User login
Therapy shows promise for PUPs with severe hemophilia A

A fourth-generation recombinant factor VIII (FVIII) therapy has demonstrated “convincing” efficacy and tolerability as well as low immunogenicity in previously untreated patients (PUPs) with severe hemophilia A, according to researchers.
The therapy, simoctocog alfa, is a B-domain-deleted recombinant FVIII product derived from a human cell line.
In the NuProtect study (also known as GENA-05), researchers are evaluating simoctocog alfa in PUPs with severe hemophilia A.
Interim results from this study were recently published in Haemophilia. The research is sponsored by Octapharma, makers of simoctocog alfa.
Thus far, NuProtect has enrolled 110 PUPs who will receive simoctocog alfa for up to 100 exposure days (EDs).
The Haemophilia article describes interim results for 66 PUPs treated for at least 20 EDs, the time by which most inhibitors arise. The patients’ median age at first treatment was 13 months (range, 3 months to 135 months).
The patients received simoctocog alfa for standard prophylaxis, surgical prophylaxis, or on-demand treatment.
Forty-five (68.2%) patients received standard prophylaxis, 13 (19.7%) received only on-demand treatment, 8 (12.1%) were initially treated on-demand but later received prophylaxis, and 13 (19.7%) patients received surgical prophylaxis (for 14 procedures).
The median number of EDs was 43.0 (range, 4-120).
Results
The primary objective of this study is to assess the immunogenicity of simoctocog alfa by determining inhibitor activity using the Nijmegen-modified Bethesda assay at a central laboratory.
After a median of 11.5 EDs (range, 6-24), 8 patients had developed high-titer anti-FVIII inhibitors, and 5 patients had developed low-titer inhibitors, 4 of them transient.
The cumulative incidence of all inhibitors was 20.8%—12.8% for high-titer and 8.4% for low-titer inhibitors.
For patients who received prophylaxis, the median annual bleeding rate, during inhibitor-free periods, was 2.40 for all bleeds and 0 for spontaneous bleeds.
When simoctocog alfa was used on-demand, 92.4% of bleeds were controlled with 1 or 2 infusions. In addition, simoctocog alfa was said to demonstrate “excellent” or “good” efficacy in 89% of surgical procedures.
Three patients experienced adverse events (other than inhibitor development) that were considered related to simoctocog alfa.
One patient developed a mild fever. Another had a mild allergic reaction after 3 consecutive infusions of simoctocog alfa (but not after subsequent infusions).
The third patient developed a rash that was described as mild but considered serious due to hospitalization. This patient continued treatment and completed the study. ![]()
A fourth-generation recombinant factor VIII (FVIII) therapy has demonstrated “convincing” efficacy and tolerability as well as low immunogenicity in previously untreated patients (PUPs) with severe hemophilia A, according to researchers.
The therapy, simoctocog alfa, is a B-domain-deleted recombinant FVIII product derived from a human cell line.
In the NuProtect study (also known as GENA-05), researchers are evaluating simoctocog alfa in PUPs with severe hemophilia A.
Interim results from this study were recently published in Haemophilia. The research is sponsored by Octapharma, makers of simoctocog alfa.
Thus far, NuProtect has enrolled 110 PUPs who will receive simoctocog alfa for up to 100 exposure days (EDs).
The Haemophilia article describes interim results for 66 PUPs treated for at least 20 EDs, the time by which most inhibitors arise. The patients’ median age at first treatment was 13 months (range, 3 months to 135 months).
The patients received simoctocog alfa for standard prophylaxis, surgical prophylaxis, or on-demand treatment.
Forty-five (68.2%) patients received standard prophylaxis, 13 (19.7%) received only on-demand treatment, 8 (12.1%) were initially treated on-demand but later received prophylaxis, and 13 (19.7%) patients received surgical prophylaxis (for 14 procedures).
The median number of EDs was 43.0 (range, 4-120).
Results
The primary objective of this study is to assess the immunogenicity of simoctocog alfa by determining inhibitor activity using the Nijmegen-modified Bethesda assay at a central laboratory.
After a median of 11.5 EDs (range, 6-24), 8 patients had developed high-titer anti-FVIII inhibitors, and 5 patients had developed low-titer inhibitors, 4 of them transient.
The cumulative incidence of all inhibitors was 20.8%—12.8% for high-titer and 8.4% for low-titer inhibitors.
For patients who received prophylaxis, the median annual bleeding rate, during inhibitor-free periods, was 2.40 for all bleeds and 0 for spontaneous bleeds.
When simoctocog alfa was used on-demand, 92.4% of bleeds were controlled with 1 or 2 infusions. In addition, simoctocog alfa was said to demonstrate “excellent” or “good” efficacy in 89% of surgical procedures.
Three patients experienced adverse events (other than inhibitor development) that were considered related to simoctocog alfa.
One patient developed a mild fever. Another had a mild allergic reaction after 3 consecutive infusions of simoctocog alfa (but not after subsequent infusions).
The third patient developed a rash that was described as mild but considered serious due to hospitalization. This patient continued treatment and completed the study. ![]()
A fourth-generation recombinant factor VIII (FVIII) therapy has demonstrated “convincing” efficacy and tolerability as well as low immunogenicity in previously untreated patients (PUPs) with severe hemophilia A, according to researchers.
The therapy, simoctocog alfa, is a B-domain-deleted recombinant FVIII product derived from a human cell line.
In the NuProtect study (also known as GENA-05), researchers are evaluating simoctocog alfa in PUPs with severe hemophilia A.
Interim results from this study were recently published in Haemophilia. The research is sponsored by Octapharma, makers of simoctocog alfa.
Thus far, NuProtect has enrolled 110 PUPs who will receive simoctocog alfa for up to 100 exposure days (EDs).
The Haemophilia article describes interim results for 66 PUPs treated for at least 20 EDs, the time by which most inhibitors arise. The patients’ median age at first treatment was 13 months (range, 3 months to 135 months).
The patients received simoctocog alfa for standard prophylaxis, surgical prophylaxis, or on-demand treatment.
Forty-five (68.2%) patients received standard prophylaxis, 13 (19.7%) received only on-demand treatment, 8 (12.1%) were initially treated on-demand but later received prophylaxis, and 13 (19.7%) patients received surgical prophylaxis (for 14 procedures).
The median number of EDs was 43.0 (range, 4-120).
Results
The primary objective of this study is to assess the immunogenicity of simoctocog alfa by determining inhibitor activity using the Nijmegen-modified Bethesda assay at a central laboratory.
After a median of 11.5 EDs (range, 6-24), 8 patients had developed high-titer anti-FVIII inhibitors, and 5 patients had developed low-titer inhibitors, 4 of them transient.
The cumulative incidence of all inhibitors was 20.8%—12.8% for high-titer and 8.4% for low-titer inhibitors.
For patients who received prophylaxis, the median annual bleeding rate, during inhibitor-free periods, was 2.40 for all bleeds and 0 for spontaneous bleeds.
When simoctocog alfa was used on-demand, 92.4% of bleeds were controlled with 1 or 2 infusions. In addition, simoctocog alfa was said to demonstrate “excellent” or “good” efficacy in 89% of surgical procedures.
Three patients experienced adverse events (other than inhibitor development) that were considered related to simoctocog alfa.
One patient developed a mild fever. Another had a mild allergic reaction after 3 consecutive infusions of simoctocog alfa (but not after subsequent infusions).
The third patient developed a rash that was described as mild but considered serious due to hospitalization. This patient continued treatment and completed the study. ![]()
Reimmunization appears safe in children with history of adverse events
(AEFI), but there are not enough data to make firm conclusions about severe AEFIs, according to a systematic review of studies that were published in English and French between 1982 and 2016 and were made available to Medline via PubMed, Embase, and the Cochrane library.
Apnea was found to be of concern only in children under the age of 1 year and usually found only in lower birth weight babies and those with ongoing hospitalization for complications related to prematurity, the investigators said. A 10-g increase in birth weight was associated with a 6% reduction in risk of apnea recurrence (odds ratio, 0.94; 95% CI, 0.89-1.00), and odds of recurrence were 23 times higher in infants hospitalized for complications related to prematurity (OR, 23; 95% CI, 2-272).
Some of the studies also evaluated for injection site reactions, Henoch-Schönlein purpura, or other AEFI, Dr. Zafack and her coauthors continued. Injection site reactions varied depending on vaccine and number of doses, but all children recovered within 19 days of immunization. Only one child in one study had a recurrence of Henoch-Schönlein purpura. Vomiting, persistent crying, decreased appetite, and drowsiness recurred in 15%, 24%, 25%, and 35% of the reimmunized patients, respectively, across all the studies.
“In a context of vaccine hesitancy and growing concerns regarding vaccine safety, evaluating the risk of recurrence of all AEFIs should become part of the standard evaluation of vaccine safety,” the researchers wrote. “Reimmunization appears to be safe for patients with mild to moderate AEFIs. However, the data are insufficient to draw firm conclusions regarding the safety of reimmunization after a severe AEFI.”
In this study, “it appears that the risk of recurrence of serious AEFIs (anaphylaxis, seizures, or apnea in term infants) was low (less than 1%). For minor to moderate AEFIs (fever, extensive limb swelling, oculorespiratory syndrome, allergic-like events, sleepiness, thrombocytopenia, decreased appetite, vomiting, or persistent crying), the risk of recurrence ranged from 4% to 48%,” the investigators concluded.
The study was funded by the Canadian Immunization Research Network, which is sponsored by the Public Health Agency of Canada and the Canadian Institutes of Health Research. Dr. Zafack reported no financial disclosures. Some of her coauthors reported financial support from GlaxoSmithKline and Pfizer.
There are few experiences more challenging for a pediatrician than trying to convince a parent to continue vaccinating his or her child after witnessing a seemingly related adverse event following immunization (AEFI). The question they want answered is, Will it happen again?
Zafack et al. have sought to answer that question by providing previously lacking estimates of the risk of recurrence. By consolidating the data available in a range of literature, they showed that the risk of a serious AEFI is less than 1%. More accurate risk assessments also are available for mild to moderate AEFI for many vaccines.
The Zafack et al. article also reinforces what vaccinologists and pediatricians have known for many years: Vaccines are incredibly safe. Vaccines are administered to millions of children every year, and the list of known adverse events still is very short. The researchers reaffirm the overwhelming value and safety of vaccines that protect infants and children from complications and death resulting from infectious diseases.
Even though this article does not address AEFI risk for all vaccines, it is impressively comprehensive and will be a useful reference for practicing pediatricians everywhere for years to come.
Sean T. O’Leary, MD, MPH , is an associate professor in the division of infectious diseases in the department of pediatrics at the University of Colorado at Denver, Aurora. Yvonne A. Maldonado, MD , is chief of the division of pediatric infectious diseases and a professor of pediatrics at Stanford (Calif.) University. She serves on a Data Safety Monitoring Board for a Pfizer vaccine trial. Both authors are members of the American Academy of Pediatrics committee on infectious disease and subcommittee on vaccine policy and vaccine hesitancy. These comments were published in an editorial accompanying the Zafack et al. article in Pediatrics (2017. doi: 10.1542/peds.2017-1760 ).
There are few experiences more challenging for a pediatrician than trying to convince a parent to continue vaccinating his or her child after witnessing a seemingly related adverse event following immunization (AEFI). The question they want answered is, Will it happen again?
Zafack et al. have sought to answer that question by providing previously lacking estimates of the risk of recurrence. By consolidating the data available in a range of literature, they showed that the risk of a serious AEFI is less than 1%. More accurate risk assessments also are available for mild to moderate AEFI for many vaccines.
The Zafack et al. article also reinforces what vaccinologists and pediatricians have known for many years: Vaccines are incredibly safe. Vaccines are administered to millions of children every year, and the list of known adverse events still is very short. The researchers reaffirm the overwhelming value and safety of vaccines that protect infants and children from complications and death resulting from infectious diseases.
Even though this article does not address AEFI risk for all vaccines, it is impressively comprehensive and will be a useful reference for practicing pediatricians everywhere for years to come.
Sean T. O’Leary, MD, MPH , is an associate professor in the division of infectious diseases in the department of pediatrics at the University of Colorado at Denver, Aurora. Yvonne A. Maldonado, MD , is chief of the division of pediatric infectious diseases and a professor of pediatrics at Stanford (Calif.) University. She serves on a Data Safety Monitoring Board for a Pfizer vaccine trial. Both authors are members of the American Academy of Pediatrics committee on infectious disease and subcommittee on vaccine policy and vaccine hesitancy. These comments were published in an editorial accompanying the Zafack et al. article in Pediatrics (2017. doi: 10.1542/peds.2017-1760 ).
There are few experiences more challenging for a pediatrician than trying to convince a parent to continue vaccinating his or her child after witnessing a seemingly related adverse event following immunization (AEFI). The question they want answered is, Will it happen again?
Zafack et al. have sought to answer that question by providing previously lacking estimates of the risk of recurrence. By consolidating the data available in a range of literature, they showed that the risk of a serious AEFI is less than 1%. More accurate risk assessments also are available for mild to moderate AEFI for many vaccines.
The Zafack et al. article also reinforces what vaccinologists and pediatricians have known for many years: Vaccines are incredibly safe. Vaccines are administered to millions of children every year, and the list of known adverse events still is very short. The researchers reaffirm the overwhelming value and safety of vaccines that protect infants and children from complications and death resulting from infectious diseases.
Even though this article does not address AEFI risk for all vaccines, it is impressively comprehensive and will be a useful reference for practicing pediatricians everywhere for years to come.
Sean T. O’Leary, MD, MPH , is an associate professor in the division of infectious diseases in the department of pediatrics at the University of Colorado at Denver, Aurora. Yvonne A. Maldonado, MD , is chief of the division of pediatric infectious diseases and a professor of pediatrics at Stanford (Calif.) University. She serves on a Data Safety Monitoring Board for a Pfizer vaccine trial. Both authors are members of the American Academy of Pediatrics committee on infectious disease and subcommittee on vaccine policy and vaccine hesitancy. These comments were published in an editorial accompanying the Zafack et al. article in Pediatrics (2017. doi: 10.1542/peds.2017-1760 ).
(AEFI), but there are not enough data to make firm conclusions about severe AEFIs, according to a systematic review of studies that were published in English and French between 1982 and 2016 and were made available to Medline via PubMed, Embase, and the Cochrane library.
Apnea was found to be of concern only in children under the age of 1 year and usually found only in lower birth weight babies and those with ongoing hospitalization for complications related to prematurity, the investigators said. A 10-g increase in birth weight was associated with a 6% reduction in risk of apnea recurrence (odds ratio, 0.94; 95% CI, 0.89-1.00), and odds of recurrence were 23 times higher in infants hospitalized for complications related to prematurity (OR, 23; 95% CI, 2-272).
Some of the studies also evaluated for injection site reactions, Henoch-Schönlein purpura, or other AEFI, Dr. Zafack and her coauthors continued. Injection site reactions varied depending on vaccine and number of doses, but all children recovered within 19 days of immunization. Only one child in one study had a recurrence of Henoch-Schönlein purpura. Vomiting, persistent crying, decreased appetite, and drowsiness recurred in 15%, 24%, 25%, and 35% of the reimmunized patients, respectively, across all the studies.
“In a context of vaccine hesitancy and growing concerns regarding vaccine safety, evaluating the risk of recurrence of all AEFIs should become part of the standard evaluation of vaccine safety,” the researchers wrote. “Reimmunization appears to be safe for patients with mild to moderate AEFIs. However, the data are insufficient to draw firm conclusions regarding the safety of reimmunization after a severe AEFI.”
In this study, “it appears that the risk of recurrence of serious AEFIs (anaphylaxis, seizures, or apnea in term infants) was low (less than 1%). For minor to moderate AEFIs (fever, extensive limb swelling, oculorespiratory syndrome, allergic-like events, sleepiness, thrombocytopenia, decreased appetite, vomiting, or persistent crying), the risk of recurrence ranged from 4% to 48%,” the investigators concluded.
The study was funded by the Canadian Immunization Research Network, which is sponsored by the Public Health Agency of Canada and the Canadian Institutes of Health Research. Dr. Zafack reported no financial disclosures. Some of her coauthors reported financial support from GlaxoSmithKline and Pfizer.
(AEFI), but there are not enough data to make firm conclusions about severe AEFIs, according to a systematic review of studies that were published in English and French between 1982 and 2016 and were made available to Medline via PubMed, Embase, and the Cochrane library.
Apnea was found to be of concern only in children under the age of 1 year and usually found only in lower birth weight babies and those with ongoing hospitalization for complications related to prematurity, the investigators said. A 10-g increase in birth weight was associated with a 6% reduction in risk of apnea recurrence (odds ratio, 0.94; 95% CI, 0.89-1.00), and odds of recurrence were 23 times higher in infants hospitalized for complications related to prematurity (OR, 23; 95% CI, 2-272).
Some of the studies also evaluated for injection site reactions, Henoch-Schönlein purpura, or other AEFI, Dr. Zafack and her coauthors continued. Injection site reactions varied depending on vaccine and number of doses, but all children recovered within 19 days of immunization. Only one child in one study had a recurrence of Henoch-Schönlein purpura. Vomiting, persistent crying, decreased appetite, and drowsiness recurred in 15%, 24%, 25%, and 35% of the reimmunized patients, respectively, across all the studies.
“In a context of vaccine hesitancy and growing concerns regarding vaccine safety, evaluating the risk of recurrence of all AEFIs should become part of the standard evaluation of vaccine safety,” the researchers wrote. “Reimmunization appears to be safe for patients with mild to moderate AEFIs. However, the data are insufficient to draw firm conclusions regarding the safety of reimmunization after a severe AEFI.”
In this study, “it appears that the risk of recurrence of serious AEFIs (anaphylaxis, seizures, or apnea in term infants) was low (less than 1%). For minor to moderate AEFIs (fever, extensive limb swelling, oculorespiratory syndrome, allergic-like events, sleepiness, thrombocytopenia, decreased appetite, vomiting, or persistent crying), the risk of recurrence ranged from 4% to 48%,” the investigators concluded.
The study was funded by the Canadian Immunization Research Network, which is sponsored by the Public Health Agency of Canada and the Canadian Institutes of Health Research. Dr. Zafack reported no financial disclosures. Some of her coauthors reported financial support from GlaxoSmithKline and Pfizer.
FROM PEDIATRICS
Key clinical point: Reimmunization appears to be safe for patients with mild to moderate adverse events following immunization.
Major finding: The risk of a serious AEFI is less than 1%. More accurate risk assessments also are available for mild to moderate AEFI.
Data source: A systematic review of 29 studies assessing the risks of AEFI.
Disclosures: The study was funded by the Canadian Immunization Research Network, which is sponsored by the Public Health Agency of Canada and the Canadian Institutes of Health Research. Dr. Zafack reported no financial disclosures. Some of her coauthors reported financial support from GlaxoSmithKline and Pfizer.
Rapid AMI rule out
Clinical Question: Can a single high-sensitivity cardiac troponin-T (hs-cTnT) reliably rule-out acute myocardial infarction (AMI) to safely enable earlier discharge?
Background: Current practice includes serial measures of hs-cTnT to rule out AMI.
Study Design: A meta-analysis of 11 prospective cohorts at various international locations
Setting: Patients presenting to emergency departments with chest pain.
Synopsis: Of 9,241, a total of 2,825 patients were classified as low risk with a single negative hs-cTnT and nonischemic EKG. The primary outcome was AMI during initial hospitalization. Of low-risk patients, 14 (0.5%) had AMI. Pooled estimated sensitivity was 98.7% and pooled negative predictive value was 99.3%. For the secondary outcome of 30-day major adverse cardiac events, pooled sensitivity was 98%. Limitations include a small number of studies, high statistical heterogeneity, variation in troponin assays, and variable prevalence of AMI across studies.
Bottom Line: A single negative hs-cTnT and nonischemic EKG after three hours of chest pain can reliably rule out AMI. Further research is, however, required to validate the unequivocal use of this early rule out strategy.
Citation: Pickering J, Than M, Cullen L, et al. Rapid rule-out of acute myocardial infarction with a single high-sensitivity cardiac troponin t measurement below the limit of detection: A collaborative meta-analysis. Ann Intern Med. 2017 May 16;166(10):715-24.
Dr. Dogra is clinical instructor of medicine in the University of Kentucky division of hospital medicine.
Clinical Question: Can a single high-sensitivity cardiac troponin-T (hs-cTnT) reliably rule-out acute myocardial infarction (AMI) to safely enable earlier discharge?
Background: Current practice includes serial measures of hs-cTnT to rule out AMI.
Study Design: A meta-analysis of 11 prospective cohorts at various international locations
Setting: Patients presenting to emergency departments with chest pain.
Synopsis: Of 9,241, a total of 2,825 patients were classified as low risk with a single negative hs-cTnT and nonischemic EKG. The primary outcome was AMI during initial hospitalization. Of low-risk patients, 14 (0.5%) had AMI. Pooled estimated sensitivity was 98.7% and pooled negative predictive value was 99.3%. For the secondary outcome of 30-day major adverse cardiac events, pooled sensitivity was 98%. Limitations include a small number of studies, high statistical heterogeneity, variation in troponin assays, and variable prevalence of AMI across studies.
Bottom Line: A single negative hs-cTnT and nonischemic EKG after three hours of chest pain can reliably rule out AMI. Further research is, however, required to validate the unequivocal use of this early rule out strategy.
Citation: Pickering J, Than M, Cullen L, et al. Rapid rule-out of acute myocardial infarction with a single high-sensitivity cardiac troponin t measurement below the limit of detection: A collaborative meta-analysis. Ann Intern Med. 2017 May 16;166(10):715-24.
Dr. Dogra is clinical instructor of medicine in the University of Kentucky division of hospital medicine.
Clinical Question: Can a single high-sensitivity cardiac troponin-T (hs-cTnT) reliably rule-out acute myocardial infarction (AMI) to safely enable earlier discharge?
Background: Current practice includes serial measures of hs-cTnT to rule out AMI.
Study Design: A meta-analysis of 11 prospective cohorts at various international locations
Setting: Patients presenting to emergency departments with chest pain.
Synopsis: Of 9,241, a total of 2,825 patients were classified as low risk with a single negative hs-cTnT and nonischemic EKG. The primary outcome was AMI during initial hospitalization. Of low-risk patients, 14 (0.5%) had AMI. Pooled estimated sensitivity was 98.7% and pooled negative predictive value was 99.3%. For the secondary outcome of 30-day major adverse cardiac events, pooled sensitivity was 98%. Limitations include a small number of studies, high statistical heterogeneity, variation in troponin assays, and variable prevalence of AMI across studies.
Bottom Line: A single negative hs-cTnT and nonischemic EKG after three hours of chest pain can reliably rule out AMI. Further research is, however, required to validate the unequivocal use of this early rule out strategy.
Citation: Pickering J, Than M, Cullen L, et al. Rapid rule-out of acute myocardial infarction with a single high-sensitivity cardiac troponin t measurement below the limit of detection: A collaborative meta-analysis. Ann Intern Med. 2017 May 16;166(10):715-24.
Dr. Dogra is clinical instructor of medicine in the University of Kentucky division of hospital medicine.

Study finds bivalirudin efficacy for PCI no better than heparin
A large study of more than 6,000 heart patients in Sweden has found that patients having percutaneous coronary intervention who received bivalirudin did not have lower rates of deleterious outcomes – death, heart attack, or major bleeding – than did patients who received heparin monotherapy, a contrast to previous trials that found that bivalirudin had a lower bleeding risk than heparin alone after PCI.
The findings were presented at the annual congress of the European Society of Cardiology and published simultaneously in the New England Journal of Medicine.
The study sought to explain the conflicting findings of previous trials investigating the efficacy of bivalirudin vs. heparin monotherapy. The VALIDATE-SWEDEHEART trial evaluated 6,006 patients who had PCI from June 2014 to September 2016, 90.3% via radial-artery access. This trial differed from previous studies because it was conducted after radial-artery access was routine and potent P2Y12 inhibitors were available, and earlier trials did not compare bivalirudin to heparin monotherapy, said David Erlinge, MD, PhD, of Lund (Sweden) University, and 38 coauthors (N Engl J Med. 2017 Aug 27. doi: 10.1056/NEJMoa1706443).
The Swedish investigators evaluated the primary endpoint – the composite of any-cause death, MI or major bleeding – during 180 days of follow-up. Among the study patients, 3,005 had ST-segment elevation MI (STEMI) and 3,001 non-STEMI (NSTEMI). All had undergone urgent PCI and most were also on P2Y inhibitors. The P2Y12 inhibitors used were ticagrelor in 5,697 patients (94.9%), prasugrel in 125 (2.1%) and cangrelor in 21 (0.3%).
Study patients with STEMI were permitted to receive up to 5,000 U of intravenous unfractionated heparin before arrival in the catheterization laboratory, and both STEMI and non-STEMI patients who had not received heparin previously could receive up to 3,000 U of intra-arterial heparin before angiography. All patients received aspirin pretreatment, and 62% received potent P2Y12 inhibitors at least one hour before PCI.
After angiography, but before PCI, patients were randomized 1:1 to receive in an open-label fashion either intravenous bivalirudin (The Medicines Company), or intra-arterial unfractionated heparin (LEO Pharma). Bivalirudin was administered as a bolus of 0.75 mg/kg of body weight followed by an infusion of 1.7 mg/kg per hour.
Research nurses contacted patients by phone 7 and 180 days after PCI. Baseline characteristics were similar between the bivalirudin and heparin groups. For example, around 31% of both groups had hyperlipidemia, and 15.2% of the bivalirudin group and 14.2% of the heparin group had a previous PCI.
“The rate of the primary endpoint did not differ significantly between the treatment groups at 30 days after PCI,” Dr. Erlinge and his coauthors noted. At 30 days, 7.2% of the bivalirudin patients and 8% of the heparin group had one of the primary endpoint outcomes, a nonsignificant difference. At 180 days, 12.3% of the bivalirudin group and 12.8% of those receiving heparin had one of the primary endpoint outcomes, also a nonsignificant difference.
Specific outcomes in the bivalirudin vs. heparin patients, respectively, at 180 days were: MI, 2% vs. 2.4%; major bleeding, 8.6% in both groups; stent thrombosis, 0.4% vs. 0.7%; and death from any cause, 2.9% vs. 2.8%, all nonsignificant differences.
“Results were consistent between patients with STEMI and those with NSTEMI and across all other prespecified subgroups,” the researchers wrote. They noted that women in the bivalirudin group had a lower, although not statistically significant, primary endpoint rate than did women in the heparin group.
In this trial, the high rate of radial-artery access and the low use of glycoprotein IIb/IIIa inhibitors may explain the low bleeding rates, the researchers said.
Among the study limitations were that patients excluded from the trial were at higher risk for a primary endpoint than those enrolled, the open-label design may have biased participating physicians in identifying outcomes, the telephone call-based follow-up may have been inherently unreliable, and the fact that most patients received a small dose of heparin before randomization may have reconciled any differences between the two drugs.
Coauthors Stefan James, MD, and Ollie Ostlund, MD, disclosed receiving grants from Astra Zeneca, and The Medicines Company. Dr. Erlinge and other coauthors had no financial relationships relevant to the work.
After considering the findings of the VALIDATE-SWEDEHEART trial, Gregg W. Stone, MD, said in an accompanying editorial, “there is no definitive answer to the question of whether to use bivalirudin or heparin during PCI.”
Dr. Stone, of New York–Presbyterian Hospital, Columbia University Medical Center, and the Cardiovascular Research Foundation, New York, noted four potential flaws in the study findings. One, the 30-day interval may be a better for evaluating procedural anticoagulation than 180 days – and at 30 days the Swedish study showed “a nonsignificant trend in favor of bivalirudin.” Two, the composite primary endpoint could bias outcomes because individual measures could essentially cancel each other out. Three, differences between treatment groups could have been further minimized because 91% of patients who received bivalirudin also received a substantial dose of heparin before and during PCI. Finally, Dr. Stone said, the study was underpowered to examine the individual components of outcomes.
The data comparing outcomes in STEMI and NSTEMI patients did not show separate results for death, bleeding, and stent thrombosis. Dr. Stone pointed to a meta-analysis of six randomized trials of 14,095 patients with STEMI, showing that bivalirudin had lower rates of major bleeding and 30-day death but higher rates of stent thrombosis than heparin, and that mortality was lower regardless of the use of femoral artery or radial-artery access or other procedural factors. By contrast, previous trials did show similar rates of death, MI, and stent thrombosis between both treatment groups, although lower bleeding rates were seen with bivalirudin.
More definitive answers may lie in investigators from the large-scale randomized trials comparing the anticoagulant agents, including the Swedish authors, combining their data on more than 36,000 patients into a single database, as they have agreed to do, Dr. Stone said. That “should provide robust evidence to guide decisions regarding anticoagulation among patients with STEMI and NSTEMI,” he concluded.
Dr. Stone had no relevant financial relationships to disclose. He made his comments in an invited editorial in the New England Journal of Medicine (doi: 10.1056/NEJMe1709247).
After considering the findings of the VALIDATE-SWEDEHEART trial, Gregg W. Stone, MD, said in an accompanying editorial, “there is no definitive answer to the question of whether to use bivalirudin or heparin during PCI.”
Dr. Stone, of New York–Presbyterian Hospital, Columbia University Medical Center, and the Cardiovascular Research Foundation, New York, noted four potential flaws in the study findings. One, the 30-day interval may be a better for evaluating procedural anticoagulation than 180 days – and at 30 days the Swedish study showed “a nonsignificant trend in favor of bivalirudin.” Two, the composite primary endpoint could bias outcomes because individual measures could essentially cancel each other out. Three, differences between treatment groups could have been further minimized because 91% of patients who received bivalirudin also received a substantial dose of heparin before and during PCI. Finally, Dr. Stone said, the study was underpowered to examine the individual components of outcomes.
The data comparing outcomes in STEMI and NSTEMI patients did not show separate results for death, bleeding, and stent thrombosis. Dr. Stone pointed to a meta-analysis of six randomized trials of 14,095 patients with STEMI, showing that bivalirudin had lower rates of major bleeding and 30-day death but higher rates of stent thrombosis than heparin, and that mortality was lower regardless of the use of femoral artery or radial-artery access or other procedural factors. By contrast, previous trials did show similar rates of death, MI, and stent thrombosis between both treatment groups, although lower bleeding rates were seen with bivalirudin.
More definitive answers may lie in investigators from the large-scale randomized trials comparing the anticoagulant agents, including the Swedish authors, combining their data on more than 36,000 patients into a single database, as they have agreed to do, Dr. Stone said. That “should provide robust evidence to guide decisions regarding anticoagulation among patients with STEMI and NSTEMI,” he concluded.
Dr. Stone had no relevant financial relationships to disclose. He made his comments in an invited editorial in the New England Journal of Medicine (doi: 10.1056/NEJMe1709247).
After considering the findings of the VALIDATE-SWEDEHEART trial, Gregg W. Stone, MD, said in an accompanying editorial, “there is no definitive answer to the question of whether to use bivalirudin or heparin during PCI.”
Dr. Stone, of New York–Presbyterian Hospital, Columbia University Medical Center, and the Cardiovascular Research Foundation, New York, noted four potential flaws in the study findings. One, the 30-day interval may be a better for evaluating procedural anticoagulation than 180 days – and at 30 days the Swedish study showed “a nonsignificant trend in favor of bivalirudin.” Two, the composite primary endpoint could bias outcomes because individual measures could essentially cancel each other out. Three, differences between treatment groups could have been further minimized because 91% of patients who received bivalirudin also received a substantial dose of heparin before and during PCI. Finally, Dr. Stone said, the study was underpowered to examine the individual components of outcomes.
The data comparing outcomes in STEMI and NSTEMI patients did not show separate results for death, bleeding, and stent thrombosis. Dr. Stone pointed to a meta-analysis of six randomized trials of 14,095 patients with STEMI, showing that bivalirudin had lower rates of major bleeding and 30-day death but higher rates of stent thrombosis than heparin, and that mortality was lower regardless of the use of femoral artery or radial-artery access or other procedural factors. By contrast, previous trials did show similar rates of death, MI, and stent thrombosis between both treatment groups, although lower bleeding rates were seen with bivalirudin.
More definitive answers may lie in investigators from the large-scale randomized trials comparing the anticoagulant agents, including the Swedish authors, combining their data on more than 36,000 patients into a single database, as they have agreed to do, Dr. Stone said. That “should provide robust evidence to guide decisions regarding anticoagulation among patients with STEMI and NSTEMI,” he concluded.
Dr. Stone had no relevant financial relationships to disclose. He made his comments in an invited editorial in the New England Journal of Medicine (doi: 10.1056/NEJMe1709247).
A large study of more than 6,000 heart patients in Sweden has found that patients having percutaneous coronary intervention who received bivalirudin did not have lower rates of deleterious outcomes – death, heart attack, or major bleeding – than did patients who received heparin monotherapy, a contrast to previous trials that found that bivalirudin had a lower bleeding risk than heparin alone after PCI.
The findings were presented at the annual congress of the European Society of Cardiology and published simultaneously in the New England Journal of Medicine.
The study sought to explain the conflicting findings of previous trials investigating the efficacy of bivalirudin vs. heparin monotherapy. The VALIDATE-SWEDEHEART trial evaluated 6,006 patients who had PCI from June 2014 to September 2016, 90.3% via radial-artery access. This trial differed from previous studies because it was conducted after radial-artery access was routine and potent P2Y12 inhibitors were available, and earlier trials did not compare bivalirudin to heparin monotherapy, said David Erlinge, MD, PhD, of Lund (Sweden) University, and 38 coauthors (N Engl J Med. 2017 Aug 27. doi: 10.1056/NEJMoa1706443).
The Swedish investigators evaluated the primary endpoint – the composite of any-cause death, MI or major bleeding – during 180 days of follow-up. Among the study patients, 3,005 had ST-segment elevation MI (STEMI) and 3,001 non-STEMI (NSTEMI). All had undergone urgent PCI and most were also on P2Y inhibitors. The P2Y12 inhibitors used were ticagrelor in 5,697 patients (94.9%), prasugrel in 125 (2.1%) and cangrelor in 21 (0.3%).
Study patients with STEMI were permitted to receive up to 5,000 U of intravenous unfractionated heparin before arrival in the catheterization laboratory, and both STEMI and non-STEMI patients who had not received heparin previously could receive up to 3,000 U of intra-arterial heparin before angiography. All patients received aspirin pretreatment, and 62% received potent P2Y12 inhibitors at least one hour before PCI.
After angiography, but before PCI, patients were randomized 1:1 to receive in an open-label fashion either intravenous bivalirudin (The Medicines Company), or intra-arterial unfractionated heparin (LEO Pharma). Bivalirudin was administered as a bolus of 0.75 mg/kg of body weight followed by an infusion of 1.7 mg/kg per hour.
Research nurses contacted patients by phone 7 and 180 days after PCI. Baseline characteristics were similar between the bivalirudin and heparin groups. For example, around 31% of both groups had hyperlipidemia, and 15.2% of the bivalirudin group and 14.2% of the heparin group had a previous PCI.
“The rate of the primary endpoint did not differ significantly between the treatment groups at 30 days after PCI,” Dr. Erlinge and his coauthors noted. At 30 days, 7.2% of the bivalirudin patients and 8% of the heparin group had one of the primary endpoint outcomes, a nonsignificant difference. At 180 days, 12.3% of the bivalirudin group and 12.8% of those receiving heparin had one of the primary endpoint outcomes, also a nonsignificant difference.
Specific outcomes in the bivalirudin vs. heparin patients, respectively, at 180 days were: MI, 2% vs. 2.4%; major bleeding, 8.6% in both groups; stent thrombosis, 0.4% vs. 0.7%; and death from any cause, 2.9% vs. 2.8%, all nonsignificant differences.
“Results were consistent between patients with STEMI and those with NSTEMI and across all other prespecified subgroups,” the researchers wrote. They noted that women in the bivalirudin group had a lower, although not statistically significant, primary endpoint rate than did women in the heparin group.
In this trial, the high rate of radial-artery access and the low use of glycoprotein IIb/IIIa inhibitors may explain the low bleeding rates, the researchers said.
Among the study limitations were that patients excluded from the trial were at higher risk for a primary endpoint than those enrolled, the open-label design may have biased participating physicians in identifying outcomes, the telephone call-based follow-up may have been inherently unreliable, and the fact that most patients received a small dose of heparin before randomization may have reconciled any differences between the two drugs.
Coauthors Stefan James, MD, and Ollie Ostlund, MD, disclosed receiving grants from Astra Zeneca, and The Medicines Company. Dr. Erlinge and other coauthors had no financial relationships relevant to the work.
A large study of more than 6,000 heart patients in Sweden has found that patients having percutaneous coronary intervention who received bivalirudin did not have lower rates of deleterious outcomes – death, heart attack, or major bleeding – than did patients who received heparin monotherapy, a contrast to previous trials that found that bivalirudin had a lower bleeding risk than heparin alone after PCI.
The findings were presented at the annual congress of the European Society of Cardiology and published simultaneously in the New England Journal of Medicine.
The study sought to explain the conflicting findings of previous trials investigating the efficacy of bivalirudin vs. heparin monotherapy. The VALIDATE-SWEDEHEART trial evaluated 6,006 patients who had PCI from June 2014 to September 2016, 90.3% via radial-artery access. This trial differed from previous studies because it was conducted after radial-artery access was routine and potent P2Y12 inhibitors were available, and earlier trials did not compare bivalirudin to heparin monotherapy, said David Erlinge, MD, PhD, of Lund (Sweden) University, and 38 coauthors (N Engl J Med. 2017 Aug 27. doi: 10.1056/NEJMoa1706443).
The Swedish investigators evaluated the primary endpoint – the composite of any-cause death, MI or major bleeding – during 180 days of follow-up. Among the study patients, 3,005 had ST-segment elevation MI (STEMI) and 3,001 non-STEMI (NSTEMI). All had undergone urgent PCI and most were also on P2Y inhibitors. The P2Y12 inhibitors used were ticagrelor in 5,697 patients (94.9%), prasugrel in 125 (2.1%) and cangrelor in 21 (0.3%).
Study patients with STEMI were permitted to receive up to 5,000 U of intravenous unfractionated heparin before arrival in the catheterization laboratory, and both STEMI and non-STEMI patients who had not received heparin previously could receive up to 3,000 U of intra-arterial heparin before angiography. All patients received aspirin pretreatment, and 62% received potent P2Y12 inhibitors at least one hour before PCI.
After angiography, but before PCI, patients were randomized 1:1 to receive in an open-label fashion either intravenous bivalirudin (The Medicines Company), or intra-arterial unfractionated heparin (LEO Pharma). Bivalirudin was administered as a bolus of 0.75 mg/kg of body weight followed by an infusion of 1.7 mg/kg per hour.
Research nurses contacted patients by phone 7 and 180 days after PCI. Baseline characteristics were similar between the bivalirudin and heparin groups. For example, around 31% of both groups had hyperlipidemia, and 15.2% of the bivalirudin group and 14.2% of the heparin group had a previous PCI.
“The rate of the primary endpoint did not differ significantly between the treatment groups at 30 days after PCI,” Dr. Erlinge and his coauthors noted. At 30 days, 7.2% of the bivalirudin patients and 8% of the heparin group had one of the primary endpoint outcomes, a nonsignificant difference. At 180 days, 12.3% of the bivalirudin group and 12.8% of those receiving heparin had one of the primary endpoint outcomes, also a nonsignificant difference.
Specific outcomes in the bivalirudin vs. heparin patients, respectively, at 180 days were: MI, 2% vs. 2.4%; major bleeding, 8.6% in both groups; stent thrombosis, 0.4% vs. 0.7%; and death from any cause, 2.9% vs. 2.8%, all nonsignificant differences.
“Results were consistent between patients with STEMI and those with NSTEMI and across all other prespecified subgroups,” the researchers wrote. They noted that women in the bivalirudin group had a lower, although not statistically significant, primary endpoint rate than did women in the heparin group.
In this trial, the high rate of radial-artery access and the low use of glycoprotein IIb/IIIa inhibitors may explain the low bleeding rates, the researchers said.
Among the study limitations were that patients excluded from the trial were at higher risk for a primary endpoint than those enrolled, the open-label design may have biased participating physicians in identifying outcomes, the telephone call-based follow-up may have been inherently unreliable, and the fact that most patients received a small dose of heparin before randomization may have reconciled any differences between the two drugs.
Coauthors Stefan James, MD, and Ollie Ostlund, MD, disclosed receiving grants from Astra Zeneca, and The Medicines Company. Dr. Erlinge and other coauthors had no financial relationships relevant to the work.
FROM THE ESC CONGRESS 2017
Key clinical point: The rates of composite death, MI, or major bleeding for patients having PCI for MI were similar regardless of whether they received bivalirudin or heparin monotherapy.
Major finding: At 180 days, 12.3% of the bivalirudin patients and 12.8% of the heparin patients had one of the primary endpoint outcomes.
Data source: VALIDATE-SWEDEHEART, a registry-based, multicenter, randomized, controlled, open-label clinical trial of 6,006 patients who had PCI between June 2014 and September 2016.
Disclosure: Coauthors Stefan James, MD, and Ollie Ostlund, MD, disclosed receiving grants from AstraZeneca, and The Medicines Company. Dr. Erlinge and other coauthors had no financial relationships relevant to the work.
VIDEO: Inflammation’s role in atherosclerosis confirmed in CANTOS
BARCELONA – The results of the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) mark the validation of many years of research on inflammation for Peter Libby, MD, Mallinckrodt Professor of Medicine, Harvard Medical School, Boston.
The CANTOS investigator said that, although some trials, most notably JUPITER, have linked reduced markers of inflammation with reduced cardiovascular events, none have been able to separate the effects of lowering LDL cholesterol from those of lowering the inflammatory marker interleukin-1B.
But using the monoclonal antibody canakinumab to target only interleukin-1B in CANTOS reduced the composite endpoint of nonfatal MI, nonfatal stroke, or cardiovascular death by 15% at the highest dosage tested, compared with placebo, while lowering high-sensitivity C-reactive protein by 39 percentage points.
Dr. Libby has been studying interleukin-1B since the 1980s. “Now, today, for the first time, in a rigorous trial, we can show that an anti-inflammatory agent that is neutral for lipids (that doesn’t lower LDL) can provide a benefit for our patients, and that’s a real step forward,” Dr. Libby said in a video interview at the annual congress of the European Society of Cardiology.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Importantly, a dividend of the investigation was that “we found a decrease in fatal cancers, particularly lung cancer. So this again opens the door toward a whole new therapeutic window in patients not just in the cardiovascular space, but also in oncology. So it’s a doubly exciting day for us.”
CANTOS was presented at the meeting by Paul Ridker, MD, also of Harvard Medical School; the results were also published online (N Engl J Med. 2017 Aug 27. doi: 10.1056/NEJMoa1707914).
BARCELONA – The results of the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) mark the validation of many years of research on inflammation for Peter Libby, MD, Mallinckrodt Professor of Medicine, Harvard Medical School, Boston.
The CANTOS investigator said that, although some trials, most notably JUPITER, have linked reduced markers of inflammation with reduced cardiovascular events, none have been able to separate the effects of lowering LDL cholesterol from those of lowering the inflammatory marker interleukin-1B.
But using the monoclonal antibody canakinumab to target only interleukin-1B in CANTOS reduced the composite endpoint of nonfatal MI, nonfatal stroke, or cardiovascular death by 15% at the highest dosage tested, compared with placebo, while lowering high-sensitivity C-reactive protein by 39 percentage points.
Dr. Libby has been studying interleukin-1B since the 1980s. “Now, today, for the first time, in a rigorous trial, we can show that an anti-inflammatory agent that is neutral for lipids (that doesn’t lower LDL) can provide a benefit for our patients, and that’s a real step forward,” Dr. Libby said in a video interview at the annual congress of the European Society of Cardiology.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Importantly, a dividend of the investigation was that “we found a decrease in fatal cancers, particularly lung cancer. So this again opens the door toward a whole new therapeutic window in patients not just in the cardiovascular space, but also in oncology. So it’s a doubly exciting day for us.”
CANTOS was presented at the meeting by Paul Ridker, MD, also of Harvard Medical School; the results were also published online (N Engl J Med. 2017 Aug 27. doi: 10.1056/NEJMoa1707914).
BARCELONA – The results of the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) mark the validation of many years of research on inflammation for Peter Libby, MD, Mallinckrodt Professor of Medicine, Harvard Medical School, Boston.
The CANTOS investigator said that, although some trials, most notably JUPITER, have linked reduced markers of inflammation with reduced cardiovascular events, none have been able to separate the effects of lowering LDL cholesterol from those of lowering the inflammatory marker interleukin-1B.
But using the monoclonal antibody canakinumab to target only interleukin-1B in CANTOS reduced the composite endpoint of nonfatal MI, nonfatal stroke, or cardiovascular death by 15% at the highest dosage tested, compared with placebo, while lowering high-sensitivity C-reactive protein by 39 percentage points.
Dr. Libby has been studying interleukin-1B since the 1980s. “Now, today, for the first time, in a rigorous trial, we can show that an anti-inflammatory agent that is neutral for lipids (that doesn’t lower LDL) can provide a benefit for our patients, and that’s a real step forward,” Dr. Libby said in a video interview at the annual congress of the European Society of Cardiology.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Importantly, a dividend of the investigation was that “we found a decrease in fatal cancers, particularly lung cancer. So this again opens the door toward a whole new therapeutic window in patients not just in the cardiovascular space, but also in oncology. So it’s a doubly exciting day for us.”
CANTOS was presented at the meeting by Paul Ridker, MD, also of Harvard Medical School; the results were also published online (N Engl J Med. 2017 Aug 27. doi: 10.1056/NEJMoa1707914).
AT THE ESC CONGRESS 2017

Make the Diagnosis - August 2017
Trichofolliculoma
Trichofolliculoma is a rare, benign skin lesion that was first described in 1944. While the etiology is largely known, it is considered to be a hamartoma with follicular differentiation that results when the pluripotent skin cells cease to differentiate into the hair follicle. The lesion lacks specific predilection for gender or race but most commonly occurs on the face of adults. Clinically, trichofolliculoma presents as a solitary, skin-colored papule or nodule. It may contain a central sebum-producing pore or have a tuft of white hair protruding from the central pore, although neither of these manifestations are mandatory. Trichofolliculomas generally are asymptomatic and are not associated with any systemic or other cutaneous diseases. Therefore, no treatment is required. However, surgical excision or curettage and electrodesiccation may be performed for cosmetic reasons.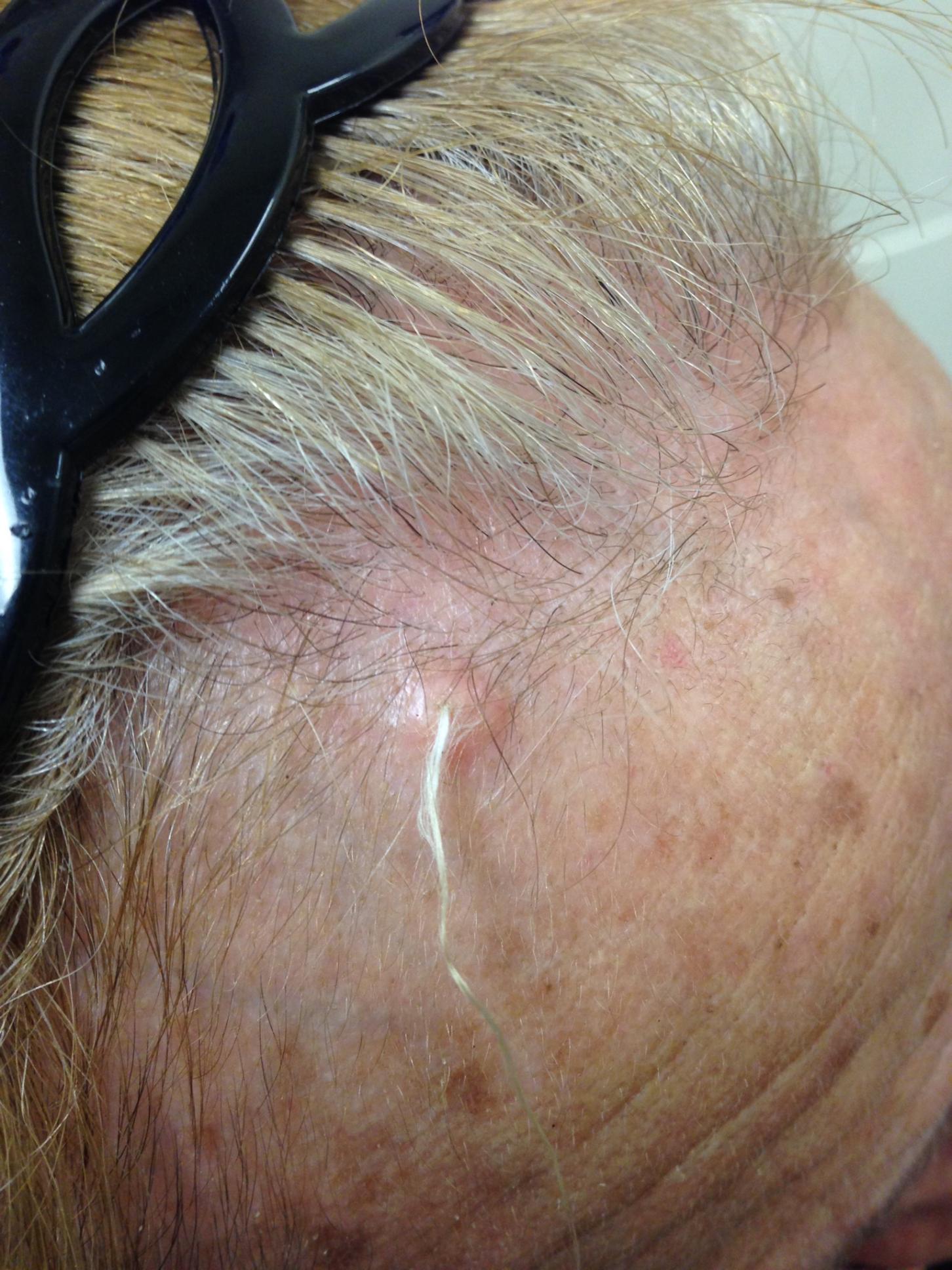
Definitive diagnosis of trichofolliculoma requires a biopsy. The histopathology shows a primary cystic structure within the dermis that may or may not be connected to the epidermis. There are multiple abnormal secondary follicles that radiate from the primary cystic structure, and the entire apparatus is surrounded by a well-circumscribed dense connective tissue. Immunohistochemical studies have revealed that trichofolliculoma is characterized by activated, aberrant cytokeratin-15 (CK15)–positive follicle stem cells that show outer root sheath differentiation while attempting to make hair. This results in disorder of the normal hair cycle.
The clinical differential diagnosis for trichofolliculoma includes dermal nevus, basal cell carcinoma, and pilar sheath acanthoma. These cutaneous entities may be ruled out by histopathology.
The histopathology of basal cell carcinoma consists of tumors of basaloid cells with little cytoplasm and hyperchromatic nuclei. There is palisading around the periphery of the tumor with stromal retraction that leads to a clefting appearance, as well as mucinous change in the stroma.
The histopathology of dermal nevus reveals small nests of melanocytes within the upper dermis that commonly localize around the pilosebaceous units. There is variability in pigmentation and cellularity of the melanocytes, and there is no junctional component.
The histopathology of pilar sheath acanthoma shows dermal proliferation of lobules comprising benign squamous epithelium that arrange themselves around small cystic spaces. The lobules themselves are surrounded by eosinophilic basement membrane.
The case and photo were submitted by Victoria Billero, MS; Adam Wulkan, MD; and Caroline Winslow, MD, all of the department of dermatology, University of Miami (Fla.).
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Trichofolliculoma
Trichofolliculoma is a rare, benign skin lesion that was first described in 1944. While the etiology is largely known, it is considered to be a hamartoma with follicular differentiation that results when the pluripotent skin cells cease to differentiate into the hair follicle. The lesion lacks specific predilection for gender or race but most commonly occurs on the face of adults. Clinically, trichofolliculoma presents as a solitary, skin-colored papule or nodule. It may contain a central sebum-producing pore or have a tuft of white hair protruding from the central pore, although neither of these manifestations are mandatory. Trichofolliculomas generally are asymptomatic and are not associated with any systemic or other cutaneous diseases. Therefore, no treatment is required. However, surgical excision or curettage and electrodesiccation may be performed for cosmetic reasons.
Definitive diagnosis of trichofolliculoma requires a biopsy. The histopathology shows a primary cystic structure within the dermis that may or may not be connected to the epidermis. There are multiple abnormal secondary follicles that radiate from the primary cystic structure, and the entire apparatus is surrounded by a well-circumscribed dense connective tissue. Immunohistochemical studies have revealed that trichofolliculoma is characterized by activated, aberrant cytokeratin-15 (CK15)–positive follicle stem cells that show outer root sheath differentiation while attempting to make hair. This results in disorder of the normal hair cycle.
The clinical differential diagnosis for trichofolliculoma includes dermal nevus, basal cell carcinoma, and pilar sheath acanthoma. These cutaneous entities may be ruled out by histopathology.
The histopathology of basal cell carcinoma consists of tumors of basaloid cells with little cytoplasm and hyperchromatic nuclei. There is palisading around the periphery of the tumor with stromal retraction that leads to a clefting appearance, as well as mucinous change in the stroma.
The histopathology of dermal nevus reveals small nests of melanocytes within the upper dermis that commonly localize around the pilosebaceous units. There is variability in pigmentation and cellularity of the melanocytes, and there is no junctional component.
The histopathology of pilar sheath acanthoma shows dermal proliferation of lobules comprising benign squamous epithelium that arrange themselves around small cystic spaces. The lobules themselves are surrounded by eosinophilic basement membrane.
The case and photo were submitted by Victoria Billero, MS; Adam Wulkan, MD; and Caroline Winslow, MD, all of the department of dermatology, University of Miami (Fla.).
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Trichofolliculoma
Trichofolliculoma is a rare, benign skin lesion that was first described in 1944. While the etiology is largely known, it is considered to be a hamartoma with follicular differentiation that results when the pluripotent skin cells cease to differentiate into the hair follicle. The lesion lacks specific predilection for gender or race but most commonly occurs on the face of adults. Clinically, trichofolliculoma presents as a solitary, skin-colored papule or nodule. It may contain a central sebum-producing pore or have a tuft of white hair protruding from the central pore, although neither of these manifestations are mandatory. Trichofolliculomas generally are asymptomatic and are not associated with any systemic or other cutaneous diseases. Therefore, no treatment is required. However, surgical excision or curettage and electrodesiccation may be performed for cosmetic reasons.
Definitive diagnosis of trichofolliculoma requires a biopsy. The histopathology shows a primary cystic structure within the dermis that may or may not be connected to the epidermis. There are multiple abnormal secondary follicles that radiate from the primary cystic structure, and the entire apparatus is surrounded by a well-circumscribed dense connective tissue. Immunohistochemical studies have revealed that trichofolliculoma is characterized by activated, aberrant cytokeratin-15 (CK15)–positive follicle stem cells that show outer root sheath differentiation while attempting to make hair. This results in disorder of the normal hair cycle.
The clinical differential diagnosis for trichofolliculoma includes dermal nevus, basal cell carcinoma, and pilar sheath acanthoma. These cutaneous entities may be ruled out by histopathology.
The histopathology of basal cell carcinoma consists of tumors of basaloid cells with little cytoplasm and hyperchromatic nuclei. There is palisading around the periphery of the tumor with stromal retraction that leads to a clefting appearance, as well as mucinous change in the stroma.
The histopathology of dermal nevus reveals small nests of melanocytes within the upper dermis that commonly localize around the pilosebaceous units. There is variability in pigmentation and cellularity of the melanocytes, and there is no junctional component.
The histopathology of pilar sheath acanthoma shows dermal proliferation of lobules comprising benign squamous epithelium that arrange themselves around small cystic spaces. The lobules themselves are surrounded by eosinophilic basement membrane.
The case and photo were submitted by Victoria Billero, MS; Adam Wulkan, MD; and Caroline Winslow, MD, all of the department of dermatology, University of Miami (Fla.).
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
A 67-year-old white female with no significant past medical history presented as a first-time patient to clinic for a full skin check. An incidental finding of a solitary, asymptomatic, non-tender 8 mm flesh-colored papule with a tuft of long white hair protruding through a central pore was observed on her right lateral forehead that had been present for longer than 10 years.
Orsiro coronary DES outperforms Xience
BARCELONA – A new model of drug-eluting coronary stent outperformed the reigning benchmark Xience stent in a head-to-head, pivotal comparison with 1,334 patients.
The results “advance a new standard for drug eluting stents,” David E. Kandzari, MD, said at the annual congress of the European Society of Cardiology. The trial tested the Orsiro stent, which features very thin, 60-micron-thick cobalt chromium struts, a bioresorbable polymer, and sirolimus as the antiproliferative drug that’s released during the first 90 days of stent placement.
“To our knowledge, this is the only trial that has demonstrated superiority [of a new stent] to the Xience drug-eluting stent in a large, randomized trial. This is a landmark in interventional cardiology that raises the bar for future comparisons” of drug eluting coronary stents, Dr. Kandzari said in an interview.
The study’s primary endpoint of target lesion failure after 12 months of follow-up – a rate that combined the incidence of cardiovascular death, target-vessel related MI, and ischemia-driven target-lesion revascularization – stood at 6.2% for the 884 patients treated with the Orsiro stent and 9.6% of the 450 randomized to treatment with the Xience stent, which has struts that are 81 microns wide, and a durable polymer that releases everolimus as the antiproliferative drug.
The difference in the primary endpoint was driven primarily by a 3.6% absolute difference in the rate of target-vessel related MIs, a statistically significant difference, plus the Orsiro-treated patients showed numerically smaller rates of cardiac death and ischemia-driven target lesion revascularization, although the between-group differences for each of these two endpoints were not statistically significant. The Orsiro stent also showed a significantly reduced rate of late stent thrombosis, occurring during day 31 through 1 year, a 0.1% rate in the Orsiro-treated patients and a 0.9% rate in those who received Xience stents.
“These are remarkable results,” said Michael Haude, MD, an interventional cardiologist at Lukas Hospital in Neuss, Germany, and a cochair of the session in which Dr. Kandzari gave his report.
These results from the Safety and Effectiveness of the Orsiro Sirolimus Eluting Coronary Stent System in Subjects With Coronary Artery Lesions (BIOFLOW-V) trial will be the centerpiece of an application for U.S. marketing approval for the Orsiro stent, Dr. Kandzari said. The BIOFLOW-V trial enrolled patients at 90 centers in 13 countries including the United States.
Concurrently with his report, the results were published online (Lancet. 2017 Aug 26. doi: 10.1016/S0140-6736[17]32249-3).
The potential advantage of a thinner-strut drug eluting stent showed up during the initial treatment phase, with a procedural success rate of 94% using the Orsiro stent and 90% with the Xience stent. This difference in procedural success seemed largely the result of an increased rate of periprocedural MIs, a difference that might be explained by the difference in strut thickness, said Dr. Kandzari, of Piedmont Heart Institute, Atlanta. Based on this success, designers of future drug-eluting stents may focus on thinner-strut models, he suggested.
An additional analysis that Dr. Kandzari reported combined results from two prior randomized comparisons of the Orsiro and Xience stents, creating a pooled analysis with 2,208 patients. This Bayesian analysis calculated a 100% probability of noninferiority of the Orsiro stent compared with the Xience stent, and a 97% probability of superiority. This 97% probability of superiority fell just short of the 97.5% threshold for establishing superiority that Dr. Kandzari and his associates had prespecified for this analysis.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
BARCELONA – A new model of drug-eluting coronary stent outperformed the reigning benchmark Xience stent in a head-to-head, pivotal comparison with 1,334 patients.
The results “advance a new standard for drug eluting stents,” David E. Kandzari, MD, said at the annual congress of the European Society of Cardiology. The trial tested the Orsiro stent, which features very thin, 60-micron-thick cobalt chromium struts, a bioresorbable polymer, and sirolimus as the antiproliferative drug that’s released during the first 90 days of stent placement.
“To our knowledge, this is the only trial that has demonstrated superiority [of a new stent] to the Xience drug-eluting stent in a large, randomized trial. This is a landmark in interventional cardiology that raises the bar for future comparisons” of drug eluting coronary stents, Dr. Kandzari said in an interview.
The study’s primary endpoint of target lesion failure after 12 months of follow-up – a rate that combined the incidence of cardiovascular death, target-vessel related MI, and ischemia-driven target-lesion revascularization – stood at 6.2% for the 884 patients treated with the Orsiro stent and 9.6% of the 450 randomized to treatment with the Xience stent, which has struts that are 81 microns wide, and a durable polymer that releases everolimus as the antiproliferative drug.
The difference in the primary endpoint was driven primarily by a 3.6% absolute difference in the rate of target-vessel related MIs, a statistically significant difference, plus the Orsiro-treated patients showed numerically smaller rates of cardiac death and ischemia-driven target lesion revascularization, although the between-group differences for each of these two endpoints were not statistically significant. The Orsiro stent also showed a significantly reduced rate of late stent thrombosis, occurring during day 31 through 1 year, a 0.1% rate in the Orsiro-treated patients and a 0.9% rate in those who received Xience stents.
“These are remarkable results,” said Michael Haude, MD, an interventional cardiologist at Lukas Hospital in Neuss, Germany, and a cochair of the session in which Dr. Kandzari gave his report.
These results from the Safety and Effectiveness of the Orsiro Sirolimus Eluting Coronary Stent System in Subjects With Coronary Artery Lesions (BIOFLOW-V) trial will be the centerpiece of an application for U.S. marketing approval for the Orsiro stent, Dr. Kandzari said. The BIOFLOW-V trial enrolled patients at 90 centers in 13 countries including the United States.
Concurrently with his report, the results were published online (Lancet. 2017 Aug 26. doi: 10.1016/S0140-6736[17]32249-3).
The potential advantage of a thinner-strut drug eluting stent showed up during the initial treatment phase, with a procedural success rate of 94% using the Orsiro stent and 90% with the Xience stent. This difference in procedural success seemed largely the result of an increased rate of periprocedural MIs, a difference that might be explained by the difference in strut thickness, said Dr. Kandzari, of Piedmont Heart Institute, Atlanta. Based on this success, designers of future drug-eluting stents may focus on thinner-strut models, he suggested.
An additional analysis that Dr. Kandzari reported combined results from two prior randomized comparisons of the Orsiro and Xience stents, creating a pooled analysis with 2,208 patients. This Bayesian analysis calculated a 100% probability of noninferiority of the Orsiro stent compared with the Xience stent, and a 97% probability of superiority. This 97% probability of superiority fell just short of the 97.5% threshold for establishing superiority that Dr. Kandzari and his associates had prespecified for this analysis.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
BARCELONA – A new model of drug-eluting coronary stent outperformed the reigning benchmark Xience stent in a head-to-head, pivotal comparison with 1,334 patients.
The results “advance a new standard for drug eluting stents,” David E. Kandzari, MD, said at the annual congress of the European Society of Cardiology. The trial tested the Orsiro stent, which features very thin, 60-micron-thick cobalt chromium struts, a bioresorbable polymer, and sirolimus as the antiproliferative drug that’s released during the first 90 days of stent placement.
“To our knowledge, this is the only trial that has demonstrated superiority [of a new stent] to the Xience drug-eluting stent in a large, randomized trial. This is a landmark in interventional cardiology that raises the bar for future comparisons” of drug eluting coronary stents, Dr. Kandzari said in an interview.
The study’s primary endpoint of target lesion failure after 12 months of follow-up – a rate that combined the incidence of cardiovascular death, target-vessel related MI, and ischemia-driven target-lesion revascularization – stood at 6.2% for the 884 patients treated with the Orsiro stent and 9.6% of the 450 randomized to treatment with the Xience stent, which has struts that are 81 microns wide, and a durable polymer that releases everolimus as the antiproliferative drug.
The difference in the primary endpoint was driven primarily by a 3.6% absolute difference in the rate of target-vessel related MIs, a statistically significant difference, plus the Orsiro-treated patients showed numerically smaller rates of cardiac death and ischemia-driven target lesion revascularization, although the between-group differences for each of these two endpoints were not statistically significant. The Orsiro stent also showed a significantly reduced rate of late stent thrombosis, occurring during day 31 through 1 year, a 0.1% rate in the Orsiro-treated patients and a 0.9% rate in those who received Xience stents.
“These are remarkable results,” said Michael Haude, MD, an interventional cardiologist at Lukas Hospital in Neuss, Germany, and a cochair of the session in which Dr. Kandzari gave his report.
These results from the Safety and Effectiveness of the Orsiro Sirolimus Eluting Coronary Stent System in Subjects With Coronary Artery Lesions (BIOFLOW-V) trial will be the centerpiece of an application for U.S. marketing approval for the Orsiro stent, Dr. Kandzari said. The BIOFLOW-V trial enrolled patients at 90 centers in 13 countries including the United States.
Concurrently with his report, the results were published online (Lancet. 2017 Aug 26. doi: 10.1016/S0140-6736[17]32249-3).
The potential advantage of a thinner-strut drug eluting stent showed up during the initial treatment phase, with a procedural success rate of 94% using the Orsiro stent and 90% with the Xience stent. This difference in procedural success seemed largely the result of an increased rate of periprocedural MIs, a difference that might be explained by the difference in strut thickness, said Dr. Kandzari, of Piedmont Heart Institute, Atlanta. Based on this success, designers of future drug-eluting stents may focus on thinner-strut models, he suggested.
An additional analysis that Dr. Kandzari reported combined results from two prior randomized comparisons of the Orsiro and Xience stents, creating a pooled analysis with 2,208 patients. This Bayesian analysis calculated a 100% probability of noninferiority of the Orsiro stent compared with the Xience stent, and a 97% probability of superiority. This 97% probability of superiority fell just short of the 97.5% threshold for establishing superiority that Dr. Kandzari and his associates had prespecified for this analysis.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
AT THE ESC CONGRESS 2017
Key clinical point:
Major finding: The target-lesion failure rate after 12 months was 6.2% with the Orsiro stent and 9.6% with the Xience stent.
Data source: BIOFLOW-V, a multicenter, randomized trial with 1,334 patients.
Disclosures: BIOFLOW-V was sponsored by Biotronik, the company that markets the Orsiro stent. Dr. Kandzari has been a consultant to and/or has received research funding from Biotronik, Boston Scientific, Medtronic, Micell Technologies, Abbott Vascular, St. Jude, Medinol, and OrbusNeich. Dr. Haude has been a consultant to and has received honoraria from Biotronik and from several other device and drug companies.
Big risk of serious falls after first episode of syncope
BARCELONA – Patients have an exorbitant 80% increased risk of hospitalization for falls resulting in fracture or head injury in the first year after discharge following a first-ever episode of syncope, according to a Danish national cohort study. One in five patients who sustained a fall resulting in hospitalization experienced a hip fracture, according to Anna-Karin Nume, MD, of the University of Copenhagen.
In this interview at the annual congress of the European Society of Cardiology, Dr. Nume highlights findings from her study, which included 125,763 Danish adults with first-time syncope.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
BARCELONA – Patients have an exorbitant 80% increased risk of hospitalization for falls resulting in fracture or head injury in the first year after discharge following a first-ever episode of syncope, according to a Danish national cohort study. One in five patients who sustained a fall resulting in hospitalization experienced a hip fracture, according to Anna-Karin Nume, MD, of the University of Copenhagen.
In this interview at the annual congress of the European Society of Cardiology, Dr. Nume highlights findings from her study, which included 125,763 Danish adults with first-time syncope.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
BARCELONA – Patients have an exorbitant 80% increased risk of hospitalization for falls resulting in fracture or head injury in the first year after discharge following a first-ever episode of syncope, according to a Danish national cohort study. One in five patients who sustained a fall resulting in hospitalization experienced a hip fracture, according to Anna-Karin Nume, MD, of the University of Copenhagen.
In this interview at the annual congress of the European Society of Cardiology, Dr. Nume highlights findings from her study, which included 125,763 Danish adults with first-time syncope.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT THE ESC CONGRESS 2017

Addition of daratumumab improves PFS in MM, company says

Interim results of a phase 3 study suggest adding daratumumab to a 3-drug regimen can improve progression-free survival (PFS) in patients with newly diagnosed multiple myeloma (MM).
In the phase 3 ALCYONE study, researchers are comparing daratumumab in combination with bortezomib, melphalan, and prednisone (VMP) to VMP alone in patients with newly diagnosed MM who are not considered candidates for autologous stem cell transplant.
Genmab A/S recently announced interim results from this trial and said additional data are expected to be submitted for presentation at an upcoming medical conference and for publication in a peer-reviewed journal.
The ALCYONE trial enrolled 706 patients who were randomized to receive 9 cycles of daratumumab plus VMP or VMP alone.
In the daratumumab arm, patients received 16 mg/kg of daratumumab once weekly for 6 weeks (cycle 1; 1 cycle = 42 days), followed by once every 3 weeks (cycles 2-9). After that, patients continued to receive 16 mg/kg of daratumumab once every 4 weeks until disease progression.
At the pre-planned interim analysis, the study’s primary endpoint was met. Treatment with daratumumab reduced the risk of disease progression or death by 50% when compared to VMP alone (hazard ratio=0.50; 95% CI 0.38-0.65; P<0.0001).
The median PFS has not been reached in the daratumumab arm but was an estimated 18.1 months for patients who received VMP alone.
Genmab said that, overall, the safety profile of daratumumab in combination with VMP is consistent with the known safety profile of the VMP regimen and the known safety profile of daratumumab.
Based on these results, an independent data monitoring committee recommended the data be unblinded. All patients will continue to be monitored for safety and overall survival.
Janssen Biotech, Inc., which licensed daratumumab from Genmab in 2012, said it will discuss with health authorities the potential for a regulatory submission for daratumumab in combination with VMP as a treatment for newly diagnosed MM.
Daratumumab is already approved in the US (as DARZALEX®) for use in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, to treat MM patients who have received at least 1 prior therapy.
Daratumumab is also approved for use in combination with pomalidomide and dexamethasone to treat MM patients who have received at least 2 prior therapies, including lenalidomide and a proteasome inhibitor (PI).
And daratumumab is approved as monotherapy for MM patients who have received at least 3 prior lines of therapy, including a PI and an immunomodulatory agent, or who are double-refractory to a PI and an immunomodulatory agent. ![]()
Interim results of a phase 3 study suggest adding daratumumab to a 3-drug regimen can improve progression-free survival (PFS) in patients with newly diagnosed multiple myeloma (MM).
In the phase 3 ALCYONE study, researchers are comparing daratumumab in combination with bortezomib, melphalan, and prednisone (VMP) to VMP alone in patients with newly diagnosed MM who are not considered candidates for autologous stem cell transplant.
Genmab A/S recently announced interim results from this trial and said additional data are expected to be submitted for presentation at an upcoming medical conference and for publication in a peer-reviewed journal.
The ALCYONE trial enrolled 706 patients who were randomized to receive 9 cycles of daratumumab plus VMP or VMP alone.
In the daratumumab arm, patients received 16 mg/kg of daratumumab once weekly for 6 weeks (cycle 1; 1 cycle = 42 days), followed by once every 3 weeks (cycles 2-9). After that, patients continued to receive 16 mg/kg of daratumumab once every 4 weeks until disease progression.
At the pre-planned interim analysis, the study’s primary endpoint was met. Treatment with daratumumab reduced the risk of disease progression or death by 50% when compared to VMP alone (hazard ratio=0.50; 95% CI 0.38-0.65; P<0.0001).
The median PFS has not been reached in the daratumumab arm but was an estimated 18.1 months for patients who received VMP alone.
Genmab said that, overall, the safety profile of daratumumab in combination with VMP is consistent with the known safety profile of the VMP regimen and the known safety profile of daratumumab.
Based on these results, an independent data monitoring committee recommended the data be unblinded. All patients will continue to be monitored for safety and overall survival.
Janssen Biotech, Inc., which licensed daratumumab from Genmab in 2012, said it will discuss with health authorities the potential for a regulatory submission for daratumumab in combination with VMP as a treatment for newly diagnosed MM.
Daratumumab is already approved in the US (as DARZALEX®) for use in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, to treat MM patients who have received at least 1 prior therapy.
Daratumumab is also approved for use in combination with pomalidomide and dexamethasone to treat MM patients who have received at least 2 prior therapies, including lenalidomide and a proteasome inhibitor (PI).
And daratumumab is approved as monotherapy for MM patients who have received at least 3 prior lines of therapy, including a PI and an immunomodulatory agent, or who are double-refractory to a PI and an immunomodulatory agent. ![]()
Interim results of a phase 3 study suggest adding daratumumab to a 3-drug regimen can improve progression-free survival (PFS) in patients with newly diagnosed multiple myeloma (MM).
In the phase 3 ALCYONE study, researchers are comparing daratumumab in combination with bortezomib, melphalan, and prednisone (VMP) to VMP alone in patients with newly diagnosed MM who are not considered candidates for autologous stem cell transplant.
Genmab A/S recently announced interim results from this trial and said additional data are expected to be submitted for presentation at an upcoming medical conference and for publication in a peer-reviewed journal.
The ALCYONE trial enrolled 706 patients who were randomized to receive 9 cycles of daratumumab plus VMP or VMP alone.
In the daratumumab arm, patients received 16 mg/kg of daratumumab once weekly for 6 weeks (cycle 1; 1 cycle = 42 days), followed by once every 3 weeks (cycles 2-9). After that, patients continued to receive 16 mg/kg of daratumumab once every 4 weeks until disease progression.
At the pre-planned interim analysis, the study’s primary endpoint was met. Treatment with daratumumab reduced the risk of disease progression or death by 50% when compared to VMP alone (hazard ratio=0.50; 95% CI 0.38-0.65; P<0.0001).
The median PFS has not been reached in the daratumumab arm but was an estimated 18.1 months for patients who received VMP alone.
Genmab said that, overall, the safety profile of daratumumab in combination with VMP is consistent with the known safety profile of the VMP regimen and the known safety profile of daratumumab.
Based on these results, an independent data monitoring committee recommended the data be unblinded. All patients will continue to be monitored for safety and overall survival.
Janssen Biotech, Inc., which licensed daratumumab from Genmab in 2012, said it will discuss with health authorities the potential for a regulatory submission for daratumumab in combination with VMP as a treatment for newly diagnosed MM.
Daratumumab is already approved in the US (as DARZALEX®) for use in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, to treat MM patients who have received at least 1 prior therapy.
Daratumumab is also approved for use in combination with pomalidomide and dexamethasone to treat MM patients who have received at least 2 prior therapies, including lenalidomide and a proteasome inhibitor (PI).
And daratumumab is approved as monotherapy for MM patients who have received at least 3 prior lines of therapy, including a PI and an immunomodulatory agent, or who are double-refractory to a PI and an immunomodulatory agent. ![]()
In T1 diabetes, CABG seems better than PCI
In patients with type 1 diabetes in need of multivessel revascularization, coronary artery bypass graft (CABG) may be a better choice than percutaneous coronary intervention (PCI), according to results from a new comparative study presented at the annual congress of the European Society of Cardiology by Martin J. Holzmann, PhD, of the Karolinska Institute, Stockholm.
The two procedures had similar mortality rates, but PCI patients fared worse with respect to mortality due to myocardial infarction and several cardiovascular outcomes.
Previous studies had also suggested better outcomes with CABG than with PCI, but they lumped together patients with type 1 and type 2 diabetes, while the current study focused only on patients with type 1 diabetes.
The study included patients in Sweden with type 1 diabetes who underwent CABG (683 patients) or PCI (1,863 patients) between 1995 and 2013. During follow-up, 44.6% of patients in the PCI group died, compared with 53.3% in the CABG group. After adjustment for between-group differences, however, there was no significant difference in mortality risk between the two groups.
However, assessments of cause-specific mortality told a different story. Subjects in the PCI group had a greater risk of death from coronary artery disease (hazard ratio, 1.45; 95% confidence interval, 1.21-1.74).
Subjects in the PCI group were also more likely to suffer myocardial infarction (HR, 1.47; 95% CI, 1.21-1.77) and were more than five times more likely to undergo repeat vascularization (adjusted HR, 5.64; 95% CI, 4.67-6.82). The CABG group had a higher 30-day stroke risk (1.9% vs. 0.8%), but there was no difference in long-term risk.
The two groups had similar risks of hospitalization for heart failure.
The researchers noted a large difference between the two groups with respect to risk during the first year of follow-up, which suggests that some patients underwent PCI because they were too ill to undergo CABG. This limitation is also suggested by the greater proportion of previous stroke, heart failure, active cancer, and end-stage renal disease in the PCI group. The researchers adjusted for these differences, but it remains possible that there were residual confounders.
No source of funding was disclosed. One of the authors has received consultancy honoraria from Actelion and Pfizer. Dr. Domanski and Dr. Farkouh report no relevant financial relationships.
In patients with aggressive multivessel CAD and stable symptoms associated with diabetes or high SYNTAX score, the mechanisms of benefit of PCI and CABG are different, and this difference likely explains the superior results of CABG.
Better stents alone cannot change the superiority of CABG, compared with PCI for patients with aggressive CAD (diabetes or high SYNTAX score), because PCI addresses only a small portion of the coronary anatomy. This does not diminish the importance of continuing advances in stent technology, but rather, it puts into appropriate perspective what can be expected from these advances.
The findings of this important study help to better inform practice, and should influence decision-making for revascularization in patients with T1DM.
These remarks were taken from an editorial by Michael J. Domanski, MD, and Michael E. Farkouh, MD (J Am Coll Cardiol. 2017. doi: 10.1016/j.jacc.2017.07.781). Dr. Domanski is with the Peter Munk Cardiac Centre, Toronto, and the Heart and Stroke Richard Lewar Centre, University of Toronto. Dr. Farokouh is the director of clinical trials at the Peter Munk Cardiac Centre, University of Toronto.
In patients with aggressive multivessel CAD and stable symptoms associated with diabetes or high SYNTAX score, the mechanisms of benefit of PCI and CABG are different, and this difference likely explains the superior results of CABG.
Better stents alone cannot change the superiority of CABG, compared with PCI for patients with aggressive CAD (diabetes or high SYNTAX score), because PCI addresses only a small portion of the coronary anatomy. This does not diminish the importance of continuing advances in stent technology, but rather, it puts into appropriate perspective what can be expected from these advances.
The findings of this important study help to better inform practice, and should influence decision-making for revascularization in patients with T1DM.
These remarks were taken from an editorial by Michael J. Domanski, MD, and Michael E. Farkouh, MD (J Am Coll Cardiol. 2017. doi: 10.1016/j.jacc.2017.07.781). Dr. Domanski is with the Peter Munk Cardiac Centre, Toronto, and the Heart and Stroke Richard Lewar Centre, University of Toronto. Dr. Farokouh is the director of clinical trials at the Peter Munk Cardiac Centre, University of Toronto.
In patients with aggressive multivessel CAD and stable symptoms associated with diabetes or high SYNTAX score, the mechanisms of benefit of PCI and CABG are different, and this difference likely explains the superior results of CABG.
Better stents alone cannot change the superiority of CABG, compared with PCI for patients with aggressive CAD (diabetes or high SYNTAX score), because PCI addresses only a small portion of the coronary anatomy. This does not diminish the importance of continuing advances in stent technology, but rather, it puts into appropriate perspective what can be expected from these advances.
The findings of this important study help to better inform practice, and should influence decision-making for revascularization in patients with T1DM.
These remarks were taken from an editorial by Michael J. Domanski, MD, and Michael E. Farkouh, MD (J Am Coll Cardiol. 2017. doi: 10.1016/j.jacc.2017.07.781). Dr. Domanski is with the Peter Munk Cardiac Centre, Toronto, and the Heart and Stroke Richard Lewar Centre, University of Toronto. Dr. Farokouh is the director of clinical trials at the Peter Munk Cardiac Centre, University of Toronto.
In patients with type 1 diabetes in need of multivessel revascularization, coronary artery bypass graft (CABG) may be a better choice than percutaneous coronary intervention (PCI), according to results from a new comparative study presented at the annual congress of the European Society of Cardiology by Martin J. Holzmann, PhD, of the Karolinska Institute, Stockholm.
The two procedures had similar mortality rates, but PCI patients fared worse with respect to mortality due to myocardial infarction and several cardiovascular outcomes.
Previous studies had also suggested better outcomes with CABG than with PCI, but they lumped together patients with type 1 and type 2 diabetes, while the current study focused only on patients with type 1 diabetes.
The study included patients in Sweden with type 1 diabetes who underwent CABG (683 patients) or PCI (1,863 patients) between 1995 and 2013. During follow-up, 44.6% of patients in the PCI group died, compared with 53.3% in the CABG group. After adjustment for between-group differences, however, there was no significant difference in mortality risk between the two groups.
However, assessments of cause-specific mortality told a different story. Subjects in the PCI group had a greater risk of death from coronary artery disease (hazard ratio, 1.45; 95% confidence interval, 1.21-1.74).
Subjects in the PCI group were also more likely to suffer myocardial infarction (HR, 1.47; 95% CI, 1.21-1.77) and were more than five times more likely to undergo repeat vascularization (adjusted HR, 5.64; 95% CI, 4.67-6.82). The CABG group had a higher 30-day stroke risk (1.9% vs. 0.8%), but there was no difference in long-term risk.
The two groups had similar risks of hospitalization for heart failure.
The researchers noted a large difference between the two groups with respect to risk during the first year of follow-up, which suggests that some patients underwent PCI because they were too ill to undergo CABG. This limitation is also suggested by the greater proportion of previous stroke, heart failure, active cancer, and end-stage renal disease in the PCI group. The researchers adjusted for these differences, but it remains possible that there were residual confounders.
No source of funding was disclosed. One of the authors has received consultancy honoraria from Actelion and Pfizer. Dr. Domanski and Dr. Farkouh report no relevant financial relationships.
In patients with type 1 diabetes in need of multivessel revascularization, coronary artery bypass graft (CABG) may be a better choice than percutaneous coronary intervention (PCI), according to results from a new comparative study presented at the annual congress of the European Society of Cardiology by Martin J. Holzmann, PhD, of the Karolinska Institute, Stockholm.
The two procedures had similar mortality rates, but PCI patients fared worse with respect to mortality due to myocardial infarction and several cardiovascular outcomes.
Previous studies had also suggested better outcomes with CABG than with PCI, but they lumped together patients with type 1 and type 2 diabetes, while the current study focused only on patients with type 1 diabetes.
The study included patients in Sweden with type 1 diabetes who underwent CABG (683 patients) or PCI (1,863 patients) between 1995 and 2013. During follow-up, 44.6% of patients in the PCI group died, compared with 53.3% in the CABG group. After adjustment for between-group differences, however, there was no significant difference in mortality risk between the two groups.
However, assessments of cause-specific mortality told a different story. Subjects in the PCI group had a greater risk of death from coronary artery disease (hazard ratio, 1.45; 95% confidence interval, 1.21-1.74).
Subjects in the PCI group were also more likely to suffer myocardial infarction (HR, 1.47; 95% CI, 1.21-1.77) and were more than five times more likely to undergo repeat vascularization (adjusted HR, 5.64; 95% CI, 4.67-6.82). The CABG group had a higher 30-day stroke risk (1.9% vs. 0.8%), but there was no difference in long-term risk.
The two groups had similar risks of hospitalization for heart failure.
The researchers noted a large difference between the two groups with respect to risk during the first year of follow-up, which suggests that some patients underwent PCI because they were too ill to undergo CABG. This limitation is also suggested by the greater proportion of previous stroke, heart failure, active cancer, and end-stage renal disease in the PCI group. The researchers adjusted for these differences, but it remains possible that there were residual confounders.
No source of funding was disclosed. One of the authors has received consultancy honoraria from Actelion and Pfizer. Dr. Domanski and Dr. Farkouh report no relevant financial relationships.
FROM THE ESC CONGRESS 2017
Key clinical point: Patients undergoing PCI had worse cardiovascular outcomes than those receiving CABG.
Major finding: The PCI group had a 45% increased risk of death due to myocardial infarction.
Data source: Observational study (n = 2,546).
Disclosures: No source of funding was disclosed. One of the authors has received consultancy honoraria from Actelion and Pfizer. Dr. Domanski and Dr. Farkouh report no relevant financial relationships.