User login
BCMA emerging as a promising target in MM
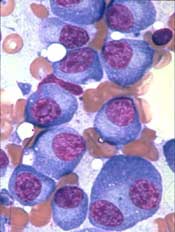
NEW YORK, NY—The B-cell maturation antigen (BCMA) is emerging as a promising target in multiple myeloma (MM), according to Adam D. Cohen, MD, of the University of Pennsylvania in Philadelphia.
BCMA is highly expressed on MM cells and, with its 2 ligands, is responsible for maintaining normal plasma cell homeostasis.
“And it’s really not expressed on any other normal tissues of the body,” Dr Cohen said.
“Importantly, BCMA is not just sitting on the cell surface as a target but actually promotes myeloma pathogenesis.”
Dr Cohen reviewed the progress being made using BCMA as a target in MM, paying particular attention to chimeric antigen receptor (CAR) T cells. He presented the update at Lymphoma & Myeloma 2017.
NCI BCMA-specific CARs
The first CAR to specifically target BCMA in MM was developed at the National Cancer Institute (NCI). It consisted of a murine single-chain variable fragment (scFv), CD3/CD28 signaling domains, and a gamma-retroviral vector.
Investigators conducted the first-in-human trial of this CAR T-cell therapy in 12 relapsed/refractory MM patients.
All patients received a lymphodepleting conditioning regimen of cyclophosphamide and fludarabine and a single infusion of 1 of 4 doses of the CAR T-cell therapy.
At higher dose levels, the BCMA-CAR produced objective responses “even in these highly refractory patients,” Dr Cohen said. “Some responses lasted 4 to 6 months.”
Patients who had the greatest degree of expansion of CAR T cells were the ones who had the best responses.
The BCMA-CAR is associated with the same toxicities as the CD19-directed CAR T-cell therapies now approved in acute lymphoblastic leukemia and non-Hodgkin lymphoma—cytokine release syndrome (CRS) and neurotoxicity.
The NCI study (NCT02215967) is ongoing.
Penn BCMA-specific CAR
A different BCMA CAR is being investigated at the University of Pennsylvania. It is a fully human CAR that consists of a human scFv, CD3/4-1BB costimulatory domains, and a lentiviral vector.
Investigators designed the first-in-human trial* (NCT02546167) with 3 different cohorts.
Patients in cohort 1 received 5 x 108 CAR T cells without any lymphodepleting chemotherapy.
The remaining patients received cyclophosphamide, followed by 5 x 107 CAR T cells in cohort 2 and 5 x 108 CAR T cells in cohort 3.
Dr Cohen reviewed current data from cohort 1, which included 9 patients. They were a median age of 57 (range, 44-70), and 67% were male. They were heavily pretreated with a median of 9 prior lines of therapy (range, 4-11).
All had high-risk cytogenetics, 67% had deletion 17p or TP53 mutation, and they had a median of 80% bone marrow plasma cells (range, 15%-95%).
“Despite this,” Dr Cohen said, “we were able to generate, successfully, CAR T cells from all patients, although 1 patient did require a second apheresis and manufacturing attempt.”
Four of the 9 patients achieved very good partial responses, and an additional 2 patients had minimal responses.
One patient had a stringent CR (sCR) for close to 2 years without having any intervening therapy.
“[The sCR] shows the potential for this [therapy] to create a durable remission in a patient without any other therapy,” Dr Cohen said. “And this patient still has circulating CAR cells detectable.”
Most of the other patients did not have as durable a response. Responses lasted a median of 3 to 5 months before the patients relapsed.
Dr Cohen noted that the Penn data confirm the NCI experience showing proof of principle.
“You can target BCMA with these cells and get objective responses that can lead to a durable one in a subset of patients,” he added.
Dr Cohen is scheduled to report the preliminary data on the cyclophosphamide cohorts (cohorts 2 and 3) at ASH 2017. Those cohorts, he said, are showing a bit more persistence and increased frequency of responses.
Other BCMA-specific CAR trials
Another BCMA CAR, bb2121, consists of a human anti-BCMA scFv, a lentiviral vector, and a CD3/4-1BB costimulatory domain.
In the first-in-human trial of bb2121* (NCT02658929), all 21 patients received cyclophosphamide and fludarabine conditioning first.
There were “very impressive response rates in this study,” Dr Cohen said.
Once patients were dosed at the higher levels of 150 million cells or greater, every patient responded.
“Many responses are still durable,” he pointed out, “some approaching a year.”
LCAR-B38M is structurally different from the other BCMA-specific CARs. It has 2 binding sites for BCMA.
Thirty-five patients enrolled on the trial of LCAR-B38M (NCT03090659), and 19 were evaluable for response.
Patients had a median of 3 or 4 lines of prior therapy. All received cyclophosphamide alone as conditioning.
All 19 evaluable patients responded, and 14 (74%) achieved an sCR.
Dr Cohen noted the differences among the 4 trials: costimulatory domains varied, the Penn study did not require a pre-existing level of BCMA on the MM cells, other studies excluded patients who didn’t meet a certain threshold level of BCMA expression, median lines of prior therapy differed, and conditioning regimens differed as well.
All trials, however, presented data on fewer than 20 patients.
“And so I think it’s a little too early to compare head-to-head between all of these, but they all are really demonstrating promising results so far,” he observed.
Toxicities
“Unfortunately, there’s no free lunch with CAR T cells,” Dr Cohen stated, “and these extraordinary responses do come at the cost of several somewhat unique toxicities that can be serious.”
Toxicities have included:
- Tumor lysis syndrome, which is expected and manageable
- B-cell aplasia, which is not as much of an issue with BCMA as with CD19-directed CAR Ts, since most B cells don’t express BCMA
- Hypogammaglobulinemia, which can be mitigated with IVIG
- CRS, which can be alleviated with tocilizumab
- Neurotoxicity and encephalopathy, which do not occur as frequently as CRS.
“These, I think, are things we still obviously need to learn more about and try to mitigate before this [BCMA-directed CAR therapy] is expanded and brought earlier to patients,” Dr Cohen said.
“The other issues with CAR T cells are logistical. Limited access (the procedure is available at only a few centers); manufacturing can take 2 to 4 weeks, during which time it is difficult to maintain disease control; and the cost is significant.”
“We are really in the early days of CAR cells for myeloma, but, certainly, this appears very bright in terms of its future.” ![]()
* Data from the presentation differ from the abstract.
NEW YORK, NY—The B-cell maturation antigen (BCMA) is emerging as a promising target in multiple myeloma (MM), according to Adam D. Cohen, MD, of the University of Pennsylvania in Philadelphia.
BCMA is highly expressed on MM cells and, with its 2 ligands, is responsible for maintaining normal plasma cell homeostasis.
“And it’s really not expressed on any other normal tissues of the body,” Dr Cohen said.
“Importantly, BCMA is not just sitting on the cell surface as a target but actually promotes myeloma pathogenesis.”
Dr Cohen reviewed the progress being made using BCMA as a target in MM, paying particular attention to chimeric antigen receptor (CAR) T cells. He presented the update at Lymphoma & Myeloma 2017.
NCI BCMA-specific CARs
The first CAR to specifically target BCMA in MM was developed at the National Cancer Institute (NCI). It consisted of a murine single-chain variable fragment (scFv), CD3/CD28 signaling domains, and a gamma-retroviral vector.
Investigators conducted the first-in-human trial of this CAR T-cell therapy in 12 relapsed/refractory MM patients.
All patients received a lymphodepleting conditioning regimen of cyclophosphamide and fludarabine and a single infusion of 1 of 4 doses of the CAR T-cell therapy.
At higher dose levels, the BCMA-CAR produced objective responses “even in these highly refractory patients,” Dr Cohen said. “Some responses lasted 4 to 6 months.”
Patients who had the greatest degree of expansion of CAR T cells were the ones who had the best responses.
The BCMA-CAR is associated with the same toxicities as the CD19-directed CAR T-cell therapies now approved in acute lymphoblastic leukemia and non-Hodgkin lymphoma—cytokine release syndrome (CRS) and neurotoxicity.
The NCI study (NCT02215967) is ongoing.
Penn BCMA-specific CAR
A different BCMA CAR is being investigated at the University of Pennsylvania. It is a fully human CAR that consists of a human scFv, CD3/4-1BB costimulatory domains, and a lentiviral vector.
Investigators designed the first-in-human trial* (NCT02546167) with 3 different cohorts.
Patients in cohort 1 received 5 x 108 CAR T cells without any lymphodepleting chemotherapy.
The remaining patients received cyclophosphamide, followed by 5 x 107 CAR T cells in cohort 2 and 5 x 108 CAR T cells in cohort 3.
Dr Cohen reviewed current data from cohort 1, which included 9 patients. They were a median age of 57 (range, 44-70), and 67% were male. They were heavily pretreated with a median of 9 prior lines of therapy (range, 4-11).
All had high-risk cytogenetics, 67% had deletion 17p or TP53 mutation, and they had a median of 80% bone marrow plasma cells (range, 15%-95%).
“Despite this,” Dr Cohen said, “we were able to generate, successfully, CAR T cells from all patients, although 1 patient did require a second apheresis and manufacturing attempt.”
Four of the 9 patients achieved very good partial responses, and an additional 2 patients had minimal responses.
One patient had a stringent CR (sCR) for close to 2 years without having any intervening therapy.
“[The sCR] shows the potential for this [therapy] to create a durable remission in a patient without any other therapy,” Dr Cohen said. “And this patient still has circulating CAR cells detectable.”
Most of the other patients did not have as durable a response. Responses lasted a median of 3 to 5 months before the patients relapsed.
Dr Cohen noted that the Penn data confirm the NCI experience showing proof of principle.
“You can target BCMA with these cells and get objective responses that can lead to a durable one in a subset of patients,” he added.
Dr Cohen is scheduled to report the preliminary data on the cyclophosphamide cohorts (cohorts 2 and 3) at ASH 2017. Those cohorts, he said, are showing a bit more persistence and increased frequency of responses.
Other BCMA-specific CAR trials
Another BCMA CAR, bb2121, consists of a human anti-BCMA scFv, a lentiviral vector, and a CD3/4-1BB costimulatory domain.
In the first-in-human trial of bb2121* (NCT02658929), all 21 patients received cyclophosphamide and fludarabine conditioning first.
There were “very impressive response rates in this study,” Dr Cohen said.
Once patients were dosed at the higher levels of 150 million cells or greater, every patient responded.
“Many responses are still durable,” he pointed out, “some approaching a year.”
LCAR-B38M is structurally different from the other BCMA-specific CARs. It has 2 binding sites for BCMA.
Thirty-five patients enrolled on the trial of LCAR-B38M (NCT03090659), and 19 were evaluable for response.
Patients had a median of 3 or 4 lines of prior therapy. All received cyclophosphamide alone as conditioning.
All 19 evaluable patients responded, and 14 (74%) achieved an sCR.
Dr Cohen noted the differences among the 4 trials: costimulatory domains varied, the Penn study did not require a pre-existing level of BCMA on the MM cells, other studies excluded patients who didn’t meet a certain threshold level of BCMA expression, median lines of prior therapy differed, and conditioning regimens differed as well.
All trials, however, presented data on fewer than 20 patients.
“And so I think it’s a little too early to compare head-to-head between all of these, but they all are really demonstrating promising results so far,” he observed.
Toxicities
“Unfortunately, there’s no free lunch with CAR T cells,” Dr Cohen stated, “and these extraordinary responses do come at the cost of several somewhat unique toxicities that can be serious.”
Toxicities have included:
- Tumor lysis syndrome, which is expected and manageable
- B-cell aplasia, which is not as much of an issue with BCMA as with CD19-directed CAR Ts, since most B cells don’t express BCMA
- Hypogammaglobulinemia, which can be mitigated with IVIG
- CRS, which can be alleviated with tocilizumab
- Neurotoxicity and encephalopathy, which do not occur as frequently as CRS.
“These, I think, are things we still obviously need to learn more about and try to mitigate before this [BCMA-directed CAR therapy] is expanded and brought earlier to patients,” Dr Cohen said.
“The other issues with CAR T cells are logistical. Limited access (the procedure is available at only a few centers); manufacturing can take 2 to 4 weeks, during which time it is difficult to maintain disease control; and the cost is significant.”
“We are really in the early days of CAR cells for myeloma, but, certainly, this appears very bright in terms of its future.” ![]()
* Data from the presentation differ from the abstract.
NEW YORK, NY—The B-cell maturation antigen (BCMA) is emerging as a promising target in multiple myeloma (MM), according to Adam D. Cohen, MD, of the University of Pennsylvania in Philadelphia.
BCMA is highly expressed on MM cells and, with its 2 ligands, is responsible for maintaining normal plasma cell homeostasis.
“And it’s really not expressed on any other normal tissues of the body,” Dr Cohen said.
“Importantly, BCMA is not just sitting on the cell surface as a target but actually promotes myeloma pathogenesis.”
Dr Cohen reviewed the progress being made using BCMA as a target in MM, paying particular attention to chimeric antigen receptor (CAR) T cells. He presented the update at Lymphoma & Myeloma 2017.
NCI BCMA-specific CARs
The first CAR to specifically target BCMA in MM was developed at the National Cancer Institute (NCI). It consisted of a murine single-chain variable fragment (scFv), CD3/CD28 signaling domains, and a gamma-retroviral vector.
Investigators conducted the first-in-human trial of this CAR T-cell therapy in 12 relapsed/refractory MM patients.
All patients received a lymphodepleting conditioning regimen of cyclophosphamide and fludarabine and a single infusion of 1 of 4 doses of the CAR T-cell therapy.
At higher dose levels, the BCMA-CAR produced objective responses “even in these highly refractory patients,” Dr Cohen said. “Some responses lasted 4 to 6 months.”
Patients who had the greatest degree of expansion of CAR T cells were the ones who had the best responses.
The BCMA-CAR is associated with the same toxicities as the CD19-directed CAR T-cell therapies now approved in acute lymphoblastic leukemia and non-Hodgkin lymphoma—cytokine release syndrome (CRS) and neurotoxicity.
The NCI study (NCT02215967) is ongoing.
Penn BCMA-specific CAR
A different BCMA CAR is being investigated at the University of Pennsylvania. It is a fully human CAR that consists of a human scFv, CD3/4-1BB costimulatory domains, and a lentiviral vector.
Investigators designed the first-in-human trial* (NCT02546167) with 3 different cohorts.
Patients in cohort 1 received 5 x 108 CAR T cells without any lymphodepleting chemotherapy.
The remaining patients received cyclophosphamide, followed by 5 x 107 CAR T cells in cohort 2 and 5 x 108 CAR T cells in cohort 3.
Dr Cohen reviewed current data from cohort 1, which included 9 patients. They were a median age of 57 (range, 44-70), and 67% were male. They were heavily pretreated with a median of 9 prior lines of therapy (range, 4-11).
All had high-risk cytogenetics, 67% had deletion 17p or TP53 mutation, and they had a median of 80% bone marrow plasma cells (range, 15%-95%).
“Despite this,” Dr Cohen said, “we were able to generate, successfully, CAR T cells from all patients, although 1 patient did require a second apheresis and manufacturing attempt.”
Four of the 9 patients achieved very good partial responses, and an additional 2 patients had minimal responses.
One patient had a stringent CR (sCR) for close to 2 years without having any intervening therapy.
“[The sCR] shows the potential for this [therapy] to create a durable remission in a patient without any other therapy,” Dr Cohen said. “And this patient still has circulating CAR cells detectable.”
Most of the other patients did not have as durable a response. Responses lasted a median of 3 to 5 months before the patients relapsed.
Dr Cohen noted that the Penn data confirm the NCI experience showing proof of principle.
“You can target BCMA with these cells and get objective responses that can lead to a durable one in a subset of patients,” he added.
Dr Cohen is scheduled to report the preliminary data on the cyclophosphamide cohorts (cohorts 2 and 3) at ASH 2017. Those cohorts, he said, are showing a bit more persistence and increased frequency of responses.
Other BCMA-specific CAR trials
Another BCMA CAR, bb2121, consists of a human anti-BCMA scFv, a lentiviral vector, and a CD3/4-1BB costimulatory domain.
In the first-in-human trial of bb2121* (NCT02658929), all 21 patients received cyclophosphamide and fludarabine conditioning first.
There were “very impressive response rates in this study,” Dr Cohen said.
Once patients were dosed at the higher levels of 150 million cells or greater, every patient responded.
“Many responses are still durable,” he pointed out, “some approaching a year.”
LCAR-B38M is structurally different from the other BCMA-specific CARs. It has 2 binding sites for BCMA.
Thirty-five patients enrolled on the trial of LCAR-B38M (NCT03090659), and 19 were evaluable for response.
Patients had a median of 3 or 4 lines of prior therapy. All received cyclophosphamide alone as conditioning.
All 19 evaluable patients responded, and 14 (74%) achieved an sCR.
Dr Cohen noted the differences among the 4 trials: costimulatory domains varied, the Penn study did not require a pre-existing level of BCMA on the MM cells, other studies excluded patients who didn’t meet a certain threshold level of BCMA expression, median lines of prior therapy differed, and conditioning regimens differed as well.
All trials, however, presented data on fewer than 20 patients.
“And so I think it’s a little too early to compare head-to-head between all of these, but they all are really demonstrating promising results so far,” he observed.
Toxicities
“Unfortunately, there’s no free lunch with CAR T cells,” Dr Cohen stated, “and these extraordinary responses do come at the cost of several somewhat unique toxicities that can be serious.”
Toxicities have included:
- Tumor lysis syndrome, which is expected and manageable
- B-cell aplasia, which is not as much of an issue with BCMA as with CD19-directed CAR Ts, since most B cells don’t express BCMA
- Hypogammaglobulinemia, which can be mitigated with IVIG
- CRS, which can be alleviated with tocilizumab
- Neurotoxicity and encephalopathy, which do not occur as frequently as CRS.
“These, I think, are things we still obviously need to learn more about and try to mitigate before this [BCMA-directed CAR therapy] is expanded and brought earlier to patients,” Dr Cohen said.
“The other issues with CAR T cells are logistical. Limited access (the procedure is available at only a few centers); manufacturing can take 2 to 4 weeks, during which time it is difficult to maintain disease control; and the cost is significant.”
“We are really in the early days of CAR cells for myeloma, but, certainly, this appears very bright in terms of its future.” ![]()
* Data from the presentation differ from the abstract.
Study reveals misperceptions among AML patients

SAN DIEGO—A study of acute myeloid leukemia (AML) patients has revealed misperceptions about treatment risks and the likelihood of cure.
Investigators surveyed 100 AML patients receiving intensive and non-intensive chemotherapy, as well as the patients’ oncologists.
The results showed that patients tended to overestimate both the risk of dying due to treatment and the likelihood of cure.
These findings were presented at the 2017 Palliative and Supportive Care in Oncology Symposium (abstract 43).
“Patients with AML face very challenging treatment decisions that are often placed upon them within days after being diagnosed,” said study investigator Areej El-Jawahri, MD, of Massachusetts General Hospital in Boston.
“Because they face a grave decision, they need to understand what the risks of treatment are versus the possibility of a cure.”
For this study, Dr El-Jawahri and her colleagues enrolled 50 patients who were receiving intensive care for AML (which usually meant hospitalization for 4 to 6 weeks) and 50 patients who were receiving non-intensive care (often given as outpatient treatment).
The patients’ median age was 71 (range, 60-100), and 92% were white. Six percent of patients had low-risk disease, 48% had intermediate-risk, and 46% had high-risk disease.
Within 3 days of starting treatment, both the patients and their physicians were given a questionnaire to assess how they perceived the likelihood of the patient dying from treatment.
One month later, patients and physicians completed a follow-up questionnaire to assess perceptions of patient prognosis. Within that time frame, most patients received laboratory results that more definitively established the type and stage of cancer.
At 24 weeks, the investigators asked patients if they had discussed their end-of-life wishes with their oncologists.
Results
Initially, most of the patient population (91.3%) thought it was “somewhat” or “extremely” likely they would die from their treatment. However, only 22% of treating oncologists said the same.
One month later, a majority of patients in both treatment groups thought it was “somewhat” or “extremely” likely they would be cured of their AML.
Specifically, 82.1% of patients receiving non-intensive chemotherapy said it was “somewhat” or “extremely” likely they would be cured, while 10% of their oncologists said the same.
Meanwhile, 97.6% of patients receiving intensive chemotherapy said it was “somewhat” or “extremely” likely they would be cured, and 42% of their oncologists said the same.
Overall, 77.8% of patients said they had not discussed their end-of-life wishes with their oncologists at 24 weeks.
“There were several very important factors we were not able to capture in our study, including what was actually discussed between patients and their oncologists and whether patients simply misunderstood or misheard the information conveyed to them,” Dr El-Jawahri said.
“Perhaps most importantly, we did not audio-record the discussions between the patients and their physicians, which could provide additional details regarding barriers to accurate prognostic understanding in these conversations.”
Related research and next steps
Prior to this study, Dr El-Jawahri and her colleagues had looked at similar perceptions in patients with solid tumor malignancies as well as in patients with hematologic malignancies who were receiving hematopoietic stem cell transplants.
The gaps in perception of treatment risk and cure for patients compared to their physicians were not as large in those cases as in the AML patients in this study. The investigators attribute this to higher levels of distress seen in AML patients due to the urgency of their treatment decisions.
Dr El-Jawahri and her colleagues have found that early consideration of palliative care in a treatment plan for patients with solid tumors improves patients’ understanding of the prognosis. The team hopes to implement a similar study in patients with AML.
“Clearly, there are important communication gaps between oncologists and their patients,” Dr El-Jawahri said. “We need to find ways to help physicians do a better job of communicating with their patients, especially in diseases like AML where stress levels are remarkably high.” ![]()
SAN DIEGO—A study of acute myeloid leukemia (AML) patients has revealed misperceptions about treatment risks and the likelihood of cure.
Investigators surveyed 100 AML patients receiving intensive and non-intensive chemotherapy, as well as the patients’ oncologists.
The results showed that patients tended to overestimate both the risk of dying due to treatment and the likelihood of cure.
These findings were presented at the 2017 Palliative and Supportive Care in Oncology Symposium (abstract 43).
“Patients with AML face very challenging treatment decisions that are often placed upon them within days after being diagnosed,” said study investigator Areej El-Jawahri, MD, of Massachusetts General Hospital in Boston.
“Because they face a grave decision, they need to understand what the risks of treatment are versus the possibility of a cure.”
For this study, Dr El-Jawahri and her colleagues enrolled 50 patients who were receiving intensive care for AML (which usually meant hospitalization for 4 to 6 weeks) and 50 patients who were receiving non-intensive care (often given as outpatient treatment).
The patients’ median age was 71 (range, 60-100), and 92% were white. Six percent of patients had low-risk disease, 48% had intermediate-risk, and 46% had high-risk disease.
Within 3 days of starting treatment, both the patients and their physicians were given a questionnaire to assess how they perceived the likelihood of the patient dying from treatment.
One month later, patients and physicians completed a follow-up questionnaire to assess perceptions of patient prognosis. Within that time frame, most patients received laboratory results that more definitively established the type and stage of cancer.
At 24 weeks, the investigators asked patients if they had discussed their end-of-life wishes with their oncologists.
Results
Initially, most of the patient population (91.3%) thought it was “somewhat” or “extremely” likely they would die from their treatment. However, only 22% of treating oncologists said the same.
One month later, a majority of patients in both treatment groups thought it was “somewhat” or “extremely” likely they would be cured of their AML.
Specifically, 82.1% of patients receiving non-intensive chemotherapy said it was “somewhat” or “extremely” likely they would be cured, while 10% of their oncologists said the same.
Meanwhile, 97.6% of patients receiving intensive chemotherapy said it was “somewhat” or “extremely” likely they would be cured, and 42% of their oncologists said the same.
Overall, 77.8% of patients said they had not discussed their end-of-life wishes with their oncologists at 24 weeks.
“There were several very important factors we were not able to capture in our study, including what was actually discussed between patients and their oncologists and whether patients simply misunderstood or misheard the information conveyed to them,” Dr El-Jawahri said.
“Perhaps most importantly, we did not audio-record the discussions between the patients and their physicians, which could provide additional details regarding barriers to accurate prognostic understanding in these conversations.”
Related research and next steps
Prior to this study, Dr El-Jawahri and her colleagues had looked at similar perceptions in patients with solid tumor malignancies as well as in patients with hematologic malignancies who were receiving hematopoietic stem cell transplants.
The gaps in perception of treatment risk and cure for patients compared to their physicians were not as large in those cases as in the AML patients in this study. The investigators attribute this to higher levels of distress seen in AML patients due to the urgency of their treatment decisions.
Dr El-Jawahri and her colleagues have found that early consideration of palliative care in a treatment plan for patients with solid tumors improves patients’ understanding of the prognosis. The team hopes to implement a similar study in patients with AML.
“Clearly, there are important communication gaps between oncologists and their patients,” Dr El-Jawahri said. “We need to find ways to help physicians do a better job of communicating with their patients, especially in diseases like AML where stress levels are remarkably high.” ![]()
SAN DIEGO—A study of acute myeloid leukemia (AML) patients has revealed misperceptions about treatment risks and the likelihood of cure.
Investigators surveyed 100 AML patients receiving intensive and non-intensive chemotherapy, as well as the patients’ oncologists.
The results showed that patients tended to overestimate both the risk of dying due to treatment and the likelihood of cure.
These findings were presented at the 2017 Palliative and Supportive Care in Oncology Symposium (abstract 43).
“Patients with AML face very challenging treatment decisions that are often placed upon them within days after being diagnosed,” said study investigator Areej El-Jawahri, MD, of Massachusetts General Hospital in Boston.
“Because they face a grave decision, they need to understand what the risks of treatment are versus the possibility of a cure.”
For this study, Dr El-Jawahri and her colleagues enrolled 50 patients who were receiving intensive care for AML (which usually meant hospitalization for 4 to 6 weeks) and 50 patients who were receiving non-intensive care (often given as outpatient treatment).
The patients’ median age was 71 (range, 60-100), and 92% were white. Six percent of patients had low-risk disease, 48% had intermediate-risk, and 46% had high-risk disease.
Within 3 days of starting treatment, both the patients and their physicians were given a questionnaire to assess how they perceived the likelihood of the patient dying from treatment.
One month later, patients and physicians completed a follow-up questionnaire to assess perceptions of patient prognosis. Within that time frame, most patients received laboratory results that more definitively established the type and stage of cancer.
At 24 weeks, the investigators asked patients if they had discussed their end-of-life wishes with their oncologists.
Results
Initially, most of the patient population (91.3%) thought it was “somewhat” or “extremely” likely they would die from their treatment. However, only 22% of treating oncologists said the same.
One month later, a majority of patients in both treatment groups thought it was “somewhat” or “extremely” likely they would be cured of their AML.
Specifically, 82.1% of patients receiving non-intensive chemotherapy said it was “somewhat” or “extremely” likely they would be cured, while 10% of their oncologists said the same.
Meanwhile, 97.6% of patients receiving intensive chemotherapy said it was “somewhat” or “extremely” likely they would be cured, and 42% of their oncologists said the same.
Overall, 77.8% of patients said they had not discussed their end-of-life wishes with their oncologists at 24 weeks.
“There were several very important factors we were not able to capture in our study, including what was actually discussed between patients and their oncologists and whether patients simply misunderstood or misheard the information conveyed to them,” Dr El-Jawahri said.
“Perhaps most importantly, we did not audio-record the discussions between the patients and their physicians, which could provide additional details regarding barriers to accurate prognostic understanding in these conversations.”
Related research and next steps
Prior to this study, Dr El-Jawahri and her colleagues had looked at similar perceptions in patients with solid tumor malignancies as well as in patients with hematologic malignancies who were receiving hematopoietic stem cell transplants.
The gaps in perception of treatment risk and cure for patients compared to their physicians were not as large in those cases as in the AML patients in this study. The investigators attribute this to higher levels of distress seen in AML patients due to the urgency of their treatment decisions.
Dr El-Jawahri and her colleagues have found that early consideration of palliative care in a treatment plan for patients with solid tumors improves patients’ understanding of the prognosis. The team hopes to implement a similar study in patients with AML.
“Clearly, there are important communication gaps between oncologists and their patients,” Dr El-Jawahri said. “We need to find ways to help physicians do a better job of communicating with their patients, especially in diseases like AML where stress levels are remarkably high.” ![]()
Drug receives fast track designation for lower-risk MDS

The US Food and Drug Administration (FDA) has granted fast track designation to the telomerase inhibitor imetelstat.
The designation is for imetelstat as a potential treatment for adults who have transfusion-dependent anemia due to low or intermediate-1 risk myelodysplastic syndromes (MDS), do not have 5q deletion, and are refractory or resistant to treatment with an erythropoiesis-stimulating agent (ESA).
Imetelstat was initially developed by Geron Corporation and exclusively licensed to Janssen Biotech, Inc.
Janssen sponsored the application for fast track designation using preliminary data from IMerge, a trial in which researchers are studying transfusion-dependent patients with low- or intermediate-1 risk MDS who have relapsed after or are refractory to treatment with an ESA.
Part 1 of IMerge is a phase 2, single-arm trial. Part 2 is a phase 3, randomized, placebo-controlled trial.
Thirty-two patients have been enrolled in part 1 of IMerge. However, this part of the trial is expanding to enroll approximately 20 additional patients who do not have 5q deletion and are naïve to treatment with a hypomethylating agent and lenalidomide.
The expansion is based on results observed in a subset of the original 32 patients who had not received prior treatment with a hypomethylating agent or lenalidomide and did not have 5q deletion.
As of May 2017, this 13-patient subset showed an increased durability and rate of red blood cell transfusion-independence compared to the overall trial population.
Results in these patients and the rest of the original 32 patients are expected to be presented at an upcoming medical conference.
About fast track designation
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the new drug application or biologics license application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA. ![]()
The US Food and Drug Administration (FDA) has granted fast track designation to the telomerase inhibitor imetelstat.
The designation is for imetelstat as a potential treatment for adults who have transfusion-dependent anemia due to low or intermediate-1 risk myelodysplastic syndromes (MDS), do not have 5q deletion, and are refractory or resistant to treatment with an erythropoiesis-stimulating agent (ESA).
Imetelstat was initially developed by Geron Corporation and exclusively licensed to Janssen Biotech, Inc.
Janssen sponsored the application for fast track designation using preliminary data from IMerge, a trial in which researchers are studying transfusion-dependent patients with low- or intermediate-1 risk MDS who have relapsed after or are refractory to treatment with an ESA.
Part 1 of IMerge is a phase 2, single-arm trial. Part 2 is a phase 3, randomized, placebo-controlled trial.
Thirty-two patients have been enrolled in part 1 of IMerge. However, this part of the trial is expanding to enroll approximately 20 additional patients who do not have 5q deletion and are naïve to treatment with a hypomethylating agent and lenalidomide.
The expansion is based on results observed in a subset of the original 32 patients who had not received prior treatment with a hypomethylating agent or lenalidomide and did not have 5q deletion.
As of May 2017, this 13-patient subset showed an increased durability and rate of red blood cell transfusion-independence compared to the overall trial population.
Results in these patients and the rest of the original 32 patients are expected to be presented at an upcoming medical conference.
About fast track designation
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the new drug application or biologics license application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA. ![]()
The US Food and Drug Administration (FDA) has granted fast track designation to the telomerase inhibitor imetelstat.
The designation is for imetelstat as a potential treatment for adults who have transfusion-dependent anemia due to low or intermediate-1 risk myelodysplastic syndromes (MDS), do not have 5q deletion, and are refractory or resistant to treatment with an erythropoiesis-stimulating agent (ESA).
Imetelstat was initially developed by Geron Corporation and exclusively licensed to Janssen Biotech, Inc.
Janssen sponsored the application for fast track designation using preliminary data from IMerge, a trial in which researchers are studying transfusion-dependent patients with low- or intermediate-1 risk MDS who have relapsed after or are refractory to treatment with an ESA.
Part 1 of IMerge is a phase 2, single-arm trial. Part 2 is a phase 3, randomized, placebo-controlled trial.
Thirty-two patients have been enrolled in part 1 of IMerge. However, this part of the trial is expanding to enroll approximately 20 additional patients who do not have 5q deletion and are naïve to treatment with a hypomethylating agent and lenalidomide.
The expansion is based on results observed in a subset of the original 32 patients who had not received prior treatment with a hypomethylating agent or lenalidomide and did not have 5q deletion.
As of May 2017, this 13-patient subset showed an increased durability and rate of red blood cell transfusion-independence compared to the overall trial population.
Results in these patients and the rest of the original 32 patients are expected to be presented at an upcoming medical conference.
About fast track designation
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the new drug application or biologics license application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA. ![]()
FDA panels support two NDAs for buprenorphine subcutaneous injections
SILVER SPRING, MD. – Two Food and Drug Administration advisory panels have recommended approval of two new drug applications (NDA) for buprenorphine subcutaneous injections for the treatment of opioid dependence.
On Nov. 1, panelists recommended approval of some of the doses proposed in the NDA submitted by Braeburn Pharmaceuticals at the joint meeting of the Psychopharmacologic Drugs Advisory and the Drug Safety and Risk Management committees. The formulation, currently known as CAM2038, is intended to be used as part of a treatment plan that can include counseling and psychosocial support. The subcutaneous depot is available weekly, in 8-, 16-, 24-, and 32-mg injections, and monthly, in 64-, 96-, 128-, and 160-mg injections.
On the previous day, Oct. 31, the panelists voted 18-1 with no abstentions to recommend an NDA submitted by Indivior. This formulation is known as RBP-6000.
Both formulations must be administered by a health care provider using a prefilled syringe with a predetermined dosage. The injection forms a biodegradable subcutaneous depot that, as it degrades, releases buprenorphine at a steady and controlled pace over the course of treatment – increasing the success of treatment for opioid use disorder.
Braeburn’s NDA was based on results of a double-blind, randomized, within-subject, inpatient laboratory study of 47 patients over 14 days. Patients were randomized into two groups: 22 patients in the 24-mg group and 25 patients in the 32-mg group. Patients were administered an initial dose on day 0 and a follow-up dose on day 7. The results of the study found a complete blockade of opioids after the first injection that was sustained over the 1-week interdosing interval.
The committees said that of most of the doses should be approved, but a majority of committee members were uncomfortable with the higher doses.
Voting on Indivior’s NDA was based, in part, on the results of a randomized, double-blind, placebo-controlled, multicenter phase 3 study. The study lasted 24 weeks and randomly assigned 504 patients into one of three groups based on monthly dosing regimen of buprenorphine: 300 mg/300 mg, 300 mg/100 mg, and placebo. After randomization, the 300 mg/300 mg group had 201 patients, the 300 mg/100 mg group had 203 patients, and the placebo group had 100 patients. The study found that the primary and secondary endpoints were met, and significantly higher percentage of abstinence with subcutaneous buprenorphine were observed. Patients in both the 300 mg/300 mg and 300 mg/100 mg groups had very similar distributions of percentage of weeks patients abstained from opioid use with more than 20% of patients achieving 80%-100% abstinence from opioids during the course of the study, a significant improvement over the placebo group.
The panels’ recommendations come against the backdrop of the opioid epidemic in the United States, which President Trump has deemed a public health emergency. Many of the panel members and speakers at both meetings expressed support for the NDAs in that context and emphasized that, unlike sublingual administration of buprenorphine, these treatments do not require daily intervention. In addition, sublingual tablets are easier to abuse or more likely to lead to overdose because the patient must self-administer the medication. Expanding the toolkit of physicians who treat opioid use disorder might help stem the tide of the epidemic, some speakers said.
Usually, the FDA follows its advisory panels’ recommendations, which are not binding.
SILVER SPRING, MD. – Two Food and Drug Administration advisory panels have recommended approval of two new drug applications (NDA) for buprenorphine subcutaneous injections for the treatment of opioid dependence.
On Nov. 1, panelists recommended approval of some of the doses proposed in the NDA submitted by Braeburn Pharmaceuticals at the joint meeting of the Psychopharmacologic Drugs Advisory and the Drug Safety and Risk Management committees. The formulation, currently known as CAM2038, is intended to be used as part of a treatment plan that can include counseling and psychosocial support. The subcutaneous depot is available weekly, in 8-, 16-, 24-, and 32-mg injections, and monthly, in 64-, 96-, 128-, and 160-mg injections.
On the previous day, Oct. 31, the panelists voted 18-1 with no abstentions to recommend an NDA submitted by Indivior. This formulation is known as RBP-6000.
Both formulations must be administered by a health care provider using a prefilled syringe with a predetermined dosage. The injection forms a biodegradable subcutaneous depot that, as it degrades, releases buprenorphine at a steady and controlled pace over the course of treatment – increasing the success of treatment for opioid use disorder.
Braeburn’s NDA was based on results of a double-blind, randomized, within-subject, inpatient laboratory study of 47 patients over 14 days. Patients were randomized into two groups: 22 patients in the 24-mg group and 25 patients in the 32-mg group. Patients were administered an initial dose on day 0 and a follow-up dose on day 7. The results of the study found a complete blockade of opioids after the first injection that was sustained over the 1-week interdosing interval.
The committees said that of most of the doses should be approved, but a majority of committee members were uncomfortable with the higher doses.
Voting on Indivior’s NDA was based, in part, on the results of a randomized, double-blind, placebo-controlled, multicenter phase 3 study. The study lasted 24 weeks and randomly assigned 504 patients into one of three groups based on monthly dosing regimen of buprenorphine: 300 mg/300 mg, 300 mg/100 mg, and placebo. After randomization, the 300 mg/300 mg group had 201 patients, the 300 mg/100 mg group had 203 patients, and the placebo group had 100 patients. The study found that the primary and secondary endpoints were met, and significantly higher percentage of abstinence with subcutaneous buprenorphine were observed. Patients in both the 300 mg/300 mg and 300 mg/100 mg groups had very similar distributions of percentage of weeks patients abstained from opioid use with more than 20% of patients achieving 80%-100% abstinence from opioids during the course of the study, a significant improvement over the placebo group.
The panels’ recommendations come against the backdrop of the opioid epidemic in the United States, which President Trump has deemed a public health emergency. Many of the panel members and speakers at both meetings expressed support for the NDAs in that context and emphasized that, unlike sublingual administration of buprenorphine, these treatments do not require daily intervention. In addition, sublingual tablets are easier to abuse or more likely to lead to overdose because the patient must self-administer the medication. Expanding the toolkit of physicians who treat opioid use disorder might help stem the tide of the epidemic, some speakers said.
Usually, the FDA follows its advisory panels’ recommendations, which are not binding.
SILVER SPRING, MD. – Two Food and Drug Administration advisory panels have recommended approval of two new drug applications (NDA) for buprenorphine subcutaneous injections for the treatment of opioid dependence.
On Nov. 1, panelists recommended approval of some of the doses proposed in the NDA submitted by Braeburn Pharmaceuticals at the joint meeting of the Psychopharmacologic Drugs Advisory and the Drug Safety and Risk Management committees. The formulation, currently known as CAM2038, is intended to be used as part of a treatment plan that can include counseling and psychosocial support. The subcutaneous depot is available weekly, in 8-, 16-, 24-, and 32-mg injections, and monthly, in 64-, 96-, 128-, and 160-mg injections.
On the previous day, Oct. 31, the panelists voted 18-1 with no abstentions to recommend an NDA submitted by Indivior. This formulation is known as RBP-6000.
Both formulations must be administered by a health care provider using a prefilled syringe with a predetermined dosage. The injection forms a biodegradable subcutaneous depot that, as it degrades, releases buprenorphine at a steady and controlled pace over the course of treatment – increasing the success of treatment for opioid use disorder.
Braeburn’s NDA was based on results of a double-blind, randomized, within-subject, inpatient laboratory study of 47 patients over 14 days. Patients were randomized into two groups: 22 patients in the 24-mg group and 25 patients in the 32-mg group. Patients were administered an initial dose on day 0 and a follow-up dose on day 7. The results of the study found a complete blockade of opioids after the first injection that was sustained over the 1-week interdosing interval.
The committees said that of most of the doses should be approved, but a majority of committee members were uncomfortable with the higher doses.
Voting on Indivior’s NDA was based, in part, on the results of a randomized, double-blind, placebo-controlled, multicenter phase 3 study. The study lasted 24 weeks and randomly assigned 504 patients into one of three groups based on monthly dosing regimen of buprenorphine: 300 mg/300 mg, 300 mg/100 mg, and placebo. After randomization, the 300 mg/300 mg group had 201 patients, the 300 mg/100 mg group had 203 patients, and the placebo group had 100 patients. The study found that the primary and secondary endpoints were met, and significantly higher percentage of abstinence with subcutaneous buprenorphine were observed. Patients in both the 300 mg/300 mg and 300 mg/100 mg groups had very similar distributions of percentage of weeks patients abstained from opioid use with more than 20% of patients achieving 80%-100% abstinence from opioids during the course of the study, a significant improvement over the placebo group.
The panels’ recommendations come against the backdrop of the opioid epidemic in the United States, which President Trump has deemed a public health emergency. Many of the panel members and speakers at both meetings expressed support for the NDAs in that context and emphasized that, unlike sublingual administration of buprenorphine, these treatments do not require daily intervention. In addition, sublingual tablets are easier to abuse or more likely to lead to overdose because the patient must self-administer the medication. Expanding the toolkit of physicians who treat opioid use disorder might help stem the tide of the epidemic, some speakers said.
Usually, the FDA follows its advisory panels’ recommendations, which are not binding.
U.S. judge orders Philips to cease AED manufacturing
A U.S. District Judge has ordered Philips North America, as well as two Philips officers, to cease manufacturing and distribution of automatic external defibrillators (AEDs) until they can comply with federal regulations in a consent decree, according to a statement from the Food and Drug Administration.
In a complaint filed with the decree, Philips North America in Andover, Mass., which operates as Philips Medical Systems and Philips Healthcare, sold compromised automatic external defibrillators and Q-CPR Meters in violation of current Federal Food, Drug and Cosmetic (FD&C) Act good manufacturing practice requirements. The injunction also applies to Carla Kriwet and Ojas Buch of the Patient Care and Monitoring Solutions business group, according to the statement.
“AEDs are life-saving tools and are designed to be used by the general public or professionals in an emergency. People rely on these devices to work when needed. By not adequately addressing corrective and preventative actions with their AEDs in a timely manner, Philips distributed adulterated products that put people at risk,” Melinda Plaisier, associate commissioner for regulatory affairs at the FDA said in the press release.
In an Oct. 11 statement, Carla Kriwet, head of Connected Care & Health Informatics at Royal Philips, said “We are committed to delivering high-quality, innovative products and solutions, and we take this matter very seriously. We are fully prepared to fulfill the terms of the decree, and we hope to resume the suspended defibrillator production in the course of 2018.”
Ms. Kriwet added that in the past several years Philips has made significant investments in its quality procedures and leadership.
The company recommends that Philips defibrillators currently in use by customers should remain in use, and should not be taken out of service as Philips has no reason to believe they pose a risk to patients.
A U.S. District Judge has ordered Philips North America, as well as two Philips officers, to cease manufacturing and distribution of automatic external defibrillators (AEDs) until they can comply with federal regulations in a consent decree, according to a statement from the Food and Drug Administration.
In a complaint filed with the decree, Philips North America in Andover, Mass., which operates as Philips Medical Systems and Philips Healthcare, sold compromised automatic external defibrillators and Q-CPR Meters in violation of current Federal Food, Drug and Cosmetic (FD&C) Act good manufacturing practice requirements. The injunction also applies to Carla Kriwet and Ojas Buch of the Patient Care and Monitoring Solutions business group, according to the statement.
“AEDs are life-saving tools and are designed to be used by the general public or professionals in an emergency. People rely on these devices to work when needed. By not adequately addressing corrective and preventative actions with their AEDs in a timely manner, Philips distributed adulterated products that put people at risk,” Melinda Plaisier, associate commissioner for regulatory affairs at the FDA said in the press release.
In an Oct. 11 statement, Carla Kriwet, head of Connected Care & Health Informatics at Royal Philips, said “We are committed to delivering high-quality, innovative products and solutions, and we take this matter very seriously. We are fully prepared to fulfill the terms of the decree, and we hope to resume the suspended defibrillator production in the course of 2018.”
Ms. Kriwet added that in the past several years Philips has made significant investments in its quality procedures and leadership.
The company recommends that Philips defibrillators currently in use by customers should remain in use, and should not be taken out of service as Philips has no reason to believe they pose a risk to patients.
A U.S. District Judge has ordered Philips North America, as well as two Philips officers, to cease manufacturing and distribution of automatic external defibrillators (AEDs) until they can comply with federal regulations in a consent decree, according to a statement from the Food and Drug Administration.
In a complaint filed with the decree, Philips North America in Andover, Mass., which operates as Philips Medical Systems and Philips Healthcare, sold compromised automatic external defibrillators and Q-CPR Meters in violation of current Federal Food, Drug and Cosmetic (FD&C) Act good manufacturing practice requirements. The injunction also applies to Carla Kriwet and Ojas Buch of the Patient Care and Monitoring Solutions business group, according to the statement.
“AEDs are life-saving tools and are designed to be used by the general public or professionals in an emergency. People rely on these devices to work when needed. By not adequately addressing corrective and preventative actions with their AEDs in a timely manner, Philips distributed adulterated products that put people at risk,” Melinda Plaisier, associate commissioner for regulatory affairs at the FDA said in the press release.
In an Oct. 11 statement, Carla Kriwet, head of Connected Care & Health Informatics at Royal Philips, said “We are committed to delivering high-quality, innovative products and solutions, and we take this matter very seriously. We are fully prepared to fulfill the terms of the decree, and we hope to resume the suspended defibrillator production in the course of 2018.”
Ms. Kriwet added that in the past several years Philips has made significant investments in its quality procedures and leadership.
The company recommends that Philips defibrillators currently in use by customers should remain in use, and should not be taken out of service as Philips has no reason to believe they pose a risk to patients.
The race is on for a Zika vaccine
WASHINGTON – A DNA vaccine developed at the National Institute of Allergy and Infectious Diseases Vaccine Research Center – one of five National Institutes of Health Zika vaccine candidates – has entered phase 2 testing in a trial underway in Brazil, Peru, Ecuador, Mexico, and Texas.
“The DNA vaccine is a simple 21st century way of developing vaccines that I think will become one of the major [methods of the future] for emerging infections, as opposed to growing a virus and inactivating or attenuating it,” Anthony S. Fauci, MD, said at the biennial meeting of the Diabetes in Pregnancy Study Group of North America. With Zika “this is the vaccine that is ahead of all the others.”
Will it be possible to test efficacy, given the declining prevalence of Zika across the Americas, and will it be too late to prevent more disease? Dr. Fauci, director of NIAID, said that’s a concern, and that an accelerated approval based on a bridging of animal efficacy data with human safety and immunogenicity data might be possible.
The Southern hemisphere is “entering their summer, so it’s conceivable there will be an uptick in Zika. … We’ll just need to wait and see,” he said.
Sexual transmission
The Zika virus is part of a “long line of arboviruses that have threatened us in the Americas,” but infection with the organism is “the first – and may be the only – arthropod-borne or mosquito-borne infection that is also sexually transmitted,” Dr. Fauci said.
Sexual contact as an important mode of viral transmission “has been documented very clearly through a number of studies in which individuals clearly had no exposure to mosquitoes but were in fact a sexual partner of someone who got infected,” he said. And recent research suggests that the “female reproductive tract is a preferentially permissive site for Zika replication, which adds to the concern about sexual transmission.”
He cited a study published in July 2017 in PLOS Pathogens in which the Zika virus was found to preferentially replicate in the reproductive tract of female rhesus macaques who received vaginal inoculations of the virus.
Zika virus was “detected in the reproductive tract before it was detected in plasma, and replication levels in the reproductive tract did not reflect viral levels in other parts of the body,” according to the author summary. The kinetics of virus replication and dissemination after intravaginal inoculation were markedly different from what was previously seen in macaques infected with the Zika virus by subcutaneous infection, the report noted (PLOS Pathogens 13[7]:e1006537).
Dr. Fauci briefly described this and several other studies and findings that he said exemplify growing knowledge of the infection. He pointed to a prospective observational study that documents episodes of oligospermia in 15 men who presented with infection in 2016 in the French Caribbean (Lancet Infect Dis. 2017;17:1200-08).
Sperm counts fell in some of the study participants by about 50% between days 7 and 60 post infection, and the counts “recovered somewhat” by day 120. “We’re still following patients in prospective studies to determine if there’s a long-term effect in men,” he said.
In the meantime, he said, research in mice has shown that “without a doubt, Zika infection damages the testes,” Dr. Fauci said, noting that the mouse model is proving to be a good model for studying Zika’s effects. “They become oligospermic and have testicular atrophy.”
Maternal-fetal transmission
Regarding maternal-fetal transmission, there’s evidence that placental trophoblasts “are exquisitely permissive for Zika virus replication,” he said.
In another recent study, primary human placental trophoblasts from nonexposed donors were found to be infected by the Zika virus ex-vivo and permissive for viral RNA replication, compared with dengue virus, a fellow flavivirus (Sci Rep. 2017;7:41389. doi: 10.1038/srep41389).
However, Yoel Sadovsky, MD, who also presented at the meeting, explained that his lab’s ex-vivo studies show that primary human trophoblasts have inherent resistance to a number of viruses – and that trophoblasts are refractory to direct infection with the Zika virus. “We don’t think the trophoblasts are very permissive at all,” he said.
Moreover, trophoblasts appear to confer their antiviral effects to other nontrophoblast cells by releasing a particular type of interferon – type III interferon IFN1 – and by delivering certain micro-RNAs (C19MC miRNAs) that are packaged within trophoblast-derived nanovesicles called exosomes, said Dr. Sadovsky, scientific director of the Magee-Womens Research Institute and professor of ob.gyn., reproductive sciences, microbiology, and molecular genetics at the University of Pittsburgh.
WASHINGTON – A DNA vaccine developed at the National Institute of Allergy and Infectious Diseases Vaccine Research Center – one of five National Institutes of Health Zika vaccine candidates – has entered phase 2 testing in a trial underway in Brazil, Peru, Ecuador, Mexico, and Texas.
“The DNA vaccine is a simple 21st century way of developing vaccines that I think will become one of the major [methods of the future] for emerging infections, as opposed to growing a virus and inactivating or attenuating it,” Anthony S. Fauci, MD, said at the biennial meeting of the Diabetes in Pregnancy Study Group of North America. With Zika “this is the vaccine that is ahead of all the others.”
Will it be possible to test efficacy, given the declining prevalence of Zika across the Americas, and will it be too late to prevent more disease? Dr. Fauci, director of NIAID, said that’s a concern, and that an accelerated approval based on a bridging of animal efficacy data with human safety and immunogenicity data might be possible.
The Southern hemisphere is “entering their summer, so it’s conceivable there will be an uptick in Zika. … We’ll just need to wait and see,” he said.
Sexual transmission
The Zika virus is part of a “long line of arboviruses that have threatened us in the Americas,” but infection with the organism is “the first – and may be the only – arthropod-borne or mosquito-borne infection that is also sexually transmitted,” Dr. Fauci said.
Sexual contact as an important mode of viral transmission “has been documented very clearly through a number of studies in which individuals clearly had no exposure to mosquitoes but were in fact a sexual partner of someone who got infected,” he said. And recent research suggests that the “female reproductive tract is a preferentially permissive site for Zika replication, which adds to the concern about sexual transmission.”
He cited a study published in July 2017 in PLOS Pathogens in which the Zika virus was found to preferentially replicate in the reproductive tract of female rhesus macaques who received vaginal inoculations of the virus.
Zika virus was “detected in the reproductive tract before it was detected in plasma, and replication levels in the reproductive tract did not reflect viral levels in other parts of the body,” according to the author summary. The kinetics of virus replication and dissemination after intravaginal inoculation were markedly different from what was previously seen in macaques infected with the Zika virus by subcutaneous infection, the report noted (PLOS Pathogens 13[7]:e1006537).
Dr. Fauci briefly described this and several other studies and findings that he said exemplify growing knowledge of the infection. He pointed to a prospective observational study that documents episodes of oligospermia in 15 men who presented with infection in 2016 in the French Caribbean (Lancet Infect Dis. 2017;17:1200-08).
Sperm counts fell in some of the study participants by about 50% between days 7 and 60 post infection, and the counts “recovered somewhat” by day 120. “We’re still following patients in prospective studies to determine if there’s a long-term effect in men,” he said.
In the meantime, he said, research in mice has shown that “without a doubt, Zika infection damages the testes,” Dr. Fauci said, noting that the mouse model is proving to be a good model for studying Zika’s effects. “They become oligospermic and have testicular atrophy.”
Maternal-fetal transmission
Regarding maternal-fetal transmission, there’s evidence that placental trophoblasts “are exquisitely permissive for Zika virus replication,” he said.
In another recent study, primary human placental trophoblasts from nonexposed donors were found to be infected by the Zika virus ex-vivo and permissive for viral RNA replication, compared with dengue virus, a fellow flavivirus (Sci Rep. 2017;7:41389. doi: 10.1038/srep41389).
However, Yoel Sadovsky, MD, who also presented at the meeting, explained that his lab’s ex-vivo studies show that primary human trophoblasts have inherent resistance to a number of viruses – and that trophoblasts are refractory to direct infection with the Zika virus. “We don’t think the trophoblasts are very permissive at all,” he said.
Moreover, trophoblasts appear to confer their antiviral effects to other nontrophoblast cells by releasing a particular type of interferon – type III interferon IFN1 – and by delivering certain micro-RNAs (C19MC miRNAs) that are packaged within trophoblast-derived nanovesicles called exosomes, said Dr. Sadovsky, scientific director of the Magee-Womens Research Institute and professor of ob.gyn., reproductive sciences, microbiology, and molecular genetics at the University of Pittsburgh.
WASHINGTON – A DNA vaccine developed at the National Institute of Allergy and Infectious Diseases Vaccine Research Center – one of five National Institutes of Health Zika vaccine candidates – has entered phase 2 testing in a trial underway in Brazil, Peru, Ecuador, Mexico, and Texas.
“The DNA vaccine is a simple 21st century way of developing vaccines that I think will become one of the major [methods of the future] for emerging infections, as opposed to growing a virus and inactivating or attenuating it,” Anthony S. Fauci, MD, said at the biennial meeting of the Diabetes in Pregnancy Study Group of North America. With Zika “this is the vaccine that is ahead of all the others.”
Will it be possible to test efficacy, given the declining prevalence of Zika across the Americas, and will it be too late to prevent more disease? Dr. Fauci, director of NIAID, said that’s a concern, and that an accelerated approval based on a bridging of animal efficacy data with human safety and immunogenicity data might be possible.
The Southern hemisphere is “entering their summer, so it’s conceivable there will be an uptick in Zika. … We’ll just need to wait and see,” he said.
Sexual transmission
The Zika virus is part of a “long line of arboviruses that have threatened us in the Americas,” but infection with the organism is “the first – and may be the only – arthropod-borne or mosquito-borne infection that is also sexually transmitted,” Dr. Fauci said.
Sexual contact as an important mode of viral transmission “has been documented very clearly through a number of studies in which individuals clearly had no exposure to mosquitoes but were in fact a sexual partner of someone who got infected,” he said. And recent research suggests that the “female reproductive tract is a preferentially permissive site for Zika replication, which adds to the concern about sexual transmission.”
He cited a study published in July 2017 in PLOS Pathogens in which the Zika virus was found to preferentially replicate in the reproductive tract of female rhesus macaques who received vaginal inoculations of the virus.
Zika virus was “detected in the reproductive tract before it was detected in plasma, and replication levels in the reproductive tract did not reflect viral levels in other parts of the body,” according to the author summary. The kinetics of virus replication and dissemination after intravaginal inoculation were markedly different from what was previously seen in macaques infected with the Zika virus by subcutaneous infection, the report noted (PLOS Pathogens 13[7]:e1006537).
Dr. Fauci briefly described this and several other studies and findings that he said exemplify growing knowledge of the infection. He pointed to a prospective observational study that documents episodes of oligospermia in 15 men who presented with infection in 2016 in the French Caribbean (Lancet Infect Dis. 2017;17:1200-08).
Sperm counts fell in some of the study participants by about 50% between days 7 and 60 post infection, and the counts “recovered somewhat” by day 120. “We’re still following patients in prospective studies to determine if there’s a long-term effect in men,” he said.
In the meantime, he said, research in mice has shown that “without a doubt, Zika infection damages the testes,” Dr. Fauci said, noting that the mouse model is proving to be a good model for studying Zika’s effects. “They become oligospermic and have testicular atrophy.”
Maternal-fetal transmission
Regarding maternal-fetal transmission, there’s evidence that placental trophoblasts “are exquisitely permissive for Zika virus replication,” he said.
In another recent study, primary human placental trophoblasts from nonexposed donors were found to be infected by the Zika virus ex-vivo and permissive for viral RNA replication, compared with dengue virus, a fellow flavivirus (Sci Rep. 2017;7:41389. doi: 10.1038/srep41389).
However, Yoel Sadovsky, MD, who also presented at the meeting, explained that his lab’s ex-vivo studies show that primary human trophoblasts have inherent resistance to a number of viruses – and that trophoblasts are refractory to direct infection with the Zika virus. “We don’t think the trophoblasts are very permissive at all,” he said.
Moreover, trophoblasts appear to confer their antiviral effects to other nontrophoblast cells by releasing a particular type of interferon – type III interferon IFN1 – and by delivering certain micro-RNAs (C19MC miRNAs) that are packaged within trophoblast-derived nanovesicles called exosomes, said Dr. Sadovsky, scientific director of the Magee-Womens Research Institute and professor of ob.gyn., reproductive sciences, microbiology, and molecular genetics at the University of Pittsburgh.
AT DPSG-NA 2017
FDA: Ultrasound surgical devices are contraindicated for uterine fibroid removal
, according to the Food and Drug Administration.
These devices have the potential to disseminate undetected tumor tissue, and there are no proven preoperative screening methods for detecting uterine sarcoma in uterine fibroids that otherwise appear to be benign, FDA officials wrote in a guidance document issued Oct. 30. The FDA is calling for new product labeling for these devices within 120 days.
The devices deliver ultrasonic energy through an oscillating tip, which leads to tissue fragmentation. This can lead to tissue dissemination that cannot be eliminated by suction/aspiration. In advanced cancers, the risk of dissemination may be outweighed by the benefits of the devices, including the debulking effect with no thermal collateral damage, as well as avoidance of the need for organ removal or resection.
The devices are currently labeled in a way that suggests they could be used in removing uterine fibroids, though the agency said that it is not aware that they are used for this purpose.
The agency recommended against their use in uterine fibroids, in part because there are alternative treatment options available. But the American College of Obstetricians and Gynecologists has challenged that assertion. When the FDA first issued a draft notice of the labeling guidance in November 2016, ACOG commented that abdominal hysterectomy is the alternative treatment option and is associated with significant morbidity and mortality beyond that seen with minimally invasive techniques. ACOG urged the FDA to prioritize informed consent and the weighing of risks and benefits.
Ultrasonic surgical aspirator devices are used for a wide range of surgical applications, but the recommendations apply specifically to laparoscopic surgery, open surgery, and gynecologic surgery.
, according to the Food and Drug Administration.
These devices have the potential to disseminate undetected tumor tissue, and there are no proven preoperative screening methods for detecting uterine sarcoma in uterine fibroids that otherwise appear to be benign, FDA officials wrote in a guidance document issued Oct. 30. The FDA is calling for new product labeling for these devices within 120 days.
The devices deliver ultrasonic energy through an oscillating tip, which leads to tissue fragmentation. This can lead to tissue dissemination that cannot be eliminated by suction/aspiration. In advanced cancers, the risk of dissemination may be outweighed by the benefits of the devices, including the debulking effect with no thermal collateral damage, as well as avoidance of the need for organ removal or resection.
The devices are currently labeled in a way that suggests they could be used in removing uterine fibroids, though the agency said that it is not aware that they are used for this purpose.
The agency recommended against their use in uterine fibroids, in part because there are alternative treatment options available. But the American College of Obstetricians and Gynecologists has challenged that assertion. When the FDA first issued a draft notice of the labeling guidance in November 2016, ACOG commented that abdominal hysterectomy is the alternative treatment option and is associated with significant morbidity and mortality beyond that seen with minimally invasive techniques. ACOG urged the FDA to prioritize informed consent and the weighing of risks and benefits.
Ultrasonic surgical aspirator devices are used for a wide range of surgical applications, but the recommendations apply specifically to laparoscopic surgery, open surgery, and gynecologic surgery.
, according to the Food and Drug Administration.
These devices have the potential to disseminate undetected tumor tissue, and there are no proven preoperative screening methods for detecting uterine sarcoma in uterine fibroids that otherwise appear to be benign, FDA officials wrote in a guidance document issued Oct. 30. The FDA is calling for new product labeling for these devices within 120 days.
The devices deliver ultrasonic energy through an oscillating tip, which leads to tissue fragmentation. This can lead to tissue dissemination that cannot be eliminated by suction/aspiration. In advanced cancers, the risk of dissemination may be outweighed by the benefits of the devices, including the debulking effect with no thermal collateral damage, as well as avoidance of the need for organ removal or resection.
The devices are currently labeled in a way that suggests they could be used in removing uterine fibroids, though the agency said that it is not aware that they are used for this purpose.
The agency recommended against their use in uterine fibroids, in part because there are alternative treatment options available. But the American College of Obstetricians and Gynecologists has challenged that assertion. When the FDA first issued a draft notice of the labeling guidance in November 2016, ACOG commented that abdominal hysterectomy is the alternative treatment option and is associated with significant morbidity and mortality beyond that seen with minimally invasive techniques. ACOG urged the FDA to prioritize informed consent and the weighing of risks and benefits.
Ultrasonic surgical aspirator devices are used for a wide range of surgical applications, but the recommendations apply specifically to laparoscopic surgery, open surgery, and gynecologic surgery.
Ulcerative Sarcoidosis: A Prototypical Presentation and Review
Sarcoidosis is a multisystem granulomatous disorder of unknown etiology that primarily affects the lungs and lymphatic system but also may involve the skin, eyes, liver, spleen, muscles, bones, and nervous system.1 Cutaneous symptoms of sarcoidosis occur in approximately 25% of patients and are classified as specific and nonspecific, with specific lesions demonstrating noncaseating granuloma formation, which is typical of sarcoidosis.2 Nonspecific lesions primarily include erythema nodosum and calcinosis cutis. Specific lesions commonly present as reddish brown infiltrated plaques that may be annular, polycyclic, or serpiginous.1,3 They also may appear as yellowish brown or violaceous maculopapular lesions. However, specific lesions may present in a wide variety of morphologies, most often papules, nodules, subcutaneous infiltrates, and lupus pernio.4 Additionally, atypical cutaneous manifestations of sarcoidosis include erythroderma; scarring alopecia; nail dystrophy; and verrucous, ichthyosiform, psoriasiform, hypopigmented, or ulcerative skin lesions.3-5 Among these many potential clinical presentations, ulcerative sarcoidosis is quite uncommon.
We report a case of a patient who presented with classic clinical and histopathological findings of ulcerative sarcoidosis to highlight the prototypical presentation of a rare condition. We also review 34 additional cases of ulcerative sarcoidosis published in the English-language literature based on a PubMed search of articles indexed for MEDLINE using the term ulcerative sarcoid.4-32 Analyzing this historical information, the scope of this unusual form of cutaneous sarcoidosis can be better understood, recognized, and treated. Although current standard-of-care treatments are most often successful, there is a paucity of definitive clinical trials to justify and verify comparative therapeutic efficacy.
Case Report
A 49-year-old black man with known pulmonary sarcoidosis, idiopathic (human immunodeficiency virus–negative) CD4 depletion syndrome, and chronic kidney disease presented with persistent bilateral ulcers of the legs of 1 month’s duration. The lesions first appeared as multiple “dark spots” on the legs. After the patient applied homemade aloe vera extract under occlusion for 1 to 2 days, the lesions became painful and began to ulcerate approximately 3 months prior to presentation. The patient applied a combination of a topical first aid antibiotic ointment, Epsom salts, and hydrogen peroxide without any improvement. A current review of systems was negative.
The patient’s medical history was notable for sarcoidosis diagnosed more than 10 years prior. During this time, he had intermittently been treated elsewhere with low-dose oral prednisone (5 mg once daily), hydroxychloroquine (200 mg twice daily), and an inhaled steroid as needed. He had a history of human immunodeficiency virus–negative, idiopathic CD4 depletion syndrome, which had been complicated by cryptococcal meningitis 7 years prior to presentation. He also had renal insufficiency, with baseline creatinine levels ranging from 1.4 to 1.7 mg/dL (reference range, 0.6–1.2 mg/dL). There was no personal or family history of known or suspected inflammatory bowel disease.
On physical examination, numerous discrete, coalescing, punched out–appearing ulcerations with foul-smelling, greenish yellow, purulent drainage were present bilaterally on the legs (Figure 1). The ulcers had a rolled border with a moderate amount of seemingly nonviable necrotic tissue. A number of hyperpigmented round papules, patches, and plaques also were present on the proximal legs. Laboratory evaluation revealed a CD4 count of 151 cc/mm3 (reference range, 500–1600 cc/mm3) and mildly elevated calcium of 10.7 mg/dL (reference range, 8.2–10.2 mg/dL).
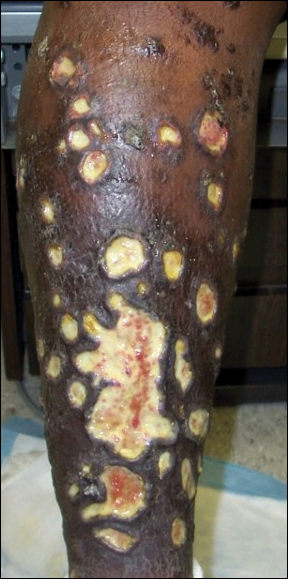
Aerobic, anaerobic, mycobacterial, and fungal cultures of the purulent exudate were obtained. Given a high suspicion for secondary infection of the exogenous wound sites, doxycycline (100 mg twice daily) and topical mupir-ocin were initiated. Gram stain revealed few to moderate polymorphonuclear cells and many gram-positive cocci in pairs, chains, and clusters, along with many gram-negative rods. Bacterial culture grew Pseudomonas aeruginosa, Enterococcus species group G streptococci, and methicillin-resistant Staphylococcus aureus–positive staphylococci. Ciprofloxacin (500 mg twice daily) was then initiated, but the ulcers showed absolutely no clinical improvement and in fact worsened both in number and depth (Figure 2) over subsequent clinic visits during the next 3 months, even after amoxicillin (500 mg 3 times daily) was added. The patient was admitted for treatment with intravenous antibiotics after additional wound cultures revealed fluoroquinolone-resistant Pseudomonas.
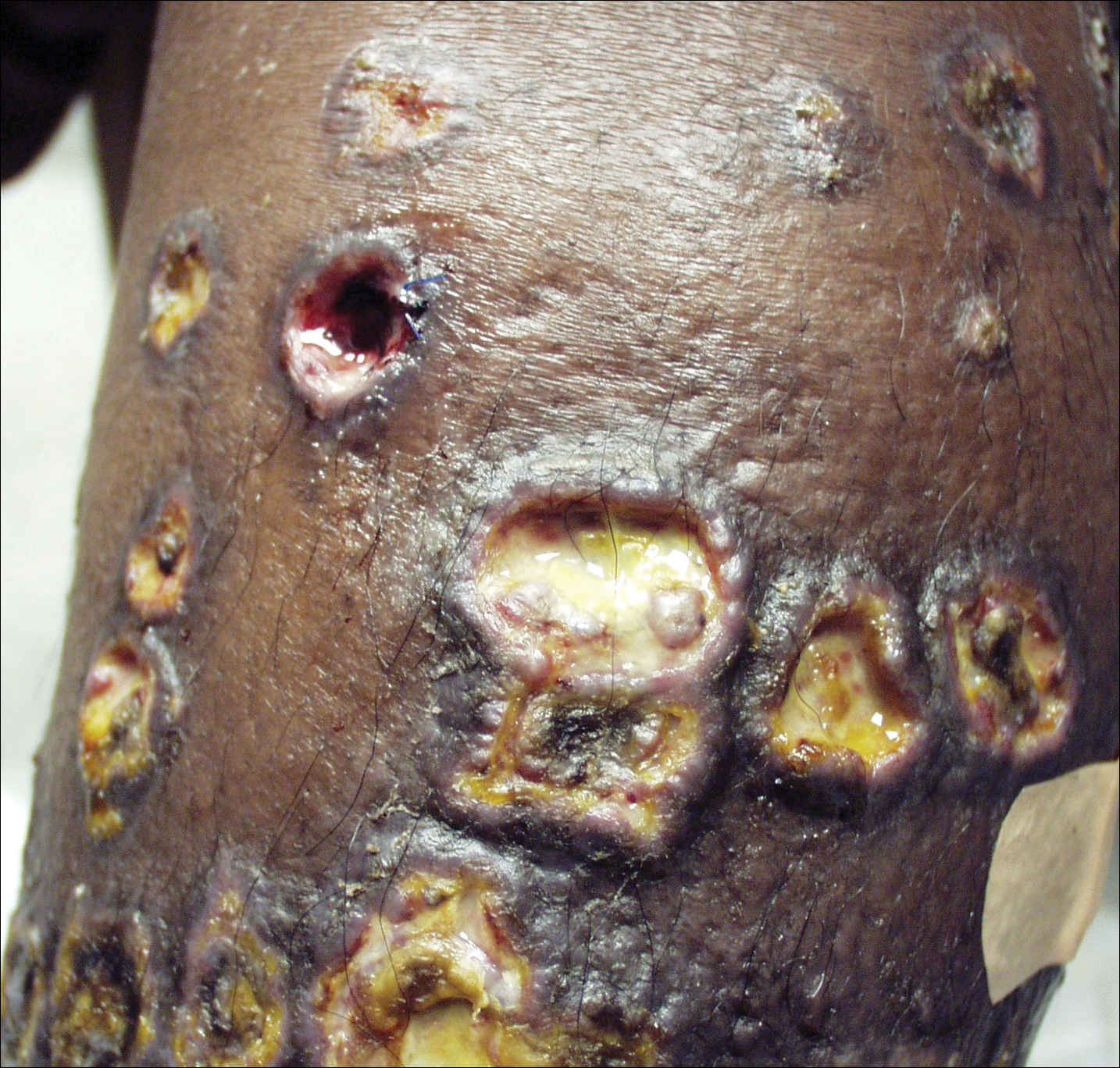
Punch biopsies of the ulcers showed nonspecific acute inflammation and tissue necrosis in the active ulcers with nonnecrotizing granulomatous inflammation extending into the deep dermis, with many Langerhans-type giant cells present in the palpable ulcer borders (Figure 3). Neither birefringent particles nor asteroid bodies were observed. Tissue Gram stains did not reveal evidence of bacterial infection. Special stains for acid-fast and fungal organisms (ie, periodic acid–Schiff, Gomori methenamine-silver, Fite, acid-fast bacilli) were similarly negative. Tissue cultures obtained on deep biopsy revealed only rare colonies of P aeruginosa and no isolates on anaerobic, mycobacterial, or fungal cultures. Polymerase chain reaction for mycobacteria and common endemic fungi also was negative. In the absence of infection and considering his history of known sarcoidosis, these histologic features were consistent with ulcerative sarcoidosis. The patient was started on prednisone (60 mg once daily) and hydroxychloroquine (200 mg twice daily). The prednisone was tapered to 20 mg once daily over a 2-year period, at which point 90% of the ulcers had healed. He was continued on hydroxychloroquine at the initial dose, and at a 3-year follow-up his ulcers had healed completely without relapse.
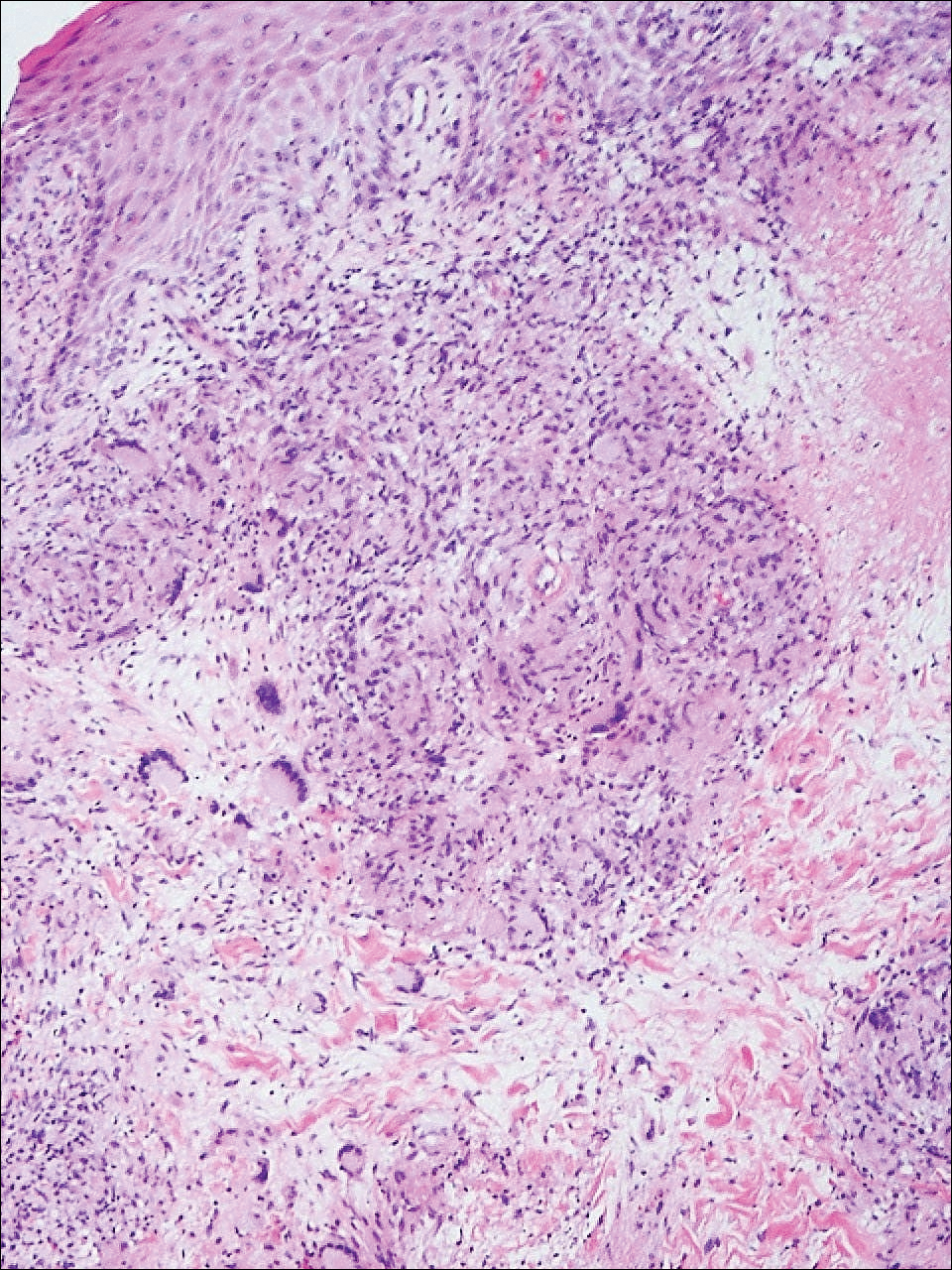
Comment
Ulcerative sarcoidosis is rare, seen worldwide in only 5% of patients with cutaneous sarcoidosis.33 However, cases have been encountered worldwide, with reports emanating from Japan, China, Germany, France, and Russia, among others.6,34-55 We reviewed 34 cases from the English-language literature based on a PubMed search of articles indexed for MEDLINE using the term ulcerative sarcoid and examined patient demographics, clinical presentation, histological findings, treatment type, and outcome. Key references are presented in the Table. Disease prevalence previously has been estimated as being 3-times more common in women than men1; in our literature review, we found a female to male ratio of 3.25 to 1. Additionally, ulcerative sarcoidosis is reported to be twice as common in black versus white individuals.33 In our literature review, when race was reported, 66% (21/32) of patients were black. Disease prevalence has been reported to peak at 20 to 40 years of age.3 In this review, the average age of presentation was 45 years (age range, 24–79 years).
Ulceration may arise de novo but more commonly arises in preexisting scars or cutaneous lesions. There are 2 distinct patterns seen in ulcerative sarcoidosis.4 The first is characterized by ulceration within necrotic yellow plaques.2 The second pattern is characterized by violaceous nodules arising in an annular confluent pattern that eventually ulcerate.4 This presentation commonly mimics or may be mimicked by multiple disease states, including sporotrichosis, tuberculosis, stasis dermatitis with venous ulceration, and even metastatic breast cancer.7,46,55,56 Regardless of presentation, the legs are the most common location of ulcer formation.1,33 In our review, 85% (29/34) of cases presented with involvement of the legs, including our own case. Other locations of ulcer formation have included the face, arms, trunk, and genital area.
On histologic examination of ulcerative sarcoidosis, epithelioid granulomas composed of multinucleated giant cells, histiocytes, and scant numbers of lymphocytes are present.1,3 These formations are the noncaseating granulomas typical of sarcoidosis (Table). All of the cases in our review of the literature were described as either a collection of epithelioid granulomas with giant cell formation or noncaseating granulomas. There also have been reports of atypical features including necrotizing granulomas and granulomatous vasculitis.4,8,9,50 The histologic differential diagnosis in this case also would primarily include an infectious granulomatous process and less so an id reaction, rosacea, a paraneoplastic phenomenon, foreign body granulomas, and metastatic Crohn disease. The presence of ulceration, the large number of lesions, and the anatomic distribution help rule out most of these alternate diagnostic considerations. Diligent extensive workup was done in our patient to insure it was not an infection.
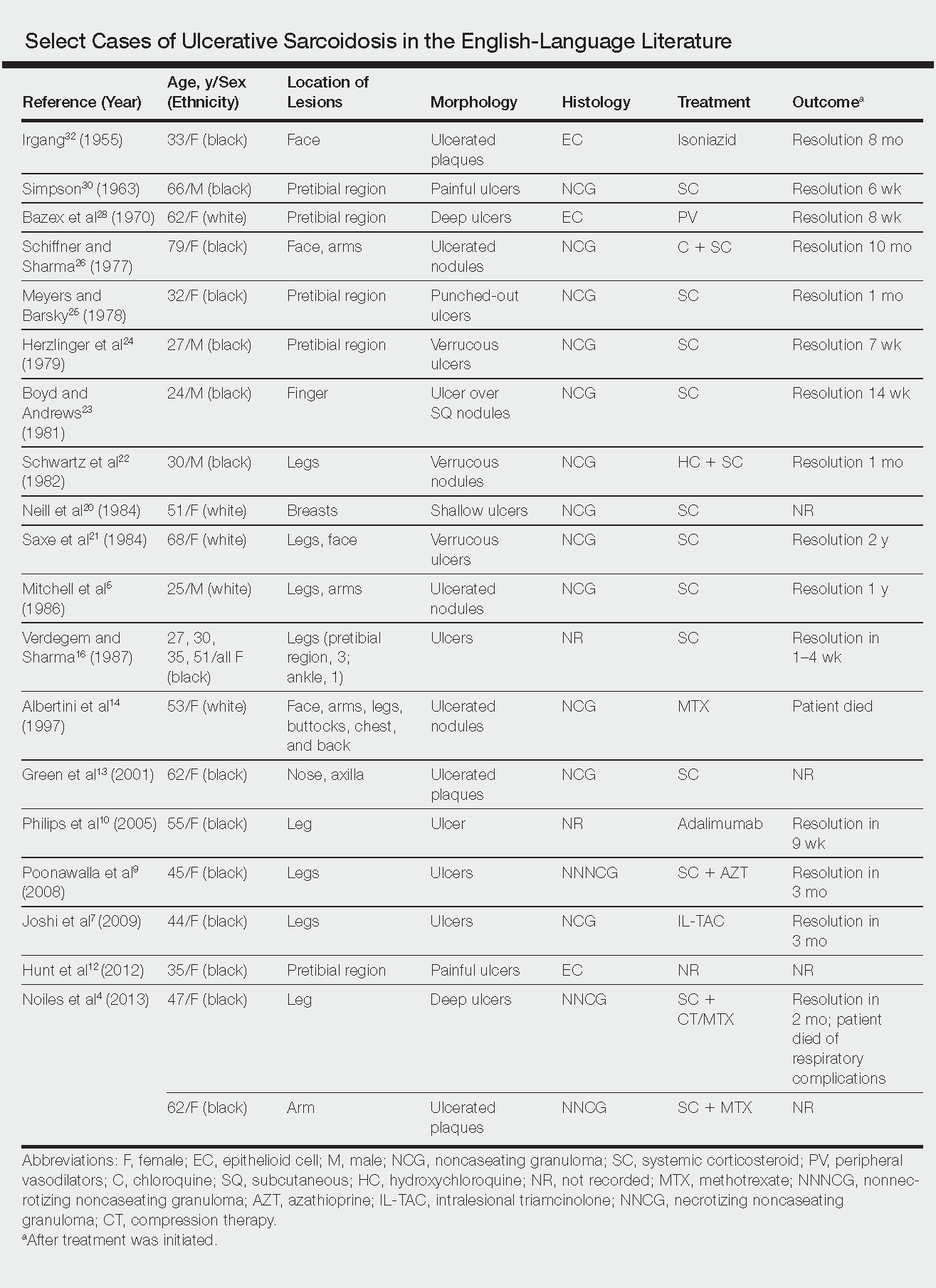
The goals of treatment include symptomatic relief, improvement in objective parameters of disease activity, and prevention of disease progression and subsequent disability.33,57 Fortunately, the majority of sarcoidosis patients with cutaneous symptoms achieve full recovery within months to years.33 Our literature review indicated that 81% (22/27) of patients with ulcerative lesions experienced full resolution within 1 year of treatment. Of those that did not (19% [5/27]), the patients were either lost to follow-up or died from other complications of sarcoidosis.
The widely accepted standard therapy for cutaneous sarcoidosis includes topical, intralesional, and systemic corticosteroids; antimalarials; and methotrexate.33,57 Steroids and methotrexate act by suppressing granuloma formation, while antimalarials prevent antigen presentation (presumably part of the pathogenesis).33 For mild to moderate disease, topical and intralesional steroids may be all that is necessary.33,57 Systemic steroids are used for disfiguring, destructive, and widespread lesions that have been refractory to local and other systemic therapies.33,57 Steroids are tapered gradually depending on the patient’s response, as it is common for patients to relapse below a certain dose.33,57 Antimalarials (chloroquine or hydroxychloroquine) and methotrexate are considered adjunct treatments for patients who are either steroid unresponsive or who are unable to tolerate corticosteroid treatment due to adverse events.33,57
Standard therapy is complicated by the side effects of treatment. Use of corticosteroids may lead to gastrointestinal tract upset, increased appetite, mood disturbances, impaired wound healing, hyperglycemia, hypertension, cushingoid features, and acne.57 Antimalarials can cause nausea, anorexia, and agranulocytosis, and chloroquine therapy in particular can lead to blurred vision, corneal deposits, and central retinopathy.33,57 Methotrexate is associated with hematologic, gastrointestinal tract, pulmonary, and hepatic toxicities well known to most practitioners.
Because of the variable clinical response of patients to standard therapy and their associated toxicities, other treatment options have been used including pentoxifylline, tetracyclines, isotretinoin, leflunomide, thalidomide, infliximab, adalimumab, allopurinol, and the pulsed dye or CO2 laser.10,33,57 In nonhealing ulcers, split-thickness grafting and a bilayered bioengineered skin substitute have been used with good results in conjunction with ongoing systemic therapy.11,47 Additionally, nanoparticle silver burn paste has been used successfully, with resolution of ulcers within 2 weeks in the Chinese literature.53
All of these treatment recommendations are based on historically accepted modalities. Controlled trials with longitudinal follow-up are needed to provide justification for the current standard of care.34
- Howard A, White CR Jr. Non-infectious granulomas. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. 2nd ed. Spain: Elsevier; 2008:1421-1435.
- Doherty CB, Rosen T. Evidence-based therapy for cutaneous sarcoidosis. Drugs. 2008;68:1361-1383.
- Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25:295-302.
- Noiles K, Beleznay K, Crawford RI, et al. Sarcoidosis can present with necrotizing granulomas histologically: two cases of ulcerated sarcoidosis and review of the literature. J Cutan Med Surg. 2013;17:377-378.
- Mitchell IC, Sweatman MC, Rustin MH, et al. Ulcerative and hypopigmented sarcoidosis. J Am Acad Dermatol. 1986;15:1062-1065.
- Yoo SS, Mimouni D, Nikolskaia OV, et al. Clinicopathologic features of ulcerative-atrophic sarcoidosis. Int J Dermatol. 2004;43:108-112.
- Joshi SS, Romanelli R, Kirsner RS. Sarcoidosis mimicking a venous ulcer: a case report. Ostomy Wound Manage. 2009;55:46-48.
- Petri M, Barr E, Cho K, et al. Overlap of granulomatous vasculitis and sarcoidosis: presentation with uveitis, eosinophilia, leg ulcers, sinusitis and past foot drop. J Rheumatol. 1988;15:1171-1173.
- Poonawalla T, Colome-Grimmer MI, Kelly B. Ulcerative sarcoidosis in the legs with granulomatous vasculitis. Clin Exp Dermatol. 2008;33:282-286.
- Philips MA, Lynch J, Azmi FH. Ulcerative sarcoidosis responding to adalimumab. J Am Acad Dermatol. 2005;53:917.
- Collison DW, Novice F, Banse L, et al. Split-thickness skin grafting in extensive ulcerative sarcoidosis. J Dermatol Surg Oncol. 1989;15:679-683.
- Hunt RD, Gonzalez ME, Robinson M, et al. Ulcerative sarcoidosis. Dermatol Online J. 2012;18:29.
- Green JJ, Lawrence N, Heymann WR. Generalized ulcerative sarcoidosis induced by therapy with the flashlamp-pumped pulsed dye. Arch Dermatol. 2001;137:507-508.
- Albertini JG, Tyler W, Miller OF. Ulcerative sarcoidosis. case report and review of the literature. Arch Dermatol. 1997;133:215-219.
- Thomas J, Williams DW. Peritoneal involvement and ulcerative skin plaques in sarcoidosis: a case report. Sarcoidosis. 1989;6:161-162.
- Verdegem TD, Sharma OP. Cutaneous ulcers in sarcoidosis. Arch Dermatol. 1987;123:1531-1534.
- Gupta AK, Haberman HF, From GL, et al. Sarcoidosis with extensive cutaneous ulceration. unusual clinical presentation. Dermatologica. 1987;174:135-139.
- Hruza GJ, Kerdel FA. Generalized atrophic sarcoidosis with ulcerations. Arch Dermatol. 1986;122:320-322.
- Muhlemann MF, Walker NP, Tan LB, et al. Elephantine sarcoidosis presenting as ulcerating lymphoedema. J R Soc Med. 1985;78:260-261.
- Neill SM, Smith NP, Eady RA. Ulcerative sarcoidosis: a rare manifestation of a common disease. Clin Exp Dermatol. 1984;9:277-279.
- Saxe N, Benatar SR, Bok L, et al. Sarcoidosis with leg ulcers and annular facial lesions. Arch Dermatol. 1984;120:93-96.
- Schwartz RA, Robertson DB, Tierney LM, et al. Generalized ulcerative sarcoidosis. Arch Dermatol. 1982;118:931-933.
- Boyd RE, Andrews BS. Sarcoidosis presenting as cutaneous ulceration, subcutaneous nodules and chronic arthritis. J Rheumatol. 1981;8:311-316.
- Herzlinger DC, Marland AM, Barr RJ. Verrucous ulcerative skin lesions in sarcoidosis. an unusual clinical presentation. Cutis. 1979;23:569-572.
- Meyers M, Barsky S. Ulcerative sarcoidosis. Arch Dermatol. 1978;114:447.
- Schiffner J, Sharma OP. Ulcerative sarcoidosis. report of an unusual case. Arch Dermatol. 1977;113:676-677.
- Williamson DM. Sarcoidosis with atrophic lesions and ulcers of the legs. Br J Dermatol. 1971;84:92-93.
- Bazex A, Dupre A, Christol B, et al. Sarcoidosis with atrophic lesions and ulcers and the presence in some sarcoid granulomata of orceinophil fibres. Br J Dermatol. 1970;83:255-262.
- Brodkin RH. Leg ulcers. a report of two cases caused by sarcoidosis. Acta Derm Venereol. 1969;49:584-587.
- Simpson JR. Sarcoidosis with erythrodermia and ulceration. Br J Dermatol. 1963;75:193-198.
- Irgang S. Ulcerative cutaneous lesion in sarcoidosis; report of a case with clinical resemblance to lupus vulgaris. Harlem Hosp Bull. 1956;8:134-139.
- Irgang S. Ulcerative cutaneous lesions in sarcoidosis; report of a case with clinical resemblance to papulonecrotic tuberculide. Br J Dermatol. 1955;67:255-260.
- Hoffman MD. Atypical ulcers. Dermatol Ther. 2013;26:222-235.
- Hopf B, Krebs A. Ulcera cruris as a rare manifestation of sarcoidosis. Dermatologica. 1974;113:55-62.
- Metz J, Hartmann A, Hautkr Z. Ulcerative form of skin sarcoidosis. Z Hautkr. 1977;52:890-896.
- Berenbeĭn BA, Malygina LA, Tiutiunnikova IA. Ulcerative form of skin sarcoidosis [in Russian]. Vestn Dermatol Venerol. 1984;4:50-53.
- Takahashi N, Hoshino M, Takase T, et al. A case of ulcerative sarcoidosis [in Japanese]. Nihon Hifuka Gakkai Zasshi. 1985;95:1049-1054.
- Schamroth JM. Sarcoidosis with severe extensive skin ulceration. Int J Dermatol. 1985;24:451-452.
- Porteau L, Dromer C, Le Guennec P, et al. Ulcer lesions in sarcoidosis: apropos of a case [in French]. Ann Med Interne (Paris). 1997;148:105-106.
- de La Blanchardière A, Bachmeyer C, Toutous L, et al. Cutaneous ulcerations in sarcoidosis [in French]. Rev Med Interne. 1995;16:927-929.
- Mitsuishi T, Nogita T, Kawashima M. Psoriasiform sarcoidosis with ulceration. Int J Dermatol. 1992;31:339-340.
- Rodionov AN, Samtsov AV. The ulcerative form of skin sarcoidosis [in Russian]. Vestn Dermatol Venerol. 1990;7:68-71.
- Jacyk WK. Cutaneous sarcoidosis in black South Africans. Int J Dermatol. 1999;38:841-845.
- Gungor E, Artuz F, Alli N, et al. Ulcerative sarcoidosis. J Eur Acad Dermatol Venereol. 1999;12:78-79.
- Schleinitz N, Luc M, Genot S, et al. Ulcerative cutaneous lesions: a rare manifestation of sarcoidosis [in French]. Rev Med Interne. 2005;26:758-759.
- Klocker J, Duckers J, Morse R, et al. Ulcerative cutaneous sarcoidosis masquerading as metastatic carcinoma of the breast. Age Ageing. 2002;31:77-79.
- Streit M, Bohlen LM, Braathen LR. Ulcerative sarcoidosis successfully treated with apligraf. Dermatology. 2001;202:367-370.
- Ichiki Y, Kitajima Y. Ulcerative sarcoidosis: case report and review of the Japanese literature. Acta Derm Venereol. 2008;88:526-528
- Meyersburg D, Schön MP, Bertsch HP, et al. Uncommon cutaneous ulcerative and systemic sarcoidosis. successful treatment with hydroxychloroquine and compression therapy [in German]. Hautarzt. 2011;62:691-695.
- Wei CH, Huang YH, Shih YC, et al. Sarcoidosis with cutaneous granulomatous vasculitis. Australas J Dermatol. 2010;51:198-201.
- Kluger N, Girard C, Durand L, et al. Leg ulcers revealing systemic sarcoidosis with splenomegaly and thrombocytopenia. Int J Dermatol. 2013;52:1425-1427.
- Jun L, Jia-Wei L, Hong-Zhong J. Ulcerative sarcoidosis. Int J Dermatol. 2014;53:E315-E316.
- Chen JH, Wang TT, Lin ZQ. Successful application of a novel dressing for the treatment of ulcerative cutaneous sarcoidosis. Chin Med J. 2013;126:3400.
- Ri G, Yoshikawa E, Shigekiyo T, et al. Takayasu artertitis and ulcerative sarcoidosis. Intern Med. 2015;54:1075-1080.
- Spiliopoulou I, Foka A, Bounas A, et al. Mycobacterium kansasii cutaneous infection in a patient with sarcoidosis treated with anti-TNF agents. Acta Clin Belg. 2014;69:229-231.
- Yang DJ, Krishnan RS, Guillen DR, et al. Disseminated sporotrichosis mimicking sarcoidosis. Int J Dermatol. 2006;45:450-453.
- Badgwell C, Rosen T. Cutaneous sarcoidosis therapy updated. J Am Acad Dermatol. 2007;56:69-83.
Sarcoidosis is a multisystem granulomatous disorder of unknown etiology that primarily affects the lungs and lymphatic system but also may involve the skin, eyes, liver, spleen, muscles, bones, and nervous system.1 Cutaneous symptoms of sarcoidosis occur in approximately 25% of patients and are classified as specific and nonspecific, with specific lesions demonstrating noncaseating granuloma formation, which is typical of sarcoidosis.2 Nonspecific lesions primarily include erythema nodosum and calcinosis cutis. Specific lesions commonly present as reddish brown infiltrated plaques that may be annular, polycyclic, or serpiginous.1,3 They also may appear as yellowish brown or violaceous maculopapular lesions. However, specific lesions may present in a wide variety of morphologies, most often papules, nodules, subcutaneous infiltrates, and lupus pernio.4 Additionally, atypical cutaneous manifestations of sarcoidosis include erythroderma; scarring alopecia; nail dystrophy; and verrucous, ichthyosiform, psoriasiform, hypopigmented, or ulcerative skin lesions.3-5 Among these many potential clinical presentations, ulcerative sarcoidosis is quite uncommon.
We report a case of a patient who presented with classic clinical and histopathological findings of ulcerative sarcoidosis to highlight the prototypical presentation of a rare condition. We also review 34 additional cases of ulcerative sarcoidosis published in the English-language literature based on a PubMed search of articles indexed for MEDLINE using the term ulcerative sarcoid.4-32 Analyzing this historical information, the scope of this unusual form of cutaneous sarcoidosis can be better understood, recognized, and treated. Although current standard-of-care treatments are most often successful, there is a paucity of definitive clinical trials to justify and verify comparative therapeutic efficacy.
Case Report
A 49-year-old black man with known pulmonary sarcoidosis, idiopathic (human immunodeficiency virus–negative) CD4 depletion syndrome, and chronic kidney disease presented with persistent bilateral ulcers of the legs of 1 month’s duration. The lesions first appeared as multiple “dark spots” on the legs. After the patient applied homemade aloe vera extract under occlusion for 1 to 2 days, the lesions became painful and began to ulcerate approximately 3 months prior to presentation. The patient applied a combination of a topical first aid antibiotic ointment, Epsom salts, and hydrogen peroxide without any improvement. A current review of systems was negative.
The patient’s medical history was notable for sarcoidosis diagnosed more than 10 years prior. During this time, he had intermittently been treated elsewhere with low-dose oral prednisone (5 mg once daily), hydroxychloroquine (200 mg twice daily), and an inhaled steroid as needed. He had a history of human immunodeficiency virus–negative, idiopathic CD4 depletion syndrome, which had been complicated by cryptococcal meningitis 7 years prior to presentation. He also had renal insufficiency, with baseline creatinine levels ranging from 1.4 to 1.7 mg/dL (reference range, 0.6–1.2 mg/dL). There was no personal or family history of known or suspected inflammatory bowel disease.
On physical examination, numerous discrete, coalescing, punched out–appearing ulcerations with foul-smelling, greenish yellow, purulent drainage were present bilaterally on the legs (Figure 1). The ulcers had a rolled border with a moderate amount of seemingly nonviable necrotic tissue. A number of hyperpigmented round papules, patches, and plaques also were present on the proximal legs. Laboratory evaluation revealed a CD4 count of 151 cc/mm3 (reference range, 500–1600 cc/mm3) and mildly elevated calcium of 10.7 mg/dL (reference range, 8.2–10.2 mg/dL).
Aerobic, anaerobic, mycobacterial, and fungal cultures of the purulent exudate were obtained. Given a high suspicion for secondary infection of the exogenous wound sites, doxycycline (100 mg twice daily) and topical mupir-ocin were initiated. Gram stain revealed few to moderate polymorphonuclear cells and many gram-positive cocci in pairs, chains, and clusters, along with many gram-negative rods. Bacterial culture grew Pseudomonas aeruginosa, Enterococcus species group G streptococci, and methicillin-resistant Staphylococcus aureus–positive staphylococci. Ciprofloxacin (500 mg twice daily) was then initiated, but the ulcers showed absolutely no clinical improvement and in fact worsened both in number and depth (Figure 2) over subsequent clinic visits during the next 3 months, even after amoxicillin (500 mg 3 times daily) was added. The patient was admitted for treatment with intravenous antibiotics after additional wound cultures revealed fluoroquinolone-resistant Pseudomonas.
Punch biopsies of the ulcers showed nonspecific acute inflammation and tissue necrosis in the active ulcers with nonnecrotizing granulomatous inflammation extending into the deep dermis, with many Langerhans-type giant cells present in the palpable ulcer borders (Figure 3). Neither birefringent particles nor asteroid bodies were observed. Tissue Gram stains did not reveal evidence of bacterial infection. Special stains for acid-fast and fungal organisms (ie, periodic acid–Schiff, Gomori methenamine-silver, Fite, acid-fast bacilli) were similarly negative. Tissue cultures obtained on deep biopsy revealed only rare colonies of P aeruginosa and no isolates on anaerobic, mycobacterial, or fungal cultures. Polymerase chain reaction for mycobacteria and common endemic fungi also was negative. In the absence of infection and considering his history of known sarcoidosis, these histologic features were consistent with ulcerative sarcoidosis. The patient was started on prednisone (60 mg once daily) and hydroxychloroquine (200 mg twice daily). The prednisone was tapered to 20 mg once daily over a 2-year period, at which point 90% of the ulcers had healed. He was continued on hydroxychloroquine at the initial dose, and at a 3-year follow-up his ulcers had healed completely without relapse.
Comment
Ulcerative sarcoidosis is rare, seen worldwide in only 5% of patients with cutaneous sarcoidosis.33 However, cases have been encountered worldwide, with reports emanating from Japan, China, Germany, France, and Russia, among others.6,34-55 We reviewed 34 cases from the English-language literature based on a PubMed search of articles indexed for MEDLINE using the term ulcerative sarcoid and examined patient demographics, clinical presentation, histological findings, treatment type, and outcome. Key references are presented in the Table. Disease prevalence previously has been estimated as being 3-times more common in women than men1; in our literature review, we found a female to male ratio of 3.25 to 1. Additionally, ulcerative sarcoidosis is reported to be twice as common in black versus white individuals.33 In our literature review, when race was reported, 66% (21/32) of patients were black. Disease prevalence has been reported to peak at 20 to 40 years of age.3 In this review, the average age of presentation was 45 years (age range, 24–79 years).
Ulceration may arise de novo but more commonly arises in preexisting scars or cutaneous lesions. There are 2 distinct patterns seen in ulcerative sarcoidosis.4 The first is characterized by ulceration within necrotic yellow plaques.2 The second pattern is characterized by violaceous nodules arising in an annular confluent pattern that eventually ulcerate.4 This presentation commonly mimics or may be mimicked by multiple disease states, including sporotrichosis, tuberculosis, stasis dermatitis with venous ulceration, and even metastatic breast cancer.7,46,55,56 Regardless of presentation, the legs are the most common location of ulcer formation.1,33 In our review, 85% (29/34) of cases presented with involvement of the legs, including our own case. Other locations of ulcer formation have included the face, arms, trunk, and genital area.
On histologic examination of ulcerative sarcoidosis, epithelioid granulomas composed of multinucleated giant cells, histiocytes, and scant numbers of lymphocytes are present.1,3 These formations are the noncaseating granulomas typical of sarcoidosis (Table). All of the cases in our review of the literature were described as either a collection of epithelioid granulomas with giant cell formation or noncaseating granulomas. There also have been reports of atypical features including necrotizing granulomas and granulomatous vasculitis.4,8,9,50 The histologic differential diagnosis in this case also would primarily include an infectious granulomatous process and less so an id reaction, rosacea, a paraneoplastic phenomenon, foreign body granulomas, and metastatic Crohn disease. The presence of ulceration, the large number of lesions, and the anatomic distribution help rule out most of these alternate diagnostic considerations. Diligent extensive workup was done in our patient to insure it was not an infection.
The goals of treatment include symptomatic relief, improvement in objective parameters of disease activity, and prevention of disease progression and subsequent disability.33,57 Fortunately, the majority of sarcoidosis patients with cutaneous symptoms achieve full recovery within months to years.33 Our literature review indicated that 81% (22/27) of patients with ulcerative lesions experienced full resolution within 1 year of treatment. Of those that did not (19% [5/27]), the patients were either lost to follow-up or died from other complications of sarcoidosis.
The widely accepted standard therapy for cutaneous sarcoidosis includes topical, intralesional, and systemic corticosteroids; antimalarials; and methotrexate.33,57 Steroids and methotrexate act by suppressing granuloma formation, while antimalarials prevent antigen presentation (presumably part of the pathogenesis).33 For mild to moderate disease, topical and intralesional steroids may be all that is necessary.33,57 Systemic steroids are used for disfiguring, destructive, and widespread lesions that have been refractory to local and other systemic therapies.33,57 Steroids are tapered gradually depending on the patient’s response, as it is common for patients to relapse below a certain dose.33,57 Antimalarials (chloroquine or hydroxychloroquine) and methotrexate are considered adjunct treatments for patients who are either steroid unresponsive or who are unable to tolerate corticosteroid treatment due to adverse events.33,57
Standard therapy is complicated by the side effects of treatment. Use of corticosteroids may lead to gastrointestinal tract upset, increased appetite, mood disturbances, impaired wound healing, hyperglycemia, hypertension, cushingoid features, and acne.57 Antimalarials can cause nausea, anorexia, and agranulocytosis, and chloroquine therapy in particular can lead to blurred vision, corneal deposits, and central retinopathy.33,57 Methotrexate is associated with hematologic, gastrointestinal tract, pulmonary, and hepatic toxicities well known to most practitioners.
Because of the variable clinical response of patients to standard therapy and their associated toxicities, other treatment options have been used including pentoxifylline, tetracyclines, isotretinoin, leflunomide, thalidomide, infliximab, adalimumab, allopurinol, and the pulsed dye or CO2 laser.10,33,57 In nonhealing ulcers, split-thickness grafting and a bilayered bioengineered skin substitute have been used with good results in conjunction with ongoing systemic therapy.11,47 Additionally, nanoparticle silver burn paste has been used successfully, with resolution of ulcers within 2 weeks in the Chinese literature.53
All of these treatment recommendations are based on historically accepted modalities. Controlled trials with longitudinal follow-up are needed to provide justification for the current standard of care.34
Sarcoidosis is a multisystem granulomatous disorder of unknown etiology that primarily affects the lungs and lymphatic system but also may involve the skin, eyes, liver, spleen, muscles, bones, and nervous system.1 Cutaneous symptoms of sarcoidosis occur in approximately 25% of patients and are classified as specific and nonspecific, with specific lesions demonstrating noncaseating granuloma formation, which is typical of sarcoidosis.2 Nonspecific lesions primarily include erythema nodosum and calcinosis cutis. Specific lesions commonly present as reddish brown infiltrated plaques that may be annular, polycyclic, or serpiginous.1,3 They also may appear as yellowish brown or violaceous maculopapular lesions. However, specific lesions may present in a wide variety of morphologies, most often papules, nodules, subcutaneous infiltrates, and lupus pernio.4 Additionally, atypical cutaneous manifestations of sarcoidosis include erythroderma; scarring alopecia; nail dystrophy; and verrucous, ichthyosiform, psoriasiform, hypopigmented, or ulcerative skin lesions.3-5 Among these many potential clinical presentations, ulcerative sarcoidosis is quite uncommon.
We report a case of a patient who presented with classic clinical and histopathological findings of ulcerative sarcoidosis to highlight the prototypical presentation of a rare condition. We also review 34 additional cases of ulcerative sarcoidosis published in the English-language literature based on a PubMed search of articles indexed for MEDLINE using the term ulcerative sarcoid.4-32 Analyzing this historical information, the scope of this unusual form of cutaneous sarcoidosis can be better understood, recognized, and treated. Although current standard-of-care treatments are most often successful, there is a paucity of definitive clinical trials to justify and verify comparative therapeutic efficacy.
Case Report
A 49-year-old black man with known pulmonary sarcoidosis, idiopathic (human immunodeficiency virus–negative) CD4 depletion syndrome, and chronic kidney disease presented with persistent bilateral ulcers of the legs of 1 month’s duration. The lesions first appeared as multiple “dark spots” on the legs. After the patient applied homemade aloe vera extract under occlusion for 1 to 2 days, the lesions became painful and began to ulcerate approximately 3 months prior to presentation. The patient applied a combination of a topical first aid antibiotic ointment, Epsom salts, and hydrogen peroxide without any improvement. A current review of systems was negative.
The patient’s medical history was notable for sarcoidosis diagnosed more than 10 years prior. During this time, he had intermittently been treated elsewhere with low-dose oral prednisone (5 mg once daily), hydroxychloroquine (200 mg twice daily), and an inhaled steroid as needed. He had a history of human immunodeficiency virus–negative, idiopathic CD4 depletion syndrome, which had been complicated by cryptococcal meningitis 7 years prior to presentation. He also had renal insufficiency, with baseline creatinine levels ranging from 1.4 to 1.7 mg/dL (reference range, 0.6–1.2 mg/dL). There was no personal or family history of known or suspected inflammatory bowel disease.
On physical examination, numerous discrete, coalescing, punched out–appearing ulcerations with foul-smelling, greenish yellow, purulent drainage were present bilaterally on the legs (Figure 1). The ulcers had a rolled border with a moderate amount of seemingly nonviable necrotic tissue. A number of hyperpigmented round papules, patches, and plaques also were present on the proximal legs. Laboratory evaluation revealed a CD4 count of 151 cc/mm3 (reference range, 500–1600 cc/mm3) and mildly elevated calcium of 10.7 mg/dL (reference range, 8.2–10.2 mg/dL).
Aerobic, anaerobic, mycobacterial, and fungal cultures of the purulent exudate were obtained. Given a high suspicion for secondary infection of the exogenous wound sites, doxycycline (100 mg twice daily) and topical mupir-ocin were initiated. Gram stain revealed few to moderate polymorphonuclear cells and many gram-positive cocci in pairs, chains, and clusters, along with many gram-negative rods. Bacterial culture grew Pseudomonas aeruginosa, Enterococcus species group G streptococci, and methicillin-resistant Staphylococcus aureus–positive staphylococci. Ciprofloxacin (500 mg twice daily) was then initiated, but the ulcers showed absolutely no clinical improvement and in fact worsened both in number and depth (Figure 2) over subsequent clinic visits during the next 3 months, even after amoxicillin (500 mg 3 times daily) was added. The patient was admitted for treatment with intravenous antibiotics after additional wound cultures revealed fluoroquinolone-resistant Pseudomonas.
Punch biopsies of the ulcers showed nonspecific acute inflammation and tissue necrosis in the active ulcers with nonnecrotizing granulomatous inflammation extending into the deep dermis, with many Langerhans-type giant cells present in the palpable ulcer borders (Figure 3). Neither birefringent particles nor asteroid bodies were observed. Tissue Gram stains did not reveal evidence of bacterial infection. Special stains for acid-fast and fungal organisms (ie, periodic acid–Schiff, Gomori methenamine-silver, Fite, acid-fast bacilli) were similarly negative. Tissue cultures obtained on deep biopsy revealed only rare colonies of P aeruginosa and no isolates on anaerobic, mycobacterial, or fungal cultures. Polymerase chain reaction for mycobacteria and common endemic fungi also was negative. In the absence of infection and considering his history of known sarcoidosis, these histologic features were consistent with ulcerative sarcoidosis. The patient was started on prednisone (60 mg once daily) and hydroxychloroquine (200 mg twice daily). The prednisone was tapered to 20 mg once daily over a 2-year period, at which point 90% of the ulcers had healed. He was continued on hydroxychloroquine at the initial dose, and at a 3-year follow-up his ulcers had healed completely without relapse.
Comment
Ulcerative sarcoidosis is rare, seen worldwide in only 5% of patients with cutaneous sarcoidosis.33 However, cases have been encountered worldwide, with reports emanating from Japan, China, Germany, France, and Russia, among others.6,34-55 We reviewed 34 cases from the English-language literature based on a PubMed search of articles indexed for MEDLINE using the term ulcerative sarcoid and examined patient demographics, clinical presentation, histological findings, treatment type, and outcome. Key references are presented in the Table. Disease prevalence previously has been estimated as being 3-times more common in women than men1; in our literature review, we found a female to male ratio of 3.25 to 1. Additionally, ulcerative sarcoidosis is reported to be twice as common in black versus white individuals.33 In our literature review, when race was reported, 66% (21/32) of patients were black. Disease prevalence has been reported to peak at 20 to 40 years of age.3 In this review, the average age of presentation was 45 years (age range, 24–79 years).
Ulceration may arise de novo but more commonly arises in preexisting scars or cutaneous lesions. There are 2 distinct patterns seen in ulcerative sarcoidosis.4 The first is characterized by ulceration within necrotic yellow plaques.2 The second pattern is characterized by violaceous nodules arising in an annular confluent pattern that eventually ulcerate.4 This presentation commonly mimics or may be mimicked by multiple disease states, including sporotrichosis, tuberculosis, stasis dermatitis with venous ulceration, and even metastatic breast cancer.7,46,55,56 Regardless of presentation, the legs are the most common location of ulcer formation.1,33 In our review, 85% (29/34) of cases presented with involvement of the legs, including our own case. Other locations of ulcer formation have included the face, arms, trunk, and genital area.
On histologic examination of ulcerative sarcoidosis, epithelioid granulomas composed of multinucleated giant cells, histiocytes, and scant numbers of lymphocytes are present.1,3 These formations are the noncaseating granulomas typical of sarcoidosis (Table). All of the cases in our review of the literature were described as either a collection of epithelioid granulomas with giant cell formation or noncaseating granulomas. There also have been reports of atypical features including necrotizing granulomas and granulomatous vasculitis.4,8,9,50 The histologic differential diagnosis in this case also would primarily include an infectious granulomatous process and less so an id reaction, rosacea, a paraneoplastic phenomenon, foreign body granulomas, and metastatic Crohn disease. The presence of ulceration, the large number of lesions, and the anatomic distribution help rule out most of these alternate diagnostic considerations. Diligent extensive workup was done in our patient to insure it was not an infection.
The goals of treatment include symptomatic relief, improvement in objective parameters of disease activity, and prevention of disease progression and subsequent disability.33,57 Fortunately, the majority of sarcoidosis patients with cutaneous symptoms achieve full recovery within months to years.33 Our literature review indicated that 81% (22/27) of patients with ulcerative lesions experienced full resolution within 1 year of treatment. Of those that did not (19% [5/27]), the patients were either lost to follow-up or died from other complications of sarcoidosis.
The widely accepted standard therapy for cutaneous sarcoidosis includes topical, intralesional, and systemic corticosteroids; antimalarials; and methotrexate.33,57 Steroids and methotrexate act by suppressing granuloma formation, while antimalarials prevent antigen presentation (presumably part of the pathogenesis).33 For mild to moderate disease, topical and intralesional steroids may be all that is necessary.33,57 Systemic steroids are used for disfiguring, destructive, and widespread lesions that have been refractory to local and other systemic therapies.33,57 Steroids are tapered gradually depending on the patient’s response, as it is common for patients to relapse below a certain dose.33,57 Antimalarials (chloroquine or hydroxychloroquine) and methotrexate are considered adjunct treatments for patients who are either steroid unresponsive or who are unable to tolerate corticosteroid treatment due to adverse events.33,57
Standard therapy is complicated by the side effects of treatment. Use of corticosteroids may lead to gastrointestinal tract upset, increased appetite, mood disturbances, impaired wound healing, hyperglycemia, hypertension, cushingoid features, and acne.57 Antimalarials can cause nausea, anorexia, and agranulocytosis, and chloroquine therapy in particular can lead to blurred vision, corneal deposits, and central retinopathy.33,57 Methotrexate is associated with hematologic, gastrointestinal tract, pulmonary, and hepatic toxicities well known to most practitioners.
Because of the variable clinical response of patients to standard therapy and their associated toxicities, other treatment options have been used including pentoxifylline, tetracyclines, isotretinoin, leflunomide, thalidomide, infliximab, adalimumab, allopurinol, and the pulsed dye or CO2 laser.10,33,57 In nonhealing ulcers, split-thickness grafting and a bilayered bioengineered skin substitute have been used with good results in conjunction with ongoing systemic therapy.11,47 Additionally, nanoparticle silver burn paste has been used successfully, with resolution of ulcers within 2 weeks in the Chinese literature.53
All of these treatment recommendations are based on historically accepted modalities. Controlled trials with longitudinal follow-up are needed to provide justification for the current standard of care.34
- Howard A, White CR Jr. Non-infectious granulomas. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. 2nd ed. Spain: Elsevier; 2008:1421-1435.
- Doherty CB, Rosen T. Evidence-based therapy for cutaneous sarcoidosis. Drugs. 2008;68:1361-1383.
- Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25:295-302.
- Noiles K, Beleznay K, Crawford RI, et al. Sarcoidosis can present with necrotizing granulomas histologically: two cases of ulcerated sarcoidosis and review of the literature. J Cutan Med Surg. 2013;17:377-378.
- Mitchell IC, Sweatman MC, Rustin MH, et al. Ulcerative and hypopigmented sarcoidosis. J Am Acad Dermatol. 1986;15:1062-1065.
- Yoo SS, Mimouni D, Nikolskaia OV, et al. Clinicopathologic features of ulcerative-atrophic sarcoidosis. Int J Dermatol. 2004;43:108-112.
- Joshi SS, Romanelli R, Kirsner RS. Sarcoidosis mimicking a venous ulcer: a case report. Ostomy Wound Manage. 2009;55:46-48.
- Petri M, Barr E, Cho K, et al. Overlap of granulomatous vasculitis and sarcoidosis: presentation with uveitis, eosinophilia, leg ulcers, sinusitis and past foot drop. J Rheumatol. 1988;15:1171-1173.
- Poonawalla T, Colome-Grimmer MI, Kelly B. Ulcerative sarcoidosis in the legs with granulomatous vasculitis. Clin Exp Dermatol. 2008;33:282-286.
- Philips MA, Lynch J, Azmi FH. Ulcerative sarcoidosis responding to adalimumab. J Am Acad Dermatol. 2005;53:917.
- Collison DW, Novice F, Banse L, et al. Split-thickness skin grafting in extensive ulcerative sarcoidosis. J Dermatol Surg Oncol. 1989;15:679-683.
- Hunt RD, Gonzalez ME, Robinson M, et al. Ulcerative sarcoidosis. Dermatol Online J. 2012;18:29.
- Green JJ, Lawrence N, Heymann WR. Generalized ulcerative sarcoidosis induced by therapy with the flashlamp-pumped pulsed dye. Arch Dermatol. 2001;137:507-508.
- Albertini JG, Tyler W, Miller OF. Ulcerative sarcoidosis. case report and review of the literature. Arch Dermatol. 1997;133:215-219.
- Thomas J, Williams DW. Peritoneal involvement and ulcerative skin plaques in sarcoidosis: a case report. Sarcoidosis. 1989;6:161-162.
- Verdegem TD, Sharma OP. Cutaneous ulcers in sarcoidosis. Arch Dermatol. 1987;123:1531-1534.
- Gupta AK, Haberman HF, From GL, et al. Sarcoidosis with extensive cutaneous ulceration. unusual clinical presentation. Dermatologica. 1987;174:135-139.
- Hruza GJ, Kerdel FA. Generalized atrophic sarcoidosis with ulcerations. Arch Dermatol. 1986;122:320-322.
- Muhlemann MF, Walker NP, Tan LB, et al. Elephantine sarcoidosis presenting as ulcerating lymphoedema. J R Soc Med. 1985;78:260-261.
- Neill SM, Smith NP, Eady RA. Ulcerative sarcoidosis: a rare manifestation of a common disease. Clin Exp Dermatol. 1984;9:277-279.
- Saxe N, Benatar SR, Bok L, et al. Sarcoidosis with leg ulcers and annular facial lesions. Arch Dermatol. 1984;120:93-96.
- Schwartz RA, Robertson DB, Tierney LM, et al. Generalized ulcerative sarcoidosis. Arch Dermatol. 1982;118:931-933.
- Boyd RE, Andrews BS. Sarcoidosis presenting as cutaneous ulceration, subcutaneous nodules and chronic arthritis. J Rheumatol. 1981;8:311-316.
- Herzlinger DC, Marland AM, Barr RJ. Verrucous ulcerative skin lesions in sarcoidosis. an unusual clinical presentation. Cutis. 1979;23:569-572.
- Meyers M, Barsky S. Ulcerative sarcoidosis. Arch Dermatol. 1978;114:447.
- Schiffner J, Sharma OP. Ulcerative sarcoidosis. report of an unusual case. Arch Dermatol. 1977;113:676-677.
- Williamson DM. Sarcoidosis with atrophic lesions and ulcers of the legs. Br J Dermatol. 1971;84:92-93.
- Bazex A, Dupre A, Christol B, et al. Sarcoidosis with atrophic lesions and ulcers and the presence in some sarcoid granulomata of orceinophil fibres. Br J Dermatol. 1970;83:255-262.
- Brodkin RH. Leg ulcers. a report of two cases caused by sarcoidosis. Acta Derm Venereol. 1969;49:584-587.
- Simpson JR. Sarcoidosis with erythrodermia and ulceration. Br J Dermatol. 1963;75:193-198.
- Irgang S. Ulcerative cutaneous lesion in sarcoidosis; report of a case with clinical resemblance to lupus vulgaris. Harlem Hosp Bull. 1956;8:134-139.
- Irgang S. Ulcerative cutaneous lesions in sarcoidosis; report of a case with clinical resemblance to papulonecrotic tuberculide. Br J Dermatol. 1955;67:255-260.
- Hoffman MD. Atypical ulcers. Dermatol Ther. 2013;26:222-235.
- Hopf B, Krebs A. Ulcera cruris as a rare manifestation of sarcoidosis. Dermatologica. 1974;113:55-62.
- Metz J, Hartmann A, Hautkr Z. Ulcerative form of skin sarcoidosis. Z Hautkr. 1977;52:890-896.
- Berenbeĭn BA, Malygina LA, Tiutiunnikova IA. Ulcerative form of skin sarcoidosis [in Russian]. Vestn Dermatol Venerol. 1984;4:50-53.
- Takahashi N, Hoshino M, Takase T, et al. A case of ulcerative sarcoidosis [in Japanese]. Nihon Hifuka Gakkai Zasshi. 1985;95:1049-1054.
- Schamroth JM. Sarcoidosis with severe extensive skin ulceration. Int J Dermatol. 1985;24:451-452.
- Porteau L, Dromer C, Le Guennec P, et al. Ulcer lesions in sarcoidosis: apropos of a case [in French]. Ann Med Interne (Paris). 1997;148:105-106.
- de La Blanchardière A, Bachmeyer C, Toutous L, et al. Cutaneous ulcerations in sarcoidosis [in French]. Rev Med Interne. 1995;16:927-929.
- Mitsuishi T, Nogita T, Kawashima M. Psoriasiform sarcoidosis with ulceration. Int J Dermatol. 1992;31:339-340.
- Rodionov AN, Samtsov AV. The ulcerative form of skin sarcoidosis [in Russian]. Vestn Dermatol Venerol. 1990;7:68-71.
- Jacyk WK. Cutaneous sarcoidosis in black South Africans. Int J Dermatol. 1999;38:841-845.
- Gungor E, Artuz F, Alli N, et al. Ulcerative sarcoidosis. J Eur Acad Dermatol Venereol. 1999;12:78-79.
- Schleinitz N, Luc M, Genot S, et al. Ulcerative cutaneous lesions: a rare manifestation of sarcoidosis [in French]. Rev Med Interne. 2005;26:758-759.
- Klocker J, Duckers J, Morse R, et al. Ulcerative cutaneous sarcoidosis masquerading as metastatic carcinoma of the breast. Age Ageing. 2002;31:77-79.
- Streit M, Bohlen LM, Braathen LR. Ulcerative sarcoidosis successfully treated with apligraf. Dermatology. 2001;202:367-370.
- Ichiki Y, Kitajima Y. Ulcerative sarcoidosis: case report and review of the Japanese literature. Acta Derm Venereol. 2008;88:526-528
- Meyersburg D, Schön MP, Bertsch HP, et al. Uncommon cutaneous ulcerative and systemic sarcoidosis. successful treatment with hydroxychloroquine and compression therapy [in German]. Hautarzt. 2011;62:691-695.
- Wei CH, Huang YH, Shih YC, et al. Sarcoidosis with cutaneous granulomatous vasculitis. Australas J Dermatol. 2010;51:198-201.
- Kluger N, Girard C, Durand L, et al. Leg ulcers revealing systemic sarcoidosis with splenomegaly and thrombocytopenia. Int J Dermatol. 2013;52:1425-1427.
- Jun L, Jia-Wei L, Hong-Zhong J. Ulcerative sarcoidosis. Int J Dermatol. 2014;53:E315-E316.
- Chen JH, Wang TT, Lin ZQ. Successful application of a novel dressing for the treatment of ulcerative cutaneous sarcoidosis. Chin Med J. 2013;126:3400.
- Ri G, Yoshikawa E, Shigekiyo T, et al. Takayasu artertitis and ulcerative sarcoidosis. Intern Med. 2015;54:1075-1080.
- Spiliopoulou I, Foka A, Bounas A, et al. Mycobacterium kansasii cutaneous infection in a patient with sarcoidosis treated with anti-TNF agents. Acta Clin Belg. 2014;69:229-231.
- Yang DJ, Krishnan RS, Guillen DR, et al. Disseminated sporotrichosis mimicking sarcoidosis. Int J Dermatol. 2006;45:450-453.
- Badgwell C, Rosen T. Cutaneous sarcoidosis therapy updated. J Am Acad Dermatol. 2007;56:69-83.
- Howard A, White CR Jr. Non-infectious granulomas. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. 2nd ed. Spain: Elsevier; 2008:1421-1435.
- Doherty CB, Rosen T. Evidence-based therapy for cutaneous sarcoidosis. Drugs. 2008;68:1361-1383.
- Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25:295-302.
- Noiles K, Beleznay K, Crawford RI, et al. Sarcoidosis can present with necrotizing granulomas histologically: two cases of ulcerated sarcoidosis and review of the literature. J Cutan Med Surg. 2013;17:377-378.
- Mitchell IC, Sweatman MC, Rustin MH, et al. Ulcerative and hypopigmented sarcoidosis. J Am Acad Dermatol. 1986;15:1062-1065.
- Yoo SS, Mimouni D, Nikolskaia OV, et al. Clinicopathologic features of ulcerative-atrophic sarcoidosis. Int J Dermatol. 2004;43:108-112.
- Joshi SS, Romanelli R, Kirsner RS. Sarcoidosis mimicking a venous ulcer: a case report. Ostomy Wound Manage. 2009;55:46-48.
- Petri M, Barr E, Cho K, et al. Overlap of granulomatous vasculitis and sarcoidosis: presentation with uveitis, eosinophilia, leg ulcers, sinusitis and past foot drop. J Rheumatol. 1988;15:1171-1173.
- Poonawalla T, Colome-Grimmer MI, Kelly B. Ulcerative sarcoidosis in the legs with granulomatous vasculitis. Clin Exp Dermatol. 2008;33:282-286.
- Philips MA, Lynch J, Azmi FH. Ulcerative sarcoidosis responding to adalimumab. J Am Acad Dermatol. 2005;53:917.
- Collison DW, Novice F, Banse L, et al. Split-thickness skin grafting in extensive ulcerative sarcoidosis. J Dermatol Surg Oncol. 1989;15:679-683.
- Hunt RD, Gonzalez ME, Robinson M, et al. Ulcerative sarcoidosis. Dermatol Online J. 2012;18:29.
- Green JJ, Lawrence N, Heymann WR. Generalized ulcerative sarcoidosis induced by therapy with the flashlamp-pumped pulsed dye. Arch Dermatol. 2001;137:507-508.
- Albertini JG, Tyler W, Miller OF. Ulcerative sarcoidosis. case report and review of the literature. Arch Dermatol. 1997;133:215-219.
- Thomas J, Williams DW. Peritoneal involvement and ulcerative skin plaques in sarcoidosis: a case report. Sarcoidosis. 1989;6:161-162.
- Verdegem TD, Sharma OP. Cutaneous ulcers in sarcoidosis. Arch Dermatol. 1987;123:1531-1534.
- Gupta AK, Haberman HF, From GL, et al. Sarcoidosis with extensive cutaneous ulceration. unusual clinical presentation. Dermatologica. 1987;174:135-139.
- Hruza GJ, Kerdel FA. Generalized atrophic sarcoidosis with ulcerations. Arch Dermatol. 1986;122:320-322.
- Muhlemann MF, Walker NP, Tan LB, et al. Elephantine sarcoidosis presenting as ulcerating lymphoedema. J R Soc Med. 1985;78:260-261.
- Neill SM, Smith NP, Eady RA. Ulcerative sarcoidosis: a rare manifestation of a common disease. Clin Exp Dermatol. 1984;9:277-279.
- Saxe N, Benatar SR, Bok L, et al. Sarcoidosis with leg ulcers and annular facial lesions. Arch Dermatol. 1984;120:93-96.
- Schwartz RA, Robertson DB, Tierney LM, et al. Generalized ulcerative sarcoidosis. Arch Dermatol. 1982;118:931-933.
- Boyd RE, Andrews BS. Sarcoidosis presenting as cutaneous ulceration, subcutaneous nodules and chronic arthritis. J Rheumatol. 1981;8:311-316.
- Herzlinger DC, Marland AM, Barr RJ. Verrucous ulcerative skin lesions in sarcoidosis. an unusual clinical presentation. Cutis. 1979;23:569-572.
- Meyers M, Barsky S. Ulcerative sarcoidosis. Arch Dermatol. 1978;114:447.
- Schiffner J, Sharma OP. Ulcerative sarcoidosis. report of an unusual case. Arch Dermatol. 1977;113:676-677.
- Williamson DM. Sarcoidosis with atrophic lesions and ulcers of the legs. Br J Dermatol. 1971;84:92-93.
- Bazex A, Dupre A, Christol B, et al. Sarcoidosis with atrophic lesions and ulcers and the presence in some sarcoid granulomata of orceinophil fibres. Br J Dermatol. 1970;83:255-262.
- Brodkin RH. Leg ulcers. a report of two cases caused by sarcoidosis. Acta Derm Venereol. 1969;49:584-587.
- Simpson JR. Sarcoidosis with erythrodermia and ulceration. Br J Dermatol. 1963;75:193-198.
- Irgang S. Ulcerative cutaneous lesion in sarcoidosis; report of a case with clinical resemblance to lupus vulgaris. Harlem Hosp Bull. 1956;8:134-139.
- Irgang S. Ulcerative cutaneous lesions in sarcoidosis; report of a case with clinical resemblance to papulonecrotic tuberculide. Br J Dermatol. 1955;67:255-260.
- Hoffman MD. Atypical ulcers. Dermatol Ther. 2013;26:222-235.
- Hopf B, Krebs A. Ulcera cruris as a rare manifestation of sarcoidosis. Dermatologica. 1974;113:55-62.
- Metz J, Hartmann A, Hautkr Z. Ulcerative form of skin sarcoidosis. Z Hautkr. 1977;52:890-896.
- Berenbeĭn BA, Malygina LA, Tiutiunnikova IA. Ulcerative form of skin sarcoidosis [in Russian]. Vestn Dermatol Venerol. 1984;4:50-53.
- Takahashi N, Hoshino M, Takase T, et al. A case of ulcerative sarcoidosis [in Japanese]. Nihon Hifuka Gakkai Zasshi. 1985;95:1049-1054.
- Schamroth JM. Sarcoidosis with severe extensive skin ulceration. Int J Dermatol. 1985;24:451-452.
- Porteau L, Dromer C, Le Guennec P, et al. Ulcer lesions in sarcoidosis: apropos of a case [in French]. Ann Med Interne (Paris). 1997;148:105-106.
- de La Blanchardière A, Bachmeyer C, Toutous L, et al. Cutaneous ulcerations in sarcoidosis [in French]. Rev Med Interne. 1995;16:927-929.
- Mitsuishi T, Nogita T, Kawashima M. Psoriasiform sarcoidosis with ulceration. Int J Dermatol. 1992;31:339-340.
- Rodionov AN, Samtsov AV. The ulcerative form of skin sarcoidosis [in Russian]. Vestn Dermatol Venerol. 1990;7:68-71.
- Jacyk WK. Cutaneous sarcoidosis in black South Africans. Int J Dermatol. 1999;38:841-845.
- Gungor E, Artuz F, Alli N, et al. Ulcerative sarcoidosis. J Eur Acad Dermatol Venereol. 1999;12:78-79.
- Schleinitz N, Luc M, Genot S, et al. Ulcerative cutaneous lesions: a rare manifestation of sarcoidosis [in French]. Rev Med Interne. 2005;26:758-759.
- Klocker J, Duckers J, Morse R, et al. Ulcerative cutaneous sarcoidosis masquerading as metastatic carcinoma of the breast. Age Ageing. 2002;31:77-79.
- Streit M, Bohlen LM, Braathen LR. Ulcerative sarcoidosis successfully treated with apligraf. Dermatology. 2001;202:367-370.
- Ichiki Y, Kitajima Y. Ulcerative sarcoidosis: case report and review of the Japanese literature. Acta Derm Venereol. 2008;88:526-528
- Meyersburg D, Schön MP, Bertsch HP, et al. Uncommon cutaneous ulcerative and systemic sarcoidosis. successful treatment with hydroxychloroquine and compression therapy [in German]. Hautarzt. 2011;62:691-695.
- Wei CH, Huang YH, Shih YC, et al. Sarcoidosis with cutaneous granulomatous vasculitis. Australas J Dermatol. 2010;51:198-201.
- Kluger N, Girard C, Durand L, et al. Leg ulcers revealing systemic sarcoidosis with splenomegaly and thrombocytopenia. Int J Dermatol. 2013;52:1425-1427.
- Jun L, Jia-Wei L, Hong-Zhong J. Ulcerative sarcoidosis. Int J Dermatol. 2014;53:E315-E316.
- Chen JH, Wang TT, Lin ZQ. Successful application of a novel dressing for the treatment of ulcerative cutaneous sarcoidosis. Chin Med J. 2013;126:3400.
- Ri G, Yoshikawa E, Shigekiyo T, et al. Takayasu artertitis and ulcerative sarcoidosis. Intern Med. 2015;54:1075-1080.
- Spiliopoulou I, Foka A, Bounas A, et al. Mycobacterium kansasii cutaneous infection in a patient with sarcoidosis treated with anti-TNF agents. Acta Clin Belg. 2014;69:229-231.
- Yang DJ, Krishnan RS, Guillen DR, et al. Disseminated sporotrichosis mimicking sarcoidosis. Int J Dermatol. 2006;45:450-453.
- Badgwell C, Rosen T. Cutaneous sarcoidosis therapy updated. J Am Acad Dermatol. 2007;56:69-83.
Practice Points
- Sarcoidosis can present as a primary ulcerative disease.
- Suspect ulcerative sarcoidosis when ulcerations are seen on the leg.
- Systemic corticosteroids may be the most effective treatment of ulcerative sarcoidosis.
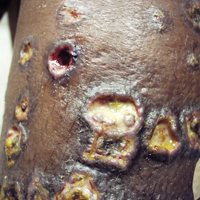
Drug-eluting balloon is as good as drug-eluting stent for in-stent restenosis
DENVER – Treatment of coronary in-stent restenosis using a paclitaxel-eluting balloon proved noninferior to an everolimus-eluting stent in terms of minimal lumen diameter at 6 months in the DARE trial, Jose P.S. Henriques, MD, reported at the Transcatheter Cardiovascular Therapeutics annual meeting.
The two forms of device therapy also yielded similar rates of adverse clinical events, including target vessel revascularization, at 12 months, he said.
The DARE (Drug-Eluting Balloon for In-Stent Restenosis) trial included 278 patients with in-stent restenosis (ISR) randomized to the SeQuent Please paclitaxel-eluting balloon or Xience everolimus-eluting stent at high-volume Dutch percutaneous coronary intervention centers. The trial was unique in that it included a mix of patients with in-stent restenosis involving DES and bare-metal stents. Indeed, 44% of participants had ISR in a bare-metal stent. These older-model stents are still used in patients who require a shorter duration of dual-antiplatelet therapy, so the DARE population reflects real-world clinical practice better than do prior studies restricted to ISR in only one stent type or the other, according to the cardiologist.
The primary outcome in this noninferiority trial was the in-segment minimal lumen diameter at 6-month angiographic follow-up. The mean diameter was 1.71 mm in the drug-eluting balloon (DEB) group and closely similar at 1.74 mm in the DES group. There was greater acute gain with the drug-eluting stent, but it was canceled out by greater late loss by 6 months.
Moreover, the 12-month composite clinical event rate composed of death, target vessel MI, and target vessel revascularization was 10.9% in the DEB recipients and 9.2% with the DES, a nonsignificant difference. Of note, target vessel revascularization occurred in 8.8% of the DEB group and was similar at 7.1% in the DES recipients, although the DARE trial wasn’t powered to detect differences in clinical events.
These results confirm the European Society of Cardiology’s class 1A recommendation for DEB as well as DES for ISR, Dr. Henriques said at the meeting sponsored by the Cardiovascular Research Foundation.
U.S. guidelines don’t address DEB for the treatment of coronary ISR. That’s because the devices, which have long been available in Europe, aren’t approved for use in the coronary tree in the United States. They are available in the United States only for treatment of peripheral vascular disease. And no U.S. clinical trials of DEBs in the coronary tree are planned.
“I wish the U.S. Food and Drug Administration was listening to the DARE results because we really would like to see this technology in the U.S.,” said Roxana Mehran, MD, who moderated a press conference where the DARE findings were highlighted.
David J. Cohen, MD, director of cardiovascular research at Saint Luke’s Mid America Heart Institute in Kansas City, Mo., commented, “This type of device, obviously with it being similar in performance to drug-eluting stents, would be a very welcome addition to our armamentarium, because one of the things I don’t like to do as a coronary interventionalist is to line up multiple stents inside each other.”
“Making club sandwiches out of patients’ arteries with stent after stent is not a good idea. We know that,” added Dr. Mehran, professor of medicine and director of interventional cardiovascular research and clinical trials at Mount Sinai School of Medicine in New York.
Cindy L. Grines, MD, chair of cardiology at the Hofstra Northwell School of Medicine in Hempstead, N.Y., said DEBs “would absolutely be welcome” if they were available to cardiologists in the United States.
“When you have repeated episodes of in-stent restenosis, you can start with a vessel that’s 3 mm in diameter; then when it restenoses and you place a second stent inside there, all of a sudden – even if you have a great stent result – you can be down to 2.25 mm. And then the next time you need to treat it for restenosis, you’re down to a very tiny lumen. That’s the big problem with trying to treat in-stent restenosis with more stents,” she explained.
The DEBs are expensive, and ISR has become so uncommon with the use of the current generation of drug-eluting stents that the device companies have little incentive to do the studies required to be able to market DEBs in the United States.
“I think the FDA should consider in-stent restenosis as an orphan disease. We really should be able to get a drug-eluting balloon approved in this country based on the data over in Europe,” Dr. Grines said.
Dr. Henriques reported receiving research grants from B. Braun, which markets the paclitaxel-eluting stent in Europe, as well as from Abbott Vascular.
DENVER – Treatment of coronary in-stent restenosis using a paclitaxel-eluting balloon proved noninferior to an everolimus-eluting stent in terms of minimal lumen diameter at 6 months in the DARE trial, Jose P.S. Henriques, MD, reported at the Transcatheter Cardiovascular Therapeutics annual meeting.
The two forms of device therapy also yielded similar rates of adverse clinical events, including target vessel revascularization, at 12 months, he said.
The DARE (Drug-Eluting Balloon for In-Stent Restenosis) trial included 278 patients with in-stent restenosis (ISR) randomized to the SeQuent Please paclitaxel-eluting balloon or Xience everolimus-eluting stent at high-volume Dutch percutaneous coronary intervention centers. The trial was unique in that it included a mix of patients with in-stent restenosis involving DES and bare-metal stents. Indeed, 44% of participants had ISR in a bare-metal stent. These older-model stents are still used in patients who require a shorter duration of dual-antiplatelet therapy, so the DARE population reflects real-world clinical practice better than do prior studies restricted to ISR in only one stent type or the other, according to the cardiologist.
The primary outcome in this noninferiority trial was the in-segment minimal lumen diameter at 6-month angiographic follow-up. The mean diameter was 1.71 mm in the drug-eluting balloon (DEB) group and closely similar at 1.74 mm in the DES group. There was greater acute gain with the drug-eluting stent, but it was canceled out by greater late loss by 6 months.
Moreover, the 12-month composite clinical event rate composed of death, target vessel MI, and target vessel revascularization was 10.9% in the DEB recipients and 9.2% with the DES, a nonsignificant difference. Of note, target vessel revascularization occurred in 8.8% of the DEB group and was similar at 7.1% in the DES recipients, although the DARE trial wasn’t powered to detect differences in clinical events.
These results confirm the European Society of Cardiology’s class 1A recommendation for DEB as well as DES for ISR, Dr. Henriques said at the meeting sponsored by the Cardiovascular Research Foundation.
U.S. guidelines don’t address DEB for the treatment of coronary ISR. That’s because the devices, which have long been available in Europe, aren’t approved for use in the coronary tree in the United States. They are available in the United States only for treatment of peripheral vascular disease. And no U.S. clinical trials of DEBs in the coronary tree are planned.
“I wish the U.S. Food and Drug Administration was listening to the DARE results because we really would like to see this technology in the U.S.,” said Roxana Mehran, MD, who moderated a press conference where the DARE findings were highlighted.
David J. Cohen, MD, director of cardiovascular research at Saint Luke’s Mid America Heart Institute in Kansas City, Mo., commented, “This type of device, obviously with it being similar in performance to drug-eluting stents, would be a very welcome addition to our armamentarium, because one of the things I don’t like to do as a coronary interventionalist is to line up multiple stents inside each other.”
“Making club sandwiches out of patients’ arteries with stent after stent is not a good idea. We know that,” added Dr. Mehran, professor of medicine and director of interventional cardiovascular research and clinical trials at Mount Sinai School of Medicine in New York.
Cindy L. Grines, MD, chair of cardiology at the Hofstra Northwell School of Medicine in Hempstead, N.Y., said DEBs “would absolutely be welcome” if they were available to cardiologists in the United States.
“When you have repeated episodes of in-stent restenosis, you can start with a vessel that’s 3 mm in diameter; then when it restenoses and you place a second stent inside there, all of a sudden – even if you have a great stent result – you can be down to 2.25 mm. And then the next time you need to treat it for restenosis, you’re down to a very tiny lumen. That’s the big problem with trying to treat in-stent restenosis with more stents,” she explained.
The DEBs are expensive, and ISR has become so uncommon with the use of the current generation of drug-eluting stents that the device companies have little incentive to do the studies required to be able to market DEBs in the United States.
“I think the FDA should consider in-stent restenosis as an orphan disease. We really should be able to get a drug-eluting balloon approved in this country based on the data over in Europe,” Dr. Grines said.
Dr. Henriques reported receiving research grants from B. Braun, which markets the paclitaxel-eluting stent in Europe, as well as from Abbott Vascular.
DENVER – Treatment of coronary in-stent restenosis using a paclitaxel-eluting balloon proved noninferior to an everolimus-eluting stent in terms of minimal lumen diameter at 6 months in the DARE trial, Jose P.S. Henriques, MD, reported at the Transcatheter Cardiovascular Therapeutics annual meeting.
The two forms of device therapy also yielded similar rates of adverse clinical events, including target vessel revascularization, at 12 months, he said.
The DARE (Drug-Eluting Balloon for In-Stent Restenosis) trial included 278 patients with in-stent restenosis (ISR) randomized to the SeQuent Please paclitaxel-eluting balloon or Xience everolimus-eluting stent at high-volume Dutch percutaneous coronary intervention centers. The trial was unique in that it included a mix of patients with in-stent restenosis involving DES and bare-metal stents. Indeed, 44% of participants had ISR in a bare-metal stent. These older-model stents are still used in patients who require a shorter duration of dual-antiplatelet therapy, so the DARE population reflects real-world clinical practice better than do prior studies restricted to ISR in only one stent type or the other, according to the cardiologist.
The primary outcome in this noninferiority trial was the in-segment minimal lumen diameter at 6-month angiographic follow-up. The mean diameter was 1.71 mm in the drug-eluting balloon (DEB) group and closely similar at 1.74 mm in the DES group. There was greater acute gain with the drug-eluting stent, but it was canceled out by greater late loss by 6 months.
Moreover, the 12-month composite clinical event rate composed of death, target vessel MI, and target vessel revascularization was 10.9% in the DEB recipients and 9.2% with the DES, a nonsignificant difference. Of note, target vessel revascularization occurred in 8.8% of the DEB group and was similar at 7.1% in the DES recipients, although the DARE trial wasn’t powered to detect differences in clinical events.
These results confirm the European Society of Cardiology’s class 1A recommendation for DEB as well as DES for ISR, Dr. Henriques said at the meeting sponsored by the Cardiovascular Research Foundation.
U.S. guidelines don’t address DEB for the treatment of coronary ISR. That’s because the devices, which have long been available in Europe, aren’t approved for use in the coronary tree in the United States. They are available in the United States only for treatment of peripheral vascular disease. And no U.S. clinical trials of DEBs in the coronary tree are planned.
“I wish the U.S. Food and Drug Administration was listening to the DARE results because we really would like to see this technology in the U.S.,” said Roxana Mehran, MD, who moderated a press conference where the DARE findings were highlighted.
David J. Cohen, MD, director of cardiovascular research at Saint Luke’s Mid America Heart Institute in Kansas City, Mo., commented, “This type of device, obviously with it being similar in performance to drug-eluting stents, would be a very welcome addition to our armamentarium, because one of the things I don’t like to do as a coronary interventionalist is to line up multiple stents inside each other.”
“Making club sandwiches out of patients’ arteries with stent after stent is not a good idea. We know that,” added Dr. Mehran, professor of medicine and director of interventional cardiovascular research and clinical trials at Mount Sinai School of Medicine in New York.
Cindy L. Grines, MD, chair of cardiology at the Hofstra Northwell School of Medicine in Hempstead, N.Y., said DEBs “would absolutely be welcome” if they were available to cardiologists in the United States.
“When you have repeated episodes of in-stent restenosis, you can start with a vessel that’s 3 mm in diameter; then when it restenoses and you place a second stent inside there, all of a sudden – even if you have a great stent result – you can be down to 2.25 mm. And then the next time you need to treat it for restenosis, you’re down to a very tiny lumen. That’s the big problem with trying to treat in-stent restenosis with more stents,” she explained.
The DEBs are expensive, and ISR has become so uncommon with the use of the current generation of drug-eluting stents that the device companies have little incentive to do the studies required to be able to market DEBs in the United States.
“I think the FDA should consider in-stent restenosis as an orphan disease. We really should be able to get a drug-eluting balloon approved in this country based on the data over in Europe,” Dr. Grines said.
Dr. Henriques reported receiving research grants from B. Braun, which markets the paclitaxel-eluting stent in Europe, as well as from Abbott Vascular.
AT TCT 2017
Key clinical point:
Major finding: The mean 6-month in-segment minimal lumen diameter following treatment of in-stent restenosis with a paclitaxel-eluting balloon was 1.71 mm and was similar at 1.74 mm in patients treated using an everolimus-eluting stent.
Data source: A prospective, multicenter Dutch randomized trial including 278 patients with in-stent restenosis.
Disclosures: The study was sponsored by the University of Amsterdam and financially supported by a research grant from B. Braun. The presenter reported receiving research grants from that company and Abbott Vascular.
In MI with cardiogenic shock, PCI of only culprit lesions is safer
In patients with acute myocardial infarction and multivessel coronary artery disease with cardiogenic shock, 30-day rates of death and renal-replacement therapy were lower when patients underwent percutaneous coronary intervention (PCI) of the culprit lesion as opposed to multivessel PCI.
The difference appeared to be driven by mortality, as the renal endpoint alone was not significant.
As many as 80% of patients with cardiogenic shock also present with multivessel coronary artery disease, and this is associated with worse mortality. It is unclear whether immediate PCI of clinically important stenoses of major nonculprit coronary arteries is of benefit, and previous randomized trials comparing the procedures did not look at patients with cardiogenic shock, Holger Thiele, MD, reported at the Transcatheter Cardiovascular Therapeutics annual educational meeting.
European guidelines suggest that PCI of nonculprit lesions should be considered in patients with cardiogenic shock, while U.S. guidelines offer no opinion, but recent appropriate use criteria recommend revascularization of a nonculprit artery if cardiogenic shock continues after the culprit artery has been repaired. It is thought that immediate revascularization of all coronary arteries with clinically important stenoses might improve overall myocardial perfusion and function in patients with cardiogenic shock, but the procedure could also have drawbacks, including additional ischemia, volume overload, and renal impairment from higher doses of contrast material.
To better understand outcomes in these patients, the Culprit Lesion Only PCI versus Multivessel PCI in Cardiogenic Shock (CULPRIT-SHOCK) trial randomized 706 patients to culprit-only PCI or multivessel PCI, in which PCI was performed on all major coronary arteries with more than 70% stenosis. Patients receiving culprit-only PCI could also undergo optional staged revascularization due to residual ischemic lesions, symptoms, or clinical or neurologic status.
At 30 days, death and/or renal-replacement therapy occurred in 45.9% of the culprit-only group, compared to 55.4% in the multivessel group (relative risk, 0.83; 95% confidence interval, 0.71-0.96; P = .01). A per-protocol analysis showed similar results (RR, 0.81; 95% CI, 0.69-0.96; P =.01), as did an analysis of the as-treated population (RR, 0.83; 95% CI, 0.72-0.97; P = .02).
All-cause mortality was lower in the culprit-only group (43.3% versus 51.6%; RR, 0.84; 95% CI, 0.72-0.98; P=.03). The rate of renal-replacement therapy was higher in the multivessel group (16.4% versus 11.6%), but this did not reach statistical significance (P = .07).
There were no statistically significant differences between the two groups with respect to recurrent myocardial infarction, rehospitalization for heart failure, bleeding, or stroke, Dr. Thiele reported at the meeting, which was sponsored by the Cardiovascular Research Foundation.
Some limitations of the study included its unblinded nature, and the fact that 75 patients originally assigned to one treatment category crossed over to the other, including 14 in the culprit-lesion only category who underwent immediate multivessel PCI. This suggests that treatment strategy may need to be adopted to a patient’s clinical circumstances.
The CULPRIT-SHOCK results were published online at the time of Dr. Thiele’s presentation (N Engl J Med. 2017 Oct 30. doi:10/056/NEJMoa1710261).
Several of the study’s authors reported financial ties to the pharmaceutical industry.
This study’s findings reinforce those of previous trials that had suggested that multivessel percutaneous coronary intervention has higher early mortality than culprit-lesion-only PCI.
The study provides compelling evidence that culprit-lesion-only PCI should be the preferred treatment choice over multivessel PCI in patients with cardiogenic shock.
A previous meta-analysis of patients with uncomplicated ST-segment elevation myocardial infarction showed lower rates of mortality or MI with initial multivessel PCI. The disagreement between the two studies suggests that patients with cardiogenic shock may experience greater risk of these adverse outcomes during multivessel PCI procedures.
Future clinical trials should test individual multivessel revascularization strategies to reduce mortality in MI patients with cardiogenic shock, such as coronary artery bypass grafting (CABG) and venoarterial extracorporeal membrane oxygenation (ECMO).
Judith Hochman, MD, and Stuart Katz, MD, of New York University Langone Health, made these comments in an accompanying editorial (N Engl J Med. 2017 Oct 30. doi: 10.1056/nejme1713341). Dr. Hochman had no relevant disclosures. Dr. Katz has consulted for Novartis, Amgen, and Regeneron, and has received funding from Amgen, American Regent, and Janssen.
This study’s findings reinforce those of previous trials that had suggested that multivessel percutaneous coronary intervention has higher early mortality than culprit-lesion-only PCI.
The study provides compelling evidence that culprit-lesion-only PCI should be the preferred treatment choice over multivessel PCI in patients with cardiogenic shock.
A previous meta-analysis of patients with uncomplicated ST-segment elevation myocardial infarction showed lower rates of mortality or MI with initial multivessel PCI. The disagreement between the two studies suggests that patients with cardiogenic shock may experience greater risk of these adverse outcomes during multivessel PCI procedures.
Future clinical trials should test individual multivessel revascularization strategies to reduce mortality in MI patients with cardiogenic shock, such as coronary artery bypass grafting (CABG) and venoarterial extracorporeal membrane oxygenation (ECMO).
Judith Hochman, MD, and Stuart Katz, MD, of New York University Langone Health, made these comments in an accompanying editorial (N Engl J Med. 2017 Oct 30. doi: 10.1056/nejme1713341). Dr. Hochman had no relevant disclosures. Dr. Katz has consulted for Novartis, Amgen, and Regeneron, and has received funding from Amgen, American Regent, and Janssen.
This study’s findings reinforce those of previous trials that had suggested that multivessel percutaneous coronary intervention has higher early mortality than culprit-lesion-only PCI.
The study provides compelling evidence that culprit-lesion-only PCI should be the preferred treatment choice over multivessel PCI in patients with cardiogenic shock.
A previous meta-analysis of patients with uncomplicated ST-segment elevation myocardial infarction showed lower rates of mortality or MI with initial multivessel PCI. The disagreement between the two studies suggests that patients with cardiogenic shock may experience greater risk of these adverse outcomes during multivessel PCI procedures.
Future clinical trials should test individual multivessel revascularization strategies to reduce mortality in MI patients with cardiogenic shock, such as coronary artery bypass grafting (CABG) and venoarterial extracorporeal membrane oxygenation (ECMO).
Judith Hochman, MD, and Stuart Katz, MD, of New York University Langone Health, made these comments in an accompanying editorial (N Engl J Med. 2017 Oct 30. doi: 10.1056/nejme1713341). Dr. Hochman had no relevant disclosures. Dr. Katz has consulted for Novartis, Amgen, and Regeneron, and has received funding from Amgen, American Regent, and Janssen.
In patients with acute myocardial infarction and multivessel coronary artery disease with cardiogenic shock, 30-day rates of death and renal-replacement therapy were lower when patients underwent percutaneous coronary intervention (PCI) of the culprit lesion as opposed to multivessel PCI.
The difference appeared to be driven by mortality, as the renal endpoint alone was not significant.
As many as 80% of patients with cardiogenic shock also present with multivessel coronary artery disease, and this is associated with worse mortality. It is unclear whether immediate PCI of clinically important stenoses of major nonculprit coronary arteries is of benefit, and previous randomized trials comparing the procedures did not look at patients with cardiogenic shock, Holger Thiele, MD, reported at the Transcatheter Cardiovascular Therapeutics annual educational meeting.
European guidelines suggest that PCI of nonculprit lesions should be considered in patients with cardiogenic shock, while U.S. guidelines offer no opinion, but recent appropriate use criteria recommend revascularization of a nonculprit artery if cardiogenic shock continues after the culprit artery has been repaired. It is thought that immediate revascularization of all coronary arteries with clinically important stenoses might improve overall myocardial perfusion and function in patients with cardiogenic shock, but the procedure could also have drawbacks, including additional ischemia, volume overload, and renal impairment from higher doses of contrast material.
To better understand outcomes in these patients, the Culprit Lesion Only PCI versus Multivessel PCI in Cardiogenic Shock (CULPRIT-SHOCK) trial randomized 706 patients to culprit-only PCI or multivessel PCI, in which PCI was performed on all major coronary arteries with more than 70% stenosis. Patients receiving culprit-only PCI could also undergo optional staged revascularization due to residual ischemic lesions, symptoms, or clinical or neurologic status.
At 30 days, death and/or renal-replacement therapy occurred in 45.9% of the culprit-only group, compared to 55.4% in the multivessel group (relative risk, 0.83; 95% confidence interval, 0.71-0.96; P = .01). A per-protocol analysis showed similar results (RR, 0.81; 95% CI, 0.69-0.96; P =.01), as did an analysis of the as-treated population (RR, 0.83; 95% CI, 0.72-0.97; P = .02).
All-cause mortality was lower in the culprit-only group (43.3% versus 51.6%; RR, 0.84; 95% CI, 0.72-0.98; P=.03). The rate of renal-replacement therapy was higher in the multivessel group (16.4% versus 11.6%), but this did not reach statistical significance (P = .07).
There were no statistically significant differences between the two groups with respect to recurrent myocardial infarction, rehospitalization for heart failure, bleeding, or stroke, Dr. Thiele reported at the meeting, which was sponsored by the Cardiovascular Research Foundation.
Some limitations of the study included its unblinded nature, and the fact that 75 patients originally assigned to one treatment category crossed over to the other, including 14 in the culprit-lesion only category who underwent immediate multivessel PCI. This suggests that treatment strategy may need to be adopted to a patient’s clinical circumstances.
The CULPRIT-SHOCK results were published online at the time of Dr. Thiele’s presentation (N Engl J Med. 2017 Oct 30. doi:10/056/NEJMoa1710261).
Several of the study’s authors reported financial ties to the pharmaceutical industry.
In patients with acute myocardial infarction and multivessel coronary artery disease with cardiogenic shock, 30-day rates of death and renal-replacement therapy were lower when patients underwent percutaneous coronary intervention (PCI) of the culprit lesion as opposed to multivessel PCI.
The difference appeared to be driven by mortality, as the renal endpoint alone was not significant.
As many as 80% of patients with cardiogenic shock also present with multivessel coronary artery disease, and this is associated with worse mortality. It is unclear whether immediate PCI of clinically important stenoses of major nonculprit coronary arteries is of benefit, and previous randomized trials comparing the procedures did not look at patients with cardiogenic shock, Holger Thiele, MD, reported at the Transcatheter Cardiovascular Therapeutics annual educational meeting.
European guidelines suggest that PCI of nonculprit lesions should be considered in patients with cardiogenic shock, while U.S. guidelines offer no opinion, but recent appropriate use criteria recommend revascularization of a nonculprit artery if cardiogenic shock continues after the culprit artery has been repaired. It is thought that immediate revascularization of all coronary arteries with clinically important stenoses might improve overall myocardial perfusion and function in patients with cardiogenic shock, but the procedure could also have drawbacks, including additional ischemia, volume overload, and renal impairment from higher doses of contrast material.
To better understand outcomes in these patients, the Culprit Lesion Only PCI versus Multivessel PCI in Cardiogenic Shock (CULPRIT-SHOCK) trial randomized 706 patients to culprit-only PCI or multivessel PCI, in which PCI was performed on all major coronary arteries with more than 70% stenosis. Patients receiving culprit-only PCI could also undergo optional staged revascularization due to residual ischemic lesions, symptoms, or clinical or neurologic status.
At 30 days, death and/or renal-replacement therapy occurred in 45.9% of the culprit-only group, compared to 55.4% in the multivessel group (relative risk, 0.83; 95% confidence interval, 0.71-0.96; P = .01). A per-protocol analysis showed similar results (RR, 0.81; 95% CI, 0.69-0.96; P =.01), as did an analysis of the as-treated population (RR, 0.83; 95% CI, 0.72-0.97; P = .02).
All-cause mortality was lower in the culprit-only group (43.3% versus 51.6%; RR, 0.84; 95% CI, 0.72-0.98; P=.03). The rate of renal-replacement therapy was higher in the multivessel group (16.4% versus 11.6%), but this did not reach statistical significance (P = .07).
There were no statistically significant differences between the two groups with respect to recurrent myocardial infarction, rehospitalization for heart failure, bleeding, or stroke, Dr. Thiele reported at the meeting, which was sponsored by the Cardiovascular Research Foundation.
Some limitations of the study included its unblinded nature, and the fact that 75 patients originally assigned to one treatment category crossed over to the other, including 14 in the culprit-lesion only category who underwent immediate multivessel PCI. This suggests that treatment strategy may need to be adopted to a patient’s clinical circumstances.
The CULPRIT-SHOCK results were published online at the time of Dr. Thiele’s presentation (N Engl J Med. 2017 Oct 30. doi:10/056/NEJMoa1710261).
Several of the study’s authors reported financial ties to the pharmaceutical industry.
FROM TCT 2017
Key clinical point: The combined rate of 30-day mortality and renal-replacement therapy was lower when the culprit lesion alone was treated.
Major finding: The culprit-only PCI group had a relative risk of death or renal-replacement therapy of 0.83.
Data source: CULPRIT-SHOCK, a randomized, controlled trial of 706 patients.
Disclosures: Some of the study’s authors reported financial ties to the pharmaceutical industry. Dr. Katz has consulted for Novartis, Amgen, and Regeneron, and has received funding from Amgen, American Regent, and Janssen.