User login
Stopping Prediabetes Before It Becomes Diabetes
In the Navajo area, 1 in 5 Native Americans has diabetes, and about 75,000 have prediabetes. That is the impetus for IHS’s new campaign, launched at the Northern Navajo Medical Center, to raise awareness and knowledge of prediabetes—and to let people know it can be reversed.
The “Do I Have Prediabetes?” campaign by the Shiprock Health Promotion Program is intended to help people assess their risk and reverse prediabetes before their blood sugar level is high enough to be type 2 diabetes. The program builds on the groundwork of a 2016 CDC national prediabetes awareness campaign. Materials include posters, an infographic that depicts the risk factors, roadside billboards, and a video for waiting rooms.
The IHS has already made progress against at least 1 diabetes-related issue: kidney failure. Through the Special Diabetes Program for Indians, innovative evidence-based interventions helped reduce kidney failure from diabetes among Native American adults by 54% between 1996 and 2013. Kidney failure from diabetes in Native Americans was the highest of any ethnic group but now has dropped the fastest. The Shiprock program will continue through early 2018.
In the Navajo area, 1 in 5 Native Americans has diabetes, and about 75,000 have prediabetes. That is the impetus for IHS’s new campaign, launched at the Northern Navajo Medical Center, to raise awareness and knowledge of prediabetes—and to let people know it can be reversed.
The “Do I Have Prediabetes?” campaign by the Shiprock Health Promotion Program is intended to help people assess their risk and reverse prediabetes before their blood sugar level is high enough to be type 2 diabetes. The program builds on the groundwork of a 2016 CDC national prediabetes awareness campaign. Materials include posters, an infographic that depicts the risk factors, roadside billboards, and a video for waiting rooms.
The IHS has already made progress against at least 1 diabetes-related issue: kidney failure. Through the Special Diabetes Program for Indians, innovative evidence-based interventions helped reduce kidney failure from diabetes among Native American adults by 54% between 1996 and 2013. Kidney failure from diabetes in Native Americans was the highest of any ethnic group but now has dropped the fastest. The Shiprock program will continue through early 2018.
In the Navajo area, 1 in 5 Native Americans has diabetes, and about 75,000 have prediabetes. That is the impetus for IHS’s new campaign, launched at the Northern Navajo Medical Center, to raise awareness and knowledge of prediabetes—and to let people know it can be reversed.
The “Do I Have Prediabetes?” campaign by the Shiprock Health Promotion Program is intended to help people assess their risk and reverse prediabetes before their blood sugar level is high enough to be type 2 diabetes. The program builds on the groundwork of a 2016 CDC national prediabetes awareness campaign. Materials include posters, an infographic that depicts the risk factors, roadside billboards, and a video for waiting rooms.
The IHS has already made progress against at least 1 diabetes-related issue: kidney failure. Through the Special Diabetes Program for Indians, innovative evidence-based interventions helped reduce kidney failure from diabetes among Native American adults by 54% between 1996 and 2013. Kidney failure from diabetes in Native Americans was the highest of any ethnic group but now has dropped the fastest. The Shiprock program will continue through early 2018.
Clinical Challenges - July 2017 What's Your Diagnosis?
The diagnosis
Answer to “What’s your diagnosis?” on page 2: Dysphagia lusoria
Dysphagia may be divided into an oropharyngeal cause or an esophageal cause. Esophageal dysphagia may be due to a luminal narrowing or a motility dysfunction. Causes of luminal narrowing include lesions within the lumen such as a foreign body, lesions within the wall of the esophagus such as a mucosal or submucosal tumor, and extrinsic lesions such as an enlarged mediastinal lymph node or mass. Esophageal dysphagia typically presents with difficulty in swallowing solids compared with liquids.
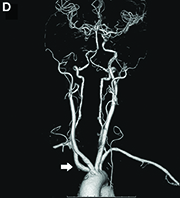
Evaluation of this condition involves a barium swallow study, CTA, or magnetic resonance angiography.2 Both CTA and MRA have largely supplanted the role of conventional angiography, which is invasive. Both CTA and magnetic resonance angiography may also diagnose any other intrathoracic pathology causing esophageal compression. The management of patients with mild to moderate dysphagia is diet modification (minced feeds to well-chewed food; eating slowly and with liquids). Vascular repair of the aberrant vessel is considered only if the patient has severe symptoms and has failed conservative management.3 Because our subject did not have significant weight loss or regurgitation, only dietary advice was offered. An interval outpatient upper endoscopy was planned upon discharge, for which she defaulted.
References
1. Bayford. An account of a singular case of obstructed degluitition. Memoirs of the Medical Society of London. 1794;2:275-86.
2. Alper, F., Akgun, M., Kantarci, M. et al. Demonstration of vascular abnormalities compressing esophagus by MDCT: special focus on dysphagia lusoria. Eur J Radiol. 2006;59:82-7.
3. Levitt, B. and Richter, J.E. Dysphagia lusoria: a comprehensive review. Dis Esophagus. 2007;20:455-60.
The diagnosis
Answer to “What’s your diagnosis?” on page 2: Dysphagia lusoria
Dysphagia may be divided into an oropharyngeal cause or an esophageal cause. Esophageal dysphagia may be due to a luminal narrowing or a motility dysfunction. Causes of luminal narrowing include lesions within the lumen such as a foreign body, lesions within the wall of the esophagus such as a mucosal or submucosal tumor, and extrinsic lesions such as an enlarged mediastinal lymph node or mass. Esophageal dysphagia typically presents with difficulty in swallowing solids compared with liquids.
Evaluation of this condition involves a barium swallow study, CTA, or magnetic resonance angiography.2 Both CTA and MRA have largely supplanted the role of conventional angiography, which is invasive. Both CTA and magnetic resonance angiography may also diagnose any other intrathoracic pathology causing esophageal compression. The management of patients with mild to moderate dysphagia is diet modification (minced feeds to well-chewed food; eating slowly and with liquids). Vascular repair of the aberrant vessel is considered only if the patient has severe symptoms and has failed conservative management.3 Because our subject did not have significant weight loss or regurgitation, only dietary advice was offered. An interval outpatient upper endoscopy was planned upon discharge, for which she defaulted.
References
1. Bayford. An account of a singular case of obstructed degluitition. Memoirs of the Medical Society of London. 1794;2:275-86.
2. Alper, F., Akgun, M., Kantarci, M. et al. Demonstration of vascular abnormalities compressing esophagus by MDCT: special focus on dysphagia lusoria. Eur J Radiol. 2006;59:82-7.
3. Levitt, B. and Richter, J.E. Dysphagia lusoria: a comprehensive review. Dis Esophagus. 2007;20:455-60.
The diagnosis
Answer to “What’s your diagnosis?” on page 2: Dysphagia lusoria
Dysphagia may be divided into an oropharyngeal cause or an esophageal cause. Esophageal dysphagia may be due to a luminal narrowing or a motility dysfunction. Causes of luminal narrowing include lesions within the lumen such as a foreign body, lesions within the wall of the esophagus such as a mucosal or submucosal tumor, and extrinsic lesions such as an enlarged mediastinal lymph node or mass. Esophageal dysphagia typically presents with difficulty in swallowing solids compared with liquids.
Evaluation of this condition involves a barium swallow study, CTA, or magnetic resonance angiography.2 Both CTA and MRA have largely supplanted the role of conventional angiography, which is invasive. Both CTA and magnetic resonance angiography may also diagnose any other intrathoracic pathology causing esophageal compression. The management of patients with mild to moderate dysphagia is diet modification (minced feeds to well-chewed food; eating slowly and with liquids). Vascular repair of the aberrant vessel is considered only if the patient has severe symptoms and has failed conservative management.3 Because our subject did not have significant weight loss or regurgitation, only dietary advice was offered. An interval outpatient upper endoscopy was planned upon discharge, for which she defaulted.
References
1. Bayford. An account of a singular case of obstructed degluitition. Memoirs of the Medical Society of London. 1794;2:275-86.
2. Alper, F., Akgun, M., Kantarci, M. et al. Demonstration of vascular abnormalities compressing esophagus by MDCT: special focus on dysphagia lusoria. Eur J Radiol. 2006;59:82-7.
3. Levitt, B. and Richter, J.E. Dysphagia lusoria: a comprehensive review. Dis Esophagus. 2007;20:455-60.
What’s your diagnosis?
By Eric Wee, MD and Ma Clarissa Buenaseda, MD. Published previously in Gastroenterology (2013;144:273,467,468).
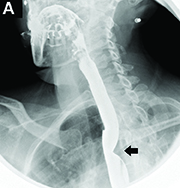
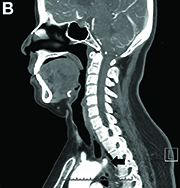
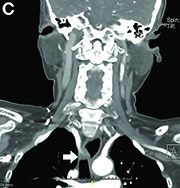
DDSEP® 8 Quick quiz - July 2017 Question 2
Q2: Answer: B
Objective: Diagnose dyssynergic defecation and treat with biofeedback therapy.
Rationale: This patient has a functional defecation disorder, or dyssynergic defecation. According to Rome III guidelines, to fulfill this diagnosis, the patient must satisfy criteria for functional constipation.
In addition, they must also have at least 2 of the following: 1) Evidence of impaired evacuation on balloon expulsion test or imaging; 2) inappropriate contraction of the pelvic floor muscles or less than 20% relaxation of basal resting sphincter pressure by manometry, imaging or EEG; and 3) inadequate propulsive forces assessed by manometry or imaging.
The treatment mainstay for functional defecation disorders is pelvic floor retraining and biofeedback. Although lubiprostone and fiber supplementation are used to treat constipation, this is not the treatment of choice for dyssynergic defecation. Amitriptyline is often used for functional gastrointestinal disorders, but is not the primary therapy for dyssynergic defection, and often can worsen constipation and therefore is not appropriate for this patient.
Finally, rectal biopsy is the gold standard for diagnosis of Hirschsprung’s disease or congenital aganglionic megacolon. This is thought to be due to the failure of neural crest cells to migrate during gestation. The manometric findings with Hirschsprung’s consist of lack of relaxation of internal anal sphincter with distention of the rectum. This is a diagnosis usually made during childhood. Adults with Hirschsprung’s disease usually describe severe constipation since birth. It is therefore not the most likely diagnosis in this patient.
Reference
1. Bharucha A.E., Wald A., Enck P., Rao S. Functional anorectal disorders. Gastroenterology 2006;130:1510-8.
Q2: Answer: B
Objective: Diagnose dyssynergic defecation and treat with biofeedback therapy.
Rationale: This patient has a functional defecation disorder, or dyssynergic defecation. According to Rome III guidelines, to fulfill this diagnosis, the patient must satisfy criteria for functional constipation.
In addition, they must also have at least 2 of the following: 1) Evidence of impaired evacuation on balloon expulsion test or imaging; 2) inappropriate contraction of the pelvic floor muscles or less than 20% relaxation of basal resting sphincter pressure by manometry, imaging or EEG; and 3) inadequate propulsive forces assessed by manometry or imaging.
The treatment mainstay for functional defecation disorders is pelvic floor retraining and biofeedback. Although lubiprostone and fiber supplementation are used to treat constipation, this is not the treatment of choice for dyssynergic defecation. Amitriptyline is often used for functional gastrointestinal disorders, but is not the primary therapy for dyssynergic defection, and often can worsen constipation and therefore is not appropriate for this patient.
Finally, rectal biopsy is the gold standard for diagnosis of Hirschsprung’s disease or congenital aganglionic megacolon. This is thought to be due to the failure of neural crest cells to migrate during gestation. The manometric findings with Hirschsprung’s consist of lack of relaxation of internal anal sphincter with distention of the rectum. This is a diagnosis usually made during childhood. Adults with Hirschsprung’s disease usually describe severe constipation since birth. It is therefore not the most likely diagnosis in this patient.
Reference
1. Bharucha A.E., Wald A., Enck P., Rao S. Functional anorectal disorders. Gastroenterology 2006;130:1510-8.
Q2: Answer: B
Objective: Diagnose dyssynergic defecation and treat with biofeedback therapy.
Rationale: This patient has a functional defecation disorder, or dyssynergic defecation. According to Rome III guidelines, to fulfill this diagnosis, the patient must satisfy criteria for functional constipation.
In addition, they must also have at least 2 of the following: 1) Evidence of impaired evacuation on balloon expulsion test or imaging; 2) inappropriate contraction of the pelvic floor muscles or less than 20% relaxation of basal resting sphincter pressure by manometry, imaging or EEG; and 3) inadequate propulsive forces assessed by manometry or imaging.
The treatment mainstay for functional defecation disorders is pelvic floor retraining and biofeedback. Although lubiprostone and fiber supplementation are used to treat constipation, this is not the treatment of choice for dyssynergic defecation. Amitriptyline is often used for functional gastrointestinal disorders, but is not the primary therapy for dyssynergic defection, and often can worsen constipation and therefore is not appropriate for this patient.
Finally, rectal biopsy is the gold standard for diagnosis of Hirschsprung’s disease or congenital aganglionic megacolon. This is thought to be due to the failure of neural crest cells to migrate during gestation. The manometric findings with Hirschsprung’s consist of lack of relaxation of internal anal sphincter with distention of the rectum. This is a diagnosis usually made during childhood. Adults with Hirschsprung’s disease usually describe severe constipation since birth. It is therefore not the most likely diagnosis in this patient.
Reference
1. Bharucha A.E., Wald A., Enck P., Rao S. Functional anorectal disorders. Gastroenterology 2006;130:1510-8.
A 36-year-old woman presented to clinic with complaints of constipation. She reported daily bowel movements, but with a sensation of rectal fullness and incomplete evacuation. She strained with bowel movements at least 50% of the time. Her symptoms have been present for most of her adult life. She denied diarrhea or blood in her stools, and has had no recent unintentional weight loss. She was sent for anorectal manometry, which revealed adequate propulsive forces, but less than 20% relaxation of the basal resting sphincter pressure. Balloon expulsion is abnormal.
DDSEP® 8 Quick quiz - July 2017 Question 1
Q1: Answer: D
Objective: Understand the role of the restrictive transfusion strategy during patient stabilization and resuscitation as the initial step in the management of a variceal bleed.
Discussion: The initial therapy for acute variceal hemorrhage is resuscitation in an intensive care unit. Blood volume restitution should be undertaken promptly but with caution, with the goals of maintaining hemodynamic stability and hemoglobin around 7–8 g/dL.
Over-transfusion or volume overexpansion can precipitate variceal re-bleeding. A randomized clinical trial found that a restrictive transfusion strategy (transfusion when the hemoglobin fell below 7 g/dL) in patients with cirrhosis significantly improved survival. Endoscopic evaluation with potential variceal band ligation is appropriate only after initial resuscitation and stabilization of the patient.
Placement of a Blakemore tube and TIPS are not first-line therapy for this patient with Childs class A cirrhosis, and could be considered for recurrent bleeding that fails endoscopic therapy.
Endoscopic variceal ligation is more effective than sclerotherapy and is associated with fewer side effects. However, in patients for whom endoscopic variceal ligation is not feasible, sclerotherapy is a reasonable alternative.
References
1. Garcia-Tsao G., Sanyal A.J., Grace N.D., Carey W. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology. 2007;46(3):922-38.
2. Garcia-Tsao G., Bosch J.. Management of varices and variceal hemorrhage in cirrhosis. N Engl J Med. 2010;362(9):823-32.
3. Villanueva C., Colomo A., Bosch A., et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med. 2013;368(1):11-21.
4. Villanueva C., Piqueras M., Aracil C., et al. A randomized controlled trial comparing ligation and sclerotherapy as emergency endoscopic treatment added to somatostatin in acute variceal bleeding. J Hepatol. 2006;45(4):560-7.
Q1: Answer: D
Objective: Understand the role of the restrictive transfusion strategy during patient stabilization and resuscitation as the initial step in the management of a variceal bleed.
Discussion: The initial therapy for acute variceal hemorrhage is resuscitation in an intensive care unit. Blood volume restitution should be undertaken promptly but with caution, with the goals of maintaining hemodynamic stability and hemoglobin around 7–8 g/dL.
Over-transfusion or volume overexpansion can precipitate variceal re-bleeding. A randomized clinical trial found that a restrictive transfusion strategy (transfusion when the hemoglobin fell below 7 g/dL) in patients with cirrhosis significantly improved survival. Endoscopic evaluation with potential variceal band ligation is appropriate only after initial resuscitation and stabilization of the patient.
Placement of a Blakemore tube and TIPS are not first-line therapy for this patient with Childs class A cirrhosis, and could be considered for recurrent bleeding that fails endoscopic therapy.
Endoscopic variceal ligation is more effective than sclerotherapy and is associated with fewer side effects. However, in patients for whom endoscopic variceal ligation is not feasible, sclerotherapy is a reasonable alternative.
References
1. Garcia-Tsao G., Sanyal A.J., Grace N.D., Carey W. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology. 2007;46(3):922-38.
2. Garcia-Tsao G., Bosch J.. Management of varices and variceal hemorrhage in cirrhosis. N Engl J Med. 2010;362(9):823-32.
3. Villanueva C., Colomo A., Bosch A., et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med. 2013;368(1):11-21.
4. Villanueva C., Piqueras M., Aracil C., et al. A randomized controlled trial comparing ligation and sclerotherapy as emergency endoscopic treatment added to somatostatin in acute variceal bleeding. J Hepatol. 2006;45(4):560-7.
Q1: Answer: D
Objective: Understand the role of the restrictive transfusion strategy during patient stabilization and resuscitation as the initial step in the management of a variceal bleed.
Discussion: The initial therapy for acute variceal hemorrhage is resuscitation in an intensive care unit. Blood volume restitution should be undertaken promptly but with caution, with the goals of maintaining hemodynamic stability and hemoglobin around 7–8 g/dL.
Over-transfusion or volume overexpansion can precipitate variceal re-bleeding. A randomized clinical trial found that a restrictive transfusion strategy (transfusion when the hemoglobin fell below 7 g/dL) in patients with cirrhosis significantly improved survival. Endoscopic evaluation with potential variceal band ligation is appropriate only after initial resuscitation and stabilization of the patient.
Placement of a Blakemore tube and TIPS are not first-line therapy for this patient with Childs class A cirrhosis, and could be considered for recurrent bleeding that fails endoscopic therapy.
Endoscopic variceal ligation is more effective than sclerotherapy and is associated with fewer side effects. However, in patients for whom endoscopic variceal ligation is not feasible, sclerotherapy is a reasonable alternative.
References
1. Garcia-Tsao G., Sanyal A.J., Grace N.D., Carey W. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology. 2007;46(3):922-38.
2. Garcia-Tsao G., Bosch J.. Management of varices and variceal hemorrhage in cirrhosis. N Engl J Med. 2010;362(9):823-32.
3. Villanueva C., Colomo A., Bosch A., et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med. 2013;368(1):11-21.
4. Villanueva C., Piqueras M., Aracil C., et al. A randomized controlled trial comparing ligation and sclerotherapy as emergency endoscopic treatment added to somatostatin in acute variceal bleeding. J Hepatol. 2006;45(4):560-7.
A 46-year-old man with a history of alcoholic cirrhosis presents to the ED with new-onset melena and hematemesis. On examination, he appears weak, but his mental status is stable with no signs of encephalopathy. His abdomen is soft, with no clinical ascites. Vitals include temperature 97.9º, BP of 83/42 mm Hg, HR 112. Labs reveal hemoglobin 6.3 g/dL, hematocrit 18%, creatinine 1.3 mg/dL, total bilirubin 1.2 mg/dL, INR 1.0, platelet count of 63 x 103/microL. You suspect that this is an esophageal variceal bleed.
Idarucizumab reverses effects of dabigatran in emergencies
BERLIN—Final results from the RE-VERSE AD trial suggest idarucizumab can reverse the anticoagulant effect of dabigatran etexilate mesylate in emergency situations.
Patients who required dabigatran reversal because they needed urgent surgery were able to have their procedure a median of 1.6 hours from idarucizumab administration.
In patients who required dabigatran reversal due to uncontrollable bleeding, the median time to bleeding cessation was 2.5 hours. However, the time to bleeding cessation could not be assessed in all patients.
“RE-VERSE AD has shown that idarucizumab reverses the anticoagulant effect of dabigatran within minutes so that treating physicians can fully focus on dealing with the emergency at hand,” said study investigator Charles Pollack, MD, of Sidney Kimmel Medical College of Thomas Jefferson University in Philadelphia, Pennsylvania.
“Prior to idarucizumab, there was no rapid, reliable, and effective method for reversing dabigatran and other orally administered blood thinners, which otherwise may take at least 12 to 24 hours to clear from the body.”
The rate of thrombotic events in RE-VERSE AD was low (6% to 7%), and researchers said there were no serious adverse safety signals related to idarucizumab.
These results were presented at the International Society on Thrombosis and Haemostasis (ISTH) 2017 Congress and published in NEJM. The research was funded by Boehringer Ingelheim Pharmaceuticals.
The phase 3 trial enrolled 503 patients who required dabigatran reversal. They were divided into 2 groups.
Group A included 301 patients with uncontrolled or life-threatening bleeding complications (eg, intracranial hemorrhage or severe trauma after a car accident).
Group B included 202 patients requiring an invasive procedure or an emergency surgery or intervention (eg, surgery for an open fracture after a fall).
Results
The study’s primary endpoint was the degree of reversal of the anticoagulant effect of dabigatran achieved by idarucizumab within 4 hours. This was measured by diluted thrombin time or ecarin clotting time.
The median maximum percentage of dabigatran reversal was 100%. Dabigatran reversal occurred independently of patients’ age, sex, renal function, and dabigatran concentration at baseline.
Group A
Roughly 68% of evaluable patients in Group A (134/203) had confirmed bleeding cessation within 24 hours of idarucizumab administration.
Bleeding cessation was not confirmed in 67 patients, bleeding stopped before treatment in 2 patients, and the time to bleeding cessation could not be assessed in the 98 patients with intracranial bleeding.
Among the 134 patients in who had confirmed bleeding cessation, the median time to hemostasis after idarucizumab administration was 2.5 hours.
Sixty-seven percent (n=201) of patients in Group A received hemostatic treatment, 62% (n=185) received whole blood/blood components, 7% (n=20) received plasma derivatives, and 23% (n=69) received volume expanders, pro-hemostatic agents, albumin, and other treatments.
At 90 days, thrombotic events had occurred in 6.3% of the patients in Group A, and the mortality rate was 18.8%.
Group B
For patients in Group B, their required procedures began a median of 1.6 hours from idarucizumab administration.
Hemostasis during the procedure was described as normal for 93.4% of the patients, mildly abnormal in 5.1%, and moderately abnormal in 1.5%.
Thirty-nine percent (n=79) of patients in Group B received hemostatic treatment, 26% (n=53) received whole blood/blood components, 4% (n=8) received plasma derivatives, and 21% (n=42) received volume expanders, pro-hemostatic agents, albumin, and other treatments.
At 90 days, thrombotic events had occurred in 7.4% of patients in Group B, and the mortality rate was 18.9%. ![]()
BERLIN—Final results from the RE-VERSE AD trial suggest idarucizumab can reverse the anticoagulant effect of dabigatran etexilate mesylate in emergency situations.
Patients who required dabigatran reversal because they needed urgent surgery were able to have their procedure a median of 1.6 hours from idarucizumab administration.
In patients who required dabigatran reversal due to uncontrollable bleeding, the median time to bleeding cessation was 2.5 hours. However, the time to bleeding cessation could not be assessed in all patients.
“RE-VERSE AD has shown that idarucizumab reverses the anticoagulant effect of dabigatran within minutes so that treating physicians can fully focus on dealing with the emergency at hand,” said study investigator Charles Pollack, MD, of Sidney Kimmel Medical College of Thomas Jefferson University in Philadelphia, Pennsylvania.
“Prior to idarucizumab, there was no rapid, reliable, and effective method for reversing dabigatran and other orally administered blood thinners, which otherwise may take at least 12 to 24 hours to clear from the body.”
The rate of thrombotic events in RE-VERSE AD was low (6% to 7%), and researchers said there were no serious adverse safety signals related to idarucizumab.
These results were presented at the International Society on Thrombosis and Haemostasis (ISTH) 2017 Congress and published in NEJM. The research was funded by Boehringer Ingelheim Pharmaceuticals.
The phase 3 trial enrolled 503 patients who required dabigatran reversal. They were divided into 2 groups.
Group A included 301 patients with uncontrolled or life-threatening bleeding complications (eg, intracranial hemorrhage or severe trauma after a car accident).
Group B included 202 patients requiring an invasive procedure or an emergency surgery or intervention (eg, surgery for an open fracture after a fall).
Results
The study’s primary endpoint was the degree of reversal of the anticoagulant effect of dabigatran achieved by idarucizumab within 4 hours. This was measured by diluted thrombin time or ecarin clotting time.
The median maximum percentage of dabigatran reversal was 100%. Dabigatran reversal occurred independently of patients’ age, sex, renal function, and dabigatran concentration at baseline.
Group A
Roughly 68% of evaluable patients in Group A (134/203) had confirmed bleeding cessation within 24 hours of idarucizumab administration.
Bleeding cessation was not confirmed in 67 patients, bleeding stopped before treatment in 2 patients, and the time to bleeding cessation could not be assessed in the 98 patients with intracranial bleeding.
Among the 134 patients in who had confirmed bleeding cessation, the median time to hemostasis after idarucizumab administration was 2.5 hours.
Sixty-seven percent (n=201) of patients in Group A received hemostatic treatment, 62% (n=185) received whole blood/blood components, 7% (n=20) received plasma derivatives, and 23% (n=69) received volume expanders, pro-hemostatic agents, albumin, and other treatments.
At 90 days, thrombotic events had occurred in 6.3% of the patients in Group A, and the mortality rate was 18.8%.
Group B
For patients in Group B, their required procedures began a median of 1.6 hours from idarucizumab administration.
Hemostasis during the procedure was described as normal for 93.4% of the patients, mildly abnormal in 5.1%, and moderately abnormal in 1.5%.
Thirty-nine percent (n=79) of patients in Group B received hemostatic treatment, 26% (n=53) received whole blood/blood components, 4% (n=8) received plasma derivatives, and 21% (n=42) received volume expanders, pro-hemostatic agents, albumin, and other treatments.
At 90 days, thrombotic events had occurred in 7.4% of patients in Group B, and the mortality rate was 18.9%. ![]()
BERLIN—Final results from the RE-VERSE AD trial suggest idarucizumab can reverse the anticoagulant effect of dabigatran etexilate mesylate in emergency situations.
Patients who required dabigatran reversal because they needed urgent surgery were able to have their procedure a median of 1.6 hours from idarucizumab administration.
In patients who required dabigatran reversal due to uncontrollable bleeding, the median time to bleeding cessation was 2.5 hours. However, the time to bleeding cessation could not be assessed in all patients.
“RE-VERSE AD has shown that idarucizumab reverses the anticoagulant effect of dabigatran within minutes so that treating physicians can fully focus on dealing with the emergency at hand,” said study investigator Charles Pollack, MD, of Sidney Kimmel Medical College of Thomas Jefferson University in Philadelphia, Pennsylvania.
“Prior to idarucizumab, there was no rapid, reliable, and effective method for reversing dabigatran and other orally administered blood thinners, which otherwise may take at least 12 to 24 hours to clear from the body.”
The rate of thrombotic events in RE-VERSE AD was low (6% to 7%), and researchers said there were no serious adverse safety signals related to idarucizumab.
These results were presented at the International Society on Thrombosis and Haemostasis (ISTH) 2017 Congress and published in NEJM. The research was funded by Boehringer Ingelheim Pharmaceuticals.
The phase 3 trial enrolled 503 patients who required dabigatran reversal. They were divided into 2 groups.
Group A included 301 patients with uncontrolled or life-threatening bleeding complications (eg, intracranial hemorrhage or severe trauma after a car accident).
Group B included 202 patients requiring an invasive procedure or an emergency surgery or intervention (eg, surgery for an open fracture after a fall).
Results
The study’s primary endpoint was the degree of reversal of the anticoagulant effect of dabigatran achieved by idarucizumab within 4 hours. This was measured by diluted thrombin time or ecarin clotting time.
The median maximum percentage of dabigatran reversal was 100%. Dabigatran reversal occurred independently of patients’ age, sex, renal function, and dabigatran concentration at baseline.
Group A
Roughly 68% of evaluable patients in Group A (134/203) had confirmed bleeding cessation within 24 hours of idarucizumab administration.
Bleeding cessation was not confirmed in 67 patients, bleeding stopped before treatment in 2 patients, and the time to bleeding cessation could not be assessed in the 98 patients with intracranial bleeding.
Among the 134 patients in who had confirmed bleeding cessation, the median time to hemostasis after idarucizumab administration was 2.5 hours.
Sixty-seven percent (n=201) of patients in Group A received hemostatic treatment, 62% (n=185) received whole blood/blood components, 7% (n=20) received plasma derivatives, and 23% (n=69) received volume expanders, pro-hemostatic agents, albumin, and other treatments.
At 90 days, thrombotic events had occurred in 6.3% of the patients in Group A, and the mortality rate was 18.8%.
Group B
For patients in Group B, their required procedures began a median of 1.6 hours from idarucizumab administration.
Hemostasis during the procedure was described as normal for 93.4% of the patients, mildly abnormal in 5.1%, and moderately abnormal in 1.5%.
Thirty-nine percent (n=79) of patients in Group B received hemostatic treatment, 26% (n=53) received whole blood/blood components, 4% (n=8) received plasma derivatives, and 21% (n=42) received volume expanders, pro-hemostatic agents, albumin, and other treatments.
At 90 days, thrombotic events had occurred in 7.4% of patients in Group B, and the mortality rate was 18.9%. ![]()
FDA grants priority review to sNDA for dasatinib

The US Food and Drug Administration (FDA) has accepted for priority review a supplemental new drug application (sNDA) for dasatinib (Sprycel).
Bristol Myers Squibb is seeking approval for dasatinib as a treatment for children with Philadelphia chromosome-positive (Ph+) chronic phase (CP) chronic myeloid leukemia (CML), as well as approval for a powder formulation of dasatinib for oral suspension.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA plans to make a decision on the dasatinib sNDA by November 9, 2017.
The sNDA includes data from CA180-226 (NCT00777036), an ongoing, phase 2 trial of dasatinib in pediatric patients with CP-CML who are resistant to or cannot tolerate imatinib and pediatric patients newly diagnosed with CP-CML.
The trial enrolled patients aged 18 and younger with newly diagnosed CML or Ph+ leukemias resistant to or intolerant of imatinib.
Cohort 1 included 29 CP-CML patients resistant to or intolerant of imatinib. Cohort 2 included patients with accelerated/blast phase CML or Ph+ acute lymphoblastic leukemia. Cohort 3 included 84 patients with newly diagnosed CP-CML.
Data from Cohorts 1 and 3 were recently presented at the 2017 ASCO Annual Meeting.
Three months into treatment with dasatinib, patients with CP-CML who were resistant to or intolerant of imatinib (Cohort 1) had a cumulative major cytogenetic response rate of 55.2%. This response rate increased over time to exceed 90% at 24 months.
Newly diagnosed patients with CP-CML (Cohort 3) received dasatinib orally or as powder for oral suspension once daily. They achieved a cumulative complete cytogenetic response rate of 64% as early as 6 months into treatment. This response rate increased to 94% at 24 months.
The median duration of response was not estimable or not yet reached in each cohort at the time of follow-up.
The estimated progression-free survival at 48 months was greater than 75% for patients in Cohort 1 and greater than 90% for patients in Cohort 3.
The safety profile of dasatinib in this study was deemed comparable to that reported in adults with CP-CML. In this study, there were no reported events of pleural/pericardial effusion, pulmonary edema/hypertension, or pulmonary arterial hypertension related to dasatinib.
Dasatinib first received FDA approval in 2006. The drug is currently approved to treat adults with:
- Newly diagnosed Ph+ CP-CML
- Chronic, accelerated, or blast phase Ph+ CML with resistance or intolerance to prior therapy including imatinib
- Ph+ acute lymphoblastic leukemia with resistance or intolerance to prior therapy.

The US Food and Drug Administration (FDA) has accepted for priority review a supplemental new drug application (sNDA) for dasatinib (Sprycel).
Bristol Myers Squibb is seeking approval for dasatinib as a treatment for children with Philadelphia chromosome-positive (Ph+) chronic phase (CP) chronic myeloid leukemia (CML), as well as approval for a powder formulation of dasatinib for oral suspension.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA plans to make a decision on the dasatinib sNDA by November 9, 2017.
The sNDA includes data from CA180-226 (NCT00777036), an ongoing, phase 2 trial of dasatinib in pediatric patients with CP-CML who are resistant to or cannot tolerate imatinib and pediatric patients newly diagnosed with CP-CML.
The trial enrolled patients aged 18 and younger with newly diagnosed CML or Ph+ leukemias resistant to or intolerant of imatinib.
Cohort 1 included 29 CP-CML patients resistant to or intolerant of imatinib. Cohort 2 included patients with accelerated/blast phase CML or Ph+ acute lymphoblastic leukemia. Cohort 3 included 84 patients with newly diagnosed CP-CML.
Data from Cohorts 1 and 3 were recently presented at the 2017 ASCO Annual Meeting.
Three months into treatment with dasatinib, patients with CP-CML who were resistant to or intolerant of imatinib (Cohort 1) had a cumulative major cytogenetic response rate of 55.2%. This response rate increased over time to exceed 90% at 24 months.
Newly diagnosed patients with CP-CML (Cohort 3) received dasatinib orally or as powder for oral suspension once daily. They achieved a cumulative complete cytogenetic response rate of 64% as early as 6 months into treatment. This response rate increased to 94% at 24 months.
The median duration of response was not estimable or not yet reached in each cohort at the time of follow-up.
The estimated progression-free survival at 48 months was greater than 75% for patients in Cohort 1 and greater than 90% for patients in Cohort 3.
The safety profile of dasatinib in this study was deemed comparable to that reported in adults with CP-CML. In this study, there were no reported events of pleural/pericardial effusion, pulmonary edema/hypertension, or pulmonary arterial hypertension related to dasatinib.
Dasatinib first received FDA approval in 2006. The drug is currently approved to treat adults with:
- Newly diagnosed Ph+ CP-CML
- Chronic, accelerated, or blast phase Ph+ CML with resistance or intolerance to prior therapy including imatinib
- Ph+ acute lymphoblastic leukemia with resistance or intolerance to prior therapy.
The US Food and Drug Administration (FDA) has accepted for priority review a supplemental new drug application (sNDA) for dasatinib (Sprycel).
Bristol Myers Squibb is seeking approval for dasatinib as a treatment for children with Philadelphia chromosome-positive (Ph+) chronic phase (CP) chronic myeloid leukemia (CML), as well as approval for a powder formulation of dasatinib for oral suspension.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA plans to make a decision on the dasatinib sNDA by November 9, 2017.
The sNDA includes data from CA180-226 (NCT00777036), an ongoing, phase 2 trial of dasatinib in pediatric patients with CP-CML who are resistant to or cannot tolerate imatinib and pediatric patients newly diagnosed with CP-CML.
The trial enrolled patients aged 18 and younger with newly diagnosed CML or Ph+ leukemias resistant to or intolerant of imatinib.
Cohort 1 included 29 CP-CML patients resistant to or intolerant of imatinib. Cohort 2 included patients with accelerated/blast phase CML or Ph+ acute lymphoblastic leukemia. Cohort 3 included 84 patients with newly diagnosed CP-CML.
Data from Cohorts 1 and 3 were recently presented at the 2017 ASCO Annual Meeting.
Three months into treatment with dasatinib, patients with CP-CML who were resistant to or intolerant of imatinib (Cohort 1) had a cumulative major cytogenetic response rate of 55.2%. This response rate increased over time to exceed 90% at 24 months.
Newly diagnosed patients with CP-CML (Cohort 3) received dasatinib orally or as powder for oral suspension once daily. They achieved a cumulative complete cytogenetic response rate of 64% as early as 6 months into treatment. This response rate increased to 94% at 24 months.
The median duration of response was not estimable or not yet reached in each cohort at the time of follow-up.
The estimated progression-free survival at 48 months was greater than 75% for patients in Cohort 1 and greater than 90% for patients in Cohort 3.
The safety profile of dasatinib in this study was deemed comparable to that reported in adults with CP-CML. In this study, there were no reported events of pleural/pericardial effusion, pulmonary edema/hypertension, or pulmonary arterial hypertension related to dasatinib.
Dasatinib first received FDA approval in 2006. The drug is currently approved to treat adults with:
- Newly diagnosed Ph+ CP-CML
- Chronic, accelerated, or blast phase Ph+ CML with resistance or intolerance to prior therapy including imatinib
- Ph+ acute lymphoblastic leukemia with resistance or intolerance to prior therapy.
Company launches automated hematology analyzers in US

Sysmex America, Inc. has launched its XN-L™ automated hematology analyzers in the US.
The company says this new, smaller XN-L line delivers the same clinical and operational value as its XN-Series™ to lower-volume hematology laboratories.
“The XN-Series has been our flagship hematology analyzer line, and, today, we expand our offering by introducing the XN-L Series to meet the demands of lower-volume facilities,” said Andy Hay, chief operating officer of Sysmex America.
“With an XN-L analyzer, clinicians can rely upon the same expanded complete blood count (CBC) featured in our XN-Series analyzers to assess and monitor patients. This expanded CBC goes well beyond the 3-part differential that is currently available to most of the low-volume segment today and can aid in the assessment of sepsis and other hematological disorders.”
The XN-L automated hematology analyzers offer a 6-part differential, including immature granulocyte analysis on each sample.
In addition, the XN-L Series offers optional software licenses for (1) a reticulocyte channel to aid in anemia management and (2) body fluid cell counts.
The XN-L analyzers will also be the first to feature BeyondCare Quality Monitor, a new approach to quality assurance.
“BeyondCare Quality Monitor, a Sysmex proprietary innovation, is a leading-edge, evidence-based QC and calibration verification management program that partners Sysmex with the laboratory to proactively monitor quality assurance metrics,” Hay said. “It will be standard on all XN-L hematology analyzers.
Additional information on XN-L analyzers is available on the Sysmex website at www.sysmex.com/XNL.
The XN-L analyzers will be available both directly through the company and through its distribution partners. Interested parties should contact their Sysmex representative or Sysmex distributor for complete details. ![]()
Sysmex America, Inc. has launched its XN-L™ automated hematology analyzers in the US.
The company says this new, smaller XN-L line delivers the same clinical and operational value as its XN-Series™ to lower-volume hematology laboratories.
“The XN-Series has been our flagship hematology analyzer line, and, today, we expand our offering by introducing the XN-L Series to meet the demands of lower-volume facilities,” said Andy Hay, chief operating officer of Sysmex America.
“With an XN-L analyzer, clinicians can rely upon the same expanded complete blood count (CBC) featured in our XN-Series analyzers to assess and monitor patients. This expanded CBC goes well beyond the 3-part differential that is currently available to most of the low-volume segment today and can aid in the assessment of sepsis and other hematological disorders.”
The XN-L automated hematology analyzers offer a 6-part differential, including immature granulocyte analysis on each sample.
In addition, the XN-L Series offers optional software licenses for (1) a reticulocyte channel to aid in anemia management and (2) body fluid cell counts.
The XN-L analyzers will also be the first to feature BeyondCare Quality Monitor, a new approach to quality assurance.
“BeyondCare Quality Monitor, a Sysmex proprietary innovation, is a leading-edge, evidence-based QC and calibration verification management program that partners Sysmex with the laboratory to proactively monitor quality assurance metrics,” Hay said. “It will be standard on all XN-L hematology analyzers.
Additional information on XN-L analyzers is available on the Sysmex website at www.sysmex.com/XNL.
The XN-L analyzers will be available both directly through the company and through its distribution partners. Interested parties should contact their Sysmex representative or Sysmex distributor for complete details. ![]()
Sysmex America, Inc. has launched its XN-L™ automated hematology analyzers in the US.
The company says this new, smaller XN-L line delivers the same clinical and operational value as its XN-Series™ to lower-volume hematology laboratories.
“The XN-Series has been our flagship hematology analyzer line, and, today, we expand our offering by introducing the XN-L Series to meet the demands of lower-volume facilities,” said Andy Hay, chief operating officer of Sysmex America.
“With an XN-L analyzer, clinicians can rely upon the same expanded complete blood count (CBC) featured in our XN-Series analyzers to assess and monitor patients. This expanded CBC goes well beyond the 3-part differential that is currently available to most of the low-volume segment today and can aid in the assessment of sepsis and other hematological disorders.”
The XN-L automated hematology analyzers offer a 6-part differential, including immature granulocyte analysis on each sample.
In addition, the XN-L Series offers optional software licenses for (1) a reticulocyte channel to aid in anemia management and (2) body fluid cell counts.
The XN-L analyzers will also be the first to feature BeyondCare Quality Monitor, a new approach to quality assurance.
“BeyondCare Quality Monitor, a Sysmex proprietary innovation, is a leading-edge, evidence-based QC and calibration verification management program that partners Sysmex with the laboratory to proactively monitor quality assurance metrics,” Hay said. “It will be standard on all XN-L hematology analyzers.
Additional information on XN-L analyzers is available on the Sysmex website at www.sysmex.com/XNL.
The XN-L analyzers will be available both directly through the company and through its distribution partners. Interested parties should contact their Sysmex representative or Sysmex distributor for complete details. ![]()
Knee Pain, Heart Strain?
ANSWER
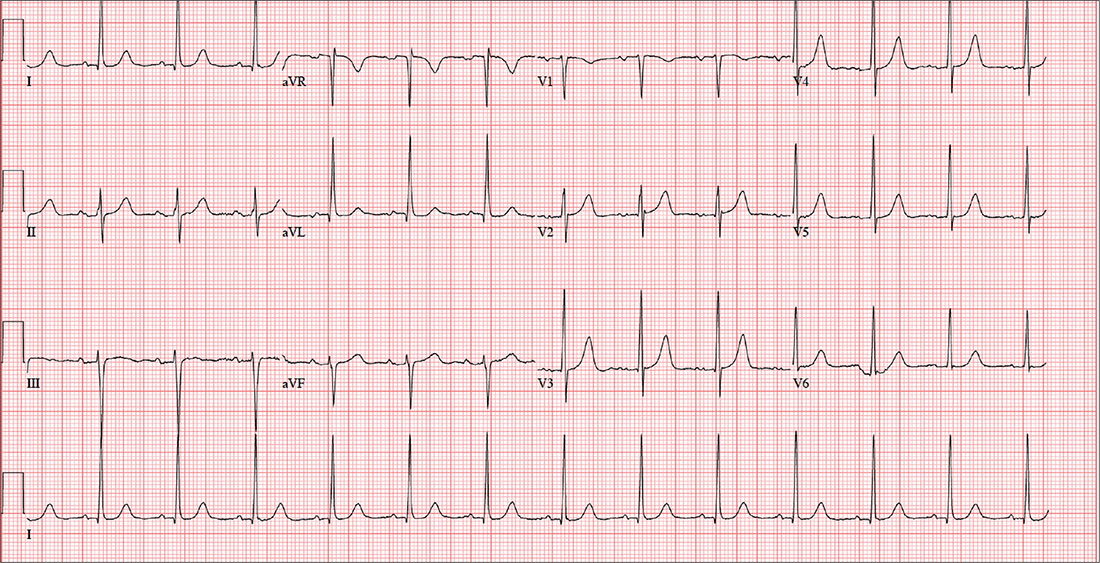
The correct interpretation includes normal sinus rhythm with left ventricular hypertrophy (LVH) and possible left atrial enlargement. Criteria for LVH include high voltages in the limb leads (R wave in lead I and S wave in lead III ≥ 25 mm) or the precordial leads (S wave in V1 and R wave in V6 ≥ 35 mm). Left atrial enlargement and ST-T wave abnormalities are often seen with LVH. The notched P wave in lead II and biphasic P wave in V1 raise suspicion for left atrial involvement. Finally, repolarization of a hypertrophic left ventricle following systole is responsible for the tall T waves seen in leads
ANSWER
The correct interpretation includes normal sinus rhythm with left ventricular hypertrophy (LVH) and possible left atrial enlargement. Criteria for LVH include high voltages in the limb leads (R wave in lead I and S wave in lead III ≥ 25 mm) or the precordial leads (S wave in V1 and R wave in V6 ≥ 35 mm). Left atrial enlargement and ST-T wave abnormalities are often seen with LVH. The notched P wave in lead II and biphasic P wave in V1 raise suspicion for left atrial involvement. Finally, repolarization of a hypertrophic left ventricle following systole is responsible for the tall T waves seen in leads
ANSWER
The correct interpretation includes normal sinus rhythm with left ventricular hypertrophy (LVH) and possible left atrial enlargement. Criteria for LVH include high voltages in the limb leads (R wave in lead I and S wave in lead III ≥ 25 mm) or the precordial leads (S wave in V1 and R wave in V6 ≥ 35 mm). Left atrial enlargement and ST-T wave abnormalities are often seen with LVH. The notched P wave in lead II and biphasic P wave in V1 raise suspicion for left atrial involvement. Finally, repolarization of a hypertrophic left ventricle following systole is responsible for the tall T waves seen in leads
A 47-year-old man presents for preoperative exam prior to right knee arthroplasty. He twisted his knee while training for a triathlon; MRI showed a bucket handle tear of the medial meniscus.
The patient has been very active throughout his life. Medical history is remarkable for essential hypertension. He has no history of chest pain, palpitations, shortness of breath, syncope, or near-syncope.
Current medications include metoprolol XL (25 mg/d)—which he hasn’t taken in five days, since he hasn’t been able to pick up his refill—and ibuprofen (600 mg tid, prn for knee pain). He denies illicit drug use.
The patient works as an accountant and is married with two children. His parents and grandparents are all alive and well. He has never smoked tobacco but does use marijuana socially on weekends. He also has one to two glasses of wine each night.
Review of systems is noncontributory: no recent colds or flu, bowel or bladder dysfunction, or weight changes. Vital signs include a blood pressure of 138/80 mm Hg; pulse, 80 beats/min; respiratory rate, 14 breaths/min-1; and temperature, 98°F. His weight is 194 lb and his height, 75 in. Pertinent physical findings include pain on palpation of the medial aspect of the right knee and a positive McMurray sign.
Bloodwork, a chest x-ray, and an ECG are obtained. The ECG shows a ventricular rate of 79 beats/min; PR interval, 184 ms; QRS duration, 76 ms; QT/QTc intervals, 382/438 ms; P axis, 48°; R axis, –29°; and T axis, 33°. What is your interpretation of this ECG?
New federal health IT leadership, same goals
WASHINGTON – Although the leadership at the Office of the National Coordinator for Health Information Technology is new, the focus of the federal office – reducing physician burden and improving interoperability of electronic heath records – remains the same.
“One priority is on the whole question of burden of [EHR] usability,” said Don Rucker, MD, the new national coordinator, at a July 11 press briefing. “The other is interoperability. We’ve obviously spent a lot of money collectively in the country on these systems, and there’s a widespread dissatisfaction with the level of interoperability.”
“We are looking at documentation and the whole quality framework around value-based purchasing,” he said. “For a lot of practices now, this has become a challenge that we just have to think about what’s the win. At some point, the expense of complying with the quality measures is a much greater expense than the innate value of the quality measures. ”
EHRs “have become symbolic of physician administrative burden, but by no means are they the whole cause,” John Flemming, MD, ONC deputy assistant secretary for health technology reform, said at the briefing. “The physician, particularly in an independent practice, must manage the practice. So he or she is the CEO. They are also on the assembly line, seeing patients. Now with EHRs, they have to be the data input person as well. It’s time consuming.”
Dr. Fleming is a family physician from Louisiana and a former Republican member of congress.
Dr. Rucker acknowledged that reducing the burden of EHRs has been discussed for quite a long time now. He recalled beginning working with them in his private practice back in 1988 and figured, based on the quick rate of technological innovation demonstrated in Silicon Valley, the issues would be solved by 1992 or 1993 at the latest.
“Right now, [EHRs] are really about documentation, about billing, but that is a funny kind of beast,” he said. “Every other industry uses their enterprise computer software to do automation, to become more efficient. We are the only business that I am aware of to have used computers to become less efficient. ... I think part of what we are trying to do ... is let some of these newer technologies that will actually reduce costs, reduce variance, have those technologies have an entrée into some of these data collections that are out there.”
WASHINGTON – Although the leadership at the Office of the National Coordinator for Health Information Technology is new, the focus of the federal office – reducing physician burden and improving interoperability of electronic heath records – remains the same.
“One priority is on the whole question of burden of [EHR] usability,” said Don Rucker, MD, the new national coordinator, at a July 11 press briefing. “The other is interoperability. We’ve obviously spent a lot of money collectively in the country on these systems, and there’s a widespread dissatisfaction with the level of interoperability.”
“We are looking at documentation and the whole quality framework around value-based purchasing,” he said. “For a lot of practices now, this has become a challenge that we just have to think about what’s the win. At some point, the expense of complying with the quality measures is a much greater expense than the innate value of the quality measures. ”
EHRs “have become symbolic of physician administrative burden, but by no means are they the whole cause,” John Flemming, MD, ONC deputy assistant secretary for health technology reform, said at the briefing. “The physician, particularly in an independent practice, must manage the practice. So he or she is the CEO. They are also on the assembly line, seeing patients. Now with EHRs, they have to be the data input person as well. It’s time consuming.”
Dr. Fleming is a family physician from Louisiana and a former Republican member of congress.
Dr. Rucker acknowledged that reducing the burden of EHRs has been discussed for quite a long time now. He recalled beginning working with them in his private practice back in 1988 and figured, based on the quick rate of technological innovation demonstrated in Silicon Valley, the issues would be solved by 1992 or 1993 at the latest.
“Right now, [EHRs] are really about documentation, about billing, but that is a funny kind of beast,” he said. “Every other industry uses their enterprise computer software to do automation, to become more efficient. We are the only business that I am aware of to have used computers to become less efficient. ... I think part of what we are trying to do ... is let some of these newer technologies that will actually reduce costs, reduce variance, have those technologies have an entrée into some of these data collections that are out there.”
WASHINGTON – Although the leadership at the Office of the National Coordinator for Health Information Technology is new, the focus of the federal office – reducing physician burden and improving interoperability of electronic heath records – remains the same.
“One priority is on the whole question of burden of [EHR] usability,” said Don Rucker, MD, the new national coordinator, at a July 11 press briefing. “The other is interoperability. We’ve obviously spent a lot of money collectively in the country on these systems, and there’s a widespread dissatisfaction with the level of interoperability.”
“We are looking at documentation and the whole quality framework around value-based purchasing,” he said. “For a lot of practices now, this has become a challenge that we just have to think about what’s the win. At some point, the expense of complying with the quality measures is a much greater expense than the innate value of the quality measures. ”
EHRs “have become symbolic of physician administrative burden, but by no means are they the whole cause,” John Flemming, MD, ONC deputy assistant secretary for health technology reform, said at the briefing. “The physician, particularly in an independent practice, must manage the practice. So he or she is the CEO. They are also on the assembly line, seeing patients. Now with EHRs, they have to be the data input person as well. It’s time consuming.”
Dr. Fleming is a family physician from Louisiana and a former Republican member of congress.
Dr. Rucker acknowledged that reducing the burden of EHRs has been discussed for quite a long time now. He recalled beginning working with them in his private practice back in 1988 and figured, based on the quick rate of technological innovation demonstrated in Silicon Valley, the issues would be solved by 1992 or 1993 at the latest.
“Right now, [EHRs] are really about documentation, about billing, but that is a funny kind of beast,” he said. “Every other industry uses their enterprise computer software to do automation, to become more efficient. We are the only business that I am aware of to have used computers to become less efficient. ... I think part of what we are trying to do ... is let some of these newer technologies that will actually reduce costs, reduce variance, have those technologies have an entrée into some of these data collections that are out there.”
Hexavalent hepatitis B vaccination mostly immunogenic 10 years later
results of an Italian study show.
The phase 3 open-label, controlled study included 732 healthy Italian children aged 11-13 years who as infants had received a two-dose primary and booster course with either Hexavac (5 mcg hepatitis B surface antigen [HBsAg]) or Infanrix hexa (10 mcg HBsAg) at 3, 5, and 11 months of age; the last dose was received at least 10 years prior to the challenge dose of a monovalent HB vaccine (HBVaxPro, 5 mcg HBsAg).
Although some of the children had HB surface antigen antibody concentrations below the seroprotection threshold, most of them had an anamnestic response when challenged with the dose of HB vaccine, “indicating the presence of specific immune memory,” the investigators said. There was no evidence of active HB disease in any of the children.
Just what the meaning of the lack of immune memory is, defined as the failure to develop an anamnestic response following an HB vaccine challenge, remains to be determined.
results of an Italian study show.
The phase 3 open-label, controlled study included 732 healthy Italian children aged 11-13 years who as infants had received a two-dose primary and booster course with either Hexavac (5 mcg hepatitis B surface antigen [HBsAg]) or Infanrix hexa (10 mcg HBsAg) at 3, 5, and 11 months of age; the last dose was received at least 10 years prior to the challenge dose of a monovalent HB vaccine (HBVaxPro, 5 mcg HBsAg).
Although some of the children had HB surface antigen antibody concentrations below the seroprotection threshold, most of them had an anamnestic response when challenged with the dose of HB vaccine, “indicating the presence of specific immune memory,” the investigators said. There was no evidence of active HB disease in any of the children.
Just what the meaning of the lack of immune memory is, defined as the failure to develop an anamnestic response following an HB vaccine challenge, remains to be determined.
results of an Italian study show.
The phase 3 open-label, controlled study included 732 healthy Italian children aged 11-13 years who as infants had received a two-dose primary and booster course with either Hexavac (5 mcg hepatitis B surface antigen [HBsAg]) or Infanrix hexa (10 mcg HBsAg) at 3, 5, and 11 months of age; the last dose was received at least 10 years prior to the challenge dose of a monovalent HB vaccine (HBVaxPro, 5 mcg HBsAg).
Although some of the children had HB surface antigen antibody concentrations below the seroprotection threshold, most of them had an anamnestic response when challenged with the dose of HB vaccine, “indicating the presence of specific immune memory,” the investigators said. There was no evidence of active HB disease in any of the children.
Just what the meaning of the lack of immune memory is, defined as the failure to develop an anamnestic response following an HB vaccine challenge, remains to be determined.
FROM VACCINE