User login
MDedge conference coverage features onsite reporting of the latest study results and expert perspectives from leading researchers.
‘Low and Slow’ hyperthermic treatment being evaluated for superficial and nodular BCCs
DENVER –
At the annual meeting of the American Society for Dermatologic Surgery, Christopher Zachary, MD, and colleagues described a novel, noninvasive standardized controlled hyperthermia and mapping protocol (CHAMP) designed to help clinicians with margin assessment and treatment of superficial and nodular basal cell cancers (BCCs). “There’s considerable interest on the part of the public in having CHAMP treatment for their BCCs,” Dr. Zachary, professor and chair emeritus, University of California, Irvine, told this news organization in advance of the meeting.
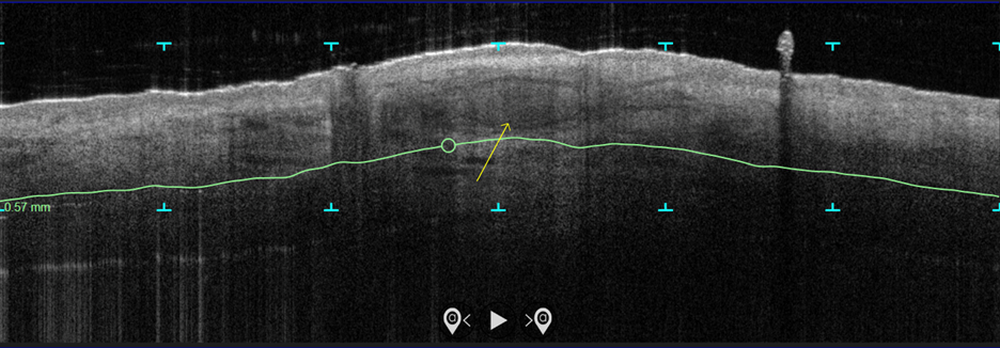
In the study, which is being conducted at three centers and plans to enroll 100 patients, more than 70 patients with biopsy-proven superficial and nodular BCCs have been scanned with the VivoSight Dx optical coherence tomography (OCT) device to map BCC tumor margins. Next, they were treated with the Sciton 1,064-nm Er:YAG laser equipped with a 4-mm beam diameter scan pattern with no overlap and an 8-millisecond pulse duration, randomized to either 120 J/cm2 pulses, until tissue graying and contraction was observed, or a novel controlled hyperthermia technique known as “Low and Slow” using repeated 25 J/cm2 pulses under thermal camera imaging to maintain a consistent temperature of 55º C for 60 seconds.
The researchers reassessed the tissue response both clinically and by OCT at 3 months and the patients were retreated with the same method if residual BCC was demonstrated. At 3-12 months post treatment, the lesion sites were saucerized and examined histologically by step sections to confirm clearance.
“In contrast to the more commonly performed ‘standard’ long-pulse 1,064-nm laser tumor coagulation, where the end point is graying and contraction of tissue, the new controlled ‘Low and Slow’ technique heats the tissue to 55º C for 60 seconds, avoids ulceration, and induces apoptotic tumor disappearance by a caspase-3 and -7 mechanism,” Dr. Zachary explained in an interview. “It’s a gentler process that allows patients an alternative to second intention wounds that occur after electrodessication and curettage or Mohs,” he added, noting that CHAMP is not intended for the treatment of more complex, large, recurrent, or infiltrative BCCs.
In both study arms, the majority of patients enrolled to date have been found to be free of tumor at 3 months by clinical and OCT examination. “The study is ongoing, but the current numbers indicate that 9 out of 10 superficial and nodular BCCs are free of tumor at 3-12 months after the last treatment,” Dr. Zachary said. The standard-treatment arm, where tissue was treated to a gray color with tissue contraction, generally resulted in more blistering and tissue necrosis with prolonged healing, compared with the Low and Slow–controlled hyperthermia arm. BCC lesions treated in the controlled hyperthermia arm had a lilac gray color with “a surprising increase” in the Doppler blood flow rate, compared with those in the standard-treatment arm, he noted.
“Blood flow following the standard technique is dramatically reduced immediately post treatment, which accounts in part for the frequent ulceration and slow healing in that group,” Dr. Zachary said.
He acknowledged certain limitations of the study, including its relatively small sample size and the fact that the optimal treatment parameters of the Low and Slow technique have yet to be realized. “It could be that we will achieve better results at 50º C for 70 seconds or similar,” he said. “While this technique will not in any way reduce the great benefits of Mohs surgery for complex BCCs, it will benefit those with simpler superficial and nodular BCCs, particularly in those who are not good surgical candidates.”
As an aside, Dr. Zachary supports the increased use of OCT scanners to improve the ability to diagnose and assess the lateral and deep margins of skin cancers. “I think that all dermatology residents should understand how to use these devices,” he said. “I’m convinced they are going to be useful in their clinical practice in the future.”
Keith L. Duffy, MD, who was asked to comment on the work, said that the study demonstrates novel ways to use existing and developing technologies in dermatology and highlights the intersection of aesthetic, surgical, and medical dermatology. “CHAMP is promising as shown by the data in the abstract and I am eager to see the final results of the study with an eye toward final cure rate and cosmesis,” said Dr. Duffy, associate professor of dermatology at the University of Utah, Salt Lake City.
“In my estimation, this technology will need to prove to be superior in one or both of these parameters in order to be considered a first- or second-line therapy,” he added. “My practice for these types of basal cell carcinomas is a simple one pass of curettage with aluminum chloride or pressure for hemostasis. The healing is fast, the cosmesis is excellent, and the cure rate is more than 90% for this simple in-office destruction. However, for those with access to this technology and proficiency with its use, CHAMP may become a viable alternative to our existing destructive methods. I look forward to seeing the published results of this multicenter trial.”
This study is being funded by Michelson Diagnostics. Sciton provided the long-pulsed 1,064-nm lasers devices being used in the trial. Neither Dr. Zachary nor Dr. Duffy reported having relevant disclosures.
DENVER –
At the annual meeting of the American Society for Dermatologic Surgery, Christopher Zachary, MD, and colleagues described a novel, noninvasive standardized controlled hyperthermia and mapping protocol (CHAMP) designed to help clinicians with margin assessment and treatment of superficial and nodular basal cell cancers (BCCs). “There’s considerable interest on the part of the public in having CHAMP treatment for their BCCs,” Dr. Zachary, professor and chair emeritus, University of California, Irvine, told this news organization in advance of the meeting.
In the study, which is being conducted at three centers and plans to enroll 100 patients, more than 70 patients with biopsy-proven superficial and nodular BCCs have been scanned with the VivoSight Dx optical coherence tomography (OCT) device to map BCC tumor margins. Next, they were treated with the Sciton 1,064-nm Er:YAG laser equipped with a 4-mm beam diameter scan pattern with no overlap and an 8-millisecond pulse duration, randomized to either 120 J/cm2 pulses, until tissue graying and contraction was observed, or a novel controlled hyperthermia technique known as “Low and Slow” using repeated 25 J/cm2 pulses under thermal camera imaging to maintain a consistent temperature of 55º C for 60 seconds.
The researchers reassessed the tissue response both clinically and by OCT at 3 months and the patients were retreated with the same method if residual BCC was demonstrated. At 3-12 months post treatment, the lesion sites were saucerized and examined histologically by step sections to confirm clearance.
“In contrast to the more commonly performed ‘standard’ long-pulse 1,064-nm laser tumor coagulation, where the end point is graying and contraction of tissue, the new controlled ‘Low and Slow’ technique heats the tissue to 55º C for 60 seconds, avoids ulceration, and induces apoptotic tumor disappearance by a caspase-3 and -7 mechanism,” Dr. Zachary explained in an interview. “It’s a gentler process that allows patients an alternative to second intention wounds that occur after electrodessication and curettage or Mohs,” he added, noting that CHAMP is not intended for the treatment of more complex, large, recurrent, or infiltrative BCCs.
In both study arms, the majority of patients enrolled to date have been found to be free of tumor at 3 months by clinical and OCT examination. “The study is ongoing, but the current numbers indicate that 9 out of 10 superficial and nodular BCCs are free of tumor at 3-12 months after the last treatment,” Dr. Zachary said. The standard-treatment arm, where tissue was treated to a gray color with tissue contraction, generally resulted in more blistering and tissue necrosis with prolonged healing, compared with the Low and Slow–controlled hyperthermia arm. BCC lesions treated in the controlled hyperthermia arm had a lilac gray color with “a surprising increase” in the Doppler blood flow rate, compared with those in the standard-treatment arm, he noted.
“Blood flow following the standard technique is dramatically reduced immediately post treatment, which accounts in part for the frequent ulceration and slow healing in that group,” Dr. Zachary said.
He acknowledged certain limitations of the study, including its relatively small sample size and the fact that the optimal treatment parameters of the Low and Slow technique have yet to be realized. “It could be that we will achieve better results at 50º C for 70 seconds or similar,” he said. “While this technique will not in any way reduce the great benefits of Mohs surgery for complex BCCs, it will benefit those with simpler superficial and nodular BCCs, particularly in those who are not good surgical candidates.”
As an aside, Dr. Zachary supports the increased use of OCT scanners to improve the ability to diagnose and assess the lateral and deep margins of skin cancers. “I think that all dermatology residents should understand how to use these devices,” he said. “I’m convinced they are going to be useful in their clinical practice in the future.”
Keith L. Duffy, MD, who was asked to comment on the work, said that the study demonstrates novel ways to use existing and developing technologies in dermatology and highlights the intersection of aesthetic, surgical, and medical dermatology. “CHAMP is promising as shown by the data in the abstract and I am eager to see the final results of the study with an eye toward final cure rate and cosmesis,” said Dr. Duffy, associate professor of dermatology at the University of Utah, Salt Lake City.
“In my estimation, this technology will need to prove to be superior in one or both of these parameters in order to be considered a first- or second-line therapy,” he added. “My practice for these types of basal cell carcinomas is a simple one pass of curettage with aluminum chloride or pressure for hemostasis. The healing is fast, the cosmesis is excellent, and the cure rate is more than 90% for this simple in-office destruction. However, for those with access to this technology and proficiency with its use, CHAMP may become a viable alternative to our existing destructive methods. I look forward to seeing the published results of this multicenter trial.”
This study is being funded by Michelson Diagnostics. Sciton provided the long-pulsed 1,064-nm lasers devices being used in the trial. Neither Dr. Zachary nor Dr. Duffy reported having relevant disclosures.
DENVER –
At the annual meeting of the American Society for Dermatologic Surgery, Christopher Zachary, MD, and colleagues described a novel, noninvasive standardized controlled hyperthermia and mapping protocol (CHAMP) designed to help clinicians with margin assessment and treatment of superficial and nodular basal cell cancers (BCCs). “There’s considerable interest on the part of the public in having CHAMP treatment for their BCCs,” Dr. Zachary, professor and chair emeritus, University of California, Irvine, told this news organization in advance of the meeting.
In the study, which is being conducted at three centers and plans to enroll 100 patients, more than 70 patients with biopsy-proven superficial and nodular BCCs have been scanned with the VivoSight Dx optical coherence tomography (OCT) device to map BCC tumor margins. Next, they were treated with the Sciton 1,064-nm Er:YAG laser equipped with a 4-mm beam diameter scan pattern with no overlap and an 8-millisecond pulse duration, randomized to either 120 J/cm2 pulses, until tissue graying and contraction was observed, or a novel controlled hyperthermia technique known as “Low and Slow” using repeated 25 J/cm2 pulses under thermal camera imaging to maintain a consistent temperature of 55º C for 60 seconds.
The researchers reassessed the tissue response both clinically and by OCT at 3 months and the patients were retreated with the same method if residual BCC was demonstrated. At 3-12 months post treatment, the lesion sites were saucerized and examined histologically by step sections to confirm clearance.
“In contrast to the more commonly performed ‘standard’ long-pulse 1,064-nm laser tumor coagulation, where the end point is graying and contraction of tissue, the new controlled ‘Low and Slow’ technique heats the tissue to 55º C for 60 seconds, avoids ulceration, and induces apoptotic tumor disappearance by a caspase-3 and -7 mechanism,” Dr. Zachary explained in an interview. “It’s a gentler process that allows patients an alternative to second intention wounds that occur after electrodessication and curettage or Mohs,” he added, noting that CHAMP is not intended for the treatment of more complex, large, recurrent, or infiltrative BCCs.
In both study arms, the majority of patients enrolled to date have been found to be free of tumor at 3 months by clinical and OCT examination. “The study is ongoing, but the current numbers indicate that 9 out of 10 superficial and nodular BCCs are free of tumor at 3-12 months after the last treatment,” Dr. Zachary said. The standard-treatment arm, where tissue was treated to a gray color with tissue contraction, generally resulted in more blistering and tissue necrosis with prolonged healing, compared with the Low and Slow–controlled hyperthermia arm. BCC lesions treated in the controlled hyperthermia arm had a lilac gray color with “a surprising increase” in the Doppler blood flow rate, compared with those in the standard-treatment arm, he noted.
“Blood flow following the standard technique is dramatically reduced immediately post treatment, which accounts in part for the frequent ulceration and slow healing in that group,” Dr. Zachary said.
He acknowledged certain limitations of the study, including its relatively small sample size and the fact that the optimal treatment parameters of the Low and Slow technique have yet to be realized. “It could be that we will achieve better results at 50º C for 70 seconds or similar,” he said. “While this technique will not in any way reduce the great benefits of Mohs surgery for complex BCCs, it will benefit those with simpler superficial and nodular BCCs, particularly in those who are not good surgical candidates.”
As an aside, Dr. Zachary supports the increased use of OCT scanners to improve the ability to diagnose and assess the lateral and deep margins of skin cancers. “I think that all dermatology residents should understand how to use these devices,” he said. “I’m convinced they are going to be useful in their clinical practice in the future.”
Keith L. Duffy, MD, who was asked to comment on the work, said that the study demonstrates novel ways to use existing and developing technologies in dermatology and highlights the intersection of aesthetic, surgical, and medical dermatology. “CHAMP is promising as shown by the data in the abstract and I am eager to see the final results of the study with an eye toward final cure rate and cosmesis,” said Dr. Duffy, associate professor of dermatology at the University of Utah, Salt Lake City.
“In my estimation, this technology will need to prove to be superior in one or both of these parameters in order to be considered a first- or second-line therapy,” he added. “My practice for these types of basal cell carcinomas is a simple one pass of curettage with aluminum chloride or pressure for hemostasis. The healing is fast, the cosmesis is excellent, and the cure rate is more than 90% for this simple in-office destruction. However, for those with access to this technology and proficiency with its use, CHAMP may become a viable alternative to our existing destructive methods. I look forward to seeing the published results of this multicenter trial.”
This study is being funded by Michelson Diagnostics. Sciton provided the long-pulsed 1,064-nm lasers devices being used in the trial. Neither Dr. Zachary nor Dr. Duffy reported having relevant disclosures.
AT ASDS 2022
Liquid injectable silicone safe for acne scarring in dark-skinned patients, study finds
DENVER – Highly , results from a recent study showed.
“Acne is pervasive, and acne scarring disproportionately affects darker skin types,” lead study author Nicole Salame, MD, told this news organization in advance of the annual meeting of the American Society for Dermatologic Surgery, where she presented the results of the study. “Treatment of acne scarring in darker skin is also particularly challenging since resurfacing can be problematic. Numerous treatment options exist but vary in effectiveness, sustainability, and side-effect profile, especially for patients with darker skin.”
Highly purified liquid injectable silicone (also known as LIS) is approved by the Food and Drug Administration for treating intraocular tamponade of retinal detachment, and has been used off label for skin augmentation. A 2005 study of LIS for five patients with acne scarring, with up to 30 years of follow-up, showed efficacy and preservation of product without complications for depressed, broad-based acne scars .
“Use of LIS as a permanent treatment for acne scarring in darker skin types has yet to be evaluated,” said Dr. Salame, a 4th-year dermatology resident at Emory University, Atlanta. “Our study is the first to retrospectively evaluate the safety and efficacy of highly purified LIS for the treatment of acne scars in all skin types.”
Dr. Salame and coauthor Harold J. Brody, MD, evaluated the charts of 96 patients with a mean age of 51 years who received highly purified LIS for the treatment of acne scars at Dr. Brody’s Atlanta-based private dermatology practice between July 2010 and March 2021. Of the 96 patients, 31 had darker skin types (20 were Fitzpatrick skin type IV and 11 were Fitzpatrick skin type V). Dr. Brody performed all treatments: a total of 206 in the 96 patients.
The average time of follow-up was 6.31 years; 19 patients had a follow-up of 1-3 years, 25 had a follow-up of 3-5 years, and 52 had a follow-up of greater than 5 years. The researchers did not observe any complications along the course of the patients’ treatments, and no patients reported complications or dissatisfaction with treatment.
“Among the most impressive findings of our study was the permanence of effectiveness of LIS for acne scarring in patients who had treatment over a decade before,” Dr. Salame said. “Our longest follow up was 12 years. These patients continued to show improvement in their acne scarring years after treatment with LIS, even as they lost collagen and volume in their face with advancing age.”
In addition, she said, none of the patients experienced complications of granulomatous reactions, migration, or extrusion of product, which were previously documented with the use of macrodroplet injectable silicone techniques. “This is likely due to the consistent use of the microdroplet injection technique in our study – less than 0.01 cc per injection at minimum 6- to 8-week intervals or more,” Dr. Salame said.
Lawrence J. Green, MD, of the department of dermatology at George Washington University, Washington, who was asked to comment on the study, said that the findings “show safety and durability of highly purified microdroplet liquid silicone to treat acne scars. The numbers of patients reviewed are small and selective (one highly skilled dermatologist), but with the right material (highly purified liquid silicone) and in a qualified and experienced physician’s hand, this treatment seems like a great option.”
Dr. Salame acknowledged certain limitations of the study, including its single-center, retrospective design. “Future prospective studies with larger patient populations of all skin types recruited from multiple centers may be needed,” she said.
The researchers reported having no relevant conflicts of interest or funding sources to disclose. Dr. Green disclosed that he is a speaker, consultant, or investigator for numerous pharmaceutical companies.
DENVER – Highly , results from a recent study showed.
“Acne is pervasive, and acne scarring disproportionately affects darker skin types,” lead study author Nicole Salame, MD, told this news organization in advance of the annual meeting of the American Society for Dermatologic Surgery, where she presented the results of the study. “Treatment of acne scarring in darker skin is also particularly challenging since resurfacing can be problematic. Numerous treatment options exist but vary in effectiveness, sustainability, and side-effect profile, especially for patients with darker skin.”
Highly purified liquid injectable silicone (also known as LIS) is approved by the Food and Drug Administration for treating intraocular tamponade of retinal detachment, and has been used off label for skin augmentation. A 2005 study of LIS for five patients with acne scarring, with up to 30 years of follow-up, showed efficacy and preservation of product without complications for depressed, broad-based acne scars .
“Use of LIS as a permanent treatment for acne scarring in darker skin types has yet to be evaluated,” said Dr. Salame, a 4th-year dermatology resident at Emory University, Atlanta. “Our study is the first to retrospectively evaluate the safety and efficacy of highly purified LIS for the treatment of acne scars in all skin types.”
Dr. Salame and coauthor Harold J. Brody, MD, evaluated the charts of 96 patients with a mean age of 51 years who received highly purified LIS for the treatment of acne scars at Dr. Brody’s Atlanta-based private dermatology practice between July 2010 and March 2021. Of the 96 patients, 31 had darker skin types (20 were Fitzpatrick skin type IV and 11 were Fitzpatrick skin type V). Dr. Brody performed all treatments: a total of 206 in the 96 patients.
The average time of follow-up was 6.31 years; 19 patients had a follow-up of 1-3 years, 25 had a follow-up of 3-5 years, and 52 had a follow-up of greater than 5 years. The researchers did not observe any complications along the course of the patients’ treatments, and no patients reported complications or dissatisfaction with treatment.
“Among the most impressive findings of our study was the permanence of effectiveness of LIS for acne scarring in patients who had treatment over a decade before,” Dr. Salame said. “Our longest follow up was 12 years. These patients continued to show improvement in their acne scarring years after treatment with LIS, even as they lost collagen and volume in their face with advancing age.”
In addition, she said, none of the patients experienced complications of granulomatous reactions, migration, or extrusion of product, which were previously documented with the use of macrodroplet injectable silicone techniques. “This is likely due to the consistent use of the microdroplet injection technique in our study – less than 0.01 cc per injection at minimum 6- to 8-week intervals or more,” Dr. Salame said.
Lawrence J. Green, MD, of the department of dermatology at George Washington University, Washington, who was asked to comment on the study, said that the findings “show safety and durability of highly purified microdroplet liquid silicone to treat acne scars. The numbers of patients reviewed are small and selective (one highly skilled dermatologist), but with the right material (highly purified liquid silicone) and in a qualified and experienced physician’s hand, this treatment seems like a great option.”
Dr. Salame acknowledged certain limitations of the study, including its single-center, retrospective design. “Future prospective studies with larger patient populations of all skin types recruited from multiple centers may be needed,” she said.
The researchers reported having no relevant conflicts of interest or funding sources to disclose. Dr. Green disclosed that he is a speaker, consultant, or investigator for numerous pharmaceutical companies.
DENVER – Highly , results from a recent study showed.
“Acne is pervasive, and acne scarring disproportionately affects darker skin types,” lead study author Nicole Salame, MD, told this news organization in advance of the annual meeting of the American Society for Dermatologic Surgery, where she presented the results of the study. “Treatment of acne scarring in darker skin is also particularly challenging since resurfacing can be problematic. Numerous treatment options exist but vary in effectiveness, sustainability, and side-effect profile, especially for patients with darker skin.”
Highly purified liquid injectable silicone (also known as LIS) is approved by the Food and Drug Administration for treating intraocular tamponade of retinal detachment, and has been used off label for skin augmentation. A 2005 study of LIS for five patients with acne scarring, with up to 30 years of follow-up, showed efficacy and preservation of product without complications for depressed, broad-based acne scars .
“Use of LIS as a permanent treatment for acne scarring in darker skin types has yet to be evaluated,” said Dr. Salame, a 4th-year dermatology resident at Emory University, Atlanta. “Our study is the first to retrospectively evaluate the safety and efficacy of highly purified LIS for the treatment of acne scars in all skin types.”
Dr. Salame and coauthor Harold J. Brody, MD, evaluated the charts of 96 patients with a mean age of 51 years who received highly purified LIS for the treatment of acne scars at Dr. Brody’s Atlanta-based private dermatology practice between July 2010 and March 2021. Of the 96 patients, 31 had darker skin types (20 were Fitzpatrick skin type IV and 11 were Fitzpatrick skin type V). Dr. Brody performed all treatments: a total of 206 in the 96 patients.
The average time of follow-up was 6.31 years; 19 patients had a follow-up of 1-3 years, 25 had a follow-up of 3-5 years, and 52 had a follow-up of greater than 5 years. The researchers did not observe any complications along the course of the patients’ treatments, and no patients reported complications or dissatisfaction with treatment.
“Among the most impressive findings of our study was the permanence of effectiveness of LIS for acne scarring in patients who had treatment over a decade before,” Dr. Salame said. “Our longest follow up was 12 years. These patients continued to show improvement in their acne scarring years after treatment with LIS, even as they lost collagen and volume in their face with advancing age.”
In addition, she said, none of the patients experienced complications of granulomatous reactions, migration, or extrusion of product, which were previously documented with the use of macrodroplet injectable silicone techniques. “This is likely due to the consistent use of the microdroplet injection technique in our study – less than 0.01 cc per injection at minimum 6- to 8-week intervals or more,” Dr. Salame said.
Lawrence J. Green, MD, of the department of dermatology at George Washington University, Washington, who was asked to comment on the study, said that the findings “show safety and durability of highly purified microdroplet liquid silicone to treat acne scars. The numbers of patients reviewed are small and selective (one highly skilled dermatologist), but with the right material (highly purified liquid silicone) and in a qualified and experienced physician’s hand, this treatment seems like a great option.”
Dr. Salame acknowledged certain limitations of the study, including its single-center, retrospective design. “Future prospective studies with larger patient populations of all skin types recruited from multiple centers may be needed,” she said.
The researchers reported having no relevant conflicts of interest or funding sources to disclose. Dr. Green disclosed that he is a speaker, consultant, or investigator for numerous pharmaceutical companies.
AT ASDS 2022
Blindness from PRP injections a rare but potentially devastating side effect
DENVER – None of the cases involved scalp injections.
“Both soft tissue fillers and [PRP] are common injection-type treatments that dermatologists perform on the head and neck area,” lead study author Sean Wu, MD, said in an interview in advance of the annual meeting of the American Society for Dermatologic Surgery, where he presented the results during an oral abstract session. “Fillers are usually used to replace volume and fill in lines while PRP is usually used for skin rejuvenation and certain forms of hair loss. We know that fillers may rarely cause blindness if accidentally injected into a facial artery.”
Certain facial areas such as the glabella, nose, and forehead are considered high risk for blindness with filler injections. But whether PRP injections in those areas may also result in blindness is not yet known, so Dr. Wu and his colleagues, Xu He, MD, and Robert Weiss, MD, at the Maryland Laser, Skin, and Vein Institute in Hunt Valley, Md., performed what is believed to be the first systematic review of the topic. In January 2022 they searched the PubMed database, which yielded 224 articles from which they selected four for full review. The results were recently published in Dermatologic Surgery.
Collectively, the four articles reported a total of seven patients with unilateral vision loss or impairment following PRP injection. They ranged in age from 41 to 63 years. Skin rejuvenation was the indication for PRP injection in six patients and temporomandibular joint (TMJ) disorder in one. Three of the cases occurred in Venezuela while one each occurred in the United States, the United Kingdom, and Malaysia. All patients had signs of arterial occlusion or ischemia on retinal examination or imaging.
Dr. Wu and colleagues found that the glabella was the most common site of injection associated with vision loss (five cases), followed by the forehead (two cases), and one case each in the lateral canthus, nasolabial fold, and the TMJ. In all but two cases, vision loss occurred immediately after injection. (The number of injections exceeded seven because two patients received PRP in more than one site.)
Associated symptoms included ocular pain, fullness, eyelid ptosis, headache, nausea, vomiting, dizziness, tinnitus, and urinary urgency. At their initial ophthalmology evaluation, six patients had no light perception in the affected eye. Only one patient reported recovery of visual acuity at 3 months but with residual deficits on eye exam. This person had been evaluated and treated by an ophthalmologist within 3 hours of symptom onset.
“The other cases reported complete blindness in one eye,” Dr. Wu said. “There is no reversing agent for PRP, unlike for many fillers, so there is no clear-cut solution for this issue.”
Based on the results of the systematic review, Dr. Wu concluded that blindness is a rare complication of PRP. “We should take the same precautions when injecting PRP on the face as we do when injecting fillers,” he advised. “This may include not injecting in high-risk areas and aspirating prior to injection to make sure we are not accidentally injecting into an artery.”
It was “notable,” he added, that no cases of blindness occurred following scalp injections of PRP for hair loss, indicating “that this use of PRP is likely very safe from a vision loss standpoint.”
Dr. Wu acknowledged certain imitations of the analysis, including the low quality of some case reports/series. “There is a notable lack of detail on the PRP injection technique, as the authors of the case reports were generally not the PRP injectors themselves,” he said. “There was also no attempt at treatment in a series of four cases.”
Asked to comment on the review, Terrence Keaney, MD, founder and director of SkinDC, in Arlington, Va., said that the analysis underscores the importance of considering blindness as a possible side effect when injecting PRP into the face. “Using techniques that can minimize intravascular injections including the use of cannulas, aspiration, and larger needle size may help reduce this rare side effect,” said Dr. Keaney, a clinical associate professor of dermatology at George Washington University, Washington.

“It is important to recognize the lack of cases of blindness when injecting the scalp, one of the most popular PRP injection locations. This reduced risk may be due to the reduced communication between the scalp vasculature and the ophthalmic vasculature,” he added.
The study authors reported having no financial disclosures. Dr. Keaney disclosed that he is a member of the advisory board for Crown Aesthetics.
DENVER – None of the cases involved scalp injections.
“Both soft tissue fillers and [PRP] are common injection-type treatments that dermatologists perform on the head and neck area,” lead study author Sean Wu, MD, said in an interview in advance of the annual meeting of the American Society for Dermatologic Surgery, where he presented the results during an oral abstract session. “Fillers are usually used to replace volume and fill in lines while PRP is usually used for skin rejuvenation and certain forms of hair loss. We know that fillers may rarely cause blindness if accidentally injected into a facial artery.”
Certain facial areas such as the glabella, nose, and forehead are considered high risk for blindness with filler injections. But whether PRP injections in those areas may also result in blindness is not yet known, so Dr. Wu and his colleagues, Xu He, MD, and Robert Weiss, MD, at the Maryland Laser, Skin, and Vein Institute in Hunt Valley, Md., performed what is believed to be the first systematic review of the topic. In January 2022 they searched the PubMed database, which yielded 224 articles from which they selected four for full review. The results were recently published in Dermatologic Surgery.
Collectively, the four articles reported a total of seven patients with unilateral vision loss or impairment following PRP injection. They ranged in age from 41 to 63 years. Skin rejuvenation was the indication for PRP injection in six patients and temporomandibular joint (TMJ) disorder in one. Three of the cases occurred in Venezuela while one each occurred in the United States, the United Kingdom, and Malaysia. All patients had signs of arterial occlusion or ischemia on retinal examination or imaging.
Dr. Wu and colleagues found that the glabella was the most common site of injection associated with vision loss (five cases), followed by the forehead (two cases), and one case each in the lateral canthus, nasolabial fold, and the TMJ. In all but two cases, vision loss occurred immediately after injection. (The number of injections exceeded seven because two patients received PRP in more than one site.)
Associated symptoms included ocular pain, fullness, eyelid ptosis, headache, nausea, vomiting, dizziness, tinnitus, and urinary urgency. At their initial ophthalmology evaluation, six patients had no light perception in the affected eye. Only one patient reported recovery of visual acuity at 3 months but with residual deficits on eye exam. This person had been evaluated and treated by an ophthalmologist within 3 hours of symptom onset.
“The other cases reported complete blindness in one eye,” Dr. Wu said. “There is no reversing agent for PRP, unlike for many fillers, so there is no clear-cut solution for this issue.”
Based on the results of the systematic review, Dr. Wu concluded that blindness is a rare complication of PRP. “We should take the same precautions when injecting PRP on the face as we do when injecting fillers,” he advised. “This may include not injecting in high-risk areas and aspirating prior to injection to make sure we are not accidentally injecting into an artery.”
It was “notable,” he added, that no cases of blindness occurred following scalp injections of PRP for hair loss, indicating “that this use of PRP is likely very safe from a vision loss standpoint.”
Dr. Wu acknowledged certain imitations of the analysis, including the low quality of some case reports/series. “There is a notable lack of detail on the PRP injection technique, as the authors of the case reports were generally not the PRP injectors themselves,” he said. “There was also no attempt at treatment in a series of four cases.”
Asked to comment on the review, Terrence Keaney, MD, founder and director of SkinDC, in Arlington, Va., said that the analysis underscores the importance of considering blindness as a possible side effect when injecting PRP into the face. “Using techniques that can minimize intravascular injections including the use of cannulas, aspiration, and larger needle size may help reduce this rare side effect,” said Dr. Keaney, a clinical associate professor of dermatology at George Washington University, Washington.
“It is important to recognize the lack of cases of blindness when injecting the scalp, one of the most popular PRP injection locations. This reduced risk may be due to the reduced communication between the scalp vasculature and the ophthalmic vasculature,” he added.
The study authors reported having no financial disclosures. Dr. Keaney disclosed that he is a member of the advisory board for Crown Aesthetics.
DENVER – None of the cases involved scalp injections.
“Both soft tissue fillers and [PRP] are common injection-type treatments that dermatologists perform on the head and neck area,” lead study author Sean Wu, MD, said in an interview in advance of the annual meeting of the American Society for Dermatologic Surgery, where he presented the results during an oral abstract session. “Fillers are usually used to replace volume and fill in lines while PRP is usually used for skin rejuvenation and certain forms of hair loss. We know that fillers may rarely cause blindness if accidentally injected into a facial artery.”
Certain facial areas such as the glabella, nose, and forehead are considered high risk for blindness with filler injections. But whether PRP injections in those areas may also result in blindness is not yet known, so Dr. Wu and his colleagues, Xu He, MD, and Robert Weiss, MD, at the Maryland Laser, Skin, and Vein Institute in Hunt Valley, Md., performed what is believed to be the first systematic review of the topic. In January 2022 they searched the PubMed database, which yielded 224 articles from which they selected four for full review. The results were recently published in Dermatologic Surgery.
Collectively, the four articles reported a total of seven patients with unilateral vision loss or impairment following PRP injection. They ranged in age from 41 to 63 years. Skin rejuvenation was the indication for PRP injection in six patients and temporomandibular joint (TMJ) disorder in one. Three of the cases occurred in Venezuela while one each occurred in the United States, the United Kingdom, and Malaysia. All patients had signs of arterial occlusion or ischemia on retinal examination or imaging.
Dr. Wu and colleagues found that the glabella was the most common site of injection associated with vision loss (five cases), followed by the forehead (two cases), and one case each in the lateral canthus, nasolabial fold, and the TMJ. In all but two cases, vision loss occurred immediately after injection. (The number of injections exceeded seven because two patients received PRP in more than one site.)
Associated symptoms included ocular pain, fullness, eyelid ptosis, headache, nausea, vomiting, dizziness, tinnitus, and urinary urgency. At their initial ophthalmology evaluation, six patients had no light perception in the affected eye. Only one patient reported recovery of visual acuity at 3 months but with residual deficits on eye exam. This person had been evaluated and treated by an ophthalmologist within 3 hours of symptom onset.
“The other cases reported complete blindness in one eye,” Dr. Wu said. “There is no reversing agent for PRP, unlike for many fillers, so there is no clear-cut solution for this issue.”
Based on the results of the systematic review, Dr. Wu concluded that blindness is a rare complication of PRP. “We should take the same precautions when injecting PRP on the face as we do when injecting fillers,” he advised. “This may include not injecting in high-risk areas and aspirating prior to injection to make sure we are not accidentally injecting into an artery.”
It was “notable,” he added, that no cases of blindness occurred following scalp injections of PRP for hair loss, indicating “that this use of PRP is likely very safe from a vision loss standpoint.”
Dr. Wu acknowledged certain imitations of the analysis, including the low quality of some case reports/series. “There is a notable lack of detail on the PRP injection technique, as the authors of the case reports were generally not the PRP injectors themselves,” he said. “There was also no attempt at treatment in a series of four cases.”
Asked to comment on the review, Terrence Keaney, MD, founder and director of SkinDC, in Arlington, Va., said that the analysis underscores the importance of considering blindness as a possible side effect when injecting PRP into the face. “Using techniques that can minimize intravascular injections including the use of cannulas, aspiration, and larger needle size may help reduce this rare side effect,” said Dr. Keaney, a clinical associate professor of dermatology at George Washington University, Washington.
“It is important to recognize the lack of cases of blindness when injecting the scalp, one of the most popular PRP injection locations. This reduced risk may be due to the reduced communication between the scalp vasculature and the ophthalmic vasculature,” he added.
The study authors reported having no financial disclosures. Dr. Keaney disclosed that he is a member of the advisory board for Crown Aesthetics.
AT ASDS 2022
Expert makes the case for not subtyping patients with rosacea
. At least they should be, according to Julie C. Harper, MD.
“How many people with papules and pustules don’t also have redness?” Dr. Harper, who practices in Birmingham, Ala., said at Medscape Live’s annual Coastal Dermatology Symposium. “If we’re not careful, and we try to classify a person into a subtype of rosacea, we end up treating only part of their rosacea; we don’t treat all of it. We have seen this in the literature,” she added.
“The idea now is to take a phenotypic approach to rosacea. What we mean by that is that you look at the patient, you document every part of rosacea that you see, and you treat according to that,” she continued. “That person with papules and pustules may also have phyma and ocular disease. They may have telangiectasia and persistent background erythema. They may also have flushing.”
Dr. Harper incorporates the mnemonic “STOP” to her visits with rosacea patients.
S stands for: Identify signs and symptoms of the condition. “Listen to the patient for symptoms,” she advised. “We’ve learned to listen to darker skinned patients for what they tell us about erythema, for example, because we may not be able to see it, yet they are experiencing it. They may also have symptomatic burning, itching, and stinging.”
T stands for: Discuss triggers. “Ask patients, ‘what is it that makes your rosacea worse?’ That’s different for everyone,” she said.
O stands for: Agree on a treatment outcome. “Ask, ‘what is it that really bothers you? Are you bothered by the bumps? The redness?’ ” she said.
“The P stands for: Develop a plan that addresses all of that,” she said.
Different treatments for different rosacea symptoms
No one-size-fits-all treatment exists for rosacea. Options that work well for papules and pustules aren’t effective for redness. Similarly, products that work for redness don’t work for telangiectasia.
“Different lesions and signs of rosacea will likely require multiple modes of treatment,” Dr. Harper said. “So, when you evaluate your rosacea patients, if they’re doing great, don’t change their regimen. But if you see somebody who is not well controlled, is there an opportunity for you to come in and add something to that regimen that may make them better? Maybe so.”
Treatment options indicated for papules and pustules include ivermectin, metronidazole, azelaic acid, sodium sulfacetamide/sulfur, modified release doxycycline, minocycline foam, and encapsulated benzoyl peroxide.
Options indicated for persistent background erythema include brimonidine and oxymetazoline, while device-based treatments include the pulsed dye laser, the KTP laser, intense pulsed light, and electrosurgery.
Anti-inflammatory action for pustules and papules
A relatively new product indicated for pustules and papules is minocycline 1.5% foam, the only minocycline that is FDA approved to treat rosacea.
“There is no oral minocycline product approved for rosacea yet,” Dr. Harper said. “There is not a known bacterial pathogen in rosacea. Tetracyclines likely work in rosacea by inhibiting neutrophil chemotaxis, inhibiting MMP and thus KLK-5 and LL-37, inhibiting pro-inflammatory cytokines, downregulating reactive oxygen species, and inhibiting angiogenesis.”
In two 12-week, phase 3 randomized studies of 1,522 patients with moderate to severe rosacea, participants were assigned to receive minocycline 5% foam or a vehicle that contained mineral oil and coconut oil.
At week 12, about 50% of patients who received minocycline 5% foam were clear, compared with about 40% of those in the vehicle arm. Also, the reduction of lesion count was about 63% for patients in the treatment group, compared with a reduction of about 54% in the vehicle arm.
Dr. Harper characterized the 63% reduction as “pretty good, but is it good enough or fast enough? I don’t think so, so even with a great drug like this, I would use something else. You can use two medications sometimes to get people better faster. There’s room to bring in something for that background erythema.”
Minocycline 1.5% foam is colored yellow and may stain fabric. “It contains coconut oil, soybean oil, and light mineral oil,” she said. “Most people prefer to use this at bedtime, but you don’t have to.”
Another treatment option is 5% microencapsulated benzoyl peroxide cream, which is FDA approved for inflammatory lesions of rosacea.
“What’s the mechanism of action? Probably not being antimicrobial,” Dr. Harper said. “I think it’s probably at least in part anti-inflammatory, because we have some data to show that it’s killing Demodex [mites]. If Demodex [are] a trigger of inflammation, and we can lessen Demodex, then we could lessen the inflammatory response after that.”
The drug’s approval was based on data from two positive, identical phase 3 randomized, double-blind, multicenter, 12-week clinical trials that evaluated its safety compared with vehicle in 733 people with inflammatory lesions of rosacea (NCT03564119 and NCT03448939).
At week 12, inflammatory lesions of rosacea were reduced by nearly 70% in both trials among those who received 5% microencapsulated benzoyl peroxide cream, compared with 38%-46% among those who received the vehicle. Also, nearly 50% of subjects in the treatment groups were clear or almost clear at 12 weeks, compared with 38%-46% of those who received the vehicle.
Dr. Harper added that about one-quarter of patients in the treatment group of the trials were clear or almost clear by week 4. “That’s pretty fast,” she said, noting that the product’s microencapsulated shell acts as a fenestrated barrier. “It has little openings, which means that it takes a while for the drug to work itself out,” she said. “I think of it as being like a speed bump for benzoyl peroxide delivery. It has to get through this little maze before it lands on the skin. We think that is what has helped with tolerability.”
Oral sarecycline, a narrow spectrum tetracycline that was FDA approved for acne in 2018, may also benefit rosacea patients. In a 12-week, investigator-blinded pilot study, 72 patients with papulopustular rosacea were assigned to receive sarecycline, while 25 received a multivitamin.
By week 12, 75% of patients in the sarecycline group were clear, compared with 16% of those in the multivitamin group, while the inflammatory lesion counts dropped from baseline by 80% and 60%, respectively. Studies of sarecycline for acne have demonstrated similar rates of vertigo, dizziness, and sunburn to those of placebo.
“There were also low rates of gastrointestinal disturbances,” Dr. Harper said. “That’s important in rosacea, because there is no bacterial pathogen.”
Dr. Harper disclosed that she serves as an advisor or consultant for Almirall, BioPharmX, Cassiopeia, Cutanea, Cutera, Dermira, EPI, Galderma, LaRoche-Posay, Ortho, Vyne, Sol Gel, and Sun. She also serves as a speaker or member of a speakers bureau for Almirall, EPI, Galderma, Ortho, and Vyne.
Medscape Live and this news organization are owned by the same parent company.
. At least they should be, according to Julie C. Harper, MD.
“How many people with papules and pustules don’t also have redness?” Dr. Harper, who practices in Birmingham, Ala., said at Medscape Live’s annual Coastal Dermatology Symposium. “If we’re not careful, and we try to classify a person into a subtype of rosacea, we end up treating only part of their rosacea; we don’t treat all of it. We have seen this in the literature,” she added.
“The idea now is to take a phenotypic approach to rosacea. What we mean by that is that you look at the patient, you document every part of rosacea that you see, and you treat according to that,” she continued. “That person with papules and pustules may also have phyma and ocular disease. They may have telangiectasia and persistent background erythema. They may also have flushing.”
Dr. Harper incorporates the mnemonic “STOP” to her visits with rosacea patients.
S stands for: Identify signs and symptoms of the condition. “Listen to the patient for symptoms,” she advised. “We’ve learned to listen to darker skinned patients for what they tell us about erythema, for example, because we may not be able to see it, yet they are experiencing it. They may also have symptomatic burning, itching, and stinging.”
T stands for: Discuss triggers. “Ask patients, ‘what is it that makes your rosacea worse?’ That’s different for everyone,” she said.
O stands for: Agree on a treatment outcome. “Ask, ‘what is it that really bothers you? Are you bothered by the bumps? The redness?’ ” she said.
“The P stands for: Develop a plan that addresses all of that,” she said.
Different treatments for different rosacea symptoms
No one-size-fits-all treatment exists for rosacea. Options that work well for papules and pustules aren’t effective for redness. Similarly, products that work for redness don’t work for telangiectasia.
“Different lesions and signs of rosacea will likely require multiple modes of treatment,” Dr. Harper said. “So, when you evaluate your rosacea patients, if they’re doing great, don’t change their regimen. But if you see somebody who is not well controlled, is there an opportunity for you to come in and add something to that regimen that may make them better? Maybe so.”
Treatment options indicated for papules and pustules include ivermectin, metronidazole, azelaic acid, sodium sulfacetamide/sulfur, modified release doxycycline, minocycline foam, and encapsulated benzoyl peroxide.
Options indicated for persistent background erythema include brimonidine and oxymetazoline, while device-based treatments include the pulsed dye laser, the KTP laser, intense pulsed light, and electrosurgery.
Anti-inflammatory action for pustules and papules
A relatively new product indicated for pustules and papules is minocycline 1.5% foam, the only minocycline that is FDA approved to treat rosacea.
“There is no oral minocycline product approved for rosacea yet,” Dr. Harper said. “There is not a known bacterial pathogen in rosacea. Tetracyclines likely work in rosacea by inhibiting neutrophil chemotaxis, inhibiting MMP and thus KLK-5 and LL-37, inhibiting pro-inflammatory cytokines, downregulating reactive oxygen species, and inhibiting angiogenesis.”
In two 12-week, phase 3 randomized studies of 1,522 patients with moderate to severe rosacea, participants were assigned to receive minocycline 5% foam or a vehicle that contained mineral oil and coconut oil.
At week 12, about 50% of patients who received minocycline 5% foam were clear, compared with about 40% of those in the vehicle arm. Also, the reduction of lesion count was about 63% for patients in the treatment group, compared with a reduction of about 54% in the vehicle arm.
Dr. Harper characterized the 63% reduction as “pretty good, but is it good enough or fast enough? I don’t think so, so even with a great drug like this, I would use something else. You can use two medications sometimes to get people better faster. There’s room to bring in something for that background erythema.”
Minocycline 1.5% foam is colored yellow and may stain fabric. “It contains coconut oil, soybean oil, and light mineral oil,” she said. “Most people prefer to use this at bedtime, but you don’t have to.”
Another treatment option is 5% microencapsulated benzoyl peroxide cream, which is FDA approved for inflammatory lesions of rosacea.
“What’s the mechanism of action? Probably not being antimicrobial,” Dr. Harper said. “I think it’s probably at least in part anti-inflammatory, because we have some data to show that it’s killing Demodex [mites]. If Demodex [are] a trigger of inflammation, and we can lessen Demodex, then we could lessen the inflammatory response after that.”
The drug’s approval was based on data from two positive, identical phase 3 randomized, double-blind, multicenter, 12-week clinical trials that evaluated its safety compared with vehicle in 733 people with inflammatory lesions of rosacea (NCT03564119 and NCT03448939).
At week 12, inflammatory lesions of rosacea were reduced by nearly 70% in both trials among those who received 5% microencapsulated benzoyl peroxide cream, compared with 38%-46% among those who received the vehicle. Also, nearly 50% of subjects in the treatment groups were clear or almost clear at 12 weeks, compared with 38%-46% of those who received the vehicle.
Dr. Harper added that about one-quarter of patients in the treatment group of the trials were clear or almost clear by week 4. “That’s pretty fast,” she said, noting that the product’s microencapsulated shell acts as a fenestrated barrier. “It has little openings, which means that it takes a while for the drug to work itself out,” she said. “I think of it as being like a speed bump for benzoyl peroxide delivery. It has to get through this little maze before it lands on the skin. We think that is what has helped with tolerability.”
Oral sarecycline, a narrow spectrum tetracycline that was FDA approved for acne in 2018, may also benefit rosacea patients. In a 12-week, investigator-blinded pilot study, 72 patients with papulopustular rosacea were assigned to receive sarecycline, while 25 received a multivitamin.
By week 12, 75% of patients in the sarecycline group were clear, compared with 16% of those in the multivitamin group, while the inflammatory lesion counts dropped from baseline by 80% and 60%, respectively. Studies of sarecycline for acne have demonstrated similar rates of vertigo, dizziness, and sunburn to those of placebo.
“There were also low rates of gastrointestinal disturbances,” Dr. Harper said. “That’s important in rosacea, because there is no bacterial pathogen.”
Dr. Harper disclosed that she serves as an advisor or consultant for Almirall, BioPharmX, Cassiopeia, Cutanea, Cutera, Dermira, EPI, Galderma, LaRoche-Posay, Ortho, Vyne, Sol Gel, and Sun. She also serves as a speaker or member of a speakers bureau for Almirall, EPI, Galderma, Ortho, and Vyne.
Medscape Live and this news organization are owned by the same parent company.
. At least they should be, according to Julie C. Harper, MD.
“How many people with papules and pustules don’t also have redness?” Dr. Harper, who practices in Birmingham, Ala., said at Medscape Live’s annual Coastal Dermatology Symposium. “If we’re not careful, and we try to classify a person into a subtype of rosacea, we end up treating only part of their rosacea; we don’t treat all of it. We have seen this in the literature,” she added.
“The idea now is to take a phenotypic approach to rosacea. What we mean by that is that you look at the patient, you document every part of rosacea that you see, and you treat according to that,” she continued. “That person with papules and pustules may also have phyma and ocular disease. They may have telangiectasia and persistent background erythema. They may also have flushing.”
Dr. Harper incorporates the mnemonic “STOP” to her visits with rosacea patients.
S stands for: Identify signs and symptoms of the condition. “Listen to the patient for symptoms,” she advised. “We’ve learned to listen to darker skinned patients for what they tell us about erythema, for example, because we may not be able to see it, yet they are experiencing it. They may also have symptomatic burning, itching, and stinging.”
T stands for: Discuss triggers. “Ask patients, ‘what is it that makes your rosacea worse?’ That’s different for everyone,” she said.
O stands for: Agree on a treatment outcome. “Ask, ‘what is it that really bothers you? Are you bothered by the bumps? The redness?’ ” she said.
“The P stands for: Develop a plan that addresses all of that,” she said.
Different treatments for different rosacea symptoms
No one-size-fits-all treatment exists for rosacea. Options that work well for papules and pustules aren’t effective for redness. Similarly, products that work for redness don’t work for telangiectasia.
“Different lesions and signs of rosacea will likely require multiple modes of treatment,” Dr. Harper said. “So, when you evaluate your rosacea patients, if they’re doing great, don’t change their regimen. But if you see somebody who is not well controlled, is there an opportunity for you to come in and add something to that regimen that may make them better? Maybe so.”
Treatment options indicated for papules and pustules include ivermectin, metronidazole, azelaic acid, sodium sulfacetamide/sulfur, modified release doxycycline, minocycline foam, and encapsulated benzoyl peroxide.
Options indicated for persistent background erythema include brimonidine and oxymetazoline, while device-based treatments include the pulsed dye laser, the KTP laser, intense pulsed light, and electrosurgery.
Anti-inflammatory action for pustules and papules
A relatively new product indicated for pustules and papules is minocycline 1.5% foam, the only minocycline that is FDA approved to treat rosacea.
“There is no oral minocycline product approved for rosacea yet,” Dr. Harper said. “There is not a known bacterial pathogen in rosacea. Tetracyclines likely work in rosacea by inhibiting neutrophil chemotaxis, inhibiting MMP and thus KLK-5 and LL-37, inhibiting pro-inflammatory cytokines, downregulating reactive oxygen species, and inhibiting angiogenesis.”
In two 12-week, phase 3 randomized studies of 1,522 patients with moderate to severe rosacea, participants were assigned to receive minocycline 5% foam or a vehicle that contained mineral oil and coconut oil.
At week 12, about 50% of patients who received minocycline 5% foam were clear, compared with about 40% of those in the vehicle arm. Also, the reduction of lesion count was about 63% for patients in the treatment group, compared with a reduction of about 54% in the vehicle arm.
Dr. Harper characterized the 63% reduction as “pretty good, but is it good enough or fast enough? I don’t think so, so even with a great drug like this, I would use something else. You can use two medications sometimes to get people better faster. There’s room to bring in something for that background erythema.”
Minocycline 1.5% foam is colored yellow and may stain fabric. “It contains coconut oil, soybean oil, and light mineral oil,” she said. “Most people prefer to use this at bedtime, but you don’t have to.”
Another treatment option is 5% microencapsulated benzoyl peroxide cream, which is FDA approved for inflammatory lesions of rosacea.
“What’s the mechanism of action? Probably not being antimicrobial,” Dr. Harper said. “I think it’s probably at least in part anti-inflammatory, because we have some data to show that it’s killing Demodex [mites]. If Demodex [are] a trigger of inflammation, and we can lessen Demodex, then we could lessen the inflammatory response after that.”
The drug’s approval was based on data from two positive, identical phase 3 randomized, double-blind, multicenter, 12-week clinical trials that evaluated its safety compared with vehicle in 733 people with inflammatory lesions of rosacea (NCT03564119 and NCT03448939).
At week 12, inflammatory lesions of rosacea were reduced by nearly 70% in both trials among those who received 5% microencapsulated benzoyl peroxide cream, compared with 38%-46% among those who received the vehicle. Also, nearly 50% of subjects in the treatment groups were clear or almost clear at 12 weeks, compared with 38%-46% of those who received the vehicle.
Dr. Harper added that about one-quarter of patients in the treatment group of the trials were clear or almost clear by week 4. “That’s pretty fast,” she said, noting that the product’s microencapsulated shell acts as a fenestrated barrier. “It has little openings, which means that it takes a while for the drug to work itself out,” she said. “I think of it as being like a speed bump for benzoyl peroxide delivery. It has to get through this little maze before it lands on the skin. We think that is what has helped with tolerability.”
Oral sarecycline, a narrow spectrum tetracycline that was FDA approved for acne in 2018, may also benefit rosacea patients. In a 12-week, investigator-blinded pilot study, 72 patients with papulopustular rosacea were assigned to receive sarecycline, while 25 received a multivitamin.
By week 12, 75% of patients in the sarecycline group were clear, compared with 16% of those in the multivitamin group, while the inflammatory lesion counts dropped from baseline by 80% and 60%, respectively. Studies of sarecycline for acne have demonstrated similar rates of vertigo, dizziness, and sunburn to those of placebo.
“There were also low rates of gastrointestinal disturbances,” Dr. Harper said. “That’s important in rosacea, because there is no bacterial pathogen.”
Dr. Harper disclosed that she serves as an advisor or consultant for Almirall, BioPharmX, Cassiopeia, Cutanea, Cutera, Dermira, EPI, Galderma, LaRoche-Posay, Ortho, Vyne, Sol Gel, and Sun. She also serves as a speaker or member of a speakers bureau for Almirall, EPI, Galderma, Ortho, and Vyne.
Medscape Live and this news organization are owned by the same parent company.
FROM MEDSCAPE LIVE COASTAL DERM
Salt pills for patients with acute decompensated heart failure?
Restriction of dietary salt to alleviate or prevent volume overload in patients with acute decompensated heart failure (ADHF) is common hospital practice, but without a solid evidence base. A trial testing whether taking salt pills might have benefits for patients with ADHF undergoing intensive diuresis, therefore, may seem a bit counterintuitive.
In just such a randomized, placebo-controlled trial, the approach made no difference to weight loss on diuresis, a proxy for volume reduction, or to serum creatinine levels in ADHF patients receiving high-dose intravenous diuretic therapy.
The patients consumed the extra salt during their intravenous therapy in the form of tablets providing 6 g sodium chloride daily on top of their hospital-provided, low-sodium meals.
During that time, serum sodium levels remained stable for the 34 patients assigned to the salt tablets but dropped significantly in the 31 given placebo pills.
They lost about the same weight, averages of 4 kg and 4.6 kg (8.8-10 lb), respectively, and their urine output was also similar. Patients who took the salt tablets showed less of an increase in blood urea nitrogen (BUN) at both 96 hours and at discharge.
The findings “challenge the routine practice of sodium chloride restriction in acute heart failure, something done thousands of times a day, millions of times a year,” Robert A. Montgomery, MD, Cleveland Clinic, said when presenting the study at the annual scientific meeting of the Heart Failure Society of America.
The trial, called OSPREY-AHF (Oral Sodium to Preserve Renal Efficiency in Acute Heart Failure), also may encourage a shift in ADHF management from a preoccupation with salt restriction to focus more on fighting fluid retention.
OSPREY-HF took on “an established practice that doesn’t have much high-quality evidentiary support,” one guided primarily by consensus and observational data, Montgomery said in an interview.
There are also potential downsides to dietary sodium restriction, including some that may complicate or block ADHF therapies.
“Low-sodium diets can be associated with decreased caloric intake and nutritional quality,” Dr. Montgomery observed. And observational studies suggest that “patients who are on a low sodium diet can develop increased neurohormonal activation. The kidney is not sensing salt, and so starts ramping up the hormones,” which promotes diuretic resistance.
But emerging evidence also suggests “that giving sodium chloride in the form of hypertonic saline can help patients who are diuretic resistant.” The intervention, which appears to attenuate the neurohormonal activation associated with high-dose intravenous diuretics, Dr. Montgomery noted, helped inspire the design of OSPREY-AHF.
Edema consists of “a gallon of water and a pinch of salt, so we really should stop being so salt-centric and think much more about water as the problem in decompensated heart failure,” said John G.F. Cleland, MD, PhD, during the question-and-answer period after Montgomery’s presentation. Dr. Cleland, of the University of Glasgow Institute of Health and Wellbeing, is not connected to OSPREY-AHF.
“I think that maybe we overinterpret how important salt is” as a focus of volume management in ADHF, offered David Lanfear, MD, Henry Ford Health System, Detroit, who is also not part of the study.
OSPREY-AHF was well conducted but applies to a “very specific” clinical setting, Dr. Lanfear said in an interview. “These people are getting aggressive diuresis, a big dose and continuous infusion. It’s not everybody that has heart failure.”
Although the study was small, “I think it will fuel interest in this area and, probably, further investigation,” he said. The trial on its own won’t change practice, “but it will raise some eyebrows.”
The trial included patients with ADHF who have been “admitted to a cardiovascular medicine floor, not the intensive care unit” and were receiving at least 10 mg per hour of furosemide. It excluded any who were “hypernatremic or severely hyponatremic,” said Dr. Montgomery when presenting the study. They were required to have an initial estimated glomerular filtration rate (eGFR) of at least 15 mL/min per 1.73 m2.
The patients were randomly assigned double blind at a single center to receive tablets providing 2 g sodium chloride or placebo pills – 34 and 31 patients, respectively – three times daily during intravenous diuresis.
At 96 hours, the two groups showed no difference in change in creatinine levels or change in weight, both primary endpoints. Nor did they differ in urine output or change in eGFR. But serum sodium levels fell further, and BUN levels went up more in those given placebo.
The two groups showed no differences in hospital length of stay, use of renal replacement therapy at 90 days, ICU time during the index hospitalization, 30-day readmission, or 90-day mortality – although the trial wasn’t powered for clinical outcomes, Dr. Montgomery reported.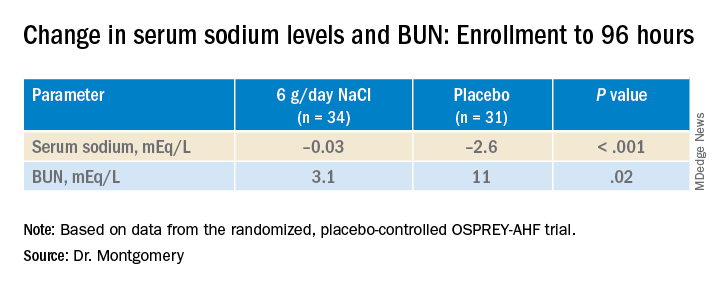
"We have patients who complain about their sodium-restricted diet, we have patients that have cachexia, who have a lot of complaints about provider-ordered meals and recommendations,” Dr. Montgomery explained in an interview.
Clinicians provide education and invest a lot of effort into getting patients with heart failure to start and maintain a low-sodium diet, he said. “But a low-sodium diet, in prior studies – and our study adds to this – is not a lever that actually seems to positively or adversely affect patients.”
Dr. Montgomery pointed to the recently published SODIUM-HF trial comparing low-sodium and unrestricted-sodium diets in outpatients with heart failure. It saw no clinical benefit from the low-sodium intervention.
Until studies show, potentially, that sodium restriction in hospitalized patients with heart failure makes a clinical difference, Dr. Montgomery said, “I’d say we should invest our time in things that we know are the most helpful, like getting them on guideline-directed medical therapy, when instead we spend an enormous amount of time counseling on and enforcing dietary restriction.”
Support for this study was provided by Cleveland Clinic Heart Vascular and Thoracic Institute’s Wilson Grant and Kaufman Center for Heart Failure Treatment and Recovery Grant. Dr. Lanfear disclosed research support from SomaLogic and Lilly; consulting for Abbott Laboratories, AstraZeneca, Janssen, Martin Pharmaceuticals, and Amgen; and serving on advisory panels for Illumina and Cytokinetics. Dr. Montgomery and Dr. Cleland disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Restriction of dietary salt to alleviate or prevent volume overload in patients with acute decompensated heart failure (ADHF) is common hospital practice, but without a solid evidence base. A trial testing whether taking salt pills might have benefits for patients with ADHF undergoing intensive diuresis, therefore, may seem a bit counterintuitive.
In just such a randomized, placebo-controlled trial, the approach made no difference to weight loss on diuresis, a proxy for volume reduction, or to serum creatinine levels in ADHF patients receiving high-dose intravenous diuretic therapy.
The patients consumed the extra salt during their intravenous therapy in the form of tablets providing 6 g sodium chloride daily on top of their hospital-provided, low-sodium meals.
During that time, serum sodium levels remained stable for the 34 patients assigned to the salt tablets but dropped significantly in the 31 given placebo pills.
They lost about the same weight, averages of 4 kg and 4.6 kg (8.8-10 lb), respectively, and their urine output was also similar. Patients who took the salt tablets showed less of an increase in blood urea nitrogen (BUN) at both 96 hours and at discharge.
The findings “challenge the routine practice of sodium chloride restriction in acute heart failure, something done thousands of times a day, millions of times a year,” Robert A. Montgomery, MD, Cleveland Clinic, said when presenting the study at the annual scientific meeting of the Heart Failure Society of America.
The trial, called OSPREY-AHF (Oral Sodium to Preserve Renal Efficiency in Acute Heart Failure), also may encourage a shift in ADHF management from a preoccupation with salt restriction to focus more on fighting fluid retention.
OSPREY-HF took on “an established practice that doesn’t have much high-quality evidentiary support,” one guided primarily by consensus and observational data, Montgomery said in an interview.
There are also potential downsides to dietary sodium restriction, including some that may complicate or block ADHF therapies.
“Low-sodium diets can be associated with decreased caloric intake and nutritional quality,” Dr. Montgomery observed. And observational studies suggest that “patients who are on a low sodium diet can develop increased neurohormonal activation. The kidney is not sensing salt, and so starts ramping up the hormones,” which promotes diuretic resistance.
But emerging evidence also suggests “that giving sodium chloride in the form of hypertonic saline can help patients who are diuretic resistant.” The intervention, which appears to attenuate the neurohormonal activation associated with high-dose intravenous diuretics, Dr. Montgomery noted, helped inspire the design of OSPREY-AHF.
Edema consists of “a gallon of water and a pinch of salt, so we really should stop being so salt-centric and think much more about water as the problem in decompensated heart failure,” said John G.F. Cleland, MD, PhD, during the question-and-answer period after Montgomery’s presentation. Dr. Cleland, of the University of Glasgow Institute of Health and Wellbeing, is not connected to OSPREY-AHF.
“I think that maybe we overinterpret how important salt is” as a focus of volume management in ADHF, offered David Lanfear, MD, Henry Ford Health System, Detroit, who is also not part of the study.
OSPREY-AHF was well conducted but applies to a “very specific” clinical setting, Dr. Lanfear said in an interview. “These people are getting aggressive diuresis, a big dose and continuous infusion. It’s not everybody that has heart failure.”
Although the study was small, “I think it will fuel interest in this area and, probably, further investigation,” he said. The trial on its own won’t change practice, “but it will raise some eyebrows.”
The trial included patients with ADHF who have been “admitted to a cardiovascular medicine floor, not the intensive care unit” and were receiving at least 10 mg per hour of furosemide. It excluded any who were “hypernatremic or severely hyponatremic,” said Dr. Montgomery when presenting the study. They were required to have an initial estimated glomerular filtration rate (eGFR) of at least 15 mL/min per 1.73 m2.
The patients were randomly assigned double blind at a single center to receive tablets providing 2 g sodium chloride or placebo pills – 34 and 31 patients, respectively – three times daily during intravenous diuresis.
At 96 hours, the two groups showed no difference in change in creatinine levels or change in weight, both primary endpoints. Nor did they differ in urine output or change in eGFR. But serum sodium levels fell further, and BUN levels went up more in those given placebo.
The two groups showed no differences in hospital length of stay, use of renal replacement therapy at 90 days, ICU time during the index hospitalization, 30-day readmission, or 90-day mortality – although the trial wasn’t powered for clinical outcomes, Dr. Montgomery reported.
"We have patients who complain about their sodium-restricted diet, we have patients that have cachexia, who have a lot of complaints about provider-ordered meals and recommendations,” Dr. Montgomery explained in an interview.
Clinicians provide education and invest a lot of effort into getting patients with heart failure to start and maintain a low-sodium diet, he said. “But a low-sodium diet, in prior studies – and our study adds to this – is not a lever that actually seems to positively or adversely affect patients.”
Dr. Montgomery pointed to the recently published SODIUM-HF trial comparing low-sodium and unrestricted-sodium diets in outpatients with heart failure. It saw no clinical benefit from the low-sodium intervention.
Until studies show, potentially, that sodium restriction in hospitalized patients with heart failure makes a clinical difference, Dr. Montgomery said, “I’d say we should invest our time in things that we know are the most helpful, like getting them on guideline-directed medical therapy, when instead we spend an enormous amount of time counseling on and enforcing dietary restriction.”
Support for this study was provided by Cleveland Clinic Heart Vascular and Thoracic Institute’s Wilson Grant and Kaufman Center for Heart Failure Treatment and Recovery Grant. Dr. Lanfear disclosed research support from SomaLogic and Lilly; consulting for Abbott Laboratories, AstraZeneca, Janssen, Martin Pharmaceuticals, and Amgen; and serving on advisory panels for Illumina and Cytokinetics. Dr. Montgomery and Dr. Cleland disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Restriction of dietary salt to alleviate or prevent volume overload in patients with acute decompensated heart failure (ADHF) is common hospital practice, but without a solid evidence base. A trial testing whether taking salt pills might have benefits for patients with ADHF undergoing intensive diuresis, therefore, may seem a bit counterintuitive.
In just such a randomized, placebo-controlled trial, the approach made no difference to weight loss on diuresis, a proxy for volume reduction, or to serum creatinine levels in ADHF patients receiving high-dose intravenous diuretic therapy.
The patients consumed the extra salt during their intravenous therapy in the form of tablets providing 6 g sodium chloride daily on top of their hospital-provided, low-sodium meals.
During that time, serum sodium levels remained stable for the 34 patients assigned to the salt tablets but dropped significantly in the 31 given placebo pills.
They lost about the same weight, averages of 4 kg and 4.6 kg (8.8-10 lb), respectively, and their urine output was also similar. Patients who took the salt tablets showed less of an increase in blood urea nitrogen (BUN) at both 96 hours and at discharge.
The findings “challenge the routine practice of sodium chloride restriction in acute heart failure, something done thousands of times a day, millions of times a year,” Robert A. Montgomery, MD, Cleveland Clinic, said when presenting the study at the annual scientific meeting of the Heart Failure Society of America.
The trial, called OSPREY-AHF (Oral Sodium to Preserve Renal Efficiency in Acute Heart Failure), also may encourage a shift in ADHF management from a preoccupation with salt restriction to focus more on fighting fluid retention.
OSPREY-HF took on “an established practice that doesn’t have much high-quality evidentiary support,” one guided primarily by consensus and observational data, Montgomery said in an interview.
There are also potential downsides to dietary sodium restriction, including some that may complicate or block ADHF therapies.
“Low-sodium diets can be associated with decreased caloric intake and nutritional quality,” Dr. Montgomery observed. And observational studies suggest that “patients who are on a low sodium diet can develop increased neurohormonal activation. The kidney is not sensing salt, and so starts ramping up the hormones,” which promotes diuretic resistance.
But emerging evidence also suggests “that giving sodium chloride in the form of hypertonic saline can help patients who are diuretic resistant.” The intervention, which appears to attenuate the neurohormonal activation associated with high-dose intravenous diuretics, Dr. Montgomery noted, helped inspire the design of OSPREY-AHF.
Edema consists of “a gallon of water and a pinch of salt, so we really should stop being so salt-centric and think much more about water as the problem in decompensated heart failure,” said John G.F. Cleland, MD, PhD, during the question-and-answer period after Montgomery’s presentation. Dr. Cleland, of the University of Glasgow Institute of Health and Wellbeing, is not connected to OSPREY-AHF.
“I think that maybe we overinterpret how important salt is” as a focus of volume management in ADHF, offered David Lanfear, MD, Henry Ford Health System, Detroit, who is also not part of the study.
OSPREY-AHF was well conducted but applies to a “very specific” clinical setting, Dr. Lanfear said in an interview. “These people are getting aggressive diuresis, a big dose and continuous infusion. It’s not everybody that has heart failure.”
Although the study was small, “I think it will fuel interest in this area and, probably, further investigation,” he said. The trial on its own won’t change practice, “but it will raise some eyebrows.”
The trial included patients with ADHF who have been “admitted to a cardiovascular medicine floor, not the intensive care unit” and were receiving at least 10 mg per hour of furosemide. It excluded any who were “hypernatremic or severely hyponatremic,” said Dr. Montgomery when presenting the study. They were required to have an initial estimated glomerular filtration rate (eGFR) of at least 15 mL/min per 1.73 m2.
The patients were randomly assigned double blind at a single center to receive tablets providing 2 g sodium chloride or placebo pills – 34 and 31 patients, respectively – three times daily during intravenous diuresis.
At 96 hours, the two groups showed no difference in change in creatinine levels or change in weight, both primary endpoints. Nor did they differ in urine output or change in eGFR. But serum sodium levels fell further, and BUN levels went up more in those given placebo.
The two groups showed no differences in hospital length of stay, use of renal replacement therapy at 90 days, ICU time during the index hospitalization, 30-day readmission, or 90-day mortality – although the trial wasn’t powered for clinical outcomes, Dr. Montgomery reported.
"We have patients who complain about their sodium-restricted diet, we have patients that have cachexia, who have a lot of complaints about provider-ordered meals and recommendations,” Dr. Montgomery explained in an interview.
Clinicians provide education and invest a lot of effort into getting patients with heart failure to start and maintain a low-sodium diet, he said. “But a low-sodium diet, in prior studies – and our study adds to this – is not a lever that actually seems to positively or adversely affect patients.”
Dr. Montgomery pointed to the recently published SODIUM-HF trial comparing low-sodium and unrestricted-sodium diets in outpatients with heart failure. It saw no clinical benefit from the low-sodium intervention.
Until studies show, potentially, that sodium restriction in hospitalized patients with heart failure makes a clinical difference, Dr. Montgomery said, “I’d say we should invest our time in things that we know are the most helpful, like getting them on guideline-directed medical therapy, when instead we spend an enormous amount of time counseling on and enforcing dietary restriction.”
Support for this study was provided by Cleveland Clinic Heart Vascular and Thoracic Institute’s Wilson Grant and Kaufman Center for Heart Failure Treatment and Recovery Grant. Dr. Lanfear disclosed research support from SomaLogic and Lilly; consulting for Abbott Laboratories, AstraZeneca, Janssen, Martin Pharmaceuticals, and Amgen; and serving on advisory panels for Illumina and Cytokinetics. Dr. Montgomery and Dr. Cleland disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM HFSA 2022
What is known about sexual dysfunction after breast cancer?
PARIS – What do doctors know about their patients’ sexual health? Not a lot. What about oncologists who treat women with breast cancer? Not much more.
To determine the extent of sexual dysfunction among women with breast cancer, Maria Alice Franzoi, MD, an oncologist at Gustave Roussy Hospital, Villejuif, France, analyzed data concerning sexuality from the CANTO cohort study. She showed that sexual dysfunction often predates the cancer diagnosis and doesn’t improve but rather worsens in the following 2 years. She presented her results at the annual meeting of the European Society for Medical Oncology.
Present at diagnosis
Dr. Franzoi, whose research projects have focused on patient monitoring post cancer, drew her conclusions from the data provided by CANTO, a longitudinal, prospective cohort study that monitors women being treated for localized breast cancer. Study participants answered the EORTC-QLQ-BR23 quality-of-life questionnaire at the time of diagnosis (T0), 1 year after diagnosis (T1), and 2 years after diagnosis (T2). Four factors were employed to better define women’s sex-related problems: poor body image, poor sexual functioning (activity and desire), lack of sexual pleasure, and a complete lack of sexual activity.
The analysis focused on the responses of 7,895 patients in the CANTO cohort study on sexual activity; 4,523 of those patients answered questions about sexual pleasure. Female respondents who reported engaging in no sexual activity did not have to answer the questions in this second section.
“Seventy-five percent of patients reported at least one of the four concerns during the study,” noted Dr. Franzoi during her presentation. This finding highlights the fact that “sexual problems are already present at the time of diagnosis in a considerable number of patients,” she said. More than a third of participants complained of at least one of the four items.
Developments after diagnosis
The proportion of women who reported no arousal or poor sexual function remained stable at around 30% over time, meaning that the sexual problems were reported in similar numbers at T0, T1, and T2. “However, after cancer, more patients are worried about a lack of sexual pleasure (38.7% at T1 and 38.1% at T2, vs. 29.1% at T0) or report having a negative body image (57.8% at T1 and 52.5% at T2, vs. 32.1% at T0),” said Dr. Franzoi.
She identified the following three variables as being associated with sexual dysfunction 2 years after diagnosis: the existence of this problem at the time of diagnosis, the use of adjuvant hormone therapy, and severe depression or a very high stress level after the first year of treatment.
Inadequate specific treatment
“Sexual dysfunction is a major unmet need with a significant impact on quality of life,” said Maryam Lustberg, MD, an oncologist at Yale School of Medicine, New Haven, Conn., who was invited to discuss the results at the conference.
Dr. Franzoi observed that most participants with sexual dysfunction that had continued 2 years after diagnosis had not been referred to a doctor for this problem. “In terms of sexual function, it’s better at T2 than at T1, but only 41% of these women have been seen by a gynecologist, and only 15% have received specific treatment,” she reported, emphasizing the need to assess and treat these issues “proactively” at the time of diagnosis and during and after treatment.
“Now we need to work out what the best treatment approach is,” commented Dr. Lustberg. She said that cancers other than breast and gynecologic cancers should also be taken into consideration. She cited the Sexual Health Assessment in Women With Lung Cancer study, which recently revealed that after being diagnosed with lung cancer, female patients experienced a drop in sexual desire (31% vs. 15% before diagnosis) and an increase in vaginal discomfort or dryness (43% vs. 13% before diagnosis). This study, presented in August to the 2022 International Association for the Study of Lung Cancer World Conference on Lung Cancer, also revealed that different parameters affect satisfaction in one’s sex life, including fatigue, sadness, relationship problems with a partner, and even breathing. Dr. Lustberg concluded from this study that a multidisciplinary approach is needed for cancer survivors.
Dr. Franzoi received research funding from Resilience Care. Dr. Lustberg has links with AstraZeneca, Pfizer, Novartis, Sanofi, and Lilly.
This article was translated from the Medscape French edition.
PARIS – What do doctors know about their patients’ sexual health? Not a lot. What about oncologists who treat women with breast cancer? Not much more.
To determine the extent of sexual dysfunction among women with breast cancer, Maria Alice Franzoi, MD, an oncologist at Gustave Roussy Hospital, Villejuif, France, analyzed data concerning sexuality from the CANTO cohort study. She showed that sexual dysfunction often predates the cancer diagnosis and doesn’t improve but rather worsens in the following 2 years. She presented her results at the annual meeting of the European Society for Medical Oncology.
Present at diagnosis
Dr. Franzoi, whose research projects have focused on patient monitoring post cancer, drew her conclusions from the data provided by CANTO, a longitudinal, prospective cohort study that monitors women being treated for localized breast cancer. Study participants answered the EORTC-QLQ-BR23 quality-of-life questionnaire at the time of diagnosis (T0), 1 year after diagnosis (T1), and 2 years after diagnosis (T2). Four factors were employed to better define women’s sex-related problems: poor body image, poor sexual functioning (activity and desire), lack of sexual pleasure, and a complete lack of sexual activity.
The analysis focused on the responses of 7,895 patients in the CANTO cohort study on sexual activity; 4,523 of those patients answered questions about sexual pleasure. Female respondents who reported engaging in no sexual activity did not have to answer the questions in this second section.
“Seventy-five percent of patients reported at least one of the four concerns during the study,” noted Dr. Franzoi during her presentation. This finding highlights the fact that “sexual problems are already present at the time of diagnosis in a considerable number of patients,” she said. More than a third of participants complained of at least one of the four items.
Developments after diagnosis
The proportion of women who reported no arousal or poor sexual function remained stable at around 30% over time, meaning that the sexual problems were reported in similar numbers at T0, T1, and T2. “However, after cancer, more patients are worried about a lack of sexual pleasure (38.7% at T1 and 38.1% at T2, vs. 29.1% at T0) or report having a negative body image (57.8% at T1 and 52.5% at T2, vs. 32.1% at T0),” said Dr. Franzoi.
She identified the following three variables as being associated with sexual dysfunction 2 years after diagnosis: the existence of this problem at the time of diagnosis, the use of adjuvant hormone therapy, and severe depression or a very high stress level after the first year of treatment.
Inadequate specific treatment
“Sexual dysfunction is a major unmet need with a significant impact on quality of life,” said Maryam Lustberg, MD, an oncologist at Yale School of Medicine, New Haven, Conn., who was invited to discuss the results at the conference.
Dr. Franzoi observed that most participants with sexual dysfunction that had continued 2 years after diagnosis had not been referred to a doctor for this problem. “In terms of sexual function, it’s better at T2 than at T1, but only 41% of these women have been seen by a gynecologist, and only 15% have received specific treatment,” she reported, emphasizing the need to assess and treat these issues “proactively” at the time of diagnosis and during and after treatment.
“Now we need to work out what the best treatment approach is,” commented Dr. Lustberg. She said that cancers other than breast and gynecologic cancers should also be taken into consideration. She cited the Sexual Health Assessment in Women With Lung Cancer study, which recently revealed that after being diagnosed with lung cancer, female patients experienced a drop in sexual desire (31% vs. 15% before diagnosis) and an increase in vaginal discomfort or dryness (43% vs. 13% before diagnosis). This study, presented in August to the 2022 International Association for the Study of Lung Cancer World Conference on Lung Cancer, also revealed that different parameters affect satisfaction in one’s sex life, including fatigue, sadness, relationship problems with a partner, and even breathing. Dr. Lustberg concluded from this study that a multidisciplinary approach is needed for cancer survivors.
Dr. Franzoi received research funding from Resilience Care. Dr. Lustberg has links with AstraZeneca, Pfizer, Novartis, Sanofi, and Lilly.
This article was translated from the Medscape French edition.
PARIS – What do doctors know about their patients’ sexual health? Not a lot. What about oncologists who treat women with breast cancer? Not much more.
To determine the extent of sexual dysfunction among women with breast cancer, Maria Alice Franzoi, MD, an oncologist at Gustave Roussy Hospital, Villejuif, France, analyzed data concerning sexuality from the CANTO cohort study. She showed that sexual dysfunction often predates the cancer diagnosis and doesn’t improve but rather worsens in the following 2 years. She presented her results at the annual meeting of the European Society for Medical Oncology.
Present at diagnosis
Dr. Franzoi, whose research projects have focused on patient monitoring post cancer, drew her conclusions from the data provided by CANTO, a longitudinal, prospective cohort study that monitors women being treated for localized breast cancer. Study participants answered the EORTC-QLQ-BR23 quality-of-life questionnaire at the time of diagnosis (T0), 1 year after diagnosis (T1), and 2 years after diagnosis (T2). Four factors were employed to better define women’s sex-related problems: poor body image, poor sexual functioning (activity and desire), lack of sexual pleasure, and a complete lack of sexual activity.
The analysis focused on the responses of 7,895 patients in the CANTO cohort study on sexual activity; 4,523 of those patients answered questions about sexual pleasure. Female respondents who reported engaging in no sexual activity did not have to answer the questions in this second section.
“Seventy-five percent of patients reported at least one of the four concerns during the study,” noted Dr. Franzoi during her presentation. This finding highlights the fact that “sexual problems are already present at the time of diagnosis in a considerable number of patients,” she said. More than a third of participants complained of at least one of the four items.
Developments after diagnosis
The proportion of women who reported no arousal or poor sexual function remained stable at around 30% over time, meaning that the sexual problems were reported in similar numbers at T0, T1, and T2. “However, after cancer, more patients are worried about a lack of sexual pleasure (38.7% at T1 and 38.1% at T2, vs. 29.1% at T0) or report having a negative body image (57.8% at T1 and 52.5% at T2, vs. 32.1% at T0),” said Dr. Franzoi.
She identified the following three variables as being associated with sexual dysfunction 2 years after diagnosis: the existence of this problem at the time of diagnosis, the use of adjuvant hormone therapy, and severe depression or a very high stress level after the first year of treatment.
Inadequate specific treatment
“Sexual dysfunction is a major unmet need with a significant impact on quality of life,” said Maryam Lustberg, MD, an oncologist at Yale School of Medicine, New Haven, Conn., who was invited to discuss the results at the conference.
Dr. Franzoi observed that most participants with sexual dysfunction that had continued 2 years after diagnosis had not been referred to a doctor for this problem. “In terms of sexual function, it’s better at T2 than at T1, but only 41% of these women have been seen by a gynecologist, and only 15% have received specific treatment,” she reported, emphasizing the need to assess and treat these issues “proactively” at the time of diagnosis and during and after treatment.
“Now we need to work out what the best treatment approach is,” commented Dr. Lustberg. She said that cancers other than breast and gynecologic cancers should also be taken into consideration. She cited the Sexual Health Assessment in Women With Lung Cancer study, which recently revealed that after being diagnosed with lung cancer, female patients experienced a drop in sexual desire (31% vs. 15% before diagnosis) and an increase in vaginal discomfort or dryness (43% vs. 13% before diagnosis). This study, presented in August to the 2022 International Association for the Study of Lung Cancer World Conference on Lung Cancer, also revealed that different parameters affect satisfaction in one’s sex life, including fatigue, sadness, relationship problems with a partner, and even breathing. Dr. Lustberg concluded from this study that a multidisciplinary approach is needed for cancer survivors.
Dr. Franzoi received research funding from Resilience Care. Dr. Lustberg has links with AstraZeneca, Pfizer, Novartis, Sanofi, and Lilly.
This article was translated from the Medscape French edition.
AT ESMO CONGRESS 2022
Long-acting naltrexone effective in alcohol use disorder
according to findings presented at the annual meeting of the American College of Emergency Physicians.
The results show the feasibility of such a program and underscore the importance of the ED in combating AUD, said the researchers, from the University of California, San Francisco.
“According to the National Institute on Alcohol Abuse and Alcoholism, 18% of ED visits had alcohol as a contributing factor – the volume of alcohol-related ED visits has been climbing every year, and it is a significant public health problem,” said Maria Raven, MD, MPH, professor of emergency medicine at UCSF. “Right now, we do very little for people who come to the ED with AUD, so it is a missed opportunity to intervene, especially given the volume of visits we see and that our patient population is one that often has significant barriers to accessing outpatient treatment.”
The findings come from a 12-week, prospective, single-arm study of ED patients who were actively drinking adults with known or suspected AUD and who had positive scores on a screening test. Of 179 patients who were approached, 32 agreed to enroll; the enrollment yield was 18%. Participants were given monthly extended-release naltrexone and case management services.
Of the 32 participants, 25 completed all their study visits and 22 (69%) continued taking naltrexone after the 12 weeks.
The researchers said the results surprised them. The average daily alcohol consumption at baseline was 7.6 drinks a day, and it fell by 7.5 drinks a day – in other words, to almost no consumption.
“The median alcohol consumption when measured over the last 2 weeks of the study was zero,” Dr. Raven said. “This doesn’t mean everyone was at zero, but this was the median and reflects that many participants stopped drinking altogether. We were pleasantly surprised by this. I don’t know that we thought so many people who participated would actually fully abstain.”
On the Kemp Quality of Life Scale – with scores from 1 to 7, with 1 being “life is very distressing,” 4 being “life is so-so,” and 7 being “life is great” – the average baseline score was 3.6. That score rose by 1.2 points by the study’s end.
Dr. Raven said she hoped more would enroll but that “a number of people actually did not want the injection or were not ready to think about stopping.” Still, the 18% enrollment is “a major improvement,” considering that no attempt was made to initiate treatment with naltrexone prior to the study. Oral naltrexone, rather than the injection, could be offered to improve participation, but oral naltrexone has to be taken daily.
She said a larger study is planned at UCSF and that other institutions are interested in starting a similar program.
“When someone is in the ED for an AUD-related issue, it can serve as a turning point for them in some cases,” she said.
Erik S. Anderson, MD, associate research director at Oakland, Calif.–based Alameda Health System, who has studied naltrexone in the ED, said the findings dovetail with what his team has found at his center. He added that psychosocial support is important as well and that his team has found that navigation services are the most important factor in connecting patients with follow-up care – even more so than providing medications.
“In my mind, this is a situation where we have treatment options and approaches that work, and it’s really about implementing these services in a novel care setting,” he said. “ED patients are at higher risk of complications for AUD simply because they are in the ED in the first place – initiating AUD treatment in this setting is the right thing to do.”
Dr. Raven and Dr. Anderson disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
according to findings presented at the annual meeting of the American College of Emergency Physicians.
The results show the feasibility of such a program and underscore the importance of the ED in combating AUD, said the researchers, from the University of California, San Francisco.
“According to the National Institute on Alcohol Abuse and Alcoholism, 18% of ED visits had alcohol as a contributing factor – the volume of alcohol-related ED visits has been climbing every year, and it is a significant public health problem,” said Maria Raven, MD, MPH, professor of emergency medicine at UCSF. “Right now, we do very little for people who come to the ED with AUD, so it is a missed opportunity to intervene, especially given the volume of visits we see and that our patient population is one that often has significant barriers to accessing outpatient treatment.”
The findings come from a 12-week, prospective, single-arm study of ED patients who were actively drinking adults with known or suspected AUD and who had positive scores on a screening test. Of 179 patients who were approached, 32 agreed to enroll; the enrollment yield was 18%. Participants were given monthly extended-release naltrexone and case management services.
Of the 32 participants, 25 completed all their study visits and 22 (69%) continued taking naltrexone after the 12 weeks.
The researchers said the results surprised them. The average daily alcohol consumption at baseline was 7.6 drinks a day, and it fell by 7.5 drinks a day – in other words, to almost no consumption.
“The median alcohol consumption when measured over the last 2 weeks of the study was zero,” Dr. Raven said. “This doesn’t mean everyone was at zero, but this was the median and reflects that many participants stopped drinking altogether. We were pleasantly surprised by this. I don’t know that we thought so many people who participated would actually fully abstain.”
On the Kemp Quality of Life Scale – with scores from 1 to 7, with 1 being “life is very distressing,” 4 being “life is so-so,” and 7 being “life is great” – the average baseline score was 3.6. That score rose by 1.2 points by the study’s end.
Dr. Raven said she hoped more would enroll but that “a number of people actually did not want the injection or were not ready to think about stopping.” Still, the 18% enrollment is “a major improvement,” considering that no attempt was made to initiate treatment with naltrexone prior to the study. Oral naltrexone, rather than the injection, could be offered to improve participation, but oral naltrexone has to be taken daily.
She said a larger study is planned at UCSF and that other institutions are interested in starting a similar program.
“When someone is in the ED for an AUD-related issue, it can serve as a turning point for them in some cases,” she said.
Erik S. Anderson, MD, associate research director at Oakland, Calif.–based Alameda Health System, who has studied naltrexone in the ED, said the findings dovetail with what his team has found at his center. He added that psychosocial support is important as well and that his team has found that navigation services are the most important factor in connecting patients with follow-up care – even more so than providing medications.
“In my mind, this is a situation where we have treatment options and approaches that work, and it’s really about implementing these services in a novel care setting,” he said. “ED patients are at higher risk of complications for AUD simply because they are in the ED in the first place – initiating AUD treatment in this setting is the right thing to do.”
Dr. Raven and Dr. Anderson disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
according to findings presented at the annual meeting of the American College of Emergency Physicians.
The results show the feasibility of such a program and underscore the importance of the ED in combating AUD, said the researchers, from the University of California, San Francisco.
“According to the National Institute on Alcohol Abuse and Alcoholism, 18% of ED visits had alcohol as a contributing factor – the volume of alcohol-related ED visits has been climbing every year, and it is a significant public health problem,” said Maria Raven, MD, MPH, professor of emergency medicine at UCSF. “Right now, we do very little for people who come to the ED with AUD, so it is a missed opportunity to intervene, especially given the volume of visits we see and that our patient population is one that often has significant barriers to accessing outpatient treatment.”
The findings come from a 12-week, prospective, single-arm study of ED patients who were actively drinking adults with known or suspected AUD and who had positive scores on a screening test. Of 179 patients who were approached, 32 agreed to enroll; the enrollment yield was 18%. Participants were given monthly extended-release naltrexone and case management services.
Of the 32 participants, 25 completed all their study visits and 22 (69%) continued taking naltrexone after the 12 weeks.
The researchers said the results surprised them. The average daily alcohol consumption at baseline was 7.6 drinks a day, and it fell by 7.5 drinks a day – in other words, to almost no consumption.
“The median alcohol consumption when measured over the last 2 weeks of the study was zero,” Dr. Raven said. “This doesn’t mean everyone was at zero, but this was the median and reflects that many participants stopped drinking altogether. We were pleasantly surprised by this. I don’t know that we thought so many people who participated would actually fully abstain.”
On the Kemp Quality of Life Scale – with scores from 1 to 7, with 1 being “life is very distressing,” 4 being “life is so-so,” and 7 being “life is great” – the average baseline score was 3.6. That score rose by 1.2 points by the study’s end.
Dr. Raven said she hoped more would enroll but that “a number of people actually did not want the injection or were not ready to think about stopping.” Still, the 18% enrollment is “a major improvement,” considering that no attempt was made to initiate treatment with naltrexone prior to the study. Oral naltrexone, rather than the injection, could be offered to improve participation, but oral naltrexone has to be taken daily.
She said a larger study is planned at UCSF and that other institutions are interested in starting a similar program.
“When someone is in the ED for an AUD-related issue, it can serve as a turning point for them in some cases,” she said.
Erik S. Anderson, MD, associate research director at Oakland, Calif.–based Alameda Health System, who has studied naltrexone in the ED, said the findings dovetail with what his team has found at his center. He added that psychosocial support is important as well and that his team has found that navigation services are the most important factor in connecting patients with follow-up care – even more so than providing medications.
“In my mind, this is a situation where we have treatment options and approaches that work, and it’s really about implementing these services in a novel care setting,” he said. “ED patients are at higher risk of complications for AUD simply because they are in the ED in the first place – initiating AUD treatment in this setting is the right thing to do.”
Dr. Raven and Dr. Anderson disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM ACEP 2022
I am not fine: The heavy toll cancer takes
PARIS – “I thought I was as exhausted, and isolated, and neglected as I could get, and then he came home.”
Those were the words of Kate Washington, PhD, from Sacramento as she gave a moving account of the immense burden she felt as caregiver to her husband with cancer.
She was taking part in the session, “I am FINE: Frustrated * Isolated * Neglected * Emotional,” at the annual meeting of the European Society for Medical Oncology. In that session,
Dr. Washington, author of “Already Toast: Caregiving and Burnout in America” (Boston: Beacon Press, 2021), explained that she cared for her husband and young family while he was “suffering through two different kinds of lymphoma and really devastating stem cell transplants.”
When her husband was first diagnosed with a rare form of lymphoma in 2015, he was placed on a watch-and-wait protocol. At that point, he seemed fine, Dr. Washington said.
A few months later, he started coughing up blood. After being rushed to the emergency department, doctors found that a slow-growing lung tumor had ruptured.
Three weeks later, he came out of the hospital with a collapsed lung – an effect of his chemotherapy, Dr. Washington said.
But that was hardly the last word. He soon experienced relapse with a “very aggressive” form of his disease, and in 2016, he underwent a stem cell transplant.
“He spent 1½ months in the hospital ... in isolation, not seeing our daughters,” Dr. Washington said. He lost his vision and developed grade 4 graft-versus-host disease, among other problems.
He was alive, just barely, Dr. Washington said.
“As you might imagine, I was pulled between the hospital and the home, taking care of our daughters, who were not seeing him during that time,” she recalled.
But every time someone asked her whether she was okay, she replied: “I am fine.”
“A total lie,” she admitted.
Dr. Washington felt frustrated, not only from the financial strain of out-of-pocket health care costs and lost earnings but also from fast evolving relationships and a feeling of being “unseen and underappreciated.”
Another jarring change: When her husband was discharged from the hospital, Dr. Washington was suddenly thrust into the role of full-time caretaker.
Her husband could not be left alone, his doctor had said. And with two young children, Dr. Washington did not know how she would manage.
The demands of being a full-time caregiver are intense. Caregivers, Dr. Washington explained, can spend 32 hours a week looking after a loved one with cancer.
Like Dr. Washington, most caregivers feel they have no choice but to take on this intense role – one for which they have little or no training or preparation. The nonstop demands leave little time for self-care and can lead to high rates of caregiver injury and illness.
Isolation often creeps in because it can be “hard to ask for help,” she said. About 30% of caregivers report having depression or anxiety, and 21% feel lonely.
“When he was very ill, I found it really difficult to connect with other people and my friends,” Dr. Washington recalled. “I didn’t feel like I could really adequately explain the kind of strain that I was under.”
Are patients fine?
Like caregivers, patients often say they are fine when they are not.
The toll cancer takes on patients is immense. Natacha Bolanos Fernandez, from the Lymphoma Coalition Europe, highlighted the physical, mental, and social strain that can affect patients with cancer.
The physical aspects can encompass a host of problems – fatigue, night sweats, weight loss, and the vomiting that accompanies many cancer treatments. Patients may face changes in their mobility and independence as well. The mental side of cancer can include anxiety, depression, and psychological distress, while the social aspects span changing, perhaps strained, relationships with family and friends.
Fatigue, in particular, is an underreported, underdiagnosed, and undertreated problem, Ms. Fernandez noted. According to recent survey data from the Lymphoma Coalition’s Global Patient Survey, 72% of patients reported fatigue. This problem worsened over time, with 59% reporting fatigue after their diagnosis and up to 82% among patients who experienced relapse two or more times.
Fatigue “may be getting worse rather than better over time,” Ms. Fernandez said, and many patients felt that their life had changed completely because of cancer-related fatigue.
To help patients manage, the Lymphoma Coalition has published a report on the impact of cancer-related fatigue and how to improve outcomes. Methods include greater awareness, regular screening, and interventions such as yoga or mindfulness-based cognitive therapy.
Are clinicians fine?
Nurses and physicians face challenges caring for patients with cancer.
Although “nurses love their jobs and are extremely committed,” the impact cancer has on a nursing career is often undervalued or “neglected,” said Lena Sharp, RN, PhD, of the Regional Cancer Centre, Stockholm-Gotland.
Burnout, in particular, remains a problem among oncologists and nurses, and it was made worse during the COVID-19 pandemic.
Fatima Cardoso, MD, explained that burnout has an impact on doctors as well as patients because it affects communication with patients and performance. Physicians can, for instance, appear detached, emotional, or tired.
Patients may then feel less inclined to tell their oncologist how they’re feeling, said Dr. Cardoso, director of the breast unit at Champalimaud Clinical Center, Lisbon.
It is important to remember to not just focus on the patient’s disease or treatment but to also ask how they are doing and what is going on in their lives.
Above all, “show that you care,” said Dr. Cardoso.
The Lymphoma Coalition Europe has relationships with Bristol-Myers Squibb, Establishment Labs, Kyowa Kirin, Novartis, Roche, Takeda. Dr. Cardoso has relationships with Amgen, Astellas/Medivation, AstraZeneca, Celgene, Daiichi Sankyo, Eisai, GE Oncology, Genentech, GlaxoSmithKline, and other companies. No other relevant financial relationships were reported.
A version of this article first appeared on Medscape.com.
PARIS – “I thought I was as exhausted, and isolated, and neglected as I could get, and then he came home.”
Those were the words of Kate Washington, PhD, from Sacramento as she gave a moving account of the immense burden she felt as caregiver to her husband with cancer.
She was taking part in the session, “I am FINE: Frustrated * Isolated * Neglected * Emotional,” at the annual meeting of the European Society for Medical Oncology. In that session,
Dr. Washington, author of “Already Toast: Caregiving and Burnout in America” (Boston: Beacon Press, 2021), explained that she cared for her husband and young family while he was “suffering through two different kinds of lymphoma and really devastating stem cell transplants.”
When her husband was first diagnosed with a rare form of lymphoma in 2015, he was placed on a watch-and-wait protocol. At that point, he seemed fine, Dr. Washington said.
A few months later, he started coughing up blood. After being rushed to the emergency department, doctors found that a slow-growing lung tumor had ruptured.
Three weeks later, he came out of the hospital with a collapsed lung – an effect of his chemotherapy, Dr. Washington said.
But that was hardly the last word. He soon experienced relapse with a “very aggressive” form of his disease, and in 2016, he underwent a stem cell transplant.
“He spent 1½ months in the hospital ... in isolation, not seeing our daughters,” Dr. Washington said. He lost his vision and developed grade 4 graft-versus-host disease, among other problems.
He was alive, just barely, Dr. Washington said.
“As you might imagine, I was pulled between the hospital and the home, taking care of our daughters, who were not seeing him during that time,” she recalled.
But every time someone asked her whether she was okay, she replied: “I am fine.”
“A total lie,” she admitted.
Dr. Washington felt frustrated, not only from the financial strain of out-of-pocket health care costs and lost earnings but also from fast evolving relationships and a feeling of being “unseen and underappreciated.”
Another jarring change: When her husband was discharged from the hospital, Dr. Washington was suddenly thrust into the role of full-time caretaker.
Her husband could not be left alone, his doctor had said. And with two young children, Dr. Washington did not know how she would manage.
The demands of being a full-time caregiver are intense. Caregivers, Dr. Washington explained, can spend 32 hours a week looking after a loved one with cancer.
Like Dr. Washington, most caregivers feel they have no choice but to take on this intense role – one for which they have little or no training or preparation. The nonstop demands leave little time for self-care and can lead to high rates of caregiver injury and illness.
Isolation often creeps in because it can be “hard to ask for help,” she said. About 30% of caregivers report having depression or anxiety, and 21% feel lonely.
“When he was very ill, I found it really difficult to connect with other people and my friends,” Dr. Washington recalled. “I didn’t feel like I could really adequately explain the kind of strain that I was under.”
Are patients fine?
Like caregivers, patients often say they are fine when they are not.
The toll cancer takes on patients is immense. Natacha Bolanos Fernandez, from the Lymphoma Coalition Europe, highlighted the physical, mental, and social strain that can affect patients with cancer.
The physical aspects can encompass a host of problems – fatigue, night sweats, weight loss, and the vomiting that accompanies many cancer treatments. Patients may face changes in their mobility and independence as well. The mental side of cancer can include anxiety, depression, and psychological distress, while the social aspects span changing, perhaps strained, relationships with family and friends.
Fatigue, in particular, is an underreported, underdiagnosed, and undertreated problem, Ms. Fernandez noted. According to recent survey data from the Lymphoma Coalition’s Global Patient Survey, 72% of patients reported fatigue. This problem worsened over time, with 59% reporting fatigue after their diagnosis and up to 82% among patients who experienced relapse two or more times.
Fatigue “may be getting worse rather than better over time,” Ms. Fernandez said, and many patients felt that their life had changed completely because of cancer-related fatigue.
To help patients manage, the Lymphoma Coalition has published a report on the impact of cancer-related fatigue and how to improve outcomes. Methods include greater awareness, regular screening, and interventions such as yoga or mindfulness-based cognitive therapy.
Are clinicians fine?
Nurses and physicians face challenges caring for patients with cancer.
Although “nurses love their jobs and are extremely committed,” the impact cancer has on a nursing career is often undervalued or “neglected,” said Lena Sharp, RN, PhD, of the Regional Cancer Centre, Stockholm-Gotland.
Burnout, in particular, remains a problem among oncologists and nurses, and it was made worse during the COVID-19 pandemic.
Fatima Cardoso, MD, explained that burnout has an impact on doctors as well as patients because it affects communication with patients and performance. Physicians can, for instance, appear detached, emotional, or tired.
Patients may then feel less inclined to tell their oncologist how they’re feeling, said Dr. Cardoso, director of the breast unit at Champalimaud Clinical Center, Lisbon.
It is important to remember to not just focus on the patient’s disease or treatment but to also ask how they are doing and what is going on in their lives.
Above all, “show that you care,” said Dr. Cardoso.
The Lymphoma Coalition Europe has relationships with Bristol-Myers Squibb, Establishment Labs, Kyowa Kirin, Novartis, Roche, Takeda. Dr. Cardoso has relationships with Amgen, Astellas/Medivation, AstraZeneca, Celgene, Daiichi Sankyo, Eisai, GE Oncology, Genentech, GlaxoSmithKline, and other companies. No other relevant financial relationships were reported.
A version of this article first appeared on Medscape.com.
PARIS – “I thought I was as exhausted, and isolated, and neglected as I could get, and then he came home.”
Those were the words of Kate Washington, PhD, from Sacramento as she gave a moving account of the immense burden she felt as caregiver to her husband with cancer.
She was taking part in the session, “I am FINE: Frustrated * Isolated * Neglected * Emotional,” at the annual meeting of the European Society for Medical Oncology. In that session,
Dr. Washington, author of “Already Toast: Caregiving and Burnout in America” (Boston: Beacon Press, 2021), explained that she cared for her husband and young family while he was “suffering through two different kinds of lymphoma and really devastating stem cell transplants.”
When her husband was first diagnosed with a rare form of lymphoma in 2015, he was placed on a watch-and-wait protocol. At that point, he seemed fine, Dr. Washington said.
A few months later, he started coughing up blood. After being rushed to the emergency department, doctors found that a slow-growing lung tumor had ruptured.
Three weeks later, he came out of the hospital with a collapsed lung – an effect of his chemotherapy, Dr. Washington said.
But that was hardly the last word. He soon experienced relapse with a “very aggressive” form of his disease, and in 2016, he underwent a stem cell transplant.
“He spent 1½ months in the hospital ... in isolation, not seeing our daughters,” Dr. Washington said. He lost his vision and developed grade 4 graft-versus-host disease, among other problems.
He was alive, just barely, Dr. Washington said.
“As you might imagine, I was pulled between the hospital and the home, taking care of our daughters, who were not seeing him during that time,” she recalled.
But every time someone asked her whether she was okay, she replied: “I am fine.”
“A total lie,” she admitted.
Dr. Washington felt frustrated, not only from the financial strain of out-of-pocket health care costs and lost earnings but also from fast evolving relationships and a feeling of being “unseen and underappreciated.”
Another jarring change: When her husband was discharged from the hospital, Dr. Washington was suddenly thrust into the role of full-time caretaker.
Her husband could not be left alone, his doctor had said. And with two young children, Dr. Washington did not know how she would manage.
The demands of being a full-time caregiver are intense. Caregivers, Dr. Washington explained, can spend 32 hours a week looking after a loved one with cancer.
Like Dr. Washington, most caregivers feel they have no choice but to take on this intense role – one for which they have little or no training or preparation. The nonstop demands leave little time for self-care and can lead to high rates of caregiver injury and illness.
Isolation often creeps in because it can be “hard to ask for help,” she said. About 30% of caregivers report having depression or anxiety, and 21% feel lonely.
“When he was very ill, I found it really difficult to connect with other people and my friends,” Dr. Washington recalled. “I didn’t feel like I could really adequately explain the kind of strain that I was under.”
Are patients fine?
Like caregivers, patients often say they are fine when they are not.
The toll cancer takes on patients is immense. Natacha Bolanos Fernandez, from the Lymphoma Coalition Europe, highlighted the physical, mental, and social strain that can affect patients with cancer.
The physical aspects can encompass a host of problems – fatigue, night sweats, weight loss, and the vomiting that accompanies many cancer treatments. Patients may face changes in their mobility and independence as well. The mental side of cancer can include anxiety, depression, and psychological distress, while the social aspects span changing, perhaps strained, relationships with family and friends.
Fatigue, in particular, is an underreported, underdiagnosed, and undertreated problem, Ms. Fernandez noted. According to recent survey data from the Lymphoma Coalition’s Global Patient Survey, 72% of patients reported fatigue. This problem worsened over time, with 59% reporting fatigue after their diagnosis and up to 82% among patients who experienced relapse two or more times.
Fatigue “may be getting worse rather than better over time,” Ms. Fernandez said, and many patients felt that their life had changed completely because of cancer-related fatigue.
To help patients manage, the Lymphoma Coalition has published a report on the impact of cancer-related fatigue and how to improve outcomes. Methods include greater awareness, regular screening, and interventions such as yoga or mindfulness-based cognitive therapy.
Are clinicians fine?
Nurses and physicians face challenges caring for patients with cancer.
Although “nurses love their jobs and are extremely committed,” the impact cancer has on a nursing career is often undervalued or “neglected,” said Lena Sharp, RN, PhD, of the Regional Cancer Centre, Stockholm-Gotland.
Burnout, in particular, remains a problem among oncologists and nurses, and it was made worse during the COVID-19 pandemic.
Fatima Cardoso, MD, explained that burnout has an impact on doctors as well as patients because it affects communication with patients and performance. Physicians can, for instance, appear detached, emotional, or tired.
Patients may then feel less inclined to tell their oncologist how they’re feeling, said Dr. Cardoso, director of the breast unit at Champalimaud Clinical Center, Lisbon.
It is important to remember to not just focus on the patient’s disease or treatment but to also ask how they are doing and what is going on in their lives.
Above all, “show that you care,” said Dr. Cardoso.
The Lymphoma Coalition Europe has relationships with Bristol-Myers Squibb, Establishment Labs, Kyowa Kirin, Novartis, Roche, Takeda. Dr. Cardoso has relationships with Amgen, Astellas/Medivation, AstraZeneca, Celgene, Daiichi Sankyo, Eisai, GE Oncology, Genentech, GlaxoSmithKline, and other companies. No other relevant financial relationships were reported.
A version of this article first appeared on Medscape.com.
AT ESMO CONGRESS 2022
52-week data show lebrikizumab atopic dermatitis effects maintained
from the phase 3 ADvocate1 and ADvocate2 trials.
“We’re focused on the responders,” said Andrew Blauvelt, MD, MBA, as he presented the positive findings at the annual congress of the European Academy of Dermatology and Venereology.
Responders were the 291 people whose atopic dermatitis greatly improved after an initial 16 weeks’ treatment with lebrikizumab in both trials and who were then randomly allocated to receive injections every 2 weeks (Q2W, n = 113) or every 4 weeks (Q4W, n = 118), or to receive placebo injections Q2W (n = 60).
“Very interestingly, for me, the Q4W maintenance dosing was just as good as the Q2W maintenance dosing,” said Dr. Blauvelt, president of Oregon Medical Research Center, Portland.
“Another highlight of these data is that the patients who went on to placebo, about 50% of the patients maintained good responses, despite no treatment from week 16 to week 52,” he added.
Most patients did not require topical steroids, and “there were no surprises here” in terms of the safety profile. Lebrikizumab, a monoclonal antibody, binds to soluble interleukin-13 and blocks IL-13 signaling.
“So, the study really shows that specific targeting of IL-13 with lebrikizumab, either Q2W or Q4W, has high maintenance of efficacy and is reasonably tolerated and safe in adolescents and adults with atopic dermatitis,” Dr. Blauvelt concluded.
“We know now that IL-13 is a critical cytokine in AD [atopic dermatitis] pathogenesis. The unique features of this drug I want to highlight is that it has high binding affinity for IL-13,” he said.
“It has a slow dissociation off rate, meaning it binds IL-13 tightly, very potently, and stays blocking and stays hold of IL-13 in a strong manner,” he added. The drug has a half-life of 25 days.
These features could be very important for long-term dosing of the drug, he argued.
Lebrikizumab phase 3 trials
ADvocate1 and ADvocate2 are two of several phase 3 trials evaluating the efficacy and safety of lebrikizumab for the treatment of atopic dermatitis.
These include the completed ADhere study, in which lebrikizumab was used in combination with topical steroids and showed positive results in skin improvement and relief of pruritus.
The ADore study, an open-label trial in adolescents, is yet to report. The ongoing ADjoin study, a long-term extension study, is actively recruiting.
ADvocate1 and ADvocate2 are two identically designed – multicenter, randomized, double-blind, placebo-controlled, parallel-group – monotherapy trials that initially pitched two dosing regimens of lebrikizumab (250 mg) against placebo with a double loading dose at baseline and week 2 and then one dose every 2 weeks. The pair of trials enrolled a total of 869 adolescents and adults.
After the 16-week induction period, all patients in the lebrikizumab arm who had responded to treatment were rerandomly assigned to receive lebrikizumab 250 mg Q2W or Q4W, or placebo Q2W during a 36-week long-term maintenance treatment period.
This brought the total treatment time to 52 weeks for those whose atopic dermatitis had initially responded to lebrikizumab, explained Blauvelt.
Responders were those who, at 16 weeks, had an Investigator’s Global Assessment score of 0 or 1 (IGA 0/1) with a 2-point improvement or who had a 75% improvement in the Eczema Area and Severity Index score (EASI 75) without the need for rescue medication, compared with baseline values.
Induction and maintenance phase results
At the end of the 16-week induction period, a greater proportion of patients who had been treated with lebrikizumab than placebo met a primary outcome of IGA 0/1 in each trial (43.1% vs. 12.7% in ADvocate1 and 33.2% vs. 10.8% in ADvocate2).
A similar result was seen for another primary outcome, EASI 75 (58.8% vs. 16.2% and 52.1% vs. 18.1%) and for a secondary outcome, improvement in pruritus using a numerical rating scale (45.9% vs. 13.0% and 39.8% vs. 11.5%).
In the maintenance phase, with respect to responders, Dr. Blauvelt reported “very similar results” between the QW2 and Q4W maintenance dosing, “and still a quite high response in [half] the patients who were randomized to placebo at week 16.”
In the ADvocate1 and ADvocate2 trials, respectively, an IGA 0/1 with at least a 2-point improvement was maintained at week 52 in 75.8% and 64.6% of patients treated with the Q2W lebrikizumab dose, 74.2% and 80.6% of those treated with the Q4W dose, and 46.5% and 49.8% of those given placebo.
EASI 75 was maintained at week 52 in a respective 79.2% and 77.4% of patients treated with the Q2W dose, 79.2% and 84.7% with the Q4W dose, and 61.3% and 72.0% with placebo.
As for maintenance of at least a 4-point improvement in pruritus score, results at 52 weeks were 81.2% and 90.3% for the 2-week dose, 80.4% and 88.1% for the 4-week dose, and 65.4% and 67.6% for placebo.
Although topical corticosteroid treatment was allowed during the maintenance phase, only about 15% of patients needed this, Dr. Blauvelt said.
Different dosing results questioned
During the discussion period, one delegate highlighted that the twice-weekly maintenance dosing schedule seemed to “do worse a little bit” than the 4-week dosing, with both “close to placebo,” although “the long-term effect is already very impressive.”
Dr. Blauvelt noted that a pooled analysis had been done and that “it’s very clear that being on lebrikizumab works better than not being on lebrikizumab.
“Now, Q2W versus Q4W. We believe that this may be due to the long half-life of the drug possibly. It could be due to the slow disassociation rate, it’s binding tightly,” he suggested.
“We also could talk about disease modification, right. So, it opens up the concept of hit hard, hit early for 16 weeks, and then maybe you can modify disease over time,” Dr. Blauvelt said.
He added: “That’s highly speculative, of course.”
Short-term safety data
The 52-week safety profile of lebrikizumab is consistent with previously published data at 16 weeks, Dr. Blauvelt said. The most common adverse events during the studies included atopic dermatitis, nasopharyngitis, conjunctivitis, conjunctivitis allergic, headache, and COVID-19.
“This drug has comparable efficacy with dupilumab and tralokinumab,” said Jashin J. Wu, MD, from the Dermatology Research and Education Foundation in Irvine, Calif., in an interview. He was not involved in the study.
“As it does not have any significant advantages with less long-term safety data, I do not see a place for it in my practice,” Dr. Wu said.
Dupilumab (Dupixent) and tralokinumab (Adbry) are monoclonal antibodies that also block IL-13. Both are already licensed for treating atopic dermatitis. Dupilumab was approved by the Food and Drug Administration in 2017, and tralokinumab was approved in 2021.
The study was funded by Dermira, a wholly owned subsidiary of Eli Lilly. Eli Lilly has exclusive rights for the development and commercialization of lebrikizumab in the United States and all countries outside Europe; European rights belong to Almirall for all dermatology indications, including atopic dermatitis. Dr. Blauvelt acts as an investigator and adviser to these companies as well as many other pharmaceutical companies that are involved in developing new dermatologic treatments. Dr. Wu has been an investigator, consultant, or speaker for multiple pharmaceutical companies.
A version of this article first appeared on Medscape.com.
from the phase 3 ADvocate1 and ADvocate2 trials.
“We’re focused on the responders,” said Andrew Blauvelt, MD, MBA, as he presented the positive findings at the annual congress of the European Academy of Dermatology and Venereology.
Responders were the 291 people whose atopic dermatitis greatly improved after an initial 16 weeks’ treatment with lebrikizumab in both trials and who were then randomly allocated to receive injections every 2 weeks (Q2W, n = 113) or every 4 weeks (Q4W, n = 118), or to receive placebo injections Q2W (n = 60).
“Very interestingly, for me, the Q4W maintenance dosing was just as good as the Q2W maintenance dosing,” said Dr. Blauvelt, president of Oregon Medical Research Center, Portland.
“Another highlight of these data is that the patients who went on to placebo, about 50% of the patients maintained good responses, despite no treatment from week 16 to week 52,” he added.
Most patients did not require topical steroids, and “there were no surprises here” in terms of the safety profile. Lebrikizumab, a monoclonal antibody, binds to soluble interleukin-13 and blocks IL-13 signaling.
“So, the study really shows that specific targeting of IL-13 with lebrikizumab, either Q2W or Q4W, has high maintenance of efficacy and is reasonably tolerated and safe in adolescents and adults with atopic dermatitis,” Dr. Blauvelt concluded.
“We know now that IL-13 is a critical cytokine in AD [atopic dermatitis] pathogenesis. The unique features of this drug I want to highlight is that it has high binding affinity for IL-13,” he said.
“It has a slow dissociation off rate, meaning it binds IL-13 tightly, very potently, and stays blocking and stays hold of IL-13 in a strong manner,” he added. The drug has a half-life of 25 days.
These features could be very important for long-term dosing of the drug, he argued.
Lebrikizumab phase 3 trials
ADvocate1 and ADvocate2 are two of several phase 3 trials evaluating the efficacy and safety of lebrikizumab for the treatment of atopic dermatitis.
These include the completed ADhere study, in which lebrikizumab was used in combination with topical steroids and showed positive results in skin improvement and relief of pruritus.
The ADore study, an open-label trial in adolescents, is yet to report. The ongoing ADjoin study, a long-term extension study, is actively recruiting.
ADvocate1 and ADvocate2 are two identically designed – multicenter, randomized, double-blind, placebo-controlled, parallel-group – monotherapy trials that initially pitched two dosing regimens of lebrikizumab (250 mg) against placebo with a double loading dose at baseline and week 2 and then one dose every 2 weeks. The pair of trials enrolled a total of 869 adolescents and adults.
After the 16-week induction period, all patients in the lebrikizumab arm who had responded to treatment were rerandomly assigned to receive lebrikizumab 250 mg Q2W or Q4W, or placebo Q2W during a 36-week long-term maintenance treatment period.
This brought the total treatment time to 52 weeks for those whose atopic dermatitis had initially responded to lebrikizumab, explained Blauvelt.
Responders were those who, at 16 weeks, had an Investigator’s Global Assessment score of 0 or 1 (IGA 0/1) with a 2-point improvement or who had a 75% improvement in the Eczema Area and Severity Index score (EASI 75) without the need for rescue medication, compared with baseline values.
Induction and maintenance phase results
At the end of the 16-week induction period, a greater proportion of patients who had been treated with lebrikizumab than placebo met a primary outcome of IGA 0/1 in each trial (43.1% vs. 12.7% in ADvocate1 and 33.2% vs. 10.8% in ADvocate2).
A similar result was seen for another primary outcome, EASI 75 (58.8% vs. 16.2% and 52.1% vs. 18.1%) and for a secondary outcome, improvement in pruritus using a numerical rating scale (45.9% vs. 13.0% and 39.8% vs. 11.5%).
In the maintenance phase, with respect to responders, Dr. Blauvelt reported “very similar results” between the QW2 and Q4W maintenance dosing, “and still a quite high response in [half] the patients who were randomized to placebo at week 16.”
In the ADvocate1 and ADvocate2 trials, respectively, an IGA 0/1 with at least a 2-point improvement was maintained at week 52 in 75.8% and 64.6% of patients treated with the Q2W lebrikizumab dose, 74.2% and 80.6% of those treated with the Q4W dose, and 46.5% and 49.8% of those given placebo.
EASI 75 was maintained at week 52 in a respective 79.2% and 77.4% of patients treated with the Q2W dose, 79.2% and 84.7% with the Q4W dose, and 61.3% and 72.0% with placebo.
As for maintenance of at least a 4-point improvement in pruritus score, results at 52 weeks were 81.2% and 90.3% for the 2-week dose, 80.4% and 88.1% for the 4-week dose, and 65.4% and 67.6% for placebo.
Although topical corticosteroid treatment was allowed during the maintenance phase, only about 15% of patients needed this, Dr. Blauvelt said.
Different dosing results questioned
During the discussion period, one delegate highlighted that the twice-weekly maintenance dosing schedule seemed to “do worse a little bit” than the 4-week dosing, with both “close to placebo,” although “the long-term effect is already very impressive.”
Dr. Blauvelt noted that a pooled analysis had been done and that “it’s very clear that being on lebrikizumab works better than not being on lebrikizumab.
“Now, Q2W versus Q4W. We believe that this may be due to the long half-life of the drug possibly. It could be due to the slow disassociation rate, it’s binding tightly,” he suggested.
“We also could talk about disease modification, right. So, it opens up the concept of hit hard, hit early for 16 weeks, and then maybe you can modify disease over time,” Dr. Blauvelt said.
He added: “That’s highly speculative, of course.”
Short-term safety data
The 52-week safety profile of lebrikizumab is consistent with previously published data at 16 weeks, Dr. Blauvelt said. The most common adverse events during the studies included atopic dermatitis, nasopharyngitis, conjunctivitis, conjunctivitis allergic, headache, and COVID-19.
“This drug has comparable efficacy with dupilumab and tralokinumab,” said Jashin J. Wu, MD, from the Dermatology Research and Education Foundation in Irvine, Calif., in an interview. He was not involved in the study.
“As it does not have any significant advantages with less long-term safety data, I do not see a place for it in my practice,” Dr. Wu said.
Dupilumab (Dupixent) and tralokinumab (Adbry) are monoclonal antibodies that also block IL-13. Both are already licensed for treating atopic dermatitis. Dupilumab was approved by the Food and Drug Administration in 2017, and tralokinumab was approved in 2021.
The study was funded by Dermira, a wholly owned subsidiary of Eli Lilly. Eli Lilly has exclusive rights for the development and commercialization of lebrikizumab in the United States and all countries outside Europe; European rights belong to Almirall for all dermatology indications, including atopic dermatitis. Dr. Blauvelt acts as an investigator and adviser to these companies as well as many other pharmaceutical companies that are involved in developing new dermatologic treatments. Dr. Wu has been an investigator, consultant, or speaker for multiple pharmaceutical companies.
A version of this article first appeared on Medscape.com.
from the phase 3 ADvocate1 and ADvocate2 trials.
“We’re focused on the responders,” said Andrew Blauvelt, MD, MBA, as he presented the positive findings at the annual congress of the European Academy of Dermatology and Venereology.
Responders were the 291 people whose atopic dermatitis greatly improved after an initial 16 weeks’ treatment with lebrikizumab in both trials and who were then randomly allocated to receive injections every 2 weeks (Q2W, n = 113) or every 4 weeks (Q4W, n = 118), or to receive placebo injections Q2W (n = 60).
“Very interestingly, for me, the Q4W maintenance dosing was just as good as the Q2W maintenance dosing,” said Dr. Blauvelt, president of Oregon Medical Research Center, Portland.
“Another highlight of these data is that the patients who went on to placebo, about 50% of the patients maintained good responses, despite no treatment from week 16 to week 52,” he added.
Most patients did not require topical steroids, and “there were no surprises here” in terms of the safety profile. Lebrikizumab, a monoclonal antibody, binds to soluble interleukin-13 and blocks IL-13 signaling.
“So, the study really shows that specific targeting of IL-13 with lebrikizumab, either Q2W or Q4W, has high maintenance of efficacy and is reasonably tolerated and safe in adolescents and adults with atopic dermatitis,” Dr. Blauvelt concluded.
“We know now that IL-13 is a critical cytokine in AD [atopic dermatitis] pathogenesis. The unique features of this drug I want to highlight is that it has high binding affinity for IL-13,” he said.
“It has a slow dissociation off rate, meaning it binds IL-13 tightly, very potently, and stays blocking and stays hold of IL-13 in a strong manner,” he added. The drug has a half-life of 25 days.
These features could be very important for long-term dosing of the drug, he argued.
Lebrikizumab phase 3 trials
ADvocate1 and ADvocate2 are two of several phase 3 trials evaluating the efficacy and safety of lebrikizumab for the treatment of atopic dermatitis.
These include the completed ADhere study, in which lebrikizumab was used in combination with topical steroids and showed positive results in skin improvement and relief of pruritus.
The ADore study, an open-label trial in adolescents, is yet to report. The ongoing ADjoin study, a long-term extension study, is actively recruiting.
ADvocate1 and ADvocate2 are two identically designed – multicenter, randomized, double-blind, placebo-controlled, parallel-group – monotherapy trials that initially pitched two dosing regimens of lebrikizumab (250 mg) against placebo with a double loading dose at baseline and week 2 and then one dose every 2 weeks. The pair of trials enrolled a total of 869 adolescents and adults.
After the 16-week induction period, all patients in the lebrikizumab arm who had responded to treatment were rerandomly assigned to receive lebrikizumab 250 mg Q2W or Q4W, or placebo Q2W during a 36-week long-term maintenance treatment period.
This brought the total treatment time to 52 weeks for those whose atopic dermatitis had initially responded to lebrikizumab, explained Blauvelt.
Responders were those who, at 16 weeks, had an Investigator’s Global Assessment score of 0 or 1 (IGA 0/1) with a 2-point improvement or who had a 75% improvement in the Eczema Area and Severity Index score (EASI 75) without the need for rescue medication, compared with baseline values.
Induction and maintenance phase results
At the end of the 16-week induction period, a greater proportion of patients who had been treated with lebrikizumab than placebo met a primary outcome of IGA 0/1 in each trial (43.1% vs. 12.7% in ADvocate1 and 33.2% vs. 10.8% in ADvocate2).
A similar result was seen for another primary outcome, EASI 75 (58.8% vs. 16.2% and 52.1% vs. 18.1%) and for a secondary outcome, improvement in pruritus using a numerical rating scale (45.9% vs. 13.0% and 39.8% vs. 11.5%).
In the maintenance phase, with respect to responders, Dr. Blauvelt reported “very similar results” between the QW2 and Q4W maintenance dosing, “and still a quite high response in [half] the patients who were randomized to placebo at week 16.”
In the ADvocate1 and ADvocate2 trials, respectively, an IGA 0/1 with at least a 2-point improvement was maintained at week 52 in 75.8% and 64.6% of patients treated with the Q2W lebrikizumab dose, 74.2% and 80.6% of those treated with the Q4W dose, and 46.5% and 49.8% of those given placebo.
EASI 75 was maintained at week 52 in a respective 79.2% and 77.4% of patients treated with the Q2W dose, 79.2% and 84.7% with the Q4W dose, and 61.3% and 72.0% with placebo.
As for maintenance of at least a 4-point improvement in pruritus score, results at 52 weeks were 81.2% and 90.3% for the 2-week dose, 80.4% and 88.1% for the 4-week dose, and 65.4% and 67.6% for placebo.
Although topical corticosteroid treatment was allowed during the maintenance phase, only about 15% of patients needed this, Dr. Blauvelt said.
Different dosing results questioned
During the discussion period, one delegate highlighted that the twice-weekly maintenance dosing schedule seemed to “do worse a little bit” than the 4-week dosing, with both “close to placebo,” although “the long-term effect is already very impressive.”
Dr. Blauvelt noted that a pooled analysis had been done and that “it’s very clear that being on lebrikizumab works better than not being on lebrikizumab.
“Now, Q2W versus Q4W. We believe that this may be due to the long half-life of the drug possibly. It could be due to the slow disassociation rate, it’s binding tightly,” he suggested.
“We also could talk about disease modification, right. So, it opens up the concept of hit hard, hit early for 16 weeks, and then maybe you can modify disease over time,” Dr. Blauvelt said.
He added: “That’s highly speculative, of course.”
Short-term safety data
The 52-week safety profile of lebrikizumab is consistent with previously published data at 16 weeks, Dr. Blauvelt said. The most common adverse events during the studies included atopic dermatitis, nasopharyngitis, conjunctivitis, conjunctivitis allergic, headache, and COVID-19.
“This drug has comparable efficacy with dupilumab and tralokinumab,” said Jashin J. Wu, MD, from the Dermatology Research and Education Foundation in Irvine, Calif., in an interview. He was not involved in the study.
“As it does not have any significant advantages with less long-term safety data, I do not see a place for it in my practice,” Dr. Wu said.
Dupilumab (Dupixent) and tralokinumab (Adbry) are monoclonal antibodies that also block IL-13. Both are already licensed for treating atopic dermatitis. Dupilumab was approved by the Food and Drug Administration in 2017, and tralokinumab was approved in 2021.
The study was funded by Dermira, a wholly owned subsidiary of Eli Lilly. Eli Lilly has exclusive rights for the development and commercialization of lebrikizumab in the United States and all countries outside Europe; European rights belong to Almirall for all dermatology indications, including atopic dermatitis. Dr. Blauvelt acts as an investigator and adviser to these companies as well as many other pharmaceutical companies that are involved in developing new dermatologic treatments. Dr. Wu has been an investigator, consultant, or speaker for multiple pharmaceutical companies.
A version of this article first appeared on Medscape.com.
FROM THE EADV CONGRESS
Ruxolitinib repigments many vitiligo-affected body areas
.
Those difficult areas include the hands and feet, said Thierry Passeron, MD, PhD, of Université Côte d’Azur and Centre Hospitalier Universitaire de Nice (France).
Indeed, a 50% or greater improvement in the Vitiligo Area Scoring Index (VASI-50) of the hands and feet was achieved with ruxolitinib cream (Opzelura) in around one-third of patients after 52 weeks’ treatment, and more than half of patients showed improvement in the upper and lower extremities.
During one of the late-breaking news sessions, Dr. Passeron presented a pooled analysis of the Topical Ruxolitinib Evaluation in Vitiligo Study 1 (TRuE-V1) and Study 2 (TruE-V2), which assessed VASI-50 data by body regions.
Similarly positive results were seen on the head and neck and the trunk, with VASI-50 being reached in a respective 68% and 48% of patients after a full year of treatment.
“VASI-50 response rates rose steadily through 52 weeks for both the head and trunk,” said Dr. Passeron. He noted that the trials were initially double-blinded for 24 weeks and that there was a further open-label extension phase through week 52.
In the latter phase, all patients were treated with ruxolitinib; those who originally received a vehicle agent as placebo crossed over to the active treatment.
First FDA-approved treatment for adults and adolescents with vitiligo
Ruxolitinib is a Janus kinase 1/2 inhibitor that has been available for the treatment for atopic dermatitis for more than a year. It was recently approved by the U.S. Food and Drug Administration for the treatment of vitiligo in adults and pediatric patients aged 12 years and older.
This approval was based on the positive findings of the TRuE-V1 and TRuE-V2 studies, which showed that after 24 weeks, 30% of patients treated with ruxolitinib had at least 75% improvement in the facial VASI, compared with 10% of placebo-treated patients.
“These studies demonstrated very nice results, especially on the face, which is the easiest part to repigment in vitiligo,” Dr. Passeron said.
“We know that the location is very important when it comes to repigmentation of vitiligo,” he added. He noted that other body areas, “the extremities, for example, are much more difficult.”
The analysis he presented specifically assessed the effect of ruxolitinib cream on repigmentation in other areas.
Pooled analysis performed
Data from the two TRuE-V trials were pooled. The new analysis included a total of 661 individuals; of those patients, 443 had been treated with topical ruxolitinib, and 218 had received a vehicle cream as a placebo.
For the first 24 weeks, patients received twice-daily 1.5% ruxolitinib cream or vehicle cream. This was followed by a 28-week extension phase in which everyone was treated with ruxolitinib cream, after which there was a 30-day final follow-up period.
Dr. Passeron reported data by body region for weeks 12, 24, and 52, which showed an increasing percentage of patients with VASI-50.
“We didn’t look at the face; that we know well, that is a very good result,” he said.
The best results were seen for the head and neck. VASI-50 was reached by 28.3%, 45.3%, and 68.1% of patients treated with ruxolitinib cream at weeks 12, 24, and 52, respectively. Corresponding rates for the placebo-crossover group were 19.8%, 23.8%, and 51%.
Repigmentation rates of the hand, upper extremities, trunk, lower extremities, and feet were about 9%-15% for both ruxolitinib and placebo at 12 weeks, but by 24 weeks, there was a clear increase in repigmentation rates in the ruxolitinib group for all body areas.
The 24-week VASI-50 rates for hand repigmentation were 24.9% for ruxolitinib cream and 14.4% for placebo. Corresponding rates for upper extremity repigmentation were 33.2% and 8.2%; for the trunk, 26.4% and 12.2%; for the lower extremities, 29.5% and 12.2%; and for the feet, 18.5% and 12.5%.
“The results are quite poor at 12 weeks,” Dr. Passeron said. “It’s very important to keep this in mind; it takes time to repigment vitiligo, it takes to 6-24 months. We have to explain to our patients that they will have to wait to see the results.”
Steady improvements, no new safety concerns
Regarding VASI-50 over time, there was a steady increase in total body scores; 47.7% of patients who received ruxolitinib and 23.3% of placebo-treated patients hit this target at 52 weeks.
“And what is also very important to see is that we didn’t reach the plateau,” Dr. Passeron reported.
Similar patterns were seen for all the other body areas. Again there was a suggestion that rates may continue to rise with continued long-term treatment.
“About one-third of the patients reached at least 50% repigmentation after 1 year of treatment in the hands and feet,” Dr. Passeron said. He noted that certain areas, such as the back of the hand or tips of the fingers, may be unresponsive.
“So, we have to also to warn the patient that probably on these areas we have to combine it with other treatment because it remains very, very difficult to treat.”
There were no new safety concerns regarding treatment-emergent adverse events, which were reported in 52% of patients who received ruxolitinib and in 36% of placebo-treated patients.
The most common adverse reactions included COVID-19 (6.1% vs. 3.1%), acne at the application site (5.3% vs. 1.3%), and pruritus at the application site (3.9% vs. 2.7%), although cases were “mild or moderate,” said Dr. Passeron.
An expert’s take-home
“The results of TRuE-V phase 3 studies are encouraging and exciting,” Viktoria Eleftheriadou, MD, MRCP(UK), SCE(Derm), PhD, said in providing an independent comment for this news organization.
“Although ruxolitinib cream is applied on the skin, this novel treatment for vitiligo is not without risks; therefore, careful monitoring of patients who are started on this topical treatment would be prudent,” said Dr. Eleftheriadou, who is a consultant dermatologist for Walsall Healthcare NHS Trust and the Royal Wolverhampton NHS Trust, Birmingham, United Kingdom.
“I would like to see how many patients achieved VASI-75 or VASI-80 score, which from patients’ perspectives is a more meaningful outcome, as well as how long these results will last for,” she added.
The study was funded by Incyte Corporation. Dr. Passeron has received grants, honoraria, or both from AbbVie, ACM Pharma, Almirall, Amgen, Astellas, Bristol-Myers Squibb, Celgene, Galderma, Genzyme/Sanofi, GlaxoSmithKline, Incyte Corporation, Janssen, LEO Pharma, Eli Lilly, Novartis, Pfizer, Sun Pharmaceuticals, and UCB. Dr. Passeron is the cofounder of YUKIN Therapeutics and has patents on WNT agonists or GSK2b antagonist for repigmentation of vitiligo and on the use of CXCR3B blockers in vitiligo. Dr. Eleftheriadou is an investigator and trial development group member on the HI-Light Vitiligo Trial (specific), a lead investigator on the pilot HI-Light Vitiligo Trial, and a medical advisory panel member of the Vitiligo Society UK. Dr. Eleftheriadou also provides consultancy services to Incyte and Pfizer.
A version of this article first appeared on Medscape.com.
.
Those difficult areas include the hands and feet, said Thierry Passeron, MD, PhD, of Université Côte d’Azur and Centre Hospitalier Universitaire de Nice (France).
Indeed, a 50% or greater improvement in the Vitiligo Area Scoring Index (VASI-50) of the hands and feet was achieved with ruxolitinib cream (Opzelura) in around one-third of patients after 52 weeks’ treatment, and more than half of patients showed improvement in the upper and lower extremities.
During one of the late-breaking news sessions, Dr. Passeron presented a pooled analysis of the Topical Ruxolitinib Evaluation in Vitiligo Study 1 (TRuE-V1) and Study 2 (TruE-V2), which assessed VASI-50 data by body regions.
Similarly positive results were seen on the head and neck and the trunk, with VASI-50 being reached in a respective 68% and 48% of patients after a full year of treatment.
“VASI-50 response rates rose steadily through 52 weeks for both the head and trunk,” said Dr. Passeron. He noted that the trials were initially double-blinded for 24 weeks and that there was a further open-label extension phase through week 52.
In the latter phase, all patients were treated with ruxolitinib; those who originally received a vehicle agent as placebo crossed over to the active treatment.
First FDA-approved treatment for adults and adolescents with vitiligo
Ruxolitinib is a Janus kinase 1/2 inhibitor that has been available for the treatment for atopic dermatitis for more than a year. It was recently approved by the U.S. Food and Drug Administration for the treatment of vitiligo in adults and pediatric patients aged 12 years and older.
This approval was based on the positive findings of the TRuE-V1 and TRuE-V2 studies, which showed that after 24 weeks, 30% of patients treated with ruxolitinib had at least 75% improvement in the facial VASI, compared with 10% of placebo-treated patients.
“These studies demonstrated very nice results, especially on the face, which is the easiest part to repigment in vitiligo,” Dr. Passeron said.
“We know that the location is very important when it comes to repigmentation of vitiligo,” he added. He noted that other body areas, “the extremities, for example, are much more difficult.”
The analysis he presented specifically assessed the effect of ruxolitinib cream on repigmentation in other areas.
Pooled analysis performed
Data from the two TRuE-V trials were pooled. The new analysis included a total of 661 individuals; of those patients, 443 had been treated with topical ruxolitinib, and 218 had received a vehicle cream as a placebo.
For the first 24 weeks, patients received twice-daily 1.5% ruxolitinib cream or vehicle cream. This was followed by a 28-week extension phase in which everyone was treated with ruxolitinib cream, after which there was a 30-day final follow-up period.
Dr. Passeron reported data by body region for weeks 12, 24, and 52, which showed an increasing percentage of patients with VASI-50.
“We didn’t look at the face; that we know well, that is a very good result,” he said.
The best results were seen for the head and neck. VASI-50 was reached by 28.3%, 45.3%, and 68.1% of patients treated with ruxolitinib cream at weeks 12, 24, and 52, respectively. Corresponding rates for the placebo-crossover group were 19.8%, 23.8%, and 51%.
Repigmentation rates of the hand, upper extremities, trunk, lower extremities, and feet were about 9%-15% for both ruxolitinib and placebo at 12 weeks, but by 24 weeks, there was a clear increase in repigmentation rates in the ruxolitinib group for all body areas.
The 24-week VASI-50 rates for hand repigmentation were 24.9% for ruxolitinib cream and 14.4% for placebo. Corresponding rates for upper extremity repigmentation were 33.2% and 8.2%; for the trunk, 26.4% and 12.2%; for the lower extremities, 29.5% and 12.2%; and for the feet, 18.5% and 12.5%.
“The results are quite poor at 12 weeks,” Dr. Passeron said. “It’s very important to keep this in mind; it takes time to repigment vitiligo, it takes to 6-24 months. We have to explain to our patients that they will have to wait to see the results.”
Steady improvements, no new safety concerns
Regarding VASI-50 over time, there was a steady increase in total body scores; 47.7% of patients who received ruxolitinib and 23.3% of placebo-treated patients hit this target at 52 weeks.
“And what is also very important to see is that we didn’t reach the plateau,” Dr. Passeron reported.
Similar patterns were seen for all the other body areas. Again there was a suggestion that rates may continue to rise with continued long-term treatment.
“About one-third of the patients reached at least 50% repigmentation after 1 year of treatment in the hands and feet,” Dr. Passeron said. He noted that certain areas, such as the back of the hand or tips of the fingers, may be unresponsive.
“So, we have to also to warn the patient that probably on these areas we have to combine it with other treatment because it remains very, very difficult to treat.”
There were no new safety concerns regarding treatment-emergent adverse events, which were reported in 52% of patients who received ruxolitinib and in 36% of placebo-treated patients.
The most common adverse reactions included COVID-19 (6.1% vs. 3.1%), acne at the application site (5.3% vs. 1.3%), and pruritus at the application site (3.9% vs. 2.7%), although cases were “mild or moderate,” said Dr. Passeron.
An expert’s take-home
“The results of TRuE-V phase 3 studies are encouraging and exciting,” Viktoria Eleftheriadou, MD, MRCP(UK), SCE(Derm), PhD, said in providing an independent comment for this news organization.
“Although ruxolitinib cream is applied on the skin, this novel treatment for vitiligo is not without risks; therefore, careful monitoring of patients who are started on this topical treatment would be prudent,” said Dr. Eleftheriadou, who is a consultant dermatologist for Walsall Healthcare NHS Trust and the Royal Wolverhampton NHS Trust, Birmingham, United Kingdom.
“I would like to see how many patients achieved VASI-75 or VASI-80 score, which from patients’ perspectives is a more meaningful outcome, as well as how long these results will last for,” she added.
The study was funded by Incyte Corporation. Dr. Passeron has received grants, honoraria, or both from AbbVie, ACM Pharma, Almirall, Amgen, Astellas, Bristol-Myers Squibb, Celgene, Galderma, Genzyme/Sanofi, GlaxoSmithKline, Incyte Corporation, Janssen, LEO Pharma, Eli Lilly, Novartis, Pfizer, Sun Pharmaceuticals, and UCB. Dr. Passeron is the cofounder of YUKIN Therapeutics and has patents on WNT agonists or GSK2b antagonist for repigmentation of vitiligo and on the use of CXCR3B blockers in vitiligo. Dr. Eleftheriadou is an investigator and trial development group member on the HI-Light Vitiligo Trial (specific), a lead investigator on the pilot HI-Light Vitiligo Trial, and a medical advisory panel member of the Vitiligo Society UK. Dr. Eleftheriadou also provides consultancy services to Incyte and Pfizer.
A version of this article first appeared on Medscape.com.
.
Those difficult areas include the hands and feet, said Thierry Passeron, MD, PhD, of Université Côte d’Azur and Centre Hospitalier Universitaire de Nice (France).
Indeed, a 50% or greater improvement in the Vitiligo Area Scoring Index (VASI-50) of the hands and feet was achieved with ruxolitinib cream (Opzelura) in around one-third of patients after 52 weeks’ treatment, and more than half of patients showed improvement in the upper and lower extremities.
During one of the late-breaking news sessions, Dr. Passeron presented a pooled analysis of the Topical Ruxolitinib Evaluation in Vitiligo Study 1 (TRuE-V1) and Study 2 (TruE-V2), which assessed VASI-50 data by body regions.
Similarly positive results were seen on the head and neck and the trunk, with VASI-50 being reached in a respective 68% and 48% of patients after a full year of treatment.
“VASI-50 response rates rose steadily through 52 weeks for both the head and trunk,” said Dr. Passeron. He noted that the trials were initially double-blinded for 24 weeks and that there was a further open-label extension phase through week 52.
In the latter phase, all patients were treated with ruxolitinib; those who originally received a vehicle agent as placebo crossed over to the active treatment.
First FDA-approved treatment for adults and adolescents with vitiligo
Ruxolitinib is a Janus kinase 1/2 inhibitor that has been available for the treatment for atopic dermatitis for more than a year. It was recently approved by the U.S. Food and Drug Administration for the treatment of vitiligo in adults and pediatric patients aged 12 years and older.
This approval was based on the positive findings of the TRuE-V1 and TRuE-V2 studies, which showed that after 24 weeks, 30% of patients treated with ruxolitinib had at least 75% improvement in the facial VASI, compared with 10% of placebo-treated patients.
“These studies demonstrated very nice results, especially on the face, which is the easiest part to repigment in vitiligo,” Dr. Passeron said.
“We know that the location is very important when it comes to repigmentation of vitiligo,” he added. He noted that other body areas, “the extremities, for example, are much more difficult.”
The analysis he presented specifically assessed the effect of ruxolitinib cream on repigmentation in other areas.
Pooled analysis performed
Data from the two TRuE-V trials were pooled. The new analysis included a total of 661 individuals; of those patients, 443 had been treated with topical ruxolitinib, and 218 had received a vehicle cream as a placebo.
For the first 24 weeks, patients received twice-daily 1.5% ruxolitinib cream or vehicle cream. This was followed by a 28-week extension phase in which everyone was treated with ruxolitinib cream, after which there was a 30-day final follow-up period.
Dr. Passeron reported data by body region for weeks 12, 24, and 52, which showed an increasing percentage of patients with VASI-50.
“We didn’t look at the face; that we know well, that is a very good result,” he said.
The best results were seen for the head and neck. VASI-50 was reached by 28.3%, 45.3%, and 68.1% of patients treated with ruxolitinib cream at weeks 12, 24, and 52, respectively. Corresponding rates for the placebo-crossover group were 19.8%, 23.8%, and 51%.
Repigmentation rates of the hand, upper extremities, trunk, lower extremities, and feet were about 9%-15% for both ruxolitinib and placebo at 12 weeks, but by 24 weeks, there was a clear increase in repigmentation rates in the ruxolitinib group for all body areas.
The 24-week VASI-50 rates for hand repigmentation were 24.9% for ruxolitinib cream and 14.4% for placebo. Corresponding rates for upper extremity repigmentation were 33.2% and 8.2%; for the trunk, 26.4% and 12.2%; for the lower extremities, 29.5% and 12.2%; and for the feet, 18.5% and 12.5%.
“The results are quite poor at 12 weeks,” Dr. Passeron said. “It’s very important to keep this in mind; it takes time to repigment vitiligo, it takes to 6-24 months. We have to explain to our patients that they will have to wait to see the results.”
Steady improvements, no new safety concerns
Regarding VASI-50 over time, there was a steady increase in total body scores; 47.7% of patients who received ruxolitinib and 23.3% of placebo-treated patients hit this target at 52 weeks.
“And what is also very important to see is that we didn’t reach the plateau,” Dr. Passeron reported.
Similar patterns were seen for all the other body areas. Again there was a suggestion that rates may continue to rise with continued long-term treatment.
“About one-third of the patients reached at least 50% repigmentation after 1 year of treatment in the hands and feet,” Dr. Passeron said. He noted that certain areas, such as the back of the hand or tips of the fingers, may be unresponsive.
“So, we have to also to warn the patient that probably on these areas we have to combine it with other treatment because it remains very, very difficult to treat.”
There were no new safety concerns regarding treatment-emergent adverse events, which were reported in 52% of patients who received ruxolitinib and in 36% of placebo-treated patients.
The most common adverse reactions included COVID-19 (6.1% vs. 3.1%), acne at the application site (5.3% vs. 1.3%), and pruritus at the application site (3.9% vs. 2.7%), although cases were “mild or moderate,” said Dr. Passeron.
An expert’s take-home
“The results of TRuE-V phase 3 studies are encouraging and exciting,” Viktoria Eleftheriadou, MD, MRCP(UK), SCE(Derm), PhD, said in providing an independent comment for this news organization.
“Although ruxolitinib cream is applied on the skin, this novel treatment for vitiligo is not without risks; therefore, careful monitoring of patients who are started on this topical treatment would be prudent,” said Dr. Eleftheriadou, who is a consultant dermatologist for Walsall Healthcare NHS Trust and the Royal Wolverhampton NHS Trust, Birmingham, United Kingdom.
“I would like to see how many patients achieved VASI-75 or VASI-80 score, which from patients’ perspectives is a more meaningful outcome, as well as how long these results will last for,” she added.
The study was funded by Incyte Corporation. Dr. Passeron has received grants, honoraria, or both from AbbVie, ACM Pharma, Almirall, Amgen, Astellas, Bristol-Myers Squibb, Celgene, Galderma, Genzyme/Sanofi, GlaxoSmithKline, Incyte Corporation, Janssen, LEO Pharma, Eli Lilly, Novartis, Pfizer, Sun Pharmaceuticals, and UCB. Dr. Passeron is the cofounder of YUKIN Therapeutics and has patents on WNT agonists or GSK2b antagonist for repigmentation of vitiligo and on the use of CXCR3B blockers in vitiligo. Dr. Eleftheriadou is an investigator and trial development group member on the HI-Light Vitiligo Trial (specific), a lead investigator on the pilot HI-Light Vitiligo Trial, and a medical advisory panel member of the Vitiligo Society UK. Dr. Eleftheriadou also provides consultancy services to Incyte and Pfizer.
A version of this article first appeared on Medscape.com.
FROM THE EADV CONGRESS