User login
Alectinib in ALK+ NSCLC is a watershed moment
CHICAGO – In what’s being hailed as practice-changing findings, the anaplastic lymphoma kinase inhibitor alectinib (Alecensa) was associated with more than doubled progression-free survival (PFS), compared with crizotinib (Xalkori), the current standard of care, in patients with treatment-naive non–small cell lung cancer (NSCLC) positive for ALK.
Additionally, in the global, phase III trial, alectinib was associated with a significantly lower risk of progression to CNS metastases, a common complication of advanced ALK+ NSCLC, reported Alice T. Shaw, MD, PhD, of the Massachusetts General Hospital Cancer Center in Boston, on behalf of investigators in the ALEX trial.
“I view this as a watershed moment for the treatment of ALK mutant–positive lung cancer,” commented ASCO expert John Heymach, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston.
Unlike other head-to-head studies of similar drugs that frequently show only incremental benefit, the ALEX results showed a dramatic difference in outcomes for patients treated with alectinib, he said.
By comparison, the median PFS difference between chemotherapy and crizotinib in the PROFILE 1014 in patients with ALK-positive NSCLC trial was 10.9 vs. 7.0 months, Dr. Heymach pointed out.
The ALEX investigators enrolled 303 patients with untreated ALK-positive NSCLC confirmed by a central immunohistochemistry lab and randomly assigned them to treatment with either oral alectinib 600 mg twice daily or crizotinib 250 mg b.i.d.
At the primary data cutoff in February 2017, median PFS, the primary endpoint, was 11.1 months for patients treated with crizotinib, versus not reached for those treated with alectinib, translating into a hazard ratio for alectinib of 0.47 (P less than .0001).
Based on an independent review, the median PFS was determined to be 10.4 months for crizotinib, vs. 25.7 months with alectinib (HR, 0.50; P not shown).
The cumulative incidence of CNS progression, a secondary endpoint, was 41.4% in the crizotinib arm, vs. 9.41% in the alectinib arm (cause-specific HR, 0.16; P not shown).
In each arm, 97% of patients had any adverse event, and the incidence of serious adverse events was similar between the arms, at 29% for crizotinib and 28% for alectinib.
Adverse events leading to treatment discontinuation, dose reduction, or dose interruption were more frequent with crizotinib.
In the question and answer portion of the briefing, Dr. Shaw was asked whether crizotinib still had a role in this population.
“Going forward, I think that it’s pretty clear, if you have a newly diagnosed patient with metastatic ALK-positive lung cancer, that likely alectinib would be the preferred first choice,” she said.
The ALEX trial is supported by Roche. Dr. Shaw disclosed consulting or an advisory role with the company, and multiple coauthors disclosed similar relationships.
CHICAGO – In what’s being hailed as practice-changing findings, the anaplastic lymphoma kinase inhibitor alectinib (Alecensa) was associated with more than doubled progression-free survival (PFS), compared with crizotinib (Xalkori), the current standard of care, in patients with treatment-naive non–small cell lung cancer (NSCLC) positive for ALK.
Additionally, in the global, phase III trial, alectinib was associated with a significantly lower risk of progression to CNS metastases, a common complication of advanced ALK+ NSCLC, reported Alice T. Shaw, MD, PhD, of the Massachusetts General Hospital Cancer Center in Boston, on behalf of investigators in the ALEX trial.
“I view this as a watershed moment for the treatment of ALK mutant–positive lung cancer,” commented ASCO expert John Heymach, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston.
Unlike other head-to-head studies of similar drugs that frequently show only incremental benefit, the ALEX results showed a dramatic difference in outcomes for patients treated with alectinib, he said.
By comparison, the median PFS difference between chemotherapy and crizotinib in the PROFILE 1014 in patients with ALK-positive NSCLC trial was 10.9 vs. 7.0 months, Dr. Heymach pointed out.
The ALEX investigators enrolled 303 patients with untreated ALK-positive NSCLC confirmed by a central immunohistochemistry lab and randomly assigned them to treatment with either oral alectinib 600 mg twice daily or crizotinib 250 mg b.i.d.
At the primary data cutoff in February 2017, median PFS, the primary endpoint, was 11.1 months for patients treated with crizotinib, versus not reached for those treated with alectinib, translating into a hazard ratio for alectinib of 0.47 (P less than .0001).
Based on an independent review, the median PFS was determined to be 10.4 months for crizotinib, vs. 25.7 months with alectinib (HR, 0.50; P not shown).
The cumulative incidence of CNS progression, a secondary endpoint, was 41.4% in the crizotinib arm, vs. 9.41% in the alectinib arm (cause-specific HR, 0.16; P not shown).
In each arm, 97% of patients had any adverse event, and the incidence of serious adverse events was similar between the arms, at 29% for crizotinib and 28% for alectinib.
Adverse events leading to treatment discontinuation, dose reduction, or dose interruption were more frequent with crizotinib.
In the question and answer portion of the briefing, Dr. Shaw was asked whether crizotinib still had a role in this population.
“Going forward, I think that it’s pretty clear, if you have a newly diagnosed patient with metastatic ALK-positive lung cancer, that likely alectinib would be the preferred first choice,” she said.
The ALEX trial is supported by Roche. Dr. Shaw disclosed consulting or an advisory role with the company, and multiple coauthors disclosed similar relationships.
CHICAGO – In what’s being hailed as practice-changing findings, the anaplastic lymphoma kinase inhibitor alectinib (Alecensa) was associated with more than doubled progression-free survival (PFS), compared with crizotinib (Xalkori), the current standard of care, in patients with treatment-naive non–small cell lung cancer (NSCLC) positive for ALK.
Additionally, in the global, phase III trial, alectinib was associated with a significantly lower risk of progression to CNS metastases, a common complication of advanced ALK+ NSCLC, reported Alice T. Shaw, MD, PhD, of the Massachusetts General Hospital Cancer Center in Boston, on behalf of investigators in the ALEX trial.
“I view this as a watershed moment for the treatment of ALK mutant–positive lung cancer,” commented ASCO expert John Heymach, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston.
Unlike other head-to-head studies of similar drugs that frequently show only incremental benefit, the ALEX results showed a dramatic difference in outcomes for patients treated with alectinib, he said.
By comparison, the median PFS difference between chemotherapy and crizotinib in the PROFILE 1014 in patients with ALK-positive NSCLC trial was 10.9 vs. 7.0 months, Dr. Heymach pointed out.
The ALEX investigators enrolled 303 patients with untreated ALK-positive NSCLC confirmed by a central immunohistochemistry lab and randomly assigned them to treatment with either oral alectinib 600 mg twice daily or crizotinib 250 mg b.i.d.
At the primary data cutoff in February 2017, median PFS, the primary endpoint, was 11.1 months for patients treated with crizotinib, versus not reached for those treated with alectinib, translating into a hazard ratio for alectinib of 0.47 (P less than .0001).
Based on an independent review, the median PFS was determined to be 10.4 months for crizotinib, vs. 25.7 months with alectinib (HR, 0.50; P not shown).
The cumulative incidence of CNS progression, a secondary endpoint, was 41.4% in the crizotinib arm, vs. 9.41% in the alectinib arm (cause-specific HR, 0.16; P not shown).
In each arm, 97% of patients had any adverse event, and the incidence of serious adverse events was similar between the arms, at 29% for crizotinib and 28% for alectinib.
Adverse events leading to treatment discontinuation, dose reduction, or dose interruption were more frequent with crizotinib.
In the question and answer portion of the briefing, Dr. Shaw was asked whether crizotinib still had a role in this population.
“Going forward, I think that it’s pretty clear, if you have a newly diagnosed patient with metastatic ALK-positive lung cancer, that likely alectinib would be the preferred first choice,” she said.
The ALEX trial is supported by Roche. Dr. Shaw disclosed consulting or an advisory role with the company, and multiple coauthors disclosed similar relationships.
AT ASCO 2017
Key clinical point: Alectinib was associated with more than double the progression-free survival of standard of care crizotinib in patients with non–small cell lung cancer positive for the anaplastic lymphoma kinase.
Major finding: Median PFS by independent review was 10.4 months with crizotinib vs. 25.7 months with alectinib.
Data source: The ALEX trial, a phase III trial of 303 patients with ALK-positive NSCLC.
Disclosures: The ALEX trial is supported by Roche. Dr. Shaw disclosed consulting or an advisory role with the company, and multiple coauthors disclosed similar relationships
Committee and chapter involvement allows SHM member to give back
Editor’s note: Each month, SHM puts the spotlight on some of our most active members who are making substantial contributions to hospital medicine. Log on to www.hospitalmedicine.org/getinvolved for more information on how you can lend your expertise to help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Paul Grant, MD, SFHM, assistant professor of medicine at the University of Michigan Medical School, Ann Arbor. Dr. Grant is the chair of SHM’s Membership Committee and an active member of SHM’s Michigan Chapter.
Why did you choose a career in hospital medicine, and how did you become an SHM member?

After residency, I completed a hospital medicine fellowship at the Cleveland Clinic. During my fellowship, I joined SHM. At that time, I knew nothing about the society, but that soon changed. My fellowship required me to attend the annual meeting and submit an abstract in the RIV competition, which was an extremely valuable experience for me. Not only was I blown away by the meeting, but my poster won the clinical vignette competition, as well! Needless to say, I’ve been an SHM member ever since.
What prompted you to join the Membership Committee? Can you discuss some of the projects the committee is currently working on?
Because SHM has done so much for my career as a hospitalist, I’ve tried to give back by volunteering on committees. After spending several years on the Early Career Hospitalist Committee, I felt the transition to the Membership Committee was a natural fit. Because SHM membership had been growing every year, our committee felt some pressure to keep this trend going. Thankfully, we have continued to see growth each year in every membership category.
Our committee has been working on several projects. One of the key demographics we have been targeting is the resident member. Residents play a significant role in the future of hospital medicine, as well as SHM membership. We are developing ways to reach out to residency program directors – particularly those running a hospital medicine track – to find ways they can benefit from SHM’s educational offerings. Additionally, our committee has been discussing ways to attract international members to SHM. Because hospital medicine is quite developed in the United States, we believe we have much to offer to hospitalists around the world.
Tell TH about your involvement with SHM’s Michigan Chapter. What does a typical chapter meeting entail?
A few years ago, at the end of SHM’s annual meeting, several of my hospital medicine colleagues in southeast Michigan happened to be on the same flight home. At the departure gate in the airport, we all agreed we should start an SHM chapter. After drawing straws, it was decided that I would be chapter president for our inaugural year. In a few short years, our chapter has grown into one of the largest in the country.
As for a typical meeting, each starts with a cocktail hour to encourage our members to network. We have a guest speaker, who presents on a hospital medicine topic, and then, we end the evening with a business meeting. We encourage students and residents to attend. More recently, we’ve been using interactive technology to broadcast our meetings to large hospital medicine groups in the western and northern parts of the state. Our chapter was thrilled to learn that we’d won the Outstanding Chapter award this year!
What value do you find in connecting with hospital medicine professionals at the local level?
Whether it’s a hospitalist working at a large, tertiary care center or one working in a small rural setting, it seems we all face similar challenges.
As a chapter, we can pull together our resources to address these issues. Furthermore, we have the ability to reach out to more trainees and show them what hospital medicine is all about. Our chapter has been able to partially fund both medical students and residents so they could attend SHM’s annual meeting. I’m always amazed at what I can learn from other hospitalists – in the state of Michigan and beyond.
Find a chapter near you and get involved at the local level at hospitalmedicine.org/chapters .
Felicia Steele is SHM’s communications coordinator.
Editor’s note: Each month, SHM puts the spotlight on some of our most active members who are making substantial contributions to hospital medicine. Log on to www.hospitalmedicine.org/getinvolved for more information on how you can lend your expertise to help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Paul Grant, MD, SFHM, assistant professor of medicine at the University of Michigan Medical School, Ann Arbor. Dr. Grant is the chair of SHM’s Membership Committee and an active member of SHM’s Michigan Chapter.
Why did you choose a career in hospital medicine, and how did you become an SHM member?
After residency, I completed a hospital medicine fellowship at the Cleveland Clinic. During my fellowship, I joined SHM. At that time, I knew nothing about the society, but that soon changed. My fellowship required me to attend the annual meeting and submit an abstract in the RIV competition, which was an extremely valuable experience for me. Not only was I blown away by the meeting, but my poster won the clinical vignette competition, as well! Needless to say, I’ve been an SHM member ever since.
What prompted you to join the Membership Committee? Can you discuss some of the projects the committee is currently working on?
Because SHM has done so much for my career as a hospitalist, I’ve tried to give back by volunteering on committees. After spending several years on the Early Career Hospitalist Committee, I felt the transition to the Membership Committee was a natural fit. Because SHM membership had been growing every year, our committee felt some pressure to keep this trend going. Thankfully, we have continued to see growth each year in every membership category.
Our committee has been working on several projects. One of the key demographics we have been targeting is the resident member. Residents play a significant role in the future of hospital medicine, as well as SHM membership. We are developing ways to reach out to residency program directors – particularly those running a hospital medicine track – to find ways they can benefit from SHM’s educational offerings. Additionally, our committee has been discussing ways to attract international members to SHM. Because hospital medicine is quite developed in the United States, we believe we have much to offer to hospitalists around the world.
Tell TH about your involvement with SHM’s Michigan Chapter. What does a typical chapter meeting entail?
A few years ago, at the end of SHM’s annual meeting, several of my hospital medicine colleagues in southeast Michigan happened to be on the same flight home. At the departure gate in the airport, we all agreed we should start an SHM chapter. After drawing straws, it was decided that I would be chapter president for our inaugural year. In a few short years, our chapter has grown into one of the largest in the country.
As for a typical meeting, each starts with a cocktail hour to encourage our members to network. We have a guest speaker, who presents on a hospital medicine topic, and then, we end the evening with a business meeting. We encourage students and residents to attend. More recently, we’ve been using interactive technology to broadcast our meetings to large hospital medicine groups in the western and northern parts of the state. Our chapter was thrilled to learn that we’d won the Outstanding Chapter award this year!
What value do you find in connecting with hospital medicine professionals at the local level?
Whether it’s a hospitalist working at a large, tertiary care center or one working in a small rural setting, it seems we all face similar challenges.
As a chapter, we can pull together our resources to address these issues. Furthermore, we have the ability to reach out to more trainees and show them what hospital medicine is all about. Our chapter has been able to partially fund both medical students and residents so they could attend SHM’s annual meeting. I’m always amazed at what I can learn from other hospitalists – in the state of Michigan and beyond.
Find a chapter near you and get involved at the local level at hospitalmedicine.org/chapters .
Felicia Steele is SHM’s communications coordinator.
Editor’s note: Each month, SHM puts the spotlight on some of our most active members who are making substantial contributions to hospital medicine. Log on to www.hospitalmedicine.org/getinvolved for more information on how you can lend your expertise to help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Paul Grant, MD, SFHM, assistant professor of medicine at the University of Michigan Medical School, Ann Arbor. Dr. Grant is the chair of SHM’s Membership Committee and an active member of SHM’s Michigan Chapter.
Why did you choose a career in hospital medicine, and how did you become an SHM member?
After residency, I completed a hospital medicine fellowship at the Cleveland Clinic. During my fellowship, I joined SHM. At that time, I knew nothing about the society, but that soon changed. My fellowship required me to attend the annual meeting and submit an abstract in the RIV competition, which was an extremely valuable experience for me. Not only was I blown away by the meeting, but my poster won the clinical vignette competition, as well! Needless to say, I’ve been an SHM member ever since.
What prompted you to join the Membership Committee? Can you discuss some of the projects the committee is currently working on?
Because SHM has done so much for my career as a hospitalist, I’ve tried to give back by volunteering on committees. After spending several years on the Early Career Hospitalist Committee, I felt the transition to the Membership Committee was a natural fit. Because SHM membership had been growing every year, our committee felt some pressure to keep this trend going. Thankfully, we have continued to see growth each year in every membership category.
Our committee has been working on several projects. One of the key demographics we have been targeting is the resident member. Residents play a significant role in the future of hospital medicine, as well as SHM membership. We are developing ways to reach out to residency program directors – particularly those running a hospital medicine track – to find ways they can benefit from SHM’s educational offerings. Additionally, our committee has been discussing ways to attract international members to SHM. Because hospital medicine is quite developed in the United States, we believe we have much to offer to hospitalists around the world.
Tell TH about your involvement with SHM’s Michigan Chapter. What does a typical chapter meeting entail?
A few years ago, at the end of SHM’s annual meeting, several of my hospital medicine colleagues in southeast Michigan happened to be on the same flight home. At the departure gate in the airport, we all agreed we should start an SHM chapter. After drawing straws, it was decided that I would be chapter president for our inaugural year. In a few short years, our chapter has grown into one of the largest in the country.
As for a typical meeting, each starts with a cocktail hour to encourage our members to network. We have a guest speaker, who presents on a hospital medicine topic, and then, we end the evening with a business meeting. We encourage students and residents to attend. More recently, we’ve been using interactive technology to broadcast our meetings to large hospital medicine groups in the western and northern parts of the state. Our chapter was thrilled to learn that we’d won the Outstanding Chapter award this year!
What value do you find in connecting with hospital medicine professionals at the local level?
Whether it’s a hospitalist working at a large, tertiary care center or one working in a small rural setting, it seems we all face similar challenges.
As a chapter, we can pull together our resources to address these issues. Furthermore, we have the ability to reach out to more trainees and show them what hospital medicine is all about. Our chapter has been able to partially fund both medical students and residents so they could attend SHM’s annual meeting. I’m always amazed at what I can learn from other hospitalists – in the state of Michigan and beyond.
Find a chapter near you and get involved at the local level at hospitalmedicine.org/chapters .
Felicia Steele is SHM’s communications coordinator.
VA to Adopt DoD Health Record System
The VA, which pioneered the electronic health record (EHR) system with the in-house development of VistA, will adopt a new EHR system based on the DoD’s MHS Genesis. “VA’s adoption of the same EHR system as DoD will ultimately result in all patient data residing in one common system and enable seamless care between the Departments without the manual and electronic exchange and reconciliation of data between two separate systems,” Secretary of Veteran Affairs David J. Shulkin, MD, explained in a press briefing.
To expedite the process, Dr. Shulkin issued a Determination and Findings, which allows the VA to circumvent the normal acquisition procedures and move forward more quickly. According to Dr. Shulkin, it took the DoD more than 2 years to complete the acquisition process. The MHS Genesis EHR system is based on the Cerner Millennium database. “It’s time to move forward, and as Secretary I was not willing to put this decision off any longer,” Dr. Shulkin said.
The move away from VistA and toward closer integration with the DoD has been a long time in coming. VistA was one of the first EHR systems, but the VA has struggled in recent years to maintain the system. It has been nearly 2 decades since Congress first asked VA and DoD to work more closely together, but integration of the 2 health systems has been expensive and incomplete.
“While we have established interoperability between VA and DoD for key aspects of the health record, seamless care is fundamentally constrained by ever-changing information sharing standards, separate chains of command, complex governance, separate implementation schedules that must be coordinated to accommodate those changes from separate program offices that have separate funding appropriations, and a host of related complexities requiring constant lifecycle maintenance,” Dr. Shulkin explained.
President Trump expressed his support for the move in a tweet. 
Even with the expedited acquisitions process, the process should still take 3 to 6 months to develop the specifications and determine the costs of the major technology overhaul. The DoD contract with Cerner is $4.3 billion, and the VA contract is expected to be larger. DoD officials involved in the MHS Genesis implementation will work with the VA, Dr. Shulkin noted, allowing the VA to leverage their experience in the EHR roll out.
The VA, which pioneered the electronic health record (EHR) system with the in-house development of VistA, will adopt a new EHR system based on the DoD’s MHS Genesis. “VA’s adoption of the same EHR system as DoD will ultimately result in all patient data residing in one common system and enable seamless care between the Departments without the manual and electronic exchange and reconciliation of data between two separate systems,” Secretary of Veteran Affairs David J. Shulkin, MD, explained in a press briefing.
To expedite the process, Dr. Shulkin issued a Determination and Findings, which allows the VA to circumvent the normal acquisition procedures and move forward more quickly. According to Dr. Shulkin, it took the DoD more than 2 years to complete the acquisition process. The MHS Genesis EHR system is based on the Cerner Millennium database. “It’s time to move forward, and as Secretary I was not willing to put this decision off any longer,” Dr. Shulkin said.
The move away from VistA and toward closer integration with the DoD has been a long time in coming. VistA was one of the first EHR systems, but the VA has struggled in recent years to maintain the system. It has been nearly 2 decades since Congress first asked VA and DoD to work more closely together, but integration of the 2 health systems has been expensive and incomplete.
“While we have established interoperability between VA and DoD for key aspects of the health record, seamless care is fundamentally constrained by ever-changing information sharing standards, separate chains of command, complex governance, separate implementation schedules that must be coordinated to accommodate those changes from separate program offices that have separate funding appropriations, and a host of related complexities requiring constant lifecycle maintenance,” Dr. Shulkin explained.
President Trump expressed his support for the move in a tweet.
Even with the expedited acquisitions process, the process should still take 3 to 6 months to develop the specifications and determine the costs of the major technology overhaul. The DoD contract with Cerner is $4.3 billion, and the VA contract is expected to be larger. DoD officials involved in the MHS Genesis implementation will work with the VA, Dr. Shulkin noted, allowing the VA to leverage their experience in the EHR roll out.
The VA, which pioneered the electronic health record (EHR) system with the in-house development of VistA, will adopt a new EHR system based on the DoD’s MHS Genesis. “VA’s adoption of the same EHR system as DoD will ultimately result in all patient data residing in one common system and enable seamless care between the Departments without the manual and electronic exchange and reconciliation of data between two separate systems,” Secretary of Veteran Affairs David J. Shulkin, MD, explained in a press briefing.
To expedite the process, Dr. Shulkin issued a Determination and Findings, which allows the VA to circumvent the normal acquisition procedures and move forward more quickly. According to Dr. Shulkin, it took the DoD more than 2 years to complete the acquisition process. The MHS Genesis EHR system is based on the Cerner Millennium database. “It’s time to move forward, and as Secretary I was not willing to put this decision off any longer,” Dr. Shulkin said.
The move away from VistA and toward closer integration with the DoD has been a long time in coming. VistA was one of the first EHR systems, but the VA has struggled in recent years to maintain the system. It has been nearly 2 decades since Congress first asked VA and DoD to work more closely together, but integration of the 2 health systems has been expensive and incomplete.
“While we have established interoperability between VA and DoD for key aspects of the health record, seamless care is fundamentally constrained by ever-changing information sharing standards, separate chains of command, complex governance, separate implementation schedules that must be coordinated to accommodate those changes from separate program offices that have separate funding appropriations, and a host of related complexities requiring constant lifecycle maintenance,” Dr. Shulkin explained.
President Trump expressed his support for the move in a tweet.
Even with the expedited acquisitions process, the process should still take 3 to 6 months to develop the specifications and determine the costs of the major technology overhaul. The DoD contract with Cerner is $4.3 billion, and the VA contract is expected to be larger. DoD officials involved in the MHS Genesis implementation will work with the VA, Dr. Shulkin noted, allowing the VA to leverage their experience in the EHR roll out.
HCV Cases Have Been Underreported, CDC Says
The number of new hepatitis C virus (HCV) infections has tripled in just 5 years—to a 15-year high—but the true scale is only now being revealed, according to the CDC.
Limited surveillance resources have led to underreporting, the CDC says. The annual number of reported HCV cases does not reflect the reality of the epidemic. Although 850 cases were reported in 2010 and 2,436 cases in 2015, the CDC estimates 34,000 actual new infections in 2015.
About three-quarters of the 3.5 million Americans with HCV are baby boomers, born between 1945 and 1965. They’re 6 times more likely to be infected with HCV and are at greater risk of death due to HCV. But HCV infections are spreading most rapidly among young adults aged 20 to 29 years, the CDC says, primarily because of injection drug use associated with opioids.
Because of that dual threat of virus spread and the opioid epidemic, HHS-recommended strategies include using comprehensive syringe service programs (SSPs). One CDC study found that 80% of young people with HCV live > 10 miles from an SSP. Another study found that only 3 states have laws that support full access to SSPs and HCV-related treatment and preventive services for people who inject drugs.
The number of new hepatitis C virus (HCV) infections has tripled in just 5 years—to a 15-year high—but the true scale is only now being revealed, according to the CDC.
Limited surveillance resources have led to underreporting, the CDC says. The annual number of reported HCV cases does not reflect the reality of the epidemic. Although 850 cases were reported in 2010 and 2,436 cases in 2015, the CDC estimates 34,000 actual new infections in 2015.
About three-quarters of the 3.5 million Americans with HCV are baby boomers, born between 1945 and 1965. They’re 6 times more likely to be infected with HCV and are at greater risk of death due to HCV. But HCV infections are spreading most rapidly among young adults aged 20 to 29 years, the CDC says, primarily because of injection drug use associated with opioids.
Because of that dual threat of virus spread and the opioid epidemic, HHS-recommended strategies include using comprehensive syringe service programs (SSPs). One CDC study found that 80% of young people with HCV live > 10 miles from an SSP. Another study found that only 3 states have laws that support full access to SSPs and HCV-related treatment and preventive services for people who inject drugs.
The number of new hepatitis C virus (HCV) infections has tripled in just 5 years—to a 15-year high—but the true scale is only now being revealed, according to the CDC.
Limited surveillance resources have led to underreporting, the CDC says. The annual number of reported HCV cases does not reflect the reality of the epidemic. Although 850 cases were reported in 2010 and 2,436 cases in 2015, the CDC estimates 34,000 actual new infections in 2015.
About three-quarters of the 3.5 million Americans with HCV are baby boomers, born between 1945 and 1965. They’re 6 times more likely to be infected with HCV and are at greater risk of death due to HCV. But HCV infections are spreading most rapidly among young adults aged 20 to 29 years, the CDC says, primarily because of injection drug use associated with opioids.
Because of that dual threat of virus spread and the opioid epidemic, HHS-recommended strategies include using comprehensive syringe service programs (SSPs). One CDC study found that 80% of young people with HCV live > 10 miles from an SSP. Another study found that only 3 states have laws that support full access to SSPs and HCV-related treatment and preventive services for people who inject drugs.
Recombinant coagulation factor IX approved for hemophilia B

Nonacog beta pegol (N9-GP, Rebinyn®), a recombinant, GlycoPEGylated coagulation factor IX, has been approved by the US Food and Drug Administration (FDA) for on-demand treatment and control of bleeding episodes in adults and children with hemophilia B.
It is also indicated for the perioperative management of bleeding in these individuals. However, it is not indicated for routine prophylaxis or for immune tolerance induction.
NovoNordisk, the manufacturer of the drug, expects the launch of the coagulation factor in the United States to take place in the first half of 2018.
N9-GP earlier this year had been recommended for marketing authorization by the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) as Refixia.® On June 6 it was actually granted marketing authorization, which covers all 28 European Union member states.
The FDA based its approval on the efficacy and safety evaluation of 115 previously treated patients across the four paradigmTM clinical trials. Results from the paradigm 4 trial were published in Thrombosis Research in May 2016.
Of 597 new bleeding episodes in 79 of 105 patients, 551 (93%) were resolved successfully with good or excellent ratings by the patients or study site investigator. Forty (7%) were rated as moderate or poor.
Most (87%) were resolved with 1 injection, 60 (10%) with 2 injections, and 16 (3%) with more than 2 injections.
The median dose to treat a bleeding episode was 42.3 IU/kg.
On-demand treatment had a success rate of 95%, with 120 (84%) of the 143 bleeds treated with one injection.
For perioperative management, N9-GP was rated as excellent or good for 13 surgeries, for a success rate of 100%.
Patients received a preoperative dose of 80 IU/kg. No patient required additional doses on the day of surgery.
During the postoperative period, patients required additional 40 IU/kg doses. The mean total consumption of factor in the pre- and postoperative period was 241 IU/kg (range, 8 – 460 IU/kg.
No unexpected postoperative bleeding occurred.
For full prescribing information, see the package insert. ![]()
Nonacog beta pegol (N9-GP, Rebinyn®), a recombinant, GlycoPEGylated coagulation factor IX, has been approved by the US Food and Drug Administration (FDA) for on-demand treatment and control of bleeding episodes in adults and children with hemophilia B.
It is also indicated for the perioperative management of bleeding in these individuals. However, it is not indicated for routine prophylaxis or for immune tolerance induction.
NovoNordisk, the manufacturer of the drug, expects the launch of the coagulation factor in the United States to take place in the first half of 2018.
N9-GP earlier this year had been recommended for marketing authorization by the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) as Refixia.® On June 6 it was actually granted marketing authorization, which covers all 28 European Union member states.
The FDA based its approval on the efficacy and safety evaluation of 115 previously treated patients across the four paradigmTM clinical trials. Results from the paradigm 4 trial were published in Thrombosis Research in May 2016.
Of 597 new bleeding episodes in 79 of 105 patients, 551 (93%) were resolved successfully with good or excellent ratings by the patients or study site investigator. Forty (7%) were rated as moderate or poor.
Most (87%) were resolved with 1 injection, 60 (10%) with 2 injections, and 16 (3%) with more than 2 injections.
The median dose to treat a bleeding episode was 42.3 IU/kg.
On-demand treatment had a success rate of 95%, with 120 (84%) of the 143 bleeds treated with one injection.
For perioperative management, N9-GP was rated as excellent or good for 13 surgeries, for a success rate of 100%.
Patients received a preoperative dose of 80 IU/kg. No patient required additional doses on the day of surgery.
During the postoperative period, patients required additional 40 IU/kg doses. The mean total consumption of factor in the pre- and postoperative period was 241 IU/kg (range, 8 – 460 IU/kg.
No unexpected postoperative bleeding occurred.
For full prescribing information, see the package insert. ![]()
Nonacog beta pegol (N9-GP, Rebinyn®), a recombinant, GlycoPEGylated coagulation factor IX, has been approved by the US Food and Drug Administration (FDA) for on-demand treatment and control of bleeding episodes in adults and children with hemophilia B.
It is also indicated for the perioperative management of bleeding in these individuals. However, it is not indicated for routine prophylaxis or for immune tolerance induction.
NovoNordisk, the manufacturer of the drug, expects the launch of the coagulation factor in the United States to take place in the first half of 2018.
N9-GP earlier this year had been recommended for marketing authorization by the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) as Refixia.® On June 6 it was actually granted marketing authorization, which covers all 28 European Union member states.
The FDA based its approval on the efficacy and safety evaluation of 115 previously treated patients across the four paradigmTM clinical trials. Results from the paradigm 4 trial were published in Thrombosis Research in May 2016.
Of 597 new bleeding episodes in 79 of 105 patients, 551 (93%) were resolved successfully with good or excellent ratings by the patients or study site investigator. Forty (7%) were rated as moderate or poor.
Most (87%) were resolved with 1 injection, 60 (10%) with 2 injections, and 16 (3%) with more than 2 injections.
The median dose to treat a bleeding episode was 42.3 IU/kg.
On-demand treatment had a success rate of 95%, with 120 (84%) of the 143 bleeds treated with one injection.
For perioperative management, N9-GP was rated as excellent or good for 13 surgeries, for a success rate of 100%.
Patients received a preoperative dose of 80 IU/kg. No patient required additional doses on the day of surgery.
During the postoperative period, patients required additional 40 IU/kg doses. The mean total consumption of factor in the pre- and postoperative period was 241 IU/kg (range, 8 – 460 IU/kg.
No unexpected postoperative bleeding occurred.
For full prescribing information, see the package insert. ![]()
Addition of ublituximab to ibrutinib improves response in r/r CLL

Ibrutinib, the Bruton’s tyrosine kinase (BTK) inhibitor, has transformed the treatment landscape for patients with relapsed or refractory (r/r) chronic lymphocytic leukemia (CLL).
Yet for patients with high-risk molecular features, such as 11q deletion, 17p deletion, or TP53 mutation, relapse remains problematic.
Investigators evaluated whether the addition of ublituximab to ibrutinib would improve the outcome of patients with genetically high-risk CLL in the GENUINE (UTX-IB-301) phase 3 study.
Jeff P. Sharman, MD, of Willamette Valley Cancer Institute and Research Center in Springfield, Oregon, reported the results at the 2017 ASCO Annual Meeting (abstract 7504).*
Ublituximab is a glycoengineered, anti-CD20 type 1 monoclonal antibody that maintains complement-dependent cytotoxicity and enhances antibody-dependent cell-mediated cytotoxicity. In a phase 2 study in combination with ibrutinib, it achieved an ORR of approximately 88%.
Protocol design
Originally, the study had co-primary endpoints of overall response rate (ORR) and progression-free survival (PFS). To adequately power for both endpoints, the target enrollment was 330 patients.
Dr Sharman explained that after 22 months of open enrollment, the trial sponsor determined that the original enrollment goal could not be met in a timely manner and elected to redesign the protocol.
In the modified protocol, ORR became the primary response rate and PFS a secondary endpoint. This allowed for a reduced target enrollment of 120. However, the study was no longer powered to detect a change in PFS.
Investigators stratified the patients by lines of prior therapy and then randomized them to receive ibrutinib or ublituximab plus ibrutinib.
The ibrutinib dose was 420 mg daily in both arms. Ublituximab dose was 900 mg on days 1, 8, and 15 of cycle 1, day 1 of cycles 2 through 6 and every third cycle thereafter.
The primary endpoint was ORR as assessed by Independent Central Review (IRC) using the iwCLL 2008 criteria.
Secondary endpoints included PFS, the complete response (CR) rate and depth of response (minimal residual disease [MRD] negativity), and safety.
The investigators assessed patients for response on weeks 8, 16, 24, and every 12 weeks thereafter.
The primary endpoint was evaluated when all enrolled patients had at least 2 efficacy evaluations.
The median follow-up was 11.4 months.
Patient characteristics
Patients with relapsed or refractory high-risk CLL had their disease centrally confirmed for the presence of deletion 17p, deletion 11q, and/or TP53 mutation.
They had measurable disease, ECOG performance status of 2 or less, no history of transformation of CLL, and no prior BTK inhibitor therapy.
The investigators randomized 126 patients, and 117 received any dose of therapy.
“The dropout was because in part ibrutinib was via commercial supply and not every patient could get access,” Dr Sharman noted.
Fifty-nine patients were treated in the combination arm and 58 in the monotherapy arm.
All patients had at least one of the specified mutations, which were relatively balanced between the 2 arms.
Patients were a mean age of 67 (range, 43 – 87), had a median of 3 prior therapies (range, 1 – 8), and more than 70% were male.
Patient characteristics were similar in each arm except for bulky disease, with 45% in the combination arm having bulky disease of 5 cm or more at baseline, compared with 26% in the monotherapy arm.
Twenty percent of the patients were considered refractory to rituximab.
Safety
Infusion reactions occurred in 54% of patients in the combination arm and 5% had grade 3/4 reactions. None occurred in the ibrutinib arm, since the latter is an orally bioavailable drug.
Other adverse events of all grades occurring in 10% of patients or more for the combination and monotherapy arms, respectively, were: diarrhea (42% and 40%), fatigue (27% and 33%), insomnia (24% and 10%), nausea (22% and 21%), headache (20% and 28%), arthralgia (19% and 17%), cough (19% and 24%), abdominal pain (15% and 9%), stomatitis (15% and 9%), upper respiratory infection (15% and 12%), dizziness (15% and 22%), contusion (15% and 29%), anemia (14% and 17%), and peripheral edema (10% and 21%).
Neutropenia was higher in the experimental arm, 22% any grade, compared with 12% in the ibrutinib arm, although grade 3 or higher neutropenia was similar in the 2 arms. Other laboratory abnormalities were similar between the arms.
Efficacy
The best ORR in the combination arm was 78%, with 7% achieving CR compared with 45% in the monotherapy arm with no CRs (P<0.001).
Nineteen percent of the combination arm achieved MRD negativity in peripheral blood compared with 2% of the monotherapy arm (P<0.01).
The reduction in lymph node size was similar between the arms.
In contrast, lymphocytosis was very different between the arms.
“As has been reported multiple times with targeted B-cell receptor signaling inhibitors,” Dr Sharman said, “patients treated with ibrutinib experienced rapid increase in their lymphocytes, returning approximately to baseline by 3 months and decreasing thereafter.”
“By contrast,” he continued, “those patients treated with the additional antibody had much more rapid resolution of their lymphocytosis. This was true whether patients were considered rituximab refractory or not.”
The investigators performed an additional analysis of ORR, this time including patients who achieved partial response with lymphocytosis (PR-L). These patients were not included in the primary endpoint because the iwCLL 2008 criteria had not yet been updated to include PR-L.
The best overall response including active PR-L patients was 83% in the experimental arm and 59% in the ibrutinib monotherapy arm (P<0.01).
PFS showed a trend toward improvement in the patients treated with the combination, with a hazard ratio of 0.559, which was not of statistical significance at the time of analysis.
The investigators concluded that the study met its primary endpoint, with a greater response rate and a greater depth of response than ibrutinib alone.
And the addition of ublituximab did not alter the safety profile of ibrutinib monotherapy.
TG Therapeutics, Inc, funded the study. ![]()
*Data in the abstract differ from the meeting presentation.
Ibrutinib, the Bruton’s tyrosine kinase (BTK) inhibitor, has transformed the treatment landscape for patients with relapsed or refractory (r/r) chronic lymphocytic leukemia (CLL).
Yet for patients with high-risk molecular features, such as 11q deletion, 17p deletion, or TP53 mutation, relapse remains problematic.
Investigators evaluated whether the addition of ublituximab to ibrutinib would improve the outcome of patients with genetically high-risk CLL in the GENUINE (UTX-IB-301) phase 3 study.
Jeff P. Sharman, MD, of Willamette Valley Cancer Institute and Research Center in Springfield, Oregon, reported the results at the 2017 ASCO Annual Meeting (abstract 7504).*
Ublituximab is a glycoengineered, anti-CD20 type 1 monoclonal antibody that maintains complement-dependent cytotoxicity and enhances antibody-dependent cell-mediated cytotoxicity. In a phase 2 study in combination with ibrutinib, it achieved an ORR of approximately 88%.
Protocol design
Originally, the study had co-primary endpoints of overall response rate (ORR) and progression-free survival (PFS). To adequately power for both endpoints, the target enrollment was 330 patients.
Dr Sharman explained that after 22 months of open enrollment, the trial sponsor determined that the original enrollment goal could not be met in a timely manner and elected to redesign the protocol.
In the modified protocol, ORR became the primary response rate and PFS a secondary endpoint. This allowed for a reduced target enrollment of 120. However, the study was no longer powered to detect a change in PFS.
Investigators stratified the patients by lines of prior therapy and then randomized them to receive ibrutinib or ublituximab plus ibrutinib.
The ibrutinib dose was 420 mg daily in both arms. Ublituximab dose was 900 mg on days 1, 8, and 15 of cycle 1, day 1 of cycles 2 through 6 and every third cycle thereafter.
The primary endpoint was ORR as assessed by Independent Central Review (IRC) using the iwCLL 2008 criteria.
Secondary endpoints included PFS, the complete response (CR) rate and depth of response (minimal residual disease [MRD] negativity), and safety.
The investigators assessed patients for response on weeks 8, 16, 24, and every 12 weeks thereafter.
The primary endpoint was evaluated when all enrolled patients had at least 2 efficacy evaluations.
The median follow-up was 11.4 months.
Patient characteristics
Patients with relapsed or refractory high-risk CLL had their disease centrally confirmed for the presence of deletion 17p, deletion 11q, and/or TP53 mutation.
They had measurable disease, ECOG performance status of 2 or less, no history of transformation of CLL, and no prior BTK inhibitor therapy.
The investigators randomized 126 patients, and 117 received any dose of therapy.
“The dropout was because in part ibrutinib was via commercial supply and not every patient could get access,” Dr Sharman noted.
Fifty-nine patients were treated in the combination arm and 58 in the monotherapy arm.
All patients had at least one of the specified mutations, which were relatively balanced between the 2 arms.
Patients were a mean age of 67 (range, 43 – 87), had a median of 3 prior therapies (range, 1 – 8), and more than 70% were male.
Patient characteristics were similar in each arm except for bulky disease, with 45% in the combination arm having bulky disease of 5 cm or more at baseline, compared with 26% in the monotherapy arm.
Twenty percent of the patients were considered refractory to rituximab.
Safety
Infusion reactions occurred in 54% of patients in the combination arm and 5% had grade 3/4 reactions. None occurred in the ibrutinib arm, since the latter is an orally bioavailable drug.
Other adverse events of all grades occurring in 10% of patients or more for the combination and monotherapy arms, respectively, were: diarrhea (42% and 40%), fatigue (27% and 33%), insomnia (24% and 10%), nausea (22% and 21%), headache (20% and 28%), arthralgia (19% and 17%), cough (19% and 24%), abdominal pain (15% and 9%), stomatitis (15% and 9%), upper respiratory infection (15% and 12%), dizziness (15% and 22%), contusion (15% and 29%), anemia (14% and 17%), and peripheral edema (10% and 21%).
Neutropenia was higher in the experimental arm, 22% any grade, compared with 12% in the ibrutinib arm, although grade 3 or higher neutropenia was similar in the 2 arms. Other laboratory abnormalities were similar between the arms.
Efficacy
The best ORR in the combination arm was 78%, with 7% achieving CR compared with 45% in the monotherapy arm with no CRs (P<0.001).
Nineteen percent of the combination arm achieved MRD negativity in peripheral blood compared with 2% of the monotherapy arm (P<0.01).
The reduction in lymph node size was similar between the arms.
In contrast, lymphocytosis was very different between the arms.
“As has been reported multiple times with targeted B-cell receptor signaling inhibitors,” Dr Sharman said, “patients treated with ibrutinib experienced rapid increase in their lymphocytes, returning approximately to baseline by 3 months and decreasing thereafter.”
“By contrast,” he continued, “those patients treated with the additional antibody had much more rapid resolution of their lymphocytosis. This was true whether patients were considered rituximab refractory or not.”
The investigators performed an additional analysis of ORR, this time including patients who achieved partial response with lymphocytosis (PR-L). These patients were not included in the primary endpoint because the iwCLL 2008 criteria had not yet been updated to include PR-L.
The best overall response including active PR-L patients was 83% in the experimental arm and 59% in the ibrutinib monotherapy arm (P<0.01).
PFS showed a trend toward improvement in the patients treated with the combination, with a hazard ratio of 0.559, which was not of statistical significance at the time of analysis.
The investigators concluded that the study met its primary endpoint, with a greater response rate and a greater depth of response than ibrutinib alone.
And the addition of ublituximab did not alter the safety profile of ibrutinib monotherapy.
TG Therapeutics, Inc, funded the study. ![]()
*Data in the abstract differ from the meeting presentation.
Ibrutinib, the Bruton’s tyrosine kinase (BTK) inhibitor, has transformed the treatment landscape for patients with relapsed or refractory (r/r) chronic lymphocytic leukemia (CLL).
Yet for patients with high-risk molecular features, such as 11q deletion, 17p deletion, or TP53 mutation, relapse remains problematic.
Investigators evaluated whether the addition of ublituximab to ibrutinib would improve the outcome of patients with genetically high-risk CLL in the GENUINE (UTX-IB-301) phase 3 study.
Jeff P. Sharman, MD, of Willamette Valley Cancer Institute and Research Center in Springfield, Oregon, reported the results at the 2017 ASCO Annual Meeting (abstract 7504).*
Ublituximab is a glycoengineered, anti-CD20 type 1 monoclonal antibody that maintains complement-dependent cytotoxicity and enhances antibody-dependent cell-mediated cytotoxicity. In a phase 2 study in combination with ibrutinib, it achieved an ORR of approximately 88%.
Protocol design
Originally, the study had co-primary endpoints of overall response rate (ORR) and progression-free survival (PFS). To adequately power for both endpoints, the target enrollment was 330 patients.
Dr Sharman explained that after 22 months of open enrollment, the trial sponsor determined that the original enrollment goal could not be met in a timely manner and elected to redesign the protocol.
In the modified protocol, ORR became the primary response rate and PFS a secondary endpoint. This allowed for a reduced target enrollment of 120. However, the study was no longer powered to detect a change in PFS.
Investigators stratified the patients by lines of prior therapy and then randomized them to receive ibrutinib or ublituximab plus ibrutinib.
The ibrutinib dose was 420 mg daily in both arms. Ublituximab dose was 900 mg on days 1, 8, and 15 of cycle 1, day 1 of cycles 2 through 6 and every third cycle thereafter.
The primary endpoint was ORR as assessed by Independent Central Review (IRC) using the iwCLL 2008 criteria.
Secondary endpoints included PFS, the complete response (CR) rate and depth of response (minimal residual disease [MRD] negativity), and safety.
The investigators assessed patients for response on weeks 8, 16, 24, and every 12 weeks thereafter.
The primary endpoint was evaluated when all enrolled patients had at least 2 efficacy evaluations.
The median follow-up was 11.4 months.
Patient characteristics
Patients with relapsed or refractory high-risk CLL had their disease centrally confirmed for the presence of deletion 17p, deletion 11q, and/or TP53 mutation.
They had measurable disease, ECOG performance status of 2 or less, no history of transformation of CLL, and no prior BTK inhibitor therapy.
The investigators randomized 126 patients, and 117 received any dose of therapy.
“The dropout was because in part ibrutinib was via commercial supply and not every patient could get access,” Dr Sharman noted.
Fifty-nine patients were treated in the combination arm and 58 in the monotherapy arm.
All patients had at least one of the specified mutations, which were relatively balanced between the 2 arms.
Patients were a mean age of 67 (range, 43 – 87), had a median of 3 prior therapies (range, 1 – 8), and more than 70% were male.
Patient characteristics were similar in each arm except for bulky disease, with 45% in the combination arm having bulky disease of 5 cm or more at baseline, compared with 26% in the monotherapy arm.
Twenty percent of the patients were considered refractory to rituximab.
Safety
Infusion reactions occurred in 54% of patients in the combination arm and 5% had grade 3/4 reactions. None occurred in the ibrutinib arm, since the latter is an orally bioavailable drug.
Other adverse events of all grades occurring in 10% of patients or more for the combination and monotherapy arms, respectively, were: diarrhea (42% and 40%), fatigue (27% and 33%), insomnia (24% and 10%), nausea (22% and 21%), headache (20% and 28%), arthralgia (19% and 17%), cough (19% and 24%), abdominal pain (15% and 9%), stomatitis (15% and 9%), upper respiratory infection (15% and 12%), dizziness (15% and 22%), contusion (15% and 29%), anemia (14% and 17%), and peripheral edema (10% and 21%).
Neutropenia was higher in the experimental arm, 22% any grade, compared with 12% in the ibrutinib arm, although grade 3 or higher neutropenia was similar in the 2 arms. Other laboratory abnormalities were similar between the arms.
Efficacy
The best ORR in the combination arm was 78%, with 7% achieving CR compared with 45% in the monotherapy arm with no CRs (P<0.001).
Nineteen percent of the combination arm achieved MRD negativity in peripheral blood compared with 2% of the monotherapy arm (P<0.01).
The reduction in lymph node size was similar between the arms.
In contrast, lymphocytosis was very different between the arms.
“As has been reported multiple times with targeted B-cell receptor signaling inhibitors,” Dr Sharman said, “patients treated with ibrutinib experienced rapid increase in their lymphocytes, returning approximately to baseline by 3 months and decreasing thereafter.”
“By contrast,” he continued, “those patients treated with the additional antibody had much more rapid resolution of their lymphocytosis. This was true whether patients were considered rituximab refractory or not.”
The investigators performed an additional analysis of ORR, this time including patients who achieved partial response with lymphocytosis (PR-L). These patients were not included in the primary endpoint because the iwCLL 2008 criteria had not yet been updated to include PR-L.
The best overall response including active PR-L patients was 83% in the experimental arm and 59% in the ibrutinib monotherapy arm (P<0.01).
PFS showed a trend toward improvement in the patients treated with the combination, with a hazard ratio of 0.559, which was not of statistical significance at the time of analysis.
The investigators concluded that the study met its primary endpoint, with a greater response rate and a greater depth of response than ibrutinib alone.
And the addition of ublituximab did not alter the safety profile of ibrutinib monotherapy.
TG Therapeutics, Inc, funded the study. ![]()
*Data in the abstract differ from the meeting presentation.
Can His Heart Handle a Hip Operation?
ANSWER
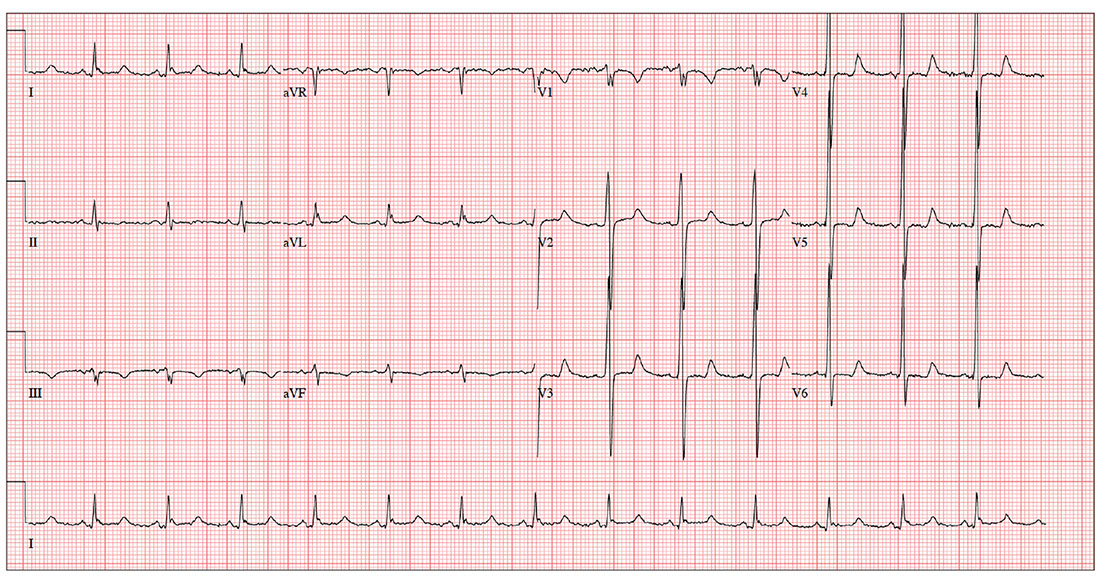
The ECG shows evidence of sinus rhythm, left ventricular hypertrophy, and a prolonged QT interval. Although it may be tempting to label the RR’ in lead V1 as right bundle branch block, recall that bundle branch block occurs with a QRS duration > 120 ms, which is not present here.
Left ventricular hypertrophy is demonstrated by high voltages in the precordial leads (S in lead V1 and R in lead V5 or V6 ≥ 35 mm); compare this with the ECG in the May issue of Clinician Reviews (2017;27(5):9, 13). A prolonged QT interval is suggested by a QTc interval of 500 m
The patient’s family history of sudden cardiac death makes these findings particularly concerning; genetic workup should be considered for both the patient and his children.
ANSWER
The ECG shows evidence of sinus rhythm, left ventricular hypertrophy, and a prolonged QT interval. Although it may be tempting to label the RR’ in lead V1 as right bundle branch block, recall that bundle branch block occurs with a QRS duration > 120 ms, which is not present here.
Left ventricular hypertrophy is demonstrated by high voltages in the precordial leads (S in lead V1 and R in lead V5 or V6 ≥ 35 mm); compare this with the ECG in the May issue of Clinician Reviews (2017;27(5):9, 13). A prolonged QT interval is suggested by a QTc interval of 500 m
The patient’s family history of sudden cardiac death makes these findings particularly concerning; genetic workup should be considered for both the patient and his children.
ANSWER
The ECG shows evidence of sinus rhythm, left ventricular hypertrophy, and a prolonged QT interval. Although it may be tempting to label the RR’ in lead V1 as right bundle branch block, recall that bundle branch block occurs with a QRS duration > 120 ms, which is not present here.
Left ventricular hypertrophy is demonstrated by high voltages in the precordial leads (S in lead V1 and R in lead V5 or V6 ≥ 35 mm); compare this with the ECG in the May issue of Clinician Reviews (2017;27(5):9, 13). A prolonged QT interval is suggested by a QTc interval of 500 m
The patient’s family history of sudden cardiac death makes these findings particularly concerning; genetic workup should be considered for both the patient and his children.
For the past three years, a now 62-year-old man has had pain in his left hip. His orthopedic surgeon recommends replacement, so he presents for preoperative assessment.
He has no history of cardiac disease but does have osteoarthritis, obesity, type 2 diabetes, hypertension, and hyperlipidemia. His surgical history is remarkable for a cholecystectomy performed when he was 48.
His medication list includes lisinopril, metoprolol, metformin, glyburide, naproxen, and atorvastatin. He is allergic to sulfa, which has caused anaphylaxis in the past.
The patient works as the warehouse supervisor of a local home improvement store. He is married with three adult children. His brother and uncle both succumbed to sudden cardiac death in their mid-40s. His mother died of a stroke, and his father of a myocardial infarction.
He reports joint pain consistent with osteoarthritis, occasional constipation, and urinary hesitancy. He is hard of hearing, wears corrective lenses, and walks with a limp.
Vital signs include a blood pressure of 148/88 mm Hg; pulse, 83 beats/min; respiratory rate, 12 breaths/min-1; and temperature, 99°F. His weight is 254 lb and his height, 69 in.
Physical exam reveals an obese man in no acute distress. Weber exam lateralizes to the left side. Dentition is in good repair, and his Mallampati score is II. The thyroid is normal, and there is no jugular venous distention. The lungs are clear bilaterally. The cardiac exam reveals a normal rhythm with a grade II/VI early systolic murmur best heard at the left upper sternal border. There are no extra heart sounds or rubs.
The abdomen is obese, with a well-healed surgical scar in the right upper quadrant. There are no masses. Peripheral pulses are strong bilaterally. There is no peripheral edema, and there are no lesions on either foot. Range of motion in the left hip is significantly limited due to pain.
An ECG reveals a ventricular rate of 83 beats/min; PR interval, 110 ms; QRS duration, 102 ms; QT/QTc interval, 426/500 ms; P axis, 4°; R axis, 5°; T axis, –21°. What is your interpretation?
Travel restrictions bar surgeon from U.S. meeting
A. Pieter Kappetein, MD, PhD, was looking forward to a busy week of meetings and activities at the annual meeting of the American Association for Thoracic Surgeons (AATS) in late April. Dr. Kappetein, a cardiothoracic surgeon at Erasmus MC in Rotterdam, the Netherlands, and secretary general for the European Association for Cardio-Thoracic Surgery, was giving two presentations at the meeting and had a full schedule of committee meetings, board meetings, and the like.
But on his way to the United States, Dr. Kappetein was pulled aside by U.S. immigration officials in Dublin and questioned about his travel background. After a long interrogation, he was informed he could not enter the United States and had to return home. The reason? His passport showed he had traveled to Iran 2 years earlier.
He was forced to give up his ticket to Boston, buy a new ticket home, and miss the work he was scheduled to do.
“I was angry,” Dr. Kappetein said. “They had to go get my luggage off the plane. I lost my ticket to Boston. It [was] a lot of money. I was very busy in Boston. I had meetings from early in the morning until late in the evening, and I had to give lectures as well. There were a lot of obligations that I could not fulfill.”
Restrictions hamper streamlined process
Dr. Kappetein’s ruined travel plans result from recent changes to the Visa Waiver Program (VWP). The program enables most citizens and nationals of participating countries to travel to the United States for tourism or business for 90 days or less without a visa; U.S. citizens and nationals get reciprocal travel access.
But changes to the program starting in 2015 mandated that most people who had traveled to or had been present in Iran, Iraq, Libya, Somalia, Sudan, Syria, or Yemen on March 1, 2011, or later, were no longer eligible to travel to under the VWP. The changes were part of the Visa Waiver Program Improvement and Terrorist Travel Prevention Act of 2015, signed by President Obama, which aimed to prevent travelers with ties to countries that pose terrorist threats from entering the country.
These new restrictions to the VWP have been enforced since January 2016, Jennifer Gabris, a spokesperson for U.S. Customs and Border Protection, said in an interview. She stressed that the eligibility requirements do not bar travel to the United States; rather, travelers who do not meet the requirements must obtain a visa.
Dr. Kappetein, however, said he has traveled to the United States from the Netherlands at least 10 times since his visit to Iran 2 years ago and encountered no problems until this year. He was under the assumption that travelers coming to the United States for business were exempt from the restrictions.
“I never had any issue at the border,” he said. “So the immigration officers, either they hadn’t seen [the passport stamp], or they had ignored it, or they didn’t care about it. I even got this prescreening [approval], so when I was in the States, I didn’t have to go in the line where you have to take off your shoes or your jacket.”
A Department of State spokeswoman referred questions about airport enforcement to the Department of Homeland Security, which declined to comment for this article.
“It takes time to implement, even after it’s signed,” Mr. Cohen said. “I don’t know to what degree it was being enforced, but the limited application means it probably just didn’t raise much attention.”
He noted there was little publicity surrounding the VWP restrictions until President Donald Trump called out the changes to justify his controversial travel ban, Mr. Cohen added. In his revised executive order released in March, the president noted that the countries named in his ban had already been “identified as presenting heightened concerns about terrorism and travel to the United States” through their exclusion from the VWP.
Harm to collaboration?
The VWP restrictions are among a number of increased security measures for foreigners and immigrants entering the country. President Trump’s March 6 Executive Order on immigration expanded uniform screening procedures for all visa classes and nationalities, while another provision suspended the Visa Interview Waiver Program. The interview program suspension means that certain applicants seeking to renew a visa must be interviewed in person by a consular officer. While a number of courts, including the 4th U.S. Circuit Court of Appeals, have blocked much of the Executive Order, the decisions did not halt the additional screening requirements or temporarily suspend the Visa Interview Waiver Program rollback. Both provisions remain in effect.
On May 4, the State Department proposed another regulation that would require more personal information from a subset of visa applicants, including 15 years of biographical information, employment history, addresses, prior passport numbers, information about family members including current and former spouses as well as travel histories and how trips were funded. Visa applicants would be required to provide phone numbers and email addresses used over the previous 5 years, according to the proposed rule. The subset of visa applicants would be determined by Department of State consular officers when resolving an applicant’s identity or when vetting for national security–related visa ineligibilities.
In a May 18 letter to the State Department, the American Association for the Advancement of Science and 17 other associations cautioned that the rule, if approved, would blunt scientific and academic collaborations, discourage foreign students from seeking to study and participate in research projects in the United States, and damage U.S. competitiveness.
“The notice, as proposed, is likely to have a chilling effect not only on those required to submit additional information, but indirectly on all international travelers to the United States,” the letter stated. “The uncertainties and confusion regarding supplemental questions will have a negative impact particularly on U.S. higher education and scientific collaborations.”
Since Dr. Kappetein’s travel mishap, he has obtained the required visa so that he may again travel to the United States. But he noted that entering this country will take at least 2 hours longer because of the visa entry protocols and additional airport screenings. The added restrictions and increased scrutiny of foreign travelers is unfortunate for U.S.-based medical conferences, he said.
“The interchange of information is really happening at those meetings,” he said. “[This] will block the exchange of information and it will block innovation. That’s a pity for [foreign physicians], but also for Americans.”
agallegos@frontlinemedcom.com
On Twitter @legal_med
A. Pieter Kappetein, MD, PhD, was looking forward to a busy week of meetings and activities at the annual meeting of the American Association for Thoracic Surgeons (AATS) in late April. Dr. Kappetein, a cardiothoracic surgeon at Erasmus MC in Rotterdam, the Netherlands, and secretary general for the European Association for Cardio-Thoracic Surgery, was giving two presentations at the meeting and had a full schedule of committee meetings, board meetings, and the like.
But on his way to the United States, Dr. Kappetein was pulled aside by U.S. immigration officials in Dublin and questioned about his travel background. After a long interrogation, he was informed he could not enter the United States and had to return home. The reason? His passport showed he had traveled to Iran 2 years earlier.
He was forced to give up his ticket to Boston, buy a new ticket home, and miss the work he was scheduled to do.
“I was angry,” Dr. Kappetein said. “They had to go get my luggage off the plane. I lost my ticket to Boston. It [was] a lot of money. I was very busy in Boston. I had meetings from early in the morning until late in the evening, and I had to give lectures as well. There were a lot of obligations that I could not fulfill.”
Restrictions hamper streamlined process
Dr. Kappetein’s ruined travel plans result from recent changes to the Visa Waiver Program (VWP). The program enables most citizens and nationals of participating countries to travel to the United States for tourism or business for 90 days or less without a visa; U.S. citizens and nationals get reciprocal travel access.
But changes to the program starting in 2015 mandated that most people who had traveled to or had been present in Iran, Iraq, Libya, Somalia, Sudan, Syria, or Yemen on March 1, 2011, or later, were no longer eligible to travel to under the VWP. The changes were part of the Visa Waiver Program Improvement and Terrorist Travel Prevention Act of 2015, signed by President Obama, which aimed to prevent travelers with ties to countries that pose terrorist threats from entering the country.
These new restrictions to the VWP have been enforced since January 2016, Jennifer Gabris, a spokesperson for U.S. Customs and Border Protection, said in an interview. She stressed that the eligibility requirements do not bar travel to the United States; rather, travelers who do not meet the requirements must obtain a visa.
Dr. Kappetein, however, said he has traveled to the United States from the Netherlands at least 10 times since his visit to Iran 2 years ago and encountered no problems until this year. He was under the assumption that travelers coming to the United States for business were exempt from the restrictions.
“I never had any issue at the border,” he said. “So the immigration officers, either they hadn’t seen [the passport stamp], or they had ignored it, or they didn’t care about it. I even got this prescreening [approval], so when I was in the States, I didn’t have to go in the line where you have to take off your shoes or your jacket.”
A Department of State spokeswoman referred questions about airport enforcement to the Department of Homeland Security, which declined to comment for this article.
“It takes time to implement, even after it’s signed,” Mr. Cohen said. “I don’t know to what degree it was being enforced, but the limited application means it probably just didn’t raise much attention.”
He noted there was little publicity surrounding the VWP restrictions until President Donald Trump called out the changes to justify his controversial travel ban, Mr. Cohen added. In his revised executive order released in March, the president noted that the countries named in his ban had already been “identified as presenting heightened concerns about terrorism and travel to the United States” through their exclusion from the VWP.
Harm to collaboration?
The VWP restrictions are among a number of increased security measures for foreigners and immigrants entering the country. President Trump’s March 6 Executive Order on immigration expanded uniform screening procedures for all visa classes and nationalities, while another provision suspended the Visa Interview Waiver Program. The interview program suspension means that certain applicants seeking to renew a visa must be interviewed in person by a consular officer. While a number of courts, including the 4th U.S. Circuit Court of Appeals, have blocked much of the Executive Order, the decisions did not halt the additional screening requirements or temporarily suspend the Visa Interview Waiver Program rollback. Both provisions remain in effect.
On May 4, the State Department proposed another regulation that would require more personal information from a subset of visa applicants, including 15 years of biographical information, employment history, addresses, prior passport numbers, information about family members including current and former spouses as well as travel histories and how trips were funded. Visa applicants would be required to provide phone numbers and email addresses used over the previous 5 years, according to the proposed rule. The subset of visa applicants would be determined by Department of State consular officers when resolving an applicant’s identity or when vetting for national security–related visa ineligibilities.
In a May 18 letter to the State Department, the American Association for the Advancement of Science and 17 other associations cautioned that the rule, if approved, would blunt scientific and academic collaborations, discourage foreign students from seeking to study and participate in research projects in the United States, and damage U.S. competitiveness.
“The notice, as proposed, is likely to have a chilling effect not only on those required to submit additional information, but indirectly on all international travelers to the United States,” the letter stated. “The uncertainties and confusion regarding supplemental questions will have a negative impact particularly on U.S. higher education and scientific collaborations.”
Since Dr. Kappetein’s travel mishap, he has obtained the required visa so that he may again travel to the United States. But he noted that entering this country will take at least 2 hours longer because of the visa entry protocols and additional airport screenings. The added restrictions and increased scrutiny of foreign travelers is unfortunate for U.S.-based medical conferences, he said.
“The interchange of information is really happening at those meetings,” he said. “[This] will block the exchange of information and it will block innovation. That’s a pity for [foreign physicians], but also for Americans.”
agallegos@frontlinemedcom.com
On Twitter @legal_med
A. Pieter Kappetein, MD, PhD, was looking forward to a busy week of meetings and activities at the annual meeting of the American Association for Thoracic Surgeons (AATS) in late April. Dr. Kappetein, a cardiothoracic surgeon at Erasmus MC in Rotterdam, the Netherlands, and secretary general for the European Association for Cardio-Thoracic Surgery, was giving two presentations at the meeting and had a full schedule of committee meetings, board meetings, and the like.
But on his way to the United States, Dr. Kappetein was pulled aside by U.S. immigration officials in Dublin and questioned about his travel background. After a long interrogation, he was informed he could not enter the United States and had to return home. The reason? His passport showed he had traveled to Iran 2 years earlier.
He was forced to give up his ticket to Boston, buy a new ticket home, and miss the work he was scheduled to do.
“I was angry,” Dr. Kappetein said. “They had to go get my luggage off the plane. I lost my ticket to Boston. It [was] a lot of money. I was very busy in Boston. I had meetings from early in the morning until late in the evening, and I had to give lectures as well. There were a lot of obligations that I could not fulfill.”
Restrictions hamper streamlined process
Dr. Kappetein’s ruined travel plans result from recent changes to the Visa Waiver Program (VWP). The program enables most citizens and nationals of participating countries to travel to the United States for tourism or business for 90 days or less without a visa; U.S. citizens and nationals get reciprocal travel access.
But changes to the program starting in 2015 mandated that most people who had traveled to or had been present in Iran, Iraq, Libya, Somalia, Sudan, Syria, or Yemen on March 1, 2011, or later, were no longer eligible to travel to under the VWP. The changes were part of the Visa Waiver Program Improvement and Terrorist Travel Prevention Act of 2015, signed by President Obama, which aimed to prevent travelers with ties to countries that pose terrorist threats from entering the country.
These new restrictions to the VWP have been enforced since January 2016, Jennifer Gabris, a spokesperson for U.S. Customs and Border Protection, said in an interview. She stressed that the eligibility requirements do not bar travel to the United States; rather, travelers who do not meet the requirements must obtain a visa.
Dr. Kappetein, however, said he has traveled to the United States from the Netherlands at least 10 times since his visit to Iran 2 years ago and encountered no problems until this year. He was under the assumption that travelers coming to the United States for business were exempt from the restrictions.
“I never had any issue at the border,” he said. “So the immigration officers, either they hadn’t seen [the passport stamp], or they had ignored it, or they didn’t care about it. I even got this prescreening [approval], so when I was in the States, I didn’t have to go in the line where you have to take off your shoes or your jacket.”
A Department of State spokeswoman referred questions about airport enforcement to the Department of Homeland Security, which declined to comment for this article.
“It takes time to implement, even after it’s signed,” Mr. Cohen said. “I don’t know to what degree it was being enforced, but the limited application means it probably just didn’t raise much attention.”
He noted there was little publicity surrounding the VWP restrictions until President Donald Trump called out the changes to justify his controversial travel ban, Mr. Cohen added. In his revised executive order released in March, the president noted that the countries named in his ban had already been “identified as presenting heightened concerns about terrorism and travel to the United States” through their exclusion from the VWP.
Harm to collaboration?
The VWP restrictions are among a number of increased security measures for foreigners and immigrants entering the country. President Trump’s March 6 Executive Order on immigration expanded uniform screening procedures for all visa classes and nationalities, while another provision suspended the Visa Interview Waiver Program. The interview program suspension means that certain applicants seeking to renew a visa must be interviewed in person by a consular officer. While a number of courts, including the 4th U.S. Circuit Court of Appeals, have blocked much of the Executive Order, the decisions did not halt the additional screening requirements or temporarily suspend the Visa Interview Waiver Program rollback. Both provisions remain in effect.
On May 4, the State Department proposed another regulation that would require more personal information from a subset of visa applicants, including 15 years of biographical information, employment history, addresses, prior passport numbers, information about family members including current and former spouses as well as travel histories and how trips were funded. Visa applicants would be required to provide phone numbers and email addresses used over the previous 5 years, according to the proposed rule. The subset of visa applicants would be determined by Department of State consular officers when resolving an applicant’s identity or when vetting for national security–related visa ineligibilities.
In a May 18 letter to the State Department, the American Association for the Advancement of Science and 17 other associations cautioned that the rule, if approved, would blunt scientific and academic collaborations, discourage foreign students from seeking to study and participate in research projects in the United States, and damage U.S. competitiveness.
“The notice, as proposed, is likely to have a chilling effect not only on those required to submit additional information, but indirectly on all international travelers to the United States,” the letter stated. “The uncertainties and confusion regarding supplemental questions will have a negative impact particularly on U.S. higher education and scientific collaborations.”
Since Dr. Kappetein’s travel mishap, he has obtained the required visa so that he may again travel to the United States. But he noted that entering this country will take at least 2 hours longer because of the visa entry protocols and additional airport screenings. The added restrictions and increased scrutiny of foreign travelers is unfortunate for U.S.-based medical conferences, he said.
“The interchange of information is really happening at those meetings,” he said. “[This] will block the exchange of information and it will block innovation. That’s a pity for [foreign physicians], but also for Americans.”
agallegos@frontlinemedcom.com
On Twitter @legal_med
Primary care involvement improves chance of meeting recommended surveillance intervals
CHICAGO – Only a small portion of patients with nondysplastic Barrett’s esophagus received appropriately timed endoscopic surveillance, a large database study showed.
But rather than being neglected, patients were more likely to be overassessed, with follow-up endoscopy performed more frequently than the recommended 3- to 5-year intervals, Anna Tavakkoli, MD, reported at the annual Digestive Disease Week.
“Very few patients entered our surveillance program with appropriate surveillance intervals,” said Dr. Tavakkoli, a gastroenterology fellow at the University of Michigan, Ann Arbor.
“We don’t have a formal program at the University of Michigan that drives coordination of care, but we do have great communication here between our primary care providers and our specialists. Our electronic medical records system makes quick messaging between providers easy, and primary care is very good about incorporating diagnoses into patients’ problem lists.”
Malignant transformation of nondysplastic Barrett’s is uncommon, with rates of no more than 4% per year. This understanding led three major societies – the American Gastroenterology Association, the American Society of Gastrointestinal Endoscopy, and the American College of Gastroenterology – to amend their surveillance recommendations in 2011 and 2012. All three societies now recommend a surveillance endoscopy every 3-5 years after the initial diagnosis of nondysplastic Barrett’s esophagus. In fact, the AGA has incorporated this suggestion into its five “Choosing Wisely” recommendations aimed at decreasing overutilization of testing and procedures.
Dr. Tavakkoli’s study examined surveillance timing in a cohort of 1,602 patients with nondysplastic Barrett’s who entered the University of Michigan Barrett’s Esophagus Registry from 1994 to 2016. All of these patents had at least three endoscopies or at least 5 years of follow-up data since their last endoscopy. The primary outcome was identification of trends in the appropriateness of surveillance of patients with nondysplastic Barrett’s esophagus at the University of Michigan. In her analysis, oversurveillance was defined as less than 3 years between the second and third endoscopy; undersurveillance was defined as more than 5 years between them. Dr. Tavakkoli and her colleagues also looked at patients who were lost to follow-up, defined as never receiving a second endoscopy after their initial diagnosis of nondysplastic Barrett’s esophagus and patients who were never surveilled, defined as never receiving their third endoscopy. All patients were compared with those who underwent appropriate surveillance, defined as 3-5 years between their second and third procedure.
The majority of patients were male, and the mean age was 59 years; 30% had long-segment Barrett’s, and 41% had a primary care provider in the university health care system. Most (90%) had their second endoscopy before 2012, when two of the three major societies issued their updated surveillance recommendations.
Of the entire cohort, 40% were lost to follow-up; 17% were never surveilled, and 3% were undersurveilled. Almost a third (31%) were oversurveilled, while just 8% had the appropriate surveillance, Dr. Tavakkoli said.
She then looked at several demographic and clinical factors associated with surveillance in each group, including sex, age, race, and income, comorbidities, length of Barrett’s, family history of esophageal cancer, and whether the patient had a University of Michigan primary care provider.
Having long-segment Barrett’s was associated with a 2.5-times increased risk of receiving a third endoscopy earlier than 3 years, which may be driven by studies that have shown that the risk of malignant transformation increases with Barrett’s length, she said.
The presence of a primary care physician significantly reduced the risk of inappropriate follow-up in every group, except patients who were undersurveilled, she said. The presence of a primary care physician at the University of Michigan decreased the risk of oversurveillance by 56%.
The positive influence of an in-system primary care physician was an important finding in this study, Dr Tavakkoli said. “The oncology data have shown us that poor coordination of care between oncologists and primary care providers contributes to avoidable patient morbidity and mortality, fragmented care, and increased costs. In 2005, the Institute of Medicine published a report emphasizing that coordination between specialists and primary care providers is one of the four key components to cancer survivorship care. There have been a number of GI studies looking at how primary care’s involvement in colorectal screening improves the rates of patients who undergo screening, but among Barrett’s patients, there have not been data showing that having a primary care physician at the center where endoscopic surveillance is done improves utilization patterns.”
Dr. Tavakkoli had no financial disclosures.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
CHICAGO – Only a small portion of patients with nondysplastic Barrett’s esophagus received appropriately timed endoscopic surveillance, a large database study showed.
But rather than being neglected, patients were more likely to be overassessed, with follow-up endoscopy performed more frequently than the recommended 3- to 5-year intervals, Anna Tavakkoli, MD, reported at the annual Digestive Disease Week.
“Very few patients entered our surveillance program with appropriate surveillance intervals,” said Dr. Tavakkoli, a gastroenterology fellow at the University of Michigan, Ann Arbor.
“We don’t have a formal program at the University of Michigan that drives coordination of care, but we do have great communication here between our primary care providers and our specialists. Our electronic medical records system makes quick messaging between providers easy, and primary care is very good about incorporating diagnoses into patients’ problem lists.”
Malignant transformation of nondysplastic Barrett’s is uncommon, with rates of no more than 4% per year. This understanding led three major societies – the American Gastroenterology Association, the American Society of Gastrointestinal Endoscopy, and the American College of Gastroenterology – to amend their surveillance recommendations in 2011 and 2012. All three societies now recommend a surveillance endoscopy every 3-5 years after the initial diagnosis of nondysplastic Barrett’s esophagus. In fact, the AGA has incorporated this suggestion into its five “Choosing Wisely” recommendations aimed at decreasing overutilization of testing and procedures.
Dr. Tavakkoli’s study examined surveillance timing in a cohort of 1,602 patients with nondysplastic Barrett’s who entered the University of Michigan Barrett’s Esophagus Registry from 1994 to 2016. All of these patents had at least three endoscopies or at least 5 years of follow-up data since their last endoscopy. The primary outcome was identification of trends in the appropriateness of surveillance of patients with nondysplastic Barrett’s esophagus at the University of Michigan. In her analysis, oversurveillance was defined as less than 3 years between the second and third endoscopy; undersurveillance was defined as more than 5 years between them. Dr. Tavakkoli and her colleagues also looked at patients who were lost to follow-up, defined as never receiving a second endoscopy after their initial diagnosis of nondysplastic Barrett’s esophagus and patients who were never surveilled, defined as never receiving their third endoscopy. All patients were compared with those who underwent appropriate surveillance, defined as 3-5 years between their second and third procedure.
The majority of patients were male, and the mean age was 59 years; 30% had long-segment Barrett’s, and 41% had a primary care provider in the university health care system. Most (90%) had their second endoscopy before 2012, when two of the three major societies issued their updated surveillance recommendations.
Of the entire cohort, 40% were lost to follow-up; 17% were never surveilled, and 3% were undersurveilled. Almost a third (31%) were oversurveilled, while just 8% had the appropriate surveillance, Dr. Tavakkoli said.
She then looked at several demographic and clinical factors associated with surveillance in each group, including sex, age, race, and income, comorbidities, length of Barrett’s, family history of esophageal cancer, and whether the patient had a University of Michigan primary care provider.
Having long-segment Barrett’s was associated with a 2.5-times increased risk of receiving a third endoscopy earlier than 3 years, which may be driven by studies that have shown that the risk of malignant transformation increases with Barrett’s length, she said.
The presence of a primary care physician significantly reduced the risk of inappropriate follow-up in every group, except patients who were undersurveilled, she said. The presence of a primary care physician at the University of Michigan decreased the risk of oversurveillance by 56%.
The positive influence of an in-system primary care physician was an important finding in this study, Dr Tavakkoli said. “The oncology data have shown us that poor coordination of care between oncologists and primary care providers contributes to avoidable patient morbidity and mortality, fragmented care, and increased costs. In 2005, the Institute of Medicine published a report emphasizing that coordination between specialists and primary care providers is one of the four key components to cancer survivorship care. There have been a number of GI studies looking at how primary care’s involvement in colorectal screening improves the rates of patients who undergo screening, but among Barrett’s patients, there have not been data showing that having a primary care physician at the center where endoscopic surveillance is done improves utilization patterns.”
Dr. Tavakkoli had no financial disclosures.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
CHICAGO – Only a small portion of patients with nondysplastic Barrett’s esophagus received appropriately timed endoscopic surveillance, a large database study showed.
But rather than being neglected, patients were more likely to be overassessed, with follow-up endoscopy performed more frequently than the recommended 3- to 5-year intervals, Anna Tavakkoli, MD, reported at the annual Digestive Disease Week.
“Very few patients entered our surveillance program with appropriate surveillance intervals,” said Dr. Tavakkoli, a gastroenterology fellow at the University of Michigan, Ann Arbor.
“We don’t have a formal program at the University of Michigan that drives coordination of care, but we do have great communication here between our primary care providers and our specialists. Our electronic medical records system makes quick messaging between providers easy, and primary care is very good about incorporating diagnoses into patients’ problem lists.”
Malignant transformation of nondysplastic Barrett’s is uncommon, with rates of no more than 4% per year. This understanding led three major societies – the American Gastroenterology Association, the American Society of Gastrointestinal Endoscopy, and the American College of Gastroenterology – to amend their surveillance recommendations in 2011 and 2012. All three societies now recommend a surveillance endoscopy every 3-5 years after the initial diagnosis of nondysplastic Barrett’s esophagus. In fact, the AGA has incorporated this suggestion into its five “Choosing Wisely” recommendations aimed at decreasing overutilization of testing and procedures.
Dr. Tavakkoli’s study examined surveillance timing in a cohort of 1,602 patients with nondysplastic Barrett’s who entered the University of Michigan Barrett’s Esophagus Registry from 1994 to 2016. All of these patents had at least three endoscopies or at least 5 years of follow-up data since their last endoscopy. The primary outcome was identification of trends in the appropriateness of surveillance of patients with nondysplastic Barrett’s esophagus at the University of Michigan. In her analysis, oversurveillance was defined as less than 3 years between the second and third endoscopy; undersurveillance was defined as more than 5 years between them. Dr. Tavakkoli and her colleagues also looked at patients who were lost to follow-up, defined as never receiving a second endoscopy after their initial diagnosis of nondysplastic Barrett’s esophagus and patients who were never surveilled, defined as never receiving their third endoscopy. All patients were compared with those who underwent appropriate surveillance, defined as 3-5 years between their second and third procedure.
The majority of patients were male, and the mean age was 59 years; 30% had long-segment Barrett’s, and 41% had a primary care provider in the university health care system. Most (90%) had their second endoscopy before 2012, when two of the three major societies issued their updated surveillance recommendations.
Of the entire cohort, 40% were lost to follow-up; 17% were never surveilled, and 3% were undersurveilled. Almost a third (31%) were oversurveilled, while just 8% had the appropriate surveillance, Dr. Tavakkoli said.
She then looked at several demographic and clinical factors associated with surveillance in each group, including sex, age, race, and income, comorbidities, length of Barrett’s, family history of esophageal cancer, and whether the patient had a University of Michigan primary care provider.
Having long-segment Barrett’s was associated with a 2.5-times increased risk of receiving a third endoscopy earlier than 3 years, which may be driven by studies that have shown that the risk of malignant transformation increases with Barrett’s length, she said.
The presence of a primary care physician significantly reduced the risk of inappropriate follow-up in every group, except patients who were undersurveilled, she said. The presence of a primary care physician at the University of Michigan decreased the risk of oversurveillance by 56%.
The positive influence of an in-system primary care physician was an important finding in this study, Dr Tavakkoli said. “The oncology data have shown us that poor coordination of care between oncologists and primary care providers contributes to avoidable patient morbidity and mortality, fragmented care, and increased costs. In 2005, the Institute of Medicine published a report emphasizing that coordination between specialists and primary care providers is one of the four key components to cancer survivorship care. There have been a number of GI studies looking at how primary care’s involvement in colorectal screening improves the rates of patients who undergo screening, but among Barrett’s patients, there have not been data showing that having a primary care physician at the center where endoscopic surveillance is done improves utilization patterns.”
Dr. Tavakkoli had no financial disclosures.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
AT DDW
Key clinical point:
Major finding: Only 8% of patients had the recommended interval of 3-5 years between surveillance endoscopies.
Data source: A retrospective database cohort of 1,602 patients.
Disclosures: Dr. Tavakkoli had no relevant financial disclosures.
Later school start tied to more sleep
BOSTON – Teenagers slept more hours when they started school later, a new study found.
With 73% of high schoolers reporting receiving less than the recommended 8 hours of sleep per night, teens are among the most sleep-deprived members of society.
In this study, the later a teenager started school, the later he or she woke up, with average wake-up times having been just after 6:00 a.m. for those starting school between 7:00 and 7:30 a.m. and about 7:00 a.m. for those starting school after 8:30 a.m. But only those teens who started after 8:30 a.m. achieved the 8-hour recommended sleep duration, said Nicole Nahmod, during a presentation at the annual meeting of the Associated Professional Sleep Societies.
The students with the later school start times averaged 32 minutes of extra sleep, when compared with their early-rising colleagues, noted Ms. Nahmod, who is one of the study’s authors, a research technician, and study coordinator at Pennsylvania State University, Hershey. Specifically, adolescents who started school after 8:30 a.m. had a mean sleep duration of 8.1 hours, while those who started school earlier slept for only 7.5 hours a night.*
Acknowledging the importance of adequate sleep on teen health, mood, and school performance, the American Academy of Pediatrics recommends that middle schools and high schools begin at 8:30 a.m. or later. However, most school days start earlier than that, as evidenced by this dataset. While 72% of the study’s participants started school between 7:00 a.m. and 8:30 a.m., as many as 15% of the study participants began school during the narrower 7:00-7:30 a.m. window.
In this study, researchers from Penn State used data from 413 adolescents (mean age, 15.4 years; 46% male), who participated in a substudy of the Fragile Families & Child Wellbeing Study conducted across 20 large American cities. The study oversampled nonmarried pregnant mothers, resulting in a racially diverse sample and a high proportion of low-income families. The household income for 29% of the sample was below the poverty line.*
For this substudy, sleep duration was calculated from app-based daily diary reports of bed times, wake times, and school start times on days when a teen attended school.
The study was limited by its cross-sectional nature, but was enriched by a diverse range of school start times sampled from 20 U.S. cities, the high proportion of at-risk teens, and the use of a daily sleep diary.
“Current literature shows associations between later school start times and academic success, mood, and health. It also shows a decrease in motor vehicle accidents, tardiness, school dropout, and daytime sleepiness,” Ms. Nahmod noted.
Her continued research in this area involves analyzing the relationships between actigraphically assessed sleep measures in students and school start times.
Ms. Nahmod reported having no financial disclosures.
*This article was updated on June 6, 2017.
BOSTON – Teenagers slept more hours when they started school later, a new study found.
With 73% of high schoolers reporting receiving less than the recommended 8 hours of sleep per night, teens are among the most sleep-deprived members of society.
In this study, the later a teenager started school, the later he or she woke up, with average wake-up times having been just after 6:00 a.m. for those starting school between 7:00 and 7:30 a.m. and about 7:00 a.m. for those starting school after 8:30 a.m. But only those teens who started after 8:30 a.m. achieved the 8-hour recommended sleep duration, said Nicole Nahmod, during a presentation at the annual meeting of the Associated Professional Sleep Societies.
The students with the later school start times averaged 32 minutes of extra sleep, when compared with their early-rising colleagues, noted Ms. Nahmod, who is one of the study’s authors, a research technician, and study coordinator at Pennsylvania State University, Hershey. Specifically, adolescents who started school after 8:30 a.m. had a mean sleep duration of 8.1 hours, while those who started school earlier slept for only 7.5 hours a night.*
Acknowledging the importance of adequate sleep on teen health, mood, and school performance, the American Academy of Pediatrics recommends that middle schools and high schools begin at 8:30 a.m. or later. However, most school days start earlier than that, as evidenced by this dataset. While 72% of the study’s participants started school between 7:00 a.m. and 8:30 a.m., as many as 15% of the study participants began school during the narrower 7:00-7:30 a.m. window.
In this study, researchers from Penn State used data from 413 adolescents (mean age, 15.4 years; 46% male), who participated in a substudy of the Fragile Families & Child Wellbeing Study conducted across 20 large American cities. The study oversampled nonmarried pregnant mothers, resulting in a racially diverse sample and a high proportion of low-income families. The household income for 29% of the sample was below the poverty line.*
For this substudy, sleep duration was calculated from app-based daily diary reports of bed times, wake times, and school start times on days when a teen attended school.
The study was limited by its cross-sectional nature, but was enriched by a diverse range of school start times sampled from 20 U.S. cities, the high proportion of at-risk teens, and the use of a daily sleep diary.
“Current literature shows associations between later school start times and academic success, mood, and health. It also shows a decrease in motor vehicle accidents, tardiness, school dropout, and daytime sleepiness,” Ms. Nahmod noted.
Her continued research in this area involves analyzing the relationships between actigraphically assessed sleep measures in students and school start times.
Ms. Nahmod reported having no financial disclosures.
*This article was updated on June 6, 2017.
BOSTON – Teenagers slept more hours when they started school later, a new study found.
With 73% of high schoolers reporting receiving less than the recommended 8 hours of sleep per night, teens are among the most sleep-deprived members of society.
In this study, the later a teenager started school, the later he or she woke up, with average wake-up times having been just after 6:00 a.m. for those starting school between 7:00 and 7:30 a.m. and about 7:00 a.m. for those starting school after 8:30 a.m. But only those teens who started after 8:30 a.m. achieved the 8-hour recommended sleep duration, said Nicole Nahmod, during a presentation at the annual meeting of the Associated Professional Sleep Societies.
The students with the later school start times averaged 32 minutes of extra sleep, when compared with their early-rising colleagues, noted Ms. Nahmod, who is one of the study’s authors, a research technician, and study coordinator at Pennsylvania State University, Hershey. Specifically, adolescents who started school after 8:30 a.m. had a mean sleep duration of 8.1 hours, while those who started school earlier slept for only 7.5 hours a night.*
Acknowledging the importance of adequate sleep on teen health, mood, and school performance, the American Academy of Pediatrics recommends that middle schools and high schools begin at 8:30 a.m. or later. However, most school days start earlier than that, as evidenced by this dataset. While 72% of the study’s participants started school between 7:00 a.m. and 8:30 a.m., as many as 15% of the study participants began school during the narrower 7:00-7:30 a.m. window.
In this study, researchers from Penn State used data from 413 adolescents (mean age, 15.4 years; 46% male), who participated in a substudy of the Fragile Families & Child Wellbeing Study conducted across 20 large American cities. The study oversampled nonmarried pregnant mothers, resulting in a racially diverse sample and a high proportion of low-income families. The household income for 29% of the sample was below the poverty line.*
For this substudy, sleep duration was calculated from app-based daily diary reports of bed times, wake times, and school start times on days when a teen attended school.
The study was limited by its cross-sectional nature, but was enriched by a diverse range of school start times sampled from 20 U.S. cities, the high proportion of at-risk teens, and the use of a daily sleep diary.
“Current literature shows associations between later school start times and academic success, mood, and health. It also shows a decrease in motor vehicle accidents, tardiness, school dropout, and daytime sleepiness,” Ms. Nahmod noted.
Her continued research in this area involves analyzing the relationships between actigraphically assessed sleep measures in students and school start times.
Ms. Nahmod reported having no financial disclosures.
*This article was updated on June 6, 2017.
AT SLEEP 2017
Key clinical point: Teenage students who start school after 8:30 a.m. are more likely to get the recommended 8 hours of sleep than are their counterparts with earlier start times.
Major finding: Adolescents who started school after 8:30 a.m. had a mean sleep duration of 8.1 hours, compared with 7.5 hours for students who started school earlier.
Data source: A national longitudinal birth cohort study of 413 adolescents.
Disclosures: Ms. Nahmod reported having no financial disclosures.