User login
The Potential Dangers of Treating Chronic Lyme Disease
“Chronic Lyme disease” is sometimes a catchall diagnosis for patients with a wide spectrum of musculoskeletal and neuropsychiatric symptoms, fatigue, and generalized pain. That, in turn, has led to a variety of treatments: courses of antibiotics lasting for months to years, IV infusions of hydrogen peroxide, immunoglobulin therapy, even stem cell transplants. Those treatments, though, may not lead to substantial long-term improvement—in fact, they can be harmful.
Clinicians, health departments, and patients have contacted the CDC reporting life-threatening complications resulting from treatment for chronic Lyme disease, including metastatic bacterial infections, septic shock, Clostridium difficile (C diff) colitis, and abscess. An article in Morbidity and Mortality Weekly Report (MMWR) described 5 cases that “highlight the severity and scope” of adverse effects caused by the use of unproven treatments for chronic Lyme disease.
One patient with fatigue and joint pain, was diagnosed with chronic Lyme disease, babesiosis, and Bartonella infection. When the symptoms worsened despite multiple courses of oral antibiotics, the patient was switched to IV ceftriaxone and cefotaxime. However, the pain did not lessen; the patient became hypotensive and tachycardic and was placed in intensive care. Her condition continued to worsen, and she died. The patient’s death was attributed to septic shock related to central venous catheter–associated bacteremia.
In another case, a woman was first diagnosed with amyotrophic lateral sclerosis, then as a second opinion, with chronic Lyme disease. After 7 months of intensive antimicrobial treatment, the pain improved but she got weaker. She also developed intractable C diff infection that required prolonged treatment. However, the patient died of complications of amyotrophic lateral sclerosis—an example, the researchers say, of a missed opportunity for appropriate treatment due to misdiagnosis.
Antibiotics and immunoglobulin therapies are effective and necessary treatments for many conditions, MMWR emphasized—“however, unnecessary antibiotic and immunoglobulin use provides no benefit to patients while putting them at risk for adverse events.”
“Chronic Lyme disease” is sometimes a catchall diagnosis for patients with a wide spectrum of musculoskeletal and neuropsychiatric symptoms, fatigue, and generalized pain. That, in turn, has led to a variety of treatments: courses of antibiotics lasting for months to years, IV infusions of hydrogen peroxide, immunoglobulin therapy, even stem cell transplants. Those treatments, though, may not lead to substantial long-term improvement—in fact, they can be harmful.
Clinicians, health departments, and patients have contacted the CDC reporting life-threatening complications resulting from treatment for chronic Lyme disease, including metastatic bacterial infections, septic shock, Clostridium difficile (C diff) colitis, and abscess. An article in Morbidity and Mortality Weekly Report (MMWR) described 5 cases that “highlight the severity and scope” of adverse effects caused by the use of unproven treatments for chronic Lyme disease.
One patient with fatigue and joint pain, was diagnosed with chronic Lyme disease, babesiosis, and Bartonella infection. When the symptoms worsened despite multiple courses of oral antibiotics, the patient was switched to IV ceftriaxone and cefotaxime. However, the pain did not lessen; the patient became hypotensive and tachycardic and was placed in intensive care. Her condition continued to worsen, and she died. The patient’s death was attributed to septic shock related to central venous catheter–associated bacteremia.
In another case, a woman was first diagnosed with amyotrophic lateral sclerosis, then as a second opinion, with chronic Lyme disease. After 7 months of intensive antimicrobial treatment, the pain improved but she got weaker. She also developed intractable C diff infection that required prolonged treatment. However, the patient died of complications of amyotrophic lateral sclerosis—an example, the researchers say, of a missed opportunity for appropriate treatment due to misdiagnosis.
Antibiotics and immunoglobulin therapies are effective and necessary treatments for many conditions, MMWR emphasized—“however, unnecessary antibiotic and immunoglobulin use provides no benefit to patients while putting them at risk for adverse events.”
“Chronic Lyme disease” is sometimes a catchall diagnosis for patients with a wide spectrum of musculoskeletal and neuropsychiatric symptoms, fatigue, and generalized pain. That, in turn, has led to a variety of treatments: courses of antibiotics lasting for months to years, IV infusions of hydrogen peroxide, immunoglobulin therapy, even stem cell transplants. Those treatments, though, may not lead to substantial long-term improvement—in fact, they can be harmful.
Clinicians, health departments, and patients have contacted the CDC reporting life-threatening complications resulting from treatment for chronic Lyme disease, including metastatic bacterial infections, septic shock, Clostridium difficile (C diff) colitis, and abscess. An article in Morbidity and Mortality Weekly Report (MMWR) described 5 cases that “highlight the severity and scope” of adverse effects caused by the use of unproven treatments for chronic Lyme disease.
One patient with fatigue and joint pain, was diagnosed with chronic Lyme disease, babesiosis, and Bartonella infection. When the symptoms worsened despite multiple courses of oral antibiotics, the patient was switched to IV ceftriaxone and cefotaxime. However, the pain did not lessen; the patient became hypotensive and tachycardic and was placed in intensive care. Her condition continued to worsen, and she died. The patient’s death was attributed to septic shock related to central venous catheter–associated bacteremia.
In another case, a woman was first diagnosed with amyotrophic lateral sclerosis, then as a second opinion, with chronic Lyme disease. After 7 months of intensive antimicrobial treatment, the pain improved but she got weaker. She also developed intractable C diff infection that required prolonged treatment. However, the patient died of complications of amyotrophic lateral sclerosis—an example, the researchers say, of a missed opportunity for appropriate treatment due to misdiagnosis.
Antibiotics and immunoglobulin therapies are effective and necessary treatments for many conditions, MMWR emphasized—“however, unnecessary antibiotic and immunoglobulin use provides no benefit to patients while putting them at risk for adverse events.”
FDA approves enasidenib to treat relapsed/refractory AML
The US Food and Drug Administration (FDA) has approved marketing of the oral IDH2 inhibitor enasidenib (IDHIFA®).
The drug is now approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation, as detected by an FDA-approved test.
Enasidenib is available in 50 mg and 100 mg tablets. The recommended dose is 100 mg once daily until disease progression or unacceptable toxicity.
The prescribing information for enasidenib includes a boxed warning that the drug may cause differentiation syndrome, and this adverse event (AE) can be fatal if not treated.
Signs and symptoms of differentiation syndrome may include fever, dyspnea, acute respiratory distress, radiographic pulmonary infiltrates, pleural or pericardial effusions, rapid weight gain, peripheral edema, and hepatic, renal, or multi-organ dysfunction. At first suspicion of symptoms, doctors should treat patients with corticosteroids and monitor patients closely until symptoms resolve.
Companion diagnostic
Enasidenib was approved concurrently with the Abbott RealTime™ IDH2 companion diagnostic test, which was approved as an aid in identifying AML patients for treatment with enasidenib.
The FDA granted approval of enasidenib to Celgene Corporation and approval of the RealTime IDH2 Assay to Abbott Laboratories.
Enasidenib is licensed from Agios Pharmaceuticals.
Trial results
The FDA’s approval of enasidenib and the companion diagnostic test was based on data from a phase 1/2 trial (Study AG221-C-001, NCT01915498).
Data from this trial were recently presented at the ASCO 2017 Annual Meeting. However, the definitive data are included in the prescribing information for enasidenib.
The prescribing information includes efficacy data for 199 adults with relapsed/refractory AML and an IDH2 mutation. IDH2 mutations were identified or confirmed by the Abbott RealTime™ IDH2 test.
The 199 patients received enasidenib at a starting dose of 100 mg daily until disease progression or unacceptable toxicity. Dose reductions were allowed to manage side effects.
The patients’ median age was 68 (range, 19 to 100). They received a median of 2 prior anticancer regimens (range, 1 to 6). More than half (52%) were refractory to previous therapy.
The rate of complete response (CR) or CR with partial hematologic improvement (CRh) was 23% (n=46). The median duration of CR/CRh was 8.2 months (range, 4.3 to 19.4).
For patients who achieved a CR/CRh, the median time to first response was 1.9 months (range, 0.5 to 7.5), and the median time to best response of CR/CRh was 3.7 months (range, 0.6 to 11.2).
Among the 157 patients who were dependent on red blood cell (RBC) and/or platelet transfusions at baseline, 53 (34%) became independent of RBC and platelet transfusions during any 56-day period post-baseline.
Of the 42 patients who were independent of both RBC and platelet transfusions at baseline, 32 (76%) remained transfusion independent during any 56-day post-baseline period.
Researchers evaluated the safety of enasidenib in 214 patients. The median duration of exposure to enasidenib was 4.3 months (range, 0.3 to 23.6).
The most common AEs of any grade (≥20%) were nausea, vomiting, diarrhea, elevated bilirubin, and decreased appetite.
Serious AEs were reported in 77.1% of patients. The most frequent serious AEs (≥2%) were leukocytosis, diarrhea, nausea, vomiting, decreased appetite, tumor lysis syndrome, and differentiation syndrome.
The 30-day and 60-day mortality rates were 4.2% (9/214) and 11.7% (25/214), respectively. ![]()
The US Food and Drug Administration (FDA) has approved marketing of the oral IDH2 inhibitor enasidenib (IDHIFA®).
The drug is now approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation, as detected by an FDA-approved test.
Enasidenib is available in 50 mg and 100 mg tablets. The recommended dose is 100 mg once daily until disease progression or unacceptable toxicity.
The prescribing information for enasidenib includes a boxed warning that the drug may cause differentiation syndrome, and this adverse event (AE) can be fatal if not treated.
Signs and symptoms of differentiation syndrome may include fever, dyspnea, acute respiratory distress, radiographic pulmonary infiltrates, pleural or pericardial effusions, rapid weight gain, peripheral edema, and hepatic, renal, or multi-organ dysfunction. At first suspicion of symptoms, doctors should treat patients with corticosteroids and monitor patients closely until symptoms resolve.
Companion diagnostic
Enasidenib was approved concurrently with the Abbott RealTime™ IDH2 companion diagnostic test, which was approved as an aid in identifying AML patients for treatment with enasidenib.
The FDA granted approval of enasidenib to Celgene Corporation and approval of the RealTime IDH2 Assay to Abbott Laboratories.
Enasidenib is licensed from Agios Pharmaceuticals.
Trial results
The FDA’s approval of enasidenib and the companion diagnostic test was based on data from a phase 1/2 trial (Study AG221-C-001, NCT01915498).
Data from this trial were recently presented at the ASCO 2017 Annual Meeting. However, the definitive data are included in the prescribing information for enasidenib.
The prescribing information includes efficacy data for 199 adults with relapsed/refractory AML and an IDH2 mutation. IDH2 mutations were identified or confirmed by the Abbott RealTime™ IDH2 test.
The 199 patients received enasidenib at a starting dose of 100 mg daily until disease progression or unacceptable toxicity. Dose reductions were allowed to manage side effects.
The patients’ median age was 68 (range, 19 to 100). They received a median of 2 prior anticancer regimens (range, 1 to 6). More than half (52%) were refractory to previous therapy.
The rate of complete response (CR) or CR with partial hematologic improvement (CRh) was 23% (n=46). The median duration of CR/CRh was 8.2 months (range, 4.3 to 19.4).
For patients who achieved a CR/CRh, the median time to first response was 1.9 months (range, 0.5 to 7.5), and the median time to best response of CR/CRh was 3.7 months (range, 0.6 to 11.2).
Among the 157 patients who were dependent on red blood cell (RBC) and/or platelet transfusions at baseline, 53 (34%) became independent of RBC and platelet transfusions during any 56-day period post-baseline.
Of the 42 patients who were independent of both RBC and platelet transfusions at baseline, 32 (76%) remained transfusion independent during any 56-day post-baseline period.
Researchers evaluated the safety of enasidenib in 214 patients. The median duration of exposure to enasidenib was 4.3 months (range, 0.3 to 23.6).
The most common AEs of any grade (≥20%) were nausea, vomiting, diarrhea, elevated bilirubin, and decreased appetite.
Serious AEs were reported in 77.1% of patients. The most frequent serious AEs (≥2%) were leukocytosis, diarrhea, nausea, vomiting, decreased appetite, tumor lysis syndrome, and differentiation syndrome.
The 30-day and 60-day mortality rates were 4.2% (9/214) and 11.7% (25/214), respectively. ![]()
The US Food and Drug Administration (FDA) has approved marketing of the oral IDH2 inhibitor enasidenib (IDHIFA®).
The drug is now approved to treat adults with relapsed or refractory acute myeloid leukemia (AML) and an IDH2 mutation, as detected by an FDA-approved test.
Enasidenib is available in 50 mg and 100 mg tablets. The recommended dose is 100 mg once daily until disease progression or unacceptable toxicity.
The prescribing information for enasidenib includes a boxed warning that the drug may cause differentiation syndrome, and this adverse event (AE) can be fatal if not treated.
Signs and symptoms of differentiation syndrome may include fever, dyspnea, acute respiratory distress, radiographic pulmonary infiltrates, pleural or pericardial effusions, rapid weight gain, peripheral edema, and hepatic, renal, or multi-organ dysfunction. At first suspicion of symptoms, doctors should treat patients with corticosteroids and monitor patients closely until symptoms resolve.
Companion diagnostic
Enasidenib was approved concurrently with the Abbott RealTime™ IDH2 companion diagnostic test, which was approved as an aid in identifying AML patients for treatment with enasidenib.
The FDA granted approval of enasidenib to Celgene Corporation and approval of the RealTime IDH2 Assay to Abbott Laboratories.
Enasidenib is licensed from Agios Pharmaceuticals.
Trial results
The FDA’s approval of enasidenib and the companion diagnostic test was based on data from a phase 1/2 trial (Study AG221-C-001, NCT01915498).
Data from this trial were recently presented at the ASCO 2017 Annual Meeting. However, the definitive data are included in the prescribing information for enasidenib.
The prescribing information includes efficacy data for 199 adults with relapsed/refractory AML and an IDH2 mutation. IDH2 mutations were identified or confirmed by the Abbott RealTime™ IDH2 test.
The 199 patients received enasidenib at a starting dose of 100 mg daily until disease progression or unacceptable toxicity. Dose reductions were allowed to manage side effects.
The patients’ median age was 68 (range, 19 to 100). They received a median of 2 prior anticancer regimens (range, 1 to 6). More than half (52%) were refractory to previous therapy.
The rate of complete response (CR) or CR with partial hematologic improvement (CRh) was 23% (n=46). The median duration of CR/CRh was 8.2 months (range, 4.3 to 19.4).
For patients who achieved a CR/CRh, the median time to first response was 1.9 months (range, 0.5 to 7.5), and the median time to best response of CR/CRh was 3.7 months (range, 0.6 to 11.2).
Among the 157 patients who were dependent on red blood cell (RBC) and/or platelet transfusions at baseline, 53 (34%) became independent of RBC and platelet transfusions during any 56-day period post-baseline.
Of the 42 patients who were independent of both RBC and platelet transfusions at baseline, 32 (76%) remained transfusion independent during any 56-day post-baseline period.
Researchers evaluated the safety of enasidenib in 214 patients. The median duration of exposure to enasidenib was 4.3 months (range, 0.3 to 23.6).
The most common AEs of any grade (≥20%) were nausea, vomiting, diarrhea, elevated bilirubin, and decreased appetite.
Serious AEs were reported in 77.1% of patients. The most frequent serious AEs (≥2%) were leukocytosis, diarrhea, nausea, vomiting, decreased appetite, tumor lysis syndrome, and differentiation syndrome.
The 30-day and 60-day mortality rates were 4.2% (9/214) and 11.7% (25/214), respectively. ![]()
Delirium linked to early death in advanced cancer patients

A diagnosis of delirium during a visit to the emergency department (ED) is a poor prognostic factor for patients with advanced cancer, according to research published in The Oncologist.
The study showed that patients with advanced cancer who were diagnosed with delirium during an ED visit were more likely to be admitted to the hospital or intensive care unit (ICU) and more likely to die earlier than patients without delirium.
This shows the importance of accurately diagnosing delirium in advanced cancer patients, said Ahmed Elsayem, MD, of the University of Texas MD Anderson Cancer Center in Houston.
Previous studies have shown that delirium is associated with poor survival in advanced cancer patients being treated in ICUs or receiving palliative care in hospices, but no one had investigated whether the same was true for patients visiting EDs.
“To the best our knowledge, this is the first study to show the poor survival of advanced cancer patients in the emergency department setting,” Dr Elsayem said.
He and his colleagues previously conducted a study in which they assessed the frequency of delirium in advanced cancer patients visiting the ED at MD Anderson. The researchers tested for delirium using 2 questionnaires—the Confusion Assessment Method (CAM) and Memorial Delirium Assessment Scale (MDAS).
Questioning 243 patients in total, the team found that 44 patients, or 18%, were suffering with delirium according to at least 1 of the questionnaires.
In the current study, Dr Elsayem and his colleagues determined how many of these cancer patients, with and without delirium, were subsequently admitted to hospital and ICUs, as well as how long the patients lived after their visit to the ED.
Results
The rate of hospitalization was 82% among patients with delirium according to CAM and/or MDAS, 77% among patients with delirium according to MDAS only, and 49% among patients without delirium (P=0.0013). Rates of ICU admission were 18%, 14%, and 2%, respectively (P=0.0004).
The median overall survival was 1.23 months for patients with delirium according to CAM and/or MDAS, 4.70 months for patients with delirium according to MDAS only, and 10.45 months for patients without delirium. The difference between the patients with and without delirium was significant (P<0.0001).
Given the influence delirium appears to have on survival, Dr Elsayem said prompt diagnosis and management in hospital EDs is essential.
He noted that, in many cases, delirium in advanced cancer patients can be resolved by simply stopping or modifying their medication and treating any associated infections.
“Treating the triggers if known—such as stopping medications—is the main treatment for an episode of delirium,” Dr Elsayem said.
He also suggested that further research needs to be done on this topic, including conducting similar studies on delirium in advanced cancer patients in other EDs and with larger groups of patients. ![]()
A diagnosis of delirium during a visit to the emergency department (ED) is a poor prognostic factor for patients with advanced cancer, according to research published in The Oncologist.
The study showed that patients with advanced cancer who were diagnosed with delirium during an ED visit were more likely to be admitted to the hospital or intensive care unit (ICU) and more likely to die earlier than patients without delirium.
This shows the importance of accurately diagnosing delirium in advanced cancer patients, said Ahmed Elsayem, MD, of the University of Texas MD Anderson Cancer Center in Houston.
Previous studies have shown that delirium is associated with poor survival in advanced cancer patients being treated in ICUs or receiving palliative care in hospices, but no one had investigated whether the same was true for patients visiting EDs.
“To the best our knowledge, this is the first study to show the poor survival of advanced cancer patients in the emergency department setting,” Dr Elsayem said.
He and his colleagues previously conducted a study in which they assessed the frequency of delirium in advanced cancer patients visiting the ED at MD Anderson. The researchers tested for delirium using 2 questionnaires—the Confusion Assessment Method (CAM) and Memorial Delirium Assessment Scale (MDAS).
Questioning 243 patients in total, the team found that 44 patients, or 18%, were suffering with delirium according to at least 1 of the questionnaires.
In the current study, Dr Elsayem and his colleagues determined how many of these cancer patients, with and without delirium, were subsequently admitted to hospital and ICUs, as well as how long the patients lived after their visit to the ED.
Results
The rate of hospitalization was 82% among patients with delirium according to CAM and/or MDAS, 77% among patients with delirium according to MDAS only, and 49% among patients without delirium (P=0.0013). Rates of ICU admission were 18%, 14%, and 2%, respectively (P=0.0004).
The median overall survival was 1.23 months for patients with delirium according to CAM and/or MDAS, 4.70 months for patients with delirium according to MDAS only, and 10.45 months for patients without delirium. The difference between the patients with and without delirium was significant (P<0.0001).
Given the influence delirium appears to have on survival, Dr Elsayem said prompt diagnosis and management in hospital EDs is essential.
He noted that, in many cases, delirium in advanced cancer patients can be resolved by simply stopping or modifying their medication and treating any associated infections.
“Treating the triggers if known—such as stopping medications—is the main treatment for an episode of delirium,” Dr Elsayem said.
He also suggested that further research needs to be done on this topic, including conducting similar studies on delirium in advanced cancer patients in other EDs and with larger groups of patients. ![]()
A diagnosis of delirium during a visit to the emergency department (ED) is a poor prognostic factor for patients with advanced cancer, according to research published in The Oncologist.
The study showed that patients with advanced cancer who were diagnosed with delirium during an ED visit were more likely to be admitted to the hospital or intensive care unit (ICU) and more likely to die earlier than patients without delirium.
This shows the importance of accurately diagnosing delirium in advanced cancer patients, said Ahmed Elsayem, MD, of the University of Texas MD Anderson Cancer Center in Houston.
Previous studies have shown that delirium is associated with poor survival in advanced cancer patients being treated in ICUs or receiving palliative care in hospices, but no one had investigated whether the same was true for patients visiting EDs.
“To the best our knowledge, this is the first study to show the poor survival of advanced cancer patients in the emergency department setting,” Dr Elsayem said.
He and his colleagues previously conducted a study in which they assessed the frequency of delirium in advanced cancer patients visiting the ED at MD Anderson. The researchers tested for delirium using 2 questionnaires—the Confusion Assessment Method (CAM) and Memorial Delirium Assessment Scale (MDAS).
Questioning 243 patients in total, the team found that 44 patients, or 18%, were suffering with delirium according to at least 1 of the questionnaires.
In the current study, Dr Elsayem and his colleagues determined how many of these cancer patients, with and without delirium, were subsequently admitted to hospital and ICUs, as well as how long the patients lived after their visit to the ED.
Results
The rate of hospitalization was 82% among patients with delirium according to CAM and/or MDAS, 77% among patients with delirium according to MDAS only, and 49% among patients without delirium (P=0.0013). Rates of ICU admission were 18%, 14%, and 2%, respectively (P=0.0004).
The median overall survival was 1.23 months for patients with delirium according to CAM and/or MDAS, 4.70 months for patients with delirium according to MDAS only, and 10.45 months for patients without delirium. The difference between the patients with and without delirium was significant (P<0.0001).
Given the influence delirium appears to have on survival, Dr Elsayem said prompt diagnosis and management in hospital EDs is essential.
He noted that, in many cases, delirium in advanced cancer patients can be resolved by simply stopping or modifying their medication and treating any associated infections.
“Treating the triggers if known—such as stopping medications—is the main treatment for an episode of delirium,” Dr Elsayem said.
He also suggested that further research needs to be done on this topic, including conducting similar studies on delirium in advanced cancer patients in other EDs and with larger groups of patients. ![]()
FDA grants acalabrutinib breakthrough designation

The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to acalabrutinib, a BTK inhibitor being developed to treat multiple B-cell malignancies.
The breakthrough designation applies to acalabrutinib as a treatment for patients with mantle cell lymphoma (MCL) who have received at least 1 prior therapy.
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
The FDA granted acalabrutinib breakthrough designation based on data from the drug’s development program, which includes data from the phase 2 ACE-LY-004 trial in patients with relapsed or refractory MCL.
Data from this trial have not yet been released to the public but are expected to be presented at an upcoming medical meeting. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to acalabrutinib, a BTK inhibitor being developed to treat multiple B-cell malignancies.
The breakthrough designation applies to acalabrutinib as a treatment for patients with mantle cell lymphoma (MCL) who have received at least 1 prior therapy.
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
The FDA granted acalabrutinib breakthrough designation based on data from the drug’s development program, which includes data from the phase 2 ACE-LY-004 trial in patients with relapsed or refractory MCL.
Data from this trial have not yet been released to the public but are expected to be presented at an upcoming medical meeting. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to acalabrutinib, a BTK inhibitor being developed to treat multiple B-cell malignancies.
The breakthrough designation applies to acalabrutinib as a treatment for patients with mantle cell lymphoma (MCL) who have received at least 1 prior therapy.
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
The FDA granted acalabrutinib breakthrough designation based on data from the drug’s development program, which includes data from the phase 2 ACE-LY-004 trial in patients with relapsed or refractory MCL.
Data from this trial have not yet been released to the public but are expected to be presented at an upcoming medical meeting. ![]()
The Ups and Downs of One Man’s Life
ANSWER
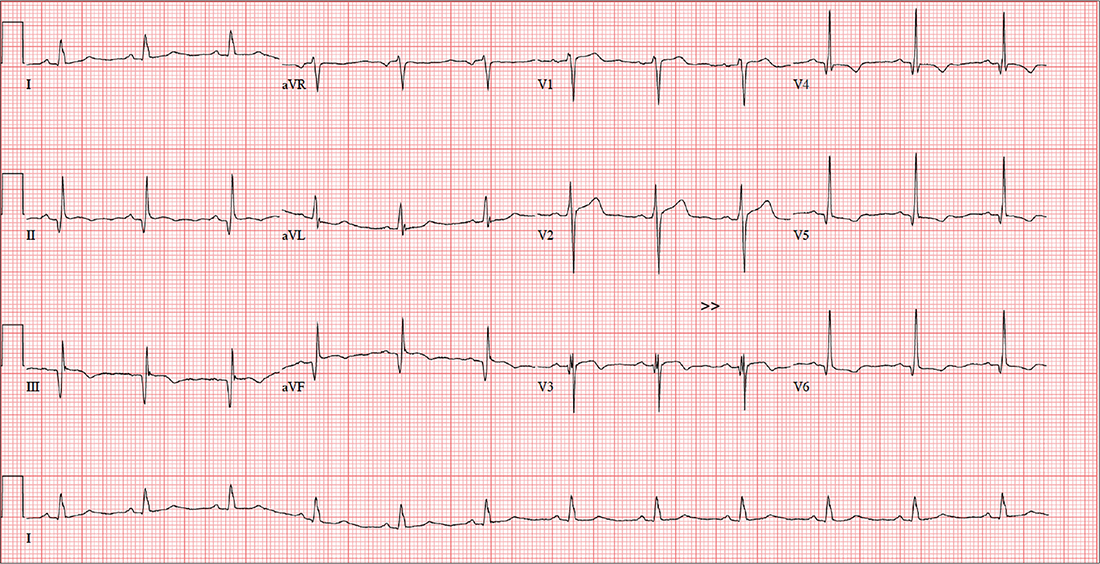
The correct interpretation includes normal sinus rhythm, evidence of a previous inferior MI, an anterior MI, and lateral ischemia (evidenced by ST- and T-wave abnormalities in the lateral chest leads).
Normal sinus rhythm is demonstrated by the presence of a P wave for every QRS complex and a QRS complex for every P wave, with a consistent PR interval of 72 beats/min. The Q waves in leads II, III, and aVF indicate an old inferior MI.
Progression of proximal LAD stenosis and an anterior MI is evidenced by the ST segment elevations in leads V1 to V3, and lateral ischemia is represented by ST- and T-wave abnormalities in leads V4 to V6. Note that there are also Q waves in leads V4 to V6, which suggest a previously undiagnosed lateral MI.
ANSWER
The correct interpretation includes normal sinus rhythm, evidence of a previous inferior MI, an anterior MI, and lateral ischemia (evidenced by ST- and T-wave abnormalities in the lateral chest leads).
Normal sinus rhythm is demonstrated by the presence of a P wave for every QRS complex and a QRS complex for every P wave, with a consistent PR interval of 72 beats/min. The Q waves in leads II, III, and aVF indicate an old inferior MI.
Progression of proximal LAD stenosis and an anterior MI is evidenced by the ST segment elevations in leads V1 to V3, and lateral ischemia is represented by ST- and T-wave abnormalities in leads V4 to V6. Note that there are also Q waves in leads V4 to V6, which suggest a previously undiagnosed lateral MI.
ANSWER
The correct interpretation includes normal sinus rhythm, evidence of a previous inferior MI, an anterior MI, and lateral ischemia (evidenced by ST- and T-wave abnormalities in the lateral chest leads).
Normal sinus rhythm is demonstrated by the presence of a P wave for every QRS complex and a QRS complex for every P wave, with a consistent PR interval of 72 beats/min. The Q waves in leads II, III, and aVF indicate an old inferior MI.
Progression of proximal LAD stenosis and an anterior MI is evidenced by the ST segment elevations in leads V1 to V3, and lateral ischemia is represented by ST- and T-wave abnormalities in leads V4 to V6. Note that there are also Q waves in leads V4 to V6, which suggest a previously undiagnosed lateral MI.
A 58-year-old man is concerned that his previously documented coronary atherosclerosis may be progressing. Three days ago, he experienced a dull heaviness in his chest but did not have pain, diaphoresis, shortness of breath, or dyspnea on exertion. Yesterday, he developed sharp substernal chest pain while walking uphill to his office from the parking lot. The pain subsided when he stopped to catch his breath but returned last night as he was pushing the recycling bin from his garage out to the curb. When he rested, it resolved, and he has not experienced any pain so far today.
For more than 30 years, the patient smoked 2.5 packs of cigarettes per day. At age 55, he had an inferior myocardial infarction (MI), prompting him to reduce his smoking habit to less than one pack per day. He was never able to quit completely, though, despite nicotine patches, medication, and hypnosis. For the past six months, he has been under considerable stress, as his business—an automobile parts distributorship he co-owns—filed for bankruptcy; he is now smoking heavily again (2 packs/d) as a result. He is married but separated. He drinks a six-pack of beer over the span of one week. He tried recreational marijuana while in college but denies previous or current illicit drug use.
Apart from the inferior MI and coronary atherosclerosis, the patient’s medical history is remarkable for hypertension, type 2 diabetes, and tobacco abuse. Surgical history is remarkable for a drug-eluting stent placed in the proximal left anterior descending (LAD) coronary artery, laparoscopic cholecystectomy, an amputated left fifth finger from a woodworking accident, and a childhood tonsillectomy.
His current medications include metoprolol XL, isosorbide dinitrate, amlodipine, and metformin. He has no known drug allergies but develops urticaria when chlorhexidine surgical scrub solution is used on his skin.
The review of systems is remarkable for recent weight gain (10 lb over the past two months) and a productive smoker’s cough that improves during the day. The patient denies any urinary or gastrointestinal symptoms, as well as any symptoms suggestive of diabetic neuropathy or endocrine dysfunction.
Vital signs include a blood pressure of 158/94 mm Hg; pulse, 70 beats/min; respiratory rate, 14 breaths/min-1; and temperature, 97.4°F. His height is 67 in and his weight, 254 lb. On physical exam, you note an obese male who smells of cigarette smoke. He wears corrective lenses, and the funduscopic exam is remarkable for arteriovenous nicking.
The thyroid is normal sized without nodules. A faint carotid bruit is present in the left carotid artery. The lung fields reveal coarse rales that change but do not disappear with vigorous coughing. The cardiac exam reveals a regular rate at 70 beats/min, with a soft early diastolic murmur heard at the left lower sternal border. There are no gallops or extra heart sounds.
The abdomen is obese but soft, with no palpable masses. Old surgical scars representing port placement for laparoscopic surgery are evident. The extremities show no evidence of swelling or edema. The fingers of the right hand are stained with nicotine, and a left fifth digit is missing beyond the proximal interphalangeal joint. Peripheral pulses are strong and equal bilaterally. The neurologic exam is grossly normal, with no focal signs. The patient has good sensation in both feet.
An ECG shows a ventricular rate of 72 beats/min; PR interval, 158 ms; QRS duration, 106 ms; QT/QTc interval, 400/438 ms; P axis, 33°; R axis, 38°; T axis, –15°. What is your interpretation?
Management of Status Epilepticus in Adults
From the Johns Hopkins Hospital, Baltimore, MD (Dr. Ramadan), and the Henry Ford Hospital, Detroit, MI (Dr. Varelas).
Abstract
- Objective: To review the management of status epilepticus (SE).
- Methods: Review of the literature.
- Results: SE is a relatively common condition that accounts for 3% to 5% of all emergency department evaluations for seizure disorders and occurs in 2% to 16% of all epilepsy patients. The 3 most common etiologies are low levels of antiepileptic drugs, remote symptomatic etiologies, and cerebrovascular accidents. The majority of SEs are convulsant, but there is growing awareness of non-convulsive SEs, which can be diagnosed only via electroencephalogram. Management, which must be initiated at the earliest possible time, has evolved to incorporate pre-hospital measures and 4 treatment stages, with supportive measures and benzodiazepine administration remaining the mainstay initially and followed by older and newer antiepileptic drugs and anesthetics for resistant cases.
- Conclusion: SE is a neurological emergency that still carries significant mortality and morbidity if not treated immediately and properly.
Key words: status epilepticus; seizures; convulsive status epilepticus; nonconvulsive status epilepticus.
Status epilepticus (SE) is a relatively common condition that accounts for 3% to 5% of all emergency department (ED) evaluations for seizure disorders and occurs in 2% to 16% of all epilepsy patients [1]. It remains a major neurological emergency that, if not properly and timely treated, leads to death or permanent neurological injury. Since most of patients with convulsive SE are admitted to the hospital via the ED and are then transferred to the intensive care unit (ICU), our focus in this review will be on the latter.
Although only a handful prospective, randomized studies have been reported, guidelines on SE have been published in Europe [2] and the US [3,4]. In this paper, we review the evolving definition and types of SE, its incidence, etiology, and pathophysiology, its diagnosis and treatment algorithms, and its outcome. Our goal is to provide the reader with a concise but thorough review of this still lethal neurological emergency.
Definitions
The International Classification of Epileptic Seizures had previously defined SE as any seizure lasting ≥ 30 minutes or intermittent seizures lasting for > 30 min without recovery of consciousness interictally [5,6]. More recently, a duration of 5 or more minutes of (a) continuous seizures or (b) 2 or more discrete seizures with incomplete recovery of consciousness in-between, proposed by Lowenstein [3,7], offers the advantage of incorporating new knowledge. The shortening of the convulsive period to 5 minutes was based on the fact that the majority of tonic-clonic seizures last for only 1 to 2 minutes, that those lasting > 5 minutes do not stop spontaneously [8], that permanent neuronal injury occurs before 30 minutes, and that refractoriness to treatment increases with longer seizure duration [9].
Refractory SE (RSE) has been defined as SE not controlled after adequate doses of an initial benzodiazepine followed by a second acceptable antiepileptic drug (AED) or SE not controlled after the initial parenteral therapy with a minimum number of standard “front-line” AEDs (either 2 or 3) or SE with a minimum duration of seizures that persist despite treatment (eg, at least or 2 hours) [3,10]. Super-refractory SE (SRSE) is defined as SE that continues or recurs 24 hours or more after the onset of anesthetic therapy or recurs on the reduction or withdrawal of anesthesia [11].
Non-convulsive SE (NCSE) is defined as the presence of altered consciousness or behavior for ≥ 30 minutes, the absence of overt clinical signs of convulsive activity during that period, and the electroencephalographic (EEG) confirmation of seizures or activity that responds to treatment together with improvement of consciousness [12–15]. Two major types of NCSE can be encountered: the one in patients with epileptic encephalopathy/coma and the one in patients with absence or complex partial seizures, who are not usually admitted to ICU and are functional yet impaired. Because of the confusion between these 2 extremes in the NCSE spectrum, working criteria for standardization of reporting, utilizing the frequency of electroencephalographic epileptiform discharges or delta/theta waveforms have been proposed [15]. A recent compendium of 123 cases of NCSE with clinical descriptions and EEG patterns following a syndromic classification approach has also been published [16].
Types of SE
Three major categories of SE have been described: generalized convulsive SE (GCSE), focal motor SE (FMSE or epilepsia partialis continua [EPC]) of Kojevnikov, and NCSE. GCSE and FMSE are easily recognized due to overt convulsions. NCSE, however, has a more obscure phenotype and can be subdivided into a spectrum encompassing typical absence and complex partial SE, atypical absence SE and tonic SE (usually in children with learning disabilities), epileptic behavioral disturbance and psychosis, including Balint–like syndrome [17], confusional states or delirium with epileptiform discharges) and SE in coma (after significant brain injuries, such as hypoxia-ischemia, most commonly encountered in ICUs) [13,18]. The 2 extremes in this NCSE spectrum have completely different prognoses, with absence SE the most benign and SE in coma the most dismal.
Lastly, SE presents either spontaneously or can be “semi-intentional” iatrogenic, encountered either in the neuro-ICU or epilepsy monitoring unit, when AEDs are withdrawn under continuous EEG recording in order for seizures to emerge and be recorded with surface or intracranial electrodes.
Incidence of SE
In a prospective population-based epidemiological study, the incidence of SE was estimated at 41–61/100,000 patients/year. For the US, this translates to 125,000 to 195,000 episodes per year [19].
The highest incidence of SE occurs during the first year of life and during the decades beyond 60 years, and is also dependent on the SE subtype. Partial SE occurs in 25% of cases of SE and NCSE accounts for another 4% to 26 % [19,20], but the incidence for the latter is considered an underestimate due to the need for continuous EEG monitoring (which is not widely available). For example, NCSE was discovered in no patient with acute stroke [21], 8% of comatose ICU patients [22], 7% of patients with intracerebral hemorrhage [23], 3% to 8% of patients with subarachnoid hemorrhage [24–26], 6% of patients with metastatic cancer [27], and 6% of patients with head trauma [28].
The incidence of RSE and SRSE is also unknown. In a recent retrospective study from a neuro-ICU in a West China hospital, the percentage of non-refractory SE, RSE, and SRSE were 67.3%, 20.4% and 12.2%, respectively [29]. Other retrospective studies have shown that 12% to 43% of SE cases become refractory [30–33] and that approximately 10% to 15% of all cases of hospital-admitted SE will become super-refractory at some point, but no prospective studies have been published.
Risk factors that have been identified for RSE are encephalitis as a cause, severe consciousness impairment, de novo episodes of SE, delay in initiation of treatment, NCSE, and focal motor seizures at onset [30,32,34,35]. In a more recent study from ICU patients in Switzerland and the US, acute SE etiology (traumatic brain injuries, cerebrovascular accidents, meningoencephalitis, brain tumors, surgical brain lesions, exposure to, or withdrawal from, recreational drugs, prescription drugs, alcohol, metabolic disturbances and fever), coma/stupor, and serum albumin < 35 g/L at SE onset were independent predictors for RSE [36].
Etiology of SE
The 3 most common etiologies for SE are low levels of antiepileptic drugs (AEDs) in 34% of the cases (usually due to noncompliance), remote symptomatic etiologies (history of neurological insults remote to the first unprovoked SE episode, 24%), and cerebrovascular accidents (ischemic and hemorrhagic strokes, 22%). These are followed by hypoxia (13%) and metabolic disturbances (15%). Because 82% of patients in the remote group have a history of cerebrovascular disease, almost 50% have either acute or remote cerebrovascular disease as etiology of SE [19].

In general ICUs, metabolic abnormalities can account for 33% of seizures, drug withdrawal for 33%, drug toxicity for 14.5%, and stroke for 9% to 39% [37,38]. In ICUs, sepsis remains a common etiology of electrographic seizures or periodic epileptiform discharges [39,40], and legal or illegal drugs, such as ciprofloxacin, levofloxacin, piperacillin/tazobactam, cefepime and carbapenems [41–43], lithium or theophylline intoxication, vigabatrin, tiagabine or crack/cocaine, are another [18] (especially when their metabolism is altered due to interactions with other drugs or when their excretion is impaired due to hepatic or renal failure).
Beyond these common causes of SE, a workup for rare etiologies should be entertained. In a systematic review of 513 papers on SE, 181 uncommon causes of SE were identified and subdivided into immunologically mediated disorders, mitochondrial diseases, rare infectious disorders, genetic disorders, and drugs or toxins [18,44].
The most recent knowledge in this category is the contribution of paraneoplastic or autoimmune conditions to a large percentage of previously cryptogenic pharmaco-resistant seizures or super-refractory SE, most in the context of limbic encephalitis. Many of these patients have never experienced seizures or SE before and a new acronym has been devised for them: new-onset refractory status epilepticus (NORSE), ie, a state of persistent seizures with no identifiable etiology in patients without preexisting epilepsy that lasts longer than 24 hour despite optimal therapy [45]. A growing array of autoantibodies against intracellular and surface or synaptic neuronal targets has been described in addition to the previous literature of Rassmussen’s encephalitis and Hashimoto’s encephalopathy [46]. The most common autoantibodies associated with seizures and SE include anti-Hu, anti-Ma2, anti-CV2/CRMP5, anti-Ri, ANNA3, anti-amphiphysin, anti-NMDA receptor, anti-LGI1 and CASPR2, anti-GABA-beta, anti-GluR3, anti-mGluR5 and alpha 3 ganglionic acetylcholine receptor [47,48]. The diagnosis frequently remains elusive due to lack of knowledge or absence of widespread availability of serologic testing (with sometimes weeks-long delay for the results to be available), but the response to treatment with removal of tumor, plasmapheresis, or immunomodulation and immunosuppression is often dramatic.
Pathophysiology of SE
Most seizures are self-terminating phenomena lasting from a few seconds to a few minutes [49]. One of the distinguishing characteristics of seizures evolving into SE, however, is the switch to a self-sustaining situation, which is time-dependent. Seizures lasting more than 30 minutes would rarely stop spontaneously compared to 47% of those lasting between 10 to 29 minutes, which are self-resolving [50]. Moreover, in one study no self-limited seizure lasted more than 11 minutes [8].
The self-limiting character of seizures is due to inhibitory circuitry that suppresses their duration and propagation in the brain. Under specific circumstances, however, the inhibitory mechanisms fail and seizures progress to SE, which leads to synaptic reorganization, blood-brain barrier disruption, inflammation, metabolic crisis, more tissue damage, and further seizures. Neuronal injury during SE is the result of increased excitotoxicity [51–53] but also stems from systemic derangements such as hypoxia, acidosis, hypotension, or multiorgan dysfunction [54]. The seminal animal studies by Meldrum have shed a light on the systemic effects: after prolonged bicuculine-induced convulsive SE in baboons, neuronal damage and cell loss was evident in the neocortex, cerebellum and hippocampus. When systemic factors were kept within normal physiological limits (paralyzed and artificially ventilated animals with adequate serum glucose levels), there was decreased but still present neocortical and hippocampal cell damage, but absent cerebellar cell injury [55,56]. These experiments showed more than 40 years ago that the seizure activity per se is responsible for the neuronal damage and the systemic derangements play an additional role.
The direct neuronal injury as a result of the ongoing seizures, the perpetuation of seizures into SE, the resistance to treatment and the refractoriness that ensues have also been elucidated at a molecular level during the last decades. Initially, the g-aminobutyric acid (GABA) inhibitory circuits may be deficient and this is the reason why benzodiazepines or barbiturates, which work through GABAergic receptor agonism, are very effective during this early period. As time passes however, GABA receptors undergo a significant shift in their ability to respond to benzodiazepines [57,58]. This is due to changes in receptor presence at the inhibitory synapse, a phenomenon that has been called “receptor trafficking” by Arancibia and Kittler in 2009 [59]. There are differences in the type of GABAA receptors found synaptically and extrasynaptically. GABAA receptors containing the γ subunit are located synaptically and mediate phasic inhibition. Conversely, the δ subunit-containing GABAA receptors are located exclusively extrasynaptically and mediate tonic inhibition [60,61]. Smith and Kittler described the highly dynamic state of receptor presence on the surface of axons and explained how receptors move laterally from extrasynaptic sites to the synapse and then out of it to be internalized and either recycled to the surface or degraded [62]. This “receptor trafficking” intensifies during SE, and the overall effect becomes a reduction in the number of functional GABAA receptors in the synapses. As GABA is the principle inhib-itory transmitter, this reduction in GABAergic activity may be an important reason for seizures to become persistent.
However, this is not all. Additional mechanisms leading to refractoriness include the following:
(a) Excessive relocation of N-methyl-D-aspartate (NMDA)type glutamate receptors to the cell surface after 1 hour of SE, leading to increase of miniature excitatory NMDA currents and NMDA neurotransmission, with potentiation of glutamate excitotoxicity [53,63]
(b) Increased brain expression of drug efflux transporters, such as P-glycoprotein at the blood-brain barrier, which may reduce concentrations of AEDs at their brain targets [64]
(c) Up- and down-regulation of specific ATP-gated ion channels (P2X receptors) inducing altered response to ATP release [65]
(d) Change in the extracellular ionic environment (for example, the normally inhibitory GABAA receptor-mediated currents may become excitatory with changes in extracellular chloride concentrations) [66]
(e) Mitochondrial insufficiency or failure, which would lead to cell necrosis and apoptosis [67]
(f) Inflammatory processes, with opening of the blood-brain barrier (BBB) contributing to perpetuation of seizures [44]. The underlying mechanism is a maladaptive response of the astrocytes to the BBB damage, leading to activation of the innate immune system and disturbed homeostasis of the extracellular potassium and glutamate [68].
(g) Large-scale changes in gene expression within the affected brain regions; these are regulated by micro-RNAs, influencing protein levels playing a role in excitability, neuronal death and neuroinflammation [69].
All of these pathophysiologic derangements may become targets for future antiepileptic treatments.
Although the direct and indirect injury from ongoing convulsive SE is not in doubt, the significance of NCSE or the ictal-interictal continuum on inflicting additional injury has been more controversial. Recent data, however, do not support a benign process in these situations. It has been shown lately that nonconvulsive seizures lead to physiologic changes in the brain, including elevated intracranial pressure, changes in the brain metabolism, and delayed increase in cerebral blood flow [25]. In addition, using microdialysis, elevated lactate/puruvate ratio, indicating metabolic crisis, has been shown during periods of nonconvulsive seizures or periodic discharges [70]. Similarly, high-frequency periodic discharges lead to inadequate increase in cerebral blood flow and tissue hypoxia [71], and lateralized periodic discharges, lateralized rhythmic delta activity, and generalized periodic discharges are associated with seizures [72].
Diagnosis of SE
The diagnosis of SE is primarily clinical and encompasses motor phenomena and alteration of mental status. Focal-onset convulsions can remain focal, follow a Jacksonian march, or immediately generalize to involve the whole body with loss of consciousness. Most of the time, this secondary generalization can only be appreciated during EEG recording. In addition, mental status alteration can differentiate simple partial SE (no change in mental status) from complex partial SE (disturbed sensorium).
The presence or absence of motor phenomena and loss of consciousness do not necessarily correlate with the EEG activity during or after SE. For example, persistent electrographic seizures or NCSE after control of convulsive SE have been demonstrated with continuous EEG [73]. Conversely, altered mental status is also a poor clinical differentiator, since 87% of patients successfully treated for convulsive SE and 100% treated for NCSE remained comatose 12 hours following the initiation of therapy [20]. In addition, only 27% of motor, seizure-like phenomena in the ICU were proven to be seizures in a retrospective study [74]. Psychogenic nonepileptic attacks, occurring in between 3.8% and 9.5% of ICU patients presenting with seizures [74,75], is another situation that may lead to confusion, inappropriate intubation, and ICU admission. Strange phenomena, such as fasciobrachial seizures (brief facial grimacing and ipsilateral arm posturing) many times preceding the onset of amnesia, confusion, or temporal lobe seizures have been described in patients who have non-paraneoplastic limbic encephalitis associated with voltage-gated potassium channel (VGKC) antibodies, especially against the leucine-rich glioma inactivated-1 (LGI1) protein [76,77].Without a continuous video EEG, these phenomena may not be captured or appreciated. Therefore, EEG monitoring is an important tool for the evaluation of these patients and criteria for its use have been published [78]. The EEG criteria for convulsive SE have been clearly delineated, but for NCSE a mix of clinical and EEG criteria should be met [14,15,79].
In addition to clinical observation and EEG, there has been interest lately in multimodality monitoring of acutely brain-injured patients for seizures or SE using electrocorticography or mini depth electrode placement, partial brain tissue oxygen tension, cerebral blood flow, and microdialysis in addition to scalp EEG. Although preliminary and limited in few academic centers, this approach has produced interesting findings. For example, in a study from Columbia University, 38% of 48 patients with subarachnoid hemorrhage and multimodality monitoring had intracortical seizures, while only 8% of them had surface seizures, all nonconvulsive [25]. In another study, 68% of seizures and 23% of periodic discharges were only captured on the depth electrodes and were missed on the surface ones [71]. Therefore, detection of SE may change in the future with use of more sensitive techniques than scalp EEG.
Treatment
Significant practice variations exist in the management of SE even among academic centers in the US [80] despite the fact that the goals of treatment are concrete. These include (1) emergent medical management, (2) termination of seizures, (3) prevention of recurrence of seizures, and (4) prevention or treatment of complications.
Management of SE must begin in a prehospital setting by the emergency medical services, because the faster the treatment is offered, the better the response. Several studies have attempted to assess the possibility of aborting SE even prior to the hospital. In a randomized, double-blinded study, lorazepam was 4.8 times and diazepam 2.3 times more effective than placebo in terminating SE on arrival in the ED when given intravenously (IV) by paramedics [81]. The RAMPART study was a double-blind, randomized, non-inferiority trial comparing the efficacy of intramuscular (IM) midazolam (10 mg followed by placebo IV) with that of IM placebo followed by intravenous lorazepam (4 mg) for children and adults in SE treated by paramedics. At the time of arrival in the ED, seizures had ceased without rescue therapy in 73.4% and 63.4%, respectively, favoring midazolam [82].

Emergent Initial Phase
During the emergent initial phase, the goals are protection of the airway, oxygenation, maintenance of blood pressure, exclusion of easily 
Urgent Control
If seizures continue, stage 2 medications should be used for benzodiazepine-refractory SE as urgent control treatment. There are some data suggesting better response rate to valproate after failure to control seizures with phenytoin than to phenytoin after failure of valproate [88]. If available, IV fosphenytoin is preferable to IV phenytoin due to potentially lower risk of side effects. Levetiracetam and phenobarbital IV are also acceptable choices. Levetiracetam can be administered as an off-label loading dose of 20–60 mg/kg IV (although the initial manufacturer was not supporting a “loading” dose; dose of up to 60 mg/kg IV up to 4500 mg maximum has been supported by the latest American Epilepsy Society guidelines [4]). This AED at an initial dose of 2–3 g/day confers an estimated success rate around 70% [89]. In a systematic review of 27 studies (798 cases of convulsive SE) comparing 5 AEDs in the treatment of benzodiazepine-resistant convulsive SE, phenobarbital and valproate had the highest efficacy (73.6% and 75.7%, respectively), followed by levetiracetam (68.5%) and phenytoin (50.2%). Lacosamide studies were excluded from the meta-analysis due to insufficient data [90], but its efficacy has been reported for patients with convulsive and NCSE [91,92]. There is not enough evidence at this point, however, to recommend its routine use for benzodiazepine refractory SE [90].
Refractory SE
When seizures continue despite the use of benzodiazepines and 2nd stage AEDs, SE becomes refractory (stage 3). Treatment of these resistant cases is frequently initiated in the ED and continued in an ICU. Outcomes were not significantly better in patients with SE admitted and managed in a neuro-ICU compared to a general medical ICU in a retrospective study, but the numbers were small (only 27% of SE were admitted to the former) [93] and this may change in the future. Intubation and mechanical ventilation is the first step, if not already present (only 21% of patients in the RAMPART study received endotracheal intubation, with 6.4% in the prehospital setting and 93.6% after admission [87]). Hemodynamic support with pressors or inotropes may be required as most anesthetic agents may decrease the blood pressure. Because of the urgency of controlling the seizures during SE, the potential aspiration risk and the questionable enteral absorption per os administration of additional AEDs is problematic, and IV formulations should be used.
Currently in the US, phenytoin, valproic acid, phenobarbital, levetiracetam, lacosamide, diazepam, lorazepam are available in IV formulations. In February 2016, the FDA also approved brivaracetam (which also is available in an IV formulation) and in October of the same year IV carbamazepine. None of these AEDs has an FDA indication for SE, although they are widely used. Parenteral lacosamide has a success rate of 33% to 67.7% (200–400 mg over 3–5 min was the most common bolus dose) depending on its use as second or third AED [94–96]. In lacosamide-naive patients with RSE on continuous EEG monitoring, the success rate for cessation of SE was 15.7, 25.5, 58.8, and 82.4 % by 4, 12, 24, and 48 hours, respectively [97]. Alternatively, topiramate in doses 300–1600 mg/day per oro/nasogastric tube can be considered [98]. In a study of 35 patients with RSE treated with topiramate as an adjunct AED, the response rate was 86% (as the third AED), and remained stable at 67% after administration as the fourth to seventh AED. Overall, RSE was terminated in 71% of patients within 72 hours after first administration of topiramate [99]. Other studies, however, adjusting for co-variates, did not prove topiramate to be effective in RSE [100]. Clobazam, a unique oral 1,5-benzodiazepine with excellent absorption, has been also used in the treatment of RSE. Seventeen patients with RSE (11 with prior epilepsy) were successfully treated with clobazam, which was introduced after a median duration of 4 days and after a median of 3 failed AEDs. Termination of RSE within 24 hours of administration, without addition or modification of concurrent AED and with successful wean of anesthetic infusions, was seen in 13 patients, whereas indeterminate response was seen in another 3. Clobazam was deemed unsuccessful in 1 patient [101]. In another recent report of 70 episodes of RSE, clobazam was used in 24 (34.3%) of them. If clobazam was the last AED added to therapy before RSE termination, the success was attributed to this drug. Based on this definition, clobazam led to 6 episodes (25%) of successful RSE resolution [102]. If primary or metastatic brain tumor is the presumed cause of SE, a combination of IV phenytoin, IV levetiracetam (median dose 3 g/d) and enterically administered pregabalin (median dose 375 mg/day) led to 70% control of SE on average 24 hours after addition of the third AED [103]. However, the major treatment options, which should not be delayed in unresponsive RSE, are propofol or midazolam infusions at high rates and under continuous EEG monitoring. These infusions should be continued for at least 24 hours and then held to reassess the situation. By that time, cocurrent metabolic derangements and low AED levels from noncompliance should have been corrected. Prolonged and high-dose propofol should be avoided because of the risk for propofol infusion syndrome, especially if pressors/inotropes are co-infused [104].
Super-refractory SE
Should seizures continue or recur, stage 4 options for SRSE are considered [105]. Pentobarbital with shorter half-life is favored to phenobarbital. The main disadvantages of barbiturates are compromised neuro-exam (which has to be assessed frequently), cardiovascular depression and hypotension, respiratory depression with need for full ventilator support, cough suppression with increased risk for atelectasis and pneumonia, immunosuppression increasing the risk for infection or sepsis, immobility increasing the risk for thromboembolism and ileus mandating parenteral nutrition [106,107]. The depth and duration of the EEG suppression that must be achieved by barbiturates is unknown. Some experts recommend instead of burst-suppression pattern complete suppression or “flat record” because of better seizure control and fewer relapses [108]. Moreover, patients with more prolonged barbiturate treatment (> 96 hours) and those receiving phenobarbital at the time of pentobarbital taper are less likely to relapse [109]. European guidelines recommend titration of propofol and barbiturate to EEG burst-suppression, and midazolam to seizure suppression, maintained for at least 24 h [2]. In recent reviews, it was found that barbiturates control refractory and super-refractory SE in 64% of patients and are ineffective in only 5% [11,110].
If SE continues or recurs after emergence from barbiturate coma, ketamine may be an option [11,83]. Ketamine offers the advantage of NMDA receptor antagonism, which may be important in the late phase of SE and lacks cardiodepressant or hypotensive properties. Early [111] or late [112] use of ketamine has been reported in small case series with various success rates. In a recent multicenter retrospective study from North America and Europe, evaluating 58 patients with 60 RSE, ketamine was likely responsible for seizure control in 12% and possibly responsible in an additional 20%. No responses were observed when infusion rate was lower than 0.9 mg/kg/h or when ketamine was introduced 8 days or more after onset of SE or after failure of seven or more drugs [113].
If all these measures have failed, stage 4.2 treatment options are available (Table 2), but these are mostly based on small case series and expert opinions (except for the recent hypothermia study). Pyridoxine hydrochloride in an IV or enteral form at a dose of 100–300 mg/day for few days can be used in stage 4 or earlier stages, as it is a cofactor in the synthesis of the inhibitory neurotransmitter GABA [114]. There are no strong data for its effectiveness, but it can be used as a cheap and safe alternative [115]. Magnesium has been successfully used in 2 girls with juvenile Alper’s syndrome [116] and is the treatment of choice for eclamptic seizures. Ketogenic diet may also be an optionfor these patients [117]. Resection of the epileptic focus after mapping with intracranial EEG electrodes may be highly effective but cannot be used in many patients due to lack of focus or eloquence location [83,106,115]. Use of steroids, plasmapheresis or IVIG, followed by immunosuppression can be tried, but one should balance risks and benefits. These immunosuppressive or immunomodulating treatments should be especially considered in patients with NORSE or suspected autoimmune or paraneoplastic encephalitides, where AEDs usually have no effect [46]. These therapies though often precede the diagnosis, since it takes time for the autoantibody panel results to return and the treating physician has to make a decision to blindly start treatment for SRSE.
There were some promising data regarding hypothermia use in these desperate situations [118,119] until the HYBERNATUS study, conducted in France, was recently published. In this study, 270 patients with convulsive SE were randomized in to hypothermia (32° to 34°C for 24 hours) in addition to standard care or to standard care alone. A Glasgow Outcome Scale score of 5 (primary outcome) occurred in 49% of patients in the hypothermia group and in 43% in the control group (a nonstatistical difference). Secondary outcomes, including mortality at 90 days, RSE on day 1, SRSE and functional sequelae on day 90 were not different except for the rate of progression to EEG-confirmed SE on the first day, which was lower in the hypothermia group (11% vs. 22% in the controls). Adverse events were more frequent in the hypothermia group than in the control group [120].
Additional anecdotal treatments are presented in Table 2, but their efficacy is questionable.
This staged management approach may change in the future to a more physiologic and rational treatment with polytherapy based on synaptic receptor trafficking during SE [63]. For example, in an animal model of severe SE, combinations of a benzodiazepine with ketamine and valproate, or with ketamine and brivaracetam, were more effective and less toxic than benzodiazepine monotherapy [121]. Allopregnalonone, a metabolite of progesterone, is an endogenous, naturally occurring neuroactive steroid produced in the ovary, the adrenal cortex and the central nervous system. It is a potent positive allosteric modulator of synaptic and extrasynaptic GABAA receptors with antiepileptic activity [122]. Neuroactive steroids, such as allopregnanolone, are currently evaluated in SE.
Outcomes
SE still carries significant mortality and morbidity. Distinct variants of SE carry different mortalities, and the range is quite broad: from zero mortality for absence or complex partial SE in ambulatory patients [12], to 19% to 27% 30-day mortality for generalized tonic-clonic SE [20,123] and to 64.7% 30-day mortality for subtle SE [20]. Variables playing an important role in the outcome are the underlying cause (regarded by most authorities the most important variable), the duration of SE (mortality 32% if persistent for > 1 hour vs 2.7% if < 1 hour), the type of SE, the treatment administered, and the age of the patient (children have better outcomes than adults) [123–125]. The etiology of SE still remains the most important prognostic factor, with alcohol and AED-withdrawal/noncompliance having the best outcomes; structural brain injuries, such as anoxia-ischemia, vascular lesions, or brain tumors, have the worst prognosis.
The most resistant cases pose significant dilemmas regarding the length of treatment using barbiturate coma and the potential for acceptable prognosis or the need to withdraw life support. For RSE, for example, in-hospital mortality is 31.7% and 76.2% of patients reach poor functional outcome. Long-term outcomes are also dismal: at 1 year post-discharge, 74% are dead or in a state of unresponsive wakefulness, 16% severely disabled, and only 10% have no or minimal disability [126]. Duration of drug-induced coma, arrhythmias requiring intervention, and pneumonia are associated with poor functional outcome, whereas prolonged mechanical ventilation with mortality and seizure control without burst-suppression or isoelectric EEG are associated with good functional outcome [127,128].
Treatment with barbiturates may contribute to these outcomes, although it is very challenging to prove causality in such a complex and prolonged ICU environment. Some data have shed light towards that direction: in a recent retrospective study of 171 patients with SE, of which 37% were treated with IV anesthetic drugs, there was a higher risk for infections and a 2.9-fold relative risk for death after adjustment for confounders in the group treated with IV anesthetics compared to the group without these agents [129].
The SE Severity Score (STESS, range 0–6) is a prognostic score for survival [130] and can be used as a scaffold for discussions with families and covariate adjustment tool for research. A favorable score of 0–2 has a negative predictive value of 0.97 for survival and likelihood to return to baseline clinical condition in survivors, although an unfavorable score (3–6) had a positive predictive value for death of only 0.39 [131].
The risk for recurrence of afebrile SE in a population-based study in Minnesota has been estimated at 31.7% over a 10-year follow-up period. The risk for recurrence was about 25% regardless of the underlying etiology, except in those patients with SE occurring in the setting of a progressive brain disorder (who had a 100% risk). Female gender, generalized (vs partial) SE and lack of response to the first AED after the initial episode of SE were independent factors for recurrence [132].
Conclusion
Despite the use of better diagnostic tools (continuous video EEG), advances in technology in the ICU, and availability of new AEDs, SE still carries significant mortality and morbidity, which depends mainly on age and etiology. The current treatment is still staged, with supportive measures and benzodiazepine administration remaining the mainstay initially and followed by older and newer AEDs and anesthetics for resistant cases. With the advance of pathophysiologic mechanisms elucidation at a molecular/receptor level, combinations of AEDs may become the foundation of future SE control.
Corresponding author: Panayiotis N. Varelas, MD, PhD, FNCS, Division Head, Neuro-Critical Care Service, Henry Ford Hospital, K-11, 2799 W. Grand Blvd., Detroit, MI 48202, pvarela1@hfhs.org.
Financial disclosures: Dr. Varelas was local principal investigator for a super-refractory status epilepticus study sponsored by Sage Therapeutics.
Author contributions: conception and design, ARR, PNV; analysis and interpretation of data, PNV; drafting of article, PNV; critical revision of the article, ARR, PNV; administrative or technical support, PNV; collection and assembly of data, ARR, PNV.
1. Hauser WA. Status epilepticus: epidemiologic considerations. Neurology 1990;40:9–13.
2. Meierkord H, Boon P, Engelsen B, et al. EFNS guideline on the management of status epilepticus. Eur J Neurology 2006;13:445–50.
3. Brophy GM, Bell R, Claassen J, et al. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care 2012;17:3–23.
4. Glauser T, Shinnar S, Gloss D, et al. Evidence-based guideline: treatment of convulsive status epilepticus in children and adults: Report of the Guideline Committee of the American Epilepsy Society. Epilepsy Curr 2016;16:48–61.
5. Gastaut H. Classification of status epilepticus. Adv Neurol 1983;34:15–35.
6. Treatment of convulsive status epilepticus. Recommendations of the Epilepsy Foundation of America’s Working Group on Status Epilepticus. JAMA 1993;270:854–9.
7. Lowenstein DH. Status epilepticus: an overview of the clinical problem. Epilepsia 1999;40 Suppl 1:S3–8; discussion S21–22.
8. Jenssen S, Gracely EJ, Sperling MR. How long do most seizures last? A systematic comparison of seizures recorded in the epilepsy monitoring unit. Epilepsia 2006;47:1499–503.
9. Goodkin HP, Kapur J. Responsiveness of status epilepticus to treatment with diazepan decreases rapidly as seizure duration increases. Epilepsy Curr 2003;3:11–2.
10. Lowenstein DH. The management of refractory status epilepticus: an update. Epilepsia 2006;47 Suppl 1:35–40.
11. Shorvon S, Ferlisi M. The treatment of super-refractory status epilepticus: a critical review of available therapies and a clinical treatment protocol. Brain 2011;134:2802–18.
12. Kaplan PW. Assessing the outcomes in patients with nonconvulsive status epilepticus: nonconvulsive status epilepticus is underdiagnosed, potentially overtreated, and confounded by comorbidity. J Clin Neurophysiol 1999;16:341–52.
13. Walker MD. Diagnosis and treatment of nonconvulsive status epilepticus. CNS Drugs 2001;15:931–9.
14. Kaplan PW. EEG criteria for nonconvulsive status epilepticus. Epilepsia 2007;48 Suppl 8:39–41.
15. Beniczky S, Hirsch LJ, Kaplan PW, et al. Unified EEG terminology and criteria for nonconvulsive status epilepticus. Epilepsia 2013;54 Suppl 6:28–9.
16. Sutter R, Kaplan PW. Electroencephalographic criteria for nonconvulsive status epilepticus: synopsis and comprehensive survey. Epilepsia 2012;53 Suppl 3:1–51.
17. Ristic AJ, Marjanovic I, Brajkovic L, et al. Balint-like syndrome as an unusual representation of non-convulsive status epilepticus. Epileptic Disord 2012;14:80–4.
18. Trinka E, Hofler J, Zerbs A. Causes of status epilepticus. Epilepsia 2012;53 Suppl 4:127–38.
19. DeLorenzo RJ, Hauser WA, Towne AR, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology 1996;46:1029–35.
20. Treiman DM, Meyers PD, Walton NY, et al. A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med 1998;339:792–8.
21. Carrera E, Michel P, Despland PA, et al. Continuous assessment of electrical epileptic activity in acute stroke. Neurology 2006;67:99–104.
22. Towne AR, Waterhouse EJ, Boggs JG, et al. Prevalence of nonconvulsive status epilepticus in comatose patients. Neurology 2000;54:340–5.
23. Claassen J, Jette N, Chum F, et al. Electrographic seizures and periodic discharges after intracerebral hemorrhage. Neurology 2007;69:1356–65.
24. Claassen J, Peery S, Kreiter KT, et al. Predictors and clinical impact of epilepsy after subarachnoid hemorrhage. Neurology 2003;60:208–14.
25. Claassen J, Perotte A, Albers D, et al. Nonconvulsive seizures after subarachnoid hemorrhage: Multimodal detection and outcomes. Ann Neurol 2013;74:53–64.
26. Lindgren C, Nordh E, Naredi S, Olivecrona M. Frequency of non-convulsive seizures and non-convulsive status epilepticus in subarachnoid hemorrhage patients in need of controlled ventilation and sedation. Neurocrit Care 2012;17:367–73.
27. Cocito L, Audenino D, Primavera A. Altered mental state and nonconvulsive status epilepticus in patients with cancer. Arch Neurol 2001;58:1310.
28. Vespa PM, Nuwer MR, Nenov V, et al. Increased incidence and impact of nonconvulsive and convulsive seizures after traumatic brain injury as detected by continuous electroencephalographic monitoring. J Neurosurg 1999;91:750–60.
29. Tian L, Li Y, Xue X, et al. Super-refractory status epilepticus in West China. Acta Neurol Scand 2015;132:1–6.
30. Holtkamp M, Othman J, Buchheim K, et al. A “malignant” variant of status epilepticus. Arch Neurol 2005;62:1428–31.
31. Lowenstein DH, Alldredge BK. Status epilepticus at an urban public hospital in the 1980s. Neurology 1993;43:483–8.
32. Mayer SA, Claassen J, Lokin J, et al. Refractory status epilepticus: frequency, risk factors, and impact on outcome. Arch Neurol 2002;59:205–10.
33. Rossetti AO, Logroscino G, Bromfield EB. Refractory status epilepticus: effect of treatment aggressiveness on prognosis. Arch Neurol 2005;62:1698–702.
34. Novy J, Logroscino G, Rossetti AO. Refractory status epilepticus: a prospective observational study. Epilepsia 2010;51:251–6.
35. Garzon E, Fernandes RM, Sakamoto AC. Analysis of clinical characteristics and risk factors for mortality in human status epilepticus. Seizure 2003;12:337–45.
36. Sutter R, Kaplan PW, Marsch S, et al. Early predictors of refractory status epilepticus: an international two-center study. Eur J Neurol 2015;22:79–85.
37. Wijdicks EF, Sharbrough FW. New-onset seizures in critically ill patients. Neurology 1993;43:1042–4.
38. Bleck TP, Smith MD, Pierre-Louis SJ, et al. Neurologic complications of critical medical illnesses. Crit Care Med 1993;21:98–103.
39. Oddo M, Carrera E, Claassen J, et al. Continuous electroencephalography in the medical intensive care unit. Crit Care Med 2009;37:2051–6.
40. Gilmore EJ, Gaspard N, Choi HA, et al. Acute brain failure in severe sepsis: a prospective study in the medical intensive care unit utilizing continuous EEG monitoring. Intensive Care Med 2015;41:686–94.
41. Misra UK, Kalita J, Patel, R. Sodium valproate vs phenytoin in status epilepticus: a pilot study. Neurology 2006;67:340–2.
42. Kim A, Kim JE, Paek YM, et al. Cefepime-induced non-convulsive status epilepticus (NCSE). J Epilepsy Res 2013;3:39–41.
43. Naeije G, Lorent S, Vincent JL, Legros B. Continuous epileptiform discharges in patients treated with cefepime or meropenem. Arch Neurol 2011;68:1303–7.
44. Tan RY, Neligan A, Shorvon SD. The uncommon causes of status epilepticus: a systematic review. Epilepsy Res 2010;91:111–22.
45. Khawaja AM, DeWolfe JL, Miller DW, Szaflarski JP. New-onset refractory status epilepticus (NORSE) - The potential role for immunotherapy. Epilepsy Behav 2015;47:17–23.
46. Davis R, Dalmau J. Autoimmunity, seizures, and status epilepticus. Epilepsia 2013;54 Suppl 6:46–9.
47. Lopinto-Khoury C, Sperling MR. Autoimmune status epilepticus. Curr Treat Options Neurol 2013;15:545–56.
48. Bansal P, Zutshi D, Suchdev K, et al. Alpha 3 ganglionic acetylcholine receptor antibody associated refractory status epilepticus. Seizure 2016;35:1–3.
49. Chen JW, Wasterlain CG. Status epilepticus: pathophysiology and management in adults. Lancet Neurol 2006;5:246–56.
50. DeLorenzo RJ, Garnett LK, Towne AR, et al. Comparison of status epilepticus with prolonged seizure episodes lasting from 10 to 29 minutes. Epilepsia 1999;40:164–9.
51. Lowenstein DH, Alldredge BK. Status epilepticus. N Engl J Med 1998;338:970–6.
52. Millikan D, Rice B, Silbergleit R. Emergency treatment of status epilepticus: current thinking. Emerg Med Clin North Am 2009;27:101–13, ix.
53. Fountain NB. Status epilepticus: risk factors and complications. Epilepsia 2000;41 Suppl 2:S23–30.
54. Aminoff MJ, Simon RP. Status epilepticus. Causes, clinical features and consequences in 98 patients. Am J Med 1980;69:657–66.
55. Meldrum BS, Brierley JB. Prolonged epileptic seizures in primates. Ischemic cell change and its relation to ictal physiological events. Arch Neurol 1973;28:10–7.
56. Meldrum BS, Vigouroux RA, Brierley JB. Systemic factors and epileptic brain damage. Prolonged seizures in paralyzed, artificially ventilated baboons. Arch Neurol 1973;29:82–7.
57. Loscher W. Mechanisms of drug resistance in status epilepticus. Epilepsia 2007;48 Suppl 8:74–7.
58. Jacob TC, Moss SJ, Jurd R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat Rev Neurosci 2008;9:331–43.
59. Arancibia-Carcamo IL, Kittler JT. Regulation of GABA(A) receptor membrane trafficking and synaptic localization. Pharmacol Ther 2009;123:17–31.
60. Bohnsack JP, Carlson SL, Morrow AL. Differential regulation of synaptic and extrasynaptic alpha4 GABA(A) receptor populations by protein kinase A and protein kinase C in cultured cortical neurons. Neuropharmacology 2016;105:124–32.
61. Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci 2005;6:215–29.
62. Smith KR, Kittler JT. The cell biology of synaptic inhibition in health and disease. Curr Opin Neurobiol 2010;20:550–6.
63. Wasterlain CG, Naylor DE, Liu H, et al. Trafficking of NMDA receptors during status epilepticus: therapeutic implications. Epilepsia 2013;54 Suppl 6:78–80.
64. Bankstahl JP, Loscher W. Resistance to antiepileptic drugs and expression of P-glycoprotein in two rat models of status epilepticus. Epilepsy Res 2008;82:70–85.
65. Henshall DC, Diaz-Hernandez M, Miras-Portugal MT, Engel T. P2X receptors as targets for the treatment of status epilepticus. Front Cell Neurosci 2013;7:237.
66. Lamsa K, Taira T. Use-dependent shift from inhibitory to excitatory GABAA receptor action in SP-O interneurons in the rat hippocampal CA3 area. J Neurophysiol 2003;90:1983–95.
67. Cock HR, Tong X, Hargreaves IP, et al. Mitochondrial dysfunction associated with neuronal death following status epilepticus in rat. Epilepsy Res 2002;48:157–68.
68. Friedman A, Dingledine R, Molecular cascades that mediate the influence of inflammation on epilepsy. Epilepsia 2011;52 Suppl 3:33–39.
69. Henshall DC. MicroRNAs in the pathophysiology and treatment of status epilepticus. Front Mol Neurosci 2013;6:37.
70. Vespa P, Tubi M, Claassen J, et al. Metabolic crisis occurs with seizures and periodic discharges after brain trauma. Ann Neurol 2016;79:579–90.
71. Witsch J, Frey HP, Schmidt JM, et al. Electroencephalographic periodic discharges and frequency-dependent brain tissue hypoxia in acute brain injury. JAMA Neurol 2017;74:301–9.
72. Rodriguez Ruiz A, Vlachy J, Lee JW, et al. Association of periodic and rhythmic electroencephalographic patterns with seizures in critically ill patients. JAMA Neurol 2017;74:181–8.
73. DeLorenzo RJ, Waterhouse EJ, Towne AR, et al. Persistent nonconvulsive status epilepticus after the control of convulsive status epilepticus. Epilepsia 1998;39:833–40.
74. Benbadis SR, Chen S, Melo M. What’s shaking in the ICU? The differential diagnosis of seizures in the intensive care setting. Epilepsia 2010;51:2338–40.
75. Pandian JD, Cascino GD, So EL, et al. Digital video-electroencephalographic monitoring in the neurological-neurosurgical intensive care unit: clinical features and outcome. Arch Neurol 2004;61:1090–4.
76. Irani SR, Vincent A, Schott JM. Autoimmune encephalitis. BMJ 2011;342:d1918.
77. Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011;69:892–900.
78. Herman ST, Abend NS, Bleck TP, et al. Consensus statement on continuous EEG in critically ill adults and children, part I: indications. J Clin Neurophysiol 2015;32:87–95.
79. Brenner RP. Is it status? Epilepsia 2002;43 Suppl 3:103–113.
80. Cook AM, Castle A, Green A, et al. Practice variations in the management of status epilepticus. Neurocrit Care 2012;17:24–30.
81. Alldredge BK, Gelb AM, Isaacs SM, et al. A comparison of lorazepam, diazepam, and placebo for the treatment of out-of-hospital status epilepticus. N Engl J Med 2001;345:631–7.
82. Silbergleit R, Durkalski V, Lowenstein D, et al. Intramuscular versus intravenous therapy for prehospital status epilepticus. N Engl J Med 2012;366:591–600.
83. Shorvon S. Super-refractory status epilepticus: an approach to therapy in this difficult clinical situation. Epilepsia 2011;52 Suppl 8:53–6.
84. Varelas P, Mirski MA. Management of status epilepticus in adults. Hosp Physician Board Rev Man 2014;2:1–15.
85. McIntyre J, Robertson S, Norris E, et al. Safety and efficacy of buccal midazolam versus rectal diazepam for emergency treatment of seizures in children: a randomised controlled trial. Lancet 2005;366:205–10.
86. Misra UK, Kalita J, Maurya PK. Levetiracetam versus lorazepam in status epilepticus: a randomized, open labeled pilot study. J Neurol 2012;259:645–8.
87. Vohra TT, Miller JB, Nicholas KS, et al. Endotracheal intubation in patients treated for prehospital status epilepticus. Neurocrit Care 2015;23:33–43.
88. Agarwal P, Kumar N, Chandra R, et al. Randomized study of intravenous valproate and phenytoin in status epilepticus. Seizure 2007;16:527–32.
89. Trinka E. What is the evidence to use new intravenous AEDs in status epilepticus? Epilepsia 2011;52 Suppl 8:35–38.
90. Yasiry Z, Shorvon SD. The relative effectiveness of five antiepileptic drugs in treatment of benzodiazepine-resistant convulsive status epilepticus: a meta-analysis of published studies. Seizure 2014;23:167–74.
91. Moreno Morales EY, Fernandez Peleteiro M, Bondy Pena EC, et al. Observational study of intravenous lacosamide in patients with convulsive versus non-convulsive status epilepticus. Clin Drug Investig 2015;35:463–9.
92. Paquette V, Culley C, Greanya ED, Ensom MH. Lacosamide as adjunctive therapy in refractory epilepsy in adults: a systematic review. Seizure 2015;25:1–17.
93. Varelas PN, Corry J, Rehman M, et al. Management of status epilepticus in neurological versus medical intensive care unit: does it matter? Neurocrit Care 2013;19:4–9.
94. Hofler J, Trinka E. Lacosamide as a new treatment option in status epilepticus. Epilepsia 2013;54:393–404.
95. Kellinghaus C, Berning S, Stogbauer F. Intravenous lacosamide or phenytoin for treatment of refractory status epilepticus. Acta Neurol Scand 2014;129:294–9.
96. Santamarina E, Toledo M, Sueiras M, et al. Usefulness of intravenous lacosamide in status epilepticus. J Neurol 2013;260:3122–8.
97. Newey CR, Le NM, Ahrens C, et al. The safety and effectiveness of intravenous lacosamide for refractory status epilepticus in the critically ill. Neurocrit Care 2017;26:273–9.
98. Towne AR, Garnett LK, Waterhouse EJ, et al. The use of topiramate in refractory status epilepticus. Neurology 2003;60:332–4.
99. Hottinger A, Sutter R, Marsch S, Ruegg S. Topiramate as an adjunctive treatment in patients with refractory status epilepticus: an observational cohort study. CNS Drugs 2012;26:761–72.
100. Madzar D, Kuramatsu JB, Gerner ST, et al. Assessing the value of topiramate in refractory status epilepticus. Seizure 2016;38:7–10.
101. Sivakumar S, Ibrahim M, Parker D Jr, et al. An effective add-on therapy in refractory status epilepticus. Epilepsia 2015;56:e83–89.
102. Madzar D, Geyer A, Knappe RU, et al. Effects of clobazam for treatment of refractory status epilepticus. BMC Neurol 2016;16:202.
103. Swisher CB, Doreswamy M, Gingrich KJ, et al. Phenytoin, levetiracetam, and pregabalin in the acute management of refractory status epilepticus in patients with brain tumors. Neurocrit Care 2012;16:109–13.
104. Smith H, Sinson G, Varelas P. Vasopressors and propofol infusion syndrome in severe head trauma. Neurocrit Care 2009;10:166–72.
105. Cuero MR, Varelas PN. Super-refractory status epilepticus. Curr Neurol Neurosci Rep 2015;15:74.
106. Varelas PN. How I treat status epilepticus in the Neuro-ICU. Neurocrit Care 2008;9:153–7.
107. Varelas PN, Spanaki MV, Mirski MA. Status epilepticus: an update. Curr Neurol Neurosci Rep 2013;13:357.
108. Krishnamurthy KB, Drislane FW. Depth of EEG suppression and outcome in barbiturate anesthetic treatment for refractory status epilepticus. Epilepsia 1999;40:759–62.
109. Krishnamurthy KB, Drislane FW. Relapse and survival after barbiturate anesthetic treatment of refractory status epilepticus. Epilepsia 1996;37:863–7.
110. Ferlisi M, Shorvon S. The outcome of therapies in refractory and super-refractory convulsive status epilepticus and recommendations for therapy. Brain 2012;135:2314–28.
111. Kramer AH. Early ketamine to treat refractory status epilepticus. Neurocrit Care 2012;16:299–305.
112. Synowiec AS, Singh DS, Yenugadhati V, et al. Ketamine use in the treatment of refractory status epilepticus. Epilepsy Res 2013;105:183–8.
113. Gaspard N, Foreman B, Judd LM, et al. Intravenous ketamine for the treatment of refractory status epilepticus: a retrospective multicenter study. Epilepsia 2013;54:1498–503.
114. Schulze-Bonhage A, Kurthen M, Walger P, Elger CE. Pharmacorefractory status epilepticus due to low vitamin B6 levels during pregnancy. Epilepsia 2004;45:81–4.
115. Shorvon S. Clinical trials in acute repetitive seizures and status epilepticus. Epileptic Disord 2012;14:138–47.
116. Visser NA, Braun KP, Leijten FS, et al. Magnesium treatment for patients with refractory status epilepticus due to POLG1-mutations. J Neurol 2011;258:218–22.
117. Thakur KT, Probasco JC, Hocker SE, et al. Ketogenic diet for adults in super-refractory status epilepticus. Neurology 2014;82:665–70.
118. Corry JJ, Dhar R, Murphy T, Diringer MN. Hypothermia for refractory status epilepticus. Neurocrit Care 2008;9:189–97.
119. Guilliams K, Rosen M, Buttram S, et al. Hypothermia for pediatric refractory status epilepticus. Epilepsia 2013;54:1586–94.
120. Legriel S, Lemiale V, Schenck M, et al. Hypothermia for neuroprotection in convulsive status epilepticus. N Engl J Med 2016;375:2457–67.
121. Wasterlain CG, Baldwin R, Naylor DE, et al. Rational polytherapy in the treatment of acute seizures and status epilepticus. Epilepsia 2011;52 Suppl 8:70–1.
122. Rogawski MA, Loya CM, Reddy K, et al. Neuroactive steroids for the treatment of status epilepticus. Epilepsia 2013;54 Suppl 6:93–8.
123. Towne AR, Pellock JM, Ko D, DeLorenzo RJ. Determinants of mortality in status epilepticus. Epilepsia 1994;35:27–34.
124. DeLorenzo RJ, Towne AR, Pellock JM, Ko D. Status epilepticus in children, adults, and the elderly. Epilepsia 1992;33 Suppl 4:S15–25.
125. Legriel S, Mourvillier B, Bele N, et al. Outcomes in 140 critically ill patients with status epilepticus. Intensive Care Med 2008;34:476–80.
126. Pugin D, Foreman B, De Marchis GM, et al. Is pentobarbital safe and efficacious in the treatment of super-refractory status epilepticus: a cohort study. Crit Care 2014;18:R103.
127. Hocker SE, Britton JW, Mandrekar JN, et al. Predictors of outcome in refractory status epilepticus. JAMA Neurol 2013;70:72–7.
128. Hocker S, Tatum WO, LaRoche S, Freeman WD. Refractory and super-refractory status epilepticus--an update. Curr Neurol Neurosci Rep 2014;14:452.
129. Sutter R, Marsch S, Fuhr P, et al. Anesthetic drugs in status epilepticus: risk or rescue? A 6-year cohort study. Neurology 2014;82:656–64.
130. Rossetti AO, Logroscino G, Bromfield EB. A clinical score for prognosis of status epilepticus in adults. Neurology 2006;66:1736–8.
131. Rossetti AO, Logroscino G, Milligan TA, et al. Status Epilepticus Severity Score (STESS): a tool to orient early treatment strategy. J Neurol 2008;255:1561–6.
132. Hesdorffer DC, Logroscino G, Cascino GD, Hauser WA. Recurrence of afebrile status epilepticus in a population-based study in Rochester, Minnesota. Neurology 2007;69:73–8.
133. Varelas PN, Mirski MA. Seizures in the adult intensive care unit. J Neurosurg Anesthesiol 2001;13:163–75.
134. Varelas PN, Mirski MA. Status epilepticus. Curr Neurol Neurosci Rep 2009;9:469–76.
From the Johns Hopkins Hospital, Baltimore, MD (Dr. Ramadan), and the Henry Ford Hospital, Detroit, MI (Dr. Varelas).
Abstract
- Objective: To review the management of status epilepticus (SE).
- Methods: Review of the literature.
- Results: SE is a relatively common condition that accounts for 3% to 5% of all emergency department evaluations for seizure disorders and occurs in 2% to 16% of all epilepsy patients. The 3 most common etiologies are low levels of antiepileptic drugs, remote symptomatic etiologies, and cerebrovascular accidents. The majority of SEs are convulsant, but there is growing awareness of non-convulsive SEs, which can be diagnosed only via electroencephalogram. Management, which must be initiated at the earliest possible time, has evolved to incorporate pre-hospital measures and 4 treatment stages, with supportive measures and benzodiazepine administration remaining the mainstay initially and followed by older and newer antiepileptic drugs and anesthetics for resistant cases.
- Conclusion: SE is a neurological emergency that still carries significant mortality and morbidity if not treated immediately and properly.
Key words: status epilepticus; seizures; convulsive status epilepticus; nonconvulsive status epilepticus.
Status epilepticus (SE) is a relatively common condition that accounts for 3% to 5% of all emergency department (ED) evaluations for seizure disorders and occurs in 2% to 16% of all epilepsy patients [1]. It remains a major neurological emergency that, if not properly and timely treated, leads to death or permanent neurological injury. Since most of patients with convulsive SE are admitted to the hospital via the ED and are then transferred to the intensive care unit (ICU), our focus in this review will be on the latter.
Although only a handful prospective, randomized studies have been reported, guidelines on SE have been published in Europe [2] and the US [3,4]. In this paper, we review the evolving definition and types of SE, its incidence, etiology, and pathophysiology, its diagnosis and treatment algorithms, and its outcome. Our goal is to provide the reader with a concise but thorough review of this still lethal neurological emergency.
Definitions
The International Classification of Epileptic Seizures had previously defined SE as any seizure lasting ≥ 30 minutes or intermittent seizures lasting for > 30 min without recovery of consciousness interictally [5,6]. More recently, a duration of 5 or more minutes of (a) continuous seizures or (b) 2 or more discrete seizures with incomplete recovery of consciousness in-between, proposed by Lowenstein [3,7], offers the advantage of incorporating new knowledge. The shortening of the convulsive period to 5 minutes was based on the fact that the majority of tonic-clonic seizures last for only 1 to 2 minutes, that those lasting > 5 minutes do not stop spontaneously [8], that permanent neuronal injury occurs before 30 minutes, and that refractoriness to treatment increases with longer seizure duration [9].
Refractory SE (RSE) has been defined as SE not controlled after adequate doses of an initial benzodiazepine followed by a second acceptable antiepileptic drug (AED) or SE not controlled after the initial parenteral therapy with a minimum number of standard “front-line” AEDs (either 2 or 3) or SE with a minimum duration of seizures that persist despite treatment (eg, at least or 2 hours) [3,10]. Super-refractory SE (SRSE) is defined as SE that continues or recurs 24 hours or more after the onset of anesthetic therapy or recurs on the reduction or withdrawal of anesthesia [11].
Non-convulsive SE (NCSE) is defined as the presence of altered consciousness or behavior for ≥ 30 minutes, the absence of overt clinical signs of convulsive activity during that period, and the electroencephalographic (EEG) confirmation of seizures or activity that responds to treatment together with improvement of consciousness [12–15]. Two major types of NCSE can be encountered: the one in patients with epileptic encephalopathy/coma and the one in patients with absence or complex partial seizures, who are not usually admitted to ICU and are functional yet impaired. Because of the confusion between these 2 extremes in the NCSE spectrum, working criteria for standardization of reporting, utilizing the frequency of electroencephalographic epileptiform discharges or delta/theta waveforms have been proposed [15]. A recent compendium of 123 cases of NCSE with clinical descriptions and EEG patterns following a syndromic classification approach has also been published [16].
Types of SE
Three major categories of SE have been described: generalized convulsive SE (GCSE), focal motor SE (FMSE or epilepsia partialis continua [EPC]) of Kojevnikov, and NCSE. GCSE and FMSE are easily recognized due to overt convulsions. NCSE, however, has a more obscure phenotype and can be subdivided into a spectrum encompassing typical absence and complex partial SE, atypical absence SE and tonic SE (usually in children with learning disabilities), epileptic behavioral disturbance and psychosis, including Balint–like syndrome [17], confusional states or delirium with epileptiform discharges) and SE in coma (after significant brain injuries, such as hypoxia-ischemia, most commonly encountered in ICUs) [13,18]. The 2 extremes in this NCSE spectrum have completely different prognoses, with absence SE the most benign and SE in coma the most dismal.
Lastly, SE presents either spontaneously or can be “semi-intentional” iatrogenic, encountered either in the neuro-ICU or epilepsy monitoring unit, when AEDs are withdrawn under continuous EEG recording in order for seizures to emerge and be recorded with surface or intracranial electrodes.
Incidence of SE
In a prospective population-based epidemiological study, the incidence of SE was estimated at 41–61/100,000 patients/year. For the US, this translates to 125,000 to 195,000 episodes per year [19].
The highest incidence of SE occurs during the first year of life and during the decades beyond 60 years, and is also dependent on the SE subtype. Partial SE occurs in 25% of cases of SE and NCSE accounts for another 4% to 26 % [19,20], but the incidence for the latter is considered an underestimate due to the need for continuous EEG monitoring (which is not widely available). For example, NCSE was discovered in no patient with acute stroke [21], 8% of comatose ICU patients [22], 7% of patients with intracerebral hemorrhage [23], 3% to 8% of patients with subarachnoid hemorrhage [24–26], 6% of patients with metastatic cancer [27], and 6% of patients with head trauma [28].
The incidence of RSE and SRSE is also unknown. In a recent retrospective study from a neuro-ICU in a West China hospital, the percentage of non-refractory SE, RSE, and SRSE were 67.3%, 20.4% and 12.2%, respectively [29]. Other retrospective studies have shown that 12% to 43% of SE cases become refractory [30–33] and that approximately 10% to 15% of all cases of hospital-admitted SE will become super-refractory at some point, but no prospective studies have been published.
Risk factors that have been identified for RSE are encephalitis as a cause, severe consciousness impairment, de novo episodes of SE, delay in initiation of treatment, NCSE, and focal motor seizures at onset [30,32,34,35]. In a more recent study from ICU patients in Switzerland and the US, acute SE etiology (traumatic brain injuries, cerebrovascular accidents, meningoencephalitis, brain tumors, surgical brain lesions, exposure to, or withdrawal from, recreational drugs, prescription drugs, alcohol, metabolic disturbances and fever), coma/stupor, and serum albumin < 35 g/L at SE onset were independent predictors for RSE [36].
Etiology of SE
The 3 most common etiologies for SE are low levels of antiepileptic drugs (AEDs) in 34% of the cases (usually due to noncompliance), remote symptomatic etiologies (history of neurological insults remote to the first unprovoked SE episode, 24%), and cerebrovascular accidents (ischemic and hemorrhagic strokes, 22%). These are followed by hypoxia (13%) and metabolic disturbances (15%). Because 82% of patients in the remote group have a history of cerebrovascular disease, almost 50% have either acute or remote cerebrovascular disease as etiology of SE [19].
In general ICUs, metabolic abnormalities can account for 33% of seizures, drug withdrawal for 33%, drug toxicity for 14.5%, and stroke for 9% to 39% [37,38]. In ICUs, sepsis remains a common etiology of electrographic seizures or periodic epileptiform discharges [39,40], and legal or illegal drugs, such as ciprofloxacin, levofloxacin, piperacillin/tazobactam, cefepime and carbapenems [41–43], lithium or theophylline intoxication, vigabatrin, tiagabine or crack/cocaine, are another [18] (especially when their metabolism is altered due to interactions with other drugs or when their excretion is impaired due to hepatic or renal failure).
Beyond these common causes of SE, a workup for rare etiologies should be entertained. In a systematic review of 513 papers on SE, 181 uncommon causes of SE were identified and subdivided into immunologically mediated disorders, mitochondrial diseases, rare infectious disorders, genetic disorders, and drugs or toxins [18,44].
The most recent knowledge in this category is the contribution of paraneoplastic or autoimmune conditions to a large percentage of previously cryptogenic pharmaco-resistant seizures or super-refractory SE, most in the context of limbic encephalitis. Many of these patients have never experienced seizures or SE before and a new acronym has been devised for them: new-onset refractory status epilepticus (NORSE), ie, a state of persistent seizures with no identifiable etiology in patients without preexisting epilepsy that lasts longer than 24 hour despite optimal therapy [45]. A growing array of autoantibodies against intracellular and surface or synaptic neuronal targets has been described in addition to the previous literature of Rassmussen’s encephalitis and Hashimoto’s encephalopathy [46]. The most common autoantibodies associated with seizures and SE include anti-Hu, anti-Ma2, anti-CV2/CRMP5, anti-Ri, ANNA3, anti-amphiphysin, anti-NMDA receptor, anti-LGI1 and CASPR2, anti-GABA-beta, anti-GluR3, anti-mGluR5 and alpha 3 ganglionic acetylcholine receptor [47,48]. The diagnosis frequently remains elusive due to lack of knowledge or absence of widespread availability of serologic testing (with sometimes weeks-long delay for the results to be available), but the response to treatment with removal of tumor, plasmapheresis, or immunomodulation and immunosuppression is often dramatic.
Pathophysiology of SE
Most seizures are self-terminating phenomena lasting from a few seconds to a few minutes [49]. One of the distinguishing characteristics of seizures evolving into SE, however, is the switch to a self-sustaining situation, which is time-dependent. Seizures lasting more than 30 minutes would rarely stop spontaneously compared to 47% of those lasting between 10 to 29 minutes, which are self-resolving [50]. Moreover, in one study no self-limited seizure lasted more than 11 minutes [8].
The self-limiting character of seizures is due to inhibitory circuitry that suppresses their duration and propagation in the brain. Under specific circumstances, however, the inhibitory mechanisms fail and seizures progress to SE, which leads to synaptic reorganization, blood-brain barrier disruption, inflammation, metabolic crisis, more tissue damage, and further seizures. Neuronal injury during SE is the result of increased excitotoxicity [51–53] but also stems from systemic derangements such as hypoxia, acidosis, hypotension, or multiorgan dysfunction [54]. The seminal animal studies by Meldrum have shed a light on the systemic effects: after prolonged bicuculine-induced convulsive SE in baboons, neuronal damage and cell loss was evident in the neocortex, cerebellum and hippocampus. When systemic factors were kept within normal physiological limits (paralyzed and artificially ventilated animals with adequate serum glucose levels), there was decreased but still present neocortical and hippocampal cell damage, but absent cerebellar cell injury [55,56]. These experiments showed more than 40 years ago that the seizure activity per se is responsible for the neuronal damage and the systemic derangements play an additional role.
The direct neuronal injury as a result of the ongoing seizures, the perpetuation of seizures into SE, the resistance to treatment and the refractoriness that ensues have also been elucidated at a molecular level during the last decades. Initially, the g-aminobutyric acid (GABA) inhibitory circuits may be deficient and this is the reason why benzodiazepines or barbiturates, which work through GABAergic receptor agonism, are very effective during this early period. As time passes however, GABA receptors undergo a significant shift in their ability to respond to benzodiazepines [57,58]. This is due to changes in receptor presence at the inhibitory synapse, a phenomenon that has been called “receptor trafficking” by Arancibia and Kittler in 2009 [59]. There are differences in the type of GABAA receptors found synaptically and extrasynaptically. GABAA receptors containing the γ subunit are located synaptically and mediate phasic inhibition. Conversely, the δ subunit-containing GABAA receptors are located exclusively extrasynaptically and mediate tonic inhibition [60,61]. Smith and Kittler described the highly dynamic state of receptor presence on the surface of axons and explained how receptors move laterally from extrasynaptic sites to the synapse and then out of it to be internalized and either recycled to the surface or degraded [62]. This “receptor trafficking” intensifies during SE, and the overall effect becomes a reduction in the number of functional GABAA receptors in the synapses. As GABA is the principle inhib-itory transmitter, this reduction in GABAergic activity may be an important reason for seizures to become persistent.
However, this is not all. Additional mechanisms leading to refractoriness include the following:
(a) Excessive relocation of N-methyl-D-aspartate (NMDA)type glutamate receptors to the cell surface after 1 hour of SE, leading to increase of miniature excitatory NMDA currents and NMDA neurotransmission, with potentiation of glutamate excitotoxicity [53,63]
(b) Increased brain expression of drug efflux transporters, such as P-glycoprotein at the blood-brain barrier, which may reduce concentrations of AEDs at their brain targets [64]
(c) Up- and down-regulation of specific ATP-gated ion channels (P2X receptors) inducing altered response to ATP release [65]
(d) Change in the extracellular ionic environment (for example, the normally inhibitory GABAA receptor-mediated currents may become excitatory with changes in extracellular chloride concentrations) [66]
(e) Mitochondrial insufficiency or failure, which would lead to cell necrosis and apoptosis [67]
(f) Inflammatory processes, with opening of the blood-brain barrier (BBB) contributing to perpetuation of seizures [44]. The underlying mechanism is a maladaptive response of the astrocytes to the BBB damage, leading to activation of the innate immune system and disturbed homeostasis of the extracellular potassium and glutamate [68].
(g) Large-scale changes in gene expression within the affected brain regions; these are regulated by micro-RNAs, influencing protein levels playing a role in excitability, neuronal death and neuroinflammation [69].
All of these pathophysiologic derangements may become targets for future antiepileptic treatments.
Although the direct and indirect injury from ongoing convulsive SE is not in doubt, the significance of NCSE or the ictal-interictal continuum on inflicting additional injury has been more controversial. Recent data, however, do not support a benign process in these situations. It has been shown lately that nonconvulsive seizures lead to physiologic changes in the brain, including elevated intracranial pressure, changes in the brain metabolism, and delayed increase in cerebral blood flow [25]. In addition, using microdialysis, elevated lactate/puruvate ratio, indicating metabolic crisis, has been shown during periods of nonconvulsive seizures or periodic discharges [70]. Similarly, high-frequency periodic discharges lead to inadequate increase in cerebral blood flow and tissue hypoxia [71], and lateralized periodic discharges, lateralized rhythmic delta activity, and generalized periodic discharges are associated with seizures [72].
Diagnosis of SE
The diagnosis of SE is primarily clinical and encompasses motor phenomena and alteration of mental status. Focal-onset convulsions can remain focal, follow a Jacksonian march, or immediately generalize to involve the whole body with loss of consciousness. Most of the time, this secondary generalization can only be appreciated during EEG recording. In addition, mental status alteration can differentiate simple partial SE (no change in mental status) from complex partial SE (disturbed sensorium).
The presence or absence of motor phenomena and loss of consciousness do not necessarily correlate with the EEG activity during or after SE. For example, persistent electrographic seizures or NCSE after control of convulsive SE have been demonstrated with continuous EEG [73]. Conversely, altered mental status is also a poor clinical differentiator, since 87% of patients successfully treated for convulsive SE and 100% treated for NCSE remained comatose 12 hours following the initiation of therapy [20]. In addition, only 27% of motor, seizure-like phenomena in the ICU were proven to be seizures in a retrospective study [74]. Psychogenic nonepileptic attacks, occurring in between 3.8% and 9.5% of ICU patients presenting with seizures [74,75], is another situation that may lead to confusion, inappropriate intubation, and ICU admission. Strange phenomena, such as fasciobrachial seizures (brief facial grimacing and ipsilateral arm posturing) many times preceding the onset of amnesia, confusion, or temporal lobe seizures have been described in patients who have non-paraneoplastic limbic encephalitis associated with voltage-gated potassium channel (VGKC) antibodies, especially against the leucine-rich glioma inactivated-1 (LGI1) protein [76,77].Without a continuous video EEG, these phenomena may not be captured or appreciated. Therefore, EEG monitoring is an important tool for the evaluation of these patients and criteria for its use have been published [78]. The EEG criteria for convulsive SE have been clearly delineated, but for NCSE a mix of clinical and EEG criteria should be met [14,15,79].
In addition to clinical observation and EEG, there has been interest lately in multimodality monitoring of acutely brain-injured patients for seizures or SE using electrocorticography or mini depth electrode placement, partial brain tissue oxygen tension, cerebral blood flow, and microdialysis in addition to scalp EEG. Although preliminary and limited in few academic centers, this approach has produced interesting findings. For example, in a study from Columbia University, 38% of 48 patients with subarachnoid hemorrhage and multimodality monitoring had intracortical seizures, while only 8% of them had surface seizures, all nonconvulsive [25]. In another study, 68% of seizures and 23% of periodic discharges were only captured on the depth electrodes and were missed on the surface ones [71]. Therefore, detection of SE may change in the future with use of more sensitive techniques than scalp EEG.
Treatment
Significant practice variations exist in the management of SE even among academic centers in the US [80] despite the fact that the goals of treatment are concrete. These include (1) emergent medical management, (2) termination of seizures, (3) prevention of recurrence of seizures, and (4) prevention or treatment of complications.
Management of SE must begin in a prehospital setting by the emergency medical services, because the faster the treatment is offered, the better the response. Several studies have attempted to assess the possibility of aborting SE even prior to the hospital. In a randomized, double-blinded study, lorazepam was 4.8 times and diazepam 2.3 times more effective than placebo in terminating SE on arrival in the ED when given intravenously (IV) by paramedics [81]. The RAMPART study was a double-blind, randomized, non-inferiority trial comparing the efficacy of intramuscular (IM) midazolam (10 mg followed by placebo IV) with that of IM placebo followed by intravenous lorazepam (4 mg) for children and adults in SE treated by paramedics. At the time of arrival in the ED, seizures had ceased without rescue therapy in 73.4% and 63.4%, respectively, favoring midazolam [82].
Emergent Initial Phase
During the emergent initial phase, the goals are protection of the airway, oxygenation, maintenance of blood pressure, exclusion of easily
Urgent Control
If seizures continue, stage 2 medications should be used for benzodiazepine-refractory SE as urgent control treatment. There are some data suggesting better response rate to valproate after failure to control seizures with phenytoin than to phenytoin after failure of valproate [88]. If available, IV fosphenytoin is preferable to IV phenytoin due to potentially lower risk of side effects. Levetiracetam and phenobarbital IV are also acceptable choices. Levetiracetam can be administered as an off-label loading dose of 20–60 mg/kg IV (although the initial manufacturer was not supporting a “loading” dose; dose of up to 60 mg/kg IV up to 4500 mg maximum has been supported by the latest American Epilepsy Society guidelines [4]). This AED at an initial dose of 2–3 g/day confers an estimated success rate around 70% [89]. In a systematic review of 27 studies (798 cases of convulsive SE) comparing 5 AEDs in the treatment of benzodiazepine-resistant convulsive SE, phenobarbital and valproate had the highest efficacy (73.6% and 75.7%, respectively), followed by levetiracetam (68.5%) and phenytoin (50.2%). Lacosamide studies were excluded from the meta-analysis due to insufficient data [90], but its efficacy has been reported for patients with convulsive and NCSE [91,92]. There is not enough evidence at this point, however, to recommend its routine use for benzodiazepine refractory SE [90].
Refractory SE
When seizures continue despite the use of benzodiazepines and 2nd stage AEDs, SE becomes refractory (stage 3). Treatment of these resistant cases is frequently initiated in the ED and continued in an ICU. Outcomes were not significantly better in patients with SE admitted and managed in a neuro-ICU compared to a general medical ICU in a retrospective study, but the numbers were small (only 27% of SE were admitted to the former) [93] and this may change in the future. Intubation and mechanical ventilation is the first step, if not already present (only 21% of patients in the RAMPART study received endotracheal intubation, with 6.4% in the prehospital setting and 93.6% after admission [87]). Hemodynamic support with pressors or inotropes may be required as most anesthetic agents may decrease the blood pressure. Because of the urgency of controlling the seizures during SE, the potential aspiration risk and the questionable enteral absorption per os administration of additional AEDs is problematic, and IV formulations should be used.
Currently in the US, phenytoin, valproic acid, phenobarbital, levetiracetam, lacosamide, diazepam, lorazepam are available in IV formulations. In February 2016, the FDA also approved brivaracetam (which also is available in an IV formulation) and in October of the same year IV carbamazepine. None of these AEDs has an FDA indication for SE, although they are widely used. Parenteral lacosamide has a success rate of 33% to 67.7% (200–400 mg over 3–5 min was the most common bolus dose) depending on its use as second or third AED [94–96]. In lacosamide-naive patients with RSE on continuous EEG monitoring, the success rate for cessation of SE was 15.7, 25.5, 58.8, and 82.4 % by 4, 12, 24, and 48 hours, respectively [97]. Alternatively, topiramate in doses 300–1600 mg/day per oro/nasogastric tube can be considered [98]. In a study of 35 patients with RSE treated with topiramate as an adjunct AED, the response rate was 86% (as the third AED), and remained stable at 67% after administration as the fourth to seventh AED. Overall, RSE was terminated in 71% of patients within 72 hours after first administration of topiramate [99]. Other studies, however, adjusting for co-variates, did not prove topiramate to be effective in RSE [100]. Clobazam, a unique oral 1,5-benzodiazepine with excellent absorption, has been also used in the treatment of RSE. Seventeen patients with RSE (11 with prior epilepsy) were successfully treated with clobazam, which was introduced after a median duration of 4 days and after a median of 3 failed AEDs. Termination of RSE within 24 hours of administration, without addition or modification of concurrent AED and with successful wean of anesthetic infusions, was seen in 13 patients, whereas indeterminate response was seen in another 3. Clobazam was deemed unsuccessful in 1 patient [101]. In another recent report of 70 episodes of RSE, clobazam was used in 24 (34.3%) of them. If clobazam was the last AED added to therapy before RSE termination, the success was attributed to this drug. Based on this definition, clobazam led to 6 episodes (25%) of successful RSE resolution [102]. If primary or metastatic brain tumor is the presumed cause of SE, a combination of IV phenytoin, IV levetiracetam (median dose 3 g/d) and enterically administered pregabalin (median dose 375 mg/day) led to 70% control of SE on average 24 hours after addition of the third AED [103]. However, the major treatment options, which should not be delayed in unresponsive RSE, are propofol or midazolam infusions at high rates and under continuous EEG monitoring. These infusions should be continued for at least 24 hours and then held to reassess the situation. By that time, cocurrent metabolic derangements and low AED levels from noncompliance should have been corrected. Prolonged and high-dose propofol should be avoided because of the risk for propofol infusion syndrome, especially if pressors/inotropes are co-infused [104].
Super-refractory SE
Should seizures continue or recur, stage 4 options for SRSE are considered [105]. Pentobarbital with shorter half-life is favored to phenobarbital. The main disadvantages of barbiturates are compromised neuro-exam (which has to be assessed frequently), cardiovascular depression and hypotension, respiratory depression with need for full ventilator support, cough suppression with increased risk for atelectasis and pneumonia, immunosuppression increasing the risk for infection or sepsis, immobility increasing the risk for thromboembolism and ileus mandating parenteral nutrition [106,107]. The depth and duration of the EEG suppression that must be achieved by barbiturates is unknown. Some experts recommend instead of burst-suppression pattern complete suppression or “flat record” because of better seizure control and fewer relapses [108]. Moreover, patients with more prolonged barbiturate treatment (> 96 hours) and those receiving phenobarbital at the time of pentobarbital taper are less likely to relapse [109]. European guidelines recommend titration of propofol and barbiturate to EEG burst-suppression, and midazolam to seizure suppression, maintained for at least 24 h [2]. In recent reviews, it was found that barbiturates control refractory and super-refractory SE in 64% of patients and are ineffective in only 5% [11,110].
If SE continues or recurs after emergence from barbiturate coma, ketamine may be an option [11,83]. Ketamine offers the advantage of NMDA receptor antagonism, which may be important in the late phase of SE and lacks cardiodepressant or hypotensive properties. Early [111] or late [112] use of ketamine has been reported in small case series with various success rates. In a recent multicenter retrospective study from North America and Europe, evaluating 58 patients with 60 RSE, ketamine was likely responsible for seizure control in 12% and possibly responsible in an additional 20%. No responses were observed when infusion rate was lower than 0.9 mg/kg/h or when ketamine was introduced 8 days or more after onset of SE or after failure of seven or more drugs [113].
If all these measures have failed, stage 4.2 treatment options are available (Table 2), but these are mostly based on small case series and expert opinions (except for the recent hypothermia study). Pyridoxine hydrochloride in an IV or enteral form at a dose of 100–300 mg/day for few days can be used in stage 4 or earlier stages, as it is a cofactor in the synthesis of the inhibitory neurotransmitter GABA [114]. There are no strong data for its effectiveness, but it can be used as a cheap and safe alternative [115]. Magnesium has been successfully used in 2 girls with juvenile Alper’s syndrome [116] and is the treatment of choice for eclamptic seizures. Ketogenic diet may also be an optionfor these patients [117]. Resection of the epileptic focus after mapping with intracranial EEG electrodes may be highly effective but cannot be used in many patients due to lack of focus or eloquence location [83,106,115]. Use of steroids, plasmapheresis or IVIG, followed by immunosuppression can be tried, but one should balance risks and benefits. These immunosuppressive or immunomodulating treatments should be especially considered in patients with NORSE or suspected autoimmune or paraneoplastic encephalitides, where AEDs usually have no effect [46]. These therapies though often precede the diagnosis, since it takes time for the autoantibody panel results to return and the treating physician has to make a decision to blindly start treatment for SRSE.
There were some promising data regarding hypothermia use in these desperate situations [118,119] until the HYBERNATUS study, conducted in France, was recently published. In this study, 270 patients with convulsive SE were randomized in to hypothermia (32° to 34°C for 24 hours) in addition to standard care or to standard care alone. A Glasgow Outcome Scale score of 5 (primary outcome) occurred in 49% of patients in the hypothermia group and in 43% in the control group (a nonstatistical difference). Secondary outcomes, including mortality at 90 days, RSE on day 1, SRSE and functional sequelae on day 90 were not different except for the rate of progression to EEG-confirmed SE on the first day, which was lower in the hypothermia group (11% vs. 22% in the controls). Adverse events were more frequent in the hypothermia group than in the control group [120].
Additional anecdotal treatments are presented in Table 2, but their efficacy is questionable.
This staged management approach may change in the future to a more physiologic and rational treatment with polytherapy based on synaptic receptor trafficking during SE [63]. For example, in an animal model of severe SE, combinations of a benzodiazepine with ketamine and valproate, or with ketamine and brivaracetam, were more effective and less toxic than benzodiazepine monotherapy [121]. Allopregnalonone, a metabolite of progesterone, is an endogenous, naturally occurring neuroactive steroid produced in the ovary, the adrenal cortex and the central nervous system. It is a potent positive allosteric modulator of synaptic and extrasynaptic GABAA receptors with antiepileptic activity [122]. Neuroactive steroids, such as allopregnanolone, are currently evaluated in SE.
Outcomes
SE still carries significant mortality and morbidity. Distinct variants of SE carry different mortalities, and the range is quite broad: from zero mortality for absence or complex partial SE in ambulatory patients [12], to 19% to 27% 30-day mortality for generalized tonic-clonic SE [20,123] and to 64.7% 30-day mortality for subtle SE [20]. Variables playing an important role in the outcome are the underlying cause (regarded by most authorities the most important variable), the duration of SE (mortality 32% if persistent for > 1 hour vs 2.7% if < 1 hour), the type of SE, the treatment administered, and the age of the patient (children have better outcomes than adults) [123–125]. The etiology of SE still remains the most important prognostic factor, with alcohol and AED-withdrawal/noncompliance having the best outcomes; structural brain injuries, such as anoxia-ischemia, vascular lesions, or brain tumors, have the worst prognosis.
The most resistant cases pose significant dilemmas regarding the length of treatment using barbiturate coma and the potential for acceptable prognosis or the need to withdraw life support. For RSE, for example, in-hospital mortality is 31.7% and 76.2% of patients reach poor functional outcome. Long-term outcomes are also dismal: at 1 year post-discharge, 74% are dead or in a state of unresponsive wakefulness, 16% severely disabled, and only 10% have no or minimal disability [126]. Duration of drug-induced coma, arrhythmias requiring intervention, and pneumonia are associated with poor functional outcome, whereas prolonged mechanical ventilation with mortality and seizure control without burst-suppression or isoelectric EEG are associated with good functional outcome [127,128].
Treatment with barbiturates may contribute to these outcomes, although it is very challenging to prove causality in such a complex and prolonged ICU environment. Some data have shed light towards that direction: in a recent retrospective study of 171 patients with SE, of which 37% were treated with IV anesthetic drugs, there was a higher risk for infections and a 2.9-fold relative risk for death after adjustment for confounders in the group treated with IV anesthetics compared to the group without these agents [129].
The SE Severity Score (STESS, range 0–6) is a prognostic score for survival [130] and can be used as a scaffold for discussions with families and covariate adjustment tool for research. A favorable score of 0–2 has a negative predictive value of 0.97 for survival and likelihood to return to baseline clinical condition in survivors, although an unfavorable score (3–6) had a positive predictive value for death of only 0.39 [131].
The risk for recurrence of afebrile SE in a population-based study in Minnesota has been estimated at 31.7% over a 10-year follow-up period. The risk for recurrence was about 25% regardless of the underlying etiology, except in those patients with SE occurring in the setting of a progressive brain disorder (who had a 100% risk). Female gender, generalized (vs partial) SE and lack of response to the first AED after the initial episode of SE were independent factors for recurrence [132].
Conclusion
Despite the use of better diagnostic tools (continuous video EEG), advances in technology in the ICU, and availability of new AEDs, SE still carries significant mortality and morbidity, which depends mainly on age and etiology. The current treatment is still staged, with supportive measures and benzodiazepine administration remaining the mainstay initially and followed by older and newer AEDs and anesthetics for resistant cases. With the advance of pathophysiologic mechanisms elucidation at a molecular/receptor level, combinations of AEDs may become the foundation of future SE control.
Corresponding author: Panayiotis N. Varelas, MD, PhD, FNCS, Division Head, Neuro-Critical Care Service, Henry Ford Hospital, K-11, 2799 W. Grand Blvd., Detroit, MI 48202, pvarela1@hfhs.org.
Financial disclosures: Dr. Varelas was local principal investigator for a super-refractory status epilepticus study sponsored by Sage Therapeutics.
Author contributions: conception and design, ARR, PNV; analysis and interpretation of data, PNV; drafting of article, PNV; critical revision of the article, ARR, PNV; administrative or technical support, PNV; collection and assembly of data, ARR, PNV.
From the Johns Hopkins Hospital, Baltimore, MD (Dr. Ramadan), and the Henry Ford Hospital, Detroit, MI (Dr. Varelas).
Abstract
- Objective: To review the management of status epilepticus (SE).
- Methods: Review of the literature.
- Results: SE is a relatively common condition that accounts for 3% to 5% of all emergency department evaluations for seizure disorders and occurs in 2% to 16% of all epilepsy patients. The 3 most common etiologies are low levels of antiepileptic drugs, remote symptomatic etiologies, and cerebrovascular accidents. The majority of SEs are convulsant, but there is growing awareness of non-convulsive SEs, which can be diagnosed only via electroencephalogram. Management, which must be initiated at the earliest possible time, has evolved to incorporate pre-hospital measures and 4 treatment stages, with supportive measures and benzodiazepine administration remaining the mainstay initially and followed by older and newer antiepileptic drugs and anesthetics for resistant cases.
- Conclusion: SE is a neurological emergency that still carries significant mortality and morbidity if not treated immediately and properly.
Key words: status epilepticus; seizures; convulsive status epilepticus; nonconvulsive status epilepticus.
Status epilepticus (SE) is a relatively common condition that accounts for 3% to 5% of all emergency department (ED) evaluations for seizure disorders and occurs in 2% to 16% of all epilepsy patients [1]. It remains a major neurological emergency that, if not properly and timely treated, leads to death or permanent neurological injury. Since most of patients with convulsive SE are admitted to the hospital via the ED and are then transferred to the intensive care unit (ICU), our focus in this review will be on the latter.
Although only a handful prospective, randomized studies have been reported, guidelines on SE have been published in Europe [2] and the US [3,4]. In this paper, we review the evolving definition and types of SE, its incidence, etiology, and pathophysiology, its diagnosis and treatment algorithms, and its outcome. Our goal is to provide the reader with a concise but thorough review of this still lethal neurological emergency.
Definitions
The International Classification of Epileptic Seizures had previously defined SE as any seizure lasting ≥ 30 minutes or intermittent seizures lasting for > 30 min without recovery of consciousness interictally [5,6]. More recently, a duration of 5 or more minutes of (a) continuous seizures or (b) 2 or more discrete seizures with incomplete recovery of consciousness in-between, proposed by Lowenstein [3,7], offers the advantage of incorporating new knowledge. The shortening of the convulsive period to 5 minutes was based on the fact that the majority of tonic-clonic seizures last for only 1 to 2 minutes, that those lasting > 5 minutes do not stop spontaneously [8], that permanent neuronal injury occurs before 30 minutes, and that refractoriness to treatment increases with longer seizure duration [9].
Refractory SE (RSE) has been defined as SE not controlled after adequate doses of an initial benzodiazepine followed by a second acceptable antiepileptic drug (AED) or SE not controlled after the initial parenteral therapy with a minimum number of standard “front-line” AEDs (either 2 or 3) or SE with a minimum duration of seizures that persist despite treatment (eg, at least or 2 hours) [3,10]. Super-refractory SE (SRSE) is defined as SE that continues or recurs 24 hours or more after the onset of anesthetic therapy or recurs on the reduction or withdrawal of anesthesia [11].
Non-convulsive SE (NCSE) is defined as the presence of altered consciousness or behavior for ≥ 30 minutes, the absence of overt clinical signs of convulsive activity during that period, and the electroencephalographic (EEG) confirmation of seizures or activity that responds to treatment together with improvement of consciousness [12–15]. Two major types of NCSE can be encountered: the one in patients with epileptic encephalopathy/coma and the one in patients with absence or complex partial seizures, who are not usually admitted to ICU and are functional yet impaired. Because of the confusion between these 2 extremes in the NCSE spectrum, working criteria for standardization of reporting, utilizing the frequency of electroencephalographic epileptiform discharges or delta/theta waveforms have been proposed [15]. A recent compendium of 123 cases of NCSE with clinical descriptions and EEG patterns following a syndromic classification approach has also been published [16].
Types of SE
Three major categories of SE have been described: generalized convulsive SE (GCSE), focal motor SE (FMSE or epilepsia partialis continua [EPC]) of Kojevnikov, and NCSE. GCSE and FMSE are easily recognized due to overt convulsions. NCSE, however, has a more obscure phenotype and can be subdivided into a spectrum encompassing typical absence and complex partial SE, atypical absence SE and tonic SE (usually in children with learning disabilities), epileptic behavioral disturbance and psychosis, including Balint–like syndrome [17], confusional states or delirium with epileptiform discharges) and SE in coma (after significant brain injuries, such as hypoxia-ischemia, most commonly encountered in ICUs) [13,18]. The 2 extremes in this NCSE spectrum have completely different prognoses, with absence SE the most benign and SE in coma the most dismal.
Lastly, SE presents either spontaneously or can be “semi-intentional” iatrogenic, encountered either in the neuro-ICU or epilepsy monitoring unit, when AEDs are withdrawn under continuous EEG recording in order for seizures to emerge and be recorded with surface or intracranial electrodes.
Incidence of SE
In a prospective population-based epidemiological study, the incidence of SE was estimated at 41–61/100,000 patients/year. For the US, this translates to 125,000 to 195,000 episodes per year [19].
The highest incidence of SE occurs during the first year of life and during the decades beyond 60 years, and is also dependent on the SE subtype. Partial SE occurs in 25% of cases of SE and NCSE accounts for another 4% to 26 % [19,20], but the incidence for the latter is considered an underestimate due to the need for continuous EEG monitoring (which is not widely available). For example, NCSE was discovered in no patient with acute stroke [21], 8% of comatose ICU patients [22], 7% of patients with intracerebral hemorrhage [23], 3% to 8% of patients with subarachnoid hemorrhage [24–26], 6% of patients with metastatic cancer [27], and 6% of patients with head trauma [28].
The incidence of RSE and SRSE is also unknown. In a recent retrospective study from a neuro-ICU in a West China hospital, the percentage of non-refractory SE, RSE, and SRSE were 67.3%, 20.4% and 12.2%, respectively [29]. Other retrospective studies have shown that 12% to 43% of SE cases become refractory [30–33] and that approximately 10% to 15% of all cases of hospital-admitted SE will become super-refractory at some point, but no prospective studies have been published.
Risk factors that have been identified for RSE are encephalitis as a cause, severe consciousness impairment, de novo episodes of SE, delay in initiation of treatment, NCSE, and focal motor seizures at onset [30,32,34,35]. In a more recent study from ICU patients in Switzerland and the US, acute SE etiology (traumatic brain injuries, cerebrovascular accidents, meningoencephalitis, brain tumors, surgical brain lesions, exposure to, or withdrawal from, recreational drugs, prescription drugs, alcohol, metabolic disturbances and fever), coma/stupor, and serum albumin < 35 g/L at SE onset were independent predictors for RSE [36].
Etiology of SE
The 3 most common etiologies for SE are low levels of antiepileptic drugs (AEDs) in 34% of the cases (usually due to noncompliance), remote symptomatic etiologies (history of neurological insults remote to the first unprovoked SE episode, 24%), and cerebrovascular accidents (ischemic and hemorrhagic strokes, 22%). These are followed by hypoxia (13%) and metabolic disturbances (15%). Because 82% of patients in the remote group have a history of cerebrovascular disease, almost 50% have either acute or remote cerebrovascular disease as etiology of SE [19].
In general ICUs, metabolic abnormalities can account for 33% of seizures, drug withdrawal for 33%, drug toxicity for 14.5%, and stroke for 9% to 39% [37,38]. In ICUs, sepsis remains a common etiology of electrographic seizures or periodic epileptiform discharges [39,40], and legal or illegal drugs, such as ciprofloxacin, levofloxacin, piperacillin/tazobactam, cefepime and carbapenems [41–43], lithium or theophylline intoxication, vigabatrin, tiagabine or crack/cocaine, are another [18] (especially when their metabolism is altered due to interactions with other drugs or when their excretion is impaired due to hepatic or renal failure).
Beyond these common causes of SE, a workup for rare etiologies should be entertained. In a systematic review of 513 papers on SE, 181 uncommon causes of SE were identified and subdivided into immunologically mediated disorders, mitochondrial diseases, rare infectious disorders, genetic disorders, and drugs or toxins [18,44].
The most recent knowledge in this category is the contribution of paraneoplastic or autoimmune conditions to a large percentage of previously cryptogenic pharmaco-resistant seizures or super-refractory SE, most in the context of limbic encephalitis. Many of these patients have never experienced seizures or SE before and a new acronym has been devised for them: new-onset refractory status epilepticus (NORSE), ie, a state of persistent seizures with no identifiable etiology in patients without preexisting epilepsy that lasts longer than 24 hour despite optimal therapy [45]. A growing array of autoantibodies against intracellular and surface or synaptic neuronal targets has been described in addition to the previous literature of Rassmussen’s encephalitis and Hashimoto’s encephalopathy [46]. The most common autoantibodies associated with seizures and SE include anti-Hu, anti-Ma2, anti-CV2/CRMP5, anti-Ri, ANNA3, anti-amphiphysin, anti-NMDA receptor, anti-LGI1 and CASPR2, anti-GABA-beta, anti-GluR3, anti-mGluR5 and alpha 3 ganglionic acetylcholine receptor [47,48]. The diagnosis frequently remains elusive due to lack of knowledge or absence of widespread availability of serologic testing (with sometimes weeks-long delay for the results to be available), but the response to treatment with removal of tumor, plasmapheresis, or immunomodulation and immunosuppression is often dramatic.
Pathophysiology of SE
Most seizures are self-terminating phenomena lasting from a few seconds to a few minutes [49]. One of the distinguishing characteristics of seizures evolving into SE, however, is the switch to a self-sustaining situation, which is time-dependent. Seizures lasting more than 30 minutes would rarely stop spontaneously compared to 47% of those lasting between 10 to 29 minutes, which are self-resolving [50]. Moreover, in one study no self-limited seizure lasted more than 11 minutes [8].
The self-limiting character of seizures is due to inhibitory circuitry that suppresses their duration and propagation in the brain. Under specific circumstances, however, the inhibitory mechanisms fail and seizures progress to SE, which leads to synaptic reorganization, blood-brain barrier disruption, inflammation, metabolic crisis, more tissue damage, and further seizures. Neuronal injury during SE is the result of increased excitotoxicity [51–53] but also stems from systemic derangements such as hypoxia, acidosis, hypotension, or multiorgan dysfunction [54]. The seminal animal studies by Meldrum have shed a light on the systemic effects: after prolonged bicuculine-induced convulsive SE in baboons, neuronal damage and cell loss was evident in the neocortex, cerebellum and hippocampus. When systemic factors were kept within normal physiological limits (paralyzed and artificially ventilated animals with adequate serum glucose levels), there was decreased but still present neocortical and hippocampal cell damage, but absent cerebellar cell injury [55,56]. These experiments showed more than 40 years ago that the seizure activity per se is responsible for the neuronal damage and the systemic derangements play an additional role.
The direct neuronal injury as a result of the ongoing seizures, the perpetuation of seizures into SE, the resistance to treatment and the refractoriness that ensues have also been elucidated at a molecular level during the last decades. Initially, the g-aminobutyric acid (GABA) inhibitory circuits may be deficient and this is the reason why benzodiazepines or barbiturates, which work through GABAergic receptor agonism, are very effective during this early period. As time passes however, GABA receptors undergo a significant shift in their ability to respond to benzodiazepines [57,58]. This is due to changes in receptor presence at the inhibitory synapse, a phenomenon that has been called “receptor trafficking” by Arancibia and Kittler in 2009 [59]. There are differences in the type of GABAA receptors found synaptically and extrasynaptically. GABAA receptors containing the γ subunit are located synaptically and mediate phasic inhibition. Conversely, the δ subunit-containing GABAA receptors are located exclusively extrasynaptically and mediate tonic inhibition [60,61]. Smith and Kittler described the highly dynamic state of receptor presence on the surface of axons and explained how receptors move laterally from extrasynaptic sites to the synapse and then out of it to be internalized and either recycled to the surface or degraded [62]. This “receptor trafficking” intensifies during SE, and the overall effect becomes a reduction in the number of functional GABAA receptors in the synapses. As GABA is the principle inhib-itory transmitter, this reduction in GABAergic activity may be an important reason for seizures to become persistent.
However, this is not all. Additional mechanisms leading to refractoriness include the following:
(a) Excessive relocation of N-methyl-D-aspartate (NMDA)type glutamate receptors to the cell surface after 1 hour of SE, leading to increase of miniature excitatory NMDA currents and NMDA neurotransmission, with potentiation of glutamate excitotoxicity [53,63]
(b) Increased brain expression of drug efflux transporters, such as P-glycoprotein at the blood-brain barrier, which may reduce concentrations of AEDs at their brain targets [64]
(c) Up- and down-regulation of specific ATP-gated ion channels (P2X receptors) inducing altered response to ATP release [65]
(d) Change in the extracellular ionic environment (for example, the normally inhibitory GABAA receptor-mediated currents may become excitatory with changes in extracellular chloride concentrations) [66]
(e) Mitochondrial insufficiency or failure, which would lead to cell necrosis and apoptosis [67]
(f) Inflammatory processes, with opening of the blood-brain barrier (BBB) contributing to perpetuation of seizures [44]. The underlying mechanism is a maladaptive response of the astrocytes to the BBB damage, leading to activation of the innate immune system and disturbed homeostasis of the extracellular potassium and glutamate [68].
(g) Large-scale changes in gene expression within the affected brain regions; these are regulated by micro-RNAs, influencing protein levels playing a role in excitability, neuronal death and neuroinflammation [69].
All of these pathophysiologic derangements may become targets for future antiepileptic treatments.
Although the direct and indirect injury from ongoing convulsive SE is not in doubt, the significance of NCSE or the ictal-interictal continuum on inflicting additional injury has been more controversial. Recent data, however, do not support a benign process in these situations. It has been shown lately that nonconvulsive seizures lead to physiologic changes in the brain, including elevated intracranial pressure, changes in the brain metabolism, and delayed increase in cerebral blood flow [25]. In addition, using microdialysis, elevated lactate/puruvate ratio, indicating metabolic crisis, has been shown during periods of nonconvulsive seizures or periodic discharges [70]. Similarly, high-frequency periodic discharges lead to inadequate increase in cerebral blood flow and tissue hypoxia [71], and lateralized periodic discharges, lateralized rhythmic delta activity, and generalized periodic discharges are associated with seizures [72].
Diagnosis of SE
The diagnosis of SE is primarily clinical and encompasses motor phenomena and alteration of mental status. Focal-onset convulsions can remain focal, follow a Jacksonian march, or immediately generalize to involve the whole body with loss of consciousness. Most of the time, this secondary generalization can only be appreciated during EEG recording. In addition, mental status alteration can differentiate simple partial SE (no change in mental status) from complex partial SE (disturbed sensorium).
The presence or absence of motor phenomena and loss of consciousness do not necessarily correlate with the EEG activity during or after SE. For example, persistent electrographic seizures or NCSE after control of convulsive SE have been demonstrated with continuous EEG [73]. Conversely, altered mental status is also a poor clinical differentiator, since 87% of patients successfully treated for convulsive SE and 100% treated for NCSE remained comatose 12 hours following the initiation of therapy [20]. In addition, only 27% of motor, seizure-like phenomena in the ICU were proven to be seizures in a retrospective study [74]. Psychogenic nonepileptic attacks, occurring in between 3.8% and 9.5% of ICU patients presenting with seizures [74,75], is another situation that may lead to confusion, inappropriate intubation, and ICU admission. Strange phenomena, such as fasciobrachial seizures (brief facial grimacing and ipsilateral arm posturing) many times preceding the onset of amnesia, confusion, or temporal lobe seizures have been described in patients who have non-paraneoplastic limbic encephalitis associated with voltage-gated potassium channel (VGKC) antibodies, especially against the leucine-rich glioma inactivated-1 (LGI1) protein [76,77].Without a continuous video EEG, these phenomena may not be captured or appreciated. Therefore, EEG monitoring is an important tool for the evaluation of these patients and criteria for its use have been published [78]. The EEG criteria for convulsive SE have been clearly delineated, but for NCSE a mix of clinical and EEG criteria should be met [14,15,79].
In addition to clinical observation and EEG, there has been interest lately in multimodality monitoring of acutely brain-injured patients for seizures or SE using electrocorticography or mini depth electrode placement, partial brain tissue oxygen tension, cerebral blood flow, and microdialysis in addition to scalp EEG. Although preliminary and limited in few academic centers, this approach has produced interesting findings. For example, in a study from Columbia University, 38% of 48 patients with subarachnoid hemorrhage and multimodality monitoring had intracortical seizures, while only 8% of them had surface seizures, all nonconvulsive [25]. In another study, 68% of seizures and 23% of periodic discharges were only captured on the depth electrodes and were missed on the surface ones [71]. Therefore, detection of SE may change in the future with use of more sensitive techniques than scalp EEG.
Treatment
Significant practice variations exist in the management of SE even among academic centers in the US [80] despite the fact that the goals of treatment are concrete. These include (1) emergent medical management, (2) termination of seizures, (3) prevention of recurrence of seizures, and (4) prevention or treatment of complications.
Management of SE must begin in a prehospital setting by the emergency medical services, because the faster the treatment is offered, the better the response. Several studies have attempted to assess the possibility of aborting SE even prior to the hospital. In a randomized, double-blinded study, lorazepam was 4.8 times and diazepam 2.3 times more effective than placebo in terminating SE on arrival in the ED when given intravenously (IV) by paramedics [81]. The RAMPART study was a double-blind, randomized, non-inferiority trial comparing the efficacy of intramuscular (IM) midazolam (10 mg followed by placebo IV) with that of IM placebo followed by intravenous lorazepam (4 mg) for children and adults in SE treated by paramedics. At the time of arrival in the ED, seizures had ceased without rescue therapy in 73.4% and 63.4%, respectively, favoring midazolam [82].
Emergent Initial Phase
During the emergent initial phase, the goals are protection of the airway, oxygenation, maintenance of blood pressure, exclusion of easily
Urgent Control
If seizures continue, stage 2 medications should be used for benzodiazepine-refractory SE as urgent control treatment. There are some data suggesting better response rate to valproate after failure to control seizures with phenytoin than to phenytoin after failure of valproate [88]. If available, IV fosphenytoin is preferable to IV phenytoin due to potentially lower risk of side effects. Levetiracetam and phenobarbital IV are also acceptable choices. Levetiracetam can be administered as an off-label loading dose of 20–60 mg/kg IV (although the initial manufacturer was not supporting a “loading” dose; dose of up to 60 mg/kg IV up to 4500 mg maximum has been supported by the latest American Epilepsy Society guidelines [4]). This AED at an initial dose of 2–3 g/day confers an estimated success rate around 70% [89]. In a systematic review of 27 studies (798 cases of convulsive SE) comparing 5 AEDs in the treatment of benzodiazepine-resistant convulsive SE, phenobarbital and valproate had the highest efficacy (73.6% and 75.7%, respectively), followed by levetiracetam (68.5%) and phenytoin (50.2%). Lacosamide studies were excluded from the meta-analysis due to insufficient data [90], but its efficacy has been reported for patients with convulsive and NCSE [91,92]. There is not enough evidence at this point, however, to recommend its routine use for benzodiazepine refractory SE [90].
Refractory SE
When seizures continue despite the use of benzodiazepines and 2nd stage AEDs, SE becomes refractory (stage 3). Treatment of these resistant cases is frequently initiated in the ED and continued in an ICU. Outcomes were not significantly better in patients with SE admitted and managed in a neuro-ICU compared to a general medical ICU in a retrospective study, but the numbers were small (only 27% of SE were admitted to the former) [93] and this may change in the future. Intubation and mechanical ventilation is the first step, if not already present (only 21% of patients in the RAMPART study received endotracheal intubation, with 6.4% in the prehospital setting and 93.6% after admission [87]). Hemodynamic support with pressors or inotropes may be required as most anesthetic agents may decrease the blood pressure. Because of the urgency of controlling the seizures during SE, the potential aspiration risk and the questionable enteral absorption per os administration of additional AEDs is problematic, and IV formulations should be used.
Currently in the US, phenytoin, valproic acid, phenobarbital, levetiracetam, lacosamide, diazepam, lorazepam are available in IV formulations. In February 2016, the FDA also approved brivaracetam (which also is available in an IV formulation) and in October of the same year IV carbamazepine. None of these AEDs has an FDA indication for SE, although they are widely used. Parenteral lacosamide has a success rate of 33% to 67.7% (200–400 mg over 3–5 min was the most common bolus dose) depending on its use as second or third AED [94–96]. In lacosamide-naive patients with RSE on continuous EEG monitoring, the success rate for cessation of SE was 15.7, 25.5, 58.8, and 82.4 % by 4, 12, 24, and 48 hours, respectively [97]. Alternatively, topiramate in doses 300–1600 mg/day per oro/nasogastric tube can be considered [98]. In a study of 35 patients with RSE treated with topiramate as an adjunct AED, the response rate was 86% (as the third AED), and remained stable at 67% after administration as the fourth to seventh AED. Overall, RSE was terminated in 71% of patients within 72 hours after first administration of topiramate [99]. Other studies, however, adjusting for co-variates, did not prove topiramate to be effective in RSE [100]. Clobazam, a unique oral 1,5-benzodiazepine with excellent absorption, has been also used in the treatment of RSE. Seventeen patients with RSE (11 with prior epilepsy) were successfully treated with clobazam, which was introduced after a median duration of 4 days and after a median of 3 failed AEDs. Termination of RSE within 24 hours of administration, without addition or modification of concurrent AED and with successful wean of anesthetic infusions, was seen in 13 patients, whereas indeterminate response was seen in another 3. Clobazam was deemed unsuccessful in 1 patient [101]. In another recent report of 70 episodes of RSE, clobazam was used in 24 (34.3%) of them. If clobazam was the last AED added to therapy before RSE termination, the success was attributed to this drug. Based on this definition, clobazam led to 6 episodes (25%) of successful RSE resolution [102]. If primary or metastatic brain tumor is the presumed cause of SE, a combination of IV phenytoin, IV levetiracetam (median dose 3 g/d) and enterically administered pregabalin (median dose 375 mg/day) led to 70% control of SE on average 24 hours after addition of the third AED [103]. However, the major treatment options, which should not be delayed in unresponsive RSE, are propofol or midazolam infusions at high rates and under continuous EEG monitoring. These infusions should be continued for at least 24 hours and then held to reassess the situation. By that time, cocurrent metabolic derangements and low AED levels from noncompliance should have been corrected. Prolonged and high-dose propofol should be avoided because of the risk for propofol infusion syndrome, especially if pressors/inotropes are co-infused [104].
Super-refractory SE
Should seizures continue or recur, stage 4 options for SRSE are considered [105]. Pentobarbital with shorter half-life is favored to phenobarbital. The main disadvantages of barbiturates are compromised neuro-exam (which has to be assessed frequently), cardiovascular depression and hypotension, respiratory depression with need for full ventilator support, cough suppression with increased risk for atelectasis and pneumonia, immunosuppression increasing the risk for infection or sepsis, immobility increasing the risk for thromboembolism and ileus mandating parenteral nutrition [106,107]. The depth and duration of the EEG suppression that must be achieved by barbiturates is unknown. Some experts recommend instead of burst-suppression pattern complete suppression or “flat record” because of better seizure control and fewer relapses [108]. Moreover, patients with more prolonged barbiturate treatment (> 96 hours) and those receiving phenobarbital at the time of pentobarbital taper are less likely to relapse [109]. European guidelines recommend titration of propofol and barbiturate to EEG burst-suppression, and midazolam to seizure suppression, maintained for at least 24 h [2]. In recent reviews, it was found that barbiturates control refractory and super-refractory SE in 64% of patients and are ineffective in only 5% [11,110].
If SE continues or recurs after emergence from barbiturate coma, ketamine may be an option [11,83]. Ketamine offers the advantage of NMDA receptor antagonism, which may be important in the late phase of SE and lacks cardiodepressant or hypotensive properties. Early [111] or late [112] use of ketamine has been reported in small case series with various success rates. In a recent multicenter retrospective study from North America and Europe, evaluating 58 patients with 60 RSE, ketamine was likely responsible for seizure control in 12% and possibly responsible in an additional 20%. No responses were observed when infusion rate was lower than 0.9 mg/kg/h or when ketamine was introduced 8 days or more after onset of SE or after failure of seven or more drugs [113].
If all these measures have failed, stage 4.2 treatment options are available (Table 2), but these are mostly based on small case series and expert opinions (except for the recent hypothermia study). Pyridoxine hydrochloride in an IV or enteral form at a dose of 100–300 mg/day for few days can be used in stage 4 or earlier stages, as it is a cofactor in the synthesis of the inhibitory neurotransmitter GABA [114]. There are no strong data for its effectiveness, but it can be used as a cheap and safe alternative [115]. Magnesium has been successfully used in 2 girls with juvenile Alper’s syndrome [116] and is the treatment of choice for eclamptic seizures. Ketogenic diet may also be an optionfor these patients [117]. Resection of the epileptic focus after mapping with intracranial EEG electrodes may be highly effective but cannot be used in many patients due to lack of focus or eloquence location [83,106,115]. Use of steroids, plasmapheresis or IVIG, followed by immunosuppression can be tried, but one should balance risks and benefits. These immunosuppressive or immunomodulating treatments should be especially considered in patients with NORSE or suspected autoimmune or paraneoplastic encephalitides, where AEDs usually have no effect [46]. These therapies though often precede the diagnosis, since it takes time for the autoantibody panel results to return and the treating physician has to make a decision to blindly start treatment for SRSE.
There were some promising data regarding hypothermia use in these desperate situations [118,119] until the HYBERNATUS study, conducted in France, was recently published. In this study, 270 patients with convulsive SE were randomized in to hypothermia (32° to 34°C for 24 hours) in addition to standard care or to standard care alone. A Glasgow Outcome Scale score of 5 (primary outcome) occurred in 49% of patients in the hypothermia group and in 43% in the control group (a nonstatistical difference). Secondary outcomes, including mortality at 90 days, RSE on day 1, SRSE and functional sequelae on day 90 were not different except for the rate of progression to EEG-confirmed SE on the first day, which was lower in the hypothermia group (11% vs. 22% in the controls). Adverse events were more frequent in the hypothermia group than in the control group [120].
Additional anecdotal treatments are presented in Table 2, but their efficacy is questionable.
This staged management approach may change in the future to a more physiologic and rational treatment with polytherapy based on synaptic receptor trafficking during SE [63]. For example, in an animal model of severe SE, combinations of a benzodiazepine with ketamine and valproate, or with ketamine and brivaracetam, were more effective and less toxic than benzodiazepine monotherapy [121]. Allopregnalonone, a metabolite of progesterone, is an endogenous, naturally occurring neuroactive steroid produced in the ovary, the adrenal cortex and the central nervous system. It is a potent positive allosteric modulator of synaptic and extrasynaptic GABAA receptors with antiepileptic activity [122]. Neuroactive steroids, such as allopregnanolone, are currently evaluated in SE.
Outcomes
SE still carries significant mortality and morbidity. Distinct variants of SE carry different mortalities, and the range is quite broad: from zero mortality for absence or complex partial SE in ambulatory patients [12], to 19% to 27% 30-day mortality for generalized tonic-clonic SE [20,123] and to 64.7% 30-day mortality for subtle SE [20]. Variables playing an important role in the outcome are the underlying cause (regarded by most authorities the most important variable), the duration of SE (mortality 32% if persistent for > 1 hour vs 2.7% if < 1 hour), the type of SE, the treatment administered, and the age of the patient (children have better outcomes than adults) [123–125]. The etiology of SE still remains the most important prognostic factor, with alcohol and AED-withdrawal/noncompliance having the best outcomes; structural brain injuries, such as anoxia-ischemia, vascular lesions, or brain tumors, have the worst prognosis.
The most resistant cases pose significant dilemmas regarding the length of treatment using barbiturate coma and the potential for acceptable prognosis or the need to withdraw life support. For RSE, for example, in-hospital mortality is 31.7% and 76.2% of patients reach poor functional outcome. Long-term outcomes are also dismal: at 1 year post-discharge, 74% are dead or in a state of unresponsive wakefulness, 16% severely disabled, and only 10% have no or minimal disability [126]. Duration of drug-induced coma, arrhythmias requiring intervention, and pneumonia are associated with poor functional outcome, whereas prolonged mechanical ventilation with mortality and seizure control without burst-suppression or isoelectric EEG are associated with good functional outcome [127,128].
Treatment with barbiturates may contribute to these outcomes, although it is very challenging to prove causality in such a complex and prolonged ICU environment. Some data have shed light towards that direction: in a recent retrospective study of 171 patients with SE, of which 37% were treated with IV anesthetic drugs, there was a higher risk for infections and a 2.9-fold relative risk for death after adjustment for confounders in the group treated with IV anesthetics compared to the group without these agents [129].
The SE Severity Score (STESS, range 0–6) is a prognostic score for survival [130] and can be used as a scaffold for discussions with families and covariate adjustment tool for research. A favorable score of 0–2 has a negative predictive value of 0.97 for survival and likelihood to return to baseline clinical condition in survivors, although an unfavorable score (3–6) had a positive predictive value for death of only 0.39 [131].
The risk for recurrence of afebrile SE in a population-based study in Minnesota has been estimated at 31.7% over a 10-year follow-up period. The risk for recurrence was about 25% regardless of the underlying etiology, except in those patients with SE occurring in the setting of a progressive brain disorder (who had a 100% risk). Female gender, generalized (vs partial) SE and lack of response to the first AED after the initial episode of SE were independent factors for recurrence [132].
Conclusion
Despite the use of better diagnostic tools (continuous video EEG), advances in technology in the ICU, and availability of new AEDs, SE still carries significant mortality and morbidity, which depends mainly on age and etiology. The current treatment is still staged, with supportive measures and benzodiazepine administration remaining the mainstay initially and followed by older and newer AEDs and anesthetics for resistant cases. With the advance of pathophysiologic mechanisms elucidation at a molecular/receptor level, combinations of AEDs may become the foundation of future SE control.
Corresponding author: Panayiotis N. Varelas, MD, PhD, FNCS, Division Head, Neuro-Critical Care Service, Henry Ford Hospital, K-11, 2799 W. Grand Blvd., Detroit, MI 48202, pvarela1@hfhs.org.
Financial disclosures: Dr. Varelas was local principal investigator for a super-refractory status epilepticus study sponsored by Sage Therapeutics.
Author contributions: conception and design, ARR, PNV; analysis and interpretation of data, PNV; drafting of article, PNV; critical revision of the article, ARR, PNV; administrative or technical support, PNV; collection and assembly of data, ARR, PNV.
1. Hauser WA. Status epilepticus: epidemiologic considerations. Neurology 1990;40:9–13.
2. Meierkord H, Boon P, Engelsen B, et al. EFNS guideline on the management of status epilepticus. Eur J Neurology 2006;13:445–50.
3. Brophy GM, Bell R, Claassen J, et al. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care 2012;17:3–23.
4. Glauser T, Shinnar S, Gloss D, et al. Evidence-based guideline: treatment of convulsive status epilepticus in children and adults: Report of the Guideline Committee of the American Epilepsy Society. Epilepsy Curr 2016;16:48–61.
5. Gastaut H. Classification of status epilepticus. Adv Neurol 1983;34:15–35.
6. Treatment of convulsive status epilepticus. Recommendations of the Epilepsy Foundation of America’s Working Group on Status Epilepticus. JAMA 1993;270:854–9.
7. Lowenstein DH. Status epilepticus: an overview of the clinical problem. Epilepsia 1999;40 Suppl 1:S3–8; discussion S21–22.
8. Jenssen S, Gracely EJ, Sperling MR. How long do most seizures last? A systematic comparison of seizures recorded in the epilepsy monitoring unit. Epilepsia 2006;47:1499–503.
9. Goodkin HP, Kapur J. Responsiveness of status epilepticus to treatment with diazepan decreases rapidly as seizure duration increases. Epilepsy Curr 2003;3:11–2.
10. Lowenstein DH. The management of refractory status epilepticus: an update. Epilepsia 2006;47 Suppl 1:35–40.
11. Shorvon S, Ferlisi M. The treatment of super-refractory status epilepticus: a critical review of available therapies and a clinical treatment protocol. Brain 2011;134:2802–18.
12. Kaplan PW. Assessing the outcomes in patients with nonconvulsive status epilepticus: nonconvulsive status epilepticus is underdiagnosed, potentially overtreated, and confounded by comorbidity. J Clin Neurophysiol 1999;16:341–52.
13. Walker MD. Diagnosis and treatment of nonconvulsive status epilepticus. CNS Drugs 2001;15:931–9.
14. Kaplan PW. EEG criteria for nonconvulsive status epilepticus. Epilepsia 2007;48 Suppl 8:39–41.
15. Beniczky S, Hirsch LJ, Kaplan PW, et al. Unified EEG terminology and criteria for nonconvulsive status epilepticus. Epilepsia 2013;54 Suppl 6:28–9.
16. Sutter R, Kaplan PW. Electroencephalographic criteria for nonconvulsive status epilepticus: synopsis and comprehensive survey. Epilepsia 2012;53 Suppl 3:1–51.
17. Ristic AJ, Marjanovic I, Brajkovic L, et al. Balint-like syndrome as an unusual representation of non-convulsive status epilepticus. Epileptic Disord 2012;14:80–4.
18. Trinka E, Hofler J, Zerbs A. Causes of status epilepticus. Epilepsia 2012;53 Suppl 4:127–38.
19. DeLorenzo RJ, Hauser WA, Towne AR, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology 1996;46:1029–35.
20. Treiman DM, Meyers PD, Walton NY, et al. A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med 1998;339:792–8.
21. Carrera E, Michel P, Despland PA, et al. Continuous assessment of electrical epileptic activity in acute stroke. Neurology 2006;67:99–104.
22. Towne AR, Waterhouse EJ, Boggs JG, et al. Prevalence of nonconvulsive status epilepticus in comatose patients. Neurology 2000;54:340–5.
23. Claassen J, Jette N, Chum F, et al. Electrographic seizures and periodic discharges after intracerebral hemorrhage. Neurology 2007;69:1356–65.
24. Claassen J, Peery S, Kreiter KT, et al. Predictors and clinical impact of epilepsy after subarachnoid hemorrhage. Neurology 2003;60:208–14.
25. Claassen J, Perotte A, Albers D, et al. Nonconvulsive seizures after subarachnoid hemorrhage: Multimodal detection and outcomes. Ann Neurol 2013;74:53–64.
26. Lindgren C, Nordh E, Naredi S, Olivecrona M. Frequency of non-convulsive seizures and non-convulsive status epilepticus in subarachnoid hemorrhage patients in need of controlled ventilation and sedation. Neurocrit Care 2012;17:367–73.
27. Cocito L, Audenino D, Primavera A. Altered mental state and nonconvulsive status epilepticus in patients with cancer. Arch Neurol 2001;58:1310.
28. Vespa PM, Nuwer MR, Nenov V, et al. Increased incidence and impact of nonconvulsive and convulsive seizures after traumatic brain injury as detected by continuous electroencephalographic monitoring. J Neurosurg 1999;91:750–60.
29. Tian L, Li Y, Xue X, et al. Super-refractory status epilepticus in West China. Acta Neurol Scand 2015;132:1–6.
30. Holtkamp M, Othman J, Buchheim K, et al. A “malignant” variant of status epilepticus. Arch Neurol 2005;62:1428–31.
31. Lowenstein DH, Alldredge BK. Status epilepticus at an urban public hospital in the 1980s. Neurology 1993;43:483–8.
32. Mayer SA, Claassen J, Lokin J, et al. Refractory status epilepticus: frequency, risk factors, and impact on outcome. Arch Neurol 2002;59:205–10.
33. Rossetti AO, Logroscino G, Bromfield EB. Refractory status epilepticus: effect of treatment aggressiveness on prognosis. Arch Neurol 2005;62:1698–702.
34. Novy J, Logroscino G, Rossetti AO. Refractory status epilepticus: a prospective observational study. Epilepsia 2010;51:251–6.
35. Garzon E, Fernandes RM, Sakamoto AC. Analysis of clinical characteristics and risk factors for mortality in human status epilepticus. Seizure 2003;12:337–45.
36. Sutter R, Kaplan PW, Marsch S, et al. Early predictors of refractory status epilepticus: an international two-center study. Eur J Neurol 2015;22:79–85.
37. Wijdicks EF, Sharbrough FW. New-onset seizures in critically ill patients. Neurology 1993;43:1042–4.
38. Bleck TP, Smith MD, Pierre-Louis SJ, et al. Neurologic complications of critical medical illnesses. Crit Care Med 1993;21:98–103.
39. Oddo M, Carrera E, Claassen J, et al. Continuous electroencephalography in the medical intensive care unit. Crit Care Med 2009;37:2051–6.
40. Gilmore EJ, Gaspard N, Choi HA, et al. Acute brain failure in severe sepsis: a prospective study in the medical intensive care unit utilizing continuous EEG monitoring. Intensive Care Med 2015;41:686–94.
41. Misra UK, Kalita J, Patel, R. Sodium valproate vs phenytoin in status epilepticus: a pilot study. Neurology 2006;67:340–2.
42. Kim A, Kim JE, Paek YM, et al. Cefepime-induced non-convulsive status epilepticus (NCSE). J Epilepsy Res 2013;3:39–41.
43. Naeije G, Lorent S, Vincent JL, Legros B. Continuous epileptiform discharges in patients treated with cefepime or meropenem. Arch Neurol 2011;68:1303–7.
44. Tan RY, Neligan A, Shorvon SD. The uncommon causes of status epilepticus: a systematic review. Epilepsy Res 2010;91:111–22.
45. Khawaja AM, DeWolfe JL, Miller DW, Szaflarski JP. New-onset refractory status epilepticus (NORSE) - The potential role for immunotherapy. Epilepsy Behav 2015;47:17–23.
46. Davis R, Dalmau J. Autoimmunity, seizures, and status epilepticus. Epilepsia 2013;54 Suppl 6:46–9.
47. Lopinto-Khoury C, Sperling MR. Autoimmune status epilepticus. Curr Treat Options Neurol 2013;15:545–56.
48. Bansal P, Zutshi D, Suchdev K, et al. Alpha 3 ganglionic acetylcholine receptor antibody associated refractory status epilepticus. Seizure 2016;35:1–3.
49. Chen JW, Wasterlain CG. Status epilepticus: pathophysiology and management in adults. Lancet Neurol 2006;5:246–56.
50. DeLorenzo RJ, Garnett LK, Towne AR, et al. Comparison of status epilepticus with prolonged seizure episodes lasting from 10 to 29 minutes. Epilepsia 1999;40:164–9.
51. Lowenstein DH, Alldredge BK. Status epilepticus. N Engl J Med 1998;338:970–6.
52. Millikan D, Rice B, Silbergleit R. Emergency treatment of status epilepticus: current thinking. Emerg Med Clin North Am 2009;27:101–13, ix.
53. Fountain NB. Status epilepticus: risk factors and complications. Epilepsia 2000;41 Suppl 2:S23–30.
54. Aminoff MJ, Simon RP. Status epilepticus. Causes, clinical features and consequences in 98 patients. Am J Med 1980;69:657–66.
55. Meldrum BS, Brierley JB. Prolonged epileptic seizures in primates. Ischemic cell change and its relation to ictal physiological events. Arch Neurol 1973;28:10–7.
56. Meldrum BS, Vigouroux RA, Brierley JB. Systemic factors and epileptic brain damage. Prolonged seizures in paralyzed, artificially ventilated baboons. Arch Neurol 1973;29:82–7.
57. Loscher W. Mechanisms of drug resistance in status epilepticus. Epilepsia 2007;48 Suppl 8:74–7.
58. Jacob TC, Moss SJ, Jurd R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat Rev Neurosci 2008;9:331–43.
59. Arancibia-Carcamo IL, Kittler JT. Regulation of GABA(A) receptor membrane trafficking and synaptic localization. Pharmacol Ther 2009;123:17–31.
60. Bohnsack JP, Carlson SL, Morrow AL. Differential regulation of synaptic and extrasynaptic alpha4 GABA(A) receptor populations by protein kinase A and protein kinase C in cultured cortical neurons. Neuropharmacology 2016;105:124–32.
61. Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci 2005;6:215–29.
62. Smith KR, Kittler JT. The cell biology of synaptic inhibition in health and disease. Curr Opin Neurobiol 2010;20:550–6.
63. Wasterlain CG, Naylor DE, Liu H, et al. Trafficking of NMDA receptors during status epilepticus: therapeutic implications. Epilepsia 2013;54 Suppl 6:78–80.
64. Bankstahl JP, Loscher W. Resistance to antiepileptic drugs and expression of P-glycoprotein in two rat models of status epilepticus. Epilepsy Res 2008;82:70–85.
65. Henshall DC, Diaz-Hernandez M, Miras-Portugal MT, Engel T. P2X receptors as targets for the treatment of status epilepticus. Front Cell Neurosci 2013;7:237.
66. Lamsa K, Taira T. Use-dependent shift from inhibitory to excitatory GABAA receptor action in SP-O interneurons in the rat hippocampal CA3 area. J Neurophysiol 2003;90:1983–95.
67. Cock HR, Tong X, Hargreaves IP, et al. Mitochondrial dysfunction associated with neuronal death following status epilepticus in rat. Epilepsy Res 2002;48:157–68.
68. Friedman A, Dingledine R, Molecular cascades that mediate the influence of inflammation on epilepsy. Epilepsia 2011;52 Suppl 3:33–39.
69. Henshall DC. MicroRNAs in the pathophysiology and treatment of status epilepticus. Front Mol Neurosci 2013;6:37.
70. Vespa P, Tubi M, Claassen J, et al. Metabolic crisis occurs with seizures and periodic discharges after brain trauma. Ann Neurol 2016;79:579–90.
71. Witsch J, Frey HP, Schmidt JM, et al. Electroencephalographic periodic discharges and frequency-dependent brain tissue hypoxia in acute brain injury. JAMA Neurol 2017;74:301–9.
72. Rodriguez Ruiz A, Vlachy J, Lee JW, et al. Association of periodic and rhythmic electroencephalographic patterns with seizures in critically ill patients. JAMA Neurol 2017;74:181–8.
73. DeLorenzo RJ, Waterhouse EJ, Towne AR, et al. Persistent nonconvulsive status epilepticus after the control of convulsive status epilepticus. Epilepsia 1998;39:833–40.
74. Benbadis SR, Chen S, Melo M. What’s shaking in the ICU? The differential diagnosis of seizures in the intensive care setting. Epilepsia 2010;51:2338–40.
75. Pandian JD, Cascino GD, So EL, et al. Digital video-electroencephalographic monitoring in the neurological-neurosurgical intensive care unit: clinical features and outcome. Arch Neurol 2004;61:1090–4.
76. Irani SR, Vincent A, Schott JM. Autoimmune encephalitis. BMJ 2011;342:d1918.
77. Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011;69:892–900.
78. Herman ST, Abend NS, Bleck TP, et al. Consensus statement on continuous EEG in critically ill adults and children, part I: indications. J Clin Neurophysiol 2015;32:87–95.
79. Brenner RP. Is it status? Epilepsia 2002;43 Suppl 3:103–113.
80. Cook AM, Castle A, Green A, et al. Practice variations in the management of status epilepticus. Neurocrit Care 2012;17:24–30.
81. Alldredge BK, Gelb AM, Isaacs SM, et al. A comparison of lorazepam, diazepam, and placebo for the treatment of out-of-hospital status epilepticus. N Engl J Med 2001;345:631–7.
82. Silbergleit R, Durkalski V, Lowenstein D, et al. Intramuscular versus intravenous therapy for prehospital status epilepticus. N Engl J Med 2012;366:591–600.
83. Shorvon S. Super-refractory status epilepticus: an approach to therapy in this difficult clinical situation. Epilepsia 2011;52 Suppl 8:53–6.
84. Varelas P, Mirski MA. Management of status epilepticus in adults. Hosp Physician Board Rev Man 2014;2:1–15.
85. McIntyre J, Robertson S, Norris E, et al. Safety and efficacy of buccal midazolam versus rectal diazepam for emergency treatment of seizures in children: a randomised controlled trial. Lancet 2005;366:205–10.
86. Misra UK, Kalita J, Maurya PK. Levetiracetam versus lorazepam in status epilepticus: a randomized, open labeled pilot study. J Neurol 2012;259:645–8.
87. Vohra TT, Miller JB, Nicholas KS, et al. Endotracheal intubation in patients treated for prehospital status epilepticus. Neurocrit Care 2015;23:33–43.
88. Agarwal P, Kumar N, Chandra R, et al. Randomized study of intravenous valproate and phenytoin in status epilepticus. Seizure 2007;16:527–32.
89. Trinka E. What is the evidence to use new intravenous AEDs in status epilepticus? Epilepsia 2011;52 Suppl 8:35–38.
90. Yasiry Z, Shorvon SD. The relative effectiveness of five antiepileptic drugs in treatment of benzodiazepine-resistant convulsive status epilepticus: a meta-analysis of published studies. Seizure 2014;23:167–74.
91. Moreno Morales EY, Fernandez Peleteiro M, Bondy Pena EC, et al. Observational study of intravenous lacosamide in patients with convulsive versus non-convulsive status epilepticus. Clin Drug Investig 2015;35:463–9.
92. Paquette V, Culley C, Greanya ED, Ensom MH. Lacosamide as adjunctive therapy in refractory epilepsy in adults: a systematic review. Seizure 2015;25:1–17.
93. Varelas PN, Corry J, Rehman M, et al. Management of status epilepticus in neurological versus medical intensive care unit: does it matter? Neurocrit Care 2013;19:4–9.
94. Hofler J, Trinka E. Lacosamide as a new treatment option in status epilepticus. Epilepsia 2013;54:393–404.
95. Kellinghaus C, Berning S, Stogbauer F. Intravenous lacosamide or phenytoin for treatment of refractory status epilepticus. Acta Neurol Scand 2014;129:294–9.
96. Santamarina E, Toledo M, Sueiras M, et al. Usefulness of intravenous lacosamide in status epilepticus. J Neurol 2013;260:3122–8.
97. Newey CR, Le NM, Ahrens C, et al. The safety and effectiveness of intravenous lacosamide for refractory status epilepticus in the critically ill. Neurocrit Care 2017;26:273–9.
98. Towne AR, Garnett LK, Waterhouse EJ, et al. The use of topiramate in refractory status epilepticus. Neurology 2003;60:332–4.
99. Hottinger A, Sutter R, Marsch S, Ruegg S. Topiramate as an adjunctive treatment in patients with refractory status epilepticus: an observational cohort study. CNS Drugs 2012;26:761–72.
100. Madzar D, Kuramatsu JB, Gerner ST, et al. Assessing the value of topiramate in refractory status epilepticus. Seizure 2016;38:7–10.
101. Sivakumar S, Ibrahim M, Parker D Jr, et al. An effective add-on therapy in refractory status epilepticus. Epilepsia 2015;56:e83–89.
102. Madzar D, Geyer A, Knappe RU, et al. Effects of clobazam for treatment of refractory status epilepticus. BMC Neurol 2016;16:202.
103. Swisher CB, Doreswamy M, Gingrich KJ, et al. Phenytoin, levetiracetam, and pregabalin in the acute management of refractory status epilepticus in patients with brain tumors. Neurocrit Care 2012;16:109–13.
104. Smith H, Sinson G, Varelas P. Vasopressors and propofol infusion syndrome in severe head trauma. Neurocrit Care 2009;10:166–72.
105. Cuero MR, Varelas PN. Super-refractory status epilepticus. Curr Neurol Neurosci Rep 2015;15:74.
106. Varelas PN. How I treat status epilepticus in the Neuro-ICU. Neurocrit Care 2008;9:153–7.
107. Varelas PN, Spanaki MV, Mirski MA. Status epilepticus: an update. Curr Neurol Neurosci Rep 2013;13:357.
108. Krishnamurthy KB, Drislane FW. Depth of EEG suppression and outcome in barbiturate anesthetic treatment for refractory status epilepticus. Epilepsia 1999;40:759–62.
109. Krishnamurthy KB, Drislane FW. Relapse and survival after barbiturate anesthetic treatment of refractory status epilepticus. Epilepsia 1996;37:863–7.
110. Ferlisi M, Shorvon S. The outcome of therapies in refractory and super-refractory convulsive status epilepticus and recommendations for therapy. Brain 2012;135:2314–28.
111. Kramer AH. Early ketamine to treat refractory status epilepticus. Neurocrit Care 2012;16:299–305.
112. Synowiec AS, Singh DS, Yenugadhati V, et al. Ketamine use in the treatment of refractory status epilepticus. Epilepsy Res 2013;105:183–8.
113. Gaspard N, Foreman B, Judd LM, et al. Intravenous ketamine for the treatment of refractory status epilepticus: a retrospective multicenter study. Epilepsia 2013;54:1498–503.
114. Schulze-Bonhage A, Kurthen M, Walger P, Elger CE. Pharmacorefractory status epilepticus due to low vitamin B6 levels during pregnancy. Epilepsia 2004;45:81–4.
115. Shorvon S. Clinical trials in acute repetitive seizures and status epilepticus. Epileptic Disord 2012;14:138–47.
116. Visser NA, Braun KP, Leijten FS, et al. Magnesium treatment for patients with refractory status epilepticus due to POLG1-mutations. J Neurol 2011;258:218–22.
117. Thakur KT, Probasco JC, Hocker SE, et al. Ketogenic diet for adults in super-refractory status epilepticus. Neurology 2014;82:665–70.
118. Corry JJ, Dhar R, Murphy T, Diringer MN. Hypothermia for refractory status epilepticus. Neurocrit Care 2008;9:189–97.
119. Guilliams K, Rosen M, Buttram S, et al. Hypothermia for pediatric refractory status epilepticus. Epilepsia 2013;54:1586–94.
120. Legriel S, Lemiale V, Schenck M, et al. Hypothermia for neuroprotection in convulsive status epilepticus. N Engl J Med 2016;375:2457–67.
121. Wasterlain CG, Baldwin R, Naylor DE, et al. Rational polytherapy in the treatment of acute seizures and status epilepticus. Epilepsia 2011;52 Suppl 8:70–1.
122. Rogawski MA, Loya CM, Reddy K, et al. Neuroactive steroids for the treatment of status epilepticus. Epilepsia 2013;54 Suppl 6:93–8.
123. Towne AR, Pellock JM, Ko D, DeLorenzo RJ. Determinants of mortality in status epilepticus. Epilepsia 1994;35:27–34.
124. DeLorenzo RJ, Towne AR, Pellock JM, Ko D. Status epilepticus in children, adults, and the elderly. Epilepsia 1992;33 Suppl 4:S15–25.
125. Legriel S, Mourvillier B, Bele N, et al. Outcomes in 140 critically ill patients with status epilepticus. Intensive Care Med 2008;34:476–80.
126. Pugin D, Foreman B, De Marchis GM, et al. Is pentobarbital safe and efficacious in the treatment of super-refractory status epilepticus: a cohort study. Crit Care 2014;18:R103.
127. Hocker SE, Britton JW, Mandrekar JN, et al. Predictors of outcome in refractory status epilepticus. JAMA Neurol 2013;70:72–7.
128. Hocker S, Tatum WO, LaRoche S, Freeman WD. Refractory and super-refractory status epilepticus--an update. Curr Neurol Neurosci Rep 2014;14:452.
129. Sutter R, Marsch S, Fuhr P, et al. Anesthetic drugs in status epilepticus: risk or rescue? A 6-year cohort study. Neurology 2014;82:656–64.
130. Rossetti AO, Logroscino G, Bromfield EB. A clinical score for prognosis of status epilepticus in adults. Neurology 2006;66:1736–8.
131. Rossetti AO, Logroscino G, Milligan TA, et al. Status Epilepticus Severity Score (STESS): a tool to orient early treatment strategy. J Neurol 2008;255:1561–6.
132. Hesdorffer DC, Logroscino G, Cascino GD, Hauser WA. Recurrence of afebrile status epilepticus in a population-based study in Rochester, Minnesota. Neurology 2007;69:73–8.
133. Varelas PN, Mirski MA. Seizures in the adult intensive care unit. J Neurosurg Anesthesiol 2001;13:163–75.
134. Varelas PN, Mirski MA. Status epilepticus. Curr Neurol Neurosci Rep 2009;9:469–76.
1. Hauser WA. Status epilepticus: epidemiologic considerations. Neurology 1990;40:9–13.
2. Meierkord H, Boon P, Engelsen B, et al. EFNS guideline on the management of status epilepticus. Eur J Neurology 2006;13:445–50.
3. Brophy GM, Bell R, Claassen J, et al. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care 2012;17:3–23.
4. Glauser T, Shinnar S, Gloss D, et al. Evidence-based guideline: treatment of convulsive status epilepticus in children and adults: Report of the Guideline Committee of the American Epilepsy Society. Epilepsy Curr 2016;16:48–61.
5. Gastaut H. Classification of status epilepticus. Adv Neurol 1983;34:15–35.
6. Treatment of convulsive status epilepticus. Recommendations of the Epilepsy Foundation of America’s Working Group on Status Epilepticus. JAMA 1993;270:854–9.
7. Lowenstein DH. Status epilepticus: an overview of the clinical problem. Epilepsia 1999;40 Suppl 1:S3–8; discussion S21–22.
8. Jenssen S, Gracely EJ, Sperling MR. How long do most seizures last? A systematic comparison of seizures recorded in the epilepsy monitoring unit. Epilepsia 2006;47:1499–503.
9. Goodkin HP, Kapur J. Responsiveness of status epilepticus to treatment with diazepan decreases rapidly as seizure duration increases. Epilepsy Curr 2003;3:11–2.
10. Lowenstein DH. The management of refractory status epilepticus: an update. Epilepsia 2006;47 Suppl 1:35–40.
11. Shorvon S, Ferlisi M. The treatment of super-refractory status epilepticus: a critical review of available therapies and a clinical treatment protocol. Brain 2011;134:2802–18.
12. Kaplan PW. Assessing the outcomes in patients with nonconvulsive status epilepticus: nonconvulsive status epilepticus is underdiagnosed, potentially overtreated, and confounded by comorbidity. J Clin Neurophysiol 1999;16:341–52.
13. Walker MD. Diagnosis and treatment of nonconvulsive status epilepticus. CNS Drugs 2001;15:931–9.
14. Kaplan PW. EEG criteria for nonconvulsive status epilepticus. Epilepsia 2007;48 Suppl 8:39–41.
15. Beniczky S, Hirsch LJ, Kaplan PW, et al. Unified EEG terminology and criteria for nonconvulsive status epilepticus. Epilepsia 2013;54 Suppl 6:28–9.
16. Sutter R, Kaplan PW. Electroencephalographic criteria for nonconvulsive status epilepticus: synopsis and comprehensive survey. Epilepsia 2012;53 Suppl 3:1–51.
17. Ristic AJ, Marjanovic I, Brajkovic L, et al. Balint-like syndrome as an unusual representation of non-convulsive status epilepticus. Epileptic Disord 2012;14:80–4.
18. Trinka E, Hofler J, Zerbs A. Causes of status epilepticus. Epilepsia 2012;53 Suppl 4:127–38.
19. DeLorenzo RJ, Hauser WA, Towne AR, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology 1996;46:1029–35.
20. Treiman DM, Meyers PD, Walton NY, et al. A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med 1998;339:792–8.
21. Carrera E, Michel P, Despland PA, et al. Continuous assessment of electrical epileptic activity in acute stroke. Neurology 2006;67:99–104.
22. Towne AR, Waterhouse EJ, Boggs JG, et al. Prevalence of nonconvulsive status epilepticus in comatose patients. Neurology 2000;54:340–5.
23. Claassen J, Jette N, Chum F, et al. Electrographic seizures and periodic discharges after intracerebral hemorrhage. Neurology 2007;69:1356–65.
24. Claassen J, Peery S, Kreiter KT, et al. Predictors and clinical impact of epilepsy after subarachnoid hemorrhage. Neurology 2003;60:208–14.
25. Claassen J, Perotte A, Albers D, et al. Nonconvulsive seizures after subarachnoid hemorrhage: Multimodal detection and outcomes. Ann Neurol 2013;74:53–64.
26. Lindgren C, Nordh E, Naredi S, Olivecrona M. Frequency of non-convulsive seizures and non-convulsive status epilepticus in subarachnoid hemorrhage patients in need of controlled ventilation and sedation. Neurocrit Care 2012;17:367–73.
27. Cocito L, Audenino D, Primavera A. Altered mental state and nonconvulsive status epilepticus in patients with cancer. Arch Neurol 2001;58:1310.
28. Vespa PM, Nuwer MR, Nenov V, et al. Increased incidence and impact of nonconvulsive and convulsive seizures after traumatic brain injury as detected by continuous electroencephalographic monitoring. J Neurosurg 1999;91:750–60.
29. Tian L, Li Y, Xue X, et al. Super-refractory status epilepticus in West China. Acta Neurol Scand 2015;132:1–6.
30. Holtkamp M, Othman J, Buchheim K, et al. A “malignant” variant of status epilepticus. Arch Neurol 2005;62:1428–31.
31. Lowenstein DH, Alldredge BK. Status epilepticus at an urban public hospital in the 1980s. Neurology 1993;43:483–8.
32. Mayer SA, Claassen J, Lokin J, et al. Refractory status epilepticus: frequency, risk factors, and impact on outcome. Arch Neurol 2002;59:205–10.
33. Rossetti AO, Logroscino G, Bromfield EB. Refractory status epilepticus: effect of treatment aggressiveness on prognosis. Arch Neurol 2005;62:1698–702.
34. Novy J, Logroscino G, Rossetti AO. Refractory status epilepticus: a prospective observational study. Epilepsia 2010;51:251–6.
35. Garzon E, Fernandes RM, Sakamoto AC. Analysis of clinical characteristics and risk factors for mortality in human status epilepticus. Seizure 2003;12:337–45.
36. Sutter R, Kaplan PW, Marsch S, et al. Early predictors of refractory status epilepticus: an international two-center study. Eur J Neurol 2015;22:79–85.
37. Wijdicks EF, Sharbrough FW. New-onset seizures in critically ill patients. Neurology 1993;43:1042–4.
38. Bleck TP, Smith MD, Pierre-Louis SJ, et al. Neurologic complications of critical medical illnesses. Crit Care Med 1993;21:98–103.
39. Oddo M, Carrera E, Claassen J, et al. Continuous electroencephalography in the medical intensive care unit. Crit Care Med 2009;37:2051–6.
40. Gilmore EJ, Gaspard N, Choi HA, et al. Acute brain failure in severe sepsis: a prospective study in the medical intensive care unit utilizing continuous EEG monitoring. Intensive Care Med 2015;41:686–94.
41. Misra UK, Kalita J, Patel, R. Sodium valproate vs phenytoin in status epilepticus: a pilot study. Neurology 2006;67:340–2.
42. Kim A, Kim JE, Paek YM, et al. Cefepime-induced non-convulsive status epilepticus (NCSE). J Epilepsy Res 2013;3:39–41.
43. Naeije G, Lorent S, Vincent JL, Legros B. Continuous epileptiform discharges in patients treated with cefepime or meropenem. Arch Neurol 2011;68:1303–7.
44. Tan RY, Neligan A, Shorvon SD. The uncommon causes of status epilepticus: a systematic review. Epilepsy Res 2010;91:111–22.
45. Khawaja AM, DeWolfe JL, Miller DW, Szaflarski JP. New-onset refractory status epilepticus (NORSE) - The potential role for immunotherapy. Epilepsy Behav 2015;47:17–23.
46. Davis R, Dalmau J. Autoimmunity, seizures, and status epilepticus. Epilepsia 2013;54 Suppl 6:46–9.
47. Lopinto-Khoury C, Sperling MR. Autoimmune status epilepticus. Curr Treat Options Neurol 2013;15:545–56.
48. Bansal P, Zutshi D, Suchdev K, et al. Alpha 3 ganglionic acetylcholine receptor antibody associated refractory status epilepticus. Seizure 2016;35:1–3.
49. Chen JW, Wasterlain CG. Status epilepticus: pathophysiology and management in adults. Lancet Neurol 2006;5:246–56.
50. DeLorenzo RJ, Garnett LK, Towne AR, et al. Comparison of status epilepticus with prolonged seizure episodes lasting from 10 to 29 minutes. Epilepsia 1999;40:164–9.
51. Lowenstein DH, Alldredge BK. Status epilepticus. N Engl J Med 1998;338:970–6.
52. Millikan D, Rice B, Silbergleit R. Emergency treatment of status epilepticus: current thinking. Emerg Med Clin North Am 2009;27:101–13, ix.
53. Fountain NB. Status epilepticus: risk factors and complications. Epilepsia 2000;41 Suppl 2:S23–30.
54. Aminoff MJ, Simon RP. Status epilepticus. Causes, clinical features and consequences in 98 patients. Am J Med 1980;69:657–66.
55. Meldrum BS, Brierley JB. Prolonged epileptic seizures in primates. Ischemic cell change and its relation to ictal physiological events. Arch Neurol 1973;28:10–7.
56. Meldrum BS, Vigouroux RA, Brierley JB. Systemic factors and epileptic brain damage. Prolonged seizures in paralyzed, artificially ventilated baboons. Arch Neurol 1973;29:82–7.
57. Loscher W. Mechanisms of drug resistance in status epilepticus. Epilepsia 2007;48 Suppl 8:74–7.
58. Jacob TC, Moss SJ, Jurd R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat Rev Neurosci 2008;9:331–43.
59. Arancibia-Carcamo IL, Kittler JT. Regulation of GABA(A) receptor membrane trafficking and synaptic localization. Pharmacol Ther 2009;123:17–31.
60. Bohnsack JP, Carlson SL, Morrow AL. Differential regulation of synaptic and extrasynaptic alpha4 GABA(A) receptor populations by protein kinase A and protein kinase C in cultured cortical neurons. Neuropharmacology 2016;105:124–32.
61. Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci 2005;6:215–29.
62. Smith KR, Kittler JT. The cell biology of synaptic inhibition in health and disease. Curr Opin Neurobiol 2010;20:550–6.
63. Wasterlain CG, Naylor DE, Liu H, et al. Trafficking of NMDA receptors during status epilepticus: therapeutic implications. Epilepsia 2013;54 Suppl 6:78–80.
64. Bankstahl JP, Loscher W. Resistance to antiepileptic drugs and expression of P-glycoprotein in two rat models of status epilepticus. Epilepsy Res 2008;82:70–85.
65. Henshall DC, Diaz-Hernandez M, Miras-Portugal MT, Engel T. P2X receptors as targets for the treatment of status epilepticus. Front Cell Neurosci 2013;7:237.
66. Lamsa K, Taira T. Use-dependent shift from inhibitory to excitatory GABAA receptor action in SP-O interneurons in the rat hippocampal CA3 area. J Neurophysiol 2003;90:1983–95.
67. Cock HR, Tong X, Hargreaves IP, et al. Mitochondrial dysfunction associated with neuronal death following status epilepticus in rat. Epilepsy Res 2002;48:157–68.
68. Friedman A, Dingledine R, Molecular cascades that mediate the influence of inflammation on epilepsy. Epilepsia 2011;52 Suppl 3:33–39.
69. Henshall DC. MicroRNAs in the pathophysiology and treatment of status epilepticus. Front Mol Neurosci 2013;6:37.
70. Vespa P, Tubi M, Claassen J, et al. Metabolic crisis occurs with seizures and periodic discharges after brain trauma. Ann Neurol 2016;79:579–90.
71. Witsch J, Frey HP, Schmidt JM, et al. Electroencephalographic periodic discharges and frequency-dependent brain tissue hypoxia in acute brain injury. JAMA Neurol 2017;74:301–9.
72. Rodriguez Ruiz A, Vlachy J, Lee JW, et al. Association of periodic and rhythmic electroencephalographic patterns with seizures in critically ill patients. JAMA Neurol 2017;74:181–8.
73. DeLorenzo RJ, Waterhouse EJ, Towne AR, et al. Persistent nonconvulsive status epilepticus after the control of convulsive status epilepticus. Epilepsia 1998;39:833–40.
74. Benbadis SR, Chen S, Melo M. What’s shaking in the ICU? The differential diagnosis of seizures in the intensive care setting. Epilepsia 2010;51:2338–40.
75. Pandian JD, Cascino GD, So EL, et al. Digital video-electroencephalographic monitoring in the neurological-neurosurgical intensive care unit: clinical features and outcome. Arch Neurol 2004;61:1090–4.
76. Irani SR, Vincent A, Schott JM. Autoimmune encephalitis. BMJ 2011;342:d1918.
77. Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011;69:892–900.
78. Herman ST, Abend NS, Bleck TP, et al. Consensus statement on continuous EEG in critically ill adults and children, part I: indications. J Clin Neurophysiol 2015;32:87–95.
79. Brenner RP. Is it status? Epilepsia 2002;43 Suppl 3:103–113.
80. Cook AM, Castle A, Green A, et al. Practice variations in the management of status epilepticus. Neurocrit Care 2012;17:24–30.
81. Alldredge BK, Gelb AM, Isaacs SM, et al. A comparison of lorazepam, diazepam, and placebo for the treatment of out-of-hospital status epilepticus. N Engl J Med 2001;345:631–7.
82. Silbergleit R, Durkalski V, Lowenstein D, et al. Intramuscular versus intravenous therapy for prehospital status epilepticus. N Engl J Med 2012;366:591–600.
83. Shorvon S. Super-refractory status epilepticus: an approach to therapy in this difficult clinical situation. Epilepsia 2011;52 Suppl 8:53–6.
84. Varelas P, Mirski MA. Management of status epilepticus in adults. Hosp Physician Board Rev Man 2014;2:1–15.
85. McIntyre J, Robertson S, Norris E, et al. Safety and efficacy of buccal midazolam versus rectal diazepam for emergency treatment of seizures in children: a randomised controlled trial. Lancet 2005;366:205–10.
86. Misra UK, Kalita J, Maurya PK. Levetiracetam versus lorazepam in status epilepticus: a randomized, open labeled pilot study. J Neurol 2012;259:645–8.
87. Vohra TT, Miller JB, Nicholas KS, et al. Endotracheal intubation in patients treated for prehospital status epilepticus. Neurocrit Care 2015;23:33–43.
88. Agarwal P, Kumar N, Chandra R, et al. Randomized study of intravenous valproate and phenytoin in status epilepticus. Seizure 2007;16:527–32.
89. Trinka E. What is the evidence to use new intravenous AEDs in status epilepticus? Epilepsia 2011;52 Suppl 8:35–38.
90. Yasiry Z, Shorvon SD. The relative effectiveness of five antiepileptic drugs in treatment of benzodiazepine-resistant convulsive status epilepticus: a meta-analysis of published studies. Seizure 2014;23:167–74.
91. Moreno Morales EY, Fernandez Peleteiro M, Bondy Pena EC, et al. Observational study of intravenous lacosamide in patients with convulsive versus non-convulsive status epilepticus. Clin Drug Investig 2015;35:463–9.
92. Paquette V, Culley C, Greanya ED, Ensom MH. Lacosamide as adjunctive therapy in refractory epilepsy in adults: a systematic review. Seizure 2015;25:1–17.
93. Varelas PN, Corry J, Rehman M, et al. Management of status epilepticus in neurological versus medical intensive care unit: does it matter? Neurocrit Care 2013;19:4–9.
94. Hofler J, Trinka E. Lacosamide as a new treatment option in status epilepticus. Epilepsia 2013;54:393–404.
95. Kellinghaus C, Berning S, Stogbauer F. Intravenous lacosamide or phenytoin for treatment of refractory status epilepticus. Acta Neurol Scand 2014;129:294–9.
96. Santamarina E, Toledo M, Sueiras M, et al. Usefulness of intravenous lacosamide in status epilepticus. J Neurol 2013;260:3122–8.
97. Newey CR, Le NM, Ahrens C, et al. The safety and effectiveness of intravenous lacosamide for refractory status epilepticus in the critically ill. Neurocrit Care 2017;26:273–9.
98. Towne AR, Garnett LK, Waterhouse EJ, et al. The use of topiramate in refractory status epilepticus. Neurology 2003;60:332–4.
99. Hottinger A, Sutter R, Marsch S, Ruegg S. Topiramate as an adjunctive treatment in patients with refractory status epilepticus: an observational cohort study. CNS Drugs 2012;26:761–72.
100. Madzar D, Kuramatsu JB, Gerner ST, et al. Assessing the value of topiramate in refractory status epilepticus. Seizure 2016;38:7–10.
101. Sivakumar S, Ibrahim M, Parker D Jr, et al. An effective add-on therapy in refractory status epilepticus. Epilepsia 2015;56:e83–89.
102. Madzar D, Geyer A, Knappe RU, et al. Effects of clobazam for treatment of refractory status epilepticus. BMC Neurol 2016;16:202.
103. Swisher CB, Doreswamy M, Gingrich KJ, et al. Phenytoin, levetiracetam, and pregabalin in the acute management of refractory status epilepticus in patients with brain tumors. Neurocrit Care 2012;16:109–13.
104. Smith H, Sinson G, Varelas P. Vasopressors and propofol infusion syndrome in severe head trauma. Neurocrit Care 2009;10:166–72.
105. Cuero MR, Varelas PN. Super-refractory status epilepticus. Curr Neurol Neurosci Rep 2015;15:74.
106. Varelas PN. How I treat status epilepticus in the Neuro-ICU. Neurocrit Care 2008;9:153–7.
107. Varelas PN, Spanaki MV, Mirski MA. Status epilepticus: an update. Curr Neurol Neurosci Rep 2013;13:357.
108. Krishnamurthy KB, Drislane FW. Depth of EEG suppression and outcome in barbiturate anesthetic treatment for refractory status epilepticus. Epilepsia 1999;40:759–62.
109. Krishnamurthy KB, Drislane FW. Relapse and survival after barbiturate anesthetic treatment of refractory status epilepticus. Epilepsia 1996;37:863–7.
110. Ferlisi M, Shorvon S. The outcome of therapies in refractory and super-refractory convulsive status epilepticus and recommendations for therapy. Brain 2012;135:2314–28.
111. Kramer AH. Early ketamine to treat refractory status epilepticus. Neurocrit Care 2012;16:299–305.
112. Synowiec AS, Singh DS, Yenugadhati V, et al. Ketamine use in the treatment of refractory status epilepticus. Epilepsy Res 2013;105:183–8.
113. Gaspard N, Foreman B, Judd LM, et al. Intravenous ketamine for the treatment of refractory status epilepticus: a retrospective multicenter study. Epilepsia 2013;54:1498–503.
114. Schulze-Bonhage A, Kurthen M, Walger P, Elger CE. Pharmacorefractory status epilepticus due to low vitamin B6 levels during pregnancy. Epilepsia 2004;45:81–4.
115. Shorvon S. Clinical trials in acute repetitive seizures and status epilepticus. Epileptic Disord 2012;14:138–47.
116. Visser NA, Braun KP, Leijten FS, et al. Magnesium treatment for patients with refractory status epilepticus due to POLG1-mutations. J Neurol 2011;258:218–22.
117. Thakur KT, Probasco JC, Hocker SE, et al. Ketogenic diet for adults in super-refractory status epilepticus. Neurology 2014;82:665–70.
118. Corry JJ, Dhar R, Murphy T, Diringer MN. Hypothermia for refractory status epilepticus. Neurocrit Care 2008;9:189–97.
119. Guilliams K, Rosen M, Buttram S, et al. Hypothermia for pediatric refractory status epilepticus. Epilepsia 2013;54:1586–94.
120. Legriel S, Lemiale V, Schenck M, et al. Hypothermia for neuroprotection in convulsive status epilepticus. N Engl J Med 2016;375:2457–67.
121. Wasterlain CG, Baldwin R, Naylor DE, et al. Rational polytherapy in the treatment of acute seizures and status epilepticus. Epilepsia 2011;52 Suppl 8:70–1.
122. Rogawski MA, Loya CM, Reddy K, et al. Neuroactive steroids for the treatment of status epilepticus. Epilepsia 2013;54 Suppl 6:93–8.
123. Towne AR, Pellock JM, Ko D, DeLorenzo RJ. Determinants of mortality in status epilepticus. Epilepsia 1994;35:27–34.
124. DeLorenzo RJ, Towne AR, Pellock JM, Ko D. Status epilepticus in children, adults, and the elderly. Epilepsia 1992;33 Suppl 4:S15–25.
125. Legriel S, Mourvillier B, Bele N, et al. Outcomes in 140 critically ill patients with status epilepticus. Intensive Care Med 2008;34:476–80.
126. Pugin D, Foreman B, De Marchis GM, et al. Is pentobarbital safe and efficacious in the treatment of super-refractory status epilepticus: a cohort study. Crit Care 2014;18:R103.
127. Hocker SE, Britton JW, Mandrekar JN, et al. Predictors of outcome in refractory status epilepticus. JAMA Neurol 2013;70:72–7.
128. Hocker S, Tatum WO, LaRoche S, Freeman WD. Refractory and super-refractory status epilepticus--an update. Curr Neurol Neurosci Rep 2014;14:452.
129. Sutter R, Marsch S, Fuhr P, et al. Anesthetic drugs in status epilepticus: risk or rescue? A 6-year cohort study. Neurology 2014;82:656–64.
130. Rossetti AO, Logroscino G, Bromfield EB. A clinical score for prognosis of status epilepticus in adults. Neurology 2006;66:1736–8.
131. Rossetti AO, Logroscino G, Milligan TA, et al. Status Epilepticus Severity Score (STESS): a tool to orient early treatment strategy. J Neurol 2008;255:1561–6.
132. Hesdorffer DC, Logroscino G, Cascino GD, Hauser WA. Recurrence of afebrile status epilepticus in a population-based study in Rochester, Minnesota. Neurology 2007;69:73–8.
133. Varelas PN, Mirski MA. Seizures in the adult intensive care unit. J Neurosurg Anesthesiol 2001;13:163–75.
134. Varelas PN, Mirski MA. Status epilepticus. Curr Neurol Neurosci Rep 2009;9:469–76.
Bed Partners May Perpetuate Insomnia
BOSTON—In an attempt to be supportive, bed partners of patients with insomnia may engage in behaviors that unintentionally perpetuate insomnia, according to preliminary results presented at the 31st Annual Meeting of the Associated Professional Sleep Societies. Bed partners also may make accommodations that affect their own sleep and life outside of work.
“It is possible that partners are unwittingly perpetuating insomnia symptoms in the patient with insomnia,” said Alix Mellor, PhD, a postdoctoral research fellow and coordinator of the Researching Effective Sleep Treatments (REST) project in the School of Psychological Sciences at Monash University in Victoria, Australia. “It is therefore important for more data to be collected to determine whether insomnia treatments may better benefit patients and their partners by proactively assessing and addressing bed partner behaviors in treatment programs.” 
Relationship problems and mismatched bedtimes and wake times within couples have been linked to insomnia. Research in disorders such as obsessive–compulsive disorder (OCD) and depression indicates that partners can reinforce maladaptive behaviors inadvertently. No data have documented the specific behaviors engaged in by partners of individuals with insomnia, however, and these behaviors might perpetuate the disorder.
Partners Encouraged Early Bedtimes
In a randomized controlled trial investigating partner-assisted interventions for insomnia, Dr. Mellor and colleagues studied 31 bed partners (14 women) of patients seeking treatment for insomnia. At baseline, partners completed the Family Accommodation Scale for OCD, Beck Anxiety Inventory, and Dyadic Adjustment Scale. Patients with insomnia completed the Insomnia Severity Index and other questionnaires at baseline and kept a sleep diary for one week prior to treatment initiation.
Approximately 74% of bed partners encouraged an early bedtime or late wake time, which directly contradicts the principles of cognitive behavioral therapy for insomnia (CBT-I). In addition, 42% of bed partners encouraged doing other things in bed, such as reading or watching TV, and 35% encouraged naps, caffeine, or reduced daytime activities. About 11% encouraged their partners to take sleep medication or alcohol to improve sleep. About 56% of bed partners reported that their partner becomes distressed when they do not provide assistance with their insomnia.
Half of bed partners adjusted their own sleep. Furthermore, 39% of bed partners adjusted their family routine, and half modified their leisure activities in response to their partners’ insomnia. Approximately 17% modified their work schedule. The bed partners who attempted to be helpful experienced more anxiety, even though the patients with insomnia perceived the relationship to be more satisfying.
Neurologists Could Explain Principles Behind Treatment
“If neurologists are using behavioral interventions like CBT-I, our findings suggest they should ask their patients about a few specific things their partner might do to unintentionally undermine the treatment,” said Sean P.A. Drummond, PhD, Professor of Clinical Neuroscience at Monash University and lead investigator of this study. “The neurologist should take the time to make sure the patient knows why those things are incompatible with treatment and encourage the patient to explain those ideas to the partner. An extra bonus would be if the patient could ask the partner for help in making sure they follow the treatment guidelines.”
Dr. Drummond and colleagues are studying a form of CBT-I that incorporates assistance from a partner. “We are explicitly incorporating the partner into the intervention in an effort to improve outcomes,” said Dr. Drummond.
—Erik Greb
Suggested Reading
Ellis JG, Deary V, Troxel WM. The role of perceived partner alliance on the efficacy of CBT-I: preliminary findings from the Partner Alliance in Insomnia Research Study (PAIRS). Behav Sleep Med. 2015;13(1):64-72.
McCall WV. Cognitive behavioral therapy for insomnia (CBT-I): What is known, and advancing the science by avoiding the pitfalls of the placebo effect. Sleep Med Rev. 2017 May 5 [Epub ahead of print].
Rogojanski J, Carney CE, Monson CM. Interpersonal factors in insomnia: a model for integrating bed partners into cognitive behavioral therapy for insomnia. Sleep Med Rev. 2013;17(1):55-64.
van Straten A, van der Zweerde T, Kleiboer A, et al. Cognitive and behavioral therapies in the treatment of insomnia: A meta-analysis. Sleep Med Rev. 2017 Feb 9 [Epub ahead of print].
BOSTON—In an attempt to be supportive, bed partners of patients with insomnia may engage in behaviors that unintentionally perpetuate insomnia, according to preliminary results presented at the 31st Annual Meeting of the Associated Professional Sleep Societies. Bed partners also may make accommodations that affect their own sleep and life outside of work.
“It is possible that partners are unwittingly perpetuating insomnia symptoms in the patient with insomnia,” said Alix Mellor, PhD, a postdoctoral research fellow and coordinator of the Researching Effective Sleep Treatments (REST) project in the School of Psychological Sciences at Monash University in Victoria, Australia. “It is therefore important for more data to be collected to determine whether insomnia treatments may better benefit patients and their partners by proactively assessing and addressing bed partner behaviors in treatment programs.”
Relationship problems and mismatched bedtimes and wake times within couples have been linked to insomnia. Research in disorders such as obsessive–compulsive disorder (OCD) and depression indicates that partners can reinforce maladaptive behaviors inadvertently. No data have documented the specific behaviors engaged in by partners of individuals with insomnia, however, and these behaviors might perpetuate the disorder.
Partners Encouraged Early Bedtimes
In a randomized controlled trial investigating partner-assisted interventions for insomnia, Dr. Mellor and colleagues studied 31 bed partners (14 women) of patients seeking treatment for insomnia. At baseline, partners completed the Family Accommodation Scale for OCD, Beck Anxiety Inventory, and Dyadic Adjustment Scale. Patients with insomnia completed the Insomnia Severity Index and other questionnaires at baseline and kept a sleep diary for one week prior to treatment initiation.
Approximately 74% of bed partners encouraged an early bedtime or late wake time, which directly contradicts the principles of cognitive behavioral therapy for insomnia (CBT-I). In addition, 42% of bed partners encouraged doing other things in bed, such as reading or watching TV, and 35% encouraged naps, caffeine, or reduced daytime activities. About 11% encouraged their partners to take sleep medication or alcohol to improve sleep. About 56% of bed partners reported that their partner becomes distressed when they do not provide assistance with their insomnia.
Half of bed partners adjusted their own sleep. Furthermore, 39% of bed partners adjusted their family routine, and half modified their leisure activities in response to their partners’ insomnia. Approximately 17% modified their work schedule. The bed partners who attempted to be helpful experienced more anxiety, even though the patients with insomnia perceived the relationship to be more satisfying.
Neurologists Could Explain Principles Behind Treatment
“If neurologists are using behavioral interventions like CBT-I, our findings suggest they should ask their patients about a few specific things their partner might do to unintentionally undermine the treatment,” said Sean P.A. Drummond, PhD, Professor of Clinical Neuroscience at Monash University and lead investigator of this study. “The neurologist should take the time to make sure the patient knows why those things are incompatible with treatment and encourage the patient to explain those ideas to the partner. An extra bonus would be if the patient could ask the partner for help in making sure they follow the treatment guidelines.”
Dr. Drummond and colleagues are studying a form of CBT-I that incorporates assistance from a partner. “We are explicitly incorporating the partner into the intervention in an effort to improve outcomes,” said Dr. Drummond.
—Erik Greb
Suggested Reading
Ellis JG, Deary V, Troxel WM. The role of perceived partner alliance on the efficacy of CBT-I: preliminary findings from the Partner Alliance in Insomnia Research Study (PAIRS). Behav Sleep Med. 2015;13(1):64-72.
McCall WV. Cognitive behavioral therapy for insomnia (CBT-I): What is known, and advancing the science by avoiding the pitfalls of the placebo effect. Sleep Med Rev. 2017 May 5 [Epub ahead of print].
Rogojanski J, Carney CE, Monson CM. Interpersonal factors in insomnia: a model for integrating bed partners into cognitive behavioral therapy for insomnia. Sleep Med Rev. 2013;17(1):55-64.
van Straten A, van der Zweerde T, Kleiboer A, et al. Cognitive and behavioral therapies in the treatment of insomnia: A meta-analysis. Sleep Med Rev. 2017 Feb 9 [Epub ahead of print].
BOSTON—In an attempt to be supportive, bed partners of patients with insomnia may engage in behaviors that unintentionally perpetuate insomnia, according to preliminary results presented at the 31st Annual Meeting of the Associated Professional Sleep Societies. Bed partners also may make accommodations that affect their own sleep and life outside of work.
“It is possible that partners are unwittingly perpetuating insomnia symptoms in the patient with insomnia,” said Alix Mellor, PhD, a postdoctoral research fellow and coordinator of the Researching Effective Sleep Treatments (REST) project in the School of Psychological Sciences at Monash University in Victoria, Australia. “It is therefore important for more data to be collected to determine whether insomnia treatments may better benefit patients and their partners by proactively assessing and addressing bed partner behaviors in treatment programs.”
Relationship problems and mismatched bedtimes and wake times within couples have been linked to insomnia. Research in disorders such as obsessive–compulsive disorder (OCD) and depression indicates that partners can reinforce maladaptive behaviors inadvertently. No data have documented the specific behaviors engaged in by partners of individuals with insomnia, however, and these behaviors might perpetuate the disorder.
Partners Encouraged Early Bedtimes
In a randomized controlled trial investigating partner-assisted interventions for insomnia, Dr. Mellor and colleagues studied 31 bed partners (14 women) of patients seeking treatment for insomnia. At baseline, partners completed the Family Accommodation Scale for OCD, Beck Anxiety Inventory, and Dyadic Adjustment Scale. Patients with insomnia completed the Insomnia Severity Index and other questionnaires at baseline and kept a sleep diary for one week prior to treatment initiation.
Approximately 74% of bed partners encouraged an early bedtime or late wake time, which directly contradicts the principles of cognitive behavioral therapy for insomnia (CBT-I). In addition, 42% of bed partners encouraged doing other things in bed, such as reading or watching TV, and 35% encouraged naps, caffeine, or reduced daytime activities. About 11% encouraged their partners to take sleep medication or alcohol to improve sleep. About 56% of bed partners reported that their partner becomes distressed when they do not provide assistance with their insomnia.
Half of bed partners adjusted their own sleep. Furthermore, 39% of bed partners adjusted their family routine, and half modified their leisure activities in response to their partners’ insomnia. Approximately 17% modified their work schedule. The bed partners who attempted to be helpful experienced more anxiety, even though the patients with insomnia perceived the relationship to be more satisfying.
Neurologists Could Explain Principles Behind Treatment
“If neurologists are using behavioral interventions like CBT-I, our findings suggest they should ask their patients about a few specific things their partner might do to unintentionally undermine the treatment,” said Sean P.A. Drummond, PhD, Professor of Clinical Neuroscience at Monash University and lead investigator of this study. “The neurologist should take the time to make sure the patient knows why those things are incompatible with treatment and encourage the patient to explain those ideas to the partner. An extra bonus would be if the patient could ask the partner for help in making sure they follow the treatment guidelines.”
Dr. Drummond and colleagues are studying a form of CBT-I that incorporates assistance from a partner. “We are explicitly incorporating the partner into the intervention in an effort to improve outcomes,” said Dr. Drummond.
—Erik Greb
Suggested Reading
Ellis JG, Deary V, Troxel WM. The role of perceived partner alliance on the efficacy of CBT-I: preliminary findings from the Partner Alliance in Insomnia Research Study (PAIRS). Behav Sleep Med. 2015;13(1):64-72.
McCall WV. Cognitive behavioral therapy for insomnia (CBT-I): What is known, and advancing the science by avoiding the pitfalls of the placebo effect. Sleep Med Rev. 2017 May 5 [Epub ahead of print].
Rogojanski J, Carney CE, Monson CM. Interpersonal factors in insomnia: a model for integrating bed partners into cognitive behavioral therapy for insomnia. Sleep Med Rev. 2013;17(1):55-64.
van Straten A, van der Zweerde T, Kleiboer A, et al. Cognitive and behavioral therapies in the treatment of insomnia: A meta-analysis. Sleep Med Rev. 2017 Feb 9 [Epub ahead of print].
Shift Work in Midlife Increases Risk of Dementia
BOSTON—Midlife shift work is associated with an increased risk of incident dementia in later life, according to research presented at the 31st Annual Meeting of the Associated Professional Sleep Societies. The association is stronger with longer duration of shift work, said Kathleen Bokenberger, a doctoral student in the Department of Medical Epidemiology and Biostatistics at the Karolinska Institutet in Stockholm. 
“To the best of our knowledge, this is the first study to find a connection between shift work and greater dementia incidence,” said Ms. Bokenberger. “As such, we will need confirmation from other studies, especially if they have more nuanced measures of shift work, and perhaps repeated measures of shift work.”
An Analysis of Two Population-Based Cohorts
Shift work interferes with the circadian and homeostatic regulation of sleep and can lead to insomnia and excessive sleepiness. A prospective Danish study found that rotating and evening shift work were associated with increased dementia mortality, but the link between shift work and dementia is still relatively unexplored, said Ms. Bokenberger.
She and her colleagues followed two population-based cohorts from the Swedish Twin Registry. The first cohort, which the researchers called STR-1973, included 13,283 people who were age 30 or older when they received a mailed questionnaire in 1973 that elicited information about shift work status and duration. The Screening Across the Lifespan Twin (SALT) sample included 41,199
The primary outcome was all-cause dementia, and dementia diagnoses were obtained from Swedish national health registers. Investigators followed participants until the date of dementia diagnosis, death, or the end of follow-up in 2015.
A Dose-Response Relationship
The mean age at baseline was 38 in the STR-1973 cohort and 58 in the SALT cohort. Median follow-up time was 41 years in the STR-73 cohort and 14 years in the SALT cohort. The proportion of participants with shift work history was 17% in the STR-1973 cohort. The proportion of people who worked nights was 30% in the SALT cohort. Dementia incidence was 7.4% in the STR-1973 cohort and 5.0% in the SALT cohort.
The researchers used Cox proportional hazard models to analyze the data and adjust for age, sex, education, cardiovascular disease, stroke, and type 2 diabetes. Having shift work versus day work was associated with an increased hazard of dementia (hazard ratio [HR], 1.40), as was having night work versus day work (HR, 1.13). Spline models indicated modest dose-response relationships in which longer duration of shift work and night work predicted greater dementia risk.
Information about APOE e4 genotype was available for subsamples of both cohorts. APOE e4 seemed to be an effect modifier for the association of night work and dementia, but not for that of shift work and dementia. APOE e4 carriers with 20 or more years of shift work had a fourfold increased risk of dementia. APOE e4 carriers with 20 or more years of night work had a twofold increased risk of dementia. The sample sizes were small, however, and “it is possible that these findings are spurious,” said Ms. Bokenberger.
“Individuals in the labor force might consider minimizing shift work exposure or managing work scheduling practices, but this is dependent on individual differences and tolerances to certain types of shift work,” she concluded.
—Erik Greb
Suggested Reading
Bokenberger K, Ström P, Dahl Aslan AK, et al. Shift work and cognitive aging: a longitudinal study. Scand J Work Environ Health. 2017 Mar 31 [Epub ahead of print].
Jørgensen JT, Karlsen S, Stayner L, et al. Shift work and overall and cause-specific mortality in the Danish nurse cohort. Scand J Work Environ Health. 2017;43(2):117-126.
BOSTON—Midlife shift work is associated with an increased risk of incident dementia in later life, according to research presented at the 31st Annual Meeting of the Associated Professional Sleep Societies. The association is stronger with longer duration of shift work, said Kathleen Bokenberger, a doctoral student in the Department of Medical Epidemiology and Biostatistics at the Karolinska Institutet in Stockholm.
“To the best of our knowledge, this is the first study to find a connection between shift work and greater dementia incidence,” said Ms. Bokenberger. “As such, we will need confirmation from other studies, especially if they have more nuanced measures of shift work, and perhaps repeated measures of shift work.”
An Analysis of Two Population-Based Cohorts
Shift work interferes with the circadian and homeostatic regulation of sleep and can lead to insomnia and excessive sleepiness. A prospective Danish study found that rotating and evening shift work were associated with increased dementia mortality, but the link between shift work and dementia is still relatively unexplored, said Ms. Bokenberger.
She and her colleagues followed two population-based cohorts from the Swedish Twin Registry. The first cohort, which the researchers called STR-1973, included 13,283 people who were age 30 or older when they received a mailed questionnaire in 1973 that elicited information about shift work status and duration. The Screening Across the Lifespan Twin (SALT) sample included 41,199
The primary outcome was all-cause dementia, and dementia diagnoses were obtained from Swedish national health registers. Investigators followed participants until the date of dementia diagnosis, death, or the end of follow-up in 2015.
A Dose-Response Relationship
The mean age at baseline was 38 in the STR-1973 cohort and 58 in the SALT cohort. Median follow-up time was 41 years in the STR-73 cohort and 14 years in the SALT cohort. The proportion of participants with shift work history was 17% in the STR-1973 cohort. The proportion of people who worked nights was 30% in the SALT cohort. Dementia incidence was 7.4% in the STR-1973 cohort and 5.0% in the SALT cohort.
The researchers used Cox proportional hazard models to analyze the data and adjust for age, sex, education, cardiovascular disease, stroke, and type 2 diabetes. Having shift work versus day work was associated with an increased hazard of dementia (hazard ratio [HR], 1.40), as was having night work versus day work (HR, 1.13). Spline models indicated modest dose-response relationships in which longer duration of shift work and night work predicted greater dementia risk.
Information about APOE e4 genotype was available for subsamples of both cohorts. APOE e4 seemed to be an effect modifier for the association of night work and dementia, but not for that of shift work and dementia. APOE e4 carriers with 20 or more years of shift work had a fourfold increased risk of dementia. APOE e4 carriers with 20 or more years of night work had a twofold increased risk of dementia. The sample sizes were small, however, and “it is possible that these findings are spurious,” said Ms. Bokenberger.
“Individuals in the labor force might consider minimizing shift work exposure or managing work scheduling practices, but this is dependent on individual differences and tolerances to certain types of shift work,” she concluded.
—Erik Greb
Suggested Reading
Bokenberger K, Ström P, Dahl Aslan AK, et al. Shift work and cognitive aging: a longitudinal study. Scand J Work Environ Health. 2017 Mar 31 [Epub ahead of print].
Jørgensen JT, Karlsen S, Stayner L, et al. Shift work and overall and cause-specific mortality in the Danish nurse cohort. Scand J Work Environ Health. 2017;43(2):117-126.
BOSTON—Midlife shift work is associated with an increased risk of incident dementia in later life, according to research presented at the 31st Annual Meeting of the Associated Professional Sleep Societies. The association is stronger with longer duration of shift work, said Kathleen Bokenberger, a doctoral student in the Department of Medical Epidemiology and Biostatistics at the Karolinska Institutet in Stockholm.
“To the best of our knowledge, this is the first study to find a connection between shift work and greater dementia incidence,” said Ms. Bokenberger. “As such, we will need confirmation from other studies, especially if they have more nuanced measures of shift work, and perhaps repeated measures of shift work.”
An Analysis of Two Population-Based Cohorts
Shift work interferes with the circadian and homeostatic regulation of sleep and can lead to insomnia and excessive sleepiness. A prospective Danish study found that rotating and evening shift work were associated with increased dementia mortality, but the link between shift work and dementia is still relatively unexplored, said Ms. Bokenberger.
She and her colleagues followed two population-based cohorts from the Swedish Twin Registry. The first cohort, which the researchers called STR-1973, included 13,283 people who were age 30 or older when they received a mailed questionnaire in 1973 that elicited information about shift work status and duration. The Screening Across the Lifespan Twin (SALT) sample included 41,199
The primary outcome was all-cause dementia, and dementia diagnoses were obtained from Swedish national health registers. Investigators followed participants until the date of dementia diagnosis, death, or the end of follow-up in 2015.
A Dose-Response Relationship
The mean age at baseline was 38 in the STR-1973 cohort and 58 in the SALT cohort. Median follow-up time was 41 years in the STR-73 cohort and 14 years in the SALT cohort. The proportion of participants with shift work history was 17% in the STR-1973 cohort. The proportion of people who worked nights was 30% in the SALT cohort. Dementia incidence was 7.4% in the STR-1973 cohort and 5.0% in the SALT cohort.
The researchers used Cox proportional hazard models to analyze the data and adjust for age, sex, education, cardiovascular disease, stroke, and type 2 diabetes. Having shift work versus day work was associated with an increased hazard of dementia (hazard ratio [HR], 1.40), as was having night work versus day work (HR, 1.13). Spline models indicated modest dose-response relationships in which longer duration of shift work and night work predicted greater dementia risk.
Information about APOE e4 genotype was available for subsamples of both cohorts. APOE e4 seemed to be an effect modifier for the association of night work and dementia, but not for that of shift work and dementia. APOE e4 carriers with 20 or more years of shift work had a fourfold increased risk of dementia. APOE e4 carriers with 20 or more years of night work had a twofold increased risk of dementia. The sample sizes were small, however, and “it is possible that these findings are spurious,” said Ms. Bokenberger.
“Individuals in the labor force might consider minimizing shift work exposure or managing work scheduling practices, but this is dependent on individual differences and tolerances to certain types of shift work,” she concluded.
—Erik Greb
Suggested Reading
Bokenberger K, Ström P, Dahl Aslan AK, et al. Shift work and cognitive aging: a longitudinal study. Scand J Work Environ Health. 2017 Mar 31 [Epub ahead of print].
Jørgensen JT, Karlsen S, Stayner L, et al. Shift work and overall and cause-specific mortality in the Danish nurse cohort. Scand J Work Environ Health. 2017;43(2):117-126.
Minimally invasive screening for Barrett’s esophagus offers cost-effective alternative
The high costs of endoscopy make screening patients with gastroesophageal reflux disease (GERD) for Barrett’s esophagus a costly endeavor. But using a minimally invasive test followed by endoscopy only if results are positive could cut costs by up to 41%, according to investigators.
The report is in the September issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2017.02.017).
The findings mirror those from a prior study (Gastroenterology. 2013 Jan;144[1]:62-73.e60) of the new cytosponge device, which tests surface esophageal tissue for trefoil factor 3, a biomarker for Barrett’s esophagus, said Curtis R. Heberle, of Massachusetts General Hospital in Boston, and his associates. In addition, two separate models found the cytosponge strategy cost effective compared with no screening (incremental cost-effectiveness ratios [ICERs], about $26,000-$33,000). However, using the cytosponge instead of screening all GERD patients with endoscopy would reduce quality-adjusted life-years (QALYs) by about 1.8-5.5 years for every 1,000 patients.
Rates of esophageal adenocarcinoma have climbed more than sixfold in the United States in 4 decades, and 5-year survival rates remain below 20%. Nonetheless, the high cost of endoscopy and 10%-20% prevalence of GERD makes screening all patients for Barrett’s esophagus infeasible. To evaluate the cytosponge strategy, the researchers fit data from the multicenter BEST2 study (PLoS Med. 2015 Jan; 12[1]: e1001780) into two validated models calibrated to high-quality Surveillance, Epidemiology and End Results (SEER) data on esophageal cancer. Both models compared no screening with a one-time screen by either endoscopy alone or cytosponge with follow-up endoscopy in the event of a positive test. The models assumed patients were male, were 60 years old, and had GERD but not esophageal adenocarcinoma.
Without screening, there were about 14-16 cancer cases and about 15,077 quality-adjusted life years (QALYs) for every 1,000 patients. The cytosponge strategy was associated with about 8-13 cancer cases and about 15,105 QALYs. Endoscopic screening produced the most benefit overall – only about 7-12 cancer cases, with more than 15,100 QALYs. “However, greater benefits were accompanied by higher total costs,” the researchers said. For every 1,000 patients, no screening cost about $704,000 to $762,000, the cytosponge strategy cost about $1.5 to $1.6 million, and population-wide endoscopy cost about $2.1 to $2.2 million. Thus, the cytosponge method would lower the cost of screening by 37%-41% compared with endoscopically screening all men with GERD. The cytosponge was also cost effective in a model of 60-year-old women with GERD.
Using only endoscopic screening was not cost effective in either model, exceeding a $100,000 threshold of willingness to pay by anywhere from $107,000 to $330,000. The cytosponge is not yet available commercially, but the investigators assumed it cost $182 based on information from the manufacturer (Medtronic) and Medicare payments for similar devices. Although the findings withstood variations in indirect costs and age at initial screening, they were “somewhat sensitive” to variations in costs of the cytosponge and its presumed sensitivity and specificity in clinical settings. However, endoscopic screening only became cost effective when the cytosponge test cost at least $225.
The models assumed perfect adherence to screening, which probably exaggerated the effectiveness of the cytosponge and endoscopic screening, the investigators said. They noted that cytosponge screening can be performed without sedation during a short outpatient visit.
The National Institutes of Health provided funding. The investigators had no relevant disclosures.
The high costs of endoscopy make screening patients with gastroesophageal reflux disease (GERD) for Barrett’s esophagus a costly endeavor. But using a minimally invasive test followed by endoscopy only if results are positive could cut costs by up to 41%, according to investigators.
The report is in the September issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2017.02.017).
The findings mirror those from a prior study (Gastroenterology. 2013 Jan;144[1]:62-73.e60) of the new cytosponge device, which tests surface esophageal tissue for trefoil factor 3, a biomarker for Barrett’s esophagus, said Curtis R. Heberle, of Massachusetts General Hospital in Boston, and his associates. In addition, two separate models found the cytosponge strategy cost effective compared with no screening (incremental cost-effectiveness ratios [ICERs], about $26,000-$33,000). However, using the cytosponge instead of screening all GERD patients with endoscopy would reduce quality-adjusted life-years (QALYs) by about 1.8-5.5 years for every 1,000 patients.
Rates of esophageal adenocarcinoma have climbed more than sixfold in the United States in 4 decades, and 5-year survival rates remain below 20%. Nonetheless, the high cost of endoscopy and 10%-20% prevalence of GERD makes screening all patients for Barrett’s esophagus infeasible. To evaluate the cytosponge strategy, the researchers fit data from the multicenter BEST2 study (PLoS Med. 2015 Jan; 12[1]: e1001780) into two validated models calibrated to high-quality Surveillance, Epidemiology and End Results (SEER) data on esophageal cancer. Both models compared no screening with a one-time screen by either endoscopy alone or cytosponge with follow-up endoscopy in the event of a positive test. The models assumed patients were male, were 60 years old, and had GERD but not esophageal adenocarcinoma.
Without screening, there were about 14-16 cancer cases and about 15,077 quality-adjusted life years (QALYs) for every 1,000 patients. The cytosponge strategy was associated with about 8-13 cancer cases and about 15,105 QALYs. Endoscopic screening produced the most benefit overall – only about 7-12 cancer cases, with more than 15,100 QALYs. “However, greater benefits were accompanied by higher total costs,” the researchers said. For every 1,000 patients, no screening cost about $704,000 to $762,000, the cytosponge strategy cost about $1.5 to $1.6 million, and population-wide endoscopy cost about $2.1 to $2.2 million. Thus, the cytosponge method would lower the cost of screening by 37%-41% compared with endoscopically screening all men with GERD. The cytosponge was also cost effective in a model of 60-year-old women with GERD.
Using only endoscopic screening was not cost effective in either model, exceeding a $100,000 threshold of willingness to pay by anywhere from $107,000 to $330,000. The cytosponge is not yet available commercially, but the investigators assumed it cost $182 based on information from the manufacturer (Medtronic) and Medicare payments for similar devices. Although the findings withstood variations in indirect costs and age at initial screening, they were “somewhat sensitive” to variations in costs of the cytosponge and its presumed sensitivity and specificity in clinical settings. However, endoscopic screening only became cost effective when the cytosponge test cost at least $225.
The models assumed perfect adherence to screening, which probably exaggerated the effectiveness of the cytosponge and endoscopic screening, the investigators said. They noted that cytosponge screening can be performed without sedation during a short outpatient visit.
The National Institutes of Health provided funding. The investigators had no relevant disclosures.
The high costs of endoscopy make screening patients with gastroesophageal reflux disease (GERD) for Barrett’s esophagus a costly endeavor. But using a minimally invasive test followed by endoscopy only if results are positive could cut costs by up to 41%, according to investigators.
The report is in the September issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2017.02.017).
The findings mirror those from a prior study (Gastroenterology. 2013 Jan;144[1]:62-73.e60) of the new cytosponge device, which tests surface esophageal tissue for trefoil factor 3, a biomarker for Barrett’s esophagus, said Curtis R. Heberle, of Massachusetts General Hospital in Boston, and his associates. In addition, two separate models found the cytosponge strategy cost effective compared with no screening (incremental cost-effectiveness ratios [ICERs], about $26,000-$33,000). However, using the cytosponge instead of screening all GERD patients with endoscopy would reduce quality-adjusted life-years (QALYs) by about 1.8-5.5 years for every 1,000 patients.
Rates of esophageal adenocarcinoma have climbed more than sixfold in the United States in 4 decades, and 5-year survival rates remain below 20%. Nonetheless, the high cost of endoscopy and 10%-20% prevalence of GERD makes screening all patients for Barrett’s esophagus infeasible. To evaluate the cytosponge strategy, the researchers fit data from the multicenter BEST2 study (PLoS Med. 2015 Jan; 12[1]: e1001780) into two validated models calibrated to high-quality Surveillance, Epidemiology and End Results (SEER) data on esophageal cancer. Both models compared no screening with a one-time screen by either endoscopy alone or cytosponge with follow-up endoscopy in the event of a positive test. The models assumed patients were male, were 60 years old, and had GERD but not esophageal adenocarcinoma.
Without screening, there were about 14-16 cancer cases and about 15,077 quality-adjusted life years (QALYs) for every 1,000 patients. The cytosponge strategy was associated with about 8-13 cancer cases and about 15,105 QALYs. Endoscopic screening produced the most benefit overall – only about 7-12 cancer cases, with more than 15,100 QALYs. “However, greater benefits were accompanied by higher total costs,” the researchers said. For every 1,000 patients, no screening cost about $704,000 to $762,000, the cytosponge strategy cost about $1.5 to $1.6 million, and population-wide endoscopy cost about $2.1 to $2.2 million. Thus, the cytosponge method would lower the cost of screening by 37%-41% compared with endoscopically screening all men with GERD. The cytosponge was also cost effective in a model of 60-year-old women with GERD.
Using only endoscopic screening was not cost effective in either model, exceeding a $100,000 threshold of willingness to pay by anywhere from $107,000 to $330,000. The cytosponge is not yet available commercially, but the investigators assumed it cost $182 based on information from the manufacturer (Medtronic) and Medicare payments for similar devices. Although the findings withstood variations in indirect costs and age at initial screening, they were “somewhat sensitive” to variations in costs of the cytosponge and its presumed sensitivity and specificity in clinical settings. However, endoscopic screening only became cost effective when the cytosponge test cost at least $225.
The models assumed perfect adherence to screening, which probably exaggerated the effectiveness of the cytosponge and endoscopic screening, the investigators said. They noted that cytosponge screening can be performed without sedation during a short outpatient visit.
The National Institutes of Health provided funding. The investigators had no relevant disclosures.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: Using a minimally invasive screen for Barrett’s esophagus and following up with endoscopy if results are positive is a cost-effective alternative to endoscopy alone in patients with gastroesophageal reflux disease.
Major finding: The two-step screening strategy cut screening costs by 37%-41% but was associated with 1.8-5.5 fewer quality-adjusted life years for every 1,000 patients with GERD.
Data source: Two validated models based on Surveillance, Epidemiology, and End Results data, and data from the multicenter BEST2 trial.
Disclosures: The National Institutes of Health provided funding. The investigators had no relevant disclosures.
Ex Vivo Confocal Microscopy: A Diagnostic Tool for Skin Malignancies
Skin cancer is diagnosed in approximately 5.4 million individuals annually in the United States, more than the total number of breast, lung, colon, and prostate cancers diagnosed per year.1 It is estimated that 1 in 5 Americans will develop skin cancer during their lifetime.2 The 2 most common forms of skin cancer are basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), accounting for 4 million and 1 million cases diagnosed each year, respectively.3 With the increasing incidence of these skin cancers, the use of noninvasive imaging tools for detection and diagnosis has grown.
Ex vivo confocal microscopy is a diagnostic imaging tool that can be used in real-time at the bedside to assess freshly excised tissue for malignancies. It images tissue samples with cellular resolution and within minutes of biopsy or excision. Ex vivo confocal microscopy is a versatile tool that can assist in the diagnosis and management of skin malignancies such as melanoma, BCC, and SCC.
Reflectance vs Fluorescence Mode
Excised lesions can be examined in reflectance or fluorescence mode in great detail but with slightly varying nuclear-to-dermis contrasts depending on the chromophore that is targeted. In reflectance mode (reflectance confocal microscopy [RCM]), melanin and keratin act as endogenous chromophores because of their high refractive index relative to water,4,5 which allows for the visualization of cellular structures of the skin at low power, as well as microscopic substructures such as melanosomes, cytoplasmic granules, and other cellular organelles at high power. Although an exogenous contrast agent is not required, acetic acid has the capability to highlight nuclei, enhancing the tumor cell-to-dermis contrast in RCM.6 Acetic acid is clinically used as a predictor for certain skin and mucosal membrane neoplasms that blanch when exposed to the solution. In the case of RCM, acetic acid increases the visibility of nuclei by inducing the compaction of chromatin. For the acetowhitening to be effective, the sample must be soaked in the solution for a specific amount of time, depending on the concentration.7 A concentration between 1% and 10% can be used, but the less concentrated the solution, the longer the time of soaking that is required to achieve sufficiently bright nuclei.6
The contrast with acetic acid, however, is quite weak when the tissue is imaged en face, or along the horizontal surface of the sample, due to the collagen in the dermal layer, which has a high reflectance index. This issue is rectified when using the confocal microscope in the fluorescence mode with an exogenous fluorescent dye as a nuclear stain. Fluorescence confocal microscopy (FCM), results in a stronger nuclear-to-dermal contrast because of the role of contrast agents.8 The 1000-fold increase in contrast between nuclei and dermis is the result of dye agents that preferentially bind to nuclear DNA, of which acridine orange is the most commonly used.5,8 Basal cell carcinoma and SCC tumor cells can be visualized with FCM because they appear hyperfluorescent when stained with acridine orange.9 The acridine orange–stained cells display bright nuclei, while the cytoplasm and collagen remains dark. A positive feature of acridine orange is that it does not alter the tissue sample during freezing or formalin fixation and thus has no effect on subsequent histopathology that may need to be performed on the sample.10
High-Resolution Images Aid in Diagnosis
After it is harvested, the tissue sample is soaked in a contrast agent or dye, if needed, depending on the confocal mode to be used. The confocal microscope is then used to take a series of high-resolution individual en face images that are then stitched together to create a final mosaic image that can be up to 12×12 mm.6,11 With a 200-µm depth visibility, confocal microscopy can capture the cellular structures in the epidermis, dermis, and (if compressed enough) subcutaneous fat in just under 3 minutes.12
The images produced through confocal microscopy have an excellent correlation to frozen histological sections and can aid in the diagnosis of many epidermal and dermal malignancies including melanoma, BCC, and SCC. New criteria have been established to aid in the interpretation of the confocal images and identify some of the more common skin cancers.5,12,13 Basal cell carcinoma samples imaged through fluorescence and reflectance in low-power mode display the distinct nodular patterns with well-demarcated edges, as seen on classical histopathology. In the case of FCM, the cells that make up the tumor display hyperfluorescent areas consistent with nucleated cells that are stained with acridine orange. The main features that identify BCC on FCM images include nuclear pleomorphism and crowding, peripheral palisading, clefting of the basaloid islands, increased nucleus-to-cytoplasm ratio, and the presence of a modified dermis surrounding the mass known as the tumoral stroma5,12 (Figure).
In addition to fluorescence and a well-defined tumor silhouette, SCC under FCM displays keratin pearls composed of keratinized squames, nuclear pleomorphism, and fluorescent scales in the stratum corneum that are a result of keratin formation.5,13 The extent of differentiation of the SCC lesion also can be determined by assessing if the silhouette is well defined. A well-defined tumor silhouette is consistent with the diagnosis of a well-differentiated SCC, and vice versa.13 Ex vivo RCM also has been shown to be useful in diagnosing malignant melanomas, with melanin acting as an endogenous chromophore. Some of the features seen on imaging include a disarranged epithelium, hyperreflective roundish and dendritic pagetoid cells, and large hyperreflective polymorphic cells in the superficial chorion.14
Comparison to Conventional Histopathology
Ex vivo confocal microscopy in both the reflectance and fluorescence mode has been shown to perform well compared to conventional histopathology in the diagnosis of biopsy specimens. Ex vivo FCM has been shown to have an overall sensitivity of 88% and specificity of 99% in detecting residual BCC at the margins of excised tissue samples and in the fraction of the time it takes to attain similar results with frozen histopathology.9 Ex vivo RCM has been shown to have a higher prognostic capability, with 100% sensitivity and specificity in identifying BCC when scanning the tissue samples en face.15
Qualitatively, the images produced by RCM and FCM are similar to histopathology in overall architecture. Both techniques enhance the contrast between the epithelium and stroma and create images that can be examined in low as well as high resolution. A substantial difference between confocal microscopy and conventional hematoxylin and eosin–stained histopathology is that the confocal microscope produces images in gray scale. One way to alter the black-and-white images to resemble hematoxylin and eosin–stained slides is through the use of digital staining,16 which could boost clinical acceptance by physicians who are accustomed to the classical pink-purple appearance of pathology slides and could potentially limit the learning curve needed to read the confocal images.
Application in Mohs Micrographic Surgery
An important clinical application of ex vivo FCM imaging that has emerged is the detection of malignant cells at the excision margins during Mohs micrographic surgery. The use of confocal microscopy has the potential to save time by eliminating the need for tissue fixation while still providing good diagnostic accuracy. Implementing FCM as an imaging tool to guide surgical excisions could provide rapid diagnosis of the tissue, expediting excisions and reconstruction or the Mohs procedure while eliminating patient wait time and the need for frozen histopathology. Ex vivo RCM also has been used to establish laser parameters for CO2 laser ablation of superficial and early nodular BCC lesions.17 Other potential uses for ex vivo RCM/FCM could include rapid evaluation of tissue during operating room procedures where rapid frozen sections are currently utilized.
Combining In Vivo and Ex Vivo Confocal Microscopy
Many of the diagnostic guidelines created with the use of ex vivo confocal microscopy have been applied to in vivo use, and therefore the use of both modalities is appealing. In vivo confocal microscopy is a noninvasive technique that has been used to map margins of skin tumors such as BCC and lentigo maligna at the bedside.5 It also has been shown to help plan both surgical and nonsurgical treatment modalities and reconstruction before the tumor is excised.18 This technique also can help the patient understand the extent of the excision and any subsequent reconstruction that may be needed.
Limitations
Ex vivo confocal microscopy used as a diagnostic tool does have some limitations. Its novelty may require surgeons and pathologists to be trained to interpret the images properly and correlate them to conventional diagnostic guidelines. The imaging also is limited to a depth of approximately 200 µm; however, the sample may be flipped so that the underside can be imaged as well, which increases the depth to approximately 400 µm. The tissue being imaged must be fixed flat, which may alter its shape. Complex tissue samples may be difficult to flatten out completely and therefore may be difficult to image. A special mount may be required for the sample to be fixed in a proper position for imaging.6
Final Thoughts
Despite some of these limitations, the need for rapid bedside tissue diagnosis makes ex vivo confocal microscopy an attractive device that can be used as an additional diagnostic tool to histopathology and also has been tested in other disciplines, such as breast cancer pathology. In the future, both in vivo and ex vivo confocal microscopy may be utilized to diagnose cutaneous malignancies, guide surgical excisions, and detect lesion progression, and it may become a basis for rapid diagnosis and detection.19
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016 [published online January 7, 2016]. CA Cancer J Clin. 2016;66:7-30.
- Robinson JK. Sun exposure, sun protection, and vitamin D. JAMA. 2005;294:1541-1543.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population, 2012. JAMA Dermatol. 2015;151:1081-1086.
- Welzel J, Kästle R, Sattler EC. Fluorescence (multiwave) confocal microscopy. Dermatol Clin. 2016;34:527-533.
- Longo C, Ragazzi M, Rajadhyaksha M, et al. In vivo and ex vivo confocal microscopy for dermatologic and Mohs surgeons. Dermatol Clin. 2016;34:497-504.
- Patel YG, Nehal KS, Aranda I, et al. Confocal reflectance mosaicing of basal cell carcinomas in Mohs surgical skin excisions. J Biomed Opt. 2007;12:034027.
- Rajadhyaksha M, Gonzalez S, Zavislan JM. Detectability of contrast agents for confocal reflectance imaging of skin and microcirculation. J Biomed Opt. 2004;9:323-331.
- Karen JK, Gareau DS, Dusza SW, et al. Detection of basal cell carcinomas in Mohs excisions with fluorescence confocal mosaicing microscopy. Br J Dermatol. 2009;160:1242-1250.
- Bennàssar A, Vilata A, Puig S, et al. Ex vivo fluorescence confocal microscopy for fast evaluation of tumour margins during Mohs surgery. Br J Dermatol. 2014;170:360-365.
- Gareau DS, Li Y, Huang B, et al. Confocal mosaicing microscopy in Mohs skin excisions: feasibility of rapid surgical pathology. J Biomed Opt. 2008;13:054001.
- Bini J, Spain J, Nehal K, et al. Confocal mosaicing microscopy of human skin ex vivo: spectral analysis for digital staining to simulate histology-like appearance. J Biomed Opt. 2011;16:076008.
- Bennàssar A, Carrera C, Puig S, et al. Fast evaluation of 69 basal cell carcinomas with ex vivo fluorescence confocal microscopy: criteria description, histopathological correlation, and interobserver agreement. JAMA Dermatol. 2013;149:839-847.
- Longo C, Ragazzi M, Gardini S, et al. Ex vivo fluorescence confocal microscopy in conjunction with Mohs micrographic surgery for cutaneous squamous cell carcinoma. J Am Acad Dermatol. 2015;73:321-322.
- Cinotti E, Haouas M, Grivet D, et al. In vivo and ex vivo confocal microscopy for the management of a melanoma of the eyelid margin. Dermatol Surg. 2015;41:1437-1440.
- , , , ‘En face’ ex vivo reflectance confocal microscopy to help the surgery of basal cell carcinoma of the eyelid [published online December 19, 2016]. Clin Exp Ophthalmol. doi:10.1111/ceo.12904.
- Gareau DS, Jeon H, Nehal KS, et al. Rapid screening of cancer margins in tissue with multimodal confocal microscopy. J Surg Res. 2012;178:533-538.
- Sierra H, Damanpour S, Hibler B, et al. Confocal imaging of carbon dioxide laser-ablated basal cell carcinomas: an ex-vivo study on the uptake of contrast agent and ablation parameters [published online September 22, 2015]. Lasers Surg Med. 2016;48:133-139.
- Hibler BP, Yélamos O, Cordova M, et al. Handheld reflectance confocal microscopy to aid in the management of complex facial lentigo maligna. Cutis. 2017;99:346-352.
- Rajadhyaksha M, Marghoob A, Rossi A, et al. Reflectance confocal microscopy of skin in vivo: from bench to bedside. Lasers Surg Med. 2017;49:7-19.
Skin cancer is diagnosed in approximately 5.4 million individuals annually in the United States, more than the total number of breast, lung, colon, and prostate cancers diagnosed per year.1 It is estimated that 1 in 5 Americans will develop skin cancer during their lifetime.2 The 2 most common forms of skin cancer are basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), accounting for 4 million and 1 million cases diagnosed each year, respectively.3 With the increasing incidence of these skin cancers, the use of noninvasive imaging tools for detection and diagnosis has grown.
Ex vivo confocal microscopy is a diagnostic imaging tool that can be used in real-time at the bedside to assess freshly excised tissue for malignancies. It images tissue samples with cellular resolution and within minutes of biopsy or excision. Ex vivo confocal microscopy is a versatile tool that can assist in the diagnosis and management of skin malignancies such as melanoma, BCC, and SCC.
Reflectance vs Fluorescence Mode
Excised lesions can be examined in reflectance or fluorescence mode in great detail but with slightly varying nuclear-to-dermis contrasts depending on the chromophore that is targeted. In reflectance mode (reflectance confocal microscopy [RCM]), melanin and keratin act as endogenous chromophores because of their high refractive index relative to water,4,5 which allows for the visualization of cellular structures of the skin at low power, as well as microscopic substructures such as melanosomes, cytoplasmic granules, and other cellular organelles at high power. Although an exogenous contrast agent is not required, acetic acid has the capability to highlight nuclei, enhancing the tumor cell-to-dermis contrast in RCM.6 Acetic acid is clinically used as a predictor for certain skin and mucosal membrane neoplasms that blanch when exposed to the solution. In the case of RCM, acetic acid increases the visibility of nuclei by inducing the compaction of chromatin. For the acetowhitening to be effective, the sample must be soaked in the solution for a specific amount of time, depending on the concentration.7 A concentration between 1% and 10% can be used, but the less concentrated the solution, the longer the time of soaking that is required to achieve sufficiently bright nuclei.6
The contrast with acetic acid, however, is quite weak when the tissue is imaged en face, or along the horizontal surface of the sample, due to the collagen in the dermal layer, which has a high reflectance index. This issue is rectified when using the confocal microscope in the fluorescence mode with an exogenous fluorescent dye as a nuclear stain. Fluorescence confocal microscopy (FCM), results in a stronger nuclear-to-dermal contrast because of the role of contrast agents.8 The 1000-fold increase in contrast between nuclei and dermis is the result of dye agents that preferentially bind to nuclear DNA, of which acridine orange is the most commonly used.5,8 Basal cell carcinoma and SCC tumor cells can be visualized with FCM because they appear hyperfluorescent when stained with acridine orange.9 The acridine orange–stained cells display bright nuclei, while the cytoplasm and collagen remains dark. A positive feature of acridine orange is that it does not alter the tissue sample during freezing or formalin fixation and thus has no effect on subsequent histopathology that may need to be performed on the sample.10
High-Resolution Images Aid in Diagnosis
After it is harvested, the tissue sample is soaked in a contrast agent or dye, if needed, depending on the confocal mode to be used. The confocal microscope is then used to take a series of high-resolution individual en face images that are then stitched together to create a final mosaic image that can be up to 12×12 mm.6,11 With a 200-µm depth visibility, confocal microscopy can capture the cellular structures in the epidermis, dermis, and (if compressed enough) subcutaneous fat in just under 3 minutes.12
The images produced through confocal microscopy have an excellent correlation to frozen histological sections and can aid in the diagnosis of many epidermal and dermal malignancies including melanoma, BCC, and SCC. New criteria have been established to aid in the interpretation of the confocal images and identify some of the more common skin cancers.5,12,13 Basal cell carcinoma samples imaged through fluorescence and reflectance in low-power mode display the distinct nodular patterns with well-demarcated edges, as seen on classical histopathology. In the case of FCM, the cells that make up the tumor display hyperfluorescent areas consistent with nucleated cells that are stained with acridine orange. The main features that identify BCC on FCM images include nuclear pleomorphism and crowding, peripheral palisading, clefting of the basaloid islands, increased nucleus-to-cytoplasm ratio, and the presence of a modified dermis surrounding the mass known as the tumoral stroma5,12 (Figure).
In addition to fluorescence and a well-defined tumor silhouette, SCC under FCM displays keratin pearls composed of keratinized squames, nuclear pleomorphism, and fluorescent scales in the stratum corneum that are a result of keratin formation.5,13 The extent of differentiation of the SCC lesion also can be determined by assessing if the silhouette is well defined. A well-defined tumor silhouette is consistent with the diagnosis of a well-differentiated SCC, and vice versa.13 Ex vivo RCM also has been shown to be useful in diagnosing malignant melanomas, with melanin acting as an endogenous chromophore. Some of the features seen on imaging include a disarranged epithelium, hyperreflective roundish and dendritic pagetoid cells, and large hyperreflective polymorphic cells in the superficial chorion.14
Comparison to Conventional Histopathology
Ex vivo confocal microscopy in both the reflectance and fluorescence mode has been shown to perform well compared to conventional histopathology in the diagnosis of biopsy specimens. Ex vivo FCM has been shown to have an overall sensitivity of 88% and specificity of 99% in detecting residual BCC at the margins of excised tissue samples and in the fraction of the time it takes to attain similar results with frozen histopathology.9 Ex vivo RCM has been shown to have a higher prognostic capability, with 100% sensitivity and specificity in identifying BCC when scanning the tissue samples en face.15
Qualitatively, the images produced by RCM and FCM are similar to histopathology in overall architecture. Both techniques enhance the contrast between the epithelium and stroma and create images that can be examined in low as well as high resolution. A substantial difference between confocal microscopy and conventional hematoxylin and eosin–stained histopathology is that the confocal microscope produces images in gray scale. One way to alter the black-and-white images to resemble hematoxylin and eosin–stained slides is through the use of digital staining,16 which could boost clinical acceptance by physicians who are accustomed to the classical pink-purple appearance of pathology slides and could potentially limit the learning curve needed to read the confocal images.
Application in Mohs Micrographic Surgery
An important clinical application of ex vivo FCM imaging that has emerged is the detection of malignant cells at the excision margins during Mohs micrographic surgery. The use of confocal microscopy has the potential to save time by eliminating the need for tissue fixation while still providing good diagnostic accuracy. Implementing FCM as an imaging tool to guide surgical excisions could provide rapid diagnosis of the tissue, expediting excisions and reconstruction or the Mohs procedure while eliminating patient wait time and the need for frozen histopathology. Ex vivo RCM also has been used to establish laser parameters for CO2 laser ablation of superficial and early nodular BCC lesions.17 Other potential uses for ex vivo RCM/FCM could include rapid evaluation of tissue during operating room procedures where rapid frozen sections are currently utilized.
Combining In Vivo and Ex Vivo Confocal Microscopy
Many of the diagnostic guidelines created with the use of ex vivo confocal microscopy have been applied to in vivo use, and therefore the use of both modalities is appealing. In vivo confocal microscopy is a noninvasive technique that has been used to map margins of skin tumors such as BCC and lentigo maligna at the bedside.5 It also has been shown to help plan both surgical and nonsurgical treatment modalities and reconstruction before the tumor is excised.18 This technique also can help the patient understand the extent of the excision and any subsequent reconstruction that may be needed.
Limitations
Ex vivo confocal microscopy used as a diagnostic tool does have some limitations. Its novelty may require surgeons and pathologists to be trained to interpret the images properly and correlate them to conventional diagnostic guidelines. The imaging also is limited to a depth of approximately 200 µm; however, the sample may be flipped so that the underside can be imaged as well, which increases the depth to approximately 400 µm. The tissue being imaged must be fixed flat, which may alter its shape. Complex tissue samples may be difficult to flatten out completely and therefore may be difficult to image. A special mount may be required for the sample to be fixed in a proper position for imaging.6
Final Thoughts
Despite some of these limitations, the need for rapid bedside tissue diagnosis makes ex vivo confocal microscopy an attractive device that can be used as an additional diagnostic tool to histopathology and also has been tested in other disciplines, such as breast cancer pathology. In the future, both in vivo and ex vivo confocal microscopy may be utilized to diagnose cutaneous malignancies, guide surgical excisions, and detect lesion progression, and it may become a basis for rapid diagnosis and detection.19
Skin cancer is diagnosed in approximately 5.4 million individuals annually in the United States, more than the total number of breast, lung, colon, and prostate cancers diagnosed per year.1 It is estimated that 1 in 5 Americans will develop skin cancer during their lifetime.2 The 2 most common forms of skin cancer are basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), accounting for 4 million and 1 million cases diagnosed each year, respectively.3 With the increasing incidence of these skin cancers, the use of noninvasive imaging tools for detection and diagnosis has grown.
Ex vivo confocal microscopy is a diagnostic imaging tool that can be used in real-time at the bedside to assess freshly excised tissue for malignancies. It images tissue samples with cellular resolution and within minutes of biopsy or excision. Ex vivo confocal microscopy is a versatile tool that can assist in the diagnosis and management of skin malignancies such as melanoma, BCC, and SCC.
Reflectance vs Fluorescence Mode
Excised lesions can be examined in reflectance or fluorescence mode in great detail but with slightly varying nuclear-to-dermis contrasts depending on the chromophore that is targeted. In reflectance mode (reflectance confocal microscopy [RCM]), melanin and keratin act as endogenous chromophores because of their high refractive index relative to water,4,5 which allows for the visualization of cellular structures of the skin at low power, as well as microscopic substructures such as melanosomes, cytoplasmic granules, and other cellular organelles at high power. Although an exogenous contrast agent is not required, acetic acid has the capability to highlight nuclei, enhancing the tumor cell-to-dermis contrast in RCM.6 Acetic acid is clinically used as a predictor for certain skin and mucosal membrane neoplasms that blanch when exposed to the solution. In the case of RCM, acetic acid increases the visibility of nuclei by inducing the compaction of chromatin. For the acetowhitening to be effective, the sample must be soaked in the solution for a specific amount of time, depending on the concentration.7 A concentration between 1% and 10% can be used, but the less concentrated the solution, the longer the time of soaking that is required to achieve sufficiently bright nuclei.6
The contrast with acetic acid, however, is quite weak when the tissue is imaged en face, or along the horizontal surface of the sample, due to the collagen in the dermal layer, which has a high reflectance index. This issue is rectified when using the confocal microscope in the fluorescence mode with an exogenous fluorescent dye as a nuclear stain. Fluorescence confocal microscopy (FCM), results in a stronger nuclear-to-dermal contrast because of the role of contrast agents.8 The 1000-fold increase in contrast between nuclei and dermis is the result of dye agents that preferentially bind to nuclear DNA, of which acridine orange is the most commonly used.5,8 Basal cell carcinoma and SCC tumor cells can be visualized with FCM because they appear hyperfluorescent when stained with acridine orange.9 The acridine orange–stained cells display bright nuclei, while the cytoplasm and collagen remains dark. A positive feature of acridine orange is that it does not alter the tissue sample during freezing or formalin fixation and thus has no effect on subsequent histopathology that may need to be performed on the sample.10
High-Resolution Images Aid in Diagnosis
After it is harvested, the tissue sample is soaked in a contrast agent or dye, if needed, depending on the confocal mode to be used. The confocal microscope is then used to take a series of high-resolution individual en face images that are then stitched together to create a final mosaic image that can be up to 12×12 mm.6,11 With a 200-µm depth visibility, confocal microscopy can capture the cellular structures in the epidermis, dermis, and (if compressed enough) subcutaneous fat in just under 3 minutes.12
The images produced through confocal microscopy have an excellent correlation to frozen histological sections and can aid in the diagnosis of many epidermal and dermal malignancies including melanoma, BCC, and SCC. New criteria have been established to aid in the interpretation of the confocal images and identify some of the more common skin cancers.5,12,13 Basal cell carcinoma samples imaged through fluorescence and reflectance in low-power mode display the distinct nodular patterns with well-demarcated edges, as seen on classical histopathology. In the case of FCM, the cells that make up the tumor display hyperfluorescent areas consistent with nucleated cells that are stained with acridine orange. The main features that identify BCC on FCM images include nuclear pleomorphism and crowding, peripheral palisading, clefting of the basaloid islands, increased nucleus-to-cytoplasm ratio, and the presence of a modified dermis surrounding the mass known as the tumoral stroma5,12 (Figure).
In addition to fluorescence and a well-defined tumor silhouette, SCC under FCM displays keratin pearls composed of keratinized squames, nuclear pleomorphism, and fluorescent scales in the stratum corneum that are a result of keratin formation.5,13 The extent of differentiation of the SCC lesion also can be determined by assessing if the silhouette is well defined. A well-defined tumor silhouette is consistent with the diagnosis of a well-differentiated SCC, and vice versa.13 Ex vivo RCM also has been shown to be useful in diagnosing malignant melanomas, with melanin acting as an endogenous chromophore. Some of the features seen on imaging include a disarranged epithelium, hyperreflective roundish and dendritic pagetoid cells, and large hyperreflective polymorphic cells in the superficial chorion.14
Comparison to Conventional Histopathology
Ex vivo confocal microscopy in both the reflectance and fluorescence mode has been shown to perform well compared to conventional histopathology in the diagnosis of biopsy specimens. Ex vivo FCM has been shown to have an overall sensitivity of 88% and specificity of 99% in detecting residual BCC at the margins of excised tissue samples and in the fraction of the time it takes to attain similar results with frozen histopathology.9 Ex vivo RCM has been shown to have a higher prognostic capability, with 100% sensitivity and specificity in identifying BCC when scanning the tissue samples en face.15
Qualitatively, the images produced by RCM and FCM are similar to histopathology in overall architecture. Both techniques enhance the contrast between the epithelium and stroma and create images that can be examined in low as well as high resolution. A substantial difference between confocal microscopy and conventional hematoxylin and eosin–stained histopathology is that the confocal microscope produces images in gray scale. One way to alter the black-and-white images to resemble hematoxylin and eosin–stained slides is through the use of digital staining,16 which could boost clinical acceptance by physicians who are accustomed to the classical pink-purple appearance of pathology slides and could potentially limit the learning curve needed to read the confocal images.
Application in Mohs Micrographic Surgery
An important clinical application of ex vivo FCM imaging that has emerged is the detection of malignant cells at the excision margins during Mohs micrographic surgery. The use of confocal microscopy has the potential to save time by eliminating the need for tissue fixation while still providing good diagnostic accuracy. Implementing FCM as an imaging tool to guide surgical excisions could provide rapid diagnosis of the tissue, expediting excisions and reconstruction or the Mohs procedure while eliminating patient wait time and the need for frozen histopathology. Ex vivo RCM also has been used to establish laser parameters for CO2 laser ablation of superficial and early nodular BCC lesions.17 Other potential uses for ex vivo RCM/FCM could include rapid evaluation of tissue during operating room procedures where rapid frozen sections are currently utilized.
Combining In Vivo and Ex Vivo Confocal Microscopy
Many of the diagnostic guidelines created with the use of ex vivo confocal microscopy have been applied to in vivo use, and therefore the use of both modalities is appealing. In vivo confocal microscopy is a noninvasive technique that has been used to map margins of skin tumors such as BCC and lentigo maligna at the bedside.5 It also has been shown to help plan both surgical and nonsurgical treatment modalities and reconstruction before the tumor is excised.18 This technique also can help the patient understand the extent of the excision and any subsequent reconstruction that may be needed.
Limitations
Ex vivo confocal microscopy used as a diagnostic tool does have some limitations. Its novelty may require surgeons and pathologists to be trained to interpret the images properly and correlate them to conventional diagnostic guidelines. The imaging also is limited to a depth of approximately 200 µm; however, the sample may be flipped so that the underside can be imaged as well, which increases the depth to approximately 400 µm. The tissue being imaged must be fixed flat, which may alter its shape. Complex tissue samples may be difficult to flatten out completely and therefore may be difficult to image. A special mount may be required for the sample to be fixed in a proper position for imaging.6
Final Thoughts
Despite some of these limitations, the need for rapid bedside tissue diagnosis makes ex vivo confocal microscopy an attractive device that can be used as an additional diagnostic tool to histopathology and also has been tested in other disciplines, such as breast cancer pathology. In the future, both in vivo and ex vivo confocal microscopy may be utilized to diagnose cutaneous malignancies, guide surgical excisions, and detect lesion progression, and it may become a basis for rapid diagnosis and detection.19
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016 [published online January 7, 2016]. CA Cancer J Clin. 2016;66:7-30.
- Robinson JK. Sun exposure, sun protection, and vitamin D. JAMA. 2005;294:1541-1543.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population, 2012. JAMA Dermatol. 2015;151:1081-1086.
- Welzel J, Kästle R, Sattler EC. Fluorescence (multiwave) confocal microscopy. Dermatol Clin. 2016;34:527-533.
- Longo C, Ragazzi M, Rajadhyaksha M, et al. In vivo and ex vivo confocal microscopy for dermatologic and Mohs surgeons. Dermatol Clin. 2016;34:497-504.
- Patel YG, Nehal KS, Aranda I, et al. Confocal reflectance mosaicing of basal cell carcinomas in Mohs surgical skin excisions. J Biomed Opt. 2007;12:034027.
- Rajadhyaksha M, Gonzalez S, Zavislan JM. Detectability of contrast agents for confocal reflectance imaging of skin and microcirculation. J Biomed Opt. 2004;9:323-331.
- Karen JK, Gareau DS, Dusza SW, et al. Detection of basal cell carcinomas in Mohs excisions with fluorescence confocal mosaicing microscopy. Br J Dermatol. 2009;160:1242-1250.
- Bennàssar A, Vilata A, Puig S, et al. Ex vivo fluorescence confocal microscopy for fast evaluation of tumour margins during Mohs surgery. Br J Dermatol. 2014;170:360-365.
- Gareau DS, Li Y, Huang B, et al. Confocal mosaicing microscopy in Mohs skin excisions: feasibility of rapid surgical pathology. J Biomed Opt. 2008;13:054001.
- Bini J, Spain J, Nehal K, et al. Confocal mosaicing microscopy of human skin ex vivo: spectral analysis for digital staining to simulate histology-like appearance. J Biomed Opt. 2011;16:076008.
- Bennàssar A, Carrera C, Puig S, et al. Fast evaluation of 69 basal cell carcinomas with ex vivo fluorescence confocal microscopy: criteria description, histopathological correlation, and interobserver agreement. JAMA Dermatol. 2013;149:839-847.
- Longo C, Ragazzi M, Gardini S, et al. Ex vivo fluorescence confocal microscopy in conjunction with Mohs micrographic surgery for cutaneous squamous cell carcinoma. J Am Acad Dermatol. 2015;73:321-322.
- Cinotti E, Haouas M, Grivet D, et al. In vivo and ex vivo confocal microscopy for the management of a melanoma of the eyelid margin. Dermatol Surg. 2015;41:1437-1440.
- , , , ‘En face’ ex vivo reflectance confocal microscopy to help the surgery of basal cell carcinoma of the eyelid [published online December 19, 2016]. Clin Exp Ophthalmol. doi:10.1111/ceo.12904.
- Gareau DS, Jeon H, Nehal KS, et al. Rapid screening of cancer margins in tissue with multimodal confocal microscopy. J Surg Res. 2012;178:533-538.
- Sierra H, Damanpour S, Hibler B, et al. Confocal imaging of carbon dioxide laser-ablated basal cell carcinomas: an ex-vivo study on the uptake of contrast agent and ablation parameters [published online September 22, 2015]. Lasers Surg Med. 2016;48:133-139.
- Hibler BP, Yélamos O, Cordova M, et al. Handheld reflectance confocal microscopy to aid in the management of complex facial lentigo maligna. Cutis. 2017;99:346-352.
- Rajadhyaksha M, Marghoob A, Rossi A, et al. Reflectance confocal microscopy of skin in vivo: from bench to bedside. Lasers Surg Med. 2017;49:7-19.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016 [published online January 7, 2016]. CA Cancer J Clin. 2016;66:7-30.
- Robinson JK. Sun exposure, sun protection, and vitamin D. JAMA. 2005;294:1541-1543.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population, 2012. JAMA Dermatol. 2015;151:1081-1086.
- Welzel J, Kästle R, Sattler EC. Fluorescence (multiwave) confocal microscopy. Dermatol Clin. 2016;34:527-533.
- Longo C, Ragazzi M, Rajadhyaksha M, et al. In vivo and ex vivo confocal microscopy for dermatologic and Mohs surgeons. Dermatol Clin. 2016;34:497-504.
- Patel YG, Nehal KS, Aranda I, et al. Confocal reflectance mosaicing of basal cell carcinomas in Mohs surgical skin excisions. J Biomed Opt. 2007;12:034027.
- Rajadhyaksha M, Gonzalez S, Zavislan JM. Detectability of contrast agents for confocal reflectance imaging of skin and microcirculation. J Biomed Opt. 2004;9:323-331.
- Karen JK, Gareau DS, Dusza SW, et al. Detection of basal cell carcinomas in Mohs excisions with fluorescence confocal mosaicing microscopy. Br J Dermatol. 2009;160:1242-1250.
- Bennàssar A, Vilata A, Puig S, et al. Ex vivo fluorescence confocal microscopy for fast evaluation of tumour margins during Mohs surgery. Br J Dermatol. 2014;170:360-365.
- Gareau DS, Li Y, Huang B, et al. Confocal mosaicing microscopy in Mohs skin excisions: feasibility of rapid surgical pathology. J Biomed Opt. 2008;13:054001.
- Bini J, Spain J, Nehal K, et al. Confocal mosaicing microscopy of human skin ex vivo: spectral analysis for digital staining to simulate histology-like appearance. J Biomed Opt. 2011;16:076008.
- Bennàssar A, Carrera C, Puig S, et al. Fast evaluation of 69 basal cell carcinomas with ex vivo fluorescence confocal microscopy: criteria description, histopathological correlation, and interobserver agreement. JAMA Dermatol. 2013;149:839-847.
- Longo C, Ragazzi M, Gardini S, et al. Ex vivo fluorescence confocal microscopy in conjunction with Mohs micrographic surgery for cutaneous squamous cell carcinoma. J Am Acad Dermatol. 2015;73:321-322.
- Cinotti E, Haouas M, Grivet D, et al. In vivo and ex vivo confocal microscopy for the management of a melanoma of the eyelid margin. Dermatol Surg. 2015;41:1437-1440.
- , , , ‘En face’ ex vivo reflectance confocal microscopy to help the surgery of basal cell carcinoma of the eyelid [published online December 19, 2016]. Clin Exp Ophthalmol. doi:10.1111/ceo.12904.
- Gareau DS, Jeon H, Nehal KS, et al. Rapid screening of cancer margins in tissue with multimodal confocal microscopy. J Surg Res. 2012;178:533-538.
- Sierra H, Damanpour S, Hibler B, et al. Confocal imaging of carbon dioxide laser-ablated basal cell carcinomas: an ex-vivo study on the uptake of contrast agent and ablation parameters [published online September 22, 2015]. Lasers Surg Med. 2016;48:133-139.
- Hibler BP, Yélamos O, Cordova M, et al. Handheld reflectance confocal microscopy to aid in the management of complex facial lentigo maligna. Cutis. 2017;99:346-352.
- Rajadhyaksha M, Marghoob A, Rossi A, et al. Reflectance confocal microscopy of skin in vivo: from bench to bedside. Lasers Surg Med. 2017;49:7-19.
Practice Points
- Confocal microscopy is an imaging tool that can be used both in vivo and ex vivo to aid in the diagnosis and management of cutaneous neoplasms, including melanoma, basal cell carcinoma, and squamous cell carcinoma, as well as inflammatory dermatoses.
- Ex vivo confocal microscopy can be used in both reflectance and fluorescent modes to render diagnosis in excised tissue or check surgical margins.
- Both in vivo and ex vivo confocal microscopy produces images with cellular resolution with a main limitation being depth of imaging.