm was run in a relatively select population of dedicated CT scans of the joints. He is also aware that a good AI algorithm needs to work well across locations and populations. “If you make something within your institution alone, it will not work in a hospital on the other side of the street.”
However, he added, the researchers used images from four different CT scanners and images from two different institutions – one in Canada and their own in Belgium, providing a case mix that makes their algorithm more refined.
Next step: Test in an unselected population
When asked to comment on the study, Mikael Boesen, MD, PhD, of Bispebjerg and Frederiksberg Hospital, Copenhagen, congratulated Dr. Jans on the work and remarked that he found the research potentially clinically useful.
“The next steps would be to test the performance of the model in an unselected population of patients who have CT scans of the abdomen for other reasons to test the model’s ability to flag potential SI joint disease to the reader, which is often overlooked, as well as [to see] how the model performs in larger datasets from other hospitals, vendors, and CT-reconstruction algorithms.”
Finally, Dr. Boesen pointed out that it would be interesting to see if the AI algorithm can detect different reasons for erosions. “Especially [for] separation between mechanical and inflammatory courses. This could potentially be done by automatically mapping the location of the erosions in the SI joints.”
Dr. Jans has now opened up the project to other radiologists to collaborate and provide images to train and test the algorithm further. “We now have 2.4 million images that have been enriched, and we will use these in the near future as we move beyond the proof-of-concept stage.
He is looking for as for as many partners as possible to help collect enriched images and develop this into a real tool for use in hospitals worldwide on clinical patients. “We have joined forces with several hospitals but continue looking for further collaborations.
“We need, just like self-driving cars, not just thousands, but tens of thousands or millions of images to develop this.”
Dr. Jans declared receiving speaker fees from UCB, AbbVie, Lilly, and Novartis, and that he is cofounder of a future spin-off of Ghent University RheumaFinder. Dr. Poddubnyy and Dr. Boesen declared no relevant disclosures.
Erosions and ankylosis in patients with sacroiliitis are detectable to a high degree of accuracy on CT images using an artificial intelligence (AI)–based algorithm, according to research presented at the 13th International Congress on Spondyloarthritides.
Lennart Jans, MD, head of clinics in musculoskeletal imaging in the department of radiology at Ghent (Belgium) University Hospital, shared data on the development and validation of the algorithm for automatic detection of erosion and ankylosis on CT images of the sacroiliac (SI) joints.
“Essentially, in terms of statistics, this AI algorithm has 95% sensitivity for picking up erosions in patients with clinical symptoms of sacroiliitis, and if this is further developed as a tool, it could aid detection in people with erosions that would otherwise go undetected and undiagnosed,” Dr. Jans said in an interview, stressing that the results were still preliminary.
“We want to move from reporting one patient at a time to a system that detects and helps to diagnose larger numbers of patients and makes a larger impact on patient outcomes.”
He stressed that, with thousands of images per patient, it is an impossible workload for any radiology department to read every image necessary to inform diagnoses, and this is only exacerbated by the shortage of rheumatologists, especially in the United States.
Denis Poddubnyy, MD, head of rheumatology at Charité University Hospital, Berlin, acknowledged that AI has potential to improve the recognition of changes indicative of spondyloarthritis (SpA) on imaging. “A standardized, valid, and reliable detection of those changes is relevant for both diagnosis, including differential diagnosis, and classification of SpA.”
Dr. Poddubnyy added that the AI-based algorithm developed by Dr. Jans and associates is designed to detect very specific SpA structural changes in the SI joints on CT. “CT is usually applied in the clinical practice after MRI ... normally in cases where MRI does not provide conclusive results,” he said. Since MRI scans have also been recently used to develop an AI-based algorithm for the detection of active inflammation – not captured by CT – and structural changes in SI joints, he noted that the “generated data on CT should be, therefore, seen in a broader context toward standardization of imaging findings detection.”
Proof-of-concept findings are due for scale-up
Dr. Jans acknowledged that the current data only establish proof of concept. Among the study’s 145 patients, 60% were used for training the AI algorithm and 40% for testing it. All patients who had clinical symptoms of sacroiliitis and had undergone a SI joint CT scan were included from two hospitals: Ghent University Hospital and the University of Alberta Hospital, Edmonton. The majority of patients were female (81 of 145). They had a mean age of 40 years, 84 had diagnosed axial SpA, 15 had mechanical back pain, and 46 did not have a final diagnosis.
CT images were examined by three independent and blinded radiologists who annotated erosions more than 1 mm and ankylosis more than 2 mm, while a type of AI algorithm known as a neural network pipeline was developed to segment the SI joints and detect structural lesions.
In the first instance, Dr. Jans explained, examination of CT images using the AI algorithm from patients who enter the hospital for other reasons, such as trauma, rheumatic diseases, kidney stones, or appendicitis, might lead to the detection of otherwise unknown erosions. “Often patients have complained of backache for years, seeing various physiotherapists and similar, but had no idea what might be causing it,” he said. “We just don’t have the time for examining all the thousands of images separately. We need some kind of aid here. We need an extra pair of eyes. This is what AI software does.”
Dr. Jans said rheumatologists who ultimately want to detect and diagnose patients with SI erosions want to reduce the false-negative findings. “They want the system to pick up all the patients who have erosions. Here, the most important parameter is sensitivity, and we find that our algorithm shows a very high sensitivity. Optimization of the AI algorithm to reduce false negatives resulted in a sensitivity of 95% for detection of erosions on CT of the sacroiliac joints on a patient level.”
While overall accuracy was over 90%, Dr. Jans acknowledged that the algorithm was run in a relatively select population of dedicated CT scans of the joints. He is also aware that a good AI algorithm needs to work well across locations and populations. “If you make something within your institution alone, it will not work in a hospital on the other side of the street.”
However, he added, the researchers used images from four different CT scanners and images from two different institutions – one in Canada and their own in Belgium, providing a case mix that makes their algorithm more refined.
Next step: Test in an unselected population
When asked to comment on the study, Mikael Boesen, MD, PhD, of Bispebjerg and Frederiksberg Hospital, Copenhagen, congratulated Dr. Jans on the work and remarked that he found the research potentially clinically useful.
“The next steps would be to test the performance of the model in an unselected population of patients who have CT scans of the abdomen for other reasons to test the model’s ability to flag potential SI joint disease to the reader, which is often overlooked, as well as [to see] how the model performs in larger datasets from other hospitals, vendors, and CT-reconstruction algorithms.”
Finally, Dr. Boesen pointed out that it would be interesting to see if the AI algorithm can detect different reasons for erosions. “Especially [for] separation between mechanical and inflammatory courses. This could potentially be done by automatically mapping the location of the erosions in the SI joints.”
Dr. Jans has now opened up the project to other radiologists to collaborate and provide images to train and test the algorithm further. “We now have 2.4 million images that have been enriched, and we will use these in the near future as we move beyond the proof-of-concept stage.
He is looking for as for as many partners as possible to help collect enriched images and develop this into a real tool for use in hospitals worldwide on clinical patients. “We have joined forces with several hospitals but continue looking for further collaborations.
“We need, just like self-driving cars, not just thousands, but tens of thousands or millions of images to develop this.”
Dr. Jans declared receiving speaker fees from UCB, AbbVie, Lilly, and Novartis, and that he is cofounder of a future spin-off of Ghent University RheumaFinder. Dr. Poddubnyy and Dr. Boesen declared no relevant disclosures.
FROM THE 2022 SPA CONGRESS
Should patients with PsA or ankylosing spondylitis with axial disease be ‘lumped’ or ‘split’?
A new study provides evidence that two conditions that fall under the umbrella of spondyloarthritis – isolated axial disease in patients with psoriatic arthritis (PsA) and isolated axial disease in patients with ankylosing spondylitis (AS) accompanied by psoriasis – are different clinical entities and may need different treatments. These relatively rare rheumatologic conditions, defined by their back involvement, have considerable clinical overlap and are often lumped together under the label axial spondyloarthritis.
This is a hot topic and current matter of debate within the scientific community: Are axial PsA and axial AS two separate diseases or just two phenotypes under the spondyloarthritis umbrella? said Fabian Proft, MD, a rheumatologist and researcher at Charité Universitätsmedizin Berlin, commenting on the new study, which was published online in Annals of the Rheumatic Diseases.

Both conditions belong to the spectrum of spondyloarthritis, but with varying viewpoints on nomenclature. They have intersections and overlaps, but not all treatments are equally effective for both. “We need to better understand their differences and similarities,” Dr. Proft said, adding that the new study is noteworthy for the size of the population included, its long-term follow-up data, and the researchers’ depth of experience treating these patients.
The researchers are based at the University of Toronto, which has separate clinics dedicated to PsA and to AS, said Dafna D. Gladman, MD, professor of medicine at the university, codirector of the PsA clinic, and corresponding author for the new study. The two clinics follow the same standardized protocols, including clinical, radiographic, genetic, and laboratory assessments. Even though the patients present quite similarly, she credits referring physicians for recognizing the distinctions by their referrals to the PsA or AS clinic.
According to previous research, pure axial PsA, without peripheral involvement, is rare, affecting about 2%-5% of patients with PsA. For this study, an observational cohort of 1,576 patients from the PsA clinic included 31% (n = 495) with axial disease, 2% (n = 32) with isolated axial PsA, and 29% (n = 463) with both axial and peripheral involvement. A total of 25 of the patients with isolated axial PsA ultimately developed peripheral disease by their most recent clinic follow-up visit. In a second cohort of 1,688 patients with AS, nearly 5% (n = 68) had isolated axial disease with psoriasis.

“In our logistic regression analysis, isolated axial PsA was found to be a different clinical entity than isolated AS with psoriasis. They are not the same patients,” Dr. Gladman said. The patients with isolated axial PsA were older at diagnosis, more likely to have psoriatic nail lesions, and less likely to have inflammatory back pain than were patients with isolated axial AS and accompanying psoriasis.
When interviewed in early September, Dr. Gladman was preparing to fly to Ghent, Belgium, to participate in a debate at the International Congress on Spondyloarthritides, taking the pro position on the thesis: Is axial inflammation in PsA distinct from axial spondyloarthritis? Taking the con position was to be Robert Landewé, MD, PhD, of Amsterdam University Medical Center in the Netherlands.
“This is an old debate, splitters versus lumpers,” Dr. Gladman told this news organization. “My message is that when you place patients in more homogeneous groups, you can learn more and perhaps find better opportunities for treating their disease.” For example, even with the similarities, do these patients need to be treated with different medications? Medications for psoriasis, including those targeting the interleukin-23 cytokine, may not be effective for AS, but patients with axial PsA may not get them because of the association with axial AS.
“Now is the opportunity to really understand what – if any – are the differences between various components of this disease group. If you lump people together, you may miss the forest for the trees,” Dr. Gladman said. “If, at the end of the day, we find out these patients essentially are the same, I will lump. But until we have proved that there are no important differences, I will split.” She added that it is important for practicing rheumatologists to make the correct diagnosis so that they know to access certain drugs.
Dr. Proft credited Dr. Gladman and colleagues’ study for adding another piece of the puzzle to better understand differences and similarities for these two axial diseases. He noted, however, that the study did not include MRI scans for every participating patient, which could have given a deeper picture.
“International efforts are being made to recruit patients for a multinational, multicenter study of axial involvement in PsA,” which will include MRI data, Dr. Gladman said. She and Dr. Proft are both part of AXIS, the Axial Involvement in Psoriatic Arthritis cohort, now recruiting patients for such a study. AXIS is a joint project of the Assessment of SpondyloArthritis international Society and the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis.
“We don’t have final answers yet, although we have given evidence to support the differences.” The proof is in the pudding, she said, and that pudding will be the clinical trials.
The University of Toronto Psoriatic Arthritis Program is supported by a grant from the Krembil Foundation. The study authors declared no competing interests. Dr. Proft reported receiving research support from Novartis, Eli Lilly, and UCB, and fees for consulting and serving on speakers bureaus from AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Eli Lilly, Hexal, Janssen, Merck Sharp & Dohme, Novartis, Pfizer, Roche, and UCB.
A new study provides evidence that two conditions that fall under the umbrella of spondyloarthritis – isolated axial disease in patients with psoriatic arthritis (PsA) and isolated axial disease in patients with ankylosing spondylitis (AS) accompanied by psoriasis – are different clinical entities and may need different treatments. These relatively rare rheumatologic conditions, defined by their back involvement, have considerable clinical overlap and are often lumped together under the label axial spondyloarthritis.
This is a hot topic and current matter of debate within the scientific community: Are axial PsA and axial AS two separate diseases or just two phenotypes under the spondyloarthritis umbrella? said Fabian Proft, MD, a rheumatologist and researcher at Charité Universitätsmedizin Berlin, commenting on the new study, which was published online in Annals of the Rheumatic Diseases.
Both conditions belong to the spectrum of spondyloarthritis, but with varying viewpoints on nomenclature. They have intersections and overlaps, but not all treatments are equally effective for both. “We need to better understand their differences and similarities,” Dr. Proft said, adding that the new study is noteworthy for the size of the population included, its long-term follow-up data, and the researchers’ depth of experience treating these patients.
The researchers are based at the University of Toronto, which has separate clinics dedicated to PsA and to AS, said Dafna D. Gladman, MD, professor of medicine at the university, codirector of the PsA clinic, and corresponding author for the new study. The two clinics follow the same standardized protocols, including clinical, radiographic, genetic, and laboratory assessments. Even though the patients present quite similarly, she credits referring physicians for recognizing the distinctions by their referrals to the PsA or AS clinic.
According to previous research, pure axial PsA, without peripheral involvement, is rare, affecting about 2%-5% of patients with PsA. For this study, an observational cohort of 1,576 patients from the PsA clinic included 31% (n = 495) with axial disease, 2% (n = 32) with isolated axial PsA, and 29% (n = 463) with both axial and peripheral involvement. A total of 25 of the patients with isolated axial PsA ultimately developed peripheral disease by their most recent clinic follow-up visit. In a second cohort of 1,688 patients with AS, nearly 5% (n = 68) had isolated axial disease with psoriasis.
“In our logistic regression analysis, isolated axial PsA was found to be a different clinical entity than isolated AS with psoriasis. They are not the same patients,” Dr. Gladman said. The patients with isolated axial PsA were older at diagnosis, more likely to have psoriatic nail lesions, and less likely to have inflammatory back pain than were patients with isolated axial AS and accompanying psoriasis.
When interviewed in early September, Dr. Gladman was preparing to fly to Ghent, Belgium, to participate in a debate at the International Congress on Spondyloarthritides, taking the pro position on the thesis: Is axial inflammation in PsA distinct from axial spondyloarthritis? Taking the con position was to be Robert Landewé, MD, PhD, of Amsterdam University Medical Center in the Netherlands.
“This is an old debate, splitters versus lumpers,” Dr. Gladman told this news organization. “My message is that when you place patients in more homogeneous groups, you can learn more and perhaps find better opportunities for treating their disease.” For example, even with the similarities, do these patients need to be treated with different medications? Medications for psoriasis, including those targeting the interleukin-23 cytokine, may not be effective for AS, but patients with axial PsA may not get them because of the association with axial AS.
“Now is the opportunity to really understand what – if any – are the differences between various components of this disease group. If you lump people together, you may miss the forest for the trees,” Dr. Gladman said. “If, at the end of the day, we find out these patients essentially are the same, I will lump. But until we have proved that there are no important differences, I will split.” She added that it is important for practicing rheumatologists to make the correct diagnosis so that they know to access certain drugs.
Dr. Proft credited Dr. Gladman and colleagues’ study for adding another piece of the puzzle to better understand differences and similarities for these two axial diseases. He noted, however, that the study did not include MRI scans for every participating patient, which could have given a deeper picture.
“International efforts are being made to recruit patients for a multinational, multicenter study of axial involvement in PsA,” which will include MRI data, Dr. Gladman said. She and Dr. Proft are both part of AXIS, the Axial Involvement in Psoriatic Arthritis cohort, now recruiting patients for such a study. AXIS is a joint project of the Assessment of SpondyloArthritis international Society and the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis.
“We don’t have final answers yet, although we have given evidence to support the differences.” The proof is in the pudding, she said, and that pudding will be the clinical trials.
The University of Toronto Psoriatic Arthritis Program is supported by a grant from the Krembil Foundation. The study authors declared no competing interests. Dr. Proft reported receiving research support from Novartis, Eli Lilly, and UCB, and fees for consulting and serving on speakers bureaus from AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Eli Lilly, Hexal, Janssen, Merck Sharp & Dohme, Novartis, Pfizer, Roche, and UCB.
A new study provides evidence that two conditions that fall under the umbrella of spondyloarthritis – isolated axial disease in patients with psoriatic arthritis (PsA) and isolated axial disease in patients with ankylosing spondylitis (AS) accompanied by psoriasis – are different clinical entities and may need different treatments. These relatively rare rheumatologic conditions, defined by their back involvement, have considerable clinical overlap and are often lumped together under the label axial spondyloarthritis.
This is a hot topic and current matter of debate within the scientific community: Are axial PsA and axial AS two separate diseases or just two phenotypes under the spondyloarthritis umbrella? said Fabian Proft, MD, a rheumatologist and researcher at Charité Universitätsmedizin Berlin, commenting on the new study, which was published online in Annals of the Rheumatic Diseases.
Both conditions belong to the spectrum of spondyloarthritis, but with varying viewpoints on nomenclature. They have intersections and overlaps, but not all treatments are equally effective for both. “We need to better understand their differences and similarities,” Dr. Proft said, adding that the new study is noteworthy for the size of the population included, its long-term follow-up data, and the researchers’ depth of experience treating these patients.
The researchers are based at the University of Toronto, which has separate clinics dedicated to PsA and to AS, said Dafna D. Gladman, MD, professor of medicine at the university, codirector of the PsA clinic, and corresponding author for the new study. The two clinics follow the same standardized protocols, including clinical, radiographic, genetic, and laboratory assessments. Even though the patients present quite similarly, she credits referring physicians for recognizing the distinctions by their referrals to the PsA or AS clinic.
According to previous research, pure axial PsA, without peripheral involvement, is rare, affecting about 2%-5% of patients with PsA. For this study, an observational cohort of 1,576 patients from the PsA clinic included 31% (n = 495) with axial disease, 2% (n = 32) with isolated axial PsA, and 29% (n = 463) with both axial and peripheral involvement. A total of 25 of the patients with isolated axial PsA ultimately developed peripheral disease by their most recent clinic follow-up visit. In a second cohort of 1,688 patients with AS, nearly 5% (n = 68) had isolated axial disease with psoriasis.
“In our logistic regression analysis, isolated axial PsA was found to be a different clinical entity than isolated AS with psoriasis. They are not the same patients,” Dr. Gladman said. The patients with isolated axial PsA were older at diagnosis, more likely to have psoriatic nail lesions, and less likely to have inflammatory back pain than were patients with isolated axial AS and accompanying psoriasis.
When interviewed in early September, Dr. Gladman was preparing to fly to Ghent, Belgium, to participate in a debate at the International Congress on Spondyloarthritides, taking the pro position on the thesis: Is axial inflammation in PsA distinct from axial spondyloarthritis? Taking the con position was to be Robert Landewé, MD, PhD, of Amsterdam University Medical Center in the Netherlands.
“This is an old debate, splitters versus lumpers,” Dr. Gladman told this news organization. “My message is that when you place patients in more homogeneous groups, you can learn more and perhaps find better opportunities for treating their disease.” For example, even with the similarities, do these patients need to be treated with different medications? Medications for psoriasis, including those targeting the interleukin-23 cytokine, may not be effective for AS, but patients with axial PsA may not get them because of the association with axial AS.
“Now is the opportunity to really understand what – if any – are the differences between various components of this disease group. If you lump people together, you may miss the forest for the trees,” Dr. Gladman said. “If, at the end of the day, we find out these patients essentially are the same, I will lump. But until we have proved that there are no important differences, I will split.” She added that it is important for practicing rheumatologists to make the correct diagnosis so that they know to access certain drugs.
Dr. Proft credited Dr. Gladman and colleagues’ study for adding another piece of the puzzle to better understand differences and similarities for these two axial diseases. He noted, however, that the study did not include MRI scans for every participating patient, which could have given a deeper picture.
“International efforts are being made to recruit patients for a multinational, multicenter study of axial involvement in PsA,” which will include MRI data, Dr. Gladman said. She and Dr. Proft are both part of AXIS, the Axial Involvement in Psoriatic Arthritis cohort, now recruiting patients for such a study. AXIS is a joint project of the Assessment of SpondyloArthritis international Society and the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis.
“We don’t have final answers yet, although we have given evidence to support the differences.” The proof is in the pudding, she said, and that pudding will be the clinical trials.
The University of Toronto Psoriatic Arthritis Program is supported by a grant from the Krembil Foundation. The study authors declared no competing interests. Dr. Proft reported receiving research support from Novartis, Eli Lilly, and UCB, and fees for consulting and serving on speakers bureaus from AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Eli Lilly, Hexal, Janssen, Merck Sharp & Dohme, Novartis, Pfizer, Roche, and UCB.
FROM ANNALS OF THE RHEUMATIC DISEASES
One fish, two fish, are good fish for you ... fish
Good news for pregnant women; bad news for fish
As soon as women find out they’re pregnant, doctors recommend they give up smoking, drinking, and eating certain types of fish. That last item may need to be reconsidered, since a recent study supports the idea that it doesn’t matter what type of fish pregnant women are eating, as long as they’re eating it.

Researchers collected data from two different studies that reviewed the mercury levels of mothers from Bristol, England, and the Seychelles, a island chain off East Africa where “fish consumption is high and prenatal mercury levels are 10 times higher than in the [United States],” they said in NeuroToxicology.
Those data showed that the mercury levels had no adverse effects on child development as long as the mother ate fish. The nutrients and vitamins in the fish – vitamin D, long-chain fatty acids, selenium, and iodine – provide protection against mercury. There’s also the already-known benefits to eyesight and intellectual abilities that have been associated with fish consumption.
This analysis goes starkly against the grain of what is commonly recommended to expectant mothers, which is to cut out fish altogether. The researchers suggested that governments should review and change those recommendations to focus on the benefits instead.
As long as women follow the researchers’ recommendation to eat “at least two portions of fish a week, one of which should be oily,” they may not have to lay off on the sushi after all.
We’ll show our gut worms the world
Never let it be said that mankind is not a generous species. Sure, we could maybe be kinder to our fellow human beings, maybe declare a little less war on each other, but for the past 50,000 years, we’ve been giving a free ride to millions upon millions to one of mankind’s closest companions: the whipworm.

This revelation into human kindness comes from Denmark, where researchers from Copenhagen conducted a genetic analysis of ancient preserved whipworm eggs found in old Viking and Norse settlements, some of which date back over 2,000 years. In normal conditions genetic material wouldn’t last very long, but these were Viking whipworms eggs with tiny little horned helmets, so the DNA within has remained unchanged. Or it may be the tough chitinous exterior of the eggs protecting the DNA from degrading, combined with their preservation in moist soil.
Once they had their Viking whipworm DNA, the researchers compared it with whipworm DNA from all over the world, tracing its history as it followed mankind from Africa. And it’s been a while: We brought whipworms with us during our initial migration into Asia and Europe over 50,000 years ago. When the Bering land bridge opened up and humanity moved into the Americas, the worms came as well.
This is all possible because the whipworm goes about its parasitic business quietly and cleverly. It mostly sits harmlessly in our digestive systems, producing thousands of eggs a day that get expelled through poop and picked up by another host (human or otherwise); whipworms only cause disease in those with compromised immune systems.
The researchers noted that their study, the first complete genetic analysis of the whipworm, could help combat the parasite, which to this day infects hundred of millions who don’t have access to modern medicine or sanitary conditions. Hopefully, though, the days of free rides will soon be over for the whipworm. After all, if we have to pay hundreds or thousands of dollars to visit other countries, it’s only fair that our parasites do as well.
From zero to vasectomy in 6.7 seconds
There’s an old saying that you’ve probably heard: When life gives you lemons, make lemonade. It’s meant to encourage optimism in the face of adversity. Then there’s the new saying we just made up: When life gives you a power outage, plug your surgical instruments into an electric pickup.

That’s what Dr. Christopher Yang did, and now we’re making the urologist from Austin, Tex., famous by sharing his surgical/electrical adventure with all 17 of LOTME’s regular readers. That’s some serious lemonade.
Dr. Yang’s tale begins when the electricity went out at his clinic, seemingly forcing him to cancel or reschedule several surgical procedures. Not so fast. Dr. Yang happens to own a Rivian R1T, an electric pickup truck that has four power outlets. A staff member suggested plugging the surgical instruments into the truck and, surprisingly, one of the day’s patients agreed to go ahead with his vasectomy.
“We were fortunate that my normal parking spot is close enough to a patient room to run an extension cord,” Dr. Yang said on TheDrive.com. That extension cord was attached to an electrocautery device, with a handheld device available as backup, and “after we were done, I told his family. We all had a good laugh together too,” Dr. Yang told radio station WGLT in Normal, Ill.
To us, anyway, this opens up all sorts of alternative energy possibilities. Can a windmill power a liposuction? Is a gerbil running in a wheel enough to do a colonoscopy? How many potatoes do you need to keep an EHR going?
Learning through random acts of not-exactly noisiness
First things first. Transcranial random noise stimulation (tRNS) is not really noise in the auditory sense of the word. For some people with learning disabilities, though, it can actually be very helpful. The technology, which uses electrodes attached to the head so a weak current can pass through specific parts of the brain, may help those with learning disabilities, perhaps even those with brain injuries and visual deficits, learn, said Dr. Onno van der Groen of Edith Cowan University in Perth, Australia.

“When you add this type of stimulation during learning, you get better performance, faster learning and better attention afterwards as well,” he said in a statement from the university.
The researchers say that tRNS can allow the brain to form new connections and pathways, which in turn help a person learn more effectively. “If you do 10 sessions of a visual perception task with the tRNS and then come back and do it again without it, you’ll find you perform better than the control group who hasn’t used it,” Dr. van der Groen noted.
Can this also work for the average person? It’s possible, but tRNS didn’t seem to improve the math skills of a top-level mathematician who underwent the process, according to a case study that Dr. van der Groen mentioned.
This line of work is still pretty new, though, so researchers don’t have all the answers yet. As always, we’re rooting for you, science!
Good news for pregnant women; bad news for fish
As soon as women find out they’re pregnant, doctors recommend they give up smoking, drinking, and eating certain types of fish. That last item may need to be reconsidered, since a recent study supports the idea that it doesn’t matter what type of fish pregnant women are eating, as long as they’re eating it.
Researchers collected data from two different studies that reviewed the mercury levels of mothers from Bristol, England, and the Seychelles, a island chain off East Africa where “fish consumption is high and prenatal mercury levels are 10 times higher than in the [United States],” they said in NeuroToxicology.
Those data showed that the mercury levels had no adverse effects on child development as long as the mother ate fish. The nutrients and vitamins in the fish – vitamin D, long-chain fatty acids, selenium, and iodine – provide protection against mercury. There’s also the already-known benefits to eyesight and intellectual abilities that have been associated with fish consumption.
This analysis goes starkly against the grain of what is commonly recommended to expectant mothers, which is to cut out fish altogether. The researchers suggested that governments should review and change those recommendations to focus on the benefits instead.
As long as women follow the researchers’ recommendation to eat “at least two portions of fish a week, one of which should be oily,” they may not have to lay off on the sushi after all.
We’ll show our gut worms the world
Never let it be said that mankind is not a generous species. Sure, we could maybe be kinder to our fellow human beings, maybe declare a little less war on each other, but for the past 50,000 years, we’ve been giving a free ride to millions upon millions to one of mankind’s closest companions: the whipworm.
This revelation into human kindness comes from Denmark, where researchers from Copenhagen conducted a genetic analysis of ancient preserved whipworm eggs found in old Viking and Norse settlements, some of which date back over 2,000 years. In normal conditions genetic material wouldn’t last very long, but these were Viking whipworms eggs with tiny little horned helmets, so the DNA within has remained unchanged. Or it may be the tough chitinous exterior of the eggs protecting the DNA from degrading, combined with their preservation in moist soil.
Once they had their Viking whipworm DNA, the researchers compared it with whipworm DNA from all over the world, tracing its history as it followed mankind from Africa. And it’s been a while: We brought whipworms with us during our initial migration into Asia and Europe over 50,000 years ago. When the Bering land bridge opened up and humanity moved into the Americas, the worms came as well.
This is all possible because the whipworm goes about its parasitic business quietly and cleverly. It mostly sits harmlessly in our digestive systems, producing thousands of eggs a day that get expelled through poop and picked up by another host (human or otherwise); whipworms only cause disease in those with compromised immune systems.
The researchers noted that their study, the first complete genetic analysis of the whipworm, could help combat the parasite, which to this day infects hundred of millions who don’t have access to modern medicine or sanitary conditions. Hopefully, though, the days of free rides will soon be over for the whipworm. After all, if we have to pay hundreds or thousands of dollars to visit other countries, it’s only fair that our parasites do as well.
From zero to vasectomy in 6.7 seconds
There’s an old saying that you’ve probably heard: When life gives you lemons, make lemonade. It’s meant to encourage optimism in the face of adversity. Then there’s the new saying we just made up: When life gives you a power outage, plug your surgical instruments into an electric pickup.
That’s what Dr. Christopher Yang did, and now we’re making the urologist from Austin, Tex., famous by sharing his surgical/electrical adventure with all 17 of LOTME’s regular readers. That’s some serious lemonade.
Dr. Yang’s tale begins when the electricity went out at his clinic, seemingly forcing him to cancel or reschedule several surgical procedures. Not so fast. Dr. Yang happens to own a Rivian R1T, an electric pickup truck that has four power outlets. A staff member suggested plugging the surgical instruments into the truck and, surprisingly, one of the day’s patients agreed to go ahead with his vasectomy.
“We were fortunate that my normal parking spot is close enough to a patient room to run an extension cord,” Dr. Yang said on TheDrive.com. That extension cord was attached to an electrocautery device, with a handheld device available as backup, and “after we were done, I told his family. We all had a good laugh together too,” Dr. Yang told radio station WGLT in Normal, Ill.
To us, anyway, this opens up all sorts of alternative energy possibilities. Can a windmill power a liposuction? Is a gerbil running in a wheel enough to do a colonoscopy? How many potatoes do you need to keep an EHR going?
Learning through random acts of not-exactly noisiness
First things first. Transcranial random noise stimulation (tRNS) is not really noise in the auditory sense of the word. For some people with learning disabilities, though, it can actually be very helpful. The technology, which uses electrodes attached to the head so a weak current can pass through specific parts of the brain, may help those with learning disabilities, perhaps even those with brain injuries and visual deficits, learn, said Dr. Onno van der Groen of Edith Cowan University in Perth, Australia.
“When you add this type of stimulation during learning, you get better performance, faster learning and better attention afterwards as well,” he said in a statement from the university.
The researchers say that tRNS can allow the brain to form new connections and pathways, which in turn help a person learn more effectively. “If you do 10 sessions of a visual perception task with the tRNS and then come back and do it again without it, you’ll find you perform better than the control group who hasn’t used it,” Dr. van der Groen noted.
Can this also work for the average person? It’s possible, but tRNS didn’t seem to improve the math skills of a top-level mathematician who underwent the process, according to a case study that Dr. van der Groen mentioned.
This line of work is still pretty new, though, so researchers don’t have all the answers yet. As always, we’re rooting for you, science!
Good news for pregnant women; bad news for fish
As soon as women find out they’re pregnant, doctors recommend they give up smoking, drinking, and eating certain types of fish. That last item may need to be reconsidered, since a recent study supports the idea that it doesn’t matter what type of fish pregnant women are eating, as long as they’re eating it.
Researchers collected data from two different studies that reviewed the mercury levels of mothers from Bristol, England, and the Seychelles, a island chain off East Africa where “fish consumption is high and prenatal mercury levels are 10 times higher than in the [United States],” they said in NeuroToxicology.
Those data showed that the mercury levels had no adverse effects on child development as long as the mother ate fish. The nutrients and vitamins in the fish – vitamin D, long-chain fatty acids, selenium, and iodine – provide protection against mercury. There’s also the already-known benefits to eyesight and intellectual abilities that have been associated with fish consumption.
This analysis goes starkly against the grain of what is commonly recommended to expectant mothers, which is to cut out fish altogether. The researchers suggested that governments should review and change those recommendations to focus on the benefits instead.
As long as women follow the researchers’ recommendation to eat “at least two portions of fish a week, one of which should be oily,” they may not have to lay off on the sushi after all.
We’ll show our gut worms the world
Never let it be said that mankind is not a generous species. Sure, we could maybe be kinder to our fellow human beings, maybe declare a little less war on each other, but for the past 50,000 years, we’ve been giving a free ride to millions upon millions to one of mankind’s closest companions: the whipworm.
This revelation into human kindness comes from Denmark, where researchers from Copenhagen conducted a genetic analysis of ancient preserved whipworm eggs found in old Viking and Norse settlements, some of which date back over 2,000 years. In normal conditions genetic material wouldn’t last very long, but these were Viking whipworms eggs with tiny little horned helmets, so the DNA within has remained unchanged. Or it may be the tough chitinous exterior of the eggs protecting the DNA from degrading, combined with their preservation in moist soil.
Once they had their Viking whipworm DNA, the researchers compared it with whipworm DNA from all over the world, tracing its history as it followed mankind from Africa. And it’s been a while: We brought whipworms with us during our initial migration into Asia and Europe over 50,000 years ago. When the Bering land bridge opened up and humanity moved into the Americas, the worms came as well.
This is all possible because the whipworm goes about its parasitic business quietly and cleverly. It mostly sits harmlessly in our digestive systems, producing thousands of eggs a day that get expelled through poop and picked up by another host (human or otherwise); whipworms only cause disease in those with compromised immune systems.
The researchers noted that their study, the first complete genetic analysis of the whipworm, could help combat the parasite, which to this day infects hundred of millions who don’t have access to modern medicine or sanitary conditions. Hopefully, though, the days of free rides will soon be over for the whipworm. After all, if we have to pay hundreds or thousands of dollars to visit other countries, it’s only fair that our parasites do as well.
From zero to vasectomy in 6.7 seconds
There’s an old saying that you’ve probably heard: When life gives you lemons, make lemonade. It’s meant to encourage optimism in the face of adversity. Then there’s the new saying we just made up: When life gives you a power outage, plug your surgical instruments into an electric pickup.
That’s what Dr. Christopher Yang did, and now we’re making the urologist from Austin, Tex., famous by sharing his surgical/electrical adventure with all 17 of LOTME’s regular readers. That’s some serious lemonade.
Dr. Yang’s tale begins when the electricity went out at his clinic, seemingly forcing him to cancel or reschedule several surgical procedures. Not so fast. Dr. Yang happens to own a Rivian R1T, an electric pickup truck that has four power outlets. A staff member suggested plugging the surgical instruments into the truck and, surprisingly, one of the day’s patients agreed to go ahead with his vasectomy.
“We were fortunate that my normal parking spot is close enough to a patient room to run an extension cord,” Dr. Yang said on TheDrive.com. That extension cord was attached to an electrocautery device, with a handheld device available as backup, and “after we were done, I told his family. We all had a good laugh together too,” Dr. Yang told radio station WGLT in Normal, Ill.
To us, anyway, this opens up all sorts of alternative energy possibilities. Can a windmill power a liposuction? Is a gerbil running in a wheel enough to do a colonoscopy? How many potatoes do you need to keep an EHR going?
Learning through random acts of not-exactly noisiness
First things first. Transcranial random noise stimulation (tRNS) is not really noise in the auditory sense of the word. For some people with learning disabilities, though, it can actually be very helpful. The technology, which uses electrodes attached to the head so a weak current can pass through specific parts of the brain, may help those with learning disabilities, perhaps even those with brain injuries and visual deficits, learn, said Dr. Onno van der Groen of Edith Cowan University in Perth, Australia.
“When you add this type of stimulation during learning, you get better performance, faster learning and better attention afterwards as well,” he said in a statement from the university.
The researchers say that tRNS can allow the brain to form new connections and pathways, which in turn help a person learn more effectively. “If you do 10 sessions of a visual perception task with the tRNS and then come back and do it again without it, you’ll find you perform better than the control group who hasn’t used it,” Dr. van der Groen noted.
Can this also work for the average person? It’s possible, but tRNS didn’t seem to improve the math skills of a top-level mathematician who underwent the process, according to a case study that Dr. van der Groen mentioned.
This line of work is still pretty new, though, so researchers don’t have all the answers yet. As always, we’re rooting for you, science!
Commentary: Better Migraine Outcomes Measures, September 2022
The theme of this month's commentary is alternative outcomes measures for future migraine studies. The traditional outcomes measures, such as headache frequency measured in headache days, have long been considered gold standards when evaluating the efficacy of preventive interventions. When headache conditions are complicated by interictal pain or other symptoms, or when medication overuse adds a higher frequency or greater severity, those traditional measures are somewhat less exact and specific. Meaningful change for patients with higher frequency of attacks, near-continuous pain, or other migraine symptoms is quite different from that for those without these complications.
Ailani and colleagues reviewed post hoc data from the CONQUER trial, a prior study evaluating the safety and efficacy of galcanezumab vs placebo in patients who had previously not benefited from two to four categories of migraine preventive medication. This refractory population was initially noted to have 4.1 fewer headache days per month than patients taking placebo, but the authors now attempted to review these data with a focus on a different measure: total pain burden (TPB). They defined daily TPB as a single composite measure assessing the frequency, duration, and severity of migraine, calculated by multiplying the number of hours of migraine by the maximum daily migraine pain severity score. The monthly TPB was calculated by adding the daily pain burden over the entire month. The Migraine Disability Assessment questionnaire (MIDAS) and Migraine-Specific Quality of Life Questionnaire (MSQ) scores were also included to compare migraine-related disability and quality of life.
The patients who received galcanezumab were noted to have a significantly lower TPB, both in episodic and chronic migraine. Significantly greater reductions in monthly TPB relative to placebo were observed at each individual month as well. The change from baseline TPB was also noted to be significantly improved in the galcanezumab group compared with the placebo group. The reduction in TPB was noted even when migraine-day reductions were accounted for as part of a sensitivity analysis.
Preventive trials for migraine treatment focus primarily on migraine-day reduction, and for many patients with higher-frequency migraine, this measure does not adequately account for their disease-related disability. This unique way of looking at pain as part of a bigger picture is much more significant and meaningful for this patient population. Migraine frequency is still a very important outcomes measure, but it would be wise to add TBP or another measure that looks more globally at disease-related disability, especially when investigating preventive options in patients with chronic migraine.
When considering whether an intervention is helpful, most patients and clinicians follow the headache frequency, severity, or quality-of-life factors. As most patients will readily report, not all "headache-free days" are created equal. Although most people with migraine will experience days with absolutely no headache pain or other migraine-associated symptoms, on many days they will still have some symptoms of migraine. Lee and colleagues attempted to quantify the difference between headache-free days and crystal-clear days.
Most headache studies use the frequency of headache days as a primary or secondary outcome. This study collected data on both headache days and crystal-clear days, using data from a questionnaire-based large South Korean nationwide population study that evaluated headache and sleep. The study questions were validated for migraine and aura, and included: "How many days have you had a headache during the previous 30 days?" and "How many days have you had crystal-clear days without headache during the previous 30 days?" The data were then analyzed and compared with the widespread pain index (criteria for fibromyalgia) as well as sleep duration, sleep quality, depression and anxiety scales, and an allodynia checklist.
A little over 3000 respondents completed the surveys; 1938 had experienced headache over the past year, 170 were classified as having a diagnosis of migraine, and 50 of those were diagnosed with aura as well. Out of the patients with migraine, 97% had "unclear days." This was higher than the rate of those with non-migraine headaches (91%). Nearly all people surveyed had some crystal-clear days (99.4%).
The number of crystal-clear days per 30 days was significantly lower in participants with migraine than in those with non-migraine headache. Participants with migraine also had higher frequencies of cutaneous allodynia, anxiety, and depression. The weekly average sleep duration in participants with migraine did not significantly differ from that in participants with non-migraine headaches. The widespread pain index rate was much higher in those with migraine as well.
Most patients will definitely understand the difference between crystal-clear and unclear headache days. Many of the newer outcomes studies in migraine have started focusing on the most bothersome symptom, as headache pain is far from the only significant or disabling symptom associated with migraine. This study makes clear that further outcomes changes are necessary, and that a potentially more meaningful result in migraine studies may actually be crystal-clear days rather than simply headache-free days.
Although there are more acute options available for headache treatment, medication overuse headache remains a major complicating factor for most clinicians who treat headache. When educating patients, there is always a strong emphasis on guidelines for acute medication use. Many patients struggle with knowing when to use an acute treatment and when to alternate with a different treatment, and often they will withhold treatment completely due to fear of medication overuse. The new class of calcitonin gene-related peptide (CGRP) antagonist medications has shown some potential benefit as a preventive option for both medication overuse headache and migraine.
The prospective study by Curone and colleagues enrolled 300 patients with confirmed medication overuse headache who did not undergo withdrawal of the overused acute medication. Patients who are already taking preventive medications were excluded, as were patients with diagnoses other than chronic migraine or medication overuse. Patients were given one of the three injectable CGRP antagonist medications for prevention and were followed up at 3, 6, 9, and 12 months. The primary outcome was MIDAS score as well as monthly headache days and analgesic consumption.
Out of 303 patients, 242 (80%) showed both a ≥50% reduction of monthly headache days and ≥50% reduction in analgesic intake at 3-month follow-up visit. At 9 months, 198 (65%) were still responders. Monthly analgesic intake decreased ≥50% in 268 of 303 patients (88%) at 3 months and in 241 of 303 patients (79%) at the 6-month follow-up.
For years there has been a debate regarding whether withdrawal of an overused medication is necessary for effective treatment of medication overuse headache. Many preventive treatments are less effective when medication overuse is ongoing. The CGRP class of medications does appear to be effective even with ongoing acute medication overuse. This class of medications should definitely be considered when withdrawing an overused medication is complicated, or when a patient needs to continue to take analgesic medications for another condition.
The theme of this month's commentary is alternative outcomes measures for future migraine studies. The traditional outcomes measures, such as headache frequency measured in headache days, have long been considered gold standards when evaluating the efficacy of preventive interventions. When headache conditions are complicated by interictal pain or other symptoms, or when medication overuse adds a higher frequency or greater severity, those traditional measures are somewhat less exact and specific. Meaningful change for patients with higher frequency of attacks, near-continuous pain, or other migraine symptoms is quite different from that for those without these complications.
Ailani and colleagues reviewed post hoc data from the CONQUER trial, a prior study evaluating the safety and efficacy of galcanezumab vs placebo in patients who had previously not benefited from two to four categories of migraine preventive medication. This refractory population was initially noted to have 4.1 fewer headache days per month than patients taking placebo, but the authors now attempted to review these data with a focus on a different measure: total pain burden (TPB). They defined daily TPB as a single composite measure assessing the frequency, duration, and severity of migraine, calculated by multiplying the number of hours of migraine by the maximum daily migraine pain severity score. The monthly TPB was calculated by adding the daily pain burden over the entire month. The Migraine Disability Assessment questionnaire (MIDAS) and Migraine-Specific Quality of Life Questionnaire (MSQ) scores were also included to compare migraine-related disability and quality of life.
The patients who received galcanezumab were noted to have a significantly lower TPB, both in episodic and chronic migraine. Significantly greater reductions in monthly TPB relative to placebo were observed at each individual month as well. The change from baseline TPB was also noted to be significantly improved in the galcanezumab group compared with the placebo group. The reduction in TPB was noted even when migraine-day reductions were accounted for as part of a sensitivity analysis.
Preventive trials for migraine treatment focus primarily on migraine-day reduction, and for many patients with higher-frequency migraine, this measure does not adequately account for their disease-related disability. This unique way of looking at pain as part of a bigger picture is much more significant and meaningful for this patient population. Migraine frequency is still a very important outcomes measure, but it would be wise to add TBP or another measure that looks more globally at disease-related disability, especially when investigating preventive options in patients with chronic migraine.
When considering whether an intervention is helpful, most patients and clinicians follow the headache frequency, severity, or quality-of-life factors. As most patients will readily report, not all "headache-free days" are created equal. Although most people with migraine will experience days with absolutely no headache pain or other migraine-associated symptoms, on many days they will still have some symptoms of migraine. Lee and colleagues attempted to quantify the difference between headache-free days and crystal-clear days.
Most headache studies use the frequency of headache days as a primary or secondary outcome. This study collected data on both headache days and crystal-clear days, using data from a questionnaire-based large South Korean nationwide population study that evaluated headache and sleep. The study questions were validated for migraine and aura, and included: "How many days have you had a headache during the previous 30 days?" and "How many days have you had crystal-clear days without headache during the previous 30 days?" The data were then analyzed and compared with the widespread pain index (criteria for fibromyalgia) as well as sleep duration, sleep quality, depression and anxiety scales, and an allodynia checklist.
A little over 3000 respondents completed the surveys; 1938 had experienced headache over the past year, 170 were classified as having a diagnosis of migraine, and 50 of those were diagnosed with aura as well. Out of the patients with migraine, 97% had "unclear days." This was higher than the rate of those with non-migraine headaches (91%). Nearly all people surveyed had some crystal-clear days (99.4%).
The number of crystal-clear days per 30 days was significantly lower in participants with migraine than in those with non-migraine headache. Participants with migraine also had higher frequencies of cutaneous allodynia, anxiety, and depression. The weekly average sleep duration in participants with migraine did not significantly differ from that in participants with non-migraine headaches. The widespread pain index rate was much higher in those with migraine as well.
Most patients will definitely understand the difference between crystal-clear and unclear headache days. Many of the newer outcomes studies in migraine have started focusing on the most bothersome symptom, as headache pain is far from the only significant or disabling symptom associated with migraine. This study makes clear that further outcomes changes are necessary, and that a potentially more meaningful result in migraine studies may actually be crystal-clear days rather than simply headache-free days.
Although there are more acute options available for headache treatment, medication overuse headache remains a major complicating factor for most clinicians who treat headache. When educating patients, there is always a strong emphasis on guidelines for acute medication use. Many patients struggle with knowing when to use an acute treatment and when to alternate with a different treatment, and often they will withhold treatment completely due to fear of medication overuse. The new class of calcitonin gene-related peptide (CGRP) antagonist medications has shown some potential benefit as a preventive option for both medication overuse headache and migraine.
The prospective study by Curone and colleagues enrolled 300 patients with confirmed medication overuse headache who did not undergo withdrawal of the overused acute medication. Patients who are already taking preventive medications were excluded, as were patients with diagnoses other than chronic migraine or medication overuse. Patients were given one of the three injectable CGRP antagonist medications for prevention and were followed up at 3, 6, 9, and 12 months. The primary outcome was MIDAS score as well as monthly headache days and analgesic consumption.
Out of 303 patients, 242 (80%) showed both a ≥50% reduction of monthly headache days and ≥50% reduction in analgesic intake at 3-month follow-up visit. At 9 months, 198 (65%) were still responders. Monthly analgesic intake decreased ≥50% in 268 of 303 patients (88%) at 3 months and in 241 of 303 patients (79%) at the 6-month follow-up.
For years there has been a debate regarding whether withdrawal of an overused medication is necessary for effective treatment of medication overuse headache. Many preventive treatments are less effective when medication overuse is ongoing. The CGRP class of medications does appear to be effective even with ongoing acute medication overuse. This class of medications should definitely be considered when withdrawing an overused medication is complicated, or when a patient needs to continue to take analgesic medications for another condition.
The theme of this month's commentary is alternative outcomes measures for future migraine studies. The traditional outcomes measures, such as headache frequency measured in headache days, have long been considered gold standards when evaluating the efficacy of preventive interventions. When headache conditions are complicated by interictal pain or other symptoms, or when medication overuse adds a higher frequency or greater severity, those traditional measures are somewhat less exact and specific. Meaningful change for patients with higher frequency of attacks, near-continuous pain, or other migraine symptoms is quite different from that for those without these complications.
Ailani and colleagues reviewed post hoc data from the CONQUER trial, a prior study evaluating the safety and efficacy of galcanezumab vs placebo in patients who had previously not benefited from two to four categories of migraine preventive medication. This refractory population was initially noted to have 4.1 fewer headache days per month than patients taking placebo, but the authors now attempted to review these data with a focus on a different measure: total pain burden (TPB). They defined daily TPB as a single composite measure assessing the frequency, duration, and severity of migraine, calculated by multiplying the number of hours of migraine by the maximum daily migraine pain severity score. The monthly TPB was calculated by adding the daily pain burden over the entire month. The Migraine Disability Assessment questionnaire (MIDAS) and Migraine-Specific Quality of Life Questionnaire (MSQ) scores were also included to compare migraine-related disability and quality of life.
The patients who received galcanezumab were noted to have a significantly lower TPB, both in episodic and chronic migraine. Significantly greater reductions in monthly TPB relative to placebo were observed at each individual month as well. The change from baseline TPB was also noted to be significantly improved in the galcanezumab group compared with the placebo group. The reduction in TPB was noted even when migraine-day reductions were accounted for as part of a sensitivity analysis.
Preventive trials for migraine treatment focus primarily on migraine-day reduction, and for many patients with higher-frequency migraine, this measure does not adequately account for their disease-related disability. This unique way of looking at pain as part of a bigger picture is much more significant and meaningful for this patient population. Migraine frequency is still a very important outcomes measure, but it would be wise to add TBP or another measure that looks more globally at disease-related disability, especially when investigating preventive options in patients with chronic migraine.
When considering whether an intervention is helpful, most patients and clinicians follow the headache frequency, severity, or quality-of-life factors. As most patients will readily report, not all "headache-free days" are created equal. Although most people with migraine will experience days with absolutely no headache pain or other migraine-associated symptoms, on many days they will still have some symptoms of migraine. Lee and colleagues attempted to quantify the difference between headache-free days and crystal-clear days.
Most headache studies use the frequency of headache days as a primary or secondary outcome. This study collected data on both headache days and crystal-clear days, using data from a questionnaire-based large South Korean nationwide population study that evaluated headache and sleep. The study questions were validated for migraine and aura, and included: "How many days have you had a headache during the previous 30 days?" and "How many days have you had crystal-clear days without headache during the previous 30 days?" The data were then analyzed and compared with the widespread pain index (criteria for fibromyalgia) as well as sleep duration, sleep quality, depression and anxiety scales, and an allodynia checklist.
A little over 3000 respondents completed the surveys; 1938 had experienced headache over the past year, 170 were classified as having a diagnosis of migraine, and 50 of those were diagnosed with aura as well. Out of the patients with migraine, 97% had "unclear days." This was higher than the rate of those with non-migraine headaches (91%). Nearly all people surveyed had some crystal-clear days (99.4%).
The number of crystal-clear days per 30 days was significantly lower in participants with migraine than in those with non-migraine headache. Participants with migraine also had higher frequencies of cutaneous allodynia, anxiety, and depression. The weekly average sleep duration in participants with migraine did not significantly differ from that in participants with non-migraine headaches. The widespread pain index rate was much higher in those with migraine as well.
Most patients will definitely understand the difference between crystal-clear and unclear headache days. Many of the newer outcomes studies in migraine have started focusing on the most bothersome symptom, as headache pain is far from the only significant or disabling symptom associated with migraine. This study makes clear that further outcomes changes are necessary, and that a potentially more meaningful result in migraine studies may actually be crystal-clear days rather than simply headache-free days.
Although there are more acute options available for headache treatment, medication overuse headache remains a major complicating factor for most clinicians who treat headache. When educating patients, there is always a strong emphasis on guidelines for acute medication use. Many patients struggle with knowing when to use an acute treatment and when to alternate with a different treatment, and often they will withhold treatment completely due to fear of medication overuse. The new class of calcitonin gene-related peptide (CGRP) antagonist medications has shown some potential benefit as a preventive option for both medication overuse headache and migraine.
The prospective study by Curone and colleagues enrolled 300 patients with confirmed medication overuse headache who did not undergo withdrawal of the overused acute medication. Patients who are already taking preventive medications were excluded, as were patients with diagnoses other than chronic migraine or medication overuse. Patients were given one of the three injectable CGRP antagonist medications for prevention and were followed up at 3, 6, 9, and 12 months. The primary outcome was MIDAS score as well as monthly headache days and analgesic consumption.
Out of 303 patients, 242 (80%) showed both a ≥50% reduction of monthly headache days and ≥50% reduction in analgesic intake at 3-month follow-up visit. At 9 months, 198 (65%) were still responders. Monthly analgesic intake decreased ≥50% in 268 of 303 patients (88%) at 3 months and in 241 of 303 patients (79%) at the 6-month follow-up.
For years there has been a debate regarding whether withdrawal of an overused medication is necessary for effective treatment of medication overuse headache. Many preventive treatments are less effective when medication overuse is ongoing. The CGRP class of medications does appear to be effective even with ongoing acute medication overuse. This class of medications should definitely be considered when withdrawing an overused medication is complicated, or when a patient needs to continue to take analgesic medications for another condition.