User login
Loss of global periods could mean $1 billion loss for dermatologists
Failing to report on global period codes this year could lead to payment changes that would cost dermatologists a collective $1 billion.
Presently, surgical procedures and follow-up visits are paid by Medicare as a single bundled payment, with the expectation that the follow-ups will occur within a 10- or 90-day period. CMS tried to eliminate these global period codes in 2014, but Congress stepped in and, as part of passage of the MACRA reform law, required the agency to study the effects of such a shift.

Whether this test will demonstrate clearly just how much dermatologists rely on global periods to cover the services they render remains to be seen.
The required reporting is resource intensive and onerous, Murad Alam, MD, of Northwestern University, Chicago, said in an interview. “No one’s really going to report them.”
“I think its definitely not going to be successful in capturing the data needed to keep the global period,” he said, adding that dermatologists alone could lose more than $1 billion if global periods were eliminated.
CMS wants to understand when follow-up visits happen. It is asking providers in those nine states to submit CPT code 99024 for each follow-up visit related to a surgical procedure and will be looking for the follow-up visit code linked to procedures reported by 100 or more physicians, that have 10,000 or more occurrences, or that have allowed charges of more than $10 million annually. The extent to which the CPT code is reported could impact whether global periods are maintained.
“The way [the test] was developed was – I would hate to think by intent but certainly by design even if not intent – it’s going to necessarily result in significant underreporting, which will inevitably result in the conclusion that … the global periods will go away,” Dr. Alam said.
One possible solution would be to simply subtract the value of the follow-up visits from the global period payments and pay them separately, but Dr. Alam said that paying them separately would not necessarily provide equal levels of payment.
“If you subtract the value of the level two follow-up visits from that code, you don’t get where you need to be,” he said. “In some cases, you actually end up with negative values for codes.”
Plus, it would take a while to properly value the codes for the follow-up visits following a surgery, particularly for those following surgical procedures in dermatology, as they tend to be resource intensive, he said.
And that does not factor into the equation the additional administrative burden of filing claims for each individual follow-up visit.
The loss of global-period billing could be huge for dermatologists, and it could cause more economic disruption than the other MACRA-based reforms, according Clifford W. Lober, MD.
“If we were to lose our global periods, it would impact us far more than [the Merit-based Incentive Payment System] will,” said Dr. Lober, a dermatologist in Kissimmee, Fla. “The worst case under MIPS will be a 9% reduction in payments several years from now. We stand to lose significantly more than 9%, particularly from highly surgical practices, if we were to lose our global periods.”
Eliminating global-period billing also could mean higher out-of-pocket costs for patients, Dr. Lober said. “If patients have pay a [copayment] when they return for surgical follow-up visits, they simply may elect not to show up.”
Failing to report on global period codes this year could lead to payment changes that would cost dermatologists a collective $1 billion.
Presently, surgical procedures and follow-up visits are paid by Medicare as a single bundled payment, with the expectation that the follow-ups will occur within a 10- or 90-day period. CMS tried to eliminate these global period codes in 2014, but Congress stepped in and, as part of passage of the MACRA reform law, required the agency to study the effects of such a shift.
Whether this test will demonstrate clearly just how much dermatologists rely on global periods to cover the services they render remains to be seen.
The required reporting is resource intensive and onerous, Murad Alam, MD, of Northwestern University, Chicago, said in an interview. “No one’s really going to report them.”
“I think its definitely not going to be successful in capturing the data needed to keep the global period,” he said, adding that dermatologists alone could lose more than $1 billion if global periods were eliminated.
CMS wants to understand when follow-up visits happen. It is asking providers in those nine states to submit CPT code 99024 for each follow-up visit related to a surgical procedure and will be looking for the follow-up visit code linked to procedures reported by 100 or more physicians, that have 10,000 or more occurrences, or that have allowed charges of more than $10 million annually. The extent to which the CPT code is reported could impact whether global periods are maintained.
“The way [the test] was developed was – I would hate to think by intent but certainly by design even if not intent – it’s going to necessarily result in significant underreporting, which will inevitably result in the conclusion that … the global periods will go away,” Dr. Alam said.
One possible solution would be to simply subtract the value of the follow-up visits from the global period payments and pay them separately, but Dr. Alam said that paying them separately would not necessarily provide equal levels of payment.
“If you subtract the value of the level two follow-up visits from that code, you don’t get where you need to be,” he said. “In some cases, you actually end up with negative values for codes.”
Plus, it would take a while to properly value the codes for the follow-up visits following a surgery, particularly for those following surgical procedures in dermatology, as they tend to be resource intensive, he said.
And that does not factor into the equation the additional administrative burden of filing claims for each individual follow-up visit.
The loss of global-period billing could be huge for dermatologists, and it could cause more economic disruption than the other MACRA-based reforms, according Clifford W. Lober, MD.
“If we were to lose our global periods, it would impact us far more than [the Merit-based Incentive Payment System] will,” said Dr. Lober, a dermatologist in Kissimmee, Fla. “The worst case under MIPS will be a 9% reduction in payments several years from now. We stand to lose significantly more than 9%, particularly from highly surgical practices, if we were to lose our global periods.”
Eliminating global-period billing also could mean higher out-of-pocket costs for patients, Dr. Lober said. “If patients have pay a [copayment] when they return for surgical follow-up visits, they simply may elect not to show up.”
Failing to report on global period codes this year could lead to payment changes that would cost dermatologists a collective $1 billion.
Presently, surgical procedures and follow-up visits are paid by Medicare as a single bundled payment, with the expectation that the follow-ups will occur within a 10- or 90-day period. CMS tried to eliminate these global period codes in 2014, but Congress stepped in and, as part of passage of the MACRA reform law, required the agency to study the effects of such a shift.
Whether this test will demonstrate clearly just how much dermatologists rely on global periods to cover the services they render remains to be seen.
The required reporting is resource intensive and onerous, Murad Alam, MD, of Northwestern University, Chicago, said in an interview. “No one’s really going to report them.”
“I think its definitely not going to be successful in capturing the data needed to keep the global period,” he said, adding that dermatologists alone could lose more than $1 billion if global periods were eliminated.
CMS wants to understand when follow-up visits happen. It is asking providers in those nine states to submit CPT code 99024 for each follow-up visit related to a surgical procedure and will be looking for the follow-up visit code linked to procedures reported by 100 or more physicians, that have 10,000 or more occurrences, or that have allowed charges of more than $10 million annually. The extent to which the CPT code is reported could impact whether global periods are maintained.
“The way [the test] was developed was – I would hate to think by intent but certainly by design even if not intent – it’s going to necessarily result in significant underreporting, which will inevitably result in the conclusion that … the global periods will go away,” Dr. Alam said.
One possible solution would be to simply subtract the value of the follow-up visits from the global period payments and pay them separately, but Dr. Alam said that paying them separately would not necessarily provide equal levels of payment.
“If you subtract the value of the level two follow-up visits from that code, you don’t get where you need to be,” he said. “In some cases, you actually end up with negative values for codes.”
Plus, it would take a while to properly value the codes for the follow-up visits following a surgery, particularly for those following surgical procedures in dermatology, as they tend to be resource intensive, he said.
And that does not factor into the equation the additional administrative burden of filing claims for each individual follow-up visit.
The loss of global-period billing could be huge for dermatologists, and it could cause more economic disruption than the other MACRA-based reforms, according Clifford W. Lober, MD.
“If we were to lose our global periods, it would impact us far more than [the Merit-based Incentive Payment System] will,” said Dr. Lober, a dermatologist in Kissimmee, Fla. “The worst case under MIPS will be a 9% reduction in payments several years from now. We stand to lose significantly more than 9%, particularly from highly surgical practices, if we were to lose our global periods.”
Eliminating global-period billing also could mean higher out-of-pocket costs for patients, Dr. Lober said. “If patients have pay a [copayment] when they return for surgical follow-up visits, they simply may elect not to show up.”
The Impact of Obesity on Simvastatin for Lowering LDL-C Among Veterans
More than one-third of Americans and > 20% of veterans have obesity with a body mass index (BMI) ≥ 30 kg/m2.1,2 It is well documented that patients with obesity have altered lipid metabolism, drug distribution, and drug clearance.3-5 As many as 8.2 million Americans may receive statin (3-hydroxymethylglutaryl coenzyme A reductase inhibitors) prescriptions if the American College of Cardiology/American Heart Association 2013 Cholesterol Guidelines are followed; therefore, it is important to examine how the efficacy of these drugs is altered in patients with obesity.6
Multiple studies have examined the benefits of statin therapy through lowering low-density lipoprotein cholesterol (LDL-C); however, few have examined the impact of obesity on statin efficacy. For example, only 18% of subjects in the Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) trial were classified as having obesity, and subjects in the Scandinavian Simvastatin Survival Study (4S) trial had a mean BMI of only 26 kg/m2.7,8 Though statins decreased mortality in both of these studies, it is unknown whether the lipid-lowering effects were the same for participants with and without obesity. The Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS) demonstrated a decrease in major cardiovascular events and all-cause mortality with atorvastatin 10 mg daily therapy in a sample where more than one-third of subjects had obesity.9 However, the mean baseline BMI of subjects in both study groups was only 28 kg/m2, and outcomes for those with and without obesity were not compared.9
Studies that have examined statin efficacy in those with and without obesity include the Heart Protection Study (HPS), a post hoc analysis of the West of Scotland Coronary Prevention Study (WOSCOPS), and a meta-analysis by Blassetto and colleagues. The HPS examined the event rate of vascular events with simvastatin 40 mg daily in patients with diabetes mellitus (DM).10 Though these subgroups were compared in HPS, no statistical difference was demonstrated between these groups for the rate of vascular events among those with and without DM.10 However, the obesity subgroup’s event rate ratios were consistently higher than were those for the nonobese group.10
A post hoc analysis of WOSCOPS examined obesity as a factor for change in LDL-C with pravastatin 40 mg therapy.11 Though the authors found that no significant difference was present between those with and those without obesity, the data supporting this claim were not disclosed, which makes drawing clinical conclusions from this analysis difficult.11 A meta-analysis by Blassetto and colleagues examined the association between rosuvastatin’s efficacy in lowering LDL-C among the subgroups of hypertension, atherosclerosis, type 2 DM, and obesity.12 Though these subgroups were not compared statistically, the obesity subgroup had the lowest mean percent change in lowering LDL-C. Moreover, patients without obesity were not examined as a subgroup.12
With the expected increase in statin therapy and a significant portion of the U.S. population having obesity, it is necessary to determine if obesity alters the efficacy of statins. This study was conducted to determine the effect of obesity on the percent change in LDL-C with statin therapy within a veteran population.
Methods
This study was a retrospective review examining follow-up data from January 1, 2009 to July 1, 2014 from the VA Midsouth Healthcare Network. This network services more than 350,000 patients each year in Tennessee, Kentucky, and West Virgin
Patients were excluded if they had received treatment for hyperlipidemia (niacin, colestyramine, colestipol, colesevelam, other statins, gemfibrozil, fenofibrate, omega-3 ethyl esters, ezetimibe) during the 6 weeks prior to the initial fill date of the statin prescription. Patients whose simvastatin therapy did not span the follow-up period from the time of filling to the follow-up lipid panel were excluded, as were those who had not filled a simvastatin prescription within 30 days of their baseline lipid panel. Also excluded were patients who were newly established at the VA, pregnant, or receiving concomitant antihyperlipidemia agents, dialysis, or interacting medications (tacrolimus, cyclosporine, atazanavir, darunavir, nelfinavir, saquinavir, ritonavir, indinavir, lopinavir, tipranavir, fosamprenavir, fluconazole, voriconazole, itraconazole, voriconazole, posaconazole, amiodarone, or colchicine). Patients with a BMI < 18 kg/m2, hepatic failure as measured by an aspartate transaminase/alanine transaminase (AST/ALT) ratio > 3 times the upper limit of normal, hepatitis, a history of alcoholism, any change in statin dose prior to follow-up cholesterol values, or no follow-up LDL-C values also were excluded.
The baseline data collected included age, sex, weight, height, BMI, hemoglobin A1c, LDL-C, ALT/AST, and serum creatinine (SCr). All other laboratory results were required to be within 270 days of the time the lipid panel was obtained. The index date was set as the date the initial prescription was filled between February 1, 2009 and April 1, 2014. Follow-up levels for LDL-C were obtained 40 to 95 days after the index date. Direct LDL-C values were preferred unless only calculated values were available. Calculated LDL-C values were determined by using the Friedewald equation. An audit of 150 patient charts was conducted to ensure the integrity of data pulled from the database.
The percent changes in LDL-C were calculated for those with and without obesity for both simvastatin 20 mg daily and simvastatin 40 mg daily. The primary outcome was the percent change in LDL-C from baseline. All laboratory values were compared using independent 2-tailed t tests with α set to .05. To have an 80% chance of detecting a 5% difference in percent change in LDL-C between the experimental and control groups, 129 patients were required. To determine whether an association was present, a correlation between BMI and percent change in LDL-C was conducted. All statistics were conducted using SAS software (Cary, North Carolina).
Results
From January 2009 through July 2014, 35,216 patients were initially screened. The majority of patients did not have a baseline LDL-C value and were excluded. A total of 1,183 patients with simvastatin 20 mg daily (BMI < 30 = 661; BMI ≥ 30 = 1,122) and 478 patients with simvastatin 40 mg daily (BMI < 30 = 259; BMI ≥ 30 = 219) met the inclusion criteria.
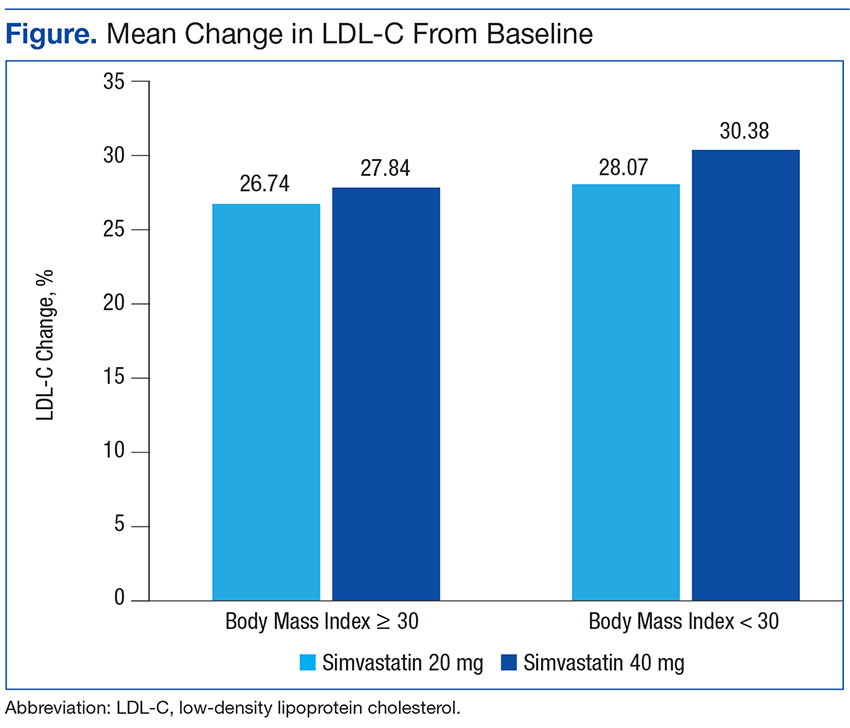
Baseline characteristics were similar between groups except for a slightly higher age in both groups without obesity (Table). Hepatic and renal serum markers indicated a baseline of adequate organ function for drug clearance for all groups. The mean baseline BMI of those without obesity was about 26 kg/m2, which is considered overweight. Baseline LDL-C values were clinically similar for those with and without obesity, though statistically different (145 mg/dL for the nonobese group and 141 mg/dL for the obese group, P < .05). The percent change in LDL-C was not statistically significant for those with and without obesity for simvastatin 20 mg daily (P = .293) or simvastatin 40 mg daily (P = .2773) (Figure). No correlation was found between the continuous percent change in LDL-C and continuous BMI for either simvastatin dosage (r2 = 0.0016 and 0.0028, respectively).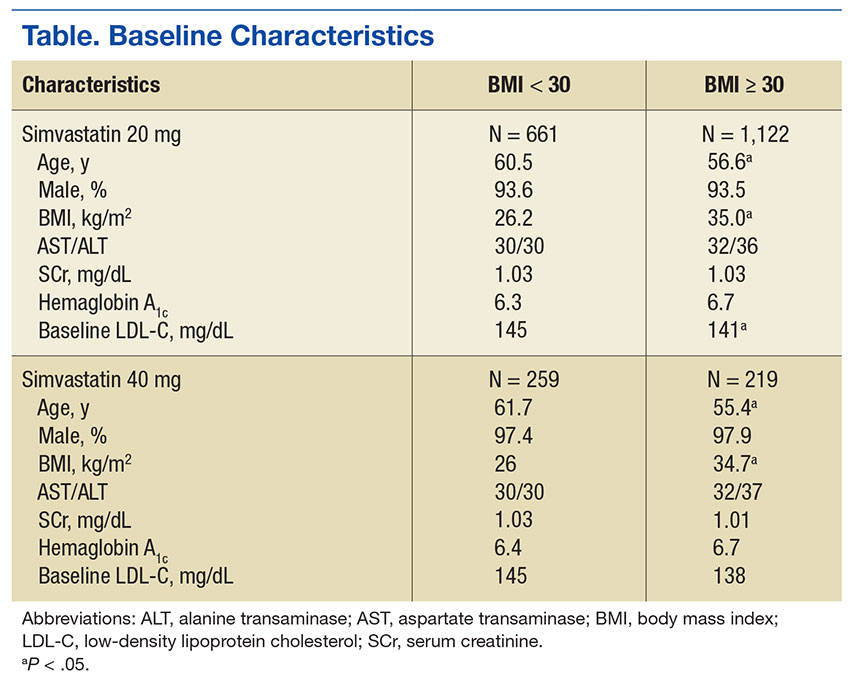
Discussion
In this retrospective chart review, it was determined that obesity did not affect the percent change in LDL-C from baseline with statin therapy. The HPS found similar results as a secondary endpoint, although that study was underpowered.10 In this study, all groups met power, and there was still no difference between those with and without obesity.
Nicholls and colleagues examined REVERSAL study data to determine whether BMI greater than the median BMI impacted inflammatory markers or lipid levels with atorvastatin 80 mg daily or pravastatin 40 mg daily. The REVERSAL study authors found no difference in percent change LDL-C between those above the median BMI compared with those below the median BMI for patients on pravastatin therapy. However, the authors did find a difference in percent change LDL-C with atorvastatin therapy.13 No difference in percent change LDL-C was present with simvastatin therapy in this study. As simvastatin is more lipophilic than is atorvastatin, lipophilicity remains an area for further study for statin therapy in patients with obesity.
The surrogate marker of percent change in LDL-C was used for the primary outcome in this study. The ACC/AHA 2013 guidelines and the National Lipid Association 2014 guidelines recommend an alternative goal of 30% to 50% change in LDL-C from baseline.14,15 Using this clinically relevant marker compensated for differences in baseline LDL-C and limited the effect of these differences on the primary outcome of this study.
Limitations
This study did not include patients who were underweight (BMI < 18 kg/m2), as these patients have previously demonstrated decreased outcomes with statin therapy.16 However, this limits these data to only those patients that have a BMI of at least 18 kg/m2. Limitations of this study also included the inability to consider adherence and lifestyle changes. These limitations were unavoidable due to the nature of a retrospective chart review.
Conclusion
The prevalence of obesity is increasing, and it is a disease that alters pharmacokinetics and lipid metabolism. Though this study did not find a difference between the LDL-C-lowering efficacy of simvastatin in those with and without obesity, continued study of the effect of obesity on the efficacy of medications is vital.
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the James H. Qullen VAMC in Mountain Home, Tennessee.
1. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA. 2014;311(8):806-814.
2. Shen Y, Sambamoorthi U, Rajan M, Miller D, Banerjea R, Pogach L. Obesity and expenditures among elderly Veterans Health Administration users with diabetes. Popul Health Manag. 2009;12(5):255-264.
3. Chan DC, Watts GF, Wang J, Hegele RA, van Bockxmeer FM, Barrett PH. Variation in Niemann-Pick C1-like 1 gene as a determinant of apolipoprotein B-100 kinetics and response to statin therapy in centrally obese men. Clin Endocrinol (Oxf). 2008;69(1):45-51.
4. Cheymol G. Effects of obesity on pharmacokinetics implications for drug therapy. Clin Pharmacokinet. 2000;39(3):215-231.
5. Hanley MJ, Abernethy DR, Greenblatt DJ. Effect of obesity on the pharmacokinetics of drugs in humans. Clin Pharmacokinet. 2010;49(2):71-87
6. Pencina MJ, Navar-Boggan AM, D’Agostino RB Sr, et al. Application of new cholesterol guidelines to a population-based sample. N Engl J Med. 2014;370(15):1422-1431.
7. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med. 1998;339(19):1349-1357.
8. Pedersen TR, Kjekshus J, Berg K, et al; Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). 1994. Atheroscler Suppl. 2004;5(3):81-87.
9. Colhoun HM, Betteridge DJ, Durrington PN, et al; CARDS investigators. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet. 2004;364(9435):685-696.
10. Collins R, Armitage J, Parish S, Sleigh P, Peto R; Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet. 2003;361(9374):2005-2016.
11. Streja L, Packard CJ, Shepherd J, Cobbe S, Ford I; WOSCOPS Group. Factors affecting low-density lipoprotein and high-density lipoprotein cholesterol response to pravastatin in the West Of Scotland Coronary Prevention Study (WOSCOPS). Am J Cardiol. 2002;90(7):731-736.
12. Blasetto JW, Stein EA, Brown WV, Chitra R, Raza A. Efficacy of rosuvastatin compared with other statins at selected starting doses in hypercholesterolemic patients and in special population groups. Am J Cardiol. 2003;91(5A):3C-10C; discussion 10C.
13. Nicholls SJ. Tuzcu EM, Sipahi I, et al. Effect of obesity on lipid-lowering, anti-inflammatory, and antiatherosclerotic benefits of atorvastatin or pravastatin in patients with coronary artery disease (from the REVERSAL Study). Am J Cardiol. 2006;97(11):1553-1557.
14. Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA Guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk on adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25, pt B):2889-2934.
15. Jacobson T, Ito M, Maki K, et al. National Lipid Association recommendation for patient-centered management of dyslipidemia: part 1-full report. J Clin Lipidol. 2015;9(2):129-169.
16. Nylén ES, Faselis C, Kheirbek R, Myers J, Panagiotakos D, Kokkinos P. Statins modulate the mortality risk associated with obesity and cardiorespiratory fitness in diabetics. J Clin Endocrinol Metab. 2013;98(8):33940-3401.
More than one-third of Americans and > 20% of veterans have obesity with a body mass index (BMI) ≥ 30 kg/m2.1,2 It is well documented that patients with obesity have altered lipid metabolism, drug distribution, and drug clearance.3-5 As many as 8.2 million Americans may receive statin (3-hydroxymethylglutaryl coenzyme A reductase inhibitors) prescriptions if the American College of Cardiology/American Heart Association 2013 Cholesterol Guidelines are followed; therefore, it is important to examine how the efficacy of these drugs is altered in patients with obesity.6
Multiple studies have examined the benefits of statin therapy through lowering low-density lipoprotein cholesterol (LDL-C); however, few have examined the impact of obesity on statin efficacy. For example, only 18% of subjects in the Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) trial were classified as having obesity, and subjects in the Scandinavian Simvastatin Survival Study (4S) trial had a mean BMI of only 26 kg/m2.7,8 Though statins decreased mortality in both of these studies, it is unknown whether the lipid-lowering effects were the same for participants with and without obesity. The Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS) demonstrated a decrease in major cardiovascular events and all-cause mortality with atorvastatin 10 mg daily therapy in a sample where more than one-third of subjects had obesity.9 However, the mean baseline BMI of subjects in both study groups was only 28 kg/m2, and outcomes for those with and without obesity were not compared.9
Studies that have examined statin efficacy in those with and without obesity include the Heart Protection Study (HPS), a post hoc analysis of the West of Scotland Coronary Prevention Study (WOSCOPS), and a meta-analysis by Blassetto and colleagues. The HPS examined the event rate of vascular events with simvastatin 40 mg daily in patients with diabetes mellitus (DM).10 Though these subgroups were compared in HPS, no statistical difference was demonstrated between these groups for the rate of vascular events among those with and without DM.10 However, the obesity subgroup’s event rate ratios were consistently higher than were those for the nonobese group.10
A post hoc analysis of WOSCOPS examined obesity as a factor for change in LDL-C with pravastatin 40 mg therapy.11 Though the authors found that no significant difference was present between those with and those without obesity, the data supporting this claim were not disclosed, which makes drawing clinical conclusions from this analysis difficult.11 A meta-analysis by Blassetto and colleagues examined the association between rosuvastatin’s efficacy in lowering LDL-C among the subgroups of hypertension, atherosclerosis, type 2 DM, and obesity.12 Though these subgroups were not compared statistically, the obesity subgroup had the lowest mean percent change in lowering LDL-C. Moreover, patients without obesity were not examined as a subgroup.12
With the expected increase in statin therapy and a significant portion of the U.S. population having obesity, it is necessary to determine if obesity alters the efficacy of statins. This study was conducted to determine the effect of obesity on the percent change in LDL-C with statin therapy within a veteran population.
Methods
This study was a retrospective review examining follow-up data from January 1, 2009 to July 1, 2014 from the VA Midsouth Healthcare Network. This network services more than 350,000 patients each year in Tennessee, Kentucky, and West Virgin
Patients were excluded if they had received treatment for hyperlipidemia (niacin, colestyramine, colestipol, colesevelam, other statins, gemfibrozil, fenofibrate, omega-3 ethyl esters, ezetimibe) during the 6 weeks prior to the initial fill date of the statin prescription. Patients whose simvastatin therapy did not span the follow-up period from the time of filling to the follow-up lipid panel were excluded, as were those who had not filled a simvastatin prescription within 30 days of their baseline lipid panel. Also excluded were patients who were newly established at the VA, pregnant, or receiving concomitant antihyperlipidemia agents, dialysis, or interacting medications (tacrolimus, cyclosporine, atazanavir, darunavir, nelfinavir, saquinavir, ritonavir, indinavir, lopinavir, tipranavir, fosamprenavir, fluconazole, voriconazole, itraconazole, voriconazole, posaconazole, amiodarone, or colchicine). Patients with a BMI < 18 kg/m2, hepatic failure as measured by an aspartate transaminase/alanine transaminase (AST/ALT) ratio > 3 times the upper limit of normal, hepatitis, a history of alcoholism, any change in statin dose prior to follow-up cholesterol values, or no follow-up LDL-C values also were excluded.
The baseline data collected included age, sex, weight, height, BMI, hemoglobin A1c, LDL-C, ALT/AST, and serum creatinine (SCr). All other laboratory results were required to be within 270 days of the time the lipid panel was obtained. The index date was set as the date the initial prescription was filled between February 1, 2009 and April 1, 2014. Follow-up levels for LDL-C were obtained 40 to 95 days after the index date. Direct LDL-C values were preferred unless only calculated values were available. Calculated LDL-C values were determined by using the Friedewald equation. An audit of 150 patient charts was conducted to ensure the integrity of data pulled from the database.
The percent changes in LDL-C were calculated for those with and without obesity for both simvastatin 20 mg daily and simvastatin 40 mg daily. The primary outcome was the percent change in LDL-C from baseline. All laboratory values were compared using independent 2-tailed t tests with α set to .05. To have an 80% chance of detecting a 5% difference in percent change in LDL-C between the experimental and control groups, 129 patients were required. To determine whether an association was present, a correlation between BMI and percent change in LDL-C was conducted. All statistics were conducted using SAS software (Cary, North Carolina).
Results
From January 2009 through July 2014, 35,216 patients were initially screened. The majority of patients did not have a baseline LDL-C value and were excluded. A total of 1,183 patients with simvastatin 20 mg daily (BMI < 30 = 661; BMI ≥ 30 = 1,122) and 478 patients with simvastatin 40 mg daily (BMI < 30 = 259; BMI ≥ 30 = 219) met the inclusion criteria.
Baseline characteristics were similar between groups except for a slightly higher age in both groups without obesity (Table). Hepatic and renal serum markers indicated a baseline of adequate organ function for drug clearance for all groups. The mean baseline BMI of those without obesity was about 26 kg/m2, which is considered overweight. Baseline LDL-C values were clinically similar for those with and without obesity, though statistically different (145 mg/dL for the nonobese group and 141 mg/dL for the obese group, P < .05). The percent change in LDL-C was not statistically significant for those with and without obesity for simvastatin 20 mg daily (P = .293) or simvastatin 40 mg daily (P = .2773) (Figure). No correlation was found between the continuous percent change in LDL-C and continuous BMI for either simvastatin dosage (r2 = 0.0016 and 0.0028, respectively).
Discussion
In this retrospective chart review, it was determined that obesity did not affect the percent change in LDL-C from baseline with statin therapy. The HPS found similar results as a secondary endpoint, although that study was underpowered.10 In this study, all groups met power, and there was still no difference between those with and without obesity.
Nicholls and colleagues examined REVERSAL study data to determine whether BMI greater than the median BMI impacted inflammatory markers or lipid levels with atorvastatin 80 mg daily or pravastatin 40 mg daily. The REVERSAL study authors found no difference in percent change LDL-C between those above the median BMI compared with those below the median BMI for patients on pravastatin therapy. However, the authors did find a difference in percent change LDL-C with atorvastatin therapy.13 No difference in percent change LDL-C was present with simvastatin therapy in this study. As simvastatin is more lipophilic than is atorvastatin, lipophilicity remains an area for further study for statin therapy in patients with obesity.
The surrogate marker of percent change in LDL-C was used for the primary outcome in this study. The ACC/AHA 2013 guidelines and the National Lipid Association 2014 guidelines recommend an alternative goal of 30% to 50% change in LDL-C from baseline.14,15 Using this clinically relevant marker compensated for differences in baseline LDL-C and limited the effect of these differences on the primary outcome of this study.
Limitations
This study did not include patients who were underweight (BMI < 18 kg/m2), as these patients have previously demonstrated decreased outcomes with statin therapy.16 However, this limits these data to only those patients that have a BMI of at least 18 kg/m2. Limitations of this study also included the inability to consider adherence and lifestyle changes. These limitations were unavoidable due to the nature of a retrospective chart review.
Conclusion
The prevalence of obesity is increasing, and it is a disease that alters pharmacokinetics and lipid metabolism. Though this study did not find a difference between the LDL-C-lowering efficacy of simvastatin in those with and without obesity, continued study of the effect of obesity on the efficacy of medications is vital.
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the James H. Qullen VAMC in Mountain Home, Tennessee.
More than one-third of Americans and > 20% of veterans have obesity with a body mass index (BMI) ≥ 30 kg/m2.1,2 It is well documented that patients with obesity have altered lipid metabolism, drug distribution, and drug clearance.3-5 As many as 8.2 million Americans may receive statin (3-hydroxymethylglutaryl coenzyme A reductase inhibitors) prescriptions if the American College of Cardiology/American Heart Association 2013 Cholesterol Guidelines are followed; therefore, it is important to examine how the efficacy of these drugs is altered in patients with obesity.6
Multiple studies have examined the benefits of statin therapy through lowering low-density lipoprotein cholesterol (LDL-C); however, few have examined the impact of obesity on statin efficacy. For example, only 18% of subjects in the Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) trial were classified as having obesity, and subjects in the Scandinavian Simvastatin Survival Study (4S) trial had a mean BMI of only 26 kg/m2.7,8 Though statins decreased mortality in both of these studies, it is unknown whether the lipid-lowering effects were the same for participants with and without obesity. The Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS) demonstrated a decrease in major cardiovascular events and all-cause mortality with atorvastatin 10 mg daily therapy in a sample where more than one-third of subjects had obesity.9 However, the mean baseline BMI of subjects in both study groups was only 28 kg/m2, and outcomes for those with and without obesity were not compared.9
Studies that have examined statin efficacy in those with and without obesity include the Heart Protection Study (HPS), a post hoc analysis of the West of Scotland Coronary Prevention Study (WOSCOPS), and a meta-analysis by Blassetto and colleagues. The HPS examined the event rate of vascular events with simvastatin 40 mg daily in patients with diabetes mellitus (DM).10 Though these subgroups were compared in HPS, no statistical difference was demonstrated between these groups for the rate of vascular events among those with and without DM.10 However, the obesity subgroup’s event rate ratios were consistently higher than were those for the nonobese group.10
A post hoc analysis of WOSCOPS examined obesity as a factor for change in LDL-C with pravastatin 40 mg therapy.11 Though the authors found that no significant difference was present between those with and those without obesity, the data supporting this claim were not disclosed, which makes drawing clinical conclusions from this analysis difficult.11 A meta-analysis by Blassetto and colleagues examined the association between rosuvastatin’s efficacy in lowering LDL-C among the subgroups of hypertension, atherosclerosis, type 2 DM, and obesity.12 Though these subgroups were not compared statistically, the obesity subgroup had the lowest mean percent change in lowering LDL-C. Moreover, patients without obesity were not examined as a subgroup.12
With the expected increase in statin therapy and a significant portion of the U.S. population having obesity, it is necessary to determine if obesity alters the efficacy of statins. This study was conducted to determine the effect of obesity on the percent change in LDL-C with statin therapy within a veteran population.
Methods
This study was a retrospective review examining follow-up data from January 1, 2009 to July 1, 2014 from the VA Midsouth Healthcare Network. This network services more than 350,000 patients each year in Tennessee, Kentucky, and West Virgin
Patients were excluded if they had received treatment for hyperlipidemia (niacin, colestyramine, colestipol, colesevelam, other statins, gemfibrozil, fenofibrate, omega-3 ethyl esters, ezetimibe) during the 6 weeks prior to the initial fill date of the statin prescription. Patients whose simvastatin therapy did not span the follow-up period from the time of filling to the follow-up lipid panel were excluded, as were those who had not filled a simvastatin prescription within 30 days of their baseline lipid panel. Also excluded were patients who were newly established at the VA, pregnant, or receiving concomitant antihyperlipidemia agents, dialysis, or interacting medications (tacrolimus, cyclosporine, atazanavir, darunavir, nelfinavir, saquinavir, ritonavir, indinavir, lopinavir, tipranavir, fosamprenavir, fluconazole, voriconazole, itraconazole, voriconazole, posaconazole, amiodarone, or colchicine). Patients with a BMI < 18 kg/m2, hepatic failure as measured by an aspartate transaminase/alanine transaminase (AST/ALT) ratio > 3 times the upper limit of normal, hepatitis, a history of alcoholism, any change in statin dose prior to follow-up cholesterol values, or no follow-up LDL-C values also were excluded.
The baseline data collected included age, sex, weight, height, BMI, hemoglobin A1c, LDL-C, ALT/AST, and serum creatinine (SCr). All other laboratory results were required to be within 270 days of the time the lipid panel was obtained. The index date was set as the date the initial prescription was filled between February 1, 2009 and April 1, 2014. Follow-up levels for LDL-C were obtained 40 to 95 days after the index date. Direct LDL-C values were preferred unless only calculated values were available. Calculated LDL-C values were determined by using the Friedewald equation. An audit of 150 patient charts was conducted to ensure the integrity of data pulled from the database.
The percent changes in LDL-C were calculated for those with and without obesity for both simvastatin 20 mg daily and simvastatin 40 mg daily. The primary outcome was the percent change in LDL-C from baseline. All laboratory values were compared using independent 2-tailed t tests with α set to .05. To have an 80% chance of detecting a 5% difference in percent change in LDL-C between the experimental and control groups, 129 patients were required. To determine whether an association was present, a correlation between BMI and percent change in LDL-C was conducted. All statistics were conducted using SAS software (Cary, North Carolina).
Results
From January 2009 through July 2014, 35,216 patients were initially screened. The majority of patients did not have a baseline LDL-C value and were excluded. A total of 1,183 patients with simvastatin 20 mg daily (BMI < 30 = 661; BMI ≥ 30 = 1,122) and 478 patients with simvastatin 40 mg daily (BMI < 30 = 259; BMI ≥ 30 = 219) met the inclusion criteria.
Baseline characteristics were similar between groups except for a slightly higher age in both groups without obesity (Table). Hepatic and renal serum markers indicated a baseline of adequate organ function for drug clearance for all groups. The mean baseline BMI of those without obesity was about 26 kg/m2, which is considered overweight. Baseline LDL-C values were clinically similar for those with and without obesity, though statistically different (145 mg/dL for the nonobese group and 141 mg/dL for the obese group, P < .05). The percent change in LDL-C was not statistically significant for those with and without obesity for simvastatin 20 mg daily (P = .293) or simvastatin 40 mg daily (P = .2773) (Figure). No correlation was found between the continuous percent change in LDL-C and continuous BMI for either simvastatin dosage (r2 = 0.0016 and 0.0028, respectively).
Discussion
In this retrospective chart review, it was determined that obesity did not affect the percent change in LDL-C from baseline with statin therapy. The HPS found similar results as a secondary endpoint, although that study was underpowered.10 In this study, all groups met power, and there was still no difference between those with and without obesity.
Nicholls and colleagues examined REVERSAL study data to determine whether BMI greater than the median BMI impacted inflammatory markers or lipid levels with atorvastatin 80 mg daily or pravastatin 40 mg daily. The REVERSAL study authors found no difference in percent change LDL-C between those above the median BMI compared with those below the median BMI for patients on pravastatin therapy. However, the authors did find a difference in percent change LDL-C with atorvastatin therapy.13 No difference in percent change LDL-C was present with simvastatin therapy in this study. As simvastatin is more lipophilic than is atorvastatin, lipophilicity remains an area for further study for statin therapy in patients with obesity.
The surrogate marker of percent change in LDL-C was used for the primary outcome in this study. The ACC/AHA 2013 guidelines and the National Lipid Association 2014 guidelines recommend an alternative goal of 30% to 50% change in LDL-C from baseline.14,15 Using this clinically relevant marker compensated for differences in baseline LDL-C and limited the effect of these differences on the primary outcome of this study.
Limitations
This study did not include patients who were underweight (BMI < 18 kg/m2), as these patients have previously demonstrated decreased outcomes with statin therapy.16 However, this limits these data to only those patients that have a BMI of at least 18 kg/m2. Limitations of this study also included the inability to consider adherence and lifestyle changes. These limitations were unavoidable due to the nature of a retrospective chart review.
Conclusion
The prevalence of obesity is increasing, and it is a disease that alters pharmacokinetics and lipid metabolism. Though this study did not find a difference between the LDL-C-lowering efficacy of simvastatin in those with and without obesity, continued study of the effect of obesity on the efficacy of medications is vital.
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the James H. Qullen VAMC in Mountain Home, Tennessee.
1. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA. 2014;311(8):806-814.
2. Shen Y, Sambamoorthi U, Rajan M, Miller D, Banerjea R, Pogach L. Obesity and expenditures among elderly Veterans Health Administration users with diabetes. Popul Health Manag. 2009;12(5):255-264.
3. Chan DC, Watts GF, Wang J, Hegele RA, van Bockxmeer FM, Barrett PH. Variation in Niemann-Pick C1-like 1 gene as a determinant of apolipoprotein B-100 kinetics and response to statin therapy in centrally obese men. Clin Endocrinol (Oxf). 2008;69(1):45-51.
4. Cheymol G. Effects of obesity on pharmacokinetics implications for drug therapy. Clin Pharmacokinet. 2000;39(3):215-231.
5. Hanley MJ, Abernethy DR, Greenblatt DJ. Effect of obesity on the pharmacokinetics of drugs in humans. Clin Pharmacokinet. 2010;49(2):71-87
6. Pencina MJ, Navar-Boggan AM, D’Agostino RB Sr, et al. Application of new cholesterol guidelines to a population-based sample. N Engl J Med. 2014;370(15):1422-1431.
7. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med. 1998;339(19):1349-1357.
8. Pedersen TR, Kjekshus J, Berg K, et al; Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). 1994. Atheroscler Suppl. 2004;5(3):81-87.
9. Colhoun HM, Betteridge DJ, Durrington PN, et al; CARDS investigators. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet. 2004;364(9435):685-696.
10. Collins R, Armitage J, Parish S, Sleigh P, Peto R; Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet. 2003;361(9374):2005-2016.
11. Streja L, Packard CJ, Shepherd J, Cobbe S, Ford I; WOSCOPS Group. Factors affecting low-density lipoprotein and high-density lipoprotein cholesterol response to pravastatin in the West Of Scotland Coronary Prevention Study (WOSCOPS). Am J Cardiol. 2002;90(7):731-736.
12. Blasetto JW, Stein EA, Brown WV, Chitra R, Raza A. Efficacy of rosuvastatin compared with other statins at selected starting doses in hypercholesterolemic patients and in special population groups. Am J Cardiol. 2003;91(5A):3C-10C; discussion 10C.
13. Nicholls SJ. Tuzcu EM, Sipahi I, et al. Effect of obesity on lipid-lowering, anti-inflammatory, and antiatherosclerotic benefits of atorvastatin or pravastatin in patients with coronary artery disease (from the REVERSAL Study). Am J Cardiol. 2006;97(11):1553-1557.
14. Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA Guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk on adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25, pt B):2889-2934.
15. Jacobson T, Ito M, Maki K, et al. National Lipid Association recommendation for patient-centered management of dyslipidemia: part 1-full report. J Clin Lipidol. 2015;9(2):129-169.
16. Nylén ES, Faselis C, Kheirbek R, Myers J, Panagiotakos D, Kokkinos P. Statins modulate the mortality risk associated with obesity and cardiorespiratory fitness in diabetics. J Clin Endocrinol Metab. 2013;98(8):33940-3401.
1. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA. 2014;311(8):806-814.
2. Shen Y, Sambamoorthi U, Rajan M, Miller D, Banerjea R, Pogach L. Obesity and expenditures among elderly Veterans Health Administration users with diabetes. Popul Health Manag. 2009;12(5):255-264.
3. Chan DC, Watts GF, Wang J, Hegele RA, van Bockxmeer FM, Barrett PH. Variation in Niemann-Pick C1-like 1 gene as a determinant of apolipoprotein B-100 kinetics and response to statin therapy in centrally obese men. Clin Endocrinol (Oxf). 2008;69(1):45-51.
4. Cheymol G. Effects of obesity on pharmacokinetics implications for drug therapy. Clin Pharmacokinet. 2000;39(3):215-231.
5. Hanley MJ, Abernethy DR, Greenblatt DJ. Effect of obesity on the pharmacokinetics of drugs in humans. Clin Pharmacokinet. 2010;49(2):71-87
6. Pencina MJ, Navar-Boggan AM, D’Agostino RB Sr, et al. Application of new cholesterol guidelines to a population-based sample. N Engl J Med. 2014;370(15):1422-1431.
7. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med. 1998;339(19):1349-1357.
8. Pedersen TR, Kjekshus J, Berg K, et al; Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). 1994. Atheroscler Suppl. 2004;5(3):81-87.
9. Colhoun HM, Betteridge DJ, Durrington PN, et al; CARDS investigators. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet. 2004;364(9435):685-696.
10. Collins R, Armitage J, Parish S, Sleigh P, Peto R; Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet. 2003;361(9374):2005-2016.
11. Streja L, Packard CJ, Shepherd J, Cobbe S, Ford I; WOSCOPS Group. Factors affecting low-density lipoprotein and high-density lipoprotein cholesterol response to pravastatin in the West Of Scotland Coronary Prevention Study (WOSCOPS). Am J Cardiol. 2002;90(7):731-736.
12. Blasetto JW, Stein EA, Brown WV, Chitra R, Raza A. Efficacy of rosuvastatin compared with other statins at selected starting doses in hypercholesterolemic patients and in special population groups. Am J Cardiol. 2003;91(5A):3C-10C; discussion 10C.
13. Nicholls SJ. Tuzcu EM, Sipahi I, et al. Effect of obesity on lipid-lowering, anti-inflammatory, and antiatherosclerotic benefits of atorvastatin or pravastatin in patients with coronary artery disease (from the REVERSAL Study). Am J Cardiol. 2006;97(11):1553-1557.
14. Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA Guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk on adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25, pt B):2889-2934.
15. Jacobson T, Ito M, Maki K, et al. National Lipid Association recommendation for patient-centered management of dyslipidemia: part 1-full report. J Clin Lipidol. 2015;9(2):129-169.
16. Nylén ES, Faselis C, Kheirbek R, Myers J, Panagiotakos D, Kokkinos P. Statins modulate the mortality risk associated with obesity and cardiorespiratory fitness in diabetics. J Clin Endocrinol Metab. 2013;98(8):33940-3401.
CMS delays start of cardiac pay bundles
The Centers for Medicare & Medicaid Services is delaying the start of three cardiac payment bundles finalized at the end of 2016.
The bundles include the Acute Myocardial Infarction (AMI) model, the Coronary Artery Bypass Graft (CABG) model, and the Cardiac Rehabilitation Incentive Payment model.
The payment bundles were schedule to go into effect on July 1, 2017, but an interim final rule published March 21 in the Federal Register delayed the start of the bundles for 3 months. The agency also is seeking comment on potentially delaying implementation of the bundles to Jan. 1, 2018.
The bundled payment models would place accountability for patient outcomes 90 days after discharge on the hospital where treatment occurred. Hospitals in 98 randomly selected metropolitan statistical areas would be placed in this model and monitored for 5 years to test whether the models lead to improved outcomes and lower costs.
For the cardiac rehabilitation model, CMS would be testing whether an incentive payment would increase the use of cardiac rehabilitation services during a care period that runs parallel with the AMI and CABG payment bundles.
Physician participation in the bundles would have been voluntary, but those participating would have been eligible for bonus payments under the Quality Payment Program as the bundles were considered advanced Alternative Payment Models.
The final rule also delays changes to the comprehensive joint replacement bundle.
CMS had announced the final rule implementing the payment bundles on Dec. 20, 2016, but it was not published in the Federal Register until Jan. 3, 2017. The March 21 interim final rule delaying the start of the bundles cites the Trump administration’s memorandum to federal agencies freezing in-process regulations to allow for review.
The Centers for Medicare & Medicaid Services is delaying the start of three cardiac payment bundles finalized at the end of 2016.
The bundles include the Acute Myocardial Infarction (AMI) model, the Coronary Artery Bypass Graft (CABG) model, and the Cardiac Rehabilitation Incentive Payment model.
The payment bundles were schedule to go into effect on July 1, 2017, but an interim final rule published March 21 in the Federal Register delayed the start of the bundles for 3 months. The agency also is seeking comment on potentially delaying implementation of the bundles to Jan. 1, 2018.
The bundled payment models would place accountability for patient outcomes 90 days after discharge on the hospital where treatment occurred. Hospitals in 98 randomly selected metropolitan statistical areas would be placed in this model and monitored for 5 years to test whether the models lead to improved outcomes and lower costs.
For the cardiac rehabilitation model, CMS would be testing whether an incentive payment would increase the use of cardiac rehabilitation services during a care period that runs parallel with the AMI and CABG payment bundles.
Physician participation in the bundles would have been voluntary, but those participating would have been eligible for bonus payments under the Quality Payment Program as the bundles were considered advanced Alternative Payment Models.
The final rule also delays changes to the comprehensive joint replacement bundle.
CMS had announced the final rule implementing the payment bundles on Dec. 20, 2016, but it was not published in the Federal Register until Jan. 3, 2017. The March 21 interim final rule delaying the start of the bundles cites the Trump administration’s memorandum to federal agencies freezing in-process regulations to allow for review.
The Centers for Medicare & Medicaid Services is delaying the start of three cardiac payment bundles finalized at the end of 2016.
The bundles include the Acute Myocardial Infarction (AMI) model, the Coronary Artery Bypass Graft (CABG) model, and the Cardiac Rehabilitation Incentive Payment model.
The payment bundles were schedule to go into effect on July 1, 2017, but an interim final rule published March 21 in the Federal Register delayed the start of the bundles for 3 months. The agency also is seeking comment on potentially delaying implementation of the bundles to Jan. 1, 2018.
The bundled payment models would place accountability for patient outcomes 90 days after discharge on the hospital where treatment occurred. Hospitals in 98 randomly selected metropolitan statistical areas would be placed in this model and monitored for 5 years to test whether the models lead to improved outcomes and lower costs.
For the cardiac rehabilitation model, CMS would be testing whether an incentive payment would increase the use of cardiac rehabilitation services during a care period that runs parallel with the AMI and CABG payment bundles.
Physician participation in the bundles would have been voluntary, but those participating would have been eligible for bonus payments under the Quality Payment Program as the bundles were considered advanced Alternative Payment Models.
The final rule also delays changes to the comprehensive joint replacement bundle.
CMS had announced the final rule implementing the payment bundles on Dec. 20, 2016, but it was not published in the Federal Register until Jan. 3, 2017. The March 21 interim final rule delaying the start of the bundles cites the Trump administration’s memorandum to federal agencies freezing in-process regulations to allow for review.
Artificial Pancreas Moves Closer to Real-Life Option
Roughly 25% of veterans have diabetes mellitus (DM) as opposed to 9% of the general public. A small percentage of veterans have type 1 DM, which according to research, can be caused by both physical and mental trauma that affects the pancreas.
“Managing type 1 diabetes currently requires a constant juggling act between checking bloodglucose levels frequently and delivering just the right amount of insulin while taking into account meals, physical activity, and other aspects of daily life, where a missed or wrong delivery could lead to potential complications,” said Dr. Andrew Bremer, of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). But that may change soon as we draw near to a functional “artificial pancreas,” a fully automated system that will sense rising glucose levels and adjust insulin automatically.
The FDA approved a hybrid model of an artificial pancreas in 2016, which still required users to adjust insulin intake. Now, 4 separate projects are designed to be the “potential last steps” toward requesting regulatory approval for permanent use of a fully automated system, according to NIDDK. The research studies beginning this year will look at safety, efficacy, user-friendliness, physical and emotional health of participants, and cost. The participants will live at home and monitor themselves with remote monitoring by study staff.
“Nearly 100 years since the discovery of insulin,” said NIDDK Director Dr. Griffin P. Rodgers, “a successful artificial pancreas would mark another huge step toward better health for people with type 1 diabetes.”
Roughly 25% of veterans have diabetes mellitus (DM) as opposed to 9% of the general public. A small percentage of veterans have type 1 DM, which according to research, can be caused by both physical and mental trauma that affects the pancreas.
“Managing type 1 diabetes currently requires a constant juggling act between checking bloodglucose levels frequently and delivering just the right amount of insulin while taking into account meals, physical activity, and other aspects of daily life, where a missed or wrong delivery could lead to potential complications,” said Dr. Andrew Bremer, of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). But that may change soon as we draw near to a functional “artificial pancreas,” a fully automated system that will sense rising glucose levels and adjust insulin automatically.
The FDA approved a hybrid model of an artificial pancreas in 2016, which still required users to adjust insulin intake. Now, 4 separate projects are designed to be the “potential last steps” toward requesting regulatory approval for permanent use of a fully automated system, according to NIDDK. The research studies beginning this year will look at safety, efficacy, user-friendliness, physical and emotional health of participants, and cost. The participants will live at home and monitor themselves with remote monitoring by study staff.
“Nearly 100 years since the discovery of insulin,” said NIDDK Director Dr. Griffin P. Rodgers, “a successful artificial pancreas would mark another huge step toward better health for people with type 1 diabetes.”
Roughly 25% of veterans have diabetes mellitus (DM) as opposed to 9% of the general public. A small percentage of veterans have type 1 DM, which according to research, can be caused by both physical and mental trauma that affects the pancreas.
“Managing type 1 diabetes currently requires a constant juggling act between checking bloodglucose levels frequently and delivering just the right amount of insulin while taking into account meals, physical activity, and other aspects of daily life, where a missed or wrong delivery could lead to potential complications,” said Dr. Andrew Bremer, of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). But that may change soon as we draw near to a functional “artificial pancreas,” a fully automated system that will sense rising glucose levels and adjust insulin automatically.
The FDA approved a hybrid model of an artificial pancreas in 2016, which still required users to adjust insulin intake. Now, 4 separate projects are designed to be the “potential last steps” toward requesting regulatory approval for permanent use of a fully automated system, according to NIDDK. The research studies beginning this year will look at safety, efficacy, user-friendliness, physical and emotional health of participants, and cost. The participants will live at home and monitor themselves with remote monitoring by study staff.
“Nearly 100 years since the discovery of insulin,” said NIDDK Director Dr. Griffin P. Rodgers, “a successful artificial pancreas would mark another huge step toward better health for people with type 1 diabetes.”
Genetically guided warfarin dosing can lower AE risk

WASHINGTON, DC—Using genetic testing to guide warfarin dosing can lower the risk of combined adverse events (AEs) after elective orthopedic surgery, according to the GIFT trial.
In this trial, investigators found that genotype-guided warfarin dosing was associated with a lower risk of combined AEs—confirmed venous thromboembolism (VTE), warfarin overdose, major bleeding, and death—when compared to clinically based warfarin dosing.
There were no deaths during this trial, so the researchers were unable to assess whether genotype-guided dosing actually reduced mortality risk.
However, they believe these findings could have implications for a broad population of patients starting warfarin therapy.
The findings were presented at the American College of Cardiology’s 66th Annual Scientific Session (abstract 411-14).
“The way we dose warfarin clinically is trial-and-error dosing,” said study investigator Brian F. Gage, MD, of Washington University School of Medicine in St. Louis, Missouri.
“We often start patients on 5 mg daily and don’t find out who is very sensitive to warfarin until their INR is 4 or more, indicating an overdose. Based on our results, as compared with optimized clinical dosing, pharmacogenetic dosing did better overall, meaning this group of patients had a lower rate of adverse events.”
Dr Gage also noted that the clinical dosing used in this trial was likely better than standard dosing used in clinical practice.
In this trial, the researchers used a computer-based, real-time interface that estimated the therapeutic dose and provided recommendations for adjusting dose based on a patient’s age, height, weight, interactions with other medications, and other clinical factors.
Trial interventions
GIFT included 1597 patients age 65 and older who were undergoing elective knee or hip replacement surgery. Most patients were female (63.8%) and Caucasian (91.1%).
The patients were genotyped for genetic variants that influence warfarin sensitivity (CYP2C9*2, CYP2C9*3), warfarin metabolism (VKORC1), and vitamin K recycling (CYP4F2).
They were randomized to receive clinical dosing or genotype-guided dosing (in addition to clinical factors being taken into account). The patients were also randomly assigned to a target international normalized ratio (INR) of either 1.8 or 2.5.
For the first 11 days of therapy, warfarin dosing in both arms was guided by a web application that incorporated clinical factors in all patients and genotype in patients randomized to genotype-guided dosing.
Most (94%) of the time, prescribers gave the dose that was recommended. After 11 days of therapy, they were free to continue the current warfarin dose or make adjustments.
Patients were monitored using standard INR testing, and most underwent screening with lower extremity Doppler ultrasound 3 to 7 weeks after arthroplasty to check for clots.
The investigators followed patients for 90 days and assessed the primary outcome through day 30, although VTEs detected through day 60 were also included in the primary outcome.
Results
The primary outcome—a composite of confirmed VTE, warfarin overdose (INR ≥ 4), major bleeding, and death—occurred in 14.7% of patients in the clinical arm and 10.8% in the genotype-guided arm (P=0.018).
The relative rate of the primary outcome was 0.73 (95% CI, 0.56 - 0.95). The relative rate was 0.24 (95% CI, 0.05 - 1.14) for major bleeding, 0.71 (95% CI, 0.51 - 0.99) for INR ≥ 4.0, and 0.85 (95% CI, 0.54 - 1.34) for VTE.
There were no deaths at the 30-day follow-up point, and 1 patient was lost to follow-up.
“Before GIFT, we had a good idea of how these genes and clinical factors affected the dose of warfarin,” Dr Gage said. “What we didn’t know is whether taking genotype into account improved outcomes. It turns out that the genes that regulate warfarin metabolism and sensitivity and vitamin K use are highly variable, so we can’t simply look at patients and predict their therapeutic warfarin dose.”
“The GIFT trial is an example of personalized medicine. If the patient stays in a safe INR range, warfarin is an incredibly effective and safe drug. By getting the dose approximately right from the get-go, we’re less likely to have the patient overdose and can lower the risk of complications.”
Dr Gage said future research could combine GIFT with prior pharmacogenetic trials in a meta-analysis and should determine what other genetic variations predict response to anticoagulants.
Additionally, as clinical and genetic factors affecting warfarin dose requirements vary by race, dosing algorithms tailored to ancestry may be beneficial.
Dr Gage also said he hopes genetic and clinical dosing algorithms will be integrated within electronic medical records.
“The hope is that when a physician starts a prescription of warfarin, electronic medical records will seamlessly give a prudent recommendation to help the doctor come up with the right dose,” he said. ![]()
WASHINGTON, DC—Using genetic testing to guide warfarin dosing can lower the risk of combined adverse events (AEs) after elective orthopedic surgery, according to the GIFT trial.
In this trial, investigators found that genotype-guided warfarin dosing was associated with a lower risk of combined AEs—confirmed venous thromboembolism (VTE), warfarin overdose, major bleeding, and death—when compared to clinically based warfarin dosing.
There were no deaths during this trial, so the researchers were unable to assess whether genotype-guided dosing actually reduced mortality risk.
However, they believe these findings could have implications for a broad population of patients starting warfarin therapy.
The findings were presented at the American College of Cardiology’s 66th Annual Scientific Session (abstract 411-14).
“The way we dose warfarin clinically is trial-and-error dosing,” said study investigator Brian F. Gage, MD, of Washington University School of Medicine in St. Louis, Missouri.
“We often start patients on 5 mg daily and don’t find out who is very sensitive to warfarin until their INR is 4 or more, indicating an overdose. Based on our results, as compared with optimized clinical dosing, pharmacogenetic dosing did better overall, meaning this group of patients had a lower rate of adverse events.”
Dr Gage also noted that the clinical dosing used in this trial was likely better than standard dosing used in clinical practice.
In this trial, the researchers used a computer-based, real-time interface that estimated the therapeutic dose and provided recommendations for adjusting dose based on a patient’s age, height, weight, interactions with other medications, and other clinical factors.
Trial interventions
GIFT included 1597 patients age 65 and older who were undergoing elective knee or hip replacement surgery. Most patients were female (63.8%) and Caucasian (91.1%).
The patients were genotyped for genetic variants that influence warfarin sensitivity (CYP2C9*2, CYP2C9*3), warfarin metabolism (VKORC1), and vitamin K recycling (CYP4F2).
They were randomized to receive clinical dosing or genotype-guided dosing (in addition to clinical factors being taken into account). The patients were also randomly assigned to a target international normalized ratio (INR) of either 1.8 or 2.5.
For the first 11 days of therapy, warfarin dosing in both arms was guided by a web application that incorporated clinical factors in all patients and genotype in patients randomized to genotype-guided dosing.
Most (94%) of the time, prescribers gave the dose that was recommended. After 11 days of therapy, they were free to continue the current warfarin dose or make adjustments.
Patients were monitored using standard INR testing, and most underwent screening with lower extremity Doppler ultrasound 3 to 7 weeks after arthroplasty to check for clots.
The investigators followed patients for 90 days and assessed the primary outcome through day 30, although VTEs detected through day 60 were also included in the primary outcome.
Results
The primary outcome—a composite of confirmed VTE, warfarin overdose (INR ≥ 4), major bleeding, and death—occurred in 14.7% of patients in the clinical arm and 10.8% in the genotype-guided arm (P=0.018).
The relative rate of the primary outcome was 0.73 (95% CI, 0.56 - 0.95). The relative rate was 0.24 (95% CI, 0.05 - 1.14) for major bleeding, 0.71 (95% CI, 0.51 - 0.99) for INR ≥ 4.0, and 0.85 (95% CI, 0.54 - 1.34) for VTE.
There were no deaths at the 30-day follow-up point, and 1 patient was lost to follow-up.
“Before GIFT, we had a good idea of how these genes and clinical factors affected the dose of warfarin,” Dr Gage said. “What we didn’t know is whether taking genotype into account improved outcomes. It turns out that the genes that regulate warfarin metabolism and sensitivity and vitamin K use are highly variable, so we can’t simply look at patients and predict their therapeutic warfarin dose.”
“The GIFT trial is an example of personalized medicine. If the patient stays in a safe INR range, warfarin is an incredibly effective and safe drug. By getting the dose approximately right from the get-go, we’re less likely to have the patient overdose and can lower the risk of complications.”
Dr Gage said future research could combine GIFT with prior pharmacogenetic trials in a meta-analysis and should determine what other genetic variations predict response to anticoagulants.
Additionally, as clinical and genetic factors affecting warfarin dose requirements vary by race, dosing algorithms tailored to ancestry may be beneficial.
Dr Gage also said he hopes genetic and clinical dosing algorithms will be integrated within electronic medical records.
“The hope is that when a physician starts a prescription of warfarin, electronic medical records will seamlessly give a prudent recommendation to help the doctor come up with the right dose,” he said. ![]()
WASHINGTON, DC—Using genetic testing to guide warfarin dosing can lower the risk of combined adverse events (AEs) after elective orthopedic surgery, according to the GIFT trial.
In this trial, investigators found that genotype-guided warfarin dosing was associated with a lower risk of combined AEs—confirmed venous thromboembolism (VTE), warfarin overdose, major bleeding, and death—when compared to clinically based warfarin dosing.
There were no deaths during this trial, so the researchers were unable to assess whether genotype-guided dosing actually reduced mortality risk.
However, they believe these findings could have implications for a broad population of patients starting warfarin therapy.
The findings were presented at the American College of Cardiology’s 66th Annual Scientific Session (abstract 411-14).
“The way we dose warfarin clinically is trial-and-error dosing,” said study investigator Brian F. Gage, MD, of Washington University School of Medicine in St. Louis, Missouri.
“We often start patients on 5 mg daily and don’t find out who is very sensitive to warfarin until their INR is 4 or more, indicating an overdose. Based on our results, as compared with optimized clinical dosing, pharmacogenetic dosing did better overall, meaning this group of patients had a lower rate of adverse events.”
Dr Gage also noted that the clinical dosing used in this trial was likely better than standard dosing used in clinical practice.
In this trial, the researchers used a computer-based, real-time interface that estimated the therapeutic dose and provided recommendations for adjusting dose based on a patient’s age, height, weight, interactions with other medications, and other clinical factors.
Trial interventions
GIFT included 1597 patients age 65 and older who were undergoing elective knee or hip replacement surgery. Most patients were female (63.8%) and Caucasian (91.1%).
The patients were genotyped for genetic variants that influence warfarin sensitivity (CYP2C9*2, CYP2C9*3), warfarin metabolism (VKORC1), and vitamin K recycling (CYP4F2).
They were randomized to receive clinical dosing or genotype-guided dosing (in addition to clinical factors being taken into account). The patients were also randomly assigned to a target international normalized ratio (INR) of either 1.8 or 2.5.
For the first 11 days of therapy, warfarin dosing in both arms was guided by a web application that incorporated clinical factors in all patients and genotype in patients randomized to genotype-guided dosing.
Most (94%) of the time, prescribers gave the dose that was recommended. After 11 days of therapy, they were free to continue the current warfarin dose or make adjustments.
Patients were monitored using standard INR testing, and most underwent screening with lower extremity Doppler ultrasound 3 to 7 weeks after arthroplasty to check for clots.
The investigators followed patients for 90 days and assessed the primary outcome through day 30, although VTEs detected through day 60 were also included in the primary outcome.
Results
The primary outcome—a composite of confirmed VTE, warfarin overdose (INR ≥ 4), major bleeding, and death—occurred in 14.7% of patients in the clinical arm and 10.8% in the genotype-guided arm (P=0.018).
The relative rate of the primary outcome was 0.73 (95% CI, 0.56 - 0.95). The relative rate was 0.24 (95% CI, 0.05 - 1.14) for major bleeding, 0.71 (95% CI, 0.51 - 0.99) for INR ≥ 4.0, and 0.85 (95% CI, 0.54 - 1.34) for VTE.
There were no deaths at the 30-day follow-up point, and 1 patient was lost to follow-up.
“Before GIFT, we had a good idea of how these genes and clinical factors affected the dose of warfarin,” Dr Gage said. “What we didn’t know is whether taking genotype into account improved outcomes. It turns out that the genes that regulate warfarin metabolism and sensitivity and vitamin K use are highly variable, so we can’t simply look at patients and predict their therapeutic warfarin dose.”
“The GIFT trial is an example of personalized medicine. If the patient stays in a safe INR range, warfarin is an incredibly effective and safe drug. By getting the dose approximately right from the get-go, we’re less likely to have the patient overdose and can lower the risk of complications.”
Dr Gage said future research could combine GIFT with prior pharmacogenetic trials in a meta-analysis and should determine what other genetic variations predict response to anticoagulants.
Additionally, as clinical and genetic factors affecting warfarin dose requirements vary by race, dosing algorithms tailored to ancestry may be beneficial.
Dr Gage also said he hopes genetic and clinical dosing algorithms will be integrated within electronic medical records.
“The hope is that when a physician starts a prescription of warfarin, electronic medical records will seamlessly give a prudent recommendation to help the doctor come up with the right dose,” he said. ![]()
FDA issues update on breast implant-associated ALCL

The US Food and Drug Administration (FDA) has issued an update on breast implant-associated anaplastic large-cell lymphoma (BIA-ALCL).
The agency said that, as of February 1, it has received 359 reports of BIA-ALCL.
However, the actual number of BIA-ALCL cases remains difficult to determine due to limitations in reporting and a lack of implant sales data.
The FDA also noted that most of the available data suggest BIA-ALCL occurs more frequently in patients who receive implants with textured surfaces rather than smooth surfaces.
The full FDA update includes background information on BIA-ALCL, a summary of medical device reports (MDRs) and the medical literature, as well as recommendations for patient care.
Background and MDRs
The FDA first identified a possible association between ALCL and breast implants in 2011.
The agency now concurs with the World Health Organization’s designation of BIA-ALCL as a rare T-cell lymphoma occurring in patients with breast implants.
The FDA continues to collect and review information about BIA-ALCL. This includes reviewing MDRs and the medical literature, as well as exchanging information with other international regulators and scientific experts.
The FDA said it has received 359 MDRs of BIA-ALCL, including 9 cases in which the patient died.
Information on the implant surface was available for 239 cases, and 203 of these cases involved textured implants.
Information on the implant filling was available in 312 cases. Of these, 186 patients had implants filled with silicone gel, and 126 had implants filled with saline.
Recommendations
The FDA said healthcare providers performing breast implant surgery should provide patients with the manufacturers’ labeling as well as any other educational materials before surgery and discuss with patients the benefits and risks of the different types of implants.
Providers should consider the possibility of BIA-ALCL when a patient presents with late-onset, persistent peri-implant seroma. The FDA noted that, in some cases, patients presented with capsular contracture or masses adjacent to the breast implant.
Patients with suspected BIA-ALCL should be referred to an appropriate specialist.
When testing for BIA-ALCL, providers should collect fresh seroma fluid and representative portions of the capsule and send these samples for pathology tests.
Diagnostic evaluation of patients with suspected BIA-ALCL should include cytological evaluation of seroma fluid with Wright Giemsa stained smears and cell block immunohistochemistry testing for cluster of differentiation and anaplastic lymphoma kinase markers.
When choosing a treatment approach for patients with BIA-ALCL, providers should consider current clinical practice guidelines, such as those from the National Comprehensive Cancer Network (included in the guidelines for T-cell lymphomas) or the Plastic Surgery Foundation.
Finally, providers should report all confirmed cases of BIA-ALCL to the FDA and to the Patient Registry and Outcomes for Breast Implants and Anaplastic Large Cell Lymphoma (ALCL) Etiology and Epidemiology (PROFILE Registry). ![]()
The US Food and Drug Administration (FDA) has issued an update on breast implant-associated anaplastic large-cell lymphoma (BIA-ALCL).
The agency said that, as of February 1, it has received 359 reports of BIA-ALCL.
However, the actual number of BIA-ALCL cases remains difficult to determine due to limitations in reporting and a lack of implant sales data.
The FDA also noted that most of the available data suggest BIA-ALCL occurs more frequently in patients who receive implants with textured surfaces rather than smooth surfaces.
The full FDA update includes background information on BIA-ALCL, a summary of medical device reports (MDRs) and the medical literature, as well as recommendations for patient care.
Background and MDRs
The FDA first identified a possible association between ALCL and breast implants in 2011.
The agency now concurs with the World Health Organization’s designation of BIA-ALCL as a rare T-cell lymphoma occurring in patients with breast implants.
The FDA continues to collect and review information about BIA-ALCL. This includes reviewing MDRs and the medical literature, as well as exchanging information with other international regulators and scientific experts.
The FDA said it has received 359 MDRs of BIA-ALCL, including 9 cases in which the patient died.
Information on the implant surface was available for 239 cases, and 203 of these cases involved textured implants.
Information on the implant filling was available in 312 cases. Of these, 186 patients had implants filled with silicone gel, and 126 had implants filled with saline.
Recommendations
The FDA said healthcare providers performing breast implant surgery should provide patients with the manufacturers’ labeling as well as any other educational materials before surgery and discuss with patients the benefits and risks of the different types of implants.
Providers should consider the possibility of BIA-ALCL when a patient presents with late-onset, persistent peri-implant seroma. The FDA noted that, in some cases, patients presented with capsular contracture or masses adjacent to the breast implant.
Patients with suspected BIA-ALCL should be referred to an appropriate specialist.
When testing for BIA-ALCL, providers should collect fresh seroma fluid and representative portions of the capsule and send these samples for pathology tests.
Diagnostic evaluation of patients with suspected BIA-ALCL should include cytological evaluation of seroma fluid with Wright Giemsa stained smears and cell block immunohistochemistry testing for cluster of differentiation and anaplastic lymphoma kinase markers.
When choosing a treatment approach for patients with BIA-ALCL, providers should consider current clinical practice guidelines, such as those from the National Comprehensive Cancer Network (included in the guidelines for T-cell lymphomas) or the Plastic Surgery Foundation.
Finally, providers should report all confirmed cases of BIA-ALCL to the FDA and to the Patient Registry and Outcomes for Breast Implants and Anaplastic Large Cell Lymphoma (ALCL) Etiology and Epidemiology (PROFILE Registry). ![]()
The US Food and Drug Administration (FDA) has issued an update on breast implant-associated anaplastic large-cell lymphoma (BIA-ALCL).
The agency said that, as of February 1, it has received 359 reports of BIA-ALCL.
However, the actual number of BIA-ALCL cases remains difficult to determine due to limitations in reporting and a lack of implant sales data.
The FDA also noted that most of the available data suggest BIA-ALCL occurs more frequently in patients who receive implants with textured surfaces rather than smooth surfaces.
The full FDA update includes background information on BIA-ALCL, a summary of medical device reports (MDRs) and the medical literature, as well as recommendations for patient care.
Background and MDRs
The FDA first identified a possible association between ALCL and breast implants in 2011.
The agency now concurs with the World Health Organization’s designation of BIA-ALCL as a rare T-cell lymphoma occurring in patients with breast implants.
The FDA continues to collect and review information about BIA-ALCL. This includes reviewing MDRs and the medical literature, as well as exchanging information with other international regulators and scientific experts.
The FDA said it has received 359 MDRs of BIA-ALCL, including 9 cases in which the patient died.
Information on the implant surface was available for 239 cases, and 203 of these cases involved textured implants.
Information on the implant filling was available in 312 cases. Of these, 186 patients had implants filled with silicone gel, and 126 had implants filled with saline.
Recommendations
The FDA said healthcare providers performing breast implant surgery should provide patients with the manufacturers’ labeling as well as any other educational materials before surgery and discuss with patients the benefits and risks of the different types of implants.
Providers should consider the possibility of BIA-ALCL when a patient presents with late-onset, persistent peri-implant seroma. The FDA noted that, in some cases, patients presented with capsular contracture or masses adjacent to the breast implant.
Patients with suspected BIA-ALCL should be referred to an appropriate specialist.
When testing for BIA-ALCL, providers should collect fresh seroma fluid and representative portions of the capsule and send these samples for pathology tests.
Diagnostic evaluation of patients with suspected BIA-ALCL should include cytological evaluation of seroma fluid with Wright Giemsa stained smears and cell block immunohistochemistry testing for cluster of differentiation and anaplastic lymphoma kinase markers.
When choosing a treatment approach for patients with BIA-ALCL, providers should consider current clinical practice guidelines, such as those from the National Comprehensive Cancer Network (included in the guidelines for T-cell lymphomas) or the Plastic Surgery Foundation.
Finally, providers should report all confirmed cases of BIA-ALCL to the FDA and to the Patient Registry and Outcomes for Breast Implants and Anaplastic Large Cell Lymphoma (ALCL) Etiology and Epidemiology (PROFILE Registry). ![]()
Adult ADHD: Pharmacologic treatment in the DSM-5 era
Attention-deficit/hyperactivity disorder (ADHD) is common; it affects 5% to 7% of children1,2 and 4% to 5% of all adults.3,4 Pediatric ADHD often persists into adulthood, as 65% of individuals diagnosed as children retain impairing symptoms by age 25.4
The prevalence of ADHD in childhood is 2 to 3 times greater among boys than girls, but more comparable between the sexes in adulthood.2 Symptoms could be more easily overlooked in women because of the greater prominence of hyperactivity and impulsivity-type symptoms in men.5
Untreated ADHD is associated with significant costs. Adults with ADHD have increased unemployment rates, poor work performance, and comparatively lower educational performance.6,7 Compared with non-ADHD adults, those with ADHD have:
- more traffic violations and accidents and a higher rate of criminal convictions and incarcerations8,9
- a mortality rate almost 2 times higher, with the greatest differences seen in deaths by suicide and accidents.10,11
Adults with ADHD also are more likely to have a comorbid psychiatric disorder—in particular, substance use11—and often are in treatment for other mental or substance use disorders. Among adults who meet diagnostic criteria for ADHD, approximately only 10% are receiving treatment for ADHD symptoms.3,12
Changes in DSM-5
Revisions within DSM-5 simplify ADHD’s diagnosis—and make it more difficult to ignore in 
DSM-5 also provides examples of behaviors more commonly found in adults, such as “feelings of restlessness,” compared with DSM-IV’s “often runs about or climbs excessively in situations in which it is inappropriate.” Finally, ADHD now may be diagnosed in a person with an autism spectrum disorder who meets diagnostic criteria for both disorders.13,14
Identifying ADHD in adults
ADHD diagnosis in adults is made through careful clinical interviewing. For example, ask about what factors motivated an individual to seek evaluation for ADHD. Often, patients present after a change in responsibility at work or at home, such as a promotion or birth/adoption of a new child.
Consider incorporating a brief screen for adult ADHD in all new outpatient evaluations (Table 2).15 Screen for other psychiatric disorders as well; comorbidity with ADHD is high, and hyperactivity and inattention symptoms may result from anxiety, depression, or substance use.
Screen for learning disorders, which can present with ADHD symptoms (such as poor concentration) when the individual attempts difficult tasks. Evaluate for risk factors associated with ADHD medications, such as a history of cardiac problems, hypertension, or tachycardia. A family history of ADHD is found in approximately 80% of cases.16,17 Determine the presence of ADHD symptoms in childhood. A careful review of the educational history often reveals long-term underachievement and struggles in school. Patients may report a chronic history of poor attention or feelings of restlessness in school. Sometimes problems do not become apparent until high school or college; some individuals, especially those with high intelligence, compensate for deficits and show fewer overt symptoms of impairment until later in their education.18Occupational history also may be revealing:
- How are they performing at work?
- Have they changed jobs multiple times in a short period?
- Do they have difficulty organizing tasks?
Subtle ADHD signs include time of arrival to appointments (eg, late or extremely early), missing data on intake paperwork, and a history of losing keys or phones.
Neuropsychological testing. Some clinicians routinely include neuropsychological testing in an adult ADHD evaluation, but these studies have shown inconsistent cognitive deficits in people with ADHD.19,20 No distinct psychometric cognitive test or profile is diagnostic of ADHD or its subtypes.21
Treatment and follow-up care
Four general categories of medications are used to treat ADHD in children and adults: 
After starting a patient on medication, at each follow-up appointment ask about new cardiac symptoms or diagnoses, new family history of cardiac problems, or new medications. Measure pulse and blood pressure every 1 to 3 months. Measure vital signs more frequently during titration and weaning periods.23
Stimulant medications
Amphetamines have dual action: they block the reuptake of dopamine and noradrenaline by competitive inhibition of the transporters and promote the release of dopamine and noradrenaline by competitive inhibition of the intraneuronal vesicular monoamine transporter.24
For most amphetamine products, including dextroamphetamine and amphetamine mixed salts, the target dosage is approximately 0.5 mg/kg. Start at a lower dosage, however, and rapidly titrate weekly so patients can adjust to the medication while not becoming frustrated with a lack of efficacy. Some patients may require short-acting forms with dosing 3 times per day, and twice daily dosing is not uncommon with extended-release (ER) formulations.
Metabolism of most amphetamine products—with the exception of lisdexamfetamine—involves the cytochrome P450 (CYP) enzyme CYP2D6, leading to the formation of the metabolite 4-hydroxyamphetamine.25 The pharmacokinetics of lisdexamfetamine in slow or ultra-rapid CYP2D6 metabolizers has not been evaluated (Shire US Inc., written communication, July 2014).
Agents that alter urinary pH can affect blood levels of amphetamine. Acidifying agents decrease amphetamine blood levels, while alkalinizing agents increase amphetamine blood levels.26
Lisdexamfetamine contains L-lysine, an essential amino acid, covalently bound to d-amphetamine via an amide linking group.27 After absorption, lisdexamfetamine is metabolized by rate-limited, enzymatic hydrolysis to yield d-amphetamine and L-lysine.24,28,29 A starting dose of 40 mg is advised; twice-daily dosing rarely is required.
A meta-analysis of 5 randomized, controlled trials in the treatment of adult ADHD showed a response rate of 70% for lisdexamfetamine compared with 37% for placebo. Trial duration ranged from 4 to 14 weeks, with dosages of 30 to 70 mg/d.30 Another analysis of data from lisdexamfetamine trials predicted an effect size of 1.07 for European adults, which is larger than the 0.8 threshold for large effect sizes.31
Methylphenidate products. Methylphenidate’s main action is through enhancement of dopamine signaling by blockade of the dopamine transporter, leading to increases in extracellular dopamine as well as norepinephrine.22,32 Optimized dosing is generally 1 mg/kg per day, and dosing up to 80 to 120 mg/d is not unusual.33
Dexmethylphenidate is the more pharmacologically active enantiomer of racemic methylphenidate and is twice as potent.34-36 Target dosing of dexmethylphenidate should be one-half as much (ie, 0.5 mg/kg per day) as other methylphenidate products.37
Managing stimulants’ side effects
Amphetamines’ side effects may include insomnia, dry mouth, decreased appetite, weight loss, headaches, and anxiety. To help minimize sleep problems, advise patients to take a second immediate-release dose at noon, rather than later in the afternoon. The longer-acting formulation taken once per day in the morning may be offered as an alternative. Some patients may experience improved sleep because of diminished bedtime ruminations.
Oral rinses, such as Biotène, could help reduce discomfort associated with dry mouth. Pilocarpine, which stimulates saliva production, is another option if rinses are not effective. To address decreased appetite, advise patients to take their medication after they eat. Switching from an immediate-release amphetamine to a longer-acting formulation also may lessen symptoms. Lisdexamfetamine might be a good choice for adults with ADHD who have undergone bariatric surgeries because it is absorbed in the small bowel.38
Methylphenidate has no interactions with CYP enzymes, making it an attractive option for patients taking CYP inhibiting or stimulating medications.39 The most common side effects of methylphenidate products include appetite loss, insomnia, irritability, and tachycardia. Some side effects will abate after 1 to 2 weeks of treatment, but persistence of insomnia and appetite loss may require a decrease in dosage. In rare cases, methylphenidate may produce tics, exacerbate an existing tic disorder, or produce mania or psychosis.40,41 Methylphenidate inhibits the metabolism of tricyclic antidepressants; use methylphenidate with caution in patients taking monoamine oxidase inhibitors.42,43Cardiovascular risks. Possible cardiovascular risks associated with stimulant use have gained widespread attention, although research has not demonstrated an increased risk of serious cardiovascular events in young and middle-aged adults receiving stimulant medications for ADHD.44 Nonetheless, obtain a thorough medical history in adult patients, including cardiac history, family history of cardiac disease, history of any cardiac symptoms, and a medication history. Baseline ECG is not required.45
Screen for a family history of sudden death in a young person, sudden death during exercise, cardiac arrhythmia, cardiomyopathies (including hypertrophic cardiomyopathy, dilated cardiomyopathy, and right ventricular cardiomyopathy), prolonged QT interval, short QT syndrome, Brugada syndrome, Wolff-Parkinson-White syndrome, Marfan syndrome, and an event requiring resuscitation in a family member younger than 35, including syncope requiring rescuscitation.23 If fainting spells, palpitations, chest pain, or other symptoms suggest preexisting cardiovascular disease, refer the patient promptly to a cardiologist.
Peripheral vasculopathy, including Raynaud’s phenomenon, is a lesser known side effect associated with stimulants.46 Symptoms are usually mild, but in rare instances stimulants are associated with digital ulceration or soft tissue breakdown.47 Advise patients to tell you if they experience any new symptoms of numbness, pain, skin color changes, or sensitivity to temperature in fingers and toes. Signs and symptoms generally improve after dosage reduction or discontinuation of the stimulant medication.46 Referral to a rheumatologist might be appropriate if symptoms persist.
A noradrenergic medication
Atomoxetine is a potent, selective inhibitor of the presynaptic noradrenaline transporter that increases the availability of extracellular noradrenaline in the prefrontal cortex.48,49 Atomoxetine may be a good alternative for adult patients with ADHD and comorbid anxiety.50
For adults, the optimal starting dosage is 40 mg in the morning for 1 week, followed by an increase to 80 mg. Insufficient dosing is common with atomoxetine, and the dosage could be increased to 100 mg/d.51 Dosing twice per day may be associated with higher rates of insomnia.
Atomoxetine’s efficacy for managing ADHD in adults has been consistently demonstrated by 6 placebo-controlled trials of 10 to 16 weeks, 3 placebo-controlled 6-month trials, and a 1-year maintenance-of-response trial.52 Atomoxetine was found to have an effect size of 0.45 (medium) (number needed to treat [NNT] = 5).53-55The most common adverse effects include nausea, dry mouth, insomnia, and erectile dysfunction. Small increases in heart rate and blood pressure have been reported, so use this medication with caution in patients for whom this might be problematic. Atomoxetine is metabolized by CYP2D6; 7% of white individuals have a genotype corresponding to a nonfunctional CYP2D6 enzyme.56-58
Alpha-2 adrenergic agonists
Clonidine and guanfacine are antihypertensive drugs that induce peripheral sympathoinhibition via the stimulation of receptors. Clonidine binds equally to adrenergic receptor subtypes α-2A, α-2B, and α-2C (as well as to α-1 and β subtypes, histamine receptors, and possibly dopamine receptors).59,60 Guanfacine binds preferentially to postsynaptic α-2A adrenoceptors in the prefrontal cortex, which have been implicated in attentional and organizational functions.61,62
ER guanfacine and ER clonidine are FDA-approved as monotherapy for ADHD in children and adolescents.
Efficacy in adults. A small (N = 17), double-blind, placebo-controlled, crossover study comparing immediate-release guanfacine and dextroamphetamine found that both medications significantly reduced adult ADHD symptoms, as measured with the DSM-IV Adult Behavior Checklist for Adults.63
No trials have been published regarding the efficacy of ER clonidine in adults with ADHD; adverse effects including sedation, bradycardia, and hypotension may limit its use. One study compared the supplemental use of ER guanfacine (1 to 6 mg/d) or a matching placebo in 26 adults with ADHD who had suboptimal response to stimulant-only treatment. After 10 weeks, both the guanfacine ER and placebo groups showed statistically significant improvements in ADHD symptoms and general functioning. The treatments did not differ in efficacy, safety, or tolerability.64
Adverse events. Compared with clonidine, guanfacine has less CNS depressant and hypotensive activity.58 A phase I trial of ER guanfacine in healthy adults found its single-dose pharmacokinetic properties in 1-, 2-, and 4-mg tablets appeared to be statistically linear. Somnolence—the most common treatment-emergent adverse effect—occurred in 33 of 52 participants (63.5%). All mean vital-sign measurements and ECG parameters remained within normal limits after dosing, and no marked changes from baseline measurements were noted.65
Antidepressants
Antidepressants used in ADHD treatment include bupropion and tricyclic antidepressants.
Bupropion is a noradrenaline and dopamine reuptake inhibitor and is considered to be a mild psychostimulant because of its amphetamine-derived chemical structure.66,67 It generally is considered a third-line medication when stimulants have not improved ADHD symptoms or are not tolerated.
A 2011 meta-analysis examined 5 randomized, controlled trials including 175 adults treated with bupropion for ADHD. Bupropion was found to be more effective than placebo (NNT = 5), although bupropion’s therapeutic benefits were not observed until weeks 5 and 6. Its effects were less pronounced than those of methylphenidate. Mean daily dosages were 362 mg for the bupropion SR trials and 393 mg for the bupropion XL trial.68
Tricyclics. Desipramine and nortriptyline have been found to be efficacious in childhood ADHD,69,70 although cardiovascular risk and toxicity in overdose limit their use.71
1. Polanczyk G, de Lima MS, Horta BL, et al. The worldwide prevalence of ADHD: a systemic review and metaregression analysis. Am J Psychiatry. 2007;164(6):942-948.
2. Simon V, Czobor P, Bálint S, et al. Prevalence and correlates of adult attention-deficit hyperactivity disorder: meta-analysis. Br J Psychiatry. 2009;194(3):204-211.
3. Kessler RC, Adler L, Barkley R, et al. The prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey Replication. Am J Psychiatry. 2006;163(4):716-723.
4. Faraone S, Biederman J, Mick E. The age-dependent decline of attention deficit hyperactivity disorder: a meta-analysis of follow-up studies. Psychol Med. 2006;36(2):159-165.
5. Gershon J. A meta-analytic review of gender differences in ADHD. J Atten Disord. 2002;5(3):143-154.
6. Halmøy A, Fasmer OB, Gillberg C, et al. Occupational outcome in adult ADHD: impact of symptom profile, comorbid psychiatric problems, and treatment: a cross-sectional study of 414 clinically diagnosed adult ADHD patients. J Atten Disord. 2009;13(2):175-187.
7. Kuriyan AB, Pelham WE Jr, Molina BS, et al. Young adult educational and vocational outcomes of children diagnosed with ADHD. J Abnorm Child Psychol. 2013;41(1):27-41.
8. Murphy K, Barkley RA. Attention deficit hyperactivity disorder in adults: comorbidities and adaptive impairment. Compr Psychiatry. 1996;37(6):393-401.
9. Mannuzza S, Klein RG, Mouton JL 3rd. Lifetime criminality among boys with attention deficit hyperactivity disorder: a prospective follow-up study into adulthood using official arrest records. Psychiatry Res. 2008;160(3):237-246.
10. Dalsgaard S, Østergaard SD, Leckman JF, et al. Mortality in children, adolescents, and adults with attention deficit hyperactivity disorder: a nationwide cohort study. Lancet. 2015;385(9983):2190-2196.
11. Barbaresi WJ, Colligan RC, Weaver AL, et al. Mortality, ADHD, and psychosocial adversity in adults with childhood ADHD: a prospective study. Pediatrics. 2013;131(4):637-644.
12. Babcock T, Ornstein CS. Comorbidity and its impact in adult patients with attention-deficit/hyperactivity disorder: a primary care perspective. Postgrad Med. 2009;121(3):73-82.
13. Attention-deficit/hyperactivity disorder. In: Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013:59-66.
14. Attention-deficit/hyperactivity disorder. In: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000:78-85.
15. Kooij JJS. Adult ADHD: diagnostic assessment and treatment. 3rd ed. Amsterdam, Netherlands: Springer; 2013:34.
16. Faraone SV, Khan SA. Candidate gene studies of attention-deficit/hyperactivity disorder. J Clin Psychiatry. 2006;67(suppl 8):13-20.
17. Neale BM, Medland SE, Ripke S, et al; Psychiatric GWAS Consortium: ADHD Subgroup. Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2010;49(9):884-897.
18. Milioni AL, Chaim TM, Cavallet M, et al. High IQ may “mask” the diagnosis of ADHD by compensating for deficits in executive functions in treatment-naïve adults with ADHD [published online October 30, 2014]. J Atten Disord. pii: 1087054714554933.
19. Rapport MD, Chung KM, Shore G, et al. Upgrading the science and technology of assessment and diagnosis: laboratory and clinic-based assessment of children with ADHD. J Clin Child Psychol. 2000;29(4):555-568.
20. Woods SP, Lovejoy DW, Ball JD. Neuropsychological characteristics of adults with ADHD: a comprehensive review of initial studies. Clin Neuropsychol. 2002;16(1):12-34.
21. Lange KW, Hauser J, Lange KM, et al. Utility of cognitive neuropsychological assessment in attention-deficit/hyperactivity disorder. Atten Defic Hyperact Disord. 2014;6(4):241-248.
22. Arnold LE. Methylphenidate vs. amphetamine: comparative review. J Atten Disord. 2000;3(4):200-211.
23. Vetter VL Elia J, Erickson, C, et al; American Heart Association Council on Cardiovascular Disease in the Young Congenital Cardiac Defects Committee; American Heart Association Council on Cardiovascular Nursing. Cardiovascular monitoring of children and adolescents with heart disease receiving medications for attention deficit/hyperactivity disorder [corrected]: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young Congenital Cardiac Defects Committee and the Council on Cardiovascular Nursing [Erratum in: Circulation. 2009;120(7):e55-e59]. Circulation. 2008;117(18):2407-2423.
24. Seiden LS, Sabol KE, Ricaurte GA. Amphetamine: effects on catecholamine systems and behavior. Annu Rev Pharmacol Toxicol. 1993;33:639-677.
25. Wu D, Otton SV, Inaba T, et al. Interactions of amphetamine analogs with human liver CYP2D6. Biochem Pharmacol. 1997;53(11):1605-1612.
26. Vyvanse [package insert]. Lexington, MA: Shire Pharmaceuticals; 2015.
27. Pennick M. Absorption of lisdexamfetamine dimesylate and its enzymatic conversion to d-amphetamine. Neuropsychiatr Dis Treat. 2010;6:317-327.
28. Heal DJ, Smith SL, Gosden J, et al. Amphetamine, past and present—a pharmacological and clinical perspective. J Psychopharmacol. 2013;27(6):479-496.
29. Krishnan SM, Pennick M, Stark JG. Metabolism, distribution and elimination of lisdexamfetamine dimesylate: open-label, single-centre, phase I study in healthy adult volunteers. Clin Drug Invest. 2008;28(12):745-755.
30. Maneeton N, Maneeton B, Suttajit S, et al. Exploratory meta-analysis on lisdexamfetamine versus placebo in adult ADHD. Drug Des Devel Ther. 2014;8:1685-1693.
31. Fridman M, Hodgkins P, Kahle JS, et al. Predicted effect size of lisdexamfetamine treatment of attention deficit/hyperactivity disorder (ADHD) in European adults: estimates based on indirect analysis using a systematic review and meta-regression analysis. Eur Psychiatry. 2015;30(4):521-527.
32. Markowitz JS, DeVane CL, Pestreich L, et al. Session 1-87-differentiation of d-, L- and dl-methylphenidate through in vitro pharmacological screening. In: Abstracts: Oral and Poster Presentations of the NCDEU 45th Annual Meeting; June 6-9, 2005; Boca Raton, FL:186.
33. Spencer T, Biederman J, Wilens T, et al. A large, double-blind, randomized clinical trial of methylphenidate in the treatment of adults with attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57(5):456-463.
34. Teo SK, Stirling DI, Thomas SD, et al. Neurobehavioral effects of racemic threo-methylphenidate and its D and L enantiomers in rats. Pharmacol Biochem Behav. 2003;74(3):747-754.
35. Ding YS, Fowler JS, Volkow ND, et al. Chiral drugs: comparison of the pharmacokinetics of [11C]d-threo and L-threo-methylphenidate in the human and baboon brain. Psychopharmacol (Berl). 1997;131(1):71-78.
36. Davids E, Zhang K, Tarazi FI, et al. Stereoselective effects of methylphenidate on motor hyperactivity in juvenile rats induced by neonatal 6-hydroxydopamine lesioning. Psychopharmacol (Berl). 2002;160(1):92-98.
37. Srinivas NR, Hubbard JW, Quinn D, et al. Enantioselective pharmacokinetics and pharmacodynamics of dl-threo-methylphenidate in children with attention deficit hyperactivity disorder. Clin Pharmacol Ther. 1992;52(5):561-568.
38. Ermer JC, Haffey MB, Doll WJ, et al. Pharmacokinetics of lisdexamfetamine dimesylate after targeted gastrointestinal release or oral administration in healthy adults. Drug Metab Dispos. 2012;40(2):290-297.
39. DeVane CL, Markowitz JS, Carson SW, et al. Single-dose pharmacokinetics of methylphenidate in CYP2D6 extensive and poor metabolizers. J Clin Psychopharmacol. 2000;20(3):347-349.
40. Graham J, Coghill D. Adverse effects of pharmacotherapies for attention-deficit hyperactivity disorder: epidemiology, prevention and management. CNS Drugs. 2008;22(3):213-237.
41. Ross RG. Psychotic and manic-like symptoms during stimulant treatment of attention deficit hyperactivity disorder. Am J Psychiatry. 2006;163(7):1149-1152.
42. Shelton Clauson A, Elliott ES, Watson BD, et al. Coadministration of phenelzine and methylphenidate for treatment-resistant depression. Ann Pharmacother. 2004;38(3):508.
43. Markowitz JS, Patrick KS. Pharmacokinetic and pharmacodynamic drug interactions in the treatment of attention-deficit hyperactivity disorder. Clin Pharmacokinet. 2001;40(10):753-772.
44. Habel LA, Cooper WO, Sox CM, et al. ADHD medications and risk of serious cardiovascular events in young and middle-aged adults. JAMA. 2011;306(24):2673-2683.
45. Graham J, Banaschewski T, Buitelaar J, et al; European Guidelines Group. European guidelines on managing adverse effects of medication for ADHD. Eur Child Adolesc Psychiatry. 2011;20(1):17-37.
46. Goldman W, Seltzer R, Reuman P. Association between treatment with central nervous system stimulants and Raynaud’s syndrome in children: a retrospective case-control study of rheumatology patients. Arthritis Rheum. 2008;58(2):563-566.
47. Syed RH, Moore TL. Methylphenidate and dextroamphetamine-induced peripheral vasculopathy. J Clin Rheum. 2008;14(1):30-33.
48. Wilens TE. Mechanism of action of agents in attention-deficit/hyperactivity disorder. J Clin Psychiatry. 2006;67(suppl 8):32-38.
49. Bymaster FP, Katner JS, Nelson DL, et al. Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology. 2002;27(5):699-711.
50. Adler LA, Liebowitz M, Kronenberger W, et al. Atomoxetine treatment in adults with attention-deficit/hyperactivity disorder and comorbid social anxiety disorder. Depress Anxiety. 2009;26(3):212-221.
51. Clemow DB. Suboptimal dosing of Strattera (atomoxetine) for ADHD patients. Postgrad Med. 2014;126(5):196-198.
52. Camporeale A, Porsdal V, De Bruyckere K, et al. Safety and tolerability of atomoxetine in treatment of attention deficit hyperactivity disorder in adult patients: an integrated analysis of 15 clinical trials. J Psychopharmacol. 2015;29(1):3-14.
53. Young JL, Sarkis E, Qiao M, et al. Once-daily treatment with atomoxetine in adults with attention-deficit/hyperactivity disorder: a 24-week, randomized, double-blind, placebo-controlled trial. Clin Neuropharmacol. 2011;34(2):51-60.
54. Bitter I, Angyalosi A, Czobor P. Pharmacological treatment of adult ADHD. Curr Opin Psychiatry. 2012;25(6):529-534.
55. Faraone SV, Glatt SJ. A comparison of the efficacy of medications for adult attention-deficit/hyperactivity disorder using meta-analysis of effect sizes. J Clin Psychiatry. 2010;71(6):754-763.
56. Ring BJ, Gillespie JS, Eckstein JA, et al. Identification of the human cytochromes P450 responsible for atomoxetine metabolism. Drug Metab Dispos. 2002;30(3):319-323.
57. Farid NA, Bergstrom RF, Ziege EA, et al. Single-dose and steady state pharmacokinetics of tomoxetine in normal subjects. J Clin Pharmacol. 1985;25(4):296-301.
58. Mizutani T. PM frequencies of major CYPs in Asians and Caucasians. Drug Metab Rev. 2003;35(2-3):99-106.
59. Jasper JR, Lesnick JD, Chang LK, et al. Ligand efficacy and potency at recombinant alpha2 adrenergic receptors: agonist-mediated [35S]GTPgammaS binding. Biochem Pharmacol. 1998;55(7):1035-1043.
60. Ruggiero S, Clavenna A, Reale L, et al. Guanfacine for attention deficit and hyperactivity disorder in pediatrics: a systematic review and meta-analysis. Eur Neuropsychopharmacol. 2014;24(10):1578-1590.
61. Arnsten AF, Pliszka SR. Catecholamine influences on prefrontal cortical function: relevance to treatment of attention deficit/hyperactivity disorder and related disorders. Pharmacol Biochem Behav. 2011;99(2):211-216.
62. Uhlén S, Wikberg JE. Delineation of rat kidney alpha 2A- and alpha 2B-adrenoceptors with [3H]RX821002 radioligand binding: computer modelling reveals that guanfacine is an alpha 2A-selective compound. Eur J Pharmacol. 1991;202(2):235-243.
63. Taylor FB, Russo J. Comparing guanfacine and dextroamphetamine for the treatment of adult attention deficit/hyperactivity disorder. J Clin Psychopharmacol. 2001;21(2):223-228.
64. Butterfield ME, Saal J, Young B, et al. Supplementary guanfacine hydrochloride as a treatment of attention deficit hyperactivity disorder in adults: a double blind, placebo-controlled study. Psychiatry Res. 2016;236:136-141.
65. Swearingen D, Pennick M, Shojaei A, et al. A phase I, randomized, open-label, crossover study of the single-dose pharmacokinetic properties of guanfacine extended-release 1-, 2-, and 4-mg tablets in healthy adults. Clin Ther. 2007;29(4):617-625.
66. Cooper BR, Wang CM, Cox RF. Evidence that the acute behavioral and electrophysiological effects of bupropion (Wellbutrin) are mediated by a noradrenergic mechanism. Neuropsychopharmacology. 1994;11(2):133-141.
67. Reimherr FW, Hedges DW, Strong RE, et al. Bupropion SR in adults with ADHD: a short-term, placebo-controlled trial. Neuropsychiatr Dis Treat. 2005;1(3):245-251.
68. Maneeton N, Maneeton B, Srisurapanont M, et al. Bupropion for adults with attention-deficit hyperactivity disorder: meta-analysis of randomized, placebo-controlled trials. Psychiatry Clin Neurosci. 2011;65(7):611-617.
69. Biederman J, Baldessarini RJ, Wright V, et al. A double-blind placebo controlled study of desipramine in the treatment of ADD: I. Efficacy. J Am Acad Child Adolesc Psychiatry. 1989;28(5):777-784.
70. Spencer T, Biederman J, Wilens T, et al. Nortriptyline treatment of children with attention-deficit hyperactivity disorder and tic disorder or Tourette’s syndrome. J Am Acad Child Adolesc Psychiatry. 1993;32(1):205-210.
71. Bond DJ, Hadjipavlou G, Lam RW, et al. The Canadian Network for Mood and Anxiety Treatments (CANMAT) task force recommendations for the management of patients with mood disorders and comorbid attention-deficit/hyperactivity disorder. Ann Clin Psychiatry. 2012;24(1):23-37.
Attention-deficit/hyperactivity disorder (ADHD) is common; it affects 5% to 7% of children1,2 and 4% to 5% of all adults.3,4 Pediatric ADHD often persists into adulthood, as 65% of individuals diagnosed as children retain impairing symptoms by age 25.4
The prevalence of ADHD in childhood is 2 to 3 times greater among boys than girls, but more comparable between the sexes in adulthood.2 Symptoms could be more easily overlooked in women because of the greater prominence of hyperactivity and impulsivity-type symptoms in men.5
Untreated ADHD is associated with significant costs. Adults with ADHD have increased unemployment rates, poor work performance, and comparatively lower educational performance.6,7 Compared with non-ADHD adults, those with ADHD have:
- more traffic violations and accidents and a higher rate of criminal convictions and incarcerations8,9
- a mortality rate almost 2 times higher, with the greatest differences seen in deaths by suicide and accidents.10,11
Adults with ADHD also are more likely to have a comorbid psychiatric disorder—in particular, substance use11—and often are in treatment for other mental or substance use disorders. Among adults who meet diagnostic criteria for ADHD, approximately only 10% are receiving treatment for ADHD symptoms.3,12
Changes in DSM-5
Revisions within DSM-5 simplify ADHD’s diagnosis—and make it more difficult to ignore in
DSM-5 also provides examples of behaviors more commonly found in adults, such as “feelings of restlessness,” compared with DSM-IV’s “often runs about or climbs excessively in situations in which it is inappropriate.” Finally, ADHD now may be diagnosed in a person with an autism spectrum disorder who meets diagnostic criteria for both disorders.13,14
Identifying ADHD in adults
ADHD diagnosis in adults is made through careful clinical interviewing. For example, ask about what factors motivated an individual to seek evaluation for ADHD. Often, patients present after a change in responsibility at work or at home, such as a promotion or birth/adoption of a new child.
Consider incorporating a brief screen for adult ADHD in all new outpatient evaluations (Table 2).15 Screen for other psychiatric disorders as well; comorbidity with ADHD is high, and hyperactivity and inattention symptoms may result from anxiety, depression, or substance use.
Screen for learning disorders, which can present with ADHD symptoms (such as poor concentration) when the individual attempts difficult tasks. Evaluate for risk factors associated with ADHD medications, such as a history of cardiac problems, hypertension, or tachycardia. A family history of ADHD is found in approximately 80% of cases.16,17 Determine the presence of ADHD symptoms in childhood. A careful review of the educational history often reveals long-term underachievement and struggles in school. Patients may report a chronic history of poor attention or feelings of restlessness in school. Sometimes problems do not become apparent until high school or college; some individuals, especially those with high intelligence, compensate for deficits and show fewer overt symptoms of impairment until later in their education.18Occupational history also may be revealing:
- How are they performing at work?
- Have they changed jobs multiple times in a short period?
- Do they have difficulty organizing tasks?
Subtle ADHD signs include time of arrival to appointments (eg, late or extremely early), missing data on intake paperwork, and a history of losing keys or phones.
Neuropsychological testing. Some clinicians routinely include neuropsychological testing in an adult ADHD evaluation, but these studies have shown inconsistent cognitive deficits in people with ADHD.19,20 No distinct psychometric cognitive test or profile is diagnostic of ADHD or its subtypes.21
Treatment and follow-up care
Four general categories of medications are used to treat ADHD in children and adults:
After starting a patient on medication, at each follow-up appointment ask about new cardiac symptoms or diagnoses, new family history of cardiac problems, or new medications. Measure pulse and blood pressure every 1 to 3 months. Measure vital signs more frequently during titration and weaning periods.23
Stimulant medications
Amphetamines have dual action: they block the reuptake of dopamine and noradrenaline by competitive inhibition of the transporters and promote the release of dopamine and noradrenaline by competitive inhibition of the intraneuronal vesicular monoamine transporter.24
For most amphetamine products, including dextroamphetamine and amphetamine mixed salts, the target dosage is approximately 0.5 mg/kg. Start at a lower dosage, however, and rapidly titrate weekly so patients can adjust to the medication while not becoming frustrated with a lack of efficacy. Some patients may require short-acting forms with dosing 3 times per day, and twice daily dosing is not uncommon with extended-release (ER) formulations.
Metabolism of most amphetamine products—with the exception of lisdexamfetamine—involves the cytochrome P450 (CYP) enzyme CYP2D6, leading to the formation of the metabolite 4-hydroxyamphetamine.25 The pharmacokinetics of lisdexamfetamine in slow or ultra-rapid CYP2D6 metabolizers has not been evaluated (Shire US Inc., written communication, July 2014).
Agents that alter urinary pH can affect blood levels of amphetamine. Acidifying agents decrease amphetamine blood levels, while alkalinizing agents increase amphetamine blood levels.26
Lisdexamfetamine contains L-lysine, an essential amino acid, covalently bound to d-amphetamine via an amide linking group.27 After absorption, lisdexamfetamine is metabolized by rate-limited, enzymatic hydrolysis to yield d-amphetamine and L-lysine.24,28,29 A starting dose of 40 mg is advised; twice-daily dosing rarely is required.
A meta-analysis of 5 randomized, controlled trials in the treatment of adult ADHD showed a response rate of 70% for lisdexamfetamine compared with 37% for placebo. Trial duration ranged from 4 to 14 weeks, with dosages of 30 to 70 mg/d.30 Another analysis of data from lisdexamfetamine trials predicted an effect size of 1.07 for European adults, which is larger than the 0.8 threshold for large effect sizes.31
Methylphenidate products. Methylphenidate’s main action is through enhancement of dopamine signaling by blockade of the dopamine transporter, leading to increases in extracellular dopamine as well as norepinephrine.22,32 Optimized dosing is generally 1 mg/kg per day, and dosing up to 80 to 120 mg/d is not unusual.33
Dexmethylphenidate is the more pharmacologically active enantiomer of racemic methylphenidate and is twice as potent.34-36 Target dosing of dexmethylphenidate should be one-half as much (ie, 0.5 mg/kg per day) as other methylphenidate products.37
Managing stimulants’ side effects
Amphetamines’ side effects may include insomnia, dry mouth, decreased appetite, weight loss, headaches, and anxiety. To help minimize sleep problems, advise patients to take a second immediate-release dose at noon, rather than later in the afternoon. The longer-acting formulation taken once per day in the morning may be offered as an alternative. Some patients may experience improved sleep because of diminished bedtime ruminations.
Oral rinses, such as Biotène, could help reduce discomfort associated with dry mouth. Pilocarpine, which stimulates saliva production, is another option if rinses are not effective. To address decreased appetite, advise patients to take their medication after they eat. Switching from an immediate-release amphetamine to a longer-acting formulation also may lessen symptoms. Lisdexamfetamine might be a good choice for adults with ADHD who have undergone bariatric surgeries because it is absorbed in the small bowel.38
Methylphenidate has no interactions with CYP enzymes, making it an attractive option for patients taking CYP inhibiting or stimulating medications.39 The most common side effects of methylphenidate products include appetite loss, insomnia, irritability, and tachycardia. Some side effects will abate after 1 to 2 weeks of treatment, but persistence of insomnia and appetite loss may require a decrease in dosage. In rare cases, methylphenidate may produce tics, exacerbate an existing tic disorder, or produce mania or psychosis.40,41 Methylphenidate inhibits the metabolism of tricyclic antidepressants; use methylphenidate with caution in patients taking monoamine oxidase inhibitors.42,43Cardiovascular risks. Possible cardiovascular risks associated with stimulant use have gained widespread attention, although research has not demonstrated an increased risk of serious cardiovascular events in young and middle-aged adults receiving stimulant medications for ADHD.44 Nonetheless, obtain a thorough medical history in adult patients, including cardiac history, family history of cardiac disease, history of any cardiac symptoms, and a medication history. Baseline ECG is not required.45
Screen for a family history of sudden death in a young person, sudden death during exercise, cardiac arrhythmia, cardiomyopathies (including hypertrophic cardiomyopathy, dilated cardiomyopathy, and right ventricular cardiomyopathy), prolonged QT interval, short QT syndrome, Brugada syndrome, Wolff-Parkinson-White syndrome, Marfan syndrome, and an event requiring resuscitation in a family member younger than 35, including syncope requiring rescuscitation.23 If fainting spells, palpitations, chest pain, or other symptoms suggest preexisting cardiovascular disease, refer the patient promptly to a cardiologist.
Peripheral vasculopathy, including Raynaud’s phenomenon, is a lesser known side effect associated with stimulants.46 Symptoms are usually mild, but in rare instances stimulants are associated with digital ulceration or soft tissue breakdown.47 Advise patients to tell you if they experience any new symptoms of numbness, pain, skin color changes, or sensitivity to temperature in fingers and toes. Signs and symptoms generally improve after dosage reduction or discontinuation of the stimulant medication.46 Referral to a rheumatologist might be appropriate if symptoms persist.
A noradrenergic medication
Atomoxetine is a potent, selective inhibitor of the presynaptic noradrenaline transporter that increases the availability of extracellular noradrenaline in the prefrontal cortex.48,49 Atomoxetine may be a good alternative for adult patients with ADHD and comorbid anxiety.50
For adults, the optimal starting dosage is 40 mg in the morning for 1 week, followed by an increase to 80 mg. Insufficient dosing is common with atomoxetine, and the dosage could be increased to 100 mg/d.51 Dosing twice per day may be associated with higher rates of insomnia.
Atomoxetine’s efficacy for managing ADHD in adults has been consistently demonstrated by 6 placebo-controlled trials of 10 to 16 weeks, 3 placebo-controlled 6-month trials, and a 1-year maintenance-of-response trial.52 Atomoxetine was found to have an effect size of 0.45 (medium) (number needed to treat [NNT] = 5).53-55The most common adverse effects include nausea, dry mouth, insomnia, and erectile dysfunction. Small increases in heart rate and blood pressure have been reported, so use this medication with caution in patients for whom this might be problematic. Atomoxetine is metabolized by CYP2D6; 7% of white individuals have a genotype corresponding to a nonfunctional CYP2D6 enzyme.56-58
Alpha-2 adrenergic agonists
Clonidine and guanfacine are antihypertensive drugs that induce peripheral sympathoinhibition via the stimulation of receptors. Clonidine binds equally to adrenergic receptor subtypes α-2A, α-2B, and α-2C (as well as to α-1 and β subtypes, histamine receptors, and possibly dopamine receptors).59,60 Guanfacine binds preferentially to postsynaptic α-2A adrenoceptors in the prefrontal cortex, which have been implicated in attentional and organizational functions.61,62
ER guanfacine and ER clonidine are FDA-approved as monotherapy for ADHD in children and adolescents.
Efficacy in adults. A small (N = 17), double-blind, placebo-controlled, crossover study comparing immediate-release guanfacine and dextroamphetamine found that both medications significantly reduced adult ADHD symptoms, as measured with the DSM-IV Adult Behavior Checklist for Adults.63
No trials have been published regarding the efficacy of ER clonidine in adults with ADHD; adverse effects including sedation, bradycardia, and hypotension may limit its use. One study compared the supplemental use of ER guanfacine (1 to 6 mg/d) or a matching placebo in 26 adults with ADHD who had suboptimal response to stimulant-only treatment. After 10 weeks, both the guanfacine ER and placebo groups showed statistically significant improvements in ADHD symptoms and general functioning. The treatments did not differ in efficacy, safety, or tolerability.64
Adverse events. Compared with clonidine, guanfacine has less CNS depressant and hypotensive activity.58 A phase I trial of ER guanfacine in healthy adults found its single-dose pharmacokinetic properties in 1-, 2-, and 4-mg tablets appeared to be statistically linear. Somnolence—the most common treatment-emergent adverse effect—occurred in 33 of 52 participants (63.5%). All mean vital-sign measurements and ECG parameters remained within normal limits after dosing, and no marked changes from baseline measurements were noted.65
Antidepressants
Antidepressants used in ADHD treatment include bupropion and tricyclic antidepressants.
Bupropion is a noradrenaline and dopamine reuptake inhibitor and is considered to be a mild psychostimulant because of its amphetamine-derived chemical structure.66,67 It generally is considered a third-line medication when stimulants have not improved ADHD symptoms or are not tolerated.
A 2011 meta-analysis examined 5 randomized, controlled trials including 175 adults treated with bupropion for ADHD. Bupropion was found to be more effective than placebo (NNT = 5), although bupropion’s therapeutic benefits were not observed until weeks 5 and 6. Its effects were less pronounced than those of methylphenidate. Mean daily dosages were 362 mg for the bupropion SR trials and 393 mg for the bupropion XL trial.68
Tricyclics. Desipramine and nortriptyline have been found to be efficacious in childhood ADHD,69,70 although cardiovascular risk and toxicity in overdose limit their use.71
Attention-deficit/hyperactivity disorder (ADHD) is common; it affects 5% to 7% of children1,2 and 4% to 5% of all adults.3,4 Pediatric ADHD often persists into adulthood, as 65% of individuals diagnosed as children retain impairing symptoms by age 25.4
The prevalence of ADHD in childhood is 2 to 3 times greater among boys than girls, but more comparable between the sexes in adulthood.2 Symptoms could be more easily overlooked in women because of the greater prominence of hyperactivity and impulsivity-type symptoms in men.5
Untreated ADHD is associated with significant costs. Adults with ADHD have increased unemployment rates, poor work performance, and comparatively lower educational performance.6,7 Compared with non-ADHD adults, those with ADHD have:
- more traffic violations and accidents and a higher rate of criminal convictions and incarcerations8,9
- a mortality rate almost 2 times higher, with the greatest differences seen in deaths by suicide and accidents.10,11
Adults with ADHD also are more likely to have a comorbid psychiatric disorder—in particular, substance use11—and often are in treatment for other mental or substance use disorders. Among adults who meet diagnostic criteria for ADHD, approximately only 10% are receiving treatment for ADHD symptoms.3,12
Changes in DSM-5
Revisions within DSM-5 simplify ADHD’s diagnosis—and make it more difficult to ignore in
DSM-5 also provides examples of behaviors more commonly found in adults, such as “feelings of restlessness,” compared with DSM-IV’s “often runs about or climbs excessively in situations in which it is inappropriate.” Finally, ADHD now may be diagnosed in a person with an autism spectrum disorder who meets diagnostic criteria for both disorders.13,14
Identifying ADHD in adults
ADHD diagnosis in adults is made through careful clinical interviewing. For example, ask about what factors motivated an individual to seek evaluation for ADHD. Often, patients present after a change in responsibility at work or at home, such as a promotion or birth/adoption of a new child.
Consider incorporating a brief screen for adult ADHD in all new outpatient evaluations (Table 2).15 Screen for other psychiatric disorders as well; comorbidity with ADHD is high, and hyperactivity and inattention symptoms may result from anxiety, depression, or substance use.
Screen for learning disorders, which can present with ADHD symptoms (such as poor concentration) when the individual attempts difficult tasks. Evaluate for risk factors associated with ADHD medications, such as a history of cardiac problems, hypertension, or tachycardia. A family history of ADHD is found in approximately 80% of cases.16,17 Determine the presence of ADHD symptoms in childhood. A careful review of the educational history often reveals long-term underachievement and struggles in school. Patients may report a chronic history of poor attention or feelings of restlessness in school. Sometimes problems do not become apparent until high school or college; some individuals, especially those with high intelligence, compensate for deficits and show fewer overt symptoms of impairment until later in their education.18Occupational history also may be revealing:
- How are they performing at work?
- Have they changed jobs multiple times in a short period?
- Do they have difficulty organizing tasks?
Subtle ADHD signs include time of arrival to appointments (eg, late or extremely early), missing data on intake paperwork, and a history of losing keys or phones.
Neuropsychological testing. Some clinicians routinely include neuropsychological testing in an adult ADHD evaluation, but these studies have shown inconsistent cognitive deficits in people with ADHD.19,20 No distinct psychometric cognitive test or profile is diagnostic of ADHD or its subtypes.21
Treatment and follow-up care
Four general categories of medications are used to treat ADHD in children and adults:
After starting a patient on medication, at each follow-up appointment ask about new cardiac symptoms or diagnoses, new family history of cardiac problems, or new medications. Measure pulse and blood pressure every 1 to 3 months. Measure vital signs more frequently during titration and weaning periods.23
Stimulant medications
Amphetamines have dual action: they block the reuptake of dopamine and noradrenaline by competitive inhibition of the transporters and promote the release of dopamine and noradrenaline by competitive inhibition of the intraneuronal vesicular monoamine transporter.24
For most amphetamine products, including dextroamphetamine and amphetamine mixed salts, the target dosage is approximately 0.5 mg/kg. Start at a lower dosage, however, and rapidly titrate weekly so patients can adjust to the medication while not becoming frustrated with a lack of efficacy. Some patients may require short-acting forms with dosing 3 times per day, and twice daily dosing is not uncommon with extended-release (ER) formulations.
Metabolism of most amphetamine products—with the exception of lisdexamfetamine—involves the cytochrome P450 (CYP) enzyme CYP2D6, leading to the formation of the metabolite 4-hydroxyamphetamine.25 The pharmacokinetics of lisdexamfetamine in slow or ultra-rapid CYP2D6 metabolizers has not been evaluated (Shire US Inc., written communication, July 2014).
Agents that alter urinary pH can affect blood levels of amphetamine. Acidifying agents decrease amphetamine blood levels, while alkalinizing agents increase amphetamine blood levels.26
Lisdexamfetamine contains L-lysine, an essential amino acid, covalently bound to d-amphetamine via an amide linking group.27 After absorption, lisdexamfetamine is metabolized by rate-limited, enzymatic hydrolysis to yield d-amphetamine and L-lysine.24,28,29 A starting dose of 40 mg is advised; twice-daily dosing rarely is required.
A meta-analysis of 5 randomized, controlled trials in the treatment of adult ADHD showed a response rate of 70% for lisdexamfetamine compared with 37% for placebo. Trial duration ranged from 4 to 14 weeks, with dosages of 30 to 70 mg/d.30 Another analysis of data from lisdexamfetamine trials predicted an effect size of 1.07 for European adults, which is larger than the 0.8 threshold for large effect sizes.31
Methylphenidate products. Methylphenidate’s main action is through enhancement of dopamine signaling by blockade of the dopamine transporter, leading to increases in extracellular dopamine as well as norepinephrine.22,32 Optimized dosing is generally 1 mg/kg per day, and dosing up to 80 to 120 mg/d is not unusual.33
Dexmethylphenidate is the more pharmacologically active enantiomer of racemic methylphenidate and is twice as potent.34-36 Target dosing of dexmethylphenidate should be one-half as much (ie, 0.5 mg/kg per day) as other methylphenidate products.37
Managing stimulants’ side effects
Amphetamines’ side effects may include insomnia, dry mouth, decreased appetite, weight loss, headaches, and anxiety. To help minimize sleep problems, advise patients to take a second immediate-release dose at noon, rather than later in the afternoon. The longer-acting formulation taken once per day in the morning may be offered as an alternative. Some patients may experience improved sleep because of diminished bedtime ruminations.
Oral rinses, such as Biotène, could help reduce discomfort associated with dry mouth. Pilocarpine, which stimulates saliva production, is another option if rinses are not effective. To address decreased appetite, advise patients to take their medication after they eat. Switching from an immediate-release amphetamine to a longer-acting formulation also may lessen symptoms. Lisdexamfetamine might be a good choice for adults with ADHD who have undergone bariatric surgeries because it is absorbed in the small bowel.38
Methylphenidate has no interactions with CYP enzymes, making it an attractive option for patients taking CYP inhibiting or stimulating medications.39 The most common side effects of methylphenidate products include appetite loss, insomnia, irritability, and tachycardia. Some side effects will abate after 1 to 2 weeks of treatment, but persistence of insomnia and appetite loss may require a decrease in dosage. In rare cases, methylphenidate may produce tics, exacerbate an existing tic disorder, or produce mania or psychosis.40,41 Methylphenidate inhibits the metabolism of tricyclic antidepressants; use methylphenidate with caution in patients taking monoamine oxidase inhibitors.42,43Cardiovascular risks. Possible cardiovascular risks associated with stimulant use have gained widespread attention, although research has not demonstrated an increased risk of serious cardiovascular events in young and middle-aged adults receiving stimulant medications for ADHD.44 Nonetheless, obtain a thorough medical history in adult patients, including cardiac history, family history of cardiac disease, history of any cardiac symptoms, and a medication history. Baseline ECG is not required.45
Screen for a family history of sudden death in a young person, sudden death during exercise, cardiac arrhythmia, cardiomyopathies (including hypertrophic cardiomyopathy, dilated cardiomyopathy, and right ventricular cardiomyopathy), prolonged QT interval, short QT syndrome, Brugada syndrome, Wolff-Parkinson-White syndrome, Marfan syndrome, and an event requiring resuscitation in a family member younger than 35, including syncope requiring rescuscitation.23 If fainting spells, palpitations, chest pain, or other symptoms suggest preexisting cardiovascular disease, refer the patient promptly to a cardiologist.
Peripheral vasculopathy, including Raynaud’s phenomenon, is a lesser known side effect associated with stimulants.46 Symptoms are usually mild, but in rare instances stimulants are associated with digital ulceration or soft tissue breakdown.47 Advise patients to tell you if they experience any new symptoms of numbness, pain, skin color changes, or sensitivity to temperature in fingers and toes. Signs and symptoms generally improve after dosage reduction or discontinuation of the stimulant medication.46 Referral to a rheumatologist might be appropriate if symptoms persist.
A noradrenergic medication
Atomoxetine is a potent, selective inhibitor of the presynaptic noradrenaline transporter that increases the availability of extracellular noradrenaline in the prefrontal cortex.48,49 Atomoxetine may be a good alternative for adult patients with ADHD and comorbid anxiety.50
For adults, the optimal starting dosage is 40 mg in the morning for 1 week, followed by an increase to 80 mg. Insufficient dosing is common with atomoxetine, and the dosage could be increased to 100 mg/d.51 Dosing twice per day may be associated with higher rates of insomnia.
Atomoxetine’s efficacy for managing ADHD in adults has been consistently demonstrated by 6 placebo-controlled trials of 10 to 16 weeks, 3 placebo-controlled 6-month trials, and a 1-year maintenance-of-response trial.52 Atomoxetine was found to have an effect size of 0.45 (medium) (number needed to treat [NNT] = 5).53-55The most common adverse effects include nausea, dry mouth, insomnia, and erectile dysfunction. Small increases in heart rate and blood pressure have been reported, so use this medication with caution in patients for whom this might be problematic. Atomoxetine is metabolized by CYP2D6; 7% of white individuals have a genotype corresponding to a nonfunctional CYP2D6 enzyme.56-58
Alpha-2 adrenergic agonists
Clonidine and guanfacine are antihypertensive drugs that induce peripheral sympathoinhibition via the stimulation of receptors. Clonidine binds equally to adrenergic receptor subtypes α-2A, α-2B, and α-2C (as well as to α-1 and β subtypes, histamine receptors, and possibly dopamine receptors).59,60 Guanfacine binds preferentially to postsynaptic α-2A adrenoceptors in the prefrontal cortex, which have been implicated in attentional and organizational functions.61,62
ER guanfacine and ER clonidine are FDA-approved as monotherapy for ADHD in children and adolescents.
Efficacy in adults. A small (N = 17), double-blind, placebo-controlled, crossover study comparing immediate-release guanfacine and dextroamphetamine found that both medications significantly reduced adult ADHD symptoms, as measured with the DSM-IV Adult Behavior Checklist for Adults.63
No trials have been published regarding the efficacy of ER clonidine in adults with ADHD; adverse effects including sedation, bradycardia, and hypotension may limit its use. One study compared the supplemental use of ER guanfacine (1 to 6 mg/d) or a matching placebo in 26 adults with ADHD who had suboptimal response to stimulant-only treatment. After 10 weeks, both the guanfacine ER and placebo groups showed statistically significant improvements in ADHD symptoms and general functioning. The treatments did not differ in efficacy, safety, or tolerability.64
Adverse events. Compared with clonidine, guanfacine has less CNS depressant and hypotensive activity.58 A phase I trial of ER guanfacine in healthy adults found its single-dose pharmacokinetic properties in 1-, 2-, and 4-mg tablets appeared to be statistically linear. Somnolence—the most common treatment-emergent adverse effect—occurred in 33 of 52 participants (63.5%). All mean vital-sign measurements and ECG parameters remained within normal limits after dosing, and no marked changes from baseline measurements were noted.65
Antidepressants
Antidepressants used in ADHD treatment include bupropion and tricyclic antidepressants.
Bupropion is a noradrenaline and dopamine reuptake inhibitor and is considered to be a mild psychostimulant because of its amphetamine-derived chemical structure.66,67 It generally is considered a third-line medication when stimulants have not improved ADHD symptoms or are not tolerated.
A 2011 meta-analysis examined 5 randomized, controlled trials including 175 adults treated with bupropion for ADHD. Bupropion was found to be more effective than placebo (NNT = 5), although bupropion’s therapeutic benefits were not observed until weeks 5 and 6. Its effects were less pronounced than those of methylphenidate. Mean daily dosages were 362 mg for the bupropion SR trials and 393 mg for the bupropion XL trial.68
Tricyclics. Desipramine and nortriptyline have been found to be efficacious in childhood ADHD,69,70 although cardiovascular risk and toxicity in overdose limit their use.71
1. Polanczyk G, de Lima MS, Horta BL, et al. The worldwide prevalence of ADHD: a systemic review and metaregression analysis. Am J Psychiatry. 2007;164(6):942-948.
2. Simon V, Czobor P, Bálint S, et al. Prevalence and correlates of adult attention-deficit hyperactivity disorder: meta-analysis. Br J Psychiatry. 2009;194(3):204-211.
3. Kessler RC, Adler L, Barkley R, et al. The prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey Replication. Am J Psychiatry. 2006;163(4):716-723.
4. Faraone S, Biederman J, Mick E. The age-dependent decline of attention deficit hyperactivity disorder: a meta-analysis of follow-up studies. Psychol Med. 2006;36(2):159-165.
5. Gershon J. A meta-analytic review of gender differences in ADHD. J Atten Disord. 2002;5(3):143-154.
6. Halmøy A, Fasmer OB, Gillberg C, et al. Occupational outcome in adult ADHD: impact of symptom profile, comorbid psychiatric problems, and treatment: a cross-sectional study of 414 clinically diagnosed adult ADHD patients. J Atten Disord. 2009;13(2):175-187.
7. Kuriyan AB, Pelham WE Jr, Molina BS, et al. Young adult educational and vocational outcomes of children diagnosed with ADHD. J Abnorm Child Psychol. 2013;41(1):27-41.
8. Murphy K, Barkley RA. Attention deficit hyperactivity disorder in adults: comorbidities and adaptive impairment. Compr Psychiatry. 1996;37(6):393-401.
9. Mannuzza S, Klein RG, Mouton JL 3rd. Lifetime criminality among boys with attention deficit hyperactivity disorder: a prospective follow-up study into adulthood using official arrest records. Psychiatry Res. 2008;160(3):237-246.
10. Dalsgaard S, Østergaard SD, Leckman JF, et al. Mortality in children, adolescents, and adults with attention deficit hyperactivity disorder: a nationwide cohort study. Lancet. 2015;385(9983):2190-2196.
11. Barbaresi WJ, Colligan RC, Weaver AL, et al. Mortality, ADHD, and psychosocial adversity in adults with childhood ADHD: a prospective study. Pediatrics. 2013;131(4):637-644.
12. Babcock T, Ornstein CS. Comorbidity and its impact in adult patients with attention-deficit/hyperactivity disorder: a primary care perspective. Postgrad Med. 2009;121(3):73-82.
13. Attention-deficit/hyperactivity disorder. In: Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013:59-66.
14. Attention-deficit/hyperactivity disorder. In: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000:78-85.
15. Kooij JJS. Adult ADHD: diagnostic assessment and treatment. 3rd ed. Amsterdam, Netherlands: Springer; 2013:34.
16. Faraone SV, Khan SA. Candidate gene studies of attention-deficit/hyperactivity disorder. J Clin Psychiatry. 2006;67(suppl 8):13-20.
17. Neale BM, Medland SE, Ripke S, et al; Psychiatric GWAS Consortium: ADHD Subgroup. Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2010;49(9):884-897.
18. Milioni AL, Chaim TM, Cavallet M, et al. High IQ may “mask” the diagnosis of ADHD by compensating for deficits in executive functions in treatment-naïve adults with ADHD [published online October 30, 2014]. J Atten Disord. pii: 1087054714554933.
19. Rapport MD, Chung KM, Shore G, et al. Upgrading the science and technology of assessment and diagnosis: laboratory and clinic-based assessment of children with ADHD. J Clin Child Psychol. 2000;29(4):555-568.
20. Woods SP, Lovejoy DW, Ball JD. Neuropsychological characteristics of adults with ADHD: a comprehensive review of initial studies. Clin Neuropsychol. 2002;16(1):12-34.
21. Lange KW, Hauser J, Lange KM, et al. Utility of cognitive neuropsychological assessment in attention-deficit/hyperactivity disorder. Atten Defic Hyperact Disord. 2014;6(4):241-248.
22. Arnold LE. Methylphenidate vs. amphetamine: comparative review. J Atten Disord. 2000;3(4):200-211.
23. Vetter VL Elia J, Erickson, C, et al; American Heart Association Council on Cardiovascular Disease in the Young Congenital Cardiac Defects Committee; American Heart Association Council on Cardiovascular Nursing. Cardiovascular monitoring of children and adolescents with heart disease receiving medications for attention deficit/hyperactivity disorder [corrected]: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young Congenital Cardiac Defects Committee and the Council on Cardiovascular Nursing [Erratum in: Circulation. 2009;120(7):e55-e59]. Circulation. 2008;117(18):2407-2423.
24. Seiden LS, Sabol KE, Ricaurte GA. Amphetamine: effects on catecholamine systems and behavior. Annu Rev Pharmacol Toxicol. 1993;33:639-677.
25. Wu D, Otton SV, Inaba T, et al. Interactions of amphetamine analogs with human liver CYP2D6. Biochem Pharmacol. 1997;53(11):1605-1612.
26. Vyvanse [package insert]. Lexington, MA: Shire Pharmaceuticals; 2015.
27. Pennick M. Absorption of lisdexamfetamine dimesylate and its enzymatic conversion to d-amphetamine. Neuropsychiatr Dis Treat. 2010;6:317-327.
28. Heal DJ, Smith SL, Gosden J, et al. Amphetamine, past and present—a pharmacological and clinical perspective. J Psychopharmacol. 2013;27(6):479-496.
29. Krishnan SM, Pennick M, Stark JG. Metabolism, distribution and elimination of lisdexamfetamine dimesylate: open-label, single-centre, phase I study in healthy adult volunteers. Clin Drug Invest. 2008;28(12):745-755.
30. Maneeton N, Maneeton B, Suttajit S, et al. Exploratory meta-analysis on lisdexamfetamine versus placebo in adult ADHD. Drug Des Devel Ther. 2014;8:1685-1693.
31. Fridman M, Hodgkins P, Kahle JS, et al. Predicted effect size of lisdexamfetamine treatment of attention deficit/hyperactivity disorder (ADHD) in European adults: estimates based on indirect analysis using a systematic review and meta-regression analysis. Eur Psychiatry. 2015;30(4):521-527.
32. Markowitz JS, DeVane CL, Pestreich L, et al. Session 1-87-differentiation of d-, L- and dl-methylphenidate through in vitro pharmacological screening. In: Abstracts: Oral and Poster Presentations of the NCDEU 45th Annual Meeting; June 6-9, 2005; Boca Raton, FL:186.
33. Spencer T, Biederman J, Wilens T, et al. A large, double-blind, randomized clinical trial of methylphenidate in the treatment of adults with attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57(5):456-463.
34. Teo SK, Stirling DI, Thomas SD, et al. Neurobehavioral effects of racemic threo-methylphenidate and its D and L enantiomers in rats. Pharmacol Biochem Behav. 2003;74(3):747-754.
35. Ding YS, Fowler JS, Volkow ND, et al. Chiral drugs: comparison of the pharmacokinetics of [11C]d-threo and L-threo-methylphenidate in the human and baboon brain. Psychopharmacol (Berl). 1997;131(1):71-78.
36. Davids E, Zhang K, Tarazi FI, et al. Stereoselective effects of methylphenidate on motor hyperactivity in juvenile rats induced by neonatal 6-hydroxydopamine lesioning. Psychopharmacol (Berl). 2002;160(1):92-98.
37. Srinivas NR, Hubbard JW, Quinn D, et al. Enantioselective pharmacokinetics and pharmacodynamics of dl-threo-methylphenidate in children with attention deficit hyperactivity disorder. Clin Pharmacol Ther. 1992;52(5):561-568.
38. Ermer JC, Haffey MB, Doll WJ, et al. Pharmacokinetics of lisdexamfetamine dimesylate after targeted gastrointestinal release or oral administration in healthy adults. Drug Metab Dispos. 2012;40(2):290-297.
39. DeVane CL, Markowitz JS, Carson SW, et al. Single-dose pharmacokinetics of methylphenidate in CYP2D6 extensive and poor metabolizers. J Clin Psychopharmacol. 2000;20(3):347-349.
40. Graham J, Coghill D. Adverse effects of pharmacotherapies for attention-deficit hyperactivity disorder: epidemiology, prevention and management. CNS Drugs. 2008;22(3):213-237.
41. Ross RG. Psychotic and manic-like symptoms during stimulant treatment of attention deficit hyperactivity disorder. Am J Psychiatry. 2006;163(7):1149-1152.
42. Shelton Clauson A, Elliott ES, Watson BD, et al. Coadministration of phenelzine and methylphenidate for treatment-resistant depression. Ann Pharmacother. 2004;38(3):508.
43. Markowitz JS, Patrick KS. Pharmacokinetic and pharmacodynamic drug interactions in the treatment of attention-deficit hyperactivity disorder. Clin Pharmacokinet. 2001;40(10):753-772.
44. Habel LA, Cooper WO, Sox CM, et al. ADHD medications and risk of serious cardiovascular events in young and middle-aged adults. JAMA. 2011;306(24):2673-2683.
45. Graham J, Banaschewski T, Buitelaar J, et al; European Guidelines Group. European guidelines on managing adverse effects of medication for ADHD. Eur Child Adolesc Psychiatry. 2011;20(1):17-37.
46. Goldman W, Seltzer R, Reuman P. Association between treatment with central nervous system stimulants and Raynaud’s syndrome in children: a retrospective case-control study of rheumatology patients. Arthritis Rheum. 2008;58(2):563-566.
47. Syed RH, Moore TL. Methylphenidate and dextroamphetamine-induced peripheral vasculopathy. J Clin Rheum. 2008;14(1):30-33.
48. Wilens TE. Mechanism of action of agents in attention-deficit/hyperactivity disorder. J Clin Psychiatry. 2006;67(suppl 8):32-38.
49. Bymaster FP, Katner JS, Nelson DL, et al. Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology. 2002;27(5):699-711.
50. Adler LA, Liebowitz M, Kronenberger W, et al. Atomoxetine treatment in adults with attention-deficit/hyperactivity disorder and comorbid social anxiety disorder. Depress Anxiety. 2009;26(3):212-221.
51. Clemow DB. Suboptimal dosing of Strattera (atomoxetine) for ADHD patients. Postgrad Med. 2014;126(5):196-198.
52. Camporeale A, Porsdal V, De Bruyckere K, et al. Safety and tolerability of atomoxetine in treatment of attention deficit hyperactivity disorder in adult patients: an integrated analysis of 15 clinical trials. J Psychopharmacol. 2015;29(1):3-14.
53. Young JL, Sarkis E, Qiao M, et al. Once-daily treatment with atomoxetine in adults with attention-deficit/hyperactivity disorder: a 24-week, randomized, double-blind, placebo-controlled trial. Clin Neuropharmacol. 2011;34(2):51-60.
54. Bitter I, Angyalosi A, Czobor P. Pharmacological treatment of adult ADHD. Curr Opin Psychiatry. 2012;25(6):529-534.
55. Faraone SV, Glatt SJ. A comparison of the efficacy of medications for adult attention-deficit/hyperactivity disorder using meta-analysis of effect sizes. J Clin Psychiatry. 2010;71(6):754-763.
56. Ring BJ, Gillespie JS, Eckstein JA, et al. Identification of the human cytochromes P450 responsible for atomoxetine metabolism. Drug Metab Dispos. 2002;30(3):319-323.
57. Farid NA, Bergstrom RF, Ziege EA, et al. Single-dose and steady state pharmacokinetics of tomoxetine in normal subjects. J Clin Pharmacol. 1985;25(4):296-301.
58. Mizutani T. PM frequencies of major CYPs in Asians and Caucasians. Drug Metab Rev. 2003;35(2-3):99-106.
59. Jasper JR, Lesnick JD, Chang LK, et al. Ligand efficacy and potency at recombinant alpha2 adrenergic receptors: agonist-mediated [35S]GTPgammaS binding. Biochem Pharmacol. 1998;55(7):1035-1043.
60. Ruggiero S, Clavenna A, Reale L, et al. Guanfacine for attention deficit and hyperactivity disorder in pediatrics: a systematic review and meta-analysis. Eur Neuropsychopharmacol. 2014;24(10):1578-1590.
61. Arnsten AF, Pliszka SR. Catecholamine influences on prefrontal cortical function: relevance to treatment of attention deficit/hyperactivity disorder and related disorders. Pharmacol Biochem Behav. 2011;99(2):211-216.
62. Uhlén S, Wikberg JE. Delineation of rat kidney alpha 2A- and alpha 2B-adrenoceptors with [3H]RX821002 radioligand binding: computer modelling reveals that guanfacine is an alpha 2A-selective compound. Eur J Pharmacol. 1991;202(2):235-243.
63. Taylor FB, Russo J. Comparing guanfacine and dextroamphetamine for the treatment of adult attention deficit/hyperactivity disorder. J Clin Psychopharmacol. 2001;21(2):223-228.
64. Butterfield ME, Saal J, Young B, et al. Supplementary guanfacine hydrochloride as a treatment of attention deficit hyperactivity disorder in adults: a double blind, placebo-controlled study. Psychiatry Res. 2016;236:136-141.
65. Swearingen D, Pennick M, Shojaei A, et al. A phase I, randomized, open-label, crossover study of the single-dose pharmacokinetic properties of guanfacine extended-release 1-, 2-, and 4-mg tablets in healthy adults. Clin Ther. 2007;29(4):617-625.
66. Cooper BR, Wang CM, Cox RF. Evidence that the acute behavioral and electrophysiological effects of bupropion (Wellbutrin) are mediated by a noradrenergic mechanism. Neuropsychopharmacology. 1994;11(2):133-141.
67. Reimherr FW, Hedges DW, Strong RE, et al. Bupropion SR in adults with ADHD: a short-term, placebo-controlled trial. Neuropsychiatr Dis Treat. 2005;1(3):245-251.
68. Maneeton N, Maneeton B, Srisurapanont M, et al. Bupropion for adults with attention-deficit hyperactivity disorder: meta-analysis of randomized, placebo-controlled trials. Psychiatry Clin Neurosci. 2011;65(7):611-617.
69. Biederman J, Baldessarini RJ, Wright V, et al. A double-blind placebo controlled study of desipramine in the treatment of ADD: I. Efficacy. J Am Acad Child Adolesc Psychiatry. 1989;28(5):777-784.
70. Spencer T, Biederman J, Wilens T, et al. Nortriptyline treatment of children with attention-deficit hyperactivity disorder and tic disorder or Tourette’s syndrome. J Am Acad Child Adolesc Psychiatry. 1993;32(1):205-210.
71. Bond DJ, Hadjipavlou G, Lam RW, et al. The Canadian Network for Mood and Anxiety Treatments (CANMAT) task force recommendations for the management of patients with mood disorders and comorbid attention-deficit/hyperactivity disorder. Ann Clin Psychiatry. 2012;24(1):23-37.
1. Polanczyk G, de Lima MS, Horta BL, et al. The worldwide prevalence of ADHD: a systemic review and metaregression analysis. Am J Psychiatry. 2007;164(6):942-948.
2. Simon V, Czobor P, Bálint S, et al. Prevalence and correlates of adult attention-deficit hyperactivity disorder: meta-analysis. Br J Psychiatry. 2009;194(3):204-211.
3. Kessler RC, Adler L, Barkley R, et al. The prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey Replication. Am J Psychiatry. 2006;163(4):716-723.
4. Faraone S, Biederman J, Mick E. The age-dependent decline of attention deficit hyperactivity disorder: a meta-analysis of follow-up studies. Psychol Med. 2006;36(2):159-165.
5. Gershon J. A meta-analytic review of gender differences in ADHD. J Atten Disord. 2002;5(3):143-154.
6. Halmøy A, Fasmer OB, Gillberg C, et al. Occupational outcome in adult ADHD: impact of symptom profile, comorbid psychiatric problems, and treatment: a cross-sectional study of 414 clinically diagnosed adult ADHD patients. J Atten Disord. 2009;13(2):175-187.
7. Kuriyan AB, Pelham WE Jr, Molina BS, et al. Young adult educational and vocational outcomes of children diagnosed with ADHD. J Abnorm Child Psychol. 2013;41(1):27-41.
8. Murphy K, Barkley RA. Attention deficit hyperactivity disorder in adults: comorbidities and adaptive impairment. Compr Psychiatry. 1996;37(6):393-401.
9. Mannuzza S, Klein RG, Mouton JL 3rd. Lifetime criminality among boys with attention deficit hyperactivity disorder: a prospective follow-up study into adulthood using official arrest records. Psychiatry Res. 2008;160(3):237-246.
10. Dalsgaard S, Østergaard SD, Leckman JF, et al. Mortality in children, adolescents, and adults with attention deficit hyperactivity disorder: a nationwide cohort study. Lancet. 2015;385(9983):2190-2196.
11. Barbaresi WJ, Colligan RC, Weaver AL, et al. Mortality, ADHD, and psychosocial adversity in adults with childhood ADHD: a prospective study. Pediatrics. 2013;131(4):637-644.
12. Babcock T, Ornstein CS. Comorbidity and its impact in adult patients with attention-deficit/hyperactivity disorder: a primary care perspective. Postgrad Med. 2009;121(3):73-82.
13. Attention-deficit/hyperactivity disorder. In: Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013:59-66.
14. Attention-deficit/hyperactivity disorder. In: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000:78-85.
15. Kooij JJS. Adult ADHD: diagnostic assessment and treatment. 3rd ed. Amsterdam, Netherlands: Springer; 2013:34.
16. Faraone SV, Khan SA. Candidate gene studies of attention-deficit/hyperactivity disorder. J Clin Psychiatry. 2006;67(suppl 8):13-20.
17. Neale BM, Medland SE, Ripke S, et al; Psychiatric GWAS Consortium: ADHD Subgroup. Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2010;49(9):884-897.
18. Milioni AL, Chaim TM, Cavallet M, et al. High IQ may “mask” the diagnosis of ADHD by compensating for deficits in executive functions in treatment-naïve adults with ADHD [published online October 30, 2014]. J Atten Disord. pii: 1087054714554933.
19. Rapport MD, Chung KM, Shore G, et al. Upgrading the science and technology of assessment and diagnosis: laboratory and clinic-based assessment of children with ADHD. J Clin Child Psychol. 2000;29(4):555-568.
20. Woods SP, Lovejoy DW, Ball JD. Neuropsychological characteristics of adults with ADHD: a comprehensive review of initial studies. Clin Neuropsychol. 2002;16(1):12-34.
21. Lange KW, Hauser J, Lange KM, et al. Utility of cognitive neuropsychological assessment in attention-deficit/hyperactivity disorder. Atten Defic Hyperact Disord. 2014;6(4):241-248.
22. Arnold LE. Methylphenidate vs. amphetamine: comparative review. J Atten Disord. 2000;3(4):200-211.
23. Vetter VL Elia J, Erickson, C, et al; American Heart Association Council on Cardiovascular Disease in the Young Congenital Cardiac Defects Committee; American Heart Association Council on Cardiovascular Nursing. Cardiovascular monitoring of children and adolescents with heart disease receiving medications for attention deficit/hyperactivity disorder [corrected]: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young Congenital Cardiac Defects Committee and the Council on Cardiovascular Nursing [Erratum in: Circulation. 2009;120(7):e55-e59]. Circulation. 2008;117(18):2407-2423.
24. Seiden LS, Sabol KE, Ricaurte GA. Amphetamine: effects on catecholamine systems and behavior. Annu Rev Pharmacol Toxicol. 1993;33:639-677.
25. Wu D, Otton SV, Inaba T, et al. Interactions of amphetamine analogs with human liver CYP2D6. Biochem Pharmacol. 1997;53(11):1605-1612.
26. Vyvanse [package insert]. Lexington, MA: Shire Pharmaceuticals; 2015.
27. Pennick M. Absorption of lisdexamfetamine dimesylate and its enzymatic conversion to d-amphetamine. Neuropsychiatr Dis Treat. 2010;6:317-327.
28. Heal DJ, Smith SL, Gosden J, et al. Amphetamine, past and present—a pharmacological and clinical perspective. J Psychopharmacol. 2013;27(6):479-496.
29. Krishnan SM, Pennick M, Stark JG. Metabolism, distribution and elimination of lisdexamfetamine dimesylate: open-label, single-centre, phase I study in healthy adult volunteers. Clin Drug Invest. 2008;28(12):745-755.
30. Maneeton N, Maneeton B, Suttajit S, et al. Exploratory meta-analysis on lisdexamfetamine versus placebo in adult ADHD. Drug Des Devel Ther. 2014;8:1685-1693.
31. Fridman M, Hodgkins P, Kahle JS, et al. Predicted effect size of lisdexamfetamine treatment of attention deficit/hyperactivity disorder (ADHD) in European adults: estimates based on indirect analysis using a systematic review and meta-regression analysis. Eur Psychiatry. 2015;30(4):521-527.
32. Markowitz JS, DeVane CL, Pestreich L, et al. Session 1-87-differentiation of d-, L- and dl-methylphenidate through in vitro pharmacological screening. In: Abstracts: Oral and Poster Presentations of the NCDEU 45th Annual Meeting; June 6-9, 2005; Boca Raton, FL:186.
33. Spencer T, Biederman J, Wilens T, et al. A large, double-blind, randomized clinical trial of methylphenidate in the treatment of adults with attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57(5):456-463.
34. Teo SK, Stirling DI, Thomas SD, et al. Neurobehavioral effects of racemic threo-methylphenidate and its D and L enantiomers in rats. Pharmacol Biochem Behav. 2003;74(3):747-754.
35. Ding YS, Fowler JS, Volkow ND, et al. Chiral drugs: comparison of the pharmacokinetics of [11C]d-threo and L-threo-methylphenidate in the human and baboon brain. Psychopharmacol (Berl). 1997;131(1):71-78.
36. Davids E, Zhang K, Tarazi FI, et al. Stereoselective effects of methylphenidate on motor hyperactivity in juvenile rats induced by neonatal 6-hydroxydopamine lesioning. Psychopharmacol (Berl). 2002;160(1):92-98.
37. Srinivas NR, Hubbard JW, Quinn D, et al. Enantioselective pharmacokinetics and pharmacodynamics of dl-threo-methylphenidate in children with attention deficit hyperactivity disorder. Clin Pharmacol Ther. 1992;52(5):561-568.
38. Ermer JC, Haffey MB, Doll WJ, et al. Pharmacokinetics of lisdexamfetamine dimesylate after targeted gastrointestinal release or oral administration in healthy adults. Drug Metab Dispos. 2012;40(2):290-297.
39. DeVane CL, Markowitz JS, Carson SW, et al. Single-dose pharmacokinetics of methylphenidate in CYP2D6 extensive and poor metabolizers. J Clin Psychopharmacol. 2000;20(3):347-349.
40. Graham J, Coghill D. Adverse effects of pharmacotherapies for attention-deficit hyperactivity disorder: epidemiology, prevention and management. CNS Drugs. 2008;22(3):213-237.
41. Ross RG. Psychotic and manic-like symptoms during stimulant treatment of attention deficit hyperactivity disorder. Am J Psychiatry. 2006;163(7):1149-1152.
42. Shelton Clauson A, Elliott ES, Watson BD, et al. Coadministration of phenelzine and methylphenidate for treatment-resistant depression. Ann Pharmacother. 2004;38(3):508.
43. Markowitz JS, Patrick KS. Pharmacokinetic and pharmacodynamic drug interactions in the treatment of attention-deficit hyperactivity disorder. Clin Pharmacokinet. 2001;40(10):753-772.
44. Habel LA, Cooper WO, Sox CM, et al. ADHD medications and risk of serious cardiovascular events in young and middle-aged adults. JAMA. 2011;306(24):2673-2683.
45. Graham J, Banaschewski T, Buitelaar J, et al; European Guidelines Group. European guidelines on managing adverse effects of medication for ADHD. Eur Child Adolesc Psychiatry. 2011;20(1):17-37.
46. Goldman W, Seltzer R, Reuman P. Association between treatment with central nervous system stimulants and Raynaud’s syndrome in children: a retrospective case-control study of rheumatology patients. Arthritis Rheum. 2008;58(2):563-566.
47. Syed RH, Moore TL. Methylphenidate and dextroamphetamine-induced peripheral vasculopathy. J Clin Rheum. 2008;14(1):30-33.
48. Wilens TE. Mechanism of action of agents in attention-deficit/hyperactivity disorder. J Clin Psychiatry. 2006;67(suppl 8):32-38.
49. Bymaster FP, Katner JS, Nelson DL, et al. Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology. 2002;27(5):699-711.
50. Adler LA, Liebowitz M, Kronenberger W, et al. Atomoxetine treatment in adults with attention-deficit/hyperactivity disorder and comorbid social anxiety disorder. Depress Anxiety. 2009;26(3):212-221.
51. Clemow DB. Suboptimal dosing of Strattera (atomoxetine) for ADHD patients. Postgrad Med. 2014;126(5):196-198.
52. Camporeale A, Porsdal V, De Bruyckere K, et al. Safety and tolerability of atomoxetine in treatment of attention deficit hyperactivity disorder in adult patients: an integrated analysis of 15 clinical trials. J Psychopharmacol. 2015;29(1):3-14.
53. Young JL, Sarkis E, Qiao M, et al. Once-daily treatment with atomoxetine in adults with attention-deficit/hyperactivity disorder: a 24-week, randomized, double-blind, placebo-controlled trial. Clin Neuropharmacol. 2011;34(2):51-60.
54. Bitter I, Angyalosi A, Czobor P. Pharmacological treatment of adult ADHD. Curr Opin Psychiatry. 2012;25(6):529-534.
55. Faraone SV, Glatt SJ. A comparison of the efficacy of medications for adult attention-deficit/hyperactivity disorder using meta-analysis of effect sizes. J Clin Psychiatry. 2010;71(6):754-763.
56. Ring BJ, Gillespie JS, Eckstein JA, et al. Identification of the human cytochromes P450 responsible for atomoxetine metabolism. Drug Metab Dispos. 2002;30(3):319-323.
57. Farid NA, Bergstrom RF, Ziege EA, et al. Single-dose and steady state pharmacokinetics of tomoxetine in normal subjects. J Clin Pharmacol. 1985;25(4):296-301.
58. Mizutani T. PM frequencies of major CYPs in Asians and Caucasians. Drug Metab Rev. 2003;35(2-3):99-106.
59. Jasper JR, Lesnick JD, Chang LK, et al. Ligand efficacy and potency at recombinant alpha2 adrenergic receptors: agonist-mediated [35S]GTPgammaS binding. Biochem Pharmacol. 1998;55(7):1035-1043.
60. Ruggiero S, Clavenna A, Reale L, et al. Guanfacine for attention deficit and hyperactivity disorder in pediatrics: a systematic review and meta-analysis. Eur Neuropsychopharmacol. 2014;24(10):1578-1590.
61. Arnsten AF, Pliszka SR. Catecholamine influences on prefrontal cortical function: relevance to treatment of attention deficit/hyperactivity disorder and related disorders. Pharmacol Biochem Behav. 2011;99(2):211-216.
62. Uhlén S, Wikberg JE. Delineation of rat kidney alpha 2A- and alpha 2B-adrenoceptors with [3H]RX821002 radioligand binding: computer modelling reveals that guanfacine is an alpha 2A-selective compound. Eur J Pharmacol. 1991;202(2):235-243.
63. Taylor FB, Russo J. Comparing guanfacine and dextroamphetamine for the treatment of adult attention deficit/hyperactivity disorder. J Clin Psychopharmacol. 2001;21(2):223-228.
64. Butterfield ME, Saal J, Young B, et al. Supplementary guanfacine hydrochloride as a treatment of attention deficit hyperactivity disorder in adults: a double blind, placebo-controlled study. Psychiatry Res. 2016;236:136-141.
65. Swearingen D, Pennick M, Shojaei A, et al. A phase I, randomized, open-label, crossover study of the single-dose pharmacokinetic properties of guanfacine extended-release 1-, 2-, and 4-mg tablets in healthy adults. Clin Ther. 2007;29(4):617-625.
66. Cooper BR, Wang CM, Cox RF. Evidence that the acute behavioral and electrophysiological effects of bupropion (Wellbutrin) are mediated by a noradrenergic mechanism. Neuropsychopharmacology. 1994;11(2):133-141.
67. Reimherr FW, Hedges DW, Strong RE, et al. Bupropion SR in adults with ADHD: a short-term, placebo-controlled trial. Neuropsychiatr Dis Treat. 2005;1(3):245-251.
68. Maneeton N, Maneeton B, Srisurapanont M, et al. Bupropion for adults with attention-deficit hyperactivity disorder: meta-analysis of randomized, placebo-controlled trials. Psychiatry Clin Neurosci. 2011;65(7):611-617.
69. Biederman J, Baldessarini RJ, Wright V, et al. A double-blind placebo controlled study of desipramine in the treatment of ADD: I. Efficacy. J Am Acad Child Adolesc Psychiatry. 1989;28(5):777-784.
70. Spencer T, Biederman J, Wilens T, et al. Nortriptyline treatment of children with attention-deficit hyperactivity disorder and tic disorder or Tourette’s syndrome. J Am Acad Child Adolesc Psychiatry. 1993;32(1):205-210.
71. Bond DJ, Hadjipavlou G, Lam RW, et al. The Canadian Network for Mood and Anxiety Treatments (CANMAT) task force recommendations for the management of patients with mood disorders and comorbid attention-deficit/hyperactivity disorder. Ann Clin Psychiatry. 2012;24(1):23-37.

AYAs struggle socially in early years after cancer diagnosis

A new study indicates that adolescent and young adult (AYA) cancer survivors continue to face social difficulties for more than 2 years after their diagnosis.
The research, published in Cancer, suggests these patients may see some improvement in their social lives during the first year after diagnosis.
However, their social functioning tends to remain constant after that, leaving them socially impaired relative to their cancer-free peers.
Previous studies have shown that AYAs with cancer experience greater challenges in social functioning than their cancer-free peers or even compared to older cancer patients.
But few studies have examined this phenomenon by following the same patients over time.
Olga Husson, PhD, of the Radboud University Medical Center in The Netherlands, and her colleagues set out to examine changes in social functioning among AYAs in the early years after a cancer diagnosis.
The researchers asked AYA cancer patients at 5 US medical institutions to complete a survey about social functioning within 4 months of their diagnosis, 12 months later, and 24 months later.
There were 141 patients (ages 14 to 39 at diagnosis) who completed the surveys.
The researchers found that, when compared to population norms, the cancer patients had inferior social functioning at all the time points studied.
Among the cancer patients, the mean social functioning score from the Medical Outcomes Study Short Form 36 Health Survey (version 2) was 52.0 around the time of cancer diagnosis, 73.1 at the 12-month follow-up, and 69.2 at the 24-month follow-up. In comparison, the population norm (for people ages 18 to 44) is 85.1 (P<0.001 for all time points).
The researchers did note that cancer patients experienced significant improvements in social functioning from baseline to the 12-month follow-up, but there was no further improvement after that.
The researchers also examined the different trajectories of social functioning over time. They found that social functioning improved over time for 47% of the cancer patients but worsened for 13%. In addition, 32% of patients had consistently low social functioning, and 9% had consistently high social functioning.
The cancer patients with consistently low social functioning were more likely to be off treatment at the time of follow-up, report more physical symptoms and higher levels of psychological distress (at both baseline and follow-up), and perceive themselves to receive less social support.
“Reducing physical symptoms and psychological distress and enhancing social support by interventions in the period after treatment may potentially help these young survivors to better reintegrate into society,” Dr Husson said. ![]()
A new study indicates that adolescent and young adult (AYA) cancer survivors continue to face social difficulties for more than 2 years after their diagnosis.
The research, published in Cancer, suggests these patients may see some improvement in their social lives during the first year after diagnosis.
However, their social functioning tends to remain constant after that, leaving them socially impaired relative to their cancer-free peers.
Previous studies have shown that AYAs with cancer experience greater challenges in social functioning than their cancer-free peers or even compared to older cancer patients.
But few studies have examined this phenomenon by following the same patients over time.
Olga Husson, PhD, of the Radboud University Medical Center in The Netherlands, and her colleagues set out to examine changes in social functioning among AYAs in the early years after a cancer diagnosis.
The researchers asked AYA cancer patients at 5 US medical institutions to complete a survey about social functioning within 4 months of their diagnosis, 12 months later, and 24 months later.
There were 141 patients (ages 14 to 39 at diagnosis) who completed the surveys.
The researchers found that, when compared to population norms, the cancer patients had inferior social functioning at all the time points studied.
Among the cancer patients, the mean social functioning score from the Medical Outcomes Study Short Form 36 Health Survey (version 2) was 52.0 around the time of cancer diagnosis, 73.1 at the 12-month follow-up, and 69.2 at the 24-month follow-up. In comparison, the population norm (for people ages 18 to 44) is 85.1 (P<0.001 for all time points).
The researchers did note that cancer patients experienced significant improvements in social functioning from baseline to the 12-month follow-up, but there was no further improvement after that.
The researchers also examined the different trajectories of social functioning over time. They found that social functioning improved over time for 47% of the cancer patients but worsened for 13%. In addition, 32% of patients had consistently low social functioning, and 9% had consistently high social functioning.
The cancer patients with consistently low social functioning were more likely to be off treatment at the time of follow-up, report more physical symptoms and higher levels of psychological distress (at both baseline and follow-up), and perceive themselves to receive less social support.
“Reducing physical symptoms and psychological distress and enhancing social support by interventions in the period after treatment may potentially help these young survivors to better reintegrate into society,” Dr Husson said. ![]()
A new study indicates that adolescent and young adult (AYA) cancer survivors continue to face social difficulties for more than 2 years after their diagnosis.
The research, published in Cancer, suggests these patients may see some improvement in their social lives during the first year after diagnosis.
However, their social functioning tends to remain constant after that, leaving them socially impaired relative to their cancer-free peers.
Previous studies have shown that AYAs with cancer experience greater challenges in social functioning than their cancer-free peers or even compared to older cancer patients.
But few studies have examined this phenomenon by following the same patients over time.
Olga Husson, PhD, of the Radboud University Medical Center in The Netherlands, and her colleagues set out to examine changes in social functioning among AYAs in the early years after a cancer diagnosis.
The researchers asked AYA cancer patients at 5 US medical institutions to complete a survey about social functioning within 4 months of their diagnosis, 12 months later, and 24 months later.
There were 141 patients (ages 14 to 39 at diagnosis) who completed the surveys.
The researchers found that, when compared to population norms, the cancer patients had inferior social functioning at all the time points studied.
Among the cancer patients, the mean social functioning score from the Medical Outcomes Study Short Form 36 Health Survey (version 2) was 52.0 around the time of cancer diagnosis, 73.1 at the 12-month follow-up, and 69.2 at the 24-month follow-up. In comparison, the population norm (for people ages 18 to 44) is 85.1 (P<0.001 for all time points).
The researchers did note that cancer patients experienced significant improvements in social functioning from baseline to the 12-month follow-up, but there was no further improvement after that.
The researchers also examined the different trajectories of social functioning over time. They found that social functioning improved over time for 47% of the cancer patients but worsened for 13%. In addition, 32% of patients had consistently low social functioning, and 9% had consistently high social functioning.
The cancer patients with consistently low social functioning were more likely to be off treatment at the time of follow-up, report more physical symptoms and higher levels of psychological distress (at both baseline and follow-up), and perceive themselves to receive less social support.
“Reducing physical symptoms and psychological distress and enhancing social support by interventions in the period after treatment may potentially help these young survivors to better reintegrate into society,” Dr Husson said. ![]()
Assessment of goals of care in nursing home reduces hospitalization for patients with dementia
CLINICAL QUESTION: For patients with advanced dementia, does a goals-of-care intervention improve communication and care outcomes?
BACKGROUND: Patients with advanced dementia are frequently admitted from nursing homes for acute conditions. Prior research demonstrates deficits in documentation of advanced directives.
STUDY DESIGN: Single-blind cluster randomized trial.
SETTING: Twenty-two nursing homes in North Carolina.
BOTTOM LINE: Goals of care discussions for patients with advanced dementia appears to reduce hospitalizations.
CITATIONS: Hanson LC, Zimmerman S, Song MK, et al. Effect of the goals of care intervention for advanced dementia: a randomized clinical trial. JAMA Intern Med. 2017 Jan;177:24-31.
Dr. Cumbler is the associate chief of hospital medicine, Division of Hospital Medicine, University of Colorado School of Medicine, Aurora.
CLINICAL QUESTION: For patients with advanced dementia, does a goals-of-care intervention improve communication and care outcomes?
BACKGROUND: Patients with advanced dementia are frequently admitted from nursing homes for acute conditions. Prior research demonstrates deficits in documentation of advanced directives.
STUDY DESIGN: Single-blind cluster randomized trial.
SETTING: Twenty-two nursing homes in North Carolina.
BOTTOM LINE: Goals of care discussions for patients with advanced dementia appears to reduce hospitalizations.
CITATIONS: Hanson LC, Zimmerman S, Song MK, et al. Effect of the goals of care intervention for advanced dementia: a randomized clinical trial. JAMA Intern Med. 2017 Jan;177:24-31.
Dr. Cumbler is the associate chief of hospital medicine, Division of Hospital Medicine, University of Colorado School of Medicine, Aurora.
CLINICAL QUESTION: For patients with advanced dementia, does a goals-of-care intervention improve communication and care outcomes?
BACKGROUND: Patients with advanced dementia are frequently admitted from nursing homes for acute conditions. Prior research demonstrates deficits in documentation of advanced directives.
STUDY DESIGN: Single-blind cluster randomized trial.
SETTING: Twenty-two nursing homes in North Carolina.
BOTTOM LINE: Goals of care discussions for patients with advanced dementia appears to reduce hospitalizations.
CITATIONS: Hanson LC, Zimmerman S, Song MK, et al. Effect of the goals of care intervention for advanced dementia: a randomized clinical trial. JAMA Intern Med. 2017 Jan;177:24-31.
Dr. Cumbler is the associate chief of hospital medicine, Division of Hospital Medicine, University of Colorado School of Medicine, Aurora.
Antipsychotics ineffective for symptoms of delirium in palliative care
CLINICAL QUESTION: Do antipsychotics provide symptomatic benefit for delirium in palliative care?
BACKGROUND: Antipsychotics are frequently used for the treatment of delirium and guideline recommended for delirium-associated distress. However, a 2016 meta-analysis found antipsychotics are not associated with change in delirium duration or severity. Antipsychotics for palliative management of delirium at end of life is not well studied.
STUDY DESIGN: Double-blind randomized controlled trial with placebo, haloperidol, and risperidone arms.
SETTING: Eleven Australian inpatient hospice or palliative care services.
SYNOPSIS: 247 patients (mean age, 74.9 years; 88.3% with cancer) with advanced incurable disease and active delirium were studied. Most had mild-moderate severity delirium. All received nonpharmacological measures and plan to address reversible precipitants. Patients were randomized to placebo (84), haloperidol (81), or risperidone (82) for 72 hours. Dose titration was allowed based on delirium symptoms. In intention to treat analysis the delirium severity scores were statistically higher in haloperidol and risperidone arms, compared with placebo. This reached statistical significance although less than the minimum clinically significant difference. Mortality, use of rescue medicines, and extrapyramidal symptoms were higher in antipsychotic groups.
BOTTOM LINE: Antipsychotics cause side effects without efficacy in palliation of symptoms of delirium.
CITATIONS: Agar MR, Lawlor PG, Quinn S, et al. Efficacy of oral risperidone, haloperidol, or placebo for symptoms of delirium among patients in palliative care: a randomized clinical trial. JAMA Intern Med. 2017 Jan;177:34-42.
Dr. Cumbler is the associate chief of hospital medicine, Division of Hospital Medicine, University of Colorado School of Medicine, Aurora.
CLINICAL QUESTION: Do antipsychotics provide symptomatic benefit for delirium in palliative care?
BACKGROUND: Antipsychotics are frequently used for the treatment of delirium and guideline recommended for delirium-associated distress. However, a 2016 meta-analysis found antipsychotics are not associated with change in delirium duration or severity. Antipsychotics for palliative management of delirium at end of life is not well studied.
STUDY DESIGN: Double-blind randomized controlled trial with placebo, haloperidol, and risperidone arms.
SETTING: Eleven Australian inpatient hospice or palliative care services.
SYNOPSIS: 247 patients (mean age, 74.9 years; 88.3% with cancer) with advanced incurable disease and active delirium were studied. Most had mild-moderate severity delirium. All received nonpharmacological measures and plan to address reversible precipitants. Patients were randomized to placebo (84), haloperidol (81), or risperidone (82) for 72 hours. Dose titration was allowed based on delirium symptoms. In intention to treat analysis the delirium severity scores were statistically higher in haloperidol and risperidone arms, compared with placebo. This reached statistical significance although less than the minimum clinically significant difference. Mortality, use of rescue medicines, and extrapyramidal symptoms were higher in antipsychotic groups.
BOTTOM LINE: Antipsychotics cause side effects without efficacy in palliation of symptoms of delirium.
CITATIONS: Agar MR, Lawlor PG, Quinn S, et al. Efficacy of oral risperidone, haloperidol, or placebo for symptoms of delirium among patients in palliative care: a randomized clinical trial. JAMA Intern Med. 2017 Jan;177:34-42.
Dr. Cumbler is the associate chief of hospital medicine, Division of Hospital Medicine, University of Colorado School of Medicine, Aurora.
CLINICAL QUESTION: Do antipsychotics provide symptomatic benefit for delirium in palliative care?
BACKGROUND: Antipsychotics are frequently used for the treatment of delirium and guideline recommended for delirium-associated distress. However, a 2016 meta-analysis found antipsychotics are not associated with change in delirium duration or severity. Antipsychotics for palliative management of delirium at end of life is not well studied.
STUDY DESIGN: Double-blind randomized controlled trial with placebo, haloperidol, and risperidone arms.
SETTING: Eleven Australian inpatient hospice or palliative care services.
SYNOPSIS: 247 patients (mean age, 74.9 years; 88.3% with cancer) with advanced incurable disease and active delirium were studied. Most had mild-moderate severity delirium. All received nonpharmacological measures and plan to address reversible precipitants. Patients were randomized to placebo (84), haloperidol (81), or risperidone (82) for 72 hours. Dose titration was allowed based on delirium symptoms. In intention to treat analysis the delirium severity scores were statistically higher in haloperidol and risperidone arms, compared with placebo. This reached statistical significance although less than the minimum clinically significant difference. Mortality, use of rescue medicines, and extrapyramidal symptoms were higher in antipsychotic groups.
BOTTOM LINE: Antipsychotics cause side effects without efficacy in palliation of symptoms of delirium.
CITATIONS: Agar MR, Lawlor PG, Quinn S, et al. Efficacy of oral risperidone, haloperidol, or placebo for symptoms of delirium among patients in palliative care: a randomized clinical trial. JAMA Intern Med. 2017 Jan;177:34-42.
Dr. Cumbler is the associate chief of hospital medicine, Division of Hospital Medicine, University of Colorado School of Medicine, Aurora.