User login
Poverty-related stress linked to aggressive head and neck cancer
A humanized mouse model suggests that head and neck cancer growth may stem from chronic stress. The study found that animals had immunophenotypic changes and a greater propensity towards tumor growth and metastasis.
Other studies have shown this may be caused by the lack of access to health care services or poor quality care. but the difference remains even after adjusting for these factors, according to researchers writing in Head and Neck.
Led by Heather A. Himburg, PhD, associate professor of radiation oncology with the Medical College of Wisconsin, Milwaukee, researchers conducted a study of head and neck cancer models in which tumor cells were implanted into a mouse with a humanized immune system.
Their theory was that psychosocial stress may contribute to the growth of head and neck tumors. The stress of poverty, social deprivation and social isolation can lead to the up-regulation of proinflammatory markers in circulating blood leukocytes, and this has been tied to worse outcomes in hematologic malignancies and breast cancer. Many such studies examined social adversity and found an association with greater tumor growth rates and treatment resistance.
Other researchers have used mouse models to study the phenomenon, but the results have been inconclusive. For example, some research linked the beta-adrenergic pathway to head and neck cancer, but clinical trials of beta-blockers showed no benefit, and even potential harm, for patients with head and neck cancers. Those results imply that this pathway does not drive tumor growth and metastasis in the presence of chronic stress.
Previous research used immunocompromised or nonhumanized mice. However, neither type of model reproduces the human tumor microenvironment, which may contribute to ensuing clinical failures. In the new study, researchers describe results from a preclinical model created using a human head and neck cancer xenograft in a mouse with a humanized immune system.
How the study was conducted
The animals were randomly assigned to normal housing of two or three animals from the same litter to a cage, or social isolation from littermates. There were five male and five female animals in each arm, and the animals were housed in their separate conditions for 4 weeks before tumor implantation.
The isolated animals experienced increased growth and metastasis of the xenografts, compared with controls. The results are consistent with findings in immunodeficient or syngeneic mice, but the humanized nature of the new model could lead to better translation of findings into clinical studies. “The humanized model system in this study demonstrated the presence of both human myeloid and lymphoid lineages as well as expression of at least 40 human cytokines. These data indicate that our model is likely to well-represent the human condition and better predict human clinical responses as compared to both immunodeficient and syngeneic models,” the authors wrote.
The researchers also found that chronic stress may act through an immunoregulatory effect, since there was greater human immune infiltrate into the tumors of stressed animals. Increased presence of regulatory components like myeloid-derived suppressor cells or regulatory T cells, or eroded function of tumor-infiltrating lymphocytes, might explain this finding. The researchers also identified a proinflammatory change in peripheral blood monocular cells in the stressed group. When they analyzed samples from patients who were low income earners of less than $45,000 in annual household income, they found a similar pattern. “This suggests that chronic socioeconomic stress may induce a similar proinflammatory immune state as our chronic stress model system,” the authors wrote.
Tumors were also different between the two groups of mice. Tumors in stressed animals had a higher percentage of cancer stem cells, which is associated with more aggressive tumors and worse disease-free survival. The researchers suggested that up-regulated levels of the chemokine SDF-1 seen in the stressed animals may be driving the higher proportion of stem cells through its effects on the CXCR4 receptor, which is expressed by stem cells in various organs and may cause migration, proliferation, and cell survival.
The study was funded by an endowment from Advancing a Healthier Wisconsin and a grant from the National Center for Advancing Translational Sciences. The authors reported no conflicts of interest.
A humanized mouse model suggests that head and neck cancer growth may stem from chronic stress. The study found that animals had immunophenotypic changes and a greater propensity towards tumor growth and metastasis.
Other studies have shown this may be caused by the lack of access to health care services or poor quality care. but the difference remains even after adjusting for these factors, according to researchers writing in Head and Neck.
Led by Heather A. Himburg, PhD, associate professor of radiation oncology with the Medical College of Wisconsin, Milwaukee, researchers conducted a study of head and neck cancer models in which tumor cells were implanted into a mouse with a humanized immune system.
Their theory was that psychosocial stress may contribute to the growth of head and neck tumors. The stress of poverty, social deprivation and social isolation can lead to the up-regulation of proinflammatory markers in circulating blood leukocytes, and this has been tied to worse outcomes in hematologic malignancies and breast cancer. Many such studies examined social adversity and found an association with greater tumor growth rates and treatment resistance.
Other researchers have used mouse models to study the phenomenon, but the results have been inconclusive. For example, some research linked the beta-adrenergic pathway to head and neck cancer, but clinical trials of beta-blockers showed no benefit, and even potential harm, for patients with head and neck cancers. Those results imply that this pathway does not drive tumor growth and metastasis in the presence of chronic stress.
Previous research used immunocompromised or nonhumanized mice. However, neither type of model reproduces the human tumor microenvironment, which may contribute to ensuing clinical failures. In the new study, researchers describe results from a preclinical model created using a human head and neck cancer xenograft in a mouse with a humanized immune system.
How the study was conducted
The animals were randomly assigned to normal housing of two or three animals from the same litter to a cage, or social isolation from littermates. There were five male and five female animals in each arm, and the animals were housed in their separate conditions for 4 weeks before tumor implantation.
The isolated animals experienced increased growth and metastasis of the xenografts, compared with controls. The results are consistent with findings in immunodeficient or syngeneic mice, but the humanized nature of the new model could lead to better translation of findings into clinical studies. “The humanized model system in this study demonstrated the presence of both human myeloid and lymphoid lineages as well as expression of at least 40 human cytokines. These data indicate that our model is likely to well-represent the human condition and better predict human clinical responses as compared to both immunodeficient and syngeneic models,” the authors wrote.
The researchers also found that chronic stress may act through an immunoregulatory effect, since there was greater human immune infiltrate into the tumors of stressed animals. Increased presence of regulatory components like myeloid-derived suppressor cells or regulatory T cells, or eroded function of tumor-infiltrating lymphocytes, might explain this finding. The researchers also identified a proinflammatory change in peripheral blood monocular cells in the stressed group. When they analyzed samples from patients who were low income earners of less than $45,000 in annual household income, they found a similar pattern. “This suggests that chronic socioeconomic stress may induce a similar proinflammatory immune state as our chronic stress model system,” the authors wrote.
Tumors were also different between the two groups of mice. Tumors in stressed animals had a higher percentage of cancer stem cells, which is associated with more aggressive tumors and worse disease-free survival. The researchers suggested that up-regulated levels of the chemokine SDF-1 seen in the stressed animals may be driving the higher proportion of stem cells through its effects on the CXCR4 receptor, which is expressed by stem cells in various organs and may cause migration, proliferation, and cell survival.
The study was funded by an endowment from Advancing a Healthier Wisconsin and a grant from the National Center for Advancing Translational Sciences. The authors reported no conflicts of interest.
A humanized mouse model suggests that head and neck cancer growth may stem from chronic stress. The study found that animals had immunophenotypic changes and a greater propensity towards tumor growth and metastasis.
Other studies have shown this may be caused by the lack of access to health care services or poor quality care. but the difference remains even after adjusting for these factors, according to researchers writing in Head and Neck.
Led by Heather A. Himburg, PhD, associate professor of radiation oncology with the Medical College of Wisconsin, Milwaukee, researchers conducted a study of head and neck cancer models in which tumor cells were implanted into a mouse with a humanized immune system.
Their theory was that psychosocial stress may contribute to the growth of head and neck tumors. The stress of poverty, social deprivation and social isolation can lead to the up-regulation of proinflammatory markers in circulating blood leukocytes, and this has been tied to worse outcomes in hematologic malignancies and breast cancer. Many such studies examined social adversity and found an association with greater tumor growth rates and treatment resistance.
Other researchers have used mouse models to study the phenomenon, but the results have been inconclusive. For example, some research linked the beta-adrenergic pathway to head and neck cancer, but clinical trials of beta-blockers showed no benefit, and even potential harm, for patients with head and neck cancers. Those results imply that this pathway does not drive tumor growth and metastasis in the presence of chronic stress.
Previous research used immunocompromised or nonhumanized mice. However, neither type of model reproduces the human tumor microenvironment, which may contribute to ensuing clinical failures. In the new study, researchers describe results from a preclinical model created using a human head and neck cancer xenograft in a mouse with a humanized immune system.
How the study was conducted
The animals were randomly assigned to normal housing of two or three animals from the same litter to a cage, or social isolation from littermates. There were five male and five female animals in each arm, and the animals were housed in their separate conditions for 4 weeks before tumor implantation.
The isolated animals experienced increased growth and metastasis of the xenografts, compared with controls. The results are consistent with findings in immunodeficient or syngeneic mice, but the humanized nature of the new model could lead to better translation of findings into clinical studies. “The humanized model system in this study demonstrated the presence of both human myeloid and lymphoid lineages as well as expression of at least 40 human cytokines. These data indicate that our model is likely to well-represent the human condition and better predict human clinical responses as compared to both immunodeficient and syngeneic models,” the authors wrote.
The researchers also found that chronic stress may act through an immunoregulatory effect, since there was greater human immune infiltrate into the tumors of stressed animals. Increased presence of regulatory components like myeloid-derived suppressor cells or regulatory T cells, or eroded function of tumor-infiltrating lymphocytes, might explain this finding. The researchers also identified a proinflammatory change in peripheral blood monocular cells in the stressed group. When they analyzed samples from patients who were low income earners of less than $45,000 in annual household income, they found a similar pattern. “This suggests that chronic socioeconomic stress may induce a similar proinflammatory immune state as our chronic stress model system,” the authors wrote.
Tumors were also different between the two groups of mice. Tumors in stressed animals had a higher percentage of cancer stem cells, which is associated with more aggressive tumors and worse disease-free survival. The researchers suggested that up-regulated levels of the chemokine SDF-1 seen in the stressed animals may be driving the higher proportion of stem cells through its effects on the CXCR4 receptor, which is expressed by stem cells in various organs and may cause migration, proliferation, and cell survival.
The study was funded by an endowment from Advancing a Healthier Wisconsin and a grant from the National Center for Advancing Translational Sciences. The authors reported no conflicts of interest.
FROM HEAD & NECK
Steroids counter ataxia telangiectasia
SEATTLE –
The disease is an autosomal recessive disorder caused by mutations in the ATM gene, which is critical to the response to cellular insults such as DNA breaks, oxidative damage, and other forms of stress. The result is clinical manifestations that range from a suppressed immune system to organ damage and neurological symptoms that typically lead patients to be wheelchair bound by their teenage years.
“It’s really multisystem and a very, very difficult disease for people to live with,” Howard M. Lederman, MD, PhD, said in an interview. Dr. Lederman is a coauthor of the study, which was presented by Stefan Zielen, PhD, professor at the University of Goethe, at the 2022 annual meeting of the American Academy of Neurology.
Various therapies have been developed to improve immunodeficiency, lung disease, and some of the other clinical aspects of the condition, but there is no treatment for its neurological effects. “There’s not really been a good animal model, which has been a big problem in trying to test drugs and design treatment trials,” said Dr. Lederman, professor of pediatrics and medicine at Johns Hopkins University, Baltimore.
The new results may change that. “In the children under the age of 9, there was really a very clear slowdown in the neurodegeneration, and specifically the time that it took for them to lose the ability to ambulate. It’s very exciting, because it’s the first time that anybody has really shown in a double-blind, placebo-controlled, large phase 3 study that any drug has been able to do this. And there were really no steroid side effects, which is the other really remarkable thing about this study,” said Dr. Lederman.
The therapy grew out of a study by researchers in Italy who treated pediatric ataxia telangiectasia patients with corticosteroids and found some transitory improvements in gross motor function, but concerns about long-term exposure to steroids limited its application. EryDel, which specializes in encapsulating therapeutics in red blood cells, became interested and developed a formulation using the patient’s own red blood cells infused with DSP. Reinfused to the patients, the red blood cells slowly release the steroid.
It isn’t clear how dexamethasone works. There are data suggesting that it might lead to transcription of small pieces of the ATM protein, “but that has really not been nailed down in any way at this point. Corticosteroids act on all kinds of cells in all kinds of ways, and so there might be a little bit of this so-called mini-ATM that’s produced, but that may or may not be related to the way in which corticosteroids have a beneficial effect on the rate of neurodegeneration,” said Dr. Lederman.
The treatment process is not easy. Children must have 50-60 cc of blood removed. Red blood cells treated to become porous are exposed to DSP, and then resealed. Then the cells are reinfused. “The whole process takes from beginning to end probably about 3 hours, with a really experienced team of people doing it. And it’s limiting because it’s not easy to put in an IV and take 50 or 60 cc of blood out of children much younger than 5 or 6. The process is now being modified to see whether we could do it with 20 to 30 cc instead,” said Dr. Lederman.
A ‘promising and impressive’ study
The study is promising, according to Nicholas Johnson, MD, who comoderated the session where the study was presented. “They were able to show a slower rate of neurological degeneration or duration on both the lower and higher dose compared with the placebo. This is promising and impressive, in the sense that it’s a really large (trial) for a rare condition,” Dr. Johnson, vice chair of research at Virginia Commonwealth University, Richmond, said in an interview.
The study included 164 patients Europe, Australia, Israel, Tunisia, India, and the United States, who received 5-10 mg dexamethasone, 14-22 mg DSP, or placebo. Mean ages in each group ranged from 9.6 to 10.4 years.
In an intention-to-treat analysis, modified International Cooperative Ataxia Rating Scale (mICARS) scores trended toward improvement in the low-dose (–1.37; P = .0847) and high-dose groups (–1.40; P = .0765) when determined by central raters during the COVID-19 pandemic. There was also a trend toward improvement when determined by local raters in the low dose group (–1.73; P = .0720) and a statistically significant change in the high dose group (–2.11; P = .0277). The researchers noted some inconsistency between local and central raters, due to inconsistency of videography and language challenges for central raters.
An intention-to-treat analysis of a subgroup of 89 patients age 6-9, who were compared with natural history data from 245 patients, found a deterioration of mICARS of 3.7 per year, compared with 0.92 in the high-dose group, for a reduction of 75% (P = .020). In the high-dose group, 51.7% had a minimal or significant improvement compared with baseline according to the Clinical Global Impression of Change, as did 29.0% on low dose, and 27.6% in the placebo group.
SEATTLE –
The disease is an autosomal recessive disorder caused by mutations in the ATM gene, which is critical to the response to cellular insults such as DNA breaks, oxidative damage, and other forms of stress. The result is clinical manifestations that range from a suppressed immune system to organ damage and neurological symptoms that typically lead patients to be wheelchair bound by their teenage years.
“It’s really multisystem and a very, very difficult disease for people to live with,” Howard M. Lederman, MD, PhD, said in an interview. Dr. Lederman is a coauthor of the study, which was presented by Stefan Zielen, PhD, professor at the University of Goethe, at the 2022 annual meeting of the American Academy of Neurology.
Various therapies have been developed to improve immunodeficiency, lung disease, and some of the other clinical aspects of the condition, but there is no treatment for its neurological effects. “There’s not really been a good animal model, which has been a big problem in trying to test drugs and design treatment trials,” said Dr. Lederman, professor of pediatrics and medicine at Johns Hopkins University, Baltimore.
The new results may change that. “In the children under the age of 9, there was really a very clear slowdown in the neurodegeneration, and specifically the time that it took for them to lose the ability to ambulate. It’s very exciting, because it’s the first time that anybody has really shown in a double-blind, placebo-controlled, large phase 3 study that any drug has been able to do this. And there were really no steroid side effects, which is the other really remarkable thing about this study,” said Dr. Lederman.
The therapy grew out of a study by researchers in Italy who treated pediatric ataxia telangiectasia patients with corticosteroids and found some transitory improvements in gross motor function, but concerns about long-term exposure to steroids limited its application. EryDel, which specializes in encapsulating therapeutics in red blood cells, became interested and developed a formulation using the patient’s own red blood cells infused with DSP. Reinfused to the patients, the red blood cells slowly release the steroid.
It isn’t clear how dexamethasone works. There are data suggesting that it might lead to transcription of small pieces of the ATM protein, “but that has really not been nailed down in any way at this point. Corticosteroids act on all kinds of cells in all kinds of ways, and so there might be a little bit of this so-called mini-ATM that’s produced, but that may or may not be related to the way in which corticosteroids have a beneficial effect on the rate of neurodegeneration,” said Dr. Lederman.
The treatment process is not easy. Children must have 50-60 cc of blood removed. Red blood cells treated to become porous are exposed to DSP, and then resealed. Then the cells are reinfused. “The whole process takes from beginning to end probably about 3 hours, with a really experienced team of people doing it. And it’s limiting because it’s not easy to put in an IV and take 50 or 60 cc of blood out of children much younger than 5 or 6. The process is now being modified to see whether we could do it with 20 to 30 cc instead,” said Dr. Lederman.
A ‘promising and impressive’ study
The study is promising, according to Nicholas Johnson, MD, who comoderated the session where the study was presented. “They were able to show a slower rate of neurological degeneration or duration on both the lower and higher dose compared with the placebo. This is promising and impressive, in the sense that it’s a really large (trial) for a rare condition,” Dr. Johnson, vice chair of research at Virginia Commonwealth University, Richmond, said in an interview.
The study included 164 patients Europe, Australia, Israel, Tunisia, India, and the United States, who received 5-10 mg dexamethasone, 14-22 mg DSP, or placebo. Mean ages in each group ranged from 9.6 to 10.4 years.
In an intention-to-treat analysis, modified International Cooperative Ataxia Rating Scale (mICARS) scores trended toward improvement in the low-dose (–1.37; P = .0847) and high-dose groups (–1.40; P = .0765) when determined by central raters during the COVID-19 pandemic. There was also a trend toward improvement when determined by local raters in the low dose group (–1.73; P = .0720) and a statistically significant change in the high dose group (–2.11; P = .0277). The researchers noted some inconsistency between local and central raters, due to inconsistency of videography and language challenges for central raters.
An intention-to-treat analysis of a subgroup of 89 patients age 6-9, who were compared with natural history data from 245 patients, found a deterioration of mICARS of 3.7 per year, compared with 0.92 in the high-dose group, for a reduction of 75% (P = .020). In the high-dose group, 51.7% had a minimal or significant improvement compared with baseline according to the Clinical Global Impression of Change, as did 29.0% on low dose, and 27.6% in the placebo group.
SEATTLE –
The disease is an autosomal recessive disorder caused by mutations in the ATM gene, which is critical to the response to cellular insults such as DNA breaks, oxidative damage, and other forms of stress. The result is clinical manifestations that range from a suppressed immune system to organ damage and neurological symptoms that typically lead patients to be wheelchair bound by their teenage years.
“It’s really multisystem and a very, very difficult disease for people to live with,” Howard M. Lederman, MD, PhD, said in an interview. Dr. Lederman is a coauthor of the study, which was presented by Stefan Zielen, PhD, professor at the University of Goethe, at the 2022 annual meeting of the American Academy of Neurology.
Various therapies have been developed to improve immunodeficiency, lung disease, and some of the other clinical aspects of the condition, but there is no treatment for its neurological effects. “There’s not really been a good animal model, which has been a big problem in trying to test drugs and design treatment trials,” said Dr. Lederman, professor of pediatrics and medicine at Johns Hopkins University, Baltimore.
The new results may change that. “In the children under the age of 9, there was really a very clear slowdown in the neurodegeneration, and specifically the time that it took for them to lose the ability to ambulate. It’s very exciting, because it’s the first time that anybody has really shown in a double-blind, placebo-controlled, large phase 3 study that any drug has been able to do this. And there were really no steroid side effects, which is the other really remarkable thing about this study,” said Dr. Lederman.
The therapy grew out of a study by researchers in Italy who treated pediatric ataxia telangiectasia patients with corticosteroids and found some transitory improvements in gross motor function, but concerns about long-term exposure to steroids limited its application. EryDel, which specializes in encapsulating therapeutics in red blood cells, became interested and developed a formulation using the patient’s own red blood cells infused with DSP. Reinfused to the patients, the red blood cells slowly release the steroid.
It isn’t clear how dexamethasone works. There are data suggesting that it might lead to transcription of small pieces of the ATM protein, “but that has really not been nailed down in any way at this point. Corticosteroids act on all kinds of cells in all kinds of ways, and so there might be a little bit of this so-called mini-ATM that’s produced, but that may or may not be related to the way in which corticosteroids have a beneficial effect on the rate of neurodegeneration,” said Dr. Lederman.
The treatment process is not easy. Children must have 50-60 cc of blood removed. Red blood cells treated to become porous are exposed to DSP, and then resealed. Then the cells are reinfused. “The whole process takes from beginning to end probably about 3 hours, with a really experienced team of people doing it. And it’s limiting because it’s not easy to put in an IV and take 50 or 60 cc of blood out of children much younger than 5 or 6. The process is now being modified to see whether we could do it with 20 to 30 cc instead,” said Dr. Lederman.
A ‘promising and impressive’ study
The study is promising, according to Nicholas Johnson, MD, who comoderated the session where the study was presented. “They were able to show a slower rate of neurological degeneration or duration on both the lower and higher dose compared with the placebo. This is promising and impressive, in the sense that it’s a really large (trial) for a rare condition,” Dr. Johnson, vice chair of research at Virginia Commonwealth University, Richmond, said in an interview.
The study included 164 patients Europe, Australia, Israel, Tunisia, India, and the United States, who received 5-10 mg dexamethasone, 14-22 mg DSP, or placebo. Mean ages in each group ranged from 9.6 to 10.4 years.
In an intention-to-treat analysis, modified International Cooperative Ataxia Rating Scale (mICARS) scores trended toward improvement in the low-dose (–1.37; P = .0847) and high-dose groups (–1.40; P = .0765) when determined by central raters during the COVID-19 pandemic. There was also a trend toward improvement when determined by local raters in the low dose group (–1.73; P = .0720) and a statistically significant change in the high dose group (–2.11; P = .0277). The researchers noted some inconsistency between local and central raters, due to inconsistency of videography and language challenges for central raters.
An intention-to-treat analysis of a subgroup of 89 patients age 6-9, who were compared with natural history data from 245 patients, found a deterioration of mICARS of 3.7 per year, compared with 0.92 in the high-dose group, for a reduction of 75% (P = .020). In the high-dose group, 51.7% had a minimal or significant improvement compared with baseline according to the Clinical Global Impression of Change, as did 29.0% on low dose, and 27.6% in the placebo group.
AT AAN 2022
Postpartum HCV treatment rare in infected mothers with opioid use disorder
Despite the availability of effective direct-acting antivirals, very few a mothers with opioid use disorder (OUD) and hepatitis C virus (HCV) during pregnancy received follow-up care or treatment for the infection within 6 months of giving birth, a retrospective study of Medicaid maternity patients found.
The study pooled data on 23,780 Medicaid-enrolled pregnant women with OUD who had a live or stillbirth during 2016-2019 and were followed for 6 months after delivery. Among these women – drawn from six states in the Medicaid Outcomes Distributed Research Network – the pooled average probability of HCV testing during pregnancy was 70.3% (95% confidence interval, 61.5%-79.1%). Of these, 30.9% (95% CI, 23.8%-38%) tested positive. At 60 days postpartum, just 3.2% (95% CI, 2.6%-3.8%) had a follow-up visit or treatment for HCV. In a subset of patients followed for 6 months, only 5.9% (95% CI, 4.9%-6.9%) had any HCV follow-up visit or medication within 6 months of delivery.

While HCV screening and diagnosis rates varied across states, postpartum follow-up rates were universally low. The results suggest a need to improve the cascade of postpartum care for HCV and, ultimately perhaps, introduce antenatal HCV treatment, as is currently given safely for HIV, if current clinical research establishes safety, according to Marian P. Jarlenski, PhD, MPH, an associate professor of public health policy and management at the University of Pittsburgh. The study was published in Obstetrics & Gynecology.
HCV infection has risen substantially in people of reproductive age in tandem with an increase in OUDs. HCV is transmitted from an infected mother to her baby in about 6% of cases, according to the Centers for Disease Control and Prevention, which in 2020 expanded its HCV screening recommendations to include all pregnant women. Currently no treatment for HCV during pregnancy has been approved.
In light of those recent recommendations, Dr. Jarlenski said in an interview that her group was “interested in looking at high-risk screened people and estimating what proportion received follow-up care and treatment for HCV. What is the promise of screening? The promise is that you can treat. Otherwise why screen?”
She acknowledged, however, that the postpartum period is a challenging time for a mother to seek health information or care for herself, whether she’s a new parent or has other children in the home. Nevertheless, the low rate of follow-up and treatment was unexpected. “Even the 70% rate of screening was low – we felt it should have been closer to 100% – but the follow-up rate was surprisingly low,” Dr. Jarlenski said.

Mishka Terplan, MD, MPH, medical director of Friends Research Institute in Baltimore, was not surprised at the low follow-up rate. “The cascade of care for hep C is demoralizing,” said Dr. Terplan, who was not involved in the study. “We know that hep C is syndemic with OUD and other opioid crises and we know that screening is effective for identifying hep C and that antiviral medications are now more effective and less toxic than ever before. But despite this, we’re failing pregnant women and their kids at every step along the cascade. We do a better job with initial testing than with the follow-up testing. We do a horrible job with postpartum medication initiation.”
He pointed to the systemic challenges mothers face in getting postpartum HCV care. “They may be transferred to a subspecialist for treatment, and this transfer is compounded by issues of insurance coverage and eligibility.” With the onus on new mothers to submit the paperwork, “the idea that mothers would be able to initiate much less continue postpartum treatment is absurd,” Dr. Terplan said.
He added that the children born to HCV-positive mothers need surveillance as well, but data suggest that the rates of newborn testing are also low. “There’s a preventable public health burden in all of this.”
The obvious way to increase eradicative therapy would be to treat women while they are getting antenatal care. A small phase 1 trial found that all pregnant participants who were HCV positive and given antivirals in their second trimester were safely treated and gave birth to healthy babies.
“If larger trials prove this treatment is safe and effective, then these results should be communicated to care providers and pregnant patients,” Dr. Jarlenski said. Otherwise, the public health potential of universal screening in pregnancy will not be realized.
This research was supported by the National Institute of Drug Abuse and by the Delaware Division of Medicaid and Medical Assistance and the University of Delaware, Center for Community Research & Service. Dr. Jarlenski disclosed no competing interests. One coauthor disclosed grant funding through her institution from Gilead Sciences and Organon unrelated to this work. Dr. Terplan reported no relevant competing interests.
Despite the availability of effective direct-acting antivirals, very few a mothers with opioid use disorder (OUD) and hepatitis C virus (HCV) during pregnancy received follow-up care or treatment for the infection within 6 months of giving birth, a retrospective study of Medicaid maternity patients found.
The study pooled data on 23,780 Medicaid-enrolled pregnant women with OUD who had a live or stillbirth during 2016-2019 and were followed for 6 months after delivery. Among these women – drawn from six states in the Medicaid Outcomes Distributed Research Network – the pooled average probability of HCV testing during pregnancy was 70.3% (95% confidence interval, 61.5%-79.1%). Of these, 30.9% (95% CI, 23.8%-38%) tested positive. At 60 days postpartum, just 3.2% (95% CI, 2.6%-3.8%) had a follow-up visit or treatment for HCV. In a subset of patients followed for 6 months, only 5.9% (95% CI, 4.9%-6.9%) had any HCV follow-up visit or medication within 6 months of delivery.
While HCV screening and diagnosis rates varied across states, postpartum follow-up rates were universally low. The results suggest a need to improve the cascade of postpartum care for HCV and, ultimately perhaps, introduce antenatal HCV treatment, as is currently given safely for HIV, if current clinical research establishes safety, according to Marian P. Jarlenski, PhD, MPH, an associate professor of public health policy and management at the University of Pittsburgh. The study was published in Obstetrics & Gynecology.
HCV infection has risen substantially in people of reproductive age in tandem with an increase in OUDs. HCV is transmitted from an infected mother to her baby in about 6% of cases, according to the Centers for Disease Control and Prevention, which in 2020 expanded its HCV screening recommendations to include all pregnant women. Currently no treatment for HCV during pregnancy has been approved.
In light of those recent recommendations, Dr. Jarlenski said in an interview that her group was “interested in looking at high-risk screened people and estimating what proportion received follow-up care and treatment for HCV. What is the promise of screening? The promise is that you can treat. Otherwise why screen?”
She acknowledged, however, that the postpartum period is a challenging time for a mother to seek health information or care for herself, whether she’s a new parent or has other children in the home. Nevertheless, the low rate of follow-up and treatment was unexpected. “Even the 70% rate of screening was low – we felt it should have been closer to 100% – but the follow-up rate was surprisingly low,” Dr. Jarlenski said.
Mishka Terplan, MD, MPH, medical director of Friends Research Institute in Baltimore, was not surprised at the low follow-up rate. “The cascade of care for hep C is demoralizing,” said Dr. Terplan, who was not involved in the study. “We know that hep C is syndemic with OUD and other opioid crises and we know that screening is effective for identifying hep C and that antiviral medications are now more effective and less toxic than ever before. But despite this, we’re failing pregnant women and their kids at every step along the cascade. We do a better job with initial testing than with the follow-up testing. We do a horrible job with postpartum medication initiation.”
He pointed to the systemic challenges mothers face in getting postpartum HCV care. “They may be transferred to a subspecialist for treatment, and this transfer is compounded by issues of insurance coverage and eligibility.” With the onus on new mothers to submit the paperwork, “the idea that mothers would be able to initiate much less continue postpartum treatment is absurd,” Dr. Terplan said.
He added that the children born to HCV-positive mothers need surveillance as well, but data suggest that the rates of newborn testing are also low. “There’s a preventable public health burden in all of this.”
The obvious way to increase eradicative therapy would be to treat women while they are getting antenatal care. A small phase 1 trial found that all pregnant participants who were HCV positive and given antivirals in their second trimester were safely treated and gave birth to healthy babies.
“If larger trials prove this treatment is safe and effective, then these results should be communicated to care providers and pregnant patients,” Dr. Jarlenski said. Otherwise, the public health potential of universal screening in pregnancy will not be realized.
This research was supported by the National Institute of Drug Abuse and by the Delaware Division of Medicaid and Medical Assistance and the University of Delaware, Center for Community Research & Service. Dr. Jarlenski disclosed no competing interests. One coauthor disclosed grant funding through her institution from Gilead Sciences and Organon unrelated to this work. Dr. Terplan reported no relevant competing interests.
Despite the availability of effective direct-acting antivirals, very few a mothers with opioid use disorder (OUD) and hepatitis C virus (HCV) during pregnancy received follow-up care or treatment for the infection within 6 months of giving birth, a retrospective study of Medicaid maternity patients found.
The study pooled data on 23,780 Medicaid-enrolled pregnant women with OUD who had a live or stillbirth during 2016-2019 and were followed for 6 months after delivery. Among these women – drawn from six states in the Medicaid Outcomes Distributed Research Network – the pooled average probability of HCV testing during pregnancy was 70.3% (95% confidence interval, 61.5%-79.1%). Of these, 30.9% (95% CI, 23.8%-38%) tested positive. At 60 days postpartum, just 3.2% (95% CI, 2.6%-3.8%) had a follow-up visit or treatment for HCV. In a subset of patients followed for 6 months, only 5.9% (95% CI, 4.9%-6.9%) had any HCV follow-up visit or medication within 6 months of delivery.
While HCV screening and diagnosis rates varied across states, postpartum follow-up rates were universally low. The results suggest a need to improve the cascade of postpartum care for HCV and, ultimately perhaps, introduce antenatal HCV treatment, as is currently given safely for HIV, if current clinical research establishes safety, according to Marian P. Jarlenski, PhD, MPH, an associate professor of public health policy and management at the University of Pittsburgh. The study was published in Obstetrics & Gynecology.
HCV infection has risen substantially in people of reproductive age in tandem with an increase in OUDs. HCV is transmitted from an infected mother to her baby in about 6% of cases, according to the Centers for Disease Control and Prevention, which in 2020 expanded its HCV screening recommendations to include all pregnant women. Currently no treatment for HCV during pregnancy has been approved.
In light of those recent recommendations, Dr. Jarlenski said in an interview that her group was “interested in looking at high-risk screened people and estimating what proportion received follow-up care and treatment for HCV. What is the promise of screening? The promise is that you can treat. Otherwise why screen?”
She acknowledged, however, that the postpartum period is a challenging time for a mother to seek health information or care for herself, whether she’s a new parent or has other children in the home. Nevertheless, the low rate of follow-up and treatment was unexpected. “Even the 70% rate of screening was low – we felt it should have been closer to 100% – but the follow-up rate was surprisingly low,” Dr. Jarlenski said.
Mishka Terplan, MD, MPH, medical director of Friends Research Institute in Baltimore, was not surprised at the low follow-up rate. “The cascade of care for hep C is demoralizing,” said Dr. Terplan, who was not involved in the study. “We know that hep C is syndemic with OUD and other opioid crises and we know that screening is effective for identifying hep C and that antiviral medications are now more effective and less toxic than ever before. But despite this, we’re failing pregnant women and their kids at every step along the cascade. We do a better job with initial testing than with the follow-up testing. We do a horrible job with postpartum medication initiation.”
He pointed to the systemic challenges mothers face in getting postpartum HCV care. “They may be transferred to a subspecialist for treatment, and this transfer is compounded by issues of insurance coverage and eligibility.” With the onus on new mothers to submit the paperwork, “the idea that mothers would be able to initiate much less continue postpartum treatment is absurd,” Dr. Terplan said.
He added that the children born to HCV-positive mothers need surveillance as well, but data suggest that the rates of newborn testing are also low. “There’s a preventable public health burden in all of this.”
The obvious way to increase eradicative therapy would be to treat women while they are getting antenatal care. A small phase 1 trial found that all pregnant participants who were HCV positive and given antivirals in their second trimester were safely treated and gave birth to healthy babies.
“If larger trials prove this treatment is safe and effective, then these results should be communicated to care providers and pregnant patients,” Dr. Jarlenski said. Otherwise, the public health potential of universal screening in pregnancy will not be realized.
This research was supported by the National Institute of Drug Abuse and by the Delaware Division of Medicaid and Medical Assistance and the University of Delaware, Center for Community Research & Service. Dr. Jarlenski disclosed no competing interests. One coauthor disclosed grant funding through her institution from Gilead Sciences and Organon unrelated to this work. Dr. Terplan reported no relevant competing interests.
FROM OBSTETRICS & GYNECOLOGY
Restless legs syndrome occurs often in X-linked adrenoleukodystrophy
Patients with X-linked adrenoleukodystrophy (ALD), a neurodegenerative disease, often experience gait and balance problems, as well as leg discomfort, sleep disturbances, and pain, wrote John W. Winkelman, MD, of Massachusetts General Hospital, Boston, and colleagues. Restless legs syndrome (RLS) has been associated with neurological conditions including Parkinson’s disease, but the prevalence of RLS in ALD patients has not been examined, they said.

In a pilot study published in Sleep Medicine, the researchers identified 21 women and 11 men with ALD who were treated at a single center. The median age of the patients was 45.9 years. Twenty-seven patients had symptoms of myelopathy, with a median age of onset of 34 years.
The researchers assessed RLS severity using questionnaires and the Hopkins Telephone Diagnostic Interview (HTDI), a validated RLS assessment tool. They also reviewed patients’ charts for data on neurological examinations, functional gait measures, and laboratory assessments. Functional gait assessments included the 25-Foot Walk test (25-FW), the Timed Up and Go test (TUG), and Six Minute Walk test (6MW).
Thirteen patients (10 women and 3 men) met criteria for RLS based on the HTDI. The median age of RLS onset was 35 years. Six RLS patients (46.2%) reported using medication to relieve symptoms, and eight RLS patients had a history of antidepressant use.
In addition, six patients with RLS reported a history of anemia or iron deficiency. Ferritin levels were available for 14 patients: 8 women with RLS and 4 women and 2 men without RLS; the mean ferritin levels were 74.0 mcg/L in RLS patients and 99.5 mcg/L in those without RLS.
Of the seven ALD patients with brain lesions, all were men, only two were diagnosed with RLS, and all seven cases were mild, the researchers noted.
Overall, patients with RLS had more neurological signs and symptoms than those without RLS; the most significant were pain and gait difficulty. However, patients with RLS also were more likely than were those without RLS to report spasticity, muscle weakness, impaired coordination, hyperreflexia, impaired sensation, and paraesthesia, as well as bladder, bowel, and erectile dysfunction.
The 40.6% prevalence of RLS in patients with ALD is notably higher than that of the general population, in which the prevalence of RLS is 5%-10%, the researchers wrote in their discussion.
“Consistent with patterns observed in the general population, risk factors for RLS in this cohort of adults with ALD included female gender, increased age, lower iron indices, and use of serotonergic antidepressants,” they said.
The study findings were limited by several factors including the small size and the possible contribution of antidepressant use to the high rate of RLS, the researchers noted.
“Awareness of RLS in patients with ALD would allow for its effective treatment, which may improve the functional impairments as well as quality of life, mood, and anxiety issues in those with ALD,” they concluded.
The study received no outside funding.
Dr. Winkelman disclosed ties with Advance Medical, Avadel, Disc Medicine, Eisai, Emalex, Idorsia, Noctrix, UpToDate, and Merck Pharmaceuticals, as well as research support from the National Institute on Drug Abuse and the Baszucki Brain Research Foundation. The study also was supported by grants from the National Institute of Neurological Disorders and Stroke, the European Leukodystrophy Association, the Arrivederci Foundation, the Leblang Foundation, and the Hammer Family Fund Journal Preproof for ALD Research and Therapies for Women.
Patients with X-linked adrenoleukodystrophy (ALD), a neurodegenerative disease, often experience gait and balance problems, as well as leg discomfort, sleep disturbances, and pain, wrote John W. Winkelman, MD, of Massachusetts General Hospital, Boston, and colleagues. Restless legs syndrome (RLS) has been associated with neurological conditions including Parkinson’s disease, but the prevalence of RLS in ALD patients has not been examined, they said.
In a pilot study published in Sleep Medicine, the researchers identified 21 women and 11 men with ALD who were treated at a single center. The median age of the patients was 45.9 years. Twenty-seven patients had symptoms of myelopathy, with a median age of onset of 34 years.
The researchers assessed RLS severity using questionnaires and the Hopkins Telephone Diagnostic Interview (HTDI), a validated RLS assessment tool. They also reviewed patients’ charts for data on neurological examinations, functional gait measures, and laboratory assessments. Functional gait assessments included the 25-Foot Walk test (25-FW), the Timed Up and Go test (TUG), and Six Minute Walk test (6MW).
Thirteen patients (10 women and 3 men) met criteria for RLS based on the HTDI. The median age of RLS onset was 35 years. Six RLS patients (46.2%) reported using medication to relieve symptoms, and eight RLS patients had a history of antidepressant use.
In addition, six patients with RLS reported a history of anemia or iron deficiency. Ferritin levels were available for 14 patients: 8 women with RLS and 4 women and 2 men without RLS; the mean ferritin levels were 74.0 mcg/L in RLS patients and 99.5 mcg/L in those without RLS.
Of the seven ALD patients with brain lesions, all were men, only two were diagnosed with RLS, and all seven cases were mild, the researchers noted.
Overall, patients with RLS had more neurological signs and symptoms than those without RLS; the most significant were pain and gait difficulty. However, patients with RLS also were more likely than were those without RLS to report spasticity, muscle weakness, impaired coordination, hyperreflexia, impaired sensation, and paraesthesia, as well as bladder, bowel, and erectile dysfunction.
The 40.6% prevalence of RLS in patients with ALD is notably higher than that of the general population, in which the prevalence of RLS is 5%-10%, the researchers wrote in their discussion.
“Consistent with patterns observed in the general population, risk factors for RLS in this cohort of adults with ALD included female gender, increased age, lower iron indices, and use of serotonergic antidepressants,” they said.
The study findings were limited by several factors including the small size and the possible contribution of antidepressant use to the high rate of RLS, the researchers noted.
“Awareness of RLS in patients with ALD would allow for its effective treatment, which may improve the functional impairments as well as quality of life, mood, and anxiety issues in those with ALD,” they concluded.
The study received no outside funding.
Dr. Winkelman disclosed ties with Advance Medical, Avadel, Disc Medicine, Eisai, Emalex, Idorsia, Noctrix, UpToDate, and Merck Pharmaceuticals, as well as research support from the National Institute on Drug Abuse and the Baszucki Brain Research Foundation. The study also was supported by grants from the National Institute of Neurological Disorders and Stroke, the European Leukodystrophy Association, the Arrivederci Foundation, the Leblang Foundation, and the Hammer Family Fund Journal Preproof for ALD Research and Therapies for Women.
Patients with X-linked adrenoleukodystrophy (ALD), a neurodegenerative disease, often experience gait and balance problems, as well as leg discomfort, sleep disturbances, and pain, wrote John W. Winkelman, MD, of Massachusetts General Hospital, Boston, and colleagues. Restless legs syndrome (RLS) has been associated with neurological conditions including Parkinson’s disease, but the prevalence of RLS in ALD patients has not been examined, they said.
In a pilot study published in Sleep Medicine, the researchers identified 21 women and 11 men with ALD who were treated at a single center. The median age of the patients was 45.9 years. Twenty-seven patients had symptoms of myelopathy, with a median age of onset of 34 years.
The researchers assessed RLS severity using questionnaires and the Hopkins Telephone Diagnostic Interview (HTDI), a validated RLS assessment tool. They also reviewed patients’ charts for data on neurological examinations, functional gait measures, and laboratory assessments. Functional gait assessments included the 25-Foot Walk test (25-FW), the Timed Up and Go test (TUG), and Six Minute Walk test (6MW).
Thirteen patients (10 women and 3 men) met criteria for RLS based on the HTDI. The median age of RLS onset was 35 years. Six RLS patients (46.2%) reported using medication to relieve symptoms, and eight RLS patients had a history of antidepressant use.
In addition, six patients with RLS reported a history of anemia or iron deficiency. Ferritin levels were available for 14 patients: 8 women with RLS and 4 women and 2 men without RLS; the mean ferritin levels were 74.0 mcg/L in RLS patients and 99.5 mcg/L in those without RLS.
Of the seven ALD patients with brain lesions, all were men, only two were diagnosed with RLS, and all seven cases were mild, the researchers noted.
Overall, patients with RLS had more neurological signs and symptoms than those without RLS; the most significant were pain and gait difficulty. However, patients with RLS also were more likely than were those without RLS to report spasticity, muscle weakness, impaired coordination, hyperreflexia, impaired sensation, and paraesthesia, as well as bladder, bowel, and erectile dysfunction.
The 40.6% prevalence of RLS in patients with ALD is notably higher than that of the general population, in which the prevalence of RLS is 5%-10%, the researchers wrote in their discussion.
“Consistent with patterns observed in the general population, risk factors for RLS in this cohort of adults with ALD included female gender, increased age, lower iron indices, and use of serotonergic antidepressants,” they said.
The study findings were limited by several factors including the small size and the possible contribution of antidepressant use to the high rate of RLS, the researchers noted.
“Awareness of RLS in patients with ALD would allow for its effective treatment, which may improve the functional impairments as well as quality of life, mood, and anxiety issues in those with ALD,” they concluded.
The study received no outside funding.
Dr. Winkelman disclosed ties with Advance Medical, Avadel, Disc Medicine, Eisai, Emalex, Idorsia, Noctrix, UpToDate, and Merck Pharmaceuticals, as well as research support from the National Institute on Drug Abuse and the Baszucki Brain Research Foundation. The study also was supported by grants from the National Institute of Neurological Disorders and Stroke, the European Leukodystrophy Association, the Arrivederci Foundation, the Leblang Foundation, and the Hammer Family Fund Journal Preproof for ALD Research and Therapies for Women.
FROM SLEEP MEDICINE
Pneumonia shows strong connection to chronic otitis media
Individuals with a prior diagnosis of pneumonia were significantly more likely to develop chronic otitis media (COM) than were those without a history of pneumonia, based on data from a nationwide cohort study of more than 100,000 patients.
“Recently, middle ear diseases, including COM, have been recognized as respiratory tract diseases beyond the pathophysiological concepts of ventilation dysfunction, with recurrent infection that occurs from anatomically adjacent structures such as the middle ear, mastoid cavity, and eustachian tube,” but the potential link between pneumonia and chronic otitis media and adults in particular has not been examined, wrote Sung Kyun Kim, MD, of Hallym University, Dongtan, South Korea, and colleagues.
In a study recently published in the International Journal of Infectious Diseases, the researchers identified 23,436 adults with COM and 93,744 controls aged 40 years and older from a Korean health insurance database between 2002 and 2015.
The overall incidence of pneumonia in the study population was significantly higher in the COM group compared with controls (9.3% vs. 7.2%, P <.001). The odds ratios of pneumonia were significantly higher in the COM group compared with controls, and a history of pneumonia increased the odds of COM regardless of sex and across all ages.
Pneumonia was defined as when a patient had a diagnosis of pneumonia based on ICD-10 codes and underwent a chest x-ray or chest CT scan. Chronic otitis media was defined as when a patient had a diagnosis based on ICD-10 codes at least two times with one of the following conditions: chronic serous otitis media, chronic mucoid otitis media, other chronic nonsuppurative otitis media, unspecified nonsuppurative otitis media, chronic tubotympanic suppurative otitis media, chronic atticoantral suppurative otitis media, other chronic suppurative otitis media, or unspecified suppurative otitis media.
Age groups were divided into 5-year intervals, and patients were classified into income groups and rural vs. urban residence.
In a further sensitivity analysis, individuals who were diagnosed with pneumonia five or more times before the index date had a significantly higher odds ratio for COM compared with those with less than five diagnoses of pneumonia (adjusted odds ratio, 1.34; P < .001).
Microbiome dysbiosis may explain part of the connection between pneumonia and COM, the researchers wrote in their discussion. Pathogens in the lungs can prompt changes in the microbiome dynamics, as might the use of antibiotics, they said. In addition, “Mucus plugging in the airway caused by pneumonia induces hypoxic conditions and leads to the expression of inflammatory markers in the eustachian tube and middle ear mucosa,” they noted.
The study findings were limited by several factors, including the retrospective design and lack of data on microbiological cultures for antibiotic susceptibility, radiologic findings on the severity of pneumonia, results of pulmonary function tests, and hearing thresholds, the researchers noted. Other limitations were the exclusion of the frequency of upper respiratory infections and antibiotic use due to lack of data, they said.
However, the results show an association between pneumonia diagnoses and increased incidence of COM, which suggests a novel perspective that “infection of the lower respiratory tract may affect the function of the eustachian tube and the middle ear to later cause COM,” they concluded.
The study received no outside funding. The researchers have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Individuals with a prior diagnosis of pneumonia were significantly more likely to develop chronic otitis media (COM) than were those without a history of pneumonia, based on data from a nationwide cohort study of more than 100,000 patients.
“Recently, middle ear diseases, including COM, have been recognized as respiratory tract diseases beyond the pathophysiological concepts of ventilation dysfunction, with recurrent infection that occurs from anatomically adjacent structures such as the middle ear, mastoid cavity, and eustachian tube,” but the potential link between pneumonia and chronic otitis media and adults in particular has not been examined, wrote Sung Kyun Kim, MD, of Hallym University, Dongtan, South Korea, and colleagues.
In a study recently published in the International Journal of Infectious Diseases, the researchers identified 23,436 adults with COM and 93,744 controls aged 40 years and older from a Korean health insurance database between 2002 and 2015.
The overall incidence of pneumonia in the study population was significantly higher in the COM group compared with controls (9.3% vs. 7.2%, P <.001). The odds ratios of pneumonia were significantly higher in the COM group compared with controls, and a history of pneumonia increased the odds of COM regardless of sex and across all ages.
Pneumonia was defined as when a patient had a diagnosis of pneumonia based on ICD-10 codes and underwent a chest x-ray or chest CT scan. Chronic otitis media was defined as when a patient had a diagnosis based on ICD-10 codes at least two times with one of the following conditions: chronic serous otitis media, chronic mucoid otitis media, other chronic nonsuppurative otitis media, unspecified nonsuppurative otitis media, chronic tubotympanic suppurative otitis media, chronic atticoantral suppurative otitis media, other chronic suppurative otitis media, or unspecified suppurative otitis media.
Age groups were divided into 5-year intervals, and patients were classified into income groups and rural vs. urban residence.
In a further sensitivity analysis, individuals who were diagnosed with pneumonia five or more times before the index date had a significantly higher odds ratio for COM compared with those with less than five diagnoses of pneumonia (adjusted odds ratio, 1.34; P < .001).
Microbiome dysbiosis may explain part of the connection between pneumonia and COM, the researchers wrote in their discussion. Pathogens in the lungs can prompt changes in the microbiome dynamics, as might the use of antibiotics, they said. In addition, “Mucus plugging in the airway caused by pneumonia induces hypoxic conditions and leads to the expression of inflammatory markers in the eustachian tube and middle ear mucosa,” they noted.
The study findings were limited by several factors, including the retrospective design and lack of data on microbiological cultures for antibiotic susceptibility, radiologic findings on the severity of pneumonia, results of pulmonary function tests, and hearing thresholds, the researchers noted. Other limitations were the exclusion of the frequency of upper respiratory infections and antibiotic use due to lack of data, they said.
However, the results show an association between pneumonia diagnoses and increased incidence of COM, which suggests a novel perspective that “infection of the lower respiratory tract may affect the function of the eustachian tube and the middle ear to later cause COM,” they concluded.
The study received no outside funding. The researchers have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Individuals with a prior diagnosis of pneumonia were significantly more likely to develop chronic otitis media (COM) than were those without a history of pneumonia, based on data from a nationwide cohort study of more than 100,000 patients.
“Recently, middle ear diseases, including COM, have been recognized as respiratory tract diseases beyond the pathophysiological concepts of ventilation dysfunction, with recurrent infection that occurs from anatomically adjacent structures such as the middle ear, mastoid cavity, and eustachian tube,” but the potential link between pneumonia and chronic otitis media and adults in particular has not been examined, wrote Sung Kyun Kim, MD, of Hallym University, Dongtan, South Korea, and colleagues.
In a study recently published in the International Journal of Infectious Diseases, the researchers identified 23,436 adults with COM and 93,744 controls aged 40 years and older from a Korean health insurance database between 2002 and 2015.
The overall incidence of pneumonia in the study population was significantly higher in the COM group compared with controls (9.3% vs. 7.2%, P <.001). The odds ratios of pneumonia were significantly higher in the COM group compared with controls, and a history of pneumonia increased the odds of COM regardless of sex and across all ages.
Pneumonia was defined as when a patient had a diagnosis of pneumonia based on ICD-10 codes and underwent a chest x-ray or chest CT scan. Chronic otitis media was defined as when a patient had a diagnosis based on ICD-10 codes at least two times with one of the following conditions: chronic serous otitis media, chronic mucoid otitis media, other chronic nonsuppurative otitis media, unspecified nonsuppurative otitis media, chronic tubotympanic suppurative otitis media, chronic atticoantral suppurative otitis media, other chronic suppurative otitis media, or unspecified suppurative otitis media.
Age groups were divided into 5-year intervals, and patients were classified into income groups and rural vs. urban residence.
In a further sensitivity analysis, individuals who were diagnosed with pneumonia five or more times before the index date had a significantly higher odds ratio for COM compared with those with less than five diagnoses of pneumonia (adjusted odds ratio, 1.34; P < .001).
Microbiome dysbiosis may explain part of the connection between pneumonia and COM, the researchers wrote in their discussion. Pathogens in the lungs can prompt changes in the microbiome dynamics, as might the use of antibiotics, they said. In addition, “Mucus plugging in the airway caused by pneumonia induces hypoxic conditions and leads to the expression of inflammatory markers in the eustachian tube and middle ear mucosa,” they noted.
The study findings were limited by several factors, including the retrospective design and lack of data on microbiological cultures for antibiotic susceptibility, radiologic findings on the severity of pneumonia, results of pulmonary function tests, and hearing thresholds, the researchers noted. Other limitations were the exclusion of the frequency of upper respiratory infections and antibiotic use due to lack of data, they said.
However, the results show an association between pneumonia diagnoses and increased incidence of COM, which suggests a novel perspective that “infection of the lower respiratory tract may affect the function of the eustachian tube and the middle ear to later cause COM,” they concluded.
The study received no outside funding. The researchers have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE INTERNATIONAL JOURNAL OF INFECTIOUS DISEASES
How common is IUD perforation, expulsion, and malposition?
The medicated intrauterine devices (IUDs), including the levonorgestrel-releasing IUD (LNG-IUD) (Mirena, Kyleena, Skyla, and Liletta) and the copper IUD (Cu-IUD; Paragard), are remarkably effective contraceptives. For the 52-mg LNG-IUD (Mirena, Liletta) the pregnancy rate over 6 years of use averaged less than 0.2% per year.1,2 For the Cu-IUD, the pregnancy rate over 10 years of use averaged 0.5% per year for the first 3 years of use and 0.2% per year over the following 7 years of use.3 IUD perforation of the uterus, expulsion, and malposition are recognized complications of IUD use. Our understanding of the prevalence and management of malpositioned IUDs is evolving and the main focus of this editorial.
Complete and partial uterus perforation
A complete uterine perforation occurs when the entire IUD is outside the walls of the uterus. A partial uterine perforation occurs when the IUD is outside the uterine cavity, but a portion of the IUD remains in the myometrium. When uterine perforation is suspected, ultrasound can determine if the IUD is properly sited within the uterus. If ultrasonography does not detect the IUD within the uterus, an x-ray of the pelvis and abdomen should be obtained to determine if the IUD is in the peritoneal cavity. If both an ultrasound and a pelvic-abdominal x-ray do not detect the IUD, the IUD was probably expelled from the patient.
Uterine perforation is uncommon and occurs once in every 500 to 1,000 insertions in non-breastfeeding women.4-8 The most common symptoms reported by patients with a perforated IUD are pain and/or bleeding.8 Investigators in the European Active Surveillance Study on Intrauterine Devices (EURAS) enrolled more than 60,000 patients who had an IUD insertion and followed them for 12 months with more than 39,000 followed for up to 60 months.7,8 The uterine perforation rate per 1,000 IUD insertions in non-breastfeeding women with 60 months of follow-up was 1.6 for the LNG-IUD and 0.8 for the Cu-IUD.8 The rate of uterine perforation was much higher in women who are breastfeeding or recently postpartum. In the EURAS study after 60 months of follow-up, the perforation rate per 1,000 insertions among breastfeeding women was 7.9 for the LNG-IUS and 4.7 for the Cu-IUD.8
Remarkably very few IUD perforations were detected at the time of insertion, including only 2% of the LNG-IUD insertions and 17% of the Cu-IUD insertions.8 Many perforations were not detected until more than 12 months following insertion, including 32% of the LNG-IUD insertions and 22% of the Cu-IUD insertions.8 Obviously, an IUD that has completely perforated the uterus and resides in the peritoneal cavity is not an effective contraceptive. For some patients, the IUD perforation was initially diagnosed after they became pregnant, and imaging studies to locate the IUD and assess the pregnancy were initiated. Complete perforation is usually treated with laparoscopy to remove the IUD and reduce the risk of injury to intra-abdominal organs.
Patients with an IUD partial perforation may present with pelvic pain or abnormal uterine bleeding.9 An ultrasound study to explore the cause of the presenting symptom may detect the partial perforation. It is estimated that approximately 20% of cases of IUD perforation are partial perforation.9 Over time, a partial perforation may progress to a complete perforation. In some cases of partial perforation, the IUD string may still be visible in the cervix, and the IUD may be removed by pulling on the strings.8 Hysteroscopy and/or laparoscopy may be needed to remove a partially perforated IUD. Following a partial or complete IUD perforation, if the patient desires to continue with IUD contraception, it would be wise to insert a new IUD under ultrasound guidance or assess proper placement with a postplacement ultrasound.
Continue to: Expulsion...
Expulsion
IUD expulsion occurs in approximately 3% to 11% of patients.10-13 The age of the patient influences the rate of expulsion. In a study of 2,748 patients with a Cu-IUD, the rate of expulsion by age for patients <20 years, 20–24 years, 25–29 years, 30–34 years, and ≥35 years was 8.2%, 3.2%, 3.0%, 2.3%, and 1.8%, respectively.10 In this study, age did not influence the rate of IUD removal for pelvic pain or abnormal bleeding, which was 4% to 5% across all age groups.10 In a study of 5,403 patients with an IUD, the rate of IUD expulsion by age for patients <20 years, 20–29 years, and 30–45 years was 14.6%, 7.3%, and 7.2%, respectively.12 In this study, the 3-year cumulative rate of expulsion was 10.2%.12 There was no statistically significant difference in the 3-year cumulative rate of expulsion for the 52-mg LNG-IUD (10.1%) and Cu-IUD (10.7%).12
The majority of patients who have an IUD expulsion recognize the event and seek additional contraception care. A few patients first recognize the IUD expulsion when they become pregnant, and imaging studies detect no IUD in the uterus or the peritoneal cavity. In a study of more than 17,000 patients using an LNG-IUD, 108 pregnancies were reported. Seven pregnancies occurred in patients who did not realize their IUD was expelled.14 Patients who have had an IUD expulsion and receive a new IUD are at increased risk for re-expulsion. For these patients, reinsertion of an IUD could be performed under ultrasound guidance to ensure and document optimal initial IUD position within the uterus, or ultrasound can be obtained postinsertion to document appropriate IUD position.
Malposition—prevalence and management
Our understanding of the prevalence and management of a malpositioned IUD is evolving. For the purposes of this discussion a malpositioned IUD is defined as being in the uterus, but not properly positioned within the uterine cavity. Perforation into the peritoneal cavity and complete expulsion of an IUD are considered separate entities. However, a malpositioned IUD within the uterus may eventually perforate the uterus or be expelled from the body. For example, an IUD embedded in the uterine wall may eventually work its way through the wall and become perforated, residing in the peritoneal cavity. An IUD with the stem in the cervix below the internal os may eventually be expelled from the uterus and leave the body through the vagina.
High-quality ultrasonography, including 2-dimensional (2-D) ultrasound with videoclips or 3-dimensional (3-D) ultrasound with coronal views, has greatly advanced our understanding of the prevalence and characteristics of a malpositioned IUD.15-18 Ultrasound features of an IUD correctly placed within the uterus include:
- the IUD is in the uterus
- the shaft is in the midline of the uterine cavity
- the shaft of the IUD is not in the endocervix
- the IUD arms are at a 90-degree angle from the shaft
- the top of the IUD is within 2 cm of the fundus
- the IUD is not rotated outside of the cornual plane, inverted or transverse.
Ultrasound imaging has identified multiple types of malpositioned IUDs, including:
- IUD embedded in the myometrium—a portion of the IUD is embedded in the uterine wall
- low-lying IUD—the IUD is low in the uterine cavity but not in the endocervix
- IUD in the endocervix—the stem is in the endocervical canal
- rotated—the IUD is rotated outside the cornual plane
- malpositioned arms—the arms are not at a 90-degree angle to the stem
- the IUD is inverted, transverse, or laterally displaced.
IUD malposition is highly prevalent and has been identified in 10% to 20% of convenience cohorts in which an ultrasound study was performed.15-18
Benacerraf, Shipp, and Bromley were among the first experts to use ultrasound to detect the high prevalence of malpositioned IUDs among a convenience sample of 167 patients with an IUD undergoing ultrasound for a variety of indications. Using 3-D ultrasound, including reconstructed coronal views, they identified 28 patients (17%) with a malpositioned IUD based on the detection of the IUD “poking into the substance of the uterus or cervix.” Among the patients with a malpositioned IUD, the principal indication for the ultrasound study was pelvic pain (39%) or abnormal uterine bleeding (36%). Among women with a normally sited IUD, pelvic pain (19%) or abnormal uterine bleeding (15%) were less often the principal indication for the ultrasound.15 The malpositioned IUD was removed in 21 of the 28 cases and the symptoms of pelvic pain or abnormal bleeding resolved in 20 of the 21 patients.15
Other investigators have confirmed the observation that IUD malposition is common.16-18 In a retrospective study of 1,748 pelvic ultrasounds performed for any indication where an IUD was present, after excluding 13 patients who were determined to have expelled their IUD (13) and 13 patients with a perforated IUD, 156 patients (8.9%) were diagnosed as having a malpositioned IUD.16 IUD malposition was diagnosed when the IUD was in the uterus but positioned in the lower uterine segment, cervix, rotated or embedded in the uterus. An IUD in the lower uterine segment or cervix was detected in 133 patients, representing 85% of cases. Among these cases, 29 IUDs were also embedded and/or rotated, indicating that some IUDs have multiple causes of the malposition. Twenty-one IUDs were near the fundus but embedded and/or rotated. Controls with a normally-sited IUD were selected for comparison to the case group. Among IUD users, the identification of suspected adenomyosis on the ultrasound was associated with an increased risk of IUD malposition (odds ratio [OR], 3.04; 95% confidence interval [CI], 1.08-8.52).16 In this study, removal of a malpositioned LNG-IUD, without initiating a highly reliable contraceptive was associated with an increased risk of pregnancy. It is important to initiate a highly reliable form of contraception if the plan is to remove a malpositioned IUD.16,19
In a study of 1,253 pelvic ultrasounds performed for any indication where an IUD was identified in the uterus, 263 IUDs (19%) were determined to be malpositioned.17 In this study the location of the malpositioned IUDs included17:
- the lower uterine segment not extending into the cervix (38%)
- in the lower uterine segment extending into the cervix (22%)
- in the cervix (26%)
- rotated axis of the IUD (12%)
- other (2%).
Among the 236 malpositioned IUDs, 24% appeared to be embedded in the uterine wall.17 Compared with patients with a normally-sited IUD on ultrasound, patients with a malpositioned IUD more frequently reported vaginal bleeding (30% vs 19%; P<.005) and pelvic pain (43% vs 30%; P<.002), similar to the findings in the Benacerraf et al. study.14
Connolly and Fox18 designed an innovative study to determine the rate of malpositioned IUDs using 2-D ultrasound to ensure proper IUD placement at the time of insertion with a follow-up 3-D ultrasound 8 weeks after insertion to assess IUD position within the uterus. At the 8-week 3-D ultrasound, among 763 women, 16.6% of the IUDs were malpositioned.18 In this study, IUD position was determined to be correct if all the following features were identified:
- the IUD shaft was in the midline of the uterine cavity
- the IUD arms were at 90 degrees from the stem
- the top of the IUD was within 3 to 4 mm of the fundus
- the IUD was not rotated, inverted or transverse.
IUD malpositions were categorized as:
- embedded in the uterine wall
- low in the uterine cavity
- in the endocervical canal
- misaligned
- perforated
- expulsed.
At the 8-week follow-up, 636 patients (83.4%) had an IUD that was correctly positioned.18 In 127 patients (16.6%) IUD malposition was identified, with some patients having more than one type of malposition. The types of malposition identified were:
- embedded in the myometrium (54%)
- misaligned, including rotated, laterally displaced, inverted, transverse or arms not deployed (47%)
- low in the uterine cavity (39%)
- in the endocervical canal (14%)
- perforated (3%)
- expulsion (0%).
Recall that all of these patients had a 2-D ultrasound at the time of insertion that identified the IUD as correctly placed. This suggests that during the 8 weeks following IUD placement there were changes in the location of the IUD or that 2-D ultrasound has lower sensitivity than 3-D ultrasound to detect malposition. Of note, at the 8-week follow-up, bleeding or pain was reported by 36% of the patients with a malpositioned IUD and 20% of patients with a correctly positioned IUD.17 Sixty-seven of the 127 malpositioned IUDs “required” removal, but the precise reasons for the removals were not delineated. The investigators concluded that 3-D ultrasonography is useful for the detection of IUD malposition and could be considered as part of ongoing IUD care, if symptoms of pain or bleeding occur.18
Continue to: IUD malposition following postplacental insertion...
IUD malposition following postplacental insertion
IUD malposition is common in patients who have had a postplacental insertion. Ultrasound imaging plays an important role in detecting IUD expulsion and malposition in these cases. Postplacental IUD insertion is defined as the placement of an IUD within 10 minutes following delivery of the placenta. Postplacental IUD insertion can be performed following a vaginal or cesarean birth and with a Cu-IUD or LNG-IUD. The good news is that postplacental IUD insertion reduces the risk of unplanned pregnancy in the years following birth. However, postplacental IUD insertion is associated with a high rate of IUD malposition.
In a study of 162 patients who had postplacental insertion of a Cu-IUD following a vaginal birth, ultrasound and physical examination at 6 months demonstrated complete IUD expulsion in 8%, partial expulsion in 16%, and malposition in 15%.20 The IUD was correctly sited in 56% of patients. Seven patients (4%) had the IUD removed, and 1 patient had a perforated IUD. Among the 25 malpositioned IUDs, 14 were not within 1 cm of the fundus, and 11 were rotated outside of the axis of the cornuas. In this study partial expulsion was defined as an IUD protruding from the external cervical os on physical exam or demonstration of the distal tip of the IUD below the internal os of the cervix on ultrasound. Malposition was defined as an IUD that was >1 cm from the fundus or in an abnormal location or axis, but not partially expelled.
In a study of 69 patients who had postplacental insertion of a Cu-IUD following a cesarean birth, ultrasound and physical examination at 6 months demonstrated complete IUD expulsion in 3%, partial expulsion (stem in the cervix below the internal os) in 4% and malposition in 30%.20 The IUD was correctly positioned in 59% of the patients.21 The IUD had been electively removed in 3%. Among the 21 patients with a malpositioned IUD, 10 were rotated within the uterine cavity, 6 were inverted (upside down), 3 were low-lying, and 2 were transverse.21 Given the relatively high rate of IUD malposition following postplacental insertion, it may be useful to perform a pelvic ultrasound at a postpartum visit to assess the location of the IUD, if ultrasonography is available.
Management of the malpositioned IUD
There are no consensus guidelines on how to care for a patient with a malpositioned IUD. Clinicians need to use their best judgment and engage the patient in joint decision making when managing a malpositioned IUD. When an IUD is malpositioned and the patient has bothersome symptoms of pelvic pain or abnormal bleeding that have not responded to standard interventions, consideration may be given to a remove and replace strategy. When the stem of the IUD is below the level of the internal os on ultrasound or visible at the external os on physical examination, consideration should be given to removing and replacing the IUD. However, if the IUD is removed without replacement or the initiation of a highly reliable contraceptive, the risk of unplanned pregnancy is considerable.16,19
IUD totally or partially within the cervix or low-lying. When an IUD is in the cervix, the contraceptive efficacy of the IUD may be diminished, especially with a Cu-IUD.22 In these cases, removing and replacing the IUD is an option. In a survey of 20 expert clinicians, >80% recommended replacing an IUD that was totally or partially in the cervical canal.23 But most of the experts would not replace an IUD that was incidentally noted on ultrasound to be low-lying, being positioned more than 2 cm below the fundus, with no portion of the IUD in the cervical canal. In the same survey, for patients with a low-lying IUD and pelvic pain or bleeding, the majority of experts reported that they would explore other causes of bleeding and pelvic pain not related to the IUD itself and not replace the IUD, but 30% of the experts reported that they would remove and replace the device.23
IUD embedded in the myometrium with pelvic pain. Based on my clinical experience, when a patient has persistent pelvic pain following the insertion of an IUD and the pain does not resolve with standard measures including medication, an ultrasound study is warranted to assess the position of the IUD. If the ultrasound demonstrates that an arm of the IUD is embedded in the myometrium, removal of the IUD may be associated with resolution of the pain. Reinsertion of an IUD under ultrasound guidance may result in a correctly-sited IUD with no recurrence of pelvic pain.
IUD rotated within the uterus with no pain or abnormal bleeding. For an IUD that is near the fundus and rotated on its axis within the uterus, if the patient has no symptoms of pain or abnormal bleeding, my recommendation to the patient would be to leave the device in situ.
Without available guidelines, engage in clinician-patient discussion
It is clear that IUD malposition is common, occurring in 10% to 20% of patients with an IUD. High-quality ultrasound imaging is helpful in detecting IUD malposition, including 2-D ultrasound with videoclips and/or 3-D ultrasound with coronal reconstruction. More data are needed to identify the best options for managing various types of malpositioned IUDs in patients with and without bothersome symptoms such as pain and bleeding. Until consensus guidelines are developed, clinicians need to engage the patient in a discussion of how to best manage the malpositioned IUD. Medicated IUDs and progestin subdermal implants are our two most effective reversible contraceptives. They are among the most important advances in health care over the past half-century. ●
- Mirena FDA approval. , 2022.
- Liletta [package insert]. Allergan USA: Irvine, California; 2019. .
- Paragard [package insert]. CooperSurgical Inc: Trumbull, Connecticut; 2019. .
- Harrison-Woolrych M, Ashton J, Coulter D. Uterine perforation on intrauterine device insertion: is the incidence higher than previously reported? Contraception. 2003;67:53-56.
- Van Houdenhoven K, van Kaam KJAF, van Grootheest AC, et al. Uterine perforation in women using a levonorgestrel-releasing intrauterine system. Contraception. 2006;73:257-260.
- van Grootheest K, Sachs B, Harrison-Woolrych M, et al. Uterine perforation with the levonorgestrel-releasing intrauterine device. Analysis of reports from four national pharmacovigilance centres. Drug Saf. 2011;34:83-88.
- Heinemann K, Reed S, Moehner S, et al. Risk of uterine perforation with levonorgestrel-releasing and copper intrauterine devices in the European Active Surveillance Study on Intrauterine Devices. Contraception. 2015;91:274-279.
- Barnett C, Moehner S, Do Minh T, et al. Perforation risk and intra-uterine devices: results of the EURAS-IUD 5-year extension study. Eur J Contracept Reprod Health Care. 2017;22:424-428.
- Zakin D, Stern WZ, Rosenblatt R. Complete and partial uterine perforation and embedding following insertion of intrauterine devices. I. Classification, complications, mechanism, incidence and missing string. Obstet Gynecol Surv. 1981;36:335-353.
- Rivera R, Chen-Mok M, McMullen S. Analysis of client characteristics that may affect early discontinuation of the TCu-380A IUD. Contraception. 1999;60:155-160.
- Aoun J, Dines VA, Stovall DW, et al. Effects of age, parity and device type on complications and discontinuation of intrauterine devices. Obstet Gynecol. 2014;123:585-592.
- Madden T, McNichols, Zhao Q, et al. Association of age and parity with intrauterine device expulsion. Obstet Gynecol. 2014;124:718-726.
- Keenahan L, Bercaw-Pratt JL, Adeyemi O, et al. Rates of intrauterine device expulsion among adolescents and young women. J Pediatr Adolesc Gynecol. 2021;34:362-365.
- Backman T, Rauramo I, Huhtala S, et al. Pregnancy during the use of levonorgestrel intrauterine system. Am J Obstet Gynecol. 2004;190:50-54.
- Benacerraf BR, Shipp TD, Bromley B. Three-dimensional ultrasound detection of abnormally located intrauterine contraceptive devices which are a source of pelvic pain and abnormal bleeding. Ultrasound Obstet Gynecol. 2009;34:110-115.
- Braaten KP, Benson CB, Maurer R, et al. Malpositioned intrauterine contraceptive devices: risk factors, outcomes and future pregnancies. Obstet Gynecol. 2011;118:1014-1020.
- Gerkowicz SA, Fiorentino DG, Kovacs AP, et al. Uterine structural abnormality and intrauterine device malposition: analysis of ultrasonographic and demographic variables of 517 patients. Am J Obstet Gynecol. 2019;220:183.e1-e8.
- Connolly CT, Fox NS. Incidence and risk factors for a malpositioned intrauterine device detected on three-dimensional ultrasound within eight weeks of placement. J Ultrasound Med. 2021 ePub Sept 27 2021.
- Golightly E, Gebbie AE. Low-lying or malpositioned intrauterine devices and systems. J Fam Plann Reprod health Care. 2014;40:108-112.
- Gurney EP, Sonalkar S, McAllister A, et al. Six-month expulsion of postplacental copper intrauterine devices placed after vaginal delivery. Am J Obstet Gynecol. 2018;219:183.e1-e9.
- Gurney EP, McAllister A, Lang B, et al. Ultrasound assessment of postplacental copper intrauterine device position 6 months after placement during cesarean delivery. Contraception. 2020;2:100040.
- Anteby E, Revel A, Ben-Chetrit A, et al. Intrauterine device failure: relation to its location with the uterine cavity. Obstet Gynecol. 1993;81:112-114.
- Golightly E, Gebbie AE. Clinicians’ views on low-lying intrauterine devices or systems. J Fam Plann Reprod Health Care. 2014;40:113-116.

Robert L. Barbieri, MD
Editor in Chief, OBG Management
Chair Emeritus, Department of Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
Dr. Barbieri reports no financial relationships relevant to this article.
Robert L. Barbieri, MD
Editor in Chief, OBG Management
Chair Emeritus, Department of Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
Dr. Barbieri reports no financial relationships relevant to this article.
Robert L. Barbieri, MD
Editor in Chief, OBG Management
Chair Emeritus, Department of Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
Dr. Barbieri reports no financial relationships relevant to this article.
The medicated intrauterine devices (IUDs), including the levonorgestrel-releasing IUD (LNG-IUD) (Mirena, Kyleena, Skyla, and Liletta) and the copper IUD (Cu-IUD; Paragard), are remarkably effective contraceptives. For the 52-mg LNG-IUD (Mirena, Liletta) the pregnancy rate over 6 years of use averaged less than 0.2% per year.1,2 For the Cu-IUD, the pregnancy rate over 10 years of use averaged 0.5% per year for the first 3 years of use and 0.2% per year over the following 7 years of use.3 IUD perforation of the uterus, expulsion, and malposition are recognized complications of IUD use. Our understanding of the prevalence and management of malpositioned IUDs is evolving and the main focus of this editorial.
Complete and partial uterus perforation
A complete uterine perforation occurs when the entire IUD is outside the walls of the uterus. A partial uterine perforation occurs when the IUD is outside the uterine cavity, but a portion of the IUD remains in the myometrium. When uterine perforation is suspected, ultrasound can determine if the IUD is properly sited within the uterus. If ultrasonography does not detect the IUD within the uterus, an x-ray of the pelvis and abdomen should be obtained to determine if the IUD is in the peritoneal cavity. If both an ultrasound and a pelvic-abdominal x-ray do not detect the IUD, the IUD was probably expelled from the patient.
Uterine perforation is uncommon and occurs once in every 500 to 1,000 insertions in non-breastfeeding women.4-8 The most common symptoms reported by patients with a perforated IUD are pain and/or bleeding.8 Investigators in the European Active Surveillance Study on Intrauterine Devices (EURAS) enrolled more than 60,000 patients who had an IUD insertion and followed them for 12 months with more than 39,000 followed for up to 60 months.7,8 The uterine perforation rate per 1,000 IUD insertions in non-breastfeeding women with 60 months of follow-up was 1.6 for the LNG-IUD and 0.8 for the Cu-IUD.8 The rate of uterine perforation was much higher in women who are breastfeeding or recently postpartum. In the EURAS study after 60 months of follow-up, the perforation rate per 1,000 insertions among breastfeeding women was 7.9 for the LNG-IUS and 4.7 for the Cu-IUD.8
Remarkably very few IUD perforations were detected at the time of insertion, including only 2% of the LNG-IUD insertions and 17% of the Cu-IUD insertions.8 Many perforations were not detected until more than 12 months following insertion, including 32% of the LNG-IUD insertions and 22% of the Cu-IUD insertions.8 Obviously, an IUD that has completely perforated the uterus and resides in the peritoneal cavity is not an effective contraceptive. For some patients, the IUD perforation was initially diagnosed after they became pregnant, and imaging studies to locate the IUD and assess the pregnancy were initiated. Complete perforation is usually treated with laparoscopy to remove the IUD and reduce the risk of injury to intra-abdominal organs.
Patients with an IUD partial perforation may present with pelvic pain or abnormal uterine bleeding.9 An ultrasound study to explore the cause of the presenting symptom may detect the partial perforation. It is estimated that approximately 20% of cases of IUD perforation are partial perforation.9 Over time, a partial perforation may progress to a complete perforation. In some cases of partial perforation, the IUD string may still be visible in the cervix, and the IUD may be removed by pulling on the strings.8 Hysteroscopy and/or laparoscopy may be needed to remove a partially perforated IUD. Following a partial or complete IUD perforation, if the patient desires to continue with IUD contraception, it would be wise to insert a new IUD under ultrasound guidance or assess proper placement with a postplacement ultrasound.
Continue to: Expulsion...
Expulsion
IUD expulsion occurs in approximately 3% to 11% of patients.10-13 The age of the patient influences the rate of expulsion. In a study of 2,748 patients with a Cu-IUD, the rate of expulsion by age for patients <20 years, 20–24 years, 25–29 years, 30–34 years, and ≥35 years was 8.2%, 3.2%, 3.0%, 2.3%, and 1.8%, respectively.10 In this study, age did not influence the rate of IUD removal for pelvic pain or abnormal bleeding, which was 4% to 5% across all age groups.10 In a study of 5,403 patients with an IUD, the rate of IUD expulsion by age for patients <20 years, 20–29 years, and 30–45 years was 14.6%, 7.3%, and 7.2%, respectively.12 In this study, the 3-year cumulative rate of expulsion was 10.2%.12 There was no statistically significant difference in the 3-year cumulative rate of expulsion for the 52-mg LNG-IUD (10.1%) and Cu-IUD (10.7%).12
The majority of patients who have an IUD expulsion recognize the event and seek additional contraception care. A few patients first recognize the IUD expulsion when they become pregnant, and imaging studies detect no IUD in the uterus or the peritoneal cavity. In a study of more than 17,000 patients using an LNG-IUD, 108 pregnancies were reported. Seven pregnancies occurred in patients who did not realize their IUD was expelled.14 Patients who have had an IUD expulsion and receive a new IUD are at increased risk for re-expulsion. For these patients, reinsertion of an IUD could be performed under ultrasound guidance to ensure and document optimal initial IUD position within the uterus, or ultrasound can be obtained postinsertion to document appropriate IUD position.
Malposition—prevalence and management
Our understanding of the prevalence and management of a malpositioned IUD is evolving. For the purposes of this discussion a malpositioned IUD is defined as being in the uterus, but not properly positioned within the uterine cavity. Perforation into the peritoneal cavity and complete expulsion of an IUD are considered separate entities. However, a malpositioned IUD within the uterus may eventually perforate the uterus or be expelled from the body. For example, an IUD embedded in the uterine wall may eventually work its way through the wall and become perforated, residing in the peritoneal cavity. An IUD with the stem in the cervix below the internal os may eventually be expelled from the uterus and leave the body through the vagina.
High-quality ultrasonography, including 2-dimensional (2-D) ultrasound with videoclips or 3-dimensional (3-D) ultrasound with coronal views, has greatly advanced our understanding of the prevalence and characteristics of a malpositioned IUD.15-18 Ultrasound features of an IUD correctly placed within the uterus include:
- the IUD is in the uterus
- the shaft is in the midline of the uterine cavity
- the shaft of the IUD is not in the endocervix
- the IUD arms are at a 90-degree angle from the shaft
- the top of the IUD is within 2 cm of the fundus
- the IUD is not rotated outside of the cornual plane, inverted or transverse.
Ultrasound imaging has identified multiple types of malpositioned IUDs, including:
- IUD embedded in the myometrium—a portion of the IUD is embedded in the uterine wall
- low-lying IUD—the IUD is low in the uterine cavity but not in the endocervix
- IUD in the endocervix—the stem is in the endocervical canal
- rotated—the IUD is rotated outside the cornual plane
- malpositioned arms—the arms are not at a 90-degree angle to the stem
- the IUD is inverted, transverse, or laterally displaced.
IUD malposition is highly prevalent and has been identified in 10% to 20% of convenience cohorts in which an ultrasound study was performed.15-18
Benacerraf, Shipp, and Bromley were among the first experts to use ultrasound to detect the high prevalence of malpositioned IUDs among a convenience sample of 167 patients with an IUD undergoing ultrasound for a variety of indications. Using 3-D ultrasound, including reconstructed coronal views, they identified 28 patients (17%) with a malpositioned IUD based on the detection of the IUD “poking into the substance of the uterus or cervix.” Among the patients with a malpositioned IUD, the principal indication for the ultrasound study was pelvic pain (39%) or abnormal uterine bleeding (36%). Among women with a normally sited IUD, pelvic pain (19%) or abnormal uterine bleeding (15%) were less often the principal indication for the ultrasound.15 The malpositioned IUD was removed in 21 of the 28 cases and the symptoms of pelvic pain or abnormal bleeding resolved in 20 of the 21 patients.15
Other investigators have confirmed the observation that IUD malposition is common.16-18 In a retrospective study of 1,748 pelvic ultrasounds performed for any indication where an IUD was present, after excluding 13 patients who were determined to have expelled their IUD (13) and 13 patients with a perforated IUD, 156 patients (8.9%) were diagnosed as having a malpositioned IUD.16 IUD malposition was diagnosed when the IUD was in the uterus but positioned in the lower uterine segment, cervix, rotated or embedded in the uterus. An IUD in the lower uterine segment or cervix was detected in 133 patients, representing 85% of cases. Among these cases, 29 IUDs were also embedded and/or rotated, indicating that some IUDs have multiple causes of the malposition. Twenty-one IUDs were near the fundus but embedded and/or rotated. Controls with a normally-sited IUD were selected for comparison to the case group. Among IUD users, the identification of suspected adenomyosis on the ultrasound was associated with an increased risk of IUD malposition (odds ratio [OR], 3.04; 95% confidence interval [CI], 1.08-8.52).16 In this study, removal of a malpositioned LNG-IUD, without initiating a highly reliable contraceptive was associated with an increased risk of pregnancy. It is important to initiate a highly reliable form of contraception if the plan is to remove a malpositioned IUD.16,19
In a study of 1,253 pelvic ultrasounds performed for any indication where an IUD was identified in the uterus, 263 IUDs (19%) were determined to be malpositioned.17 In this study the location of the malpositioned IUDs included17:
- the lower uterine segment not extending into the cervix (38%)
- in the lower uterine segment extending into the cervix (22%)
- in the cervix (26%)
- rotated axis of the IUD (12%)
- other (2%).
Among the 236 malpositioned IUDs, 24% appeared to be embedded in the uterine wall.17 Compared with patients with a normally-sited IUD on ultrasound, patients with a malpositioned IUD more frequently reported vaginal bleeding (30% vs 19%; P<.005) and pelvic pain (43% vs 30%; P<.002), similar to the findings in the Benacerraf et al. study.14
Connolly and Fox18 designed an innovative study to determine the rate of malpositioned IUDs using 2-D ultrasound to ensure proper IUD placement at the time of insertion with a follow-up 3-D ultrasound 8 weeks after insertion to assess IUD position within the uterus. At the 8-week 3-D ultrasound, among 763 women, 16.6% of the IUDs were malpositioned.18 In this study, IUD position was determined to be correct if all the following features were identified:
- the IUD shaft was in the midline of the uterine cavity
- the IUD arms were at 90 degrees from the stem
- the top of the IUD was within 3 to 4 mm of the fundus
- the IUD was not rotated, inverted or transverse.
IUD malpositions were categorized as:
- embedded in the uterine wall
- low in the uterine cavity
- in the endocervical canal
- misaligned
- perforated
- expulsed.
At the 8-week follow-up, 636 patients (83.4%) had an IUD that was correctly positioned.18 In 127 patients (16.6%) IUD malposition was identified, with some patients having more than one type of malposition. The types of malposition identified were:
- embedded in the myometrium (54%)
- misaligned, including rotated, laterally displaced, inverted, transverse or arms not deployed (47%)
- low in the uterine cavity (39%)
- in the endocervical canal (14%)
- perforated (3%)
- expulsion (0%).
Recall that all of these patients had a 2-D ultrasound at the time of insertion that identified the IUD as correctly placed. This suggests that during the 8 weeks following IUD placement there were changes in the location of the IUD or that 2-D ultrasound has lower sensitivity than 3-D ultrasound to detect malposition. Of note, at the 8-week follow-up, bleeding or pain was reported by 36% of the patients with a malpositioned IUD and 20% of patients with a correctly positioned IUD.17 Sixty-seven of the 127 malpositioned IUDs “required” removal, but the precise reasons for the removals were not delineated. The investigators concluded that 3-D ultrasonography is useful for the detection of IUD malposition and could be considered as part of ongoing IUD care, if symptoms of pain or bleeding occur.18
Continue to: IUD malposition following postplacental insertion...
IUD malposition following postplacental insertion
IUD malposition is common in patients who have had a postplacental insertion. Ultrasound imaging plays an important role in detecting IUD expulsion and malposition in these cases. Postplacental IUD insertion is defined as the placement of an IUD within 10 minutes following delivery of the placenta. Postplacental IUD insertion can be performed following a vaginal or cesarean birth and with a Cu-IUD or LNG-IUD. The good news is that postplacental IUD insertion reduces the risk of unplanned pregnancy in the years following birth. However, postplacental IUD insertion is associated with a high rate of IUD malposition.
In a study of 162 patients who had postplacental insertion of a Cu-IUD following a vaginal birth, ultrasound and physical examination at 6 months demonstrated complete IUD expulsion in 8%, partial expulsion in 16%, and malposition in 15%.20 The IUD was correctly sited in 56% of patients. Seven patients (4%) had the IUD removed, and 1 patient had a perforated IUD. Among the 25 malpositioned IUDs, 14 were not within 1 cm of the fundus, and 11 were rotated outside of the axis of the cornuas. In this study partial expulsion was defined as an IUD protruding from the external cervical os on physical exam or demonstration of the distal tip of the IUD below the internal os of the cervix on ultrasound. Malposition was defined as an IUD that was >1 cm from the fundus or in an abnormal location or axis, but not partially expelled.
In a study of 69 patients who had postplacental insertion of a Cu-IUD following a cesarean birth, ultrasound and physical examination at 6 months demonstrated complete IUD expulsion in 3%, partial expulsion (stem in the cervix below the internal os) in 4% and malposition in 30%.20 The IUD was correctly positioned in 59% of the patients.21 The IUD had been electively removed in 3%. Among the 21 patients with a malpositioned IUD, 10 were rotated within the uterine cavity, 6 were inverted (upside down), 3 were low-lying, and 2 were transverse.21 Given the relatively high rate of IUD malposition following postplacental insertion, it may be useful to perform a pelvic ultrasound at a postpartum visit to assess the location of the IUD, if ultrasonography is available.
Management of the malpositioned IUD
There are no consensus guidelines on how to care for a patient with a malpositioned IUD. Clinicians need to use their best judgment and engage the patient in joint decision making when managing a malpositioned IUD. When an IUD is malpositioned and the patient has bothersome symptoms of pelvic pain or abnormal bleeding that have not responded to standard interventions, consideration may be given to a remove and replace strategy. When the stem of the IUD is below the level of the internal os on ultrasound or visible at the external os on physical examination, consideration should be given to removing and replacing the IUD. However, if the IUD is removed without replacement or the initiation of a highly reliable contraceptive, the risk of unplanned pregnancy is considerable.16,19
IUD totally or partially within the cervix or low-lying. When an IUD is in the cervix, the contraceptive efficacy of the IUD may be diminished, especially with a Cu-IUD.22 In these cases, removing and replacing the IUD is an option. In a survey of 20 expert clinicians, >80% recommended replacing an IUD that was totally or partially in the cervical canal.23 But most of the experts would not replace an IUD that was incidentally noted on ultrasound to be low-lying, being positioned more than 2 cm below the fundus, with no portion of the IUD in the cervical canal. In the same survey, for patients with a low-lying IUD and pelvic pain or bleeding, the majority of experts reported that they would explore other causes of bleeding and pelvic pain not related to the IUD itself and not replace the IUD, but 30% of the experts reported that they would remove and replace the device.23
IUD embedded in the myometrium with pelvic pain. Based on my clinical experience, when a patient has persistent pelvic pain following the insertion of an IUD and the pain does not resolve with standard measures including medication, an ultrasound study is warranted to assess the position of the IUD. If the ultrasound demonstrates that an arm of the IUD is embedded in the myometrium, removal of the IUD may be associated with resolution of the pain. Reinsertion of an IUD under ultrasound guidance may result in a correctly-sited IUD with no recurrence of pelvic pain.
IUD rotated within the uterus with no pain or abnormal bleeding. For an IUD that is near the fundus and rotated on its axis within the uterus, if the patient has no symptoms of pain or abnormal bleeding, my recommendation to the patient would be to leave the device in situ.
Without available guidelines, engage in clinician-patient discussion
It is clear that IUD malposition is common, occurring in 10% to 20% of patients with an IUD. High-quality ultrasound imaging is helpful in detecting IUD malposition, including 2-D ultrasound with videoclips and/or 3-D ultrasound with coronal reconstruction. More data are needed to identify the best options for managing various types of malpositioned IUDs in patients with and without bothersome symptoms such as pain and bleeding. Until consensus guidelines are developed, clinicians need to engage the patient in a discussion of how to best manage the malpositioned IUD. Medicated IUDs and progestin subdermal implants are our two most effective reversible contraceptives. They are among the most important advances in health care over the past half-century. ●
The medicated intrauterine devices (IUDs), including the levonorgestrel-releasing IUD (LNG-IUD) (Mirena, Kyleena, Skyla, and Liletta) and the copper IUD (Cu-IUD; Paragard), are remarkably effective contraceptives. For the 52-mg LNG-IUD (Mirena, Liletta) the pregnancy rate over 6 years of use averaged less than 0.2% per year.1,2 For the Cu-IUD, the pregnancy rate over 10 years of use averaged 0.5% per year for the first 3 years of use and 0.2% per year over the following 7 years of use.3 IUD perforation of the uterus, expulsion, and malposition are recognized complications of IUD use. Our understanding of the prevalence and management of malpositioned IUDs is evolving and the main focus of this editorial.
Complete and partial uterus perforation
A complete uterine perforation occurs when the entire IUD is outside the walls of the uterus. A partial uterine perforation occurs when the IUD is outside the uterine cavity, but a portion of the IUD remains in the myometrium. When uterine perforation is suspected, ultrasound can determine if the IUD is properly sited within the uterus. If ultrasonography does not detect the IUD within the uterus, an x-ray of the pelvis and abdomen should be obtained to determine if the IUD is in the peritoneal cavity. If both an ultrasound and a pelvic-abdominal x-ray do not detect the IUD, the IUD was probably expelled from the patient.
Uterine perforation is uncommon and occurs once in every 500 to 1,000 insertions in non-breastfeeding women.4-8 The most common symptoms reported by patients with a perforated IUD are pain and/or bleeding.8 Investigators in the European Active Surveillance Study on Intrauterine Devices (EURAS) enrolled more than 60,000 patients who had an IUD insertion and followed them for 12 months with more than 39,000 followed for up to 60 months.7,8 The uterine perforation rate per 1,000 IUD insertions in non-breastfeeding women with 60 months of follow-up was 1.6 for the LNG-IUD and 0.8 for the Cu-IUD.8 The rate of uterine perforation was much higher in women who are breastfeeding or recently postpartum. In the EURAS study after 60 months of follow-up, the perforation rate per 1,000 insertions among breastfeeding women was 7.9 for the LNG-IUS and 4.7 for the Cu-IUD.8
Remarkably very few IUD perforations were detected at the time of insertion, including only 2% of the LNG-IUD insertions and 17% of the Cu-IUD insertions.8 Many perforations were not detected until more than 12 months following insertion, including 32% of the LNG-IUD insertions and 22% of the Cu-IUD insertions.8 Obviously, an IUD that has completely perforated the uterus and resides in the peritoneal cavity is not an effective contraceptive. For some patients, the IUD perforation was initially diagnosed after they became pregnant, and imaging studies to locate the IUD and assess the pregnancy were initiated. Complete perforation is usually treated with laparoscopy to remove the IUD and reduce the risk of injury to intra-abdominal organs.
Patients with an IUD partial perforation may present with pelvic pain or abnormal uterine bleeding.9 An ultrasound study to explore the cause of the presenting symptom may detect the partial perforation. It is estimated that approximately 20% of cases of IUD perforation are partial perforation.9 Over time, a partial perforation may progress to a complete perforation. In some cases of partial perforation, the IUD string may still be visible in the cervix, and the IUD may be removed by pulling on the strings.8 Hysteroscopy and/or laparoscopy may be needed to remove a partially perforated IUD. Following a partial or complete IUD perforation, if the patient desires to continue with IUD contraception, it would be wise to insert a new IUD under ultrasound guidance or assess proper placement with a postplacement ultrasound.
Continue to: Expulsion...
Expulsion
IUD expulsion occurs in approximately 3% to 11% of patients.10-13 The age of the patient influences the rate of expulsion. In a study of 2,748 patients with a Cu-IUD, the rate of expulsion by age for patients <20 years, 20–24 years, 25–29 years, 30–34 years, and ≥35 years was 8.2%, 3.2%, 3.0%, 2.3%, and 1.8%, respectively.10 In this study, age did not influence the rate of IUD removal for pelvic pain or abnormal bleeding, which was 4% to 5% across all age groups.10 In a study of 5,403 patients with an IUD, the rate of IUD expulsion by age for patients <20 years, 20–29 years, and 30–45 years was 14.6%, 7.3%, and 7.2%, respectively.12 In this study, the 3-year cumulative rate of expulsion was 10.2%.12 There was no statistically significant difference in the 3-year cumulative rate of expulsion for the 52-mg LNG-IUD (10.1%) and Cu-IUD (10.7%).12
The majority of patients who have an IUD expulsion recognize the event and seek additional contraception care. A few patients first recognize the IUD expulsion when they become pregnant, and imaging studies detect no IUD in the uterus or the peritoneal cavity. In a study of more than 17,000 patients using an LNG-IUD, 108 pregnancies were reported. Seven pregnancies occurred in patients who did not realize their IUD was expelled.14 Patients who have had an IUD expulsion and receive a new IUD are at increased risk for re-expulsion. For these patients, reinsertion of an IUD could be performed under ultrasound guidance to ensure and document optimal initial IUD position within the uterus, or ultrasound can be obtained postinsertion to document appropriate IUD position.
Malposition—prevalence and management
Our understanding of the prevalence and management of a malpositioned IUD is evolving. For the purposes of this discussion a malpositioned IUD is defined as being in the uterus, but not properly positioned within the uterine cavity. Perforation into the peritoneal cavity and complete expulsion of an IUD are considered separate entities. However, a malpositioned IUD within the uterus may eventually perforate the uterus or be expelled from the body. For example, an IUD embedded in the uterine wall may eventually work its way through the wall and become perforated, residing in the peritoneal cavity. An IUD with the stem in the cervix below the internal os may eventually be expelled from the uterus and leave the body through the vagina.
High-quality ultrasonography, including 2-dimensional (2-D) ultrasound with videoclips or 3-dimensional (3-D) ultrasound with coronal views, has greatly advanced our understanding of the prevalence and characteristics of a malpositioned IUD.15-18 Ultrasound features of an IUD correctly placed within the uterus include:
- the IUD is in the uterus
- the shaft is in the midline of the uterine cavity
- the shaft of the IUD is not in the endocervix
- the IUD arms are at a 90-degree angle from the shaft
- the top of the IUD is within 2 cm of the fundus
- the IUD is not rotated outside of the cornual plane, inverted or transverse.
Ultrasound imaging has identified multiple types of malpositioned IUDs, including:
- IUD embedded in the myometrium—a portion of the IUD is embedded in the uterine wall
- low-lying IUD—the IUD is low in the uterine cavity but not in the endocervix
- IUD in the endocervix—the stem is in the endocervical canal
- rotated—the IUD is rotated outside the cornual plane
- malpositioned arms—the arms are not at a 90-degree angle to the stem
- the IUD is inverted, transverse, or laterally displaced.
IUD malposition is highly prevalent and has been identified in 10% to 20% of convenience cohorts in which an ultrasound study was performed.15-18
Benacerraf, Shipp, and Bromley were among the first experts to use ultrasound to detect the high prevalence of malpositioned IUDs among a convenience sample of 167 patients with an IUD undergoing ultrasound for a variety of indications. Using 3-D ultrasound, including reconstructed coronal views, they identified 28 patients (17%) with a malpositioned IUD based on the detection of the IUD “poking into the substance of the uterus or cervix.” Among the patients with a malpositioned IUD, the principal indication for the ultrasound study was pelvic pain (39%) or abnormal uterine bleeding (36%). Among women with a normally sited IUD, pelvic pain (19%) or abnormal uterine bleeding (15%) were less often the principal indication for the ultrasound.15 The malpositioned IUD was removed in 21 of the 28 cases and the symptoms of pelvic pain or abnormal bleeding resolved in 20 of the 21 patients.15
Other investigators have confirmed the observation that IUD malposition is common.16-18 In a retrospective study of 1,748 pelvic ultrasounds performed for any indication where an IUD was present, after excluding 13 patients who were determined to have expelled their IUD (13) and 13 patients with a perforated IUD, 156 patients (8.9%) were diagnosed as having a malpositioned IUD.16 IUD malposition was diagnosed when the IUD was in the uterus but positioned in the lower uterine segment, cervix, rotated or embedded in the uterus. An IUD in the lower uterine segment or cervix was detected in 133 patients, representing 85% of cases. Among these cases, 29 IUDs were also embedded and/or rotated, indicating that some IUDs have multiple causes of the malposition. Twenty-one IUDs were near the fundus but embedded and/or rotated. Controls with a normally-sited IUD were selected for comparison to the case group. Among IUD users, the identification of suspected adenomyosis on the ultrasound was associated with an increased risk of IUD malposition (odds ratio [OR], 3.04; 95% confidence interval [CI], 1.08-8.52).16 In this study, removal of a malpositioned LNG-IUD, without initiating a highly reliable contraceptive was associated with an increased risk of pregnancy. It is important to initiate a highly reliable form of contraception if the plan is to remove a malpositioned IUD.16,19
In a study of 1,253 pelvic ultrasounds performed for any indication where an IUD was identified in the uterus, 263 IUDs (19%) were determined to be malpositioned.17 In this study the location of the malpositioned IUDs included17:
- the lower uterine segment not extending into the cervix (38%)
- in the lower uterine segment extending into the cervix (22%)
- in the cervix (26%)
- rotated axis of the IUD (12%)
- other (2%).
Among the 236 malpositioned IUDs, 24% appeared to be embedded in the uterine wall.17 Compared with patients with a normally-sited IUD on ultrasound, patients with a malpositioned IUD more frequently reported vaginal bleeding (30% vs 19%; P<.005) and pelvic pain (43% vs 30%; P<.002), similar to the findings in the Benacerraf et al. study.14
Connolly and Fox18 designed an innovative study to determine the rate of malpositioned IUDs using 2-D ultrasound to ensure proper IUD placement at the time of insertion with a follow-up 3-D ultrasound 8 weeks after insertion to assess IUD position within the uterus. At the 8-week 3-D ultrasound, among 763 women, 16.6% of the IUDs were malpositioned.18 In this study, IUD position was determined to be correct if all the following features were identified:
- the IUD shaft was in the midline of the uterine cavity
- the IUD arms were at 90 degrees from the stem
- the top of the IUD was within 3 to 4 mm of the fundus
- the IUD was not rotated, inverted or transverse.
IUD malpositions were categorized as:
- embedded in the uterine wall
- low in the uterine cavity
- in the endocervical canal
- misaligned
- perforated
- expulsed.
At the 8-week follow-up, 636 patients (83.4%) had an IUD that was correctly positioned.18 In 127 patients (16.6%) IUD malposition was identified, with some patients having more than one type of malposition. The types of malposition identified were:
- embedded in the myometrium (54%)
- misaligned, including rotated, laterally displaced, inverted, transverse or arms not deployed (47%)
- low in the uterine cavity (39%)
- in the endocervical canal (14%)
- perforated (3%)
- expulsion (0%).
Recall that all of these patients had a 2-D ultrasound at the time of insertion that identified the IUD as correctly placed. This suggests that during the 8 weeks following IUD placement there were changes in the location of the IUD or that 2-D ultrasound has lower sensitivity than 3-D ultrasound to detect malposition. Of note, at the 8-week follow-up, bleeding or pain was reported by 36% of the patients with a malpositioned IUD and 20% of patients with a correctly positioned IUD.17 Sixty-seven of the 127 malpositioned IUDs “required” removal, but the precise reasons for the removals were not delineated. The investigators concluded that 3-D ultrasonography is useful for the detection of IUD malposition and could be considered as part of ongoing IUD care, if symptoms of pain or bleeding occur.18
Continue to: IUD malposition following postplacental insertion...
IUD malposition following postplacental insertion
IUD malposition is common in patients who have had a postplacental insertion. Ultrasound imaging plays an important role in detecting IUD expulsion and malposition in these cases. Postplacental IUD insertion is defined as the placement of an IUD within 10 minutes following delivery of the placenta. Postplacental IUD insertion can be performed following a vaginal or cesarean birth and with a Cu-IUD or LNG-IUD. The good news is that postplacental IUD insertion reduces the risk of unplanned pregnancy in the years following birth. However, postplacental IUD insertion is associated with a high rate of IUD malposition.
In a study of 162 patients who had postplacental insertion of a Cu-IUD following a vaginal birth, ultrasound and physical examination at 6 months demonstrated complete IUD expulsion in 8%, partial expulsion in 16%, and malposition in 15%.20 The IUD was correctly sited in 56% of patients. Seven patients (4%) had the IUD removed, and 1 patient had a perforated IUD. Among the 25 malpositioned IUDs, 14 were not within 1 cm of the fundus, and 11 were rotated outside of the axis of the cornuas. In this study partial expulsion was defined as an IUD protruding from the external cervical os on physical exam or demonstration of the distal tip of the IUD below the internal os of the cervix on ultrasound. Malposition was defined as an IUD that was >1 cm from the fundus or in an abnormal location or axis, but not partially expelled.
In a study of 69 patients who had postplacental insertion of a Cu-IUD following a cesarean birth, ultrasound and physical examination at 6 months demonstrated complete IUD expulsion in 3%, partial expulsion (stem in the cervix below the internal os) in 4% and malposition in 30%.20 The IUD was correctly positioned in 59% of the patients.21 The IUD had been electively removed in 3%. Among the 21 patients with a malpositioned IUD, 10 were rotated within the uterine cavity, 6 were inverted (upside down), 3 were low-lying, and 2 were transverse.21 Given the relatively high rate of IUD malposition following postplacental insertion, it may be useful to perform a pelvic ultrasound at a postpartum visit to assess the location of the IUD, if ultrasonography is available.
Management of the malpositioned IUD
There are no consensus guidelines on how to care for a patient with a malpositioned IUD. Clinicians need to use their best judgment and engage the patient in joint decision making when managing a malpositioned IUD. When an IUD is malpositioned and the patient has bothersome symptoms of pelvic pain or abnormal bleeding that have not responded to standard interventions, consideration may be given to a remove and replace strategy. When the stem of the IUD is below the level of the internal os on ultrasound or visible at the external os on physical examination, consideration should be given to removing and replacing the IUD. However, if the IUD is removed without replacement or the initiation of a highly reliable contraceptive, the risk of unplanned pregnancy is considerable.16,19
IUD totally or partially within the cervix or low-lying. When an IUD is in the cervix, the contraceptive efficacy of the IUD may be diminished, especially with a Cu-IUD.22 In these cases, removing and replacing the IUD is an option. In a survey of 20 expert clinicians, >80% recommended replacing an IUD that was totally or partially in the cervical canal.23 But most of the experts would not replace an IUD that was incidentally noted on ultrasound to be low-lying, being positioned more than 2 cm below the fundus, with no portion of the IUD in the cervical canal. In the same survey, for patients with a low-lying IUD and pelvic pain or bleeding, the majority of experts reported that they would explore other causes of bleeding and pelvic pain not related to the IUD itself and not replace the IUD, but 30% of the experts reported that they would remove and replace the device.23
IUD embedded in the myometrium with pelvic pain. Based on my clinical experience, when a patient has persistent pelvic pain following the insertion of an IUD and the pain does not resolve with standard measures including medication, an ultrasound study is warranted to assess the position of the IUD. If the ultrasound demonstrates that an arm of the IUD is embedded in the myometrium, removal of the IUD may be associated with resolution of the pain. Reinsertion of an IUD under ultrasound guidance may result in a correctly-sited IUD with no recurrence of pelvic pain.
IUD rotated within the uterus with no pain or abnormal bleeding. For an IUD that is near the fundus and rotated on its axis within the uterus, if the patient has no symptoms of pain or abnormal bleeding, my recommendation to the patient would be to leave the device in situ.
Without available guidelines, engage in clinician-patient discussion
It is clear that IUD malposition is common, occurring in 10% to 20% of patients with an IUD. High-quality ultrasound imaging is helpful in detecting IUD malposition, including 2-D ultrasound with videoclips and/or 3-D ultrasound with coronal reconstruction. More data are needed to identify the best options for managing various types of malpositioned IUDs in patients with and without bothersome symptoms such as pain and bleeding. Until consensus guidelines are developed, clinicians need to engage the patient in a discussion of how to best manage the malpositioned IUD. Medicated IUDs and progestin subdermal implants are our two most effective reversible contraceptives. They are among the most important advances in health care over the past half-century. ●
- Mirena FDA approval. , 2022.
- Liletta [package insert]. Allergan USA: Irvine, California; 2019. .
- Paragard [package insert]. CooperSurgical Inc: Trumbull, Connecticut; 2019. .
- Harrison-Woolrych M, Ashton J, Coulter D. Uterine perforation on intrauterine device insertion: is the incidence higher than previously reported? Contraception. 2003;67:53-56.
- Van Houdenhoven K, van Kaam KJAF, van Grootheest AC, et al. Uterine perforation in women using a levonorgestrel-releasing intrauterine system. Contraception. 2006;73:257-260.
- van Grootheest K, Sachs B, Harrison-Woolrych M, et al. Uterine perforation with the levonorgestrel-releasing intrauterine device. Analysis of reports from four national pharmacovigilance centres. Drug Saf. 2011;34:83-88.
- Heinemann K, Reed S, Moehner S, et al. Risk of uterine perforation with levonorgestrel-releasing and copper intrauterine devices in the European Active Surveillance Study on Intrauterine Devices. Contraception. 2015;91:274-279.
- Barnett C, Moehner S, Do Minh T, et al. Perforation risk and intra-uterine devices: results of the EURAS-IUD 5-year extension study. Eur J Contracept Reprod Health Care. 2017;22:424-428.
- Zakin D, Stern WZ, Rosenblatt R. Complete and partial uterine perforation and embedding following insertion of intrauterine devices. I. Classification, complications, mechanism, incidence and missing string. Obstet Gynecol Surv. 1981;36:335-353.
- Rivera R, Chen-Mok M, McMullen S. Analysis of client characteristics that may affect early discontinuation of the TCu-380A IUD. Contraception. 1999;60:155-160.
- Aoun J, Dines VA, Stovall DW, et al. Effects of age, parity and device type on complications and discontinuation of intrauterine devices. Obstet Gynecol. 2014;123:585-592.
- Madden T, McNichols, Zhao Q, et al. Association of age and parity with intrauterine device expulsion. Obstet Gynecol. 2014;124:718-726.
- Keenahan L, Bercaw-Pratt JL, Adeyemi O, et al. Rates of intrauterine device expulsion among adolescents and young women. J Pediatr Adolesc Gynecol. 2021;34:362-365.
- Backman T, Rauramo I, Huhtala S, et al. Pregnancy during the use of levonorgestrel intrauterine system. Am J Obstet Gynecol. 2004;190:50-54.
- Benacerraf BR, Shipp TD, Bromley B. Three-dimensional ultrasound detection of abnormally located intrauterine contraceptive devices which are a source of pelvic pain and abnormal bleeding. Ultrasound Obstet Gynecol. 2009;34:110-115.
- Braaten KP, Benson CB, Maurer R, et al. Malpositioned intrauterine contraceptive devices: risk factors, outcomes and future pregnancies. Obstet Gynecol. 2011;118:1014-1020.
- Gerkowicz SA, Fiorentino DG, Kovacs AP, et al. Uterine structural abnormality and intrauterine device malposition: analysis of ultrasonographic and demographic variables of 517 patients. Am J Obstet Gynecol. 2019;220:183.e1-e8.
- Connolly CT, Fox NS. Incidence and risk factors for a malpositioned intrauterine device detected on three-dimensional ultrasound within eight weeks of placement. J Ultrasound Med. 2021 ePub Sept 27 2021.
- Golightly E, Gebbie AE. Low-lying or malpositioned intrauterine devices and systems. J Fam Plann Reprod health Care. 2014;40:108-112.
- Gurney EP, Sonalkar S, McAllister A, et al. Six-month expulsion of postplacental copper intrauterine devices placed after vaginal delivery. Am J Obstet Gynecol. 2018;219:183.e1-e9.
- Gurney EP, McAllister A, Lang B, et al. Ultrasound assessment of postplacental copper intrauterine device position 6 months after placement during cesarean delivery. Contraception. 2020;2:100040.
- Anteby E, Revel A, Ben-Chetrit A, et al. Intrauterine device failure: relation to its location with the uterine cavity. Obstet Gynecol. 1993;81:112-114.
- Golightly E, Gebbie AE. Clinicians’ views on low-lying intrauterine devices or systems. J Fam Plann Reprod Health Care. 2014;40:113-116.
- Mirena FDA approval. , 2022.
- Liletta [package insert]. Allergan USA: Irvine, California; 2019. .
- Paragard [package insert]. CooperSurgical Inc: Trumbull, Connecticut; 2019. .
- Harrison-Woolrych M, Ashton J, Coulter D. Uterine perforation on intrauterine device insertion: is the incidence higher than previously reported? Contraception. 2003;67:53-56.
- Van Houdenhoven K, van Kaam KJAF, van Grootheest AC, et al. Uterine perforation in women using a levonorgestrel-releasing intrauterine system. Contraception. 2006;73:257-260.
- van Grootheest K, Sachs B, Harrison-Woolrych M, et al. Uterine perforation with the levonorgestrel-releasing intrauterine device. Analysis of reports from four national pharmacovigilance centres. Drug Saf. 2011;34:83-88.
- Heinemann K, Reed S, Moehner S, et al. Risk of uterine perforation with levonorgestrel-releasing and copper intrauterine devices in the European Active Surveillance Study on Intrauterine Devices. Contraception. 2015;91:274-279.
- Barnett C, Moehner S, Do Minh T, et al. Perforation risk and intra-uterine devices: results of the EURAS-IUD 5-year extension study. Eur J Contracept Reprod Health Care. 2017;22:424-428.
- Zakin D, Stern WZ, Rosenblatt R. Complete and partial uterine perforation and embedding following insertion of intrauterine devices. I. Classification, complications, mechanism, incidence and missing string. Obstet Gynecol Surv. 1981;36:335-353.
- Rivera R, Chen-Mok M, McMullen S. Analysis of client characteristics that may affect early discontinuation of the TCu-380A IUD. Contraception. 1999;60:155-160.
- Aoun J, Dines VA, Stovall DW, et al. Effects of age, parity and device type on complications and discontinuation of intrauterine devices. Obstet Gynecol. 2014;123:585-592.
- Madden T, McNichols, Zhao Q, et al. Association of age and parity with intrauterine device expulsion. Obstet Gynecol. 2014;124:718-726.
- Keenahan L, Bercaw-Pratt JL, Adeyemi O, et al. Rates of intrauterine device expulsion among adolescents and young women. J Pediatr Adolesc Gynecol. 2021;34:362-365.
- Backman T, Rauramo I, Huhtala S, et al. Pregnancy during the use of levonorgestrel intrauterine system. Am J Obstet Gynecol. 2004;190:50-54.
- Benacerraf BR, Shipp TD, Bromley B. Three-dimensional ultrasound detection of abnormally located intrauterine contraceptive devices which are a source of pelvic pain and abnormal bleeding. Ultrasound Obstet Gynecol. 2009;34:110-115.
- Braaten KP, Benson CB, Maurer R, et al. Malpositioned intrauterine contraceptive devices: risk factors, outcomes and future pregnancies. Obstet Gynecol. 2011;118:1014-1020.
- Gerkowicz SA, Fiorentino DG, Kovacs AP, et al. Uterine structural abnormality and intrauterine device malposition: analysis of ultrasonographic and demographic variables of 517 patients. Am J Obstet Gynecol. 2019;220:183.e1-e8.
- Connolly CT, Fox NS. Incidence and risk factors for a malpositioned intrauterine device detected on three-dimensional ultrasound within eight weeks of placement. J Ultrasound Med. 2021 ePub Sept 27 2021.
- Golightly E, Gebbie AE. Low-lying or malpositioned intrauterine devices and systems. J Fam Plann Reprod health Care. 2014;40:108-112.
- Gurney EP, Sonalkar S, McAllister A, et al. Six-month expulsion of postplacental copper intrauterine devices placed after vaginal delivery. Am J Obstet Gynecol. 2018;219:183.e1-e9.
- Gurney EP, McAllister A, Lang B, et al. Ultrasound assessment of postplacental copper intrauterine device position 6 months after placement during cesarean delivery. Contraception. 2020;2:100040.
- Anteby E, Revel A, Ben-Chetrit A, et al. Intrauterine device failure: relation to its location with the uterine cavity. Obstet Gynecol. 1993;81:112-114.
- Golightly E, Gebbie AE. Clinicians’ views on low-lying intrauterine devices or systems. J Fam Plann Reprod Health Care. 2014;40:113-116.
COMMENT & CONTROVERSY
UTIs IN PREGNANCY: MANAGING URETHRITIS, ASYMPTOMATIC BACTERIURIA, CYSTITIS, AND PYELONEPHRITIS
PATRICK DUFF, MD (JANUARY 2022)
Clarification on UTI issues
Regarding the article on urinary tract infections (UTIs) in pregnancy, I have 3 points of clarification. First, in 27 years of practice in which I universally performed screening urine cultures on prenatal patients plus all of those with symptoms, I have seen a total of 2 cultures with Staphylococcus saprophyticus. I see this organism listed in references as a major UTI causative, but is that the case? Second, the clinical case and symptoms discussed are accurate, but costovertebral angle tenderness or fever of 101 °F or higher indicate pyelonephritis and should be treated aggressively. Many of these patients will have nausea and vomiting and will be dehydrated. This decreases urine flow, allowing progressive bacterial growth in renal parenchyma. An initial bolus of intravenous fluids, at least 2 L wide open through a large-bore catheter, rapidly decreases fever, flushes the urinary tract, and improves nausea, headaches, and malaise. Finally, nitrofurantoin is excreted in the urine so rapidly that it does not achieve adequate tissue levels, and it should never be used to treat pyelonephritis or, for that matter, any infection other than uncomplicated cystitis/urethritis.
David Janowitz, MD
Houston, Texas
Dr. Duff responds
I appreciate Dr. Janowitz’s interest and thoughtful comments. The patient presented in the case study has acute cystitis, characterized by a low-grade fever, suprapubic pain, dysuria, frequency, and hesitancy. Patients with pyelonephritis typically have a higher fever and significant costovertebral angle pain and tenderness. I agree completely with Dr. Janowitz’s observations about the seriousness of pyelonephritis in pregnancy. Pyelonephritis is an important cause of preterm labor, bacteremia, and even septic shock. As I point out in the article, women with moderate to severe kidney infections should be hospitalized and treated with intravenous fluids, antipyretics, antiemetics, and intravenous antibiotics. My usual recommendation is ceftriaxone. Intravenous antibiotics should be continued until the patient has been afebrile and asymptomatic for 24 to 48 hours. Once patients improve, they can be transitioned to oral antibiotics to complete a 10-day course of therapy. Again, I agree with Dr. Janowitz’s statement that nitrofurantoin is not an appropriate drug for treatment of pyelonephritis because it does not reach acceptable concentrations in either the blood or the renal parenchyma. Rather, amoxicillin-clavulanate and trimethoprim-sulfamethoxazole are much better choices for oral therapy. However, once the infection is cleared, nitrofurantoin is an excellent agent for suppression of recurrent infection.
Finally, there is no doubt that the principal pathogens that cause UTIs in pregnant women are Escherichia coli, Klebsiella pneumoniae, and Proteus species. However, 3 aerobic Gram-positive cocci do, in fact, cause a small percentage of infections: group B streptococci, enterococci, and Staphylococcus saprophyticus. When the latter bacterium is identified as a single organism in high colony count, particularly in a catheterized urine specimen, it should be considered a true pathogen and not simply a contaminant.
CAN WE RETURN TO THE ABCs OF CRAFTING A MEDICAL RECORD NOTE?
ROBERT L. BARBIERI, MD (OCTOBER 2021)
Another suggestion for reducing note bloat in the EMR
Thank you for picking up a topic that is important for all physicians and one that has been annoying me since the introduction of electronic medical records (EMRs). I like the APSO approach, that works well. My idea for reducing “note bloat” is to eliminate all normal and routine findings and to hide them behind a hyperlink or behind a QR code. This would give you a truly short note and, should you need or want more details, you could always scan the QR code for access to the complete (and bloated) note. I would also recommend hiding all details that do not contribute to the immediate pressing issue at hand (for example, routine depression screening) behind a hyperlink or QR code. The same principle should apply to sending faxes to other physicians’ offices. I “love” receiving a chart an inch thick, only to discover that the whole pile of paper could be reduced to a single page of true information. Too few people speak up about this major time and productivity thief. Thank you!
Matthias Muenzer, MD
Rochester, New Hampshire
Dr. Barbieri responds
I thank Dr. Muenzer for his innovative suggestions for improving medical record notes. We spend many hours per week crafting notes in the medical record. Yet, very little attention is given to the development of best practices for improving the value and effectiveness of our notes for our patients and colleagues.
UTIs IN PREGNANCY: MANAGING URETHRITIS, ASYMPTOMATIC BACTERIURIA, CYSTITIS, AND PYELONEPHRITIS
PATRICK DUFF, MD (JANUARY 2022)
Clarification on UTI issues
Regarding the article on urinary tract infections (UTIs) in pregnancy, I have 3 points of clarification. First, in 27 years of practice in which I universally performed screening urine cultures on prenatal patients plus all of those with symptoms, I have seen a total of 2 cultures with Staphylococcus saprophyticus. I see this organism listed in references as a major UTI causative, but is that the case? Second, the clinical case and symptoms discussed are accurate, but costovertebral angle tenderness or fever of 101 °F or higher indicate pyelonephritis and should be treated aggressively. Many of these patients will have nausea and vomiting and will be dehydrated. This decreases urine flow, allowing progressive bacterial growth in renal parenchyma. An initial bolus of intravenous fluids, at least 2 L wide open through a large-bore catheter, rapidly decreases fever, flushes the urinary tract, and improves nausea, headaches, and malaise. Finally, nitrofurantoin is excreted in the urine so rapidly that it does not achieve adequate tissue levels, and it should never be used to treat pyelonephritis or, for that matter, any infection other than uncomplicated cystitis/urethritis.
David Janowitz, MD
Houston, Texas
Dr. Duff responds
I appreciate Dr. Janowitz’s interest and thoughtful comments. The patient presented in the case study has acute cystitis, characterized by a low-grade fever, suprapubic pain, dysuria, frequency, and hesitancy. Patients with pyelonephritis typically have a higher fever and significant costovertebral angle pain and tenderness. I agree completely with Dr. Janowitz’s observations about the seriousness of pyelonephritis in pregnancy. Pyelonephritis is an important cause of preterm labor, bacteremia, and even septic shock. As I point out in the article, women with moderate to severe kidney infections should be hospitalized and treated with intravenous fluids, antipyretics, antiemetics, and intravenous antibiotics. My usual recommendation is ceftriaxone. Intravenous antibiotics should be continued until the patient has been afebrile and asymptomatic for 24 to 48 hours. Once patients improve, they can be transitioned to oral antibiotics to complete a 10-day course of therapy. Again, I agree with Dr. Janowitz’s statement that nitrofurantoin is not an appropriate drug for treatment of pyelonephritis because it does not reach acceptable concentrations in either the blood or the renal parenchyma. Rather, amoxicillin-clavulanate and trimethoprim-sulfamethoxazole are much better choices for oral therapy. However, once the infection is cleared, nitrofurantoin is an excellent agent for suppression of recurrent infection.
Finally, there is no doubt that the principal pathogens that cause UTIs in pregnant women are Escherichia coli, Klebsiella pneumoniae, and Proteus species. However, 3 aerobic Gram-positive cocci do, in fact, cause a small percentage of infections: group B streptococci, enterococci, and Staphylococcus saprophyticus. When the latter bacterium is identified as a single organism in high colony count, particularly in a catheterized urine specimen, it should be considered a true pathogen and not simply a contaminant.
CAN WE RETURN TO THE ABCs OF CRAFTING A MEDICAL RECORD NOTE?
ROBERT L. BARBIERI, MD (OCTOBER 2021)
Another suggestion for reducing note bloat in the EMR
Thank you for picking up a topic that is important for all physicians and one that has been annoying me since the introduction of electronic medical records (EMRs). I like the APSO approach, that works well. My idea for reducing “note bloat” is to eliminate all normal and routine findings and to hide them behind a hyperlink or behind a QR code. This would give you a truly short note and, should you need or want more details, you could always scan the QR code for access to the complete (and bloated) note. I would also recommend hiding all details that do not contribute to the immediate pressing issue at hand (for example, routine depression screening) behind a hyperlink or QR code. The same principle should apply to sending faxes to other physicians’ offices. I “love” receiving a chart an inch thick, only to discover that the whole pile of paper could be reduced to a single page of true information. Too few people speak up about this major time and productivity thief. Thank you!
Matthias Muenzer, MD
Rochester, New Hampshire
Dr. Barbieri responds
I thank Dr. Muenzer for his innovative suggestions for improving medical record notes. We spend many hours per week crafting notes in the medical record. Yet, very little attention is given to the development of best practices for improving the value and effectiveness of our notes for our patients and colleagues.
UTIs IN PREGNANCY: MANAGING URETHRITIS, ASYMPTOMATIC BACTERIURIA, CYSTITIS, AND PYELONEPHRITIS
PATRICK DUFF, MD (JANUARY 2022)
Clarification on UTI issues
Regarding the article on urinary tract infections (UTIs) in pregnancy, I have 3 points of clarification. First, in 27 years of practice in which I universally performed screening urine cultures on prenatal patients plus all of those with symptoms, I have seen a total of 2 cultures with Staphylococcus saprophyticus. I see this organism listed in references as a major UTI causative, but is that the case? Second, the clinical case and symptoms discussed are accurate, but costovertebral angle tenderness or fever of 101 °F or higher indicate pyelonephritis and should be treated aggressively. Many of these patients will have nausea and vomiting and will be dehydrated. This decreases urine flow, allowing progressive bacterial growth in renal parenchyma. An initial bolus of intravenous fluids, at least 2 L wide open through a large-bore catheter, rapidly decreases fever, flushes the urinary tract, and improves nausea, headaches, and malaise. Finally, nitrofurantoin is excreted in the urine so rapidly that it does not achieve adequate tissue levels, and it should never be used to treat pyelonephritis or, for that matter, any infection other than uncomplicated cystitis/urethritis.
David Janowitz, MD
Houston, Texas
Dr. Duff responds
I appreciate Dr. Janowitz’s interest and thoughtful comments. The patient presented in the case study has acute cystitis, characterized by a low-grade fever, suprapubic pain, dysuria, frequency, and hesitancy. Patients with pyelonephritis typically have a higher fever and significant costovertebral angle pain and tenderness. I agree completely with Dr. Janowitz’s observations about the seriousness of pyelonephritis in pregnancy. Pyelonephritis is an important cause of preterm labor, bacteremia, and even septic shock. As I point out in the article, women with moderate to severe kidney infections should be hospitalized and treated with intravenous fluids, antipyretics, antiemetics, and intravenous antibiotics. My usual recommendation is ceftriaxone. Intravenous antibiotics should be continued until the patient has been afebrile and asymptomatic for 24 to 48 hours. Once patients improve, they can be transitioned to oral antibiotics to complete a 10-day course of therapy. Again, I agree with Dr. Janowitz’s statement that nitrofurantoin is not an appropriate drug for treatment of pyelonephritis because it does not reach acceptable concentrations in either the blood or the renal parenchyma. Rather, amoxicillin-clavulanate and trimethoprim-sulfamethoxazole are much better choices for oral therapy. However, once the infection is cleared, nitrofurantoin is an excellent agent for suppression of recurrent infection.
Finally, there is no doubt that the principal pathogens that cause UTIs in pregnant women are Escherichia coli, Klebsiella pneumoniae, and Proteus species. However, 3 aerobic Gram-positive cocci do, in fact, cause a small percentage of infections: group B streptococci, enterococci, and Staphylococcus saprophyticus. When the latter bacterium is identified as a single organism in high colony count, particularly in a catheterized urine specimen, it should be considered a true pathogen and not simply a contaminant.
CAN WE RETURN TO THE ABCs OF CRAFTING A MEDICAL RECORD NOTE?
ROBERT L. BARBIERI, MD (OCTOBER 2021)
Another suggestion for reducing note bloat in the EMR
Thank you for picking up a topic that is important for all physicians and one that has been annoying me since the introduction of electronic medical records (EMRs). I like the APSO approach, that works well. My idea for reducing “note bloat” is to eliminate all normal and routine findings and to hide them behind a hyperlink or behind a QR code. This would give you a truly short note and, should you need or want more details, you could always scan the QR code for access to the complete (and bloated) note. I would also recommend hiding all details that do not contribute to the immediate pressing issue at hand (for example, routine depression screening) behind a hyperlink or QR code. The same principle should apply to sending faxes to other physicians’ offices. I “love” receiving a chart an inch thick, only to discover that the whole pile of paper could be reduced to a single page of true information. Too few people speak up about this major time and productivity thief. Thank you!
Matthias Muenzer, MD
Rochester, New Hampshire
Dr. Barbieri responds
I thank Dr. Muenzer for his innovative suggestions for improving medical record notes. We spend many hours per week crafting notes in the medical record. Yet, very little attention is given to the development of best practices for improving the value and effectiveness of our notes for our patients and colleagues.
Uterine incision closure: Is it the culprit in the cesarean scar niche and related complications?
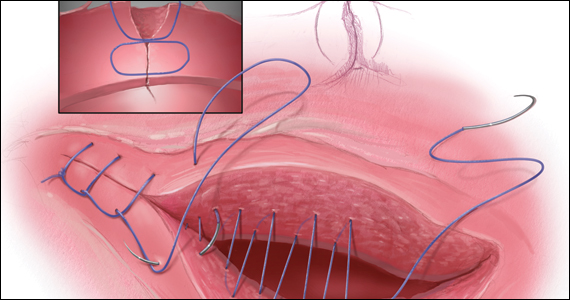
While its etiology remains uncertain, cesarean scar niche (CSN) is well publicized, as are its pathological clinical manifestations. In a future pregnancy, they include cesarean scar pregnancy (CSP), which in turn can lead to placenta accreta spectrum, and possible uterine rupture/dehiscence of a residual thin myometrial layer. CSP refers to the implantation of an early pregnancy on the scar or in the niche at the site of a prior cesarean delivery (CD); it has an incidence of 1 per 1,000 pregnancies. An estimated 52% of CSPs occur after even just one CD.1 CSP has been linked to placenta accreta spectrum and has been shown to be its precursor.2 Both CSP and placenta accreta spectrum can be consequences of CD and share a common histology of villous or placental attachment/invasion into the cesarean scar.3 The incidence of placenta accreta spectrum has risen from about 1 in 4,000 live births in the 1970s to 1 in 2,500 in the 1980s; in 2016, the incidence of placenta accreta spectrum was reported as 1 per 272 live births.4
Placenta accreta spectrum denotes the attachment of the placenta into and through the myometrium,5 and it can result in severe complications, including hemorrhage, hysterectomy, and intensive care treatment. The increasing rate of placenta accreta spectrum parallels the increasing CD rate, which rose from 5.8% in 1970 to 31.9% in 2016.6 Multiple repeat CDs are increasing in frequency as well. At the beginning of the century, placenta accreta spectrum mainly occurred after manual removal of the placenta, uterine curettage, or endometritis. Recently, experts are in agreement that the main determinant of placenta accreta spectrum is the uterine scar and niche formation after a previous CD.5 Larger niches are associated with an increased incidence of uterine rupture or dehiscence in a subsequent pregnancy.7
In the nonpregnant state, such niches are associated with intermenstrual bleeding, pelvic pain, painful intercourse, painful menses, and subfertility, becoming increasingly more severe in women with greater numbers of CDs.8-10 Conception rate with assisted reproductive treatment is notably reduced.11
Understanding its etiology
Monteagudo and colleagues first described a “niche” in 100% of 44 women evaluated for postmenopausal bleeding who had a prior CD.12 CSN has been the subject of well over 3,000 publications over the past 30 years. While the topic generates much interest among researchers, it is garnering little traction among practicing obstetricians. Such “niches,” also referred to as isthmocele, cesarean scar defect, or a diverticulum, was first described in 196113 and later defined on ultrasonography as a hypoechoic triangular-shaped uterine defect outlined by saline instillation sonohysterogram (SIS), reflecting a discontinuation of the myometrium at the site of a previous CD.12 In 2019, a European task force further defined a CSN as an “indentation at the site in the cesarean section scar with a depth of at least 2 mm” and extended the classification to include branches as extensions toward the anterior uterine serosa.14 Using this criterion, sonographic postoperative evaluation after one CD revealed a CSN in 68.9% of women with one single-layer uterine closure and in 73.6% of women after a double-layer closure.15 Larger niche sizes with thinner residual myometrial thickness appeared more frequently when a single-layer closure technique was used, without closure of the peritoneum. Its prevalence varies from 56% to 84%.16,17
Etiology of CSN formation: Our hypotheses
The precise pathophysiology of CSN remains elusive. Speculations attributed niche formation to numerous factors: timing of surgery, cervical incision, incomplete closure of the uterine incision, adhesion formation between the CD scar and the abdominal wall, and inherent maternal conditions which may impair healing, such as smoking, obesity, diabetes, maternal age, and labor status.18-20 Retroflexion of the uterus is reportedly associated with increased incidence and size of the niche, with CSN 50% more likely to develop in women with a retroflexed versus an anteverted uterus.21 We demonstrated the origin of niche formation in real-time from the start to the completion of uterine closure by a video capture of a single-layer closure followed by an immediate SIS of the ex vivo hysterectomized uterus, and histopathologic proof of the presence of endometrial cells defining the “niche.”22 This case exposes the misalignment of the uterine wall, while including the endometrium in the closure (FIGURE 1). Similarly, pathologic studies of hysteroscopy-resected isthmocele ridges of symptomatic women with niche-related subfertility revealed the tissue edges lined by endocervical, endometrial, or isthmic mucosa either combined or isolated in the scar.23 The presence of endometrial/cervical tissue in the myometrial closure has been debated for over a century.24,25
Continue to: Uterine closure techniques...
Uterine closure techniques: Historical perspective
In 1882, Max Sanger introduced a vertical uterine closure of a classical cesarean operation in response to hysterectomy as the contemporaneous alternative to prevent infection, bleeding, and death.24 Dr. Sanger emphasized layer approximation, suturing, and the avoidance of decidua in the first layer (FIGURE 2). This became the teaching of the classical CD until the 1970s. In 1926, Munro Kerr addressed uterine rupture with labor after a classical CD by introducing the lower uterine segment transverse incision. He cautioned to maintain the decidua inside the uterine 2-layer closure of the cavity.25 These pioneers were joined by others to rally for endometrium exclusion while promoting layer approximation. These techniques became universally standard and were taught across teaching medical centers in the United States and abroad until about 50 years ago.
In the 1970s, newer developments brought significant changes to uterine closure techniques. Initiated by Joel-Cohen,26 blunt dissection of the abdominal incision was adapted by Michael Stark, creating what came to be known as the Misgav-Ladach cesarean technique.27 Stark emphasized blunt dissection and introduced single-layer closure. Thereby the exclusion of the endometrium, used for more than 70 years, was abandoned by the present-day single- or double-layer uterine closure in favor of cost and time savings. Systematic reviews and meta-analyses comparing the two contrasting techniques were inconclusive, noting that the niche prevalence and size were similar in both groups. These studies did not take into account the variety of individual techniques or the position of the endometrium in the final closures.28
Endometrium and uterine closure
Our recent study examining uterine scar defect in women after one primary CD by SIS concluded that a specific endometrium-free closure technique (EFCT) (FIGURE 3) is associated with fewer and less severe defects and a thicker residual myometrial thickness when compared with closures with unknown or endometrium inclusion.29 The study found non-specific closure techniques to be 6 times more likely to form a niche of 2-mm deep or greater than the EFCT.
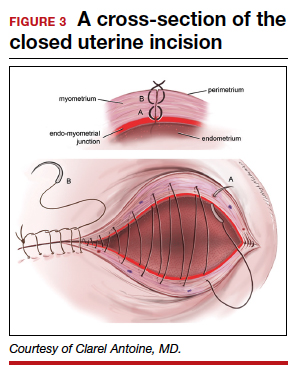
Furthermore, we surveyed the diversity of uterine closures and the location of the endometrium among obstetricians in one institution.30 Presence of endometrium on the surface of the final uterine closure was reported by 20% of respondents (see Figure 1). When asked for their opinion on the impact of CD techniques on placenta accreta spectrum, without available evidence 80% of the survey respondents reported no relationship to techniques, and only 20% suggested an association. This particular study demonstrates that the surgical techniques just described are random, unfettered, and applied without consideration of clinical outcomes.
Our recent retrospective study that spanned 30 years and examined the EFCT—performed anywhere between 3 to 9 consecutive CDs—revealed no abnormal placentation in any subsequent pregnancies.31 This was one of the few clinical studies of the long-term consequences of a uterine closure technique. In this study, the endometrium was excluded during the uterine closure, allowing its free edges to abut and heal. This step avoids scarring the endometrial-myometrial (EM) interface and unintentional inclusion of endometrium in the closed uterine wall. In this context, Jauniaux and colleagues cited the destruction of the EM interface as the main factor for placenta-adherent disorders.32 Sholapurkar and others highlight the need to further examine intrinsic details of uterine closure beyond single- and double-layer techniques to better understand the etiology of cesarean scar formation.19 The search for the pathophysiology of CSN continues to present significant challenges imposed by the variety of currently practiced uterine closures.
Continue to: Focus on prevention...
Research: Focus on prevention
Our research aims to address the endometrium, a specific layer that was the topic of concern in nascent CD techniques, as a renewed and contemporary one. The presence of the endometrium in ectopic locations or its destruction from intrauterine surgeries or infections has been implicated in abnormal placentation.13,24 Our approach, in theory, is to limit the position of the endometrium to its innermost location and avoid its iatrogenic suturing and inclusion into the uterine wall closure. The rationale of sparing the endometrium in a layer-by-layer approximation is to allow for a closer restoration to normal anatomy and physiology than a random “en masse” uterine wall closure would permit. For this reason, the EM junction, the perimetrium, and the serosa must be identified and realigned for a more effective closure that incorporates the entire myometrial thickness. As evidence supports technical impact on the development of uterine scar defect in women after one CD, future studies are needed to evaluate uterine integrity by saline infusion sonohysterography in multiparous women with a prior random closure technique or a prior EFCT.
The potential long-term risks of blunt dissection for opening the uterus have not been studied. There are no physiologic lines in the uterine wall to facilitate a regular-bordered uterine stretch. The tissue stretch, which depends on the individual surgeon’s strength applied during the procedure and patient’s labor status, may result in an irregular tear and a difficult repair. The EFCT technique shows a more optimized risk-benefit ratio for an anatomical repair and is replicable. The safety of uterine layer re-approximation has been demonstrated and can be studied in large populations using strict uniform criteria.
Current and future challenges
Residency training
Most recently, teachers of resident trainees are mostly familiar with blunt dissection, techniques of which are passed on unchallenged from resident to resident. The endometrium and peritoneum are neither identified nor treated as separate layers, thus becoming obsolete as surgical and anatomical landmarks.
Standardization of CD techniques
Front-line obstetricians are persuaded to practice a standardized approach that relies on the benefits of cost related to operating room turnover as well as surgeons’ time savings without consideration of outcomes in subsequent pregnancies. Sholapurkar has warned that “wrong standardization” is far worse than no standardization, worse for the training of junior obstetricians, as it can inhibit critical reasoning about safe surgical techniques that can optimize outcomes of the condition of the lower uterine segment.33
Emergence of cost and time savings in clinical practice
A time-cost savings argument is relatively negligeable in an estimated 40-minute CD. By contrast, deliberate surgical technique and carrying out the appropriate steps for the particular condition at hand to achieve the best outcomes assume more weight.32 Furthermore, this short-term cost benefit is challenged by the comparatively larger costs associated with the diagnosis, the treatment of post-CD adverse consequences (outlined above), as well as the emotional impact on women and their families. Additionally, the emphasis on time savings creates a generation of surgeons fixated with total operative time without consideration of long-term risks and adverse maternal outcomes.
Physician autonomy has led to the unmonitored freedom of obstetricians to choose their own technique for a CD, with some employing the commonly practiced culture of fastest turnaround even in nonurgent circumstances.
Documentation and terminology
Current documenting systems are not detail-oriented enough to assist in a thorough correlation between surgical techniques and outcomes. The use of single- or double-layer closure terminology is insufficient and has proven to be flawed, without describing the handling of the endometrium in terms of its inclusion or exclusion in the closure.
Quality improvement feedback
Long-term post-CD complications are often not reported to the physician or institution involved in the prior CD. In our opinion, some sort of registry would be of value. Perhaps then subsequent CD outcomes could be traced back and reported to the prior institution and surgeon. Feedback is critical to understanding the correlation between techniques and outcomes and more specifically to gathering learning points and using data for quality improvement of future cases.
Patient education
While women continue to have complications following the presently used surgical techniques, they often have expectations not discussed with their obstetricians. Women should be educated and empowered to realize the different approaches to all aspects and consequences of CDs.
Conclusion
The technique of excluding the endometrium in closing the uterine incision appears to reduce subsequent abnormal placentation and diminish the frequency and size of post-CD scar defect. The revival of the endometrium-free closure technique may allow significant change in the postoperative results. Currently, standardization of CD technique is being promoted on the basis of time- and cost-savings rather than clinical outcomes. Simultaneously, inroads are being made to better understand the risks and consequences of CD.
Emerging evidence suggests that a post-CD niche is the result of poor layer approximation as well as inclusion of the endometrium, which prevent healing of the uterine wall and often enables faulty implantation of the fertilized oocyte in the next pregnancy, potentially giving rise to placenta accreta spectrum. The prevalence and size of the defect can be minimized by techniques aimed at restoring the anatomy of the uterine wall and the physiology of the endometrium. Specialized training and education are necessary to stress the importance of anatomical assessment and decision making at the time of uterine closure. ●
- Rotas MA, Haberman S, Levgur M. Cesarean scar ectopic pregnancies: etiology, diagnosis, and management. Obstet Gynecol. 2006;107:1373-1381.
- Timor-Tritsch IE, Monteagudo A, Calì G, et al. Cesarean scar pregnancy is a precursor of morbidly adherent placenta. Ultrasound Obstet Gynecol. 2014;44:346-353. doi:10.1002/ uog.13426.
- Timor-Tritsch IE, Monteagudo A, Cali G, et al. Cesarean scar pregnancy and early placenta accreta share common histology. Ultrasound Obstet Gynecol. 2014;43:383-395. doi: 10.1002/uog.13282.
- Mogos MF, Salemi JL, Ashley M, et al. Recent trends in placenta accreta in the United States and its impact on maternal-fetal morbidity and healthcare-associated costs, 1998-2011. J Matern Fetal Neonatal Med. 2016;29:1077-1082.
- Jauniaux E, Collins S, Burton GJ. Placenta accreta spectrum: pathophysiology and evidence-based anatomy for prenatal ultrasound imaging. Am J Obstet Gynecol. 2018;218:75-87.
- Martin JA, Hamilton BE, Osterman MJK. Births in the United States, 2016. NCHS Data Brief. 2017(287):1-8.
- Vikhareva Osser O, Valentin L. Clinical importance of appearance of cesarean hysterotomy scar at transvaginal ultrasonography in nonpregnant women. Obstet Gynecol. 2011;117:525-532.
- Chen YY, Tsai CC, Kung FT, et al. Association between hysteroscopic findings of previous cesarean delivery scar defects and abnormal uterine bleeding. Taiwanese J Obstet Gynecol. 2019;58:541-544.
- Stegwee SI, Beij A, de Leeuw RA, et al. Niche-related outcomes after caesarean section and quality of life: a focus group study and review of literature. Qual Life Res. 2020;29:1013-1025.
- Vissers J, Hehenkamp W, Lambalk CB, et al. Post-caesarean section niche-related impaired fertility: hypothetical mechanisms. Hum Reprod. 2020;35:1484-1494.
- Vissers J, Sluckin TC, van Driel-Delprat CCR, et al. Reduced pregnancy and live birth rates after in vitro fertilization in women with previous caesarean section: a retrospective cohort study. Hum Reprod. 2020;35:595-604.
- Monteagudo A, Carreno C, Timor-Tritsch IE. Saline infusion sonohysterography in nonpregnant women with previous cesarean delivery: the “niche” in the scar. J Ultrasound Med. 2001;20:1105-1115.
- Poidevin LO. The value of hysterography in the prediction of cesarean section wound defects. Am J Obstet Gynecol. 1961;81:67-71.
- Jordans IPM, de Leeuw RA, Stegwee SI, et al. Sonographic examination of uterine niche in non-pregnant women: a modified Delphi procedure. Ultrasound Obstet Gynecol. 2019;53:107-115.
- Stegwee SI, van der Voet LF, Ben AJ, et al. Effect of single- versus double-layer uterine closure during caesarean section on postmenstrual spotting (2Close): multicentre, double-blind, randomised controlled superiority trial. BJOG. 2021;128:866-878.
- Bij de Vaate AJ, van der Voet LF, Naji O, et al. Prevalence, potential risk factors for development and symptoms related to the presence of uterine niches following cesarean section: systematic review. Ultrasound Obstet Gynecol. 2014;43:372-382.
- van der Voet LF, Bij de Vaate AM, Veersema S, et al. Long-term complications of caesarean section. The niche in the scar: a prospective cohort study on niche prevalence and its relation to abnormal uterine bleeding. BJOG. 2014;121:236-244.
- Vervoort AJ, Uittenbogaard LB, Hehenkamp WJ, et al. Why do niches develop in caesarean uterine scars? Hypotheses on the aetiology of niche development. Hum Reprod. 2015;30:2695-2702.
- Sholapurkar SL. Etiology of cesarean uterine scar defect (niche): detailed critical analysis of hypotheses and prevention strategies and peritoneal closure debate. J Clin Med Res. 2018;10:166-173.
- Kamel R, Eissa T, Sharaf M, et al. Position and integrity of uterine scar are determined by degree of cervical dilatation at time of cesarean section. Ultrasound Obstet Gynecol. 2021;57:466-470.
- Sanders RC, Parsons AK. Anteverted retroflexed uterus: a common consequence of cesarean delivery. AJR Am J Roentgenol. 2014;203:W117-124.
- Antoine C, Pimentel RN, Timor-Tritsch IE, et al. Origin of a post-cesarean delivery niche: diagnosis, pathophysiologic characteristics, and video documentation. J Ultrasound Med. 2021;40:205-208.
- AbdullGaffar B, Almulla A. A histopathologic approach to uterine niche: what to expect and to report in hysteroscopy-resected isthmocele specimens. Int J Surg Pathol. 2021:10668969211039415. doi: 10.1177/10668969211039415.
- Nagy S, Papp Z. Global approach of the cesarean section rates. J Perinatal Med. 2020;49:1-4.
- Kerr JM. The technic of cesarean section, with special reference to the lower uterine segment incision. Am J Obstet Gynecol. 1926;12:729-734.
- Joel-Cohen S. Abdominal and vaginal hysterectomy: new techniques based on time and motion studies. Lippincott Williams & Wilkins; 1977.
- Holmgren G, Sjoholm L, Stark M. The Misgav Ladach method for cesarean section: method description. Acta Obstet Gynecol Scand. 1999;78:615-621.
- Abalos E, Addo V, Brocklehurst P, et al. Caesarean section surgical techniques: 3-year follow-up of the CORONIS fractional, factorial, unmasked, randomised controlled trial. Lancet. 2016;388:62-72.
- Antoine C, Meyer JA, Silverstein JS, et al. The impact of uterine incision closure techniques on post-cesarean delivery niche formation and size: sonohysterographic examination of nonpregnant women. J Ultrasound Med. 2021. doi: 10.1002/ jum.15859.
- Antoine C AJ, Yaghoubian Y, Harary J. Variations in uterine closure technique: an institutional survey of obstetricians and implications for patient counseling and prevention of adverse sequelae [Abstract]. 2021.
- Antoine C, Pimentel RN, Reece EA, et al. Endometrium-free uterine closure technique and abnormal placental implantation in subsequent pregnancies. J Matern-Fetal Neonatal Med. 2019:1-9.
- Jauniaux E, Jurkovic D. Placenta accreta: pathogenesis of a 20th century iatrogenic uterine disease. Placenta. 2012;33:244-251.
- Sholapurkar S. Review of unsafe changes in the practice of cesarean section with analysis of flaws in the interpretation of statistics and the evidence. Surgical Case Reports. 2021;4:2-6.
While its etiology remains uncertain, cesarean scar niche (CSN) is well publicized, as are its pathological clinical manifestations. In a future pregnancy, they include cesarean scar pregnancy (CSP), which in turn can lead to placenta accreta spectrum, and possible uterine rupture/dehiscence of a residual thin myometrial layer. CSP refers to the implantation of an early pregnancy on the scar or in the niche at the site of a prior cesarean delivery (CD); it has an incidence of 1 per 1,000 pregnancies. An estimated 52% of CSPs occur after even just one CD.1 CSP has been linked to placenta accreta spectrum and has been shown to be its precursor.2 Both CSP and placenta accreta spectrum can be consequences of CD and share a common histology of villous or placental attachment/invasion into the cesarean scar.3 The incidence of placenta accreta spectrum has risen from about 1 in 4,000 live births in the 1970s to 1 in 2,500 in the 1980s; in 2016, the incidence of placenta accreta spectrum was reported as 1 per 272 live births.4
Placenta accreta spectrum denotes the attachment of the placenta into and through the myometrium,5 and it can result in severe complications, including hemorrhage, hysterectomy, and intensive care treatment. The increasing rate of placenta accreta spectrum parallels the increasing CD rate, which rose from 5.8% in 1970 to 31.9% in 2016.6 Multiple repeat CDs are increasing in frequency as well. At the beginning of the century, placenta accreta spectrum mainly occurred after manual removal of the placenta, uterine curettage, or endometritis. Recently, experts are in agreement that the main determinant of placenta accreta spectrum is the uterine scar and niche formation after a previous CD.5 Larger niches are associated with an increased incidence of uterine rupture or dehiscence in a subsequent pregnancy.7
In the nonpregnant state, such niches are associated with intermenstrual bleeding, pelvic pain, painful intercourse, painful menses, and subfertility, becoming increasingly more severe in women with greater numbers of CDs.8-10 Conception rate with assisted reproductive treatment is notably reduced.11
Understanding its etiology
Monteagudo and colleagues first described a “niche” in 100% of 44 women evaluated for postmenopausal bleeding who had a prior CD.12 CSN has been the subject of well over 3,000 publications over the past 30 years. While the topic generates much interest among researchers, it is garnering little traction among practicing obstetricians. Such “niches,” also referred to as isthmocele, cesarean scar defect, or a diverticulum, was first described in 196113 and later defined on ultrasonography as a hypoechoic triangular-shaped uterine defect outlined by saline instillation sonohysterogram (SIS), reflecting a discontinuation of the myometrium at the site of a previous CD.12 In 2019, a European task force further defined a CSN as an “indentation at the site in the cesarean section scar with a depth of at least 2 mm” and extended the classification to include branches as extensions toward the anterior uterine serosa.14 Using this criterion, sonographic postoperative evaluation after one CD revealed a CSN in 68.9% of women with one single-layer uterine closure and in 73.6% of women after a double-layer closure.15 Larger niche sizes with thinner residual myometrial thickness appeared more frequently when a single-layer closure technique was used, without closure of the peritoneum. Its prevalence varies from 56% to 84%.16,17
Etiology of CSN formation: Our hypotheses
The precise pathophysiology of CSN remains elusive. Speculations attributed niche formation to numerous factors: timing of surgery, cervical incision, incomplete closure of the uterine incision, adhesion formation between the CD scar and the abdominal wall, and inherent maternal conditions which may impair healing, such as smoking, obesity, diabetes, maternal age, and labor status.18-20 Retroflexion of the uterus is reportedly associated with increased incidence and size of the niche, with CSN 50% more likely to develop in women with a retroflexed versus an anteverted uterus.21 We demonstrated the origin of niche formation in real-time from the start to the completion of uterine closure by a video capture of a single-layer closure followed by an immediate SIS of the ex vivo hysterectomized uterus, and histopathologic proof of the presence of endometrial cells defining the “niche.”22 This case exposes the misalignment of the uterine wall, while including the endometrium in the closure (FIGURE 1). Similarly, pathologic studies of hysteroscopy-resected isthmocele ridges of symptomatic women with niche-related subfertility revealed the tissue edges lined by endocervical, endometrial, or isthmic mucosa either combined or isolated in the scar.23 The presence of endometrial/cervical tissue in the myometrial closure has been debated for over a century.24,25
Continue to: Uterine closure techniques...
Uterine closure techniques: Historical perspective
In 1882, Max Sanger introduced a vertical uterine closure of a classical cesarean operation in response to hysterectomy as the contemporaneous alternative to prevent infection, bleeding, and death.24 Dr. Sanger emphasized layer approximation, suturing, and the avoidance of decidua in the first layer (FIGURE 2). This became the teaching of the classical CD until the 1970s. In 1926, Munro Kerr addressed uterine rupture with labor after a classical CD by introducing the lower uterine segment transverse incision. He cautioned to maintain the decidua inside the uterine 2-layer closure of the cavity.25 These pioneers were joined by others to rally for endometrium exclusion while promoting layer approximation. These techniques became universally standard and were taught across teaching medical centers in the United States and abroad until about 50 years ago.
In the 1970s, newer developments brought significant changes to uterine closure techniques. Initiated by Joel-Cohen,26 blunt dissection of the abdominal incision was adapted by Michael Stark, creating what came to be known as the Misgav-Ladach cesarean technique.27 Stark emphasized blunt dissection and introduced single-layer closure. Thereby the exclusion of the endometrium, used for more than 70 years, was abandoned by the present-day single- or double-layer uterine closure in favor of cost and time savings. Systematic reviews and meta-analyses comparing the two contrasting techniques were inconclusive, noting that the niche prevalence and size were similar in both groups. These studies did not take into account the variety of individual techniques or the position of the endometrium in the final closures.28
Endometrium and uterine closure
Our recent study examining uterine scar defect in women after one primary CD by SIS concluded that a specific endometrium-free closure technique (EFCT) (FIGURE 3) is associated with fewer and less severe defects and a thicker residual myometrial thickness when compared with closures with unknown or endometrium inclusion.29 The study found non-specific closure techniques to be 6 times more likely to form a niche of 2-mm deep or greater than the EFCT.
Furthermore, we surveyed the diversity of uterine closures and the location of the endometrium among obstetricians in one institution.30 Presence of endometrium on the surface of the final uterine closure was reported by 20% of respondents (see Figure 1). When asked for their opinion on the impact of CD techniques on placenta accreta spectrum, without available evidence 80% of the survey respondents reported no relationship to techniques, and only 20% suggested an association. This particular study demonstrates that the surgical techniques just described are random, unfettered, and applied without consideration of clinical outcomes.
Our recent retrospective study that spanned 30 years and examined the EFCT—performed anywhere between 3 to 9 consecutive CDs—revealed no abnormal placentation in any subsequent pregnancies.31 This was one of the few clinical studies of the long-term consequences of a uterine closure technique. In this study, the endometrium was excluded during the uterine closure, allowing its free edges to abut and heal. This step avoids scarring the endometrial-myometrial (EM) interface and unintentional inclusion of endometrium in the closed uterine wall. In this context, Jauniaux and colleagues cited the destruction of the EM interface as the main factor for placenta-adherent disorders.32 Sholapurkar and others highlight the need to further examine intrinsic details of uterine closure beyond single- and double-layer techniques to better understand the etiology of cesarean scar formation.19 The search for the pathophysiology of CSN continues to present significant challenges imposed by the variety of currently practiced uterine closures.
Continue to: Focus on prevention...
Research: Focus on prevention
Our research aims to address the endometrium, a specific layer that was the topic of concern in nascent CD techniques, as a renewed and contemporary one. The presence of the endometrium in ectopic locations or its destruction from intrauterine surgeries or infections has been implicated in abnormal placentation.13,24 Our approach, in theory, is to limit the position of the endometrium to its innermost location and avoid its iatrogenic suturing and inclusion into the uterine wall closure. The rationale of sparing the endometrium in a layer-by-layer approximation is to allow for a closer restoration to normal anatomy and physiology than a random “en masse” uterine wall closure would permit. For this reason, the EM junction, the perimetrium, and the serosa must be identified and realigned for a more effective closure that incorporates the entire myometrial thickness. As evidence supports technical impact on the development of uterine scar defect in women after one CD, future studies are needed to evaluate uterine integrity by saline infusion sonohysterography in multiparous women with a prior random closure technique or a prior EFCT.
The potential long-term risks of blunt dissection for opening the uterus have not been studied. There are no physiologic lines in the uterine wall to facilitate a regular-bordered uterine stretch. The tissue stretch, which depends on the individual surgeon’s strength applied during the procedure and patient’s labor status, may result in an irregular tear and a difficult repair. The EFCT technique shows a more optimized risk-benefit ratio for an anatomical repair and is replicable. The safety of uterine layer re-approximation has been demonstrated and can be studied in large populations using strict uniform criteria.
Current and future challenges
Residency training
Most recently, teachers of resident trainees are mostly familiar with blunt dissection, techniques of which are passed on unchallenged from resident to resident. The endometrium and peritoneum are neither identified nor treated as separate layers, thus becoming obsolete as surgical and anatomical landmarks.
Standardization of CD techniques
Front-line obstetricians are persuaded to practice a standardized approach that relies on the benefits of cost related to operating room turnover as well as surgeons’ time savings without consideration of outcomes in subsequent pregnancies. Sholapurkar has warned that “wrong standardization” is far worse than no standardization, worse for the training of junior obstetricians, as it can inhibit critical reasoning about safe surgical techniques that can optimize outcomes of the condition of the lower uterine segment.33
Emergence of cost and time savings in clinical practice
A time-cost savings argument is relatively negligeable in an estimated 40-minute CD. By contrast, deliberate surgical technique and carrying out the appropriate steps for the particular condition at hand to achieve the best outcomes assume more weight.32 Furthermore, this short-term cost benefit is challenged by the comparatively larger costs associated with the diagnosis, the treatment of post-CD adverse consequences (outlined above), as well as the emotional impact on women and their families. Additionally, the emphasis on time savings creates a generation of surgeons fixated with total operative time without consideration of long-term risks and adverse maternal outcomes.
Physician autonomy has led to the unmonitored freedom of obstetricians to choose their own technique for a CD, with some employing the commonly practiced culture of fastest turnaround even in nonurgent circumstances.
Documentation and terminology
Current documenting systems are not detail-oriented enough to assist in a thorough correlation between surgical techniques and outcomes. The use of single- or double-layer closure terminology is insufficient and has proven to be flawed, without describing the handling of the endometrium in terms of its inclusion or exclusion in the closure.
Quality improvement feedback
Long-term post-CD complications are often not reported to the physician or institution involved in the prior CD. In our opinion, some sort of registry would be of value. Perhaps then subsequent CD outcomes could be traced back and reported to the prior institution and surgeon. Feedback is critical to understanding the correlation between techniques and outcomes and more specifically to gathering learning points and using data for quality improvement of future cases.
Patient education
While women continue to have complications following the presently used surgical techniques, they often have expectations not discussed with their obstetricians. Women should be educated and empowered to realize the different approaches to all aspects and consequences of CDs.
Conclusion
The technique of excluding the endometrium in closing the uterine incision appears to reduce subsequent abnormal placentation and diminish the frequency and size of post-CD scar defect. The revival of the endometrium-free closure technique may allow significant change in the postoperative results. Currently, standardization of CD technique is being promoted on the basis of time- and cost-savings rather than clinical outcomes. Simultaneously, inroads are being made to better understand the risks and consequences of CD.
Emerging evidence suggests that a post-CD niche is the result of poor layer approximation as well as inclusion of the endometrium, which prevent healing of the uterine wall and often enables faulty implantation of the fertilized oocyte in the next pregnancy, potentially giving rise to placenta accreta spectrum. The prevalence and size of the defect can be minimized by techniques aimed at restoring the anatomy of the uterine wall and the physiology of the endometrium. Specialized training and education are necessary to stress the importance of anatomical assessment and decision making at the time of uterine closure. ●
While its etiology remains uncertain, cesarean scar niche (CSN) is well publicized, as are its pathological clinical manifestations. In a future pregnancy, they include cesarean scar pregnancy (CSP), which in turn can lead to placenta accreta spectrum, and possible uterine rupture/dehiscence of a residual thin myometrial layer. CSP refers to the implantation of an early pregnancy on the scar or in the niche at the site of a prior cesarean delivery (CD); it has an incidence of 1 per 1,000 pregnancies. An estimated 52% of CSPs occur after even just one CD.1 CSP has been linked to placenta accreta spectrum and has been shown to be its precursor.2 Both CSP and placenta accreta spectrum can be consequences of CD and share a common histology of villous or placental attachment/invasion into the cesarean scar.3 The incidence of placenta accreta spectrum has risen from about 1 in 4,000 live births in the 1970s to 1 in 2,500 in the 1980s; in 2016, the incidence of placenta accreta spectrum was reported as 1 per 272 live births.4
Placenta accreta spectrum denotes the attachment of the placenta into and through the myometrium,5 and it can result in severe complications, including hemorrhage, hysterectomy, and intensive care treatment. The increasing rate of placenta accreta spectrum parallels the increasing CD rate, which rose from 5.8% in 1970 to 31.9% in 2016.6 Multiple repeat CDs are increasing in frequency as well. At the beginning of the century, placenta accreta spectrum mainly occurred after manual removal of the placenta, uterine curettage, or endometritis. Recently, experts are in agreement that the main determinant of placenta accreta spectrum is the uterine scar and niche formation after a previous CD.5 Larger niches are associated with an increased incidence of uterine rupture or dehiscence in a subsequent pregnancy.7
In the nonpregnant state, such niches are associated with intermenstrual bleeding, pelvic pain, painful intercourse, painful menses, and subfertility, becoming increasingly more severe in women with greater numbers of CDs.8-10 Conception rate with assisted reproductive treatment is notably reduced.11
Understanding its etiology
Monteagudo and colleagues first described a “niche” in 100% of 44 women evaluated for postmenopausal bleeding who had a prior CD.12 CSN has been the subject of well over 3,000 publications over the past 30 years. While the topic generates much interest among researchers, it is garnering little traction among practicing obstetricians. Such “niches,” also referred to as isthmocele, cesarean scar defect, or a diverticulum, was first described in 196113 and later defined on ultrasonography as a hypoechoic triangular-shaped uterine defect outlined by saline instillation sonohysterogram (SIS), reflecting a discontinuation of the myometrium at the site of a previous CD.12 In 2019, a European task force further defined a CSN as an “indentation at the site in the cesarean section scar with a depth of at least 2 mm” and extended the classification to include branches as extensions toward the anterior uterine serosa.14 Using this criterion, sonographic postoperative evaluation after one CD revealed a CSN in 68.9% of women with one single-layer uterine closure and in 73.6% of women after a double-layer closure.15 Larger niche sizes with thinner residual myometrial thickness appeared more frequently when a single-layer closure technique was used, without closure of the peritoneum. Its prevalence varies from 56% to 84%.16,17
Etiology of CSN formation: Our hypotheses
The precise pathophysiology of CSN remains elusive. Speculations attributed niche formation to numerous factors: timing of surgery, cervical incision, incomplete closure of the uterine incision, adhesion formation between the CD scar and the abdominal wall, and inherent maternal conditions which may impair healing, such as smoking, obesity, diabetes, maternal age, and labor status.18-20 Retroflexion of the uterus is reportedly associated with increased incidence and size of the niche, with CSN 50% more likely to develop in women with a retroflexed versus an anteverted uterus.21 We demonstrated the origin of niche formation in real-time from the start to the completion of uterine closure by a video capture of a single-layer closure followed by an immediate SIS of the ex vivo hysterectomized uterus, and histopathologic proof of the presence of endometrial cells defining the “niche.”22 This case exposes the misalignment of the uterine wall, while including the endometrium in the closure (FIGURE 1). Similarly, pathologic studies of hysteroscopy-resected isthmocele ridges of symptomatic women with niche-related subfertility revealed the tissue edges lined by endocervical, endometrial, or isthmic mucosa either combined or isolated in the scar.23 The presence of endometrial/cervical tissue in the myometrial closure has been debated for over a century.24,25
Continue to: Uterine closure techniques...
Uterine closure techniques: Historical perspective
In 1882, Max Sanger introduced a vertical uterine closure of a classical cesarean operation in response to hysterectomy as the contemporaneous alternative to prevent infection, bleeding, and death.24 Dr. Sanger emphasized layer approximation, suturing, and the avoidance of decidua in the first layer (FIGURE 2). This became the teaching of the classical CD until the 1970s. In 1926, Munro Kerr addressed uterine rupture with labor after a classical CD by introducing the lower uterine segment transverse incision. He cautioned to maintain the decidua inside the uterine 2-layer closure of the cavity.25 These pioneers were joined by others to rally for endometrium exclusion while promoting layer approximation. These techniques became universally standard and were taught across teaching medical centers in the United States and abroad until about 50 years ago.
In the 1970s, newer developments brought significant changes to uterine closure techniques. Initiated by Joel-Cohen,26 blunt dissection of the abdominal incision was adapted by Michael Stark, creating what came to be known as the Misgav-Ladach cesarean technique.27 Stark emphasized blunt dissection and introduced single-layer closure. Thereby the exclusion of the endometrium, used for more than 70 years, was abandoned by the present-day single- or double-layer uterine closure in favor of cost and time savings. Systematic reviews and meta-analyses comparing the two contrasting techniques were inconclusive, noting that the niche prevalence and size were similar in both groups. These studies did not take into account the variety of individual techniques or the position of the endometrium in the final closures.28
Endometrium and uterine closure
Our recent study examining uterine scar defect in women after one primary CD by SIS concluded that a specific endometrium-free closure technique (EFCT) (FIGURE 3) is associated with fewer and less severe defects and a thicker residual myometrial thickness when compared with closures with unknown or endometrium inclusion.29 The study found non-specific closure techniques to be 6 times more likely to form a niche of 2-mm deep or greater than the EFCT.
Furthermore, we surveyed the diversity of uterine closures and the location of the endometrium among obstetricians in one institution.30 Presence of endometrium on the surface of the final uterine closure was reported by 20% of respondents (see Figure 1). When asked for their opinion on the impact of CD techniques on placenta accreta spectrum, without available evidence 80% of the survey respondents reported no relationship to techniques, and only 20% suggested an association. This particular study demonstrates that the surgical techniques just described are random, unfettered, and applied without consideration of clinical outcomes.
Our recent retrospective study that spanned 30 years and examined the EFCT—performed anywhere between 3 to 9 consecutive CDs—revealed no abnormal placentation in any subsequent pregnancies.31 This was one of the few clinical studies of the long-term consequences of a uterine closure technique. In this study, the endometrium was excluded during the uterine closure, allowing its free edges to abut and heal. This step avoids scarring the endometrial-myometrial (EM) interface and unintentional inclusion of endometrium in the closed uterine wall. In this context, Jauniaux and colleagues cited the destruction of the EM interface as the main factor for placenta-adherent disorders.32 Sholapurkar and others highlight the need to further examine intrinsic details of uterine closure beyond single- and double-layer techniques to better understand the etiology of cesarean scar formation.19 The search for the pathophysiology of CSN continues to present significant challenges imposed by the variety of currently practiced uterine closures.
Continue to: Focus on prevention...
Research: Focus on prevention
Our research aims to address the endometrium, a specific layer that was the topic of concern in nascent CD techniques, as a renewed and contemporary one. The presence of the endometrium in ectopic locations or its destruction from intrauterine surgeries or infections has been implicated in abnormal placentation.13,24 Our approach, in theory, is to limit the position of the endometrium to its innermost location and avoid its iatrogenic suturing and inclusion into the uterine wall closure. The rationale of sparing the endometrium in a layer-by-layer approximation is to allow for a closer restoration to normal anatomy and physiology than a random “en masse” uterine wall closure would permit. For this reason, the EM junction, the perimetrium, and the serosa must be identified and realigned for a more effective closure that incorporates the entire myometrial thickness. As evidence supports technical impact on the development of uterine scar defect in women after one CD, future studies are needed to evaluate uterine integrity by saline infusion sonohysterography in multiparous women with a prior random closure technique or a prior EFCT.
The potential long-term risks of blunt dissection for opening the uterus have not been studied. There are no physiologic lines in the uterine wall to facilitate a regular-bordered uterine stretch. The tissue stretch, which depends on the individual surgeon’s strength applied during the procedure and patient’s labor status, may result in an irregular tear and a difficult repair. The EFCT technique shows a more optimized risk-benefit ratio for an anatomical repair and is replicable. The safety of uterine layer re-approximation has been demonstrated and can be studied in large populations using strict uniform criteria.
Current and future challenges
Residency training
Most recently, teachers of resident trainees are mostly familiar with blunt dissection, techniques of which are passed on unchallenged from resident to resident. The endometrium and peritoneum are neither identified nor treated as separate layers, thus becoming obsolete as surgical and anatomical landmarks.
Standardization of CD techniques
Front-line obstetricians are persuaded to practice a standardized approach that relies on the benefits of cost related to operating room turnover as well as surgeons’ time savings without consideration of outcomes in subsequent pregnancies. Sholapurkar has warned that “wrong standardization” is far worse than no standardization, worse for the training of junior obstetricians, as it can inhibit critical reasoning about safe surgical techniques that can optimize outcomes of the condition of the lower uterine segment.33
Emergence of cost and time savings in clinical practice
A time-cost savings argument is relatively negligeable in an estimated 40-minute CD. By contrast, deliberate surgical technique and carrying out the appropriate steps for the particular condition at hand to achieve the best outcomes assume more weight.32 Furthermore, this short-term cost benefit is challenged by the comparatively larger costs associated with the diagnosis, the treatment of post-CD adverse consequences (outlined above), as well as the emotional impact on women and their families. Additionally, the emphasis on time savings creates a generation of surgeons fixated with total operative time without consideration of long-term risks and adverse maternal outcomes.
Physician autonomy has led to the unmonitored freedom of obstetricians to choose their own technique for a CD, with some employing the commonly practiced culture of fastest turnaround even in nonurgent circumstances.
Documentation and terminology
Current documenting systems are not detail-oriented enough to assist in a thorough correlation between surgical techniques and outcomes. The use of single- or double-layer closure terminology is insufficient and has proven to be flawed, without describing the handling of the endometrium in terms of its inclusion or exclusion in the closure.
Quality improvement feedback
Long-term post-CD complications are often not reported to the physician or institution involved in the prior CD. In our opinion, some sort of registry would be of value. Perhaps then subsequent CD outcomes could be traced back and reported to the prior institution and surgeon. Feedback is critical to understanding the correlation between techniques and outcomes and more specifically to gathering learning points and using data for quality improvement of future cases.
Patient education
While women continue to have complications following the presently used surgical techniques, they often have expectations not discussed with their obstetricians. Women should be educated and empowered to realize the different approaches to all aspects and consequences of CDs.
Conclusion
The technique of excluding the endometrium in closing the uterine incision appears to reduce subsequent abnormal placentation and diminish the frequency and size of post-CD scar defect. The revival of the endometrium-free closure technique may allow significant change in the postoperative results. Currently, standardization of CD technique is being promoted on the basis of time- and cost-savings rather than clinical outcomes. Simultaneously, inroads are being made to better understand the risks and consequences of CD.
Emerging evidence suggests that a post-CD niche is the result of poor layer approximation as well as inclusion of the endometrium, which prevent healing of the uterine wall and often enables faulty implantation of the fertilized oocyte in the next pregnancy, potentially giving rise to placenta accreta spectrum. The prevalence and size of the defect can be minimized by techniques aimed at restoring the anatomy of the uterine wall and the physiology of the endometrium. Specialized training and education are necessary to stress the importance of anatomical assessment and decision making at the time of uterine closure. ●
- Rotas MA, Haberman S, Levgur M. Cesarean scar ectopic pregnancies: etiology, diagnosis, and management. Obstet Gynecol. 2006;107:1373-1381.
- Timor-Tritsch IE, Monteagudo A, Calì G, et al. Cesarean scar pregnancy is a precursor of morbidly adherent placenta. Ultrasound Obstet Gynecol. 2014;44:346-353. doi:10.1002/ uog.13426.
- Timor-Tritsch IE, Monteagudo A, Cali G, et al. Cesarean scar pregnancy and early placenta accreta share common histology. Ultrasound Obstet Gynecol. 2014;43:383-395. doi: 10.1002/uog.13282.
- Mogos MF, Salemi JL, Ashley M, et al. Recent trends in placenta accreta in the United States and its impact on maternal-fetal morbidity and healthcare-associated costs, 1998-2011. J Matern Fetal Neonatal Med. 2016;29:1077-1082.
- Jauniaux E, Collins S, Burton GJ. Placenta accreta spectrum: pathophysiology and evidence-based anatomy for prenatal ultrasound imaging. Am J Obstet Gynecol. 2018;218:75-87.
- Martin JA, Hamilton BE, Osterman MJK. Births in the United States, 2016. NCHS Data Brief. 2017(287):1-8.
- Vikhareva Osser O, Valentin L. Clinical importance of appearance of cesarean hysterotomy scar at transvaginal ultrasonography in nonpregnant women. Obstet Gynecol. 2011;117:525-532.
- Chen YY, Tsai CC, Kung FT, et al. Association between hysteroscopic findings of previous cesarean delivery scar defects and abnormal uterine bleeding. Taiwanese J Obstet Gynecol. 2019;58:541-544.
- Stegwee SI, Beij A, de Leeuw RA, et al. Niche-related outcomes after caesarean section and quality of life: a focus group study and review of literature. Qual Life Res. 2020;29:1013-1025.
- Vissers J, Hehenkamp W, Lambalk CB, et al. Post-caesarean section niche-related impaired fertility: hypothetical mechanisms. Hum Reprod. 2020;35:1484-1494.
- Vissers J, Sluckin TC, van Driel-Delprat CCR, et al. Reduced pregnancy and live birth rates after in vitro fertilization in women with previous caesarean section: a retrospective cohort study. Hum Reprod. 2020;35:595-604.
- Monteagudo A, Carreno C, Timor-Tritsch IE. Saline infusion sonohysterography in nonpregnant women with previous cesarean delivery: the “niche” in the scar. J Ultrasound Med. 2001;20:1105-1115.
- Poidevin LO. The value of hysterography in the prediction of cesarean section wound defects. Am J Obstet Gynecol. 1961;81:67-71.
- Jordans IPM, de Leeuw RA, Stegwee SI, et al. Sonographic examination of uterine niche in non-pregnant women: a modified Delphi procedure. Ultrasound Obstet Gynecol. 2019;53:107-115.
- Stegwee SI, van der Voet LF, Ben AJ, et al. Effect of single- versus double-layer uterine closure during caesarean section on postmenstrual spotting (2Close): multicentre, double-blind, randomised controlled superiority trial. BJOG. 2021;128:866-878.
- Bij de Vaate AJ, van der Voet LF, Naji O, et al. Prevalence, potential risk factors for development and symptoms related to the presence of uterine niches following cesarean section: systematic review. Ultrasound Obstet Gynecol. 2014;43:372-382.
- van der Voet LF, Bij de Vaate AM, Veersema S, et al. Long-term complications of caesarean section. The niche in the scar: a prospective cohort study on niche prevalence and its relation to abnormal uterine bleeding. BJOG. 2014;121:236-244.
- Vervoort AJ, Uittenbogaard LB, Hehenkamp WJ, et al. Why do niches develop in caesarean uterine scars? Hypotheses on the aetiology of niche development. Hum Reprod. 2015;30:2695-2702.
- Sholapurkar SL. Etiology of cesarean uterine scar defect (niche): detailed critical analysis of hypotheses and prevention strategies and peritoneal closure debate. J Clin Med Res. 2018;10:166-173.
- Kamel R, Eissa T, Sharaf M, et al. Position and integrity of uterine scar are determined by degree of cervical dilatation at time of cesarean section. Ultrasound Obstet Gynecol. 2021;57:466-470.
- Sanders RC, Parsons AK. Anteverted retroflexed uterus: a common consequence of cesarean delivery. AJR Am J Roentgenol. 2014;203:W117-124.
- Antoine C, Pimentel RN, Timor-Tritsch IE, et al. Origin of a post-cesarean delivery niche: diagnosis, pathophysiologic characteristics, and video documentation. J Ultrasound Med. 2021;40:205-208.
- AbdullGaffar B, Almulla A. A histopathologic approach to uterine niche: what to expect and to report in hysteroscopy-resected isthmocele specimens. Int J Surg Pathol. 2021:10668969211039415. doi: 10.1177/10668969211039415.
- Nagy S, Papp Z. Global approach of the cesarean section rates. J Perinatal Med. 2020;49:1-4.
- Kerr JM. The technic of cesarean section, with special reference to the lower uterine segment incision. Am J Obstet Gynecol. 1926;12:729-734.
- Joel-Cohen S. Abdominal and vaginal hysterectomy: new techniques based on time and motion studies. Lippincott Williams & Wilkins; 1977.
- Holmgren G, Sjoholm L, Stark M. The Misgav Ladach method for cesarean section: method description. Acta Obstet Gynecol Scand. 1999;78:615-621.
- Abalos E, Addo V, Brocklehurst P, et al. Caesarean section surgical techniques: 3-year follow-up of the CORONIS fractional, factorial, unmasked, randomised controlled trial. Lancet. 2016;388:62-72.
- Antoine C, Meyer JA, Silverstein JS, et al. The impact of uterine incision closure techniques on post-cesarean delivery niche formation and size: sonohysterographic examination of nonpregnant women. J Ultrasound Med. 2021. doi: 10.1002/ jum.15859.
- Antoine C AJ, Yaghoubian Y, Harary J. Variations in uterine closure technique: an institutional survey of obstetricians and implications for patient counseling and prevention of adverse sequelae [Abstract]. 2021.
- Antoine C, Pimentel RN, Reece EA, et al. Endometrium-free uterine closure technique and abnormal placental implantation in subsequent pregnancies. J Matern-Fetal Neonatal Med. 2019:1-9.
- Jauniaux E, Jurkovic D. Placenta accreta: pathogenesis of a 20th century iatrogenic uterine disease. Placenta. 2012;33:244-251.
- Sholapurkar S. Review of unsafe changes in the practice of cesarean section with analysis of flaws in the interpretation of statistics and the evidence. Surgical Case Reports. 2021;4:2-6.
- Rotas MA, Haberman S, Levgur M. Cesarean scar ectopic pregnancies: etiology, diagnosis, and management. Obstet Gynecol. 2006;107:1373-1381.
- Timor-Tritsch IE, Monteagudo A, Calì G, et al. Cesarean scar pregnancy is a precursor of morbidly adherent placenta. Ultrasound Obstet Gynecol. 2014;44:346-353. doi:10.1002/ uog.13426.
- Timor-Tritsch IE, Monteagudo A, Cali G, et al. Cesarean scar pregnancy and early placenta accreta share common histology. Ultrasound Obstet Gynecol. 2014;43:383-395. doi: 10.1002/uog.13282.
- Mogos MF, Salemi JL, Ashley M, et al. Recent trends in placenta accreta in the United States and its impact on maternal-fetal morbidity and healthcare-associated costs, 1998-2011. J Matern Fetal Neonatal Med. 2016;29:1077-1082.
- Jauniaux E, Collins S, Burton GJ. Placenta accreta spectrum: pathophysiology and evidence-based anatomy for prenatal ultrasound imaging. Am J Obstet Gynecol. 2018;218:75-87.
- Martin JA, Hamilton BE, Osterman MJK. Births in the United States, 2016. NCHS Data Brief. 2017(287):1-8.
- Vikhareva Osser O, Valentin L. Clinical importance of appearance of cesarean hysterotomy scar at transvaginal ultrasonography in nonpregnant women. Obstet Gynecol. 2011;117:525-532.
- Chen YY, Tsai CC, Kung FT, et al. Association between hysteroscopic findings of previous cesarean delivery scar defects and abnormal uterine bleeding. Taiwanese J Obstet Gynecol. 2019;58:541-544.
- Stegwee SI, Beij A, de Leeuw RA, et al. Niche-related outcomes after caesarean section and quality of life: a focus group study and review of literature. Qual Life Res. 2020;29:1013-1025.
- Vissers J, Hehenkamp W, Lambalk CB, et al. Post-caesarean section niche-related impaired fertility: hypothetical mechanisms. Hum Reprod. 2020;35:1484-1494.
- Vissers J, Sluckin TC, van Driel-Delprat CCR, et al. Reduced pregnancy and live birth rates after in vitro fertilization in women with previous caesarean section: a retrospective cohort study. Hum Reprod. 2020;35:595-604.
- Monteagudo A, Carreno C, Timor-Tritsch IE. Saline infusion sonohysterography in nonpregnant women with previous cesarean delivery: the “niche” in the scar. J Ultrasound Med. 2001;20:1105-1115.
- Poidevin LO. The value of hysterography in the prediction of cesarean section wound defects. Am J Obstet Gynecol. 1961;81:67-71.
- Jordans IPM, de Leeuw RA, Stegwee SI, et al. Sonographic examination of uterine niche in non-pregnant women: a modified Delphi procedure. Ultrasound Obstet Gynecol. 2019;53:107-115.
- Stegwee SI, van der Voet LF, Ben AJ, et al. Effect of single- versus double-layer uterine closure during caesarean section on postmenstrual spotting (2Close): multicentre, double-blind, randomised controlled superiority trial. BJOG. 2021;128:866-878.
- Bij de Vaate AJ, van der Voet LF, Naji O, et al. Prevalence, potential risk factors for development and symptoms related to the presence of uterine niches following cesarean section: systematic review. Ultrasound Obstet Gynecol. 2014;43:372-382.
- van der Voet LF, Bij de Vaate AM, Veersema S, et al. Long-term complications of caesarean section. The niche in the scar: a prospective cohort study on niche prevalence and its relation to abnormal uterine bleeding. BJOG. 2014;121:236-244.
- Vervoort AJ, Uittenbogaard LB, Hehenkamp WJ, et al. Why do niches develop in caesarean uterine scars? Hypotheses on the aetiology of niche development. Hum Reprod. 2015;30:2695-2702.
- Sholapurkar SL. Etiology of cesarean uterine scar defect (niche): detailed critical analysis of hypotheses and prevention strategies and peritoneal closure debate. J Clin Med Res. 2018;10:166-173.
- Kamel R, Eissa T, Sharaf M, et al. Position and integrity of uterine scar are determined by degree of cervical dilatation at time of cesarean section. Ultrasound Obstet Gynecol. 2021;57:466-470.
- Sanders RC, Parsons AK. Anteverted retroflexed uterus: a common consequence of cesarean delivery. AJR Am J Roentgenol. 2014;203:W117-124.
- Antoine C, Pimentel RN, Timor-Tritsch IE, et al. Origin of a post-cesarean delivery niche: diagnosis, pathophysiologic characteristics, and video documentation. J Ultrasound Med. 2021;40:205-208.
- AbdullGaffar B, Almulla A. A histopathologic approach to uterine niche: what to expect and to report in hysteroscopy-resected isthmocele specimens. Int J Surg Pathol. 2021:10668969211039415. doi: 10.1177/10668969211039415.
- Nagy S, Papp Z. Global approach of the cesarean section rates. J Perinatal Med. 2020;49:1-4.
- Kerr JM. The technic of cesarean section, with special reference to the lower uterine segment incision. Am J Obstet Gynecol. 1926;12:729-734.
- Joel-Cohen S. Abdominal and vaginal hysterectomy: new techniques based on time and motion studies. Lippincott Williams & Wilkins; 1977.
- Holmgren G, Sjoholm L, Stark M. The Misgav Ladach method for cesarean section: method description. Acta Obstet Gynecol Scand. 1999;78:615-621.
- Abalos E, Addo V, Brocklehurst P, et al. Caesarean section surgical techniques: 3-year follow-up of the CORONIS fractional, factorial, unmasked, randomised controlled trial. Lancet. 2016;388:62-72.
- Antoine C, Meyer JA, Silverstein JS, et al. The impact of uterine incision closure techniques on post-cesarean delivery niche formation and size: sonohysterographic examination of nonpregnant women. J Ultrasound Med. 2021. doi: 10.1002/ jum.15859.
- Antoine C AJ, Yaghoubian Y, Harary J. Variations in uterine closure technique: an institutional survey of obstetricians and implications for patient counseling and prevention of adverse sequelae [Abstract]. 2021.
- Antoine C, Pimentel RN, Reece EA, et al. Endometrium-free uterine closure technique and abnormal placental implantation in subsequent pregnancies. J Matern-Fetal Neonatal Med. 2019:1-9.
- Jauniaux E, Jurkovic D. Placenta accreta: pathogenesis of a 20th century iatrogenic uterine disease. Placenta. 2012;33:244-251.
- Sholapurkar S. Review of unsafe changes in the practice of cesarean section with analysis of flaws in the interpretation of statistics and the evidence. Surgical Case Reports. 2021;4:2-6.
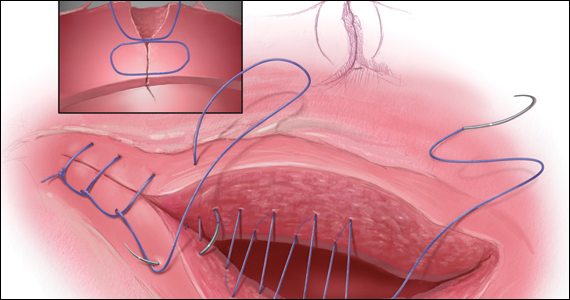
About 19% of COVID-19 headaches become chronic
Approximately one in five patients who presented with headache during the acute phase of COVID-19 developed chronic daily headache, according to a study published in Cephalalgia. The greater the headache’s intensity during the acute phase, the greater the likelihood that it would persist.
The research, carried out by members of the Headache Study Group of the Spanish Society of Neurology, evaluated the evolution of headache in more than 900 Spanish patients. Because they found that headache intensity during the acute phase was associated with a more prolonged duration of headache, the team stressed the importance of promptly evaluating patients who have had COVID-19 and who then experience persistent headache.
Long-term evolution unknown
Headache is a common symptom of COVID-19, but its long-term evolution remains unknown. The objective of this study was to evaluate the long-term duration of headache in patients who presented with this symptom during the acute phase of the disease.
Recruitment for this multicenter study took place in March and April 2020. The 905 patients who were enrolled came from six level 3 hospitals in Spain. All completed 9 months of neurologic follow-up.
Their median age was 51 years, 66.5% were women, and more than half (52.7%) had a history of primary headache. About half of the patients required hospitalization (50.5%); the rest were treated as outpatients. The most common headache phenotype was holocranial (67.8%) of severe intensity (50.6%).
Persistent headache common
In the 96.6% cases for which data were available, the median duration of headache was 14 days. The headache persisted at 1 month in 31.1% of patients, at 2 months in 21.5%, at 3 months in 19%, at 6 months in 16.8%, and at 9 months in 16.0%.
“The median duration of COVID-19 headache is around 2 weeks,” David García Azorín, MD, PhD, a member of the Spanish Society of Neurology and one of the coauthors of the study, said in an interview. “However, almost 20% of patients experience it for longer than that. When still present at 2 months, the headache is more likely to follow a chronic daily pattern.” Dr. García Azorín is a neurologist and clinical researcher at the headache unit of the Hospital Clínico Universitario in Valladolid, Spain.
“So, if the headache isn’t letting up, it’s important to make the most of that window of opportunity and provide treatment in that period of 6-12 weeks,” he continued. “To do this, the best option is to carry out preventive treatment so that the patient will have a better chance of recovering.”
Study participants whose headache persisted at 9 months were older and were mostly women. They were less likely to have had pneumonia or to have experienced stabbing pain, photophobia, or phonophobia. They reported that the headache got worse when they engaged in physical activity but less frequently manifested as a throbbing headache.
Secondary tension headaches
On the other hand, Jaime Rodríguez Vico, MD, head of the headache unit at the Jiménez Díaz Foundation Hospital in Madrid, said in an interview that, according to his case studies, the most striking characteristics of post–COVID-19 headaches “in general are secondary, with similarities to tension headaches that patients are able to differentiate from other clinical types of headache. In patients with migraine, very often we see that we’re dealing with a trigger. In other words, more migraines – and more intense ones at that – are brought about.”
He added: “Generally, post–COVID-19 headache usually lasts 1-2 weeks, but we have cases of it lasting several months and even over a year with persistent daily headache. These more persistent cases are probably connected to another type of pathology that makes them more susceptible to becoming chronic, something that occurs in another type of primary headache known as new daily persistent headache.”
Primary headache exacerbation
Dr. García Azorín pointed out that it’s not uncommon that among people who already have primary headache, their condition worsens after they become infected with SARS-CoV-2. However, many people differentiate the headache associated with the infection from their usual headache because after becoming infected, their headache is predominantly frontal, oppressive, and chronic.
“Having a prior history of headache is one of the factors that can increase the likelihood that a headache experienced while suffering from COVID-19 will become chronic,” he noted.
This study also found that, more often than not, patients with persistent headache at 9 months had migraine-like pain.
As for headaches in these patients beyond 9 months, “based on our research, the evolution is quite variable,” said Dr. Rodríguez Vico. “Our unit’s numbers are skewed due to the high number of migraine cases that we follow, and therefore our high volume of migraine patients who’ve gotten worse. The same thing happens with COVID-19 vaccines. Migraine is a polygenic disorder with multiple variants and a pathophysiology that we are just beginning to describe. This is why one patient is completely different from another. It’s a real challenge.”
Infections are a common cause of acute and chronic headache. The persistence of a headache after an infection may be caused by the infection becoming chronic, as happens in some types of chronic meningitis, such as tuberculous meningitis. It may also be caused by the persistence of a certain response and activation of the immune system or to the uncovering or worsening of a primary headache coincident with the infection, added Dr. García Azorín.
“Likewise, there are other people who have a biological predisposition to headache as a multifactorial disorder and polygenic disorder, such that a particular stimulus – from trauma or an infection to alcohol consumption – can cause them to develop a headache very similar to a migraine,” he said.
Providing prognosis and treatment
Certain factors can give an idea of how long the headache might last. The study’s univariate analysis showed that age, female sex, headache intensity, pressure-like quality, the presence of photophobia/phonophobia, and worsening with physical activity were associated with headache of longer duration. But in the multivariate analysis, only headache intensity during the acute phase remained statistically significant (hazard ratio, 0.655; 95% confidence interval, 0.582-0.737; P < .001).
When asked whether they planned to continue the study, Dr. García Azorín commented, “The main questions that have arisen from this study have been, above all: ‘Why does this headache happen?’ and ‘How can it be treated or avoided?’ To answer them, we’re looking into pain: which factors could predispose a person to it and which changes may be associated with its presence.”
In addition, different treatments that may improve patient outcomes are being evaluated, because to date, treatment has been empirical and based on the predominant pain phenotype.
In any case, most doctors currently treat post–COVID-19 headache on the basis of how similar the symptoms are to those of other primary headaches. “Given the impact that headache has on patients’ quality of life, there’s a pressing need for controlled studies on possible treatments and their effectiveness,” noted Patricia Pozo Rosich, MD, PhD, one of the coauthors of the study.
“We at the Spanish Society of Neurology truly believe that if these patients were to have this symptom correctly addressed from the start, they could avoid many of the problems that arise in the situation becoming chronic,” she concluded.
Dr. García Azorín and Dr. Rodríguez Vico disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Approximately one in five patients who presented with headache during the acute phase of COVID-19 developed chronic daily headache, according to a study published in Cephalalgia. The greater the headache’s intensity during the acute phase, the greater the likelihood that it would persist.
The research, carried out by members of the Headache Study Group of the Spanish Society of Neurology, evaluated the evolution of headache in more than 900 Spanish patients. Because they found that headache intensity during the acute phase was associated with a more prolonged duration of headache, the team stressed the importance of promptly evaluating patients who have had COVID-19 and who then experience persistent headache.
Long-term evolution unknown
Headache is a common symptom of COVID-19, but its long-term evolution remains unknown. The objective of this study was to evaluate the long-term duration of headache in patients who presented with this symptom during the acute phase of the disease.
Recruitment for this multicenter study took place in March and April 2020. The 905 patients who were enrolled came from six level 3 hospitals in Spain. All completed 9 months of neurologic follow-up.
Their median age was 51 years, 66.5% were women, and more than half (52.7%) had a history of primary headache. About half of the patients required hospitalization (50.5%); the rest were treated as outpatients. The most common headache phenotype was holocranial (67.8%) of severe intensity (50.6%).
Persistent headache common
In the 96.6% cases for which data were available, the median duration of headache was 14 days. The headache persisted at 1 month in 31.1% of patients, at 2 months in 21.5%, at 3 months in 19%, at 6 months in 16.8%, and at 9 months in 16.0%.
“The median duration of COVID-19 headache is around 2 weeks,” David García Azorín, MD, PhD, a member of the Spanish Society of Neurology and one of the coauthors of the study, said in an interview. “However, almost 20% of patients experience it for longer than that. When still present at 2 months, the headache is more likely to follow a chronic daily pattern.” Dr. García Azorín is a neurologist and clinical researcher at the headache unit of the Hospital Clínico Universitario in Valladolid, Spain.
“So, if the headache isn’t letting up, it’s important to make the most of that window of opportunity and provide treatment in that period of 6-12 weeks,” he continued. “To do this, the best option is to carry out preventive treatment so that the patient will have a better chance of recovering.”
Study participants whose headache persisted at 9 months were older and were mostly women. They were less likely to have had pneumonia or to have experienced stabbing pain, photophobia, or phonophobia. They reported that the headache got worse when they engaged in physical activity but less frequently manifested as a throbbing headache.
Secondary tension headaches
On the other hand, Jaime Rodríguez Vico, MD, head of the headache unit at the Jiménez Díaz Foundation Hospital in Madrid, said in an interview that, according to his case studies, the most striking characteristics of post–COVID-19 headaches “in general are secondary, with similarities to tension headaches that patients are able to differentiate from other clinical types of headache. In patients with migraine, very often we see that we’re dealing with a trigger. In other words, more migraines – and more intense ones at that – are brought about.”
He added: “Generally, post–COVID-19 headache usually lasts 1-2 weeks, but we have cases of it lasting several months and even over a year with persistent daily headache. These more persistent cases are probably connected to another type of pathology that makes them more susceptible to becoming chronic, something that occurs in another type of primary headache known as new daily persistent headache.”
Primary headache exacerbation
Dr. García Azorín pointed out that it’s not uncommon that among people who already have primary headache, their condition worsens after they become infected with SARS-CoV-2. However, many people differentiate the headache associated with the infection from their usual headache because after becoming infected, their headache is predominantly frontal, oppressive, and chronic.
“Having a prior history of headache is one of the factors that can increase the likelihood that a headache experienced while suffering from COVID-19 will become chronic,” he noted.
This study also found that, more often than not, patients with persistent headache at 9 months had migraine-like pain.
As for headaches in these patients beyond 9 months, “based on our research, the evolution is quite variable,” said Dr. Rodríguez Vico. “Our unit’s numbers are skewed due to the high number of migraine cases that we follow, and therefore our high volume of migraine patients who’ve gotten worse. The same thing happens with COVID-19 vaccines. Migraine is a polygenic disorder with multiple variants and a pathophysiology that we are just beginning to describe. This is why one patient is completely different from another. It’s a real challenge.”
Infections are a common cause of acute and chronic headache. The persistence of a headache after an infection may be caused by the infection becoming chronic, as happens in some types of chronic meningitis, such as tuberculous meningitis. It may also be caused by the persistence of a certain response and activation of the immune system or to the uncovering or worsening of a primary headache coincident with the infection, added Dr. García Azorín.
“Likewise, there are other people who have a biological predisposition to headache as a multifactorial disorder and polygenic disorder, such that a particular stimulus – from trauma or an infection to alcohol consumption – can cause them to develop a headache very similar to a migraine,” he said.
Providing prognosis and treatment
Certain factors can give an idea of how long the headache might last. The study’s univariate analysis showed that age, female sex, headache intensity, pressure-like quality, the presence of photophobia/phonophobia, and worsening with physical activity were associated with headache of longer duration. But in the multivariate analysis, only headache intensity during the acute phase remained statistically significant (hazard ratio, 0.655; 95% confidence interval, 0.582-0.737; P < .001).
When asked whether they planned to continue the study, Dr. García Azorín commented, “The main questions that have arisen from this study have been, above all: ‘Why does this headache happen?’ and ‘How can it be treated or avoided?’ To answer them, we’re looking into pain: which factors could predispose a person to it and which changes may be associated with its presence.”
In addition, different treatments that may improve patient outcomes are being evaluated, because to date, treatment has been empirical and based on the predominant pain phenotype.
In any case, most doctors currently treat post–COVID-19 headache on the basis of how similar the symptoms are to those of other primary headaches. “Given the impact that headache has on patients’ quality of life, there’s a pressing need for controlled studies on possible treatments and their effectiveness,” noted Patricia Pozo Rosich, MD, PhD, one of the coauthors of the study.
“We at the Spanish Society of Neurology truly believe that if these patients were to have this symptom correctly addressed from the start, they could avoid many of the problems that arise in the situation becoming chronic,” she concluded.
Dr. García Azorín and Dr. Rodríguez Vico disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Approximately one in five patients who presented with headache during the acute phase of COVID-19 developed chronic daily headache, according to a study published in Cephalalgia. The greater the headache’s intensity during the acute phase, the greater the likelihood that it would persist.
The research, carried out by members of the Headache Study Group of the Spanish Society of Neurology, evaluated the evolution of headache in more than 900 Spanish patients. Because they found that headache intensity during the acute phase was associated with a more prolonged duration of headache, the team stressed the importance of promptly evaluating patients who have had COVID-19 and who then experience persistent headache.
Long-term evolution unknown
Headache is a common symptom of COVID-19, but its long-term evolution remains unknown. The objective of this study was to evaluate the long-term duration of headache in patients who presented with this symptom during the acute phase of the disease.
Recruitment for this multicenter study took place in March and April 2020. The 905 patients who were enrolled came from six level 3 hospitals in Spain. All completed 9 months of neurologic follow-up.
Their median age was 51 years, 66.5% were women, and more than half (52.7%) had a history of primary headache. About half of the patients required hospitalization (50.5%); the rest were treated as outpatients. The most common headache phenotype was holocranial (67.8%) of severe intensity (50.6%).
Persistent headache common
In the 96.6% cases for which data were available, the median duration of headache was 14 days. The headache persisted at 1 month in 31.1% of patients, at 2 months in 21.5%, at 3 months in 19%, at 6 months in 16.8%, and at 9 months in 16.0%.
“The median duration of COVID-19 headache is around 2 weeks,” David García Azorín, MD, PhD, a member of the Spanish Society of Neurology and one of the coauthors of the study, said in an interview. “However, almost 20% of patients experience it for longer than that. When still present at 2 months, the headache is more likely to follow a chronic daily pattern.” Dr. García Azorín is a neurologist and clinical researcher at the headache unit of the Hospital Clínico Universitario in Valladolid, Spain.
“So, if the headache isn’t letting up, it’s important to make the most of that window of opportunity and provide treatment in that period of 6-12 weeks,” he continued. “To do this, the best option is to carry out preventive treatment so that the patient will have a better chance of recovering.”
Study participants whose headache persisted at 9 months were older and were mostly women. They were less likely to have had pneumonia or to have experienced stabbing pain, photophobia, or phonophobia. They reported that the headache got worse when they engaged in physical activity but less frequently manifested as a throbbing headache.
Secondary tension headaches
On the other hand, Jaime Rodríguez Vico, MD, head of the headache unit at the Jiménez Díaz Foundation Hospital in Madrid, said in an interview that, according to his case studies, the most striking characteristics of post–COVID-19 headaches “in general are secondary, with similarities to tension headaches that patients are able to differentiate from other clinical types of headache. In patients with migraine, very often we see that we’re dealing with a trigger. In other words, more migraines – and more intense ones at that – are brought about.”
He added: “Generally, post–COVID-19 headache usually lasts 1-2 weeks, but we have cases of it lasting several months and even over a year with persistent daily headache. These more persistent cases are probably connected to another type of pathology that makes them more susceptible to becoming chronic, something that occurs in another type of primary headache known as new daily persistent headache.”
Primary headache exacerbation
Dr. García Azorín pointed out that it’s not uncommon that among people who already have primary headache, their condition worsens after they become infected with SARS-CoV-2. However, many people differentiate the headache associated with the infection from their usual headache because after becoming infected, their headache is predominantly frontal, oppressive, and chronic.
“Having a prior history of headache is one of the factors that can increase the likelihood that a headache experienced while suffering from COVID-19 will become chronic,” he noted.
This study also found that, more often than not, patients with persistent headache at 9 months had migraine-like pain.
As for headaches in these patients beyond 9 months, “based on our research, the evolution is quite variable,” said Dr. Rodríguez Vico. “Our unit’s numbers are skewed due to the high number of migraine cases that we follow, and therefore our high volume of migraine patients who’ve gotten worse. The same thing happens with COVID-19 vaccines. Migraine is a polygenic disorder with multiple variants and a pathophysiology that we are just beginning to describe. This is why one patient is completely different from another. It’s a real challenge.”
Infections are a common cause of acute and chronic headache. The persistence of a headache after an infection may be caused by the infection becoming chronic, as happens in some types of chronic meningitis, such as tuberculous meningitis. It may also be caused by the persistence of a certain response and activation of the immune system or to the uncovering or worsening of a primary headache coincident with the infection, added Dr. García Azorín.
“Likewise, there are other people who have a biological predisposition to headache as a multifactorial disorder and polygenic disorder, such that a particular stimulus – from trauma or an infection to alcohol consumption – can cause them to develop a headache very similar to a migraine,” he said.
Providing prognosis and treatment
Certain factors can give an idea of how long the headache might last. The study’s univariate analysis showed that age, female sex, headache intensity, pressure-like quality, the presence of photophobia/phonophobia, and worsening with physical activity were associated with headache of longer duration. But in the multivariate analysis, only headache intensity during the acute phase remained statistically significant (hazard ratio, 0.655; 95% confidence interval, 0.582-0.737; P < .001).
When asked whether they planned to continue the study, Dr. García Azorín commented, “The main questions that have arisen from this study have been, above all: ‘Why does this headache happen?’ and ‘How can it be treated or avoided?’ To answer them, we’re looking into pain: which factors could predispose a person to it and which changes may be associated with its presence.”
In addition, different treatments that may improve patient outcomes are being evaluated, because to date, treatment has been empirical and based on the predominant pain phenotype.
In any case, most doctors currently treat post–COVID-19 headache on the basis of how similar the symptoms are to those of other primary headaches. “Given the impact that headache has on patients’ quality of life, there’s a pressing need for controlled studies on possible treatments and their effectiveness,” noted Patricia Pozo Rosich, MD, PhD, one of the coauthors of the study.
“We at the Spanish Society of Neurology truly believe that if these patients were to have this symptom correctly addressed from the start, they could avoid many of the problems that arise in the situation becoming chronic,” she concluded.
Dr. García Azorín and Dr. Rodríguez Vico disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM CEPHALALGIA
JIA disease activity, disability linked to social factors
For children with polyarticular juvenile idiopathic arthritis (pJIA), functional disability lasts longer and disease activity is increased among those who belong to a racial/ethnic minority or come from homes with low household income or low family education, according to a study published online in Pediatric Rheumatology. The findings also initially revealed a higher likelihood of functional disability among those living in a poorer community, but that association lost statistical significance after adjustment for confounders.
“We chose community poverty level as the primary predictor for outcomes in pJIA because the socioeconomic context of communities and neighborhoods affects the characteristics of the social, service, and physical environments to which all residents are exposed regardless of their own socioeconomic position and may have a greater negative impact on those with fewer individual resources,” the authors write. “While community poverty level was not associated with an increase in odds of moderate-to-severe disease activity, those with high community poverty level did have higher disease activity scores (0.33 points greater on average than those with low community poverty level, in adjusted analysis).”
Nayimisha Balmuri, MD, an assistant professor of pediatrics at Johns Hopkins Medicine and study coauthor, told this news organization that anecdotal experience from everyday practice has shown that “patients with myriad social determinants of health stacked against them present sicker, take longer to present, and require far more aggressive therapies and follow-up,” which wreaks havoc in terms of disease activity. “It’s really difficult, then, to play catch-up to other cohorts of patients,” Dr. Balmuri added.
Disparities in outcomes persist
A key clinical take-home message from these findings is that the differences in clinical outcomes are relevant throughout the entire year of therapy, Dr. Balmuri said. “Patients get better; however, they don’t get better the same,” she said, and this is because of a variety of reasons. “Getting in the door is one of [those reasons] but then continuing to follow-up care is another.” For general practitioners, it’s especially important to refer patients who complain of joint pains to a specialist and to then follow up to be sure they’re improving and they’re getting the care they need.
For pediatric rheumatologists and subspecialists, “it’s important for us to realize that the disparity doesn’t end when patients come into your door to begin with,” Dr. Balmuri said. “It continues over the short term and far past that into adulthood.”
Candace Feldman, MD, MPH, ScD, an assistant professor of medicine in the Division of Rheumatology, Inflammation, and Immunity at Brigham and Women’s Hospital, Boston, told this news organization that the research “provides an important foundation to the study of the impact of social determinants of health on disease activity and disability among children with JIA. Individuals with rheumatic conditions should be screened for social determinants of health–related needs, and infrastructure should exist within the rheumatology clinic to help address the needs uncovered.” Dr. Feldman was not involved in the study.
In addition to the results’ clinical significance, Dr. Feldman also noted the policy implications of these findings. “Physicians should advocate for efforts to dismantle structural racism, to address income inequality, and to mitigate the effects of climate change, which also disproportionately affect historically marginalized populations,” Dr. Feldman said. Although this study focused predominantly on poverty, she noted that financial insecurity, food insecurity, homelessness, or housing instability were other social determinants of health to consider in future research.
Dr. Balmuri and William Daniel Soulsby, MD, a clinical fellow in pediatric rheumatology at the University of California, San Francisco, who is the study’s lead author, said they focused on poverty in this study not only because it’s so understudied in patients with pJIA but also because research in adults with lupus has found that leaving poverty was associated with a reversal of accrued disease damage.
Interactions of social determinants
The authors analyzed retrospective data from 1,684 pediatric patients in the Childhood Arthritis and Rheumatology Research Alliance (CARRA) registry covering the period of April 2015 to February 2020. All study participants had been diagnosed with pJIA. Symptom onset occurred before age 16, and at least five joints were involved. The authors excluded patients who had been diagnosed with other systemic inflammatory or autoimmune diseases.
The authors defined exposure to a high level of community poverty as living in a ZIP code where at least 20% of residents lived at or below the federal poverty level. The authors also collected data on household income, although these data were missing for more than a quarter of participants (27%) and were therefore included only in sensitivity analyses. They used the clinical Juvenile Arthritis Disease Activity Score–10 (cJADAS-10) and the Child Health Assessment Questionnaire (CHAQ) to assess disease activity and disability at baseline and 6 and 12 months later. A cutoff of 2.5 on the cJADAS-10 distinguished mild disease activity from moderate to high disease activity, and a CHAQ score of 0.25 was the cutoff for having functional disability.
Among those who reported household income, just over half the cohort had an income of at least $50,000. The study population was 74% White, and more non-White patients lived in high-poverty communities (36.4%) than did White patients (21.3%). Patients whose families had no more than a high school education (23.1% vs. 13.7%) and those with public insurance (43.0% vs. 21.5%) were also over-represented in poorer communities.
The median cJADAS-10 scores declined overall during patients’ first year of therapy. However, those with public insurance, a lower family education level, or residency in poorer communities made up the greatest proportion of patients who continued to have moderate to severe disease activity a year after diagnosis.
The unadjusted calculations showed that children living in high community poverty had 1.8 times greater odds of functional disability (odds ratio, 1.82; P < .001). However, after adjustment for age, sex, race/ethnicity, insurance status, family education, rheumatoid factor, and cyclic citrullinated peptide antibody, the association lost statistical significance (P = .3). Community poverty level was not associated with disease activity before or after adjustment.
“Race was adjusted for as a confounder; however, the association between race/ethnicity and social determinants of health is likely more complex,” Dr. Feldman said. “Interactions, for example, between individual race and area-level poverty could be investigated.”
Odds of persistent function disability were 1.5 times greater for children with public insurance (adjusted OR, 1.56; P = .023) and 1.9 times greater for those whose families had a lower education level (aOR, 1.89; P = .013). Children whose race/ethnicity was indicated as being other than White had more than double the odds of higher disease activity (aOR, 2.48; P = .002) and were nearly twice as likely to have persistent functional disability (aOR, 1.91; P = .031).
Future directions
Dr. Soulsby was struck by the difference in statistical significance between individual-level poverty, as measured by household income, and community-level poverty. “It’s interesting because it may suggest that both of these forms of poverty are different and have different impacts on disease,” he said. Dr. Balmuri elaborated on the nuances and interactions that exist with social determinants of health and how objective outcomes, such as disease activity as measured by clinical tools, can differ from subjective outcomes, such as patients’ reports of pain, daily disability, and social experiences.
“The human condition is far more complicated, unfortunately, than any dataset could have on their own collected,” Dr. Balmuri said. She said she plans to expand her pJIA research into other social determinants of health. “It’s first about getting people’s eyes and minds open to something we see every day that, for some reason, sometimes people are blinded to, [using] the data that we do have, and then our hope is to build upon that.”
Dr. Feldman noted that ZIP codes, which were used as a proxy for community poverty, may not provide the best perspective regarding a patient’s neighborhood, because significant variation may exist within a single ZIP code, which is something the authors noted as well. The investigators were limited in the data available from the registry, and Dr. Balmuri and Dr. Soulsby suggested that 9-digit ZIP codes or census tracts might better capture neighborhood deprivation.
The research was funded by the Arthritis Foundation and the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Dr. Feldman has received research support from Pfizer and the Bristol-Myers Squibb Foundation. Dr. Soulsby and Dr. Balmuri have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
For children with polyarticular juvenile idiopathic arthritis (pJIA), functional disability lasts longer and disease activity is increased among those who belong to a racial/ethnic minority or come from homes with low household income or low family education, according to a study published online in Pediatric Rheumatology. The findings also initially revealed a higher likelihood of functional disability among those living in a poorer community, but that association lost statistical significance after adjustment for confounders.
“We chose community poverty level as the primary predictor for outcomes in pJIA because the socioeconomic context of communities and neighborhoods affects the characteristics of the social, service, and physical environments to which all residents are exposed regardless of their own socioeconomic position and may have a greater negative impact on those with fewer individual resources,” the authors write. “While community poverty level was not associated with an increase in odds of moderate-to-severe disease activity, those with high community poverty level did have higher disease activity scores (0.33 points greater on average than those with low community poverty level, in adjusted analysis).”
Nayimisha Balmuri, MD, an assistant professor of pediatrics at Johns Hopkins Medicine and study coauthor, told this news organization that anecdotal experience from everyday practice has shown that “patients with myriad social determinants of health stacked against them present sicker, take longer to present, and require far more aggressive therapies and follow-up,” which wreaks havoc in terms of disease activity. “It’s really difficult, then, to play catch-up to other cohorts of patients,” Dr. Balmuri added.
Disparities in outcomes persist
A key clinical take-home message from these findings is that the differences in clinical outcomes are relevant throughout the entire year of therapy, Dr. Balmuri said. “Patients get better; however, they don’t get better the same,” she said, and this is because of a variety of reasons. “Getting in the door is one of [those reasons] but then continuing to follow-up care is another.” For general practitioners, it’s especially important to refer patients who complain of joint pains to a specialist and to then follow up to be sure they’re improving and they’re getting the care they need.
For pediatric rheumatologists and subspecialists, “it’s important for us to realize that the disparity doesn’t end when patients come into your door to begin with,” Dr. Balmuri said. “It continues over the short term and far past that into adulthood.”
Candace Feldman, MD, MPH, ScD, an assistant professor of medicine in the Division of Rheumatology, Inflammation, and Immunity at Brigham and Women’s Hospital, Boston, told this news organization that the research “provides an important foundation to the study of the impact of social determinants of health on disease activity and disability among children with JIA. Individuals with rheumatic conditions should be screened for social determinants of health–related needs, and infrastructure should exist within the rheumatology clinic to help address the needs uncovered.” Dr. Feldman was not involved in the study.
In addition to the results’ clinical significance, Dr. Feldman also noted the policy implications of these findings. “Physicians should advocate for efforts to dismantle structural racism, to address income inequality, and to mitigate the effects of climate change, which also disproportionately affect historically marginalized populations,” Dr. Feldman said. Although this study focused predominantly on poverty, she noted that financial insecurity, food insecurity, homelessness, or housing instability were other social determinants of health to consider in future research.
Dr. Balmuri and William Daniel Soulsby, MD, a clinical fellow in pediatric rheumatology at the University of California, San Francisco, who is the study’s lead author, said they focused on poverty in this study not only because it’s so understudied in patients with pJIA but also because research in adults with lupus has found that leaving poverty was associated with a reversal of accrued disease damage.
Interactions of social determinants
The authors analyzed retrospective data from 1,684 pediatric patients in the Childhood Arthritis and Rheumatology Research Alliance (CARRA) registry covering the period of April 2015 to February 2020. All study participants had been diagnosed with pJIA. Symptom onset occurred before age 16, and at least five joints were involved. The authors excluded patients who had been diagnosed with other systemic inflammatory or autoimmune diseases.
The authors defined exposure to a high level of community poverty as living in a ZIP code where at least 20% of residents lived at or below the federal poverty level. The authors also collected data on household income, although these data were missing for more than a quarter of participants (27%) and were therefore included only in sensitivity analyses. They used the clinical Juvenile Arthritis Disease Activity Score–10 (cJADAS-10) and the Child Health Assessment Questionnaire (CHAQ) to assess disease activity and disability at baseline and 6 and 12 months later. A cutoff of 2.5 on the cJADAS-10 distinguished mild disease activity from moderate to high disease activity, and a CHAQ score of 0.25 was the cutoff for having functional disability.
Among those who reported household income, just over half the cohort had an income of at least $50,000. The study population was 74% White, and more non-White patients lived in high-poverty communities (36.4%) than did White patients (21.3%). Patients whose families had no more than a high school education (23.1% vs. 13.7%) and those with public insurance (43.0% vs. 21.5%) were also over-represented in poorer communities.
The median cJADAS-10 scores declined overall during patients’ first year of therapy. However, those with public insurance, a lower family education level, or residency in poorer communities made up the greatest proportion of patients who continued to have moderate to severe disease activity a year after diagnosis.
The unadjusted calculations showed that children living in high community poverty had 1.8 times greater odds of functional disability (odds ratio, 1.82; P < .001). However, after adjustment for age, sex, race/ethnicity, insurance status, family education, rheumatoid factor, and cyclic citrullinated peptide antibody, the association lost statistical significance (P = .3). Community poverty level was not associated with disease activity before or after adjustment.
“Race was adjusted for as a confounder; however, the association between race/ethnicity and social determinants of health is likely more complex,” Dr. Feldman said. “Interactions, for example, between individual race and area-level poverty could be investigated.”
Odds of persistent function disability were 1.5 times greater for children with public insurance (adjusted OR, 1.56; P = .023) and 1.9 times greater for those whose families had a lower education level (aOR, 1.89; P = .013). Children whose race/ethnicity was indicated as being other than White had more than double the odds of higher disease activity (aOR, 2.48; P = .002) and were nearly twice as likely to have persistent functional disability (aOR, 1.91; P = .031).
Future directions
Dr. Soulsby was struck by the difference in statistical significance between individual-level poverty, as measured by household income, and community-level poverty. “It’s interesting because it may suggest that both of these forms of poverty are different and have different impacts on disease,” he said. Dr. Balmuri elaborated on the nuances and interactions that exist with social determinants of health and how objective outcomes, such as disease activity as measured by clinical tools, can differ from subjective outcomes, such as patients’ reports of pain, daily disability, and social experiences.
“The human condition is far more complicated, unfortunately, than any dataset could have on their own collected,” Dr. Balmuri said. She said she plans to expand her pJIA research into other social determinants of health. “It’s first about getting people’s eyes and minds open to something we see every day that, for some reason, sometimes people are blinded to, [using] the data that we do have, and then our hope is to build upon that.”
Dr. Feldman noted that ZIP codes, which were used as a proxy for community poverty, may not provide the best perspective regarding a patient’s neighborhood, because significant variation may exist within a single ZIP code, which is something the authors noted as well. The investigators were limited in the data available from the registry, and Dr. Balmuri and Dr. Soulsby suggested that 9-digit ZIP codes or census tracts might better capture neighborhood deprivation.
The research was funded by the Arthritis Foundation and the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Dr. Feldman has received research support from Pfizer and the Bristol-Myers Squibb Foundation. Dr. Soulsby and Dr. Balmuri have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
For children with polyarticular juvenile idiopathic arthritis (pJIA), functional disability lasts longer and disease activity is increased among those who belong to a racial/ethnic minority or come from homes with low household income or low family education, according to a study published online in Pediatric Rheumatology. The findings also initially revealed a higher likelihood of functional disability among those living in a poorer community, but that association lost statistical significance after adjustment for confounders.
“We chose community poverty level as the primary predictor for outcomes in pJIA because the socioeconomic context of communities and neighborhoods affects the characteristics of the social, service, and physical environments to which all residents are exposed regardless of their own socioeconomic position and may have a greater negative impact on those with fewer individual resources,” the authors write. “While community poverty level was not associated with an increase in odds of moderate-to-severe disease activity, those with high community poverty level did have higher disease activity scores (0.33 points greater on average than those with low community poverty level, in adjusted analysis).”
Nayimisha Balmuri, MD, an assistant professor of pediatrics at Johns Hopkins Medicine and study coauthor, told this news organization that anecdotal experience from everyday practice has shown that “patients with myriad social determinants of health stacked against them present sicker, take longer to present, and require far more aggressive therapies and follow-up,” which wreaks havoc in terms of disease activity. “It’s really difficult, then, to play catch-up to other cohorts of patients,” Dr. Balmuri added.
Disparities in outcomes persist
A key clinical take-home message from these findings is that the differences in clinical outcomes are relevant throughout the entire year of therapy, Dr. Balmuri said. “Patients get better; however, they don’t get better the same,” she said, and this is because of a variety of reasons. “Getting in the door is one of [those reasons] but then continuing to follow-up care is another.” For general practitioners, it’s especially important to refer patients who complain of joint pains to a specialist and to then follow up to be sure they’re improving and they’re getting the care they need.
For pediatric rheumatologists and subspecialists, “it’s important for us to realize that the disparity doesn’t end when patients come into your door to begin with,” Dr. Balmuri said. “It continues over the short term and far past that into adulthood.”
Candace Feldman, MD, MPH, ScD, an assistant professor of medicine in the Division of Rheumatology, Inflammation, and Immunity at Brigham and Women’s Hospital, Boston, told this news organization that the research “provides an important foundation to the study of the impact of social determinants of health on disease activity and disability among children with JIA. Individuals with rheumatic conditions should be screened for social determinants of health–related needs, and infrastructure should exist within the rheumatology clinic to help address the needs uncovered.” Dr. Feldman was not involved in the study.
In addition to the results’ clinical significance, Dr. Feldman also noted the policy implications of these findings. “Physicians should advocate for efforts to dismantle structural racism, to address income inequality, and to mitigate the effects of climate change, which also disproportionately affect historically marginalized populations,” Dr. Feldman said. Although this study focused predominantly on poverty, she noted that financial insecurity, food insecurity, homelessness, or housing instability were other social determinants of health to consider in future research.
Dr. Balmuri and William Daniel Soulsby, MD, a clinical fellow in pediatric rheumatology at the University of California, San Francisco, who is the study’s lead author, said they focused on poverty in this study not only because it’s so understudied in patients with pJIA but also because research in adults with lupus has found that leaving poverty was associated with a reversal of accrued disease damage.
Interactions of social determinants
The authors analyzed retrospective data from 1,684 pediatric patients in the Childhood Arthritis and Rheumatology Research Alliance (CARRA) registry covering the period of April 2015 to February 2020. All study participants had been diagnosed with pJIA. Symptom onset occurred before age 16, and at least five joints were involved. The authors excluded patients who had been diagnosed with other systemic inflammatory or autoimmune diseases.
The authors defined exposure to a high level of community poverty as living in a ZIP code where at least 20% of residents lived at or below the federal poverty level. The authors also collected data on household income, although these data were missing for more than a quarter of participants (27%) and were therefore included only in sensitivity analyses. They used the clinical Juvenile Arthritis Disease Activity Score–10 (cJADAS-10) and the Child Health Assessment Questionnaire (CHAQ) to assess disease activity and disability at baseline and 6 and 12 months later. A cutoff of 2.5 on the cJADAS-10 distinguished mild disease activity from moderate to high disease activity, and a CHAQ score of 0.25 was the cutoff for having functional disability.
Among those who reported household income, just over half the cohort had an income of at least $50,000. The study population was 74% White, and more non-White patients lived in high-poverty communities (36.4%) than did White patients (21.3%). Patients whose families had no more than a high school education (23.1% vs. 13.7%) and those with public insurance (43.0% vs. 21.5%) were also over-represented in poorer communities.
The median cJADAS-10 scores declined overall during patients’ first year of therapy. However, those with public insurance, a lower family education level, or residency in poorer communities made up the greatest proportion of patients who continued to have moderate to severe disease activity a year after diagnosis.
The unadjusted calculations showed that children living in high community poverty had 1.8 times greater odds of functional disability (odds ratio, 1.82; P < .001). However, after adjustment for age, sex, race/ethnicity, insurance status, family education, rheumatoid factor, and cyclic citrullinated peptide antibody, the association lost statistical significance (P = .3). Community poverty level was not associated with disease activity before or after adjustment.
“Race was adjusted for as a confounder; however, the association between race/ethnicity and social determinants of health is likely more complex,” Dr. Feldman said. “Interactions, for example, between individual race and area-level poverty could be investigated.”
Odds of persistent function disability were 1.5 times greater for children with public insurance (adjusted OR, 1.56; P = .023) and 1.9 times greater for those whose families had a lower education level (aOR, 1.89; P = .013). Children whose race/ethnicity was indicated as being other than White had more than double the odds of higher disease activity (aOR, 2.48; P = .002) and were nearly twice as likely to have persistent functional disability (aOR, 1.91; P = .031).
Future directions
Dr. Soulsby was struck by the difference in statistical significance between individual-level poverty, as measured by household income, and community-level poverty. “It’s interesting because it may suggest that both of these forms of poverty are different and have different impacts on disease,” he said. Dr. Balmuri elaborated on the nuances and interactions that exist with social determinants of health and how objective outcomes, such as disease activity as measured by clinical tools, can differ from subjective outcomes, such as patients’ reports of pain, daily disability, and social experiences.
“The human condition is far more complicated, unfortunately, than any dataset could have on their own collected,” Dr. Balmuri said. She said she plans to expand her pJIA research into other social determinants of health. “It’s first about getting people’s eyes and minds open to something we see every day that, for some reason, sometimes people are blinded to, [using] the data that we do have, and then our hope is to build upon that.”
Dr. Feldman noted that ZIP codes, which were used as a proxy for community poverty, may not provide the best perspective regarding a patient’s neighborhood, because significant variation may exist within a single ZIP code, which is something the authors noted as well. The investigators were limited in the data available from the registry, and Dr. Balmuri and Dr. Soulsby suggested that 9-digit ZIP codes or census tracts might better capture neighborhood deprivation.
The research was funded by the Arthritis Foundation and the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Dr. Feldman has received research support from Pfizer and the Bristol-Myers Squibb Foundation. Dr. Soulsby and Dr. Balmuri have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM PEDIATRIC RHEUMATOLOGY